Результат интеллектуальной деятельности: ПО СУЩЕСТВУ, ЧИСТЫЙ ФЛУОРЕСЦЕИН
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение направлено на создание композиций, содержащих по существу чистый флуоресцеин, способов получения по существу чистого флуоресцеина, по существу чистого флуоресцеина, полученного такими способами, аналитических методик определения чистоты флуоресцеина и фармацевтических композиций для применения в ангиографии.
Предпосылки создания изобретения
Флуоресцеин представляет собой оранжево-красное соединение, C20H12O5, которое демонстрирует интенсивную флуоресценцию в щелочном растворе и которое применяют как средство для диагностических целей, индикатор в океанографии и как текстильный краситель.
Флуоресцеин был впервые синтезирован германским химиком Адольфом фон Байером в 1871 г. из выделенных из нефти резорцина (1,3-дигидроксибензола) и фталевого ангидрида. Пауль Эрлих, германский бактериолог, применял этот флуоресцентный краситель (в виде натриевой соли флуоресцеина), известный в то время как «уранин», для прослеживания путей секреции водянистой жидкости в глазах. Считают, что это было первым примером применения флуоресцентного красителя in vivo в физиологическом исследовании.
Ангиография с применением флуоресцеина является важным диагностическим инструментом, позволяющим исследовать состояние кровеносных сосудов глазного дна. Эти сосуды являются важным фактором во многих заболеваниях, затрагивающих сетчатку. Ангиографию проводят, инъецируя флуоресцеин в вену в руке пациента. Спустя короткий промежуток времени (обычно через несколько секунд) краситель попадает в сосуды глазного дна, и с помощью камеры со специальными фильтрами регистрируют циркуляцию красителя в кровеносных сосудах глаза. Исследуя изображения, полученные таким образом, можно оценить нарушения кровотока, например, проницаемость сосудов, отеки, аномальные или новообразованные сосуды и т.д.
Флуоресцеин поглощает синий свет с пиком поглощения и возбуждения при длинах волн 465-490 нм. Флуоресценция имеет место в желто-зеленой области при длинах волн 520-530 нм. Хотя обычно его называют просто флуоресцеином, краситель, применяемый в ангиографии, является натрий-флуоресцеином, растворимой динатриевой солью флуоресцеина.
Нормальная доза флуоресцеина для взрослых составляет 500 мг, инъецируемых внутривенно. Обычно его расфасовывают в дозах по 5 мл 10%-ного раствора или по 2 мл 25%-ного раствора. После введения в кровоток приблизительно 80% молекул красителя связываются с белками сыворотки. Оставшиеся несвязанными свободные молекулы флуоресцеина флуоресцируют при возбуждении светом с соответствующей длиной волны. Краситель метаболизируется печенью с образованием моноглюкуронида флуоресцеина и выводится с мочой в течение 24-36 часов после введения.
Сообщали, что чистота флуоресцеина в его готовых формах может коррелировать с побочными эффектами и переносимостью инъекций. («Effective differences in the formulation of intravenous fluorescein and related side effects» (Эффективные различия в готовых формах внутривенного флуоресцеина и ассоциированных побочных эффектах) Yannuzi et al. в Am. J. Ophthalmol. 1974, 78 (2), страницы 217-221). Поэтому главной целью настоящего изобретения является устранение всех или по существу всех примесей из композиций флуоресцеина, применяемых для ангиографии.
За дополнительной информацией о композициях флуоресцеина и способах получения и очистки флуоресцеина можно обращаться к следующим публикациям.
Германский патент № 136498 (Friedrich и др.) под заголовком «Process for Preparing Highly Purified Fluorescein for Injection Purposes» (Способ получения высокоочищенного флуоресцеина для инъекций) описывает способ получения флуоресцеина с использованием пиридина.
Опубликованная заявка на патент США № 2006/0106234A1 (Tran-Guyon и др.) под заголовком «High Purity Phthalein Derivatives и Method for Preparing Same» (Производные фталеина высокой чистоты и способ их получения) описывает способ получения флуоресцеина с использованием безводного растворителя.
За дополнительными сведениями о предпосылках настоящего изобретения можно также обратиться за консультацией к следующим патентам или публикациям: патент США № 5637733 (Sujeeth) под заголовком «Synthesis of Fluorescein Compounds with Excess Resorcinol as a Solvent» (Синтез соединений флуоресцеина с избытком резорцина в качестве растворителя) и патент США № 1965842 (Kranz) под заголовком «Production of Hydroxybenzene-Phthaleins» (Производство гидроксибензолфталеинов).
Высокоочищенный флуоресцеин необходим для получения растворов для инъекционных целей. Применяемый очищенный флуоресцеин в идеале должен быть: (i) свободным от примесей, которые могут быть токсичными и/или нефлуоресцирующими; (ii) с низким содержанием соли, которая может приводить к неприемлемо высокой осмоляльности или гипертоничности инъецируемого продукта флуоресцеина; и (iii) слабоокрашенным. Определенные примеси являются сильноокрашенными. Поэтому отсутствие окраски при определенных частотах может указывать на отсутствие таких примесей. Поэтому цветовой профиль композиций флуоресцеина считают важной качественной характеристикой и визуальным показателем чистоты.
Необходим способ идентификации и количественного определения очень низких уровней содержания примесей, которые могут присутствовать в композициях флуоресцеина. Такой способ должен быть способным разделять, идентифицировать и количественно определять примеси, которые могут присутствовать.
Таким образом, имеется потребность в композиции флуоресцеина, которая является высоко чистой, слабоокрашенной и с низким содержанием хлорида натрия, и в способе получения такого флуоресцеина, не требующем применения пиридина или другого неводного (и потенциально вредного) растворителя, а также в способе определения чистоты такого флуоресцеина. Настоящее изобретение направлено на удовлетворение этой потребности.
Сущность изобретения
Настоящее изобретение направлено на создание композиций, содержащих по существу чистый флуоресцеин, новых и усовершенствованных способов получения очищенного флуоресцеина и композиций флуоресцеина, полученного этими способами. Настоящее изобретение также направлено на создание фармацевтической композиции для применения в ангиографии, содержащей по существу чистый флуоресцеин, и способа определения чистоты композиции флуоресцеина. Высокоочищенный флуоресцеин, полученный способом согласно настоящему изобретению, имеет более низкий уровень примесей родственных соединений, чем ранее известные композиции флуоресцеина. Флуоресцеин, полученный этими новыми способами, является также менее окрашенным (при 590 нм), чем другие известные композиции, что предоставляет отчетливо видимый показатель чистоты. Флуоресцеин согласно настоящему изобретению имеет также более низкое содержание хлорида натрия и поэтому его легче подготавливать для фармацевтического применения, чем другие известные композиции. Способы согласно настоящему изобретению превосходят другие известные способы тем, что они устраняют применение пиридина в процессе очистки, не требуют применения безводного растворителя, снижают количество уксусного ангидрида, требующегося для ацетилирования неочищенного флуоресцеина, и улучшают выход высокоочищенного флуоресцеина. Настоящее изобретение также повышает уровень техники в данной области, предоставляя надежный способ отделения и количественного определения примесей родственных соединений в композициях флуоресцеина и, тем самым, определения чистоты композиций флуоресцеина.
Настоящее изобретение можно осуществлять в различных областях применения, включая (но, не ограничиваясь ими) те, которые приведены ниже:
Один вариант осуществления настоящего изобретения направлен на создание композиции, содержащей по существу чистый флуоресцеин; более конкретно, флуоресцеин, по существу свободный от пиридина.
Другой вариант осуществления настоящего изобретения направлен на получение по существу чистого флуоресцеина, не содержащего никаких примесей родственных соединений в концентрациях, больших чем примерно 0,1% по массе; более предпочтительно 0,01% по массе.
Другой вариант осуществления настоящего изобретения направлен на получение по существу чистого флуоресцеина, имеющего цветовой индекс от примерено 0,015 до примерно 0,050 единиц оптической плотности.
Другой вариант осуществления настоящего изобретения направлен на получение по существу чистого флуоресцеина, имеющего остаточное содержание хлорида, меньшее чем примерно 0,25% по массе.
Другой вариант осуществления настоящего изобретения направлен на получение по существу чистого флуоресцеина, в котором общее количество примесей родственных соединений составляет менее чем примерно 0,6% по массе; предпочтительно менее чем 0,06% по массе.
Другой вариант осуществления настоящего изобретения направлен на создание способа получения по существу чистого флуоресцеина. Этот способ включает гидролиз диацетилфлуоресцеина с образованием флуоресцеина, обработку флуоресцеина активированным углем, фильтрование, добавление этанола к фильтрату, регулирование рН с использованием кислого раствора для образования осадка, фильтрование и промывание. В одном аспекте этого варианта осуществления уровень рН доводят от примерно 1,0 до примерно 2,5. В других аспектах при регулировании уровня рН охлаждающую температуру поддерживают от примерно 20°C до примерно 25°C и уровень рН регулируют в течение примерно 2-4 часов. Настоящее изобретение также направлено на получение композиций по существу чистого флуоресцеина, полученного такими способами.
Другой вариант осуществления настоящего изобретения предоставляет метод ВЭЖХ количественного определения уровней содержания примесей соединений, родственных флуоресцеину. Этот метод включает получение жидкостной хроматограммы высокого давления для данной композиции; идентификацию пиков на хроматограмме, соответствующих примесям родственных веществ, и измерения площадей этих пиков для определения их относительных концентраций. В одном аспекте этого варианта осуществления пики имеют относительные времена удерживания при ВЭЖХ, равные примерно 0,75, 1,19, 1,23, 1,68 и 1,71. Другой вариант осуществления предоставляет методику ВЭЖХ/МС для идентификации примесей родственных веществ во флуоресцеине.
Предпочтительный вариант осуществления настоящего изобретения направлен на создание фармацевтической композиции для применения в ангиографии, содержащей по существу чистый флуоресцеин; более конкретно, композиции, в которой флуоресцеин по существу свободен от пиридина.
Другой предпочтительный вариант осуществления настоящего изобретения направлен на создание фармацевтической композиции для применения в ангиографии, содержащей по существу чистый флуоресцеин, которая не содержит никаких примесей родственных веществ в концентрациях, больших чем примерно 0,1% по массе; более предпочтительно 0,01% по массе.
Другой предпочтительный вариант осуществления настоящего изобретения направлен на создание фармацевтической композиции для применения в ангиографии, содержащей по существу чистый флуоресцеин, в которой общее количество присутствующих примесей родственных веществ составляет менее чем примерно 0,6% по массе; более предпочтительно менее чем примерно 0,06% по массе.
Другой предпочтительный вариант осуществления настоящего изобретения направлен на создание фармацевтической композиции для применения в ангиографии, содержащей по существу чистый флуоресцеин, в которой этот флуоресцеин имеет цветовой индекс от примерно 0,015 до примерно 0,050 единиц оптической плотности.
Другой предпочтительный вариант осуществления настоящего изобретения направлен на создание фармацевтической композиции флуоресцеина для применения в ангиографии, в которой остаточное содержание хлорида составляет менее чем примерно 0,25% по массе.
Настоящее изобретение более подробно обсуждается с помощью следующих фигур и подробного описания.
Краткое описание чертежей
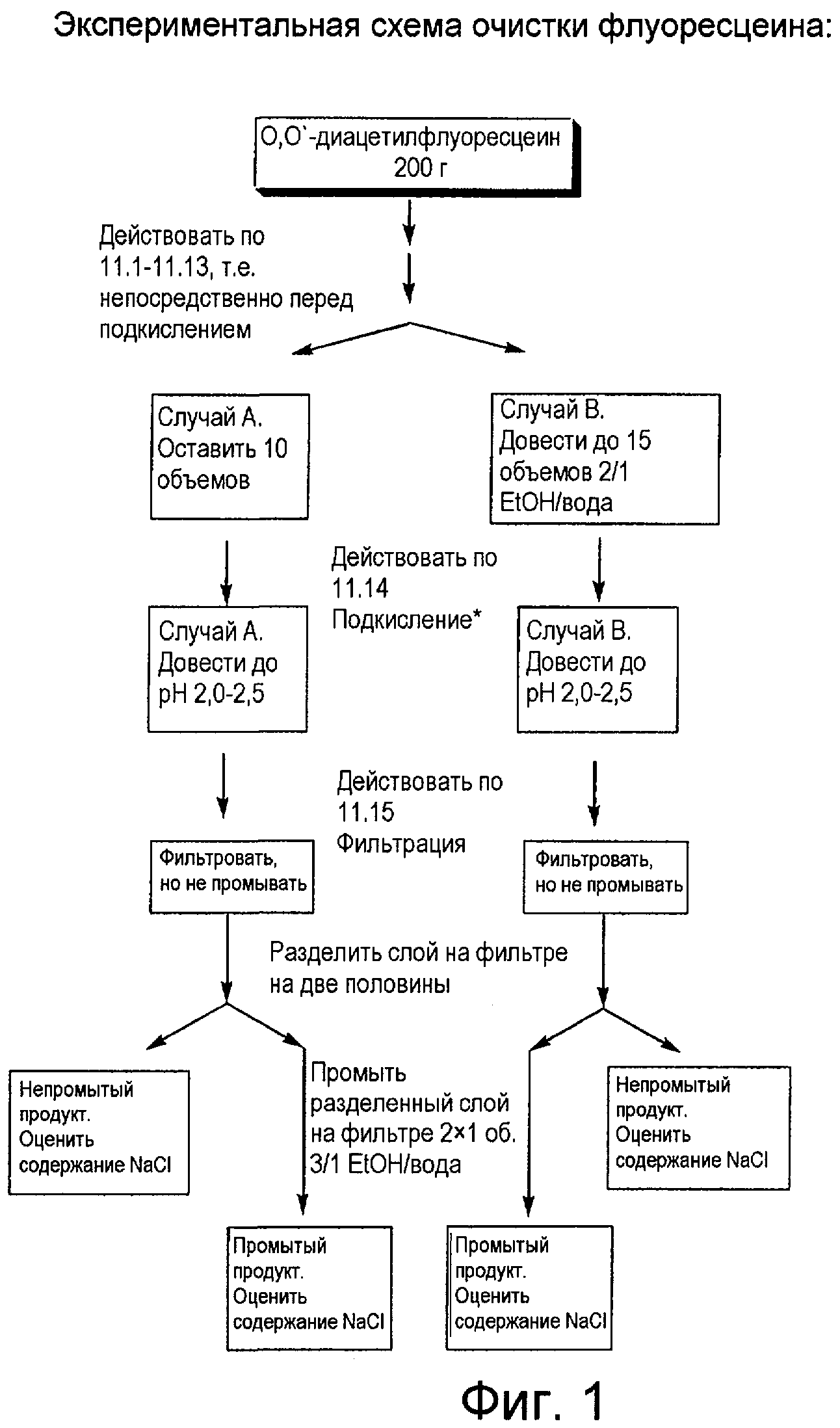
Фигура 1 представляет собой схему проведения эксперимента для промывки флуоресцеина.
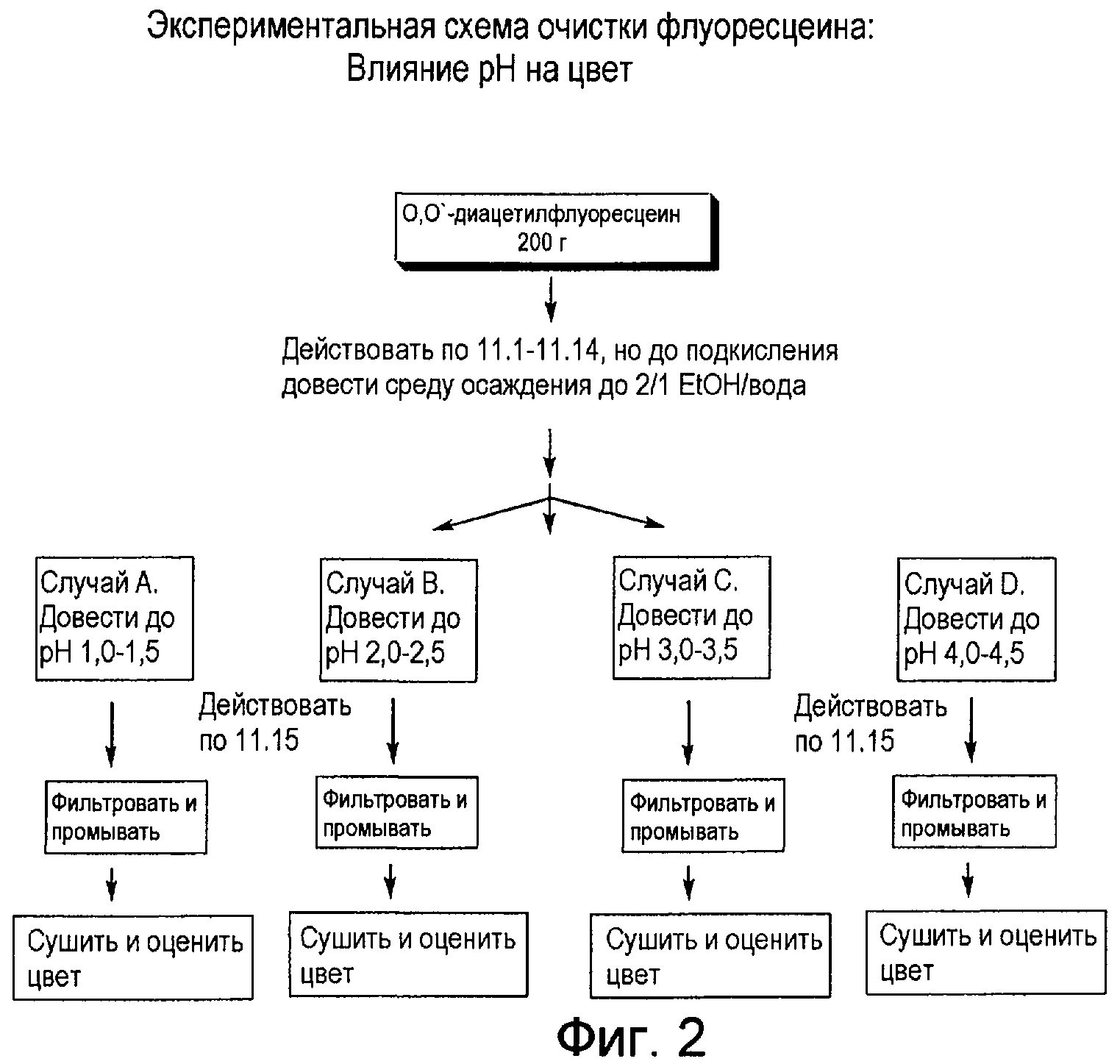
Фигура 2 представляет собой схему проведения эксперимента для рН-осаждения флуоресцеина.
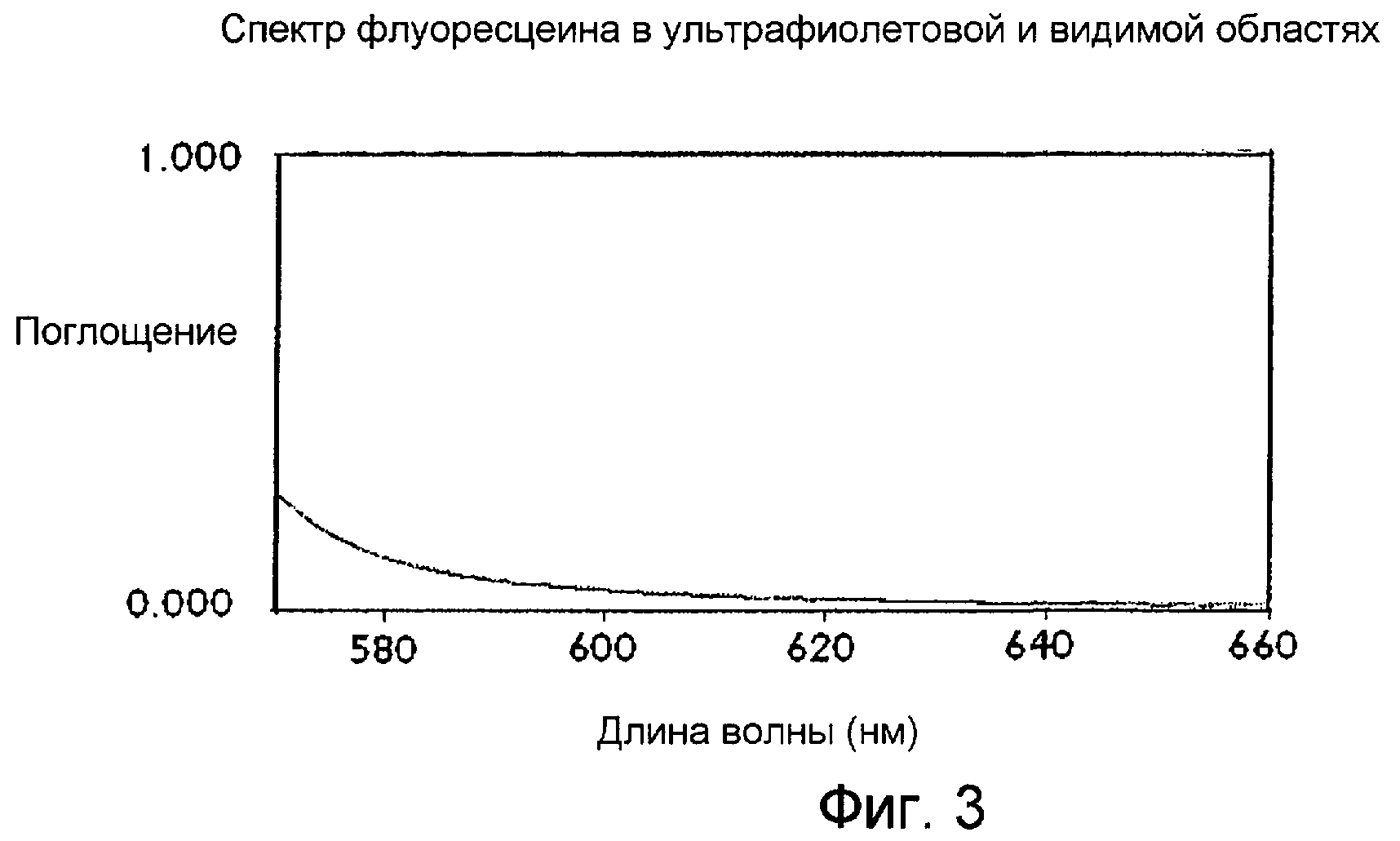
Фигура 3 представляет собой спектр в ультрафиолетовой и видимой областях, показывающий интенсивность окраски лекарственного вещества флуоресцеина, как описано в Примере 3.
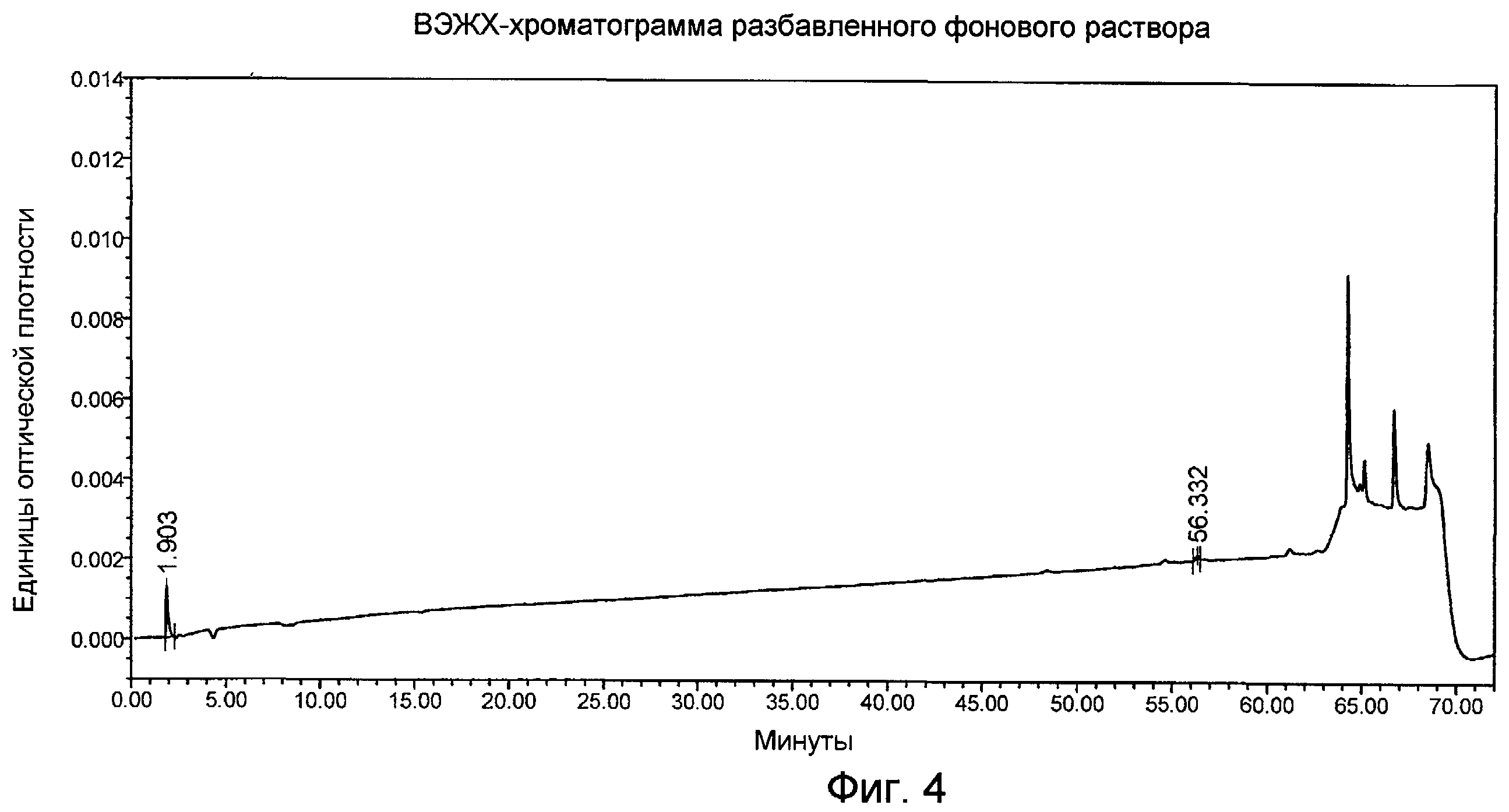
Фигура 4 представляет собой ВЭЖХ-хроматограмму фонового разбавителя, как описано в Примере 5.
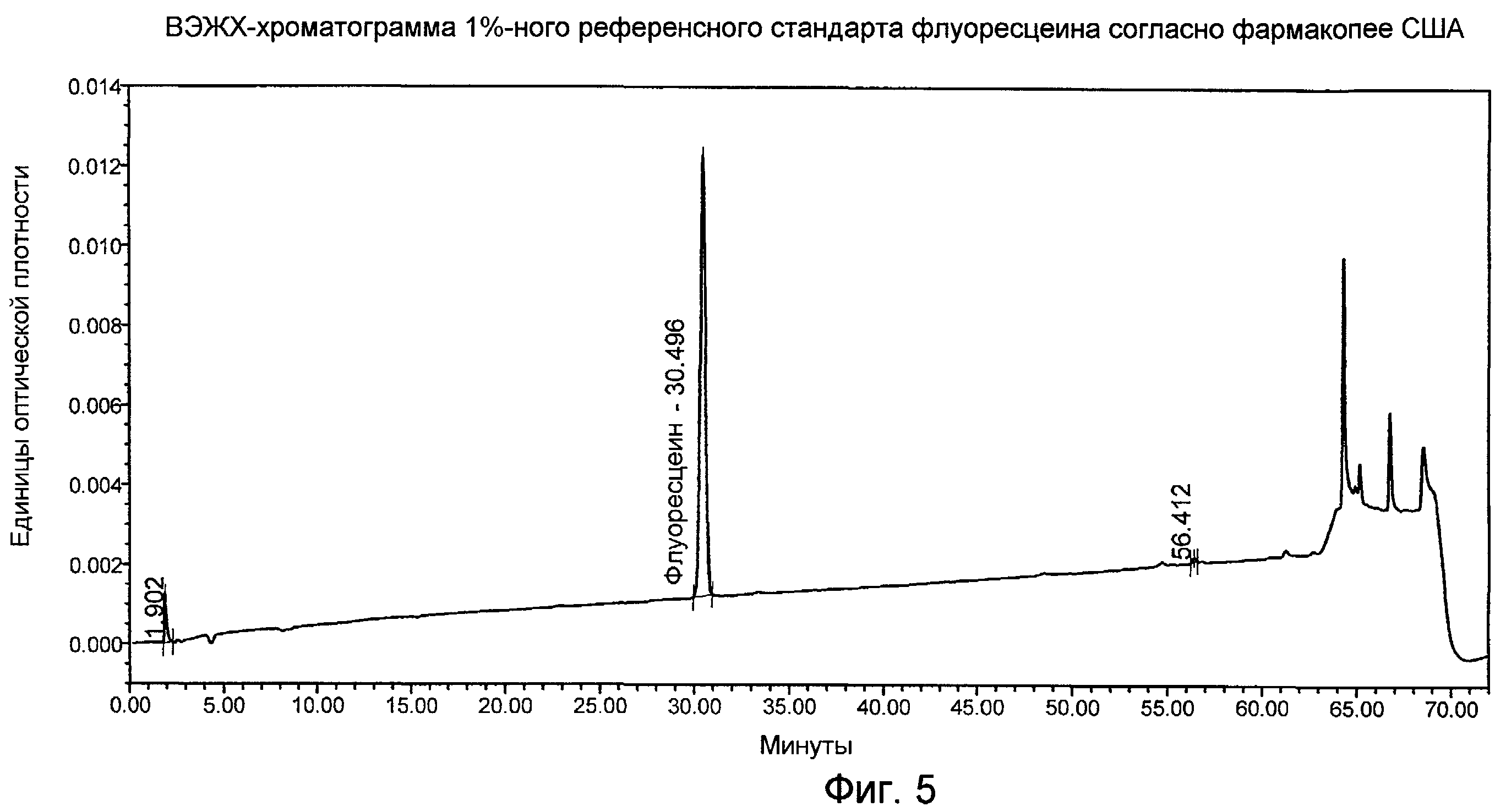
Фигура 5 представляет собой ВЭЖХ-хроматограмму 1%-ного референсного стандарта флуоресцеина согласно Фармакопее США (USP), как описано в Примере 5.
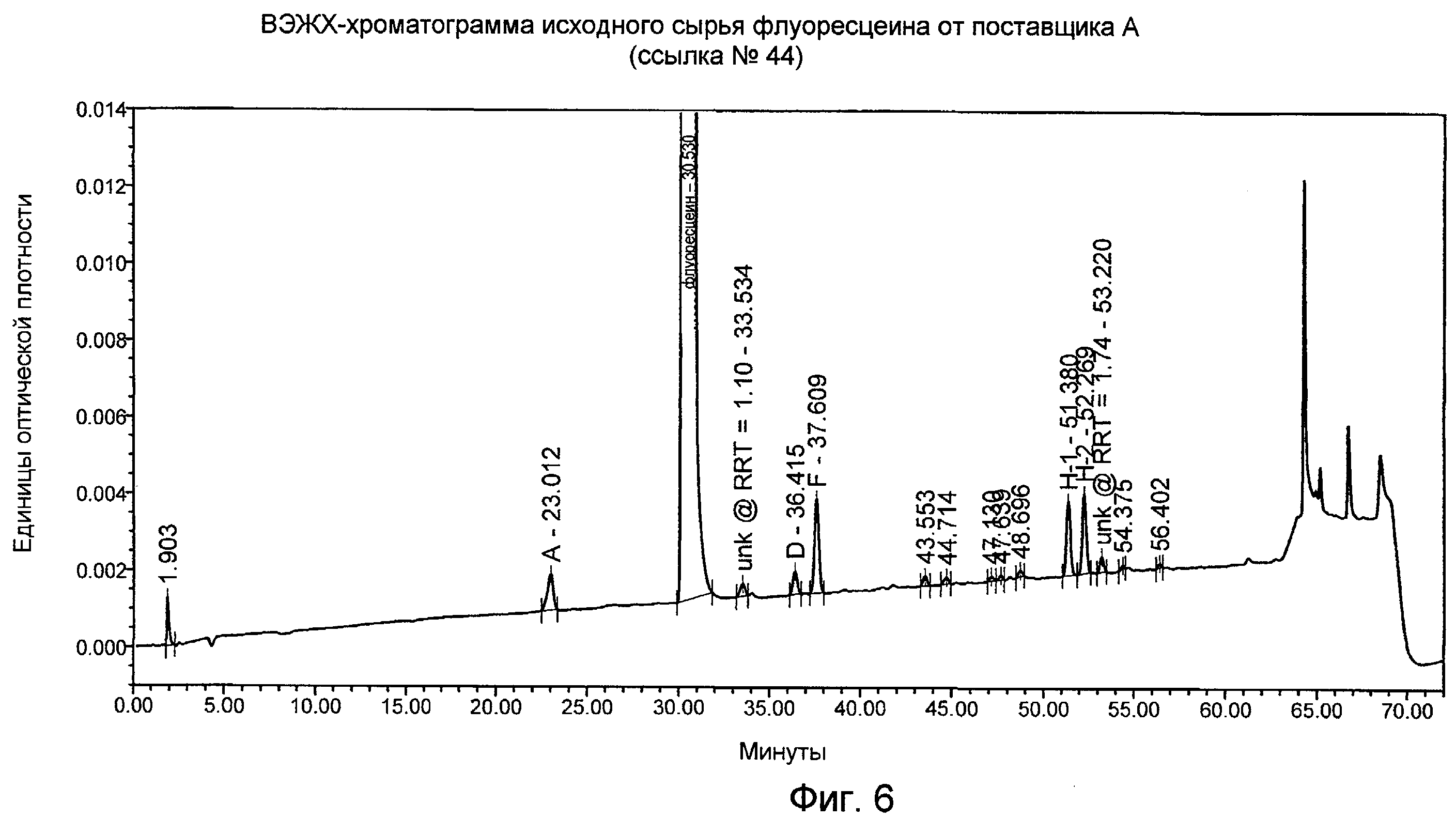
Фигура 6 представляет собой ВЭЖХ-хроматограмму исходного сырья флуоресцеина от поставщика А, как описано в Примере 5.
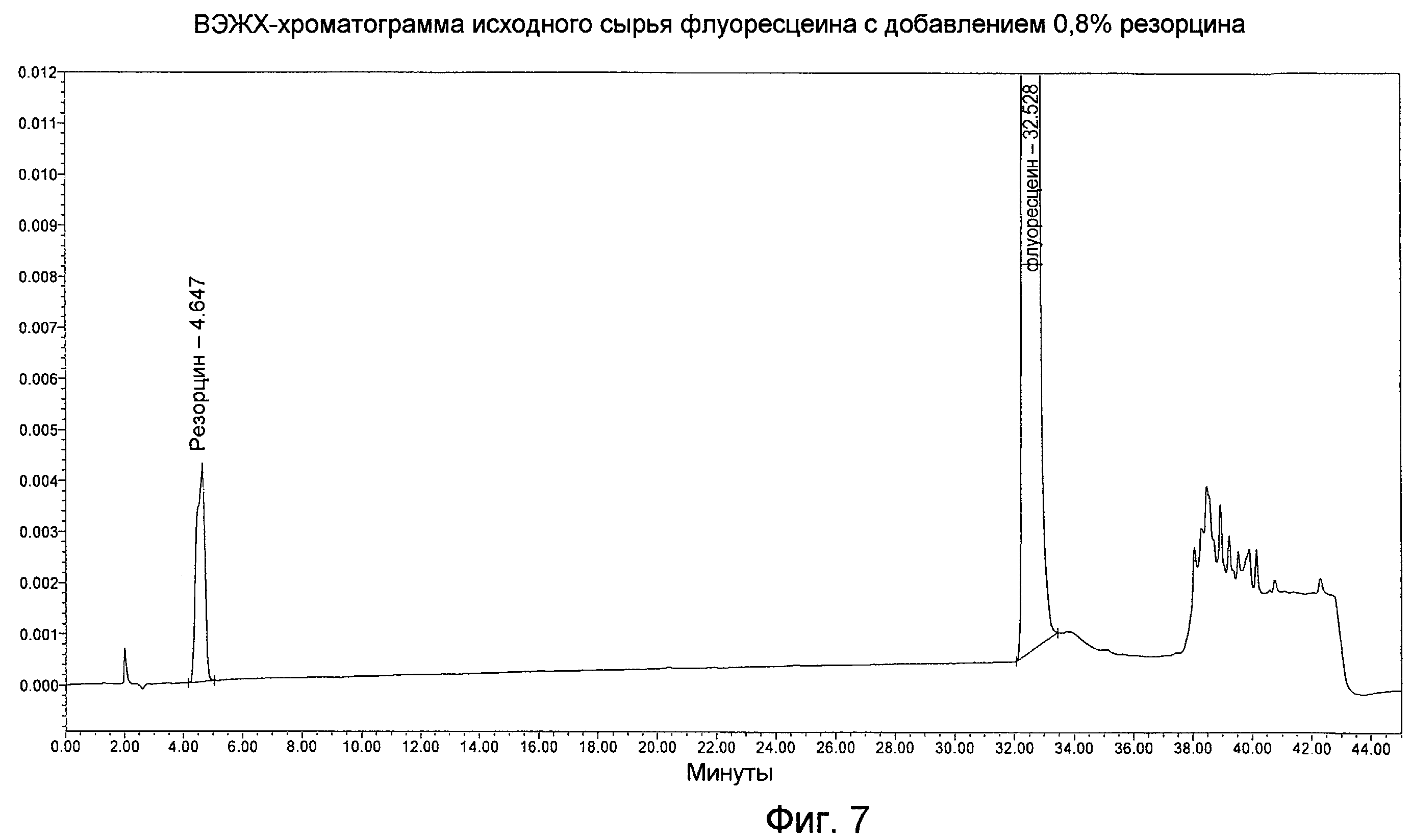
Фигура 7 представляет собой ВЭЖХ-хроматограмму исходного сырья с 0,8% резорцина, как описано в Примере 5.
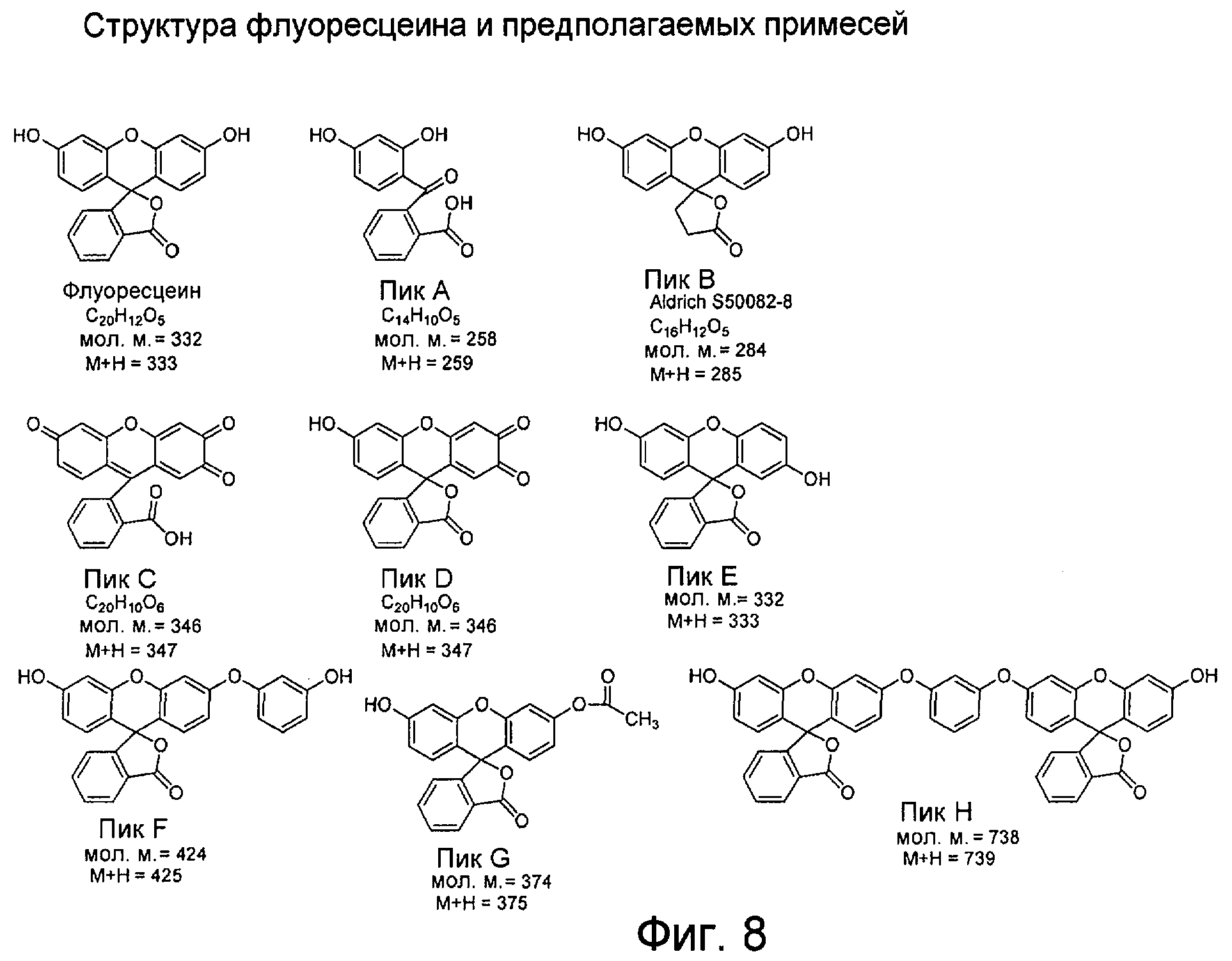
Фигура 8 представляет собой диаграмму «Структура флуоресцеина и предложенные структуры примесей родственных веществ», как описано в Примерах 5 и 6.
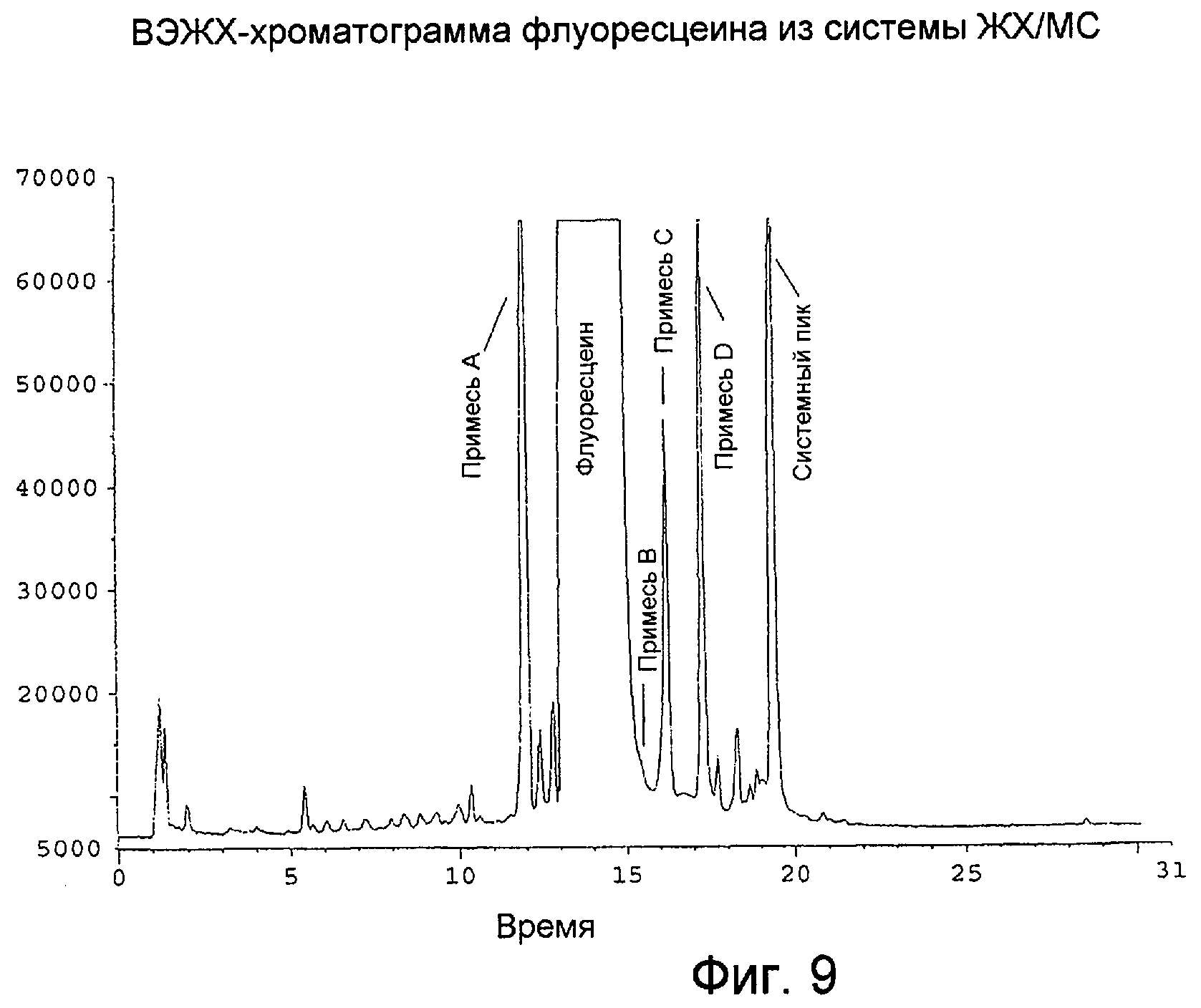
Фигура 9 представляет собой типичную ВЭЖХ-хроматограмму флуоресцеина, полученную с помощью системы ЖХ/МС.
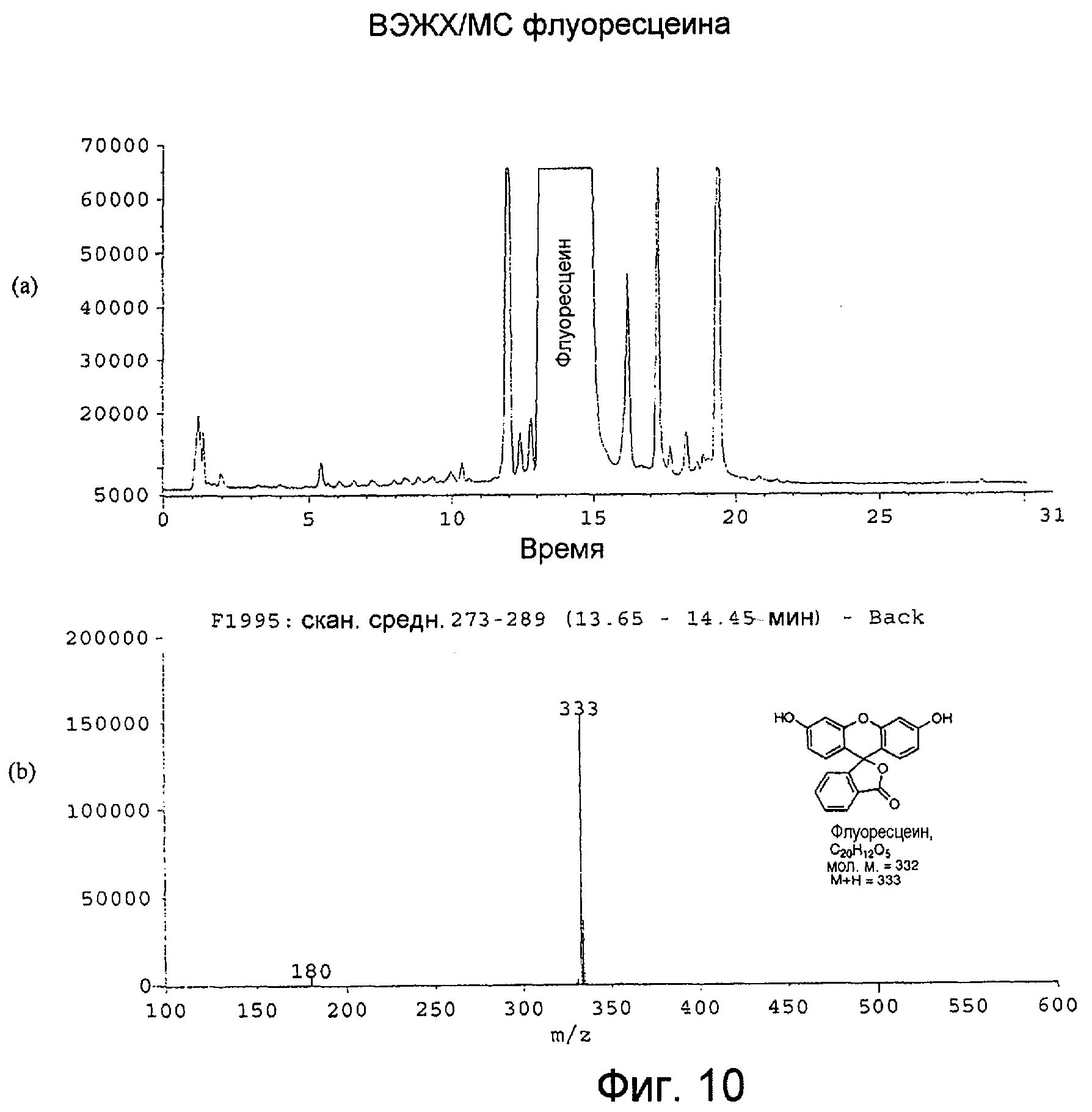
Фигура 10 представляет собой: (а) хроматограмму флуоресцеина c УФ-детектором; и (b) масс-спектр с термораспылением для пика ВЭЖХ-хроматограммы флуоресцеина, собранного в интервале 13,65-14,45 минут.
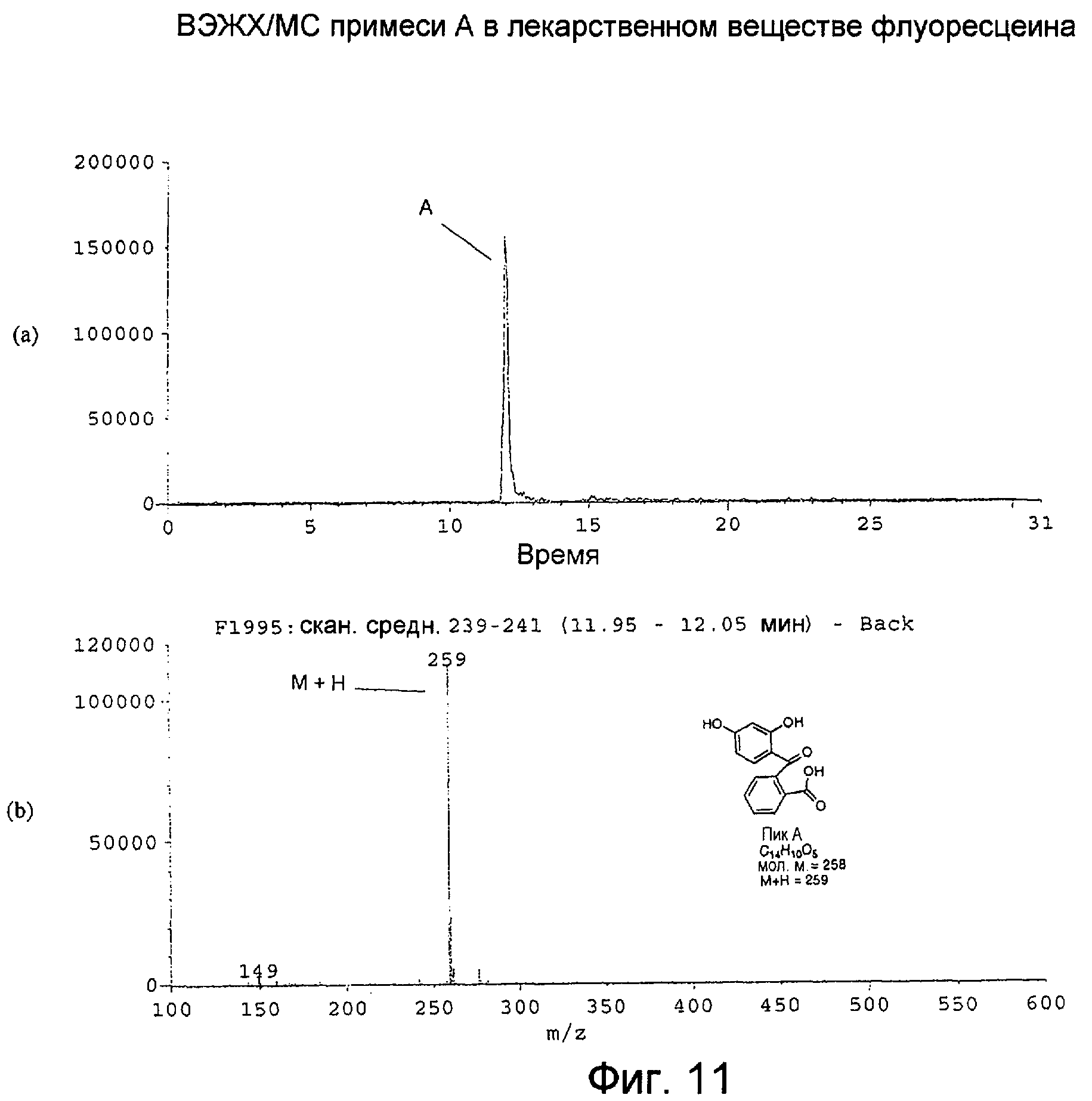
Фигура 11 представляет собой: (а) хроматограмму примеси А во флуоресцеине с масс-селективным детектированием при m/z 259 при использовании масс-спектрометра; и (b) масс-спектр с термораспылением для ВЭЖХ-пика примеси А, собранного в интервале 11,95-12,05 минут.
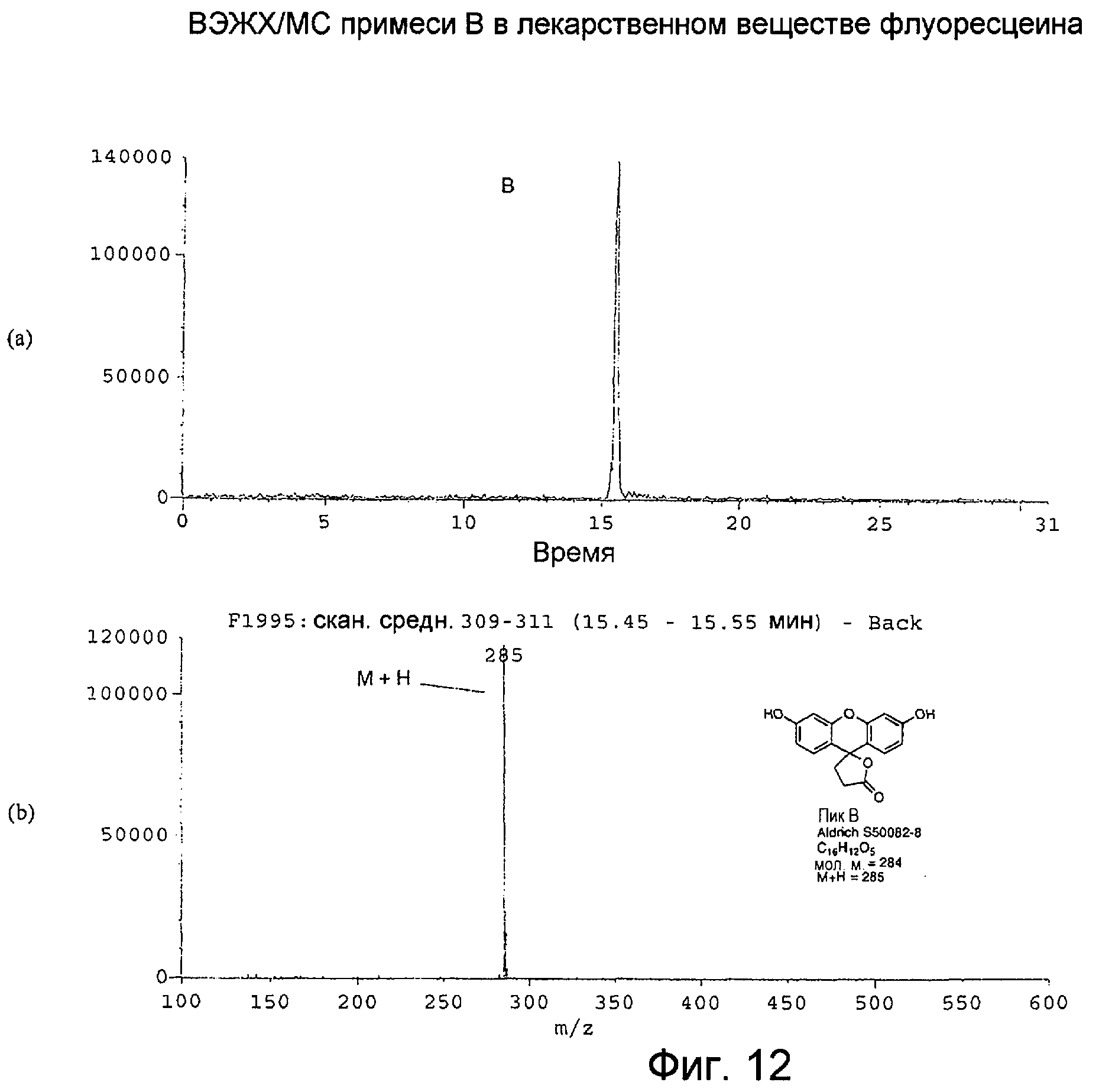
Фигура 12 представляет собой: (а) хроматограмму примеси В во флуоресцеине с масс-селективным детектированием при m/z 285 при использовании масс-спектрометра; и (b) масс-спектр с термораспылением для ВЭЖХ-пика примеси В, собранного в интервале 15,45-15,55 минут.
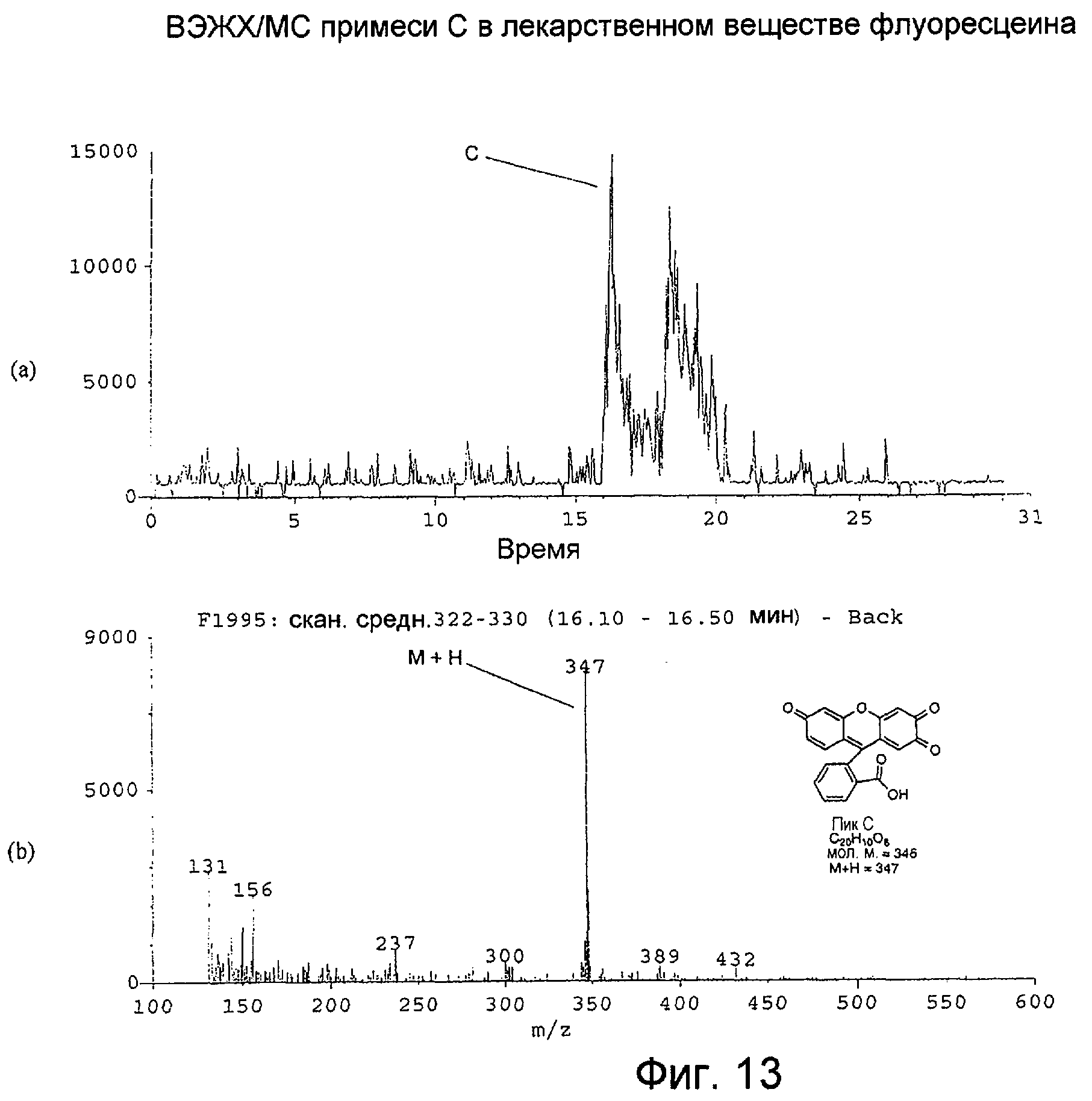
Фигура 13 представляет собой: (а) хроматограмму примеси С во флуоресцеине с масс-селективным детектированием при m/z 347 при использовании масс-спектрометра; и (b) масс-спектр с термораспылением для ВЭЖХ-пика примеси С, собранного в интервале 16,10-16,50 минут.
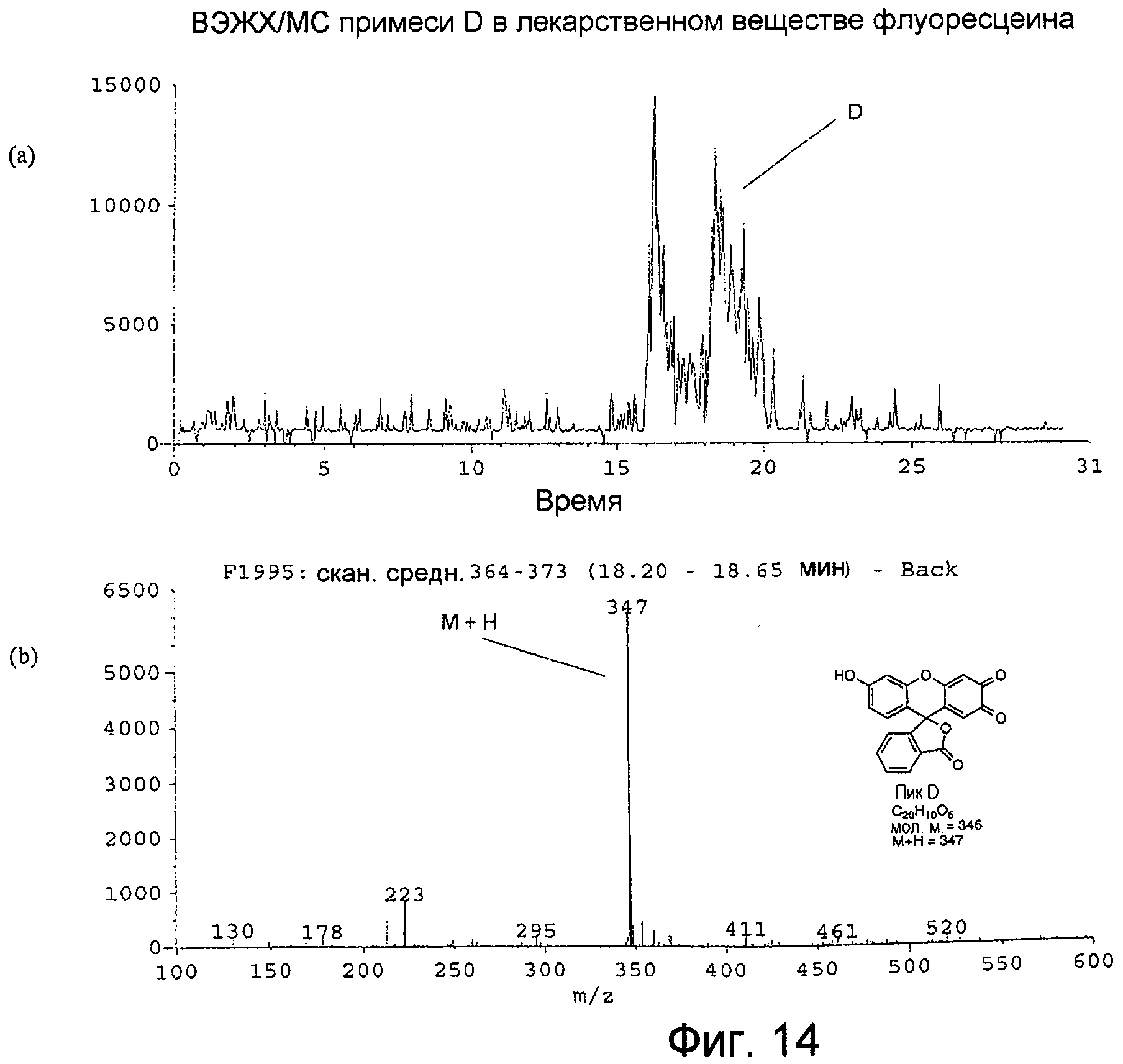
Фигура 14 представляет собой: (а) хроматограмму примеси D во флуоресцеине с масс-селективным детектированием при m/z 347 при использовании масс-спектрометра; и (b) масс-спектр с термораспылением для ВЭЖХ-пика примеси D, собранного в интервале 18,20-18,65 минут.
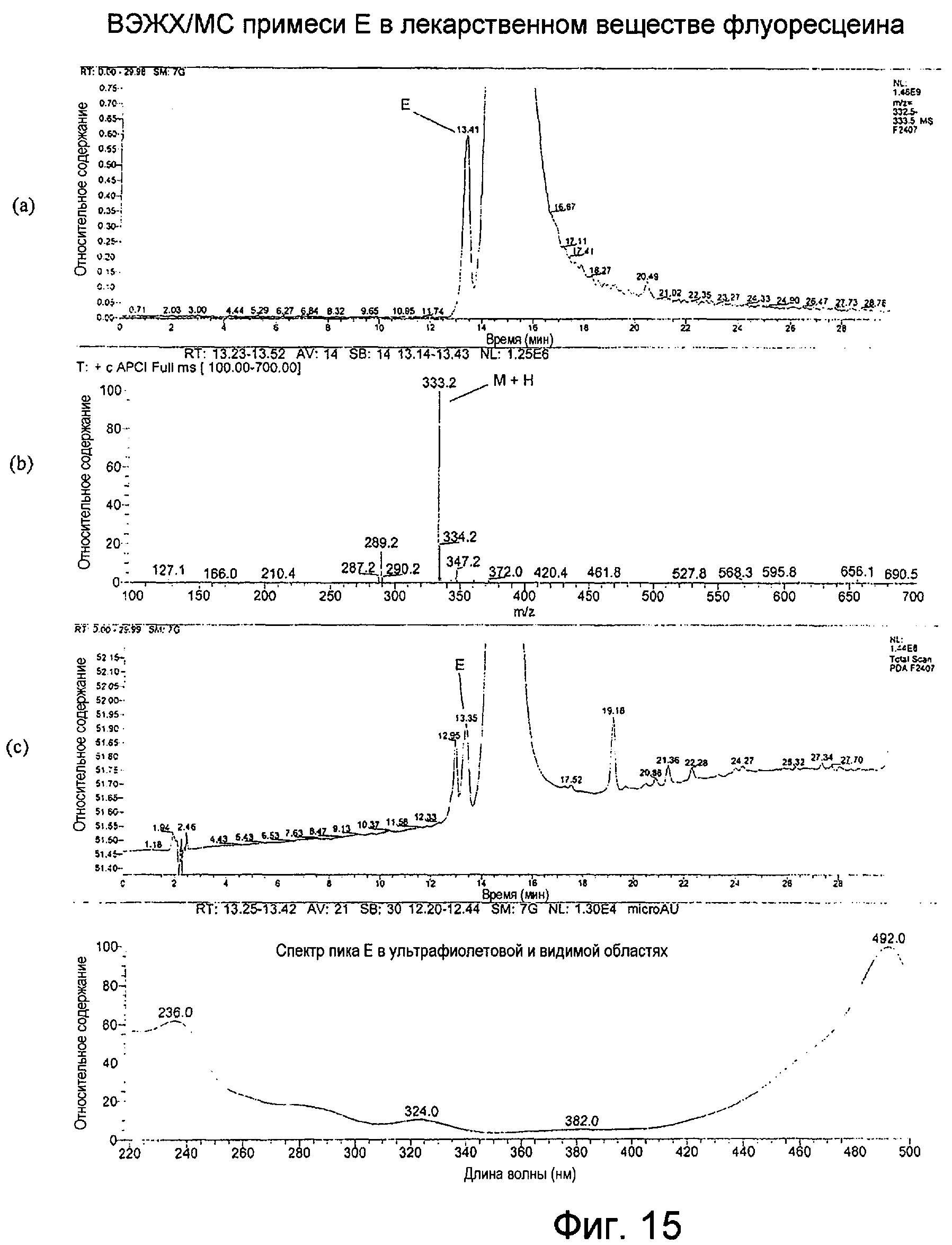
Фигура 15 представляет собой: (а) хроматограмму примеси Е во флуоресцеине с масс-селективным детектированием при m/z 333 при использовании масс-спектрометра; (b) APCI-масс-спектр для ВЭЖХ-пика примеси Е, собранного в интервале 13,23-13,52 минут; (с) хроматограмму примеси Е во флуоресцеине с детектированием посредством общего сканирования поглощения от 220 до 500 нм; и (d) спектр пика примеси Е в ультрафиолетовой и видимой области.
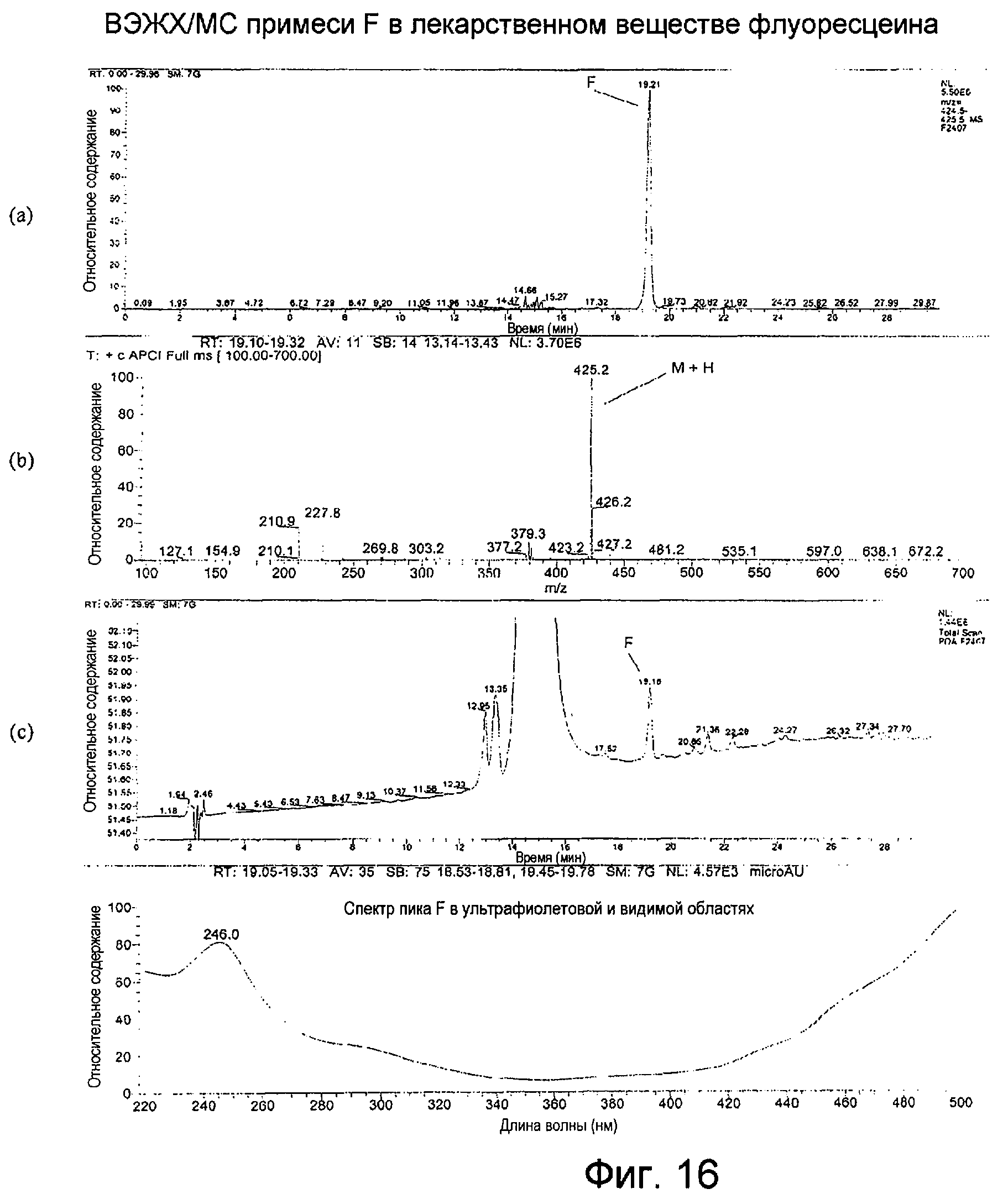
Фигура 16 представляет собой: (а) хроматограмму примеси F во флуоресцеине с масс-селективным детектированием при m/z 425 при использовании масс-спектрометра; (b) APCI-масс-спектр для ВЭЖХ-пика примеси F, собранного в интервале 19,10-19,32 минут; (с) хроматограмму примеси F во флуоресцеине с детектированием посредством общего сканирования поглощения от 220 до 500 нм; и (d) спектр пика примеси F в ультрафиолетовой и видимой области.
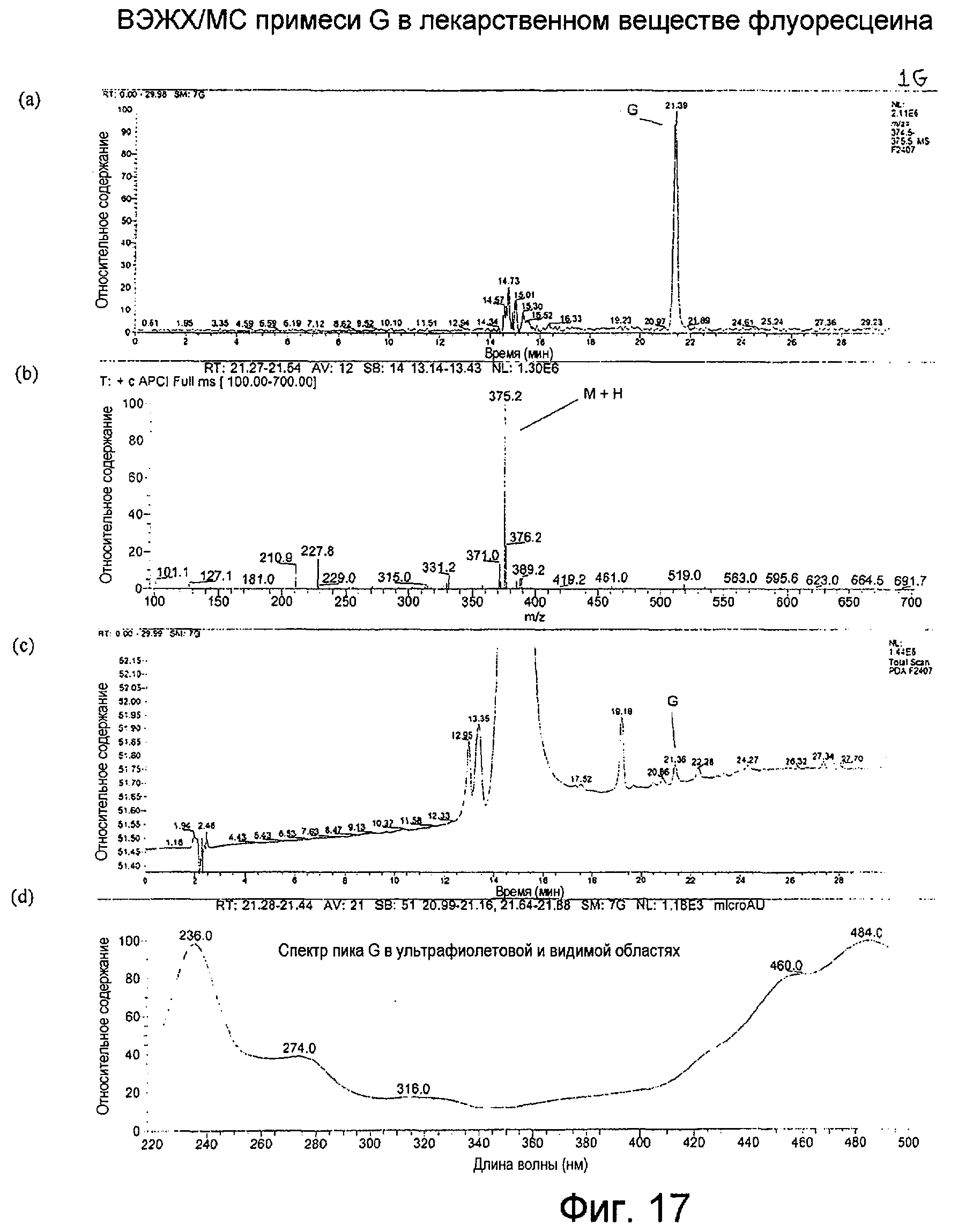
Фигура 17 представляет собой: (а) хроматограмму примеси G во флуоресцеине с масс-селективным детектированием при m/z 375 при использовании масс-спектрометра; (b) APCI-масс-спектр для ВЭЖХ-пика примеси G, собранного в интервале 21,27-21,54 минут; (с) хроматограмму примеси G во флуоресцеине с детектированием посредством общего сканирования поглощения от 220 до 500 нм; и (d) спектр пика примеси G в ультрафиолетовой и видимой области.
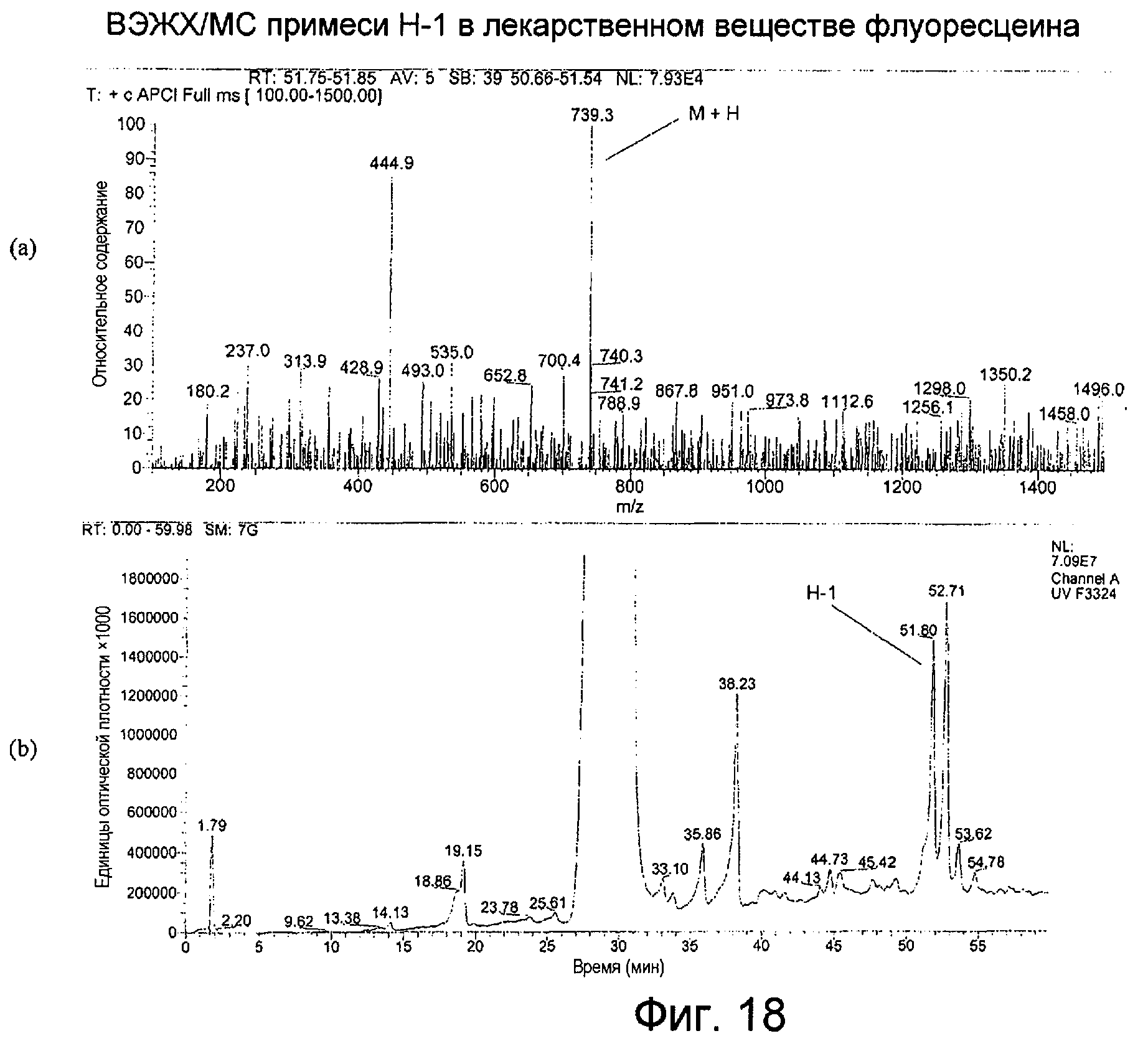
Фигура 18 представляет собой: (а) APCI-масс-спектр для ВЭЖХ-пика примеси H-1, собранного в интервале 51,75-51,85 минут; (b) хроматограмму примеси H-1 во флуоресцеине с детектированием посредством общего сканирования поглощения от 220 до 500 нм.
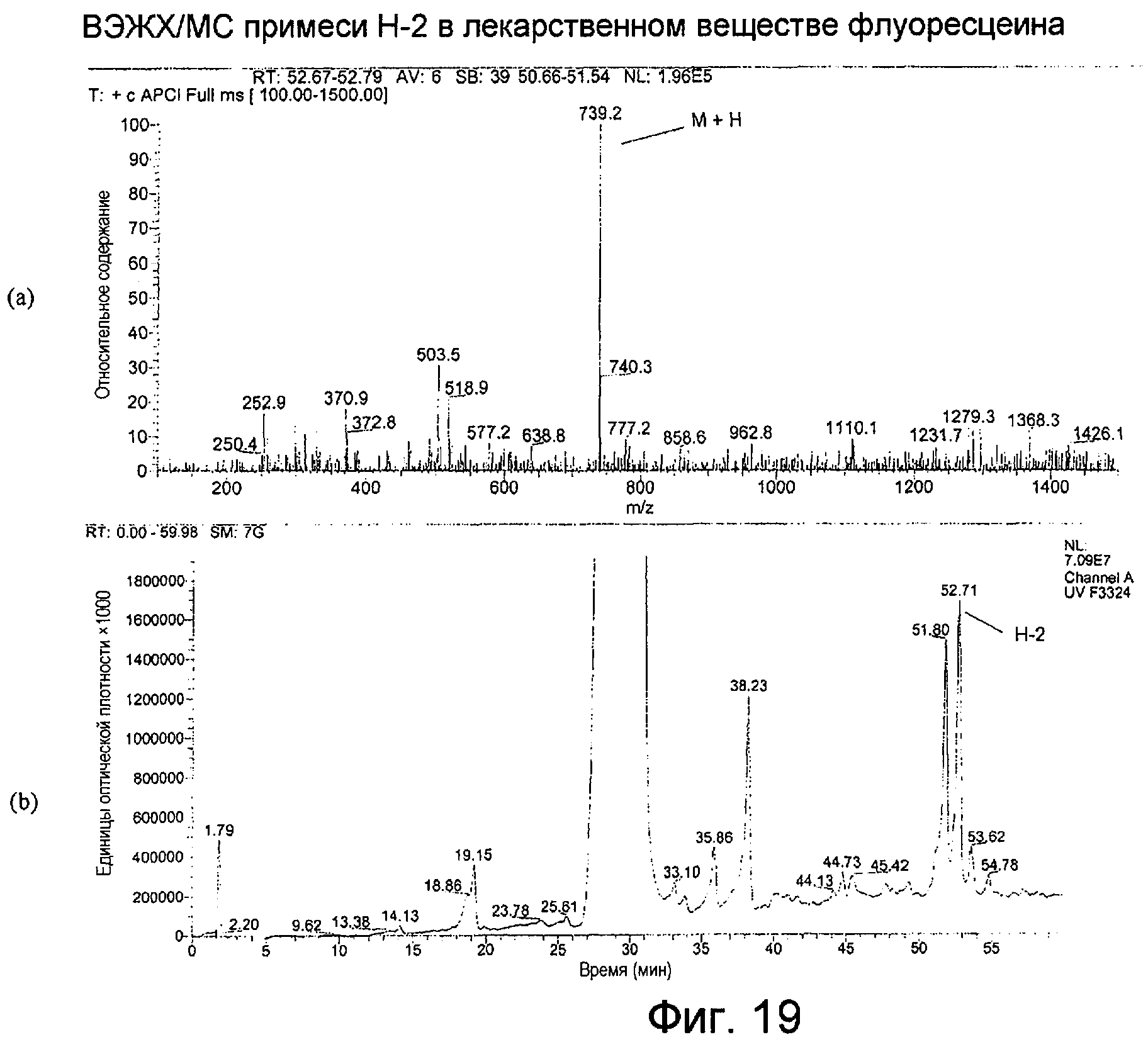
Фигура 19 представляет собой: (а) APCI-масс-спектр для ВЭЖХ-пика примеси H-2, собранного в интервале 52,67-52,79 минут; (b) хроматограмму примеси H-2 во флуоресцеине с детектированием посредством общего сканирования поглощения от 220 до 500 нм.
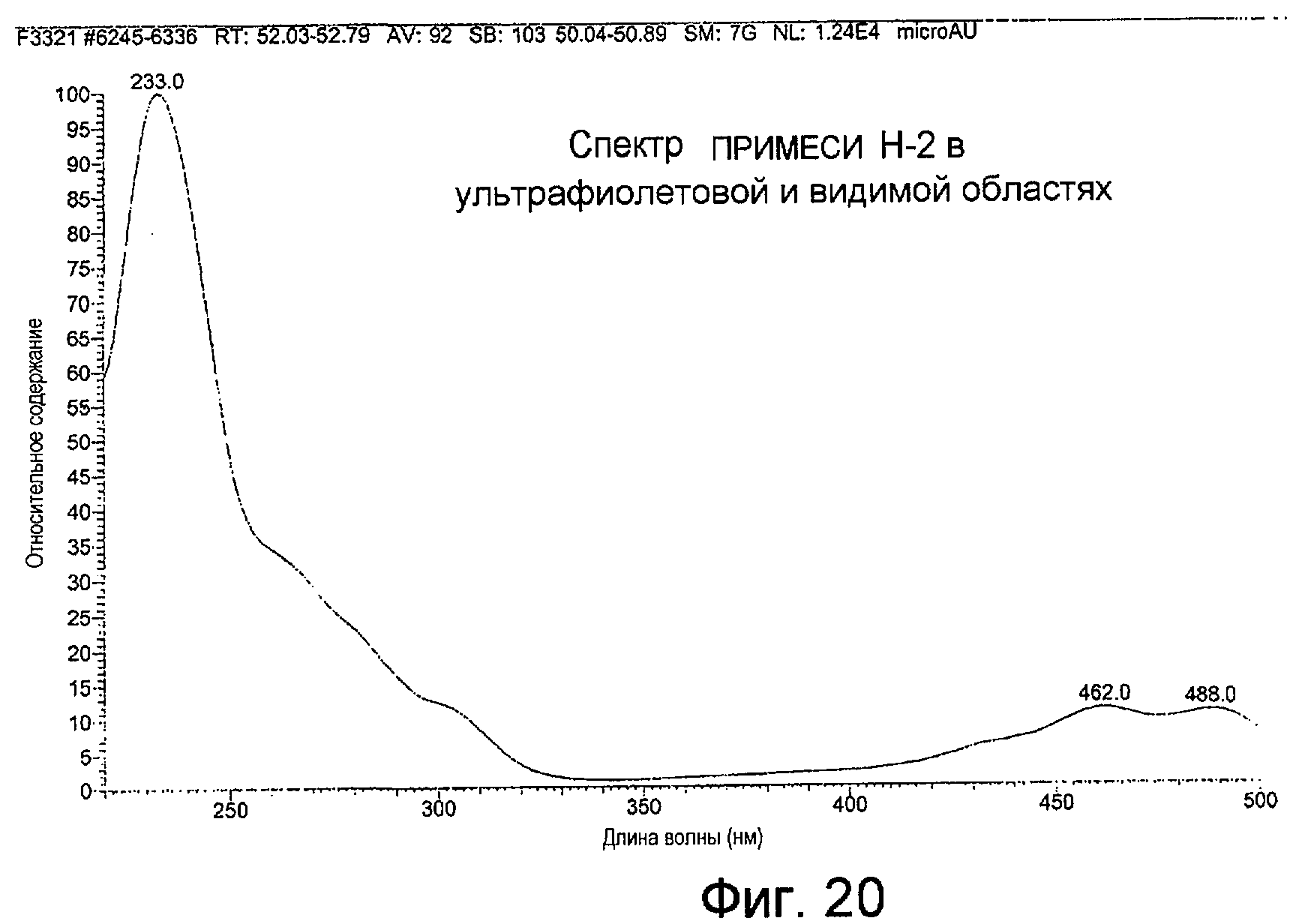
Фигура 20 представляет собой спектр поглощения примеси Н-2 в ультрафиолетовой и видимой области.
Подробное описание изобретения
Использованные в настоящем документе следующие аббревиатуры и термины, если не указано иначе, следует понимать в следующих значениях:
Аббревиатура «APCI» означает «atmospheric pressure chemical ionization» (химическая ионизация при атмосферном давлении).
Аббревиатура «M/S» (М/С) или «MS» (МС) означает масс-спектрометр или масс-спектрометрию.
Аббревиатура «ВЭЖХ» означает высокоэффективную жидкостную хроматографию.
Аббревиатура «UV-Vis» означает ультрафиолетовую и видимую область.
Аббревиатура «ЖХ/МС» означает жидкостную хроматографию с масс-спектрометрометром.
Термин «активированный уголь» охватывает активированные угольные агенты, эффективные для снижения цветового индекса. Типичные агенты включают, но не ограничиваются ими, Norit® SA Plus и Norit® SX Ultra, коммерчески доступные у поставщика Univar USA, Dallas, Texas. Формы активированного угля, способные снижать цветовой индекс можно определить посредством обычного экспериментирования. (Например, было определено, что другая коммерчески доступная форма активированного угля, Darco® KB, для снижения цветового индекса неэффективна.)
Термин «цветовой индекс» означает поглощение света 1,0%-ным раствором исходного сырья флуоресцеина, приготовленного в водном растворе гидроксида натрия и бикарбоната натрия при рН 9,4, измеренное при 590 нм.
Термины «лекарственное вещество флуоресцеина» и «исходное сырье флуоресцеина» в настоящем документе применяются взаимозаменяемо.
Термин «родственная примесь» охватывает синтетические примеси, изомеры, продукты окисления, продукты димеризации и продукты разложения флуоресцеина и/или веществ, реагирующих с флуоресцеином. Типичные структуры таких примесей родственных веществ показаны на Фигуре 8.
Термин «по существу свободный от пиридина» означает, что композиция флуоресцеина свободна от пиридина по меньшей мере на 99%. Более предпочтительно, когда аналитическая чистота составляет по меньшей мере 99,9%; даже еще более предпочтительно, когда композиция флуоресцеина полностью свободна от пиридина.
Термин «по существу чистый флуоресцеин» относится к полному отсутствию или почти полному отсутствию примесей, таких как примеси родственных веществ. Например, когда говорят, что композиция флуоресцеина является по существу чистой, это означает, что либо нет детектируемых примесей родственных веществ, либо, если была детектирована единственная примесь родственного вещества, она присутствует в количестве, не более чем 0,1% по массе, либо, если было детектировано множество примесей родственных веществ, они присутствуют в общем количестве, не более чем 0,6% по массе.
Способы согласно настоящему изобретению позволяют получить флуоресцеин в виде продуктов с низким содержанием примесей родственных веществ. Общеизвестно, что даже очищенный флуоресцеин может содержать низкие уровни определенных примесей, например резорцин и 2-(2',4'-дигидроксибензоил)бензойную кислоту. Однако ранее не было известно, что коммерческие образцы флуоресцеина, одновременно с резорцином и 2-(2',4'-дигидроксибензоил)бензойной кислотой, могут содержать и ряд других примесей. Количество таких потенциальных примесей по существу понижают способами согласно настоящему изобретению. Такие примеси в настоящем документе объединенно называют «примесями родственных веществ».
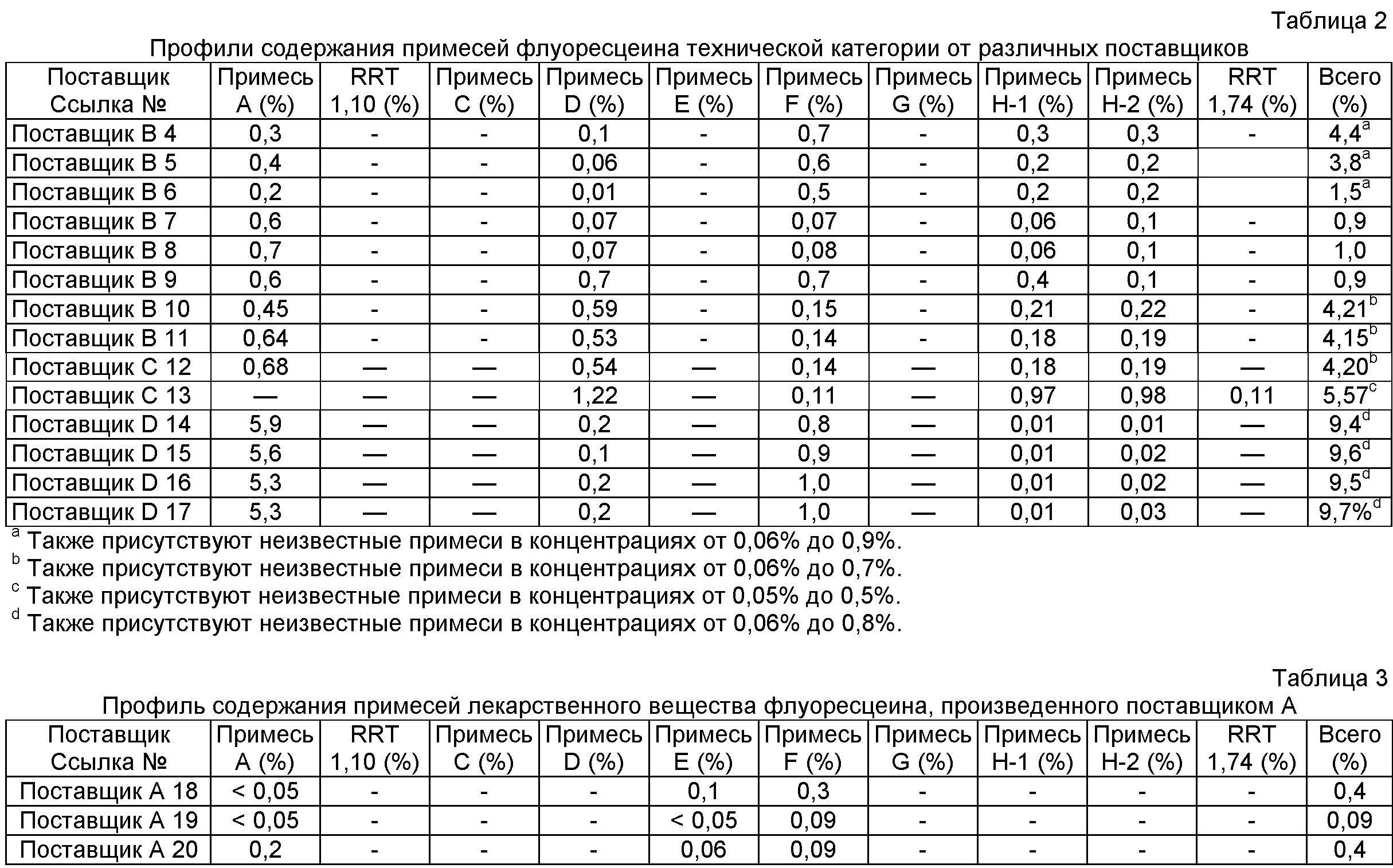
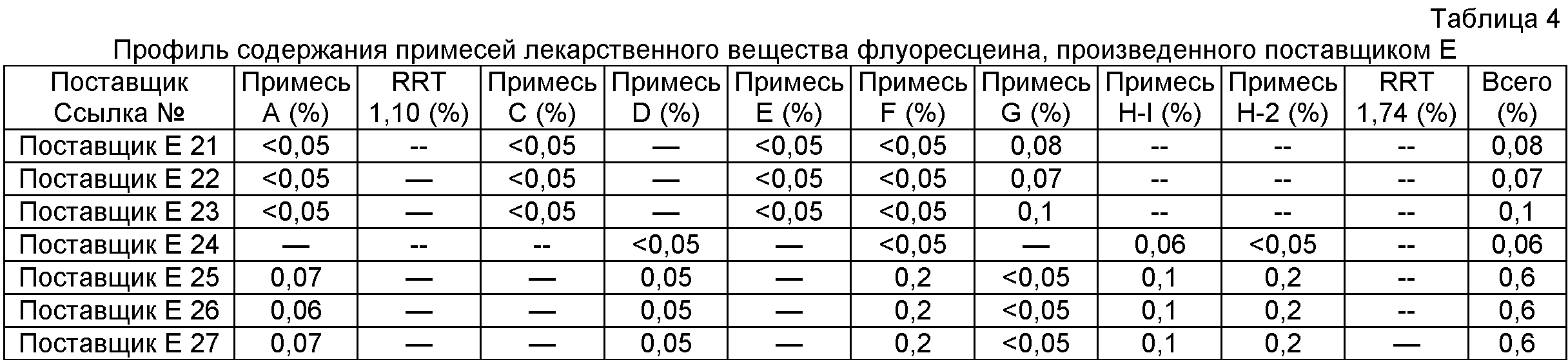
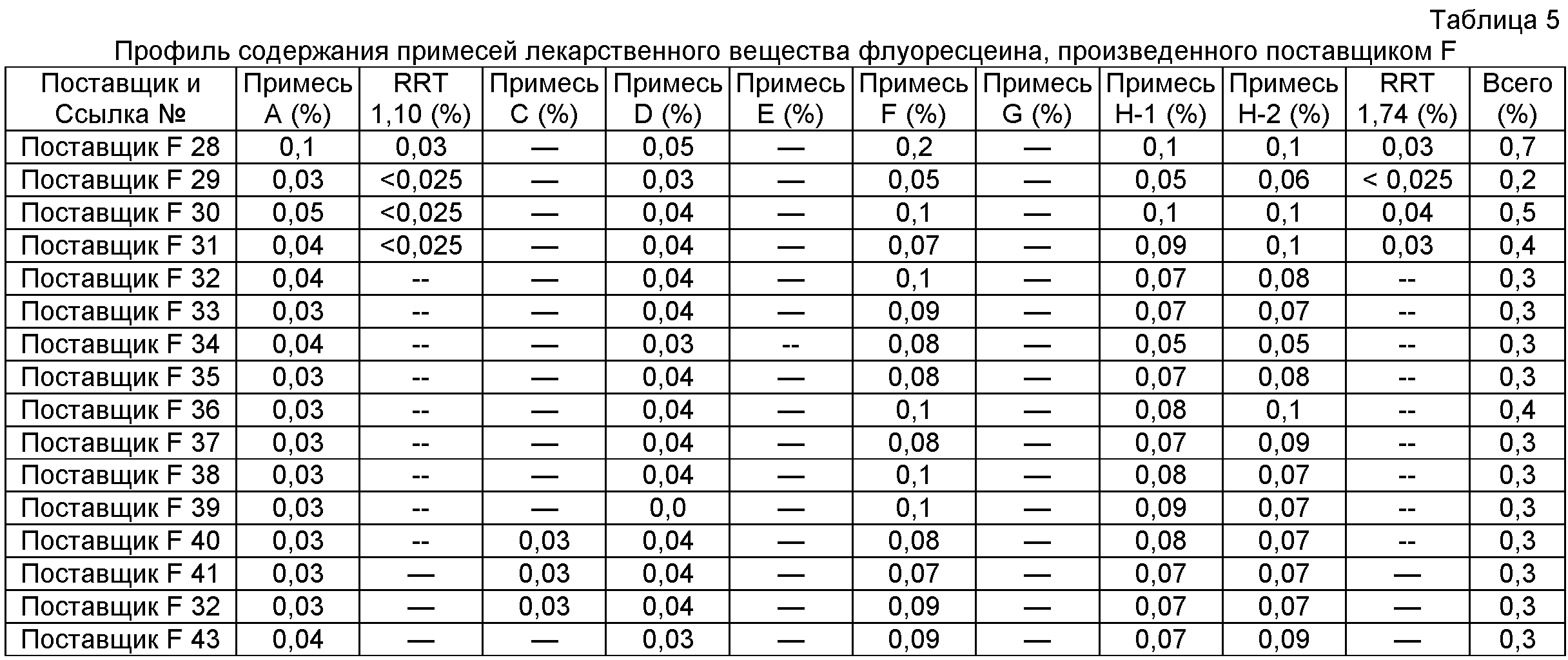
Проводили эксперименты для определения молекулярных масс этих примесей родственных веществ с помощью ЖХ/МС; хотя и без теоретического обоснования, авторы настоящего изобретения предполагают для этих примесей структуры, показанные на Фигуре 8. Был открыт подробно описанный ниже способ разделения и количественного определения низких уровней примесей родственных веществ, которые могут присутствовать даже в композициях очищенного флуоресцеина. Было открыто, что способы согласно настоящему изобретению предоставляют высокоочищенный флуоресцеин, который имеет по существу сниженные уровни примесей родственных веществ. Это можно видеть на профилях содержания примесей лекарственного вещества флуоресцеина, очищенного согласно настоящему изобретению, как показано ниже в Таблице 1, в сравнении с профилем содержания примесей флуоресцеина технической категории от различных производителей, как показано ниже в Таблице 2, и с профилем содержания примесей лекарственного вещества флуоресцеина от различных производителей, как показано ниже в Таблицах 3-5.

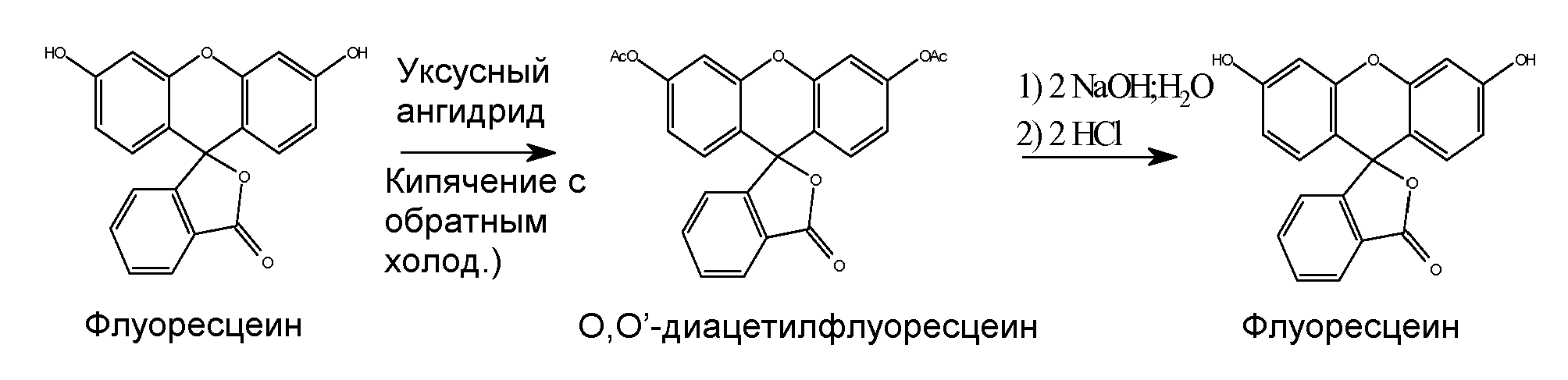
Ниже проиллюстрирована схема общего технологического процесса согласно настоящему изобретению. Флуоресцеин коммерческой категории диацетилируют по реакции с уксусным ангидридом при температурах кипения с обратным холодильником. Полученный таким образом диацетилированный флуоресцеин выделяют и затем проводят его реакцию с основанием для получения деацетилированного флуоресцеина, который затем обрабатывают активированным углем для получения слабоокрашенного флуоресцеина высокой чистоты и с низким содержанием хлорида. Схема реакции проиллюстрирована ниже:
Конкретные растворители, времена и температуры реакций и значения рН, применяемые для получения чистого флуоресцеина согласно настоящему изобретению, определяли на основании ряда экспериментов. Целью этих экспериментов было получение высоко чистого слабоокрашенного флуоресцеина фармацевтической категории с низким содержанием соли без применения вредного растворителя, применяемого в предыдущих известных способах, а именно, пиридина. Дополнительной целью была минимизация материальных затрат и затрат времени при проведении процессов посредством, например, применения минимальных количеств растворителей или уменьшения времени реакционных циклов на необходимых стадиях.
Процесс очистки флуоресцеина начинают с преобразования флуоресцеина в О,O'-диацетилфлуоресцеин. Для этой цели уксусный ангидрид применяют одновременно в качестве растворителя и реагента, полностью избегая применения пиридина, вредного растворителя, применяемого в прототипном способе. В частности, смесь флуоресцеина и уксусного ангидрида перемешивают в течение нескольких часов при кипячении с обратным холодильником и полученной суспензии дают охладиться. Дальнейшее охлаждение до температуры замерзания или несколько ниже ее обеспечивает полную кристаллизацию. Кристаллизованное вещество собирают и промывают сначала холодным уксусным ангидридом, затем холодным ацетоном. После этого вещество ресуспендируют в ацетоне при перемешивании и осторожном нагревании. После охлаждения собирают белое кристаллическое вещество, которое промывают холодным ацетоном и сушат на воздухе, что дает высокочистый О,O'-диацетилфлуоресцеин.
Затем O,О'-диацетилфлуоресцеин преобразовывают назад во флуоресцеин с образованием натриевой соли и с удалением конечных примесей. Для осуществления этого преобразования ацетильные группы O,О'-диацетилфлуоресцеина гидролизуют с использованием щелочного раствора. Для этого O,О'-диацетилфлуоресцеин и метанол загружают в подходящий сосуд, добавляют приготовленный раствор гидроксида натрия в деионизованной воде и при перемешивании смесь нагревают до кипения. Затем смесь охлаждают, фильтруют, применяя вспомогательное средство, и затем промывают метанолом. Объем фильтрата затем уменьшают отгонкой в вакууме, добавляют воду и реакционную смесь охлаждают. После этого устанавливают рН реакционной смеси в диапазоне 8,5-8,7. При перемешивании в течение одного часа добавляют подходящий активированный уголь, например, Norit® SX Ultra. Если необходимо, стадию с активированным углем повторяют. Затем следуют критические стадии осаждения. Сначала к фильтрату добавляют этанол, получая соотношение этанола к воде, равное 2:1. Это соотношение основано на неожиданном открытии, что повышенное отношение органического растворителя к воде в процедуре осаждения приводит к меньшему содержанию хлорида в флуоресцеине-продукте. Эксперименты, проведенные для выявления этого эффекта, описаны ниже. Затем для подкисления флуоресцеина добавляют разбавленный раствор хлористоводородной кислоты, устанавливая калиброванный диапазон рН. Этот диапазон основан на неожиданном открытии, что пониженные значения рН обеспечивают получение продукта с более желаемым цветом. Более конкретно, было экспериментально определено, что оптимальный диапазон рН фильтрата должен быть между 1,0 и 2,5 и что подкисление следует проводить медленно, например в течение 2-4 часов, во избежание агрегации продукта и при охлаждении. После дальнейшего перемешивания и охлаждения флуоресцеин отделяют фильтрованием. Продукт промывают раствором этанола в воде и сушат, что дает 80-90%-ный выход флуоресцеина очень высокого качества. Высокочистый флуоресцеин можно применять для получения флуоресцеина для инъекций. Для этой цели флуоресцеин преобразуют в форму растворимой динатриевой соли с использованием гидроксида натрия и разливают в ампулы с последующей стерилизацией.
Примером эксперимента, проведенного для разработки способа согласно настоящему изобретению является калибровка диапазона рН, при котором осаждают флуоресцеин-продукт. Этот диапазон устанавливали от более высоких до более низких диапазонов, основываясь в частности на эмпирических наблюдениях цветового спектра образующегося продукта. Таким образом было определено, что оптимальный диапазон рН, при котором следует осаждать флуоресцеин-продукт для получения слабоокрашенного продукта составляет от примерно pH 1,0 до примерно pH 2,5.
Было также неожиданно открыто, что высокие отношения органического растворителя к водному растворителю в процедуре осаждения приводят к более низкому содержанию хлорида во флуоресцеине-продукте. Этот результат был неожиданным, поскольку предполагалось, что для снижения уровня хлорида натрия необходимо низкое отношение органики к воде. Применение более высоких концентраций органического растворителя имело дополнительную выгоду в том, что оно улучшало скорость фильтрации, тем самым, сокращая время, требуемое для переработки материала.
Дальнейшие эксперименты по осаждению, которые проводили для разработки способов для настоящего изобретения, описаны ниже; они показаны на Фигурах 1 и 2. В частности, проводили эксперименты по снижению уровня хлорида, меняя способ осаждения, как показано ниже в Таблице 6.
|
Таблица 6 показывает, что изменение соотношения растворителей в среде осаждения влияет на количество присутствующего хлорида. В частности, увеличение отношения этанола к воде в среде осаждения производит более низкое содержание хлорида. Эксперимент А показывает слабое уменьшение содержания хлорида, когда объем среды осаждения с соотношением вода:этанол, равным 1:1 увеличивали от 10 объемов (референсная система) до 15 объемов (см. Таблицу 6). Эксперимент В сравнивает осаждение из среды с соотношением вода:этанол, равным 1:1 (10 объемов, референсная система), с осаждением из среды с соотношением вода:этанол, равным 2:1 (15 объемов). Результат этого эксперимента является неожиданным в том, что референсная реакция, имеющая более низкое содержание воды и меньший объем, производит более низкое содержание хлорида.
Эксперимент С сравнивает осаждение из среды с соотношением вода:этанол, равным 1:1 (10 объемов), с осаждением из среды с соотношением вода:этанол, равным 1:2 (15 объемов). Результаты эксперимента С показывают, что среда осаждения с более высоким содержанием органического растворителя производит более низкое содержание хлорида.
Тренд, по которому более высокое содержание органического растворителя в среде осаждения производит более низкое содержание хлорида, воспроизведен в экспериментах D, E, F и G и экспериментах H, I, J и K как для непромытого, так и для промытого продукта. Хотя эти результаты представляются малопонятными, полагают, без теоретического обоснования, что более высокое содержание органического растворителя делает промывку слоя продукта более быстрой и эффективной.
Другим рассмотренным аспектом была зависимость окраски флуоресцеина от рН среды осаждения; см. ниже Таблицу 7, в которой в качестве среды осаждения применяли смесь этанола с водой в отношении 2:1.
|
Результаты свидетельствуют, что цвет флуоресцеина чувствителен к изменениям рН. Например, при рН около 3,0, внешний вид продукта начинает принимать красно-коричневый оттенок, что считают нежелательным.
Примеры 1-8 приведены ниже, чтобы дополнительно проиллюстрировать определенные варианты осуществления настоящего изобретения. Репрезентативные данные, полученные из Примеров 5, 6, 7 и/или 8, показаны на Фигурах 9-19.
Данные для Фигур 9-14 были получены с помощью ЖХ/МС с использованием масс-спектрометра с термораспылением, сопряженного с ВЭЖХ. Пики наблюдали с использованием УФ-детектора (280 нм) и масс-спектрометра с термораспылением. Экспериментальные условия: инструмент = масс-спектрометр с термораспылением Vestec модели 201В, сопряженный с системой ВЭЖХ Waters модели 600 MS и УФ-детектором Waters модели 486MS (280 нм); колонка = Waters Symmetry C-8, 5 мкм, 3,9 × 150 мм; подвижная фаза = линейный градиент, запрограммированный от 0% В до 100% В за 25 минут; подвижная фаза А = 0,1 М ацетат аммония в смеси метанола с водой (10:90); подвижная фаза В = 0,1 М ацетат аммония в метаноле; скорость потока = 1,0 мл/мин; концентрация образца = без разбавления; инъецируемый объем = 20 мкл.
Данные для Фигур 15-20 были получены с помощью ЖХ/МС с использованием масс-спектрометра, сопряженного с ВЭЖХ. Применяли масс-спектрометр с химической ионизацией при атмосферном давлении (atmospheric pressure chemical ionization, APCI); масс-спектрометр работал в режиме детектирования положительных ионов. Пики наблюдали с использованием детектора для УФ- и видимой области с мониторингом общего поглощения в диапазоне 220-500 нм и с использованием масс-спектрометра. Колонку Waters Symmetry C-8 (3,9 × 150 мм) применяли при скорости потока 0,6 мл/мин по программе от 0% подвижной фазы В до 100% подвижной фазы В за 30 минут. Подвижная фаза В представляла собой 0,01 М ацетат аммония в метаноле, и мобильная фаза А представляла собой 0,01 М ацетат аммония в смеси метанол:вода (10:90).
ПРИМЕР 1
Образование диацетата флуоресцеина
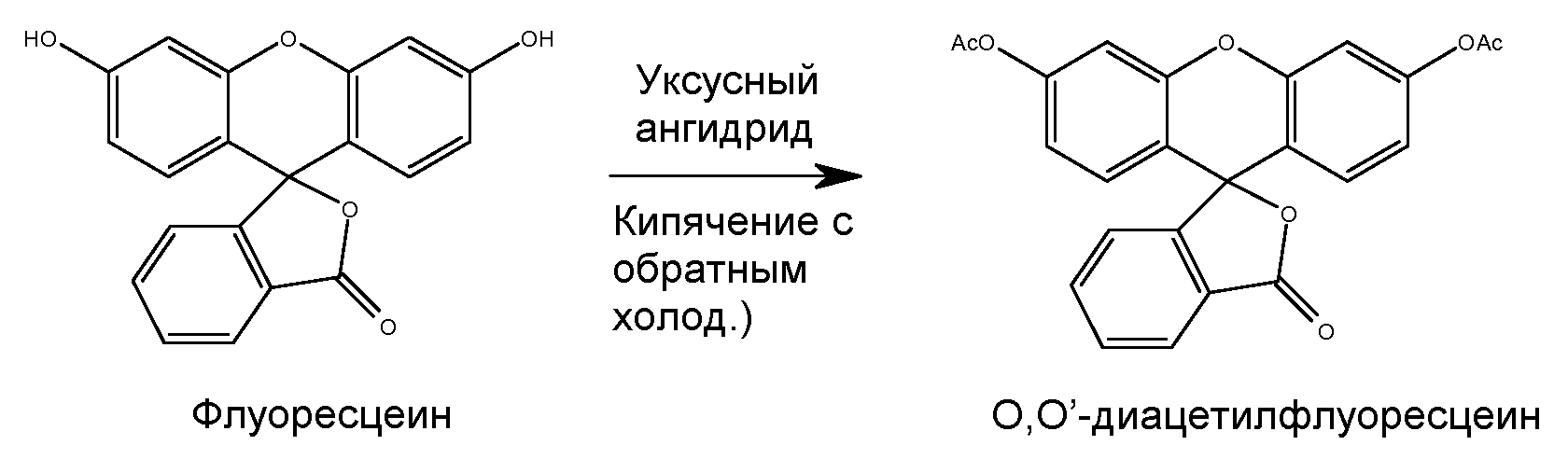
В 5-литровую 3-горлую круглодонную колбу добавляли флуоресцеин (1000 г, 3,01 моль) и уксусный ангидрид (1622 г, 15,9 моль). Полученную смесь перемешивали в течение 3-5 часов при кипячении с обратным холодильником и полученной суспензии давали охладиться до комнатной температуры. При непрерывном перемешивании реакционную смесь далее охлаждали до температуры от -5 до 3°C для полной кристаллизации. Кристаллизованное вещество собирали на воронке Бюхнера, промывали холодным уксусным ангидридом (2 × 500 мл), затем холодным ацетоном (1 × 600 мл). Вещество частично сушили и ресуспендировали в ацетоне (1000 мл) при перемешивании и осторожном нагревании. После охлаждения собирали на фильтре белое кристаллическое вещество, промывали холодным ацетоном (2 × 700 мл) и сушили на воздухе. Выход: ~75%-85%; единственное пятно при ТСХ; т. пл. = 203-205,5°C; чистота 99,7%.
ПРИМЕР 2
Образование флуоресцеина из диацетилфлуоресцеина, образование натриевой соли и удаление конечных примесей
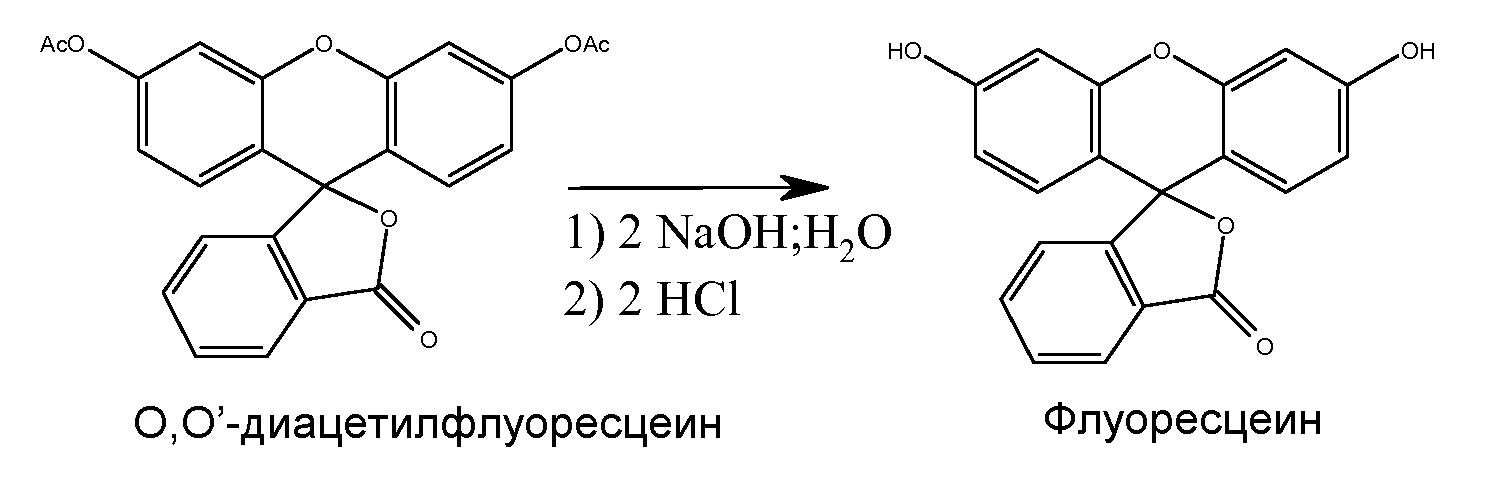
O,О'-диацетилфлуоресцеин (1000 г) и метанол (4000 мл) загружали в подходящий реактор. Отдельно готовили раствор гидроксида натрия (480 г, 50% щелочи) в деионизованной воде (620 мл). Раствор гидроксида натрия загружали в реактор, содержащий O,О'-диацетилфлуоресцеин и метанол. Смесь нагревали до кипения с обратным холодильником и перемешивали при кипячении с обратным холодильником в течение 90 минут. Реакционную смесь охлаждали до 20-25°С. Смесь фильтровали с использованием вспомогательного средства (100 г) и затем промывали метанолом (500 мл). Фильтрат (5000 мл) отгоняли в вакууме до остаточного объема 1400-1700 мл и затем реакционную смесь охлаждали до 20-25°С. К концентрату после отгонки добавляли деионизованную воду (5000 мл). Отдельно готовили раствор гидроксида натрия (56 г, 50% щелочи) в деионизованной воде (72 г). Свежеприготовленный раствор гидроксида натрия (100 мл) применяли для приведения рН реакции к 8,5-8,7. При комнатной температуре в реакцию загружали Norit SX Ultra (100 г), вспомогательное средство для фильтрования (100 г) и деионизованную воду (500 мл) и затем смесь перемешивали в течение 1 часа. Эту партию фильтровали и в фильтрат загружали добавочный Norit SX Ultra (100 г) и деионизованную воду (500 мл) и затем смесь перемешивали в течение 1 часа. Партию фильтровали и промывали деионизованной водой (2000 мл). В фильтрат загружали этанол (10000 мл). Отдельно готовили раствор хлористоводородной кислоты, растворяя соляную кислоту (32%, 820 г) в деионизованной воде (320 мл). Разбавленный раствор кислоты применяли для приведения рН фильтрата к 1,0-2,5, поддерживая температуру партии при 20-25°С. Партию перемешивали при 20-25°С в течение 1 часа и затем выделяли фильтрованием. Слой на фильтре промывали раствором этанола в воде для инъекций (1:3) (2 × 1000 мл). Продукт сушили, получая типичный 80-90%-ный выход флуоресцеина очень высокого качества.
ПРИМЕР 3
Интенсивность цвета лекарственного вещества флуоресцеина
Оборудование
Спектрофотометр, способный принимать 1-см кюветы и проводить сканирование от 660 нм до 570 нм.
Спектрофотометрические кюветы (с длиной пути, равной 1 см) из материала, подходящего для длин волн 660-570 нм, такого как кварц.
Интенсивность цвета лекарственного вещества флуоресцеина измеряли как описано ниже. Процедуру применяли для определения цвета 1,0%-ного раствора исходного сырья флуоресцеина, полученного в водном растворе гидроксида натрия и бикарбоната натрия при рН 9,4, измеряя поглощение при 590 нм. Это значение можно также называть «цветовым индексом». Увеличение поглощения, измеренного при 590 нм, соответствует увеличению интенсивности видимого цвета конечного продукта лекарственного средства.
Флуоресцеин (250 мг ± 5 мг, точно взвешенный) и бикарбонат натрия (50 мг) отвешивали в 25-мл стакан. Добавляли гидроксид натрия (5 мл 1%). Раствор осторожно нагревали при перемешивании. Гидроксид натрия (1%, до 1 дополнительного мл, всего 6,0 мл) добавляли до растворения всего материала и получения прозрачного раствора. Реакцию охлаждали до комнатной температуры. Величину рН доводили до 9,4, при необходимости добавляя по каплям 1%-ный гидроксид натрия. Если значение рН было выше 9,4, раствор отбрасывали и готовили новый, применяя меньше гидроксида натрия. Раствор количественно переносили в мерную колбу (25 мл) и добавляли очищенную воду QS до 25,0 мл. Конечная концентрация составляла 10 мг/мл, или 1%.
Способ
Спектрофотометр обнуляли, устанавливая фоновую базовую линию пользователя с очищенной водой в кювете образца и в кювете сравнения при сканировании от 660 нм до 570 нм со скоростью 100 нм/мин. В кювету образца добавляли раствор флуоресцеина (1%). Раствор образца сканировали от 660 нм до 570 нм при скорости 100 нм/мин. Значение поглощения записывали при 590 нм. Дублирующее измерение проводили на отдельной аликвоте образца. Таблица 1 показывает типичные результаты измерений цветового индекса для некоторых испытаний образцов исходного сырья флуоресцеина с применением этого способа. Результаты были исправлены с использованием уравнения, показанного в разделе, приведенном ниже. Фигура 3 показывает типичный спектр, полученный для образца исходного сырья флуоресцеина.
Расчеты
В значения поглощения вносили поправку на концентрацию образца следующим образом:
Исправленное поглощение = Поглощение × (Целевая масса)/(Действительная масса)
Результаты измерения интенсивности цвета в типичных партиях флуоресцеина получали, как описано в Примере 2; они приведены ниже в Таблице 8.
|
ПРИМЕР 4
Определение остаточного хлорида с применением потенциометрического титрования
Оборудование
Автоматический титратор Brinkman 716 DMS Titrino или эквивалент
Устройство для смены образцов Brinkman 730 и автосэмплер модели 759 с поворотной головкой или эквивалент
Электрод Ag Titrode
5-разрядные аналитические весы или эквивалент
Ультразвуковой аппарат
Нагревательная плитка
Центрифуга на 3000 об/мин
Культуральные пробирки, 16 × 125 мм, VWR Кат. № 47729-578 или эквивалент
Белые крышки для 16-мм культуральных пробирок, VWR Кат. № 60828-760 или эквивалент
Фильтры сыворотки крови, 6 × 16 мм, VWR Кат. № 28295-556 или эквивалент
Следующий способ применяли для количественного определения содержания остаточного хлорида в флуоресцеине с использованием потенциометрического титрования нитратом серебра, автоматического титратора и серебряного электрода.
(1) Получение раствора реагента, гидроксида аммония (5 н.)
К очищенной воде (~600 мл) добавляли концентрированный гидроксид аммония (~338 мл) и разбавляли до 1000 мл очищенной водой. Этот раствор применяли для промывания автотитратора и хранения серебряного электрода.
(2) Получение стандартизованного раствора, нитрата серебра, 0,10 н., водного
Предпочтительно применение 0,10 н. нитрата серебра, получаемого коммерчески в виде раствора с аналитическим сертификатом. Если, однако, коммерческий сертифицированный раствор нитрата серебра недоступен, можно отвесить нитрат серебра (17,0 г), растворить его в очищенной воде (1000 мл) и стандартизовать. Референсный стандарт хлорида калия (сухой, 50 мг) отвешивали и растворяли в очищенной воде (~30 мл) в стакане для титрования. К этому раствору добавляли азотную кислоту (1 мл). Раствор титровали потенциометрически, применяя серебряный стержневой электрод. Каждый мл 0,10 н нитрата серебра эквивалентен 7,455 мг хлорида калия. Нормальность нитрата серебра рассчитывали, применяя следующее уравнение: N AgNO3 = (мг KCl × Чистота KCl)/(мл AgNO3 × 74,55).
(3) Подготовка образца и титрование
Исходное сырье флуоресцеина (2 г) отвешивали в культуральные пробирки (16 × 125 мм). Добавляли очищенную воду (горячую, 10 мл). Добавляли азотную кислоту (1 мл) и все пробирки закрывали крышками, встряхивали в течение 2 минут и обрабатывали ультразвуком в течение 15 минут. Все пробирки центрифугировали в течение 30 минут (~3000 об/мин). Осадок отделяли от надосадочной жидкости, применяя фильтры сыворотки крови. Раствор декантировали в стакан для титрования. (Фильтры) сыворотки крови ополаскивали и фильтровали с использованием очищенной воды (порции по 5 мл). Смывы выливали в стакан для титрования. Фильтры сыворотки крови удаляли и выбрасывали. Вышеописанную процедуру повторяли второй раз, за исключением того, что все пробирки закрывали крышками и встряхивали 1 минуту и все пробирки центрифугировали 20 минут (~3000 об/мин). Растворы из первого и второго экстрактов объединяли и сывороточные фильтры ополаскивали очищенной водой. Объединенный раствор титровали 0,10 н AgNO3 до его потенциометрической конечной точки. Параметры титрования включают применение цикла промывки (5 н. гидроксидом аммония) и цикл ополаскивания (5 н. гидроксидом аммония) после каждого образца. Список параметров показан ниже.
(4) Расчеты
Процент хлорида = (V) × (N) × 35,453 × 100/W
V = объем титрованного 0,10 N AgNO3
N = нормальность титранта AgNO3
35,453 = молекулярная масса хлорида
W = масса взятого образца флуоресцеина
|
ПРИМЕР 5
В этой процедуре получали раствор исходного сырья флуоресцеина в метаноле и отделяли от его родственных веществ, применяя систему высокоэффективной жидкостной хроматографии (ВЭЖХ), программирование градиентной подвижной фазы и колонку С-18. Примеси родственных веществ определяли количественно против 1%-ного раствора флуоресцеина в качестве сравнительного стандарта. Ультрафиолетовый ВЭЖХ-детектор применяли для измерения пиковых ответов при длине волны 280 нм. Мобильная фаза А представляет собой 0,01 М раствор ацетата аммония в смеси 10% метанола и 90% воды с 0,5% уксусной кислоты. Мобильная фаза В представляет собой 0,01 М раствор ацетата аммония в 100% метанола с 0,5% уксусной кислоты. Сравнительным стандартом являлся раствор (0,5 мг/мл) флуоресцеина в метаноле, разбавленный в метаноле до конечной концентрации 0,005 мг/мл или 1% конечной теоретической концентрации образца с концентрацией флуоресцеина, равной 0,5 мг/мл, полученного подобным образом. Применяли систему высокоэффективной жидкостной хроматографии, допускающую программированные градиентные операции, с ВЭЖХ-детектором для УФ- и видимой области, допускающую мониторирование при 280 нм. Колонка: Waters Symmetry C-18, 3,9 × 150 мм, 5 мкм (или эквивалент), способная обеспечивать для флуоресцеина не менее 20000 тарелок на колонку. Скорость потока: 0,6 мл/мин. Градиент программировали следующим образом:
|
Как показано ниже в разделе о расчетах, процентную концентрацию известных и неизвестных примесей, равную или большую 0,025%, рассчитывали, используя площади пиков. Хотя предел количественного определения для этого способа составляет 0,025%, обычно сообщали о концентрациях примесей, составлявших ≥ 0,05%. В анализе флуоресцеина нашли девять примесей, молекулярные массы которых определяли посредством ЖХ/МС. Их предполагаемые структуры представлены на Фигуре 8. Примесь Н было найдено в виде двух диастереомеров, Н-1 и Н-2. Типичные относительные времена удерживания (RRT), факторы емкости (k') и состав градиента во время элюции (% B) для примесей, идентифицированных в партиях флуоресцеина, указанных в настоящей процедуре, определенные с использованием этого хроматографического способа, являются следующими:
|
Пик на хроматограмме флуоресцеина можно идентифицировать как родственное вещество A, D, F, H-1 или H-2, если относительные времена удерживания, фактор емкости и приблизительный состав подвижной фазы данного пика соответствуют родственным соединениям, приведенным выше. Однако каждое из значений относительного времени удерживания может различаться приблизительно на 0,02 в различных хроматографических системах. Модификации в хроматографических системах могут также влиять на значения, приведенные выше. Хотя примеси B, C, E и G не были идентифицированы в четырех партиях флуоресцеина, проанализированных в настоящем изобретении, прежний ЖХ/МС-анализ флуоресцеина поставщика А свидетельствует о том, что примеси B, C, E и G могут иметь приблизительные значения RRT, равные 1,09, 1,11, 1,20 и 1,44. Две известных примеси с RRT, равными 1,10 и 1,74 соответственно, присутствовали в четырех партиях исходного сырья флуоресцеина, указанных в настоящем документе. Их концентрации были между 0,025% и 0,05%. Возможно, что неизвестный пик с RRT = 1,10 мог быть либо примесью В, либо примесью С. Хроматограмма образца исходного сырья флуоресцеина показана на Фигуре 6. В лекарственном веществе флуоресцеина наблюдали диацетилфлуоресцеин, который появляется с временем удерживания относительно флуоресцеина, равным 1,35.
Резорцин является известной обычной примесью для флуоресцеина. Резорцин применяют как исходное сырье в синтезе флуоресцеина; он является и потенциальным продуктом разложения. Было найдено, что резорцин элюируется при RRT, равном 0,14, с k', равным 2,9, и % В, равным 24,6, как показано выше. Иногда можно наблюдать, что резорцин элюируется в виде неразделенного дуплета. Хроматограмма лекарственного вещества флуоресцеина, содержащего резорцин, представлена на Фигуре 7.
Все неродственные пики (т.е. фронт растворителя, системные пики) плюс резорцин были идентифицированы и устранены из следующих расчетов. Резорцин элюировался с RRT, равным примерно 0,14 (см. Фигуру 7). Процентные концентрации для каждого родственного вещества рассчитывали, как показано ниже.
% родственных веществ = [(площадь родственного вещества)/площадь стандарта)] × [(концентрация стандарта)/(концентрация образца)] × 100
% суммы родственных соединений = Σ(индивидуальных примесей ≥ 0,025%)
Рассчитывали сумму примесей, суммируя примеси с концентрациями, равными или большими 0,025%.
Относительное удерживание рассчитывали согласно следующей формуле:
RRT = t i /t f
t i = время удерживания пика примеси
t f = время удерживания пика флуоресцеина
Предел количественного определения флуоресцеина для данного способа был установлен при 0,127 мкг/мл (0,025% концентрации препарата образца). Было определено, что предел детектирования флуоресцеина составляет 0,05 мкг/мл (0,01% концентрации препарата образца).
Данным способом проанализировали четыре партии исходного сырья флуоресцеина от поставщика А. Детектировали семь примесей и пять примесей идентифицировали (A, D, F, H-1 и H-2). Общий процент указываемых примесей (≥ 0,025%) составлял от 0,2% до 0,7%. Результаты приведены ниже в Таблице 10.
В альтернативе этой процедуре заменили разбавитель для препаратов образцов и стандарта, чтобы сделать возможным одновременный анализ резорцина и других примесей родственных веществ. Для приготовления разбавителя сначала растворяли 0,77 г ацетата аммония в 1000 мл воды, уксусной кислотой доводили рН до 3,9 и затем добавляли равные объемы аммоний-ацетатного буфера и метанола. После начального растворения флуоресцеина в метаноле в отношении 50 мг на 15 мл этот разбавитель затем применяли вместо метанола для разбавления стандартов и образцов и в процедуре, описанной в Примере 5; в бланке также заменяли метанол этим разбавителем. После разбавления разбавителем стандарт флуоресцеина и препараты образов защищали от света. Типичные относительные времена удерживания (RRT) для резорцина и фталевой кислоты были следующими:
|
В расчет примесей можно также добавить параметр RRF (Relative Response Factor, относительный фактор ответа, или относительная чувствительность), который представляет собой относительный ответ детектора в сравнении с флуоресцеином.
|
|
Процентные концентрации для каждого родственного вещества можно рассчитать, как показано ниже:
% родственных веществ = [(площадь родственного вещества)/(площадь стандарта)] × [(концентрация стандарта)/(концентрация образца)] × 100
|
ПРИМЕР 6
Проводили исследование для идентификации примесей веществ, родственных флуоресцеину. Образцы флуоресцеина анализировали на присутствие и концентрацию примесей. Идентификационный анализ проводили посредством высокоэффективной жидкостной хроматографии с масс-спектрометрией (ЖХ/МС).
Репрезентативная ВЭЖХ-хроматограмма из системы ЖХ/МС воспроизведена на Фигуре 9. Флуоресцеин производил главный пик на этой хроматограмме. Масс-спектр с термораспылением, показанный на Фигуре 10(b), производил молекулярный ион M+H при m/z 333, что согласуется с молекулярной массой, равной 332.
Масс-спектр с термораспылением для примеси А, как показано на Фигуре 11(b), производил молекулярный ион М+Н при m/z 259, свидетельствуя о молекулярной массе, равной 258. Структура, предложенная на Фигуре 8 для примеси А, [2-(2',4'-дигидроксибензоил)бензойная кислота], была ранее описана как примесь в препаратах флуоресцеина.
Масс-спектр с термораспылением для примеси В, как показано на Фигуре 12(b), демонстрирует молекулярный ион M+H при m/z 285, свидетельствуя о молекулярной массе, равной 284. Молекулярная масса 284 может соответствовать элементным формулам C15H8O6, C16H12O5 или C17H18O4. Предложенная структура, показанная для примеси В на фигуре 8, может образовываться при реакции резорцина с янтарной кислотой (примесь фталевой кислоты как предшественника флуоресцеина).
Масс-спектр с термораспылением для примеси С, как показано на Фигуре 13(b), демонстрирует молекулярный ион М+Н при m/z 347. Это представляет молекулярную массу, равную 346, и соответствует увеличению молекулярной массы на 14 единиц молекулярной массы по сравнению с флуоресцеином.
Масс-спектр с термораспылением для примеси D, как показано на Фигуре 14(b), также производил молекулярный ион М+Н при m/z 347, который, по-видимому, является изомером примеси С. Структуры, предложенные для примесей С и D, являются взаимно таутомерными продуктами окисления флуоресцеина хининового типа.
APCI примеси Е, как показано на Фигуре 15(b), производила молекулярный ион М+Н при m/z 333, свидетельствуя о молекулярной массе, равной 332; УФ-спектр имел максимум при 492. Таким образом, примесь Е, по-видимому, может представлять собой позиционный изомер флуоресцеина.
APCI примеси F, как показано на Фигуре 16(b), производила молекулярный ион М+Н при m/z 425, свидетельствуя о молекулярной массе, равной 424; УФ-спектр имел максимум при длине волны, большей 400 нм. Спектры, по-видимому, согласуются с дополнительной молекулой резорцина, добавленной к исходному соединению. Таким образом, соединение F может образовываться из трех резорцинов, тогда как исходное вещество может образовываться из двух резорцинов.
APCI примеси G, как показано на Фигуре 17(b), производила молекулярный ион М+Н при m/z 375, свидетельствуя о молекулярной массе, равной 374; УФ-спектр имел максимум при 484 нм. Как спектры, так и липофильность, по-видимому, согласуются с ацетатным сложным эфиром флуоресцеина.
APCI примесей H-1 и H-2, как показано на Фигурах 18(а) и 19(а), производила молекулярный ион М+Н при m/z 739, свидетельствуя о молекулярной массе, равной 738 для каждого из них. Спектры поглощения в ультрафиолетовой и видимой областях обоих соединений были одинаковыми, с максимумом при 233 нм и более слабыми максимумами поглощения при 462 и 488 нм. Спектр поглощения примеси Н-2 показан на Фигуре 20.
Пример 7
Получение флуоресцеина для инъекций, или Флуоресцита 25%
Необходимое количество гидроксида натрия отвешивали в емкости для смешивания и растворяли в 60% требуемой воды для инъекций. Добавляли и растворяли флуоресцеин. Добавляли добавочную воду для инъекций, если это требовалось для растворения, но объем не превышал 90% общего объема. Если после 30 минут перемешивания флуоресцеин полностью не растворялся, переходили к следующей стадии регулирования рН. Значение рН доводили до 9,4 гидроксидом натрия (3 н.) и/или хлористоводородной кислотой (1 н.). Смесь перемешивали в течение 30 минут при 180 об/мин. Повторно проверяли значение рН. Если оно было меньше 9,3 или больше 9,5, его повторно доводили до 9,4 с помощью 3 н. гидроксида натрия и/или 1 н. хлористоводородной кислоты. Раствор натрий-флуоресцеина доводили до нужного объема дополнительной водой для инъекций и перемешивали в течение 15 минут. Повторно проверяли рН, как указано выше. Применяя азотный резервуар, раствор фильтровали под давлением через серию из трех мембранных фильтров с порами размером 5 микрон, 0,8 микрон и 0,45 микрон в стерильную емкость для розлива. Значение рН у продукта опять проверяли, применяя вышеуказанную процедуру. Образец асептически отбирали для лабораторных испытаний. Продуктом наполняли предварительно стерилизованные ампулы. В каждую ампулу добавляли 2,15-2,25 мл. Непосредственно после заполнения образцы стандартными способами запаивали, оплавляя концы или заплавляя при вытяжении. При стерилизации испытывали каждую ампулу. Ампулы стерилизовали автоклавированием при 121°С в течение 20 минут или более продолжительно в зависимости от размера партии. Тщательно проверяли на герметичность. Каждую ампулу индивидуально проверяли на наличие нерастворенных частиц в условиях оптимального освещения.
ПРИМЕР 8
Получение флуоресцеина для инъекций, или Флуоресцита 10%
В качестве альтернативной процедуры получения флуоресцеина для инъекций, в подходящую емкость из нержавеющей стали добавляли холодную воду для инъекций (приблизительно 30°С) в объеме, составлявшем приблизительно 70-75% от количества партии. Добавляли флуоресцеин при перемешивании до полного суспендирования. Регистрировали начальное значение рН. Добавляли достаточное количество (приблизительно 7,5% от общего объема) 7 н. гидроксида натрия, затем добавляли дополнительные количества 7 н. гидроксида натрия, повторно проверяя значение рН после выдерживания в течение приблизительно 15 минут между добавлениями, пока значение рН не становилось равным 9,3-9,5 при целевом рН 9,4. Если значение рН было больше 9,5, его подводили до 9,3-9,5, добавляя 1 н. хлористоводородную кислоту. После достижения этого диапазона рН перемешивание продолжали не менее 15 минут. Водой для инъекций партию доводили до конечной массы и перемешивали не менее 30 минут. Проверяли рН и при необходимости его подводили гидроксидом натрия или хлористоводородной кислотой. Продуктом асептически заполняли стерильные флаконы с последующей проверкой и испытаниями согласно стандартным рабочим процедурам.
Конкретные варианты осуществления, описанные в настоящем документе, не предназначены для того, чтобы быть каталогом всех вариантов осуществления настоящего изобретения. Кроме того, специалисты в данной области признают или будут способны определить, применяя только обычные эксперименты, многие эквиваленты вариантов осуществления настоящего изобретения. Такие эквиваленты считают охваченными следующими пунктами формулы изобретения.
Водные фармацевтические композиции, содержащие борат-полиольные комплексы
Низковязкие высокофлокулированные суспензии триамцинолона ацетонида для интравитреальных инъекций
Автоматическое инжекторное устройство для введения интраокулярной линзы
Ультразвуковая рукоятка со смещением
Фармацевтические композиции, обладающие желаемой биодоступностью
Инжектор интраокулярной линзы постоянной силы
Выравнивание интраокулярной линзы с использованием центра роговой оболочки
Устройство для введения интраокулярной линзы, содержащее картридж с внутренним покрытием
Стабилизированные фармацевтические субмикронные суспензии и способы их получения
Офтальмологическая эндоиллюминация с использованием света, генерируемого волокном
Водные фармацевтические композиции, содержащие борат-полиольные комплексы
Низковязкие высокофлокулированные суспензии триамцинолона ацетонида для интравитреальных инъекций
Автоматическое инжекторное устройство для введения интраокулярной линзы
Ультразвуковая рукоятка со смещением
Фармацевтические композиции, обладающие желаемой биодоступностью
Инжектор интраокулярной линзы постоянной силы
Выравнивание интраокулярной линзы с использованием центра роговой оболочки
Устройство для введения интраокулярной линзы, содержащее картридж с внутренним покрытием
Стабилизированные фармацевтические субмикронные суспензии и способы их получения
Офтальмологическая эндоиллюминация с использованием света, генерируемого волокном

