Результат интеллектуальной деятельности: СПИРО-КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛИЕВОГО КАНАЛА НАРУЖНОГО МЕДУЛЛЯРНОГО СЛОЯ
Вид РИД
Изобретение
Уровень техники
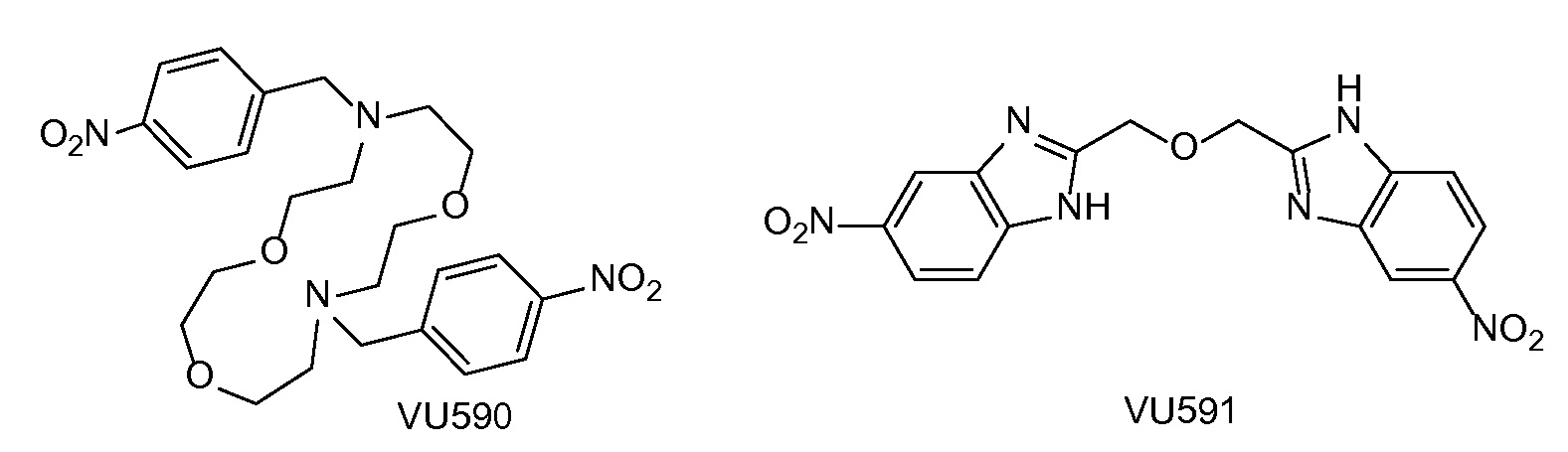
Калиевый (ROMK) канал (Kir1.1) наружного медуллярного слоя (см., например, публикацию Ho K. и др. "Cloning and expression of an inwardly rectifying ATP-regulated potassium channel", Nature, 1993, 362(6415): стр. 31-8.1, 2; и публикацию Shuck M.E. и др. "Cloning and characterization of multiple forms of the human kidney ROM-K potassium channel", J. Biol. Chem., 1994, 269(39): стр. 24261-70) является членом семейства калиевых каналов входящего выпрямления, экспрессированных в двух областях почек: толстой восходящей части петли Генле (TALH) и кортикальном собирающем протоке (CCD) (см. публикацию Hebert S.C. и др. "Molecular diversity and regulation of Renal potassium channels", Physiol. Rev., 2005, 85(1): стр. 319-713). В TALH (толстой восходящей части петли Генле) ROMK участвует в рециркуляции калия через люминальную мембрану, которая является решающей для функционирования котранспортера Na+/K+/2C1- стадией, определяющей скорость обратного захвата соли в этой части нефрона. В CCD ROMK обеспечивает путь секреции калия, который тесно связан с поглощением натрия через амилорид-чувствительный натриевый канал (см. публикацию Reinalter S.C. и др. "Pharmacotyping of hypokalaemic salt-losing tubular disorders", Acta Physiol. Scand., 2004, 181(4): стр. 513-21; и публикацию Wang W. "Renal potassium channels: recent developments", Curr Opin Nephrol Hypertens, 2004, 13(5): стр. 549-55). Как ожидается, селективные ингибиторы канала ROMK (также упоминаемые здесь как ингибиторы ROMK или ROMK-ингибиторы) представляют собой новые диуретические средства для лечения гипертонии и других состояний, лечению которых могли бы способствовать диуретические средства с потенциально меньшими недостатками (то есть, для лечения гипокалиемии или гиперкалиемии, впервые выявленного диабета, дислипидемии), по сравнению с применяемыми в настоящее время клиническим средствами (см. публикацию Lifton R.P., A.G. Gharavi и D.S. Geller "Molecular mechanisms of human hypertension", Cell, 2001, 104(4): стр. 545-56). Такие ожидания опираются на генетику человека (см. публикацию Ji W. и др. "Rare independent mutations in renal salt handling genes contribute to blood pressure variation", Nat Genet, 2008, 40(5): стр. 592-9; и публикацию Tobin M.D. и др. "Common variants in genes underlying monogenic hypertension and hypotension and blood pressure in the general population", Hypertension, 2008, 51(6): стр. 1658-64) и генетическую абляцию ROMK у грызунов (см. публикацию Lorenz J.N. и др. "Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter's syndrome", J. Biol. Chem., 2002, 277(40): стр. 37871-80 и публикацию Lu M. и др. "Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter's) knockout mice", J. Biol. Chem., 2002, 277(40): стр. 37881-7). К сведению, впервые об открытии селективных низкомолекулярных ингибиторов ROMK, включая VU590, публично сообщалось в работе, осуществленной в университете Вандербилта, как описано в публикации Lewis L.M. и др., "High-Throughput Screening Reveals a Small-Molecule Inhibitor of the renal Outer Medullary potassium channel and Kir7.1", Mol. Pharmacol., 2009, 76(5): стр. 1094-1103. Позже о соединении VU591 сообщалось в публикации Bhave G. и др. "Development of a Selective Small-Molecule Inhibitor of Kir1.1, the Renal Outer Medullary Potassium Channel", Mol. Pharmacol., 2011, 79(1), стр. 42-50, в тексте которой утверждается, что "ROMK (Kir1.1) является мишенью для гипотетического лекарственного средства нового класса петлевых диуретиков, которые могут снижать кровяное давление, не вызывая гипокалиемии."
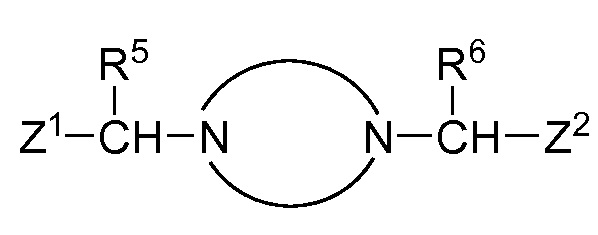
В публикации патентной заявки WO 2010/129379, опубликованной 11 ноября 2010, компанией Merck Sharp & Dohme Corp. в качестве представителя (также опубликована как патентная заявка США 2010/0286123 на ту же дату), описаны ингибиторы ROMK общей формулы:
и, например, вариант осуществления изобретения
 ,
,
в котором R5 и R6 независимо представляют собой -H, -Cl-6-алкил, -C3-6- циклоалкил, -CF3, -CHF2, -CH2F или -CH2OH; X представляет собой -H, -OH, -OCl-3-алкил, -F, оксо, NH2 или -CH3; и X1 представляет собой -H или -CH3.
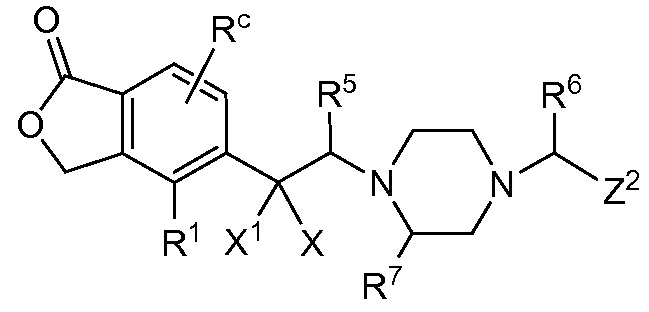
В публикации патентной заявки WO 2012/058134, опубликованной 3 мая 2012 г, компанией Merck Sharp & Dohme Corp. в качестве общего представителя описаны ингибиторы ROMK общей формулы:
 ,
,
в которой A и B представляют собой моно и/или бициклические ароматические группы; R2 представляет собой -H, -Cl-6-алкил, -C3-6-циклоалкил, CF3, -CH2OH или -CO2R; или R2 может объединяться с R1 или R10а с образованием кольца; R3 представляет собой -H, -Cl-6-алкил, -C3-6-циклоалкил, -OH, -F, -OCl-3-алкил или -CH2OH; или R3 может объединяться с R10b с образованием кольца.
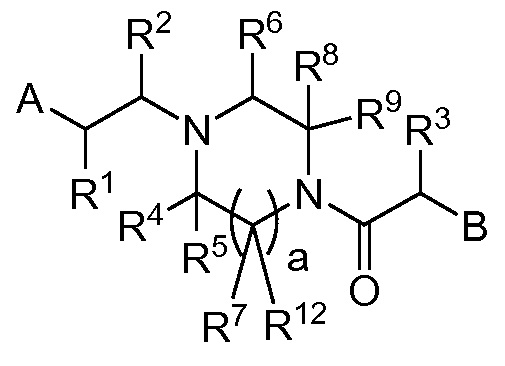
В публикации патентной заявки WO 2012/058116, опубликованной 3 мая 2012 г., компанией Merck Sharp & Dohme Corp. в качестве общего представителя описаны ингибиторы ROMK общей формулы:
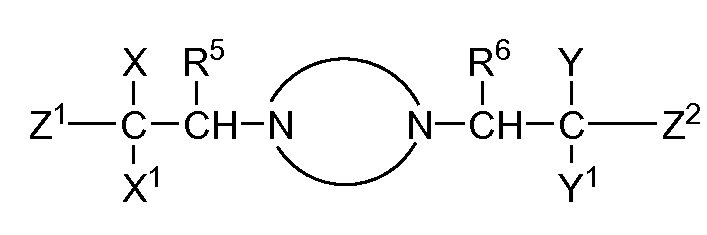
и, например, вариант осуществления изобретения
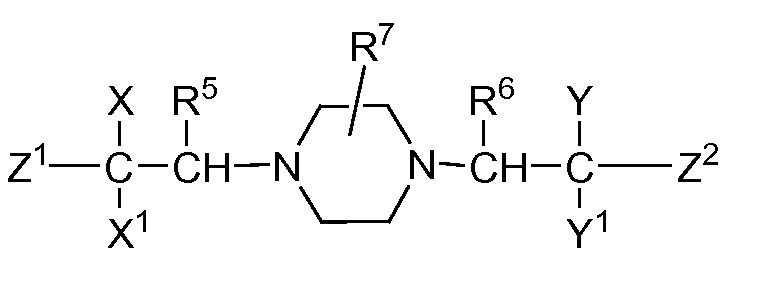 ,
,
в которых
R5 и R6 независимо представляют собой -H, -Cl-6-алкил или -C(О)OC1-3-алкил; и X, X1, Y и Y1 независимо представляют собой -H или -Cl-6-алкил; или Yl может объединяться с Z2 с образованием конденсированной циклической системы.
Однако продолжающееся исследование селективных низкомолекулярных ингибиторов ROMK все еще необходимо для разработки новых способов лечения гипертонии, сердечной недостаточности, отечных состояний и связанных расстройств. Соединения формулы I и их соли согласно настоящему изобретению являются селективными ингибиторами ROMK-канала и могут применяться для лечения гипертонии, сердечной недостаточности и других состояний, лечению которых способствуют диуретические или натрийуретические средства.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает соединения формулы I
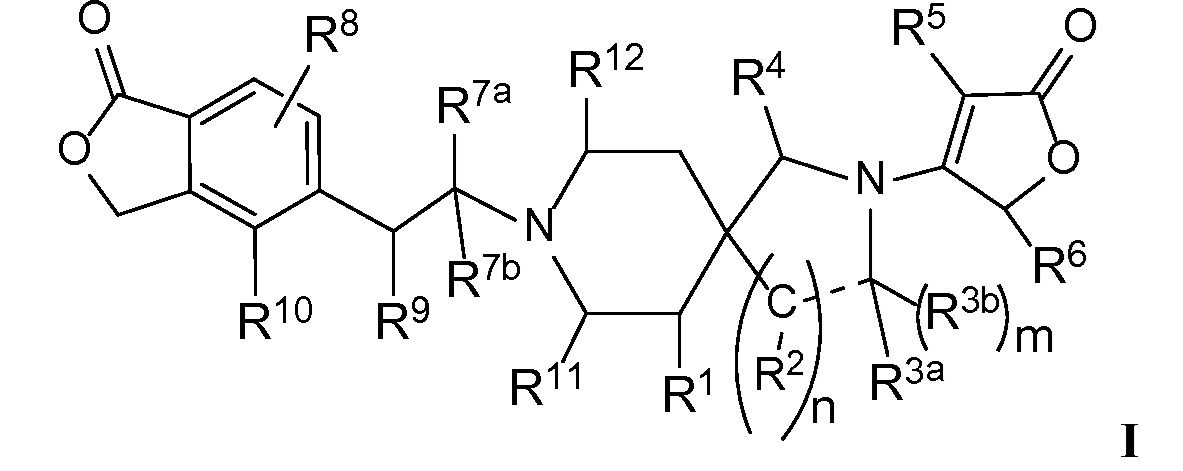
и их фармацевтически приемлемые соли. Соединения формулы I являются ингибиторами ROMK-канала (Kir1.1). В результате соединения формулы I могут применяться в способах лечения, ингибирования или улучшения одного или нескольких болезненных состояний, которые могут улучшаться благодаря ингибированию ROMK. Соединения согласно настоящему изобретению могут применяться в способах лечения, которые включают введение терапевтически или профилактически эффективного количества соединения формулы I пациенту, нуждающемуся в диуретическом и/или натрийуретическом средстве. Следовательно, соединения формулы I могут представлять собой полезные фармацевтически активные соединения для терапии, профилактики (или того и другого) медицинских состояний, включая, но не ограничиваясь перечисленным, сердечно-сосудистые заболевания, такие как гипертония и сердечная недостаточность, а также хроническое заболевание почек и состояния, ассоциированные с избытком соли и задержкой жидкости в организме. Соединения согласно настоящему изобретению дополнительно можно применять в комбинации с другими терапевтически эффективными средствами, включая но, не ограничиваясь перечисленным, другие лекарственные средства, которые применимы для лечения гипертонии, сердечной недостаточности и состояний, ассоциированных с избытком соли и задержкой жидкости в организме. Кроме того, изобретение относится к способам получения соединений формулы I и фармацевтических композиций, которые содержат соединения формулы I. Эти и другие аспекты изобретения будут очевидны из содержащегося в настоящем документе описания.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям следующей структурной формулы I:
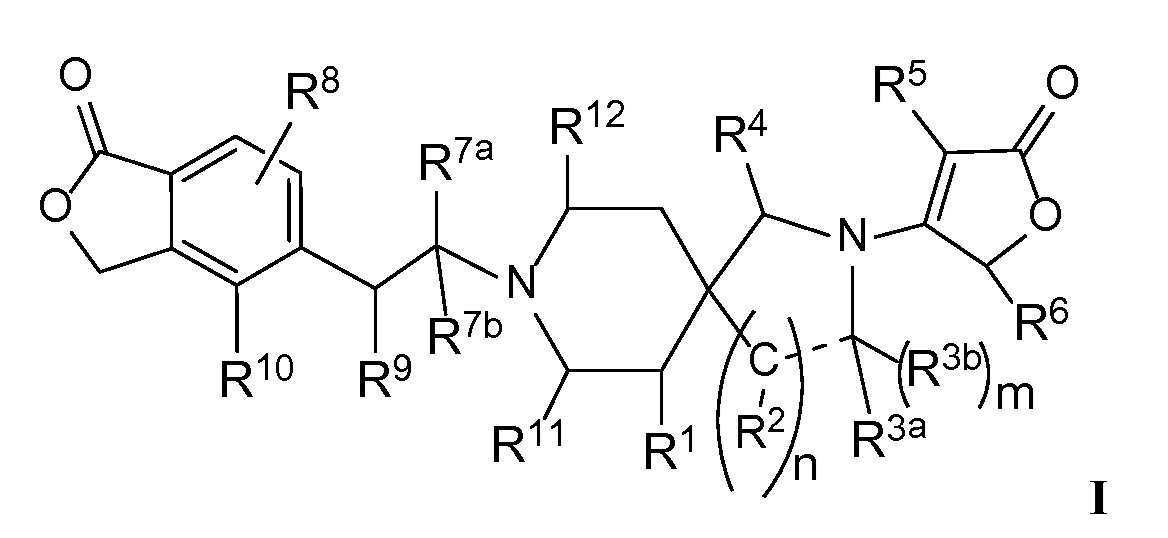
или их фармацевтически приемлемым солям, в которой:
R1 представляет собой -H, галоген, в частности -F, -OH или -OC1-3-алкил, в частности -OCH3;
m равно целому числу, выбранному из нуля (в случае отсутствия R3b) и 1 (в случае присутствия R3b);
n равно целому числу, выбранному из 1 или 2;
R2 в каждом случае независимо выбран из -H, =О (оксо), -OH, -Cl-3-алкила или -OCl-3-алкила, при условии, что когда n равно 2, то, по меньшей мере, один R2 представляет собой -H;
R3a представляет собой -H, =О, -C3-4-циклоалкил или -Cl-3-алкил, необязательно замещенный -OCH3 или 1 - 3 атомами -F, при условии, что только один из R2 или R3a может представлять собой =О;
R3b представляет собой -H или -Cl-3-алкил, или R3b отсутствует, когда R3а представляет собой =О или, когда обозначенная пунктиром связь представляет собой двойную связь или ароматическую связь;
или R3a и R3b объединяются вместе с атомом углерода, к которому они оба присоединены, с образованием циклопропила или циклобутила;
или когда n равно 1, R2 и R3a могут объединяться вместе с атомами углерода, к которым каждый из них присоединен, с образованием (1) фенильного кольца, которое конденсировано с пирролидиновым кольцом, и m, равным нулю, или с образованием (2) циклопропильного кольца, конденсированного с пирролидиновым кольцом, и m, равным 1;
R4 представляет собой -H или =О;
R5 представляет собой (a) -H, (b) галоген, в частности -Cl или -F, (c) -Cl-3-алкил, необязательно замещенный -О-Cl-3-алкилом, (d) -C3-6-циклоалкил или (e) гетероцикл, необязательно замещенный -Cl-3-алкилом или галогеном, в частности -F или -Cl;
R6 представляет собой -H или -Cl-3-алкил;
R7а представляет собой -H или -Cl-3-алкил, необязательно замещенный -OH, -OCH3 или 1-3 атомами -F;
R7b представляет собой -H или -Cl-3-алкил;
или R7a и R7b объединяются вместе с атомом углерода, к которому они оба присоединены, с образованием -C3-4-циклоалкила;
R8 представляет собой -H, галоген, в частности -F, или -Cl-3-алкил;
R9 представляет собой -H, -F, -OH, -ОCl-3-алкил, -CН2OH, -NH- R13 или 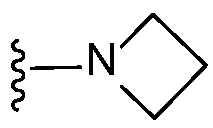 ;
;
R10 представляет собой -H, галоген, -CN, -C3-4-циклоалкил, или -Cl-3-алкил, необязательно замещенный 1-3 атомами -F;
или R9 представляет собой -O- и объединяется вместе с R10, чтобы представлять собой -CH2-CH2-O-;
R11 представляет собой -H, -CH2OH, -CH2OCH3 или -Cl-3-алкил, необязательно замещенный 1-3 атомами-F;
Rl2 представляет собой -H, -CH2OH, -CH2OCH3 или -Cl-3-алкил, необязательно замещенный 1-3 атомами-F;
или Rll и Rl2 объединяются вместе, чтобы представлять собой -CH2-CH2-, -CH2-N(CH3)-CH2- или -CH2OCH2-;
Rl3 представляет собой -H, -(CH2)0-2-C3-6-циклоалкил, -(CH2)1-2-OC3-6-циклоалкил, -(CH2)1-2-OC1-3-алкил, -(CH2)1-2-CN, -C(О)OC1-3-алкил, -SO2CH3 или -Cl-3-алкил, необязательно замещенный одним-тремя атомами -F; и
обозначенная пунктиром связь ("--") представляет собой одинарную, двойную или ароматическую связь, при условии, что
(A) когда n равно 2, то обозначенная пунктиром связь представляет собой одинарную связь, и m равно 1; и
(B) когда n равно 1 и
(i) m равно 1 (включая, но не ограничиваясь соединениями, в которых R2 и R3a объединяются, чтобы представлять собой циклопропил, конденсированный с пирролидиновым кольцом), или
(ii) R3a представляет собой =О, и m равно нулю,
то обозначенная пунктиром связь представляет собой одинарную связь; и
(C) когда n равно 1, m равно нулю, R2 не представляет собой =О и R3a не представляет собой =О, обозначенная пунктиром связь представляет собой
(i) двойную связь или
(ii) ароматическую связь, когда R2 и R3a объединяются вместе с образованием фенильного кольца, конденсированного с пирролидиновым кольцом.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, имеющим структурную формулу II, и их фармацевтически приемлемым солям:
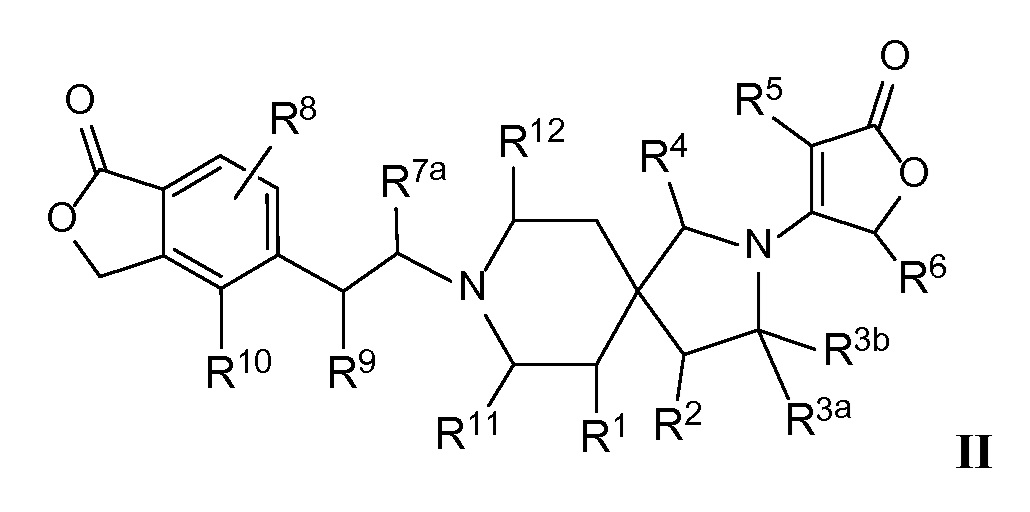
в которой n равно 1, и m равно 1, и каждая из переменных Rl, R2, R3a, R3b, R4, R5, R6, R7a, R8, R9, R10, Rl1, Rl2 и Rl3 и все другие переменные в данной формуле имеют такие же значения, которые указаны в формуле I.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, имеющим структурную формулу III, и их фармацевтически приемлемым солям:
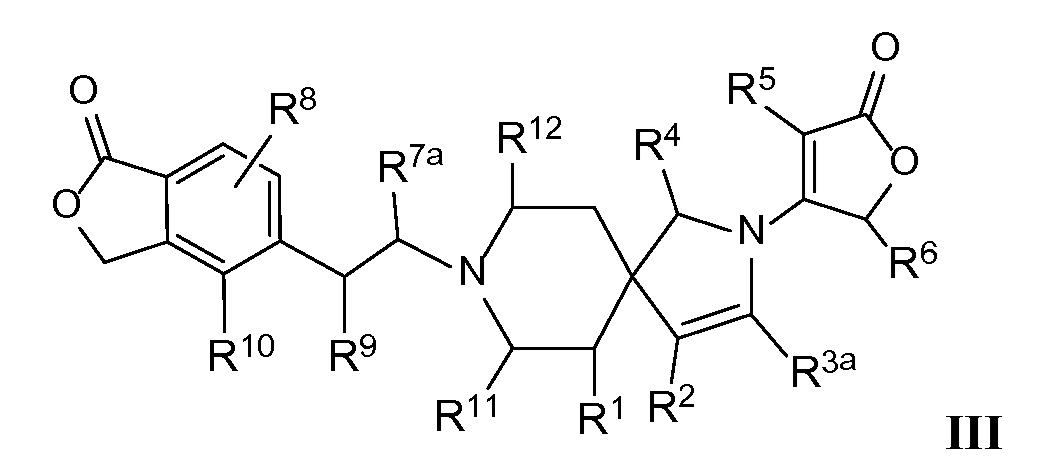
в которой n равно 1, и m равно нулю, и каждая из переменных Rl, R2, R3a, R4, R5, R6, R7a, R8, R9, R10, R11, Rl2 и Rl3 и все другие переменные в данной формуле имеют такие же значения, которые указаны в формуле I; и в которой двойная связь между R2 и R3a представляет собой неароматическую двойную связь или ароматическую связь, когда R2 и R3a объединяются вместе с атомами углерода, к которым каждый из них присоединен, с образованием фенильного кольца.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, имеющим структурную формулу IV и их фармацевтически приемлемым солям:
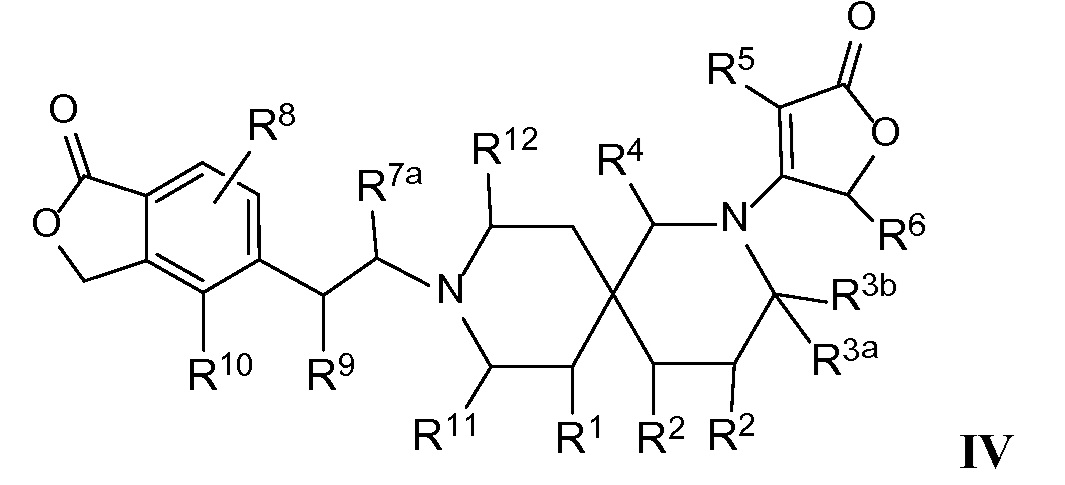
в которой n равно 2, и m равно 1, и каждая из переменных Rl, R2, R3a, R3b, R4, R5, R6, R7a, R8, R9, R10, Rl1, Rl2 и Rl3 и все другие переменные в данной формуле имеют такие же значения, которые указаны в формуле I.
Еще один вариант осуществления настоящего изобретения относится к соединениям формулы I, имеющим структурную формулу V:
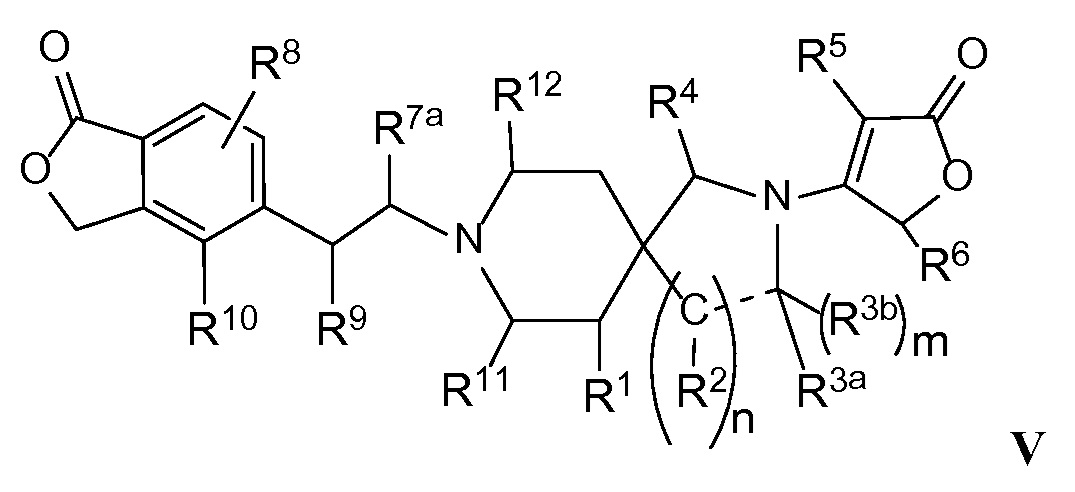
или их фармацевтически приемлемым солям,
в которой:
Rl представляет собой -H, -F, -OH или -OCH3;
m равно целому числу, выбранному из нуля (в случае отсутствия R3b) и 1 (в случае присутствия R3b);
n равно целому числу, выбранному из 1 или 2;
R2 в каждом случае независимо выбран из -H, =О (оксо), -OH, -Cl-3-алкила или -OCl-3-алкила, при условии, что когда n равно 2, то, по меньшей мере, один R2 представляет собой -H;
R3a представляет собой -H, =О, -C3-4-циклоалкил или -Cl-3-алкил, необязательно замещенный -OCH3 или 1-3 атомами -F, при условии, что только один из R2 или R3a может представлять собой =О;
R3b представляет собой -H или -Cl-3-алкил, или R3b отсутствует, когда R3a представляет собой =О, или когда обозначенная пунктиром связь представляет собой двойную связь или ароматическую связь;
или R3a и R3 объединяются вместе с атомом углерода, к которому они оба присоединены с образованием циклопропила или циклобутила;
или когда n равно 1, R2 и R3a могут объединяться вместе с атомами углерода, к которым каждый из них присоединен, с образованием (1) фенильного кольца, которое конденсировано с пирролидиновым кольцом, и m равно нулю; или с образованием (2) циклопропильного кольца, конденсированного с пирролидиновым кольцом, и m равно 1;
R4 представляет собой -H или =О;
R5 представляет собой -H, -Cl, -F, -Cl-3-алкил, -C3-6-циклоалкил или гетероцикл, необязательно замещенный -F, -Cl или -Cl-3-алкилом;
R6 представляет собой -H или -Cl-3-алкил;
R7a представляет собой -H или -Cl-3-алкил, необязательно замещенный -OH, -OCH3 или 1-3 атомами -F;
R8 представляет собой -H, -F или -Cl-3-алкил;
R9 представляет собой -H, -F, -OH, -ОCl-3-алкил, -CН2OH, -NH-R13 или 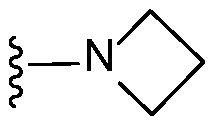 ;
;
R10 представляет собой -H, галоген, -CN, -C3-4-циклоалкил, или -Cl-3-алкил, необязательно замещенный 1-3 атомами -F;
или R9 представляет собой -O- и объединяется вместе с R10, чтобы представлять собой -CH2-CH2-O-;
Rll представляет собой -H, -CH2OH, -CH2OCH3 или -Cl-3-алкил, необязательно замещенный 1-3 атомами -F;
Rl2 представляет собой -H, -CH2OH, -CH2OCH3 или -Cl-3-алкил, необязательно замещенный 1-3 атомами -F;
или Rll и Rl2 объединяются вместе, чтобы представлять собой -СH2-CH2-, -CH2-N(CH3)-CH2- или -CH2OCH2-;
Rl3 представляет собой -H, -(CH2)0-2-C3-6-циклоалкил, -(CH2)1-2-OC3-6-циклоалкил, -(CH2)1-2-OCl-3-алкил, -(CH2)1-2-CN, -C(О)OCl-3-алкил, -SO2CH3 или -Cl-3-алкил, необязательно замещенный одним-тремя атомами-F; и
обозначенная пунктиром связь ("---") представляет собой одинарную, двойную или ароматическую связь, при условии, что
когда n равно 2, то обозначенная пунктиром связь представляет собой одинарную связь, и m равно 1, и
когда n равно 1, и m равно 1, то обозначенная пунктиром связь представляет собой одинарную связь, и
когда n равно 1, и m равно нулю, то обозначенная пунктиром связь представляет собой
(a) двойную связь или
(b) ароматическую связь, когда R2 и R3a объединяются вместе с образованием фенильного кольца.
Еще один вариант осуществления настоящего изобретения относится к соединениям формулы I, в которых: n равно 2, и обозначенная пунктиром связь представляет собой одинарную связь, и m равно 1; или n равно 1, и обозначенная пунктиром связь представляет собой одинарную связь, и m равно 2, или обозначенная пунктиром связь представляет собой двойную связь, и m равно 1; R1 представляет собой -H, -F, -OH или -Cl-3-алкил; R4 представляет собой =О; R5 представляет собой -H или -Cl-3-алкил; R8 представляет собой -H или -Cl-3-алкил; R9 представляет собой -OH, -OCl-3-алкил или - NHR13; R10 - как указано в формуле I; Rll и Rl2 представляют собой -H; и все другие переменные имеют такие же значения, которые указаны в формуле I.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, в которой обозначенная пунктиром связь представляет собой двойную связь или ароматическую связь. Формулы II и IV иллюстрируют примеры вариантов осуществления изобретения, в которых обозначенная пунктиром связь представляет собой одинарную связь.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R1 представляет собой H или F, и более конкретно представляет собой -H.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R2 в каждом случае представляет собой - H.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R3a представляет собой -H, -C1-3-алкил, циклопропил и более конкретно представляет собой -H или -CH3.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, IV или V, в которых R3b представляет собой -H или -C1-3-алкил, и более конкретно представляет собой -H, или R3b отсутствует, когда обозначенная пунктиром связь представляет собой двойную связь или ароматическую связь.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R4 представляет собой =О.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R5 представляет собой (a) -H, (b) галоген, и в частности -Cl или -F, (c) -Cl-3-алкил (d) -C3-6-циклоалкил, и более конкретно представляет собой -H или -CH3.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R6 представляет собой -H или -CH3, и более конкретно представляет собой -H.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III или IV, в которых R7a представляет собой -H или -Cl-3-алкил, необязательно замещенный -OH, -OCH3 или 1-3 атомами -F, и более конкретно, представляет собой -H или -CH3.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III или IV, в которых R7b представляет собой -H или -C1-3-алкил, и более конкретно представляет собой -H.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R8 представляет собой -H.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R9 представляет собой -H, -OH, -OCH3 или -NH2 и, в частности, представляет собой -OH.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R10 представляет собой -H, -C1-3-алкил, -C3-4-циклоалкил, -F, и в частности, представляет собой -CH3.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R11 представляет собой -H или -C1-3-алкил, и более конкретно представляет собой -H.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых Rl2 представляет собой -H или -C1-3-алкил, и более конкретно представляет собой -H; или R11 и Rl2 объединяются вместе, чтобы представлять собой -CH2-CH2-, -CH2-N(CH3)-CH2- или -CH2OCH2-.
Вариант осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R13 представляет собой -H или -C1-3-алкил, и более конкретно представляет собой -H.
Вариант A осуществления настоящего изобретения относится к соединениям формулы I, II, III, IV или V, в которых R3a представляет собой -H или -CH3; R4 представляет собой =О; R5 представляет собой -H или -CH3; R7a представляет собой -H или CH3; R9 представляет собой -H, -OH, -OCH3 или -NH2, и в частности, представляет собой -OH; и R10 представляет собой -H или -CH3, и в частности, представляет собой -CH3. В таком классе соединений представлены соединения по варианту осуществления A, упоминаемому как вариант В осуществления настоящего изобретения, в котором R1 представляет собой -H; R2 в каждом случае представляет собой -H; R3b представляет собой -H (для соединений формулы I, II или IV); R6 представляет собой -H; R8 представляет собой -H; Rll представляет собой -H; и Rl2 представляет собой -H.
Все структурные формулы и описанные здесь варианты их осуществления включают фармацевтически приемлемые соли соединений, определяемых такой формулой.
В данном контексте, кроме случаев, когда указано иначе, подразумевается, что "алкил" включает насыщенные алифатические углеводородные группы как с разветвленной, так и прямой цепью, содержащие конкретное число атомов углерода. На протяжении всего описания для алкильных групп применяются общепринятые аббревиатуры. Например, термин "Cl-6-алкил" (или "C1-C6-алкил") означает алкильные группы с линейной или разветвленной цепью, включая все изомеры, содержащие конкретное число атомов углерода, и включает все гексильные изомеры и пентильные изомеры, а также н-, изо-, втор- и трет-бутил (бутил, втор-бутил, изобутил, т-бутил; Bu = бутил), n-пропил и изопропил (Pr = пропил), этил (Et) и метил (Me). "Циклоалкил" представляет собой циклизованное алкильное кольцо, содержащее указанное число атомов углерода. Примеры циклоалкилов включают циклопропил, циклобутил, циклопентил и циклогексил.
"Галоген" означает -F, -Cl, -Br или -I.
Подразумевается, что "гетероцикл" включает пиридил (все изомеры), пиразинил, пиридазинил или пиримидинил.
Как хорошо известно в данной области, термин "двойная связь" относится к ковалентной связи, где две пары электронов поделены между двумя атомами. Понятие ароматичности также хорошо известно в данной области; примером ароматичности являются бензол и фенил, которые обычно изображаются в виде соединений, содержащих 3 чередующиеся двойные связи, но также могут рассматриваться как соединения, содержащие углерод-углеродные связи, каждая из которых представляет собой гибрид одинарной связи и двойной связи. В данном контексте "ароматическая связь" относится к ароматической природе двойной связи между -C(R2)- и -C(R3a), когда R2 и R3a объединяются с образованием фенильного кольца, конденсированного с пирролидинильным кольцом, как указано в формулах I, III и V.
Если не оговорено или не описано иное, переменным, показанным в структурной формуле с "плавающей" связью, таким как R8, разрешается находиться на любом имеющемся атоме углерода в кольце, к которому переменная присоединяется.
Соединения формулы I могут иметь один или несколько хиральных (асимметричных) центров. Настоящее изобретение охватывает все стереоизомерные формы соединений формулы I. Все центры асимметрии, которые присутствуют в соединениях формулы I, могут независимо друг от друга иметь (R)- или (S)-конфигурацию. Когда в структурных формулах согласно изобретению связи с хиральным атомом углерода показаны в виде прямых линий, или когда название соединения приводится без упоминания обозначения хиральности (R) или (S) для хирального атома углерода, следует понимать, что как (R), так и (S)-конфигурации каждого такого хирального атома углерода и, следовательно, каждый энантиомер или диастереомер и их смеси включены в формулу или название соединения. Получение конкретных стереоизомеров или их смесей можно обнаружить в примерах, где такие стереоизомеры или их смеси получены, однако это ни в коей мере не ограничивает включение всех стереоизомеров и их смесей в пределы объема настоящего изобретения.
Изобретение включает все возможные энантиомеры и диастереомеры и смеси двух или более стереоизомеров, например, смеси энантиомеров и/или диастереомеров во всех соотношениях. Таким образом, энантиомеры являются предметом изобретения в энантиомерно чистой форме, как в виде левовращающего антипода, так и в виде правовращающего антипода, в форме рацематов и в форме смесей двух энантиомеров во всех их соотношениях. В случае цис/транс-изомерии изобретение включает как цис-форму, так и транс-форму, а также смеси таких форм во всех их соотношениях. Получение индивидуальных стереоизомеров можно осуществлять, если требуется, путем разделения смеси общепринятыми способами, например, с помощью хроматографии или кристаллизации, путем применения стереохимически однородных исходных материалов для синтеза или с помощью стереоселективного синтеза. Необязательно перед разделением стереоизомеров можно осуществлять дериватизацию. Разделение смеси стереоизомеров можно осуществлять на промежуточной стадии во время синтеза соединения формулы I, или разделение можно выполнять для конечного рацемического продукта. Абсолютную стереохимию можно определять с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые дериватизируют, если необходимо, с помощью реагента, содержащего стереогенный центр известной конфигурации. Альтернативно абсолютную стереохимию можно определять с помощью спектроскопии колебательного циркулярного дихроизма (VCD). Когда соединения согласно настоящему изобретению способны к таутомеризации, все индивидуальные таутомеры, а также их смеси включены в объем настоящего изобретения. Настоящее изобретение включает все такие изомеры, а также соли, сольваты (которые включают гидраты) и сольватированные соли таких рацематов, энантиомеров, диастереомеров и таутомеров и их смеси.
Ссылка на описанные здесь соединения формулы I охватывает соединения формул I, II, III, IV и V и все варианты их осуществления. Подразумевается, что ссылка на соединения согласно настоящему изобретению в виде соединений конкретной формулы или варианта осуществления, например, на соединения формулы I, II, III, IV или V или варианты их осуществления, или на любую другую общую структурную формулу или конкретное соединение, описанное или заявленное в настоящем документе, охватывает конкретное соединение или соединения, попадающие в объем формулы или вариант осуществления, включая их соли, в частности, фармацевтически приемлемые соли, сольваты (включая гидраты) таких соединений и сольватированные формы их солей, когда такие формы возможны, если не указано иначе.
В соединениях формулы I атомы могут проявлять свой природный изотопный состав, или один или несколько атомов могут быть искусственно обогащены конкретным изотопом, имеющим тот же самый атомный номер, но атомную массу или массовое число, отличающиеся от атомной массы или массового числа, преимущественно обнаруживаемых в природе. Подразумевается, что настоящее изобретение включает все подходящие изотопные варианты соединений формулы I. Например, разные изотопные формы водорода (H) включают протий (1Н) и дейтерий (2Н). Протий представляет собой преобладающий изотоп водорода, обнаруживаемый в природе. Обогащение дейтерием может давать определенные терапевтические преимущества, такие как увеличение in vivo периода полувыведения или уменьшение рекомендуемой дозировки, или может обеспечивать соединение, применимое в качестве стандарта для характеристики биологических образцов. Изотопно-обогащенные соединения формулы I можно получать без излишнего экспериментального исследования с помощью общеизвестных способов, хорошо известных специалисту в данной области техники, или с помощью способов, аналогичных способам, описанным здесь на схемах и в примерах с применением подходящих изотопно-обогащенных реагентов и/или промежуточных соединений.
Когда соединения формулы I содержат одну или несколько кислотных или основных групп, изобретение также включает соответствующие фармацевтически приемлемые соли. Таким образом, соединения формулы I, которые содержат кислотные группы, можно применять согласно изобретению, например, но, не ограничиваясь перечисленным, в виде солей щелочных металлов, солей щелочно-земельных металлов или солей аммония. Примеры таких солей включают в себя, но не ограничиваются перечисленным, соли натрия, соли калия, соли кальция, соли магния или соли с аммиаком или органическими аминами, например, такими как этиламин, этаноламин, триэтаноламин или аминокислоты. Соединения формулы I, которые содержат одну или несколько основных групп, то есть групп, которые можно протонировать, можно применять согласно изобретению в форме их кислотно-аддитивных солей с неорганическими или органическими кислотами, например, но, не ограничиваясь перечисленным, таких как кислотно-аддитивные соли с хлористым водородом, бромистым водородом, фосфорной кислотой, серной кислотой, азотной кислотой, бензолсульфоновой кислотой, метансульфоновой кислотой, п-толуолсульфоновой кислотой, нафталиндисульфоновыми кислотами, щавелевой кислотой, уксусной кислотой, трифторуксусной кислотой, винной кислотой, молочной кислотой, салициловой кислотой, бензойной кислотой, муравьиной кислотой, пропионовой кислотой, пивалевой кислотой, диэтилуксусной кислотой, малоновой кислотой, янтарной кислотой, пимелиновой кислотой, фумаровой кислотой, малеиновой кислотой, яблочной кислотой, сульфаминовой кислотой, фенилпропионовой кислотой, глюконовой кислотой, аскорбиновой кислотой, изоникотиновой кислотой, лимонной кислотой, адипиновой кислотой и т.д. Если соединения формулы I одновременно содержат в молекуле кислотные и основные группы, в дополнение к упомянутым формам в виде солей изобретение также включает внутренние соли или бетаины (цвиттерионы). Соли можно получать из соединений формулы I общепринятыми способами, которые известны специалисту в данной области, например, путем объединения с органической или неорганической кислотой или основанием в растворителе или диспергирующей фазе или с помощью анионного обмена или катионного обмена из других солей. Настоящее изобретение также включает все соли соединений формулы I, которые из-за низкой физиологической совместимости не подходят непосредственно для применения в фармацевтических композициях, но которые можно применять, например, в качестве промежуточных соединений для химических реакций или для получения фармацевтически приемлемых солей.
Кроме того, соединения согласно настоящему изобретению могут существовать в аморфной форме и/или в одной или нескольких кристаллических формах, и подразумевается, что все такие аморфные и кристаллических формы и смеси соединений формулы I должны быть включены в объем настоящего изобретения. Кроме того, некоторые из соединений согласно настоящему изобретению могут образовывать сольваты с водой (то есть гидраты) или обычными органическими растворителями. Такие сольваты и гидраты, в частности фармацевтически приемлемые сольваты и гидраты соединений по изобретению также попадают в объем настоящего изобретения наряду с несольватированными и безводными формами.
Любая фармацевтически приемлемая пролекарственная модификация соединения согласно настоящему изобретению, которая приводит к превращению in vivo в соединение, попадающее в объем настоящего изобретения, также попадает в объем настоящего изобретения. Например, сложные эфиры необязательно могут быть получены путем этерификации содержащейся в соединении группы карбоновой кислоты или путем образования сложного эфира на месте содержащейся в соединении гидрокси-группы. Точно так же можно получать легко разлагающиеся амиды. Фармацевтически приемлемые сложные эфиры или амиды соединений согласно настоящему изобретению можно получать в качестве пролекарств, которые можно подвергать гидролизу до кислоты (или -COO- в зависимости от значения pH флюида или ткани, где такое превращение происходит) или до гидрокси-формы, в частности in vivo, и в таком качестве они охвачены объемом настоящего изобретения. Примеры фармацевтически приемлемых пролекарственных модификаций включают в себя, но не ограничиваются перечисленным, сложные -C1-6-алкиловые эфиры и сложные -C1-6-алкиловые эфиры, замещенные фенилом.
Соответственно соединения общих структурных формул, варианты их осуществления и конкретные соединения, описанные и заявленные в настоящем описании, охватывают соли, все возможные стереоизомеры и таутомеры, их физические формы (например, аморфные и кристаллические формы), сольватные и гидратные формы и любую комбинацию таких форм, а также их солей, пролекарственных форм и солей пролекарственных форм, когда такие формы возможны, если не указано иначе.
Соединения формулы I согласно изобретению являются ингибиторами ROMK и, следовательно, могут применяться в качестве диуретических и/или натрийуретических средств. Ингибиторы ROMK можно применять для содействия увеличению мочеиспускания и увеличению объема мочи, а также для предотвращения или уменьшения реабсорбции натрия в почках, что приводит к повышенной экскреции натрия и воды. Следовательно, соединения могут применяться для лечения или профилактики (или того и другого) расстройств, которые улучшаются при повышенной экскреции воды и натрия из организма. Соответственно соединения согласно настоящему изобретению могут применяться в способе ингибирования ROMK, включающем введение соединения формулы I пациенту, нуждающемся в этом, в эффективном для ингибирования ROMK количестве. Способ также охватывает применение соединений для ингибирования ROMK у пациента, включающее введение терапевтически эффективного количества соединения по п. 1 пациенту, нуждающемуся в диурезе, натрийурезе или в том и другом. Ингибирование ROMK соединениями формулы I можно исследовать, например, с помощью описанного ниже анализа транспорта таллия и/или электрофизиологического анализа. Кроме того, настоящее изобретение также относится к применению соединений формулы I или их солей для оценки в анализах in vitro, например, но, не ограничиваясь перечисленным, в описанных здесь анализе транспорта таллия и/или электрофизиологическом анализе.
Соединения согласно настоящему изобретению могут применяться в способе вызывания диуреза, натрийуреза или того и другого, включающем введение терапевтически эффективного количества соединения формулы I пациенту, нуждающемся в этом. Следовательно, соединения формулы I согласно настоящему изобретению могут применяться в способах лечения, предотвращения или уменьшения риска развития медицинских состояний, которые улучшаются при повышенной экскреции воды и натрия, таких как (но, не ограничиваясь перечисленным) одно или несколько состояний, выбранных из гипертонии, такой как эссенциальная гипертензия (также известная как первичная гипертензия или идиопатическая гипертензия), которая представляет собой форму гипертонии, которая может обнаруживаться без видимых причин, из сердечной недостаточности (которая включает как острую сердечную недостаточность, так и хроническую сердечную недостаточность, последняя из которых также известна как застойная сердечная недостаточность) и/или других состояний, ассоциированных с избытком соли и задержкой жидкости в организме. Соединения также можно применять для лечения гипертонии, которая ассоциирована с любым из нескольких основных заболеваний, таких как почечные, легочные, эндокринные и сосудистые заболевания, включая лечение пациентов с медицинскими состояниями, такими как сердечная недостаточность и/или хроническое заболевание почек. Кроме того, соединения формулы I могут применяться в способах лечения, предотвращения или уменьшения риска развития одного или нескольких расстройств, таких как легочная гипертензия, в частности, легочной артериальной гипертензии (PAH), сердечно-сосудистого заболевания, отечных состояний, сахарного диабета, несахарного диабета, объемной перегрузки в послеоперационном периоде, эндотелиальной дисфункции, диастолической дисфункции, систолической дисфункции, стабильной и нестабильной стенокардии, тромбозов, рестеноза, инфаркта миокарда, апоплексического удара, сердечной недостаточности, легочной гипертонии, атеросклероза, цирроза печени, асцита, преэклампсии, отека головного мозга, нефропатии, гломерулонефрита, нефротического синдрома, острой почечной недостаточности, хронической почечной недостаточности (также упоминаемой как хроническое заболевание почек или более обобщенно как нарушение функции почек), острого табулярного некроза, гиперкальциемии, идиопатической отечной болезни, болезни Дента, болезни Меньера, глаукомы, идиопатической внутричерепной гипертензии и других состояний, при которых диуретические или натрийуретические средства или те и другие могут давать терапевтическое или профилактическое улучшение. Соединения согласно изобретению можно вводить пациенту, имеющему одно или несколько состояний или при риске наличия одного или нескольких состояний, при которых диуретические или натрийуретические средства или те и другие могут давать терапевтическое или профилактическое улучшение, такие как описанные здесь соединения.
Соединения формулы I потенциально могут обладать меньшими недостатками (например, при гипокалиемии или гиперкалиемии, выявленном впервые диабете, дислипидемии и т.д.) по сравнению с применяемыми в настоящее время клиническими средствами. Также соединения могут обладать меньшим риском привыкания к диуретическим средствам, которое может представлять собой проблему при долговременном применении петлевых диуретиков.
В общем случае соединения, которые являются ингибиторами ROMK, можно идентифицировать как такие соединения, которые в испытании имеют значение IC50 5 мкМ или менее, предпочтительно 1 мкМ или менее и более предпочтительно - 0,25 мкМ или менее, по меньшей мере, при проведении одного из следующих анализов: 1) анализа транспорта таллия (Flux Assay), 2) электрофизиологического анализа. Такие анализы более подробно дополнительно описаны ниже.
Величина дозы соединения, подлежащего введению, зависит от конкретного случая и по обыкновению для достижения оптимального эффекта должна адаптироваться для индивидуальных обстоятельств. Таким образом, она зависит от природы и степени тяжести расстройства, подлежащего лечению, а также от пола, возраста, массы и индивидуальной ответной реакции человека или животного, подлежащего лечению, от эффективности и продолжительности действия применяемых соединений, от того применяется ли терапия для лечения острого или хронического заболевания или является профилактической, или от того, вводятся ли другие активные соединения в дополнение к соединениям формулы I. Рассмотрение таких факторов хорошо известно в рамках деятельности врача-клинициста с целью определения терапевтически эффективной или профилактически эффективной по величине дозы, необходимой для предотвращения, противодействия или остановки развития состояния. Ожидается, что соединение будет вводиться постоянно, изо дня в день в течение промежутка времени, подходящего для лечения или предотвращения медицинского состояния, имеющего значение для пациента, включая курс терапии, длящийся дни, месяцы, года или всю жизнь пациента.
В общем случае суточная доза, составляющая приблизительно от 0,001 до 100 мг/кг, предпочтительно от 0,001 до 30 мг/кг, в частности, от 0,001 до 10 мг/кг (в каждом случае мг на кг массы тела), подходит для введения взрослому человеку с весом приблизительно 75 кг, чтобы получить при этом требуемые результаты. Суточная доза предпочтительно вводится в виде одной дозы или может делиться на несколько, например, на две, три или четыре отдельных дозы, и может составлять, например, но не ограничиваясь перечисленным, 0,1 мг, 0,25 мг, 0,5 мг, 0,75 мг, 1 мг, 1,25 мг, 2 мг, 2,5 мг, 5 мг, 10 мг, 20 мг, 40 мг, 50 мг, 75 мг, 100 мг, 125 мг, 150 мг, 175 мг, 200 мг и т.д. ежедневно. В некоторых случаях в зависимости от активности соединения или индивидуальной реакции может потребоваться отклонение в сторону повышения или в сторону понижения от заданной суточной дозы. Кроме того, соединение может быть получено для незамедлительного или модифицированного высвобождения, такого как пролонгированное или контролируемое высвобождение.
Термин "пациент" включает животных, предпочтительно млекопитающих и, в частности, человека, кто применяет настоящие активные средства для профилактики или лечения медицинского состояния.
Введение лекарственного средства пациенту включает как самостоятельное введение, так и введение пациенту лекарственного средства другим человеком. Пациент может нуждаться в лечении существующего заболевания или медицинского состояния, или может желать профилактического лечения для предотвращения или уменьшения риска развития упомянутого заболевания или медицинского состояния, или развития длительных осложнений от заболевания или медицинского состояния.
Подразумевается, что термин "терапевтически эффективное количество" означает то количество лекарственного средства или фармацевтического средства, которое добьется биологического или медицинского ответа ткани, системы, животного или человека, к которому стремится исследователь, ветеринар, врач или другой клиницист. Подразумевается, что профилактически эффективное количество означает то количество фармацевтического лекарственного средства, которое будет предотвращать или уменьшать риск появления биологического или медицинского осложнения, которое стремится предотвратить исследователь, ветеринар, врач или другой клиницист в ткани, системе, у животного или человека. Термины "предотвращающее", "предотвращение," "профилактическое" и производные от таких терминов в данном контексте относятся к введению пациенту соединения до возникновения клинических симптомов состояния, пока еще не присутствующего у пациента. Понятно, что конкретная величина суточной дозировки одновременно может представлять собой как терапевтически эффективное количество, например, для лечения гипертонии, так и профилактически эффективное количество, например, для предотвращения или уменьшения риска инфаркта миокарда или предотвращения или уменьшения риска осложнений, связанных с гипертонией.
В способах лечения согласно настоящему изобретению ингибиторы ROMK можно вводить с помощью любого подходящего способа введения, например, такого как оральный, парентеральный или ректальный способ введения стандартных лекарственных препаратов, содержащих общеизвестные нетоксичные фармацевтически приемлемые носители, вспомогательные вещества и наполнители. Термин парентеральный в данном контексте включает подкожные инъекции, внутривенную (IV), внутримышечную, интрастернальную инъекцию или способы вливания. Препараты для орального введения являются предпочтительными для лечения хронических показаний, таких как гипертония или хроническая сердечная недостаточность, в частности, предпочтительными являются твердые стандартные лекарственные формы для орального введения, такие как пилюли, таблетки или капсулы, и более конкретно - таблетки. IV внутривенное введение доз является предпочтительным для неотложного лечения, например для лечения острой сердечной недостаточности.
Настоящее изобретение также обеспечивает фармацевтические композиции, содержащие соединения формулы I и фармацевтически приемлемый носитель, который состоит из одного или нескольких инертных наполнителей или добавок. Инертный наполнитель или добавка представляет собой инертное вещество, применяемое для объединения с активным ингредиентом лекарственного средства. Фармацевтические композиции для орального введения согласно настоящему изобретению, содержащие активный ингредиент, могут быть в таких формах, как пилюли, таблетки, пастилки, леденцы, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы, или сиропы, или эликсиры. Композиции, предполагаемые для орального введения, можно получать согласно любому способу, известному в данной области для производства фармацевтических композиций. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми инертными наполнителями, которые подходят для производства таблеток. Инертные наполнители, например, могут представлять собой инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, маннит, фосфат кальция или фосфат натрия; гранулирующие средства и вещества для улучшения распадаемости таблеток, например, кукурузный крахмал, или альгиновая кислота; связующие, например, крахмал, желатин или камедь; и смазки, например, стеарат магния, стеариновая кислота или тальк.
Фармацевтические композиции также могут содержать другие общепринятые добавки, например, но не ограничиваясь перечисленным, увлажняющие средства, стабилизаторы, эмульгаторы, диспергирующие присадки, консерванты, подсластители, красители, корригенты, ароматизаторы, загустители, буферные вещества, растворители, солюбилизаторы, средства для обеспечения депо-эффекта (замедленного всасывания), соли для изменения осмотического давления, вещества для покрытия или антиоксиданты. Можно использовать стандартные лекарственные формы незамедлительного высвобождения и контролируемого по времени высвобождения для орального введения, а также стандартные лекарственные формы с кишечнорастворимой оболочкой для орального введения. Таблетки могут быть без покрытия или их можно покрывать известными способами для эстетических целей, для маскировки вкуса или по другим причинам. Также можно применять покрытия для замедления распада и поглощения в желудочно-кишечном тракте и тем самым обеспечивать пролонгированное действие на протяжении более долгого периода. Например, можно использовать материал для задержки высвобождения, такой как глицерилмоностеарат или глицерилдистеарат.
Препараты для орального введения также могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с твердым инертным разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активные ингредиенты смешаны с водой или способными смешиваться растворителями, такими как пропилeнгликоль, ПЭГи и этанол, или с масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии содержат активный материал в смеси с инертными наполнителями, подходящими для производства водных суспензий. Масляные суспензии можно готовить путем суспендирования активного ингредиента в растительном масле, например, арахисовом масле, оливковом масле, кунжутном масле, кокосовом масле или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Можно добавлять подсластители и корригенты, получая при этом приятный на вид и запах препарат для орального введения. Такие композиции можно сохранять путем добавления антиоксиданта, такого как аскорбиновая кислота. Сиропы и эликсиры можно готовить с подсластителями, например, глицерином, пропиленгликолем, сорбитом или сахарозой.
Настоящее изобретение также охватывает способ получения фармацевтической композиции, включающий объединение соединения формулы I с фармацевтически приемлемым носителем. Также охвачена фармацевтическая композиция, которая изготовлена путем объединения соединения формулы I с фармацевтически приемлемым носителем. Кроме того, терапевтически эффективное количество соединения согласно настоящему изобретению можно применять для получения лекарственного препарата, применимого для ингибирования ROMK, для вызывания диуреза и/или натрийуреза, и/или для лечения, предотвращения или уменьшения риска любых описанных здесь медицинских состояний при описанных здесь величинах доз.
Количество активного соединения формулы I и/или его фармацевтически приемлемых солей в фармацевтической композиции может, например, составлять, но не ограничивается перечисленным, приблизительно от 0,1 мг до 1 г, в частности, от 0,1 мг до приблизительно 200 мг, более конкретно приблизительно от 0,1 мг до приблизительно 100 мг, и еще более конкретно приблизительно от 0,1 до приблизительно 50 мг на дозу, в расчете на массу свободной кислоты/свободного основания, хотя в зависимости от типа фармацевтической композиции, активности активного ингредиента и/или медицинского состояния, подвергаемого лечению, количество активного соединения также может быть ниже или выше. Фармацевтические композиции обычно содержат приблизительно от 0,5 до приблизительно 90% масс. активного соединения в расчете на массу свободной кислоты/свободного основания.
Соединения формулы I ингибируют ROMK. Благодаря такому свойству, наряду с применением в качестве фармацевтически активных соединений в медицине и ветеринарии их также можно использовать в качестве научного средства или в качестве вспомогательного средства при биохимических исследованиях, в которых предполагается такое влияние на ROMK, а также для диагностических целей, например, для диагностики in vitro клеточных образцов или образцов тканей. Соединения формулы I также можно использовать в качестве промежуточных соединений для получения других фармацевтически активных соединений.
В комбинации с соединением формулы I можно вводить одно или несколько дополнительных фармакологически активных средств. Подразумевается, что "дополнительное активное средство" (или средства) означает лекарственное соединение, которое отличается от соединения формулы I, и которое является фармацевтически активным средством (или средствами) и активно в организме, включая пролекарства, например, сложноэфирные формы, которые после введения превращаются в фармацевтически активную форму, а также включая свободную кислоту, свободное основание и фармацевтически приемлемые соли упомянутых дополнительных активных средств, когда такие формы поступают в продажу или химически возможны по другим причинам. В общем случае можно применять любое подходящее, дополнительное, активное средство или средства, включая но не ограничиваясь перечисленным, антигипертензивные средства, дополнительные диуретические средства, анти-атеросклеротические средства, такие как липид-модифицирующее соединение, антидиабетические средства и/или средства против ожирения в любой комбинации с соединением формулы I в виде препарата с однократной дозировкой (фиксированная комбинация лекарственных средств), или пациенту можно вводить одну или несколько дозированных лекарственных форм, которые позволяют осуществлять одновременное или последовательное введение активных средств (совместное введение отдельных активных средств). Примеры одного или нескольких дополнительных активных средств, которые можно использовать, включают но не ограничиваются перечисленным, диуретики типа тиазида, например, гидрохлортиазид (HCTZ или HCT); ингибиторы ангиотензинпревращающего фермента (например, алацеприл, беназеприл, каптоприл, церонаприл, цилазаприл, делаприл, эналаприл, эналаприлат, фозиноприл, имидаприл, лизиноприл, мовелтиприл, периндоприл, хинаприл, рамиприл, спираприл, темокаприл или трандолаприл); двойные ингибиторы ангиотензинпревращающего фермента (ACE) и нейтральной эндопептидазы (NEP), такие как омапатрилат, сампатрилат и фазидотрил; антагонисты рецепторов ангиотензина II, также известные как блокаторы рецепторов ангиотензина или ARB, которые могут находиться в форме свободного основания, свободной кислоты, соли или пролекарства, такие как азилсартан, например, азилсартана медоксомил калия (EDARBI®), кандесартан, например, кандесартана цилексетил (ATACAND®), эпрозартан, например, эпрозартана мезилат (TEVETAN®), ирбесартан (AVAPRO®), лозартан, например, лозартан калия (COZAAR®), олмезартан, например, олмезартана медоксомил (BENICAR®), телмисартан (MICARDIS®), валсартан (DIOVAN®) и любые из таких лекарственных средств, применяемых в комбинации с диуретиком типа тиазида, таким как гидрохлортиазид (например, HYZAAR®, DIOVAN HCT®, ATACAND HCT®) и т.д.); умеренные калиевые диуретики, такие как амилорид HCl, спиронолактон, эплеренон, триамтерен, каждый с HCTZ или без него; ингибиторы карбоангидразы, такие как ацетазоламид; ингибиторы нейтральной эндопептидазы (например, тиорфан и фосфорамидон); антагонисты альдостерона; ингибиторы альдостеронсинтазы; ингибиторы ренина (например, производные ди- и трипептидов мочевинного типа (см. патент США № 5116835), аминокислоты и производные (патенты США №№ 5095119 и 5104869), аминокислотные цепочки, соединенные непептидными связями (патент США № 5114937), ди- и трипептидные производные (патент США № 5106835), пептидиламинодиолы (патенты США №№ 5063208 и 4845079) и карбаматы пептидил-бета-аминоациламинодиолов (патент США № 5089471); также ряд других пептидных аналогов, которые описаны в следующих патентах США №№ 5071837; 5064965; 5063207; 5036054; 5036053; 5034512 и 4894437; и низкомолекулярные ингибиторы ренина (включая диолсульфонамиды и сульфинилы (патент США № 5098924), N-морфолинопроизводные (патент США № 5055466), N-гетероциклические спирты (патент США № 4885292) и пиролимидазолоны (патент США № 5075451); также производные пепстатина (патент США № 4980283) и фтор- и хлорпроизводные статон-содержащих пептидов (патент США № 5066643); эналкреин; RO 42-5892; A 65317; CP 80794; ES 1005; ES 8891; SQ 34017; алискирен (гемифумарат 2(S),4(S),5(S),7(S)-N-(2-карбамоил-2-метилпропил)-5-амино-4-гидрокси-2,7-диизопропил-8-[4-метокси-3-(3-метоксипропокси)фенил]октанамида) SPP600, SPP630 и SPP635); антагонисты эндотелиновых рецепторов; вазодилататоры (например, нитропруссид); блокаторы кальциевых каналов (например, aмлoдипин, нифедипин, верапамил, дилтиазем, фелодипин, галлопамил, нилудипин, нимодипин, никардипин, бепридил, низолдипин); активаторы калиевых каналов (например, никорандил, пинацидил, кромакалим, миноксидил, априлкалим, лопразолам); симпатолитики; лекарственные средства, явлющиеся бета-адреноблокаторами (например, ацебутолол, атенолол, бетаксолол, бисопролол, карведилол, метопролол, метопролол-тартрат, надолол, пропранолол, соталол, тимолол); лекарственные средства, явлющиеся альфа-адреноблокаторами (например, доксазозин, празоцин или альфа-метилдопа); агонисты центральных альфа-адренергических рецепторов; периферические вазодилататоры (например, гидралазин); соединения, выступающие в качестве доноров нитратов или оксида азота, например, мононитрат изосорбида; гиполипидемические средства, например, ингибиторы HMG-CoA-редуктазы, такие как симвастатин и ловастатин, которые поступают на рынок в виде препаратов ZOCOR® и MEVACOR® в лактонной форме пролекарства и действуют в качестве ингибиторов после введения; и ингибиторы HMG-CoA-редуктазы в форме фармацевтически приемлемых солей дигидроксикислот с разомкнутым кольцом, такие как аторвастатин (в частности, кальциевая соль, продаваемая под торговой маркой LIPITOR®), розувастатин (в частности, кальциевая соль, продаваемая под торговой маркой CRESTOR®), правастатин (в частности, натриевая соль, продаваемая под торговой маркой PRAVACHOL®) и флувастатин (в частности, натриевая соль, продаваемая под торговой маркой LESCOL®); ингибитор абсорбции холестерина, такой как эзетимиб (ZETIA®), и эзетимиб в комбинации с любыми другими гиполипидемическими средствами, такими как упомянутые выше ингибиторы HMG-CoA-редуктазы и, в частности, в комбинации с симвастатином (VYTORIN®) или аторвастатином-кальция; ниацин в лекарственных формах незамедлительного высвобождения или контролируемого высвобождения и, в частности, ниацин в комбинации с DP-антагонистом, таким как ларопипрант (TREDAPTIVE®), и/или в комбинации с ингибитором HMG-CoA-редуктазы; агонисты ниациновых рецепторов, такие как аципимокс и ацифран, а также частичные агонисты ниациновых рецепторов; средства, изменяющие метаболизм, включающие инсулин-сенсибилизирующие средства и родственные соединения для лечения диабета, такие как бигуаниды (например, метформин), меглитиниды (например, репаглинид, натеглинид), сульфонилмочевины (например, хлорпропамид, глимепирид, глипизид, глибурид, толазамид, толбутамид), тиазолидиндионы, также упоминаемые как глитазоны (например, пиоглитазон, розиглитазон), ингибиторы альфа-глюкозидаз (например, акарбоза, миглитол), ингибиторы дипептидилпептидазы (например, ситаглиптин (JANUVIA®), алоглиптин, вилдаглиптин, саксаглиптин, линаглиптин, дутоглиптин, гемиглиптин), алкалоиды спорыньи (например, бромкриптин), комбинированные лекарственные препараты, такие как JANUMET® (ситаглиптин с метформином), и лекарственные препараты, инъецируемые при диабете, такие как эксенатид и прамлинтида ацетат; ингибиторы фосфодиэстеразы-5 (PDE5), такие как силденафил (Revatio, Viagra), тадалафил (Cialis, Adcirca), варденафил HCl (Levitra); или в комбинации с другими лекарственными средствами, способствующими предотвращению или лечению упомянутых выше заболеваний, включая, но не ограничиваясь перечисленным, диазоксид; и включая свободную кислоту, свободное основание и фармацевтически приемлемые соли, пролекарственные формы (включая, но не ограничиваясь перечисленным, сложные эфиры) и соли пролекарств упомянутых выше лекарственных препаратов, где они химически возможны. Торговые марки фармацевтических лекарственных средств, указанные выше, обеспечивают для иллюстрации поставляемой на рынок формы активного средства (средств); такие фармацевтические лекарственные средства могут применяться в дозированной лекарственной форме для одновременного или последовательного введения с соединением формулы I или активным средством (средствами), и могут применяться в фиксированной комбинации лекарственных средств, включающей соединение формулы I.
Несколько способов получения соединений согласно настоящему изобретению описаны на следующих схемах и в примерах. Исходные материалы и промежуточные соединения приобретают, получают согласно известным процедурам или иным способом, как проиллюстрировано. Некоторые, часто применяемые способы получения соединений формулы I также описаны с помощью указанных ниже схем. В некоторых случаях порядок осуществления стадий на реакционных схемах можно менять, чтобы облегчить проведение реакции или избежать нежелательных продуктов реакции. Заместители "R" на схемах соответствуют заместителям, указанным в формуле I и в тех же положениях химических структур.
Соединение IA, которое замещено группой -OH в бензильном положении, можно получать, следуя последовательности, подробно описанной на схеме 1. Связывание эпоксида 1 со спироциклическими аминами 2 при повышенных температурах приводит к образованию спиртов IA (Nomura Y. и др. Chemical & Pharmaceutical Bulletin, 1995, 43(2), 241-6). Реакцию можно осуществлять с помощью традиционного нагревания или путем нагревания с применением микроволновой печи. При проведении такой реакции можно применять ряд растворителей, например, этанол и 2-пропанол. Спироциклические амины могут представлять собой свободные основания или они могут представлять собой (аддитивные) соли, в случае которых можно добавлять основание, такое как триэтиламин или N,N-диизопропилэтиламин. Следует отметить, что когда используются энантиочистые хиральные эпоксиды (такие как (R)-1 на схеме 1), раскрытие эпоксида происходит с сохранением стереохимии в бензильном положении, и можно получать индивидуальный изомер (R)-IA (и если используется (S)-эпоксид, получаемый спирт будет иметь стереохимию, противоположную той, которая показана). Альтернативно можно осуществлять разделение энантиомеров или диастереомеров IA с помощью хиральной ВЭЖХ, чтобы обеспечить индивидуальные энантиомеры или диастереомеры.
СХЕМА 1
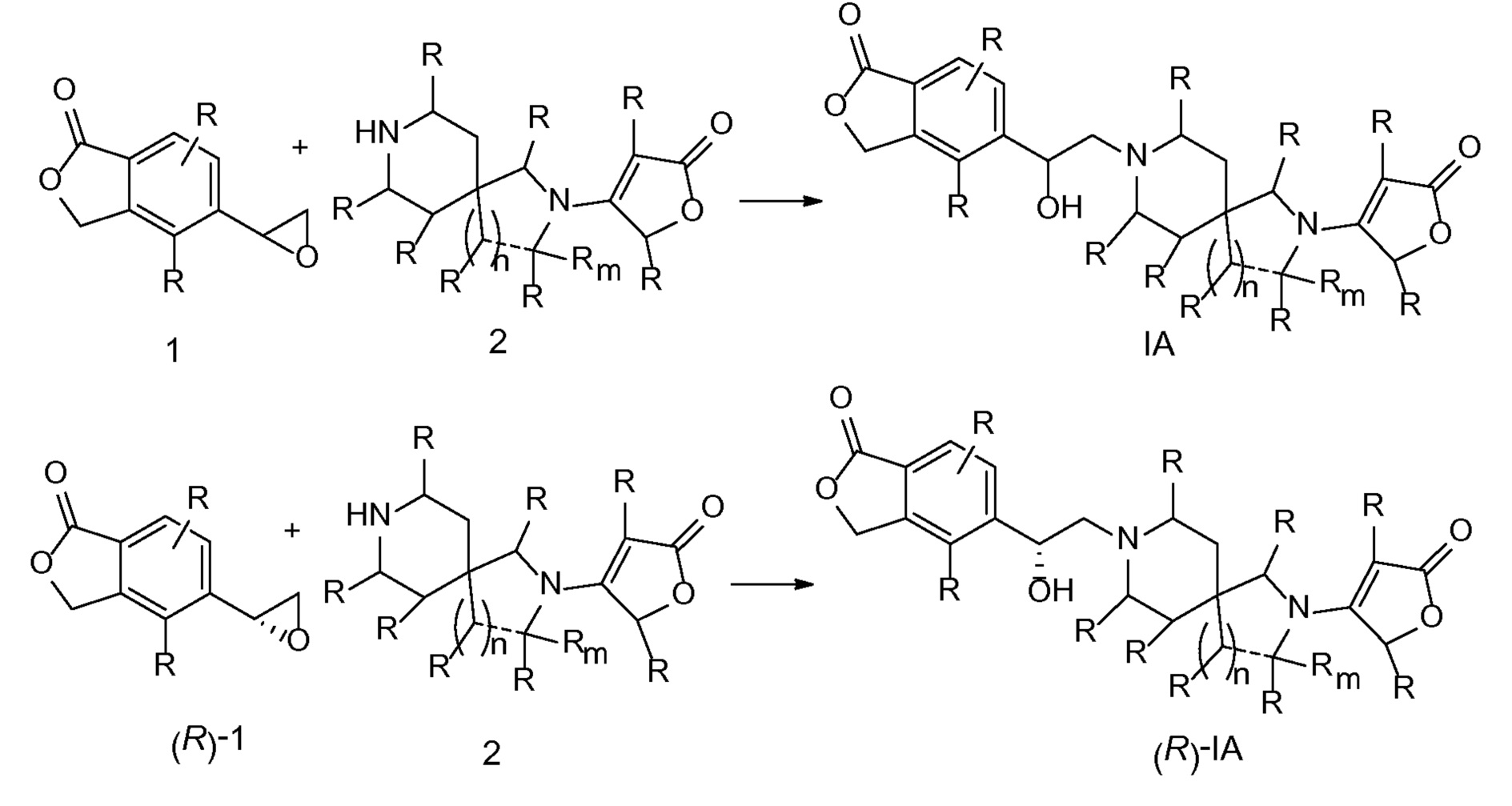
Соединения формулы IB можно получать с помощью последовательности, подробно описанной на схеме 2. Альдегиды или кетоны 3 можно применять в реакциях восстановительного алкилирования спироциклических аминов 2, получая при этом ингибиторы ROMK формулы IB, с применением различных условий восстановительного аминирования (например, с применением цианоборгидрида натрия, триацетоксиборгидрида натрия или тетраизопропоксида титана c последующим применением боргидрида натрия или цианоборгидрида натрия).
СХЕМА 2

Эпоксиды 1 (и индивидуальные энантиомеры (R)-1 и (S)-1) можно получать следующим способом, подробно описанным на схеме 3. Обработка 4 (где X представляет собой хлорид, бромид, иодид или трифторметансульфонат) имеющимся в продаже винилтрифторборатом калия (Molander, G.; Luciana, A. Journal of Organic Chemistry, 2005, 70(10), 3950-3956) в условиях реакции связывания, катализируемой палладием с подходящим фосфиновым лигандом, приводит к стиролу 5 (Molander, G.; Brown, A. Journal of Organic Chemistry, 2006, 71(26), 9681-9686). Альтернативно можно использовать другие способы, например, можно применять в качестве реагентов винилстаннаны и катализ в присутствии палладия. Полученные стиролы 5 можно превращать в соответствующие эпоксиды 1 в различных условиях эпоксидирования, например, с помощью mCPBA (Fringuelli F. и др. Organic Preparations and Procedures International, 1989, 21(6), 757-761). Рацемические эпоксиды 1 можно разделять в условиях хиральной хроматографии (ВЭЖХ), получая при этом их энантиомеры, которые можно применять вместо 1 согласно схеме 1.
СХЕМА 3
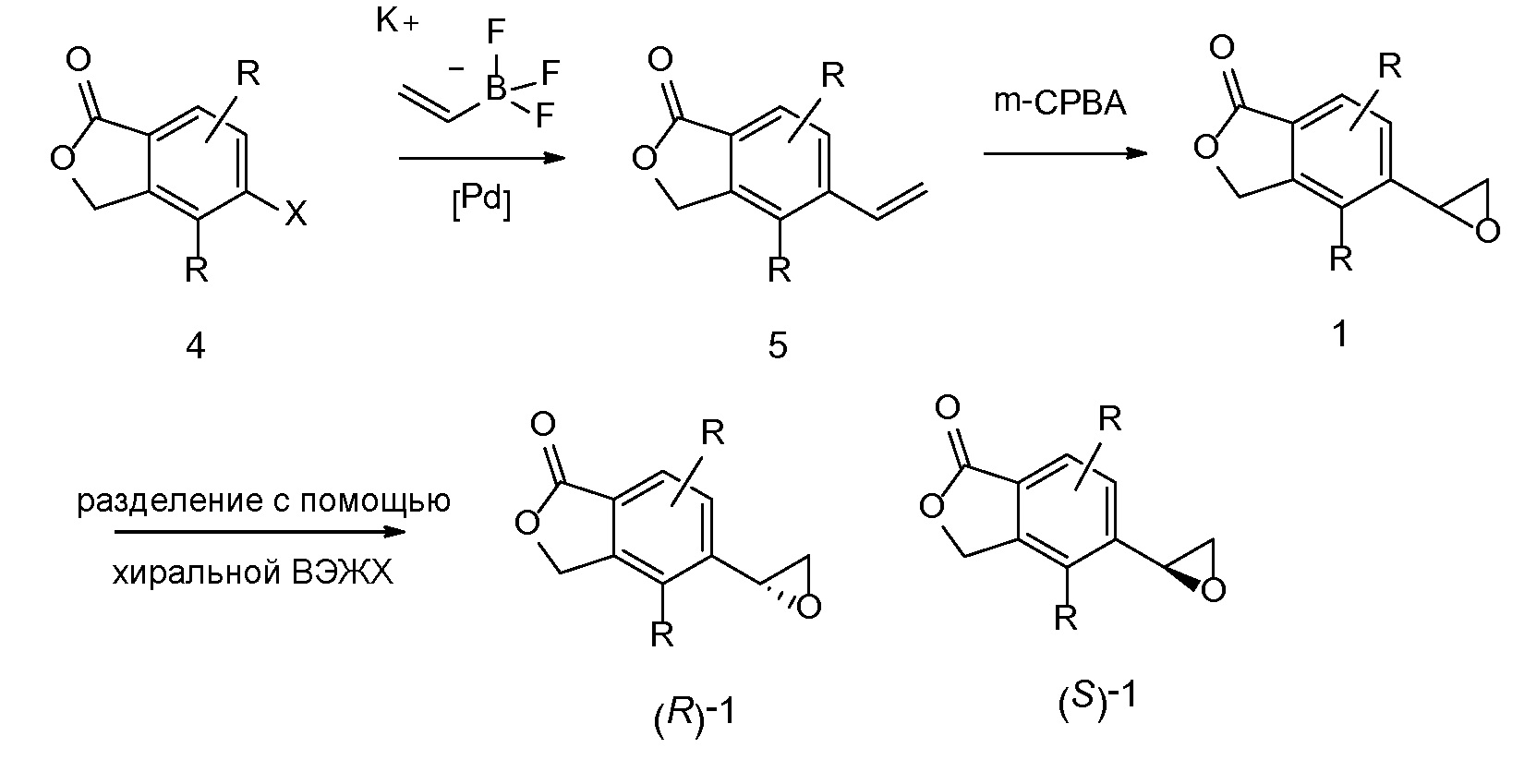
Альтернативно энантиочистые эпоксиды (R)-1 или (S)-1 можно получать, как показано на схеме 4. Обработка 4 (где X представляет собой бромид, иодид или трифторметансульфонат) имеющимся в продаже простым винилбутиловым эфиром 6 в условиях катализа с применением палладиевого катализатора с подходящим лигандом (например, Pd(OAc)2, DPPP) может обеспечивать простые енольные эфиры 7. Простые енольные эфиры можно получать с применением других способов, известных специалисту-химику. Обработка полученных простых енольных эфиров 7 с помощью NBS или других подобных реагентов приводит к соответствующим бромметилкетонам 8. Их можно подвергать различным условиям асимметричного восстановления кетонов, например, с помощью фермента, который может влиять на такое транс-образование с высокой энантиоселективностью. Последующая обработка основанием, таким как триэтиламин, приводит к циклизации, давая при этом энантиообогащенные эпоксиды (R)-1 или (S)-1 (в зависимости от асимметричного восстановителя).
СХЕМА 4
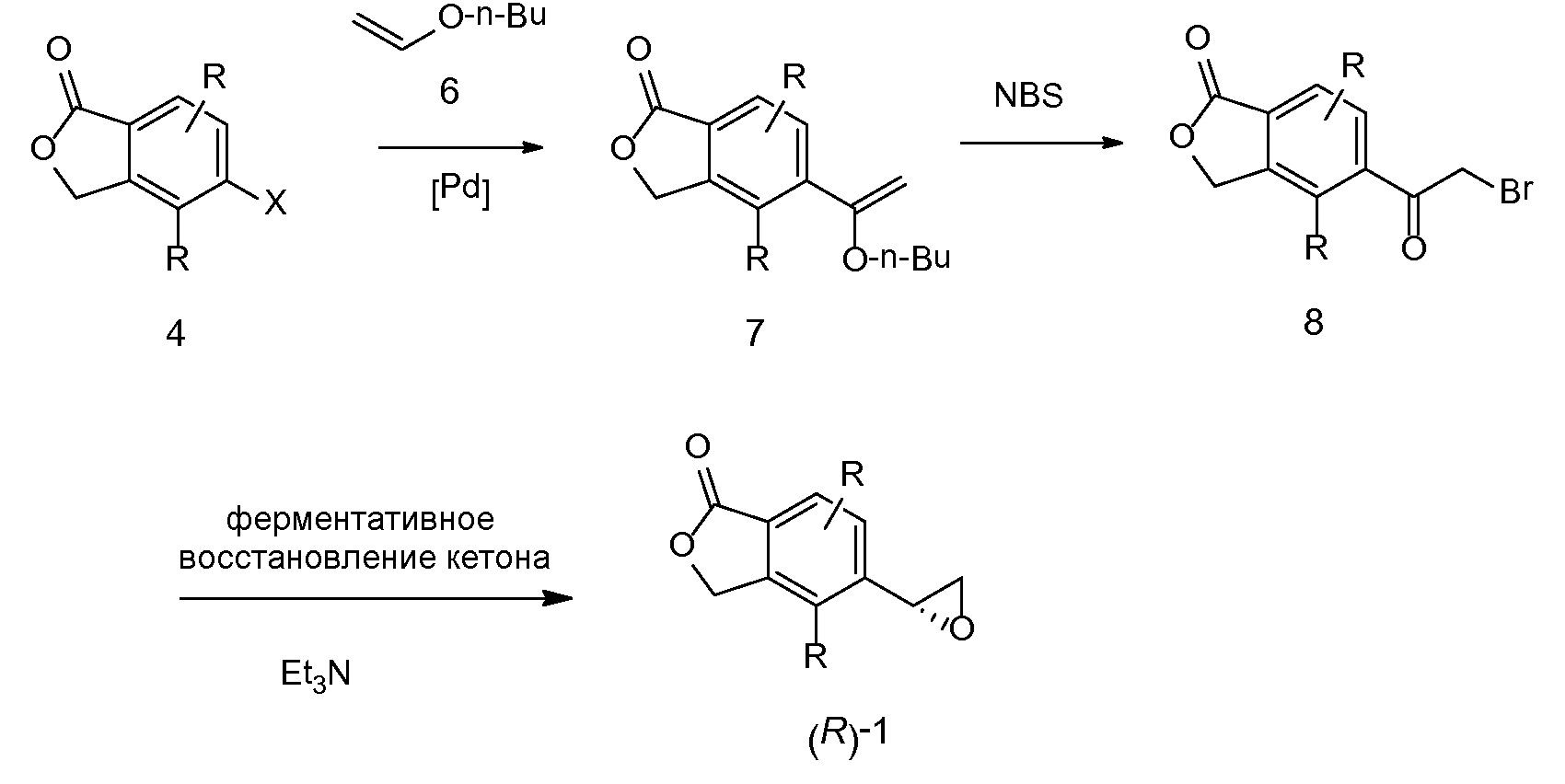
Альдегиды 3A можно получать многочисленными способами, причем два таких подхода описаны на схеме 5. Обработка 4 (где X представляет собой бромид, иодид или трифторметансульфонат) с бром-(1,3-диоксолан-2-илметил)цинком в присутствии подходящего палладиевого катализатора и лиганда, таких как ацетат палладия (II) и комплекс три-т-бутилфосфин-BF4, обеспечивает соответствующее арил-1,3-диоксолан-2-илметильное производное 9. Затем можно получать альдегиды 3A путем обработки HCl в присутствии воды и органического растворителя. Альтернативно реакция 4 (где X представляет собой бромид, иодид или трифторметансульфонат) с аллилтрибутилстаннаном в присутствии палладиевого катализатора дает аллильный продукт 10. Окисление, например озоном, с последующей обработкой диметилсульфидом обеспечивает альдегиды 3A.
СХЕМА 5
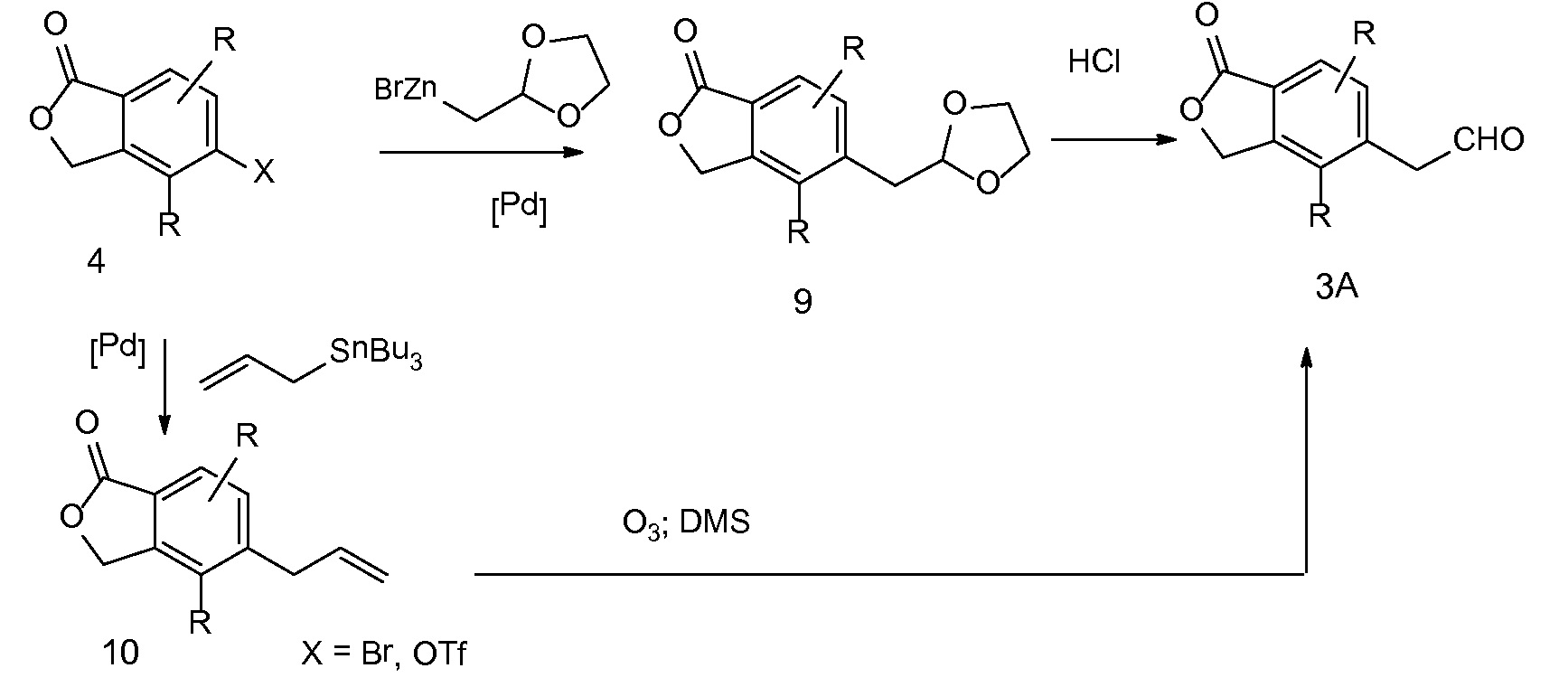
Спироциклические аминофураноны 2 можно получать, как описано на схеме 6. Спироциклические диамины или аминолактамы (где R11 и R12 вместе представляют собой карбонильную группу) 11, защищенные соответствующим образом (Greene T., Wuts, P. G. M. Protective Groups in Organic Synthesis, John Wiley и Sons, Inc., New York, NY 1991), можно связывать с фуранонтрифлатами или бромидами 12 в присутствии палладиевого катализатора и лиганда, например, ацетата палладия и 4,5-бис(дифенилфосфино)-9,9-диметилксантена. Некоторые из описанных здесь спироциклических диаминов или аминолактамов 11 имеются в продаже; другие можно получать, как описано ниже в экспериментальной части. 4-бромфуран-2(5H)-он имеется в продаже; другие фураноны 12 можно получать, как описано ниже в примерах. Промежуточные соединения 13 превращают в спироциклические аминофураноны 2 путем удаления защитной группы, например, трет-бутоксикарбонильную группу можно удалять с помощью TFA или HCl.
СХЕМА 6
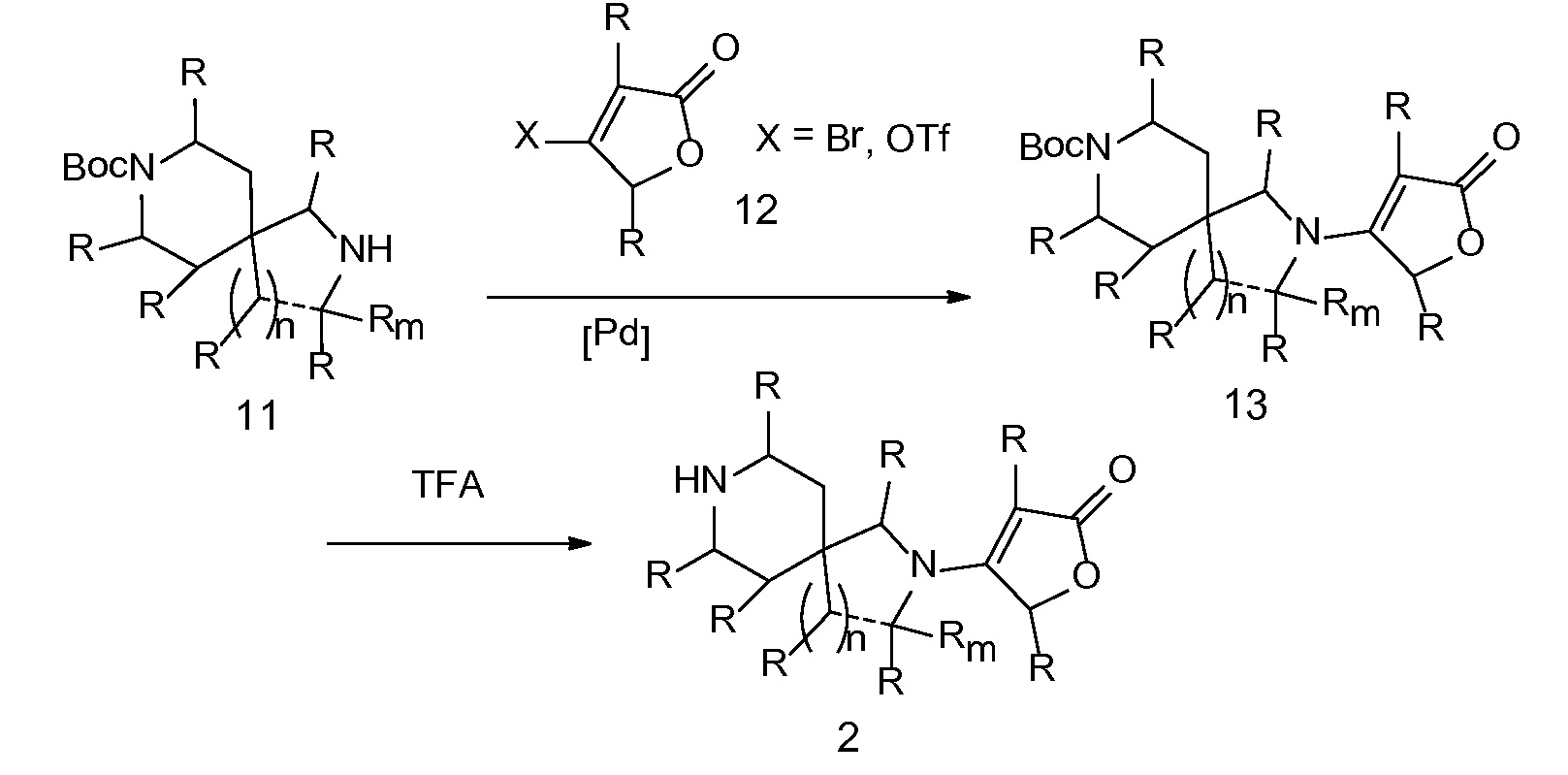
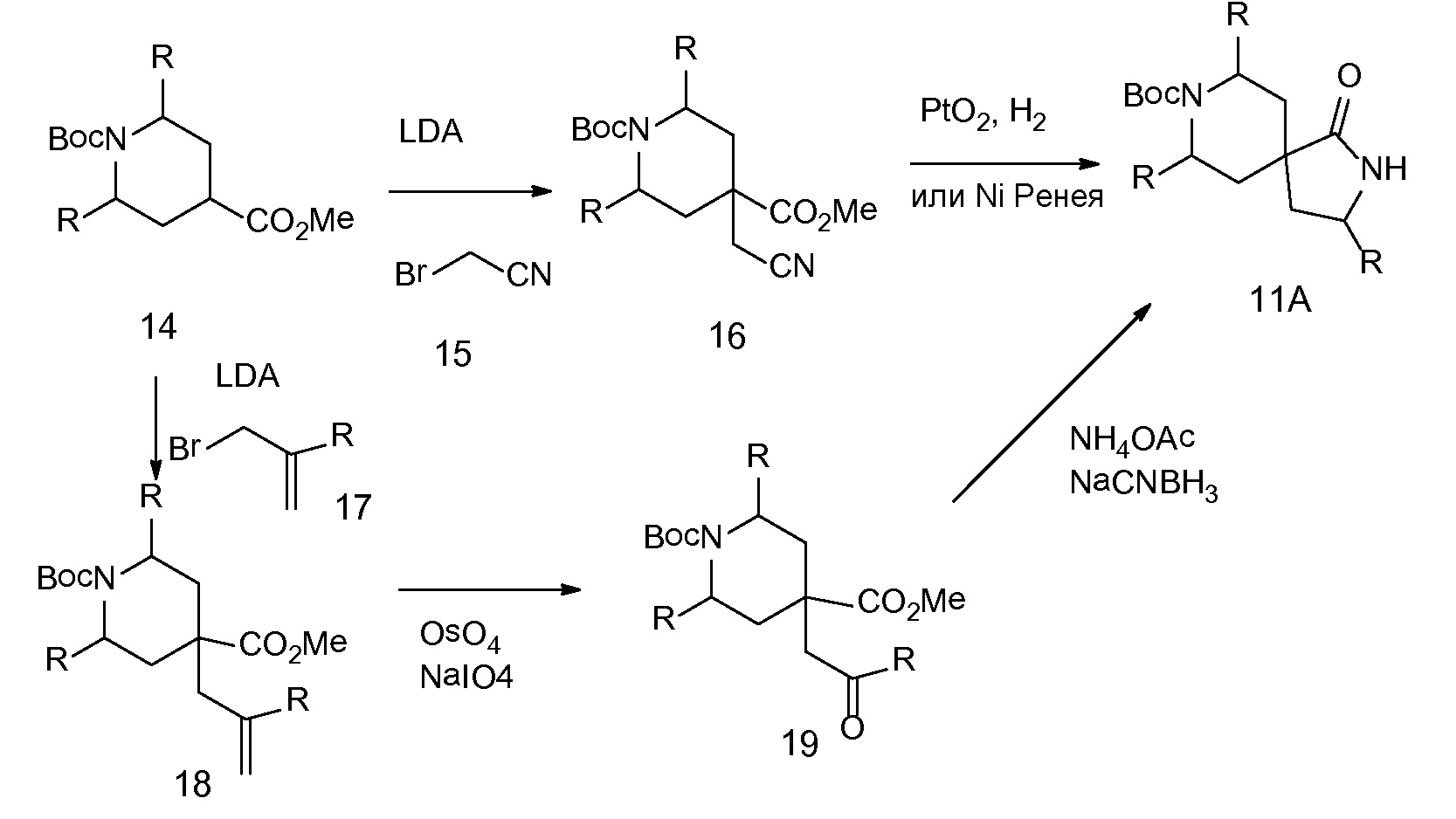
Спироциклические аминолактамы 11A можно получать многочисленными способами, включая способы, описанные на схеме 7. Имеющиеся в продаже сложные аминоэфиры 14 можно алкилировать с помощью бромацетонитрила 15 с применением основания, такого как диизопропиламид лития, получая при этом нитрильные промежуточные соединения 16. Восстановление, например, с применением оксида платины и водорода или никеля Ренея, приводит к получению лактамов 11A. Альтернативно сложные аминоэфиры можно алкилировать с помощью аллилгалогенидов 17 с применением основания, такого как диизопропиламид лития, чтобы обеспечить аллильные промежуточные соединения 18. Окислительное расщепление с применением, например, тетраоксида осмия и периодата натрия обеспечивает кетоны и альдегиды 19. Восстановительное аминирование с последовательной лактамной циклизацией до 11A можно осуществлять несколькими способами, включая обработку ацетатом аммония и цианоборгидридом натрия в растворителе, таком как метанол, как показано (на схеме 7).
СХЕМА 7
Независимый синтез диастереомеров и энантиомеров или их хроматографические разделения можно обеспечивать, как известно в данной области техники, путем подходящей модификации описанной здесь процедуры. Их абсолютную стереохимию можно определять с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые дериватизируют, если необходимо, реагентом, содержащим асимметричный центр с известной абсолютной стереохимией, или с помощью спектроскопии вибрационного циркулярного дихроизма (VCD).
Заявленные соединения можно получать путем модификации процедур, описанных в разделе "Примеры", в зависимости от конкретного случая. Исходные материалы имеются в продаже или их получают с помощью известных процедур или как проиллюстрировано. Следующие примеры предлагаются только с целью дополнительной иллюстрации, и не подразумевается, что они должны ограничивать описываемое изобретение.
Реакции, чувствительные к влаге или воздуху, осуществляли в атмосфере азота или аргона с применением безводных растворителей и реагентов. Степень протекания реакций определяли либо с помощью аналитической тонкослойной хроматографии (ТСХ), обычно осуществляемой на пластинах для ТСХ (E. Merck), предварительно покрытых силикагелем 60F-254 с толщиной слоя 0,25 мм, либо с помощью способа жидкостной хроматографии - масс-спектрометрии (ЖХ-МС).
Обычно применяемая аналитическая система ЖХ-МС состояла из платформы Waters ZQ с электрораспылительной ионизацией в режиме детектирования положительных ионов с применением системы ВЭЖХ серии Agilent 1100 с автосамплером. Колонка обычно представляла собой колонку Waters Xterra MS C18, 3,0×50 мм, 5 мкм. Скорость потока составляла 1 мл/мин, и объем впрыска составлял 10 мкл. Диапазон детектирования в УФ-области составлял 210-400 нм. Подвижная фаза состояла из растворителя A (вода + 0,06% TFA) и растворителя B (ацетонитрил + 0,05% TFA) с градиентом от 100% растворителя A в течение 0,7 мин, меняющимся до 100% растворителя B на протяжении 3,75 мин и поддерживаемым в течение 1,1 мин, с последующим возвращением к 100% растворителя A на протяжении 0,2 мин.
Очистку веществ с помощью препаративной ВЭЖХ обычно осуществляли с применением управляемой масс-спектрометрической системы. Обычно очистку проводили на лабораторном оборудовании для хроматографии Waters Chromatography Workstation, оснащенном ЖХ-МС-системой, состоящей из одного квадрупольного масс-спектрометра Waters ZQ с электрораспылительной ионизацией, градиентного насоса Waters 2525, инжектора/коллектора Waters 2767, детектора Waters 996 PDA; условия МС: 150-750 а.е.м., электрораспылительная ионизация в режиме положительных ионов (Positive Electrospray), сборник фракций с управлением от МС и колонка Waters Sunfire C-18, 5 микрон, 30 мм (вн.д.) × 100 мм. Подвижные фазы состояли из смесей ацетонитрила (10-100%) и воды, содержащей 0,1% TFA. Скорости потока поддерживали при 50 мл/мин, объем впрыска составлял 1800 мкл, и диапазон детектирования в УФ-области составлял 210-400 нм. Градиенты подвижных фаз оптимизировали для индивидуальных соединений.
Реакции, проводимые с применением микроволнового облучения, обычно осуществляли с применением микроволнового реактора Emrys Optimizer фирмы Personal Chemistry или микроволнового реактора Initiator фирмы Biotage.
Концентрирование растворов осуществляли на ротационном испарителе при пониженном давлении. Флэш-хроматографию обычно проводили с применением оборудования для флэш-хроматографии фирмы Biotage (Dyax Corp.) на силикагеле (32-63 мМ, размер пор 60 Å) в предварительно заполненных картриджах известного размера. 1HЯМР-спектры регистрировали на спектрометрах с рабочей частотой 500 MГц с использованием растворов образцов в CDCl3, если не указано иначе. Химические сдвиги приведены в миллионных долях (м.д.). В качестве внутреннего стандарта в растворах CDCl3 применяли тетраметилсилан (TMS), и в качестве внутреннего стандарта в растворах образцов в CD3OD применяли пик остаточного CH3OH или TMS. Константы взаимодействия (J) приведены в герцах (Hz).
Хиральную аналитическую хроматографию обычно осуществляли на одной из хиральных колонок Chiralpak AS, Chiralpak AD, Chiralcel OD, Chiralcel IA или Chiralcel OJ (250×4,6 мм) (Daicel Chemical Industries, Ltd.) с указанием процентного содержания либо этанола в гексане (%Et/гексан), либо изопропанола в гептане (%IPA/гептан) в качестве систем растворителей для изократического режима элюирования. Хиральную препаративную хроматографию иногда проводили на одной из хиральных колонок Chiralpak AS, Chiralpak AD, Chiralcel OD, Chiralcel IA или Chiralcel OJ (20×250 мм) (Daicel Chemical Industries, Ltd.) с требуемыми системами растворителей для изократического режима элюирования, указанными для хиральной аналитической хроматографии или условий сверхкритической флюидной хроматографии (СФХ). Альтернативно хиральную препаративную хроматографию осуществляли в условиях сверхкритической флюидной хроматографии (СФХ) с применением одной из хиральных колонок Chiralpak AS, Chiralpak AD-Н, Chiralcel OD-Н, Chiralpak IС или Chiralcel OJ-H (250×21,2 мм) (Daicel Chemical Industries, Ltd.). Когда в примерах и таблицах приводятся времена удерживания, подразумевается, что они не должны представлять собой исчерпывающую характеристику конкретного соединения, поскольку, как известно специалисту в данной области, времена удерживания будут меняться, и время и/или порядок пиков элюирования может меняться в зависимости от условий осуществления хроматографии, таких как применяемая колонка, состояние колонки и системы растворителей и применяемое оборудование.
Концентрирование растворов обычно осуществляют на ротационном испарителе при пониженном давлении. Флэш-хроматографию осуществляли на силикагеле (230-400 меш). Спектры ЯМР получали для раствора образцов в CDCl3, если не указано иначе. Константы взаимодействия (J) приведены в герцах (Гц).
Аббревиатуры и сокращения, применяемые в описании, включают в себя: -C(О)CH3 (Ac); -OC(О)CH3 (OAc); уксусную кислоту (AcOH; HOAc); 1-хлорэтилхлорформиат (ACE-Cl); 2,2'-бис(дифенилфосфино)-1,1'- бинафтил (BINAP); т-бутилоксикарбонил (Boc или BOC); ди-т-бутилдикарбонат ((BOС)2O, BОC2O); бензилоксикарбонил (Cbz); простой циклопентилметиловый эфир (CPME); карбонилдиимидазол (CDI); трифторид диэтиламиносеры (DAST); дибензилиденацетон (dba); 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU); 1,2-дихлорэтан (DCE); дихлорметан (ДХМ); диметоксиэтан (DME); гидрид диизобутилалюминия (DIBAL-H); N,N-диизопропилэтиламин (DIEA, DIPEA, основание Хунига); диизопропиламин (DIPA); 1,1'-бис(дифенилфосфино)ферроцен (dppf, DPPF); периодинан Десса-Мартина (DMP; 1,1,1-триацетокси-1,1-дигидро-1,2-бензиодоксол-3(1H)-он); диметилсульфид (DMS); диметилсульфоксид (ДМСО); N,N-диметилформамид (ДМФА); 4-диметиламинопиридин (DMAP); диметилацетамид (DMA; DMAC); 1,3-бис(дифенилфосфино)пропан (DPPP); этилацетат (EtOAc); простой диэтиловый эфир (простой эфир или Et2О); 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC, EDAC или EDCI); гексафторфосфат 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU); гексан (гес.); гескаметилфосфорамид (HMPA); гидрат 1-гидроксибензотриазола (HOBt); изопропанол (IPA); изопропилацетат (IPAc); бис(триметилсилил)амид калия (KHMDS); алюмогидрид лития (LAH); диизопропиламид лития (LDA); 3-хлорпероксибензойная кислота (mCPBA); метанол (MeOH); CH3SO2- (мезил или Ms); метансульфонилхлорид или мезилхлорид (MsCl); метансульфоновая кислота (MsOH); простой метил-трет-бутиловый эфир (MTBE); никотинамидадениндинуклеотидфосфат (NADP); N-бромсукцинимид (NBS); N-хлорсукцинимид (NCS); N-йодсукцинимид (NIS); N-метилморфолин-N-оксид (NMO); N-метилморфолин (NMP); гексаметилдисилазид натрия (NaHMDS); триацетоксиборгидрид натрия (NaBH(OAc)3); хлорхромат пиридиния (PCC); фенил (Ph); петролейный эфир (PE или петролейный эфир); тетракис(трифенилфосфин)палладий (Pd(PPh3)4); трис(дибензилидинацетон)дипалладий (Pd2(dba)3);
Pd(dppf)Cl2 или PdCl2(dppf) представляет собой 1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), который может образовывать комплекс с CH2Cl2; фторид тетра-н-бутиламмония (TBAF); трет-бутилдиметилсилилхлорид (TBS-Cl); триэтиламин (TEA); трифторуксусная кислота (TFA); -SO2CF3 (Tf); трифторметансульфоновая кислота (трифлатная кислота, TfOH); трифторметансульфоновый ангидрид (ангидрид трифлатной кислоты, (Tf)2О); 2-тетрагидрофуран (ТГФ); N,N,N',N'-тетраметилэтилендиамин (TMEDA); п-толуолсульфоновая кислота (TsOH); дициклогексилфосфино-2',4',6'-триизопропилбифенил (X-Phos); тетрафторборат диэтиламинодифторсульфиния (XtalFluor-E®); 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Ксантфос). Дополнительные аббревиатуры и сокращения: исходный материал (SM); круглодонная колба (RB или RBF); водный (aq); насыщенный водный раствор (sat'd); насыщенный водный раствор хлорида натрия (насыщенный раствор соли); жидкостная хроматография среднего давления (ЖХСД); высокоэффективная жидкостная хроматография (ВЭЖХ); препаративная ВЭЖХ (преп-ВЭЖХ); флэш-хроматография (FC); жидкостная хроматография (LC); сверхкритическая флюидная хроматография (СФХ); тонкослойная хроматография (ТСХ); препаративная ТСХ (преп-ТСХ); масс-спектр (ms или МС); жидкостная хроматография - масс-спектрометрия (ЖХ-МС, ЖХМС или ЖХ/МС); объем колонки (CV); комнатная температура (rt, r.t. или RT); час (часы) (ч или час); минута (минуты) (мин); время удерживания (Rt); грамм (граммы) (г); миллиграмм (миллиграммы) (мг); миллилитр (миллилитры) (мл); микролитр (микролитры) (мкм); миллимоль (ммоль); объем:объем (V/V). CELITE® представляет собой торговую марку диатомовой земли, и SOLKA FLOC® представляет собой торговую марку порошкообразной целлюлозы. X или x можно применять для обозначения числа повторений действия (например, промывали 2×200 мл 1Н HCl) или для обозначения размера (например, размер колонки составляет 30×250 мм).
Далее представлены типичные процедуры получения промежуточных соединений, применяемых для получения конечных продуктов, описанных в примерах, которые следуют далее. Такие примеры приведены только с целью дополнительной иллюстрации и не подразумевают каких-либо ограничений описанного изобретения.
Понятно, что хиральный центр в соединении может существовать в виде "S" или "R" - стереоконфигурации, или в виде смеси обеих конфигураций. Во многих примерах соединения, содержащие хиральный центр, разделяли на отдельные стереоизомеры (например, упоминаемые как изомер A и изомер B или как быстро/медленно элюирующие изомеры), или каждый изомер синтезировали из отдельного изомера промежуточного соединения. За исключением определенного хирального центра в исходной смеси абсолютную стереохимию (R или S) каждого из отдельных изомеров не определяли, если специально не указано иначе.
Описанные ниже промежуточные соединения могут быть упомянуты в настоящем описании под их номером, перед которым стоит цифра "I-". Например, промежуточное соединение 4 A сокращенно обозначается как I-4 A.
Промежуточное соединение 1
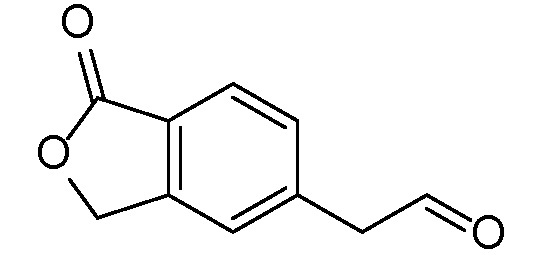
(1-оксо-1,3-дигидро-2-бензофуран-5-ил)ацетальдегид
Стадия A
5-(1,3-диоксолан-2-ил)-метил)-2-бензофуран-1(3H)-он
В трехгорлую 5 л круглодонную колбу, оборудованную магнитной мешалкой для перемешивания, клапаном Файрстона, термопарой, холодильником и колбонагревателем, загружали тетрафторборат три-т-бутилфосфония (500 мг, 1,72 ммоль), ацетат палладия (II) (250 мг, 1,1 ммоль) и 5-бром-2-бензофуран-1(3H)-он (100 г, 470 ммоль). В колбу добавляли ДМФА (1,88 л) и трижды дегазировали смесь обработкой в вакууме, чередующейся с продувкой азотом. С помощью канюли добавляли раствор имеющегося в продаже бром-(1,3-диоксолан-2-илметил)цинка (1,03 л, 516 ммоль) и смесь снова трижды дегазировали. Затем смесь нагревали при 85°C в течение 5 часов. Анализ с помощью ВЭЖХ-МС показывал, что реакция не завершилась. Смесь перемешивали при 85°C в течение 5 более часов. Затем смеси давали охладиться до комнатной температуры в течение ночи. Добавляли 2-метил-ТГФ (2 л) и насыщенный раствор соли и перемешивали смесь в течение 5 мин. Слои разделяли и водный слой снова экстрагировали 2-метил-ТГФ. Органические слои объединяли, трижды промывали насыщенным раствором соли (каждый раз по 4 л), сушили над MgSО4, фильтровали, и концентрировали. Неочищенный продукт очищали флэш-хроматографией (картридж с 1,5 кг диоксида кремния) путем элюирования раствором 0-20% этилацетата в дихлорметане, получая при этом 5-(1,3-диоксолан-2-илметил)-2-бензофуран-1(3H)-он. ЖХ-МС (IE, m/z): 221 [M+1]+.
Стадия B
(1-оксо-l,3-дигидро-2-бензофуран-5-ил)ацетальдегид
5-(1,3-диоксолан-2-илметил)-2-бензофуран-1(3H)-он (61 г, 280 ммоль) объединяли с водой (2,2 л) в 5 л круглодонной колбе, оборудованной насадкой Кляйзена, термопарой, магнитной мешалкой и барботером азота. Добавляли водный раствор HCl (2M, 1,14 л, 2,29 моль) и нагревали полученную смесь при 40°C в течение 8 часов. Затем смесь перемешивали в течение ночи при комнатной температуре. Смесь трижды экстрагировали 2 л этилацетата. Объединенные органические слои концентрировали, получая при этом (1-оксо-l,3-дигидро-2-бензофуран-5-ил)ацетальдегид. ЖХ-МС (IE, m/z): 177 (M+1)+.
Промежуточное соединение 2
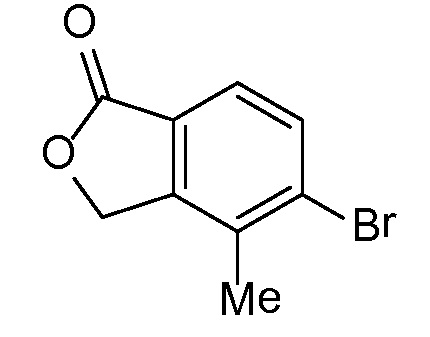
5-бром-4-метил-2-бензофуран-1(3H)-он
Стадия A
(3-бром-2-метилфенил)метанол
К раствору 3-бром-2-метилбензойной кислоты (35 г, 163 ммоль) в ТГФ (200 мл) добавляли комплекс боран-ТГФ (1,0 M, 212 мл, 212 ммоль). Смесь оставляли перемешиваться в течение 24 часов. ТСХ-анализ показывал наличие одного пятна одного продукта. Реакционную смесь гасили водой. Растворитель ТГФ удаляли при пониженном давлении. Полученное твердое вещество растворяли в этилацетате (500 мл), промывали 1Н раствором HCl, раствором карбоната натрия и насыщенным раствором соли. Органический слой сушили над сульфатом натрия и концентрировали, получая при этом (3-бром-2-метилфенил)метанол. 1H ЯМР (500 МГц, CDCl3) δ 7,76 (д, J=8,0 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), 5,30 (с, 2H), 2,42 (с, 3H).
Стадия B
5-бром-4-метил-2-бензофуран-1(3H)-он
В колбу с (3-бром-2-метилфенил)метанолом (6,0 г, 30 ммоль) добавляли 1M раствор трифторацетата таллия (16,2 г, 29,8 ммоль) в TFA. Смесь перемешивали при комнатной температуре в течение ночи. ТСХ-анализ показывал отсутствие остатков исходного материала. Растворитель удаляли в вакууме, и остаток откачивали в высоком вакууме в течение 30 минут, чтобы гарантировать полное удаление TFA. Затем к остатку добавляли хлорид палладия (II) (529 мг, 2,98 ммоль), хлорид лития (2,53 г, 59,7 ммоль), оксид магния (2,41 г, 59,7 ммоль) и MeOH (150 мл). Через реакционную смесь дважды продували CO и выдерживали в атмосфере CO при комнатной температуре. ЖХ-анализ показывал большое пятно продукта не позднее, чем через 2 часа. К полученному раствору добавляли этилацетат, получая при этом осадок солей. Раствор черного цвета фильтровали через рыхлый слой CELITE®, промывали EtOAc, адсорбировали на диоксиде кремния и очищали хроматографией на силикагеле, получая при этом указанное в заголовке соединение. 1H-ЯМР (500 МГц, CDCl3) δ м.д. 7,71 (д, J=8,0 Гц, 1H), 7,58 (д, J=8,0 Гц, 1H), 5,25 (с, 2H), 2,37 (с, 3H).
Промежуточное соединение 3
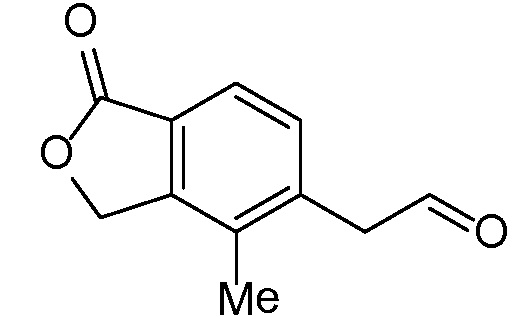
(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)ацетальдегид
Стадия A
4-метил-5-проп-2-ен-1-ил-2-бензофуран-1(3H)-он
В колбу загружали 5-бром-4-метил-2-бензофуран-1(3H)-он (320 мг, 1,409 ммоль) и при перемешивании с помощью магнитной мешалки добавляли аллил-три-н-бутилолово (0,655 мл, 2,11 ммоль), Pd(PPh3)4 (244 мг, 0,211 ммоль), хлорид лития (179 мг, 4,23 ммоль) и толуол (15 мл). Реакционную смесь 2 раза продували азотом, затем нагревали с обратным холодильником в течение 4 часов. Продукт выделяли хроматографией на силикагеле, получая при этом 4-метил-5-проп-2-ен-1-ил-2-бензофуран-1(3H)-он.
Стадия B
(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)ацетальдегид
Раствор полученного выше олефина (220 мг, 1,2 ммоль) в MeOH (20 мл) охлаждали до -78°C. Через полученный раствор барботировали озон до тех пор, пока реакционная смесь не посинеет. Затем через реакционную смесь барботировали азот, чтобы удалить избыток озона, с последующим добавлением DMS (0,870 мл; 11,7 ммоль). Реакционной смеси давали нагреться до комнатной температуры. Неочищенный продукт очищали флэш-хроматографией, получая при этом указанное в заголовке соединение. 1H-ЯМР (500 МГц, CDCl3) δ м.д. 9,78 (с, 1H), 7,75 (д, J=7,5 Гц, 1H), 7,34 (д, J=7,5 Гц, 1H), 5,27 (с, 2H), 3,90 (с, 2H), 2,23 (с, 3H).
Промежуточное соединение 4
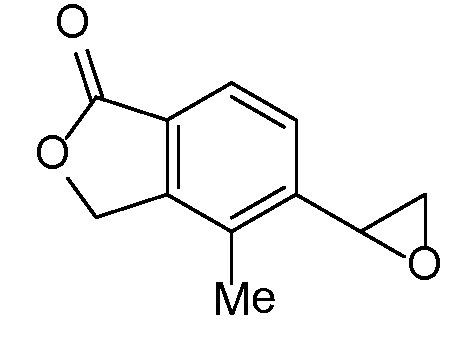
4-метил-5-оксиран-2-ил-2-бензофуран-1(3H)-он
Стадия A
5-этенил-4-метил-2-бензофуран-1(3H)-он
Смесь 5-бром-4-метил-2-бензофуран-1(3H)-она (598 мг, 4,47 ммоль), винилтрифторбората калия (507 мг, 2,23 ммоль), аддукта PdCl2(dppf)-CH2Cl2 (182 мг, 0,223 ммоль) и TEA (0,622 мл, 4,47 ммоль) добавляли к 10 мл этанола в 20 мл трубке для микроволновой печи. Трубку герметизировали и дегазировали, затем нагревали до 140°C в течение 20 мин. ЖХ-МС-анализ показывал появление пика продукта. Реакционную смесь разбавляли этилацетатом, дважды промывали насыщенным раствором соли, сушили и выпаривали досуха. Неочищенный продукт очищали ЖХСД-хроматографией на колонке Redi-sep размером 120 г с применением системы растворителей 0-80% EtOAc/гексан, получая при этом 5-этенил-4-метил-2-бензофуран-1(3H)-он. 1H-ЯМР (500 МГц, CDCl3): δ м.д. 7,76 (д, J=8 Гц, 1H), 7,03 (дд, J=11, 17 Гц, 1H), 5,84 (д, J=17 Гц, 1H), 5,55 (д, J=11 Гц, 1H), 5,29 (с, 2H), 2,34 (с, 3H); ЖХ-МС: M+1= 175.
Стадия B
4-метил-5-оксиран-2-ил-2-бензофуран-1(3H)-он
5-этенил-4-метил-2-бензофуран-1(3H)-он (1,46 г, 8,38 ммоль) добавляли к ДХМ (25 мл) при 0°C, затем добавляли mCPBA (2,89 г, 16,8 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. Реакционную смесь по очереди однократно промывали насыщенным водным раствором Na2S2О3, NaHCО3 и насыщенным раствором соли. Органический слой сушили над Na2SО4, фильтровали и выпаривали досуха. Неочищенный материал очищали ЖХСД-хроматографией на колонке Redi-sep размером 120 г путем элюирования системы растворителей 0-80% EtOAc/гексан, получая при этом требуемый 4-метил-5-оксиран-2-ил-2-бензофуран-1(3H)-он. 1H-ЯМР (500 МГц, CDCl3): δ м.д. 7,77 (д, J=8 Гц, 1H), 7,43 (д, J=8 Гц, 1H), 5,30 (с, 2H), 4,12 (с, 1H), 3,27 (т, J=4 Гц, 1H), 2,735 (дд, J=2,2, 5,5 Гц, 1H) , 2,43 (с, 3H). ЖХ-МС: M+1=191.
Промежуточные соединения 4A и 4B (способ 1)
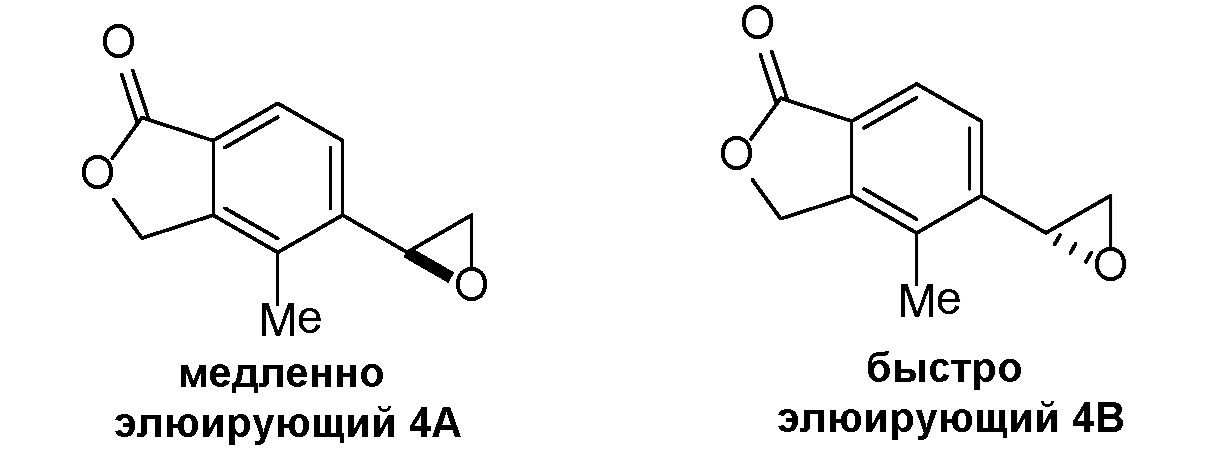
4A: 4-метил-5-[(2S)-оксиран-2-ил]-2-бензофуран-1(3H)-он;
4B: 4-метил-5-[(2R)-оксиран-2-ил]-2-бензофуран-1(3H)-он
Рацемическую смесь 4-метил-5-оксиран-2-ил-2-бензофуран-1(3H)-она разделяли на хиральной колонке ChiralPak® AD-H (5×25 см) в условиях сверхкритической флюидной хроматографии (СФХ) на оборудовании для препаративной СФХ Berger MGIII. Рацемат разбавляли до 50 мг/мл смесью 1:1 ДХМ:MeOH. Разделение осуществляли с применением 10% EtOH/CО2, скорость потока 200 мл/мин, 100 бар, 25°C. Впрыскивания по 500 мкл осуществляли с промежутком в 2,12 минут. Первым вымывался быстро элюирующий эпоксид (4-метил-5-[(2R)-оксиран-2-ил]-2-бензофуран-1(3H)-он, 4B), и вторым вымывался медленно элюирующий эпоксид (4-метил-5-[(2S)-оксиран-2-ил]-2-бензофуран-1(3H)-он, 4A).
Альтернативно разделение также можно обеспечивать с применением подвижной фазы 8% MeOH/98% CО2 при скорости потока 100 мл/мин. В таком случае образец получали путем растворения в метаноле до концентрации 20 мг/мл и с применением объема впрыска 1 мл за одно впрыскивание. После разделения фракции сушили на ротационном испарителе при температуре бани 40°C.
Заключение об абсолютной стереохимии каждого энантиомера делали на основе рентгеновского определения кристаллической структуры конечного соединения (эфира Мошера и эфира Троста), полученного с использованием соединения 4B и реагента Мошера и реагента Троста и анализа HNMR-спектров сложных эфиров, полученных из исходного соединения 4B. Оба эпоксидных изомера находят применение в настоящем изобретении.
Промежуточное соединение 4B (способ 2)
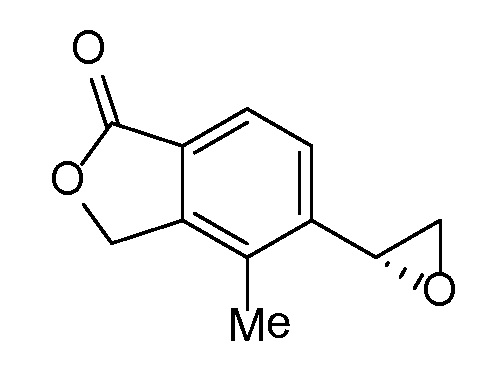
4-метил-5-[(2R)-оксиран-2-ил]-2-бензофуран-1(3H)-он
Стадия A
3-гидроксиметил-2-метилфенол
В 5 л трехгорлую круглодонную колбу (RB), оборудованную верхнеприводной мешалкой, загружали NaBH4 (87,0 г; 2,30 моль) и ТГФ (3,0 л) и охлаждали полученную взвесь до 10°C. Затем к взвеси порциями добавляли 3-гидрокси-2-метилбензойную кислоту (175 г; 1,15 моль) на протяжении 20 мин (Tmax 17°C). Образовавшуюся при перемешивании взвесь выдерживали в течение дополнительных 45 мин при 10-15°C, после чего медленно добавляли BF3-OEt2(321 мл, 2,53 моль) на протяжении 1,5 часов. Взвесь выдерживали при 10°C -15°C в течение 2 часов, затем проводили анализ на завершение реакции (степень превращения 98,5%). Взвесь охлаждали до температуры <10°C и медленно гасили с помощью 931 мл MeOH на протяжении 1,5 часов (выделение газа). Полученную взвесь выдерживали в течение ночи при комнатной температуре. Смесь охлаждали до температуры <10°C, затем гасили 1Н раствором HCl (1,5 л), получая при этом гомогенный раствор (pH раствора ~ 1), который выдерживали в течение 30 мин и затем удаляли органические растворители путем ротационного выпаривания приблизительно до 1,8 л общего объема реакционной смеси (температуру бани устанавливали на 50°C; температура внутри концентрата после ротационного выпаривания составляла ~ 40°C). Взвесь выдерживали при 45°C в течение 30 мин, затем медленно охлаждали до 15°C. Твердые вещества отфильтровывали и промывали холодной (15°C) водой (2×300 мл), получая при этом 3-гидроксиметил-2-метилфенол. 1H-ЯМР (400 МГц, DMSO-d6): δ 9,11 (с, 1H), 6,95 (т, J=7,8 Гц, 1H), 6,82 (д, J=7,4 Гц, 1H), 6,71 (д, J=7,8 Гц, 1H), 4,93 (т, J=5,5 Гц, 1H), 4,44 (д, J=5,5 Гц, 2H), 2,06 (с, 3H).
Стадия B
4-бром-3-гидроксиметил-2-метилфенол
3-Гидроксиметил-2-метилфенол (113,9 г; 824,0 ммоль) растворяли в смеси ацетонитрила (850 мл) и трифторуксусной кислоты (750,0 мл; 9,735 ммоль) в трехгорлой 5 л колбе в атмосфере азота. Реакционную смесь охлаждали до -33°C. На протяжении 15 минут добавляли N-бромсукцинимид (141 г; 791 ммоль), поддерживая во время добавления температуру в диапазоне от -35 до -33°C. Реакционную смесь оставляли перемешиваться в течение дополнительных 15 мин, во время которых температура снижалась до -40°C. Охлаждающую баню убирали и добавляли карбонат калия (741,0 г; 5,358 ммоль), разбавленный водой до общего объема 1,0 л. Наблюдали газовыделение и повышали температуру до 25°C. Добавляли MTBE (1,5 л) и переносили реакционную смесь в делительную воронку. Слои разделяли. Водный слой разбавляли водой (500 мл) и экстрагировали смесью MTBE (1 л) + EtOAc (500 мл) и затем MTBE (500 мл) + EtOAc (250 мл). Объединенные органические слои промывали водой (240 мл) и сушили над сульфатом натрия. Сульфат натрия удаляли фильтрованием, промывали дополнительным количеством MTBE и концентрировали при пониженном давлении. Добавляли MTBE (684 мл, 2 объема) и нагревали суспензию до 40°C, получая при этом гомогенный раствор. Раствору давали охладиться до комнатной температуры. Добавляли шесть объемов гептана и перемешивали суспензию в течение ночи. Фильтровали суспензию и промывали кристаллы смесью гептан: MTBE (500 мл) 4:1 с последующим промыванием гептаном (500 мл). Твердое вещество сушили в вакууме, получая при этом 4-бром-3-гидроксиметил-2-метилфенол. 1H ЯМР (400 МГц, DMSO-d6): δ 9,52 (с, 1H), 7,21 (д, J=8,6 Гц, 1H), 6,71 (д, J=8,6 Гц, 1H), 4,88 (т, J=5,1 Гц, 1H), 4,59 (д, J=5,1 Гц, 2H), 2,23 (с, 3H).
Стадия C
5-гидрокси-4-метил-3H-изобензофуран-1-он
В 2 л 3-горлую колбу, оборудованную верхнеприводной мешалкой, входным отверстием для N2 и холодильником загружали 4-бром-3-гидроксиметил-2-метилфенол (100 г; 461 ммоль), CuCN (83,0 г, 921 ммоль) и ДМФА (500 мл). Раствор барботировали N2 в течение 15 мин, затем нагревали до 145°C, получая при этом гомогенный раствор. Раствор выдерживали при 145°C в течение 2 часов, затем реакционную смесь охлаждали до 95°C. Добавляли 41,5 мл воды (барботированную N2) и выдерживали реакционную смесь в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры, затем твердые вещества отфильтровывали через фильтр SOLKA FLOC® и осадок на фильтре промывали 50 мл ДМФА. В 3 л колбу, содержащую 1 л EtOAc, добавляли фильтрат с ДМФА. На дне колбы образовывался слой осадка. Суспензию в ДМФА/EtOAc фильтровали через фильтр SOLKA FLOC® и промывали осадок на фильтре 250 мл EtOAc. Полученный фильтрат промывали 5%-ным раствором соли (3×500 мл). Водные слои экстрагировали 500 мл EtOAc, сушили объединенную органику над MgSО4, фильтровали и выпаривали. Твердые вещества суспендировали в 250 мл MTBE при комнатной температуре, затем фильтровали и промывали 100 мл MTBE. Твердые вещества сушили в вакууме при комнатной температуре, получая при этом 5-гидрокси-4-метил-3H-изобензофуран-1-он. 1H ЯМР (400 МГц, DMSO-d6): δ 10,52 (с, 1H), 7,51 (д, J=8,3 Гц, 1H), 6,99 (д, J=8,3 Гц, 1H), 5,28 (с, 2H), 2,07 (с, 3H).
Стадия D
4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил трифторметансульфонат
5-Гидрокси-4-метил-3H-изобензофуран-1-он (46,8 г, 285 ммоль) суспендировали в дихлорметане (935 мл) в 2 л круглодонной колбе, оборудованной верхнеприводной мешалкой, в атмосфере азота. Добавляли триэтиламин (59,5 мл; 427 ммоль) и реакционную смесь охлаждали на ледяной бане до 3,8°C. На протяжении 50 мин с помощью капельной воронки добавляли трифторметансульфоновый ангидрид (67,4 мл, 399 ммоль), поддерживая температуру <10°C. После перемешивания реакционной смеси в течение дополнительных 15 мин реакционную смесь гасили водой (200 мл), затем перемешивали с DARCO® KB (активированный уголь, 25 г) в течение 15 мин. Двухфазную смесь фильтровали через фильтр SOLKA FLOC®, промывая дополнительным количеством дихлорметана, и переносили в делительную воронку, где ее разбавляли дополнительным количеством воды (300 мл). Слои разделяли, и органический слой промывали водой (500 мл) и 10% раствором соли (200 мл). Дихлорметановый раствор сушили над сульфатом натрия, фильтровали и выпаривали. Твердый остаток адсорбировали на силикагеле (27,5 г) и элюировали через рыхлый слой силикагеля (271 г) смесь 25% этилацетат/гексаны. Полученный раствор концентрировали в вакууме с кристаллизацией продукта во время концентрирования. Суспензию фильтровали, твердое вещество промывали гептаном и сушили в вакууме и атмосфере азота, получая при этом сложный 4-метил-1-оксо-1,3-дигидроизобензофуран-5-иловый эфир трифторметансульфоновой кислоты. 1H ЯМР (400 МГц, CDCl3): δ 7,87 (д, J=8,4 Гц, 1H), 7,47 (д, J=8,4 Гц, 1H), 5,32 (с, 2H), 2,41 (с, 3H).
Стадия E
5-(1-бутоксивинил)-4-метил-3H-изобензофуран-1-он
В 1 л 3-горлую колбу загружали сложный 4-метил-1-оксо-1,3-дигидроизобензофуран-5-иловый эфир трифторметансульфоновой кислоты (63,0 г, 213 ммоль), ДМФА (315 мл), простой бутилвиниловый эфир (138 мл, 1063 ммоль), затем Et3N (35,6 мл, 255 ммоль). В раствор в течение 20 мин барботировали N2. К раствору добавляли Pd(OAc)2 (1,19 г, 5,32 ммоль) и DPPP (2,41 г, 5.85 ммоль) и барботировали N2 в течение дополнительных 10 мин, затем нагревали до 80°C. После выдержки в течение 1 часа раствор охлаждали до <10°C, затем гасили 630 мл EtOAc и промывали 5% NH4Cl (2×315 мл), 10%-ным раствором соли (2×315 мл), сушили над MgSО4, фильтровали, концентрировали путем ротационного выпаривания и промывали EtOAc (3×100 мл), чтобы удалить избыток простого бутилвинилового эфира, получая при этом неочищенный 5-(1-бутоксивинил)-4-метил-3H-изобензофуран-1-он. 1H ЯМР (400 МГц, DMSO-d6): δ 7,67 (д, J=7,7 Гц, 1H), 7,48 (д, J=7,7 Гц, 1H), 5,42 (с, 2H), 4,54 (д, J=2,3 Гц, 1H), 4,27 (д, J=2,3 Гц, 1H), 3,85 (т, J=6,4 Гц, 2H), 2,27 (с, 3H), 1,71-1,64 (м, 2H), 1,46-1,37 (м, 2H), 0,92 (т, J=7,4 Гц, 3H).
Стадия F
5-(2-бром-aцетил)-4-метил-3H-изобензофуран-1-он
В 1 л 3-горлую колбу, оборудованную верхнеприводной мешалкой, добавляли неочищенный 5-(1-бутоксивинил)-4-метил-3H-изобензофуран-1-он (55,8 г) и ТГФ (315 мл). Раствор охлаждали до <5°C, после чего добавляли воду (79 мл) и поддерживали раствор при <5°C. Затем порциями добавляли NBS (41,6 г), поддерживая при этом Tmax = 19°C. Затем раствор нагревали до комнатной температуры в течение 30 минут. Добавляли HBr (48%, 0,241 мл) и выдерживали реакционную смесь при комнатной температуре в течение приблизительно 1 часа, после чего к партии добавляли 236 мл воды. Для поддержания температуры при 20°C применяли водяную баню. Добавляли дополнительные 315 мл воды (состав растворителя ТГФ:вода в отношении 1:2) и охлаждали взвесь до 15°C. Полученные твердые вещества отфильтровывали и промывали в 150 мл холодной смеси ТГФ:вода в отношении 1:2 (15°C) путем промывки вытеснением, за которым следует промывка 100 мл взвеси. Твердые вещества сушили в вакууме при комнатной температуре, получая при этом 5-(2-бромaцетил)-4-метил-3H-изобензофуран-1-она. 1H ЯМР (400 МГц, DMSO-d6): δ 7,99 (д, J=7,8 Гц, 1H), 7,82 (д, J=7,8 Гц, 1H), 5,49 (с, 2H), 4,92 (с, 2H), 2,33 (с, 3H).
Стадия G
4-метил-5-[(2R)-оксиран-2-ил]-2-бензофуран-1(3H)-он
5-(2-бромaцетил)-4-метил-3H-изобензофуран-1-он (48,8 г; 181 ммоль) загружали в 5 л 3-горлую круглодонную колбу, оборудованную верхнеприводной мешалкой, термопарой и колбонагревателем. Добавляли 2-пропанол (1,22 л) с последующим добавлением 610 мл 0,1 M буферного раствора фосфата калия со значением pH=7. В 1,0 л колбу Эрленмейера загружали буферный раствор (610 мл), добавляли в колбу Эрленмейера 2,44 г NADP и перемешивали путем вращения сосуда до растворения. В колбу Эрленмейера добавляли восстанавливающий фермент KRED MIF-20 (2,44 г) (поставляемый компанией Codexis, Inc., 200 Penobscot Drive, Redwood City, CA 94063, www.codexis.com, т. 1-650-421-8100) и перемешивали смесь путем вращения сосуда до растворения твердых веществ. Полученный раствор добавляли в 5 л круглодонную колбу, которую затем нагревали до 28°C и выдерживали в течение 6 часов; полученную при этом реакционную смесь охлаждали до комнатной температуры и добавляли триэтиламин (50,2 мл; 360 ммоль). Полученный раствор выдерживали при 40°C в течение 1 часа. Раствор, содержащий легкую взвесь, охлаждали до комнатной температуры, после чего добавляли 122 г NaCl. Раствор выдерживали при комнатной температуре, затем экстрагировали 1,22 л изопропилацетата (IPAc). Водный слой повторно экстрагировали 400 мл IPAc и промывали объединенную органику 400 мл 20%-ного раствора соли, сушили над MgSО4, фильтровали и концентрировали ротационным выпариванием. Полученные твердые вещества помещали в 100 мл IPAc (густая взвесь). Добавляли гексаны (400 мл) и выдерживали суспензию при комнатной температуре, затем фильтровали и промывали раствором гексаны:IPAc (150 мл) в отношении 5:1. Кристаллические твердые вещества сушили в вакууме при комнатной температуре, получая при этом 4-метил-5-[(2R)-оксиран-2-ил]-2-бензофуран-1(3H)-он. 1H ЯМР (400 МГц, CDCl3): δ 7,75 (д, J=8,1 Гц, 1H), 7,42 (д, J=8,1 Гц, 1H), 5,28 (с, 2H), 4,10 (дд, J=4,0, 2,8, 1H), 3,26 (дд, J=5,6, 4,0, 1H), 2,72 (дд, J=5,6, 2,8, 1H), 2,42 (с, 3H).
Промежуточное соединение 5
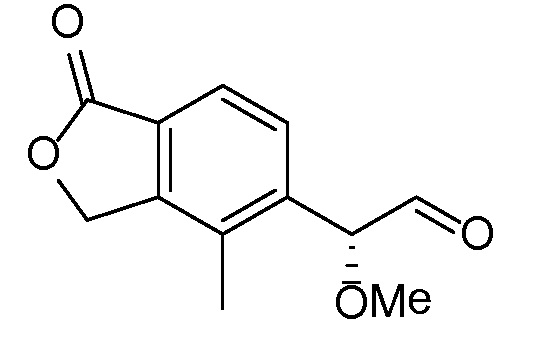
(R)-2-метокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)ацетальдегид
Стадия A
(R)-5-(2-гидрокси-1-метоксиэтил)-4-метилизобензофуран-1(3H)-он
К раствору (S)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-она (2,00 г, 10,5 ммоль) в MeOH (20 мл) добавляли моногидрат п-толуолсульфоновой кислоты (0,100 г, 0,526 ммоль). После нагревания при 80°C в течение 48 часов реакционную смесь охлаждали до комнатной температуры и затем концентрировали. Неочищенный продукт очищали колоночной хроматографией путем элюирования смесью 0-45% EtOAc/гексан. ЖХ-МС (IE, m/z): 223,2 (M+1)+.
Стадия B
(R)-2-метокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)ацетальдегид
(R)-5-(2-гидрокси-1-метоксиэтил)-4-метилизобензофуран-1(3H)-он (500 мг, 2,25 ммоль) растворяли в ДХМ (10 мл) и обрабатывали периодинаном Десса-Мартина (954 мг, 2,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение ночи. Неочищенный продукт применяли без очистки. ЖХ-МС (IE, m/z): 239,2 (M+H2О+1)+.
Промежуточное соединение 6
5-(1,2-дигидроксипропил)-4-метилизобензофуран-1(3H)-он
Стадия A
(E)-4-метил-5-(проп-1-ен-1-ил)изобензофуран-1(3H)-он
К Pd(dppf)Cl2 (0,220 г, 0,338 ммоль), K3PО4 (6,75 мл, 1 M в воде, 6,75 ммоль) в ТГФ (22 мл) добавляли (E)-трифтор(проп-1-ен-1-ил)борат калия (0,749 г, 5,06 ммоль) и 4-метил-1-оксо-1,3- дигидроизобензофуран-5-ил трифторметансульфонат (I-4B, способ 2, стадия D, 1,0 г, 3,38 ммоль). Реакционную смесь дегазировали в течение 10 мин и перемешивали полученную смесь в течение ночи при 70°C. Реакционную смесь охлаждали до комнатной температуры и разбавляли EtOAc и водой. После разделения слоев водный слой экстрагировали EtOAc и промывали объединенные органические слои насыщенным раствором соли, сушили над безводным MgSО4, фильтровали, концентрировали и очищали колоночной хроматографией на силикагеле с применением в качестве подвижной фазы смеси (0-50%) ацетон-гексаны, получая при этом указанное в заголовке соединение. ЖХ/МС:[(M+1)]+ = 189.
Стадия B
5-(1,2-дигидроксипропил)-4-метилизобензофуран-1(3H)-он
К (E)-4-метил-5-(проп-1-ен-1-ил)изобензофуран-1(3H)-ону (300 мг, 1,59 ммоль) в смеси ацетонитрил/вода (10/1, 18 мл) добавляли NMO (243 мг, 2,07 ммоль) и дигидрат осмата(VI) калия (29,4 мг, 0,080 ммоль) при 0°C. Реакционной смеси давали нагреться до комнатной температуры и перемешивали при комнатной температуре в течение 2 часов. ТСХ показывала, что реакция завершилась. Реакционную смесь фильтровали через рыхлый слой силикагеля, промывали смесью 10% MeOH/ДХМ. Неочищенный продукт очищали колоночной хроматографией (0-10% MeOH/ДХМ), получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1)]+ = 223.
Промежуточное соединение 7
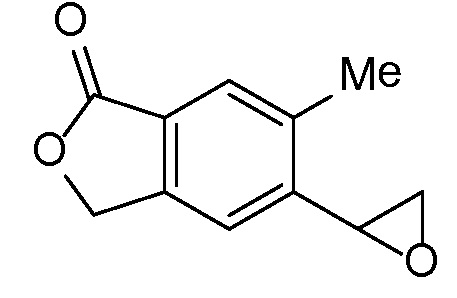
6-метил-5-оксиран-2-ил-2-бензофуран-1(3H)-он
Стадия A
5-проп-2-ен-1-ил-2-бензофуран-1(3H)-он
Смесь 5-бром-2-бензофуран-1(3H)-она (15,0 г, 70,4 ммоль), аллилтрибутилстаннана (25,6 г, 77,5 ммоль), LiCl (11,8 г, 282 ммоль) и Pd(PPh3)4 (1,2 г, 1,0 ммоль) в 100 мл толуола нагревали в атмосфере N2 при 90-100°C в течение ночи. После охлаждения до комнатной температуры смесь разбавляли 250 мл EtOAc и фильтровали. Фильтрат промывали водой и насыщенным раствором соли, сушили над безводным Na2SО4 и концентрировали досуха. Остаток очищали на колонке (ДХМ/петролейный эфир = 1:5), получая при этом указанное в заголовке соединение.
Стадия B
5-(2-гидроксиэтил)-2-бензофуран-1(3H)-он
В раствор 5-проп-2-ен-1-ил-2-бензофуран-1(3H)-она (13,5 г, 45,2 ммоль) в 200 мл ДХМ/MeOH (об./об.=1:1) барботировали О3 при -78°C в течение 30 мин и барботировали N2 в течение еще 15 мин при -78°C. Затем добавляли 20 мл Me2S и перемешивали смесь при комнатной температуре в течение ночи перед тем, как концентрировать досуха. Остаток растворяли в MeOH (100 мл) и затем охлаждали до 0°C. Порциями добавляли NaBH4 (5,90 г, 155 ммоль). Полученную смесь перемешивали при 0°C в течение 1 часа, затем гасили лимонной кислотой (водн.) и трижды экстрагировали EtOAc. Объединенные органические слои промывали NaHCО3 (водн.) и насыщенным раствором соли, сушили над безводным Na2SО4 и концентрировали досуха. Остаток очищали колоночной хроматографией (EtOAc/петролейный эфир=1:5), получая при этом указанное в заголовке соединение. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 7,86 (д, J=7,8 Гц, 1H), 7,41 (д, J=7,8 Гц, 1H), 7,38 (с, 1H), 5,29 (с, 2H), 3,92~3,98 (м, 2H), 3,01 (т, J=6,4 Гц, 2H).
Стадия C
5-(2-гидроксиэтил)-6-йод-2-бензофуран-1(3H)-он
К охлажденному (0°C) раствору 5-(2-гидроксиэтил)-2-бензофуран-1(3H)-она (9,00 г, 50,6 ммоль) в 100 мл TfOH добавляли NIS (12,5 г, 55,6 ммоль), затем смесь перемешивали при 0°C в течение 2 часов и выливали в ледяную воду (500 мл). Раствор экстрагировали три раза 500 мл EtOAc и промывали объединенные органические слои насыщенным раствором NaHCО3 и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией (EtOAc/петролейный эфир=1:5), получая при этом требуемый 5-(2-гидроксиэтил)-6-йод-2-бензофуран-1(3H)-он и региоизомер 5-(2-гидроксиэтил)-4-йод-2-бензофуран-1(3H)-он в качестве побочного продукта. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 7,84 (д, J=7,8 Гц, 1H), 7,46 (д, J=7,8 Гц, 1H), 5,09 (с, 2H), 3,93 (кв., J=6,3 Гц, 2H), 3,16 (т, J=6,3 Гц, 2H), 1,45 (т, J=5,5 Гц, 1H).
Стадия D
5-(2-гидроксиэтил)-6-метил-2-бензофуран-1(3H)-он
В колбу загружали 5-(2-гидроксиэтил)-6-йод-2-бензофуран-1(3H)-он (6,00 г, 19,7 ммоль) и при перемешивании с помощью магнитной мешалки добавляли Pd2(dba)3 (452 мг, 0,493 ммоль), PPh3 (1 г, 4 ммоль) и NMP (50 мл). Смесь продували N2 и нагревали до 50°C в течение 10 мин с последующим добавлением CuI (375 мг, 1,97 ммоль). Затем смесь нагревали в течение еще 10 мин, добавляли в реакционную смесь Sn(CH3)4 (5,30 г, 29,6 ммоль) и нагревали реакционную смесь до 120°C в течение 2 часов. После охлаждения до комнатной температуры смесь разбавляли насыщенным раствором NH4Cl (200 мл) и экстрагировали EtOAc (3 раза по 200 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 7,72 (с, 1H), 7,33 (с, 1H), 5,27 (с, 2H), 3,93 (т, J=6,3 Гц, 2H), 3,01 (т, J=6,3 Гц, 2H), 2,44 (с, 3H).
Стадия E
2-(6-метил-1-оксо-l,3-дигидро-2-бензофуран-5-ил)этилметансульфонат
К раствору 5-(2-гидроксиэтил)-6-метил-2-бензофуран-1(3H)-она (1,20 г, 6,25 ммоль) и TEA (2,5 г, 25 ммоль) в ДХМ (100 мл) добавляли MsCl (1,40 г, 12,5 ммоль) при 0°C. Смесь перемешивали при температуре окружающей среды в течение ночи, затем промывали водой и насыщенным раствором соли. Органический слой сушили и концентрировали досуха. Полученное, указанное в заголовке соединение применяли на следующей стадии без какой либо очистки.
Стадия F
5-этенил-6-метил-2-бензофуран-1(3H)-он
К смеси 2-(6-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этилметансульфоната (2,00 г, 7,41 ммоль) и TEA (5 мл) в ДХМ (50 мл) медленно добавляли DBU (5 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение ночи и затем разбавляли 50 мл ДХМ, три раза промывали 2Н раствором HCl и насыщенным раствором соли. Органический слой сушили и концентрировали досуха. Остаток очищали препаративной ТСХ, получая при этом 5-этенил-6-метил-2-бензофуран-1(3H)-он.
Стадия G
6-метил-5-оксиран-2-ил-2-бензофуран-1(3H)-он
К раствору 5-этенил-6-метил-2-бензофуран-1(3H)-она (1,00 г, 5,75 ммоль) в 50 мл ДХМ медленно добавляли mCPBA (3,50 г, 17,4 ммоль) в 50 мл ДХМ при 0°C. Смесь нагревали до комнатной температуры и перемешивали в течение 2 дней. Смесь промывали водным раствором Na2SО3 до тех пор, пока не происходило изменения окраски индикаторной бумаги с KI. Органический слой промывали насыщенным раствором соли и затем концентрировали. Остаток очищали на колонке с диоксидом кремния, получая при этом указанное в заголовке соединение. ЖХ-МС M+1 (рассчитано 191, обнаружено 191).
Промежуточные соединения 7A и 7B
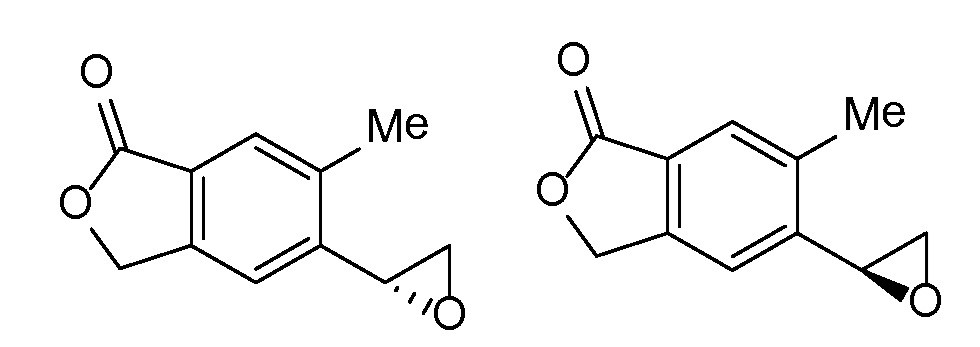
(R)-6-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-он и
(S)6-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-он
Указанные в заголовке соединения получали хиральным СФХ- разделением рацемической смеси 6-метил-5-оксиран-2-ил-2-бензофуран-1(3H)-онов (I-7) с применением хиральной колонки (250 мм × 50 мм, 10 мкм), chiralpac AD; подвижная фаза: A:сверхкритический CО2, B: MeOH, A:B =85:15 при скорости 250 мл/мин. Первый пик при элюировании (изомер 7A). 1H ЯМР 400 МГц CDCl3, δ 7,68 (с, 1H), 7,36 (с, 1H), 5,24 (д, J=3,6 Гц,2H), 4,05 (дд, J=2,8 Гц, 3,6 Гц, 1H), 3,24 (дд, J=4,0 Гц, 6,4 Гц, 1H), 2,63 (дд, J=2,8 Гц, 6,4 Гц, 1H), 2,50 (с, 3H); второй пик при элюировании (изомер 7В): 400 МГц CDCl3, δ 7,68 (с, 1H), 7,35 (с, 1H), 5,24 (д, J=3,6 Гц,2H), 4,05 (дд, J=2,8 Гц, 3,6 Гц, 1H), 3,24 (дд, J=4,0 Гц, 6,4 Гц, 1H), 2,63 (дд, J=2,8 Гц, 6,4 Гц, 1H), 2,50 (с, 3H).
Промежуточное соединение 8
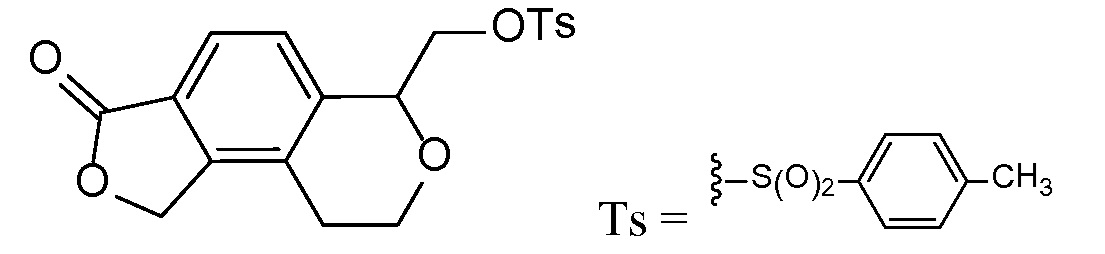
(3-оксо-3,6,8,9-тетрагидро-1H-фуро[3,4-f]изохромен-6-ил)метил-4-метилбензолсульфонат
Стадия A
5-бром-4-йод-2-бензофуран-1(3H)-он
К раствору 5-бром-2-бензофуран-1(3H)-она (5,00 г, 23,5 ммоль) в TfOH (100 мл) добавляли NIS (5,55 г, 24,6 ммоль) при 0°C. Смесь перемешивали при комнатной температуре в течение ночи; анализ реакционной смеси с помощью ЖХ показывал полное завершение реакции. Затем реакционную смесь медленно выливали в ледяную воду (1 л) при перемешивании. Затем к раствору добавляли EtOAc (500 мл) и затем перемешивали в течение 10 мин. Смесь фильтровали и отделяли органический слой. Водный слой экстрагировали EtOAc (2×200 мл). Объединенные органические слои промывали водой (500 мл), насыщенным раствором соли (500 мл), сушили над Na2SО4, фильтровали и концентрировали досуха; полученный материал абсорбировали на силикагеле и выделяли с помощью системы растворителей (гексаны/EtOAc=1/1), получая при этом 5-бром-4-йод-2-бензофуран-l(3H)-он.
Стадия B
5-бром-4-проп-2-ен-1-ил-2-бензофуран-1(3H)-он
Смесь 5-бром-4-йод-2-бензофуран-1(3H)-она (2,42 г, 7,13 ммоль), аллилтрибутилолова (2,36 г, 7,13 ммоль), LiCl (1,50 г, 35,7 ммоль) и Pd(PPh3)4 (200 г, 0,173 ммоль) в толуоле (50 мл) нагревали при 90-100°C в атмосфере N2 в течение ночи; Когда ЖХ-анализ показывал, что реакция прошла полностью, к раствору приливали EtOAc (100 мл) и промывали насыщенным раствором соли. Органический слой сушили над Na2SО4, фильтровали и концентрировали досуха, абсорбировали на силикагеле и затем выделяли на колонке с силикагелем, получая при этом указанное в заголовке соединение.
Стадия C
5-бром-4-(2-гидроксиэтил)-2-бензофуран-1(3H)-он
В раствор 5-бром-4-проп-2-ен-1-ил-2-бензофуран-1(3H)-она (1,27 г, 5,02 ммоль) в MeOH (50 мл) и ДХМ (50 мл) барботировали O3 при -78°C до тех пор, пока раствор не посинеет; избыток озона удаляли в высоком вакууме. После изменения окраски раствора на бесцветную к реакционной смеси добавляли NaBH4 (0,8 г, 20 ммоль) и затем перемешивали реакционную смесь при комнатной температуре в течение 30 мин. ЖХ-анализ и ТСХ показывали, что реакция прошла полностью. Растворитель удаляли в высоком вакууме; затем остаток повторно растворяли в EtOAc и промывали водой, сушили над Na2SО4, фильтровали и концентрировали досуха. Органический остаток абсорбировали на силикагеле и выделяли на колонке с силикагелем, получая при этом указанное в заголовке соединение.
Стадия D
5-этенил-4-(2-гидроксиэтил)-2-бензофуран-1(3H)-он
Смесь 5-бром-4-(2-гидроксиэтил)-2-бензофуран-1(3H)-она (0,460 г, 1,78 ммоль), трибутил(винил)олова (0,676 г, 2,13 ммоль), LiCl (0,224 г, 5,33 ммоль) и Pd(PPh3)4 (0,10 г, 0,087 ммоль) в толуоле (50 мл) нагревали при 100-110°C в атмосфере N2 в течение ночи, после чего ТСХ показывала, что реакция прошла полностью. Затем к раствору приливали EtOAc (100 мл) и промывали его насыщенным раствором соли, затем водой, затем сушили над Na2SО4, фильтровали и концентрировали досуха. Затем остаток абсорбировали на силикагеле и выделяли на колонке с диоксидом кремния, получая при этом указанное в заголовке соединение.
Стадия E
4-(2-гидроксиэтил)-5-оксиран-2-ил-2-бензофуран-1(3H)-он
5-этенил-4-(2-гидроксиэтил)-2-бензофуран-1(3H)-он (1,2 г, 5,9 ммоль) добавляли в колбу, содержащую якорь магнитной мешалки. В колбу добавляли дихлорметан (20 мл). Колбу помещали на охлаждающую баню при 0°C; в колбу выливали mCPBA (1,5 г, 8,8 ммоль) и полученную смесь перемешивали при комнатной температуре в течение ночи; ЖХ-анализ, а также ТСХ (гексаны/EtOAc=1/1) показывал, что реакция прошла полностью. Раствор обрабатывали дихлорметаном и промывали NaHCО3, Na2S2О3 и водой, органический слой сушили над Na2SО4, фильтровали и концентрировали досуха, затем обрабатывали AcOH (20 мл) и перемешивали в течение ночи; ЖХ-анализ показывал образование циклизованного продукта. Растворитель удаляли, абсорбировали полученный остаток на силикагеле и выделяли указанное в заголовке соединение с помощью системы растворителей гексаны/EtOAc (1/1).
Стадия F
(3-оксо-3,6,8,9-тетрагидро-1H-фуро[3,4-f]изохромен-6-ил)метил-4-метилбензолсульфонат
6-гидроксиметил-8,9-дигидро-1H-фуро[3,4-f]изохромен-3(6H)-он, в ДХМ (10 мл) обрабатывали п-толуолсульфонилхлоридом (0,40 г, 2,3 ммоль); к смеси добавляли пиридин (2 мл) и перемешивали полученную смесь при комнатной температуре в течение 12 часов. ТСХ (гексаны/EtOAc=1/0,5) и ЖХ-анализ показывали расход исходного материала и образование требуемого продукта. К реакционной смеси добавляли дихлорметан и промывали ее раствором NaCl, водой и сушили над Na2SО4, фильтровали и концентрировали досуха, абсорбировали на силикагеле и затем подвергали очистке на силикагеле; указанное в заголовке соединение выделяли с помощью системы растворителей гексаны/EtOAc (1/0,5). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 7,781 (д, J=8 Гц, 1H), 7,727 (д, J=8 Гц, 1H), 7,367 (д, J=8 Гц, 1H), 7,257 (д, J=8,5 Гц, 1H), 7,206 (д, J=8 Гц, 1H), 5,253 (с, 2H), 5,110 (с, 1H), 4,481-4,452 (м, 2H), 4,419-4,385 (м, 2H), 4,196-4,153 (м, 2H), 2,495 (с, 3H).
Промежуточное соединение 9
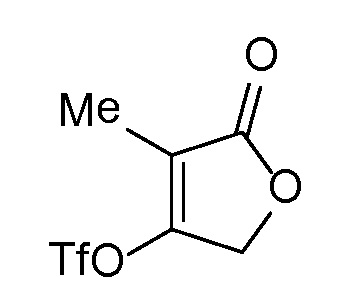
4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат
Стадия A
Этил-4-бром-2-метил-3-оксобутаноат
К раствору этил-2-метил-3-оксобутаноата (5,05 г, 35,0 ммоль) в воде (10 мл) при 0°C по каплям добавляли бром (1,81 мл, 35,0 ммоль) на протяжении 2 часов. Полученный раствор перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь экстрагировали этилацетатом, органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом этил-4-бром-2-метил-3-оксобутаноат. 1H-ЯМР (500 МГц, CDCl3), δ 4,322-4,274 (м, 2H), 2,455 (с, 2H), 1,991 (с, 3H), 1,337-1,309 (т, 3H).
Стадия B
4-гидрокси-3-метилфуран-2(5H)-он
Этил-4-бром-2-метил-3-оксобутаноат (7,81 г, 35 ммоль) обрабатывали бромистым водородом (0,040 мл, 48%, 0,35 ммоль) и нагревали смесь при 100°C в течение 6 часов. Осадок собирали фильтрованием с последующим промыванием этилацетатом, получая при этом 4-гидрокси-3-метилфуран-2(5H)-он. 1H-ЯМР (500 МГц, CDCl3), δ 4,595 (с, 2H), 3,314 (с, 1H), 1,668 (с, 3H).
Стадия C
4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат
К раствору 4-гидрокси-3-метилфуран-2(5H)-она (400 мг, 3,51 ммоль) в дихлорметане (10 мл) при -78°C по каплям добавляли 2,6-лутидин (0,612 мл, 5,26 ммоль) и трифлатный ангидрид (0,711 мл, 4,21 ммоль). Поддерживали температуру реакции при -78°C в течение 0,5 часа перед нагреванием до комнатной температуры в течение 1 часа. Смесь разбавляли ДХМ (100 мл) и промывали 1Н раствором хлористого водорода (3 раза 100 мл), затем разбавленным раствором бикарбоната натрия, сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 247,0.
Промежуточное соединение 10
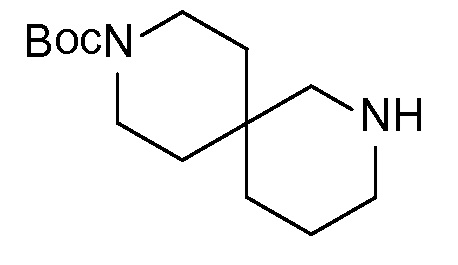
трет-Бутил-3,8-диазаспиро[5,5]ундекан-3-карбоксилат
Стадия A
трет-Бутил-4-(гидроксиметил)пиперидинкарбоксилат
Смесь 70 г LiAlH4 в 1500 мл ТГФ охлаждали до 0°C, затем по каплям добавляли 180 г 1-трет-бутил-4-метилпиперидин-l,4-дикарбоксилата в ТГФ. Когда реакция завершалась, добавляли 200 мл этилацетата и твердый безводный Na2SО4. Добавляли воду, пока раствор не становился прозрачным. Смесь фильтровали и выпаривали фильтрат, получая при этом указанное в заголовке соединение.
Стадия B
трет-Бутил-4-формилпиперидинкарбоксилат
Раствор 200 мл ДМСО в CH2Cl2 охлаждали до -78°C, по каплям добавляли 118 мл (COCl)2. Затем также по каплям добавляли 255 г трет-бутил 4-(гидроксиметил)пиперидинкарбоксилата. Смесь перемешивали в течение 4 часов. После завершения реакции при -78°C добавляли 638 мл Et3N. Органический слой промывали насыщенным раствором соли, сушили и очищали колоночной хроматографией, получая при этом указанное в заголовке соединение.
Стадия C
трет-бутил-4-формил-4-пропилпиперидинкарбоксилат
трет-бутил-4-формилпиперидинкарбоксилат растворяли в 66 мл акрилонитрила и добавляли 5 г 50% водного раствора гидроксида натрия, затем смесь нагревали до 50°C, пока реакция полностью не завершится согласно результатам ТСХ. Затем смесь выливали в 700 мл простого эфира, промывали насыщенным раствором соли и очищали колоночной хроматографией, получая при этом указанное в заголовке соединение.
Стадия D
трет-Бутил-3,8-диазаспиро[5.5]ундекан-3-карбоксилат
трет-Бутил-4-формил-4-пропилпиперидинкарбоксилат (30 г) растворяли в насыщенном растворе аммиака в метаноле и добавляли 15 г Ni Ренея. Реакционную смесь нагревали до 110°C при давлении 80 атмосфер в 2 л автоклаве высокого давления. Смесь фильтровали, чтобы удалить катализатор, и концентрировали фильтрат, получая при этом остаток, который очищали колоночной хроматографией, получая при этом трет-бутил-3,8-диазаспиро[5.5]ундекан-3-карбоксилат.
Промежуточное соединение 11
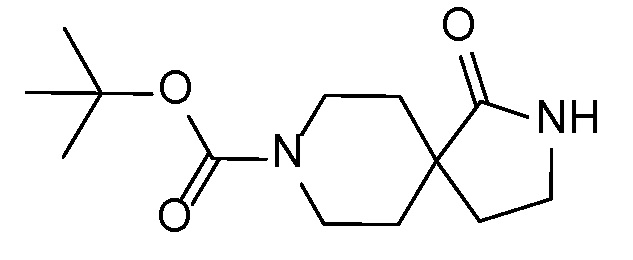
трет-Бутил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
Указанное в заголовке соединение поступает в продажу от ряда поставщиков, например, от компании Shanghai AQ BioPharma Co., Ltd, каталог #ABP1882. Альтернативно его можно получать различными способами, включая описанную ниже процедуру.
Стадия A
1-трет-бутил-4-метил-4-(цианометил)пиперидин-1,4-дикарбоксилат
К раствору имеющегося в продаже 1-трет-бутил-4-метилпиперидин-1,4-дикарбоксилата (200 г, 0,82 моль) в безводном ТГФ (2 л) при -65°C по каплям добавляли LDA (2 M в ТГФ, 575 мл, 1,15 моль) в атмосфере N2. Смесь перемешивали при -65°C в течение 1,5 часов. К смеси при -65°C добавляли бромацетонитрил (148 г, 1,23 моль) в безводном ТГФ (500 мл). Смесь перемешивали при -65°C в течение 1 часа, затем нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь гасили водой (800 мл) при 0°C и концентрировали объединенную реакционную смесь в вакууме, получая при этом неочищенный продукт, который экстрагировали этилацетатом (три раза по 1 л). Объединенные органические фазы промывали насыщенным раствором соли (1 л) и сушили над Na2SО4. Органический слой фильтровали, и фильтрат концентрировали в вакууме, получая при этом неочищенный продукт, который очищали колоночной хроматография на силикагеле путем элюирования смесью петролейный эфир/этилацетат (от петролейного эфира до соотношения 2/1), получая при этом указанное в заголовке соединение. 1H-ЯМР (400 МГц, CDCl3) δ 3,900-3,750 (м, 5H), 3,120-3,000 (м, 2H), 2,612-2,562 (м, 2H), 2,190-2,111 (м, 2H), 1,590-1,502 (м, 2H), 1,402 (с, 9H).
Стадия B
трет-Бутил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
Суспензию 1-трет-бутил-4-метил-4-(цианометил)пиперидин-1,4-дикарбоксилата (70,0 г, 247,9 ммоль) и Ni Ренея (60 г) в MeOH (1500 мл) и NН3.Н2О (80 мл) перемешивали при 50°C и давлении водорода 2 МПа в течение 18 часов. Реакционную смесь фильтровали через рыхлый слой CELITE®, и фильтрат концентрировали в вакууме, получая при этом неочищенный продукт, который промывали этилацетатом (200 мл), получая при этом указанное в заголовке соединение. 1H-ЯМР (400 МГц, CDCl3) δ 6,05 (с, 1H), 4,0 (с, 2H), 3,37-3,34 (м, 2H), 3,02-2,96 (м, 2H), 2,08-2,05 (м, 2H), 1,88-1,87 (м, 2H), 1,51-1,41 (м, 11H).
Промежуточное соединение 12
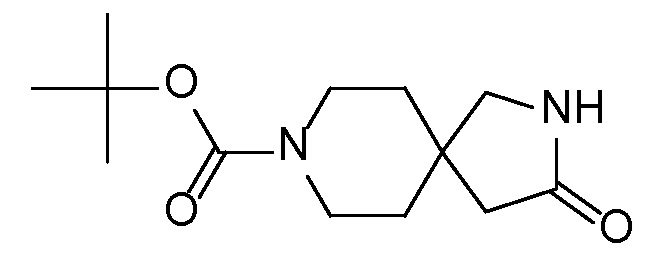
Сложный трет-бутиловый эфир 3-оксо-2,8-диазаспиро[4,5]декан-8-карбоновой кислоты
Стадия A
трет-Бутил-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилат
В 10 л 4-горлую круглодонную колбу, продутую азотом и поддерживаемую с инертной атмосферой азота, при 0°C помещали суспензию NaH (74,0 г, 2,16 моль, 1,05 экв., 70%) в тетрагидрофуране (2000 мл), затем при 0°C и перемешивании добавляли по каплям этил-2-(диэтоксифосфорил)ацетат (514 г, 2,06 моль, 1,05 экв., 98%). После этого при 0°C и перемешивании по каплям добавляли раствор трет-бутил-4-оксопиперидин-1-карбоксилата (400 г, 1,97 моль, 1,00 экв., 98%) в тетрагидрофуране (1200 мл). Полученный раствор перемешивали в течение 60 мин при комнатной температуре, затем гасили путем добавления 2000 мл воды. Полученный раствор экстрагировали (2×1000 мл) этилацетатом. Органические слои объединяли, сушили над безводным сульфатом магния и концентрировали в вакууме. Остаток промывали гексаном (1×1000 мл) и сушили, получая при этом трет-бутил-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилат.
Стадия B
трет-Бутил-4-(2-этокси-2-оксоэтил)-4-(нитрометил)пиперидин-1-карбоксилат
В 3000 мл 4-горлую круглодонную колбу помещали карбонат калия (93,2 г, 662 ммоль, 0,50 экв.) и ДМСО (2000 мл). Полученный раствор нагревали до 80°C. После этого медленно добавляли трет-бутил-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилат (368 г, 1,30 моль, 1,00 экв., 95%) и CH3NO2 (417 г, 6,70 моль, 5,00 экв., 98%). Полученный раствор перемешивали в течение 120 мин при 90°C. После охлаждения до комнатной температуры рН реакционной смеси доводили до значение рН=5 с помощью HCl (0,5 моль/л) и разбавляли 2000 мл воды. Полученный раствор экстрагировали простым эфиром (3×1500 мл). Органические слои объединяли, промывали водой (1×2000 мл) и насыщенным раствором соли (1×2000 мл), сушили и концентрировали в вакууме. Остаток вносили в колонку с силикагелем и элюировали смесью этилацетат/петролейный эфир (1:20~1:15~1:10), получая при этом указанное в заголовке соединение.
Стадия C
Сложный трет-бутиловый эфир 3-оксо-2,8-диазаспиро[4,5]декан-8-карбоновой кислоты
Смесь трет-бутил-4-(2-этокси-2-оксоэтил)-4-(нитрометил)пиперидин-1-карбоксилата (330 г, 990 ммоль, 1,00 экв., 99%) и Ni (40 г, 0,15 экв.) в этаноле (1200 мл) перемешивали в течение 24 часов в атмосфере водорода при комнатной температуре. Твердое вещество отфильтровывали. Фильтрат концентрировали в вакууме. Неочищенный продукт очищали перекристаллизацией из простого эфира, получая при этом указанное в заголовке соединение. ЖХ-МС (ES, m/z): 199 [M+H]+; ЖХ-МС (ES, m/z): 199 [M+H]+; 1H-ЯМР (400 МГц, CDCl3, м.д.): 1,447-1,476 (9H, с), 1,597-1,673 (4H, м, J=30,4 Гц), 2,235 (2H, с), 3,226 (2H, с), 3,284-3,348 (2H, м, J=25,6 Гц), 3,507-3,567 (2H, m, J=24 Гц), 6,048 (1H, с).
Промежуточное соединение 13
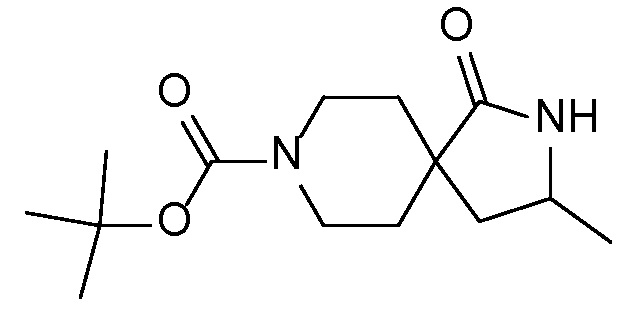
трет-Бутил-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
Стадия A
1-трет-бутил-4-метил 4-(2-метилаллил)пиперидин-1,4-дикарбоксилат
Раствор сложного метилового эфира N-Boc-пиперидин-4-карбоновой кислоты (2,00 г, 8,22 ммоль) в ТГФ (40 мл) охлаждали до -78°C. В атмосфере азота по каплям добавляли 2,0 M раствор LDA (6,17 мл, 12,3 ммоль) в ТГФ. Реакционную смесь перемешивали при -78°C в течение 30 минут перед добавлением раствора 3-бром-2-метилпропена (1,60 г, 11,9 ммоль) в ТГФ (2 мл). Затем реакционную смесь перемешивали в течение 1 часа при указанной температуре, брали образец для проведения ЖХ-МС-анализа, который показывал, что реакция полностью завершилась. Реакционную смесь гасили путем добавления насыщенного раствора (5 мл) хлорида аммония, и давали смеси нагреться до комнатной температуры. Затем смесь экстрагировали EtOAc (дважды по 50 мл). Объединенные органические слои промывали насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтраты концентрировали и очищали неочищенный продукт колоночной хроматографией путем элюирования смесью 0-30% этилацетат/гексан, получая при этом указанное в заголовке соединение.
ЖХ-МС (IE, m/z): 242,21 [M-56+1]+.
Стадия B
1-трет-бутил-4-метил-4-(2-оксопропил)пиперидин-1,4-дикарбоксилат
К раствору 1-трет-бутил-4-метил-4-(2-метилаллил)пиперидин-1,4-дикарбоксилата (2,2 г, 7,4 ммоль) в смеси диоксан/вода (60 мл, 1/1) в атмосфере азота добавляли тетраоксид осмия (0,038г, 0,15 ммоль) и периодат натрия (2,88 г, 13,5 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Затем смесь разбавляли дихлорметаном (50 мл) и промывали 20% раствором Na2S2О3 (20 мл). Органические слои объединяли и промывали насыщенным раствором соли (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтраты концентрировали и остаток очищали колоночной хроматография путем элюирования смесью 0-60% этилацетат/гексан, получая при этом 1-трет-бутил-4-метил-4-(2-оксопропил)пиперидин-1,4-дикарбоксилат. ЖХ-МС (IE, m/z): 322,26 (M+23)+.
Стадия C
трет-Бутил-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
Раствор 1-трет-бутил-4-метил-4-(2-оксопропил)пиперидин-1,4-дикарбоксилата (1,15 г, 3,84 ммоль) в метаноле (25 мл) обрабатывали ацетатом аммония (3,85 г, 49,9 ммоль), цианоборгидридом натрия (0,681 г, 10,83 ммоль) и сульфатом магния (2,54 г, 21,1 ммоль). Смесь нагревали при 80°C в герметично закрывающейся трубке в течение 12 часов. Реакционную смесь фильтровали через рыхлый слой CELITE®, и осадок на фильтре промывали метанолом. Затем фильтраты концентрировали и очищали остаток колоночной хроматографией путем элюирования смесью 0-10% метанол/этилацетат, получая при этом указанное в заголовке соединение. ЖХ-МС (IE, m/z): 291,27 (M+23)+.
Промежуточные соединения 13A и 13B
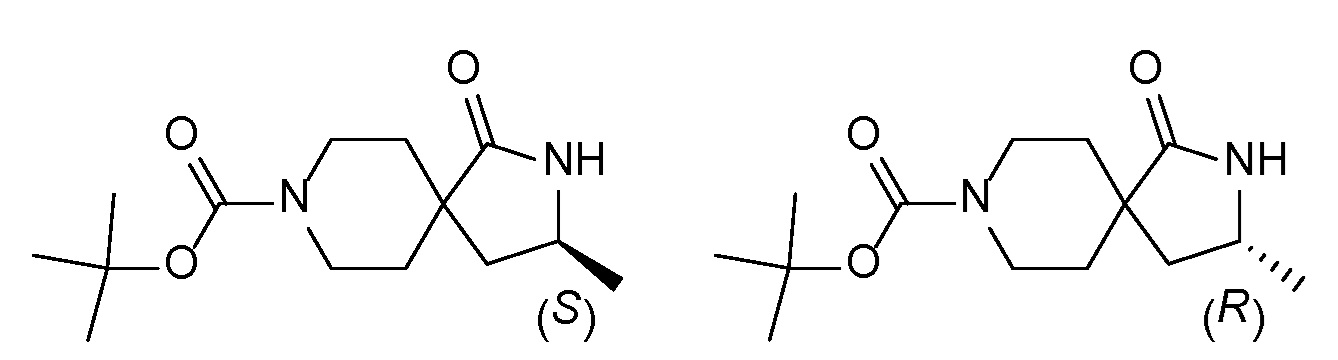
(S)-трет-бутил-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат и (R)-трет-бутил-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
трет-Бутил-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат подвергали СФХ-очистке. Два энантиомера разделяли на хиральной колонке Chiralcel IA путем элюирования смесью 30% MeOH:MeCN (2:1)/CО2 (100 бар, 35°C). На основе данных спектроскопии вибрационного циркулярного дихроизма (VCD) быстро элюирующий изомер определяли как (S)-трет-бутил-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат, и медленного элюирующий изомер определяли как (R)-трет-бутил-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат. ЖХ-МС (IE, m/z): 291 (M+23)+.
Промежуточное соединение 14
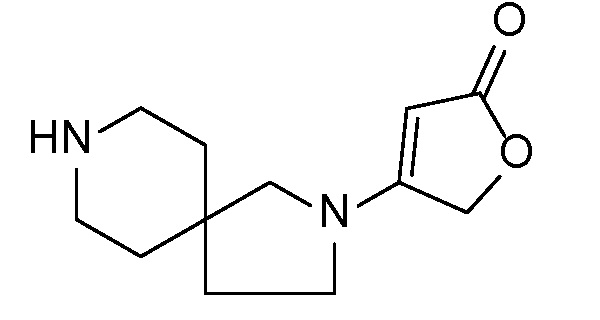
4-(2,8-диазаспиро[4.5]декан-2-ил)фуран-2(5H)-он
Имеющийся в продаже фуран-2,4(3H,5H)-дион (0,433 г, 4,33 ммоль) и трет-бутил-2,8-диазаспиро[4.5]декан-8-карбоксилат (поступающая в продажу от многих поставщиков соль уксусной кислоты, 0,65 г, 2,2 ммоль) в 20 мл i-PrOH нагревали в герметично закрывающейся трубке при 110°C в течение ночи. Твердые вещества отфильтровывали, и фильтрат концентрировали в вакууме. Остаток растворяли в 30 мл ДХМ и 5 мл TFA и перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали и очищали остаток препаративной ТСХ (10% 2Н NH3 в смеси метанол-ДХМ). ЖХ/МС: [(M+1)]+ = 223.
Промежуточное соединение 15
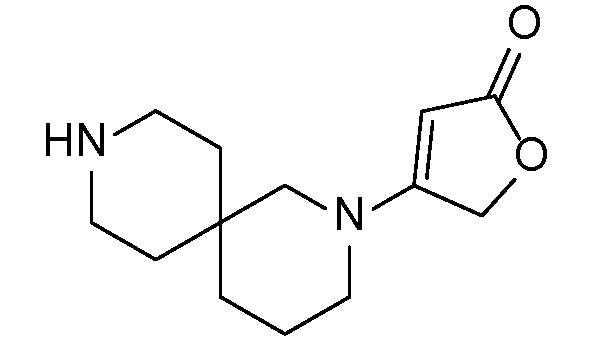
4-(2,9-диазаспиро[5.5]ундекан-2-ил)фуран-2(5H)-он
Стадия A
трет-Бутил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-9-карбоксилат
К имеющемуся в продаже 4-бромфуран-2(5H)-ону (128 мг, 0,786 ммоль) в ТГФ (4 мл) добавляли основание Хунига (275 мкл, 1,57 ммоль) и трет-бутил-3,8-диазаспиро[5,5]ундекан-3-карбоксилат (200 мг, 0,786 ммоль). Реакционную смесь перемешивали при 76°C в течение ночи, концентрировали и очищали колоночной хроматографией (0-10% MeOH в ДХМ), получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1)]+ = 337.
Стадия B
4-(2,9-диазаспиро[5.5]ундекан-2-ил)фуран-2(5H)-он
К раствору трет-бутил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-9-карбоксилата (266 мг, 0,792 ммоль) в ДХМ (2 мл) при 0°C добавляли TFA (2 мл, 26,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали при пониженном давлении и помещали в высокий вакуум. Остаток растворяли в MeOH и загружали в SCX-колонку (предварительно промытую MeOH) с 2 г сорбента Bond Elut. Остаток промывали MeOH и элюировали продукт с помощью 2M раствора NН3 в MeOH, получая при этом 4-(2,9-диазаспиро[5.5]ундекан-2-ил)фуран-2(5H)-он. ЖХ/МС: [(M+1)]+ = 237.
Промежуточное соединение 16
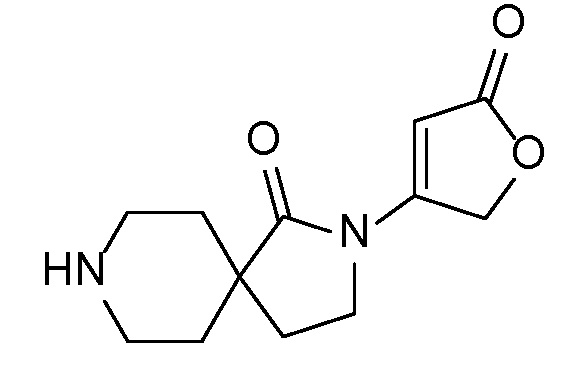
2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
трет-Бутил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилат
Смесь трет-бутил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (1,83 г, 7,20 ммоль), имеющегося в продаже 4-бромфуран-2(5H)-она (1,41 г, 8,63 ммоль), ксантфоса (0,416 г, 0,720 ммоль), воды (0,389 мл, 21,6 ммоль) и карбоната калия (1,989 г, 14,39 ммоль) в толуоле (50 мл) дегазировали азотом с последующим добавлением ацетата палладия (0,081 г, 0,36 ммоль). Полученную смесь нагревали при 65°C в течение 16 часов. После фильтрования через CELITE® фильтрат концентрировали, и остаток очищали на колонке с силикагелем с применением смеси EtOAc/гексан в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 337,18.
Стадия B
2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору трет-бутил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилата (5,70 г, 16,9 ммоль) в метиленхлориде (10 мл) добавляли трифторуксусную кислоту (26,1 мл, 339 ммоль) и перемешивали полученный раствор при комнатной температуре в течение 1 часа. После концентрирования остаток ощелачивали на ионообменной колонке с последующим промыванием 1Н раствором аммиака в метаноле, получая при этом 2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он. ЖХ/МС: (M+1)+: 237,06.
Промежуточное соединение 17
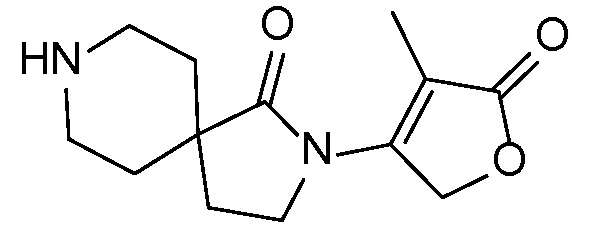
2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
трет-Бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
К смеси трет-бутил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (I-11, 80,0 г, 315 ммоль) и 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (I-9, 85,2 г, 346 ммоль), ксантфоса (13,6 г, 23,6 ммоль), Cs2CO3 (153,7 г, 471,8 ммоль) в толуоле (1200 мл) добавляли Pd2(dba)3 (7,20 г, 7,86 ммоль) в атмосфере N2. Полученную реакционную смесь нагревали до 90°C и перемешивали в атмосфере N2 в течение 18 часов. Смесь фильтровали через рыхлый слой CELITE® и концентрировали фильтрат. Остаток очищали осаждением, получая при этом трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат. 1H ЯМР (400 МГц, CDCl3) δ 5,23 (с, 2H), 4,02-3,99 (м, 4H), 3,06-3,05 (м, 2H), 2,15-2,11 (м, 2H), 2,02 (с, 3H), 1,87-1,81 (м, 2H), 1,51-1,41 (м, 11H).
Стадия B
2-(4-Метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К смеси трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (57,0 г, 163 ммоль) в EtOAc (180 мл) при 0°C добавляли насыщенный раствор HCl (г)/EtOAc (712 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Смесь фильтровали и концентрировали фильтрат, получая при этом кислотно-аддитивную соль HCl. К смеси кислотно-аддитивной соли HCl (54,2 г, 189 ммоль) в MeOH (550 мл) при 0°C добавляли NaHCО3 (31,8 г, 378 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 3 часов до значения pH=8. Смесь фильтровали и концентрировали фильтрат. Остаток повторно растворяли в MeOH и концентрировали до тех пор, пока не появлялся осадок. Смесь фильтровали и концентрировали фильтрат, получая при этом 2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он в виде свободного амина. 1H ЯМР (400 МГц, CD3OD) δ 5,24 (с, 2H), 4,10-4,07 (м, 2H), 3,22-3,16 (м, 2H), 2,93-2,87 (м, 2H), 2,22-2,19 (м, 2H), 2,0 (с, 3H), 1,94-1,87 (м, 2H), 1,67-1,61 (м, 2H).
Промежуточное соединение 18
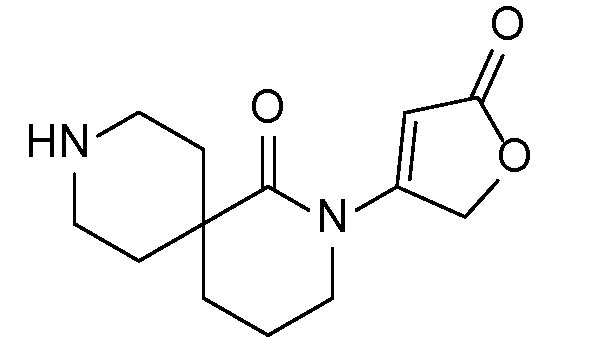
2-(5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-1-он
Стадия A
трет-бутил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-9-карбоксилат
В ампулу для микроволновой печи загружали имеющийся в продаже трет-бутил-1-оксо-2,9-диазаспиро[5.5]ундекан-9-карбоксилат (Shanghai AQ BioPharma Co., Ltd, каталог # ABP3640, 100 мг, 0,373 ммоль), 4-бромфуран-2(5H)-он (72,9 мг, 0,447 ммоль), Pd2(dba)3 (17,06 мг, 0,019 ммоль), ксантфос (32,3 мг, 0,056 ммоль) и карбонат цезия (182 мг, 0,559 ммоль). Ампулу герметизировали, дегазировали и заполняли толуолом (1,5 мл). Реакционную смесь нагревали при 90°C в течение ночи и фильтровали через CELITE®. Фильтрат выпаривали, получая при этом неочищенный продукт, которые очищали колоночной хроматографией (0-10% MeOH/ДХМ), получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1-56)]+ = 295.
Стадия B
2-(5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-1-он
трет-Бутил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-9-карбоксилат (100 мг, 0,285 ммоль) в ДХМ (2 мл) обрабатывали TFA (660 мкл, 8,56 ммоль) при 0°C, получая при этом соль TFA. Затем SCX-колонку с 2 г сорбента Bond Elut сначала промывали MeOH, в колонку с MeOH загружали образец, картридж промывали, добавляя MeOH по каплям, чтобы удалить TFA, и в конце промывали 2Н раствором NH3/MeOH, получая при этом указанное в заголовке соединение в виде свободного амина. ЖХ/МС: [(M+1)]+ = 251.
Промежуточное соединение 19
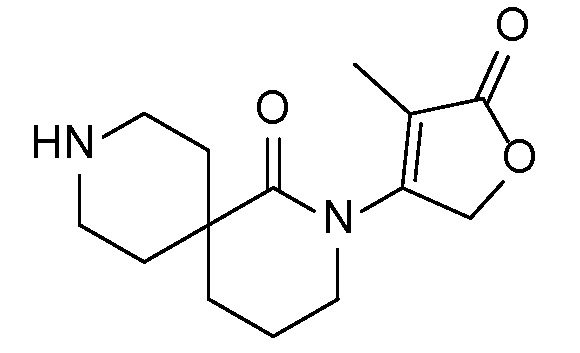
2-(4-Метил-5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-1-он
Стадия A
трет-Бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,9-диазаспиро[5.5]ундекан-9-карбоксилат
В ампулу для микроволновой печи загружали имеющийся в продаже трет-бутил-1-оксо-2,9-диазаспиро[5.5]ундекан-9-карбоксилат (Shanghai AQ BioPharma Co., Ltd, каталог # ABP3640, 100 мг, 0,373 ммоль), 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат (110 мг, 0,447 ммоль), Pd2(dba)3 (17 мг, 0,019 ммоль), ксантфос (32 мг, 0,056 ммоль) и карбонат цезия (182 мг, 0,559 ммоль). Ампулу герметизировали, дегазировали и заполняли толуолом (1,5 мл). Реакционную смесь нагревали при 90°C в течение ночи и фильтровали через CELITE®. Фильтрат выпаривали, получая при этом неочищенный продукт, который очищали колоночной хроматографией (0-10% MeOH/ДХМ), получая при этом указанное в заголовке соединение. ЖХ/МС:[(M+1-56)]+ = 309.
Стадия B
2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-1-он
трет-Бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,9-диазаспиро[5.5]ундекан-9-карбоксилат (130 мг, 0,357 ммоль) в ДХМ (2 мл) обрабатывали TFA (824 мкл, 10,7 ммоль) при 0°C, получая при этом соль TFA. Затем SCX-колонку с 2 г сорбента Bond Elut (ионообменный картридж) сначала промывали MeOH, загружали образец в колонку с MeOH, картридж промывали, добавляя MeOH по каплям, чтобы удалить TFA и в конце промывали 2Н раствором NH3/MeOH, получая при этом указанное в заголовке соединение в виде свободного амина. ЖХ/МС: [(M+1)]+ = 265.
Промежуточное соединение 20
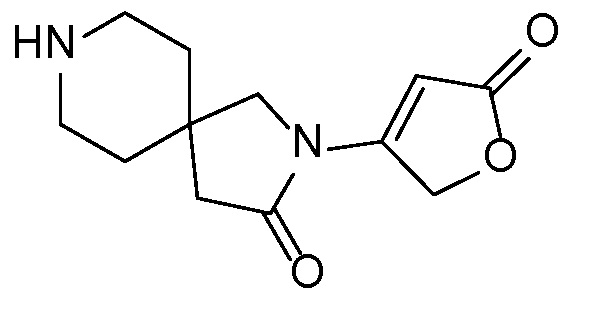
2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-3-он
Указанное в заголовке соединение получали из сложного трет-бутилового эфира 3-оксо-2,8-диазаспиро[4,5]декан-8-карбоновой кислоты и 4-бромфуран-2(5H)-она в две стадии способом, аналогичным способу, описанному выше для получения 2-(5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-1-она (I-18). ЖХ/МС: [(M+1)]+ = 237.
Промежуточное соединение 21
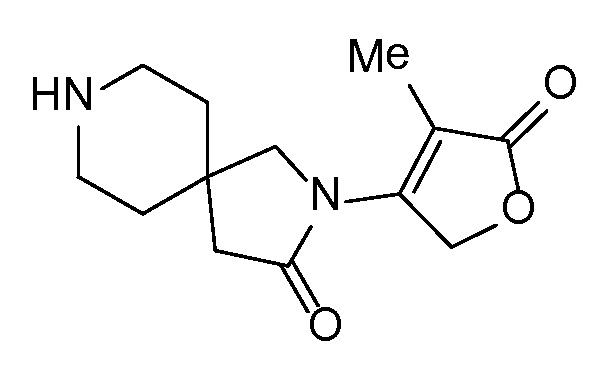
2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-3-он
Указанное в заголовке соединение получали из сложного трет-бутилового эфира 3-оксо-2,8-диазаспиро[4,5]декан-8-карбоновой кислоты и 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната в две стадии способом, аналогичным способу, описанному выше для получения 2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-1-она (I-19).
Промежуточное соединение 22
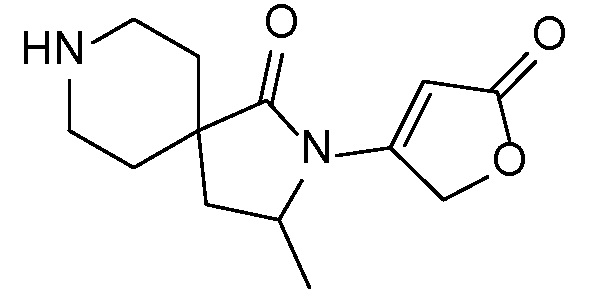
3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
трет-бутил-3-метил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилат
Смесь трет-бутил-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (I-13) (505 мг, 1,88 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (109 мг, 0,188 ммоль), ацетата палладия(II) (21 мг, 0,094 ммоль), карбоната калия (520 мг, 3,76 ммоль), воды (102 мкл, 5,65 ммоль) и имеющегося в продаже 4-бромфуран-2-она (368 мг, 2,26 ммоль) в толуоле (13 мл) дегазировали и затем нагревали при 60°C в течение 16 часов. Смесь фильтровали через рыхлый слой CELITE®, и осадок на фильтре промывали этилацетатом. Фильтраты концентрировали, и остаток очищали колоночной хроматографией путем элюирования с градиентом 10-100% этилацетат/гексан, получая при этом указанное в заголовке соединение. ЖХ-МС (IE, m/z): 373,3 (M+23)+.
Стадия B
3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
трет-Бутил-3-метил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилат (90 мг, 0,257 ммоль) растворяли в дихлорметане (2 мл) и обрабатывали TFA (1 мл). После перемешивания при комнатной температуре в течение 1,5 часов реакционную смесь концентрировали, чтобы удалить избыток реагента, и три раза выпаривали совместно с дихлорметаном, получая при этом указанное в заголовке соединение. ЖХ-МС (IE, m/z): 251 (M+1)+.
Промежуточное соединение 22А
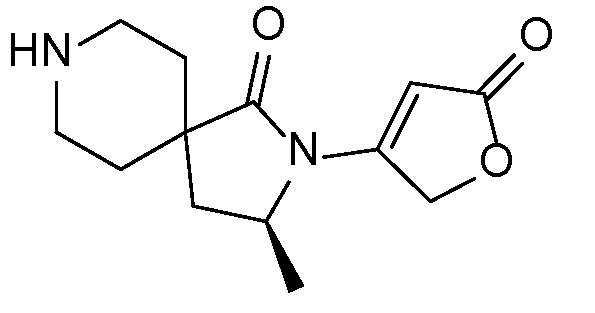
(S)-3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Указанное в заголовке соединение получали в две стадии из (S)-трет-бутил-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата способом, аналогичным способу, описанному непосредственно выше для получения рацемата 3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (I-22). ЖХ-МС (IE, m/z): 251 (M+1)+.
Промежуточное соединение 23
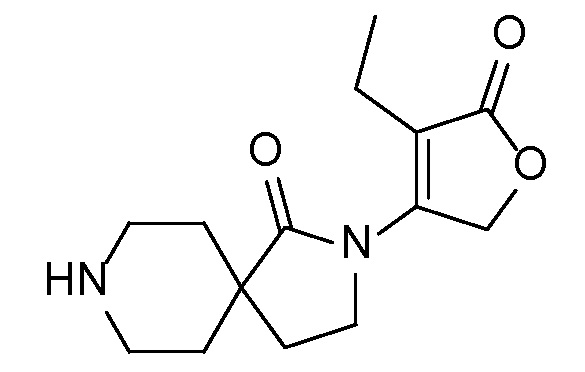
2-(4-этил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
Этил-4-бром-2-этил-3-оксобутаноат
К раствору этил-2-этил-3-оксобутаноата (5,17 г, 32,7 ммоль) в воде (10 мл) при 0°C по каплям добавляли бром (1,684 мл, 32,7 ммоль) на протяжении 2 часов. Полученный раствор перемешивали при комнатной температуре в течение 16 часов. Смесь экстрагировали этилацетатом (100 мл), и органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом этил-4-бром-2-этил-3-оксобутаноат. 1H-ЯМР (500 МГц, CDCl3), δ 4,327-4,284 (м, 2H), 2,412 (с, 2H), 2,320-2,212 (кв., 2H), 1,338-10309 (м, 3H), 1,042-1,013 (т, J=7,3 Гц, 3H).
Стадия B
3-этил-4-гидроксифуран-2(5H)-он
Смесь этил-4-бром-2-этил-3-оксобутаноата и бромистого водорода (48%, 0,037 мл, 0,327 ммоль) нагревали при 100°C в течение 20 часов. После охлаждения до комнатной температуры твердые вещества выделяли фильтрованием с последующим промыванием простым диэтиловым эфиром, получая при этом 3-этил-4-гидроксифуран-2(5H)-он. ЖХ/МС: (M+1)+: 129,05.
Стадия C
4-этил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат
К раствору 3-этил-4-гидроксифуран-2(5H)-она (400 мг, 3,12 ммоль) в дихлорметане (10 мл) при -78°C по каплям добавляли 2,6-лутидин (0,545 мл, 4,68 ммоль) и трифлатный ангидрид (0,633 мл, 3,75 ммоль). Реакционный раствор перемешивали при -78°C в течение 1 часа перед его нагреванием до комнатной температуры в течение 2 часов. Смесь разбавляли дихлорметаном (100 мл) и промывали 1Н раствором хлористого водорода (3×100 мл), раствором бикарбоната натрия, сушили над сульфатом натрия и концентрировали, получая при этом 4-этил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат. ЖХ/МС:(M+1)+: 261,01.
Стадия D
трет-Бутил-2-(4-этил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
Смесь трет-бутил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (200 мг, 0,786 ммоль), 4-этил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (246 мг, 0,944 ммоль), ксантфоса (45,5, 0,079 ммоль), ацетата палладия (II) (8,8 мг, 0,039 ммоль), воды (0,043 мл, 2,4 ммоль) и карбоната калия (217 мг, 1,57 ммоль) в толуоле (20 мл) нагревали при 60°C в течение 16 часов. После фильтрования через CELITE® фильтрат концентрировали и очищали остаток на силикагеле с применением смеси этилацетат/гексан, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 365,19.
Стадия E
2-(4-этил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору трет-бутил-2-(4-этил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (0,34 г, 0,93 ммоль) в дихлорметане (1 мл) добавляли трифторуксусную кислоту (2,16 мл, 28 ммоль) и перемешивали полученный раствор при комнатной температуре в течение 1 часа. После удаления летучих веществ остаток растворяли в дихлорметане (2 мл) и обрабатывали раствором хлористого водорода (2 мл, 4Н в диоксане), смесь концентрировали, получая при этом 2-(4-этил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он. ЖХ/МС: (M+1)+: 265,16.
Промежуточное соединение 24
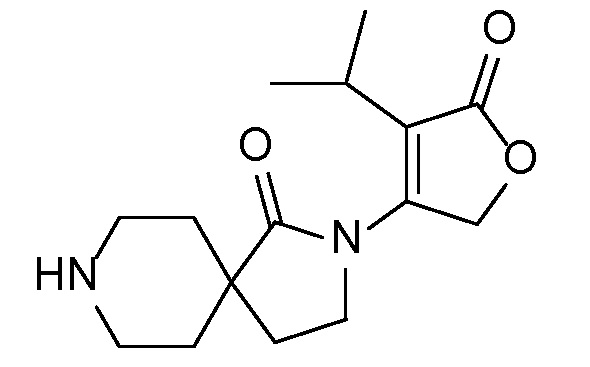
2-(4-изопропил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
Этил-4-бром-2-изопропил-3-оксобутаноат
К раствору этил-2-aцетил-3-метилбутаноата (5,10 г, 29,6 ммоль) в воде (10 мл) при 0°C по каплям добавляли бром (1,53 мл, 29,6 ммоль) на протяжении 2 часов. Добавляли хлороформ (30 мл) и полученный раствор перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли этилацетат (300 мл), и органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. 1H-ЯМР (500 МГц, CDCl3), δ 4,300-4,282 (м, 2H), 2,601-2,575 (м, 1H), 2,406 (с, 2H), 1,331-1,303 (т, J=7,1 Гц, 3H), 1,085-1,072 (д, J=6,5 Гц, 3H), 1,048-1,035 (д, J=6,7 Гц, 3H).
Стадия B
4-гидрокси-3-изопропилфуран-2(5H)-он
Смесь этил-4-бром-2-изопропил-3-оксобутаноата (7,1 г, 28 ммоль) и бромистого водорода (48%, 0,032 мл, 0,28 ммоль) нагревали при 100°C в течение 8 часов. После охлаждения до комнатной температуры твердое вещество выделяли фильтрованием с последующим промыванием простым диэтиловым эфиром, получая при этом 4-гидрокси-3-изопропилфуран-2(5H)-он. ЖХ/МС: (M+1)+:143,09.
Стадия C
4-изопропил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат
К раствору 4-гидрокси-3-изопропилфуран-2(5H)-она (400 мг, 2,81 ммоль) в метиленхлориде (10 мл) при -78°C по каплям добавляли 2,6-лутидин (0,492 мл, 4,22 ммоль) и трифлатный ангидрид (0,570 мл, 3,38 ммоль) и поддерживали температуру реакции при -78°C в течение 1 часа перед нагреванием до комнатной температуры в течение 2 часов. Смесь распределяли между метиленхлоридом и 1Н раствором хлористого водорода. Органическую фазу промывали 1Н раствором хлористого водорода, затем разбавленным раствором бикарбоната натрия, сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 275,07.
Стадия D
трет-бутил-2-(4-изопропил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
Смесь трет-бутил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (I-11) (200 мг, 0,786 ммоль), 4-изопропил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (259 мг, 0,944 ммоль), ксантфоса (46 мг, 0,079 ммоль), ацетата палладия (II) (8,8 мг, 0,039 ммоль), воды (0,043 мл, 2,4 ммоль) и карбоната калия (217 мг, 1,57 ммоль) в толуоле (20 мл) нагревали при 66°C в течение 16 часов. После фильтрования через CELITE® фильтрат концентрировали и остаток очищали на колонке с силикагелем, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1): 379,21.
Стадия E
2-(4-изопропил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору трет-бутил-2-(4-изопропил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (195 мг, 0,515 ммоль) в метиленхлориде добавляли трифторуксусную кислоту при комнатной температуре и перемешивали полученный раствор при комнатной температуре в течение 1 часа. После удаления летучих веществ остаток растворяли в метиленхлориде и обрабатывали раствором хлористого водорода (2 мл, 4Н в диоксане) и снова концентрировали, получая при этом 2-(4-изопропил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он в виде (аддитивной) соли с хлористым водородом. ЖХ/МС: (M+1)+: 279,16.
Промежуточное соединение 25
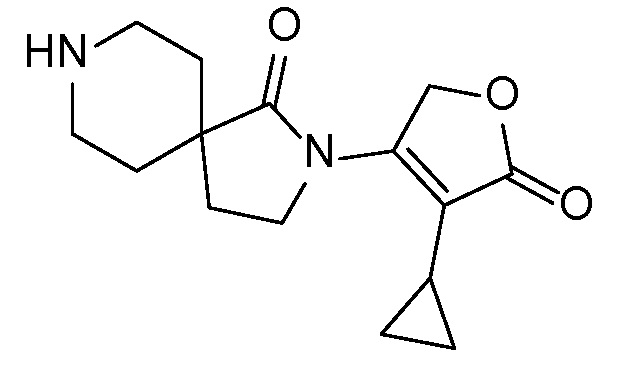
2-(4-циклопропил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
трет-Бутил-2-(4-бром-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
трет-Бутил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилат (I-16, стадия A) (784 мг, 2,33 ммоль) растворяли в ДХМ (20 мл) и обрабатывали NBS (498 мг, 2,80 ммоль) при 25°C в течение 12 часов. Реакционную смесь разбавляли ДХМ (20 мл), промывали водой (20 мл) и насыщенным раствором соли (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтраты концентрировали, и неочищенный продукт очищали колоночной хроматографией (колонка ISCO с 40 г силикагеля) путем элюирования с градиентом 50-100% этилацетат/гексан, получая при этом указанное в заголовке соединение.
Стадия B
трет-Бутил-2-(4-циклопропил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
В ампуле для микроволновой печи трет-бутил-2-(4-бром-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (90 мг, 0,22 ммоль) растворяли в толуоле (2 мл) и воде (0,2 мл). Добавляли фосфат калия (138 мг, 0,650 ммоль), трициклогексилфосфин (18 мг, 0,065 ммоль), циклопропилбороновую кислоту (74,5 мг, 0,867 ммоль) и ацетат палладия (4,87 мг, 0,022 ммоль). Реакционную смесь дегазировали и нагревали при 100°C в течение 12 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали, и полученный остаток растворяли в EtOAc (50 мл), промывали водой (30 мл) и насыщенным раствором соли (30 мл), сушили над сульфатом натрия и фильтровали. При удалении растворителя получали неочищенный продукт, который очищали колоночной хроматографией путем элюирования с градиентом 0-100% EtOAc/гексан, получая при этом указанное в заголовке соединение. 1H ЯМР (500 МГц, CDCl3) δ 5,20 (с, 2H), 4,22 (м, 2H), 3,99 (м, 2H), 3,05 (м, 2H), 2,09 (м, 2H), 1,92 (м, 2H), 1,60 (м, 2H), 1,56 (м, 10H), 1,02 (м, 2H), 0,82 (м, 2H).
Стадия C
2-(4-циклопропил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Указанное в заголовке соединение можно получать способом, аналогичным способу, описанному выше для получения 2-(4-изопропил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (I-24) с применением TFA.
Промежуточное соединение 26
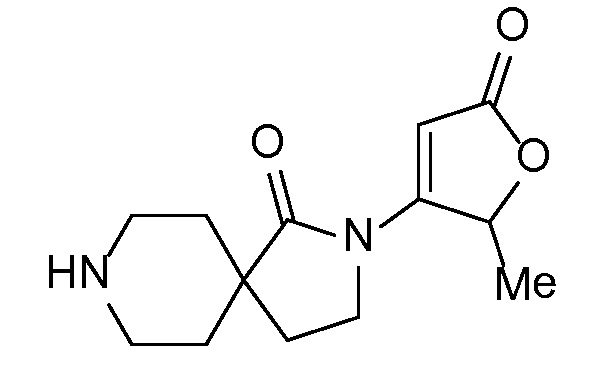
2-(2-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
Этил-4-бром-3-оксопентаноат
К раствору этил-3-оксопентаноата (5,00 г, 34,7 ммоль) в хлороформе (27 мл) при 0°C по каплям добавляли бром (1,79 мл, 34,7 ммоль) в хлороформе (10 мл). Полученный раствор перемешивали при комнатной температуре в течение 16 часов. Раствор промывали водой, сушили над сульфатом натрия, концентрировали, получая при этом этил-4-бром-3-оксопентаноат. 1H-ЯМР (500 МГц, CDCl3), δ 4,670-4,630 (дд, J=6,7 Гц, 1H), 4,251-4,208 (дд, J=7,2 Гц, 2H), 3,883-3,851 (д, J=16 Гц, 1H), 3,687-3,655 (д, J=16 Гц, 1H), 1,804-1,791 (д, J=6,8 Гц, 3H), 1,323-1,295 (м, J=7,2 Гц, 3H).
Стадия B
4-гидрокси-5-метилфуран-2(5H)-он
Этил-4-бром-3-оксопентаноат (7,49 г, 33,6 ммоль) обрабатывали гидроксидом калия (5,03 г, 90 ммоль) в воде (36 мл) при 0°C. Полученную смесь энергично перемешивали при 0°C в течение 4 часов. Реакционную смесь экстрагировали метиленхлоридом (дважды по 100 мл). Щелочную фазу подкисляли до pH <1 с применением 6Н раствора хлористого водорода. Кислую фазу экстрагировали метиленхлоридом (3×100 мл). Последнюю объединенную органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. 1H-ЯМР (500 МГц, CDCl3), δ 5,064 (с, 1H), 4,949-4,878 (м, 1H), 3,251-3,239 (м, 1H), 1,566-1,547 (м, 3H).
Стадия C
2-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат
К раствору 4-гидрокси-5-метилфуран-2(5H)-она в метиленхлориде (10 мл) при -78°C по каплям добавляли 2,6-лутидин (0,612 мл, 5,26 ммоль) и трифлатный ангидрид (0,711 мл, 4,21 ммоль). Температуру реакции поддерживали при -78°C в течение 0,5 часа перед нагреванием реакционной смеси до комнатной температуры в течение 1 часа. Смесь промывали раствором хлористого водорода (1 Н, три раза по 100 мл), разбавленным раствором бикарбоната натрия и сушили над сульфатом натрия, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 247,00.
Стадия D
трет-Бутил-2-(2-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
Смесь трет-бутил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (200 мг, 0,786 ммоль), 2-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (232 мг, 0,944 ммоль), ксантфоса (45,5 мг, 0,079 ммоль), ацетата палладия (II) (8,83 мг, 0,039 ммоль), воды (0,043 мл, 2,359 ммоль) и карбоната калия (217 мг, 1,573 ммоль) в толуоле (20 мл) дегазировали азотом и нагревали при 65°C в течение 16 часов. После фильтрования через CELITE® фильтрат концентрировали, и остаток очищали на колонке с силикагелем с применением этилацетата и гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 351,15.
Стадия E
2-(2-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору трет-бутил-2-(2-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (127 мг, 0,362 ммоль) в метиленхлориде (1 мл) добавляли трифторуксусную кислоту (1,396 мл, 18,12 ммоль) и перемешивали полученный раствор при комнатной температуре в течение 1 часа. Остаток после концентрирования обрабатывали метиленхлоридом (1 мл) и хлористым водородом (1 мл, 4Н в диоксане). Полученную смесь концентрировали, получая при этом указанное в заголовке соединение в виде (аддитивной) соли с хлористым водородом. ЖХ/МС: (M+1)+: 251,19.
Промежуточное соединение 27
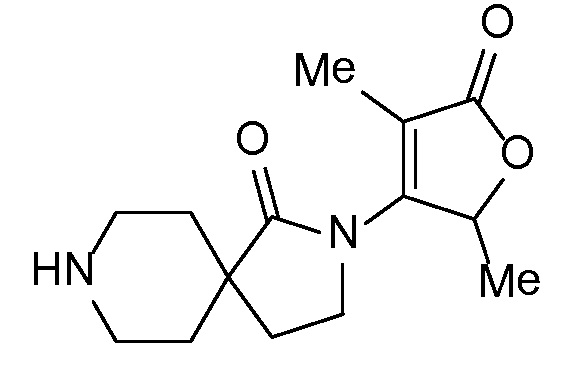
2-(2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
Этил-4-бром-2-метил-3-оксопентаноат
К раствору этил-2-метил-3-оксопентаноата (5,0 г, 34,7 ммоль) в хлороформе при 0°C по каплям добавляли бром (1,79 мл, 34,7 ммоль) в хлороформе (10 мл). Полученный раствор перемешивали при комнатной температуре в течение 16 часов. Раствор промывали водой, сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. 1H-ЯМР (500 МГц, CDCl3), δ 4,781-4,740 (дд, J=6,6 Гц, 1H), 4,140-4,098 (дд, J=7,0 Гц,1H), 3,770 (с, 3H), 1,797-1,783 (д, J=6,6 Гц, 3H), 1,455-1,442 (д, J=7,0 Гц, 3H).
Стадия B
4-гидрокси-3,5-диметилфуран-2(5H)-он
К этил-4-бром-2-метил-3-оксопентаноату (7,49 г, 31,6 ммоль) добавляли охлажденный гидроксид калия (4,7 г, 84 ммоль) в воде (36 мл) при 0°C; полученную смесь энергично перемешивали при 0°C в течение 4 часов. Реакционную смесь экстрагировали метиленхлоридом (2×100 мл), щелочную фазу подкисляли до значения рН 1 с применением 6Н раствора хлористого водорода с последующей экстракцией метиленхлоридом (3 раза по 100 мл). Последнюю объединенную органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом 4-гидрокси-3,5-диметилфуран-2(5H)-он. ЖХ/МС: (M+23)+: 151,10, 1H-ЯМР (500 МГц, CDCl3), δ 4,882-4,842 (дд, J=6,8 Гц, 1H), 3,744 (с, 1H), 1,759 (с, 3H), 1,526-1,513 (д, J=6,8 Гц, 3H).
Стадия C
2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат
К раствору 4-гидрокси-3,5-диметилфуран-2(5H)-она (400 мг, 3,12 ммоль) в метиленхлориде (10 мл) при -78°C по каплям добавляли 2,6-лутидин (0,545 мл, 4,68 ммоль) и трифлатный ангидрид (0,633 мл, 3,75 ммоль). Температуру реакции поддерживали при -78°C в течение 1 часа перед нагреванием реакционной смеси до комнатной температуры в течение 2 часов. Смесь разбавляли метиленхлоридом (100 мл), промывали 1Н раствором хлористого водорода (3×100 мл) и разбавленным раствором бикарбоната натрия, затем сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 261,00.
Стадия D
трет-бутил-2-(2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
Смесь трет-бутил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (200 мг, 0,786 ммоль), 2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната, ксантфоса (45,5 мг, 0,079 ммоль), воды (0,043 мл, 2,4 ммоль) и карбоната калия (217 мг, 1,57 ммоль) в толуоле (20 мл) дегазировали азотом в течение 20 мин с последующим добавлением ацетата палладия (8,8 мг, 0,039 ммоль). Полученную смесь нагревали при 65°C в течение 16 часов. После фильтрования фильтрат концентрировали и остаток очищали на колонке с силикагелем путем элюирования смесью EtOAc/гексан получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 365,20.
Стадия E
2-(2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору трет-бутил-2-(2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (195 мг, 0,535 ммоль) в метиленхлориде (1 мл) добавляли трифторуксусную кислоту (2,06 мл, 26,8 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 1 часа. После удаления летучих веществ остаток растворяли в метиленхлориде (1 мл), обрабатывали раствором хлористого водорода (4 мл, 1Н в простом диэтиловом эфире) и концентрировали, получая при этом 2-(2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он. ЖХ/МС: (M+1)+: 265,19.
Промежуточное соединение 28
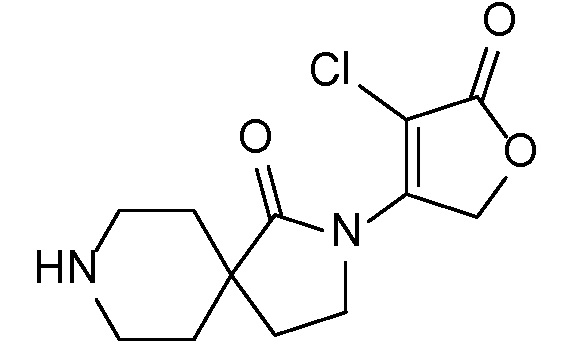
2-(4-хлор-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
трет-Бутил-2-(4-хлор-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
К раствору трет-бутил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилата (I-16, стадия A) (2,1 г, 6,2 ммоль) в хлороформе (50 мл) при комнатной температуре добавляли NCS (1,00 г, 7,49 ммоль), и полученный раствор нагревали при 60°C в течение ночи. После удаления летучих веществ остаток очищали на колонке с силикагелем с применением EtOAc/гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 371,11; 372,99.
Стадия B
2-(4-хлор-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору трет-бутил-2-(4-хлор-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (2,26 г, 6,09 ммоль) в метиленхлориде (10 мл) добавляли трифторуксусную кислоту (9,39 мл, 122 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 1 часа. После удаления летучих веществ остаток распределяли между метиленхлоридом (100 мл) и 1Н раствором гидроксида натрия (100 мл). Щелочную фазу экстрагировали метиленхлоридом (2×100 мл). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом 2-(4-хлор-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он. ЖХ/МС: (M+1)+: 271,07; 272,96.
Промежуточное соединение 29
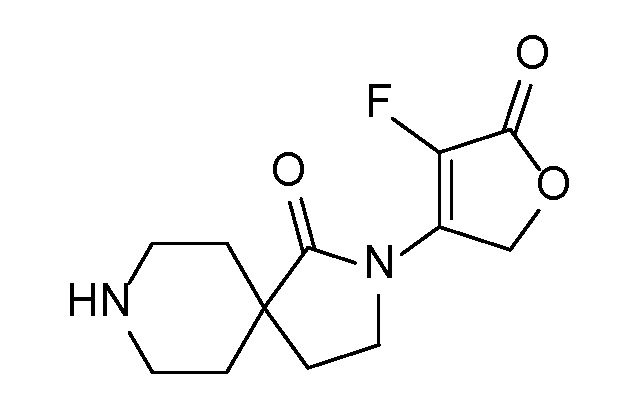
2-(4-фтор-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
3-бром-4-этокси-3-фтор-4-гидроксидигидрофуран-2(3H)-он
К раствору 4-гидроксифуран-2(5H)-она (2,25 г, 22,5 ммоль) в этаноле (20 мл) добавляли NBS (4.00 г, 22,5 ммоль) и перемешивали полученный раствор при комнатной температуре в течение 40 мин. Затем добавляли 1-хлорметил-4-фтор-1,4-диазонийбицикло[2,2.2]октан-бис(тетрафторборат) (7,97 г, 22,5 ммоль) и перемешивали полученную смесь при комнатной температуре в течение ночи. После фильтрования и концентрирования остаток очищали на колонке с силикагелем с применением EtOAc/гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. 1H-ЯМР (500 МГц, CDCl3), δ 4,322-4,303 (м, J=7,0 Гц, 2H), 3,879-3,788 (м, 2H), 1,356-1,328 (т, J=7,0 Гц, 3H).
Стадия B
3-фтор-4-гидроксифуран-2(5H)-он
К раствору 3-бром-4-этокси-3-фтор-4-гидроксидигидрофуран-2(3H)-она (4,39 г, 18,1 ммоль) в тетрагидрофуране (20 мл) добавляли гидрид три-н-бутилолова (9,39 мл, 35,0 ммоль) при 0°C в атмосфере N2. Полученный раствор перемешивали при комнатной температуре в течение ночи. После удаления летучих веществ остаток перемешивали в 30 мл 50% уксусной кислоты и 30 мл гексана при комнатной температуре в течение 30 мин. Кислотную фазу перед концентрированием промывали гексаном (3×30 мл). Остаток растворяли в растворе карбоната натрия (50 мл, 2Н), экстрагировали смесью 40% EtOAc/гексан (4×50 мл), щелочную фазу подкисляли до значения pH <1 с помощью 1Н раствора хлористого водорода. Затем кислотную фазу экстрагировали этилацетатом (8 раз 60 мл). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом 3-фтор-4-гидроксифуран-2(5H)-он. 1H-ЯМР (500 МГц, DMSO-d6), δ 4,694-4,687 (д, J=3,9 Гц, 2H).
Стадия C
4-фтор-2-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат
К раствору 3-фтор-4-гидроксифуран-2(5H)-она (400 мг, 3,39 ммоль) в метиленхлориде (10 мл) при -78°C по каплям добавляли 2,6-лутидин (0,592 мл, 5,08 ммоль) и трифлатный ангидрид (0,687 мл, 4,07 ммоль), температуру реакции поддерживали при -78°C в течение 1 часа перед нагреванием до комнатной температуры в течение 2 часов. Смесь промывали 1Н раствором хлористого водорода (3 раза по 100 мл) и разбавленным раствором бикарбоната натрия, сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. 1H-ЯМР (500 МГц, CDCl3), δ 4,986-4,974 (д, J=6,0 Гц, 2H).
Стадия D
трет-Бутил-2-(4-фтор-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
Смесь трет-бутил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (150 мг, 0,590 ммоль), 4-фтор-2-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (148 мг, 0,590 ммоль), ксантфоса (34,1 мг, 0,059 ммоль), воды (0,032 мл, 1,77 ммоль) в толуоле(20 мл) дегазировали азотом с последующим добавлением ацетата палладия (6,6 мг, 0,029 ммоль). Полученную смесь нагревали при 65°C в течение ночи. После фильтрования через CELITE® фильтрат концентрировали, и остаток очищали на колонке с силикагелем с применением этилацетата и гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 355,15.
Стадия E
2-(4-фтор-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору трет-бутил-2-(4-фтор-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (109 мг, 0,308 ммоль) в метиленхлориде (1 мл) добавляли трифторуксусную кислоту (1,896 мл, 24,61 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 1 часа. После удаления летучих веществ остаток растворяли в метаноле и загружали в ионообменную колонку. После промывания метанолом продукт элюировали 2Н раствором аммиака в метаноле, затем концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 255,09.
Промежуточное соединение 30
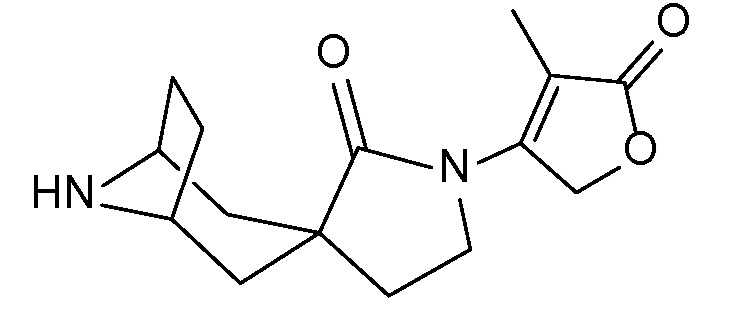
(1R,3r,5S)-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-2'-он
Стадия A
(1R,3s,5S)-8-трет-бутил-3-метил-8-азабицикло[3.2.1]октан-3,8-дикарбоксилат
К раствору (1R,3s,5S)-8-(трет-бутоксикарбонил)-8-азабицикло[3.2.1]октан-3-карбоновой кислоты (5,00 г, 19,6 ммоль) в смеси растворителей, состоящей из безводного MeOH (60 мл) и ДХМ (60,0 мл), добавляли (триметилсилил)диазометан (19,6 мл, 39,2 ммоль). Смесь перемешивали в течение 0,5 часа. Добавляли AcOH (5 мл). Летучие вещества удаляли при пониженном давлении. Остаток растворяли в EtOAc, промывали раствор насыщенным раствором NaHCО3 и насыщенным раствором соли и сушили над MgSО4. Растворитель удаляли, получая при этом твердое вещество, которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС MS: 214,10 (M+1-56).
Стадия B
(1R,3r,5S)-8-трет-бутил-3-метил-3-(цианометил)-8-азабицикло[3.2.1]октан-3,8-дикарбоксилат
К раствору (1R,3r,5S)-8-трет-бутил-3-метил-8-азабицикло[3.2.1]октан-3,8-дикарбоксилата (5,00 г, 18,6 ммоль) в ТГФ (100 мл) добавляли LDA (13,9 мл, 27,8 ммоль) при -78°C. Смесь перемешивали при той же температуре в течение 30 мин, затем путем впрыскивания добавляли бромацетонитрил (1,94 мл, 27,8 ммоль) в ТГФ (15 мл). Смесь перемешивали при -78°C в течение 15 мин, гасили насыщенным раствором KHSO4 при -78°C, нагревали до комнатной температуры и разбавляли простым эфиром (100 мл). Органический слой отделяли, водный слой экстрагировали простым эфиром (50 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили (MgSО4) и концентрировали. Остаток очищали на колонке (силикагель 120 г, градиент EtOAc-гексан 0-50%, затем 50% EtOAc). 1H-ЯМР (500 МГц, CDCl3): δ м.д. 4,18 (1H, м), 4,27 (1H, м), 3,83 (3H, с), 2,58 (2H, м), 2,43 (2H, м), 1,55-1,95 (6H, м), 1,50 (9H, с). ЖХ-МС 209,23 (M+1-100).
Стадия С
(1R,3r,5S)-8-трет-бутил-3-метил-3-(2-аминоэтил)-8-азабицикло[3.2.1]октан-3,8-дикарбоксилат
К раствору (1R,3r,5S)-8-трет-бутил-3-метил-3-(цианометил)-8-азабицикло[3.2.1]октан-3,8-дикарбоксилата (4,0 г, 12,97 ммоль) в этаноле (20 мл) и AcOH (20 мл) добавляли оксид платины(IV) (0,295 г, 1,30 ммоль). Смесь подвергали гидрированию в шейкере (при давлении водорода 45 фунт/кв.дюйм) в течение 24 часов. Катализатор отфильтровывали через рыхлый слой CELITE® и концентрировали фильтрат. Неочищенный материал применяли на следующей стадии без дополнительной очистки. ЖХ-МС: 313,20 (M+1), 257,18 (M+1-56).
Стадия D
(1R,3r,5S)-трет-бутил-2'-оксо-8-азаспиро[бицикло [3.2.1]октан -3,3'-пирролидин]-8-карбоксилат
Смесь (1R,3r,5S)-трет-бутил-3-метил-3-(2-аминоэтил)-8-азабицикло[3.2.1]октан-3,8-дикарбоксилата (4,2 г, 13,4 ммоль) и карбоната калия (9,29 г, 67,2 ммоль) в MeOH (50 мл) нагревали при 60°C в течение 1 часа. Смесь концентрировали и добавляли ДХМ (50 мл). Суспензию фильтровали через рыхлый слой силикагеля. Фильтрат концентрировали, получая при этом твердое вещество, которое применяли непосредственно на следующей стадии. 1H-ЯМР (500 МГц, CDCl3): δ м.д. 5,95 (1H, шир.с), 4,30 (1H, м), 4,20 (1H, м), 3,26 (2H, т, J=7,0 Гц), 1,75-2,15 (6H, м), 1,47 (9H, с). ЖХМС: 281,15 (+1), 225,14 (M+1-56).
Стадия E
(1R,3r,5S)-трет-бутил-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилат
Смесь 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (1,861 г, 7,56 ммоль), (1R,3r,5S)-трет-бутил-2'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилата (1,63 г, 5,81 ммоль), ацетата палладия(II) (0,065 г, 0,291 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (0,336 г, 0,581 ммоль), карбоната калия (2,411 г, 17,44 ммоль) и воды (0,314 мл, 17,4 ммоль) в толуоле (150 мл) нагревали при 60°C в атмосфере N2 в течение ночи. Смесь разбавляли EtOAc. Твердые вещества отфильтровывали через рыхлый слой CELITE® и концентрировали фильтрат. Остаток очищали на колонке (80 г силикагеля, 0-100% EtOAc в гексане, затем 100% EtOAc). ЖХ-МС: 377,12 (M+1).
Стадия F
(1R,3r,5S)-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-2'-он
Раствор (1R,3r,5S)-трет-бутил-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилата в ДХМ (50 мл) и TFA (10 мл) перемешивали при комнатной температуре в течение 1 часа. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в метаноле и загружали в ионообменную колонку Bond Elut SCX, после чего колонку промывали 20 мл метанола. Колонку с требуемым соединением элюировали метанолом, чтобы удалить TFA (~20 мл), и требуемое соединение в форме свободного основания элюировали 2Н раствором NH3 в метаноле (~20 мл). Раствор концентрировали, получая при этом свободное основание (в твердой форме). 1H-ЯМР (500 МГц, CDCl3): δ м.д. 5,25 (2H, с), 3,95 (2H, т, J=6,9 Гц), 3,73 (2H, м), 2,15 (4H, м), 2,07 (3H, с), 2,05 (4H, м), 1,85 (2H, м). ЖХ/МС 277,10 (M+1).
Промежуточное соединение 31
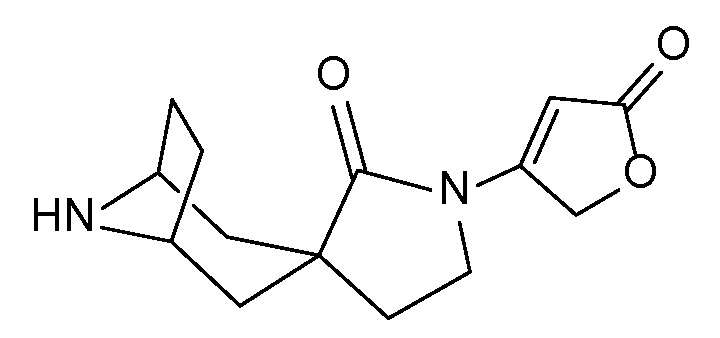
(1R,3r,5S)-1'-(5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-2'-он
Указанное в заголовке соединение получали способом, аналогичным способу, описанному для (1R,3r,5S)-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-2'-она, за исключением того, что на стадии E вместо 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната применяли 4-бромфуран-2-он. ЖХМС: 263 (M+1).
Промежуточное соединение 32
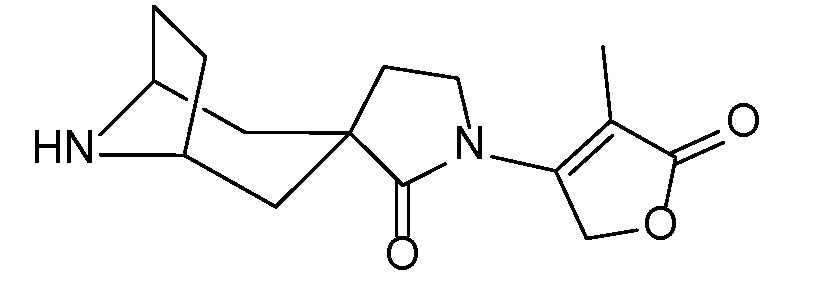
(1R,3r,5S)-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-2'-он
Стадия A
(1R,5S,Z)-трет-бутил-3-(1-циано-2-метокси-2-оксоэтилиден)-8-азабицикло[3.2.1]октан-8-карбоксилат
Метил-2-цианоацетат (3,63 г, 36,6 ммоль), имеющийся в продаже (1R,5S)-трет-бутил-3-оксо-8-азабицикло[3.2.1]октан-8-карбоксилат (5,5 г, 24,41 ммоль), ацетат аммония (2,51 мл, 36,6 ммоль), уксусную кислоту (5,59 мл, 98 ммоль) и толуол (100 мл) помещали в 500 мл круглодонную колбу, присоединенную к водоотделителю непрерывного действия Дина-Старка, который соединяли с обратным холодильником. Колбу нагревали на масляной бане при 150°C, и воду, которая отгонялась из смеси при ее нагревании с толуолом и обратным холодильником, удаляли из водоотделителя с некоторой периодичностью (в течение ночи). Растворитель удаляли при пониженном давлении. Остаток растворяли в EtOAc (150 мл). Раствор промывали насыщенным раствором соли, сушили (MgSО4) и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (градиент гексана в EtOAc 0-60%). 1H-ЯМР (500 МГц, CDCl3): δ м.д. 4,40 (2H, м), 3,86 (3H, м), 2,75 (2H, м), 2,55 (2H, м), 2,06 (2H, м), 1,57 (2H, м), 1,50 (9H, с). ЖХ-МС-250,99,12 (M+1-56).
Стадия B
(1R,3r,5S)-трет-бутил-3-(1-циано-2-метокси-2-оксоэтил)-3-винил-8-азабицикло[3.2.1]октан-8-карбоксилат
К суспензии (1R,,5S,Z)-трет-бутил-3-(1-циано-2-метокси-2- оксоэтилиден)-8-азабицикло[3.2.1]октан-8-карбоксилата (7,17 г, 23,4 ммоль) и иодида меди(I) (2,229 г, 11,70 ммоль) в ТГФ (150 мл) добавляли бромид винилмагния (35,1 мл, 35,1 ммоль) путем впрыскивания при -10°C. Смесь перемешивали при той же температуре в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором ацетата аммония и разбавляли EtOAc (100 мл). Органический слой отделяли и экстрагировали водный слой EtOAc (100 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили над MgSО4 и концентрировали. Неочищенный материал очищали колоночной хроматографией на силикагеле (элюирование 0-50% EtOAc, затем 50% EtOAc в гексане). 1H-ЯМР (500 МГц, CDCl3): δ м.д. 5,82 (1H, дд, J1 = 17,3 Гц, J2 = 10,7 Гц), 5,15 (1H, д, J=10,7 Гц), 5,10 (1H, д, J=17,3 Гц), 4,20-4,50 (2H, м), 3,85 (1H, с), 3,81 (3H, с), 2,17-2,35 (2H, м), 1,90-2,15 (4H, м), 1,58-1,72 (2H, м), 1,47 (9H, с). ЖХ-МС: 278,92 (M+1-56).
Стадия C
(1R,3s,5S)-трет-бутил-3-(цианометил)-3-винил-8-азабицикло[3.2.1]октан-8-карбоксилат
Суспензию (1R,3r,5S)-трет-бутил-3-(1-циано-2-метокси-2-оксоэтил)-3-винил-8-азабицикло[3.2.1]октан-8-карбоксилата (7,2 г, 21,53 ммоль) и хлорида натрия (1,258 г, 21,53 ммоль) в ДМСО (40 мл) и воде (4 мл) нагревали при 160°C на масляной бане в течение 2 часов, затем охлаждали до комнатной температуры. Добавляли воду (50 мл) и экстрагировали смесь простым этиловым эфиром (дважды по 50 мл). Объединенные эфирные слои промывали насыщенным раствором соли, сушили над MgSО4 и концентрировали при пониженном давлении. Неочищенный материал очищали колоночной хроматографией (0-70% этилацетата в гексане). 1H-ЯМР (500 МГц, CDCl3): δ м.д. 5,73 (1H, дд, J1=17,7 Гц, J2=11,0 Гц), 5,10 (1H, д, J=11 Гц), 5,08 (1H, д, J=17,7 Гц), 5,30 (2H, м), 2,70 (2H, с), 1,95-2,12 (4H, м), 1,70 (4H, м), 1,45 (9H, с). ЖХ-МС: 221,11 (M+1-56).
Стадия D
(1R,3s,5S)-трет-бутил-3-(цианометил)-3-формил-8-азабицикло[3.2.1]октан-8-карбоксилат
К суспензии (1R,3s,5S)-трет-бутил-3-(цианометил)-3-винил-8-азабицикло[3.2.1]октан-8-карбоксилата (4,00 г, 14,5 ммоль, воды (15 мл) и периодата натрия (12,4 г, 57,9 ммоль) в диоксане (45 мл) добавляли тетроксид осмия (0,184 г, 0,724 ммоль). Суспензию перемешивали в течение 17 часов при комнатной температуре. Смесь подкисляли 1Н раствором хлористоводородной кислоты и разбавляли EtOAc (50 мл). Органический слой отделяли, промывали насыщенным раствором соли, сушили над MgSО4 и концентрировали при пониженном давлении. Остаток применяли без дополнительной очистки. 1H-ЯМР (500 МГц, CDCl3): δ м.д. 9,14 (1H, с), 4,17 (2H, м), 3,80 (3H, с), 2,32 (2H, м), 2,15 (2H, м), 1,73 (2H, м), 1,55 (2H, м). ЖХ-МС-223,16 (M+1-56).
Стадия E
(1R,3s,5S)-8-(трет-бутоксикарбонил)-3-(цианометил)-8-азабицикло[3.2.1]октан-3-карбоновая кислота
К раствору (1R,3s,5S)-трет-бутил-3-(цианометил)-3-формил-8- азабицикло[3.2.1]октан-8-карбоксилата (5,10 г, 18,3 ммоль) в т-BuOH/H2О (2:1) добавляли гидрат дигидрофосфата натрия (7,59 г, 55,0 ммоль) и 2-метилбут-2-ен (9,7 мл, 92 ммоль). Суспензию охлаждали до 0°C, и порциями добавляли хлорит натрия (4,97 г, 55,0 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 часа, подкисляли 1M раствором HCl, экстрагировали смесью CHCl3:2-пропанол (3:1), сушили (Na2SО4) и концентрировали. Неочищенный материал нагревали досуха с обратным холодильником в присутствии толуола и аппарата Дина-Старка. Горячий толуольный раствор отделяли от твердого вещества и концентрировали раствор, получая при этом указанное в заголовке соединение. ЖХ/МС 239,23 (M+1-56), 295,23 (+1).
Стадия F
(1R,3s,5S)-8-трет-бутил-3-метил-3-(цианометил)-8-азабицикло[3.2.1]октан-3,8-дикарбоксилат
К раствору (1R,3s,5S)-8-(трет-бутоксикарбонил)-3-(цианометил)-8-азабицикло[3.2.1]октан-3-карбоновой кислоты (5,20 г, 17,7 ммоль) в смеси с MeOH (30 мл) и ДХМ (30 мл) медленно добавляли (триметилсилил)диазометан (13,3 мл, 26,5 ммоль) при комнатной температуре. Смесь перемешивали при той же температуре в течение 0,5 часа. Добавляли уксусную кислоту (~5 мл), чтобы удалить избыток (триметилсилил)диазометана. Раствор концентрировали, и остаток применяли непосредственно на следующей стадии. 1H-ЯМР (500 МГц, CDCl3): δ м.д. 4,32 (2H, м), 3,74 (3H, с), 2,85 (2H, с), 2,65 (2H, м), 2,08 (2H, м), 1,65 (4H, м), 1,42 (9H, с). ЖХМС: 253 (M+1-56).
Стадия G
(1R,3s,5S)-8-трет-бутил-3-метил-3-(2-аминоэтил)-8-азабицикло[3.2.1]октан-3,8-дикарбоксилат
Смесь (1R,3s,5S)-8-трет-бутил-3-метил-3-(цианометил)-8- азабицикло[3.2.1]октан-3,8-дикарбоксилата (3,5 г, 11,4 ммоль) и оксида платины (IV) (0,258 г, 1,14 ммоль) в этаноле (20 мл) и AcOH (20 мл) подвергали гидрированию в шейкере (при давлении водорода 45 фунт/кв.дюйм) при комнатной температуре в течение 48 часов. Катализатор отфильтровывали через рыхлый слой CELITE®. Фильтрат концентрировали, и остаток применяли непосредственно на следующей стадии. ЖХ/MS 313,25 (+1), 257,25 (+1-56).
Стадия H
(1R,3s,5S)-трет-бутил-2'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилат
Смесь (1R,3s,5S)-8-трет-бутил-3-метил-3-(2-аминоэтил)-8- азабицикло[3.2.1]октан-3,8-дикарбоксилата (3,50 г, 11,2 ммоль) и карбоната калия (7,74 г, 56,0 ммоль) в MeOH (50 мл) нагревали при 60°C в течение 1 часа. Смесь концентрировали и добавляли ДХМ (50 мл). Суспензию фильтровали через рыхлый слой силикагеля, и фильтрат концентрировали, получая при этом твердое вещество, которое применяли непосредственно на следующей стадии. 1H-ЯМР (500 МГц, CDCl3): δ м.д. 6,43 (1H, шир.с), 4,25 (1H, м), 4,37 (1H, м), 3,32 (2H, т, J=7,0 Гц), 2,24 (2H, м), 2,35 (2H, м), 1,97 (2H, м), 1,82 (2H, м), 1,45 (9H, с), 1,46 (2H, м). ЖХМС: 281 (M+1), 225 (M+1-56).
Стадия I: (1R,3s,5S)-трет-бутил-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилат
Смесь (1R,3s,5S)-трет-бутил-2'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилата (2,00 г, 7,13 ммоль), 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (1,93 г, 7,85 ммоль), диацетоксипалладия (0,080 г, 0,36 ммоль), (9,9-диметил-9H-ксантен-4,5-диил)бис(дифенилфосфина) (0,413 г, 0,713 ммоль), карбоната калия (2,96 г, 21,4 ммоль) и воды (0,386 г, 21,40 ммоль) в толуоле (100 мл) нагревали при 60°C в атмосфере N2 в течение 4 часов. Смесь разбавляли EtOAc (50 мл). Твердое вещество отфильтровывали через рыхлый слой CELITE®, и концентрировали фильтрат. Остаток очищали колоночной хроматографией (0-100% EtOAc в гексане, затем 100% EtOAc), получая при этом указанное в заголовке соединение. ЖХ-МС 321,03 (+1-56), 377,03 (+1).
Стадия J
(1R,3s,5S)-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-2'-он
(1R,3s,5S)-трет-бутил-2'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилат перемешивали с TFA (5 мл) в ДХМ (30 мл) при комнатной температуре в течение 1 часа. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в метаноле и загружали в колонку Bond Elut SCX (ионообменный картридж), после чего колонку промывали 20 мл метанола. Колонку с требуемым соединением элюировали метанолом, чтобы удалить TFA (~20 мл), требуемое соединение в форме свободного основания элюировали 2Н раствором NH3 в метаноле (~20 мл). Раствор концентрировали, получая при этом указанное в заголовке соединение в виде свободного основания (в твердой форме). 1H-ЯМР (500 МГц, CDCl3): δ м.д. 5,25 (1H, с), 4,00 (2H, т, J=6,9 Гц), 3,67 (2H, м), 2,42 (2H, т, J=6,9 Гц), 2,06 (3H, с), 2,04 (2H, м), 1,97 (2H, м), 1,82 (2H, м). ЖХМС 277,03 (M+1), 321,03 (M+1-56).
Промежуточное соединение 33А и 33В
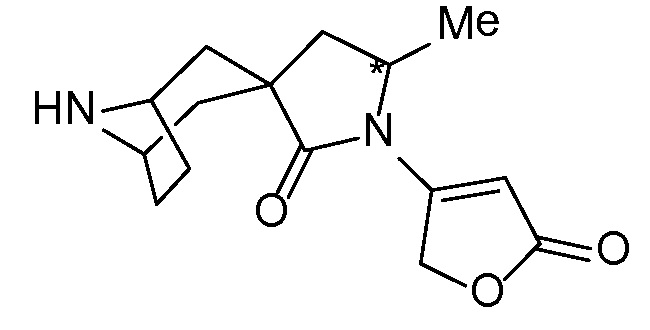
(1R,3r,5S)-5'-метил-1'-(5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-2'-он (изомеры A и B)
Стадия A
(1R,3r,5S)-8-трет-бутил-3-метил-8-азабицикло[3.2.1]октан-3,8-дикарбоксилат
К раствору имеющейся в продаже (1R,3r,5S)-8-(трет-бутоксикарбонил)-8-азабицикло[3.2.1]октан-3-карбоновой кислоты (10,0 г, 39,2 ммоль) и метанола (4,75 мл, 118 ммоль) в метиленхлориде (200 мл) добавляли EDC (11,3 г, 58,8 ммоль), диизопропилэтиламин (13,7 мл, 78 ммоль) и DMAP (0,479 г, 3,92 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 16 часов. Реакционный раствор промывали бисульфатом калия (1Н, 200 мл), водой (200 мл), насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 270,1.
Стадия B
(1R,3r,5S)-8-трет-бутил-3-метил 3-(2-метилаллил)-8- азабицикло[3.2.1]октан-3,8-дикарбоксилат
К раствору диизопропиламина (5,94 мл, 41,7 ммоль) в тетрагидрофуране (5 мл) при 0°C по каплям добавляли н-бутиллитий (16,7 мл, 41,7 ммоль) и перемешивали полученный раствор при 0°C в течение 0,5 часа. Полученный раствор при -78°C добавляли по каплям к раствору соединения со стадии A (7,48 г, 27,8 ммоль) в тетрагидрофуране (90 мл). Полученный раствор перемешивали при -78°C в течение 1 часа и затем по каплям добавляли 3-бром-2-метилпропен (4,03 мл, 40,0 ммоль). После перемешивания при -78°C в течение 1,5 часов реакционную смесь гасили путем добавления насыщенного раствора ацетата аммония. Смесь разбавляли этилацетатом, дважды промывали насыщенным раствором ацетата аммония, сушили над сульфатом натрия, концентрировали и остаток очищали на колонке с силикагелем с применением этилацетата/гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 324,3.
Стадия C
(1R,3r,5S)-8-трет-бутил-3-метил-3-(2-оксопропил)-8-азабицикло[3.2.1]октан-3,8-дикарбоксилат
К раствору (1R,3r,5S)-8-трет-бутил-3-метил-3-(2-метилаллил)-8-азабицикло[3.2.1]октан-3,8-дикарбоксилата (7,99 г, 24,7 ммоль) в диоксане (100 мл) и воде (50 мл) добавляли периодат натрия (10,6 г, 49,4 ммоль) и тетроксид осмия (0,126 г, 0,494 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 18 часов и затем добавляли тиосульфат натрия (1 г). После перемешивания при комнатной температуре в течение 0,5 часа смесь фильтровали и концентрировали фильтрат. Остаток растворяли в этилацетате (300 мл) и промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Полученный остаток очищали на силикагеле с применением этилацетата и гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 326,2.
Стадия D
(1R,3r,5S)-трет-бутил-5'-метил-2'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилат
К раствору (1R,3r,5S)-8-трет-бутил-3-метил-3-(2-оксопропил)-8-азабицикло[3.2.1]октан-3,8-дикарбоксилата (7,90 г, 24,3 ммоль) в метаноле (50 мл) добавляли сульфат магния (5,84 г, 48,6 ммоль), ацетат аммония (3,74 г, 48,6 ммоль) и цианоборгидрид натрия (3,05 г, 48,6 ммоль). Полученную смесь нагревали при 80°C в герметично закрывающейся трубке в течение 18 часов. После охлаждения до комнатной температуры смесь фильтровали, фильтрат концентрировали, и остаток распределяли между метиленхлоридом и насыщенным раствором бикарбоната натрия. Водную фазу экстрагировали метиленхлоридом, объединенную органическую фазу сушили над сульфатом натрия, концентрировали и остаток очищали на силикагеле с применением этилацетата в качестве элюирующего растворителя, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 295,3.
Стадия E
(1R,3r,5S)-трет-бутил-5'-метил-2'-оксо-1'-(5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилат, изомер (A) и изомер (B)
К раствору (1R,3r,5S)-трет-бутил-5'-метил-2'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилата (2,41 г, 8,19 ммоль) в толуоле (30 мл) добавляли 3-бромфуранон (1,60 г, 9,82 ммоль), ксантфос (0,474 г, 0,819 ммоль), карбонат калия (2,263 г, 16,37 ммоль), воду (0,442 г, 24,6 ммоль) и ацетат палладия (0,092 г, 0,409 ммоль). Полученную смесь продували азотом в течение 30 мин, затем нагревали при 65°C в течение 16 часов. После охлаждения до комнатной температуры смесь фильтровали, фильтрат концентрировали, и остаток очищали на силикагеле с применением этилацетата и гексана в качестве элюирующих растворителей, получая при этом рацемат (1R,3r,5S)-трет-бутил-5'-метил-2'-оксо-1'-(5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилата (ЖХ/МС: (M+1)+: 377,05). Рацемат разделяли на хиральной колонке AS (30×250 мм) с применением смеси метанол/ацетонитрил/диоксид углерода, получая при этом изомер (A) - быстро элюирующий энантиомер; ЖХ/МС: (M+1)+: 377,04; и изомер (B) - медленно элюирующий энантиомер; ЖХ/МС: (M+1)+: 377,03.
Стадия F
Изомер A и изомер B (1R,3r,5S)-5'-метил-1'-(5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-2'-она
TFA (3,27 мл, 42,5 ммоль) добавляли к раствору изомера (A) (1R,3r,5S)-трет-бутил-5'-метил-2'-оксо-1'-(5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилата (0,80 г, 2,1 ммоль) в метиленхлориде (5 мл) и перемешивали полученный раствор при комнатной температуре в течение 1 часа. После удаления летучих веществ остаток растворяли в метаноле (5 мл) и ощелачивали до свободного основания на ионообменной колонке путем промывания сначала метанолом с последующим промыванием 1Н раствором аммиака в метаноле, получая при этом изомер (A) указанного в заголовке соединения: ЖХ/МС: (M+1)+: 277,07.
К раствору изомера (B) (1R,3r,5S)-трет-бутил-5'-метил-2'-оксо-1'-(5-оксо-2,5-дигидрофуран-3-ил)-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-8-карбоксилата (0,8 г, 2,13 ммоль) в метиленхлориде (5 мл) добавляли TFA (3,27 мл, 42,5 ммоль) и перемешивали полученный раствор при комнатной температуре в течение 1 часа. После удаления летучих веществ остаток растворяли в метаноле (5 мл) и ощелачивали до свободного основания на ионообменной колонке путем промывания сначала метанолом с последующим промыванием 1Н раствором аммиака в метаноле, получая при этом изомер B указанного в заголовке соединения: ЖХ/МС:(M+1)+: 377,07.
Промежуточное соединение 34
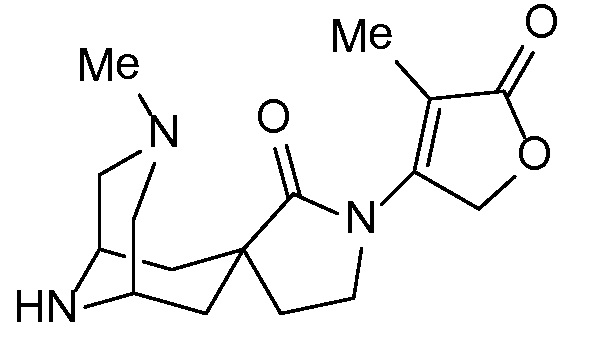
(1R,3's,5S)-3-метил-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-3,9-диазаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-2'-он
Стадия A
(1R,5S)-трет-бутил-3-метил-7-(((трифторметил)сульфонил)окси)-3,9-диазабицикло[3.3.1]нон-6-ен-9-карбоксилат
К раствору имеющегося в продаже (1R,5S)-трет-бутил-3-метил-7-оксо-3,9-диазабицикло[3.3.1]нонан-9-карбоксилата (10,0 г, 39,3 ммоль) в тетрагидрофуране (100 мл) при -78°C по каплям добавляли раствор LDA (23,6 мл, 47,2 ммоль). Полученный раствор перемешивали при -78°C в течение 0,5 часа перед тем, как добавлять к раствору 2-[N,N-бис(трифторметансульфонил)амино]-5-хлорпиридина (18,5 г, 47,2 ммоль) в тетрагидрофуране (25 мл). Полученный раствор перемешивали при -78°C в течение 2 часов, затем нагревали до комнатной температуры в течение 20 минут перед гашением путем добавления насыщенного раствора хлорида аммония и этилацетата. Органическую фазу промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали, остаток очищали на силикагеле с применением этилацетата и гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 386,94.
Стадия B
(1R,5S)-9-трет-бутил-7-метил-3-метил-3,9-диазабицикло[3.3.1]нон-6-ен-7,9-дикарбоксилат
К раствору (1R,5S)-трет-бутил-3-метил-7-(((трифторметил)сульфонил)окси)-3,9-диазабицикло[3.3.1]нон-6-ен-9-карбоксилата (14 г, 36 ммоль) и диизопропилэтиламина (9,47 мл, 54,3 ммоль) в метаноле (100 мл) и ДМФА (100 мл) добавляли трифенилфосфин (0,95 г, 3,62 ммоль) и ацетат палладия(II) (0,407 г, 1,81 ммоль). Смесь перемешивали в атмосфере моноксида углерода в течение 24 часов. Смесь концентрировали и затем распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли, сушили над сульфатом натрия, концентрировали и остаток очищали на силикагеле с применением этилацетата и гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 297,2.
Стадия C
(1R,5S)-9-трет-бутил-7-метил-3-метил-3,9-диазабицикло[3.3.1]нонан-7,9-дикарбоксилат
К раствору (1R,5S)-9-трет-бутил-7-метил-3-метил-3,9-диазабицикло[3.3.1]нон-6-ен-7,9-дикарбоксилата (4,69 г, 15,8 ммоль) в метаноле (50 мл) добавляли палладий на угле (10%, 1,684 г, 1,583 ммоль) и подвергали полученную смесь гидрированию под давлением 40 фунт/кв.дюйм в течение трех дней. После фильтрования через CELITE® в атмосфере азота фильтрат концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 299,1.
Стадия D
(1R,5S,7s)-9-трет-бутил-7-метил-7-(цианометил)-3-метил-3,9- диазабицикло[3.3.1]нонан-7,9-дикарбоксилат
К раствору диизопропиламина (3,28 мл, 23,02 ммоль) в тетрагидрофуране (5 мл) при 0°C по каплям добавляли н-бутиллитий (11,51 мл, 23,02) и перемешивали полученный раствор при 0°C в течение 0,5 часа. К раствору (1R,5S)-9-трет-бутил-7-метил 3-метил-3,9-диазабицикло[3.3.1]нонан-7,9-дикарбоксилата (4,58 г, 15,35 ммоль) в тетрагидрофуране (50 мл) при -78°C по каплям добавляли полученный выше раствор LDA. После перемешивания при -78°C в течение 1 часа добавляли по каплям бромацетонитрил (1,54 мл, 22,1 ммоль) и полученный раствор перемешивали при -78°C в течение 1 часа перед тем, как гасить смесь путем добавления насыщенного раствора хлорида аммония. Смесь экстрагировали этилацетатом. Объединенную органическую фазу сушили над сульфатом натрия, концентрировали и остаток очищали на силикагеле с применением метанола и дихлорметана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 338,2.
Стадия E
(1R,5S,7s)-9-трет-бутил-7-метил-7-(2-аминоэтил)-3-метил-3,9-диазабицикло[3.3.1]нонан-7,9-дикарбоксилат
К раствору (1R,5S,7s)-9-трет-бутил-7-метил-7-(цианометил)-3-метил-3,9-диазабицикло[3.3.1]нонан-7,9-дикарбоксилата (4,39 г, 13,0 ммоль) в метаноле (30 мл) добавляли оксид платины (IV) (0,207 г, 0,911 ммоль) и полученную смесь подвергали гидрированию под давлением 40 фунт/кв.дюйм в течение 16 часов. После фильтрования через CELITE® в атмосфере азота фильтрат концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 342,2.
Стадия F
(1R,3's,5S)-трет-бутил-3-метил-2'-оксо-3,9-диазаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-9-карбоксилат
К раствору (1R,5S,7s)-9-трет-бутил-7-метил-7-(2-аминоэтил)-3-метил-3,9-диазабицикло[3.3.1]нонан-7,9-дикарбоксилата (4,44 г, 13,0 ммоль) в метаноле (100 мл) добавляли карбонат калия (10,8 г, 78 ммоль) и нагревали полученный раствор с обратным холодильником в течение 8 часов. После охлаждения до комнатной температуры смесь фильтровали и концентрировали фильтрат, остаток растворяли в метиленхлориде (200 мл), сушили над сульфатом натрия, и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 310,26.
Стадия G
(1R,3's,5S)-трет-бутил-3-метил-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2'-оксо-3,9-диазаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-9-карбоксилат
Смесь (1R,3's,5S)-трет-бутил-3-метил-2'-оксо-3,9-диазаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-9-карбоксилата (4,0 г, 12,9 ммоль), 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (3,82 г, 15,5 ммоль), Ксантфоса (0,748 г, 1,29 ммоль) и карбоната калия (3,57 г, 25,9 ммоль) в толуоле (100 мл) дегазировали азотом в течение 20 минут с последующим добавлением ацетата палладия (II) (0,145 г, 0,646 ммоль). Полученную смесь нагревали при 65°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через CELITE®, фильтрат концентрировали и остаток очищали на силикагеле, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 406,21.
Стадия H
(1R,3's,5S)-3-метил-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-3,9-диазаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-2'-он
К раствору соединения, полученного на стадии G (2,63 г, 6,49 ммоль), в дихлорметане (5 мл) добавляли трифторуксусную кислоту (10 мл, 130 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 1 часа. После удаления летучих веществ остаток ощелачивали на ионообменной колонке, промывали метанолом с последующим промыванием 1Н раствором аммиак/метанол, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 306,09.
Промежуточное соединение 35
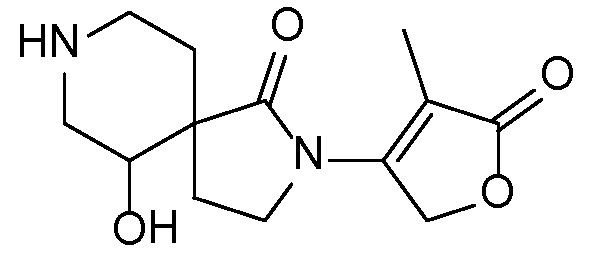
6-Гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
Этил-1-бензил-4-(цианометил)-3-оксопиперидин-4-карбоксилат
В колбу загружали этил-1-бензил-3-оксопиперидин-4-карбоксилат (1,0 г, 3,8 ммоль) и при перемешивании с помощью магнитной мешалки добавляли K2CO3 (1,06 г, 7,6 ммоль), бромацетонитрил (0,92 г, 7,6 ммоль) и ацетон (15 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 часов. ЖХ-анализ показывал медленное взаимодействие. Затем реакционную смесь нагревали до 45°C в течение 3 часов. ЖХ-анализ на данном этапе показывал полное завершение реакции. Реакционную смесь гасили с помощью NH4Cl, экстрагировали EtOAc, сушили над Na2SО4, фильтровали и концентрировали. Неочищенный продукт очищали с помощью ЖХСД, чтобы обеспечить указанное в заголовке соединение. ЖХ/МС: m/z 301 (M+H)+.
Стадия B
8-бензил-6-гидрокси-2,8-диазаспиро[4.5]декан-1-он
В колбу загружали этил-1-бензил-4-(цианометил)-3-оксопиперидин-4-карбоксилат (900 мг, 3,0 ммоль) и при перемешивании с помощью магнитной мешалки добавляли оксид платины (100 мг, 0,44 ммоль), MeOH (20 мл) и уксусную кислоту (20 мл). Смесь оставляли энергично перемешиваться в атмосфере водорода в течение 24 часов. ЖХ-анализ на данном этапе показывал полное завершение реакции. Катализатор удаляли фильтрованием через рыхлый слой CELITE®, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в EtOH (100 мл) и добавляли K2CO3 (2,1 г, 15 ммоль). Смесь нагревали до 90°C в течение 4 часов. Реакционную смесь охлаждали, и к осадку твердых веществ добавляли ДХМ (200 мл). Затем твердые вещества удаляли фильтрованием, и неочищенную реакционную смесь адсорбировали на силикагеле и промывали с помощью ДХМ и 10% MeOH (смешанного с 10% NH4OH), получая при этом указанное в заголовке соединение. ЖХ/МС: m/z 261 (M+H)+.
Стадия C
8-бензил-6-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
В колбу загружали 8-бензил-6-гидрокси-2,8-диазаспиро[4.5]декан-1-он (520 мг, 2,0 ммоль) и при перемешивании с помощью магнитной мешалки добавляли ацетат палладия (22 мг, 0,10 ммоль), K2CO3 (550 мг, 4,00 ммоль), Ксантфос (120 мг, 0,20 ммоль), 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат (640 мг, 2,6 ммоль) и воду (110 мг, 6,0 ммоль). Смесь нагревали до 60°C в течение 2 часов. ЖХ-анализ на данном этапе показывал полное завершение реакции. Реакционную смесь разбавляли EtOAc, промывали водой, и разделяли фазы. Неочищенный раствор сушили над Na2SО4, фильтровали и концентрировали до образования масла. Полученное масло загружали в колонку с силикагелем и очищали с помощью ЖХСД гексаном и EtOAc. Два пятна с требуемой молекулярной массой разделялись в отношении приблизительно 1 к 7. Пятно более полярного вещества представляло собой основной продукт. ЖХ/МС: m/z 357 (M+H)+.
Стадия D
6-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору 8-бензил-6-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (150 мг, 0,42 ммоль) в MeOH (2 мл) добавляли палладий на угле (45 мг, 0,42 ммоль) и несколько капель HOAc. Смесь оставляли перемешиваться в атмосфере водорода в течение 16 часов. ЖХ-анализ показывал полное завершение реакции. Катализатор отфильтровывали и применяли неочищенный материал без дополнительной очистки. ЖХ/МС: m/z 267 (M+H)+.
Промежуточные соединения 36A и 36B (два изомера)
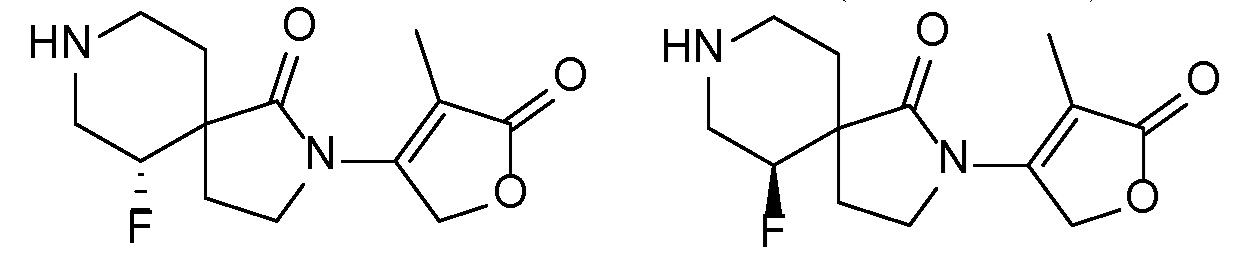
6-Фтор-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он, изомеры A и B
Стадия A
8-бензил-6-фтор-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (изомеры A и B)
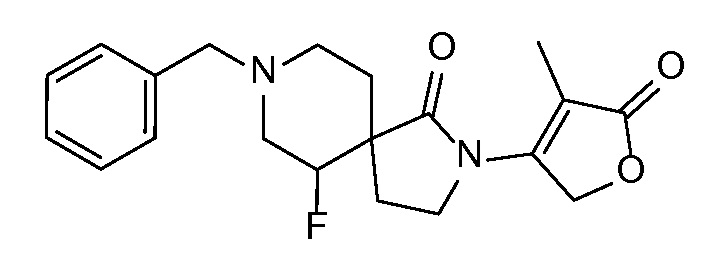
В колбу загружали ДХМ (5 мл) и при перемешивании с помощью магнитной мешалки при -78°C добавляли DAST (0,092 мл, 0,69 ммоль), с последующим добавлением 8-бензил-6-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (I-35, стадия C) (165 мг, 0,46 ммоль) в ДХМ. Смесь перемешивали при -78°C в течение 15 минут, и затем позволяли ей медленно нагреваться до комнатной температуры. Реакционную смесь гасили водным раствором NaHCО3 и после 3 часов при комнатной температуре экстрагировали ее ДХМ, сушили над сульфатом натрия и очищали ЖХСД с применением гексана и EtOAc. Получали два пятна с требуемой молекулярной массой в отношении 1:4. Менее полярное соединение, которое представляет собой менее распространенный изомер, обозначали как изомер A, и более полярный основной изомер обозначали как изомер B. ЖХ/МС: m/z 359 (M+H)+.
Стадия B-1
6-фтор-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (изомер B, I-36B)
К раствору изомера B со стадии A (100 мг, 0,28 ммоль) в DCE (2 мл) добавляли ACE-Cl (0,15 мл, 1,4 ммоль). Смесь нагревали до 80°C в течение 3 часов. Затем растворитель удаляли, и остаток откачивали в высоком вакууме в течение 15 минут. Затем остаток растворяли в MeOH (5 мл) и нагревали с обратным холодильником в течение 30 минут. ЖХ-анализ показывал образование требуемого продукта. Реакционную смесь концентрировали, и неочищенный продукт очищали с помощью ЖХСД с применением системы ДХМ и MeOH, получая при этом изомер B указанного в заголовке соединения (которое представляет собой I-36B). ЖХМС: m/z 269 (M+H)+.
Стадия B-2
6-фтор-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (изомер A, I-36 A)
Указанное в заголовке промежуточное соединение (которое представляет собой I-36A) получали по такому же способу, который описан на стадии B-1, однако исходя из изомера A, полученного на стадии A). ЖХ/МС: m/z 269 (M+H)+.
Промежуточные соединения 37A и 37B
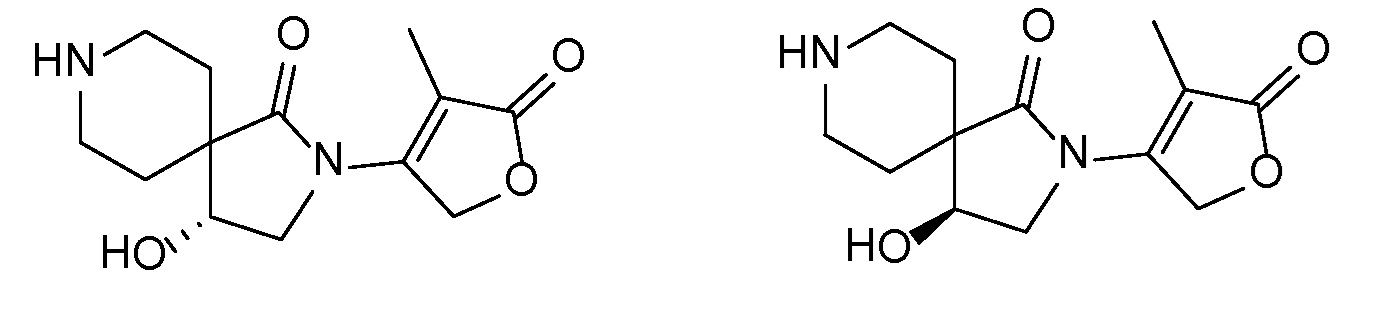
4-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он. Изомер A и Изомер B
Стадия A
1-трет-бутил-4-метил-4-(2-((трет-бутоксикарбонил)амино)-1-гидроксиэтил)пиперидин-l,4-дикарбоксилат
К раствору LDA (полученному путем добавления н-бутиллития (20,0 мл, 49,3 ммоль) к диизопропиламину (5,16 мг, 51,0 ммоль) в ТГФ (40 мл) при 0°C и перемешивании в течение 30 минут) по каплям добавляли 1-трет-бутил-4-метилпиперидин-1,4-дикарбоксилат (4,00 г, 16,4 ммоль) в TMEDA (15 мл, 99 ммоль) с помощью поршневого насоса при -78°C в течение 10 мин. Смесь перемешивали при той же температуре в течение 30 мин, затем медленно с помощью поршневого насоса в течение 15 мин добавляли трет-бутил-(2-оксоэтил)карбамат (8,11 г, 51,0 ммоль) в ТГФ (20 мл). Смесь перемешивали при -78°C в течение 30 мин, гасили насыщенным раствором NH4Cl при -78°C, нагревали до комнатной температуры и разбавляли EtOAc (200 мл). Органический слой отделяли, водный слой экстрагировали EtOAc (100 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили (MgSО4) и концентрировали. Остаток очищали колоночной хроматографией (80 г, силикагель, MeOH/ДХМ, градиент 0-10%, отслеживание при 210 нм), получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1)]+ = 403.
Стадия B
трет-бутил-4-гидрокси-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
К раствору 1-трет-бутил-4-метил-4-(2-((трет-бутоксикарбонил)амино)-1-гидроксиэтил)пиперидин-1,4-дикарбоксилата (4000 мг, 9,94 ммоль) в ДХМ (100 мл) при 0°C добавляли TFA (23 мл, 298 ммоль) и полученный раствор перемешивали в течение 2 часов. После удаления летучих веществ к раствору в течение короткого периода применяли высокий вакуум, чтобы удалить избыток TFA, остаток растворяли в MeOH (100 мл) и добавляли карбонат калия (13,7 г, 99 ммоль). Реакционную смесь нагревали при 60°C в течение 2 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли водный раствор NaHCО3 (50 мл), (BOC)2О (6,51 г, 29,8 ммоль) и перемешивали реакционную смесь в течение ночи. Реакционную смесь экстрагировали ДХМ, сушили с помощью MgSО4 и концентрировали, получая при этом неочищенный продукт, который очищали колоночной хроматографией (0-20% MeOH/ДХМ, отслеживание при 210 нм), получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1)]+ = 271.
Стадия C
трет-бутил-4-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат, изомер A и изомер B
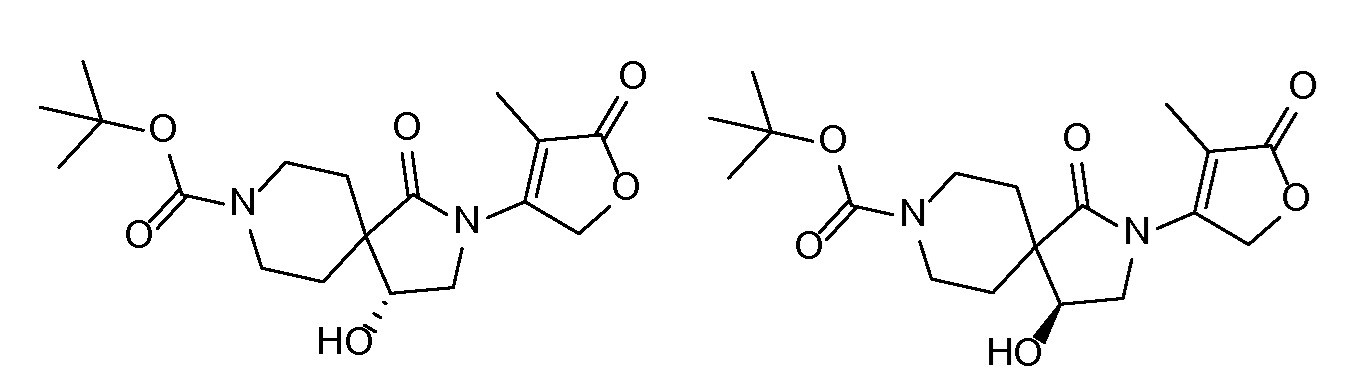
В круглодонную колбу загружали трет-бутил-4-гидрокси-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (400 мг, 1,48 ммоль), 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфонат (546 мг, 2,22 ммоль), Pd2(dba)3 (33,9 мг, 0,037 ммоль), Ксантфос (64,2 мг, 0,111 ммоль) и карбонат цезия (964 мг, 2,96 ммоль). Колбу оборудовали холодильником, вакуумировали, заполняли N2 и добавляли диоксан (6 мл). Реакционную смесь нагревали при 90°C в течение ночи и фильтровали через CELITE®. Фильтрат выпаривали, получая при этом неочищенный продукт, который очищали колоночной хроматографией (0-10% MeOH/ДХМ), получая при этом указанное в заголовке соединение в виде рацемической смеси. ЖХ/МС: [(M+1)]+ = 367. Рацемическую смесь разделяли с помощью СФХ-ВЭЖХ с применением следующих условий: хиральная колонка Chiralcel OJ, 21×250 мм, 10% MeOH + 0,2 DEA, 50 мл/мин, получая при этом изомер A (быстро элюирующий энантиомер) ЖХ/МС: [(M+1)]+ = 367, и изомер B (медленно элюирующий энантиомер) ЖХ/МС: [(M+1)]+ = 367.
Стадия D
4-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он, изомер A и изомер B
Указанные в заголовке соединения получали из изомеров A и B трет-бутил-4-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата, соответственно, с применением TFA по способу, аналогичному описанному ранее для I-19. Изомер A: ЖХ/МС: [(M+1)]+ = 267; изомер B: ЖХ/МС: [(M+1)]+ = 267.
Промежуточное соединение 38
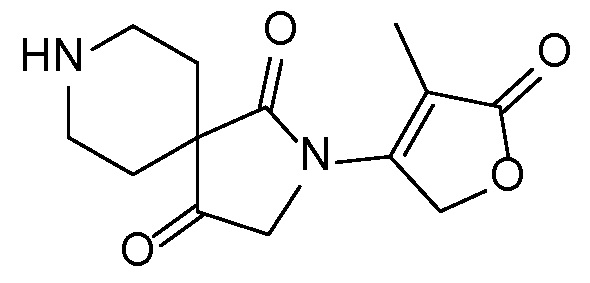
2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1,4-дион
Стадия A
трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1,4-диоксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
К раствору трет-бутил-4-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (I-37, стадия C) (200 мг, 0,546 ммоль) в ДХМ (2,8 мл) добавляли бикарбонат натрия (68,8 мг, 0,819 ммоль) и периодинан Десса-Мартина (347 мг, 0,819 ммоль). Реакционную смесь энергично перемешивали в течение 1,5 часов, затем гасили 10%-ным раствором Na2S2О3, NaHCO3 и перемешивали в течение 20 мин. Водный слой экстрагировали ДХМ, а органические слои промывали насыщенным раствором соли, сушили и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1)]+ = 365.
Стадия B
2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1,4-дион
Указанное в заголовке соединение получали из трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1,4-диоксо-2,8-диазаспиро[4.5]декан-8-карбоксилата по способу, аналогичному описанному для I-19. ЖХ/МС: [(M+1)]+ = 265
Промежуточные соединения 39A и 39B
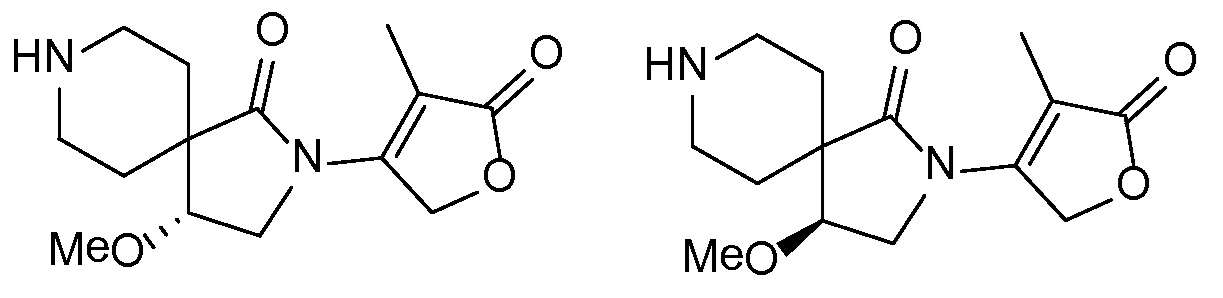
4-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он, изомер A и изомер B
Стадия A
трет-бутил-4-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (энантиомер A и энантиомер B)
К раствору трет-бутил-4-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (100 мг, 0,273 ммоль) в ацетонитриле (1 мл) добавляли йодметан (171 мкл, 2,73 ммоль) и оксид серебра (69,6 мг, 0,300 ммоль). Ампулу герметизировали, заворачивали в алюминиевую фольгу и перемешивали реакционную смесь при 58°C в течение 15 часов. Реакционную смесь фильтровали через CELITE® и концентрировали, получая при этом неочищенный продукт, который очищали колоночной хроматографией (0-10% MeOH/ДХМ), получая при этом указанное в заголовке соединение в виде смеси энантиомеров. ЖХ/МС: [(M+1)]+ = 381. Рацемическую смесь разделяли с помощью СФХ-ВЭЖХ с применением следующих условий: хиральная колонка Chiralcel AD-H, 2×25 см, 15% MeOH, 60 мл/мин, получая при этом быстро элюирующий энантиомер A: ЖХ/МС: [(M+1)]+ = 381; и медленно элюирующий энантиомер B: ЖХ/МС: [(M+1)]+ = 381.
Стадия B
4-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он, изомер A и изомер B
Индивидуальные изомеры указанного в заголовке соединения получали из каждого отдельного энантиомера с предыдущей стадии по способу, аналогичному описанному для I-19, стадия B, с применением TFA. Изомер A (полученный из энантиомера A, стадия A): ЖХ/МС: [(M+1)]+ = 281. Изомер B (полученный из энантиомера B, стадия A): ЖХ/МС: [(M+1)]+ = 281.
Промежуточное соединение 40
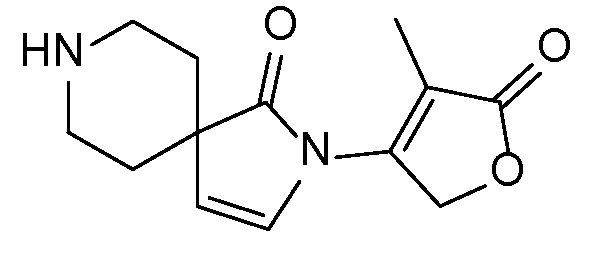
2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]дец-3-ен-1-он
Стадия A
трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]дец-3-ен-8-карбоксилат
К раствору трет-бутил-4-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (300 мг, 0,819 ммоль) в ДХМ (8,2 мл) при 0°C добавляли DBU (370 мкл, 2,46 ммоль) и XtalFluor-E® (562 мг, 2,46 ммоль). Смесь перемешивали в течение ночи по мере нагревания до комнатной температуры, и гасили водным раствором NaHCО3. Органический слой отделяли, и водный слой экстрагировали ДХМ (30 мл). Объединенные органические слои сушили (MgSО4) и очищали колоночной хроматографией (0-100%) EtOAc/гексан), получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1)]+ = 349.
Стадия B
2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]дец-3-ен-1-он
Указанное в заголовке соединение получали из трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]дец-3-ен-8-карбоксилата с применением TFA по способу, аналогичному описанному для получения I-19, стадия B. ЖХ/МС: [(M+1)]+ = 249.
Промежуточное соединение 41
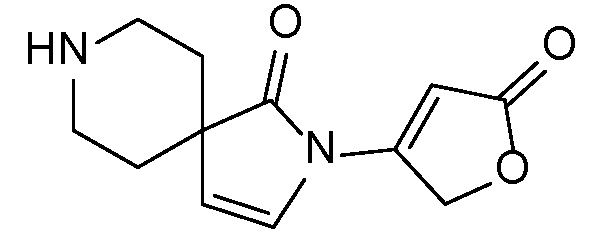
2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]дец-3-ен-1-он
Стадия A
трет-бутил-4-гидрокси-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилат
В круглодонную колбу загружали трет-бутил-4-гидрокси-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (300 мг, 1,11 ммоль), 4-бромфуран-2(5H)-он (271 мг, 1,66 ммоль), Pd(OAc)2 (24,9 мг, 0,111 ммоль), Ксантфос (96 мг, 0,166 ммоль) и K2CО3 (307 мг, 2,22 ммоль). Колбу герметизировали, вакуумировали, заполняли N2 и загружали диоксан (4,5 мл) и H2О (60,0 мкл, 3.33 ммоль). Реакционную смесь нагревали при 90°C в течение ночи, затем фильтровали через CELITE® и выпаривали, получая при этом неочищенный продукт, который очищали колоночной хроматографией (0-10% MeOH/ДХМ), получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1)]+ = 353.
Стадия B
трет-бутил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]дец-3-ен-8-карбоксилат
К раствору трет-бутил-4-гидрокси-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилата (210 мг, 0,596 ммоль) в ДХМ (6 мл) при 0°C добавляли DBU (269 мкл, 1,79 ммоль) и XtalFluor-E® (409 мг, 1,79 ммоль). Реакционную смесь перемешивали в течение ночи по мере нагревания до комнатной температуры, затем гасили водным раствором NaHCО3. Органический слой отделяли, и водный слой экстрагировали ДХМ (30 мл). Объединенные органические слои сушили (MgSО4) и очищали колоночной хроматографией (0-100% EtO Ac/гексан), получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1)]+ = 335.
Стадия C
2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]дец-3-ен-1-он
Указанное в заголовке соединение получали из трет-бутил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]дец-3-ен-8-карбоксилата с применением TFA по способу, аналогичному описанному для получения I-19, стадия B. ЖХ/МС: [(M+1)]+ = 235.
Промежуточное соединение 42
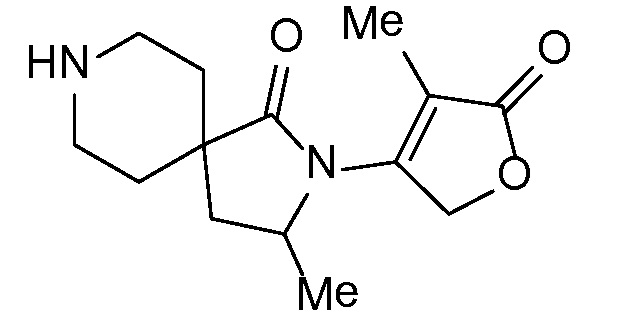
3-метил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Указанное в заголовке соединение получали в две стадии по способу, аналогичному описанному для получения 3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (I-22), за исключением того, что исходили из 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (I-9). ЖХ-МС (IE, m/z): 265 (M+1)+.
Промежуточное соединение 43
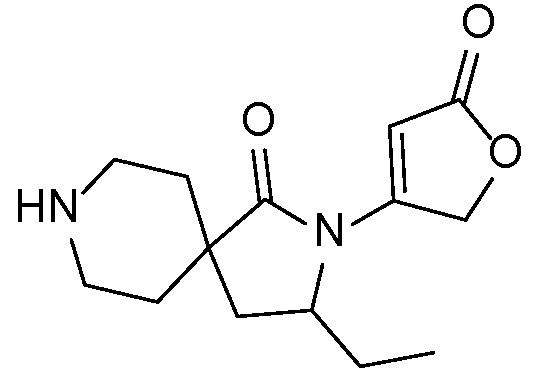
3-этил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
1-трет-бутил-4-этил-4-(2-бромаллил)пиперидин-1,4-дикарбоксилат
Диизопропиламид лития (29,1 мл, 58,3 ммоль) при -78°C добавляли по каплям к раствору 1-трет-бутил-4-этилпиперидин-1,4-дикарбоксилата (9,56 мл, 38,9 ммоль) в ТГФ (200 мл) и перемешивали при данной температуре в течение 50 минут. В реакционную смесь медленно добавляли раствор 2,3-дибромпроп-1-ена (5,47 мл, 56,0 ммоль) в ТГФ (10 мл) и перемешивали полученную смесь в течение 1,5 часов при -78°C. Реакционную смесь гасили раствором NH4Cl (15 мл), давали нагреться до комнатной температуры и экстрагировали водный слой EtOAc (30 мл ×3). Объединенную органику промывали насыщенным раствором соли (30 мл), сушили над MgSО4 и концентрировали, получая при этом неочищенный продукт, который очищали колоночной хроматографией на силикагеле (220 г, колонка RediSep Gold) с применением смеси (0-30)% EtO Ac/гексаны в качестве подвижной фазы, получая при этом указанное в заголовке соединение.
Стадия B
1-трет-бутил-4-этил-4-(2-метиленбутил)пиперидин-1,4-дикарбоксилат
К раствору 1-трет-бутил-4-этил-4-(2-бромаллил)пиперидин-1,4-дикарбоксилата (5,0 г, 13,3 ммоль) в ТГФ (80 мл) в герметично закрывающейся трубке добавляли BINAP (3,31 г, 5,32 ммоль), диэтилцинк (15,95 мл, 15,95 ммоль) и Pd(OAc)2 (0,597 г, 2,66 ммоль), дегазировали полученную смесь и нагревали в течение 16 часов при 100°C. Реакционную смесь упаривали при пониженном давлении, чтобы удалить растворитель, и неочищенный продукт очищали колоночной хроматографией на силикагеле (220 г, колонка RediSep Gold) с применением смеси (0-30)% EtOAc/гексаны в качестве подвижной фазы, и выделяли указанное в заголовке соединение.
Стадия C
1-трет-бутил-4-этил-4-(2-оксобутил)пиперидин-1,4-дикарбоксилат
К раствору 1-трет-бутил-4-этил-4-(2-метиленбутил)пиперидин-l,4-дикарбоксилата (1,0 г, 3,07 ммоль) в ацетоне (20 мл) и воде (20 мл) добавляли тетрагидроксодиоксоосмат калия (0,041 г, 0,111 ммоль) и перемешивали в течение 10 мин. Добавляли твердый периодат натрия (2,62 г, 12,26 ммоль) четырьмя порциями на протяжении 1 часа и поддерживали температуру реакции ниже 40°C с применением ледяной бани. Полученную смесь перемешивали в течение 1 часа при комнатной температуре. На данном этапе ЖХ/МС-анализ показывал завершенную реакцию. Добавляли еще 0,036 экв. тетрагидроксодиоксоосмата калия (0,041 г, 0,111 ммоль) и затем перемешивали смесь при комнатной температуре в течение 2 часов. После 2 часов ЖХ/МС-анализ показывал полное завершение реакции. Суспензию фильтровали, и фильтрат концентрировали, чтобы удалить ацетон; водный слой экстрагировали ДХМ (15 мл ×3). Объединенные органические слои промывали 10%-ным раствором Na2S2О3 (20 мл ×2), сушили над безводным Na2SО4, фильтровали, затем концентрировали, получая при этом неочищенный продукт, который очищали колоночной хроматографией на силикагеле (80 г, колонка RediSep Gold) с применением смеси (0-35)% EtOAc/гексаны в качестве подвижной фазы, и выделяли указанное в заголовке соединение.
Стадия D
трет-бутил-3-этил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
К раствору 1-трет-бутил-4-этил-4-(2-оксобутил)пиперидин-1,4-дикарбоксилата (0,78 г, 2,38 ммоль) в этаноле (24 мл) в герметично закрывающейся трубке при перемешивании добавляли ацетат аммония (2,39 г, 31,0 ммоль), цианоборгидрид натрия (0,422 г, 6,72 ммоль) и сульфат магния (1,577 г, 13,10 ммоль), и нагревали полученную смесь в течение 16 часов при 80°C. Реакционную смесь фильтровали через CELITE®, чтобы удалить MgSО4, и концентрировали фильтрат. Остаток снова растворяли в ДХМ (20 мл) и промывали насыщенным раствором NaHCО3 (10 мл), водой (10 мл) и насыщенным раствором соли (10 мл). Органический слой сушили над безводным MgSO4, фильтровали, концентрировали, затем очищали колоночной хроматографией на силикагеле (40 г, колонка RediSep Gold) с применением смеси (0-10)% MeOH/EtOAc, получая при этом указанное в заголовке соединение.
Стадии E и F
3-этил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Указанное в заголовке соединение получали в две стадии из трет-бутил-3-этил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата по способу, аналогичному описанному для получения 3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (I-22).
Промежуточное соединение 44
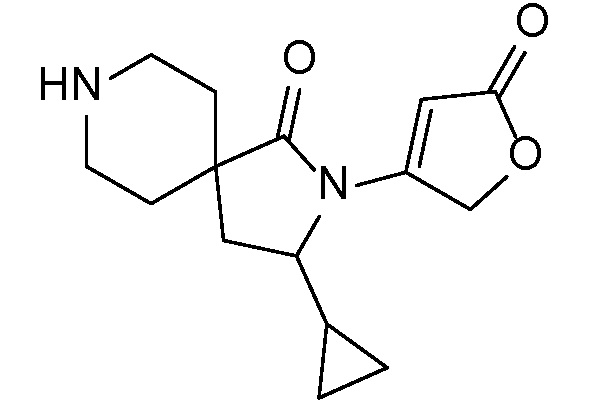
3-циклопропил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
1-трет-бутил-4-этил-4-(2-циклопропилаллил)пиперидин-1,4-дикарбоксилат
1-трет-бутил-4-этил-4-(2-бромаллил)пиперидин-1,4-дикарбоксилат (1,0 г, 2,66 ммоль), циклопропилтрифторборат калия (0,413 г, 2,79 ммоль), карбонат цезия (2,60 г, 7,97 ммоль) и [1,1'- бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (0,058 г, 0,080 ммоль) в толуоле (14 мл) и воде (1,39 мл) помещали в ампулу для микроволновой печи, дегазировали и нагревали при 80°C в течение ночи. Реакционную смесь охлаждали, и использовали ЖХ/МС-анализ, который показывал почти полное завершение реакции. Реакционную смесь разбавляли EtOAc и водой. Водный слой экстрагировали EtOAc (×2), объединенные органические слои сушили над безводным MgSО4, фильтровали, концентрировали и очищали колоночной хроматографией на силикагеле (80 г, колонка RediSep Gold) с применением смеси (0-30)%) EtOAc/гексаны в качестве подвижной фазы, получая при этом указанное в заголовке соединение.
3-циклопропил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Указанное в заголовке соединение получали из 1-трет-бутил-4-этил-4-(2-циклопропилаллил)пиперидин-1,4-дикарбоксилата в четыре стадии по способу, аналогичному описанному для получения 3-этил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (I-43).
Промежуточное соединение 45
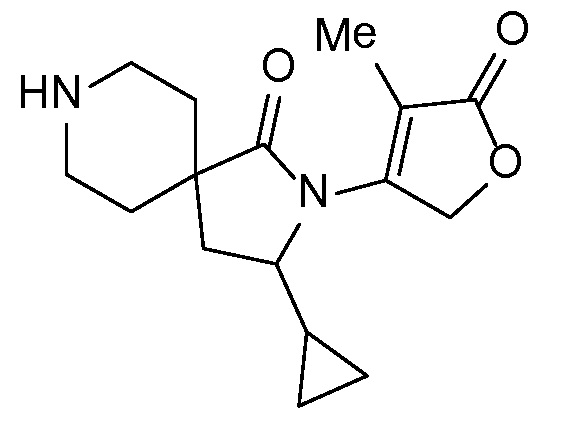
3-циклопропил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Указанное в заголовке соединение получали по способу, аналогичному описанному для получения I-44, но с применением 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната.
Промежуточное соединение 46
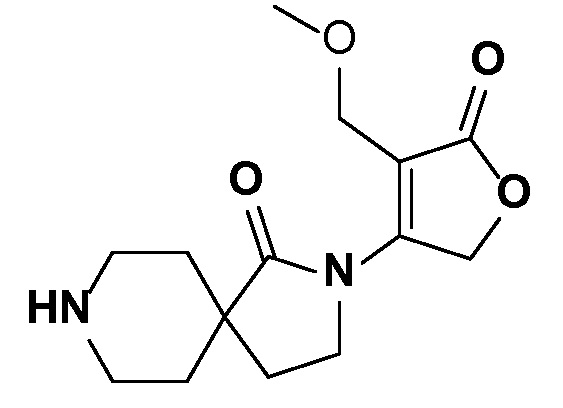
2-(4-(метоксиметил)-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
трет-бутил-2-(4-бром-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
трет-бутил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилат (I-16, стадия A) (2,73 г, 8,12 ммоль) растворяли в ДХМ (70 мл) и обрабатывали N-бромсукцинимидом (1,73 г, 9,74 ммоль) при 25°C, полученную смесь перемешивали в течение ночи при комнатной температуре. На следующий день реакционную смесь разбавляли ДХМ, промывали водой и насыщенным раствором соли, затем сушили над Na2SО4. После удаления растворителя получали неочищенный продукт, который очищали колоночной хроматографией на силикагеле (80 г, колонка RediSep Gold) с применением смеси (25-80)% EtOAc/гексаны в качестве подвижной фазы, получая при этом указанное в заголовке соединение.
Стадия B
трет-бутил-1-оксо-2-(5-оксо-4-винил-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилат
трет-бутил-2-(4-бром-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (2,2 г, 5,30 ммоль), трифтор(винил)борат калия (1,06 г, 7,95 ммоль), [1,1'- бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (0,345 г, 0,530 ммоль) и трехосновный фосфат калия (10,60 мл, 10,60 ммоль) в ТГФ (44,1 мл) помещали в герметично закрываемую трубку, дегазировали и нагревали полученную смесь в течение ночи при 70°C. Реакционную смесь охлаждали до комнатной температуры и затем разбавляли EtOAc и водой. После разделения слоев водный слой экстрагировали EtOAc (×2). Объединенные органические слои сушили над безводным MgSO4, фильтровали, концентрировали и очищали колоночной хроматографией на силикагеле с применением смеси (30-100)% EtOAc/гексаны в качестве подвижной фазы, получая при этом указанное в заголовке соединение.
Стадия C
трет-бутил-2-(4-формил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
трет-бутил-1-оксо-2-(5-оксо-4-винил-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилат (1,6 г, 4,4 ммоль) растворяли в ацетоне (36 мл) и воде (36 мл), затем добавляли K2OsO4.2H2O и перемешивали смесь в течение ~5 минут. Добавляли твердый периодат натрия (3,77 г, 17,6 ммоль) 4 порциями на протяжении 1 часа, поддерживая температуру реакции ниже 40°C с помощью ледяной бани. Полученную смесь перемешивали в течение 1 часа при комнатной температуре. ЖХ/МС-анализ после 2 часов показывал полный расход исходного материала. Суспензию фильтровали и концентрировали фильтрат, чтобы удалить ацетон. Водный слой экстрагировали ДХМ (×3). Объединенные органические слои промывали 10%-ным раствором Na2S2О3 (×2), сушили над безводным Na2SО4, фильтровали и концентрировали, получая при этом указанное в заголовке соединение, которое использовали на следующей стадии без дополнительной очистки.
Стадия D
трет-бутил-2-(4-(гидроксиметил)-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
трет-бутил-2-(4-формил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (1,18 г, 3,24 ммоль) растворяли в ТГФ (13 мл) и MeOH (13 мл), охлаждали смесь до -78°C и перемешивали в течение 15 мин. Добавляли боргидрид натрия (0,147 г, 3,89 ммоль) в виде двух равных порций и перемешивали полученную смесь в течение ~15 минут при -78°C. ЖХ-МС-анализ после 15 минут перемешивания при -78°C показывал полный расход исходного материала. Реакционную смесь разбавляли этилацетатом и гасили водным раствором хлорида аммония при -78°C. Водный слой экстрагировали EtOAc (×2), объединенные органические слои промывали водой, насыщенным раствором соли, затем сушили (MgSО4) и фильтровали, выпаривали растворитель при пониженном давлении, получая при этом продукт, который использовали на следующей стадии без очистки.
Стадия E
трет-бутил-2-(4-(метоксиметил)-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
трет-бутил-2-(4-(гидроксиметил)-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (0,42 г, 1,15 ммоль), оксид серебра (0,292 г, 1,26 ммоль) и метилиодид (0,358 мл, 5,73 ммоль) помещали в ДХМ (5 мл) и перемешивали в течение ночи при комнатной температуре в атмосфере азота. К смеси с DCE (8 мл) добавляли дополнительное количество оксида серебра (0,292 г, 1,26 ммоль) и метилиодида (0,358 мл, 5,73 ммоль) и нагревали полученную смесь при 54°C в течение ночи. Реакционную смесь фильтровали через CELITE®, чтобы удалить оксид серебра, концентрировали и очищали колоночной хроматографией на силикагеле (40 г, колонка RediSep Gold) с применением смеси (20-80)% EtOAc/ДХМ в качестве подвижной фазы, получая при этом указанное в заголовке соединение.
Стадия F
2-(4-(метоксиметил)-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Указанное в заголовке соединение получали из трет-бутил-2-(4-(метоксиметил)-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата по способу, аналогичному описанному для получения 3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (I-22, последняя стадия).
Промежуточное соединение 47
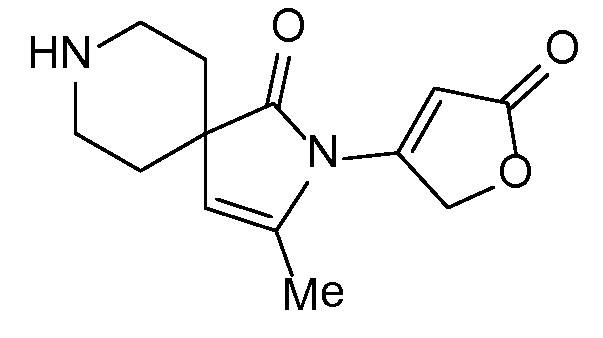
3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]дец-3-ен-1-он
Стадия A
1-трет-бутил-4-метил-4-(2-((трет-бутоксикарбонил)амино)-1-гидроксипропил)пиперидин-1,4-дикарбоксилат
К раствору LDA (полученному путем добавления н-BuLi (27,7 мл, 55,5 ммоль) к диизопропиламину (8,04 мл, 57,3 ммоль) в ТГФ (40 мл) при 0°C и перемешивании в течение 30 минут) при -78°C по каплям добавляли 1-трет-бутил-4-метилпиперидин-1,4-дикарбоксилат (4500 мг, 18,5 ммоль) в TMEDA (16,6 мл, 111 ммоль) с помощью поршневого насоса в течение 20 минут. Смесь перемешивали при той же температуре в течение 30 минут и с помощью поршневого насоса в течение 20 минут медленно добавляли (S)-трет-бутил-(1-оксопропан-2-ил)карбамат (9931 мг, 57,3 ммоль) в ТГФ (20 мл). Смесь перемешивали при -78°C в течение 30 минут, гасили насыщенным раствором NH4Cl при -78°C, нагревали до комнатной температуры и разбавляли EtOAc (200 мл). Органический слой отделяли, водный слой экстрагировали EtOAc (100 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили (MgSО4) и концентрировали. Остаток очищали колоночной хроматографией (MeOH/ДХМ, градиент 0-10%), получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1)]+ = 417.
Стадия B
трет-бутил-4-гидрокси-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
К раствору 1-трет-бутил-4-метил 4-(2-((трет-бутоксикарбонил)амино)-1-гидроксипропил)пиперидин-1,4-дикарбоксилата (8000 мг, 19,2 ммоль) в ДХМ (190 мл) добавляли TFA (44,4 мл, 576 ммоль) при 0°C и перемешивали полученный раствор в течение 2 часов. После удаления летучих веществ в течение короткого периода применяли высокий вакуум, чтобы удалить избыток TFA, остаток растворяли в MeOH (190 мл), и добавляли K2CO3 (26,5 г, 192 ммоль). Реакционную смесь нагревали при 60°C в течение 2 часов. После охлаждения до комнатной температуры добавляли насыщенный раствор NaHCО3 (60 мл) с последующим добавлением (BOC)2О (12,6 г, 57,6 ммоль). Реакционную смесь перемешивали в течение ночи и экстрагировали ДХМ. Органический слой сушили с помощью MgSО4 и концентрировали, получая при этом неочищенный продукт, который очищали колоночной хроматографией (0-20% MeOH/ДХМ, отслеживание при 210 нм), получая при этом указанное в заголовке соединение. ЖХ/МС:[(M+1)]+ =285.
Стадия C
трет-бутил-4-гидрокси-3-метил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилат
В круглодонную колбу загружали трет-бутил-4-гидрокси-3-метил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (1000 мг, 3,52 ммоль), 4-бромфуран-2(5H)-он (860 мг, 5,28 ммоль), Pd(OAc)2 (79 мг, 0,352 ммоль), Ксантфос (305 мг, 0,528 ммоль) и K2CO3 (972 мг, 7,03 ммоль). Колбу герметизировали, вакуумировали, заполняли N2 и загружали диоксан (14 мл) и воду (190 мкл, 10,6 ммоль). Реакционную смесь нагревали при 90°C в течение ночи и фильтровали через CELITE®. Фильтрат выпаривали, получая при этом неочищенный продукт, который очищали колоночной хроматографией (0-10% MeOH/ДХМ, появляется при 6% MeOH/ДХМ с последующим элюированием смесью 0-100% EtO Ac/гексан на другой колонке), получая при этом указанное в заголовке соединение. ЖХ/МС:[(M+1)]+ = 367.
Стадия D
трет-бутил-4-йод-3-метил-1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-8-карбоксилат
К раствору соединения, полученного на стадии C (370 мг, 1,01 ммоль), в толуоле (20 мл) при комнатной температуре добавляли PPh3 (397 мг, 1,515 ммоль), имидазол (137 мг, 2,02 ммоль) и I2 (384 мг, 1,515 ммоль). Смесь перемешивали при 100°C в течение 10 часов и гасили водным раствором NaHCО3. Органический слой разбавляли ДХМ, отделяли, и водный слой экстрагировали ДХМ. Объединенные органические слои сушили (MgSО4) и очищали колоночной хроматографией (0-100% EtO Ac/гексан), получая при этом указанное в заголовке соединение. ЖХ/МС: [(M+1-56)]+ = 421.
Стадия E
4-йод-3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Соединение, полученное на стадии D (100 мг, 0,210 ммоль), в ДХМ (1,1 мл) обрабатывали TFA (485 мкл, 6,30 ммоль) при 0°C, чтобы удалить Boc-группу, что обеспечивало соль TFA после выпаривания растворителя. Затем колонку с 2 г картриджем Bond Elut SCX сначала промывали MeOH, загружали образец с MeOH, по каплям промывали MeOH, чтобы удалить TFA, и в конце промывали 2Н раствором NH3/MeOH, получая при этом указанное в заголовке соединение в виде свободного основания. ЖХ/МС:[(M+1)]+ = 377.
Стадия F
3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]дец-3-ен-1-он
К раствору соединения, полученного на стадии E (180 мг, 0,478 ммоль), в ТГФ (4,8 мл) при комнатной температуре добавляли 10-этил-2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (полимерсвязанный, 1,15 ммоль/г, 2,5 г смолы). Реакционную смесь нагревали при 60°C в течение 5 часов на шейкере, затем смолу отфильтровывали с промыванием MeOH и полученную смесь упаривали, получая при этом указанное в заголовке соединение. ЖХ/МС:[(M+1)]+ = 249.
Промежуточное соединение 48
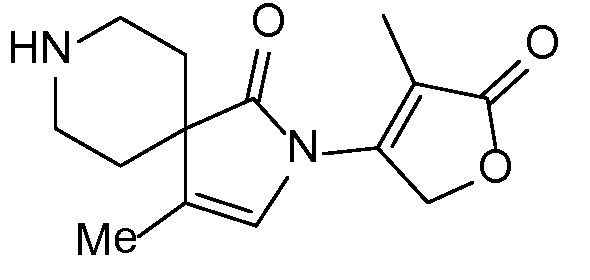
4-метил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]дец-3-ен-1-он
Стадия A
трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1,4-диоксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
К трет-бутил-4-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилату (см. пример I-37A и I-37B, стадия C, рацемат перед разделением с помощью хиральной ВЭЖХ)(200 мг, 0,546 ммоль) в ДХМ (2,8 мл) добавляли бикарбонат натрия (68,8 мг, 0,819 ммоль) и периодинан Десса-Мартина (347 мг, 0,819 ммоль). Реакционную смесь энергично перемешивали в течение 1,5 часов, затем гасили 10%-ными водными растворами Na2S2О3 и NaHCО3 и перемешивали в течение 20 мин. Водный слой экстрагировали ДХМ, а органические слои промывали насыщенным раствором соли, сушили и концентрировали, получая при этом трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1,4-диоксо-2,8-диазаспиро[4.5]декан-8-карбоксилат. ЖХ/МС:[(M+1)]+ = 365.
Стадия B
трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-4-(((трифторметил)сульфонил)окси)-2,8-диазаспиро[4.5]дец-3-ен-8-карбоксилат
В колбу, содержащую трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1,4-диоксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (200 мг, 0,549 ммоль) в ТГФ (3 мл), при -78°C по каплям добавляли NaHMDS (1,10 мл, 1 M в ТГФ, 1,10 ммоль) на протяжении 5 минут. Раствор перемешивали в течение 2 часов, затем по каплям на протяжении 5 мин добавляли N-фенилбис(трифторметансульфонимид) (314 мг, 0,878 ммоль) в ТГФ (2 мл). Реакционную смесь перемешивали при -78°C в течение 2 часов, затем нагревали до комнатной температуры и перемешивали в течение ночи. Реакционный раствор гасили путем добавления насыщенного водного раствора NH4Cl и экстрагировали EtOAc. Объединенные органические слои сушили (MgSО4) и концентрировали. Неочищенный материал очищали колоночной хроматографией (0-10% MeOH/ДХМ), получая при этом указанное в заголовке соединение. ЖХ/МС:[(M+1)]+ = 497.
Стадия C
трет-бутил-4-метил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]дец-3-ен-8-карбоксилат
трет-бутил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-4-(((трифторметил)сульфонил)окси)-2,8-диазаспиро[4.5]дец-3-ен-8-карбоксилат (180 мг, 0,363 ммоль) растворяли в ТГФ (3,6 мл) и добавляли Pd(Ph3P)4 (209 мг, 0,181 ммоль) с последующим добавлением триметилалюминия (3,6 мл, 7,25 ммоль) при 0°C. Такую реакционную смесь перемешивали при комнатной температуре в течение 2 часов перед ее гашением насыщенным водным раствором NaHCО3 при 0°C (высоко экзотермический процесс). Полученную смесь разбавляли EtOAc, сушили над MgSО4 и концентрировали при пониженном давлении. Неочищенный материал очищали колоночной хроматографией (0-10% MeOH/ДХМ), получая при этом указанное в заголовке соединение. ЖХ/МС:[(M+1)]+ = 363.
Стадия D
4-метил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]дец-3-ен-1-он
трет-бутил-4-метил-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]дец-3-ен-8-карбоксилат (130 мг, 0,359 ммоль) в ДХМ (1,8 мл) обрабатывали TFA (0,8 мл, 10,7 ммоль) при 0°C, чтобы удалить Boc-группу, что обеспечивало соль TFA. Соль TFA загружали с MeOH в ионообменную колонку с 2 г картриджем Bond Elut SCX, затем дополнительно по каплям промывали MeOH, чтобы удалить TFA, и в конце промывали 2Н раствором NH3/MeOH, получая при этом указанное в заголовке соединение в виде свободного амина. ЖХ/МС:[(M+1)]+ = 263.
Промежуточное соединение 49
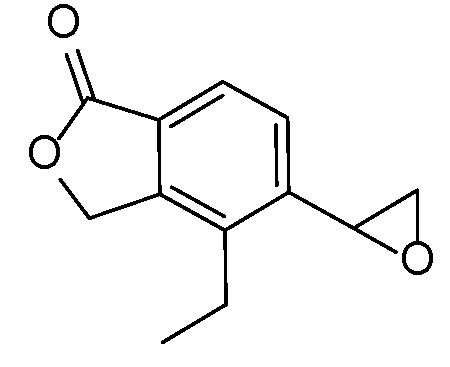
4-этил-5-оксиран-2-ил-2-бензофуран-1(3H)-он
Стадия A
5-бром-4-этил-2-бензофуран-1(3H)-он
Смесь 5-бром-4-винил-2-бензофуран-1(3H)-он (2,0 г, 8,37 ммоль) и Pd/C (400 мг) в 50 мл MeOH перемешивали при комнатной температуре в атмосфере H2 (1 атм.) в течение ночи и затем фильтровали. Фильтрат концентрировали. Полученное масло очищали колоночной хроматографией, получая при этом 5-бром-4-этил-2-бензофуран-1(3H)-он.
Стадия B
4-этил-5-винил-2-бензофуран-1(3H)-он
Смесь 5-бром-4-этил-2-бензофуран-1(3H)-она (1,81 г, 7,51 ммоль), винилтрифторбората калия (1,21 г, 9,01 ммоль), Pd(dppf)Cl2 (200 мг) в 20 мл TEA и 20 мл EtOH нагревали в течение ночи с обратным холодильником в атмосфере N2 и затем концентрировали. Полученный материал очищали колоночной хроматографией, получая при этом 4-этил-5-винил-2-бензофуран-1(3H)-он.
Стадия C
4-этил-5-оксиран-2-ил-2-бензофуран-1(3H)-он
К раствору 4-этил-5-винил-2-бензофуран-1(3H)-она (1,1 г, 5,85 ммоль) в 50 мл ДХМ при 0°C медленно добавляли mCPBA (3,60 г, степень чистоты 85%, 17,6 ммоль) в 50 мл ДХМ. Смесь нагревали до комнатной температуры и затем перемешивали в течение 3 дней. Смесь промывали водным раствором Na2SО3 до тех пор, пока не происходило изменения окраски индикаторной бумаги с KI. Органические слои объединяли, промывали насыщенным раствором соли и концентрировали. Остаток очищали колоночной хроматографией, получая при этом 4-этил-5-оксиран-2-ил-2-бензофуран-1(3H)-он. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 7,75 (д, J=8,6 Гц, 1H), 7,41 (д, J=7,8 Гц, 1H), 5,30 (с, 2H), 4,11-4,13 (м, 1H), 3,23-3,25 (м, 1H), 2,75-2,82 (м, 2H), 2,70-2,72 (м, 1H), 1,27 (т, J=7,4 Гц, 3H).
Промежуточное соединение 50
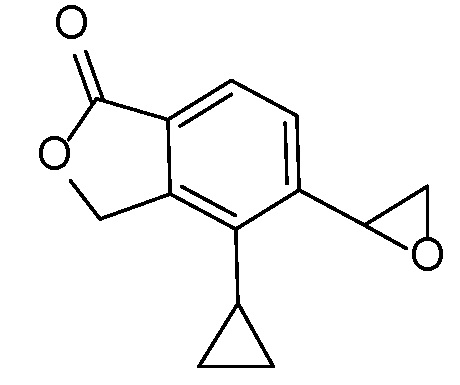
4-циклопропил-5-оксиран-2-ил-2-бензофуран-1(3H)-он
Стадия A
5-бром-4-йод-2-бензофуран-1(3H)-он
К охлажденному (0°C) раствору 5-бром-2-бензофуран-1(3H)-она (50 г, 0,235 моль) в трифторметансульфоновой кислоте (400 мл) добавляли N-йодсукцинимид (55,5 г, 0,247 моль). Полученную смесь перемешивали при комнатной температуре в течение ночи, затем медленно выливали в ледяную воду (2 л), фильтровали и фильтрат экстрагировали EtOAc. Объединенные органические слои промывали водой и насыщенным раствором соли, сушили и концентрировали, получая при этом 5-бром-4-йод-2-бензофуран-1(3H)-он.
Стадия B
5-бром-4-винил-2-бензофуран-l(3H)-он
Смесь 5-бром-4-йод-2-бензофуран-1(3H)-она (1 г, 2,95 ммоль), винилтрифторбората калия (474 мг, 3,54 ммоль), Pd(dppf)Cl2 (200 мг) в 20 мл TEA и 20 мл EtOH нагревали в атмосфере N2 с обратным холодильником в течение 2 часов. TСХ-анализ показывал полное завершение реакции. Большую часть растворителя удаляли, и остаток растворяли в EtOAc (100 мл). Раствор промывали 0,1Н раствором HCl, раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом 5-бром-4-винил-2-бензофуран-1(3H)-он.
Стадия C
5-бром-4-циклопропил-2-бензофуран-1(3H)-он:
К охлажденной (0°C) смеси 5-бром-4-винил-2-бензофуран-l(3H)-она (2,2 г, 9,21 моль) и Pd(OAc)2 (100 мг) в EtOAc (50 мл) медленно добавляли раствор CH2N2 в простом эфире (100 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи, затем гасили уксусной кислотой, фильтровали и промывали фильтрат водой и насыщенным раствором соли, сушили и концентрировали, получая при этом указанное в заголовке соединение.
Стадия D
4-циклопропил-5-винил-2-бензофуран-1(3H)-он
Смесь 5-бром-4-циклопропил-2-бензофуран-1(3H)-она (760 мг, 3,004 ммоль), винилтрифторбората калия (805 мг, 6,01 ммоль) и Pd(dppf)Cl2 (100 мг) в 20 мл TEA и 20 мл EtOH нагревали в атмосфере N2 с обратным холодильником в течение 8 часов. После того, как TСХ-анализ показал полное завершение реакции, большую часть растворителя удаляли и остаток растворяли в EtOAc (100 мл). Раствор промывали 0,1Н раствором HCl, раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали. Полученное масло очищали колоночной хроматографией, получая при этом указанное в заголовке соединение.
Стадия E
4-циклопропил-5-оксиран-2-ил-2-бензофуран-1(3H)-он
К раствору 4-циклопропил-5-винил-2-бензофуран-1(3H)-она (440 мг, 2,2 ммоль) в 50 мл ДХМ при 0°C медленно добавляли mCPBA (1,14 г, 6,6 ммоль) в 50 мл ДХМ. После нагревания до комнатной температуры смесь перемешивали в течение 12 часов. Смесь промывали водным раствором Na2SО3 до тех пор, пока не происходило изменения окраски индикаторной бумаги с KI. Органические слои объединяли, промывали насыщенным раствором соли и затем концентрировали. Остаток очищали препаративной ТСХ, получая при этом указанное в заголовке соединение. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 7,77 (д, J=8,6 Гц, 1H), 7,39 (д, J=7,8 Гц, 1H), 5,39 (с, 2H), 4,43-4,45 (м, 1H), 3,26-3,28 (м, 1H), 2,68-2,70 (м, 1H), 1,94-2,01 (м, 1H), 1,08-1,12 (м, 2H), 0,65-0,75 (м, 2H).
Промежуточное соединение 51
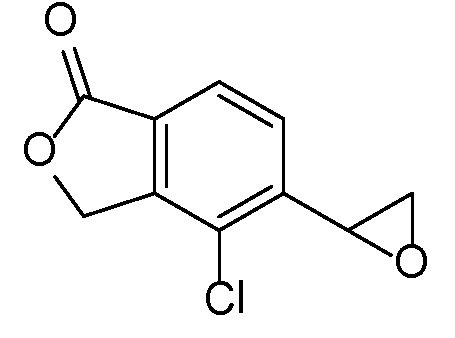
4-хлор-5-оксиран-2-ил-2-бензофуран-1(3H)-он
Стадия A
2-хлор-3-(гидроксиметил)фенол
К раствору 2-хлор-3-гидроксибензальдегида (8,10 г, 51,7 ммоль) в MeOH при 0°C добавляли NaBH4 (1,96 г, 51,7 ммоль). Реакционную смесь оставляли перемешиваться в течение 30 минут. Затем реакционную смесь разбавляли EtOAc (400 мл), промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Неочищенный продукт применяли на стадии B без дополнительной очистки.
Стадия B
4-бром-2-хлор-3-(гидроксиметил)фенол
В колбу загружали 2-хлор-3-(гидроксиметил)фенол и при перемешивании с помощью магнитной мешалки добавляли NBS (10,8 г, 60,5 ммоль) и TFA (50 мл). Реакционную смесь оставляли перемешиваться в течение 16 часов при комнатной температуре, затем растворитель удаляли в вакууме. Остаток повторно растворяли в EtOAc, промывали водой и очищали флэш-хроматографией на силикагеле. После разделения получали пару региоизомеров. Пятно менее полярного вещества согласно ЯМР-анализу соответствовало требуемому 4-бром-2-хлор-3-(гидроксиметил)фенолу.
Стадия C
4-хлор-5-гидрокси-2-бензофуран-1(3H)-он
В колбу загружали 4-бром-2-хлор-3-(гидроксиметил)фенол (2,44 г, 10,3 ммоль) и при перемешивании с помощью магнитной мешалки добавляли CuCN (2,76 г, 30,8 ммоль) и ДМФА (25 мл). Колбу оснащали холодильником и три раза продували азотом. Затем раствор нагревали до 145°C в течение 2 часов. На данном этапе к реакционной смеси добавляли воду (0,555 мл, 30,8 ммоль) с помощью шприца и поддерживали реакционную смесь при 100°C в течение дополнительных 24 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли ДХМ (100 мл) и фильтровали через рыхлый слой CELITE®, чтобы удалить твердые вещества. Фильтрат промывали насыщенным раствором NH4OAc, сушили над сульфатом натрия, концентрировали и очищали флэш-хроматографией на силикагеле, получая при этом указанное в заголовке соединение.
Стадия D
4-хлор-5-этенил-2-бензофуран-1(3H)-он
К охлажденному раствору 4-хлор-5-гидрокси-2-бензофуран-1(3H)-она (1,39 г, 7,53 ммоль) в ДХМ (25 мл) добавляли основание Хунига (3,29 мл, 18,8 ммоль) и трифторметансульфоновый ангидрид (2,54 мл, 15,1 ммоль). Смесь оставляли перемешиваться в течение 16 часов. Анализ с помощью TСХ показывал полный расход всех исходных материалов. Реакционную смесь разбавляли гексаном и промывали водой. Раствор сушили с помощью сульфата натрия, концентрировали и очищали флэш-хроматографией на колонке с диоксидом кремния. Растворитель удаляли при пониженном давлении, получая при этом трифлат промежуточного соединения. ЖХ-МС (M+1 = 317). К трифлату при перемешивании с помощью магнитной мешалки добавляли винилтрифторборат калия (1,33 г, 9,90 ммоль), PdCl2(dppf) (0,243 г, 0,332 ммоль), триэтиламин (1,89 мл, 13,3 ммоль) и изопропанол (50 мл). Смесь три раза продували азотом и нагревали до 60°C в течение 2 часов. TСХ-анализ на данном этапе показывал полное завершение реакции. Большую часть растворителя удаляли в вакууме. Неочищенный остаток разбавляли EtOAc (200 мл), промывали насыщенным раствором соли, сушили над сульфатом натрия, адсорбировали на силикагеле и очищали флэш-хроматографией, получая при этом указанное в заголовке соединение.
Стадия E
4-хлор-5-оксиран-2-ил-2-бензофуран-1(3H)-он
К раствору 4-хлор-5-этенил-2-бензофуран-1(3H)-она (1,1 г, 5,7 ммоль) в ДХМ (40 мл) добавляли mCPBA (1,9 г, 8,5 ммоль). Раствор оставляли перемешиваться при комнатной температуре в течение 16 часов. Анализ с помощью ТСХ и ЖХ показывал образование требуемого продукта. Реакционную смесь разбавляли ДХМ (200 мл), промывали водными растворами Na2S2О3 и Na2CО3, сушили над сульфатом натрия, концентрировали и очищали флэш-хроматографией на силикагеле, получая при этом указанное в заголовке соединение. 1H-ЯМР (500 МГц, CDCl3) δ м.д. 7,86 (д, J=8,0 Гц, 1H), 7,48 (д, J=8,0 Гц, 1H), 5,34 (с, 2H), 4,33 (м, 1H), 3,33 (м, 1H), 2,75 (м, 1H).
Промежуточное соединение 52
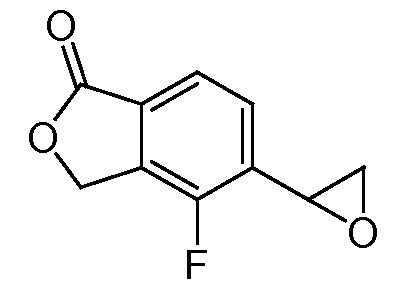
4-фтор-5-оксиран-2-ил-2-бензофуран-1(3H)-он
Стадия A
5-бром-4-фтор-2-бензофуран-1(3H)-он
Раствор н-BuLi (40 мл, 100 ммоль) при -70°C по каплям добавляли к раствору диизопропиламина (10,6 г, 105 ммоль) в 150 мл ТГФ. Смесь перемешивали при 0°C в течение 15 минут и затем снова охлаждали до -70°C. По каплям добавляли раствор 4-бром-3-фторбензойной кислоты (10 г, 45,7 ммоль в 50 мл ТГФ). Полученную смесь перемешивали при -70°C в течение 1 часа, затем через смесь барботировали газообразный CH2О (полученный путем нагревания 5,1 г пара-формальдегида до 200°C). Полученную смесь перемешивали при -70°C в течение 1 часа, затем ей давали нагреться до комнатной температуры и перемешивали в течение еще 2 часов. Через суспензию в течение 15 минут барботировали газообразный HCl, получая при этом раствор. Смесь разбавляли 1 л EtOAc и затем промывали водой, насыщенным раствором Na2CО3 и насыщенным раствором соли, сушили над безводным Na2SО4 и концентрировали, получая при этом 5-бром-4-фтор-2-бензофуран-1(3H)-он. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 7,72-7,75 (м, 1H), 7,58 (д, J=8,0 Гц, 1H), 5,36 (с, 2H).
Стадия B
4-фтор-5-винил-3H-изобензофуран-1-он
Смесь 5-бром-4-фтор-2-бензофуран-1(3H)-она (5,0 г, 21,6 ммоль), винилтрифторбората калия (4,4 г, 32,5 ммоль), Pd(dppf)Cl2 (500 мг) в 100 мл TEA и 100 мл EtOH нагревали с обратным холодильником в атмосфере N2 в течение 4 часов и затем концентрировали. Полученное масло очищали колоночной хроматографией, получая при этом 4-фтор-5-винил-3H-изобензофуран-1-он. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 7,67-7,68 (м, 2H), 6,90-6,97 (м, 1H), 6,00 (д, J=17,2 Гц, 1H), 5,60 (д, J=11,0 Гц, 1H), 5,35 (с, 2H).
Стадия C
4-фтор-5-оксиранил-3H-изобензофуран-1-он
К раствору 4-фтор-5-винил-3H-изобензофуран-1-она (4,0 г, 17,3 ммоль) в 100 мл ДХМ при 0°C медленно добавляли mCPBA (6,0 г, степень чистоты 85%, 34,6 ммоль) в 50 мл ДХМ. После нагревания до комнатной температуры смесь перемешивали в течение ночи. Смесь промывали водным раствором Na2SО3 до тех пор, пока не происходило изменения окраски индикаторной бумаги с KI. Органические слои промывали насыщенным раствором соли и затем концентрировали. Остаток очищали колоночной хроматографией, получая при этом 4-фтор-5-оксиранил-3H-изобензофуран-1-он. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 7,71 (д, J=7,8 Гц, 1H), 7,37-7,40 (м, 1H), 5,37 (с, 2H), 4,21-4,22 (м, 1H), 3,25-3,27 (м, 1H), 2,80-2,82 (м, 1H).
Промежуточное соединение 53
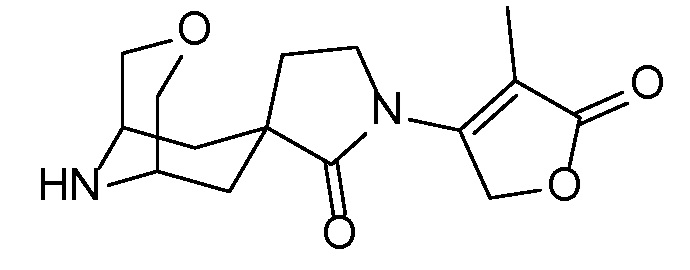
(1R,3'r,5S)-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-3-окса-9-азаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-2'-он
Стадия A
(1R,5S,E)-трет-бутил-7-(1-циано-2-метокси-2-оксоэтилиден)-3-окса-9-азабицикло[3.3.1]нонан-9-карбоксилат
Раствор метил-2-цианоацетата (4,39 мл, 49,7 ммоль), (1R,5S)-трет-бутил-7-оксо-3-окса-9-азабицикло[3.3.1]нонан-9-карбоксилата (8 г, 33,2 ммоль), ацетата аммония (3,83 г, 49,7 ммоль) и уксусной кислоты (7,59 мл, 133 ммоль) в толуоле (100 мл) нагревали при 150°C в течение ночи и образовавшуюся воду отделяли с помощью ловушки Дина-Старка. После выпаривания растворителя остаток растворяли в этилацетате (200 мл) и промывали раствор насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия, концентрировали и очищали остаток на колонке с силикагелем с применением этилацетата/гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1-100)+: 223,01.
Стадия B
(1R,5S,7s)-трет-бутил-7-(1-циано-2-метокси-2-оксоэтил)-7-винил-3-окса-9-азабицикло[3.3.1]нонан-9-карбоксилат
К суспензии (1R,5S,E)-трет-бутил-7-(1-циано-2-метокси-2-оксоэтилиден)-3-окса-9-азабицикло[3.3.1]нонан-9-карбоксилата (10,82 г, 33,6 ммоль) и иодида меди(I) (6,39 г, 33,6 ммоль) в тетрагидрофуране при 0°C медленно на протяжении 2 часов добавляли бромид винилмагния (50,3 мл, 50,3 ммоль). Полученную смесь перемешивали при температуре от 0°C до комнатной температуры в течение 2 часов. Реакционную смесь гасили насыщенным раствором хлорида аммония (300 мл), смесь трижды экстрагировали этилацетатом, объединенную органическую фазу сушили над сульфатом натрия, затем концентрировали и очищали остаток на силикагеле с применением этилацетата/гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+23)+: 372,94.
Стадия C
2-((1R,5S,7s)-9-(трет-бутоксикарбонил)-7-винил-3-окса-9-азабицикло[3.3.1]нонан-7-ил)-2-цианоуксусная кислота
К раствору (1R,5S,7s)-трет-бутил-7-(1-циано-2-метокси-2-оксоэтил)-7-винил-3-окса-9-азабицикло[3.3.1]нонан-9-карбоксилата (1,3 г, 3,7 ммоль) в смеси тетрагидрофурана (10 мл), метанола (3 мл) и воды (3 мл) добавляли гидроксид лития (18,55 мл, 18,55 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. После удаления летучих веществ щелочную фазу при 0°C подкисляли 1Н раствором HCl до pH 4. Затем смесь экстрагировали смесью 30% изопропанол/метиленхлорид (3×100 мл), сушили объединенную органическую фазу над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС:(M-100+1)+: 237,04.
Стадия D
(1R,5S,7r)-трет-бутил-7-(цианометил)-7-винил-3-окса-9-азабицикло[3.3]нонан-9-карбоксилат
Раствор 2-((1R,5S,7s)-9-(трет-бутоксикарбонил)-7-винил-3-окса-9-азабицикло[3.3.1]нонан-7-ил)-2-цианоуксусной кислоты (1,10 г, 3,27 ммоль) в ДМФА (10 мл) нагревали при 130°C в течение 20 минут. После охлаждения до комнатной температуры реакционную смесь распределяли между этилацетатом (200 мл) и насыщенным раствором соли, органическую фазу трижды промывали насыщенным раствором соли, сушили над сульфатом натрия, затем концентрировали и очищали остаток на колонке с силикагелем с применением этилацетатата/гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M-100+1)+: 193,08.
Стадия E
(1R,5S,7r)-трет-бутил-7-(цианометил)-7-формил-3-окса-9-азабицикло[3.3.1]нонан-9-карбоксилат
К раствору (1R,5S,7r)-трет-бутил-7-(цианометил)-7-винил-3-окса-9-азабицикло[3.3.1]нонан-9-карбоксилата (0,66 г, 2,257 ммоль) в диоксане (10 мл) и воде (3 мл) добавляли периодат натрия (1,931 г, 9,03 ммоль) и тетраоксид осмия (0,035 мл, 0,113 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли тиосульфат (2 г) и перемешивали смесь при комнатной температуре в течение 0,5 часа. Реакционную смесь фильтровали, фильтрат распределяли между метиленхлоридом и насыщенным раствором бикарбоната натрия и экстрагировали щелочную фазу метиленхлоридом (3×100 мл). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M-100+1)+: 195,09.
Стадия F
(1R,5S,7r)-9-(трет-бутоксикарбонил)-7-(цианометил)-3-окса-9- азабицикло[3.3.1]нонан-7-карбоновая кислота
К раствору (1R,5S,7r)-трет-бутил-7-(цианометил)-7-формил-3-окса-9-азабицикло[3.3.1]нонан-9-карбоксилата (613 мг, 2,083 ммоль) в трет-BuOH (10 мл) и воде (5 мл) добавляли дигидрофосфат натрия (750 мг, 6,25 ммоль) и 2-метил-2-бутен (1,098 мл, 10,41 ммоль). Раствор охлаждали до 0°C и порциями добавляли хлорит натрия (565 мг, 6,25 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа. Добавляли дополнительное количество дигидрофосфата натрия (750 мг, 6,25 ммоль), 2-метил-2-бутена (1,098 мл, 10,41 ммоль) и хлорита натрия (565 мг, 6,25 ммоль). Реакционную смесь оставляли перемешиваться при 0°C в течение 2 часов перед ее гашением путем добавления 1Н раствора HCl до значения pH 4. Затем смесь разбавляли водой (100 мл) и экстрагировали метиленхлоридом (3×100 мл), поддерживая pH 4 с помощью 1Н раствора HCl. Объединенную органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+23)+: 333,03.
Стадия G
(1R,5S,7r)-9-трет-бутил-7-метил-7-(цианометил)-3-окса-9-азабицикло[3.3.1]нонан-7,9-дикарбоксилат
К раствору (1R,5S,7r)-9-(трет-бутоксикарбонил)-7-(цианометил)-3-окса-9-азабицикло[3.3.1]нонан-7-карбоновой кислоты (646 мг, 2,082 ммоль) в метаноле (10 мл) по каплям добавляли ТМС-диазометан (5,20 мл, 10,41 ммоль) до тех пор, пока не образовывались пузырьки, и затем гасили реакционную смесь путем добавления уксусной кислоты (несколько капель). Смесь концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+23)+: 346,98.
Стадия H
(1R,5S,7r)-9-трет-бутил-7-метил-7-(2-аминоэтил)-3-окса-9-азабицикло[3.3.1]нонан-7,9-дикарбоксилат
Смесь (1R,5S,7r)-9-трет-бутил-7-метил-7-(цианометил)-3-окса-9-азабицикло[3.3.1]нонан-7,9-дикарбоксилата (0,58 г, 1,788 ммоль) и оксида платины (IV) (0,082 г, 0,358 ммоль) в метаноле (10 мл) и уксусной кислоте (10 мл) подвергали гидрированию под давлением 45 фунт/кв.дюйм в течение уикенда. После фильтрования через CELITE® в атмосфере азота фильтрат концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 329,04.
Стадия I
(1R3'r,5S)-трет-бутил-2'-оксо-3-окса-9-азаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-9-карбоксилат
Смесь (1R,5S,7r)-9-трет-бутил-7-метил-7-(2-аминоэтил)-3-окса-9-азабицикло[3.3.1]нонан-7,9-дикарбоксилата (0,59 г, 1,797 ммоль) и карбоната калия (1,490 г, 10,78 ммоль) в метаноле (50 мл) нагревали с обратным холодильником в течение 4 часов. После выпаривания растворителя остаток распределяли между метиленхлоридом и водой и пять раз экстрагировали водную фазу метиленом. Объединенную органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+23)+: 319,03.
Стадия J
(1R3'r,5S)-трет-бутил-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2'-оксо-3-окса-9-азаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-9-карбоксилат
К смеси (1R,3'r,5S)-трет-бутил-2'-оксо-3-окса-9-азаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-9-карбоксилата (0,42 г, 1,417 ммоль) и 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (0,454 г, 1,842 ммоль) в толуоле (15 мл) добавляли карбонат калия (0,588 г, 4,25 ммоль), Ксантфос (0,328 г, 0,567 ммоль) и воду (0,077 мл, 4,25 ммоль). Перед добавлением ацетата палладия (II) (0,064 г, 0,283 ммоль) смесь продували азотом в течение 20 минут. Полученную смесь нагревали при 70°C в течение ночи. После фильтрования остаток очищали на колонке с силикагелем с применением смеси этилацетат/гексан, получая при этом указанное в заголовке соединение. ЖХ/МС:(M+1)+: 393,24.
Стадия K
(1R,3'r,5S)-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-3-окса-9-азаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-2'-он
К раствору (1R,3'r,5S)-трет-бутил-1'-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2'-оксо-3-окса-9-азаспиро[бицикло[3.3.1]нонан-7,3'-пирролидин]-9-карбоксилата (0,22 г, 0,561 ммоль) в метиленхлориде (4 мл) добавляли трифторуксусную кислоту (4 мл, 51,9 ммоль) и перемешивали полученный раствор при комнатной температуре в течение 2 часов. После удаления летучих веществ остаток ощелачивали на ионообменной колонке, промывая метанолом с последующим элюированием 1Н раствором аммиака в метаноле, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 293,21.
Промежуточное соединение 54
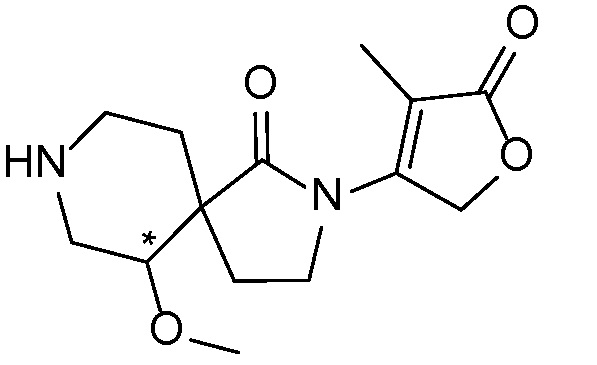
6-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (быстро элюирующий цис-изомер)
Стадия A
этил-1-бензил-4-(цианометил)-3-оксопиперидин-4-карбоксилат
В 250-мл 3-горлую круглодонную колбу помещали раствор этил-1-бензил-3-оксопиперидин-4-карбоксилата (10 г, 38,27 ммоль, 1,00 экв.) в тетрагидрофуране (80 мл). Затем к раствору при перемешивании и 0°C по каплям добавляли раствор KHMDS в тетрагидрофуране (42 мл, 1M) и 30 минут спустя при 0°C по каплям добавляли 2-бромацетонитрил (6,89 г, 57,44 ммоль, 1,50 экв.) при перемешивании. Полученный раствор перемешивали в течение 4 часов при комнатной температуре. Полученную смесь концентрировали в вакууме. Остаток помещали в колонку с силикагелем и элюировали смесью этилацетат/петролейный эфир, получая при этом указанное в заголовке соединение.
Стадия B
этил-4-(2-аминоэтил)-1-бензил-3-гидроксипиперидин-4-карбоксилат
В 10000 мл 4-горлую круглодонную колбу помещали раствор этил-1-бензил-4-(цианометил)-3-оксопиперидин-4-карбоксилата (180 г, 599,30 ммоль) в смеси уксусная кислота/метанол (1:1, 5,4 л) и оксид платины (27 г, 118,90 ммоль). Затем колбу продували и загружали водородом и перемешивали содержимое в течение ночи при комнатной температуре. После фильтрования через CELITE® в атмосфере азота фильтрат концентрировали в вакууме, получая при этом указанное в заголовке соединение.
Стадия C
8-бензил-6-гидрокси-2,8-диазаспиро[4.5]декан-1-он
В 10000 мл 3-горлую круглодонную колбу помещали раствор этил-4-(2-аминоэтил)-1-бензил-3-гидроксипиперидин-4-карбоксилата (220 г, 718,02 ммоль, 1,00 экв.) в метаноле (2000 мл) и аммиаке (2000 мл). Полученный раствор перемешивали в течение ночи при 45°C. Полученную смесь концентрировали в вакууме. Остаток помещали в колонку с силикагелем и элюировали смесью дихлорметан/метанол, получая при этом указанное в заголовке соединение.
Стадия D
цис-трет-бутил-6-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат
В продутую азотом 2000 мл 3-горлую круглодонную колбу с поддерживаемой инертной атмосферой азота помещали раствор 8-бензил-6-гидрокси-2,8-диазаспиро[4.5]декан-1-она (46,3 г, 177,85 ммоль, 1,00 экв.), Cs2CО3 (115,9 г, 354,62 ммоль, 1,99 экв.), Ксантфоса (6,17 г, 10,66 ммоль, 0,06 экв.), Pd2(dba)3 (5,52 г, 6,03 ммоль, 0,03 экв.) и 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (56,92 г, 231,23 ммоль, 1,30 экв.) в диоксане (900 мл). Полученный раствор перемешивали в течение ночи при 80°C. Полученную смесь охлаждали и концентрировали в вакууме. Остаток разбавляли в 1000 мл воды, затем экстрагировали этилацетатом. Органические слои объединяли, промывали 1000 мл воды и 1000 мл насыщенного раствора соли, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток помещали в колонку с силикагелем и элюировали этилацетатом, получая при этом транс-трет-бутил-6-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат. ЖХ/МС: (M+1)+: 357; H-ЯМР (300 МГц, CDCl3, м.д.): δ 7,33-7,26 (5H, м), 5,27-5,25 (2H, м), 4,04-3,96 (2H, м), 3,75-3,72 (1H, м), 3,58 (2H, д, J=2,4 Гц), 2,88-2,75 (3H, м), 2,55-2,50 (1H, м), 2,40-2,12 (3H, м), 2,09-2,00 (4H, м), 1,57-1,55 (1H, м), и цис-трет-бутил-6-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат. ЖХ/МС: (M+1)+: 357; H-ЯМР (300МГц, CDCl3, м.д.): δ 7,33-7,26 (5H, м), 5,27-5,25 (2H, м), 4,06-3,94 (3H, м), 3,61-3,50 (2H, м), 2,93-2,81 (1H, м), 2,79-2,74 (1H, м), 2,57-2,48 (1H, м), 2,19-2,12 (2H, м), 2,03-2,02 (3H, м), 1,97-1,85 (2H, м), 1,68-1,58 (2H, м).
Стадия D
трет-бутил-6-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (цис-изомер)
К раствору 8-бензил-6-гидрокси-2-(2-метил-3-оксоциклопент-1-ен-1-ил)-2,8-диазаспиро[4.5]декан-1-она (цис-изомер) (10 г, 28,2 ммоль) и ди-трет-бутилкарбоната (7,21 мл, 31,0 ммоль) в метаноле (50 мл) добавляли палладий на угле (1,501 г, 1,411 ммоль) и подвергали полученную смесь гидрированию при давлении 45 фунт/кв.дюйм при комнатной температуре в течение уикенда. Суспензию фильтровали через CELITE® в атмосфере азота, фильтрат концентрировали и очищали остаток на силикагеле с применением этилацетата/гексана в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 367,17.
Стадия E
трет-бутил-6-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (быстро элюирующий цис-изомер) и трет-бутил-6-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (медленно элюирующий цис-изомер).
К смеси трет-бутил-6-гидрокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (цис-изомер) (2 г, 5,46 ммоль) и оксида серебра (1,391 г, 6,00 ммоль) в ацетонитриле (50 мл) добавляли метилиодид (3,41 мл, 54,6 ммоль). Смесь нагревали при 60°C в герметично закрытой трубке в течение ночи. После охлаждения до комнатной температуры смесь фильтровали, концентрировали фильтрат и очищали остаток хроматографией на силикагеле с применением смеси этилацетат/гексан, получая при этом трет-бутил-6-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (цис-изомер, рацемат). ЖХ/МС:(M+1)+: 380,99. Рацемат дополнительно разделяли на хиральной колонке AD, получая при этом трет-бутил-6-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (быстро элюирующий цис-изомер). ЖХ/МС:(M+1)+: 381,05; и трет-бутил-6-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилат (медленно элюирующий цис-изомер). ЖХ/МС:(M+1)+: 381,01.
Стадия F
6-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (быстро элюирующий цис-изомер)
К раствору трет-бутил-6-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (быстро элюирующий цис-изомер) (2,17 г, 5,70 ммоль) в метиленхлориде (7 мл) добавляли трифторуксусную кислоту (7 мл, 91 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 2 часов. После концентрирования остаток ощелачивали на ионообменной колонке путем промывания сначала метанолом и затем путем элюирования 1Н раствором аммиака в метаноле, получая при этом 6-метокси-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (быстро элюирующий цис-изомер). ЖХ/МС: (M+1)+: 281,11.
В следующих примерах и упомянутых выше промежуточных соединениях изомер "A" и изомер "B" (например, изомер 6A и 6B и т.п.), относятся к быстро элюирующим и медленно элюирующим диастереомерам, соответственно, исходя из наблюдаемого порядка элюирования индивидуальных диастереомеров после их выделения из смеси изомеров. За исключением некоторого хирального центра в исходной смеси изомеров абсолютную стереохимию каждого из выделенных изомеров не определяли, если не указано иначе.
В примерах, где абсолютную стереохимию каждого из выделенных изомеров не определяли, на рисунке, связанном с химической структурой, можно применять звездочку (*), которая указывает место не определенного хирального центра.
Пример 1
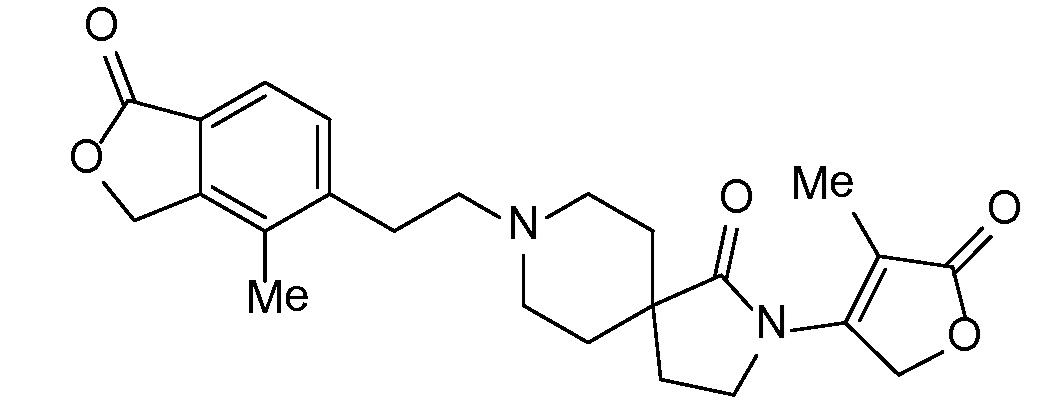
8-(2-(4-метил-1-оксо-l,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Смесь (4-метил-1-оксо-l,3-дигидро-2-бензофуран-5-ил)ацетальдегида (I-3) (100 мг, неочищенный) и 2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (I-16) (145 мг, 0,58 ммоль) в ТГФ (5 мл) перемешивали при комнатной температуре в течение 2 часов, затем к смеси добавляли NaBH(OAc)3 (167 мг, 0,79 ммоль) и дополнительно перемешивали при 50°C в течение 3 часов. После гашения смеси насыщенным водным раствором NH4Cl смесь экстрагировали EtOAc и сушили органические слой над Na2SО4. Растворитель удаляли при пониженном давлении, и остаток очищали препаративной ТСХ (EtOAc/MeOH: 1/1) и препаративной ВЭЖХ, получая при этом указанный в заголовке продукт. МС (ESI) m/z: 425(M+H+); 1H ЯМР (400 МГц, MeOD): δ 7,69 (д, J=7,6 Гц, 1H), 7,50 (д, J=7,6 Гц, 1H), 5,38 (с, 2H), 5,27-5,25 (м, 2H), 4,20-4,14 (м, 2H), 3,83-3,80 (м, 1H), 3,68-3,66 (м, 1H), 3,42-3,38 (м, 2H), 3,33-3,23 (м, 4H), 2,41 (с, 3H), 2,39-2,30 (м, 2H), 2,24-2,17 (м, 2H), 2,10-2,03 (м, 5H).
Следующие соединения в таблице 1 получали по способу, аналогичному примеру 1, исходя из промежуточных пиперидиновых и альдегидных соединений, полученных, как описано выше.
|
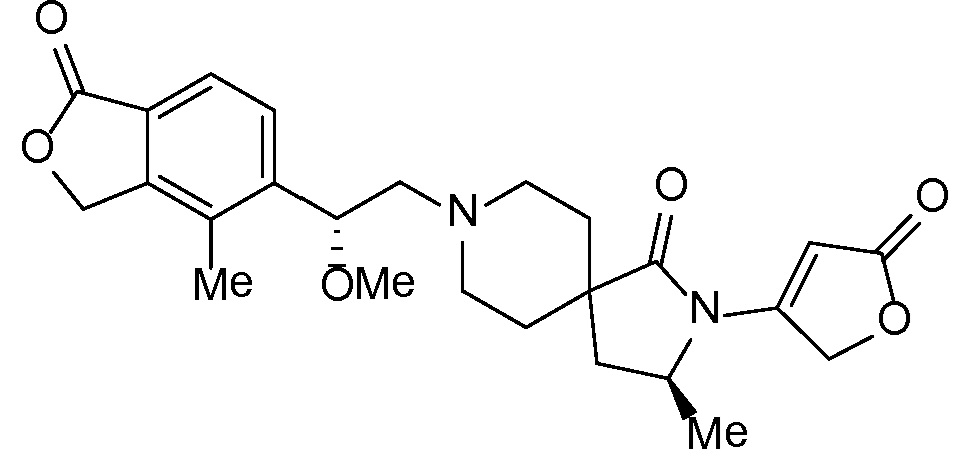
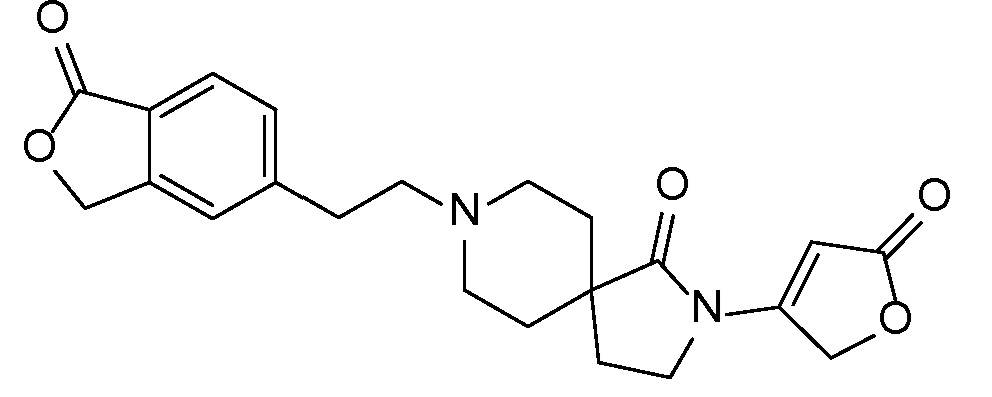
|
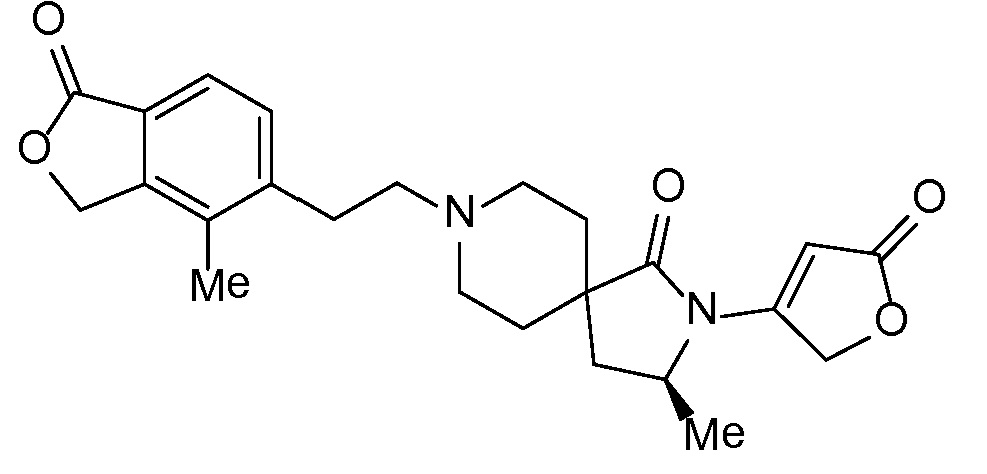
Пример 5
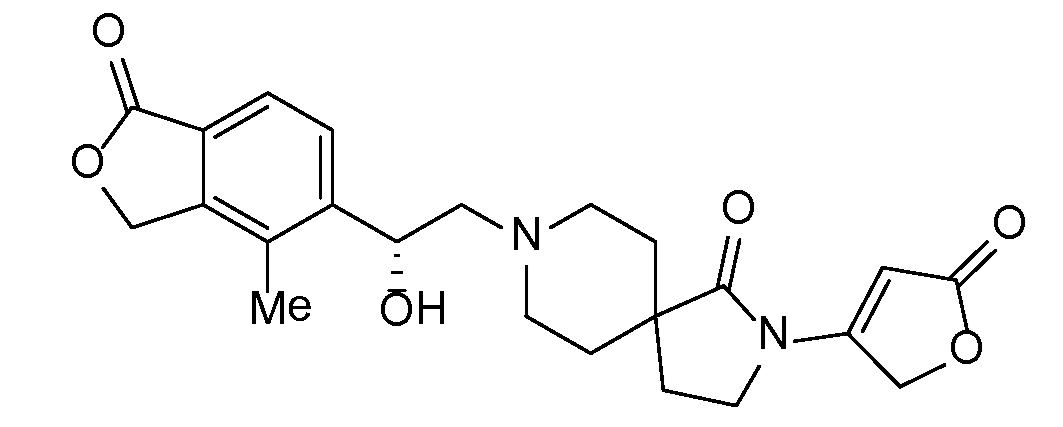
8-[(2R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (I-16, 200 мг, 0,846 ммоль) объединяли с 4-метил-5-[(2R)-оксиран-2-ил]-2-бензофуран-1(3H)-оном (I-4B) (177 мг, 0,931 ммоль) в этаноле (5 мл) и нагревали в микроволновой печи при 145°C в течение 3 часов. Растворитель удаляли и очищали остаток препаративной ТСХ путем элюирования смесью 25% метанол/EtOAc, получая при этом указанное в заголовке соединение. ЖХ-МС (IE, m/z): 427(M+1)+.
Пример 6
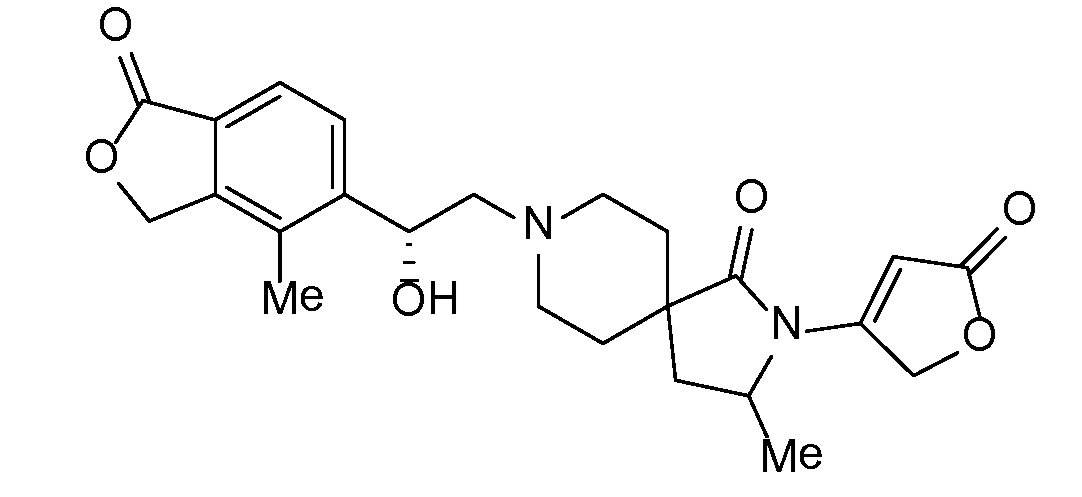
8-((R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (I-22, 0,257 ммоль) растворяли в этаноле (3 мл) и обрабатывали диизопропилэтиламином (135 мкл, 0,771 ммоль) с последующим добавлением (R)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-она (I-4B, 73,3 мг, 0,385 ммоль). Смесь нагревали при 80°C в герметично закрывающейся трубке в течение 12 часов. Затем реакционную смесь охлаждали до комнатной температуры и концентрировали. Полученный неочищенный продукт очищали колоночной хроматографией путем элюирования смесью с градиентом 0-20% MeOH/EtOAc, получая при этом указанный в заголовке продукт в виде смеси диастереомеров. ЖХ-МС (IE, m/z): 441,3 (M+1)+.
Пример 6A и 6B (выделенные отдельные изомеры)
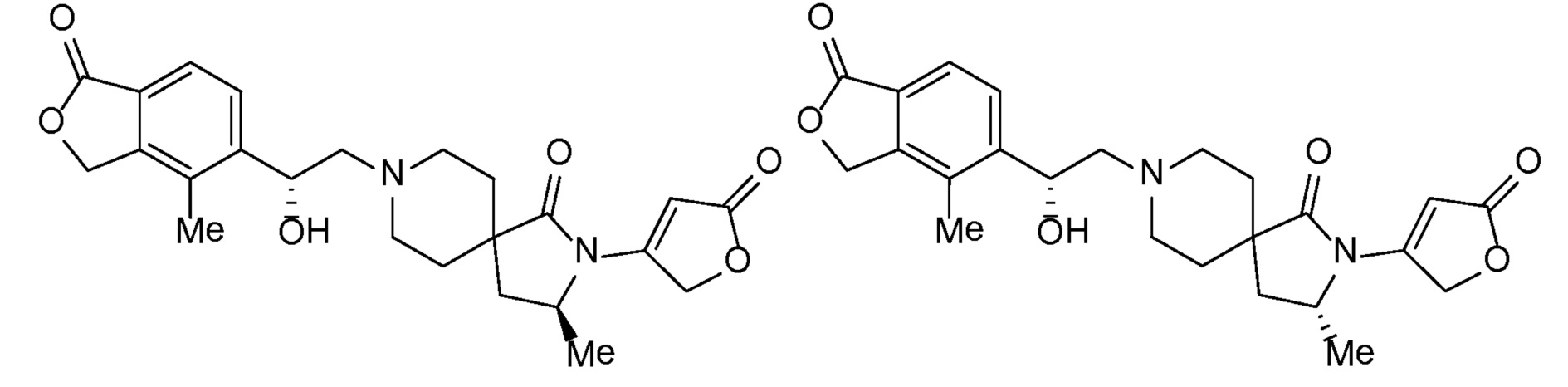
(R)-8-((R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он;
(S)-8-((R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-3-метил-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (выделенные изомеры A и B)
Продукт по примеру 6 разделяли с применением СФХ- элюирования смесью 30% MeOH (0,2% DEA)/CО2 на хиральной колонке Chiralcel OD, получая при этом изомер 6A (быстро элюирующий): ЖХ-МС (IE, m/z): 441,3; и изомер 6B (медленно элюирующий): ЖХ-МС (IE, m/z): 441,3 (M+1)+.
Пример 7
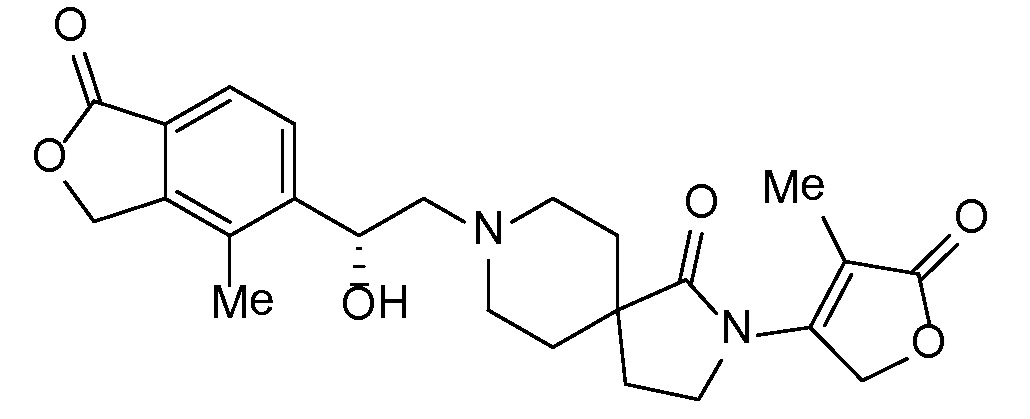
8-[(2R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Раствор 2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (I-17, 6,00 г, 24,0 ммоль) и 4-метил-5-[(2R)-оксиран-2-ил]-2-бензофуран-1(3H)-она (I-4B, 5,93 г, 31,2 ммоль) в этаноле (20 мл) нагревали в герметично закрытой трубке при 95°C в течение ночи. После концентрирования остаток очищали на колонке с силикагелем с применением смеси метанол/дихлорметан, затем осаждали из метанола, получая при этом указанное в заголовке соединение. ЖХ/МС, (M+1)+: 440,93. 1H-ЯМР (500 МГц, CDCl3), δ 7,845-7,809 (м, 2H), 5,290-5,278 (м, 4H), 5,139-5,112 (м, 1H), 4,072-4,044 (т, J=7,1 Гц, 2H), 3,190-3,167 (м, 1H), 2,876-2,859 (м, 1H), 2,636-2,604 (м, 2H), 2,468-2,421 (м, 1H), 2,311 (с, 3H),2,350-2,328 (м, 1H), 2,193-2,165 (т, J=7,1 Гц, 2H), 2,101-2,039 (м, 5H), 1,668-1,618 (м, 2H).
Пример 8
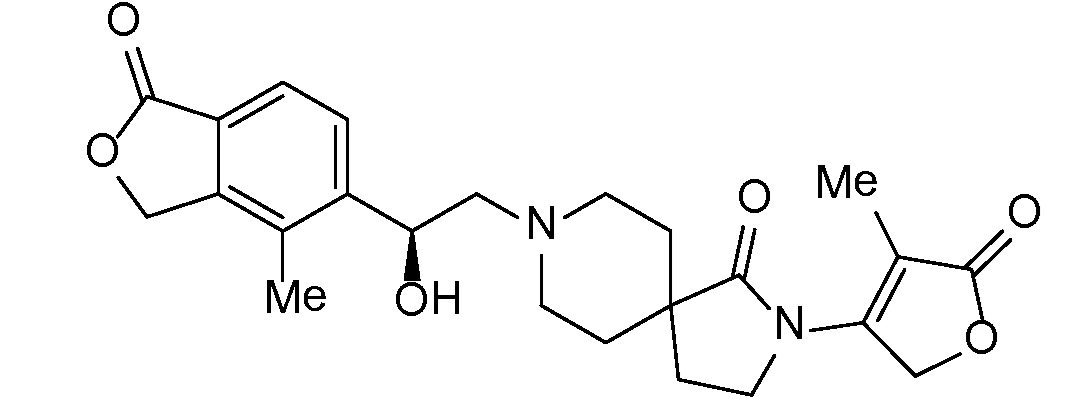
8-[(2S)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору 2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (I-17, 10 мг, 0,040 ммоль) в этаноле (2 мл), находящемуся в трубке для микроволновой печи, добавляли (S)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-он (I-4A, 9,9 мг, 0,052 ммоль) и нагревали полученный раствор при 140°C в микроволновой печи в течение 3 часов. После концентрирования остаток очищали препаративной ТСХ с применением смеси 30% метанол/этилацетат, получая при этом указанное в заголовке соединение. ЖХ/МС, (M+1)+: 441,00, 1H-ЯМР (500 МГц, CDCl3), δ 7,840-7,804 (м, 2H), 5,304-5,240 (м, 4H), 5,134-5,107 (м, 1H), 4,066-4,038 (т, J=7,1 Гц, 2H), 3,185-3,161 (м, 1H), 2,8885-2,862 (м, 1H), 2,629-2,568 (м, 2H), 2,462-2,416 (м, 1H), 2,305 (с, 3H), 2,344-2,323 (м, 1H), 2,197-2,158 (т, J=7,1 Гц, 2H), 2,108-2,033 (м, 5H), 1,661-1,604 (м, 2H).
Пример 9
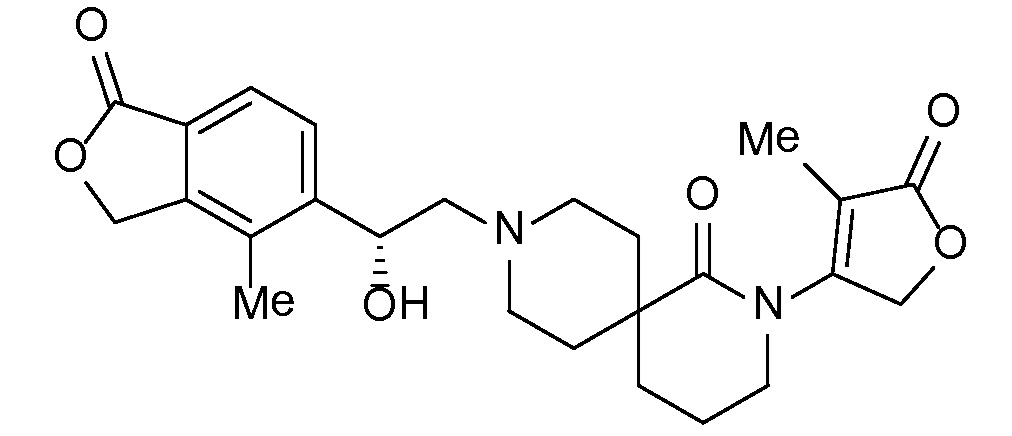
9-[(2R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2-аза-9-азонийспиро[5.5]ундекан;
К раствору 2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,9-диазаспиро[5.5]ундекан-1-она (I-19, 50 мг, 0,19 ммоль) в этаноле (2 мл), находящемуся в трубке для микроволновой печи, добавляли (S)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-он (I-4B, 43,2 мг, 0,227 ммоль)) и нагревали полученный раствор при 145°C в микроволновой печи в течение 35 мин. После концентрирования остаток очищали препаративной TСХ (2000 Mм, 8% MeOH/EtOAc), получая при этом указанное в заголовке соединение в виде свободного основания. Свободное основание превращали в кислотно-аддитивную соль HCl путем добавления 1 M раствора HCl в простом эфире (189 мл, 0,189 ммоль) и концентрирования досуха. ЖХ/МС, (M+1)+: 455
Следующие соединения в таблице 2 получали по способу, аналогичному описанному в примерах 5 - 9, исходя из указанных промежуточных пиперидиновых и эпоксидных соединений, полученных, как описано выше.
|
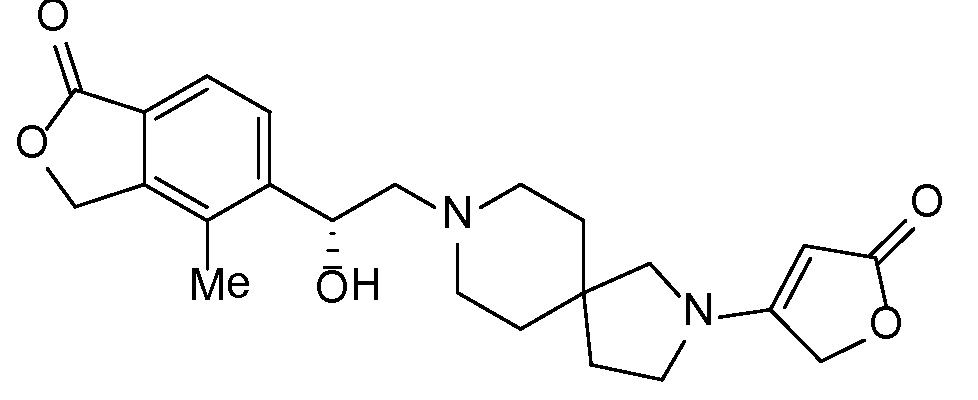
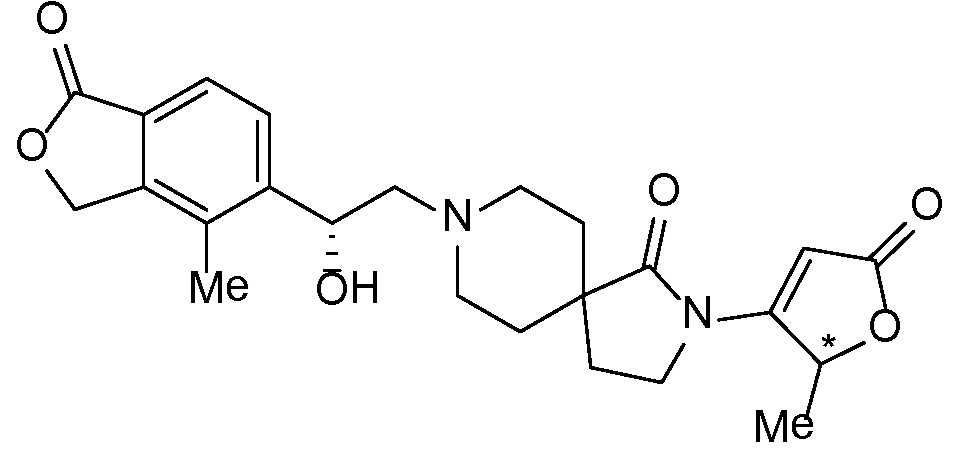
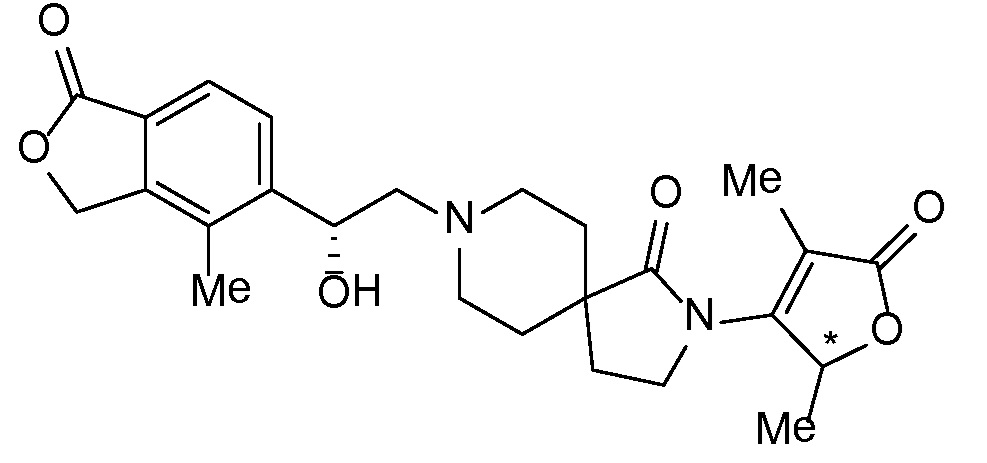
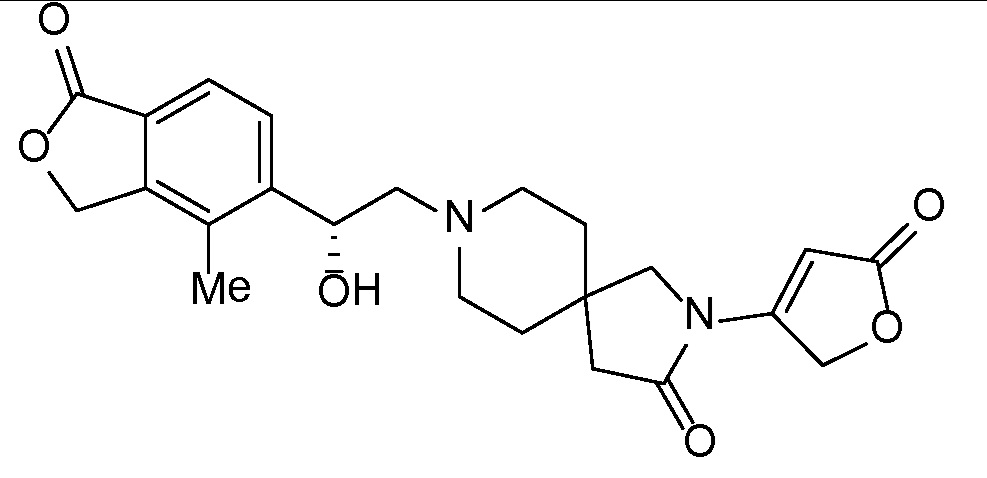
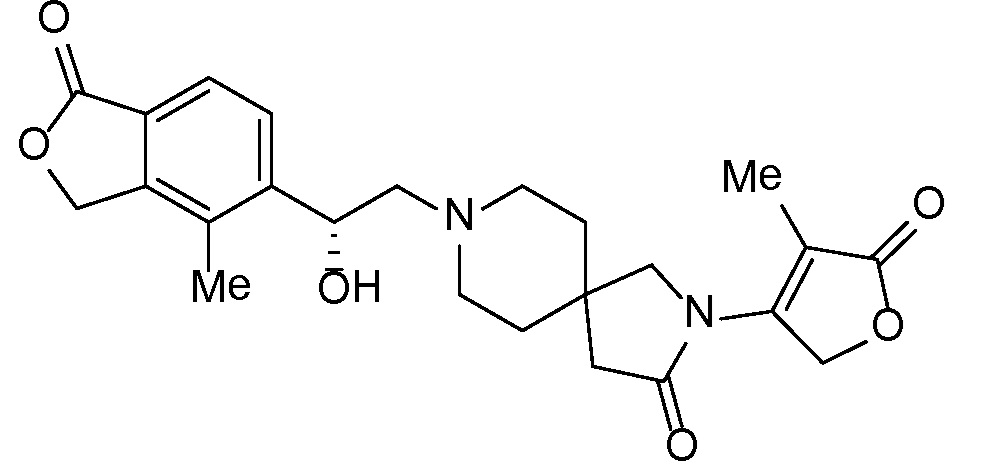
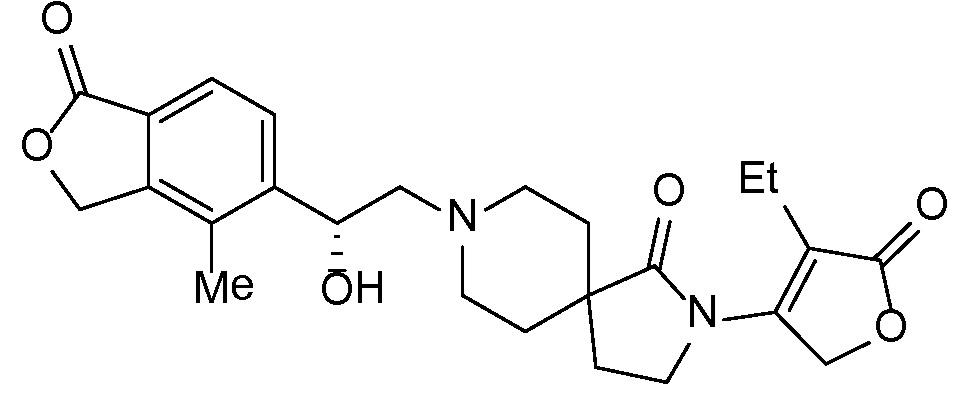
|
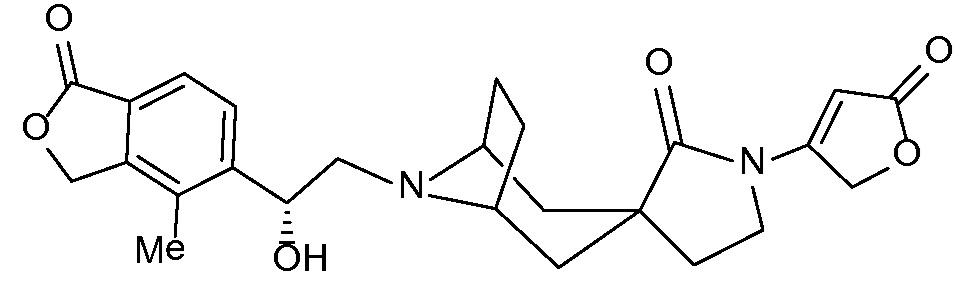
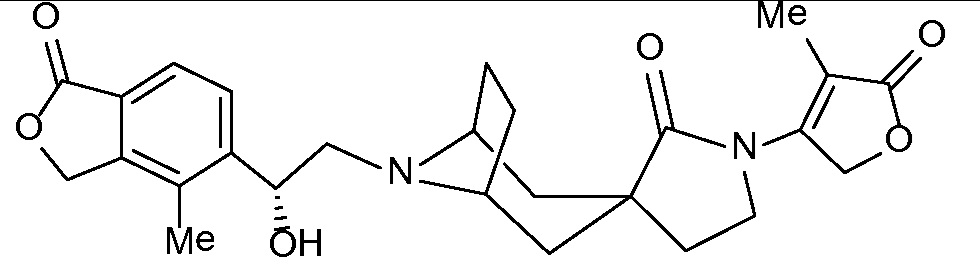

|
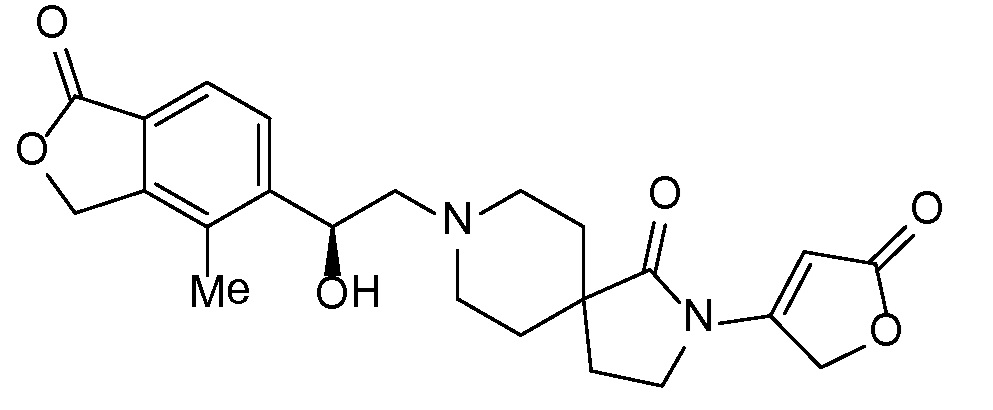
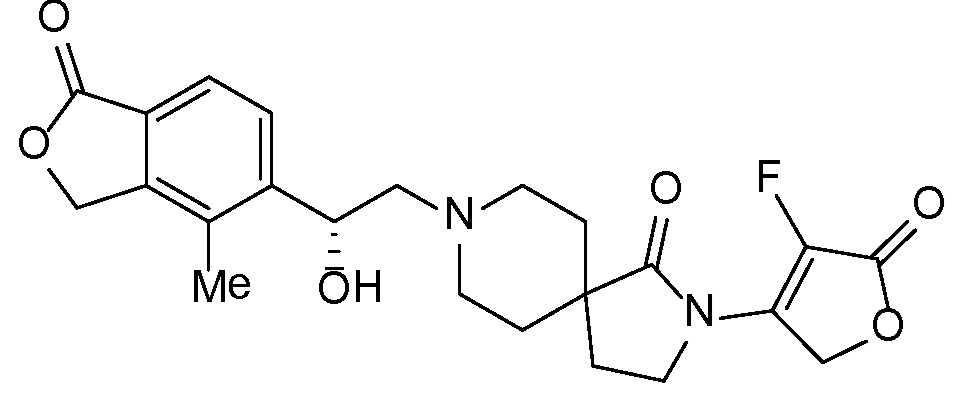
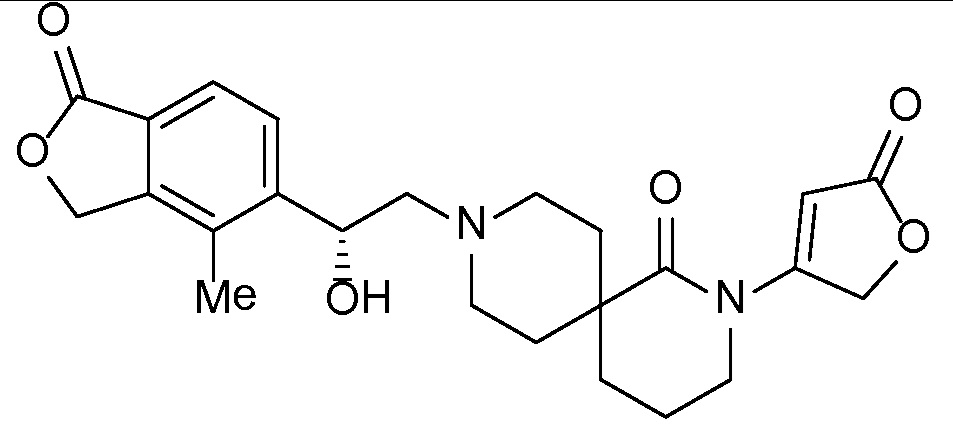
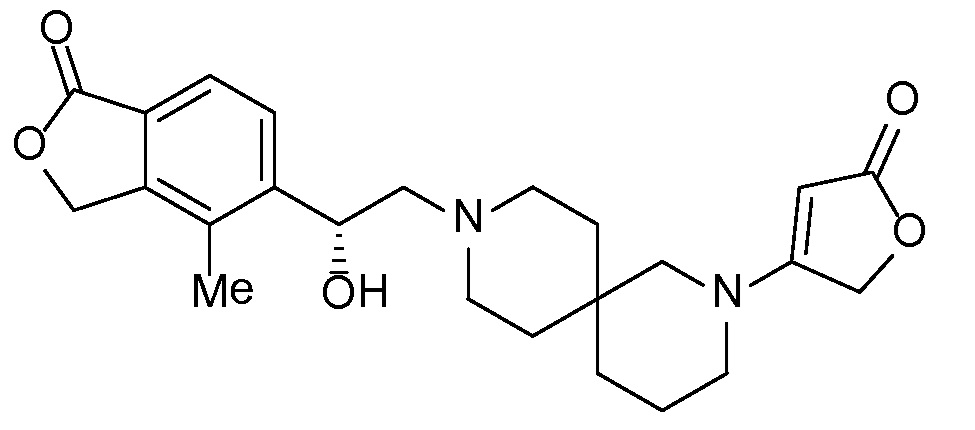
|
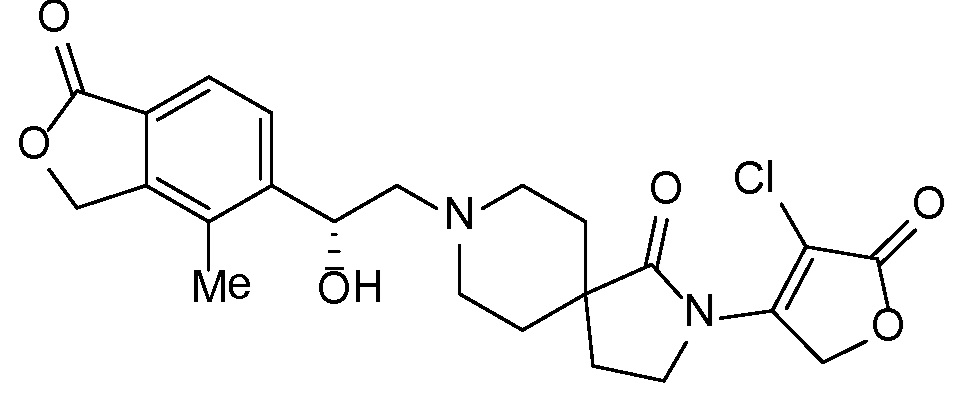
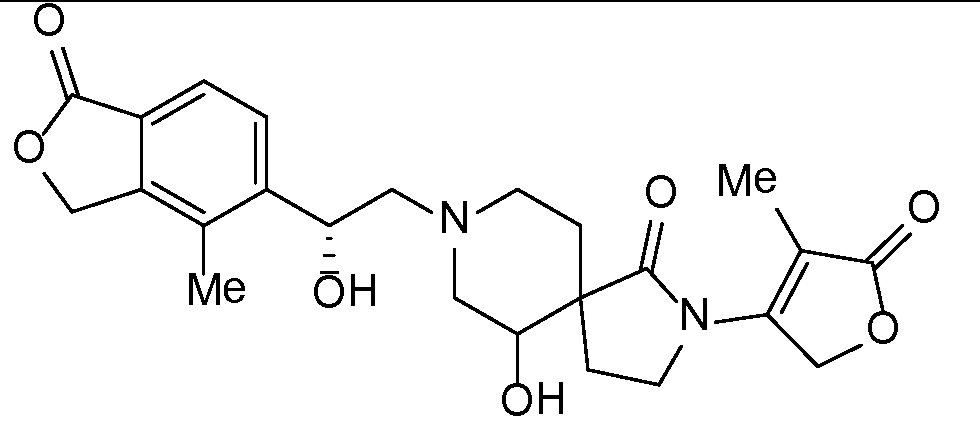
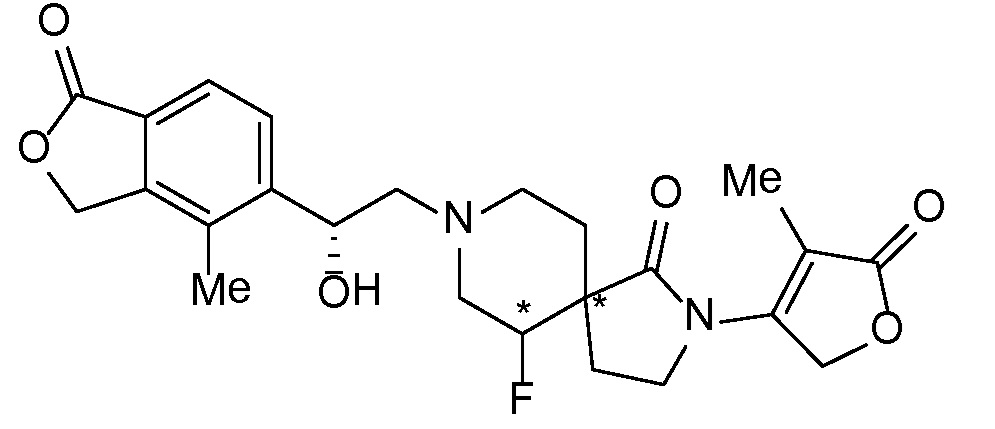
|
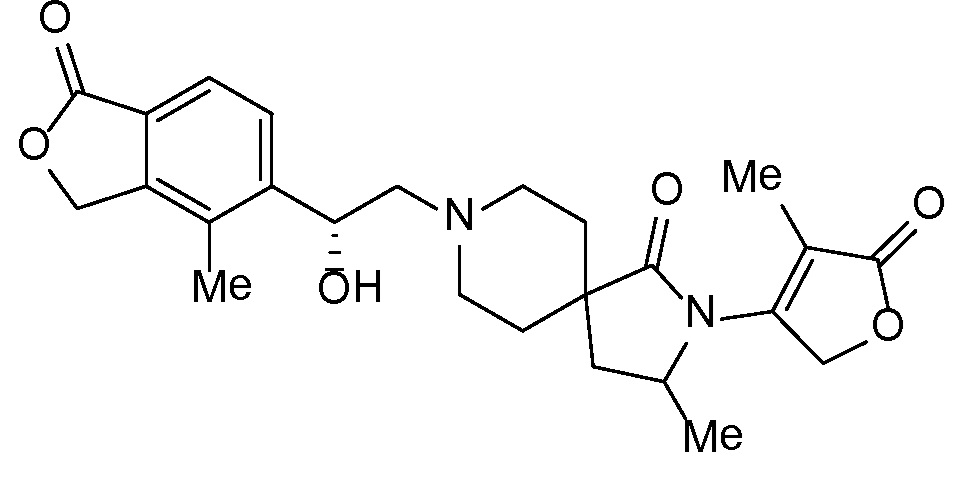
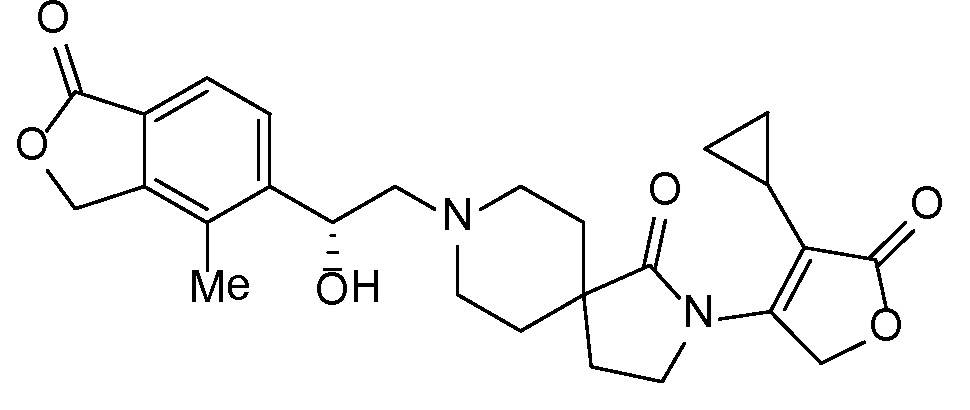
|
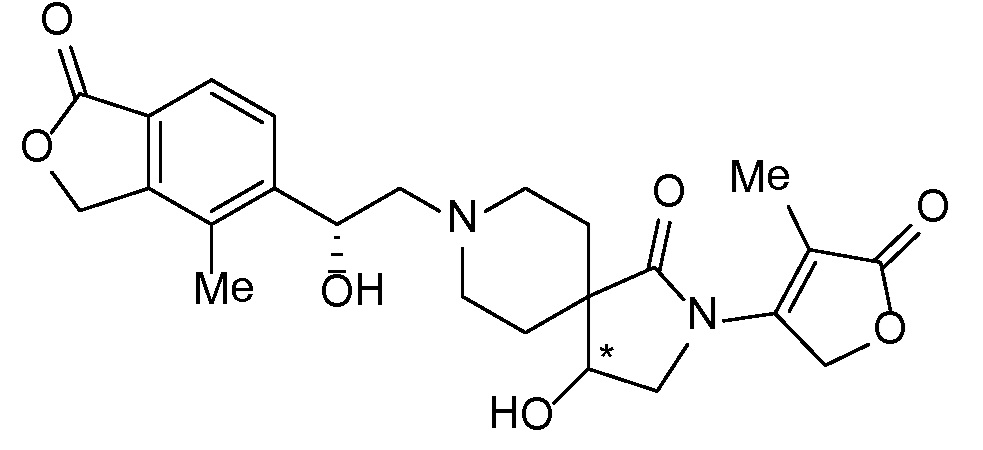
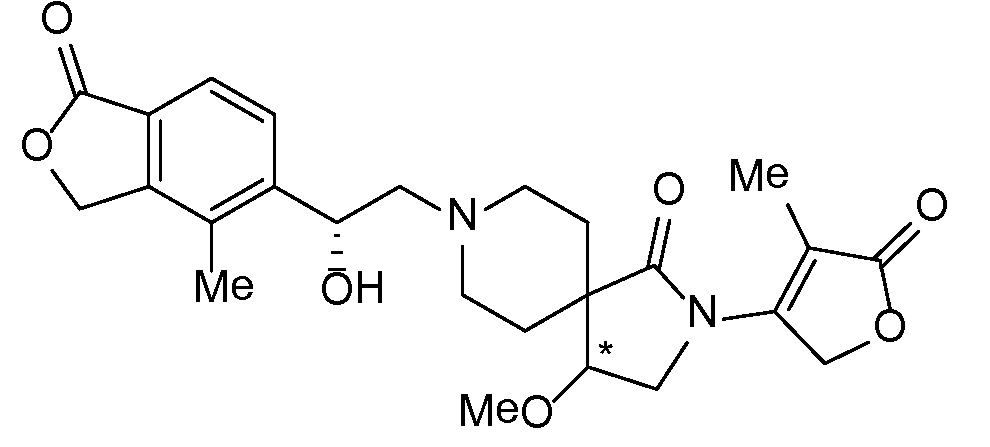
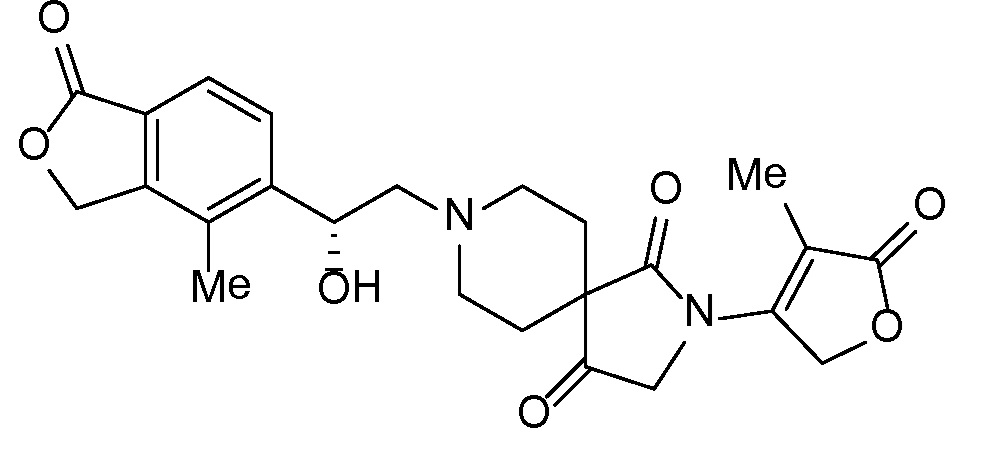
|
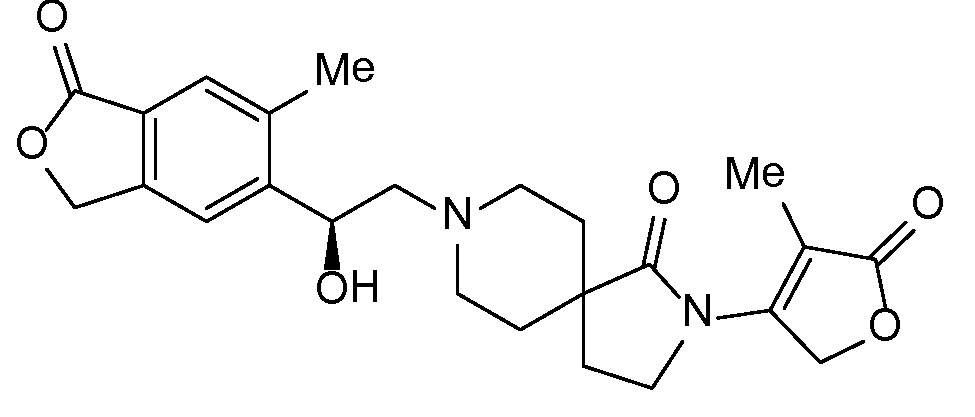
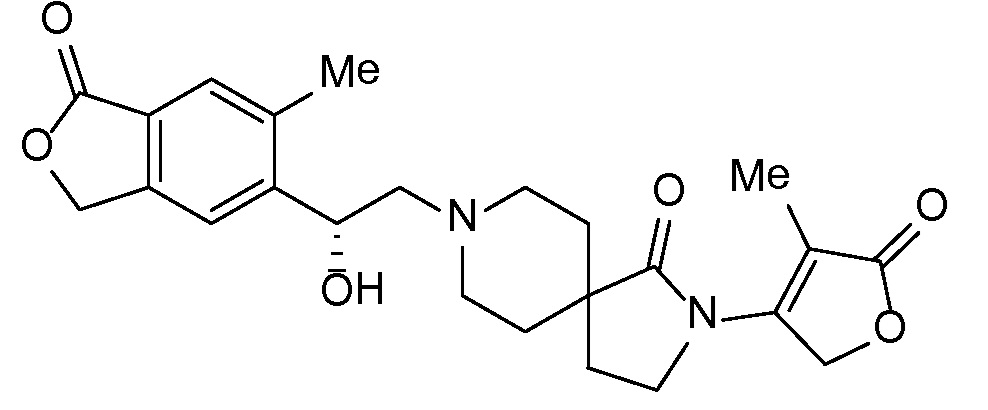
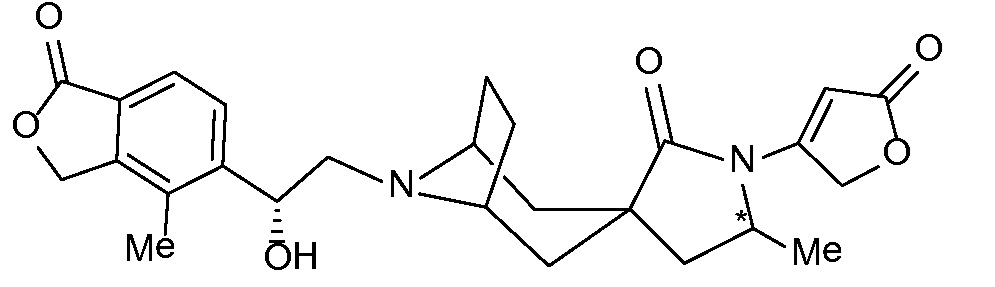
|
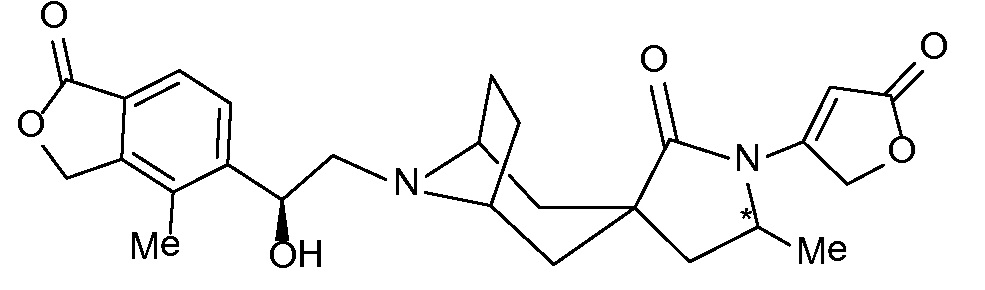
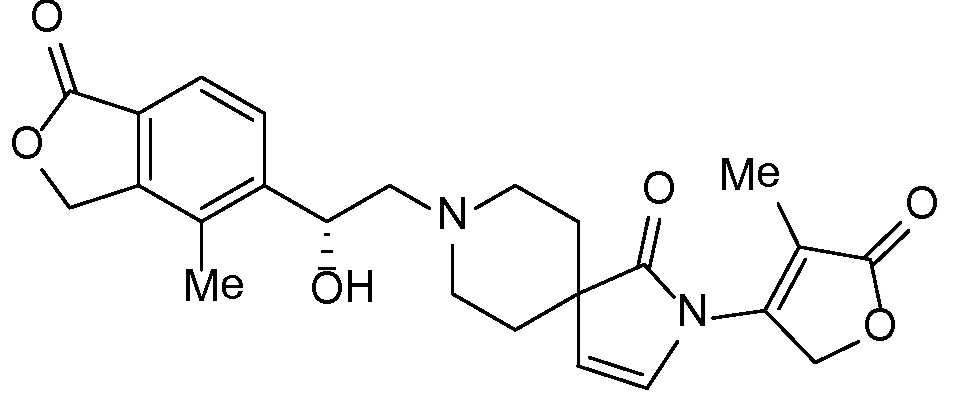
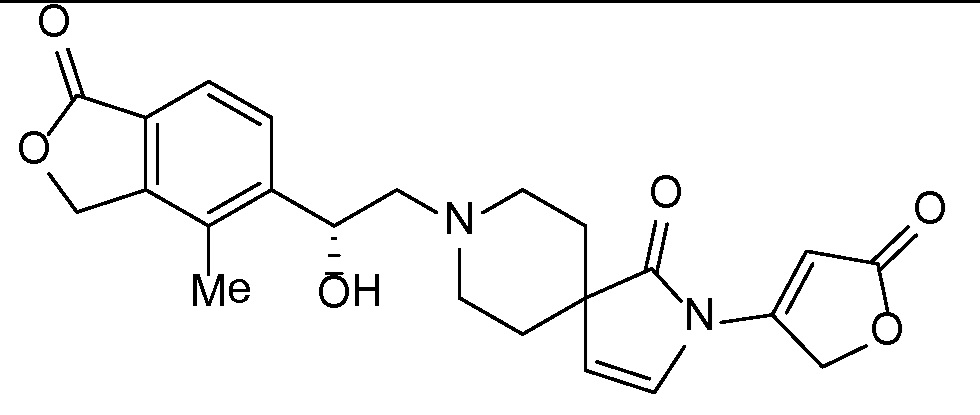
|
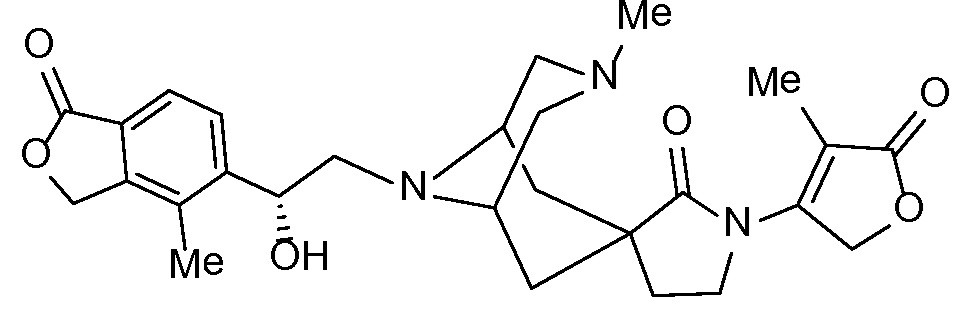
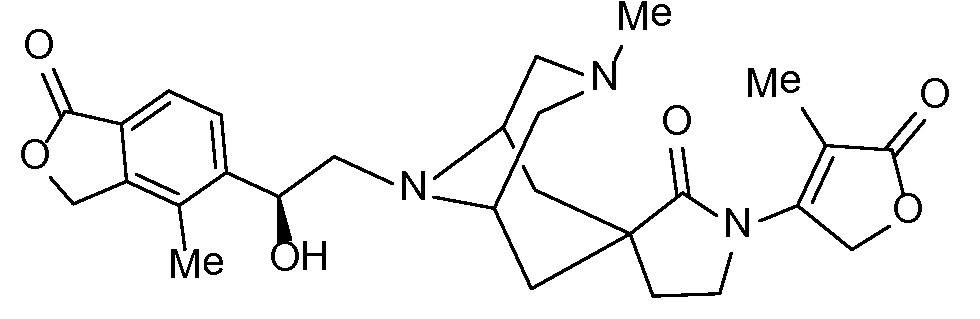
Пример 47
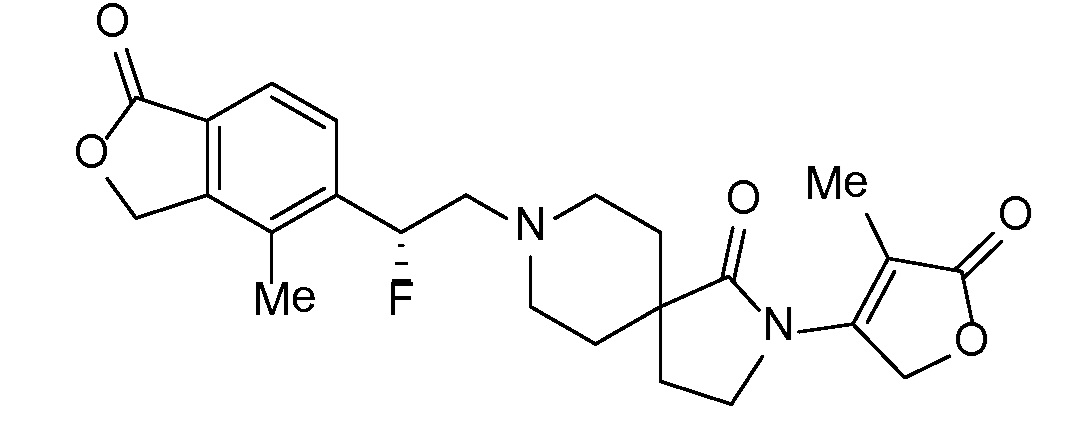
8-[(2R)-2-фтор-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
8-[(2S)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К суспензии 4-метил-5-[(2S)-оксиран-2-ил]-2-бензофуран-1(3H)-она (I-4A) (200 мг, 1,05 ммоль) и 2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (300 мг, 1,05 ммоль) в 10 мл EtOH добавляли DIPEA (271 мг, 2,10 ммоль). Полученную смесь перемешивали при 80°C в течение ночи и затем охлаждали до комнатной температуры; растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (этилацетат:MeOH=20:1), получая при этом указанное в заголовке соединение. МС-ESI (m/z): 441 (M+1)+.
Стадия B
8-[(2R)-2-фтор-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору продукта, полученного на стадии A (170 мг, 0,39 ммоль), в 5 мл ДХМ при -78°C добавляли Et3N.3HF (10 капель) и DAST (5 капель). Смесь перемешивали в течение ночи и гасили водным раствором NaHCО3. Органический слой отделяли, и водный слой экстрагировали ДХМ. Объединенные органические слои сушили над безводным Na2SО4, фильтровали и удаляли растворитель в вакууме; остаток очищали препаративной ТСХ (EtOAc:MeOH=1:1), получая при этом указанное в заголовке соединение. 1H ЯМР (400 МГц, CDCl3): δ 7,81 (д, J=7,8 Гц, 1H), 7,61 (д, J=7,8 Гц, 1H), 6,02-5,88 (м, 1H), 5,28-5,22 (м, 4H), 4,01 (т, J=7,0 Гц, 2H), 3,06-2,85 (м, 3H), 2,73-2,61 (м, 1H), 2,46-2,38 (м, 2H), 2,31 (с, 3H), 2,11 (т, J=7,0 Гц, 2H), 2,05-1,96 (м, 5H), 1,55-1,50 (м, 2H). MS-ESI (m/z): 443 (M+1) +.
Пример 48
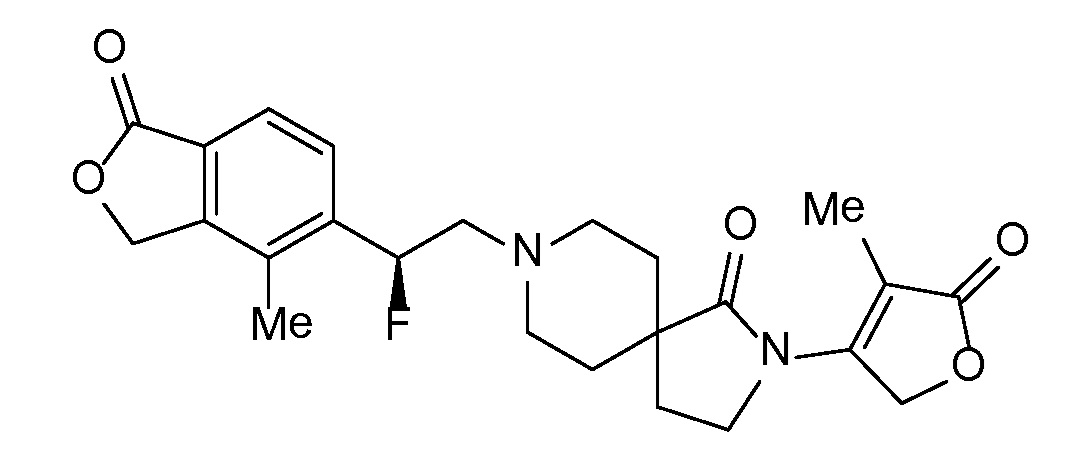
8-[(2S)-2-фтор-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Указанное в заголовке соединение получали по способу, аналогичному описанному непосредственно выше для синтеза соединения по примеру 47, за исключением того, что исходили из 4-метил-5-[(2R)-оксиран-2-ил]-2-бензофуран-1(3H)-она (I-4B). 1H ЯМР (400 МГц, CDCl3): δ 7,81 (д, J=7,8 Гц, 1H), 7,61 (д, J=7,8 Гц, 1H), 6,02-5,88 (м, 1H), 5,28-5,23 (м, 4H), 4,01 (т, J=7,0 Гц, 2H), 3,05-2,86 (м, 3H), 2,73-2,61 (м, 1H), 2,46-2,38 (м, 2H), 2,31 (с, 3H), 2,13 (т, J=7,0 Гц, 2H), 2,05-1,96 (м, 5H), 1,55-1,50 (м, 2H). MS-ESI (m/z): 443 (M+1) +.
Пример 49
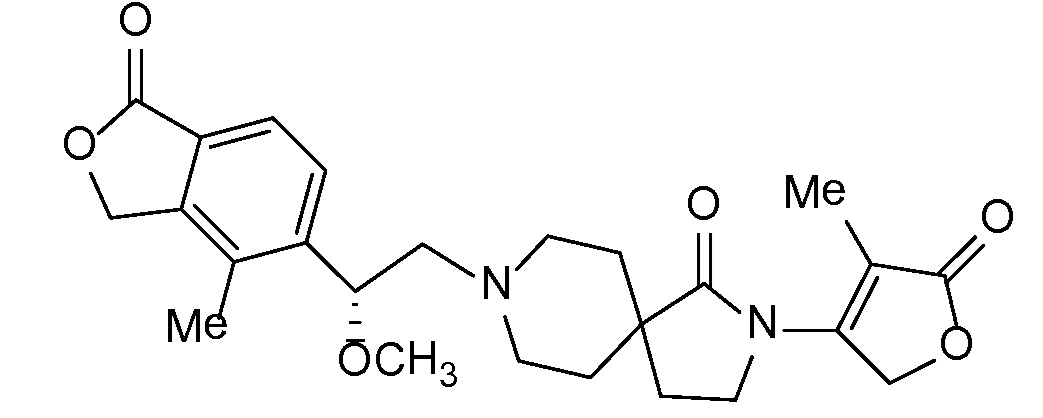
8-[(2R)-2-метокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2-аза-8-азонийспиро[4.5]декан
Стадия A
(S)-8-(2-хлор-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору 8-[(2R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (пример 7) (1,1 г, 2,5 ммоль) и метансульфонилхлорида (0,584 мл, 7,49 ммоль) в ДХМ (30 мл) добавляли триэтиламин (1,22 мл, 8,74 ммоль) и N,N-диметилпиридин-4-амин (0,031 г, 0,250 ммоль) при температуре от -10 до -15°C (лед-NaCl). Смесь перемешивали при той же температуре в течение 20 минут и гасили водным раствором NH4Cl. Органический слой отделяли, а водный слой экстрагировали ДХМ (30 мл). Объединенные органические слои сушили (MgSО4) и концентрировали при пониженном давлении, получая при этом продукт, который применяли на следующей стадии без дополнительной очистки. ЖХМС: 459,05 (M+1).
Стадия B
8-[(2R)-2-метокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2-аза-8-азонийспиро[4.5]декан
Один небольшой кусок металлического натрия в метаноле (20 мл) перемешивали до тех пор, пока натрий не исчезал. К раствору добавляли продукт, полученный на стадии A (45 мг, 0,098 ммоль). Смесь перемешивали при комнатной температуре в течение 20 мин, добавляли 3 мл 1Н раствора HCl и концентрировали смесь. Остаток растворяли в ДМСО, и очищали с помощью ВЭЖХ с обращенной фазой (компания Gilson, 3%-45% смеси 0,1% TFA-вода в 0,1% TFA-AcCN), получая при этом указанное в заголовке соединение. 1H-ЯМР (500 МГц), δ 7,82 (1H, д, J=7,6 Гц), 7,70 (1H, д, J=7,8 Гц), 5,42 (2H, с), 5,28 (2H, с), 5,20 (1H, м), 4,15 (3H, с), 3,50-3,90 (4H, м), 3,12-3,50 (4H, м), 2,45 (3H, с), 2,15-2,37 (4H, м), 2,07 (3H, с), 1,95-2,05 (2H, м). ЖХ-МС 455,1 (M+1), 477,09 (M+23).
Пример 50
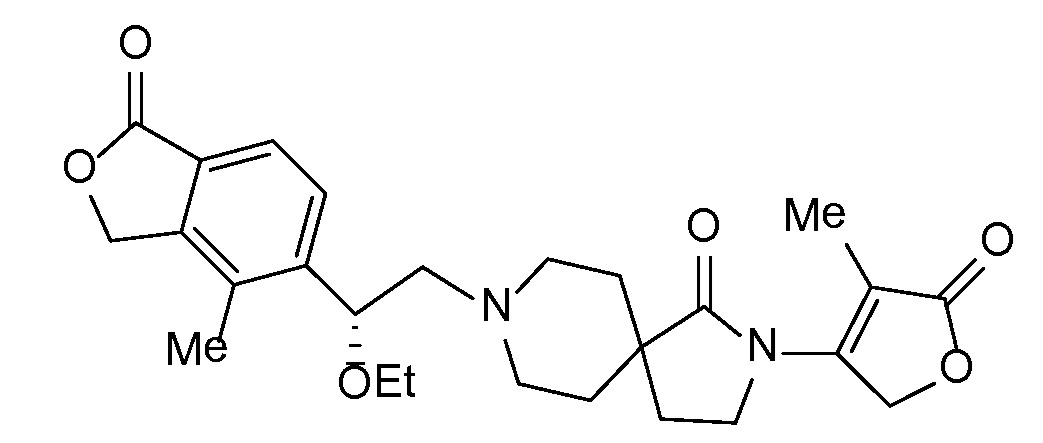
8-[(2R)-2-этокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Указанное в заголовке соединение получали с применением по существу процедуры по примеру 49 за исключением применения этанола вместо метанола в качестве реакционного растворителя на стадии B. ЖХ-МС: 469,13 (M+1).
Пример 51
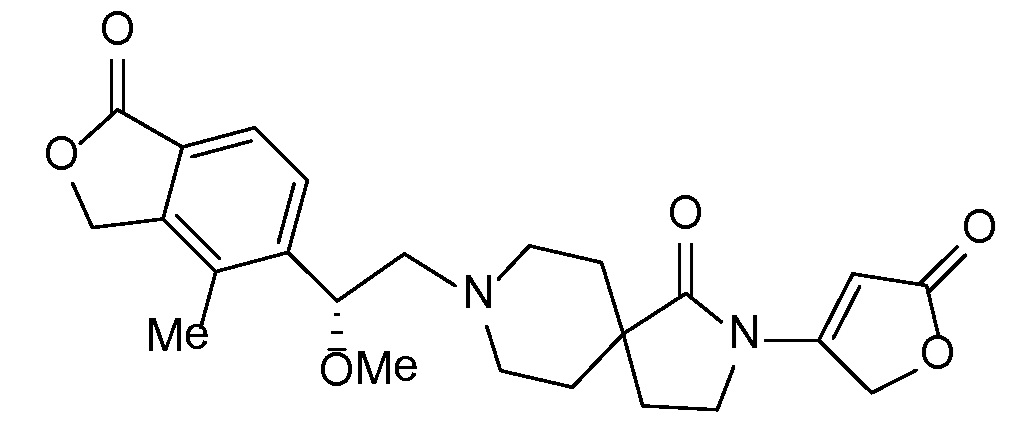
8-[(2R)-2-метокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Указанное в заголовке соединение получали с применением по-существу процедуры по примеру 49, за исключением того, что исходили из 8-[(2R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она. ЖХ-МС: 441 (M+1).
Пример 52
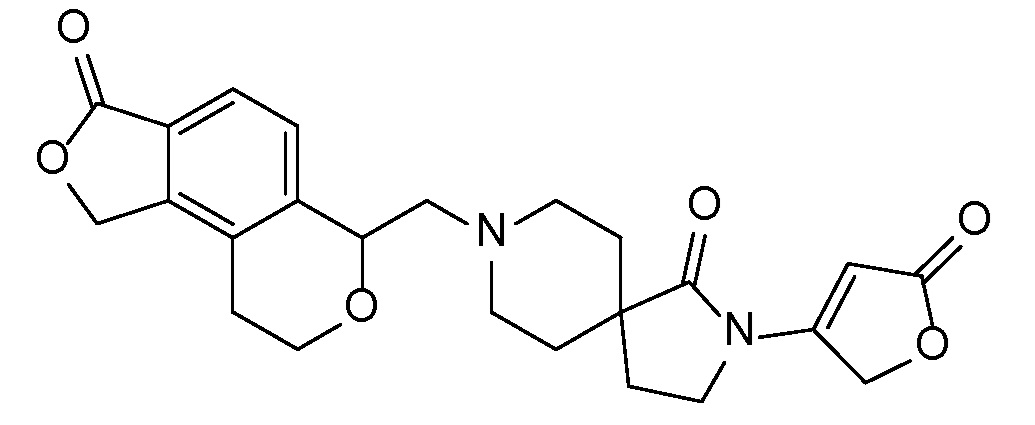
1-оксо-2-(5-оксо-2,5-дигидрофуран-3-ил)-8-[(3-оксо-3,6,8,9-тетрагидро-1Н-фуро[3,4-f]изохромен-6-ил)метил]-2-аза-8-азонийспиро[4.5]декан
(3-оксо-3,6,8,9-тетрагидро-1H-фуро[3,4-f]изохромен-6-ил)метил-4-метилбензолсульфонат (I-8) (80 мг, 0,21 ммоль) объединяли с 2-(5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-оном (I-16) (50,5 мг, 0,214 ммоль) и триэтиламином (30 мкл, 0,21 ммоль) в ацетонитриле (2 мл) в трубке для микроволновой печи и нагревали при 140°C. Растворитель удаляли, и остаток очищали ЖХСД путем элюирования смесью 5% метанол/ДХМ, получая при этом указанное в заголовке соединение в виде смеси двух энантиомеров. ЖХ-МС: 439 (M+1).
Примеры 53A, 53B, 53C, 53D (четыре индивидуальных изомера)
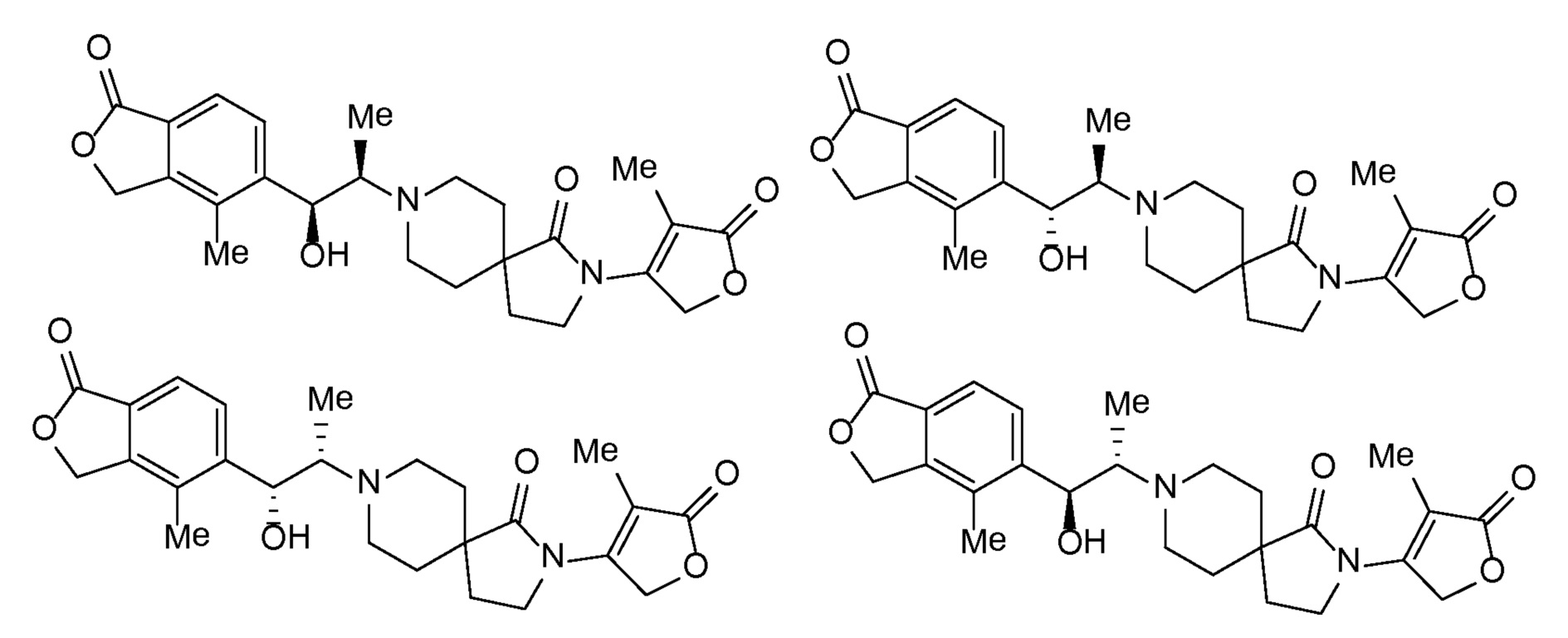
8-((1S,2R)-1-гидрокси-1-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он;
8-((1R,2R)-1-гидрокси-1-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он;
8-((1R,2S)-1-гидрокси-1-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он и
8-((1S,2S)-1-гидрокси-1-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (четыре отдельных выделенных изомера).
К 5-(l,2-дигидроксипропил)-4-метилизобензофуран-1(3H)-ону (I-6) (300 мг, 1,35 ммоль) и 2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-ону (I-17) (405 мг, 1,62 ммоль) в т-амиловом спирте (2,7 мл) добавляли 2-(дициклогексилфосфино)-1-фенил-1H-пиррол (82 мг, 0,243 ммоль) и карбонил рутения (51,8 мг, 0,081 ммоль). Реакционную смесь дегазировали и нагревали при 140°C в течение двух дней. Реакционную смесь концентрировали и очищали колоночной хроматографией (0-10% MeOH/ДХМ) для разделения на син- и анти-изомеры. Затем полученные син- и анти-изомеры соответственно очищали с помощью СФХ-ВЭЖХ с применением следующих условий: колонка Chiralpak AD, 30×250 мм, 65% MeOH+0,2% DEA, 70 мл/мин, получая при этом четыре отдельных изомера указанного в заголовке соединения. В настоящее время абсолютная стереохимия каждого изомера не определена со стопроцентной вероятностью. Изомеры обозначают следующим образом. Изомер 53A: быстро элюирующий при колоночной хроматографии с MeOH, быстро элюирующий при СФХ-ВЭЖХ; ЖХ/МС:[(M+1)]+ = 455. Изомер 53B: быстро элюирующий при колоночной хроматографии с MeOH, медленно элюирующий при СФХ-ВЭЖХ; ЖХ/МС:[(M+1)]+ = 455. Изомер 53C: медленно элюирующий при колоночной хроматографии с MeOH, быстро элюирующий при СФХ-ВЭЖХ; ЖХ/МС: [(M+1)]+ = 455. Изомер 53D: медленно элюирующий при колоночной хроматографии с MeOH, медленно элюирующий при СФХ-ВЭЖХ; ЖХ/МС: [(M+1)]+ = 455.
Пример 54
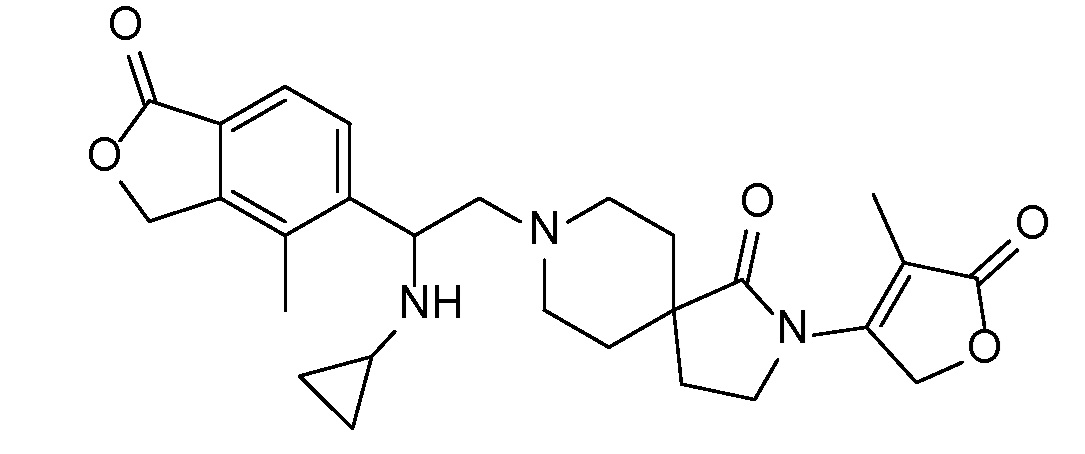
8-(2-(циклопропиламино)-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
8-(2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)-2-оксоэтил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (I-17) (2 г, 7,99 ммоль), 5-(2-бромaцетил)-4-метил-3H-изобензофуран-1-он (I-4B, способ 2, стадия F) (2,150 г, 7,99 ммоль) и N-этил-N-изопропилпропан-2-амин (3,48 мл, 19,98 ммоль) в ДХМ (50 мл) перемешивали в течение ночи при комнатной температуре. Смесь промывали насыщенным водным раствором хлорида аммония и насыщенным раствором соли, сушили над MgSО4 и концентрировали при пониженном давлении. Неочищенный материал применяли на следующей стадии без дополнительной очистки. 1H-ЯМР (500 МГц, CDCl3): δ м.д. 7,83 (1H, д, J=7,8 Гц), 7,79 (1H, д, J=7,8 Гц), 5,34 (2H, с), 5,32 (2H, с), 5,26 (2H, с), 4,03 (2H, т, J=7,0 Гц), 3,71 (1H, м), 3,15 (1H, м), 2,94 (2H, м), 2,47 (2H, т, J=7,0 Гц), 2,42 (3H, с), 2,17 (2H, м), 2,07 (3H, с), 2,02 (2H, м), 1,58 (2H, м). ЖХМС 438,9 (M+1).
Стадия B
8-(2-(циклопропиламино)-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К смеси 8-(2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)-2-оксоэтил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (25 мг, 0,057 ммоль) и циклопропанамина (6,5 мг, 0,114 ммоль) в безводной смеси 5% HOAc/ТГФ (1 мл) добавляли NaCNBH3 (18 мг, 0,28 ммоль). Реакционную смесь герметизировали и перемешивали при встряхивании при температуре окружающей среды в течение 16 часов. ЖХМС-анализ показывал, что образовался требуемый продукт. Реакционную смесь гасили водой (0,5 мл) и выпаривали растворитель при пониженном давлении. Остаток растворяли в ДМСО (1,5 мл) и фильтровали. Неочищенный продукт очищали с применением ВЭЖХ с обращенной фазой (ацетонитрил с 0,1% TFA: вода с 0,1% TFA от 10% до 60%), получая при этом указанное в заголовке соединение. ЖХ-МС (IE, m/z): 480 [M+1]+.
Примеры в приведенной ниже таблице 3 получали по способу, аналогичному получению упомянутого выше 8-(2-(циклопропиламино)-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (пример 54, стадия B), исходя из 8-(2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)-2-оксоэтил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (пример 54, стадия B) и указанных аминов.
|
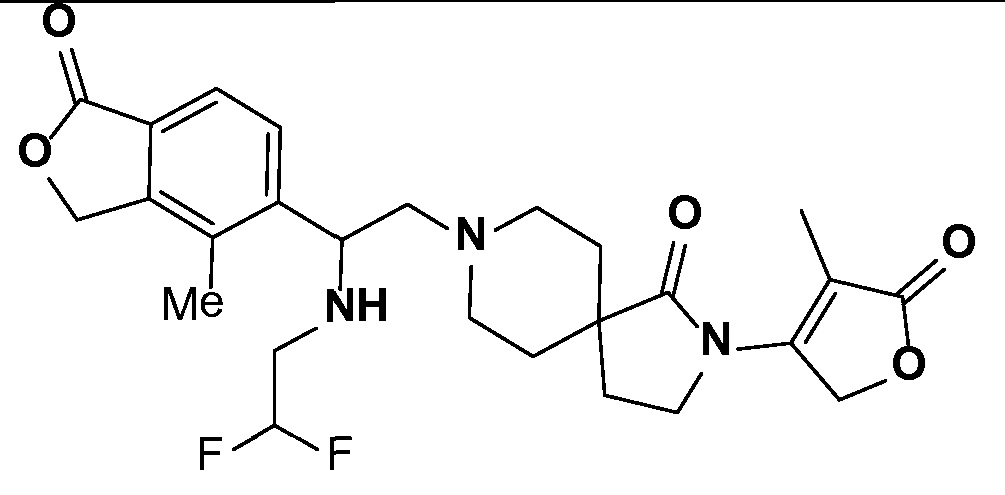
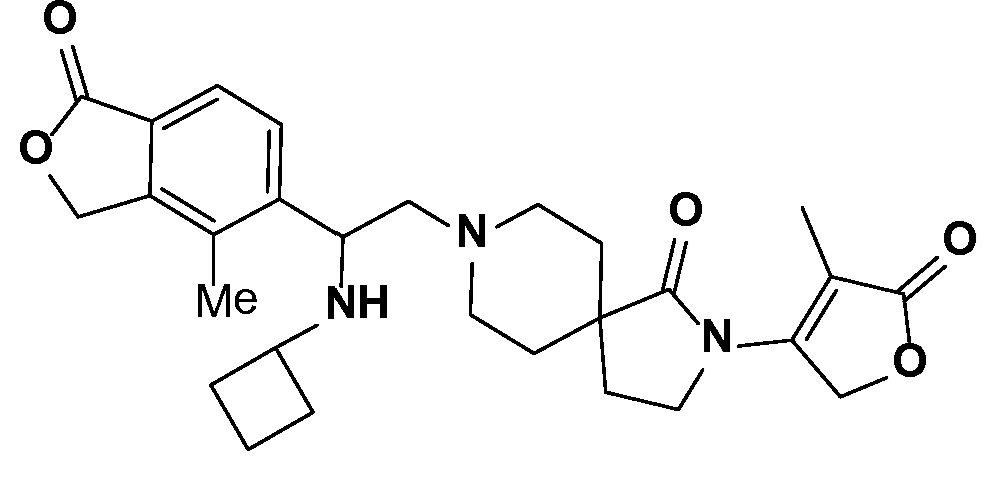
|
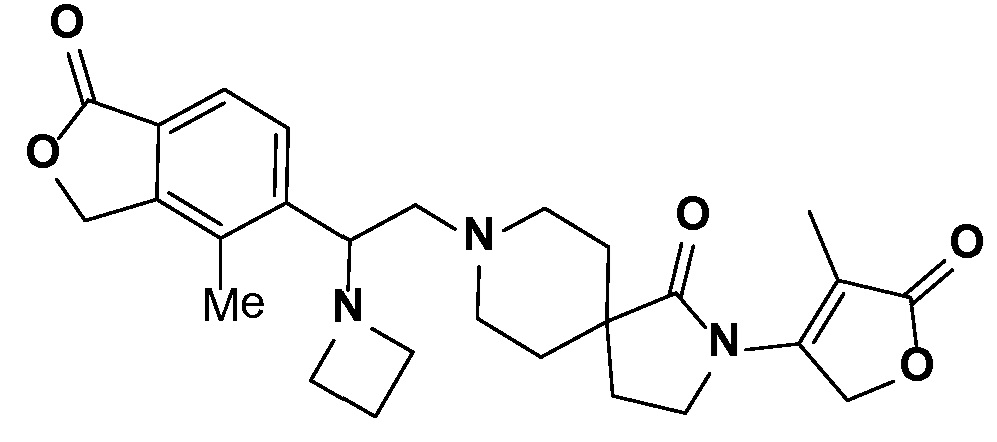
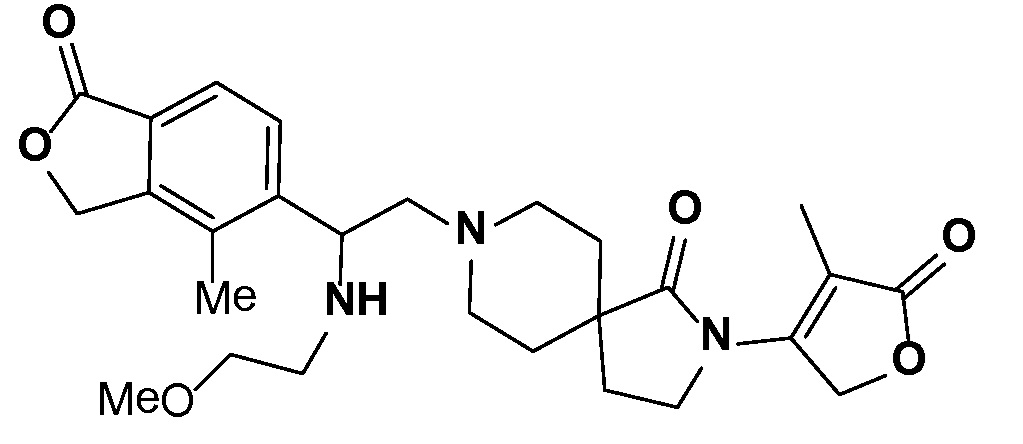
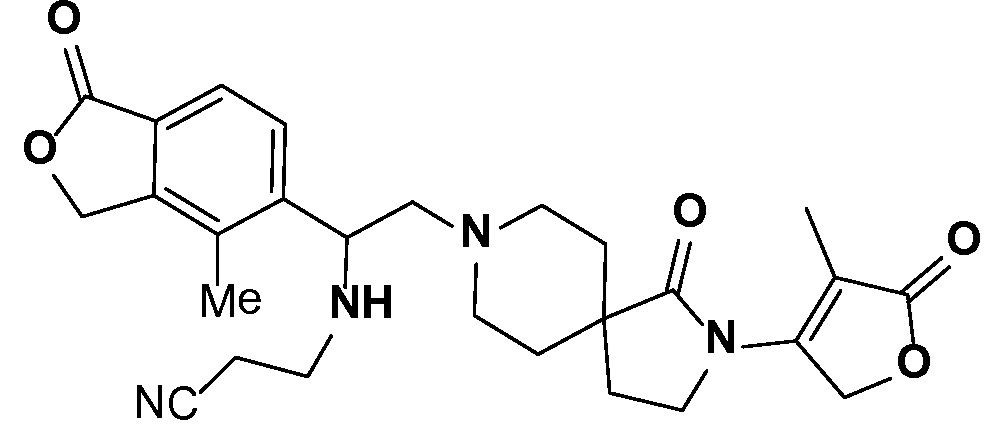
Пример 60
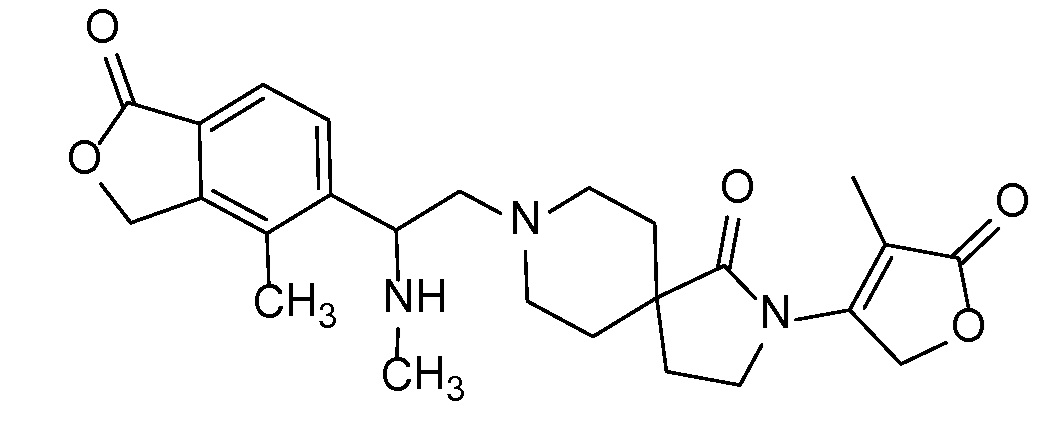
8-[2-(метиламино)-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К смеси 8-(2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)-2-оксоэтил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (25 мг, 0,057 ммоль) и метиламина (4 мг, 0,114 ммоль) в безводном ТГФ добавляли Ti(О-iPr)4 (35 мкл, 0,114 ммоль). Реакционную смесь герметизировали и перемешивали при встряхивании при температуре окружающей среды в течение 16 часов. К полученной реакционной смеси добавляли EtOH (200 градусов крепости (proof), 0,5 мл) с последующим добавлением порциями NaBH4 (11 мг, 0,171 ммоль) в течение 30 мин. Реакционную смесь перемешивали при встряхивании в течение 3 часов и гасили водой (0,5 мл). Реакционную смесь распределяли между EtOAc (4 мл ×2) и аммиаком (аммонием) (2 Н, 1,5 мл). Органическую фазу объединяли и выпаривали при пониженном давлении. Остаток растворяли в ДМСО (1,5 мл) и фильтровали. Неочищенный продукт очищали с применением ВЭЖХ с обращенной фазой (ацетонитрил со смесью 0,1% TFA: вода с 0,1% TFA, от 10% до 60%), получая при этом указанное в заголовке соединение. ЖХ-МС (IE, m/z): 454 [M+1]+.
Пример 61
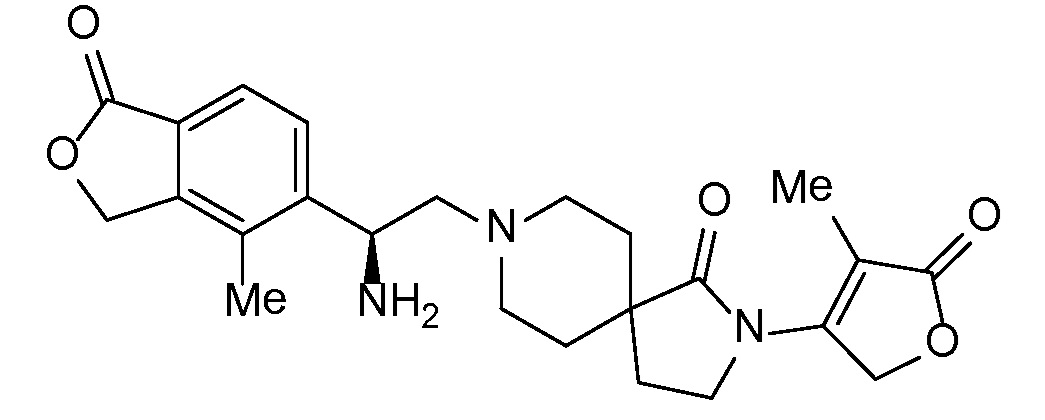
(S)-8-(2-амино-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
(S)-8-(2-азидо-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К суспензии 8-[(2R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (пример 7, 300 мг, 0,68 ммоль) и дифенилфосфорилазида (578 мг, 1,36 ммоль) в смешанном растворителе, состоящем из толуола и дихлорметана (об.:об. 10:1; 11 мл) добавляли DBU (310 мг, 1,36 ммоль). Реакционную смесь перемешивали при 35°C в течение 20 часов. Растворитель удаляли выпариванием, и остаток очищали колоночной флэш-хроматографией (градиент 0-100% этилацетата в петролейном эфире), получая при этом указанное в заголовке соединение. 1H ЯМР (400 МГц, CDCl3): δ 7,32 (д, J=8,0 Гц, 1H), 7,50 (д, J=8,0 Гц, 1H), 5,20 (с, 2H), 5,18 (с, 2H), 4,94 (дд, J=4,0 Гц, 9,2 Гц, 1H), 3,94 (т, J=6,8 Гц, 2H), 2,88-2,85 (м, 2H), 2,72-2,67 (м, 1H), 2,54-2,50 (м, 1H), 2,42-2,39 (м, 1H), 2,33-2,27 (м, 4H), 2,04 (т, J=6,8 Гц, 2H), 1,95 (с, 3H), 1,94-1,87 (м, 2H), 1,51-1,47 (м, 2H). MS-ESI (m/z): 466 (M+1)+.
Стадия B
(S)-8-(2-амино-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору (S)-8-(2-азидо-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (200 мг, 0,43 ммоль) в смешанном растворителе, состоящем из тетрагидрофурана и воды (об.:об.; 12:1; 20 мл) добавляли трифенилфосфин (225 мг, 0,86 ммоль) в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Растворитель удаляли путем выпаривания и очищали остаток препаративной ТСХ (дихлорметан:метанол=10:1), получая при этом указанное в заголовке соединение в виде твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,86 (д, J=7,6 Гц, 1H), δ 7,53 (д, J=7,6 Гц, 1H), 5,24 (с, 2H), 5,22 (с, 2H), 4,50 (дд, J=4,0 Гц, 8,8 Гц, 1H), 3,98 (т, J=6,8 Гц, 2H), 3,04-3,01 (м, 1H), 2,81-2,78 (м, 1H), 2,40-2,34 (м, 2H), 2,30 (с, 3H), 2,18-2,08 (м, 4H), 2,01 (с, 3H), 2,00-1,95 (м, 2H), 1,51-1,47 (м, 2H). MS-ESI (m/z): 440 (M+1)+.
Пример 62
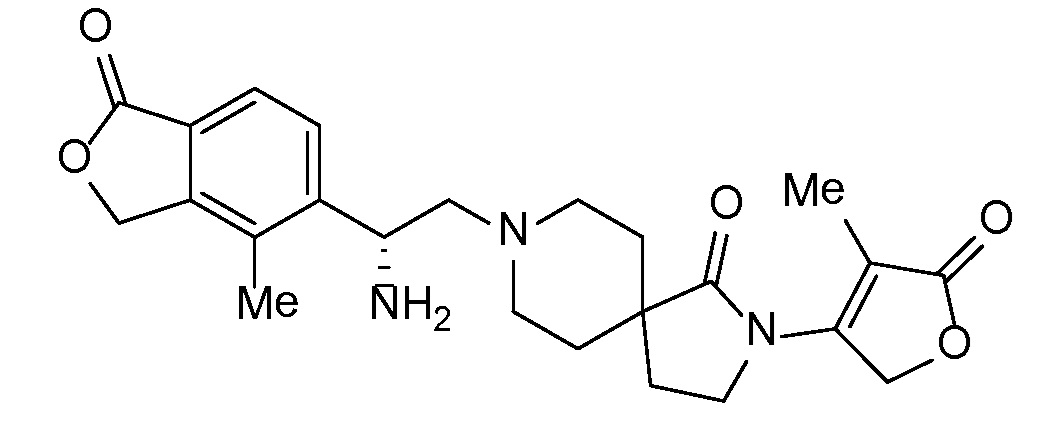
(R)-8-(2-амино-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
(R)-8-(2-азидо-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К суспензии 8-[(2S)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (пример 8, 500 мг, 1,13 ммоль) и дифенилфосфорилазида (643 мг, 1.36 ммоль) в смешанном растворителе из толуола и дихлорметана (об.:об.; 10:1; 11 мл) добавляли DBU (343 мг, 2,26 ммоль). Реакционную смесь перемешивали при 35°C в течение 20 часов. Растворитель удаляли выпариванием, и остаток очищали колоночной флэш-хроматографией (0-100% этилацетата в петролейном эфире), получая при этом указанное в заголовке соединение. 1H ЯМР (400 МГц, CDCl3): δ 7,78 (д, J=8,0 Гц, 1H), 7,55 (д, J=8,0 Гц, 1H), 5,26 (с, 2H), 5,23 (с, 2H), 4,99 (дд, J=4,0 Гц, 9,2 Гц, 1H), 4,00 (т, J=6,8 Гц, 2H), 2,95-2,87 (м, 2H), 2,78-2,73 (м, 1H), 2,60-2,57 (м, 1H), 2,48-2,43 (м, 1H), 2,33 (с, 3H), 2,10 (т, J=6,8 Гц, 2H), 2,03-1,86 (м, 6H), 1,51-1,48 (м, 2H). MS-ESI (m/z): 466 (M+1)+.
Стадия C
(R)-8-(2-амино-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору (R)-8-(2-азидо-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (250 мг, 0,54 ммоль) в смешанном растворителе из тетрагидрофурана и воды (об.:об.; 12:1; 20 мл) добавляли трифенилфосфин (283 мг, 1,08 ммоль) в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Растворитель удаляли выпариванием, и остаток очищали препаративной ТСХ (дихлорметан:метанол=10:1), получая при этом указанное в заголовке соединение. 1H ЯМР (400 МГц, Метанол-d4): δ 7,80 (д, J=8,0 Гц, 1H), δ 7,66 (д, J=8,0 Гц, 1H), 5,40 (с, 2H), 5,24 (с, 2H), 4,73 (дд, J=4,0 Гц, 8,8 Гц, 1H), 4,09 (т, J=6,8 Гц, 2H), 3,30-3,21 (м, 1H), 2,98-2,91 (м, 1H), 2,87-2,81 (м, 1H), 2,70-2,66 (м, 1H), 2,55-2,47 (м, 2H), 2,40 (с, 3H), 2,18-2,07 (м, 4H), 2,00 (с, 3H), 1,74-1,68 (м, 2H). MS-ESI (m/z): 440 (M+1)+.
Пример 63
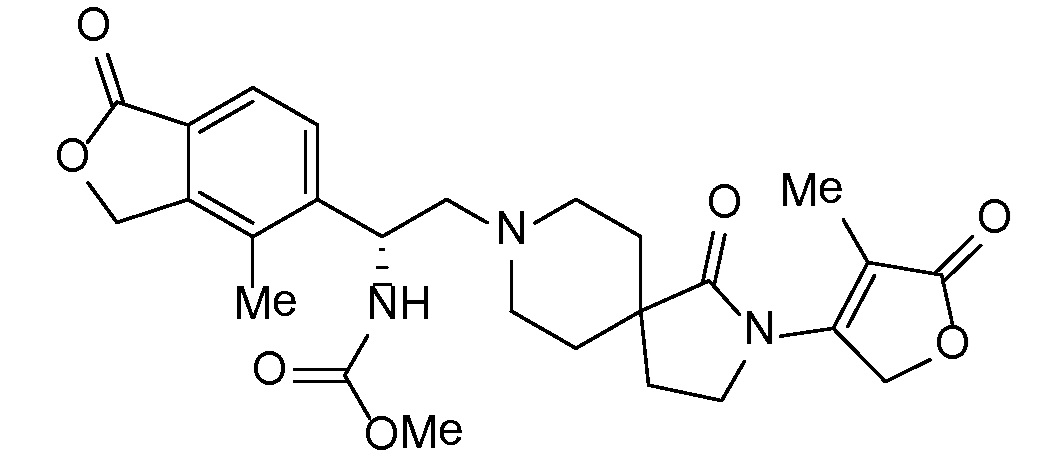
метил{(1R)-1-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)-2-[2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]дец-8-ил]этил}карбамат
К раствору (R)-8-(2-амино-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (пример 62) (35 мг, 0,08 ммоль) и триэтиламина (17 мкл, 0,12 ммоль) в дихлорметане (2 мл) добавляли метилхлорформиат (11 мг, 0,12 ммоль) при 0°C в атмосфере азота. Реакционной смеси давали нагреться до комнатной температуры при перемешивании в течение 2 часов. ТСХ-анализ показывал, что оставалась половина исходного материала. Реакционную смесь повторно охлаждали до 0°C и добавляли к ней триэтиламин (17 мкл, 0,12 ммоль) с последующим добавлением метилхлорформиата (11 мг, 0,12 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение еще 2 часов. ТСХ-анализ показывал полное завершение реакции. Полученную смесь концентрировали в вакууме. Остаток помещали в небольшое количество ДХМ и очищали препаративной ТСХ (дихлорметан:метанол=15:1), получая при этом указанное в заголовке соединение. МС-ESI (m/z): 498 (M+1)+.
Пример 64
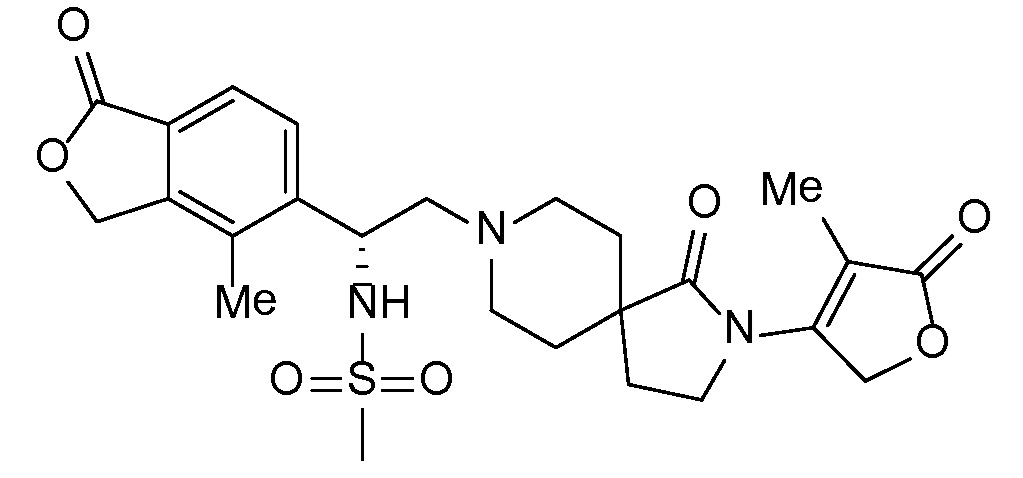
(R)-N-(1-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)-2-(2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-1-оксо-2,8-диазаспиро[4.5]декан-8-ил)этил)метансульфонамид;
К раствору (R)-8-(2-амино-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (30 мг, 0,068 ммоль) и триэтиламина (29 мкл, 0,20 ммоль) в ДХМ (2 мл) в атмосфере азота при 0°C добавляли метансульфонилхлорид (5,3 мкм, 0,068 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 3 часов. ТСХ-анализ показывал полное завершение реакции. При выпаривании растворителя получали остаток, который помещали в небольшое количество ДХМ и очищали препаративной ТСХ (дихлорметан:метанол=10:1), получая при этом указанное в заголовке соединение. 1H ЯМР (400 МГц, CDCl3): δ 7,63 (дд, J=8,0 Гц, J=8,8 Гц, 2H), 5,30 (с, 2H), 5,15 (с, 2H), 4,95-4,92 (м, 1H), 4,00 (т, J=7,2 Гц, 2H), 2,94-2,91 (м, 1H), 2,82-2,78 (м, 1H), 2,72 (с, 3H), 2,65-2,59 (м, 1H), 2,42-2,39 (м, 1H), 2,38-2,24 (м, 4H), 2,21-2,18 (м, 1H), 2,07 (т, J=7,2 Гц, 2H), 1,92 (с, 3H), 1,86-1,72 (м, 2H), 1,52-1,47 (м, 2H). MS-ESI (m/z): 518 (M+1)+.
Пример 65
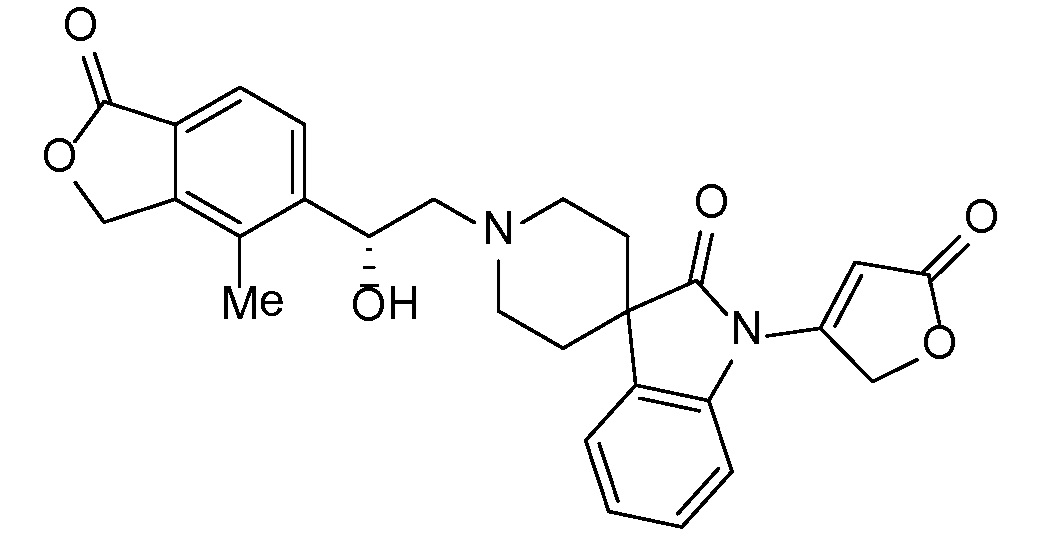
1'-[(2R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-оксо-1-(5-оксо-2,5-дигидрофуран-3-ил)-1,2-дигидроспиро[индол-3,4'-пиперидиний]
Стадия A
(R)-1'-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)спиро[индолин-3,4'-пиперидин]-2-он
Имеющийся в продаже гидрохлорид спиро[индолин-3,4'-пиперидин]-2-она (500 мг, 2,10 ммоль) объединяли с 4-метил-5-[(2S)-оксиран-2-ил]-2-бензофуран-1(3H)-оном (I-4A) (398 мг, 2,10 ммоль) и DIEA (439 мкл, 2,51 ммоль) в этаноле (7 мл) и нагревали при 80°C в течение 3 часов. Реакционную смесь концентрировали и очищали ЖХСД путем элюирования сначала смесью 30% этилацетат/гексаны и затем смесью 10% метанол/ДХМ, получая при этом указанное в заголовке соединение, которое представляло собой смесь региоизомеров. МС-ESI (m/z): 393 (M+1)+;
Стадия B
1'-[(2R)-2-гидрокси-2-(4-метил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)этил]-2-оксо-1-(5-оксо-2,5-дигидрофуран-3-ил)-1,2-дигидроспиро[индол-3,4'-пиперидиний]
(R)-1'-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)спиро[индолин-3,4'-пиперидин]-2-он (502 мг, 1,28 ммоль), 4-бромфуран-2-он (250 мг, 1,53 ммоль), карбонат цезия (625 мг, 1,9 2 ммоль), Pd(dba)2 (37 мг, 0,064 ммоль), Ксантфос (111 мг, 0,192 ммоль) объединяли в ампуле для микроволновой печи в 5 мл толуола. Суспензию продували азотом и нагревали при 90°C в течение ночи. Реакционную смесь фильтровали через CELITE®, промывали этилацетатом и концентрировали фильтрат. Неочищенный продукт очищали ЖХСД с применением сначала градиента этилацетат/гексаны и затем смеси 10% метанол/ДХМ для элюирования продукта. Чтобы отделить требуемый продукт от примесей и региоизомеров, остаток дополнительно очищали препаративной ТСХ (смесь 10% метанол/ДХМ) и СФХ-ВЭЖХ с применением хиральной колонки Chiralcel OD. МС-ESI (m/z): 475 (M+1)+.
Следующие соединения в таблице 4 получали способом, аналогичным способам по примерам 5-9, исходя из указанных промежуточных пиперидиновых и эпоксидных соединений, полученных, как описано выше. В примерах, где получали смеси изомеров, использовали хиральную СФХ-ВЭЖХ для разделения изомеров с применением указанной колонки для ВЭЖХ.
|
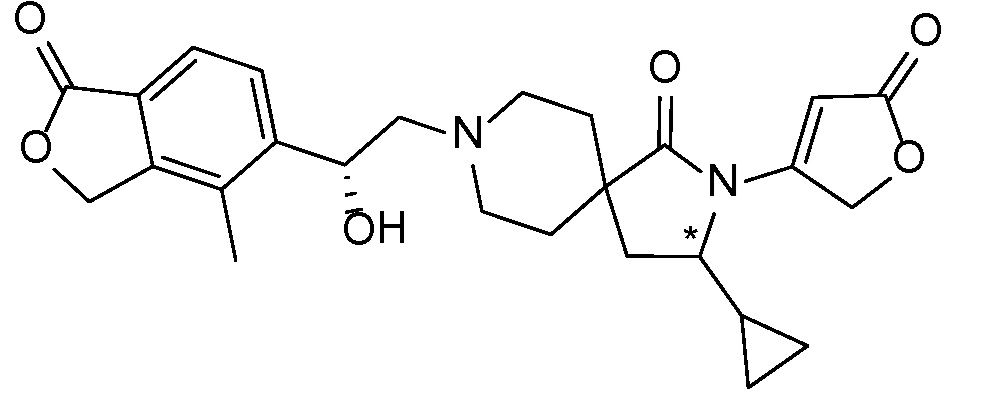
|
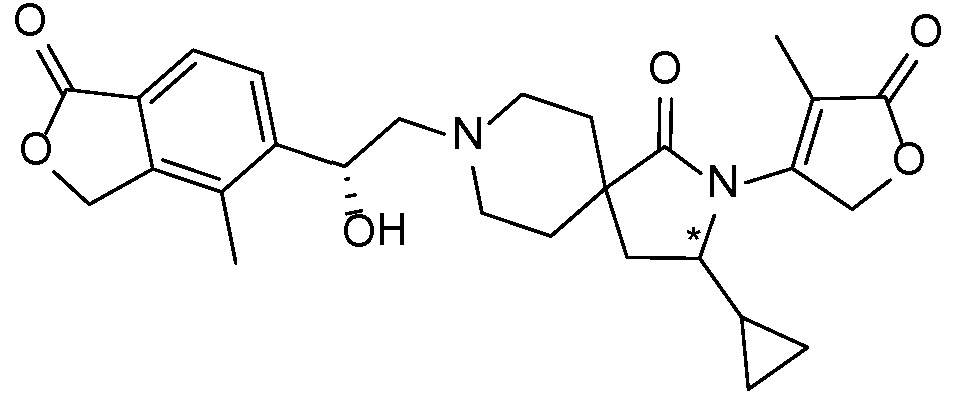
|
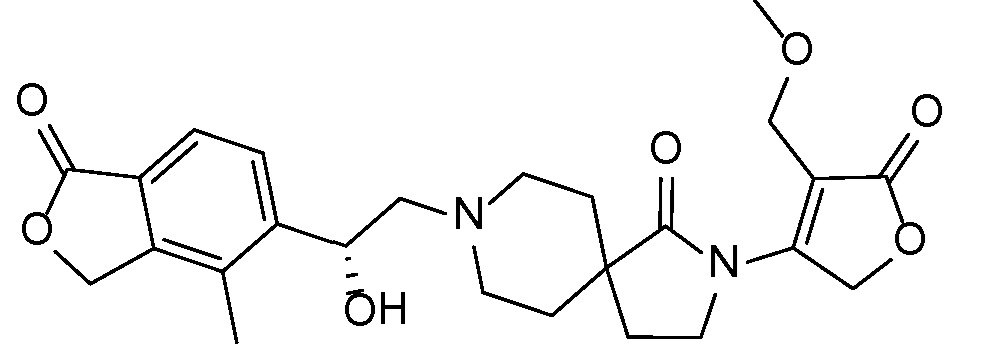
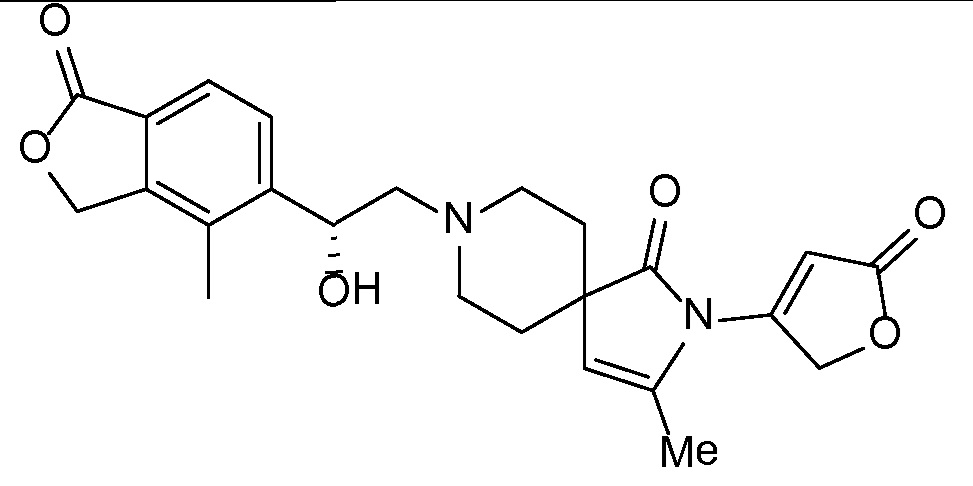
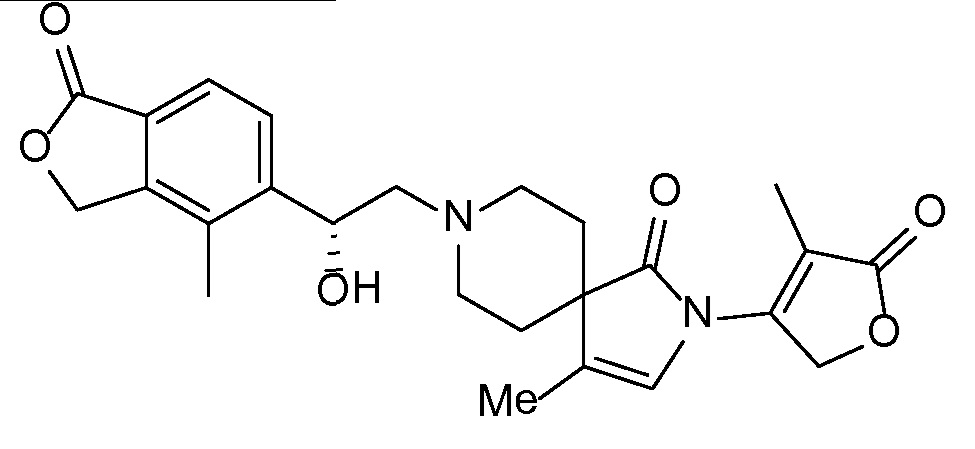
|
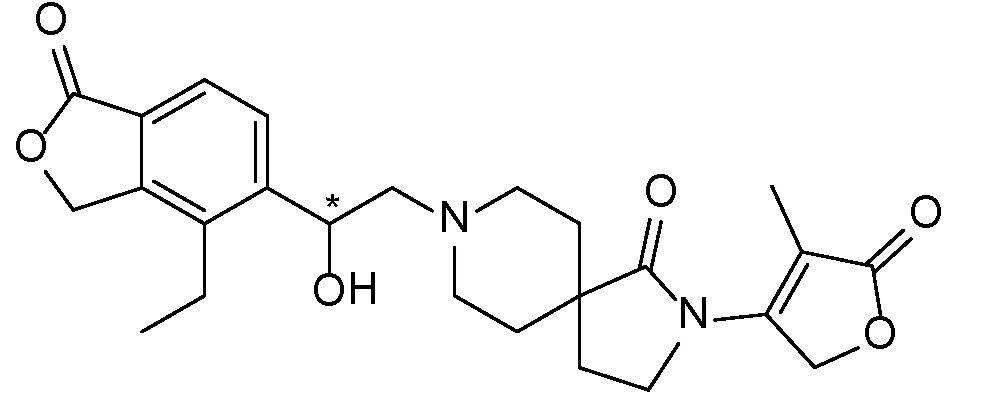
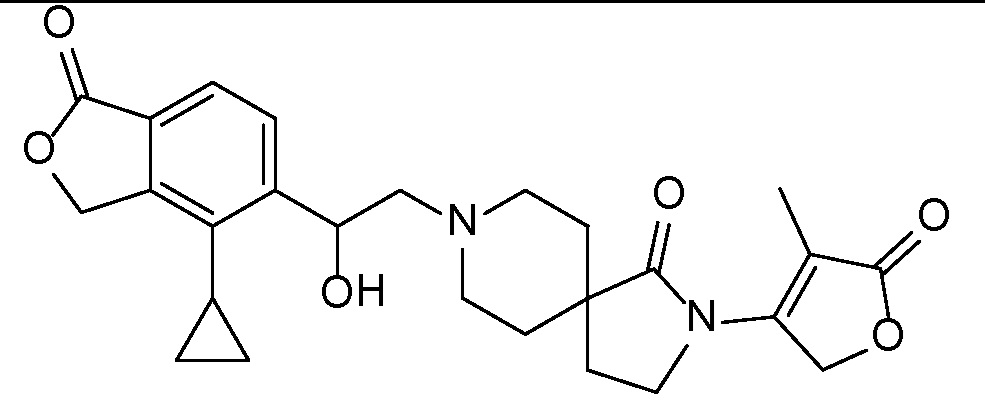
|
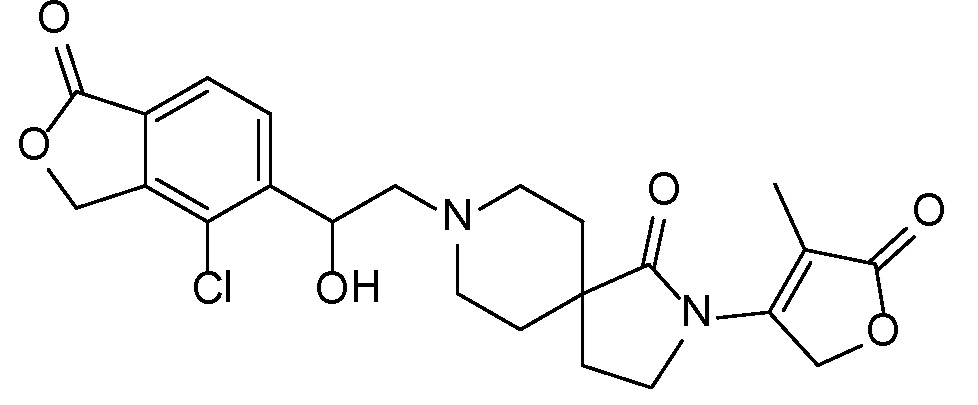
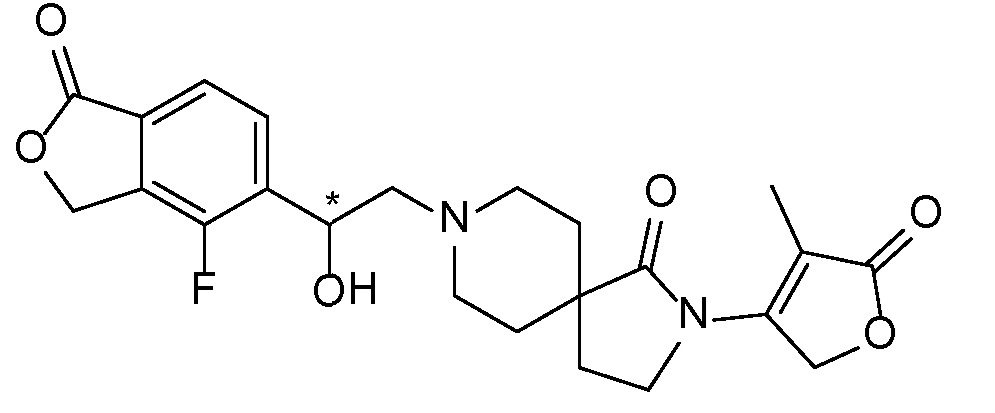
|
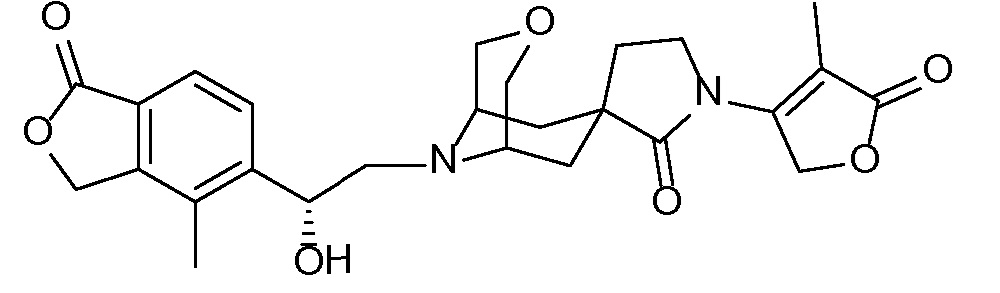
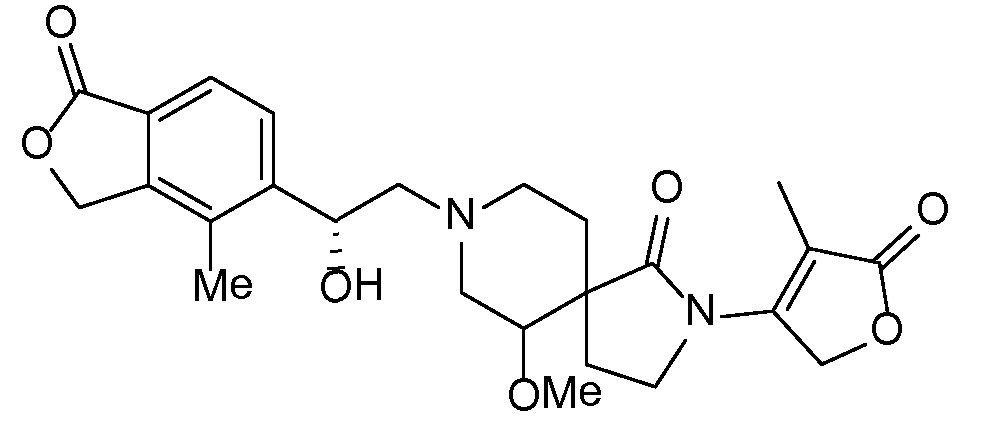
Пример 84А и 84В (отдельные выделенные изомеры)
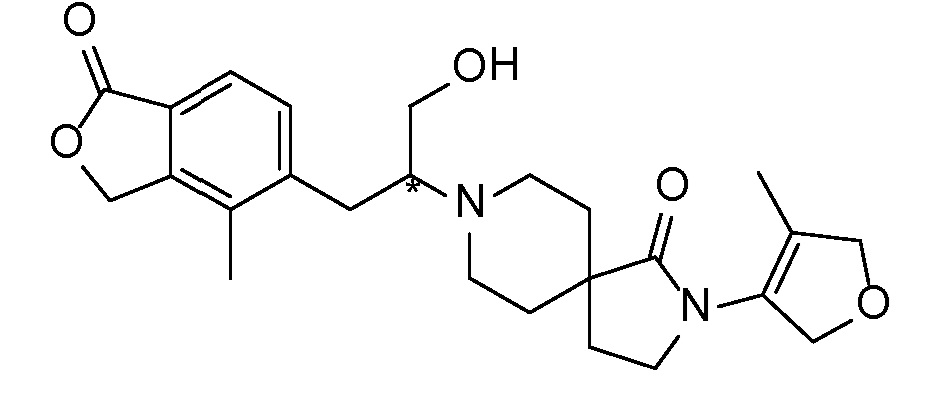
(S)-8-(1-гидрокси-3-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он и
(R)-8-(1-гидрокси-3-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
5-(2,3-дигидроксипропил)-4-метилизобензофуран-1(3H)-он
К раствору 4-метил-5-проп-2-ен-1-ил-2-бензофуран-1(3H)-она (см. стадия A для I-3) (2,00 г, 10,6 ммоль) в ацетоне (30 мл) и воде (10 мл) добавляли OsО4 (0,27 г, 1,06 ммоль) и NMO (6,70 г, 11,7 ммоль) и перемешивали раствор при 25°C в течение 18 часов. Реакционную смесь разбавляли водой (30 мл) и водный слой экстрагировали CH2Cl2 (3×40 мл). Органические слои объединяли, промывали насыщенным раствором соли (2×40 мл), сушили (MgSО4), фильтровали и концентрировали. Остаток очищали колоночной флэш-хроматографией (0-10% метанола в дихлорметане), получая при этом указанное в заголовке соединение.
Стадия B
5-(3-((трет-бутилдиметилсилил)окси)-2-гидроксипропил)-4-метилизобензофуран-1(3H)-он
К раствору 5-(2,3-дигидроксипропил)-4-метилизобензофуран-1(3H)-она (2,00 г, 9,0 ммоль) в сухом ДМФА (20 мл) добавляли имидазол (1,20 г, 18,0 ммоль) и TBSCl (1,50 г, 9,9 ммоль). После перемешивания при 25°C в течение 2,5 часов реакционную смесь разбавляли CH2Cl2 (50 мл) и промывали H2O (3×20 мл) и насыщенным раствором NaHCО3 (3×20 мл). Объединенные водные слои пять раз экстрагировали CH2Cl2 (20 мл). Объединенные органические слои промывали насыщенным раствором соли (3×30 мл), сушили над Na2SО4, фильтровали и концентрировали. Остаток очищали колоночной флэш-хроматографией (0-60% этилацетата в петролейном эфире), получая при этом указанное в заголовке соединение.
Стадия C
5-(3-((трет-бутилдиметилсилил)окси)-2-оксопропил)-4-метилизобензофуран-1(3H)-он
К раствору 5-(3-((трет-бутилдиметилсилил)окси)-2-гидроксипропил)-4-метилизобензофуран-1(3H)-она (1,00 г, 3,0 ммоль) в CH2Cl2 (30 мл) добавляли периодинан Десса-Мартина (6,30 г, 15,0 ммоль). После перемешивания при 25°C в течение 12 часов реакционную смесь фильтровали через тонкий рыхлый слой SiO2 с CH2Cl2 (100 мл) в качестве элюирующего растворителя. После концентрации остаток очищали колоночной флэш-хроматографией (0-60% этилацетата в петролейном эфире), получая при этом указанное в заголовке соединение.
Стадия D
8-(1-((трет-бутилдиметилсилил)окси)-3-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору 5-(3-((трет-бутилдиметилсилил)окси)-2-оксопропил)-4-метилизобензофуран-1(3H)-она (0,50 г, 1,5 ммоль) в метаноле (20 мл) добавляли 2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (I-17) (0,45 г, 1,8 ммоль) и изопропоксид титана (IV) (2,10 г, 7,5 ммоль). После перемешивания при комнатной температуре в течение 48 часов добавляли цианоборогидрид натрия (190 мг, 3.0 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 20 часов. Реакционную смесь гасили водой (10 мл), полученный неорганический осадок отфильтровывали и промывали метанолом (50 мл). Затем фильтрат концентрировали и неочищенный продукт растворяли в этилацетате, фильтровали, чтобы удалить остающиеся неорганические твердые вещества, и концентрировали. Остаток очищали колоночной флэш-хроматографией (0-10% метанола в дихлорметане), получая при этом указанное в заголовке соединение. ЖХ-МС (ESI, m/z): 569 [M+1]+.
Стадия E
8-(1-гидрокси-3-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору 8-(1-((трет-бутилдиметилсилил)окси)-3-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-она (0,10 г, 0,18 ммоль) в ДХМ (3 мл) при 0°C добавляли трифторуксусную кислоту (3 мл). После перемешивания при комнатной температуре в течение 24 часов растворитель удаляли при пониженном давлении и остаток очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение, которое выделяли хиральной СФХ-хроматографией в виде двух отдельных энантиомеров: изомер A (быстро элюирующий) и изомер B (медленно элюирующий). Колонка: Chiralpak AD-3, 250×30 мм I.D., 20 мкм; подвижная фаза: сверхкритический CО2/EtOH (0,2% NH3.Н2О)=45/55; Скорость потока: 80 мл/мин. Для обоих изомеров: ЖХ-МС (ESI, m/z): 455 [M+1]+.
Пример 85A и 85B (отдельные выделенные изомеры)
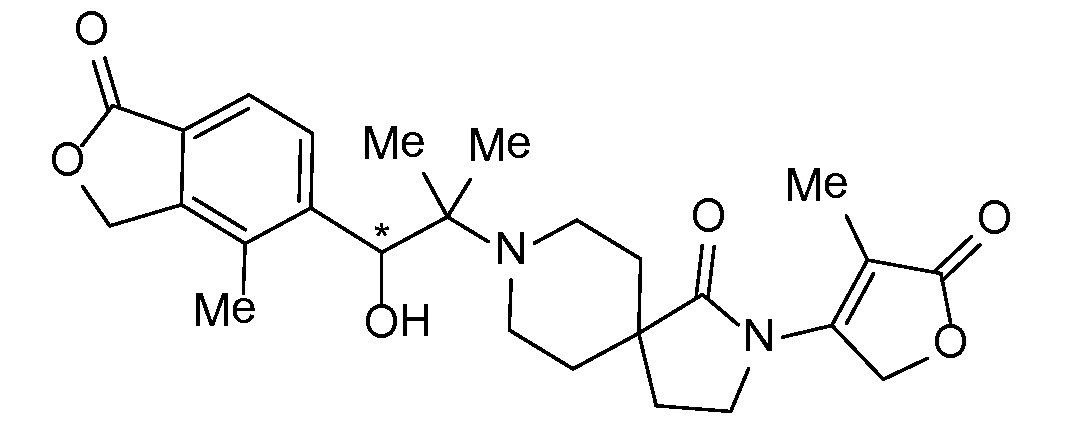
(S)-8-(1-гидрокси-2-метил-1-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он и
(R)-8-(1-гидрокси-2-метил-1-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
Стадия A
2,8-диазаспиро[4.5]декан-1-он
К раствору трет-бутил-1-оксо-2,8-диазаспиро[4.5]декан-8-карбоксилата (I-11) (5,0 г, 19,7 ммоль) в метиленхлориде (10 мл) добавляли трифторуксусную кислоту (15,2 мл, 197 ммоль) и перемешивали полученный раствор при комнатной температуре в течение 1 часа. После выпаривания летучих веществ остаток ощелачивали на ионообменной колонке, промывали метанолом с последующим промыванием 1Н раствора аммиака в метаноле, получая при этом 2,8-диазаспиро[4.5]декан-1-он. ЖХ/МС: (M+1)+: 155,11.
Стадия B
Этил-2-метил-2-(1-оксо-2,8-диазаспиро[4.5]декан-8-ил)пропаноат
Смесь 2,8-диазаспиро[4.5]декан-1-она (3,03 г, 19,65 ммоль), триэтиламина (5,48 мл, 39,3 ммоль) и этил-2-бром-2-метилпропаноата (5,77 мл, 39,3 ммоль) нагревали при 80°C в течение ночи. Реакционную смесь распределяли между метиленхлоридом (200 мл) и насыщенным раствором бикарбоната натрия, щелочную фазу экстрагировали метиленхлоридом (3×100 мл). Объединенную органическую фазу сушили над сульфатом магния, концентрировали и очищали остаток на силикагеле с применением смеси метанол/метиленхлорид, получая при этом указанное в заголовке соединение. ЖХ/МС (M+1)+: 269,4.
Стадия C
2-метил-2-(1-оксо-2,8-диазаспиро[4.5]декан-8-ил)пропанaль
К раствору этил-2-метил-2-(1-оксо-2,8-диазаспиро[4.5]декан-8-ил)пропаноата (3,77 г, 14,1 ммоль) в толуоле (100 мл) при -78°C по каплям добавляли DIBAL-H (45,0 мл, 45,0 ммоль). Спустя 2 часа реакционную смесь гасили путем добавления по каплям метанола (10 мл), затем нагревали до комнатной температуры, добавляли 30 мл насыщенного раствора сульфата натрия и энергично перемешивали смесь при комнатной температуре в течение 1 часа. После фильтрования фильтрат и фильтрационный осадок распределяли между ДХМ и насыщенным раствором бикарбоната натрия, щелочную фазу трижды экстрагировали ДХМ, объединенную органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС (M+1+18)+: 243,3; (M+1+32)+: 257,4.
Стадия D
4-метил-5-(триметилстаннил)изобензофуран-1(3H)-он
Раствор 4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил трифторметансульфоната (5,0 г, 16,9 ммоль), хлорида лития (4,29 г, 101 ммоль) и тетракис(трифенилфосфин)палладия(0) (1,951 г, 1,688 ммоль) в диоксане (30 мл) дегазировали азотом в течение 0,5 часа в герметично закрывающейся трубке перед добавлением гексаметилдиолова (5,25 мл, 25,3 ммоль), трубку герметизировали и нагревали при 100°C в течение ночи. После фильтрования через CELITE® фильтрат концентрировали и остаток очищали на колонке с силикагелем с применением смеси этилацетат/гексан, получая при этом 4-метил-5-(триметилстаннил)изобензофуран-1(3H)-он. ЖХ/МС: (M+1)+: 308,99; 310,87; 312,78.
Стадия E
5-бром-4-метилизобензофуран-1(3H)-он:
К раствору 4-метил-5-(триметилстаннил)изобензофуран-1(3H)-она (4,37 г, 14,05 ммоль) в ДХМ (20 мл) при 0°C добавляли бром (0,796 мл, 15,46 ммоль). Реакционную смесь перемешивали при 0°C в течение 0,5 часа. Добавляли насыщенный раствор тиосульфата и экстрагировали смесь метиленхлоридом (2×100 мл), объединенную органическую фазу сушили над сульфатом натрия и концентрировали, получая при этом 5-бром-4-метилизобензофуран-1(3H)-он. ЖХ/МС: (M+1)+: 226,89; 228,89.
Стадия F
5-бром-4-метил-1,3-дигидроизобензофуран-1-ол
К раствору 5-бром-4-метилизобензофуран-1(3H)-она (3,45 г, 15,19 ммоль) в толуоле (100 мл) при -78°C по каплям добавляли DIBAL-H (21,27 мл, 21,27 ммоль). После перемешивания при -78°C в течение 2 часов реакционную смесь гасили метанолом при -78°C, затем нагревали до комнатной температуры, добавляли 20 мл насыщенного раствора сульфата натрия и энергично перемешивали смесь при комнатной температуре в течение 30 мин. Затем смесь фильтровали и промывали этилацетатом. Фильтрат промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия, затем концентрировали, получая при этом указанное в заголовке соединение. ЖХ/МС: (M-17)+: 210,92; 212,91.
Стадия G
((5-бром-4-метил-1,3-дигидроизобензофуран-1-ил)окси)(трет-бутил)диметилсилан
Раствор TBS-Cl (3,63 г, 24,10 ммоль) в метиленхлориде (15 мл) при 0°C добавляли к раствору 5-бром-4-метил-1,3-дигидроизобензофуран-1-ола (2,76 г, 12,1 ммоль) и имидазола (1,72 г, 25,3 ммоль) в метиленхлориде (80 мл); полученный раствор перемешивали при комнатной температуре в течение ночи. Смесь распределяли между ДХМ и водой, водную фазу экстрагировали метиленхлоридом и объединенную органическую фазу сушили над сульфатом натрия, затем концентрировали, и остаток очищали на колонке с силикагелем с применением смеси этилацетат/гексан, получая при этом указанное в заголовке соединение. ЖХ/МС: (M-131)+: 210,91; 212,91.
Стадия H
8-(1(1-((трет-бутилдиметилсилил)окси)-4-метил-1,3-дигидроизобензофуран-5-ил)-1-гидрокси-2-метилпропан-2-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору ((5-бром-4-метил-1,3-дигидроизобензофуран-1-ил)окси)(трет-бутил)диметилсилана (3,9 г, 11,36 ммоль) в тетрагидрофуране (50 мл) при -78°C добавляли N-бутиллитий (5,00 мл, 12,50 ммоль). Спустя 15 минут добавляли 2-метил-2-(1-оксо-2,8-диазаспиро[4.5]декан-8-ил)пропаналь (0,892 г, 3,98 ммоль) в виде одной порции. Полученный раствор перемешивали при -78°C в течение 3,5 часов перед гашением раствора путем добавления метанола (6 мл) и насыщенного раствора бикарбоната натрия (100 мл). Смесь экстрагировали смесью 30% изопропанол/метиленхлорид (3×100 мл). Объединенную органическую фазу сушили над сульфатом натрия, концентрировали и остаток очищали на силикагеле с применением метанола/метиленхлорида, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 489,15.
Стадия I
8-(1-(1-((трет-бутилдиметилсилил)окси)-4-метил-1,3-дигидроизобензофуран-5-ил)-1-гидрокси-2-метилпропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К смеси 8-(1-(1-((трет-бутилдиметилсилил)окси)-4-метил-1,3- дигидроизобензофуран-5-ил)-1-гидрокси-2-метилпропан-2-ил)-2,8-диазаспиро[4.5]декан-1-она (1,35 г, 2,76 ммоль), 4-метил-5-оксо-2,5-дигидрофуран-3-ил трифторметансульфоната (I-9) (0,884 г, 3,59 ммоль) в толуоле (40 мл) добавляли карбонат калия (0,764 г, 5,52 ммоль), Ксантфос (0,320 г, 0,552 ммоль) и воду (0,149 мл, 8,29 ммоль). Смесь продували азотом в течение 20 минут перед добавлением ацетата палладия(II) (0,062 г, 0,276 ммоль). Полученную смесь нагревали при 70°C в течение ночи. После фильтрования фильтрат концентрировали, и остаток очищали на колонке с силикагелем с применением метанола/метиленхлорида в качестве элюирующих растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1-114)+: 471,22.
Стадия J
8-(1-гидрокси-1-(1-гидрокси-4-метил-1,3-дигидроизобензофуран-5-ил)-2-метилпропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору продукта, полученного на стадии 1 (1,282 г, 2,192 ммоль), в тетрагидрофуране (50 мл) добавляли TBAF (2,63 мл, 2,63 ммоль) при 0°C и перемешивали полученный раствор при 0°C в течение 2 часов. После концентрирования, остаток очищали ТСХ путем элюирования 10% MeOH/ДХМ, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 471,20.
Стадия K
8-(1-гидрокси-2-метил-1-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он
К раствору продукта, полученного на стадии J (340 мг, 0,723 ммоль), в метиленхлориде (30 мл) при 0°C добавляли PCC (311 мг, 1,45 ммоль) и полученный раствор перемешивали при 0°C в течение 2 часов. Реакционную смесь распределяли между метиленхлоридом и насыщенным раствором бикарбоната и экстрагировали щелочную фазу метиленхлоридом (3×50 мл). Объединенную органическую фазу сушили над сульфатом натрия, концентрировали, и остаток очищали ТСХ с применением смеси 10% метанол/метиленхлорид в качестве проявляющей системы растворителей, получая при этом указанное в заголовке соединение. ЖХ/МС: (M+1)+: 469,20.
Стадия L
8-(1-гидрокси-2-метил-1-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)пропан-2-ил)-2-(4-метил-5-оксо-2,5-дигидрофуран-3-ил)-2,8-диазаспиро[4.5]декан-1-он (отдельные выделенные изомеры)
Указанное в заголовке соединение (в виде смеси изомеров) (100 мг, 0,213 ммоль) разделяли с помощью СФХ на колонке OJ, получая при этом два изомера. Изомер A (быстро элюирующий): ЖХ/МС: (M+1)+: 469,25; 1H-ЯМР (500 МГц, CDCl3), δ 7,766-7,289 (м, 2H), 5,287 (с, 2H), 5,261 (с, 2H), 5,139 (с, 1H), 4,072-4,044 (т, J=7,1 Гц, 2H), 3,190-3,088 (м, 2H), 2,545-2,506 (м, 2H), 2,341 (с, 3H), 2,191-2,162 (м, 2H), 2,066-2,037 (м, 2H), 1,695-1,670 (м, 2H), 0,985 (с, 3H), 0,956 (с, 3H). Изомер B (медленно элюирующий). ЖХ/МС: (M+1)+: 469,25. ЖХ/МС: (M+1)+: 469,25, 1H-ЯМР (500 МГц, CDCl3), δ 7,825-7,768 (м, 2H), 5,289 (с, 2H), 5,262 (с, 2H), 5,138 (с, 1H), 4,073-4,044 (т, J=6,9 Гц, 2H), 3,191-3,086 (м, 2H), 2,592-2,502 (м, 2H), 2,341 (с, 3H), 2,191-2,162 (м, 2H), 2,066-2,037 (м, 2H), 1,695-1,670 (м, 2H), 0,985 (с, 3H), 0,956 (с, 3H).
Для каждого из конечных соединений, полученных по примерам, проводили следующий анализ транспорта таллия (Flux Assay) и/или электрофизиологический анализ, если в примере не указано иначе.
Анализ транспорта таллия (Flux Assay)
Условия культивирования клеток: HEK-293-клетки, стабильно экспрессирующие hROMK (hKir1.1), выращивали при 37°C во влажной камере с 10% CO2 в готовых питательных средах: среда Игла, модифицированная по способу Дульбекко, с добавлением неэссенциальных аминокислот, пенициллина/стрептомицина/глутамина, G418 и FBS. Для сбора клеток при достижении конфлюентности >80% из колбы аспирировали среду и промывали клетки 10 мл фосфатного солевого буфера (ФСБ), не содержащего кальция/магния. Добавляли 5 мл 1х трипсина (приготовленного в ФСБ, не содержащем Ca/Mg) в T-225 колбу и возвращали колбу в камеру (37°C/CО2) на 2-3 минуты. Для выделения клеток осторожно похлопывали по стороне колбы рукой. Полностью растирали клетки и затем переносили клетки в 25 мл готовых сред. Центрифугировали при 1500 об./мин в течение 6 минут с последующим повторным суспендированием в готовых питательных средах и определяли концентрацию клеток. При обычном повторном пересеве 4E6-клетки/колба T-225 >80% конфлюентность будет достигаться через 4 дня. В условиях идеального роста и подходящих практических методик культивирования клеток тканей, такая клеточная линия стабильна в течение 40-45 пассажей.
Компоненты набора FluxOR (Invitrogen F10017):
- реагент FluxOR™ (компонент A);
- буфер для анализа FluxOR™ (компонент B), концентрат (10 х);
- концентрат PowerLoad™ (компонент C) - концентрат (100 х);
- пробенецид (компонент D) - лиофилизованный образец хранят при -20°C; водорастворимый, (100 х) после солюбилизации в 1 мл воды; хранят при 4°C;
- буфер FluxOR™, не содержащий хлорида (компонент E) - концентрат (5 х);
- концентрат сульфата калия (K2SО4) (компонент F) - 125 мМ в воде; хранят при 4°C;
- концентрат сульфата таллия (Tl2SО4) (компонент G) - 50 мМ в воде; хранят при 4°C;
-ДМСО (диметилсульфоксид, компонент H)-1 мл (100%).
Получение реагента: рабочие растворы FluxOR
- 1000X реагент FluxOR™: разбавляют содержимое ампулы с компонентом A в 100 мкл ДМСО; хорошо перемешивают; хранят аликвоты 10 мкл при -20°C.
- Буфер для анализа 1X FluxOR™: разбавляют компонент B 10-кратно водой; доводят pH до значения 7,4 с помощью Hepes/NaOH; фильтруют и хранят при 4°C.
- Пробенецид/буфер для анализа: 100 мл буфера для анализа IX FluxOR™; 1 мл растворенного компонента D; хранят при 4°C.
- Загрузочный буфер (на микропланшет): 10 мкл 1000 × реагент FluxOR™; 100 мкл компонента C; 10 мл смеси пробенецид/буфер для анализа.
- Буфер с соединением (на микропланшет): 20 мл пробенецид/буфер для анализа; 0,3 мМ уабаина (10 мМ уабаина в воде можно хранить в склянке из темного стекла/алюминиевой фольге при комнатной температуре); тестируемое соединение.
- Буфер FluxOR™ (1х), не содержащий хлорида: готовят рабочий раствор (1х) в воде; можно хранить при комнатной температуре.
- Стимулирующий буфер (получают при 5х конечной концентрации в буфере IX FluxOR™, не содержащем хлорида): 7,5 мМ сульфата таллия и 0,75 мМ сульфата калия (чтобы получить конечную концентрацию для анализа 3 мМ таллия/0,3 мМ калия). Когда не используют, хранят при 4°C. Если поддерживать стерильность, такой раствор остается пригодным в течение месяцев.
Протокол анализа.
Анализ транспорта таллия через функциональный канал ROMK осуществляли в 384-луночных планшетах с применением оборудования FLIPR-Tetra. HEK-hKir1.1-клетки высевали в микропланшеты, покрытые поли-D-лизином, и выдерживали при 37°C в камере (10% CO2) в течение ночи. В день эксперимента питательные среды заменяли загрузочным буфером с реагентом FluxOR™ и инкубировали, защищая от света, при температуре окружающей среды (23-25°C) в течение 90 мин. Загрузочный буфер заменяли буфером для анализа ± тестируемое соединение с последующим 30 минутным инкубированием при температуре окружающей среды, затем в микропланшет добавляли таллий/калиевый стимулятор.
Пошаговый протокол
1. Высевают клетки HEK-hKir1.1 (50 мкл в количестве 20000 клеток/лунка) в 384-луночных микропланшетах с PDL-покрытием.
2. Оставляют клетки для адгезии в течение ночи во влажной камере 37°C/10%CO2.
3. Полностью удаляют питательные среды для роста клеток из микропланшета и заменяют их 25 мкл загрузочного буфера.
4. Инкубируют защищенный от света микропланшет при комнатной температуре в течение 90 мин.
5. Удаляют загрузочный буферный и заменяют его 25 мкл смеси (1x) буфер для анализа ± тестируемое соединение.
6. Инкубируют защищенный от света микропланшет при комнатной температуре в течение 30 мин.
7. Микропланшет FLIPR-Tetra 384: в микропланшет добавляют раствор стимулятора (таллий/калий) и отслеживают флуоресценцию; длина волны возбуждения = 400 нм; длина волны испускания = 460 & 580 нм. Собирают данные в течение ~ 10 мин.
Расчет данных. В настоящем изобретении для определения ROMK-чувствительного компонента транспорта таллия использовали интенсивность флуоресценции лунок, содержащих 3 мкМ стандартного контрольного ROMK-ингибитора. Флуоресценцию в присутствии тестируемых соединений нормализовали до контрольных значений, получая при этом % изменения флуоресценции. Значения IC50 представляют собой концентрацию соединения, которая ингибирует 50% ROMK сигнала транспорта таллия.
Стандарт для проведения анализа. Обычно было включено контрольное соединение, чтобы подтвердить, что анализ дает согласующиеся результаты по сравнению с предыдущими измерениями, хотя для получения результатов относительно тестируемых соединений контрольное соединение не требуется. Контрольное соединение может представлять собой любое соединение формулы I согласно настоящему изобретению, предпочтительно с активностью IC50 менее 1 мкМ при таком анализе. Альтернативно контрольное соединение может представлять собой другое соединение (не входящее в объем формулы I), которое обладает активностью IC50 при таком анализе менее 1 мкМ.
Электрофизиологический анализ
Блокаду калиевых токов (Kir1.1 (ROMK1)) исследовали на вольтклампе для целой клетки (Hamill и др. Pfluegers Archives 391:85-100 (1981)) с применением автоматизированной платформы Ion Works Quattro (Molecular Devices, Sunnyvale, CA) для исследования электрофизиологии. Яйцеклетки китайского хомячка, стабильно экспрессирующие Kir1.1-каналы, выдерживали в питательных культуральных средах в колбах T-75 во влажной камере при 10% CО2 и 37°C. Перед началом эксперимента экспрессию Kir1.1 индуцировали инкубированием с 1 мМ бутирата натрия в течение ночи. В день эксперимента клетки отделяли с 2,5 мл Versene (Invitrogen 15040-066) приблизительно в течение 6 минут при 37°C и суспендировали в 10 мл промывающего раствора, содержащего (в мМ): 150 NaCl, 10 KCl, 2,7 CaCl2, 0,5 MgCl2, 5 HEPES, при pH 7,4. После центрифугирования дебрис ресуспендировали приблизительно в 4,0 мл промывающего раствора и помещали в измерительный прибор Ion Works. Интрацеллюлярный раствор состоял из (в мМ): 80 K глюконата, 40 KC1, 20 KF, 3,2 MgCl2, 3 EGTA, 5 Hepes, pH 7,4. Электрический доступ к цитоплазме обеспечивали путем перфорации в 0,13 мг/мл амфотерицина B в течение 4 мин. Амфотерицин B (Sigma A-4888) получали в виде 40 мг/мл раствора в ДМСО.
Протоколы напряжения и регистрации тока осуществляли с применением системы аппаратного/программного обеспечения Ion Works HT. Токи испытывали при 1 кГц. Потенциалы на границе раздела жидкостей применяли без поправок. Перед 6 минутным периодом инкубации с соединением и после нее прикладывали 100 мсек пробный импульс до 0 мВ от исходного потенциала -70 мВ с последующим 100 мсек линейным изменением напряжения от -70 мВ до +70 мВ. Испытуемые соединения получали путем разбавления промывающим раствором маточных растворов в ДМСО до конечной концентрации (3x) и помещали в измерительный прибор в полипропиленовых 96-луночных планшетах. Амплитуды тока измеряли с применением программного обеспечения Ion Works. Чтобы оценить активность соединения, в программе Microsoft Excel (Microsoft, Redmond, CA) рассчитывали частичную блокаду во время перепада напряжений до 0 мВ, и кривые дозовой зависимости согласовывали с Igor Pro 4.0 (WaveMetrics, Lake Oswego, OR). Обычно было включено контрольное соединение, чтобы Обычно было включено контрольное соединение, чтобы подтвердить, что анализ дает согласующиеся результаты по сравнению с предыдущими измерениями, хотя для получения результатов относительно тестируемых соединений контрольное соединение не требуется. Контрольное соединение может представлять собой любое соединение формулы I-V согласно настоящему изобретению, предпочтительно с активностью IC50 менее 1 мкМ при таком анализе. Альтернативно контрольное соединение может представлять собой другое соединение (не входящее в объем формулы I-V), которое обладает активностью IC50 при таком анализе менее 1 мкМ.
Данные, полученные для соединений по примерам настоящего изобретения с применением анализа транспорта таллия (Flux Assay) и электрофизиологического анализа приведены ниже в таблице 5. Все тестируемые соединения, представляющие собой конечные продукты согласно примерам (диастереомерные смеси и индивидуальные диастереомеры) имели активности IC50 менее 1 мкM в одном или в обоих испытаниях: при анализе транспорта таллия (Flux Assay) и электрофизиологическом анализе.
|
Анализ с использованием модели спонтанно-гипертензивных крыс (SHR)
Спонтанно-гипертензивная крыса (SHR) имеет возрастозависимую гипертонию, поэтому для повышения кровяного давления не требуется введения экзогенных средств, и не требуется диеты с повышенным содержанием соли для повышения кровяного давления. Таким образом, ее эссенциальная гипертония мало чем отличается от эссенциальной гипертонии человека и обеспечивает возможность оценки способности новых средств снижать кровяное давление в зависимости от дозы.
Экспериментальные протоколы для проведения оценки эффективности снижения кровяного давления соединениями согласно настоящему изобретению у спонтанно-гипертензивных крыс (SHR):
Спонтанно-гипертензивным крысам (SHR, самец, 6 месяцев, Charles River) имплантировали телеметрическое устройство DSI TA11PA-C40 (Data Sciences, Inc., St. Paul, MN) в условиях изофлурановой или кетамин/метомидиновой анестезии. Катетер с телеметрическим блоком вставляли в нисходящий отдел аорты через бедренную артерию и подкожно имплантировали телеметрическое устройство с левого бока. Перед началом любых исследований животным давали возможность оправиться после хирургической операции в течение 14 дней. Кровяное давление, частоту сердечных сокращений и сигналы от деятельности находящихся в сознании, свободно передвигающихся крыс непрерывно регистрировали в течение 30 секунд каждые 10 минут. HCTZ (25 мг/кг/день, PO) использовали в качестве контрольного диуретика в дозе, приводящей приблизительно к его максимальной эффективности для SHR. Эффективность соединений согласно настоящему изобретению в отношении способности снижать кровяное давление по сравнению с плацебо (наполнителем) оценивали после каждого орального введения при принудительном питании каждый день при продолжительности от трех до четырнадцати дней. Данные получали как среднечасовые, и рассчитывали изменения кровяного давления за вычетом данных, относящихся к плацебо (наполнителю) на почасовой основе. Соединения по примерам №№ 5, 7, 9 и 43 оценивали при PO, QD-дозировании в виде одной или нескольких доз в диапазоне от 0,3 до 10 мг/кг, и они приводили к среднестатистическим уменьшениям ежедневного (24 часа) значения систолического кровяного давления в диапазоне от 6 мм рт. ст. до 24 мм рт.ст при употреблении доз до дня окончания исследований.
Исследование на спонтанно-гипертензивных крысах хорошо известно и часто применяется в данной области в качестве экспериментальной модели, воспроизводящей гипертонию человека (см., например, публикацию Lerman L.O. и др., J. Lab. Clin. Med., 2005; 146:160-173).
Несмотря на то, что изобретение описано со ссылкой на некоторые конкретные варианты его осуществления, многочисленные альтернативные варианты осуществления будут очевидны специалисту в данной области техники из описанных здесь методик. Объем формулы изобретения не должен ограничиваться предпочтительными вариантами осуществления изобретения, изложенными в примерах, но должен иметь самую широкую интерпретацию, соответствующую описанию в целом. Подразумевается, что перечисление или описание конкретного соединения в формуле изобретения (то есть в видовых пунктах формулы изобретения) без обозначения конкретной стереоконфигурации или с таким обозначением не для всех хиральных центров охватывает рацематы, рацемические смеси, каждый индивидуальный энантиомер, смесь диастереоизомеров и каждый индивидуальный диастереомер соединения, когда такие формы возможны благодаря присутствию одного или нескольких асимметричных центров. Все процитированные здесь патенты, патентные заявки и публикации включены в данный документ путем ссылки в их полном объеме.
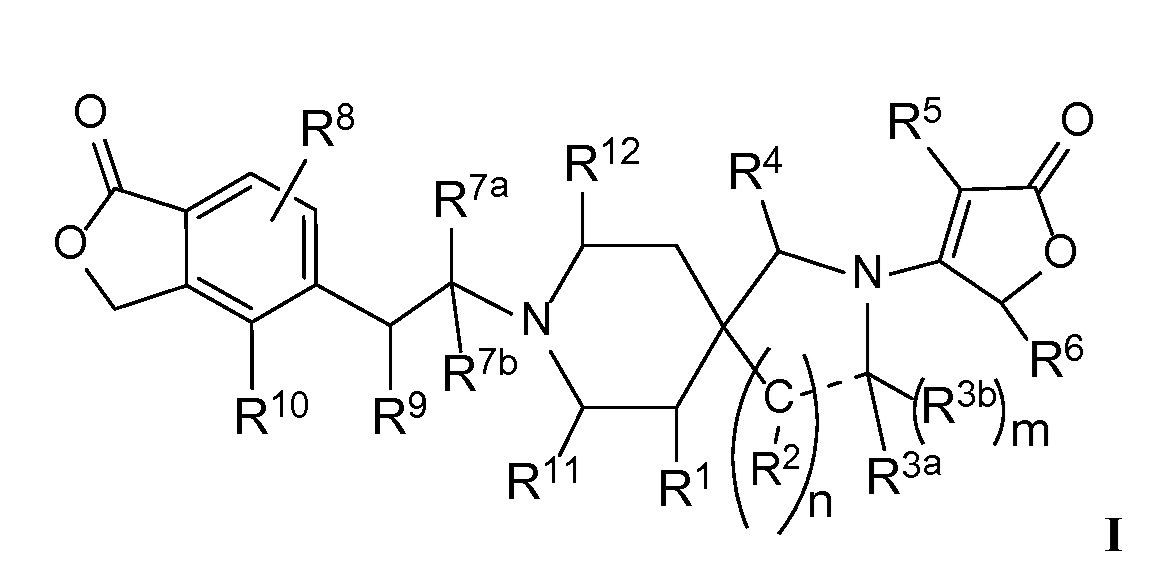
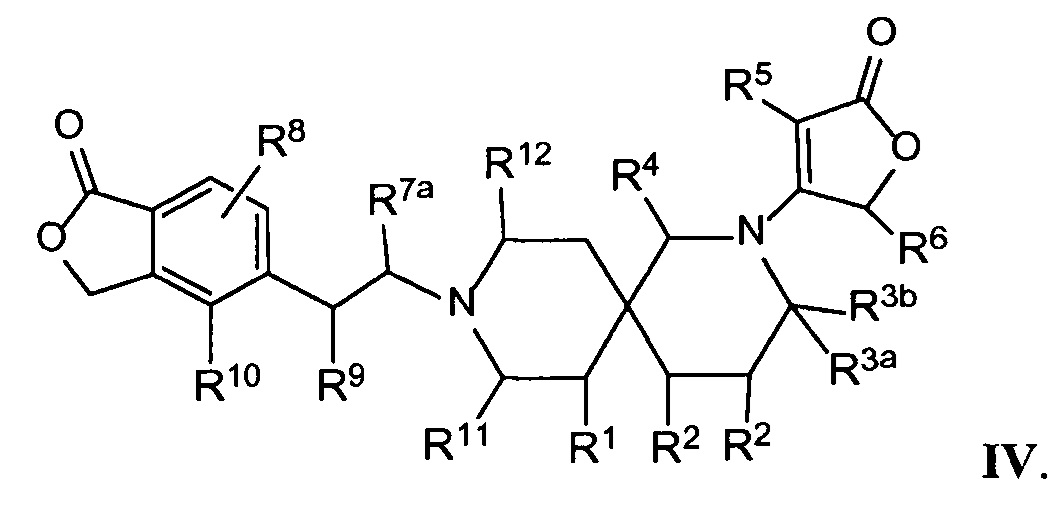
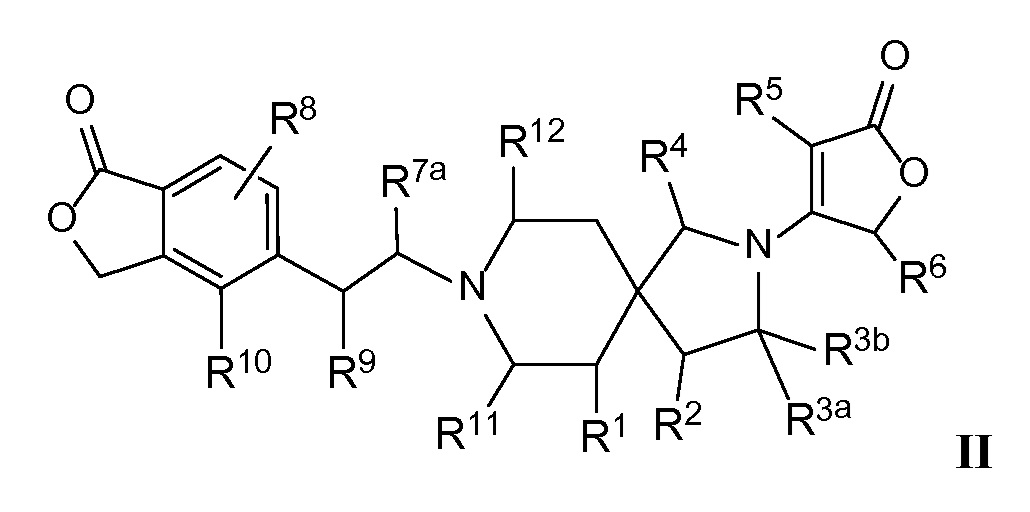
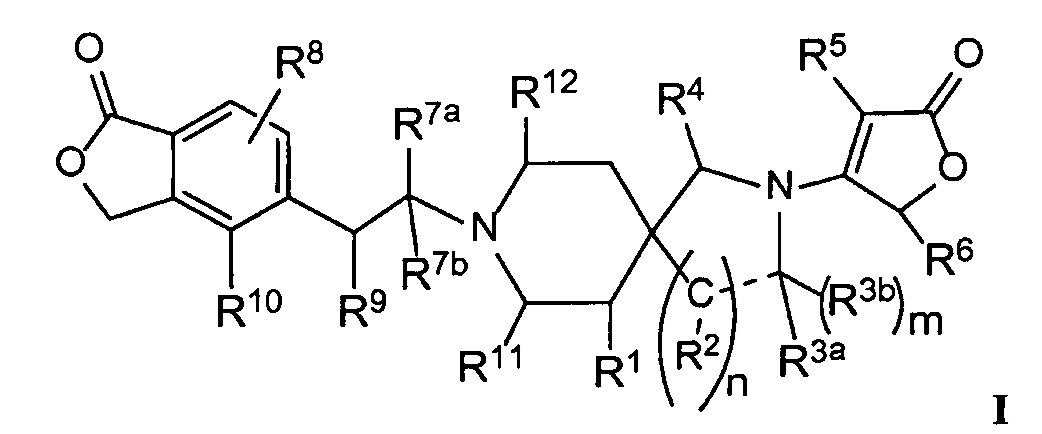
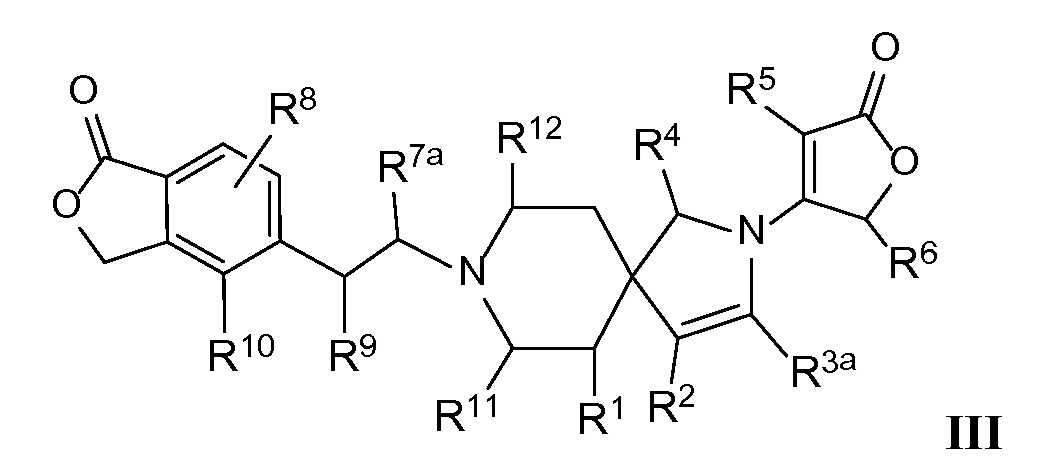
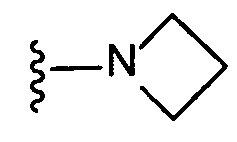
Фармацевтически приемлемые соли 2-{4-[(3s)-пиперидин-3-ил]фенил}-2н-индазол-7-карбоксамида
Гетероциклические ингибиторы аспартильной протеазы
Положительные аллостерические модуляторы м1-рецепторов на основе пираниларилметилбензохиназолинона
Ингибиторы сетр
Соединения, которые являются ингибиторами erk
Агенты, ингибирующие р38 киназу
Аминотетрагидропираны в качестве ингибиторов дипептидилпептидазы-iv для лечения или предупреждения диабета
Соединения замещенных диазепанов в качестве антагонистов орексиновых рецепторов
Стабильные составы антител против рецептора программируемой смерти pd-1 человека и относящиеся к ним способы лечения
Экстракты kibdelos porangium в качестве антибактериальных средств

