Результат интеллектуальной деятельности: СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, КОТОРЫЕ МОЖНО ИСПОЛЬЗОВАТЬ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ ATR
Вид РИД
Изобретение
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Киназа ATR («связанная с ATM и Rad3») представляет собой протеинкиназу, участвующую в клеточных ответах на повреждение ДНК. Киназа ATR действует с киназой ATM («мутация при атаксии-телеангиэктазии») и многими другими белками, регулируя ответ клетки на повреждение ДНК, который часто называют «ответом на повреждение ДНК» (DDR). DDR стимулирует репарацию ДНК, способствует выживанию и останавливает последовательность клеточного цикла путем активации контрольных точек клеточного цикла, обеспечивая клетке время на репарацию. Без DDR клетки гораздо в большей степени подвержены повреждению ДНК и легко погибают от нарушений ДНК, вызванных эндогенными клеточными процессами, такими как репликация ДНК, или экзогенными повреждающими ДНК агентами, которые часто используются в терапии рака.
Здоровые клетки для репарации ДНК могут использовать множество различных белков, включая DDR-киназу ATR. В некоторых случаях эти белки могут компенсировать друг друга путем активации функционально избыточных процессов репарации ДНК. С другой стороны, многие раковые клетки сохраняют дефекты в некоторых из своих процессов репарации ДНК, таких как ATM-сигнализация, и поэтому демонстрируют большую зависимость от своих оставшихся неповрежденными белков для репарации ДНК, к которым относится ATR.
Кроме того, многие раковые клетки экспрессируют активированные онкогены или у них отсутствуют ключевые супрессоры опухоли, в результате чего такие раковые клетки склонны к нарушению регуляции фаз репликации ДНК, что, в свою очередь, приводит к повреждению ДНК. Предполагается, что ATR является очень важным компонентом в ответе DDR на нарушение репликации ДНК. В результате этого выживание таких раковых клеток в большей степени зависит от активности ATR, чем у здоровых клеток. Соответственно, ингибиторы ATR можно использовать для лечения рака, либо отдельно, либо в комбинации с агентами, повреждающими ДНК, поскольку они отключают механизм репарации ДНК, который более важен для выживания многих раковых клеток, чем для нормальных здоровых клеток.
В действительности было показано, что нарушение функции ATR (например, путем делеции гена) способствует гибели раковой клетки как в отсутствие, так и при наличии агентов, повреждающих ДНК. Это свидетельствует о том, что ингибиторы ATR могут быть эффективны как в качестве одиночных агентов, так и в качестве мощных сенсибилизаторов в лучевой терапии или генотоксической химиотерапии.
В силу всех этих причин существует потребность в разработке мощных селективных ингибиторов ATR для лечения рака, используемых либо в качестве одиночных агентов, либо в комплексной терапии с лучевой терапией или генотоксической химиотерапией. Более того, было бы желательно разработать такой вариант синтеза ингибиторов ATR, который позволял бы проводить крупномасштабный синтез и обеспечивал бы усовершенствование известных в настоящее время способов.
Пептид ATR можно экспрессировать и выделить с использованием различных способов, известных в литературе (см., например, Ünsal-Kaçmaz et al., PNAS 99: 10, стр. 6673–6678, 14 мая 2002 г.; см. также Kumagai et al. Cell 124, стр. 943–955, 10 марта 2006 г.; Unsal-Kacmaz et al. Molecular and Cellular Biology, февраль 2004 г., стр. 1292–1300; и Hall-Jackson et al. Oncogene 1999 г., 18, 6707–6713).
ОПИСАНИЕ РИСУНКОВ
РИСУНОК 1a: XRPD свободного основания соединения I-2
РИСУНОК 2a: TGA свободного основания соединения I-2
РИСУНОК 3a: DSC свободного основания соединения I-2
РИСУНОК 4a: модель ORTEP монокристаллической структуры асимметричного звена свободной формы соединения I-2
РИСУНОК 1b: XRPD соединения I-2 • HCl
РИСУНОК 2b: TGA соединения I-2 • HCl
РИСУНОК 3b: DSC соединения I-2 • HCl
РИСУНОК 4b: модель ORTEP асимметричного звена безводной структуры соединения I-2 • HCl
РИСУНОК 1c: XRPD соединения I-2 • 2HCl
РИСУНОК 2c: TGA соединения I-2 • 2HCl
РИСУНОК 3c: DSC соединения I-2 • 2HCl
РИСУНОК 4c: 13C ЯМР твердого тела соединения I-2 • 2HCl
РИСУНОК 1d: XRPD моногидрата соединения I-2 • HCl
РИСУНОК 2d: TGA моногидрата соединения I-2 • HCl
РИСУНОК 3d: DSC моногидрата соединения I-2 • HCl
РИСУНОК 1e: XRPD соединения I-2 • HCl • 2H2O
РИСУНОК 2e: TGA соединения I-2 • HCl • 2H2O
РИСУНОК 3e: DSC соединения I-2 • HCl • 2H2O
РИСУНОК 5a: 13C ЯМР твердого тела свободного основания соединения I-2
РИСУНОК 5b: 13C ЯМР твердого тела соединения I-2 • HCl
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам и промежуточным соединениям для получения соединений, которые можно использовать в качестве ингибиторов киназы ATR, таких как производные аминопиразинизоксазола и связанные молекулы. Производные аминопиразинизоксазола можно использовать в качестве ингибиторов ATR, а также можно использовать для получения ингибиторов ATR. Настоящее изобретение также относится к твердым формам ингибиторов ATR, а также к дейтерированным ингибиторам ATR.
Один аспект настоящего изобретения представляет способ получения соединения формулы I:
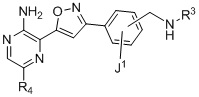
I
содержащий получение соединения формулы 4 :
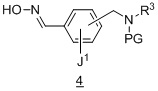
из соединения формулы 3:
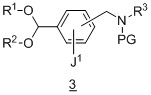
в подходящих условиях образования оксима.
Другой аспект содержит получение соединения формулы 4:
из соединения формулы 3:
в подходящих условиях образования оксима.
Другой аспект настоящего изобретения содержит соединение формулы II:
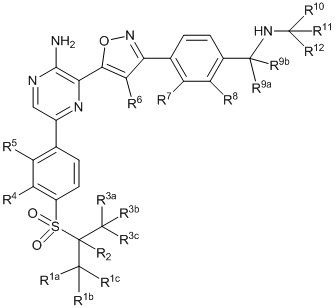
II
или его фармацевтически приемлемую соль, где каждый R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a, R9b, R10, R11, R12 и R13 по отдельности представляет собой водород или дейтерий, а по меньшей мере один из R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a, R9b, R10, R11, R12 и R13 представляет собой дейтерий.
Еще один аспект настоящего изобретения представляет твердые формы соединения формулы I-2:
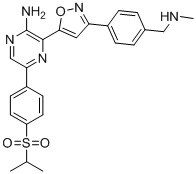
I-2
Другие аспекты настоящего изобретения описаны в настоящем документе.
Настоящее изобретение имеет несколько преимуществ по сравнению с ранее известными способами. Во-первых, настоящий способ имеет меньшее число этапов полного синтеза по сравнению с ранее описанными способами. Во-вторых, настоящий способ отличается более высокими выходами по сравнению с ранее описанными способами. В-третьих, настоящий способ эффективен для соединений, в которых R3 представлен самыми различными группами, например алкильными группами или объемным, стерически затрудненным фрагментом, таким как кольцо. В-четвертых, настоящий способ содержит промежуточные соединения, которые являются более устойчивыми и имеют более продолжительный срок хранения. В некоторых вариантах осуществления образование оксимной группы без участия кислоты в настоящем способе позволяет сохранить в ходе синтеза чувствительные к кислоте защитные группы, такие как Boc или CBz. В других вариантах осуществления способ гораздо проще масштабировать для получения больших количеств в результате исключения хроматографии в качестве этапа очистки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Один аспект настоящего изобретения представляет способ получения соединения для получения соединения формулы 4:
из соединения формулы 3:
в подходящих условиях образования оксима;
где
R1 представляет собой C1–6-алкил;
R2 представляет собой C1–6-алкил;
или R1 и R2 вместе с атомами кислорода, к которым они прикреплены, образуют необязательно замещенное 5- или 6-членное насыщенное гетероциклическое кольцо, имеющее два атома кислорода;
R3 представляет собой водород, C`1–6-алкил или 3–6-членный насыщенный или частично ненасыщенный гетероциклил, имеющий 1–2 гетероатома, выбранных из группы, состоящей из кислорода, азота и серы; причем гетероциклил необязательно замещен 1 экземпляром галогена или C1–3-алкила;
J1 представляет собой галоген, C1–4-алкил или C1–4-алкокси;
PG представляет собой защитную группу карбамата.
Другой аспект представляет способ получения соединения формулы I:
I
содержащий следующие этапы:
получение соединения формулы 4:
из соединения формулы 3:
в подходящих условиях образования оксима;
где
R1 представляет собой C1–6-алкил;
R2 представляет собой C1–6-алкил;
или R1 и R2 вместе с атомами кислорода, к которым они прикреплены, образуют необязательно замещенное 5- или 6-членное насыщенное гетероциклическое кольцо, имеющее два атома кислорода;
R3 представляет собой водород, C`1–6-алкил или 3–6-членный насыщенный или частично ненасыщенный гетероциклил, имеющий 1–2 гетероатома, выбранных из группы, состоящей из кислорода, азота и серы; причем гетероциклил необязательно замещен 1 экземпляром галогена или C1–3-алкила;
R4 представляет собой 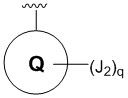 ;
;
Q представляет собой фенил, пиридил или N-алкилированный пиридин;
J1 представляет собой H, галоген, C1–4-алкил или C1–4-алкокси;
J2 представляет собой галоген; CN; фенил; оксазолил; или C1–6-алифатическую группу, где максимум 2 метиленовых звена необязательно заменены на O, NR”, C(O), S, S(O) или S(O)2; указанная C1–6-алифатическая группа необязательно замещена 1–3 фторами или CN;
q равно 0, 1 или 2;
PG представляет собой защитную группу карбамата.
Другой вариант осуществления дополнительно содержит этап введения защиты соединения формулы 2:
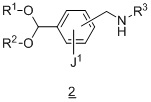
в подходящих условиях введения защиты с образованием соединения формулы 3.
Другой вариант осуществления дополнительно содержит этап взаимодействия соединения формулы 1:
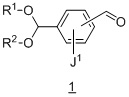
с подходящим амином в подходящих условиях восстановительного аминирования с образованием соединения формулы 2.
В некоторых вариантах осуществления подходящий амин представляет собой NHCH3. В других вариантах осуществления подходящий амин представляет собой 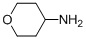 .
.
Другой вариант осуществления дополнительно содержит этап взаимодействия соединения формулы 4 :
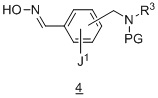
в подходящих условиях образования изоксазола с образованием соединения формулы 5:
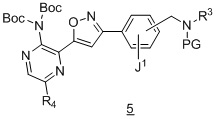 .
.
Другой вариант осуществления дополнительно содержит этап взаимодействия соединения формулы 5 в подходящих условиях сочетания, а затем в подходящих условиях снятия защиты с образованием соединения формулы I.
В некоторых вариантах осуществления PG представляет собой Boc или Cbz. В некоторых вариантах осуществления PG представляет собой Boc.
В других вариантах осуществления R1 представляет собой этил и R2 представляет собой этил.
В других вариантах осуществления R3 представляет собой CH3 или 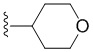 .
.
В некоторых вариантах осуществлений R4 представляет собой
; где Q представляет собой фенил. В некоторых вариантах осуществления Q замещен J2 в параположении, где q равно 1.
В некоторых вариантах осуществления J1 представляет собой H или галоген. В некоторых вариантах осуществления J1 представляет собой H. В других вариантах осуществления J1 представляет собой галоген.
В других вариантах осуществления J2 представляет собой C1–6-алифатическую группу, где максимум 1 метиленовое звено необязательно заменено на S(O)2. В некоторых вариантах осуществления J2 представляет собой -S(O)2-(C1–5-алкил). В некоторых вариантах осуществления q равно 1.
В соответствии с другим вариантом осуществления
R1 представляет собой этил;
R2 представляет собой этил;
R3 представляет собой CH3 или ;
PG представляет собой Boc или Cbz;
J1 представляет собой H;
R4 представляет собой , где Q представляет собой фенил; J2 представляет собой -S(O)2-CH(CH3)2;
q равно 1.
В некоторых вариантах осуществления R3 представляет собой CH3. В некоторых вариантах осуществления R3 представляет собой CH3. В других вариантах осуществления R3 представляет собой CH3 или .
В соответствии с другим вариантом осуществления
R1 представляет собой этил;
R2 представляет собой этил;
R3 представляет собой ;
PG представляет собой Boc;
J1 представляет собой H;
R4 представляет собой , где Q представляет собой пиридил; J2 представляет собой 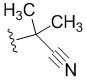 ;
;
q равно 1.
В некоторых вариантах осуществления R4 представляет собой 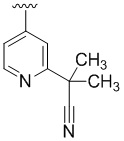 .
.
Условия реакций
В некоторых вариантах осуществления подходящие условия образования оксима состоят либо из одноэтапной последовательности, либо из двухэтапной последовательности.
В некоторых вариантах осуществления двухэтапная последовательность состоит, во-первых, из снятия защиты с кетальной группы в соединении формулы 3 с образованием альдегида в подходящих условиях снятия защиты, а затем образования оксима формулы 4 в подходящих условиях образования оксима. В некоторых вариантах осуществления подходящие условия снятия защиты содержат добавление каталитических количеств паратолуолсульфоновой кислоты (pTSA), ацетона и воды; а подходящие условия образования оксима содержат смешивание вместе гидроксиламина, каталитического количества кислоты, дегидрирующего агента и спиртосодержащего растворителя. В других вариантах осуществления кислота представляет собой pTSA или HCl, дегидратирующий агент представляет собой молекулярные сита или диметоксиацетон, а спиртосодержащим растворителем является метанол или этанол.
В других вариантах осуществлениях одноэтапная последовательность содержит добавление NH2OH.HCl и смеси THF и воды. В других вариантах осуществления последовательность содержит добавление NH2OH.HCl со смесью 2-метилтетрагидрофурана и воды, необязательно буферизованной Na2SO4. В некоторых вариантах осуществления 1 эквивалент соединения формулы 3 объединяют с 1,1 эквивалента NH2OH.HCl в смеси THF и воды в соотношении 10:1 об./об. В некоторых вариантах осуществления 1 эквивалент соединения формулы 3 объединяют с 1,1 эквивалента NH2OH.HCl в смеси 2-метилтетрагидрофурана и воды, необязательно буферизованной Na2SO4, в соотношении 10:1 об./об.
В других вариантах осуществления условия введения защиты выбраны из группы, состоящей из
● R-OCOCl, подходящего третичного азотсодержащего основания и подходящего растворителя; где R представляет собой C1–6-алкил, необязательно замещенный фенилом;
● R(CO2)OR’, подходящего растворителя и необязательно каталитического количества основания, где R и R’ каждый по отдельности представляют собой C1–6-алкил, необязательно замещенный фенилом;
● [RO(C=O)]2O, подходящего основания и подходящего растворителя.
В некоторых вариантах осуществления подходящее основание представляет собой Et3N, диизопропиламин и пиридин; а подходящий растворитель выбран из хлорированного растворителя, эфира или ароматического углеводорода. В других вариантах осуществления подходящее основание представляет собой Et3N, подходящий растворитель представляет собой хлорированный растворитель, выбранный из DCM. В других вариантах осуществления условия введения защиты содержат добавление 1,20 эквивалента (Boc)2O и 1,02 эквивалента Et3N в DCM.
В соответствии с другим вариантом осуществления подходящие условия сочетания содержат добавление подходящего металла и подходящего основания в подходящем растворителе. В других вариантах осуществления подходящий металл представляет собой Pd[P(tBu)3]2; подходящий растворитель представляет собой смесь ацетонитрила и воды; а подходящее основание представляет собой карбонат натрия. В других вариантах осуществления подходящие условия сочетания содержат добавление 0,1 эквивалента Pd[P(tBu)3]2; 1 эквивалента бороновой кислоты или эфира; и 2 эквивалентов карбоната натрия в соотношении 2:1 об./об. ацетонитрил/вода при 60–70 °C.
В соответствии с другим вариантом осуществления подходящие условия снятия защиты содержат объединение соединения формулы 5 с подходящей кислотой в подходящем растворителе. В некоторых вариантах осуществления подходящая кислота выбрана из паратолуолсульфоновой кислоты (pTSA), HCl, TBAF, H3PO4 или TFA, а подходящий растворитель выбран из ацетона, метанола, этанола, CH2Cl2, EtOAc, THF, 2-MeTHF, диоксана, толуола или диэтилового эфира.
В соответствии с другим вариантом осуществления подходящие условия образования изоксазола состоят из двух этапов, причем первый этап содержит взаимодействие соединения формулы 4 в подходящих условиях образования хлороксима с образованием хлороксимного промежуточного соединения; второй этап содержит взаимодействие хлороксимного промежуточного соединения с ацетиленом в подходящих условиях циклоприсоединения с образованием соединения формулы 5.
В соответствии с другим вариантом осуществления подходящие условия образования хлороксима выбраны из
▪ N-хлорсукцинимида и подходящего растворителя или
▪ пероксимоносульфата калия, HCl и диоксана.
В некоторых вариантах осуществления подходящий растворитель выбран из апротонного растворителя, ароматического углеводорода или алкилацетата. В соответствии с другим вариантом осуществления подходящие условия образования хлороксима представляют собой 1,05 эквивалента N-хлорсукцинимида в изопропилацетате при 40–50 °C.
В соответствии с другим вариантом осуществления подходящие условия циклоприсоединения состоят из подходящего основания и подходящего растворителя. В некоторых вариантах осуществления подходящее основание выбрано из пиридина, DIEA, TEA, t-BuONa и K2CO3, а подходящий растворитель выбран из ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, MTBE, EtOAc, i-PrOAc, DCM, толуола, DMF и метанола. В других вариантах осуществления подходящее основание выбрано из Et3N, а подходящий растворитель выбран из DCM.
В соответствии с другим вариантом осуществления второй этап содержит взаимодействие 1 эквивалента ацетилена с 1,2 эквивалента хлороксимного промежуточного соединения и 1,3 эквивалента Et3N в DCM при комнатной температуре.
В соответствии с другим вариантом осуществления подходящие условия образования изоксазола содержат объединение соединения формулы 4 с окислителем в подходящем растворителе. В некоторых вариантах осуществления указанным окислителем является [бис(трифторацетокси)йод]бензол, а указанным растворителем является смесь метанола, воды и диоксана в соотношении 1:1:1.
Синтез соединений I-2 и I-3
Один вариант осуществления представляет способ получения соединения формулы I-2:
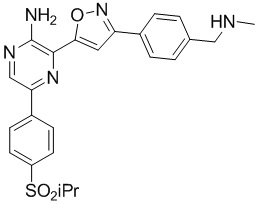 ,
,
I-2
содержащий один или более из следующих этапов:
a) взаимодействие соединения формулы 1b:
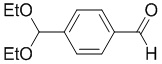
1b
с метиламином в подходящих условиях восстановительного аминирования с образованием соединения формулы 2b:
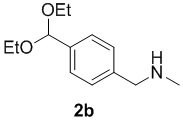 ;
;
b) взаимодействие соединения формулы 2b в подходящих условиях введения защиты Boc с образованием соединения формулы 3b:
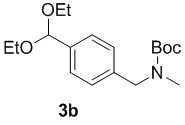 ;
;
c) взаимодействие соединения формулы 3b в подходящих условиях образования оксима с образованием соединения формулы 4-i:
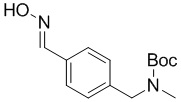
4-i
d) взаимодействие соединения формулы 4-i в подходящих условиях образования хлороксима с образованием соединения формулы 4-ii:
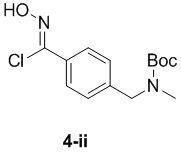
e) взаимодействие соединения формулы 4-ii с соединением формулы 4-iii:
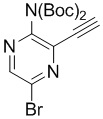
4-iii
в подходящих условиях циклоприсоединения с образованием соединения формулы 4-iv:
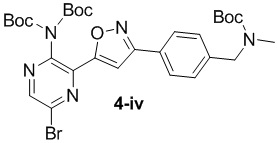
f) взаимодействие соединения формулы 4-iv с соединением формулы A-5-i:
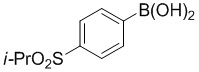
A-5-i
в подходящих условиях сочетания с образованием соединения формулы 5-i:
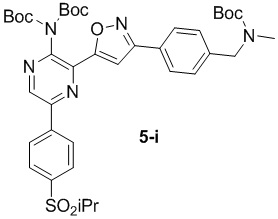
g) снятие защиты с соединения формулы 5-i в подходящих условиях снятия Boc-защиты необязательно с последующей обработкой в условиях водного раствора основания с образованием соединения формулы I-2.
Другой вариант осуществления представляет способ получения соединения
формулы I-3:
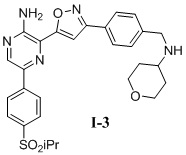
содержащий один или более из следующих этапов:
a) взаимодействие соединения формулы A-1:
A-1
с тетрагидро-2H-пиран-4-амином в подходящих условиях восстановительного аминирования с образованием соединения формулы A-2:
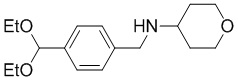
A-2
b) взаимодействие соединения формулы A-2 в подходящих условиях введения Boc-защиты с образованием соединения формулы A-3:
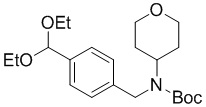
A-3
c) взаимодействие соединения формулы A-3 в подходящих условиях образования оксима с образованием соединения формулы A-4:
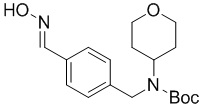
A-4
d) взаимодействие соединения формулы A-4:
A-4
в подходящих условиях образования хлороксима с образованием соединения формулы A-4-i:
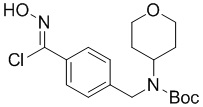
A-4-i
e) взаимодействие соединения формулы A-4-i с соединением формулы A-4-ii:
A-4-ii
в подходящих условиях циклоприсоединения с образованием соединения формулы A-5:
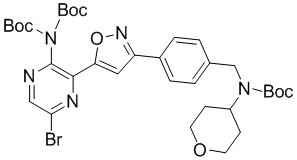
A-5
f) взаимодействие соединения формулы A-5 с соединением формулы A-5-i:
A-5-i
в подходящих условиях сочетания с образованием соединения формулы A-6:
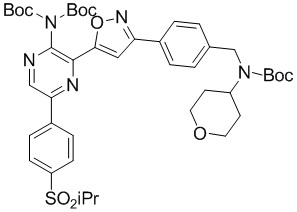
A-6
g) снятие защиты с соединения формулы А-6 в подходящих условиях снятия Boc-защиты необязательно с последующей обработкой в условиях водного раствора основания с образованием соединения формулы I-3.
Подходящие условия сочетания содержат объединение подходящего палладиевого катализатора с подходящим основанием в подходящем растворителе. Подходящий палладиевый катализатор включает в себя, без ограничений, Pd[P(tBu)3]2, Pd(dtbpf)Cl2, Pd(PPh3)2Cl2, Pd(PCy3)2Cl2 , Pd(dppf)Cl2 и Pd(dppe)Cl2. Подходящие растворители включают в себя, без ограничений, толуол, MeCN, воду, EtOH, IPA, 2-Me-THF или IPAc. Подходящие основания включают в себя, без ограничений, K2CO3, Na2CO3 или K3PO4.
Подходящие условия образования оксима состоят либо из одноэтапной последовательности, либо из двухэтапной последовательности. Двухэтапная последовательность состоит, во-первых, из снятия защиты с кетальной группы в соединении формулы А-3 с образованием альдегида в подходящих условиях снятия защиты, а затем образования оксима формулы А-4 в подходящих условиях образования оксима.
Одноэтапная последовательность содержит, например, смешивание вместе гидроксиламина, кислоты, органического растворителя и воды. В некоторых вариантах осуществления NH2OH.HCl добавляют к смеси THF и воды. В некоторых вариантах осуществления 1 эквивалент соединения формулы А-3 объединяют с 1,1 эквивалента NH2OH.HCl в смеси THF/вода в соотношении 10:1 об./об.
Подходящие условия снятия защиты содержат добавление кислоты, ацетона и воды. Подходящие кислоты включают в себя pTSA или HCl, подходящие органические растворители включают в себя хлорированные растворители (например, дихлорметан (DCM), дихлорэтан (DCE), CH2Cl2 и хлороформ); эфир (например, THF, 2-MeTHF и диоксан); ароматические углеводороды (например, толуол и ксилолы или другие апротонные растворители).
Подходящие условия циклоприсоединения содержат подходящее основание (например, пиридин, DIEA, TEA, t-BuONa или K2CO3) и подходящий растворитель (например, ацетонитрил, тетрагидрофуран, 2-метилтетрагидрофуран, MTBE, EtOAc, i-PrOAc, DCM, толуол, DMF и метанол).
Подходящие условия образования хлороксима содержат добавление HCl в диоксане к раствору оксима в присутствии NCS в подходящем растворителе, выбранном из апротонных растворителей (DCM, DCE, THF и диоксан), ароматических углеводородов (например, толуол, ксилолы) и алкилацетатов (например, изопропилацетат, этилацетат).
Подходящие условия снятия Boc-защиты содержат добавление подходящего агента для снятия Boc-защиты (например, TMS-Cl, HCl, TBAF, H3PO4 или TFA) и подходящего растворителя (например, ацетон, толуол, метанол, этанол, 1-пропанол, изопропанол, CH2Cl2, EtOAc, изопропилацетат, тетрагидрофуран, 2-метилтетрагидрофуран, диоксан и диэтиловый эфир). В некоторых вариантах осуществления подходящие условия снятия Boc-защиты содержат добавление подходящего агента для снятия Boc-защиты, выбранного из HCl, TFA, и подходящего растворителя, выбранного из ацетона, толуола, изопропилацетата, тетрагидрофурана или 2-метилтетрагидрофурана.
Подходящие условия введения Boc-защиты включают в себя (Boc)2O, подходящее основание и подходящий растворитель. Подходящие основания включают в себя, без ограничений, Et3N, диизопропиламин и пиридин. Подходящие растворители включают в себя, без ограничений, хлорированные растворители (например, дихлорметан (DCM), дихлорэтан (DCE), CH2Cl2 и хлороформ); эфир (например, THF, 2-MeTHF и диоксан); ароматические углеводороды (например, толуол и ксилолы или другие апротонные растворители). В некоторых вариантах осуществления подходящее основание представляет собой Et3N, подходящий растворитель представляет собой DCM, тетрагидрофуран или 2-метилтетрагидрофуран. В некоторых вариантах осуществления условия введения защиты содержат добавление 1,05 эквивалента (Boc)2O в 2-метилтетрагидрофуране или DCM.
Подходящие условия восстановительного аминирования содержат добавление восстанавливающего агента, выбранного из NaBH4, NaBH4, NaBH3CN или NaBH(OAc)3, в присутствии растворителя, выбранного из дихлорметана (DCM), дихлорэтана (DCE), спиртосодержащего растворителя, выбранного из метанола, этанола, 1-пропанола, изопропанола, или апротонного растворителя, выбранного из диоксана, тетрагидрофурана или 2-метилтетрагидрофурана, и необязательно основания, выбранного из Et3N или диизопропилэтиламина. В некоторых вариантах осуществления подходящие условия восстановительного аминирования содержат добавление 1,2 эквивалента капсул NaBH4 в присутствии Et3N в MeOH.
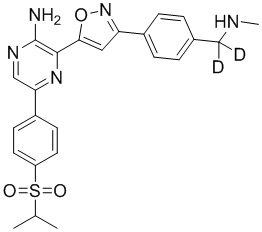
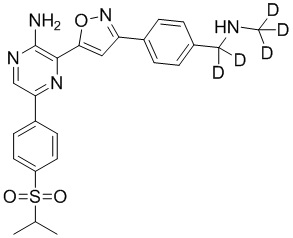
Другой аспект настоящего изобретения представляет соединение формулы II:
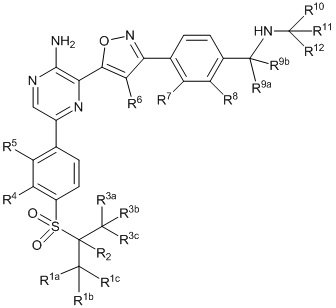
II
или его фармацевтически приемлемую соль,
где каждый R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a, R9b, R10a, R10b и R10c по отдельности представляет собой водород или дейтерий, и
по меньшей мере один из R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляет собой дейтерий.
В некоторых вариантах осуществления R9a и R9b представляют собой одно и то же. В других вариантах осуществления R9a и R9b представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R10, R11a, R11b, R12a, R12b, R13a, R13b, R14a и R14b представляют собой дейтерий или водород. В еще одном варианте осуществления R9a и R9b представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R10, R11a, R11b, R12a, R12b, R13a, R13b, R14a и R14b представляют собой водород.
В одном варианте осуществления R9a, R9b, R10a, R10b и R10c представляют собой одно и то же. В другом варианте осуществления R9a, R9b, R10a, R10b и R10c представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7 и R8 представляют собой дейтерий или водород. В некоторых вариантах осуществления R9a, R9b, R10a, R10b и R10c представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7 и R8 представляют собой водород.
В других вариантах осуществления R10a, R10b и R10c представляют собой одно и то же. В одном варианте осуществления R9a, R9b, R10a, R10b и R10c представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7 и R8 представляют собой дейтерий или водород. В другом варианте осуществления R10a, R10b и R10c представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a и R9b представляют собой водород.
В некоторых вариантах осуществления R1a, R1b, R1c, R2, R3a, R3b и R3c представляют собой одно и то же. В другом варианте осуществления R1a, R1b, R1c, R2, R3a, R3b и R3c представляют собой дейтерий, а R4, R5, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий или водород. В еще одном варианте осуществления R1a, R1b, R1c, R2, R3a, R3b и R3c представляют собой дейтерий, и R4, R5, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий.
В другом варианте осуществления R6 представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий или водород. В еще одном варианте осуществления R6 представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой водород.
В других вариантах осуществления R2 представляет собой дейтерий, а R1a, R1b, R1c, R3a, R3b и R3c, R4, R5, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий или водород. В другом варианте осуществления R2 представляет собой дейтерий, а R1a, R1b, R1c, R3a, R3b и R3c, R4, R5, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой водород.
В другом варианте осуществления R7 представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R8, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий или водород. В других вариантах осуществления R7 представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R8, R9a, R9b, R10a, R10b, R10c представляют собой водород.
В еще одном варианте осуществления R8 представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий или водород. В другом варианте осуществления R8 представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R9a, R9b, R10a, R10b, R10c представляют собой водород.
В некоторых вариантах осуществления по меньшей мере один из R10a, R10b или R10c представляет собой одно и то же. В другом варианте осуществления по меньшей мере один из R10a, R10b или R10c представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a и R9b представляют собой дейтерий или водород. В еще одном варианте осуществления по меньшей мере один из R10a, R10b или R10c представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a и R9b представляют собой водород.
В некоторых вариантах осуществления по меньшей мере два из R10a, R10b или R10c представляют собой одно и то же. В другом варианте осуществления по меньшей мере два из R10a, R10b или R10c представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a и R9b представляют собой дейтерий или водород. В еще одном варианте осуществления по меньшей мере два из R10a, R10b или R10c представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a и R9b представляют собой водород.
В другом варианте осуществления R1a, R1b, R1c, R3a, R3b и R3c представляют собой одно и то же. В некоторых вариантах осуществления R1a, R1b, R1c, R3a, R3b и R3c представляют собой дейтерий, а R2, R4, R5, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий или водород. В еще одном варианте осуществления R1a, R1b, R1c, R3a, R3b и R3c представляют собой дейтерий, а R2, R4, R5, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой водород.
В еще одном варианте осуществления R4 представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R5, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий или водород. В других вариантах осуществления R4 представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R5, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой водород.
В другом варианте осуществления R5 представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий или водород. В еще одном варианте осуществления R5 представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой водород.
В другом варианте осуществления по меньшей мере один из R9a или R9b представляет собой одно и то же. В других вариантах осуществления по меньшей мере один из R9a или R9b представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R10a, R10b и R10c представляют собой дейтерий или водород. В некоторых вариантах осуществления по меньшей мере один из R9a и R9b представляет собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R7, R8, R10a, R10b, R10c представляют собой водород.
В одном варианте осуществления R6, R9a и R9b представляют собой одно и то же. В некоторых вариантах осуществления R6, R9a и R9b представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R7, R8, R10a, R10b, R10c представляют собой дейтерий или водород. В других вариантах осуществления R6, R9a и R9b представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R7, R8, R10a, R10b и R10c представляют собой водород.
В некоторых вариантах осуществления R2, R10a, R10b и R10c представляют собой одно и то же. В другом варианте осуществления R2, R10a, R10b и R10c представляют собой дейтерий, а R1a, R1b, R1c, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a и R9b представляют собой дейтерий или водород. В еще одном варианте осуществления R2, R10a, R10b и R10c представляют собой дейтерий, а R1a, R1b, R1c, R3a, R3b, R3c, R4, R5, R6, R7, R8, R9a и R9b представляют собой водород.
В некоторых вариантах осуществления R7 и по меньшей мере два из R10a, R10b или R10c представляют собой одно и то же. В другом варианте осуществления R7 и по меньшей мере два из R10a, R10b или R10c представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R8, R9a и R9b представляют собой дейтерий или водород. В еще одном варианте осуществления R7 и по меньшей мере два из R10a, R10b или R10c представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R4, R5, R6, R8, R9a и R9b представляют собой водород.
В некоторых вариантах осуществления R1a, R1b, R1c, R2, R3a, R3b, R3c и по меньшей мере один из R10a, R10b или R10c представляют собой одно и то же. В другом варианте осуществления R1a, R1b, R1c, R2, R3a, R3b, R3c и по меньшей мере один из R10a, R10b или R10c представляют собой дейтерий, а R4, R5, R6, R7, R8, R9a и R9b представляют собой дейтерий или водород. В еще одном варианте осуществления R1a, R1b, R1c, R2, R3a, R3b, R3c и по меньшей мере один из R10a, R10b или R10c представляют собой дейтерий, а R4, R5, R6, R7, R8, R9a и R9b представляют собой водород.
В некоторых вариантах осуществления R1a, R1b, R1c, R3a, R3b, R3c и R5 представляют собой одно и то же. В другом варианте осуществления R1a, R1b, R1c, R3a, R3b, R3c и R5 представляют собой дейтерий, а R2, R4, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий или водород. В еще одном варианте осуществления R1a, R1b, R1c, R3a, R3b, R3c и R5 представляют собой дейтерий, а R2, R4, R6, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой водород.
В других вариантах осуществления R4 и R6 представляют собой одно и то же. В другом варианте осуществления R4 и R6 представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R5, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий или водород. В еще одном варианте осуществления R4 и R6 представляют собой дейтерий, а R1a, R1b, R1c, R2, R3a, R3b, R3c, R5, R7, R8, R9a, R9b, R10a, R10b и R10c представляют собой водород.
В одном варианте осуществления R2, R5, R9a и R9b представляют собой одно и то же. В некоторых вариантах осуществления R2, R5, R9a и R9b представляют собой дейтерий, а R1a, R1b, R1c, R3a, R3b, R3c, R4, R6, R7, R8, R10a, R10b и R10c представляют собой дейтерий или водород. В другом варианте осуществления R2, R5, R9a и R9b представляют собой дейтерий, а R1a, R1b, R1c, R3a, R3b, R3c, R4, R6, R7, R8, R10a, R10b и R10c представляют собой водород.
В еще одном варианте осуществления R1a, R1b, R1c, R2, R3a, R3b, R3c, R5, R6, R9a, R9b, R10a, R10b и R10c представляют собой одно и то же. В некоторых вариантах осуществления R1a, R1b, R1c, R2, R3a, R3b, R3c, R5, R6, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий, а R4, R7 и R8 представляют собой дейтерий или водород. В других вариантах осуществления R1a, R1b, R1c, R2, R3a, R3b, R3c, R5, R6, R9a, R9b, R10a, R10b и R10c представляют собой дейтерий, а R4, R7 и R8 представляют собой водород.
В некоторых вариантах осуществления переменные соответствуют переменным, показанным в соединениях настоящего описания, включая соединения, указанные в приведенных ниже таблицах.
Таблица I
|
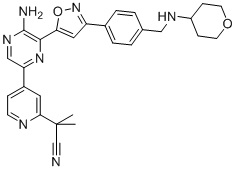
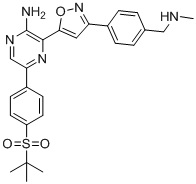
Таблица II
|
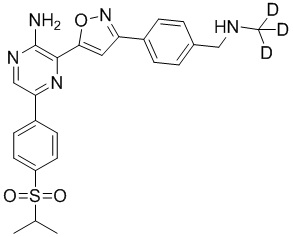
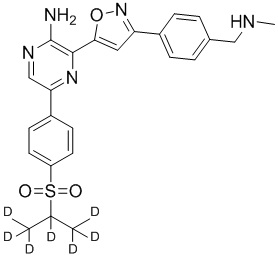
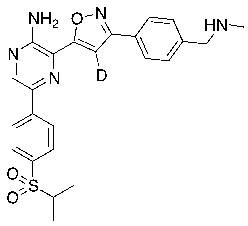
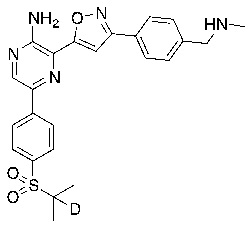
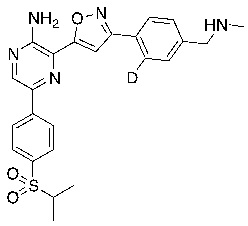
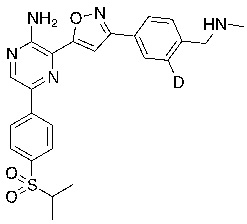
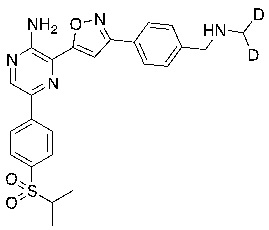
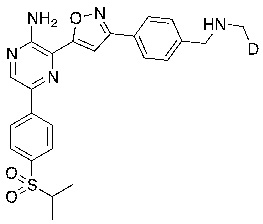
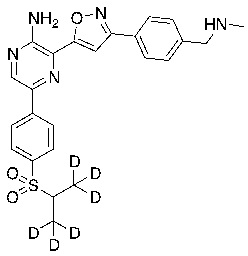
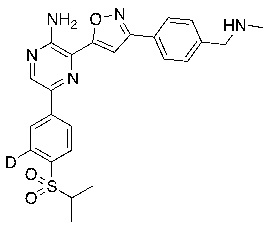
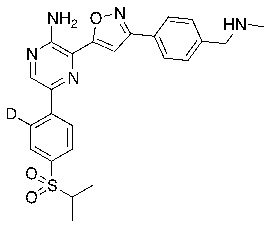
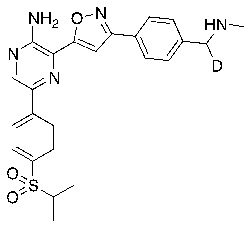
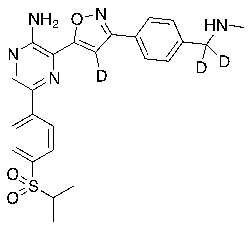
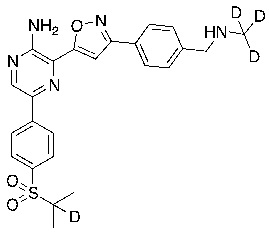
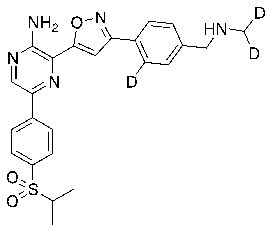
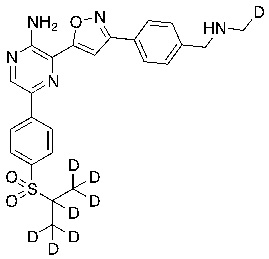
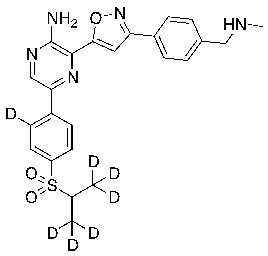
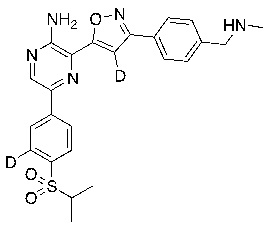
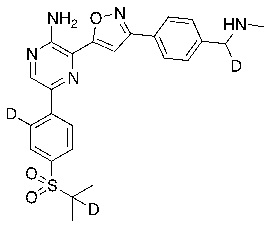
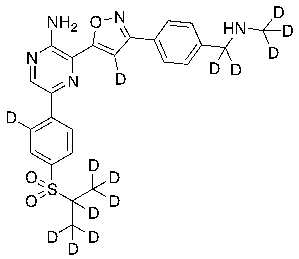
Соединения настоящего изобретения включают в себя соединения, описанные по существу в настоящем документе и дополнительно проиллюстрированные классами, подклассами и видами, описанными в настоящем документе. В настоящем документе в отсутствие иных указаний будут применимы следующие определения. Для целей настоящего изобретения химические элементы идентифицируются в соответствии с периодической таблицей элементов, версия CAS, Handbook of Chemistry and Physics, 75th Ed. Кроме того, общие принципы органической химии описаны в Organic Chemistry, Thomas Sorrell, University Science Books, Sausalito: 1999 г., и March’s Advanced Organic Chemistry, 5th Ed., Ed.: Smith, M.B. and March, J., John Wiley & Sons, New York: 2001 г., содержание которых полностью включено в настоящий документ путем ссылки.
В настоящем документе указанный числовой диапазон атомов включает в себя любое целое число в нем. Например, группа, имеющая 1–4 атома, может иметь 1, 2, 3 или 4 атома.
Описанные в настоящем документе соединения настоящего изобретения могут быть необязательно замещены одним или более заместителями, такими как представленные по существу в настоящем документе или представленные в примерах конкретных классов, подклассов и форм изобретения. Следует понимать, что фраза «необязательно замещенный» взаимозаменяема с фразой «замещенный или незамещенный». По существу термин «замещенный», которому предшествует или не предшествует термин «необязательно», относится к замене водородных радикалов в заданной структуре на радикал указанного заместителя. Если не указано иное, необязательно замещенная группа может иметь заместитель в каждом замещаемом положении группы, и, когда в заданную структуру более чем в одном положении вводятся несколько заместителей, выбранных из указанной группы, заместитель в каждом положении может быть либо таким же, либо другим. Объединения заместителей, предусмотренных настоящим изобретением, предпочтительно приводят к образованию устойчивых или химически возможных соединений.
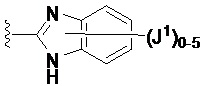
Если не указано иное, соединение заместителя связью, изображенной выходящей из центра кольца, означает, что заместитель может быть связан с любым положением в кольце. В приведенном ниже примере i, например, J1 может быть связан с любым положением на пиридиловом кольце. В случае бициклических колец связь, изображенная проходящей через оба кольца, указывает на то, что заместитель может быть связан с любым положением бициклического кольца. В приведенном ниже примере ii, например, J1 может быть связан с 5-членным кольцом (например, у атома азота) и 6-членным кольцом.
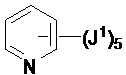
i ii
Используемый в настоящем документе термин «устойчивый» относится к соединениям, которые по существу не изменяются в условиях, которые позволяют их получить, обнаружить, выделить, очистить и использовать для одной или более целей, описанных в настоящем документе. В некоторых вариантах осуществления устойчивое соединение или химически возможное соединение представляет собой такое соединение, которое по существу не изменяется при хранении при температуре 40 °C или ниже в отсутствие влаги или других химически активных условий в течение по меньшей мере недели.
Используемый в настоящем документе термин «алифатический» или «алифатическая группа» означает линейную (т. е. неразветвленную), разветвленную или циклическую, замещенную или незамещенную углеводородную цепь, которая является полностью насыщенной или которая содержит одно или более ненасыщенных звеньев, имеющих одну точку соединения с остальной молекулой.
Если не указано иное, алифатические группы содержат 1–20 алифатических атомов углерода. В некоторых вариантах осуществления алифатические группы содержат
1–10 алифатических атомов углерода. В других вариантах осуществления алифатические группы содержат 1–8 алифатических атомов углерода. В других вариантах осуществления алифатические группы содержат 1–6 алифатических атомов углерода, а в еще других вариантах осуществления алифатические группы содержат
1–4 алифатических атома углерода. Алифатические группы могут представлять собой линейные или разветвленные, замещенные или незамещенные алкильные, алкенильные или алкинильные группы. Конкретные примеры, без ограничений, включают в себя метил, этил, изопропил, н-пропил, втор-бутил, винил, н-бутенил, этинил и трет-бутил. Алифатические группы также могут быть циклическими или иметь комбинацию линейных, разветвленных или циклических групп. Примеры таких типов алифатических групп включают в себя, без ограничений, циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил, -CH2-циклопропил, CH2CH2CH(CH3)-циклогексил.
Термин «циклоалифатический» (или «карбоцикл» или «карбоциклил») относится к моноциклическому C3–C8-углеводороду или бициклическому C8–C12-углеводороду, который полностью насыщен или который содержит одно или более ненасыщенных звеньев, не являющихся ароматическими, и который имеет одну точку соединения с остальной молекулой, причем любое отдельное кольцо указанной бициклической кольцевой системы имеет 3–7 членов. Примеры циклоалифатических групп включают в себя, без ограничений, циклоалкильную и циклоалкенильную группы. Конкретные примеры включают в себя, без ограничений, циклогексил, циклопропенил и циклобутил.
В настоящем документе термины «гетероцикл», «гетероциклил» или «гетероциклический» означают неароматические моноциклические, бициклические или трициклические кольцевые системы, в которых один или более членов кольца представляют собой независимо выбранный гетероатом. В некоторых вариантах осуществления «гетероцикл», «гетероциклил» или «гетероциклическая» группа имеет от трех до четырнадцати членов кольца, в которых один или более членов кольца представляют собой гетероатом, независимо выбранный из кислорода, серы, азота или фосфора, и каждое кольцо в системе содержит от 3 до 7 членов кольца.
Примеры гетероциклов включают в себя, без ограничений, 3-1H-бензимидазол-2-он, 3-(1-алкил)-бензимидазол-2-он, 2-тетрагидрофуранил, 3-тетрагидрофуранил,
2-тетрагидротиофенил, 3-тетрагидротиофенил, 2-морфолино, 3-морфолино, 4-морфолино, 2-тиоморфолино, 3-тиоморфолино, 4-тиоморфолино, 1-пирролидинил, 2-пирролидинил, 3-пирролидинил, 1-тетрагидропиперазинил, 2-тетрагидропиперазинил, 3-тетрагидропиперазинил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 1-пиразолинил, 3-пиразолинил, 4-пиразолинил, 5-пиразолинил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-пиперидинил, 2-тиазолидинил, 3-тиазолидинил, 4-тиазолидинил, 1-имидазолидинил, 2-имидазолидинил, 4-имидазолидинил, 5-имидазолидинил, индолинил, тетрагидрохинолинил, тетрагидроизохинолинил, бензотиолан, бензодитиан и 1,3-дигидроимидазол-2-он.
Циклические группы (например, циклоалифатические или гетероциклы) могут быть линейно конденсированными, мостиковыми или спироциклическими.
Термин «гетероатом» означает один или более из кислорода, серы, азота, фосфора или кремния (включая любую окисленную форму азота, серы, фосфора или кремния; кватернизированную форму любого основного азота; или замещаемый азот гетероциклического кольца, например, N (как в 3,4-дигидро-2H-пирролиле), NH (как в пирролидиниле) или NR+ (как в N-замещенном пирролидиниле)).
В настоящем документе термин «ненасыщенный» означает, что фрагмент имеет одно или более ненасыщенных звеньев. Как известно специалистам в данной области, ненасыщенные группы могут быть частично ненасыщенными или полностью ненасыщенными. Примеры частично ненасыщенных групп включают в себя, без ограничений, бутен, циклогексен и тетрагидропиридин. Полностью ненасыщенные группы могут быть ароматическими, антиароматическими или неароматическими. Примеры полностью ненасыщенных групп включают в себя, без ограничений, фенил, циклооктатетраен, пиридил, тиенил и 1-метилпиридин-2(1H)-он.
В настоящем документе термин «алкокси» или «тиоалкил» означает алкильную группу в соответствии с приведенным ранее определением, присоединенную через атом кислорода («алкокси») или серы («тиоалкил»).
Термины «галогеналкил», «галогеналкенил», «галогеналифатический» и «галогеналкокси» означают алкил, алкенил или алкокси, в зависимости от ситуации, замещенные одним или более атомами галогена. Данный термин включает в себя перфторированные алкильные группы, такие как –CF3 и –CF2CF3.
Термины «галоген», «галогено» и «галоид» означают F, Cl, Br или I.
Термин «арил», используемый отдельно или в составе более крупного фрагмента, например, «аралкила», «арилалкокси» или «арилоксиалкила», означает моноциклическую, бициклическую или трициклическую кольцевые системы, имеющие общее количество членов кольца от пяти до четырнадцати, причем по меньшей мере одно кольцо в системе является ароматическим и причем каждое кольцо в системе содержит от 3 до 7 членов кольца. Термин «арил» можно использовать взаимозаменяемо с термином «арильное кольцо».
Термин «гетероарил», используемый отдельно или в составе более крупного фрагмента, такого как «гетероаралкил» или «гетероарилалкокси», означает моноциклическую, бициклическую или трициклическую кольцевые системы, имеющие общее количество членов кольца от пяти до четырнадцати, причем по меньшей мере одно кольцо в системе является ароматическим, по меньшей мере одно кольцо в системе содержит один или более гетероатомов, и причем каждое кольцо в системе содержит от 3 до 7 членов кольца. Термин «гетероарил» можно использовать взаимозаменяемо с термином «гетероарильное кольцо» или термином «гетероароматический». Примеры гетероарильных колец включают в себя, без ограничений, 2-фуранил, 3-фуранил, N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил, бензимидазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, N-пирролил, 2-пирролил, 3-пирролил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, пиридазинил (например, 3-пиридазинил), 2-тиазолил, 4-тиазолил, 5-тиазолил, тетразолил (например, 5-тетразолил), триазолил (например, 2-триазолил и 5-триазолил), 2-тиенил, 3-тиенил, бензофурил, бензотиофенил, индолил (например, 2-индолил), пиразолил (например, 2-пиразолил), изотиазолил, 1,2,3-оксадиазолил, 1,2,5-оксадиазолил, 1,2,4-оксадиазолил, 1,2,3-триазолил, 1,2,3-тиадиазолил, 1,3,4-тиадиазолил, 1,2,5-тиадиазолил, пуринил, пиразинил, 1,3,5-триазинил, хинолинил (например, 2-хинолинил, 3-хинолинил, 4-хинолинил) и изохинолинил (например, 1-изохинолинил, 3-изохинолинил или 4-изохинолинил).
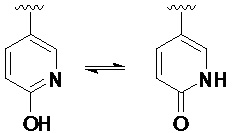
Следует понимать, что термин «гетероарил» включает в себя некоторые типы гетероарильных колец, существующих в равновесии между двумя различными формами. Более конкретно, например, варианты, такие как гидропиридин и пиридинон (и, аналогичным образом, гидроксипиримидин и пиримидинон) должны подпадать под определение «гетероарила».
В настоящем документе термины «защитная группа» и «защищающая группа» являются взаимозаменяемыми и обозначают агент, используемый для временного блокирования одной или более необходимых функциональных групп в соединении с множеством реакционноспособных участков. В некоторых вариантах осуществления защитная группа имеет одну или более или предпочтительно все следующие характеристики: а) избирательно добавляется в функциональную группу с хорошим выходом с получением защищенного субстрата, который b) обладает устойчивостью к реакциям, протекающим по одному или более другим реакционноспособным участкам; и c) может быть избирательно удалена с хорошим выходом с помощью реагентов, не атакующих восстановленную функциональную группу со снятой защитой. Как может быть понятно специалисту в данной области, в некоторых случаях реагенты не атакуют другие реакционноспособные группы в соединении. В других случаях реагенты также могут взаимодействовать с другими реакционноспособными группами в соединении. Примеры защитных групп подробно описаны в публикации Greene, T.W., Wuts, P. G, Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons, New York: 1999 г. (и в других изданиях книги), полное содержание которой включено в настоящий документ путем ссылки. В настоящем документе термин «защитная группа азота» означает агент, используемый для временного блокирования одного или более необходимых реакционноспособных участков азота в многофункциональном соединении. Предпочтительные защитные группы азота также обладают характеристиками, приведенными в качестве примера для указанной выше защитной группы, и определенные примеры защитных групп азота также подробно описаны в главе 7 публикации Greene, T.W., Wuts, P. G, Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons, New York: 1999 г., полное содержание которой включено в настоящий документ путем ссылки.
В некоторых вариантах осуществления метиленовое звено алкильной или алифатической цепи необязательно замещено другим атомом или группой. Примеры таких атомов или групп включают в себя, без ограничений, азот, кислород, серу,
-C(O)-, -C(=N-CN)-, -C(=NR)-, -C(=NOR)-, -SO- и -SO2-. Эти атомы или группы могут быть объединены с образованием групп большего размера. Примеры таких групп большего размера включают в себя, без ограничений, -OC(O)-, -C(O)CO-, -CO2-,
-C(O)NR-, -C(=N-CN), -NRCO-, -NRC(O)O-, -SO2NR-, -NRSO2-, -NRC(O)NR-, -OC(O)NR- и -NRSO2NR-, где R представляет собой, например, H или C1–6-алифатическую группу. Следует понимать, что эти группы могут быть связаны с метиленовыми звеньями алифатической цепи одинарными, двойными или тройными связями. Примером необязательной замены (в данном случае атома азота), связанной с алифатической цепью двойной связью, может быть –CH2CH=N-CH3. В некоторых случаях, в особенности на терминальном конце, необязательная замена может быть связана с алифатической группой тройной связью. Одним примером этого может быть CH2CH2CH2C N. Следует понимать, что в данной ситуации концевой азот не связан с другим атомом.
N. Следует понимать, что в данной ситуации концевой азот не связан с другим атомом.
Также следует понимать, что термин «метиленовое звено» также может относиться к разветвленным или замещенным метиленовым звеньям. Например, в изопропиловом фрагменте [-CH(CH3)2] атом азота (например, NR), замещающий первое упомянутое «метиленовое звено», приведет к образованию диметиламина [-N(CH3)2]. В таких случаях специалисту в данной области будет понятно, что с атомом азота не будут связаны никакие дополнительные атомы, а «R» в «NR» в данном случае будет отсутствовать.
Если не указано иное, необязательные замены образуют химически устойчивое соединение. Необязательные замены могут происходить как внутри цепи, так и/или на любом конце цепи; т. е. как в точке присоединения, так и/или на конце цепи. Две необязательные замены также могут быть смежны друг с другом внутри цепи, при условии что это приводит к химически устойчивому соединению. Например, C3-алифатическая группа может быть необязательно заменена 2 атомами азота с образованием –C–NN. Необязательные замены также могут полностью заменить все атомы углерода в цепи. Например, C3-алифатическая группа может быть необязательно заменена -NR-, -C(O)- и -NR- с образованием -NRC(O)NR- (мочевина).
Если не указано иное, когда замена происходит на терминальном конце, атом замены связывается с атомом водорода на терминальном конце. Например, если метиленовое звено -CH2CH2CH3 необязательно заменено на -O-, полученное соединение может представлять собой -OCH2CH3, -CH2OCH3 или -CH2CH2OH. Следует понимать, что если концевой атом не содержит никаких свободных валентных электронов, то атом водорода на терминальном конце не является обязательным (например, -CH2CH2CH=O или -CH2CH2CN).
Если не указано иное, также считается, что показанные в настоящем документе структуры включают в себя все изомерные (например, энантиомерные, диастереомерные, геометрические, конформационные и вращательные) формы структуры. Например, в настоящее изобретение включены конфигурации R и S для каждого асимметричного центра, изомеры по двойной связи (Z) и (E) и конформационные изомеры (Z) и (E). Как может быть понятно специалисту в данной области, заместитель может свободно вращаться вокруг любых вращаемых связей. Например, заместитель, показанный как 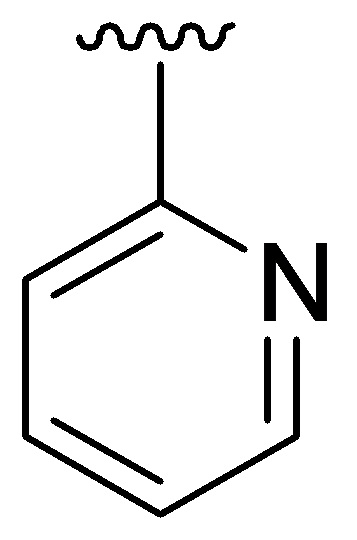 , также представляет собой
, также представляет собой 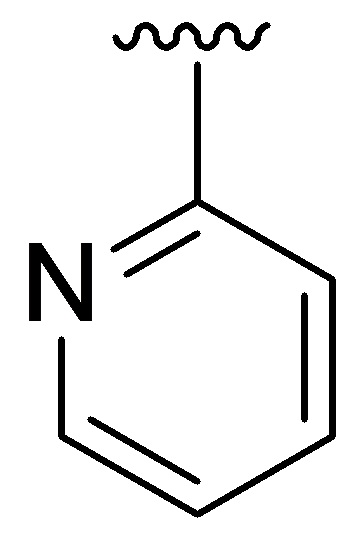 .
.
Поэтому отдельные стереохимические изомеры, а также энантиомерные, диастереомерные, геометрические, конформационные и вращательные смеси настоящих соединений не выходят за пределы объема настоящего изобретения.
Если не указано иное, все таутомерные формы соединений настоящего изобретения не выходят за пределы настоящего изобретения.
В соединениях настоящего изобретения любой атом, конкретно не обозначенный как конкретный изотоп, предназначен для представления любого стабильного изотопа этого атома. Если не указано иное, когда положение обозначено конкретно как «H» или «водород», имеется в виду, что в этом положении в распространенной в природе изотопной композиции находится водород. Кроме того, если не указано иное, когда положение обозначено конкретно как «D» или «дейтерий», имеется в виду, что в этом положении в композиции наиболее распространен дейтерий, что по меньшей мере в 3340 раз больше распространенности дейтерия в природе, которое составляет 0,015% (т. е. включение дейтерия составляет по меньшей мере 50,1%).
Оба из «D» и «d» обозначают дейтерий.
Кроме того, если не указано иное, также считается, что структуры, показанные в настоящем документе, включают в себя соединения, которые отличаются только наличием одного или более изотопно обогащенных атомов. Например, соединения, имеющие настоящие структуры, за исключением замены водорода на дейтерий или тритий или замены углерода на 13C- или 14C-обогащенный углерод, не выходят за пределы объема настоящего изобретения. Такие соединения можно использовать, например, в качестве аналитических инструментов или зондов в биологических анализах.
Способы
Способы и соединения, описанные в настоящем документе, можно использовать для получения ингибиторов ATR, которые содержат аминопиразинизоксазольное ядро. Общие процедуры синтеза, показанные на схемах в настоящем документе, можно использовать для создания широкого набора химических соединений, которые можно использовать в производстве фармацевтических соединений.
СХЕМА А
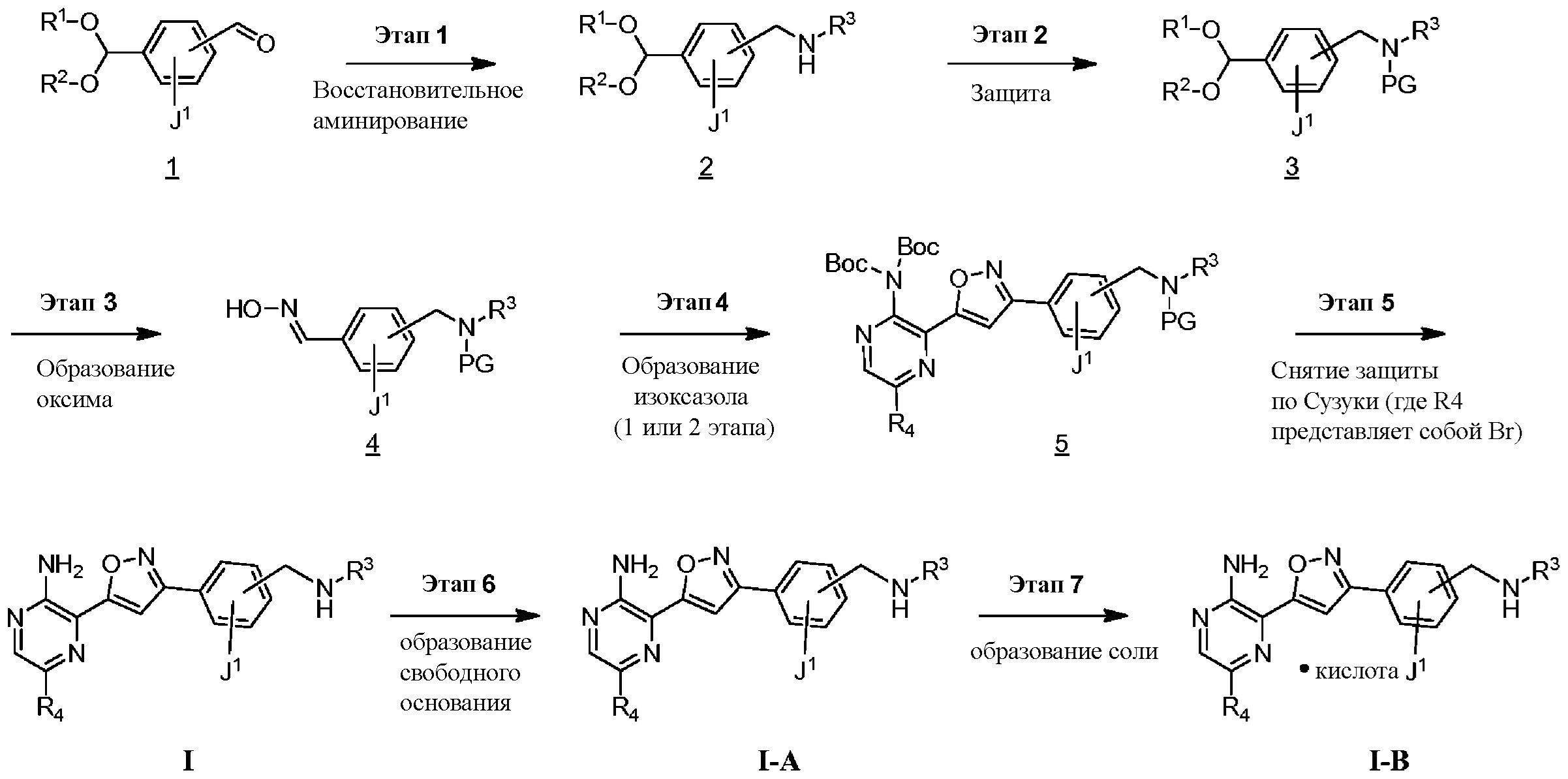
Этап 1
Соединение формулы I можно получить в соответствии с этапами, показанными на схеме A. На этапе 1 описано применение доступного альдегида/кеталя в качестве исходного материала для получения соединений формулы I, I-A и I-B. Восстановительное аминирование между соединением 1 и подходящим первичным амином в условиях, известных специалистам в данной области, позволяет получить соединение 2, в которое был введен бензиламиновый фрагмент. Например, имины могут быть образованы путем объединения амина и альдегида в подходящем растворителе, таком как дихлорметан (DCM), дихлорэтан (DCE), спиртосодержащий растворитель (например, метанол, этанол) или апротонный растворитель (например, диоксан или тетрагидрофуран (THF)). Затем такие имины могут быть восстановлены с использованием известных восстанавливающих агентов, включая, без ограничений, NaBH4, NaBH3CN и NaBH(OAc)3 (см. JOC 1996 г., 3849). В некоторых вариантах осуществления 1,05 эквивалента амина объединяют с 1 эквивалентом альдегида в метаноле. В других вариантах осуществления 1,2 эквивалента амина объединяют с 1 эквивалентом альдегида в метаноле. После этого этапа следует восстановление от 0,6 до 1,4 (например, 1,2) эквивалента NaBH4. В некоторых случаях при использовании соли амина также может быть добавлено основание (например, Et3N или диизопропилэтиламин).
Этап 2
На этапе 2 показано введение защиты полученного выше бензиламина 1 с использованием защитной группы на основе карбамата в подходящих условиях введения защиты, известных специалистам в данной области. Могут использоваться различные защитные группы, такие как Cbz и Boc. Условия введения защиты включают в себя, без ограничений, следующее:
a) R-OCOCl, подходящее третичное азотсодержащее основание и подходящий растворитель; где R представляет собой C1–6-алкил, необязательно замещенный фенилом;
b) R(CO2)OR’, подходящий растворитель и необязательно каталитическое количество основания, где каждый из R и R’ по отдельности представляют собой C1–6-алкил, необязательно замещенный фенилом;
c) [RO(C=O)]2O, подходящее основание и подходящий растворитель.
Примеры подходящих оснований включают в себя, без ограничений, Et3N, диизопропиламин и пиридин. Примеры подходящих растворителей включают в себя хлорированные растворители (например, дихлорметан (DCM), дихлорэтан (DCE), CH2Cl2 и хлороформ), эфиры (например, THF, 2-MeTHF и диоксан), ароматические углеводороды (например, толуол, ксилолы) и другие апротонные растворители.
В некоторых вариантах осуществления защита может быть реализована путем взаимодействия бензиламина с (Boc)2O и Et3N в DCM. В некоторых вариантах осуществления используется 1,02 эквивалента (Boc)2O и 1,02 эквивалента Et3N 1,02. В другом варианте осуществления защита может быть реализована путем взаимодействия бензиламина с (Boc)2O в 2-MeTHF. В некоторых вариантах осуществления используется 1,05 эквивалента (Boc)2O.
Этап 3
На этапе 3 показано, каким образом функциональная группа кеталя в 3 может быть преобразована в оксим 4 за один этап. Такое непосредственное преобразование кеталя в оксим недостаточно широко описано в литературе, и следует понимать, что данный этап также может быть реализован в двухэтапной последовательности с промежуточным альдегидом после снятия защиты с кеталя с использованием методик, известных специалистам в данной области.
Условия образования оксима содержат смешивание вместе гидроксиламина, кислоты, необязательно дегидрирующего агента и спиртосодержащего растворителя. В некоторых вариантах осуществления кислоту добавляют в каталитических количествах. В некоторых вариантах осуществления кислота представляет собой pTSA или HCl, дегидрирующий агент представляет собой молекулярные сита или диметоксиацетон, а спиртосодержащий растворитель представляет собой метанол или этанол. В некоторых вариантах осуществления используется гидрохлорид гидроксиламина, и в этом случае дополнительная кислота не требуется. В других вариантах осуществления желаемый продукт выделяют путем разделения на две фазы с необязательным осаждением или кристаллизацией. При разделении на две фазы дегидратирующий агент не требуется.
В другом варианте осуществления условия образования оксима содержат смешивание вместе гидроксиламина, кислоты, органического растворителя и воды. Примеры подходящих органических растворителей включают в себя хлорированные растворители (например, дихлорметан (DCM), дихлорэтан (DCE), CH2Cl2 и хлороформ), эфиры (например, THF, 2-MeTHF и диоксан), ароматические углеводороды (например, толуол, ксилолы) и другие апротонные растворители. В некоторых вариантах осуществления используются 1,5 эквивалента гидрохлорида гидроксиламина, органический растворитель представляет собой 2-MeTHF, а водный раствор буферизован Na2SO4. В другом варианте осуществления используется 1,2 эквивалента гидрохлорида гидроксиламина, а органический растворитель представляет собой THF.
В некоторых вариантах осуществления подходящие условия снятия защиты содержат добавление каталитических количеств паратолуолсульфоновой кислоты (pTSA), ацетона и воды; а затем образование оксима с использованием условий, известных специалистам в данной области. В других вариантах осуществления используется одноэтапная последовательность. В некоторых вариантах осуществления одноэтапная последовательность содержит добавление NH2OH.HCl и смеси THF и воды. В некоторых вариантах осуществления 1 эквивалент соединения формулы 3 объединяют с 1,1 эквивалента NH2OH.HCl в смеси THF/вода в соотношении 10:1 об./об.
Этап 4
На этапе 4 показано, как оксим 4 преобразуется и вступает в реакцию циклоприсоединения [3+2] с образованием изоксазола 5. Такое преобразование можно провести в одном сосуде, однако она требует двух отдельных этапов. Первый этап представляет собой окисление оксимной функциональной группы в нитрон или аналогичное промежуточное соединение с той же степенью окисления, например, хлороксим. Затем эти реакционноспособные формы взаимодействуют с алкином в циклоприсоединении [3+2] с образованием изоксазолового аддукта.
В некоторых вариантах осуществления подходящие условия образования изоксазола состоят из двух этапов, причем первый этап содержит взаимодействие соединения формулы 4 в подходящих условиях образования хлороксима с образованием хлороксимного промежуточного соединения; второй этап содержит взаимодействие хлороксимного промежуточного соединения с ацетиленом в подходящих условиях циклоприсоединения с образованием соединения формулы 5.
В некоторых вариантах осуществления условия образования хлороксима выбраны из
a) N-хлорсукцинимида и подходящего растворителя;
b) пероксимоносульфата калия, HCl и диоксана; и
c) гипохлорита натрия и подходящего растворителя.
К примерам подходящих растворителей относятся, без ограничений, апротонные растворители (например, DCM, DCE, THF, 2-MeTHF, MTBE и диоксан), ароматические углеводороды (например, толуол, ксилолы) и алкилацетаты (например, изопропилацетат, этилацетат).
Выделения продукта можно достичь путем добавления антирастворителя к раствору соединения формулы 5. К примерам подходящих растворителей для выделения хлороксимного промежуточного соединения относятся смеси подходящих растворителей (EtOAc, IPAC) с углеводородами (например, гексаны, гептан, циклогексан) или ароматическими углеводородами (например, толуол, ксилолы). В некоторых вариантах осуществления гептан добавляют к раствору хлороксима в IPAC.
Подходящие условия циклоприсоединения состоят из объединения хлороксима с ацетиленом с подходящим основанием и подходящим растворителем. К подходящим растворителям относятся протонные растворители, апротонные растворители, полярные растворители и неполярные растворители. К примерам подходящих растворителей относятся, без ограничений, ацетонитрил, тетрагидрофуран, 2-метилтетрагидрофуран, MTBE, EtOAc, i-PrOAc, DCM, толуол, DMF и метанол. Подходящие основания включают в себя, без ограничений, пиридин, DIEA, TEA, t-BuONa и K2CO3. В некоторых вариантах осуществления подходящие условия циклоприсоединения содержат добавления 1,0 эквивалента хлороксима, 1,0 эквивалента ацетилена, 1,1 эквивалента Et3N в DCM.
Выделения продукта можно достичь путем добавления антирастворителя к раствору соединения формулы 5. К примерам подходящих растворителей для выделения хлороксима относятся смеси подходящих растворителей (EtOAc, IPAC) с углеводородами (например, гексаны, гептан, циклогексан) или ароматическими углеводородами (например, толуол, ксилолы). В некоторых вариантах осуществления гептан добавляют к раствору хлороксима в IPAC.
Этап 5
На этапе 5 показан (-ы) заключительный (-ые) этап (-ы) получения соединений формулы I. Если группа R4 представляет собой бром, промежуточное соединение 5 может вступать в реакцию перекрестного сочетания Сузуки с бороновой кислотой или эфирами в условиях, известных специалистам в данной области, с образованием соединений, где R4 представляет собой арил, гетероарил или альтернативные фрагменты, полученные в результате реакции сочетания с участием металла. При введении подходящих функциональных групп в промежуточное соединение 5 можно провести этап снятия защиты для удаления защитных групп и получения соединений формулы I.
Специалистам в данной области известны реакции сочетания с участием металла (см., например, Org.Proc. Res. Dev. 2010 г., 30–47). В некоторых вариантах осуществления подходящие условия сочетания содержат добавление 0,1 эквивалента Pd[P(tBu)3]2; 1 эквивалента бороновой кислоты или эфира; и 2 эквивалентов карбоната натрия в соотношении 2:1 об./об. ацетонитрил/вода при 60–70 °C. В других вариантах осуществления подходящие условия сочетания содержат добавление 0,010–0,005 эквивалента Pd(dtbpf)Cl2, 1 эквивалента бороновой кислоты или эфира и 2 эквивалентов карбоната калия в смеси толуола и воды в соотношении 7:2 об./об. при 70 °C.
Конечный продукт можно обработать поглотителем металла (силикагель, функционализированные смолы, древесный уголь) (см., например, Org. Proc. Res. Dev. 2005 г., 198–205). В некоторых вариантах осуществления раствор продукта обрабатывают смолой Biotage MP-TMT.
Продукт также можно выделить путем кристаллизации из спиртосодержащего раствора (например, метанола, этанола, изопропанола). В некоторых вариантах осуществления растворитель представляет собой этанол. В других вариантах осуществления растворитель представляет собой изопропанол.
Снятие защиты с Boc-групп известно в данной области (см., например, Protecting Groups in Organic Synthesis, Greene and Wuts). В некоторых вариантах осуществления подходящие условия снятия защиты представляют собой соляную кислоту в ацетоне при 35–45 °C. В некоторых вариантах осуществления подходящие условия снятия защиты представляют собой TFA в DCM.
Этап 6
На этапе 6 показано, каким образом соединения формулы I преобразуются в соединения формулы I-A с использованием основания в подходящих условиях, известных специалистам в данной области. В некоторых вариантах осуществления выделения соединений формулы I в форме свободного основания можно достичь путем добавления подходящего основания, такого как NaOH, к спиртосодержащему кислотному раствору соединений формулы I для осаждения продукта.
Этап 7
На этапе 7 показано, каким образом соединения формулы I-А преобразуются в соединения формулы I-В с использованием кислоты в подходящих условиях, известных специалистам в данной области.
В некоторых вариантах осуществления подходящие условия включают добавление водной HCl к суспензии соединений формулы I-A в ацетоне при 35 °C с последующим нагреванием при 50 °C.
СХЕМА B: образование d1-бороната
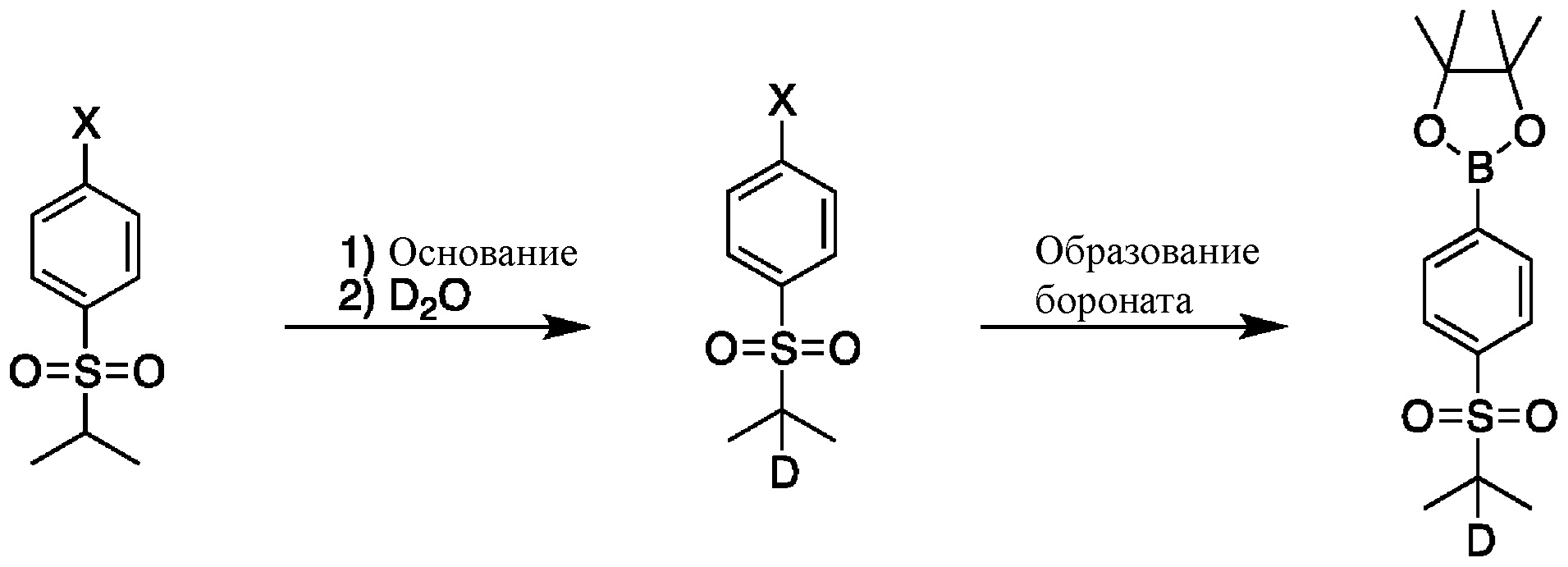
На схеме B показан общий способ синтеза для получения промежуточных соединений d1-бороната. Подходящий 1-галоген-(изопропилсульфонил)бензол обрабатывают основанием, таким как, без ограничений, NaH, LiHMDS или KHMDS, с последующим гашением аниона источником дейтерия, таким как D2O. Затем галоген преобразуют в соответствующее производное бороната путем, например, металл-опосредованного перекрестного сочетания, катализированного, например, Pd(tBu3)2 или Pd(dppf)Cl2∙DCM.
СХЕМА C: образование d6-бороната
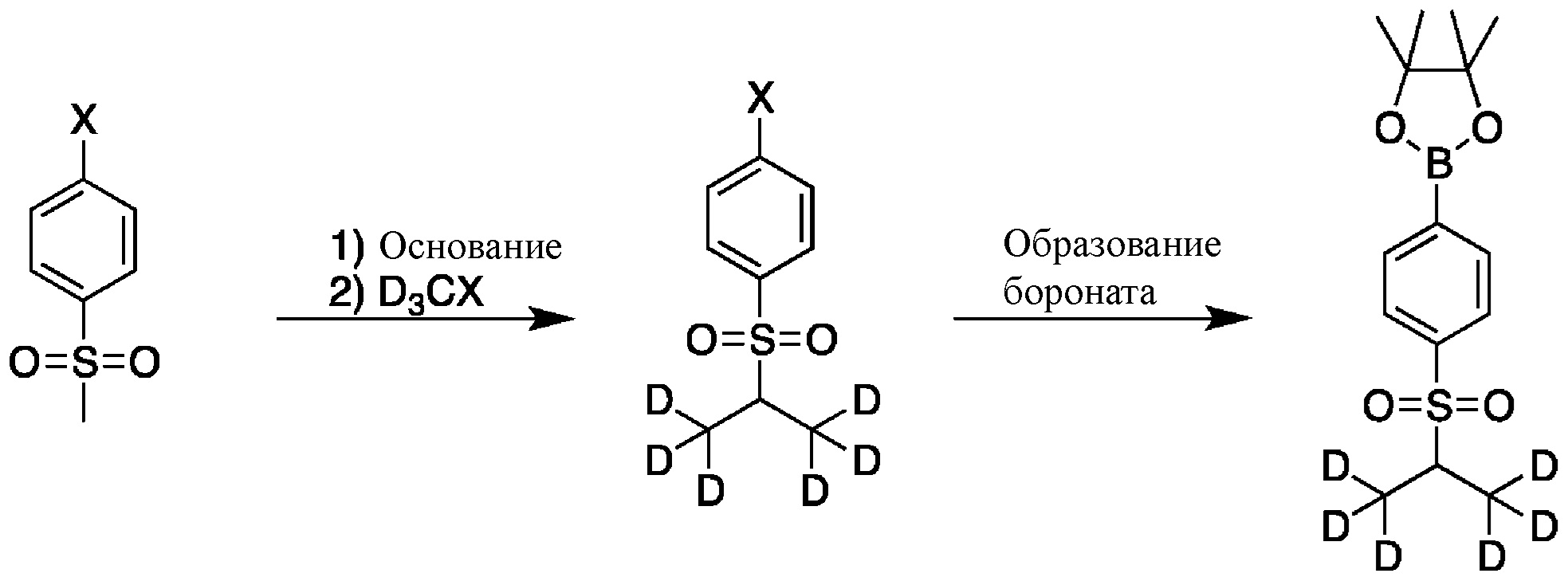
На схеме C показан общий способ синтеза для получения промежуточных соединений d6-бороната. Подходящий 1-галоген-(метилсульфонил)бензол обрабатывают основанием, таким как, без ограничений, NaH, LiHMDS или KHMDS, с последующим гашением аниона источником дейтерия, таким как D3CI. Эту реакцию повторяют до тех пор, пока в молекулу не будет включено желаемое количество дейтерия. Затем галоген преобразуют в соответствующее производное бороната путем, например, металл-опосредованного перекрестного сочетания, катализированного, например, Pd(tBu3)2 или Pd(dppf)Cl2∙DCM.
СХЕМА D: образование d7-бороната
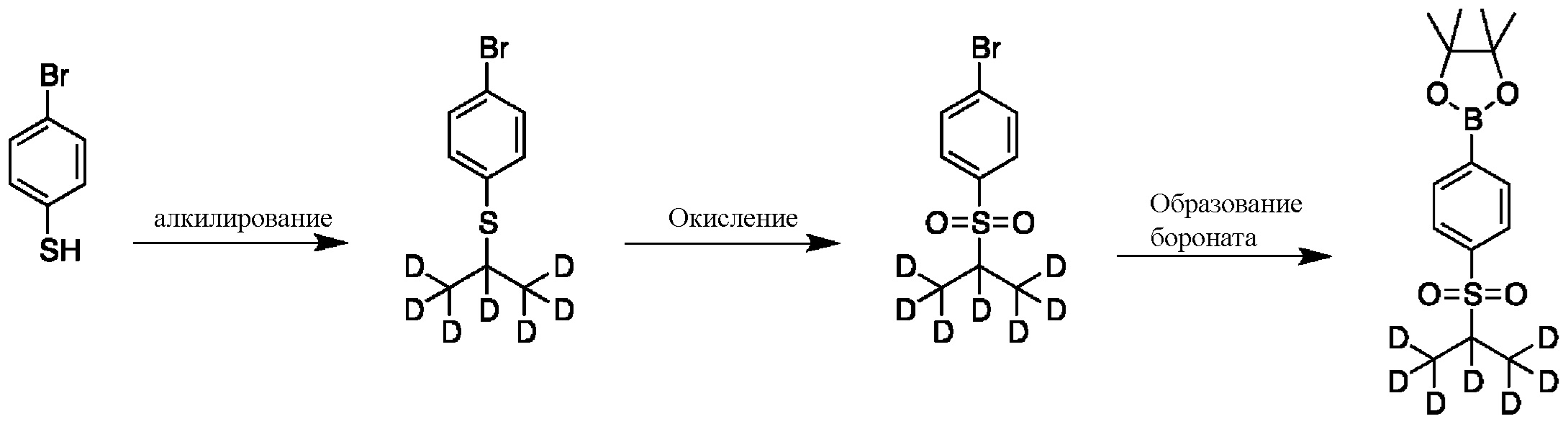
На схеме D показан общий способ синтеза для получения промежуточных соединений d7-бороната. 4-Бромбензолтиол обрабатывают основанием, таким как, без ограничений, NaH, LiHMDS или KHMDS, с последующим гашением аниона источником дейтерия, таким как 1,1,1,2,3,3,3-гептадейтерио-2-йод-пропан. Затем сульфид окисляют до соответствующего сульфона с использованием, например, mCPBA или Oxone. Затем галоген преобразуют в соответствующее производное бороната путем, например, металл-опосредованного перекрестного сочетания, катализированного, например, Pd(tBu3)2 или Pd(dppf)Cl2∙DCM.
СХЕМА E: образование дейтерированного бороната с арильным кольцом
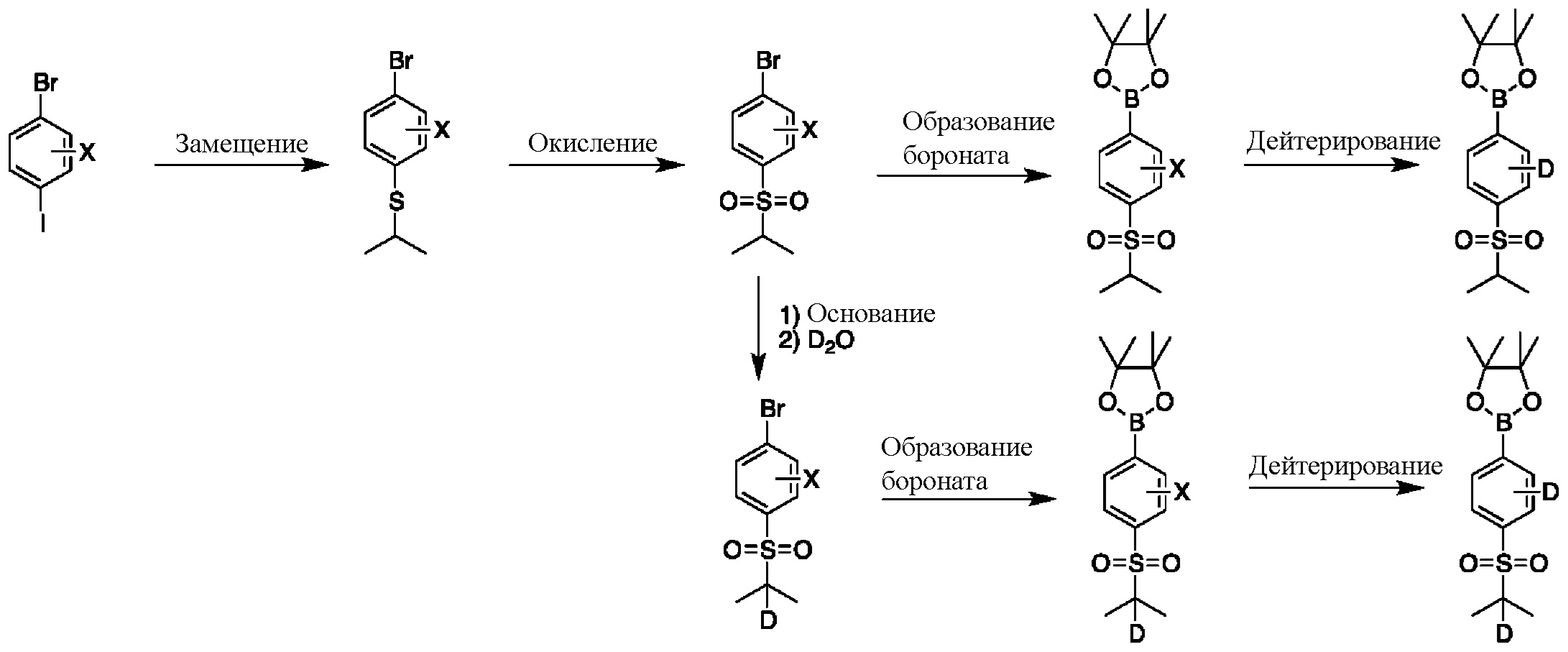
На схеме E показан общий способ синтеза для получения промежуточных соединений бороната, в котором арильное кольцо замещено дейтерием. Подходящее производное 1-йод-4-бромарил обрабатывают замещенным тиолом, таким как пропан-2-тиол, в условиях металл-катализируемого сочетания с использованием катализатора, такого как CuI. Затем сульфид окисляют до соответствующего сульфона с использованием, например, mCPBA или Oxone. После этого бромид преобразуют в подходящее производное бороната путем, например, металл-опосредованного перекрестного сочетания, катализированного, например, Pd(tBu3)2 или Pd(dppf)Cl2∙DCM. Затем остальные заместители преобразуют в дейтерий путем, например, металл-катализированного обмена галоген-дейтерий с использованием подходящего металлического катализатора, такого как Pd на С, в атмосфере газа дейтерия. Кроме того, 1-бром-(изопропилсульфонил)бензол можно обработать основанием, таким как, без ограничений, NaH, LiHMDS или KHMDS, с последующим гашением аниона источником дейтерия, таким как D2O. Затем бромид преобразуют в подходящее производное бороната путем, например, металл-опосредованного перекрестного сочетания, катализированного, например, Pd(tBu3)2 или Pd(dppf)Cl2∙DCM. Затем остальные заместители преобразуют в дейтерий путем, например, металл-катализированного обмена галоген-дейтерий с использованием подходящего металлического катализатора, такого как Pd на С, в атмосфере газа дейтерия.
СХЕМА F: образование дейтерированного бороната с арильным кольцом
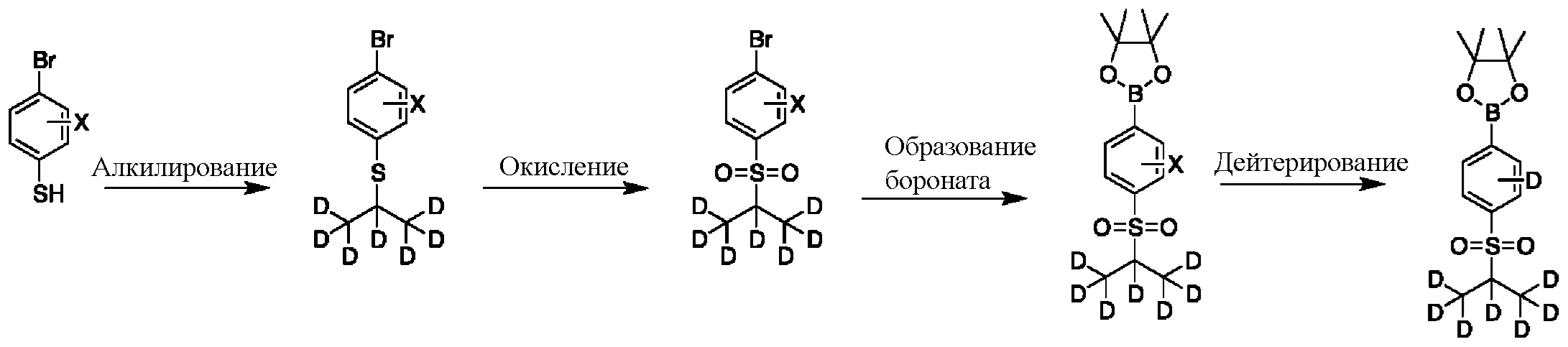
На схеме F показан другой общий способ синтеза для получения промежуточных соединений бороната, в которых арильное кольцо замещено дейтерием. Замещенный 4-бромбензолтиол обрабатывают основанием, таким как, без ограничений, NaH, LiHMDS или KHMDS, с последующим гашением аниона источником дейтерия, таким как 1,1,1,2,3,3,3-гептадейтерио-2-йод-пропан. Затем сульфид окисляют до соответствующего сульфона с использованием, например, mCPBA или Oxone. Затем галоген преобразуют в соответствующее производное бороната путем, например, металл-опосредованного перекрестного сочетания, катализированного, например, Pd(tBu3)2 или Pd(dppf)Cl2∙DCM. Затем остальные заместители преобразуют в дейтерий путем, например, металл-катализированного обмена галоген-дейтерий с использованием подходящего металлического катализатора, такого как Pd на С, в атмосфере газа дейтерия.
СХЕМА G: образование дейтерированного бороната с арильным кольцом
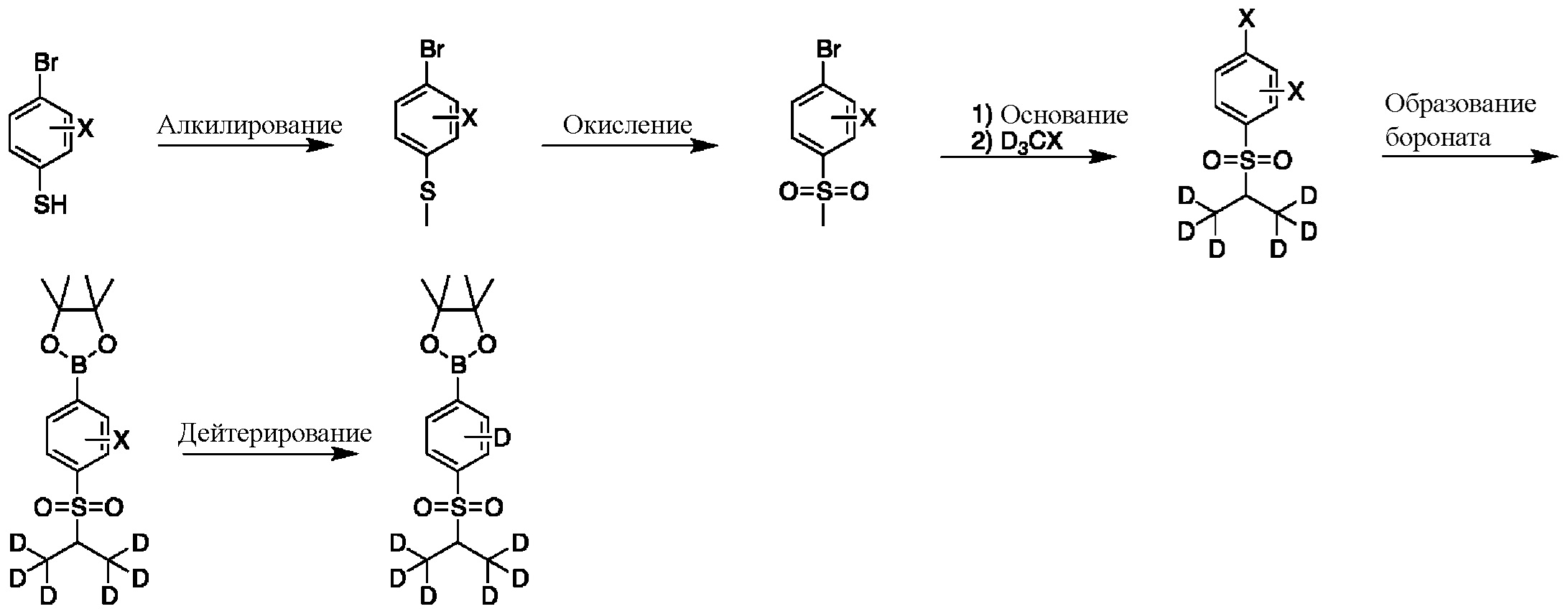
На схеме G показан другой общий способ синтеза для получения промежуточных соединений бороната, в которых арильное кольцо замещено дейтерием. Замещенный 4-бромбензолтиол обрабатывают основанием, таким как, без ограничений, NaH, LiHMDS или KHMDS, с последующим гашением аниона, например, с помощью MeI. Затем сульфид окисляют до соответствующего сульфона с использованием, например, mCPBA или Oxone. Сульфон обрабатывают основанием, таким как, без ограничений, NaH, LiHMDS или KHMDS, с последующим гашением аниона источником дейтерия, таким как D3CI. Эту реакцию повторяют до тех пор, пока в молекулу не будет включено желаемое количество дейтерия. Затем галоген преобразуют в соответствующее производное бороната путем, например, металл-опосредованного перекрестного сочетания, катализированного, например, Pd(tBu3)2 или Pd(dppf)Cl2∙DCM. Затем остальные заместители преобразуют в дейтерий путем, например, металл-катализированного обмена галоген-дейтерий с использованием подходящего металлического катализатора, такого как Pd на С, в атмосфере газа дейтерия.
СХЕМА H: образование дейтерированных промежуточных соединений оксима с арильным кольцом
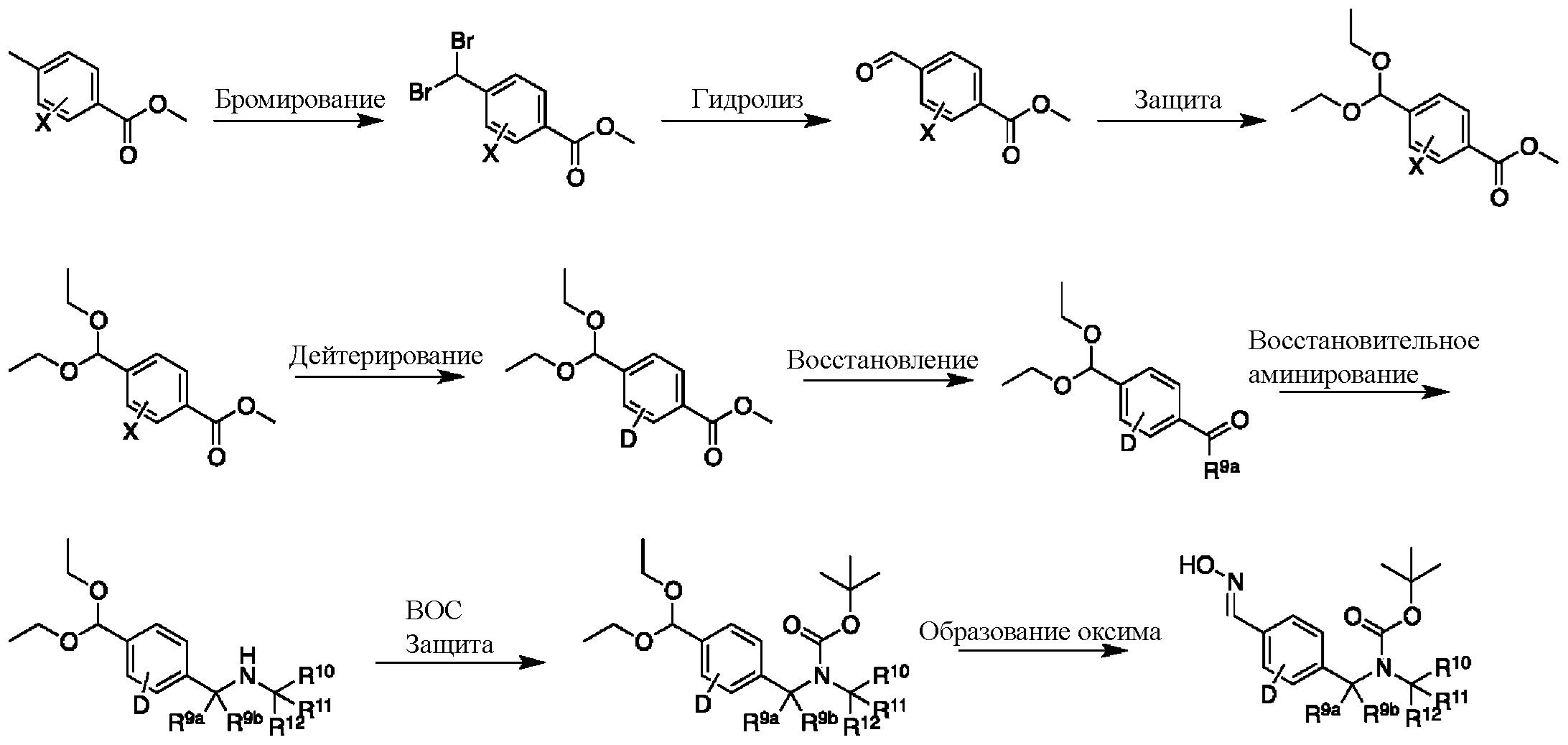
На схеме H показан общий способ синтеза для получения промежуточных соединений оксима, в которых арильное кольцо замещено дейтерием. Метильную группу надлежащим образом замещенного производного метилового эфира 4-метилбензойной кислоты можно преобразовать в соответствующий дибромид в таких условиях, как бромирование, катализируемое AIBN, с NBS. Затем такой дибромид гидролизуют до соответствующего альдегида, например, с использованием AgNO3 в ацетоне/воде. Защита альдегида в виде подходящего ацеталя, например, диэтилацеталя, и последующее преобразование остальных заместителей в дейтерий, например, металл-катализируемым обменом галоген-дейтерий с использованием подходящего металлического катализатора, такого как Pd на С, в атмосфере газа дейтерия, приводят к образованию промежуточного соединения дейтерированного эфира. Эфирная функциональная группа может быть восстановлена с использованием таких реагентов, как LiAlH4, NaBH4, NaBD4 или LiAlD4, с образованием соответствующего альдегида. Это соединение может вступать в реакцию в условиях восстановительного аминирования с применением подходящего амина, такого как метиламин или d3-метиламин, с использованием восстанавливающего агента, такого как NaBH4 или NaBD4, с получением соответствующего производного амина. Соединение может быть защищено, например, Boc-группой и ацеталем, преобразованным в оксим, с использованием, например, гидрохлорида гидроксиламина в THF/воде.
СХЕМА I: образование дейтерированных промежуточных соединений оксима с арильным кольцом
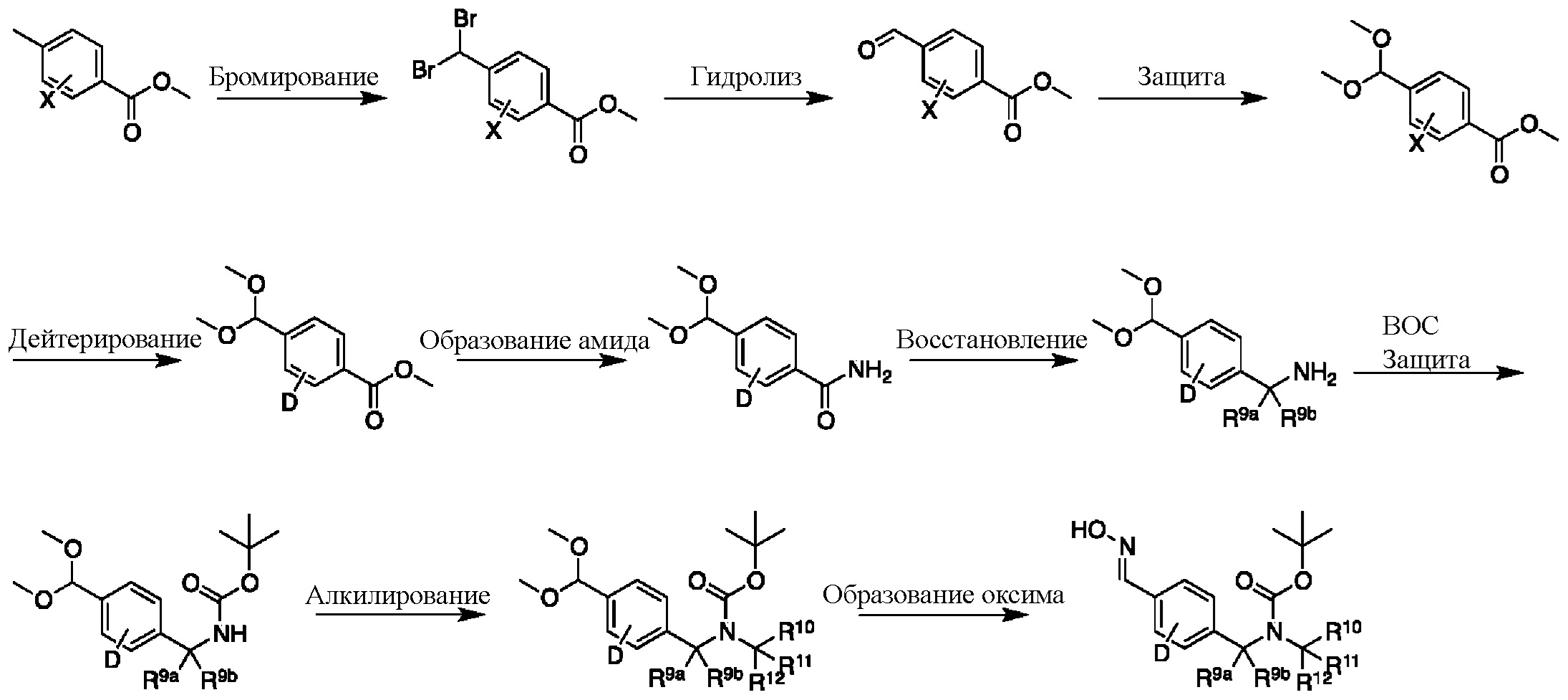
На схеме I показан другой общий способ синтеза для получения промежуточных соединений оксима, в которых арильное кольцо замещено дейтерием. Метильную группу надлежащим образом замещенного производного метилового эфира 4-метилбензойной кислоты можно преобразовать в соответствующий дибромид в таких условиях, как бромирование, катализируемое AIBN, с NBS. Затем такой дибромид гидролизуют до соответствующего альдегида, например, с использованием AgNO3 в ацетоне/воде. Защита альдегида в виде подходящего ацеталя, например, диметилацеталя, и последующее преобразование остальных заместителей в дейтерий, например, металл-катализируемым обменом галоген-дейтерий с использованием подходящего металлического катализатора, такого как Pd на С, в атмосфере газа дейтерия, приводят к образованию промежуточного соединения дейтерированного эфира. Эфирная функциональная группа может быть преобразована в соответствующий первичный амид в стандартных условиях, таких как нагрев с раствором аммиака в метаноле. Амид может быть восстановлен до соответствующего амина с помощью реагентов, без ограничений, LiAlH4 или LiAlD4. Соединение может быть защищено, например, Boc-группой. Карбамат NH можно алкилировать в щелочной среде с использованием, например, NaH, LiHMDS или KHMDS, с последующим гашением аниона источником дейтерия, таким как MeI или D3CI. Ацеталь можно преобразовать в оксим с использованием, например, гидрохлорида гидроксиламина в THF/воде.
СХЕМА J: образование дейтерированных промежуточных соединений оксима с арильным кольцом
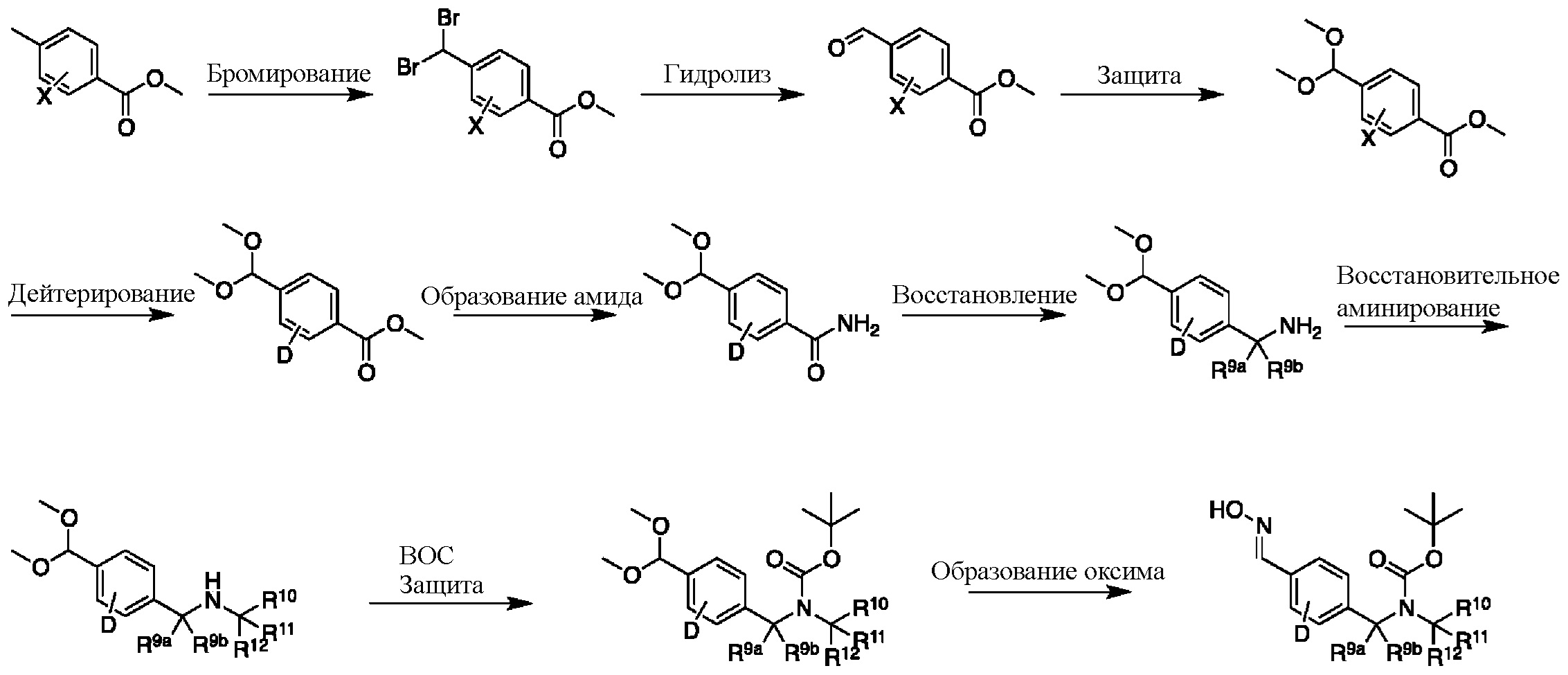
На схеме J показан другой общий способ синтеза для получения промежуточных соединений оксима, в которых арильное кольцо замещено дейтерием. Метильную группу надлежащим образом замещенного производного метилового эфира 4-метилбензойной кислоты можно преобразовать в соответствующий дибромид в таких условиях, как бромирование, катализируемое AIBN, с NBS. Затем такой дибромид гидролизуют до соответствующего альдегида, например, с использованием AgNO3 в ацетоне/воде. Защита альдегида в виде подходящего ацеталя, например, диметилацеталя, и последующее преобразование остальных заместителей в дейтерий, например, металл-катализируемым обменом галоген-дейтерий с использованием подходящего металлического катализатора, такого как Pd на С, в атмосфере газа дейтерия, приводят к образованию промежуточного соединения дейтерированного эфира. Эфирная функциональная группа может быть преобразована в соответствующий первичный амид в стандартных условиях, таких как нагрев с раствором аммиака в метаноле. Амид может быть восстановлен до соответствующего амина с помощью реагентов, без ограничений, LiAlH4 или LiAlD4. Это соединение может вступать в реакцию в условиях восстановительного аминирования с использованием подходящего амина, такого как метиламин, d3-метиламин, формальдегид или d2-формальдегид, с использованием восстанавливающего агента, такого как NaBH4 или NaBD4, с получением соответствующего производного амина. Соединение может быть защищено, например, Boc-группой. Ацеталь можно преобразовать в оксим с использованием, например, гидрохлорида гидроксиламина в THF/воде.
СХЕМА K: образование дейтерированных промежуточных соединений оксима с арильным кольцом
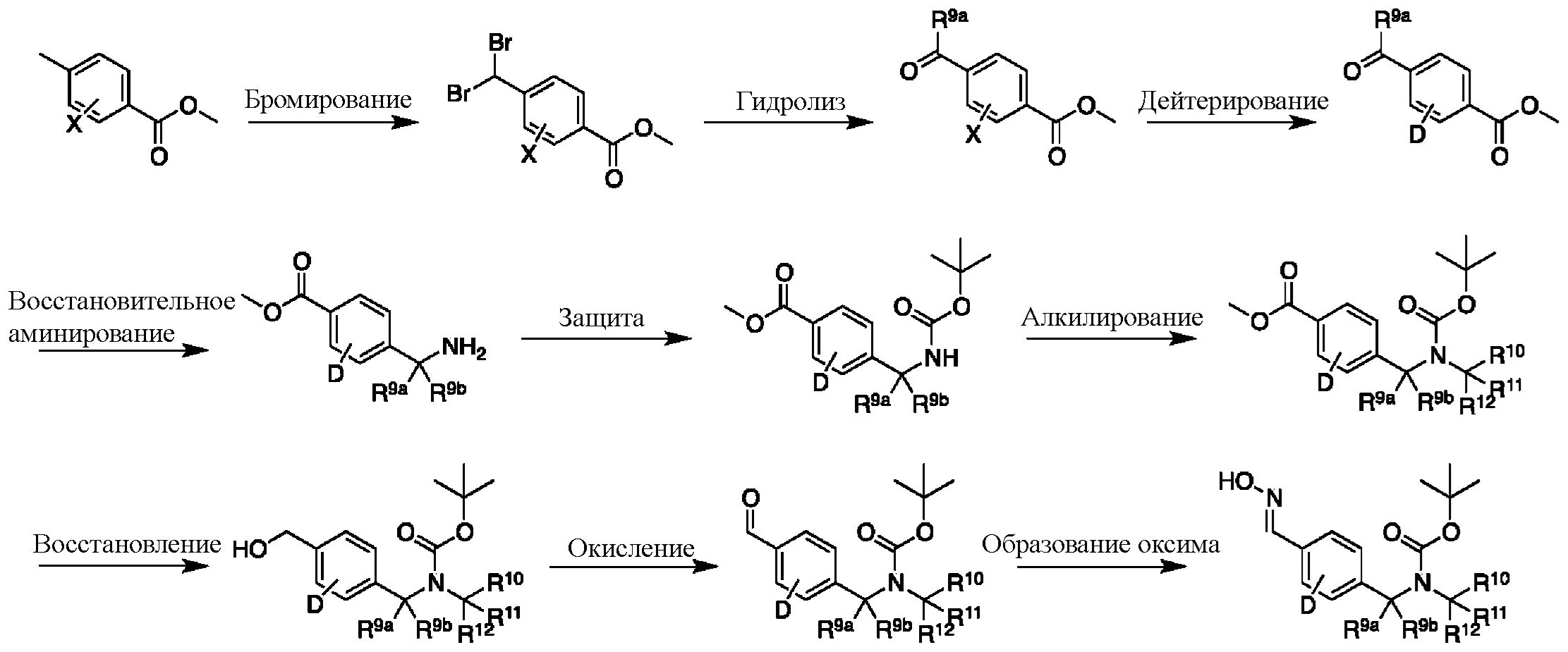
На схеме К показан другой общий способ синтеза для получения промежуточных соединений оксима, в которых арильное кольцо замещено дейтерием. Метильную группу надлежащим образом замещенного производного метилового эфира 4-метилбензойной кислоты можно преобразовать в соответствующий дибромид в таких условиях, как бромирование, катализируемое AIBN, с NBS. Затем такой дибромид гидролизуют до соответствующего альдегида, например, с использованием AgNO3 в ацетоне/воде. Защита альдегида в виде подходящего ацеталя, например, диметилацеталя, и последующее преобразование остальных заместителей в дейтерий, например, металл-катализируемым обменом галоген-дейтерий с использованием подходящего металлического катализатора, такого как Pd на С, в атмосфере газа дейтерия, приводят к образованию промежуточного соединения дейтерированного эфира. Это соединение может вступать в реакцию в условиях восстановительного аминирования с использованием подходящего амина, такого как гидроксид аммиака, с использованием восстанавливающего агента, такого как NaBH4 или NaBD4, с получением соответствующего производного амина. Соединение может быть защищено, например, Boc-группой и карбаматом NH, алкилированным в щелочной среде с использованием, например, NaH, LiHMDS или KHMDS, с последующим гашением аниона источником дейтерия, таким как MeI или D3CI. Эфир может быть восстановлен до соответствующего спирта с использованием подходящего восстанавливающего агента, такого как LiBH4 или NaBH4. Спирт можно окислить до альдегида с помощью реагентов, таких как MnO2 или периодинан Десса — Мартина. Ацеталь может быть преобразован в оксим с использованием, например, водного раствора гидроксиламина.
СХЕМА L: образование дейтерированных промежуточных соединений оксима
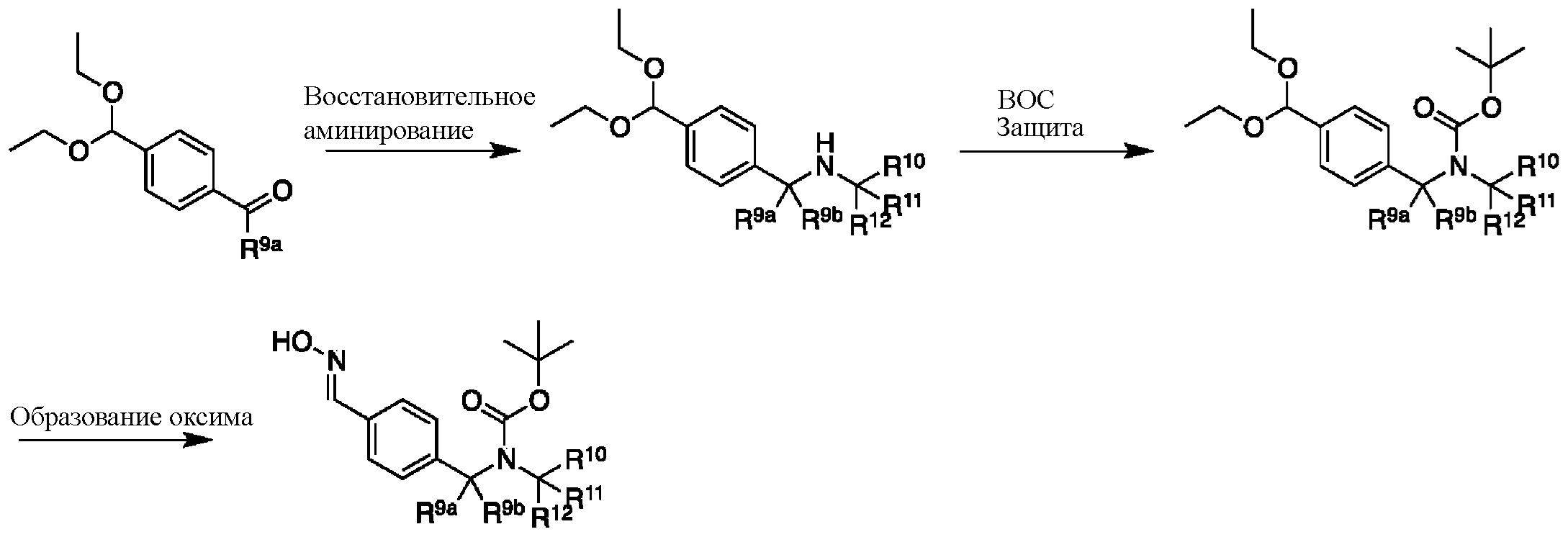
На схеме L показан общий способ синтеза для получения промежуточных соединений дейтерированного оксима. 4-(Диэтоксиметил)бензальдегид может вступить в реакцию в условиях восстановительного аминирования с использованием подходящего амина, такого как метиламин или d3-метиламин, с использованием восстанавливающего агента, такого как NaBH4 или NaBD4, с получением соответствующего производного амина. Соединение может быть защищено, например, Boc-группой и ацеталем, преобразованным в оксим, с использованием, например, гидрохлорида гидроксиламина в THF/воде.
СХЕМА M: образование дейтерированных промежуточных соединений оксима
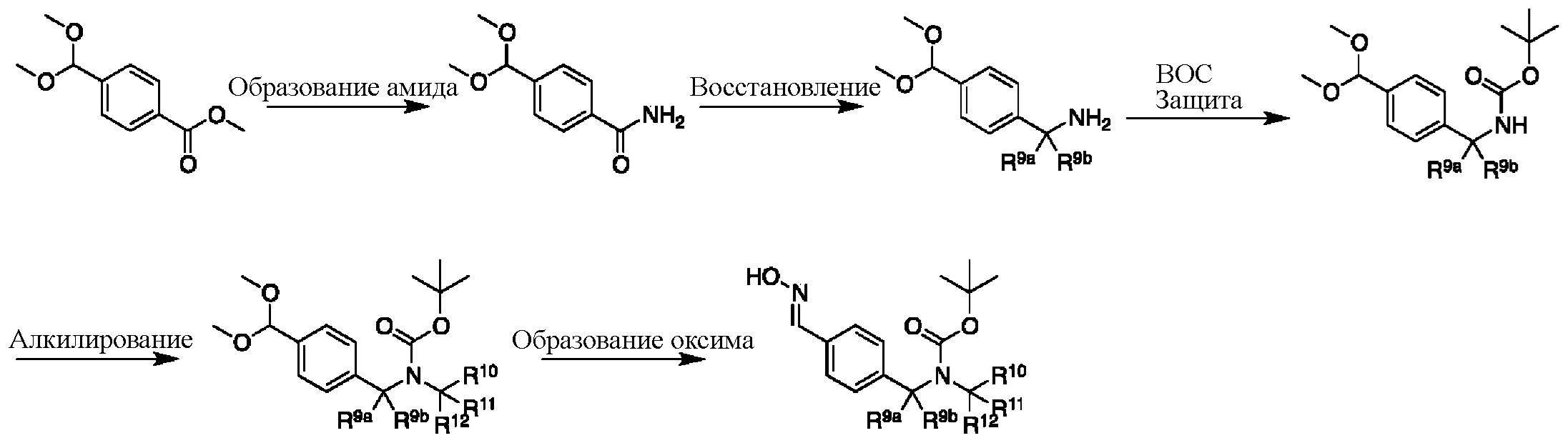
На схеме M показан другой общий способ синтеза для получения промежуточных соединений дейтерированного оксима. Эфирная функциональная группа метилового эфира 4-(диметоксиметил)бензоата может быть преобразована в соответствующий первичный амид в стандартных условиях, таких как нагрев с раствором аммиака в метаноле. Амид может быть восстановлен до соответствующего амина с помощью реагентов, без ограничений, LiAlH4 или LiAlD4. Соединение может быть защищено, например, Boc-группой. Карбамат NH можно алкилировать в щелочной среде с использованием, например, NaH, LiHMDS или KHMDS, с последующим гашением аниона источником дейтерия, таким как MeI или D3CI. Ацеталь можно преобразовать в оксим с использованием, например, гидрохлорида гидроксиламина в THF/воде.
СХЕМА N: образование дейтерированных промежуточных соединений оксима
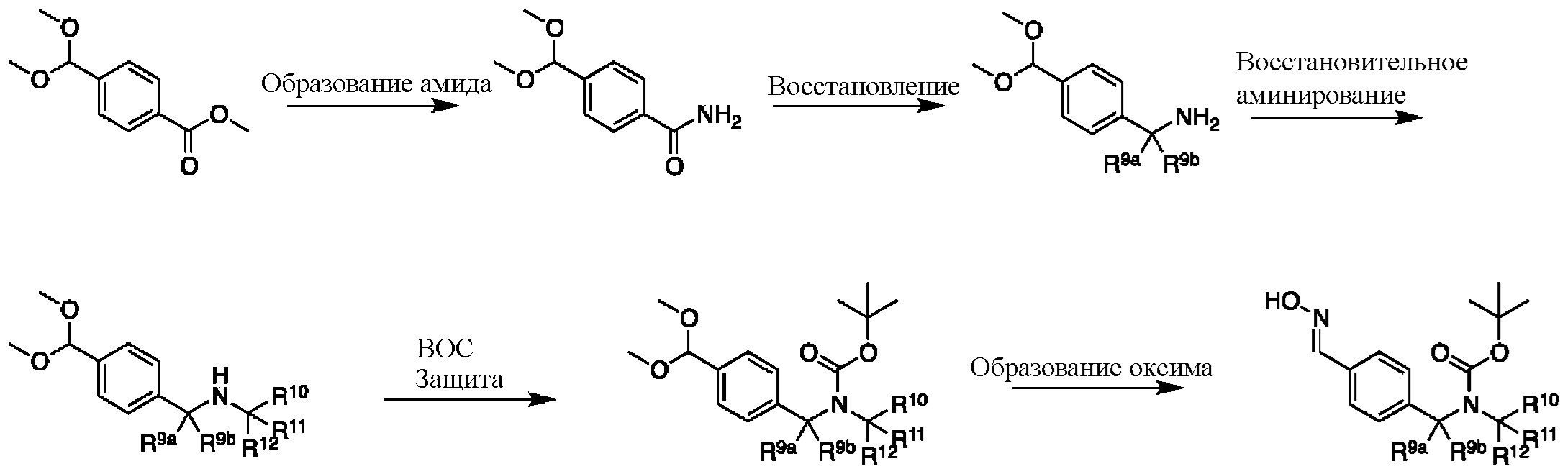
На схеме N показан другой общий способ синтеза для получения промежуточных соединений дейтерированного оксима. Эфирная функциональная группа метилового эфира 4-(диметоксиметил)бензоата может быть преобразована в соответствующий первичный амид в стандартных условиях, таких как нагрев с раствором аммиака в метаноле. Амид может быть восстановлен до соответствующего амина с помощью реагентов, без ограничений, LiAlH4 или LiAlD4. Это соединение может вступать в реакцию в условиях восстановительного аминирования с использованием подходящего амина, такого как метиламин, d3-метиламин, формальдегид или d2-формальдегид, с использованием восстанавливающего агента, такого как NaBH4 или NaBD4, с получением соответствующего производного амина. Соединение может быть защищено, например, Boc-группой. Ацеталь можно преобразовать в оксим с использованием, например, гидрохлорида гидроксиламина в THF/воде.
СХЕМА O: образование дейтерированных промежуточных соединений оксима
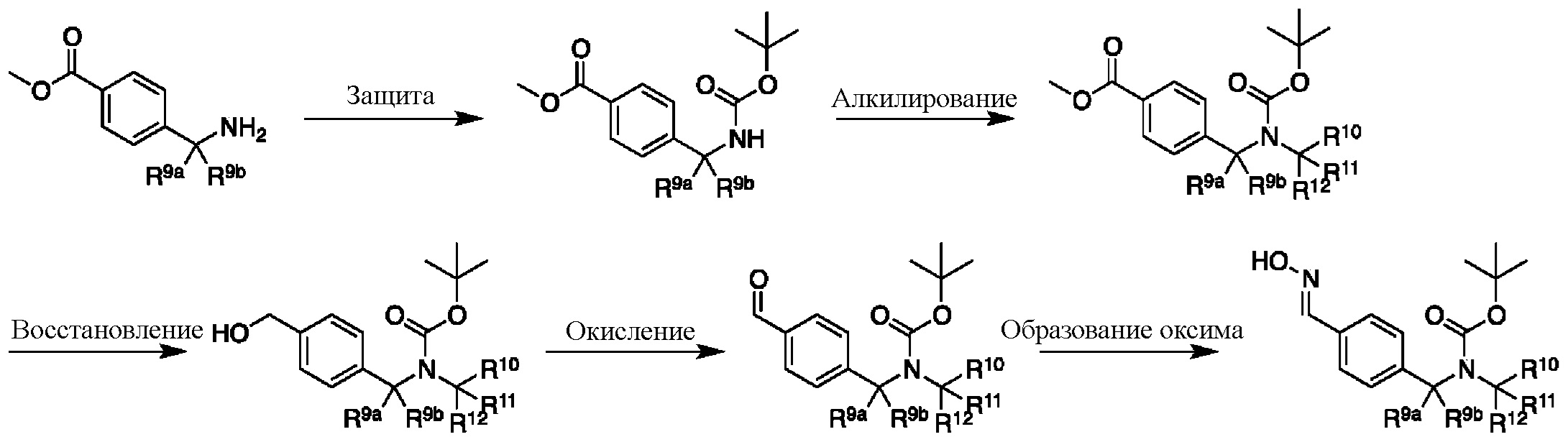
На схеме О показан другой общий способ синтеза для получения промежуточных соединений дейтерированного оксима. 4-Замещенный бензиламин можно защитить, например, Boc-группой. Карбамат NH можно алкилировать в щелочной среде с использованием, например, NaH, LiHMDS или KHMDS, с последующим гашением аниона источником дейтерия, таким как MeI или D3CI. Эфир может быть восстановлен до соответствующего спирта с использованием подходящего восстанавливающего агента, такого как LiBH4 или NaBH4. Спирт можно окислить до альдегида с помощью реагентов, таких как MnO2 или периодинан Десса-Мартина. Ацеталь может быть преобразован в оксим с использованием, например, водного раствора гидроксиламина.
СХЕМА P: образование производных изоксазола
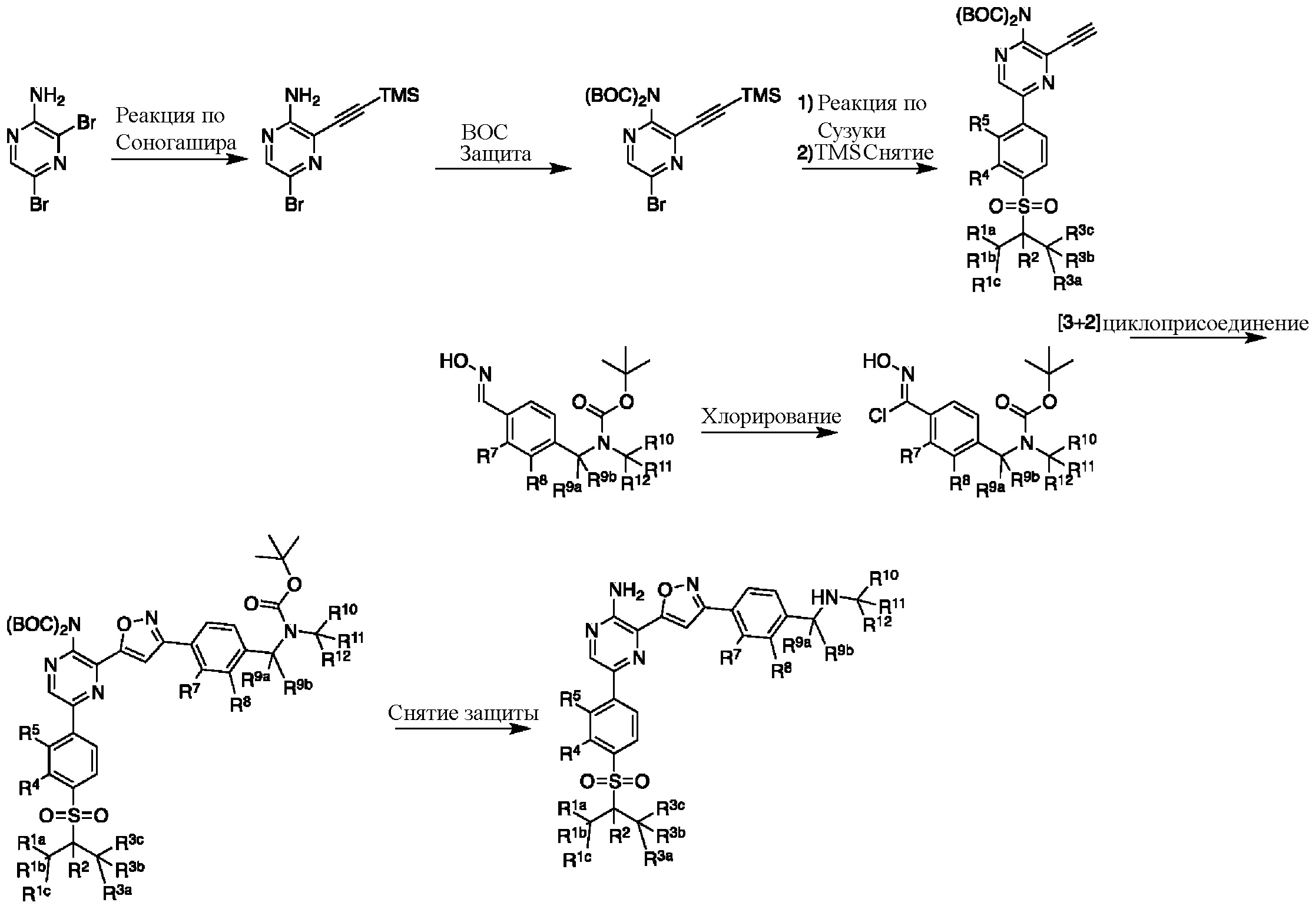
На схеме P показан общий способ синтеза для получения дейтерированных производных пиразинизоксазола. 3,5-Дибромпиразин-2-амин преобразовали в соответствующий силил-защищенный алкин в стандартных условиях реакции Соногашира, используя в качестве катализаторов, например, Pd(PPh3)4 и CuI. Затем пиразин NH2 можно защитить так же, как, например, производное ди-Boc. Сочетание бромида пиразина с боронатом, например, как кратко описано в схемах 1–6 выше, в стандартных условиях перекрестного сочетания Сузуки с последующим удалением защитных групп силила с образованием желаемого промежуточного соединения алкина. Оксимы, например, которые кратко описаны в схемах 7–14, можно преобразовать в соответствующие хлороксимы с использованием, например, NCS. Промежуточные соединения алкина и хлороксима можно подвергнуть реакции циклоприсоединения [3+2] с образованием соответствующего изоксазола в стандартных условиях, например, путем добавления Et3N. Защитные Boc-группы можно удалить в кислотной среде, такой как TFA в DCM или HCl в смеси MeOH/DCM, с образованием производных дейтерированного пиразинизоксазола.
СХЕМА Q: образование дейтерированных производных изоксазола
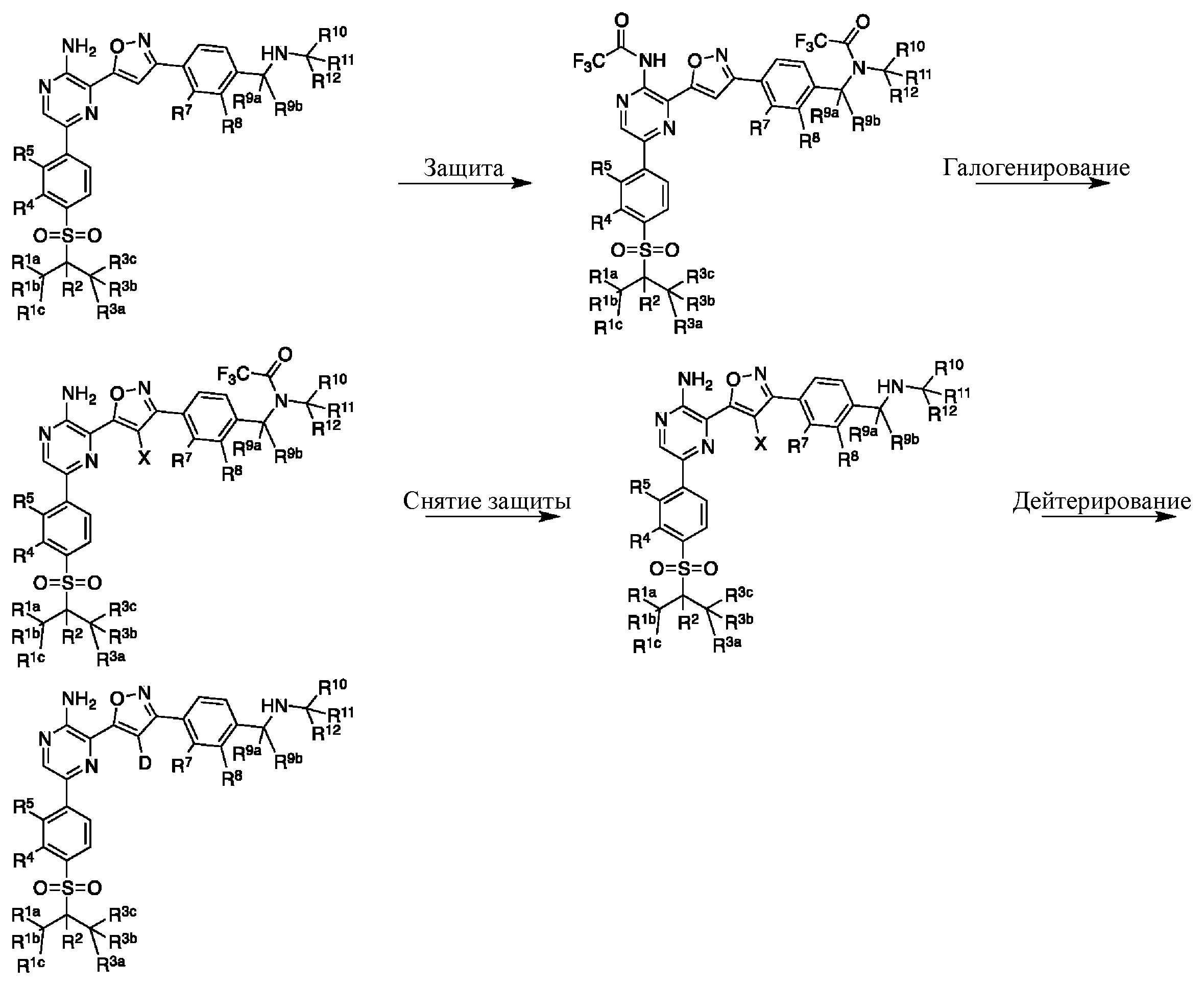
На схеме Q показан общий способ синтеза для получения дейтерированных производных изоксазола. Пиразин NH2 и бензиламинамин NH можно защитить в стандартных условиях с использованием трифторуксусного ангидрида. Галогенирование изоксазольного кольца, например, с помощью NIS, с последующим удалением трифторуксусной защитной группы в щелочной среде обеспечивает желаемые галогензамещенные промежуточные соединения. Затем галоген можно преобразовать в дейтерий путем, например, металл-катализируемого обмена галоген-дейтерий с использованием подходящего металлического катализатора, такого как Pd на С, в атмосфере газа дейтерия.
Сокращения
Приняты следующие сокращения:
АТФ аденозинтрифосфат
Boc трет-бутилкарбамат
Cbz карбоксибензил
DCM дихлорметан
DMSO диметилсульфоксид
Et3N триэтиламин
2-MeTHF 2-метилтетрагидрофуран
NMM N-метилморфолин
DMAP 4-диметиламинопиридин
TMS триметилсилил
MTBE метил-трет-бутиловый эфир
EtOAc этилацетат
i-PrOAc изопропилацетат
IPAC изопропилацетат
DMF диметилформамид
DIEA диизопропилэтиламин
TEA триэтиламин
t-BuONa трет-бутоксид натрия
K2CO3 карбонат калия
PG защитная группа
pTSA пара-толуолсульфоновая кислота
TBAF фторид тетра-н-бутиламмония
1H ЯМР протонный ядерный магнитный резонанс
ВЭЖХ высокоэффективная жидкостная хроматография
ЖХМС жидкостная хроматография-масс-спектрометрия
ТСХ тонкослойная хроматография
Rt время удержания
СХЕМЫ И ПРИМЕРЫ
Соединения настоящего описания можно получить в свете технического описания с использованием этапов, по существу известных специалистам в данной области. Такие соединения можно анализировать известными способами, включая, без ограничений, ЖХМС (жидкостная хроматография-масс-спектрометрия) и ЯМР (ядерный магнитный резонанс). Приведенные ниже общие схемы и примеры иллюстрируют способы получения соединений настоящего описания. Примеры приведены только в целях иллюстрации и не призваны ограничить каким-либо образом объем настоящего изобретения. Спектры 1H ЯМР регистрировали на 400 МГц с использованием прибора Bruker DPX 400. Образцы для масс-спектрометрии анализировали на масс-спектрометре MicroMass Quattro Micro, работающем в одиночном МС-режиме с ионизацией электрораспылением.
Пример 1. Синтез 2-(4-(5-амино-6-(3-(4-((тетрагидро-2 H -пиран-4-иламино)метил)фенил)изоксазол-5-ил)пиразин-2-ил)пиридин-2-ил)-2-метилпропаннитрила (соединение I-1)
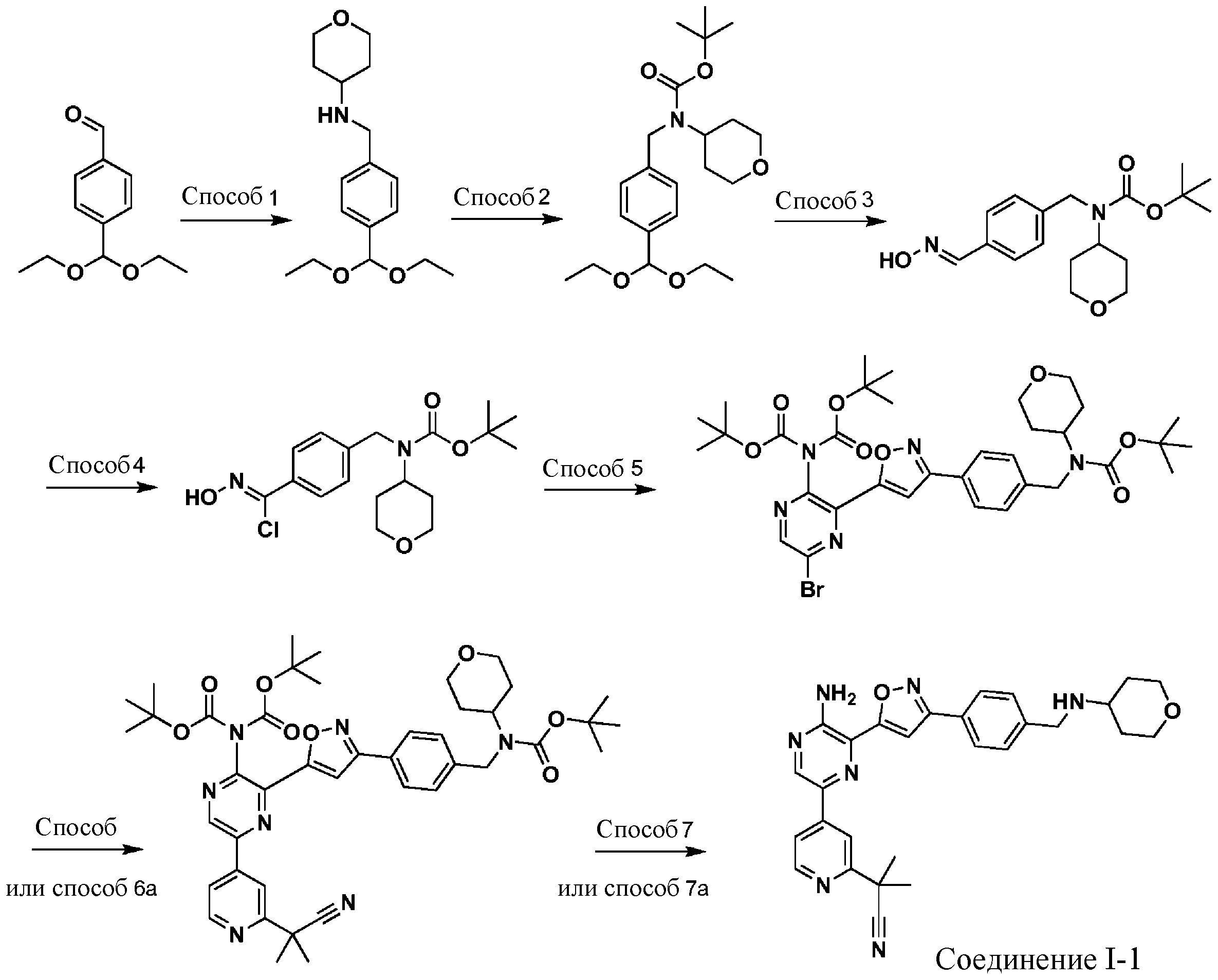
Способ 1
К раствору тетрагидропиран-4-амина (100 г, 988,7 ммоль) в MeOH (3,922 л) в течение более 2 минут при комнатной температуре добавляли 4-(диэтоксиметил)бензальдегид (196,1 г, 941,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 80 минут до завершения образования альдимина (по результатам ЯМР). В течение более 45 минут осторожно добавляли NaBH4 (44,49 г, 1,176 моль), поддерживая температуру от 24 °C до 27 °C с помощью ледяной бани. Через 75 минут при комнатной температуре реакция была завершена. Реакционную смесь гасили 1 M NaOH (1 л). Реакционную смесь разделяли между соляным раствором (2,5 л) и TBDME (4 л и затем 2 × 1 л). Органическую фазу промывали соляным раствором (500 мл) и концентрировали in vacuo. Сырую смесь растворяли в DCM (2 л). Водную фазу отделяли, органическую фазу высушивали над MgSO4, фильтровали и концентрировали in vacuo с получением заявляемого соединения в виде желтого масла (252,99 г, 91%).
Способ 2
Раствор N-[[4-(диэтоксиметил)фенил]метил]тетрагидропиран-4-амина (252,99 г, 862,3 ммоль) и Boc-ангидрида (191,9 г, 202,0 мл, 879,5 ммоль) в DCM (2,530 л) охлаждали до 3,3 °C. Добавляли Et3N (89,00 г, 122,6 мл, 879,5 ммоль) в течение более 4 минут, поддерживая внутреннюю температуру ниже 5 °C. Через 45 минут после окончания добавления ледяную баню снимали, а реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь последовательно промывали 0,5 M лимонной кислоты (1 л), насыщенным раствором NaHCO3 (1 л) и соляным раствором (1 л). Органическую фазу высушивали (MgSO4), фильтровали и концентрировали in vacuo с получением бесцветного масла (372,38 г, 110%). 1H ЯМР (400,0 МГц, DMSO); МС (ES+).
Способ 3
Трет-бутиловый эфир N-[[4-(диэтоксиметил)фенил]метил]-N-тетрагидропиран-4-илкарбамата (372,38 г, 946,3 ммоль) растворяли в THF (5 л) и воде (500 мл). Одной порцией добавляли гидрохлорид гидроксиламина (72,34 г, 1,041 моль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь разделяли между DCM (5 л) и водой. Объединенный органический экстракт промывали водой (1 л × 2). Органическую фазу концентрировали in vacuo до объема приблизительно 2 л. Органический слой высушивали над MgSO4, фильтровали и концентрировали in vacuo с получением липкого бесцветного масла, которое кристаллизовали при отстаивании в условиях вакуума (334,42 г, 106%). 1H ЯМР (400,0 МГц, CDCl3); МС (ES+).
Способ 4
Трет-бутиловый эфир N-[[4-[(E)-гидроксииминометил]фенил]метил]-N-тетрагидропиран-4-илкарбамата (334,13 г, 999,2 ммоль) растворяли в изопропилацетате (3,0 л) (смесь нагревали до 40 °C для обеспечения перехода всех твердых веществ в раствор). В течение более 5 минут порциями добавляли N-хлорсукцинимид (140,1 г, 1,049 моль) и реакционную смесь нагревали до 55 °C (температура внешнего блока). Через 45 минут при 55 °C реакция была завершена. Реакционную смесь охлаждали до комнатной температуры. Твердые вещества фильтровали и промывали изопропилацетатом (1 л). Объединенный органический экстракт последовательно промывали водой (1,5 л, 5 раз) и соляным раствором, высушивали над MgSO4, фильтровали и концентрировали in vacuo с получением вязкого желтого масла (355,9 г; 96%). 1H ЯМР (400,0 МГц, CDCl3); МС (ES+).
Способ 5
В течение более 20 минут добавляли Et3N (76,97 г, 106,0 мл, 760,6 ммоль) к раствору трет-бутилового эфира N-(5-бром-3-этинилпиразин-2-ил)-N-трет-бутоксикарбонилкарбамата (233,0 г, 585,1 ммоль) и трет-бутилового эфира N-[[4-[(Z)-C-хлор-N-гидроксикарбонимидоил]фенил]метил]-N-тетрагидропиран-4-илкарбамата (269,8 г, 731,4 ммоль) в DCM (2,330 л) при комнатной температуре. Во время добавления триэтиламина экзотермическую реакцию стабилизировали путем охлаждения смеси на ледяной бане, затем реакционную смесь постепенно прогревали до комнатной температуры и перемешивали в течение ночи при комнатной температуре. Реакционную смесь последовательно промывали водой (1,5 л, 3 раза) и соляным раствором. Органический экстракт высушивали над MgSO4, фильтровали и частично концентрировали in vacuo. Добавляли гептан (1,5 л) и продолжали концентрирование с получением 547,63 г твердого желто-оранжевого вещества.
542,12 г отбирали в ~2 об. (1 л) этилацетата. Смесь нагревали до внутренней температуры 74–75 °C и перемешивали до полного перехода твердого вещества в раствор. К горячему раствору через капельную воронку медленно добавляли гептан (3,2 л), поддерживая внутреннюю температуру от 71 °C до 72 °C. В конце добавления к темно-коричневом раствору добавляли в виде зародышей некоторое количество перекристаллизованного продукта. Реакционной смеси давали остыть до комнатной температуры без перемешивания для кристаллизации в течение ночи. Твердое вещество фильтровали и промывали гептаном (2 × 250 мл), а затем высушивали in vacuo с получением 307,38 г заявляемого продукта (72%). 1H ЯМР (400,0 МГц, CDCl3); МС (ES+).
Способ 6
Трет-бутиловый эфир N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-бромпиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-тетрагидропиран-4-илкарбамата (303 г, 414,7 ммоль) и 2-метил-2-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]пропаннитрил (112,9 г, 414,7 ммоль) суспендировали в MeCN (2 л) и H2O (1 л). Добавляли Na2CO3 (414,7 мл 2 M раствора, 829,4 ммоль), а затем Pd[P(tBu)3]2 (21,19 г, 41,47 ммоль), и реакционную смесь дегазировали N2 в течение 1 ч. Реакционную смесь помещали в атмосферу азота и нагревали при 70 °C (температура блока) в течение 4 ч (внутренняя температура варьировалась от 60 °C до 61 °C). Реакционную смесь охлаждали до комнатной температуры и перемешивали в течение ночи при комнатной температуре. Реакционную смесь разделяли между EtOAc (2 л) и водой (500 мл). Объединенный органический экстракт промывали соляным раствором (500 мл), фильтровали через небольшой слой целита и концентрировали при пониженном давлении до объема приблизительно 3 л. Раствор высушивали над MgSO4, фильтровали и частично концентрировали in vacuo. Добавляли iPrOH (1,5 л) и удаляли растворитель in vacuo с получением желаемого продукта в виде пены светло-коричневого цвета (405 г).
400 г пены отбирали в ~5 об. (2 л) iPrOH и нагревали смесь до 80 °C до полного перехода всего твердого вещества в раствор. В темно-коричневый раствор помещали зародыши и реакционной смеси давали медленно остыть до комнатной температуры в течение ночи. Твердый осадок отфильтровывали и промывали iPrOH (2 × 250 мл) и петролейным эфиром (2 × 200 мл). Полученное твердое вещество суспендировали в петролейном эфире (2,5 л), отфильтровывали и высушивали in vacuo. Полученное твердое вещество растворяли в DCM (2,5 л) и медленно перемешивали в течение 1 ч с 30 г SPM32 (3-меркаптопропилэтилсульфидкремнезем). Кремнезем фильтровали через слой флоризила и промывали DCM. Процедуру повторяли дважды, а затем раствор DCM концентрировали in vacuo с получением 238,02 г твердого светло-желтого вещества.
Способ 7
Трет-бутиловый эфир N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-[2-(1-циано-1-метилэтил)-4-пиридил]пиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-тетрагидропиран-4-илкарбамата (238 г, 299,0 ммоль) растворяли в DCM (2,380 л). При комнатной температуре в течение более 3 минут добавляли TFA (500 мл, 6,490 моль). Реакционную смесь перемешивали при комнатной температуре в течение 3,5 ч. Реакционную смесь концентрировали при пониженном давлении и получали азеотроп с помощью гептана (2 × 300 мл). Масло суспендировали в абс. EtOH (2,5 L) и фильтровали. Твердое вещество растворяли в смеси этанола (1,190 л) и воды (1,190 л). К смеси добавляли раствор карбоната калия (124,0 г, 897,0 ммоль) в воде (357,0 мл) и перемешивали при комнатной температуре в течение ночи.
Твердое вещество отфильтровывали, промывали водой (2,5 л) и высушивали при 50 °C in vacuo с получением 108,82 г заявляемого соединения (соединение I-1) в виде желтого порошка (73%).
Способы 6a и 7a
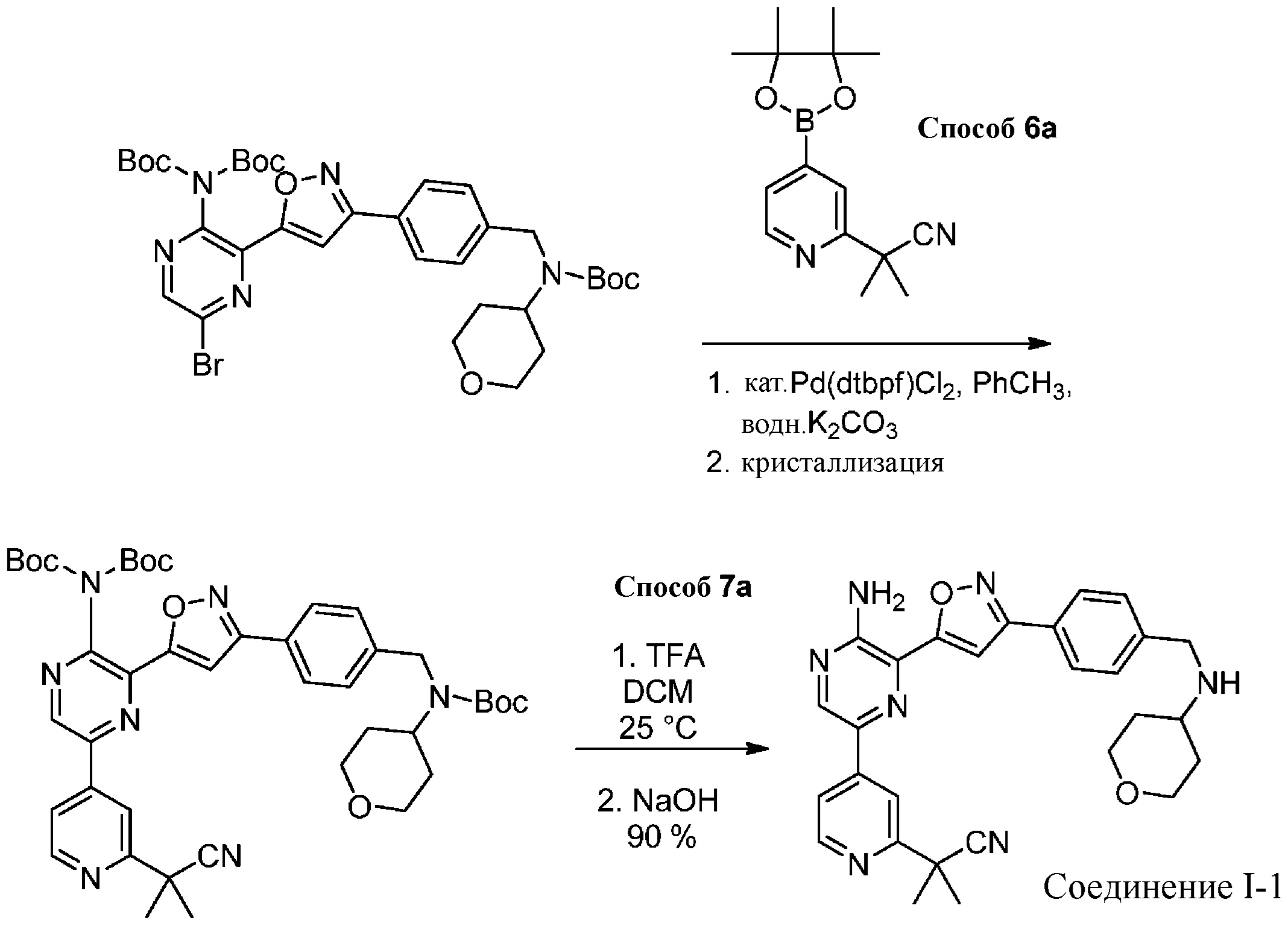
Смесь трет-бутилового эфира N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-бромпиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-тетрагидропиран-4-илкарбамата (110,0 г, 151 ммоль), K2CO3 (41,6 г, 301 ммоль) и 2-метил-2-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]пропаннитрила (41,0 г, 151 ммоль) в толуоле (770 мл) и воде (220 мл) перемешивают и дегазируют N2 в течение 30 мин при 20 °C. Добавляют катализатор Pd(dtbpf)Cl2 (1,96 г, 3,01 ммоль) и смесь дегазируют дополнительно 10 мин. Смесь нагревают при 70 °C до тех пор, пока реакция не будет завершена. Смесь охлаждают до температуры окружающей среды, разбавляют водой (220 мл) и фильтруют через слой целита. Органическую фазу концентрируют для удаления большей части растворителя. Концентрат разбавляют с помощью i-PrOH (550 мл). Полученную суспензию перемешивают в течение по меньшей мере 1 ч, а затем твердое вещество собирают фильтрованием с получением коричневого порошка.
Твердое вещество растворяют в толуоле (990 мл) и перемешивают со смолой Biotage MP-TMT (18,6 г) в течение 2 ч при температуре окружающей среды. Смолу удаляют фильтрованием. Фильтрат концентрируют, а затем разбавляют с помощью i-PrOH (550 мл), после чего повторно концентрируют. Добавляют i-PrOH (550 мл) и перемешивают в течение 1 ч при температуре окружающей среды. Суспензию охлаждают до 5 °C и твердое вещество собирают фильтрованием, после чего высушивают с получением трет-бутилового эфира N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-[2-(1-циано-1-метилэтил)-4-пиридил]пиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-тетрагидропиран-4-илкарбамата (соединение I-1) (81,9 г; выход 68%, чистота 98,7% по площади по ВЭЖХ) в виде порошка кремового цвета.
Изменение формы соединения I-1•HCl•1,5 H2O
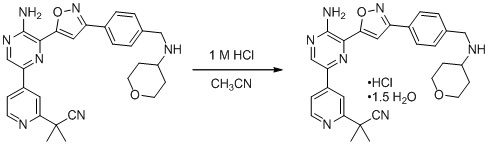
Суспензию трет-бутилового эфира N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-[2-(1-циано-1-метилэтил)-4-пиридил]пиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-тетрагидропиран-4-илкарбамата (соединение I-1) (36,0 г, 72,6 ммоль) в CH3CN (720 мл) перемешивают при температуре окружающей среды (20 °C) в колбе, оснащенной механической мешалкой. Добавляют 1 M водного раствора HCl (72,6 мл; 72,6 ммоль). Суспензию перемешивают при температуре окружающей среды в течение 20 ч. Твердое вещество собирают фильтрованием. Фильтровальный осадок промывают CH3CN (3 × 50 мл), затем высушивают в условиях вакуума с высокой влажностью в течение 2 ч с получением соединения I-1•HCl•1,5 H2O (30,6 г; выход 74%, чистота 98,8% по площади по ВЭЖХ) в виде желтого порошка. 1H ЯМР (400 МГц, DMSO) δ 9,63 (д, J = 4,7 Гц, 2H), 9,05 (с, 1H), 8,69 (д, J = 5,2 Гц, 1H), 8,21 (с, 1H), 8,16–8,03 (м, 3H), 7,84 (т, J = 4,1 Гц, 3H), 7,34 (уш. с, 2H), 4,40–4,18 (м, 2H), 3,94 (дд, J = 11,2, 3,9 Гц, 2H), 3,32 (т, J = 11,2 Гц, 3H), 2,17–2,00 (м, 2H), 1,81 (с, 6H), 1,75 (дд, J = 12,1, 4,3 Гц, 2H).
Пример 2. Синтез 3-[3-[4-[дидейтерио(метиламино)метил]фенил]изоксазол-5-ил]-5-(4-изопропилсульфонилфенил)пиразин-2-амина (соединение II-1)
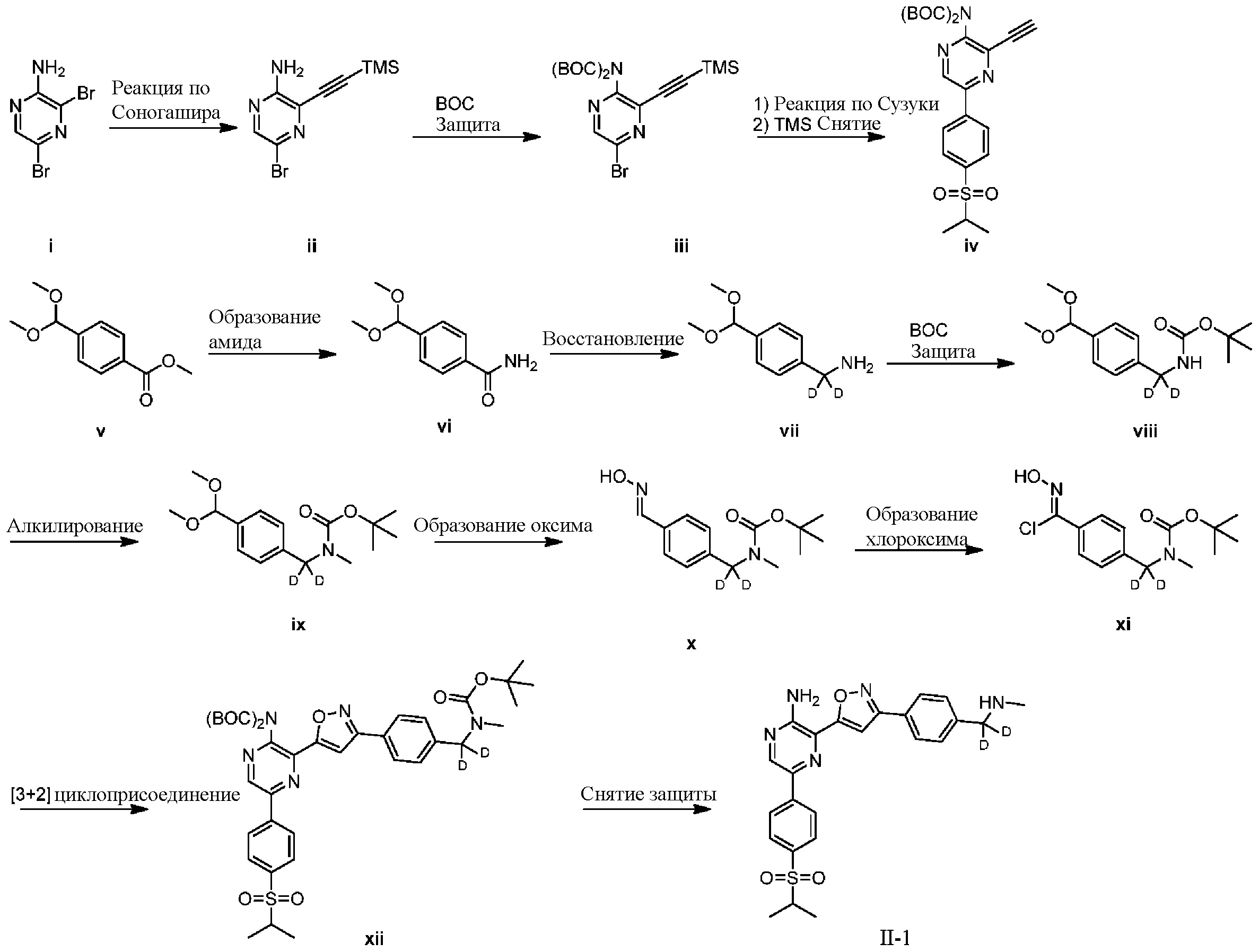
Этап 1. 5-Бром-3-((триметилсилил)этинил)пиразин-2-амин
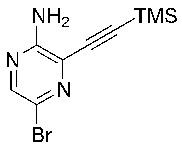
ii
(Триметилсилил)ацетилен (1,845 г, 2,655 мл, 18,78 ммоль) по каплям добавляли в раствор 3,5-дибромпиразин-2-амина (соединение i) (5 г, 19,77 ммоль) в DMF (25 мл). Затем добавляли триэтиламин (10,00 г, 13,77 мл, 98,85 ммоль), йодид меди (I) (451,7 мг, 2,372 ммоль) и Pd(PPh3)4 (1,142 г, 0,9885 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь разбавляли EtOAc и водой и отделяли слои. Водный слой дополнительно экстрагировали с помощью EtOAc и объединенные органические слои промывали водой, высушивали (MgSO4) и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, элюируя 15% EtOAc/петролейного эфира, с образованием продукта в виде желтого твердого вещества (3,99 г, выход 75%). 1H ЯМР (400,0 МГц, DMSO) δ 0,30 (9H, с), 8,06 (IH, с); МС (ES+) 271,82.
Этап 2. Трет-бутиловый эфир N-трет-бутоксикарбонил-N-[5-бром-3-((триметилсилил)этинил)пиразин-2-ил]карбамата
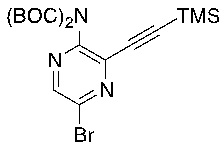
iii
5-Бром-3-(2-триметилсилилэтинил)пиразин-2-амин (2,85 г, 10,55 ммоль) растворяли в DCM (89,06 мл) и обрабатывали Boc-ангидридом (6,908 г, 7,272 мл, 31,65 ммоль), а затем DMAP (128,9 мг, 1,055 ммоль). Реакцию оставляли для перемешивания при температуре окружающей среды в течение 2 часов. Затем смесь разбавляли с помощью DCM и NaHCO3 и отделяли слои. Затем водный слой экстрагировали дополнительно с помощью DCM, высушивали (MgSO4), фильтровали и концентрировали in vacuo. Полученный остаток очищали с помощью колоночной хроматографии, элюируя дихлорметаном, с образованием желаемого продукта в виде бесцветного масла (4,95 г, выход 99%). 1H ЯМР (400,0 МГц, DMSO) δ 0,27 (9H, с), 1,42 (18H, с), 8,50 (1H, с); МС (ES+) 472,09.
Этап 3. Трет-бутиловый эфир N-(3-этинил-5-(4-(изопропилсульфонил)фенил)пиразин-2-ил)N-трет-бутоксикарбонилкарбамата
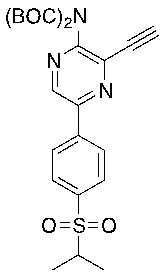
iv
N-[5-Бром-3-(2-триметилсилилэтинил)пиразин-2-ил]-N-трет-бутоксикарбонилкарбамат (3 г, 6,377 ммоль) и (4-изопропилсульфонилфенил)бороновую кислоту (1,491 г, 6,536 ммоль) растворяли в MeCN/воде (60/12 мл). Добавляли K3PO4 (2,706 г, 12,75 ммоль) и реакционную смесь дегазировали в потоке азота (5 циклов). Добавляли Pd[P(tBu)3]2 (162,9 мг, 0,3188 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь быстро выливали в смесь этилацетата (500 мл), воды (90 мл) и 1%-го водного метабисульфита натрия при 4 °С, хорошо встряхивали и отделяли слои. Органическую фракцию сушили над MgSO4, фильтровали, и фильтрат обрабатывали 3-меркаптопропилэтилсульфидом на кремнеземе (0,8 ммоль/г, 1 г), предварительно абсорбированном на силикагеле, а затем очищали с помощью колоночной хроматографии на силикагеле, элюируя 30–40% смесью EtOAc/петролейного эфира. Растворители концентрировали in vacuo, оставляя продукт в виде вязкого масла желтого цвета, которое растирали с петролейным эфиром для получения продукта в виде бежевых кристаллов (1,95 г, выход 61%); 1H ЯМР (400 МГц, DMSO) δ 1,20 (м, 6H), 1,39 (с, 18H), 3,50 (м, 1H), 5,01 (с, 1H), 8,03 (м, 2H), 8,46 (м, 2H) и 9,37 (с, 1H).
Этап 4. 4-(Диметоксиметил)бензамид
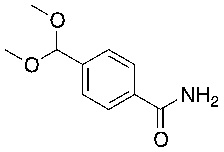
vi
Смесь метилового эфира 4-(диметоксиметил)бензоата (3,8 г, 18,08 ммоль) и 7 M NH3 в MeOH (30 мл 7 M, 210,0 ммоль) в запаянной трубке нагревали при 110 °C в течение 22 часов. Добавляли дополнительную порцию 7 M NH3 в MeOH (20 мл 7 M, 140,0 ммоль) и реакцию нагревали при 135 °C в течение 23 часов. Реакцию охлаждали до температуры окружающей среды и растворитель удаляли in vacuo. Остаток повторно вводили в условия реакции (7 M NH3 в MeOH (30 мл 7 M, 210,0 ммоль) при 115 °C) дополнительно на 16 часов. Растворитель удаляли in vacuo, и остаток растирали из Et2O. Полученный осадок выделяли фильтрованием с получением указанного в подзаголовке соединения в виде белого твердого вещества (590 мг, выход 17%). Фильтрат очищали с помощью колоночной хроматографии (ISCO Companion, колонка 40 г, элюирование 0–100% смесью EtOAc/петролейного эфира до 10% MeOH/EtOAc, загружено в EtOAc/MeOH) с получением дополнительной порции указанного в подзаголовке продукта в виде белого твердого вещества (225 мг, выход 6%). Всего выделяли (815 мг, выход 23%); 1H ЯМР (400 МГц, DMSO) δ 3,26 (с, 6H), 5,44 (с, 1H), 7,37 (с, 1H), 7,46 (д, J = 8,0 Гц, 2H), 7,84–7,91 (м, 2H) и 7,98 (с, 1H) м.д.; МС (ES+) 196,0.
Этап 5. Дидейтерио-[4-(диметоксиметил)фенил]метанамин
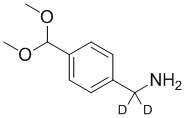
vii
LiDH4 (12,52 мл, 1 M, 12,52 ммоль) добавляли по каплям в перемешанный раствор 4-(диметоксиметил)бензамида (815 мг, 4,175 ммоль) в THF (20 мл) при 0 °C в атмосфере азота. Реакцию нагревали с обратным холодильником в течение 16 часов, а затем охлаждали до температуры окружающей среды. Реакцию гасили последовательным добавлением D2O (1 мл), 15% NaOH в D2O (1 мл) и D2O (4 мл). Полученное твердое вещество удаляли фильтрованием и промывали EtOAc. Фильтрат концентрировали in vacuo, и остаток сушили азеотропной перегонкой с толуолом (× 3) с получением указанного в подзаголовке соединения в виде желтого масла (819 мг), которое использовали без дополнительной очистки; 1H ЯМР (400 МГц, DMSO) δ 3,23 (с, 6H), 5,36 (с, 1H) и 7,30–7,35 (м, 4H) м.д.; МС (ES+) 167,0.
Этап 6. Трет-бутиловый эфир N-[дидейтерио-[4-(диметоксиметил)фенил]метил]карбамата
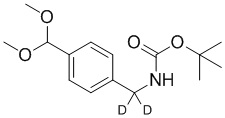
viii
Et3N (633,7 мг, 872,9 мкл, 6,262 ммоль) добавляли к перемешанной суспензии дидейтерио-[4-(диметоксиметил)фенил]метанамина (765 мг, 4,175 ммоль) в THF (15 мл) при 0 °C. Реакцию оставляли для перемешивания при данной температуре в течение 30 минут, затем порциями добавляли Boc2O (956,8 мг, 1,007 мл, 4,384 ммоль). Реакции давали нагреться до температуры окружающей среды, и перемешивали в течение 18 часов. Растворитель удаляли in vacuo, и остаток очищали с помощью колоночной хроматографии (ISCO Companion, колонка 120 г, элюирование 0–50% смесью EtOAc/петролейного эфира, загружено в DCM) с получением указанного в подзаголовке продукта в виде бесцветного масла (1,04 г, выход 88%); 1H ЯМР (400 МГц, DMSO) δ 1,40 (с, 9H), 3,23 (с, 6H), 5,36 (с, 1H), 7,24 (д, J = 8,2 Гц, 2H), 7,33 (д, J = 8,0 Гц, 2H) и 7,38 (с, 1H) м.д.
Этап 7. Трет-бутиловый эфир N-[дидейтерио-[4-(диметоксиметил)фенил]метил]-N-метилкарбамата
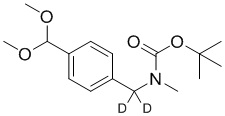
ix
LiHMDS (1 M в THF) (1,377 мл, 1 M, 1,377 ммоль) добавляли по каплям в перемешанный раствор трет-бутилового эфира N-[дидейтерио-[4-(диметоксиметил)фенил]метил]карбамата (300 мг, 1,059 ммоль) в THF (5 мл) при -78 °C. Раствор перемешивали при этой температуре в течение 10 минут, затем по каплям добавляли йодметан (225,4 мг, 98,86 мкл, 1,588 ммоль) и давали смеси нагреться до температуры окружающей среды в течение более 1 часа. Реакцию снова охлаждали до -78 °C и добавляли LiHMDS (1 M в THF) (635,4 мкл, 1 M, 0,6354 ммоль). Через 10 минут добавляли йодметан (105,2 мг, 46,14 мкл, 0,7413 ммоль), и реакции давали нагреться до температуры окружающей среды в течение более 6 часов. Смесь разбавляли с помощью EtOAc, и органический слой промывали насыщенным водным раствором NaHCO3 (× 2), соляным раствором (× 1), высушивали (MgSO4), фильтровали и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии (ISCO Companion, колонка 24 г, элюирование 0–30% смесью EtOAc/петролейного эфира, загружено в DCM) с получением указанного в подзаголовке продукта в виде бесцветного масла (200 мг, выход 63%); 1H ЯМР (400 МГц, DMSO) δ 1,41 (д, J = 27,7 Гц, 9H), 2,76 (с, 3H), 3,24 (с, 6H), 5,37 (с, 1H), 7,23 (д, J = 7,9 Гц, 2H) и 7,37 (д, J = 8,0 Гц, 2H) м.д.
Этап 8. Трет-бутиловый эфир N-[дидейтерио-[4-гидроксииминометил]фенил]метил]-N-(метил)карбамата
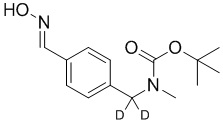
x
Гидрохлорид гидроксиламина (51,15 мг, 0,7361 ммоль) добавляли в перемешанный раствор трет-бутилового эфира N-[дидейтерио-[4-(диметоксиметил)фенил]метил]-N-метилкарбамата (199 мг, 0,6692 ммоль) в THF (10 мл)/вода (1,000 мл) и реакцию оставляли перемешиваться при температуре окружающей среды в течение 4 часов. Реакцию разделяли между DCM и соляным раствором и отделяли слои. Водный слой экстрагировали с помощью DCM (× 2), и объединенные органические экстракты промывали соляным раствором (× 1), высушивали (MgSO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения в виде белого твердого вещества (180 мг, выход 100%). 1H ЯМР (400 МГц, DMSO) δ 1,41 (д, J = 24,6 Гц, 9H) 2,76 (с, 3H), 7,25 (д, J = 8,1 Гц, 2H), 7,58 (д, J = 8,0 Гц, 2H), 8,13 (с, 1H) и 11,20 (с, 1H) м.д.; МС (ES+) 211,0 (M-Boc).
Этап 9. Трет-бутиловый эфир N-[[4-[хлор-N-гидроксикарбонимидоил]фенил]дидейтериометил]-N-метилкарбамата
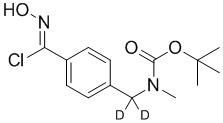
xi
Трет-бутиловый эфир N-[дидейтерио-[4-[гидроксииминометил]фенил]метил]-N-(метил)карбамата (178 мг, 0,6683 ммоль) в DMF (2 мл) обрабатывали с помощью NCS (89,24 мг, 0,6683 ммоль), и реакцию нагревали до 65 °C в течение 1 часа. Реакцию охлаждали до температуры окружающей среды и разбавляли водой. Смесь экстрагировали EtOAc (× 2), и объединенные органические экстракты промывали соляным раствором (× 4), сушили (MgSO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения в виде белого твердого вещества (188 мг, выход 94%); 1H ЯМР (400 МГц, DMSO) δ 1,42 (д, J = 24,7 Гц, 9H), 2,78 (с, 3H), 7,32 (д, J = 8,4 Гц, 2H), 7,78 (д, J = 8,2 Гц, 2H) и 12,36 (с, 1H) м.д.
Этап 10. Трет-бутиловый эфир N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-(4-изопропилсульфонилфенил)пиразин-2-ил]изоксазол-3-ил]фенил]-дидейтериометил]-N-метилкарбамата
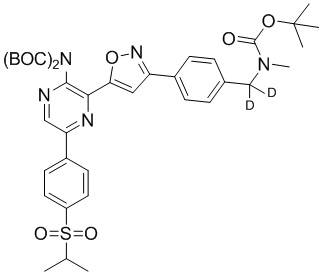
xii
Et3N (36,31 мг, 50,01 мкл, 0,3588 ммоль) добавляли по каплям в перемешанный раствор трет-бутилового эфира N-трет-бутоксикарбонил-N-[3-этинил-5-(4-изопропилсульфонилфенил)пиразин-2-ил]карбамата (150 мг, 0,2990 ммоль) и трет-бутилового эфира N-[[4-[хлор-N-гидроксикарбонимидоил]фенил]-дидейтериометил]-N-метилкарбамата (89,93 мг, 0,2990 ммоль) в безводном THF (3 мл), и реакционную смесь нагревали при 65 °C в течение 3 часов. Реакционную смесь охлаждали до температуры окружающей среды и разбавляли смесью EtOAc/соляного раствора. Воду добавляли до прозрачности водного слоя и отделения слоев. Водный слой экстрагировали с помощью EtOAc (×1), и объединенные органические экстракты промывали соляным раствором (× 1), высушивали (MgSO4), фильтровали и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии (ISCO Companion, колонка 40 г, элюирование 0–30% смесью EtOAc/петролейного эфира, загружено в DCM) с получением указанного в подзаголовке продукта в виде белого твердого вещества (134 мг, выход 59%); 1H ЯМР (400 МГц, DMSO) δ 1,22 (д, J = 6,8 Гц, 6H) 1,32 (с, 18H), 1,43 (д, J = 23,1 Гц, 9H), 2,82 (с, 3H), 3,56 (пент, 1H), 7,43 (д, J = 8,3 Гц, 3H), 8,02–8,03 (м, 3H), 8,06–8,11 (м, 2H), 8,62–8,67 (м, 2H) и 9,51 (с, 1H) м.д.; МС (ES+) 666,2 (M-Boc).
Этап 11. 3-[3-[4-[Дидейтерио(метиламино)метил]фенил]изоксазол-5-ил]-5-(4-изопропилсульфонилфенил)пиразин-2-амин (соединение II-1)
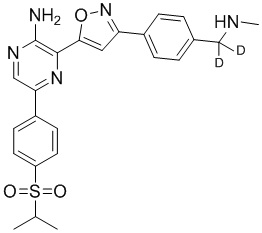
II-1
3 M HCl в MeOH (1,167 мл 3 M, 3,500 ммоль) добавляли в перемешанный раствор трет-бутилового эфира N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-(4-изопропилсульфонилфенил)пиразин-2-ил]изоксазол-3-ил]фенил]-дидейтериометил]-N-метилкарбамата (134 мг, 0,1750 ммоль) в DCM (5 мл), и реакцию нагревали с обратным холодильником в течение 16 часов. Реакцию охлаждали до температуры окружающей среды, и полученный осадок выделяли фильтрованием и сушили под вакуумом при 40 °C с получением дигидрохлорида заявляемого соединения в виде твердого вещества желтого цвета (58,8 мг, выход 62%); 1H ЯМР (400 МГц, DMSO) δ 1,20 (д, J = 6,8 Гц, 6H), 2,60 (т, J = 5,4 Гц, 3H), 3,48 (гепт, J = 6,8 Гц, 1H), 7,22 (уш. с, 2H), 7,69–7,75 (м, 2H), 7,85 (с, 1H), 7,92–7,99 (м, 2H), 8,08–8,15 (м, 2H) 8,37–8,42 (м, 2H), 8,97 (с, 1H) и 9,10 (д, J = 5,8 Гц, 2H) м.д.; МС (ES+) 466,2.
Пример 3. Синтез 3-[3-[4-[дидейтерио-(тридейтериометиламино)метил]фенил]изоксазол-5-ил]-5-(4-изопропилсульфонилфенил)пиразин-2-амина (соединение II-2)
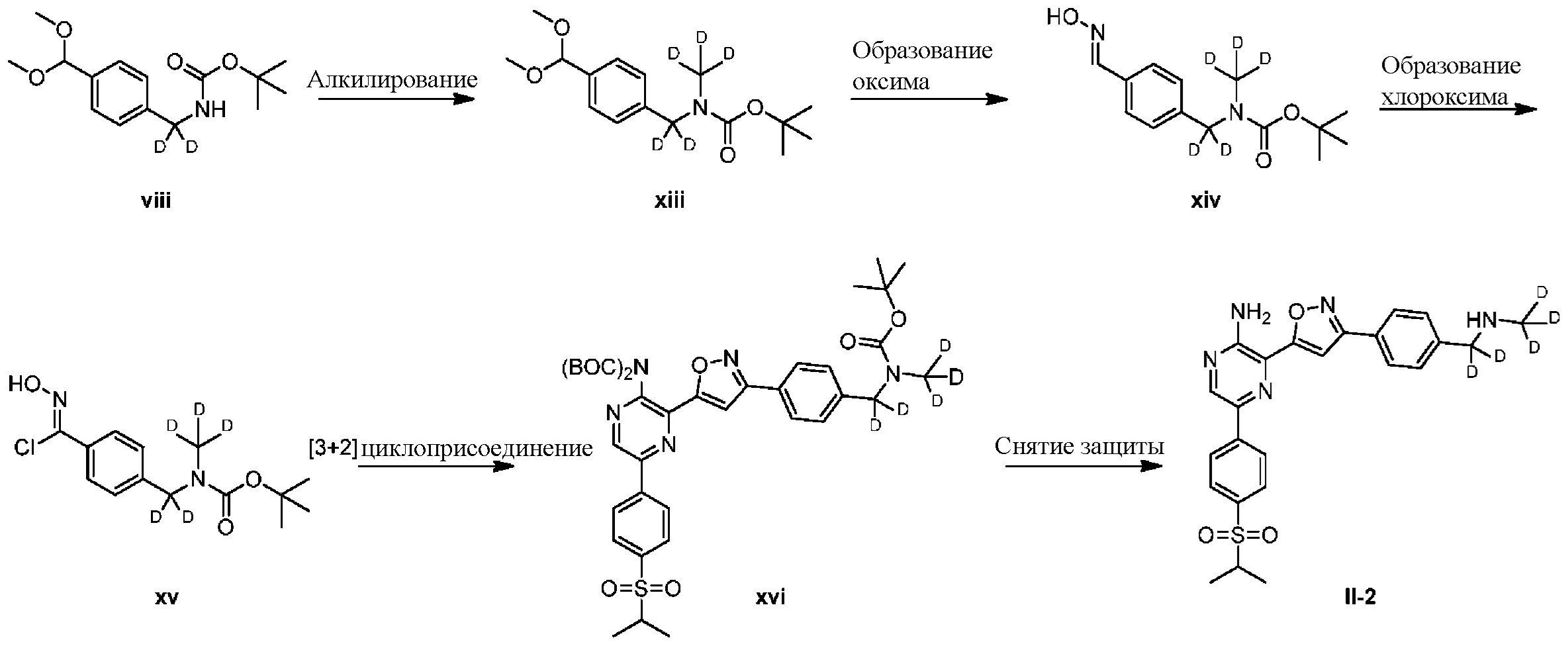
Этап 1. Трет-бутиловый эфир N-[дидейтерио-[4-(диметоксиметил)фенил]метил]-I-(тридейтериометил)карбамата
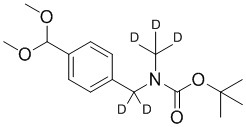
xiii
LiHMDS (1 M в THF) (1,181 мл 1 M, 1,181 ммоль) добавляли по каплям в перемешанный раствор трет-бутилового эфира N-[дидейтерио-[4-(диметоксиметил)фенил]метил]карбамата (300 мг, 1,059 ммоль) в THF (5 мл) при
-78 °C. Раствор перемешивали при этой температуре в течение 30 минут, затем по каплям добавляли тридейтериойодметан (198,0 мг, 84,98 мкл, 1,366 ммоль), и смеси давали нагреться до температуры окружающей среды в течение более 21 часа. Реакцию повторно охлаждали до -78 °C и добавляли дополнительную порцию LiHMDS (1 M в THF) (635,4 мкл, 1 M, 0,6354 ммоль). Через 15 минут добавляли еще тридейтериойодметан (76,75 мг, 32,94 мкл, 0,5295 ммоль), и реакции давали нагреться до температуры окружающей среды в течение более 5 часов. Смесь разбавляли с помощью EtOAc, и органический слой промывали насыщенным водным раствором NaHCO3 (× 2), соляным раствором (× 1), высушивали (MgSO4), фильтровали и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии (ISCO Companion, колонка 24 г, элюирование 0–30% смесью EtOAc/петролейного эфира, загружено в DCM) с получением указанного в подзаголовке продукта в виде бесцветного масла (213 мг, выход 67%); 1H ЯМР (400 МГц, DMSO) δ 1,36–1,42 (м, 9H) 3,22 (с, 6H), 5,35 (с, 1H), 7,21 (д, J = 7,8 Гц, 2H) и 7,35 (д, J = 7,7 Гц, 2H) м.д.
Этап 2. Трет-бутиловый эфир N-[дидейтерио-[4-гидроксииминометил]фенил]метил]-N-(тридейтериометил)карбамата
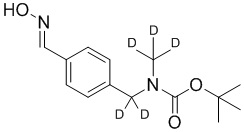
xiv
Гидрохлорид гидроксиламина (53,95 мг, 0,7763 ммоль) добавляли в перемешанный раствор трет-бутилового эфира N-[дидейтерио-[4-(диметоксиметил)фенил]метил]-N-(тридейтериометил)карбамата (212 мг, 0,7057 ммоль) в THF (10 мл)/вода (1,000 мл), и реакцию оставляли перемешиваться при температуре окружающей среды в течение 22 часов. Реакцию разделяли между DCM и соляным раствором и отделяли слои. Водный слой экстрагировали с помощью DCM (×2), и объединенные органические экстракты промывали соляным раствором (× 1), высушивали (MgSO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения в виде белого твердого вещества (190 мг, выход 100%). 1H ЯМР (400 МГц, DMSO) δ 1,41 (д, J = 24,2 Гц, 9H), 7,25 (д, J = 8,1 Гц, 2H), 7,58 (д, J = 8,0 Гц, 2H), 8,13 (с, 1H) и 11,20 (с, 1H) м.д.
Этап 3. Трет-бутиловый эфир N-[[4-[хлор-N-гидроксикарбонимидоил]фенил]-дидейтериометил]-N-(тридейтериометил)карбамата
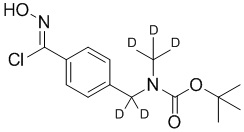
xv
Трет-бутиловый эфир N-[дидейтерио-[4-[гидроксииминометил]фенил]метил]-N-(тридейтериометил)карбамата (190,0 мг, 0,7054 ммоль) в DMF (2 мл) обрабатывали с помощью NCS (94,19 мг, 0,7054 ммоль), и реакцию нагревали до 65 °C в течение 1 часа. Реакцию охлаждали до температуры окружающей среды и разбавляли водой. Смесь экстрагировали с помощью EtOAc (×2), и объединенные органические экстракты промывали соляным раствором (× 4), высушивали (MgSO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения в виде белого твердого вещества (198 мг, выход 93%); 1H ЯМР (400 МГц, DMSO) δ 1,41 (д, J = 26,0 Гц, 9H), 7,32 (д, J = 8,3 Гц, 2H), 7,78 (д, J = 8,2 Гц, 2H) и 12,36 (с, 1H) м.д.
Этап 4. Трет-бутиловый эфир N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-(4-изопропилсульфонилфенил)пиразин-2-ил]изоксазол-3-ил]фенил]-дидейтериометил]-N-(тридейтериометил)карбамата
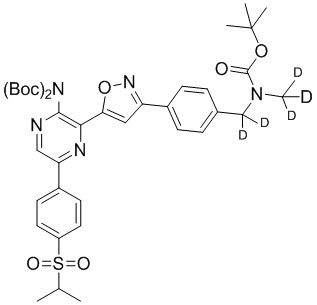
xvi
Et3N (36,31 мг, 50,01 мкл, 0,3588 ммоль) добавляли по каплям в перемешанный раствор трет-бутилового эфира N-трет-бутоксикарбонил-N-[3-этинил-5-(4-изопропилсульфонилфенил)пиразин-2-ил]карбамата (150 мг, 0,2990 ммоль) и трет-бутилового эфира N-[[4-[хлор-N-гидроксикарбонимидоил]фенил]-дидейтериометил]-N-(тридейтериометил)карбамата (90,84 мг, 0,2990 ммоль) в безводном THF (3 мл), и реакционную смесь нагревали при температуре 65 °C в течение 3,5 часов. Реакционную смесь охлаждали до температуры окружающей среды и разбавляли смесью EtOAc/соляного раствора. Воду добавляли до прозрачности водного слоя и отделения слоев. Водный слой экстрагировали с помощью EtOAc (×1), и объединенные органические экстракты промывали соляным раствором (× 1), высушивали (MgSO4), фильтровали и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии (ISCO Companion, колонка 40 г, элюирование 0–35% смесью EtOAc/петролейного эфира, загружено в DCM) с получением указанного в подзаголовке продукта в виде белого твердого вещества (158 мг, выход 69%); 1H ЯМР (400 МГц, DMSO) δ 1,22 (д, J = 6,8 Гц, 6H), 1,44 (д, J = 22,0 Гц, 9H), 3,56 (дт, J = 13,5, 6,7 Гц, 2H), 7,43 (д, J = 8,2 Гц, 3H), 8,02 (д, J = 6,9 Гц, 2H), 8,08 (д, J = 8,7 Гц, 2H), 8,65 (д, J = 8,8 Гц, 2H) и 9,51 (с, 1H) м.д.; МС (ES+) 669,3 (M-Boc).
Этап 5. 3-[3-[4-[Дидейтерио-(тридейтериометиламино)метил]фенил]изоксазол-5-ил]-5-(4-изопропилсульфонилфенил)пиразин-2-амин (соединение II-2)
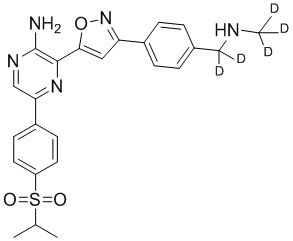
II-2
3 M HCl в MeOH (1,361 мл 3 M, 4,084 ммоль) добавляли в перемешанный раствор трет-бутилового эфира N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-(4-изопропилсульфонилфенил)пиразин-2-ил]изоксазол-3-ил]фенил]-дидейтериометил]-N-(тридейтериометил)карбамата (157 мг, 0,2042 ммоль) в DCM (5 мл), и реакцию нагревали с обратным холодильником в течение 16 часов. Реакцию охлаждали до температуры окружающей среды и полученный осадок выделяли фильтрованием и сушили под вакуумом при 40 °C с получением дигидрохлорида заявляемого соединения в виде желтого твердого вещества (72,5 мг, выход 66%); 1H ЯМР (400 МГц, DMSO) δ 1,20 (д, J = 6,8 Гц, 6H), 3,48 (дв. кв, J = 13,6, 6,7 Гц, 1H), 7,21 (с, 2H), 7,68–7,78 (м, 2H), 7,85 (с, 1H), 7,91–7,99 (м, 2H), 8,08–8,13 (м, 2H), 8,36–8,42 (м, 2H), 8,96 (с, 1H) и 9,14 (с, 2H) м.д.; МС (ES+) 469,1.
Пример 4. Синтез 5-(4-изопропилсульфонилфенил)-3-[3-[4-[(тридейтериометиламино)метил]фенил]изоксазол-5-ил]пиразин-2-амина
(соединение II-3)
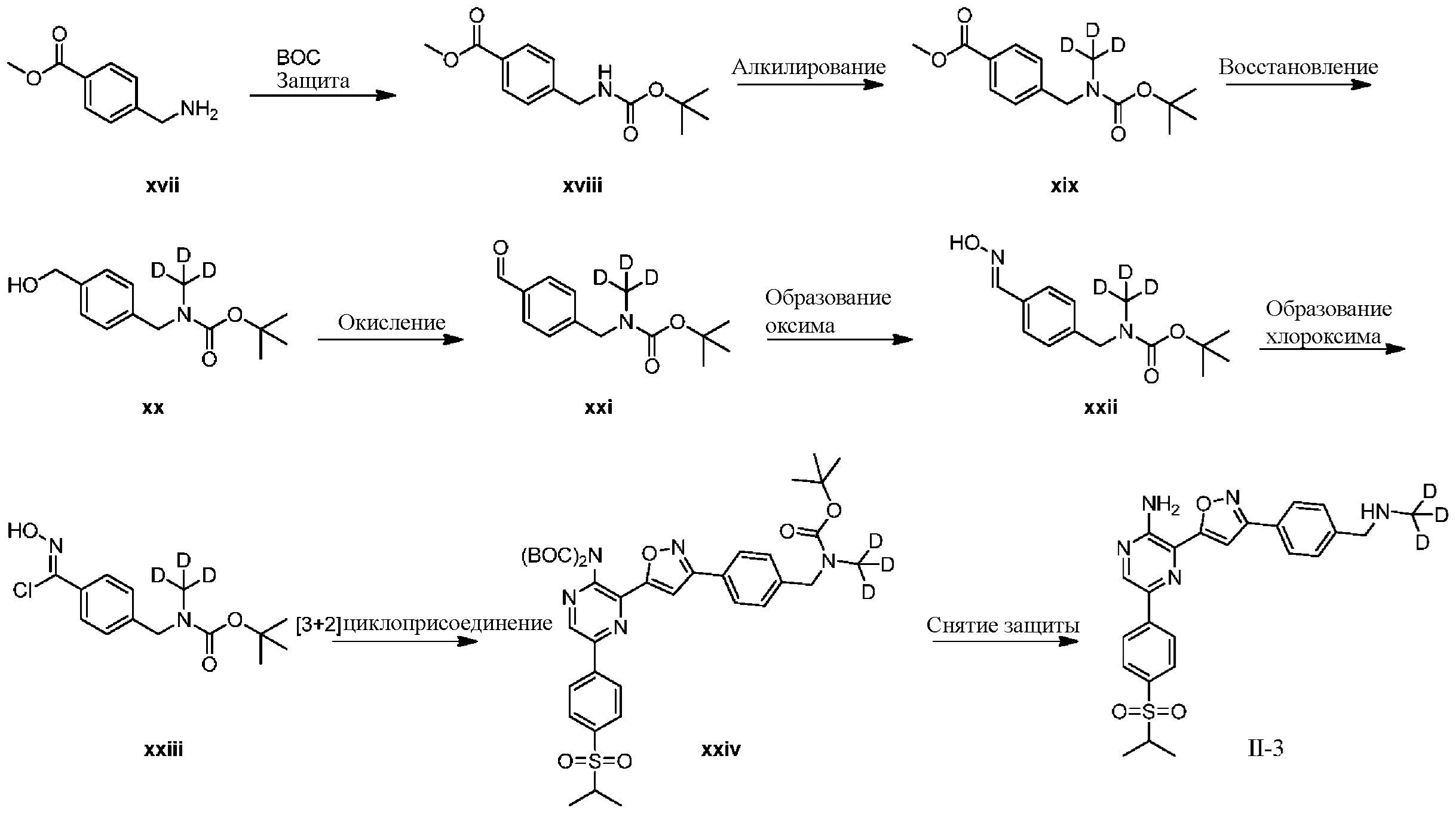
Этап 1. Метиловый эфир 4-[(трет-бутоксикарбониламино)метил]бензоата
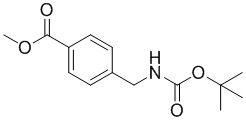
xviii
Et3N (1,882 г, 2,592 мл, 18,60 ммоль) добавляли в перемешанную суспензию метилового эфира 4-(аминометил)бензоата (соляная кислота (1)) (1,5 г, 7,439 ммоль) в THF (20 мл) при 0 °C. Реакцию оставляли перемешиваться при данной температуре в течение 30 минут, затем порциями добавляли Boc2O (1,705 г, 1,795 мл, 7,811 ммоль). Реакции давали нагреться до температуры окружающей среды и перемешивали в течение 18 часов. Смесь разбавляли с помощью EtOAc. Органический слой промывали 1 M водного раствора HCl (× 2), насыщенным водным раствором NaHCO3 (× 2) и соляным раствором (× 1). Органический слой высушивали (MgSO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения в виде белого твердого вещества, которое использовали без дополнительной очистки (1,93 г, выход 98%); 1H ЯМР (400 МГц, DMSO) δ 1,40 (с, 9H), 3,85 (с, 3H), 4,20 (д, J = 6,1 Гц, 2H), 7,38 (д, J = 8,2 Гц, 2H), 7,49 (т, J = 6,1 Гц, 1H) и 7,92 (д, J = 8,2 Гц, 2H) м.д.; МС (ES+) 251,1 (M-Me).
Этап 2. Метиловый эфир 4-[[трет-бутоксикарбонил(тридейтериометил)амино]метил]бензоата
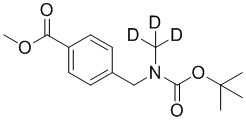
xix
LiHMDS (1 M в THF) (8,112 мл, 1 M, 8,112 ммоль) по каплям добавляли в перемешанный раствор метилового эфира 4-[(трет-бутоксикарбониламино)метил]бензоата (1,93 г, 7,275 ммоль) в THF (10 мл) при -78 °C. Раствор перемешивали при этой температуре в течение 30 минут, а затем по каплям добавляли тридейтериойодметан (1,360 г, 9,385 ммоль) и смеси давали нагреться до температуры окружающей среды в течение более 3 часов. Реакцию охлаждали до
-78 °C и добавляли дополнительную порцию LiHMDS (1 M в THF) (2,182 мл 1 M, 2,182 ммоль). Через 10 минут добавляли дополнительную порцию тридейтериойодметана (527,4 мг, 3,638 ммоль) и реакции давали нагреться до температуры окружающей среды в течение более 17 часов. Смесь разбавляли с помощью EtOAc, и органический слой промывали насыщенным водным раствором NaHCO3 (× 2), соляным раствором (× 1), высушивали (MgSO4), фильтровали и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии (ISCO Companion, колонка 120 г, элюирование 0–30% смесью EtOAc/петролейного эфира, загружено в DCM) с получением указанного в подзаголовке продукта в виде бледно-желтого масла (1,37 г, выход 67%); 1H ЯМР (400 МГц, DMSO) δ 1,38 (д, J = 44,2 Гц, 9H), 3,83 (с, 3H), 4,43 (с, 2H), 7,33 (д, J = 8,2 Гц, 2H) и 7,94 (д, J = 8,1 Гц, 2H) м.д.; МС (ES+) 268,1 (M-Me).
Этап 3. Трет-бутиловый эфир N-[[4-(гидроксиметил)фенил]метил]-N-(тридейтериометил)карбамата
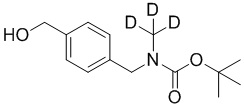
xx
LiBH4 (158,5 мг, 7,278 ммоль) добавляли в перемешанный раствор метилового эфира 4-[[трет-бутоксикарбонил(тридейтериометил)амино]метил]бензоата (1,37 г, 4,852 ммоль) в THF (10 мл) и нагревали реакцию до 85 °C в течение 15 часов. Добавляли дополнительную порцию LiBH4 (158,5 мг, 7,278 ммоль) и перемешивали реакцию при температуре 65 °C в течение дополнительных 7 часов. Реакционную смесь охлаждали до температуры окружающей среды, а затем выливали на колотый лед и при перемешивании по каплям добавляли 1 M HCl до окончания наблюдаемого выделения газа. Смесь перемешивали в течение 10 минут, а затем добавляли насыщенный водный раствор NaHCO3 до достижения смесью pH 8. Водный слой экстрагировали с помощью EtOAc (× 3), и объединенные органические экстракты высушивали (MgSO4), фильтровали и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии (ISCO Companion, колонка 120 г, элюирование 0–100% смесью EtOAc/петролейного эфира, загруженного в DCM) с получением указанного в подзаголовке продукта в виде бесцветного масла (1,03 г, выход 84%); 1H ЯМР (400 МГц, DMSO) δ 1,42 (д, J = 14,6 Гц, 9H), 4,35 (с, 2H), 4,48 (д, J = 5,7 Гц, 2H), 5,15 (т, J = 5,7 Гц, 1H), 7,18 (д, J = 7,9 Гц, 2H) и 7,30 (д, J = 7,7 Гц, 2H) м.д.; МС (ES+) 181,1 (M-OtBu).
Этап 4. Трет-бутиловый эфир N-[(4-формилфенил)метил]-N-(тридейтериометил)карбамата
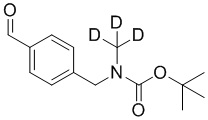
xxi
MnO2 (5,281 г, 1,051 мл, 60,75 ммоль) добавляли в перемешанный раствор трет-бутилового эфира N-[[4-(гидроксиметил)фенил]метил]-N-(тридейтериометил)карбамата (1,03 г, 4,050 ммоль) в DCM (10 мл) и перемешивали реакцию при температуре окружающей среды в течение 20 часов. Реакцию фильтровали через слой целита и промывали DCM. Фильтрат концентрировали in vacuo с получением указанного в подзаголовке соединения в виде бесцветного масла (891 мг, выход 88%); 1H ЯМР (400 МГц, DMSO) δ 1,40 (д, J = 43,4 Гц, 9H), 4,48 (с, 2H), 7,43 (д, J = 8,0 Гц, 2H), 7,91 (д, J = 7,9 Гц, 2H) и 10,00 (с, 1H), м.д.
Этап 5. Трет-бутиловый эфир N-[[4-[гидроксииминометил]фенил]метил]-N-(тридейтериометил)карбамата
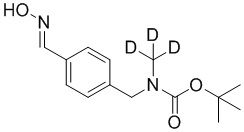
xxii
Гидроксиламин (466,0 мкл, 50% (в./об.), 7,054 ммоль) добавляли в перемешанный раствор трет-бутилового эфира N-[(4-формилфенил)метил]-N-(тридейтериометил)карбамата (890 мг, 3,527 ммоль) в этаноле (5 мл) и перемешивали реакционную смесь при температуре окружающей среды в течение 45 минут. Реакционную смесь концентрировали in vacuo, остаток отбирали в воду и экстрагировали с помощью EtOAc (× 3). Объединенные органические экстракты промывали соляным раствором (× 1), высушивали (MgSO4), фильтровали и концентрировали in vacuo. Остаток растирали с петролейным эфиром и выделяли осадок фильтрованием с получением указанного в подзаголовке продукта в виде белого твердого вещества (837 мг, выход 89%); 1H ЯМР (400 МГц, DMSO) δ 1,41 (д, J = 25,8 Гц, 9H), 4,38 (с, 2H), 7,24 (д, J = 8,0 Гц, 2H), 7,58 (д, J = 8,0 Гц, 2H), 8,13 (с, 1H) и 11,20 (с, 1H) м.д.; МС (ES+) 212,0 (M-tBu).
Этап 6. Трет-бутиловый эфир N-[[4-[хлор-N-гидроксикарбонимидоил]фенил]метил]-N-(тридейтериометил)карбамата
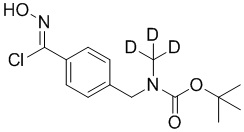
xxiii
Трет-бутиловый эфир N-[[4-[гидроксииминометил]фенил]метил]-N-(тридейтериометил)карбамата (250 мг, 0,9351 ммоль) в DMF (2,5 мл) обрабатывали с помощью NCS (124,9 мг, 0,9351 ммоль) и нагревали реакцию до 65 °C в течение 1 часа. Реакцию охлаждали до температуры окружающей среды и разбавляли водой. Смесь экстрагировали с помощью EtOAc (×2), и объединенные органические экстракты промывали соляным раствором (× 4), высушивали (MgSO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения в виде белого твердого вещества (259 мг, выход 92%); 1H ЯМР (400 МГц, DMSO) δ 1,41 (д, J = 29,6 Гц, 9H), 4,42 (с, 2H), 7,31 (д, J = 8,3 Гц, 2H), 7,78 (д, J = 8,0 Гц, 2H) и 12,38 (с, 1H), м.д.
Этап 7. Трет-бутиловый эфир N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-(4-изопропилсульфонилфенил)пиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-(тридейтериометил)карбамата
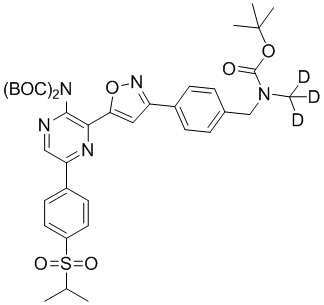
xxiv
Et3N (48,41 мг, 66,68 мкл, 0,4784 ммоль) добавляли по каплям в перемешанный раствор трет-бутилового эфира N-трет-бутоксикарбонил-N-[3-этинил-5-(4-изопропилсульфонилфенил)пиразин-2-ил]карбамата (200 мг, 0,3987 ммоль) и трет-бутилового эфира N-[[4-[хлор-N-гидроксикарбоимидоил]фенил]метил]-N-(тридейтериометил)карбамата (120,3 мг, 0,3987 ммоль) в безводном THF (5 мл) и нагревали реакционную смесь при 65 °C в течение 2,5 часов. Реакционную смесь охлаждали до температуры окружающей среды и разбавляли смесью EtOAc/соляного раствора. Воду добавляли до прозрачности водного слоя и отделения слоев. Водный слой экстрагировали с помощью EtOAc (×1), и объединенные органические экстракты промывали соляным раствором (× 1), высушивали (MgSO4), фильтровали и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии (ISCO Companion, колонка 40 г, элюирование 0–20% смесью EtOAc/петролейного эфира, загружено в DCM) с получением указанного в подзаголовке продукта в виде белого твердого вещества (213,5 мг, выход 70%); 1H ЯМР (400 МГц, DMSO) δ 1,22 (д, J = 6,8 Гц, 6H), 1,31 (с, 18H), 1,43 (д, J = 26,2 Гц, 9H), 3,51–3,60 (м, 1H), 4,47 (с, 2H), 7,42 (д, J = 8,1 Гц, 2H), 8,03 (д, J = 5,2 Гц, 3H), 8,08 (д, J = 8,6 Гц, 2H), 8,65 (д, J = 8,6 Гц, 2H) и 9,52 (с, 1H) м.д.; МС (ES+) 667,4 (M-Boc).
Этап 8. 5-(4-Изопропилсульфонилфенил)-3-[3-[4-[(тридейтериометиламино)метил]фенил]изоксазол-5-ил]пиразин-2-амин (соединение II-3)
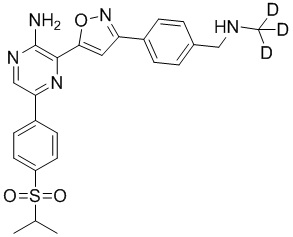
II-3
3 M HCl в MeOH (1,5 мл 3 M, 4,500 ммоль) добавляли в перемешанный раствор трет-бутилового эфира N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-(4-изопропилсульфонилфенил)пиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-(тридейтериометил)карбамата (213 мг, 0,2777 ммоль) в DCM (6 мл) и нагревали реакцию с обратным холодильником в течение 15 часов. Добавляли дополнительную порцию 3 M HCl в MeOH (0,5 мл, 3 M, 1,500 ммоль) и нагревали реакцию с обратным холодильником в течение дополнительных 7 часов. Реакцию охлаждали до температуры окружающей среды и полученный осадок выделяли фильтрованием и сушили под вакуумом при 40 °C с получением дигидрохлорида заявляемого соединения в виде желтого твердого вещества (97,6 мг, выход 65%); 1H ЯМР (400 МГц, DMSO) δ 1,20 (д, J = 6,8 Гц, 6H), 3,47 (тт, J = 14,0, 6,9 Гц, 1H), 4,19–4,25 (м, 2H), 7,23 (с, 2H), 7,72 (д, J = 8,4 Гц, 2H), 7,85 (с, 1H), 7,95 (д, J = 8,7 Гц, 2H), 8,11 (д, J = 8,4 Гц, 2H), 8,39 (д, J = 8,7 Гц, 2H), 8,97 (с, 1H) и 9,11 (с, 2H) м.д.; МС (ES+) 467,2.
Пример 5. Синтез 3-[3-[4-(метиламинометил)фенил]изоксазол-5-ил]-5-[4-[1,2,2,2-тетрадейтерио-1-(тридейтериометил)этил]сульфонилфенил]пиразин-2-амина (соединение II-4)
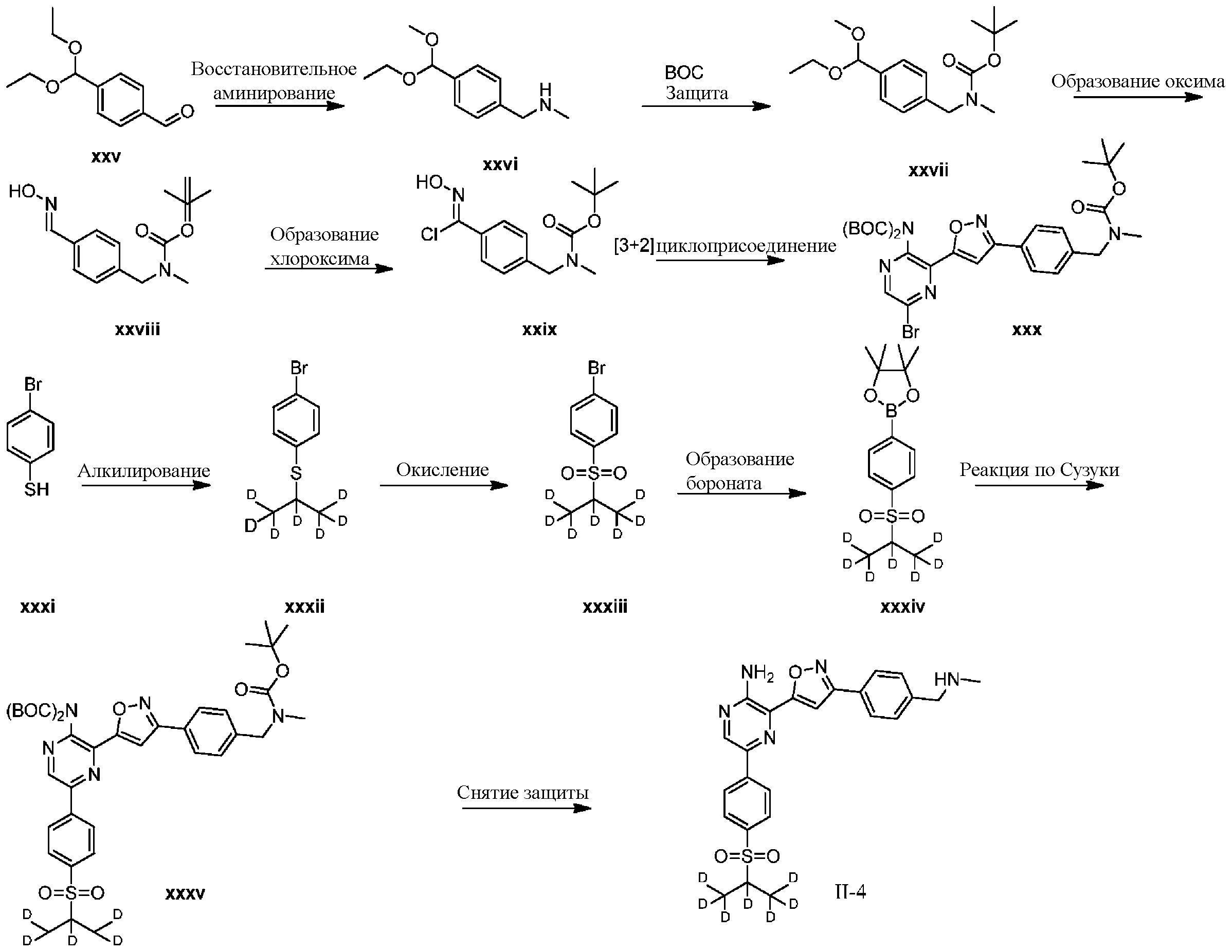
Этап 1. 1-[4-(Диэтоксиметил)фенил]-N-метилметанамин
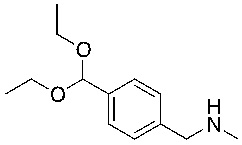
xxvi
2 M метиламина в MeOH (288,1 мл, 576,2 ммоль) разбавляли метанолом (1,000 л) и перемешивали при ~20 °C. По каплям в течение более 1 минуты добавляли 4-(диэтоксиметил)бензальдегид (100 г, 480,2 ммоль) и перемешивали реакцию при температуре окружающей среды в течение 1,25 часа. Порциями в течение более 20 минут добавляли боргидрид натрия (29,07 г, 30,76 мл, 768,3 ммоль), поддерживая температуру в диапазоне от 20 до 30 °C с помощью водно-ледяной бани. Реакционный раствор перемешивали при температуре окружающей среды в течение ночи, а затем гасили путем добавления по каплям NaOH (960,4 мл, 1,0 M, 960,4 ммоль) в течение более 20 минут. Реакцию перемешивали в течение 30 минут и концентрировали in vacuo для удаления MeOH. Реакцию разделяли с MTBE (1,200 л) и отделяли фазы. Органическую фазу промывали водой (300,0 мл), высушивали (Na2SO4) и концентрировали in vacuo с получением заявляемого соединения в форме желтого масла (102,9 г, выход 96%); 1H ЯМР (400 МГц, CDCl3) δ 1,25 (т, 6H), 2,46 (с, 3H), 3,45–3,65 (м, 4H), 3,75 (с, 2H), 5,51 (с, 1H), 7,32 (д, 2H) и 7,44 (д, 2H) м.д.
Этап 2. Трет-бутиловый эфир N-[[4-(диэтоксиметил)фенил]метил]-N-метилкарбамата
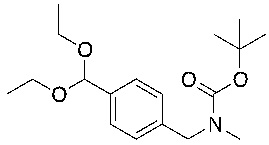
xxvii
В стеклянный реактор с рубашкой объемом 1 л устанавливали верхнеприводную мешалку, термопару и холодильник. Раствор 1-[4-(диэтоксиметил)фенил]-N-метилметанамина (80,0 г, 358,2 ммоль) в DCM (480,0 мл) перемешивали при 18 °C. Раствор ангидрида Boc (79,75 г, 83,95 мл, 365,4 ммоль) в DCM (160,0 мл) добавляли в течение более 10 минут и раствор перемешивали при 20–25 °C в течение ночи. Реакционную смесь фильтровали, промывали DCM (3 × 50 мл), и фильтрат концентрировали in vacuo с получением заявляемого соединения в виде бледно-желтой жидкости (116,6 г, количественный выход); 1H ЯМР (400 МГц, CDCl3) δ 1,25 (т, 6H), 1,49–1,54 (2 × с, 9H), 2,78–2,83 (2 × с, 3H), 3,50–3,66 (м, 4H), 4,42 (с, 2H), 5,49 (с, 1H), 7,22 (д, 2H) и 7,45 (д, 2H) м.д.
Этап 3. Трет-бутиловый эфир N-[[4-гидроксииминометил]фенил]метил]-N-метилкарбамата
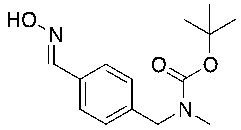
xxviii
Двухфазный раствор трет-бутилового эфира N-[[4-(диэтоксиметил)фенил]метил]-N-метилкарбамата (50,0 г, 154,6 ммоль) в 2-MeTHF (400,0 мл) и Na2SO4 (100,0 мл, 10% в./об., 70,40 ммоль) перемешивали при 8–10 °C в стеклянном реакторе с рубашкой объемом 1 л. Добавляли гидрохлорид гидроксиламина (46,38 мл 5,0 M, 231,9 ммоль), и двухфазный раствор перемешивали при 30 °C в течение 16 часов. Реакцию разбавляли с помощью MTBE (200,0 мл) и отделяли слои. Органическую фазу промывали водой (200,0 мл), высушивали (Na 2SO4), фильтровали и концентрировали in vacuo. Остаток разбавляли гептаном (200,0 мл) и полученную суспензию перемешивали при температуре окружающей среды в течение 30 минут. Твердое вещество собирали фильтрованием с получением заявляемого соединения в виде белого твердого вещества (36,5 г, выход 89%); 1H ЯМР (400 МГц, CDCl3) δ 1,50 (с, 9H), 2,88 (уш. с, 3H), 4,60 (с, 2H), 7,26 (д, 2H), 7,52 (д, 2H) и 8,15 (с, 1H) м.д.
Этап 4. Трет-бутиловый эфир N-[[4-[хлор-N-гидроксикарбонимидоил]фенил]метил]-N-метилкарбамата
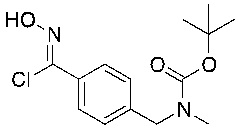
xxix
Суспензию трет-бутилового эфира N-[[4-[гидроксииминометил]фенил]метил]-N-метилкарбамата (100 г, 378,3 ммоль) в изопропилацетате (1,000 л) перемешивали при температуре окружающей среды. Добавляли N-хлорсукцинимид (53,04 г, 397,2 ммоль) и перемешивали при температуре окружающей среды в течение 16 часов. Реакцию разделяли с водой (500,0 мл) и отделяли фазы. Органическую фазу промывали водой (500,0 мл) (2 x), высушивали (Na2SO4), фильтровали и концентрировали in vacuo для удаления большей части растворителя. Добавляли гептан (1,000 л) и смесь концентрировали in vacuo для удаления большей части растворителя. Добавляли гептан (1,000 л) и полученный осадок выделяли фильтрованием. Фильтровальный осадок промывали гептаном (500 мл) и сушили на воздухе с получением заявляемого соединения в виде грязно-белого порошка (105,45 г, выход 93%); 1H ЯМР (400 МГц, CDCl3) δ 1,48 (2 × с, 9H), 2,90 (2 × с, 3H), 4,47 (с, 2H), 7,26 (д, 2H), 7,77 (д, 2H) и 8,82 (с, 1H) м.д.
Этап 5. Трет-бутиловый эфир N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-бромпиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-метилкарбамата
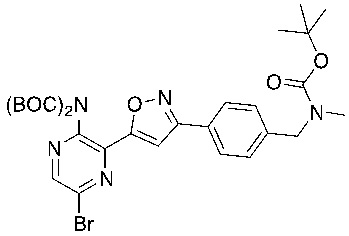
xxx
Суспензию трет-бутилового эфира N-[[4-[хлор-N-гидроксикарбонимидоил]фенил]метил]-N-метилкарбамата (100,0 г, 334,7 ммоль) и трет-бутилового эфира N-трет-бутоксикарбонил-N-[3-этинил-5-(4-изопропилсульфонилфенил)пиразин-2-ил]карбамата (121,2 г, 304,3 ммоль) в DCM (1,212 л) перемешивали при температуре окружающей среды. Одной порцией добавляли триэтиламин (33,87 г, 46,65 мл, 334,7 ммоль) и реакцию перемешивали при температуре окружающей среды в течение 16 часов. Реакцию разделяли с водой (606,0 мл) и отделяли фазы. Органическую фазу промывали водой (606,0 мл), высушивали (Na 2SO4), фильтровали и концентрировали in vacuo до практически сухого состояния. Добавляли гептан (363,6 мл) и смесь концентрировали до приблизительно 300 мл. Добавляли дополнительный гептан (1,212 л) и смесь нагревали до 90 °C при перемешивании. Смесь медленно охлаждали до температуры окружающей среды и перемешивали при этой температуре в течение 1 часа. Полученный осадок выделяли фильтрованием и фильтровальный осадок промывали гептаном (2 × 363,6 мл) и сушили на воздухе с получением заявляемого соединения в виде бежевого порошка (181,8 г, выход 90%); 1H ЯМР (400 МГц, CDCl3) δ 1,41 (с, 18H), 1,51 (с, 9H), 2,88 (2 × с, 3H), 4,50 (с, 2H), 7,36–7,38 (м, 3H), 7,86 (д, 2H) и 8,65 (с, 1H) м.д.
Этап 6. 1-Бром-4-[1,2,2,2-тетрадейтерио-1-(тридейтериометил)этил]сульфанилбензол
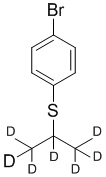
xxxii
Гидрид натрия (246,5 мг, 6,163 ммоль) добавляли порциями в перемешанный раствор 4-бромбензолтиола (соединение xxxi) (970,9 мг, 5,135 ммоль) в DMF (10 мл) при 0 °С. После перемешивания при этой температуре в течение 15 минут добавляли 1,1,1,2,3,3,3-гептадейтерио-2-йодпропан (1 г, 5,649 ммоль) и реакционную смесь оставляли нагреваться до температуры окружающей среды в течение более 18 часов. Реакцию гасили добавлением воды и смесь перемешивали в течение 10 минут. Смесь экстрагировали диэтиловым эфиром (×3), и объединенные органические экстракты промывали водой (×2), соляным раствором (× 2), высушивали (MgSO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения, которое использовали непосредственно без дополнительной очистки, предполагая 100%-й выход и чистоту; 1H ЯМР (500 МГц, DMSO) δ 7,25–7,37 (м, 2H) и 7,48–7,55 (м, 2H) м.д.
Этап 7. 1-Бром-4-[1,2,2,2-тетрадейтерио-1-(тридейтериометил)этил]сульфонилбензол
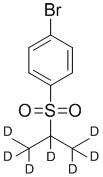
xxxiii
mCPBA (2,875 г, 12,83 ммоль) добавляли порциями в перемешанный раствор 1-бром-4-[1,2,2,2-тетрадейтерио-1-(тридейтериометил)этил]сульфанилбензола (1,223 г, 5,134 ммоль) в DCM (20 мл) при 0 °С и реакционной смеси давали нагреться до температуры окружающей среды в течение более 17 часов. Смесь промывали 1 М водного раствора NaOH (2 х), насыщенным водным раствором Na2S2O3 (× 3), соляным раствором (× 1), высушивали (MgSO4), фильтровали и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии (ISCO Companion, колонка 80 г, элюирование 0–40% смесью EtOAc/петролейного эфира, загружено в DCM) с получением указанного в подзаголовке соединения в виде бесцветного масла (1,19 г, выход 86%); 1Н ЯМР (500 МГц, DMSO) δ 7,77–7,81 (м, 2H) и 7,88–7,92 (м, 2H) м.д.
Этап 8. 4,4,5,5-Тетраметил-2-[4-[1,2,2,2-тетрадейтерио-1-(тридейтериометил)этил]сульфонилфенил]-1,3,2-диоксаборолан
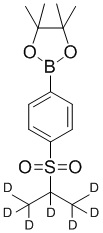
xxxiv
Pd(dppf)Cl2.DCM (179,8 мг, 0,2202 ммоль) добавляли к перемешанной суспензии 1-бром-4-[1,2,2,2-тетрадейтерио-1-(тридейтериометил)этил]сульфонилбензола (1,19 г, 4,404 ммоль), бис(дипинаколато)дибора (1,342 г, 5,285 ммоль) и KOAc (1,296 г, 13,21 ммоль) в диоксане (10 мл). Реакцию помещали в атмосферу азота с помощью 5 × азотных/вакуумных циклов, и смесь нагревали при 80 °С в течение 4,5 часов. Реакцию охлаждали до температуры окружающей среды и растворитель удаляли in vacuo. Остаток распределяли между Et2O и водой и отделяли слои. Органический слой высушивали (MgSO4), фильтровали и концентрировали in vacuo. Остаток растворяли в 30% смеси EtOAc/петролейного эфира (35 мл) и добавляли 1,2 г Florosil. Смесь перемешивали в течение 30 минут, затем фильтровали, промывали твердые вещества дополнительными аликвотами 30% смеси EtOAc/бензина (×3). Фильтрат концентрировали in vacuo и растирали из 10% EtOAc/петролейного эфира. Полученное твердое вещество выделяли фильтрованием, промывали петролейным эфиром и сушили in vacuo с получением указанного в подзаголовке соединения в виде грязно-белого твердого вещества (1052,1 мг, выход 75%); 1H ЯМР (400 МГц, DMSO) δ 1,33 (с, 12H), 7,87 (д, J = 8,4 Гц, 2H) и 7,94 (д, J = 8,4 Гц, 2H) м.д.
Этап 9. Трет-бутиловый эфир N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-[4-[1,2,2,2-тетрадейтерио-1-(тридейтериометил)этил]сульфонилфенил]пиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-метилкарбамата
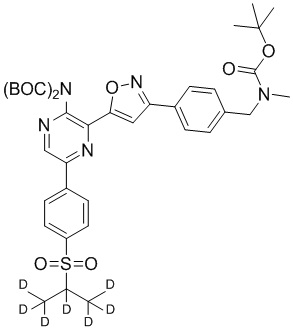
xxxv
[1,1′-Бис(ди-трет-бутилфосфино)ферроцен]дихлорпалладий (II) (106,8 мг, 0,1639 ммоль) добавляли к смеси 4,4,5,5-тетраметил-2-[4-[1,2,2,2-тетрадейтерио-1-(тридейтериометил)этил]сульфонилфенил]-1,3,2-диоксоборолана (1,3 г, 4,098 ммоль), трет-бутиловый эфир N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-бромпиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-метилкарбамата (2,707 г, 4,098 ммоль) и K2CO3 (1,133 г, 8,200 ммоль) в толуоле (9,100 мл), EtOH (2,600 мл) и воде (2,600 мл) и реакционную смесь дегазировали в потоке азота (5 циклов).
Полученную смесь нагревали при 75 °C в течение 1,5 часов. Реакцию охлаждали до температуры окружающей среды и добавляли воду (5,2 мл). После перемешивания слои отделяли, а органический слой высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Остаток растирали с IPA и полученный осадок выделяли фильтрованием, промывали с помощью IPA (3 × 4 мл) и высушивали in vacuo при 50 °C с получением заявляемого соединения в виде белого твердого вещества (2,4 г, выход 76%); 1H ЯМР (400 МГц, CDCl3) δ 1,41 (с, 18H), 1,50 (с, 9H), 2,85–2,89 (м, 3H), 4,50 (с, 2H), 7,36–7,38 (м, 3H), 7,87 (д, 2H), 8,09 (д, 2H), 8,35 (д, 2H) и 9,06 (с, 1H) м.д.
Этап 10. 3-[3-[4-(Метиламинометил)фенил]изоксазол-5-ил]-5-[4-[1,2,2,2-тетрадейтерио-1-(тридейтериометил)этил]сульфонилфенил]пиразин-2-амин (соединение II-4)
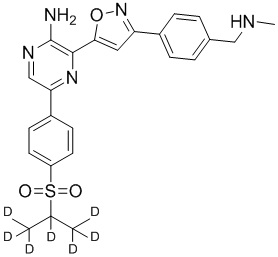
II-4
Концентрированную HCl (3,375 г, 2,812 мл, 37% вес./вес., 34,25 ммоль) добавляли в раствор трет-бутилового эфира N-[[4-[5-[3-[бис(трет-бутоксикарбонил)амино]-6-[4-[1,2,2,2-тетрадейтерио-1-(тридейтериометил)этил]сульфонилфенил]пиразин-2-ил]изоксазол-3-ил]фенил]метил]-N-метилкарбамата (2,2 г, 2,854 ммоль) в ацетоне (28,60 мл) и реакцию нагревали с обратным холодильником в течение 7 часов. Реакцию охлаждали до температуры окружающей среды и полученный осадок выделяли фильтрованием, промывали ацетоном (2 × 4,5 мл) и высушивали in vacuo при 50 °C с получением дигидрохлорида заявляемого соединения в виде желтого твердого вещества (1,42 г, выход 92%); 1H ЯМР (400 МГц, DMSO) δ 2,58 (т, 3H), 4,21 (т, 2H), 5,67 (уш. с, 2H), 7,74 (д, 2H), 7,85 (с, 1H), 7,94 (д, 2H), 8,10 (д, 2H), 8,38 (д, 2H), 8,96 (с, 1H) и 9,33 (уш. с, 2H) м.д.; МС (ES+) 471,8.
Пример 6. Синтез 5-(4-(трет-бутилсульфонил)фенил)-3-(3-(4-((метиламино)метил)фенил)изоксазол-5-ил)пиразин-2-амина (соединение I-2)
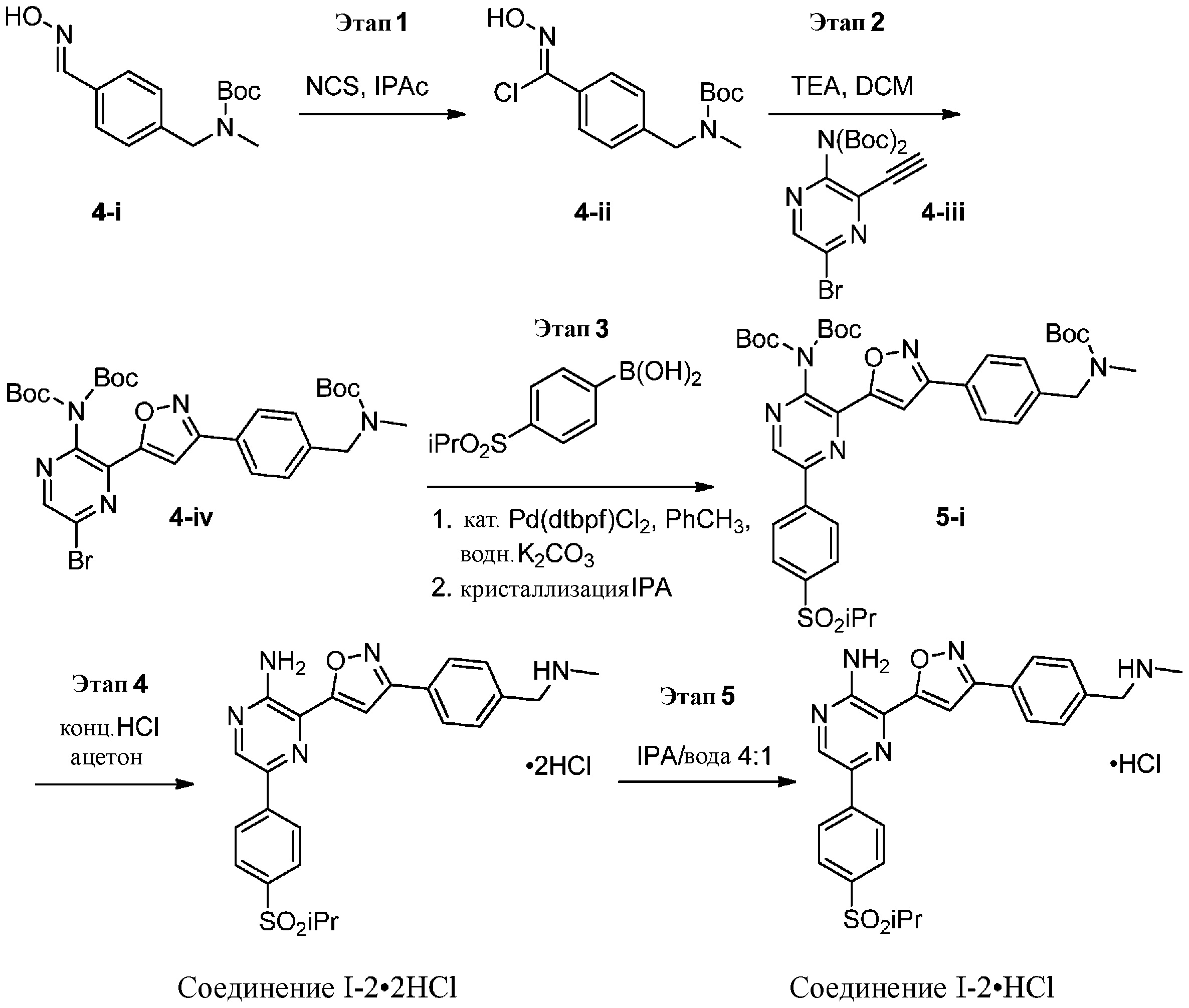
Этап 1. Получение соединения 4-ii
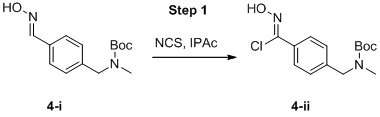
Суспензию трет-бутилового эфира 4-((гидроксимино)метил)бензил(метил)карбамата (соединение 4-i) (650 г, 2,46 моль) в изопропилацетате (6,5 л) перемешивают при температуре окружающей среды. Добавляют N-хлорсукцинимид (361 г, 2,71 моль) и реакционную смесь выдерживают в течение ночи при 20–28 °C для обеспечения полноты реакции. Реакционную смесь разбавляют водой (3,25 л) и EtOAc (1,3 л) и отделяют фазы. Органическую фазу промывают водой (2 × 3,25 л), высушивают (Na2SO4) и концентрируют до влажного осадка. Концентрат разбавляют гептаном (9,1 л), удаляют ~2 л растворителя, а затем перемешивают при температуре окружающей среды в течение 2–20 ч. Твердое вещество собирают фильтрованием. Фильтровальный осадок промывают гептаном (2 × 975 мл) и высушивают с получением соединения 4-ii (692 г; выход 94%, чистота 99,2% по площади по ВЭЖХ) в виде бесцветного порошка.
Этап 2. Получение трет -бутилового эфира (5-бром-3-(3-(4-((( трет -бутоксикарбонил)(метил)амино)метил)фенил)изоксазол-5-ил)пиразин-2-ил) ( трет -бутоксикарбонил)карбамата ( соединение 4-iv)
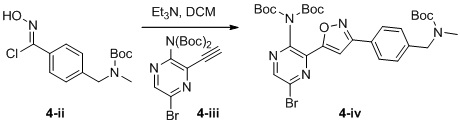
Суспензию трет-бутилового эфира N-(5-бром-3-этинилпиразин-2-ил)-N-трет-бутоксикарбонилкарбамата (соединение 4-iii) (1,59 кг, 3,99 моль) и трет-бутилового эфира 4-(хлор(гидроксиимино)метил)бензил(тетрагидро-2H-пиран-4-ил)карбамата (1,31 кг, 4,39 моль; 1,10 экв.) в CH2Cl2 (12,7 л) перемешивают при температуре окружающей среды. Триэтиламин (444 г, 611 мл, 4,39 моль) добавляют к суспензии и температуру реакции поддерживают в диапазоне 20–30 °C в течение 20–48 ч, чтобы обеспечить завершение реакции. Реакционную смесь разбавляют водой (8 л) и тщательно перемешивают, после чего отделяют фазы. Органическую фазу промывают водой (8 л), высушивают (Na2SO4), а затем концентрируют до приблизительно 1 л CH2Cl2. Концентрат разбавляют гептаном (3,2 л) и повторно концентрируют при 40 °C/20,7 кПа (200 Торр) до прекращения образования дистиллята. Концентрат перемешивают и дополнительно разбавляют гептаном (12,7 л) для осаждения твердого вещества. Суспензию перемешивают в течение ночи. Твердое вещество собирают фильтрованием, промывают гептаном (2 × 3 л), а затем высушивают с получением соединения 4-iv (2,42 кг; выход 92%, чистота 100% по площади по ВЭЖХ) в виде светло-коричневого порошка. 1H ЯМР (400 МГц, CDCl3) δ 8,61 (с, 1H), 7,82 (д, J = 8,2 Гц, 2H), 7,31 (м, 3H), 4,46 (уш. с, 2H), 2,84 (уш. д, 3H), 1,57 (с, 2H), 1,44 (уш. с, 9H), 1,36 (с, 18H).
Этап 3. Получение соединения 5-i
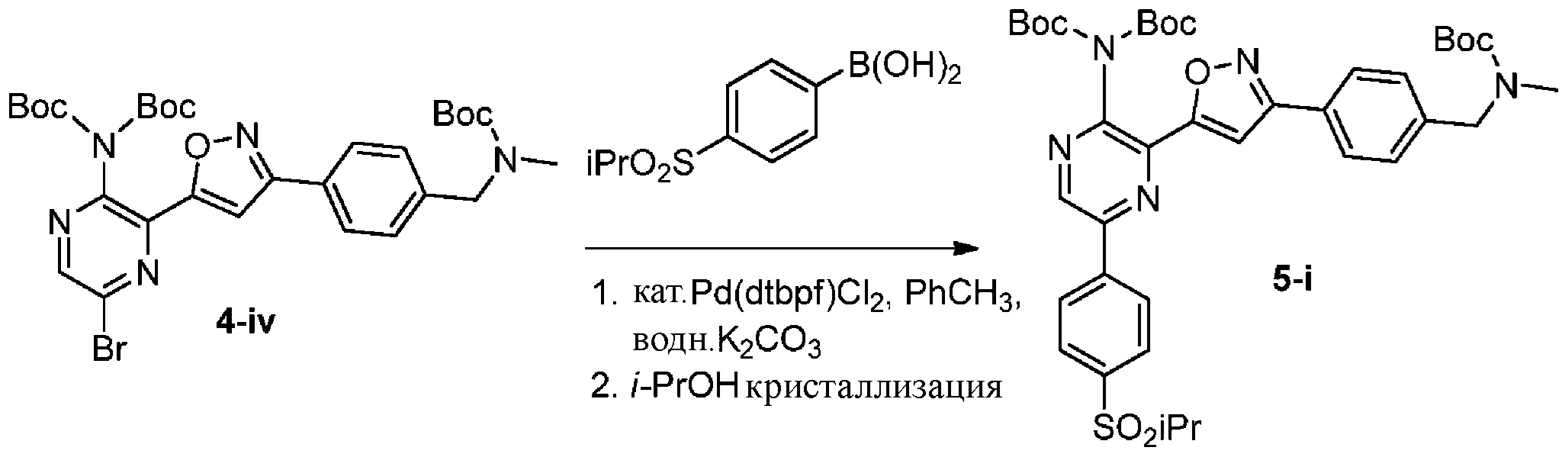
Смесь трет-бутилового эфира (5-бром-3-(3-(4-(((трет-бутоксикарбонил)(метил)амино)метил)фенил)изоксазол-5-ил)пиразин-2-ил)(трет-бутоксикарбонил)карбамата (соединение 4-iv) (1,00 кг, 1,51 моль), K2CO3 (419 г, 3,02 моль) и (4-(изопропилсульфонил)фенил)бороновой кислоты (345 г, 1,51 моль) в толуоле (7,0 л) и воде (2,0 л) перемешивают и дегазируют с помощью N2 в течение 30 мин. Затем добавляют 1,1’-бис(ди-трет-бутилфосфино)ферроцендихлорпалладий (II) [Pd(dtbpf)Cl2; 19,7 г, 30,3 ммоль] и дегазируют еще дополнительно 20 мин. Реакционную смесь нагревают при 70 °C в течение по меньшей мере 1 ч, чтобы обеспечить завершение реакции. Реакционную смесь охлаждают до температуры окружающей среды, затем фильтруют через целит. Реакционный сосуд и фильтровальный слой промывают толуолом (2 × 700 мл). Фильтраты объединяют и фазы отделяют. Органическую фазу перемешивают со смолой Biotage MP-TMT (170 г) в течение 4–20 ч. Смолу удаляют фильтрованием через целит и фильтровальный слой промывают толуолом (2 × 700 мл). Фильтрат и смывы объединяют и концентрируют практически до сухого состояния, после чего разбавляют i-PrOH (5,75 л) и повторно концентрируют. Концентрат вновь растворяют в теплом (45 °C) i-PrOH (5,75 л), после чего охлаждают до температуры окружающей среды при перемешивании, чтобы инициировать кристаллизацию, а затем продолжают перемешивание в течение приблизительно 16–20 ч. Твердое вещество собирают фильтрованием, промывают i-PrOH (2 × 1 л) и высушивают с получением VRT-1018729 (967 г; 84%) в виде бежевого порошка. 1H ЯМР (400 МГц, CDCl3) δ 9,04 (с, 1H), 8,33 (д, J = 8,6 Гц, 2H), 8,06 (д, J = 8,5 Гц, 2H), 7,85 (д, J = 8,1 Гц, 2H), 7,34 (м, 3H), 4,47 (уш. с, 2H), 3,25 (гепт, J = 7,0 Гц, 1H), 2,85 (уш. д, 3H), 1,47 (с, 9H), 1,38 (с, 18H), 1,33 (д, J = 6,9 Гц, 6H).
Этап 4. Получение соединения I-2 • 2HCl
Раствор соединения 5-i (950 г, 1,24 моль) в ацетоне (12,35 л) нагревают до 40 °C, затем добавляют концентрированную HCl (1,23 кг, 1,02 л, 37% вес./вес., 12,4 моль) с такой скоростью, чтобы температура реакции поддерживалась в диапазоне 40–45 °C в течение по меньшей мере 5 ч для завершения реакции. Суспензию охлаждают до температуры ниже 30 °C и твердое вещество собирают фильтрованием. Фильтровальный осадок промывают ацетоном (2 × 950,0 мл), а затем высушивают с получением соединения I-2 • 2HCl (578 г; выход 87%, чистота 99,5% по площади по ВЭЖХ) в виде желтого порошка. 1H ЯМР (400 МГц, DMSO) δ 9,53 (уш. д, J = 4,8 Гц, 2H), 8,93 (с, 1H), 8,37 (д, J = 8,5 Гц, 2H), 8,07 (д, J = 8,3 Гц, 2H), 7,92 (д, J = 8,6 Гц, 2H), 7,84 (с, 1H), 7,75 (д, J = 8,3 Гц, 2H), 4,23–4,15 (м, 2H), 3,43 (гепт, J = 6,8 Гц, 1H), 2,55 (т, J = 5,3 Гц, 3H), 1,17 (д, J = 6,8 Гц, 6H).
Этап 5. Получение соединения I-2 • HCl из соединения I-2 • 2HCl
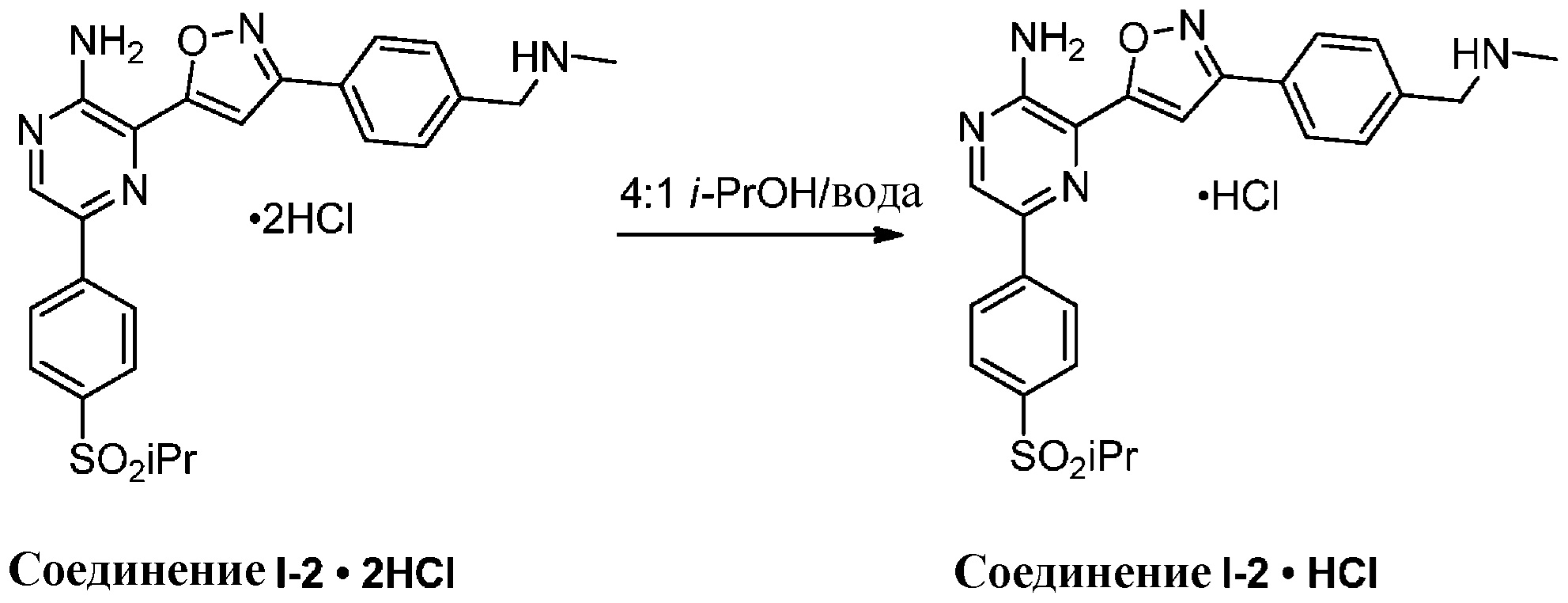
Процесс, проходящий в двух сосудах
Перемешанную суспензию соединения I-2 • 2HCl (874 г, 1,63 моль) в i-PrOH (3,50 л) и воде (0,87 л) нагревают при 50 °C в течение 1–2 ч, охлаждают до температуры окружающей среды и перемешивают в течение 1–20 ч. XRPD проводят на небольшом образце, чтобы убедиться в преобразовании соединения I-2 • 2HCl в другую форму. Суспензию охлаждают до 5 °C и перемешивают в течение 1 ч. Твердое вещество собирают фильтрованием, затем фильтровальный осадок промывают смесью i-PrOH/вода в соотношении 80/20 (2 × 874 мл) и быстро высушивают.
Если XRPD показывает соединение I-2 • HCl/безводная форма, твердое вещество высушивают с получением соединения I-2 • HCl/безводная форма (836 г, выход 99%, чистота 99,2% по площади по ВЭЖХ) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO) δ 9,38 (с, 2H), 8,96 (с, 1H), 8,46–8,34 (м, 2H), 8,10 (д, J = 8,3 Гц, 2H), 7,94 (д, J = 8,6 Гц, 2H), 7,85 (с, 1H), 7,75 (д, J = 8,3 Гц, 2H), 7,23 (уш. с, 2H), 4,21 (с, 2H), 3,47 (гепт, J = 6,7 Гц, 1H), 2,58 (с, 3H), 1,19 (д, J = 6,8 Гц, 6H).
Если XRPD показывает соединение I-2 • HCl/водная форма, твердое вещество перемешивают в свежеприготовленном i-PrOH (3,50 л) и воде (0,87 л) при 50 °C в течение по меньшей мере 2 ч до тех пор, пока XRPD не покажет полное преобразование в соединение I-2 • HCl/безводная форма. Затем суспензию охлаждают до 5 °C и перемешивают в течение 1 ч. Твердое вещество собирают фильтрованием, затем фильтровальный осадок промывают смесью i-PrOH/вода в соотношении 80/20 (2 × 874 мл), после чего высушивают с получением соединения I-2 • HCl/безводная форма.
Используемая альтернативная процедура (один сосуд)
Соединение I-2 • 2HCl (392 г) загружают в реактор. В реактор загружают смесь IPA/вода в соотношении 4:1 (8 л) и перемешивают при температуре окружающей среды в течение ночи. XRPD используют для подтверждения преобразования в моногидрохлорид в форме моногидрата. Смесь нагревают до 50 °C. Добавляют зародыши соединения I-2 • HCl/безводная форма (16 г) и смесь нагревают при 50 °C до подтверждения с помощью XRPD полного преобразования в желаемую безводную форму. Смесь охлаждают до комнатной температуры, фильтруют, и твердое вещество промывают смесью IPA/вода в соотношении 4:1 (2 × 800 мл), а затем высушивают с получением соединения I-2 • HCl/безводная форма (343 г, выход 94%).
Этап 4. Альтернативный способ 1. Получение свободного основания соединения I-2
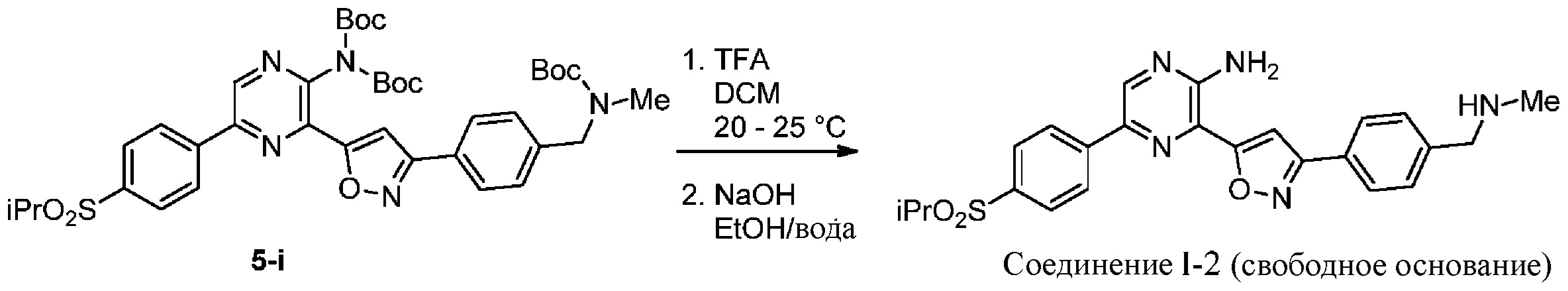
Раствор соединения 5-i (100 г, 131 ммоль) в DCM (200 мл) перемешивали при температуре окружающей среды, затем добавляли TFA (299 г, 202 мл, 2,62 моль). Через 2 ч реакционный раствор охлаждали до 5 °C. Реакционную смесь разбавляли EtOH (1,00 л) в течение более приблизительно 5 минут с получением ярко-желтой суспензии. Суспензию охлаждали до 10 °C, а затем в течение более 30 мин добавляли NaOH (1,64 л 2,0 M, 3,28 моль), после чего перемешивали при температуре окружающей среды в течение ночи. Твердое вещество собирали фильтрованием, затем промывали водой (2 × 400 мл), EtOH (2 × 200 мл), после чего сушили с получением свободного основания соединения I-2 (57,0 г, выход 94%, чистота 99,7% по площади по ВЭЖХ) в виде тонкодисперсного желтого порошка. 1H ЯМР (400 МГц, DMSO) δ 8,95 (с, 1H), 8,39 (д, J = 8,5 Гц, 2H), 7,95 (дд, J = 11,6, 8,4 Гц, 4H), 7,78 (с, 1H), 7,51 (д, J = 8,2 Гц, 2H), 7,21 (уш. с, 2H), 3,72 (с, 2H), 3,47 (гепт, J = 6,8 Гц, 1H), 2,29 (с, 3H), 1,19 (д, J = 6,8 Гц, 6H).
Этап 4. Альтернативный способ 2. Получение соединения I-2 • HCl
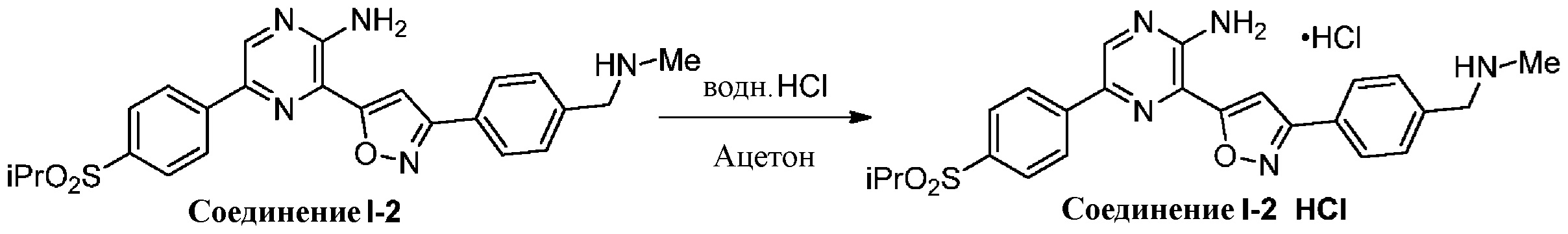
Суспензию свободного основания соединения I-2 (10,0 г, 21,6 ммоль) в ацетоне (80 мл) перемешивали и нагревали до 35 °C. Добавляли водный раствор HCl (11,9 мл 2,0 M, 23,8 ммоль), разбавленный водой (8,0 мл), и смесь нагревали при 50 °C в течение 4 ч. Суспензии давали охладиться до температуры окружающей среды, после чего ее перемешивали в течение ночи. Твердое вещество собирали фильтрованием. Фильтровальный осадок промывали ацетоном (2 × 20 мл), затем сушили с получением 10,2 г гидрохлорида соединения I-2 (выход 95%) в виде желтого порошка.
Пример 7. Синтез 5-(4-(изопропилсульфонил)фенил)-3-(3-(4-(тетрагидропиран-4-иламино)метил)фенил)изоксазол-5-ил)пиразин-2-амина (соединение I-3)
Схема. Пример синтеза соединения I-3
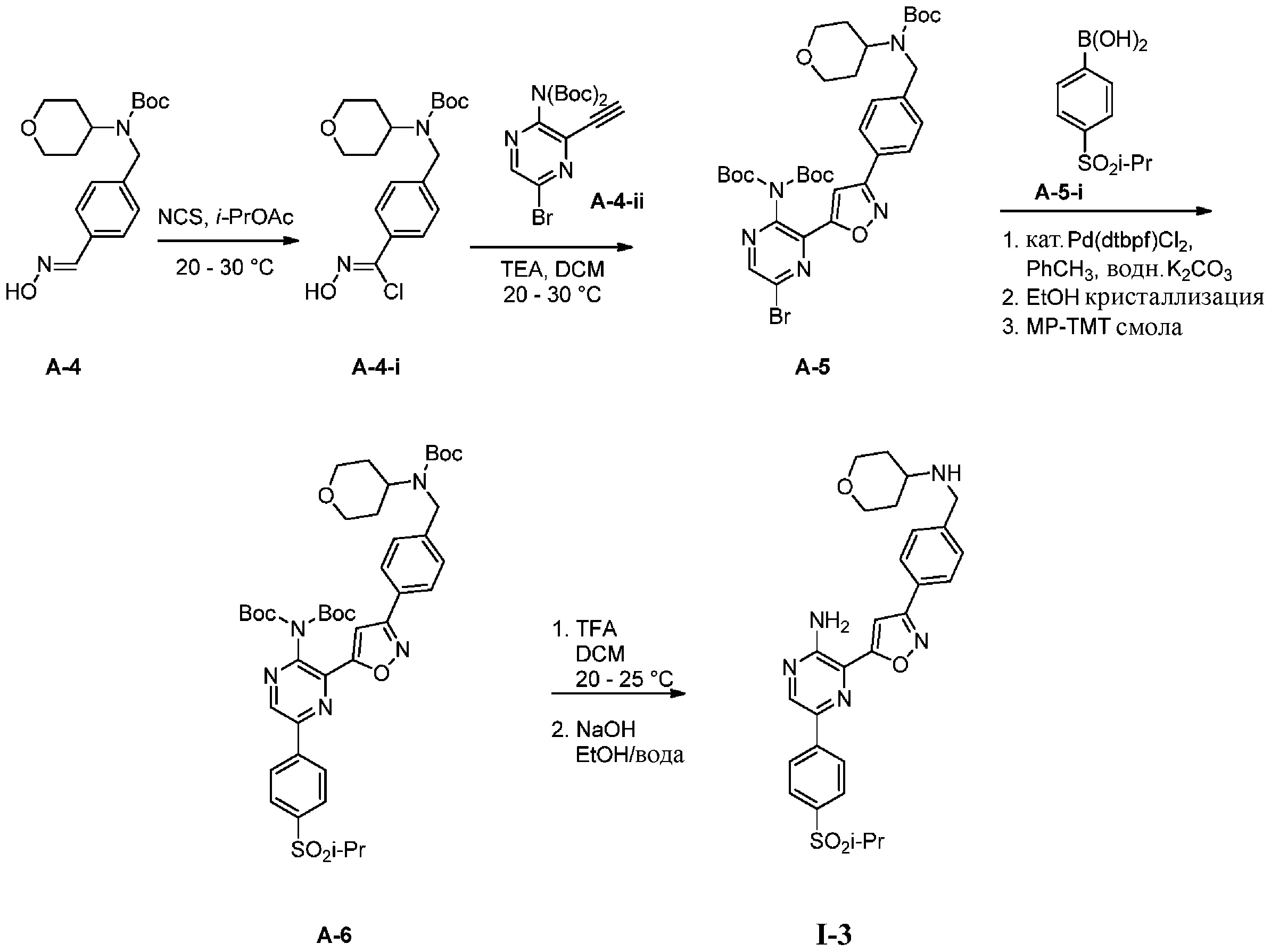
Этап 1. Получение N -(4-(диэтоксиметил)бензил)тетрагидро-2 H -пиран-4-амина (A-2)
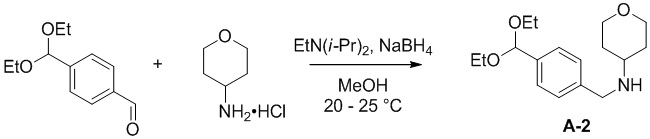
Раствор гидрохлорида тетрагидро-2H-пиран-4-амина (1,13 кг, 8,21 моль) в MeOH (14,3 л) перемешивают при приблизительно 20 °C, а затем добавляют Et3N (1,06 кг, 1,43 л, 8,21 моль). Смесь перемешивают в течение по меньшей мере 5 мин, затем добавляют диэтилацеталь терефталевого альдегида (1,43 кг, 6,84 моль) при поддержании температуры реакции в диапазоне 20–25 °C. Смесь перемешивают в течение по меньшей мере 45 мин для образования имина. Гранулы NaBH4 (414 г, 11,0 моль) добавляют при поддержании температуры реакции ниже приблизительно 25 °С. Смесь перемешивают в течение 1 ч после завершения добавления. Реакционную смесь гасят путем добавления 1 М NaOH (13,7 л), затем экстрагируют с помощью МТВЕ. Органический раствор промывают соляным раствором (7,13 л), затем высушивают (Na2SO4) и концентрируют с получением соединения A-2 (2197 г; выход 109%, чистота 94,4% по площади по ВЭЖХ) в виде мутного масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,43 (д, J = 8,1 Гц, 2H), 7,31 (д, J = 8,1 Гц, 2H), 5,49 (с, 1H), 4,66 (уш. с, 1H), 4,03–3,91 (м, 2H), 3,82 (с, 2H), 3,69–3,47 (м, 4H), 3,38 (тд, J = 11,6, 2,1 Гц, 2H), 2,78–2,65 (м, 1H), 1,90–1,81 (м, 2H), 1,53–1,37 (м, 2H), 1,23 (т, J = 7,1 Гц, 6H).
Этап 2. Получение трет- бутилового эфира 4-(диэтоксиметил)бензил(тетрагидро-2 H -пиран-4-ил)карбамата (A-3)
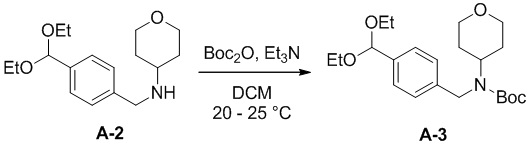
Смесь N-(4-(диэтоксиметил)бензил)тетрагидро-2H-пиран-4-амина (A-2) (2195 г, 7,48 моль) в CH2Cl2 (22,0 л) перемешивают при 25 °C и затем добавляют ди-трет-бутилдикарбонат (1,71 кг, 7,86 моль). Затем добавляют Et3N (795 г, 1,10 л) при поддержании температуры реакции в диапазоне 20–25 °C. Реакционную смесь перемешивают при приблизительно 25 °C в течение 12–20 ч. После завершения реакции смесь охлаждают до приблизительно 20 °C и гасят 0,5 M водного раствора лимонной кислоты (7,48 л, 3,74 моль) при поддержании температуры реакции в диапазоне 20–25 °C. Органическую фазу собирают, промывают насыщ. NaHCO3 (6,51 л, 7,48 моль), промывают соляным раствором (6,59 л) и высушивают (Na2SO4), а затем концентрируют с получением трет-бутилового эфира 4-(диэтоксиметил)бензил(тетрагидро-2H-пиран-4-ил)карбамата (A-3) (2801 г; выход 95%, чистота 98,8% по площади по ВЭЖХ) в виде густого масла янтарного цвета. 1Н ЯМР (400 МГц, CDCl3) δ 7,40 (д, J = 8,1 Гц, 2H), 7,21 (д, J = 7,9 Гц, 2H), 5,49 (с, 1H), 4,39 (уш. с, 3H), 3,93 (уш. дд, J = 10,8, 3,8 Гц, 2H), 3,67–3,47 (м, 4H), 3,40 (уш. м, 2H), 1,68–1,59 (м, 4H), 1,39 (уш. с, 9H), 1,23 (т, J = 7,1 Гц, 6H).
Этап 3. Получение трет- бутилового эфира 4-((гидроксиимино)метил)бензил(тетрагидро-2 H -пиран-4-ил)карбамата (A-4)
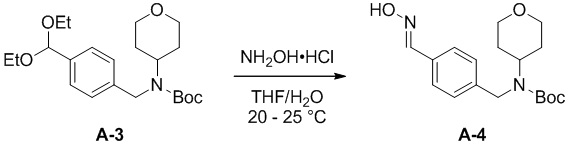
Раствор трет-бутилового эфира 4-(диэтоксиметил)бензил(тетрагидро-2H-пиран-4-ил)карбамата (A-3) (2,80 кг, 7,12 моль) в THF (28,0 л) и воде (2,80 л) перемешивают при приблизительно 20 °C. Добавляют гидрохлорид гидроксиламина (593 г, 8,54 моль), поддерживая температуру реакции в диапазоне 20–25 °С. Реакционную смесь перемешивают при приблизительно 20 °C в течение 16–20 ч, затем разбавляют с помощью CH2Cl2 (8,4 л) и 50%-го соляного раствора (11,2 л) и перемешивают в течение по меньшей мере 5 мин. Фазы отделяют, затем органическую фазу промывают 50%-м соляным раствором (2 × 2,8 л), высушивают (Na2SO4) и концентрируют. Концентрат разбавляют MeOH (1,4 л) и повторно концентрируют. Концентрат разбавляют МеОН (14,0 л) и переносят в реакционный сосуд. Раствор нагревают до приблизительно 25 ºC, затем добавляют воду (14,0 л) в течение приблизительно более 1–1,5 ч; после добавления приблизительно 10 л воды в смесь вносят зародыши и наблюдают мутную суспензию. Добавляют дополнительное количество воды (8,4 л) в течение более 1,5 ч для дополнительного осаждения продукта. После выдерживания твердое вещество собирают путем фильтрования. Фильтровальный осадок промывают гептаном (5,6 л) и высушивают с получением трет-бутилового эфира 4-((гидроксиимино)метил)бензил(тетрагидро-2H-пиран-4-ил)карбамата (A-4) (1678 г; выход 71%, чистота 91,5% по площади по ВЭЖХ) в виде грязно-белого порошка. 1H NMR (400 МГц, CDCl3) δ 8,12 (с, 1H), 7,51 (д, J = 8,2 Гц, 2H), 7,24 (д, J = 7,9 Гц, 2H), 4,40 (уш. с, 3H), 3,96 (дд, J = 10,4, 3,6 Гц, 2H), 3,41 (уш. м, 2H), 1,69–1,61 (м, 4H), 1,39 (уш. с, 9H).
Этап 4. Получение
трет
-бутилового эфира 4-(хлор(гидроксиимино)метил)бензил(тетрагидро-2
H
-пиран-4-ил)карбамата (A-4-i)
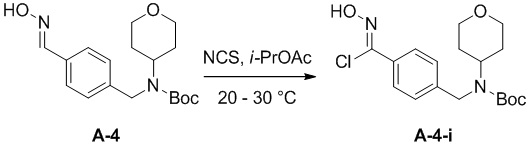
Суспензию (E)-трет-бутилового эфира 4-((гидроксиимино)метил)бензил(тетрагидро-2H-пиран-4-ил)карбамата (A-4) (1662 г, 4,97 моль) в i-PrOAc (16,6 л) перемешивают при 20 °C в реакторе. Добавляют N-хлорсукцинимид (730 г, 5,47 моль) при поддержании температуры приблизительно 20 °C. Суспензию перемешивают при приблизительно 20 °C для завершения реакции. Суспензию разбавляют водой (8,3 л) и перемешивают для растворения твердого вещества. Фазы отделяют и органическую фазу промывают водой (8,3 л). Органическую фазу концентрируют, а затем разбавляют с помощью i-PrOAc (831 мл). Медленно добавляют гептан (13,3 л; 8 об.) для инициации кристаллизации. Затем густую суспензию перемешивают в течение 1 ч. Твердое вещество собирают фильтрованием; фильтровальный осадок промывают гептаном (2 × 1,6 л; 2 × 1 об.) и высушивают с получением (Z)-трет-бутилового эфира 4-(хлор(гидроксиимино)метил)бензил(тетрагидро-2H-пиран-4-ил)карбамата (A-4-i) (1628 г; 89%, чистота 98,0% по площади по ВЭЖХ) в виде белого порошка.
Этап 5. Получение трет -бутилового эфира (5-бром-3-(3-(4-((( трет -бутоксикарбонил)(тетрагидро-2 H -пиран-4-ил)амино)метил)фенил)изоксазол-5-ил)пиразин-2-ил)( трет -бутоксикарбонил)карбамата (A-5)
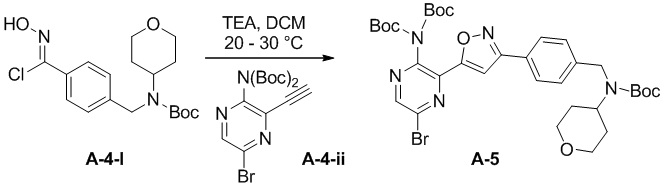
Раствор трет-бутилового эфира 4-(хлор(гидроксиимино)метил)бензил(тетрагидро-2H-пиран-4-ил)карбамата (A-4-i) (1,60 кг, 4,34 моль) и трет-бутилового эфира N-(5-бром-3-этинилпиразин-2-ил)-N-трет-бутоксикарбонилкарбамата (соединение A-4-ii) (1,73 кг, 4,34 моль) в CH2Cl2 (12,8 л) перемешивают при 20 °C. Добавляют Et3N (483 г, 665 мл; 4,77 моль) и поддерживают температуру реакции на уровне ниже 30 °C. Суспензию перемешивают при 20 °C для завершения реакции, а затем разбавляют водой (8,0 л) и встряхивают. Фазы отделяют и органическую фазу промывают водой (8,0 л), после чего концентрируют. Добавляют i-PrOAc (1,6 л) и смесь нагревают до 50 °C. Медленно добавляют гептан (4,0 л), затем суспензии дают остыть до температуры окружающей среды и перемешивают в течение ночи. К суспензии добавляют дополнительный гептан (7,2 л) и перемешивают в течение 1 ч. Твердое вещество собирают фильтрованием. Фильтровальный осадок промывают гептаном (2 × 1,6 л) и высушивают с получением трет-бутилового эфира (5-бром-3-(3-(4-(((трет-бутоксикарбонил)(тетрагидро-2H-пиран-4-ил)амино)метил)фенил)изоксазол-5-ил)пиразин-2-ил)(трет-бутоксикарбонил)карбамата (A-5) (2,478 кг; выход 78%, чистота 97,8% по площади по ВЭЖХ) в виде тонкодисперсного коричневого порошка. 1Н ЯМР (400 МГц, CDCl3) δ 8,60 (с, 1H), 7,78 (д, J = 8,3 Гц, 2H), 7,31 (м, 3H), 4,42 (уш. м, 3H), 4,03–3,82 (м, 2H), 3,38 (уш. c, 2H), 1,60 (м, 4H), 1,36 (с, 27H).
Этап 6. Получение трет -бутилового эфира трет -бутоксикарбонил(3-(3-(4-((( трет -бутоксикарбонил)(тетрагидро-2 H -пиран-4-ил)амино)метил)фенил)изоксазол-5-ил)-5-(4-(изопропилсульфонил)фенил)пиразин-2-ил)карбамата
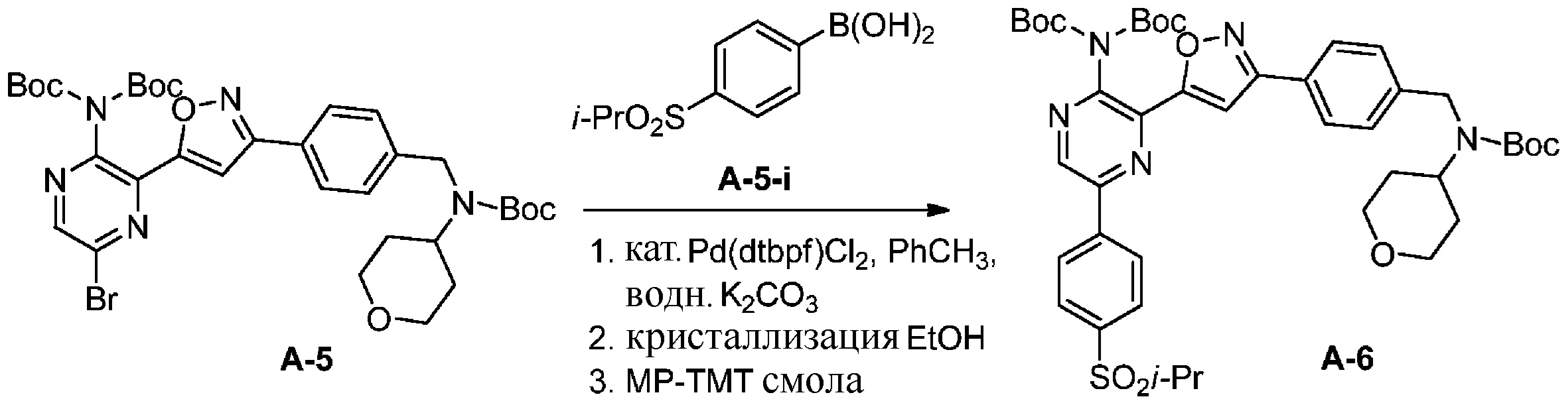
Смесь трет-бутилового эфира (5-бром-3-(3-(4-(((трет-бутоксикарбонил)(тетрагидро-2H-пиран-4-ил)амино)метил)фенил)изоксазол-5-ил)пиразин-2-ил)(трет-бутоксикарбонил)карбамата (A-5) (425 г, 582 ммоль), K2CO3 (161 г, 1,16 моль; 2,0 экв.) и (4-(изопропилсульфонил)фенил)бороновой кислоты (133 г, 582 ммоль) в толуоле (2,98 л) и воде (850 мл) перемешивают и дегазируют с помощью N2 при температуре окружающей среды. Добавляют катализатор [1,1′-бис(ди-трет-бутилфосфин)ферроцен]дихлорпалладий (II), (Pd(dtbpf)Cl2; 1,90 г, 2,91 ммоль) и дегазируют смесь в течение дополнительно 10 мин. Смесь нагревают при 70 °C до тех пор, пока реакция не будет завершена. Смесь охлаждают до 50 °C, разбавляют водой (850 мл) и фильтруют через слой целита. Фазы отделяют. Органическую фазу концентрируют, затем остаток разбавляют с помощью EtOH (1,70 л) и повторно концентрируют. При перемешивании при 40 °C концентрат разбавляют с помощью EtOH (1,70 л), чтобы инициировать кристаллизацию. Суспензию охлаждают до 20 °C и перемешивают в течение 4 ч. Твердое вещество собирают фильтрованием. Фильтровальный осадок промывают с помощью EtOH (2 × 425 мл) и высушивают на воздухе с получением трет-бутилового эфира трет-бутоксикарбонил(3-(3-(4-(((трет-бутоксикарбонил)(тетрагидро-2H-пиран-4-ил)амино)метил)фенил)изоксазол-5-ил)-5-(4-(изопропилсульфонил)фенил)пиразин-2-ил)карбамата (A-6) в виде бежевого порошка. Твердое вещество растворяют в THF (2,13 л) и суспендируют смолой Biotage MP-TMT (48 г) при температуре окружающей среды. Смолу удаляют фильтрованием и фильтрат концентрируют для удаления большей части THF. Концентрат разбавляют с помощью EtOH (970 мл) и концентрируют повторно до приблизительно половины исходного объема. Концентрат снова разбавляют EtOH (970 мл) и смешивают в течение 1 ч при 40 °C. Суспензию охлаждают до температуры окружающей среды и твердое вещество собирают фильтрованием, затем высушивают с получением трет-бутилового эфира трет-бутоксикарбонил(3-(3-(4-(((трет-бутоксикарбонил)(тетрагидро-2H-пиран-4-ил)амино)метил)фенил)изоксазол-5-ил)-5-(4-(изопропилсульфонил)фенил)пиразин-2-ил)карбамата (A-6) (416 г; выход 86%, чистота 99,3% по площади по ВЭЖХ) в виде белого порошка. 1H ЯМР (400 МГц, CDCl3) δ 9,04 (с, 1H), 8,38–8,28 (м, 2H), 8,10–8,01 (м, 2H), 7,82 (д, J = 8,2 Гц, 2H), 7,34 (м, 3H), 4,44 (уш. с, 2H), 3,94 (дд, J = 10,5, 3,5 Гц, 2H), 3,40 (уш. с, 2H), 3,25 (гепт, J = 6,8 Гц, 1H), 1,65 (м, 4H), 1,38 (уш. с, 27H), 1,33 (д, J = 6,9 Гц, 6H).
Этап 7. Получение 5-(4-(изопропилсульфонил)фенил)-3-(3-(4-(((тетрагидро-2 H -пиран-4-ил)амино)метил)фенил)изоксазол-5-ил)пиразин-2-амина (I-3) в форме свободного основания
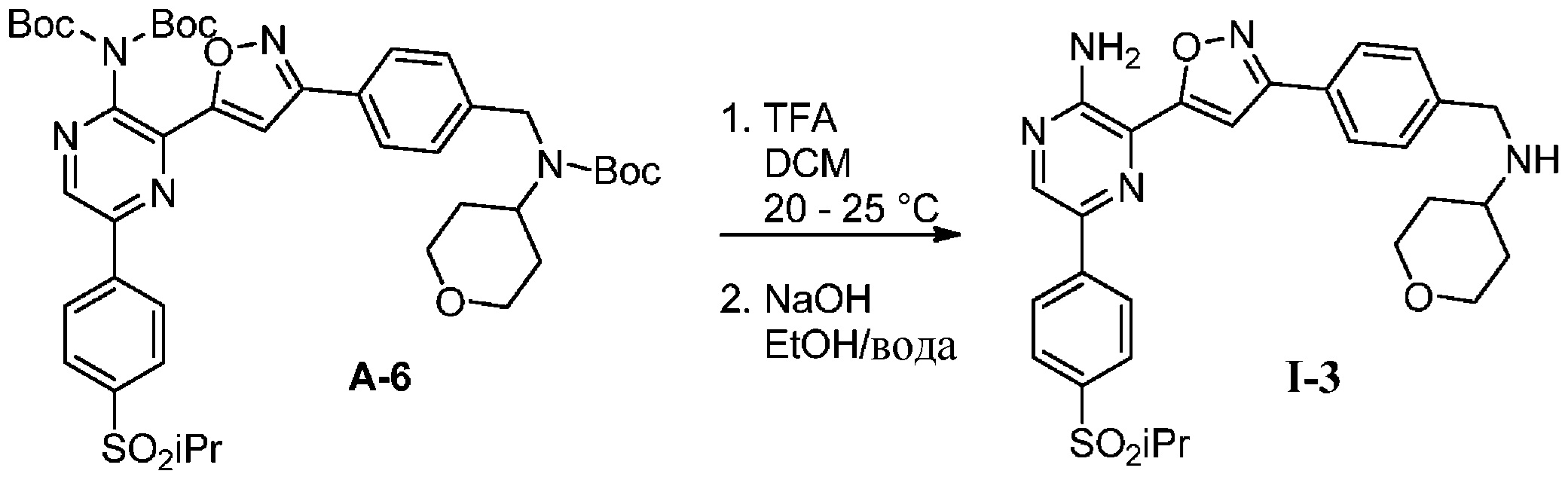
Суспензию трет-бутилового эфира трет-бутоксикарбонил(3-(3-(4-(((трет-бутоксикарбонил)(тетрагидро-2H-пиран-4-ил)амино)метил)фенил)изоксазол-5-ил)-5-(4-(изопропилсульфонил)фенил)пиразин-2-ил)карбамата (A-6) (410 г; 492 ммоль) в CH2Cl2 (410 мл) перемешивают в колбе при температуре окружающей среды. Добавляют TFA (841 г, 568 мл; 7,4 моль), поддерживая температуру реакции в диапазоне 20–25 °C. Раствор перемешивают при температуре окружающей среды в течение приблизительно 3 ч до тех пор, пока анализ не покажет завершение реакции. Раствор охлаждают до приблизительно 5–10 °C и разбавляют с помощью EtOH (3,3 л), поддерживая температуру ниже 20 °C. Добавляют 5,0 M водного раствора NaOH (1,77 л; 8,85 моль), позволяя температуре реакции подняться от приблизительно 14 °C до приблизительно 42 °C. Суспензию нагревают при 70–75 °C в течение 6 ч, удаляя дистиллят. Суспензии дают остыть до температуры окружающей среды. Твердое вещество собирают фильтрованием и фильтровальный осадок промывают водой (4 × 1,64 л). Фильтровальный осадок промывают с помощью EtOH (2 × 820 мл) и высушивают с получением 5-(4-(изопропилсульфонил)фенил)-3-(3-(4-(((тетрагидро-2H-пиран-4-ил)амино)метил)фенил)изоксазол-5-ил)пиразин-2-амина (соединение I-1) (257 г; выход 98%, чистота 99,5% по площади по ВЭЖХ) в виде желтого порошка.
1H ЯМР (400 МГц, DMSO) δ 8,94 (с, 1H), 8,44–8,33 (м, 2H), 7,94 (т, J = 8,2 Гц, 4H), 7,76 (с, 1H), 7,53 (д, J = 8,2 Гц, 2H), 7,20 (с, 2H), 3,83 (м, 1H), 3,80 (с, 3H), 3,46 (гепт, J = 6,8 Гц, 1H), 3,25 (тд, J = 11,4, 2,1 Гц, 2H), 2,66–2,54 (м, 1H), 1,79 (уш. дд, 2H), 1,36–1,22 (м, 2H), 1,19 (д, J = 6,8 Гц, 6H). 13C ЯМР (101 МГц, DMSO) δ 167,57, 151,76, 141,07, 137,58, 135,75, 129,16, 128,53, 126,57, 126,41, 125,69, 124,52, 102,13, 65,83, 54,22, 52,60, 49,19, 33,18, 15,20.
Аналитические данные о соединении
|
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ
Пример 8. Получение оксима 5a
СХЕМА BB.
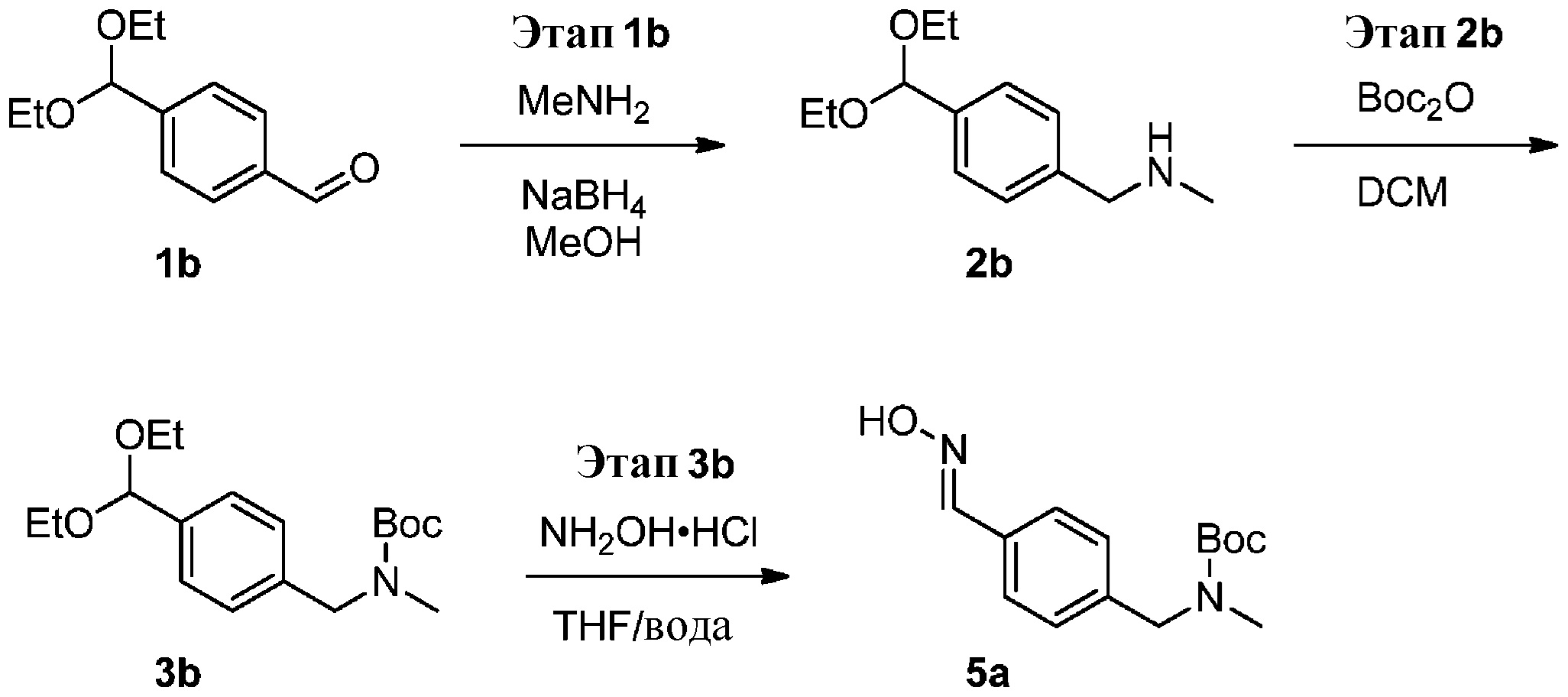
Этап 1b
В реактор добавляют MeOH (28,00 л) и 4-(диэтоксиметил)бензальдегид (соединение 1b) (3500 г, 16,81 моль) при 20 °C. Добавляют метиламин, 33% в EtOH (1,898 кг, 2,511 л 33% вес./вес., 20,17 моль), поддерживая 20–30 °C, затем перемешивают в течение 1,5 ч с образованием имина. Добавляют гранулы NaBH4 (381,7 г, 10,09 моль), поддерживая температуру в диапазоне 20–30 °C. Перемешивают при комнатной температуре в течение по меньшей мере 30 мин, чтобы обеспечить завершение реакции. Добавляют водный раствор NaOH (16,81 л, 2,0 M, 33,62 моль), поддерживая приблизительно 20 °C. Добавляют MTBE (17,50 л) и соляной раствор (7,0 л), перемешивают в течение по меньшей мере 5 мин, затем дают фазам отделиться. Водный слой экстрагируют с помощью MTBE (7,0 л), затем объединяют органические фазы и промывают соляным раствором (3,5 л), после чего высушивают (Na2SO4) и концентрируют до 6 л. Двухфазную смесь переносят в делительную воронку и удаляют водную фазу. Органическую фазу концентрируют с получением 1-(4-(диэтоксиметил)фенил)-N-метилметанамина (соединение 2b) (3755 г, 16,82 моль, выход 100%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 7,43 (д, J = 8,1 Гц, 2H), 7,31 (д, J = 8,1 Гц, 2H), 5,49 (с, 1H), 3,75 (с, 2H), 3,68–3,46 (м, 4H), 2,45 (с, 3H), 1,23 (т, J = 7,1 Гц, 6H).
Этапы 2b и 3b
В реактор добавляют 2-MeTHF (15,00 л) и 1-(4-(диэтоксиметил)фенил)-N-метилметанамин (соединение 2b) (3750 г, 16,79 моль) при 20 °C. Добавляют раствор ангидрида Boc (3,848 кг, 4,051 л, 17,63 моль) в 2-MeTHF (7,500 л), поддерживая приблизительно 25 °C. Перемешивают в течение по меньшей мере 30 мин, чтобы обеспечить полное преобразование в трет-бутиловый эфир 4-(диэтоксиметил)бензил(метил)карбамата (соединение 3b), затем добавляют раствор Na2SO4 (1,192 кг, 8,395 моль) в воде (11,25 л). Нагревают до 35 °C, после чего добавляют раствор гидрохлорида гидроксиламина (1,750 кг, 25,18 моль) в воде (3,75 л), затем перемешивают в течение по меньшей мере 6 ч, чтобы обеспечить завершение реакции. Охлаждают до 20 °C, останавливают перемешивание и удаляют водную фазу. Промывают органический слой соляным раствором (3,75 л), высушивают (Na2SO4), фильтруют и концентрируют до приблизительно 9 л. Добавляют гептан (15,00 л) и кристаллический трет-бутиловый эфир 4-((гидроксимино)метил)бензил(метил)карбамата (соединение 5a) (порциями по 1,0 г каждые 10 мин) до очевидного зародышеобразования, затем концентрируют с получением твердой суспензии. Добавляют гептан (3,75 л), затем охлаждают до комнатной температуры и фильтруют. Промывают гептаном (5,625 л), затем высушивают с получением трет-бутилового эфира 4-((гидроксимино)метил)бензил(метил)карбамата (соединение 5a) (4023 г, 15,22 моль, выход 91%, чистота 97,2% по площади по ВЭЖХ) в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,13 (с, 1H), 7,54 (д, J = 8,1 Гц, 2H), 7,25 (уш. д, 2H), 4,44 (уш. с, 2H), 2,83 (уш. д, 3H), 1,47 (уш. с, 9H).
Схема CC. Синтез промежуточного соединения A-4-ii
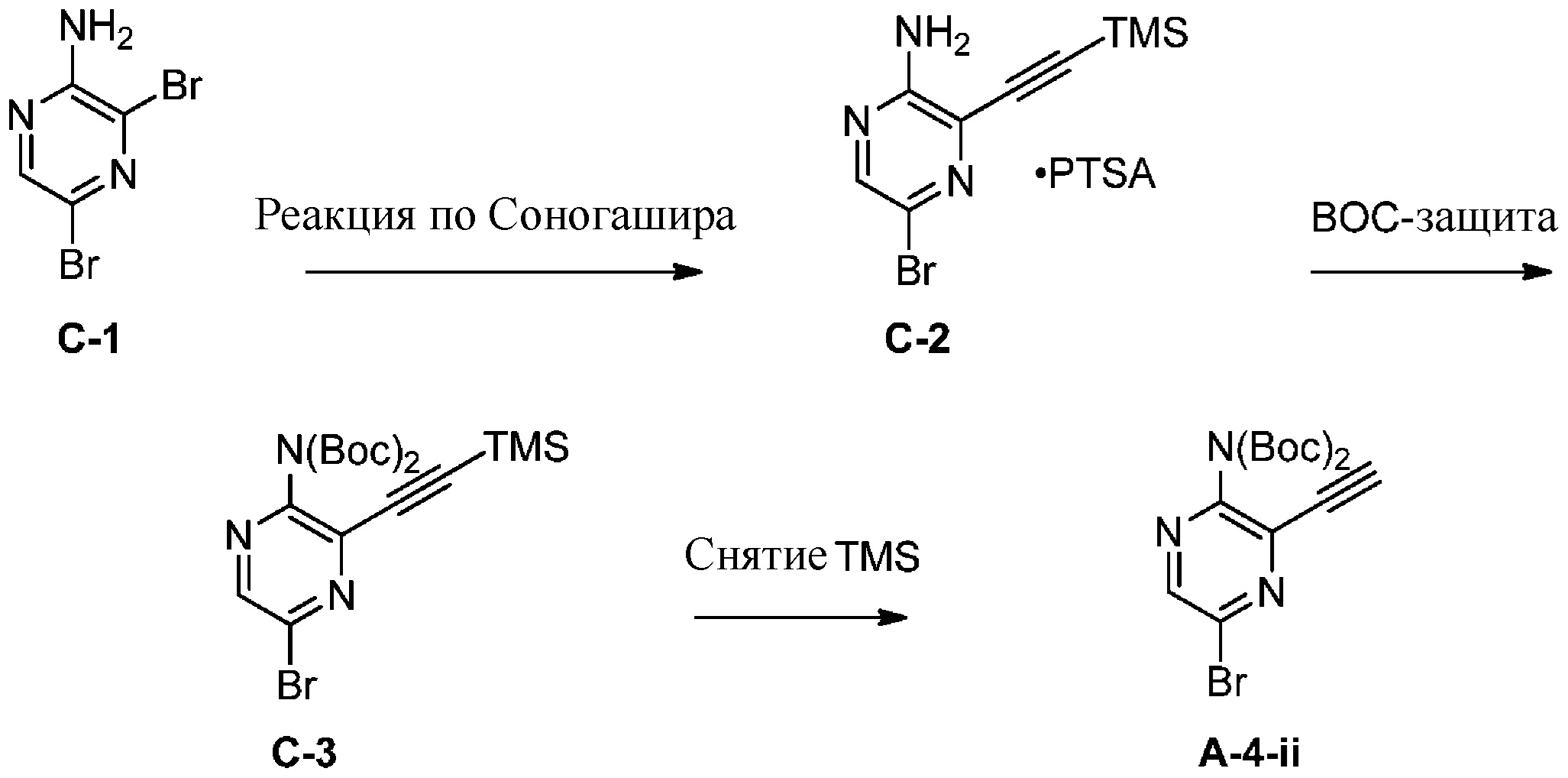
Соединение формулы A-4-ii может быть получено в соответствии с этапами, кратко описанными в схеме С. Реакции сочетания по Соногашира известны в данной области (см. например, Chem. Rev. 2007 г., 874–922). В некоторых вариантах осуществления подходящие условия реакции сочетания по Соногашира содержат добавление 1 эквивалента соединения формулы C-1, 1 эквивалента ТМS-ацетилена, 0,010 эквивалента Pd(PPh3)2Cl2, 0,015 эквивалента CuI и 1,20 эквивалента NMM в изопропаноле. Продукт можно выделить путем добавления воды в спиртосодержащую реакционную смесь.
Аминные соли продукта могут быть образованы путем растворения амина в распространенном органическом растворителе и добавления кислоты. Примеры подходящих растворителей включают в себя хлорированные растворители (например, дихлорметан (DCM), дихлорэтан (DCE), CH2Cl2 и хлороформ), простые эфиры (например, THF, 2-MeTHF и диоксан), сложные эфиры (например, EtOAc, IPAC) и другие апротонные растворители. Примеры подходящих кислот включают в себя, без ограничений, HCl, H3PO4, H2SO4, MSA и PTSA. В некоторых вариантах осуществления растворителем является IPAC, а кислотой — pTSA. В некоторых вариантах осуществления кислотно-аддитивную соль преобразуют снова в свободное аминное основание в присутствии подходящего растворителя и подходящего основания. Подходящие растворители включают в себя EtOAc, IPAC, дихлорметан (DCM), дихлорэтан (DCE), CH2Cl2, хлороформ, 2-MeTHF, а подходящие основания включают в себя NaOH, NaHCO3, Na2CO3, KOH, KHCO3, K2CO3 и Cs2CO3. В некоторых вариантах осуществления подходящий растворитель представляет собой EtOAc, а подходящее основание представляет собой KHCO3.
Амин соединения С-2 может быть защищен различными аминными защитными группами, такими как Boc(трет-бутоксикарбонил). Введение защитных групп Boc известно в данной области (см., например, Protecting Groups in Organic Synthesis, Greene and Wuts). В некоторых вариантах осуществления подходящие условия включают добавление 1,00 эквивалента амина, 2,10 эквивалента ди-трет-бутилдикарбоната и 0,03 эквивалента DMAP в EtOAc.
Восстановления Pd достигают путем обработки металлическим поглотителем (силикагель, функционализированные смолы, древесный уголь). В некоторых вариантах осуществления подходящие условия включают добавление древесного угля.
Защитная группа TMS (триметилсилил) может быть удалена с соединения C-3 в условиях, известных специалисту в данной области. В некоторых вариантах осуществления условия удаления TMS содержат взаимодействие TMS-защищенного соединения с подходящим основанием в подходящем растворителе. Примеры подходящих растворителей включают в себя хлорированные растворители (например, дихлорметан (DCM), дихлорэтан (DCE), CH2Cl2 и хлороформ), простые эфиры (например, THF, 2-MeTHF и диоксан), сложные эфиры (например, EtOAc, IPAC), другие апротонные растворители и спиртовые растворители (например, MeOH, EtOH, iPrOH). Примеры подходящих оснований включают в себя, без ограничений (например, NaOH, KOH, K2CO3, Na2CO3). В некоторых вариантах осуществления подходящие условия содержат добавление 1,00 эквивалента TMS-защищенного ацетилена, 1,10 эквивалента K2CO3, EtOAc и EtOH. В некоторых вариантах осуществления спиртосодержащий растворитель, такой как EtOH, добавляют в реакцию последним. В некоторых вариантах осуществления полученный ацетилен выделяют путем добавления воды.
Схема DD. Пример синтеза соединения A-4-ii
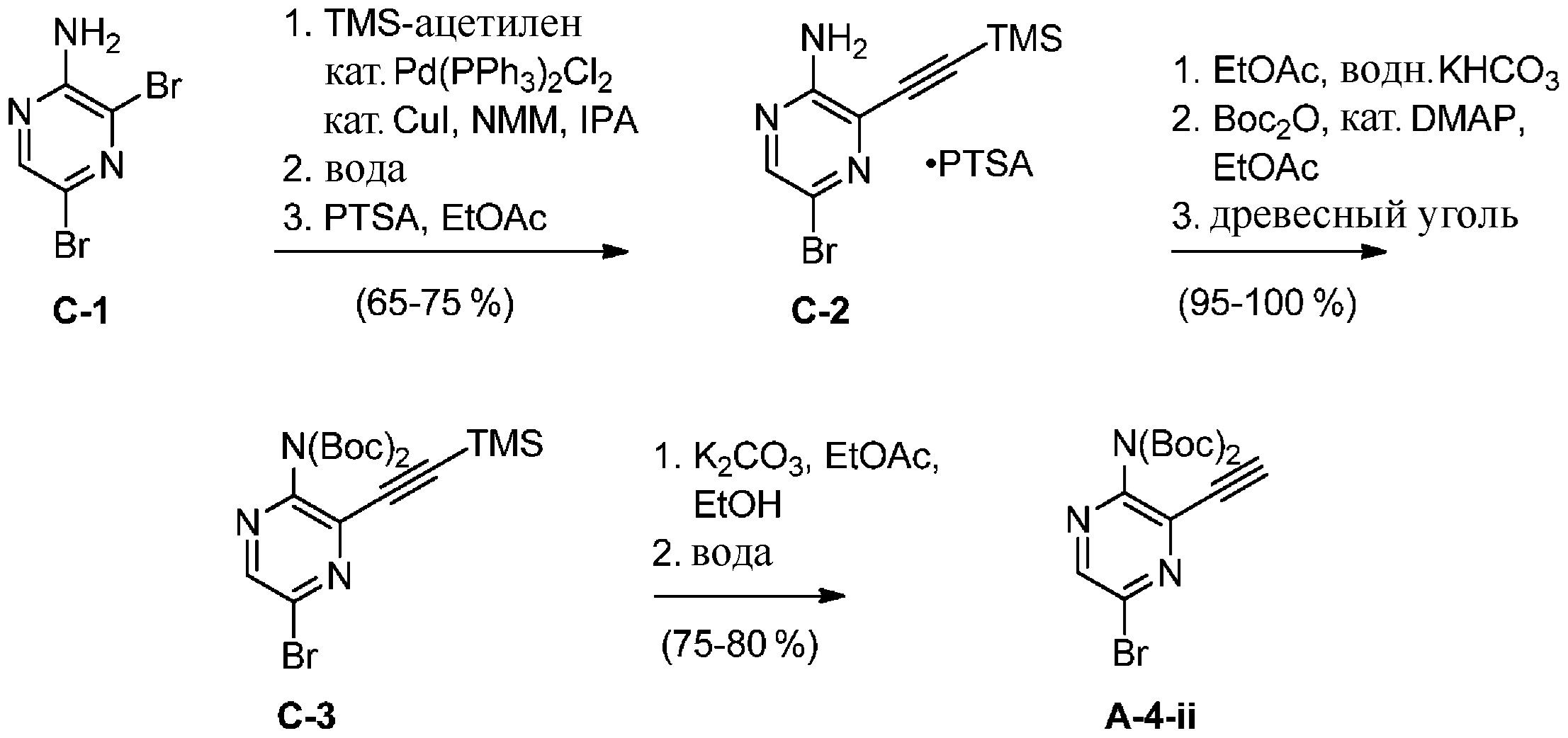
Пример 9. Синтез соединения A-4-ii
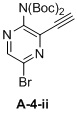
Этап 1. Получение 5-бром-3-((триметилсилил)этинил)пиразин-2-амина (соединение C-2)
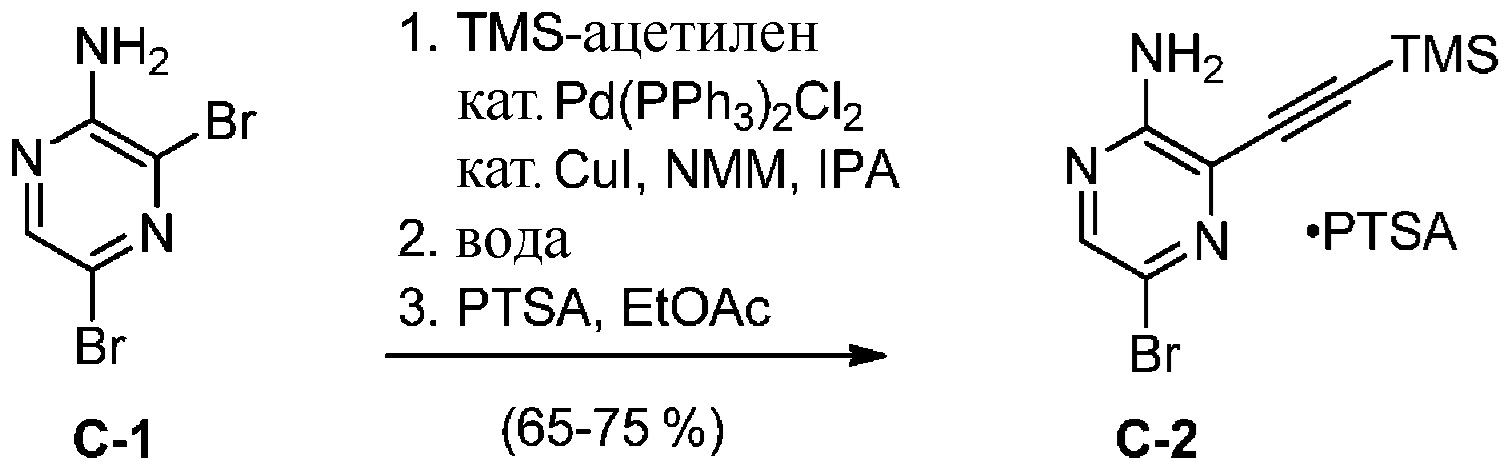
Изопропанол (8,0 л) загружают в реактор, затем перемешивают и барботируют потоком N2. 3,5-Дибромпиразин-2-амин (соединение C-1) (2000 г, 7,91 моль), Pd(PPh3)2Cl2 (56 г, 0,079 моль), CuI (23 г, 0,119 моль) и NMM (1043 мл, 9,49 моль) добавляют в реактор в атмосфере N2. Температуру реакции регулируют до 25 °С. Реактор продувают с помощью N2, используя по меньшей мере три цикла продувки вакуумом/N2. К реакционной смеси добавляют TMS-ацетилен (1,12 л, 7,91 моль) и поддерживают температуру реакции ниже 30 °С. Когда реакция завершена, температуру реакционной смеси снижают до 15 °С, затем добавляют воду (10 л) и перемешивают в течение по меньшей мере 2 ч. Твердое вещество собирают фильтрованием, промывая твердое вещество смесью IPA/вода в соотношении 1:1 (2 × 6 л). Фильтровальный осадок высушивают в вакууме, затем загружают в реактор и растворяют в EtOAc (12,5 л). Гидрат pTSA (1,28 кг, 6,72 моль) загружают в реактор в виде твердого вещества. Смесь перемешивают при температуре окружающей среды в течение по меньшей мере 5 ч, затем твердое вещество собирают фильтрованием, промывают смесью гептан/EtOAc в соотношении 1:1 (3,5 л), а затем гептаном (3,5 л). Фильтровальный осадок высушивают с получением 5-бром-3-((триметилсилил)этинил)пиразин-2-амина (соединение C-2) в виде соли pTSA (2356 г, выход 67%, чистота 98,9% по площади по ВЭЖХ). 1Н ЯМР (400 МГц, DMSO) δ 8,12 (с, 1H), 7,48 (д, J = 8,1 Гц, 2H), 7,12 (д, J = 8,0 Гц, 2H), 2,29 (с, 3H), 0,26 (с, 9H).
Этапы 2 и 3
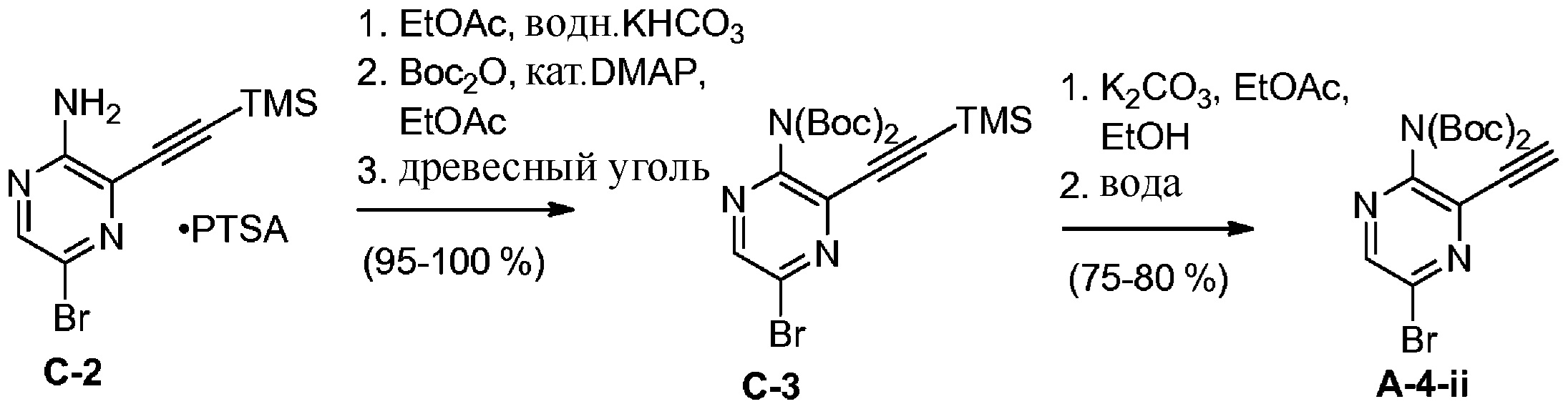
Этап 2. Получение трет -бутилового эфира N - трет -бутоксикарбонил- N -[5-бром-3-((триметилсилил)этинил)пиразин-2-ил]карбамата (соединение C-3)
Раствор 5-бром-3-((триметилсилил)этинил)пиразин-2-аминной соли pTSA (соединение C-2) (2350 г, 5,31 моль) в EtOAc (11,5 л) перемешивают с 20% вес./вес. водн. раствора KHCO3 (4,5 кг, 1,5 экв.) в течение по меньшей мере 30 мин. Слои отделяют и органический слой концентрируют, а затем растворяют в EtOAc (7 л) и добавляют в реактор. Добавляют DMAP (19,5 г, 0,16 моль) после медленного добавления раствора Boc2O (2436 г, 11,16 моль) в EtOAc (3 л). Реакционную смесь перемешивают в течение по меньшей мере 30 мин, чтобы обеспечить завершение реакции, а затем добавляют активированный древесный уголь (Darco G-60, 720 г) и целит (720 г) и перемешивают в течение по меньшей мере 2 ч. Смесь фильтруют, промывая твердый слой с помощью EtOAc (2 × 1,8 л). Фильтрат концентрируют с получением трет-бутилового эфира N-трет-бутоксикарбонил-N-[5-бром-3-((триметилсилил)этинил)пиразин-2-ил]карбамата (соединение C-3), который непосредственно используется на следующем этапе.
Этап 3. Получение трет -бутилового эфира N -(5-бром-3-этинилпиразин-2-ил)- N -трет-бутоксикарбонилкарбамата (соединение A-4-ii)
K2CO3 (811 г, 5,87 моль) загружают в реактор с последующим растворением раствора соединения С-3 (2300 г, 4,89 моль) в EtOAc (4,6 л), начинают встряхивание. Медленно добавляют EtOH (9,2 л) и смесь перемешивают в течение по меньшей мере 1 ч, чтобы обеспечить завершение реакции. Затем добавляют воду (4,6 л) и перемешивают в течение по меньшей мере 2 ч. Твердое вещество собирают фильтрованием и промывают смесью EtOH/вода в соотношении 1:1 (4,6 л, а затем 2,3 л) и затем EtOH (2,3 л). Фильтровальный осадок высушивают с получением трет-бутилового эфира N-(5-бром-3-этинилпиразин-2-ил)-N-трет-бутоксикарбонилкарбамата (соединение A-4-ii) (1568 г, выход 78%, чистота 97,5% по площади по ВЭЖХ). 1Н ЯМР (400 МГц, CDCl3) δ 8,54 (с, 1H), 3,52 (с, 1H), 1,42 (с, 18H).
Твердые формы соединения I-2
Соединение I-2 было приготовлено в различных твердых формах, включая соли и сосольваты. Твердые формы настоящего изобретения можно использовать в производстве лекарственных препаратов для лечения рака. В одном варианте осуществления предложено использование твердой формы, описанной в настоящем документе, для лечения рака. В некоторых вариантах осуществления рак включает в себя рак поджелудочной железы или немелкоклеточный рак легких. В другом варианте осуществления предложена фармацевтическая композиция, содержащая твердую форму, описанную в настоящем документе, и фармацевтически приемлемый носитель.
В настоящем документе авторы заявки описывают пять инновационных твердых форм соединения I-2. Названия и стехиометрия для каждой из этих твердых форм приведены в таблице S-1 ниже:
Таблица S-1
|
Спектры ЯМР твердого состояния получали на спектрометре Bruker-Biospin 400 МГц Advance III с широким отверстием, оснащенном зондом Bruker-Biospin 4 мм HFX. Образцы загружали в турбинки ZrO2 4 мм (приблизительно 70 мг или менее, в зависимости от доступного количества образца). Скорость вращения под магическим углом (MAS) составляла, как правило, 12,5 кГц. Температуру головки зонда устанавливали на уровне 275K, чтобы свести к минимуму эффект нагревания от трения в процессе вращения. Время протонной релаксации измеряли в эксперименте насыщение-восстановление с использованием 1H MAS T1, чтобы установить точную задержку рециркуляции в эксперименте 13C кросс-поляризации (CP) MAS. Задержка рециркуляции в эксперименте 13C CPMAS была настроена на уровне по меньшей мере в 1,2 раза больше, чем измеренное время релаксации 1H T1, чтобы добиться максимального соотношения сигнал-шум в углеродном спектре. Время контакта CP в эксперименте 13C CPMAS устанавливали равным 2 мс. Использовали протонный импульс CP c линейно нарастающим фронтом (от 50% до 100%). Условия соответствия Хартмана-Хана оптимизировали по внешнему эталонному образцу (глицин). Углеродные спектры получали с развязкой SPINAL 64 с напряженностью поля приблизительно 100 кГц. Химический сдвиг определяли относительно внешнего стандарта адамантана, причем его резонансный сигнал в сильном поле имел сдвиг 29,5 м.д.
Данные XRPD в примерах 13–14 были получены на Bruker D8 Advance System (Asset V014333), оснащенном герметичной трубкой с источником Cu и детектором Vantec-1 (Bruker AXS, г. Мэдисон, штат Висконсин, США), при комнатной температуре. Рентгеновский генератор работал при напряжении 40 кВ и силе тока 40 мА. Образец порошка помещали в неглубокий кремниевый держатель. Данные регистрировали в режиме сканирования отражения (фиксированный связанный режим) в диапазоне 3–40° 2 тета с шагом 0,0144° и выдержкой 0,25 с (105 с на шаг). Использовали изменяемые щели расходимости.
Пример 10. Соединение I-2 (свободное основание)
Свободное основание соединения I-2 может быть получено в соответствии со способами, описанными в примере 6, этап 4. Альтернативный способ 1.
XRPD соединения I-2 (свободное основание)
На рисунке 1a показана рентгеновская порошковая дифрактограмма образца, которая характерна для кристаллического вещества лекарственного средства.
Типичные пики XRPD свободного основания соединения I-2:
|
Термический анализ свободного основания соединения I-2
Термогравиметрический анализ свободного основания соединения I-2 выполняли для определения процентной потери массы в зависимости от времени. Образец нагревали от температуры окружающей среды до 350 °C со скоростью 10 °C/мин на приборе TA Instrument TGA Q5000 (Asset V014258). На рисунке 2a показан результат TGA с одноэтапной потерей массы до испарения или термического разложения. Потеря массы при нагревании от температуры окружающей среды (приблизительно 25 °C) до 215 °C составляла ~1,9%.
Дифференциальная сканирующая калориметрия свободного основания соединения I-2
Термические свойства свободного основания соединения I-2 измеряли с помощью инструмента TA Instrument DSC Q2000 (Asset V014259). Образец свободного основания соединения I-2 (1,6900 мг) взвешивали в алюминиевой герметической чашке с предварительно пробитым микроотверстием и нагревали от температуры окружающей среды до 350 °C при 10 °C/мин. Один эндотермический пик наблюдали при 210 °C, причем температура начала составляла 201 °C (рисунок 3a). Энтальпия, соответствующая данному эндотермическому пику, составляет 78 Дж/г.
ЯМР твердого тела свободного основания соединения I-2
13C CPMAS на свободном основании соединения I-2
275K; 1H T1 = 1,30 с
вращение 12,5 кГц; эт. адамантан 29,5 м.д.
Весь спектр см. на рисунке 4a.
Типичные пики
|
Кристаллическая структура свободного основания соединения I-2
Свободную форму соединения I-2 получали из соли соединения I-2 HCl. 200 мг соли соединения I-2 HCl добавляли к 1 мл 6 н. раствора NaOH. 20 мл дихлорметана использовали для экстрагирования свободной формы. Слой дихлорметана сушили над K2CO3. Раствор отфильтровывали и добавляли к нему 5 мл н-гептана. Кристаллы получали медленным выпариванием раствора при комнатной температуре в течение ночи.
Большинство полученных кристаллов представляли собой тонкие пластинки. Среди них встречались кристаллы призматической формы.
Был выбран желтый призматический кристалл с размерами 0,2 × 0,1 × 0,1 мм3, который устанавливали на MicroMount и центрировали в дифрактометре Bruker APEX II. Получали три группы по 40 кадров, разделенных в обратном пространстве для обеспечения ориентационной матрицы и получения начальных параметров ячейки. Конечные параметры ячейки получали и уточняли после окончания сбора данных на основе полного набора данных.
Набор дифракционных данных в обратном пространстве получали с разрешением 116,96° угла 2θ с использованием шагов 0,5° с выдержкой 10 с для каждого кадра. Данные собирали c использованием альфа-излучения Cu K при температуре 100 (2) K с криосистемой в потоке азота. Интеграцию интенсивностей и уточнение параметров ячейки проводили с использованием программного обеспечения APEXII.
ПАРАМЕТРЫ КРИСТАЛЛА
|
Пример 11. Соединение I-2 • HCl
Соединение I-2 • HCl может быть образовано в соответствии со способами, описанными в примере 6, этап 4. Альтернативный способ 2 и пример 6, этап 5.
XRPD соединения I-2 • HCl
На рисунке 1b показана рентгеновская порошковая дифрактограмма образца, которая характерна для кристаллического вещества лекарственного средства.
Типичные пики XRPD соединения I-2 • HCl
|
Термический анализ соединения I-2 • HCl
Термогравиметрический анализ соединения I-2 • HCl выполняли для определения процентной потери массы в зависимости от времени. Образец нагревали от температуры окружающей среды до 350 °C со скоростью 10 °C/мин на приборе TA Instrument TGA Q5000 (Asset V014258). На рисунке 2b показан результат TGA с двухэтапной потерей массы до испарения или термического разложения. Потеря массы от температуры окружающей среды (приблизительно 25 °C) до 100 °C составляла ~1,1%, а потеря массы от 110 °C до 240 °C составляла ~0,8%.
Дифференциальная сканирующая калориметрия соединения I-2 • HCl
Термические свойства соединения I-2 • HCl измеряли с помощью инструмента TA Instrument DSC Q2000 (Asset V014259). Образец соединения I-2 • HCl (3,8110 мг) взвешивали в алюминиевой герметической чашке с предварительно пробитым микроотверстием и нагревали от температуры окружающей среды до 350 °C при 10 °C/мин. Один эндотермический пик наблюдали при температуре 293 °C, причем температура начала составляла 291 °C (рисунок 3b). Энтальпия, соответствующая эндотермическому пику, составляла 160,3 Дж/г. Второй эндотермический пик наблюдали при приблизительно 321 °C. Оба пика были связаны с испарением образца и разложением.
ЯМР твердого тела соединения I-2 • HCl
15 CPMAS на соединении I-2 • HCl
275K; вращение 12,5 кГц; эт. адамантан 29,5 м.д.
Весь спектр см. на рисунке 4b.
Типичные пики
|
Кристаллическая структура соединения I-2 • HCl
180 мг соединения I-2 • HCl добавляли в сосуд с 0,8 мл 2-пропанола и 0,2 мл воды. Герметичный сосуд выдерживали в печи при 70 ºC в течение двух недель. Наблюдали образование кристаллов дифракционного качества.
Выбрали желтый кристалл игольчатой формы с размерами 0,15 × 0,02 × 0,02 мм3, который устанавливали на MicroMount и центрировали в дифрактометре Bruker APEX II (V011510). Получали три группы по 40 кадров, разделенных в обратном пространстве для обеспечения ориентационной матрицы и получения начальных параметров ячейки. Конечные параметры ячейки собирали и уточняли на основе полного набора данных.
Набор данных дифракции в обратном пространстве получали с разрешением 106º угла 2θ с использованием шагов 0,5° со временем выдержки 20 с для каждого кадра среди кадров с малыми углами и 60 с для каждого кадра среди кадров с большими углами. Данные собирали при комнатной температуре c использованием альфа-излучения Cu K.
Чтобы получить данные в таблице 1, кристалл обдували сухим азотом со скоростью 6 литров/мин, чтобы исключить влагу из окружающей среды. Данные в таблице 2 получали без азота. Интеграцию интенсивностей и уточнение параметров ячейки проводили с использованием программного обеспечения APEXII. Доля воды может меняться в диапазоне от 0 до 1.
|
Элементный анализ CHN
Элементный анализ CHN соединения I-2 • HCl свидетельствует об образовании монохлорида.
|
Пример 12. Соединение I-2 • 2HCl
Соединение I-2 • 2HCl может быть получено в соответствии со способами, описанными в примере 6, этап 4.
XRPD соединения I-2 • 2HCl
Схемы XRPD получают при комнатной температуре в режиме отражения с использованием системы Bruker D8 Discover (Asset Tag V012842), оснащенной источником в запаянной трубке и зонным детектором Hi-Star (Bruker AXS, г. Мэдисон, штат Висконсин, США). Рентгеновский генератор работает при напряжении 40 кВ и силе тока 35 мА. Образец порошка размещают в никелевом держателе. Регистрируют два кадра со временем экспозиции 120 с каждый. Впоследствии данные интегрируют в диапазоне 4,5–39° 2 тета с шагом 0,02° и объединяют в один непрерывный профиль.
На рисунке 1с показана рентгеновская порошковая дифрактограмма образца, которая характерна для кристаллического вещества лекарственного средства.
Типичные пики XRPD соединения I-2 • 2HCl
|
Термический анализ соединения I-2 • 2HCl
Термогравиметрический анализ соединения I-2 • 2HCl проводили на приборе TA Instruments TGA, модель Q5000. Соединение I-2 • 2HCl помещали в платиновую чашку для образца и нагревали при 10 °C/мин до 350 °C от комнатной температуры. На рисунке 2c приведены результаты TGA, которые указывают на потерю массы, равную 7,0 %, от комнатной температуры до 188 °C, что соответствует потере 1 эквивалента HCl (6,8 %). Температура начала разложения/плавления составляет 263 °C.
Дифференциальная сканирующая калориметрия соединения I-2 • 2HCl
Термограмму DSC для лекарственного вещества соединения I-2 • 2HCl партии 3 получали с использованием прибора TA Instruments DSC Q2000. Соединение
I-2 • 2HCl нагревали при 2 °C/мин до 275 °C от -20 °C и модулировали при ± 1 °C каждые 60 с. На термограмме DSC (рисунок 3c) показан эндотермический пик ниже 200 °C, который может соответствовать потере 1 эквивалента HCl. Плавление/перекристаллизация происходят в диапазоне 215–245 °C с последующим разложением.
ЯМР твердого тела соединения I-2 • 2HCl
13C CPMAS на соединении I-2 • 2HCl
275K; 1H T1 = 1,7 с
вращение 12,5 кГц; эт. адамантан 29,5 м.д.
Весь спектр см. на рисунке 4с.
|
Кристаллическая структура соединения I-2 • 2HCl
180 мг соединения I-2 • HCl добавляли в сосуд с 0,8 мл 2-пропанола и 0,2 мл воды. Герметичный сосуд выдерживали в печи при 70 ºC в течение двух недель. Наблюдали образование кристаллов дифракционного качества.
Выбрали желтый кристалл игольчатой формы с размерами 0,15 x 0,02 x 0,02 мм3, который устанавливали на MicroMount и центрировали в дифрактометре Bruker APEX II (V011510). Получали три группы по 40 кадров, разделенных в обратном пространстве для обеспечения ориентационной матрицы и получения начальных параметров ячейки. Конечные параметры ячейки собирали и уточняли на основе полного набора данных.
Набор данных дифракции в обратном пространстве получали с разрешением 106º угла 2θ с использованием шагов 0,5° со временем выдержки 20 с для каждого кадра среди кадров с малыми углами и 60 с для каждого кадра среди кадров с большими углами. Данные собирали при комнатной температуре. Кристалл обдували сухим азотом со скоростью 6 литров/мин, чтобы исключить влагу из окружающей среды. Интеграцию интенсивностей и уточнение параметров ячейки проводили с использованием программного обеспечения APEXII.
ПАРАМЕТРЫ КРИСТАЛЛА
|
Пример 13. Соединение I-2 • HCl • H2O
Соединение I-2 • HCl • H2O может быть получено из соединения I-2 • 2HCl. (E29244-17). Суспензию соединения I-2 • 2HCl (10,0 г, 18,6 ммоль) в изопропиловом спирте (40 мл) и воде (10 мл) нагревают при 50 °C в течение приблизительно 1 ч, а затем охлаждают до ниже 10 °C. Твердое вещество собирают фильтрованием. Фильтровальный осадок промывают смесью изопропиловый спирт/вода в соотношении 80/20 (2 x 10 мл) и высушивают на воздухе для получения соединения I-2 • HCl• 2H2O в виде желтого порошка.
XRPD соединения I-2 • HCl • H2O
Схемы XRPD получают при комнатной температуре в режиме отражения с использованием системы Bruker D8 Discover (Asset Tag V012842), оснащенной источником в запаянной трубке и зонным детектором Hi-Star (Bruker AXS, г. Мэдисон, штат Висконсин, США). Рентгеновский генератор работает при напряжении 40 кВ и силе тока 35 мА. Образец порошка размещают в никелевом держателе. Регистрируют два кадра со временем экспозиции 120 с каждый. Впоследствии данные интегрируют в диапазоне 4,5–39° 2 тета с шагом 0,02° и объединяют в один непрерывный профиль.
На рисунке 1d показана рентгеновская порошковая дифрактограмма образца, которая характерна для кристаллического вещества лекарственного средства.
Типичные пики XRPD соединения I-2 • HCl • H2O
|
Термический анализ соединения I-2 • HCl • H2O
Термогравиметрический анализ (TGA) для соединения I-2 • HCl • H2O проводили на приборе TA Instruments TGA, модель Q5000. Соединение I-2 • HCl • H2O помещали в платиновую чашку для образца и нагревали при 10 °C/мин до 400 °C от комнатной температуры. Термограмма (рисунок 2d) показывает потерю массы, равную 2,9 %, от комнатной температуры до 100 °C и потерю массы, равную 0,6 %, от 100 °C до 222 °C, что соответствует теоретическому моногидрату (3,5 %).
Дифференциальная сканирующая калориметрия соединения I-2 • HCl • H2O
Термограмму DSC для соединения I-2 • HCl • H2O получали с использованием прибора TA Instruments DSC Q2000. Соединение I-2 • HCl • H2O нагревали при 2 °C/мин до 275 °C от -20 °C и модулировали при ± 1 °C каждые 60 с. На термограмме DSC (рисунок 3d) показан эндотермический пик ниже 200 °C, который может соответствовать потере 1 эквивалента HCl. Плавление/перекристаллизация происходят в диапазоне 215–245 °C с последующим разложением.
Пример 14. Соединение I-2 • HCl • 2H2O
Соединение I-2 • HCl• 2H2O может быть получено из соединения I-2 • 2HCl (E29244-17). Суспензию соединения I-2 • 2HCl (10,0 г, 18,6 ммоль) в изопропиловом спирте (40 мл) и воде (10 мл) нагревают при 50 °C в течение приблизительно 1 ч, а затем охлаждают до ниже 10 °C. Твердое вещество собирают фильтрованием. Фильтровальный осадок промывают смесью изопропиловый спирт/вода в соотношении 80/20 (2 x 10 мл) и высушивают на воздухе для получения соединения I-2 • HCl• 2H2O в виде желтого порошка.
XRPD соединения I-2 • HCl • 2H2O
Измерения порошковой рентгеновской дифракции проводили с использованием дифрактометра PANalytical X-pert Pro при комнатной температуре с излучением меди (1,54060 ˚A). Оптика падающего луча включала переменную щель расходимости для гарантии постоянства длины излучения образца и дифракционного луча, использовали быстрый линейный твердотельный детектор с активной длиной 2,12 градуса 2 тета, измерения проводили в режиме сканирования. Образец порошка помещали в углубленную зону кремниевого держателя с нулевым фоном и выполняли вращение, чтобы добиться более высоких статистических результатов. Симметричное сканирование измеряли от 4–40 градусов 2 тета с шагом 0,017 градуса и временем сканирования шага 15,5 с.
На рисунке 1d показана рентгеновская порошковая дифрактограмма образца, которая характерна для кристаллического вещества лекарственного средства.
Типичные пики XRPD соединения I-2 • HCl • 2H2O
|
Термический анализ соединения I-2 • HCl • 2H2O
Термограммы TGA (термогравиметрический анализ) получали с использованием прибора TA TGA Q500 соответственно со скоростью сканирования 10 °C/мин в диапазоне температур 25–300 °C. Для анализа TGA образцы помещали в открытую чашку. Термограмма свидетельствует о потере массы на ~6 % от комнатной температуры до 100 °C, что соответствует теоретическому дигидрату (6,7 %).
Дифференциальная сканирующая калориметрия соединения I-2 • HCl • 2H2O
Термограммы DSC (дифференциальная сканирующая калориметрия) получали с использованием прибора TA instrument DSC Q2000 со скоростью сканирования 10 °C/мин в диапазоне температур 25–300 °C. Для анализа DSC образцы отвешивали в алюминиевые герметичные чашки T-zero, которые герметизировали и в которых выполняли одно отверстие. На термограмме DSC наблюдается дегидратация в диапазоне от комнатной температуры до 120 °C с последующим плавлением/перекристаллизацией в диапазоне 170–250 °C.
Кристаллическая структура соединения I-2 • HCl с водой
180 мг соединения I-2 • HCl добавляли в сосуд с 0,8 мл 2-пропанола и 0,2 мл воды. Герметичный сосуд выдерживали в печи при 70 ºC в течение двух недель. Наблюдали образование кристаллов дифракционного качества.
Выбрали желтый кристалл игольчатой формы с размерами 0,15 x 0,02 x 0,02 мм3, который устанавливали на MicroMount и центрировали в дифрактометре Bruker APEX II (V011510). Затем отверстие закрывали каптоновой трубкой с водой. Трубку герметизировали, чтобы обеспечить равновесие кристалла с водой, и выдерживали в течение двух дней до дифракционных экспериментов. Получали три группы по 40 кадров, разделенных в обратном пространстве для обеспечения ориентационной матрицы и получения начальных параметров ячейки. Конечные параметры ячейки собирали и уточняли на основе полного набора данных.
Набор данных дифракции в обратном пространстве получали с разрешением 106º угла 2θ с использованием шагов 0,5° со временем выдержки 20 с для каждого кадра среди кадров с малыми углами и 60 с для каждого кадра среди кадров с большими углами. Данные собирали при комнатной температуре. Интеграцию интенсивностей и уточнение параметров ячейки проводили с использованием программного обеспечения APEXII.
ПАРАМЕТРЫ КРИСТАЛЛА
|
Пример 15. Анализ ингибирования клеточной ATR
Можно провести скрининг соединений на их способность ингибировать внутриклеточную ATR, используя иммунофлуоресцентный микроскопический анализ для обнаружения фосфорилирования субстратного гистона ATR H2AX в клетках, обработанных гидроксимочевиной. Клетки HT29 высевают по 14 000 клеток на лунку в 96-луночные черные планшеты для визуализации (BD 353219) в среде Маккоя 5A (Sigma M8403) с добавлением 10 % эмбриональной бычьей сыворотки (JRH Biosciences 12003), раствора пенициллин/стрептомицин, разведенного в соотношении 1:100 (Sigma P7539), и 2 мM раствора L-глутамина (Sigma G7513) и оставляют для прикрепления в течение ночи при 37 °C в 5 % CO2. Затем к клеточной среде добавляют соединения с итоговой концентрации 25 мкМ в 3-кратных последовательных разведениях и инкубируют клетки при 37 °C в 5 % CO2. Через 15 минут добавляют гидроксимочевину (Sigma H8627) в итоговой концентрации 2 мM.
Через 45 мин обработки гидроксимочевиной клетки промывают в ФСБ (фосфатно-солевой буфер), фиксируют в течение 10 мин в 4 % формальдегиде, разведенном в ФСБ (Polysciences Inc 18814), промывают в 0,2 % Tween-20 в ФСБ (промывочный буфер) и пермеабилизируют в течение 10 мин в 0,5 % растворе Triton X-100 в ФСБ. Все операции проводят при комнатной температуре. Затем клетки однократно промывают в промывочном буфере и блокируют в течение 30 мин при комнатной температуре в 10 % козьей сыворотке (Sigma G9023), разбавленной в промывочном буфере (блокирующий буфер). Затем для обнаружения уровней фосфорилирования H2AX клетки инкубируют в течение 1 ч при комнатной температуре в первичном антителе (мышиное моноклональное антитело к фосфорилированному гистону H2AX Ser139; Upstate 05-636), разведенном в блокирующем буфере 1:250. После этого клетки пять раз промывают в промывочном буфере до инкубации в течение 1 ч при комнатной температуре в темноте в смеси вторичных антител (козьи антимышиные антитела, конъюгированные с Alexa Fluor 488; Invitrogen A11029) и красителя Hoechst (Invitrogen H3570); разбавленной 1:500 и 1:5000, соответственно, в промывочном буфере. После этого клетки пять раз промывают в промывочном буфере и, наконец, в каждую лунку добавляют 100 мкл ФСБ до визуализации.
Клетки визуализируют на интенсивность окрашивания Alexa Fluor 488 и Hoechst с использованием BD Pathway 855 Bioimager и программного обеспечения Attovision (BD Biosciences, версия 1.6/855) для количественного определения фосфорилированного H2AX Ser139 и окрашивания ДНК соответственно. Затем для каждой лунки вычисляют процент положительных на фосфорилированный H2AX ядер в монтаже 9 изображений при увеличении 20x с использованием программного обеспечения BD Image Data Explorer (BD Biosciences, версия 2.2.15). Положительные на фосфорилированный H2AX ядра определяли как Hoechst-положительные участки, в которых интенсивность окрашивания Alexa Fluor 488 составляла 1,75 средней интенсивности окрашивания Alexa Fluor 488 в клетках, не обработанных гидроксимочевиной. Наконец, строили графики зависимости процента положительных на H2AX ядер от концентрации для каждого соединения и определяли значения IC50 для ингибирования внутриклеточной ATR с использованием программного обеспечения Prism (GraphPad Prism, версия 3.0cx, для Macintosh, GraphPad Software, г. Сан-Диего, штат Калифорния, США).
Описанные в настоящем документе соединения также можно протестировать в соответствии с другими способами, известными в данной области (см. Sarkaria et al., Inhibition of ATM and ATR Kinase Activities by the Radiosensitizing Agent, Caffeine: Cancer Research 59: 4375–5382 (1999 г.); Hickson et al., Identification and Characterization of a Novel and Specific Inhibitor of the Ataxia-Telangiectasia Mutated Kinase ATM, Cancer Research, 64: 9152–9159 (2004 г.); Kim et al., Substrate Specificities and Identification of Putative Substrates of ATM Kinase Family Members, The Journal of Biological Chemistry, 274(53): 37538–37543 (1999 г.); и Chiang et al., Determination of the Catalytic Activities of mTOR and Other Members of the Phosphoinositide-3-Kinase-related Kinase Family, Methods Mol. Biol. 281:125–41 (2004 г.)).
Пример 16. Анализ ингибирования ATR
Можно провести скрининг соединений на их способность ингибировать киназу ATR с использованием анализа на включение радиоактивного фосфата. Анализы проводят в смеси 50 мM Tris/HCl (pH 7,5), 10 мM MgCl2 и 1 мM DTT. Итоговые концентрации субстрата составляют 10 мкM [γ-33P]АТФ (3 мКи 33P АТФ/ммоль АТФ, Perkin Elmer) и 800 мкM пептида-мишени (ASELPASQPQPFSAKKK).
Анализы проводят при 25 °C в присутствии 5 нM ATR с полной длиной. Готовят базовый буферный раствор для анализа, содержащий все указанные выше реагенты, за исключением АТФ и тестируемого соединения. 13,5 мкл базового раствора помещают в 96-луночный планшет с последующим добавлением 2 мкл базового раствора DMSO, содержащего последовательные разведения тестируемого соединения (как правило, с итоговой концентрации 15 мкM с 3-кратными последовательными разведениями) в двух повторностях (итоговая концентрация DMSO 7 %). Планшет предварительно инкубируют в течение 10 минут при 25 °C и инициируют реакцию путем добавления 15 мкл [γ-33P]АТФ (итоговая концентрация 10 мкM).
Реакцию останавливали через 24 часа путем добавления 30 мкл 0,1 M фосфорной кислоты, содержащей 2 мM АТФ. Многоэкранный 96-луночный планшет с фосфоцеллюлозным фильтром (Millipore, номер по каталогу MAPHN0B50) предварительно обрабатывали 100 мкл 0,2 M фосфорной кислоты до добавления 45 мкл аналитической смеси с остановленной реакцией. Планшет промывают 5 x 200 мкл 0,2 M фосфорной кислоты. После высушивания в лунку добавляют 100 мкл жидкой смеси для сцинтилляционного счета Optiphase ‘SuperMix’ (Perkin Elmer) и проводят сцинтилляционные измерения (1450 Microbeta Liquid Scintillation Counter, Wallac).
После удаления средних фоновых значений для всех точек данных рассчитывают данные Ki(app) с использованием нелинейного регрессионного анализа данных по начальной скорости, используя программное обеспечение Prism (GraphPad Prism, версия 3.0cx, для Macintosh, GraphPad Software, г. Сан-Диего, штат Калифорния, США).
В целом соединения настоящего изобретения являются эффективными при ингибировании ATR. Соединения I-1, I-2, II-1, II-2, II-3 и II-4 ингибируют ATR со значениями Ki ниже 0,001 мкМ.
Пример 17. Анализ сенсибилизации к цисплатину
Можно провести скрининг соединений на их способность сенсибилизировать клетки колоректального рака HCT116 к цисплатину с использованием 96-часового анализа на жизнеспособность клеток (MTS). Клетки HCT116, которые имеют дефект сигнального пути ATM к цисплатину (см. Kim et al.; Oncogene 21:3864 (2002 г.); см. также Takemura et al.; JBC 281:30814 (2006 г.)), высевают по 470 клеток на лунку в 96-луночные полистирольные планшеты (Costar 3596) в 150 мкл среды Маккоя 5A (Sigma M8403) с добавлением 10 % эмбриональной бычьей сыворотки (JRH Biosciences 12003), раствора пенициллин/стрептомицин, разведенного 1:100 (Sigma P7539), и 2 мM раствора L-глутамина (Sigma G7513) и оставляют для прикрепления в течение ночи при 37 °C в 5 % CO2. Затем к клеточной среде одновременно добавляют соединения и цисплатин в 2-кратных последовательных разведениях с максимальной итоговой концентрации 10 мкM в виде полной матрицы концентраций в итоговом клеточном объеме 200 мкл, а затем клетки инкубируют при 37 °C в 5 % CO2. Через 96 ч в каждую лунку добавляют 40 мкл реагента MTS (Promega G358a) и затем инкубируют клетки в течение 1 ч при 37 °C в 5 % CO2. Наконец, определяют поглощение на длине волны 490 нм с использованием прибора для считывания SpectraMax Plus 384 (Molecular Devices) и сообщают концентрацию соединения, требуемую для снижения IC50 цисплатина по меньшей мере в 3 раза (до 1 знака после запятой).
Пример 18. Активность одиночного агента против HCT116
Можно провести скрининг соединений на их способность проявлять активность в качестве одиночного агента против клеток колоректального рака HCT116 с использованием 96-часового анализа на жизнеспособность клеток (MTS). Клетки HCT116 высевают по 470 клеток на лунку в 96-луночные полистирольные планшеты (Costar 3596) в 150 мкл среды Маккоя 5A (Sigma M8403) с добавлением 10 % эмбриональной бычьей сыворотки (JRH Biosciences 12003), раствора пенициллин/стрептомицин, разведенного 1:100 (Sigma P7539), и 2 мM раствора L-глутамина (Sigma G7513) и оставляют для прикрепления в течение ночи при 37 °C в 5 % CO2. Затем к клеточной среде добавляют соединения в 2-кратных последовательных разведениях с максимальной итоговой концентрации 10 мкM в виде полной матрицы концентраций в итоговом клеточном объеме 200 мкл и инкубируют клетки при 37 °C в 5 % CO2. Через 96 ч в каждую лунку добавляют 40 мкл реагента MTS (Promega G358a) и затем инкубируют клетки в течение 1 ч при 37 °C в 5 % CO2. Наконец, определяют поглощение на длине волны 490 нм с использованием прибора для считывания SpectraMax Plus 384 (Molecular Devices) и рассчитывают значения IC50.
Данные для примеров 18–21
|
Хотя в документе описан ряд вариантов осуществления настоящего изобретения, очевидно, что базовые примеры могут быть изменены для обеспечения других вариантов осуществления, в которых используются соединения, способы и процессы настоящего изобретения. Таким образом, следует понимать, что объем настоящего изобретения определяется приложенными пунктами формулы изобретения, а не конкретными вариантами осуществления изобретения, которые были представлены в настоящем документе в качестве примеров.
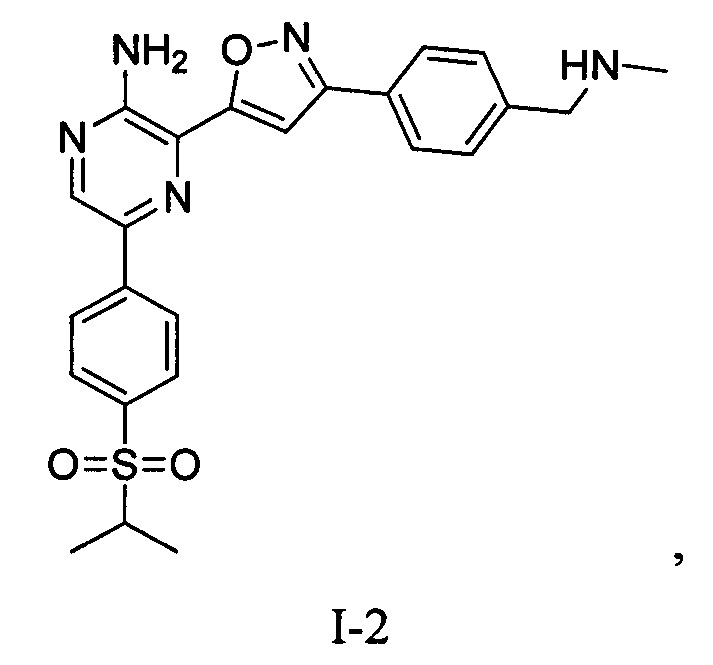
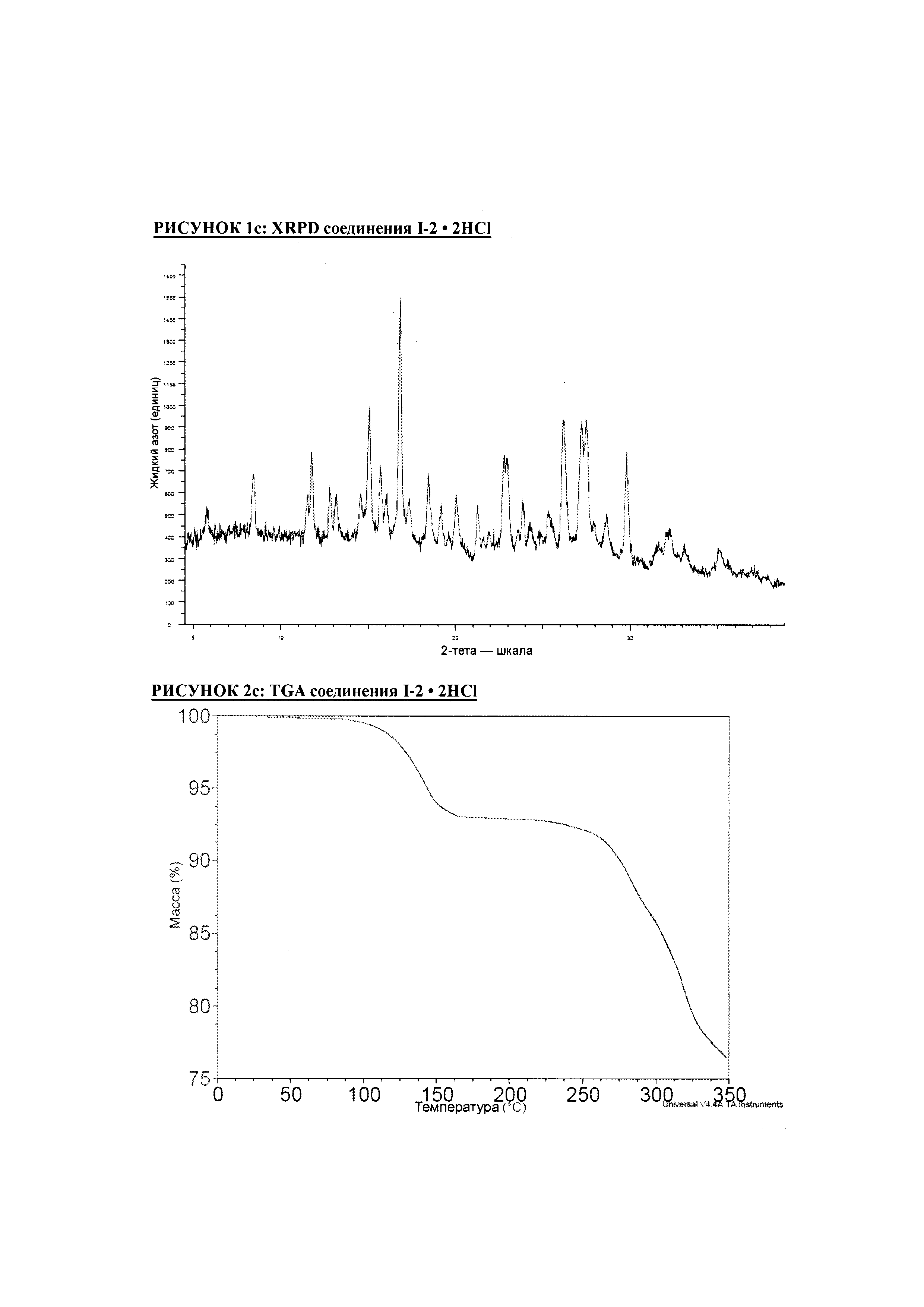
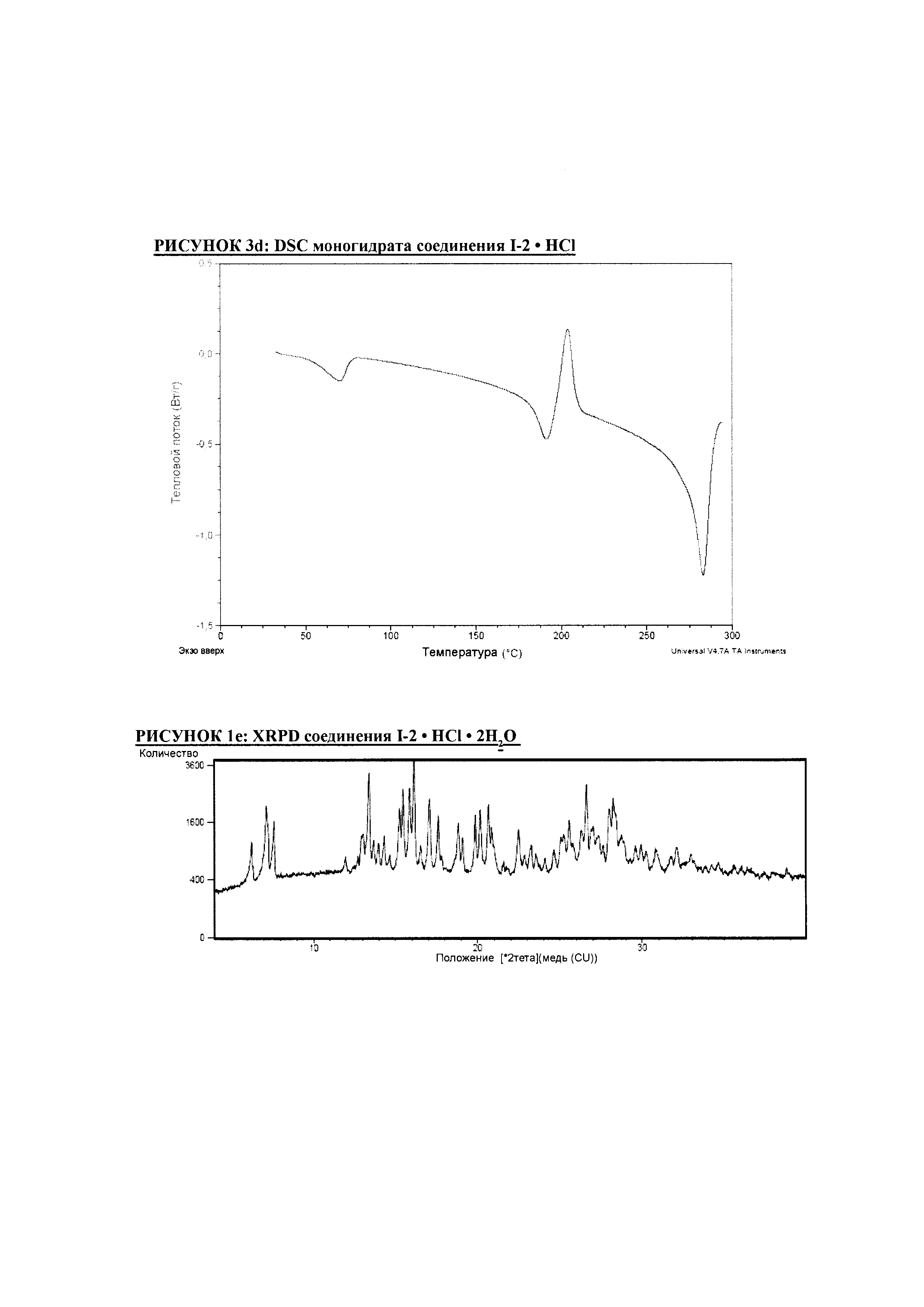
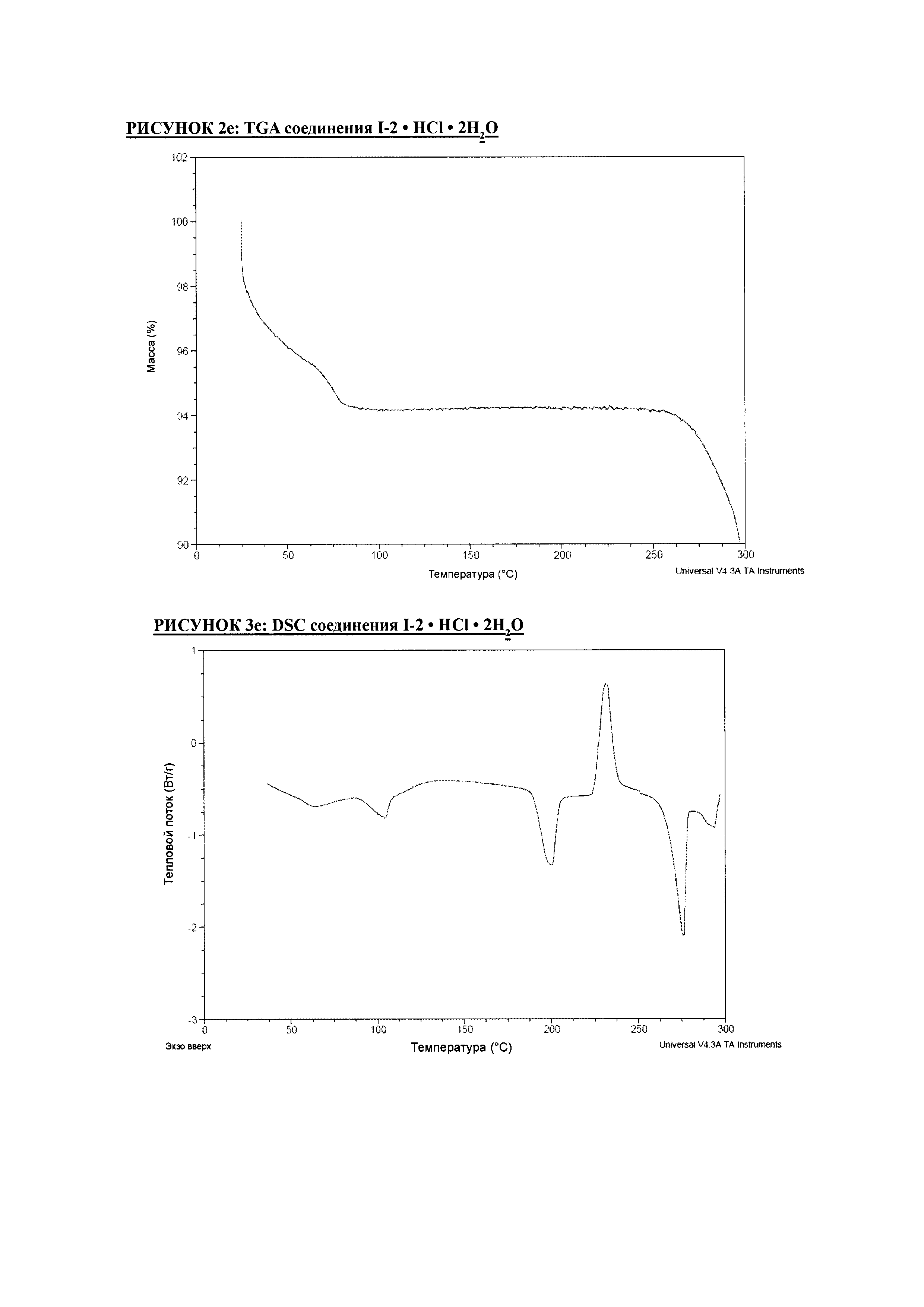
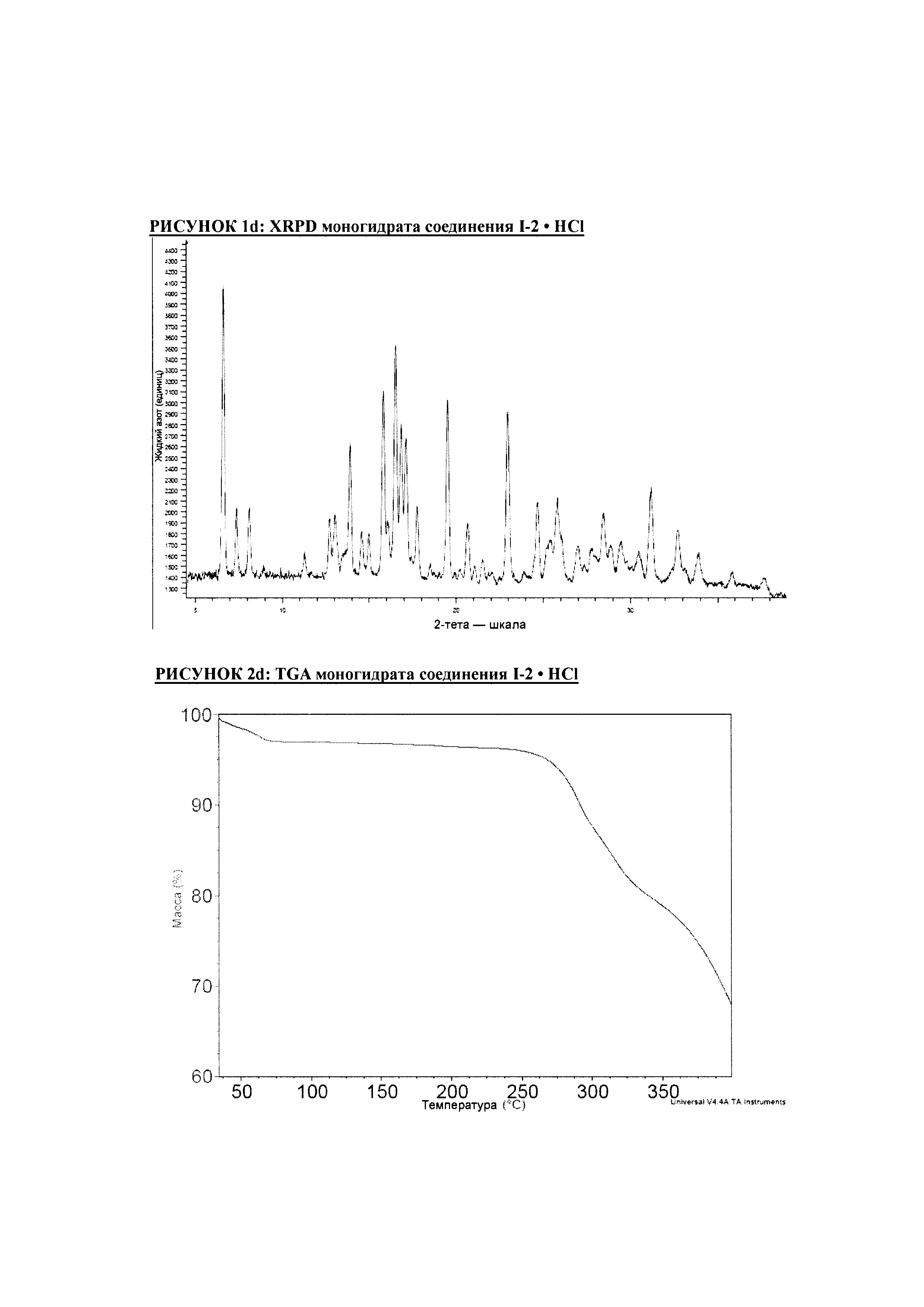
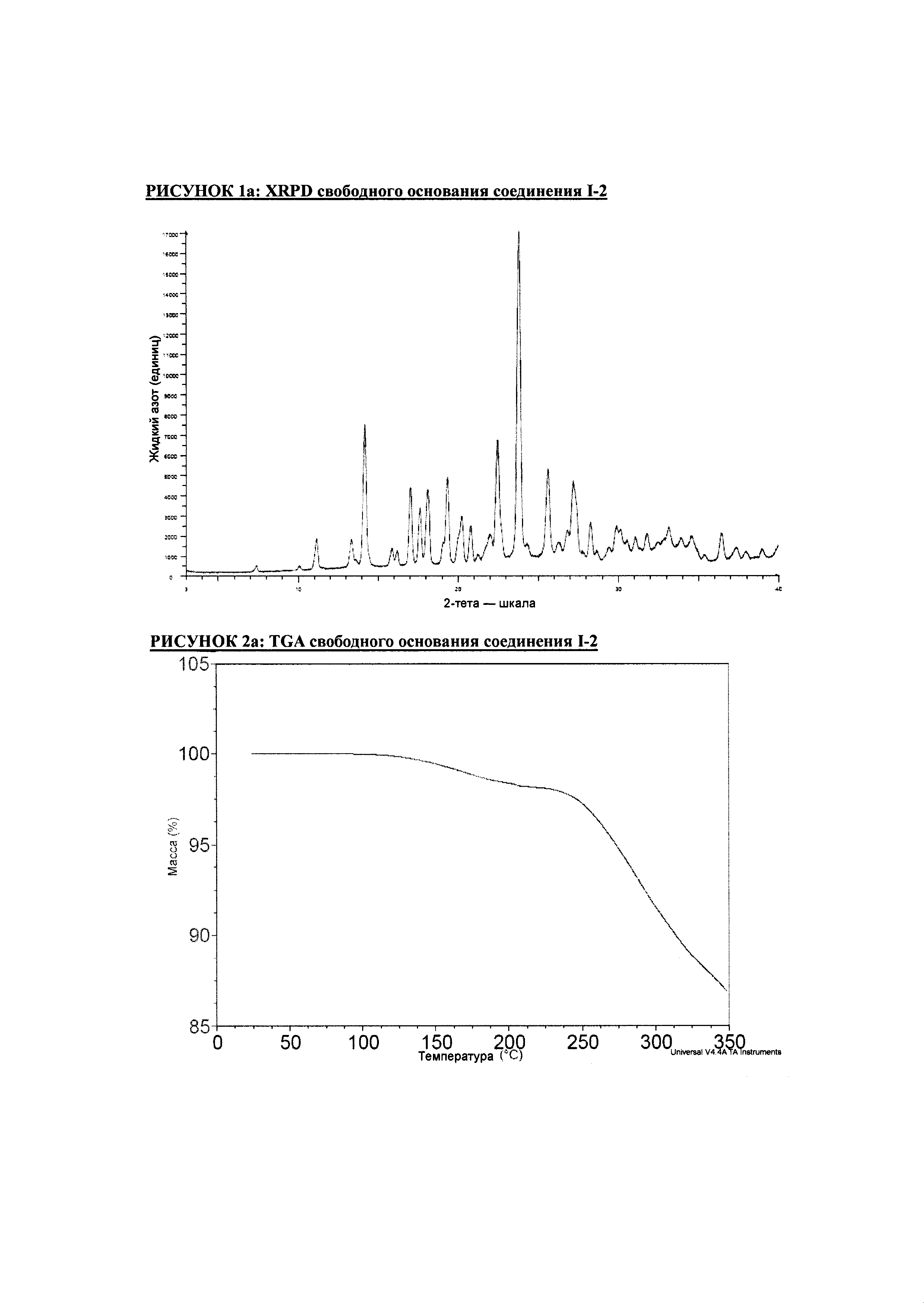
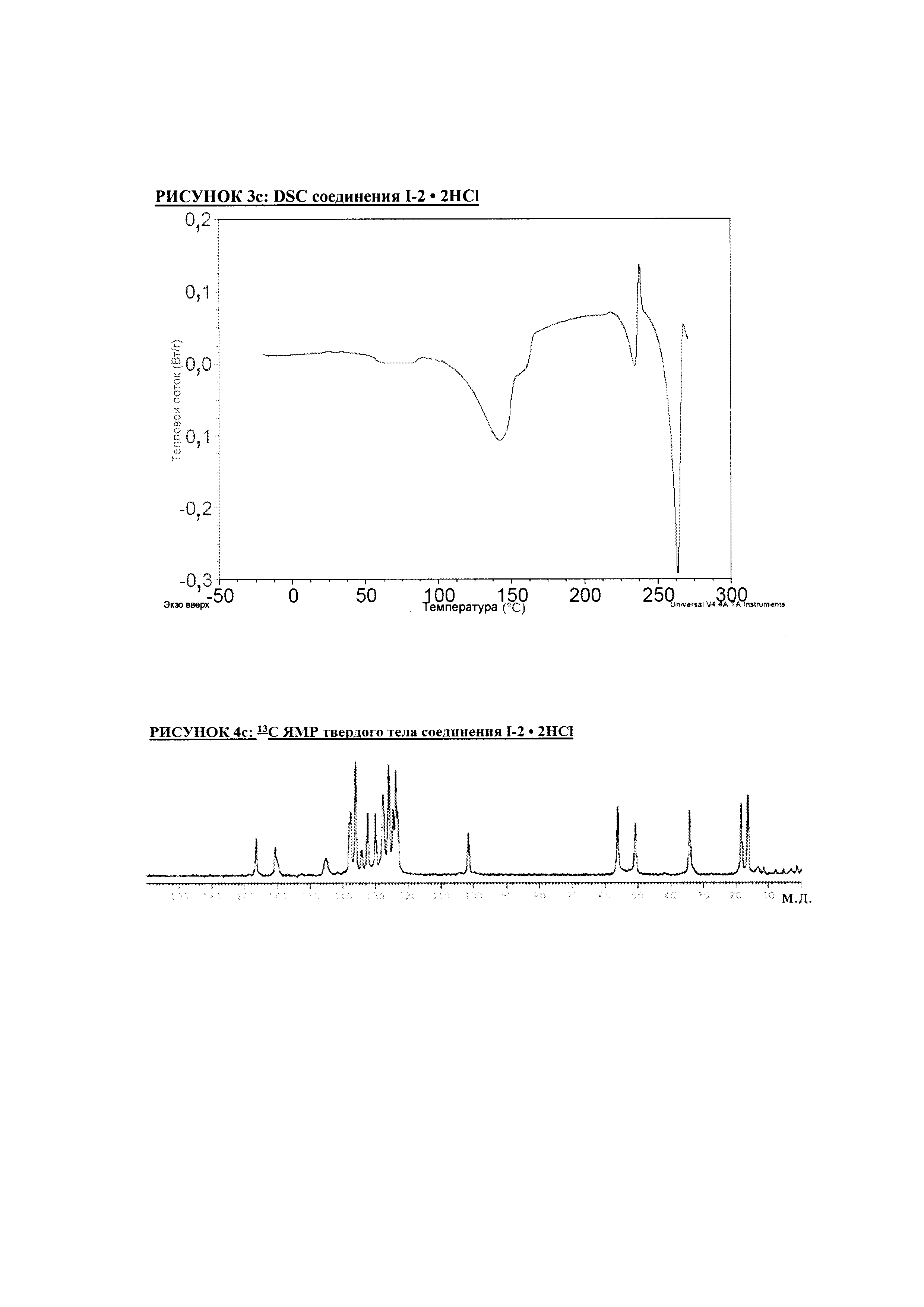

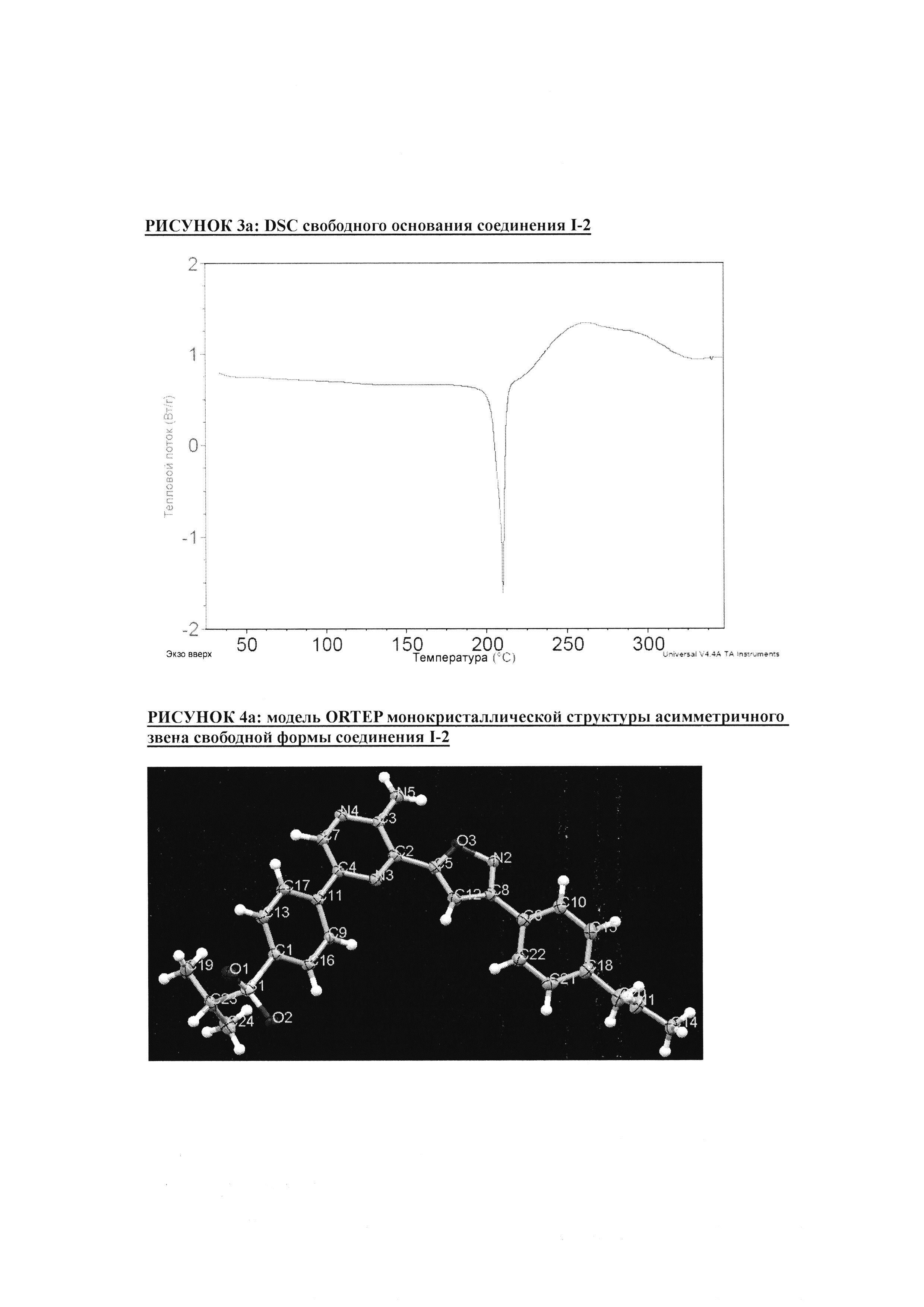
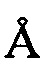
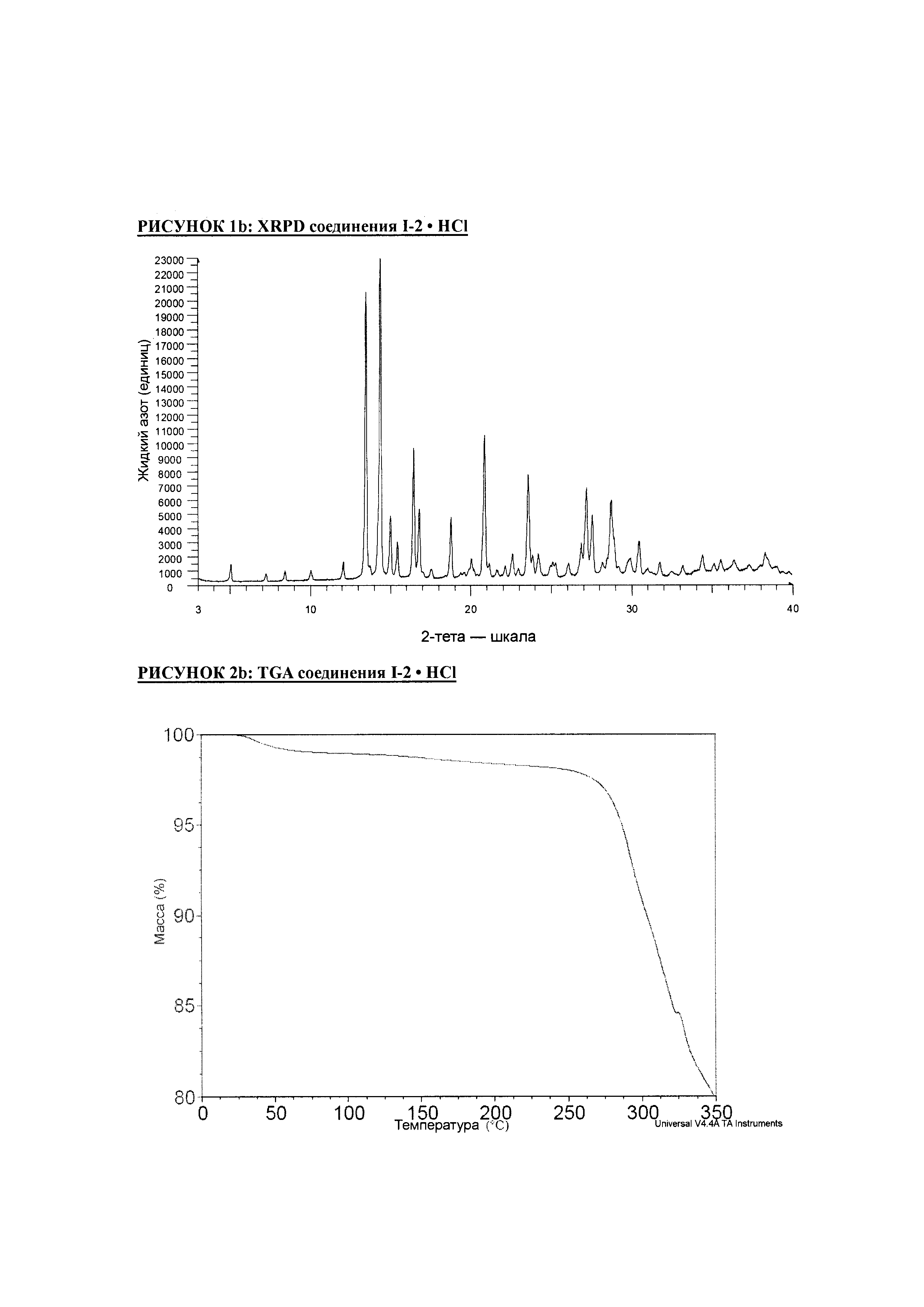
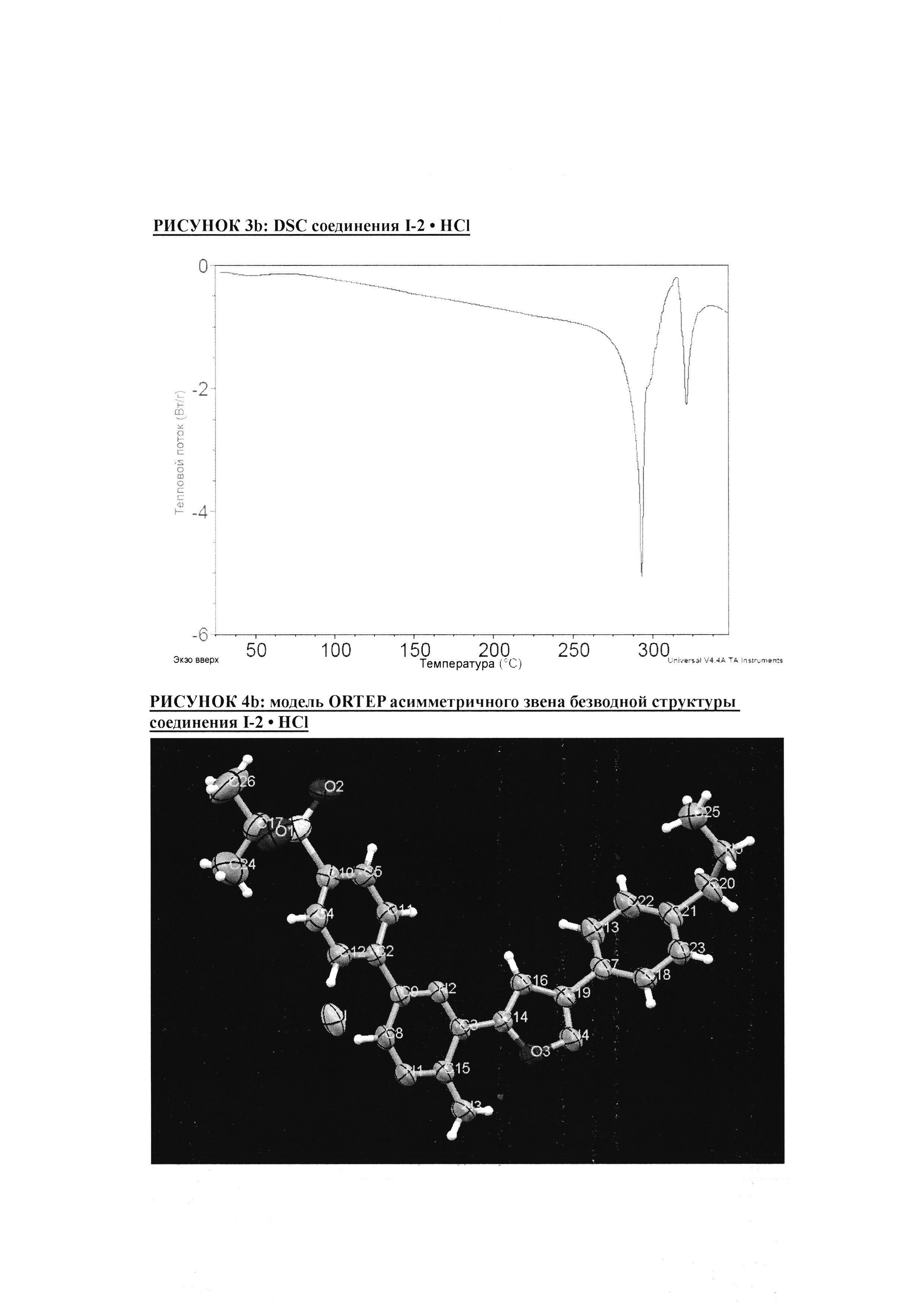
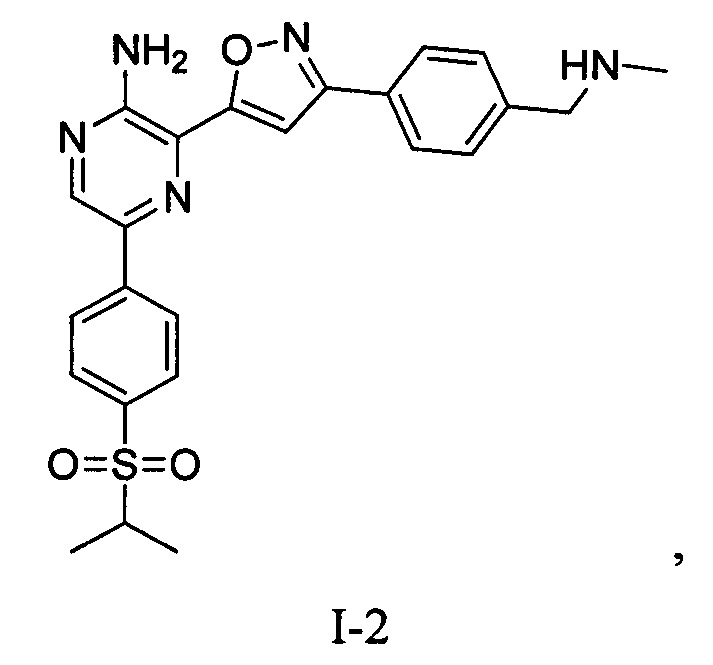
Дигидродиазепины, которые можно использовать в качестве ингибиторов протеинкиназ
Способы и промежуточные соединения для получения стерических соединений
Модуляторы атф-связывающих кассетных транспортеров
Твердые формы n-(7-азабицикло[2.2.1]гептан-7-ил-)-2-(трифторметил)фенил)-4-оксо-5-(трифторметил)-1,4-дигидрохинолин-3-карбоксамида
Производные 3-карбоксамида-4-оксохинолина, полезные в качестве модуляторов регулятора трансмембранной проводимости кистозного фиброза
Способ модуляции транспортеров атф-связывающей кассеты
Модуляторы транспортеров атф-связывающей кассеты
Способы и промежуточные соединения
Способ получения азабициклических соединений
Способ получения модуляторов регулятора трансмембранной проводимости кистозного фиброза
Дигидродиазепины, которые можно использовать в качестве ингибиторов протеинкиназ
Способ получения циклоалкилкарбоксамидо-индольных соединений
Модуляторы транспортеров атф-связывающей кассеты
Азаиндолы, полезные в качестве ингибиторов jak и других протеинкиназ
Соединения, пригодные для использования в качестве ингибиторов atr киназы
Соединения, используемые в качестве ингибиторов киназы atr
Пролекарства пиридонамидов, применяемые в качестве модуляторов натриевых каналов
Ингибиторы репликации вирусов гриппа
Радиоактивно меченные производные 2-амино-6-фтор-n-[5-фтор-пиридин-3-ил]-пиразоло[1, 5-а]пиримидин-3-карбоксамида, используемые в качестве ингибитора atr киназы, препараты на основе этого соединения и его различные твердые формы
Способ получения циклоалкилкарбоксамидо-индольных соединений

