Результат интеллектуальной деятельности: КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТНОГО ИНГИБИТОРА JAK-КИНАЗЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к кристаллической форме II бисульфата (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида и ее получению и применению. Соединение формулы (I), полученное согласно способу по настоящему изобретению, можно использовать для лечения артрита.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Артрит представляет собой наиболее распространенное хроническое заболевание в мире, существует много причин артрита, и причины повреждения суставов также различны. В настоящее время Тофацитиниб (CP-690550) представляет собой новый пероральный ингибитор JAK (Янус-киназа) пути, разработанный Pfizer Inc., и Тофацитиниб представляет собой первое в своей группе лекарственное средство, разработанное для лечения ревматоидного артрита. На основе структуры тофацитиниба был разработан целый ряд соединений-ингибиторов JAK, которые являются активными in vitro и in vivo и сильно поглощаемыми, см. WO 2013091539. Соединения, описанные в WO 2013091539, подвергались скринингу и были получены в форме солей, среди которых получали бисульфат (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида формулы (I), способ его получения описан в патентной заявке PCT PCT/CN2014/076794 (заявка, ранее поданная заявителем).
Хорошо известно, что кристаллическая структура фармацевтически активного ингредиента часто влияет на химическую стабильность лекарственного средства. Разные условия кристаллизации и условия хранения могут приводить к изменению в кристаллической структуре соединения и иногда сопутствующему образованию других форм кристаллической формы. Соединение формулы (I) растворяли в метаноле, и затем часть растворителя упаривали с осаждением кристалла, который был назван кристаллической формой I, см. китайскую патентную заявку 201410529863.8. В последующем исследовании авторами изобретения неожиданно обнаружено, нагревали с обратным холодильником в системе из многих растворителей с достижением превращения кристаллов или кристаллизуется в различных растворителях, получают другой кристалл, который в данном документе определен как кристаллическая форма II. Посредством факторов, влияющих на кристаллическую форму, и специального тестирования стабильности обнаружено, что стабильность кристаллической формы II является хорошей, и данную кристаллическую форму проще всего получать, и она является стабильной. Способ ее получения является контролируемым и воспроизводимым, таким образом он больше подходит для промышленного производства.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является обеспечение стабильной кристаллической формы бисульфата (соединение формулы (I)) в качестве ингибитора JAK и способа ее получения.
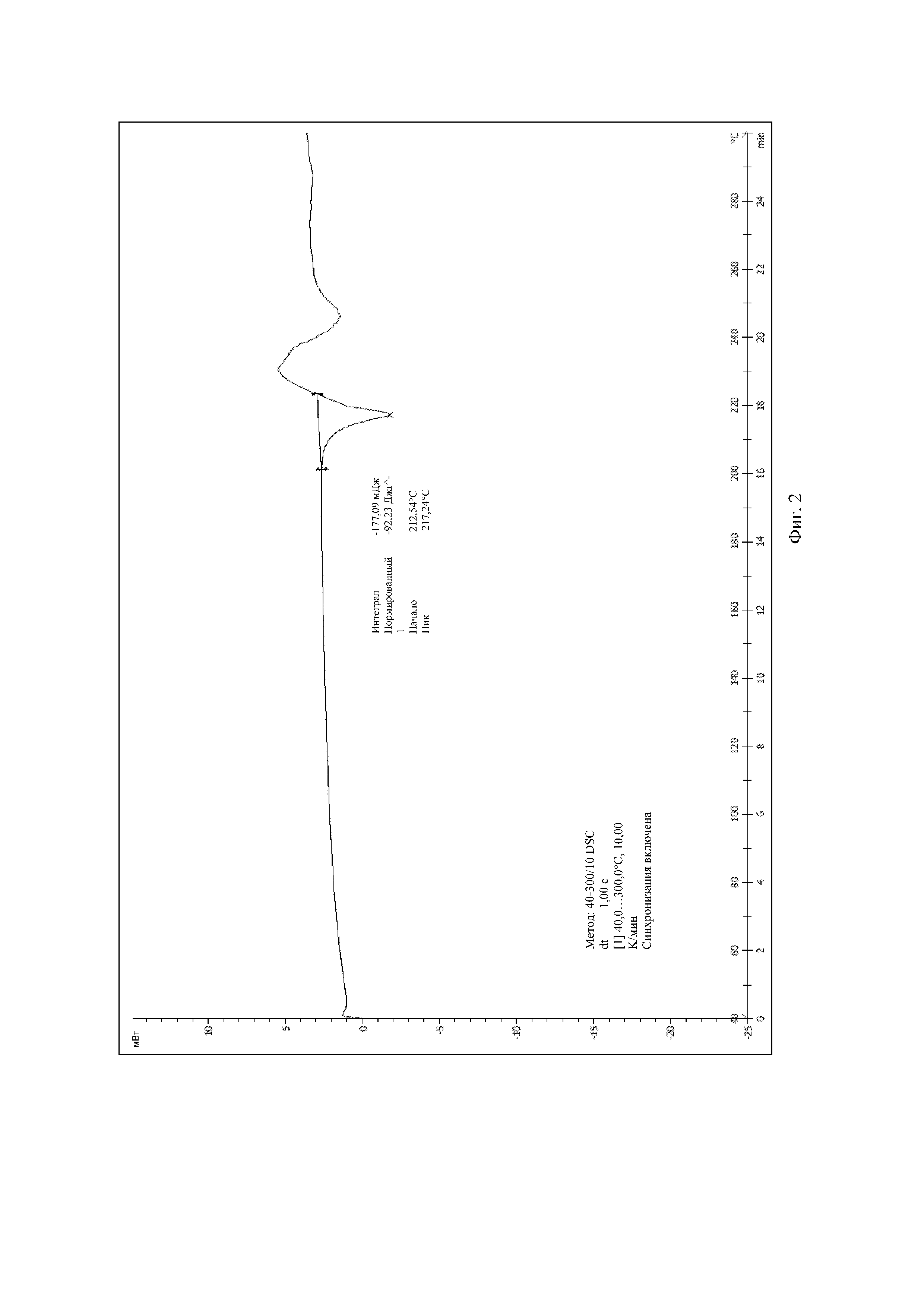
Авторы изобретения протестировали целый ряд кристаллических продуктов соединения формулы (I), полученных в разных условиях, посредством рентгеновской дифракции и метода дифференциальной сканирующей калориметрии (DSC). Обнаружено, что стабильная кристаллическая форма соединения формулы (I), которая названа кристаллической формой II, может быть получена в условиях кристаллизации согласно настоящему изобретению. На спектре DSC кристаллической формы II соединения формулы (I) согласно настоящей заявке показан эндотермический пик плавления при приблизительно 218°C. Спектр рентгеновской порошковой дифракции, который получен с использованием Cu-Kα-излучения и представлен с помощью угла 2θ и межплоскостного расстояния (величина d), показан на Фиг. 1, где приведены характеристические пики при 8,96 (9,87), 11,80 (7,50), 13,12 (6,74), 13,53 (6,54), 13,89 (6,37), 14,42 (6,14), 14,98 (5,91), 16,52 (5,36), 18,20 (4,87), 18,75 (4,73), 19,15 (4,63), 19,72 (4,50), 20,82 (4,26), 22,05 (4,03), 22,52 (3,95), 22,92 (3,88), 23,58 (3,77) и 27,04 (3,30).
В способе получения кристаллической формы II по настоящему изобретению существующая форма соединения формулы (I), используемая в качестве исходного вещества, конкретно не ограничена, и может быть использована любая кристаллическая форма или аморфное твердое вещество. Способ получения кристаллической формы II соединения формулы (I) по настоящему изобретению, включающий применение некоторых низших органических растворителей, предпочтительно полярных органических растворителей с меньшим количеством атомов углерода и более высокой летучестью, которые могут быть использованы в качестве кристаллизующих растворителей, таких как спирты, кетоны, сложные эфиры, галогенированные углеводороды или их смесь; более предпочтительно, метанол, этанол, изопропанол, этилацетат, ацетон, дихлорметан или их смесь в качестве кристаллизующих растворителей. Кристаллизующий растворитель может представлять собой простой растворитель или смешанный растворитель, содержащий упомянутые выше растворители.
Конкретно, согласно настоящему изобретению предложен способ получения кристаллической формы II соединения формулы (I), включающий следующие стадии:
(1) растворение (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида в органическом растворителе, затем добавление по каплям концентрированной серной кислоты, фильтрование раствора и осаждение кристалла; или растворение твердой фазы соединения формулы (I) в соответствующем количестве органического растворителя при нагревании, затем охлаждение раствора с осаждением кристалла;
(2) фильтрование кристалла, затем его промывание и высушивание.
В предпочтительном воплощении настоящего изобретения органический растворитель на стадии (1) выбран из любого из спиртов, кетонов, сложных эфиров, имеющих 3 или меньше атомов углерода, или смешанного растворителя одного или более чем одного растворителя, упомянутого выше, и галогенированного углеводорода, имеющего 3 или меньше атомов углерода. Еще более предпочтительно органический растворитель выбран из метанола, этанола, изопропанола, ацетона, этилацетата или системы смешанного растворителя, такой как дихлорметан/метанол, дихлорметан/метанол/этанол, дихлорметан/метанол/изопропанол, дихлорметан/метанол/этилацетат, дихлорметан/метанол/ацетон.
Наиболее предпочтительно, простой растворитель представляет собой метанол.
В другом предпочтительном воплощении настоящего изобретения смешанный растворитель представляет собой дихлорметан/метанол/этанол, и соотношение данных трех растворителей конкретно не ограничено. В предпочтительном воплощении настоящего изобретения соотношение данных трех растворителей составляет 12:3:10.
Способ перекристаллизации конкретно не ограничен, и может быть осуществлен традиционным способом перекристаллизации. Например, любая форма соединения формулы (I) может быть растворена в органическом растворителе при нагревании, и затем раствор медленно охлаждают и перемешивают с осаждением кристалла. После завершения кристаллизации полученный продукт фильтруют и сушат с получением желаемого кристалла. Кристалл, полученный посредством фильтрации, обычно сушат в вакууме при пониженном давлении при температуре нагревания примерно 30 - 100°C, предпочтительно 40 - 60°C для удаления растворителя перекристаллизации.
Полученную кристаллическую форму соединения формулы (I) определяют посредством спектра DSC и рентгеновской дифракции. Между тем, также определяют остаточный растворитель полученного кристалла.
Кристаллическая форма II соединения формулы (I), полученная согласно настоящему способу, не содержит или содержит только относительно низкое содержание остаточного растворителя, что удовлетворяет требованию национальной фармакопеи в отношении ограничения остаточного растворителя лекарственных средств. Таким образом, кристалл по настоящему изобретению подходит для применения в качестве фармацевтически активного ингредиента.
Результаты исследования показывают, что стабильность кристаллической формы II соединения формулы (I), полученной посредством настоящего изобретения, значительно лучше, чем стабильность аморфного образца в условиях высокой температуры и высокой влажности. Кроме того, кристаллическая форма II обладает хорошей стабильностью в условиях истирания, давления и нагревания, что удовлетворяет требованиям получения, транспортировки и хранения лекарственных средств. Способ получения кристаллической формы II является стабильным, воспроизводимым и контролируемым, и подходит для промышленного производства.
Согласно настоящему изобретению дополнительно предложена фармацевтическая композиция, содержащая кристаллическую форму II соединения формулы (I) и по меньшей мере один фармацевтически приемлемый носитель. Фармацевтически приемлемый носитель выбран по меньшей мере из одного из лактозы, маннита, микрокристаллической целлюлозы, кроскармеллозы натрия, карбоксиметилкрахмала натрия, гидроксипропилметилцеллюлозы, повидона и стеарата магния. Содержание кристаллической формы II в фармацевтической композиции по настоящему изобретению составляет 0,5 мг-200 мг.
Настоящее изобретение дополнительно относится к применению кристаллической формы II соединения формулы (I) или фармацевтической композиции по настоящему изобретению в получении лекарственного средства для лечения заболевания, связанного с JAK, предпочтительно ревматического и ревматоидного артрита.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 показан спектр рентгеновской порошковой дифракции кристаллической формы II соединения формулы (I) (на фигуре представлена символом SHR0302).
На Фиг. 2 показан спектр DSC кристаллической формы II соединения формулы (I).
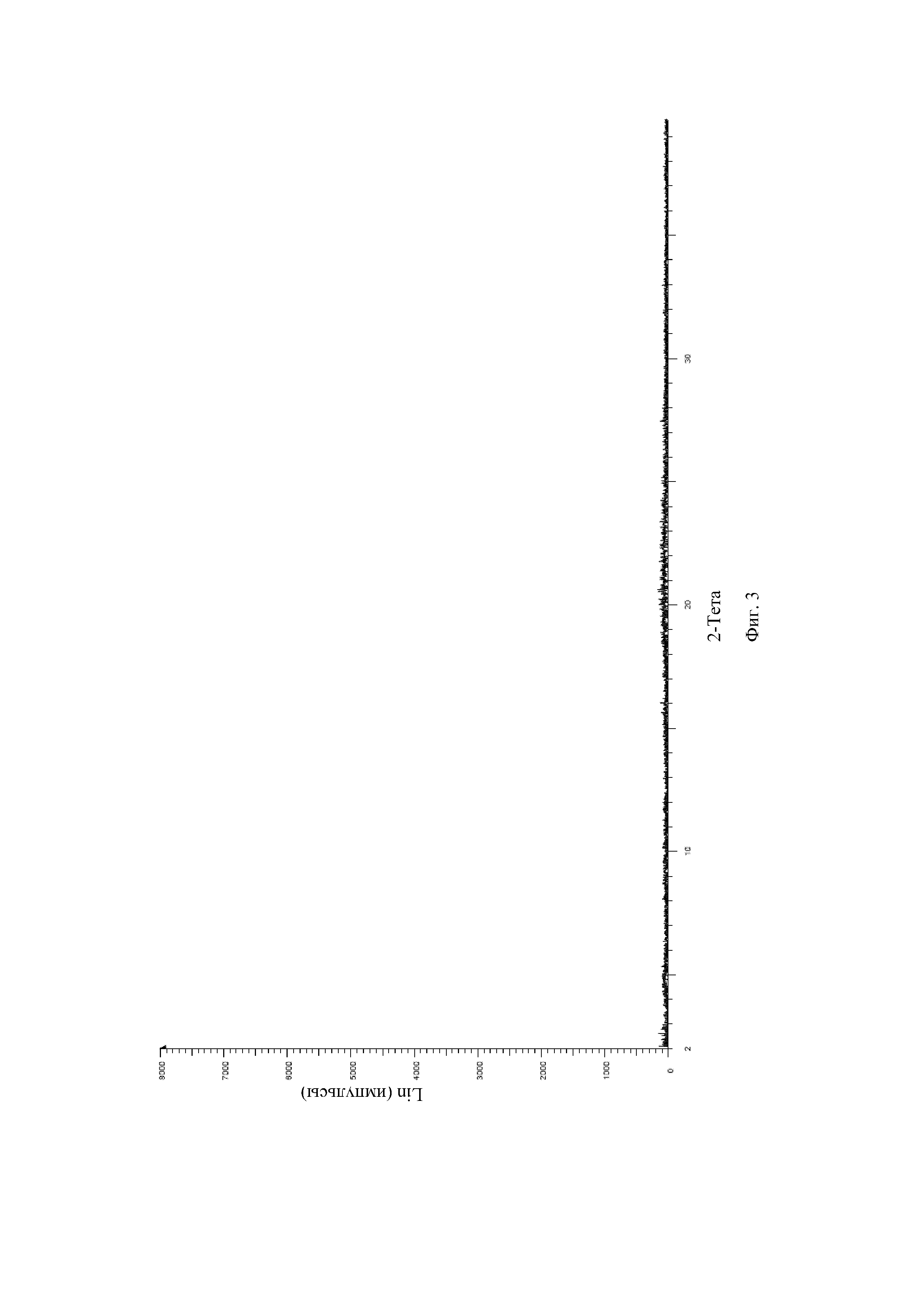
На Фиг. 3 показан спектр рентгеновской порошковой дифракции аморфного твердого вещества соединения формулы (I).
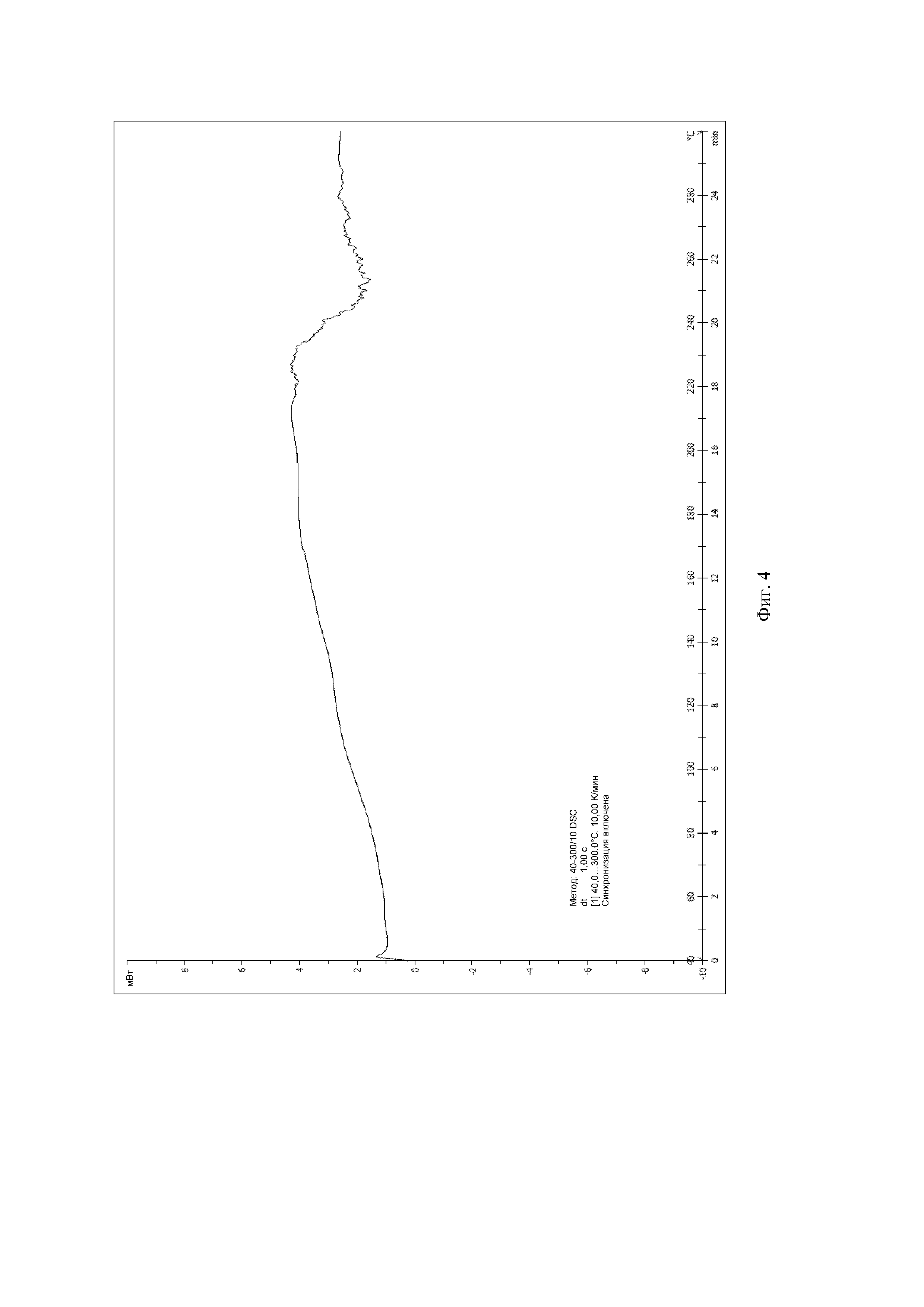
На Фиг. 4 показан спектр DSC аморфного твердого вещества соединения формулы (I).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение подробно проиллюстрировано следующими примерами, но примеры изобретения только предназначены для описания технического решения изобретения, и их не следует рассматривать ограничивающими объем настоящего изобретения.
Испытательные приборы, используемые в экспериментах:
1. Спектр DSC
Тип прибора: Mettler Toledo DSC 1 Staree System
Продувочный газ: Азот
Скорость нагрева: 10,0°C/мин
Диапазон температуры: 40 - 300°C
2. Спектр рентгеновской дифракции
Тип прибора: рентгеновский порошковый дифрактометр D/Max-RA Japan Rigaku
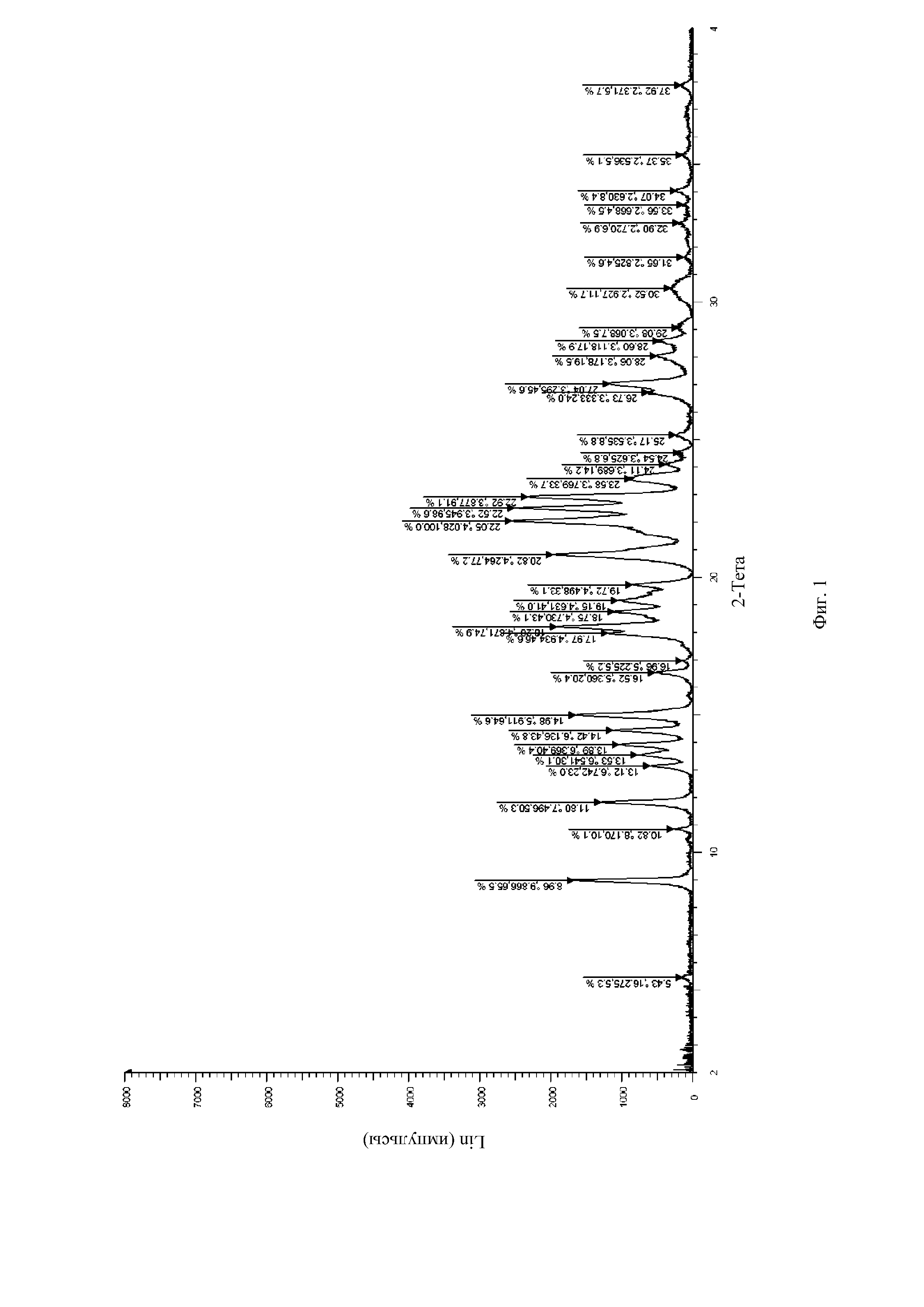
Лучи: монохроматические Cu-Kα лучи (λ= 1,5418 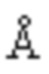 )
)
Режим сканирования: θ/2θ, Диапазон углов: 2 - 40°
Напряжение: 40 кВСила тока: 40 мА
Пример 1:
1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида добавляли в 50 мл колбу Эрленмейера с последующим добавлением 12 мл дихлорметана и 3 мл безводного метанола. Реакционную смесь перемешивали при комнатной температуре, затем по каплям добавляли 0,25 г (2,5 ммоль) концентрированной серной кислоты. После того, как суспензия становилась прозрачной, нерастворимые вещества удаляли посредством фильтрации. После того, как фильтрат перемешивали в течение 6 часов, осаждения твердого вещества не происходило. Добавляли 10 мл изопропанола, затем осаждали большое количество белого твердого вещества. Реакционную смесь перемешивали еще в течение 18 часов, фильтровали и сушили с получением 1,138 г белого твердого вещества с выходом 92,1%. Спектр рентгеновской дифракции данного кристаллического образца показан на Фиг. 1, где представлены характеристические пики при 8,96 (9,87), 11,80 (7,50), 13,12 (6,74), 13,53 (6,54), 13,89 (6,37), 14,42 (6,14), 14,98 (5,91), 16,52 (5,36), 18,20 (4,87), 18,75 (4,73), 19,15 (4,63), 19,72 (4,50), 20,82 (4,26), 22,05 (4,03), 22,52 (3,95), 22,92 (3,88), 23,58 (3,77) и 27,04 (3,30). На Фиг. 2 показан спектр DSC, имеющий эндометрический пик плавления при 217,24°C. Кристаллическую форму определяли как кристаллическая форма II.
Пример 2:
1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида добавляли в 50 мл колбу Эрленмейера с последующим добавлением 12 мл дихлорметана и 3 мл безводного метанола. Реакционную смесь перемешивали при комнатной температуре, затем по каплям добавляли 0,25 г (2,5 ммоль) концентрированной серной кислоты. После того, как суспензия становилась прозрачной, нерастворимые вещества удаляли посредством фильтрации, и фильтрат концентрировали досуха с получением соединения формулы (I) в аморфной форме. Спектр рентгеновской дифракции твердого образца показан на Фиг. 3, где отсутствуют характеристические пики поглощения кристалла. На Фиг. 4 показан спектр DSC твердого образца.
Пример 3:
1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида добавляли в 50 мл колбу Эрленмейера с последующим добавлением 12 мл дихлорметана и 3 мл безводного метанола. Реакционную смесь перемешивали при комнатной температуре, затем по каплям добавляли 0,25 г (2,5 ммоль) концентрированной серной кислоты. После того, как суспензия становилась прозрачной, нерастворимые вещества удаляли посредством фильтрации, и фильтрат перемешивали в течение 24 часов с осаждением кристалла, фильтровали и сушили с получением 1,15 г белого твердого вещества с выходом 93,2%. Продукт идентифицировали как кристаллическую форму II после изучения и сравнения спектров рентгеновской дифракции и DSC.
Пример 4:
1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида добавляли в 50 мл колбу Эрленмейера с последующим добавлением 12 мл дихлорметана и 3 мл безводного метанола. Реакционную смесь перемешивали при комнатной температуре, затем по каплям добавляли 0,25 г (2,5 ммоль) концентрированной серной кислоты. После того, как суспензия становилась прозрачной, нерастворимые вещества удаляли посредством фильтрации. После того, как фильтрат перемешивали в течение 6 часов, осаждения твердого вещества не происходило. Добавляли 10 мл этанола, затем осаждали большое количество белого твердого вещества. Реакционную смесь перемешивали еще в течение 18 часов, фильтровали и сушили с получением 1,17 г белого твердого вещества с выходом 94,9%. Продукт идентифицировали как кристаллическую форму II после изучения и сравнения спектров рентгеновской дифракции и DSC.
Пример 5:
1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида добавляли в 50 мл колбу Эрленмейера с последующим добавлением 12 мл дихлорметана и 3 мл безводного метанола. Реакционную смесь перемешивали при комнатной температуре, затем по каплям добавляли 0,25 г (2,5 ммоль) концентрированной серной кислоты. После того, как суспензия становилась прозрачной, нерастворимые вещества удаляли посредством фильтрации. После того, как фильтрат перемешивали в течение 6 часов, осаждения твердого вещества не происходило. Добавляли 5 мл этилацетата, затем осаждали большое количество белого твердого вещества. Реакционную смесь перемешивали еще в течение 18 часов, фильтровали и сушили с получением 1,16 г белого твердого вещества с выходом 94,2%. Продукт идентифицировали как кристаллическую форму II после изучения и сравнения спектров рентгеновской дифракции и DSC.
Пример 6:
1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида добавляли в 50 мл колбу Эрленмейера с последующим добавлением 12 мл дихлорметана и 3 мл безводного метанола. Реакционную смесь перемешивали при комнатной температуре, затем по каплям добавляли 0,25 г (2,5 ммоль) концентрированной серной кислоты. После того, как суспензия становилась прозрачной, нерастворимые вещества удаляли посредством фильтрации. После того, как фильтрат перемешивали в течение 6 часов, осаждения твердого вещества не происходило. Добавляли 5 мл ацетона, затем осаждали большое количество белого твердого вещества. Реакционную смесь перемешивали еще в течение 18 часов, фильтровали и сушили с получением 1,14 г белого твердого вещества с выходом 92,3%. Продукт идентифицировали как кристаллическую форму II после изучения и сравнения спектров рентгеновской дифракции и DSC.
Пример 7:
1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида добавляли в 50 мл колбу Эрленмейера с последующим добавлением 12 мл дихлорметана и 3 мл безводного метанола. Реакционную смесь перемешивали при комнатной температуре, затем по каплям добавляли 0,25 г (2,5 ммоль) концентрированной серной кислоты. После того, как суспензия становилась прозрачной, нерастворимые вещества удаляли посредством фильтрации, и фильтрат концентрировали досуха с получением соединения формулы (I) в аморфной форме. Полученный образец добавляли в 5 мл метанола, затем смесь нагревали до температуры флегмообразования в течение 20 минут, охлаждали, перемешивали в течение 2 часов с достижением превращения кристаллов, фильтровали и сушили с получением 942 мг белого твердого вещества с выходом 76,2%. Продукт идентифицировали как кристаллическую форму II после изучения и сравнения спектров рентгеновской дифракции и DSC.
Пример 8
Соединение формулы (I) в аморфой форме получали из 1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида согласно экспериментальному способу из Примера 2. Полученный образец добавляли в 5 мл этанола, затем смесь нагревали до температуры флегмообразования до тех пор, пока раствор не становился прозрачным. Раствор нагревали с обратным холодильником в течение еще 20 минут, охлаждали, перемешивали в течение 2 часов с достижением превращения кристаллов, фильтровали и сушили с получением 1,08 г белого твердого вещества с выходом 82,3%. Продукт идентифицировали как кристаллическую форму II после изучения и сравнения спектров рентгеновской дифракции и DSC.
Пример 9
Соединение формулы (I) в аморфной форме получали из 1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида согласно экспериментальному способу из Примера 2. Полученный образец добавляли к 5 мл изопропанола, затем смесь нагревали до температуры флегмообразования до тех пор, пока раствор не становился прозрачным. Раствор нагревали с обратным холодильником в течение еще 20 минут, охлаждали, перемешивали в течение 2 часов с достижением превращения кристаллов, фильтровали и сушили с получением 1,05 г белого твердого вещества с выходом 85,0%. Продукт идентифицировали как кристаллическую форму II после изучения и сравнения спектров рентгеновской дифракции и DSC.
Пример 10
Соединение формулы (I) в аморфной форме получали из 1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида согласно экспериментальному способу из Примера 2. Полученный образец добавляли к 5 мл этилацетата, затем смесь нагревали до температуры флегмообразования до тех пор, пока раствор не становился прозрачным. Раствор нагревали с обратным холодильником в течение еще 20 минут, охлаждали, перемешивали в течение 2 часов с достижением превращения кристаллов, фильтровали и сушили с получением 1,11 г белого твердого вещества с выходом 90,1%. Продукт идентифицировали как кристаллическую форму II после изучения и сравнения спектров рентгеновской дифракции и DSC.
Пример 11
Соединение формулы (I) в аморфной форме получали из 1,0 г (2,4 ммоль) (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1H)-формамида согласно экспериментальному способу из Примера 2. Полученный образец добавляли к 5 мл ацетона, затем смесь нагревали до температуры флегмообразования до тех пор, пока раствор не становился прозрачным. Раствор нагревали с обратным холодильником в течение еще 20 минут, охлаждали, перемешивали в течение 2 часов с достижением превращения кристаллов, фильтровали и сушили с получением 1,13 г белого твердого вещества с выходом 91,5%. Продукт идентифицировали как кристаллическую форму II после изучения и сравнения спектров рентгеновской дифракции и DSC.
Пример 12
1,0 г (2,4 ммоль) соединения формулы (I) (кристаллическая форма I), которое получали согласно способу по китайской патентной заявке 201410529863.8, и 100 мл метанола добавляли в 250 мл одногорлую колбу и нагревали до температуры флегмообразования до тех пор, пока раствор не становился прозрачным, затем раствор нагревали с обратным холодильником в течение еще 10 минут и охлаждали. Посредством упаривания удаляли приблизительно 90 мл метанола при пониженном давлении, затем смесь перемешивали при комнатной температуре в течение 4 часов, фильтровали и сушили с получением 842 мг белого твердого вещества с выходом 84,2%. Продукт идентифицировали как кристаллическую форму II после изучения и сравнения спектров рентгеновской дифракции и DSC.
Пример 13
Кристаллическую форму II, полученную в Примере 1, и аморфный образец, полученный в Примере 2, распределяли в виде плоского слоя в воздухе для тестирования их стабильности в условиях освещенности (4500 лк), нагревания (40°C, 60°C) и высокой влажности (относительная влажность 75%, относительная влажность 90%). Изучали моменты времени отбора образцов 5 суток и 10 суток, и чистота, детектированная посредством HPLC (высокоэффективная жидкостная хроматография), показана в Таблице 1.
Таблица 1. Сравнение стабильности кристаллической формы II и аморфного образца соединения формулы (I)
|
После того, как кристаллическую форму II и аморфный образец соединения формулы (I) распределяли в виде плоского слоя в воздухе для тестирования их стабильности в условиях освещенности, высокой температуры, высокой влажности, результаты исследования стабильности показали, что высокая влажность не оказывает большого влияния на данные два образца, а в условиях освещенности и высокой температуры стабильность кристаллической формы II значительно лучше, чем стабильность аморфного образца.
Пример 14
Кристаллическую форму II соединения формулы (I), полученную согласно способу из Примера 1, растирали, нагревали и прессовали. Результаты показали, что кристаллическая форма является стабильной, и подробные экспериментальные данные показаны ниже в Таблице 2.
Таблица 2. Специальное исследование стабильности кристаллической формы II соединения формулы (I)
|
Соединения 2-(2-оксоиндолин-3-илиден)метил-5-(2-гидрокси-3-морфолин-4-илпропил)-6,7 дигидро-1-н-пиррол[3,2-с]пиридин-4(5н)-она и их применение в качестве ингибиторов протеинкиназы
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Бициклозамещенные азопроизводные пиразолона, способ их получения и фармацевтическое применение
Фармацевтически приемлемая соль (е)-n-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2r)-1-метилпирролидин-2-ил]проп-2-енамида, способ ее получения и применение при лечении рака
Производные с-арилглюкозидов, способ их получения и фармацевтическое применение
Производные пирролопиримидина, полезные в качестве ингибиторов jak-киназы
Полициклические производные, способ их получения и их фармацевтическое применение
Производные оксотиоимидазолидина, способы их получения и их применение в медицине в качестве ингибиторов андрогенного рецептора
Кристаллическая форма i дималеата ингибитора тирозинкиназы и способ ее получения
Бисульфат ингибитора янус-киназы (jak) и способ его получения
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли n-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Фармацевтическая композиция для лечения диабета 2 типа
Промежуточное соединение для синтеза каспофунгина и способ его получения
Производные оксотиоимидазолидина, способы их получения и их применение в медицине в качестве ингибиторов андрогенного рецептора
Кристаллическая форма i дималеата ингибитора тирозинкиназы и способ ее получения
Бисульфат ингибитора янус-киназы (jak) и способ его получения
Производные пиразолопиримидона или пирролотриазона, способ их получения и их фармацевтические применения
Амидные производные и их фармацевтически приемлемые соли, способ их получения и медицинское применение

