Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ (4S)-4-(4-ЦИАНО-2-МЕТОКСИФЕНИЛ)-5-ЭТОКСИ-2,8-ДИМЕТИЛ-1,4-ДИГИДРО-1,6-НАФТИРИДИН-3-КАРБОКСАМИДА И ВЫДЕЛЕНИЯ (4S)-4-(4-ЦИАНО-2-МЕТОКСИФЕНИЛ)-5-ЭТОКСИ-2,8-ДИМЕТИЛ-1,4-ДИГИДРО-1,6-НАФТИРИДИН-3-КАРБОКСАМИДА ПОСРЕДСТВОМ ЭЛЕКТРОХИМИЧЕСКИХ МЕТОДОВ
Вид РИД
Изобретение
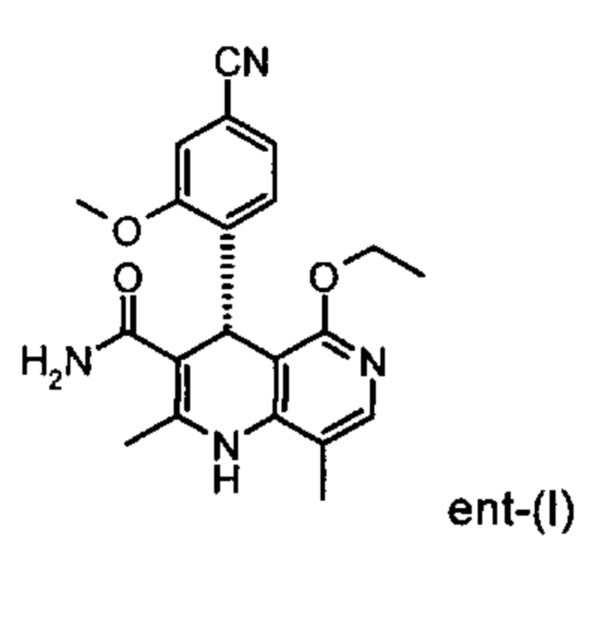
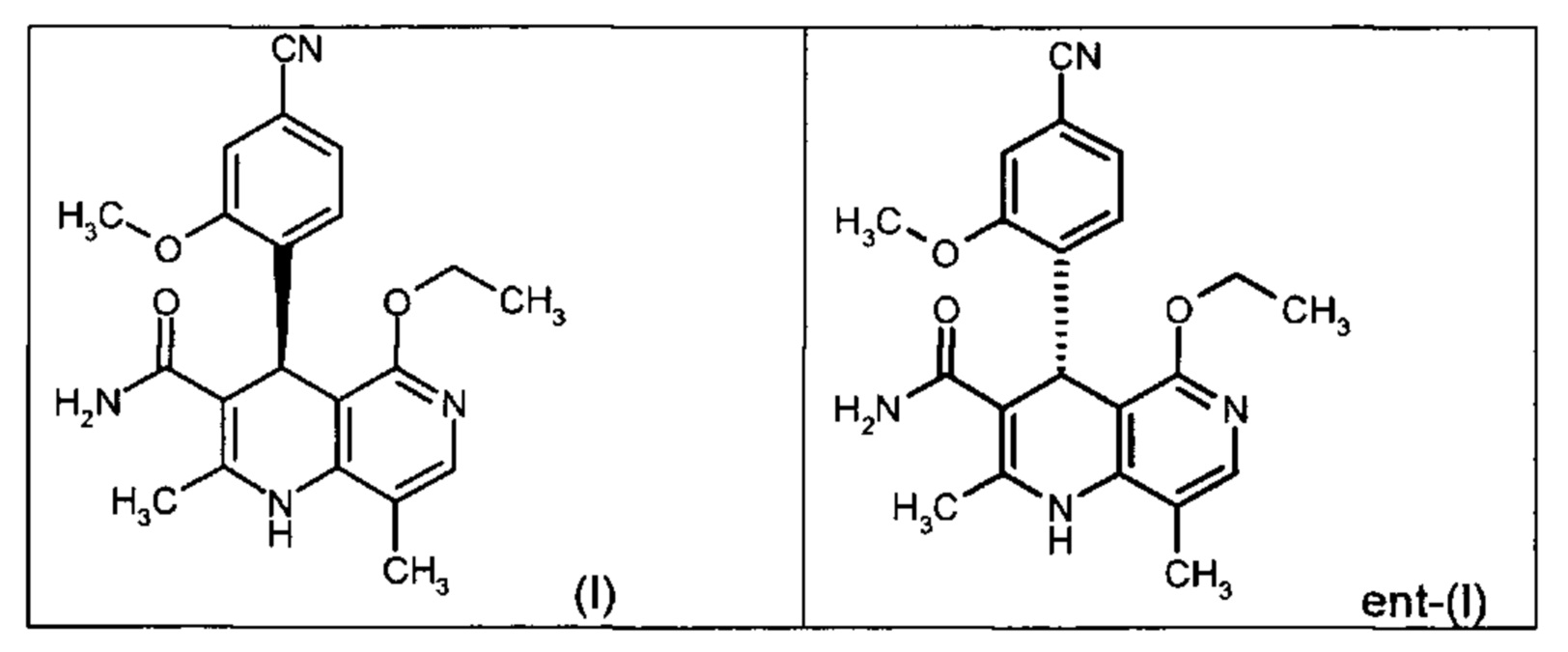
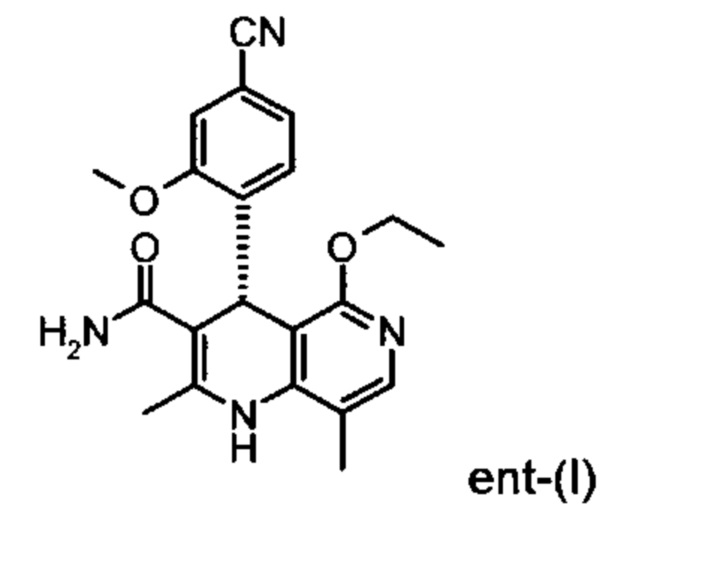
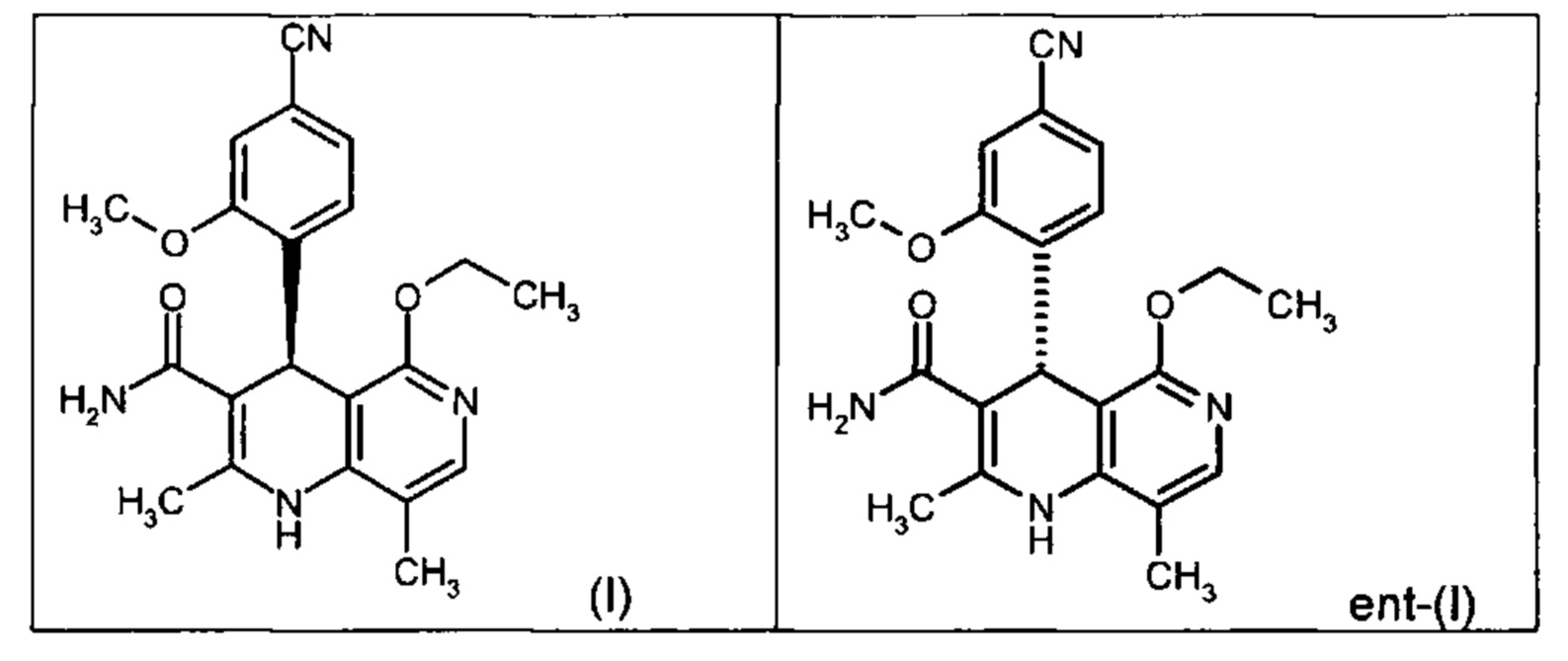
Настоящее изобретение касается нового способа получения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I) и выделения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I)
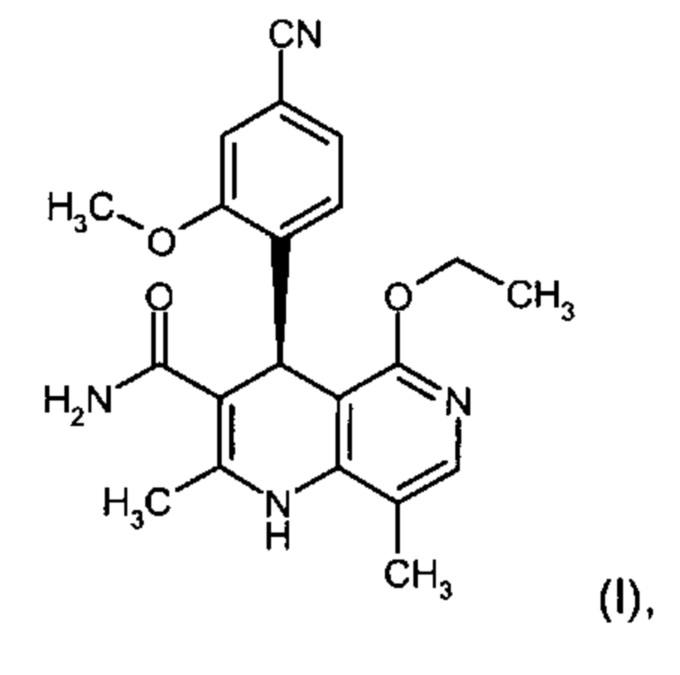
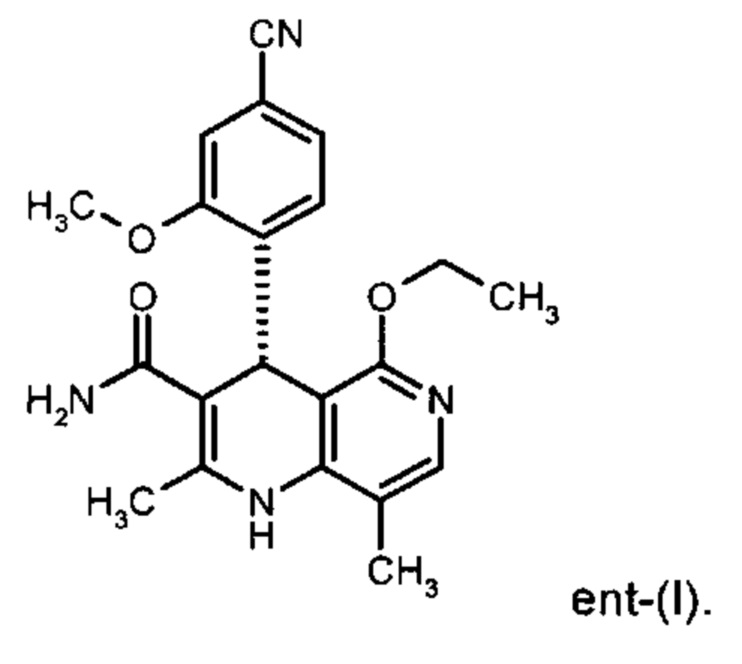
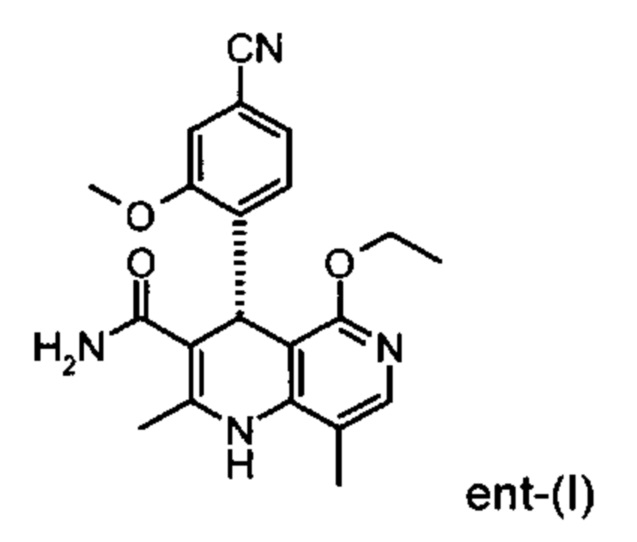
исходя из (4R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы ent-(I)
Соединение формулы (I) действует как нестероидный антагонист минералкортикоидного рецептора и может быть использовано в качестве средства для профилактики и/или лечения сердечно-сосудистых и почечных заболеваний, таких как сердечная недостаточность и диабетическая нефропатия.
Соединение формулы (I) и процесс его получения описан в международной заявке WO 2008/104306 и статье ChemMedChem 2012, 7, 1385, причем в обеих публикациях раскрывается обстоятельное обсуждение научно-исследовательских синтезов. Недостатком описанных там синтезов является тот факт, что эти синтезы не пригодны для промышленных способов, так как большинство стадий осуществляют при очень сильном разбавлении, с очень большими избытками реагентов, но при этом с относительно низким общим выходом. Кроме того требуется много хроматографических промежуточных очисток, которые как правило являются технически очень трудоемкими, требуют высокого расхода растворителей, и следовательно являются дорогостоящими, и которые по возможности по указанной причине следует избегать. Некоторые стадии не могут быть реализованы по причине сложностей, связанных с технологическим исполнением и безопасностью труда.
Поэтому существовала потребность в реализуемом на практике, промышленном синтезе, который воспроизводимо давал бы соединение формулы (I) с высоким общим выходом, низкими производственными расходами и высокой чистотой, и соответствовал бы всем нормативным требованиям, чтобы обеспечить клинические испытания действующего вещества и применение для последующей государственной регистрации.
Был найден очень эффективный синтез, который позволил выполнить выше указанные требования.
В публикации ChemMedChem 2012, 7, 1385, в которой раскрыт научно-исследовательский синтез соединения формулы (I), получают соединение формулы (I) в 10 стадий, исходя из ванилина, с общим выходом 3,76% от теоретического. Соединение формулы (I) получали в виде аморфного твердого вещества посредством упаривания хроматографической фракции, до сих пор не было описано конкретного способа кристаллизации на конечной стадии для регулирования полиморфизма.
Далее следующая схема 1 показывает известный способ получения соединения формулы (I).
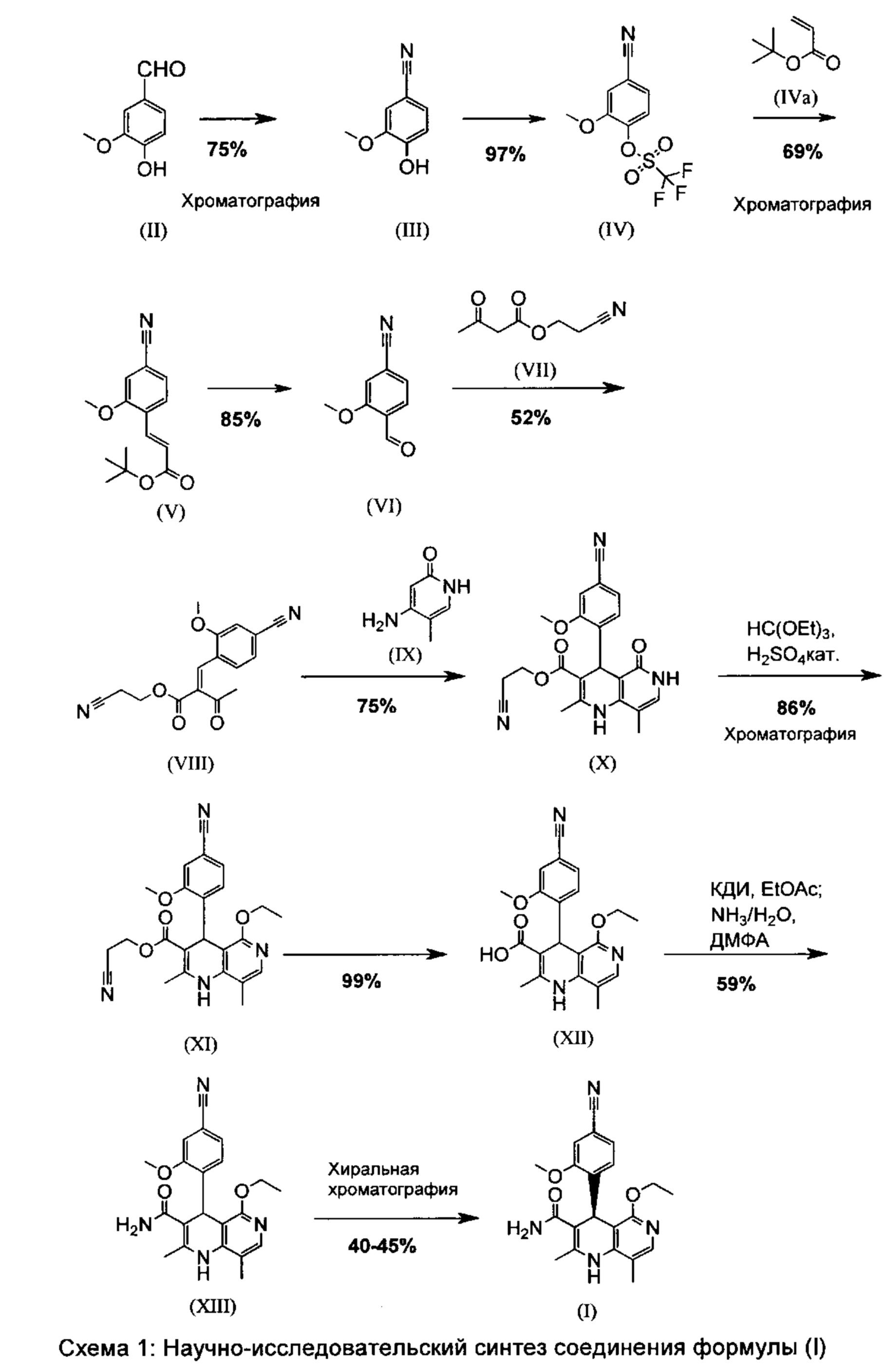
Используют 3 хроматографических очистки, а также одну хиральную хроматографическую стадию для разделения энантиомеров рацемата формулы (XIII). Стадии проводят частично при очень сильном разбавлении и с использованием очень большого количества реагентов.
Так в частности с точки зрения атомной эффективности является не приемлемой последовательность получения промежуточных соединений нитрил-альдегид (VI), которая играет ключевую роль в этом синтезе.
Кроме того указанный способ не может быть перенесен на промышленный масштаб, так как применяют очень дорогие реагенты, такие как ангидрид трифторметансульфоновой кислоты [(III)=>(IV)] и избыток трет-бутилового эфира акриловой кислоты. При масштабировании реакции Хека (IV)=>(V) в реакционном сосуде образуется полимероподобный осадок, который является результатом полимеризации трет-бутилового эфира акриловой кислоты, использованного в избытке. Это не приемлемо при промышленном проведении процесса, так как возникает опасность в том, что это может привести к поломке перемешивающего устройства и к трудно удаляемым осадкам в смесителе.
Также необходимо избегать последующего разрыва двойной связи периодатом натрия и высоко токсичным тетраоксидом осмия, так как при описанных условиях проведения опыта это приводит к замедлению реакции и тем самым обуславливает сильную экзотерму и связанный с этим неуправляемый разогрев.
Схема 2 наглядно показывает новый способ, который дает соединение формулы (I) в 9 стадий с общим выходом 27,7% от теоретического без хроматографической очистки промежуточных соединений.
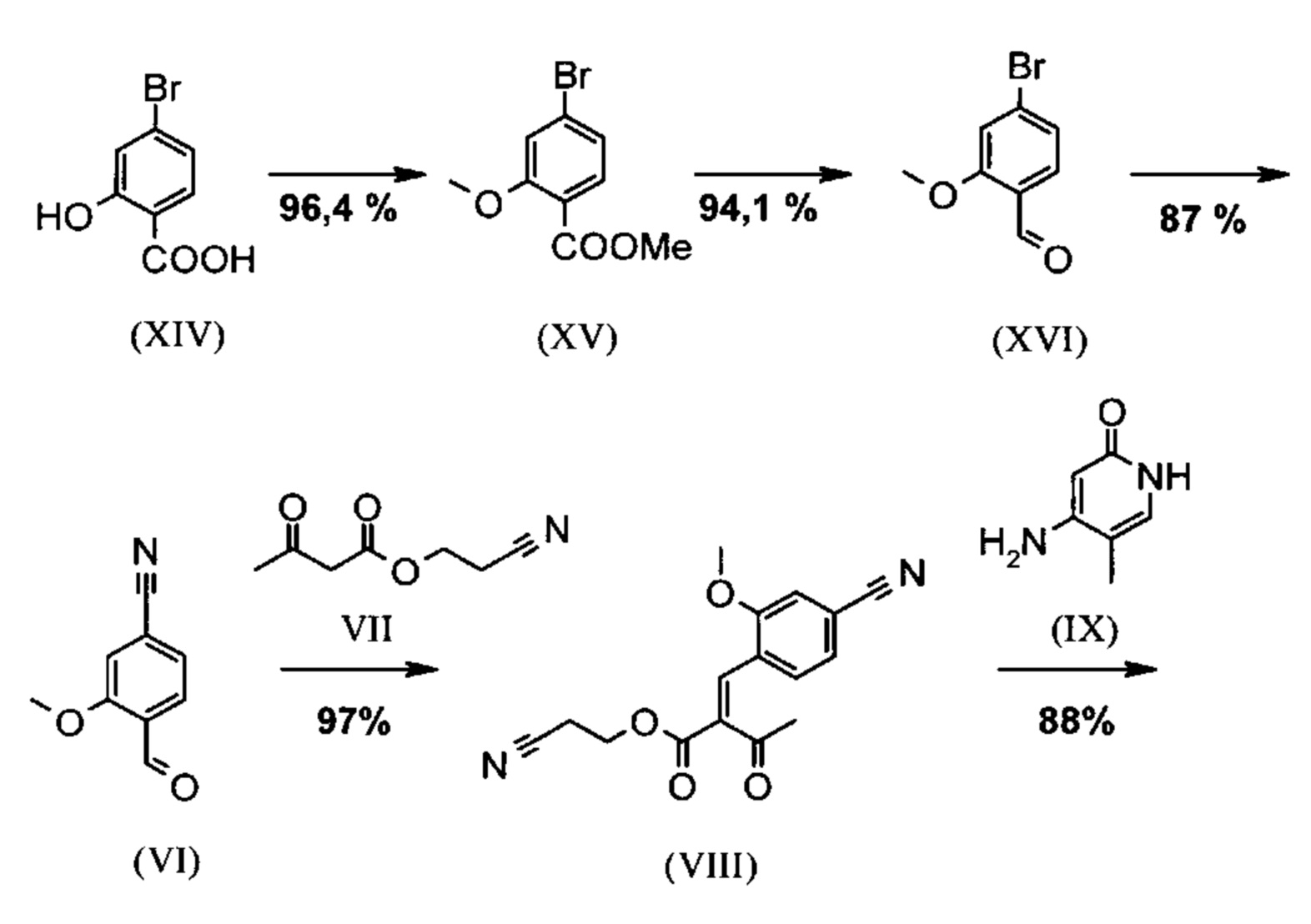
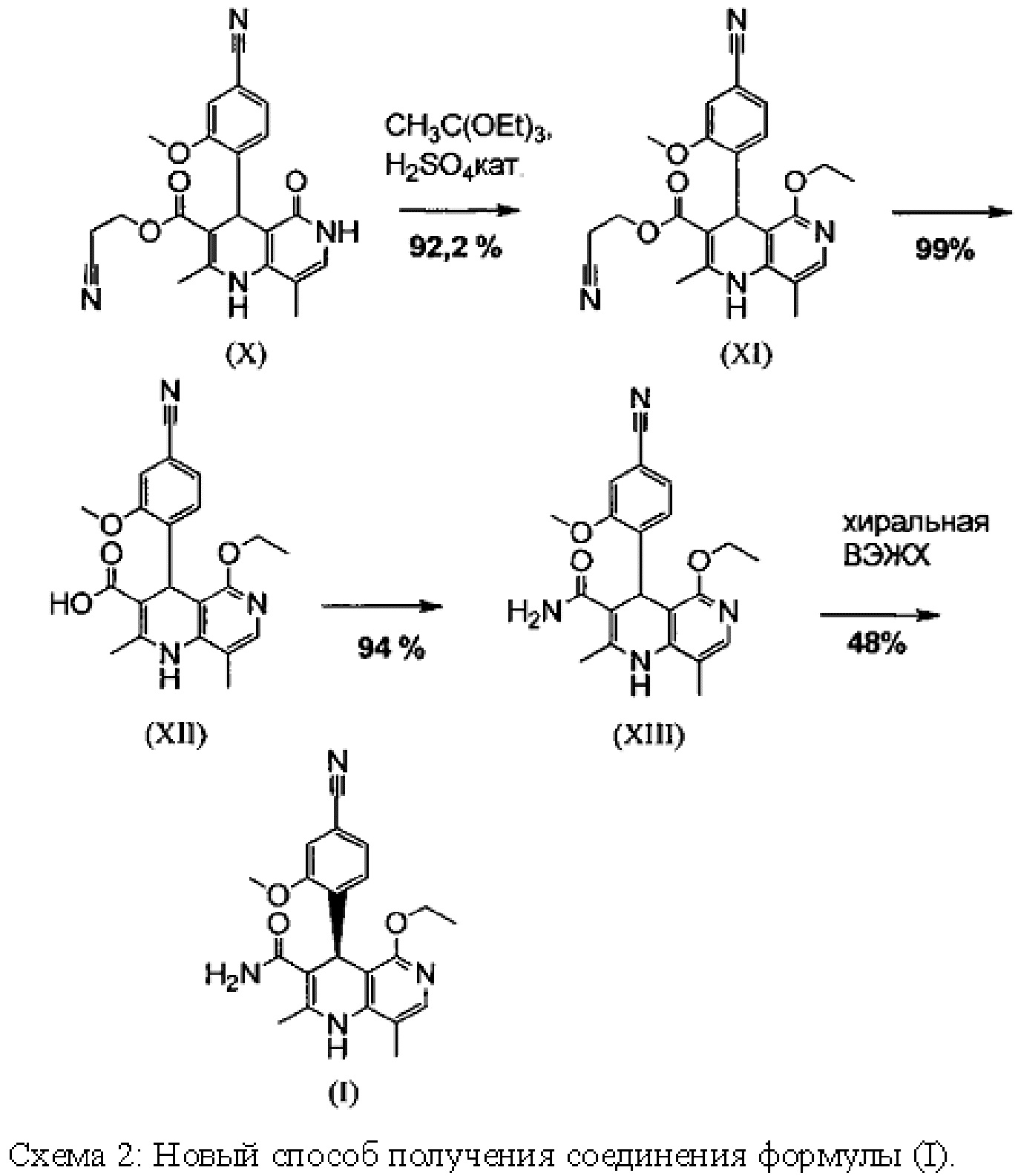
Метиловый сложный эфир (XV), а также альдегид (XVI) не выделяют, а далее подвергают превращению непосредственно в растворе, причем тем самым обеспечивают только 7 выделяемых промежуточных продуктов. Для разделения энантиомеров применяют методы препаративной хиральной ВЭЖХ (например, SMB-технология (хроматография с псевдодвижущимся слоем адсорбента), Varicol).
Альдегид (VI) известен в литературе (J. Med. Chem. 2007, 50, 2468-2485) и представляет собой важный промежуточный продукт в указанном синтезе. Сейчас существует также возможность купить данное соединение.
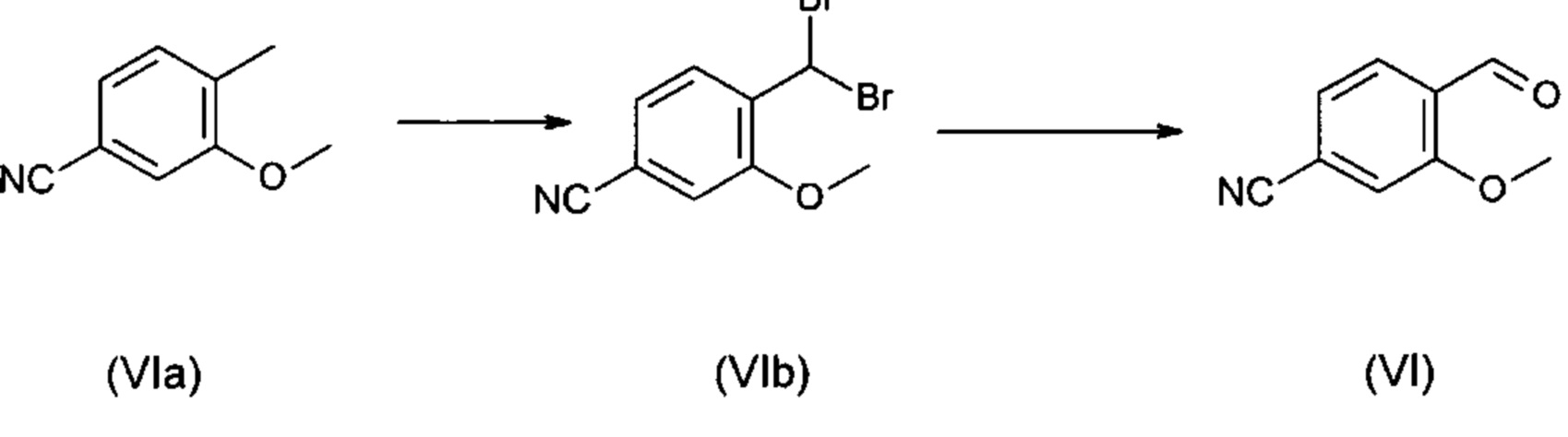
Исходя из 4-циано-2-метокситолуола (VIa) при помощи NBS (N-бромсукцинимид) получают дибромид (VIb), который в этаноле превращают с 2,46 экв. нитрата серебра (в воде) в желаемый альдегид (VI). Этот описанный в литературе синтез, а также описанный в научно-исследовательском синтезе способ совершенно не подходит для масштабирования в мульти-тоннажный размер, так что существует высокая потребность в новом эффективном и экономически выгодном синтезе.
Галогенбензойные кислоты (XIV) и (XIVa)
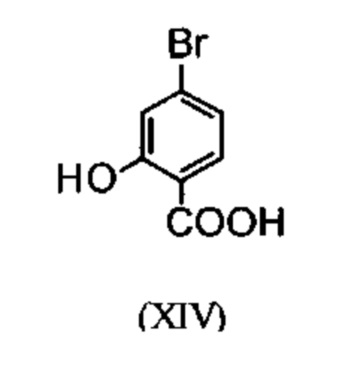
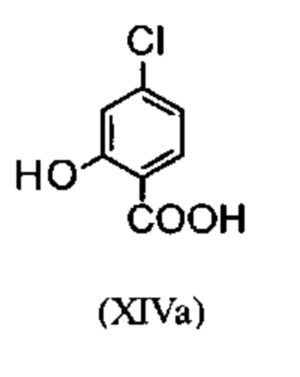
коммерчески доступны в больших количествах. Был разработан очень эффективный и низкозатратный процесс, при котором промежуточные продукты (XV) и (XVI)
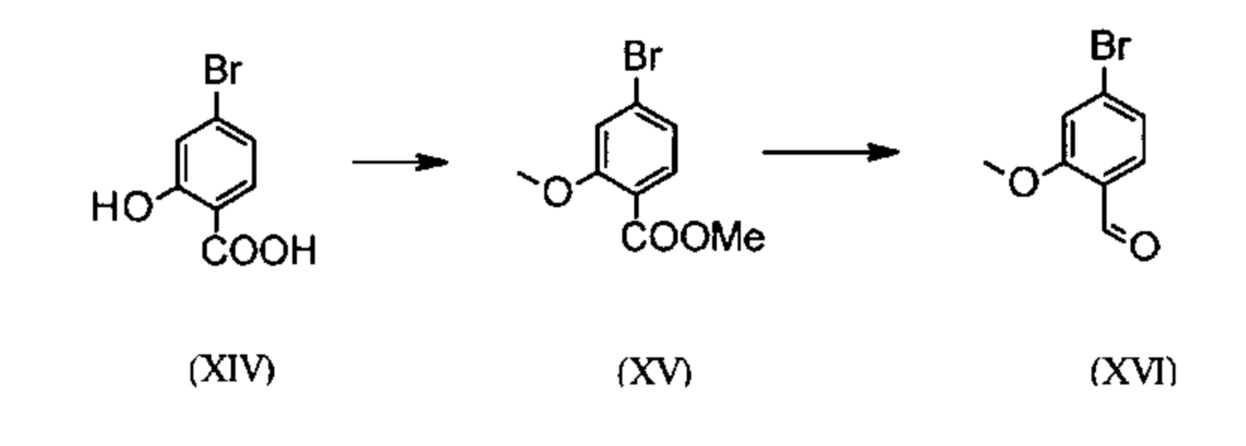
не выделяют, а в растворенном виде далее подвергают превращению в растворе. Это возможно только потому, что выход и чистота соответствующего превращения очень высоки (>95% от теории). Сложный эфир простого метилового эфира (XV) известен в литературе (Journal of Medicinal Chemistry, 1992, том. 35, с. 734-740) и доступен посредством взаимодействия с очень легколетучим, дорогостоящим и вредным для здоровья метилйодидом.
При помощи нового способа может быть продемонстрировано, что аналогично может быть использован малолетучий, недорогой диметилсульфат. Исходя из кислоты (XIV), которую в растворителе, таком как ацетон, 2-бутанон, ТГФ, 2-метил-ТГФ, ДМФА, ДМА и N-метил-2-пирролидон подвергают превращению с диметилсульфатом при помощи вспомогательного основания, такого как карбонат калия, карбонат натрия, карбонат кальция, карбонат лития, N-метилимидазол, триэтиламин, пиридин, 2,6-лутидин при температуре от 50 до 100°С в сложный эфир простого метилового эфира (XV). При этом речь идет о известных специалисту в данной области методах эстерификации кислот и этерификации фенолов (Tetrahedron, 2013, том 69, с. 2807-2815, Journal of the American Chemical Society, 2013, том 135, с. 5656-5668). В качестве особо предпочтительного выявлено превращение в ацетоне при кипечении с возвратом флегмы (56°С) с использованием диметилсульфата и карбоната калия. К тому же диметилсульфат дозируют в течение 4 часов к кипящей реакционной смеси. Ацетон отгоняют и заменяют на толуол (редистилляция). Для обработки добавляют воду (разложение избыточного диметилсульфата), отделяют толуольную фазу и промывают водой и насыщенным раствором поваренной соли, а затем толуольный раствор подвергают дистилляции до определенного объема (служит для азеотропной сушки, т.е. удаления воды для следующих стадий). Определение содержания раствора показывает близкую к полной конверсию (>96% от теории). Вместо бром-соединения может быть аналогично использовано хлор-соединение, полученная при этом конверсия является идентичной с бром-соединением.
Получение альдегида (XVI) описано в литературе, для примера можно назвать: Glaxo Group Limited US 2008/312209 А1, 2008, European Journal of Medicinal Chemistry, 1986, том 21, с. 397-402, Journal of Medicinal Chemistry, 1992, том 35, с. 734-740, Journal of Materials Chemistry, 2011, том 21, с. 9523-9531. Однако применяемые в данном превращении исходные реагенты являются очень дорогими и не могут быть получены в больших количествах, поэтому новый способ был разработан исходя из сложного эфира простого метилового эфира (XV). Превращение из (XV) в альдегид (XVI) происходит при помощи REDAL (натрий-бис(2-метоксиэтокси)алюминий-дигидрид) в толуоле при добавлении N-метилпиперазина. Этот метод описан в литературе (Synthesis 2003, No. 6, 823-828 и Tetrahedron 57 (2001) 2701-2710). Если указанную реакцию проводят в аналогичной приведенной в литературе стехиометрии, то в смеси помимо альдегида находят также и другое соединение. Оказалось, что речь идет о соответствующем бензиловом спирте, которого образуется до 10% в результате перевосстановления. Оказалось, что важно точно отрегулировать стехиометрию REDAL и N-метилпиперазина, 1,21 экв. REDAL + 1,28 экв. N-метилпиперазина, тогда удается указанный побочный продукт, который мешает на следующей стадии кристаллизации, сократить до <1%. К тому же 65%-ный раствор REDAL в толуоле приготавливают при 0-5°С (предпочтительно 1,21 экв.) и добавляют 1,28 экв. N-метилпиперазина. Полученный таким образом раствор REDAL с N-метилпиперазином дозируют в течение ок. 30 минут к приготовленному раствору бромметилового сложного эфира (XIV) в толуоле и затем перемешивают один час при 0°С. Реакционный раствор резко выливают в смесь вода/кислота, предпочтительно водную серную кислоту, отделяют толуольную фазу и промывают водой и насыщенным раствором поваренной соли. Отгоняют толуол и редистиллируют на ДМФА (растворитель следующей стадии). Выход превращения составляет как правило >94% от теории. Соответствующее превращение с хлор-соединением протекает аналогично, выход также является аналогичным. Диметилформамидный раствор напрямую используют в следующей реакции.
Далее по ходу синтеза бромальдегид (XVI) известным как таковым образом в соответствии с общепринятыми специалистами методами (Synth. Commun. 1994, 887-890, Angew. Chemie 2003, 1700-1703, Tetrahedron Lett. 2007, 2555-2557, Tetrahedron Lett. 2004, 1441-1444, JACS 2003, 125, 2890-2891, Journal of Organometallic Chemistry 689 (2004), 4576-4583) переводят в нитрил, при этом получают нитрилальдегид (VI). В случае бром-соединения оказалось особенно выгодным проводить катализируемое палладием превращение в присутствии тригидрата гексацианоферрата калия в качестве источника цианида (Tetrahedron Lett. 48 (2007), 1087-1090). Для этого загружают бромальдегид (XVI) в ДМФА (8-10 кратное количество) 0,22 экв. тригидрата гексацианоферрата калия и 1 экв. карбоната натрия, а затем добавляют 0,005 экв. ацетата палладия. Нагревают 3 часа до 120°С. Раствор охлаждают до 20°С, затем добавляют воду и этилацетат. Этилацетатную фазу отделяют, водную фазу вновь промывают этилацетатом и затем объединенную этилацетатную фазу редистиллируют на изопропанол. Продукт высаживают посредством осаждения водой при кипячении. После разделения сушат в вакууме. В некоторых случаях осаждали продукт непосредственно из ДМФА посредством добавления воды и после разделения и сушки напрямую использовали в следующей стадии. Выход указанного превращения составляет как правило >85% от теоретического. Для превращения хлор-соединения ацетата палладия было не достаточно, здесь оказалось особенно выгодным использовать известные специалисту в данной области палладиевые катализаторы, такие как описано в Tetrahedron Lett. 48 (2007), 1087-1090, выход был несколько ниже, чем в случае бром-соединения, как правило 80-85% от теоретического.
Эфир коричной кислоты (VIII а, b) получают исходя из альдегида формулы (VI) по реакции Кнёвенагеля со сложным цианоэфиром (III) в виде цис/транс-смеси.
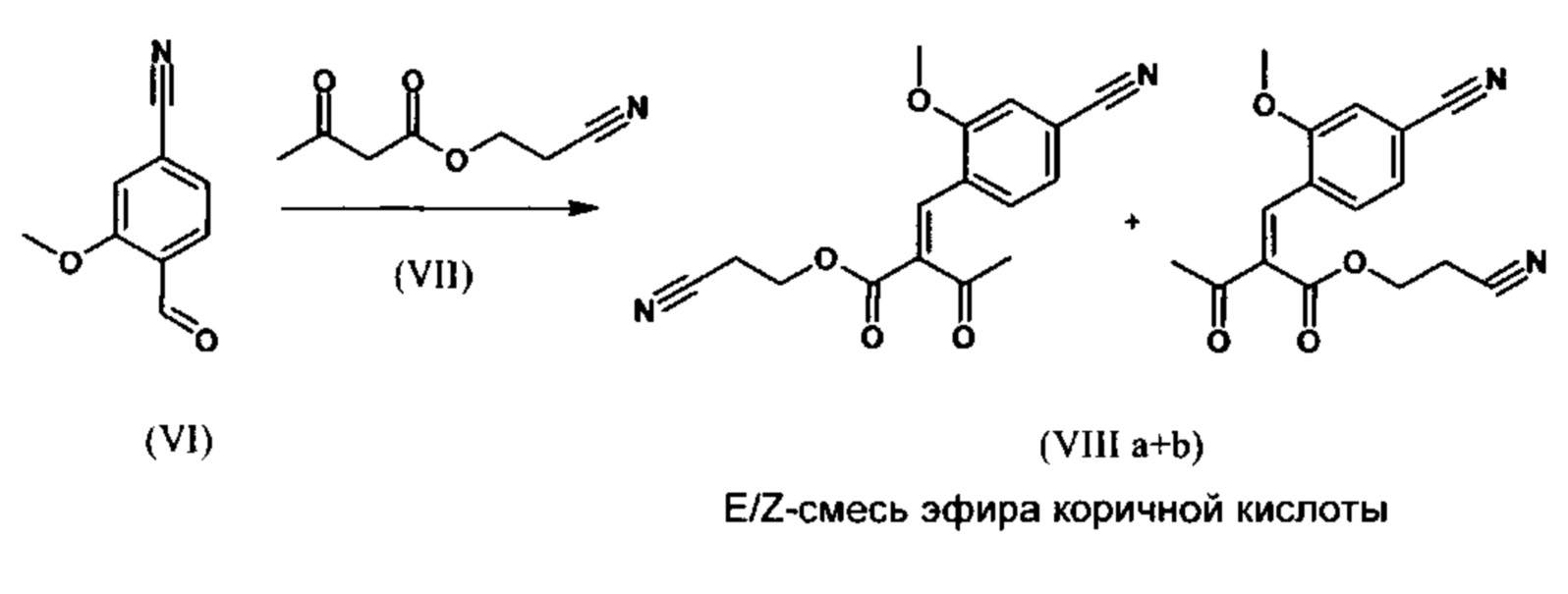
В научно-исследовательской методике нагревали в 16,6 кратном количестве дихлорметана и 0,2 экв. пиперидина/0,2 экв. ледяной уксусной кислоты в течение 20 часов в водоотделителе. После водной обработки и выпаривания растворителя кристаллизуют из метанола, и получают целевое соединение с выходом 52% от теоретического.
Реакция протекает предпочтительно в кипящем дихлорметане (10-кратное количество) с добавлением 5-20% мол. пиперидина, предпочтительно 10% мол., и 5-20% мол. ледяной уксусной кислоты, предпочтительно 5-10% мол., в водоотделителе. Время реакции составляет 4-12 ч, предпочтительно однако 5-6 ч, особо предпочтительно 6 ч. Сложный цианоэфир (VII) добавляют в количестве 1,0-1,5 экв., предпочтительно однако от 1,1 до 1,35 экв. Особо предпочтительно 1,1 экв. Получение сложного цианоэфира (VII) описано в Pharmazie, 2000, том 55, с. 747-750 и Bioorg. Med. Chem. Lett. 16, 798-802 (2006). После окончания реакции охлаждают до 20°С и органическую фазу 2 раза промывают водой. Промытую органическую фазу редистиллируют на 2-бутанол и цис/транс-смесь эфира коричной кислоты (VIII а+b) без промежуточного выделения используют напрямую в следующей реакции с гетероциклом (IX) с получением дигидропиридина (X):
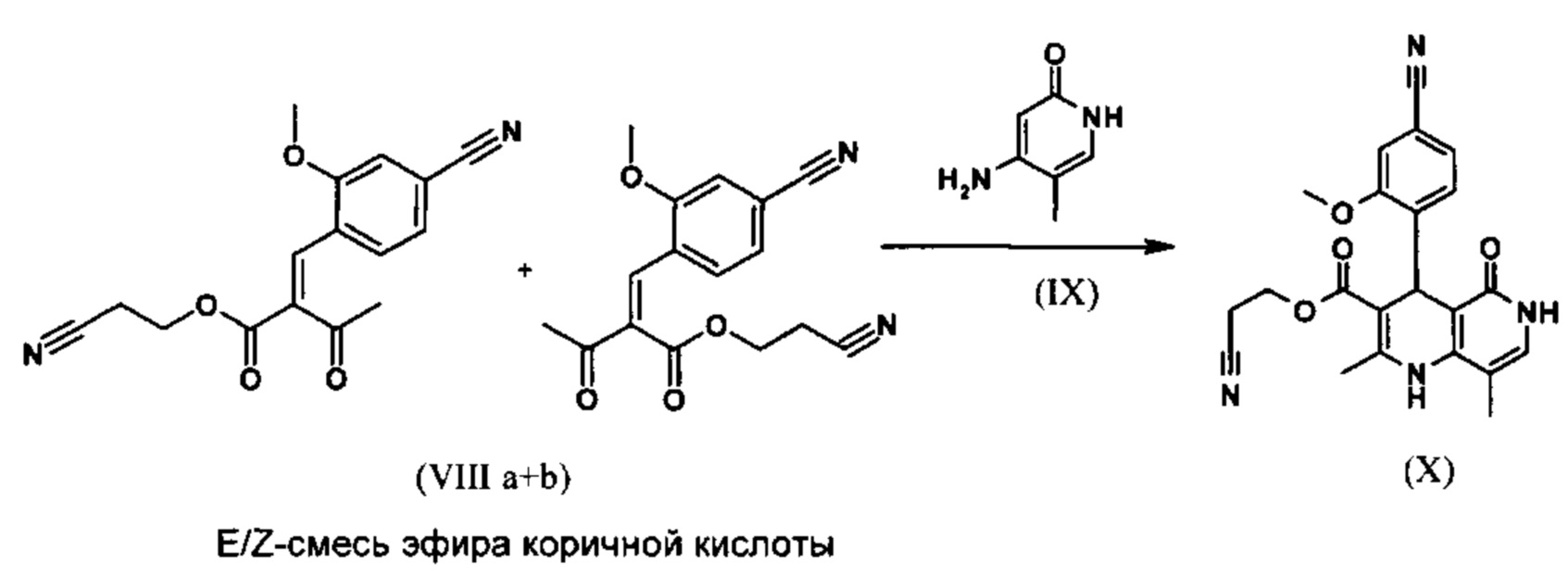
В научно-исследовательском синтезе для дальнейшего превращения с гетероциклом (IX) в изопропаноле в течение 40 часов нагревали до кипения с возвратом флегмы.
Было найдено, что реакция может быть проведена предпочтительно во вторичном спирте, таком как изопропанол, изобутанол, 2-амиловый спирт, циклогексанол, при температуре 80-160°С, при нормальном давлении, а также в автоклаве (2-10 бар), времени реакции 8-40 ч, предпочтительно однако 20-25 ч, в кипящем 2-бутаноле при нормальном давлении или в изопропаноле в автоклаве (100°С, 2-10 бар, предпочтительно 3-5 бар, 8-24 ч). Для обработки охлаждают до 0°С - 20°С, кристаллы отфильтровывают и повторно промывают изопропанолом, затем сушат (в вакууме, 60°С).
Если из-за экологических причин следует отказаться от применения дихлорметана, оказалось выгодным получать эфир коричной кислоты (VIII а, b) в изопропаноле, для этого загружают альдегид (VI) в изопропанол (3-9 кратное количество, предпочтительно 5-7 кратное) и добавляют 5-20% мол. пиперидина, предпочтительно 5-10% мол., и 5-20% мол. ледяной уксусной кислоты, предпочтительно 5-10% мол. При 30°С 1,0-1,5 экв., предпочтительно 1,1-1,35 экв., особо предпочтительно 1,1 экв. сложного цианоэфира (VII), при необходимости растворенного в небольшом количестве изопропанола, добавляют в течение 3 часов и перемешивают 1 час при 30°С. Эфир коричной кислоты (VIII а, b) выкристаллизовывается в течение реакции. Затем продукт, при необходимости после охлаждения, отфильтровывают при 0°С, промывают небольшим количеством изопропанола (охлажденного до 0°С) и влажным используют в следующей реакции, как описано ранее. Выход составляет >96% от теории. Предпочтительно в следующем превращении в течение 20-24 часов обрабатывают в 10-15 кратном количестве (соответственно на альдегид (VI)), предпочтительно 11-12 кратном количестве изопропанола при 100°С под давлением. Продукт после окончания реакции и охлаждения выделяют посредством фильтрации или центрифугирования. Затем сушат при 40-90°С в вакууме. Так как превращение эфира коричной кислоты является почти количественным, процесс можно легко стандартизировать для следующей стадии, без необходимости корректировать количество гетероцикла (IX), поэтому продукт можно использовать влажным по изопропанолу. Выход находится около >87% от теории. Гетероцикл (IX) может быть получен известными в литературе методами, например, таким как описан в Synthesis 1984, 765-766.
Исходя из дигидропиридина (X) этиловый простой эфир (XI) получают превращением при кислотном катализе сложным ортоэфиром, причем R означает -Н и метил:
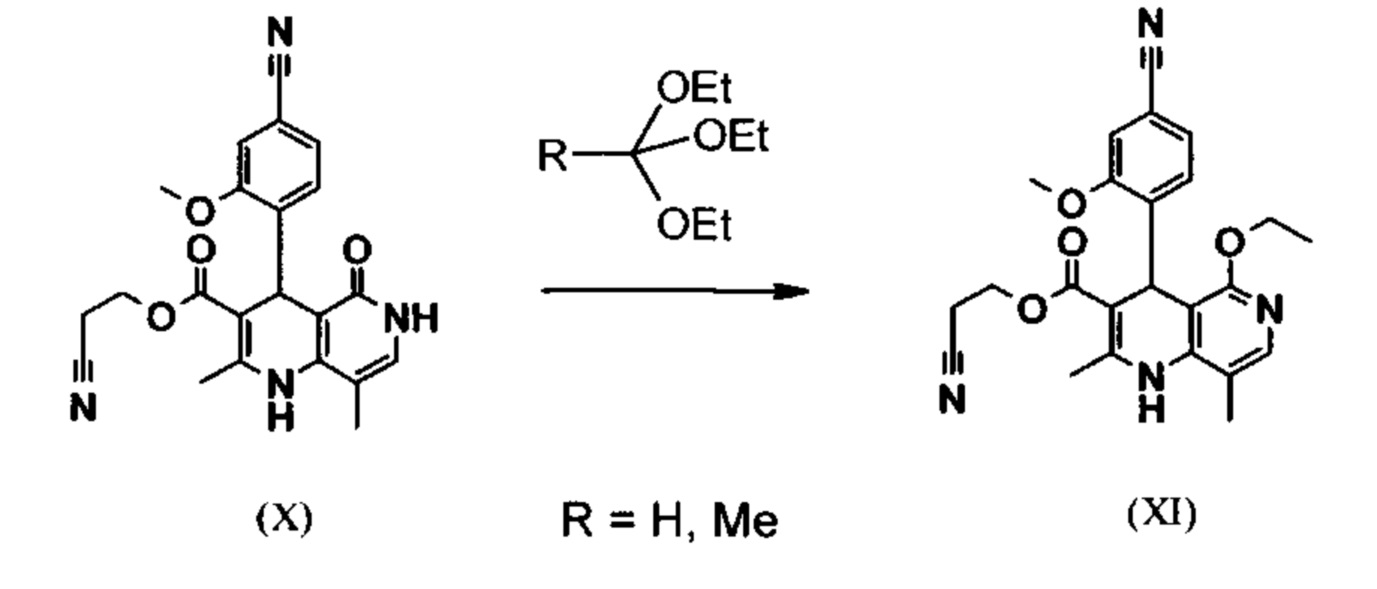
В научно-исследовательском синтезе для этого подвергали превращению в 25 кратном количестве ДМФА с 20,2 экв. триэтилортоформиата, каталитическим количеством концентрированной серной кислоты при 135°С. Концентрировали досуха и остаток очищали хроматографией, выход 86% от теории. Этот способ по причине сильного разбавления и применения легко воспламеняемого при низкой температуре триэтилортоформиата, который используют в очень большом избытке, и последующей хроматографии не подходит для промышленного проведения.
Неожиданно было обнаружено, что реакция может быть проведена в более концентрированных условиях (до 1,5 г растворителя на 1 г исходного продукта), в растворителе, таком как диметилацетамид, N-метил-2-пирролидон (1-метил-2-пирролидон), ДМФА (диметилформамид), при добавлении 4-10% масс. концентрированной серной кислоты, предпочтительно 6-8% масс. Реакция тогда неожиданным образом протекает уже с 2,5-5 экв. сложного ортоэфира. Было найдено, что существенно выгоднее использовать в реакции соответствующий триэтилортоацетат, так как он реагирует намного чище и значительно менее огнеопасен, что делает его особенно подходящим для промышленного проведения. Превращение протекает предпочтительно в ДМА (диметилацетамиде) и N-метил-2-пирролидоне (1-метил-2-пирролидоне), при температуре 100-120°C, предпочтительно 115°С. Оказалось выгодным перед стартом данной реакции часть растворителя (ДМА или N-метил-2-пирролидона) отогнать при повышенной температуре (100-120°С в вакууме), чтобы удалить при необходимости присутствующий остаток изопропанола с предыдущей стадии, так как в противном случае образуется нежелательный побочный продукт. Превращение: перемешивают 1,5-3 часа, предпочтительно 2 часа. Для обработки добавляют воду непосредственно в реакционную смесь, при этом выкристаллизовывается продукт. Чтобы иметь в распоряжении особо стабильный и воспроизводимый способ, сначала добавляют частичное количество воды (например, 1/3), потом вносят кристаллическую затравку и добавляют оставшееся количество воды. Эти действия гарантируют, что всегда будет получаться одинаковая модификация кристаллов, которая демонстрирует самые лучшие свойства для выделения. Продукт повторно промывают водой и высушивают. Выход находится около >92% от теории.
Исходя из простого этилового эфира (XI) посредством щелочного омыления и последующей кислотной обработки получают кислоту (XII):
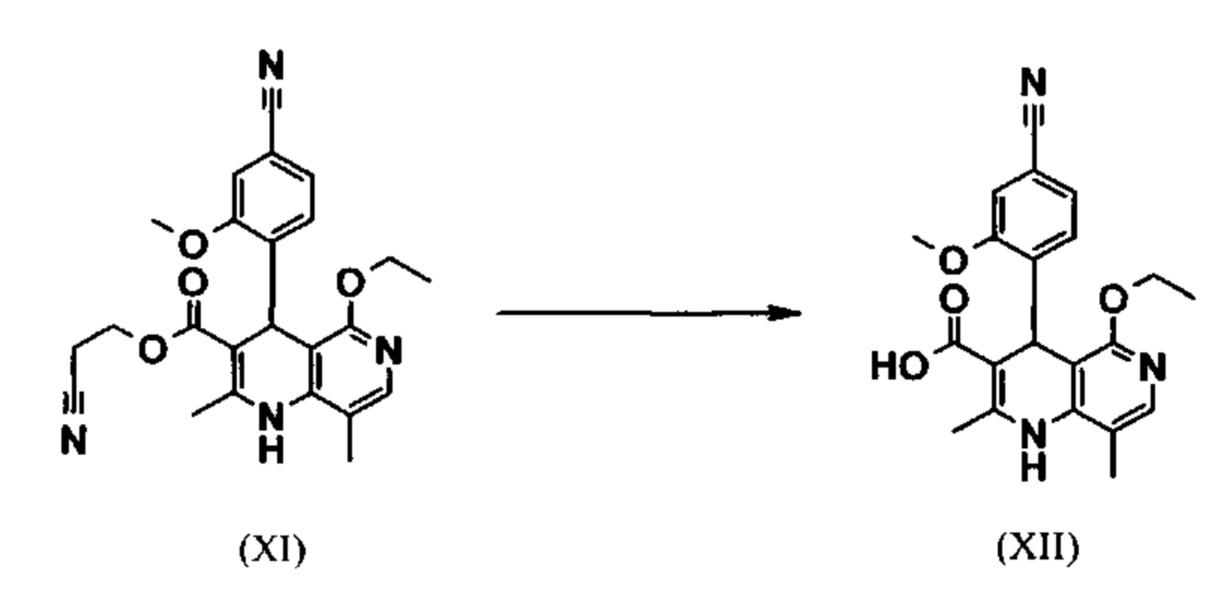
В научно-исследовательском синтезе омыление проводили при сильном разбавлении (33,9 кратном) в смеси ДМЭ/вода 3:1. Здесь прежде всего требовалось увеличить производительность и заменить используемый ДМЭ (диметоксиэтан), который имеет очень низкую температуру вспышки и, следовательно, оценивается как особо опасный для промышленного применения. Неожиданно было найдено, что реакция может быть проведена также очень легко в смеси ТГФ/вода при более концентрированных условиях. Для этого работают предпочтительно в смеси ТГФ/вода 2:1 (9 кратное количество), добавляют раствор едкого натра при 0-5°С, а затем перемешивают 1-2 часа при 0-5°С. Можно также использовать водный раствор едкого калия, однако предпочтительно применять NaOH. Для обработки экстрагируют МТБЭ (метил-трет-бутиловый эфир) и этилацетатом и для выделения посредством минеральной кислоты, такой как соляная кислота, серная кислота или фосфорная кислота, но предпочтительно соляной кислоты, устанавливают рН 6,5-7,0. Затем смешивают с насыщенным раствором аммониевой соли соответствующей кислоты, предпочтительно однако с раствором хлорида аммония, при этом продукт количественно выкристаллизовывается. После выделения повторно промывают водой и этилацетатом или ацетонитрилом или ацетоном, предпочтительно однако ацетонитрилом, и сушат в вакууме при 40-50°С. Выход является почти количественным (99%). Альтернативная предпочтительная обработка: в качестве альтернативы для обработки смешивают с толуолом, добавляют ацетат натрия и перемешивают при 20°С, затем фазы разделяют и в водной фазе при 0°С посредством 10%ной водной соляной кислоты устанавливают рН 6,5-7,0 (при рН 9,5-10,0 может быть внесена кристаллическая затравка). Оставляют ненадолго перемешиваться и отфильтровывают продукт, повторно промывают небольшим количеством воды и толуола и сушат в вакууме при 40-50°С. Также в этом случае полученные выходы являются количественными.
Следующее превращение кислоты в амид (XIII) в научном исследовании проводили, как показано далее: Кислоту (XII) растворяли в примерно 10 кратном количестве ДМФА, добавляли 1,25 экв. 1,1'-карбодиимидазола и 0,1 экв. ДМАП (4-(диметиламино)-пиридин) и перемешивали 4 часа при комнатной температуре. Затем примешивали 20 экв. аммиака в форме 25%-ного водного раствора и эту смесь переносили в предварительно нагретую до 110°С масляную баню. При этой процедуре моментально образуется большое количество газообразного аммиака, который выделяется из системы и дополнительно способствует сильному увеличению давления. Добавляли примерно 90 кратное количество воды и устанавливали рН 7 посредством добавления ацетата натрия. Выпавший продукт отфильтровывали и высушивали (выход: 59% от теории). Из маточного раствора посредством затратной экстракции (примерно 100 кратное количество этилацетата) выделяли дополнительное количество, которое вымешивали из легковоспламеняющегося диэтилового эфира и которое содержало примерно 14% ДМФА. Очевидно, что такой процесс не может быть реализован в производственных рамках, а значит существует большая потребность в альтернативном технологическом подходе. Усилия по выделению этого частичного количества непропорциональны выделяемому при этом количеству.
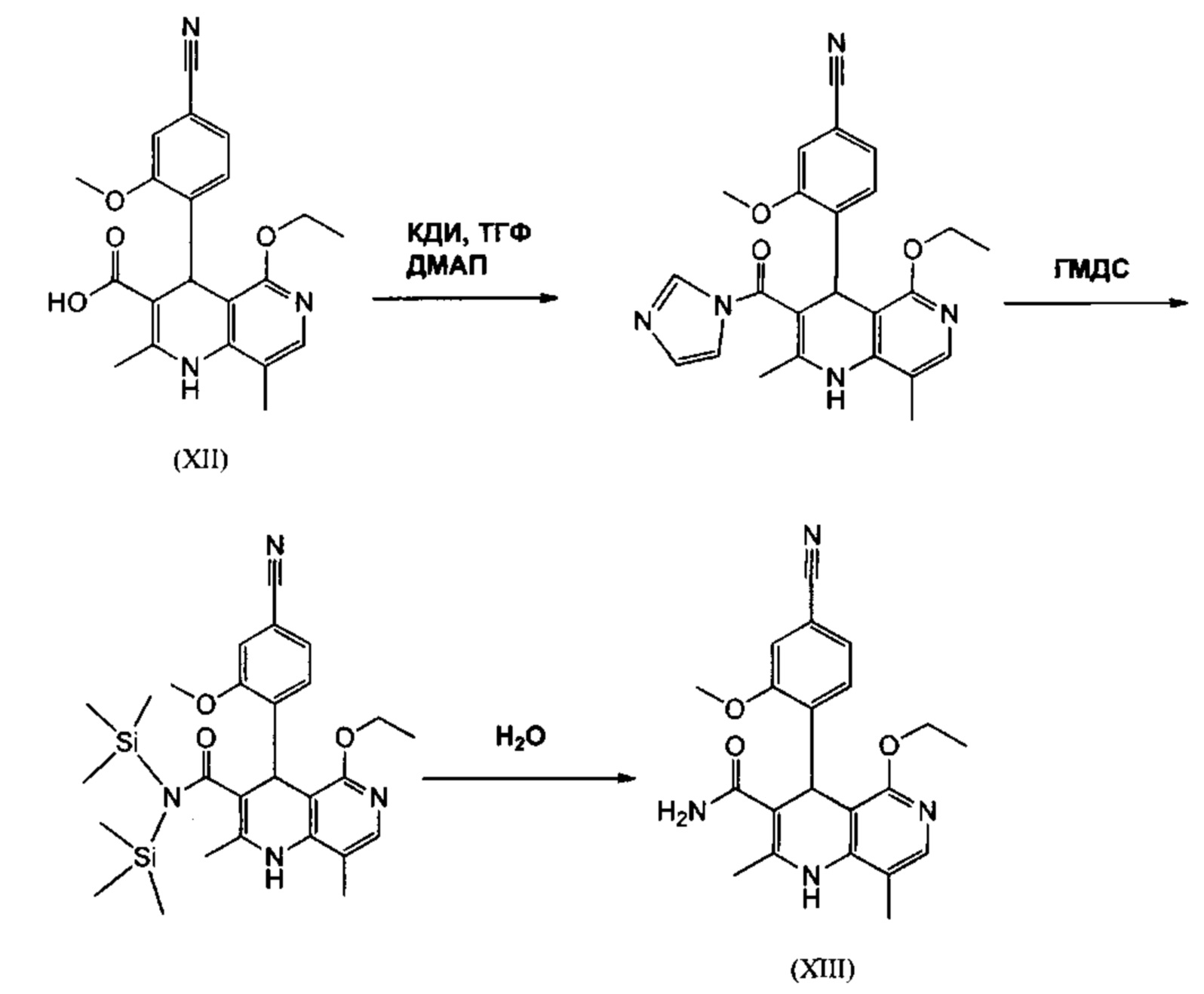
Неожиданно было обнаружено, что при превращении кислоты (XII) в ТГФ амид (XIII) кристаллизуется непосредственно из раствора и может быть получен с высоким выходом и чистотой. Для этого карбоновую кислоту (XII) с 1,1-1,6 экв., предпочтительно 1,3-1,4 экв. 1,1'-карбодиимидазола при катализе ДМАП (5-15% мол., предпочтительно 10% мол.) в ТГФ подвергают превращению в имидазолид, превращение проводят при температурах между 20-50°С, предпочтительной оказалась процедура согласно которой, сначала следует начинать при 20°С, затем перемешивать в течение 1-2 часов при этой температуре, а затем в течение 2-3 часов далее перемешивать при 50°С. После окончания активирования добавляют 3-8 экв., предпочтительно 4,5 экв. гексаметилдисилазана и кипятят 16-24 часов, предпочтительно однако 16 часов, с возвратом флегмы. Полученное при этом дисилиламидное соединение может быть при необходимости выделено, но было обнаружено, что более выгодно продолжить дальнейшую реакцию по однореакторной технологии (нем. Eintopfreaktion). Поэтому после окончания реакции охлаждают до 0-3°С и добавляют водную смесь или смесь воды и ТГФ, оказалось более выгодным использовать воду в 0,5 -0,7 кратном количестве (соответственно на исходный продукт), особо выгодно 0,52 кратное количество воды. Вода может быть добавлена непосредственно или в смеси с ТГФ, взятым в примерно от однократного до двойного объемном количестве. После окончания гашения, нагревают в общей сложности 1-3 часа, предпочтительно 1 час, до кипения с возвратом флегмы. Охлаждают до 0°С и далее перемешивают в течение 1-5 часов, предпочтительно в течение 3 часов при указанной температуре, затем продукт выделяют фильтрованием или центрифугированием. Промывают ТГФ и водой и сушат в вакууме при повышенной температуре (от 30 до 100°С, предпочтительно от 60°С до 90°С). Выход является очень высоким и составляет как правило >93% от теории. Чистота обычно составляет >99% (ВЭЖХ, 100%-ный метод). Соединение (XIII) также может быть получено непосредственно превращением с газообразным аммиаком в автоклаве (примерно от 25 до 30 бар). Для этой цели проводят предварительное активирование, описанное выше, а затем нагревают в газообразном аммиаке под давлением. После окончания реакции охлаждают и отфильтровывают продукт. Полученные таким образом выходы и чистота сопоставимы.
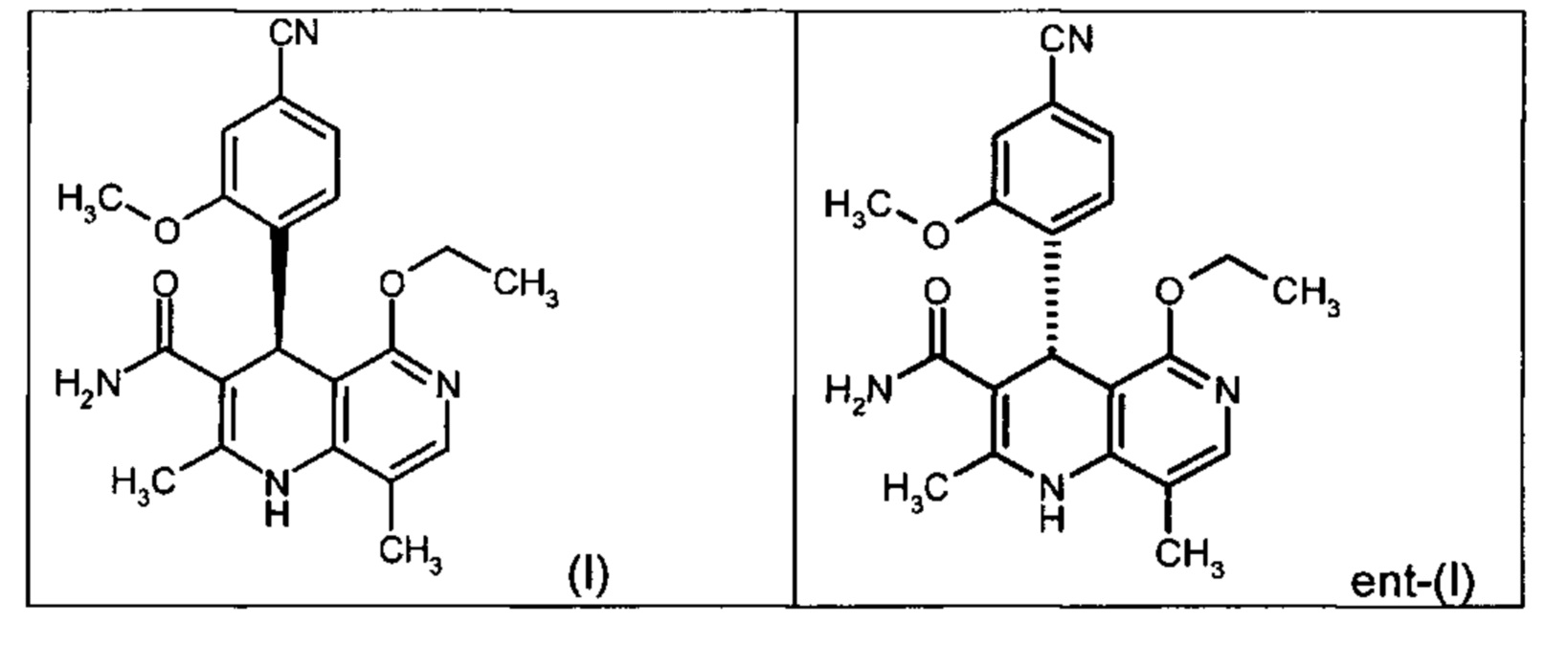
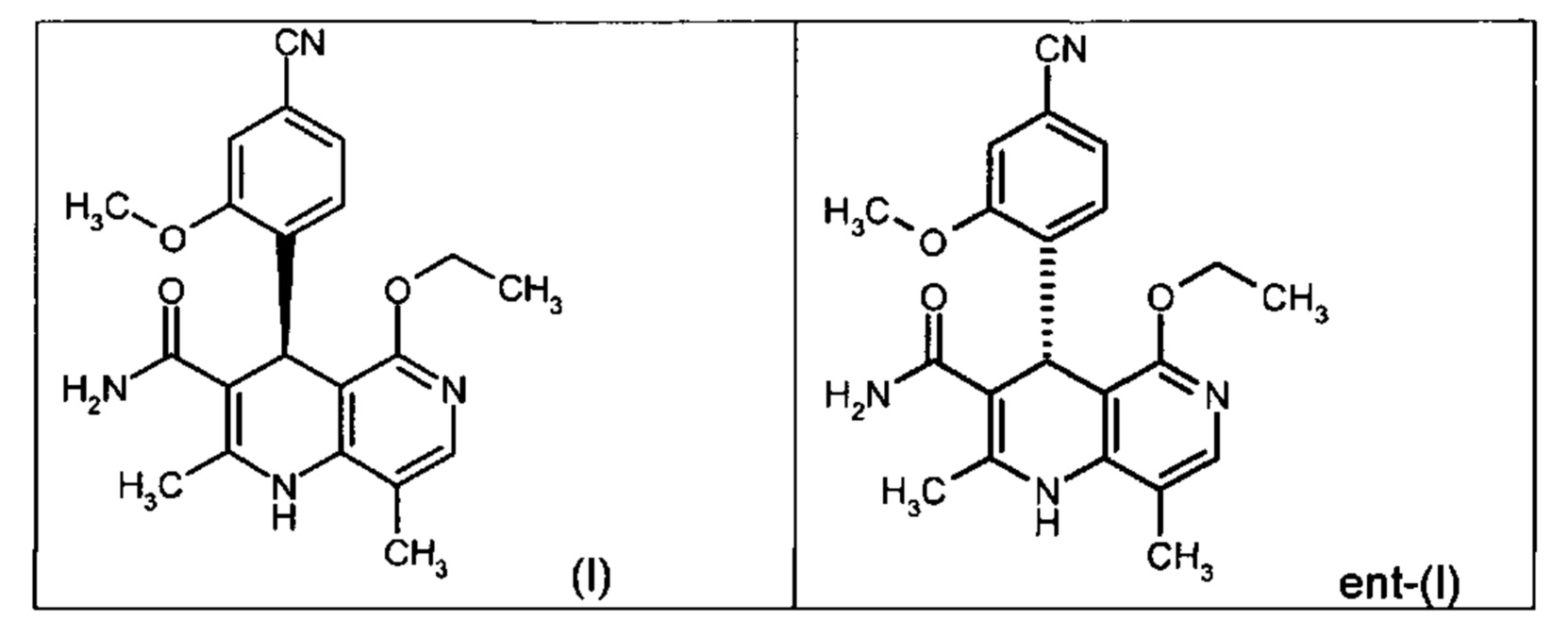
Чтобы получить соединение формулы (I), рацемическая смесь амидов формулы (XIII) должна быть разделена на энантиомеры. В опубликованном научно-исследовательском синтезе для этой цели была использована специально синтезированная хиральная фаза (собственное производство), которая содержала N-(дициклопропилметил)-N2-метакрилоил-D-лейцинамид в качестве хирального селектора. Этот селектор получают на многоступенчатой стадии и затем полимеризуют на специальном силикагеле. В качестве элюента служит метанол/этилацетат. Основным недостатком этого метода была очень низкая загрузка, 30 мг за одно разделение на хроматографической колонке 500*63 мм, поэтому возникла высокая потребность в поиске более эффективного метода разделения, который позволил бы разделять энантиомеры в многотонном масштабе. Неожиданно было найдено, что разделение также можно проводить на коммерчески легко доступной фазе. Речь здесь идет о фазе Chiralpak AS-V, 20 мкм. В качестве элюента была использована смесь метанол/ацетонитрил 60:40. Эта смесь имеет большое преимущество в том, что ее после дистилляционной обработки можно опять регенерировать в качестве элюента с идентичным составом (60:40 соответствует азеотропу). Таким образом достигается очень эффективный процесс, при котором выход разделения составляет >47% от теории (теоретически возможным является 50%). Оптическая чистота составляет при этом >93% ее, предпочтительно однако >98.5% ее. В этом случае хроматографию можно проводить на коммерческой хроматографической колонке, но предпочтение отдается методам, известным специалистам в данной области, таким как SMB или Varicol (Computers and Chemical Engineering 27 (2003) 1883-1901). Так, например, на SMB-установке было разделено около 500 кг рацемического амида формулы (XIII), причем был получен выход 48%. Продукт производится в виде 3-8%-ного, предпочтительно 5-7%-ного раствора в смеси метанол/ацетонитрил 60:40 и может быть использован непосредственно в «финальной обработке». Возможны и другие соотношения растворителей в смеси между ацетонитрилом и метанолом (90:10 - 10:90). Альтернативно, однако, для SMB-разделения могут быть использованы другие смеси растворителей, такие как ацетонитрил/этанол, в соотношениях смеси от 10:90 до 90:10. Конкретное соотношение растворителей частично зависит от технических характеристик SMB-установки и должно быть при необходимости приведено в соответствие (например, различная скорость потока, рециркуляция растворителя на тонкопленочном испарителе).
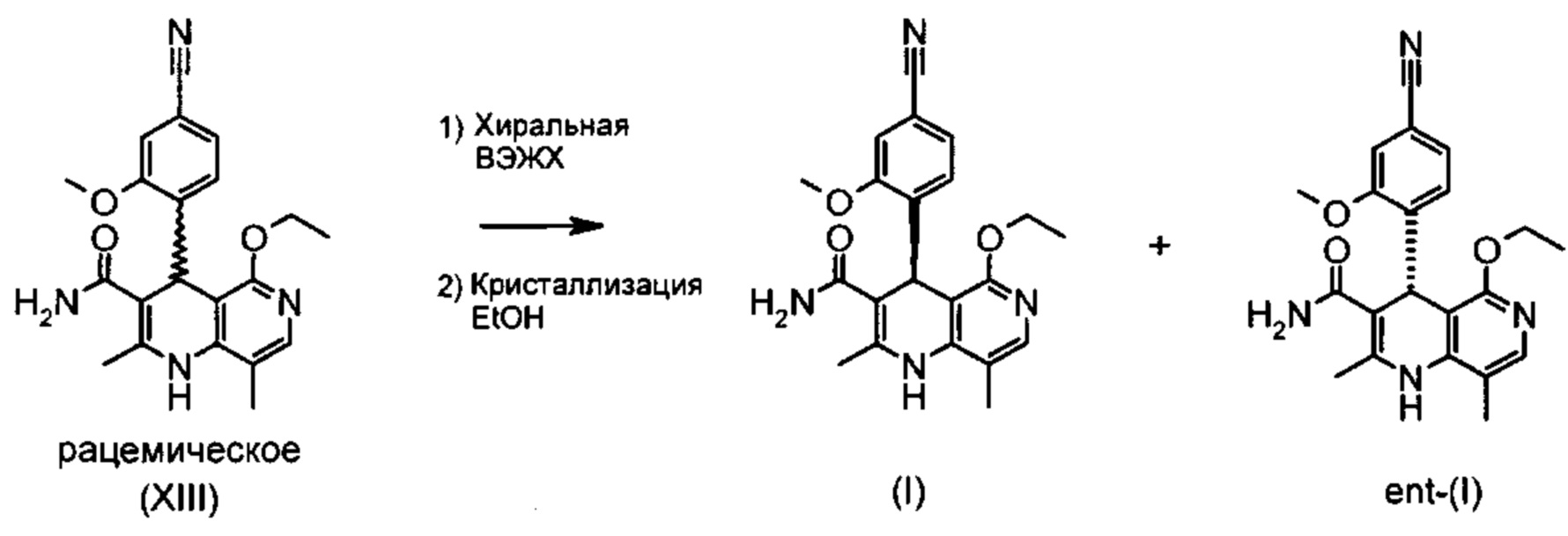
Помимо целевого соединения формулы (I) также получают энантиомерное соединение формулы ent-(I) практически с тем же выходом. Из-за финансово-экономических аспектов существовала потребность не разрушать указанный энантиомер формулы ent-(I), а изобрести способ, который позволяет превратить соединение формулы ent-(I) в рацемическую смесь формулы (XIII), чтобы снова подвергнуть ее энантиомерному разделению с помощью SMB.
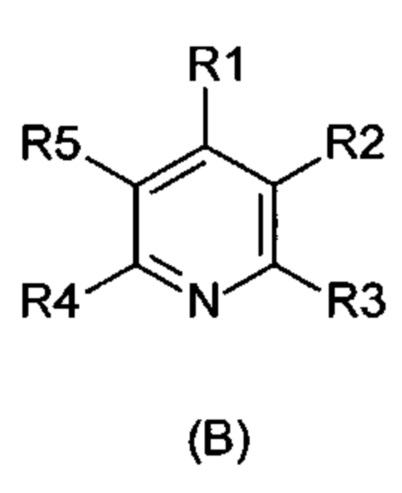
Указанная задача была решена посредством применения непрямого электрохимического способа согласно изобретению для окисления 1,4-дигидропиридиновых производных формулы (А) в пиридиновые аналоги формулы (В)
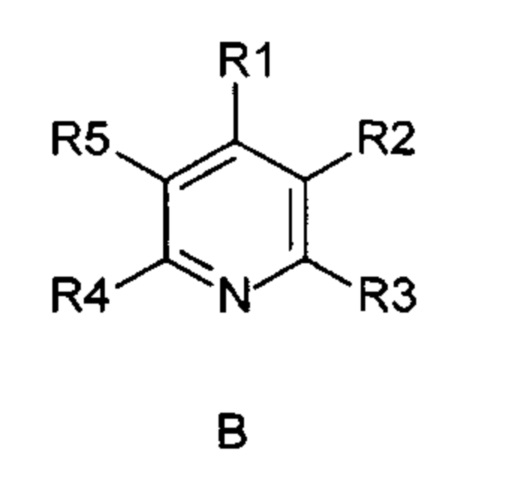
и последующего восстановления.
Для синтеза фармацевтических активных веществ окисление 1,4-дигидропиридиновых производных, таких как описаны в формуле (А), в пиридиновые аналоги (В)
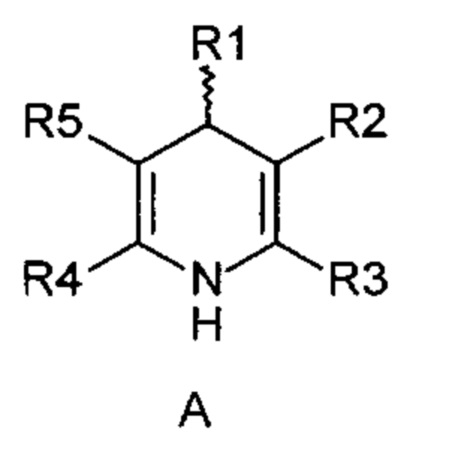
 причем
причем
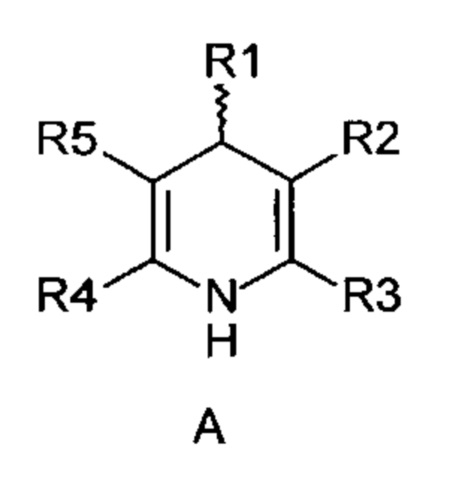
R1-R5 независимо друг от друга означают водород, фтор, хлор, бром, йод, карбоксил, сложный карбоксиловый эфир, гидроксил, гидроксиэфир, циано, нитро, замещенный и незамещенный амид, алкил с 1-6 атомами углерода, галогеналкил с 1-6 атомами углерода, формил, замещенный и незамещенный фенил, замещенный и незамещенный бензил, замещенный и незамещенный нафтил, замещенный и незамещенный 5- или 6-членный гетероцикл, имеющий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, S, О, бензоконденсированный 5- или 6-членный гетероцикл,
проводят с использованием химических окислительных средств.
Han и др. [Org. Lett. 2014, 16, 4142-4145] описывают стадию окисления 1,4-дигидропиридинового производного (С) [метилового эфира 4- (3,6-дигидро-2Н-пиран-4-ил)-7,7-диметил-5-оксо-2-(пропан-2-ил)-1,4,5,6,7,8-гексагидро-хинолин-3-карбоновой кислоты] с использованием 1,2 эквивалента DDQ [2,3-дихлор-5,6-дициано-1,4-бензохинона]. При этом получают выделенное пиридиновое производное (D) [метиловый эфир 4-(3,6-дигидро-2Н-пиран-4-ил)-7,7-диметил-5-оксо-2-(пропан-2-ил)-5,6,7,8-тетрагидрохинолин-3-карбоновой кислоты] с выходом 93,5 мас. %.
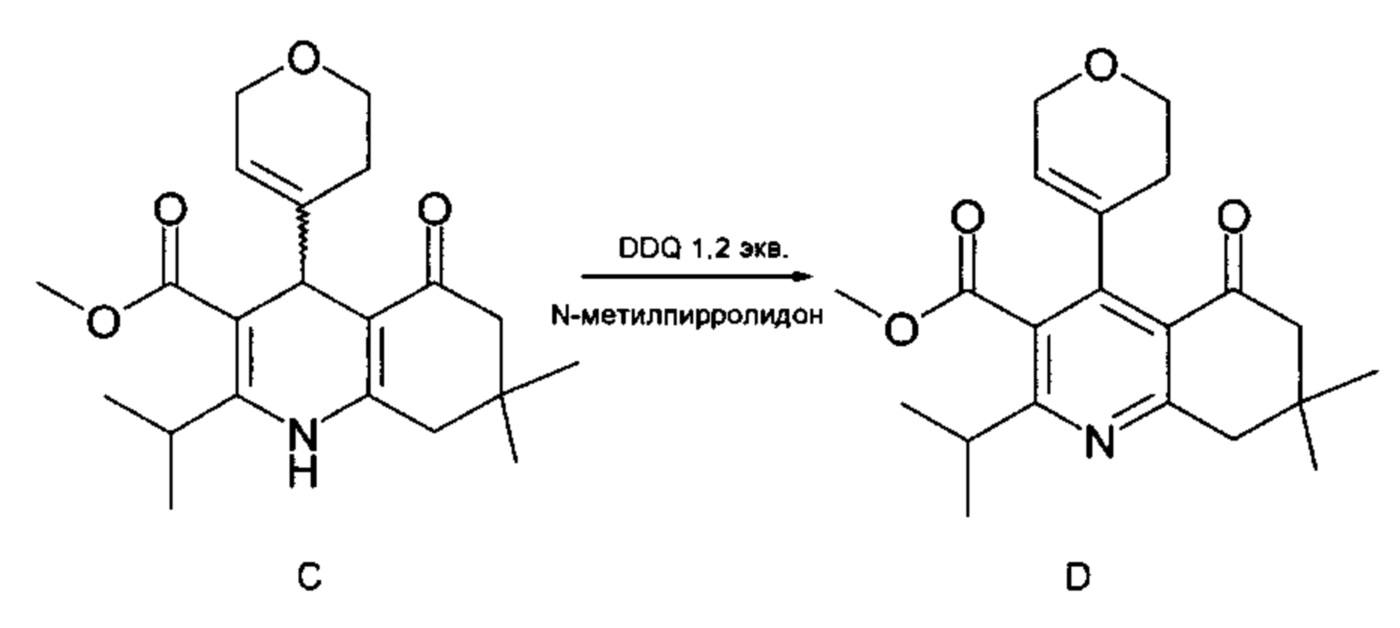
Недостатком указанного способа является большое количество окислительного средства (DDQ), требуемое для отделения двух протонов и двух электронов от субстрата. В лучшем случае для завершения реакции необходимы стехиометрические количества химических окислительных средств. В большинстве случаев применяют избыток химических реагентов для обеспечения полной конверсии и максимального выхода. Таким образом, образуется много отходов, и, кроме того, в результате использования больших количеств окислительного средства также увеличивается стоимость производства.
По аналогии с работой Han и др. можно предположить, что этот метод применим для всех 1,4-дигидропиридиновых производных (DHP) формулы А и соответствующих пиридинов (PYR) формулы (В).
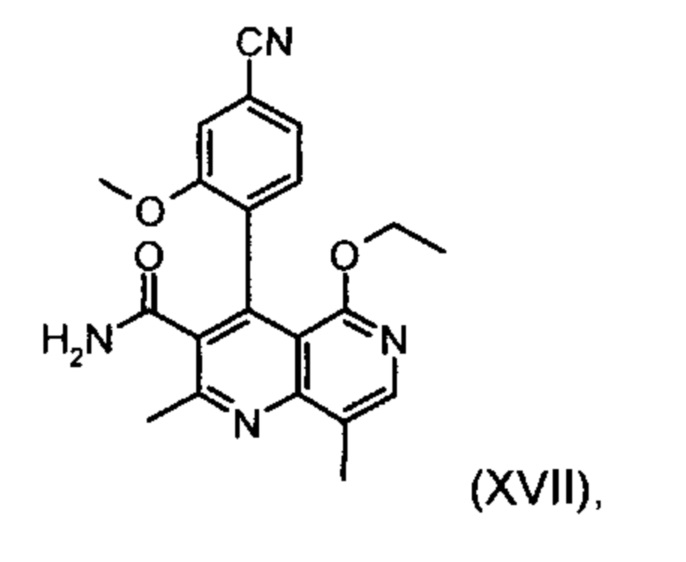
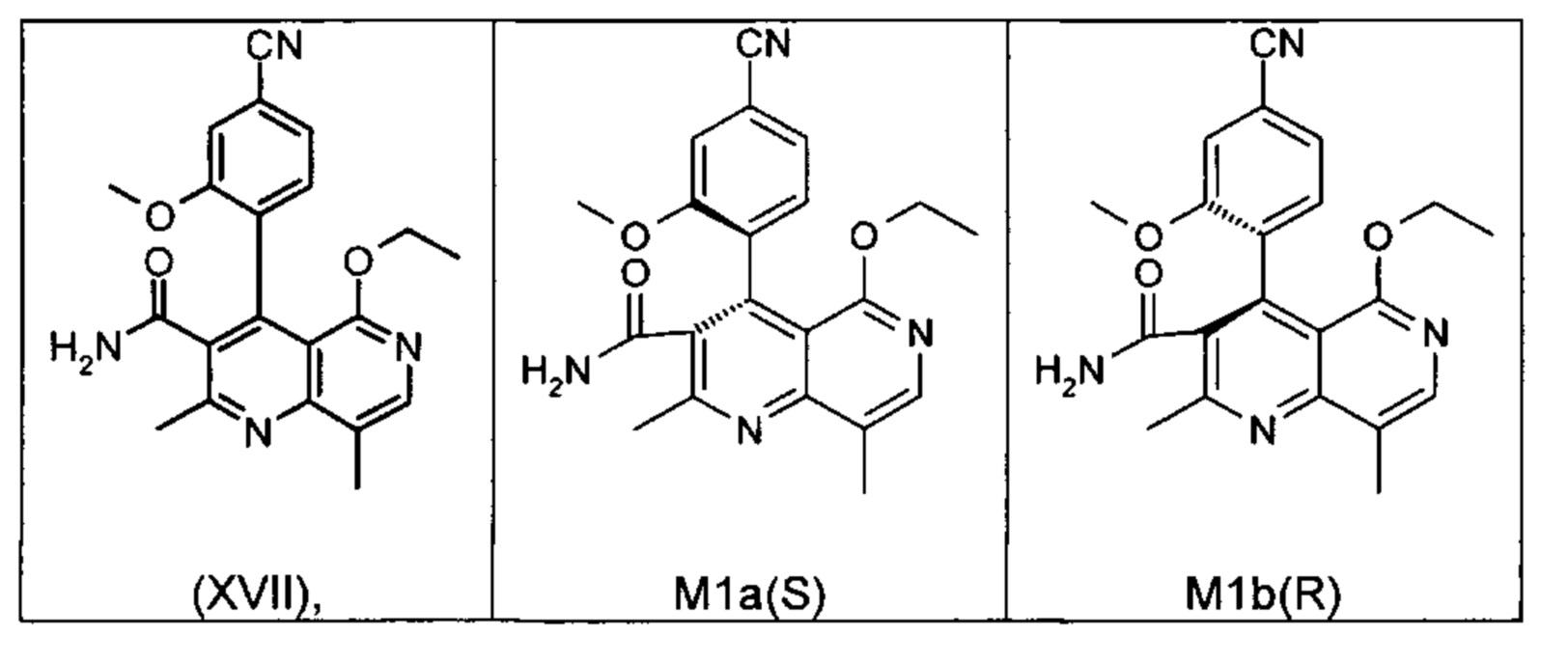
Применение согласно изобретению вышеописанного способа окисления представляет собой новый способ получения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (I) посредством рециркуляционного процесса из энантиомера формулы ent-(I), образующегося в процессе получения соединения (I).
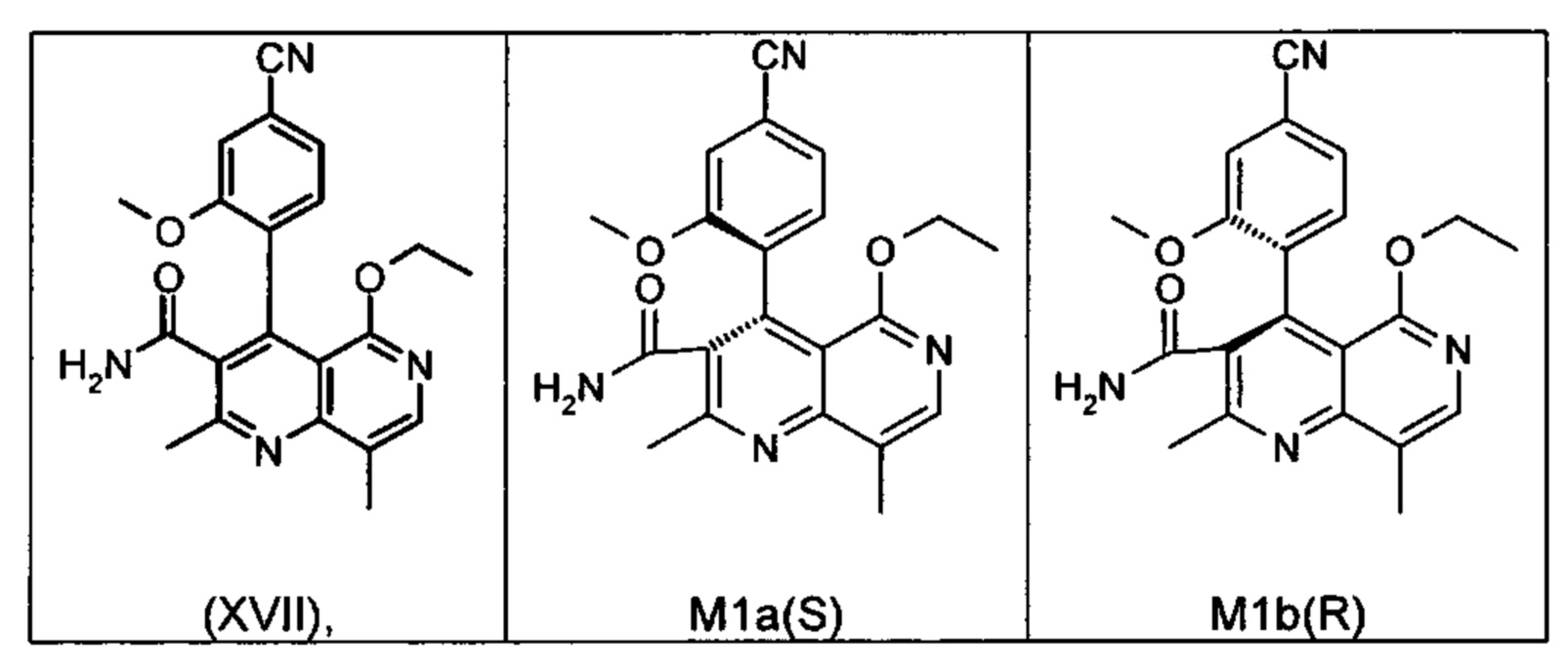
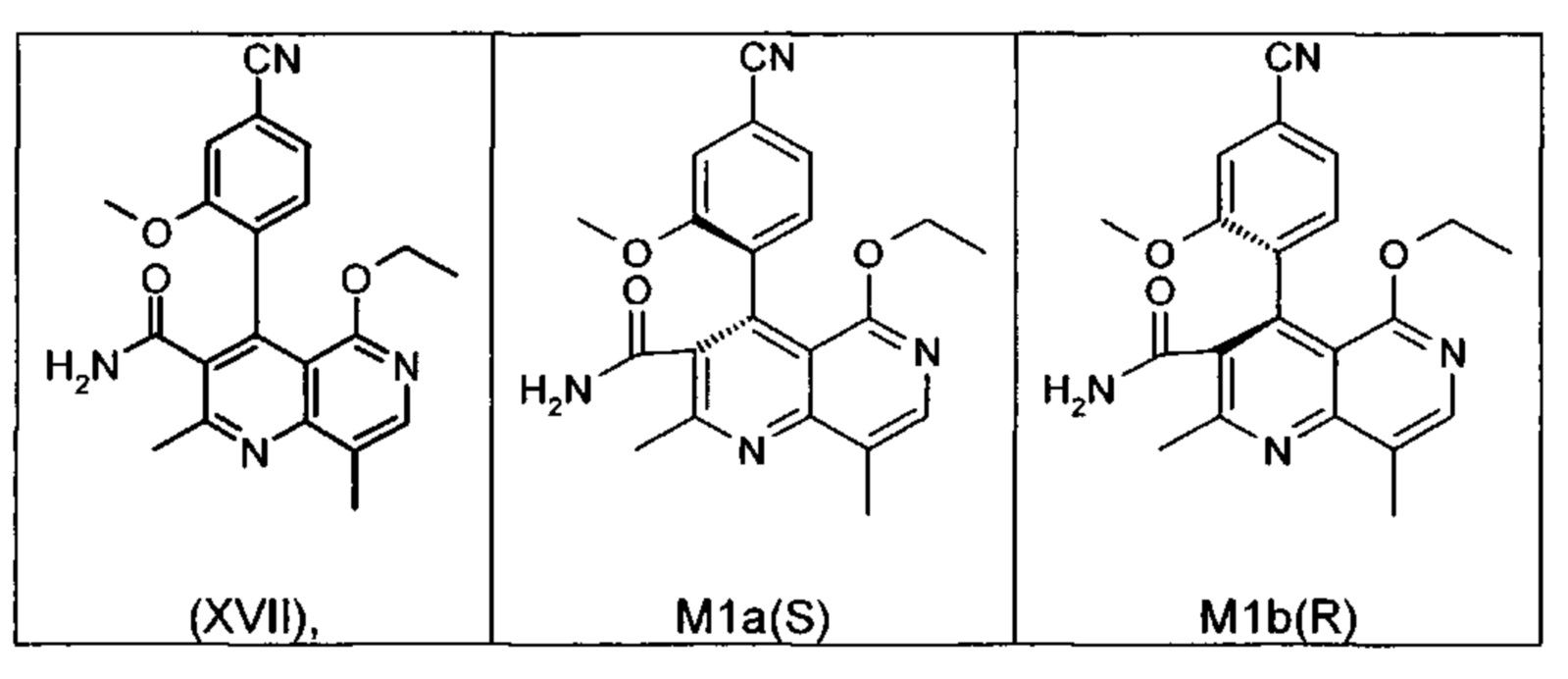
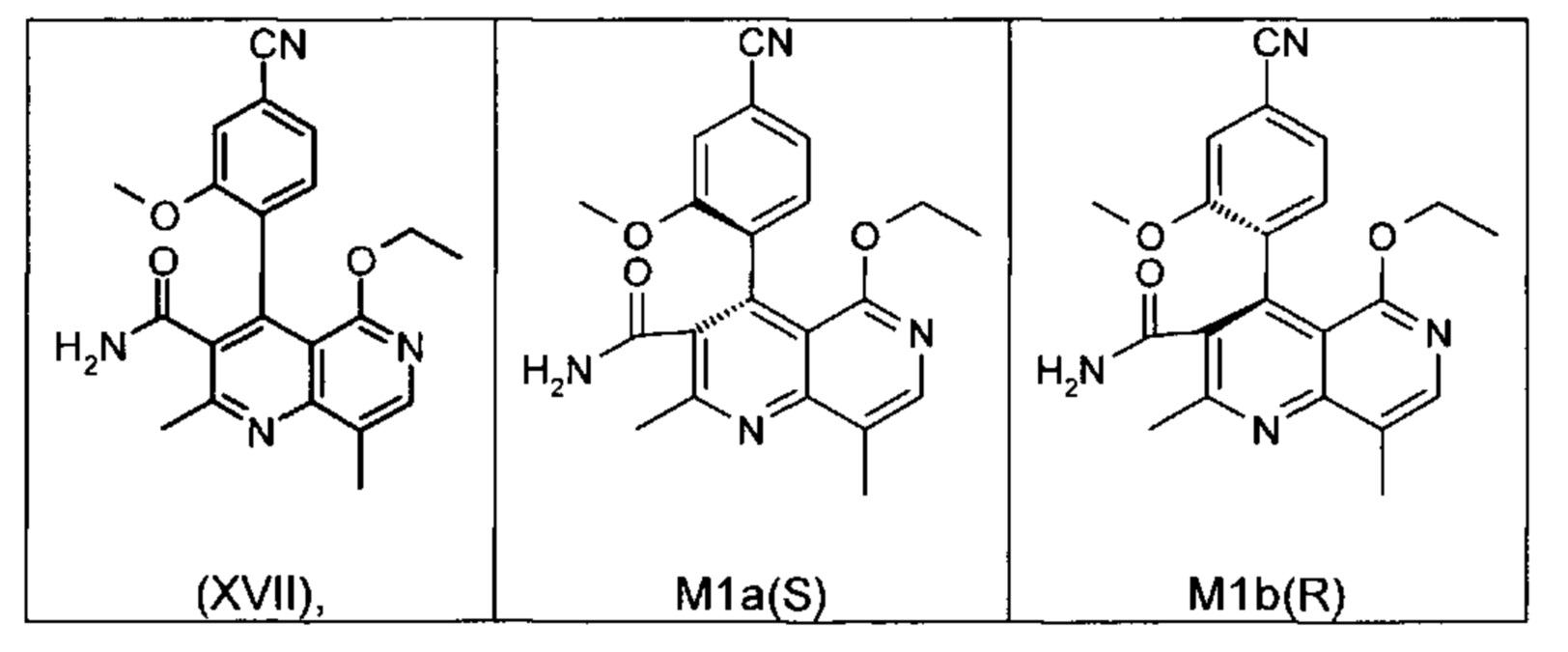
Это достигается сначала окислением (ароматизацией) ненужного энантиомера формулы ent-(I) в пиридин формулы (XVII), а затем его электрохимическим восстановлением:
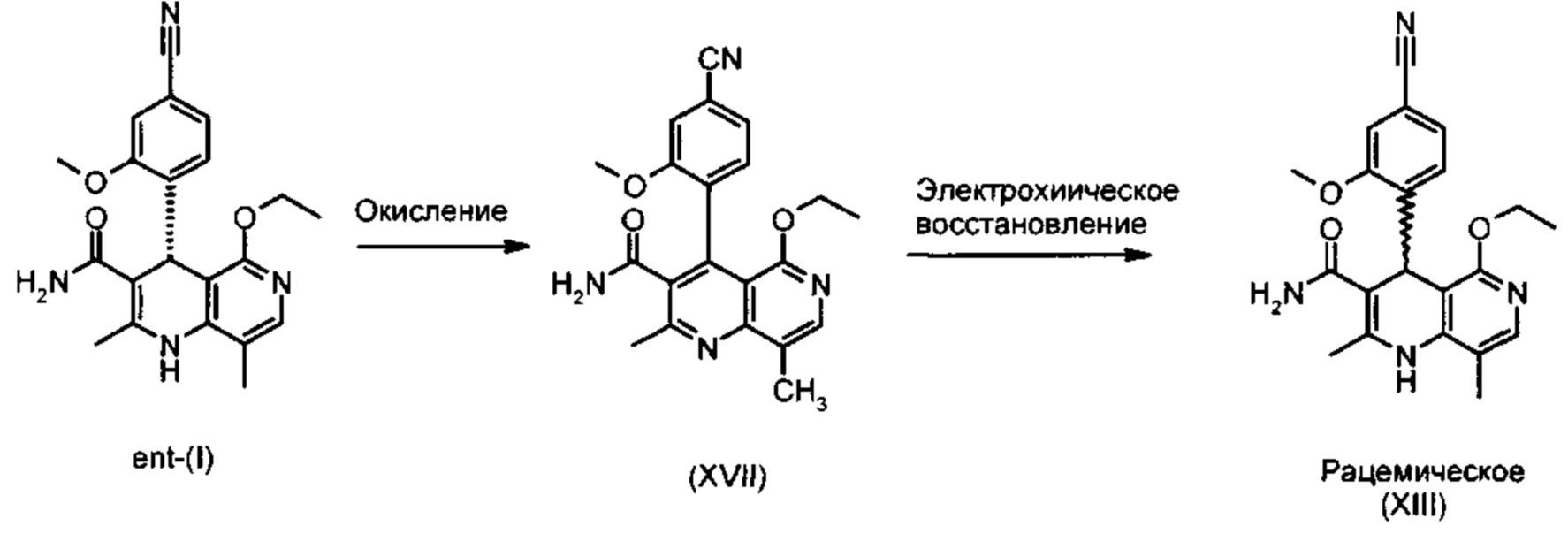
Следующее далее описание разъясняет новый изобретательский способ:
На первой стадии проводят окисление (ароматизацию) соединения формулы ent-(I);
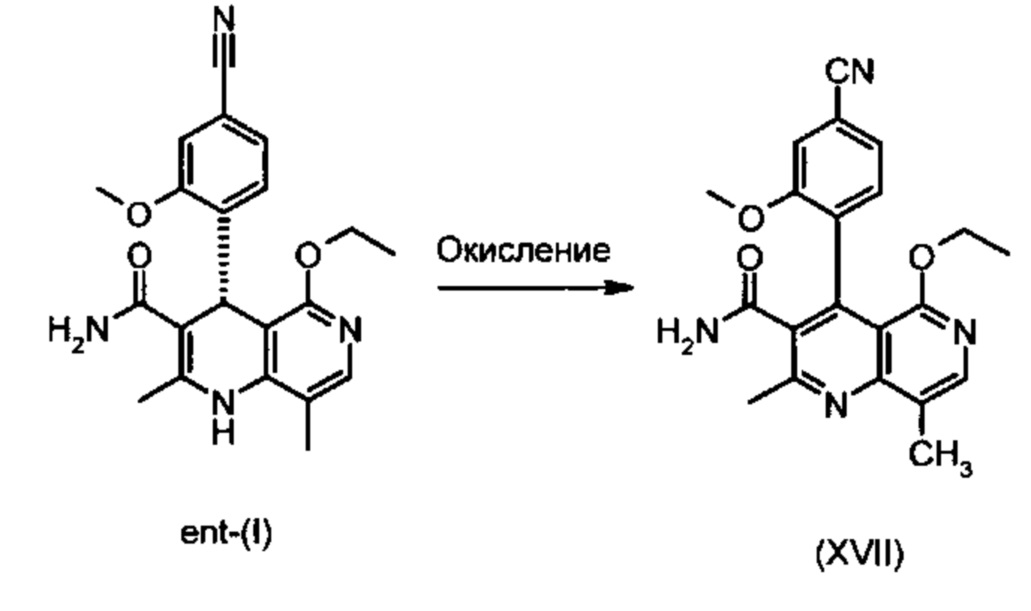
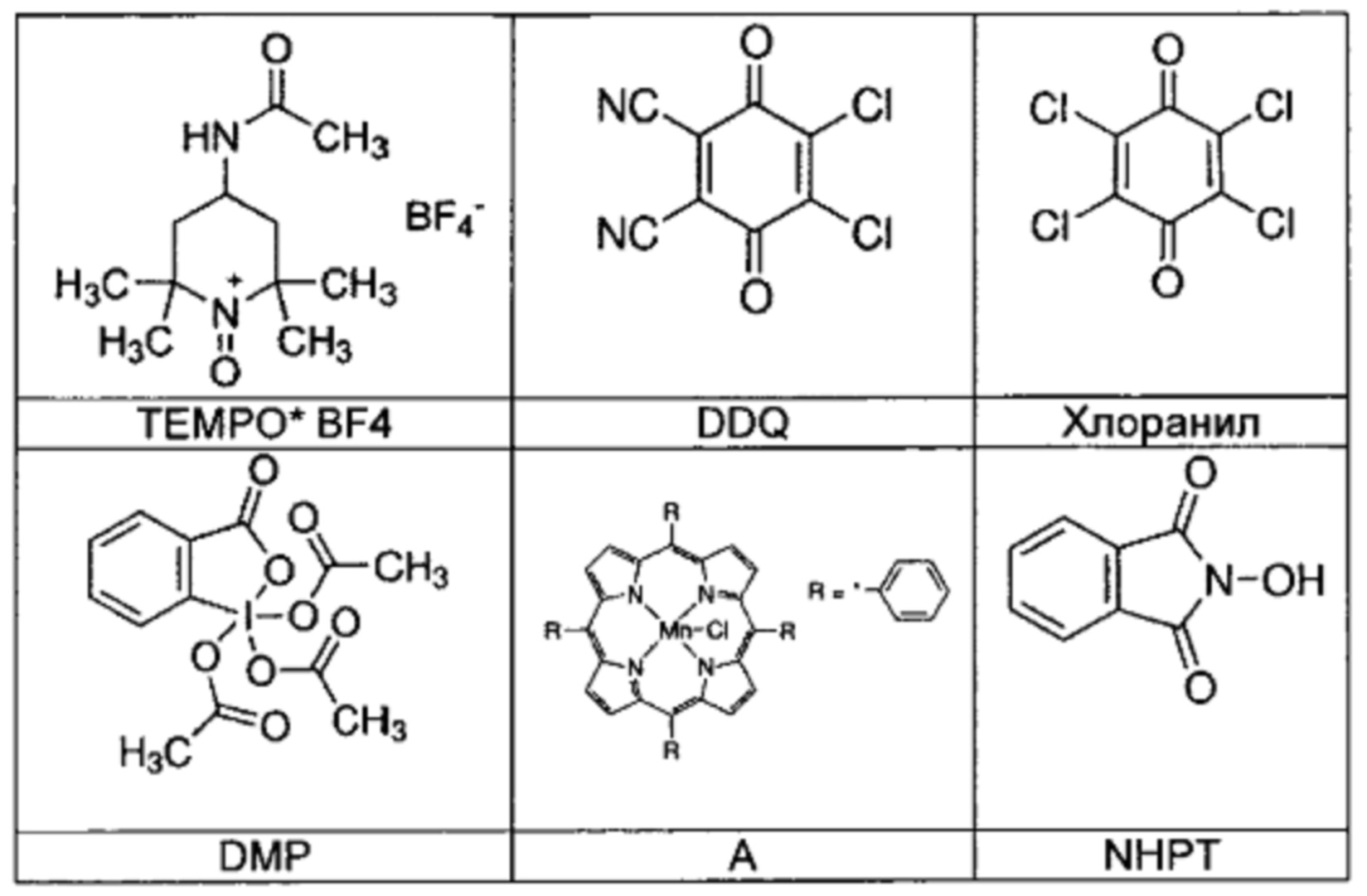
В качестве окислительных средств могут быть использованы известные специалисту в данной области окислительные средства для ароматизации пиперидинов и дигидропиридинов, которые например описаны в книге: Pyridines: From Lab to Production; под ред. Eric F.V. Scriven, издательство Elsevier 2013, часть 8, страницы 116-144. В качестве примера могут быть названы DDQ в дихлорметане, хлоранил в дихлорметане, двуокись марганца в дихлорметане, перманганат калия в ацетоне, ацетат марганца(III) в ледяной уксусной кислоте, ацетат аммонийцерия в ацетонитриле, хлорхромат пиридиния в дихлорметане, концентрированная азотная кислота в дихлорметане, йод в метаноле. Особо предпочтительным является DDQ или концентрированная азотная кислота в дихлорметане. Выходы в основном очень высокие, как правило >86% от теории.
В более ранних работах (A. Straub, Tetrahedron Asymmetry 12 (2001) 341-345) были свидетельства того, что окисленные дигидропиридины, в частности пиридиларилы показывают затрудненное вращение. Вращательный барьер настолько высок, что можно разделить антиподы при комнатной температуре (осевая хиральность → атропоизомерия).
Поэтому, исходя из рацематов, были разработаны препаративные методы хиральной хроматографии, чтобы разделить их на антиподы. Это неожиданным образом удалось и в данном случае
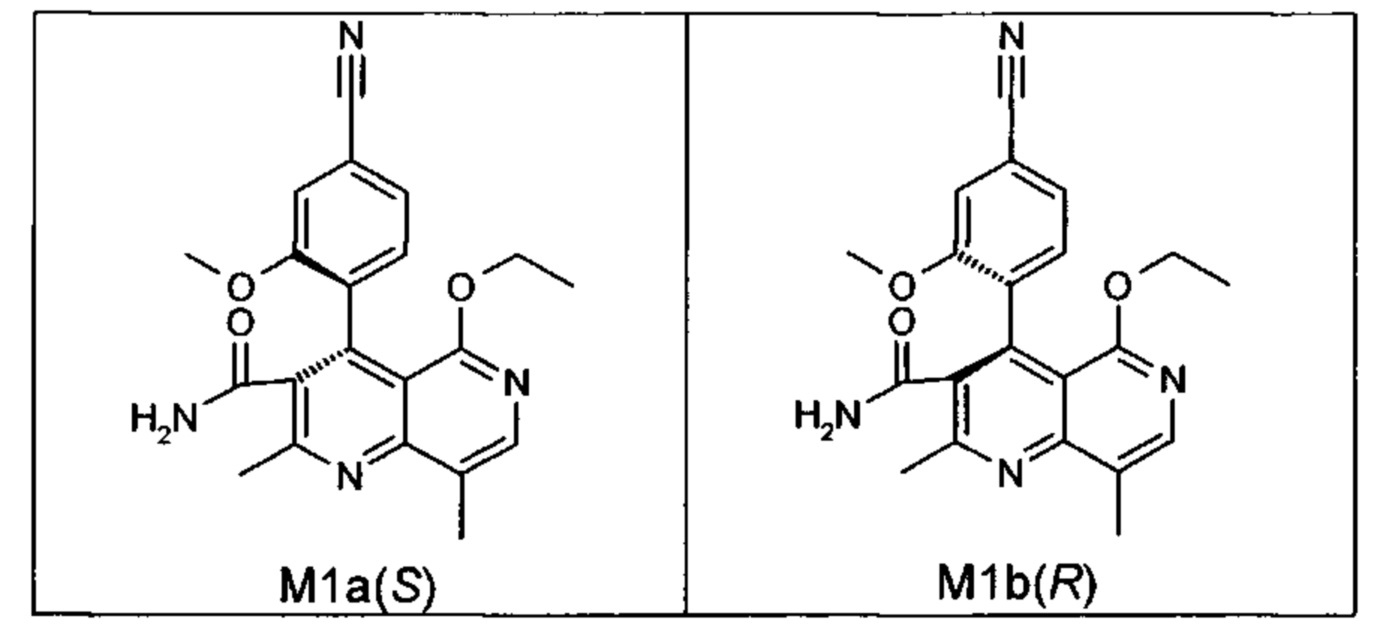
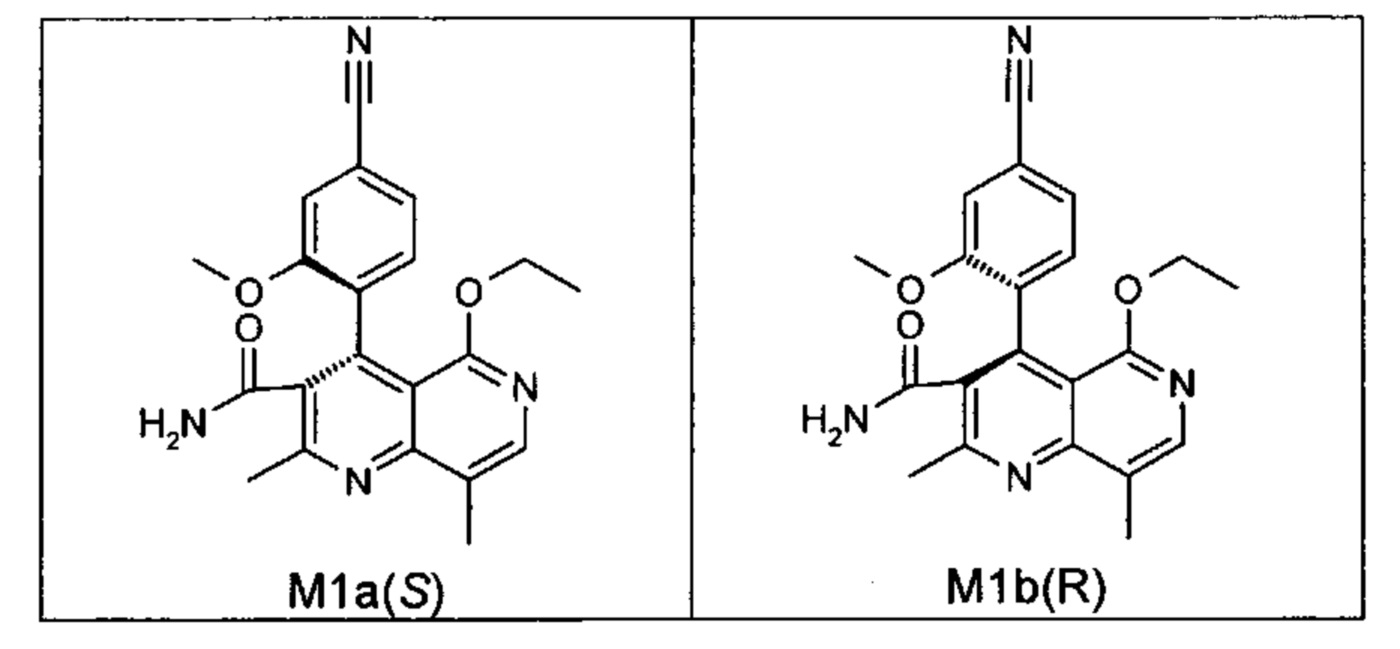
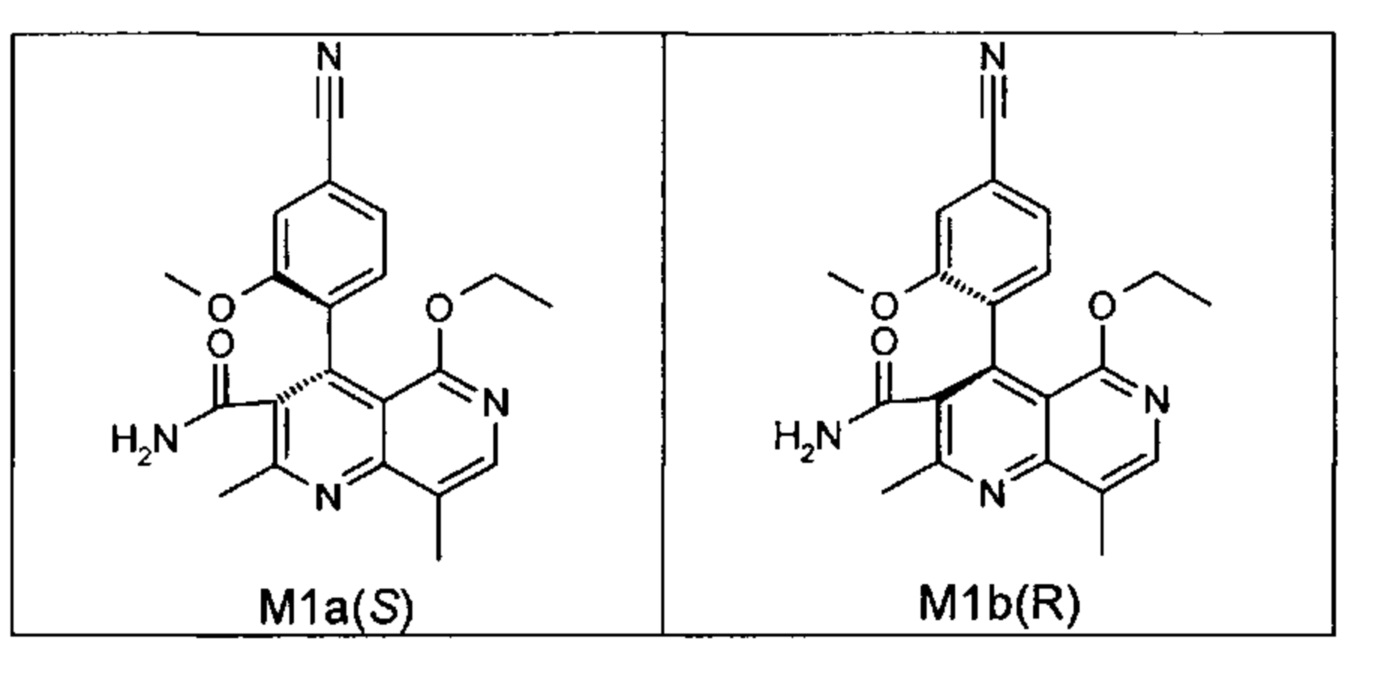
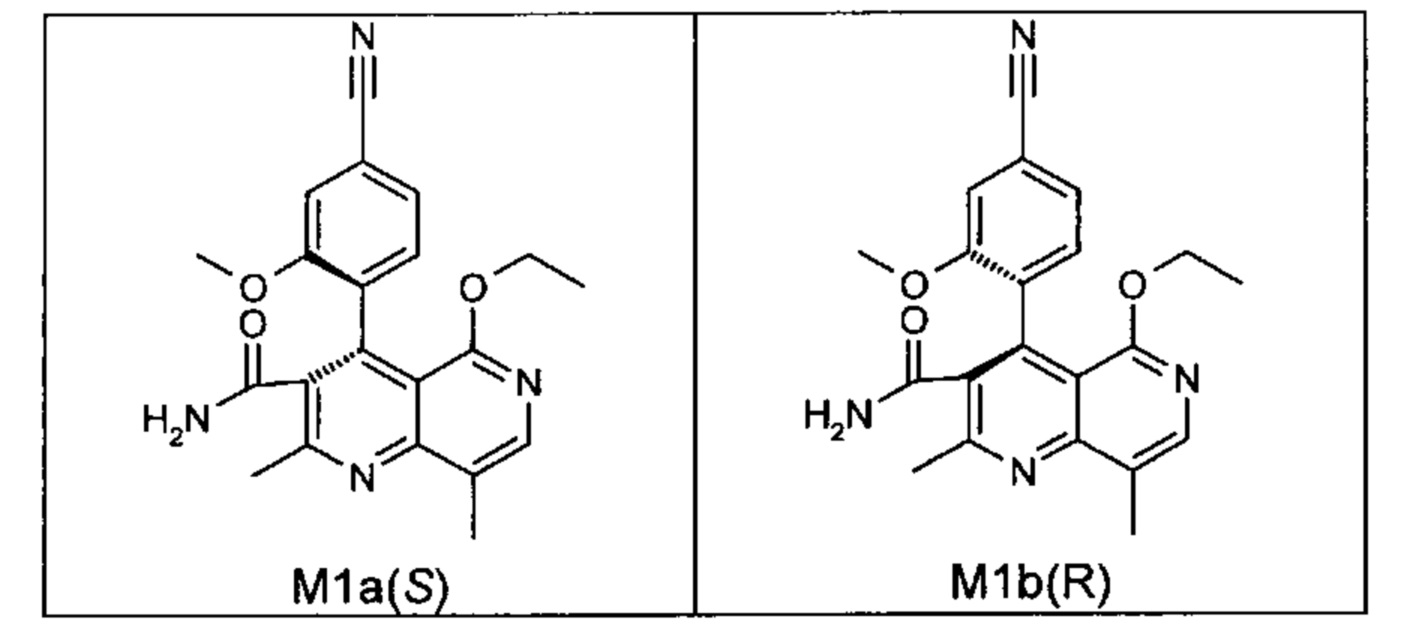
Оба полученных атропоизомера также являются основными метаболитами (соединения формул M1a(S) и M1b(R)), которые наблюдаются in vivo после введения соединения формулы (I). Их абсолютная конфигурация может быть определена посредством рентгеноструктурного анализа кристаллов (см. экспериментальную часть)
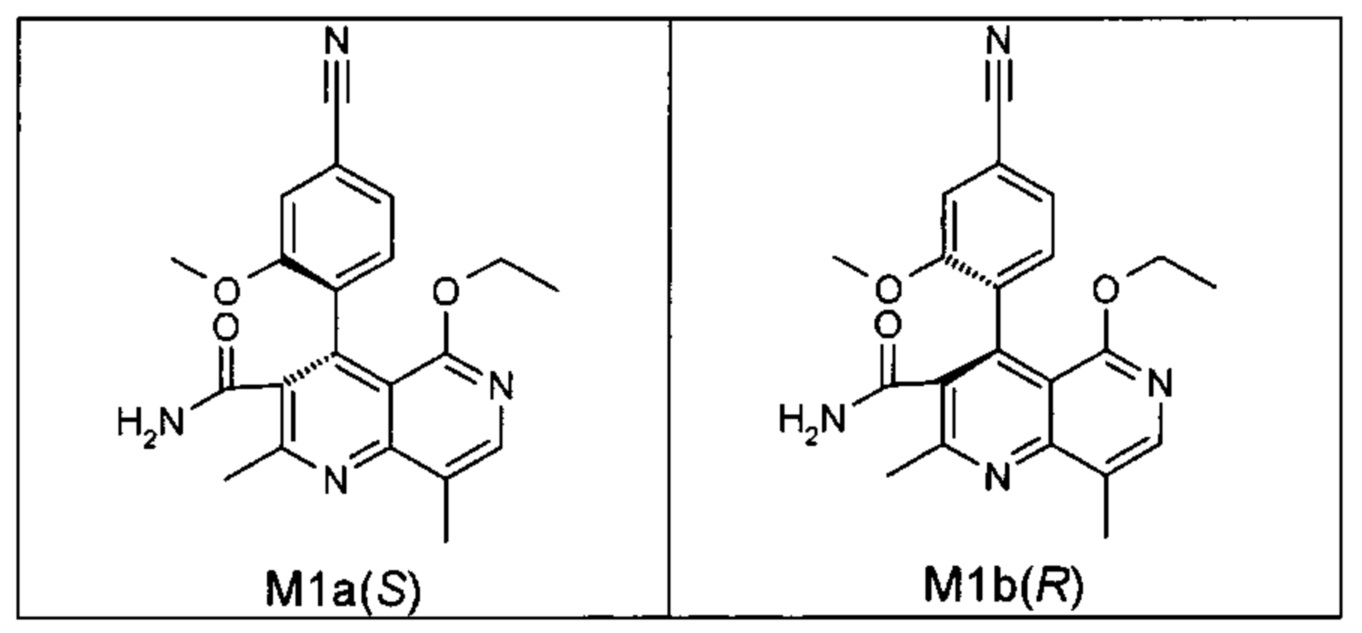
Удивительным является то, что оптически активное титульное соединение формулы (I) в S-конфигурации у грызунов и млекопитающих, а также людей (собака, крыса, мышь, человек) в основном метаболизируется до M1a(S). Если применяют R-энантиомер формулы ent-(I),
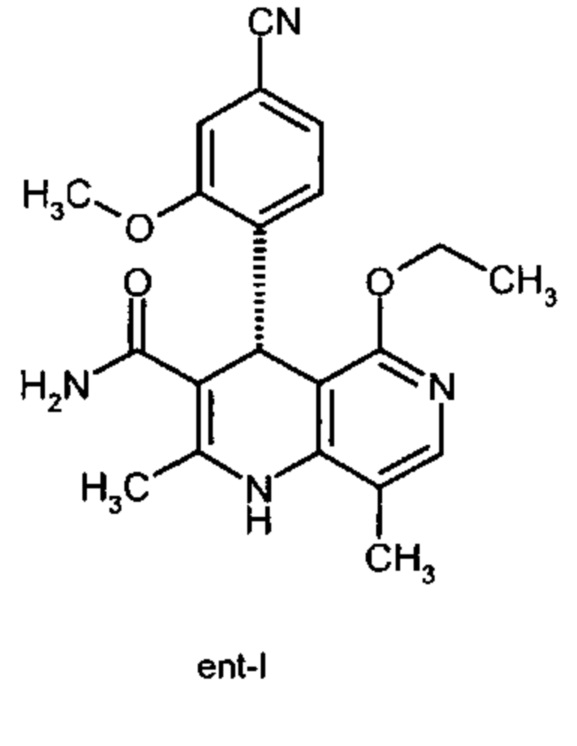
то в основном образуется метаболит формулы M1b(R).
Если, например, окисление проводят с помощью химических окислительных средств, преимущественно получают метаболит других серий, из титульного соединения формулы (I) (S-конфигурация) образуется преимущественно соединение формулы M1b(R), а из соединения формулы ent-(I) (R-конфигурация) образуется преимущественно соединение формулы M1a(S).
Если оптически активное соединение формулы (I) подвергают превращению с различными известными специалисту в данной области окислительными средствами, то получают следующие результаты:
В случае если растворитель не указан, то в качестве растворителя использовали стандартный дихлорметан. Соотношение определяли методом хиральной ВЭЖХ, M1a(S) / M1b(R) нормировали к 100%. Выход определяли по конверсии с помощью ВЭЖХ (ахиральный метод)
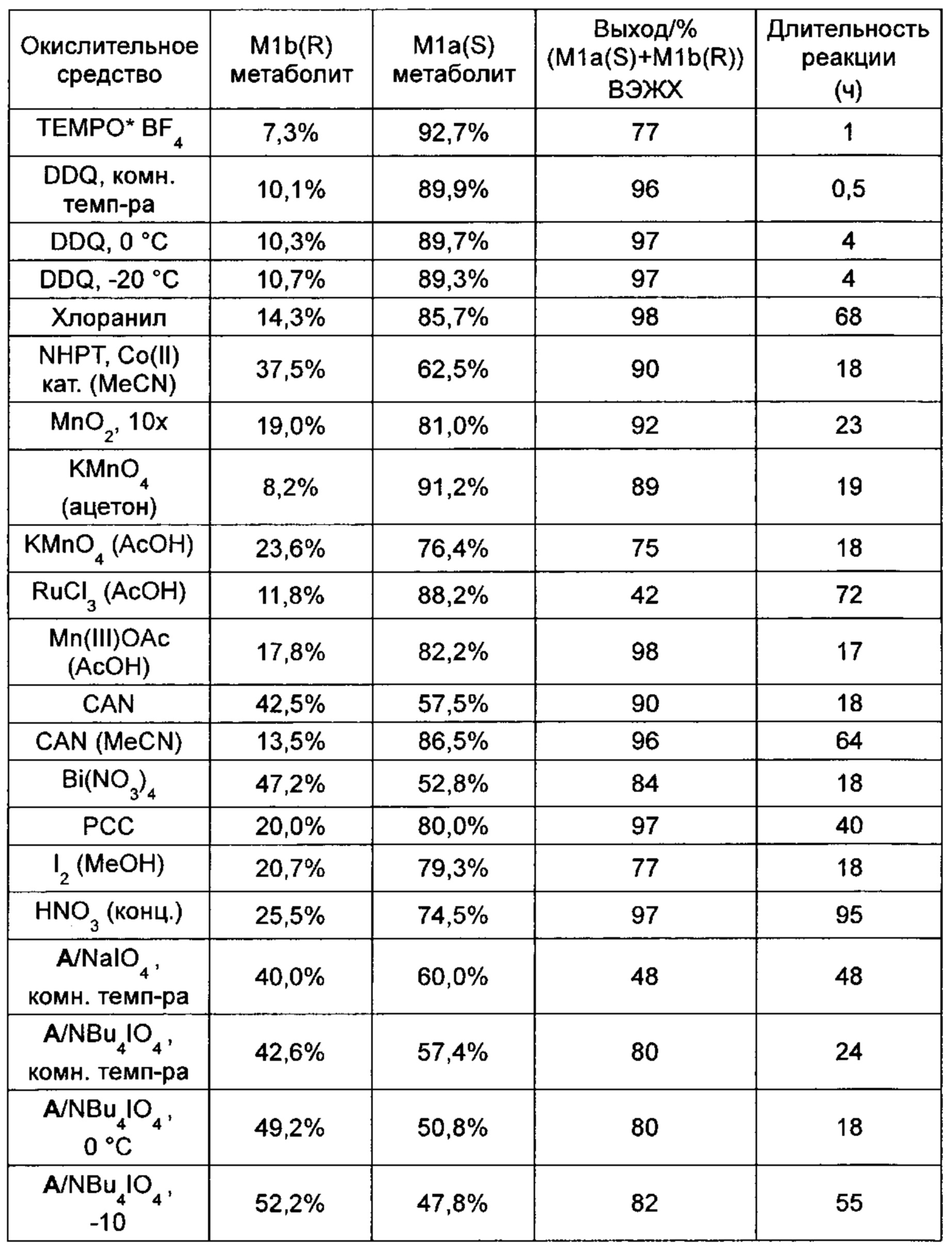
Использованные реагенты приводятся в таблице ниже:
Могло быть также продемонстрировано, что отдельные антиподы могут быть термически рацемизированы, для этого их нагревают в растворителе с высокой температурой кипения >70°С, однако также можно работать в низкокипящем растворителе, но тогда все необходимо выполнять под давлением. Подходящими растворителями являются все распространенные растворители, такие как этанол, метанол, пропанол, изопропанол, ТГФ, диоксан, метиленхлорид (под давлением), ДМФА, ДМА, N-метил-2-пирролидон, этилацетат, 2-Метил-ТГФ. Предпочтительно работают в 1-бутаноле и этаноле.
В качестве примера необходимо упомянуть термическую рацемизацию в 1-бутаноле (примерно 20 кратное растворение). Для этой цели энантиомерный избыток в ее % соединения формулы M1a(S) в бутаноле определяли при 3 разных температурах (см. фиг. 1). Можно видеть, что полная рацемизация происходит в течение 1 часа при 105°С. Скорость рацемизации можно ускорить с помощью добавления кислоты (каталитические количества метансульфоновой кислоты в 1-бутаноле) (см. фиг. 2).
В результате добавления каталитического количества кислоты термическая рацемизация также может быть проведена при более низких температурах. Подходящими кислотами являются метансульфоновая кислота, серная кислота, соляная кислота, п-толуолсульфоновая кислота, а также большинство ароматических сульфоновых кислот. Предпочтительно однако используют сульфоновые кислоты, особо предпочтительно метансульфоновую кислоту.
Основным недостатком приведенных способов окисления является то, что необходимо использовать стехиометрические или соответственно превышающие стехиометрические количества окислительного средства, и таким образом образуется много отходов. Поэтому было желание удерживать количество отходов окислительного средства на как можно более низком уровне. Это удается при помощи настоящего изобретения. Использование каталитических количеств DDQ заметно снижает количество отходов до минимума, что является значительным преимуществом нового способа согласно изобретения.
Лучшей альтернативой химическому окислению было бы электрохимическое окисление с заменой химических окислительных средств электронами. Посредством использования электрохимии, можно точно установить окислительный потенциал и отказаться от применения химических реагентов. Arguello и др. [Electrochemica Acta 49 (2004), стр. 4849-4856] и Lopez-Alarcon и др. [Electrochimica Acta 48 (2003), стр. 2505-2516] описывают вольтамперометрическое окисление 1,4-дигидропиридинов, полученных по методу Ганча, в протонной и апротонной средах. Однако они сообщили о высоких потенциалах окисления, изменяющихся от +915 мВ до +1093 мВ относительно эталонного электрода Ag/AgCl в апротонной среде. При данном высоком окислительном потенциале, как известно, обнаруживаются окисления функциональных групп, например, аминогрупп или фенольных групп, [а) Handbook of electrochemistry, Elsevier, издатель C.G. Zoski, 2007; 6) Fundamentals and Applications of organic Electrochemistry; Fuchigami и др., 2015 John Wiley & Sons, Ltd.; c) David и др., Tetrahedron 51 (1995) 3181-3196]. Таким образом, прямое электрохимическое окисление дигидропиридиновых производных имеет ограниченную применимость.
В качестве альтернативы прямому электрохимическому окислению Francke и Little описывают использование непрямых электрохимических реакций с применением различных типов медиаторов в общем случае [Chem. Soc. Rev. 43 (8) 2014, стр. 2492-2521]. Однако не приводятся примеры, в которых дигидропиридины могут быть успешно окислены до их пиридиновых аналогов. Описано использование DDQ в непрямом электросинтезе, но, согласно комментарию авторов, оно еще не полностью изучено. Примеры ограничиваются бензильным окислением, т.е. функционализацией боковой цепи в водной уксусной кислоте. В случае применения сухих апротонных растворителей реакция не была успешной.
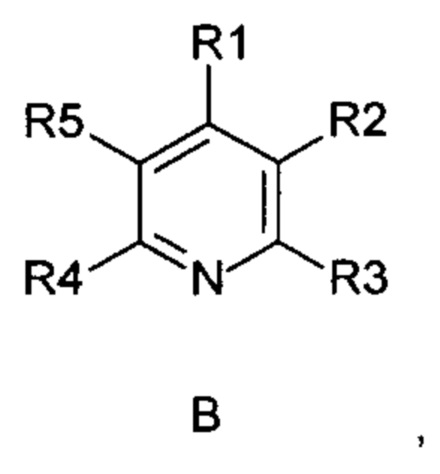
Задача изобретения состояла в разработке способа окисления дигидропиридинов (А) в пиридиновые аналоги (В),
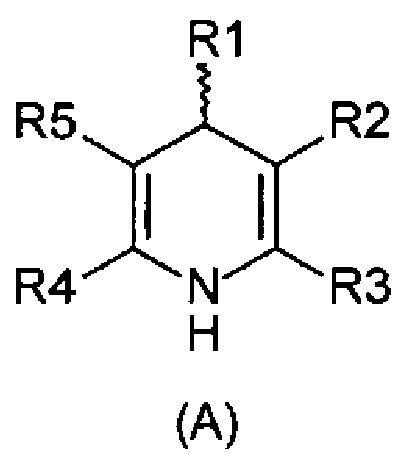
 , причем
, причем
R1-R5 независимо друг от друга означают водород, фтор, хлор, бром, йод, карбоксил, сложный карбоксиловый эфир, гидроксил, гидроксиэфир, циано, нитро, замещенный и незамещенный амид, алкил с 1-6 атомами углерода, галогеналкил с 1-6 атомами углерода, формил, замещенный и незамещенный фенил, замещенный и незамещенный бензил, замещенный и незамещенный нафтил, замещенный и незамещенный 5- или 6-членный гетероцикл, имеющий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, S, О, бензоконденсированный 5- или 6-членный гетероцикл,
отличающийся тем, что
i) используют субстехиометрические окислительные реагенты и способ
ii) является допустимым для боковых цепей и ряда заместителей в мягких условиях.
Для решения этой задачи неожиданным образом было обнаружено, что дигидропиридиновые производные могут быть успешно окислены в их пиридиновые аналоги с высокими выходами с использованием непрямого электрохимического окисления с субстехиометрическими количествами медиаторов.
Оптимальными условиями реакции для способа согласно изобретению являются температура 1-100°С, предпочтительно 10-50°С, более предпочтительно 20-30°С, при нормальном давлении и потенциалах окисления от -0,1 В до +0,6 В относительно Ag/Ag+-эталонного электрода (10 ммоль/л), предпочтительно от 0,0 до +0,5 В, и особенно предпочтительно от 0,1 до 0,4 В, относительно Ag/Ag+-эталонного электрода (10 ммоль/л) (измерено в апротонных органических растворителях).
В очень мягких условиях, т.е. комнатная температура (25°С), нормальное давление и малые потенциалы окисления (+0,4 В относительно Ag/Ag+ 10 ммоль/л) по сравнению с прямым электрохимическим окислением (>+1 V относительно Ag/Ag+ 10 ммоль/л), достигали высоких выходов пиридиновых производных. Не было признаков окисления боковой цепи, и реакцию можно было также проводить в апротонных растворителях. Насколько нам известно, об этом ранее никогда не сообщалось в литературе. Наиболее близким примером из литературы является бензильное окисление. Оно было успешным в водной уксусной кислоте, но не протекало при использовали сухих апротонных растворителей (таких как применялись нами). [Chem. Soc. Rev. 43 (8) 2014, стр. 2492-2521].
Количество медиатора, например DDQ, можно было снизить до менее чем 10% мол. (в идеале около 2% при сохранении выхода продукта >95%), а потенциала окисления только 0,3-0,4 В относительно Ag/Ag+-эталонного электрода было достаточно для высокой конверсии, высокого выхода, а также для высокой электрической эффективности.
Подходящими медиаторами, которыми однако изобретение не должно быть ограничено, являются: триарамины (тип Ar3N), TEMPO и другие N-оксильные радикалы, соли галогенидов (типа НХ с X=Cl, Br, I), соли металлов (Cr(VI) / Cr(III), Fe(III) / Fe(II), V(IV) / V(III), Ce(IV) /Се (III), Co(III) / Co(II), Ru(VIII) / Ru(IV), Os(VIII) / Os(VI), Mn(III) / Mn(II)), йодбензол и производные йодбензола, нитратные соли и триарилимидазол, процитированные в Chem. Soc. Rev. 43 (8) 2014, стр. 2492-2521.
Для электроорганического синтеза используют известные специалисту в данной области электролизные аппараты, так называемые «трехэлектродные системы» [Handbook of Electrochemistry; издатель C.G. Zoski; 2007 Elsevier B.V. & Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices, первое издание, Т. Fuchigami, M. Atobe и S. Inagi; 2015 John Wiley & Sons, Ltd]. При этом для трех электродов используют названия: рабочий электрод, контрэлектрод и эталонный электрод. Существует множество эталонных электродов, причем для неводных электролитов, т.е. органических растворителей, предпочтительным является эталонный электрод серебро/катион серебра (Ag/Ag+) из-за его стабильности и высокой воспроизводимости измерений. При этом серебряную проволоку погружают в 10 мМ или 0,1 М раствор AgNO3. В качестве растворителя можно использовать ацетонитрил, диметилформамид или диметилсульфоксид. В качестве токопроводящей соли стандартно используется перхлорат тетрабутиламмония (Bu4NClO4). В качестве альтернативы, однако, могут быть использованы другие токопроводящие соли: Et4NBF4, Bu4NBF4, Bu4NPF6, Bu4NX(c X=I, Br) или перхлораты (NaClO4, LiClO4, Et4NClO4).
Пространственное разделение между рабочим электродом и контрэлектродом или соответственно обеими так называемыми полуячейками является в большинстве случаев преимущественным, чтобы предотвратить попадание как исходных материалов, так и целевого продукта на контрэлектрод и инициирование там нежелательных побочных реакций, приводящих к снижению выхода.
Для пространственного разделения рабочего и контрэлектрода используют разделители, которые посредством ограниченной пористости и/или также в результате их химической структуры или функциональности предотвращают любой обмен между обеими полуячейками. Известные разделители представляют собой спеченные стеклянные фритты, фильтрующие мембраны из ПТФЭ, катионообменные мембраны, поливинилиденфторидные или полипропиленовые фильтрующие мембраны, а также неперечисленные здесь материалы, которые являются устойчивыми к органическим растворителям и чьи размеры пор достаточно малы, чтобы ограничить или полностью предотвратить прохождение исходного материала и продукта к другой полуячейке.
Для электрохимического окисления дигидропиридина (А) рабочий электрод подсоединяют в качестве анода, а контрэлектрод - в качестве катода. На катоде ожидается и наблюдается выделение водорода.
Известными электродными материалами являются платина, палладий, золото, графит, стеклоуглерод, алмаз, легированный бором, цинк, медь, никель, олово, самарий, сталь, ртуть, свинец или сплавы, состоящие из меди, олова и свинца, так называемые свинцовые бронзы. Кроме того, специалисту в данной области знакомы дополнительные металлические и металлоксидные электроды, которые также легированы или используются в сплавах: Ru/RuO2, Ti/TiO2, RuO2/TiO2, Ir/IrO2, Pt/Ti, платина/иридий.
В частности, специалисту в данной области техники известно катодное образование газообразного водорода в качестве конкурирующей реакции. Поэтому предпочтительными являются катодные материалы, которые имеют высокое перенапряжение относительно образования водорода. Таким образом, перенапряжение для образования Н2 возрастает в следующем порядке: Pd<Au<Pt<Ni<Cu<Sn<Pb<Zn<Hg.
Типичными и также описанными для электро-органического синтеза растворителями являются ацетонитрил, этанол, тетрагидрофуран (ТГФ), ацетон, N,N-диметилформамид (ДМФА), метанол, дихлорметан, диметилсульфоксид (ДМСО), гексаметилфосфорамиды ([(CH3)2N]3PO; CAS: 680-31-9). Растворителями, известными в основном специалисту в данной области, являются также N-метил-2-пирролидон, N,N-диметилацетамид, пропанол, изопропанол, метиленхлорид, этилацетат.
Токопроводящими солями, которые добавляют к органическим растворителям для повышения проводимости, являются: Et4NBF4, Bu4NBF4, Bu4NPF6, Bu4NX(c X=I, Br) или перхлораты (NaClO4, LiClO4, Et4NClO4, Bu4NClO4).
Подробно описанные и широко распространенные «трехэлектродные системы» как правило используются в известных специалистам в данной области техники ячейках в виде лабораторных стаканов, Н-ячейках или других сосудах. С помощью магнитных мешалок реакционные смеси могут непрерывно перемешиваться. В большинстве случаев речь идет о периодических экспериментах, в которых смесь растворитель/токопроводящая соль вводят в обе полуячейки. Исходный продукт помещают только в ту полуячейку, в которой он также должен быть подвергнут электрохимическому превращению.
При постоянной циркуляции реакционной смеси с помощью циркуляционных насосов такие ячейки также могут работать в потоке. Кроме того, в литературе описаны очень специфические геометрии для проточных ячеек [Handbook of Electrochemistry; издатель C.G. Zoski; 2007 Elsevier B.V. & Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices, первое издание, Т. Fuchigami, M. Atobe и S. Inagi; 2015 John Wiley & Sons, Ltd]. См. фигуру 3. Особенно предпочтительными являются проточные ячейки в конструкции фильтровального пресса с точки зрения масштабирования синтеза. Начиная с очень малых площадей поперечного сечения (10 см2), масштабирование может быть реализовано с одной стороны за счет увеличения площади поперечного сечения до 0,4 м2 на модуль (доступны в продаже от фирмы Electrocell, модель MFC до 0,001 м2, модель МРС от 0,01 до 0,2 м2, модель ESC от 0,04 до 1,04 м2, модель ЕРС от 0,4 до 16,0 м2), а с другой стороны за счет мультиплицирования, то есть соединения нескольких идентичных модулей в пучок. Риск такого процесса масштабирования является предсказуемым, поскольку другие геометрические размеры, например, расстояние между электродами, электродный материал (для анода и катода), а также рабочие параметры (в частности, плотность тока) не нуждаются в изменении.
С помощью регулируемой скорости потока можно контролировать время пребывания в ячейке. Типичные времена пребывания находятся в диапазоне 0,1-100 с за один проход («Single pass»). Для способа согласно изобретению в случае использования в электрохимическом восстановлении проточных ячеек время пребывания предпочтительно составляет от 0,5 до 50 с, и особо предпочтительным является время пребывания за один проход от 1 до 10 с.
Выбор плотности тока зависит как от времени пребывания, так и от кинетики целевой реакции, а также от нежелательных побочных реакций. Слишком высокая плотность тока при одновременном длительном времени пребывания и газообразовании (например, Н2) приводила бы к экранированию поверхности электрода путем образования газовой подушки в ячейке. Для электрохимического окисления (XIII) в (XVII) с DDQ в качестве медиатора допустимыми являются плотности тока 1-100 мА/см2. Однако для достижения максимальной селективности при удовлетворительной объемной производительности предпочтительными являются плотности тока в диапазоне 5-50 мА/см2 и особо предпочтительными - в диапазоне 10-30 мА/см2. В принципе возможно использование различных растворителей из приведенного выше списка. Предпочтительными растворителями являются ДМФА, ДМА, N-метил-2-пирролидон, ацетонитрил и их смеси.
Для осуществления способа согласно изобретению в случае соединения ent-(I) оказалась пригодной следующая процедура:
Окисление соединения ent-(I) в соответствующее производное (XVII) с DDQ в качестве медиатора происходит согласно следующей схеме на фигуре 4, она выглядит как ent-(I) → (XVII) + Н2 при использовании напряжения и электрического тока (см. фргуру 4).
Для исследования и лучшего понимания системы проводили циклическую вольтамперометрию в неразделенной ячейке в виде лабораторного стакана диаметром 5 см с сетчатым Pt-электродом (рабочий электрод) на внешней стороне и проволочным Pt-электродом (контрэлектрод) по середине. Рядом с рабочим электродом помещали эталонный Ag/Ag+-электрод (10 ммоль/л в ацетонитриле). Ячейку заполняют 100 мл ацетонитрила, в котором растворяют 2,17 г (10 ммоль) тетрафторбората тетраэтиламмония (Et4NBF4) вместе с 22,7 мг (0,1 ммоль) DDQ и 378,4 мг (1 ммоль) соединения формулы ent-(I). Для циклической вольтамперометрии без соединения формулы ent-(I) или без DDQ соответствующее количество не добавляли. Циклическую вольтамперометрию регистрировали с использованием потенциостата модели Gamry Interface 1000 со скоростью развертки потенциала 250 мВ/с и 100 мВ/с в течение 10 циклов между -0,5 и +1 В по сравнению с эталонным электродом. После исключения первого и последнего циклов результат был усреднен. Циклическая вольтамперография известна специалисту в данной области как возможность для исследования электрохимических реакций на поверхности электрода.
Результаты цикловольтамперометрического исследования представлены на рисунке 5. Отчетливо видно, что в случае DDQ без субстрата ent-(I) - пунктирная линия - хорошо различимы 2 пика. Пик восстановления (отрицательный) при около +0,1 В относительно Ag/Ag+, связан с реакцией DDQ→H2DDQ, и пик окисления (положительный) при около +0,3 В относительно Ag/Ag+, связан с реакцией H2DDQ→DDQ. Кроме того, циклическая вольтамперограмма полностью симметрична, что означает, что реакции полностью обратимы.
При рассмотрении циклической вольтамперограммы субстрата ent-(I) без DDQ (штриховая линия), то есть в режиме прямого окисления, можно увидеть, что ent-(I) может быть окислен только при превышении 0,6 В относительно Ag/Ag+, а для получения приемлемой конверсии требуется по меньшей мере 1 В (см. фигуру 5). Спустя 1 час электролиза при +1,0 В относительно Ag/Ag+ наблюдалось обесцвечивание раствора и наличие нескольких побочных компонентов при ВЭЖХ. Точная идентификация и количественная оценка была невозможна. Этого следовало ожидать, потому что из литературы [a] Handbook of electrochemistry, Elsevier, издатель C.G. Zoski, 2007; б) Fundamentals and Applications of organic Electrochemistry; Fuchigami и др., 2015 John Wiley & Sons, Ltd.] известно, что амины и амиды (которые например присутствуют в молекуле) могут быть окислены между +0,5 и +1,0 В относительно насыщенного каломельного электрода (GKE) (Handbook of Electrochemistry, стр. 819).
При сравнении с электролизом в присутствии медиатора, то есть субстрат +10 мол. % DDQ (сплошная линия), можно увидеть образование очень эффективного комплекса переноса заряда, причем субстрат может быть окислен при том же потенциале DDQ (около 0,3 В) и способ является очень эффективным и, таким образом, показывает самый высокий ток. Кроме того, можно видеть, что обратная реакция (пик восстановления) полностью исчезла, поскольку DDQ теперь может реагировать только с субстратом и больше не доступен для электрода.
Следует отметить, что после протекания реакции системы с потенциалом +0,4 В относительно Ag/Ag+ в течение 2 часов была достигнута конверсия субстрата около 98% и не наблюдалось никаких побочных компонентов, а только желаемый компонент. См. также примеры. Это можно сравнить с прямым преобразованием, в котором была достигнута исключительно низкая селективность.
Таким образом, можно определить идеальное рабочее окно (потенциал между 0,3 и 0,5 В), в котором регенерация DDQ (то есть окисление H2DDQ→DDQ) максимальна (>0,3 В), а прямая неселективная реакция субстрата с электродом (<0,5 V) полностью исключена. Указанное идеальное рабочее окно обеспечивает максимальные выход и селективность.
Следует также отметить, что в таких условиях (т.е. +0,4 В относительно Ag/Ag+ в качестве эталона) может быть достигнут высокий ток 65 мА и, следовательно, высокая скорость реакции 1,2 ммоль/ч. Для поддержания той же скорости в прямой электрохимической системе без медиаторов требуется напряжение +1,0 В, которое может нанести вред молекуле.
По аналогии с циклической вольтамперометрией проводили производственные испытания в такой же серийной ячейке с темже раствором и конфигурацией. В соответствии с этим раствор подвергали электролизу при постоянном потенциале +0,4 В в течение 2 часов и каждые 15 минут отбирали пробу и анализировали с помощью ВЭЖХ. По истечении 2 часов конверсия неожиданным образом достигла 98%, выход продукта составил >97,5% (селективность >99%), и еще более удивительно, что пропускали заряд только 2,1 Ф. Учитывая, что требуемое стехиометрическое (минимальное) количество электроэнергии составляет 2 F, электрическая эффективность составляла более 95%. Если низкая электрическая эффективность не указывает на неселективную реакцию, то селективные реакции являются необходимым условием для высокой электрической эффективности. Вместе с анализом ВЭЖХ становится ясно, что окисление в присутствии медиаторов соединения (I) в (XVII), или более обобщенно (А) в (В), является гораздо более благоприятным процессом с точки зрения селективности и выхода, чем химическое и прямое электрохимическое окисление (см. фигуру 6).
Соединение (XIII) существует в двух энантиомерных формах: (I) и ent-(I). Продукт (XVII) существует в двух формах осевой хиральности, известных как M1a(S) и M1b(R). Неожиданно было обнаружено, что в результате преобразования посредством электрохимического восстановления в присутствии медиаторов, согласно выше приведенному описанию, соединение формулы (I) превращают предпочтительно в M1b(R), причем соотношение M1a(S): M1b(R) составляет 13:87 (пример 27), и ent-(I) превращают предпочтительно в M1a(S), причем соотношение M1a(S): M1b(R) составляет 90:10 (пример 26). Эти результаты сопоставимы с результатами, полученными химическим окислением стехиометрическими количествами DDQ (примеры 11а и 11b). Неожиданным образом соединение формулы (I) в клетках животных (включая человека) предпочтительно метаболизируется в соединение формулы M1a(S), тогда как соединение формулы ent-(I) напротив - в соединение формулы M1b(R).
При использовании рацемической смеси соединения формулы (I), то есть соединения формулы (XIII), электрохимическое окисление приводит, как и ожидалось, к рацемической смеси (XVII) с соотношением M1a(S): M1b(R) 50:50 (пример 28).
Учитывая наблюдаемую высокую селективность и электрическую эффективность, описанная выше процедура также может быть выполнена без особых трудностей в проточных ячейках описанного выше типа (т.е. фирмы Electrocell). Это позволяет повысить объемную производительность и увеличить производство до промышленных масштабов.
Выделение (XVII): После проведения электрохимической реакции (исходного материала (I), как правило, <1%), реакционный раствор обрабатывают. Реакция протекает с высокими выходами (>98%) и с неожиданной чистотой, практически без примесей. Оказалось выгодным, что растворитель сначала в значительной степени отгоняют, а затем продукт переводят в осадок посредством водного осаждения (добавление воды), отфильтровывают и сушат. Полученный таким образом продукт может быть перекристаллизован из этанола или изопропанола или 1-бутанола или 2-бутанола.
На следующем этапе проводят электрохимическое восстановление пиридина формулы (XVII) до дигидропиридина:
Уровень техники для (электро)химического восстановления пиридинов
Straub и Goehrt [Alexander Straub und Axel Goehrt, Angew. Chem., 108 (1996), 2832-2834 (Название: Inversion optisch aktiver Dihydropyridine durch Oxidation und Elektroreduktion)] описывают электрохимическое восстановление на ртутных электродах пиридиновых производных, которые помимо прочего все характеризуются наличием сложноэфирной группы (-CO2Et). Также применяемые Kita и др. [Yoshio Kita, Hirofumi Maekawa, Yasuhiro Yamasaki and Ikuzo Nishiguchi, Tetrahedron Letters 40 (1999) 8587-8590 (Название: Selective and facile electroreductive synthesis of dihydro- and tetrahydropyridine dicarboxylic acid derivatives), Yoshio Kita, Hirofumi Maekawa, Yasuhiro Yamasaki und Ikuzo Nishiguchi, Tetrahedron 57 (2001) 2095-2102 (Название: Highly selective and facile synthesis of dihydro-and tetrahydropyridine dicarboxylic acid derivatives using electroreduction as a key step)] пиридины имеют даже два сложноэфирных заместителя (-CO2Me). Straub и Goehrt приводят выход 83% для очень маленькой лабораторной загрузки в 0,72 ммоль пиридинового производного.
Кита и др. описывают в качестве продуктов, как 1,2-, так и 1,4-дигидропиридин. Эксперименты на С- и Pb-электродах давали 0%-ную конверсию. На Pt-катодах достигали выходов 36%. Только при использовании хлорида аммония и температурах значительно ниже комнатной температуры (5-10°С) на Pt-катодах могут быть достигнуты выходы более 83%.
Eisner и Kuthan [Ulli Eisner и Josef Kuthan, Chem. Rev. (1972), 72, 1-42 (Название: The Chemistry of Dihydropyridines)] описывают химическое восстановление пиридинов при помощи NaBH4 или посредством каталитического гидрирования. В обоих случаях снижение выхода является результатом неселективного восстановления заместителей (например, сложноэфирной группы в спирт) или результатом восстановления нитрильной группы. Кроме того, здесь также наблюдали 1,2-дигиропиридины в качестве основных продуктов.
Таким образом, в предшествующем уровне техники не раскрывается общеприменимый способ селективного восстановления пиридиновых производных, в частности, пиридиновых производных, которые не имеют заместителей сложного метилового эфира или сложного этилового эфира. Ртутные электроды также являются непригодными для синтеза фармацевтических агентов вследствие их токсичного характера. Кроме того, следует избегать образования 1,2-дигидропиридиновых производных, как описано в уровне техники, поскольку это также приводит к снижению выхода.
Нижеследующее описание объясняет вторую стадию способа согласно изобретению, электрохимическое восстановление пиридина (XVII) до амида (XIII):
Для электроорганического синтеза используют известные специалисту в данной области электролизные аппараты, так называемые «трехэлектродные системы» [Handbook of Electrochemistry; под редакцией C.G. Zoski; 2007 Elsevier B.V. & Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices, первое издание, Т. Fuchigami, M. Atobe и S. Inagi; 2015 John Wiley & Sons, Ltd]. При этом для трех электродов используют названия: рабочий электрод, контрэлектрод и эталонный электрод. Существует множество эталонных электродов, [Handbook of Electrochemistry; под редакцией C.G. Zoski; 2007 Elsevier B.V.], причем для неводных электролитов, т.е. органических растворителей, предпочтительным является эталонный электрод серебро/катион серебра (Ag/Ag+) из-за его стабильности и высокой воспроизводимости измерений. При этом серебряную проволоку погружают в 10 мМ или 0,1 М раствор AgNO3. В качестве растворителя можно использовать ацетонитрил, диметилформамид или диметилсульфоксид. В качестве токопроводящей соли стандартно используется перхлорат тетрабутиламмония (Bu4NClO4). В качестве альтернативы, однако, могут быть использованы другие токопроводящие соли: Et4NBF4, Bu4NBF4, Bu4NPF6, Bu4NX(c X=I, Br) или перхлораты (NaClO4, LiClO4, Et4NClО4).
Пространственное разделение между рабочим электродом и контрэлектродом или соответственно обеими так называемыми полуячейками является в большинстве случаев преимущественным, чтобы предотвратить попадание как исходных материалов, так и целевого продукта на контрэлектрод и инициирование там нежелательных побочных реакций, приводящих к снижению выхода.
Для пространственного разделения рабочего и контрэлектрода используют разделители, которые посредством ограниченной пористости и/или также в результате их химической структуры или функциональности предотвращают любой обмен между обеими полуячейками. Известные разделители представляют собой спеченные стеклянные фритты, фильтрующие мембраны из ПТФЭ, катионообменные мембраны, поливинилиденфторидные или полипропиленовые фильтрующие мембраны, а также неперечисленные здесь материалы, которые являются устойчивыми к органическим растворителям и чьи размеры пор достаточно малы, чтобы ограничить или полностью предотвратить прохождение исходного материала и продукта к другой полуячейке.
Для электрохимического восстановления пиридина (XVII) рабочий электрод подсоединяют в качестве катода, а контрэлектрод - в качестве анода.
Известными электродными материалами являются платина, палладий, золото, графит, стеклоуглерод, алмаз, легированный бором, цинк, медь, никель, олово, самарий, сталь, ртуть, свинец или сплавы, состоящие из меди, олова и свинца, так называемые свинцовые бронзы. Кроме того, специалисту в данной области знакомы дополнительные металлические и металлоксидные электроды, которые также легированы или используются в сплавах: Ru/RuO2, Ti/TiO2, RuO2/TiO2, Ir/IrO2, Pt/Ti, платина/иридий.
В частности в водных электролитах специалисту в данной области техники известно катодное образование газообразного водорода в качестве конкурирующей реакции. Поэтому предпочтительными являются катодные материалы, которые имеют высокое перенапряжение относительно образования водорода. Таким образом, перенапряжение для образования Н2 возрастает в следующем порядке: Pd<Au<Pt<Ni<Cu<Sn<Pb<Zn<Hg.
В неводных электролитах электрохимическая стабильность растворителя и токопроводящей соли влияет на возникновение и масштаб побочных реакций на электродах.
Так называемое электрохимическое окно для выбранных смесей растворителей/электролитов является табличным значением [Handbook of Electrochemistry; под редакцией C.G. Zoski; 2007 Elsevier B.V. & Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices, первое издание, Т. Fuchigami, M. Atobe и S. Inagi; 2015 John Wiley & Sons, Ltd]. Например, можно назвать такие комбинации, как ацетонитрил/0,1 М Bu4NPF6, тетрагидрофуран/0,1 М Bu4NPF6, ацетонитрил/ 0,1 М Et4NBF4, ДМФА/0,1М Bu4NClO4, которые также при более отрицательных потенциалах, чем -2,0 В (относительно насыщенного каломельного электрода) по-прежнему считаются электрохимически стабильными. Использование других растворителей не ограничивают или в принципе не исключают.
Типичными и также описанными для электро-органического синтеза растворителями являются ацетонитрил, этанол, тетрагидрофуран (ТГФ), ацетон, N,N-диметилформамид (ДМФА), метанол, дихлорметан, диметилсульфоксид (ДМСО), гексаметилфосфорамиды ([(СН3)2N]3PO; CAS: 680-31-9). Растворителями, в основном известными специалисту в данной области, являются также N-метил-2-пирролидон, N,N-диметилацетамид, пропанол, изопропанол, метиленхлорид, этилацетат.
Токопроводящими солями, которые добавляют к органическим растворителям для повышения проводимости, являются: Et4NBF4, Bu4NBF4, Bu4NPF6, Bu4NX(c X=I, Br) или перхлораты (NaClO4, LiClO4, Et4NClO4, Bu4NClO4).
Подробно описанные и широко распространенные «трехэлектродные системы» как правило используются в известных специалистам в данной области техники ячейках в виде лабораторных стаканов, Н-ячейках или других сосудах. С помощью магнитных мешалок реакционные смеси могут непрерывно перемешиваться. В большинстве случаев речь идет о периодических экспериментах, в которых смесь растворитель/токопроводящая соль вводят в обе полуячейки. Исходный продукт помещают только в ту полуячейку, в которой он также должен быть подвергнут электрохимическому превращению.
При постоянной циркуляции реакционной смеси с помощью циркуляционных насосов такие ячейки также могут работать в потоке. Кроме того, в литературе описаны очень специфические геометрии для проточных ячеек [Handbook of Electrochemistry; под редакцией C.G. Zoski; 2007 Elsevier В.V.]. Особенно предпочтительными являются проточные ячейки в конструкции фильтровального пресса с точки зрения масштабирования синтеза. Начиная с очень малых площадей поперечного сечения (10 см2), масштабирование может быть реализовано с одной стороны за счет увеличения площади поперечного сечения до 0,4 м2 на модуль (в качестве модульной единицы „Electro Prod Cell”, доступные в продаже от фирмы Electrocell), а с другой стороны за счет мультиплицирования, то есть соединения нескольких идентичных модулей в пучок. Риск такого процесса масштабирования является предсказуемым, поскольку другие геометрические размеры, например, расстояние между электродами, электродный материал (для анода и катода), а также рабочие параметры (в частности, плотность тока) не нуждаются в изменении. Для способа согласно изобретению помимо простых ячеек в виде лабораторного стакана успешно использовались проточные ячейки, такие как микропотоковая ячейка (Micro-Flow Cell) с активной площадью поперечного сечения электрода 10 см2 и многоцелевая ячейка (Multipurpose Cell) с активной площадью поперечного сечения электрода 100 см2 фирмы Electrocell.
С помощью регулируемой скорости потока можно контролировать время пребывания в ячейке. Типичные времена пребывания находятся в диапазоне 0,1-100 с за один проход («Single pass»). Для способа согласно изобретению в случае использования в электрохимическом восстановлении проточных ячеек время пребывания предпочтительно составляет от 0,5 до 50 с, и особо предпочтительным является время пребывания за один проход от 1 до 10 с.
Выбор плотности тока зависит как от времени пребывания, так и от кинетики целевой реакции, и от нежелательных побочных реакций. Слишком высокая плотность тока при одновременном длительном времени пребывания и газообразовании (например, Н2) приводила бы к экранированию поверхности электрода путем образования газовой подушки в ячейке. Для электрохимического восстановления рацемата М1 допустимыми являются плотности тока 1-100 мА/см2. Однако для достижения максимальной селективности при удовлетворительной объемной производительности предпочтительными являются плотности тока в диапазоне 5-50 мА/см2 и особо предпочтительными - в диапазоне 10-30 мА/см2, поскольку было неожиданно установлено, что слишком высокие плотности тока приводят к нежелательным побочным реакциям, и, следовательно, уменьшается выход.
В принципе возможно использование различных растворителей из приведенного выше списка. Предпочтительными растворителями являются метанол, ДМФА, ДМА, N-метил-2-пирролидон, ацетонитрил и их смеси.
Неожиданно было обнаружено, что использование метанола в качестве растворителя в ячейках в виде лабораторного стакана дает выход целевого продукта более 97%. Неожиданно было обнаружено, что комбинация апротонного растворителя и протонного растворителя в проточной ячейке показала улучшенную электрическую эффективность по сравнению с чистым метанолом. В проточной ячейке можно было достигнуть конверсии и выходы выше 94%, причем обе полуячейки были отделены друг от друга катионообменной мембраной. Успешный перенос электрохимического восстановления пиридина формулы (XVII) в амид формулы (XIII) из ячейки в виде лабораторного стакана в проточную ячейку обеспечивает масштабируемость способа и, следовательно, промышленное применение.
Особое предпочтение отдается смесям, имеющим равную или большую долю апротонного растворителя и равную или соответственно меньшую долю протонного растворителя. Апротонные растворители в основном известны специалисту в данной области. Предпочтительными являются в частности ДМФА, ДМА и ацетонитрил. Протонные растворители также в основном известны специалисту в данной области. Предпочтительными протонными растворителями являются метанол, муравьиная кислота, этанол и уксусная кислота. Особо предпочтительной является комбинация из метанола и ДМФА. Доля метанола при этом должна находится между 0,1-50% масс. Предпочтительно доля метанола составляет 0,5-25% масс. и особо предпочтительно 1-10% масс. В этой смеси помимо метанола предпочтительно находится также этанол. Также особо предпочтительной является комбинация из этанола и ДМФА. Доля этанола при этом должна находится между 0,1-50% масс. Предпочтительно доля этанола составляет 0,5-25% масс и особо предпочтительно 1-10% масс. Использование этанола предотвращает реакцию переэтерификации, при которой этиловый эфир может быть переэтерифицирован в метиловый эфир.
Приведенные ниже примеры свидетельствуют о том, что, исходя из рацемического пиридина формулы (XVII), целевой продукт, а именно рацемический амид формулы (XIII), получают посредством электрохимического восстановления и, следовательно, способ согласно изобретению при помощи следующей стадии (разделение двух энантиомеров формул (I) и ent-(I), например, на установке SMB) приводит к чистому целевому соединению формулы (I). Неожиданно было также обнаружено, что при использовании чистых атропизомеров M1b(R) и M1a(S) электрохимическое восстановление в ячейках в виде лабораторного стакана на электродах из платиново-иридиевой сетки не приводит к рацемическому продукту формулы (XIII). При восстановлении соединения формулы M1b(R) желаемый энантиомер (целевой продукт) формулы (I) предпочтительно образуется в соотношении примерно 78:22 [(I): ent-(I)]. Исходя из атропизомера формулы M1a(S), в избытке получают нежелательный энантиомер формулы ent-(I): соотношение [(I): ent-(I)]=22:78. Это наблюдение открывает возможность посредством селективного окисления соединения формулы ent-(I) в соединение формулы M1b(R) дополнительно увеличить выход рециркулирования целевого продукта формулы (I) за цикл (окисление-восстановление-хиральная ВЭЖХ).
Выделение соединения формулы (XVII) После проведения электрохимической реакции (исходного продукта формулы (XVII), как правило, <1%), реакционный раствор обрабатывают. Реакция протекает с высокими выходами (>98%) и с неожиданной чистотой, практически без примесей. Оказалось выгодным, что растворитель сначала в значительной степени отгоняют, а затем продукт переводят в осадок посредством водного осаждения (добавление воды), отфильтровывают и сушат. Полученный таким образом продукт может быть перекристаллизован из этанола или ТГФ и вновь подвергнут разделению на энантиомеры при помощи SMB.
Таким образом, процесс проводят следующим образом: Сначала осуществляют окисление нежелательного энантиомера формулы ent-(I), в результате чего получают в избытке соединение формулы M1a(S), и в рамках обработки проводят термическую рацемизацию (при необходимости катализируемую кислотой). Затем выделенное рацемическое соединение формулы (XVII) подвергают электрохимическому восстановлению. После обработки рацемическое соединение формулы (XIII) выделяют и перекристаллизовывают. Полученный таким образом продукт формулы (XIII) обладает высокой степенью чистоты и подается в SMB-процесс.
Описанный здесь процесс окисления/восстановления может быть осуществлен несколько раз подряд и, таким образом, дает возможность при производстве в почти непрерывном режиме работы превратить нежелательный энантиомер формулы ent-(I) в правильный продукт формулы (I), что имеет большие преимущества с точки зрения стоимости производства. После нескольких технологических циклов достигают почти полной переработки нежелательного энантиомера формулы ent-(I).
В качестве особо важного преимущества нового способа рекуперации соединения формулы (XIII) следует учитывать его высокую химическую чистоту. Поскольку речь идет о фармацевтическом активном веществе, то все операции выполняют согласно стандарту GMP и требуют высокой чистоты промежуточных продуктов.
С новым синтезом удалось получить соединение формулы (I) очень эффективным образом. Этот способ дает значительные преимущества по сравнению с уровнем техники с точки зрения масштабируемости и технической реализации. Общий выход значительно выше по сравнению с опубликованными данными и кроме того достигается превосходная чистота активного вещества. Новый способ делает возможным воспроизводимое, экономичное получение конкретного соединения формулы (I). Посредством представленного здесь способа уже было успешно получено 200 кг материала для клинических испытаний.
Объектом настоящего изобретения является способ получения соединений формулы (В):
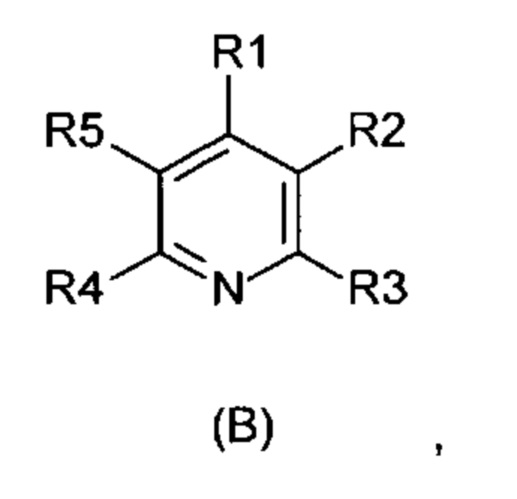
отличающийся тем, что соединения формулы (А)
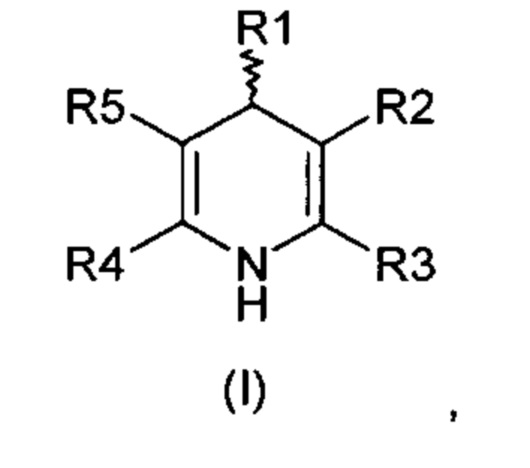
причем
R1-R5 независимо друг от друга означают водород, фтор, хлор, бром, йод, карбоксил, сложный карбоксиловый эфир, гидроксил, гидроксиэфир, циано, нитро, замещенный и незамещенный амид, алкил с 1-6 атомами углерода, галогеналкил с 1-6 атомами углерода, формил, замещенный и незамещенный фенил, замещенный и незамещенный бензил, замещенный и незамещенный нафтил, замещенный и незамещенный 5- или 6-членный гетероцикл, имеющий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, S, О, бензоконденсированный 5- или 6-членный гетероцикл,
электрохимически окисляют в результате непрямого электрохимического окисления.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при температуре 1-100°С и нормальном давлении.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при потенциале окисления от -0.1 V до +0,6 V относительно эталонного Ag/Ag+-электрода.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят с использованием DDQ в качестве медиатора.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при температуре 1-100°С и нормальном давлении при потенциале окисления от -0.1 V до +0.6 V относительно эталонного Ag/Ag+-электрода и с использованием DDQ в качестве медиатора.
Другим объектом настоящего изобретения является способ получения соединения формулы (XVII):
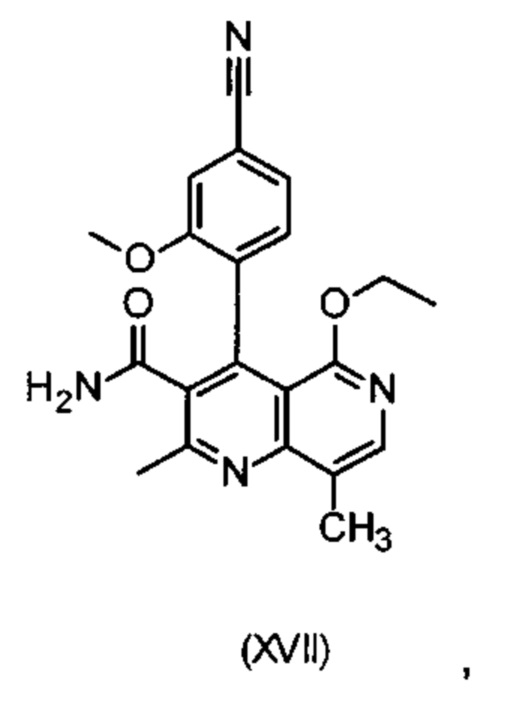
отличающийся тем, что соединения формулы ent-(I)
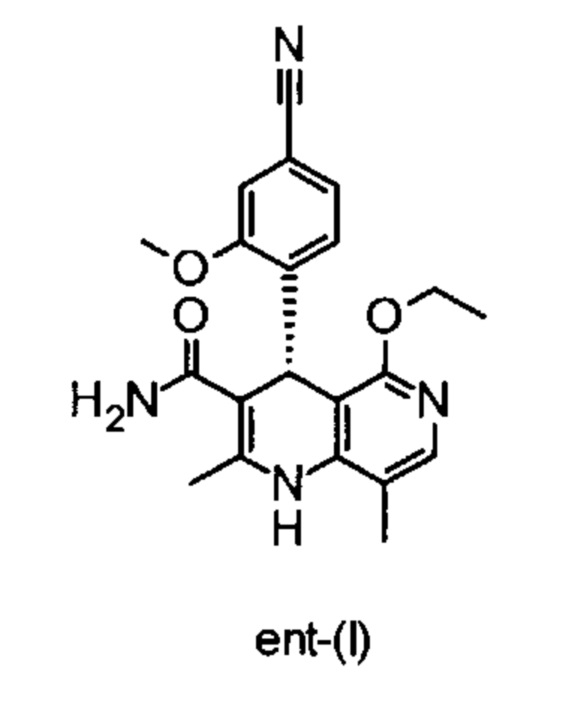
электрохимически окисляют в результате непрямого электрохимического окисления.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при температуре 1-100°С и нормальном давлении.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при потенциале окисления от -0.1 V до +0.6 V относительно эталонного Ag/Ag+-электрода.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят с использованием DDQ в качестве медиатора.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при температуре 1-100°С и нормальном давлении при потенциале окисления от -0.1 V до +0.6 V относительно эталонного Ag/Ag+-электрода и с использованием DDQ в качестве медиатора.
Другим объектом настоящего изобретения является способ получения соединения формулы (XVII):
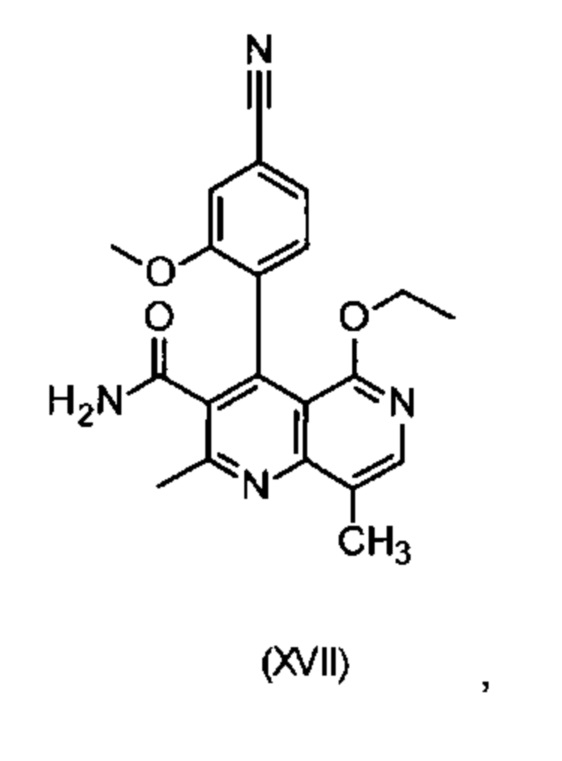
отличающийся тем, что соединения формулы (XIII)
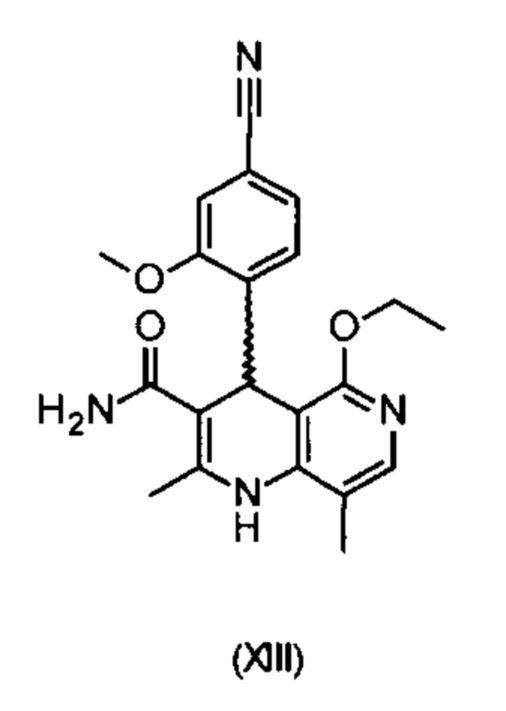
электрохимически окисляют в результате непрямого электрохимического окисления.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при температуре 1-100°С и нормальном давлении.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при потенциале окисления от -0.1 V до +0.6 V относительно эталонного Ag/Ag+-электрода.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят с использованием DDQ в качестве медиатора.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при температуре 1-100°С и нормальном давлении при потенциале окисления от -0.1 V до +0.6 V относительно эталонного Ag/Ag+-электрода и с использованием DDQ в качестве медиатора.
Другим объектом настоящего изобретения является способ получения соединения формулы (XVII):
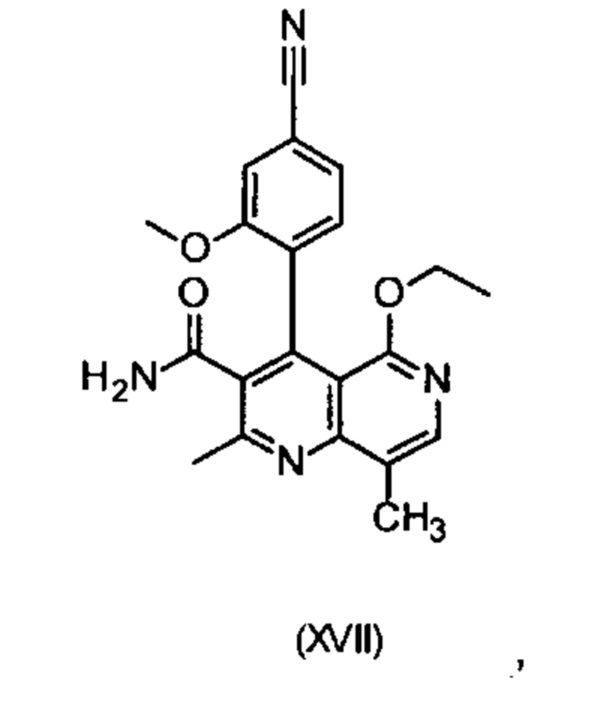
отличающийся тем, что соединения формулы (I)
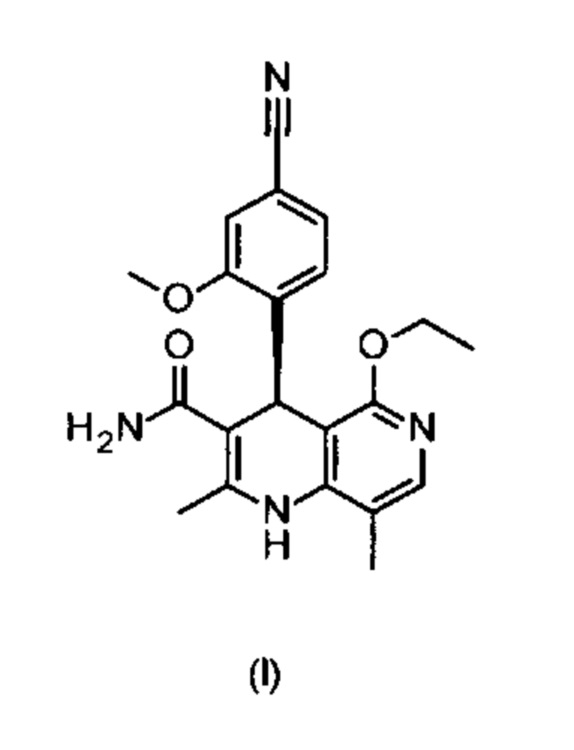
электрохимически окисляют в результате непрямого электрохимического окисления.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при температуре 1-100°С и нормальном давлении.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при потенциале окисления от -0.1 V до +0,6 V относительно эталонного Ag/Ag+-электрода.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят с использованием DDQ в качестве медиатора.
Другим объектом настоящего изобретения является способ, как показано выше, отличающийся тем, что непрямое электрохимическое окисление проводят при температуре 1-100°С и нормальном давлении при потенциале окисления от -0.1 V до +0.6 V относительно эталонного Ag/Ag+-электрода и с использованием DDQ в качестве медиатора.
Объектом настоящего изобретения является способ получения соединений формул M1a(S) и M1b(R)
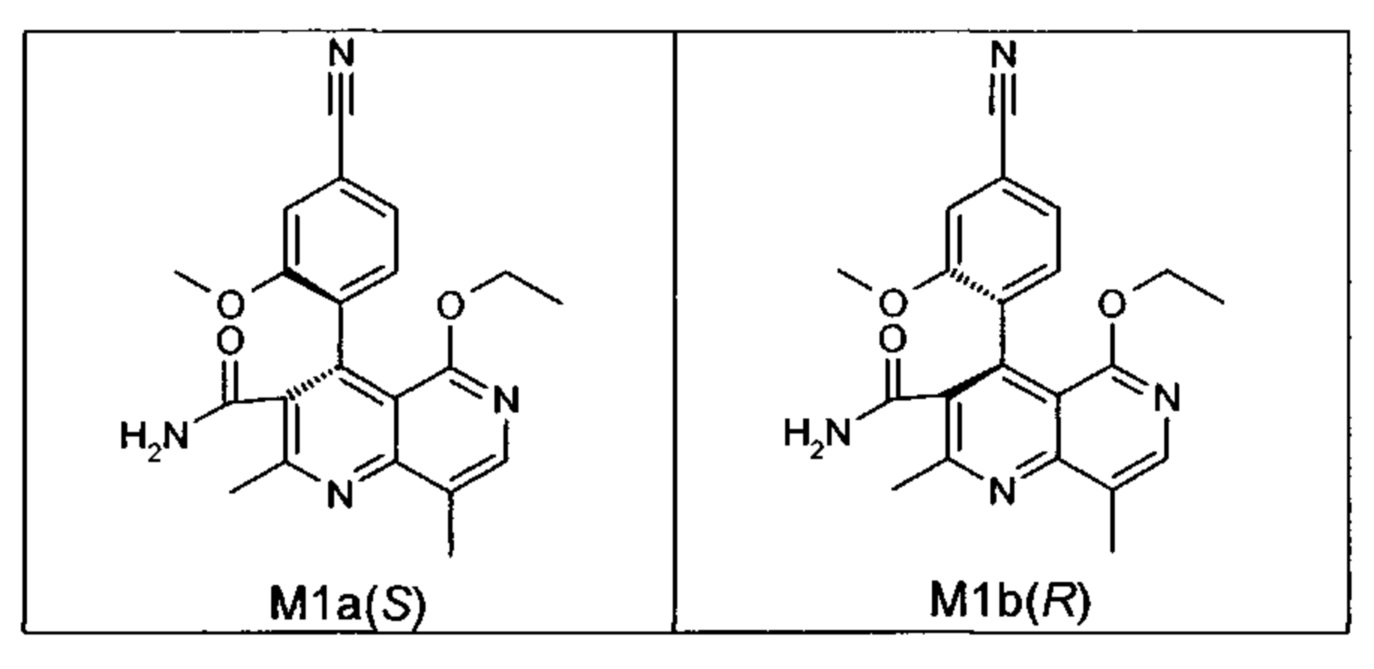
отличающийся тем, что соединения формулы ent-(I)
окисляют.
Объектом настоящего изобретения является способ получения соединений формул M1a(S) и M1b(R), как описано выше, отличающийся тем, что окисление проводят химическими окислительными средствами.
Другим объектом настоящего изобретения является способ получения рацемического соединения формулы (XVII)
отличающийся тем, что смесь соединений формул M1a(S) и M1b(R)
рацемизируют термически.
Объектом настоящего изобретения является способ получения соединения формулы (XVII), как описано выше, отличающийся тем, что смесь соединений формул M1a(S) и M1b(R) рацемизируют при температуре от 70 до 110°С с добавлением или без добавления кислоты.
Другим объектом настоящего изобретения является способ получения соединений формул (I) и ent-(I)
отличающийся тем, что соединения формул (XVII) или M1a(S) или M1b(R) или смесь M1a(S) и M1b(R)
подвергают электрохимическому восстановлению.
Объектом настоящего изобретения является способ получения соединений формул (I) и ent-(I), как описано выше, отличающийся тем, что электрохимическое восстановление проводят в ячейке в виде лабораторного стакана или проточной ячейке в присутствии метанола.
Объектом настоящего изобретения является способ получения соединений формул (I) и ent-(I), как описано выше, отличающийся тем, что электрохимическое восстановление проводят в ячейке в виде лабораторного стакана или проточной ячейке в присутствии этанола.
Другим объектом настоящего изобретения является способ получения соединений формул (I) и ent-(I), как описано выше,
отличающийся тем, что соединения формул (XVII) или M1a(S) или M1b(R) или смесь M1a(S) и M1b(R)
подвергают электрохимическому восстановлению,
и отличающийся тем, что соединения формул (XVII), M1a(S) и M1b(R) получают посредством термической изомеризации соединений формул M1a(S) и M1b(R)
и отличающийся тем, что соединение формулы ent-(I)
окисляют.
Объектом настоящего изобретения является способ получения соединений формул (I) и ent-(I), как описано выше,
отличающийся тем, что соединения формул (XVII) или M1a(S) или M1b(R) или смесь M1a(S) и M1b(R)
подвергают электрохимическому восстановлению в ячейке в виде лабораторного стакана или проточной ячейке в присутствии метанола,
и отличающийся тем, что соединения формул (XVII), M1a(S) и M1b(R) получают посредством термической изомеризации соединений формул M1a(S) и M1b(R)
и отличающийся тем, что соединение формулы ent-(I)
окисляют химическими окислительными средствами.
Объектом настоящего изобретения является способ получения соединений формул (I) и ent-(I), как описано выше,
отличающийся тем, что соединения формул (XVII) или M1a(S) или M1b(R) или смесь M1a(S) и M1b(R)
подвергают электрохимическому восстановлению в ячейке в виде лабораторного стакана или проточной ячейке в присутствии этанола,
и отличающийся тем, что соединения формул (XVII), M1a(S) и M1b(R) получают посредством термической изомеризации соединений формул M1a(S) и M1b(R)
и отличающийся тем, что соединение формулы ent-(I)
окисляют химическими окислительными средствами.
Экспериментальная часть
Сокращения и аббревиатуры:
MS: Масса согласно масс-спектрометрии
ВЭЖХ: Высокоэффективная жидкостная хроматография
ДМФА: Диметилформамид
Раствор Red-Al в толуоле: Бис-(2-метоксиэтокси)-алюминийдигидрид натрия в толуоле
ТГФ: тетрагидрофуран
Водн. HCl: водная соляная кислота
ДМАП: 4-(диметиламино)-пиридин
Примеры
Пример 1
Метил-4-бром-2-метоксибензоат (XV)
3,06 кг (22,12 моль) карбоната калия загружали в 3,6 л ацетона и нагревали до кипения с возвратом флегмы. К этой суспензии добавляли 1,2 кг 4-бром-2-гидроксибензойной кислоты (5,53 моль), суспендированной в 7,8 л ацетона, и дополнительно промывали 0,6 л ацетона. Нагревали в течение часа при температуре кипения с возвратом флегмы (сильное выделение газа!). Затем при кипячении добавляли 2,65 кг (21,01 моль) диметилсульфата в течение 4 часов. Затем дополнительно перемешивали при кипении с возвратом флегмы в течение 2,5 часов. Растворитель в значительной степени отгоняли (до состояния пригодного к перемешиванию) и добавляли 12 л толуола, затем при 110°С отгоняли оставшийся ацетон. Отгоняли около 3 литров дистиллята, для этого пополняли реакционную смесь добавлением еще 3 л толуола. Смеси давали остыть до 20°С, добавляли 10,8 л воды и энергично перемешивали. Органическую фазу отделяли, а водную фазу еще раз дополнительно экстрагировали 6,1 л толуола. Объединенные органические фазы промывали 3 л насыщенного раствора хлорида натрия и толуольную фазу концентрировали до примерно 4 л. Определение содержания посредством выпаривания частичного количества давало в пересчете выход 1,306 кг (96,4% от теории). Раствор напрямую использовали в следующей стадии.
ВЭЖХ-метод A: RT: около 11,9 мин.
MS (Elpos): m/z=245 [М+Н]+
1Н ЯМР (400 MHz, CD2Cl2): δ=3.84 (с, 3Н), 3.90 (с, 3Н), 7.12-7.20 (м, 2Н), 7.62 (д, 1Н).
Пример 2
4-бром-2-метоксибензальдегид (XVI)
1,936 кг (6,22 моль) 65%-ного раствора Red-AI в толуоле загружали с 1,25 л толуола при -5°С. К этому раствору дозировали 0,66 кг (6,59 моль) 1-метилпиперазина и дополнительно смывали 150 мл толуола, при этом поддерживали температуру от -7 до -5°С. Смесь оставляли дополнительно перемешиваться в течение 30 минут при 0°С. Затем этот раствор дозировали к раствору 1,261 кг (5,177 моль) метил-4-бром-2-метоксибензоата (XV), растворенного в 4 л толуола, температуру поддерживали от -8 до 0°С. Дважды дополнительно смывали 0,7 л толуола и дополнительно перемешивали в течение 1,5 часов при 0°С. Для обработки добавляли холодную (0°С) водную серную кислоту (12,5 л воды + 1,4 кг концентрированной серной кислоты). Температура не должна была превышать 10°С (медленное дозирование). Значение рН при необходимости доводили до 1 посредством добавления дополнительной серной кислоты. Органическую фазу отделяли, а водную фазу дополнительно экстрагировали 7,6 л толуола. Объединенные органические фазы промывали 5,1 л воды и затем в значительной степени концентрировали и остаток растворяли 10 л ДМФА. Вновь концентрировали до объема примерно 5 л. Определение содержания посредством выпаривания частичного количества давало в пересчете выход 1,041 кг (94,1% от теории). Раствор напрямую использовали в следующей стадии.
ВЭЖХ-метод A: RT: около 12,1 мин.
MS (Elpos): m/z=162 [M+H]+
1Н-ЯМР (CDCl3, 400MHz): δ=3.93 (3Н, с), 7.17 (2H, м), 7.68 (1Н, д), 10.40 (1Н, с)
Пример 3
4-формил-3-метоксибензонитрил (VI)
719 г (3,34 моль) 4-бром-2-метоксибензальдегида (XVI) в виде раствора в 4,5 л ДМФА загружали с 313 г (0,74 моль) гексацианоферрата калия (K4[Fe(CN)6]) и 354 г (3,34 моль) карбоната натрия и дополнительными 1,2 л ДМФА и добавляли 3,8 г (0,017 моль) ацетата палладия. Перемешивали 3 часа при 120°С. Давали остыть до 20°С и к загрузке добавляли 5,7 л воды. Экстрагировали 17 л этилацетата и водную фазу еще раз дополнительно промывали 17 л этилацетата. Органические фазы объединяли и концентрировали в значительной степени, растворяли с 5 л изопропанола и концентрировали до примерно 2 л. Нагревали до кипения и прибавляли по каплям 2 л воды. Охлаждали до 50°С и снова добавляли 2 литра воды. Охлаждали до 3°С и перемешивали в течение одного часа при этой температуре. Продукт отфильтровывали и дополнительно промывали водой (2 раза по 1,2 л). Высушивали при 40°С в вакууме.
Выход: 469 г (87% от теории) твердого вещества бежевого цвета.
ВЭЖХ-метод A: RT: около 8,3 мин.
MS (Elpos): m/z=162 [М+Н]+
1Н-ЯМР (300 MHz, ДМСО-d6): δ=3.98 (с, 3Н), 7.53 (д, 1Н), 7.80 (с, 1Н), 7.81 (д, 1Н), 10.37 (с, 1Н).
Пример 4
2-цианоэтил-4-(4-циано-2-метоксифенил)-2,8-диметил-5-оксо-1,4,5,6-тетрагидро-1,6-наФтиридин-3-карбоксилат (X)
Вариант А
1,035 кг (6,422 моль) 4-формил-3-метоксибензонитрила (VI), 1,266 кг (8,028 моль) 2-цианоэтил-3-оксобутаноата, 54,6 г (0,642 моль) пиперидина и 38,5 г (0,642 моль) ледяной уксусной кислоты нагревали в 10 л дихлорметана в течение 6,5 часов при кипячении с возвратом флегмы на водоотделителе. Оставляли охлаждаться до комнатной температуры и 2 раза промывали органическую фазу 5 л воды в каждом случае. Затем дихлорметановую фазу концентрировали при нормальном давлении и еще перемешиваемый остаток растворяли в 15,47 кг 2-бутанола и добавляли 0,717 кг (5,78 моль) 4-амино-5-метилпиридона. Оставшийся дихлорметан отгоняли до тех пор, пока не была достигнута внутренняя температура 98°С. Затем нагревали в течение 20 часов при кипении с возвратом флегмы. Охлаждали до 0°С, оставляли далее перемешиваться при указанной температуре в течение 4 часов, и отфильтровывали продукт. Затем сушили при 40°С в вакууме в присутствии газа-носителя.
Выход: 2,049 кг (87,6% от теории, в пересчете на 4-амино-5-метилпиридон, поскольку этот компонент используется в недостатке) твердого вещества слегка желтого цвета.
ВЭЖХ-метод A: RT: около 9,7 мин.
MS (Elpos): m/z=405 [М+Н]+
1Н ЯМР (300 MHz, ДМСО-d6): δ=2.03 (с, 3Н), 2.35 (с, 3Н), 2.80 (м, 2Н), 3.74 (с, 3Н), 4.04 (м, 1Н), 4.11 (м, 1Н), 5.20 (с, 1Н), 6.95 (с, 1Н), 7.23 (дд, 1Н), 7.28-7.33 (м, 2Н), 8.18 (с, 1Н), 10.76 (с, 1Н).
Вариант В
1,344 кг (8,34 моль) 4-формил-3-метоксибензонитрила (VI), 71 г (0,834 моль) пиперидина и 50,1 г (0,834 моль) ледяной уксусной кислоты загружали в 6 л изопропанола и при 30°С в течение 3 часов добавляли раствор 1,777 кг (11,26 моль) 2-цианоэтил-3-оксобутаноата в 670 мл изопропанола. Дополнительно перемешивали один час при 30°С. Охлаждали до 0-3°С и дополнительно перемешивали в течение 0,5 часа. Продукт отфильтровывали и дважды дополнительно промывали 450 мл холодного изопропанола в каждом случае. Для определения выхода сушили в вакууме при 50°С (2,413 кг, 97% от теории); однако как правило из-за высокого выхода далее работали напрямую с сырым по изопропанолу продуктом. Для этого продукт растворяли в 29 л изопропанола и добавляли 1,277 кг (7,92 моль) 4-амино-5-метилпиридона и затем нагревали в течение 24 ч при внутренней температуре 100°С при избыточном давлении около 1,4 бар в закрытом сосуде. Охлаждали на рампе до 0°С в течение 5 часов и дополнительно перемешивали в течение 3 часов при 0°С. Продукт отфильтровывали и дополнительно промывали 2,1 л холодного изопропанола. Сушили в вакууме при 60°С.
Выход: 2,819 кг (88% от теории, в пересчете на 4-амино-5-метилпиридон, поскольку этот компонент используется в недостатке) твердого вещества слегка желтого цвета.
ВЭЖХ-метод A: RT: около 9,7 мин.
MS (Elpos): m/z=405 [М+Н]+
1Н ЯМР (300 MHz, ДМСО-d6): δ=2.03 (с, 3Н), 2.35 (с, 3Н), 2.80 (м, 2Н), 3.74 (с, 3Н), 4.04 (м, 1Н), 4.11 (м, 1Н), 5.20 (с, 1Н), 6.95 (с, 1Н), 7.23 (дд, 1Н), 7.28-7.33 (м, 2Н), 8.18 (с, 1Н), 10.76 (с, 1Н).
Пример 5
2-цианоэтил-4-(4-циано-2-метоксиФенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксилат (XI)
2,142 кг (5,3 моль) 2-цианоэтил-4-(4-циано-2-метоксифенил)-2,8-диметил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилата (X) и 4,70 кг (29 моль) триэтилортоацетата растворяли в 12,15 л диметилацетамида и добавляли 157,5 г концентрированной серной кислоты. Нагревали в течение 1,5 часов при 115°С и затем охлаждали до 50°С. При 50°С по каплям добавляли 12,15 л воды в течение 30 минут. После завершения добавления вводили 10 г титульного соединения (XI) в качестве затравки и добавляли по каплям дополнительные 12,15 л воды в течение 30 минут при 50°С. Охлаждали до 0°С (рампа, 2 часа) и дополнительно перемешивали в течение 2 часов при 0°С. Отфильтровывали продукт, два раза промывали 7,7 л воды в каждом случае и сушили в вакууме при 50°С.
Выход: 2114,2 г (92,2% от теории) твердого вещества слегка желтоватого цвета.
ВЭЖХ-метод В: RT: около 10,2 мин.
MS (Elpos): m/z=433 [М+Н]+
1Н ЯМР (300 MHz, ДМСО-d6): δ=1.11 (т, 3Н), 2.16 (с, 3Н), 2.42 (с, 3Н), 2.78 (м, 2Н), 3.77 (с, 3Н), 4.01-4.13 (м, 4Н), 5.37 (с, 1Н), 7.25 (д, 1Н), 7.28-7.33 (м, 2Н), 7.60 (с, 1Н), 8.35 (с, 1Н).
В качестве альтернативы, реакцию можно проводить в N-метил-2-пирролидоне.
2-цианоэтил-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксилат (XI)
2,142 кг (5,3 моль) 2-цианоэтил-4-(4-циано-2-метоксифенил)-2,8-диметил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилата (X) и 2,35 кг (14,5 моль) триэтилортоацетата растворяли в 3,21 кг N-метил-2-пирролидона и добавляли 157,5 г концентрированной серной кислоты. Нагревали в течение 1,5 часов при 115°С и затем охлаждали до 50°С. При 50°С по каплям добавляли 2,2 л воды в течение 30 минут. После завершения добавления вводили 10 г титульного соединения (XI) в качестве затравки и добавляли по каплям дополнительные 4,4 л воды в течение 30 минут при 50°С. Охлаждали до 0°С (рампа, 2 часа) и дополнительно перемешивали в течение 2 часов при 0°С. Отфильтровывали продукт, два раза промывали 4 л воды в каждом случае и сушили в вакууме при 50°С.
Выход: 2180,7 г (95,1% от теории) твердого вещества слегка желтоватого цвета.
ВЭЖХ-метод В: RT: около 10,2 мин.
Пример 6
2-цианоэтил4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота (XII)
2,00 кг (4,624 моль) 2-цианоэтил-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксилата (XI) растворяли в смеси 12 л ТГФ и 6 л воды и охлаждали до 0°С. К этому раствору добавляли по каплям в течение 15 минут при 0°С раствор гидроксида натрия (полученный из 0,82 кг 45%-ного водного раствора NaOH (9,248 моль) и 4,23 л воды) и дополнительно перемешивали в течение 1,5 часов при 0°С. Дважды экстрагировали 4,8 литрами метил-трет-бутилового эфира в каждом случае и один раз 4,8 литрами этилацетата. Водный раствор при 0°С доводили до значения рН 7 разбавленной соляной кислотой (полученной из 0,371 кг 37%-ной HCl и 1,51 л воды). Давали нагреться до 20°С и добавляли водный раствор 2,05 кг хлорида аммония в 5,54 л воды. Дополнительно перемешивали в течение 1 часа при 20°С, продукт фильтровали и дважды промывали 1,5 литрами воды в каждом случае и один раз 4 литрами ацетонитрила. Сушили при 40°С в вакууме в присутствии газа-носителя.
Выход: 1736,9 г (99% от теории) почти бесцветного порошка (очень слабый желтоватый оттенок).
ВЭЖХ-метод С: RT: около 6,8 мин.
MS (Elpos): m/z=380 [М+Н]+
1Н ЯМР (300 MHz, ДМСО-d6): δ=1.14 (т, 3Н), 2.14 (с, 3Н), 2.37 (с, 3Н), 3.73 (с, 3Н), 4.04 (м, 2Н), 5.33 (с, 1Н), 7.26 (м, 2Н), 7.32 (с, 1Н), 7.57 (с, 1Н), 8.16 (с, 1Н), 11.43 (уш. с,1Н).
В качестве альтернативы для экстракции обрабатывали толуолом:
2-цианоэтил4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота (XII)
2,00 кг (4,624 моль) 2-цианоэтил-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксилата (XI) растворяли в смеси 12 л ТГФ и 6 л воды и охлаждали до 0°С. К этому раствору добавляли по каплям в течение 15 минут при 0°С раствор гидроксида натрия (полученный из 0,82 кг 45%-ного водного раствора NaOH (9,248 моль) и 4,23 л воды) и дополнительно перемешивали в течение 1,5 часов при 0°С. Добавляли 5 л воды и 381,3 г ацетата натрия и дополнительно энергично перемешивали. Фазам давали осесть и отделяли органическую фазу. Водную фазу доводили до рН 6,9 с помощью 10%-ной соляной кислоты (при рН около 9,5 вносили 10 г титульного соединения в качестве затравки). После завершения осаждения продукта дополнительно перемешивали в течение одного часа при 0°С, и затем отфильтровывали и два раза промывали 4 л воды в каждом случае и два раза 153 мл толуола в каждом случае. Сушили при 40°С в вакууме в присутствии газа-носителя (азот, 200 мбар). Выход: 1719,5 г (98% от теории) почти бесцветного порошка (очень слабый желтоватый оттенок).
ВЭЖХ-метод С: RT: около 6,8 мин.
Пример 7
4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (XIII)
1,60 кг (4,22 моль) 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоновой кислоты (XII) и 958 г (5,91 моль) 1,1-карбодиимидазола помещали в 8 л ТГФ и добавляли при 20°С 51 г (0,417 моль) 4-диметиламинопиридина (ДМАП). Перемешивали в течение одного часа при 20°С (выделение газа!), а затем нагревали в течение 2,5 часов при 50°С. К этому раствору добавляли 2,973 кг (18,42 моль) гексаметилдисилазана и кипятили с возвратом флегмы в течение 22 часов. Добавляли еще 1,8 л ТГФ и охлаждали до 5°С. Смесь 1,17 л ТГФ и 835 г воды добавляли в течение 3 часов, так чтобы температура оставалась между 5 и 20°С. После кипятили с возвратом флегмы в течение одного часа, затем охлаждали до 0°С на рампе (3 часа) и дополнительно перемешивали при этой температуре в течение одного часа. Отфильтровывали продукт и два раза дополнительно промывали 2,4 л ТГФ в каждом случае и два раза 3,2 л воды в каждом случае. Сушили в вакууме при 70°С в присутствии газа-носителя.
Выход: 1,501 кг (94% от теории) почти бесцветного порошка (очень слабый желтоватый оттенок).
ВЭЖХ-метод В: RT: около 6,7 мин.
MS (Elpos): m/z=379 [М+Н]+
1Н ЯМР (300 MHz, ДМСО-d6): δ=1.05 (т, 3Н), 2.12 (с, 3Н), 2.18 (с, 3Н), 3.82 (с, 3Н), 3.99-4.07 (м, 2Н), 5.37 (с, 1Н), 6.60-6.84 (м, 2Н), 7.14 (д, 1Н), 7.28 (дд, 1Н), 7.37 (д, 1Н), 7.55 (с, 1Н), 7.69 (с, 1Н).
Пример 8
(4S)-4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (I) в виде раствора в ацетонитриле/метаноле 40:60
Разделение на энантиомеры на SMB-установке
В качестве питающего раствора был использован раствор, соответствующей концентрации, состоящий из 50 г рацемата 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (XIII), растворенного в 1 литре смеси метанола/ацетонитрила 60:40.
Его хроматографировали в SMB-установке на неподвижной фазе: Chiralpak AS-V, 20 мкм. Давление составляло 30 бар, в качестве элюента использовали смесь метанол/ацетонитрил 60:40.
9,00 кг 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (XII) растворяли в 180 л смеси, состоящей из метанола/ацетонитрила 60:40 и хроматографировали с помощью SMB. После концентрирования содержащих продукт фракций получили 69,68 л 6,2%-ного раствора (соответствует 4,32 кг (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (I) в виде раствора в ацетонитриле/метаноле 40:60).
Выход: 4,32 кг (S)-энантиомера (48% от теории), растворенного в 69,68 л ацетонитрила/метанола 40:60 в виде бесцветной фракции.
Энантиомерная чистота: >98,5% ее (ВЭЖХ, метод D)
Пробу концентрируют в вакууме и анализируют: MS (Elpos): m/z=379 [М+Н]+
1Н ЯМР (300 MHz, ДМСО-d6): δ=1.05 (т, 3Н), 2.12 (с, 3Н), 2.18 (с, 3Н), 3.82 (с, 3Н), 3.99-4.07 (м, 2Н), 5.37 (с, 1Н), 6.60-6.84 (м, 2Н), 7.14 (д, 1Н), 7.28 (дд, 1Н), 7.37 (д, 1Н), 7.55 (с, 1Н), 7.69 (с, 1Н).
Аналогичным образом выделяли (R)-энантиомер ent-(I)
Выход: 4,41 кг (R)-энантиомера (48% от теории), растворенного в 71,00 л ацетон итри л а/метанол а 40:60 в виде бесцветной фракции.
Энантиомерная чистота: >98,5% ее (ВЭЖХ, метод D)
Пробу концентрируют в вакууме, и анализируют: MS (Elpos): m/z=379 [М+Н]+
1Н-ЯМР (300 MHz, ДМСО-d6): δ=1.05 (т, 3Н), 2.12 (с, 3Н), 2.18 (с, 3Н), 3.82 (с, 3Н), 3.99-4.07 (м, 2Н), 5.37 (с, 1Н), 6.60-6.84 (м, 2Н), 7.14 (д, 1Н), 7.28 (дд, 1Н), 7.37 (д, 1Н), 7.55 (с, 1Н), 7.69 (с, 1Н).
Пример 9
(4S)-4-(4-ииано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (I)
Кристаллизация и регулирование полиморфизма
64,52 л 6,2%-ного раствора из примера 8 в смеси ацетонитрил/метанол 40:60 (соответствует 4,00 кг соединения (I) фильтровали через фильтрующий патрон (1,2 мкм) и затем концентрировали при 250 мбар, так что раствор все еще был пригоден к перемешиванию. Добавляли 48 л этанола, денатурировали толуолом и снова отгоняли при 250 мбар до предела перемешиваемости (передистилляция на этанол). Добавляли дополнительные 48 л этанола, денатурировали толуолом и затем отгоняли при нормальном давлении до общего объема примерно 14 л (температура кожуха 98°С). Охлаждали на рампе до 0°С (4 часа), дополнительно перемешивали в течение 2 часов при 0°С и отфильтровывали продукт. Два раза дополнительно промывали 4 л холодного этанола в каждом случае и затем сушили в вакууме при 50°С.
Выход: 3,64 кг (91% от теории) почти бесцветного кристаллического порошка.
Энантиомерная чистота: >> 99% ее (ВЭЖХ, метод D); время удерживания(RT)/относительное время удерживания (RRT): (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (1) около 11 мин. RRT: 1,00; (4R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (1) около 9 мин. RRT: 0,82
Чистота: >99,8% (ВЭЖХ, метод В), RT: около 6,7 мин.
Содержание: 99,9% (относительно внешнего стандарта)
Удельный вращение (хлороформ, 589 нм, 19,7°С, с=0,388600 г/100 мл): -148.8°.
MS (Elpos): m/z=379 [М+Н]+
1Н ЯМР (300 MHz, ДМСО-d6): δ=1.05 (т, 3Н), 2.12 (с, 3Н), 2.18 (с, 3Н), 3.82 (с, 3Н), 3.99-4.07 (м, 2Н), 5.37 (с, 1Н), 6.60-6.84 (м, 2Н), 7.14 (д, 1Н), 7.28 (дд, 1Н), 7.37 (д, 1Н), 7.55 (с, 1Н), 7.69 (с, 1Н).
Температура плавления: 252°С (соединение формулы ent-(I) в кристаллической форме модификации I)
Аналогичным образом выделяют (R)-энантиомер ent-(I). Однако концентрируют еще больше для минимизации потерь выхода:
71,00 л примерно 6,2%-ного раствора из примера 8 в смеси ацетонитрил/метанол 40:60 (соответствует 4,00 кг соединения ent-(I)) фильтровали через фильтрующий патрон (1,2 мкм) и затем концентрировали при 250 мбар, так что раствор все еще был пригоден к перемешиванию. Добавляли 48 л этанола, денатурировали толуолом и снова отгоняли при 250 мбар до предела перемешиваемости (передистилляция на этанол). Добавляли дополнительные 48 л этанола, денатурировали толуолом и затем отгоняли при нормальном давлении до общего объема примерно 10 л (температура кожуха 98°С). Охлаждали на рампе до 0°С (4 часа), дополнительно перемешивали в течение 2 часов при 0°С и отфильтровывали продукт. Два раза дополнительно промывали 2 л холодного этанола в каждом случае и затем сушили в вакууме при 50°С.
Выход: 3,88 г (97% от теории) почти бесцветного кристаллического порошка.
Энантиомерная чистота: >> 99% ее (ВЭЖХ, метод D); время удерживания(RT)/относительное время удерживания (RRT): (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (1) около 11 мин. RRT: 1,00; (4R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (1) около 9 мин. RRT: 0,82
Чистота: >99,8% (ВЭЖХ, метод В), RT: около 6,7 мин.
Содержание: 99,9% (относительно внешнего стандарта)
Удельный вращение (хлороформ, 589 нм, 19,7°С, с=0,388600 г/100 мл): +148.8°.
MS (Elpos): m/z=379 [М+Н]+
1Н-ЯМР (300 MHz, ДМСО-d6):=1.05 (т, 3Н), 2.12 (с, 3Н), 2.18 (с, 3Н), 3.82 (с, 3Н), 3.99-4.07 (м, 2Н), 5.37 (с, 1Н), 6.60-6.84 (м, 2Н), 7.14 (д, 1Н), 7.28 (дд, 1Н), 7.37 (д, 1Н), 7.55 (с, 1Н), 7.69 (с, 1Н).
Температура плавления: 252°С
Химическое окисление
Пример 10
Получение рацемического (XVII) из рацемического (XIII) химическими методами
Rac-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,6-нафтиридин-3-карбоксамид
100,00 г (264,25 ммоль) 4(R,S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (XIII) загружали в 4 кг дихлорметана и добавляли 68,98 г (303,88 ммоль) 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ) при 20°С. Дополнительно перемешивали один час при 20°С. Выпавший осадок отфильтровывали и дважды промывали 400 г дихлорметана в каждом случае. Концентрировали досуха в вакууме и остаток растворяли в 1200 г этанола. Нагревали до кипения с возвратом флегмы и отгоняли около 800 г этанола. Давали охладиться до комнатной температуры и дополнительно перемешивали в течение 1 часа при 20°С. Продукт отфильтровывали и промывали небольшим количеством этанола (около 80 г), сушили в течение ночи в вакууме (50°С).
Выход: 87,30 г (87,54% от теории) твердого вещества бежевого цвета.
MS (Elpos): m/z=378 [М+Н]+
1Н-ЯМР (500 MHz, ДМСО-d6): δ=0.72 (т, 3Н), 2.50 (с, 3Н), 2.70 (с, 3Н), 3.65 (с, 1Н), 4.00 (м (уш.), 2Н), 7.30 (д, 1Н), 7.45 (д, 1Н), 7.50 (с, 2Н), 7.69 (с, 1Н), 8.05 (с, 1Н)
Пример 11а
Получение M1a(S) из ent-(I) химическими методами
(S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,6-нафтиридин-3-карбоксамид (M1a(S))
100,00 г (264,25 ммоль) 4(R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (ent-(I)) загружали в 4 кг дихлорметана и добавляли 68,98 г (303,88 ммоль) 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ) при 20°С. Дополнительно перемешивали один час при 20°С. Выпавший осадок отфильтровывали и дважды промывали 400 г дихлорметана в каждом случае. Концентрировали досуха в вакууме и остаток растворяли в 1200 г этанола. Нагревали до кипения с возвратом флегмы и отгоняли около 800 г этанола. Давали охладиться до комнатной температуры и дополнительно перемешивали в течение 1 часа при 20°С. Продукт отфильтровывали и промывали небольшим количеством этанола (около 80 г), сушили в течение ночи в вакууме (50°С).
Выход: 85,80 г (86,04% от теории) твердого вещества бежевого цвета.
ВЭЖХ: RT: около 6,08 мин. (хиральная фаза: Chiralpak AS-H (250×4 мм), элюент: изогексан: этанол = 50:50)
MS (Elpos): m/z=378 [М+Н]+
1Н-ЯМР (500 MHz, ДМСО-d6): δ=0.72 (т, 3Н), 2.50 (с, 3Н), 2.70 (с, 3Н), 3.65 (с, 1Н), 4.00 (м (уш.), 2Н), 7.30 (д, 1Н), 7.45 (д, 1Н), 7.50 (с, 2Н), 7.69 (с, 1Н), 8.05 (с, 1Н)
Пример 11b
Получение M1b(R) из (I)
(R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,6-нафтиридин-3-карбоксамид (M1bR))
100,00 г (264,25 ммоль) 4(S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (I) загружали в 4 кг дихлорметана и добавляли 68,98 г (303,88 ммоль) 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ) при 20°С. Дополнительно перемешивали один час при 20°С. Выпавший осадок отфильтровывали и дважды промывали 400 г дихлорметана в каждом случае. Концентрировали досуха в вакууме и остаток растворяли в 1200 г этанола. Нагревали до кипения с возвратом флегмы и отгоняли около 800 г этанола. Давали охладиться до комнатной температуры и дополнительно перемешивали в течение 1 часа при 20°С. Продукт отфильтровывали и промывали небольшим количеством этанола (около 80 г), сушили в течение ночи в вакууме (50°С).
Выход: 85,80 г (86,04% от теории) твердого вещества бежевого цвета.
ВЭЖХ: RT: около 9,03 мин. (хиральная фаза: Chiralpak AS-H (250×4 мм), элюент: изогексан: этанол = 50:50)
MS (Elpos): m/z=378 [М+Н]+
1Н-ЯМР (500 MHz, ДМСО-d6): δ=0.72 (т, 3Н), 2.50 (с, 3Н), 2.70 (с, 3Н), 3.65 (с, 1Н), 4.00 (м (уш.), 2Н), 7.30 (д, 1Н), 7.45 (д, 1Н), 7.50 (с, 2Н), 7.69 (с, 1Н), 8.05 (с, 1Н)
Пример 12а
Получение рацемического (XVII) из ent-(I)
4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,6-нафтиридин-3-карбоксамид
100,00 г (264,25 ммоль) 4(R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (ent-(I)) загружали в 4 кг дихлорметана и добавляли 68,98 г (303,88 ммоль) 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ) при 20°С. Дополнительно перемешивали один час при 20°С. Выпавший осадок отфильтровывали и дважды промывали 400 г дихлорметана в каждом случае. Концентрировали досуха в вакууме и остаток растворяли в 1200 г этанола. Нагревали в автоклаве в течение 3 часов при 120°С под давлением и затем отгоняли примерно 900 г этанола. Давали охладиться до комнатной температуры и дополнительно перемешивали в течение 1 часа при 20°С. Продукт отфильтровывали и промывали небольшим количеством этанола (около 40 г), сушили в течение ночи в вакууме (50°С).
Выход: 92,47 g г (92,73% от теории) твердого вещества бежевого цвета.
MS (Elpos): m/z=378 [М+Н]+
1Н-ЯМР (500 MHz, ДМСО-d6): δ=0.72 (т, 3Н), 2.50 (с, 3Н), 2.70 (с, 3Н), 3.65 (с, 1Н), 4.00 (м (уш.), 2Н), 7.30 (д, 1Н), 7.45 (д, 1Н), 7.50 (с, 2Н), 7.69 (с, 1Н), 8.05 (с, 1Н)
Пример 12b
Получение M1a(S) из ent-(I) окислением HNO3
Выполняется в атмосфере N2. 75,0 г 4(R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (ent (I)) суспендировали в 1000 г ацетонитрила (ACN) и охлаждали до 9°С. Туда же по каплям в течение 10 мин добавляли 12,68 г дымящейся азотной кислоты. Твердое вещество ненадолго скомковалось, а затем немедленно растворилось. Оставляли до достижения комнатной температуры (продолжительность 1 час) и получали светло-желтый, прозрачный раствор. Перемешивали в течение 4 часов при комнатной температуре, примерно через 30 минут образовывался темно-оранжевый раствор, затем грязно-желтая суспензия. Через 4 ч смесь охлаждали до 10°С и гасили 50 мл воды. Затем с помощью 80 мл насыщенного раствора NaHCO3 доводили рН до 7,2 (получали желтую суспензию). Выделяли твердое вещество (первая часть) и промывали водой. Фильтрат концентрировали на роторном испарителе при 40°С до примерно 1/3 от исходного объема, перемешивали в течение 1,5 ч на ледяной бане (5°С), выделяли осажденное твердое вещество (вторая часть) и промывали 100 мл холодной воды. Полученные твердые вещества сушили в вакууме в течение ночи.
Выход: 59,7 г = 86,7% от теории
Аналитические данные 1 части
ЕЕ: М1а: 83,6% M1b: 16,4%
Содержание: 98,9%
Аналитические данные 2 части
ЕЕ: М1а: 77,4% M1b: 22,6%
Чистота: 99,2% по площади пиков
Содержание: 94,5%
Получение рацемического М1 (XVII) из обогащенного M1a(S)
100 г обогащенного М1а (ЕЕ: М1а: 83,6% M1b: 16,4%) загружали в 1000 мл н-бутанола и нагревали до 135°С температуры бани. Дальше перемешивали в течение 6 ч при кипячении с возвратом флегмы (желтая, разбавленная суспензия). Охлаждали и оставляли перемешиваться в течение ночи при комнатной температуре. Раствор концентрировали на роторном испарителе при температуре около 50°С (до еще перемешиваемой суспензии) и затем перемешивали при 5°С в течение 1 часа. Кристаллы отфильтровывали, промывали небольшим количеством холодного бутанола и сушили в течение ночи в вакууме <200 мбар при 40°С.
Выход: 85,9 г = 85,9% от теории (в пересчете на исходный продукт: 90,9% от теории).
ЕЕ: 50,5% М1а, 49,5% M1b
Электрохимическое окисление
Пример 24
Циклическая вольтамперометрия (I) в отсутствие 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ)
2,17 г (10 ммоль) тетрафторбората тетраэтиламмония (Et4NBF4) растворяют в 100 мл ацетонитрила. Затем добавляют 378,4 мг (1 ммоль) (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (I).
Циклическую вольтамперометрию проводят с помощью Pt-сетки в качестве рабочего электрода и Pt-проволоки в качестве контрэлектрода и Ag/Ag+ (10 ммоль/л) в ацетонитриле в качестве эталонного электрода в течение 10 циклов со скоростью развертки потенциала 250 или соответственно 100 мВ/с.
Пример 25
Циклическая вольтамперометрия (I) в присутствии DDQ
2,17 г (10 ммоль) тетрафторбората тетраэтиламмония (Et4NBF4) растворяют в 100 мл ацетонитрила. Затем добавляют 22,7 мг (0,1 ммоль) DDQ и 378,4 мг (1 ммоль) соединения формулы (I). Молярное соотношение DDQ : дигидропиридин составляет, таким образом, 1:10.
Циклическую вольтамперометрию проводят с помощью Pt-сетки в качестве рабочего электрода и Pt-проволоки в качестве контрэлектрода и Ag/Ag+ (10 ммоль/л) в ацетонитриле в качестве эталонного электрода в течение 10 циклов со скоростью развертки потенциала 250 или соответственно 100 мВ/с.
Пример 26:
Окисление ent-(I) в присутствии DDQ (10% мол.)
2,17 г (10 ммоль) тетрафторбората тетраэтиламмония (Et4NBF4) растворяют в 100 мл ацетонитрила. Затем добавляют 22,7 мг (0,1 ммоль) DDQ и 378,4 мг (1 ммоль) соединения формулы ent-(I) (10 ммоль). Молярное соотношение DDQ : ent-(I) составляет, таким образом, 1:10.
Затем раствор подвергают электролизу с постоянным потенциалом, причем на аноде (рабочий электрод) поддерживают потенциал +300 мВ относительно Ag/Ag+ (10 ммоль/л). После прохождения заряда 180 Кл (эквивалентно 2,1Ф) (в течение примерно 2 часов) реакцию останавливают. В этот момент выход (XVII) составил 94% с атропизомерным соотношением M1a(S): M1b(R)=90:10.
За профилем реакции следили посредством частого отбора проб и анализа с помощью ВЭЖХ. Профиль представлен на фигуре 6. Количество продукта (XVII) со временем возрастает, а исходного реагента - уменьшается. Образование продукта согласуется с переносом электрического заряда, что указывает на высокую электрическую эффективность.
Пример 27:
Окисление (I) в присутствии DDQ (1% мол.)
2,17 г (10 ммоль) тетрафторбората тетраэтиламмония (Et4NBF4) растворяют в 100 мл ацетонитрила. Затем добавляют 2,3 мг (0,01 ммоль) DDQ и 378,4 мг (1 ммоль) соединения формулы (I). Молярное соотношение DDQ: (I) составляет, таким образом, 1:100.
Затем раствор подвергают электролизу с постоянным потенциалом, причем на аноде (рабочий электрод) поддерживают потенциал +300 мВ относительно Ag/Ag+ (10 ммоль/л). После прохождения заряда 180 Кл (2,1Ф) (в течение примерно 4 часов) реакцию останавливают. На указанный момент выход М1 согласно анализу ВЭЖХ составил 89% (М1а: M1b=13:87). В результате последующего добавления 2,3 мг (0,01 ммоль) DDQ (и, таким образом, увеличения его доли до 2 мол. %) и последующего электролиза в течение 1 часа, выход повышается согласно анализу ВЭЖХ до 96% соединения формулы (XVII) (М1а (S): M1b (R)=13:87).
Пример 28:
Прямое электрохимическое окисление (XIII)
2,17 г (10 ммоль) тетрафторбората тетраэтиламмония (Et4NBF4) растворяют в 100 мл ацетонитрила. Затем добавляют 378,4 мг (1 ммоль) соединения формулы (XIII).
Затем раствор подвергают электролизу с постоянным потенциалом, причем на аноде (рабочий электрод) поддерживают потенциал +1000 мВ относительно Ag/Ag+ (10 ммоль/л). После прохождения заряда 180 Кл (2,1 Ф) (в течение примерно 2 часов) реакцию останавливают. На указанный момент выход (XVII) составил <50%
Пример 29:
Рацемизация и выделение (XVII) после электрохимического окисления в присутствии медиаторов.
Раствор из примера 26 помещали в 200 г этанола. Нагревали в автоклаве в течение 3 часов при 120°С под давлением и отгоняли примерно 150 г этанола. Давали охладиться до комнатной температуры и дополнительно перемешивали в течение 1 часа при 20°С. Продукт отфильтровывали и промывали небольшим количеством этанола (около 80 г), сушили в течение ночи в вакууме (50°С).
Электро-химическое восстановление
В качестве исходных реагентов для электрохимического восстановления использовали полученные путем окисления соединения формулы ent-I, соединения формулы (XIII) или соответственно из соединения формулы (I) атропизомеры соединения 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,6-нафтиридин-3-карбоксамида, т.е. соединения M1a(S) или M1b(R) и также их смесь (rac.М1).
Конверсию указанных исходных реагентов, а также выход целевого продукта определяли посредством непрерывного отбора проб во время электрохимического восстановления и последующего ВЭЖХ-анализа [ВЭЖХ-метод Е]. Нормированное к 100 соотношение энантиомеров I к ent-(I) дополнительно определяли один раз в конце эксперимента с помощью хирального ВЭЖХ-метода [ВЭЖХ-метод F].
Пример 13
Восстановление соединения M1b(R) (загрузка 0,2 г)
В качестве установки служила трехэлектродная система, состоящая из рабочего электрода [сетчатый электрод Винклера, состоящий из платины/иридия 90/10% (225 меш/см2, диаметр проволоки = 0,12 мм, цилиндрическая геометрия), контрэлектрод [фирмы ALS: витая платиновая проволока, длиной 23 см и диаметром 0,5 мм] и эталонный электрод [фирмы ALS: Ag/Ag+ тип; неводный эталонный электрод с 0,01 М AgNO3 и 0,1 М перхлоратом тетрабутиламмония в ацетонитриле]. Контрэлектрод расположен в стеклянной трубке, закрытой снизу с помощью мембраны. В качестве мембраны использовали ПТФЭ-фильтр [фирмы: Sartorius Stedim Biotech GmbH] с размером пор 0,45 мкм. Источником тока и напряжения был потенциостат фирмы Gamry [Тип: Interface 1000].
0,2 г соединения M1b(R) (0,53 ммоль), полученного в примере 11b, растворяли в 75 г метанола. Кроме того, в качестве токопроводящей соли добавляли 3,2 г тетрафторбората тетраэтиламмония (14,74 ммоль). Химический стакан заполнили указанным раствором. К отделенной мембраной камере контрэлектрода добавляли не содержащий субстрат раствор, состоящий из 0,16 М тетрафторбората тетраэтиламмония в метаноле.
В течение 2 часов регулировали ток на значении -30 мА. Затем ток устанавливали на значении -180 мА. После дальнейших 4 часов определили конверсию >99% и выход in-situ >97%. Соотношение энантиомеров (I): ent (I) было определено как 79:21.
Пример 14
Восстановление соединения M1b(R) (загрузка 0,2 г)
В качестве установки служила трехэлектродная система, описанная в примере 13.
1,0 г соединения M1b(R) (2,66 ммоль), полученного в примере 11b, суспендировали в 80 г метанола, причем субстрат растворялся почти полностью. Кроме того, в качестве токопроводящей соли добавляли 4,5 г тетрафторбората тетраэтиламмония (20,73 ммоль). Химический стакан заполнили указанным раствором. К отделенной мембраной камере контрэлектрода добавляли не содержащий субстрат раствор, состоящий из 0,21 М тетрафторбората тетраэтиламмония в метаноле.
Эксперимент проводили потенциостатически, причем целевой потенциал -3 В относительно эталонного электрода не достигался. В течение всего времени испытания поддерживали максимально возможное напряжение ячейки, которое может быть установлено посредством Interface 1000 («предписанное максимальное напряжение» согласно изготовителю: 22 В). После 6-часового экспериментального периода и прохождения заряда 2650 Кулон (соответствует среднему току 122 мА) определяли конверсию >99% и выход целевого продукта in-situ >97%. Соотношение энантиомеров (I): ent (I) было определено как 76:24.
Пример 15
Восстановление соединения M1b(R) (загрузка 0,2 г)
В качестве установки служила трехэлектродная система, описанная в примере 13.
1,0 г соединения M1b(R) (2,66 ммоль), полученного в примере 11b, суспендировали в 80 г метанола, причем субстрат растворялся почти полностью. Кроме того, в качестве токопроводящей соли добавляли 3,5 г тетрафторбората тетраэтиламмония (16,12 ммоль). Химический стакан заполнили указанным раствором. К отделенной мембраной камере контрэлектрода добавляли не содержащий субстрат раствор, состоящий из 0,16 М тетрафторбората тетраэтиламмония в метаноле.
Эксперимент проводили потенциостатически аналогично примеру 14.
После 4-часового экспериментального периода и прохождения заряда 2193 Кулон (соответствует среднему току 152 мА) эксперимент завершали. К этому моменту конверсия составляла 79%, а выход целевого продукта in situ был равен 79%. Соотношение энантиомеров (I): ent-(I) было определено как 78:22.
Пример 16
Восстановление соединения M1a(S) (загрузка 0,5 г)
В качестве установки служила трехэлектродная система, описанная в примере 13.
0,5 г соединения M1a(S) (1,33 ммоль), полученного в примере 11а, растворяли в 80 г метанола. Кроме того, в качестве токопроводящей соли добавляли 3,5 г тетрафторбората тетраэтиламмония (16,12 ммоль). Химический стакан заполнили указанным раствором. К отделенной мембраной камере контрэлектрода добавляли не содержащий субстрат раствор, состоящий из 0,16 М тетрафторбората тетраэтиламмония в метаноле.
Эксперимент проводили потенциостатически аналогично примеру 14.
После 5-часового экспериментального периода и прохождения заряда 2132 Кулон (соответствует среднему току 118 мА) эксперимент завершали. К этому моменту конверсия составляла 73%, а выход целевого продукта in situ был равен 73%. Соотношение энантиомеров (I): ent-(I) было определено как 22:78.
Пример 17
Получение рацемического (XIII) из рацемического M1(XVII): Восстановление смеси антропизомеров, состоящей из 50% масс. M1b(R) и 50% масс. M1a(S) (загрузка рацемата 0,5 г)
В качестве установки служила трехэлектродная система, описанная в примере 13.
0,5 г рацемата M1a(S) / M1b(R) (1,33 ммоль), полученного в примере 12, растворяли в 80 г метанола. Кроме того, в качестве токопроводящей соли добавляли 3,5 г тетрафторбората тетраэтиламмония (16,12 ммоль). Химический стакан заполнили указанным раствором. К отделенной мембраной камере контрэлектрода добавляли не содержащий субстрат раствор, состоящий из 0,16 М тетрафторбората тетраэтиламмония в метаноле.
Эксперимент проводили потенциостатически аналогично примеру 14.
После 4,5-часового экспериментального периода и прохождения заряда 2500 Кулон (соответствует среднему току 154 мА) эксперимент завершали. К этому моменту конверсия составляла 79%, а выход целевого продукта in situ был равен 79%. Соотношение энантиомеров (I): ent-(I) было определено как 50:50.
Пример 18
Восстановление соединения M1b(R) (загрузка 0,6 г)
В качестве установки служила трехэлектродная система, описанная в примере 13.
0,6 г соединения M1b(R) (1,59 ммоль), полученного в примере 11b, растворяли в смеси растворителей, состоящей из 50 г метанола и 50 г N,N-диметилформамида. Кроме того, в качестве токопроводящей соли добавляли 6 г тетрафторбората тетраэтиламмония (27,64 ммоль). Химический стакан заполнили указанным раствором. К отделенной мембраной камере контрэлектрода добавляли не содержащий субстрат раствор, состоящий из 0,24 М тетрафторбората тетраэтиламмония в метаноле.
Эксперимент проводили потенциостатически аналогично примеру 14.
После 4,5-часового экспериментального периода и прохождения заряда 1187 Кулон (соответствует среднему току 74 мА) конверсия составляла 98% и выход целевого продукта in-situ был равен 95%. Соотношение энантиомеров (I): ent (I) было определено как 83:17.
Пример 19
Восстановление соединения M1b(R) (загрузка 0.6 г)
Установку и условия эксперимента выбирали аналогично примеру 18, за исключением того, что в качестве рабочего электрода использовали пористый углеродный электрод (фирмы: ALS).
Эксперимент проводили потенциостатически аналогично примеру 14.
После 3-часов и 10 минут экспериментального периода и прохождения заряда 494 Кулон (соответствует среднему току 43 мА) конверсия составляла 100% и выход целевого продукта in-situ был равен 97%. Соотношение энантиомеров (I): ent-(I) было определено как 52:48.
Пример 20
Восстановление соединения M1b(R) (проточная ячейка)
В далее следующих примерах вместо ячейки в виде лабораторного стакана использовали проточную ячейку (Micro Flow Cell) фирмы Electrocell. В качестве рабочего электрода использовали титановый электрод с платиновым покрытием. В качестве контрэлектрода служил графит.Анолитную и католитную камеру отделяли друг от друга катионообменной мембраной (тип Fumapem F-9100-PK фирмы Fumatech). Мембрану предварительно погружали в деминерализованную воду и устанавливали влажной. После осуществления сборки ячейки ее промывали метанолом. С помощью шланговых перистальтических насосов [тип: Sci-Q 323; фирмы: Watson Marlow], через обе полуячейки могли быть непрерывно поданы сначала промывочный метанол, а затем реакционные растворы (в каждом случае 6 л/ч).
Источником тока и напряжения был потенциостат фирмы Gamry [Тип: Reference 3000].
1 г соединения M1b(R) (2,66 ммоль), полученного в примере 11b, растворяли в смеси растворителей, состоящей из 4 г метанола и 190 г ДМФА. Кроме того, в качестве токопроводящей соли добавляли 4,5 г тетрафторбората тетраэтиламмония (20,73 ммоль). Этим раствором заполняли контур католита через интегрированный в контур резервуар-накопитель. Аналогичный раствор без соединения M1b(R) заполняли в контур анолита.
В данном эксперименте ток был ограничен макс. 300 мА. После прохождения заряда примерно 1000 Кл (соответствует 4 Ф) конверсия составляла 63%, а после суммарных 3000 Кл (12Ф) конверсия составила >94%. Никаких значимых количеств побочных продуктов не наблюдалось.
Пример 21
Получение рацемического (XIII) из рацемического М1(XVII)
В качестве электролизной ячейки вновь использовали описанную в примере 20 ячейку Micro Flow Cell фирмы Electrocell. В отличие от примера 20 в этом случае растворяли 10 г (26,6 ммоль) соединения rac-M1b, полученного в примере 12, в смеси растворителей, состоящей из 4 г метанола и 190 г ДМФА. Кроме того, в качестве токопроводящей соли добавляли 4,5 г тетрафторбората тетраэтиламмония (20,73 ммоль). Этим раствором заполняли контур католита. Аналогичный раствор без исходного реагента заполняли в контур анолита.
После прохождения заряда 30000 Кл (12 Ф) электрохимическое восстановление останавливали. Выход in situ rac-(XIII), определенный ВЭЖХ (метод Е), составлял 95%. Затем раствор католита подавали на обработку.
Выделение rac-(XIII): Сначала в значительной степени отгоняют растворитель, а затем продукт переводят в осадок посредством водного осаждения (добавление воды), отфильтровывают и сушат.
Полученный таким образом сырой продукт может быть перекристаллизован из этанола или ТГФ и вновь подвергнут разделению на энантиомеры при помощи SMB.
Пример 30
Получение рацемического (XIII) из рацемического M1(XVII): Восстановление смеси антропизомеров, состоящей из 50% масс. M1b(R) и 50% масс. M1a(S) (загрузка рацемата 10 г)
В далее следующих примерах вместо ячейки в виде лабораторного стакана использовали проточную ячейку (Micro Flow Cell: площадь поверхности электрода 10 см2) фирмы Electrocell.
В качестве рабочего электрода использовали титановый электрод с платиновым покрытием. В качестве контрэлектрода служил графит. Анолитную и католитную камеру отделяли друг от друга катионообменной мембраной (тип Nafion® N-424 фирмы DUPONT). Мембрану предварительно погружали в деминерализованную воду и устанавливали влажной. После осуществления сборки ячейки ее промывали смесью растворителей, состоящей из 20% масс. метанола и 80% масс. ДМФА. С помощью шланговых перистальтических насосов [тип: Sci-Q 323; фирмы: Watson Marlow], через обе полуячейки могли быть непрерывно поданы сначала промывочный раствор метанол/ДМФА, а затем реакционные растворы (в каждом случае 5 кг/ч). Посредством отдельно подключенного контура охлаждения в обоих растворах электролитов (контур анолита и католита) поддерживали температуру 20°С. Источником тока и напряжения был потенциостат фирмы Gamry [Тип: Reference 3000].
10 г соединения rac.M1(R) (2,66 ммоль), полученного в примере 12b, растворяли в смеси растворителей, состоящей из 21,4 г метанола и 85,6 г ДМФА. Кроме того, добавляли 1,25 г тетрафторбората тетраэтиламмония (5,76 ммоль) в качестве токопроводящей соли и 1,45 г уксусной кислоты (24,17 ммоль). Этим раствором заполняли контур католита через интегрированный в контур резервуар-накопитель. Аналогичный раствор без соединения rac.M1(R) заполняли в контур анолита (масса анолита в начале эксперимента = 358,7 г). Эксперимент проводили гальваностатически. Постоянный ток составлял 350 мА. Через 20 часов эксперимент останавливали и сливали оба контура полуячеек. Конверсия соединения rac.М1 составляла 99%. Выход in situ целевого компонента (XIII) через 20 часов составил >98%. Никаких значимых количеств побочных продуктов не наблюдалось. Селективность продукта (XIII) составляет около 99%. Концентрация продукта после окончания эксперимента составляла около 37 мг/г. Разбавление происходит из-за переноса растворителя из анолита в католит (общая масса католита и анолита после опустошения ячеек = 264 и 214 г соответственно).
Выделение rac-(XIII): После удаления смеси растворителей (ДМФА/МеОН) и токопроводящей соли продукт получали с высоким выходом и чистотой.
Пример 31
Получение рацемического (XIII) из рацемического M1(XVII): Восстановление смеси антропизомеров, состоящей из 50% масс. M1b(R) и 50% масс. M1a(S) (загрузка рацемата 10 г)
Использовали описанную в примере 30 электролизную ячейку, а также описанную там экспериментальную установку.
В отличие от примера 30 растворяли 10 г соединения rac.M1(R) (26,6 ммоль), полученного в примере 12b, в смеси растворителей, состоящей из 16,6 г метанола и 66,4 г ДМФА. Кроме того, добавляли 0,97 г тетрафторбората тетраэтиламмония (4,47 ммоль) в качестве токопроводящей соли и 1,09 г уксусной кислоты (18,1 ммоль). Этим раствором заполняли контур католита через интегрированный в контур резервуар-накопитель. Аналогичный раствор без соединения rac.M1(R) заполняли в контур анолита (масса анолита в начале эксперимента = 282 г).
Эксперимент проводили гальваностатически. Постоянный ток составлял 400 мА.
Примерно через 6 ч экспериментального периода католит становился мутным, а в резервуаре-накопителе наблюдалось образование белого осадка. Через 10 часов эксперимент останавливали. Образовавшийся осадок отфильтровывали (1,7 г) и без дальнейших стадий очистки исследовали с помощью ВЭЖХ-анализа. Анализ рацемического (XIII) показал >99,6% по площади пиков. В оставшемся фильтрате соотношение между целевым продуктом (XIII) и субстратом (rac.M1 (XVII)) составляло 89:10% по площади пиков. Никаких значимых количеств побочных продуктов не наблюдалось. Селективность продукта (XIII) составляет около 99%.
Концентрация продукта (соединения XIII) в фильтрате составляла около 43 мг/г. Общая масса католита и анолита после опустошения ячеек составляла 174 и 197 г соответственно.
Пример 32
Получение рацемического (XIII) из рацемического M1(XVII): Восстановление смеси антропизомеров, состоящей из 50% масс. M1b(R) и 50% масс. M1a(S) (загрузка рацемата 36 г)
В качестве электролизной ячейки в данном примере использовали многоцелевую ячейку (МРС, площадь поверхности электрода 100 см2) фирмы Electrocell. В качестве рабочего электрода использовали титановый электрод с платиновым покрытием. В качестве контрэлектрода служил графит. Анолитную и католитную камеру отделяли друг от друга катионообменной мембраной (тип Nafion® N-424 фирмы DUPONT). Мембрану предварительно погружали в деминерализованную воду и устанавливали влажной. После осуществления сборки ячейки ее промывали смесью растворителей, состоящей из 20% масс. метанола и 80% масс. ДМФА. С помощью циркуляционных насосов [тип: Лабораторный реакционный смеситель HMR 050; фирмы: Fink] и расходомера Cori-Flow (фирмы Bronkhorst) через обе полуячейки могли быть непрерывно поданы сначала промывочный раствор метанол/ДМФА, а затем реакционные растворы (в каждом случае 50 кг/ч). Многоцелевая ячейка также была подключена к контуру охлаждения, который регулировался посредством криостата (фирмы Julabo, тип: FP45). Температура анолита и католита составляла 22°С.
Источником тока и напряжения был потенциостат фирмы Delta Elektronika [Тип: ES030-10].
После промывки контуров анолита и католита в течение по меньшей мере 15 минут в резервуар-накопитель помещали следующие растворы:
Католит: 36 г (95,7 ммоль) соединения rac.M1 (XVII), полученного в примере 12b, растворяли в смеси растворителей, состоящей из 100 г метанола, 400 г ДМФА, 6 г (27,64 ммоль) тетрафторбората тетраэтиламмония (Et4NBF4) и 5 г (83,3 ммоль) уксусной кислоты.
Анолит: Здесь использовали не содержащий субстрат раствор, состоящий из 250 г метанола, 1000 г ДМФА, 15 г (69,1 ммоль) токопроводящей соли (Et4NBF4) и 12,5 г (208,3 ммоль) уксусной кислоты.
Эксперимент проводили гальваностатически. Постоянный ток составлял 3 А. Через 10 часов эксперимент останавливали и сливали оба контура полуячеек. Конверсия соединения rac.М1 составляла 95,7% (ВЖЭХ, % по площади пиков). Выход in situ целевого компонента (XIII) через 10 часов составил 95,3% (ВЖЭХ, % по площади пиков). Никаких значимых количеств побочных продуктов не наблюдалось (селективность целевого продукта >99,5%). Концентрация продукта после окончания эксперимента составляла около >2,6% масс. Разбавление происходит из-за переноса растворителя из анолита в католит. (общая масса католита и анолита после опустошения ячеек составляла 1296 г и 482 г соответственно).
Выделение rac-(XIII): После удаления смеси растворителей (ДМФА/МеОН) и токопроводящей соли продукт получали с высоким выходом и чистотой. Полученный сырой продукт может быть дополнительно перекристаллизован из этанола или ТГФ и вновь подвергнут разделению на энантиомеры при помощи SMB.
Пример 22
Монокристаллический ренгеноструктурный анализ соединения формулы M1b(R): (R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,6-нафтиридин-3-карбоксамид
Аналитический метод: Монокристаллический ренгеноструктурный анализ
Измеренный кристалл: бесцветный блок, 0,40×0,20×0,20 мм3
Экспериментальная часть:
Определение кристаллической структуры проводили с использованием дифрактометра (Oxford Diffraction, серия Xcalibur), оборудованного CCD плоскопанельным детектором (модель Ruby), так называемой «sealed tube» рентгеновской трубкой с CuKa-излучением, осмиевым отражателем в качестве монохроматора и устройством охлаждения Cryojet для измерений при низкой температуре (Т=100 K).
Угол сбора данных 360° (Омега и Пси сканирование). Использованные программы: Сбор данных и их обработка с помощью Crysalis (Oxford Diffraction 2007). Расчет кристаллической структуры был реализован прямыми методами, как описано в SHELXTL Версии 6.10 (Sheldrick, Университет Гёттингена (Германия), 2000) и визуализирован с использованием ХР-программы. Затем отсутствующие атомы были локализованы при помощи дифференциального синтеза Фурье и добавлены в список атомов. Уточнение по методу наименьших квадратов на F2 выполнялось со всеми измеренными интенсивностями и с использованием программы SHELXTL Версия 6.10 (Sheldrick, Университет Геттингена (Германия), 2000). Все атомы, отличающиеся от водорода, были уточнены, включая параметры анизотропного смещения.
Параметры кристалла и уточнение структуры соединения формулы M1b (R): (R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,6-нафтиридин-3-карбоксамид
Идентифицирующий код: M1b
Общая формула: С21 Н20 N4 O3
Молекулярная масса: 376.41
Температура: 100 K
Длина волны: 1.54178 Å
Кристаллическая система: Орторомбическая
Пространственная группа: Р2(1)2(1)2(1)
Параметры кристаллической решетки: а=9.70950 (10) Å решетка
b=10.67390(10) Å β=90°.
с=18.9480(2) Å γ=90°.
Объемы: 1963.74(3) Å3
Z 4
Удельная плотность (рассчитанная): 1,273 мг/м3
Коэффициент поглощения: 0,714 мм-1
F(000) 792
Размеры кристалла: 0,40×0,20×0,20 мм3
Тэта-область для сбор данных: от 4.67 до 5.66°.
Диапазон индексов: -11≤h≤9, -12≤k≤12, -19≤I≤22
Зафиксированные отражения: 15493
Независимые отражения: 3367 [R(int)=0.0230]
Полнота сбора данных при тета = 65.66° 99.5%
Поправка на поглощение: Crysalis
Метод уточнения: Полноматричный метод наименьших квадратов по F2
Данные/ограничения/параметры: 3367/0/257
Критерий согласия по F2: 1,048
Конечные значения R: [I>2сигма(I)] R1=0.0242, wR2=0.0636
Значения R (все данные): R1=0,0249, wR2=0,0641
Абсолютный структурный параметр: -0.18(13)
Максимальная и минимальная деформационная плотность: 0.142 и -0.139 е.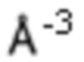
Ренгеноструктурный анализ:
Рентгеноструктурный анализ показал, что если кольцевая система 1,6-нафтиридин-3-карбоксамида лежит на плоскости изображения, 4-циано-2-метоксифенильный заместитель находится перпендикулярно к ней, причем метоксигруппа находится за плоскостью изображения.
Определение абсолютной конфигурации
Н.D. Flack, Acta Cryst., 1983, А39, 876-881
Н.D. Flack, G. Bernardinelli, Acta Cryst., 1999, A55, 908-915
H.D. Flack, G. Bernardinelli, J. Appl. Cryst., 2000, 33, 1143-1148.
Таким образом, соединение формулы M1b(R) имеет абсолютную конфигурацию R (Ra)
Абсолютная конфигурация названа в соответствии с правилами Кана - Ингольда - Прелога для аксиально хиральных соединений
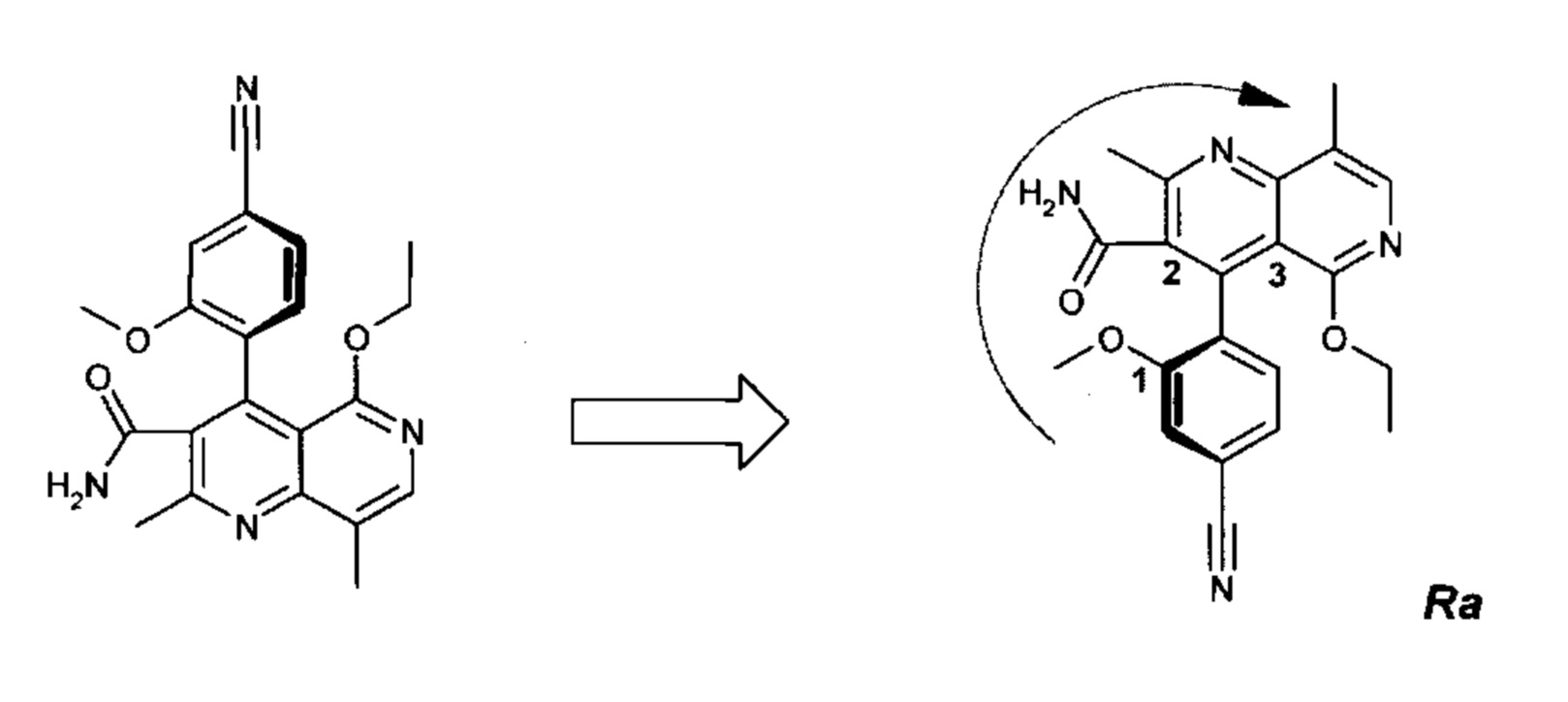
Пример 23
Определение абсолютной конфигурации Mb-серии путем корреляции СР-спектров
(См. фигуру 7)
Физико-химические характеристики соединения формулы (I) в кристаллической форме модификации I
Соединение формулы (I) в кристаллической форме модификации I плавится при 252°С, ΔН=95-113 Дж*г-1 (скорость нагрева 20 Kмин-1).
Снижение температуры плавления наблюдалось в зависимости от скорости нагрева.
Температура плавления снижается при меньшей скорости нагрева (например, 2 Kмин-1), так как происходит разложение.
Никаких дополнительных фазовых переходов не наблюдалось. До температуры 175°С наблюдали потерю массы примерно 0,1%.
Стабильность и хранение в присутствии влаги
Образцы соединения формулы (I) в кристаллической форме модификации I хранили при 85% и 97% относ. влажности воздуха (25°С). Образцы через 12 месяцев оценивали с помощью дифференциальной сканирующей калориметрии (DSC), термогравиметрического анализа (TGA) и порошковой рентгенодифрактометрии (XRPD). Через 12 месяцев в обоих случаях обнаруживается изменение массы <0,1%. Это означает, что соединение формулы (I) в кристаллической форме модификации I не показывает значительного поглощения воды в указанных условиях хранения. Согласно DSC, TGA и XRPD, нет различий с соединением формулы (I) в кристаллической форме модификации I.
ВЭЖХ-условия/методы
Метод А
YMC Hydrosphere С18
150*4,6 мм, 3,0 мкм
25°С, 1 мл/мин, 270 нм, 4 нм
0' : 70% трифторуксусная к-та 0,1%*; 30% ацетонитрил
17' : 20% трифторуксусная к-та 0,1%*; 80% ацетонитрил
18 ' : 70% трифторуксусная к-та 0,1%*; 30% ацетонитрил
*: трифторуксусная к-та в воде
Метод В
YMC Hydrosphere С18
150*4,6 мм, 3,0 мкм
25°С, 1 мл/мин, 255 нм, 6 нм
0' : 90% трифторуксусная к-та 0,1%*; 10% ацетонитрил
20' : 10% трифторуксусная к-та 0,1%*; 90% ацетонитрил
18' : 10% трифторуксусная к-та 0,1%*; 90% ацетонитрил
Метод С
Nucleodur Gravity С18
150*2 мм, 3,0 мкм
35°С, 0,22 мл/мин, 255 нм, 6 нм
Раствор А: 0,58 г гидрофосфата аммония и 0,66 г дигидрофосфата
аммония в 1 л воды (аммоний-фосфатный буфер, рН 7,2)
Раствор В: Ацетонитрил
0' : 30% В; 70% А
15' : 80% В; 20% А
25' : 80% В; 20% А
Метод D
Длина колонки: 25 см
Внутренний диаметр: 4,6 мм
Наполнитель: Chiralpak IA, 5 мкм.
Реагенты: 1. Ацетонитрил, для ВЭЖХ
2. Метил-трет-бутиловый эфир (МТБЭ), ч.д.а.
Испытуемый раствор: Образец растворяют в ацетонитриле в концентрации 1,0 мг/мл.
(например, около 25 мг образца точно взвешивают, и растворяют в ацетонитриле до 25,0 мл).
Элюент А. Ацетонитрил
В. Метил-трет-бутиловый эфир (МТБЭ), ч.д.а.
Расход 0,8 мл/мин
Температура термостата колонки 25°С
Длина волны детектирования: 255 нм
Ширина полосы: 6 нм
Объем пробы 5 мкл
Смесь элюентов А и В готовят в объемном соотношении 90:10.
Продолжительность хроматограммы 30 мин
Время удерживания(RT)/относительное время удерживания (RRT):
(4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (1) около 11 мин. RRT: 1,00
(4R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (1) около 9 мин. RRT: 0,82
Метод Е
YMC Hydrosphere С18
150*4,6 мм, размер частиц 3 мкм
25°С, 1 мл/мин, обычное начальное давление: ок. 160 бар
Измерительная длина волны: 255 нм, Ширина полосы: 6 нм
Градиент:
0' : 90% муравьиная к-та 0,1%*; 10% ацетонитрил
20' : 10% муравьиная к-та 0,1%*; 90% ацетонитрил
25' : 90% муравьиная к-та 0,1%*; 10% ацетонитрил
*: муравьиная к-та в воде
Время удерживания:
Соединение (I) или соответственно ent-(I): ок. 9,9 мин
Соединение М1а или соответственно M1b: ок. 15,5 мин
Метод F
Chiralpak IA
150*4,6 мм, размер частиц 5 мкм
25°С, 0,8 мл/мин
Измерительная длина волны: 255 нм, Ширина полосы: 6 нм
Подвижная фаза: Смешанные в объемном соотношении 90:10
ацетонитрил + метил-трет-бутиловый эфир (МТБЭ)
Время удерживания:
Соединение формулы M1b(R): ок. 5,1 мин
Соединение формулы M1a(S): ок. 5,5 мин
Соединение формулы (I): ок. 8,6 мин
Соединение формулы ent-(I): ок. 10,8 мин
Описание чертежей:
Фигура 1: Термическая рацемизация соединения формулы ent-(I) в 1-бутаноле без добавления каталитического количества кислоты.
Фигура 2: Термическая рацемизация соединения формулы ent-(I) в 1-бутаноле с добавлением и без добавления каталитического количества кислоты.
Фигура 3: Общие типы электрохимических ячеек. Ячейка в виде лабораторного стакана, "Н"-ячейка и проточная ячейка в виде фильтр-пресса.
Фигура 4: Схема реакции электрохимического окисления в присутствии медиатора соединения ent-(I) в (XVII) посредством DDQ.
Фигура 5: Циклическая вольтамперометрия DDQ, дигидропиридина (DHP) и соединения формулы ent-(I) и смеси DDQ : дигидропиридин (V) 1:10 согласно примерам 24 и 25
Фигура 6: Изменение измеренного методом ВЭЖХ исходного реагента DHP ent-(I) и продукта PYR (XVII) в зависимости от времени в соответствии с примером 26. Линии представляют собой расчетное значение, основанное только на потоке электронов и 100% электрической эффективности.
Фигура 7: Спектр кругового дихроизма (CD-спектр) соединения формулы M1b(R) (в ацетонитриле)
Фигура 8: Кристаллическая структура соединения формулы M1b (R): (R)-4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,6-нафтиридин-3-карбоксамида
Фигура 9: Кристаллическая структура соединения формулы M1b (R): (R)-4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,6-нафтиридин-3-карбоксамида
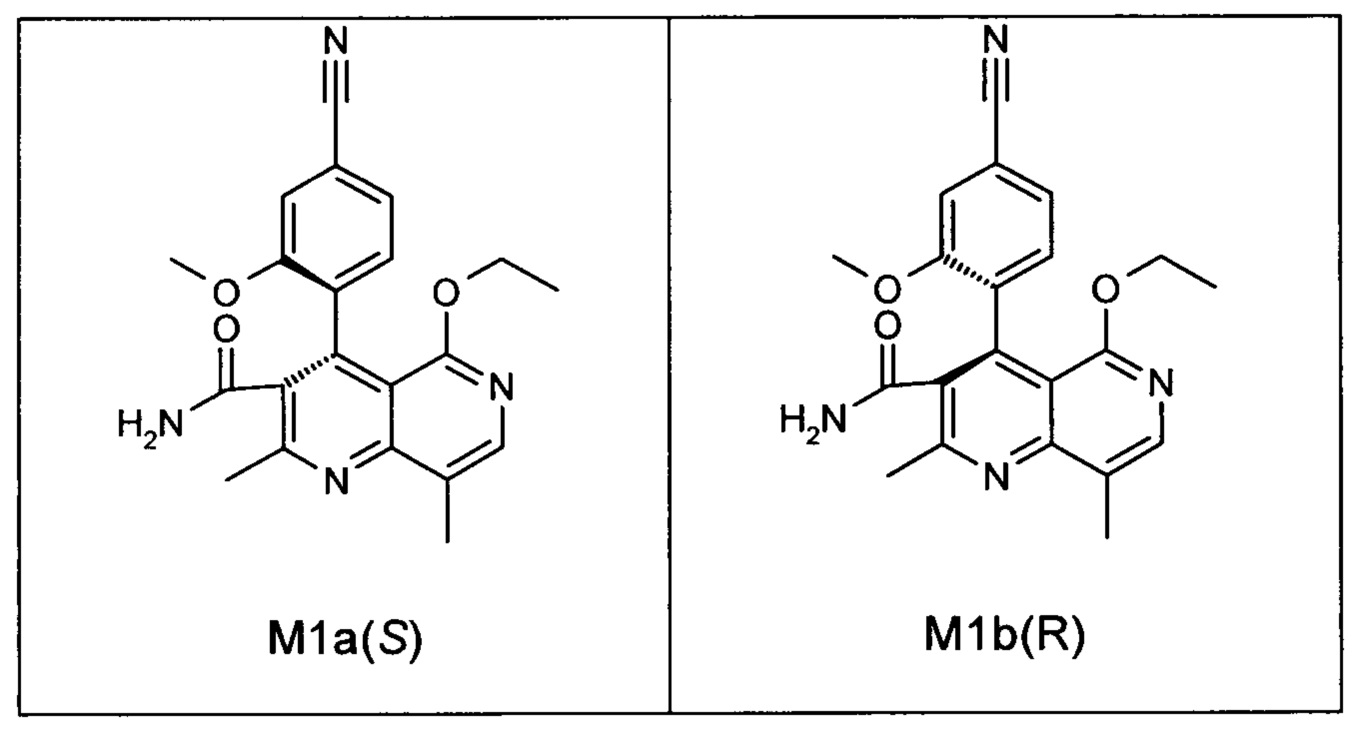
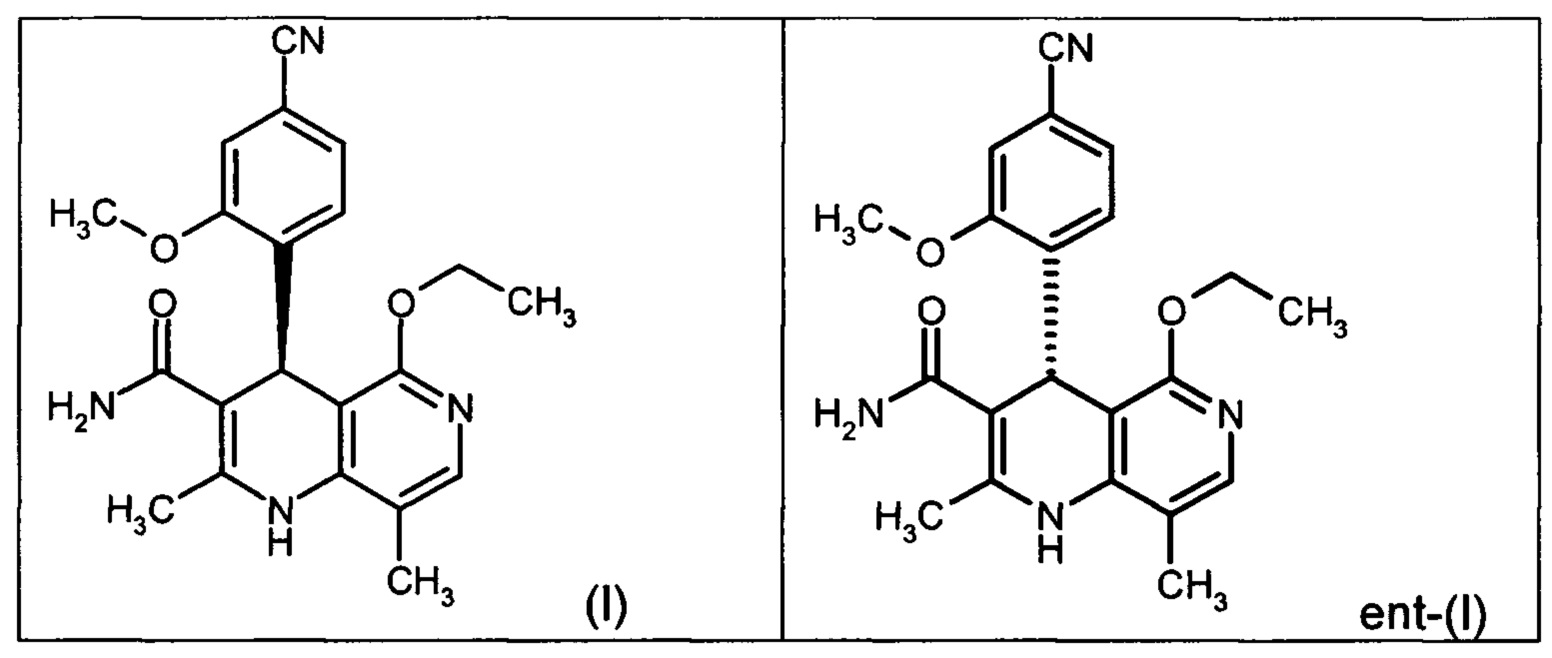
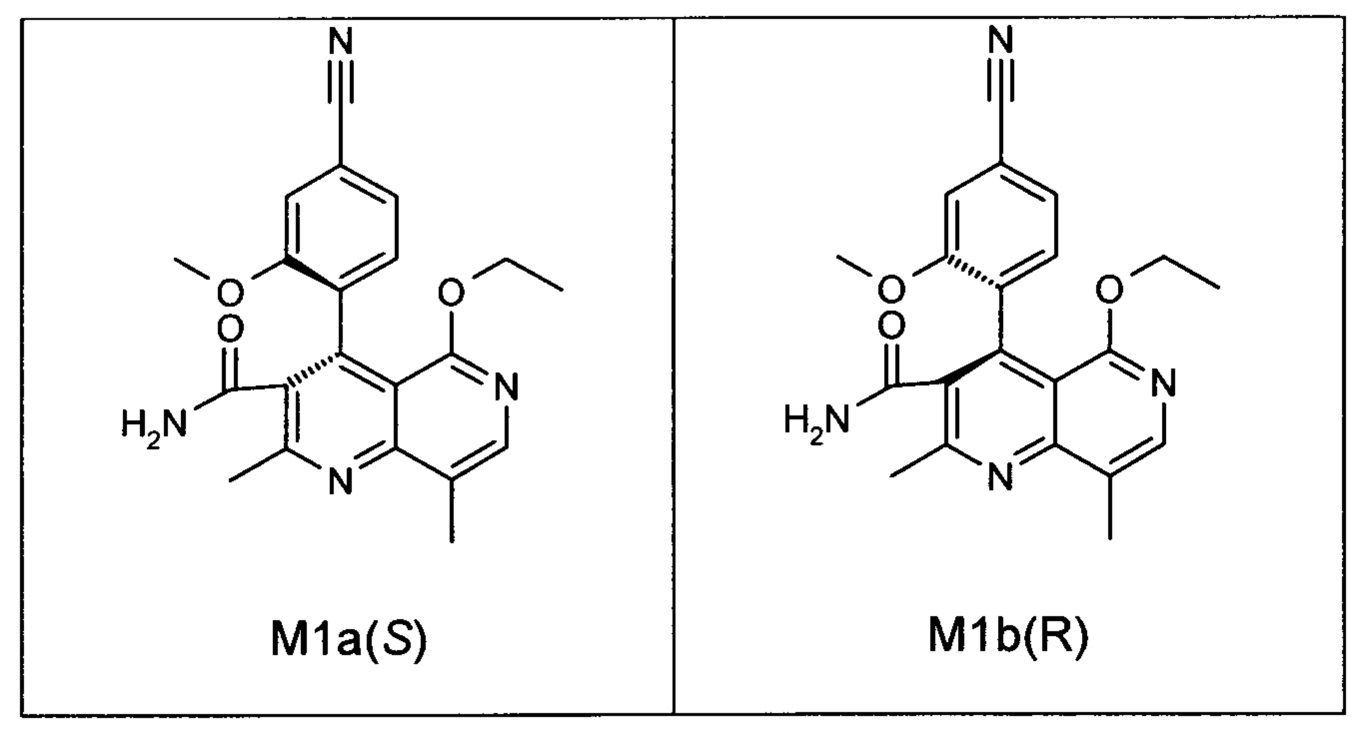

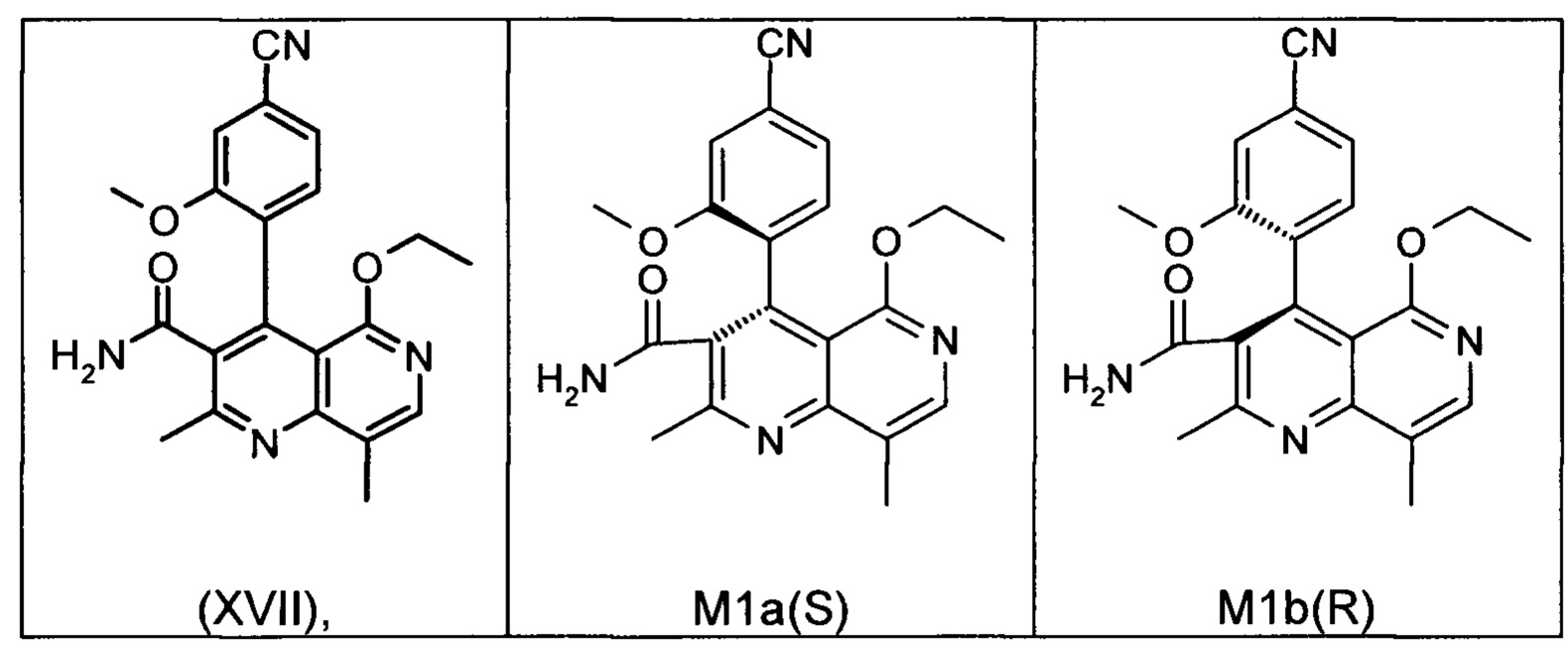
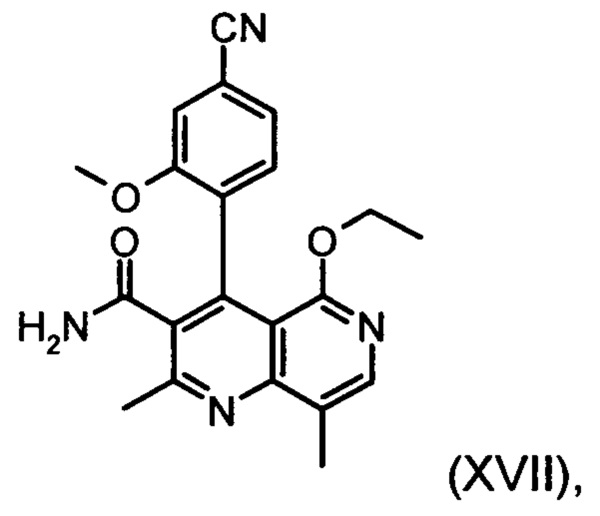
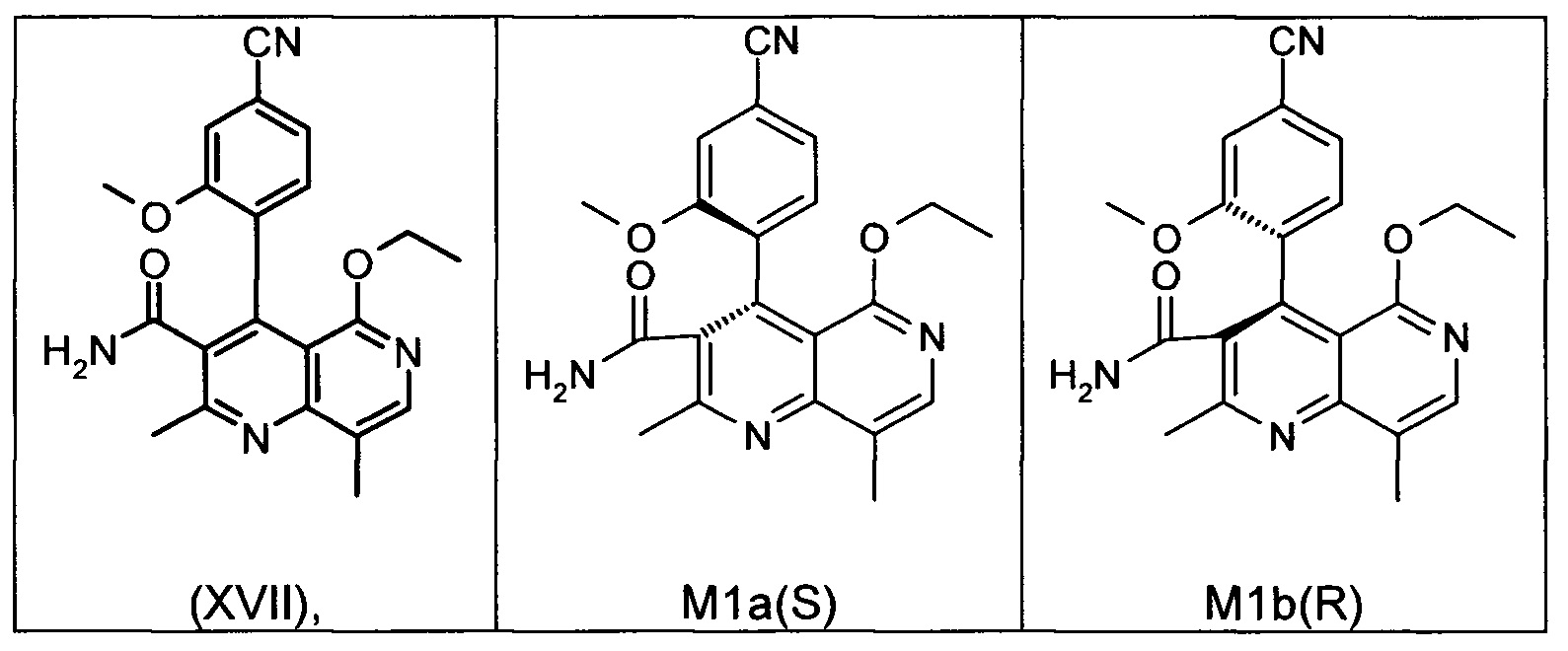
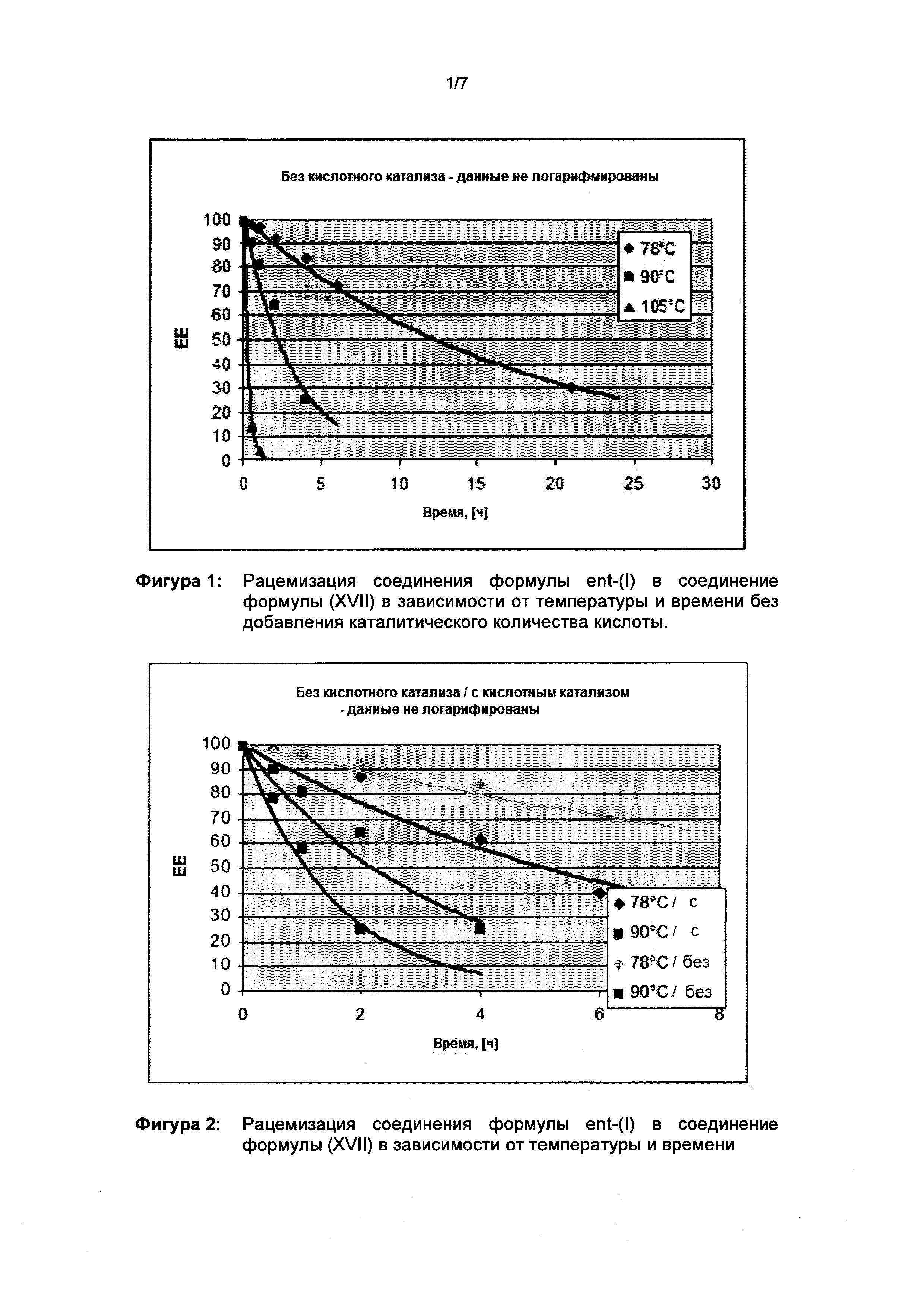
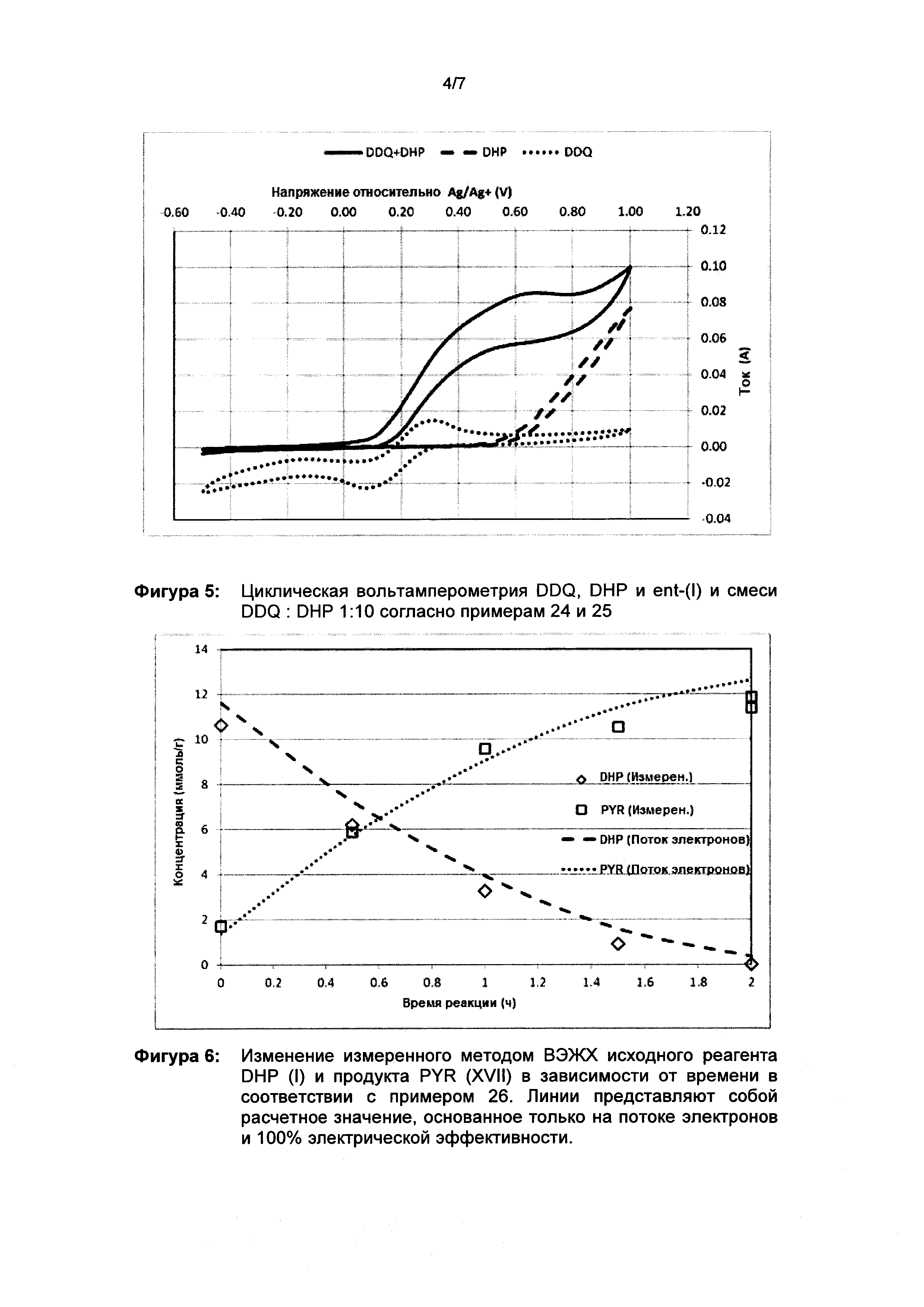
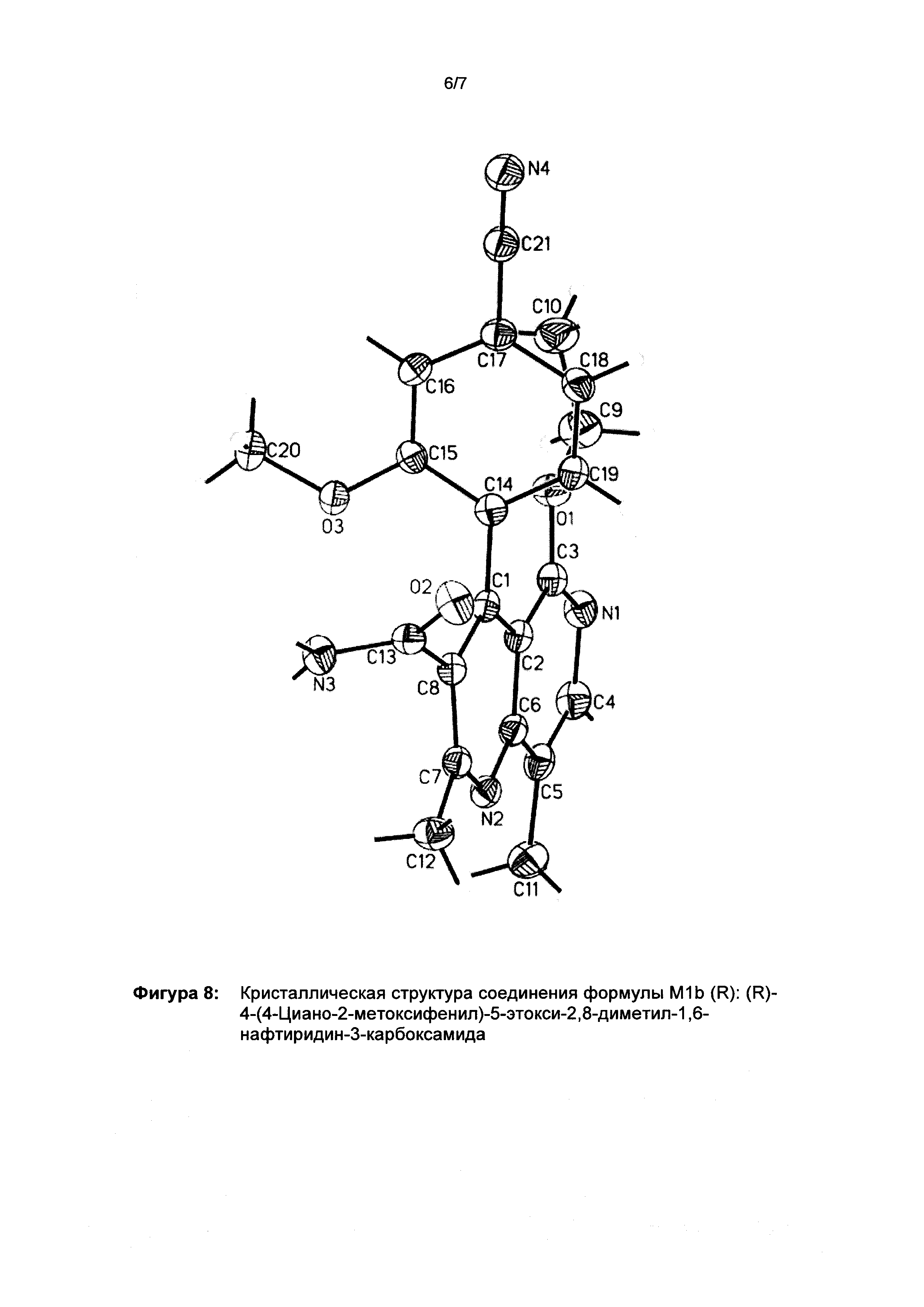
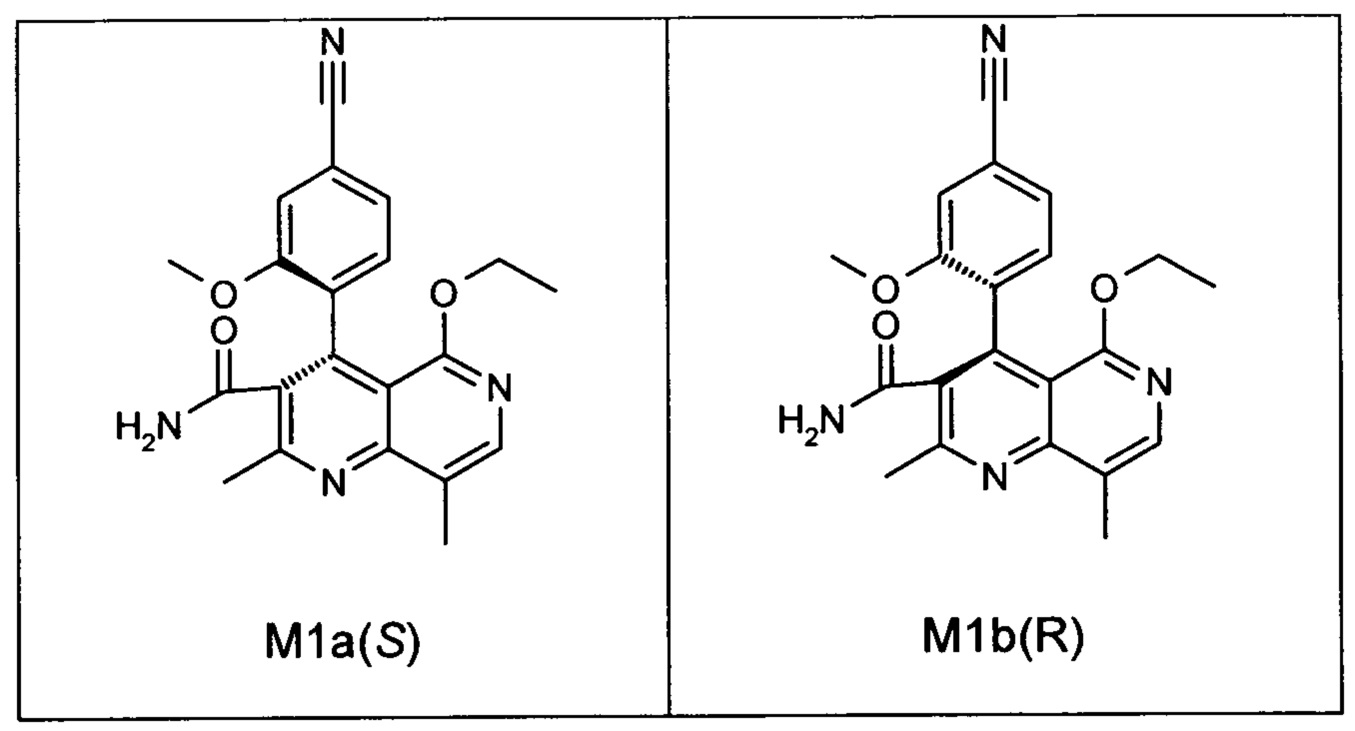
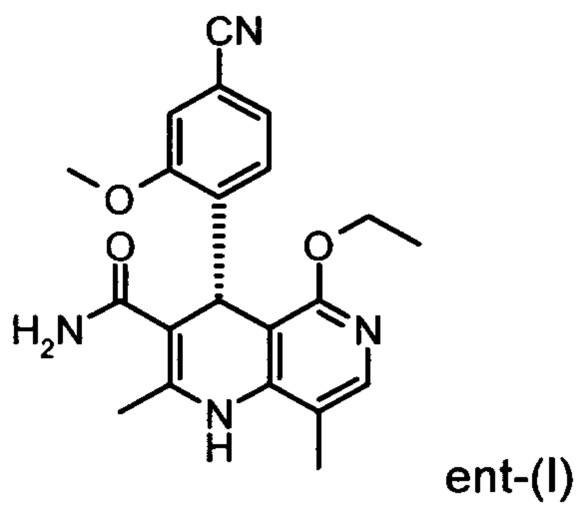
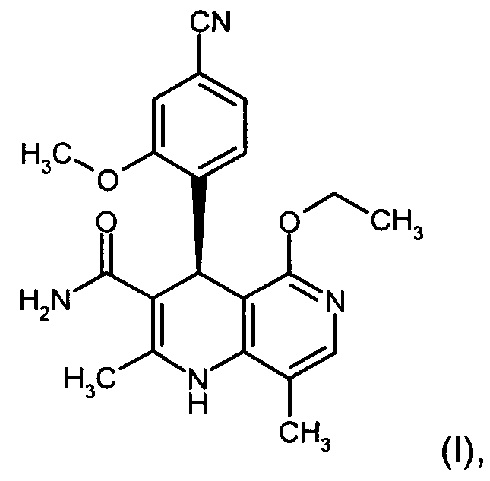
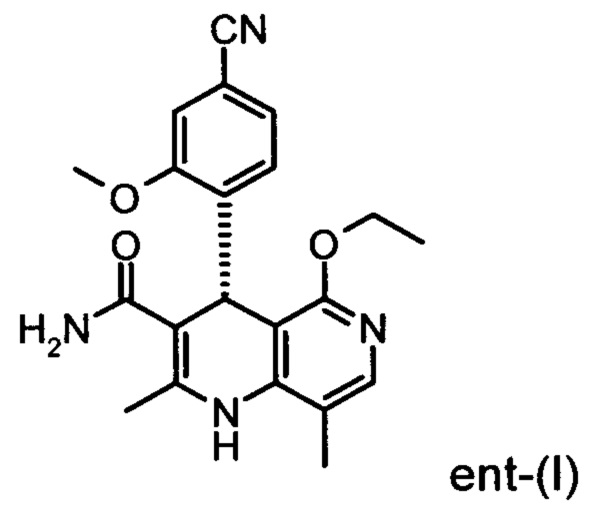
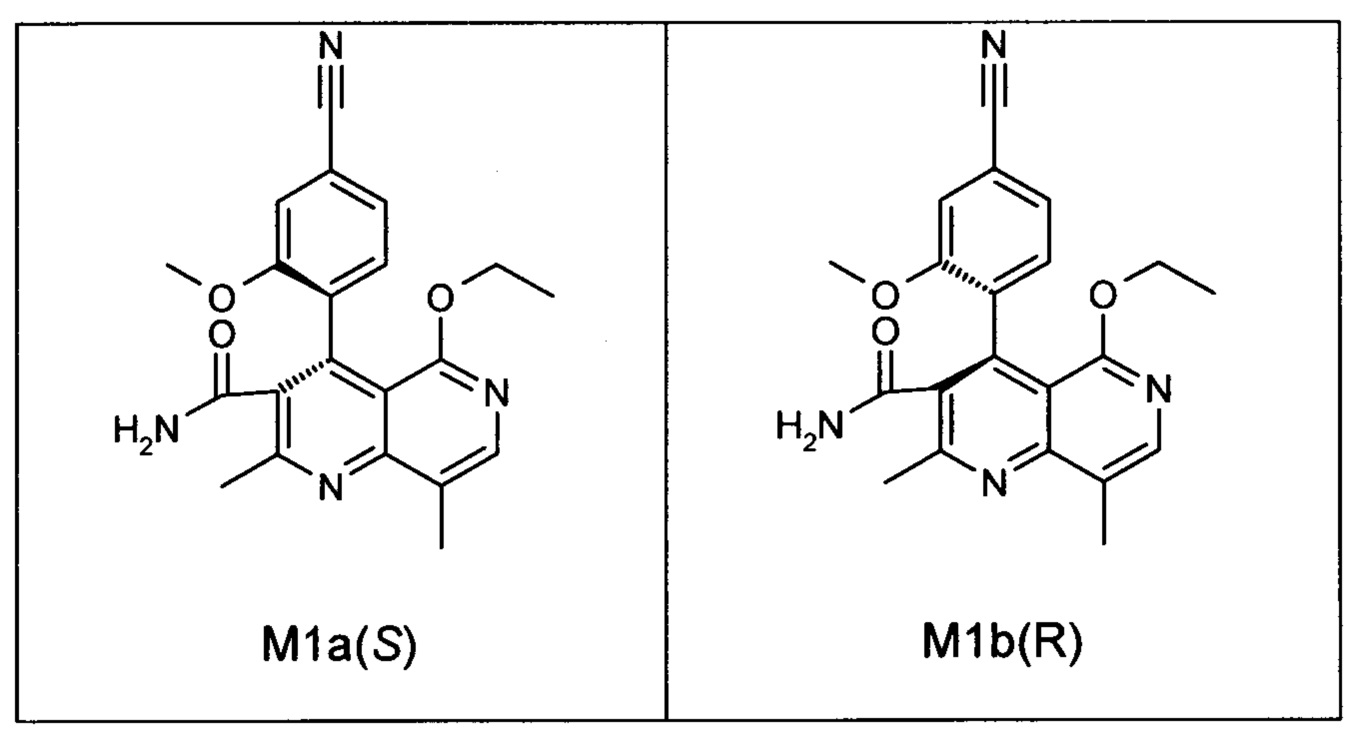
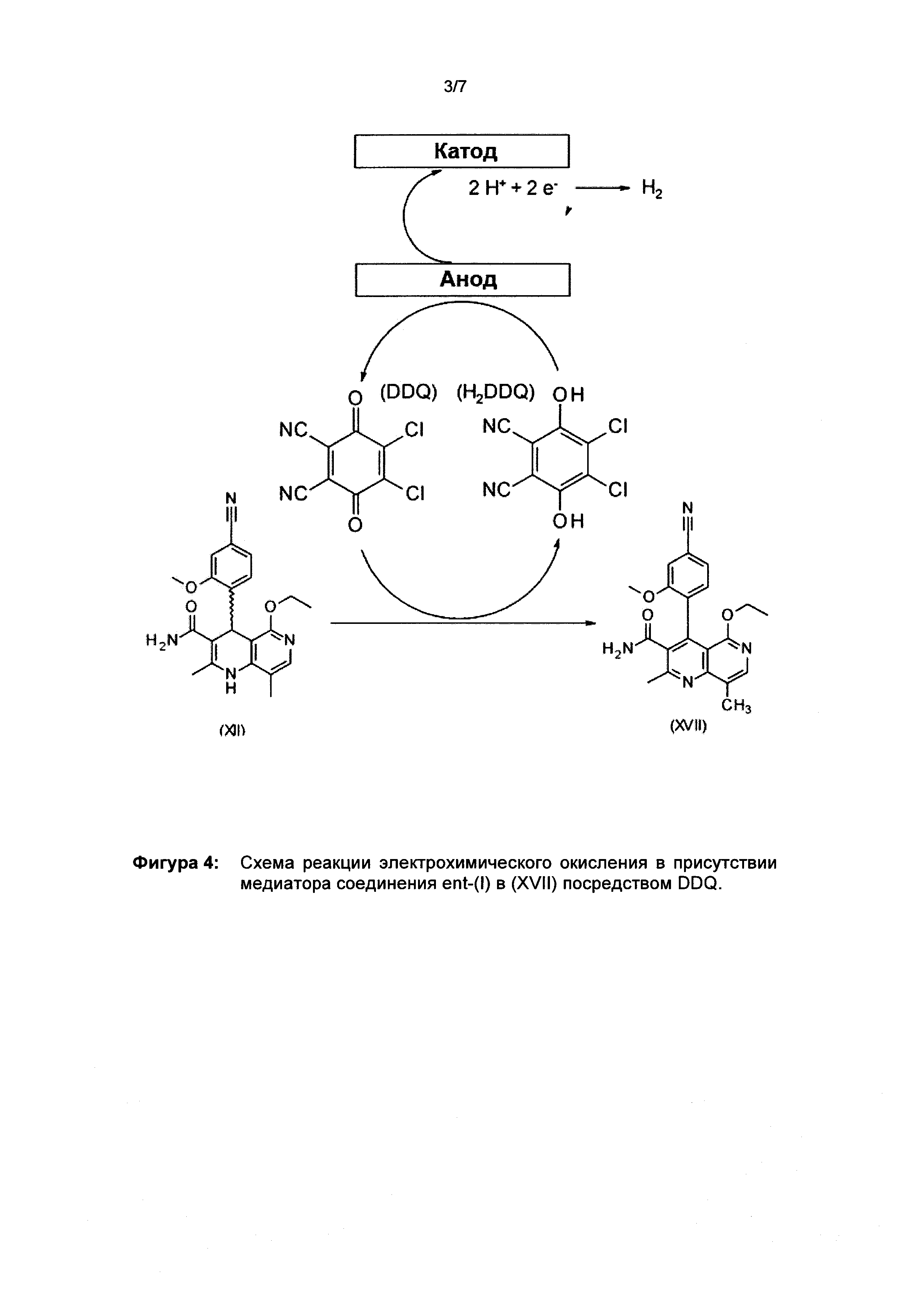
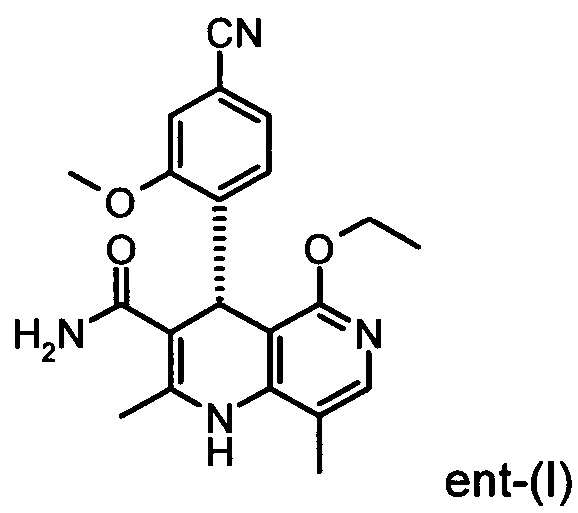
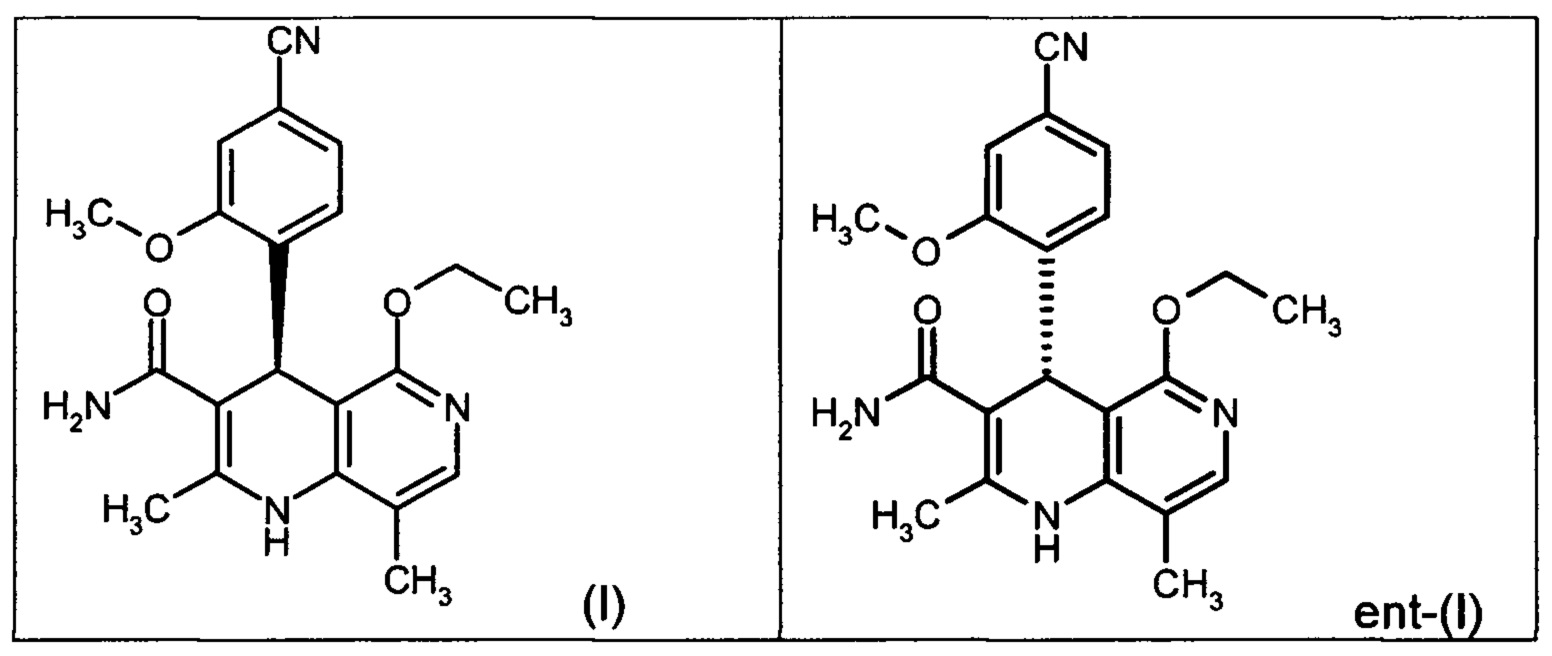
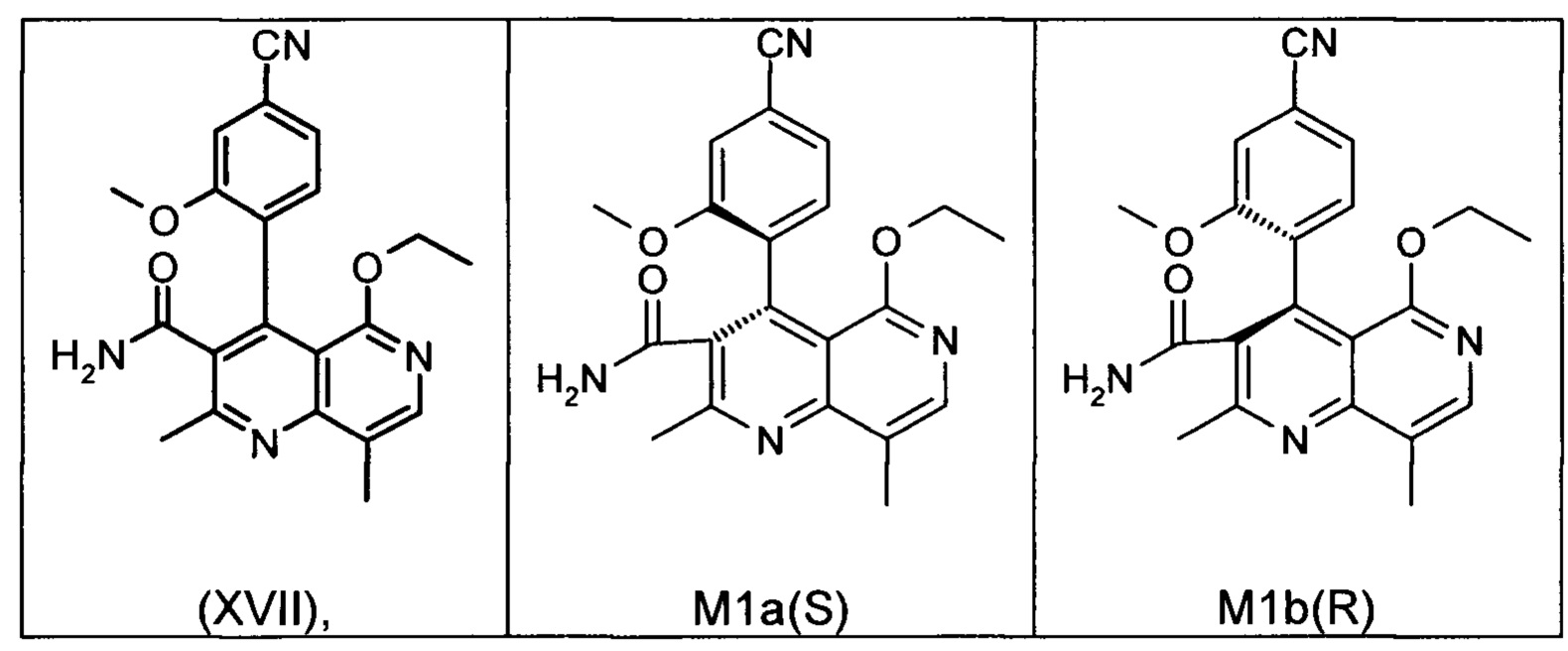
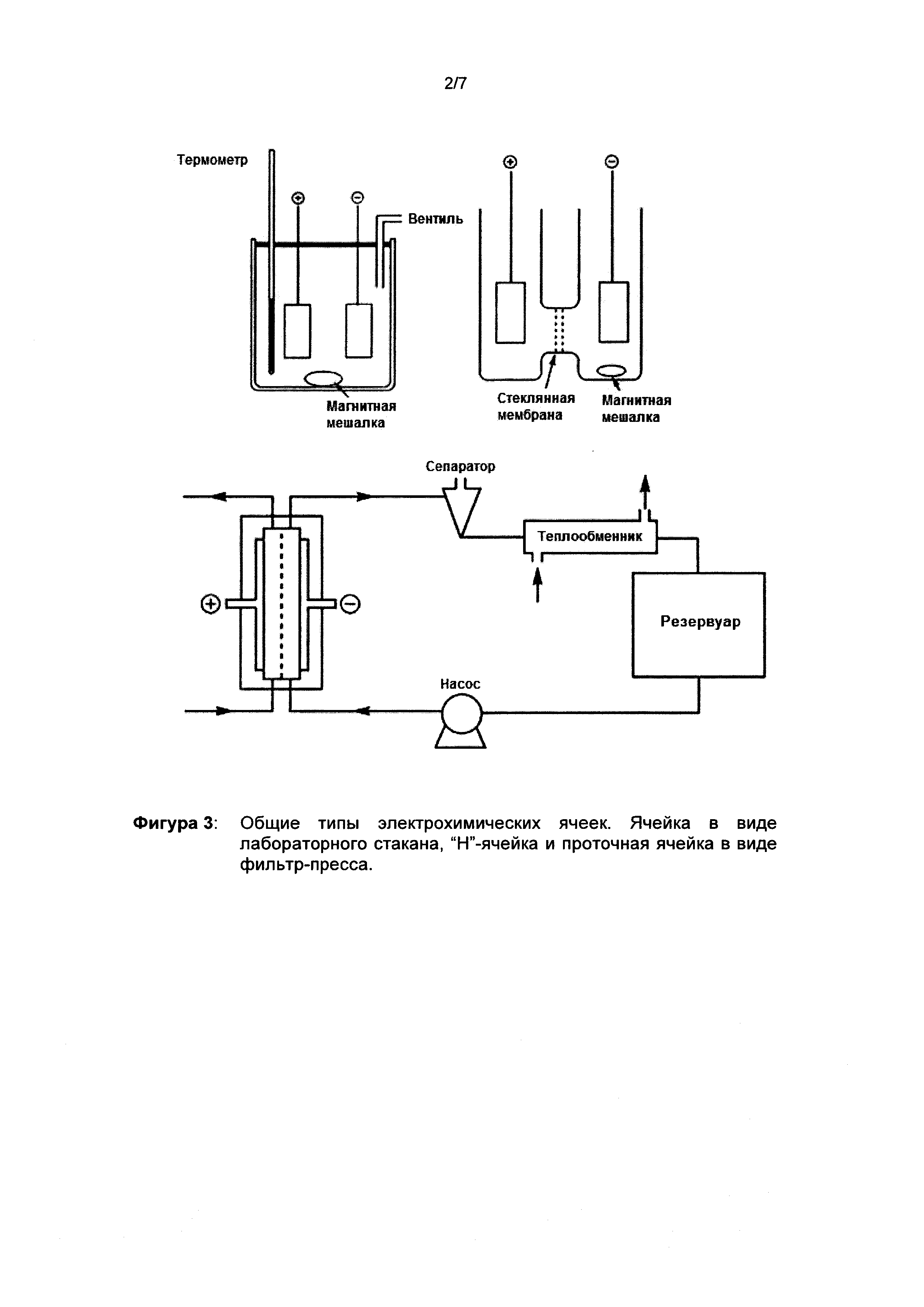
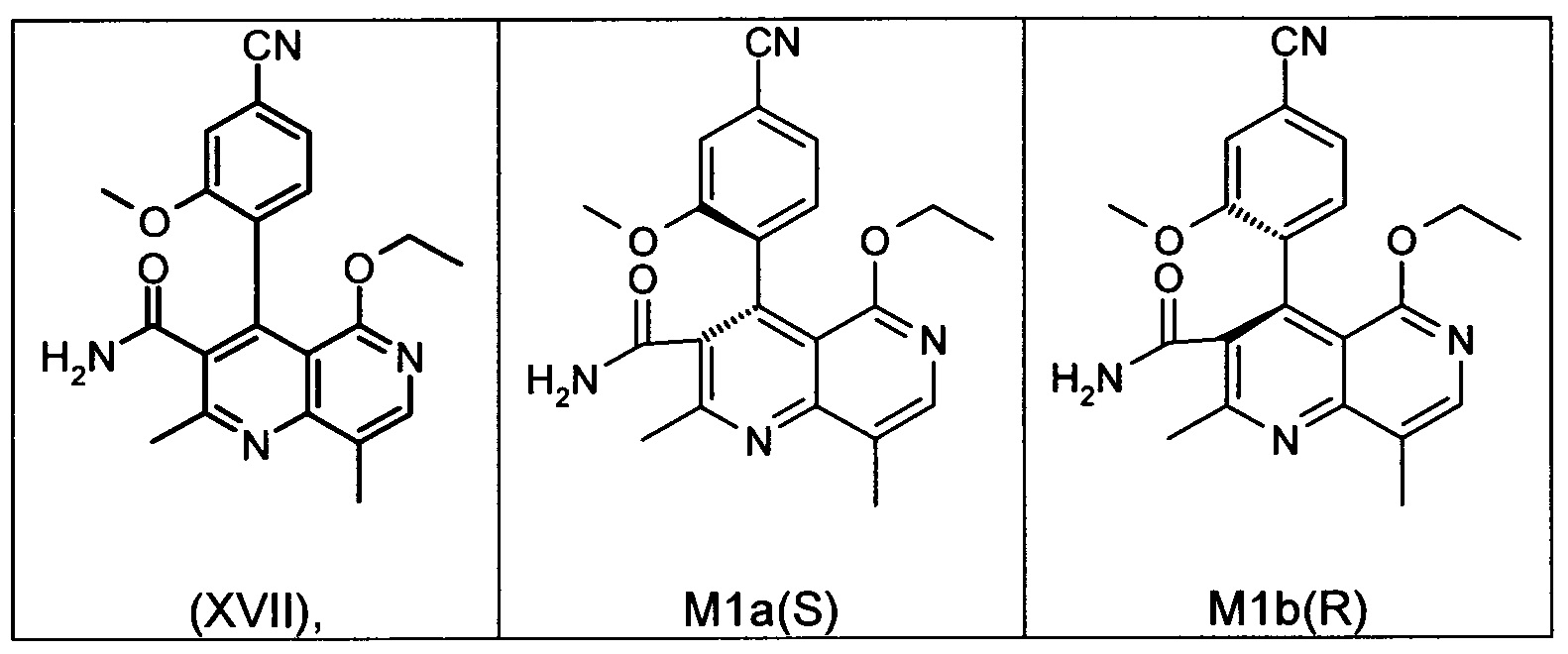
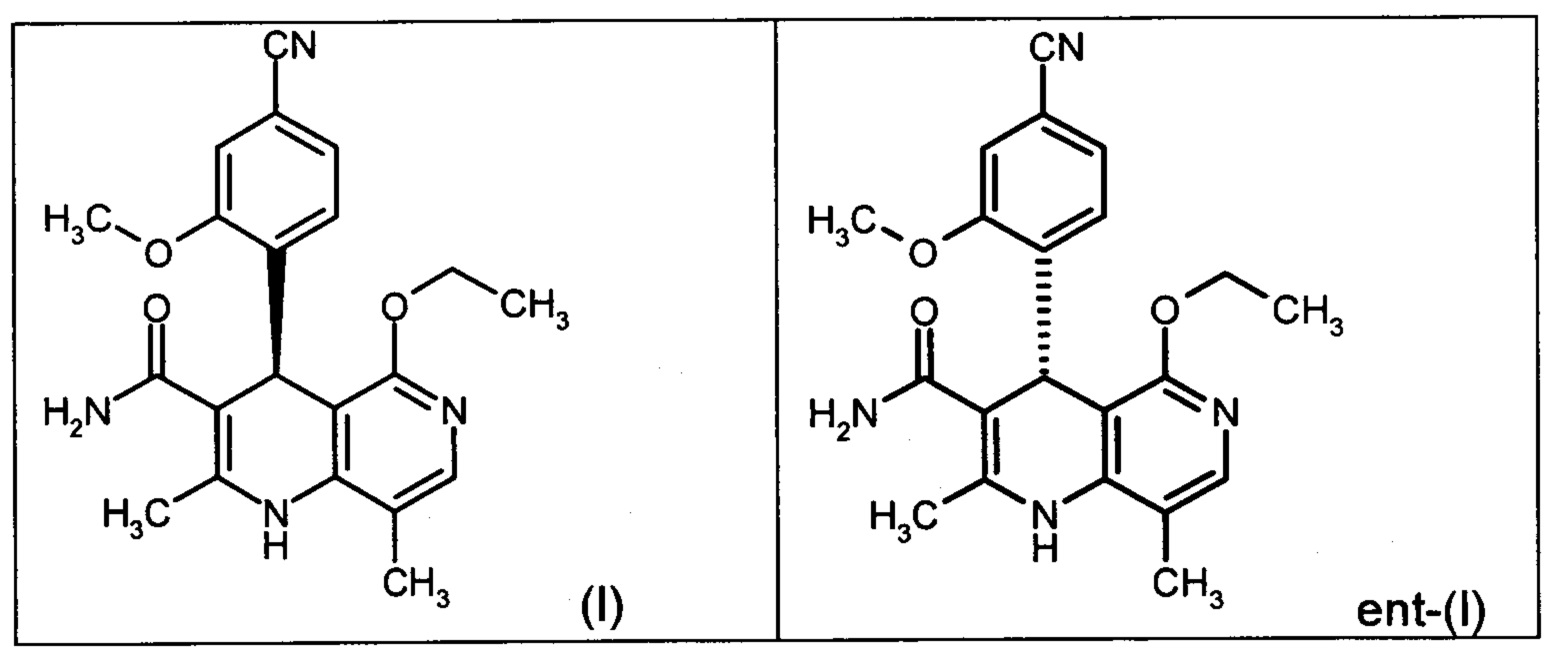
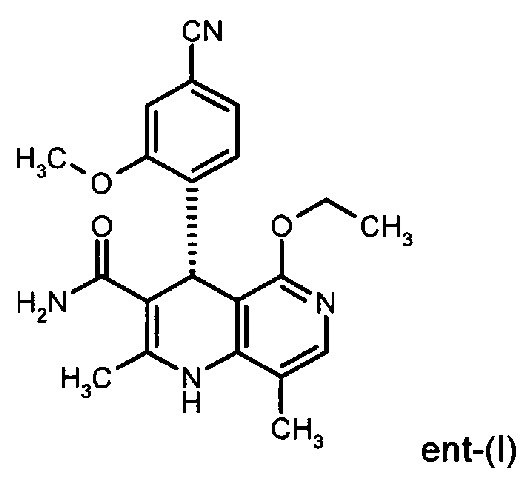
Анти-с4.4а антитела и их применение
Дипептидное пролекарство, его применение и лекарственное средство
Замещенные триазолопиридины и их применение в качестве ингибиторов тирозин треонин киназы (ттк)
Фиксирующий механизм устройства для инъекции лекарственного препарата
Препарат для биспецифических активаторов т-клеток (bite)
Конъюгаты связующего (adc) с ингибиторами ksp
Пиперидинилпиразолопиримидиноны и их применение
Способ получения 5-гидроксиалкилзамещенных производных 1-фенил-1,2,4-триазола
Способ получения 5-гидроксиалкилзамещенных производных 1-фенил-1,2,4-триазола
Способ получения 5-гидроксиалкилзамещенных производных 1-фенил-1,2,4-триазола

