Результат интеллектуальной деятельности: АЗОТСОДЕРЖАЩИЕ КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОДУКЦИИ БЕТА-АМИЛОИДА
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к фармацевтическому веществу, более конкретно к азотсодержащему конденсированному гетероциклическому соединению, эффективному в лечении нейродегенеративного заболевания, вызываемого амилоидом-β (далее называемым как Aβ), такого как болезнь Альцгеймера или синдром Дауна, и к лекарственному средству, в частности лекарственному средству для лечения заболевания, вызываемого Aβ, содержащему данное соединение в качестве активного компонента.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Болезнь Альцгеймера является заболеванием, характеризующимся дегенерацией и потерей нейронов, а также образованием старческих бляшек и нейрофибриллярной дегенерацией. В настоящее время болезнь Альцгеймера лечат только методами терапии симптомов с использованием средства для улучшения симптомов, типичным представителем которого является ингибитор ацетилхолинэстеразы, а фундаментальное лекарственное средство, ингибирующее прогрессирование болезни все еще не создано. Необходимо разработать способ контроля причины возникновения патологии, чтобы создать фундаментальное лекарственное средство для лечения болезни Альцгеймера.
Считается, что Aβ-белки как метаболиты амилоидных белков-предшественников (далее называемых как APP) сильно вовлечены в процессы дегенерации и потери нейронов и возникновения симптомов деменции (см., например, НЕПАТЕНТНЫЕ ДОКУМЕНТЫ 1 и 2). Основными составляющими молекулы Aβ-белка являются Aβ40, состоящий из 40 аминокислот, и Aβ42 с двумя дополнительными аминокислотами, присоединенными на С-конце. Известно, что Aβ40 и Aβ42 обладают высокой агрегационной способностью (см., например, НЕПАТЕНТНЫЙ ДОКУМЕНТ 3) и являются основными составляющими старческих бляшек (см., например, НЕПАТЕНТНЫЕ ДОКУМЕНТЫ 3, 4 и 5). Известно также, что Aβ40 и Aβ42 отличаются увеличенной мутацией в генах APP и пресенилина, которая наблюдается при болезни Альцгеймера семейного типа (см., например, НЕПАТЕНТНЫЕ ДОКУМЕНТЫ 6, 7 и 8). Поэтому ожидается, что соединение, понижающее продукцию Aβ40 и Aβ42, будет ингибитором прогрессирования или профилактическим средством при болезни Альцгеймера.
Aβ получают расщеплением APP β-секретазой и затем γ-секретазой. Поэтому были предприняты попытки создания ингибиторов β-секретазы и γ-секретазы, чтобы уменьшить продукцию Aβ. Многие из указанных ингибиторов секретаз уже известны, например пептиды и миметики пептидов, такие как L-685458 (см., например, НЕПАТЕНТНЫЙ ДОКУМЕНТ 9), LY-411575 (см., например, НЕПАТЕНТНЫЕ ДОКУМЕНТЫ 10, 11 и 12) и LY-450139 (см. НЕПАТЕНТНЫЕ ДОКУМЕНТЫ 13, 14 и 15). Непептидными соединениями являются, например, MRK-560 (см. НЕПАТЕНТНЫЕ ДОКУМЕНТЫ 16 и 17) и соединения, содержащие многочисленные ароматические циклы, как раскрыто в ПАТЕНТНЫХ ДОКУМЕНТАХ 1 и 2. Однако соединение, представленное формулой (VI) на странице 17 описания ПАТЕНТНОГО ДОКУМЕНТА 1, отличается от соединения по настоящему изобретению тем, что оно ограничивается соединением, содержащим 2-аминотиазолильную группу в качестве основной структуры. А соединение, представленное формулой (I), как раскрыто на странице 6 описания ПАТЕНТНОГО ДОКУМЕНТА 2, отличается от соединения по настоящему изобретению тем, что оно ограничивается соединением, содержащим этиниленовый, этениленовый или метиновый линкер, описанный как X1.
ДОКУМЕНТЫ ИЗВЕСТНОГО УРОВНЯ ТЕХНИКИ
ПАТЕНТНЫЕ ДОКУМЕНТЫ
ПАТЕНТНЫЙ ДОКУМЕНТ 1: WO 2004/110350
ПАТЕНТНЫЙ ДОКУМЕНТ 2: WO 2007/102580
НЕПАТЕНТНЫЕ ДОКУМЕНТЫ
НЕПАТЕНТНЫЙ ДОКУМЕНТ 1: Klein W.L., and seven others, Alzheimer′s disease-affected brain: Presence of oligomeric Aβ ligands (ADDLs) suggests molecular basis for reversible memory loss, Proceeding of the National Academy of Science USA, 2003, Sep., 2; 100 (18), p. 10417-10422.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 2: Nitsch R.M., and sixteen others, Antibodies against β-amyloid slow cognitive decline in Alzheimer′s disease, Neuron, 2003, May 22; 38, p. 547-554.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 3: Jarrett J.T., and two others, The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimers′ disease, Biochemistry, 1993, 32 (18), p. 4693-4697.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 4: Glenner G.G., and one other, Alzheimer′s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein, Biochemical and Biophysical Research Communications, 1984, May 16, 120 (3), p.885-890.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 5: Masters C.L., and five others, Amyloid plaque core protein in Alzheimer disease and Down Syndrome, Proceeding of the National Academy of Science USA, 1985, Jun., 82 (12), p.4245-4249.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 6: Gouras G.K., and eleven others, Intraneuronal Aβ42 accumulation in human brain, American Journal of Pathology, 2000, Jan., 156 (1), p.15-20.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 7: Scheuner D., and twenty others, Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer′s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer′s disease, Nature Medicine, 1996, Aug., 2 (8), p.864-870.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 8: Forman M.S., and four others, Differential effects of the swedish mutant amyloid precursor protein on β-amyloid accumulation and secretion in neurons and nonneuronal cells, The Journal of Biological Chemistry, 1997, Dec., 19, 272 (51), p.32247-32253.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 9: Shearman M.S., and nine others, L-685, 458, Aspartyl Protease Transition State Mimic, Is Potent Inhibitor of Amyloid β-Protein Precursor γ-Secretase Activity, Biochemistry, 2000, Aug., 1, 39 (30), p.8698-8704.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 10: Shearman M.S., and six others, Catalytic Site-Directed γ-Secretase Complex Inhibitors Do Not Discriminate Pharmacologically between Notch S3 and β-APP Clevages, Biochemistry, 2003, Jun., 24, 42 (24), p.7580-7586.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 11: Lanz T.A., and three others, Studies of Aβ pharmacodynamics in the brain, cerebrospinal fluid, and plasma in young (plaque-free) Tg2576 mice using the γ-secretase inhibitor N2-[(2S)-2-(3,5-difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-dihydro-5H-dibenzo[b,d]azepin-7-yl]-L-alaninamide (LY-411575), The Journal of Pharmacology and Experimental Therapeutics, 2004, Apr., 309 (1), p.49-55.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 12: Wong G.T., and twelve others, Chronic trea™ent with the γ-secretase inhibitor LY-411, 575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation, The Journal of Biological Chemistry, 2004, Mar., 26, 279 (13), p.12876-12882.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 13: Gitter B.D., and ten others, Stereoselective inhibition of amyloid beta peptide secretion by LY450139, a novel functional gamma secretase inhibitor, Neurology of Aging 2004, 25, sup2, p.571.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 14: Lanz T.A., and eighteen others, Concentration-dependent modulation of amyloid-β in vivo and in vitro using the γ-secretase inhibitor, LY-450139, The Journal of Pharmacology and Experimantal Therapeutics, 2006, Nov., 319 (2) p.924-933.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 15: Siemers E.R., and thirteen others, Effects of γ-secretase inhibitor in randamized study of patients with Alzheimer disease, Neurology, 2006, 66, p.602-604.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 16: Best J.D., and nine others, In vivo characterization of Aβ (40) changes in brain and cerebrospinal fluid using the novel γ-secretase inhibitor N-[cis-4-[(4- chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1,1-trifluoromethanesulphonylamide (MK-560) in the rat, The Journal of Pharmacology and Experimental Therapeutics, 2006, May 317 (2) p.786-790.
НЕПАТЕНТНЫЙ ДОКУМЕНТ 17: Best J.D., and thirteen others, The novel γ-secretase inhibitor N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1,1-trifluoromethanesulphonylamide (MK-560) reduces amylid plaque deposition without evidence notch-related pathology in the Tg2576 mouse, The Journal of Pharmacology and Experimental Therapeutics, 2007, Feb., (2) p.552-558.
КРАТКОЕ ОПИСАНИЕ СУЩЕСТВА ИЗОБРЕТЕНИЯ
ЗАДАЧА, РЕШАЕМАЯ ИЗОБРЕТЕНИЕМ
Как описано выше, уже ожидалось соединение, ингибирующее продукцию Aβ из APP, в качестве терапевтического или профилактического средства от болезни, вызываемой Aβ, типичным представителем которой является болезнь Альцгеймера. Однако непептидное соединение, с высокой эффективностью ингибирующее продукцию Aβ, все еще не известно. Следовательно, существует потребность в новом низкомолекулярном соединении, которое бы ингибировало продукцию Aβ.
СПОСОБ РЕШЕНИЯ УКАЗАННОЙ ЗАДАЧИ
В результате обширных исследований авторы настоящего изобретения нашли непептидное полициклическое соединение, которое ингибирует продукцию Aβ из APP, и, следовательно, нашли терапевтическое средство от болезни, вызываемой Aβ, типичным представителем которой является болезнь Альцгеймера. Данная находка привела к созданию настоящего изобретения.
В частности, настоящее изобретение относится к следующим предложениям 1)-15):
1) соединение, представленное формулой (I):
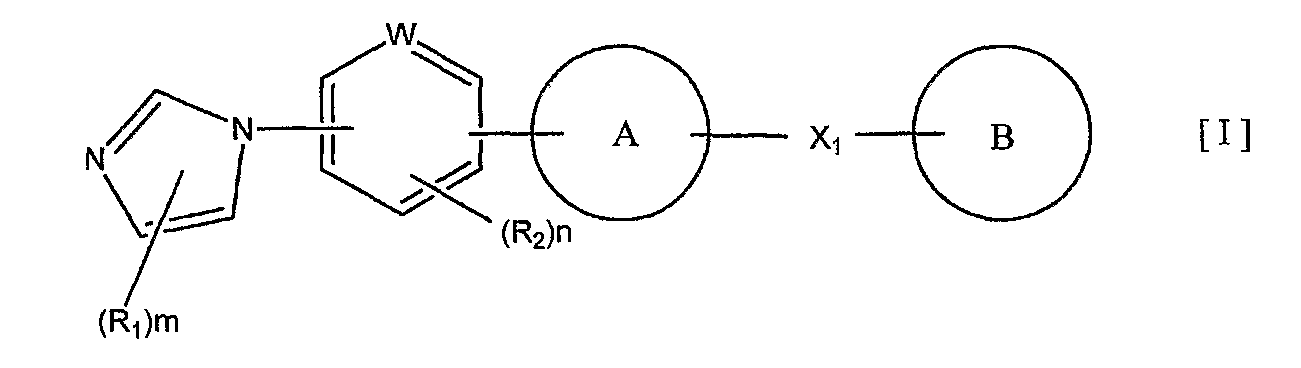
или его фармакологически приемлемые соль или сложный эфир,
где R1 и R2 являются одинаковыми или разными и каждый представляет собой заместитель, выбранный из следующей группы а1 заместителей;
m представляет собой целое число от 0 до 3;
n представляет собой целое число от 0 до 2;
W представляет собой атом азота или атом углерода;
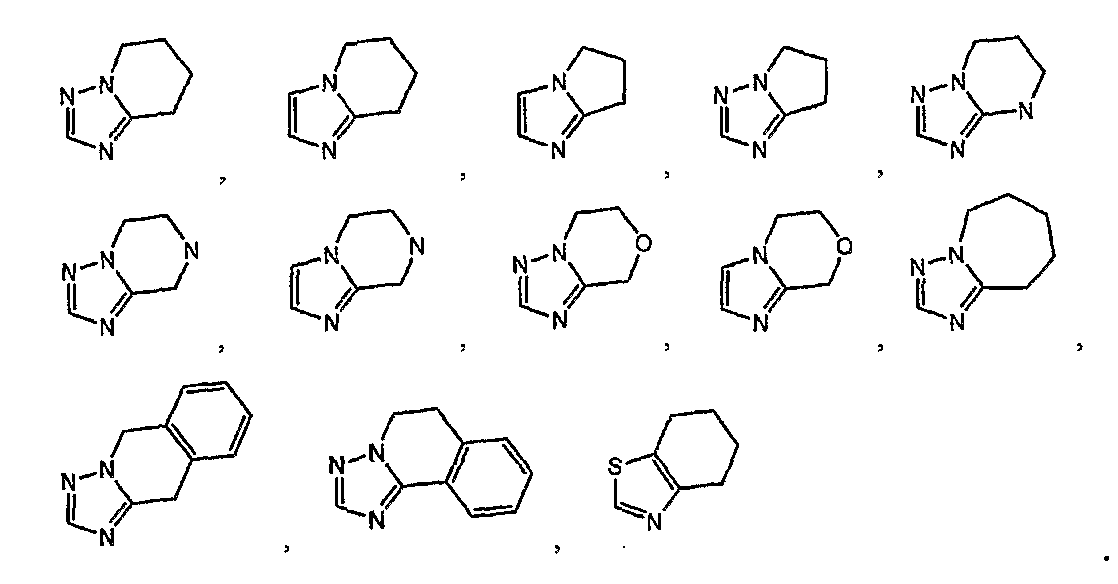
кольцо A представляет собой пятичленную ароматическую гетероциклическую группу, конденсированную с 5-14-членной неароматической кольцевой группой, которая содержит два или более атомов азота и может содержать 1-3 заместителя, выбранных из следующей группы b1 заместителей (где 5-14-членная неароматическая гетероциклическая группа может иметь структуру с поперечными связями);
X1 представляет собой i) одинарную связь, ii) C1-6 алкиленовую группу, iii) виниленовую группу, которая может содержать 1-2 C1-6 алкильные группы, или iv) -X2- (где X2 представляет собой -NR3-, -NR3C(O)-, -C(O)NR3-, -O-, -S-, -S(O)- или -S(O)2- и R3 представляет собой атом водорода, C1-6 алкильную группу, C3-6 циклоалкильную группу, C2-6 алканоильную группу или C1-6 алкилсульфонильную группу); и
кольцо B представляет собой моноциклическую или конденсированную циклическую ароматическую кольцевую группу, выбранную из формул [2]-[19]:
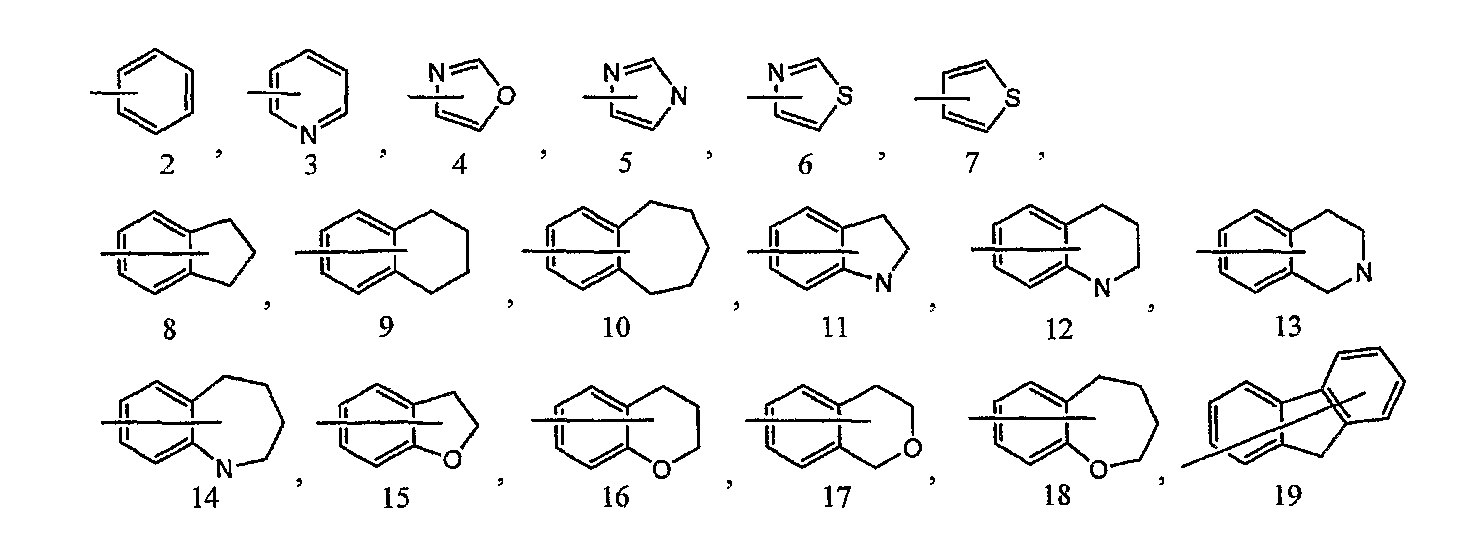
каждая из которых может содержать 1-3 заместителя, выбранных из следующей группы c1 заместителей;
[группа a1 заместителей: C1-6 алкильная группа, C3-8 циклоалкильная группа, C2-6 алкенильная группа, C1-6 алкоксигруппа, C2-6 алкенилоксигруппа, C3-8 циклоалкилоксигруппа, аминогруппа (где аминогруппа может содержать одну C2-6 алканоильную группу или C1-6 алкилсульфонильную группу или 1-2 C1-6 алкильные группы или C3-8 циклоалкильные группы), цианогруппа, формильная группа, атом галогена, гидроксильная группа и нитрогруппа;
группа b1 заместителей: C1-6 алкильная группа (где алкильная группа может быть замещенной 1-3 атомами галогена), C2-6 алкенильная группа, C3-8 циклоалкильная группа, C6-14 арильная группа, C6-14 арил-C1-6 алкильная группа, C1-6 алкоксигруппа, C2-6 алкенилоксигруппа, C3-8 циклоалкилоксигруппа, C2-6 алканоильная группа, C4-9 циклоалкилкарбонильная группа, C7-15 ароильная группа, C1-6 алкилсульфонильная группа, C2-6 алкенилсульфонильная группа, C3-8 циклоалкилсульфонильная группа, C6-14 арилсульфонильная группа, C1-6 алкилтиогруппа, C2-6 алкенилтиогруппа, C3-8 циклоалкилтиогруппа, аминосульфонильная группа (где аминосульфонильная группа может содержать 1-2 C1-6 алкильные группы, C2-6 алкенильные группы или C3-8 циклоалкильные группы), аминогруппа (где аминогруппа может содержать одну C2-6 алканоильную группу, C1-6 алкилсульфонильную группу или C3-8 циклоалкилсульфонильную группу или 1-2 C1-6 алкильные группы или C3-8 циклоалкильные группы), цианогруппа, формильная группа, атом галогена, гидроксильная группа, нитрогруппа, оксогруппа, 1-пирролидинильная группа, 1-пиперидинильная группа, 1-гомопиперидинильная группа, индолин-1-ильная группа, 1,2,3,4-тетрагидрохинолин-1-ильная группа и 4-морфолинильная группа;
группа c1 заместителей: i) аминогруппа (где аминогруппа может содержать одну C2-6 алканоильную группу, C1-6 алкилсульфонильную группу или C3-8 циклоалкилсульфонильную группу или 1-2 C1-6 алкильные группы или C3-8 циклоалкильные группы), ii) цианогруппа, iii) атом галогена, iv) гидроксильная группа и v) v-i) C1-6 алкильная группа, v-ii) C2-6 алкенильная группа, v-iii) C2-6 алкинильная группа, v-iv) C1-6 алкоксигруппа, v-v) C1-6 алкиламинокарбонильная группа, v-vi) C1-6 алкиламиносульфонильная группа, v-vii) C1-6 алкилсульфонильная группа, v-viii) C1-6 алкилтиогруппа, v-ix) C2-6 алканоильная группа, v-x) фенильная группа, v-xi) пиридильная группа, v-xii) пиридазинильная группа, v-xiii) пиримидинильная группа, v-xiv) 1-пирролидинильная группа, v-xv) 1-пиперидинильная группа, v-xvi) 1-гомопиперидинильная группа и v-xvii) 4-морфолинильная группа, каждая из которых может содержать 1-3 заместителя, выбранных из группы, состоящей из C1-6 алкильной группы и атома галогена];
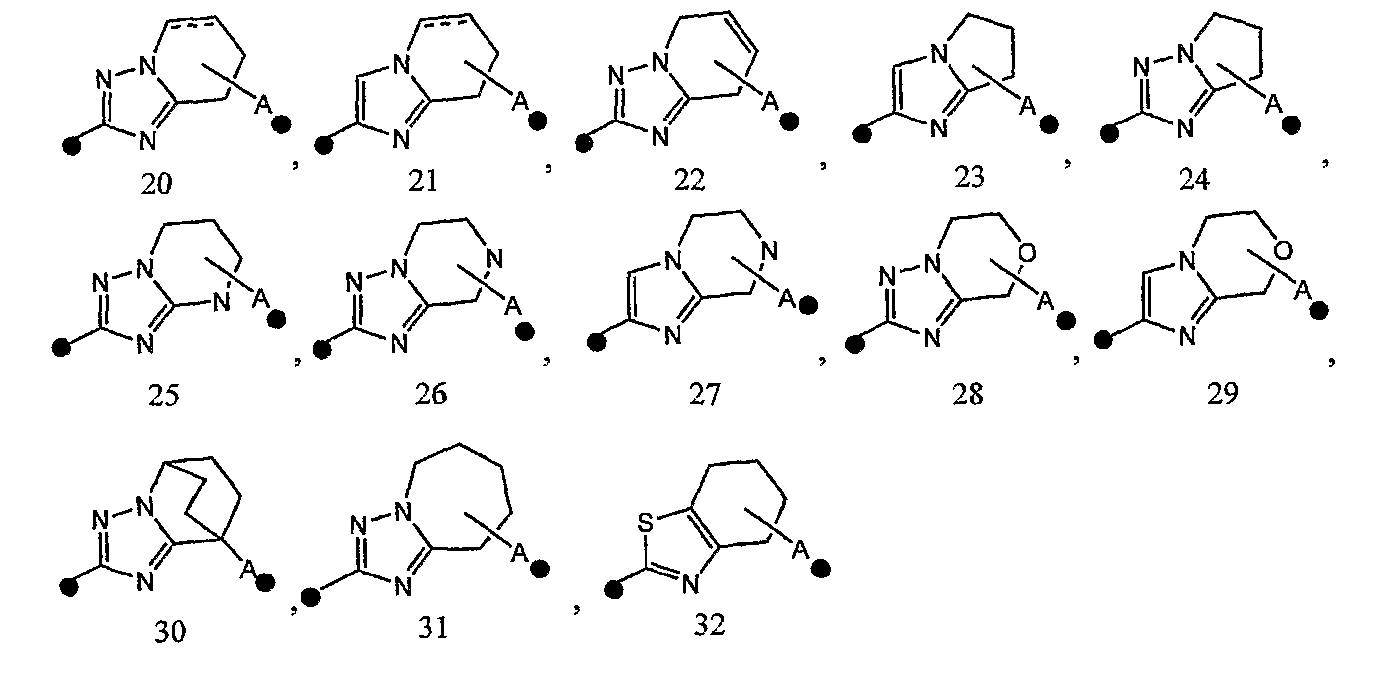
2) соединение или его фармакологически приемлемые соль или сложный эфир по п.1), где кольцо A представляет собой любое одно кольцо, выбранное из группы, состоящей из формул [20]-[32]:
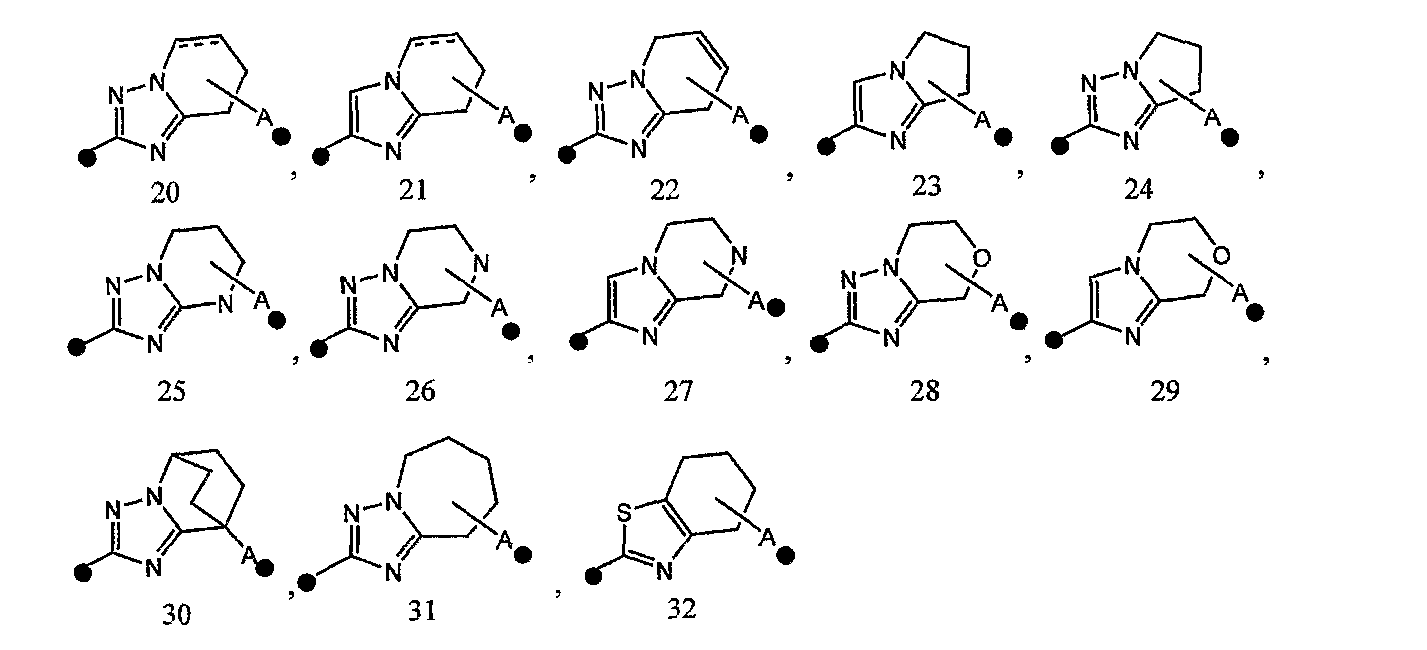
каждое из которых может содержать 1-3 заместителя, выбранных из группы b1 заместителей,
где • представляет собой место связывания с формулой [33]:
A• представляет собой место связывания с X1, и
 представляет собой одинарную связь или двойную связь;
представляет собой одинарную связь или двойную связь;
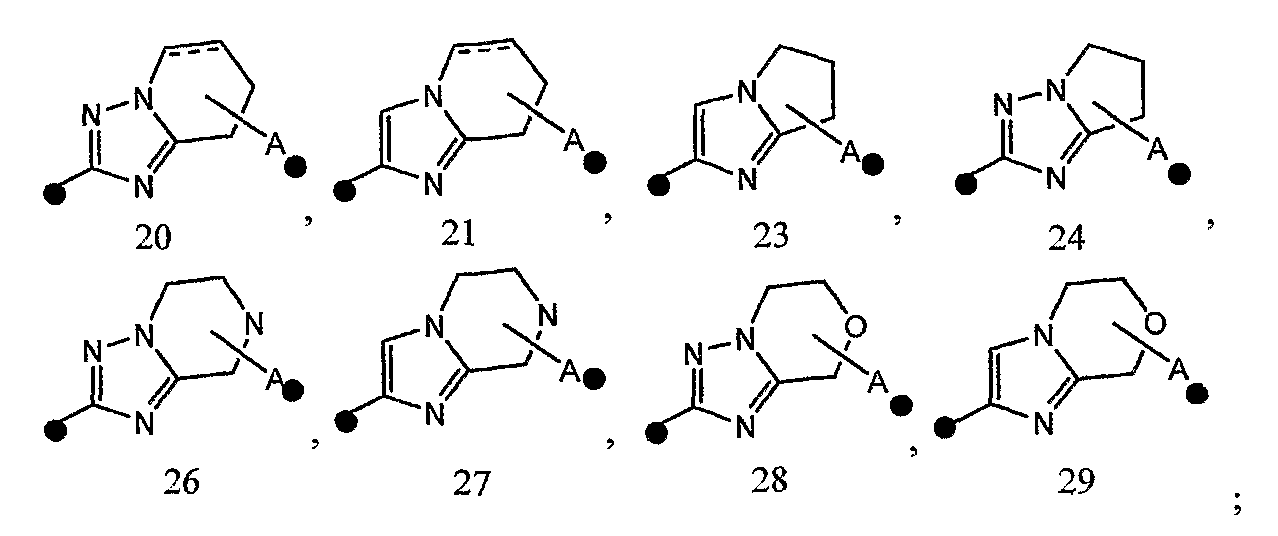
3) соединение или его фармакологически приемлемые соль или сложный эфир по п.2), где кольцо A представляет собой любое одно кольцо, выбранное из группы, состоящей из формул [20], [21], [23], [24] и [26]-[29]:
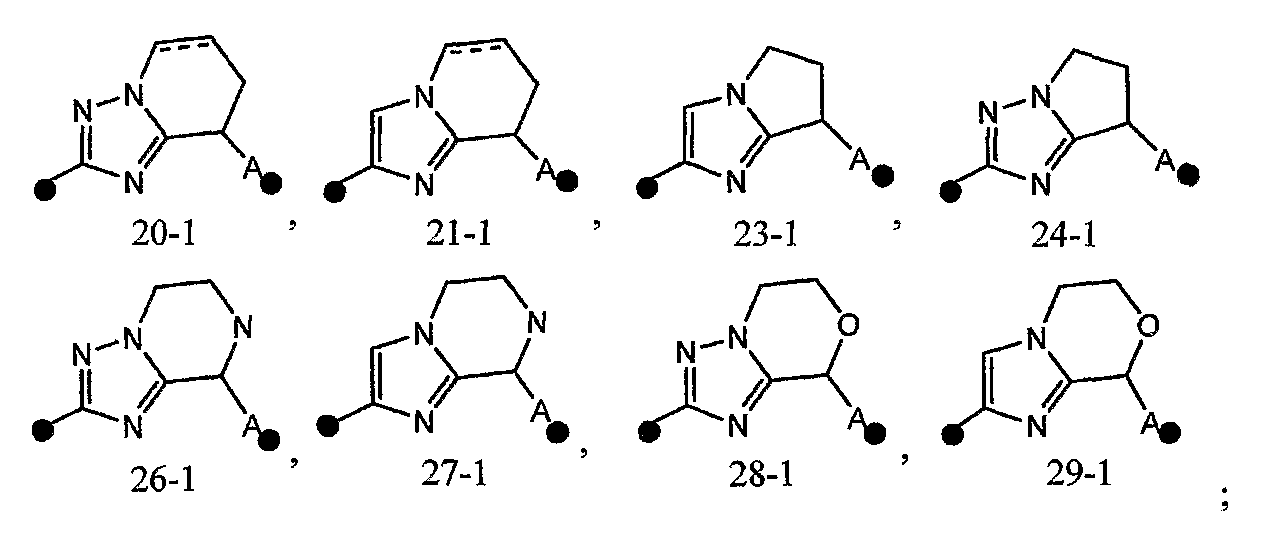
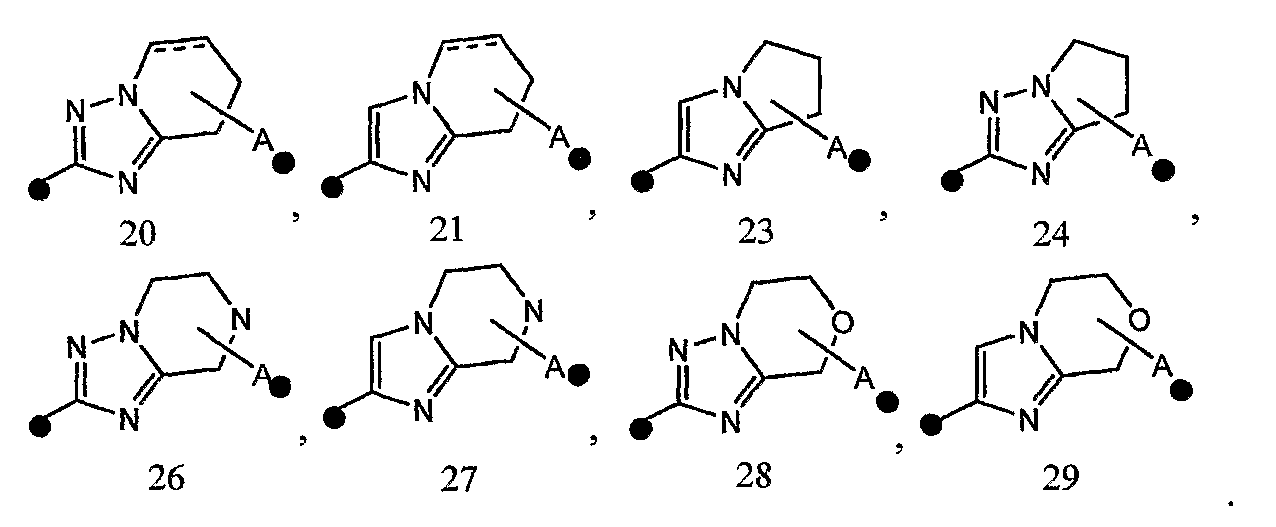
4) соединение или его фармакологически приемлемые соль или сложный эфир по п.2), где кольцо A представляет собой любое одно кольцо, выбранное из группы, состоящей из формул [20-1], [21-1], [23-1], [24-1] и [26-1]-[29-1]:
5) соединение или его фармакологически приемлемые соль или сложный эфир по п.1), где кольцо B представляет собой фенильную группу, пиридильную группу, оксазолильную группу, имидазолильную группу, тиазолильную группу, дигидробензофуранильную группу или тиенильную группу;
6) соединение или его фармакологически приемлемые соль или сложный эфир по п.1), где X1 представляет собой i) одинарную связь или ii) C1-6 алкиленовую группу;
7) соединение или его фармакологически приемлемые соль или сложный эфир по п.1), где W представляет собой атом углерода;
8) соединение или его фармакологически приемлемые соль или сложный эфир по п.1), где W представляет собой атом азота;
9) соединение или его фармакологически приемлемые соль или сложный эфир по п.1), где R1 представляет собой C1-6 алкильную группу или атом галогена и m представляет собой 1-2;
10) соединение или его фармакологически приемлемые соль или сложный эфир по п.1), где R2 представляет собой C1-6 алкоксигруппу и n представляет собой 1;
11) соединение или его фармакологически приемлемые соль или сложный эфир по п.1), где заместитель для кольца A выбран из группы, состоящей из C1-6 алкильной группы (где алкильная группа может быть замещенной 1-3 атомами галогена), C3-8 циклоалкильной группы, C6-14 арильной группы, C6-14 арил-C1-6 алкильной группы, C1-6 алкоксигруппы, C3-8 циклоалкилоксигруппы, C2-6 алканоильной группы, C7-15 ароильной группы, C1-6 алкилсульфонильной группы, C3-8 циклоалкилсульфонильной группы, C6-14 арилсульфонильной группы, цианогруппы, формильной группы, атома галогена, гидроксильной группы и оксогруппы;
12) соединение или его фармакологически приемлемые соль или сложный эфир по п.1), где заместитель для кольца B выбран из группы, состоящей из i) аминогруппы (где аминогруппа может содержать одну C2-6 алканоильную группу, C1-6 алкилсульфонильную группу или C3-8 циклоалкилсульфонильную группу или 1-2 C1-6 алкильные группы или C3-8 циклоалкильные группы), ii) цианогруппы, iii) атома галогена, iv) гидроксильной группы и v) v)-i) C1-6 алкильной группы, v)-ii) C1-6 алкоксигруппы, v)-iii) C1-6 алкилтиогруппы и v)-iv) фенильной группы, каждая из которых может содержать 1-3 заместителя, выбранных из группы, состоящей из C1-6 алкильной группы и атома галогена;
13) одно соединение, выбранное из группы, состоящей из соединений следующих формул [A-1]-[A-7]:
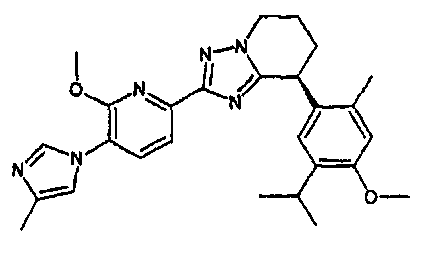
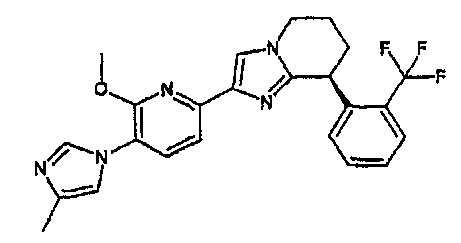
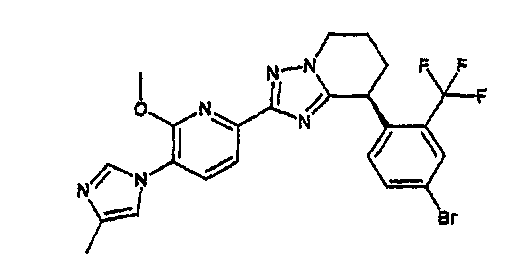
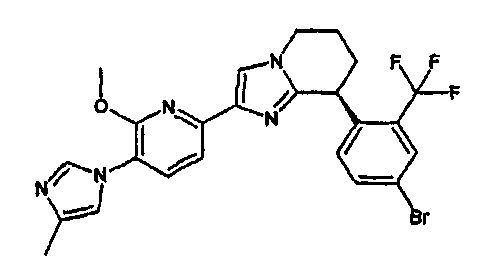
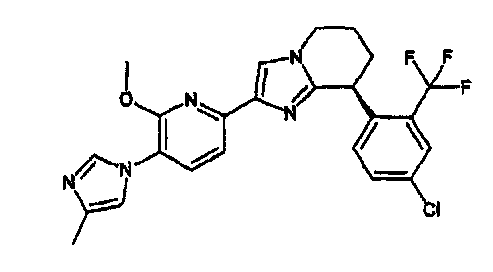
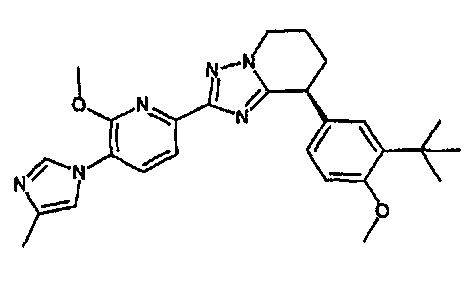
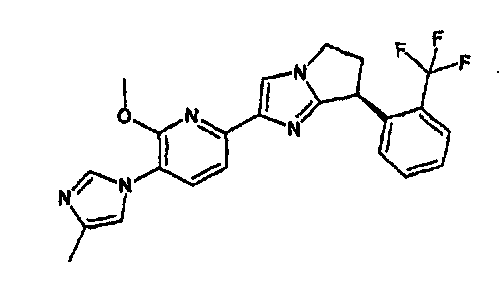
или его фармакологически приемлемые соль или сложный эфир;
14) лекарственное средство, содержащее соединение или его фармакологически приемлемые соль или сложный эфир по любому из пунктов 1)-13) в качестве активного компонента; и
15) лекарственное средство по п.14) для лечения заболевания, выбранного из болезни Альцгеймера, деменции, синдрома Дауна и амилоидоза.
СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Соединение общей формулы (I) или его фармакологически приемлемая соль по настоящему изобретению и терапевтическое средство для лечения заболевания, вызываемого Aβ, по настоящему изобретению являются новыми изобретениями, которые еще не описаны ни в каких документах.
Соединение по настоящему изобретению может быть превращено в химический зонд для захвата белка-мишени в биоактивном низкомолекулярном соединении. В частности, соединение по настоящему изобретению может быть превращено в зонд для аффинной хроматографии, фотоаффинный зонд или тому подобное введением метящей группы, линкера или тому подобного в фрагмент, отличающийся от структурного фрагмента, важного для экспрессии активности соединения, по методике, описанной, например, в J. Mass Spectrum. Soc. Jpn. Vol. 51, No. 5, 2003, p. 492-498 или WO 2007/139149.
Примеры метящей группы, линкера или тому подобного, используемых для химического зонда, включают группы, показанные в следующей группе, состоящей из (1)-(5):
(1) белок-метящие группы, такие как фотоаффинные метящие группы (такие как бензоильная группа, бензофеноновая группа, азидогруппа, карбонилазидогруппа, диазиридиновая группа, еноновая группа, диазогруппа и нитрогруппа) и группы химического сродства (такие как кетоновая группа, замещенная у α-атома углерода атомом галогена, карбамоильная группа, сложноэфирная группа, алкилтиогруппа, акцепторы Михаэля, такие как α,β-ненасыщенные кетоны и сложные эфиры, и оксирановая группа),
(2) расщепляемые линкеры, такие как -S-S-, -O-Si-O-, моносахариды (такие как глюкозная группа и галактозная группа) и дисахариды (такие как лактоза), и ферментативно расщепляемые олигопептидные линкеры,
(3) группы, которые создают улавливающую как рыболовный крючок метку (fishing tag), такие как биотин и 3-(4,4-дифтор-5,7-диметил-4H-3a,4a-диаза-4-бора-s-индацен-3-ил)пропионил,
(4) обнаруживаемые маркеры, такие как радиоактивные метящие группы, такие как 125I, 32P, 3H и 14C; флуоресцентные метящие группы, такие как флуоресцеин, родамин, данзил, умбеллиферон, 7-нитрофуразанил и 3-(4,4-дифтор-5,7-диметил-4H-3a,4a-диаза-4-бора-s-индацен-3-ил)пропионил; хемилюминесцентные группы, такие как люциферин и люминол; и ионы тяжелых металлов, такие как ионы лантаноидных металлов и ионы радия, и
(5) группы, связанные с твердофазными носителями, такими как стеклянные шарики, стеклянные слои, пластины микротитратора, агарозные шарики, агарозные слои, полистироловые шарики, полистироловые слои, найлоновые шарики и найлоновые слои.
Когда зонд получают введением в соединение по настоящему изобретению метящей группы или тому подобного, выбранных из группы, состоящей из описанных выше (1)-(5), в соответствии с методом, описанным в указанных выше документах или тому подобном, зонд может быть использован как химический зонд для идентификации меченых белков, полезных, например, для поиска новых мишеней лекарственных средств.
Ниже объясняются значения символов, терминов и тому подобного, использованных в данном описании, и дается более подробное описание настоящего изобретения.
В данном описании структурная формула соединения может для удобства представлять какой-либо изомер. Но настоящее изобретение включает все изомеры и изомерные смеси, такие как геометрические изомеры, которые могут образовываться в связи со структурой соединения, оптические изомеры, образование которых основано на наличии асимметрического атома углерода, стереоизомеры и таутомеры. Настоящее изобретение не ограничивается описанием выбранной из соображений удобства химической формулы и может охватывать любой из изомеров или их смесей. Так, соединение по настоящему изобретению может иметь в молекуле асимметрический углеродный атом и существовать в виде оптически активного соединения или рацемата, и настоящее изобретение включает без ограничений как оптически активное соединение, так и рацемат. Могут существовать кристаллические полиморфы соединения, но соединение не ограничивается ими тоже и может существовать в виде монокристалла или смеси монокристаллов. Соединение может быть ангидридом или гидратом.
Настоящее изобретение также включает изотопно меченные соединения, которые идентичны соединениям формулы (I), кроме того, что один или несколько атомов заменены атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, обычно находящихся в природе. Примеры изотопов, которые могут быть введены в соединения по настоящему изобретению включают изотопы водорода, углерода, азота, кислорода, фосфороа, фтора, йода и хлора, такие как 2H, 3H, 11C, 14C, 18F, 35S, 123I и 125I.
Соединения по настоящему изобретению и фармацевтически приемлемые производные (например, соли) соединений, которые содержат вышеуказанные изотопы и/или другие изотопы других атомов, входят в объем настоящего изобретения. Изотопно меченные соединения по настоящему изобретению, например те, в которые введены радиоактивные изотопы, такие как 3H и/или 14C, полезны в исследованиях распределения лекарственного средства и/или субстрата в тканях. 3H и 14C считают полезными из-за легкости их получения и обнаруживаемости. Изотопы 11C и 18F считают полезными в PET (эмиссионная позитронная томография) и изотопы 125I считают полезными в SPECT (однофотонная эмиссионная компьютерная томография), все они применимы в визуализации головного мозга. Замещение более тяжелыми изотопами, такими как 2H, может дать некоторые терапевтические преимущества, обусловленные большей метаболической устойчивостью, например, увеличенный полупериод существования in vivo или пониженные требования к дозированию, и поэтому в некоторых случаях считается полезным. Изотопно меченные соединения формулы (I) по настоящему изобретению обычно могут быть получены осуществлением методик, раскрытых в схемах и/или примерах, описанных ниже, с заменой неизотопно меченного реагента легко доступным изотопно меченным реагентом.
Термин “заболевания, вызываемые Aβ” относится к широкому кругу состояний, таких как болезнь Альцгеймера (см., например, Klein W.L., and 7 others, Alzheimer′s disease-affected brain: Presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss, Proceeding National Academy of Science USA, 2003, Sep. 2, 100(18), p.10417-10422; Nitsch R.M., and 16 others, Antibodies against β-amyloid slow cognitive decline in Alzheimer′s disease, Neuron, 2003, May 22, 38(4), p.547-554; Jarrett J.T., and 2 others, The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer′s disease, Biochemistry, 1993, May 11, 32(18), p.4693-4697; Glenner G.G., and another, Alzheimer′s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein, Biochemical and biophysical research communications, 1984, May 16, 120(3), p.885-890; Masters C.L., and 6 others, Amyloid plaque core protein in Alzheimer disease and Down syndrome, Proceeding National Academy of Science USA, 1985, June, 82(12), p.4245-4249; Gouras G.K., and 11 others, Intraneuronal Aβ42 accumulation in human brain, American journal of pathology, 2000, Jan., 156(1), p.15-20; Scheuner D., and 20 others, Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer′s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer′s disease, Nature Medicine, 1996, Aug., 2(8), p.864-870; Forman M.S., and 4 others, Differential effects of the swedish mutant amyloid precursor protein on β-amyloid accumulation and secretion in neurons and nonneuronal cells, The journal of biological chemistry, 1997, Dec. 19, 272(51), p.32247-32253), старческая деменция (см., например, Blass J.P., Brain metabolism and brain disease: Is metabolic deficiency the proximate cause of Alzheimer dementia? Journal of Neuroscience Research, 2001, Dec. 1, 66(5), p.851-856), лобно-височная деменция (см., например, Evin G., and 11 others, Alternative transcripts of presenilin-1 associated with frontotemporal dementia, Neuroreport, 2002, Apr. 16, 13(5), p.719-723), болезнь Пика (см., например, Yasuhara O., and 3 others, Accumulation of amyloid precursor protein in brain lesions of patients with Pick disease, Neuroscience Letters, 1994, Apr. 25, 171(1-2), p.63-66), синдром Дауна (см., например, Teller J.K., and 10 others, Presence of soluble amyloid β-peptide precedes amyloid plaque formation in Down′s syndrome, Nature Medicine, 1996, Jan., 2(1), p.93-95; Tokuda T., and 6 others, Plasma levels of amyloid β proteins Aβ1-40 and Aβ1-42(43) are elevated in Down′s syndrome, Annals of Neurology, 1997, Feb., 41(2), p.271-273), церебральная ангиопатия (см., например, Hayashi Y., and 9 others, Evidence for presenilin-1 involvement in amyloid angiopathy in the Alzheimer′s disease-affected brain, Brain Research, 1998, Apr. 13, 789(2), p.307-314; Barelli H., and 15 others, Characterization of new polyclonal antibodies specific for 40 and 42 amino acid-long amyloid β peptides: their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer′s disease and cerebral amyloid angiopathy cases, Molecular Medicine, 1997, Oct., 3(10), p.695-707; Calhoun M.E., and 10 others, Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid, Proceeding National Academy of Science USA, 1999, Nov. 23, 96(24), p.14088-14093; and Dermaut B., and 10 others, Cerebral amyloid angiopathy is a pathogenic lesion in Alzheimer′s Disease due to a novel presenilin-1 mutation, Brain, 2001, Dec., 124(12), p.2383-2392), наследственная церебральная геморрагия с амилоидозом (голландского типа) (см., например, Cras P., and 9 others, Presenile Alzheimer dementia characterized by amyloid angiopathy and large amyloid core type senile plaques in the APP 692Ala → Gly mutation, Acta Neuropathologica (Berl), 1998, Sep., 96(3), p.253-260; Herzig M.C., and 14 others, Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis, Nature Neuroscience, 2004, Sep., 7(9), p.954-960; van Duinen S.G., and 5 others, Hereditary cerebral hemorrhage with amyloidosis in patients of Dutch origin is related to Alzheimer disease, Proceeding National Academy of Science USA, 1987, Aug., 84(16), p.5991-5994; and Levy E., and 8 others, Mutation of the Alzheimer′s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type, Science, 1990, Jun. 1, 248(4959), p.1124-1126), когнитивное нарушение (см., например, Laws S.M., and 7 others, Association between the presenilin-1 mutation Glu318Gly and complaints of memory impairment, Neurobiology of Aging, 2002, Jan.-Feb., 23(1), p.55-58), нарушение памяти и неспособность к обучению (см., например, Vaucher E., and 5 others, Object recognition memory and cholinergic parameters in mice expressing human presenilin 1 transgenes, Experimental Neurology, 2002, Jun., 175(2), p.398-406; Morgan D., and 14 others, Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer′s disease, Nature, 2000, Dec. 21-28, 408(6815), p.982-985; Moran P.M., and 3 others, Age-related learning deficits in transgenic mice expressing the 751-amino acid isoform of human β-amyloid precursor protein, Proceeding National Academy of Science USA, 1995, June 6, 92(12), p.5341-5345), амилоидоз, церебральная ишемия (см., например, Laws S.M., and 7 others, Association between the presenilin-1 mutation Glu318Gly and complaints of memory impairment, Neurobiology of Aging, 2002, Jan.-Feb., 23(1), p.55-58; Koistinaho M., and 10 others, β-amyloid precursor protein transgenic mice that harbor diffuse Aβ deposits but do not form plaques show increased ischemic vulnerability: Role of inflammation, Proceeding National Academy of Science USA, 2002, Feb. 5, 99(3), p.1610-1615; Zhang F., and 4 others, Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein, The journal of neuroscience, 1997, Oct. 15, 17(20), p.7655-7661), сосудистая деменция (см., например, Sadowski M., and 6 others, Links between the pathology of Alzheimer′s disease and vascular dementia, Neurochemical Research, 2004, Jun., 29(6), p.1257-1266), офтальмоплегия (см., например, O′Riordan S., and 7 others, Presenilin-1 mutation (E280G), spastic paraparesis, and cranial MRI white-matter abnormalities, Neurology, 2002, Oct. 8, 59(7), p.1108-1110), рассеянный склероз (см., например, Gehrmann J., and 4 others, Amyloid precursor protein (APP) expression in multiple sclerosis lesions, Glia, 1995, Oct., 15(2), p.141-51; Reynolds W.F., and 6 others, Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer′s disease, Experimental Neurology, 1999, Jan., 155(1), p.31-41), травма головы, краниальная травма (см., например, Smith D.H., and 4 others, Protein accumulation in traumatic brain injury, NeuroMolecular Medicine, 2003, 4(1-2), p.59-72), апраксия (см., например, Matsubara-Tsutsui M., and 7 others, Molecular evidence of presenilin 1 mutation in familial early onset dementia, American journal of Medical Genetics, 2002, Apr. 8, 114(3), p.292-298), первичное заболевание, семейная амилоидная нейропатия, болезнь повтора триплетов (см., например, Kirkitadze M.D., and 2 others, Paradigm shifts in Alzheimer′s disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies, Journal of Neuroscience Research, 2002, Sep. 1, 69(5), p.567-577; Evert B.O., and 8 others, Inflammatory genes are upreglulated in expanded ataxin-3-expressing cell lines and spinocerebellar ataxia type 3 brains, The Journal of Neuroscience, 2001, Aug. 1, 21(15), p.5389-5396; and Mann D.M., and another, Deposition of amyloid(A4) protein within the brains of persons with dementing disorders other than Alzheimer′s disease and Down′s syndrome, Neuroscience Letters, 1990, Feb. 5, 109(1-2), p.68-75), болезнь Паркинсона (см., например, Primavera J., and 4 others, Brain accumulation of amyloid-β in Non-Alzheimer Neurodegeneration, Journal of Alzheimer′s Disease, 1999, Oct., 1(3), p.183-193), деменция с тельцами Леви (см., например, Giasson B.I., and 2 others, Interactions of amyloidogenic proteins. NeuroMolecular Medicine, 2003, 4(1-2), p.49-58; Masliah E., and 6 others, β-amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a trancgenic mouse model linking Alzheimer′s disease and Parkinson′s disease, Proceeding National Academy of Science USA, 2001, Oct. 9, 98(21), p.12245-12250; Barrachina M., and 6 others, Amyloid-β deposition in the cerebral cortex in Dementia with Lewy bodies is accompanied by a relative increase in AβPP mRNA isoforms containing the Kunitz protease inhibitor, Neurochemistry International, 2005, Feb., 46(3), p.253-260; Primavera J., and 4 others, Brain accumulation of amyloid-β in Non-Alzheimer Neurodegeneration, Journal of Alzheimer′s Disease, 1999, Oct., 1(3), p.183-193), комплекс паркинсонизм-деменция (см., например, Schmidt M.L., and 6 others, Amyloid plaques in Guam amyotrophic lateral sclerosis/parkinsonism-dementia complex contain species of Aβ similar to those found in the amyloid plaques of Alzheimer′s disease and pathological aging, Acta Neuropathologica (Berl), 1998, Feb., 95(2), p.117-122; Ito H., and 3 others, Demonstration of β amyloid protein-containing neurofibrillary tangles in parkinsonism-dementia complex on Guam, Neuropathology and applied neurobiology, 1991, Oct., 17(5), p.365-373), лобно-височная деменция и паркинсонизм, связанный с хромосомой 17 (см., например, Rosso S.M., and 3 others, Coexistent tau and amyloid pathology in hereditary frontotemporal dementia with tau mutations, Annals of the New York academy of sciences, 2000, 920, p.115-119), деменция с аргирофильными зернами (см., например, Tolnay M., and 4 others, Low amyloid (Aβ) plaque load and relative predominance of diffuse plaques distinguish argyrophilic grain disease from Alzheimer′s disease, Neuropathology and applied neurobiology, 1999, Aug., 25(4), p.295-305), болезнь Ниманна-Пика (см., например, Jin L.W., and 3 others, Intracellular accumulation of amyloidogenic fragments of amyloid-β precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities, American Journal of Pathology, 2004, Mar., 164(3), p.975-985), боковой амиотрофический склероз (см., например, Sasaki S., and another, Immunoreactivity of β-amyloid precursor protein in amyotrophic lateral sclerosis, Acta Neuropathologica(Berl), 1999, May, 97(5), p.463-468; Tamaoka A., and 4 others, Increased amyloid β protein in the skin of patients with amyotrophic lateral sclerosis, Journal of neurology, 2000, Aug., 247(8), p.633-635; Hamilton R.L., and another, Alzheimer disease pathology in amyotrophic lateral sclerosis, Acta Neuropathologica, 2004, Jun., 107(6), p.515-522; Turner B.J., and 6 others, Brain β-amyloidaccumulation in transgenic mice expressing mutant superoxide dismutase 1, Neurochemical Research, 2004, Dec., 29(12), p.2281-2286), гидроцефалия (см., например, Weller R.O., Pathology of cerebrospinal fluid and interstitial fluid of the CNS: Significance for Alzheimer disease, prior disorders and multiple sclerosis, Journal of Neuropathology and Experimental Neurology, 1998, Oct., 57(10), p.885-894; Silverberg G.D., and 4 others, Alzheimer′s disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: a hypothesis, Lancet neurology, 2003, Aug., 2(8), p.506-511; Weller R.O., and 3 others, Cerebral amyloid angiopathy: Accumulation of Aβ in interstitial fluid drainage pathways in Alzheimer′s disease, Annals of the New York academy of sciences, 2000, Apr., 903, p.110-117; Yow H.Y., and another, A role for cerebrovascular disease in determining the pattern of β-amyloid deposition in Alzheimer′s disease, Neurology and applied neurobiology, 2002, 28, p.149; Weller R.O., and 4 others, Cerebrovascular disease is a major factor in the failure of elimination of Aβ from the aging human brain, Annals of the New York academy of sciences, 2002, Nov., 977, p.162-168), парапарез (см., например, O′Riordan S., and 7 others, Presenilin-1 mutation (E280G), spastic paraparesis, and cranial MRI white-matter abnormalities, Neurology, 2002, Oct. 8, 59(7), p.1108-1110; Matsubara-Tsutsui M., and 7 others, Molecular evidence of presenilin 1 mutation in familial early onset dementia, American journal of Medical Genetics, 2002, Apr. 8, 114(3), p.292-298; Smith M.J., and 11 others, Variable phenotype of Alzheimer′s disease with spastic paraparesis, Annals of Neurology, 2001, 49(1), p.125-129; Crook R., and 17 others, A variant of Alzheimer′s disease with spastic pararesis and unusual plaques due to deletion of exon 9 of presenilin 1, Nature Medicine, 1998, Apr.; 4(4), p.452-455), прогрессивный супрануклеарный паралич (см., например, Barrachina M., and 6 others, Amyloid-β deposition in the cerebral cortex in Dementia with Lewy bodies is accompanied by a relative increase in AβPP mRNA isoforms containing the Kunitz protease inhibitor, Neurochemistry International, 2005, Feb., 46(3), p.253-260; Primavera J., and 4 others, Brain accumulation of amyloid-β in Non-Alzheimer Neurodegeneration, Jornal of Alzheimer′s Disease, 1999, Oct., 1(3), p.183-193), внутримозговое кровоизлияние (см., например, Atwood C.S., and 3 others, Cerebrovascular requirement for sealant, anti-coagulant and remodeling molecules that allow for the maintenance of vascular integrity and blood supply, Brain Research Reviews, 2003, Sep., 43(1), p.164-78; Lowenson J.D., and 2 others, Protein aging: Extracellular amyloid formation and intracellular repair, Trends in cardiovascular medicine, 1994, 4(1), p.3-8), конвульсия (см., например, Singleton A.B., and 13 others, Pathology of early-onset Alzheimer′s disease cases bearing the Thr113-114ins presenilin-1 mutation, Brain, 2000, Dec., 123(Pt12), p.2467-2474), легкое когнитивное нарушение (см., например, Gattaz W.F., and 4 others, Platelet phospholipase A2 activity in Alzheimer′s disease and mild cognitive impairment, Journal of Neural Transmission, 2004, May, 111(5), p.591-601; Assini A., and 14 others, Plasma levels of amyloid β-protein 42 are increased in women with mild cognitive impariment, Neurology, 2004, Sep. 14, 63(5), p.828-831), артериосклероз (см., например, De Meyer G.R., and 8 others, Platelet phagocytosis and processing of β-amyloid precursor protein as a mechanism of macrophage activation in atherosclerosis, Circulation Reserach, 2002, Jun. 14, 90(11), p.1197-1204).
В соответствии с настоящим изобретением "группа a1 заместителей", "группа b1 заместителей" и "группа c1 заместителей" имеют следующие значения в соединении, представленном формулой (I), эффективном в лечении или профилактике заболевания, вызываемого Aβ.
"Группа a1 заместителей" относится к группе, состоящей из C1-6 алкильной группы, C3-8 циклоалкильной группы, C2-6 алкенильной группы, C1-6 алкоксигруппы, C2-6 алкенилоксигруппы, C3-8 циклоалкилоксигруппы, аминогруппы (где аминогруппа может содержать одну C2-6 алканоильную группу или C1-6 алкилсульфонильную группу или 1-2 C1-6 алкильные группы или C3-8 циклоалкильные группы), цианогруппы, формильной группы, атома галогена, гидроксильной группы и нитрогруппы.
"Группа b1 заместителей" относится к группе, состоящей из C1-6 алкильной группы (где алкильная группа может быть замещенной 1-3 атомами галогена), C2-6 алкенильной группы, C3-8 циклоалкильной группы, C6-14 арильной группы, C6-14 арил-C1-6 алкильной группы, C1-6 алкоксигруппы, C2-6 алкенилоксигруппы, C3-8 циклоалкилоксигруппы, C2-6 алканоильной группы, C4-9 циклоалкилкарбонильной группы, C7-15 ароильной группы, C1-6 алкилсульфонильной группы, C2-6 алкенилсульфонильной группы, C3-8 циклоалкилсульфонильной группы, C6-14 арилсульфонильной группы, C1-6 алкилтиогруппы, C2-6 алкенилтиогруппы, C3-8 циклоалкилтиогруппы, аминосульфонильной группы (где аминосульфонильная группа может содержать 1-2 C1-6 алкильные группы, C2-6 алкенильные группы или C3-8 циклоалкильные группы), аминогруппы (где аминогруппа может содержать одну C2-6 алканоильную группу, C1-6 алкилсульфонильную группу или C3-8 циклоалкилсульфонильную группу или 1-2 C1-6 алкильные группы или C3-8 циклоалкильные группы), цианогруппы, формильной группы, атома галогена, гидроксильной группы, нитрогруппы, оксогруппы, 1-пирролидинильной группы, 1-пиперидинильной группы, 1-гомопиперидинильной группы, индолин-1-ильной группы, 1,2,3,4-тетрагидрохинолин-1-ильной группы и 4-морфолинильной группы.
"Группа c1 заместителей" относится к i) аминогруппе (где аминогруппа может содержать одну C2-6 алканоильную группу, C1-6 алкилсульфонильную группу или C3-8 циклоалкилсульфонильную группу или 1-2 C1-6 алкильные группы или C3-8 циклоалкильные группы), ii) цианогруппе, iii) атому галогена, iv) гидроксильной группе и v) v-i) C1-6 алкильной группе, v-ii) C2-6 алкенильной группе, v-iii) C2-6 алкинильной группе, v-iv) C1-6 алкоксигруппе, v-v) C1-6 алкилтиогруппе, v-vi) C1-6 алкиламинокарбонильной группе, v-vii) C1-6 алкилсульфонильной группе, v-viii) C1-6 алкиламиносульфонильной группе, v-ix) C2-6 алканоильной группе, v-x) фенильной группе, v-xi) пиридильной группе, v-xii) пиридазинильной группе, v-xiii) пиримидинильной группе, v-xiv) 1-пирролидинильной группе, v-xv) 1-пиперидинильной группе, v-xvi) 1-гомопиперидинильной группе и v-xvii) 4-морфолинильной группе, каждая из которых может содержать 1-3 заместителя, выбранных из группы, состоящей из C1-6 алкильной группы и атома галогена.
"Атом галогена" относится к атому фтора, атому хлора, атому брома, атому йода или тому подобному и предпочтительно является атомом фтора, атомом хлора или атомом брома.
“C1-6 алкильная группа" относится к алкильной группе, содержащей 1-6 углеродных атомов. Предпочтительные примеры группы включают неразветвленные или разветвленные алкильные группы, такие как метильная группа, этильная группа, н-пропильная группа, изопропильная группа, н-бутильная группа, изобутильная группа, трет-бутильная группа, н-пентильная группа, изопентильная группа, неопентильная группа, н-гексильная группа, 1-метилпропильная группа, 1,2-диметилпропильная группа, 1-этилпропильная группа, 1-метил-2-этилпропильная группа, 1-этил-2-метилпропильная группа, 1,1,2-триметилпропильная группа, 1-метилбутильная группа, 2-метилбутильная группа, 1,1-диметилбутильная группа, 2,2-диметилбутильная группа, 2-этилбутильная группа, 1,3-диметилбутильная группа, 2-метилпентильная группа и 3-метилпентильная группа.
"C1-6 алкиленовая группа" относится к алкиленовой группе, содержащей 1-6 углеродных атомов. Предпочтительные примеры группы включают неразветвленные или разветвленные алкиленовые группы, такие как метиленовая группа, этиленовая группа, метилметиленовая группа, пропиленовая группа, метилэтиленовая группа, этилметиленовая группа, диметилметиленовая группа, бутиленовая группа, метилпропиленовая группа, этилэтиленовая группа, диметилэтиленовая группа, пропилметиленовая группа, пентиленовая группа и гексиленовая группа. Из них предпочтительными являются, например, метиленовая группа, этиленовая группа, метилметиленовая группа, пропиленовая группа, метилэтиленовая группа, этилметиленовая группа и диметилметиленовая группа.
“C3-8 циклоалкильная группа” относится к циклической алкильной группе, содержащей 3-8 атомов углерода. Предпочтительные примеры группы включают циклопропильную группу, циклобутильную группу, циклопентильную группу, циклогексильную группу, циклогептильную группу и циклооктильную группу.
"C2-6 алкенильная группа" относится к алкенильной группе, содержащей 2-6 атомов углерода. Предпочтительные примеры группы включают неразветвленные или разветвленные алкенильные группы, такие как винильная группа, аллильная группа, 1-пропенильная группа, изопропенильная группа, 1-бутен-1-ильная группа, 1-бутен-2-ильная группа, 1-бутен-3-ильная группа, 2-бутен-1-ильная группа и 2-бутен-2-ильная группа.
"C2-6 алкинильная группа" относится к алкинильной группе, содержащей 2-6 атомов углерода. Предпочтительные примеры группы включают неразветвленные или разветвленные алкинильные группы, такие как этинильная группа, 1-пропинильная группа, 2-пропинильная группа, бутинильная группа, пентинильная группа и гексинильная группа.
"C3-8 циклоалкилоксигруппа" относится к циклической алкильной группе, содержащей 3-8 углеродных атомов, в которой один атом водорода заменен атомом кислорода. Предпочтительные примеры группы включают циклопропоксигруппу, циклобутоксигруппу, циклопентоксигруппу, циклогексоксигруппу, циклогептилоксигруппу и циклооктилоксигруппу.
"C3-8 циклоалкилтиогруппа" относится к циклической алкильной группе, содержащей 3-8 углеродных атомов, в которой один атом водорода заменен атомом серы. Предпочтительные примеры группы включают циклопропилтиогруппу, циклобутилтиогруппу, циклопентилтиогруппу, циклогексилтиогруппу, циклогептилтиогруппу и циклооктилтиогруппу.
"C1-6 алкоксигруппа" относится к алкильной группе, содержащей 1-6 углеродных атомов, в которой атом водорода заменен атомом кислорода. Предпочтительные примеры группы включают метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу, трет-бутоксигруппу, н-пентоксигруппу, изопентоксигруппу, втор-пентоксигруппу, трет-пентоксигруппу, н-гексоксигруппу, изогексоксигруппу, 1,2-диметилпропоксигруппу, 2-этилпропоксигруппу, 1-метил-2-этилпропоксигруппу, 1-этил-2-метилпропоксигруппу, 1,1,2-триметилпропоксигруппу, 1,1,2-триметилпропоксигруппу, 1,1-диметилбутоксигруппу, 2,2-диметилбутоксигруппу, 2-этилбутоксигруппу, 1,3-диметилбутоксигруппу, 2-метилпентоксигруппу, 3-метилпентоксигруппу и гексилоксигруппу.
"C1-6 алкилтиогруппа" относится к алкильной группе, содержащей 1-6 углеродных атомов, в которой один атом водорода заменен атомом серы. Предпочтительные примеры группы включают метилтиогруппу, этилтиогруппу, н-пропилтиогруппу, изопропилтиогруппу, н-бутилтиогруппу, изобутилтиогруппу, трет-бутилтиогруппу, н-пентилтиогруппу, изопентилтиогруппу, неопентилтиогруппу, н-гексилтиогруппу и 1-метилпропилтиогруппу.
"C2-6 алканоильная группа" относится к алкильной группе, содержащей 1-6 углеродных атомов, в которой один атом водорода заменен карбонильной группой. Предпочтительные примеры группы включают ацетильную группу, пропионильную группу и бутирильную группу.
"C1-6 алкилсульфонильная группа" относится к алкильной группе, содержащей 1-6 углеродных атомов, в которой один атом водорода заменен сульфонильной группой. Предпочтительные примеры группы включают метансульфонильную группу и этансульфонильную группу.
"C2-6 алкенилоксигруппа" относится к алкенильной группе, содержащей 2-6 углеродных атомов, в которой один атом водорода заменен атомом кислорода. Предпочтительные примеры группы включают неразветвленные или разветвленные алкенилоксигруппы, такие как винилоксигруппа, аллилоксигруппа, 1-пропенилоксигруппа, изопропенилоксигруппа, 1-бутен-1-илоксигруппа, 1-бутен-2-илоксигруппа, 1-бутен-3-илоксигруппа, 2-бутен-1-илоксигруппа и 2-бутен-2-илоксигруппа.
"C2-6 алкенилтиогруппа" относится к алкенильной группе, содержащей 2-6 углеродных атомов, в которой один атом водорода заменен атомом серы. Предпочтительные примеры группы включают неразветвленные или разветвленные алкенилсульфонильные группы, такие как винилтиогруппа, аллилтиогруппа, 2-пропенилтиогруппа, 1-бутен-1-илтиогруппа, 1-бутен-2-илтиогруппа, 1-бутен-3-илтиогруппа, 2-бутен-1-илтиогруппа и 2-бутен-2-илтиогруппа.
"C2-6 алкенилсульфонильная группа" относится к алкенильной группе, содержащей 2-6 углеродных атомов, в которой один атом водорода заменен сульфонильной группой. Предпочтительные примеры группы включают винилсульфонильную группу, аллилсульфонильную группу, 2-пропенилсульфонильную группу, 1-бутен-1-илсульфонильную группу, 1-бутен-2-илсульфонильную группу и 1-бутен-3-илсульфонильную группу.
"C3-8 циклоалкилсульфонильная группа" относится к циклической алкильной группе, содержащей 3-8 углеродных атомов, в которой один атом водорода заменен сульфонильной группой. Предпочтительные примеры группы включают циклопропилсульфонильную группу, циклобутилсульфонильную группу, циклопентилсульфонильную группу, циклогексилсульфонильную группу, циклогептилсульфонильную группу и циклооктилсульфонильную группу.
"C6-14 арильная группа" относится к моноциклической, бициклической или трициклической ароматической углеводородной кольцевой группе, содержащей 6-14 углеродных атомов. Предпочтительные примеры группы включают 6-14-членную моноциклическую, бициклическую или трициклическую ароматическую углеводородную кольцевую группу, такую как фенильная группа, инденильная группа, нафтильная группа, азуленильная группа, гепталенильная группа, бифенильная группа, флуоренильная группа, феналенильная группа, фенантрильная группа и антрильная группа.
"C7-15 ароильная группа" относится к вышеуказанной C6-14 арильной группе, в которой один атом водорода заменен карбонильной группой. Предпочтительные примеры группы включают бензоильную группу, инденкарбонильную группу, нафтоильную группу, бифенилкарбонильную группу, флуоренилкарбонильную группу, фенантрилкарбонильную группу и антрилкарбонильную группу.
"C6-14 арил-C1-6 алкильная группа" относится к вышеуказанной C1-6 алкильной группе, в которой один атом водорода заменен вышеуказанной C6-14 арильной группой. Предпочтительные примеры группы включают бензильную группу, фенетильную группу, фенилпропильную группу, нафтилметильную группу и бифенилметильную группу.
"C6-14 арилсульфонильная группа" относится к вышеуказанной C6-14 арильной группе, в которой один атом водорода заменен сульфонильной группой. Предпочтительные примеры группы включают бензолсульфонильную группу, нафталинсульфонильную группу и бифенилсульфонильную группу.
"C1-6 алкиламинокарбонильная группа" относится к алкильной группе, содержащей 1-6 углеродных атомов, в которой один атом водорода заменен аминокарбонильной группой. Предпочтительные примеры группы включают метиламинокарбонильную группу, этиламинокарбонильную группу, пропиламинокарбонильную группу, бутиламинокарбонильную группу и гексиламинокарбонильную группу.
"C1-6 алкиламиносульфонильная группа" относится к алкильной группе, содержащей 1-6 углеродных атомов, в которой один атом водорода заменен аминокарбонильной группой. Предпочтительные примеры группы включают метиламиносульфонильную группу, этиламиносульфонильную группу, пропиламиносульфонильную группу, бутиламиносульфонильную группу и гексиламиносульфонильную группу.
Когда W представляет собой атом азота и R2 представляет собой гидроксильную группу, соединение включает, например, таутомер, представленный формулой:
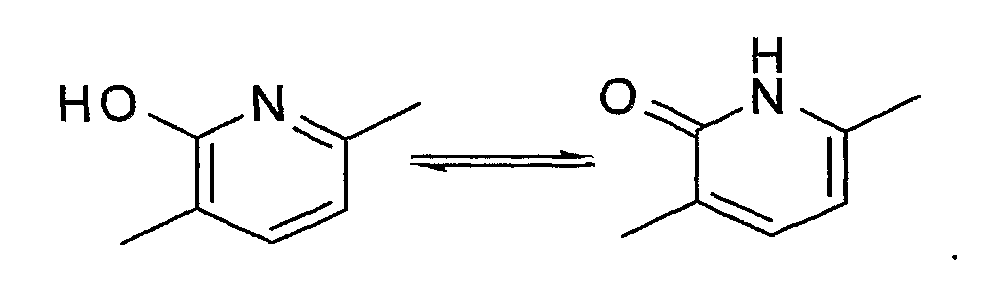
"Пятичленная ароматическая гетероциклическая группа, конденсированная с 5-14-членной неароматической кольцевой группой, которая содержит один или несколько атомов азота и может содержать 1-3 заместителя, выбранных из следующей группы b1 заместителей", в определении кольца A относится к пятичленному ароматическому гетероциклу, содержащему один или несколько атомов азота, такому как пиразол, имидазол, триазол, тетразол, оксазол, тиазол, оксадиазол или тиадиазол, конденсированному с 5-14-членным неароматическим кольцом, таким как кольцо, представленное следующей формулой:
Один-три заместителя, выбранных из группы b1 заместителей, могут находиться в любом замещаемом положении в кольце.
Предложение "5-14-членная неароматическая группа может иметь структуру с поперечными связями" в определении кольца A относится к тому факту, что два углеродных атома в неароматической кольцевой группе вместе могут образовывать структуру с поперечными связями. Например, кольцо, имеющее структуру с поперечными связями, представленное формулой:
вместе с вышеуказанной пятичленной ароматической гетероциклической группой, такой как следующее триазолильное кольцо:
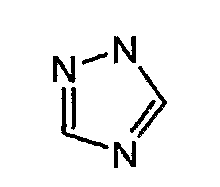
могут образовывать конденсированное кольцо, представленное следующей формулой:
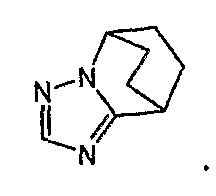
Кольцо A может быть присоединено к X1 в замещаемом положении в кольце, отличном от пятичленного ароматического гетероцикла, образующего конденсированное кольцо. Например, когда соединение между кольцом A и X1 представлено формулой [34]:
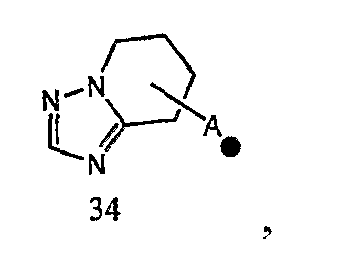
кольцо A может быть присоединено к X1 в замещаемом положении, указанном в одной из следующих формул [34-1]-[34-4]:
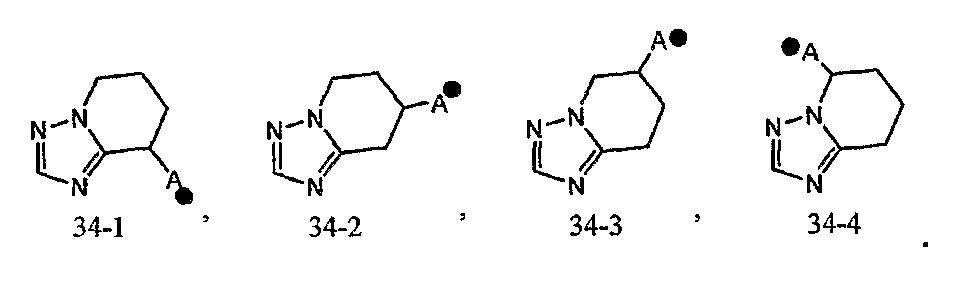
Когда соединение между кольцом A и X1 представлено формулой [35]:
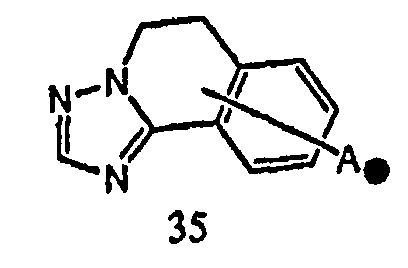 ,
,
кольцо A может быть соединено с X1 в замещаемом положении, указанном в одной из следующих формул [35-1]-[35-6]:
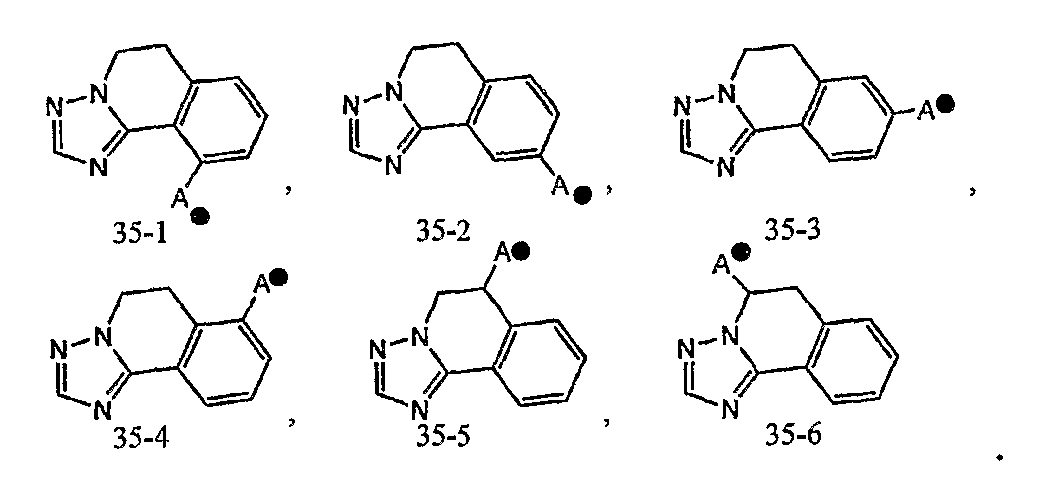
Один-три заместителя, выбранных из группы c1 заместителей, в определении кольца B могут находиться в любом замещаемом положении в кольце. Кольцо B может быть соединено с X1 в любом замещаемом положении в кольце.
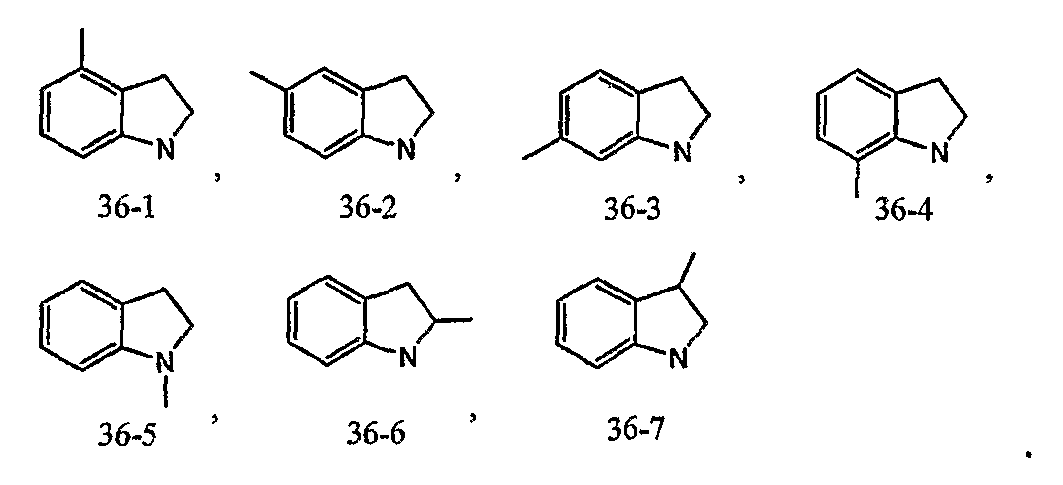
Например, когда кольцо B представлено формулой [36]:
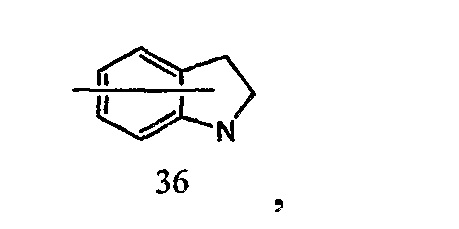
кольцо B может быть соединено с X1 в любом замещаемом положении, указанном в одной из следующих формул [36-1]-[36-7]:
В настоящем изобретении "фармакологически приемлемую соль" особо не ограничивают, лишь бы она была фармакологически приемлемой солью, образованной соединением общей формулы (I), которое является терапевтическим средством при заболевании, вызываемом Aβ.
Предпочтительные конкретные примеры соли включают гидрогалогениды (такие как гидрофториды, гидрохлориды, гидробромиды и гидроиодиды), соли неорганических кислот (такие как сульфаты, нитраты, перхлораты, фосфаты, карбонаты и бикарбонаты), органические карбоксилаты (такие как ацетаты, оксалаты, малеаты, тартраты, фумараты и цитраты), органические сульфонаты (такие как метансульфонаты, трифторметансульфонаты, этансульфонаты, бензолсульфонаты, толуолсульфонаты и камфорсульфонаты), соли аминокислот (такие как аспартаты и глутаматы), соли четвертичных аминов, соли щелочных металлов (такие как натриевые соли и калиевые соли) и соли щелочноземельных металлов (такие как магниевые соли и кальциевые соли).
Далее описано соединение формулы [I] по настоящему изобретению.
В соединении формулы [I] или его фармакологически приемлемой соли, предпочтительно, R1 представляет собой C1-6 алкильную группу или атом галогена и m представляет собой целое число от 1 до 2; особенно предпочтительно, R1 представляет собой C1-6 алкильную группу и m представляет собой целое число от 1 до 2; и наиболее предпочтительно, R1 представляет собой метильную группу и m представляет собой 1.
В соединении формулы [I] или его фармакологически приемлемой соли, предпочтительно, R2 представляет собой атом галогена, гидроксильную группу или C1-6 алкоксигруппу и n представляет собой целое число от 1-2; более предпочтительно, R2 представляет собой C1-6 алкоксигруппу и n представляет собой целое число 1-2; и особенно предпочтительно, R2 представляет собой метоксигруппу и n представляет собой 1.
В соединении формулы [I] или его фармакологически приемлемой соли X1 представляет собой предпочтительно i) одинарную связь или ii) C1-6 алкиленовую группу.
В соединении формулы [I] или его фармакологически приемлемой соли кольцо A представлено предпочтительно любой из следующих формул 20-32:
и особенно предпочтительно любой из следующих формул:
В соединении формулы [I] или его фармакологически приемлемой соли кольцо B представлено предпочтительно любой из следующих формул:
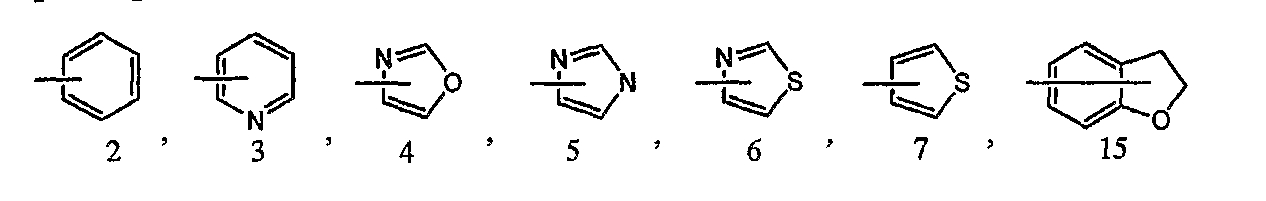
каждая из которых может быть замещенной 1-3 заместителями, выбранными из группы c1 заместителей.
Группа b1 заместителей является предпочтительно группой заместителей, состоящей из (1) C1-6 алкильной группы (где алкильная группа может быть замещенной 1-3 атомами галогена), (2) C3-8 циклоалкильной группы, (3) C6-14 арильной группы, (4) C6-14 арил-C1-6 алкильной группы, (5) C1-6 алкоксигруппы, (6) C3-8 циклоалкилоксигруппы, (7) C2-6 алканоильной группы, (8) C7-15 ароильной группы, (9) C1-6 алкилсульфонильной группы, (10) C3-8 циклоалкилсульфонильной группы, (11) C6-14 арилсульфонильной группы, (12) цианогруппы, (13) формильной группы, (14) атома галогена, (15) гидроксильной группы и (16) оксогруппы.
Группа c1 заместителей является предпочтительно группой заместителей, состоящей из (1) аминогруппы (где аминогруппа может содержать одну C2-6 алканоильную группу, C1-6 алкилсульфонильную группу или C3-8 циклоалкилсульфонильную группу или 1-2 C1-6 алкильные группы или C3-8 циклоалкильные группы), (2) цианогруппы, (3) атома галогена, (4) гидроксильной группы и (5) (5)-1) C1-6 алкильной группы, (5)-2) C1-6 алкоксигруппы, (5)-3) C1-6 алкилтиогруппы и (5)-4) фенильной группы, каждая из которых может содержать 1-3 заместителя, выбранных из группы, состоящей из C1-6 алкильной группы и атома галогена.
Например, по меньшей мере одно соединение, выбранное из группы, состоящей из следующих формул [A-1]-[A-7]:
или его фармакологически приемлемая соль является особенно подходящим(ей) и полезно(а) в качестве терапевтического средства от заболевания, вызываемого амилоидом-β, такого как болезнь Альцгеймера, старческая деменция, синдром Дауна или амилоидоз.
Ниже описаны способы получения соединения общей формулы (I) по настоящему изобретению.
Соединение, представленное общей формулой (I):
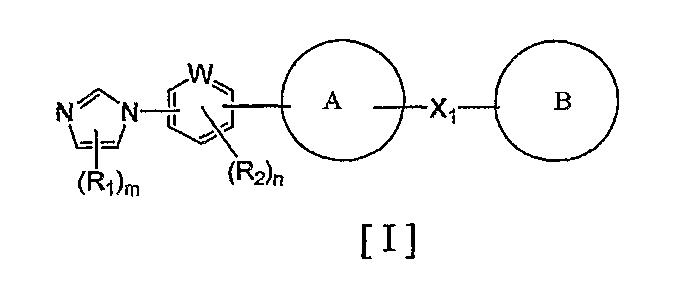
где R1, R2, m, n, W, кольцо A, X1 и кольцо B такие, как определено выше, синтезируют, например, способом, таким как следующие общие способы получения 1-8. Очевидно, что для удобства получения соединения по настоящему изобретению способ содержит подходящие ствдию реакции введения защиты и стадию реакции снятия защиты с использованием известной специалисту в данной области техники защитной группы, которую подходящим образом выбирают для каждой стадии (см. T. Greene et al., "Protective Groups in Organic Synthesis", John Wiley & Sons, Inc., New York, 1981). Очевидно, что для удобства получения соединения по настоящему изобретению способ содержит конверсию заместителей, введение заместителей и тому подобное подходящим образом для каждой стадии, известным специалисту в данной области техники. Очевидно также, что для удобства получения соединения по настоящему изобретению все изомеры и смеси изомеров, таких как геометрические изомеры, которые могут быть образованы в зависимости от структуры соединения, оптические изомеры, основанные на асимметрическом атоме углерода, стереоизомеры и таутомеры, могут быть получены в виде отдельного соединения известным специалисту в данной области техники методом, подходящим для каждой стадии, таким как фракционная кристаллизация или колоночная хроматография.
Общий способ получения 1
Ниже описан обычно используемый общий способ получения 1 для соединения общей формулы (I) в соответствии с настоящим изобретением.
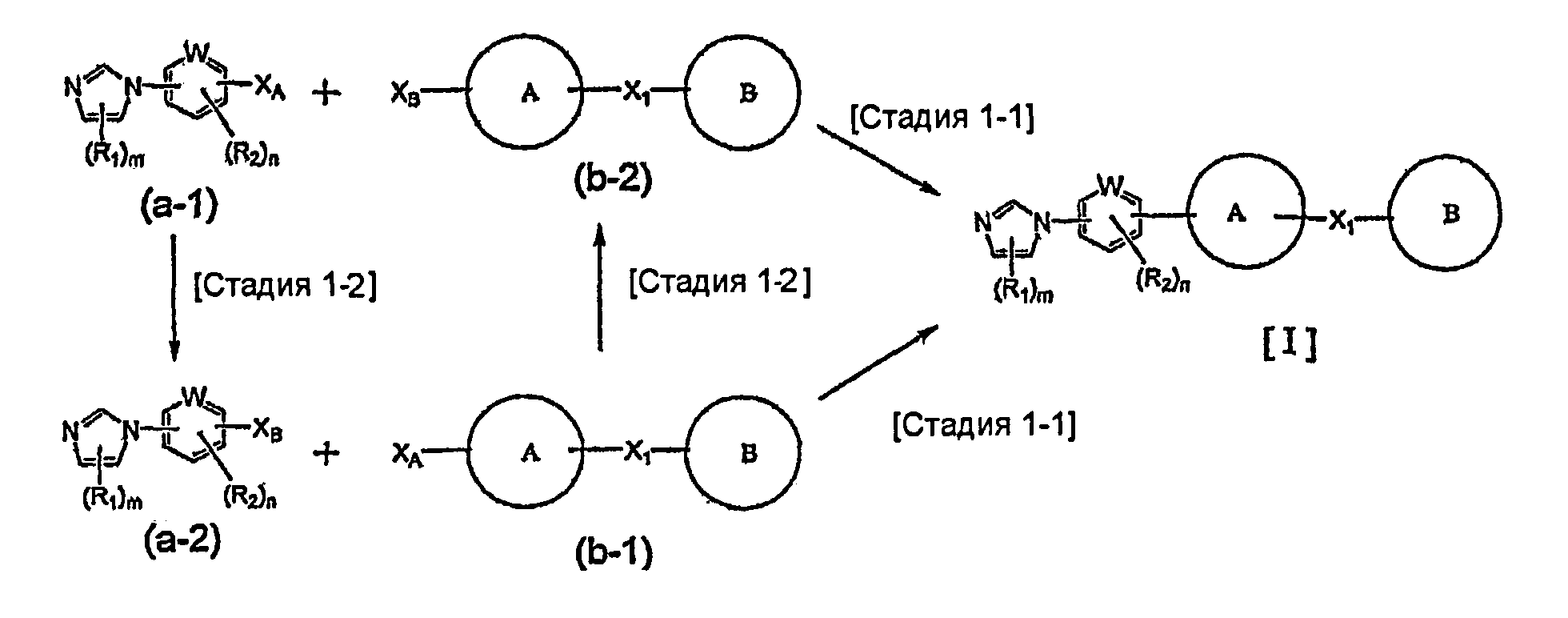
На схеме R1, R2, m, n, W, кольцо A, X1 и кольцо B такие, как определено выше; XA представляет собой атом галогена, такой как атом хлора, атом брома или атом йода, или сульфонатную группу, такую как метансульфонатная группа, п-толуолсульфонатная группа или трифторметансульфонатная группа; и XB представляет собой триалкилстаннильную группу, группу бороновой кислоты или боронатную группу, такую как пинаколборонатная группа.
Указанный выше общий способ получения 1 является способом получения соединения общей формулы [I] подверганием реакции связывания на стадии 1-1 соединения общей формулы (a-1) и соединения общей формулы (b-2) или способом получения соединения общей формулы [I] подверганием реакции связывания на стадии 1-1 соединения общей формулы (a-2) и соединения общей формулы (b-1), где заместители XA и XB заменены один на другой.
Реакция связывания на стадии 1-1 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Предпочтительные примеры методики включают реакцию Сузуки-Мияуры (см., например, A. Suzuki, "Chem. Rev.", 1995, vol. 95, p. 2457) и реакцию связывания по Стилле (см., например, J.K. Stille, "Angew. Chem. Int. Ed. Engl.", 1986, vol. 25, p.508).
В реакции Сузуки-Мияуры, например, галогенсодержащее соединение или трифторметансульфонатное соединение общей формулы (a-1) предпочтительно связывают с 1,0-5,0 эквивалентами соединения общей формулы (b-2) (где XB представляет собой предпочтительно группу бороновой кислоты, боронатную группу, такую как пинаколборонатная группа, алкилборалкенильная группа или тому подобное) относительно соединения общей формулы (a-1) в присутствии 0,01-0,5 эквивалента катализатора на основе переходного металла относительно соединения общей формулы (a-1). Из-за удобства и эффективности перемешивания данную реакцию предпочтительно осуществляют в присутствии растворителя. Используемый растворитель меняют в соответствии с исходным материалом и используемым катализатором на основе переходного металла и особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают ацетонитрил, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол, 1-метил-2-пирролидон, N,N-диметилформамид, воду и состоящий из них смешанный растворитель. Температура реакции должна быть такой, которая позволяет закончить реакцию связывания и предпочтительно находится в интервале от комнатной температуры до 200°C. Данную реакцию проводят предпочтительно в атмосфере инертного газа и более предпочтительно в атмосфере азота или аргона. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии. Катализатором на основе переходного металла является предпочтительно известный палладиевый комплекс и более предпочтительно известный палладиевый комплекс, такой как ацетат палладия(II), дихлорбис(трифенилфосфин)палладий(II), тетракис(трифенилфосфин)палладий(0) или трис(дибензилиденацетон)дипалладий(0). Подходящим образом может быть добавлен фосфорный лиганд (предпочтительно, например, трифенилфосфин, три-о-толилфосфин, трициклогексилфосфин или три-трет-бутилфосфин), чтобы реакция протекала эффективно. Может быть также подходящим образом добавлена соль четвертичного аммония, предпочтительно, например, тетрабутиламмонийхлорид или тетрабутиламмонийбромид, чтобы реакция протекала эффективно. В данной реакции предпочтительный результат может быть получен в присутствии основания. Используемое при этом основание меняют в соответствии с исходным материалом, используемым растворителем и тому подобным и особо не ограничивают. Предпочтительные примеры основания включают гидроксид натрия, гидроксид бария, фторид калия, фторид цезия, карбонат натрия, карбонат калия, карбонат цезия и фосфат калия. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии.
В реакции связывания по Стилле, например, галогенсодержащее соединение или содержащее трифторметансульфонатную группу соединение общей формулы (a-1) предпочтительно связывают с 1,0-5,0 эквивалентами соединения общей формулы (b-2) (где XB представляет собой предпочтительно триалкилстаннильную группу) относительно соединения общей формулы (a-1) в присутствии 0,01-0,2 эквивалента катализатора на основе переходного металла относительно соединения общей формулы (a-1). Является предпочтительным подходящим образом использовать в данной реакции 0,1-5,0 эквивалентов галогенида меди(I) и/или хлорида лития, чтобы реакция протекала эффективно. Предпочтительные примеры растворителя, используемого в данной реакции, включают толуол, ксилол, N,N-диметилформамид, N,N-диметилацетамид, 1-метил-2-пирролидон и диметилсульфоксид. Температура реакции должна быть такой, которая позволяет закончить реакцию связывания, и предпочтительно находится в интервале от комнатной температуры до 150°C. Предпочтительным катализатором на основе переходного металла является палладиевый комплекс, предпочтительно, например, известный палладиевый комплекс, такой как ацетат палладия(II), дихлорбис(трифенилфосфин)палладий(II), тетракис(трифенилфосфин)палладий(0) или трис(дибензилиденацетон)дипалладий(0), и более предпочтительно, например, ацетат палладия(II), тетракис(трифенилфосфин)палладий(0) или трис(дибензилиденацетон)дипалладий(0). Подходящим образом может быть добавлен, например, фосфорный лиганд (предпочтительно, например, трифенилфосфин, три-о-толилфосфин, 1,3-бис(дифенилфосфино)пропан или три-трет-бутилфосфин), чтобы реакция протекала эффективно. Данную реакцию проводят предпочтительно в атмосфере инертного газа и более предпочтительно в атмосфере азота или аргона. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии.
Стадия 1-2 является примером способа получения соединения общей формулы (a-2) и соединения общей формулы (b-2), где заместители XA и XB заменены один на другой. Данная стадия изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Могут быть использованы способы, подобные способам получения, таким как реакция Сузуки-Мияуры (см., например, A. Suzuki, "Chem. Rev.", 1995, vol. 95, p.2457) и реакция связывания по Стилле (см., например, J.K. Stille, "Angew. Chem. Int. Ed. Engl.", 1986, vol. 25, p.508).
Получение соединения общей формулы (a-1)
На следующей схеме показан пример получения соединения общей формулы (a-1).
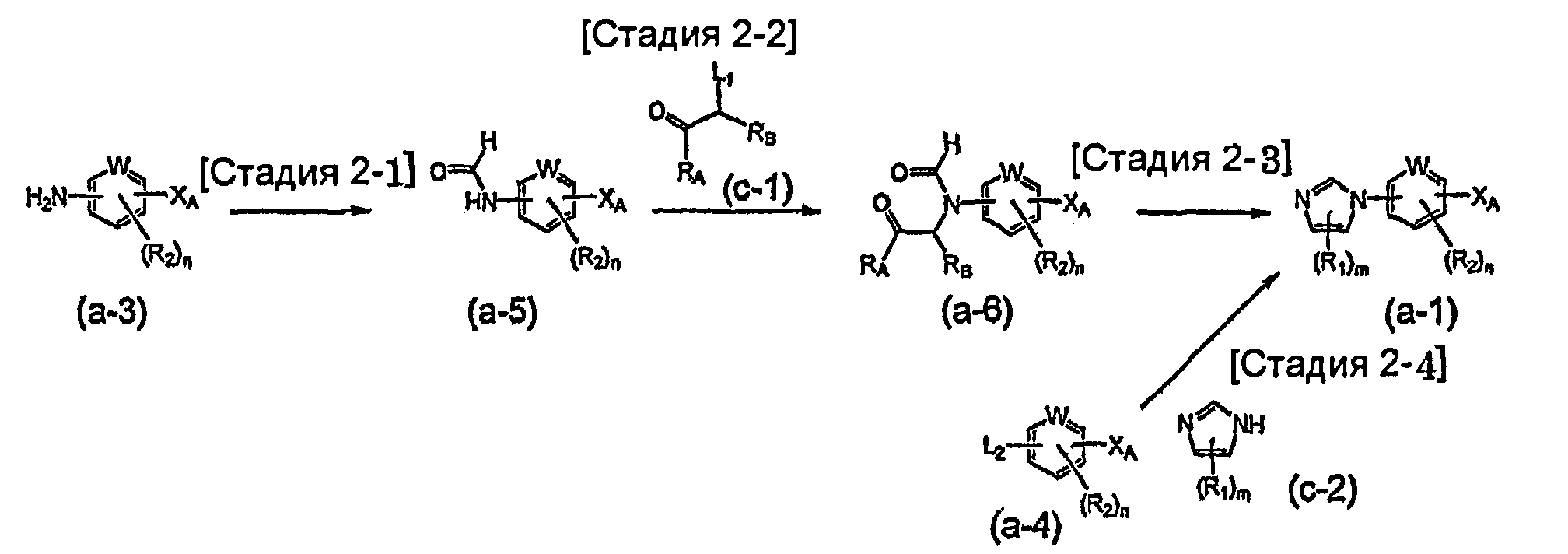
На схеме R1, R2, m, n, W и XA такие, как определено выше; RA и RB такие, как определено выше для R1; L1 представляет собой атом галогена, такой как атом хлора, атом брома или атом йода, или сульфонатную группу, такую как метансульфонатная группа, п-толуолсульфонатная группа или трифторметансульфонатная группа; и L2 представляет собой атом галогена, такой как атом фтора, атом хлора, атом брома или атом йода, сульфонатную группу, такую как метансульфонатная группа, п-толуолсульфонатная группа или трифторметансульфонатная группа, или группу бороновой кислоты.
Соединение общей формулы (a-1) может быть получено из аминосоединения (a-3) как исходного материала через формилирование на стадии 2-1, реакцию алкилирования на стадии 2-2 и образование имидазольного кольца на стадии 2-3, или может быть получено из соединения общей формулы (a-4) в качестве исходного материала осуществлением реакции связывания на стадии 2-4.
Стадия 2-1 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах, или тому подобное (T. Greene et al., "Protective Groups in Organic Synthesis", John Wiley & Sons, Inc., New York, 1981, например).
Стадия 2-2 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Предпочтительные примеры методики включают способ перемешивания соединения общей формулы (a-5) и 1,0-10,0 эквивалентов соединения общей формулы (c-1) относительно соединения общей формулы (a-5) в растворителе в присутствии 1,0-10,0 эквивалентов основания относительно соединения общей формулы (a-5). Используемое основание изменяют в соответствии с исходным материалом и особо не ограничивают. Предпочтительные примеры основания включают гидриды щелочных металлов (такие как гидрид натрия и гидрид лития), соли щелочных металлов (такие как карбонат калия, карбонат натрия и карбонат цезия) и алкоксиды металлов (такие как метоксид натрия и трет-бутоксид калия). Используемый растворитель меняют в соответствии с исходным материалом и особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают растворители в виде простых эфиров, таких как тетрагидрофуран, 1,4-диоксан и диэтиловый эфир; галогенированные растворители, такие как метиленхлорид, 1,2-дихлорэтан и хлороформ; полярные растворители, такие как N,N-диметилформамид и N-метилпирролидон; неполярные растворители, такие как толуол и бензол; и смеси указанных растворителей. Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и находится в интервале предпочтительно от 0°C до 200°C, например. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии. Нежелательный побочный продукт может быть удален методом, известным специалисту в данной области техники, таким как традиционный метод хроматографии, экстракция и/или кристаллизация.
Стадия 2-3 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в The Chemistry of Heterocyclic Compounds. Imidazole and Derivatives, Part I, p.33, Inters. Publish. 1953). Предпочтительные примеры методики включают способ получения соединения общей формулы (a-1) образованием имидазольного кольца из соединения общей формулы (a-6) и аммония, аммониевой соли, формамида или тому подобного в качестве источника азота. Используемый растворитель особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают неполярные растворители, такие как толуол и бензол; спиртовые растворители, такие как метанол и этанол; органические кислоты, такие как уксусная кислота или трифторуксусная кислота, сульфоновые кислоты, такие как п-толуолсульфоновая кислота и трифторметансульфоновая кислота; воду и смеси указанных растворителей. Формамид может быть необязательно использован в качестве источника атома азота и в качестве растворителя. Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и предпочтительно находится в интервале от комнатной температуры до 250°C, например. Выход может быть повышен, когда реакцию осуществляют, используя герметичный сосуд. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии. Нежелательный побочный продукт может быть удален методом, известным специалисту в данной области техники, таким как традиционный метод хроматографии, экстракция и/или кристаллизация.
Реакция связывания на стадии 2-4 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в D.D. Davey et al., "J. Med. Chem.", 1991, vol. 34, p.2671-2677). Примеры способа включают способ перемешивания соединения общей формулы (a-4) (где L2 представляет собой предпочтительно атом галогена или тому подобное) и 1,0-5,0 эквивалентов имидазольного соединения (c-2) относительно соединения общей формулы (a-4) в растворителе в присутствии или в отсутствие 1,0-5,0 эквивалентов основания относительно соединения общей формулы (a-4). Предпочтительные примеры используемого основания включают гидрид натрия, гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия, карбонат цезия, карбонат бария, пиридин, лутидин и триэтиламин. Используемый растворитель меняют в соответствии с исходным материалом и особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают ацетонитрил, тетрагидрофуран, диметилсульфоксид, N,N-диметилформамид и N-метилпирролидон. Основание может быть необязательно использовано в качестве растворителя. Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и предпочтительно находится в интервале от комнатной температуры до 150°C, например. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии. Нежелательный побочный продукт может быть удален методом, известным специалисту в данной области техники, таким как традиционный метод хроматографии и/или кристаллизация.
Примеры реакции связывания на стадии 2-4 включают способ перемешивания соединения общей формулы (a-4) (где L2 представляет собой предпочтительно группу бороновой кислоты или тому подобное) в растворителе в присутствии медного катализатора (такого как описанный в J.P. Collman et al., "Org. Letters.", 2000, vol. 2, p. 1233-1236). Предпочтительные примеры методики включают способ перемешивания соединения общей формулы (a-4) и 0,1-10,0 эквивалентов имидазольного соединения (c-2) относительно соединения общей формулы (a-4) в растворителе в присутствии 0,01-1,0 эквивалента медного реагента, такого как медь, бромид меди или иодид меди, относительно соединения общей формулы (a-4). Используемый медный реагент изменяют в соответствии с исходным материалом и особо не ограничивают. Предпочтительные примеры медного реагента включают галогенид меди(I), ацетат меди(II), нитрат меди(II) и хлорид ди-µ-гидроксо-бис[(N,N,N′,N′-тетраметилэтилендиамин)меди(II)]. Используемый растворитель меняют в соответствии с исходным материалом, реагентом и тому подобным и особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают растворители в виде простых эфиров, таких как тетрагидрофуран, 1,4-диоксан и диэтиловый эфир; галогенированные растворители, такие как метиленхлорид, 1,2-дихлорэтан и хлороформ; полярные растворители, такие как этилацетат, N,N-диметилформамид и N-метилпирролидон; неполярные растворители, такие как толуол, бензол и дихлорбензол; и смеси указанных растворителей. Основание может быть использовано в зависимости от исходного материала, реагента и тому подобного. Предпочтительные примеры основания включают органические основания, такие как триэтиламин, пиридин и тетраметилэтилендиамин; соли щелочных металлов, такие как карбонат калия, карбонат натрия, ацетат калия, ацетат натрия и карбонат цезия; и алкоксиды металлов, такие как метоксид натрия и трет-бутоксид калия. Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и предпочтительно находится в интервале от комнатной температуры до 200°C, например. Хорошие результаты, такие как уменьшение времени реакции и повышение выхода, могут быть получены, когда реакцию осуществляют в атмосфере кислорода или струе воздуха. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии. Нежелательный побочный продукт может быть удален методом, известным специалисту в данной области техники, таким как традиционный метод хроматографии, экстракция и/или кристаллизация.
Соединение формулы (a-3), соединение формулы (a-4), соединение формулы (c-1) и соединение формулы (c-2) являются известными или коммерчески доступными соединениями или являются соединениями, которые могут быть получены из указанных соединений традиционным способом.
Получение соединения общей формулы (b-1)
На следующей схеме показан пример получения соединения общей формулы (b-1).
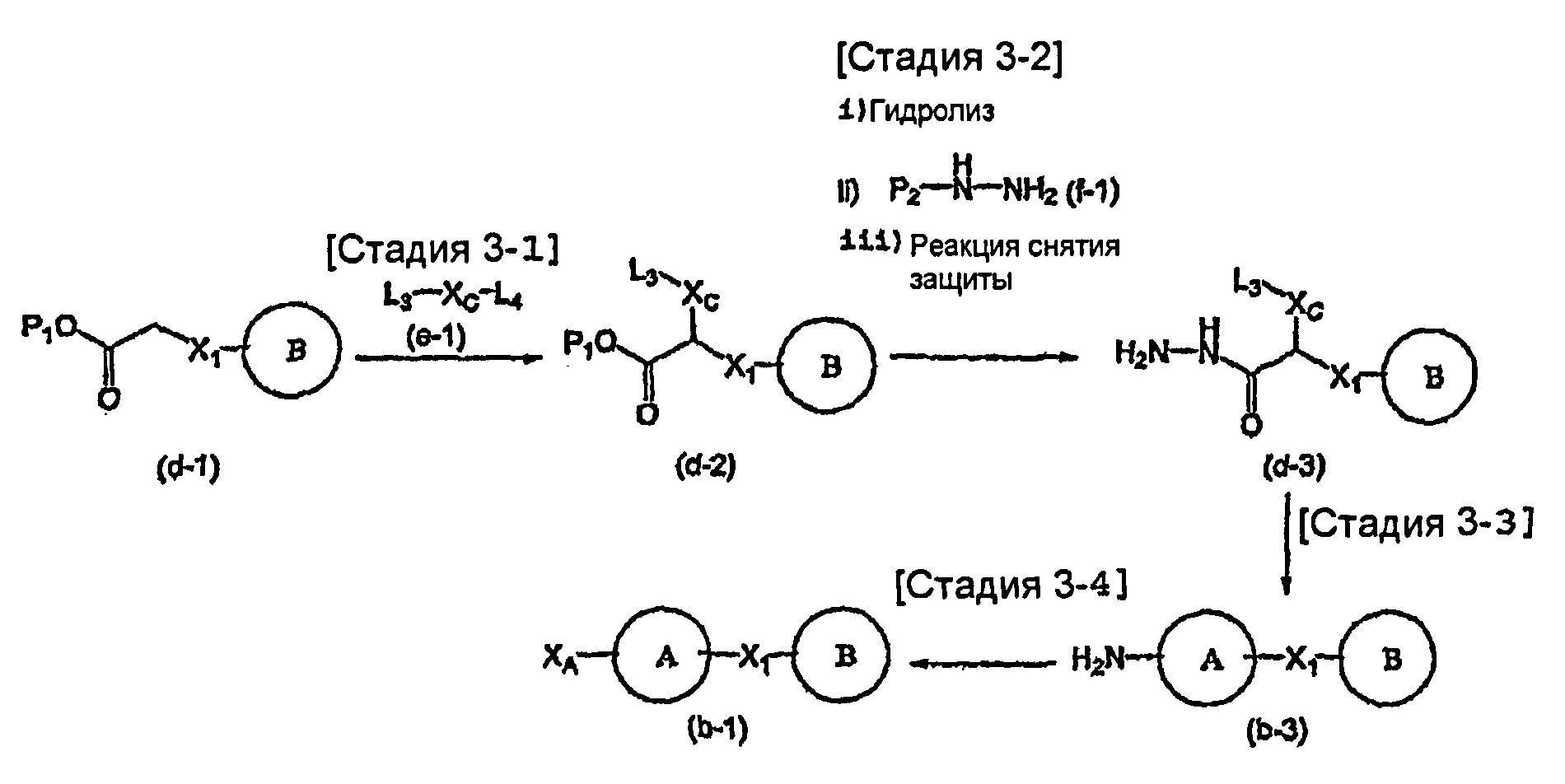
На схеме X1, XA, кольцо A и кольцо B такие, как определено выше; L3 и L4 такие, как определено выше для L1; XC представляет собой C2-4 алкиленовую группу или C2-3 алкиленовую группу, в которой одна метиленовая группа заменена атомом кислорода или атомом азота (где атом азота может содержать заместитель, такой как C1-6 алкильная группа или бензильная группа); P1 представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, аллильная группа, трифенилметильная группа, трет-бутильная группа или трет-бутилдиметилсилильная группа, или атом водорода; и P2 представляет собой азотзащитную группу, такую как трет-бутоксикарбонильная группа или бензилоксикарбонильная группа.
Соединение общей формулы (b-1) может быть получено из соединения общей формулы (d-1) как исходного материала через алкилирование на стадии 3-1, гидролиз сложного эфира, гидразидирование и реакцию снятия защиты на стадии 3-2, образование кольца A на стадии 3-3 и реакцию Сандмейера на стадии 3-4.
Стадия 3-1 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Предпочтительные примеры методики включают способ перемешивания соединения общей формулы (d-1) и 1,0-10,0 эквивалентов соединения общей формулы (e-1) относительно соединения общей формулы (d-1) в растворителе в присутствии 1,0-10,0 эквивалентов основания относительно соединения общей формулы (d-1). Используемое основание изменяют в соответствии с исходным материалом и особо не ограничивают. Предпочтительные примеры основания включают гидриды щелочных металлов (такие как гидрид натрия и гидрид лития), соли щелочных металлов (такие как карбонат калия, карбонат натрия и карбонат цезия), алкоксиды металлов (такие как метоксид натрия и трет-бутоксид калия) и металлоорганические основания (такие как бутиллитий, диизопропиламид лития и бистриметилсилиламид лития). Используемый растворитель меняют в соответствии с исходным материалом и особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают растворители в виде простых эфиров, таких как тетрагидрофуран, 1,4-диоксан и диэтиловый эфир; галогенированные растворители, такие как метиленхлорид, 1,2-дихлорэтан и хлороформ; полярные растворители, такие как N,N-диметилформамид и N-метилпирролидон; неполярные растворители, такие как толуол и бензол; и смеси указанных растворителей. Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и предпочтительно находится в интервале от -100°C до 100°C, например. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии. Нежелательный побочный продукт может быть удален методом, известным специалисту в данной области техники, таким как традиционный метод хроматографии, экстракция и/или кристаллизация.
Реакция гидролиза сложного эфира как первый этап стадии 3-2 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Реакция снятия защиты, известная специалисту в данной области техники, может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (см., например, T. Greene et al., "Protective Groups in Organic Synthesis", John Wiley & Sons, Inc., New York, 1981). Реакция гидразидирования как второй этап изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Реакция амидирования, известная специалисту в данной области техники, может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в The Chemical Society of Japan (ed.), Jikken Kagaku Koza (Courses in Experimental Chemistry), 4th edition (vol. 22) Yuki Gosei (Organic Synthesis) [IV], Maruzen Co., Ltd., November 1992, p.137-144). Реакция снятия защиты как третий этап изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Реакция снятия защиты, известная специалисту в данной области техники, может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (см., например, T. Greene et al., "Protective Groups in Organic Synthesis", John Wiley & Sons, Inc., New York, 1981).
Реакция образования кольца A на стадии 3-3 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Предпочтительные примеры методики включают способ нагревания соединения общей формулы (d-3) и 1,0-10,0 эквивалентов аминогуанидина, изотиомочевины, цианамида или тому подобного относительно соединения общей формулы (d-3) в растворителе в щелочных или кислых условиях. Используемые основание или кислоту изменяют в соответствии с исходным материалом и особо не ограничивают. Примеры основания или кислоты включают основания, такие как гидриды щелочных металлов (такие как гидрид натрия и гидрид лития), соли щелочных металлов (такие как карбонат калия, карбонат натрия и карбонат цезия), алкоксиды металлов (такие как метоксид натрия и трет-бутоксид калия) и органические основания (такие как триэтиламин, пиридин и 1,8-диазабицикло[5.4.0]ундец-7-ен); и кислоты, такие как хлористоводородная кислота, серная кислота, п-толуолсульфоновая кислота и камфорсульфоновая кислота. Используемый растворитель меняют в соответствии с исходным материалом и особо не ограничивают, лишь бы растворитель не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают спиртовые растворители, такие как метанол, этанол и трет-бутанол; растворители в виде простых эфиров, таких как тетрагидрофуран, 1,4-диоксан и диэтиловый эфир; галогенированные растворители, такие как метиленхлорид, 1,2-дихлорэтан и хлороформ; полярные растворители, такие как ацетонитрил, N,N-диметилформамид и N-метилпирролидон; неполярные растворители, такие как ксилол, толуол и бензол; и смеси указанных растворителей. Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и предпочтительно находится в интервале от -100°C до 100°C, например. В предпочтительных условиях реакции реакция заканчивается за 1-48 часов, и ход реакции может быть проконтролирован известным методом хроматографии. Нежелательный побочный продукт может быть удален методом, известным специалисту в данной области техники, таким как традиционный метод хроматографии, экстракция и/или кристаллизация.
Реакция Сандмейера на стадии 3-4 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в The Chemical Society of Japan (ed.), Jikken Kagaku Koza (Courses in Experimental Chemistry), 4th edition (vol. 19) Yuki Gosei (Organic Synthesis) [I], Maruzen Co., Ltd., November 1992, p.450-453).
Соединение формулы (d-1), соединение формулы (e-1) и соединение формулы (f-1) являются известными или коммерчески доступными соединениями или являются соединениями, которые могут быть получены из указанных соединений традиционным способом.
Общий способ получения 2
Ниже описан обычно используемый общий способ получения 2 для соединения общей формулы [I] по настоящему изобретению.
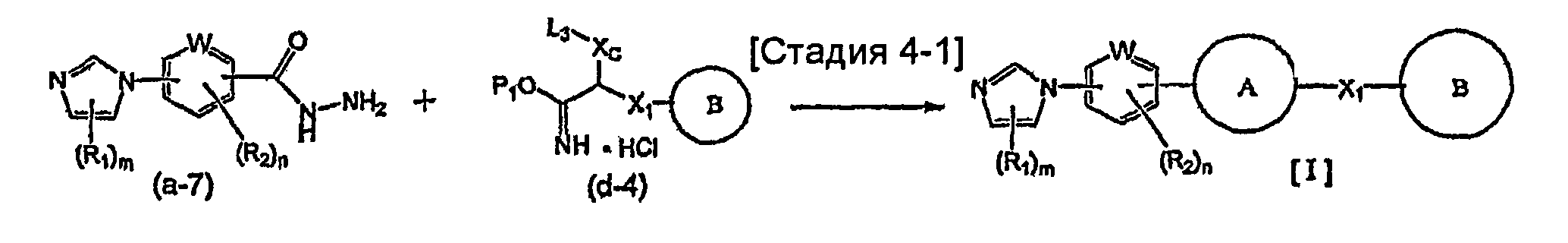
На схеме R1, R2, X1, XC, P1, L3, m, n, W, кольцо A и кольцо B такие, как определено выше.
Указанный выше общий способ получения 2 является примером способа получения соединения общей формулы [I] подверганием соединения общей формулы (a-7) и соединения общей формулы (d-4) реакции циклизации на стадии 4-1.
Реакция образования кольца A на стадии 4-1 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Предпочтительные примеры методики включают способ перемешивания соединения общей формулы (a-7) и 1,0-5,0 эквивалентов соединения общей формулы (d-4) относительно соединения общей формулы (a-7) в растворителе в присутствии 1,0-10,0 эквивалентов основания относительно соединения общей формулы (a-7). Из-за удобства и эффективности перемешивания данную реакцию предпочтительно осуществляют в присутствии растворителя. Используемый растворитель меняют в соответствии с исходным материалом и особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают спиртовые растворители, такие как метанол, этанол и трет-бутанол; растворители в виде простых эфиров, таких как тетрагидрофуран, 1,4-диоксан и диэтиловый эфир; галогенированные растворители, такие как метиленхлорид, 1,2-дихлорэтан и хлороформ; полярные растворители, такие как ацетонитрил, пропионитрил, N,N-диметилформамид и N-метилпирролидон; неполярные растворители, такие как толуол и бензол; и смеси указанных растворителей. Используемое основание изменяют в соответствии с исходным материалом и особо не ограничивают. Предпочтительные примеры основания включают гидриды щелочных металлов (такие как гидрид натрия и гидрид лития), соли щелочных металлов (такие как карбонат калия, карбонат натрия и карбонат цезия), алкоксиды металлов (такие как метоксид натрия и трет-бутоксид калия) и органические основания (такие как триэтиламин, N,N-диизопропилэтиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен и имидазол). Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и предпочтительно находится в интервале от комнатной температуры до 200°C, например. В предпочтительных условиях реакции реакция заканчивается за 1-7 суток, и ход реакции может быть проконтролирован известным методом хроматографии. Нежелательный побочный продукт может быть удален методом, известным специалисту в данной области техники, таким как традиционный метод хроматографии, экстракция и/или кристаллизация.
Получение соединения общей формулы (a-7)
На следующей схеме показан пример получения соединения общей формулы (a-7).
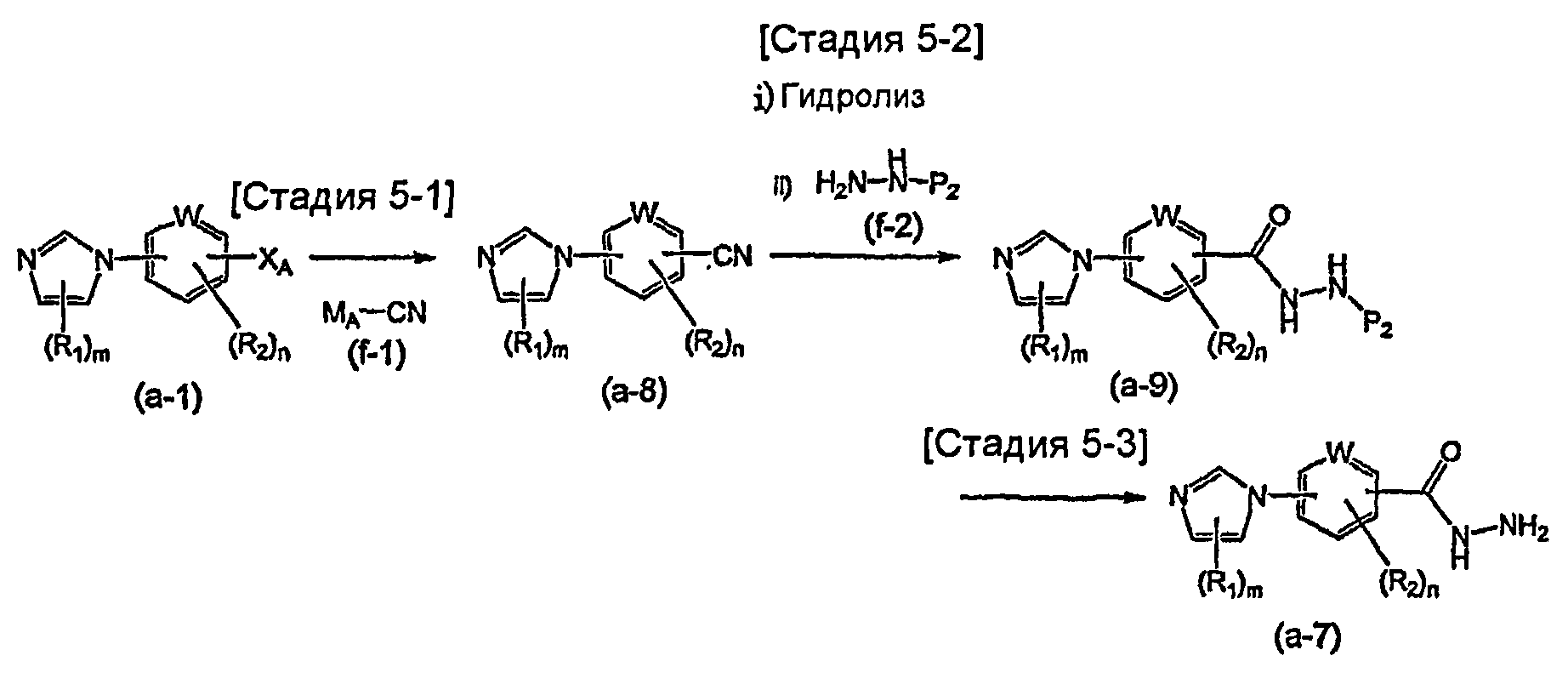
На схеме R1, R2, m, n, XA, W и P2 такие, как определено выше; и MA представляет собой металл, такой как цинк или медь.
Соединение общей формулы (a-7) может быть получено из соединения общей формулы (a-1) как исходного материала через реакцию связывания на стадии 5-1, реакцию гидролиза и гидразидирование на стадии 5-2 и реакцию снятия защиты на стадии 5-3.
Реакция связывания на стадии 5-1 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Галогенсодержащее соединение или трифторметансульфонатное соединение общей формулы (a-1) предпочтительно связывают с 1,0-5,0 эквивалентами цианида металла, такого как цианид цинка(II), представленный общей формулой (f-1), относительно соединения общей формулы (a-1) в присутствии 0,01-0,2 эквивалента катализатора на основе переходного металла относительно соединения общей формулы (a-1), например. Из-за удобства и эффективности перемешивания данную реакцию предпочтительно осуществляют в присутствии растворителя. Используемый растворитель меняют в соответствии с исходным материалом и используемым катализатором на основе переходного металла и особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают ацетонитрил, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол, 1-метил-2-пирролидон и N,N-диметилформамид. Температура реакции должна быть такой, которая позволяет закончить реакцию связывания, и предпочтительно находится в интервале от комнатной температуры до 150°C. Данную реакцию проводят предпочтительно в атмосфере инертного газа и более предпочтительно в атмосфере азота или аргона. Катализатором на основе переходного металла, например, является предпочтительно палладиевый комплекс и более предпочтительно известный палладиевый комплекс, такой как ацетат палладия(II), дихлорбис(трифенилфосфин)палладий(II), тетракис(трифенилфосфин)палладий(0) или трис(дибензилиденацетон)дипалладий(0). Является также предпочтительным подходящим образом добавлять фосфорный лиганд (предпочтительно трифенилфосфин, три-о-толилфосфин, три-трет-бутилфосфин или 2-(ди-трет-бутилфосфино)бифенил, например), чтобы реакция протекала эффективно. Предпочтительный результат может быть получен в присутствии основания. Используемое основание особо не ограничивают, лишь бы оно было применяемым в реакции связывания, подобной данной реакции. Предпочтительные примеры основания включают триэтиламин, N,N-диизопропилэтиламин, N,N-дициклогексилметиламин и тетрабутиламмонийхлорид. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии.
Реакция гидролиза как первый этап стадии 5-2 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах (такой как описанный в The Chemical Society of Japan (ed.), Jikken Kagaku Koza (Courses in Experimental Chemistry), 4th edition (vol. 22) Yuki Gosei (Organic Synthesis) [IV], Maruzen Co., Ltd., November 1992, p.12-13). Реакция гидразидирования как второй этап изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Реакция амидирования, известная специалисту в данной области техники, может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в The Chemical Society of Japan (ed.), Jikken Kagaku Koza (Courses in Experimental Chemistry), 4th edition (vol. 22) Yuki Gosei (Organic Synthesis) [IV], Maruzen Co., Ltd., November 1992, p.137-144).
Реакция снятия защиты на стадии 5-3 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Реакция снятия защиты, известная специалисту в данной области техники, может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (см., например, T. Greene et al., "Protective Groups in Organic Synthesis", John Wiley & Sons, Inc., New York, 1981).
Соединение формулы (f-2) является известным или коммерчески доступным соединением или является соединением, которое может быть получено из таких соединений традиционным способом.
Получение соединения общей формулы (d-4)
На следующей схеме показан пример получения соединения общей формулы (d-4).
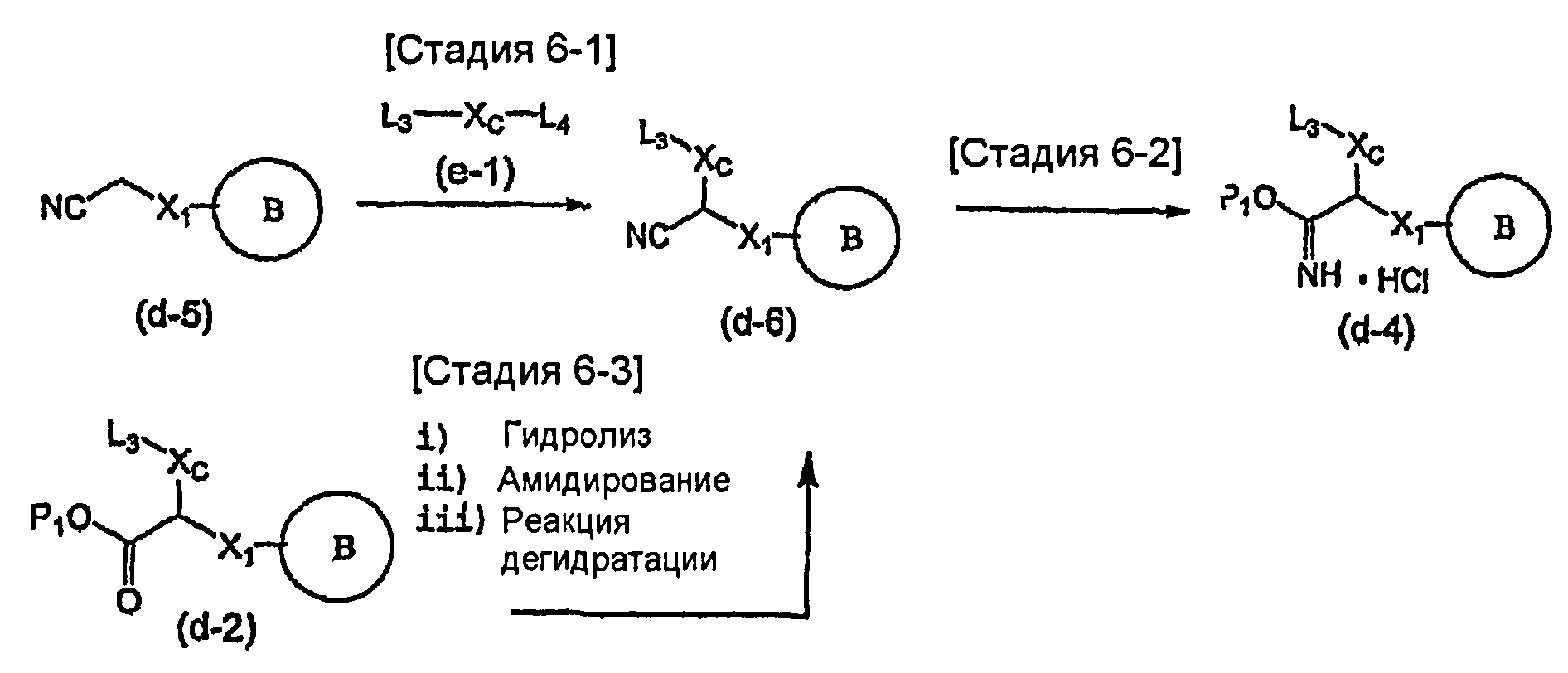
На схеме X1, XC, кольцо B, P1, L3 и L4 такие, как определено выше.
Соединение общей формулы (d-4) может быть получено из соединения общей формулы (d-5) как исходного материала через реакцию алкилирования на стадии 6-1 и имидирование на стадии 6-2.
Соединение общей формулы (d-6) может также быть получено из соединения общей формулы (d-2) как исходного материала через реакцию гидролиза, амидирование и реакцию дегидратации на стадии 6-3.
Стадию 6-1 осуществляют таким же способом, как на описанной выше стадии 3-1, с получением соединения общей формулы (d-6) из соединения общей формулы (d-5).
Стадия 6-2 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Предпочтительные примеры методики включают способ перемешивания соединения общей формулы (d-6) в спиртовом растворителе в присутствии 5,0-100,0 эквивалентов кислоты относительно соединения общей формулы (d-6). Используемую кислоту изменяют в соответствии с исходным материалом и особо не ограничивают. Предпочтительные примеры кислоты включают газообразный хлороводород и ацетилхлорид. Используемый растворитель меняют в соответствии с исходным материалом и особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают спиртовые растворители, такие как метанол, этанол и трет-бутанол. Предпочтительные примеры растворителя также включают галогенированные растворители, такие как метиленхлорид, 1,2-дихлорэтан и хлороформ; полярные растворители, такие как N,N-диметилформамид и N-метилпирролидон; неполярные растворители, такие как толуол и бензол; и состоящие из них смешанные растворители. Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и находится в интервале предпочтительно от 0°C до 100°C, например. В предпочтительных условиях реакции реакция заканчивается за 1-7 суток, и ход реакции может быть проконтролирован известным методом хроматографии. Нежелательный побочный продукт может быть удален методом, известным специалисту в данной области техники, таким как традиционный метод хроматографии, экстракция и/или кристаллизация.
Гидролиз сложного эфира как первый этап стадии 6-3 осуществляют таким же способом, как на описанной выше стадии 3-2. Реакцию амидирования как второй этап особо не ограничивают, пока условия подобны условиям в данной реакции. Реакция амидирования, известная специалисту в данной области техники, может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в The Chemical Society of Japan (ed.), Jikken Kagaku Koza (Courses in Experimental Chemistry), 4th edition (vol. 22) Yuki Gosei (Organic Synthesis) [IV], Maruzen Co., Ltd., November 1992, p.137-144). Реакция дегидратации от амида до нитрила как третий этап изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Реакция дегидратации известным специалисту в данной области техники способом может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в The Chemical Society of Japan (ed.), Jikken Kagaku Koza (Courses in Experimental Chemistry), 4th edition (vol. 20) Yuki Gosei (Organic Synthesis) [IV], Maruzen Co., Ltd., November 1992, p.449-450).
Соединение формулы (d-2) и соединение формулы (d-5) являются известными или коммерчески доступными соединениями или являются соединениями, которые могут быть получены из указанных соединений традиционным способом.
Общий способ получения 3
Ниже описывается обычно используемый общий способ получения 3 для соединения общей формулы (I) по настоящему изобретению.
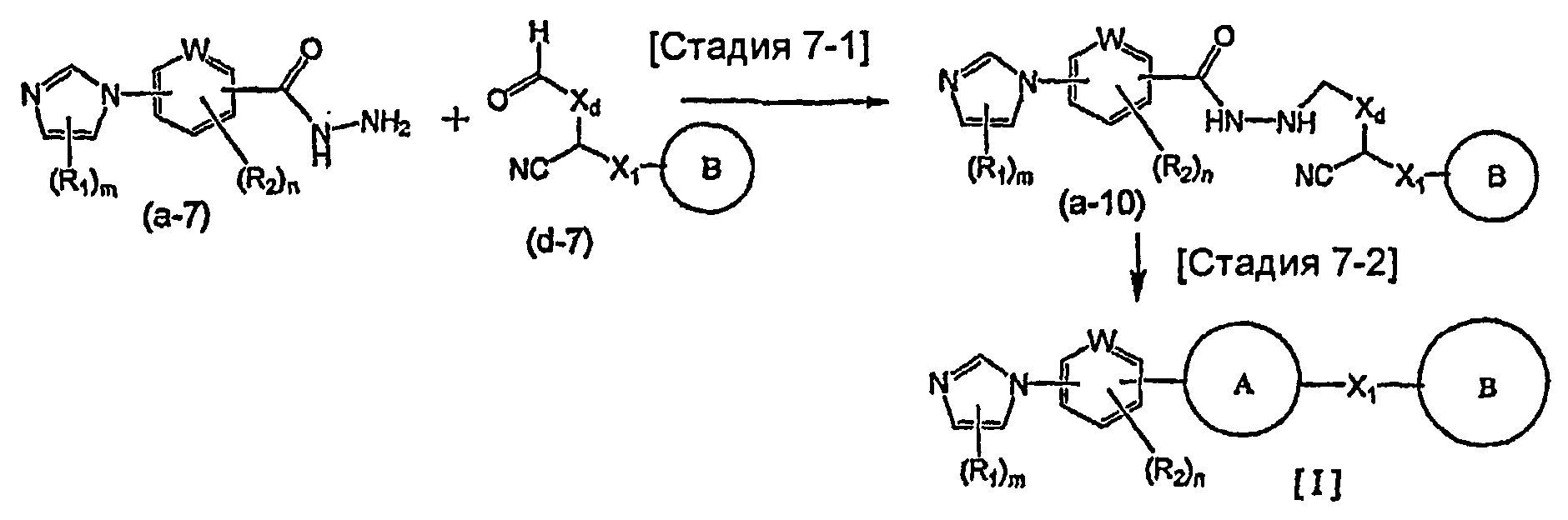
На схеме R1, R2, m, n, W, кольцо A, X1 и кольцо B такие, как определено выше; и Xd представляет собой C1-3 алкиленовую группу или C1-2 алкиленовую группу, в которой одна метиленовая группа заменена атомом кислорода или атомом азота (где атом азота может содержать заместитель, такой как C1-6 алкильная группа или бензильная группа).
Указанный выше общий способ получения 3 является примером способа получения соединения [I] конденсированием гидразидного соединения (a-7) и альдегидного соединения (d-7) на стадии 7-1 и затем осуществлением внутримолекулярной циклизации на стадии 7-2.
Хотя соединение (a-10) может быть получено алкилированием гидразидного соединения (a-7) и соединения (d-6), но трудно контролировать положения и число алкилированных мест. Соединение (a-10) предпочтительно получают конденсированием гидразидного соединения (a-7) с альдегидным соединением (d-7).
Реакция конденсации на стадии 7-1 изменяется в соответствии с материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Предпочтительные примеры методики включают способ с использованием боргидридного реагента и способ восстановительного алкилирования с использованием каталитического восстановления.
В способе с использованием боргидридного реагента является предпочтительным осуществлять взаимодействие гидразидного соединения (a-7) и 1,0-1,5 эквивалента альдегидного соединения (d-7) относительно соединения (a-7) с 1,0-4,0 эквивалентами триацетоксигидробората натрия относительно соединения (a-7) в растворителе в виде простого эфира, таком как тетрагидрофуран или диоксан, галогенированном растворителе, таком как дихлорметан, или состоящем из них смешанном растворителе в присутствии 2,0-6,0 эквивалентов уксусной кислоты относительно соединения (a-7), например. Температура реакции находится в интервале предпочтительно от 0°C до комнатной температуры.
Является также предпочтительным осуществлять взаимодействие гидразидного соединения (a-7) и 1,0-1,5 эквивалентов альдегидного соединения (d-7) относительно соединения (a-7) с 1,0-4,0 эквивалентами цианотригидробората натрия относительно соединения (a-7) в спиртовом растворителе, таком как метанол или этанол, растворителе в виде простого эфира, таком как тетрагидрофуран или диоксан, или состоящем из них смешанном растворителе, например. При необходимости может быть добавлен кислотный катализатор, такой как уксусная кислота. Температура реакции находится в интервале предпочтительно от 0°C до комнатной температуры.
Является также предпочтительным использовать способ подвергания гидразидного соединения (a-7) и альдегидного соединения (d-7) реакции дегидратации в присутствии кислоты, такой как уксусная кислота или п-толуолсульфоновая кислота, с образованием имина и затем восстановления имина боргидридным реагентом.
При восстановительном алкилировании с использованием каталитического восстановления является предпочтительным каталитически восстанавливать гидразидное соединение (a-7) и 1,0-1,5 эквивалента альдегидного соединения (d-7) относительно соединения (a-7) в спиртовом растворителе, таком как метанол или этанол, растворителе в виде простого эфира, таком как тетрагидрофуран или диоксан, или состоящем из них смешанном растворителе в присутствии катализатора, такого как оксид платины или палладий-на-углероде, в атмосфере водорода при 1-4 атм., например. При необходимости может быть добавлен кислотный катализатор, такой как уксусная кислота или хлористоводородная кислота.
Является также предпочтительным использовать способ подвергания гидразидного соединения (a-7) и альдегидного соединения (d-7) реакции дегидратации в присутствии кислоты, такой как уксусная кислота или п-толуолсульфоновая кислота, с образованием имина и затем восстановительного алкилирования имина с использованием каталитического восстановления.
Стадия 7-2 является реакцией внутримолекулярной циклизации и является примером образования при этом триазольного кольца, конденсированного с неароматической кольцевой группой, например. Данная реакция изменяется в соответствии с материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Растворитель для реакции особо не ограничивают, лишь бы он не замедлял реакцию. Примеры растворителя включают ароматические углеводородные растворители, такие как растворитель в виде уксусной кислоты, толуол и ксилол; растворители в виде простых эфиров, такие как тетрагидрофуран и диоксан; спиртовые растворители, такие как метанол и этанол; и состоящие из них смешанные растворители. При необходимости могут быть добавлены кислоты, такие как уксусная кислота, п-толуолсульфоновая кислота или хлористоводородная кислота. Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и предпочтительно находится в интервале от комнатной температуры до температуры кипения растворителя, например. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии. Нежелательный побочный продукт может быть удален методом, известным специалисту в данной области техники, таким как традиционный метод хроматографии, экстракция и/или кристаллизация.
Получение соединения общей формулы (d-7)
На следующей схеме показан пример получения соединения общей формулы (d-7).
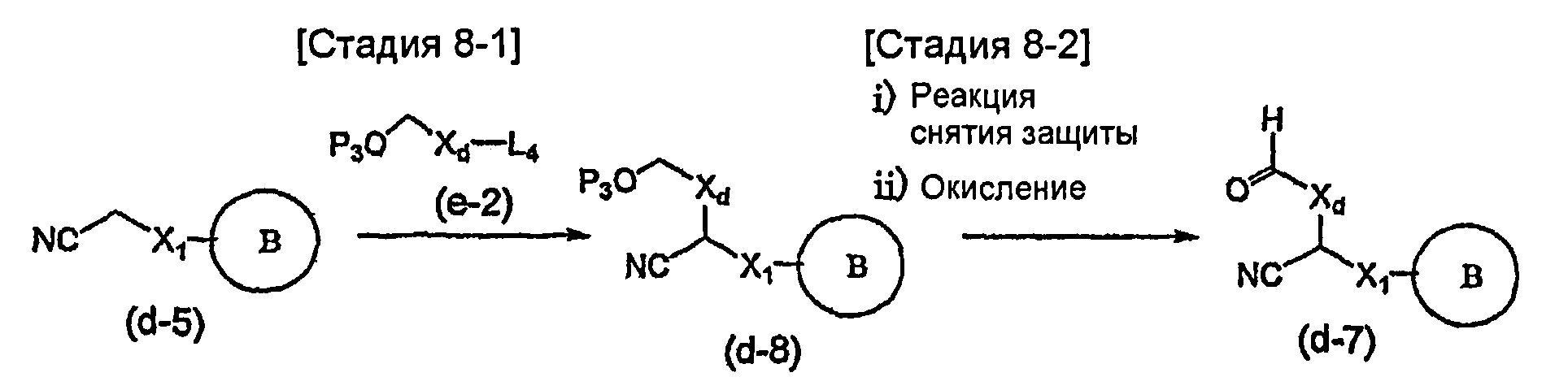
На схеме X1, L4, Xd и кольцо B такие, как определено выше; и P3 представляет собой спиртзащитную группу, такую как трет-бутилдиметилсилильная группа или бензильная группа.
Альдегидное соединение (d-7) может быть получено из нитрильного соединения (d-5) как исходного материала через реакцию алкилирования на стадии 8-1 и реакцию снятия защиты и реакцию окисления спирта на стадии 8-2.
Стадию 8-1 осуществляют таким же способом, как на описанной выше стадии 3-1, с получением соединения общей формулы (d-8) из соединения общей формулы (d-5). Спиртзащитную группу P3 в соединении общей формулы (e-2) особо не ограничивают, лишь бы защитная группа была устойчивой в условиях реакции и могла быть легко удалена. Предпочтительные примеры ее включают трет-бутилдиметилсилильную группу и бензильную группу. Соединение (e-2) является коммерчески доступным или может быть легко синтезировано защитой спиртового соединения способом, известным специалисту в данной области техники (см., например, T. Greene et al., "Protective Groups in Organic Synthesis", John Wiley & Sons, Inc., New York, 1981).
Реакция снятия защиты как первый этап стадии 8-2 особо не ограничивается, пока условия подобны условиям в данной реакции. Реакция снятия защиты, известная специалисту в данной области техники, может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (см., например, T. Greene et al., "Protective Groups in Organic Synthesis", John Wiley & Sons, Inc., New York, 1981). Например, когда защитная группа представляет собой трет-бутилдиметилсилильную группу, является предпочтительным использовать кислоту, такую как хлористоводородная кислота, трифторуксусная кислота, муравьиная кислота или уксусная кислота, или использовать тетра-н-бутиламмонийфторид или тому подобное. Когда защитная группа представляет собой бензильную группу, является предпочтительным каталитический гидрогенолиз. Реакция окисления до альдегида как второй этап изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Реакция окисления, известная специалисту в данной области техники, может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в The Chemical Society of Japan (ed.), Jikken Kagaku Koza (Courses in Experimental Chemistry), 4th edition (vol. 21) Yuki Gosei (Organic Synthesis) [IV], Maruzen Co., Ltd., November 1992, p.2-23). Предпочтительные примеры методики включают окисление с использованием хлорхромата пиридиния, окисление по Сверну, окисление по Пфитцнеру-Моффатту и окисление по Дессу-Мартину.
Получение соединения общей формулы (d-8)
На следующей схеме показан пример получения соединения общей формулы (d-8).
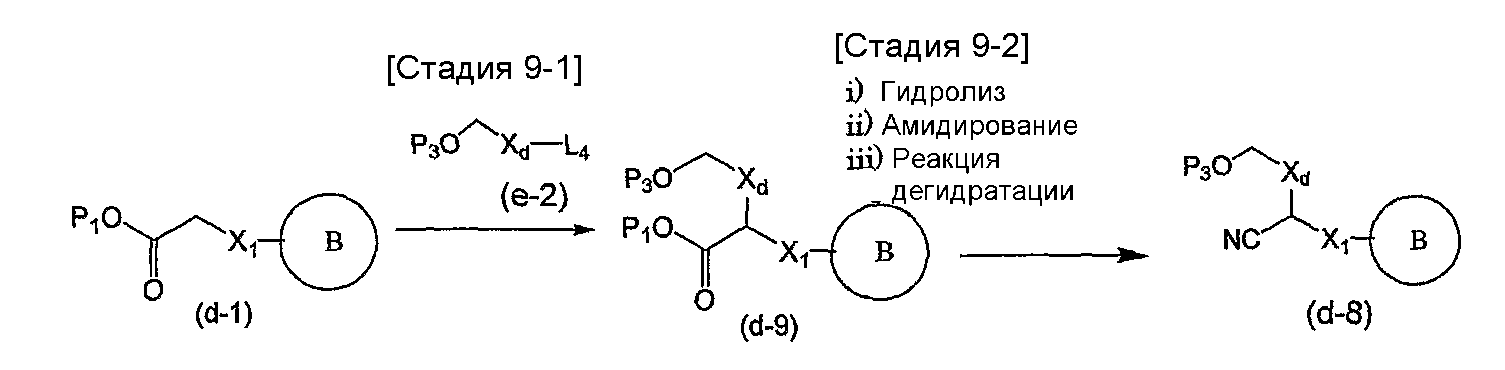
На схеме X1, Xd, L4, P1, P3 и кольцо B такие, как определено выше.
Нитрильное соединение (d-8) может быть получено из соединения (d-1) как исходного материала через алкилирование на стадии 9-1 и гидролиз сложного эфира, амидирование и реакцию дегидратации на стадии 9-2.
Стадию 9-1 осуществляют таким же способом, как описанную выше стадию 3-1, с получением соединения (d-9) из соединения (d-1).
Стадию 9-2 осуществляют таким же способом, как описанную выше стадию 6-3, с получением нитрильного соединения (d-8) из соединения (d-9).
Общий способ получения 4
Ниже описывается обычно используемый общий способ получения 4 для соединения общей формулы (I) по настоящему изобретению.
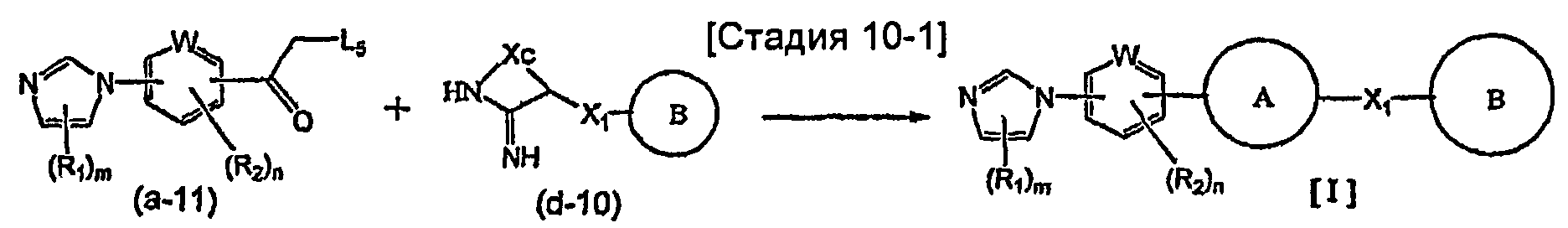
На схеме R1, R2, m, n, W, Xc, кольцо A, X1 и кольцо B такие, как определено выше; и L5 представляет собой атом галогена, такой как хлор, бром или йод.
Указанный выше общий способ получения 4 является примером способа получения соединения [I] осуществлением взаимодействия соединения общей формулы (a-11) с циклическим амидиновым соединением (d-10).
Стадия 10-1 является примером образования имидазольного кольца, конденсированного с неароматической кольцевой группой, например, алкилированием и последующим осущуествлением реакции дегидратации циклического амидинового соединения (d-10). Алкилирование протекает региоселективно, в результате чего образуется отдельное кольцо A (как описано в L. Langlois et al., "J. Heterocyclic Chem.", 1982, vol. 19, p.193-200). Предпочтительные примеры методики включают способ осуществления взаимодействия соединения (a-11) с 1,0-2,0 эквивалентами соединения (d-10) относительно соединения (a-11) в растворителе. Растворитель особо не ограничивают, лишь бы он не замедлял реакцию. Примеры растворителя включают растворители в виде простых эфиров, такие как тетрагидрофуран и диоксан; спиртовые растворители, такие как метанол и этанол; воду; и состоящие из них смешанные растворители. Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и предпочтительно находится в интервале от комнатной температуры до температуры кипения растворителя, например. В предпочтительных условиях реакции реакция заканчивается за 1-24 часа, и ход реакции может быть проконтролирован известным методом хроматографии. Нежелательный побочный продукт может быть удален методом, известным специалисту в данной области техники, таким как традиционный метод хроматографии, экстракция и/или кристаллизация.
Получение соединения общей формулы (a-11)
На следующей схеме показан пример получения соединения общей формулы (a-11).
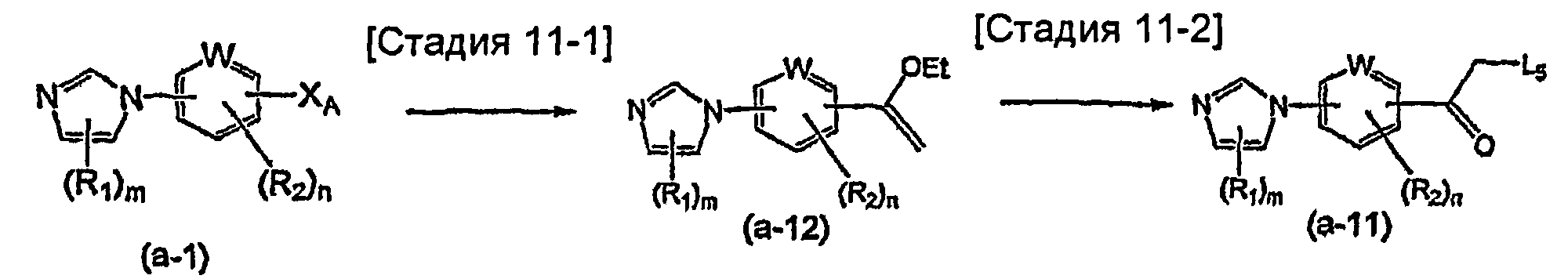
На схеме R1, R2, m, n, W, Xa и L5 такие, как определено выше.
Соединение общей формулы (a-11) может быть получено из соединения (a-1) как исходного материала через реакцию связывания на стадии 11-1 и галогенирование на стадии 11-2.
Реакция связывания на стадии 11-1 изменяется в соответствии с исходным материалом и особо не ограничивается, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Предпочтительные примеры методики включают способ осуществления взаимодействия соединения (a-1) с 1,0-1,2 эквивалента трибутил(1-этоксивинил)олова относительно соединения (a-1) в растворителе в присутствии палладиевого катализатора. Палладиевым катализатором является предпочтительно 0,02-0,1 эквивалента хлорида бис(трифенилфосфин)палладия(II) относительно соединения (a-1), например. Используемый растворитель меняют в соответствии с исходным материалом, реагентом и тому подобным и особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают неполярные растворители, такие как толуол и ксилол; растворители в виде простых эфиров, таких как тетрагидрофуран и диоксан; полярные растворители, такие как N,N-диметилформамид; и смеси указанных растворителей. Температура реакции предпочтительно находится в интервале от комнатной температуры до температуры кипения растворителя.
Стадия 11-2 является стадией превращения соединения в виде простого винилового эфира (a-12) в α-галогенкетоновое соединение (a-11). Данную реакцию изменяют в соответствии с исходным материалом и особо не ограничивают, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Например, предпочтительным является способ с использованием брома, йода, N-бромсукцинимида или тому подобного.
В способе с использованием N-бромсукцинимида является предпочтительным осуществлять взаимодействие соединения в виде простого винилового эфира (a-12) с 1,0-1,1 эквивалента N-бромсукцинимида относительно соединения (a-12) в растворителе в виде простого эфира, таком как тетрагидрофуран или диоксан, галогенированном растворителе, таком как дихлорметан, спиртовом растворителе, таком как метанол или этанол, состоящем из них смешанном растворителе или смешанном растворителе, состоящем из указанных растворителей и воды, например. Температура реакции находится в интервале предпочтительно от 0°C до комнатной температуры. Соединение (a-11) может быть взято в виде соли или получено перед применением и использовано как есть способом, известным специалисту в данной области техники.
Получение соединения общей формулы (d-10)
На следующей схеме показан пример получения соединения общей формулы (d-10).

На схеме P1, L3, Xc, X1 и кольцо B такие, как определено выше.
Циклическое амидиновое соединение (d-10) может быть получено из соединения (d-4) как исходного материала через амидинирование и затем осуществление реакции циклизации на стадии 12-1.
Образование циклического амидина на стадии 12-1 особо не ограничивают, пока условия подобны условиям в данной реакции. Примеры его включают способ перемешивания соединения в виде имидата (d-4) в насыщенном спиртовом растворе аммония. Температура реакции предпочтительно является комнатной температурой. Ход реакции может быть проконтролирован ЖХ-МС.
Общий способ получения 5
Ниже описывается обычно используемый общий способ получения 5 для соединения общей формулы (I) по настоящему изобретению.
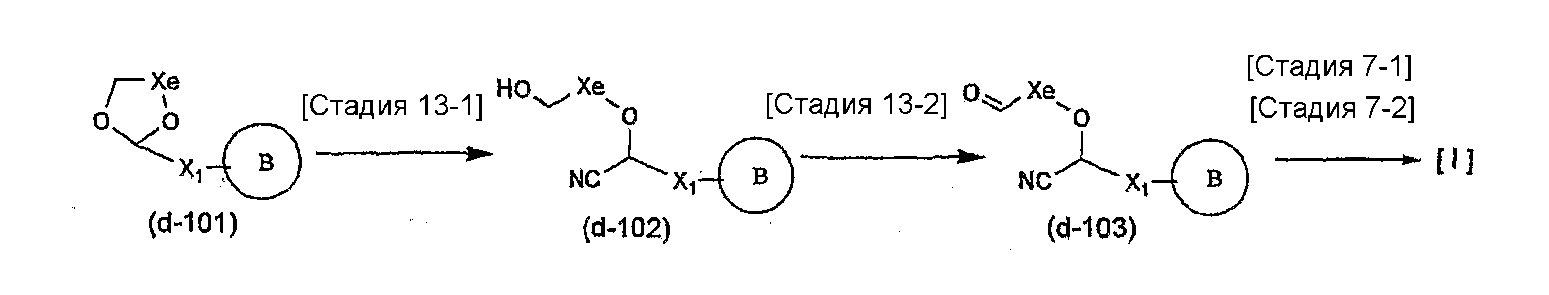
На схеме X1 и кольцо B такие, как определено выше; и Xe представляет собой C1-2 алкиленовую группу.
Указанный выше общий способ получения 5 представляет собой общий способ получения соединения общей формулы (I), содержащего атом кислорода в β-положении X1.
Соединение общей формулы (I) может быть получено подверганием ацетального соединения (d-101) как исходного материала следующему за циангидринированием расщеплению ацеталя на стадии 13-1 и осуществлением реакции окисления на стадии 13-2 и подверганием полученного соединения общей формулы (d-103) реакциям, проводимым на стадиях 7-1 и 7-2 вышеописанного общего способа получения 3.
Следующее за циангидринированием расщепление ацеталя на стадии 13-1 изменяют в соответствии с исходным материалом и особо не ограничивают, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в Synthesis, p.498, 1983; Tetrahedron Lett., vol. 31, p.5343, 1990). Предпочтительно ацетальное соединение (d-101) подвергают взаимодействию с 1,0-2,0 эквивалентами триметилсилилцианида относительно соединения (d-101) в растворителе или без растворителя в присутствии 0,01-0,5 эквивалента кислоты Льюиса относительно соединения (d-101) в безводных условиях, например. Примеры используемой кислоты Льюиса включают иодид цинка(II), тетрахлорид титана, тетрахлорид олова, хлорид железа(III), хлорид цинка(II), и бромид цинка(II). Используемый растворитель меняют в соответствии с исходным материалом и особо не ограничивают, лишь бы растворитель не замедлял реакцию и позволял исходному материалу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают галогенированные растворители, такие как метиленхлорид, 1,2-дихлорэтан и хлороформ; полярные растворители, такие как N,N-диметилформамид, нитрометан и N-метилпирролидон; неполярные растворители, такие как толуол и бензол; растворители в виде простых эфиров, такие как тетрагидрофуран, 1,4-диоксан и диэтиловый эфир; и смеси указанных растворителей. Температура реакции должна быть такой, которая позволяет закончить реакцию, не способствуя образованию нежелательного побочного продукта, и находится в интервале предпочтительно от 0°C до температуры кипения растворителя, например. Продукт в реакционном растворе представляет собой триметилсилиловый эфир спирта и его превращают в спирт последующей обработкой водой.
Стадию 13-2 осуществляют таким же способом, как в реакции окисления как второго этапа вышеописанной стадии 8-2, с получением соединения общей формулы (d-103) из спиртового соединения (d-102).
Соединение общей формулы (I) может быть получено подверганием альдегидного соединения (d-103) реакциям, проводимым на стадиях 7-1 и 7-2 вышеописанного общего способа получения 3.
Соединение (d-101) является известным или коммерчески доступным соединением или является соединением, которое может быть получено из таких соединений традиционным способом.
Общий способ получения 6
Ниже описывается обычно используемый общий способ получения 6 для соединения общей формулы (I) по настоящему изобретению.
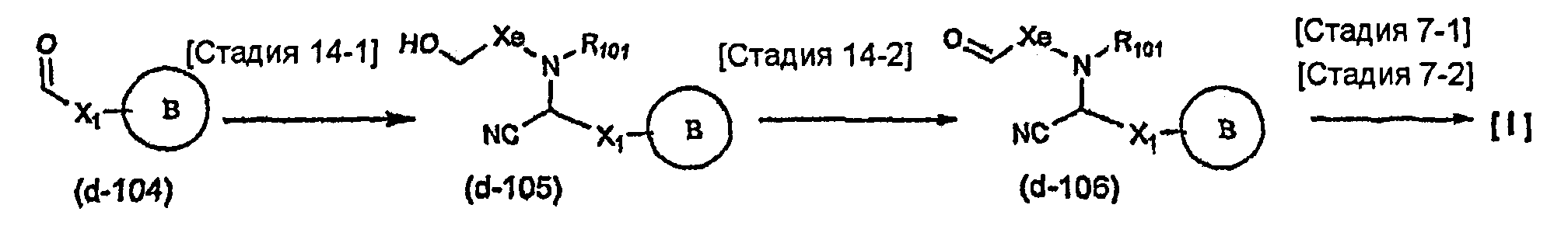
На схеме X1 и кольцо B такие, как определено выше; R101 представляет собой заместитель для азота (такой как метильная группа, изопропильная группа, фенильная группа или бензильная группа); и Xe представляет собой C1-2 алкиленовую группу.
Указанный выше общий способ получения 6 является общим способом получения соединения общей формулы (I), содержащего атом азота в β-положении X1.
Общий способ получения 6 является примером способа получения соединения общей формулы (I) подверганием альдегидного соединения (d-104) как исходного материала образованию аминонитрила на стадии 14-1 и реакции окисления на стадии 14-2 и подверганием полученного соединения общей формулы (d-106) реакциям, проводимым на стадиях 7-1 и 7-2 вышеописанного общего способа получения 3.
Образование аминонитрила на стадии 14-1 изменяют в соответствии с исходным материалом и особо не ограничивают, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в Tetrahedron Lett., vol. 48, p. 8001, 2007; Synthesis, p.109, 1983).
Стадию 14-2 осуществляют таким же способом, как в реакции окисления как второго этапа вышеописанной стадии 8-2 с получением соединения общей формулы (d-106) из спиртового соединения (d-105).
Соединение [I] может быть получено подверганием альдегидного соединения (d-106) реакциям, проводимым на стадиях 7-1 и 7-2 вышеописанного общего способа получения 3.
Соединение (d-104) является известным или коммерчески доступным соединением или является соединением, которое может быть получено из таких соединений традиционным способом.
Общий способ получения 7
Ниже описывается обычно используемый общий способ получения 7 для соединения общей формулы (I) по настоящему изобретению.
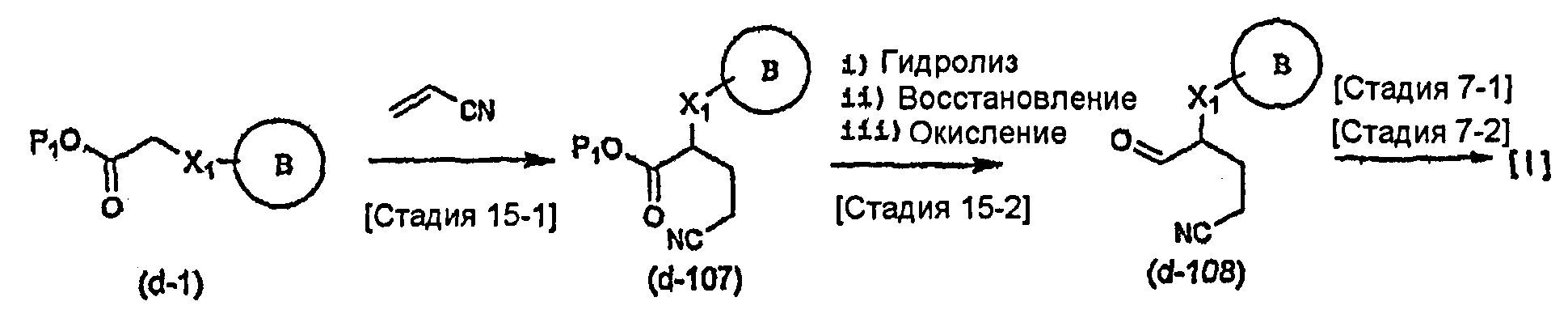
На схеме X1, P1 и кольцо B такие, как определено выше.
Указанный выше общий способ получения 7 является примером общего способа получения соединения общей формулы (I), имеющего положение присоединения X1, отличное от положение присоединения X1 соединения общей формулы (I), произведенного от соединения общей формулы (d-7).
Общий способ получения 7 является примером способа получения соединения общей формулы (I) подверганием соединения в виде сложного эфира (d-1) как исходного материала реакции присоединения по Михаэлю на стадии 15-1 и превращением из сложного эфира в альдегид на стадии 15-2 и подверганием полученного соединения общей формулы (d-108) реакциям, проводимым на стадиях 7-1 и 7-2 вышеописанного общего способа получения 3.
Реакцию присоединения по Михаэлю на стадии 15-1 изменяют в соответствии с исходным материалом и особо не ограничивают, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в "Modern Synthetic Reactions House 2nd edition" (W.A. Benjamin, Inc., California, 1972) p.595-623; J. Med. Chem., vol. 36, p.2416, 1993).
Превращение из сложного эфира в альдегид на стадии 15-2 осуществляют гидролизом сложного эфира, восстановлением карбоновой кислоты до спирта и окислением спирта до альдегида. Гидролиз сложного эфира как первый этап осуществляют таким же способом, как на первом этапе вышеописанной стадии 3-2. Восстановление как второй этап особо не ограничивают, пока условия подобны условиям в данной реакции. Реакция восстановления, известная специалисту в данной области техники, может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в The Chemical Society of Japan (ed.), Jikken Kagaku Koza (Courses in Experimental Chemistry), 4th edition (vol. 20) Yuki Gosei (Organic Synthesis) [IV], Maruzen Co., Ltd., November 1992, p.10-14); предпочтительным является борановое восстановление. Окисление как третий этап осуществляют таким же способом, как окисление на втором этапе вышеописанной стадии 8-2, с получением соединения общей формулы (d-108) из сложноэфирного соединения (d-107).
Соединение [I] может быть получено подверганием альдегидного соединения (d-108) реакциям, проводимым на стадиях 7-1 и 7-2 вышеописанного общего способа получения 3.
Общий способ получения 8
Ниже описывается обычно используемый общий способ получения 8 для соединения общей формулы (I) по настоящему изобретению.
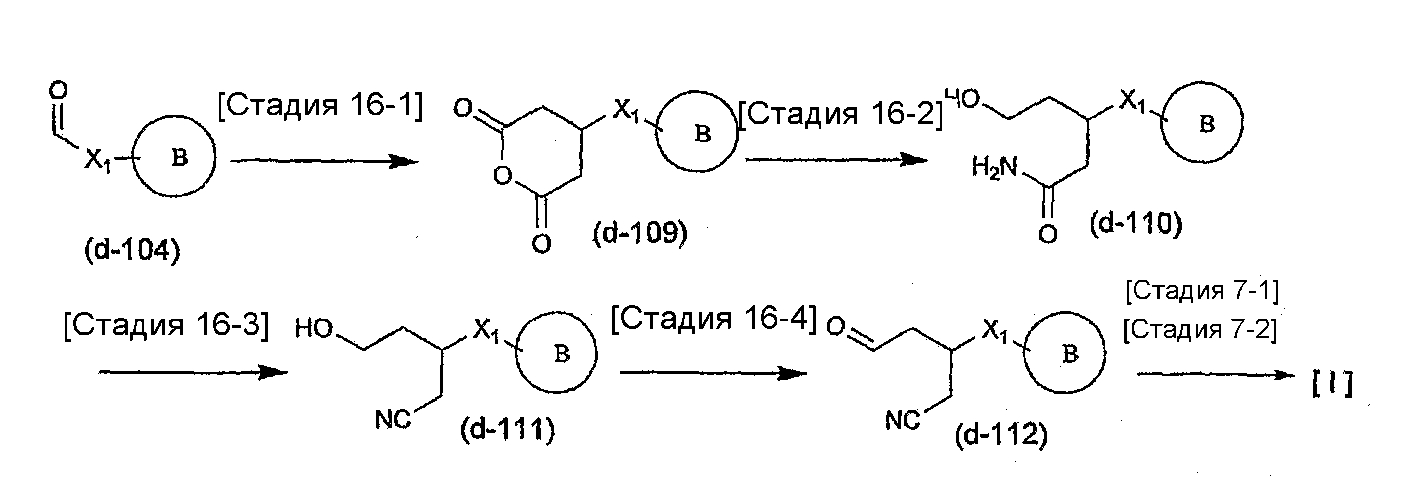
На схеме X1, P1 и кольцо B такие, как определено выше.
Указанный выше общий способ получения 8 является примером общего способа получения соединения общей формулы (I), имеющего положение присоединения X1, отличное от положение присоединения X1 соединения общей формулы (I), произведенного от соединения общей формулы (d-7).
Общий способ получения 8 является примером способа получения соединения общей формулы (I) подверганием альдегидного соединения (d-104) как исходного материала превращению в глутаровый ангидрид на стадии 16-1, превращению в амидоспирт на стадии 16-2, реакции дегидратации амида на стадии 16-3 и реакции окисления до альдегида на стадии 16-4 и подверганием полученного соединения общей формулы (d-112) реакциям, проводимым на стадиях 7-1 и 7-2 вышеописанного общего способа получения 3.
Превращение в глутаровый ангидрид на стадии 16-1 изменяют в соответствии с исходным материалом и особо не ограничивают, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в Tetrahedron Asymmetry, vol. 16, p.2475, 2005).
Стадия 16-2 включает расшепление глутарового ангидрида аммонием и затем реакцию восстановления карбоновой кислоты до спирта. Расшепление глутарового ангидрида аммонием как первый этап изменяют в соответствии с исходным материалом и особо не ограничивают, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах, или тому подобное (такой как описанный в J. Med. Chem., vol. 34, p.1162, 1991). Восстановление как второй этап осуществляют таким же способом, как в реакции восстановления на втором этапе вышеописанной стадии 15-2.
Стадия 16-3 включает установление защиты спирта, реакцию дегидратации амида до нитрила и реакцию снятия защиты спирта. Установление защиты спирта как первый этап изменяют в соответствии с исходным материалом и особо не ограничивают, пока условия подобны условиям в данной реакции. Для данной реакции может быть использована методика, известная специалисту в данной области техники. Может быть использован способ, описанный во многих документах, или тому подобное (T. Greene et al., "Protective Groups in Organic Synthesis", John Wiley & Sons, Inc., New York, 1981, например). Защитной группой является предпочтительно трет-бутилдифенилсилильная группа, обеспечивающая устойчивость защитной группы на последующей стадии. Реакцию дегидратации как второй этап осуществляют таким же способом, как реакцию дегидратации на третьем этапе вышеописанной стадии 6-3. Реакция снятия защиты как третий этап изменяют в соответствии с исходным материалом и особо не ограничивают, пока условия подобны условиям в данной реакции. Реакция снятия защиты, известная специалисту в данной области техники, может быть использована для данной реакции. Может быть использован способ, описанный во многих документах, или тому подобное (T. Greene et al., "Protective Groups in Organic Synthesis", John Wiley & Sons, Inc., New York, 1981, например).
Окисление на стадии 16-4 осуществляют таким же способом, как в реакции окисления на втором этапе вышеописанной стадии 8-2, с получением соединения общей формулы (d-112) из спиртового соединения (d-111).
Соединение [I] может быть получено подверганием альдегидного соединения (d-112) реакциям, проводимым на стадиях 7-1 и 7-2 вышеописанного общего способа получения 3.
Как подробно описано выше, соединение общей формулы (I) может быть получено в соответствии с общими способами получения 1-8 соединения по настоящему изобретению, и может также быть получено другим способом, хорошо известным специалисту в данной области техники. Примеры, описанные далее, имеют отношение к указанным способам получения, а на основе этих примеров соединение общей формулы (I) может быть легко получено способом, самим по себе известным специалисту в данной области техники.
Соединение общей формулы (I) или его фармакологически приемлемая соль по настоящему изобретению полезно(а) для лечения заболевания, вызываемого Aβ, и превосходно с точки зрения фармакокинетики, токсичности, устойчивости, всасывания и тому подобного.
Терапевтическое средство для лечения болезни, вызываемой Aβ, содержащее соединение формулы (I) или его фармакологически приемлемую соль по настоящему изобретению в качестве активного компонента, может быть получено традиционным способом. Предпочтительные примеры лекарственной формы включают таблетки, порошки, микрогранулы, гранулы, таблетки с покрытием, капсулы, сиропы, пастилки, лекарственные формы для ингаляции, суппозитории, растворы для инъекций, мази, глазные растворы, глазные мази, капли в нос, ушные капли, припарки и лосьоны. Средство может быть получено с использованием обычно используемых компонентов, таких как наполнитель, связывающее вещество, смазывающее вещество, красящее вещество и корригент, и компонентов, используемых по потребности, таких как стабилизатор, эмульгатор, вещество, облегчающее абсорбцию, поверхностно-активное вещество, регулятор pH, консервант и антиоксидант, и может быть получено смешиванием компонентов, обычно используемых в качестве материалов для изготовления фармацевтических препаратов. Примеры таких компонентов включают животные и растительные масла, такие как соевое масло, говяжий жир и синтетические глицериды; углеводороды, такие как жидкий парафин, сквалан и твердый парафин; сложноэфирные масла, такие как октилдодецилмиристат и изопропилмиристат; высшие спирты, такие как цетостеариловый спирт и бегениловый спирт; силиконовую смолу; силиконовое масло; поверхностно-активные вещества, такие как полиоксиэтиленовый сложный эфир жирной кислоты, сорбитановый сложный эфир жирной кислоты, глицериновый сложный эфир жирной кислоты, полиоксиэтиленсорбитановый сложный эфир жирной кислоты, полиоксиэтиленгидрогенизированное касторовое масло и блок-сополимер полиоксиэтилена и полиоксипропилена; водорастворимые полимеры, такие как гидроксиэтилцеллюлоза, полиакриловая кислота, карбоксивиниловый полимер, полиэтиленгликоль, поливинилпирролидон и метилцеллюлоза; низшие спирты, такие как этанол и изопропанол; многоатомные спирты, такие как глицерин, пропиленгликоль, дипропиленгликоль и сорбит; сахара, такие как глюкоза и сахароза; неорганические порошки, такие как кремниевый ангидрид, алюмосиликат магния и силикат алюминия, и дистиллированную воду. Примеры используемого наполнителя включают лактозу, кукурузный крахмал, сахарозу, глюкозу, маннит, сорбит, кристаллическую целлюлозу и диоксид кремния. Примеры используемого связывающего вещества включают поливиниловый спирт, простой поливиниловый эфир, метилцеллюлозу, этилцеллюлозу, аравийскую камедь, трагакант, желатин, шеллак, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, поливинилпирролидон, блок-сополимер полипропиленгликоля и полиоксиэтилена и меглумин. Примеры используемого разрыхлителя включают крахмал, агар, желатиновый порошок, кристаллическую целлюлозу, карбонат кальция, бикарбонат натрия, цитрат кальция, декстрин, пектин и кальций-карбоксиметилцеллюлозу. Примеры используемого смазывающего вещества включают стеарат магния, тальк, полиэтиленгликоль, диоксид кремния и гидрогенизированное растительное масло. Примеры используемого красящего вещества включают красящие вещества, приемлемые для фармацевтических препаратов. Примеры корригента включают порошок какао, ментол, эмпазм (empasm), мятное масло, борнеол и порошок корицы.
Например, пероральный препарат получают смешиванием служащего в качестве активного компонента соединения или его соли или гидрата соединения или соли, наполнителя и, когда требуется, например, связывающего вещества, разрыхлителя, смазывающего вещества, красящего вещества и корригента и затем превращением смеси, например, в порошок, мелкие гранулы, гранулы, таблетки, таблетки с покрытием или капсулы традиционным способом. Очевидно, что таблетки или гранулы при необходимости могут быть снабжены подходящим покрытием, например сахарным покрытием. Сироп или препарат для инъекций получают смешиванием, например, регулятора рН, солюбилизатора и изотонизирующего средства и, когда требуется, способствующего растворению вспомогательного средства, стабилизатора и тому подобного традиционным способом. Наружный препарат может быть изготовлен любым традиционным способом без особых ограничений. В качестве материала для основы может быть использован любой из многочисленных материалов, обычно применяемых для получения фармацевтического препарата, квазилекарственного средства (quasi drug), косметического средства или тому подобного. Примеры материала для основы включают такие материалы, как животные и растительные масла, минеральные масла, эфирные масла, воски, высшие спирты, жирные кислоты, силиконовые масла, поверхностно-активные вещества, фосфолипиды, спирты, многоатомные спирты, водорастворимые полимеры, глинистые минералы и дистиллированная вода. При необходимости могут быть добавлены регулятор рН, антиоксидант, хелатообразователь, консервант и фунгицид, красящее вещество, вкусовое вещество или тому подобное. Кроме того, при необходимости может быть подмешан компонент, обладающий эффектом индуцирования дифференциации, такой как усилитель кровотока, бактерицид, противовоспалительное средство, активатор клеток, витамин, аминокислота, увлажнитель или кератолитическое средство.
Дозу терапевтического средства по настоящему изобретению изменяют, например, в соответствии со степенью проявления симптомов, возрастом, полом, массой тела, способом введения, типом соли и конкретным видом болезни. Соединение формулы (I) или его фармакологически приемлемую соль вводят перорально взрослому обычно в количестве примерно от 30 мкг до 10 г, предпочтительно от 100 мкг до 5 г и более предпочтительно от 100 мкг до 100 мг в сутки или вводят взрослому путем инъекции примерно от 30 мкг до 1 г, предпочтительно от 100 мкг до 500 мг и более предпочтительно от 100 мкг до 30 мг в сутки разовой дозой или разделенными дозами, соответственно.
Для лечения заболевания, вызываемого амилоидом-β, такого как болезнь Альцгеймера, старческая деменция, синдром Дауна или амилоидоз, соединение формулы (I) или его фармакологически приемлемую соль по настоящему изобретению можно использовать в комбинации с соединениями, обладающими следующими механизмами.
Например, такие соединения включают ингибиторы холинэстеразы (например, донепезил, хуперзин A, такрин, ривастигмин, галантамин); антагонисты рецепторов AMPA (например, 1,2-дигидропиридиновые соединения, такие как 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-он); антагонисты рецептороа NMDA (например, мемантин); стимуляторы высвобождения ацетилхолина (например, прамирацетам; анирацетам); агонисты кальциевых каналов (например, нефирацетам); акцепторы свободных радикалов (например, EGb 761); антагонисты фактора активации тромбоцитов (например, EGb 761); антагонисты агрегации тромбоцитов (например, EGb 761, трифлузал); сенсибилизаторы инсулина (например, розиглитазон); агонисты рецепторов, активируемых пероксисомным пролифератором (например, розиглитазон); агонисты гамма-рецепторов, активируемых пероксисомным пролифератором (например, розиглитазон); ингибиторы моноаминоксидазы B (например, разагилин, селегилин, прокаин); стимуляторы карнитинацетилтрансферазы (например, левацекарнин); NSAID (НСПВС) (например, трифлузал, ингибиторы циклооксигеназы-2, такие как целекоксиб); агонисты фактора роста нерва (например, ксалипроден, FPF 1070); ингибиторы бета-амилоида (например, таренфлурбил, трамипрозат, лейпрорелин-D); иммуномодуляторы (например, таренфлурбил, иммуноглобулин, сложный икозапентэтиловый эфир); ингибиторы NF-каппа B (например, таренфлурбил); тиротропинвысвобождающий гормон (например, талтирелин); антагонисты допаминовых D2 рецепторов (например, рисперидон); антагонисты рецепторов серотонина-2 (например, рисперидон); агонисты мускариновых M1 рецепторов (например, цевимелин); агонисты альфа 1 адренорецепторов (например, модафинил); антагонисты рецепторов серотонина-3 (например, алозетрон); агонисты допаминовых D2 рецепторов (например, арипипразол); антагонисты допаминовых D2 рецепторов (например, арипипразол); агонисты серотониновых 1A рецепторов (например, арипипразол); антагонисты серотониновых 2A рецепторов (например, арипипразол); антагонисты глюкокортикоида (например, мифепристон); антагонисты прогестерона (например, мифепристон); ингибиторы HMG-CoA редуктазы (например, аторвастатин, симвастатин); ингибиторы поглощения аденозина (например, пропентофиллин); ингибиторы фосфодиэстеразы (например, пропентофиллин); агонисты ацетилхолиновых рецепторов (например, холиналфосцерат); усилители мембранной проницаемости (например, холиналфосцерат); антагонисты рецепторов каннабиноида 1 (например, римонабант); агонисты каннабиноидных рецепторов (например, дронабинол); ингибиторы ангиогенеза (например, паклитаксел); иммунодепрессанты (например, паклитаксел); антагонисты тубулина (например, паклитаксел); ингибиторы тромбоксан A-синтазы (например, трифлузал); антиоксиданты (например, идебинон); антагонисты альфа-адренорецепторов (например, ницерголин); антагонисты эстрогена (например, конъюгированные эстрогены, трилостан); ингибиторы 3-бета гидроксистероидной дегидрогеназы (например, трилостан); ингибиторы пути передачи сигнала (например, трилостан); агонисты мелатониновых рецепторов (например, рамелтеон); иммуностимуляторы (например, иммуноглобулин, сложный икозапентэтиловый эфир, прокаин); ингибиторы проникновения ВИЧ (например, прокаин); антагонисты натриевых каналов (например, прокаин); ингибитор микротрубок (например, CPH 82); агонисты глицинового сайта NMDA (например, циклосерин); антагонисты рецепторов аденозина A1 (например, KW 3902); стимуляторы АТФазы (например, триацетилуридин); усилители митохондриальной функции (например, триацетилуридин); агонисты фактора высвобождения гормона роста (например, тезаморелин); ингибитор бутилхолинэстеразы (например, биснорцимсерин); антагонисты альфа-адренергических рецепторов (например, ницерголин); ингибиторы NO синтазы типа II (например, арундовая кислота); хелатирующие вещества (например, PBT 2); ингибиторы амилоидного фибриллогенеза (например, TTP488, PF 4494700); агонисты рецепторов серотонин-4 (например, PRX 03140); антагонисты рецепторов серотонин-6 (например, SB 742457); обратные агонисты бензодиазепиновых рецепторов (например, радехинил); антагонисты Ca каналов (например, сафинамид); агонисты никотиновых рецепторов (например, испрониклин) и ингибитор BACE (например, CTS 21166).
Кроме того, указанные выше соединения включают, например, донепезил, хуперзин A, такрин, ривастигмин, галантамин, прамирацетам, анирацетам, нефирацетам, EGb 761, розиглитазон, разагилин, левацекарнин, целекоксиб, 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-он, талампанел, бекампанел, мемантин, ксалипроден, таренфлурбил, трамипросат, лейпрорелин-D, талтирелин, рисперидон, цевимелин, модафинил, алосетрон, арипипразол, мифепристон, аторвастатин, пропентофиллин, холиналфосцерат, FPF 1070 (CAS №143637-01-8), римонабант, дронабинол, докозагексаеновую кислоту, паклитаксел, трифлузал, идебенон, ницерголин, конъюгированные эстрогены, трилостан, симвастатин, селегилин, рамелтеон, иммуноглобулин, сложный икозапентэтиловый эфир, прокаин, CPH 82, циклосерин, KW 3902 (CAS №136199-02-5), триацетилуридин, эстрогенные терапевтические средства для лечения деменции (например, MIGENIX, Vancouver, Canada), тезаморелин, бисноруимсерин, ницерголин, арундовую кислоту, PBT 2, TTP488, PF 4494700, PRX 03140, SB 742457, радехинил, сафинамид, испрониклин, CTS 21166, Бапинейцумаб, NP 031112, (2S,3aS,7aS)-1{[(R,R)-2-фенилциклопропил]карбонил}-2-[(тиазолидин-3-ил)карбонил]октагидро-1H-индол, циталопрам, венлафаксин, левпрорелин, прастерон, пептид T (CAS №53-43-0), бесипиридин, лексипафант, стакофиллин, SGS 742 (CAS №123690-78-8), T 588 (CAS №142935-03-3), нериспиридин, дексанабинол, сабкомелин, GTS 21 (CAS №156223-05-1), CX 516 (CAS №154235-83-3), ABT 089 (CAS №161417-03-4), анапсос, тезофензин, SIB 1553A (т.е. 4-[[2-(1-метил-2-пирролидинил)этил]тио]фенол), ладостигил, радхинил, GPI 1485, изпрониклин, арундовую кислоту, MEM 1003 (т.е. 3-изопропил 5-(2-метоксил) 4-(2-хлор-3-цианофенил)-2,6-диметилпиридин-3,5-дикарбоксилат), V 3381 (т.е. гидрохлорид 2-(2,3-дигидро-1H-инден-3-иламино)ацетамида), фарампатор, палироден, прастерон-паладин, урокортин, DP b99 (т.е. 2,2′-(этилендиокси)бис(2,1-фенилен)бис[N-[2-[2-(октилокси)этокси]-2-оксоэтил]имино]бис(уксусную кислоту)), капсерод, DU 125530, бапинейцумаб, AL 108 (т.е. L-аспарагинил-L-аланил-L-пролил-L-валил-L-серил-L-изолейцил-L-пролил-L-глутамин), DAS 431, DEBIO 9902, DAR 100, митохинон, IPL 455903 (т.е. 5(S)-[3-(циклопентилокси)-4-метоксифенил]-3(S)-(3-метилбензил)пиперидин-2-он), E2CDS, PYM 50028, PBT 2, лекозотан, SB 742457, CX 717, AVE 1625 (т.е. 1-(бис(4-хлорфенил)метил)-3-((3,5-дифторфенил)(метилсульфонил)метилен)азетидин), LY 450139 (т.е. N2-[2(s)-гидрокси-3-метилбутирил]-N1-[3-метил-2-оксо-2,3,4,5-тетрагидро-1H-3-бензазепин-1(S)-ил]-L-аланинамид), EM 1421 (т.е. 4,4′-[(2R,3S)-2,3-диметилбутан-1,4-диил]бис(1,2-диметоксибензол), SRN 001, TTP 488, PRX 03140, димеболин, глицин-пролин-глутамат, C105, AL 208, MEM 3454, AC 1202, L 830982, LY 451395 (т.е. (R)-N-[2-[4′-(метилсульфонамидометил)бифенил-4-ил]пропил]пропан-2-сульфонамид), MK 0249, LY 2062430, диэтилнорспермин, небогламин, S 18986, SA 4503 (CAS №165377-44-6), GRI 1, S 17092 (т.е. (2S,3aS,7aS)-1{[(R,R)-2-фенилциклопропил]карбонил}-2-[(тиазолидин-3-ил)карбонил]октагидро-1H-индол), SL 251188, EUK 189, R 1450, 6,6-диметил-3-(2-гидроксиэтил)тио-1-(тиазол-2-ил)-6,7-дигидро-2-бензотиофен-4(5H)-он, CERE 110, дексефароксан, CAD 106, HF 0220, HF 0420, EHT 0202, VP 025, MEM 1414, BGC 201259 (т.е. 4-[1(S)-(метиламино)-3-(4-нитрофенокси)пропил]фениловый эфир N,N-диметилкарбаминовой кислоты), EN 100, ABT 834, ABT 239 (т.е. 4-[2-[2-[(2R)-2-метилпирролидинил]этил]бензофуран-5-ил]бензонитрил), SGS 518, R 1500, C 9138, SSR 180711, альфатрадиол, R 1577, T 817MA (т.е. 1-[3-[2-(1-бензотиен-5-ил)этокси]пропил]азетидин-3-олмалеат), CNP 1061 (т.е. 4-метил-5-(2-нитрооксиэтил)тиазол), KTX 0101 (т.е. бета-гидроксибутират натрия), GSK 189254 (т.е. 6-[3-циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси]-N-метилникотинамид), AZD 1080, ACC 001, PRX 07034, мидазолам, R-фенсерин, AZD 103 (CAS №488-59-5), SN 522, NGX 267 (CAS №503431-81-0), N-PEP-12, RN 1219, FGLL, AVE 8112, EVT 101, NP 031112, MK 0752, MK 0952, LX 6171, PAZ 417, AV 965, PF 3084014, SYN 114, GSI 953, SAM 315, SAM 531, D-серин, летеприним калий, BR 16A (CAS №149175-77-9), RPR 107393 (CAS № 190841-57-7), NXD 2858, REN 1654, CDD 0102, NC 1900 (CAS №132925-74-7), циклоспорин, NCX 2216 (т.е. (E)-4-(нитроокси)бутил 3-[4-[2-(2-фторбифенил-4-ил)пропаноилокси]-3-метоксифенил]акрилат), NXD 3109, NXD 1191, ZSET 845 (т.е. 3,3-дифенилимидазо[1,2-a]пиридин-2-(3H)-он), ET 002, NT 13, RO 638695 (т.е. [1,6-(1,6-диоксогексил)]дипирролидин-(2R)-карбоновую кислоту), биснорцимсерин, BA 1016, XD 4241, EUK 207 (т.е. (SP-5-13)-(ацетато-κO)[13,16,19,22-тетраокса-3,6-диазатрицикло[21,3,18,12]октакоза-1(27),2,6,8,10,12(28),23,25-октаен-27,28-диолато(2-)-κN3,κN6,κO27,κO28]марганец), ингибиторы LG 617, ZSET 1446, PAN 811, F 14413 (т.е. 2-[5-фтор-2(S)-метокси-2,3-дигидро-1,4-бензодиоксин-2-ил]-4,5-дигидро-1H-имидазол), FP 7832 (т.е. N-[2-(5-метокси-1-нитрозо-1H-индол-3-ил)этил]ацетамид), ARA 014418 (т.е. N-(4-метоксибензил)-N′-(5-нитро-1,3-тиазол-2-ил)мочевину), AZD 3102, KP 544 (т.е. 2-амино-5-(4-хлорфенилэтинил)-4-(4-транс-гидроксициклогексиламино)пиримидин), DP 155, 5-хлор-N-[3-[2-(диметиламино)этил]-1H-индол-5-ил]нафталин-2-сульфонамид, TAK 070, хуперзин, гидрохлорид N-[2-(3,5-диметиладамант-1-ил)этил]ацетамидина, 6-[4-[(диметиламино)метил]-5-этил-2-метоксифенил]пиридин-2-амин, 4,6-дифенил-3-(4-(пиримидин-2-ил)пиперазин-1-ил)пиридазин, гидрохлорид N-[(1S,2R)-3-(3,5-дифторфенил)-1-гидрокси-1-[(5S,6R)-5-метил-6-(неопентилокси)морфолин-3-ил]пропан-2-ил]ацетамида, N-[(1R,2S)-3-(3,5-дифторфенил)-1-гидрокси-1-[(2R,4R)-4-феноксипирролидин-2-ил]пропан-2-ил]-3-[(R)-2-(метоксиметил)пирролидин-1-карбонил]-5-метилбензамид, R 1589, мидафотел, фенсерин, колурацетам, физостигмин, кипрализант, нитрофлурбипрофен, PPI 1019 (т.е. (3α, 5β, 7α, 12α)-тригидроксихолан-24-оил-L-лейцил-L-валил-L-фенилаланил-L-фенилаланил-L-аланин), дапсон, MDL 100453 (CAS №129938-34-7), NS 377, мидаксифиллин, пропофолфосфат, метрифонат, церонаприл, тенилсетам, суфоксазин, сеглитид, эбиратид, небрацетам, милацемид, йоддоксорубицин, SM 10888 (CAS №129297-21-8), U 80816 (CAS №138554-11-7), YM 954 (CAS №132041-85-1), SUT 8701 (CAS №123577-73-1), аповинкамин, FR 121196 (CAS №133920-65-7), LY 274614 (CAS №136109-04-1), CL 275838 (CAS №115931-65-2), игмезин, K 7259 (CAS №133667-88-6), винконат, итазетрон, CL 287663 (CAS №125109-98-0), WAY 100289 (CAS №136013-69-9), SR 46559A (CAS №137733-33-6), GYKI 46903 (CAS №142999-59-5), L 670548 (CAS №121564-89-4), Y 29794 (CAS №129184-48-1), AF 125 (CAS №7631-86-9), KFM 19 (CAS №133058-72-7), ST 796 (т.е. (S)-3-[3-(трифторметил)бензоил)амино]гексагидроазепин-2-он), RU 33965 (CAS №122321-05-5), SDZ 210086 (т.е. (-)-1′,2(S)-диметилспиро[1,3-диоксан-4,4′-пиперидин]), L 689660 (CAS №144860-79-7), L 689560 (CAS №139051-78-8), ST 618 (т.е. 1-(6,7-диметокси-1,2,3,4-тетрагидро-2-нафтил)-4-гидроксипирролидин-2-он), U 74500A (CAS №110101-65-0), GEA 857 (CAS №120493-42-7), BIBN 99 (CAS №145301-48-0), DX 9366, ONO 1603 (CAS №114668-76-7), MDL 102234 (CAS №137766-81-5), P 9939 (CAS №157971-37-4), PD 140532 (CAS №157971-39-6), азетирелин, MR 16728 (CAS №147614-21-9), дабелотин, MDL 102503 (т.е. 8-[1(R)-метил-2-фенилэтил]-1,3-дипропил-7H-ксантин), PD 141606 (т.е. (±)-(Z)-3-(3-фенил-2-пропинилоксиимино)-1-азабицикло[2.2.1]гептан), SNK 882 (CAS №152221-12-0), L 696986 (CAS №141553-45-9), тазомелин, LY 235959 (CAS №137433-06-8), 2-(2-тиооксопирролидин-1-ил)ацетамид, AK 30 NGF, ABT 418 (CAS №147402-53-7), итамелин, HUP 13, сибопирдин, KST 5452 (CAS №157998-88-4), TJ 54, U 92798 (т.е. 7-[4-[бис(4-фторфенил)метил]пергидро-1,4-диазепин-1-илметил]-4-изопропил-2-метокси-2,4,6-циклогептатриен-1-он), U 92032 (CAS №142223-92-5), 3-(сульфамоилокси)эстра-1,3,5(10)-триен-17-он, P 11012 (CAS №164723-36-8), A 82695 (CAS №147388-86-1), FR 76659 (CAS №116904-25-7), апаксифиллин, CX 417, 7 MEOTA (CAS №5778-80-3), BU 4514N (CAS №151013-39-7), прегненолон, мексидол, ST 857 (CAS №154755-63-2), RU 49041 (CAS №123828-80-8), RU 35929 (CAS №111711-47-8), P 878184, P 128 (CAS №157716-52-4), эвристатин A, эвристатин B, LK 12, NBI 108, NBI 107, NBI 117, L 705106, бакозид A+B, клаузенамид, SM 21 (CAS №155156-22-2), алаптид, RS 17017 (т.е. гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-(1-пиперидинил)-1-пентанона), AF 150(S) (т.е. (S)-[1-метил-пиперидин-4-спиро-(2′-метилтиазолин)]), RO 153505 (CAS №78771-13-8), PV 113 (т.е. 1,2,3,4-тетрагидропиррол-[1,2-a]пиразин), арисугацин, A 98284 (т.е. 2(R)-(3-метилоксазол-5-ил)хинуклидин), AP 5 (CAS №136941-85-0), BD 1054, SDZ NDD 094 (т.е. бис-(2-(2-метилимидазол-1-ил]метил)пиридин-трис(гидрофумарат), AZ 36041 (CAS №173324-76-0), хилостигмин, A 84543 (т.е. 3-[1-метилпирролидин-2-(S)-илметокси]пиридинфумарат), BTG 4247 (т.е. (2-[2-хлорэтокси[4-(диметиламино)фенил]фосфорил]ацетогидразин), CGP 50068 (CAS №158647-49-5), цереброкраст, десферринорданоксамин, изолихенан, MHP 133 (т.е. 3-(N,N-диметилкарбамоилокси)-1-метил-2-(4-фенилсемикарбазонометил)пиридинийхлорид), FR 152558 (CAS №151098-08-7), GVS 111 (CAS №157115-85-0), P 11149 (CAS №164724-79-2), PDC 008004, KST 2818 (CAS №158623-26-8), KST 5410 (CAS №158623-27-9), RU 52583 (CAS №123829-33-4), PD 151832 (CAS №149929-39-5), UCL 1199 (т.е. 4-[2-[(5-нитропиридин-2-илсульфанил)этил]-1H-имидазол), изованихуперзин A, SIB 1765F (CAS №179120-52-6), JWS USC 751X (т.е. 3-[[[2-[[(5-диметиламиноэтил)-2-фуранил]метил]тио]этил]амино]-4-нитропиридазин), GR 175737 (т.е. 3-(4-хлорбензил)-5-[2-(1H-имидазол-4-ил)этил]-1,2,4-оксадиазол), KS 505A (CAS №131774-53-3), ZTTA 1 (т.е. N-бензилоксикарбонил-тиопропил-тиопропиналь-диметилацеталь), AGN 190837 (CAS №136527-40-7), P 10358 (188240-59-7), WAY 131256 (CAS №174001-71-9), DBO 83 (т.е. моногидрат дигидрохлорида 3-(6-хлорпиразин-3-ил)диазабицикло[3.2.1]октана), FUB 181 (CAS №152029-80-6), RJR 2557, WSU 2088, LVV-хаеморфин-7, M 40 (т.е. галанин[1-12]-Pro3-(Ala-Leu)2-Ala-NH2), SIB 1757, SKF 74652 (т.е. [5-хлор-2-(4-метоксифенил)-3-бензофуранил][4-[3-(диметиламино)пропокси]фенил]метанон), CGP 71982, SCH 57790 (т.е. 4-циклогексил-альфа-[4-[[4-метоксифенил]сульфинил]фенил]-1-пиперазинацетонитрил), Путресцин-D-YiAbetal1, DU 14 (т.е. п-O-(сульфамоил)-N-тетрадеканоилтирамин), CLZ 4, SL 340026, PPRT 424, ципроксифан, UR 1827 (т.е. 2-(1-бензилпиперидин-4-ил)-1-[4-(5-метилпиримидин-4-иламино)фенил]-1-этанон), капроктамин, TGS 20 (т.е. L-пироглутамил-D-аланинамид), PG 9 (т.е. альфа-тропанил 2-[(4-бром)фенил]пропионат), TEI 3356 (т.е. (16S)-15-дезокси-16-гидрокси-16-метил-9-(O)-метано-дельта6(9-альфа)-простагландин I1), LY 392098 (т.е. 3-[(2-метилэтил-2)сульфониламинопропил-2]фенил-4-илтиофен), PG 1000, DM 232, NEPP 11 (т.е. сложный метиловый эфир 12-изо-15-дезокси-18-(4-метил)фенил-13,14-дигидро-дельта7-простагландина A1), VA 100 (т.е. (2,3-дигидро-2-[[(4-фторбензоил)амино]этил]-1-метил-5-фенил-1H-1,4-бензодиазепин), VA 101 (т.е. (2,3-дигидро-2-[[(2-тиенилкарбонил)амино]этил]-1-метил-5-фенил-1H-1,4-бензодиазепин), NC 111585 (т.е. (3S)-1,3-бис-[3-[(3-азабицикло[2.2.2]октанил)-1,2,5-тиадиазол-4-илокси]-1-пропин-1-ил]бензол, 2L-(+)-тартрат), IN 201, имопроксифан, канокодиол, пикрозид I, пикрозид II, DM 235 (т.е. 1-(4-бензоилпиперазин-1-ил)пропан-1-он), моноклональное антитело 10D5, JLK2, JLK 6, JLK 7, DAPT (т.е. сложный трет-бутиловый эфир N-[N-(3,5-дифторфенацетил)-L-аланил]-S-фенилглицина), хуперин X, SGS 111 (т.е. (S)-этил 2-[1-(2-фенилацетил)пирролидин-2-карбоксамидо]ацетат), NP 7557, C 9136, C 7617, R 1485, рофекоксиб, веллнакрин, монтирелин, лазабемид, ORG 2766 (CAS №50913-82-1), сабелузол, адафеноксат, CAS №9061-61-4, ипидакрин, бемезетрон, идазоксан, линопирдин, селфотел, суритозол, миламелин, ксаномелин, TJ 960, фазорацетам, эптастигмин, энзакулин, занапезил, позатирелин, закоприд, RS 86 (CAS №3576-73-6), ORG 5667 (CAS №37552-33-3), RX 77368 (CAS №76820-40-1), BMS 181168 (CAS №123259-91-6), BY 1949 (CAS №90158-59-1), AWD 5239 (CAS №109002-93-9), YM 796 (171252-79-2), алорацетам, CI 933 (CAS №91829-95-7), ST 793 (CAS №99306-37-3), цебарацетам, зифросилон, талзаклидин, алвамелин, JTP 2942 (148152-77-6), OPC 14117 (CAS №103233-65-4), элциверин, AP 521 (т.е. гидрохлорид N-(1,3-бензодиоксол-5-илметил)-1,2,3,4-тетрагидро[1]бензотиено[2,3-c]пиридин-3(R)-карбоксамида), S 8510 (CAS №151466-23-8), JTP 4819 (CAS №162203-65-8), икопезил, SC 110, FK 960 (CAS №133920-70-4), DMP 543 (CAS №160588-45-4), ганстигмин, CI 1017 (т.е. O-(3-(3′-метоксифенил)-2-пропионил)оксиммалеат (R)-(-)-(Z)-1-азабицикло[2.2.1]гептан-3-она), T 82 (т.е. гемифумарат 2-[2-(1-бензилпиперидин-4-ил)этил]-2,3-дигидро-9-метокси-1H-пирроло[3,4-b]хинолин-1-она), NGD 971, вакцина аспартил-аланил-глутамил-фенилаланил-аргинил-гистидил-аспартил-серил-глицил-тирозил-глутамил-валил-гистидил-гистидил-глутаминил-лизил-лейцил-валил-фенилаланил-фенилаланил-аланил-глутамил-аспартил-валил-глицил-серил-аспарагинил-лизил-глицил-аланил-изолейцил-изолейцил-глицил-лейцил-метионил-валил-глицил-глицил-валил-валил-изолейцил-аланин, PBT 1 (CAS №130-26-7), TCH 346, FK 962 (т.е. N-(1-ацетилпиперидин-4-ил)-4-фторбензамид), воксерголид, KW 6055 (CAS №63233-46-5), тиопилокарпин, ZK 93426 (CAS №89592-45-0), SDZ NVI 085 (CAS №104195-17-7), CI 1002 (CAS №149028-28-4), Z 321 (CAS №130849-58-0), миризетрон, CHF 2060 (т.е. 2,4a,9-триметил-2,3,4,4a,9,9a-гексагидро-1,2-оксазино[6,5-b]индол-6-иловый сложный эфир-L-тартрат N-гептилкарбаминовой кислоты), гедокарнил, тербехинил, HOE 065 (CAS №123060-44-6), SL 650102, GR 253035, ALE 26015, SB 271046 (т.е. 5-хлор-N-(4-метокси-3-пиперазин-1-илфенил)-3-метил-2-бензотиофенсульфонамид), iAbeta5, SCH 211803 (т.е. 1-[1-(3-метил-2-аминофенил)карбонилпиперидин-4-ил]-4-[(3-хлорфенил)сульфонилфенил-4]метилпиперидин), EVT 301, альфа-линоленовая кислота/линолевая кислота, Kamikihi-To, сиагозид, FG 7142 (CAS №78538-74-6), RU 47067 (CAS №111711-92-3), RU 35963 (CAS №139886-03-6), FG 7080 (CAS №100332-18-1), E 2030 (CAS №142007-70-3), трансформирующий фактор роста бета-1, A 72055 (т.е. 2′,1-диметилспиро[пиперидин-4,5′оксазолидин]-3′-карбоксальдегид), NS 626, димирацетам, GT 3001, GT 2501, GT 2342, GT 2016 (CAS №152241-24-2), ORG 20091 (CAS №141545-50-8), BCE 001 (CAS №95678-81-2), CGP 35348 (CAS №123690-79-9), WAY 100635 (CAS №146714-97-8), E 4804 (CAS №162559-34-4), LIGA 20 (CAS №126586-85-4), NG 121 (т.е. 2-[4,8-диметил-3(E),7(E)-моноадиенил]-3,5-дигидрокси-2-метил-3,4,7,9-тетрагидро-2H-фтор[3,4-h]-1-бензопиран-7-он), MF 247 (т.е. (3aS,8aR)-1,3a,8-триметил-1,2,3,3a,8,8a-гексагидропирроло[2,3-b]индол-5-иловый сложный эфир N-[10-(диэтиламино)децил]карбаминовой кислоты), JTP 3399 (т.е. N-бензил-2(S)-[2(S)-(феноксиацетил)пирролидин-1-илкарбонил]пирролидин-1-карбоксамид), KF 17329, тиоперамид, F 3796 (т.е. 1-[2-(1-бензилпиперидин-4-ил)этил]-3-[3,4-(метилендиокси)бензоил]тиомочевину), GT 4001, GT 4002, FPL 14995 (CAS №123319-03-9), RU 34332 (CAS №137157-58-5), SR 96777A (CAS №115767-94-7), SIB T1980, NS 649 (CAS №146828-02-6), PD 142505 (CAS №149929-08-8), GYKI 52466 (CAS №102771-26-6), RO 246173 (CAS №159723-57-6), SCH 50911 (CAS №160415-07-6), Z 4105 (CAS №119737-52-9), RS 67333 (CAS №168986-60-5), NS 1546, ZM 241385 (CAS №139180-30-6), RO 249975 (т.е. [1S,3S(2′S),5R]-3-(1-бензил-5-оксопирролидин-2-илметил)-5-(1H-имидазол-5-илметил)циклогексан-1-ацетамид), AF 185 (т.е. 8-метил-3-(2-пропинил)-1,3,8-триазаспиро[4,5]декан-2,4-дион), CEP 427, CX 423, CX 438, CX 480, CDP-этаноламин, GT 4003, GT 4011, GT 5011, MS 430 (CAS №122113-44-4), MBF 379 (т.е. [3,3-бис(гидроксиметил)-8-гидрокси-3,4-дигидро-2H-1,4-бензоксазин-5-ил][3′,5′-дигидрокси-4′-(2-оксо-2-фенилэтокси)фенил]метанон), NGD 187 (CAS №163565-48-8), DUP 856, MR 3066, MF 8615 (т.е. 5-амино-6-хлор-4-гидрокси-3,4-дигидро-1H-тиопирано-[3,4-b]хинолинон), химбацин, ABS 300, RJR 2403 (CAS №538-79-4), MF 268 (CAS №174721-00-7), RO 465934 (т.е. 3-(2-циклогексил)-2,3,3a,4,5,9b-гексагидро-1H-бензо[e]индол-6-иловый сложный эфир N,N-диметилкарбаминовой кислоты), NS 393, RGH 2716 (CAS №134069-68-4), WIN 678702 (12,12-бис(3-фурил)-6,11-дигидро-6,11-этанобензо[b]хинолизинийхлорид), RS 66252 (т.е. 1-бутил-2-[(2′-(2H-тетразол-5-ил)бифенил-4-ил)метил]-1H-индол-3-карбоновую кислоту), AIT 034 (CAS №138117-48-3), NG 012 (CAS №131774-53-3), PD 142012 (CAS №5778-84-7), GT 4054, GT 4077, GT 4035, P 26 (CAS №152191-74-7), RGH 5279 (т.е. 2-ацетоксиэтиловый сложный эфир (-)-(13aR,13bS)-13a-этил-2,3,5,6,13a,13b-гексагидро-1H-индоло[3.2.1-de]пиридо[3.2.1-ij][1,5]нафтиридин-12-карбоновой кислоты), AIT 083, CeNeS, эстрадиол (т.е. 1,3,5(10)-эстратриен-3,17-бета-диол), WAY 132983 (гидрохлорид (3R,4R)-3-(3-гексасульфанилпиразин-2-илокси)-1-азабицикло[2.2.1]гептана), ABS 205, ABS 401, SX 3507 (т.е. 3-(3-пропил-1,2,4-оксадиазол-5-ил)хиноксалин-2(1H)-он), ARR 17779 (т.е. (-)-спиро[1-азабицикло[2.2.2]октаен-3,5-оксазолидин]-2-он), XE 991 (т.е. 10,10-бис(4-пиридилметил)антрацен-10(9H)-он), фенетилнорцимсерин, RO 657199, RJR 1781 (т.е. R(+)-2-(3-пиридил)-1-азабицикло[2.2.2.]октан), RJR 1782 (т.е. S(-)-2-(3-пиридил)-1-азабицикло[2.2.2.]октан), гилатид, толсерин, TC 2559 (т.е. (E)-N-метил-4-[3-(5-этоксипиридин)ил]-3-бутен-1-амин), ER 127528 (т.е. гидрохлорид 1-(3-фторбензил)-4-[(2-фтор-5,6-диметокси-1-инданон-2-ил)метил]пиперидина), тиатолсерин, таргацепт, аксоникс, цимсерин, тиацимсерин, моноклональное антитело 266, Apan-CH, DP 103, SPI 339 (т.е. сложный этиловый эфир 4-[3-(4-оксо-4,5,6,7-тетрагидроиндол-1-ил)пропиониламино]бензойной кислоты), S 37245 (т.е. 4-(1,4-бензодиоксан-5-ил)-1-[3(S)-гидрокси-5-нитроиндан-2-ил]пиперазин), LLG 88, AZD 2858, трометамол, AN 240, NG 002 (т.е. 5-гидрокси-5-(2-гидрокси-1-метилэтил)-4-метоксифуран-2(5H)-он), UCB 29427 (т.е. 2-циклопропил-4-(циклопропиламино)-6-(морфолино)-1,3,5-триазин), TRH-SR, RO 401641 (CAS №122199-02-4), MPV 1743AIII (CAS №150586-64-4), IDRA 21 (CAS №22503-72-6), CEP 431, ACPD (CAS №67684-64-4), CT 3577 (т.е. 3,7-диметил-1-[11-(3,4,5-триметоксибензиламино)-11-оксоундецил]ксантин), CT 2583, NXD 9062, десферри-норданоксамин, DP b99, PBT 1, T 817MA, Альфатрадиол (CAS №57-91-0), AL 108, SL 650102, RS 67333 (CAS №168986-60-5), RS 17017, SGS 518, SYN 114, SB 271046, RO 657199, PRX 07034, суритозол, (CAS №110623-33-19), тербехинил (CAS №113079-82-6), FG 7142 (CAS №78538-74-6). RU 34332 (CAS №137157-58-5), SX 3507, RO 153505 (CAS №78771-13-8), RU 33965 (CAS №122321-05-5), S 8510 (CAS №151466-23-8), сабелузол (CAS №104383-17-7), цереброкраст (CAS №118790-71-9), NS 626, NS 649 (CAS №146828-02-6), U 92032 (CAS №142223-92-5), MEM 1003, U 92798, RGH 2716 (CAS №134069-68-4), сафинамид (CAS №133865-89-1), AZD 0328, MEM 63908, ABT 418 (CAS №147402-53-7), ARR 17779, RJR 2403 (CAS №538-79-4), TC 2559, A 82695 (CAS №147388-86-1), A 84543, A 98284, DBO 83, RJR 2557, SIB 1765F (CAS №179120-52-6), GTS 21 (CAS №156223-05-1), MEM 3454, SIB 1553A, EVP 6124, SSR 180711, ABT 089 (CAS №161417-03-4), ABT 107, ABT 560, TC 5619, TAK 070, гидрохлорид N-[(1S,2R)-3-(3,5-дифторфенил)-1-гидрокси-1-[(5S,6R)-5-метил-6-(неопентилокси)морфолин-3-ил]пропан-2-ил]ацетамида, 6-фтор-5-(2-фтор-5-метилфенил)-3,4-дигидропиридин, 2-амино-6-[2-(3′-метоксибифенил-3-ил)этил]-3,6-диметил-5,6-гидроксипиримидин-4(3H)-он, AZD 1080, ARA 014418, XD 4241, Z 321 (CAS №130849-58-0), ONO 1603 (CAS №114668-76-7), JTP 3399, эвристатин A (CAS №137563-63-4), эвристатин B (CAS №137563-64-5), P 128 (CAS №157716-52-4), Y 29794(CAS №129184-48-1), ZTTA 1, JTP 4819 (CAS №162203-65-8), моноклональное антитело 266, дулоксетин, эсциталопрам оксалат, флуоксетин, флувоксамин малеат, пароксетин, сертралин, дапоксетин, десвенлафаксин, сибутрамин, нефазодон, милнаципран, дезипрамин, дулоксетин и бицифадин.
Далее настоящее изобретение описано более подробно со ссылками на примеры, но примеры даны только для иллюстрации. Профилактическое или терапевтическое средство по настоящему изобретению для лечения болезни, вызываемой Aβ, ни в коем случае не ограничивается следующими ниже конкретными примерами. Специалист в данной области может в полной мере реализовать настоящее изобретение, внося различные изменения не только в следующие ссылочные примеры и примеры, но и в формулу изобретения данного описания в пределах объема формулы изобретения, приложенной к данному описанию.
Когда соединения примеров имеют стереоизомеры, названия соединений с оптическим вращением, необязательно, могут соответствовать структурным формулам, показанным далее в следующих примерах, если абсолютная конфигурация не определена.
В следующих далее примерах использованы следующие сокращения:
|
Хроматографию проводили, используя в качестве носителя BW-300 производства Fuji Silysia Chemical Ltd., если не указано иное.
Препаративные колонки, использованные для анализа и препаративного разделения хиральных соединений, CHIRALPAK™ AD-H, CHIRALPAK™ IA, CHIRALPAK™ IB, CHIRALCEL™ OJ-H и CHIRALCEL™ OD-H, были изготовлены Daicel Chemical Industries, Ltd.
ПРИМЕРЫ
Примеры 1 и 2
Синтез (+)-8-(5-изопропил-4-метокси-2-метилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-(5-изопропил-4-метокси-2-метилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
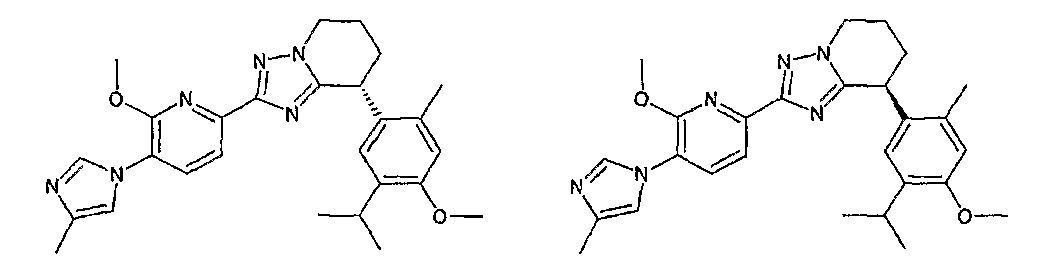
2-Метокси-3-(4-метилимидазол-1-ил)-6-трибутилстаннилпиридин, полученный в примере получения 1-2 (315 мг), 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин, полученный в примере получения 2-1 (120 мг), 1,3-бис(дифенилфосфино)пропан (54 мг) и оксид меди (I) (94 мг) суспендировали в N-метил-2-пирролидиноне (5 мл). Ацетат палладия(II) (15 мг) добавляли и смесь нагревали при перемешивании при 120°C в атмосфере азота в течение трех часов. Смесь охлаждали, добавляли этилацетат и воду. Реакционный раствор фильтровали через целит и органический слой отделяли. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем NH с получением рацемата указанного в заголовке соединения (103 мг). Полученный рацемат отделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: гексан:этанол = 5:5) с получением указанного в заголовке оптически активного соединения с положительным оптическим вращением (41 мг, >99% ee) и указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (40 мг, 99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 237 [1/2M++H], 473 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,06 (д, J=6,8 Гц, 3H), 1,10 (д, J=6,8 Гц, 3H), 1,90-2,38 (м, 4H), 2,30 (с, 3H), 2,34 (с, 3H), 3,19 (кв.кв., J=6,8, 6,8 Гц, 1H), 3,81 (с, 3H), 4,17 (с, 3H), 4,39-4,53 (м, 3H), 6,59 (с, 1H), 6,68 (с, 1H), 7,00 (с, 1H), 7,57 (д, J=8,0 Гц, 1H), 7,79 (д, J=8,0 Гц, 1H), 7,80-7,82 (м, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 237 [1/2M++H], 473 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,06 (д, J=6,8 Гц, 3H), 1,10 (д, J=6,8 Гц, 3H), 1,90-2,38 (м, 4H), 2,30 (с, 3H), 2,34 (с, 3H), 3,19 (кв.кв., J=6,8, 6,8 Гц, 1H), 3,81 (с, 3H), 4,17 (с, 3H), 4,39-4,53 (м, 3H), 6,59 (с, 1H), 6,68 (с, 1H), 7,00 (с, 1H), 7,57 (д, J=8,0 Гц, 1H), 7,79 (д, J=8,0 Гц, 1H), 7,81 (м, 1H).
Примеры 3 и 4
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
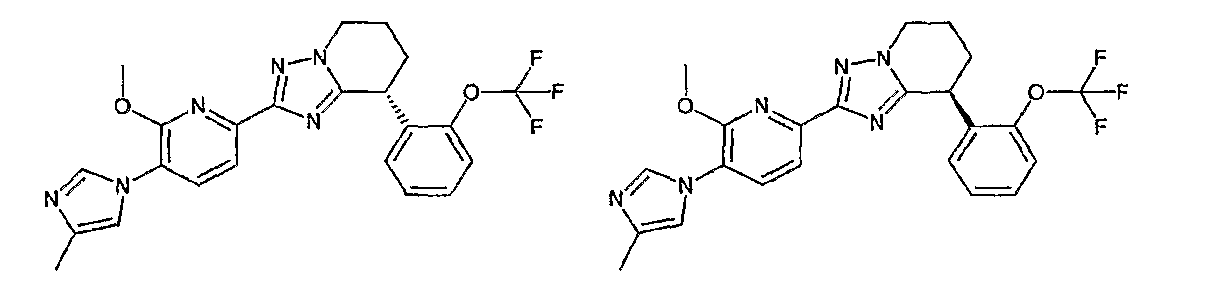
Рацемат указанного в заголовке соединения (43 мг) получали в соответствии со способом примеров 1 и 2 из 2-бром-8-(2-трифторметоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примере получения 2-2 (120 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-]a]пиридина. Полученный рацемат разделяли с помощью CHIRALPAK™ AD-H (2 см × 25 см, подвижная фаза: этанол, скорость потока: 10 мл/мин) с получением указанного в заголовке соединения с временем удерживания 11,6 минуты и положительным оптическим вращением (8 мг) и указанного в заголовке соединения с временем удерживания 14,5 минуты и отрицательным оптическим вращением (6,8 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 11,6 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,00-2,26 (м, 3H), 2,29 (с, 3H), 2,34-2,42 (м, 1H), 4,16 (с, 3H), 4,40 (дд, J=5,5, 5,5 Гц, 2H), 4,70 (дд, J=7,0, 5,9 Гц, 1H), 6,95 (д, J=7,4 Гц, 1H), 6,98-7,01 (м, 1H), 7,17-7,23 (м, 1H), 7,29-7,36 (м, 2H), 7,57 (д, J=7,8 Гц, 1H), 7,76 (д, J=7,8 Гц, 1H), 7,80-7,84 (м, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 14,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,00-2,26 (м, 3H), 2,29 (с, 3H), 2,34-2,42 (м, 1H), 4,16 (с, 3H), 4,40 (дд, J=5,5, 5,5 Гц, 2H), 4,70 (дд, J=7,0, 5,9 Гц, 1H), 6,95 (д, J=7,4 Гц, 1H), 6,98-7,01 (м, 1H), 7,17-7,23 (м, 1H), 7,29-7,36 (м, 2H), 7,57 (д, J=7,8 Гц, 1H), 7,76 (д, J=7,8 Гц, 1H), 7,80-7,84 (м, 1H).
Примеры 5 и 6
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
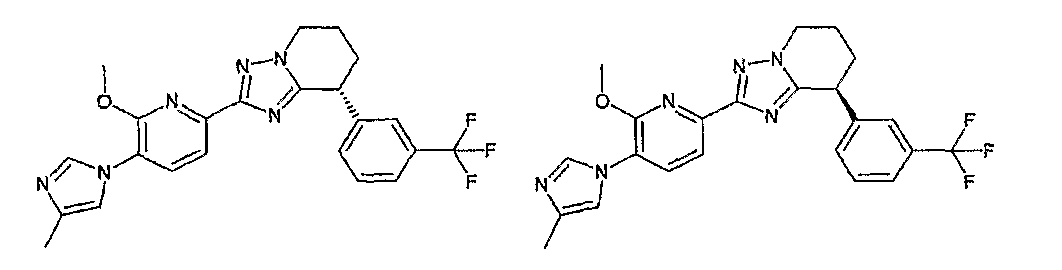
Рацемат указанного в заголовке соединения (125 мг) получали в соответствии со способом примеров 1 и 2 из 2-метокси-3-(4-метилимидазол-1-ил)-6-трибутилстаннилпиридина, полученного в примере получения 1-2 (415 мг) и из 2-бром-8-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примере получения 2-4 (150 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; подвижная фаза: гексан:этанол = 5:5) с получением указанного в заголовке оптически активного соединения с положительным оптическим вращением (51 мг, >99% ee) и указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (51 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 228 [1/2M++H], 455 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,05-2,28 (м, 3H), 2,30 (с, 3H), 2,39-2,48 (м, 1H), 4,16 (с, 3H), 4,39-4,51 (м, 3H), 7,00 (м, 1H), 7,32 (д, J=6,8 Гц, 1H), 7,44-7,49 (м, 2H), 7,55 (д, J=7,6 Гц, 1H), 7,59 (д, J=7,6 Гц, 1H), 7,77 (д, J=7,6 Гц, 1H), 7,82-7,83 (м, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 228 [1/2M++H], 455 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,05-2,28 (м, 3H), 2,30 (с, 3H), 2,39-2,48 (м, 1H), 4,16 (с, 3H), 4,39-4,51 (м, 3H), 7,00 (м, 1H), 7,32 (д, J=6,8 Гц, 1H), 7,44-7,49 (м, 2H), 7,55 (д, J=7,6 Гц, 1H), 7,59 (д, J=7,6 Гц, 1H), 7,77 (д, J=7,6 Гц, 1H), 7,82-7,83 (м, 1H).
Примеры 7 и 8
Синтез (-)-2-[5-метокси-6-(4-метилимидазол-1-ил)пиридин-3-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (+)-2-[5-метокси-6-(4-метилимидазол-1-ил)пиридин-3-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
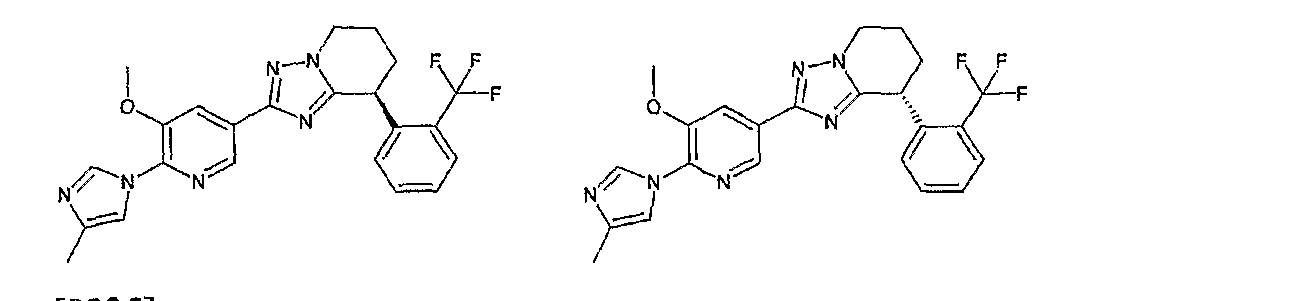
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 1 и 2 из раствора, 3-метокси-2-(4-метил-1H-имидазол-1-ил)-5-трибутилстаннилпиридина, полученного в примере получения 1-3 (100 мг), в N,N-диметилформамиде (2 мл) и из 2-бром-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примере получения 2-2 (60 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина. Полученный рацемат разделяли с помощью CHIRALCEL™ AD-H (2 см × 25 см; подвижная фаза: этанол) с получением указанного в заголовке оптически активного соединения с временем удерживания 11 минут и отрицательным оптическим вращением (13,5 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 17 минут и положительным оптическим вращением (29,9 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 455 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,94-2,04 (м, 1H), 2,10-2,36 (м, 2H), 2,29 (с, 3H), 2,45-2,53 (м, 1H), 3,99 (с, 3H), 4,32-4,45 (м, 2H), 4,73 (дд, J=8,0, 6,0 Гц, 1H), 7,01 (д, J=7,6 Гц, 1H), 7,41 (т, J=7,6 Гц, 1H), 7,49 (т, J=7,6 Гц, 1H), 7,55 (ушир.с, 1H), 7,75 (д, J=7,6 Гц, 1H), 7,95 (д, J=2,0 Гц, 1H), 8,37 (ушир.с, 1H), 8,70 (д, J=2,0 Гц, 1H).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 455 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,94-2,04 (м, 1H), 2,10-2,36 (м, 2H), 2,29 (с, 3H), 2,45-2,53 (м, 1H), 3,99 (с, 3H), 4,32-4,45 (м, 2H), 4,73 (дд, J=8,0, 6,0 Гц, 1H), 7,01 (д, J=7,6 Гц, 1H), 7,41 (т, J=7,6 Гц, 1H), 7,49 (т, J=7,6 Гц, 1H), 7,55 (ушир.с, 1H), 7,75 (д, J=7,6 Гц, 1H), 7,95 (д, J=2,0 Гц, 1H), 8,37 (ушир.с, 1H), 8,70 (д, J=2,0 Гц, 1H).
Примеры 9 и 10
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилбензил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилбензил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
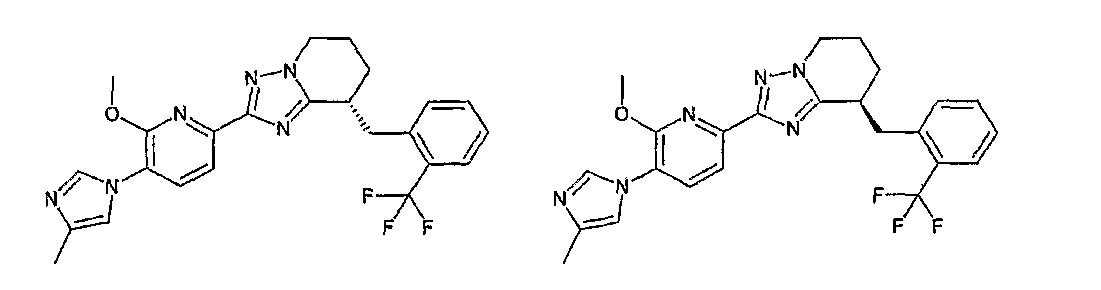
Рацемат указанного в заголовке соединения (223 мг) получали в соответствии со способом примеров 1 и 2 из 2-метокси-3-(4-метилимидазол-1-ил)-6-трибутилстаннилпиридина, полученного в примере получения 1-2 (377 мг), и из 2-бром-8-(2-трифторметилбензил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примере получения 2-7 (237 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина. Полученный рацемат разделяли с помощью CHIRALPAK™ AD-H (2 см × 25 см; подвижная фаза: гексан:этанол = 5:5) с получением указанного в заголовке оптически активного соединения с положительным оптическим вращением (74 мг, >99% ee) и указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (74 мг, 99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 235 [1/2M++H], 469 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,58-1,70 (м, 1H), 1,89-2,03 (м, 2H), 2,15-2,23 (м, 1H), 2,31 (с, 3H), 3,00-3,10 (м, 1H), 3,36-3,44 (м, 1H), 3,83 (дд, J=14,6, 4,6 Гц, 1H), 4,18 (с, 3H), 4,19-4,27 (м, 1H), 4,30-4,38 (м, 1H), 7,03 (с, 1H), 7,37 (дд, J=7,6, 7,2 Гц, 1H), 7,46-7,55 (м, 2H), 7,64 (д, J=7,6 Гц, 1H), 7,69 (д, J=8,0 Гц, 1H), 7,82-7,86 (м, 2H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 235 [1/2M++H], 469 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,58-1,70 (м, 1H), 1,89-2,03 (м, 2H), 2,15-2,23 (м, 1H), 2,31 (с, 3H), 3,00-3,10 (м, 1H), 3,36-3,44 (м, 1H), 3,83 (дд, J=14,6, 4,6 Гц, 1H), 4,18 (с, 3H), 4,19-4,27 (м, 1H), 4,30-4,38 (м, 1H), 7,03 (с, 1H), 7,37 (дд, J=7,6, 7,2 Гц, 1H), 7,46-7,55 (м, 2H), 7,64 (д, J=7,6 Гц, 1H), 7,69 (д, J=8,0 Гц, 1H), 7,82-7,86 (м, 2H).
Примеры 11 и 12
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-4-(4-трифторметилфенил)-4,5,6,7-тетрагидробензотиазола и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-4-(4-трифторметилфенил)-4,5,6,7-тетрагидробензотиазола
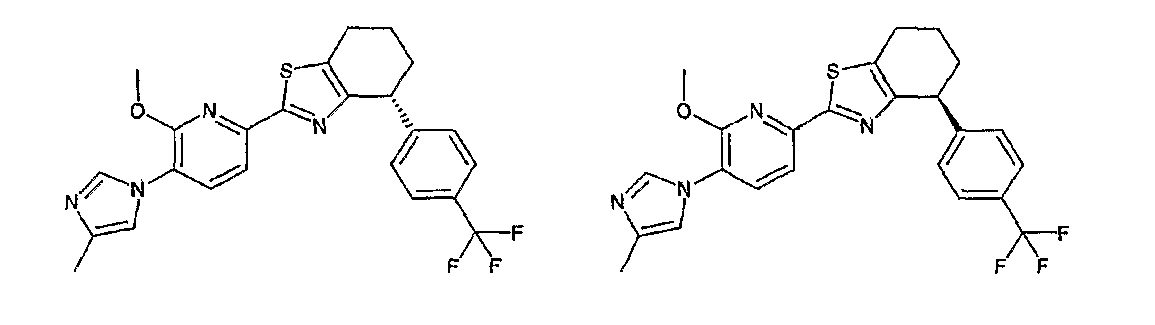
Рацемат указанного в заголовке соединения (79 мг) получали в соответствии со способом примеров 1 и 2 из 2-метокси-3-(4-метилимидазол-1-ил)-6-трибутилстаннилпиридина, полученного в примере получения 1-2 (198 мг), и из 2-бром-4-(4-трифторметилфенил)-4,5,6,7-тетрагидробензотиазола, полученного в примере получения 2-8 (150 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; подвижная фаза: гексан:этанол = 9:1) с получением указанного в заголовке оптически активного соединения с положительным оптическим вращением (27 мг, >99% ee) и указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (28 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 471 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,85-2,00 (м, 3H), 2,25-2,33 (м, 4H), 2,89-3,04 (м, 2H), 4,10 (с, 3H), 4,36 (т, J=5,6 Гц, 1H), 6,97 (д, J=1,2 Гц, 1H), 7,22 (д, J=7,6 Гц, 2H), 7,51-7,56 (м, 3H), 7,68 (д, J=8,0 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 471 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,85-2,00 (м, 3H), 2,25-2,33 (м, 4H), 2,89-3,04 (м, 2H), 4,10 (с, 3H), 4,36 (т, J=5,6 Гц, 1H), 6,97 (д, J=1,2 Гц, 1H), 7,22 (д, J=7,6 Гц, 2H), 7,51-7,56 (м, 3H), 7,68 (д, J=8,0 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Примеры 13 и 14
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-фтор-2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-фтор-2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
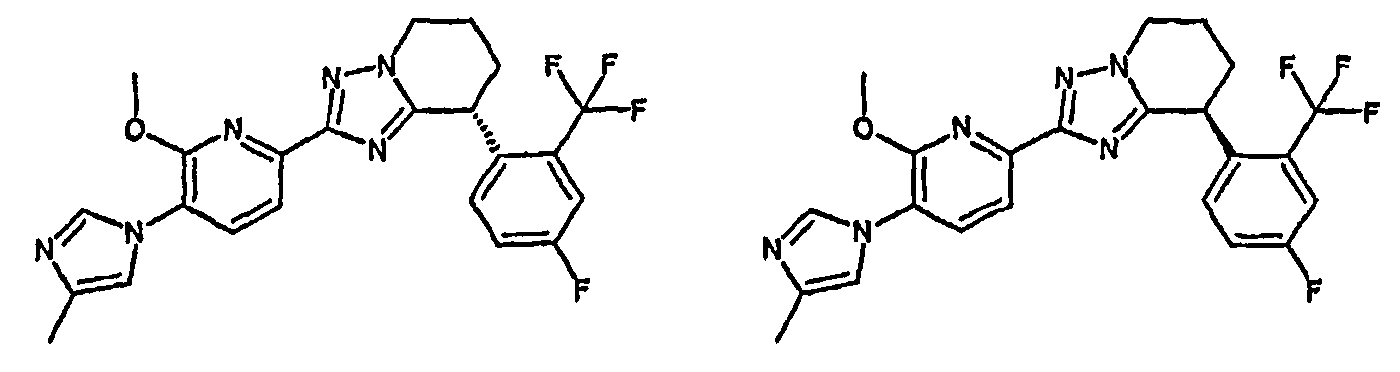
Рацемат указанного в заголовке соединения (66 мг) получали в соответствии со способом примеров 1 и 2 из 2-бром-8-(4-фтор-2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примере получения 2-5 (250 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-]a]пиридина. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: этанол:гексан = 3:7, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 16 минут и положительным оптическим вращением (12,6 мг) и указанного в заголовке соединения с временем удерживания 18,7 минуты и отрицательным оптическим вращением (15,1 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 16 минут были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,88-2,00 (м, 1H), 2,10-2,23 (м, 1H), 2,23-2,34 (м, 1H), 2,29 (с, 3H), 2,42-2,52 (м, 1H), 4,15 (с, 3H), 4,30-4,50 (м, 2H), 4,70 (дд, J=8,2, 6,2 Гц, 1H), 6,96-7,01 (м, 2H), 7,14-7,20 (м, 1H), 7,42-7,47 (м, 1H), 7,56 (д, J=8,2 Гц, 1H), 7,73 (д, J=8,2 Гц, 1H), 7,80-7,84 (м, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 18,7 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,88-2,00 (м, 1H), 2,10-2,23 (м, 1H), 2,23-2,34 (м, 1H), 2,29 (с, 3H), 2,42-2,52 (м, 1H), 4,15 (с, 3H), 4,30-4,50 (м, 2H), 4,70 (дд, J=8,2, 6,2 Гц, 1H), 6,96-7,01 (м, 2H), 7,14-7,20 (м, 1H), 7,42-7,47 (м, 1H), 7,56 (д, J=8,2 Гц, 1H), 7,73 (д, J=8,2 Гц, 1H), 7,80-7,84 (м, 1H).
Примеры 15 и 16
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
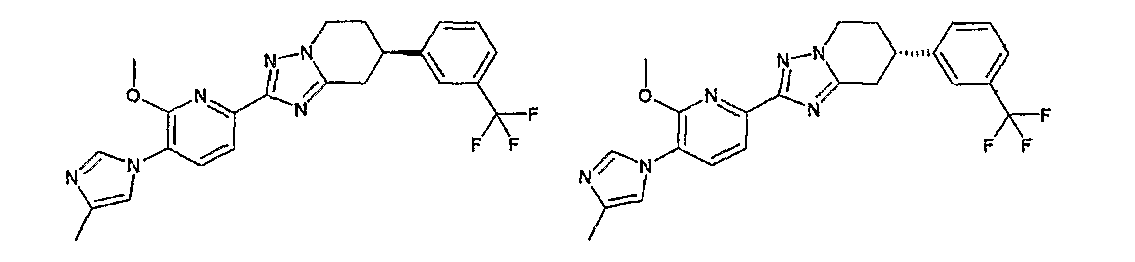
Рацемат указанного в заголовке соединения (25 мг) получали в соответствии со способом примеров 1 и 2 из 2-метокси-3-(4-метилимидазол-1-ил)-6-трибутилстаннилпиридина, полученного в примере получения 1-2 (190 мг), и из 2-бром-7-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примере получения 2-10 (115 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: этанол:гексан = 3:7, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 27 минут и положительным оптическим вращением (2,0 мг) и указанного в заголовке соединения с временем удерживания 38,5 минуты и отрицательным оптическим вращением (1,2 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 27 минут были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 2,33-2,49 (м, 2H), 3,11 (дд, J=16,0, 9,4 Гц, 1H), 3,33-3,47 (м, 2H), 4,18 (с, 3H), 4,28-4,37 (м, 1H), 4,46 (ддд, J=8,6, 5,1, 3,1 Гц, 1H), 7,02-7,04 (м, 1H), 7,44-7,61 (м, 4H), 7,64 (д, J=7,8 Гц, 1H), 7,83 (д, J=7,8 Гц, 1H), 7,85 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 38,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 2,33-2,49 (м, 2H), 3,11 (дд, J=16,0, 9,4 Гц, 1H), 3,33-3,47 (м, 2H), 4,18 (с, 3H), 4,28-4,37 (м, 1H), 4,46 (ддд, J=8,6, 5,1, 3,1 Гц, 1H), 7,02-7,04 (м, 1H), 7,44-7,61 (м, 4H), 7,64 (д, J=7,8 Гц, 1H), 7,83 (д, J=7,8 Гц, 1H), 7,85 (д, J=1,2 Гц, 1H).
Примеры 17 и 18
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(5-фтор-2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(5-фтор-2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
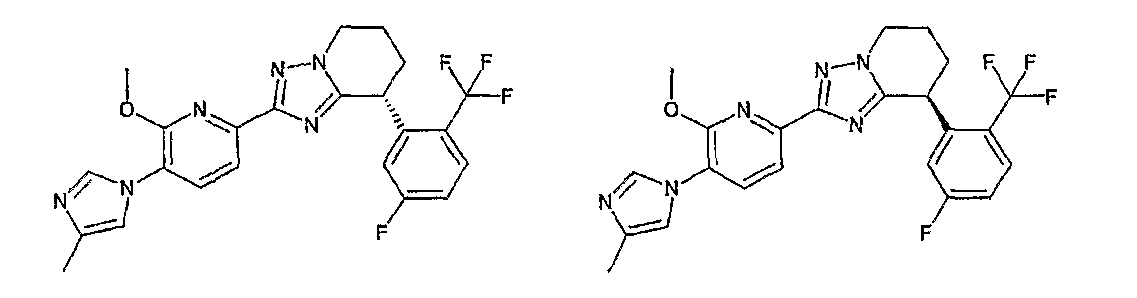
Рацемат указанного в заголовке соединения (10,2 мг) получали в соответствии со способом примеров 1 и 2 из 2-бром-8-(5-фтор-2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примере получения 2-6 (250 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина. Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: этанол:гексан = 3:7, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 12,5 минуты и положительным оптическим вращением (2,1 мг) и указанного в заголовке соединения с временем удерживания 22 минуты и отрицательным оптическим вращением (1,41 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 12,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,90-2,01 (м, 1H), 2,10-2,23 (м, 1H), 2,23-2,35 (м, 1H), 2,29 (с, 3H), 2,43-2,53 (м, 1H), 4,16 (с, 3H), 4,38-4,50 (м, 2H), 4,75 (дд, J=7,4, 6,6 Гц, 1H), 6,67-6,71 (м, 1H), 6,98-7,01 (м, 1H), 7,05-7,11 (м, 1H), 7,57 (д, J=7,8 Гц, 1H), 7,71-7,76 (м, 1H), 7,74 (д, J=7,8 Гц, 1H), 7,79-7,82 (м, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 22 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,90-2,01 (м, 1H), 2,10-2,23 (м, 1H), 2,23-2,35 (м, 1H), 2,29 (с, 3H), 2,43-2,53 (м, 1H), 4,16 (с, 3H), 4,38-4,50 (м, 2H), 4,75 (дд, J=7,4, 6,6 Гц, 1H), 6,67-6,71 (м, 1H), 6,98-7,01 (м, 1H), 7,05-7,11 (м, 1H), 7,57 (д, J=7,8 Гц, 1H), 7,71-7,76 (м, 1H), 7,74 (д, J=7,8 Гц, 1H), 7,79-7,82 (м, 1H).
Примеры 19 и 20
Синтез (+)-(7,8-анти)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-метил-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-(7,8-анти)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-метил-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
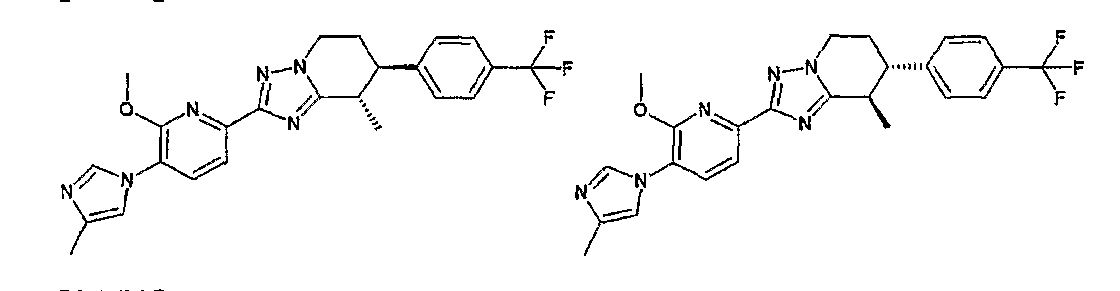
Рацемат указанного в заголовке соединения (80 мг) получали в соответствии со способом примеров 1 и 2 из 2-бром-8-метил-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примере получения 2-11, вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-]a]пиридина. Полученный рацемат разделяли с помощью CHIRALCEL™ OD-H (2 см × 25 см, подвижная фаза: этанол:гексан = 2:8, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 28,0 минуты и отрицательным оптическим вращением (4,8 мг) и указанного в заголовке соединения с временем удерживания 37,2 минутыы и положительным оптическим вращением (6,1 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 28,0 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,39 (д, J=7,0 Гц, 3H), 2,31 (с, 3H), 2,31-2,45 (м, 2H), 2,91 (ддд, J=11,3, 11,3, 3,1 Гц, 1H), 3,18-3,29 (м, 1H), 4,18 (с, 3H), 4,32 (ддд, J=11,7, 11,7, 3,9 Гц, 1H), 4,45-4,51 (м, 1H), 6,91-6,93 (м, 1H), 7,39 (д, J=7,8 Гц, 2H), 7,63 (д, J=7,8 Гц, 1H), 7,63-7,68 (м, 2H), 7,84 (д, J=7,8 Гц, 1H), 7,85 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 37,2 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,39 (д, J=7,0 Гц, 3H), 2,31 (с, 3H), 2,31-2,45 (м, 2H), 2,91 (ддд, J=11,3, 11,3, 3,1 Гц, 1H), 3,18-3,29 (м, 1H), 4,18 (с, 3H), 4,32 (ддд, J=11,7, 11,7, 3,9 Гц, 1H), 4,45-4,51 (м, 1H), 6,91-6,93 (м, 1H), 7,39 (д, J=7,8 Гц, 2H), 7,63 (д, J=7,8 Гц, 1H), 7,63-7,68 (м, 2H), 7,84 (д, J=7,8 Гц, 1H), 7,85 (д, J=1,2 Гц, 1H).
Примеры 21 и 22
Синтез (+)-2-[3-метокси-4-(4-метил-1H-имидазол-1-ил)фенил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[3-метокси-4-(4-метил-1H-имидазол-1-ил)фенил]8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
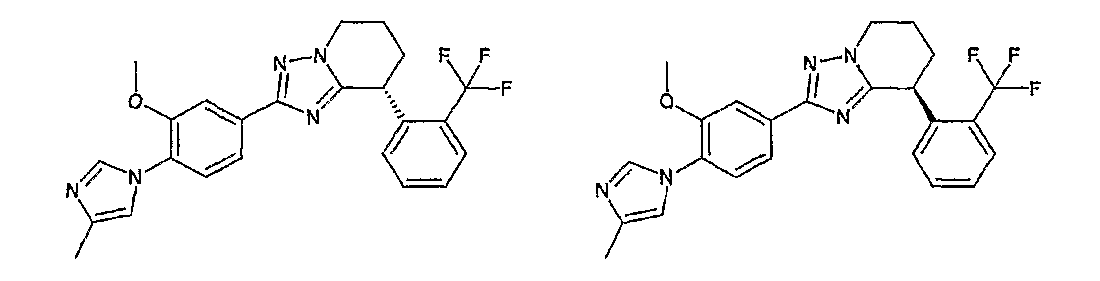
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 1 и 2 из 1-(2-метокси-4-трибутилстаннилфенил)-4-метил-1H-имидазола, полученного в примере получения 1-4 (276 мг), и из 2-бром-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примере получения 2-3 (100 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина. Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: гексан:этанол = 80:20) с получением указанного в заголовке оптически активного соединения с временем удерживания 5,8 минуты и положительным оптическим вращением (35,6 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 7,2 минуты и отрицательным оптическим вращением (40,6 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 454 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,93-2,01 (м, 1H), 2,10-2,20 (м, 1H), 2,24-2,30 (м, 1H), 2,29 (д, J=1,2 Гц, 3H), 2,43-2,51 (м, 1H), 3,90 (с, 3H), 4,36-4,39 (м, 2H), 4,74 (дд, J=6,4 Гц, 8,0 Гц, 1H), 6,94 (т, J=1,2 Гц, 1H), 7,00 (д, J=7,6 Гц, 1H), 7,25 (д, J=8,8 Гц, 1H), 7,39 (т, J=7,6 Гц, 1H), 7,47 (т, J=7,6 Гц, 1H), 7,69 (дд, J=1,6 Гц, 8,8 Гц, 1H), 7,70 (д, J=1,6 Гц, 1H), 7,72 (д, J=1,2 Гц, 1H), 7,73 (д, J=7,6 Гц, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 454 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,93-2,01 (м, 1H), 2,10-2,20 (м, 1H), 2,24-2,30 (м, 1H), 2,29 (д, J=1,2 Гц, 3H), 2,43-2,51 (м, 1H), 3,90 (с, 3H), 4,36-4,39 (м, 2H), 4,74 (дд, J=6,4 Гц, 8,0 Гц, 1H), 6,94 (т, J=1,2 Гц, 1H), 7,00 (д, J=7,6 Гц, 1H), 7,25 (д, J=8,8 Гц, 1H), 7,39 (т, J=7,6 Гц, 1H), 7,47 (т, J=7,6 Гц, 1H), 7,69 (дд, J=1,6 Гц, 8,8 Гц, 1H), 7,70 (д, J=1,6 Гц, 1H), 7,72 (д, J=1,2 Гц, 1H), 7,73 (д, J=7,6 Гц, 1H).
Примеры 23 и 24
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]5-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]5-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
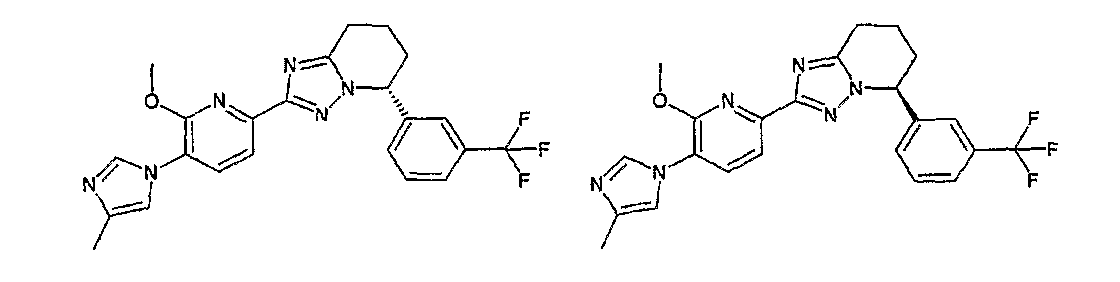
Рацемат указанного в заголовке соединения (38,7 мг) получали в соответствии со способом примеров 1 и 2 из 2-метокси-3-(4-метилимидазол-1-ил)-6-трибутилстаннилпиридина, полученного в примере получения 1-2 (62,2 мг), и из 2-бром-5-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примере получения 2-12 (32 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина. Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: гексан:этанол = 60:40) с получением указанного в заголовке оптически активного соединения с временем удерживания 3,8 минуты и положительным оптическим вращением (13,4 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 6,3 минуты и отрицательным оптическим вращением (10,4 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 455 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,94-2,00 (м, 2H), 2,16-2,23 (м, 1H), 2,29 (д, J=1,2 Гц, 3H), 2,46-2,55 (м, 1H), 3,07-3,24 (м, 2H), 4,13 (с, 3H), 5,66 (т, J=5,2 Гц, 1H), 7,00 (т, J=1,2 Гц, 1H), 7,14 (д, J=7,6 Гц, 1H), 7,36 (с, 1H), 7,48 (т, J=7,6 Гц, 1H), 7,58 (д, J=7,6 Гц, 2H), 7,75 (д, J=7,6 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 455 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,94-2,00 (м, 2H), 2,16-2,23 (м, 1H), 2,29 (д, J=1,2 Гц, 3H), 2,46-2,55 (м, 1H), 3,07-3,24 (м, 2H), 4,13 (с, 3H), 5,66 (т, J=5,2 Гц, 1H), 7,00 (т, J=1,2 Гц, 1H), 7,14 (д, J=7,6 Гц, 1H), 7,36 (с, 1H), 7,48 (т, J=7,6 Гц, 1H), 7,58 (д, J=7,6 Гц, 2H), 7,75 (д, J=7,6 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Пример 25
Синтез 4-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-7-(4-трифторметилфенил)2,3,5-триазатрицикло[5,2,2,0*2,6*]ундека-3,5-диен
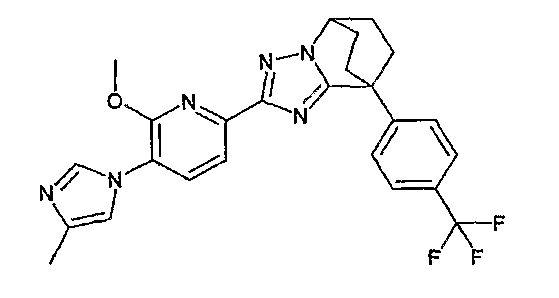
Указанное в заголовке соединение (0,35 мг) получали в соответствии со способом примеров 1 и 2 из 2-метокси-3-(4-метилимидазол-1-ил)-6-трибутилстаннилпиридина, полученного в примере получения 1-2 (10,8 мг), и из 4-бром-7-(4-трифторметилфенил)-2,3,5-триазатрицикло[5,2,2,0*2,6*]ундека-3,5-диена, полученного в примере получения 2-13 (2,8 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина. Показатели свойств соединения были следующими.
ESI-MS; m/z 481 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,03-2,17 (м, 6H), 2,30 (с, 3H), 2,30-2,35 (м, 2H), 4,17 (с, 3H), 5,13 (с, 1H), 7,01 (с, 1H), 7,59 (д, J=8,0 Гц, 1H), 7,73 (д, J=8,4 Гц, 2H), 7,80-7,87 (м, 4H).
Примеры 26, 27, 28 и 29
Синтез (+)-(6,8-син)-6-метокси-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-(6,8-син)-6-метокси-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (+)-(6,8-анти)-6-метокси-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (+)-(6,8-анти)-6-метокси-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
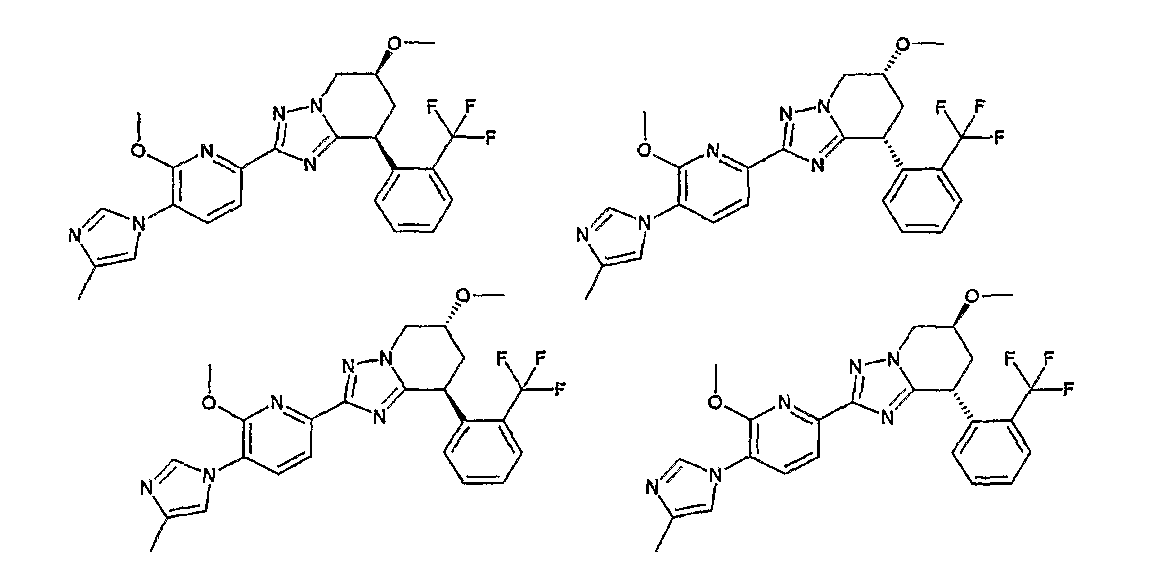
Синтез 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-6-ола
Указанное в заголовке соединение (910 мг) получали в виде желтого твердого вещества в соответствии со способом примеров 1 и 2 из 2-метокси-3-(4-метилимидазол-1-ил)-6-трибутилстаннилпиридина, полученного в примере получения 1-2 (1,31 г), и из 2-бром-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина-6-ола, полученного в примере получения 2-9 (900 мг), вместо 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина.
ESI-МС; m/z 471 [M++H].
Синтез (+)-(6,8-син)-6-метокси-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-(6,8-син)-6-метокси-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (+)-(6,8-анти)-6-метокси-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (+)-(6,8-анти)-6-метокси-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
2-[6-Метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина-6-ол (100 мг) растворяли в тетрагидрофуране (2 мл), и затем гидрид натрия (21,3 мг) и метилиодид (19,9 мкл) добавляли при 0°C. После перемешивания при комнатной температуре в течение 24 часов, реакционный раствор распределяли между насыщенным раствором бикарбоната натрия и этилацетатом. Органический слой промывали насыщенным раствором соли, сушили над сульфатом магния и затем концентрировали при пониженном давлении. Рацемическое антисоединение, очищенное хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-100%) (16,9 мг), разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: этанол:гексан = 4:6, скорость потока: 13 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 13,5 минуты и положительным оптическим вращением (3,3 мг) и указанного в заголовке оптически активного соединения с временем удерживания 19,5 минуты и отрицательным оптическим вращением (5,4 мг). С другой стороны, рацемическое син-соединение (25,9 мг) разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: этанол:гексан = 2:8, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 22,6 минуты и положительным оптическим вращением (2,5 мг) и указанного в заголовке оптически активного соединения с временем удерживания 38,5 минуты и отрицательным оптическим вращением (1,4 мг).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из антисоединения, с временем удерживания 13,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,95 (дд, J=13,5, 11,3 Гц, 1H), 2,29 (с, 3H), 2,72-2,80 (м, 1H), 3,47 (с, 3H), 4,03-4,09 (м, 1H), 4,15 (с, 3H), 4,43 (дд, J=14,1, 3,2 Гц, 1H), 4,64 (дд, J=14,1, 1,5 Гц, 1H), 4,84 (дд, J=11,3, 4,9 Гц, 1H), 6,96-6,99 (м, 1H), 7,11 (д, J=7,8 Гц, 1H), 7,38-7,45 (м, 1H), 7,46-7,51 (м, 1H), 7,54 (д, J=7,8 Гц, 1H), 7,71 (д, J=7,8 Гц, 1H), 7,74 (д, J=7,8 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из антисоединения, с временем удерживания 19,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,95 (дд, J=13,5, 11,3 Гц, 1H), 2,29 (с, 3H), 2,72-2,80 (м, 1H), 3,47 (с, 3H), 4,03-4,09 (м, 1H), 4,15 (с, 3H), 4,43 (дд, J=14,1, 3,2 Гц, 1H), 4,64 (дд, J=14,1, 1,5 Гц, 1H), 4,84 (дд, J=11,3, 4,9 Гц, 1H), 6,96-6,99 (м, 1H), 7,11 (д, J=7,8 Гц, 1H), 7,38-7,45 (м, 1H), 7,46-7,51 (м, 1H), 7,54 (д, J=7,8 Гц, 1H), 7,71 (д, J=7,8 Гц, 1H), 7,74 (д, J=7,8 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из син-соединения, с временем удерживания 22,6 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,02 (ддд, J=12,9, 9,8, 9,8 Гц, 1H), 2,29 (с, 3H), 2,69-2,78 (м, 1H), 3,41 (с, 3H), 3,96-4,04 (м, 1H), 4,21 (с, 3H), 4,24 (дд, J=12,9, 8,6 Гц, 1H), 4,68-4,76 (м, 2H), 6,97-6,99 (м, 1H), 7,12 (д, J=7,8 Гц, 1H), 7,37-7,43 (м, 1H), 7,45-7,51 (м, 1H), 7,55 (д, J=7,8 Гц, 1H), 7,72 (д, J=7,8 Гц, 1H), 7,73 (д, J=7,8 Гц, 1H), 7,81 (д, J=1,7 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из син-соединения, с временем удерживания 38,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,02 (ддд, J=12,9, 9,8, 9,8 Гц, 1H), 2,29 (с, 3H), 2,69-2,78 (м, 1H), 3,41 (с, 3H), 3,96-4,04 (м, 1H), 4,21 (с, 3H), 4,24 (дд, J=12,9, 8,6 Гц, 1H), 4,68-4,76 (м, 2H), 6,97-6,99 (м, 1H), 7,12 (д, J=7,8 Гц, 1H), 7,37-7,43 (м, 1H), 7,45-7,51 (м, 1H), 7,55 (д, J=7,8 Гц, 1H), 7,72 (д, J=7,8 Гц, 1H), 7,73 (д, J=7,8 Гц, 1H), 7,81 (д, J=1,7 Гц, 1H).
Соединения примеров 30-41 получали таким же способом, как в примерах 1 и 2 (таблица 1).
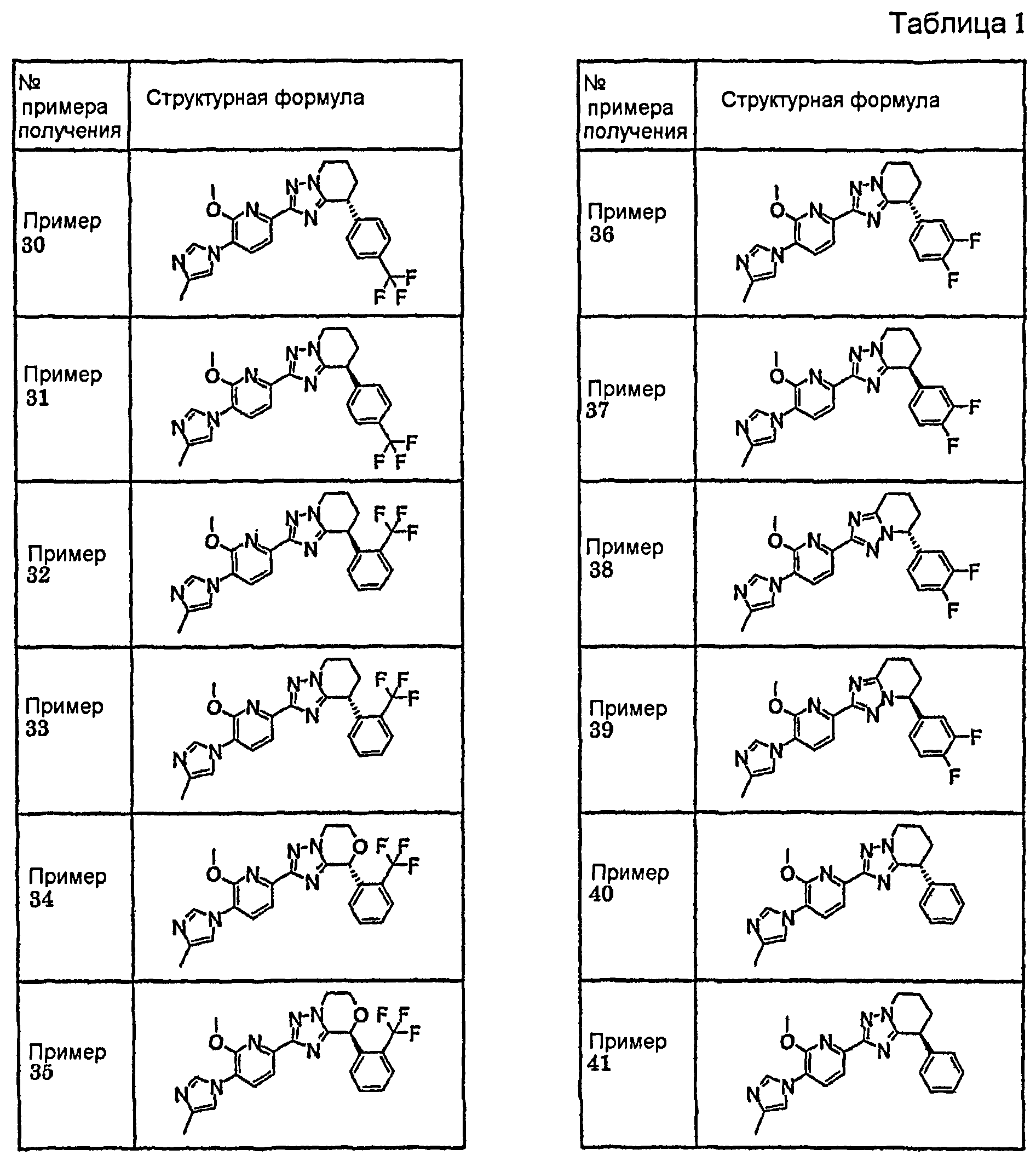
Примеры 42 и 43
Синтез (-)-8-(4-бром-2-трифторметилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (+)-8-(4-бром-2-трифторметилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
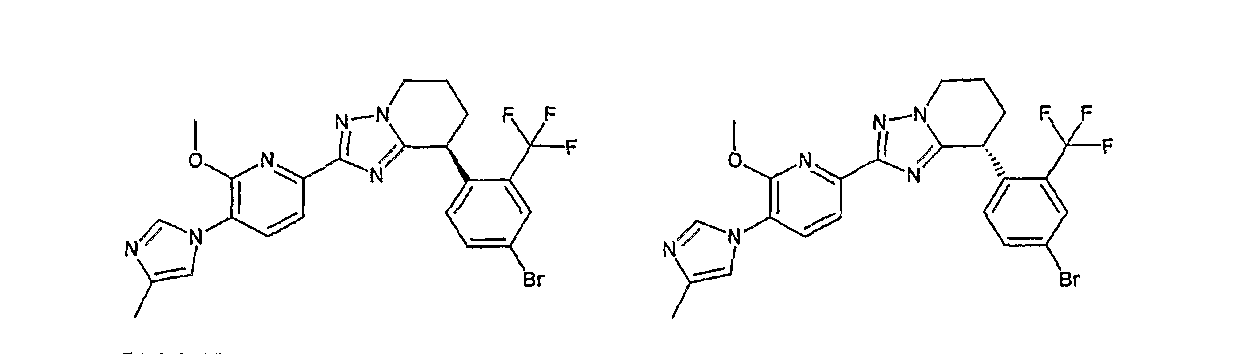
Гидрохлорид этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата, полученного в примере получения 3-1 (100 мг), добавляли к суспензии гидрохлорида гидразида 6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-карбоновой кислоты, полученного в примере получения 1-6 (50 мг), и имидазола (83 мг) в N,N-диметилформамиде (1 мл) и смесь перемешивали при комнатной температуре в течение трех часов. К реакционному раствору добавляли этанол (1 мл) с последующим перемешиванием при комнатной температуре в течение пяти суток. Затем реакционный раствор перемешивали при 70°C в течение двух часов. Реакционный раствор охлаждали до комнатной температуры. Затем добавляли этилацетат, воду и 2 н. хлористоводородную кислоту (0,3 мл) и отделяли органический слой. Полученный органический слой промывали последовательно водой и насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Chromatorex™ NH; элюирующий растворитель: этилацетат:гептан = 1:3 → 1:1 → 1:0) с получением рацемата указанного в заголовке соединения. Полученный рацемат разделяли с помощью CHIRALPAK™ AD-H (2 см × 25 см; подвижная фаза: этанол) с получением указанного в заголовке оптически активного соединения с временем удерживания 18 минут и отрицательным оптическим вращением (31,3 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 29 минут и положительным оптическим вращением (29,9 мг, >98% ee).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 535 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,87-1,98 (м, 1H), 2,10-2,35 (м, 2H), 2,30 (с, 3H), 2,42-2,53 (м, 1H), 4,15 (с, 3H), 4,35-4,51 (м, 2H), 4,69 (дд, J=8,4, 6,0 Гц, 1H), 6,88 (д, J=8,4 Гц, 1H), 6,99 (ушир.с, 1H), 7,56 (д, J=8,0 Гц, 1H), 7,59 (дд, J=8,4, 2,0 Гц, 1H), 7,72 (д, J=8,0 Гц, 1H), 7,81 (д, J=1,6 Гц, 1H), 7,87 (д, J=2,0 Гц, 1H).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 533 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,87-1,98 (м, 1H), 2,10-2,35 (м, 2H), 2,30 (с, 3H), 2,42-2,53 (м, 1H), 4,15 (с, 3H), 4,35-4,51 (м, 2H), 4,69 (дд, J=8,4, 6,0 Гц, 1H), 6,88 (д, J=8,4 Гц, 1H), 6,99 (ушир.с, 1H), 7,56 (д, J=8,0 Гц, 1H), 7,59 (дд, J=8,4, 2,0 Гц, 1H), 7,72 (д, J=8,0 Гц, 1H), 7,81 (д, J=1,6 Гц, 1H), 7,87 (д, J=2,0 Гц, 1H).
Примеры 44 и 45
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(2-трифторметилфенил)-6,7-дигидро-5H-пирроло[1,2-b][1,2,4]триазола и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(2-трифторметилфенил)-6,7-дигидро-5H-пирроло[1,2-b][1,2,4]триазола
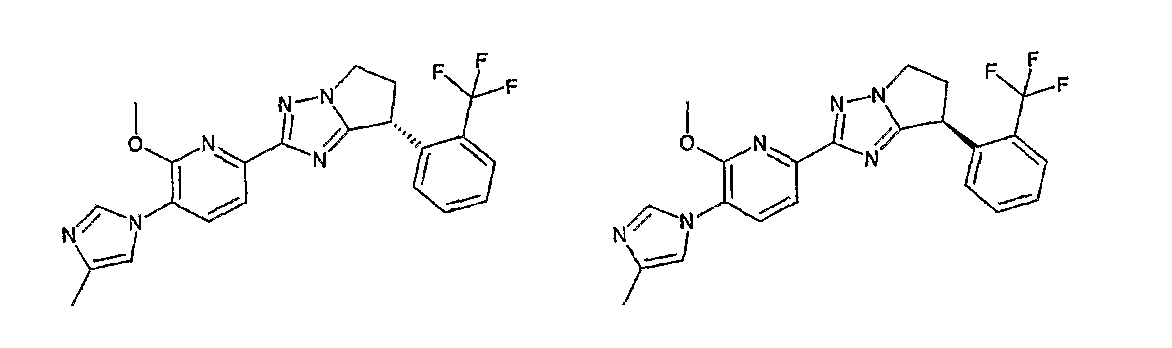
Рацемат указанного в заголовке соединения (81 мг) получали в соответствии со способом примеров 42 и 43 из гидрохлорида гидразида 6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-карбоновой кислоты, полученного в примере получения 1-6 (250 мг), и из гидрохлорида этил 4-хлор-2-(2-трифторметилфенил)бутилимидата, полученного в примере получения 3-14 (367 мг), вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; подвижная фаза: гексан:этанол = 7:3) с получением указанного в заголовке оптически активного соединения с положительным оптическим вращением (10 мг, >99% ee) и указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (10 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 441 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,32 (с, 3H), 2,67-2,78 (м, 1H), 3,34-3,43 (м, 1H), 4,10 (с, 3H), 4,46-4,54 (м, 1H), 4,64-4,72 (м, 1H), 4,97 (дд, J=8,4, 8,0 Гц, 1H), 7,03 (д, J=1,2 Гц, 1H), 7,24 (д, J=8,0 Гц, 1H), 7,41 (дд, J=8,0, 7,6 Гц, 1H), 7,52 (дд, J=7,6, 6,8 Гц, 1H), 7,71-7,75 (м, 2H), 7,87 (д, J=1,2 Гц, 1H), 8,08 (д, J=8,0 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 441 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,32 (с, 3H), 2,67-2,78 (м, 1H), 3,34-3,43 (м, 1H), 4,10 (с, 3H), 4,46-4,54 (м, 1H), 4,64-4,72 (м, 1H), 4,97 (дд, J=8,4, 8,0 Гц, 1H), 7,03 (д, J=1,2 Гц, 1H), 7,24 (д, J=8,0 Гц, 1H), 7,41 (дд, J=8,0, 7,6 Гц, 1H), 7,52 (дд, J=7,6, 6,8 Гц, 1H), 7,71-7,75 (м, 2H), 7,87 (д, J=1,2 Гц, 1H), 8,08 (д, J=8,0 Гц, 1H).
Примеры 46 и 47
Синтез (+)-8-(бифенил-4-ил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-(бифенил-4-ил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
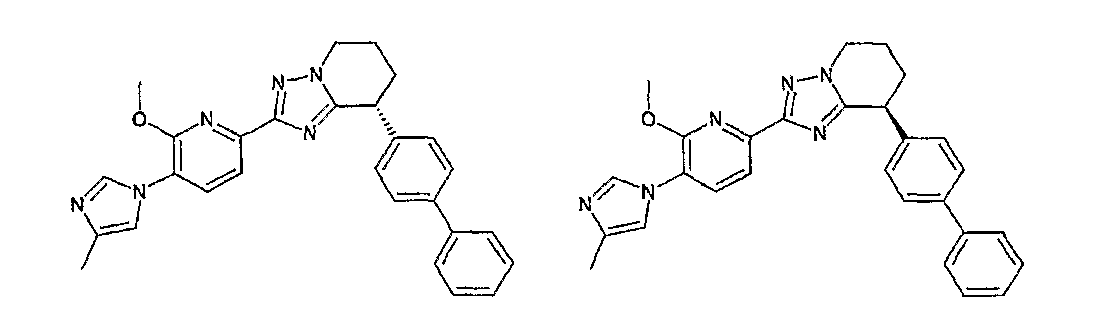
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-5, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: 50% этанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 13 минут и положительным оптическим вращением (21,9 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 20 минут и отрицательным оптическим вращением (20,9 мг, >98% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 463 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,05-2,33 (м, 3H), 2,30 (с, 3H), 2,35-2,46 (м, 1H), 4,17 (с, 3H), 4,35-4,51 (м, 3H), 7,00 (ушир.с, 1H), 7,20 (д, J=8,0 Гц, 2H), 7,34 (т, J=8,0 Гц, 1H), 7,43 (т, J=8,0 Гц, 2H), 7,54-7,61 (м, 5H), 7,79-7,85 (м, 2H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 463 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,05-2,33 (м, 3H), 2,30 (с, 3H), 2,35-2,46 (м, 1H), 4,17 (с, 3H), 4,35-4,51 (м, 3H), 7,00 (ушир.с, 1H), 7,20 (д, J=8,0 Гц, 2H), 7,34 (т, J=8,0 Гц, 1H), 7,43 (т, J=8,0 Гц, 2H), 7,54-7,61 (м, 5H), 7,79-7,85 (м, 2H).
Примеры 48 и 49
Синтез (-)-8-(бифенил-2-ил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (+)-8-(бифенил-2-ил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
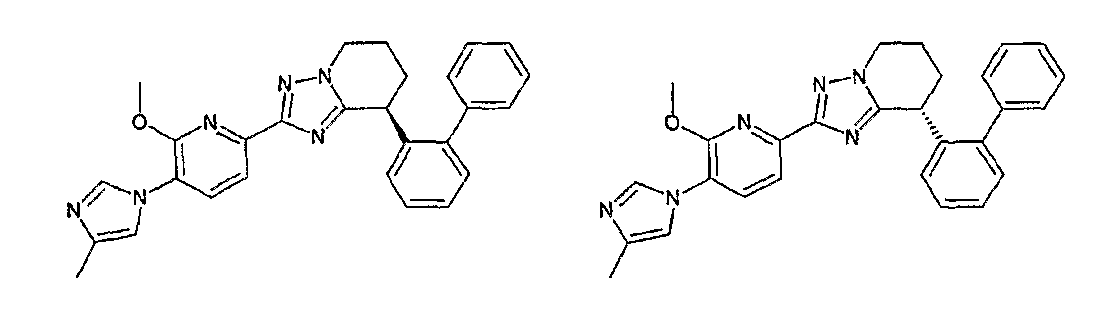
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-6, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: 50% этанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 9 минут и отрицательным оптическим вращением (12,6 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 12 минут и положительным оптическим вращением (11,8 мг, >98% ee).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 463 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,83-1,97 (м, 2H), 2,04-2,21 (м, 2H), 2,30 (с, 3H), 4,16 (с, 3H), 4,25-4,40 (м, 2H), 4,48-4,56 (м, 1H), 6,95 (д, J=7,2 Гц, 1H), 7,00 (ушир.с, 1H), 7,20-7,50 (м, 8H), 7,58 (д, J=8,0 Гц, 1H), 7,80 (д, J=8,0 Гц, 1H), 7,82 (ушир.с, 1H).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 463 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,83-1,97 (м, 2H), 2,04-2,21 (м, 2H), 2,30 (с, 3H), 4,16 (с, 3H), 4,25-4,40 (м, 2H), 4,48-4,56 (м, 1H), 6,95 (д, J=7,2 Гц, 1H), 7,00 (ушир.с, 1H), 7,20-7,50 (м, 8H), 7,58 (д, J=8,0 Гц, 1H), 7,80 (д, J=8,0 Гц, 1H), 7,82 (ушир.с, 1H).
Примеры 50 и 51
Синтез (+)-8-(бифенил-3-ил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-(бифенил-3-ил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
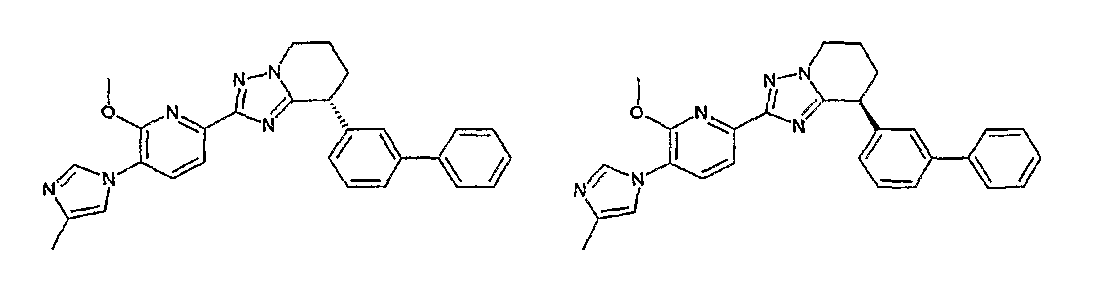
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-7, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: 50% этанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 9 минут и положительным оптическим вращением (40,9 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 19 минут и отрицательным оптическим вращением (39,8 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 463 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,05-2,34 (м, 3H), 2,30 (д, J=1,2 Гц, 3H), 2,37-2,47 (м, 1H), 4,17 (с, 3H), 4,35-4,55 (м, 3H), 7,00 (т, J=1,2 Гц, 1H), 7,09 (д, J=8,0 Гц, 1H), 7,31-7,37 (м, 2H), 7,37-7,44 (м, 3H), 7,48-7,56 (м, 3H), 7,58 (д, J=8,0 Гц, 1H), 7,81 (д, J=8,0 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 463 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,05-2,34 (м, 3H), 2,30 (д, J=1,2 Гц, 3H), 2,37-2,47 (м, 1H), 4,17 (с, 3H), 4,35-4,55 (м, 3H), 7,00 (т, J=1,2 Гц, 1H), 7,09 (д, J=8,0 Гц, 1H), 7,31-7,37 (м, 2H), 7,37-7,44 (м, 3H), 7,48-7,56 (м, 3H), 7,58 (д, J=8,0 Гц, 1H), 7,81 (д, J=8,0 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H).
Примеры 52 и 53
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-фторфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-фторфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
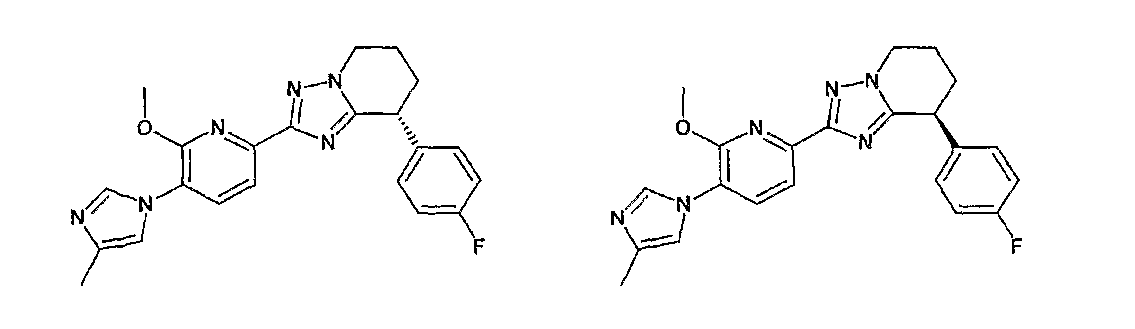
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-15, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; подвижная фаза: гексан:этанол = 5:5) с получением указанного в заголовке оптически активного соединения с положительным оптическим вращением (74 мг, >99% ee) и указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (75 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 405 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,03-2,26 (м, 3H), 2,30 (с, 3H), 2,33-2,40 (м, 1H), 4,16 (с, 3H), 4,34-4,45 (м, 3H), 7,00-7,06 (м, 3H), 7,08-7,13 (м, 2H), 7,59 (д, J=8,0 Гц, 1H), 7,78 (д, J=8,0 Гц, 1H), 8,82 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 405 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,03-2,26 (м, 3H), 2,30 (с, 3H), 2,33-2,40 (м, 1H), 4,16 (с, 3H), 4,34-4,45 (м, 3H), 7,00-7,06 (м, 3H), 7,08-7,13 (м, 2H), 7,59 (д, J=8,0 Гц, 1H), 7,78 (д, J=8,0 Гц, 1H), 8,82 (д, J=1,2 Гц, 1H).
Примеры 54 и 55
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
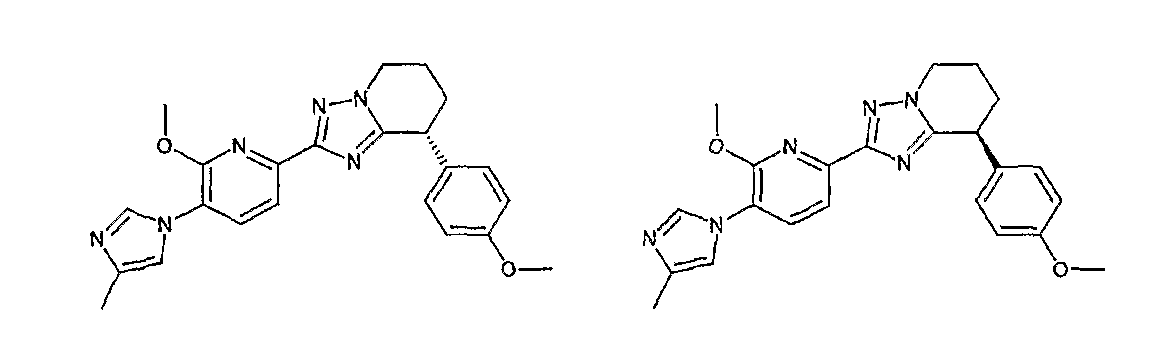
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-16, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; подвижная фаза: гексан:этанол = 5:5) с получением указанного в заголовке оптически активного соединения с положительным оптическим вращением (64 мг, >99% ee) и указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (55 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 417 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,03-2,12 (м, 2H), 2,15-2,26 (м, 1H), 2,30 (с, 3H), 2,30-2,38 (м, 1H), 3,79 (с, 3H), 4,16 (с, 3H), 4,33-4,45 (м, 3H), 6,84-6,89 (м, 2H), 6,99-7,08 (м, 3H), 7,58 (д, J=8,0 Гц, 1H), 7,79 (д, J=8,0 Гц, 1H), 7,82 (д, J=0,8 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 417 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,03-2,12 (м, 2H), 2,15-2,26 (м, 1H), 2,30 (с, 3H), 2,30-2,38 (м, 1H), 3,79 (с, 3H), 4,16 (с, 3H), 4,33-4,45 (м, 3H), 6,84-6,89 (м, 2H), 6,99-7,08 (м, 3H), 7,58 (д, J=8,0 Гц, 1H), 7,79 (д, J=8,0 Гц, 1H), 7,82 (д, J=0,8 Гц, 1H).
Примеры 56 и 57
Синтез (+)-8-(5-трет-бутил-2-метоксифенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-(5-трет-бутил-2-метоксифенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
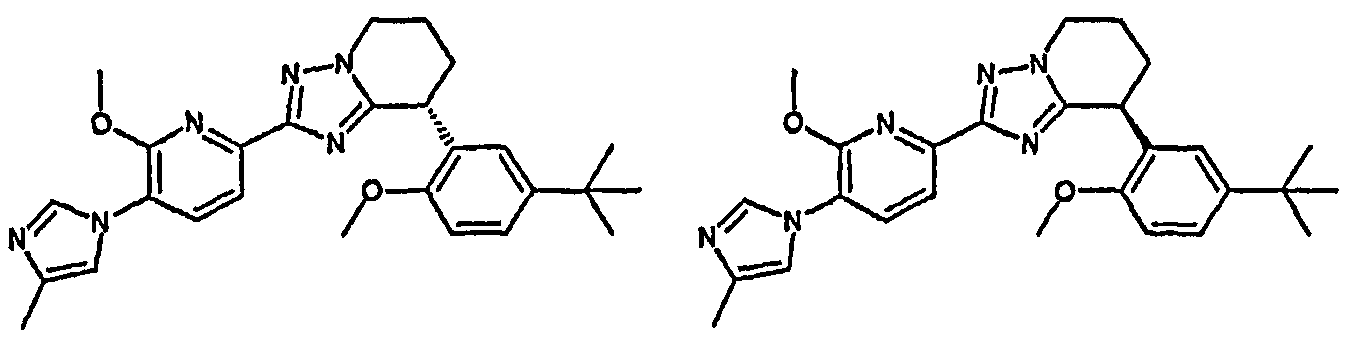
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-3, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ AD-H (2 см × 25 см; подвижная фаза: 25% этанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 10 минут и положительным оптическим вращением (41,8 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 17 минут и отрицательным оптическим вращением (33,5 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 473 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,23 (с, 9H), 2,00-2,35 (м, 4H), 2,30 (д, J=1,2 Гц, 3H), 3,75 (с, 3H), 4,17 (с, 3H), 4,39 (т, J=5,6 Гц, 2H), 4,62 (т, J=6,4 Гц, 1H), 6,83 (д, J=8,4 Гц, 1H), 6,91 (д, J=2,4 Гц, 1H), 6,99 (т, J=1,2 Гц, 1H), 7,23-7,29 (м, 1H), 7,57 (д, J=8,0 Гц, 1H), 7,78 (д, J=8,0 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 473 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,23 (с, 9H), 2,00-2,35 (м, 4H), 2,30 (д, J=1,2 Гц, 3H), 3,75 (с, 3H), 4,17 (с, 3H), 4,39 (т, J=5,6 Гц, 2H), 4,62 (т, J=6,4 Гц, 1H), 6,83 (д, J=8,4 Гц, 1H), 6,91 (д, J=2,4 Гц, 1H), 6,99 (т, J=1,2 Гц, 1H), 7,23-7,29 (м, 1H), 7,57 (д, J=8,0 Гц, 1H), 7,78 (д, J=8,0 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Примеры 58 и 59
Синтез (+)-8-(3-трет-бутил-4-метоксифенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-(3-трет-бутил-4-метоксифенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
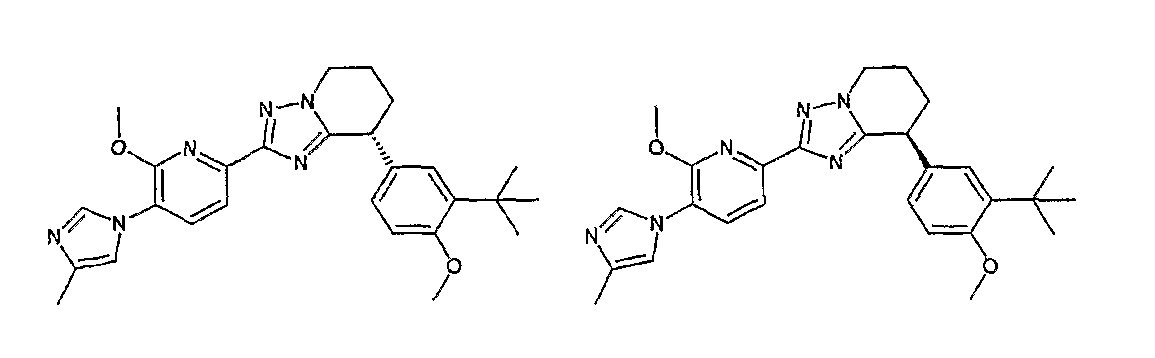
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-8, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ AD-H (2 см × 25 см; подвижная фаза: 30% этанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 11 минут и положительным оптическим вращением (43,5 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 18 минут и отрицательным оптическим вращением (42,8 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 473 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,35 (с, 9H), 2,00-2,40 (м, 4H), 2,30 (с, 3H), 3,81 (с, 3H), 4,17 (с, 3H), 4,30-4,46 (м, 3H), 6,79 (д, J=8,4 Гц, 1H), 6,82 (дд, J=8,4, 2,4 Гц, 1H), 7,00 (ушир.с, 1H), 7,13 (J=2,4 Гц, 1H), 7,58 (д, J=8,0 Гц, 1H), 7,81 (д, J=8,0 Гц, 1H), 7,82 (с, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 473 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,35 (с, 9H), 2,00-2,40 (м, 4H), 2,30 (с, 3H), 3,81 (с, 3H), 4,17 (с, 3H), 4,30-4,46 (м, 3H), 6,79 (д, J=8,4 Гц, 1H), 6,82 (дд, J=8,4, 2,4 Гц, 1H), 7,00 (ушир.с, 1H), 7,13 (J=2,4 Гц, 1H), 7,58 (д, J=8,0 Гц, 1H), 7,81 (д, J=8,0 Гц, 1H), 7,82 (с, 1H).
Примеры 60 и 61
Синтез (+)-8-(3,3-диметил-2,3-дигидробензофуран-5-ил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-(3,3-диметил-2,3-дигидробензофуран-5-ил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
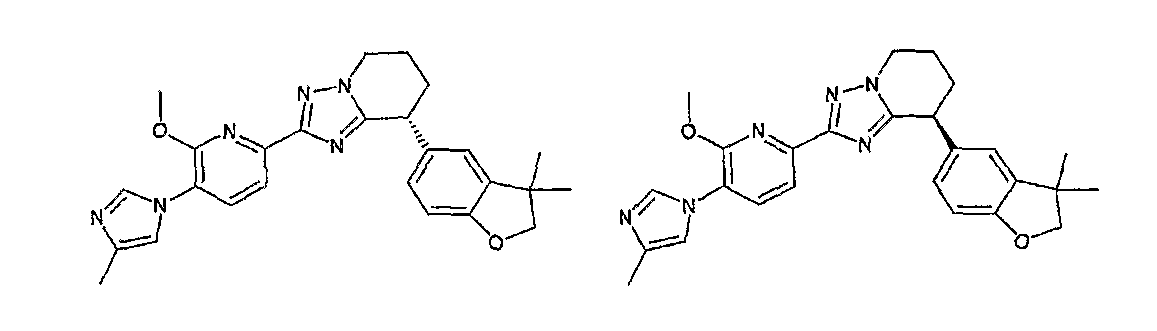
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-2, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ AD-H (2 см × 25 см; подвижная фаза: 50% этанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 10 минут и положительным оптическим вращением (54,0 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 18 минут и отрицательным оптическим вращением (47,4 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 457 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,30 (с, 3H), 1,32 (с, 3H), 2,00-2,13 (м, 2H), 2,16-2,40 (м, 2H), 2,30 (с, 3H), 4,16 (с, 3H), 4,23 (с, 2H), 4,30-4,47 (м, 3H), 6,72 (д, J=8,0 Гц, 1H), 6,83 (дд, J=8,0, 1,6 Гц, 1H), 6,87 (д, J=1,6 Гц, 1H), 7,00 (ушир.с, 1H), 7,58 (д, J=8,0 Гц, 1H), 7,80 (д, J=8,0 Гц, 1H), 7,82 (с, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 457 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,30 (с, 3H), 1,32 (с, 3H), 2,00-2,13 (м, 2H), 2,16-2,40 (м, 2H), 2,30 (с, 3H), 4,16 (с, 3H), 4,23 (с, 2H), 4,30-4,47 (м, 3H), 6,72 (д, J=8,0 Гц, 1H), 6,83 (дд, J=8,0, 1,6 Гц, 1H), 6,87 (д, J=1,6 Гц, 1H), 7,00 (ушир.с, 1H), 7,58 (д, J=8,0 Гц, 1H), 7,80 (д, J=8,0 Гц, 1H), 7,82 (с, 1H).
Примеры 62 и 63
Синтез (-)-8-(3-трет-бутил-2-метоксифенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (+)-8-(3-трет-бутил-2-метоксифенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
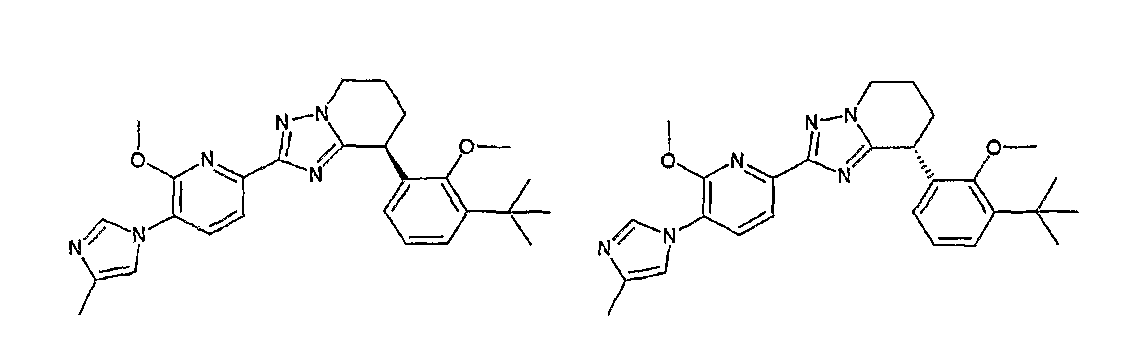
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-9, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; подвижная фаза: 10% этанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 23 минуты и отрицательным оптическим вращением (48,5 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 38 минут и положительным оптическим вращением (53,8 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 473 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,44 (с, 9H), 1,98-2,16 (м, 2H), 2,18-2,40 (м, 2H), 2,30 (д, J=1,2 Гц, 3H), 3,98 (с, 3H), 4,16 (с, 3H), 4,36-4,46 (м, 2H), 4,73 (т, J=6,8 Гц, 1H), 6,71 (дд, J=8,0, 1,6 Гц, 1H), 6,96 (т, J=8,0, 1H), 6,99 (ушир.с, 1H), 7,25-7,29 (м, 1H), 7,56 (д, J=7,6 Гц, 1H), 7,76 (д, J=7,6 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 473 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,44 (с, 9H), 1,98-2,16 (м, 2H), 2,18-2,40 (м, 2H), 2,30 (д, J=1,2 Гц, 3H), 3,98 (с, 3H), 4,16 (с, 3H), 4,36-4,46 (м, 2H), 4,73 (т, J=5,6 Гц, 1H), 6,71 (дд, J=8,0, 1,6 Гц, 1H), 6,96 (т, J=8,0, 1H), 6,99 (ушир.с, 1H), 7,25-7,29 (м, 1H), 7,56 (д, J=7,6 Гц, 1H), 7,76 (д, J=7,6 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Примеры 64 и 65
Синтез (+)-8-(3-бромфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина и (-)-8-(3-бромфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина
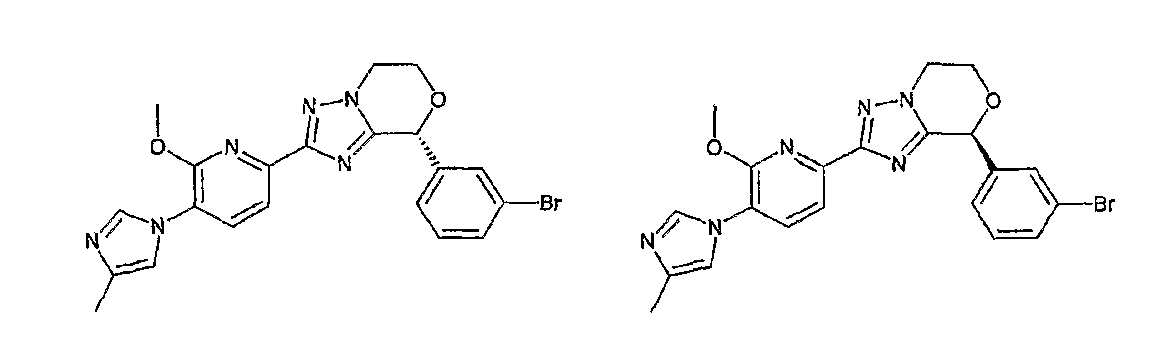
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-17, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 5,27 минуты, полученным из анализа с помощью CHIRALPAK™ IB (Lot. IB00CD-LG026, гексан:этанол = 8:2, 1,0 мл/мин), и положительным оптическим вращением и указанного в заголовке оптически активного соединения с временем удерживания 7,98 минуты, полученным из такого же анализа, и отрицательным оптическим вращением, соответственно.
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 4,11-4,21 (м, 1H), 4,17 (с, 3H), 4,31 (дт, J=4,8, 12,4 Гц, 1H), 4,34-4,51 (м, 2H), 5,98 (с, 1H), 7,01 (т, J=0,8 Гц, 1H), 7,29 (т, J=8,0 Гц, 1H), 7,45-7,47 (м, 1H), 7,51-7,54 (м, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,67 (д, J=1,6 Гц, 1H), 7,82 (д, J=8,0 Гц, 1H), 7,84 (д, J=1,6 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 4,11-4,21 (м, 1H), 4,17 (с, 3H), 4,31 (дт, J=4,8, 12,4 Гц, 1H), 4,34-4,51 (м, 2H), 5,98 (с, 1H), 7,01 (т, J=0,8 Гц, 1H), 7,29 (т, J=8,0 Гц, 1H), 7,45-7,47 (м, 1H), 7,51-7,54 (м, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,67 (д, J=1,6 Гц, 1H), 7,82 (д, J=8,0 Гц, 1H), 7,84 (д, J=1,6 Гц, 1H).
Примеры 66 и 67
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(3-нитрофенил)-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(3-нитрофенил)-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина
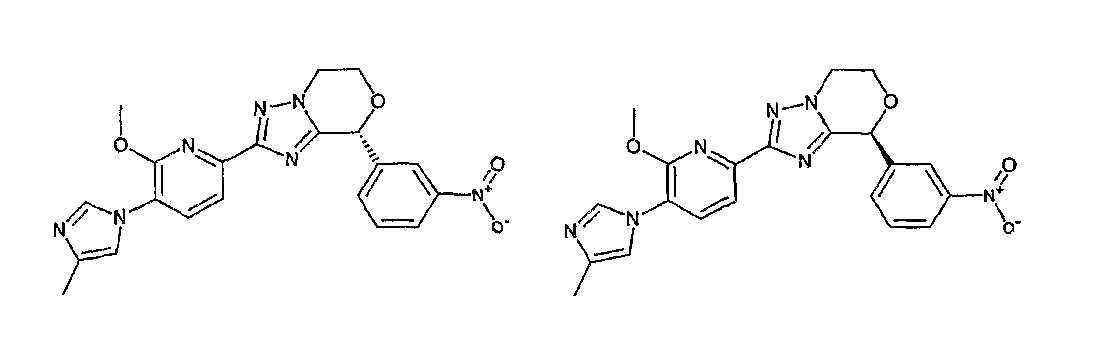
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-18, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IB производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 10,6 минуты, полученным из анализа с помощью CHIRALPAK™ IB (Lot. IB00CD-FD026, гексан:этанол = 7:3, 1,0 мл/мин), и положительным оптическим вращением и указанного в заголовке оптически активного соединения с временем удерживания 15,0 минуты, полученным из такого же анализа, и отрицательным оптическим вращением, соответственно.
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 4,16 (с, 3H), 4,25 (ддд, J=4,4, 8,0, 12,4 Гц, 1H), 4,40 (дт, J=4,0, 12,4 Гц, 1H), 4,45-4,56 (м, 2H), 6,08 (с, 1H), 7,01 (т, J=0,8 Гц, 1H), 7,62 (т, J=7,6 Гц, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,80 (д, J=7,6 Гц, 1H), 7,84 (д, J=1,2 Гц, 1H), 7,94-7,96 (м, 1H), 8,25-8,27 (м, 1H), 8,48 (м, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 4,16 (с, 3H), 4,25 (ддд, J=4,4, 8,0, 12,4 Гц, 1H), 4,40 (дт, J=4,0, 12,4 Гц, 1H), 4,45-4,56 (м, 2H), 6,08 (с, 1H), 7,01 (т, J=0,8 Гц, 1H), 7,62 (т, J=7,6 Гц, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,80 (д, J=7,6 Гц, 1H), 7,84 (д, J=1,2 Гц, 1H), 7,94-7,96 (м, 1H), 8,25-8,27 (м, 1H), 8,48 (м, 1H).
Примеры 68 и 69
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-бром-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-бром-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
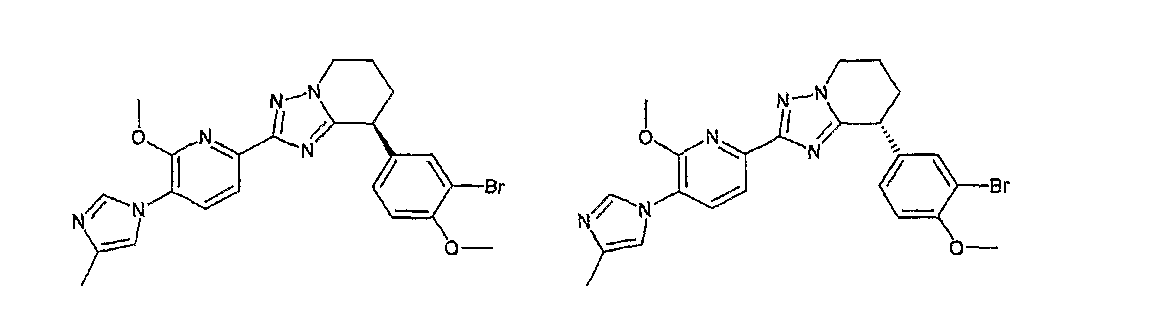
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-4, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: этанол:гексан = 3:7, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 37,5 минуты и положительным оптическим вращением (3,52 мг) и указанного в заголовке соединения с временем удерживания 44 минуты и отрицательным оптическим вращением (2,53 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 37,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,12-2,17 (м, 2H), 2,28-2,29 (м, 1H), 2,31-2,41 (м, 1H), 2,41 (с, 3H), 3,89 (с, 3H), 4,19 (с, 3H), 4,32-4,42 (м, 3H), 6,86 (д, J=8,6 Гц, 1H), 7,03-7,08 (м, 2H), 7,33 (д, J=1,9 Гц, 1H), 7,65 (д, J=7,8 Гц, 1H), 7,87 (д, J=7,8 Гц, 1H), 8,25 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 44 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,12-2,17 (м, 2H), 2,28-2,29 (м, 1H), 2,31-2,41 (м, 1H), 2,41 (с, 3H), 3,89 (с, 3H), 4,19 (с, 3H), 4,32-4,42 (м, 3H), 6,86 (д, J=8,6 Гц, 1H), 7,03-7,08 (м, 2H), 7,33 (д, J=1,9 Гц, 1H), 7,65 (д, J=7,8 Гц, 1H), 7,87 (д, J=7,8 Гц, 1H), 8,25 (д, J=1,2 Гц, 1H).
Примеры 70 и 71
(+)-8-(6-Бромпиридин-2-ил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-(6-бромпиридин-2-ил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
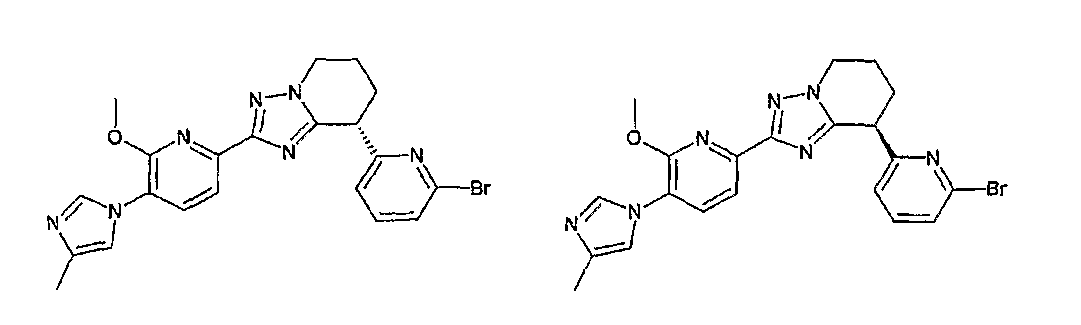
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-10, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат (37,2 мг) отделяли с помощью CHIRALPAK™ IA производства Daicel Chemical Industries, Ltd. (2 см × 25 см; подвижная фаза: гексан:этанол = 50:50) с получением указанного в заголовке оптически активного соединения с временем удерживания 5,1 минуты и положительным оптическим вращением (11 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 8,9 минуты и отрицательным оптическим вращением (9,1 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 466 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,08-2,27 (м, 2H), 2,30 (д, J=1,2 Гц, 3H), 2,38-2,43 (м, 2H), 4,16 (с, 3H), 4,31-4,45 (м, 2H), 4,54 (т, J=6,4 Гц, 1H), 7,00 (т, J=1,2 Гц, 1H), 7,14 (дд, J=0,8 Гц, 8,0 Гц, 1H), 7,39 (дд, J=0,8 Гц, 8,0 Гц, 1H), 7,50 (т, J=8,0 Гц, 1H), 7,60 (д, J=7,6 Гц, 1H), 7,78 (д, J=7,6 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 466 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,08-2,27 (м, 2H), 2,30 (д, J=1,2 Гц, 3H), 2,38-2,43 (м, 2H), 4,16 (с, 3H), 4,31-4,45 (м, 2H), 4,54 (т, J=6,4 Гц, 1H), 7,00 (т, J=1,2 Гц, 1H), 7,14 (дд, J=0,8 Гц, 8,0 Гц, 1H), 7,39 (дд, J=0,8 Гц, 8,0 Гц, 1H), 7,50 (т, J=8,0 Гц, 1H), 7,60 (д, J=7,6 Гц, 1H), 7,78 (д, J=7,6 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H).
Пример 72
Синтез 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-7,8-дигидро-6H-[1,2,4]триазоло[1,5-a]пиридина-5-она
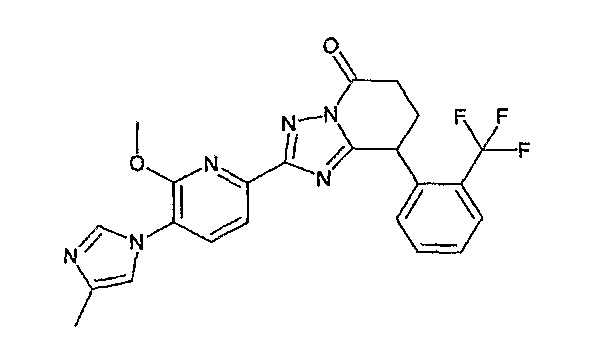
Синтез этил 4-[5-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-2H-[1,2,4]триазол-3-ил]-4-(2-трифторметилфенил)бутирата
Указанное в заголовке соединение (370 мг) получали в соответствии с примерами 42 и 43 из гидрохлорида этил 4-этоксикарбонимидоил-4-(2-трифторметилфенил)бутирата, полученного в примере получения 3-19 (244 мг). Полученное соединение использовали непосредственно для следующей стадии в виде неочищенного продукта.
Синтез 4-[5-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-2H-[1,2,4]триазол-3-ил]-4-(2-трифторметилфенил)масляной кислоты
Этил 4-[5-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-2H-[1,2,4]триазол-3-ил]-4-(2-трифторметилфенил)бутират (370 мг) растворяли в тетрагидрофуране (1 мл). Добавляли воду (1 мл) и 5 н. гидроксид натрия (1 мл) и начинали реакцию. Через три часа реакционный раствор распределяли между диэтиловым эфиром и водой. Полученный водный слой нейтрализовали 5 н. хлористоводородной кислотой и экстрагировали метиленхлоридом два раза. Концентрирование при пониженном давлении дало оранжевое масло (208 мг), которое использовали для следующей стадии.
ESI-МС; m/z 487 [M++H].
Синтез 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-7,8-дигидро-6H-[1,2,4]триазоло[1,5-a]пиридина-5-она
4-[5-[6-Метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-2H-[1,2,4]триазол-3-ил]-4-(2-трифторметилфенил)масляную кислоту (208 мг) растворяли в тетрагидрофуране (4,3 мл). Диизопропилэтиламин (220 мкл), 1-гидроксибензотриазол (86,8 мг) и гексафторфосфат бензотриазол-1-илокситрис(пирролидино)фосфония (334 мг) добавляли при комнатной температуре и начинали реакцию. Через 19 часов реакцию останавливали водой с последующим разбавлением реакционной смеси этилацетатом. Органический слой промывали насыщенным раствором хлорида аммония и насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-100%) с получением указанного в заголовке соединения (128 мг) в виде оранжевого твердого вещества.
1H-ЯМР (CDCl3) δ (ppm): 2,17 (с, 3H), 2,18-2,34 (м, 2H), 2,89-2,97 (м, 1H), 3,23-3,34 (м, 1H), 4,09 (с, 3H), 4,87 (дд, J=11,8, 5,5 Гц, 1H), 7,31-7,35 (м, 1H), 7,40-7,48 (м, 1H), 7,53-7,59 (м, 1H), 7,66-7,75 (м, 2H), 7,82 (д, J=7,8 Гц, 1H), 7,93 (д, J=7,8 Гц, 1H), 7,97-8,01 (м, 1H).
Соединения примеров 74-83 получали таким же способом, как в примерах 42 и 43 (таблица 2).
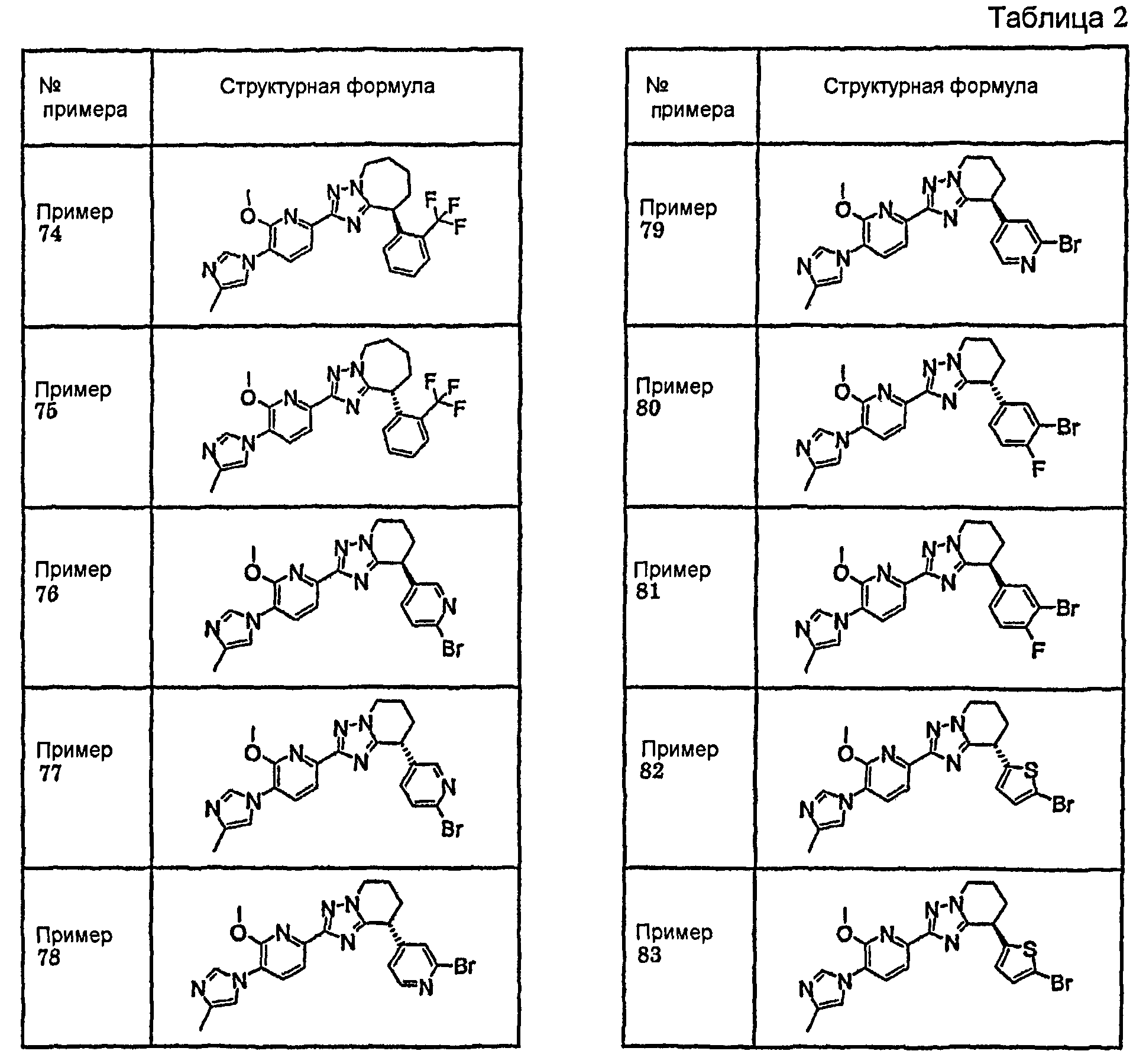
Примеры 84 и 85
Синтез (+)-7-бензил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина и (-)-7-бензил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
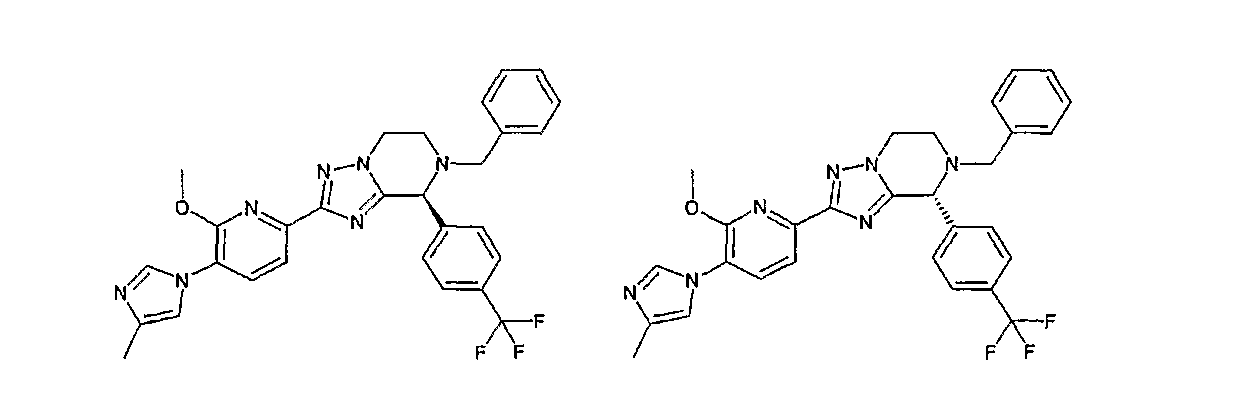
Синтез [бензил(2-оксоэтил)амино]-(4-трифторметилфенил)ацетонитрила
Диметилсульфоксид (689 мкл) медленно добавляли по каплям к раствору оксалилхлорида (584 мкл) в дихлорметане (15 мл) при -78°C. После перемешивания при той же самой температуре в течение 10 минут, к реакционному раствору добавляли раствор [бензил-(2-гидроксиэтил)амино]-(4-трифторметилфенил)ацетонитрила, полученного в примере получения 4-1 (1,08 г), в дихлорметане (5 мл) и смесь перемешивали при той же самой температуре в течение 10 минут. Триэтиламин (1,8 мл) добавляли к реакционному раствору и смесь перемешивали при той же самой температуре в течение 10 минут и затем при 0°C в течение 20 минут. Реакционный раствор распределяли между насыщенным раствором бикарбоната натрия и этилацетатом. Полученный органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: гептан:этилацетат = 9:1 → 8:1) с получением 959 мг указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 3,37-3,47 (м, 2H), 3,62 (д, J=13,2 Гц, 1H), 3,91 (д, J=12,8 Гц, 1H), 5,08 (с, 1H), 7,30-7,43 (м, 5H), 7,69 (д, J=8,0 Гц, 1H), 7,76 (д, J=8,0 Гц, 1H) 9,52 (м, 1H).
Синтез N′-(2-{бензил[циано(4-трифторметилфенил)метил]амино}этил)гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты
Гидрохлорид гидразида 6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-карбоновой кислоты, полученный в примере получения 1-6 (852 мг), метанол (10 мл) и уксусную кислоту (1 мл) последовательно добавляли к раствору [бензил(2-оксоэтил)амино]-(4-трифторметилфенил)ацетонитрила (950 мг) в дихлорметане (50 мл) и смесь перемешивали при комнатной температуре в течение пяти минут. Цианоборгидрид натрия (359 мг) добавляли к реакционному раствору с последующим перемешиванием при комнатной температуре в течение 16 часов. Реакционный раствор распределяли между насыщенным раствором бикарбоната натрия и этилацетатом. Органический слой сушили над безводным сульфатом магния, давали ему пройти через рыхлый слой диоксида кремния и затем концентрировали его при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: этилацетат:метанол = 1:0 → 9:1) с получением 1,31 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,30 (с, 3H), 2,74-2,96 (м, 3H), 3,03-3,35 (м, 1H), 3,45 (д, J=13,2 Гц, 1H), 4,02 (с, 3H), 4,07 (д, J=12,8 Гц, 1H), 5,01 (м, 1H), 5,23 (с, 1H), 7,01 (т, J=1,2 Гц, 1H), 7,30-7,32 (м, 1H), 7,35-7,39 (м, 2H), 7,44 (м, 2H), 7,68 (д, J=8,4 Гц, 2H), 7,70 (д, J=7,6 Гц, 1H), 7,79 (д, J=8,4 Гц, 2H), 7,86 (с, 1H), 7,87 (д, J=8,0 Гц, 1H), 8,86 (д, J=7,2 Гц, 1H).
Синтез (+)-7-бензил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина и (-)-7-бензил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
Уксусную кислоту (1,37 мл) добавляли к раствору N′-(2-{бензил[циано(4-трифторметилфенил)метил]амино}этил)гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты (1,3 г) в толуоле (13,7 мл) и смесь перемешивали при 80°C в течение двух суток. Реакционный раствор концентрировали и распределяли между насыщенным раствором бикарбоната натрия и этилацетатом. Органический слой сушили над безводным сульфатом магния, давали ему пройти через рыхлый слой диоксида кремния и затем концентрировали при пониженном давлении. Полученный остаток перекристаллизовывали из смеси гептан-этилацетат с получением 308 мг рацемата указанного в заголовке соединения. Полученный рацемат указанного в заголовке соединения (30 мг) разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: гексан:этанол = 7:3, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 13,0 минут, полученным из анализа с помощью CHIRALPAK™ IB производства ф. Daicel Chemical Industries, Ltd. (Lot. IB00CD-LG026, гексан:этанол = 7:3, 1,0 мл/мин) (8,6 мг) и указанного в заголовке оптически активного соединения с временем удерживания 14,9 минуты, полученным из такого же анализа, (7,4 мг), соответственно.
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 13,0 минуты в условиях анализа были следующими.
ESI-MS; m/z 546 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 2,93 (м, 1H), 3,36 (дт, J=4,4, 8,4 Гц, 1H), 3,49 (д, J=13,6 Гц, 1H), 3,87 (д, J=13,2 Гц, 1H), 4,14 (с, 3H), 4,33-4,44 (м, 2H), 4,97 (с, 1H), 6,99 (с, 1H), 7,29-7,38 (м, 5H), 7,57 (д, J=7,6 Гц, 1H), 7,63 (д, J=8,0 Гц, 2H), 7,67 (д, J=8,4 Гц, 2H), 7,73 (д, J=7,6 Гц, 1H), 7,83 (с, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 14,9 минуты в условиях анализа были следующими.
ESI-MS; m/z 546 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 2,93 (м, 1H), 3,36 (дт, J=4,4, 8,4 Гц, 1H), 3,49 (д, J=13,6 Гц, 1H), 3,87 (д, J=13,2 Гц, 1H), 4,14 (с, 3H), 4,33-4,44 (м, 2H), 4,97 (с, 1H), 6,99 (с, 1H), 7,29-7,38 (м, 5H), 7,57 (д, J=7,6 Гц, 1H), 7,63 (д, J=8,0 Гц, 2H), 7,67 (д, J=8,4 Гц, 2H), 7,73 (д, J=7,6 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Примеры 86 и 87
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-7-фенил-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-7-фенил-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
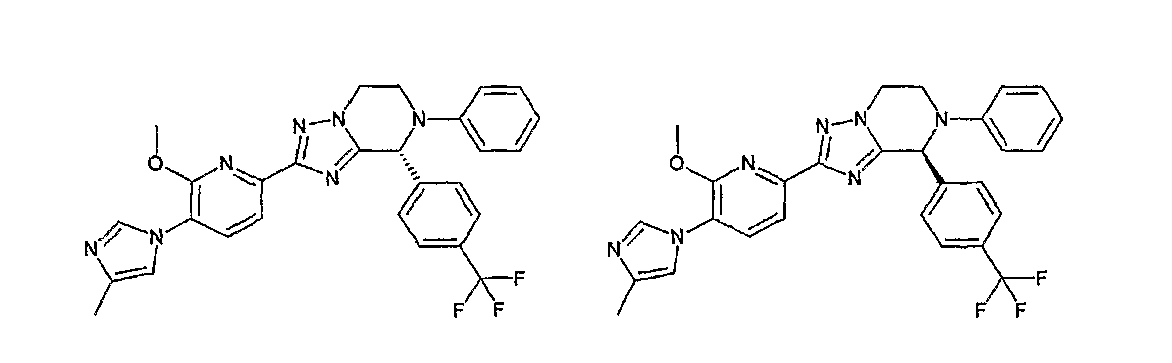
Рацемат указанного в заголовке соединения (48 мг) получали в соответствии со способом примеров 84 и 85 из [(2-гидроксиэтил)фениламино]-(4-трифторметилфенил)ацетонитрила, полученного в примере получения 4-2, вместо [бензил(2-оксоэтил)амино]-(4-трифторметилфенил)ацетонитрила. Полученный рацемат разделяли с помощью CHIRALPAK™ IB производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 8,78 минуты, полученным из анализа с помощью CHIRALPAK™ IB (Lot. IB00CD-FD026, гексан:этанол = 8:2, 1,0 мл/мин) и отрицательным оптическим вращением (14 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 11,27 минуты, полученным из такого же анализа, и положительным оптическим вращением (14 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 532 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 3,69-3,76 (м, 1H), 4,01-4,05 (м, 1H), 4,17 (с, 3H), 4,33-4,38 (м, 1H), 4,42-4,49 (м, 1H), 6,21 (с, 1H), 6,95-7,03 (м, 4H), 7,28-7,33 (м, 2H), 7,57-7,62 (м, 4H), 7,65 (д, J=7,6 Гц, 1H), 7,86 (д, J=1,2 Гц, 1H), 7,88 (д, J=8,0 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 532 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 3,69-3,76 (м, 1H), 4,01-4,05 (м, 1H), 4,17 (с, 3H), 4,33-4,38 (м, 1H), 4,42-4,49 (м, 1H), 6,21 (с, 1H), 6,95-7,03 (м, 4H), 7,28-7,33 (м, 2H), 7,57-7,62 (м, 4H), 7,65 (д, J=7,6 Гц, 1H), 7,86 (д, J=1,2 Гц, 1H), 7,88 (д, J=8,0 Гц, 1H).
Примеры 88 и 89
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-метил-8-фенил-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-метил-8-фенил-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
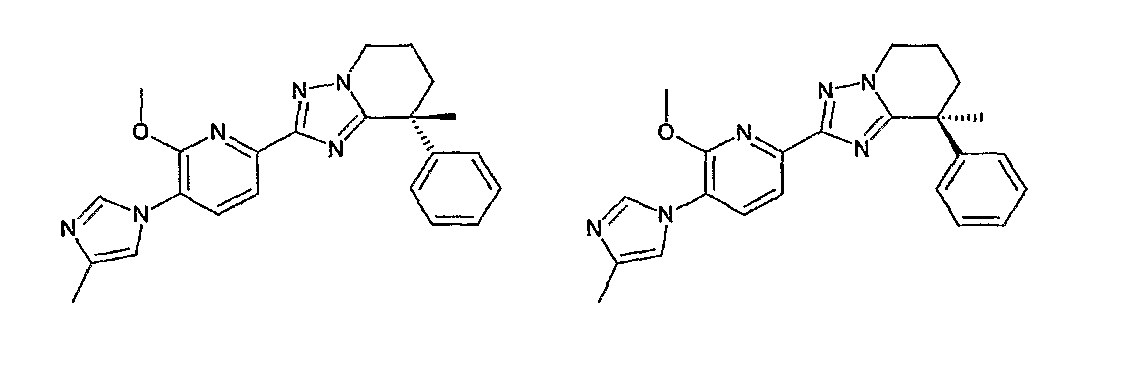
Рацемат указанного в заголовке соединения получали в соответствии с примерами 84 и 85 из 5-гидрокси-2-метил-2-фенилпентаннитрила (CAS #745054-73-3) в качестве исходного материала, вместо [бензил(2-оксоэтил)амино]-(4-трифторметилфенил)ацетонитрила. Полученный рацемат разделяли с помощью CHIRALCEL™ IB производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 5,4 минуты, полученным из анализа с помощью CHIRALPAK™ IB производства Daicel Chemical Industries, Ltd. (Lot. IB00CD-LG026, гексан:этанол = 8:2, 1,0 мл/мин) и отрицательным оптическим вращением и указанного в заголовке оптически активного соединения с временем удерживания 7,1 минуты, полученным из такого же анализа, и положительным оптическим вращением, соответственно.
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,87 (с, 3H), 1,89-2,11 (м, 3H), 2,31 (с, 3H), 2,41 (м, 1H), 4,18 (с, 3H), 4,24 (м, 1H), 4,40 (м, 1H), 7,02 (м, 1H), 7,09-7,12 (м, 2H), 7,20-7,24 (м, 1H), 7,27-7,31 (м, 2H), 7,62 (д, J=7,6 Гц, 1H), 7,84 (д, J=1,2 Гц, 1H), 7,89 (д, J=8,0 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,87 (с, 3H), 1,89-2,11 (м, 3H), 2,31 (с, 3H), 2,41 (м, 1H), 4,18 (с, 3H), 4,24 (м, 1H), 4,40 (м, 1H), 7,02 (м, 1H), 7,09-7,12 (м, 2H), 7,20-7,24 (м, 1H), 7,27-7,31 (м, 2H), 7,62 (д, J=7,6 Гц, 1H), 7,84 (д, J=1,2 Гц, 1H), 7,89 (д, J=8,0 Гц, 1H).
Примеры 90 и 91
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-6-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-6-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
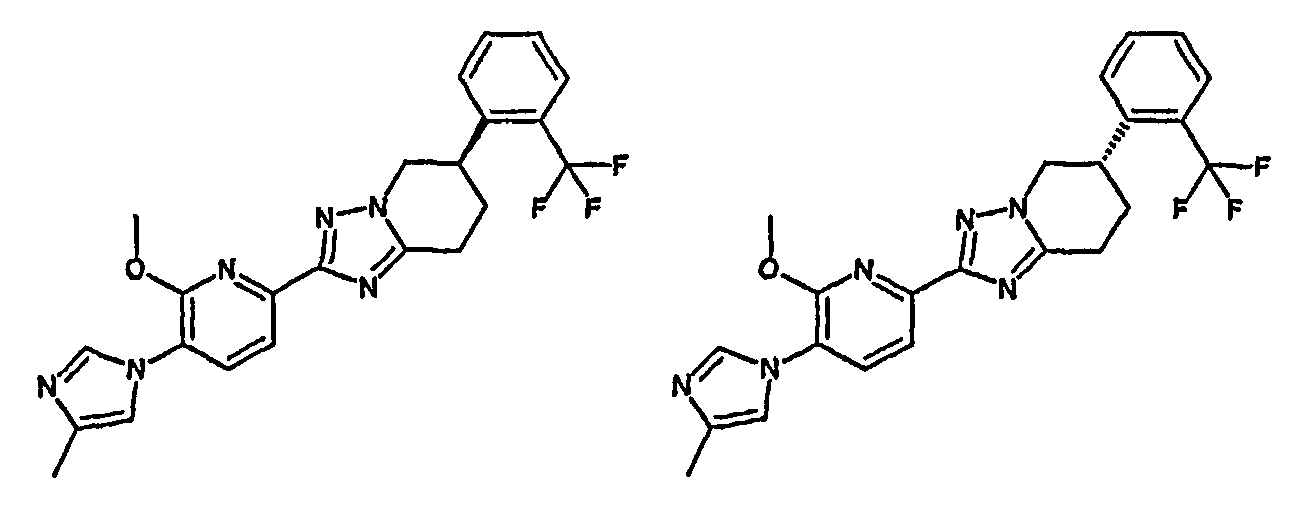
Рацемат указанного в заголовке соединения (186 мг) получали в соответствии с примерами 84 и 85 из 5-гидрокси-4-(2-трифторметилфенил)пентаннитрила, полученного в примере получения 4-3, вместо [бензил(2-оксоэтил)амино]-(4-трифторметилфенил)ацетонитрила. Полученный рацемат разделяли с помощью CHIRALPAK™ IB производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: этанол:гексан = 3:7, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 13,0 минуты и положительным оптическим вращением (42 мг) и указанного в заголовке соединения с временем удерживания 22,5 минуты и отрицательным оптическим вращением (49,7 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 13,0 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,20-2,34 (м, 2H), 2,31 (с, 3H), 3,09 (ддд, J=15,2, 13,0, 6,2 Гц, 1H), 3,30 (ддд, J=15,2, 5,1, 2,7 Гц, 1H), 3,72-3,82 (м, 1H), 4,17 (с, 3H), 4,23 (дд, J=11,7, 11,7 Гц, 1H), 4,60 (дд, J=15,2, 5,1 Гц, 1H), 7,02-7,04 (м, 1H), 7,43-7,52 (м, 2H), 7,60-7,65 (м, 1H), 7,64 (д, J=7,8 Гц, 1H), 7,75 (д, J=7,8 Гц, 1H), 7,83 (д, J=7,8 Гц, 1H), 7,85 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 22,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,20-2,34 (м, 2H), 2,31 (с, 3H), 3,09 (ддд, J=15,2, 13,0, 6,2 Гц, 1H), 3,30 (ддд, J=15,2, 5,1, 2,7 Гц, 1H), 3,72-3,82 (м, 1H), 4,17 (с, 3H), 4,23 (дд, J=11,7, 11,7 Гц, 1H), 4,60 (дд, J=15,2, 5,1 Гц, 1H), 7,02-7,04 (м, 1H), 7,43-7,52 (м, 2H), 7,60-7,65 (м, 1H), 7,64 (д, J=7,8 Гц, 1H), 7,75 (д, J=7,8 Гц, 1H), 7,83 (д, J=7,8 Гц, 1H), 7,85 (д, J=1,2 Гц, 1H).
Примеры 92 и 93
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
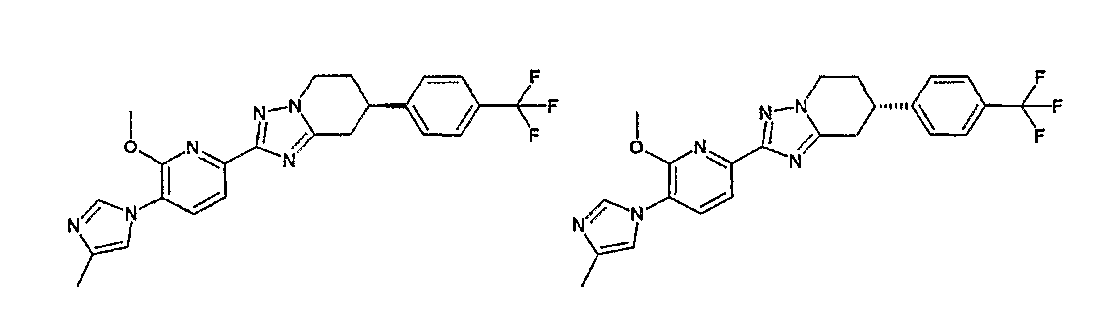
Рацемат указанного в заголовке соединения (184,1 мг) получали в соответствии с примерами 84 и 85 из 5-гидрокси-3-(4-трифторметилфенил)пентаннитрила, полученного в примере получения 4-4, вместо [бензил(2-оксоэтил)амино]-(4-трифторметилфенил)ацетонитрила. Полученный рацемат разделяли с помощью CHIRALPAK™ IB производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: этанол:гексан = 5:5, скорость потока: 13 мл/мин) с получением указанного в заголовке соединения с временем удерживания 16,5 минуты и положительным оптическим вращением (43,0 мг) и указанного в заголовке соединения с временем удерживания 23,0 минуты и отрицательным оптическим вращением (44,0 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 16,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 2,33-2,49 (м, 2H), 3,11 (дд, J=16,7, 9,4 Гц, 1H), 3,34-3,47 (м, 2H), 4,18 (с, 3H), 4,28-4,37 (м, 1H), 4,39-4,47 (м, 1H), 7,01-7,05 (м, 1H), 7,40 (д, J=8,2 Гц, 2H), 7,62-7,68 (м, 3H), 7,83 (д, J=7,8 Гц, 1H), 7,84-7,87 (м, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 23,0 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 2,33-2,49 (м, 2H), 3,11 (дд, J=16,7, 9,4 Гц, 1H), 3,34-3,47 (м, 2H), 4,18 (с, 3H), 4,28-4,37 (м, 1H), 4,39-4,47 (м, 1H), 7,01-7,05 (м, 1H), 7,40 (д, J=8,2 Гц, 2H), 7,62-7,68 (м, 3H), 7,83 (д, J=7,8 Гц, 1H), 7,84-7,87 (м, 1H).
Примеры 94 и 95
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
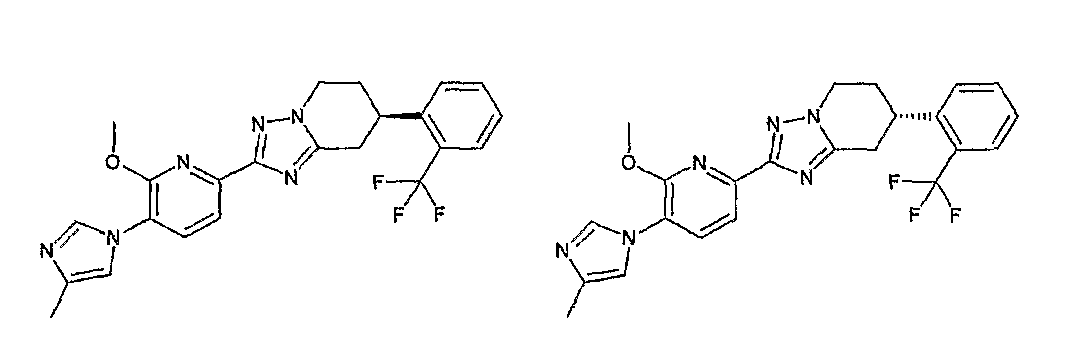
Рацемат указанного в заголовке соединения (222 мг) получали в соответствии со способом примеров 84 и 85 из 5-гидрокси-3-(2-трифторметилфенил)пентаннитрила, полученного в примере получения 4-5, вместо [бензил(2-оксоэтил)амино]-(4-трифторметилфенил)ацетонитрила. Полученный рацемат разделяли с помощью CHIRALCEL™ OD-H (2 см × 25 см, подвижная фаза: этанол:гексан = 2:8, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 22,5 минуты и положительным оптическим вращением (25,1 мг) и указанного в заголовке соединения с временем удерживания 49,5 минуты и отрицательным оптическим вращением (35,8 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 22,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,30 (с, 3H), 2,31-2,42 (м, 2H), 3,08 (дд, J=17,1, 11,7 Гц, 1H), 3,41 (дд, J=17,1, 5,1 Гц, 1H), 3,62-3,73 (м, 1H), 4,18 (с, 3H), 4,27-4,37 (м, 1H), 4,52 (ддд, J=13,3, 5,1, 2,3 Гц, 1H), 6,92-6,94 (м, 1H), 7,40-7,45 (м, 1H), 7,48 (д, J=7,8 Гц, 1H), 7,60 (д, J=7,8 Гц, 1H), 7,64 (д, J=7,8 Гц, 1H), 7,73 (д, J=7,8 Гц, 1H), 7,82 (д, J=7,8 Гц, 1H), 7,86 (д, J=1,0 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 49,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,30 (с, 3H), 2,31-2,42 (м, 2H), 3,08 (дд, J=17,1, 11,7 Гц, 1H), 3,41 (дд, J=17,1, 5,1 Гц, 1H), 3,62-3,73 (м, 1H), 4,18 (с, 3H), 4,27-4,37 (м, 1H), 4,52 (ддд, J=13,3, 5,1, 2,3 Гц, 1H), 6,92-6,94 (м, 1H), 7,40-7,45 (м, 1H), 7,48 (д, J=7,8 Гц, 1H), 7,60 (д, J=7,8 Гц, 1H), 7,64 (д, J=7,8 Гц, 1H), 7,73 (д, J=7,8 Гц, 1H), 7,82 (д, J=7,8 Гц, 1H), 7,86 (д, J=1,0 Гц, 1H).
Примеры 96 и 97
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-фтор-4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-фтор-4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
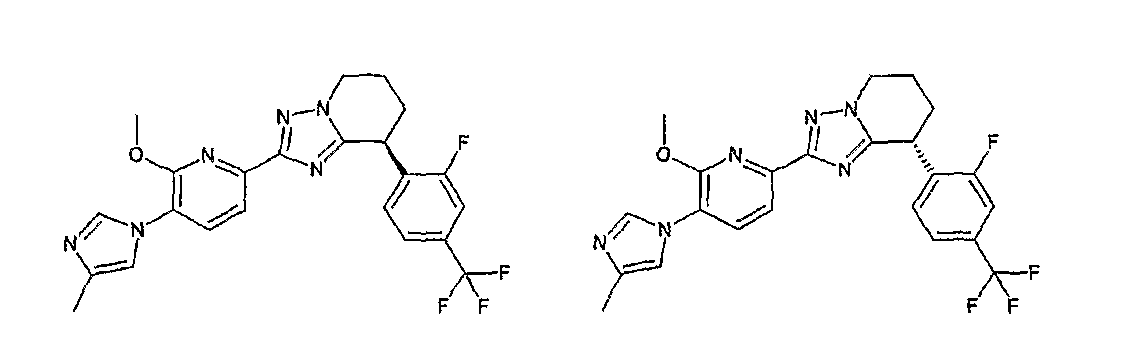
Рацемат указанного в заголовке соединения (52,7 мг) получали в соответствии со способом примеров 84 и 85 из 2-(2-фтор-4-трифторметилфенил)-5-гидроксипентаннитрила, полученного в примере получения 4-6, вместо [бензил(2-оксоэтил)амино]-(4-трифторметилфенил)ацетонитрила. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: этанол:гексан = 2:8, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 48 минут и положительным оптическим вращением (1,81 мг) и указанного в заголовке соединения с временем удерживания 56,5 минуты и отрицательным оптическим вращением (2,15 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 48 минут были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,04-2,25 (м, 3H), 2,30 (с, 3H), 2,38-2,48 (м, 1H), 4,16 (с, 3H), 4,42 (дд, J=7,4, 5,5 Гц, 2H), 4,69 (дд, J=7,0, 7,0 Гц, 1H), 6,98-7,01 (м, 1H), 7,08-7,14 (м, 1H), 7,37 (д, J=9,0 Гц, 1H), 7,59 (д, J=7,8 Гц, 1H), 7,76 (д, J=7,8 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 56,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,04-2,25 (м, 3H), 2,30 (с, 3H), 2,38-2,48 (м, 1H), 4,16 (с, 3H), 4,42 (дд, J=7,4, 5,5 Гц, 2H), 4,69 (дд, J=7,0, 7,0 Гц, 1H), 6,98-7,01 (м, 1H), 7,08-7,14 (м, 1H), 7,37 (д, J=9,0 Гц, 1H), 7,59 (д, J=7,8 Гц, 1H), 7,76 (д, J=7,8 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Соединения примеров 98-99 получали таким же способом, как в примерах 84 и 85 (таблица 3).
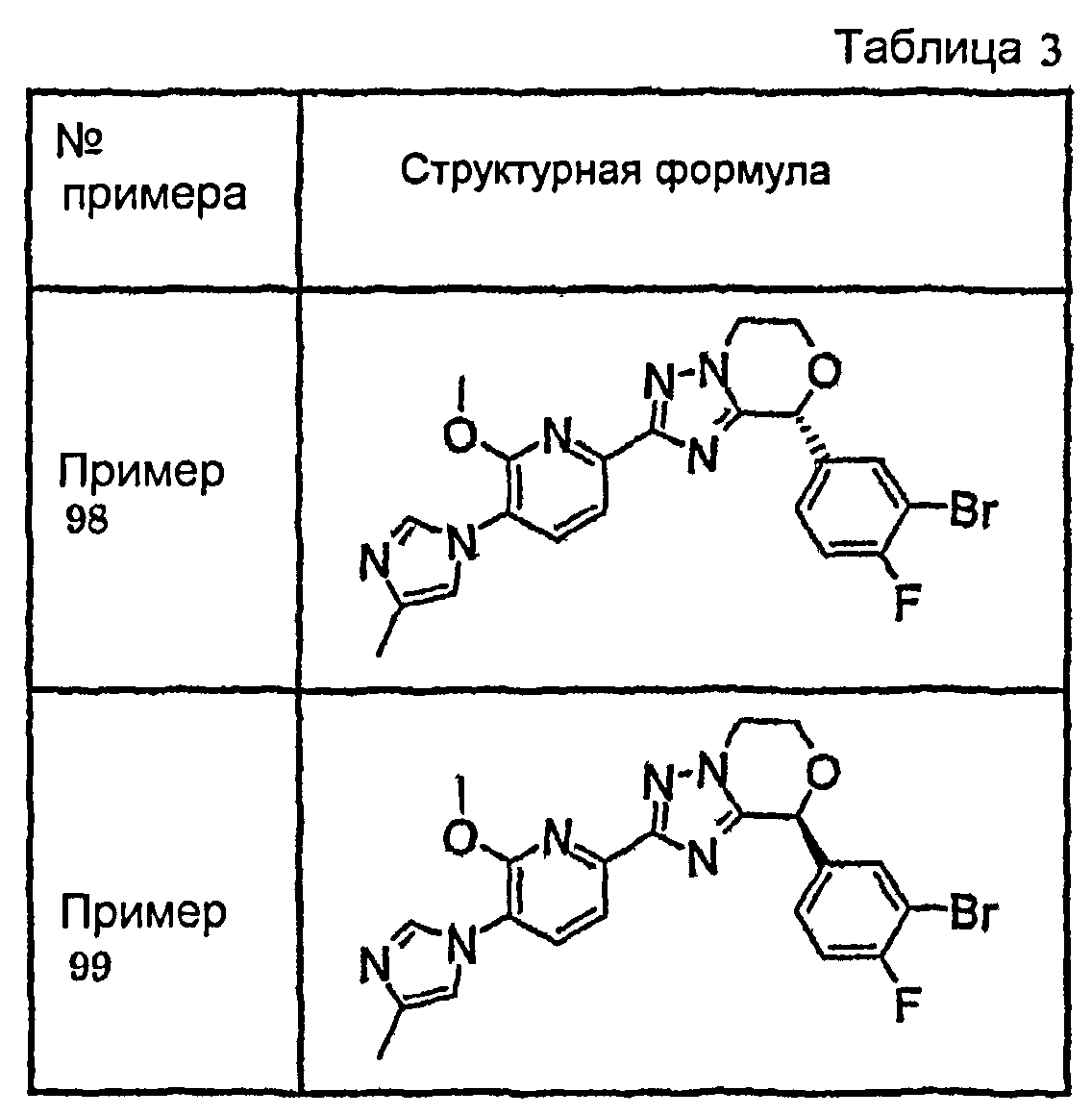
Примеры 100 и 101
Синтез (-)-8-(4-бром-2-трифторметилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина и (+)-8-(4-бром-2-трифторметилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина
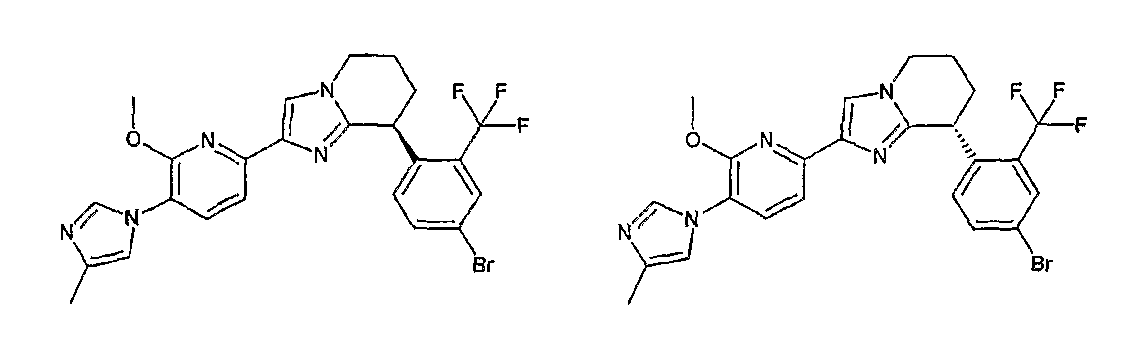
Синтез 3-(4-бром-2-трифторметилфенил)пиперидин-2-илиденамина
Насыщенный этаноловый раствор аммония (2,5 мл) добавляли к гидрохлориду этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата, полученного в примере получения 3-1 (500 мг), и смесь перемешивали при комнатной температуре в течение восьми суток. Нерастворимый материал удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении. Метиленхлорид и 1 н. раствор гидроксида натрия добавляли к полученному остатку и отделяли органический слой. Водный слой повторно экстрагировали метиленхлоридом. Объединенные органические слои сушили над безводным карбонатом калия и концентрировали при пониженном давлении с получением 320 мг указанного в заголовке соединения. Показатели свойств соединения были следующими.
ESI-MS; m/z 321 [M++H].
1H-ЯМР (ДМСО-d6) δ (ppm): 1,42-1,65 (м, 3H), 1,95-2,05 (м, 1H), 3,35-3,45 (м, 2H), 3,68 (д, J=6,8 Гц, 1H), 5,55 (ушир.с, 2H), 7,32 (д, J=8,4 Гц, 1H), 7,85-7,92 (м, 2H).
Синтез (-)-8-(4-бром-2-трифторметилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина и (+)-8-(4-бром-2-трифторметилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина
N-бромсукцинимид (98 мг) добавляли к раствору 6-(1-этоксивинил)-2-метокси-3-(4-метилимидазол-1-ил)пиридина, полученного в примере получения 1-7 (130 мг), в смеси тетрагидрофуран (5 мл)-вода (0,2 мл) с последующим перемешиванием при комнатной температуре в течение 15 минут. Раствор 3-(4-бром-2-трифторметилфенил)пиперидин-2-илиденамина (318 мг) в этаноле (2 мл) добавляли к реакционному раствору с последующим перемешиванием при комнатной температуре в течение двух часов и 15 минут. Затем реакционный раствор нагревали при кипячении с обратным холодильником в течение одного часа и 30 минут. Реакционному раствору давали охладиться до комнатной температуры. Этилацетат, воду и насыщенный раствор бикарбоната натрия добавляли к реакционному раствору и отделяли органический слой. Полученный органический слой промывали последовательно с полунасыщенным раствором соли и насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Chromatorex™ NH; элюирующий растворитель: этилацетат:гептан = 1:9 → 1:1) с получением рацемата указанного в заголовке соединения.
Полученный рацемат разделяли с помощью CHIRALCEL™ OD-H (2 см × 25 см; подвижная фаза: 20% изопропанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 14 минут и отрицательным оптическим вращением (36,1 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 20 минут и положительным оптическим вращением (36,3 мг, >97% ee).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 534 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,82-1,94 (м, 1H), 2,00-2,22 (м, 2H), 2,29 (с, 3H), 2,35-2,45 (м, 1H), 4,03 (с, 3H), 4,17 (дд, J=6,8, 5,6 Гц, 2H), 4,65 (дд, J=8,4, 6,0 Гц, 1H), 6,92 (д, J=8,4 Гц, 1H), 6,93 (с, 1H), 7,46 (д, J=8,0 Гц, 1H), 7,50 (д, J=8,0 Гц, 1H), 7,54 (с, 1H), 7,55 (дд, J=8,4, 2,0 Гц, 1H), 7,74 (д, J=1,2 Гц, 1H), 7,84 (д, J=2,0 Гц, 1H).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 534 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,82-1,94 (м, 1H), 2,00-2,22 (м, 2H), 2,29 (с, 3H), 2,35-2,45 (м, 1H), 4,03 (с, 3H), 4,17 (дд, J=6,8, 5,6 Гц, 2H), 4,65 (дд, J=8,4, 6,0 Гц, 1H), 6,92 (д, J=8,4 Гц, 1H), 6,93 (с, 1H), 7,46 (д, J=8,0 Гц, 1H), 7,50 (д, J=8,0 Гц, 1H), 7,54 (с, 1H), 7,55 (дд, J=8,4, 2,0 Гц, 1H), 7,74 (д, J=1,2 Гц, 1H), 7,84 (д, J=2,0 Гц, 1H).
Примеры 102 и 103
Синтез (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина и (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина
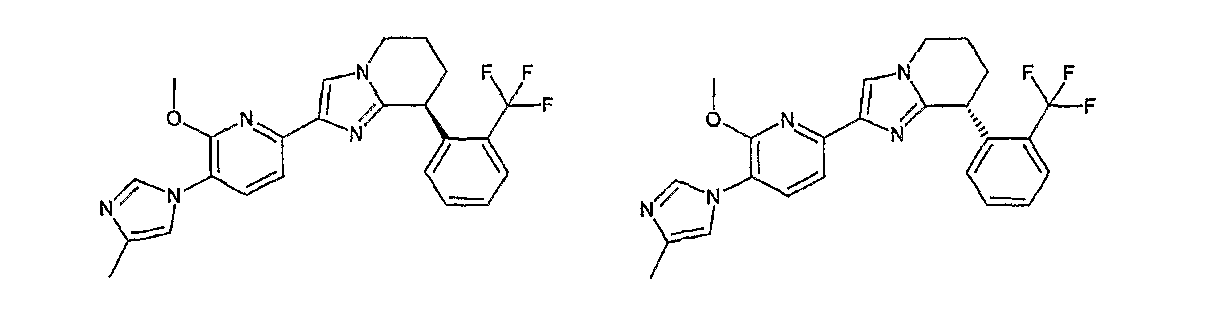
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 100 и 101 из гидрохлорида этил 2-(2-трифторметилфенил)-5-хлорпентанимидата, полученного в примере получения 3-12. Полученный рацемат разделяли с помощью CHIRALCEL™ OD-H (2 см × 25 см; подвижная фаза: 30% изопропанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 10 минут и отрицательным оптическим вращением (12,2 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 22 минуты и положительным оптическим вращением (11,8 мг, >97% ee).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 454 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,85-2,23 (м, 3H), 2,28 (с, 3H), 2,35-2,45 (м, 1H), 4,04 (с, 3H), 4,10-4,25 (м, 2H), 4,73 (т, J=6,8 Гц, 1H), 6,92 (ушир.с, 1H), 7,01 (д, J=7,6 Гц, 1H), 7,35 (т, J=7,6 Гц, 1H), 7,43 (т, J=7,6 Гц, 1H), 7,45 (д, J=8,0 Гц, 1H), 7,51 (д, J=8,0 Гц, 1H), 7,55 (с, 1H), 7,71 (д, J=7,6 Гц, 1H), 7,74 (ушир.с, 1H).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 454 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,85-2,23 (м, 3H), 2,28 (с, 3H), 2,35-2,45 (м, 1H), 4,04 (с, 3H), 4,10-4,25 (м, 2H), 4,73 (т, J=6,8 Гц, 1H), 6,92 (ушир.с, 1H), 7,01 (д, J=7,6 Гц, 1H), 7,35 (т, J=7,6 Гц, 1H), 7,43 (т, J=7,6 Гц, 1H), 7,45 (д, J=8,0 Гц, 1H), 7,51 (д, J=8,0 Гц, 1H), 7,55 (с, 1H), 7,71 (д, J=7,6 Гц, 1H), 7,74 (ушир.с, 1H).
Примеры 104 и 105
Синтез (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(2-трифторметилфенил)-6,7-дигидро-5H-пирроло[1,2-a]имидазола и (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(2-трифторметилфенил)-6,7-дигидро-5H-пирроло[1,2-a]имидазола
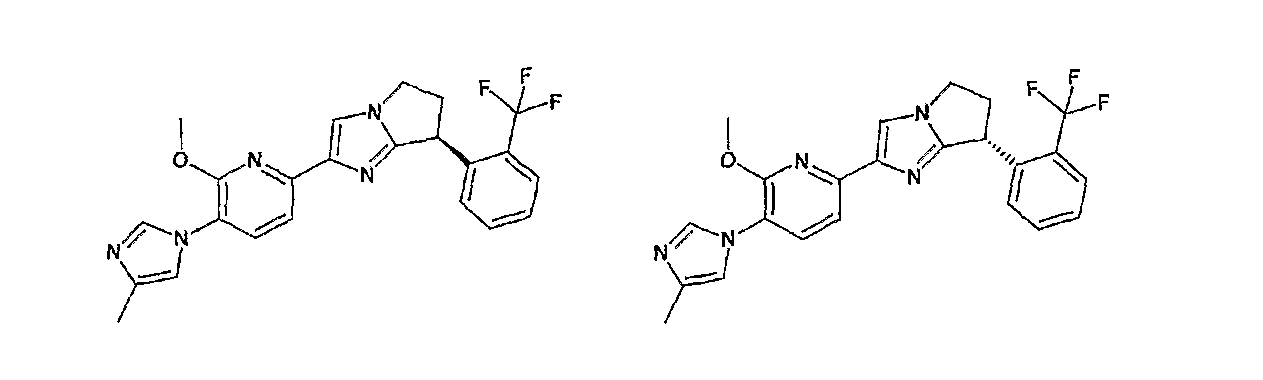
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 100 и 101 из гидрохлорида этил 4-хлор-2-(2-трифторметилфенил)бутилимидата, полученного в примере получения 3-14, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат очищали с помощью CHIRALPAK™ IB (2 см × 25 см; подвижная фаза: 30% изопропанол-гексан) и затем разделяли с помощью CHIRALCEL™ OD-H (2 см × 25 см; подвижная фаза: 30% изопропанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 10 минут и отрицательным оптическим вращением (2,43 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 16 минут и положительным оптическим вращением (2,00 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 440 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,29 (д, J=1,2 Гц, 3H), 2,40-2,52 (м, 1H), 3,17-3,28 (м, 1H), 4,06 (с, 3H), 4,07-4,21 (м, 2H), 4,82 (дд, J=6,0, 8,4 Гц, 1H), 6,96 (ушир.с, 1H), 7,05 (д, J=7,6 Гц, 1H), 7,37 (т, J=7,6 Гц, 1H), 7,47 (т, J=7,6 Гц, 1H), 7,51 (д, J=8,0 Гц, 1H), 7,59 (д, J=8,0 Гц, 1H), 7,68 (с, 1H), 7,71 (д, J=7,6 Гц, 1H), 7,76 (д, J=1,6 Гц, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 440 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,29 (д, J=1,2 Гц, 3H), 2,40-2,52 (м, 1H), 3,17-3,28 (м, 1H), 4,06 (с, 3H), 4,07-4,21 (м, 2H), 4,82 (дд, J=6,0, 8,4 Гц, 1H), 6,96 (ушир.с, 1H), 7,05 (д, J=7,6 Гц, 1H), 7,37 (т, J=7,6 Гц, 1H), 7,47 (т, J=7,6 Гц, 1H), 7,51 (д, J=8,0 Гц, 1H), 7,59 (д, J=8,0 Гц, 1H), 7,68 (с, 1H), 7,71 (д, J=7,6 Гц, 1H), 7,76 (д, J=1,6 Гц, 1H).
Примеры 106 и 107
Синтез (-)-8-(4-фтор-2-трифторметилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина и (+)-8-(4-фтор-2-трифторметилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина
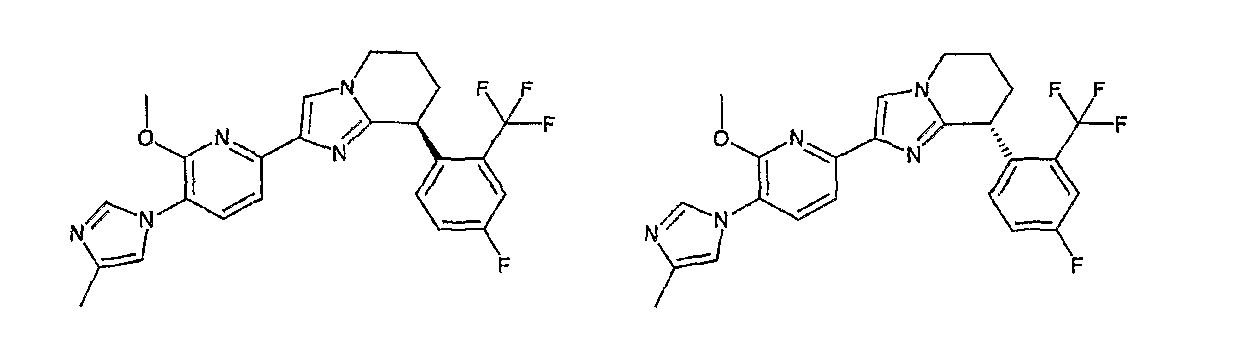
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 100 и 101 из гидрохлорида этил 2-(4-фтор-2-трифторметилфенил)-5-хлорпентанимидата, полученного в примере получения 3-13, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: 30% изопропанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 11 минут и отрицательным оптическим вращением (14,8 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 21 минута и положительным оптическим вращением (22,7 мг, >98% ee).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 472 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,84-1,94 (м, 1H), 1,98-2,22 (м, 2H), 2,28 (с, 3H), 2,35-2,46 (м, 1H), 4,03 (с, 3H), 4,17 (дд, J=7,2, 4,8 Гц, 2H), 4,67 (дд, J=8,0, 6,0 Гц, 1H), 6,93 (ушир.с, 1H), 7,02 (дд, J=8,4, 5,6 Гц, 1H), 7,14 (тд, J=8,4, 2,4 Гц, 1H), 7,41 (дд, J=8,8, 2,4 Гц, 1H), 7,46 (д, J=8,0 Гц, 1H), 7,50 (д, J=8,0 Гц, 1H), 7,54 (с, 1H), 7,74 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 472 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,84-1,94 (м, 1H), 1,98-2,22 (м, 2H), 2,28 (с, 3H), 2,35-2,46 (м, 1H), 4,03 (с, 3H), 4,17 (дд, J=7,2, 4,8 Гц, 2H), 4,67 (дд, J=8,0, 6,0 Гц, 1H), 6,93 (ушир.с, 1H), 7,02 (дд, J=8,4, 5,6 Гц, 1H), 7,14 (тд, J=8,4, 2,4 Гц, 1H), 7,41 (дд, J=8,8, 2,4 Гц, 1H), 7,46 (д, J=8,0 Гц, 1H), 7,50 (д, J=8,0 Гц, 1H), 7,54 (с, 1H), 7,74 (д, J=1,2 Гц, 1H).
Примеры 108 и 109
Синтез (-)-8-(4-хлор-2-трифторметилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина и (+)-8-(4-хлор-2-трифторметилфенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина
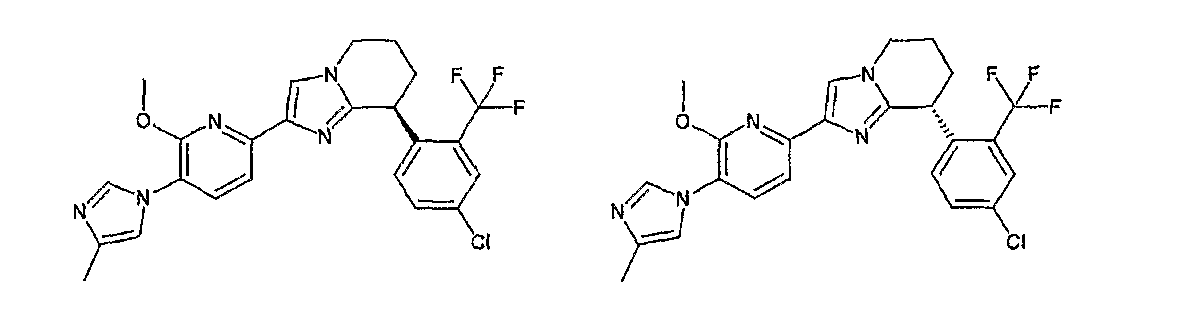
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 100 и 101 из гидрохлорида этил 2-(4-хлор-2-трифторметилфенил)-5-хлорпентанимидата, полученного в примере получения 3-20, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: 40% изопропанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 12 минут и отрицательным оптическим вращением (43,0 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 22 минуты и положительным оптическим вращением (41,2 мг, >98% ee).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 488 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,82-1,95 (м, 1H), 2,00-2,22 (м, 2H), 2,29 (с, 3H), 2,35-2,45 (м, 1H), 4,03 (с, 3H), 4,17 (дд, J=6,8, 5,2 Гц, 2H), 4,67 (дд, J=8,0, 6,0 Гц, 1H), 6,93 (ушир.с, 1H), 6,98 (д, J=8,4 Гц, 1H), 7,40 (дд, J=8,4, 2,4 Гц, 1H), 7,46 (д, J=8,0 Гц, 1H), 7,49 (д, J=8,0 Гц, 1H), 7,54 (с, 1H), 7,69 (д, J=2,4 Гц, 1H), 7,74 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 488 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,82-1,95 (м, 1H), 2,00-2,22 (м, 2H), 2,29 (с, 3H), 2,35-2,45 (м, 1H), 4,03 (с, 3H), 4,17 (дд, J=6,8, 5,2 Гц, 2H), 4,67 (дд, J=8,0, 6,0 Гц, 1H), 6,93 (ушир.с, 1H), 6,98 (д, J=8,4 Гц, 1H), 7,40 (дд, J=8,4, 2,4 Гц, 1H), 7,46 (д, J=8,0 Гц, 1H), 7,49 (д, J=8,0 Гц, 1H), 7,54 (с, 1H), 7,69 (д, J=2,4 Гц, 1H), 7,74 (д, J=1,2 Гц, 1H).
Примеры 110 и 111
Синтез (+)-8-(5-трет-бутил-2-метоксифенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина и (-)-8-(5-трет-бутил-2-метоксифенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина
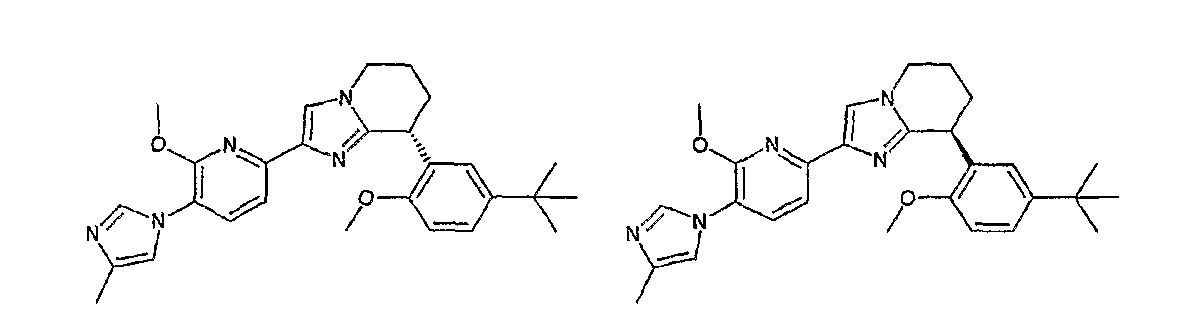
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 100 и 101 из гидрохлорида этил 2-(5-трет-бутил-2-метоксифенил)-5-хлорпентанимидата, полученного в примере получения 3-3, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: 15% изопропанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 14 минут и положительным оптическим вращением и указанного в заголовке оптически активного соединения с временем удерживания 26 минут и отрицательным оптическим вращением.
Соответственные оптически активные соединения очищали с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: 15% этанол-гексан) с получением указанного в заголовке оптически активного соединения с положительным оптическим вращением (14,5 мг, >99% ee) и указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (14,5 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 472 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,21 (с, 9H), 1,90-2,35 (м, 4H), 2,29 (с, 3H), 3,78 (с, 3H), 4,00-4,20 (м, 2H), 4,05 (с, 3H), 4,63 (т, J=6,0 Гц, 1H), 6,81 (д, J=8,4 Гц, 1H), 6,86 (д, J=2,4 Гц, 1H), 6,94 (с, 1H), 7,22 (дд, J=8,4, 2,4 Гц, 1H), 7,47 (д, J=8,0 Гц, 1H), 7,54 (д, J=8,0 Гц, 1H), 7,55 (с, 1H), 7,74 (с, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 472 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,21 (с, 9H), 1,90-2,35 (м, 4H), 2,29 (с, 3H), 3,78 (с, 3H), 4,00-4,20 (м, 2H), 4,05 (с, 3H), 4,63 (т, J=6,0 Гц, 1H), 6,81 (д, J=8,4 Гц, 1H), 6,86 (д, J=2,4 Гц, 1H), 6,94 (с, 1H), 7,22 (дд, J=8,4, 2,4 Гц, 1H), 7,47 (д, J=8,0 Гц, 1H), 7,54 (д, J=8,0 Гц, 1H), 7,55 (с, 1H), 7,74 (с, 1H).
Примеры 112 и 113
Синтез (-)-8-(3-трет-бутил-4-метоксифенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина и (+)-8-(3-трет-бутил-4-метоксифенил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина
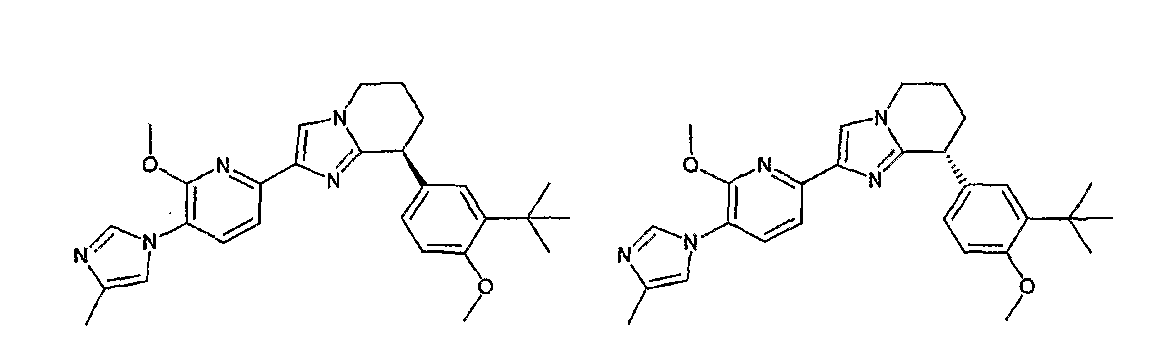
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 100 и 101 из гидрохлорида имидата, полученного в примере получения 3-8, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат грубо очищали с помощью CHIRALPAK™ IB (2 см × 25 см; подвижная фаза: 20% этанол-гексан). Затем рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; подвижная фаза: 10% этанол-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 16 минут и отрицательным оптическим вращением и указанного в заголовке оптически активного соединения с временем удерживания 18 минут и положительным оптическим вращением (22,7 мг, >99% ee).
Указанное в заголовке оптически активное соединение с отрицательным оптическим вращением опять очищали с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: 25% изопропанол-гексан) с получением указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (18,5 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 472 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,35 (с, 9H), 1,90-2,15 (м, 3H), 2,25-2,35 (м, 1H), 2,29 (д, J=1,2 Гц, 3H), 3,80 (с, 3H), 4,03-4,20 (м, 2H), 4,04 (с, 3H), 4,33 (м, J=5,6 Гц, 1H), 6,77 (д, J=8,4 Гц, 1H), 6,80 (дд, J=8,4, 2,4 Гц, 1H), 6,94 (т, J=1,2 Гц, 1H), 7,13 (д, J=2,4 Гц, 1H), 7,48 (д, J=8,0 Гц, 1H), 7,54 (с, 1H), 7,58 (д, J=8,0 Гц, 1H), 7,74 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 472 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,35 (с, 9H), 1,90-2,15 (м, 3H), 2,25-2,35 (м, 1H), 2,29 (д, J=1,2 Гц, 3H), 3,80 (с, 3H), 4,03-4,20 (м, 2H), 4,04 (с, 3H), 4,33 (м, J=5,6 Гц, 1H), 6,77 (д, J=8,4 Гц, 1H), 6,80 (дд, J=8,4, 2,4 Гц, 1H), 6,94 (т, J=1,2 Гц, 1H), 7,13 (д, J=2,4 Гц, 1H), 7,48 (д, J=8,0 Гц, 1H), 7,54 (с, 1H), 7,58 (д, J=8,0 Гц, 1H), 7,74 (д, J=1,2 Гц, 1H).
Примеры 114 и 115
Синтез (-)-8-(3,3-диметил-2,3-дигидробензофуран-5-ил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина и (+)-8-(3,3-диметил-2,3-дигидробензофуран-5-ил)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидроимидазо[1,2-a]пиридина
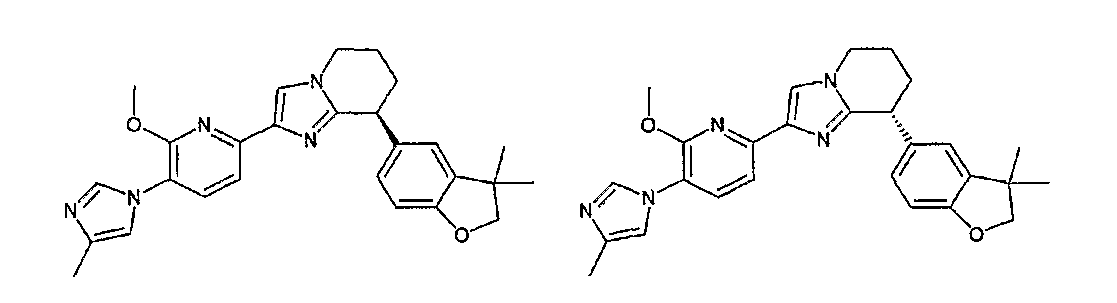
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 100 и 101 из гидрохлорида имидата, полученного в примере получения 3-2, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Полученный рацемат разделяли с помощью CHIRALCEL™ OD-H (2 см × 25 см; подвижная фаза: 25% изопропиловый спирт-гексан) с получением указанного в заголовке оптически активного соединения с временем удерживания 10 минут и отрицательным оптическим вращением и указанного в заголовке оптически активного соединения с временем удерживания 16 минут и положительным оптическим вращением (22,5 мг, >99% ee).
Указанное в заголовке оптически активное соединение с отрицательным оптическим вращением опять очищали с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: 30% этанол-гексан) с получением указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (19,8 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 456 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,31 (с, 6H), 1,90-2,16 (м, 3H), 2,25-2,36 (м, 1H), 2,29 (с, 3H), 4,04 (с, 3H), 4,05-4,21 (м, 2H), 4,22 (с, 2H), 4,34 (т, J=6,0 Гц, 1H), 6,69 (д, J=8,4 Гц, 1H), 6,88 (дд, J=8,4, 1,6 Гц, 1H), 6,91 (д, J=1,6 Гц, 1H), 6,94 (ушир.с, 1H), 7,49 (д, J=8,0 Гц, 1H), 7,55 (с, 1H), 7,57 (д, J=8,0 Гц, 1H), 7,75 (ушир.с, 1H).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 456 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,31 (с, 6H), 1,90-2,16 (м, 3H), 2,25-2,36 (м, 1H), 2,29 (с, 3H), 4,04 (с, 3H), 4,05-4,21 (м, 2H), 4,22 (с, 2H), 4,34 (т, J=6,0 Гц, 1H), 6,69 (д, J=8,4 Гц, 1H), 6,88 (дд, J=8,4, 1,6 Гц, 1H), 6,91 (д, J=1,6 Гц, 1H), 6,94 (ушир.с, 1H), 7,49 (д, J=8,0 Гц, 1H), 7,55 (с, 1H), 7,57 (д, J=8,0 Гц, 1H), 7,75 (ушир.с, 1H).
Примеры 116 и 117
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
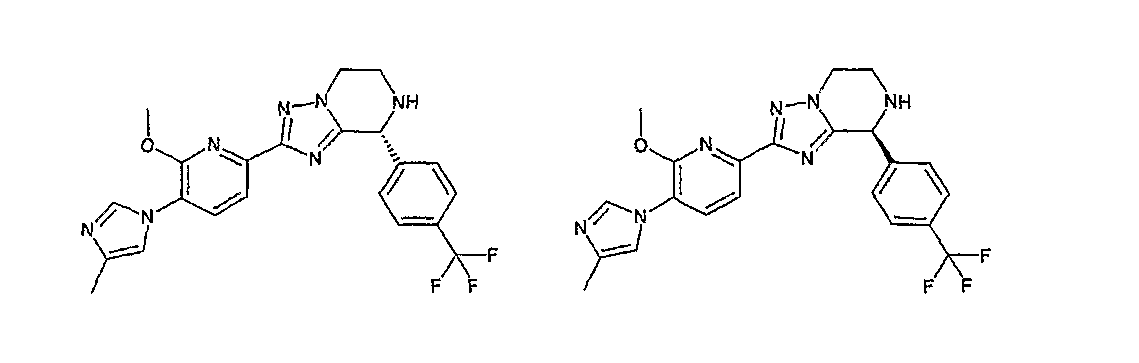
Уксусную кислоту (1 мл) и 20% гидроксид палладия-на-углероде (400 мг) добавляли к раствору 7-бензил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина, полученного в примерах 84 и 85 (411 мг), в метаноле (40 мл) и смесь перемешивали при комнатной температуре в атмосфере водорода в течение двух суток. После того как реакционную атмосферу вытесняли азотом, к реакционному раствору добавляли этилацетат. Смесь фильтровали через целит и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Chromatorex™ NH, элюирующий растворитель: этилацетат) с получением 290 мг рацемата указанного в заголовке соединения. Полученный рацемат указанного в заголовке соединения (36 мг) разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: гексан:этанол = 1:1, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 8,5 минуты, полученным из анализа с помощью CHIRALPAK™ IA (Lot. IA00CE-FA020, гексан:этанол = 1:1, 1,0 мл/мин), и положительным оптическим вращением (10 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 16,0 минуты, полученным из такого же анализа, и отрицательным оптическим вращением (11 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 456 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,30 (с, 3H), 3,36-3,49 (м, 2H), 4,16 (с, 3H), 4,34-4,46 (м, 2H), 5,42 (с, 1H), 7,00 (с, 1H), 7,58-7,66 (м, 5H), 7,78 (д, J=7,6 Гц, 1H), 7,83 (с, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 456 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,30 (с, 3H), 3,36-3,49 (м, 2H), 4,16 (с, 3H), 4,34-4,46 (м, 2H), 5,42 (с, 1H), 7,00 (м, J=1,2 Гц, 1H), 7,58-7,66 (м, 5H), 7,78 (д, J=7,6 Гц, 1H), 7,83 (д, J=1,6 Гц, 1H).
Примеры 118 и 119
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-7-метил-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-7-метил-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
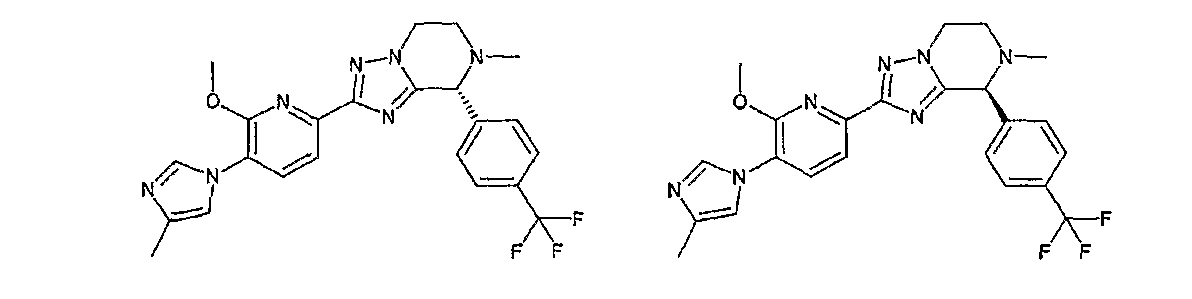
37% Формальдегидный раствор (2 мл) добавляли к раствору 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина, полученного в примерах 116 и 117 (54 мг), в муравьиной кислоте (2 мл) и смесь перемешивали при кипячении с обратным холодильником в течение 3,5 часов. Реакционный раствор нейтрализовали насыщенным раствором бикарбоната натрия при 0°C и распределяли этилацетатом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Chromatorex™ NH, элюирующий растворитель: гептан ацетат:этилацетат = 1:1 → 0:1) с получением 24 мг рацемата указанного в заголовке соединения. Полученный рацемат указанного в заголовке соединения разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: гексан:этанол = 6:4, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 6,6 минуты, полученным из анализа с помощью CHIRALPAK™ IA (Lot. IA00CE-FA020, гексан:этанол = 1:1, 1,0 мл/мин), и положительным оптическим вращением (6,5 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 15,2 минуты, полученным из такого же анализа, и отрицательным оптическим вращением (7,5 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 470 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 2,38 (с, 3H), 3,03 (ддд, J=4,4, 9,6, 12,8 Гц, 1H), 3,34 (ддд, J=2,8, 4,4, 12,4 Гц, 1H), 4,14 (с, 3H), 4,41-4,54 (м, 2H), 4,59 (с, 1H), 6,98 (т, J=1,2 Гц, 1H), 7,53 (д, J=9,2 Гц, 2H), 7,55 (д, J=8,0 Гц, 1H), 7,65 (д, J=8,4 Гц, 2H), 7,70 (д, J=8,0 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 470 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 2,38 (с, 3H), 3,03 (ддд, J=4,4, 9,6, 12,8 Гц, 1H), 3,34 (ддд, J=2,8, 4,4, 12,4 Гц, 1H), 4,14 (с, 3H), 4,41-4,54 (м, 2H), 4,59 (с, 1H), 6,98 (т, J=1,2 Гц, 1H), 7,53 (д, J=9,2 Гц, 2H), 7,55 (д, J=8,0 Гц, 1H), 7,65 (д, J=8,4 Гц, 2H), 7,70 (д, J=8,0 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H).
Примеры 120 и 121
Синтез (+)-1-[2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6-дигидро-8H-[1,2,4]триазоло[1,5-a]пиразин-7-ил]этанона и (-)-1-[2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6-дигидро-8H-[1,2,4]триазоло[1,5-a]пиразин-7-ил]этанона
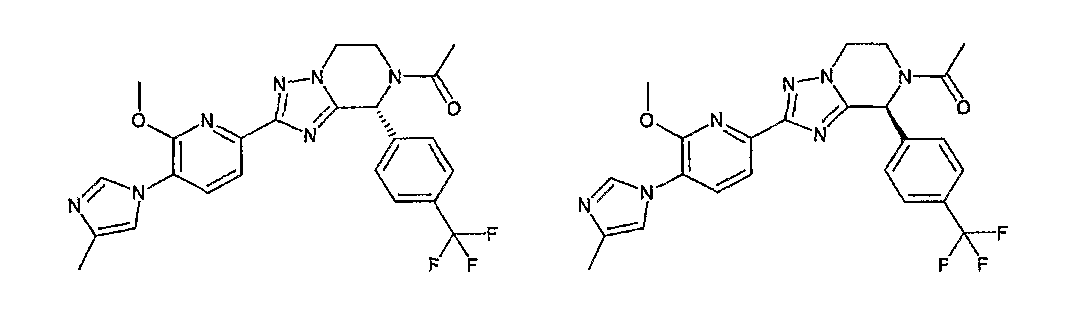
Пиридин (31,3 мкл) и уксусный ангидрид (14,5 мкл) добавляли к раствору 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина, полученного в примерах 116 и 117 (35 мг), в дихлорметане (1 мл) при 0°C и смесь перемешивали при комнатной температуре в течение 1,5 часов. Добавляли к реакционному раствору воду с последующим экстрагированием хлороформом. Органический слой концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Chromatorex™ NH; элюирующий растворитель: этилацетат:метанол = 1:0 → 9:1) с получением рацемата указанного в заголовке соединения. Полученный рацемат указанного в заголовке соединения разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: гексан:этанол = 65:35, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 6,95 минуты, полученным из анализа с помощью CHIRALPAK™ IB (Lot. IB00CD-FD025, гексан:этанол = 8:2, 1,0 мл/мин), и положительным оптическим вращением (6,3 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 8,25 минуты, полученным из такого же анализа, и отрицательным оптическим вращением (7,4 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 498 [M++H].
1H-ЯМР (CD3OD) δ (ppm): 2,25 (с, 3H), 2,33 (с, 3H), 3,74-3,81 (м, 1H), 4,13 (с, 3H), 4,37-4,51 (м, 3H), 7,16 (с, 1H), 7,22 (с, 1H), 7,60 (д, J=8,0 Гц, 2H), 7,71 (д, J=8,4 Гц, 2H), 7,85 (д, J=8,0 Гц, 1H), 7,89 (д, J=8,0 Гц, 1H), 7,98 (д, J=1,6 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 498 [M++H].
1H-ЯМР (CD3OD) δ (ppm): 2,25 (с, 3H), 2,33 (с, 3H), 3,74-3,81 (м, 1H), 4,13 (с, 3H), 4,37-4,51 (м, 3H), 7,16 (с, 1H), 7,22 (с, 1H), 7,60 (д, J=8,0 Гц, 2H), 7,71 (д, J=8,4 Гц, 2H), 7,85 (д, J=8,0 Гц, 1H), 7,89 (д, J=8,0 Гц, 1H), 7,98 (д, J=1,6 Гц, 1H).
Примеры 122 и 123
Синтез (+)-[2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6-дигидро-8H-[1,2,4]триазоло[1,5-a]пиразин-7-ил]фенилметанона и (-)-[2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6-дигидро-8H-[1,2,4]триазоло[1,5-a]пиразин-7-ил]фенилметанона
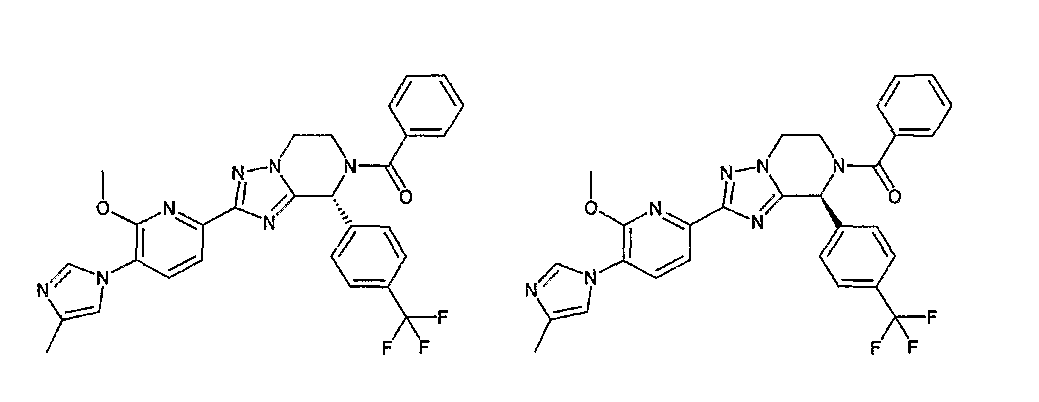
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 120 и 121 из 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина, полученного в примерах 116 и 117. Полученный рацемат указанного в заголовке соединения разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: этанол, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 7,5 минуты, полученным из анализа с помощью CHIRALPAK™ IA (Lot. IA00CE-FA020, этанол, 1,0 мл/мин) и отрицательным оптическим вращением (6,5 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 14,4 минуты, полученным из такого же анализа, и положительным оптическим вращением (10,9 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 560 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 3,53-3,65 (м, 2H), 4,17 (с, 3H), 4,40-4,42 (м, 2H), 7,03 (с, 1H), 7,46-7,23 (м, 6H), 7,64-7,68 (м, 5H), 7,85-7,86 (м, 2H).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 560 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 3,53-3,65 (м, 2H), 4,17 (с, 3H), 4,40-4,42 (м, 2H), 7,03 (с, 1H), 7,46-7,23 (м, 6H), 7,64-7,68 (м, 5H), 7,85-7,86 (м, 2H).
Примеры 124 и 125
Синтез (+)-7-метансульфонил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина и (-)-7-метансульфонил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
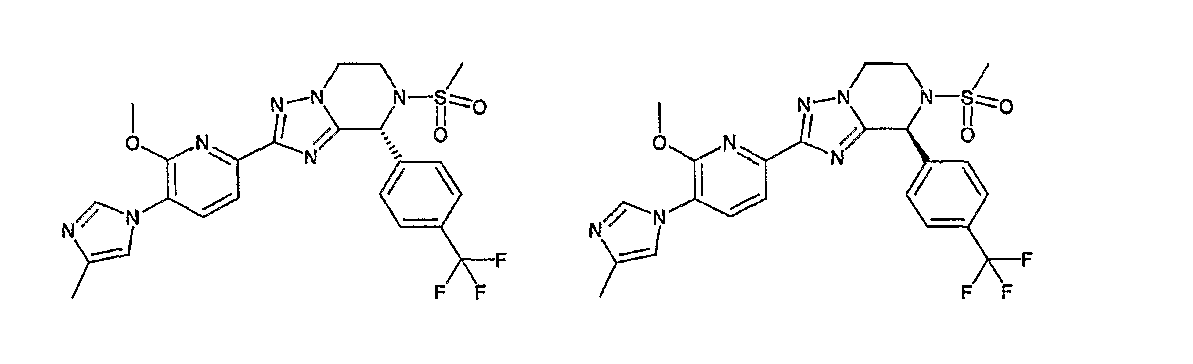
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 120 и 121 из 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина, полученного в примерах 116 и 117. Полученный рацемат указанного в заголовке соединения разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: гексан:этанол = 65:35, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 7,95 минуты, полученным из анализа с помощью CHIRALPAK™ IB (Lot. IB00CD-FD025, гексан:этанол = 8:2, 1,0 мл/мин), и положительным оптическим вращением (16 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 8,99 минуты, полученным из такого же анализа, и отрицательным оптическим вращением (14,8 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 534 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 2,91 (с, 3H), 3,59 (ддд, J=5,2, 11,2, 14,8 Гц, 1H), 4,17 (с, 3H), 4,25-4,30 (м, 1H), 4,45-4,52 (м, 2H), 6,52 (с, 1H), 7,03 (т, J=1,2 Гц, 1H), 7,56 (д, J=8,4 Гц, 2H), 7,65 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,4 Гц, 2H), 7,85 (д, J=8,0 Гц, 1H), 7,87 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 534 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 2,91 (с, 3H), 3,59 (ддд, J=5,2, 11,2, 14,8 Гц, 1H), 4,17 (с, 3H), 4,25-4,30 (м, 1H), 4,45-4,52 (м, 2H), 6,52 (с, 1H), 7,03 (т, J=1,2 Гц, 1H), 7,56 (д, J=8,4 Гц, 2H), 7,65 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,4 Гц, 2H), 7,85 (д, J=8,0 Гц, 1H), 7,87 (д, J=1,2 Гц, 1H).
Примеры 126 и 127
Синтез (+)-7-бензолсульфонил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина и (-)-7-бензолсульфонил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
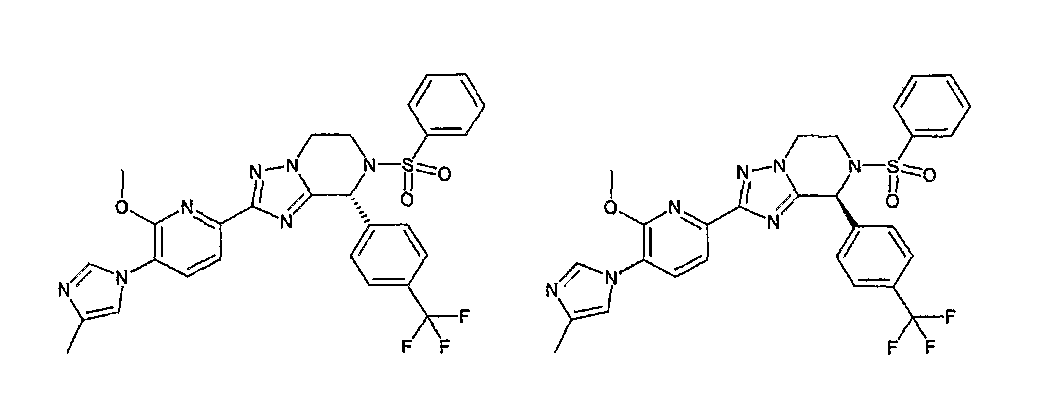
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 120 и 121 из 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина, полученного в примерах 116 и 117. Полученный рацемат указанного в заголовке соединения разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: гексан:этанол = 6:4, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 9,5 минуты, полученным из анализа с помощью CHIRALPAK™ IA (Lot. IA00CE-FA020, гексан:этанол = 1:1, 1,0 мл/мин) (11,4 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 12,9 минуты, полученным из такого же анализа, (16,6 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 9,5 минуты в условиях анализа были следующими.
ESI-MS; m/z 596 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 3,52 (ддд, J=4,4, 12,0, 15,2 Гц, 1H), 3,98 (дт, J=5,2, 12,8 Гц, 1H), 4,15 (с, 3H), 4,21 (дд, J=4,0, 131,2 Гц, 1H), 4,30 (дд, J=5,2, 15,2 Гц, 1H), 6,62 (с, 1H), 7,02 (т, J=1,2 Гц, 1H), 7,45-7,49 (м, 4H), 7,57-7,62 (м, 3H), 7,64 (д, J=8,0 Гц, 1H), 7,82-7,86 (м, 4H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 12,9 минуты в условиях анализа были следующими.
ESI-MS; m/z 596 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,31 (с, 3H), 3,52 (ддд, J=4,4, 12,0, 15,2 Гц, 1H), 3,98 (дт, J=5,2, 12,8 Гц, 1H), 4,15 (с, 3H), 4,21 (дд, J=4,0, 131,2 Гц, 1H), 4,30 (дд, J=5,2, 15,2 Гц, 1H), 6,62 (с, 1H), 7,02 (т, J=1,2 Гц, 1H), 7,45-7,49 (м, 4H), 7,57-7,62 (м, 3H), 7,64 (д, J=8,0 Гц, 1H), 7,82-7,86 (м, 4H).
Примеры 128, 129, 130, 131, 132 и 133
Синтез (+)-(6,8-син)-6-фтор-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-(6,8-син)-6-фтор-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-7,8-дигидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-7,8-дигидро[1,2,4]триазоло[1,5-a]пиридина и (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,8-дигидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,8-дигидро[1,2,4]триазоло[1,5-a]пиридина
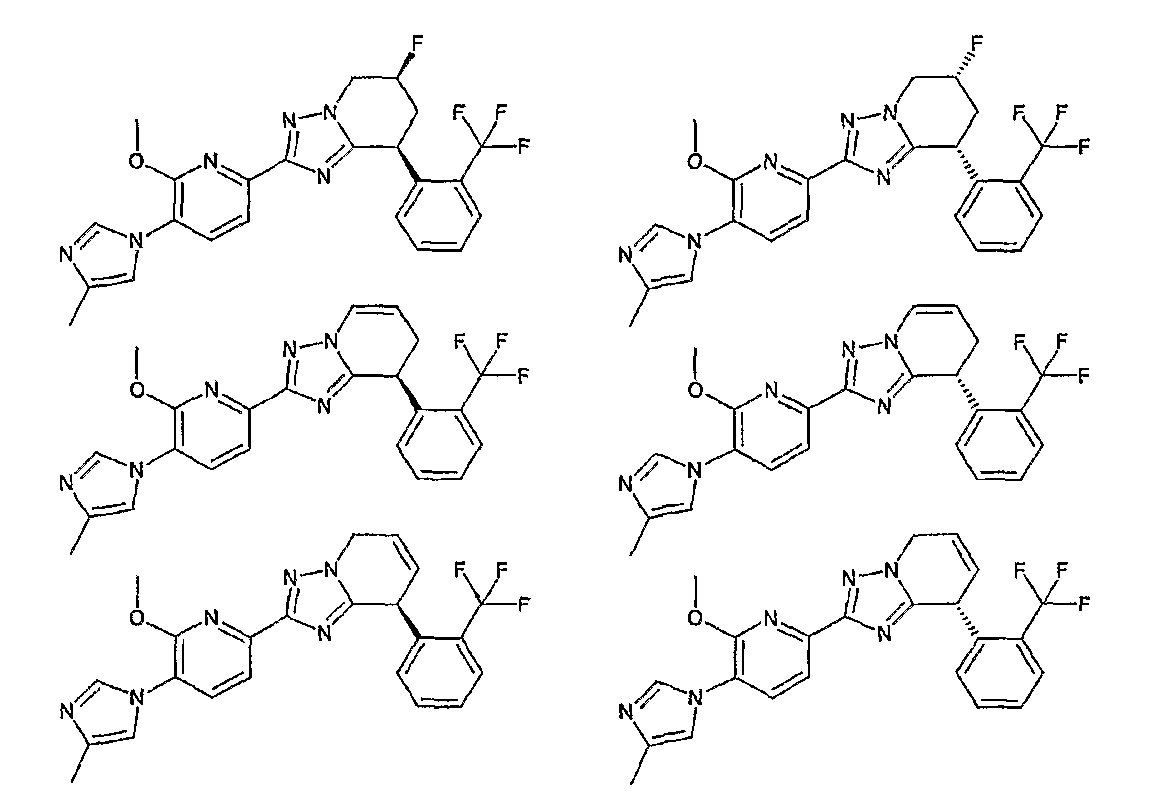
2-[6-Метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-6-ол, полученный в примере 26 (100 мг), растворяли в метиленхлориде (1 мл) и [бис(2-метоксиэтил)амино]сератрифторид (78,5 мкл) добавляли при комнатной температуре. После перемешивания в течение 24 часов, реакционный раствор распределяли между насыщенным раствором бикарбоната натрия и этилацетатом. Органический слой промывали насыщенным раствором соли, сушили над сульфатом магния и затем концентрировали при пониженном давлении. Очистка хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-100%) дала смесь фторированного соединения и двух олефиновых соединений (48,7 мг). Смесь разделяли с помощью CHIRALPAK™ AD-H (2 см × 25 см, подвижная фаза: этанол:гексан = 3:7, скорость потока: 15 мл/мин) с получением 7,8-дигидросоединения с временем удерживания 11,8 минуты и положительным оптическим вращением (1,6 мг) и 7,8-дигидросоединения с временем удерживания 14,1 минуты и отрицательным оптическим вращением (1,7 мг) и фторированного соединения с временем удерживания 23,0 минуты и положительным оптическим вращением (2,7 мг) и фторированного соединения с временем удерживания 26,3 минуты и отрицательным оптическим вращением (3,9 мг). При этом было извлечено 5,8-дигидросоединение в виде рацемата с временем удерживания 15,5 минуты. Рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: этанол:гексан = 4:6, скорость потока: 13 мл/мин) с получением 5,8-дигидросоединения с временем удерживания 13,4 минуты и положительным оптическим вращением (3,3 мг) и 5,8-дигидросоединения с временем удерживания 16,1 минуты и отрицательным оптическим вращением (2,3 мг).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из фторированного соединения с временем удерживания 23,0 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 2,52 (ддд, J=14,8, 14,8, 7,4 Гц, 1H), 2,64-2,80 (м, 1H), 4,15 (с, 3H), 4,59-4,75 (м, 2H), 4,91 (дд, J=7,0, 7,0 Гц, 1H), 5,25-5,43 (м, 1H), 6,98-7,00 (м, 1H), 7,03 (д, J=6,6 Гц, 1H), 7,37-7,42 (м, 1H), 7,43-7,48 (м, 1H), 7,57 (д, J=7,8 Гц, 1H), 7,73 (д, J=6,6 Гц, 1H), 7,75 (д, J=7,8 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из фторированного соединения с временем удерживания 26,3 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 2,52 (ддд, J=14,8, 14,8, 7,4 Гц, 1H), 2,64-2,80 (м, 1H), 4,15 (с, 3H), 4,59-4,75 (м, 2H), 4,91 (дд, J=7,0, 7,0 Гц, 1H), 5,25-5,43 (м, 1H), 6,98-7,00 (м, 1H), 7,03 (д, J=6,6 Гц, 1H), 7,37-7,42 (м, 1H), 7,43-7,48 (м, 1H), 7,57 (д, J=7,8 Гц, 1H), 7,73 (д, J=6,6 Гц, 1H), 7,75 (д, J=7,8 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из 7,8-дигидросоединения с временем удерживания 11,8 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 2,59-2,69 (м, 1H), 2,95-3,05 (м, 1H), 4,16 (с, 3H), 4,95 (дд, J=9,0, 9,0 Гц, 1H), 5,72 (ддд, J=7,8, 3,9, 3,9 Гц, 1H), 6,90-7,05 (м, 1H), 7,26-7,35 (м, 2H), 7,40-7,45 (м, 1H), 7,49-7,56 (м, 1H), 7,58 (д, J=8,2 Гц, 1H), 7,74 (д, J=6,6 Гц, 1H), 7,78 (д, J=8,2 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из 7,8-дигидросоединения с временем удерживания 14,1 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 2,59-2,69 (м, 1H), 2,95-3,05 (м, 1H), 4,16 (с, 3H), 4,95 (дд, J=9,0, 9,0 Гц, 1H), 5,72 (ддд, J=7,8, 3,9, 3,9 Гц, 1H), 6,90-7,05 (м, 1H), 7,26-7,35 (м, 2H), 7,40-7,45 (м, 1H), 7,49-7,56 (м, 1H), 7,58 (д, J=8,2 Гц, 1H), 7,74 (д, J=6,6 Гц, 1H), 7,78 (д, J=8,2 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из 5,8-дигидросоединения с временем удерживания 13,4 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 4,16 (с, 3H), 5,00 (дд, J=17,5, 6,6 Гц, 1H), 5,10 (дд, J=17,5, 4,7 Гц, 1H), 5,35-5,41 (м, 1H), 6,06 (с, 2H), 6,91 (д, J=7,4 Гц, 1H), 6,97-7,05 (м, 1H), 7,34-7,46 (м, 2H), 7,57 (д, J=7,8 Гц, 1H), 7,73 (д, J=7,4 Гц, 1H), 7,78 (д, J=7,8 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из 5,8-дигидросоединения с временем удерживания 16,1 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 4,16 (с, 3H), 5,00 (дд, J=17,5, 6,6 Гц, 1H), 5,10 (дд, J=17,5, 4,7 Гц, 1H), 5,35-5,41 (м, 1H), 6,06 (с, 2H), 6,91 (д, J=7,4 Гц, 1H), 6,97-7,05 (м, 1H), 7,34-7,46 (м, 2H), 7,57 (д, J=7,8 Гц, 1H), 7,73 (д, J=7,4 Гц, 1H), 7,78 (д, J=7,8 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H.
Примеры 134 и 135
Синтез (+)-6,6-дифтор-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-6,6-дифтор-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
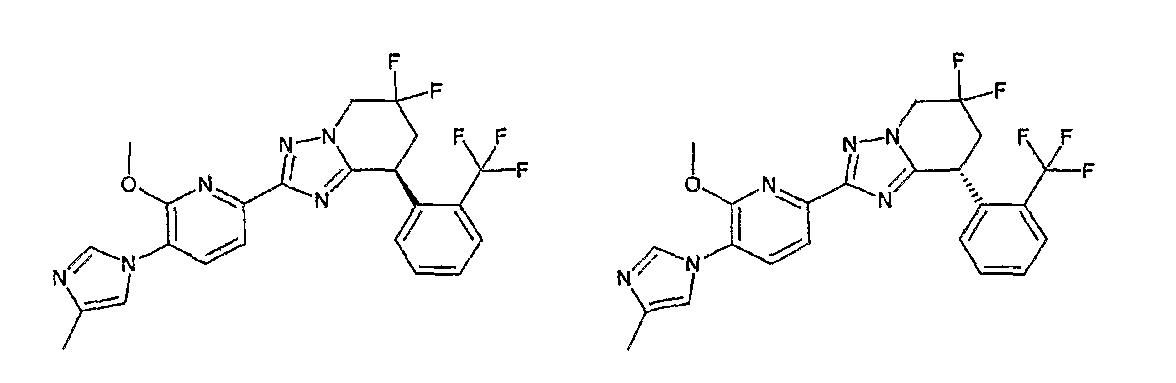
Синтез 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-7,8-дигидро-[1,2,4]триазоло[1,5-a]пиридин-6-она
2-[6-Метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-6-ол, полученный в примере 26 (125 мг), растворяли в метиленхлориде (3 мл) и добавляли при комнатной температуре реактив Деса-Мартина (226 мг). После перемешивания в течение 30 минут реакционный раствор распределяли между насыщенным раствором бикарбоната натрия и этилацетатом. Органический слой промывали насыщенным раствором соли, сушили над сульфатом магния и затем концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-100% → метанол/этилацетат = 10%) с получением указанного в заголовке соединения (28 мг).
ESI-МС; m/z 469 [M++H].
Синтез (+)-6,6-дифтор-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-6,6-дифтор-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 128, 129, 130, 131, 132 и 133 из 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-7,8-дигидро[1,2,4]триазоло[1,5-a]пиридин-6-она (20,6 мг). Полученный рацемат (2,5 мг) разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: этанол:гексан = 3:7, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 22,5 минуты и положительным оптическим вращением (1,4 мг) и указанного в заголовке соединения с временем удерживания 28,5 минуты и отрицательным оптическим вращением (0,7 мг). Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 22,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,19-2,38 (м, 1H), 2,30 (с, 3H), 2,54-2,68 (м, 1H), 2,74-2,85 (м, 2H), 4,15 (с, 3H), 4,81 (дд, J=7,8, 6,6 Гц, 1H), 6,99-7,06 (м, 1H), 7,25 (д, J=7,8 Гц, 1H), 7,43-7,48 (м, 1H), 7,49-7,55 (м, 1H), 7,57 (д, J=8,2 Гц, 1H), 7,77 (д, J=7,8 Гц, 1H), 7,79 (д, J=8,2 Гц, 1H), 7,84 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 28,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,19-2,38 (м, 1H), 2,30 (с, 3H), 2,54-2,68 (м, 1H), 2,74-2,85 (м, 2H), 4,15 (с, 3H), 4,81 (дд, J=7,8, 6,6 Гц, 1H), 6,99-7,06 (м, 1H), 7,25 (д, J=7,8 Гц, 1H), 7,43-7,48 (м, 1H), 7,49-7,55 (м, 1H), 7,57 (д, J=8,2 Гц, 1H), 7,77 (д, J=7,8 Гц, 1H), 7,79 (д, J=8,2 Гц, 1H), 7,84 (д, J=1,2 Гц, 1H).
Примеры 136 и 137
Синтез (+)-5,5-дифтор-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-5,5-дифтор-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
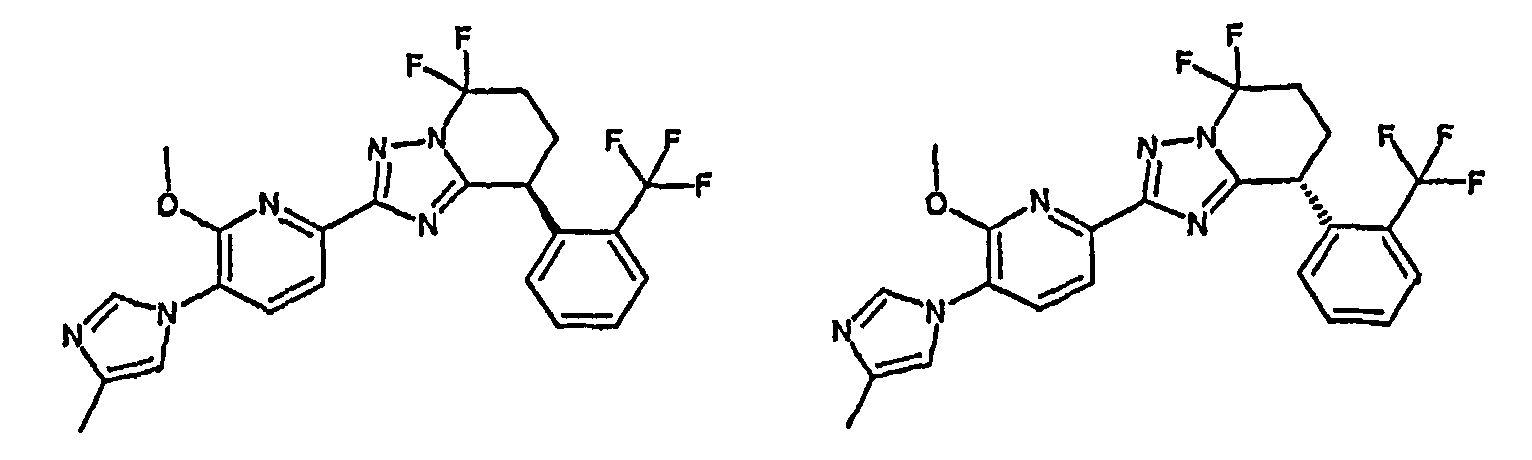
Рацемат указанного в заголовке соединения (15 мг) получали в соответствии со способом примеров 134 и 135 из 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(2-трифторметилфенил)-7,8-дигидро-6H-[1,2,4]триазоло[1,5-a]пиридин-5-она, полученного в примере 72 (100 мг). Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: этанол:гексан = 2:8, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 24,6 минуты и положительным оптическим вращением (1,4 мг) и указанного в заголовке соединения с временем удерживания 28,5 минуты и отрицательным оптическим вращением (1,6 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 24,6 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,22-2,28 (м, 1H), 2,30 (с, 3H), 2,52-2,67 (м, 2H), 2,74-2,85 (м, 1H), 4,15 (с, 3H), 4,23 (дд, J=11,7, 11,7 Гц, 1H), 4,82 (дд, J=7,8, 6,6 Гц, 1H), 6,95-7,01 (м, 1H), 7,05 (д, J=7,8 Гц, 1H), 7,43-7,48 (м, 1H), 7,49-7,54 (м, 1H), 7,58 (д, J=7,8 Гц, 1H), 7,77 (д, J=7,8 Гц, 1H), 7,79 (д, J=7,8 Гц, 1H), 7,84 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 28,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,22-2,28 (м, 1H), 2,30 (с, 3H), 2,52-2,67 (м, 2H), 2,74-2,85 (м, 1H), 4,15 (с, 3H), 4,23 (дд, J=11,7, 11,7 Гц, 1H), 4,82 (дд, J=7,8, 6,6 Гц, 1H), 6,95-7,01 (м, 1H), 7,05 (д, J=7,8 Гц, 1H), 7,43-7,48 (м, 1H), 7,49-7,54 (м, 1H), 7,58 (д, J=7,8 Гц, 1H), 7,77 (д, J=7,8 Гц, 1H), 7,79 (д, J=7,8 Гц, 1H), 7,84 (д, J=1,2 Гц, 1H).
Примеры 138, 139, 140 и 141
Синтез (+)-(7,8-син)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-фтор-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-(7,8-син)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-фтор-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (+)-(7,8-анти)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-фтор-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-(7,8-анти)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-фтор-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
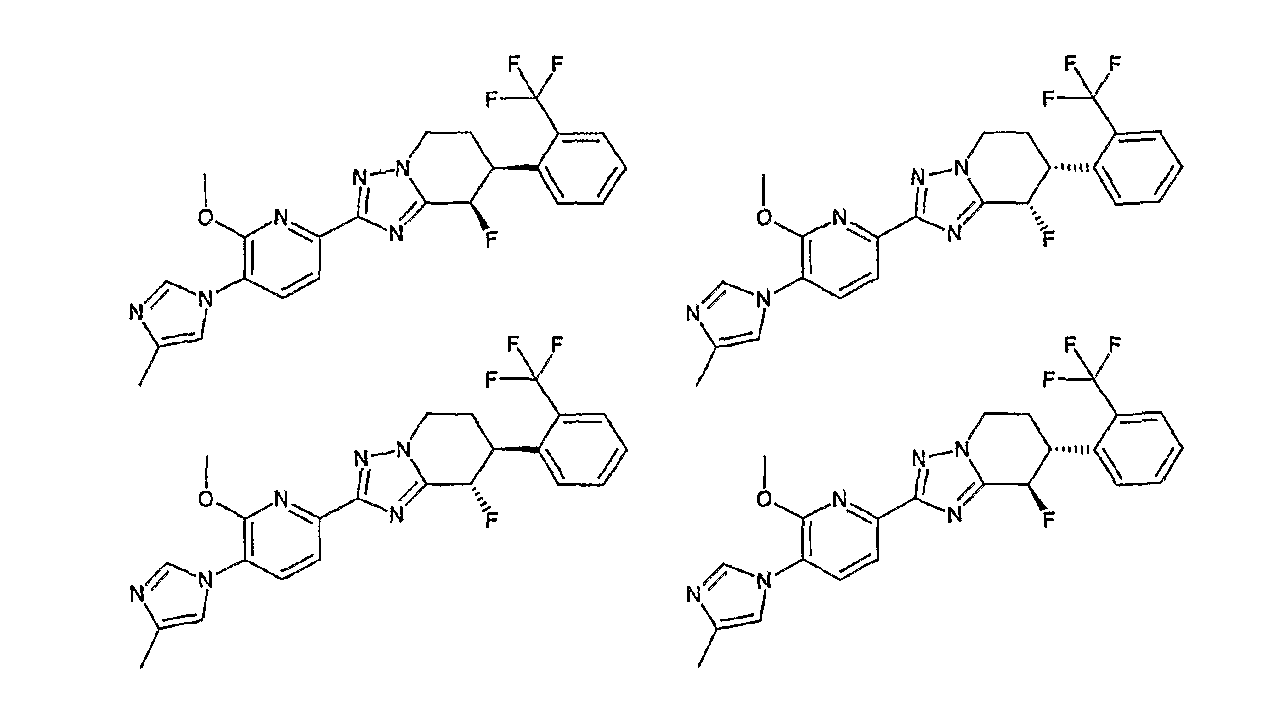
Раствор 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-7-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примерах 94 и 95 (50 мг), в тетрагидрофуране (1 мл) добавляли при 0°C к раствору диизопропиламина лития, приготовленного из н-бутиллития (93,1 мкл) и диизопропиламина (35 мкл), в тетрагидрофуране (1 мл). После перемешивания при той же самой температуре в течение 30 минут добавляли N-фтор-N′-хлорметилтриэтилендиамин бис(тетрафторборат) (90,2 мг). После перемешивания при 0°C в течение 50 минут температуру реакции повышали до комнатной температуры. Еще через 50 минут реакционный раствор распределяли между водой и этилацетатом. Органический слой промывали насыщенным раствором соли и затем сушили над сульфатом магния. Масло, полученное удалением растворителя при пониженном давлении, растворяли в метиленхлориде (1,1 мл), после чего добавляли [бис(2-метоксиэтил)амино]сератрифторид (40,6 мкл). Побочный гидроксид превращали во фторированное соединение.
Рацемическое син-соединение, очищенное хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-100%) (10,6 мг), разделяли с помощью CHIRALPAK™ AD-H (2 см × 25 см, подвижная фаза: этанол:гексан = 2:8, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 33,0 минуты и отрицательным оптическим вращением (1,1 мг) и указанного в заголовке оптически активного соединения с временем удерживания 46,0 минуты и положительным оптическим вращением (0,6 мг). С другой стороны, рацемическое анти-соединение (7,1 мг) разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: этанол:гексан = 2:8, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 42,5 минуты и положительным оптическим вращением (0,68 мг) и указанного в заголовке оптически активного соединения с временем удерживания 61,0 минуты и отрицательным оптическим вращением (0,44 мг). Показатели свойств указанного в заголовке оптически активного соединения, полученного из син-соединения, с временем удерживания 33,0 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,20-2,30 (м, 1H), 2,31 (с, 3H), 2,95-3,05 (м, 1H), 3,65-3,80 (м, 1H), 4,19 (с, 3H), 4,30-4,40 (м, 1H), 4,65-4,72 (м, 1H), 5,75 (дд, J=49,2, 5,5 Гц, 1H), 6,92-6,94 (м, 1H), 7,46-7,51 (м, 1H), 61-7,71 (м, 2H), 7,66 (д, J=7,8 Гц, 1H), 7,76 (д, J=7,8 Гц, 1H), 7,84-7,87 (м, 1H), 7,87 (д, J=7,8 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из син-соединения, с временем удерживания 46,0 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,20-2,30 (м, 1H), 2,31 (с, 3H), 2,95-3,05 (м, 1H), 3,65-3,80 (м, 1H), 4,19 (с, 3H), 4,30-4,40 (м, 1H), 4,65-4,72 (м, 1H), 5,75 (дд, J=49,2, 5,5 Гц, 1H), 6,92-6,94 (м, 1H), 7,46-7,51 (м, 1H), 61-7,71 (м, 2H), 7,66 (д, J=7,8 Гц, 1H), 7,76 (д, J=7,8 Гц, 1H), 7,84-7,87 (м, 1H), 7,87 (д, J=7,8 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из антисоединения, с временем удерживания 42,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,25-2,40 (м, 1H), 2,31 (с, 3H), 2,49-2,59 (м, 1H), 3,96-4,08 (м, 1H), 4,19 (с, 3H), 4,30-4,45 (м, 2H), 5,95 (дд, J=50,0, 7,0 Гц, 1H), 7,03-7,05 (м, 1H), 7,20-7,23 (м, 1H), 7,44-7,49 (м, 1H), 7,56-7,61 (м, 1H), 7,66 (д, J=7,8 Гц, 1H), 7,78 (д, J=7,8 Гц, 1H), 7,86-7,88 (м, 1H), 7,92 (д, J=7,8 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения, полученного из антисоединения, с временем удерживания 61,0 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,25-2,40 (м, 1H), 2,31 (с, 3H), 2,49-2,59 (м, 1H), 3,96-4,08 (м, 1H), 4,19 (с, 3H), 4,30-4,45 (м, 2H), 5,95 (дд, J=50,0, 7,0 Гц, 1H), 7,03-7,05 (м, 1H), 7,20-7,23 (м, 1H), 7,44-7,49 (м, 1H), 7,56-7,61 (м, 1H), 7,66 (д, J=7,8 Гц, 1H), 7,78 (д, J=7,8 Гц, 1H), 7,86-7,88 (м, 1H), 7,92 (д, J=7,8 Гц, 1H).
Примеры 142 и 143
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-метокси-3-пиримидин-2-илфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-метокси-3-пиримидин-2-илфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
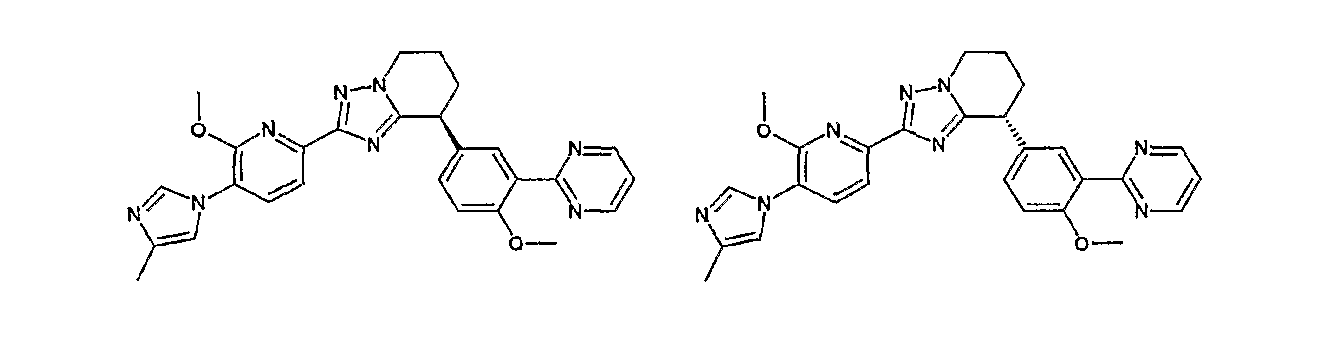
2-[6-Метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-бром-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин, полученный в примерах 68 и 69 (61 мг), растворяли в N-метилпирролидоне (1,5 мл). Добавляли ацетат палладия (5,52 мг), 1,3-бис(дифенилфосфино)пропан (20,3 мг), 2-трибутилстаннилпиримидин (45,4 мг) и оксид меди (I) (26,4 мг) и начинали реакцию при 120°C. Через 19 часов реакционный раствор распределяли между этилацетатом и водой. Органический слой промывали водным аммиаком (два раза) и насыщенным раствором соли. Концентрирование при пониженном давлении дало масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-100% → метанол/этилацетат = 10%) с получением рацемата указанного в заголовке соединения (33 мг). Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: этанол:гексан = 8:2, скорость потока: 10 мл/мин) с получением указанного в заголовке соединения с временем удерживания 38 минут и положительным оптическим вращением (8,2 мг) и указанного в заголовке соединения с временем удерживания 57,6 минуты и отрицательным оптическим вращением (7,8 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 38 минут были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,02-2,28 (м, 3H), 2,30 (с, 3H), 2,34-2,43 (м, 1H), 3,87 (с, 3H), 4,16 (с, 3H), 4,34-4,46 (м, 3H), 6,96-7,13 (м, 2H), 7,16-7,25 (м, 2H), 7,55 (д, J=2,3 Гц, 1H), 7,58 (д, J=7,8 Гц, 1H), 7,80 (д, J=7,8 Гц, 1H), 7,82 (д, J=2,3 Гц, 1H), 8,84 (д, J=5,0 Гц, 2H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 57,6 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,02-2,28 (м, 3H), 2,30 (с, 3H), 2,34-2,43 (м, 1H), 3,87 (с, 3H), 4,16 (с, 3H), 4,34-4,46 (м, 3H), 6,96-7,13 (м, 2H), 7,16-7,25 (м, 2H), 7,55 (д, J=2,3 Гц, 1H), 7,58 (д, J=7,8 Гц, 1H), 7,80 (д, J=7,8 Гц, 1H), 7,82 (д, J=2,3 Гц, 1H), 8,84 (д, J=5,0 Гц, 2H).
Примеры 144 и 145
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-метокси-3-пиридазин-3-илфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-метокси-3-пиридазин-3-илфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
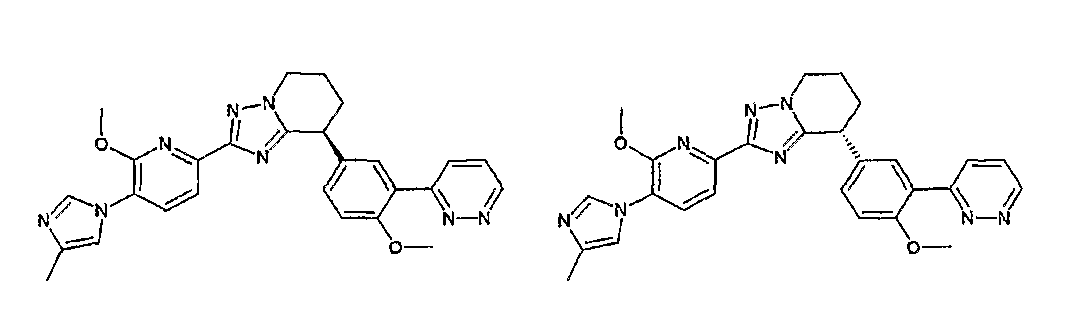
Рацемат указанного в заголовке соединения (37,7 мг) получали в соответствии со способом примеров 142 и 143 из 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-бром-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примерах 68 и 69. Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: этанол:гексан = 9:1, скорость потока: 10 мл/мин) с получением указанного в заголовке соединения с временем удерживания 20,5 минуты и положительным оптическим вращением (7,4 мг) и указанного в заголовке соединения с временем удерживания 65,5 минуты и отрицательным оптическим вращением (9,9 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 20,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,04-2,28 (м, 3H), 2,30 (с, 3H), 2,37-2,45 (м, 1H), 3,85 (с, 3H), 4,16 (с, 3H), 4,37-4,45 (м, 3H), 6,88-6,93 (м, 2H), 7,17-7,24 (м, 2H), 7,59 (д, J=8,2 Гц, 1H), 7,62 (дд, J=5,2, 2,2 Гц, 1H), 7,78 (д, J=8,2 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H), 9,17 (дд, J=5,2, 1,2 Гц, 1H), 9,37 (дд, J=1,3, 1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 65,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,04-2,28 (м, 3H), 2,30 (с, 3H), 2,37-2,45 (м, 1H), 3,85 (с, 3H), 4,16 (с, 3H), 4,37-4,45 (м, 3H), 6,88-6,93 (м, 2H), 7,17-7,24 (м, 2H), 7,59 (д, J=8,2 Гц, 1H), 7,62 (дд, J=5,2, 2,2 Гц, 1H), 7,78 (д, J=8,2 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H), 9,17 (дд, J=5,2, 1,2 Гц, 1H), 9,37 (дд, J=1,3, 1,2 Гц, 1H).
Примеры 146 и 147
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-фтор-3-пиримидин-2-илфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-фтор-3-пиримидин-2-илфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
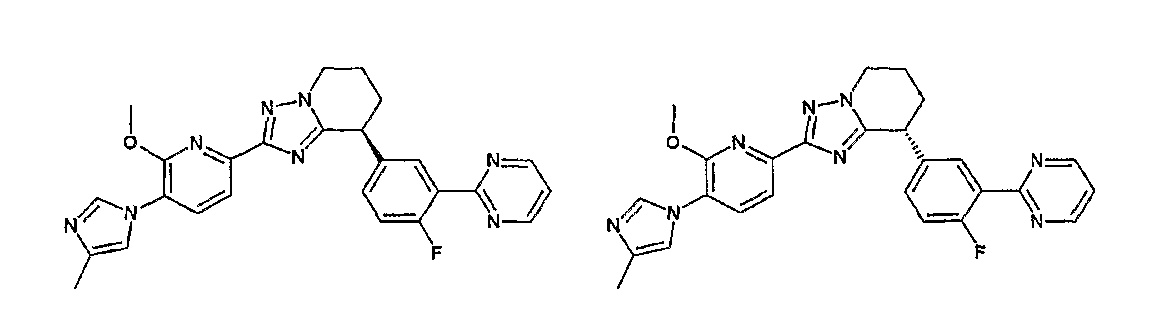
Рацемат указанного в заголовке соединения (32 мг) получали в соответствии со способом примеров 142 и 143 из 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-бром-4-фторфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примерах 80 и 81 (70 мг). Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: этанол:гексан = 75:25, скорость потока: 10 мл/мин) с получением указанного в заголовке соединения с временем удерживания 25 минут и положительным оптическим вращением (3,8 мг) и указанного в заголовке соединения с временем удерживания 34,5 минуты и отрицательным оптическим вращением (3 мг). Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 25 минут были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,10-2,18 (м, 2H), 2,20-2,25 (м, 1H), 2,30 (с, 3H), 2,37-2,48 (м, 1H), 4,16 (с, 3H), 4,42 (дд, J=11,7, 6,2 Гц, 2H), 4,48 (дд, J=6,6, 6,6 Гц, 1H), 6,91-6,98 (м, 1H), 7,15-7,26 (м, 3H), 7,58 (д, J=7,8 Гц, 1H), 7,78 (д, J=7,8 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H), 7,91 (дд, J=7,0, 1,2 Гц, 1H), 8,85 (д, J=5,1 Гц, 2H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 34,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,10-2,18 (м, 2H), 2,20-2,25 (м, 1H), 2,30 (с, 3H), 2,37-2,48 (м, 1H), 4,16 (с, 3H), 4,42 (дд, J=11,7, 6,2 Гц, 2H), 4,48 (дд, J=6,6, 6,6 Гц, 1H), 6,91-6,98 (м, 1H), 7,15-7,26 (м, 3H), 7,58 (д, J=7,8 Гц, 1H), 7,78 (д, J=7,8 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H), 7,91 (дд, J=7,0, 1,2 Гц, 1H), 8,85 (д, J=5,1 Гц, 2H).
Примеры 148 и 149
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(3-пиразин-2-илфенил)-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(3-пиразин-2-илфенил)-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина
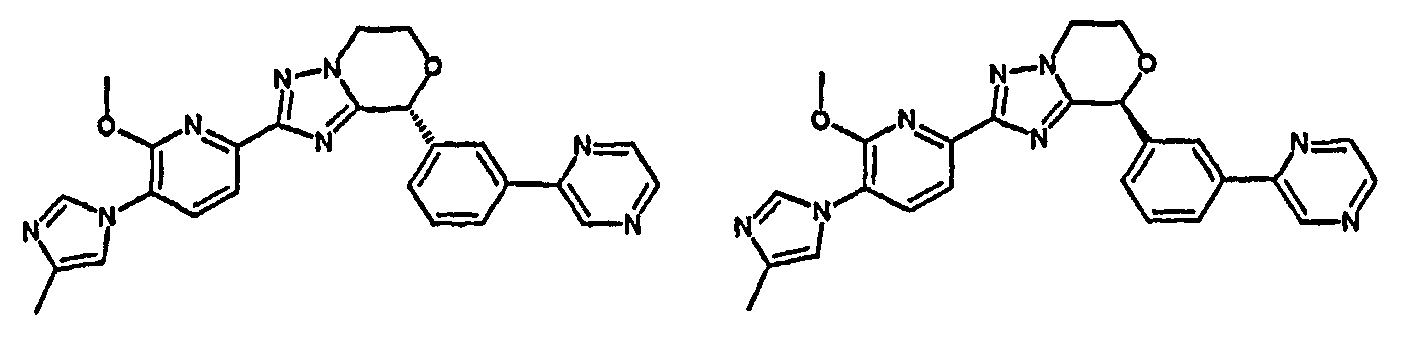
Рацемат указанного в заголовке соединения получали в соответствии со способом примеров 142 и 143 из 8-(3-бромфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина, полученного в примерах 64 и 65 (40 мг). Полученный рацемат указанного в заголовке соединения разделяли с помощью CHIRALCEL™ IB (2 см × 25 см, подвижная фаза: гексан:этанол = 1:1, скорость потока: 15 мл/мин) с получением указанного в заголовке оптически активного соединения с временем удерживания 14,16 минуты, полученным из анализа с помощью CHIRALPAK™ IB (Lot. IB00CD-FD026, гексан:этанол = 7:3, 1,0 мл/мин), и положительным оптическим вращением (1,6 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 18,25 минуты, полученным из такого же анализа, и отрицательным оптическим вращением (3,5 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,30 (с, 3H), 4,17 (с, 3H), 4,18-4,25 (м, 1H), 4,38 (дт, J=4,4, 12,0 Гц, 1H), 4,41-4,56 (м, 2H), 6,11 (с, 1H), 7,00 (д, J=2,4 Гц, 1H), 7,56-7,63 (м, 3H), 7,81-7,84 (м, 2H), 8,05 (дт, J=1,6, 7,6 Гц, 1H), 8,19 (т, J=2,0 Гц, 1H), 8,53 (д, J=2,4 Гц, 1H), 8,64 (дд, J=1,6, 3,6 Гц, 1H), 9,04 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,30 (с, 3H), 4,17 (с, 3H), 4,18-4,25 (м, 1H), 4,38 (дт, J=4,4, 12,0 Гц, 1H), 4,41-4,56 (м, 2H), 6,11 (с, 1H), 7,00 (д, J=2,4 Гц, 1H), 7,56-7,63 (м, 3H), 7,81-7,84 (м, 2H), 8,05 (дт, J=1,6, 7,6 Гц, 1H), 8,19 (т, J=2,0 Гц, 1H), 8,53 (д, J=2,4 Гц, 1H), 8,64 (дд, J=1,6, 3,6 Гц, 1H), 9,04 (д, J=1,2 Гц, 1H).
Примеры 150 и 151
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-метокси-3-(1-метил-1H-пиразол-4-ил)фенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-метокси-3-(1-метил-1H-пиразол-4-ил)фенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
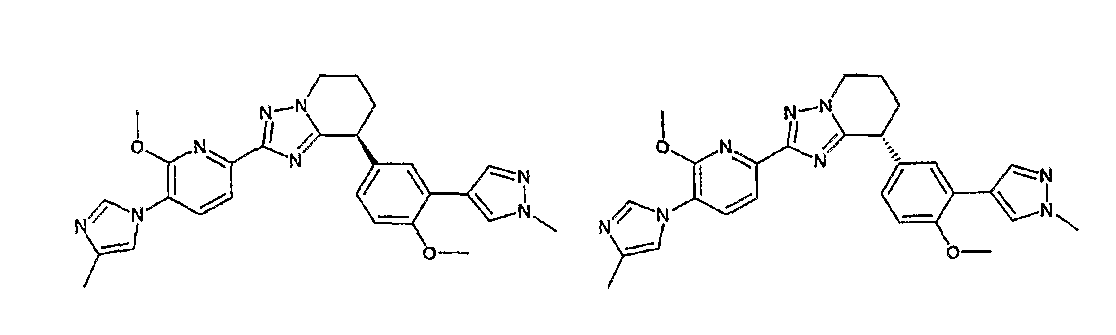
2-[6-Метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-бром-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин, полученный в примерах 68 и 69 (100 мг), растворяли в толуоле (3 мл) и метаноле (600 мкл). Добавляли 2 М раствор карбоната натрия (101 мкл), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (63 мг) и тетракистрифенилфосфинпалладий (23,4 мг) и смесь перемешивали под микроволновым излучением при 120°C в течение трех часов. Реакционный раствор распределяли между этилацетатом и водой. Затем органический слой промывали водой и насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-100% → метанол/этилацетат = 20%) с получением рацемата указанного в заголовке соединения (50,6 мг). Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: этанол:гексан = 5:5, скорость потока: 12 мл/мин) с получением указанного в заголовке соединения с временем удерживания 29,5 минуты и положительным оптическим вращением (13,2 мг) и указанного в заголовке соединения с временем удерживания 39,0 минуты и отрицательным оптическим вращением (11,6 мг). Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 29,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,04-2,16 (м, 2H), 2,18-2,27 (м, 1H), 2,30 (с, 3H), 2,34-2,43 (м, 1H), 3,89 (с, 3H), 3,93 (с, 3H), 4,17 (с, 3H), 4,33-4,47 (м, 3H), 6,87-6,94 (м, 2H), 6,99-7,01 (м, 1H), 7,31 (д, J=2,0 Гц, 1H), 7,58 (д, J=7,8 Гц, 1H), 7,80 (д, J=7,8 Гц, 1H), 7,81-7,83 (м, 3H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 39,0 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,04-2,16 (м, 2H), 2,18-2,27 (м, 1H), 2,30 (с, 3H), 2,34-2,43 (м, 1H), 3,89 (с, 3H), 3,93 (с, 3H), 4,17 (с, 3H), 4,33-4,47 (м, 3H), 6,87-6,94 (м, 2H), 6,99-7,01 (м, 1H), 7,31 (д, J=2,0 Гц, 1H), 7,58 (д, J=7,8 Гц, 1H), 7,80 (д, J=7,8 Гц, 1H), 7,81-7,83 (м, 3H).
Примеры 152 и 153
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-метокси-3-пиридин-3-илфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(4-метокси-3-пиридин-3-илфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
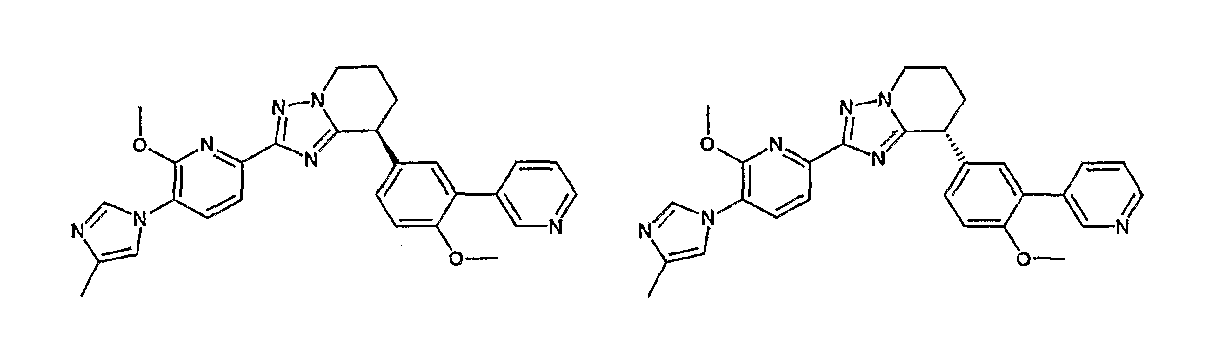
Рацемат указанного в заголовке соединения (56 мг) получали в соответствии со способом примеров 150 и 151 из 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-бром-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примерах 68 и 69 (100 мг). Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: этанол:гексан = 3:7, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 41,5 минуты и положительным оптическим вращением (17,5 мг) и указанного в заголовке соединения с временем удерживания 53,5 минуты и отрицательным оптическим вращением (18 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 41,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,06-2,16 (м, 2H), 2,19-2,28 (м, 1H), 2,30 (с, 3H), 2,35-2,44 (м, 1H), 3,81 (с, 3H), 3,99 (с, 3H), 4,16 (с, 3H), 4,34-4,47 (м, 3H), 6,95 (д, J=9,4 Гц, 1H), 6,95-7,05 (м, 1H), 7,09-7,14 (м, 2H), 7,31 (дд, J=7,8, 5,7 Гц, 1H), 7,59 (д, J=7,8 Гц, 1H), 7,78-7,84 (м, 3H), 7,87 (д, J=2,0 Гц, 1H), 8,54 (дд, J=5,7, 2,0 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 53,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,06-2,16 (м, 2H), 2,19-2,28 (м, 1H), 2,30 (с, 3H), 2,35-2,44 (м, 1H), 3,81 (с, 3H), 3,99 (с, 3H), 4,16 (с, 3H), 4,34-4,47 (м, 3H), 6,95 (д, J=9,4 Гц, 1H), 6,95-7,05 (м, 1H), 7,09-7,14 (м, 2H), 7,31 (дд, J=7,8, 5,7 Гц, 1H), 7,59 (д, J=7,8 Гц, 1H), 7,78-7,84 (м, 3H), 7,87 (д, J=2,0 Гц, 1H), 8,54 (дд, J=5,7, 2,0 Гц, 1H).
Примеры 154 и 155
Синтез (+)-8-[3-(6-хлорпиридин-3-ил)-4-метоксифенил]-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-[3-(6-хлорпиридин-3-ил)-4-метоксифенил]-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
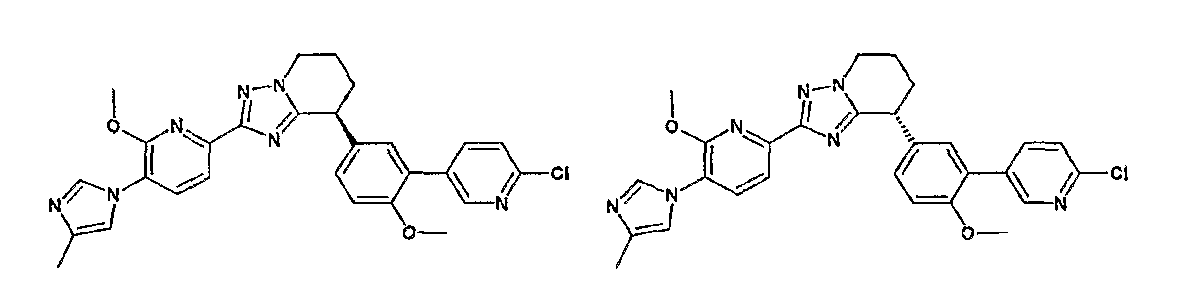
Рацемат указанного в заголовке соединения (90,3 мг) получали в соответствии со способом примеров 150 и 151 из 2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-бром-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примерах 68 и 69 (100 мг). Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: этанол:гексан = 2:8, скорость потока: 15 мл/мин) с получением указанного в заголовке соединения с временем удерживания 42 минуты и положительным оптическим вращением (22 мг) и указанного в заголовке соединения с временем удерживания 58,5 минуты и отрицательным оптическим вращением (20 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 42 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,04-2,17 (м, 2H), 2,18-2,28 (м, 1H), 2,30 (с, 3H), 2,35-2,45 (м, 1H), 3,81 (с, 3H), 3,99 (с, 3H), 4,16 (с, 3H), 4,34-4,47 (м, 3H), 6,95 (д, J=8,2 Гц, 1H), 6,98-7,03 (м, 1H), 7,07-7,16 (м, 2H), 7,34 (д, J=8,2 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,75-7,85 (м, 3H), 8,49 (д, J=2,0 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 58,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,04-2,17 (м, 2H), 2,18-2,28 (м, 1H), 2,30 (с, 3H), 2,35-2,45 (м, 1H), 3,81 (с, 3H), 3,99 (с, 3H), 4,16 (с, 3H), 4,34-4,47 (м, 3H), 6,95 (д, J=8,2 Гц, 1H), 6,98-7,03 (м, 1H), 7,07-7,16 (м, 2H), 7,34 (д, J=8,2 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,75-7,85 (м, 3H), 8,49 (д, J=2,0 Гц, 1H).
Пример 156
Синтез (+)-8-бифенил-3-ил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина
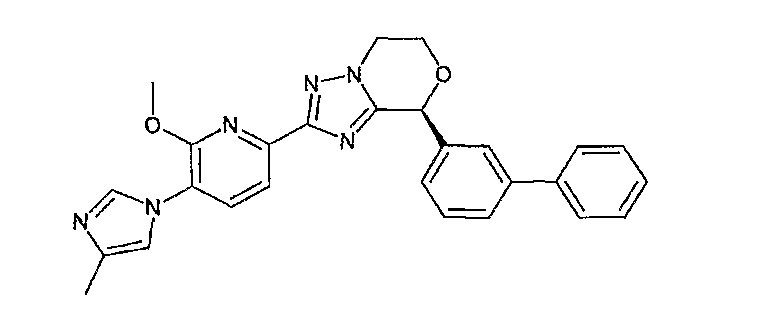
Указанное в заголовке соединение (22 мг) получали в соответствии со способом примеров 150 и 151 из (+)-8-(3-бромфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина, полученного в примере 64 (42,6 мг). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,30 (с, 3H), 4,17 (с, 3H), 4,18-4,22 (м, 1H), 4,36 (дт, J=4,4, 12,4 Гц, 1H), 4,44-4,52 (м, 2H), 6,08 (с, 1H), 7,01 (т, J=1,2 Гц, 1H), 7,35 (тт, J=2,0, 7,2 Гц, 1H), 7,41-7,50 (м, 4H), 7,57-7,63 (м, 4H), 7,70 (т, J=1,6 Гц, 1H), 7,82 (д, J=9,2 Гц, 1H), 7,83 (с, 1H).
Пример 157
Синтез (-)-8-бифенил-3-ил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазин
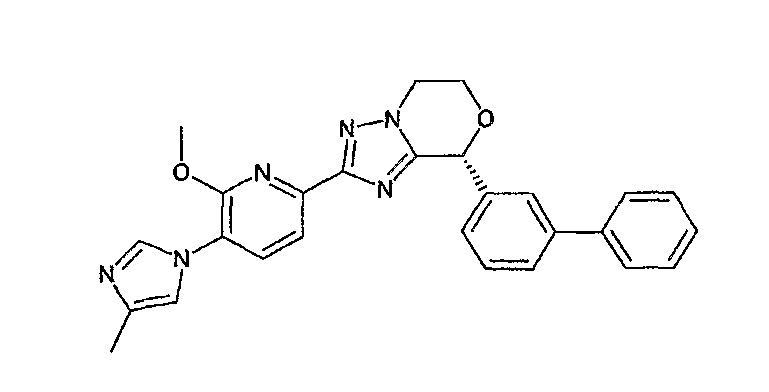
Указанное в заголовке соединение (17 мг) получали в соответствии со способом примеров 150 и 151 из (-)-8-(3-бромфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина, полученного в примере 65 (22,7 мг). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,30 (с, 3H), 4,17 (с, 3H), 4,18-4,22 (м, 1H), 4,36 (дт, J=4,4, 12,4 Гц, 1H), 4,44-4,52 (м, 2H), 6,08 (с, 1H), 7,01 (т, J=1,2 Гц, 1H), 7,35 (тт, J=2,0, 7,2 Гц, 1H), 7,41-7,50 (м, 4H), 7,57-7,63 (м, 4H), 7,70 (т, J=1,6 Гц, 1H), 7,82 (д, J=9,2 Гц, 1H), 7,83 (с, 1H).
Примеры 158 и 159
(+)-2-[6-Метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(6-фенилпиридин-2-ил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(6-фенилпиридин-2-ил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
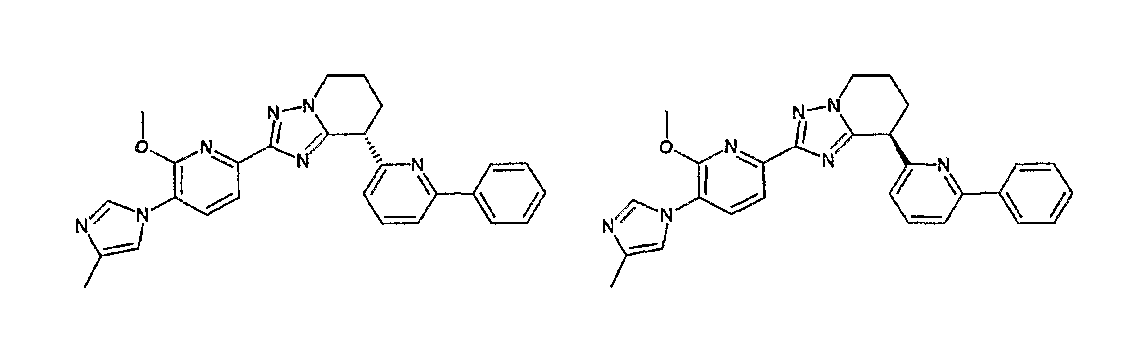
Рацемат указанного в заголовке соединения (19 мг) получали в соответствии со способом примеров 150 и 151 из 8-(6-бромпиридин-2-ил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина, полученного в примерах 70 и 71 (41,7 мг). Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: гексан:этанол = 50:50) с получением указанного в заголовке оптически активного соединения с временем удерживания 4,9 минуты и положительным оптическим вращением (7,1 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 7,6 минуты и отрицательным оптическим вращением (6,3 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 464 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,09-2,14 (м, 1H), 2,30 (д, J=1,2 Гц, 3H), 2,31-2,53 (м, 2H), 2,54-2,59 (м, 1H), 4,17 (с, 3H), 4,34-4,39 (м, 1H), 4,44-4,50 (м, 1H), 4,65 (т, J=6,0 Гц, 1H), 7,00 (т, J=1,2 Гц, 1H), 7,14 (дд, J=0,8 Гц, 7,6 Гц, 1H), 7,38-7,47 (м, 3H), 7,58 (д, J=8,0 Гц, 1H), 7,63 (дд, J=0,8 Гц, 7,6 Гц, 1H), 7,71 (т, J=7,6 Гц, 1H), 7,81 (д, J=8,0 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H), 7,96-7,98 (м, 2H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 464 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,09-2,14 (м, 1H), 2,30 (д, J=1,2 Гц, 3H), 2,31-2,53 (м, 2H), 2,54-2,59 (м, 1H), 4,17 (с, 3H), 4,34-4,39 (м, 1H), 4,44-4,50 (м, 1H), 4,65 (т, J=6,0 Гц, 1H), 7,00 (т, J=1,2 Гц, 1H), 7,14 (дд, J=0,8 Гц, 7,6 Гц, 1H), 7,38-7,47 (м, 3H), 7,58 (д, J=8,0 Гц, 1H), 7,63 (дд, J=0,8 Гц, 7,6 Гц, 1H), 7,71 (т, J=7,6 Гц, 1H), 7,81 (д, J=8,0 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H), 7,96-7,98 (м, 2H).
Примеры 160 и 161
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-метансульфонил-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-метансульфонил-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
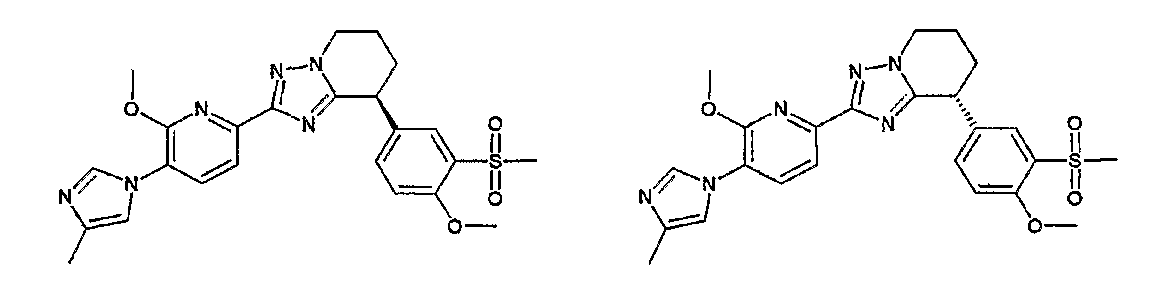
2-[6-Метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-8-(3-бром-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин, полученный в примерах 68 и 69 (100 мг), растворяли в диметилсульфоксиде (2 мл). Добавляли метансульфинат натрия (165 мг), L-пролин (18,6 мг), гидроксид натрия (12,8 мг) и иодид меди (30,8 мг). Реакцию инициировали под микроволновым излучением при 140°C. Через три часа реакционный раствор распределяли между этилацетатом и водой. Затем органический слой промывали водой и насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-100% → метанол/этилацетат = 15%) с получением рацемата указанного в заголовке соединения (58,9 мг). Полученный рацемат разделяли с помощью CHIRALCEL™ OJ-H (2 см × 25 см, подвижная фаза: этанол:гексан = 5:5, скорость потока: 12 мл/мин) с получением указанного в заголовке соединения с временем удерживания 16,0 минуты и положительным оптическим вращением (19 мг) и указанного в заголовке соединения с временем удерживания 22,0 минуты и отрицательным оптическим вращением (16,5 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 16,0 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,03-2,18 (м, 2H), 2,18-2,28 (м, 1H), 2,30 (с, 3H), 2,35-2,44 (м, 1H), 3,24 (с, 3H), 3,99 (с, 3H), 4,16 (с, 3H), 4,36-4,45 (м, 3H), 6,89-6,92 (м, 1H), 6,92 (д, J=9,0 Гц, 1H), 7,34-7,38 (м, 1H), 7,59 (д, J=9,0 Гц, 1H), 7,75 (д, J=9,0 Гц, 1H), 7,81-7,85 (м, 1H), 7,87 (д, J=2,3 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 22,0 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,03-2,18 (м, 2H), 2,18-2,28 (м, 1H), 2,30 (с, 3H), 2,35-2,44 (м, 1H), 3,24 (с, 3H), 3,99 (с, 3H), 4,16 (с, 3H), 4,36-4,45 (м, 3H), 6,89-6,92 (м, 1H), 6,92 (д, J=9,0 Гц, 1H), 7,34-7,38 (м, 1H), 7,59 (д, J=9,0 Гц, 1H), 7,75 (д, J=9,0 Гц, 1H), 7,81-7,85 (м, 1H), 7,87 (д, J=2,3 Гц, 1H).
Пример 162
Фениламид 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоновой кислоты
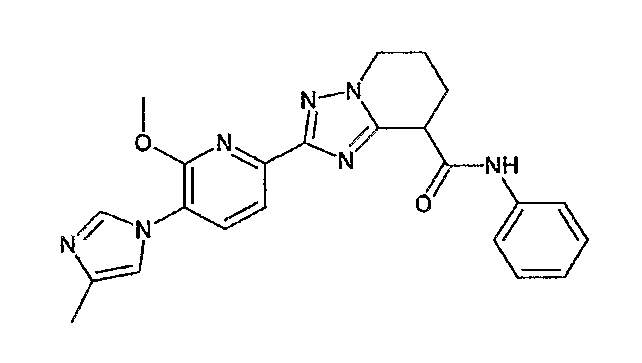
2-[6-Метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоновую кислоту, полученную в примере получения 5-1 (25 мг), растворяли в N,N-диметилформамиде (0,5 мл). Последовательно добавляли анилин (7,2 мг), гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (40,6 мг), 1-гидроксибензотриазол (28,6 мг) и N,N-диизопропилэтиламин (75,7 мкл) и смесь перемешивали при комнатной температуре в течение пяти часов. Добавляли этилацетат и воду к реакционному раствору и органический слой и водный слой разделяли. Водный слой дополнительно экстрагировали этилацетатом. Полученные органические слои объединяли, распыляли газообразным азотом и концентрировали. Остаток очищали хроматографией на колонке с силикагелем NH с получением указанного в заголовке соединения (14,1 мг). Соединение затем разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: гексан:этанол = 50:50) с получением указанного в заголовке соединения с временем удерживания 7,6 минуты (4,5 мг). Показатели свойств соединения были следующими.
ESI-MS; m/z 430 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,14-2,34 (м, 2H), 2,32 (д, J=1,2 Гц, 3H), 2,38-2,54 (м, 2H), 4,05 (т, J=6,4 Гц, 1H), 4,19 (с, 3H), 4,27-4,37 (м, 2H), 7,04 (т, J=1,2 Гц, 1H), 7,12 (тт, J=0,8 Гц, 7,6 Гц, 1H), 7,33 (дт, J=0,8 Гц, 7,6 Гц, 2H), 7,58 (дд, J=0,8 Гц, 7,6 Гц, 2H), 7,69 (д, J=8,0 Гц, 1H), 7,85 (д, J=8,0 Гц, 1H), 7,86 (д, J=1,2 Гц, 1H), 10,20 (с, 1H).
Соединения примеров 163-170 получали таким же способом, как в примере 162 (таблица 4).
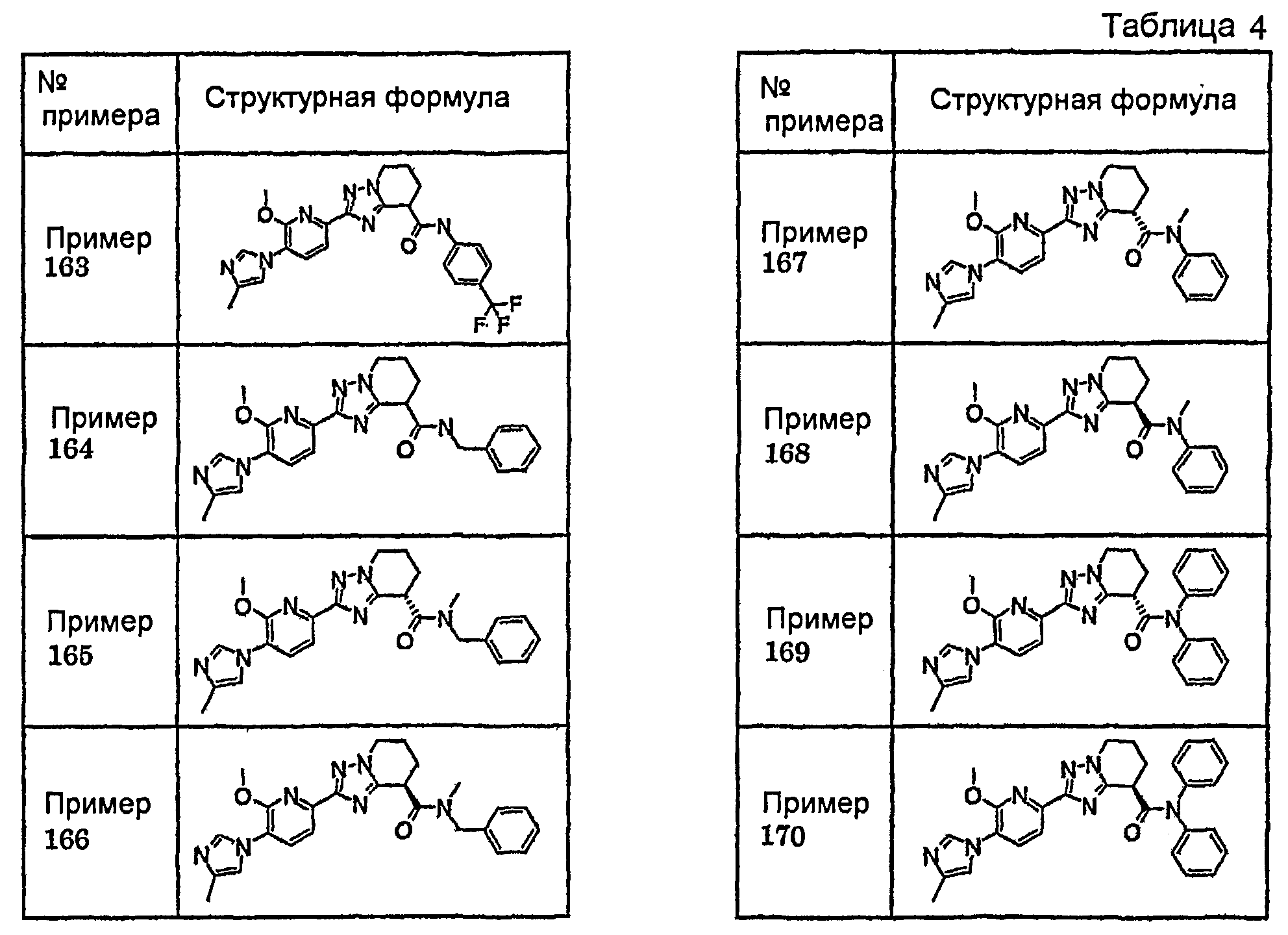
Примеры 171 и 172
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-фенил-1H-имидазол-2-ил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-фенил-1H-имидазол-2-ил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
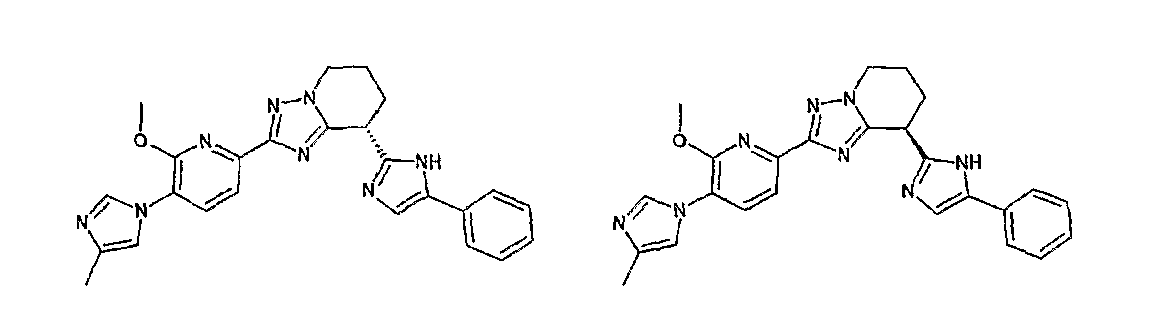
Синтез (2-оксо-2-фенилэтил)амида 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоновой кислоты
Указанное в заголовке соединение (25,6 мг) получали в соответствии со способом примера 162 из 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоновой кислоты, полученной в примере получения 5-1 (33,2 мг), и α-аминоацетофенонгидрохлорида (17,7 мг). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,12-2,45 (м, 4H), 2,32 (д, J=1,2 Гц, 3H), 3,97 (т, J=6,4 Гц, 1H), 4,22 (с, 3H), 4,25-4,36 (м, 2H), 4,79 (дд, J=4,0 Гц, 19,6 Гц, 1H), 4,87 (дд, J=4,0 Гц, 19,6 Гц, 1H), 7,04 (т, J=1,2 Гц, 1H), 7,51 (дт, J=1,2 Гц, 7,6 Гц, 2H), 7,63 (тт, J=1,2 Гц, 7,6 Гц, 1H), 7,71 (д, J=8,0 Гц, 1H), 7,86 (д, J=1,2 Гц, 1H), 7,99 (дд, J=1,2 Гц, 7,6 Гц, 2H), 8,05 (д, J=8,0 Гц, 1H), 9,07 (т, J=4,0 Гц, 1H).
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-фенил-1H-имидазол-2-ил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-фенил-1H-имидазол-2-ил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
(2-Оксо-2-фенилэтил)амид 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоновой кислоты (25 мг) растворяли в ледяной уксусной кислоте (0,5 мл). Добавляли ацетат аммония (20,4 мг) и смесь перемешивали при кипячении с обратным холодильником в течение восьми часов. Реакционный раствор охлаждали льдом. Затем добавляли этилацетат, воду и концентрированный водный аммиак (1 мл) и отделяли органический слой. Полученный органический слой промывали последовательно насыщенным раствором бикарбоната натрия и насыщенным раствором соли и сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем NH с получением рацемата указанного в заголовке соединения (16,8 мг). Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: гексан:этанол = 50:50) с получением указанного в заголовке оптически активного соединения с временем удерживания 4,2 минуты и положительным оптическим вращением (6,3 мг, >99% ee) и указанного в заголовке оптически активного соединения с временем удерживания 6,1 минуты и отрицательным оптическим вращением (6,9 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 453 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,22-2,76 (м, 4H), 2,31 (с, 3H), 4,11 (с, 3H), 4,25-4,44 (м, 2H), 4,49 (т, J=6,0 Гц, 1H), 7,01 (с, 1H), 7,01-7,39 (м, 5H), 7,63-7,65 (м, 1H), 7,74 (ушир.с, 1H), 7,84 (ушир.с, 2H), 11,09 (ушир.с, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 453 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,22-2,76 (м, 4H), 2,31 (с, 3H), 4,11 (с, 3H), 4,25-4,44 (м, 2H), 4,49 (т, J=6,0 Гц, 1H), 7,01 (с, 1H), 7,01-7,39 (м, 5H), 7,63-7,65 (м, 1H), 7,74 (ушир.с, 1H), 7,84 (ушир.с, 2H), 11,09 (ушир.с, 1H).
Пример 173
Синтез 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-фенилоксазол-2-ил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
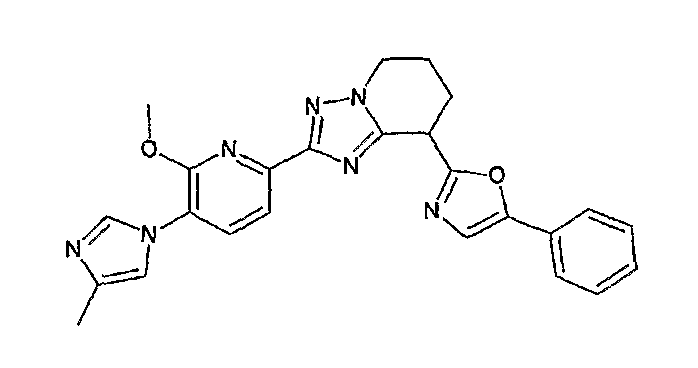
(2-Оксо-2-фенилэтил)амид 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоновой кислоты, полученный в примере 172 (35,7 мг), суспендировали в толуоле (0,5 мл). Оксалилхлорид (20,4 мг) добавляли и смесь перемешивали при кипячении с обратным холодильником в течение двух часов. Добавляли толуол (3 мл) и фосфорилхлорид (60 мкл) с последующим дополнительным перемешиванием в течение двух часов и 30 минут. Реакционный раствор охлаждали льдом. Затем добавляли хлороформ и насыщенный раствор бикарбоната натрия и отделяли органический слой. Полученный органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем NH с получением указанного в заголовке соединения (4,4 мг). Соединение затем очищали с помощью CHIRALPAK™ IA (2 см × 25 см; подвижная фаза: гексан:этанол = 50:50) с получением указанного в заголовке соединения (1,2 мг, рацемат). Показатели свойств соединения были следующими.
ESI-MS; m/z 454 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,18-2,24 (м, 1H), 2,30 (д, J=1,2 Гц, 3H), 2,40-2,55 (м, 3H), 4,16 (с, 3H), 4,32-4,38 (м, 1H), 4,44-4,50 (м, 1H), 4,75 (т, J=5,6 Гц, 1H), 7,00 (т, J=1,2 Гц, 1H), 7,29 (с, 1H), 7,33 (тт, J=1,2 Гц, 7,6 Гц, 1H), 7,41 (дт, J=1,2 Гц, 7,6 Гц, 2H), 7,59 (д, J=8,0 Гц, 1H), 7,62 (дд, J=1,2 Гц, 7,6 Гц, 2H), 7,81 (д, J=8,0 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Пример 174
Синтез 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-фенилтиазол-2-ил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
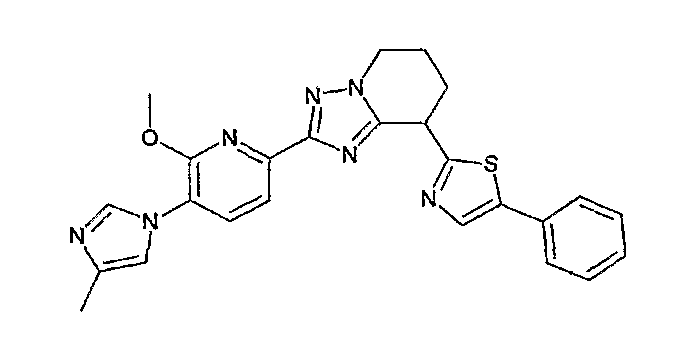
(2-Оксо-2-фенилэтил)амид 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоновой кислоты, полученный в примере 172 (26,6 мг), растворяли в ТГФ (1 мл). Добавляли реактив Лавессона (45,6 мг) и смесь перемешивали при кипячении с обратным холодильником в течение 11 часов. Реакционный раствор охлаждали льдом. Затем добавляли хлороформ и насыщенный раствор бикарбоната натрия и отделяли органический слой. Полученный органический слой сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем NH с получением указанного в заголовке соединения (2,8 мг). Соединение затем очищали с помощью CHIRALCEL™ IB (2 см × 25 см; подвижная фаза: гексан:этанол = 30:70) с получением указанного в заголовке соединения (1,5 мг, рацемат). Показатели свойств соединения были следующими.
ESI-MS; m/z 470 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,19-2,22 (м, 1H), 2,31 (с, 3H), 2,34-2,38 (м, 1H), 2,51-2,59 (м, 2H), 4,17 (с, 3H), 4,33-4,45 (м, 2H), 4,81 (т, J=6,0 Гц, 1H), 7,01 (с, 1H), 7,32 (тт, J=1,2 Гц, 7,6 Гц, 1H), 7,39 (дт, J=1,2 Гц, 7,6 Гц, 2H), 7,52 (дд, J=1,2 Гц, 7,6 Гц, 2H), 7,62 (д, J=8,0 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H), 7,86 (д, J=8,0 Гц, 1H), 7,89 (с, 1H).
Примеры 175 и 176
(+)-2-[6-Метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-пиридин-2-ил-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-пиридин-2-ил-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
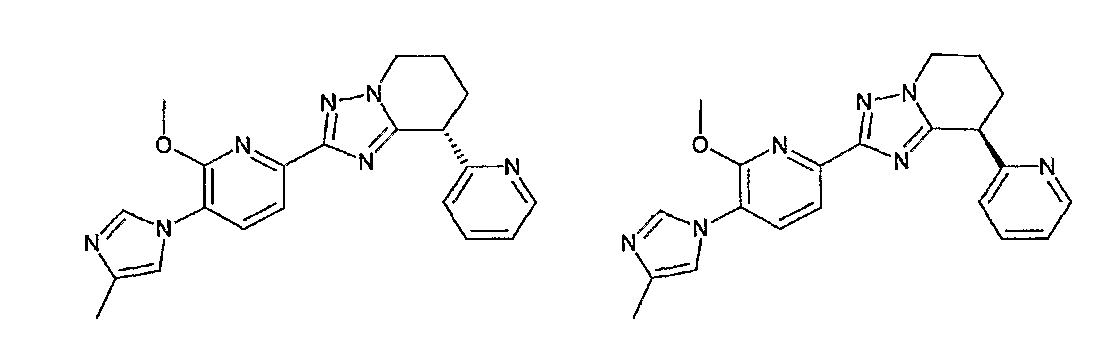
8-(6-Бромпиридин-2-ил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин, полученный в примерах 70 и 71 (35 мг), растворяли в этаноле (2 мл). Добавляли 10% палладий-на-углероде (10 мг) и смесь перемешивали в атмосфере водорода при 1 атм при комнатной температуре в течение семи часов. Палладий-на-углероде удаляли фильтрованием через целит с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем NH с получением рацемата указанного в заголовке соединения (16,6 мг). Полученный рацемат (16,6 мг) разделяли с помощью CHIRALPAK™ AD-H (2 см × 25 см; подвижная фаза: этанол) с получением указанного в заголовке оптически активного соединения с временем удерживания 4,3 минуты и положительным оптическим вращением (7,3 мг, 70% ee) и указанного в заголовке оптически активного соединения с временем удерживания 7,7 минуты и отрицательным оптическим вращением (5,2 мг, 94% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими.
ESI-MS; m/z 388 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,08-2,28 (м, 2H), 2,30 (д, J=1,2 Гц, 3H), 2,34-2,45 (м, 2H), 4,16 (с, 3H), 4,32-4,46 (м, 2H), 4,57 (т, J=6,0 Гц, 1H), 7,00 (т, J=1,2 Гц, 1H), 7,17-7,21 (м, 2H), 7,58 (д, J=7,6 Гц, 1H), 7,65 (дт, J=2,0 Гц, 8,0 Гц, 1H), 7,78 (д, J=7,6 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H), 8,57-8,58 (м, 1H).
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими.
ESI-MS; m/z 388 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,08-2,28 (м, 2H), 2,30 (д, J=1,2 Гц, 3H), 2,34-2,45 (м, 2H), 4,16 (с, 3H), 4,32-4,46 (м, 2H), 4,57 (т, J=6,0 Гц, 1H), 7,00 (т, J=1,2 Гц, 1H), 7,17-7,21 (м, 2H), 7,58 (д, J=7,6 Гц, 1H), 7,65 (дт, J=2,0 Гц, 8,0 Гц, 1H), 7,78 (д, J=7,6 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H), 8,57-8,58 (м, 1H).
Соединения примеров 177-180 получали таким же способом, как в примерах 175 и 176 (таблица 5).
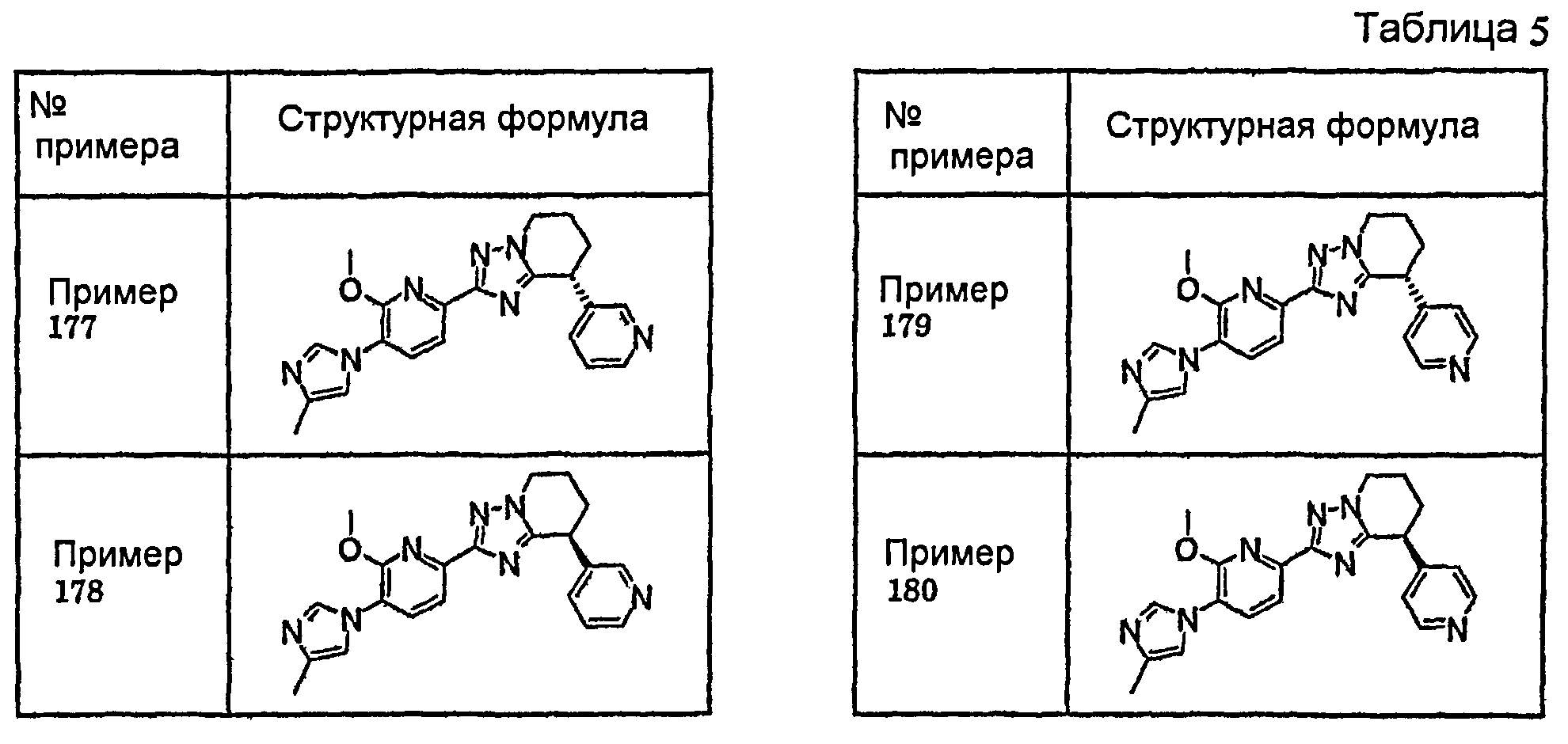
Пример 181
Синтез 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-9-(2-трифторметилфенил)-6,7,8,9-тетрагидро-5H-[1,2,4]триазоло[1,5-a]азепин-9-ола
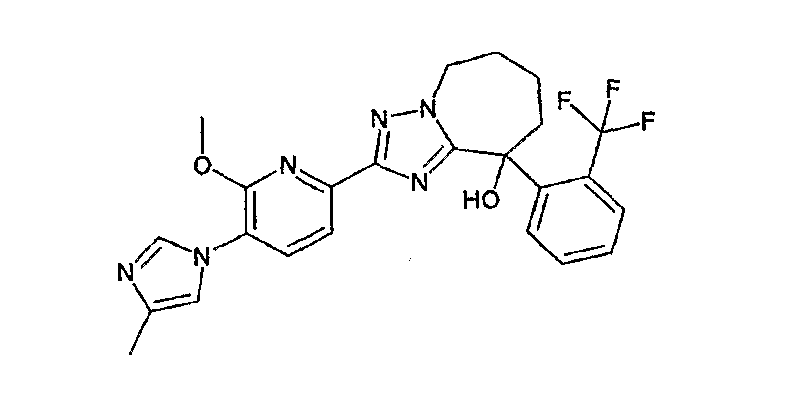
Гидрид натрия (60%) добавляли к раствору 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-9-(2-трифторметилфенил)-6,7,8,9-тетрагидро-5H-[1,2,4]триазоло[1,5-a]азепина, полученного в примерах 74 и 75 (83 мг), в ДМФА (5 мл) с перемешиванием при охлаждении льдом, пока не прекращалось выделение пузырьков. После этого смесь перемешивали при комнатной температуре в атмосфере кислорода. Через четверо суток к реакционному раствору добавляли ледяную воду, хлороформ и насыщенный раствор тиосульфата натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Остаток разбавляли толуолом и концентрировали при пониженном давлении. Остаток снова разбавляли толуолеом и концентрировали при пониженном давлении. Выпавшее в осадок твердое вещество собирали фильтрованием, промывали хлороформом и затем сушили на воздухе с получением 36,8 мг указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,78-1,82 (м, 1H), 1,90-2,04 (м, 1H), 2,08-2,30 (м, 3H), 2,28 (с, 3H), 2,55-2,64 (м, 1H), 4,10 (с, 3H), 4,45-4,61 (м, 2H), 4,93 (ушир.с, 1H), 6,93 (с, 1H), 7,41 (д, J=8,0 Гц, 1H), 7,43-7,54 (м, 3H), 7,60 (д, J=8,0 Гц, 1H), 7,64 (с, 1H), 7,79 (д, J=7,6 Гц, 1H).
Пример 182
Синтез 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-9-(2-трифторметилфенил)-6,7-дигидро-5H-[1,2,4]триазоло[1,5-a]азепина
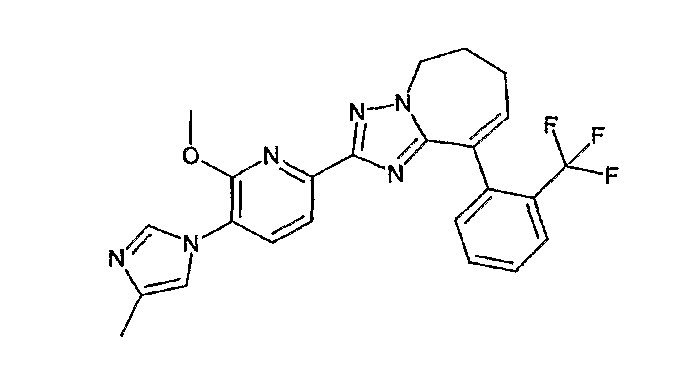
Смесь 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-9-(2-трифторметилфенил)-6,7,8,9-тетрагидро-5H-[1,2,4]триазоло[1,5-a]азепин-9-ола, полученного в примере 181 (49 мг), п-толуолсульфоновой кислоты (76 мг) и толуола (7 мл) кипятили с обратным холодильником в течение двух часов. К реакционному раствору добавляли этилацетат и насыщенный бикарбонат натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Твердое вещество удаляли фильтрованием. Маточный раствор концентрировали при пониженном давлении и затем остаток очищали хроматографией на колонке с силикагелем (хлороформ-метаноловая система) с получением 3,46 мг указанного в заголовке соединения в виде масла. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,20-2,31 (м, 2H), 2,29 (с, 3H), 2,73-2,77 (м, 2H), 4,12 (с, 3H), 4,58-4,61 (м, 2H), 6,25 (дд, J=5,2, 5,2 Гц, 1H), 6,96 (с, 1H), 7,44-7,50 (м, 2H), 7,49 (д, J=7,2 Гц, 1H), 7,59 (т, J=7,2 Гц, 1H), 7,61 (д, J=7,2 Гц, 1H), 7,69 (д, J=7,2 Гц, 1H), 7,78 (с, 1H).
Примеры 183 и 184
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-4-(2-трифторметилфенил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-4-(2-трифторметилфенил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиридина
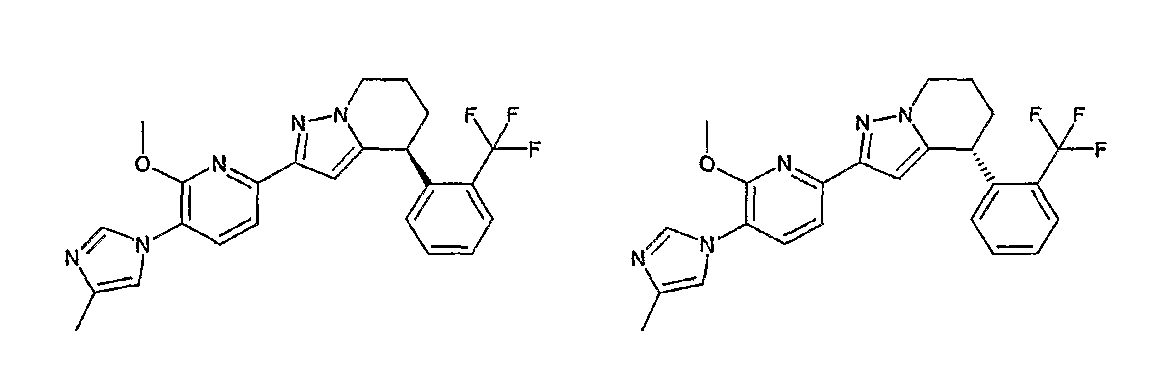
Синтез хлорида 5-хлор-2-(2-трифторметилфенил)валериановой кислоты
5-Хлор-2-(2-трифторметилфенил)валериановую кислоту, полученную в примере получения 2-3 (500 мг), растворяли в тетрагидрофуране (9 мл). Диметилформамид (0,1 мл) и оксалилхлорид (184 мкл) добавляли при 0°C и начинали реакцию при комнатной температуре. Через 1,5 часа концентрирование при пониженном давлении дало масло (532 мг), которое использовали непосредственно для следующей реакции в виде неочищенного продукта.
Синтез 7-хлор-1-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-4-(2-трифторметилфенил)гептан-1,3-диона
Раствор 1-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]этанона, полученного в примере получения 1-8 (412 мг), в тетрагидрофуране (10 мл) добавляли при -30°C к раствору диизопропиламина лития, приготовленному из н-бутиллития (1,4 мл) и диизопропиламина (528 мкл), в тетрагидрофуране (30 мл). После перемешивания при той же самой температуре в течение 30 минут добавляли хлорид 5-хлор-2-(2-трифторметилфенил)валериановой кислоты (532 мг) и смесь перемешивали в течение 1,5 часов. После дополнительного перемешивания при комнатной температуре в течение 30 минут реакцию останавливали водой с последующим разбавлением реакционной смеси этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над сульфатом магния. Масло после концентрирования при пониженном давлении очищали хроматографией на силикагеле (подвижная фаза: этилацетат/гептан = 0-100%) с получением указанного в заголовке соединения (90 мг).
ESI-МС; m/z 494 [M++H].
Синтез (+)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-4-(2-трифторметилфенил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-4-(2-трифторметилфенил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиридина
7-Хлор-1-[6-метокси-5-(4-метилимидазол-1-ил)пиридин-2-ил]-4-(2-трифторметилфенил)гептан-1,3-дион (90 мг) растворяли в этаноле (2 мл) и добавляли при комнатной температуре моногидрат гидразина (26,5 мкл). Через 1,5 часа смесь кипятили с обратным холодильником. Затем этанол (1 мл) и моногидрат гидразина (13,3 мкл) добавляли 3,5 часа после нагревания. После кипячения с обратным холодильником в течение 18 часов реакционный раствор распределяли между этилацетатом и водой. Органический слой промывали насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-100%) с получением рацемата указанного в заголовке соединения (31,2 мг). Полученный рацемат разделяли с помощью CHIRALPAK™ AD-H (2 см × 25 см, подвижная фаза: этанол:гексан = 3:7, скорость потока: 13 мл/мин) с получением указанного в заголовке соединения с временем удерживания 23,5 минуты и положительным оптическим вращением (9,0 мг) и указанного в заголовке соединения с временем удерживания 34,2 минуты и отрицательным оптическим вращением (8,2 мг).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 23,5 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,89-1,95 (м, 1H), 2,05-2,40 (м, 3H), 2,29 (с, 3H), 3,99 (с, 3H), 4,25 (ддд, J=12,9, 10,9, 5,1 Гц, 1H), 4,42 (ддд, J=12,9, 9,3, 5,4 Гц, 1H), 4,59 (дд, J=10,2, 5,5 Гц, 1H), 6,28 (с, 1H), 6,95-6,97 (м, 1H), 7,27-7,30 (м, 1H), 7,36-7,41 (м, 1H), 7,48-7,50 (м, 1H), 7,54 (д, J=7,8 Гц, 1H), 7,60 (д, J=7,8 Гц, 1H), 7,72 (д, J=7,8 Гц, 1H), 7,77 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с временем удерживания 34,2 минуты были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,89-1,95 (м, 1H), 2,05-2,40 (м, 3H), 2,29 (с, 3H), 3,99 (с, 3H), 4,25 (ддд, J=12,9, 10,9, 5,1 Гц, 1H), 4,42 (ддд, J=12,9, 9,3, 5,4 Гц, 1H), 4,59 (дд, J=10,2, 5,5 Гц, 1H), 6,28 (с, 1H), 6,95-6,97 (м, 1H), 7,27-7,30 (м, 1H), 7,36-7,41 (м, 1H), 7,48-7,50 (м, 1H), 7,54 (д, J=7,8 Гц, 1H), 7,60 (д, J=7,8 Гц, 1H), 7,72 (д, J=7,8 Гц, 1H), 7,77 (д, J=1,2 Гц, 1H).
Пример 185
Синтез 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-4-(2-трифторметилфенил)-4,5,6,7-тетрагидро[1,2,4]триазоло[1,5-a]пиримидина
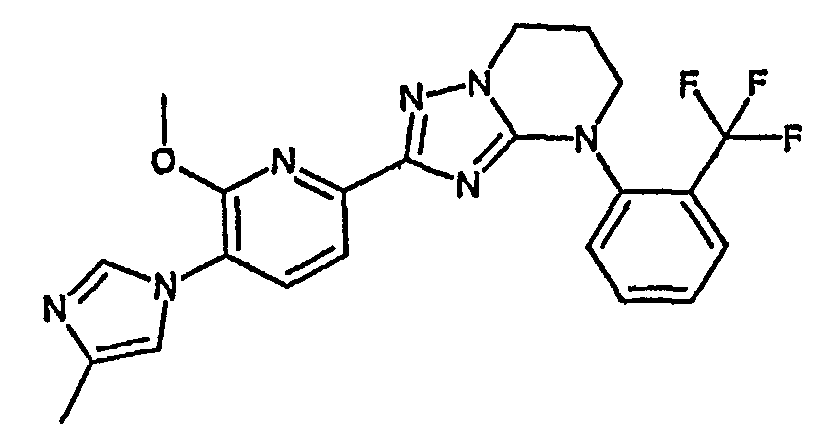
Синтез фенил (2-трифторметилфенил)карбамата
2-Аминобензотрифторид (5 мл) и пиридин (6,44 мл) растворяли в тетрагидрофуране (100 мл) и фенилхлороформиат (5,5 мл) добавляли по каплям при охлаждении льдом. После добавления реагента по каплям смесь доводили снова до комнатной температуры и перемешивали в течение 4,5 часов. Добавляли к реакционному раствору воду и смесь концентрировали при пониженном давлении с последующим экстрагированием этилацетатом. Полученный органический слой промывали 2 н. хлористоводородной кислотой и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (12,1 г). Показатели свойств соединения были следующими.
ESI-МС; m/z 282 [M++H].
Синтез гидразида (2-трифторметилфенил)карбаминовой кислоты
Фенил (2-трифторметилфенил)карбамат (12,1 г) растворяли в этаноле (150 мл). Добавляли моногидрат гидразина (16,7 мл) и смесь кипятили с обратным холодильником в течение 2,25 часов. Реакционный раствор концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (7,5 г). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 3,90 (ушир.с, 2H), 6,29 (ушир.с, 1H), 7,13 (дд, J=7,6, 6,4 Гц, 1H), 7,52 (дд, J=8,4, 6,4 Гц, 1H), 7,58 (д, J=7,6 Гц, 1H), 8,28 (д, J=8,4 Гц, 1H), 8,87 (ушир.с, 1H).
Синтез {5-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-[1,3,4]оксазол-2-ил}-(2-трифторметилфенил)амина
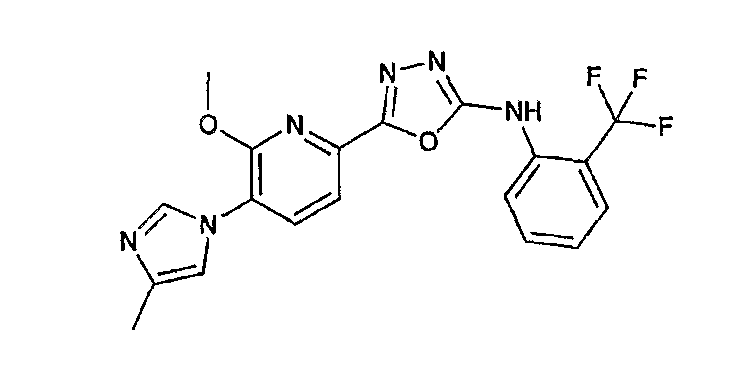
6-Метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновую кислоту, полученную в примере получения 1-5 (581 мг), гидразид (2-трифторметилфенил)карбаминовой кислоты (230 мг) и N,N-диизопропилэтиламин (914 мкл) растворяли в дихлорметане (10 мл), после чего добавляли хлорид бис(2-оксо-3-оксазолидинил)фосфиновой кислоты (321 мг). После добавления реагента смесь перемешивали при комнатной температуре в течение ночи. Добавляли к реакционному раствору раствор бикарбоната натрия и смешанный растворитель 5% метанол/этилацетат и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением конденсата (349 мг).
Фосфороксихлорид (30 мл) добавляли к конденсату (2,06 г) и смесь нагревали при перемешивании при 120°C в течение 1,6 часа. Смесь охлаждали, реакционный раствор концентрировали при пониженном давлении. Раствор гидроксида натрия и смешанный растворитель 10% метанол/хлороформ добавляли и отделяли органический слой. Полученный органический слой промывали 2 н. хлористоводородной кислотой и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Полученное остаточное твердое вещество промывали трет-бутилметиловым эфиром с получением указанного в заголовке соединения (754 мг). Показатели свойств соединения были следующими.
ESI-МС; m/z 417 [M++H].
Синтез (3-хлорпропил)-{5-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-[1,3,4]оксазол-2-ил}-(2-трифторметилфенил)амина
{5-[6-Метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-[1,3,4]оксазол-2-ил}-(2-трифторметилфенил)амин (657 мг) и 1-хлор-3-йодпропан (1,66 мл) растворяли в N,N-диметилформамиде (15 мл) и добавляли при охлаждении льдом 60% гидрид натрия (172 мг). После добавления реагента смесь доводили обратно до комнатной температуры и перемешивали в течение двух часов. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем NH с получением указанного в заголовке соединения (216 мг). Показатели свойств соединения были следующими.
ESI-МС; m/z 493 [M++H].
Синтез 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-4-(2-трифторметилфенил)-4,5,6,7-тетрагидро[1,2,4]триазоло[1,5-a]пиримидина
(3-Хлорпропил)-{5-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-[1,3,4]оксазол-2-ил}-(2-трифторметилфенил)амин (239 мг) и аммонийацетат, высушенный при пониженном давлении (1,12 г), суспендировали в уксусной кислоте (10 мл). Суспензию нагревали при перемешивании при 180°C в течение двух часов, используя микроволновый реактор. Реакционный раствор концентрировали при пониженном давлении. Добавляли раствор бикарбоната натрия и этилацетат и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем NH и затем еще очищали с помощью CHIRALPAK™ IB (2 см × 25 см; подвижная фаза: гексан:этанол = 4:6) с получением указанного в заголовке соединения (30 мг). Показатели свойств соединения были следующими.
ESI-MS; m/z 456 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,28 (с, 3H), 2,30-2,50 (м, 2H), 3,64-3,74 (м, 2H), 4,12 (с, 3H), 4,30-7,41 (м, 2H), 6,94-6,98 (м, 1H), 7,47-7,54 (м, 3H), 7,59 (д, J=7,6 Гц, 1H), 7,67 (ддд, J=7,6, 7,6, 1,2 Гц, 1H), 7,76-7,82 (м, 2H).
Пример получения 1-1
Синтез 6-бром-2-метокси-3-(4-метил-1H-имидазол-1-ил)пиридина
Синтез N-(6-бром-2-метоксипиридин-3-ил)формамида
Уксусный ангидрид (203 мл) добавляли по каплям к муравьиной кислоте (204 мл) при охлаждении льдом и смесь перемешивали при той же самой температуре в течение 25 минут. В реакционную смесь вносили в течение 10 минут порошок 6-бром-2-метоксипиридин-3-амина (CAS #89466-18-2, 146 г) и затем реакционный раствор перемешивали при той же самой температуре в течение 30 минут. Водяную баню убирали. трет-Бутилметиловый эфир (300 мл) и н-гептан (500 мл) последовательно добавляли по каплям к реакционному раствору и затем реакционный раствор перемешивали в течение 30 минут. Осажденный порошок собирали фильтрованием. Полученный порошок измельчали в ступке, промывали трет-бутилметиловым эфиром и затем сушили при пониженном давлении с получением 137,4 г указанного в заголовке соединения.
Затем объединенный фильтрат и промывной раствор концентрировали при пониженном давлении. Остаток растирали в трет-бутилметиловом эфире и сушили при пониженном давлении с получением 21,9 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 4,03 (с, 3H), 7,08 (д, J=8,0 Гц, 1H), 7,61 (ушир.с, 1H), 8,47-8,51 (м, 2H).
Синтез N-(6-бром-2-метоксипиридин-3-ил)-N-(2-оксопропил)формамида
Хлорацетон (82 мл) добавляли по каплям к суспензии N-(6-бром-2-метоксипиридин-3-ил)формамида (159,3 г), карбоната цезия (359 г) и иодида калия (11,4 г) в N,N-диметилформамиде (800 мл) в течение семи минут. Затем реакционный раствор перемешивали при комнатной температуре в течение одного часа и 20 минут.
Реакционный раствор концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат и воду и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением 215,2 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,17 (с, 3H), 4,00 (с, 3H), 4,47 (с, 2H), 7,13 (д, J=7,6 Гц, 1H), 7,48 (д, J=7,6 Гц, 1H), 8,22 (с, 1H).
Синтез 6-бром-2-метокси-3-(4-метил-1H-имидазол-1-ил)пиридина
Суспензию аммонийацетата (267 г) и N-(6-бром-2-метоксипиридин-3-ил)-N-(2-оксопропил)формамида (199 г) в ледяной уксусной кислоте (400 мл) перемешивали при 130°C в течение одного часа и 10 минут. Реакционный раствор доводили обратно до комнатной температуры. К реакционному раствору добавляли этилацетат и ледяную воду и смесь охлаждали льдом. Затем концентрированный водный аммиак (500 мл) добавляли по каплям, после чего отделяли органический слой. Полученный органический слой промывали последовательно водой и насыщенным раствором соли и сушили над безводным сульфатом магния. Затем органический слой очищали хроматографией на короткой колонке с силикагелем (носитель: Wakogel™ C-200 производства Wako Pure Chemical Industries, Ltd.; элюирующий растворитель: этилацетат). Элюированную фракцию концентрировали. Полученный остаток растирали в этилацетате и трет-бутилметиловом эфире и сушили при пониженном давлении с получением 107,7 г указанного в заголовке соединения.
Затем маточную жидкость для растирания концентрировали. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel™ C-200; элюирующий растворитель: толуол-этилацетатная система). Целевую фракцию концентрировали. Полученный остаток растирали в трет-бутилметиловом эфире и сушили при пониженном давлении с получением 12,9 г указанного в заголовке соединения.
Показатели свойств соединения были следующими.ESI-MS; m/z 268 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,29 (д, J=0,8 Гц, 3H), 4,03 (с, 3H), 6,92 (дд, J=1,2, 0,8 Гц, 1H), 7,16 (д, J=8,0 Гц, 1H), 7,40 (д, J=8,0 Гц, 1H), 7,73 (д, J=1,2 Гц, 1H).
Пример получения 1-2
Синтез 2-метокси-3-(4-метил-1H-имидазол-1-ил)-6-трибутилстаннилпиридина
6-Бром-2-метокси-3-(4-метил-1H-имидазол-1-ил)пиридин (10 г) и гекса-н-бутилдиолово (31,8 мл) растворяли в толуоле (300 мл). Тетракис(трифенилфосфин)палладий (2,2 г) добавляли и смесь кипятили с обратным холодильником в атмосфере азота в течение четырех часов. Смесь охлаждали, нерастворимый материал удаляли из реакционного раствора фильтрованием через целит с последующим концентрированием при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем NH и затем хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,4 г). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 0,90 (т, J=7,2 Гц, 9H), 1,03-1,22 (м, 6H), 1,30-1,40 (м, 6H), 1,49-1,70 (м, 6H), 2,29 (с, 3H), 4,01 (с, 3H), 6,96 (с, 1H), 7,10 (д, J=7,2 Гц, 1H), 7,63 (д, J=7,2 Гц, 1H), 7,75-7,77 (м, 1H).
Пример получения 1-3
Синтез 3-метокси-2-(4-метил-1H-имидазол-1-ил)-5-трибутилстаннилпиридина
Синтез N-(5-бром-3-метоксипиридин-2-ил)формамида
Железо (67,3 г) и аммонийхлорид (129 г) добавляли к раствору 5-бром-3-метокси-2-нитропиридина (56,0 г, CAS #152684-26-9) в этаноле (500 мл) и воде (200 мл). Реакционный раствор перемешивали при 80-90°C в течение одного часа и затем охлаждали до комнатной температуры. Реакционный раствор фильтровали через целит и промывали этанолом. Затем фильтрат концентрировали при пониженном давлении. Остаток разбавляли этилацетатом и водой и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Остаток разбавляли ТГФ (84 мл). ТГФ раствор добавляли по каплям к смешанному раствору из муравьиной кислоты (78,1 мл) и уксусного ангидрида (78,3 мл) при комнатной температуре. Затем реакционный раствор перемешивали в течение одного часа. Добавляли к реакционному раствору ледяную воду (500 мл) и осажденные кристаллы собирали фильтрованием. Кристаллы промывали водой и затем сушили на воздухе. Кристаллы перекристаллизовывали из толуола с получением 34,1 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
ESI-МС; m/z 231 [M++H].
Синтез N-(5-бром-3-метоксипиридин-2-ил)-N-(2-оксопропил)формамида
Карбонат цезия (96 г), иодид калия (2,45 г) и хлорацетон (23,5 мл) добавляли к раствору N-(5-хлор-3-метоксипиридин-2-ил)формамида (34,1 г) в ДМФА (200 мл) и смесь перемешивали при 80°C в течение 45 минут. К реакционному раствору добавляли ледяную воду и этилацетат и отделяли органический слой. Органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 52,8 г неочищенного указанного в заголовке соединения. Показатели свойств соединения были следующими.
ESI-МС; m/z 287 [M++H].
Синтез 5-бром-3-метокси-2-(4-метил-1H-имидазол-1-ил)пиридина
Смесь неочищенного N-(5-бром-3-метоксипиридин-2-ил)-N-(2-оксопропил)формамида (26,4 г), уксусной кислоты (52,8 мл) и аммонийацетата (35,5 г) перемешивали при 130°C в течение одного часа. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли ледяной водой, этилацетатом и водным аммиаком и отделяли органический слой. Органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: гептан-этилацетатная система) с получением 5,69 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
ESI-MS; m/z 268 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 2,29 (с, 3H), 3,97 (с, 3H), 7,48 (ушир.с, 1H), 7,49 (д, J=2,0 Гц, 1H), 8,12 (д, J=2,0 Гц, 1H), 8,30 (ушир.с, 1H).
Синтез 3-метокси-2-(4-метил-1H-имидазол-1-ил)-5-трибутилстаннилпиридина
Гекса-н-бутилдиолово (2,07 мл) и тетракис(трифенилфосфин)палладий(0) (319 мг) добавляли к суспензии 5-бром-3-метокси-2-(4-метил-1H-имидазол-1-ил)пиридина (740 мг) в ксилоле (11 мл) и смесь перемешивали при 155°C в течение двух часов и 30 минут. Реакционный раствор очищали хроматографией на колонке с силикагелем (носитель: Wakogel C-200; элюирующий растворитель: гептан:этилацетат = 3:1) с получением 325 мг указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 0,90 (т, J=7,2 Гц, 9H), 1,13 (т, J=7,2 Гц, 6H), 1,30-1,40 (м, 6H), 1,50-1,60 (м, 6H), 2,30 (д, J=1,2 Гц, 3H), 3,95 (с, 3H), 7,38 (д, J=1,2 Гц, 1H), 7,52 (т, J=1,2 Гц, 1H), 8,03 (д, J=1,2 Гц, 1H), 8,33 (д, J=1,2 Гц, 1H).
Пример получения 1-4
Синтез 1-(2-метокси-4-трибутилстаннилфенил)-4-метил-1H-имидазола
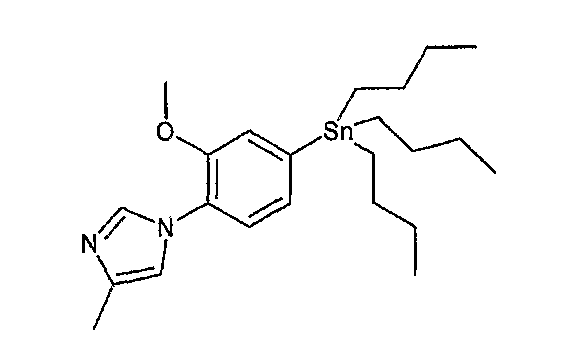
1-(4-Бром-2-метоксифенил)-4-метил-1H-имидазол (CAS No. 870838-56-5, 900 мг) и гекса-н-бутилдиолово (3,91 г) растворяли в толуоле (20 мл). Добавляли тетракистрифенилфосфинпалладий (195 мг) и смесь перемешивали при кипячении с обратным холодильником в течение семи часов и 20 минут. Смесь охлаждали, нерастворимый материал удаляли фильтрованием через целит с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем NH с получением указанного в заголовке соединения (893 мг). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 0,90 (т, J=7,2 Гц, 9H), 1,09-1,11 (м, 6H), 1,32-1,40 (м, 6H), 1,53-1,59 (м, 6H), 2,30 (д, J=1,2 Гц, 3H), 3,85 (с, 3H), 6,92 (т, J=1,2 Гц, 1H), 7,09 (дд, J=1,2 Гц, 7,6 Гц, 1H), 7,10 (д, J=1,2 Гц, 1H), 7,21 (д, J=7,6 Гц, 1H), 7,69 (д, J=1,2 Гц, 1H).
Пример получения 1-5
Синтез 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты
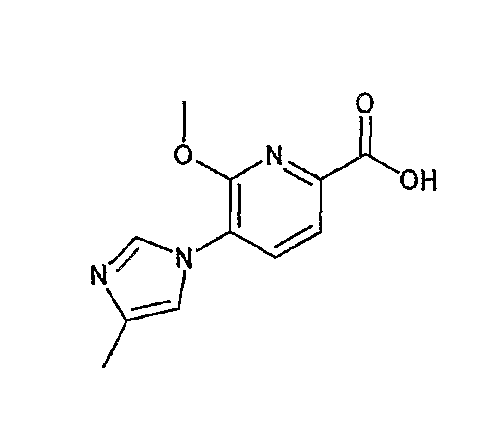
Синтез 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбонитрила
6-Бром-2-метокси-3-(4-метил-1H-имидазол-1-ил)пиридин, полученный в примере получения 1-1 (50 г), и цианид цинка (II) (35 г) суспендировали в N-метилпирролидоне (400 мл). Добавляли тетракис(трифенилфосфин)палладий (0) (8,5 г) и смесь перемешивали при 100°C в течение одного часа и 10 минут. Реакционный раствор добавляли по каплям к раствору ледяной воды (1,5 л) и концентрированного водного аммиака (150 мл), смешанных перемешиванием. Осажденный порошок отфильтровывали. Полученный порошок промывали водой и затем сушили на воздухе в течение ночи с получением 56,5 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,30 (с, 3H), 4,09 (с, 3H), 7,02 (ушир.с, 1H), 7,44 (д, J=8,0 Гц, 1H), 7,64 (д, J=8,0 Гц, 1H), 7,90 (ушир.с, 1H).
Синтез 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты
Порошок гидроксида лития (13 г) добавляли к суспензии 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбонитрила (52,4 г) в воде (464 мл) и смесь кипятили с обратным холодильником в течение трех часов. Реакционный раствор охлаждали до комнатной температуры. Реакционный раствор фильтровали через целит и целит промывали водой (100 мл × 4). К фильтрату добавляли концентрированную хлористоводородную кислоту при охлаждении льдом, чтобы довести pH до 4-5. Осажденный порошок собирали фильтрованием. Полученный порошок промывали водой и затем сушили на воздухе в течение трех суток с получением 51,9 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (ДМСО-d6) δ (ppm): 2,17 (с, 3H), 4,01 (с, 3H), 7,33 (ушир.с, 1H), 7,76 (д, J=7,6 Гц, 1H), 7,99 (д, J=7,6 Гц, 1H), 8,02 (ушир.с, 1H).
Пример получения 1-6
Синтез гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты и гидрохлорида гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты
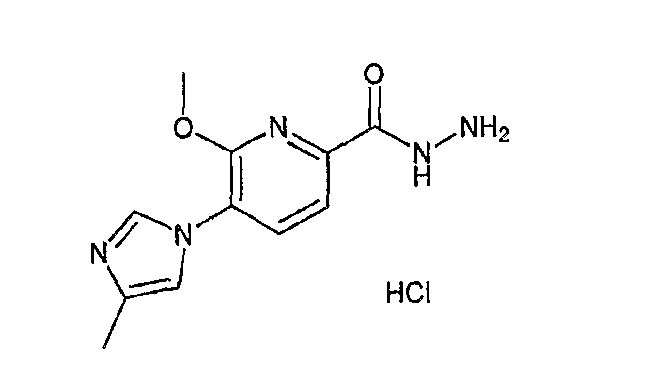
Синтез бензил N′-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбонил]гидразинкарбоксилата
Бензилкарбазат (27,8 г), 1-гидроксибензтриазол (24,8 г) и гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (35,4 г) последовательно добавляли к раствору 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты, полученной в примере получения 1-5 (51,9 г), и IPEA (44 мл) в N,N-диметилформамиде (184 мл) и смесь перемешивали при комнатной температуре в течение шести часов и 30 минут. Добавляли к реакционному раствору этилацетат, ледяную воду и насыщенный раствор бикарбоната натрия и отделяли органический слой. Полученный органический слой сушили над безводным сульфатом магния и затем фильтровали через рыхлый слой силикагеля NH. Полученный фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат и порошок собирали фильтрованием. Полученный порошок сушили на воздухе с получением 28,4 г указанного в заголовке соединения.
Затем уже экстрагированный водный слой повторно экстрагировали этилацетатом. Полученный органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом магния и затем фильтровали через рыхлый слой силикагеля NH. Полученный фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат и порошок собирали фильтрованием. Полученный порошок сушили на воздухе с получением 9,15 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,31 (д, J=1,2 Гц, 3H), 4,08 (с, 3H), 5,23 (с, 2H), 6,87 (ушир.с, 1H), 7,01 (т, J=1,2 Гц, 1H), 7,32-7,45 (м, 5H), 7,71 (д, J=8,0 Гц, 1H), 7,87 (д, J=1,2 Гц, 1H), 7,91 (д, J=8,0 Гц, 1H), 9,17 (ушир.с, 1H).
Синтез гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты и гидрохлорида гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты
10% палладий-на-углероде (влажность 50%, 2,84 г) добавляли к раствору бензил N′-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбонил]гидразинкарбоксилата (28,4 г) в метаноле (300 мл). Смесь гидрировали при умеренном давлении (2-3 атм.) в течение пяти часов. Хлороформ (600 мл) добавляли к реакционному раствору и затем палладий-на-углероде удаляли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении с получением 19,5 мг указанного в заголовке соединения в свободной форме. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,30 (д, J=1,2 Гц, 3H), 4,06 (с, 3H), 4,10 (с, 1H), 4,11 (с, 1H), 7,01 (т, J=1,2 Гц, 1H), 7,70 (д, J=8,0 Гц, 1H), 7,86 (д, J=1,2 Гц, 1H), 7,89 (д, J=8,0 Гц, 1H), 8,69 (ушир.с, 1H).
Гидрохлорид указанного в заголовке соединения получали такой же операцией при условии, что реакцию гидрирования осуществляли в смешанном растворителе хлороформ-метанол. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,31 (д, J=1,2 Гц, 3H), 4,06 (с, 3H), 7,01 (т, J=1,2 Гц, 1H), 7,71 (д, J=7,6 Гц, 1H), 7,88 (д, J=1,2 Гц, 1H), 7,89 (д, J=7,6 Гц, 1H), 8,69 (ушир.с, 1H).
Пример получения 1-7
Синтез 6-(1-этоксивинил)-2-метокси-3-(4-метил-1H-имидазол-1-ил)пиридина
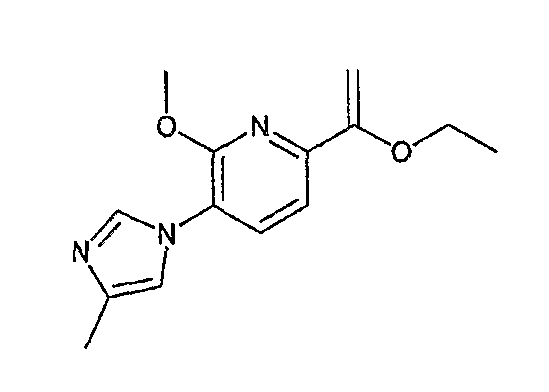
1-Этоксивинилтри-н-бутилолово (3,7 мл) добавляли к суспензии 6-бром-2-метокси-3-(4-метил-1H-имидазол-1-ил)пиридина, полученного в примере получения 1-1 (2,66 г), и хлорида бис(трифенилфосфин)палладия (II) (350 мг) в диоксане (25 мл) и смесь перемешивали при 100°C в течение пяти часов и 45 минут. Реакционному раствору давали охладиться до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel™ C-200; элюирующий растворитель: гептан:этилацетат = 1:0 → 9:1 → 3:1 → 1:1). Целевую фракцию концентрировали. Полученный порошок растирали в смеси диэтиловый эфир-н-гексан и сушили при пониженном давлении с получением 1,57 г указанного в заголовке соединения. Затем маточную жидкость для растирания концентрировали с получением 858 мг указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,45 (т, J=7,2 Гц, 3H), 2,30 (с, 3H), 3,98 (кв., J=7,2 Гц, 2H), 4,04 (с, 3H), 4,38 (д, J=1,6 Гц, 1H), 5,48 (д, J=1,6 Гц, 1H), 6,97 (с, 1H), 7,38 (д, J=8,0 Гц, 1H), 7,52 (д, J=8,0 Гц, 1H), 7,78 (с, 1H).
Пример получения 1-8
Синтез 1-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]этанона
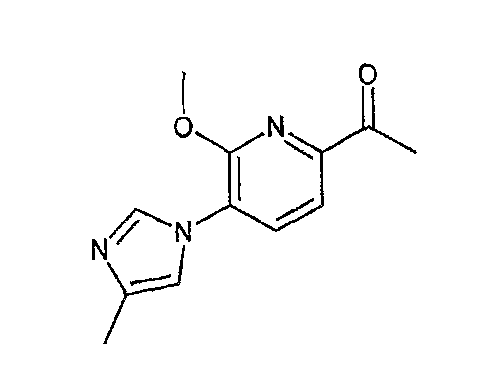
6-Бром-2-метокси-3-(4-метил-1H-имидазол-1-ил)пиридин, полученный в примере получения 1-1 (2,5 г), растворяли в N-метилпирролидоне (93 мл). Добавляли ацетат палладия (418 мг), 1,3-бис(дифенилфосфино)пропан (1,54 г), 1-этоксивинилтри-н-бутилолово (3,15 мл) и оксид меди (I) (2 г) с последующим перемешиванием при 120°C. Через 16 часов реакционный раствор распределяли между водой и этилацетатом. Органический слой промывали водным аммиаком (два раза) и насыщенным раствором соли. После сушки над сульфатом магния концентрирование при пониженном давлении дало масло. Масло растворяли в метиленхлориде (20 мл), после чего добавляли трифторуксусную кислоту (20 мл). После перемешивания в течение одного часа концентрирование при пониженном давлении дало масло, которое распределяли между водой и метиленхлоридом. Органический слой промывали 1 н. гидроксидом натрия, водой (четыре раза) и насыщенным раствором соли и сушили над сульфатом магния. Масло после концентрирования при пониженном давлении очищали хроматографией на силикагеле (подвижная фаза: этилацетат/гептан = 0-100%) с получением указанного в заголовке соединения (1 г) в виде желтого твердого вещества.
ESI-МС; m/z 232 [M++H].
Пример получения 2-1
Синтез 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
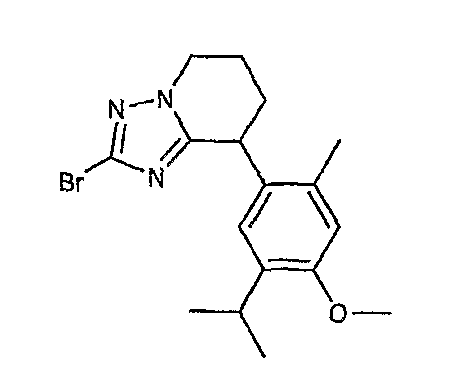
Синтез метил (5-изопропил-4-метокси-2-метилфенил)ацетата
(5-Изопропил-4-метокси-2-метилфенил)уксусную кислоту (CAS No. 81354-65-6, 5,5 г) растворяли в метаноле (50 мл), и тионилхлорид (3,5 мл) добавляли по каплям при охлаждении льдом. После добавления реагента по каплям смесь доводили обратно до комнатной температуры и перемешивали в течение двух часов. Реакционный раствор концентрировали при пониженном давлении. Добавляли насыщенный раствор бикарбоната натрия и трет-бутилметиловый эфир и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,0 г). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,19 (д, J=7,2 Гц, 3H), 1,19 (д, J=7,2 Гц, 3H), 2,28 (с, 3H), 3,25 (кв.кв., J=7,2, 7,2 Гц, 1H), 3,58 (с, 2H), 3,68 (с, 3H), 3,80 (с, 3H), 6,66 (с, 1H), 7,00 (с, 1H).
Синтез метил 5-хлор-2-(5-изопропил-4-метокси-2-метилфенил)пентаноата
60% Гидрид натрия (928 мг) суспендировали в безводном N,N-диметилформамиде (50 мл). Добавляли раствор метил (5-изопропил-4-метокси-2-метилфенил)ацетата (5 г) в безводном N,N-диметилформамиде (30 мл) в атмосфере азота с поддержанием внутренней температуры на уровне 4-6°C. После перемешивания при той же самой температуре в течение пяти минут, добавляли 1-хлор-3-йодпропан (4,5 мл) с поддержанием внутренней температуры на уровне 4-6°C. После добавления реагента смесь доводили обратно до комнатной температуры и перемешивали в течение четырех часов. К реакционному раствору добавляли этилацетат с последующей промывкой водой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (7,7 г). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,17 (д, J=7,2 Гц, 3H), 1,19 (д, J=7,2 Гц, 3H), 1,64-1,97 (м, 3H), 2,14-2,25 (м, 1H), 2,35 (с, 3H), 3,24 (кв.кв., J=7,2, 7,2 Гц, 1H), 3,48-3,56 (м, 2H), 3,64 (с, 3H), 3,77 (т, 7,6 Гц, 1H), 3,08 (с, 3H), 6,64 (с, 1H), 7,08 (с, 1H).
Синтез трет-бутил N′-[5-хлор-2-(5-изопропил-4-метокси-2-метилфенил)пентаноил]гидразинкарбоксилата
Метил 5-хлор-2-(5-изопропил-4-метокси-2-метилфенил)пентаноат (7,7 г) растворяли в смешанном растворителе из метанола (25 мл) и тетрагидрофурана (25 мл). Добавляли 5 н. раствор гидроксида натрия (22 мл) и смесь перемешивали при комнатной температуре в течение четырех часов. Реакционный раствор концентрировали при пониженном давлении и добавляли воду с последующей промывкой гептаном. Водный слой подкисляли 5 н. хлористоводородной кислотой с последующим экстрагированием трет-бутилметиловым эфиром. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и органический слой концентрировали при пониженном давлении с получением 5-хлор-2-(5-изопропил-4-метокси-2-метилфенил)пентановой кислоты (6,2 г).
5-Хлор-2-(5-изопропил-4-метокси-2-метилфенил)пентановую кислоту (6,2 г), трет-бутилкарбазат (4,1 г) и N,N-диизопропилэтиламин (10,8 мл) растворяли в дихлорметане (120 мл). Хлорид бис(2-оксо-3-оксазолидинил)фосфиновой кислоты (7,9 г) добавляли при охлаждении льдом. После добавления реагента смесь доводили обратно до комнатной температуры и перемешивали в течение ночи. Реакционный раствор концентрировали при пониженном давлении. Добавляли раствор бикарбоната натрия и трет-бутилметилового эфира и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (6,1 г). Показатели свойств соединения были следующими.
ESI-МС; m/z 435 [M++Na].
Синтез гидразида 5-хлор-2-(5-изопропил-4-метокси-2-метилфенил)пентановой кислоты
трет-Бутил N′-[5-хлор-2-(5-изопропил-4-метокси-2-метилфенил)пентаноил]гидразинкарбоксилат (6,1 г) растворяли в этилацетате (50 мл). Добавляли 4 н. этилацетатный раствор хлористоводородной кислоты (50 мл) и смесь перемешивали при комнатной температуре в течение 3,5 часов. Реакционный раствор подщелачивали 5 н. раствором гидроксида натрия при охлаждении льдом и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,4 г). Показатели свойств соединения были следующими.
ESI-MS; m/z 313 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,186 (д, J=6,8 Гц, 3H), 1,190 (д, J=7,2 Гц, 3H), 1,61-1,85 (м, 2H), 1,93-2,04 (м, 1H), 2,26-2,88 (м, 1H), 2,27 (с, 3H), 3,25 (кв.кв., J=7,2, 6,8 Гц, 1H), 3,48-3,59 (м, 3H), 3,75-3,89 (м, 5H), 6,49 (ушир.с, 1H), 6,65 (с, 1H), 7,08 (с, 1H).
Синтез 8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-2-иламина
Гидразид 5-хлор-2-(5-изопропил-4-метокси-2-метилфенил)пентановой кислоты (4,4 г) и цианамид (3,6 г) растворяли в этаноле (150 мл). Добавляли моногидрат п-толуолсульфоновой кислоты (4 г) и смесь кипятили с обратным холодильником при 80°C в течение двух часов. После охлаждения до комнатной температуры добавляли триэтиламин (9,8 мл) и смесь еще кипятили с обратным холодильником при 80°C в течение трех суток. Реакционный раствор концентрировали при пониженном давлении. Добавляли раствор бикарбоната натрия и этилацетат и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (2,4 г). Показатели свойств соединения были следующими.
ESI-MS; m/z 301 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,12 (д, J=6,8 Гц, 3H), 1,14 (д, J=6,8 Гц, 3H), 1,86-2,26 (м, 4H), 2,27 (с, 3H), 3,19 (кв.кв., J=6,8, 6,8 Гц, 1H), 3,79 (с, 3H), 4,02 (ушир.с, 2H), 4,06-4,12 (м, 2H), 4,19-4,24 (м, 1H), 6,64 (с, 1H), 6,69 (с, 1H).
Синтез 2-бром-8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
8-(5-изопропил-4-метокси-2-метилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-2-иламин (1,5 г) и бромид меди(II) (1,7 г) растворяли в ацетонитриле (50 мл). Добавляли изоамилнитрит (1 мл) и смесь нагревали при перемешивании при 70°C в течение 45 минут. К реакционному раствору добавляли этилацетат с последующей промывкой водным аммиаком. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,4 г). Показатели свойств соединения были следующими.
ESI-MS; m/z 364 [M++H].
1Н-ЯМР (CDCl3) δ (ppm): 1,11 (д, J=7,2 Гц, 3Н), 1,12 (д, J=7,2 Гц, 3Н), 1,90-2,30 (м, 4Н), 2,26 (с, 3Н), 3,19 (кв.кв., J=7,2, 7,2 Гц, 1Н), 3,80 (с, 3Н), 4,24-4,29 (м, 2Н), 4,30-4,36 (м, 1Н), 6,59 (с, 1Н), 6,65 (с, 1Н).
Соединения примеров получения 2-2 - 2-6 получали таким же способом, как в примере 2-1 (таблица 6).
|
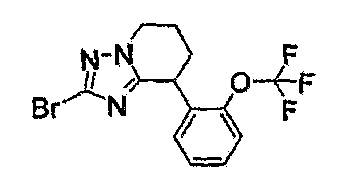
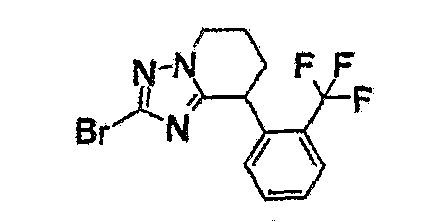
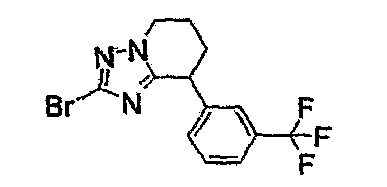
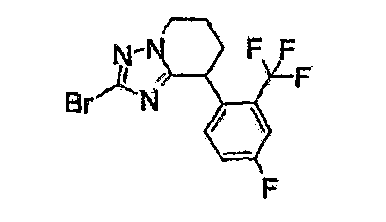
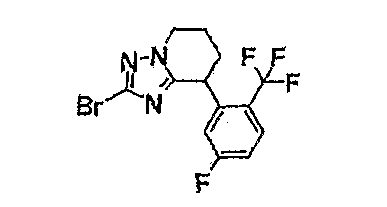
Пример получения 2-7
Синтез 2-бром-8-(2-трифторметилбензил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
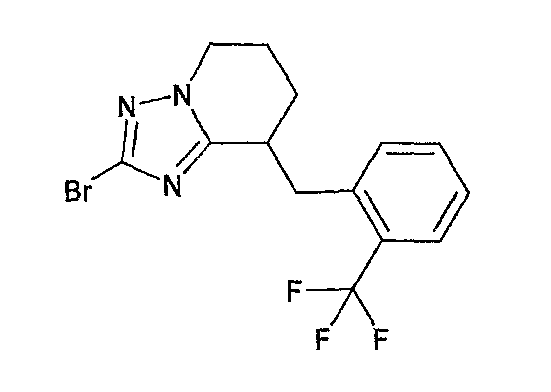
Синтез этил 5-хлор-2-[1-(2-трифторметилфенил)метилиден]пентаноата
2-(трифторметил)бензальдегид (1 г) и этил 5-хлор-2-(диэтоксифосфорил)пентаноат (CAS №870843-20-2, 2,1 г) растворяли в смешанном растворителе из тетрагидрофурана (30 мл) и этанола (10 мл). Добавляли гидроксид лития (412 мг) и смесь перемешивали при комнатной температуре в течение ночи. Добавляли к реакционному раствору воду с последующим экстрагированием этилацетатом. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением смеси геометрических изомеров указанного в заголовке соединения (1,4 г).
Синтез 1-амино-3-(2-трифторметилбензил)пиперидин-2-она
Смесь геометрических изомеров этил 5-хлор-2-[1-(2-трифторметилфенил)метилиден]пентаноата (1,4 г) растворяли в этаноле (30 мл) и каталитически гидрировали в течение двух часов, используя картридж с 10% палладием-на-углероде в системе H-Cube™ (производства THALES Nanotechnology Inc.). Реакционный раствор концентрировали при пониженном давлении, чтобы уменьшить количество катализатора наполовину. Добавляли моногидрат гидразина (4,4 мл) и смесь кипятили с обратным холодильником в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении и добавляли воду с последующим экстрагированием этилацетатом. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем NH с получением указанного в заголовке соединения (916 мг). Показатели свойств соединения были следующими.
ESI-MS; m/z 273 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,43-1,53 (м, 1H), 1,65-1,80 (м, 2H), 1,88-1,98 (м, 1H), 2,67-2,76 (м, 1H), 2,80-2,89 (м, 1H), 3,45-3,55 (м, 2H), 3,65 (дд, J=14,6, 4,2 Гц, 1H), 4,53 (ушир.с, 2H), 7,31 (дд, J=8,0, 7,2 Гц, 1H), 7,40 (д, J=8,0 Гц, 1H), 7,48 (дд, J=7,6, 7,2 Гц, 1H), 7,64 (д, J=7,6 Гц, 1H).
Синтез 8-(2-трифторметилбензил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-2-иламина
1-амино-3-(2-трифторметилбензил)пиперидин-2-он (916 мг) и цианамид (849 мг) растворяли в этаноле (15 мл). Добавляли моногидрат п-толуолсульфоновой кислоты (961 мг) и смесь кипятили с обратным холодильником при 80°C в течение 3,5 часов. После охлаждения до комнатной температуры добавляли триэтиламин (2,3 мл) и смесь еще кипятили с обратным холодильником при 80°C в течение четырех суток. Добавляли к реакционному раствору раствор бикарбоната натрия и этилацетат и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Полученное твердое вещество промывали этилацетатом с получением указанного в заголовке соединения (244 мг). Показатели свойств соединения были следующими.
ESI-MS; m/z 297 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,48-1,60 (м, 1H), 1,75-1,94 (м, 2H), 2,04-2,13 (м, 1H), 2,92 (ддд, J=14,4, 10,8, 1,2 Гц, 1H), 3,13-3,22 (м, 1H), 3,67 (дд, J=14,6, 4,2 Гц, 1H), 3,88-4,12 (м, 4H), 7,34 (дд, J=7,6, 7,2 Гц, 1H), 7,41 (д, J=7,6 Гц, 1H), 7,51 (дд, J=8,0, 7,2 Гц, 1H), 7,67 (д, J=8,0 Гц, 1H).
2-Бром-8-(2-трифторметилбензил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Указанное в заголовке соединение получали таким же способом, как в примере получения 2-1, из 8-(2-трифторметилбензил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-2-иламина.
ESI-МС; m/z 360, 362 [M++H].
Пример получения 2-8
Синтез 2-бром-4-(4-трифторметилфенил)-4,5,6,7-тетрагидробензотиазола
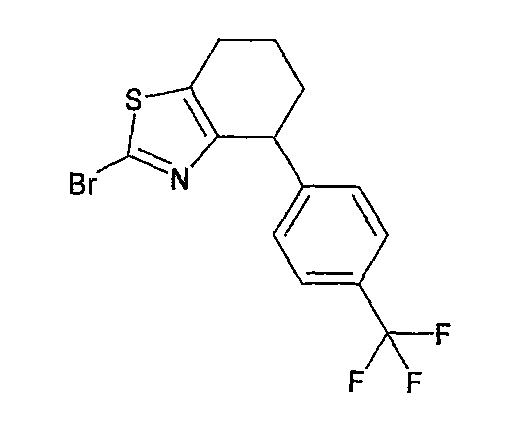
Синтез 2-(4-трифторметилфенил)циклогексанона
1-Бром-4-трифторметилбензол (5 мл), циклогексанон (7,4 мл) и трет-бутоксид калия (8 г) суспендировали в тетрагидрофуране (100 мл). Добавляли трис(дибензилиенацетон)дипалладий (1,3 г) и 2-дициклогексилфосфино-2′-(N,N-диметиламино)бифенил (843 мг) и смесь нагревали при перемешивании при 70°C в атмосфере азота в течение 1,5 часов. Смесь охлаждали, добавляли трет-бутилметиловый эфир с последующим фильтрованием через целит. Полученный органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем три раза. Полученное твердое вещество промывали гептаном с получением указанного в заголовке соединения (2,1 г). Показатели свойств соединения были следующими.
ESI-MS; m/z 243 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,80-1,93 (м, 2H), 1,96-2,08 (м, 2H), 2,16-2,24 (м, 1H), 2,26-2,34 (м, 1H), 2,44-2,60 (м, 2H), 3,65-3,77 (м, 1H), 7,25 (д, J=8,0 Гц, 2H), 7,59 (д, J=8,0 Гц, 2H).
Синтез 4-(4-трифторметилфенил)-4,5,6,7-тетрагидробензотиазол-2-иламина
2-(4-Трифторметилфенил)циклогексанон (1,2 г) растворяли в хлороформе (20 мл). Добавляли по каплям раствор брома в тетрагидрофуране, приготовленный при 0,3 М (20 мл), с поддержанием внутренней температуры на уровне 5°C или ниже. После этого смесь снова доводили до комнатной температуры и перемешивали в течение трех часов. Добавляли к реакционному раствору раствор бикарбоната натрия и хлороформ и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением диастереомерной смеси 2-бром-6-(4-трифторметилфенил)циклогексанона (1,2 г).
Диастереомерную смесь 2-бром-6-(4-трифторметилфенил)циклогексанона (500 мг) растворяли в этаноле (10 мл). Добавляли тиомочевину (130 мг) и смесь перемешивали при комнатной температуре в течение ночи. Добавляли к реакционному раствору раствор бикарбоната натрия и этилацетат и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (394 мг). Показатели свойств соединения были следующими.
ESI-MS; m/z 243 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,74-1,88 (м, 3H), 2,13-2,22 (м, 1H), 2,62-2,77 (м, 2H), 4,00-4,05 (м, 1H), 4,72 (ушир.с, 2H), 7,22 (д, J=8,4 Гц, 2H), 7,53 (д, J=8,4 Гц, 2H).
Синтез 2-бром-4-(4-трифторметилфенил)-4,5,6,7-тетрагидробензотиазола
Указанное в заголовке соединение получали таким же способом, как в примере получения 2-1, из 4-(4-трифторметилфенил)-4,5,6,7-тетрагидробензотиазол-2-иламина. Показатели свойств соединения были следующими.
ESI-MS; m/z 364 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,78-1,93 (м, 3H), 2,18-2,28 (м, 1H), 2,75-2,90 (м, 2H), 4,23-4,28 (м, 1H), 7,17 (д, J=8,2 Гц, 2H), 7,54 (д, J=8,2 Гц, 2H).
Пример получения 2-9
Синтез 2-бром-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-6-ола
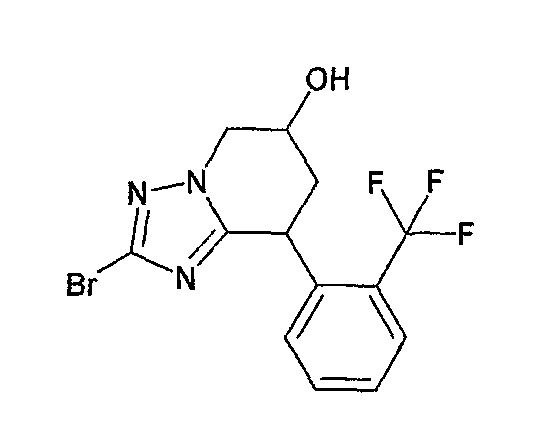
Синтез метил 3-оксиранил-2-(2-трифторметилфенил)пропионата
Метил (2-трифторметилфенил)ацетат (5,0 г) растворяли в тетрагидрофуране (10 мл) и раствор добавляли при -78°C к раствору, содержащему бистриметилсилиламид калия (50 мл) в тетрагидрофуране (190 мл). После перемешивания в течение 30 минут добавляли эпибромгидрин (1,96 мл) при той же самой температуре. Еще через 10 минут добавляли комплекс трифторид бора-диэтиловый эфир (3,16 мл). После перемешивания при -78°C в течение одного часа реакцию останавливали насыщенным водным раствором бикарбоната натрия. Органический слой, разбавленный этилацетатом, промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало остаток, который растворяли в метаноле (50 мл) и метиленхлориде (50 мл) и обрабатывали карбонатом калия (15,8 г). Через один час осаждали карбонат калия добавлением метиленхлорида (200 мл) и удаляли его фильтрованием через целит. Полученный раствор промывали насыщенным водным раствором хлорида аммония и насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало неочищенный продукт. Неочищенный продукт очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-30%) с получением диастереомерной смеси (2,61 г).
Синтез 1-амино-5-гидрокси-3-(2-трифторметилфенил)пиперидин-2-она
Метил 3-оксиранил-2-(2-трифторметилфенил)пропионат (2,61 г) растворяли в этаноле (20 мл). Моногидрат гидразина (2,3 мл) добавляли при комнатной температуре и осуществляли взаимодействие смеси в течение 24 часов. Концентрирование при пониженном давлении дало масло, которое распределяли между метиленхлоридом и водой. Водный слой экстрагировали метиленхлоридом четыре раза. После сушки над сульфатом магния концентрирование при пониженном давлении дало указанное в заголовке соединение (1,8 г) в виде бледно-желтого твердого вещества.
ESI-МС; m/z 275 [M++H].
Синтез 2-бром-8-(2-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-6-ола
Указанное в заголовке соединение получали в виде белого твердого вещества таким же способом, как в примере получения 2-7, из 1-амино-5-гидрокси-3-(2-трифторметилфенил)пиперидин-2-она.
ESI-МС; m/z 362 [M++H].
Пример получения 2-10
Синтез 2-бром-7-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
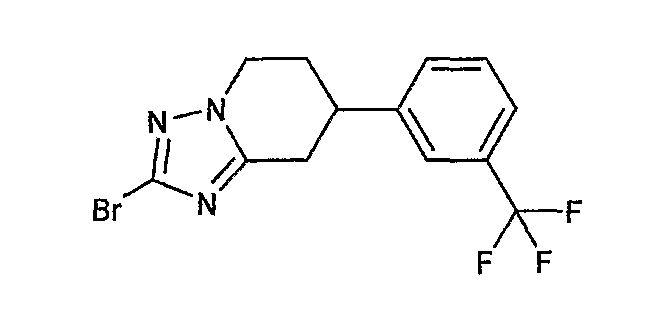
Синтез 4-(3-трифторметилфенил)тетрагидропиран-2-она
(Ацетилацетонато)бис(этилен)родий(I) (180 мг) растворяли в 1,4-диоксане (144 мл). Рацемический BINAP (2,2′-бис(дифенилфосфино)-1,1′-бинафталин) (867 мг) добавляли при комнатной температуре с последующим перемешиванием в течение 30 минут. Затем добавляли воду (14,4 мл) и 3-(трифторметил)фенилбороновую кислоту (5,29 г) с последующим перемешиванием в течение 10 минут. Затем добавляли 5,6-дигидро-2H-пиран-2-он (2 мл) и осуществляли взаимодействие смеси при 100°C в течение 12 часов. Реакционный раствор распределяли между этилацетатом и водой. Затем органический слой промывали насыщенным раствором бикарбоната натрия и сушили над сульфатом магния. Концентрирование при пониженном давлении дало масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-60%) с получением указанного в заголовке соединения (2,5 г).
ESI-MS; m/z 245 [M++H].
Синтез гидразида 5-гидрокси-3-(3-трифторметилфенил)пентановой кислоты
4-(3-Трифторметилфенил)тетрагидропиран-2-он (2,5 г) растворяли в этаноле (10 мл) и добавляли моногидрат гидразина (4,96 мл) при комнатной температуре. Смесь перемешивали при 80°C в течение 4,5 часов и затем концентрировали при пониженном давлении. Полученное масло распределяли между метиленхлоридом и водой. Водный слой экстрагировали метиленхлоридом пять раз. После сушки над сульфатом магния концентрирование при пониженном давлении и очистка хроматографией на колонке с силикагелем (аминосиликагель, подвижная фаза: этилацетат/гептан = 0-50% → метанол/этилацетат = 10%) дали указанное в заголовке соединение (1,55 г).
ESI-MS; m/z 277 [M++H]
Синтез трет-бутил N′-[5-гидрокси-3-(3-трифторметилфенил)пентаноил]гидразинкарбоксилат
Гидразид 5-гидрокси-3-(3-трифторметилфенил)пентановой кислоты (1,55 г) растворяли в тетрагидрофуране (26 мл) и добавляли ди-трет-бутилдикарбонат (1,35 г) при комнатной температуре. После перемешивания в течение 15 часов реакционный раствор разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли. После сушки над сульфатом магния концентрирование при пониженном давлении дало указанное в заголовке соединение (2,0 г). Полученное соединение использовали непосредственно для следующей реакции в виде неочищенного продукта.
Синтез трет-бутил N′-[5-бром-3-(3-трифторметилфенил)пентаноил]гидразинкарбоксилата
трет-Бутил N′-[5-гидрокси-3-(3-трифторметилфенил)пентаноил]гидразинкарбоксилат (2,0 г) растворяли в метиленхлориде (53 мл). Добавляли пиридин (1,29 мл), тетрабромид углерода (2,64 г) и трифенилфосфин (2,1 г) при комнатной температуре с последующим перемешиванием в течение 30 минут. Реакционный раствор распределяли между водой и этилацетатом. Затем органический слой промывали насыщенным водным раствором хлорида аммония и насыщенным водным раствором бикарбоната натрия и сушили над сульфатом магния. Концентрирование при пониженном давлении дало масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-50%) с получением указанного в заголовке соединения (1,6 г).
Синтез гидразида 5-бром-3-(3-трифторметилфенил)пентановой кислоты
трет-Бутил N′-[5-бром-3-(3-трифторметилфенил)пентаноил]гидразинкарбоксилат (1,6 г) растворяли в метиленхлориде (8 мл) и добавляли при 0°C трифторуксусную кислоту (10 мл). Смесь перемешивали при комнатной температуре в течение двух часов и затем концентрировали при пониженном давлении. Полученное масло разбавляли этилацетатом и затем промывали 1 н. водным раствором гидроксида натрия и насыщенным раствором соли и сушили над сульфатом магния. Масло после концентрирования при пониженном давлении (990 мг) использовали непосредственно для следующей реакции в виде неочищенного продукта.
Синтез 2-бром-7-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Указанное в заголовке соединение получали таким же способом, как в примере получения 2-1, из гидразида 5-бром-3-(3-трифторметилфенил)пентановой кислоты.
ESI-MS; m/z 348 [M++H].
Пример получения 2-11
Синтез 2-бром-8-метил-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
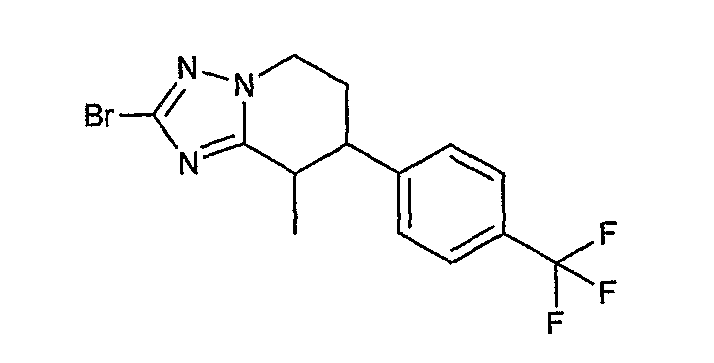
Синтез 3-метил-4-(4-трифторметилфенил)тетрагидропиран-2-она
4-(4-Трифторметилфенил)тетрагидропиран-2-он (995 мг), полученный таким же способом, как в примере получения 2-10, из 4-(трифторметил)фенилбороновой кислоты (5,29 г) и 5,6-дигидро-2H-пиран-2-он растворяли в тетрагидрофуране (10 мл). Раствор добавляли по каплям при -78°C в раствор диизопропиламида лития, ранее приготовленного из н-бутиллития (3,91 мл) и диизопропиламина (1,46 мл), в тетрагидрофуране (30 мл). После перемешивания при той же самой температуре в течение 30 минут добавляли йодметан (760 мкл) и гексаметилфосфорамид (1,77 мл) и продолжали перемешивание. После перемешивания в течение 30 минут реакционный раствор постепенно нагревали до комнатной температуры в течение трех часов и 20 минут. Реакцию останавливали насыщенным водным раствором хлорида аммония. Органический слой промывали насыщенным раствором соли и затем сушили над сульфатом магния. Удаление при пониженном давлении дало масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-50%) с получением указанного в заголовке соединения (500 мг).
ESI-MS; m/z 259 [M++H].
Синтез 2-бром-8-метил-7-(4-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Указанное в заголовке соединение получали таким же способом, как в примере получения 2-10, из 3-метил-4-(4-трифторметилфенил)тетрагидропиран-2-она.
Пример получения 2-12
Синтез 2-бром-5-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
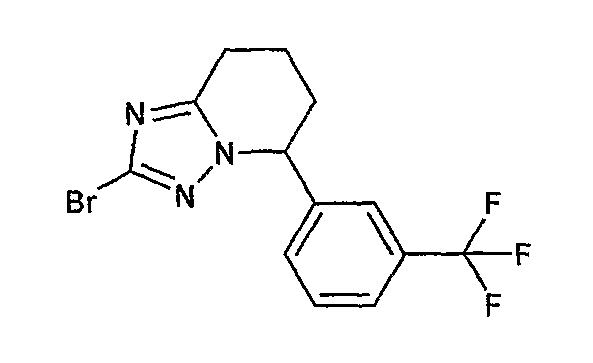
Синтез этил 5-(N′-трет-бутоксикарбонилгидразино)-5-(3-трифторметилфенил)пентаноата
Этил 5-оксо-5-(3-трифторметилфенил)пентаноат (CAS №898777-75-8, 330 мг) и трет-бутилкарбазат (227 мг) растворяли в смешанном растворителе из ТГФ (6 мл) и уксусной кислоты (3 мл). После перемешивания при комнатной температуре в течение 25 минут, добавляли цианоборгидрид натрия (108 мг). Еще через один час и 30 минут добавляли цианоборгидрид натрия (108 мг) и смесь перемешивали при комнатной температуре в течение одного часа. Добавляли к реакционному раствору насыщенный раствор бикарбоната натрия и диэтиловый эфир и отделяли органический слой. Полученный органический слой промывали насыщенным раствором бикарбоната натрия и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (324 мг). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,22 (т, J=7,2 Гц, 3H), 1,43 (с, 9H), 1,47-1,84 (м, 4H), 2,27 (т, J=6,8 Гц, 2H), 4,09 (кв., J=7,2 Гц, 2H), 4,14 (т, J=7,2 Гц, 1H), 4,25 (ушир.с, 1H), 5,88 (ушир.с, 1H), 7,43-7,62 (м, 4H).
Синтез трет-бутил [2-оксо-6-(3-трифторметилфенил)пиперидин-1-ил]карбамата
Этил 5-(N′-трет-бутоксикарбонилгидразино)-5-(3-трифторметилфенил)пентаноат (323 мг) растворяли в метаноле (7 мл). Добавляли 1 н. раствор гидроксида натрия (5 мл) и смесь перемешивали при комнатной температуре в течение двух часов и 30 минут. К реакционному раствору добавляли 1 н. хлористоводородную кислоту (5 мл) и этилацетат и органический слой отделяли и сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток растворяли в диметилформамиде (8 мл). Добавляли гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (317 мг) и 1-гидроксибензотриазол (223 мг) и смесь перемешивали при комнатной температуре в течение 13 часов. Добавляли к реакционному раствору гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (317 мг) и смесь еще перемешивали в течение одного часа. Добавляли к реакционному раствору диэтиловый эфир и воду и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (214 мг). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,43 (с, 9H), 1,86-1,94 (м, 3H), 2,26-2,39 (м, 1H), 2,59-2,69 (м, 2H), 4,99 (ушир.с, 1H), 6,39 (ушир.с, 1H), 7,39 (д, J=7,6 Гц, 1H), 7,44 (с, 1H), 7,50 (т, J=7,6 Гц, 1H), 7,57 (д, J=7,6 Гц, 1H).
Синтез 1-амино-6-(3-трифторметилфенил)пиперидин-2-она
трет-Бутил [2-оксо-6-(3-трифторметилфенил)пиперидин-1-ил]карбамат (210 мг) растворяли в хлороформе (4 мл), после чего добавляли трифторуксусную кислоту (2 мл). После перемешивания при комнатной температуре в течение четырех часов концентрирование при пониженном давлении дало указанное в заголовке соединение (119 мг). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,72-1,92 (м, 3H), 2,23-2,59 (м, 1H), 2,59 (дт, J=2,4 Гц, 6,4 Гц, 2H), 4,36 (с, 2H), 4,79 (т, J=6,4 Гц, 1H), 7,37 (д, J=7,6 Гц, 1H), 7,44 (с, 1H), 7,52 (т, J=7,6 Гц, 1H), 7,58 (д, J=7,6 Гц, 1H).
Синтез 2-бром-5-(3-трифторметилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Указанное в заголовке соединение получали в соответствии со способом примера получения 2-7 из 1-амино-6-(3-трифторметилфенил)пиперидин-2-она. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,89-2,04 (м, 2H), 2,07-2,15 (м, 1H), 2,41-2,49 (м, 1H), 3,03 (дт, J=3,2 Гц, 6,8 Гц, 2H), 5,46 (т, J=6,0 Гц, 1H), 7,14 (д, J=7,6 Гц, 1H), 7,33 (с, 1H), 7,49 (т, J=7,6 Гц, 1H), 7,59 (д, J=7,6 Гц, 1H).
Пример получения 2-13
Синтез 4-бром-7-(4-трифторметилфенил)-2,3,5-триаза-трицикло[5.2.2.0*2,6*]ундека-3,5-диена
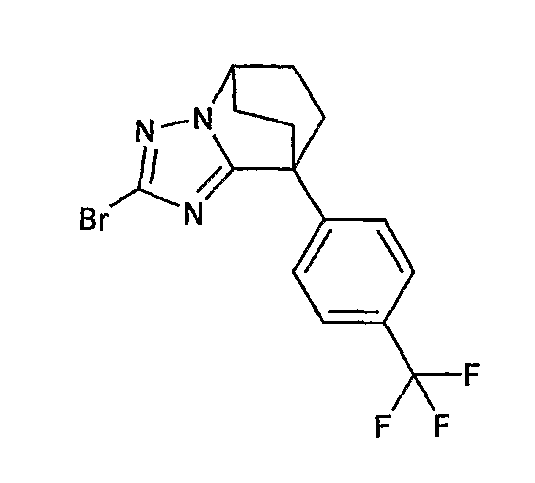
Синтез диметил 4-гидрокси-1-(4-трифторметилфенил)-3-циклогексен-1,3-дикарбоксилата
Метил (4-трифторфенил)ацетат (CAS №135325-18-7, 1 г) и метилакрилат (789 мг) растворяли в ТГФ (13 мл). Добавляли трет-бутоксид калия (617 мг) и смесь перемешивали при комнатной температуре в течение 14 часов и 30 минут. Добавляли к реакционному раствору воду с последующим перемешиванием при 85°C в течение пяти часов и 30 минут. Смесь охлаждали, к реакционному раствору добавляли этилацетат, после чего смесь нейтрализовали 1 н. хлористоводородной кислотой. Органический слой отделяли, промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,13 г). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,13-2,60 (м, 4H), 2,75 (д, J=16,4 Гц, 1H), 3,08 (д, J=16,4 Гц, 1H), 3,66 (с, 3H), 3,82 (с, 3H), 7,47 (д, J=8,8 Гц, 2H), 7,60 (д, J=8,8 Гц, 2H), 12,11 (с, 1H).
Синтез метил 4-оксо-1-(4-трифторметилфенил)циклогексанкарбоксилата
Диметил 4-гидрокси-1-(4-трифторметилфенил)-3-циклогексен-1,3-дикарбоксилат (1,13 г) растворяли в смешанном растворителе из ТГФ (3 мл) и метанола (3 мл). Добавляли 1 н. раствор гидроксида калия (1,89 мл) и смесь перемешивали при 100°C в течение восьми часов. Смесь охлаждали, добавляли к реакционному раствору этилацетат и воду, после чего смесь нейтрализовали 1 н. хлористоводородной кислотой. Органический слой отделяли, промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (272 мг). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,21-2,28 (м, 2H), 2,41-2,47 (м, 2H), 2,52-2,60 (м, 2H), 2,77-2,83 (м, 2H), 3,74 (с, 3H), 7,54 (д, J=8,4 Гц, 2H), 7,64 (д, J=8,4 Гц, 2H).
Синтез метил 4-(N′-трет-бутоксикарбонилгидразино)-1-(4-трифторметилфенил)циклогексанкарбоксилата
Метил 4-оксо-1-(4-трифторметилфенил)циклогексанкарбоксилат (100 мг) и трет-бутилкарбазат (66 мг) растворяли в смешанном растворителе из ТГФ (6 мл) и уксусной кислоты (3 мл). После перемешивания при комнатной температуре в течение 50 минут, добавляли цианоборгидрид натрия (31,4 мг). Еще через два часа добавляли цианоборгидрид натрия (31,4 мг) и смесь перемешивали при комнатной температуре в течение одного часа. Диэтиловый эфир и насыщенный раствор бикарбоната натрия добавляли к реакционному раствору и отделяли органический слой. Полученный органический слой промывали насыщенным раствором бикарбоната натрия и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (74,5 мг). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,28-1,36 (м, 2H), 1,47 (с, 9H), 1,60-1,66 (м, 2H), 1,94-1,96 (м, 2H), 2,66-2,69 (м, 2H), 2,87 (ушир.с, 1H), 3,66 (с, 3H), 4,05 (ушир.с, 1H), 6,08 (ушир.с, 1H), 7,48 (д, J=8,4 Гц, 2H), 7,57 (д, J=8,4 Гц, 2H).
Синтез трет-бутил [3-оксо-4-(4-трифторметилфенил)-2-аза-бицикло[2,2,2]окт-2-ил]карбамат
Метил 4-(N′-трет-бутоксикарбонилгидразино)-1-(4-трифторметилфенил)циклогексанкарбоксилат (74,5 мг) растворяли в метаноле (1,5 мл), после чего добавляли 1 н. раствор гидроксида натрия (1 мл). Смесь перемешивали при комнатной температуре в течение 15 часов и затем перемешивали при кипячении с обратным холодильником в течение 11 часов и 30 минут. Смесь охлаждали, добавляли к реакционному раствору диэтиловый эфир, после чего смесь нейтрализовали 1 н. хлористоводородной кислотой. Органический слой отделяли и затем промывали насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (35,7 мг). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,48 (с, 9H), 1,80-1,85 (м, 2H), 2,02-2,11 (м, 2H), 2,22-2,32 (м, 4H), 3,90 (с, 1H), 6,77 (ушир.с, 1H), 7,48 (д, J=8,4 Гц, 2H), 7,60 (д, J=8,4 Гц, 2H).
Синтез 4-бром-7-(4-трифторметилфенил)-2,3,5-триаза-трицикло[5.2.2.0*2,6*]ундека-3,5-диена
Указанное в заголовке соединение получали в соответствии со способом примера получения 2-12, из трет-бутил [3-оксо-4-(4-трифторметилфенил)-2-азабицикло[2,2,2]окт-2-ил]карбамата. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,93-2,12 (м, 6H), 2,24-2,30 (м, 2H), 4,97 (тт, J=1,6 Гц, 3,2 Гц, 1H), 7,69 (д, J=8,8 Гц, 2H), 7,72 (д, J=8,8 Гц, 2H).
Пример получения 3-1
Синтез гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата
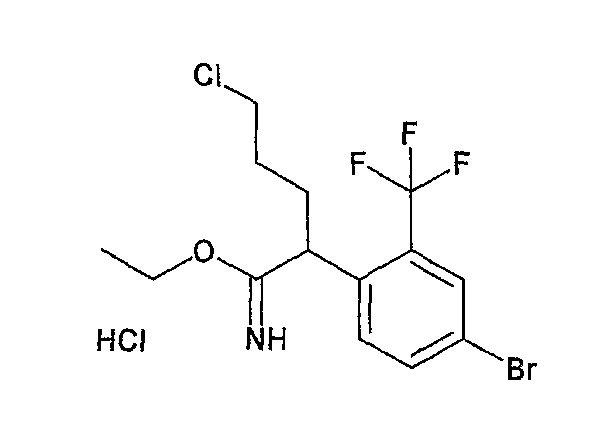
Синтез 4-бром-1-бромметил-2-трифторметилбензола
N-Бромсукцинимид (2,6 г) и 2,2′-азобис(изобутиронитрил) (35 мг) добавляли к раствору 5-бром-2-метилбензотрифторида (CAS #86845-27-4, 3,5 г) в тетрахлориде углерода (35 мл) и смесь кипятили с обратным холодильником в течение пяти часов. Реакционный раствор охлаждали льдом и затем удаляли фильтрованием нерастворимый материал. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel C-200; элюирующий растворитель: гептан) с получением 3,86 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 4,58 (с, 2H), 7,47 (д, J=8,4 Гц, 1H), 7,69 (дд, J=8,4, 2,0 Гц, 1H), 7,79 (д, J=2,0 Гц, 1H).
Синтез (4-бром-2-трифторметилфенил)ацетонитрил
Цианид калия (630 мг) добавляли к эмульсии 4-бром-1-бромметил-2-трифторметилбензола (3,86 г) в смеси этанол (15 мл)-вода (5 мл) и смесь перемешивали при 70°C в течение трех часов и 15 минут. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel C-200; элюирующий растворитель: гептан:этилацетат = 9:1) с получением 2,28 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 3,91 (с, 2H), 7,57 (д, J=8,4 Гц, 1H), 7,76 (дд, J=8,4, 2,0 Гц, 1H), 7,85 (д, J=2,0 Гц, 1H).
Синтез 2-(4-бром-2-трифторметилфенил)-5-хлорпентаннитрила
Раствор (4-бром-2-трифторметилфенил)ацетонитрила (2,28 г) в тетрагидрофуране (6 мл) добавляли к суспензии гидрида натрия (60% содержание в минеральном масле, 362 мг) в тетрагидрофуране (12 мл) при охлаждении льдом. Смесь перемешивали при той же самой температуре в течение 20 минут. 1-хлор-3-йодпропан (0,94 мл) добавляли по каплям к реакционному раствору и затем реакционный раствор перемешивали при комнатной температуре в течение 40 минут. Добавляли к реакционному раствору ледяную воду и гептан и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel™ C-200; элюирующий растворитель: гептан:этилацетат = 49:1 → 19:1) с получением 2,67 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,88-2,17 (м, 4H), 3,58 (м, J=6,0 Гц, 2H), 4,13 (м, J=7,2 Гц, 1H), 7,59 (д, J=8,4 Гц, 1H), 7,78 (дд, J=8,4, 2,0 Гц, 1H), 7,84 (д, J=2,0 Гц, 1H).
Синтез гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата
Раствор 2-(4-бром-2-трифторметилфенил)-5-хлорпентаннитрила (2,67 г) в этаноле (7,5 мл) барботировали газообразным хлороводородом при охлаждении льдом в течение 15 минут. Реакционный раствор перемешивали при комнатной температуре в течение суток. Реакционный раствор концентрировали при пониженном давлении. К остатку добавляли диэтиловый эфир и полученный порошок собирали фильтрованием. Собранный фильтрованием продукт сушили при пониженном давлении с получением 2,96 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
ESI-MS; m/z 388 [M++H-HCl].
1H-ЯМР (ДМСО-d6) δ (ppm): 1,27 (т, J=7,2 Гц, 3H), 1,49-1,75 (м, 2H), 2,10-2,35 (м, 2H), 3,64 (т, J=6,4 Гц, 2H), 4,26 (т, J=7,6 Гц, 1H), 4,34-4,50 (м, 2H), 7,73 (д, J=8,4 Гц, 1H), 7,97 (д, J=2,0 Гц, 1H), 8,02 (дд, J=8,4, 2,0 Гц, 1H), 11,69 (ушир.с, 1H).
Пример получения 3-2
Синтез гидрохлорида этил 5-хлор-2-(3,3-диметил-2,3-дигидробензофуран-5-ил)пентанимидата
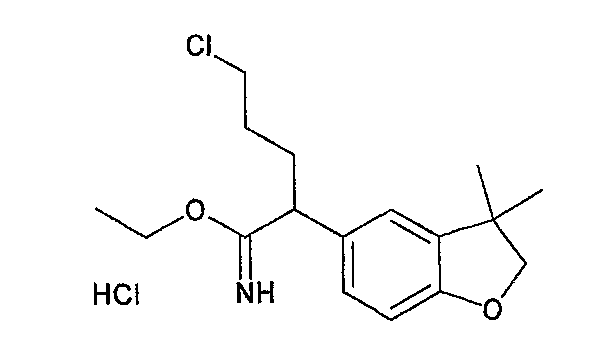
Синтез 3,3,5-триметил-2,3-дигидробензофурана
3-Хлор-2-метилпропен (9,7 мл) добавляли по каплям к смешанному раствору из п-крезола (CAS #106-44-5, 10,8 г) и концентрированной серной кислоты (2,5 г) и смесь перемешивали при комнатной температуре в течение одного часа и 20 минут. Реакционный раствор охлаждали льдом и добавляли 5 н. водный раствор гидроксида натрия (20 мл) с последующим перемешиванием при 90°C в течение 45 минут. Добавляли к реакционному раствору 5 н. водный раствор гидроксида натрия (10 мл) и смесь перемешивали при 90°C в течение двух часов и 45 минут. Реакционный раствор охлаждиали до комнатной температуры. Добавляли к реакционному раствору трет-бутилметиловый эфир и отделяли органический слой. Полученный органический слой промывали последовательно водой (три раза) и насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Merck силикагель 60 (230-400 меш); элюирующий растворитель: гептан:этилацетат = 1:0 → 49:1). Целевую фракцию концентрировали и полученный остаток снова очищали хроматографией на колонке с силикагелем (носитель: Chromatorex™ NH; элюирующий растворитель: гептан). Целевую фракцию концентрировали и полученный остаток дистиллировали при пониженном давлении с получением 1,87 г целевого соединения (т. кип. 102°C/20 мм рт.ст.). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,33 (с, 6H), 2,30 (с, 3H), 4,20 (с, 2H), 6,68 (д, J=7,6 Гц, 1H), 6,88-6,95 (м, 2H).
Синтез гидрохлорида этил 5-хлор-2-(3,3-диметил-2,3-дигидробензофуран-5-ил)пентанимидата
Указанное в заголовке соединение получали в соответствии со способом примера получения 3-1, из 3,3,5-триметил-2,3-дигидробензофурана. Показатели свойств соединения были следующими.
ESI-MS; m/z 310 [M++H-HCl].
Пример получения 3-3
Синтез гидрохлорида этил 2-(5-трет-бутил-2-метоксифенил)-5-хлорпентанимидата
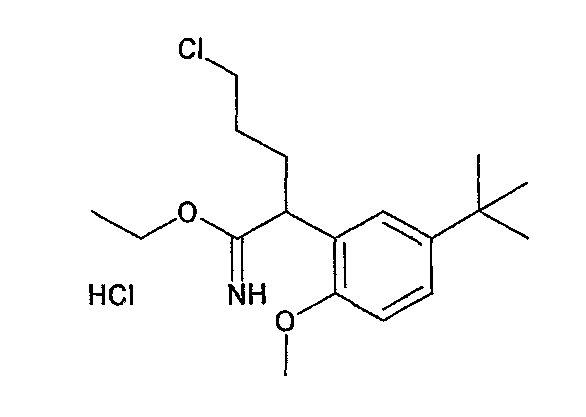
Синтез 4-трет-бутил-1-метокси-2-метилбензола
Карбонат калия (8,4 г) и метилиодид (2,9 мл) добавляли к раствору 4-трет-бутил-2-метилфенола (CAS #98-27-1, 5,0 г) в N,N-диметилформамиде (25 мл) и смесь перемешивали при комнатной температуре в течение трех суток. Добавляли к реакционному раствору ледяную воду и гексан и отделяли органический слой. Полученный органический слой промывали последовательно водой и насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением 5,16 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1Н-ЯМР (CDCl3) δ (ppm): 1,30 (с, 9Н), 2,22 (с, 3Н), 3,81 (с, 3Н), 6,76 (д, J=9,2 Гц, 1Н), 7,15-7,19 (м, 2Н).
Синтез 2-бромметил-4-трет-бутил-1-метоксибензола
N-Бромсукцинимид (5,66 г) и 2,2′-азобис(изобутиронитрил) (71 мг) добавляли к раствору 4-трет-бутил-1-метокси-2-метилбензола (5,16 г) в тетрахлориде углерода (25 мл) и смесь кипятили с обратным холодильником в течение 1,5 часов. Реакционный раствор охлаждали льдом и затем удаляли фильтрованием нерастворимый материал. Фильтрат концентрировали при пониженном давлении с получением 7,78 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,30 (с, 9H), 3,88 (с, 3H), 4,58 (с, 2H), 6,81 (д, J=8,4 Гц, 1H), 7,31 (дд, J=8,4, 2,4 Гц, 1H), 7,34 (д, J=2,4 Гц, 1H).
Синтез (5-трет-бутил-2-метоксифенил)ацетонитрила
Цианид калия (2,96 г) добавляли к раствору 2-бромметил-4-трет-бутил-1-метоксибензола (7,78 г) в диметилсульфоксиде (50 мл) и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли лед и трет-бутилметиловый эфир и отделяли органический слой. Водный слой повторно экстрагировали трет-бутилметиловым эфиром. Объединенные органические слои промывали последовательно водой (два раза) и насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Chromatorex™ NH; элюирующий растворитель: гептан → этилацетат:гептан = 1:49). Целевую фракцию концентрировали. Полученный остаток растирали в гексане с получением 1,90 г указанного в заголовке соединения. Маточную жидкость для растирания концентрировали. Затем полученный остаток растирали в гексане с получением 0,47 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,31 (с, 9H), 3,68 (с, 2H), 3,85 (с, 3H), 6,82 (д, J=8,4 Гц, 1H), 7,32 (дд, J=8,4, 2,4 Гц, 1H), 7,36 (д, J=2,4 Гц, 1H).
Синтез 2-(5-трет-бутил-2-метоксифенил)-5-хлорпентаннитрила
Раствор н-бутиллития в гексане (2,69 M, 3,0 мл) добавляли к раствору N,N-диизопропиламина (1,2 мл) в ТГФ (15 мл) при охлаждении льдом и смесь перемешивали при той же самой температуре в течение 10 минут. Раствор охлаждали до -78°C и раствор (5-трет-бутил-2-метоксифенил)ацетонитрила (1,5 г) в ТГФ (6,5 мл) добавляли по каплям. Раствор перемешивали при -30°C в течение 25 минут и затем опять охлаждали до -78°C. 1-хлор-3-йодпропан (1,2 мл) добавляли по каплям к раствору и затем реакционный раствор постепенно нагревали до комнатной температуры. После охлаждения реакционного раствора льдом добавляли к нему раствор гексаметилдисилазида лития в тетрагидрофуране (1,0 M, 4,4 мл). Затем к реакционному раствору добавляли раствор диизопропиламида лития, приготовленный из N,N-диизопропиламина (0,6 мл) и раствора н-бутиллития в гексане (2,69 M, 1,5 мл). Добавляли к реакционному раствору насыщенный водный раствор хлорида аммония. Затем добавляли этилацетат и воду и отделяли органический слой. Полученный органический слой промывали последовательно 1 н. хлористоводородной кислотой, водой, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Merck силикагель 60 (230-400 меш); элюирующий растворитель: гептан:этилацетат = 1:49) с получением 864 мг 1:7 смеси (5-трет-бутил-2-метоксифенил)ацетонитрила и указанного в заголовке соединения. Показатели свойств указанного в заголовке соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,31 (с, 9H), 1,86-2,12 (м, 4H), 3,54-3,60 (м, 2H), 3,84 (с, 3H), 4,18-4,24 (м, 1H), 6,83 (д, J=8,4 Гц, 1H), 7,31 (дд, J=8,4, 2,4 Гц, 1H), 7,40 (д, J=2,4 Гц, 1H).
Синтез гидрохлорида этил 2-(5-трет-бутил-2-метоксифенил)-5-хлорпентанимидат и гидрохлорида этил 2-(5-трет-бутил-2-метоксифенил)ацетимидат
Раствор 1:7 смеси (5-трет-бутил-2-метоксифенил)ацетонитрила и 2-(5-трет-бутил-2-метоксифенил)-5-хлорпентаннитрила (864 мг) в этаноле (8 мл) барботировали газообразным хлороводородом при охлаждении льдом в течение 15 минут. Реакционный раствор перемешивали при комнатной температуре в течение суток. Реакционный раствор концентрировали при пониженном давлении с получением смеси указанного в заголовке соединения.
Показатели свойств гидрохлорида этил 2-(5-трет-бутил-2-метоксифенил)-5-хлорпентанимидата были следующими.
ESI-MS; m/z 326 [M++H-HCl].
Показатели свойств гидрохлорида этил 2-(5-трет-бутил-2-метоксифенил)ацетимидата были следующими.
ESI-MS; m/z 250 [M++H-HCl].
Пример получения 3-4
Синтез гидрохлорида этил 2-(3-бром-4-метоксифенил)-5-хлорпентанимидата
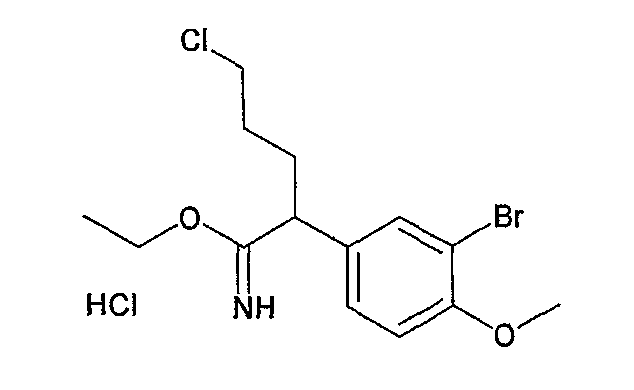
Указанное в заголовке соединение (3,62 г) получали в виде белого твердого вещества из 3-бром-4-метоксифенилацетонитрила (3,0 г) в соответствии со способом примера 3-1.
ESI-MS; m/z 350 [M++H-HCl].
Соединения примеров получения 3-5 - 3-13 получали таким же способом, как в примере 3-1 (таблица 7).
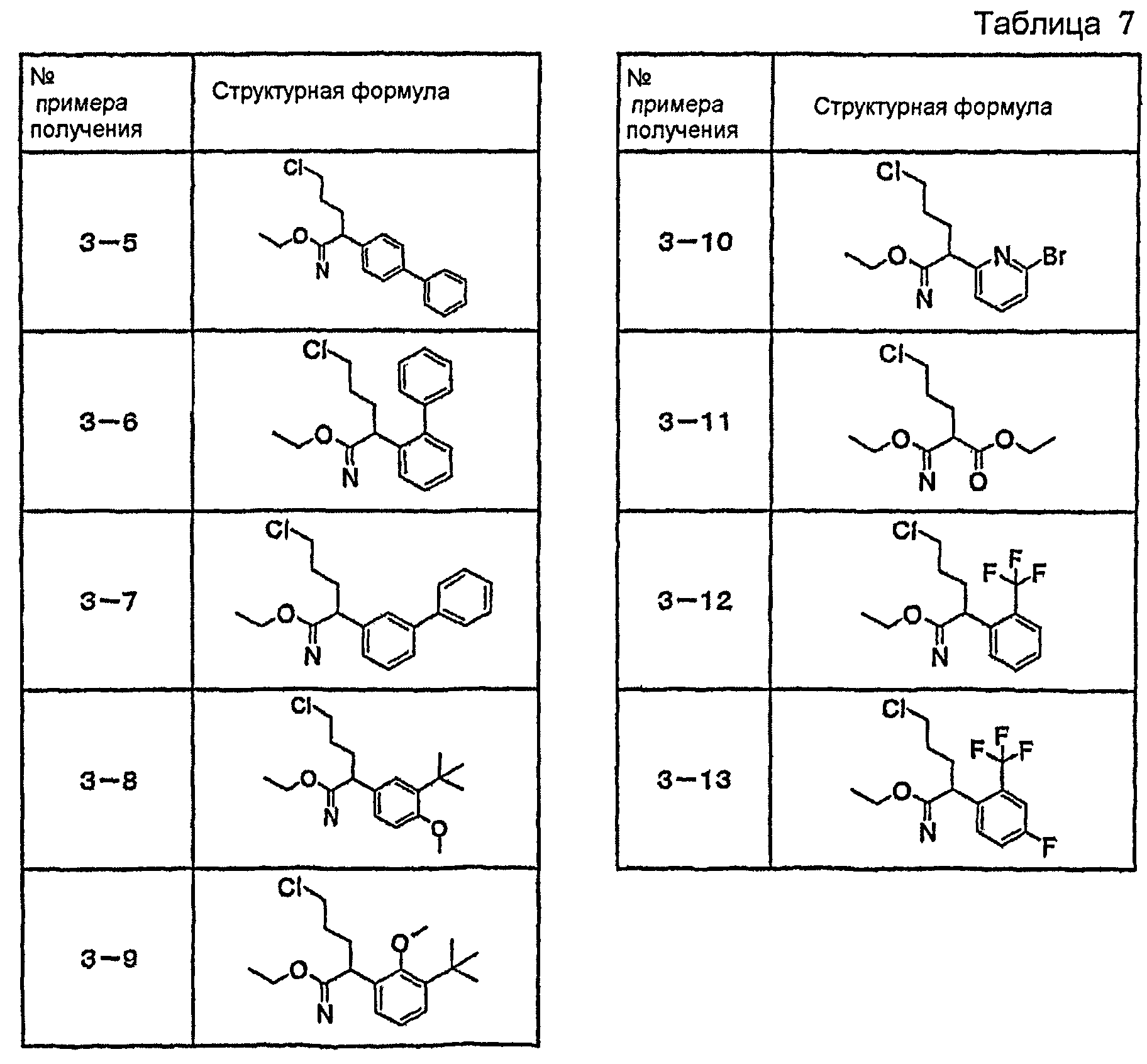
Пример получения 3-14
Синтез гидрохлорида этил 4-хлор-2-(2-трифторметилфенил)бутанимидата
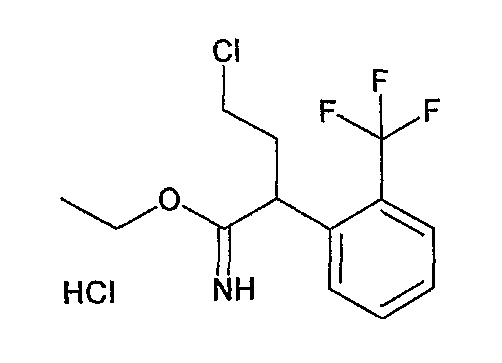
Синтез 4-(трет-бутилдиметилсилилокси)-2-(2-трифторметилфенил)бутиронитрила
(2-Трифторметилфенил)ацетонитрил (3 г) и бисульфат тетрабутиламмония (550 мг) суспендировали в толуоле (30 мл) и 50% водный раствор гидроксида натрия (15 мл) добавляли по каплям при охлаждении льдом. После перемешивания в течение 10 минут (2-бромэтокси)-трет-бутилдиметилсилан (5,2 мл) добавляли при той же самой температуре. Смесь доводили обратно до комнатной температуры и перемешивали в течение ночи. Добавляли к реакционному раствору воду с последующим экстрагированием трет-бутилметиловым эфиром. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5 г). Показатели свойств соединения были следующими.
ESI-MS; m/z 344 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 0,08 (с, 3H), 0,11 (с, 3H), 0,92 (с, 9H), 1,98-2,14 (м, 2H), 3,80-3,95 (м, 2H), 4,52 (дд, J=10,2, 6,2 Гц, 1H), 7,42-7,48 (м, 1H), 7,59-7,65 (м, 1H), 7,68-7,74 (м, 2H).
Синтез 4-гидрокси-2-(2-трифторметилфенил)бутиронитрила
4-(трет-Бутилдиметилсилилокси)-2-(2-трифторметилфенил)бутиронитрил (5 г) растворяли в тетрагидрофуране (70 мл). Добавляли тетрабутиламмонийфторид (1 M раствор в тетрагидрофуране) (18 мл) и смесь перемешивали при комнатной температуре в течение 4,5 часов. Реакционный раствор концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,1 г). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,11-2,19 (м, 2H), 3,86-3,97 (м, 2H), 4,49 (дд, J=8,6, 7,0 Гц, 1H), 7,45-7,51 (м, 1H), 7,62-7,68 (м, 1H), 7,69-7,76 (м, 2H).
Синтез 4-хлор-2-(2-трифторметилфенил)бутиронитрила
4-гидрокси-2-(2-трифторметилфенил)бутиронитрил (2,6 г) растворяли в пиридине (10 мл). Осторожно добавляли тионилхлорид (2,1 мл) и смесь нагревали при перемешивании при 60°C в течение четырех часов. Добавляли к реакционному раствору воду с последующим экстрагированием этилацетатом. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,3 г). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,27-2,44 (м, 2H), 3,72-3,79 (м, 2H), 4,53 (дд, J=9,8, 5,0 Гц, 1H), 7,46-7,53 (м, 1H), 7,62-7,66 (м, 1H), 7,70-7,76 (м, 2H).
Синтез гидрохлорида этил 4-хлор-2-(2-трифторметилфенил)бутанимидата
Указанное в заголовке соединение (1,3 г) получали таким же способом, как в примере получения 3-1, из 4-хлор-2-(2-трифторметилфенил)бутиронитрила (1,5 г). Показатели свойств соединения были следующими.
ESI-MS; m/z 294 [M++H-HCl].
1H-ЯМР (CDCl3) δ (ppm): 1,41 (м, J=7,0 Гц, 3H), 2,56-2,66 (м, 1H), 3,03-3,14 (м, 1H), 3,56-3,68 (м, 2H), 4,59-4,75 (м, 3H), 7,50 (дд, J=7,6, 7,6 Гц, 1H), 7,68 (дд, J=7,6, 7,6 Гц, 1H), 7,74 (д, J=7,6 Гц, 1H), 7,93 (д, J=7,6 Гц, 1H).
Соединения примеров получения 3-15 - 3-16 получали таким же способом, как в примере 3-14 (таблица 8).
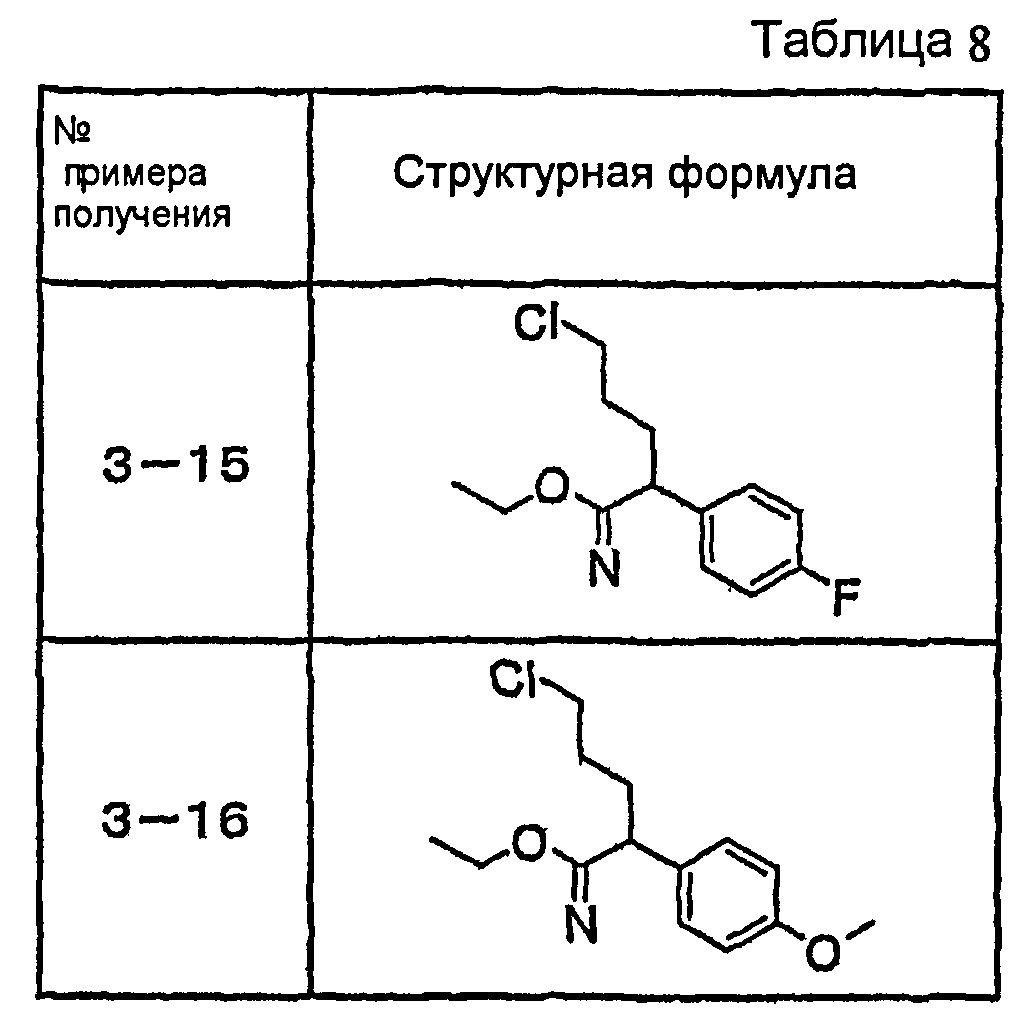
Пример получения 3-17
Синтез гидрохлорида этил 2-(3-бромфенил)-2-(2-хлорэтокси)ацетимидата
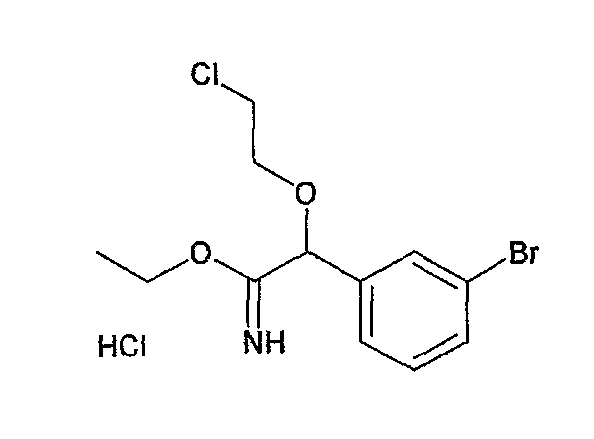
Синтез (3-бромфенил)-(2-гидроксиэтокси)ацетонитрил
Иодид цинка (50,6 мг) добавляли к смеси 2-(3-бромфенил)-1,3-диоксолана (3 мл) и триметилсилилцианида (2,49 мл) с последующим перемешиванием при комнатной температуре в течение 1,5 часов. Насыщенный водный раствор бикарбоната натрия добавляли к реакционному раствору с последующим экстрагированием диэтиловым эфиром. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Затем полученный остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: гептан:этилацетат = 7:3 → 1:1) с получением 3,93 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,83 (ушир., 1H), 3,73-3,78 (м, 1H), 3,84-3,90 (м, 3H), 5,33 (с, 1H), 7,35 (т, J=7,6 Гц, 1H), 7,44-7,47 (м, 1H), 7,56-7,59 (м, 1H), 7,67 (дт, J=0,4, 1,2 Гц, 1H).
Синтез (3-бромфенил)-(2-хлорэтокси)ацетонитрила
Тионилхлорид (1,23 мл) добавляли по каплям к раствору (3-бромфенил)-(2-гидроксиэтокси)ацетонитрила (3,62 г) в пиридине (12,1 мл) при 50°C и смесь перемешивали при 60°C в течение двух часов. Добавляли к реакционному раствору воду с последующим экстрагированием этилацетатом. Органический слой промывали водой. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Затем полученный остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: гептан:этилацетат = 9:1 → 8:2) с получением 3,21 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 3,70-3,73 (м, 2H), 3,87 (ддд, J=5,2, 6,4, 10,4 Гц, 1H), 4,01 (ддд, J=5,6, 5,6, 10,0 Гц, 1H), 5,36 (с, 1H), 7,33 (т, J=7,6 Гц, 1H), 7,45-7,48 (м, 1H), 7,57-7,59 (м, 1H), 7,67 (дт, J=0,4, 1,6 Гц, 1H).
Синтез гидрохлорида этил 2-(3-бромфенил)-2-(2-хлорэтокси)ацетимидата
Указанное в заголовке соединение (2,71 г) получали таким же способом, как в примере получения 3-1, из (3-бромфенил)-(2-хлорэтокси)ацетонитрила (3,21 г). Показатели свойств соединения были следующими.
1H-ЯМР (ДМСО-d6) δ (ppm): 1,24 (т, J=7,2 Гц, 3H), 3,76-3,90 (м, 4H), 4,41-4,43 (м, 2H), 5,50 (с, 1H), 7,40-7,47 (м, 2H), 7,63-7,68 (м, 2H).
Соединение примера получения 3-18 получали таким же способом, как в примере 3-17 (таблица 9).
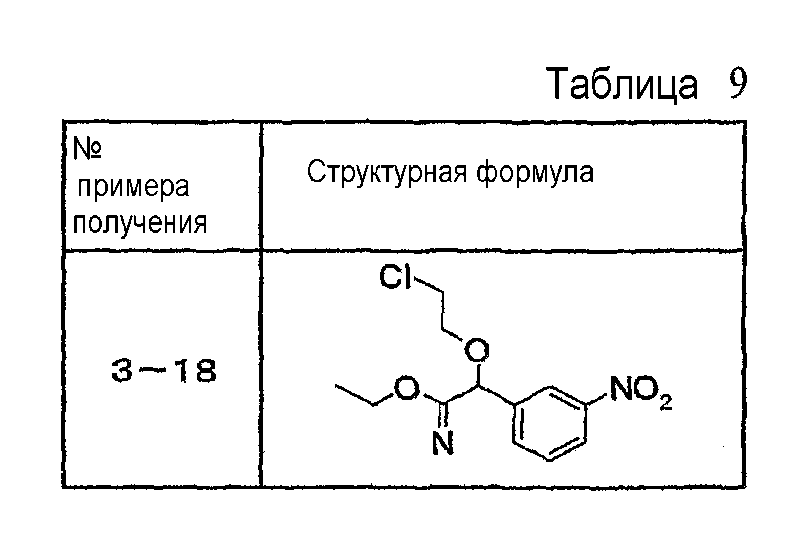
Пример получения 3-19
Синтез этил 4-этоксикарбонимидоил-4-(2-трифторметилфенил)бутаноата
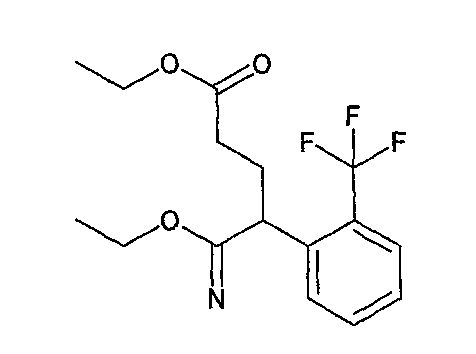
Синтез метил 4-циано-4-(2-трифторметилфенил)бутаноата
2-(Трифторметил)фенилацетонитрил (2,0 г) растворяли в тетрагидрофуране (50 мл). Трет-бутоксид калия (60,6 мг), 18-краун-6 (2,85 г) и метилакрилат (973 мкл) добавляли при 0°C и смесь перемешивали при той же самой температуре в течение 40 минут. Реакцию останавливали насыщенным раствором хлорида аммония с последующим разбавлением реакционной смеси этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-30%) с получением указанного в заголовке соединения (2,2 г).
1H-ЯМР (CDCl3) δ (ppm): 2,25 (дд, J=14,9, 7,4 Гц, 2H), 2,51-2,65 (м, 2H), 3,70 (с, 3H), 4,31 (т, J=7,8 Гц, 1H), 7,46-7,51 (м, 1H), 7,65-7,67 (м, 1H), 7,69-7,73 (м, 2H).
Синтез гидрохлорида этил 4-этоксикарбонимидоил-4-(2-трифторметилфенил)бутаноата
Метил 4-циано-4-(2-трифторметилфенил)бутаноат (200 мг) растворяли в этаноле (1,03 мл) и добавляли при 0°C ацетилхлорид (838 мкл). Смесь нагревали до комнатной температуры и начинали реакцию. После перемешивания в течение 24 часов концентрирование при пониженном давлении дало неочищенный продукт указанного в заголовке соединения в виде масла (300 мг).
ESI-MS; m/z 332 [M++H-HCl].
Пример получения 3-20
Синтез гидрохлорида этил 2-(4-хлор-2-трифторметилфенил)-5-хлорпентанимидата
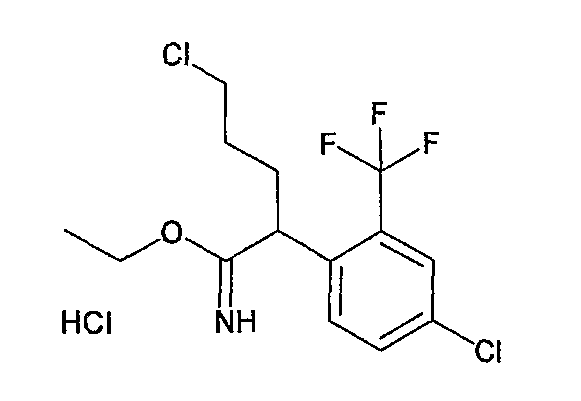
Синтез метил (4-хлор-2-трифторметилфенил)ацетата
Тионилхлорид (9,5 мл) добавляли по каплям к раствору 4-хлор-2-(трифторметил)фенилуксусной кислоты (CAS #601513-31-9, 10,5 г) в метаноле (50 мл) при охлаждении льдом. Затем реакционный раствор кипятили с обратным холодильником в течение трех часов. Реакционный раствор концентрировали при пониженном давлении. К полученному остатку добавляли насыщенный раствор бикарбоната натрия и этилацетат и отделяли органический слой. Полученный органический слой промывали последовательно насыщенным раствором бикарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением 10,96 г указанного в заголовке соединения.
1H-ЯМР (CDCl3) δ (ppm): 3,71 (с, 3H), 3,80 (с, 2H), 7,35 (д, J=8,4 Гц, 1H), 7,50 (дд, J=8,4, 2,0 Гц, 1H), 7,65 (д, J=2,0 Гц, 1H).
Синтез метил 5-хлор-2-(4-хлор-2-трифторметилфенил)пентаноата
Раствор метил (4-хлор-2-трифторметилфенил)ацетата (5,5 г) и 3-хлор-1-йодпропана (4,6 мл) в тетрагидрофуране (15 мл) добавляли по каплям к суспензии гидрида натрия (60% содержание в минеральном масле, 955 мг) в тетрагидрофуране (50 мл) при охлаждении льдом. Реакционный раствор перемешивали при той же самой температуре в течение 10 минут и затем при комнатной температуре в течение трех часов и 20 минут. К реакционному раствору добавляли ледяную воду и этилацетат и отделяли органический слой. Водный слой повторно экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором соли и сушили над безводным сульфатом магния с последующим концентрированием при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel™ C-200; элюирующий растворитель: гептан:этилацетат = 19:1) с получением 5,30 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,59-1,71 (м, 1H), 1,76-1,96 (м, 2H), 2,17-2,28 (м, 1H), 3,51 (т, J=6,4 Гц, 2H), 3,68 (с, 3H), 3,99 (т, J=7,2 Гц, 1H), 7,52 (дд, J=8,4, 2,0 Гц, 1H), 7,55 (д, J=8,4 Гц, 1H), 7,65 (д, J=2,0 Гц, 1H).
Синтез 5-хлор-2-(4-хлор-2-трифторметилфенил)пентановой кислоты
5 н. водный раствор гидроксида натрия (13 мл) добавляли к смешанному раствору метил 5-хлор-2-(4-хлор-2-трифторметилфенил)пентаноата (5,3 г) в смеси тетрагидрофуран (15 мл)-метанол (15 мл) и смесь перемешивали при комнатной температуре в течение двух часов и 10 минут. После добавления к реакционному раствору ледяной воды органический растворитель выпаривали из реакционного раствора при пониженном давлении. Полученный водный слой промывали гептаном. Водный слой подкисляли 5 н. хлористоводородной кислотой при охлаждении льдом с последующим экстрагированием этилацетатом два раза. Объединенные этилацетатные слои промывали насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением 4,75 г указанного в заголовке соединения.
1H-ЯМР (CDCl3) δ (ppm): 1,59-1,71 (м, 1H), 1,79-2,01 (м, 2H), 2,19-2,30 (м, 1H), 3,51 (т, J=6,4 Гц, 2H), 4,03 (т, J=7,6 Гц, 1H), 7,54 (ушир.с, 2H), 7,67 (ушир.с, 1H).
Синтез амида 5-хлор-2-(4-хлор-2-трифторметилфенил)пентановой кислоты
Аммонийхлорид (3,23 г), 1-гидроксибензотриазол (2,45 г), IPEA (13,7 мл) и гексафторфосфат бензотриазол-1-илокситрис(пирролидино)фосфония (9,43 г) последовательно добавляли к раствору 5-хлор-2-(4-хлор-2-трифторметилфенил)пентановой кислоты (4,75 г) в N,N-диметилформамиде (45 мл). Смесь перемешивали при комнатной температуре в течение ночи. К реакционному раствору добавляли ледяную воду и трет-бутилметиловый эфир и отделяли органический слой. Водный слой повторно экстрагировали трет-бутилметиловым эфиром. Объединенные органические слои промывали последовательно 1 н. хлористоводородной кислотой, водой, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel™ C-200; элюирующий растворитель: гептан:этилацетат = 4:1 → 3:1) с получением 3,89 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,60-1,67 (м, 1H), 1,80-1,97 (м, 2H), 2,20-2,31 (м, 1H), 3,45-3,56 (м, 2H), 3,77 (т, J=7,2 Гц, 1H), 5,30 (ушир.с, 1H), 5,41 (ушир.с, 1H), 7,54 (дд, J=8,4, 2,0 Гц, 1H), 7,66 (д, J=2,0 Гц, 1H), 7,69 (д, J=8,4 Гц, 1H).
Синтез 5-хлор-2-(4-хлор-2-трифторметилфенил)пентаннитрила
Метилдихлорфосфат (1,9 мл) добавляли по каплям при 20°C или ниже к охлаждаемому льдом раствору амида 5-хлор-2-(4-хлор-2-трифторметилфенил)пентановой кислоты (3,0 г) и 1,8-диазабицикло[5.4.0]ундец-7-ена (6,4 мл) в метиленхлориде (30 мл). По окончании добавления по каплям реакционный раствор перемешивали при комнатной температуре в течение одного часа и 25 минут. Добавляли к реакционному раствору насыщенный водный раствор бикарбоната натрия. После перемешивания в течение пяти минут отделяли органический слой. Водный слой повторно экстрагировали метиленхлоридом. Объединенные органические слои промывали последовательно 1 н. хлористоводородной кислотой, водой, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel C-200; элюирующий растворитель: гептан:этилацетат = 49:1 → 19:1) с получением 2,25 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,89-2,18 (м, 4H), 3,58 (т, J=6,0 Гц, 2H), 4,15 (дд, J=8,0, 6,4 Гц, 1H), 7,63 (дд, J=8,4, 2,0 Гц, 1H), 7,66 (д, J=8,4 Гц, 1H), 7,70 (д, J=2,0 Гц, 1H).
Синтез гидрохлорида этил 2-(4-хлор-2-трифторметилфенил)-5-хлорпентанимидата
Указанное в заголовке соединение получали таким же способом, как в примере получения 3-1, из 5-хлор-2-(4-хлор-2-трифторметилфенил)пентаннитрила.
ESI-MS; m/z 342 [M++H-HCl].
Пример получения 4-1
Синтез [бензил(2-гидроксиэтил)амино]-(4-трифторметилфенил)ацетонитрила
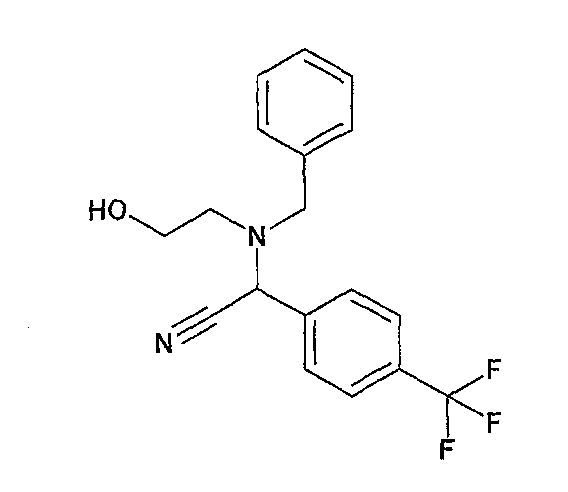
4-(трифторметил)бензальдегид (CAS #455-19-6, 2 г) и метанол (1 мл) добавляли к раствору бисульфита натрия (1,33 г) в воде (40 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 10 минут. Цианид натрия (1,33 г) добавляли к реакционному раствору и смесь перемешивали при той же самой температуре в течение 40 минут. N-бензилэтаноламин (1,83 г) и метанол (3 мл) добавляли к реакционному раствору и смесь перемешивали при той же самой температуре в течение 16 часов. Реакционный раствор разделяли этилацетатом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: гептан:этилацетат = 9:1 → 1:1) с получением 1,79 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,66 (ушир.с, 1H), 2,70-2,81 (м, 2H), 3,55 (д, J=13,6 Гц, 1H), 3,58-3,61 (м, 1H), 3,75-3,82 (м, 1H), 4,02 (д, J=13,2 Гц, 1H), 5,06 (с, 1H), 7,30-7,40 (м, 5H), 7,64-7,71 (м, 4H).
Пример получения 4-2
Синтез [(2-гидроксиэтил)фениламино]-(4-трифторметилфенил)ацетонитрила
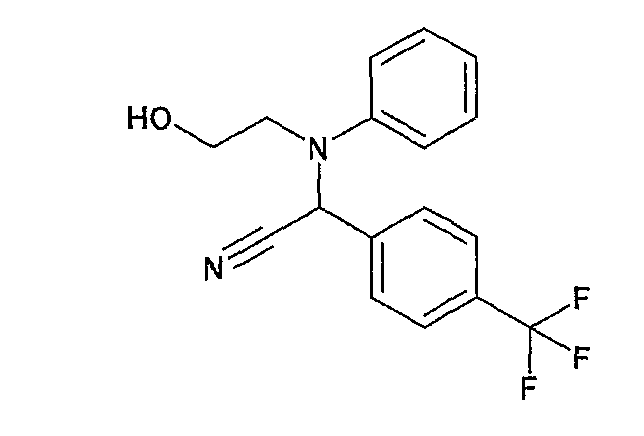
Триметилсилилцианид (723 мкл) добавляли к смеси 4-(трифторметил)бензальдегида (CAS #455-19-6, 1 г), N-фенилэтаноламина (870 мг) и сульфаминовой кислоты (28 мг) и смесь перемешивали при комнатной температуре в течение 20 часов. Реакционный раствор распределяли между водой и хлороформом. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: гептан:этилацетат = 9:1 → 1:1) с получением 775 мг указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,59 (ушир.с, 1H), 3,30 (ддд, J=5,6, 7,2, 12,4 Гц, 1H), 3,53 (ддд, J=4,8, 7,2, 7,2 Гц, 1H), 3,64-3,69 (м, 2H), 5,70 (с, 1H), 7,01-7,06 (м, 3H), 7,31-7,35 (м, 2H), 7,69-7,72 (м, 4H).
Пример получения 4-3
Синтез 5-гидрокси-4-(2-трифторметилфенил)пентаннитрила
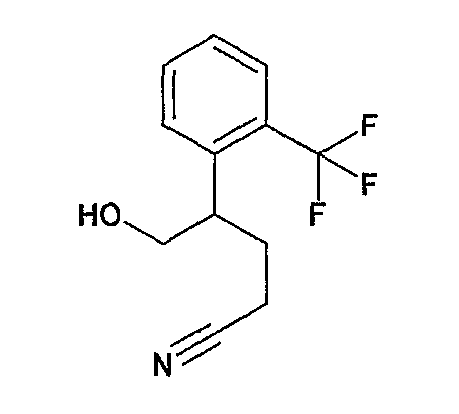
Синтез трет-бутил (2-трифторметилфенил)ацетата
(2-Трифторметил)фенилуксусную кислоту (10,5 г) растворяли в тетрагидрофуране (150 мл) и добавляли при 0°C трет-бутил 2,2,2-трихлорацетимидат (18,4 мл) и комплекс трифторид бора-диэтиловый эфир (323 мкл). После перемешивания при комнатной температуре в течение 22 часов реакцию останавливали насыщенным водным раствором бикарбоната натрия с последующим разбавлением реакционной смеси этилацетатом. Органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало белое твердое вещество. Твердое вещество очищали лишь с помощью рыхлого слоя аминосиликагеля (подвижная фаза: этилацетат:гептан = 1:15) с получением указанного в заголовке соединения (13,0 г).
Синтез трет-бутил 4-циано-2-(2-трифторметилфенил)бутаноата
трет-Бутил (2-трифторметилфенил)ацетат (12,9 г) растворяли в толуоле (129 мл). Гидроксид бензилтриметиламмония (782 мкл) добавляли при комнатной температуре с последующим перемешиванием в течение 10 минут. После этого добавляли при 0°C акрилонитрил (6,49 мл) и начинали реакцию. После перемешивания при комнатной температуре в течение 12 часов реакцию останавливали 2 н. хлористоводородной кислотой с последующим разбавлением реакционной смеси диэтиловым эфиром. Органический слой промывали водой (два раза) и насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало желтое масло (17 г), который использовали непосредственно для следующей реакции в виде неочищенного продукта.
Синтез 4-циано-2-(2-трифторметилфенил)бутановой кислоты
трет-Бутил 4-циано-2-(2-трифторметилфенил)бутаноат (7,2 г) растворяли в метиленхлориде (40 мл). Трифторуксусную кислоту (50 мл) добавляли при 0°C и начинали реакцию. После перемешивания при комнатной температуре в течение одного часа концентрирование при пониженном давлении дало масло. Полученное масло растворяли в диэтиловом эфире, промывали водой (три раза) и насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало желтое масло. Масло снова растворяли в диэтиловом эфире и распределяли между диэтиловым эфиром и 5 н. гидроксидом натрия (15 мл). Полученный водный слой подкисляли 5 н. хлористоводородной кислотой (16 мл) и экстрагировали метиленхлоридом два раза с получением указанного в заголовке соединения (6,76 г) в виде бесцветного прозрачного масла.
Синтез 5-гидрокси-4-(2-трифторметилфенил)пентаннитрила
Тионилхлорид (8,25 мл) добавляли к 4-циано-2-(2-трифторметилфенил)бутановой кислоте (2,9 г) и смесь кипятили с обратным холодильником. Через один час концентрирование при пониженном давлении дало масло. Масло растворяли в тетрагидрофуране (50 мл) и этаноле (20 мл) и добавляли при 0°C боргидрид натрия (855 мг). После перемешивания при комнатной температуре в течение одного часа реакцию останавливали водой с последующим разбавлением реакционной смеси этилацетатом. Органический слой промывали 1 н. хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало бледно-желтое масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-50%) с получением указанного в заголовке соединения (2,14 г).
ESI-MS: m/z 244 [M++H].
Пример получения 4-4
Синтез 5-гидрокси-3-(4-трифторметилфенил)пентаннитрила
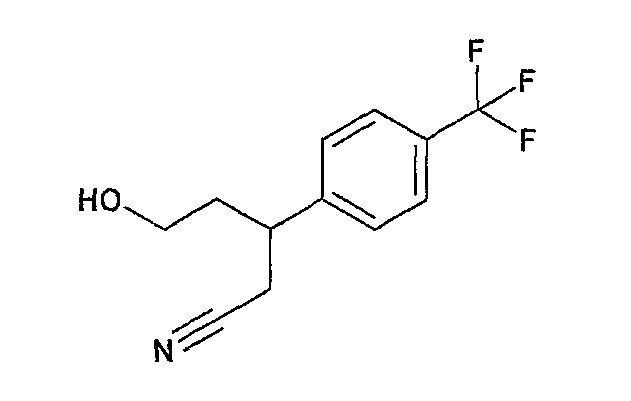
Синтез амида 5-гидрокси-3-(4-трифторметилфенил)пентановой кислоты
Насыщенный метаноловый раствор аммония (10 мл) добавляли к 4-(3-трифторметилфенил)тетрагидропиран-2-ону, полученному в соответствии со способом примера получения 2-10 (1,0 г), и смесь перемешивали при 60°C в течение 23 часов. Для завершения реакции еще добавляли насыщенный метаноловый раствор аммония (5 мл) и продолжали перемешивание при той же самой температуре. Спустя 24 часа концентрирование при пониженном давлении и очистка хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-100%) дали указанное в заголовке соединение (850 мг).
ESI-MS: m/z 262 [M++H].
Синтез амида 5-(трет-бутилдифенилсиланилокси)-3-(4-трифторметилфенил)пентановой кислоты
Амид 5-гидрокси-3-(4-трифторметилфенил)пентановой кислоты (350 мг) растворяли в диметилформамиде (13 мл) и имидазол (365 мг) и трет-бутилдифенилсилилхлорид (697 мкл) добавляли при комнатной температуре. После перемешивания в течение одного часа реакционный раствор распределяли между этилацетатом и водой. Органический слой промывали водой (два раза), 1 н. хлористоводородной кислотой и насыщенным раствором соли. После сушки над сульфатом магния концентрирование при пониженном давлении и очистка хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-90%) дали указанное в заголовке соединение (680 мг).
ESI-MS: m/z 500 [M++H].
Синтез 5-(трет-бутилдифенилсиланилокси)-3-(4-трифторметилфенил)пентаннитрила
Амид 5-(трет-бутилдифенилсиланилокси)-3-(4-трифторметилфенил)пентановой кислоты (640 мг) растворяли в метиленхлориде (13 мл) и 1,8-диазабицикло[5,4,0]ундец-7-ен (862 мкл) и метилдихлорфосфат (301 мкл) добавляли при 0°C. После перемешивания при комнатной температуре в течение 1,5 часа реакцию останавливали насыщенным водным раствором бикарбоната натрия. Органический слой промывали насыщенным раствором соли и затем сушили над сульфатом магния. Концентрирование при пониженном давлении дало масло. Масло пропускали через рыхлый слой силикагеля (подвижная фаза: этилацетат:гептан = 1:2) с получением указанного в заголовке соединения (600 мг).
Синтез 5-гидрокси-3-(4-трифторметилфенил)пентаннитрила
5-(трет-Бутилдифенилсиланилсиланилокси)-3-(4-трифторметилфенил)пентаннитрил (600 мг) растворяли в тетрагидрофуране (6 мл) и добавляли при комнатной температуре фторид тетрабутиламмония (1,5 мл). Через два часа смесь распределяли между водой и этилацетатом. Органический слой промывали насыщенным раствором соли и затем сушили над сульфатом магния. Концентрирование при пониженном давлении дало масло. Масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 70%) с получением указанного в заголовке соединения (306 мг).
ESI-MS: m/z 244 [M++H].
Пример получения 4-5
Синтез 5-гидрокси-3-(2-трифторметилфенил)пентаннитрила
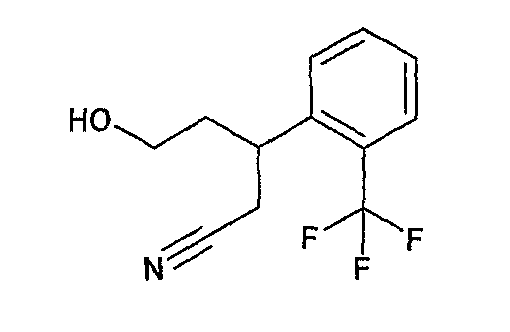
Синтез 3-(2-трифторметилфенил)пентандиовой кислоты
Пиперидин (1,0 мл) добавляли к 2-трифторметилбензальдегиду (5,0 г) и этилацетоацетату (7,27 мл) при 0°C. Затем смесь доводили до комнатной температуры и перемешивали в течение четырех суток. Этанол (50 мл) добавляли к полученной реакционной смеси, которую перемешивали при кипячении с обратным холодильником в течение пяти часов. Концентрирование при пониженном давлении дало масло. Полученное масло очищали хроматографией на колонке с силикагелем (подвижная фаза: этилацетат/гептан = 0-40%). Хотя масло (7,81 г) содержало большое количество примесей, масло растворяли в этаноле (18,5 мл) и воде (24,9 мл), чтобы использовать его непосредственно для следующей реакции. Добавляли гидроксид калия (32,7 г) и начинали реакцию нагреванием с обратным холодильником. Через 1,5 часа реакционную смесь распределяли между водой и диэтиловым эфиром. Полученный водный слой нейтрализовали концентрированной хлористоводородной кислотой (20 мл). После экстрагирования метиленхлоридом два раза сушка над сульфатом магния и концентрирование при пониженном давлении дали желтое твердое вещество. Полученный неочищенный продукт перекристаллизовывали из смеси этилацетата (30 мл) и гептана (65 мл) с получением указанного в заголовке соединения (1,2 г) в виде белого порошка.
ESI-MS: m/z 277 [M++H].
Синтез 4-(2-трифторметилфенил)дигидропиран-2,6-диона
3-(2-трифторметилфенил)пентандиовую кислоту (1,2 г) растворяли в уксусном ангидриде (12 мл) и смесь перемешивали при 110°C в течение двух часов. Масло после концентрирования при пониженном давлении растворяли в этилацетате и промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли. После сушки над сульфатом магния концентрирование при пониженном давлении дало оранжевое твердое вещество. Твердое вещество перекристаллизовывали из смеси этилацетата (10 мл) и гептана (70 мл) с получением указанного в заголовке соединения (636 мг) в виде белого порошка.
1H-ЯМР (CDCl3) δ (ppm): 2,19-2,35 (м, 2H), 2,91 (ддд, J=18,3, 12,5, 6,2 Гц, 1H), 3,10 (ддд, J=18,3, 4,3, 2,7 Гц, 1H), 4,2 (дд, J=12,0, 5,9 Гц, 1H), 7,35 (д, J=7,8 Гц, 1H), 7,45 (м, 1H), 7,60-7,65 (м, 1H), 7,75 (д, J=7,8 Гц, 1H).
Синтез 4-карбамоил-3-(2-трифторметилфенил)бутановой кислоты
Насыщенный метаноловый раствор аммония (10 мл) добавляли к 4-(2-трифторметилфенил)дигидропиран-2,6-диону (636 мг) и смесь перемешивали при комнатной температуре в течение 15 часов. Концентрирование при пониженном давлении дало неочищенный продукт (716 мг), который использовали для следующей реакции без очистки.
Синтез амида 5-гидрокси-3-(2-трифторметилфенил)пентановой кислоты
4-карбамоил-3-(2-трифторметилфенил)бутановую кислоту (646 мг) растворяли в метиленхлориде (32 мл) и добавляли при 0°C тетрагидрофурановый раствор диборана (7,12 мл). После перемешивания при комнатной температуре в течение двух часов реакционный раствор охлаждали до 0°C. Реакцию останавливали 1 н. хлористоводородной кислотой с последующим разбавлением реакционной смеси этилацетатом. Органический слой промывали 1 н. гидроксидом натрия и насыщенным раствором соли и сушили над сульфатом магния. Концентрирование при пониженном давлении дало неочищенный продукт (388 мг), который использовали для следующей реакции без очистки.
Синтез 5-гидрокси-3-(2-трифторметилфенил)пентаннитрила
Указанное в заголовке соединение получали в соответствии со способом примера получения 4-4 из амида 5-гидрокси-3-(2-трифторметилфенил)пентановой кислоты.
Пример получения 4-6
Синтез 2-(2-фтор-4-трифторметилфенил)-5-гидроксипентаннитрила
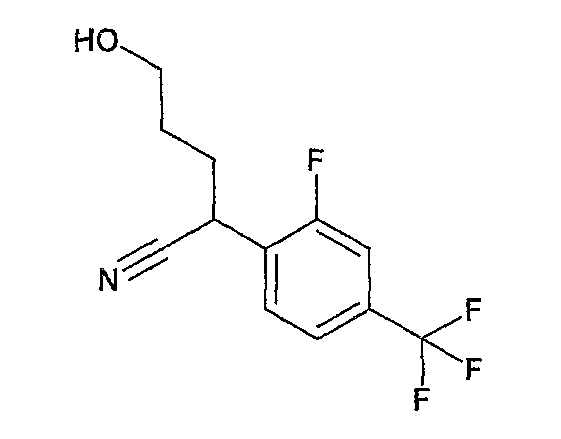
Указанное в заголовке соединение (1,3 г) получали в соответствии со способом примера 4-1 из 2-фтор-4-(трифторметил)фенилацетонитрила (2 г).
1H-ЯМР (CDCl3) δ (ppm): 1,70-1,84 (м, 2H), 1,98-2,14 (м, 2H), 3,66-3,78 (м, 2H), 4,25 (т, J=6,8 Гц, 1H), 7,35 (д, J=9,8 Гц, 1H), 7,49 (д, J=9,8 Гц, 1H), 7,61-7,66 (м, 1H).
Пример получения 5-1
Синтез 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоновой кислоты
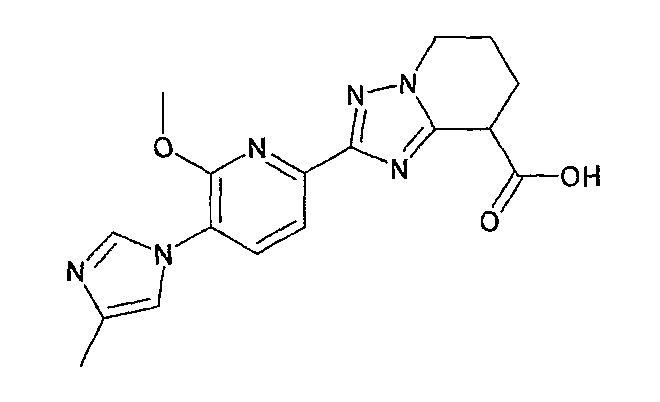
Синтез этил 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоксилата
Указанное в заголовке соединение (695 мг) получали в соответствии со способом примеров 42 и 43 из гидрохлорида имидата, полученного в примере получения 3-11, вместо гидрохлорида этил 2-(4-бром-2-трифторметилфенил)-5-хлорпентанимидата. Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,30 (т, J=7,2 Гц, 3H), 2,10-2,40 (м, 4H), 2,30 (с, 3H), 4,11 (т, J=6,0 Гц, 1H), 4,16 (с, 3H), 4,20-4,39 (м, 4H), 7,01 (с, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,82 (д, J=8,0 Гц, 1H), 7,83 (с, 1H).
Синтез 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоновой кислоты
Этил 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-карбоксилат (331 мг) растворяли в смешанном растворителе из ТГФ (5 мл) и метанола (5 мл). Добавляли 5 н. водный раствор гидроксида натрия (693 мкл) и смесь перемешивали при комнатной температуре в течение двух суток. Реакционный раствор нейтрализовали 5 н. хлористоводородной кислотой (693 мкл) и затем концентрировали при пониженном давлении для удаления органического растворителя. К остатку добавляли метанол (2 мл) и выпавшую в осадок соль отделяли фильтрованием. После этого фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения.
ESI-MS: m/z 355 [M++H].
Пример получения 1-9
Синтез гидразида 3-метокси-4-(4-метилимидазол-1-ил)бензойной кислоты
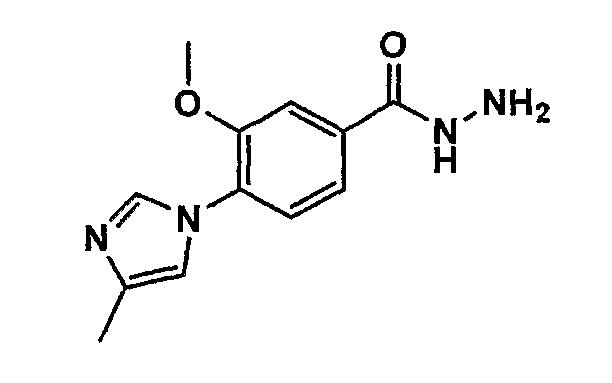
Синтез бензил N′-[3-метокси-4-(4-метилимидазол-1-ил)бензоил]гидразинкарбоксилата
IPEA (1,05 мл) и гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (1,16 г) последовательно добавляли к раствору 3-метокси-4-(4-метилимидазол-1-ил)бензойной кислоты (CAS#937026-26-1, 933 мг), бензилкарбазата (737 мг) и HOBT (817 мг) в ДМФА (15 мл) и смесь перемешивали при комнатной температуре в течение ночи. Добавляли к реакционному раствору этилацетат, ледяную воду и насыщенный раствор бикарбоната натрия и отделяли органический слой. Полученный органический слой промывали последовательно водой и насыщенным раствором соли. Объединенный водный слой экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат и NH силикагель и затем смесь концентрировали при пониженном давлении.
Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Chromatorex™ NH; элюирующий растворитель: этилацетат:гептан = 1:1 → 10:1 → 10% метанол в этилацетате). Целевую фракцию концентрировали. Полученный порошок растирали в этилацетате и трет-бутилметиловом эфире с получением 1,30 г указанного в заголовке соединения. Показатели свойств соединения были следующими.
1H-ЯМР (ДМСО-d6) δ (ppm): 2,16 (с, 3H), 3,90 (с, 3H), 5,14 (с, 2H), 7,22 (с, 1H), 7,25-7,45 (м, 5H), 7,52 (д, J=8,0 Гц, 1H), 7,56 (д, J=8,0 Гц, 1H), 7,67 (с, 1H), 7,88 (с, 1H), 9,43 (ушир.с, 1H), 10,46 (ушир.с, 1H).
Синтез гидразида 3-метокси-4-(4-метилимидазол-1-ил)бензойной кислоты
К раствору бензил N′-[3-метокси-4-(4-метилимидазол-1-ил)бензоил] гидразинкарбоксилата (1,25 г) в метаноле (60 мл) добавляли 20% гидроксид палладия-на-углероде (50% влажность, 250 мг) и смесь гидрировали при умеренном давлении (3,8 атм.) в течение двух часов и 30 минут. Палладий-на-углероде удаляли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении с получением 826 мг указанного в заголовке соединения. Показатели свойств соединения были следующими.
ESI-MS; m/z 247 [M++H].
1H-ЯМР (ДМСО-d6) δ (ppm): 2,16 (д, J=1,2 Гц, 3H), 3,89 (с, 3H), 4,55 (ушир.с, 2H), 7,20 (с, 1H), 7,46 (д, J=8,0 Гц, 1H), 7,52 (дд, J=8,0, 1,6 Гц, 1H), 7,64 (д, J=1,6 Гц, 1H), 7,84 (д, J=1,2 Гц, 1H), 9,90 (ушир.с, 1H).
Пример получения 1-10
Синтез 1-[4-(1-этоксивинил)-2-метоксифенил]-4-метил-1H-имидазола
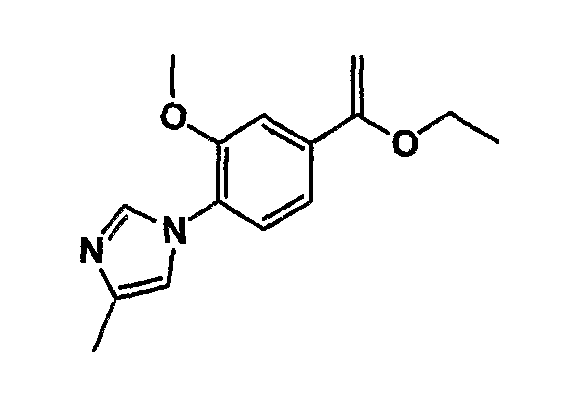
В соответствии со способом примера получения 1-7 575 мг указанного в заголовке соединения получали из 1-(4-бром-2-метоксифенил)-4-метил-1H-имидазола (CAS #87038-56-5, 1,5 г). Показатели свойств соединения были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,45 (т, J=7,2 Гц, 3H), 2,49 (с, 3H), 3,95 (кв., J=7,2 Гц, 2H), 3,96 (с, 3H), 4,27 (д, J=2,8 Гц, 1H), 4,70 (д, J=2,8 Гц, 1H), 7,33 (д, J=1,6 Гц, 1H), 7,34 (дд, J=8,8, 1,6 Гц, 1H), 7,73 (д, J=8,8 Гц, 1H), 8,65 (с, 1H).
Пример получения 1-11
Синтез 6-бром-3-имидазол-1-ил-2-метоксипиридина
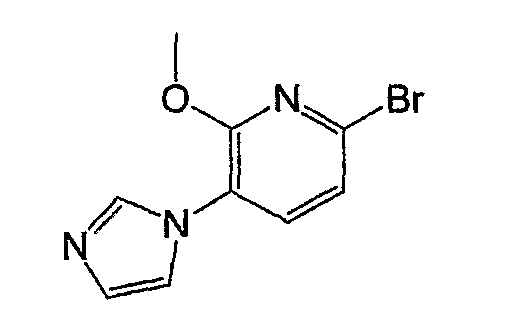
В соответствии со способом примера получения 1-1 257 мг указанного в заголовке соединения получали из N-(6-бром-2-метоксипиридин-3-ил)формамида.
ESI-MS: m/z 254 [M++H].
Пример получения 1-12
Синтез 6-бром-2-метокси-3-(2-метил-1H-имидазол-1-ил)пиридина
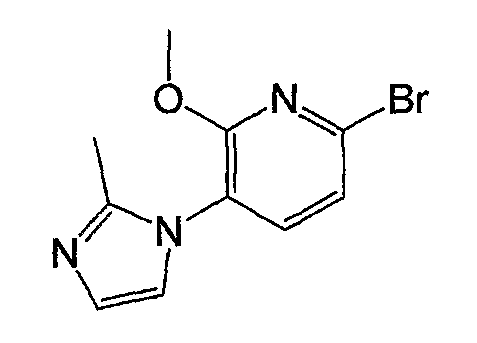
Синтез 6-бром-2-метоксипиридил-3-бороновой кислоты
К охлаждаемому льдом раствору диизопропиламина (777 мкл) в ТГФ (20 мл) добавляли н-бутиллитий (2,6 M гексановый раствор) и смесь перемешивали при той же температуре в течение 10 минут. Раствор охлаждали до -78°C, после чего добавляли по каплям 2-бром-6-метоксипиридин (CAS #40473-07-2, 799 мг) и триизопропилборат (1,27 мл). Температуру реакции повышали до комнатной температуры и перемешивали в течение часа. К реакционной смеси добавляли раствор хлорида аммония и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом натрия. Осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Оставшиеся кристаллы собирали фильтрованием и промывали диэтиловым эфиром с получением указанного в заголовке соединения (545 мг).
Синтез 6-бром-2-метокси-3-(2-метил-1H-имидазол-1-ил)пиридина
К раствору 6-бром-2-метоксипиридил-3-бороновой кислоты (1 г) и 2-метил-1H-имидазола (354 мг) в дихлорметане (20 мл) добавляли хлорид ди-µ-гидроксо-бис[(N,N,N′,N′-тетраметилэтилендиамин)меди(II)] (300 мг). Реакционную смесь перемешивали в атмосфере кислорода при комнатной температуре в течение 3 суток. Смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (311 мг).
Соединения от примера получения 1-13 до примера получения 1-15 получали в соответствии со способами получения примера 1-7. (Таблица 11)
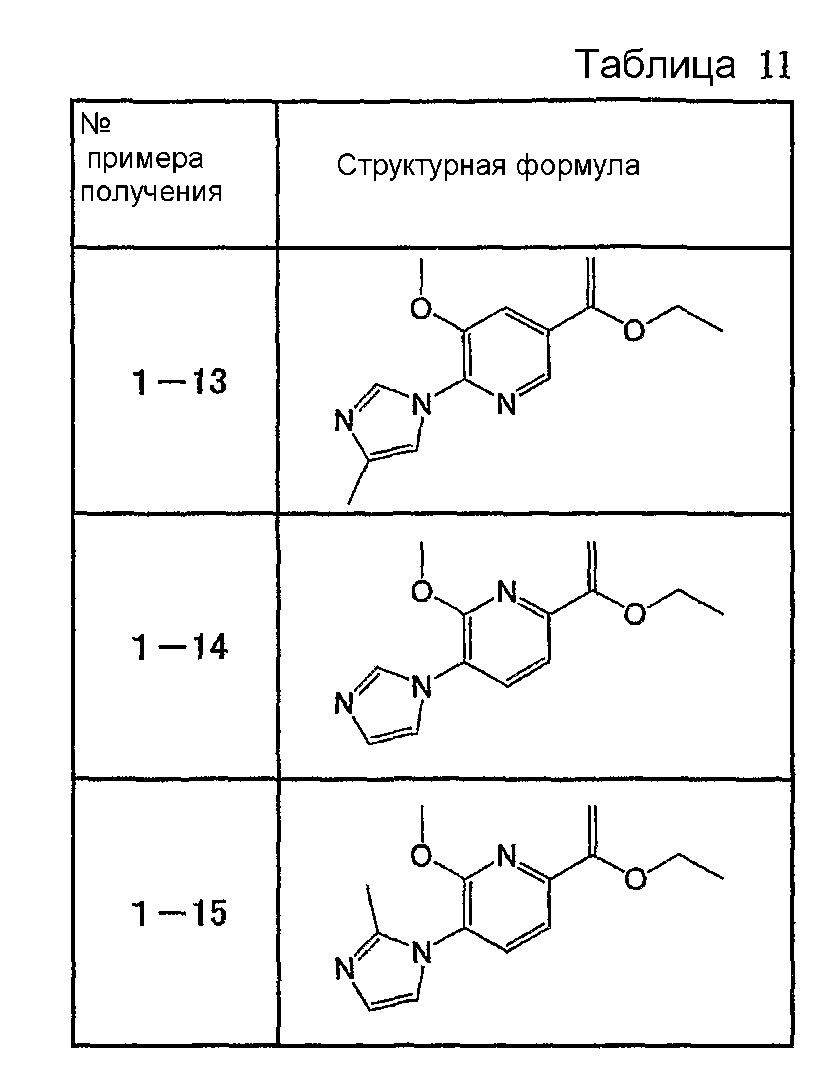
Пример получения 3-21
Синтез гидрохлорида сложного этилового эфира 2-[2-бром-5-(цианодиметилметил)фенил-5-хлорпентанимидной кислоты
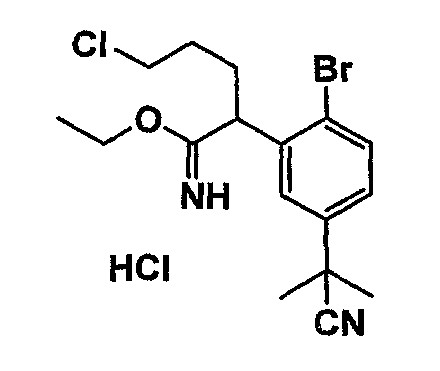
Синтез 2-(4-бром-3-метилфенил)-2-метилпропионитрила
К охлаждаемой льдом суспензии гидрида натрия (60% в масле, 524 мг) в ТГФ (5 мл) и N-метилпирролидоне (5 мл) добавляли раствор (4-бром-3-метилфенил)ацетонитрила (CAS #215800-25-2, 1,1 г) и йодметана (1 мл) в ТГФ (3 мл) и N-метилпирролидоне (3 мл). После перемешивания в течение 40 минут при комнатной температуре добавляли воду и трет-бутилметиловый эфир. Отделяли органический слой и водный слой экстрагировали трет-бутилметиловым эфиром. Объединенный органический слой промывали водой (два раза) и насыщенным раствором соли. После сушки над безводным сульфатом магния осушающий агент отделяли фильтрованием и затем органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel™ C-200; элюирующий растворитель: гептан:этилацетат = 19:1) с получением указанного в заголовке соединения (1,06 г). Показатели свойств соединения были следующими.
1Н-ЯМР (CDCl3) δ (ppm): 1,71 (с, 6Н), 2,43 (с, 3Н), 7,14 (дд, J=8,4, 2,4 Гц, 1Н), 7,34 (д, J=2,4 Гц, 1Н), 7,53 (д, J=8,4 Гц, 1Н).
Синтез 2-(4-бром-3-цианометилфенил)-2-метилпропионитрила
N-бромсукцинимид (1,04 г) и 2,2′-азобис(изобутилнитрил) (30 мг) добавляли к тетрахлоридуглеродному раствору (7 мл) 2-(4-бром-3-метилфенил)-2-метилпропионитрила (1,26 г) и затем раствор кипятили с обратным холодильником в течение часа. Раствор охлаждали на льду, после чего, удалив нерастворимые вещества фильтрованием, фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в этаноле (10 мл) и воде (1 мл) и к раствору добавляли раствор цианида калия (0,41 г). Реакционную смесь перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли охлаждаемую льдом воду и этилацетат и отделяли органический слой. Полученный органический слой промывали последовательно водой (два раза) и насыщенным раствором соли. После сушки над безводным сульфатом магния органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel™ C-200; элюирующий растворитель: гептан:этилацетат, 19:1 → 5:1) с получением указанного в заголовке соединения (917 мг). Показатели свойств полученного соединения были следующими:
1H-ЯМР (CDCl3) δ (ppm): 1,74 (с, 6H), 3,87 (с, 2H), 7,38 (дд, J=8,4, 2,4 Гц, 1H), 7,57 (д, J=2,4 Гц, 1H), 7,64 (д, J=8,4 Гц, 1H).
Синтез 2-[2-бром-5-(цианодиметилметил)фенил]-5-хлорпентанитрила
В условиях охлаждения льдом к смешанному раствору 2-(4-бром-3-цианометилфенил)-2-метилпропионитрила в ТГФ (5 мл) и ДМФА (5 мл) добавляли гидрид натрия (60% в масле, 144 мг) и реакционную смесь перемешивали в течение 10 минут при той же самой температуре. К реакционной смеси добавляли 1-бром-3- хлорпропан (0,44 мл) и смесь перемешивали в течение 25 минут при той же самой температуре. Добавляли к реакционной смеси охлаждаемую льдом воду и трет-бутилметиловый эфир и затем отделяли органический слой. Водный слой повторно экстрагировали трет-бутилметиловым эфиром. Объединенный органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния.
После концентрирования полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel™ C-200, элюирующий растворитель: гептан:этилацетат, 19:1 → 5:1) с получением указанного в заголовке соединения (941 мг). Показатели свойств полученного соединения были следующими:
1Н-ЯМР (CDCl3) δ (ppm): 1,75 (с, 6Н), 1,87-2,18 (м, 4Н), 3,61 (т, J=6,0 Гц, 2Н), 4,31-4,36 (м, 1Н), 7,38 (дд, J=8,4, 2,4 Гц, 1Н), 7,60 (д, J=2,4 Гц, 1Н), 7,63 (д, J=8,4 Гц, 1Н).
Синтез гидрохлорида этил 2-[2-бром-5-(цианодиметилметил)фенил]-5-хлорпентанимидата
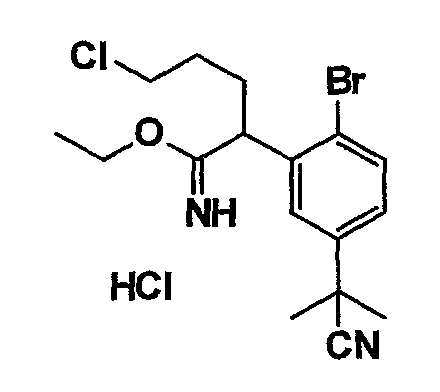
В условиях охлаждения льдом раствор 2-[2-бром-5-(цианодиметилфенил)]-5-хлорпентаннитрила (100 мг) в этаноле (4 мл) барботировали газообразным хлороводородом в течение 10 минут. После того как реакционный раствор перемешивали при комнатной температуре в течение 2 часов, раствор концентрировали при пониженном давлении. К остатку добавляли трет-бутилметиловый эфир и раствор концентрировали при пониженном давлении с получением указанного в заголовке соединения (124 мг). Показатель свойств полученного соединения был следующим:
ESI-MS: m/z 387 [M++H-HCl].
Соединения от примера получения 3-22 до примера получения 3-32 получали в соответствии со способами, описанными в примере получения 3-21.
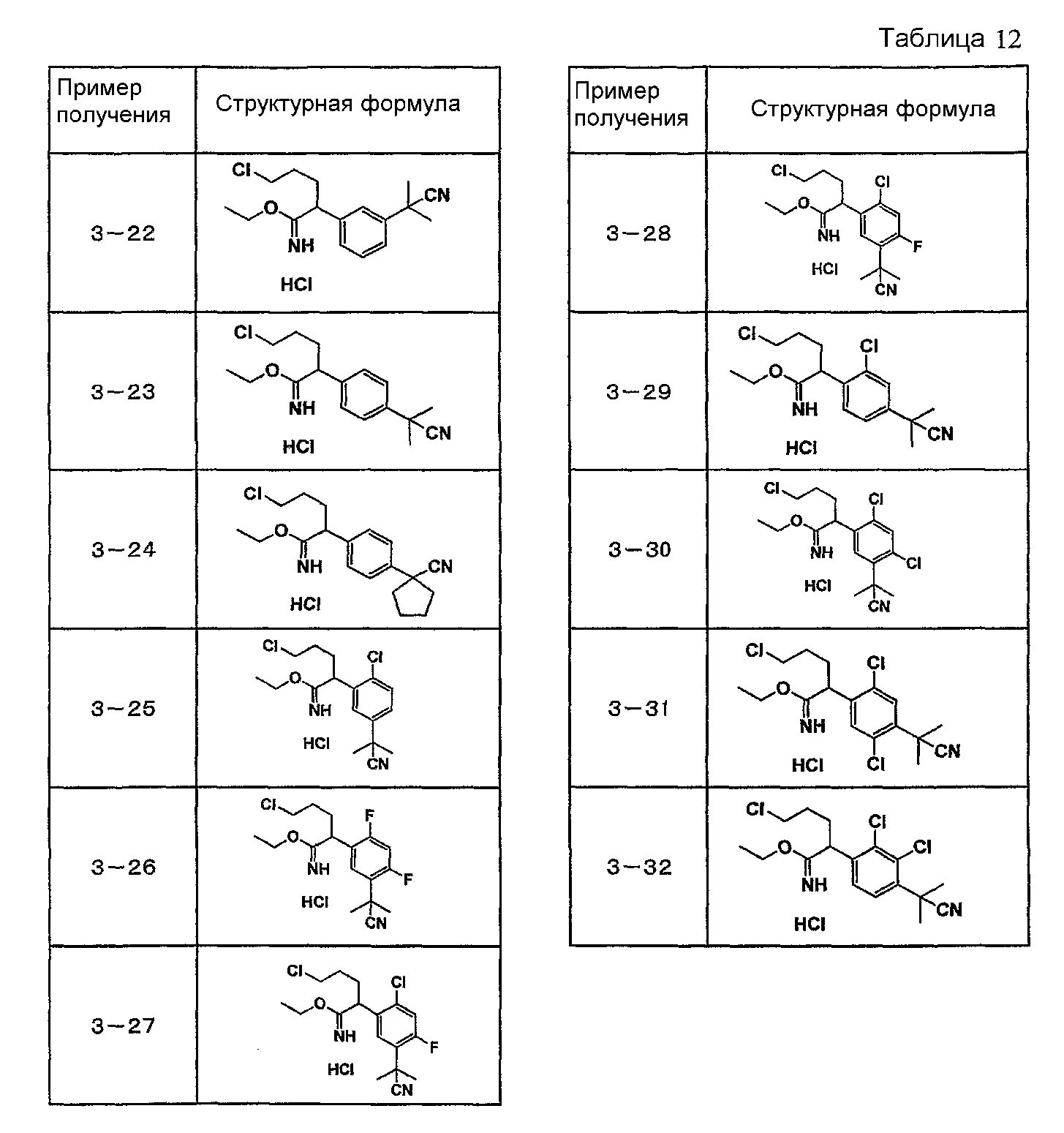
Соединения от примера получения 3-33 до примера получения 3-37 получали в соответствии со способами, описанными в примере получения 3-3.
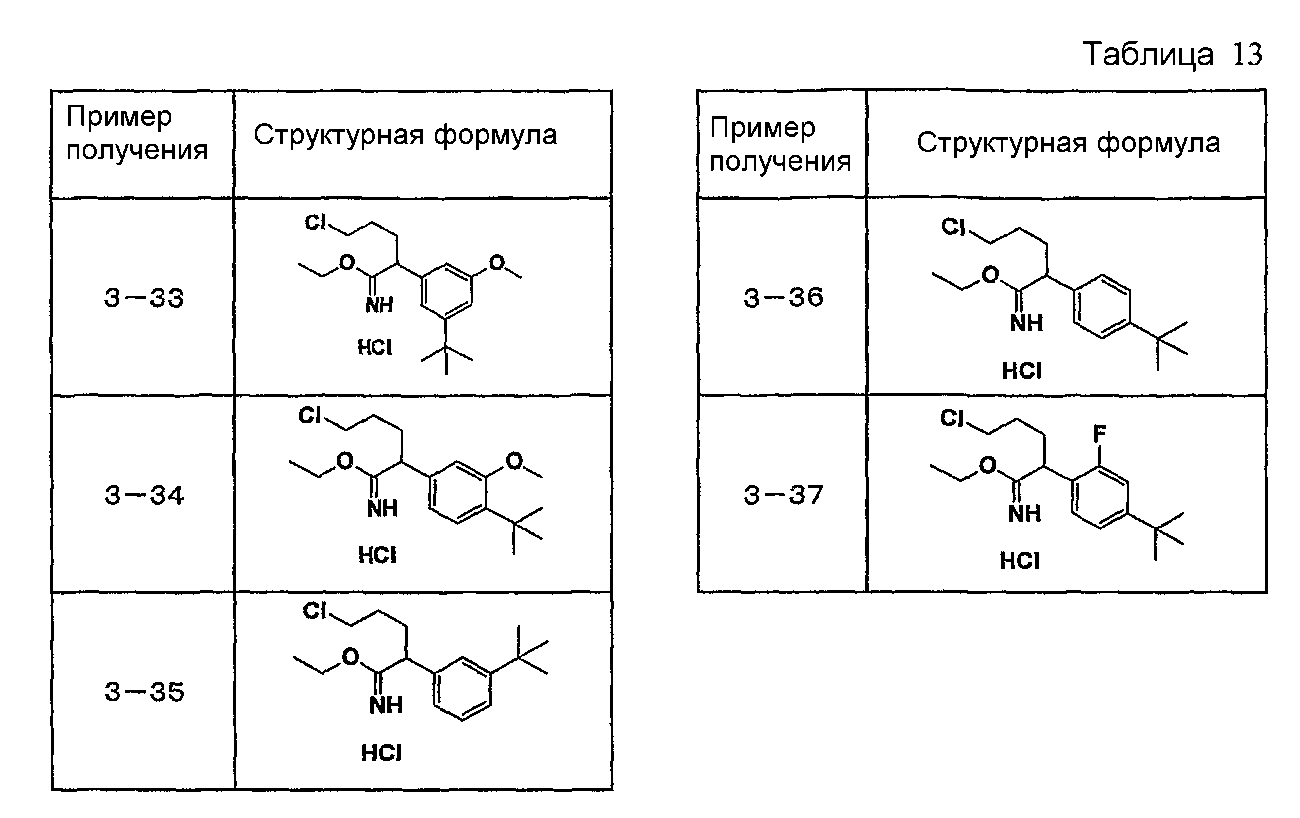
Соединения от примера получения 3-38 до примера получения 3-45 получали в соответствии со способами, описанными в примере получения 3-1.
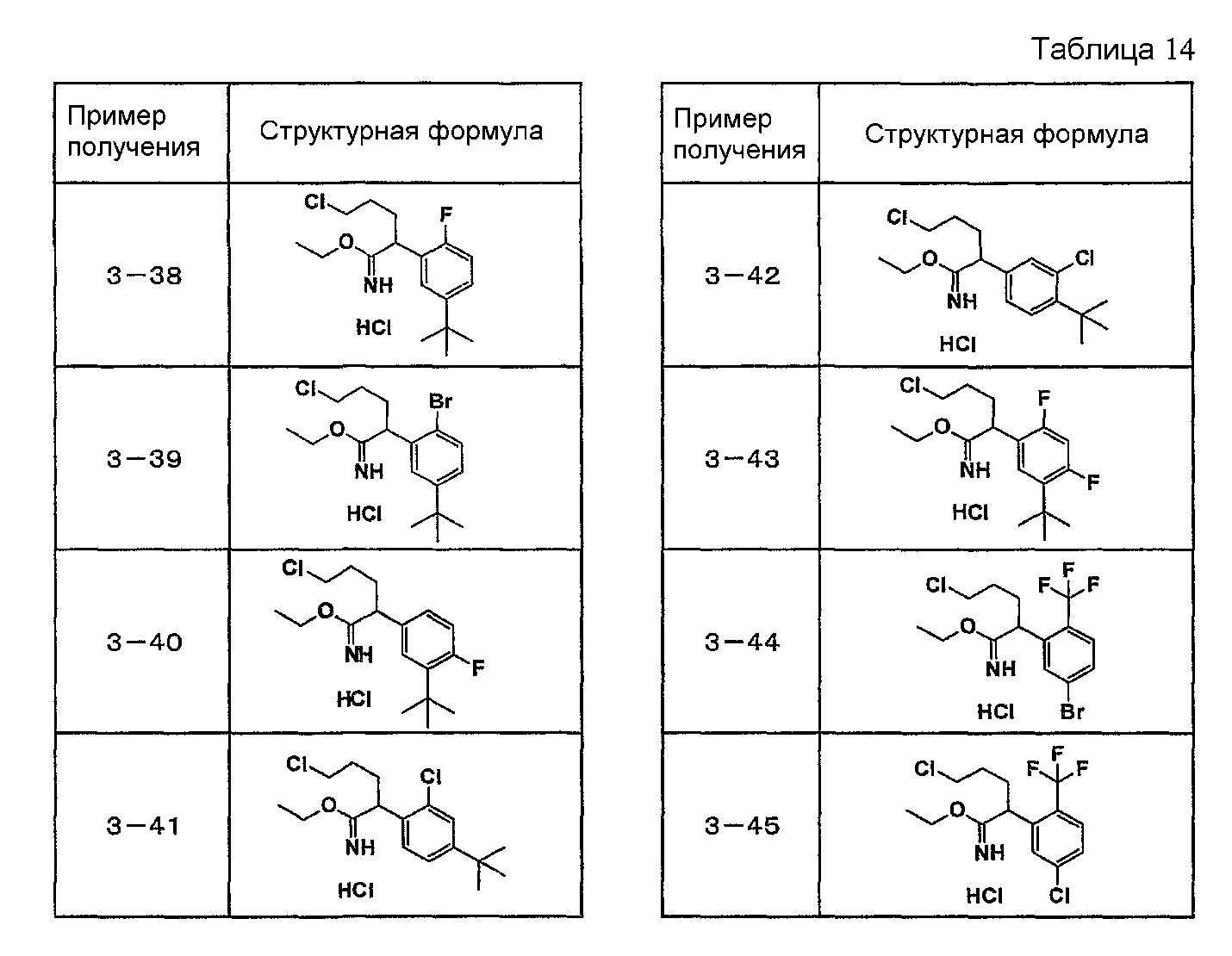
Пример получения соединения 3-46
Синтез 1-[2,4-дихлор-5-(4-хлор-1-цианобутил)фенил] циклопропанкарбонитрила
Синтез (2,4-дихлор-5-метилфенил)ацетонитрила
К раствору 1,5-дихлор-2-хлорметил-4-
метилбензола (CAS #101349-87-5, 5,22 г) в диметилсульфоксиде (20 мл) добавляли цианид калия (1,36 г) и реакционный раствор перемешивали при комнатной температуре в течение 6 часов. Добавляли к реакционному раствору цианид калия (0,68 г) и раствор еще перемешивали при комнатной температуре в течение ночи. К реакционному раствору добавляли охлаждаемую льдом воду и трет-бутилметиловый эфир и отделяли органический слой. Водный слой повторно экстрагировали трет-бутилметиловым эфиром. Объединенный органический слой промывали последовательно водой (два раза) и насыщенным раствором соли. После сушки над безводным сульфатом магния органический слой концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel™ C-200; элюирующий растворитель: гептан:этилацетат 49:1 → 19:1) с получением указанного в заголовке соединения (1,88 г). Показатели свойств полученного соединения были следующими:
1H-ЯМР (CDCl3) δ (ppm): 2,38 (с, 3H), 3,78 (с, 2H), 7,38 (с, 1H), 7,43 (с, 1H).
Синтез 1-(2,4-дихлор-5-метилфенил)циклопропанкарбонитрила
К смеси (2,4-дихлор-5-метилфенил)ацетонитрила (385 мг) и 1,2-дибромэтана (1,0 г) добавляли бензилтриэтиламмонийхлорид (44 мг) и 50% раствор гидроксида натрия (0,8 мл) и смесь перемешивали в течение ночи при комнатной температуре. Добавляли к реакционной смеси охлаждаемую льдом воду и трет-бутилметиловый эфир и отделяли органический слой. Полученный органический слой промывали последовательно водой (два раза) и насыщенным раствором соли. После сушки над безводным сульфатом магния органический слой концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (носитель: Wakogel™ C-200; элюирующий растворитель: гептан:этилацетат 49:1 → 19:1) с получением указанного в заголовке соединения (317 мг). Показатели свойств полученного соединения были следующими:
1H-ЯМР (CDCl3) δ (ppm): 1,32 (дд, J=7,6, 5,2 Гц, 2H), 1,72 (дд, J=7,6, 5,2 Гц, 2H), 2,34 (с, 3H), 7,20 (с, 1H), 7,43 (с, 1H).
Синтез 1-[2,4-дихлор-5-(4-хлор-1-цианобутил)фенил]циклопропанкарбонитрила
Подобно способам примера получения 3-21 указанное в заголовке соединение (113 мг) получали из 1-(2,4-дихлор-5-метилфенил)циклопропанкарбонитрила (317 мг). Показатели свойств полученного соединения были следующими:
1H-ЯМР (CDCl3) δ (ppm): 1,30-1,34 (м, 1H), 1,40-1,45 (м, 1H), 1,77-1,85 (м, 2H), 1,93-2,15 (м, 4H), 3,61 (т, J=5,6 Гц, 2H), 4,23-4,29 (м, 1H), 7,54 (с, 1H).
Пример получения 3-47
Синтез этил 5-хлор-2-[2-метокси-5-(2,2,2-трифтор-1,1-диметилэтил)фенил]пентанимидата
Синтез 1-(4-метокси-3-метилфенил)этанона
К раствору 4-гидрокси-3-метилацетофенона (CAS #876-02-8, 5 г) в ДМФА (30 мл) добавляли карбонат калия (9,21 г) и йодметан (6,14 г). Реакционный раствор перемешивали при комнатной температуре в течение 3 суток. Добавляли к реакционной смеси воду и гептан и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. После фильтрования для удаления осушающего агента органический слой концентрировали при пониженном давлении. Остаток растворяли в растворе в виде смеси гептана и этилацетата (гептан:этилацетат, 3:1) и раствор подвергали колоночной хроматографии на силикагеле. Раствор концентрировали при пониженном давлении с получением указанного в заголовке соединения (5,7 г). Показатели свойств соединения были следующими:
1H-ЯМР (CDCl3) δ (ppm): 2,25 (с, 3H), 2,55 (с, 3H), 3,90 (с, 3H), 6,85 (д, J=8,4 Гц, 1H), 7,78 (д, J=2,0 Гц, 1H), 7,83 (дд, J=8,4, 2,0 Гц, 1H).
Синтез триметил-[2,2,2-трифтор-1-(4-метокси-3-метилфенил)-1-метилэтокси]силана
К охлаждаемому льдом раствору 1-(4-метокси-3-метилфенил)этанона (1 г) в ДМФА (6 мл) добавляли ацетат лития (20,1 мг) и (трифторметил)триметилсилан (1,04 г). При той же самой температуре раствор перемешивали в течение 1,5 часа и затем перемешивали в течение 3,5 часа при комнатной температуре. Добавляли к реакционному раствору воду и гептан и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. После удаления осушающего агента органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: гептан → гептан:этилацетат 7:3) с получением указанного в заголовке соединения (711 мг). Показатели свойств полученного соединения были следующими:
1H-ЯМР (CDCl3) δ (ppm): 0,13 (с, 3H), 1,79 (с, 3H), 2,23 (с, 3H), 3,84 (с, 3H), 6,80 (д, J=8,8 Гц, 1H), 7,27 (д, J=2,4 Гц, 1H), 7,32 (дд, J=8,8, 2,4 Гц, 1H).
Синтез 1,1,1-трифтор-2-(4-метокси-3-метилфенил)пропан-2-ола
Триметил-[2,2,2-трифтор-1-(4-метокси-3-
метилфенил)-1-метилэтокси]силан (711 мг) растворяли в ТГФ (20 мл) и добавляли 1 н. HCl раствор (20 мл). После перемешивания при комнатной температуре в течение 6 суток добавляли к реакционной смеси воду и этилацетат и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. После удаления осушающего агента органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: гептан → гептан:этилацетат 7:3) с получением указанного в заголовке соединения (442 мг). Показатели свойств полученного соединения были следующими:
1H-ЯМР (CDCl3) δ (ppm): 1,76 (с, 3Н), 2,24 (с, 3Н), 3,84 (с, 3Н), 6,83 (д, J=8,4 Гц, 1Н), 7,34-7,37 (м, 2Н).
Синтез 1-метокси-2-метил-4-(2,2,2-трифтор-1,1-диметилэтил)бензола
К охлаждаемому льдом раствору 1,1,1-трифтор-2-(4-метокси-3-метилфенил)пропан-2-ола (440 мг) в дихлорметане (20 мл) добавляли тетрахлорид титана (357 мг). При той же самой температуре смесь перемешивали в течение 2 часов. Добавляли к органическому слою охлаждаемую льдом воду и дихлорметан и органический слой отделяли. Полученный органический слой промывали насыщенным раствором бикарбоната натрия и сушили над безводным карбонатом калия. После удаления осушающего агента органический слой концентрировали при пониженном давлении. После того как остаток растворяли в дихлорметане (15 мл), раствор охлаждали при -70°С и добавляли тетрахлорид титана (302 мг) и диметилцинк (1 М, н-гексановый раствор, 3,18 мл). Смесь доводили обратно до комнатной температуры и перемешивали в течение 3 часов. Добавляли к раствору охлаждаемую льдом воду и дихлорметан и отделяли органический слой. Полученный органический слой промывали насыщенным раствором бикарбоната натрия и затем сушили над безводным сульфатом магния. После удаления осушающего агента органический слой концентрировали при пониженном давлении.
Остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: гептан → гептан:этилацетат 9:1) с получением указанного в заголовке соединения (203 мг). Показатели свойств полученного соединения были следующими:
1H-ЯМР (CDCl3) δ (ppm): 1,54 (с, 6H), 2,23 (с, 3H), 3,83 (с, 3H), 6,80 (д, J=8,4 Гц, 1H), 7,25-7,29 (м, 2H).
Синтез гидрохлорида этил 5-хлор-2-[2-метокси-5-(2,2,2-трифтор-1-метокси-1-метилэтил)фенил]пентанимидата
В соответствии со способом примера получения 3-1 указанное в заголовке соединение получали из 1-метокси-2-метил-4-(2,2,2-трифтор-1,1-диметилэтил)бензола.
Пример получения 3-48
Синтез гидрохлорида этил 5-хлор-2-[2-метокси-5-(2,2,2-трифтор-1-метокси-1-метилэтил)фенил]пентанимидата
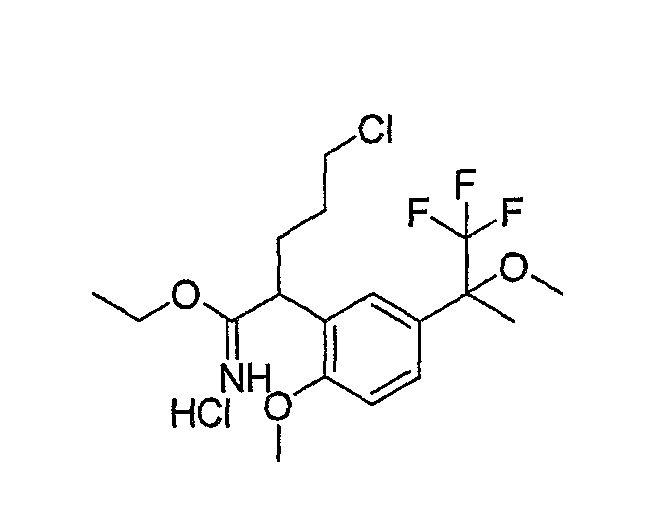
Синтез 1-метокси-2-метил-4-(2,2,2-трифтор-1-метокси-1-метилэтил)бензола
К раствору 1,1,1-трифтор-2-(4-метокси-3-метилфенил)пропан-2-ола (1 г) в ДМФА (20 мл) добавляли гидрид натрия (60% в минеральном масле, 205 мг) и йодметан (727 мг). После перемешивания в течение 5 часов при комнатной температуре реакционную смесь охлаждали льдом, после чего добавляли к реакционной смеси воду и этилацетат. Органический слой отделяли и промывали насыщенным раствором соли и сушили над безводным сульфатом магния. После удаления осушающего агента органический слой концентрировали при пониженном давлении.
Остаток очищали хроматографией на колонке с силикагелем (элюирующий растворитель: гептан → гептан:этилацетат 9:1) с получением указанного в заголовке соединения (747 мг). Показатели свойств полученного соединения были следующими:
1H-ЯМР (CDCl3) δ (ppm): 1,74 (с, 3H), 2,24 (с, 3H), 3,21 (с, 3H), 3,85 (с, 3H), 6,83 (д, J=8,4 Гц, 1H), 7,25-7,30 (м, 2H).
Синтез гидрохлорида этил 5-хлор-2-[2-метокси-5-(2,2,2-трифтор-1-метокси-1-метилэтил)фенил]пентанимидата
В соответствии со способом примера получения 3-1 указанное в заголовке соединение получали из 1-метокси-2-метил-4-(2,2,2-трифтор-1-метокси-1-метилэтил)бензола.
Пример получения 6-1
Синтез (2-фтор-5-пиперидин-1-илфенил)метанола
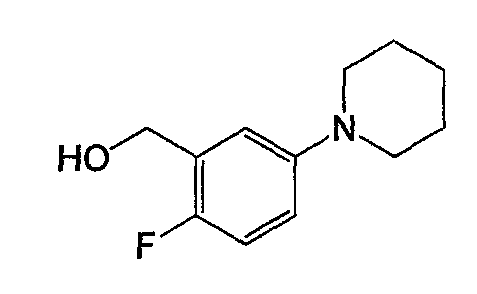
Синтез метил 2-фтор-5-нитробензоата
К охлаждаемому льдом раствору 2-фтор-5-нитробензойной кислоты (3 г) в метаноле (30 мл) добавляли по каплям тионилхлорид (2,32 мл). После перемешивания в течение ночи при комнатной температуре реакционную смесь концентрировали при пониженном давлении. К остатку добавляли этилацетат и водный раствор бикарбоната натрия и отделяли органический слой. Полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. После удаления осушающего агента органический слой концентрировали при пониженном давлении с получением указанного в заголовке соединения (3,28 г).
Синтез метил 5-амино-2-фторбензоата
К раствору метил 2-фтор-5-нитробензоата (2 г) в метаноле (80 мл) добавляли 6 гидратный дихлорид никеля(II) (475 мг) и реакционную смесь охлаждали до -30°С. При той же самой температуре добавляли порциями боргидрид натрия (1,13 г). Раствор подогревали до -20°С и перемешивали в течение 20 минут, после чего добавляли водный раствор карбоната калия. После удаления нерастворимого вещества фильтрованием через целит фильтрат концентрировали при пониженном давлении для удаления метанола. Полученный остаток экстрагировали этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом натрия. После удаления осушающего агента фильтрованием органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,5 г).
Синтез метил 2-фтор-5-(пиперидин-1-ил)бензоата
Метил 5-амино-2-фторбензоат (1,5 г), 1,5-дибромпентан (5,93 мл) и IPEA (7,66 мл) растворяли в толуоле (30 мл) и смесь перемешивали при 100°С в течение 15 часов. После охлаждения добавляли к раствору этилацетат и отделяли органический слой. Органический слой промывали водой, насыщенным раствором соли и сушили над безводным сульфатом натрия. После удаления осушающего агента фильтрованием органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (2,12 г). Показатель свойств полученного соединения был следующим
ESI-MS: m/z 238 [M++H].
Синтез (2-фтор-5-пиперидин-1-ил)фенилметанола
Метил 2-фтор-5-(пиперидин-1-ил)бензоат (2,12 г) добавляли по каплям к суспензии литийалюминийгидрида (372 мг) в ТГФ (30 мл) при охлаждении льдом. По окончании добавления смесь подогревали до комнатной температуры и перемешивали в течение 1 часа. Добавляли к смеси последовательно воду (372 мл), 5 н. водный раствор гидроксида натрия (372 мл) и воду (1,12 мл) при охлаждении льдом. Осадок удаляли фильтрованием через целит и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,88 г).
ESI-MS: m/z 210 [M++H].
Соединения от примера получения 6-2 до примера получения 6-10 получали в соответствии со способом примера получения 6-1 (таблица 15).
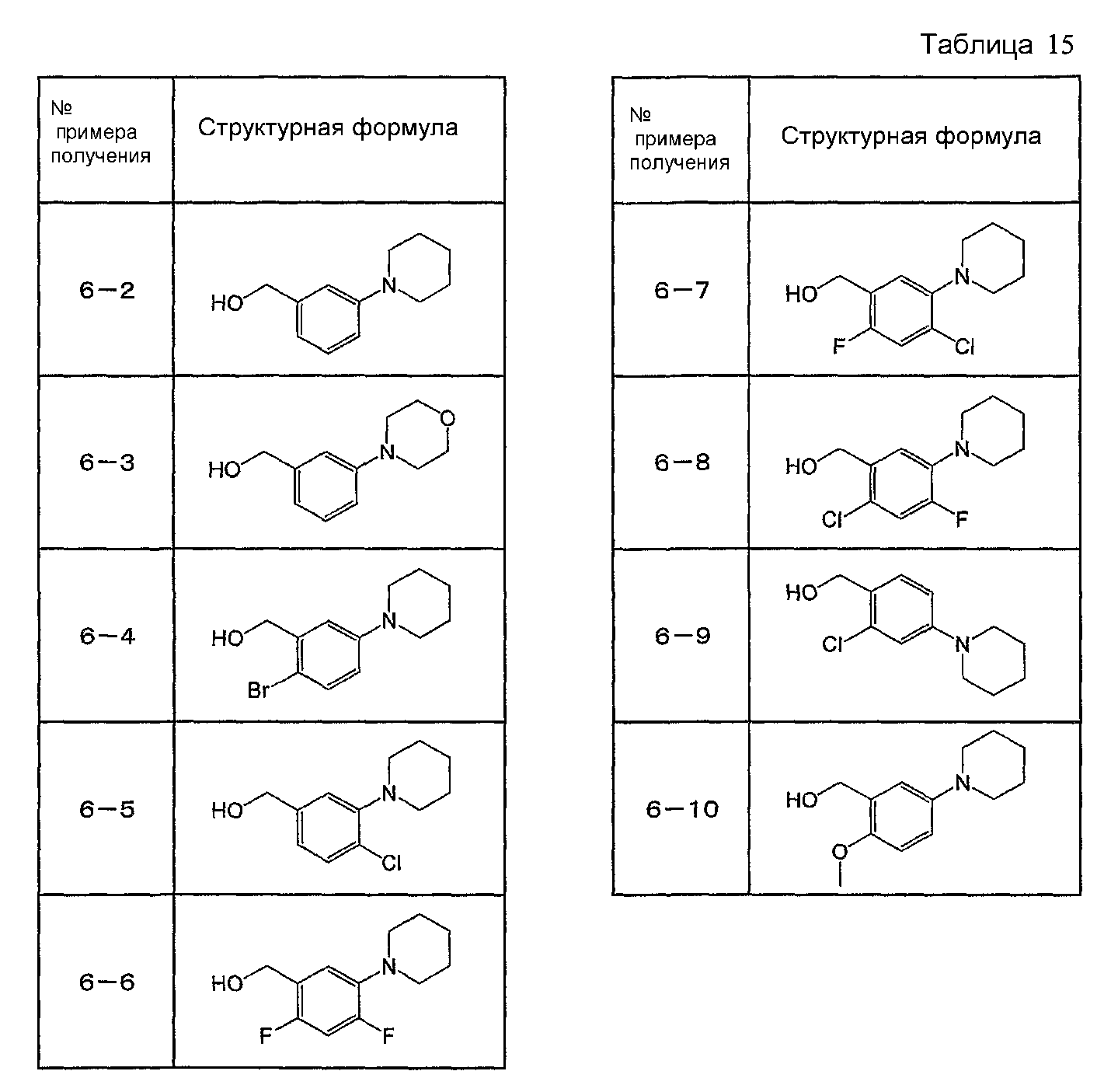
Пример получения 6-11
Синтез (3-пирролидин-1-ил)фенилметанола
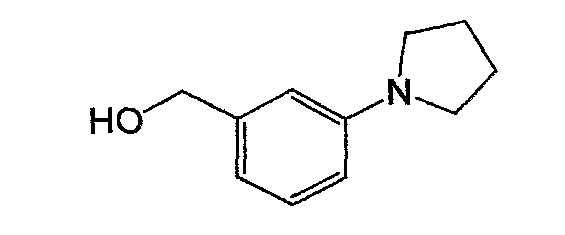
Синтез 1-[3-(трет-бутилдиметилсилилоксиметил)фенил]пирролидина
Трис(дибензилиденацетон)дипалладий (152 мг) добавляли к суспензии (3-бромбензилокси)-трет-бутилдиметилсилана (1,00 г), полученного в примере получения 7-2, пирролидина (416 мкл), BINAP (310 мг) и трет-бутоксида натрия (348 мг) в толуоле (20 мл) в атмосфере азота. Смесь нагревали при 80°C при перемешивании в течение 3 часов и затем охлаждали до комнатной температуры. Смесь разбавляли этилацетатом, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (913 мг).
ESI-MS: m/z 292 [M++H].
Синтез (3-пирролидин-1-ил)фенилметанола
Тетрабутиламмонийфторид (1,0 М раствор в ТГФ, 3,8 мл) добавляли к раствору 1-[3-(трет-бутилдиметилсилилоксиметил)фенил]пирролидина в ТГФ (20 мл) и смесь перемешивали при комнатной температуре в течение 3,5 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (557 мг).
ESI-MS: m/z 178 [M++H].
Соединения примеров получения 6-12 - 6-16 получали в соответствии со способом примера получения 6-11 (таблица 16).
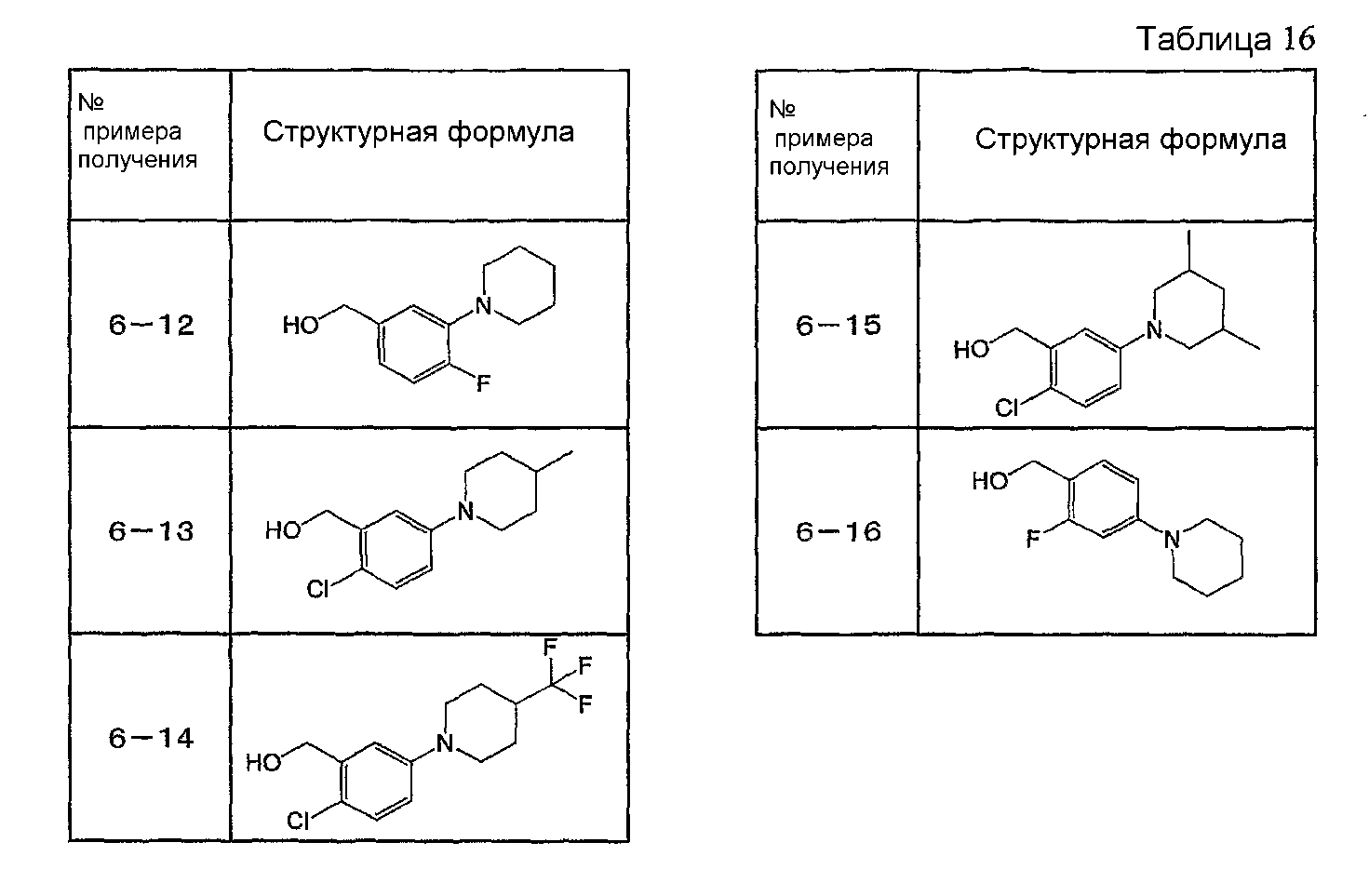
Пример получения 6-17
Синтез [5-(4,4-дифторпиперидин-1-ил)-2-фторфенил]метанола
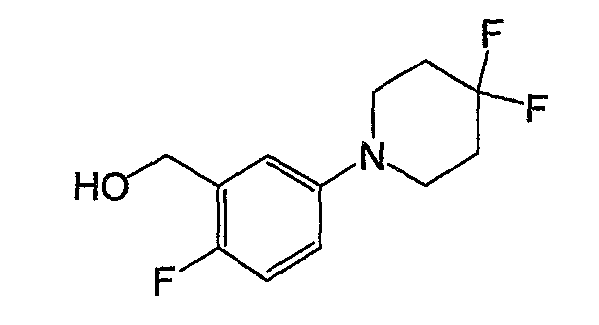
Синтез иодида 1-бензил-1-метил-4-оксопиперидиния
Йодметан (20,2 мл) добавляли к раствору 1-бензил-4-пиперидона (50 мл) в ацетоне (300 мл) и смесь перемешивали при комнатной температуре в течение ночи. Выпавшие в осадок кристаллы собирали и промывали ацетоном с получением указанного в заголовке соединения (77,1 г).
Синтез метил 2-фтор-5-(4-оксопиперидин-1-ил)бензоата
Карбонат калия (149 мг) и иодид 1-бензил-1-метил-4-оксопиперидиния (2,87 г) последовательно добавляли к раствору метил 5-амино-2-фторбензоата (1,22 г), полученного в примере получения 6-1, в смешанном растворителе из этанола (10 мл) и воды (5 мл). Реакционную смесь кипятили с обратным холодильником в течение 2,5 часов. Смесь разбавляли водой и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,22 г).
ESI-MS: m/z 252 [M++H].
Синтез метил 5-(4,4-дифторпиперидин-1-ил)-2-фторбензоата
Диэтиламиносератрифторид (1,72 мл) добавляли к раствору метил 2-фтор-5-(4-оксопиперидин-1-ил)бензоата (1,22 г) в дихлорметане (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часа. Добавляли к реакционной смеси насыщенный водный раствор бикарбоната натрия и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (878 мг).
ESI-MS: m/z 274 [M++H].
Синтез [5-(4,4-дифторпиперидин-1-ил)-2-фторфенил]метанола
В соответствии со способом примера получения 6-1 указанное в заголовке соединение (761 мг) получали из метил 5-(4,4-дифторпиперидин-1-ил)-2-фторбензоата (878 мг).
ESI-MS: m/z 246 [M++H].
Соединения примеров получения 6-18 и 6-19 получали в соответствии со способом примера получения 6-17 (таблица 17).
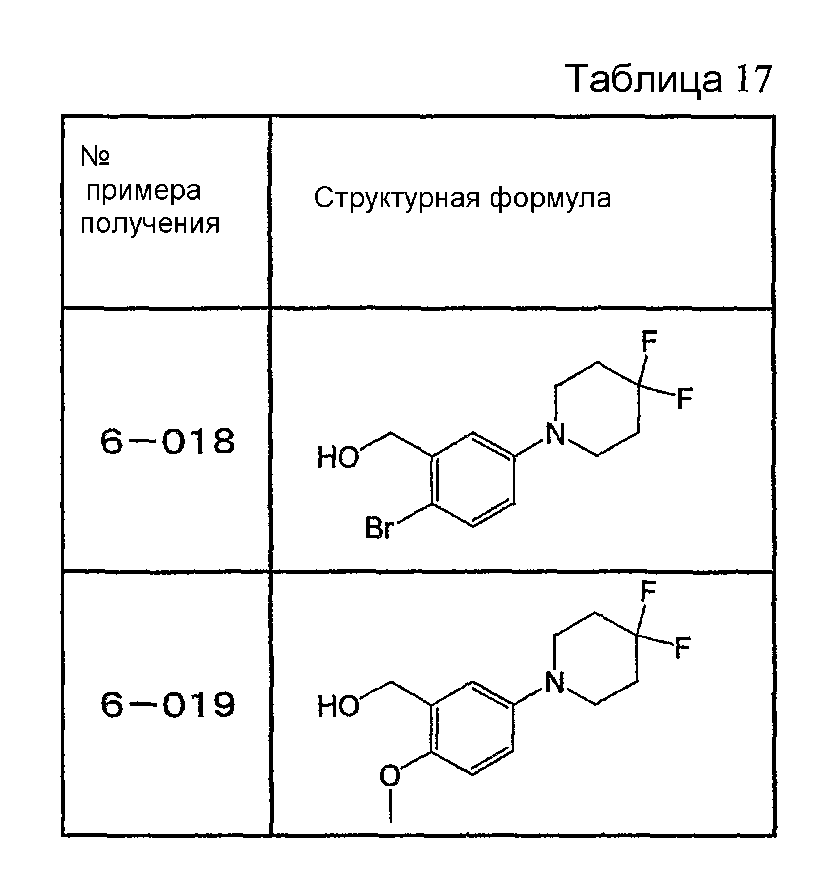
Пример получения 6-20
Синтез [(3-бром-4-пиперидин-1-ил)фенил]метанола
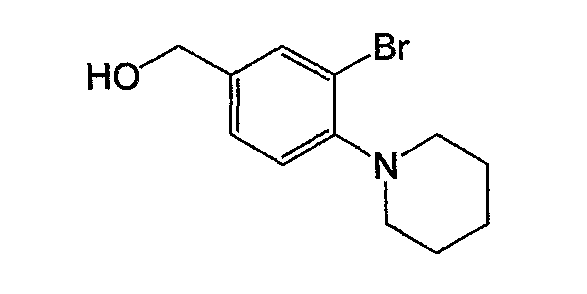
Синтез метил 4-фтор-3-нитробензоата
В соответствии со способом примера получения 6-1 указанное в заголовке соединение (2,19 г) получали из 4-фтор-3-нитробензойной кислоты (2,00 г).
Синтез метил 3-нитро-4-(пиперидин-1-ил)бензоата
Пиперидин (1,24 мл) добавляли к раствору метил 4-фтор-3-нитробензоата (1,00 г) в ДМФА (20 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь разбавляли этилацетатом, промывали последовательно водой и насыщенным раствором соли и сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,50 г).
ESI-MS: m/z 265 [M++H].
Синтез метил 3-амино-4-(пиперидин-1-ил)бензоата
Используя картридж с 10% палладием-на-углероде, раствор метил 3-нитро-4-(пиперидин-1-ил)бензоата (3,07 г) в метаноле (50 мл) прокачивали через реактор H-Cube™ (ThalesNano Inc.). После того как всю реакционную смесь пропускали через реактор H-Cube™, реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH силикагелем с получением указанного в заголовке соединения (2,32 г).
ESI-MS: m/z 235 [M++H].
Синтез метил 3-бром-4-(пиперидин-1-ил)бензоата
Изоамилнитрит (439 мкл) добавляли к смеси метил 3-амино-4-(пиперидин-1-ил)бензоата (500 мг) и бромида меди (II) (714 мг) в ацетонитриле (25 мл). Реакционную смесь перемешивали при 70°C в течение 30 минут. Смесь разбавляли этилацетатом, промывали последовательно раствором аммония и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (158 мг).
Синтез (3-бром-4-пиперидин-1-ил)фенилметанола
В соответствии со способом примера получения 6-1 указанное в заголовке соединение (488 мг) получали из метил 3-бром-4-(пиперидин-1-ил)бензоата (546 мг).
Соединения примера получения 6-21 и примера получения 6-22 получали в соответствии со способом примера получения 6-20 (таблица 18).
|
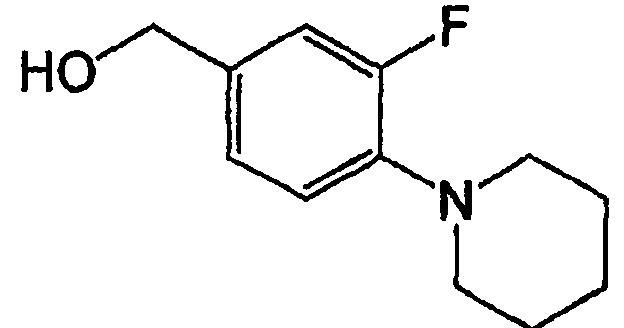
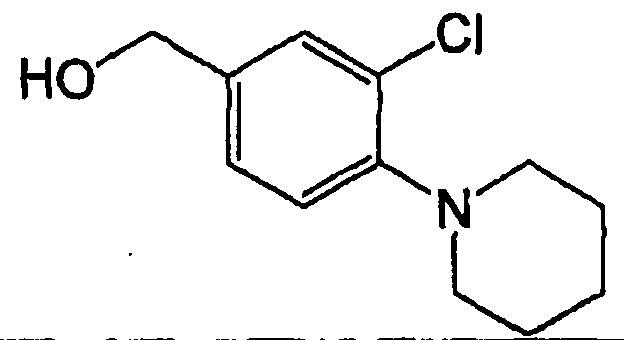
Пример получения 6-23
Синтез {2-хлор-4-[циклопропилметил(2,2,2-трифторэтил)амино]фенил}метанола
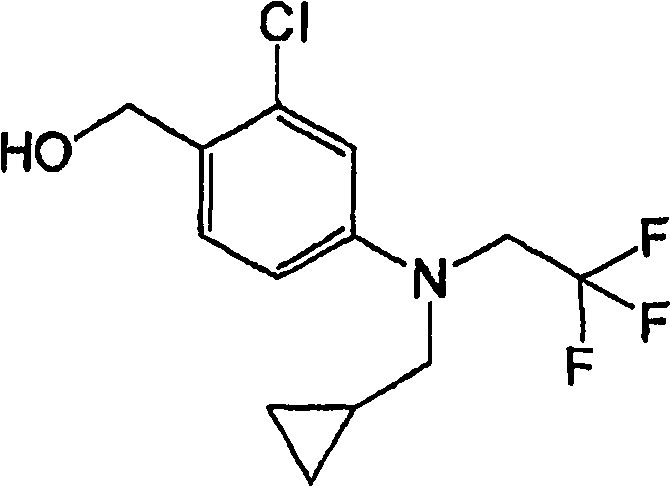
Синтез метил 2-хлор-4-фторбензоата
В соответствии со способом примера получения 6-1 указанное в заголовке соединение (4,37 г) получали из 2-хлор-4-фторбензойной кислоты (4,00 г).
Синтез метил 2-хлор-4-(циклопропилметиламино)бензоата
Циклопропилметиламин (1,51 г) и карбонат калия (1,46 г) добавляли к раствору метил 2-хлор-4-фторбензоата (1,00 г). Реакционную смесь перемешивали при комнатной температуре в течение 6 часов и затем нагревали при перемешивании при 100°C в течение 6 часов под действием микроволнового реактора. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,23 г).
ESI-MS: m/z 240 [M++H].
Синтез метил 2-хлор-4-[циклопропилметил(2,2,2-трифторацетил)амино]бензоата
Трифторуксусный ангидрид (1,18 мл) добавляли к раствору метил 2-хлор-4-(циклопропилметиламино)бензоата (1,00 г) и пиридина (686 мкл) в дихлорметане (40 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,42 г).
Синтез {2-хлор-4-[циклопропилметил(2,2,2-трифторэтил)амино]фенил}метанола
Комплекс боран-ТГФ (1,0 М раствор в ТГФ, 13,7 мл) добавляли при охлаждении льдом к раствору метил 2-хлор-4-[циклопропилметил(2,2,2-трифторацетил)амино]бензоата (500 мг) в дихлорметане (10 мл). Реакционную смесь затем кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры смесь разбавляли водой и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом натрия.
Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (257 мг).
ESI-MS: m/z 294 [M++H].
Соединение примера получения 6-24 получали в соответствии со способом примера получения 6-23 (таблица 19).
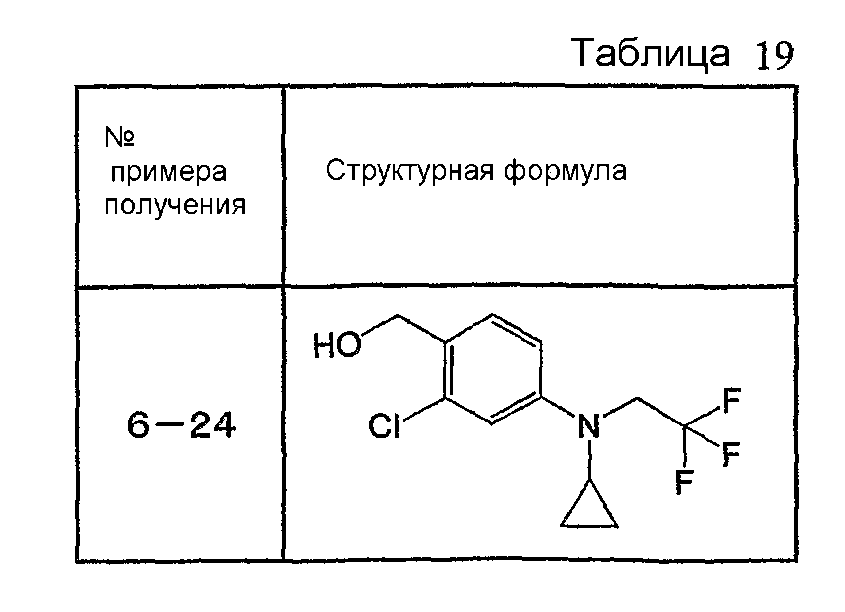
Пример получения 6-25
Синтез [5-(1-фторциклопропил)-2-метоксифенил]метанола
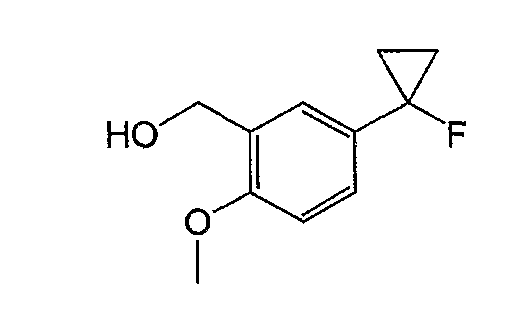
Синтез 1-(3-бром-4-метоксифенил)циклопропанола
Этилмагнийбромид (3 М раствор в диэтиловом эфире, 54,4 мл) добавляли при комнатной температуре к раствору метил 3-бром-4-метоксибензоата (CAS №35450-37-4, 10 г) и изопропоксида титана(IV) (23,9 мл) в ТГФ (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли к реакционной смеси этилацетат и воду и осадок удаляли через целит. Органический слой отделяли, промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (7,77 г).
Синтез [1-(3-бром-4-метоксифенил)циклопропилокси]триметилсилана
Триметилсилилхлорид (6,54 мл) добавляли к раствору 1-(3-бром-4-метоксифенил)циклопропанола (5,0 г) и триэтиламина (8,61 мл) в ТГФ (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 суток. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,84 г).
Синтез 2-бром-4-(1-фторциклопропил)-1-метоксибензола
Диэтиламиносератрифторид (2,4 мл) добавляли к раствору [1-(3-бром-4-метоксифенил)циклопропилокси]триметилсилана (3,84 г) в дихлорметане (68 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2,5 часов. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,88 г).
Синтез 5-(1-фторциклопропил)-2-метоксибензальдегида
н-Бутиллитий (2,7 М раствор в гексане, 3,42 мл) добавляли при -78°C к раствору 2-бром-4-(1-фторциклопропил)-1-метоксибензола (1,88 г) в ТГФ (62 мл), после чего добавляли раствор ДМФА (650 мкл) в ТГФ. После того как реакционную смесь перемешивали при -78°C в течение 30 минут, добавляли к реакционной смеси насыщенный водный раствор бикарбоната натрия и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,00 г).
Синтез [5-(1-фторциклопропил)-2-метоксифенил]метанола
Боргидрид натрия (194 мг) добавляли при охлаждении льдом к раствору 5-(1-фторциклопропил)-2-метоксибензальдегида (500 мг) в метаноле (10 мл). После того как реакционную смесь перемешивали при комнатной температуре в течение 2 часов, добавляли к реакционной смеси насыщенный водный раствор бикарбоната натрия и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (400 мг).
Пример получения 7-1
Синтез (5-бром-2-хлорбензилокси)трет-бутилметилсилана
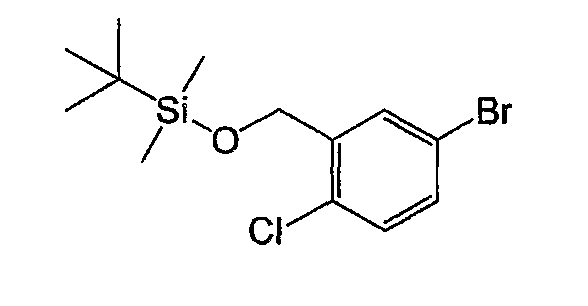
Синтез метил 5-бром-2-хлорбензоата
В соответствии со способом примера получения 6-1 указанное в заголовке соединение (9,23 г) получали из 5-бром-2-хлорбензойной кислоты (10,0 г).
Синтез (5-бром-2-хлорфенил)метанола
Раствор метил 5-бром-2-хлорбензоата (9,23 г) в ТГФ (50 мл) добавляли по каплям при охлаждении льдом к суспензии литийалюминийгидрида (1,42 г) в ТГФ (100 мл). По окончании добавления реакционную смесь перемешивали при комнатной температуре в течение 45 минут. Добавляли к реакционной смеси при охлаждении льдом последовательно воду (1,4 мл), 5 н. раствор гидроксида натрия (1,4 мл) и воду (4,2 мл). Осадок удаляли через целит и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (7,56 г).
Синтез (5-бром-2-хлорбензилокси)трет-бутилдиметилсилана
Имидазол (5,22 г) и трет-бутилдиметилсилилхлорид (8,64 г) добавляли к раствору (5-бром-2-хлорфенил)метанола (8,47 г) в ДМФА (80 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли этилацетатом и промывали два раза водой. Водный слой опять экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (12,6 г).
Соединения примеров получения 7-2 - 7-4 получали в соответствии со способом примера получения 7-1 (таблица 20).
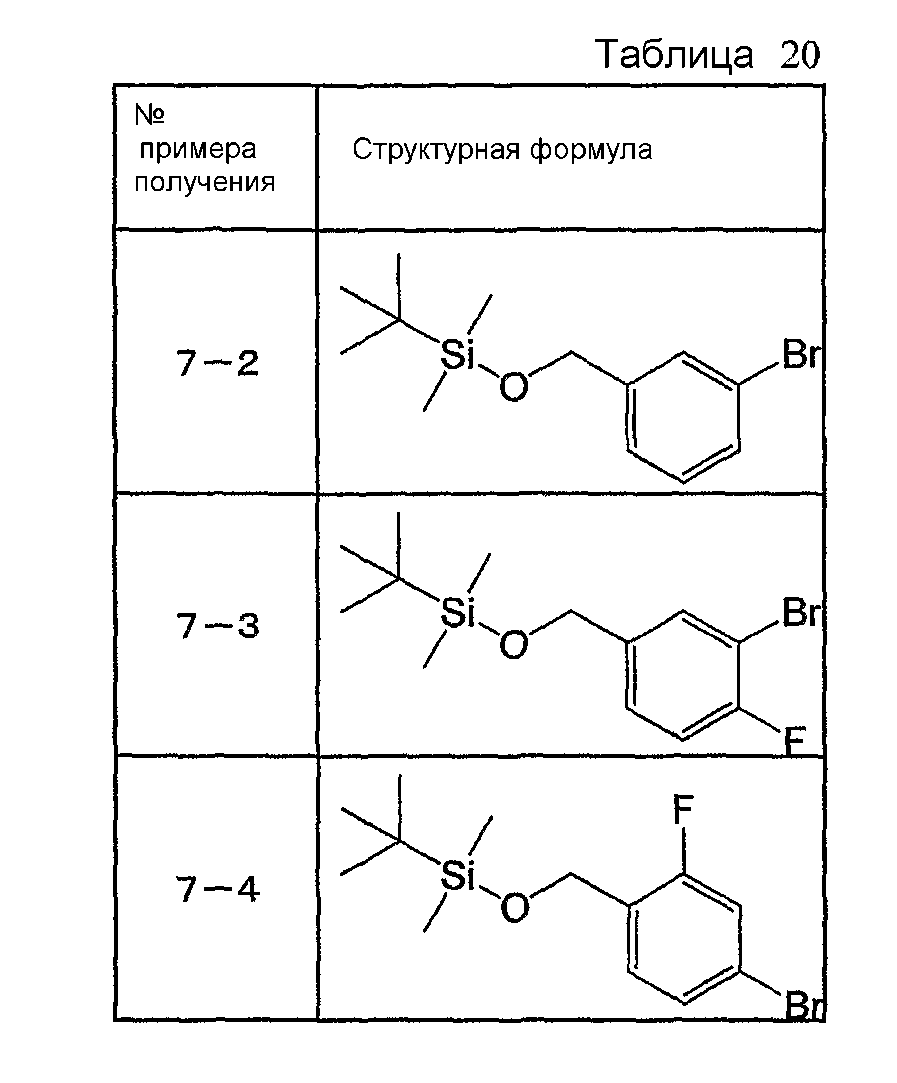
Пример получения 8-1
Синтез [(2-фтор-5-пиперидин-1-ил)фенил]ацетонитрила
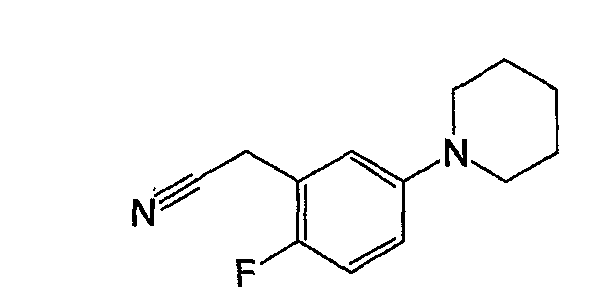
Тионилхлорид (1,05 мл) добавляли по каплям к раствору (2-фтор-5-пиперидин-1-ил)фенилметанола (1,0 г), полученного в примере получения 6-1, в толуоле (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 8,5 часов. Реакционную смесь разбавляли трет-бутилметиловым эфиром и промывали с 1 н. раствором гидроксида натрия и насыщенным водным раствором бикарбоната натрия. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и затем фильтрат концентрировали при пониженном давлении. К раствору полученного остатка в ДМФА (10 мл) добавляли цианид калия (623 мг). После перемешивания при комнатной температуре в течение 12 часов к реакционной смеси добавляли трет-бутилметиловый эфир и воду. Органический слой отделяли, промывали водой и насыщенным раствором соли и сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (908 мг).
ESI-MS: m/z 219 [M++H].
Соединения примеров получения 8-2 - 8-19 получали в соответствии со способом примера получения 8-1 (таблица 21).
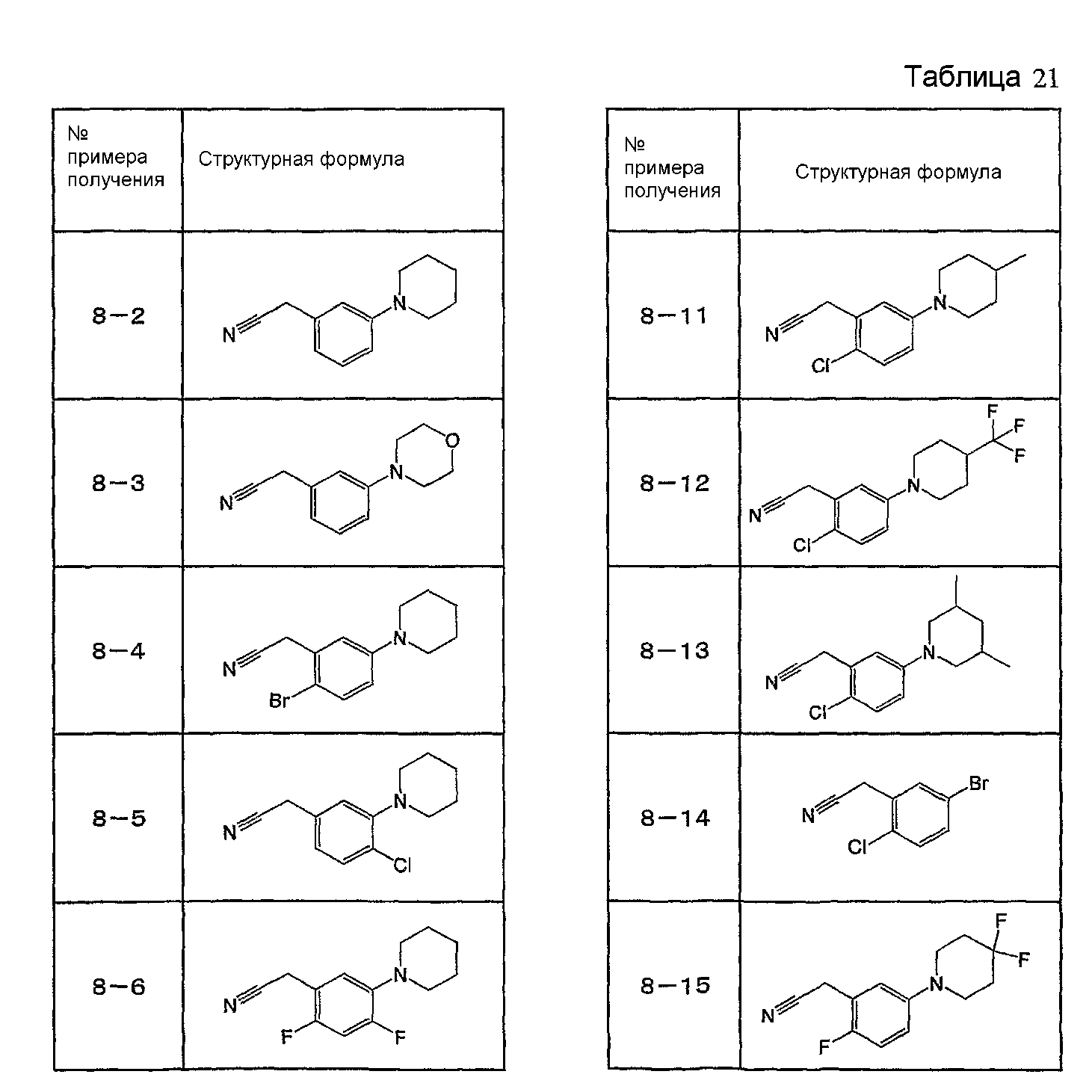
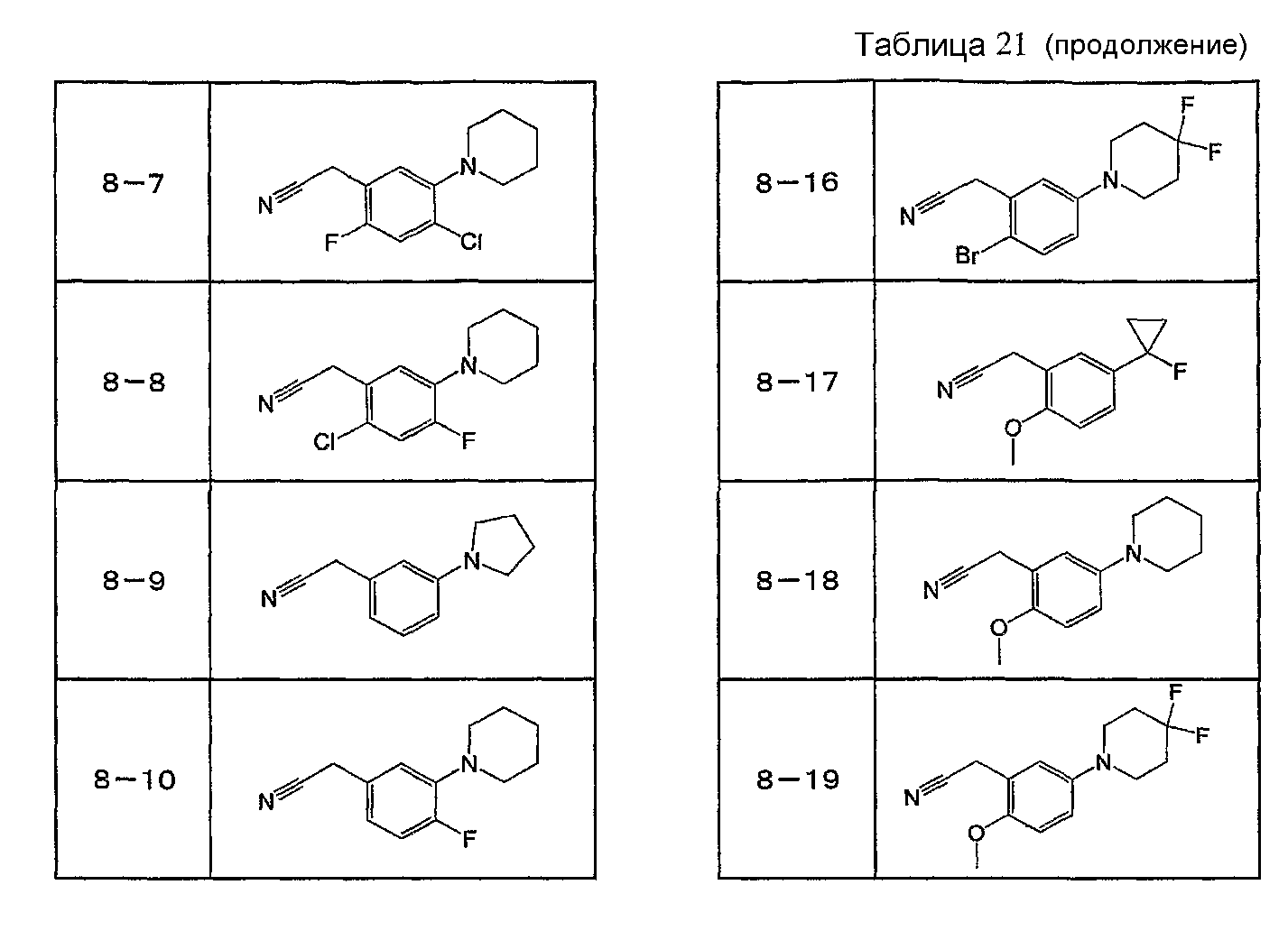
Пример получения 8-20
Синтез (3-бром-4-пиперидин-1-ил)фенилацетонитрила
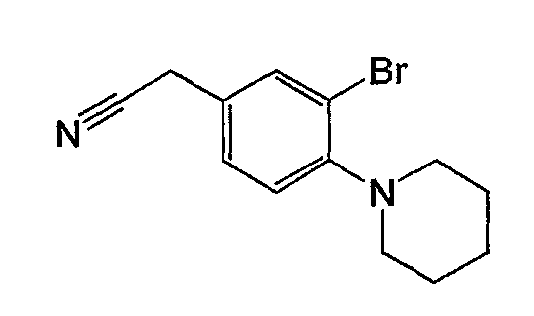
Синтез (3-бром-4-пиперидин-1-ил)бензальдегида
Диоксид марганца (1,26 г) добавляли к раствору (3-бром-4-пиперидин-1-ил)фенилметанола (488 мг), полученного в примере получения 6-20, в хлороформе (20 мл). Реакционную смесь нагревали при перемешивании при 90°C в течение 2 часов. Реакционную смесь фильтровали через фильтровальную бумагу и затем через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (386 мг).
Синтез [(3-бром-4-пиперидин-1-ил)фенил]ацетонитрила
Раствор п-толуолсульфонилизоцианида (309 мл) в 1,2-диметоксиэтане (8 мл) добавляли к суспензии трет-бутоксида калия (355 мг) в 1,2-диметоксиэтане (8 мл) при -30°C. После перемешивания в течение 10 минут добавляли к смеси раствор (3-бром-4-пиперидин-1-ил)бензальдегида (386 мг) в 1,2-диметоксиэтане (8 мл). Смесь перемешивали при -30°C в течение 1,5 часа. Добавляли к реакционной смеси этанол (5 мл) и смесь кипятили с обратным холодильником в течение 20 минут. Смесь охлаждали до комнатной температуры, после чего добавляли воду и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (255 мг).
ESI-MS: m/z 279, 281 [M++H].
Соединения примеров получения 8-21 - 8-26 получали в соответствии со способом примера получения 8-20 (таблица 22).
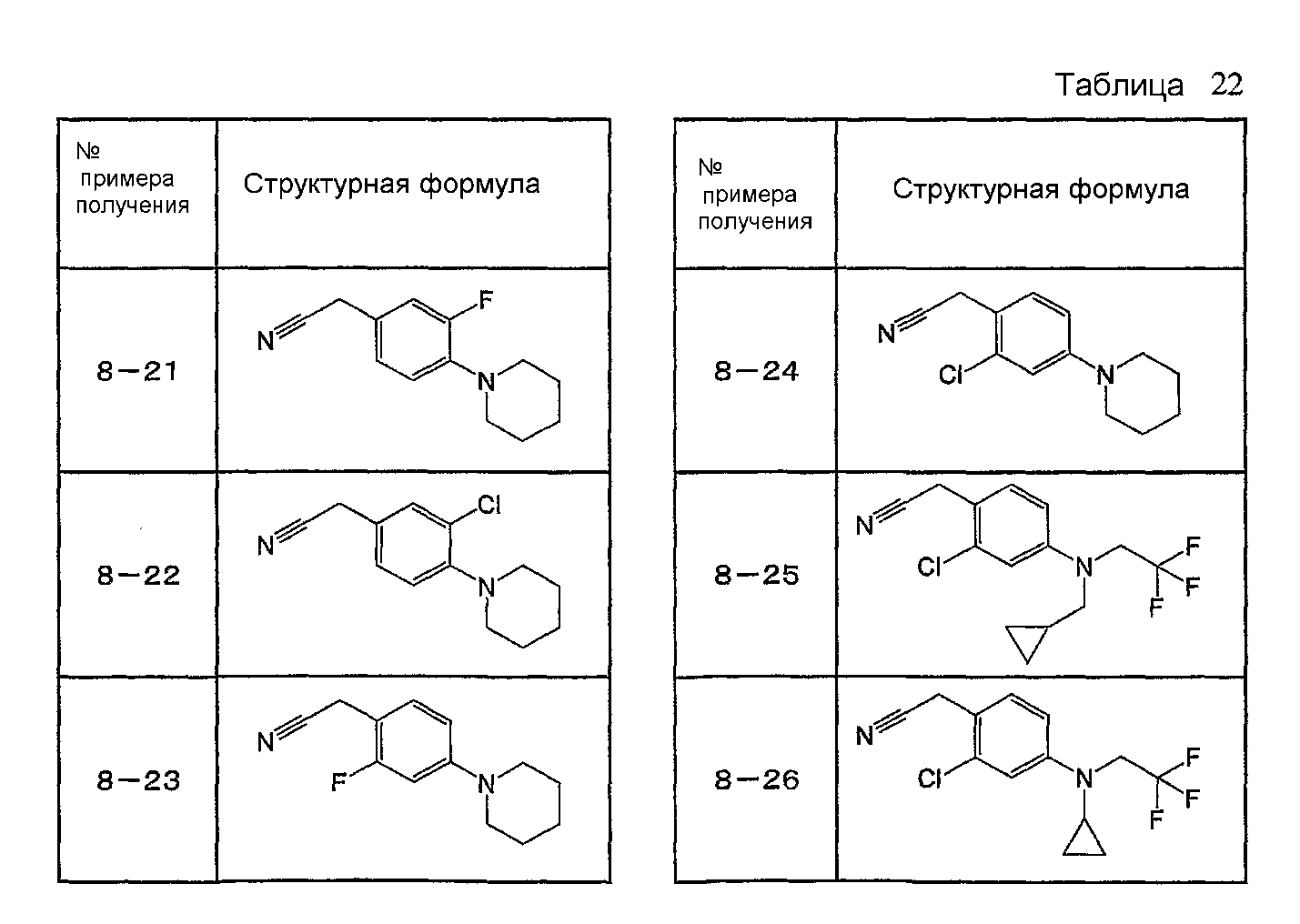
Пример получения 3-51
Синтез гидрохлорида этил [5-хлор-2-(2-фтор-5-пиперидин-1-ил)фенил]пентанимидата
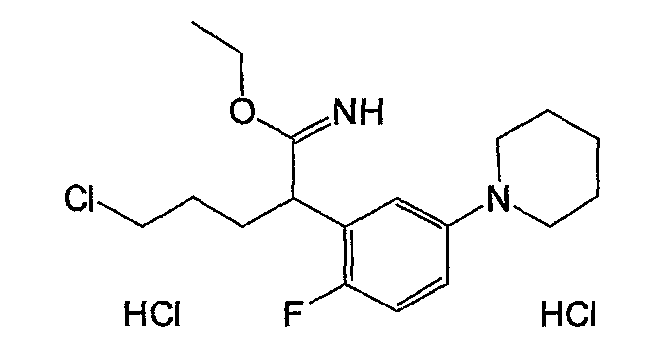
Синтез гидрохлорида этил [5-хлор-2-(2-фтор-5-пиперидин-1-ил)фенил]пентанимидата
В соответствии со способом примера получения 3-1 указанное в заголовке соединение (1,54 г) получали из [(2-фтор-5-пиперидин-1-ил)фенил]ацетонитрила (908 мг), полученного в примере получения 8-1.
Соединения примеров получения 3-52 - 3-84 получали в соответствии со способом примера получения 3-51 (таблица 23).
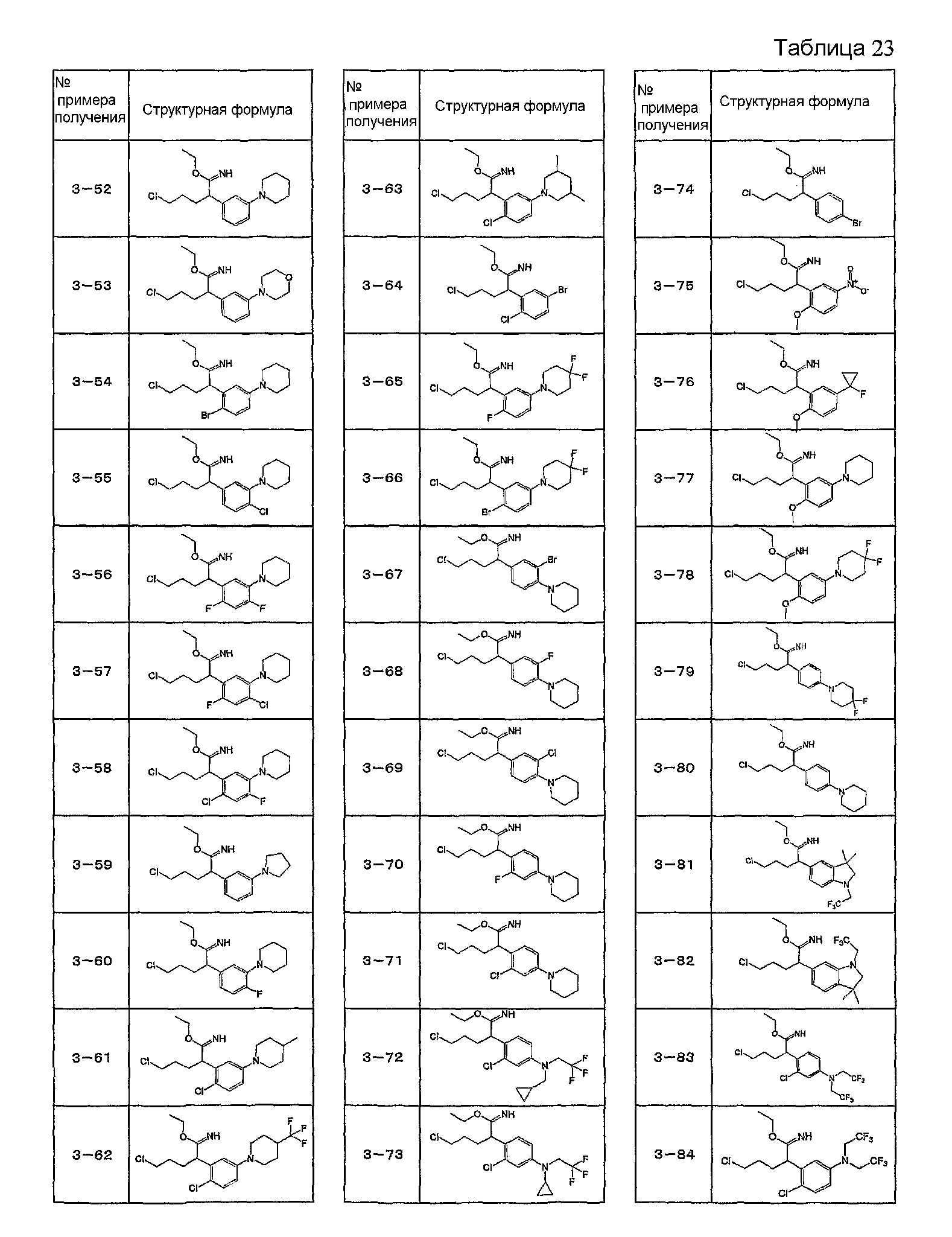
Пример получения 3-85
Синтез гидрохлорида этил 5-хлор-2-{2-хлор-5-[циклопропилметил(2,2,2-трифторэтил)амино]фенил}пентанимидата
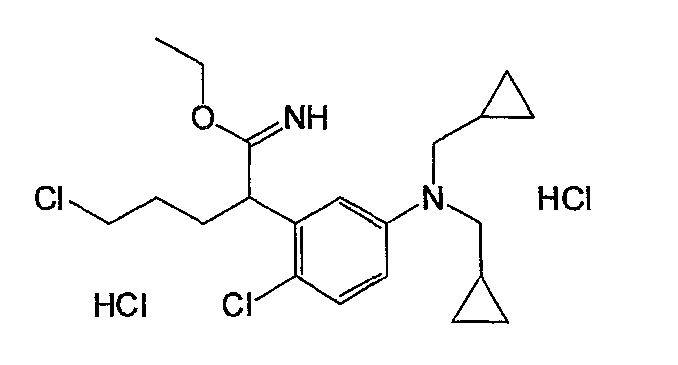
Синтез этил 5-амино-2-хлорбензоата
В соответствии со способом примера получения 6-1 указанное в заголовке соединение (85,9 г) получали из 5-амино-2-хлорбензойной кислоты (5 г).
ESI-MS: m/z 200 [M++H]
Синтез этил 2-хлор-5-(циклопропилметиламино)бензоата и этил 5-[бис(циклопропилметил)амино]-2-хлорбензоата
К смеси этил 5-амино-2-хлорбензоата, циклопропанкарбоксальдегида (458 мкл) и уксусной кислоты (1,43 мл), растворенной в метаноле (20 мл), добавляли при температуре охлаждения льдом α-пиколинборан (804 мг) и смесь перемешивали в атмосфере азота в течение ночи при комнатной температуре. После того как реакционную смесь концентрировали при пониженном давлении, к остатку добавляли водный раствор гидрокарбоната натрия и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением этил 2-хлор-5-(циклопропилметиламино)бензоата (881 мг) и этил 5-[бис(циклопропилметил)амино]-2-хлорбензоата (423 мг).
Показатель свойств этил 2-хлор-5-(циклопропилметиламино)бензоата был следующим.
ESI-MS: m/z 254 [M++H]
Показатель свойств этил 5-[бис(циклопропилметил)амино]-2-хлорбензоата был следующим.
ESI-MS: m/z 308 [M++H]
Синтез гидрохлорида этил 5-хлор-2-{2-хлор-5-[циклопропилметил-(2,2,2-трифторэтил)амино]фенил}пентанимидата
В соответствии со способом примера получения 6-23 и примера получения 3-51 указанное в заголовке соединение (501 мг) получали из этил 2-хлор-5-(циклопропилметиламино)бензоата (881 мг)
ESI-MS: m/z 425 [M++H-HCl]
Пример получения 3-86
Синтез гидрохлорида этил 2-{5-[бис(циклопропилметил)амино]-2-хлорфенил}-5-хлорпентанимидата
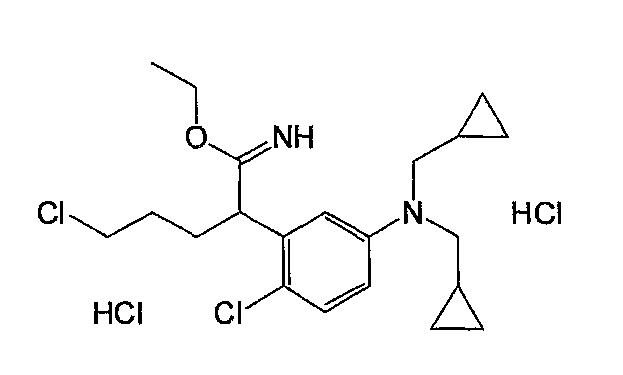
В соответствии со способом примера получения 3-51 указанное в заголовке соединение (578 мг) получали из этил 5-[бис(циклопропилметил)амино]-2-хлорбензоата (423 мг).
ESI-MS: m/z 397 [M++H-HCl]
Примеры 186-233
В соответствии со способами примеров 42 и 43, соединения примеров 186-233 получали из гидрохлоридов имидата, полученных в примерах получения от 3-21 до 3-44, и гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты, полученного в примере получения 1-6. (Таблица 24)
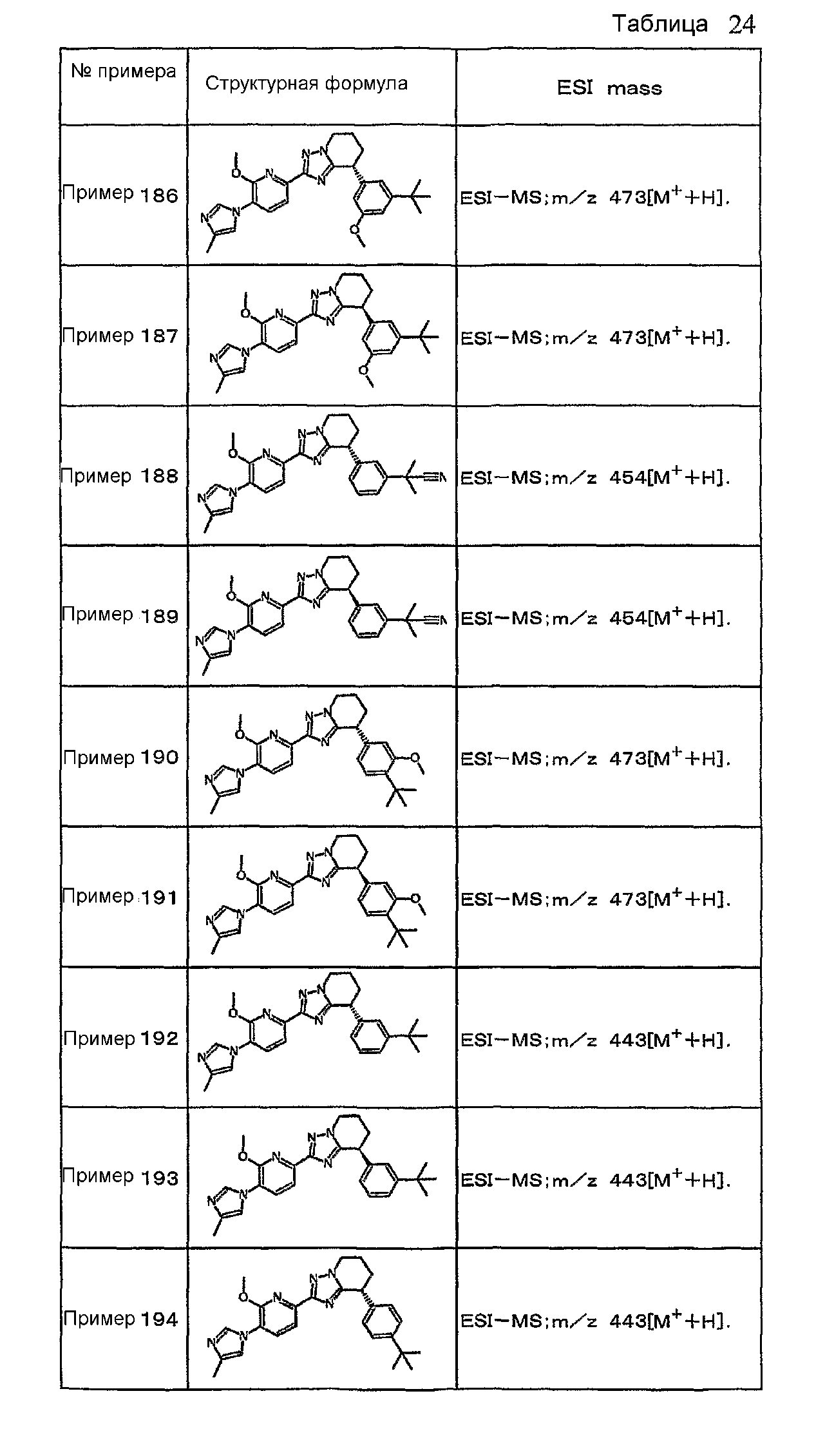
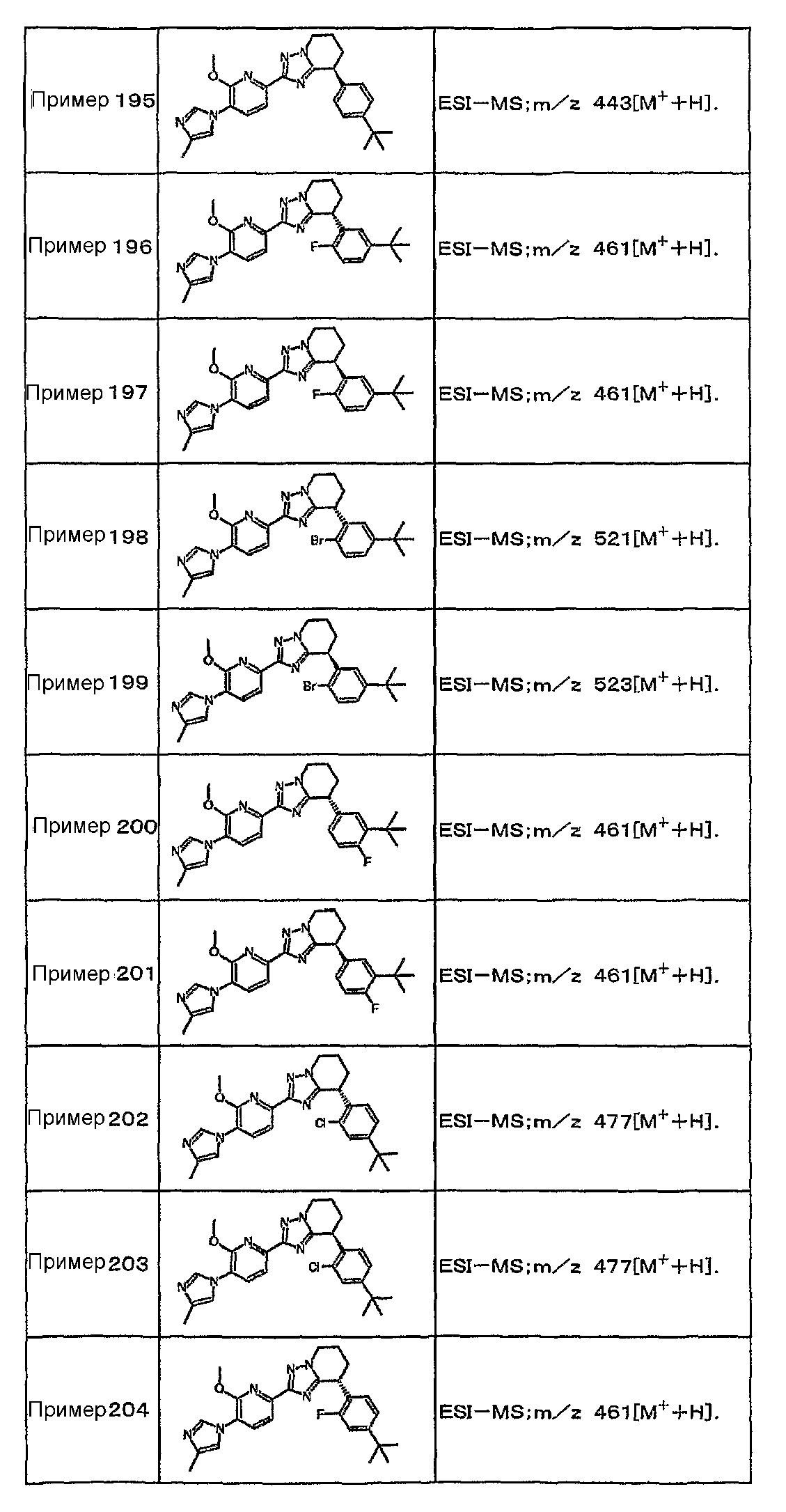
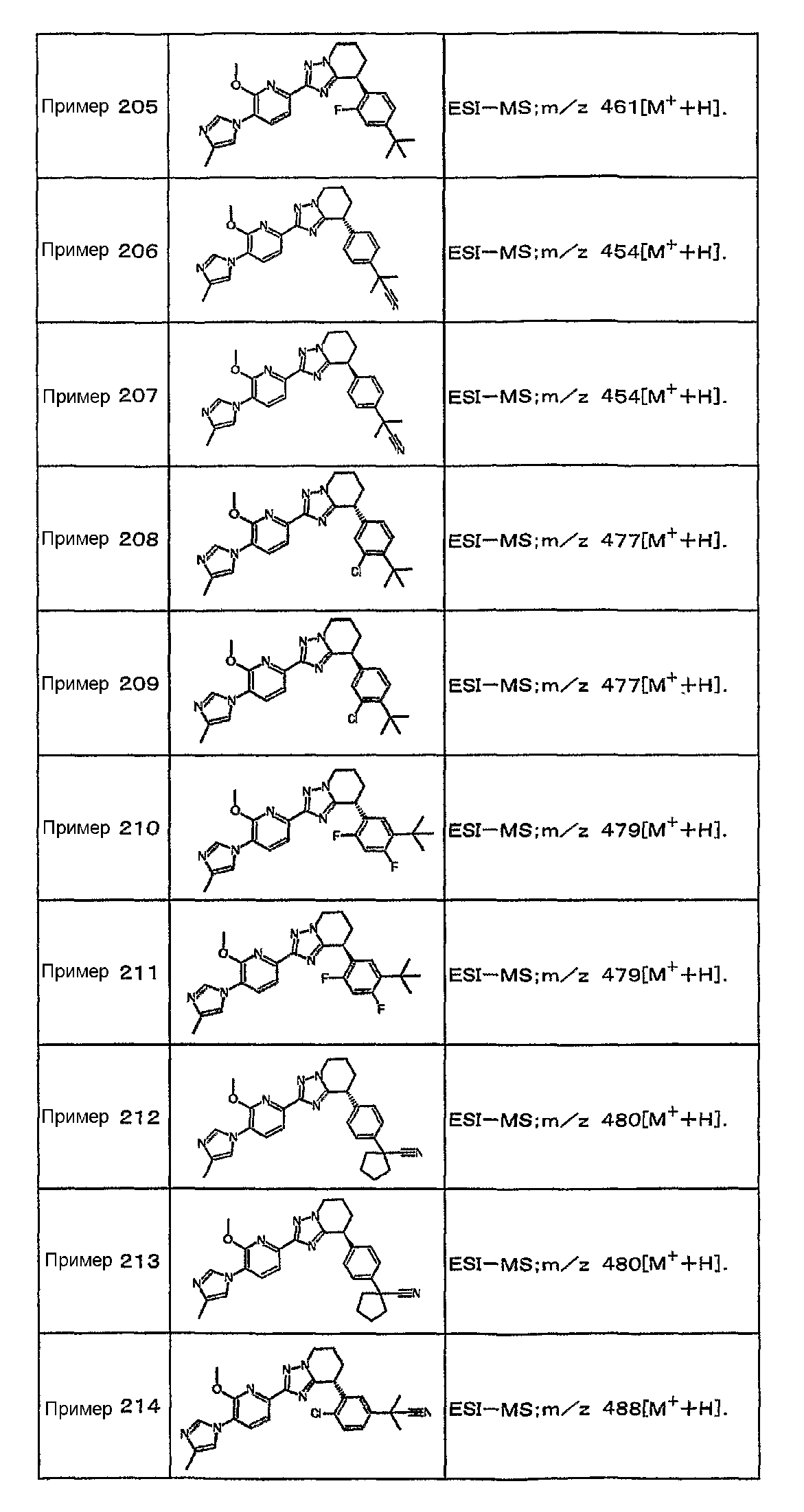
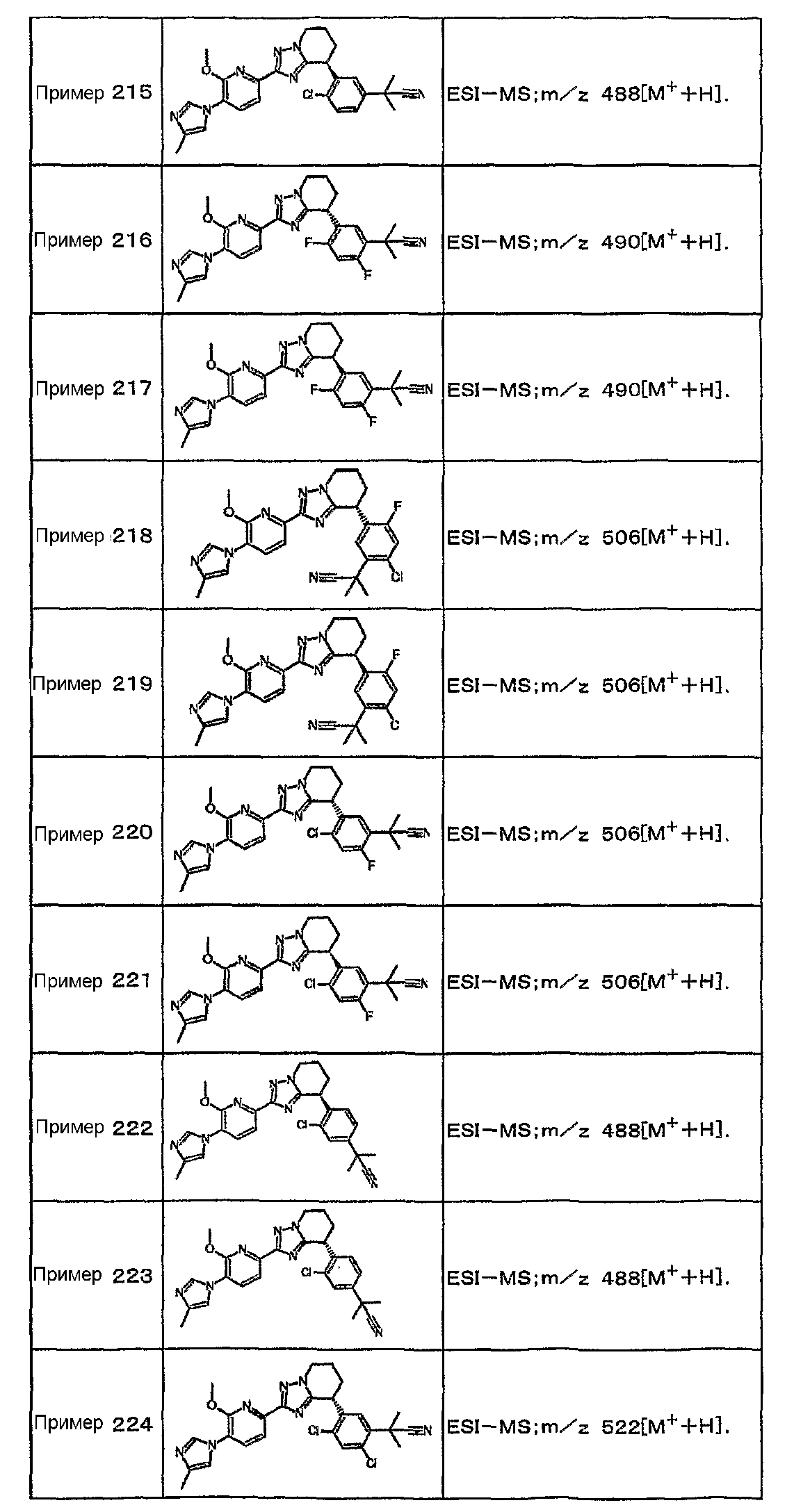
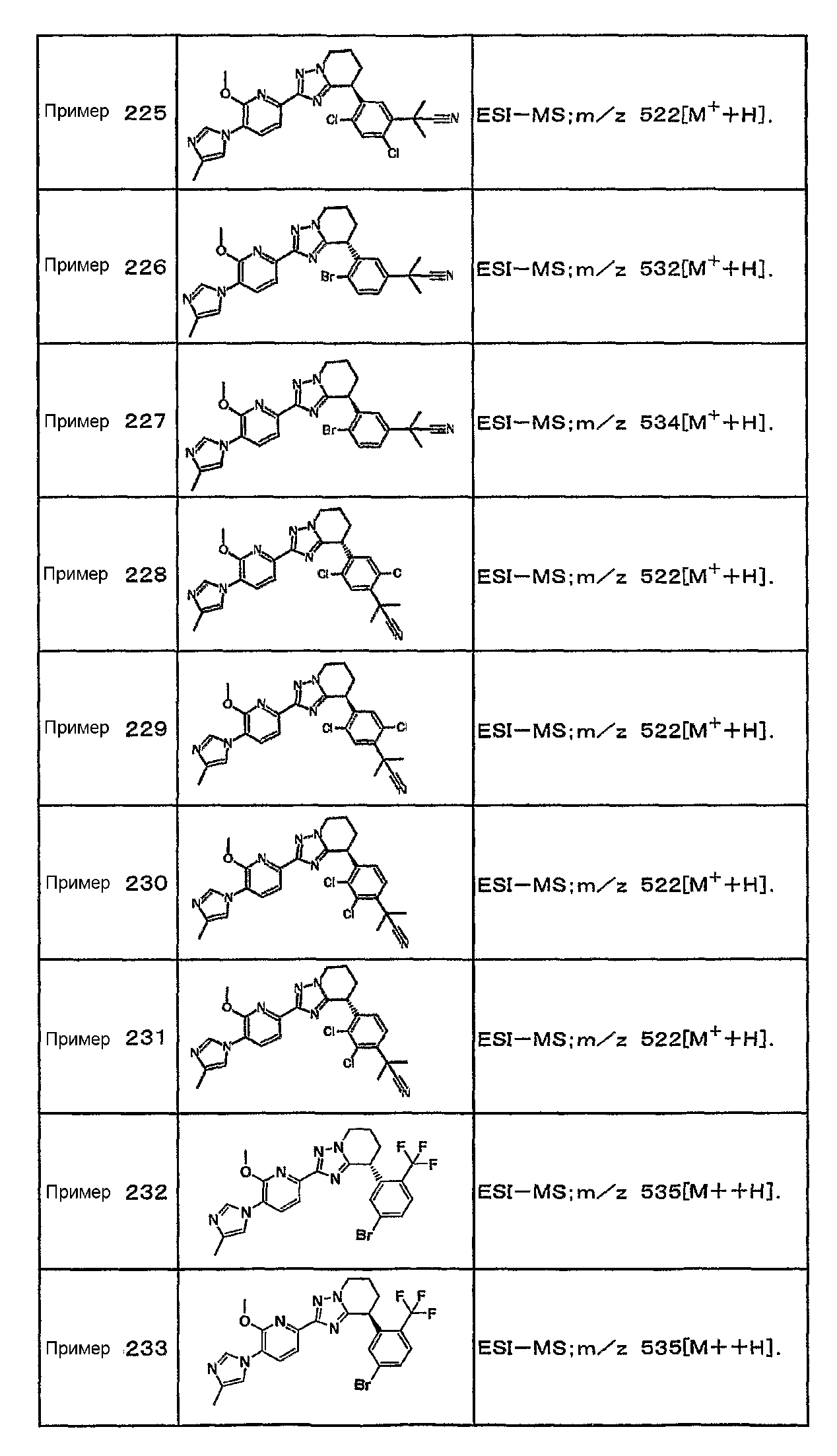
Пример 234, пример 235, пример 236 и пример 237
Синтез этил 1-(2,4-дихлор-5-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}фенил)циклопропанкарбоксилата, амида 1-(2,4-дихлор-5-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}фенил)циклопропанкарбоновой кислоты, (+)-1-(2,4-дихлор-5-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}фенил)циклопропанкарбонитрила и (-)-1-(2,4-дихлор-5-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}фенил)циклопропанкарбонитрила.
В этаноловый (4 мл) раствор 1-[2,4-дихлор-5-(4-хлор-1-цианобутил)фенил]циклопропанкарбонитрила (113 мг), полученного в примере получения 3-46, вдували газообразный хлороводород в течение 10 минут. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре и затем концентрировали при пониженном давлении. Остаток растворяли в трет-бутилметиловом эфире и раствор опять концентрировали при пониженном давлении. Полученный остаток растворяли в ДМФА (2 мл) и к раствору добавляли последовательно имидазол (125 мг) и гидразид 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты, полученный в примере получения 1-6, реакционную смесь перемешивали в течение ночи при комнатной температуре и затем в течение 3,5 часов при 110°C. К реакционной смеси после охлаждения добавляли этилацетат, воду и 1 н. хлористоводородную кислоту (0,5 мл) и отделяли органический слой. Органический слой промывали последовательно водой и насыщенным раствором соли, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали препаративной тонкослойной хроматографией на силикагеле (носитель: Chromatorex™ NH; элюирующий растворитель: этилацетат) с получением рацемического сложного этилового эфира 1-(2,4-дихлор-5-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}фенил)циклопропанкарбоновой кислоты (11,7 мг). Показатели свойств соединения были следующими.
ESI-MS; m/z 589 [M++Na].
1H-ЯМР (CDCl3) δ (ppm): 1,00-1,30 (м, 2H), 1,15 (т, J=7,2 Гц, 3H), 1,50-1,75 (м, 2H), 2,00-2,26 (м, 3H), 2,31 (с, 3H), 2,36-2,50 (м, 1H), 4,03-4,15 (м, 2H), 4,16 (с, 3H), 4,41 (т, J=6,0 Гц, 2H), 4,75 (т, J=6,0 Гц, 1H), 6,86 (с, 1H), 7,01 (ушир.с, 1H), 7,46 (с, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,75 (д, J=8,0 Гц, 1H), 7,85 (ушир.с, 1H).
Получали также рацемический амид 1-(2,4-дихлор-5-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}фенил)циклопропанкарбоновой кислоты (11,9 мг). Показатели свойств соединения были следующими.
ESI-MS; m/z 560 [M++Na].
1H-ЯМР (CDCl3) δ (ppm): 1,01 (ушир.с, 2H), 1,97-2,35 (м, 3H), 2,30 (с, 3H), 2,40-2,50 (м, 1H), 4,13 (с, 3H), 4,35-4,51 (м, 2H), 4,65-4,75 (м, 1H), 5,27 (ушир.с, 1H), 5,69 (ушир.с, 1H), 7,00 (с, 1H), 7,09 (с, 1H), 7,52 (с, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,71 (д, J=8,0 Гц, 1H), 7,84 (ушир.с, 1H).
Получали также рацемический 1-(2,4-дихлор-5-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}фенил)циклопропанкарбонитрил (15,7 мг). Полученные рацемические соединения очищали с помошью CHIRALCEL™ IA (Daicel Chemical Industries, Ltd.; 2 см × 25 см; элюирующий растворитель: 70% этанол/гексан), выделив поворотный (+)-оптический изомер, время удерживания которого было 13 минут (2,0 мг, >99% ee.), и поворотный (-)-оптический изомер, время удерживания которого было 43 минуты (1,4 мг, >99% ee.).
Показатели свойств поворотного (+)-оптического изомера были следующими.
ESI-MS; m/z 520 [M++H].
1H-ЯМР (CDCl3) δ (ppm): 1,15-1,30 (м, 2H), 1,60-1,75 (м, 2H), 1,95-2,35 (м, 3H), 2,31 (с, 3H), 2,40-2,50 (м, 1H), 4,16 (с, 3H), 4,35-4,50 (м, 2H), 4,65-4,75 (м, 1H), 6,94 (с, 1H), 7,01 (ушир.с, 1H), 7,53 (с, 1H), 7,60 (д, J=7,6 Гц, 1H), 7,74 (д, J=7,6 Гц, 1H), 7,86 (ушир.с, 1H).
Показатели свойств поворотного (-)-оптического изомера были одинаковы с (+)-изомером.
В соответствии с примером 100 и примером 101 получали соединения примеров 238-251 (таблица 25) из гидрохлоридов имидата, полученных в примере получения 3-9, примере получения 3-25, примере получения 3-29, примере получения 3-34, примере получения 3-36, примере получения 3-44 и примере получения 3-45, и 6-(1-этоксивинил)-2-метокси-3-(4-метил-1H-имидазол-1-ил)пиридина, полученного в примере получения 1-7.
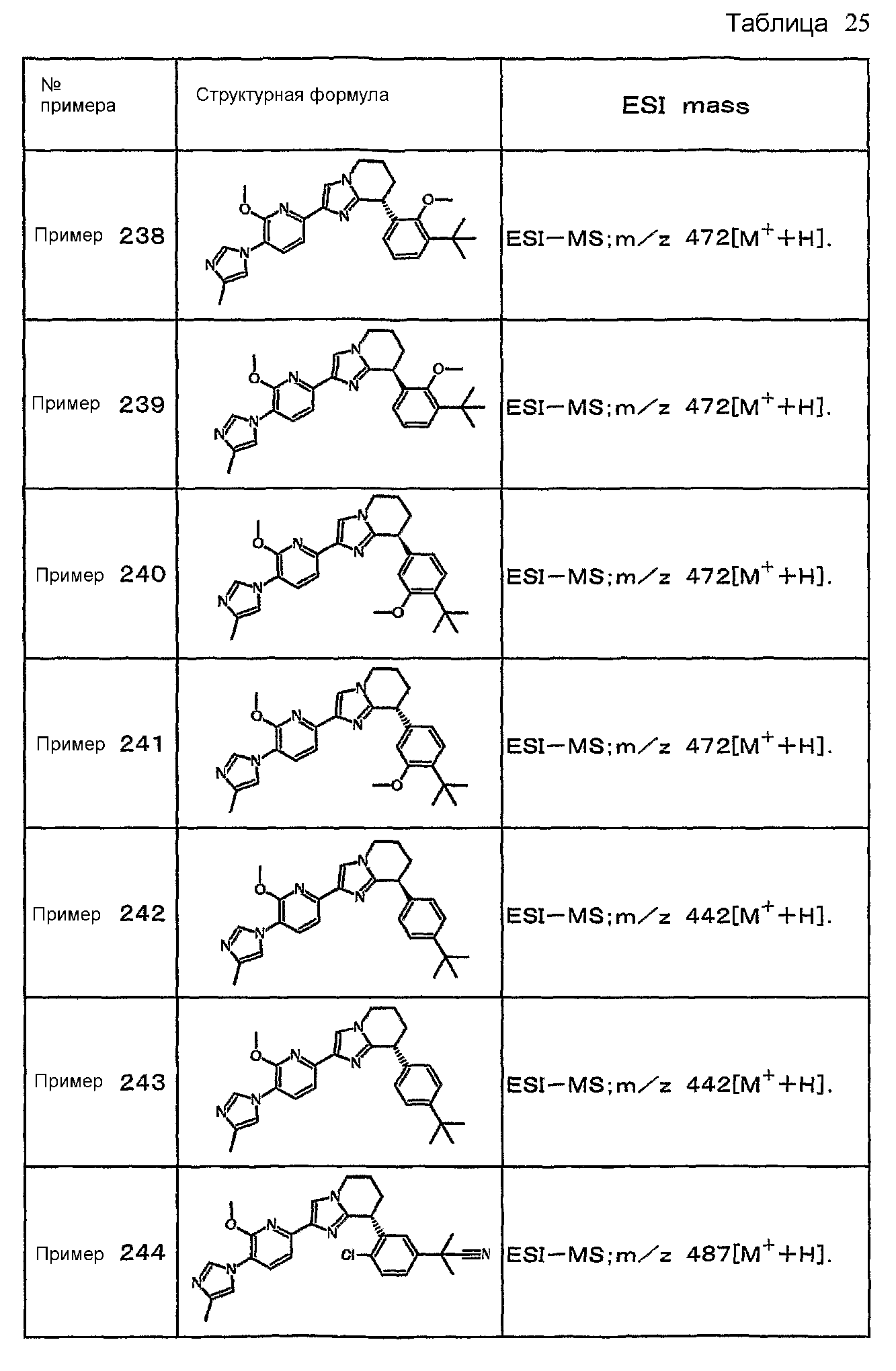
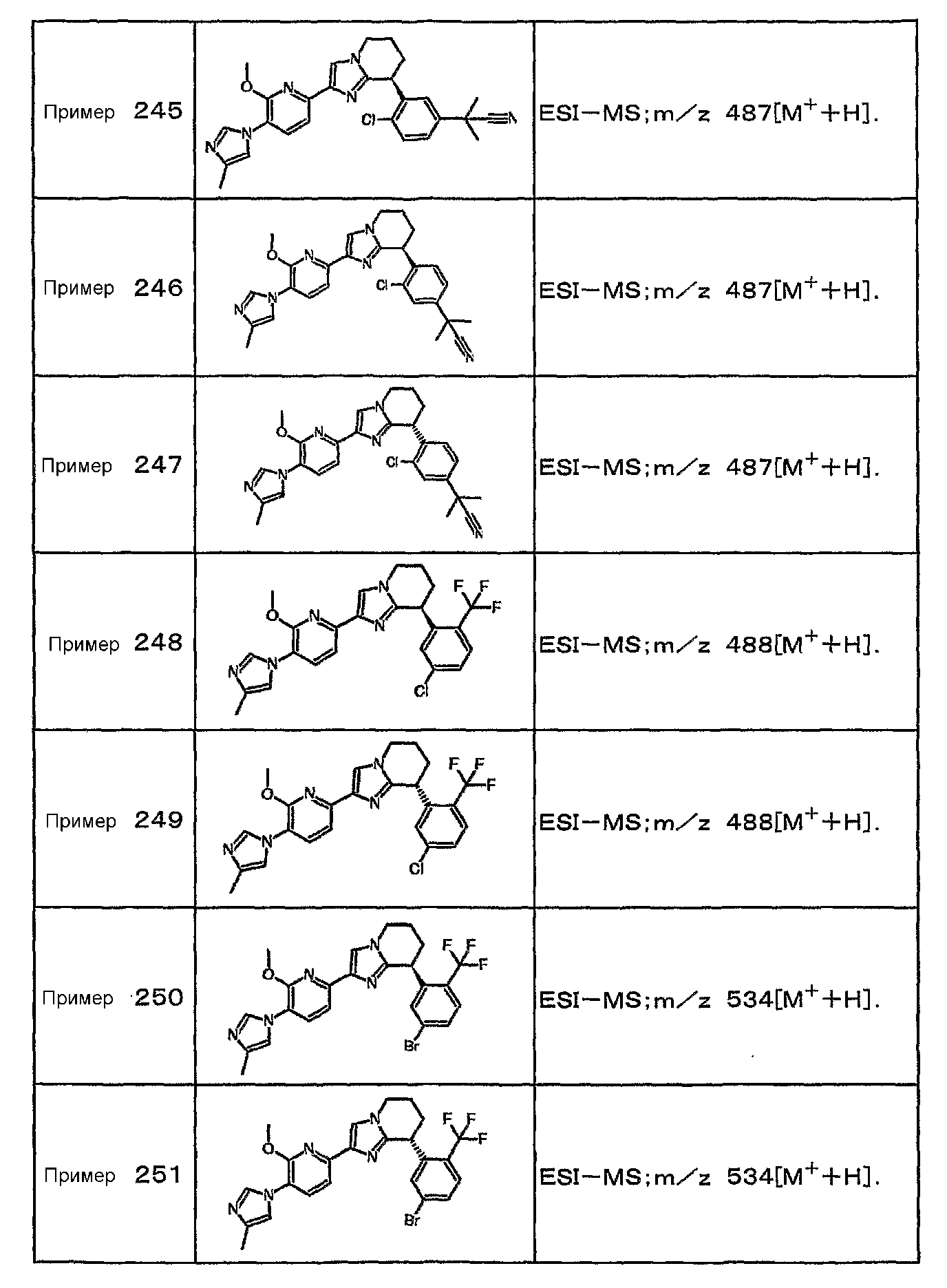
В соответствии со способом примеров 42 и 43 соединения примеров 252-259 (таблица 26) получали из гидрохлоридов имидата, полученных в примере получения 3-1, примере получения 3-21, примере получения 3-25 и примере получения 3-29, и гидразида 3-метокси-4-(4-метил-1H-имидазол-1-ил)бензойной кислоты, полученного в примере получения 1-9.
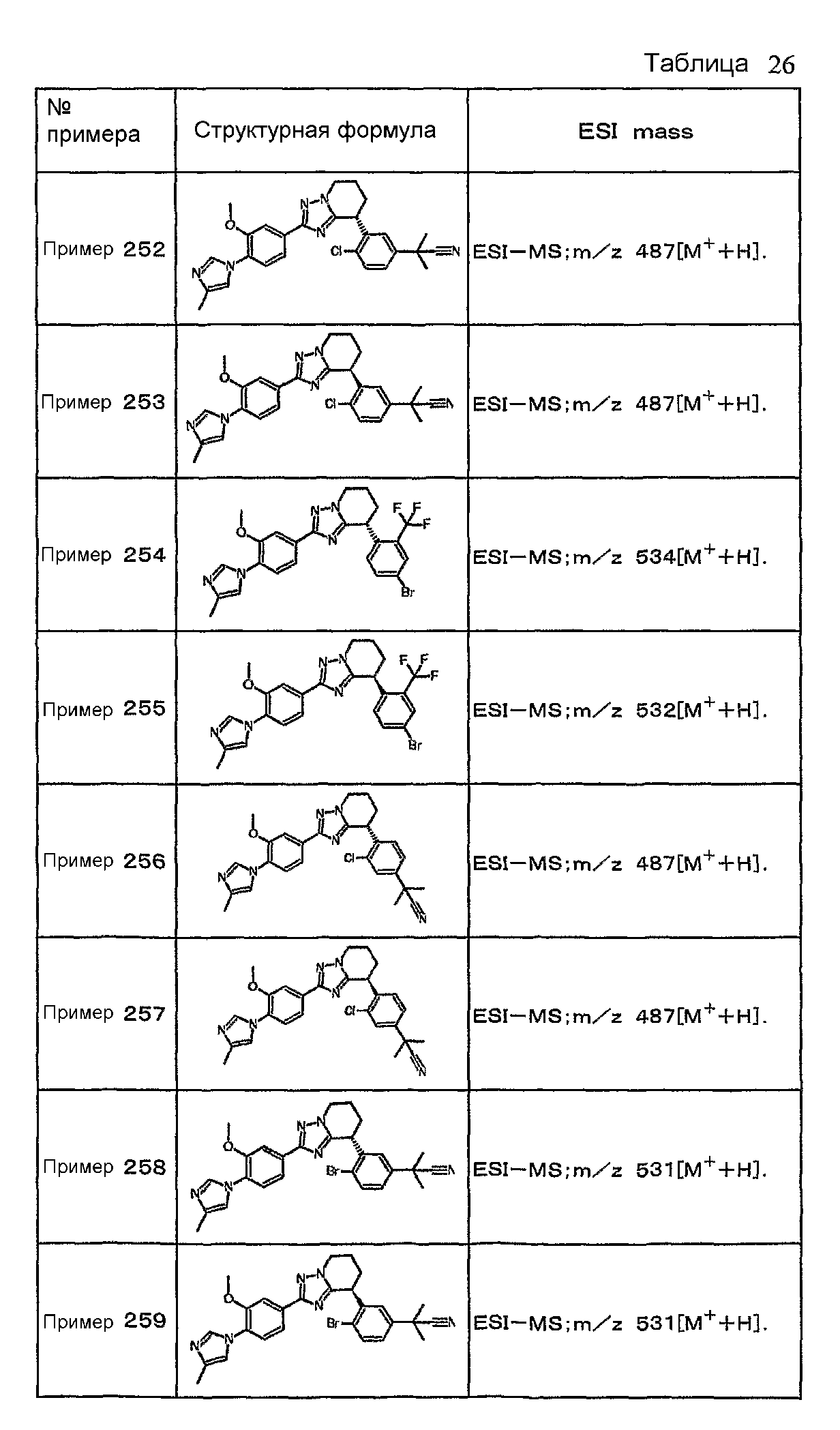
Пример 260 и пример 261
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(3-циклопропил-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(3-циклопропил-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
В соответствии с примером 150 и примером 151 рацемическое указанное в заголовке соединение (65,1 мг) получали из 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(3-бром-4-метоксифенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина. Полученное рацемическое соединение очищали с помощью CHIRALCEL™ IA (Daicel Chemical Industries, Ltd.; 2 см × 25 см; элюирующий растворитель: 30% этанол/гексан), выделив поворотный (+)-оптический изомер, время удерживания которого было 16 минут (4,2 мг), и поворотный (-)-оптический изомер, время удерживания которого было 28,5 минуты (3,7 мг). Показатель свойств указанных в заголовке соединений был следующим.
ESI-MS: m/z 457 [M++H]
Пример 262 и пример 263
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-илпиридин-2-ил)-8-[3-(1-фтор-1-метилэтил)фенил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-илпиридин-2-ил)-8-[3-(1-фтор-1-метилэтил)фенил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Синтез метил 3-(4-хлор-1-этоксикарбонимидоилбутил)бензоата
В соответствии со способом примера получения 3-1 указанное в заголовке соединение (1,47 г) получали из метил 3-цианометил-бензоата (999 мг). Показатель свойств указанных в заголовке соединений был следующим.
ESI-MS: m/z 298 [M++H]
Синтез метил 3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}бензоата
В соответствии с способом примеров 42 и 43 указанное в заголовке соединение (1,64 г) получали из гидрохлорида метил 3-(4-хлор-1-этоксикарбонимидоилбутил)бензоата (1,47 г) и гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты (1,09 г), полученного в примере получения 1-6. Показатель свойств указанных в заголовке соединений был следующим.
ESI-MS: m/z 445 [M++H]
Синтез 2-(3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил})фенилпропан-2-ола
К ТГФ (19,5 мл) раствору метил 3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}бензоата (866 мг) добавляли при 10°C метилмагнийбромид (10,1 мл) и реакционную смесь перемешивали при той же самой температуре. Реакционную смесь гасили добавлением насыщенного водного раствора аммонийхлорида и экстрагировали этилацетатом. Полученную органическую фазу промывали насыщенным раствором соли, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаточный маслянистый материал очищали хроматографией на колонке с силикагелем (носитель: Yamazen силикагель, элюирующий растворитель: этилацетат/гептан = 0~100% и затем метанол/этилацетат = 15%) с получением указанного в заголовке соединения (785 мг). Показатели свойств указанных в заголовке соединений были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,59 (с, 6H), 2,06-2,16 (м, 2H), 2,16-2,27 (м, 1H), 2,30 (с, 3H), 2,34-2,44 (м, 1H), 4,16 (с, 3H), 4,32-4,47 (м, 3H), 6,94-6,98 (м, 1H), 6,99-7,01 (м, 1H), 7,30 (д, J=7,8 Гц, 1H), 7,35-7,38 (м, 2H), 7,58 (д, J=7,8 Гц, 1H), 7,78 (д, J=7,8 Гц, 1H), 7,82 (д, J=1,5 Гц, 1H).
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-илпиридин-2-ил)-8-[3-(1-фтор-1-метилэтил)фенил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-илпиридин-2-ил)-8-[3-(1-фтор-1-метилэтил)фенил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
2-(3-{6-Метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил}-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил)фенилпропан-2-ол (159 мг) растворяли в метиленхлориде (3,6 мл) и к смеси добавляли при комнатной температуре [бис(2-метоксиэтил)амино]сератрифторид (79,6 мкл). Через 18 часов перемешивания реакционную смесь гасили добавлением насыщенного водного раствора гидрокарбоната натрия и экстрагировали этилацетатом. Органическую фазу промывали последовательно насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаточный маслянистый материал очищали хроматографией на колонке с силикагелем (носитель: Yamazen силикагель, элюирующий растворитель: хлороформ/метанол = 25/1) с получением указанного в заголовке соединения (115,5 мг) в виде смеси с экзо-олефиновым соединением. Затем полученный материал растворяли в ТГФ (2,7 мл) и к смеси добавляли при комнатной температуре 9-борабицикло[3,3,1]нонан (644 мкл) и реакционную смесь перемешивали при 80°C в течение 17 часов. К реакционной смеси добавляли, после охлаждения до 0°C, этанол (0,9 мл), 2 н. водный раствор гидроксида натрия (0,9 мл) и водный раствор пероксида водорода (0,75 мл) и затем реакционную смесь перемешивали в течение 2 часов при 80°C. Реакционную смесь гасили добавлением насыщенного водного раствора тиосульфата натрия и экстрагировали этилацетатом. Органический слой промывали последовательно водой и насыщенным раствором соли, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаточный маслянистый материал очищали хроматографией на колонке с силикагелем (носитель: Yamazen силикагель, элюирующий растворитель: этилацетат/гептан = 0~100% и затем метанол/этилацетат = 10%) с получением указанного в заголовке соединения (52 мг).
Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: 20% этанол-гексан) с получением указанного в заголовке соединения с временем удерживания 28 минут и положительным оптическим вращением (14,4 мг) и указанного в заголовке соединения с временем удерживания 54 минуты и отрицательным оптическим вращением (12,4 мг).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,64 (с, 3H), 1,69 (с, 3H), 2,04-2,16 (м, 2H), 2,20 (ддд, J=8,1, 8,1, 6,3 Гц, 1H), 2,30 (с, 3H), 2,34-2,44 (м, 1H), 4,16 (с, 3H), 4,32-4,48 (м, 3H), 6,96-7,01 (м, 2H), 7,23-7,33 (м, 3H), 7,58 (д, J=7,8 Гц, 1H), 7,78 (д, J=7,8 Гц, 1H), 7,81-7,83 (м, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,64 (с, 3H), 1,69 (с, 3H), 2,04-2,16 (м, 2H), 2,20 (ддд, J=8,1, 8,1, 6,3 Гц, 1H), 2,30 (с, 3H), 2,34-2,44 (м, 1H), 4,16 (с, 3H), 4,32-4,48 (м, 3H), 6,96-7,01 (м, 2H), 7,23-7,33 (м, 3H), 7,58 (д, J=7,8 Гц, 1H), 7,78 (д, J=7,8 Гц, 1H), 7,81-7,83 (м, 1H).
Примеры 264 и 265
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(3-(1-метокси-1-метилэтил)фенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-((3-(1-метокси-1-метилэтил)фенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
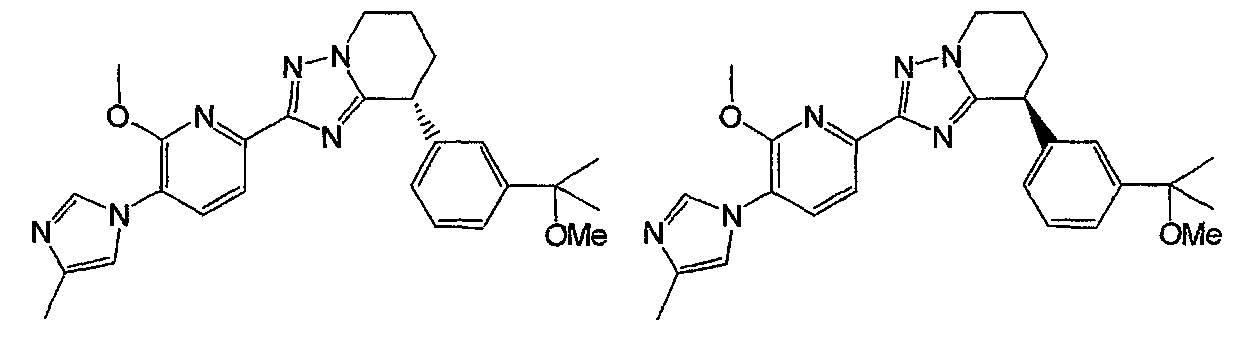
2-(3-{2-[6-Метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}фенил-2-ол (60 мг) растворяли в метаноле (1,3 мл) и затем DOWEX™ 50X8 (6 мг) добавляли при комнатной температуре. После перемешивания при комнатной температуре в течение 24 часов еще добавляли DOWEX™ 50X8 (20 мг) и смесь перемешивали в течение 6 часов при той же самой температуре. Триметилортоформиат (44,3 мкл) добавляли к реакционному раствору с последующим перемешиванием в течение 19 часов.
Моногидрат п-толуолсульфоновой кислоты (5,1 мг) добавляли к реакционному раствору с последующим перемешиванием в течение 24 часов. Еще добавляли к реакционному раствору моногидрат п-толуолсульфоновой кислоты (5,1 мг) с последующим перемешиванием в течение 4 суток и реакцию гасили насыщенным раствором карбоната калия с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным раствором карбоната калия и сушили над сульфатом натрия. Растворитель удаляли и полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (носитель: Yamazen силикагель; элюирующий растворитель: этилацетат:гептан = 0:100 → 100:0, затем 15% метанол-этилацетат) с получением указанного в заголовке соединения.
Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см: подвижная фаза: 20% этанол-гексан) с получением указанного в заголовке соединения с временем удерживания 23,5 минуты и положительным оптическим вращением (21,9 мг) и указанного в заголовке соединения с временем удерживания 47 минут и отрицательным оптическим вращением (15,1 мг).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,50 (с, 6H), 2,04-2,16 (м, 2H), 2,16-2,27 (м, 1H), 2,30 (с, 3H), 2,34-2,44 (м, 1H), 3,07 (с, 3H), 4,16 (с, 3H), 4,32-4,48 (м, 3H), 6,94-6,98 (м, 1H), 6,99-7,01 (м, 1H), 7,24-7,27 (м, 1H), 7,29 (д, J=5,1 Гц, 1H), 7,58 (д, J=7,8 Гц, 1H), 7,77 (д, J=7,8 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,50 (с, 6H), 2,04-2,16 (м, 2H), 2,16-2,27 (м, 1H), 2,30 (с, 3H), 2,34-2,44 (м, 1H), 3,07 (с, 3H), 4,16 (с, 3H), 4,32-4,48 (м, 3H), 6,94-6,98 (м, 1H), 6,99-7,01 (м, 1H), 7,24-7,27 (м, 1H), 7,29 (д, J=5,1 Гц, 1H), 7,58 (д, J=7,8 Гц, 1H), 7,77 (д, J=7,8 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H).
Примеры 266 и 267
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(2-хлор-5-(1-метокси-1-метилэтил)фенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(2-хлор-5-(1-метокси-1-метилэтил)фенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
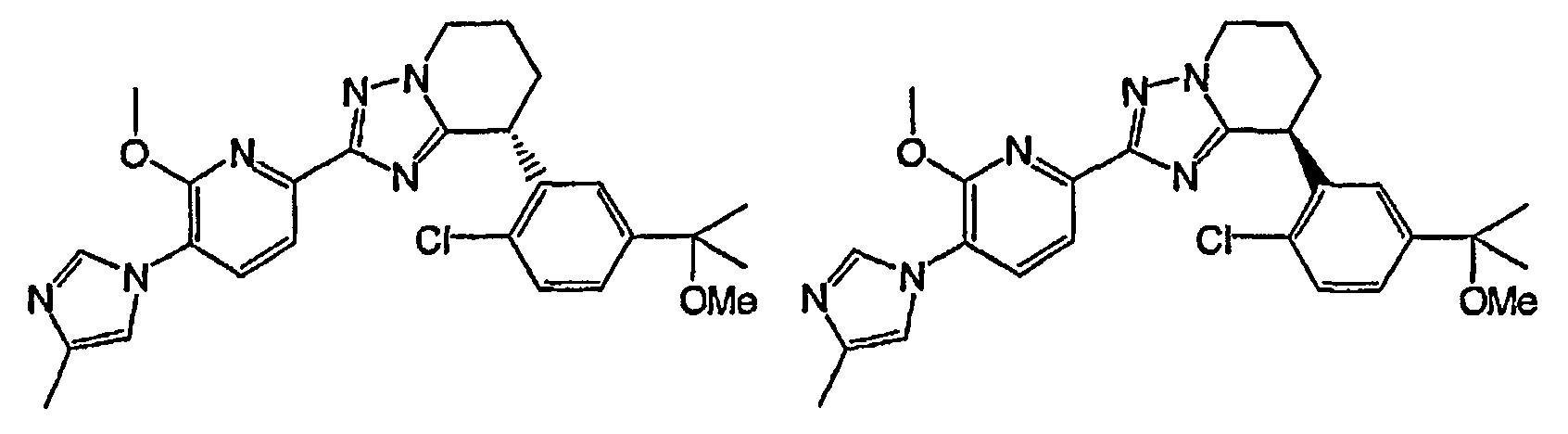
Синтез гидрохлорида этил 4-хлор-3-(4-хлор-1-этоксикарбонимидолилбутил)бензоата
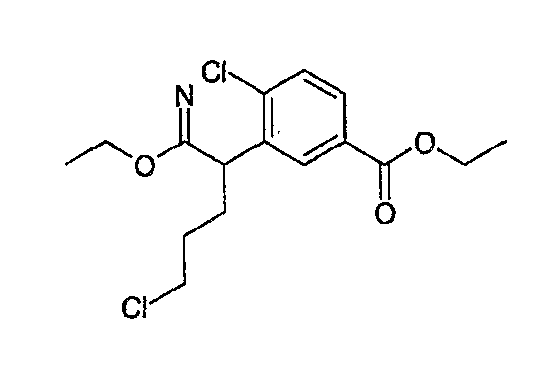
Указанное в заголовке соединение (1,46 г) получали в соответствии со способом примера получения 3-1 из этил 4-хлор-3-цианометилбензоата (1,32 г). Показатель свойств соединения был следующим.
ESI-MS: m/z 346 [M++H]
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(2-хлор-5-(1-метокси-1-метилэтил)фенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(2-хлор-5-(1-метокси-1-метилэтил)фенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Рацемат указанного в заголовке соединения (65 мг) получали в соответствии со способом примеров 262 и 263 из 2-(4-хлор-3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}фенилпропан-2-ола (76 мг) и этил 4-хлор-3-(4-хлор-1-этоксикарбонимидолилбутил)бензоат хлористоводородной кислоты. Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: 20% этанол-гексан) с получением указанного в заголовке соединения с временем удерживания 19,5 минуты и положительным оптическим вращением (24 мг) и указанного в заголовке соединения с временем удерживания 30 минут и отрицательным оптическим вращением (22 мг).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,41 (с, 6H), 1,41 (с, 6H), 2,00-2,25 (м, 3H), 2,30 (с, 3H), 2,38-2,48 (м, 1H), 3,00 (с, 3H), 4,15 (с, 3H), 4,36-4,45 (м, 2H), 4,78 (дд, J=7,4, 5,9 Гц, 1H), 6,97-7,11 (м, 2H), 7,21-7,25 (м, 1H), 7,37 (д, J=8,2 Гц, 1H), 7,57 (д, J=7,8 Гц, 1H), 7,74 (д, J=7,8 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 1,41 (с, 6H), 1,41 (с, 6H), 2,00-2,25 (м, 3H), 2,30 (с, 3H), 2,38-2,48 (м, 1H), 3,00 (с, 3H), 4,15 (с, 3H), 4,36-4,45 (м, 2H), 4,78 (дд, J=7,4, 5,9 Гц, 1H), 6,97-7,11 (м, 2H), 7,21-7,25 (м, 1H), 7,37 (д, J=8,2 Гц, 1H), 7,57 (д, J=7,8 Гц, 1H), 7,74 (д, J=7,8 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H).
Примеры 268 и 269
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-бром-2-фторфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-бром-2-фторфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
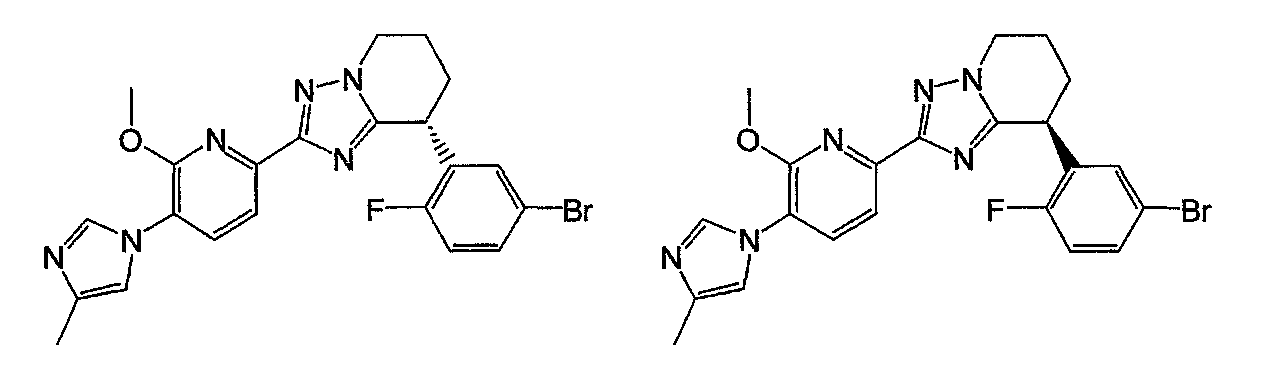
Синтез этил 2-(5-бром-2-фторфенил)-5-хлорпентанимидата
Указанное в заголовке соединение (3,1 г) получали в соответствии со способом примера получения 3-1 из (5-бром-2-фторфенил)ацетонитрила. Показатель свойств соединения был следующим.
ESI-MS: m/z 336 [M++H]
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-бром-2-фторфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-бром-2-фторфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Рацемат указанного в заголовке соединения (1,80 г) получали в соответствии со способом примеров 42 и 43 из гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоксилата (2,05 г), полученного в примере получения 1-6, и этил 2-(5-бром-2-фторфенил)-5-хлорпентанимидата (3,1 г).
Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: 15% этанол-гексан) с получением указанного в заголовке соединения с временем удерживания 24,5 минуты и положительным оптическим вращением (21 мг) и указанного в заголовке соединения с временем удерживания 43 минуты и отрицательным оптическим вращением (22,5 мг).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,01-2,26 (м, 3H), 2,30 (с, 3H), 2,34-2,42 (м, 1H), 4,17 (с, 3H), 4,36-4,43 (м, 2H), 4,60 (дд, J=6,6, 6,2 Гц, 1H), 6,96-7,02 (м, 2H), 7,07 (дд, J=16,6, 2,3 Гц, 1H), 7,36-7,41 (м, 1H), 7,60 (д, J=7,8 Гц, 1H), 7,77 (д, J=7,8 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
1H-ЯМР (CDCl3) δ (ppm): 2,01-2,26 (м, 3H), 2,30 (с, 3H), 2,34-2,42 (м, 1H), 4,17 (с, 3H), 4,36-4,43 (м, 2H), 4,60 (дд, J=6,6, 6,2 Гц, 1H), 6,96-7,02 (м, 2H), 7,07 (дд, J=16,6, 2,3 Гц, 1H), 7,36-7,41 (м, 1H), 7,60 (д, J=7,8 Гц, 1H), 7,77 (д, J=7,8 Гц, 1H), 7,83 (д, J=1,2 Гц, 1H).
Примеры 270 и 271
Синтез (+)-8-(2-фтор-5-пиперидин-1-илфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-(2-фтор-5-пиперидин-1-илфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
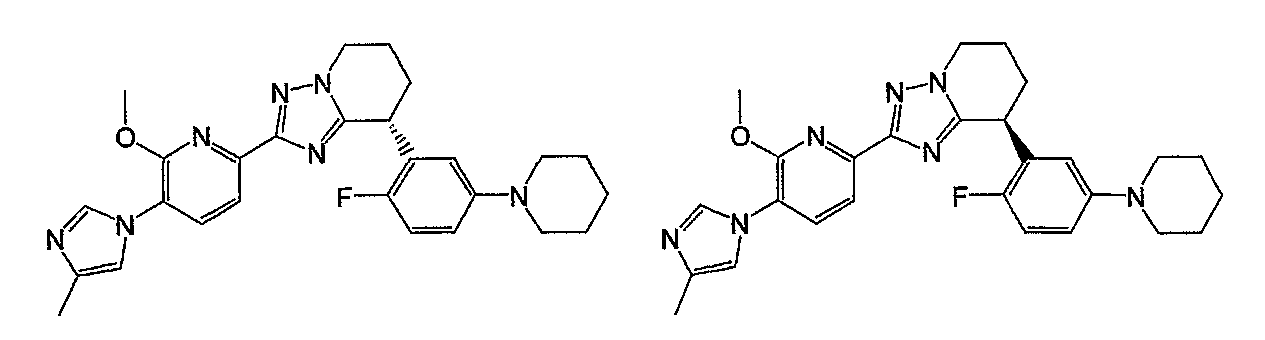
Рацемат указанного в заголовке соединения (164 мг) получали в соответствии со способом примеров 42 и 43 из гидрохлорида гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты (200 мг), полученного в примере получения 1-6, и гидрохлорида этил 5-хлор-2-(2-фтор-5-пиперидин-1-илфенил)пентанимидата (350 мг), полученного в примере получения 3-51.
Полученный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, подвижная фаза: 50% этанол-гексан) с получением указанного в заголовке соединения с положительным оптическим вращением (59 мг, >99% ee) и указанного в заголовке соединения с отрицательным оптическим вращением (56 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS: m/z 245 [1/2M++H], 488 [M++H].
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS: m/z 245 [1/2M++H], 488 [M++H].
Примеры 272-349
Соединения примеров 272-349 были получены в соответствии со способом примеров 270 и 271 и показаны в таблицах 27-39.
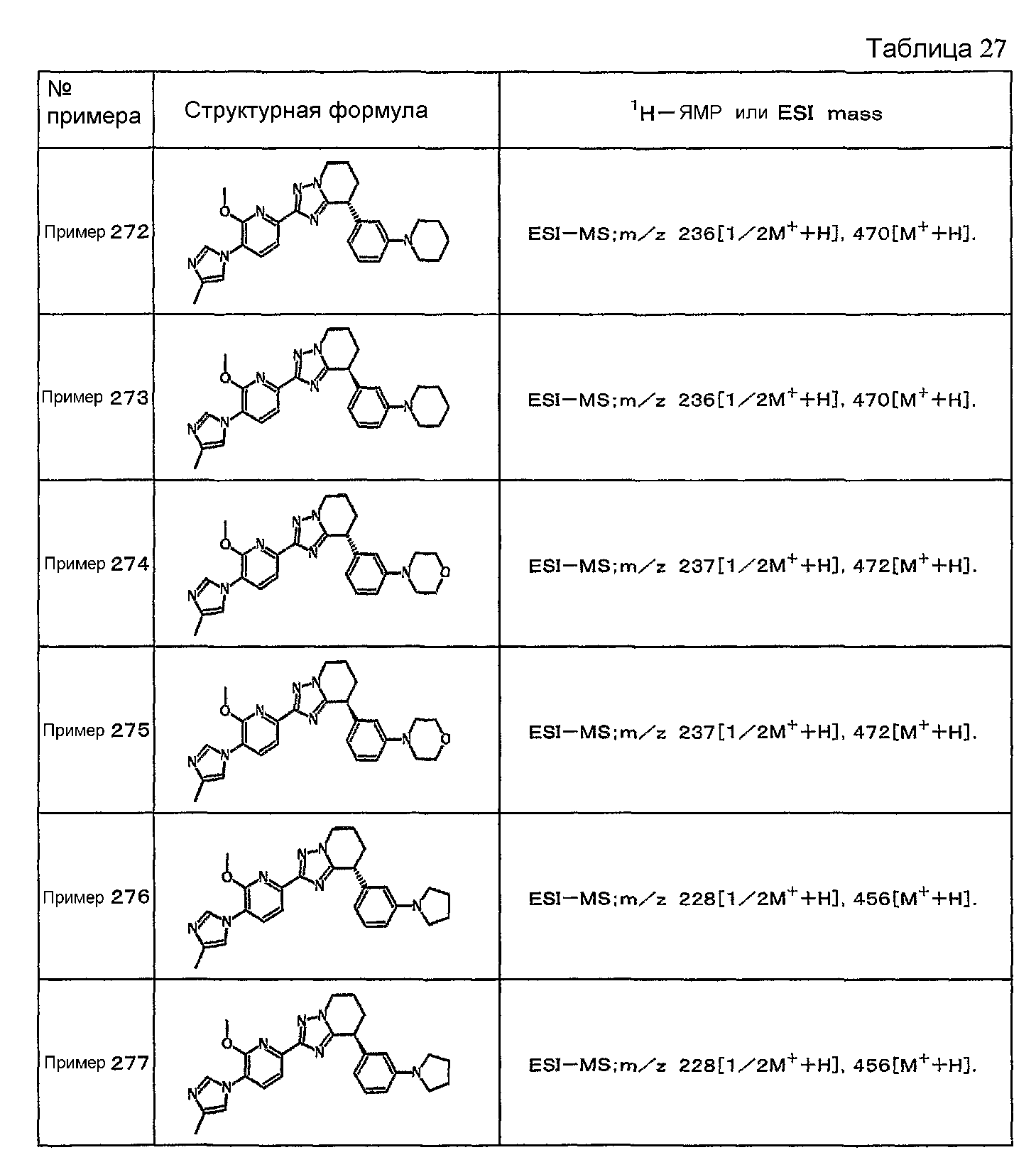
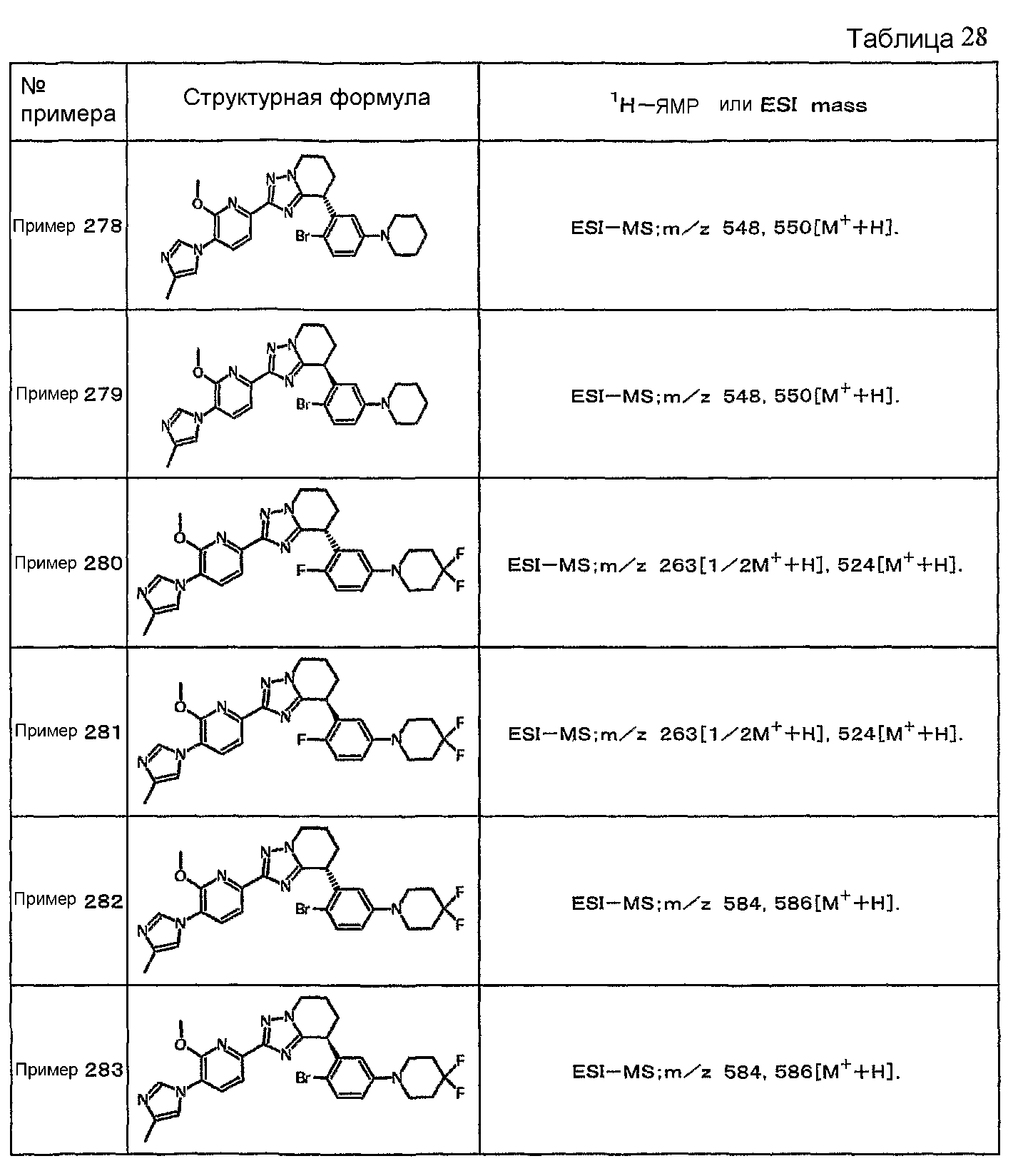
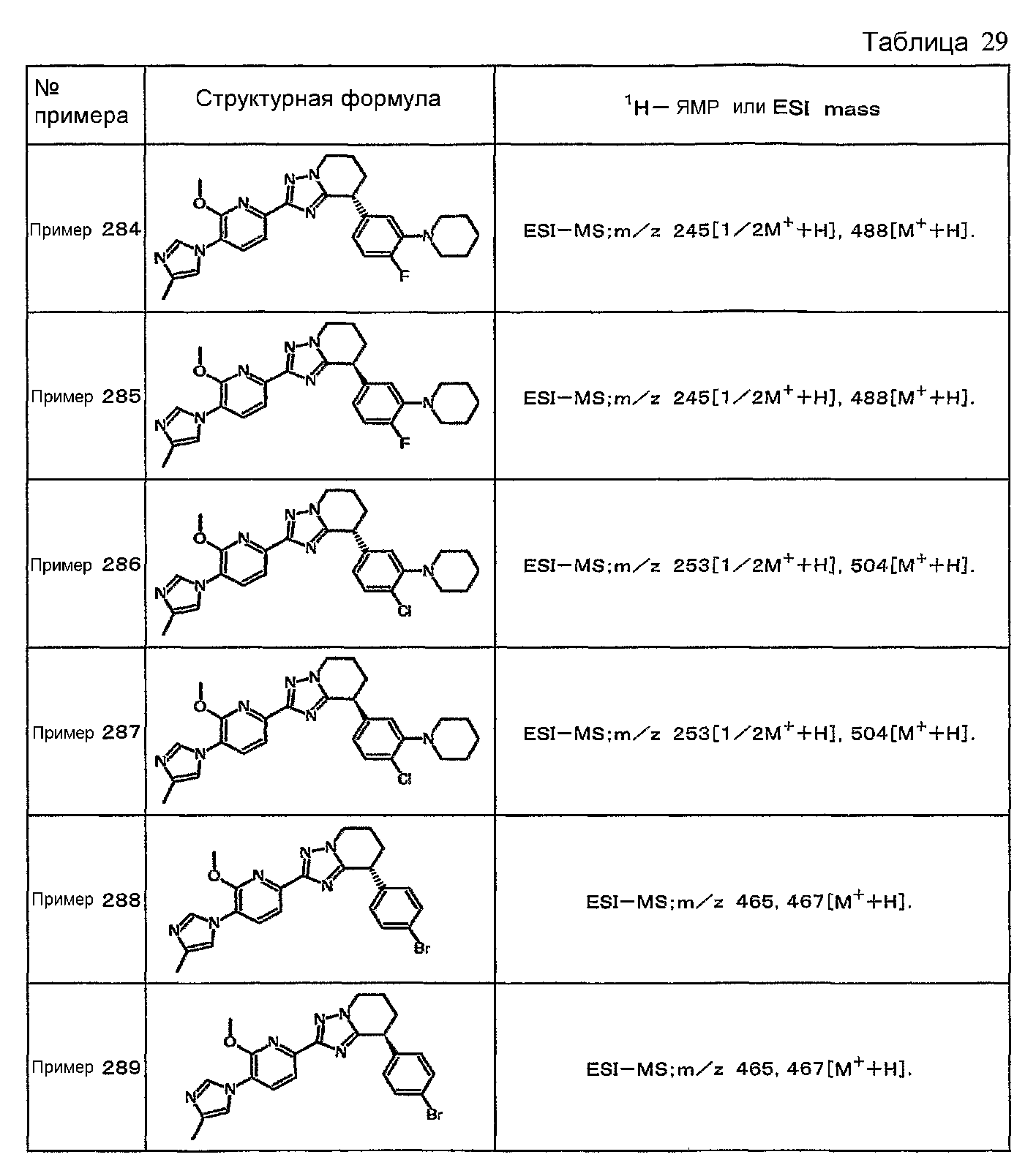
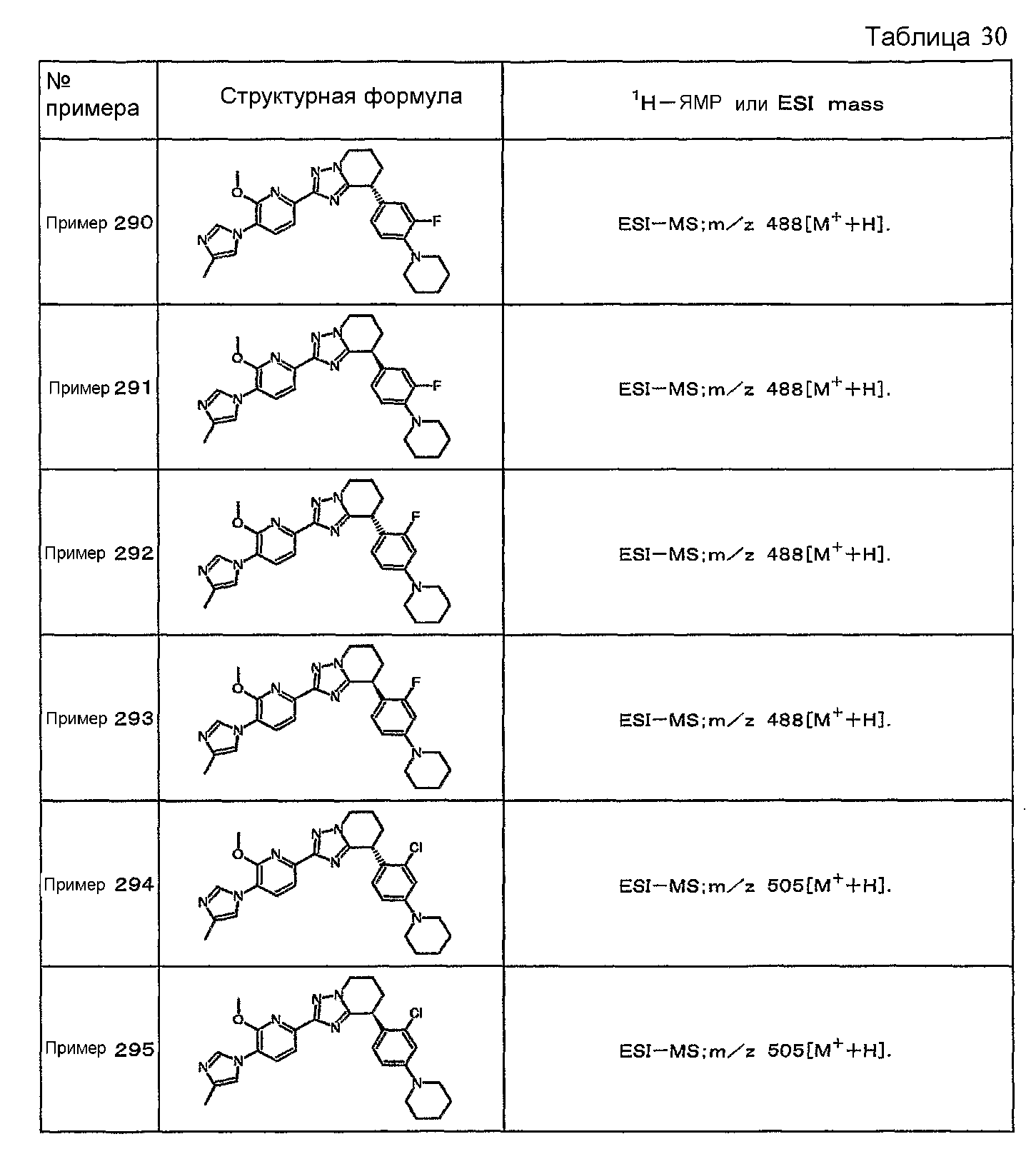
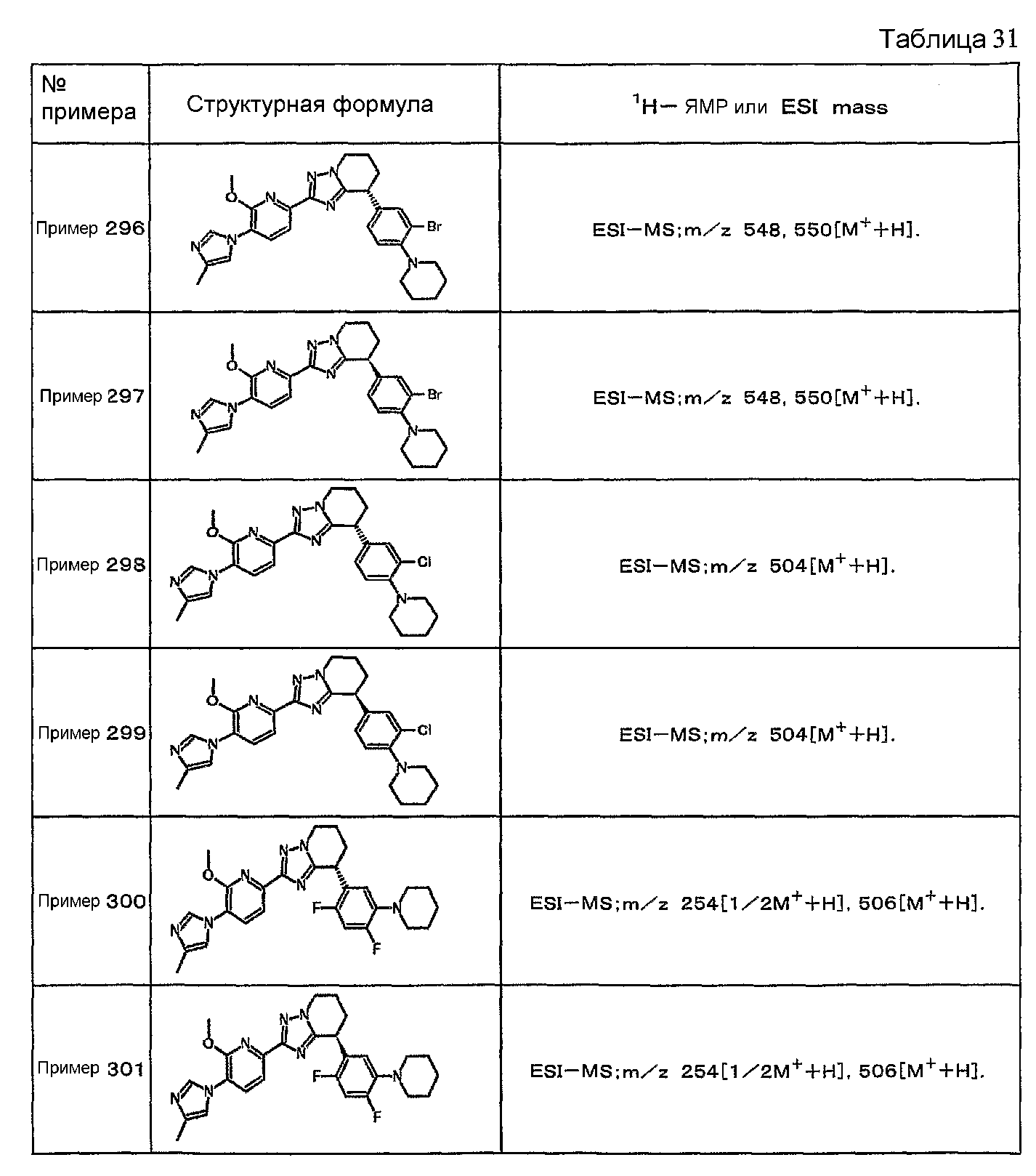
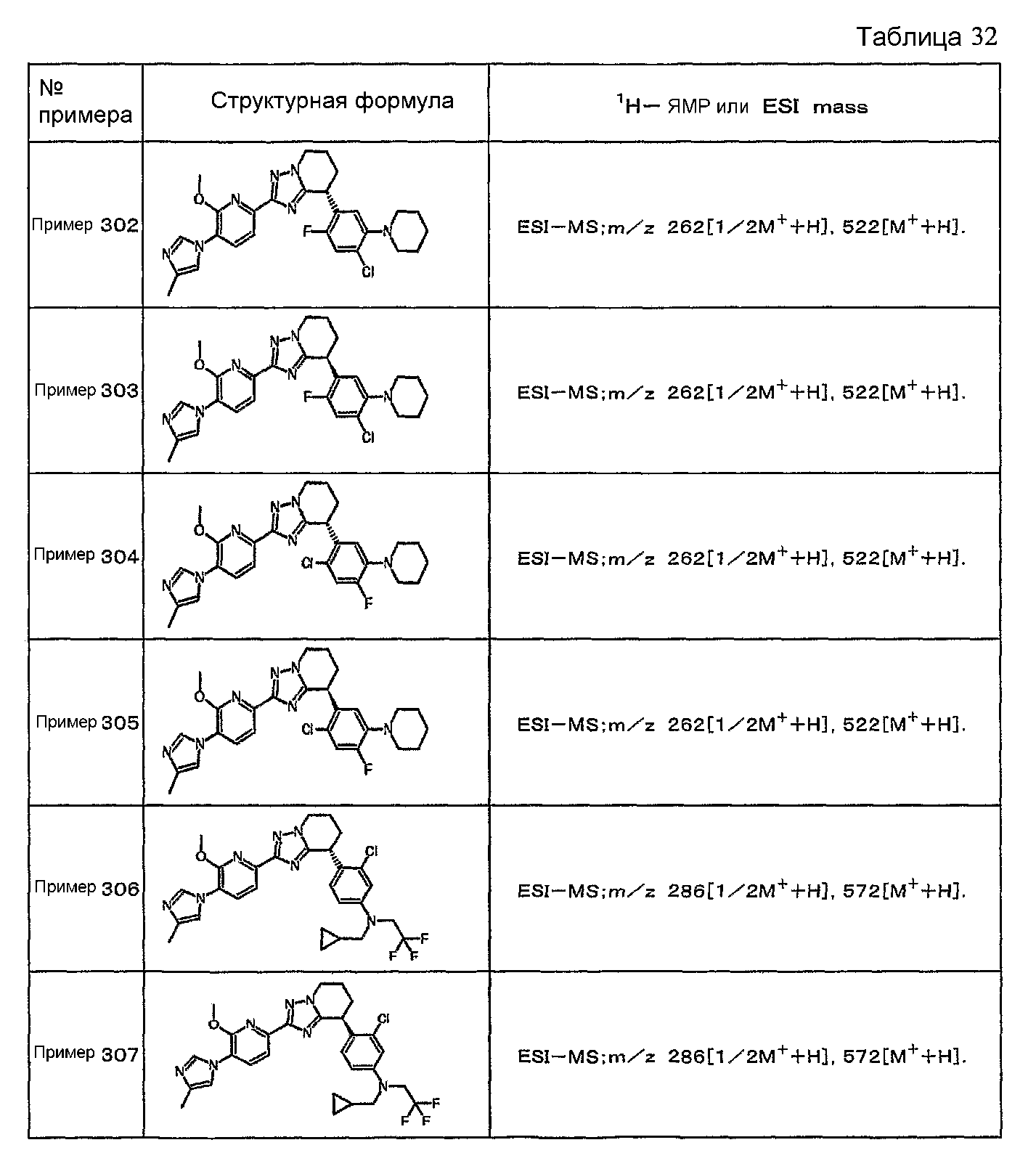
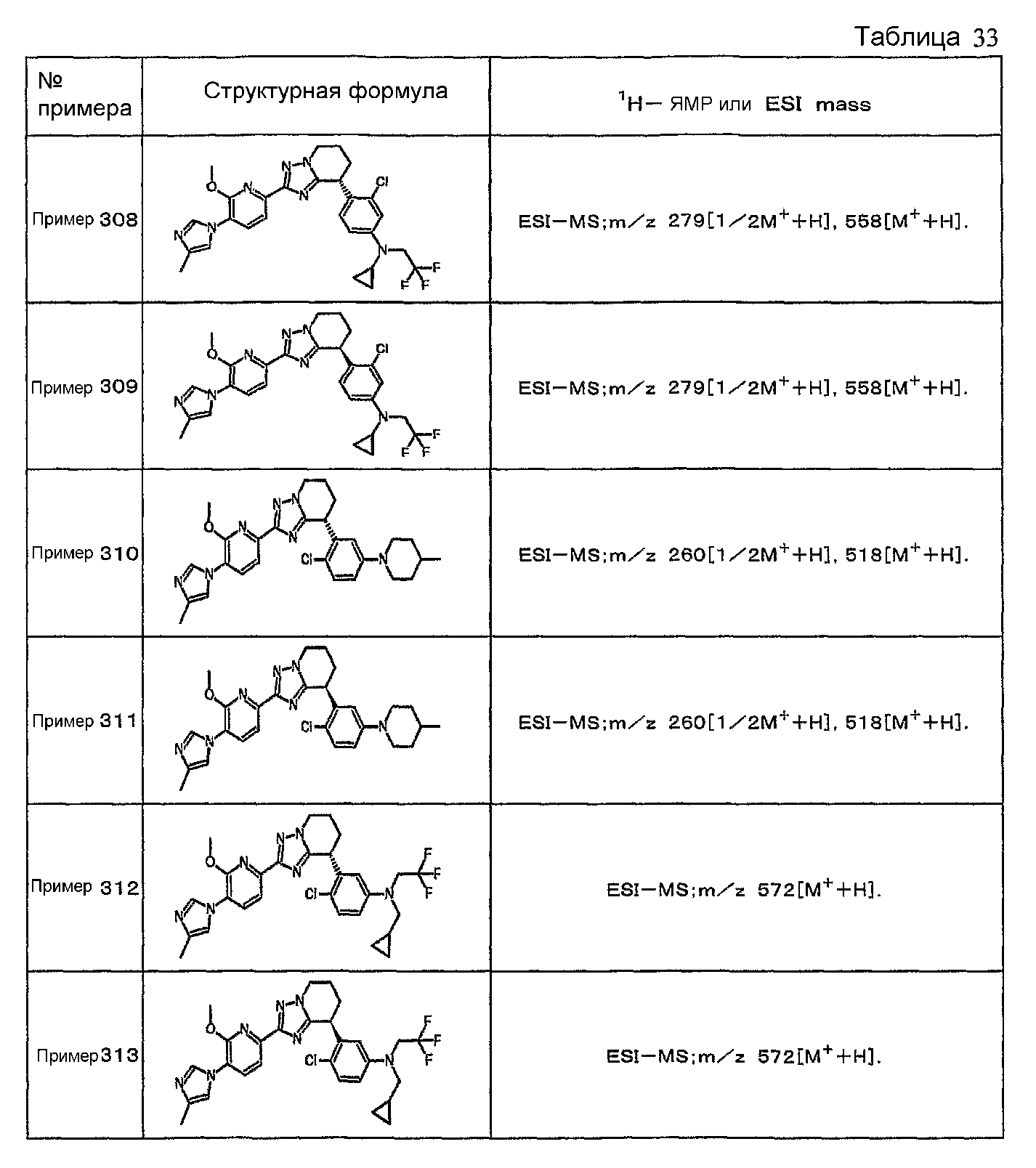
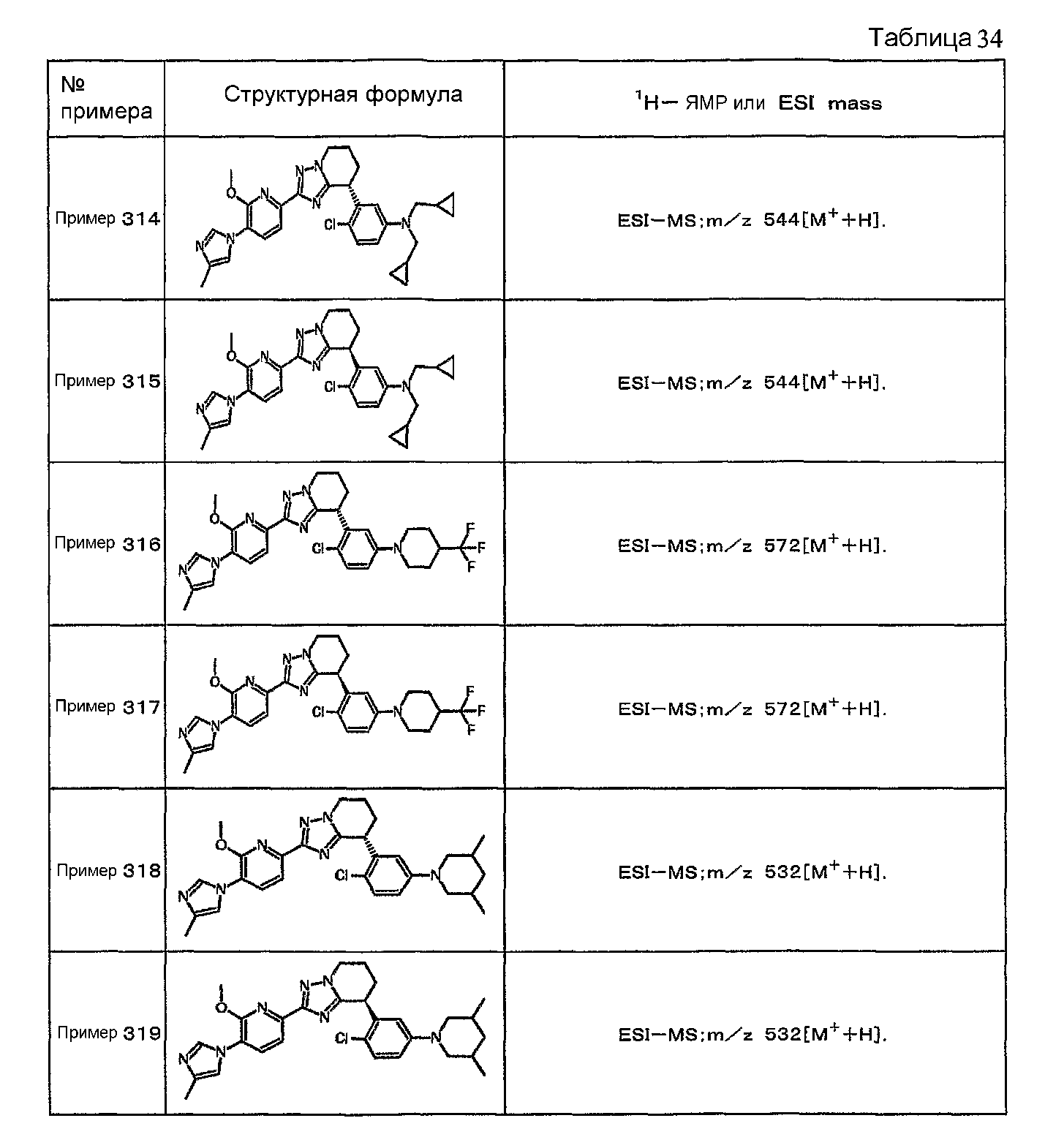
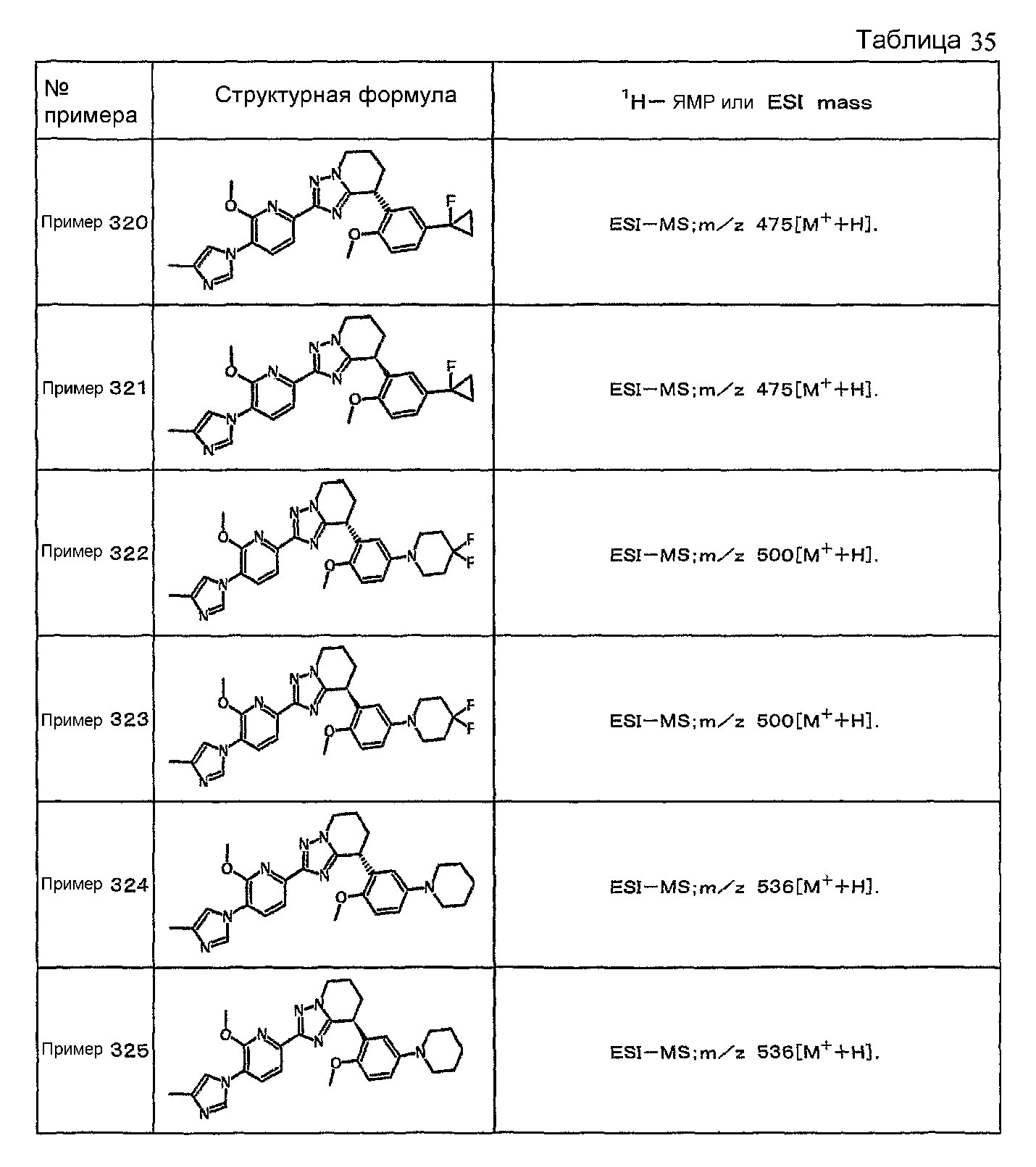
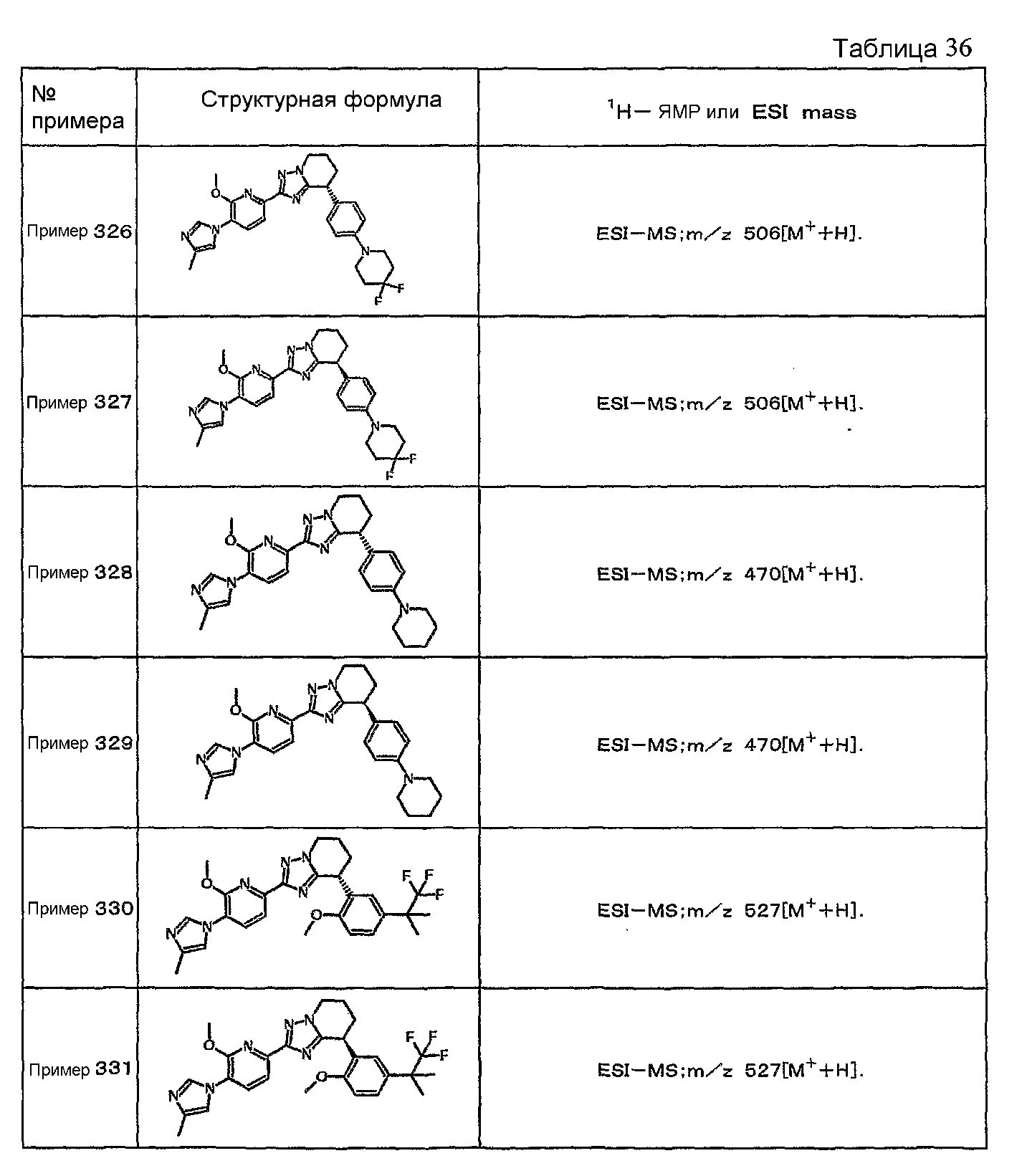
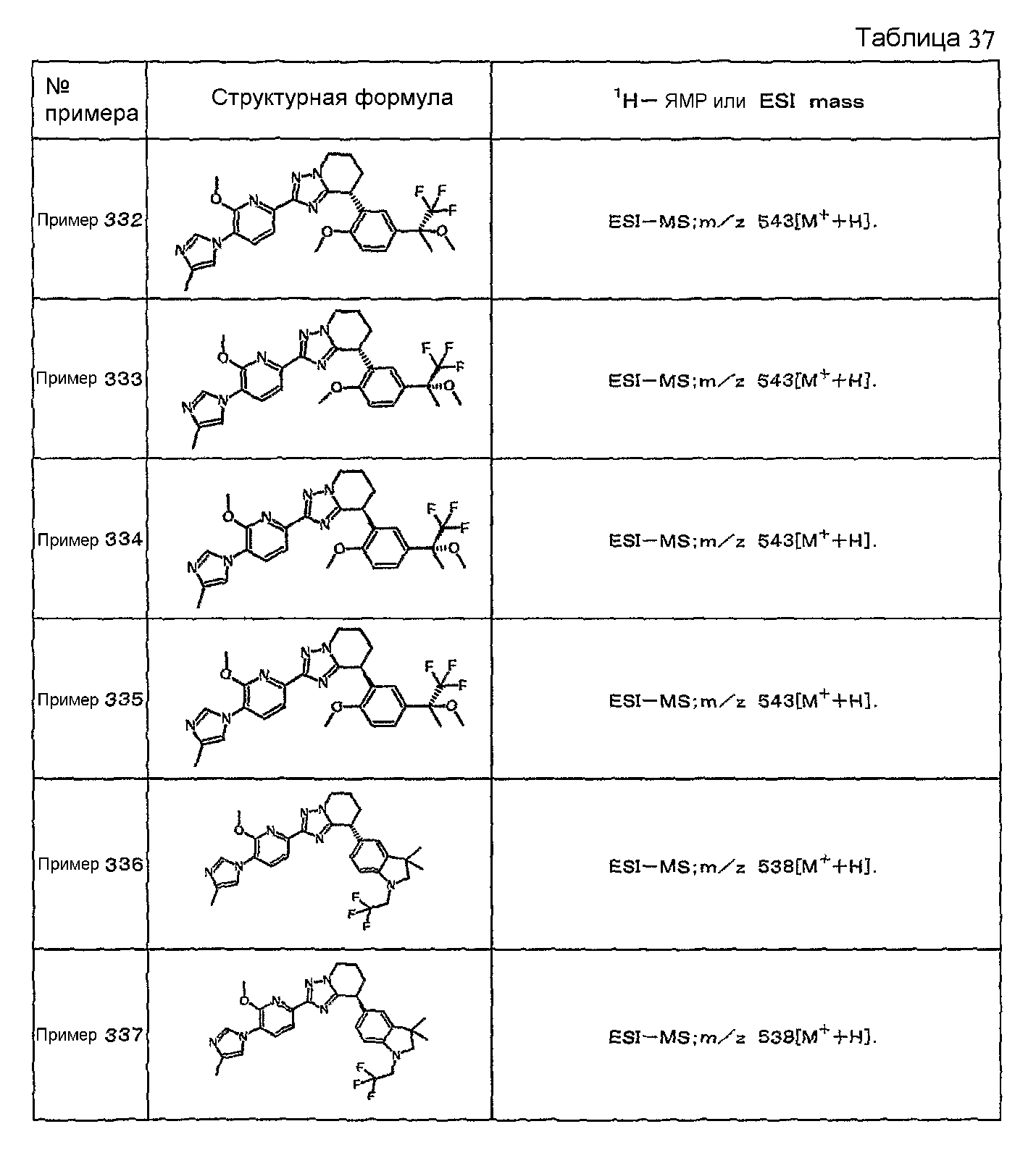
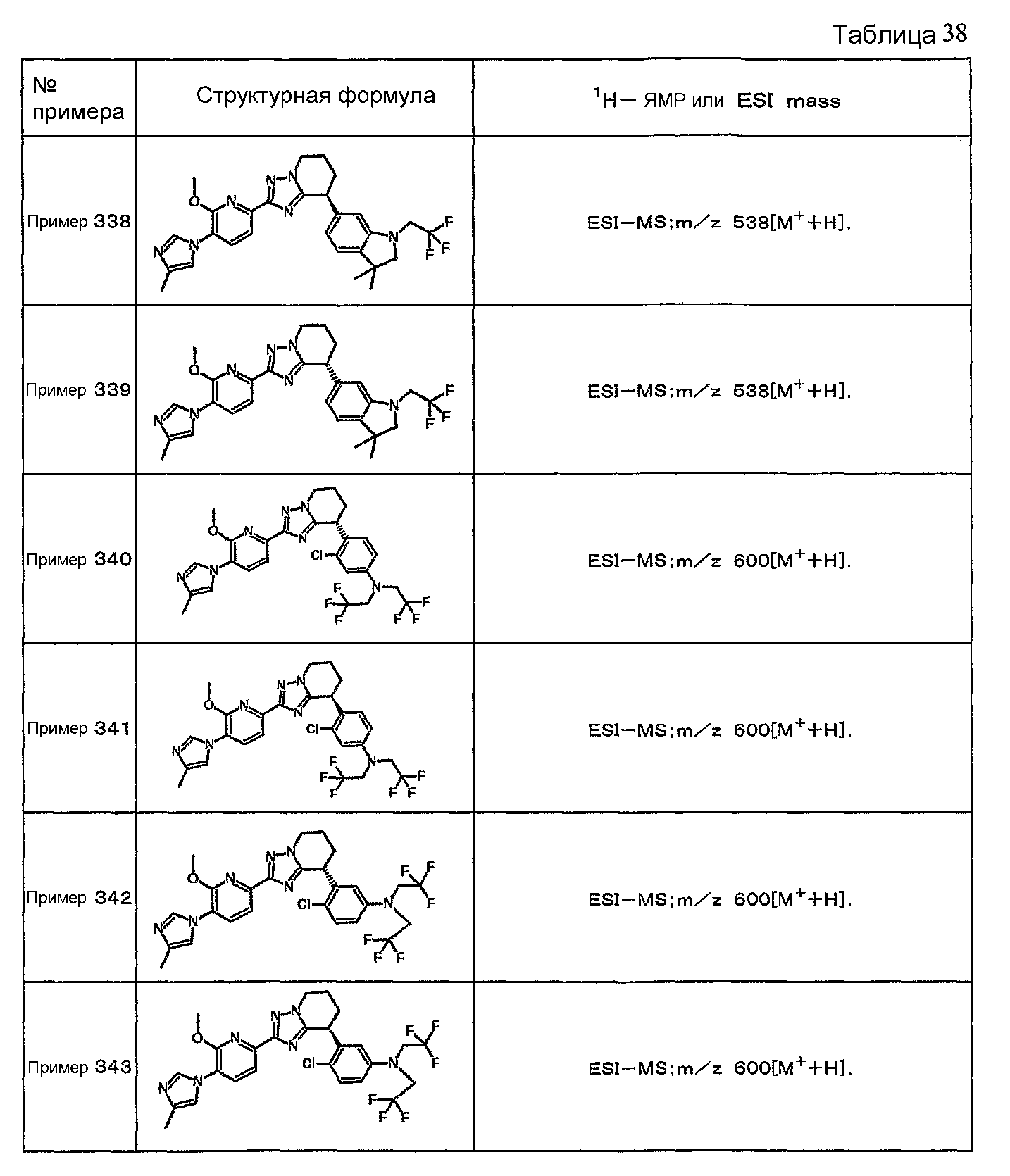
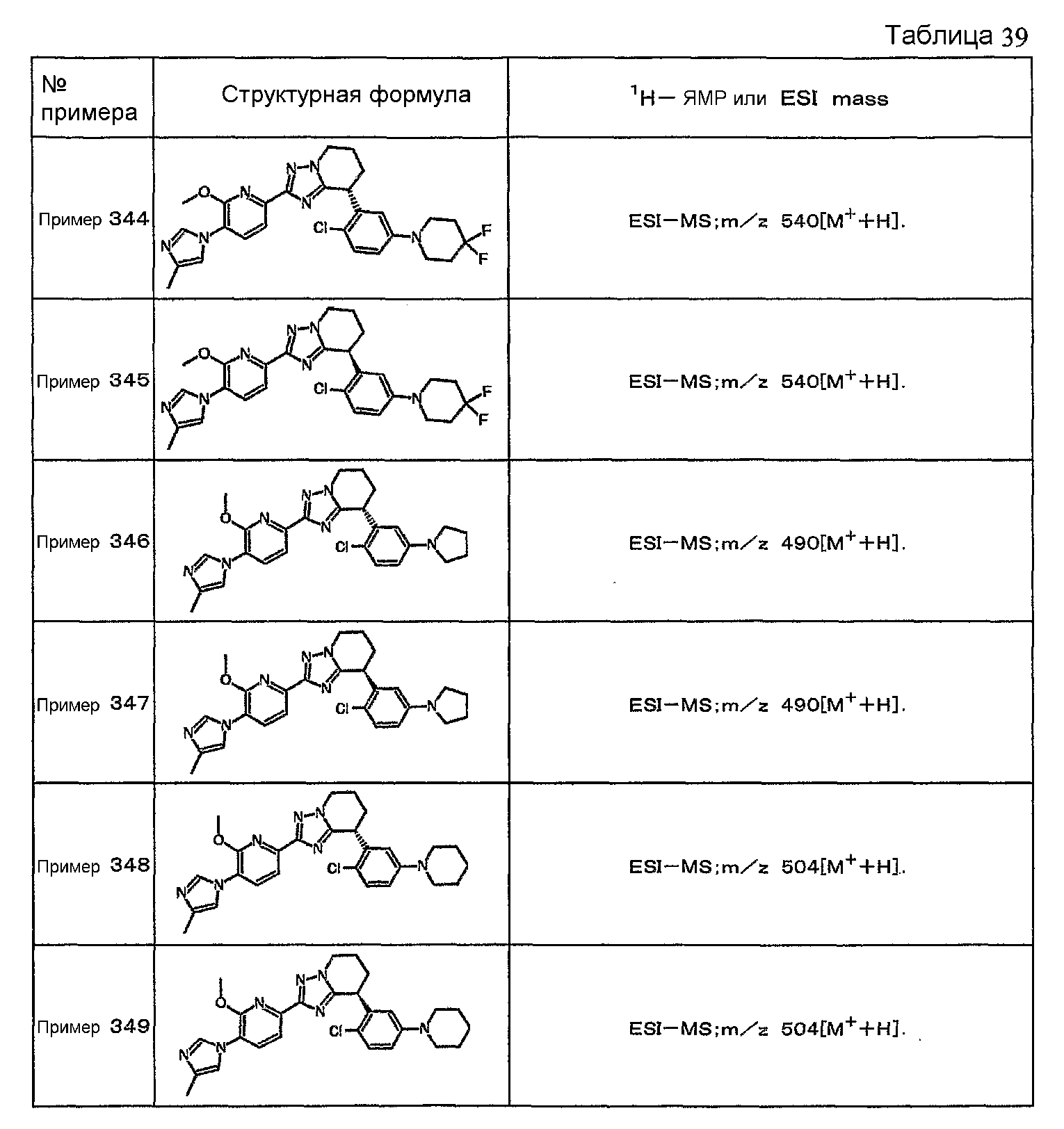
Примеры 350 и 351
Синтез (+)-8-(2-хлор-5-пиридин-3-ил)фенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-(2-хлор-5-пиридин-3-ил)фенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
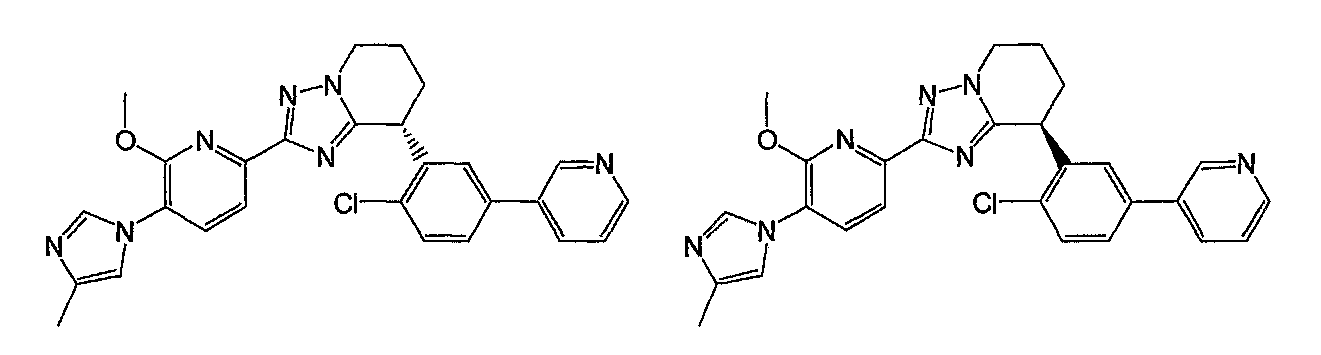
8-(5-Бром-2-фторфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин
Рацемат указанного в заголовке соединения (3,22 г) получали в соответствии со способом примеров 42 и 43 из гидрохлорида гидразида 6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-карбоновой кислоты (2,48 г), полученного в примере получения 1-6, и гидрохлорида этил 2-(5-бром-2-хлорфенил)-5-хлорпентанимидата (3,9 г), полученного в примере получения 3-64.
ESI-MS: m/z 501 [M++H].
Синтез (+)-8-(2-хлор-5-пиридин-3-ил)фенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-(2-хлор-5-пиридин-3-ил)фенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Рацемат указанного в заголовке соединения (181 мг) получали в соответствии со способом примеров 1 и 2 из 8-(5-бром-2-хлорфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина (200 мг) и 3-трибутилстаннилпиридина (221 мг).
Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см, подвижная фаза: 50% этанол-гексан) с получением указанного в заголовке соединения с положительным оптическим вращением (80 мг, >99% ee) и указанного в заголовке соединения с отрицательным оптическим вращением (78 мг, >99% ee).
Показатели свойств указанного в заголовке оптически активного соединения с положительным оптическим вращением были следующими.
ESI-MS: m/z 250 [1/2M++H], 498 [M++H].
Показатели свойств указанного в заголовке оптически активного соединения с отрицательным оптическим вращением были следующими.
ESI-MS: m/z 250 [1/2M++H], 498 [M++H].
Примеры 352-359
Соединения примеров 352-359 получали в соответствии со способом примеров 350 и 351, показанных в таблице 40.
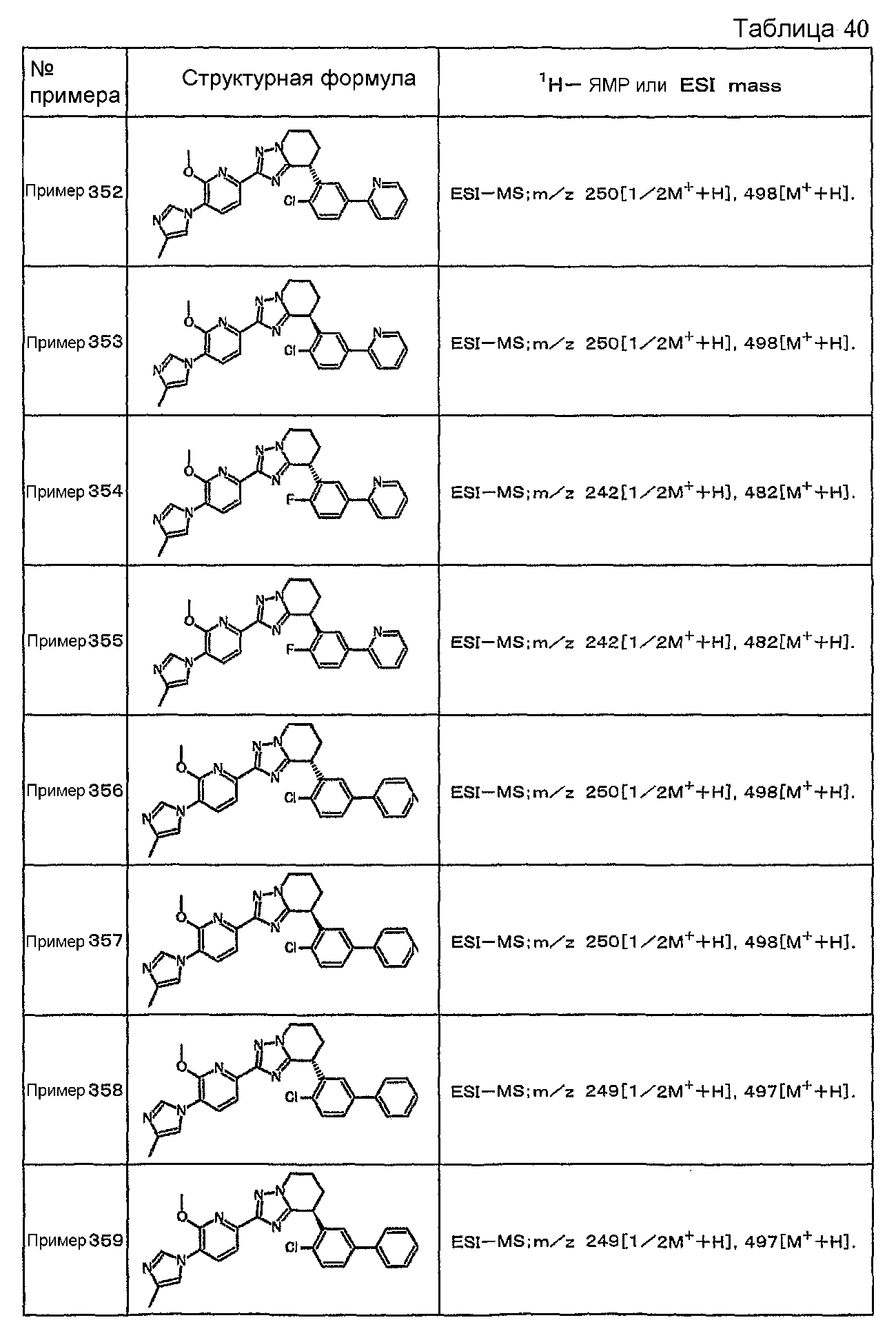
Примеры 360-361
Синтез (+)-8-[2-хлор-5-(5-трифторметилпиридин-2-ил)фенил]-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-[2-хлор-5-(5-трифторметилпиридин-2-ил)фенил]-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
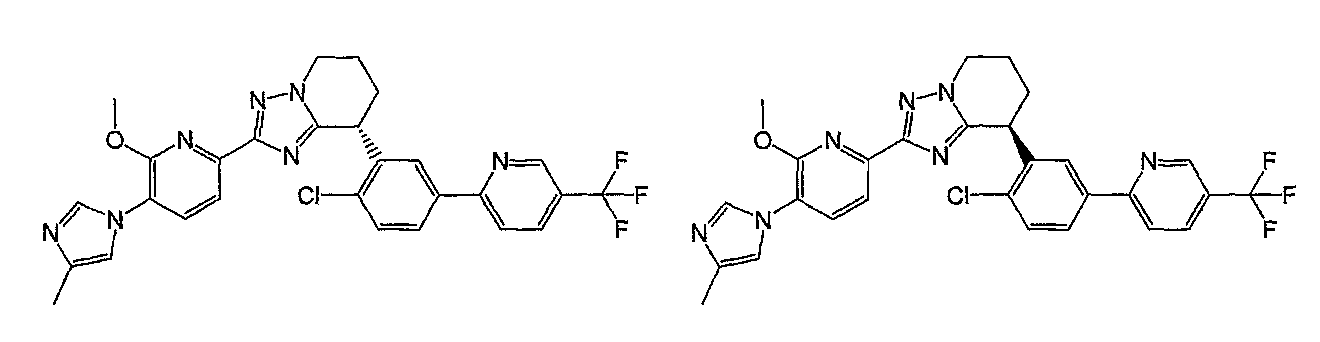
Синтез 8-(2-хлор-5-трибутилстаннилфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
К раствору 8-(5-бром-2-хлорфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина (500 мг), полученного в примерах 350 и 351, и гекса-н-бутилдиолова (633 мкл) в N-метил-2-пирролидине (10 мл) добавляли тетракис(трифенилфосфин)палладий (57,8 мг) и смесь перемешивали при 120°С в течение 7 ч в атмосфере азота. После охлаждения реакционной смеси добавляли этилацетат и смесь промывали водой. Полученный органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом натрия. После отфильтровывания осушающего агента органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH силикагелем с получением указанного в заголовке соединения (507 мг).
Синтез (+)-8-[2-хлор-5-(5-трифторметилпиридин-2-ил)фенил]-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-8-[2-хлор-5-(5-трифторметилпиридин-2-ил)фенил]-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Рацемат (253 мг) указанного в заголовке соединения получали в соответствии со способами примеров 1 и 2 из 8-(2-хлор-5-три-н-бутилстаннилфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина (507 мг) и 2-бром-5-(трифторметил)пиридина (242 мг). Полученный рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; элюирующий растворитель: гексан:этанол = 5:5, Daicel Co., Ltd.) с получением указанного в заголовке оптически активного соединения с положительным оптическим вращением (83 мг, >99% ee) и указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (84 мг, >99% ee).
Показатели свойств указанного в заголовке соединения с положительным оптическим вращением были следующими:
ESI-MS: m/z 284 [1/2M++H], 566 [M++H].
Показатели свойств указанного в заголовке соединения с отрицательным оптическим вращением были следующими:
ESI-MS: m/z 284 [1/2M++H], 566 [M++H].
Соединения примеров 362-363 получали в соответствии со способом примеров 361-362 (Таблица 29).
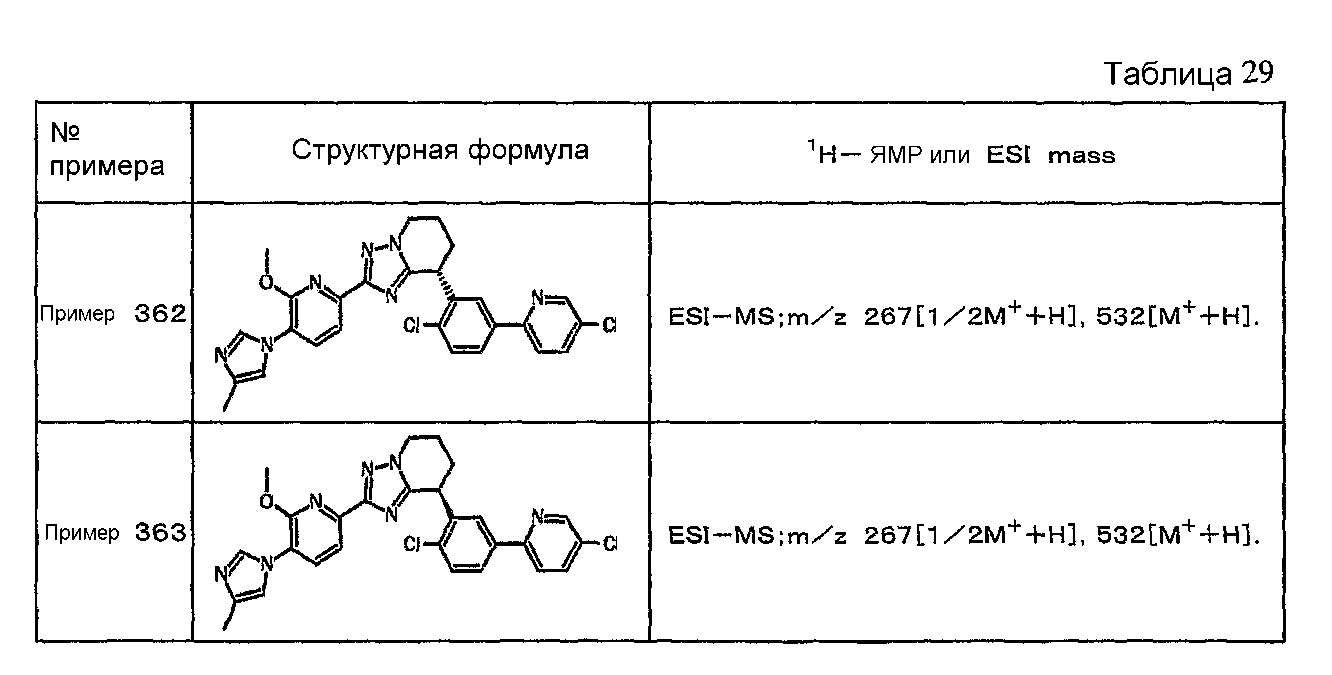
Соединения в примерах 364-371 получали такими же способами синтеза, как в примерах 100-101 (Таблица 30).
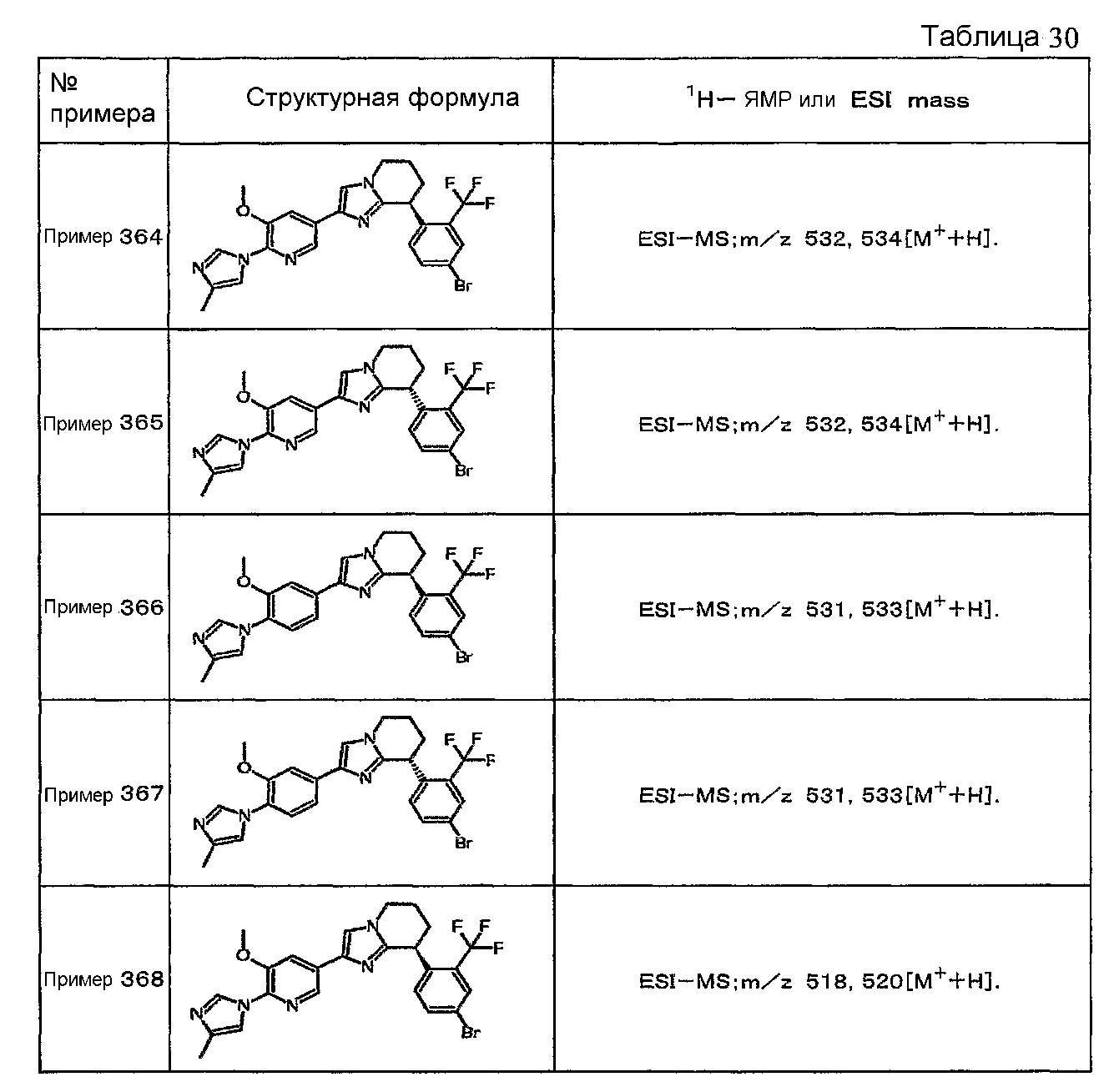
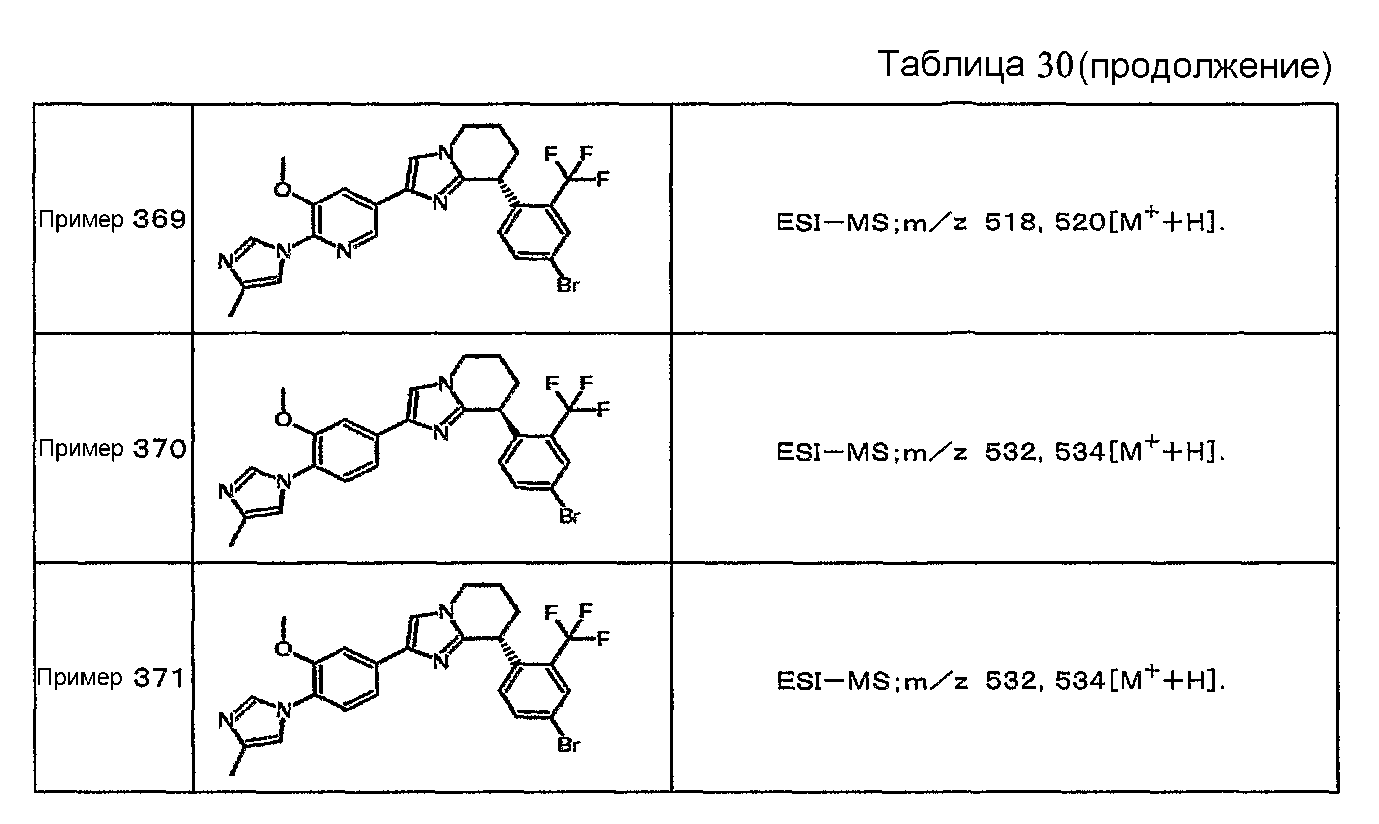
Примеры 372-373
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-пиперидин-1-илтиофен-2-ил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(5-пиперидин-1-илтиофен-2-ил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
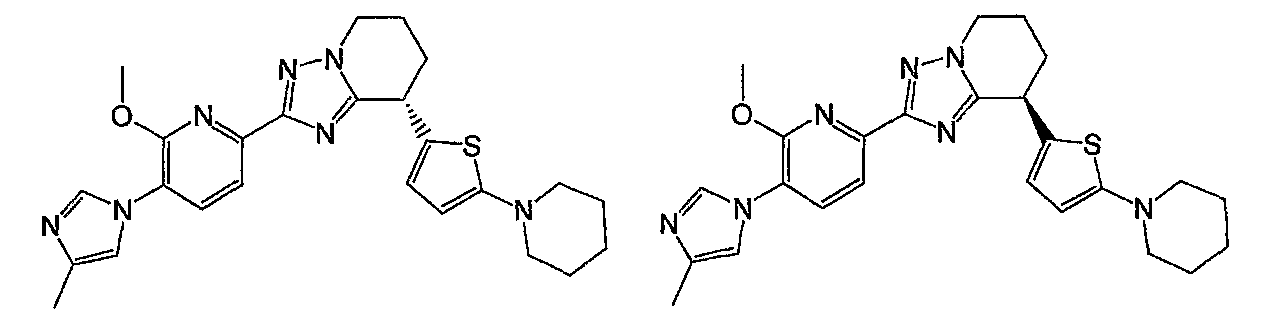
К раствору 8-(5-бромтиофен-2-ил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина (43,8 мг), полученного в примерах 82 и 83, и пиперидина (9,49 мг) в диоксане (2,0 мл) добавляли бис(дибензилиденацетон)палладий (2,67 мг), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (5,38 мг) и трет-бутоксид натрия (35,7 мг) и смесь перемешивали при 120°С в течение 3 ч в атмосфере азота. После охлаждения реакционную смесь фильтровали через рыхлый слой NH силикагеля и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на NH силикагеле с получением рацемата указанного в заголовке соединения (12,8 мг). Рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; элюирующий растворитель: гексан:этанол = 8:2, Daicel Co., Ltd.) с получением указанного в заголовке оптически активного соединения с положительным оптическим вращением (1,2 мг, >99% ee) и указанного в заголовке оптически активного соединения с отрицательным оптическим вращением (1,3 мг, >99% ee).
Показатель свойств указанного в заголовке соединения с положительным оптическим вращением был следующим:
ESI-MS: m/z 476 [M++H].
Показатель свойств указанного в заголовке соединения с отрицательным оптическим вращением был следующим:
ESI-MS: m/z 476 [M++H].
Соединения примеров 374-383 получали такими же способами, как в примерах 372-373 (Таблица 31).
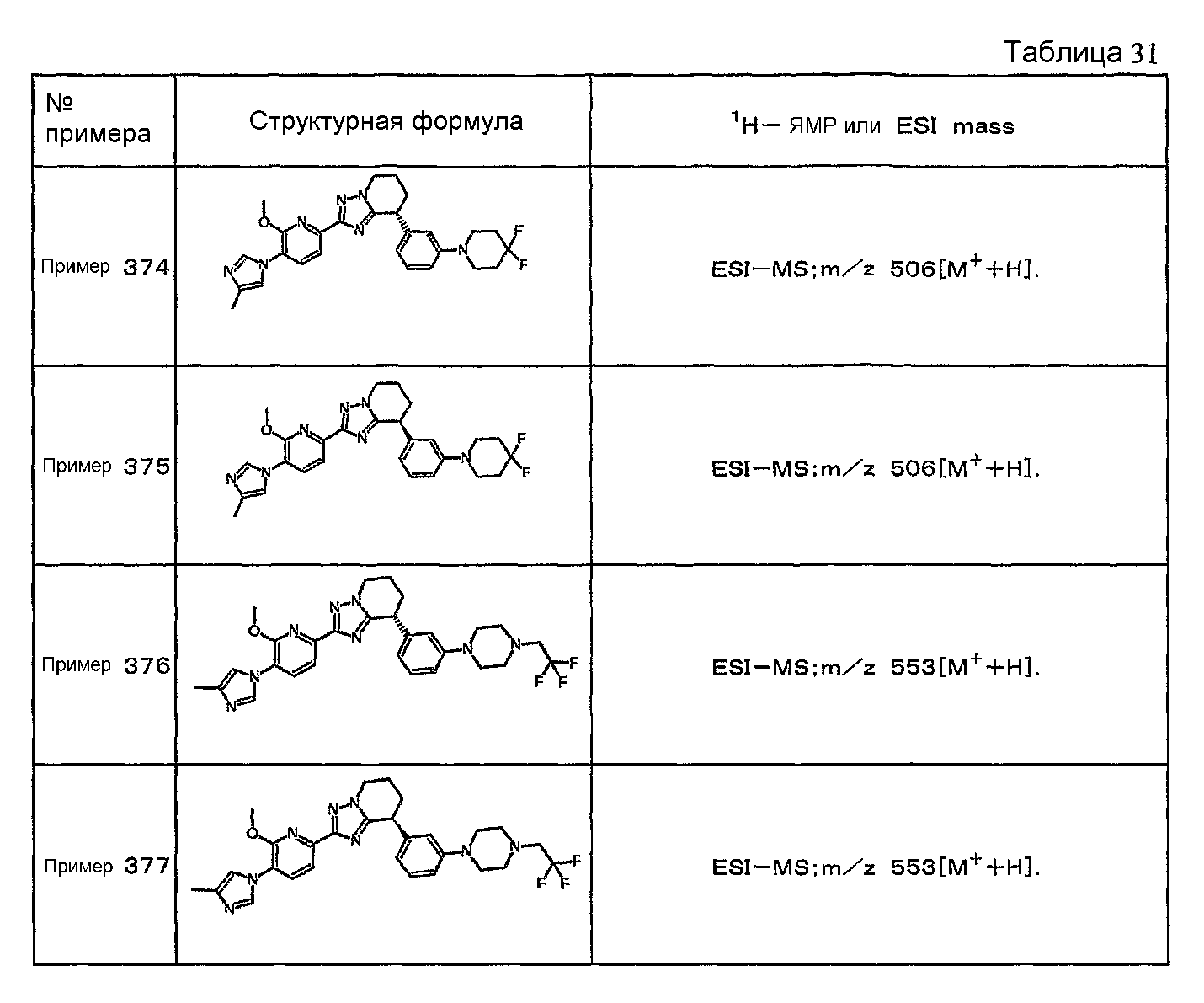
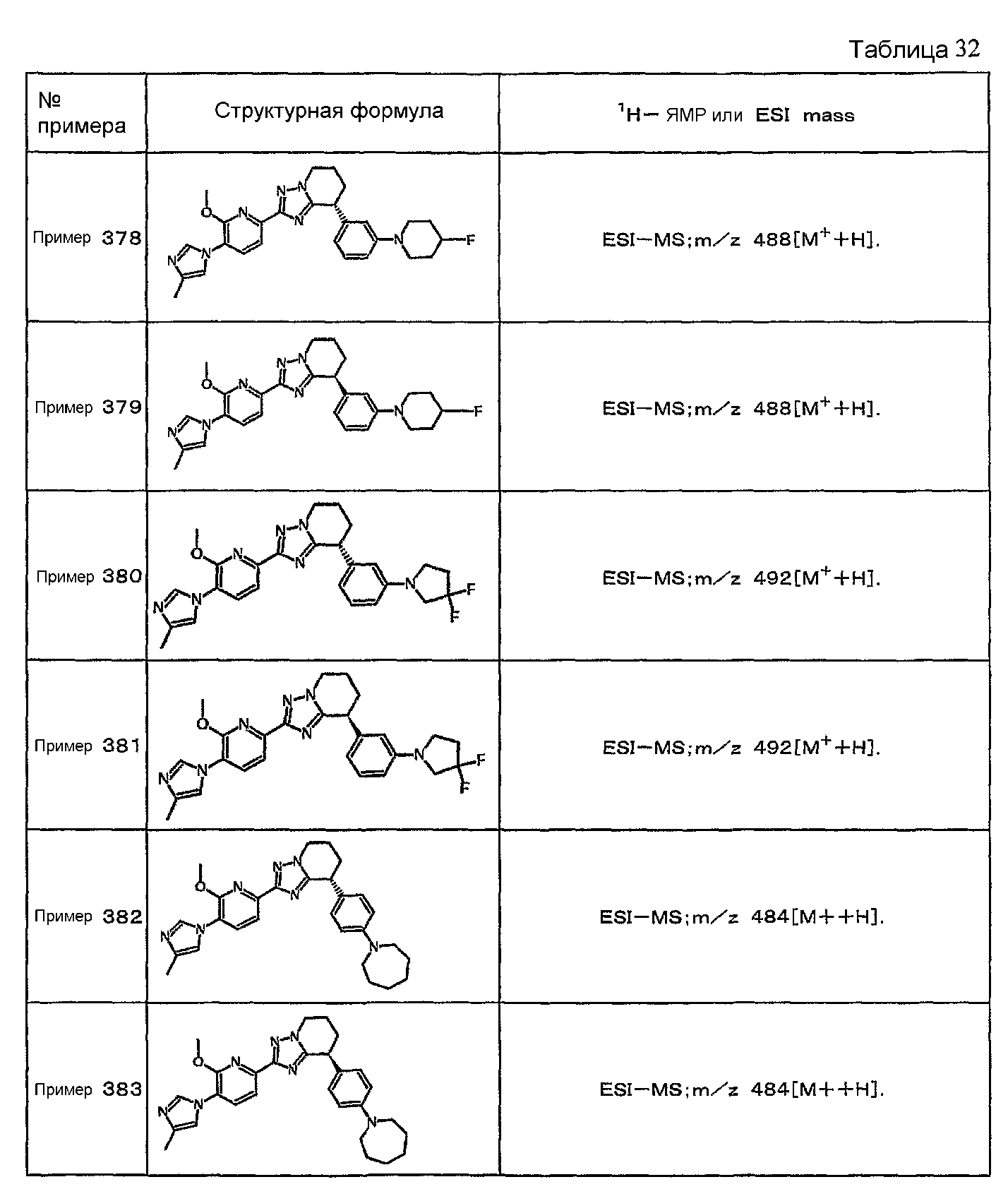
Соединения в примерах 384-385 получали такими же способами синтеза, как в примерах 150-151 (таблица 33).
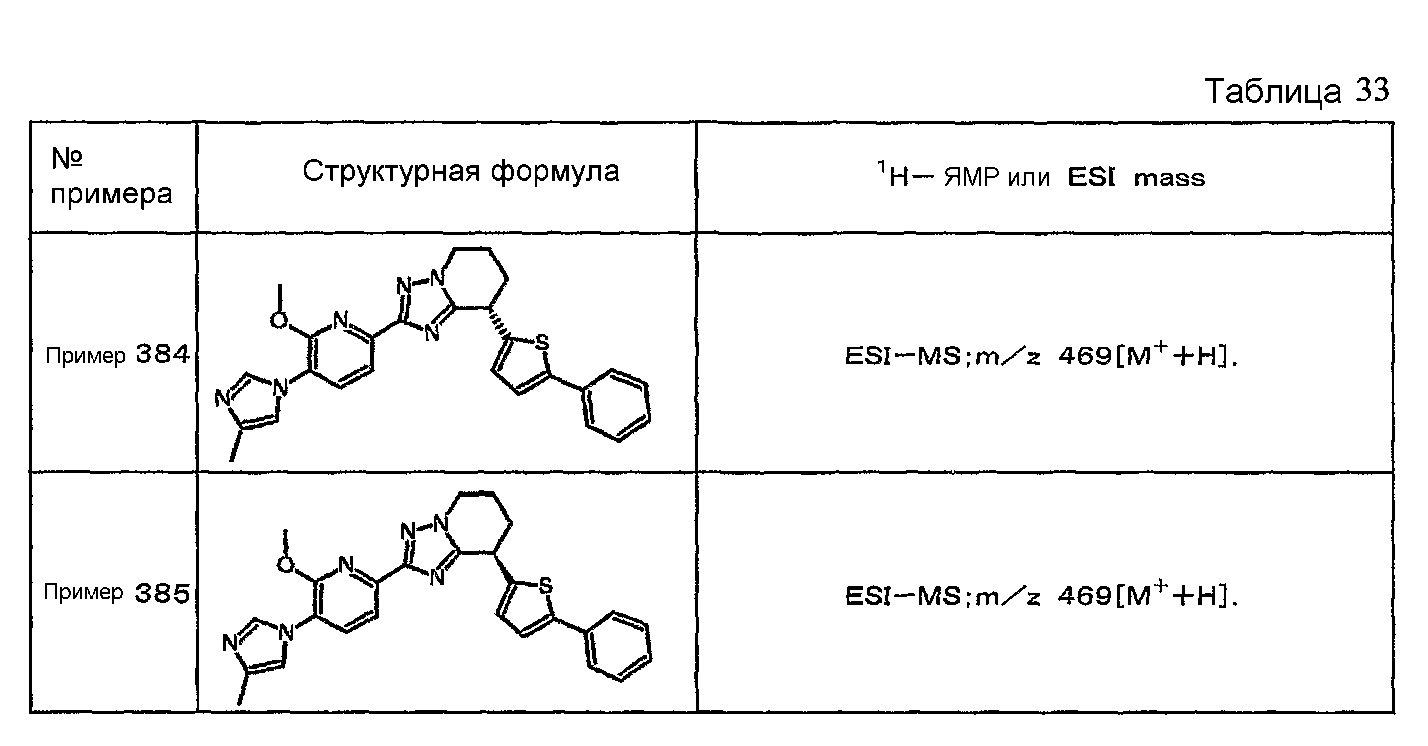
Примеры 386-387
Синтез (-)-2-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}-4-пиперидин-1-илбензонитрила и (+)-2-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-8-ил}-4-пиперидин-1-илбензонитрила
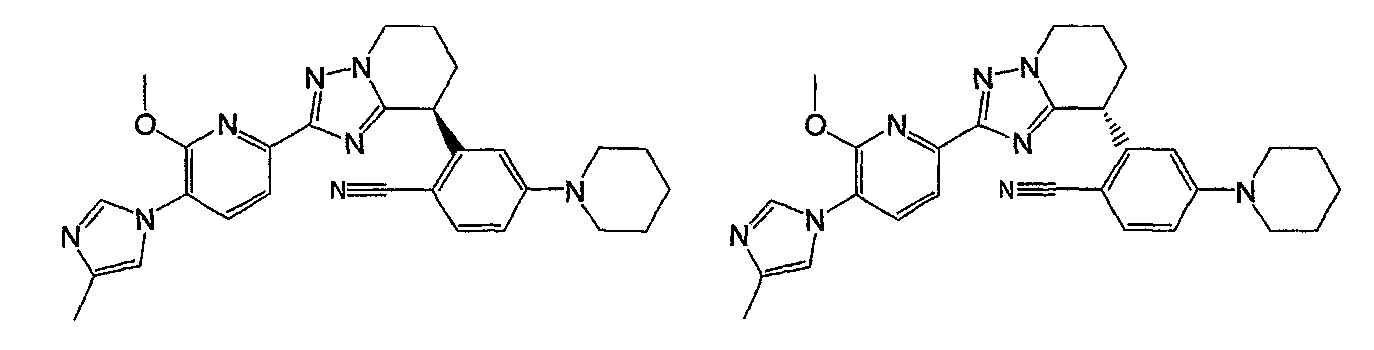
К раствору 8-(2-бром-5-пиперидин-1-илфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина (29 мг), полученного в примерах 278 и 279, и цианида цинка (13 мг) в N-метил-2-пирролидиноне (3 мл) добавляли тетракис(трифенилфосфин)палладий (6 мг) и реакционную смесь перемешивали при 130°С в течение 1 ч в микроволновом реакторе. Затем добавляли воду и реакционную смесь экстрагировали этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом натрия. После отфильтровывания осушающего агента фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH силикагелем с получением рацемата указанного в заголовке соединения (20 мг). Рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; элюирующий растворитель: гексан:этанол = 4:6, Daicel Co., Ltd.). Получали указанное в заголовке оптически активное соединение с отрицательным оптическим вращением (6 мг, >99% ee) и указанное в заголовке оптически активное соединение с положительным оптическим вращением (7 мг, >99% ee).
Показатель свойств указанного в заголовке соединения с отрицательным оптическим вращением был следующим:
ESI-MS: m/z 248 [1/2M++H], 495 [M++H].
Показатель свойств указанного в заголовке соединения с положительным оптическим вращением был следующим:
ESI-MS: m/z 248 [1/2M++H], 495 [M++H].
Соединения в примерах 388-389 получали такими же способами синтеза, как в примерах 386-387 (таблица 34).
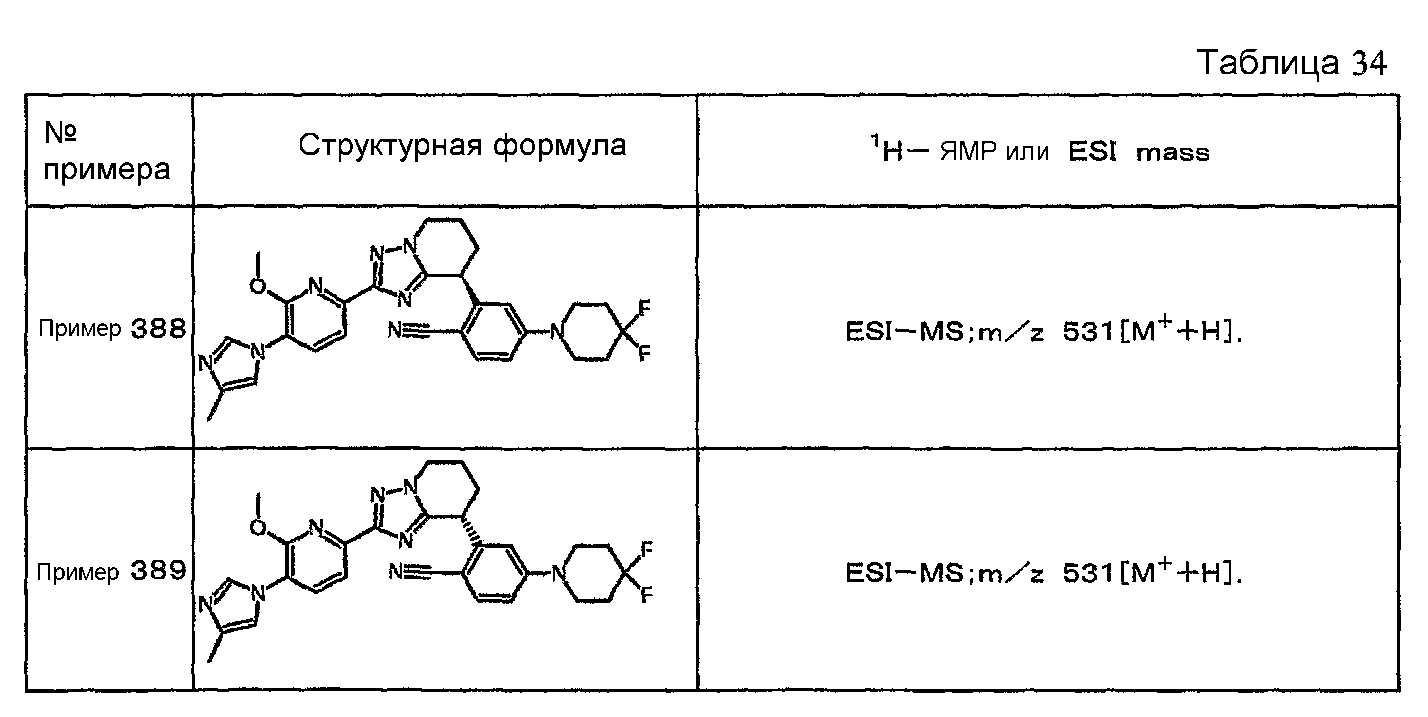
Примеры 390-391
Синтез (+)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-триметилсиланилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина и (-)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(4-триметилсиланилфенил)-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина
Суспензию 8-[4-бромфенил]-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридина (40 мг), полученного в примерах 288 и 289, трис(дибензилиденацетон)дипалладия (1,6 мг), 2-(ди-трет-бутилфосфино)бифенила (2,6 мг) и фторида калия (25 мг) в 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидиноне (3 мл) перемешивали при комнатной температуре в течение 5 мин. Добавляли к реакционной смеси тетраметилдисилан (21 мкл) и воду (28 мкл) и смесь перемешивали при 150°С в течение 1 ч в микроволновом реакторе. Добавляли к реакционной смеси воду и смесь экстрагировали этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом натрия. После отфильтровывания осушающего агента фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH силикагелем с получением рацемата указанного в заголовке соединения (22 мг). Неочищенный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; элюирующий растворитель: гексан:этанол = 6:4, Daicel Co., Ltd.). Получали указанное в заголовке оптически активное соединение с положительным оптическим вращением (1,7 мг, >99% ee) и указанное в заголовке оптически активное соединение с отрицательным оптическим вращением (1 мг, >99% ee).
Показатель свойств указанного в заголовке соединения с положительным оптическим вращением был следующим:
ESI-MS: m/z 459 [M++H].
Показатель свойств указанного в заголовке соединения с отрицательным оптическим вращением был следующим:
ESI-MS: m/z 459 [M++H].
Примеры 392-393
Синтез (+)-1-(3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазин-8-ил}фенил)пирролидин-2-она и (-)-1-(3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазин-8-ил}фенил)пирролидин-2-она
Синтез 3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазин-8-ил}фениламина
Суспензию 2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-8-(3-нитрофенил)-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазина (703 мг), полученного в примерах 66 и 67, железного порошка (362 мг) и аммонийхлорида (693 мг) в смеси этанола (25 мл) и воды (5 мл) перемешивали при 90°С в течение 5,3 ч и затем при комнатной температуре в течение ночи. Нерастворимый продукт удаляли фильтрованием через Целит и вытекающий поток концентрировали при пониженном давлении. К остатку добавляли насыщенный водный раствор бикарбоната натрия и смесь экстрагировали этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом магния. После отфильтровывания осушающего агента органический слой концентрировали при пониженном давлении с получением указанного в заголовке соединения (403 мг).
Синтез (+)-1-(3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазин-8-ил}фенил)пирролидин-2-она и (-)-1-(3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазин-8-ил}фенил)пирролидин-2-она
К раствору 3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазин-8-ил}фениламина (49 мг) и триэтиламина (67,6 мкл) в дихлорметане (4,3 мл) добавляли 4-хлорбутирилхлорид (15,2 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Насыщенный водный раствор бикарбоната натрия добавляли к реакционной смеси и смесь экстрагировали хлороформом. Экстракт концентрировали при пониженном давлении и полученный остаток растворяли в ТГФ (5 мл). К раствору добавляли трет-бутоксид калия (13,6 мг) и реакционную смесь перемешивали при комнатной температуре в течение 3,5 ч. После добавления насыщенного водного раствора бикарбоната натрия реакционную смесь экстрагировали этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом магния. После отфильтровывания осушающего агента фильтрат концентрировали при пониженном давлении с получением неочищенного рацемата указанного в заголовке соединения. Рацемат разделяли с помощью CHIRALPAK™ IB (2 см × 25 см; элюирующий растворитель: гексан:этанол = 5:5, Daicel Co., Ltd.). Получали указанное в заголовке оптически активное соединение с положительным оптическим вращением (6,6 мг) и указанное в заголовке оптически активное соединение с отрицательным оптическим вращением (6 мг).
Показатель свойств указанного в заголовке соединения с положительным оптическим вращением был следующим:
ESI-MS: m/z 472 [M++H].
Показатель свойств указанного в заголовке соединения с отрицательным оптическим вращением был следующим:
ESI-MS: m/z 472 [M++H].
Примеры 394-395
Синтез N-(+)-(3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазин-8-ил}фенил)циклогексанкарбоксамида и N-(-)-(3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазин-8-ил}фенил)циклогексанкарбоксамида
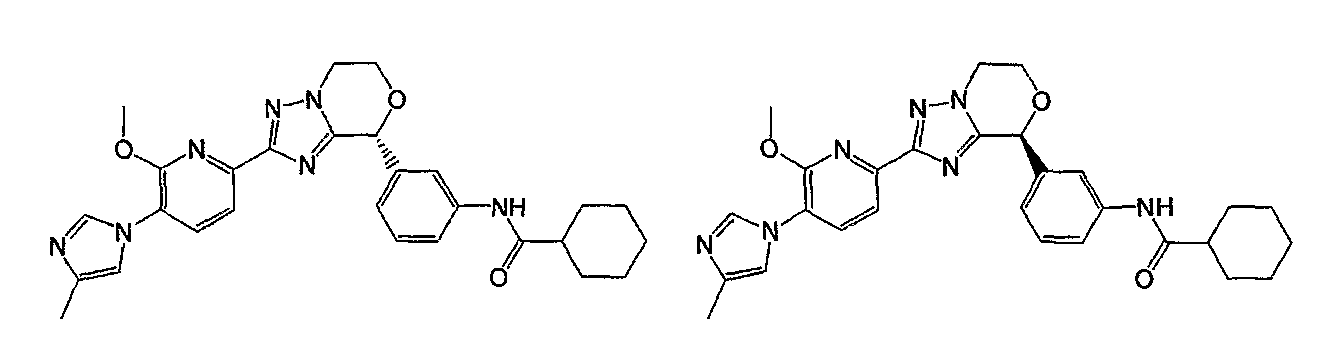
К раствору 3-{2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6-дигидро-8H-[1,2,4]триазоло[1,5-c][1,4]оксазин-8-ил}фениламина (40 мг), полученного в примерах 392 и 393, и триэтиламина (41,4 мкл) в дихлорметане (3 мл) добавляли циклогексанкарбонилхлорид (16,1 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 4 суток. Насыщенный водный раствор бикарбоната натрия добавляли к реакционной смеси и смесь экстрагировали хлороформом. Экстракт концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с NH силикагелем с получением неочищенного рацемата указанного в заголовке соединения (35 мг). Неочищенный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см; элюирующий растворитель: гексан:этанол = 6,5:3,5, Daicel Co., Ltd.). Получали указанное в заголовке оптически активное соединение с положительным оптическим вращением (17 мг) и указанное в заголовке оптически активное соединение с отрицательным оптическим вращением (12 мг).
Показатель свойств указанного в заголовке соединения с положительным оптическим вращением был следующим:
ESI-MS: m/z 514 [M++H].
Показатель свойств указанного в заголовке соединения с отрицательным оптическим вращением был следующим:
ESI-MS: m/z 514 [M++H].
Соединения в примерах 396-397 получали такими же способами синтеза, как в примерах 394-395 (таблица 35).
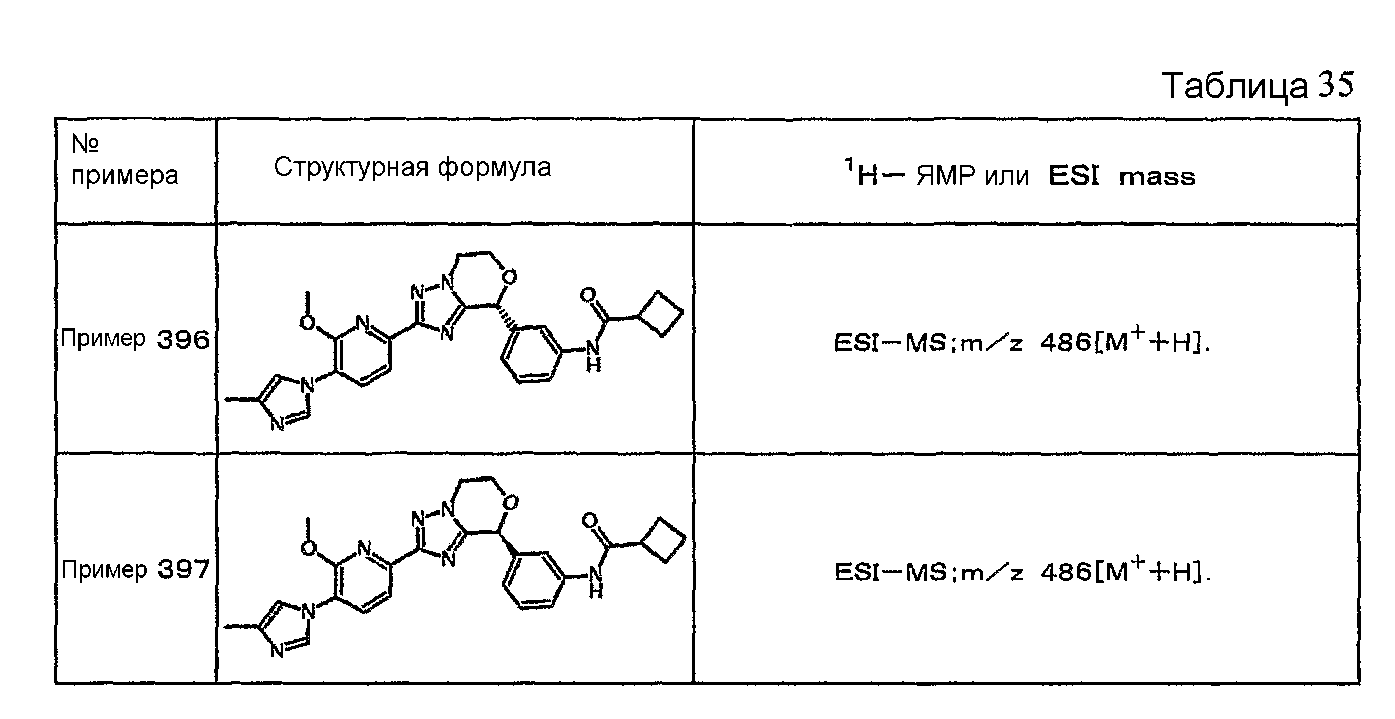
Примеры 398-399
Синтез (+)-8-бифенил-3-ил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-7-метил-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина и (-)-8-бифенил-3-ил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-7-метил-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
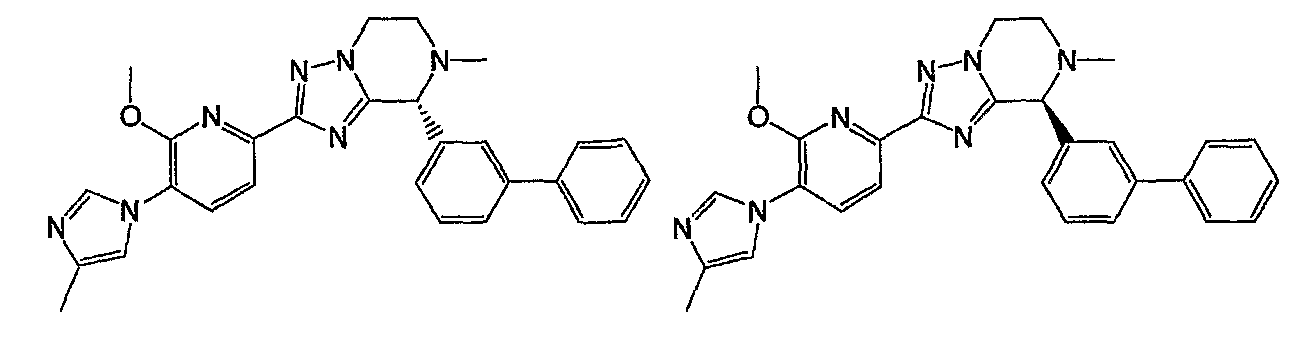
Синтез 7-бензил-8-бифенил-3-ил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
К раствору 7-бензил-8-(3-бромфенил)-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина (400 мг), полученного в примере 84 и 85, в толуоле (18 мл) и метаноле (4 мл) добавляли 2 М водный раствор карбоната натрия (395 мкл), фенилбороновую кислоту (219 мг) и тетракис(трифенилфосфин)палладий (83,1 мг). Смесь перемешивали при 120°С в течение 7 ч. Добавляли к реакционной смеси воду и смесь и экстрагировали этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом магния. После отфильтровывания осушающего агента фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (541 мг).
Синтез 8-бифенил-3-ил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
К раствору 7-бензил-8-бифенил-3-ил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина (541 мг) и уксусной кислоты (1,32 мл) в метаноле (52 мл) добавляли 20% гидроксид палладия (743 мг) и реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 4 суток. После того как реакционную атмосферу вытесняли азотом, добавляли к реакционной смеси этилацетат. Катализатор удаляли фильтрованием через Целит. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с NH силикагелем с получением указанного в заголовке соединения (45 мг) и исходного материала (291 мг).
Синтез (+)-8-бифенил-3-ил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-7-метил-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина и (-)-8-бифенил-3-ил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-7-метил-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина
Смесь 8-бифенил-3-ил-2-[6-метокси-5-(4-метил-1H-имидазол-1-ил)пиридин-2-ил]-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина (45 мг), формальдегида (37% раствор) (2 мл) и муравьиной кислоты (2 мл) кипятили с обратным холодильником в течение 3 ч. Реакционную смесь охлаждали на льду и нейтрализовали насыщенным водным раствором бикарбоната натрия, после чего экстрагировали этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом магния. После отфильтровывания осушающего агента фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH силикагелем с получением неочищенного рацемата указанного в заголовке соединения (41 мг). Полученный неочищенный рацемат разделяли с помощью CHIRALPAK™ IA (2 см × 25 см, Daicel Co., Ltd.). Получали указанное в заголовке оптически активное соединение с положительным оптическим вращением (17 мг) и указанное в заголовке оптически активное соединение с отрицательным оптическим вращением (14 мг).
Показатель свойств указанного в заголовке соединения с положительным оптическим вращением был следующим:
ESI-MS: m/z 478 [M++H].
Показатель свойств указанного в заголовке соединения с отрицательным оптическим вращением был следующим:
ESI-MS: m/z 478 [M++H].
Пример 1 испытания
Количественное определение Aβ пептида в культуре нейронов из головного мозга плода крысы
Авторы настоящего изобретения провели следующие испытания, чтобы показать полезность соединения общей формулы (I) по настоящему изобретению.
(1) Первичная культура нервных клеток крысы
Первичные культуры нейронов получали из коры головного мозга 18-дневных эмбрионов крыс Wistar (Charles River Japan, Yokohama, Japan). В частности, эмбрионы были асептически извлечены из беременных крыс под эфирной анестезией. Мозг извлекали из эмбриона и погружали в охлаждаемую льдом среду L-15 (например, Invitrogen Corp. Cat. #11415-064, Carlsbad, CA, USA, или SIGMA L1518). Кору головного мозга брали из извлеченного мозга под стереоскопическим микроскопом. Собранные фрагменты коры головного мозга ферментативно обрабатывали в ферментном растворе, содержащем 0,25% трипсин (Invitrogen Corp. Cat. #15050-065, Carlsbad, CA, USA) и 0,01% DNase (Sigma D5025, St. Louis, MO, USA), при 37°C в течение 30 минут для диспергирования клеток. После этого ферментативную реакцию останавливали добавлением к раствору инактивированной лошадиной сыворотки. Ферментативно обработанный раствор центрифугировали при 1500 об./мин в течение пяти минут для удаления супернатанта. К полученной клеточной массе добавляли 5-10 мл среды. В качестве среды (Neurobasal/B27/2-ME) использовали нейробазальную среду (Invitrogen Corp. Cat. #21103-049, Carlsbad, CA, USA), дополненную 2% B27 добавкой (Invitrogen Corp. Cat #17504-044, Carlsbad, CA, USA), 25 мкМ 2-меркаптоэтанолом (2-ME, WAKO Cat. #139-06861, Osaka, Japan), 0,5 мМ L-глутамином (Invitrogen Corp. Cat. #25030-081, Carlsbad, CA, USA) и антибиотиками с антимикотиками (Invitrogen Corp. Cat. #15240-062, Carlsbad, CA, USA). Однако для анализа использовали указанную выше нейробазальную среду, не дополненную 2-ME (Neurobasal/B27). Клетки повторно диспергировали умеренной обработкой с помощью пипетки клеточной массы, к которой добавляли указанную среду. Клеточную дисперсию фильтровали через 40-мкм найлоновую сетку (Cell Strainer, Cat. #35-2340, Becton Dickinson Labware, Franklin Lakes, NJ, USA) для удаления оставшейся клеточной массы и в результате получали суспензию нервных клеток. Суспензию нервных клеток разбавляли описанной средой и затем высевали в объеме 100 мкл/лунка при начальной плотности клеток 5×105 клеток/см2 на 96-луночный полистироловый культуральный планшет, предварительно покрытый поли-L или D-лизином (Falcon Cat. #35-3075, Becton Dickinson Labware, Franklin Lakes, NJ, USA, покрытый поли-L-лизином способом, описанным ниже, или BIOCOAT™ cell environments poly-D-lysine cell ware 96-well plate, Cat. #35-6461, Becton Dickinson Labware, Franklin Lakes, NJ, USA). Покрытие поли-L-лизином осуществляли следующим образом. Асептически приготавливали, используя 0,15 M боратный буфер (pH 8,5), 100 мкг/мл раствор поли-L-лизина (SIGMA P2636, St. Louis, MO, USA). По 100 мкг/лунка полученного раствора вносили в лунки 96-луночного полистиролового культурального планшета и инкубировали при комнатной температуре в течение одного или более часов или при 4°C в течение ночи или более. Затем снабженный покрытием 96-луночный полистироловый культуральный планшет промывали стерильной водой четыре раза или более, после чего сушили или прополаскивали стерильной PBS или средой и использовали для посева клеток. Посеянные клетки выращивали на культуральном планшете при 37°C в атмосфере 5% CO2-95% воздуха в течение одних суток. Затем всю среду заменяли свежей средой Neurobasal/B27/2-ME, после чего клетки культивировали еще трое суток.
Внесение соединения
На день 4 культивирования в лунки планшета вносили лекарственное средство следующим образом. Всю среду удаляли из лунок и вносили в них по 180 мкл/лунка нейробазальной среды, не содержащей 2-ME и содержащей 2% B-27 (Neurobasal/B27). Раствор испытуемого соединения в диметилсульфоксиде (далее называемом как ДМСО) разбавляли средой Neurobasal/B27 до концентрации в 10 раз более высокой, чем конечная концентрация. В находящуюся в лунках среду добавляли по 20 мкл/лунка полученного разбавленного раствора и в достаточной мере смешивали его со средой. Конечная концентрация ДМСО составляла 1% или менее. В контрольную группу добавляли только ДМСО.
Взятие образца
После добавления соединения клетки культивировали в течение трех суток и всю среду собирали. Полученную среду использовали в качестве образца для ELISA.
Оценка выживаемости клеток
Выживаемость клеток оценивали MTT анализом по следующей методике. После сбора среды в лунки вносили по 100 мкл/лунка предварительно подогретой среды. Затем в лунки добавляли по 8 мкл/лунка раствора 8 мг/мл MTT (SIGMA M2128, St. Louis, MO, USA) в D-PBS(-) (Dulbecco′s phosphate buffered Saline, SIGMA D8537, St. Louis, MO, USA). 96-луночный полистироловый культуральный планшет инкубировали в инкубаторе при 37°C в атмосфере 5% CO2-95% воздуха в течение 20 минут. Добавляли по 100 мкл/лунка буфера для MTT лизиса и кристаллы MTT формазана в достаточной мере растворяли в буфере в инкубаторе при 37°C в атмосфере 5% CO2-95% воздуха. Затем измеряли оптическую плотность при 550 нм в каждой лунке. Буфер для MTT лизиса получали следующим образом. 100 г SDS (додецилсульфат натрия (лаурилсульфат натрия), WAKO 191-07145, Osaka, Japan) растворяли в смешанном растворе 250 мл N,N-диметилформамида (WAKO 045-02916, Osaka, Japan) с 250 мл дистиллированной воды. Затем к раствору добавляли по 350 мкл концентрированной хлористоводородной кислоты и уксусной кислоты для обеспечения конечного pH около 4,7.
При измерении лунки, не содержащие посеянных клеток и содержащие только среду и раствор MTT, считали фоном (bkg). Измеренные значения соответственно применяли в следующей ниже формуле, вычтя из них bkg значения. Так, для сравнения и оценки активности по отношению к выживаемости клеток вычисляли процентное отношение к контрольной группе (группа, не обработанная лекарственным веществом, CTRL) (% CTRL).
% CTRL=(A550_sample-A550_bkg)/(A550_CTRL-bkg)×100
(A550_sample: оптическая плотность при 550 нм лунки с образцом, A550_bkg: оптическая плотность при 550 нм фоновой лунки, A550_CTRL: оптическая плотность при 550 нм лунки контрольной группы)
Aβ ELISA
Для определения Aβ методом ELISA использовали набор Human/Rat β Amyloid (42) ELISA Kit Wako (#290-62601) от Wako Pure Chemical Industries, Ltd., или набор Human Amyloid beta (1-42) Assay Kit (#27711) от IBL Co., Ltd. Анализ Aβ ELISA проводили в соответствии с инструкциями, рекомендованными производителями (методики, описанные в прилагаемых документах). Однако Aβ калибрационную кривую получали, используя бета-амилоидный пептид 1-42, крысиный (Calbiochem, #171596 [Aβ42]).
(2) Результаты измерений показаны в таблице 10 в виде процентного отношения к концентрации Aβ в среде контрольной группы (% CTRL).
|
|
Результаты в таблице 10 подтвердили, что соединение по настоящему изобретению обладает эффектом уменьшения продукции Aβ42.
Таким образом, соединение общей формулы (I) или его фармакологически приемлемая соль по настоящему изобретению обладает эффектом уменьшения продукции Aβ42. Следовательно, настоящее изобретение может, в частности, дать терапевтическое средство для лечения нейродегенеративного заболевания, вызываемого Aβ, такого как болезнь Альцгеймера или синдром Дауна.
Промышленная применимость
Соединение общей формулы (I) по настоящему изобретению обладает эффектом уменьшения продукции Aβ и потому особенно полезно как терапевтическое средство для лечения нейродегенеративного заболевания, вызываемого Aβ, такого как болезнь Альцгеймера или синдром Дауна.

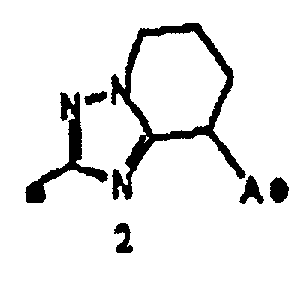
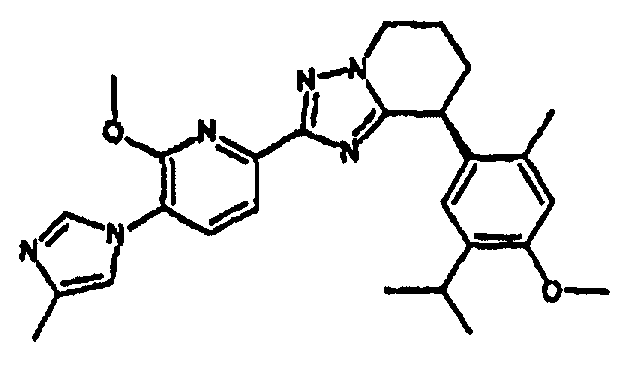
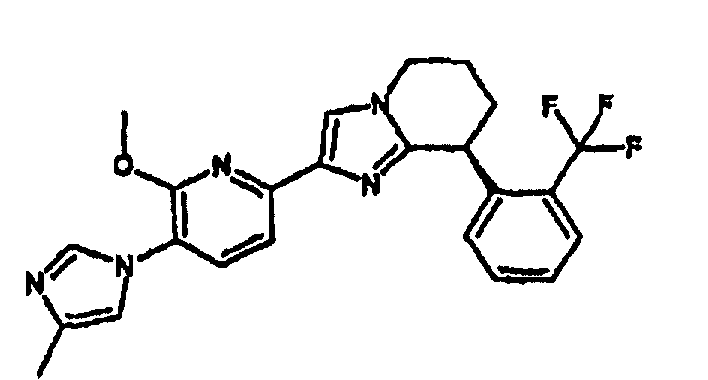
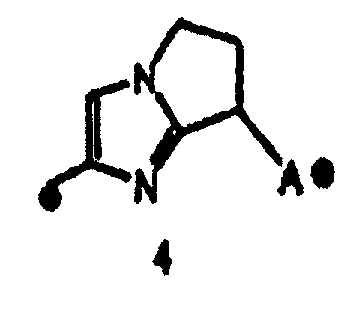

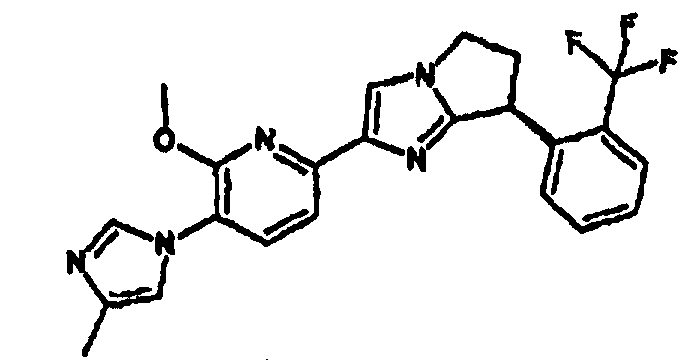
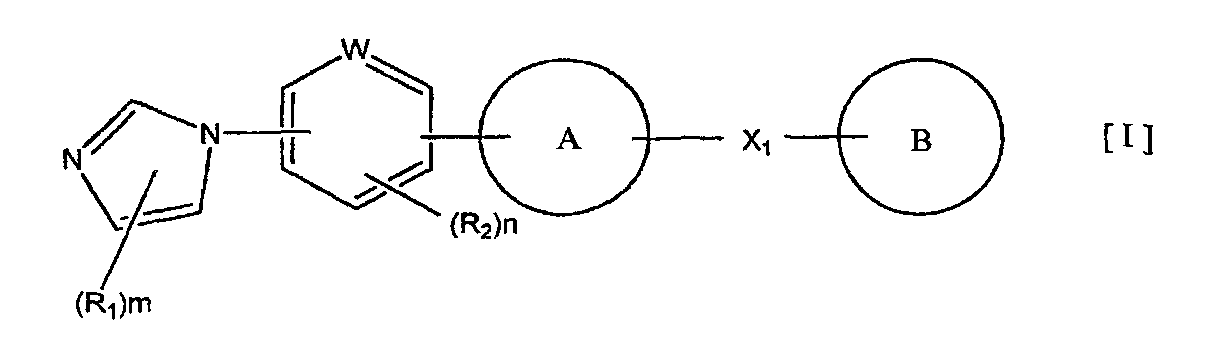
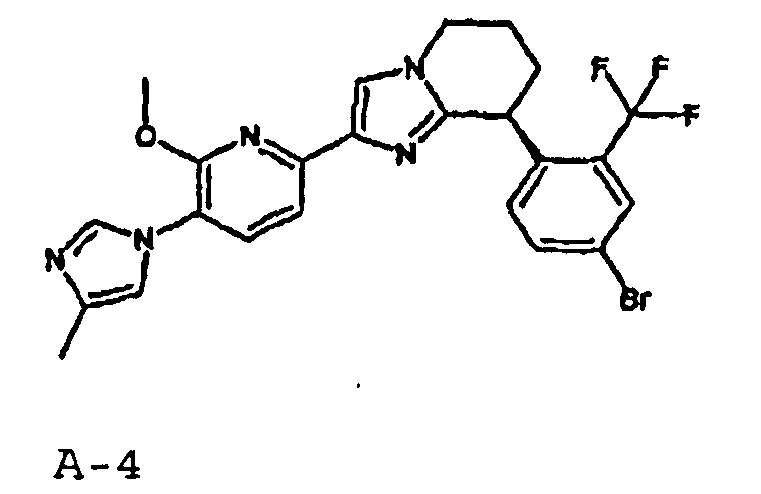
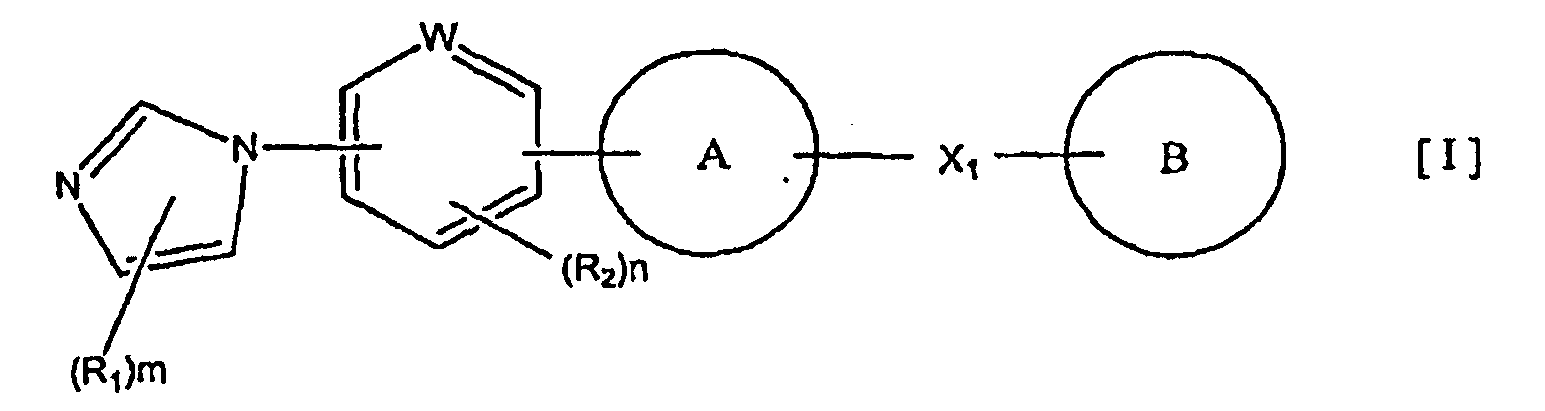
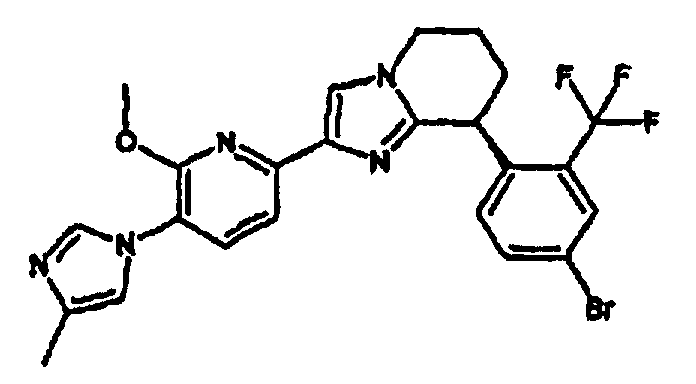
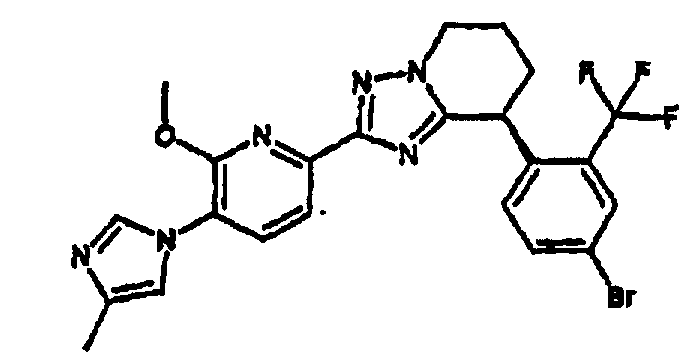
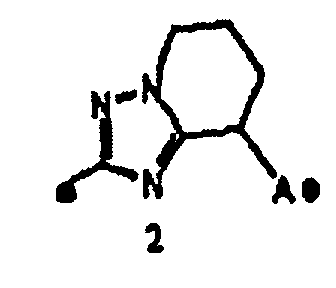
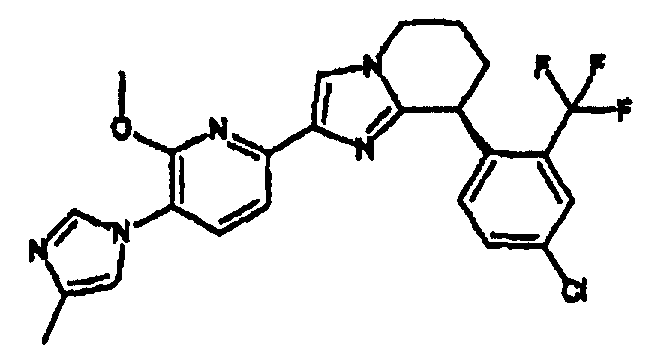
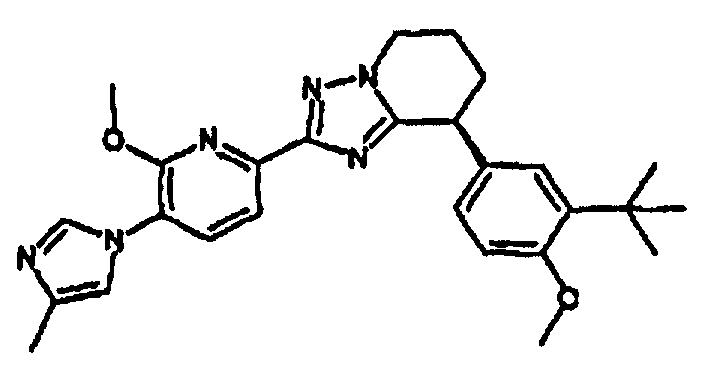
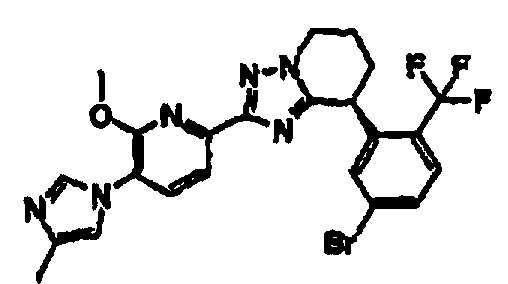
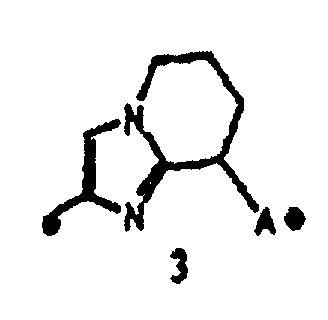
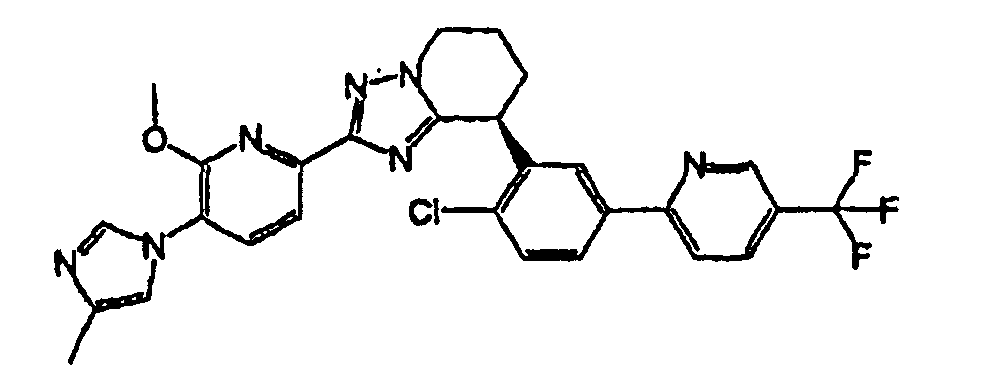
Липосомальная композиция
Конденсированное производное аминодигидротиазина
Производные пиридина, замещенные гетероциклическим кольцом и фосфоноксиметильной группой и содержащие их противогрибковые средства
Промежуточные соединения и способы синтеза аналогов галихондрина в
Реагенты и способы для бета-кетоамидного синтеза синтетического предшественника иммунологического адъюванта е6020
Новое конденсированное производное аминодигидротиазина
Аналоги галихондрина в
Противоопухолевое средство, задействующее соединения с ингибирующим эффектом к киназам в комбинации
Биомаркер для болезни альцгеймера или умеренного когнитивного расстройства
Композиции и способы для лечения воспалительных нарушений
Способ получения замещенных гетероциклом производных пиридина
Липосомальная композиция
Конденсированное производное аминодигидротиазина
Производные пиридина, замещенные гетероциклическим кольцом и фосфоноксиметильной группой и содержащие их противогрибковые средства
Промежуточные соединения и способы синтеза аналогов галихондрина в
Реагенты и способы для бета-кетоамидного синтеза синтетического предшественника иммунологического адъюванта е6020
Новое конденсированное производное аминодигидротиазина
Аналоги галихондрина в
Противоопухолевое средство, задействующее соединения с ингибирующим эффектом к киназам в комбинации
Биомаркер для болезни альцгеймера или умеренного когнитивного расстройства

