Результат интеллектуальной деятельности: 6,7-ДИГИДРО-5Н-ПИРРОЛО[3,4-b]ПИРИДИН-5-ОН, ПОЛЕЗНЫЙ В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ-ГАММА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
[0001] Настоящее изобретение относится к соединению, применяемому в качестве ингибитора гамма-изоформы фосфатидилинозитол-3-киназы (PI3Kγ). Настоящее изобретение также относится к фармацевтически приемлемым композициям, содержащим соединение настоящего изобретения, и способам применения этого химического соединения и композиций для лечения различных патологий.
Уровень техники изобретения
[0002] PI3Ks представляют собой семейство липидкиназ, которые катализируют фосфорилирование мембранного липида фосфатидилинозитола (PI) в положении 3’-OH инозитольного кольца с образованием PI 3-фосфата [PI(3)P, PIP], PI 3,4-бисфосфата [PI(3,4)P2, PIP2] и PI 3,4,5-трифосфата [PI(3,4,5)P3, PIP3]. PI(3,4)P2 и PI(3,4,5)P3 действуют как сайты рекрутирования для различных внутриклеточных сигнальных белков, которые в свою очередь образуют сигнальные комплексы для передачи внеклеточных сигналов на цитоплазматическую сторону плазматической мембраны.
[0003] К настоящему времени было идентифицировано восемь PI3K млекопитающих, включая четыре PI3K класса I. Класс Ia включает PI3Kα, PI3Kβ и PI3Kδ. Все ферменты класса Ia представляют собой гетеродимерные комплексы, содержащие каталитическую субъединицу (p110α, p110β или p110δ), связанную с содержащей SH2 домен адаптерной субъединицей p85. PI3K класса Ia активируются сигнальным путем с участием тирозинкиназ, и они принимают участие в клеточной пролиферации и выживании. PI3Kα и PI3Kβ также вовлечены в онкогенез различных видов рака у человека. Таким образом, фармакологические ингибиторы PI3Kα и PI3Kβ пригодны для лечения различных видов рака.
[0004] PI3Kγ является единственным членом PI3Ks класса Ib, она состоит из каталитической субъединицы p110γ, которая связана с регуляторной субъединицей p101. PI3Kγ регулируется сопряженными с G-белком рецепторами путем связывания βγ субъединиц гетеродимерных G-белков. PI3Kγ экспрессируется преимущественно в гемопоэтических клетках и кардиомиоцитах и принимает участие в воспалении и функции тучных клеток. Таким образом, фармакологические ингибиторы PI3Kγ пригодны для лечения различных воспалительных заболеваний, аллергий и сердечно-сосудистых заболеваний.
[0005] Несмотря на то, что был получен ряд ингибиторов PI3K-гамма, существует потребность в дополнительных химических соединениях, ингибирующих PI3K-гамма, для лечения различных патологий и заболеваний. Особенно предпочтительными являются ингибиторы PI3K-гамма с улучшенными фармакокинетическими/фармакодинамическими характеристиками in vivo, такие как, например, ингибиторы, которые увеличивают воздействие лекарственного средства на целевую ткань, минимизируя при этом побочные эффекты. Более сильное воздействие на единицу дозы снижает побочное воздействие по отношению к воздействию на целевую ткань. Часто дозолимитирующая токсичность имеет место в органах, принимающих участие в выведении лекарственного средства из кровотока, или в случае перорально вводимого препарата в желудочно-кишечном тракте (ЖКТ). Пониженный клиренс и улучшенная биодоступность повышает Cmax в плазме крови, но ограничивает Cmax в органах выведения, таких как почки, печень и ЖКТ. Кроме того, повышенная абсорбция и пониженный клиренс (улучшенная биодоступность) часто приводит к уменьшению различий между пациентами по показателю воздействия, что также улучшает профиль безопасности вводимого агента. Кроме того, также необходимы агенты, обладающими улучшенными физическими свойствами, таким как, например, более высокая растворимость в воде.
Сущность изобретения
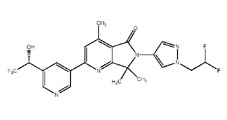
[0006] Было обнаружено, что соединение настоящего изобретения (R)-6-(1-(2,2-дифторэтил)-1Н-пиразол-4-ил)-4,7,7-триметил-2-(5-(2,2,2-трифтор-1-гидроксиэтил)пиридин-3-ил)-6,7-дигидро-5Н-пирроло[3,4-b]пиридин-5-он и его фармацевтически приемлемые композиции являются эффективными и селективными ингибиторами PI3Kγ, обладающими улучшенным фармакокинетическим/фармакодинамическим профилем по сравнению с другими ингибиторами PI3Kγ. Таким образом, настоящее изобретение относится к химическому соединению, имеющему следующую структурную формулу:
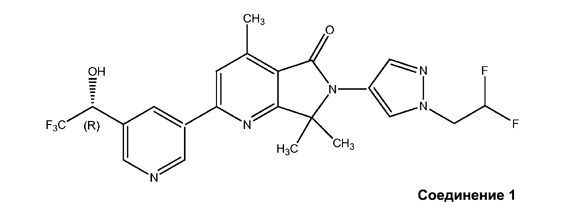
[0007] Настоящее изобретение также относится к фармацевтическим композициям, которые включают соединение 1 и фармацевтически приемлемый носитель, адъювант или наполнитель. Эти соединения и фармацевтические композиции являются пригодными для лечения или уменьшения тяжести различных патологий, включая воспалительные и иммунорегуляторные патологии, такие как астма, атопический дерматит, ринит, аллергические заболевания, хроническая обструктивная болезнь легких (ХОБЛ), септический шок, идиопатический легочный фиброз, инсульт, ожог, заболевания суставов, ревматоидный артрит, системная красная волчанка, атеросклероз, острый панкреатит, псориаз, воспалительное заболевание кишечника (ВЗК), язвенный колит, болезнь Крона и болезнь Грейвcа.
[0008] Химическое соединение и композиции настоящего изобретения также пригодны для изучения роли PI3K в биологических и патологических явлениях; изучения внутриклеточных путей передачи сигнала с участием таких киназ; и сравнительной оценки новых ингибиторов киназ.
Краткое описание чертежей
[0009] На Фигуре 1 показаны результаты испытания соединения 1 в терапевтической мышиной модели коллаген-индуцированного артрита (КИА) в количестве 2,5 мг/кг два раза в сутки, 5 мг/кг два раза в сутки и 10 мг/кг два раза в сутки (20 мг/кг/сутки).
[0010] На Фигуре 2 показаны результаты испытания соединения 1 в модели КИА в количестве 2,5 мг/кг два раза в сутки, 5 мг/кг два раза в сутки и 10 мг/кг два раза в сутки (20 мг/кг/сутки). Результаты показаны только для тех лап, которые имели клинические признаки артрита при включении в эксперимент.
[0011] На Фигуре 3 показаны результаты испытания Соединения 1 в модели КИА в количестве 2,5 мг/кг два раза в сутки, 5 мг/кг два раза в сутки и 10 мг/кг два раза в сутки (20 мг/кг/сутки). Результаты показаны только для тех лап, у которых отсутствовали клинические признаки артрита при включении в эксперимент.
[0012] На Фигуре 4 показаны результаты испытания Соединения 1 в модели ВЗК в количестве 5 мг/кг два раза в сутки и 10 мг/кг два раза в сутки.
Подробное описание изобретения
Определения и Общая терминология
[0013] Используемые следующие определения будут применяться, если не указано иное. Для целей настоящего изобретения химические элементы идентифицируются в соответствии с Периодической таблицей элементов, регистрационным номером Химической реферативной службы (CAS) и Handbook of Chemistry and Physics, 75th Ed. 1994. Кроме того, общие принципы органической химии описаны в «Organic Chemistry» Thomas Sorrell, University Science Books, Sausalito: 1999 и «March's Advanced Organic Chemistry» 5th Ed., Smith, M.B. and March, J., eds. John Wiley & Sons, New York: 2001, полное содержание которых включено в настоящее изобретение путем ссылки.
[0014] Химические соединения, которые изображены со стереохимическими центрами, определяются как стереохимически чистые, но абсолютная стереохимия которых все еще не определена. Такие химические соединения могут иметь или R- или S-конфигурацию. В тех случаях, когда такая абсолютная конфигурация была определена, хиральный центр(ы) на схеме обозначается буквами R или S. Когда Соединение 1 настоящего изобретения изображено в R-конфигурации, настоящее изобретение включает в себя варианты осуществления в S-конфигурации и рацемические смеси R- и S-конфигураций.
[0015] Настоящее изобретение также относится к фармацевтической композиции, содержащей любое соединение настоящего изобретения и фармацевтически приемлемый носитель, адъювант или наполнитель.
[0016] В одном варианте осуществления настоящее изобретение относится к способу ингибирования киназной активности PI3K у пациента путем введения этому пациенту соединения 1 или его фармацевтической композиции. В дополнительном варианте осуществления изобретения PI3K-гамма селективно ингибируется сильнее, чем PI3K-альфа, PI3K-бета или PI3K-дельта.
[0017] В другом варианте осуществления настоящее изобретение относится к способу лечения или уменьшения тяжести заболевания или патологического состояния, выбранного из воспалительных и иммунорегуляторных патологий, таких как астма, атопический дерматит, ринит, аллергические заболевания, хроническая обструктивная болезнь легких (ХОБЛ), септический шок, идиопатический легочный фиброз, инсульт, ожог, заболевания суставов, ревматоидный артрит, системная красная волчанка, атеросклероз, острый панкреатит, псориаз, воспалительное заболевание кишечника, язвенный колит, болезнь Крона и болезнь Грейвcа, у пациента путем введения этому пациенту Соединения 1 или его фармацевтической композиции.
[0018] Настоящее изобретение также относится к нетерапевтическому способу ингибирования киназной активности PI3K-гамма в биологическом образце in vitro, включающему в себя приведение указанного биологического образца в контакт с Соединением 1 или композицией, содержащей указанное соединение.
Композиции, препараты и введение химических соединений настоящего изобретения
[0019] В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей Соединение 1. Например, настоящее изобретение относится к композиции, содержащей соединение 1 или его фармацевтически приемлемое производное и фармацевтически приемлемый носитель, адъювант или наполнитель. В одном варианте осуществления изобретения количество соединения в композиции настоящего изобретения является таким, которое является эффективным для значительного ингибирования PI3Kγ в биологическом образце или у пациента. В одном варианте осуществления изобретения композиция настоящего изобретения приготовлена для введения пациенту, нуждающемуся в такой композиции. В еще одном варианте осуществления изобретения композиция настоящего изобретения приготовлена для перорального введения пациенту. Используемый термин «пациент» означает животное, предпочтительно млекопитающее, и более предпочтительно человека.
[0020] Кроме того, следует понимать, что некоторые химические соединения настоящего изобретения могут существовать в свободной форме для лечения или, когда это необходимо, в виде их фармацевтически приемлемых производных. В соответствии с настоящим изобретением фармацевтически приемлемое производное включает в себя без ограничений фармацевтически приемлемые пролекарства, соли, сложные эфиры, соли таких сложных эфиров или любой другой аддукт или производное, которое после введения нуждающемуся в этом пациенту способно обеспечить прямо или опосредованно образование ранее описанного соединения или его метаболита или остатка. Используемый термин «обладающий ингибирующей активностью его метаболит или остаток» означает такой его метаболит или остаток, который также является ингибитором PI3K-гамма.
[0021] Используемый здесь термин «фармацевтически приемлемая соль» относится к таким солям, которые в пределах медицинской оценки являются пригодными для использования в контакте с тканями человек и низших животных, не вызывая чрезмерной токсичности, раздражения, аллергической реакции и тому подобного.
[0022] Фармацевтически приемлемые соли хорошо известны в данной области техники. Например, S. M. Berge et al. подробно описывают фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 66: 1-19, 1977, который включен в настоящее изобретение путем ссылки. Фармацевтически приемлемые соли химических соединений настоящего изобретения включают в себя соли, полученные из подходящих неорганических и органических кислот и оснований. Примерами фармацевтически приемлемых нетоксичных солей присоединения кислоты являются соли аминогруппы, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и хлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или при помощи других способов, применяемых в данной области техники, таких как ионный обмен. Другие фармацевтически приемлемые соли включают в себя адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентилпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептаноат, гексаноат, гидроиодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканоат, валерат и тому подобное. Соли полученные из соответствующих оснований включают в себя соли щелочных металлов, щелочноземельных металлов, аммония и N+(C1-4 алкил)4. Настоящее изобретение также предусматривает кватернизацию любых основных азотсодержащих групп раскрытых здесь химических соединений. При помощи такой кватернизации могут быть получены растворимые или диспергируемые в воде или в масле продукты. Типичные соли щелочного или щелочноземельного металла включают в себя соли натрия, лития, калия, кальция, магния и тому подобное. Кроме того, при необходимости фармацевтически приемлемые соли включают в себя нетоксичные катионы аммония, четвертичного аммония или амина, образованные с использованием противоионов, таких как галид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, C1-8 сульфонат и арилсульфонат.
[0023] Как описано выше, фармацевтически приемлемые композиции настоящего изобретения дополнительно содержат фармацевтически приемлемый носитель, адъювант или наполнитель, который, как предусматривается в настоящем изобретении, включает в себя любой и все растворители, разбавители или другой жидкий носитель, вещества, способствующие диспергированию или суспендированию, поверхностно-активные агенты, изотонические агенты, загустители или эмульгаторы, консерванты, твердые связующие вещества, смазывающие вещества и тому подобное, которые являются подходящими для конкретной желаемой лекарственной формы. Различные носители, используемые при изготовлении фармацевтически приемлемых композиций, и известные способы их получения описаны в Remington: The Science and Practice of Pharmacy, 21st edition, 2005, ed. D.B. Troy, Lippincott Williams & Wilkins, Philadelphia, и Encyclopedia of Pharmaceutical Technology, eds. J. Swarbrick and J. C. Boylan, 1988-1999, Marcel Dekker, New York, содержание которых включено в настоящее изобретение путем ссылки. За исключением тех случаев, когда любая стандартно используемая среда-носитель является несовместимой с химическими соединениями настоящего изобретения, например, из-за возникновения любого нежелательного биологического эффекта или другого вредного взаимодействия с любым другим компонентом(ами) фармацевтически приемлемой композиции, предполагается, что ее использование находится в пределах объема настоящего изобретения.
[0024] Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают в себя без ограничений ионообменники, окись алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота, или сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, двузамещенный фосфат натрия, двузамещенный фосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, полиакрилаты, воски, полиэтилен-полиоксипропилен-блок-сополимеры, ланолин, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетатцеллюлоза; порошкообразный трагакант; солод; желатин; тальк; вспомогательные вещества, такие как какао-масло и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло; сафлоровое масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли; такие как пропиленгликоль или полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; свободная от пирогенов вода; изотонический солевой раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые лубриканты, такие как лаурилсульфат натрия и стеарат магния, а также красители, высвобождающие агенты, покрывающие агенты, подсластители, вкусовые и ароматизирующие агенты, консерванты и антиоксиданты также могут присутствовать в композиции по усмотрению разработчика рецептуры.
[0025] Композиции настоящего изобретения могут вводиться перорально, парентерально, путем ингаляции, местно, ректально, назально, трансбуккально, вагинально или через имплантированный резервуар. Используемый здесь термин «парентерально» включает в себя подкожное, внутривенное, внутримышечное, внутрисуставное, внутрисиновиальное, внутригрудинное, интратекальное, внутриглазное, внутрипеченочное, внутриочаговое, эпидуральное, интраспинальное и внутричерепное введение при помощи инъекции или инфузии. Предпочтительно, композиции вводятся перорально, внутрибрюшинно или внутривенно. Стерильные инъекционные формы композиций настоящего изобретения могут представлять собой водные или масляные суспензии. Эти суспензии могут быть приготовлены способами, известными из уровня техники, с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильные инъекционные препараты также могут представлять собой стерильный инъекционный раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3- бутандиоле. К числу приемлемых наполнителей и растворителей, которые могут быть использованы, относятся вода раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно применяются стерильные нелетучие масла.
[0026] Для этой цели может быть использовано любое легкое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, являются пригодными для получения инъекционных препаратов, как и натуральные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Эти масляные растворы или суспензии также могут содержать длинноцепочечный спиртовой разбавитель или диспергирующий агент, такой как карбоксиметилцеллюлоза или аналогичные диспергирующие агенты, которые традиционно используются при изготовлении фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Другие традиционно используемые поверхностно-активные вещества, такие как Твины, Спаны и другие эмульгирующие агенты или усилители биодоступности, которые обычно используются в производстве фармацевтически приемлемых твердых, жидких или других лекарственных форм, также могут быть использованы для составления рецептуры.
[0027] Фармацевтические композиции настоящего изобретения могут вводиться перорально в виде любой приемлемой для перорального введения лекарственной формы, включая без ограничений капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения, обычно используемые носители включают лактозу и кукурузный крахмал. Смазывающие агенты, такие как стеарат магния, также обычно добавляют. Для перорального введения в виде капсулы подходящие разбавители включают в себя лактозу и высушенный кукурузный крахмал. Когда для перорального применения необходимы водные суспензии, активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. При необходимости также могут быть добавлены некоторые подсластители вкусоароматические агенты или красители.
[0028] В альтернативном варианте фармацевтически приемлемые композиции настоящего изобретения могут вводиться в форме суппозиториев для ректального введения. Они могут быть получены путем смешивания агента с подходящим нераздражающим вспомогательным веществом, которое является твердым при комнатной температуре, но жидким при ректальной температуре, и следовательно будет плавиться в прямой кишке с высвобождением лекарственного средства. Такие материалы включают какао-масло, пчелиный воск и полиэтиленгликоли.
[0029] Жидкие лекарственные формы для перорального введения включают в себя без ограничений фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным химическим соединениям жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области техники, таких как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, масло зародышей пшеницы, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбита, и их смеси. Кроме инертных разбавителей, композиции для перорального применения могут также включать в себя адъюванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, вкусовые и ароматизирующие агенты.
[0030] Инъекционные препараты, например стерильные инъекционные водные или маслянистые суспензии, могут быть приготовлены способами, известными в данной области техники, с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъекционный препарат может представлять собой стерильный инъекционный раствор, суспензию или эмульсию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. К числу приемлемых наполнителей и растворителей, которые могут быть использованы, относятся вода раствор Рингера Фармакопея США и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно применяются стерильные нелетучие масла. Для этой цели может быть использовано любое легкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, используются для приготовления инъекционных препаратов.
[0031] Инъекционные препараты могут быть стерилизованы, например, путем фильтрации через задерживающий бактерии фильтр или путем включения стерилизующих агентов в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или другой стерильной инъекционной среде перед использованием.
[0032] Для того чтобы продлить действие химического соединения настоящего изобретения, часто желательно замедлить абсорбцию этого химического соединения из подкожной или внутримышечной инъекции. Это может быть осуществлено путем использования жидкой суспензии кристаллического или аморфного материала с плохой растворимостью в воде. Скорость абсорбции химического соединения в этом случае зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и формы кристаллов. В альтернативном варианте, растворение или суспендирование химического соединения в масляном наполнителе осуществляет замедление абсорбции парентерально вводимой формы химического соединения. Инъекционные депо-формы изготавливают путем получения микроинкапсулирующих матриц химического соединения в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения между химическим соединением и полимером и от природы конкретного используемого полимера, можно регулировать скорость высвобождения химического соединения. Примеры других биоразлагаемых полимеров включают в себя поли(ортоэфиры) и поли(ангидриды). Инъекционные депо-препараты также получают путем заключения химического соединения в липосомы или микроэмульсии, которые являются совместимыми с тканями организма. Химические соединения настоящего изобретения могут быть включены в другие общеизвестные препараты с замедленным высвобождением, для обеспечения их контролируемого высвобождения в течение необходимого периода времени от нескольких часов до нескольких судок и до месяцев.
[0033] Композиции для ректального или вагинального введения предпочтительно являются суппозиториями, которые могут быть получены путем смешивания химических соединений настоящего изобретения с подходящими нераздражающими вспомогательными веществами или носителями, такими как какао-масло, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но жидкими при температуре тела и, следовательно, расплавляются в прямой кишке или вагинальной полости и высвобождают активное химическое соединение.
[0034] Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное химическое соединение смешивают с, по меньшей мере, одним инертным, фармацевтически приемлемым вспомогательным веществом или носителем, таким как цитрат натрия или дикальцийфосфат и/или с a) наполнителями или сухими разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связующими веществами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и камедь, c) увлажнителями, такими как глицерин, d) дезинтегрирующими агентами, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, e) замедляющими растворение агентами, такими как парафин, f) ускорителями абсорбции, такими как четвертичные аммониевые соединения, g) смачивающими агентами, такими как, например, цетиловый спирт и глицеролмоностеарат, h) абсорбентами, такими как каолин и бентонитовая глина, и i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль лекарственная форма также может содержать буферные агенты.
[0035] Твердые композиции подобного типа также могут использоваться в качестве наполнителей в мягких и твердых заполненных желатиновых капсулах с использованием таких вспомогательных веществ, как лактоза или молочный сахар, а также полиэтиленгликоли с высокой молекулярной массой и тому подобное. Твердые лекарственные формы таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие оболочки, хорошо известные в области получения фармацевтических препаратов. При необходимости они могут содержать рентгеноконтрастные агенты, и могут также представлять собой композицию, которая высвобождает активный ингредиент(ы) только, или предпочтительно, в определенной части кишечного тракта, при необходимости, замедленным способом. Примеры образующих матрицу композиций, которые могут быть использованы, включают полимерные вещества и воски. Твердые композиции подобного типа также могут использоваться в качестве наполнителей в мягких и твердых заполненных желатиновых капсулах с использованием таких вспомогательных веществ, как лактоза или молочный сахар, а также полиэтиленгликоли с высокой молекулярной массой и тому подобное.
[0036] Активные химические соединения также могут находиться в микроинкапсулированной форме с одним или несколькими вспомогательными веществами, как указано выше. Твердые лекарственные формы таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, покрытия с контролируемым высвобождением и другие оболочки, хорошо известные в области получения фармацевтических препаратов. В таких твердых лекарственных формах активное соединение может быть смешано, по меньшей мере, с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы помимо инертных разбавителей также могут включать, как и в обычной практике, дополнительные вещества, например, смазывающие вещества для таблетирования и другие облегчающие таблетирование вещества, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль лекарственные формы также могут содержать буферные агенты. При необходимости они могут содержать рентгеноконтрастные агенты и могут также представлять собой композицию, которая высвобождает активный ингредиент(ы) только, или предпочтительно, в определенной части кишечного тракта, при необходимости, замедленным способом. Примеры образующих матрицу композиций, которые могут быть использованы, включают в себя полимерные вещества и воски.
[0037] Лекарственные формы для местного или трансдермального введения химического соединения настоящего изобретению включают в себя мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, средства для ингаляции или пластыри. Активный компонент смешивается в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, которые могут потребоваться. Офтальмологические препараты, ушные капли и глазные капли также рассматриваются как находящиеся в пределах объема настоящего изобретения. Кроме того, настоящее изобретение предполагает использование трансдермальных пластырей, которые обладают дополнительным преимуществом обеспечения контролируемой доставки химического соединения в организм. Такие лекарственные формы могут быть получены путем растворения или диспергирования химического соединения в соответствующей среде. Усилители абсорбции также могут быть использованы для увеличения тока химического соединения через кожу. Скорость может контролироваться или при помощи регулирующей скорость мембраны, или путем диспергирования химического соединения в полимерной матрице или геле.
[0038] Химические соединения настоящего изобретения предпочтительно получают в виде дозированной лекарственной формы для простоты введения и однородности дозы. Используемый термин «дозированная лекарственная форма» относится к физически дискретной единице агента, подходящей для получающего лечение пациента. Однако следует понимать, что общая суточная доза химических соединений и композиций настоящего изобретения будет определяться лечащим врачом на основании тщательной медицинской оценки. Конкретный уровень эффективной дозы для любого конкретного пациента или организма будет зависеть от множества факторов, включая подлежащую лечению патологию и тяжесть заболевания; активность конкретного используемого химического соединения; конкретную используемую композицию; возраст, массу тела, общее состояние здоровья, пол и рацион питания пациента; время введения, путь введения и скорость экскреции конкретного используемого химического соединения; продолжительность лечения; лекарственные препараты, используемые в комбинации или совместно с конкретным используемым химическим соединением, и тому подобные факторы, хорошо известные в области медицины.
[0039] Количество химических соединений настоящего изобретения, которое может быть объединено с материалами-носителями для получения композиции в виде единицы дозы, будет изменяться в зависимости от получающего лечение субъекта, конкретного способа введения. Предпочтительно композиции должны быть составлены таким образом, чтобы пациенту, получающему эти композиции, могли быть введены дозы ингибитора в интервале от 0,01 до 100 мг/кг, например, от 0,1 до 100 мг/кг и от 0,1 до 10 мг/кг массы тела/ в сутки.
[0040] В зависимости от конкретного состояния или заболевания, подлежащего лечению или предотвращению, дополнительные терапевтические агенты, которые обычно вводят для лечения или предотвращения этого состояния, также могут присутствовать в композициях настоящего изобретения. Используемые здесь дополнительные терапевтические агенты, которые обычно вводят для лечения или предотвращения конкретного заболевания или состояния, известны как «подходящие для заболевания или состояния, подлежащего лечению». Примеры дополнительных терапевтических агентов приводятся ниже.
[0041] Количество дополнительного терапевтического агента, присутствующее в композициях настоящего изобретения, не будет превышать количество, которое обычно вводят в композицию, содержащую этот терапевтический агент в качестве единственного активного агента. Предпочтительно количество дополнительного терапевтического агента в описанных здесь композициях будет находиться в диапазоне от примерно 50% до 100% от количества, которое обычно присутствует композиции, содержащей этот агент в качестве единственного терапевтически активного агента.
Применения химических соединений и композиций настоящего изобретения
[0042] В одном аспекте настоящего изобретения данное изобретение относится к способу лечения или уменьшения тяжести опосредованного PI3K-гамма патологического состояния или заболевания. Используемый термин «опосредованное PI3K-гамма заболевание» означает любое заболевание или другое патологическое состояние, для которого известно, что изоформа PI3K-гамма играет в нем определенную роль.
[0043] Таким образом, в дополнительном варианте осуществления изобретения химическое соединение настоящего изобретения является селективным для ингибирования изоформы PI3K-гамма. В одном варианте осуществления изобретения химическое соединение или композиция настоящего изобретения селективно ингибирует изоформу PI3K-гамма, по меньшей мере, в 20 раз сильнее, чем изоформу PI3K-альфа в анализе in vitro. В другом варианте осуществления изобретения PI3Kγ-селективное химическое соединение настоящего изобретения ингибирует гамма изоформу, по меньшей мере, в 20 раз сильнее, чем каждую из изоформ альфа, бета и дельта в анализе in vitro. Настоящее изобретение также включает в себя применение селективности химических соединений и композиций настоящего изобретения in vivo для лечения пациентов, нуждающихся в такой терапии.
[0044] Соединения или композиции настоящего изобретения могут вводиться с одним или несколькими дополнительными терапевтическими средствами, где это дополнительное терапевтическое средство является подходящим для заболевания, подлежащего лечению, и это дополнительное терапевтическое средство вводится вместе с химическим соединением или композицией настоящего изобретения в виде единой лекарственной формы или отдельно от соединения или композиции, как часть множественной лекарственной формы. Дополнительный терапевтический агент может вводиться в то же самое время, что и соединение настоящего изобретения, или в другое время. В последнем случае введение может проводиться с интервалом, например, 6 часов, 12 часов, 1 сутки, 2 суток, 3 суток, 1 неделя, 2 недели, 3 недели, 1 месяц или 2 месяца.
[0045] Настоящее изобретение относится к способу ингибирования киназной активности PI3K-гамма в биологическом образце, который включает контактирование биологического образца с соединением или композицией настоящего изобретения. Используемый термин «биологический образец» означает образец вне живого организма и включает без ограничений культуры клеток или их экстракты; биопсийный материал, полученный от млекопитающего или его экстракт; и кровь, слюну, мочу, фекалии, сперму, слезы или другие жидкости организма или их экстракты. Ингибирование киназной активности, в частности, киназной активности PI3K-гамма в биологическом образце применимо для различных целей, известных специалисту в данной области техники. Примеры таких целей включают без ограничений хранение биологического образца и биологические анализы. В одном варианте осуществления изобретения способ ингибирования киназной активности PI3K-гамма в биологическом образце ограничен нетерапевтическими методами.
Получение химического соединения настоящего изобретения
[0046] В данном описании все сокращения, символы и условные обозначения соответствуют тем, которые используются в современной научной литературе. См., например, Janet S. Dodd, ed., The ACS Style Guide: A Manual for Authors and Editors, 2nd Ed., Washington, D.C.: American Chemical Society, 1997. Следующие определения описывают термины и сокращения, используемые в настоящем изобретении:
|
Методика синтеза
[0047] Как правило, соединение 1 может быть получено описанными способами или другими способами, известными специалистам в данной области техники.
Пример 1. Получение 2-хлор-6-(1-(2,2-дифторэтил)-1Н-пиразол-4-ил)-4,7,7-триметил-6,7-дигидро-5Н-пирроло[3,4-b]пиридин-5-она (Соединение[1006]).
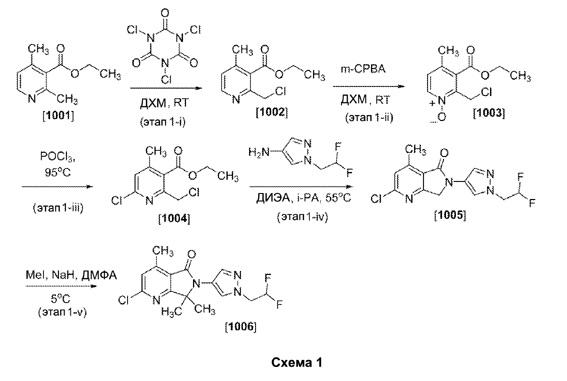
[0048] Как показано на этапе 1-i Схемы 1, к раствору этил-2,4-диметилпиридин-3-карбоксилата (Соединение 1001, 20,2 г, 112,5 ммоль) в дихлорметане (100 мл) в атмосфере азота порциями добавляли 1,3,5-трихлор-1,3,5-триазин-2,4,6-трион (31,4 г, 135,0 ммоль) в течение 15 минут. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Полученный белый осадок отфильтровывли и затем фильтрат последовательно промывали насыщенным водным раствором NaHCO3 (2×100 мл) и рассолом (100 мл). Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением этил-2-(хлорметил)-4-метилникотината (22,9 г, Соединение 1002) в виде желтого масла: МСЭРИ (M+H)=213,96. Этот материал использовали на следующем этапе без дополнительной очистки.
[0049] Как показано на этапе 1-ii Схемы 1, к раствору этил-2-(хлорметил)-4-метилникотината, (Соединение 1002, 112,0 г, 524,2 ммоль) в дихлорметане (484 мл) добавляли 3-хлорпероксибензойную кислоту (141,0 г, 629,0 ммоль). Реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение ночи. Смесь разбавляли дихлорметаном (200 мл) и последовательно промывали насыщенным водным раствором NaHCO3 (100 мл), насыщенным водным раствором Na2CO3 (100 мл 2М раствора) и рассолом. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением 2-(хлорметил)-3-(этоксикарбонил)-4-метилпиридин-1-оксида (Соединение 1003): МСЭРИ (M+H)=230,25. Этот материал использовали на следующем этапе без дополнительной очистки.
[0050] Как показано на этапе 1-iii Схемы 1, раствор 2-(хлорметил)-3-(этоксикарбонил)-4-метилпиридин-1-оксида (Соединение 1003, 69,3 г, 301,8 ммоль) в оксихлориде фосфора (450,1 мл, 4,8 моль) перемешивали в атмосфере азота и нагревали при температуре 95°C в течение 60 часов. Реакционную смесь охлаждали до комнатной температуры и оксихлорид фосфора отгоняли в вакууме. Полученный темно-окрашенный остаток растворяли в дихлорметане (100 мл) и выливали на лед (500 г) в 1 л лабораторном стакане. Полученную смесь перемешивали в течение 10 минут. Значение рН смеси доводили до значения немного выше 7 при помощи насыщенного водного раствора NaHCO3. Органическую фазу отделяли и водную фазу снова экстрагировали дополнительным количеством дихлорметана. Объединенные органические фазы последовательно промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток пропускали через слой силикагеля, используя смесь EtOAc/гексаны (1:3), с получением продукта с чистотой 80% (по данным 1Н-ЯМР анализа) после удаления летучих веществ при пониженном давлении. Этот материал далее очищали при помощи хроматографии на силикагеле при среднем давлении (10-25% EtOAc/гексаны, 330 г колонка Teledyne ISCO) с получением этил-6-хлор-2-(хлорметил)-4-метилникотината (Соединение 1004, 36,5 г): МСЭРИ (М+Н)=248,04.
[0051] Как показано на этапе 1-iv Схемы 1, к раствору 1-(2,2-дифторэтил)пиразол-4-амина (48,2 г, 327,5 ммоль) в изопропаноле (1,7 л) добавляли этил-6-хлор-2-(хлорметил)-4-метилникотинат (Соединение 1004, 65,0 г, 262,0 ммоль), а затем N,N-диизопропилэтиламин (45,6 мл, 262,0 ммоль). Реакционную смесь нагревали при температуре 55°C в течение 72 часов. Полученную густую белую суспензию охлаждали до комнатной температуры, фильтровали и промывали дополнительным количеством изопропанола (200 мл) и диэтиловым эфиром (500 мл). Полученное твердое вещество сушили при температуре 50°C в течение ночи в вакуумной печи, получая 56 г 2-хлор-6-(1-(2,2-дифторэтил)-1H-пиразол-4-ил)-4-метил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (Соединение 1005): 1Н ЯМР (300 МГц, ДМСО-d6) δ 8,28 (s, 1Н), 7,87 (d, J=0,5 Гц, 1H), 7,52 (s, 1H), 6,37 (tt, J=54,9, 3,7 Гц, 1H), 4,85 (s, 2H), 4,67 (td, J=15,2, 3,7 Гц, 2H), 2,65 (s, 3H). Этот материал содержал 7% нециклизированного побочного продукта [этил-6-хлор-2-(((1-(2,2-дифторэтил)-1H-пиразол-4-ил)амино)метил)-4-метилникотинат] и был использован в этом виде в последующих реакциях.
[0052] Как показано на этапе 1-v Схемы 1, к раствору 2-хлор-6-(1-(2,2- дифторэтил)-1H-пиразол-4-ил)-4-метил-6,7-дигидро-5Н-пирроло[3,4-b]пиридин-5-она (Соединение 1005, 46,0 г, 147,1 ммоль) в ДМФА (782,0 мл) добавляли метилиодид (20,1 мл, 323,6 ммоль). Смесь охлаждали до температуры 5°C и порциями в течение 15 минут добавляли гидрид натрия (12,9 г, 323,6 ммоль 60%-ная дисперсия в минеральном масле). Реакционную смесь перемешивали при температуре 3°C в течение 45 минут. ВЭЖХ-анализ показал наличие смеси продуктов монометилирования (10%), бис-метилирования (74%) и три-метилирования (15%), вместе с расходованием исходного материала. Дополнительно добавляли гидрид натрия (1,29 г, 32,36 ммоль 60%-ная дисперсия в минеральном масле), и через час после этого, ВЭЖХ-анализ показал наличие 84% целевого продукта бис-метилирования и 16% три-метилированного побочного продукта. Реакцию останавливали путем добавления насыщенного водного раствора NH4Cl (1 л), тиосульфата натрия (400 мл) и воды (1 л). Водную фазу дважды экстрагировали EtOAc (800 мл). Объединенные органические фазы сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали при помощи хроматографии на силикагеле при среднем давлении (с градиентом 0-40% EtOAc/гексаны, используя 800 г колонку Teledyne ISCO), с получением 2-хлор-6-(1-(2,2-дифторэтил)-1H-пиразол-4-ил)-4,7,7-триметил-6,7-дигидро-5Н-пирроло[3,4-b]пиридин-5-она (Соединение 1006, 24 г) в виде белого твердого вещества: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,21 (s, 1Н), 7,83 (s, 1H), 7,54 (s, 1H), 6.58-6.25 (m, 1Н), 4,67 (td, J=15,1, 3,7 Гц, 2H), 2,65 (s, 3H), 1 0,51 (s, 6H).
Пример 2. Получение (R)-6-(1-(2,2-дифторэтил)-1H-пиразол-4-ил)-4,7,7-триметил-2-(5-(2,2,2-трифтор-1-гидроксиэтил)пиридин-3-ил)-6,7-дигидро-5Н-пирроло [3,4-b]пиридин-5-она (Соединение 1).
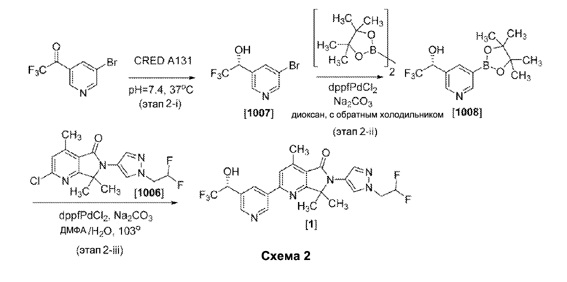
[0053] Как показано на этапе 2-i Схемы 2, в 20-литровый реактор добавляли водe (10 л), с последующим добавлением KH2PO4 (136 г). Смесь перемешивали до достижения однородного состояния с получением 0,1 М KH2PO4 буфера. Значение pH этого буфера доводили до 7,5 путем добавления 2М NaOH. Внутреннюю температуру доводили до 37°C. Примерно 500 мл буфера удаляли, чтобы использоваться для последующего добавления НАД и CRED клеточной пасты A131 (Almac Group, Ltd.). Соответственно, раствор НАД (20 г) в буфере (100 мл) добавляли к оставшимся 9,5 л буфера с последующим добавлением раствора 1-(5-бромпиридин-3-ил)-2,2,2-трифторэтана-1-она (1020 г) в МТБЭ (1 0,5 л). Изопропиловый спирт (1 л) использовали для промывки дополнительно колбы для проведения реакции. Восстановление инициировали путем добавления суспензии CRED клеточной пасты A131 (100 г) в буфере (400 мл) к перемешиваемой реакционной смеси. В течение реакции, двухфазные образцы реакции (~2 мл) отбирали из перемешивания смеси, экстрагировали EtOAc (10 мл), сушили над MgSO4, упаривали и анализировали при помощи 1H ЯМР в ДМСО-d6 для мониторинга превращения исходного материала в продукт. Через 1 ч наблюдалось превращение 50%. Через 3 часа наблюдалось превращение >99% конверсии. Значение рН реакционной смеси доводили до 11 при помощи 2М NaOH и перемешивали в течение 30 минут для денатурации фермента. Значение рН доводили до 9 с помощью 2М HCl. К реакционной смеси добавляли EtOAc (5 л) и диатомовую землю (650 г) и продолжали перемешивание в течение 10 минут. Полученную эмульсию фильтровали через слой диатомовой земли для разделения органической и водной фазы. Слой промывали EtOAc (2 л) и отделяли органическую фазу. Водную фазу повторно экстрагировали EtOAc (6 л) и объединенные органические фазы промывали рассолом (4 л) и сушили путем добавления MgSO4 (200 г) и перемешивали в течение 30 минут. После фильтрования и удаления летучих веществ в вакууме получали бледно-желтое масло, которое быстро затвердевало при отстаивании. Твердое вещество разделяли на куски и получали взвесь в циклогексане (2 л). К смеси добавляли EtOAc (~500 мл), и затем перемешивали при температуре 45°C для растворения твердых веществ. Затем эту смесь повторно концентрировали в вакууме до тех пор, пока чистое белое твердое вещество не начинало выпадать в осадок из раствора. Добавляли дополнительное количество циклогексана (2 л), и раствор охлаждали до температуры 0°C на ледяной бане. После перемешивания в течение 30 мин, твердое вещество собирали фильтрацией, промывали циклогексаном (500 мл) и сушили в вакуумной печи с получением 812 г (R)-1)-1-(5-бромпиридин-3-ил)-2,2,2 трифторэтан-1-ола (Соединение 1007, 99,95% э.и. по данным ВЭЖХ-анализа) в виде гранул твердого вещества белого цвета: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,72 (s, 1 Н), 8,61 (s, 1H), 8,08 (s, 1H), 5.09-5.15 (m, 1Н).
[0054] Как показано на этапе 2-ii на схеме 2, раствор (1R)-1-(5-бром-3-пиридил)-2,2,2-трифтор-этанола (Cоединение 1007, 49,5 г, 193,3 ммоль), 4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолана (58,9 г , 232,0 ммоль) и КОАс (37,9 г, 386,6 ммоль) в диоксане (1,2 л) продували азотом в течение 20 минут. К реакционной смеси добавляли dppfPdCl2*ДХМ (7,8 г, 9,7 ммоль). Смесь продували азотом в течение еще 20 минут и нагревали с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры смесь фильтровали через слой флоризила (400 мл) и осадок на фильтре промывали 50% EtOAc/CH2Cl2 (1,5 л). Полученный фильтрат концентрировали при пониженном давлении с получением желтого масла, которое разбавляли гексаном (800 мл) и концентрировали в вакууме с получением пенистого твердого вещества желтого цвета. Перемешивание этого желтого твердого вещество с гексаном (800 мл) в течение 2 ч приводило к образованию белого твердого осадка. Белое твердое вещество собирали фильтрованием и сушили с получением (1R)-2,2,2-трифтор-1-[5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3-пиридил]этанола (Соединение 1008, 48,4 г): 1Н ЯМР (400 МГц, CDCl3) δ 8,97 (s, 1Н), 8,78 (s, 1H), 8,29 (s, 1H), 5,10-5,21 (m, 1H); 1,38 (s, 6H), 1,29 (s, 6Н).
[0055] Как показано на этапе 2-iii Схемы 2, раствор 2-хлор-6-(1-(2,2-дифторэтил)-1H-пиразол-4-ил)-4,7,7-триметил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (Соединение 1006, 42,0 г, 123,3 ммоль), (1R)-2,2,2-трифтор-1-[5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил) -3-пиридил]этанола (Соединение 1008, 46,7 г, 148,0 ммоль) и Na2CO3 ( 28,8 г, 271,3 ммоль) в ДМФА (630 мл) и воды (210 мл) продували азотом в течение 30 минут. К этой смеси добавляли dppfPdCl2*ДХМ (2,99 г, 3,699 ммоль) и полученную смесь продували азотом в течение еще 30 минут. Реакционную смесь нагревали до 103°C и перемешивали в течение 2 часов. Смесь охлаждали до комнатной температуры и разбавляли водой (2 л). Водную фазу дважды экстрагировали EtOAc (1 л). Объединенные органические фазы концентрировали в глубоком вакууме для удаления ДМФА. Остаток разбавляли EtOAc и промывали водой с последующей промывкой рассолом. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный остаток очищали с помощью хроматографии на силикагеле при среднем давлении (с градиентом 0-100% EtOAc/гексаны с использованием 1500 г колонки Teledyne ISCO) с получением 54 г целевого продукта в виде светло-красного пенообразного твердого вещества. Твердое вещество растворяли в дихлорметане, пропускали через слой флоризила (200 мл), который последовательно промывали смесями EtOAc/CH2Cl2, [первая 40% (1 л), затем 60% (1 л) и затем 80% (1 л)]. Фильтраты объединяли и концентрировали в вакууме. Остаток дважды разбавляли гептанами (400 мл) и концентрировали в вакууме с получением осадка, который затем был промывали МТБЭ для удаления светло-желтого цвета и сушили в вакуумной печи при температуре 60°C в течение 4 суток для получения (R)-6-(1-(2,2-дифторэтил)-1Н-пиразол-4-ил)-4,7,7-триметил-2-(5-(2,2,2-трифтор-1-гидроксиэтил)пиридин-3-ил)-6,7-дигидро-5Н-пирроло[3,4-b]пиридин-5-она (Соединение 1, 47 г): 1Н ЯМР (400 МГц, CDCl3) δ 9,33 (s, 1H), 8,75 (s, 1H), 8,54 (s, 1H), 8,01 (s, 1H), 7,75 (s, 1H), 7,62 (s, 1Н), 6,14 (tt, J=55,4, 3,6 Гц, 1H), 5,28-5,11 (m, 1Н), 4,51 (td, J=13,5, 4,2 Гц, 2H), 4,33 (d, J=4,6 Гц, 1Н), 2,82 (s, 3H), 1,68 (s, 6H).
Активность in vitro в отношении липидкиназы PI3K-гамма
Пример 3. Анализ ингибирования PI3K
[0056] При помощи станции Biomek FX (Beckman Coulter) 1,5 мкл каждого из десяти 2,5-кратных серийных разведений химического соединения настоящего изобретения в 100% ДМСО добавляли в отдельные лунки (далее «тест-лунки») в 96-луночном полистирольном планшете [Corning, Costar артикул No.3697]. Одна тест-лунка также содержала 1,5 мкл ДМСО без химического соединения. Другая лунка содержала ингибитор в ДМСО в концентрации, о которой известно, что она полностью ингибирует фермент (далее «фоновая лунка»). При помощи дозатора Titertek Multidrop в каждую лунку добавляли 50 мкл реакционной смеси [100 мМ HEPES pH 7,5, 50 мМ NaCl, 10 mM ДТТ, 0,2 мг/мл БСА, 60 мкМ фосфатидилинозитол(4,5)-бисфосфат diC16 (PI(4,5)P2; Avanti Polar Lipids, Кат. No.840046P) и соответствующая изоформа PI3K (см. Таблицу 1 для концентраций изоформ)]. Для инициирования реакции в каждую лунку добавляли 50 мкл смеси АТФ [20 мМ MgCl2, 6 мкМ АТФ (100 мкКи/мкмоль 33P-АТФ)] с последующей инкубацией лунок в течение 30 минут при температуре 25°C. Конечная концентрация в каждой лунке составляла 50 мМ HEPES 7,5, 10 мМ MgCl2, 25 мМ NaCl, 5 mM ДТТ, 0,1 мг/мл БСА, 30 мкМ PI(4,5)P2, 3 мкМ АТФ и и соответствующая изоформа PI3K (см. Таблицу 2). Конечная концентрация химического соединения в каждой лунке находилась в диапазоне от 10 мкМ до 1 нМ.
|
[0057] После инкубации реакции в каждой лунке останавливали путем добавления 50 мкл останавливающего раствора [30% ТХУ/Вода, 10мМ АТФ]. Каждую остановленную реакционную смесь затем переносили в 96-луночный планшет со стекловолоконным фильтром [Corning, Costar артикул No.3511]. Планшет подвергали вакуумной фильтрации и три раза промывали 150 мкл 5% ТХУ/Вода в модифицированном автоматическом устройстве для отмывки планшетов ELX-405 (Bio-Tek). В каждую лунку добавляли по 50 мкл сцинтилляционной жидкости и анализировали планшет на жидкостном сцинтилляционном счетчике Perkin-Elmer TopCount™ NXT для получения 33P-значений, соответствующих значениям ингибирования.
[0058] Значение, полученное для фоновой лунки, вычитали из значения, полученного для каждой тест-лунки, и данные использовали в уравнении конкурентной прочности связывания Ki, описанном в Morrison and Stone, Comments Mol. Cell Biophys. 2: 347-368, 1985. Степень ингибирования PI3K-гамма для Соединения 1 изменялась линейно с концентрацией АТФ, демонстрируя конкурентное ингибирование с значение Ki 8±4 нМ. Кроме того, селективность в отношении изоформы также наблюдалась, когда Соединение 1 тестировалось в отношении изоформ PI3K-альфа, PI3K-бета и PI3K-дельта, и было показано, что это химическое соединение обладает в 15 раз большей селективностью по отношению к PI3K-гамма, как показано в Таблице 2.
|
Клеточная активность
[0059] PI3Ks представляют собой мультисубъединичные комплексы, состоящие из регуляторной субъединицы и каталитической субъединицы. Этот класс ферментов катализирует фосфорилирование фосфатидилинозитол-4,5-бифосфата (PIP2) с образованием второй мессенджера фосфатидилинозитол-3,4,5-трифосфата (PIP3). Активация рецепторов приводит к транзиторному повышению уровней PIP3. PIP3 выступает в качестве сайта прикрепления на плазматической мембране, рекрутинга и активации содержащих гомологичный плекстрину (PH) домен белков, таких как Akt, PDK-1, Tek киназы и т.д. Затем они регулируют ключевые клеточные функции, такие как рост, метаболизм, миграция, окислительный взрыв и т.д. Так как нижележащие эффекторы являются общими в PI3K сигнальном пути, то эти рецепторы определяют какая изоформа PI3K рекрутируется после активации. Исходя из этого, ряд основанных на биохимии pAkt и функциональных анализов был использован для анализа активности и селективности ингибиторов PI3K-гамма по сравнению с другими изоформами PI3K.
Пример 4. MCP-1 стимулированный pAkt в клетках THP-1
[0060] Хемокины, такие как MCP-1, связываются со своими рецепторами, что приводит к активации PI3K-гамма сигнального пути. PI3K-гамма вызывает образование PIP3 и активации нижележащих молекул, таких как PDK-1 и Akt. Фосфорилирование Akt является показателем активности PI3K в клетке. В этом анализе клетки ТНР-1 (клеточная линия моноцитов человека) выдерживали на обедненной среде в течение ночи, чтобы истощить уровень pAkt. Последующая стимуляция MCP-1 в течение 3 минут приводила к индуцированному PI3K-гамма фосфорилированию Akt в положениях треонин 308 и серин 473. Клетки фиксировали и окрашивали для выявления внутриклеточного фосфорилированного Akt (Сер473), а затем анализировали с использованием цитометра BD FACSCalibur™, в результате получая показатель клеточной активности PI3K-гамма. Соединение 1 имело в этом анализе ИК50 0,24±0,07 мкМ. См. Таблицу 3.
|
Пример 5. Анализ окислительного взрыва в цельной крови и лейкоцитах
[0061] Нейтрофилы, моноциты и макрофаги являются ключевыми медиаторами врожденного иммунитета. Они высвобождают активные формы кислорода (АФК), как часть врожденного иммунного воспалительного ответа. Синтез АФК индуцируется медиаторами воспаления, такими как хемокины, бактериальные пептиды и комплемент, которые рекрутируют клетки врожденного иммуннитета к месту воспаления. Эти медиаторы воспаления вызывают активацию PI3K-гамма и инициируют нисходящий сигнальный каскад, который приводит к сборке полного НАДФH оксидазного комплекса на плазматической мембране, синтезирующего АФК. Функциональную активность PI3K-гамма измеряли в цельной крови и в нейтрофилах и моноцитах лейкоцитарной пленки по образованию АФК. Клетки стимулировали ФНО-альфа в течение 10 минут, а затем полученным из бактериальной клеточной стенки хемотактическим пептидом fMLP в течение 20 минут, и нагружали нефлуоресцентным красителем дигидрородамин 1,2,3 (ДГР). Продуцируемые клетками АФК окисляли ДГР с образованием флуоресцентного родамина, вызывая увеличение содержания флуоресцентного родамина в клетках. Функциональную активность PI3K-гамма измеряли по способности нейтрофилов и моноцитов синтезировать АФК после стимуляции fMLP. Лейкоциты и образцы цельной крови анализировали при помощи цитометра BD FACSCalibur™ для количественной оценки клеток, положительных по флуоресцентному родамину. ИК50s для клеток лейкоцитарной пленки определяли в отсутствии сыворотки, и ИК50s в анализе цельной крови определяли в присутствии сыворотки. Соединение 1 имело ИК50 0,22 мкМ в анализе лейкоцитов лейкоцитарной пленки и 0,57 мкМ в анализе цельной крови. См. Таблицу 3.
Пример 6. КСФ-1 стимулированный pAkt в клетках THP-1.
[0062] Факторы роста, цитокины и другие лиганды рецепторных тирозинкиназ, такие как КСФ-1, связываются со своими рецепторами, приводя к активации класса сигнального пути PI3K класса Ia. Активация PI3K приводит к образованию PIP3 и активации нижележащих молекул, таких как PDK-1 и Akt. Фосфорилирование Akt является показателем активности PI3K в клетке. Клетки ТНР-1 (клеточная линия моноцитов человека) выдерживали на обедненной среде в течение ночи, чтобы истощить уровень pAkt. Стимуляция КСФ-1 в течение 5 минут приводила к индуцированному PI3Kα/β/δ фосфорилированию Akt в положениях треонин 308 и серин 473. Клетки фиксировали и окрашивали для выявления внутриклеточного фосфорилированного Akt (Сер473), а затем анализировали с использованием цитометра BD FACSCalibur™. Это является показателем клеточного ингибирования PI3K класса Ia. Соединение 1 имело в этом анализе ИК50>9,7 мкМ, демонстрируя селективность в отношении PI3Ks класса Ia. См. Таблицу 4.
Пример 7. Анализ пролиферации B-клеток человека
[0063] В своем развитии и активности B-клетки сильно зависят от PI3Kδ. PI3K-гамма принадлежит существенная и и недублированная роль в передаче сигнала через комплекс B-клеточного рецептора (BCR). И индуцированный IgM поток ионов Сa+ и пролиферация являются ослабленными у мышей с отсутствующей PI3K-δ или с ингибиторами PI3K-δ. PI3K-гамма не играет никакой роли в какой-либо активности B-клеток. Специфичность комплекса BCR по отношению к изоформе PI3K-гамма делает его идеальным для использования в анализе PI3K-гамма для оценки степени ингибирования PI3K-гамма в клетках. Очищенные B-клетки человека стимулировали анти-IgM в присутствии исследуемых химических соединений. Через четверо суток жизнеспособность/пролиферацию измеряли при помощи набора для определения жизнеспособности клеток CellTiter-Glo, измеряя содержание АТФ в клетках в лунке. Отсутствующая или уменьшенная пролиферация являлась показателем степени ингибирования PI3K-гамма. Соединение 1 имело ИК50 3,05±0,44 мкМ, демонстрируя 11-кратный интервал селективности в отношении PI3K-δ. См. Таблицу 4.
Пример 8. Анализ пролиферации ЭКПВЧ
[0064] PI3Ks являются важными сигнальными молекулами, расположенными в сигнальном каскаде ниже ряда факторов роста, которые регулируют выживание клеток, рост и фазы клеточного цикла. Обе изоформы PI3K-альфа и PI3K-бета регулируют фазы клеточного цикла; ингибирование любой из этих изоформ приводит к уменьшенному клеточному росту. ЭКПВЧ представляют собой первичные эндотелиальные клетки пупочной вены человека, которые экспрессируют обе изоформы PI3Kα и PI3Kβ. Ингибирование одной или обеих изоформ PI3K-альфа или PI3K-бета будет ингибировать клеточный рост ЭКПВЧ. Химические соединения наносили на ЭКПВЧ, клеточную пролиферацию/жизнеспособность измеряли через 96 часа после обработки химическим соединением при помощи набора для определения жизнеспособности клеток CellTiter-Glo, измеряя содержание АТФ в клетках в лунке. Отсутствующая или уменьшенная пролиферация являлась показателем степени ингибирования PI3K-альфа и/или PI3K-бета. Соединение 1 в этом анализе ИК50>19 мкМ, демонстрируя >74-кратную селективность в отношении PI3K-альфа/бета. См. Таблицу 4.
Пример 9. Анализ пролиферации MSF7
[0065] Путь передачи сигнала PI3K-Akt регулирует многие нормальные клеточные процессы, включая клеточную пролиферацию, выживание и рост, которые имеют решающее значение для онкогенеза. Нарушение регуляции сигнального пути PI3K, как правило, происходит у человека при раке. MCF7 представляет собой клеточную линию карциномы молочной железы человека, который имеет активирующую E545K гетерозиготную мутацию PI3Kα в спиральном домене белка p110α, приводящую к гиперактивности пути PI3K-альфа. Ингибирование сигнального пути PI3K-альфа в клетках MCF7 ингибирует рост клеток и, таким образом, ингибиторы PI3K-альфа можно оценивать в анализе пролиферации MCF7. Химические соединения добавляли к клеткам MCF7, клеточную пролиферацию/жизнеспособность измеряли через 96 часа после обработки химическим соединением при помощи набора для определения жизнеспособности клеток CellTiter-Glo, измеряя содержание АТФ в клетках в лунке. Отсутствующая или уменьшенная пролиферация являлась показателем степени ингибирования PI3K-альфа. Соединение 1 в этом анализе ИК50>17 мкМ, демонстрируя, таким образом, >67-кратную селективность в отношении PI3K-альфа. См. Таблицу 4.
[0066] Как показано выше, Соединение 1 обладает отличной эффективностью, 0,25 мкМ в PI3K-гамма ассоциированных клеточных анализах, имеет 12-кратную селективность по отношению к PI3K-дельта, и 40-80 кратную селективность по отношению к PI3K-альфа и PI3K-бета в анализах с использованием клеток.
|
Метаболизм лекарственных средств
Пример 10. Метаболизм Соединения 1 рекомбинантным ферментами CYP
[0067] Микросомы, содержащие рекомбинантные ферменты CYP (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 и 3A4) использовали для определения того, какие CYP катализирует окисление Соединения 1. Соответственно, Соединение 1 (1 мкМ) инкубировали с отдельными рекомбинантными CYPs в присутствии кофактора НАДФH. Процент исходного лекарственного вещества, оставшегося в конце периода инкубации, определяли с помощью ЖХМС/МС и сравнивали с процентом в начале. Результаты приведены в Таблице 5. Метаболизм Соединения 1 был обнаружен только при инкубации с CYP3A4, хотя с низкой эффективной скоростью.
|
Пример 11. Ингибирование ферментов CYP Соединением 1
[0068] Оценивали способность Соединения 1 (от 0,01 до 100 мкМ) обратимо ингибировать ферменты CYP (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 и 3A4) в микросомах печени человека. Анализ ингибирования проводили с селективными зондами CYP для каждого фермента CYP при определенных концентрациях субстрата (близких к значению его константы диссоциации (Km)). Данные приведены в Таблице 6. Соединение 1 оказалось умеренным ингибитором CYP1А2, 2B6 и 2C8; Значения ИК50 находились в диапазоне от 4 до 7 мкМ.
|
[0069] Зависимое от времени ингибирование CYP3A4 соединением 1 также оценивали в микросомах печени человека с использованием анализа сдвига ИК50. В этом исследовании Соединение 1 в количестве от 0,1 до 50 мкМ предварительно инкубировали с микросомами печени человека в присутствии и в отсутствие НАДФH в течение периода времени 0 и 30 минут. Инкубируемую смесь затем разбавляли в 10 раз буфером и остаточную активность CYP3A4 (тестостерон-6-бета-гидроксилаза) измеряли в течение 10-минутного периода времени. Не было отмечено никаких существенных изменений (ИК50 сдвиг = 1) в ИК50 Соединения 1 в присутствии или в отсутствии НАДФH.
[0070] Этот результат был подтвержден в дополнительном исследовании, в котором оценивали время-зависимое ингибирование CYP3A4 в микросомах печени человека после предварительной инкубации с 10 или 50 мкМ Соединения 1 в течение периода времени 0, 5, 10, 15 и 30 минут. Наблюдаемая константа скорости для потери активности CYP3A4 в этом исследовании (Kobs) составляла 0,0039/мин при обеих испытанных концентрациях по сравнению с Kobs=0,053/мин для мифепристона, используемого в качестве положительного контроля. Данные, полученные в обоих этих исследованиях, показывают, что соединение 1 не ингибирует CYP3A4 время-зависимым образом.
Пример 12. Индукция ферментов
[0071] Способность соединения 1 активировать прегнан-Х-рецептор (PXR) оценивали в диапазоне шести концентраций от 0,1 до 30 мкМ в клетках DPX2 клеточной линии гепатомы человека, которая стабильно сверхэкспрессирует ген PXR человека и репортерный ген люциферазы, связанный с двумя промоторами в гене CYP3A4 человека. Рифампицин использовали в качестве положительного контроля. В этом анализе Соединение 1 показало незначительную степень активации, обеспечивая ответ, который составил 5% от положительного контроля со значением ЭК50>30 мкМ.
[0072] Способность соединения 1 индуцировать CYP1A2 и CYP3A4 также оценивали в гепатоцитах. Соединение 1 в концентрации 0,1-30 мкМ инкубировали с культивируемыми криоконсервированными первичными гепатоцитами человека от трех различных доноров в течение 48 часов и сравнивали с действием положительного контроля (т.е. индукторов CYP: 50 мкМ омепразола для CYP1A2 и 10 мкМ рифампицина для CYP3A4). Активность CYP определяли путем мониторинга образования метаболитов специфических субстратов-зондов CYP (фенацетин для CYP1A2 и тестостерона для CYP3A4) при помощи ЖХМС/МС. Матричную рибонуклеиновую кислоту CYP (мРНК) анализировали при помощи ОТ-ПЦР для подтверждения CYP-индуцирующей активности. Результаты показаны в Таблице 7.
[0073] Изменение активности CYP1A2 или CYP3A4 и уровней мРНК во всех трех образцах гепатоцитов после воздействия Соединения 1 составляло менее 20% от положительного контроля. Эти данные in vitro свидетельствуют о том, что Соединение 1 обладает низкой способностью индуцировать CYP1A2 или CYP3A4 в гепатоцитах человека после 48-часовой экспозиции.
|
Пример 13. Проницаемость потенциала выведения
[0074] Проницаемость Соединения 1 оценивали с использованием клеточных линий дикого типа Сасо-2 и клеток Мадин-Дарби почек собак (MDCK). Клетки подвергали воздействию препарата в буфере на апикальную сторону (измерение проницаемости в направлении от А к B) или базолатеральную сторону (измерение проницаемости в направлении от В к A), и инкубировали при температуре 37°C в течение одного часа. Результаты приведены в Таблице 7. Проницаемость была высокой в направлении от А к B в обеих клеточных линиях MDCK и Сасо-2 (33 и 18×106 см/сек соответственно).
[0075] Оценку того, является ли Соединение 1 субстратом эффлюксных транспортеров, проводили с использованием клеточной линии MDCK, сверхэкспрессирующей P-гликопротеин человека (MDR). В этой клеточной линии был обнаружен векторный транспорт (коэффициент выведения=35,1), указывающий на то, что Соединение 1 является субстратом гликопротеина-Р. См. Таблицу 8.
|
Фармакокинетика in vivo
Пример 14. Внутривенное болюсное введение
[0076] После внутривенного введения однократной болюсной дозы, Соединение 1 имело низкий системный клиренс и длительный период полувыведения у всех подопытных видов. См. Таблицу 9. Значения клиренса Соединения 1 составляют приблизительно 5,4%, 3,6%, 13% и 26% печеночного кровотока у мыши, крысы, собаки и обезьяны. Объем распределения был больше, чем общий объем воды в организме, что указывает на доставку Соединения 1 к тканям.
|
Пример 15. Пероральная биодоступность
[0077] Как показано в Таблице 10, пероральная биодоступность Соединения 1 была высокой (>80%) после введения однократной дозы Соединения 1 мыши, крысе, собаке и обезьяне. Биодоступность у обезьяны (более 100%) может быть объяснена десятикратной разницей между внутривенной и пероральной дозами. Соединение 1 быстро всасывается у всех видов с максимальной системной концентрацией, наблюдаемой в течение приблизительно от одного до трех часов после введения дозы.
|
[0078] В таблице 11 приведены фармакокинетические данные для ингибиторов PI3K, описанных в публикации международной патентной заявки No.WO2011/087776 (далее «заявка 776»), каждый из которых имеет ту же самую основную структуру фармакофора, что и Соединение 1. См. соединения 705, 709, 735 и 772 на страницах 229, 229, 234 и 242 заявки 776, соответственно. Системный плазменный клиренс Соединения 1 после внутривенного введения был значительно (от 2,5 раз до 8 раз) ниже, чем наблюдалось в исследованиях других химических соединений. Данные плазменного клиренса согласуются с данными внутривенного введения перорального введения, значениями ППК и Cmax. Эти данные показывают, что Соединение 1 имеет неожиданно благоприятный фармакокинетический профиль по сравнению с этими химическими соединениями.
|
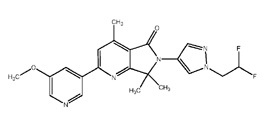
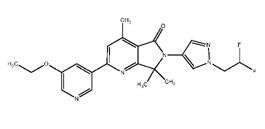
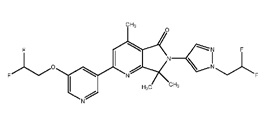
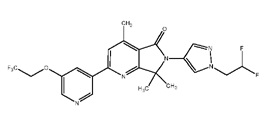
Пример 16. Распределение в тканях у крыс
[0079] Соединение 1 хорошо распределялось в большинстве тканей после перорального введения самцам крыс в дозе 5 мг/кг. См. Таблицу 12. Головной мозг и спинномозговая жидкость (СМЖ) оказались органами, испытавшими очень низкое воздействие Соединения 1 с Cmax 111 нг/г и 27 нг/мл, соответственно (<0,1% от концентрации в плазме крови). Соединение 1 хорошо распределялось в печени и почках с Cmax 11400 и 4770 нг/г, соответственно. Отношение концентрации в ткани к концентрации в плазме крови имело следующую тенденцию: печень>почки>сердце>легкие>селезенка>головной мозг>СМЖ. Кинетика выведения Соединения 1 в каждом исследуемом органе следовала за кинетикой в плазме. Не было получено никаких данных, свидетельствующих о накоплении Соединения 1 в тканях после введения однократной дозы.
|
Пример 17. Соединение 1 в мышиной модели КИА
[0080] Соединение 1 испытывали в терапевтической мышиной модели коллаген-индуцированного артрита (КИА) в количестве 2,5 мг/кг два раза в сутки (5 мг/кг/сутки), 5 мг/кг два раза в сутки (10 мг/кг/сутки) или 10 мг/кг два раза в сутки (20 мг/кг/сутки). Фостаматиниб, который представляет собой низкомолекулярный ингибитор Syk, используемый в качестве эталонного стандарта, перорально вводили в количестве 30 мг/кг два раза в сутки (60 мг/кг/сутки). Химические соединения вводили в течение 10 суток по схеме 12/12 часов два раза в сутки до конца исследования. Соединение 1 вводили с наполнителем 0,2% МКЦ, 1%ЛСН. Объем дозы составлял 10 мл/кг. Конечные образцы плазмы отбирали через 2, 4 и 12 часов после последней дозы. Все четыре лапы и оба колена брали и обрабатывали для гистопатологии.
[0081] Лечение Соединением 1 оказало значительное благотворное действие в модели КИА, что было установлено путем оценки клинических показателей артрита и гистопатологии суставов. Ежедневно измеряемые показатели артрита были значительно уменьшены в сторону нормальных значений у мышей, получавших 2,5 мг/кг Соединения 1 два раза в сутки (*d2-11), 5 мг/кг Соединения 1 два раза в сутки (*d2-11), 10 мг/кг Соединения 1 два раза в сутки (*d2-11) или 30 мг/кг Фостаматиниба два раза в сутки (*d2-11), по сравнению с вводимым в качестве контроля наполнителем. См. Фигуру 1. При рассмотрении только тех лап, которые имели клинические признаки артрита при включении в эксперимент (подлежащие лечению лапы), клинические показатели артрита были значительно уменьшены у мышей, получавших 5 мг/кг Соединения 1 два раза в сутки (*d5-11) или 10 мг/кг Соединения 1 два раза в сутки (*d2-11), но не в группе, получавшей 30 мг/кг Фостаматиниба два раза в сутки. См. Фигуру 2. При рассмотрении только тех лап, которые не имели клинических признаков артрита при включении в эксперимент (подлежащие профилактике лапы), клинические показатели артрита были значительно уменьшены у мышей, получавших 2,5 мг/кг Соединения 1 два раза в сутки (*d2, 4-11), 5 мг/кг Соединения 1 два раза в сутки (*d2-11), 10 мг/кг Соединения 1 два раза в сутки (*d2-11) или 30 мг/кг Фостаматиниба два раза в сутки (*d5-9), по сравнению с вводимым в качестве контроля наполнителем. См. Фигуру 3.
[0082] Как показано в Таблице 13, клинические показатели артрита, выраженные в виде площади под кривой (ППК), были значительно уменьшены в сторону нормальных значений у мышей, получавших 30 мг/кг Фостаматиниба два раза в сутки (25%), 2,5 мг/кг Соединения 1 два раза в сутки (28%), 5 мг/кг Соединения 1 два раза в сутки (63%) или 10 мг/кг Соединения 1 два раза в сутки (89%), по сравнению с вводимым в качестве контроля наполнителем. При рассмотрении только подлежащих лечению лап, ППК клинических показателей артрита были значительно уменьшены у мышей, получавших 10 мг/кг Соединения 1 два раза в сутки (79%). При рассмотрении только подлежащих профилактике лап, ППК клинических показателей артрита были значительно уменьшены у мышей, получавших 30 мг/кг Фостаматиниба два раза в сутки (32%), 2,5 мг/кг Соединения 1 два раза в сутки (42%), 5 мг/кг Соединения 1 два раза в сутки (81%) и 10 мг/кг Соединения 1 два раза в сутки (98%), по сравнению с вводимым в качестве контроля наполнителем.
|
Пример 18. Соединение 1 в мышиной модели ВЗК
[0083] Ингибитор ΡΙ3Κγ Соединение 1 испытывали в модели CD40 индуцированного колита, чтобы определить его влияние на течение этого заболевания. CD40 модель ВЗК индуцируется путем введения анти-CD40 моноклональных антител (агонистов, т.е. активирующих антител против нейтрализующих антител) мышам с дефицитом T- и B-клеток (Rag 1-/- мыши), чтобы индуцировать как системное, так и кишечное воспаление, приводящее к колитам и изнуряющей болезни через путь врожденного иммунитета. (См., например, Immunity 25, 309-318, August 2006). В кратком изложении, нокаутным мышам Rag1 внутрибрюшинно вводили анти-CD40 моноклональное антитело FGK45. Начиная с дня 0 мыши получали ФСБ, наполнитель или Соединение 1 в количестве 5 мг/кг два раза в сутки или 10 мк/кг два раза в сутки. Соединение 1 вводили внутрибрюшинно в течение 7 дней по схеме введения 10/14 часов два раза в сутки. Соединение 1 вводили в составе композиции, содержащей Соединение 1/5%, НМП/15%, ПЭГ400/80% в 0,5% растворе ГПМЦ-E50 в воде. По окончании исследования образцы сыворотки крови и толстой кишки отбирали через 2 часа после введения последней дозы для анализа концентрации лекарственного препарата. Массу тела измеряли ежедневно в ходе исследования. Концентрации Соединения 1 анализировали в образцах плазмы крови, отобранных через 2 часа после введения последней дозы в конце исследования. Концентрации определяли при помощи метода высокоэффективной жидкостной хроматографии с тандемной масс-спектрометрией (ВЭЖХ/МС/МС).
[0084] У мышей, которым вводили анти-CD40 моноклональные антитела, и которые получали носитель или ФСБ, наблюдалась потеря массы тела (измеряемая как процентное изменение относительно исходного значения), которая начиналась в день 1 и достигала своего пика в день 3-4 перед восстановлением в направлении исходных значений. Как показано на Фигуре 4, вызванная болезнью потеря массы тела составляла 17,2% и 17% в дни 3 и 4, соответственно, в группе, получавшей ФСБ, и 12,8% и 12,6% в дни 3 и 4, соответственно, в группе, получавшей наполнитель. На Фигуре 4 также показано, что потеря массы тела значительно ингибировалась у мышей, получавших Соединение 1 в количестве 5 мг/кг два раза в сутки (*p<0,05 d3, **p<0,01 d4) или 10 мг/кг два раза в сутки (***p<0,001 d3-d4), по сравнению с вводимым в качестве контроля наполнителем.
|
[0085] Несмотря на то, что настоящее изобретение было подробно описано путем приведения иллюстраций и примеров для ясности понимания, специалистам в данной области техники в свете идеи настоящего изобретения будет очевидно, что в него могут быть внесены определенные изменения и модификации без отступления от сущности и объема прилагаемой формулы изобретения.
![6,7-ДИГИДРО-5Н-ПИРРОЛО[3,4-b]ПИРИДИН-5-ОН, ПОЛЕЗНЫЙ В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ-ГАММА](https://fips.edrid.ru/images/rid/44/3d/7f/40ff7fc981c34a0d526e74b564edd40c.png)
![6,7-ДИГИДРО-5Н-ПИРРОЛО[3,4-b]ПИРИДИН-5-ОН, ПОЛЕЗНЫЙ В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ-ГАММА](https://fips.edrid.ru/images/rid/44/3d/7f/4d4bb3107f22027c90b6acc5b6c4f3ec.png)
![6,7-ДИГИДРО-5Н-ПИРРОЛО[3,4-b]ПИРИДИН-5-ОН, ПОЛЕЗНЫЙ В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ-ГАММА](https://fips.edrid.ru/images/rid/44/3d/7f/50ed7f7c4b8e4f4427b9436ade3bd52c.png)
![6,7-ДИГИДРО-5Н-ПИРРОЛО[3,4-b]ПИРИДИН-5-ОН, ПОЛЕЗНЫЙ В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ-ГАММА](https://fips.edrid.ru/images/rid/44/3d/7f/ce187a4d1060275bc0f992f623d893b7.png)
![6,7-ДИГИДРО-5Н-ПИРРОЛО[3,4-b]ПИРИДИН-5-ОН, ПОЛЕЗНЫЙ В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ-ГАММА](https://fips.edrid.ru/images/rid/44/3d/7f/62512a3a8e6cc471baea9e24ca76b46b.jpg)
![6,7-ДИГИДРО-5Н-ПИРРОЛО[3,4-b]ПИРИДИН-5-ОН, ПОЛЕЗНЫЙ В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ-ГАММА](https://fips.edrid.ru/images/rid/44/3d/7f/61cfb9b7636e116634a97dae464b09b7.jpg)
Дигидродиазепины, которые можно использовать в качестве ингибиторов протеинкиназ
Способы и промежуточные соединения для получения стерических соединений
Модуляторы атф-связывающих кассетных транспортеров
Твердые формы n-(7-азабицикло[2.2.1]гептан-7-ил-)-2-(трифторметил)фенил)-4-оксо-5-(трифторметил)-1,4-дигидрохинолин-3-карбоксамида
Производные 3-карбоксамида-4-оксохинолина, полезные в качестве модуляторов регулятора трансмембранной проводимости кистозного фиброза
Способ модуляции транспортеров атф-связывающей кассеты
Модуляторы транспортеров атф-связывающей кассеты
Способы и промежуточные соединения
Способ получения азабициклических соединений
Способ получения модуляторов регулятора трансмембранной проводимости кистозного фиброза
Ингибиторы фосфатидилинозитол-3-киназы на основе изоиндолинона
Пиримидиновые ингибиторы гиразы и топоизомеразы iv
Ингибиторы днк-пк
Азаиндолы, полезные в качестве ингибиторов jak и других протеинкиназ

