Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ СОЕДИНЕНИЯ 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к производным соединениям 1,3,4-оксадиазола, имеющим ингибирующее действие на гистондеацетилазу 6 (HDAC6), их стереоизомерам их фармацевтически приемлемым солям, их применению в получении лекарственного средства, фармацевтической композиции, содержащей их, терапевтическому способу с применением композиции и способу их получения.
Уровень техники
В клетках, пост-трансляционная модификация, такая как ацетилирование, служит в качестве очень важного регулирующего модуля на концентраторе биологических процессов, а также сильно контролируется множеством ферментов. В качестве корового белка, составляющего хроматин, гистон функционирует в качестве оси, вокруг которой оборачивается ДНК, и, таким образом, помогает конденсации ДНК. Также, баланс между ацетилированием и деацетилирование гистона играет очень важную роль в экспрессии гена.
Известно, что в качестве фермента для удаления ацетильной группы из лизинового остатка гистонового белка, который составляет хроматин, гистондеацетилаза (HDAC) ассоциирована с выключением генов и индуцированием остановки клеточного цикла, ангиогенным ингибированием, иммунорегуляцией, апоптозом и т.д. (Hassig et al., Curr. Opin. Chem. Biol. 1997, 1, 300-308). Также сообщается, что ингибирование функций фермента HDAC индуцирует раковые клетки совершать апоптоз для самих себя за счет снижения активности факторов, связанных с выживанием раковых клеток, и активации факторов, связанных с гибелью раковых клеток в организме (Warrell et al., J. Natl. Cancer Inst. 1998, 90, 1621-1625).
У людей, 18 HDAC известны и классифицируются на четыре класса согласно гомологии с HDAC дрожжей. В этом случае, одиннадцать HDAC с применением цинка в качестве кофактора могут быть поделены на три группы: класс I (HDAC1, 2, 3, 8), класс II (IIa: HDAC4, 5, 7, 9; IIb: HDAC6, 10) и класс IV (HDAC11). Далее, семь HDAC класса III (SIRT 1-7) применяют NAD+ в качестве кофактора вместо цинка (Bolden et al., Nat. Rev. Drug Discov. 2006, 5(9), 769-784).
Разные ингибиторы HDAC в настоящее время находятся на стадии предклинической или клинической разработки, но только не селективные ингибиторы HDAC известны в качестве противоракового агента на данный момент. Вориностат (SAHA) и ромидепсин (FK228) получили одобрение в качестве терапевтического агента для кожной Т-клеточной лимфомы, в то время как панобиностат (LBH-589) получил одобрение в качестве терапевтического агента для множественной миеломы. Однако, известно, что не селективные ингибиторы HDAC обычно являются причиной побочных эффектов, таких как общая слабость, тошнота и подобные в высоких дозах (Piekarz et al., Pharmaceuticals 2010, 3, 2751-2767). Сообщалось, что побочные эффекты вызываются ингибированием класса I HDAC. Из-за побочных эффектов, и т.д., не селективные ингибиторы HDAC были подвергнуты ограничениям на разработку лекарственных средств в других областях, кроме противоракового агента (Witt et al., Cancer Letters 277 (2009) 8.21).
Между тем, сообщали, что селективное ингибирование HDAC класса II не проявляет токсичности, которая имеет место при ингибировании HDAC класса I. В случае разработки селективных ингибиторов HDAC, вероятно, будут устранены побочные эффекты, такие как токсичность и т. д., вызванные неселективным ингибированием HDAC. Соответственно, существует вероятность того, что селективные ингибиторы HDAC могут быть разработаны в качестве эффективных терапевтических агентов при различных заболеваниях (Matthias et al., Mol. Cell. Biol. 2008, 28, 1688-1701).
Известно, что HDAC6, один из класса IIb HDAC, в основном присутствует в цитоплазме и содержит белок тубулин, таким образом вовлекаясь в деацетилирование множества не гистоновых субстратов (HSP90, кортактин, и т.д.) (Yao et al., Mol. Cell 2005, 18, 601-607). HDAC6 имеет два каталитических домена, в которых цинк пальцевый домен C-конца может связываться с убиквитинированным белком. Известно, что HDAC6 имеет множество не гистоновых белков в качестве субстрата и, таким образом, играет важную роль в разных заболеваниях, таких как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания, нейродегенеративные заболевания и подобные (Santo et al., Blood 2012 119: 2579-2589; Vishwakarma et al., International Immunopharmacology 2013, 16, 72-78; Hu et al., J. Neurol. Sci. 2011, 304, 1-8).
Структурная особенность, которая является общей для различных ингибиторов HDAC, состоит из кэп-группы, линкера и цинк-связывающей группы (ZBG), как показано на следующей структуре вориностата. Многие исследователи провели исследование ингибирующей активности в отношении ферментов и селективности через структурную модификацию кэп-группы и линкера. Среди групп, известно, что цинк- связывающая группа играет более важную роль в ингибирующей активности и селективности ферментов (Wiest et al., J. Org. Chem. 2013 78: 5051-5065; Methot et al., Bioorg. Med. Chem. Lett. 2008, 18, 973-978).
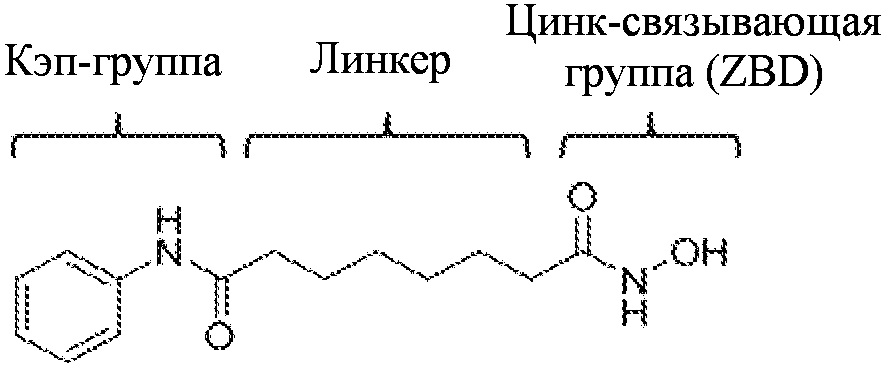
Большая часть указанной цинк-связывающей группы состоит из гидроксаминовой кислоты или бензамида, из которых производные гидроксамовой кислоты проявляют сильное HDAC ингибирующее действие, но имеют проблему низкой биодоступности и серьезной нецелевой активности. Бензамид содержит анилин и, следовательно, имеет проблему, заключающуюся в том, что он может продуцировать токсичные метаболиты in vivo (Woster et al., Med. Chem. Commun. 2015, онлайн публикация).
Соответственно, для лечения рака, воспалительных заболеваний, аутоиммунных заболеваний, неврологических заболеваний, нейродегенеративных нарушений и подобных существует необходимость в разработке селективного ингибитора HDAC6, который имеет цинка-связывающую группу с улучшенной биодоступностью, не вызывая при этом побочных эффектов, в отличие от не селективных ингибиторов, имеющих побочные эффекты.
[Ссылки известного уровня техники]
[Патентный документ]
Международная патентная публикация № WO 2011/091213 (опубликована 28 июля 2011): ACY-1215
Международная патентная публикация № WO 2011/011186 (опубликована 27 января 2011): Tubastatin
Международная патентная публикация № WO 2013/052110 (опубликована 11 апреля 2013): Sloan-K
Международная патентная публикация № WO 2013/041407 (опубликована 28 марта 2013): Cellzome
Международная патентная публикация № WO 2013/134467 (опубликована 12 сентября 2013): Kozi
Международная патентная публикация № WO 2013/008162 (опубликована 17 января 2013): Novartis
Международная патентная публикация № WO 2013/080120 (опубликована 06 июня 2013): Novartis
Международная патентная публикация № WO 2013/066835 (опубликована 10 мая 2013): Tempero
Международная патентная публикация № WO 2013/066838 (опубликована 10 мая 2013): Tempero
Международная патентная публикация № WO 2013/066833 (опубликована 10 мая 2013): Tempero
Международная патентная публикация № WO 2013/066839 (опубликована 10 мая 2013): Tempero
Подробное описание изобретения
Техническая проблема
Объектом настоящего изобретения является предоставление производных 1,3,4-оксадиазола, обладающих селективной ингибирующей активностью в отношении HDAC6, их стереоизомеров или их фармацевтически приемлемых солей.
Другим объектом настоящего изобретения является предоставление фармацевтической композиции, содержащей производные 1,3,4-оксадиазола, обладающие селективной ингибирующей активностью в отношении HDAC6, их стереоизомеры или их фармацевтически приемлемые соли.
Еще одной целью настоящего изобретения является предоставление способа его получения.
Еще одной целью настоящего изобретения является предоставление фармацевтической композиции, содержащей указанные соединения, для профилактики или лечения заболеваний, связанных с активностью HDAC6, включая рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания или нейродегенеративные нарушения.
Еще одной целью настоящего изобретения является предоставление его применения для профилактики или лечения заболеваний, связанных с активностью HDAC6.
Еще одна цель настоящего изобретения состоит в том, чтобы предоставить его применение для приготовления лекарственного средства для профилактики или лечения заболеваний, связанных с активностью HDAC6.
Еще одна цель настоящего изобретения состоит в том, чтобы предоставить способ лечения заболеваний, связанных с активностью HDAC6, включающий введение терапевтически эффективного количества фармацевтической композиции, содержащей указанные соединения.
Техническое решение
Авторы настоящего изобретения обнаружили производные соединения 1,3,4-оксадиазола, обладающие ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6) и применение их в профилактике или лечении заболеваний, тем самым завершив настоящее изобретение.
Производные соединения 1,3,4-оксадиазола
Производные соединения 1,3,4-оксадиазола настоящего изобретения, их стереоизомеры или их фармацевтически приемлемые соли представлены следующей химической формулой I:
[Химическая формула I]
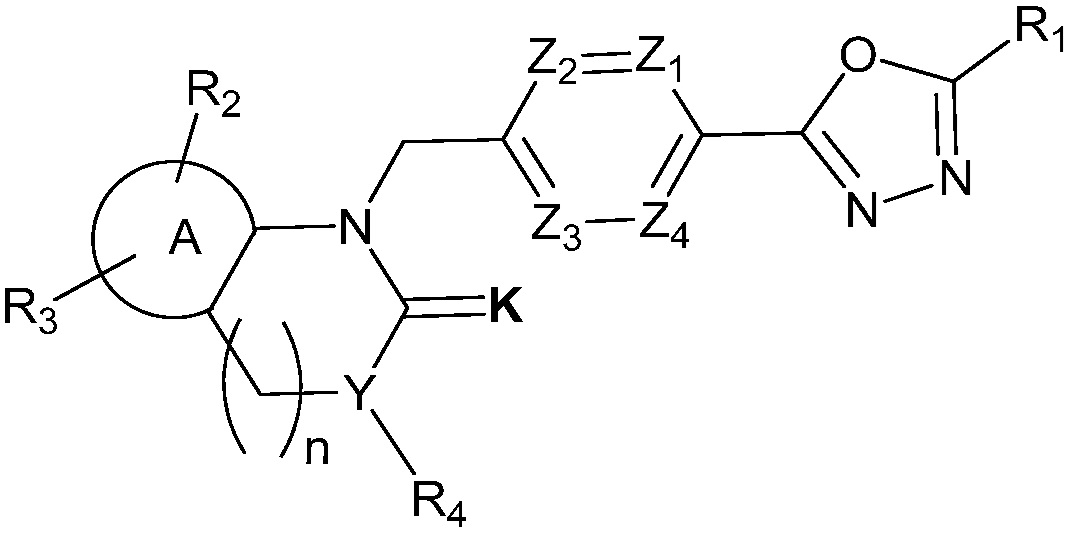
где,
Z1 - Z4 каждый независимо является N или CRa, где Ra является H, X, C1-C4 алкилом или O-(C1-C4 алкилом) и Ra могут отличаться друг от друга, когда CRa в количестве 2 или более;
K является O или S;
R1 является CX3 или CX2H;
 является C6-C12 ариленом или C2-C10 гетероариленом;
является C6-C12 ариленом или C2-C10 гетероариленом;
R2 и R3 каждый независимо является H, X, C1-C4 алкилом, C1-C4 галоалкилом, C6-C12 арилом, C2-C10 гетероарилом, C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, C2-C10 гетероциклоалкенилом, N(Rb)(Rc), NH-(C1-C4 алкил)-N(Rb)(Rc), NH-O-(C1-C4 алкилом) или NHC(=O)-R5,
где, по меньшей мере, один H из C1-C4 алкила, C6-C12 арила, C2-C10 гетероарила, C2-C10 гетероциклоалкенила или C2-C10 гетероциклоалкила каждый может быть независимо замещен X, C1-C4 алкилом, C1-C4 галоалкилом, C3-C10 циклоалкилом, C6-C12 арилом, C2-C10 гетероарилом, C2-C10 гетероциклоалкилом, (C1-C4 алкил)-(C2-C10 гетероарилом), (C1-C4 алкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкилом), (C2-C10 гетероциклоалкил)-(C1-C4 галоалкилом), (C2-C10 гетероциклоалкил)-(C3-C10 циклоалкилом), (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкил)-(C3-C10 галоциклоалкилом), (C1-C4 алкил)-(C3-C10 циклоалкилом), S(O2)-(C1-C4 алкилом), C(=O)-N(Rb)(Rc), C(=O)-R6, -N(Rb)(Rc), (C1-C4 алкил)-N(Rb)(Rc), O-(C1-C4 алкилом), (C1-C4 алкил)-O-(C1-C4 алкилом), C(=O)O-(C2-C10 гетероциклоалкилом), (C1-C4 алкил)-C(=O)-R7 или (C2-C10 гетероциклоалкил)-C(=O)-R8;
Y является CH, N, O или S {где R4 равен нулю, если Y является O или S};
R4 является H, C1-C4 алкилом, C3-C7 циклоалкилом, C2-C10 гетероциклоалкилом, (C1-C4 алкил)-(C2-C10 гетероциклоалкилом), C6-C12 арилом, C2-C10 гетероарилом или C(=O)-R9,
где, по меньшей мере, один H из C1-C4 алкила, C2-C10 гетероциклоалкила или (C1-C4 алкил)-(C2-C10 гетероциклоалкила) каждый может быть независимо замещен X, C1-C4 алкила C1-C4 галоалкила, O-(C1-C4 алкила), -N(Rb)(Rc), C3-C10 циклоалкила, C2-C10 гетероциклоалкила, (C2-C10 гетероциклоалкил)-C(=O)-R10, S(O2)-(C1-C4 алкила), C(=O)-R11, (C1-C4 алкил)-O-(C1-C4 алкила), C6-C12 арила, C2-C10 гетероарила, (C1-C4 алкил)-(C2-C10 гетероарила), (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкила) или C(=O)-(C1-C4 алкил)-O-(C1-C4 алкила);
R5, R6, R7, R8, R9, R10 и R11 каждый независимо является H, C1-C4 алкилом, (C1-C4 алкил)-OH, (C1-C4 алкил)-O-(C1-C4 алкилом), C2-C10 гетероциклоалкилом, C6-C12 арилом, C2-C10 гетероарилом, O-(C1-C4 алкилом), C3-C7 циклоалкилом или (C1-C4 алкил)-N(Rb)(Rc),
где, по меньшей мере, один H из C6-C12 арила или C2-C10 гетероарила каждый может быть независимо замещен C1-C4 алкилом, X или C1-C4 галоалкилом;
Rb и Rc каждый независимо является H, C1-C4 алкилом, C6-C12 арилом, C2-C10 гетероарилом, C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C1-C4 алкил)NH(C1-C4 алкилом), (C1-C4 алкил)N(C1-C4 алкилом)2, (C1-C4 алкил)-O-(C1-C4 алкилом), C(=O)-(C1-C4 алкилом), C(=O)-(C2-C10 гетероарилом), C(=O)-(C2-C10 гетероциклоалкилом) или C(=O)-(C3-C10 циклоалкилом);
X является атомом галогена; и
n равен любому целому числу, выбранному из 0, 1, 2 и 3.
В одном варианте осуществления, в вышеуказанной химической формуле I,
Z1 - Z4 каждый независимо является N или CRa, где Ra является H, X, C1-C4 алкилом или O-(C1-C4 алкилом) и Ra могут отличаться друг от друга, когда CRa в количестве 2 или более;
K является O или S;
R1 является CX3 или CX2H;
является C6-C12 ариленом или C2-C10 гетероариленом,
где C2-C10 гетероарилен может содержать по меньшей мере, один N;
R2 и R3 каждый независимо является H, X, C1-C4 галоалкилом, C6-C12 арилом, C2-C10 гетероциклоалкилом, C2-C10 гетероциклоалкенилом, N(Rb)(Rc) или C2-C10 гетероарилом,
где, по меньшей мере, один H из C6-C12 арила, C2-C10 гетероарила, C2-C10 гетероциклоалкенила или C2-C10 гетероциклоалкила, где каждый может быть независимо замещен X, C1-C4 алкилом, C1-C4 галоалкилом, C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C1-C4 алкил)-(C2-C10 гетероарилом), (C1-C4 алкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкилом), (C2-C10 гетероциклоалкил)-(C1-C4 галоалкилом), (C2-C10 гетероциклоалкил)-(C3-C10 циклоалкилом), (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкил)-(C3-C10 галоциклоалкилом), S(O2)-(C1-C4 алкилом), C(=O)-R6, O-(C1-C4 алкилом) или (C2-C10 гетероциклоалкил)-C(=O)-R8;
Y является CH, N, O или S {где R4 равен нулю, если Y является O или S};
R4 является C1-C4 алкилом, C2-C10 гетероциклоалкилом, C2-C10 гетероарилом или (C1-C4 алкил)-(C2-C10 гетероциклоалкилом),
где, по меньшей мере, один H из C1-C4 алкила, C2-C10 гетероциклоалкила или (C1-C4 алкил)-(C2-C10 гетероциклоалкила) каждый может быть независимо замещен C1-C4 алкилом, C1-C4 галоалкилом, O-(C1-C4 алкилом), -N(Rb)(Rc), C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C2-C10 гетероциклоалкил)-C(=O)-R10, S(O2)-(C1-C4 алкилом), C(=O)-R11, (C1-C4 алкил)-O-(C1-C4 алкилом), C6-C12 арилом, (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом) или (C1-C4 алкил)-(C2-C10 гетероарилом);
R6, R8, R10 и R11 каждый независимо является C1-C4 алкилом, (C1-C4 алкил)-OH, (C1-C4 алкил)-O-(C1-C4 алкилом), C2-C10 гетероциклоалкилом, C6-C12 арилом, C2-C10 гетероарилом или O-(C1-C4 алкилом),
где, по меньшей мере, один H из C6-C12 арила или C2-C10 гетероарила каждый может быть независимо замещен C1-C4 алкилом или C1-C4 галоалкилом;
Rb и Rc каждый независимо является H, C1-C4 алкилом или C(=O)-(C1-C4 алкилом);
X является атомом галогена; и
n равен целому числу, выбранному из 0, 1, 2 и 3.
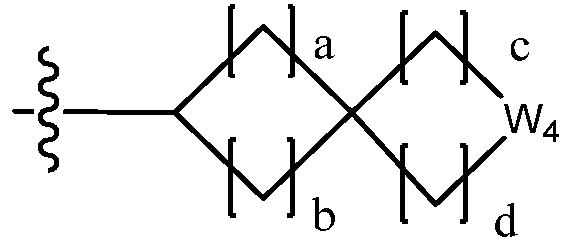
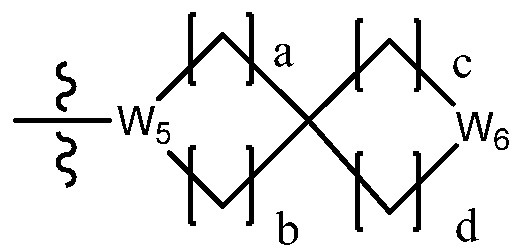
В одном варианте осуществления, в вышеуказанной химической формуле I, C2-C10 гетероциклоалкил является
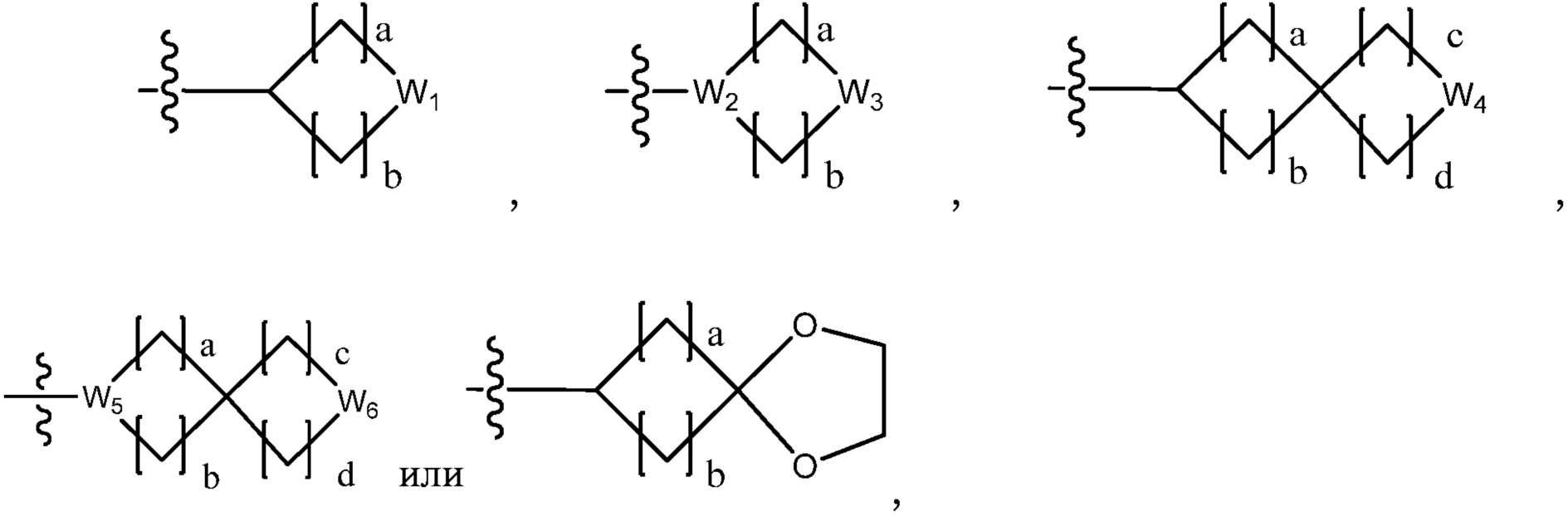
где W1 - W6 каждый независимо является N, NH, O, S или SO2, и
a - d каждый независимо является целым числом 1, 2 или 3.
В одном варианте осуществления, соединения, представленные вышеуказанной химической формулой I, могут содержать соединения, представленные следующей химической формулой II:
[Химическая формула II]
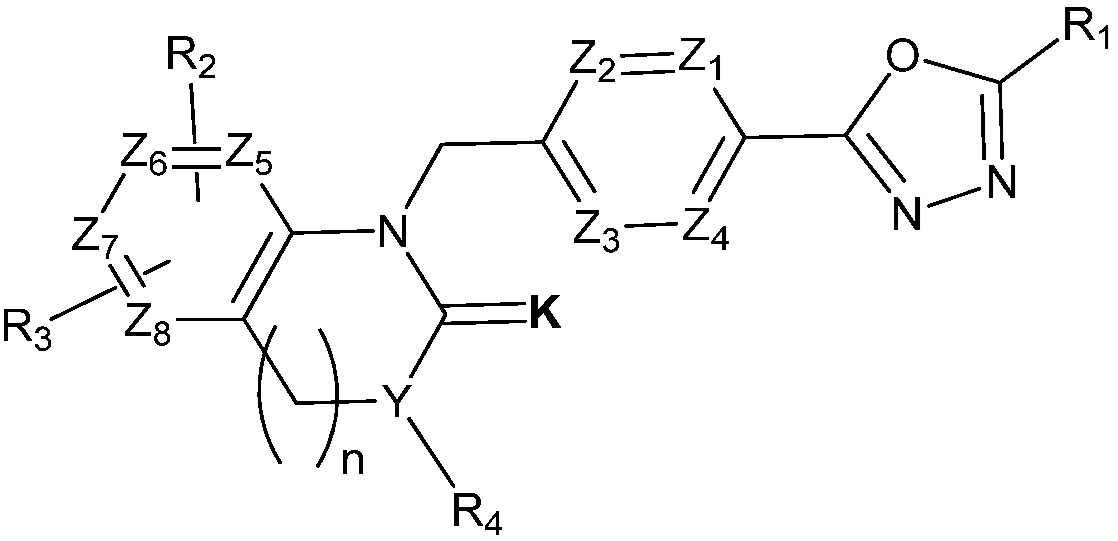
где,
Z1 - Z4 каждый независимо является N или CRa, где Ra является H, X, C1-C4 алкилом или O-(C1-C4 алкилом) и Ra могут отличаться друг от друга, когда CRa в количестве 2 или более;
Z5 - Z8 каждый независимо является CR2, CR3, CH или N, где Z5 - Z8 содержит CR2, CR3, CH и N, содержит CR2, CR3 и два N, или содержит CR2, CR3 и два CH;
K является O или S;
R1 является CX3 или CX2H;
R2 и R3 каждый независимо является H, X, C1-C4 алкилом, C1-C4 галоалкилом, C6-C12 арилом, C2-C10 гетероарилом, C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, C2-C10 гетероциклоалкенилом, N(Rb)(Rc), NH-(C1-C4 алкил)-N(Rb)(Rc), NH-O-(C1-C4 алкилом) или NHC(=O)-R5,
где, по меньшей мере, один H из C1-C4 алкила C6-C12 арила, C2-C10 гетероарила, C2-C10 гетероциклоалкенила или C2-C10 гетероциклоалкила каждый может быть независимо замещен X, C1-C4 алкилом, C1-C4 галоалкилом, C3-C10 циклоалкилом, C6-C12 арилом, C2-C10 гетероарилом, C2-C10 гетероциклоалкилом, (C1-C4 алкил)-(C2-C10 гетероарилом), (C1-C4 алкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкилом), (C2-C10 гетероциклоалкил)-(C1-C4 галоалкилом), (C2-C10 гетероциклоалкил)-(C3-C10 циклоалкилом), (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкил)-(C3-C10 галоциклоалкилом), (C1-C4 алкил)-(C3-C10 циклоалкилом), S(O2)-(C1-C4 алкилом), C(=O)-N(Rb)(Rc), C(=O)-R6, -N(Rb)(Rc), (C1-C4 алкил)-N(Rb)(Rc), O-(C1-C4 алкилом), (C1-C4 алкил)-O-(C1-C4 алкилом), C(=O)O-(C2-C10 гетероциклоалкилом), (C1-C4 алкил)-C(=O)-R7 или (C2-C10 гетероциклоалкил)-C(=O)-R8;
Y является CH, N, O или S {где R4 равен нулю, если Y является O или S};
R4 является H, C1-C4 алкилом, C3-C7 циклоалкилом, C2-C10 гетероциклоалкилом, (C1-C4 алкил)-(C2-C10 гетероциклоалкилом), C6-C12 арилом, C2-C10 гетероарилом или C(=O)-R9,
где, по меньшей мере, один H из C1-C4 алкила, C2-C10 гетероциклоалкила или (C1-C4 алкил)-(C2-C10 гетероциклоалкила) каждый может быть независимо замещен X, C1-C4 алкилом, C1-C4 галоалкилом, O-(C1-C4 алкилом), -N(Rb)(Rc), C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C2-C10 гетероциклоалкил)-C(=O)-R10, S(O2)-(C1-C4 алкилом), C(=O)-R11, (C1-C4 алкил)-O-(C1-C4 алкилом), C6-C12 арилом, C2-C10 гетероарилом, (C1-C4 алкил)-(C2-C10 гетероарилом), (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом) или C(=O)-(C1-C4 алкил)-O-(C1-C4 алкилом);
R5, R6, R7, R8, R9, R10 и R11 каждый независимо является H, C1-C4 алкилом, (C1-C4 алкил)-OH, (C1-C4 алкил)-O-(C1-C4 алкилом), C2-C10 гетероциклоалкилом, C6-C12 арилом, C2-C10 гетероарилом, O-(C1-C4 алкилом), C3-C7 циклоалкилом или (C1-C4 алкил)-N(Rb)(Rc),
где, по меньшей мере, один H из C6-C12 арила или C2-C10 гетероарила каждый может быть независимо замещен C1-C4 алкилом, X или C1-C4 галоалкилом;
Rb и Rc каждый независимо является H, C1-C4 алкилом, C6-C12 арилом, C2-C10 гетероарилом, C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C1-C4 алкил)NH(C1-C4 алкилом), (C1-C4 алкил)N(C1-C4 алкилом)2, (C1-C4 алкил)-O-(C1-C4 алкилом), C(=O)-C1-C4 алкилом, C(=O)-C2-C10 гетероарилом, C(=O)-(C2-C10 гетероциклоалкилом) или C(=O)-(C3-C10 циклоалкилом);
X является атомом галогена; и
n равен любому целому числу, выбранному из 0, 1, 2 и 3.
В одном варианте осуществления, в вышеуказанной химической формуле II,
Z1 - Z4 каждый независимо является N или CRa, где Ra является H, X, C1-C4 алкилом или O-(C1-C4 алкилом) и Ra могут отличаться друг от друга, когда CRa в количестве 2 или более;
Z5 - Z8 каждый независимо является CR2, CR3, CH или N, где Z5 - Z8 содержит CR2, CR3, CH и N или содержит CR2, CR3 и два CH (однако, Z6 является N, когда Z5 - Z8 содержит CR2, CR3, CH и N);
K является O или S;
R1 является CX3 или CX2H;
R2 и R3 каждый независимо является H, X, C1-C4 галоалкилом, C6-C12 арилом, C2-C10 гетероциклоалкилом, C2-C10 гетероциклоалкенилом, N(Rb)(Rc) или C2-C10 гетероарилом,
где, по меньшей мере, один H из C6-C12 арила, C2-C10 гетероарила, C2-C10 гетероциклоалкенила или C2-C10 гетероциклоалкила каждый может быть независимо замещен X, C1-C4 алкилом, C1-C4 галоалкилом, C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C1-C4 алкил)-(C2-C10 гетероарилом), (C1-C4 алкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкилом), (C2-C10 гетероциклоалкил)-(C1-C4 галоалкилом), (C2-C10 гетероциклоалкил)-(C3-C10 циклоалкилом), (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкил)-(C3-C10 галоциклоалкилом), S(O2)-(C1-C4 алкилом), C(=O)-R6, O-(C1-C4 алкилом) или (C2-C10 гетероциклоалкил)-C(=O)-R8;
Y является CH, N, O или S {где R4 равен нулю, если Y является O или S};
R4 является C1-C4 алкилом, C2-C10 гетероциклоалкилом, C2-C10 гетероарилом или (C1-C4 алкил)-(C2-C10 гетероциклоалкилом),
где, по меньшей мере, один H из C1-C4 алкила, C2-C10 гетероциклоалкила или (C1-C4 алкил)-(C2-C10 гетероциклоалкила) каждый может быть независимо замещен C1-C4 алкилом, C1-C4 галоалкилом, O-(C1-C4 алкилом), -N(Rb)(Rc), C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C2-C10 гетероциклоалкил)-C(=O)-R10, S(O2)-(C1-C4 алкилом), C(=O)-R11, (C1-C4 алкил)-O-(C1-C4 алкилом), C6-C12 арилом, (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом) или (C1-C4 алкил)-(C2-C10 гетероарилом);
R6, R8, R10 и R11 каждый независимо является C1-C4 алкилом, (C1-C4 алкил)-OH, (C1-C4 алкил)-O-(C1-C4 алкилом), C2-C10 гетероциклоалкилом, C6-C12 арилом, C2-C10 гетероарилом или O-(C1-C4 алкилом),
где, по меньшей мере, один H из C6-C12 арила или C2-C10 гетероарила каждый может быть независимо замещен C1-C4 алкилом или C1-C4 галоалкилом;
Rb и Rc каждый независимо является H, C1-C4 алкилом или C(=O)-(C1-C4 алкилом);
X является атомом галогена; и
n равен любому целому числу, выбранному из 0, 1, 2 и 3.
В одном варианте осуществления, в вышеуказанной химической формуле II,
Z1 - Z4 каждый независимо является N или CRa, где Ra является H, X, C1-C4 алкилом или O-(C1-C4 алкилом) и Ra могут отличаться друг от друга, когда CRa в количестве 2 или более;
Z5 - Z8 каждый независимо является CR2, CR3, CH или N, где Z5 - Z8 содержит CR2, CR3, CH и N или содержит CR2, CR3 и два CH (однако, Z6 является N, если Z5 - Z8 содержит CR2, CR3, CH и N);
K является O или S;
R1 является CX3 или CX2H;
R2 и R3 каждый независимо является H, X, C1-C4 галоалкилом, C6-C12 арилом, C2-C10 гетероциклоалкилом, C2-C10 гетероциклоалкенилом, N(Rb)(Rc) или C2-C10 гетероарилом,
где, по меньшей мере, один Н из C6-C12 арила, C2-C10 гетероарила, C2-C10 гетероциклоалкенила или C2-C10 гетероциклоалкила каждый может быть независимо замещен X, C1-C4 алкилом, C1-C4 галоалкилом, C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C1-C4 алкил)-(C2-C10 гетероарилом), (C1-C4 алкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкилом), (C2-C10 гетероциклоалкил)-(C1-C4 галоалкилом), (C2-C10 гетероциклоалкил)-(C3-C10 циклоалкилом), (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкил)-(C3-C10 галоциклоалкилом), S(O2)-(C1-C4 алкилом), C(=O)-R6, O-(C1-C4 алкилом) или (C2-C10 гетероциклоалкил)-C(=O)-R8;
Y является N, O или S {где R4 равен нулю, если Y является O или S};
R4 является C1-C4 алкилом, C2-C10 гетероциклоалкилом, C2-C10 гетероарилом или (C1-C4 алкил)-(C2-C10 гетероциклоалкилом),
где, по меньшей мере, один Н из C1-C4 алкила, C2-C10 гетероциклоалкила или (C1-C4 алкил)-(C2-C10 гетероциклоалкила) каждый может быть независимо замещен C1-C4 алкилом, C1-C4 галоалкилом, O-(C1-C4 алкилом), -N(Rb)(Rc), C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C2-C10 гетероциклоалкил)-C(=O)-R10, S(O2)-(C1-C4 алкилом), C(=O)-R11, (C1-C4 алкил)-O-(C1-C4 алкилом), C6-C12 арилом, (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом) или (C1-C4 алкил)-(C2-C10 гетероарилом);
R6, R8, R10 и R11 каждый независимо является C1-C4 алкилом, (C1-C4 алкил)-OH, (C1-C4 алкил)-O-(C1-C4 алкилом), C2-C10 гетероциклоалкилом, C6-C12 арилом, C2-C10 гетероарилом или O-(C1-C4 алкилом),
где, по меньшей мере, один Н из C6-C12 арила или C2-C10 гетероарила каждый может быть независимо замещен C1-C4 алкилом или C1-C4 галоалкилом;
Rb и Rc каждый независимо является H, C1-C4 алкилом или C(=O)-(C1-C4 алкилом);
X является атомом галогена; и
n равен любому целому числу, выбранному из 0, 1, 2 и 3.
В одном варианте осуществления, в вышеуказанной химической формуле I,
Z1 - Z4 каждый независимо является N или CRa, где Ra является H или X, и Ra могут отличаться друг от друга, когда CRa в количестве 2 или более;
K является O;
R1 является CF3 или CF2H;
является фениленом или пиридиниленом,
R2 и R3 каждый независимо является H, X, C1-C4 галоалкилом, C6-C12 арилом, C2-C10 гетероциклоалкилом, C2-C10 гетероциклоалкенилом, N(Rb)(Rc) или C2-C5 гетероарилом,
где, по меньшей мере, один Н из C6-C12 арила, C2-C5 гетероарила, C2-C10 гетероциклоалкенила или C2-C10 гетероциклоалкила каждый может быть независимо замещен X, C1-C4 алкилом, C1-C4 галоалкилом, C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C1-C4 алкил)-(C2-C10 гетероарилом), (C1-C4 алкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкилом), (C2-C10 гетероциклоалкил)-(C1-C4 галоалкилом), (C2-C10 гетероциклоалкил)-(C3-C10 циклоалкилом), (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом), (C2-C10 гетероциклоалкил)-(C1-C4 алкил)-(C3-C10 галоциклоалкилом), S(O2)-(C1-C4 алкилом), C(=O)-R6, O-(C1-C4 алкилом) или (C2-C10 гетероциклоалкил)-C(=O)-R8;
Y является N, O или S {где R4 равен нулю, если Y является O или S};
R4 является C1-C4 алкилом, C2-C10 гетероциклоалкилом, C2-C10 гетероарилом или (C1-C4 алкил)-(C2-C10 гетероциклоалкилом),
где, по меньшей мере, один Н из C1-C4 алкила, C2-C10 гетероциклоалкила или (C1-C4 алкил)-(C2-C10 гетероциклоалкила) каждый может быть независимо замещен C1-C4 алкилом, C1-C4 галоалкилом, O-(C1-C4 алкилом), -N(Rb)(Rc), C3-C10 циклоалкилом, C2-C10 гетероциклоалкилом, (C2-C10 гетероциклоалкил)-C(=O)-R10, S(O2)-(C1-C4 алкилом), C(=O)-R11, (C1-C4 алкил)-O-(C1-C4 алкилом), C6-C12 арилом, (C2-C10 гетероциклоалкил)-(C2-C10 гетероциклоалкилом) или (C1-C4 алкил)-(C2-C10 гетероарилом);
R6 является C1-C4 алкил, O-(C1-C4 алкил) или (C1-C4 алкил)-OH;
R8 является О-(C1-C4 алкилом);
R10 является С1-C4 алкилом;
R11 является С1-C4 алкилом, (C1-C4 алкил)-OH, (C1-C4 алкил)-O-(C1-C4 алкилом), C2-C10 гетероциклоалкилом, C6-C12 арилом, C2-C10 гетероарилом или O-(C1-C4 алкилом), где, по меньшей мере, один Н из C6-C12 арила или C2-C10 гетероарила каждый может быть независимо замещен C1-C4 алкилом или C1-C4 галоалкилом;
Rb и Rc каждый независимо является H, C1-C4 алкилом или C(=O)-(C1-C4 алкилом);
X является F, Cl или Br; и
n равно 0 или 1.
Преимущественные эффекты
Согласно настоящему изобретению, производные соединения 1,3,4-оксадиазола, представленные указанной выше химической формулой I, их стереоизомеры или их фармацевтически приемлемые соли обладают не только ингибирующей активностью HDAC6, но также замечательно превосходным эффектом профилактики или лечения связанных с активностью HDAC6 заболеваний через селективное ингибирование HDAC6.
Кроме того, производные соединения 1,3,4-оксадиазола по изобретению, обладающие селективной ингибирующей активностью HDAC6, их стереоизомеры или их фармацевтически приемлемые соли могут быть использованы для профилактики или лечения заболеваний, связанных с активностью HDAC6, таких как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания или нейродегенеративные нарушения и т.д.
Наилучший способ осуществления изобретения
Термины, используемые в настоящей заявке, используют только для описания определенного типового варианта осуществления и не предназначены для ограничения настоящего изобретения. Формы единственного числа должны включать формы множественного числа, если иное явно не указано контекстом. В настоящей заявке, такие термины, как «содержит», «имеет» или подобные предназначены для обозначения наличия признаков, стадий, структур или их комбинаций, описанных в настоящем документе, и не должны толковаться как заранее исключающие возможное присутствие или добавление одного или несколько других признаков, стадий, структур или их комбинаций.
Все используемые здесь термины, включая технические или научные термины, имеют то же значение, которое обычно понимается специалистами в данной области техники, к которой относится настоящее изобретение, если не указано иное. Такие термины, как те, которые определены в обычно используемом словаре, должны интерпретироваться как имеющие значения, равные контекстуальным значениям в соответствующей области техники, и не должны интерпретироваться как имеющие идеальные или чрезмерно формальные значения, если они четко не определены в настоящей заявке.
В настоящем изобретении термин «замещенный» представляет группу, имеющую заместитель, который замещает, по меньшей мере, один водород на атоме углерода основной цепи. «Замещение», «замещаемый~» или «замещенный~» определены как включающие неявные условия, где замещение следует разрешенной валентности замещенного атома и заместителя и дает соединение, стабилизированное замещением, например, соединение которое не подвергается естественным изменениям при перегруппировке, циклизации, удалении и т. д.
В настоящем изобретении «Cx-y» означает наличие атомов углерода в диапазоне от x до y.
В настоящем изобретении «алкил» означает линейную (или прямую) насыщенную углеводородную группу или разветвленную (или боковую) насыщенную углеводородную группу, и включает метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил и т. д.
В настоящем изобретении «галоалкил» означает функциональную группу, где, по меньшей мере, один водород алкила, определенного выше, замещен атомом галогена F, Cl, Br или I.
В настоящем изобретении «алкилен» означает двухвалентную функциональную группу, которая образована из алкильной группы, как определено выше.
В настоящем изобретении «арил» включает моноциклическую ароматическую структуру или полициклическую ароматическую структуру, а также структуру, в которой насыщенное углеводородное кольцо конденсировано с моноциклической ароматической или полициклической ароматической группой. Арил включает фенил, бифенил, нафталенил, тетрагидронафталенил, антраценил, фенантренил, пиренил и т. д.
В настоящем изобретении «гетероарил» означает моноциклическое или полициклическое гетерокольцо, где, по меньшей мере, один атом углерода арила, определенного выше, замещен азотом (N), кислородом (O) или серой (S). Гетероарил включает пиридинил, тиофенил, триазолил, тетразолил, бензодиоксолил, бензотиазолил, бензотиофенил, хинолинил, индолил, изоиндолил, бензофуранил, бензопирролил, фуранил, пирролил, тиазолил, изотиазолил, имидазолил, пиразолил, оксазолил, изоксазолил, пиразинил, пиридазинил, пиримидинил, изохинолинил, карбазолил, бензоксазолил, бензодиоксазолил, бензодиоксинил, бензимидазолил, дигидробензотиофенил, дигидробензофуранил, пуринил, индолизинил, хроманил, хроменил, дигидробензодиоксинил и т.д., но не ограничиваются ими.
В настоящем изобретении «циклоалкил» означает насыщенное углеводородное кольцо, обычно имеющее определенное число атомов углерода, и насыщенное углеводородное кольцо в совокупности относится к моноциклическим и полициклическим структурам, и кольцевой структуре, в которой, по меньшей мере, два кольца имеют, по меньшей мере, один общий атом углерода (например, спиро кольцо, мостиковое кольцо и т. д.). Циклоалкил включает циклогексил, циклогептанил, циклооктанил, тетрагидронафталенил и т.д., но не ограничивается ими.
В настоящем изобретении «галоциклоалкил» означает функциональную группу, где, по меньшей мере, один водород циклоалкила, определенного выше, замещен атомом галогена F, Cl, Br или I.
В настоящем изобретении «гетероциклоалкил» включает насыщенные моноциклические и полициклические гетеро кольца, содержащие от одного до четырех гетероатомов, независимо выбранных из азота (N), кислорода (O) и серы (S), и кольцевую структуру, где, по меньшей мере, два кольца имеют, по меньшей мере, один общий атом углерода (например, спиро кольцо, мостиковое кольцо и т. д.).
В настоящем изобретении, гетероциклоалкил может содержать 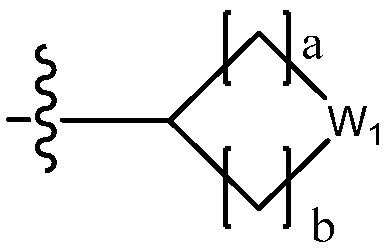 ,
, 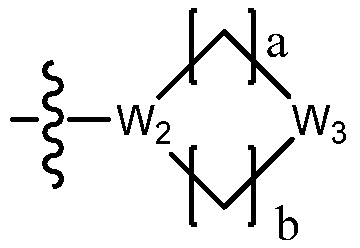 ,
,  ,
,  или
или 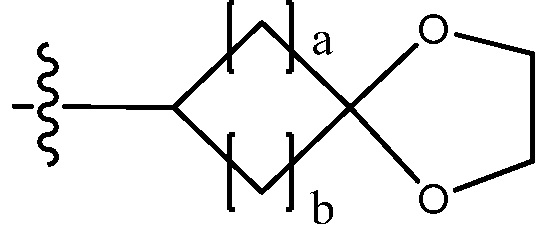 . В этом случае, W1 - W6 каждый независимо является N, NH, O, S или SO2 и a - d каждый независимо является целым числом 1, 2 или 3.
. В этом случае, W1 - W6 каждый независимо является N, NH, O, S или SO2 и a - d каждый независимо является целым числом 1, 2 или 3.
В настоящем изобретении, конкретные примеры гетероциклоалкила могут включать оксиранил, оксетанил, морфолинил, тиетанил, азетидин, пирролидинил, пиперидинил, тетрагидрофуранил, тетрагидротиофенил, тетрагидропиранил, тетрагидротиопиранил, тетрагидротиопиран, 1,1-диоксид, 6-азабицикло[3.2.1]октанил, 2-окса-6-азаспиро[3.3]гептанил, 2-оксаспиро[3.3]гептанил, 1,4-диоксаспиро[4.5]деканил, и т.д, но особенно ими не ограничен.
В настоящем изобретении «гетероциклоалкенил» означает структуру, включающую, по меньшей мере, одну двойную связь углерод-углерод или двойную связь углерод-азот из моноциклических и полициклических гетероколец, содержащих от одного до четырех гетероатомов, независимо выбранных из азота (N), кислорода (О) и серы (S). Гетероциклоалкенил включает тетрагидропиридинил, дигидропиранил, дигидротиопиранил, дигидропирролил, дигидрофуранил, дигидротиофенил и т.д., но не ограничивается этим.
В настоящем изобретении, 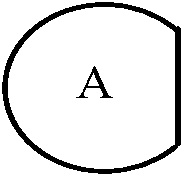 представляет структуру, конденсированную через два общие атома углерода с другим кольцом и два общих/конденсированных атома углерода означает два, расположенных в ряд. Что касается , «арилен» означает фенилен, нафталенилен и т.д., и «гетероарилен» означает пиримидилен, пиридилен и т.д., содержащий, по меньшей мере, один гетероатом, например, N, O или S в указанном арилене. В этом случае, указанный арилен и указанный гетероарилен конденсированы через два общих атома углерода с другим кольцом (кольцом, содержащим Y химической формулы I, имеющим структуру, представленную
представляет структуру, конденсированную через два общие атома углерода с другим кольцом и два общих/конденсированных атома углерода означает два, расположенных в ряд. Что касается , «арилен» означает фенилен, нафталенилен и т.д., и «гетероарилен» означает пиримидилен, пиридилен и т.д., содержащий, по меньшей мере, один гетероатом, например, N, O или S в указанном арилене. В этом случае, указанный арилен и указанный гетероарилен конденсированы через два общих атома углерода с другим кольцом (кольцом, содержащим Y химической формулы I, имеющим структуру, представленную  ). В этом случае, два атома углерода, конденсированные через общие арилен или гетероарилен, расположены в ряд от атомов углерода, составляющих другое кольцо (кольцо, содержащее Y химической формулы I). В качестве одного примера, если является фениленом, химическая формула I может содержать структуру
). В этом случае, два атома углерода, конденсированные через общие арилен или гетероарилен, расположены в ряд от атомов углерода, составляющих другое кольцо (кольцо, содержащее Y химической формулы I). В качестве одного примера, если является фениленом, химическая формула I может содержать структуру 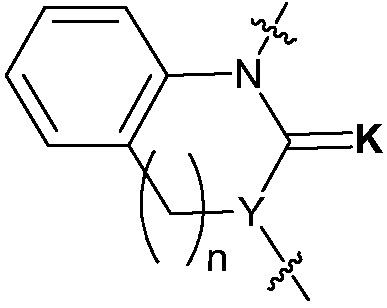 .
.
В настоящем изобретении, «стереоизомеры» включают диастереомер и оптический изомер, где оптический изомер включает не только энантиомер, но также смесь энантиомера и даже рацемат.
В настоящем изобретении «фармацевтически приемлемые соли» означают соли, обычно используемые в фармацевтической промышленности. Например, есть соли неорганических ионов, полученные из кальция, калия, натрия, магния и т.д.; соли неорганических кислот, полученные из хлористоводородной кислоты, азотной кислоты, фосфорной кислоты, бромной кислоты, йодной кислоты, хлорной кислоты, серной кислоты, йодистоводородной кислоты и т.д.; соли органических кислот, полученные из уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, янтарной кислоты, щавелевой кислоты, бензойной кислоты, винной кислоты, фумаровой кислоты, миндальной кислоты, пропионовой кислоты, молочной кислоты, гликолевой кислоты, глюконовой кислоты, галактуроновой кислоты, глутаминовой кислоты, глутаровой кислоты, глюкуроновой кислоты, аспарагиновой кислоты, аскорбиновой кислоты, карбоновой кислоты, ванилиновой кислоты и т.д.; соли сульфоновой кислоты, полученные из метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты, нафталинсульфоновой кислоты и т.д.; соли аминокислот, полученные из глицина, аргинина, лизина и т.д.; соли амина, полученные из триметиламина, триэтиламина, аммиака, пиридина, пиколина и т.д.; и подобные, но типы солей, подразумеваемые в настоящем изобретении, не ограничиваются перечисленными солями.
В настоящем изобретении, предпочтительные соли могут включать гидрохлорид, фосфат, сульфат, трифторацетат, цитрат, бромат, малеат или тартрат.
Композиция, содержащая производные соединения 1,3,4-оксадиазола, их применение и терапевтический способ с их применением
Настоящее изобретение представляет фармацевтическую композицию, содержащую производные соединения 1,3,4-оксадиазола, представленные приведенной выше химической формулой I, их стереоизомеры или их фармацевтически приемлемые соли в качестве эффективного компонента.
Настоящее изобретение представляет фармацевтическую композицию для профилактики или лечения заболеваний, опосредованных гистондеацетилазой (HDAC), содержащую производные соединения 1,3,4-оксадиазола, представленные вышеуказанной химической формулой I, их стереоизомеры или их фармацевтически приемлемые соли, в качестве эффективного компонента. Предпочтительно, настоящее изобретение представляет фармацевтическую композицию для профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6 (HDAC6). Приведенная выше химическая формула I такая же, как определено выше.
Фармацевтическая композиция по настоящему изобретению селективно ингибирует HDAC6, тем самым демонстрируя замечательное действие по профилактике или лечению заболеваний, связанных с активностью HDAC6.
Заболевания, связанные с активностью HDAC6, включают, по меньшей мере, одно выбранное из группы, состоящей из инфекционных заболеваний, таких как прионная болезнь; новообразования, такого как доброкачественная опухоль (например, миелодиспластический синдром) или злокачественная опухоль (например, множественная миелома, лимфома, лейкоз, рак легких, колоректальный рак, рак толстой кишки, рак простаты, уротелиальная карцинома, рак груди, меланома, рак кожи, печени рак, рак мозга, рак желудка, рак яичников, рак поджелудочной железы, рак головы и шеи, рак полости рта или глиома); эндокринопатии, нарушений питания и обмена веществ, таких как болезнь Вильсона, амилоидоз или диабет; психических и поведенческих расстройств, таких как депрессия или синдром ретта; неврологических заболеваний, таких как атрофия центральной нервной системы (например, болезнь Хантингтона, спинальная мышечная атрофия (SMA), спиноцеребеллярная атаксия (SCA)), нейродегенеративные заболевания (например, болезнь Альцгеймера), двигательные расстройства (например, болезнь Паркинсона), невропатии (например, наследственная невропатия (болезнь Шарко-Мари-Тута), спорадическая невропатия, воспалительная невропатия, лекарственная невропатия, моторная невропатия (например, боковой амиотрофический склероз (ALS)), демиелинизирующее заболевание центральной нервной системы (например, рассеянный склероз (MC)) или подобные; заболеваний глаз и придатков глаза, таких как увеит; заболевания системы кровообращения, такие как фибрилляция предсердий, инсульт или подобные; респираторных заболеваний, таких как астма; заболеваний пищеварительной системы, таких как алкогольная болезнь печени, воспалительное заболевание кишечника, болезнь Крона, язвенная болезнь кишечника или подобные; заболеваний кожи и подкожной ткани, таких как псориаз; заболевания опорно-двигательного аппарата и заболеваний соединительной ткани, таких как ревматоидный артрит, остеоартрит, системная красная волчанка (SLE) или подобные; или тератоза, деформаций и хромосомных аберраций, таких как аутосомно-доминантный поликистоз почек, и также включают другие симптомы или заболевания, связанные с аномальными функциями гистондеацетилазы.
Указанные фармацевтически приемлемые соли являются такими же, как описано в фармацевтически приемлемых солях производных соединений 1,3,4-оксадиазола по настоящему изобретению.
Для введения фармацевтическая композиция по настоящему изобретению может дополнительно содержать, по меньшей мере, один тип фармацевтически приемлемого носителя в дополнение к указанным производным соединениям 1,3,4-оксадиазола, их стереоизомерам или их фармацевтически приемлемым солям. Используемый фармацевтически приемлемый носитель может включать солевой раствор, стерилизованную воду, раствор Рингера, забуференный солевой раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и смесь, по меньшей мере, одного из его компонентов и других обычных добавок, таких как антиоксиданты, буферные растворы, при необходимости к ним могут быть добавлены бактериостатические агенты и т.д. Также, такая фармацевтическая композиция может быть составлена в виде дозированных форм для инъекций, таких как водные растворы, суспензии, эмульсии и т.д., пилюли, капсулы, гранулы или таблетки, таким образом, что к ним дополнительно добавляют разбавители, диспергирующие агенты, поверхностно-активные вещества, связующие и смазывающие агенты. Таким образом, композиция по настоящему изобретению может быть пластырями, жидкостями и растворами, пилюлями, капсулами, гранулами, таблетками, суппозиториями и т.д. Такие препараты могут быть получены обычным способом, применяемым для составления в данной области техники, или способом, описанным в Remington's Pharmaceutical Science (latest edition), Mack Publishing Company, Easton PA, и такая композиция может быть составлена в виде различных препаратов в зависимости от каждого заболевания или компонента.
Композиция по настоящему изобретению может вводиться перорально или парентерально (например, применяться внутривенно, подкожно, внутрибрюшинно или местно) в соответствии с таргетным способом, где ее дозировка варьируется в определенном диапазоне в зависимости от веса, возраста, пола пациента, состояния здоровья, диеты, времени введения, способа введения, скорости выведения, тяжести заболевания и подобных. Суточная доза производных соединений 1,3,4-оксадиазола по настоящему изобретению, их стереоизомеров или их фармацевтически приемлемых солей может составлять примерно от 1 до 1000 мг/кг, предпочтительно от 5 до 100 мг/кг, и их можно вводить один раз в сутки или несколько раз в сутки, разделив суточную дозировку соединений.
В дополнение к указанным производным соединениям 1,3,4-оксадиазола, их стереоизомерам или их фармацевтически приемлемым солям, указанная фармацевтическая композиция по настоящему изобретению может дополнительно содержать, по меньшей мере, один эффективный компонент, который проявляет лечебный эффект, такой же или аналогичный ему.
Настоящее изобретение представляет способ профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6 (HDAC6), включающий введение терапевтически эффективного количества указанных производных соединений 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей.
Используемый в настоящем документе термин «терапевтически эффективное количество» относится к количеству указанных производных соединений 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей, которые эффективны для профилактики или лечения заболеваний, связанных с активностью HDAC6.
Кроме того, настоящее изобретение представляет способ селективного ингибирования HDAC6 путем введения указанных производных соединений 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей млекопитающим, включая человека.
Способ профилактики или лечения заболеваний, связанных с активностью HDAC6, в соответствии с настоящим изобретением включает не только лечение самих заболеваний до проявления их симптомов, но также ингибирование или профилактику таких симптомов введением указанных производных соединений 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей. При лечении заболевания профилактическая или терапевтическая доза определенного активного компонента может варьироваться в зависимости от природы и тяжести заболевания или состояния и пути введения активного компонента. Доза и ее частота могут варьироваться в зависимости от возраста, веса и реакций конкретного пациента. Подходящая доза и применение могут быть легко выбраны специалистами в данной области техники, естественно, с учетом таких факторов. Кроме того, способ профилактики или лечения заболеваний, связанных с активностью HDAC6, в соответствии с настоящим изобретением, может дополнительно включать введение терапевтически эффективного количества дополнительного активного агента, который полезен при лечении заболеваний, наряду с соединениями, представленными вышеуказанной химической формула. I, и дополнительный активный агент может проявлять синергетический эффект или аддитивный эффект вместе с указанными производными соединениями 1,3,4-оксадиазола, их стереоизомерами или их фармацевтически приемлемыми солями.
Настоящее изобретение также представляет применение соединений, представленных вышеуказанной химической формулой I, их стереоизомеров или их фармацевтически приемлемых солей для профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6 (HDAC6). Для профилактики или лечения заболеваний, связанных с активностью HDAC6, указанные производные соединения 1,3,4-оксадиазола, их стереоизомеры или их фармацевтически приемлемые соли могут быть объединены с приемлемыми адъювантами, разбавителями, носителями и т. д., и могут быть приготовлены в виде комплексного препарата вместе с другими активными агентами и, таким образом, обладают синергетическим действием активных компонентов.
Настоящее изобретение также представляет применение соединений, представленных указанной выше химической формулой I, их стереоизомеров или их фармацевтически приемлемых солей для получения лекарственного средства для лечения заболеваний, связанных с активностью гистондеацетилазы 6 (HDAC6). Для приготовления лекарственного средства, указанные производные соединения 1,3,4-оксадиазола, их стереоизомеры или их фармацевтически приемлемые соли могут быть объединены с приемлемыми адъювантами, разбавителями, носителями и т.д. и могут быть приготовлены в виде комплексного препарата вместе с другими активными агентами. и таким образом обладают синергетическим действием активных компонентов.
Вопросы, упомянутые в применении, составе и терапевтическом способе настоящего изобретения, применяются одинаково, если не противоречат друг другу.
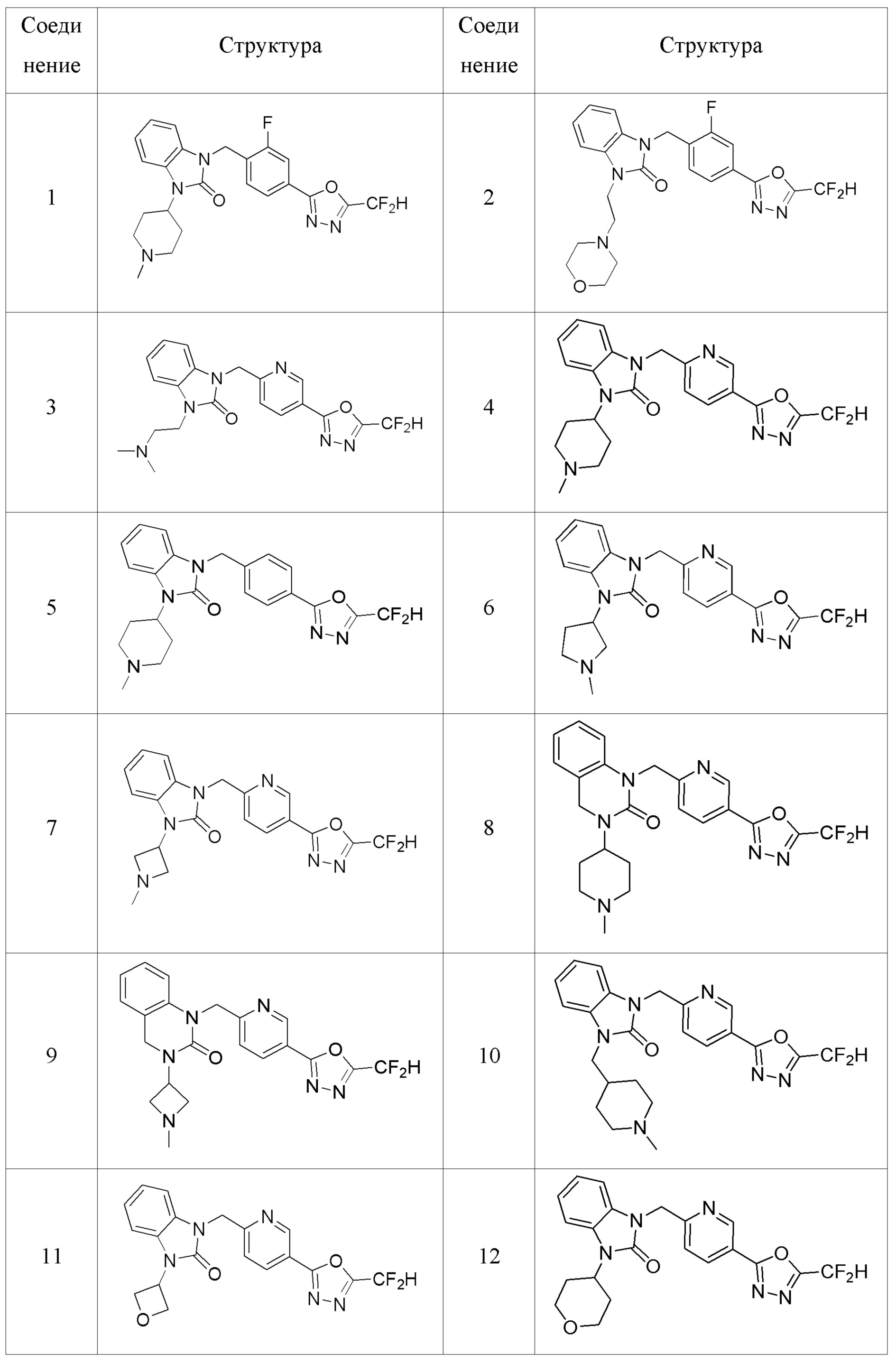
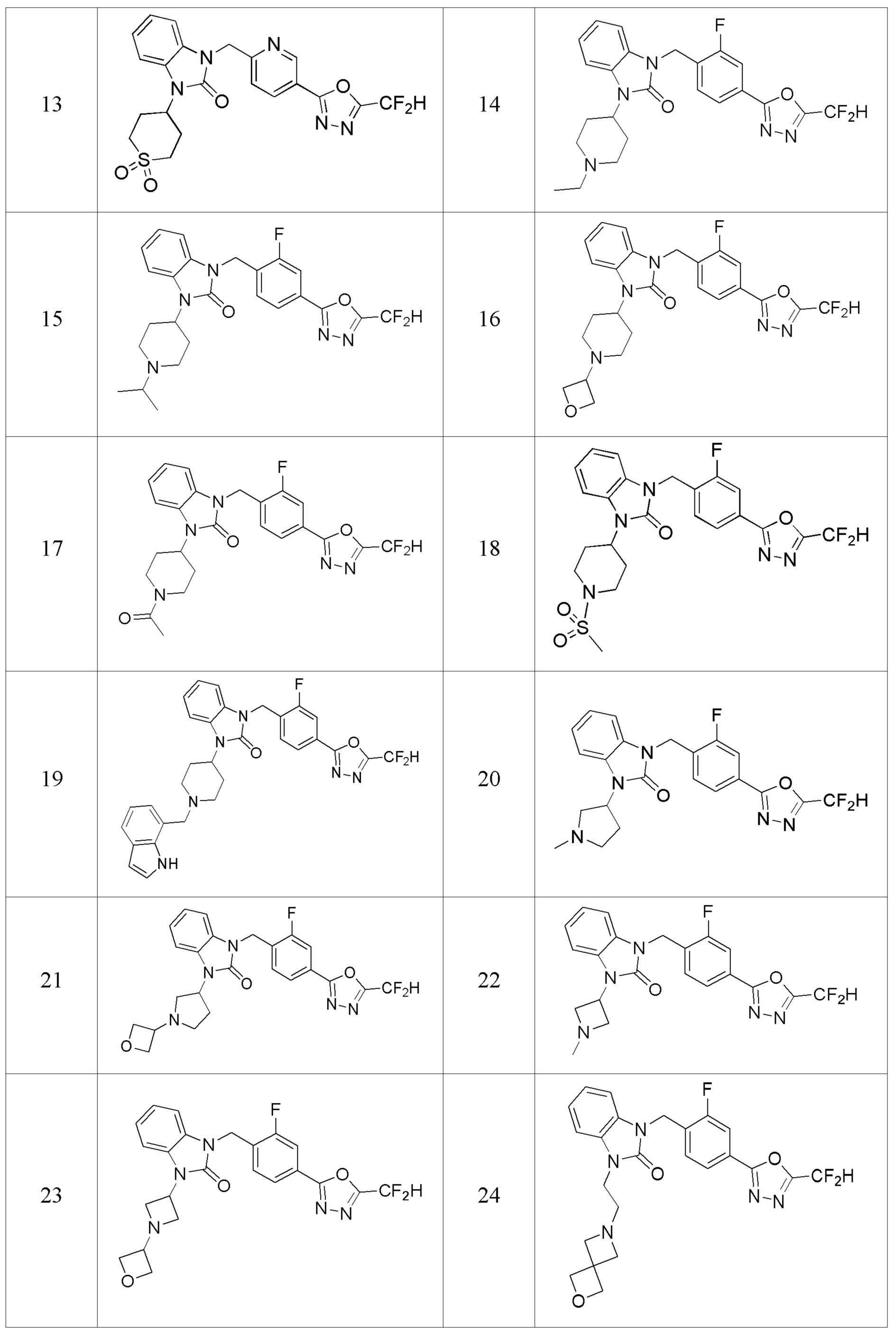
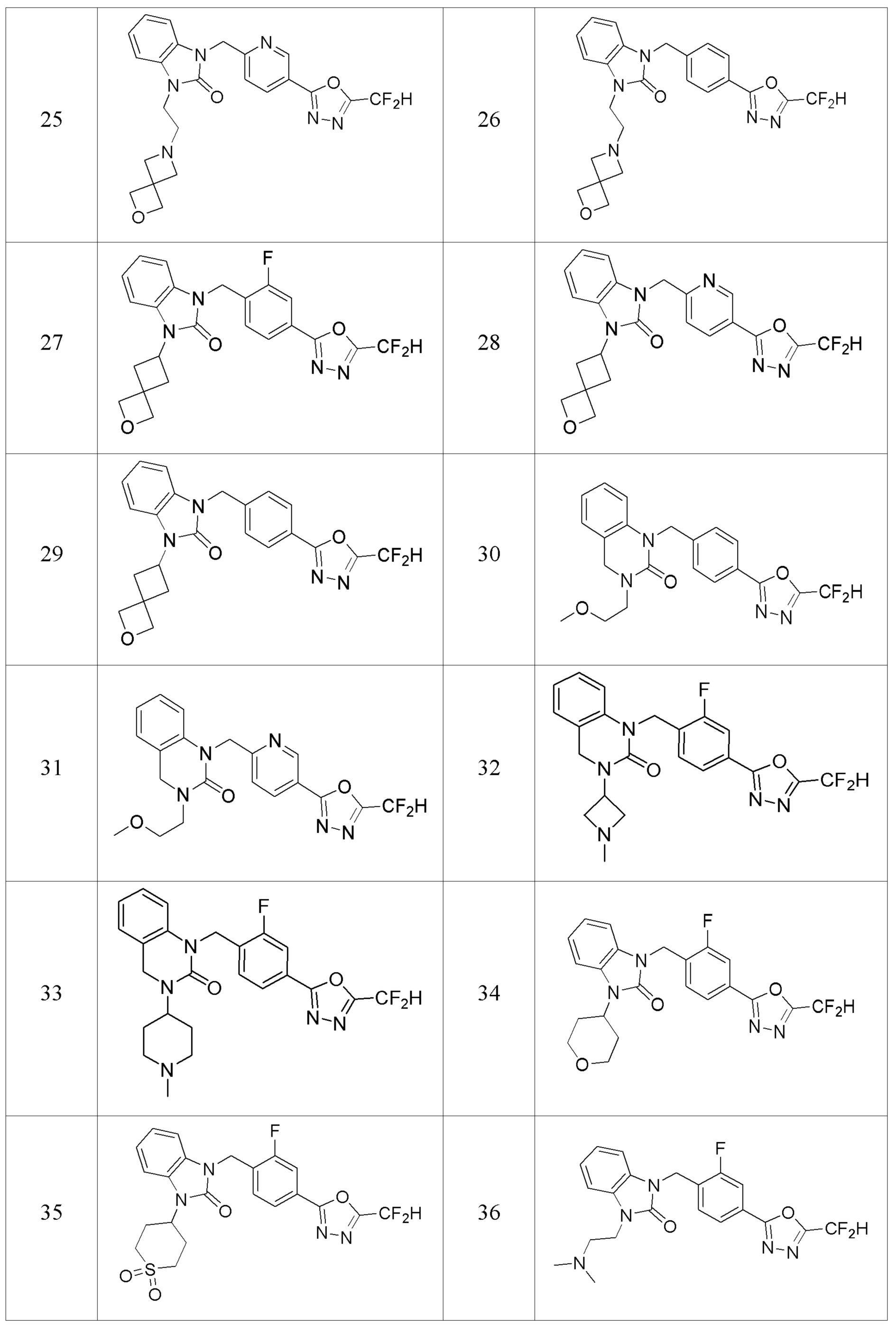
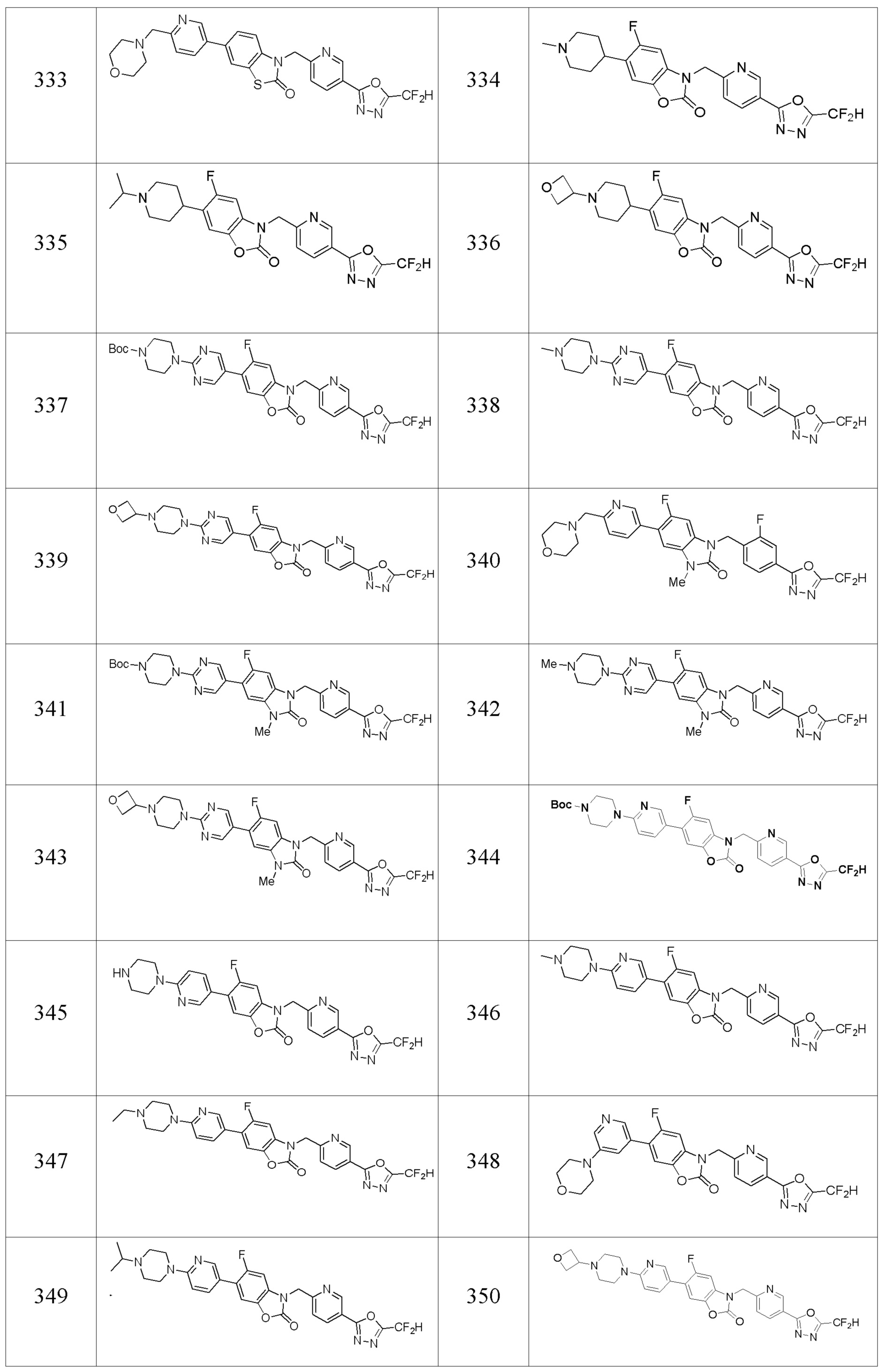
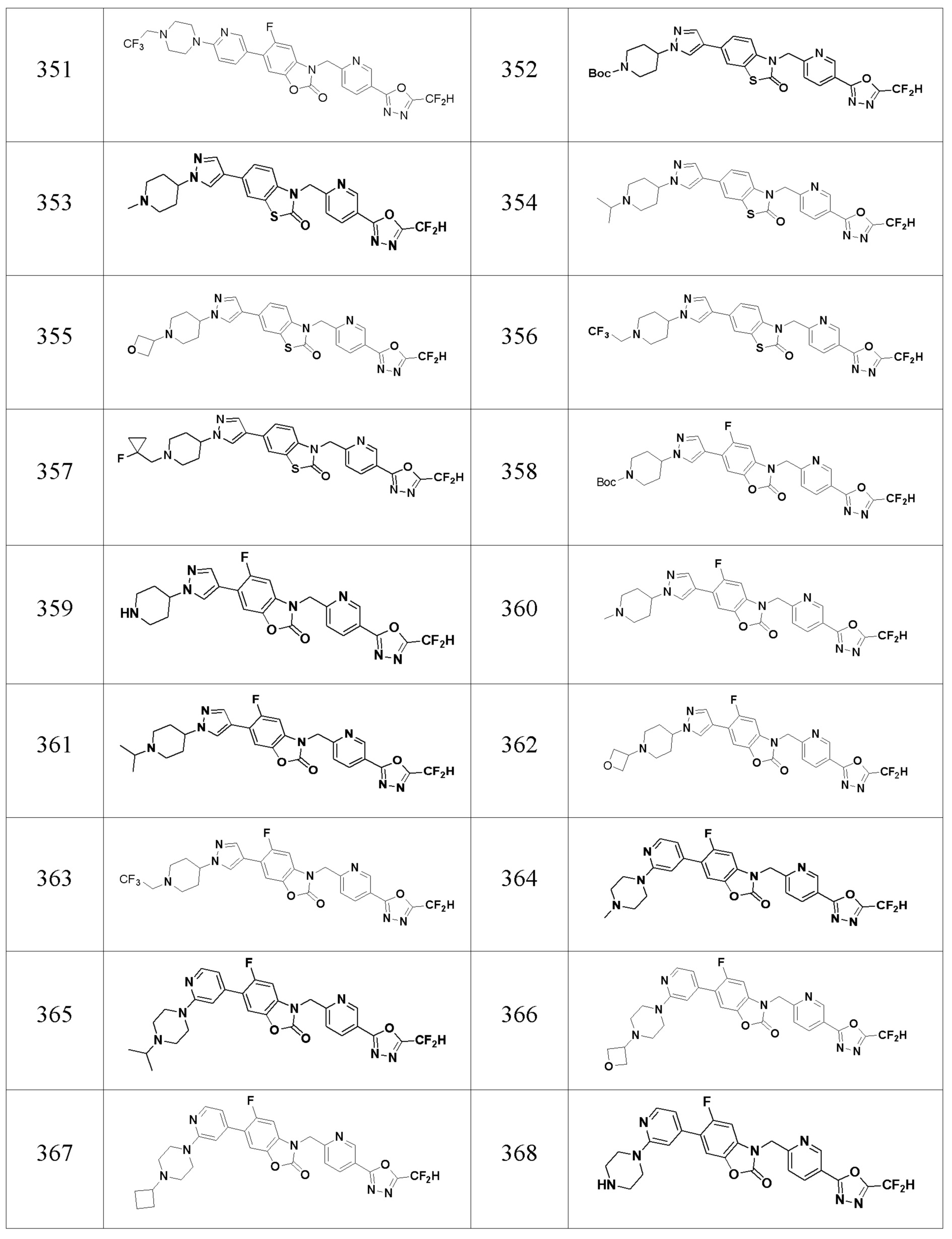
В одном варианте осуществления, производные соединения 1,3,4-оксадиазола, представленные химической формулой I по настоящему изобретению, их стереоизомеры или их фармацевтически приемлемые соли включают соединения, показанные в следующей таблице 1.
Таблица 1
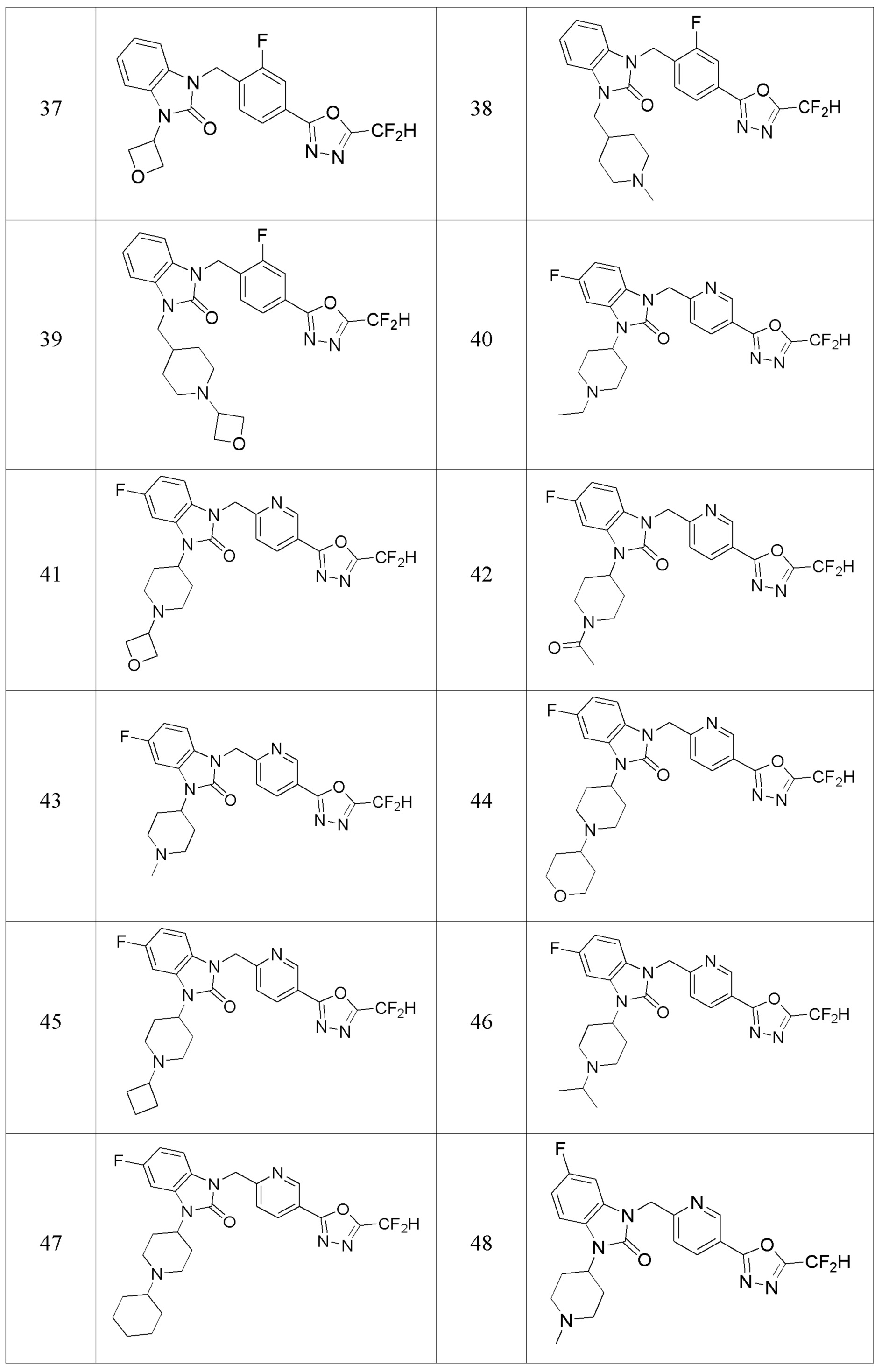
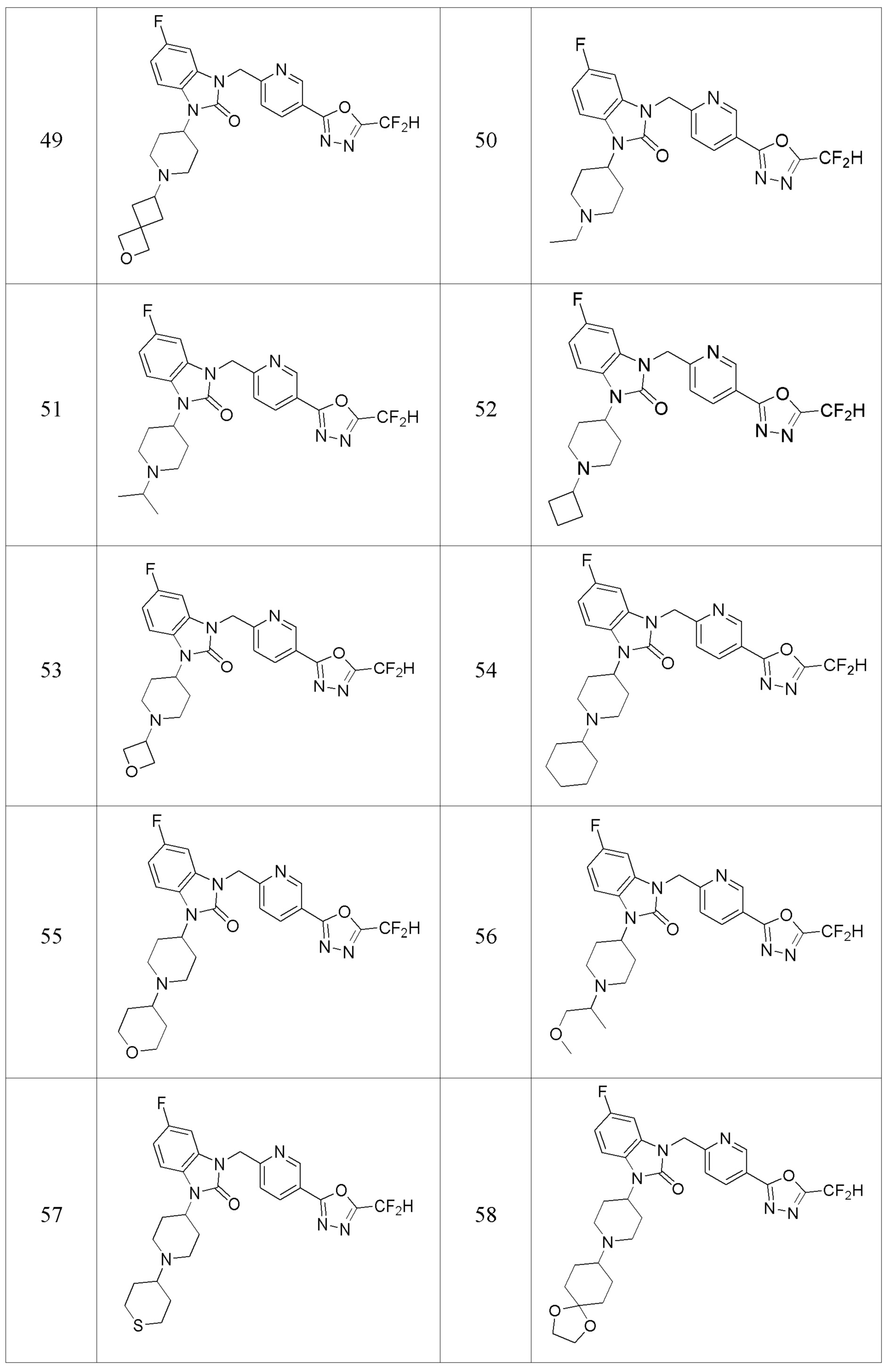
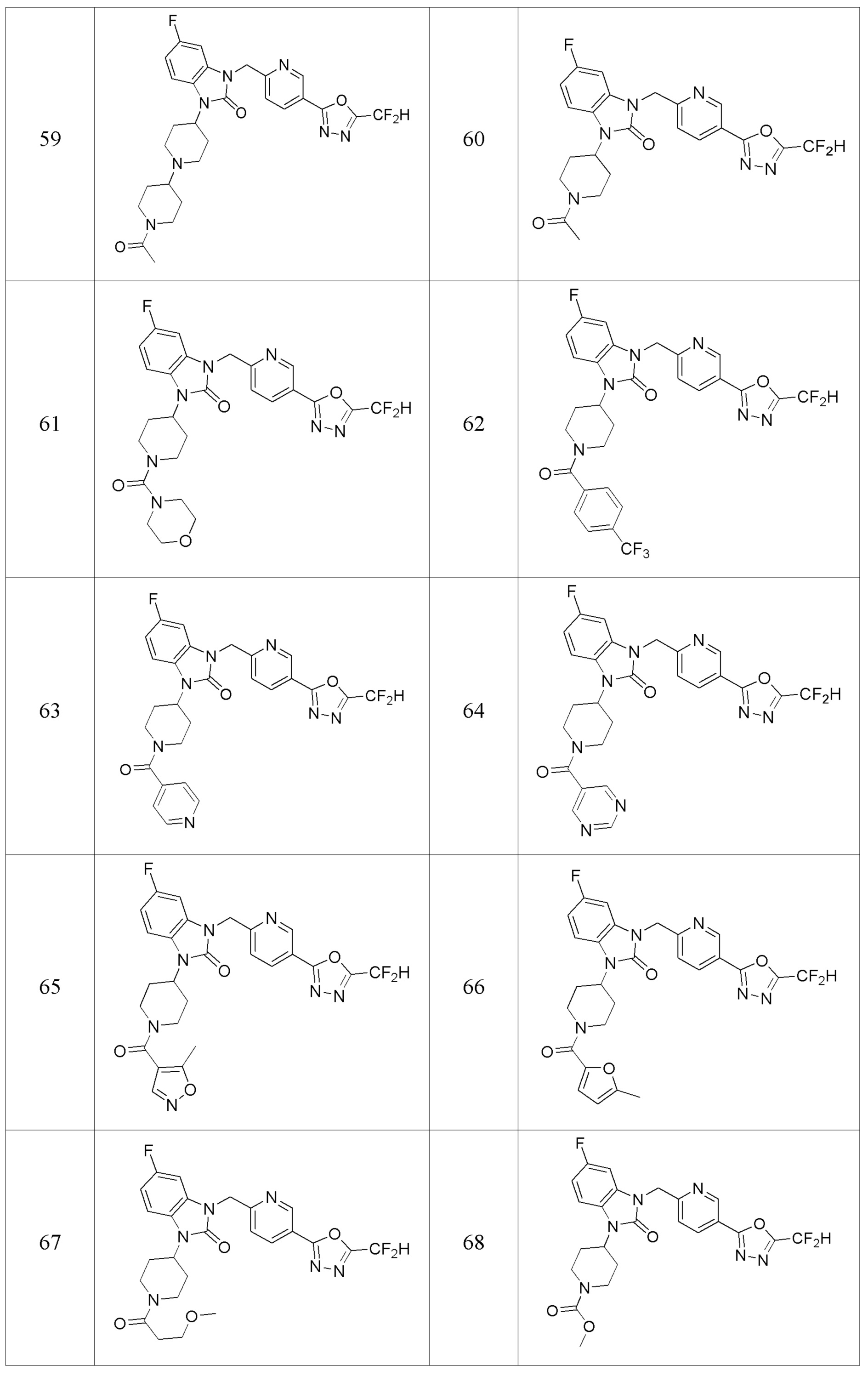
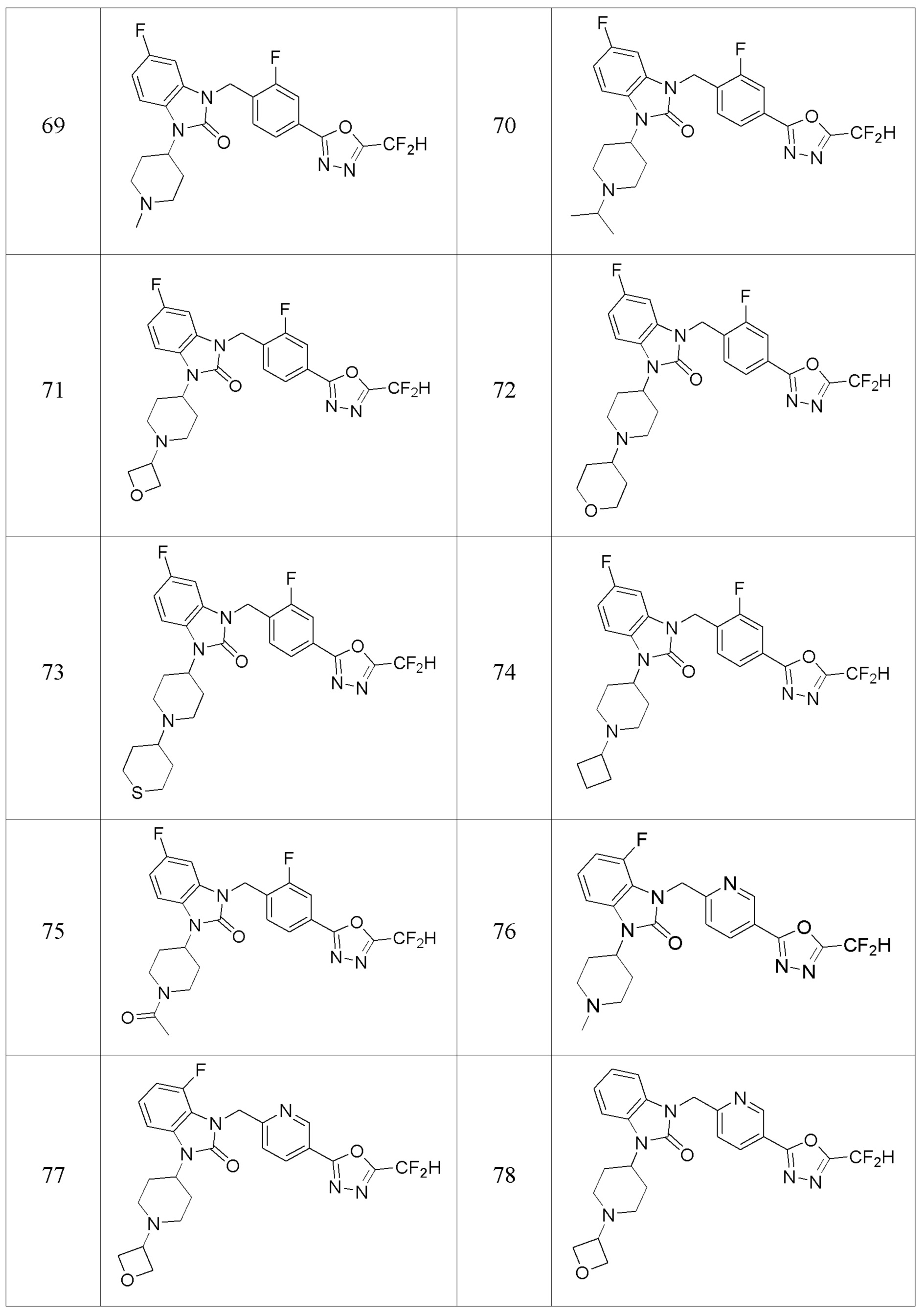
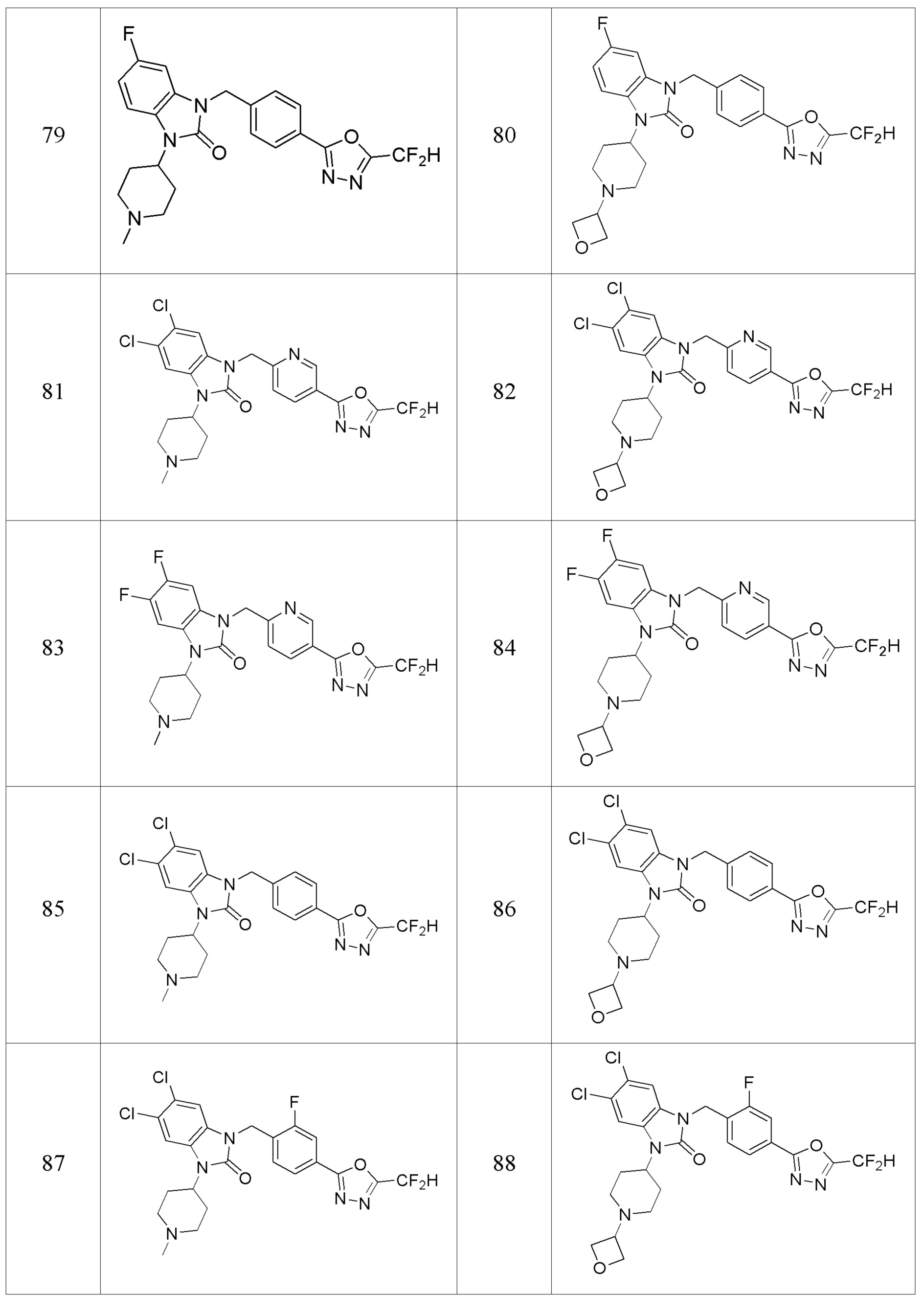
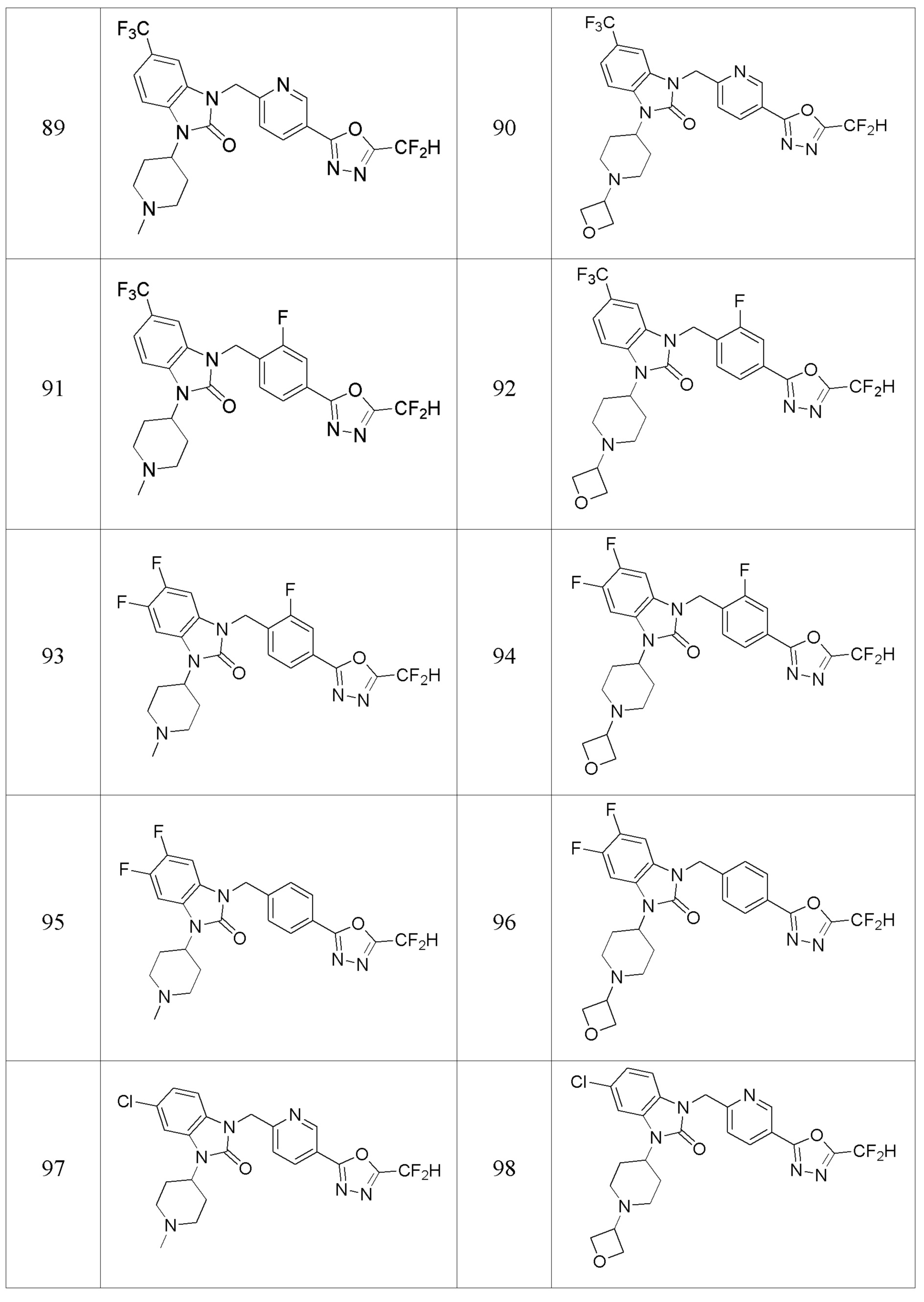
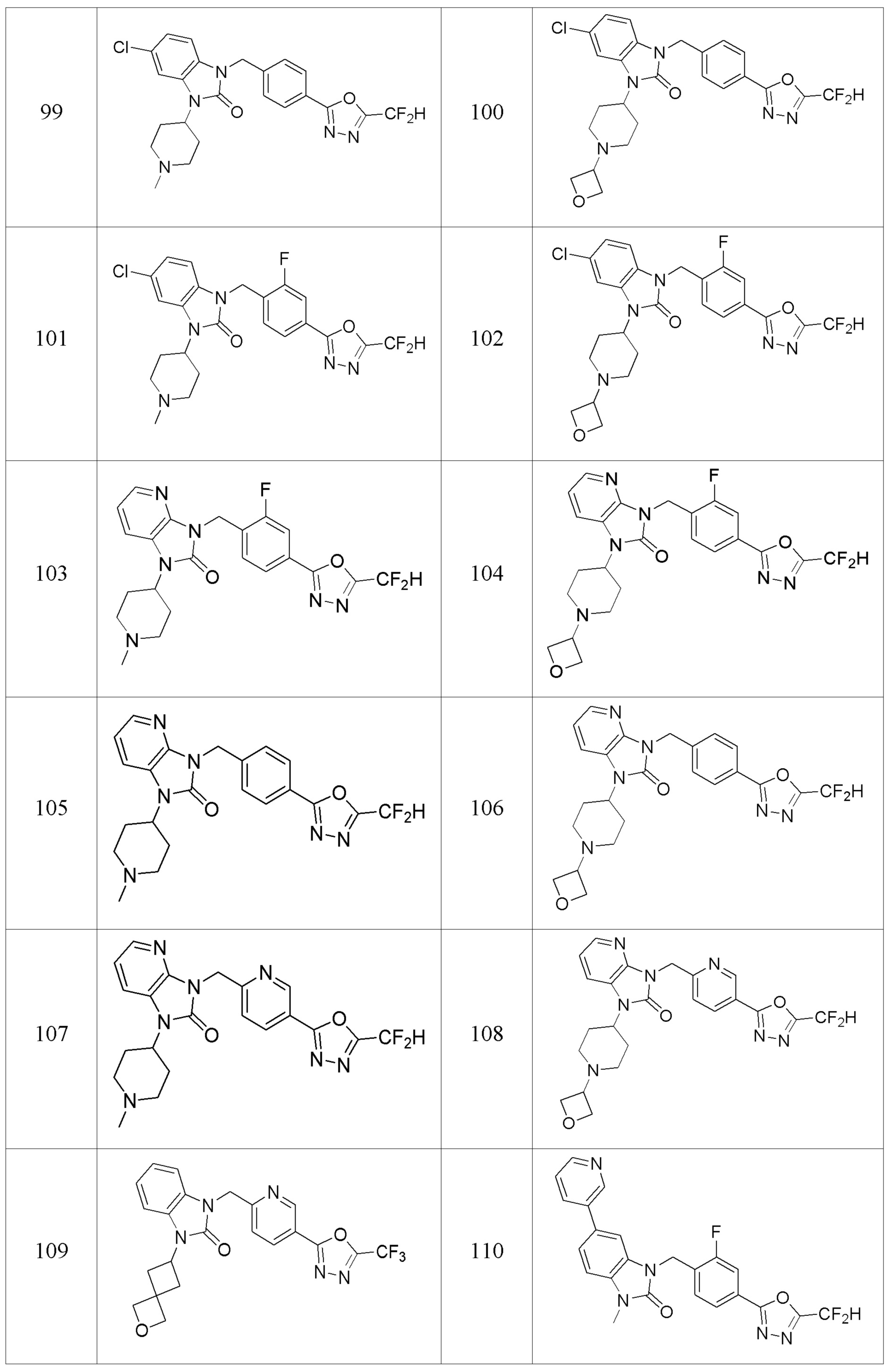
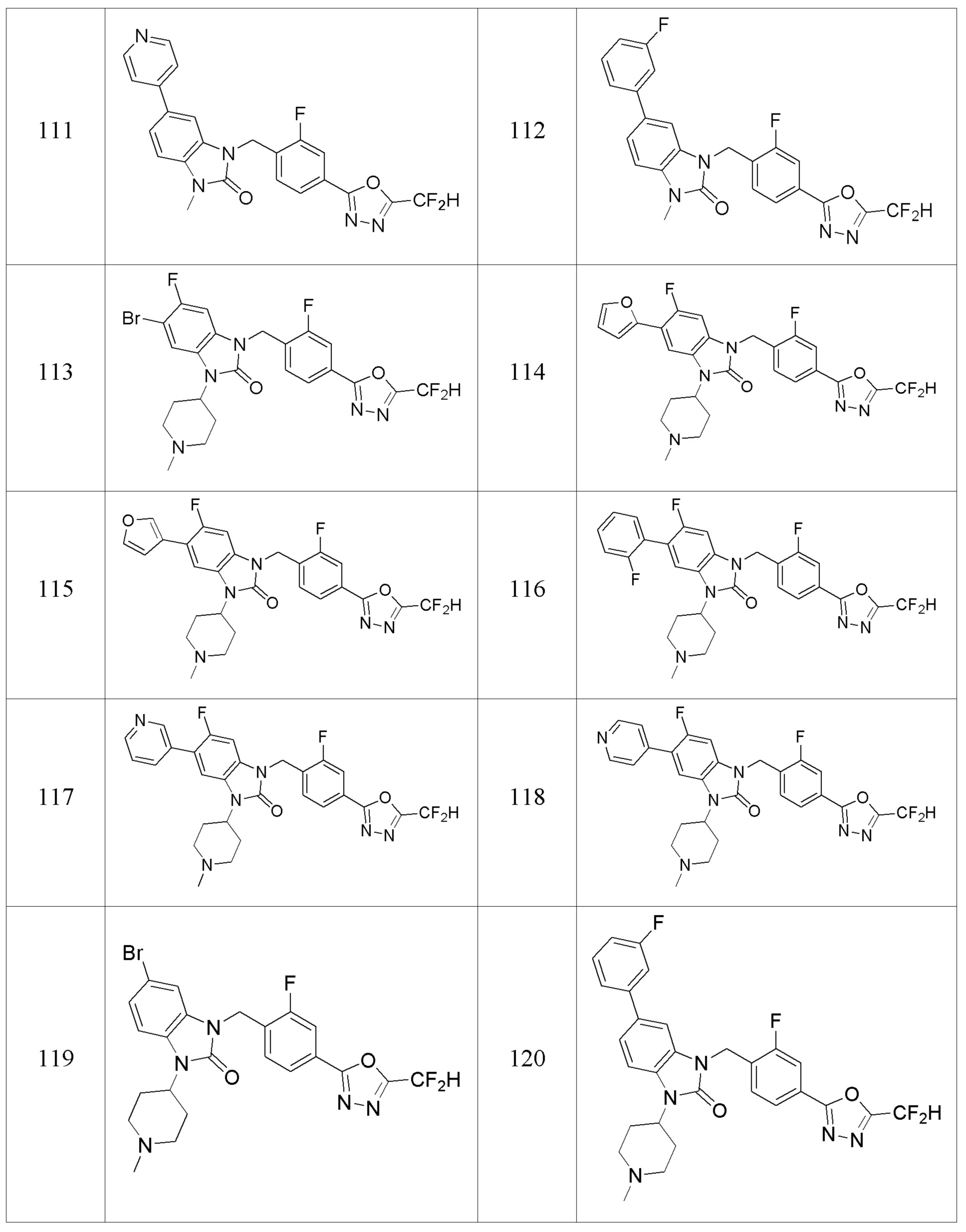
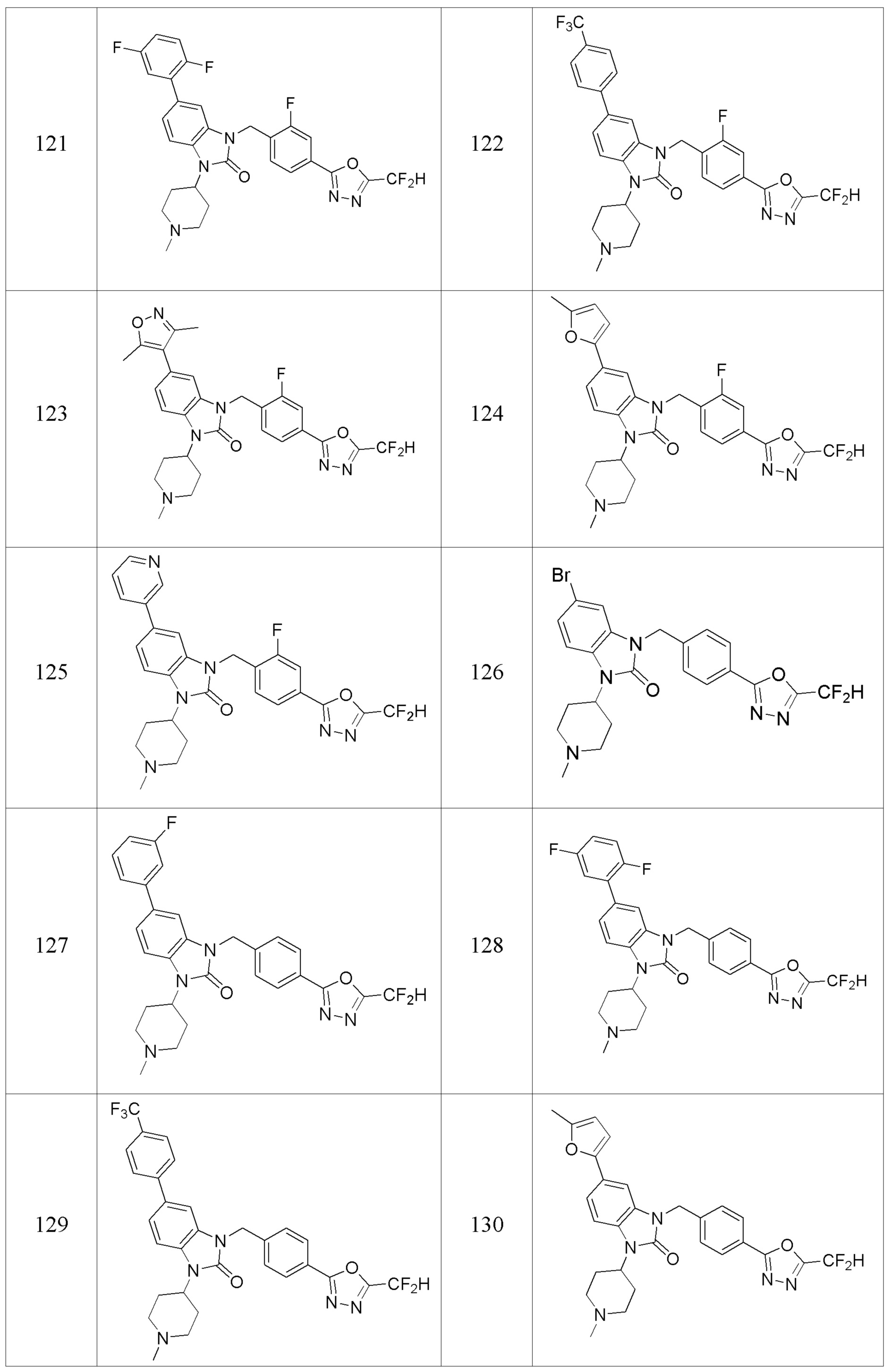
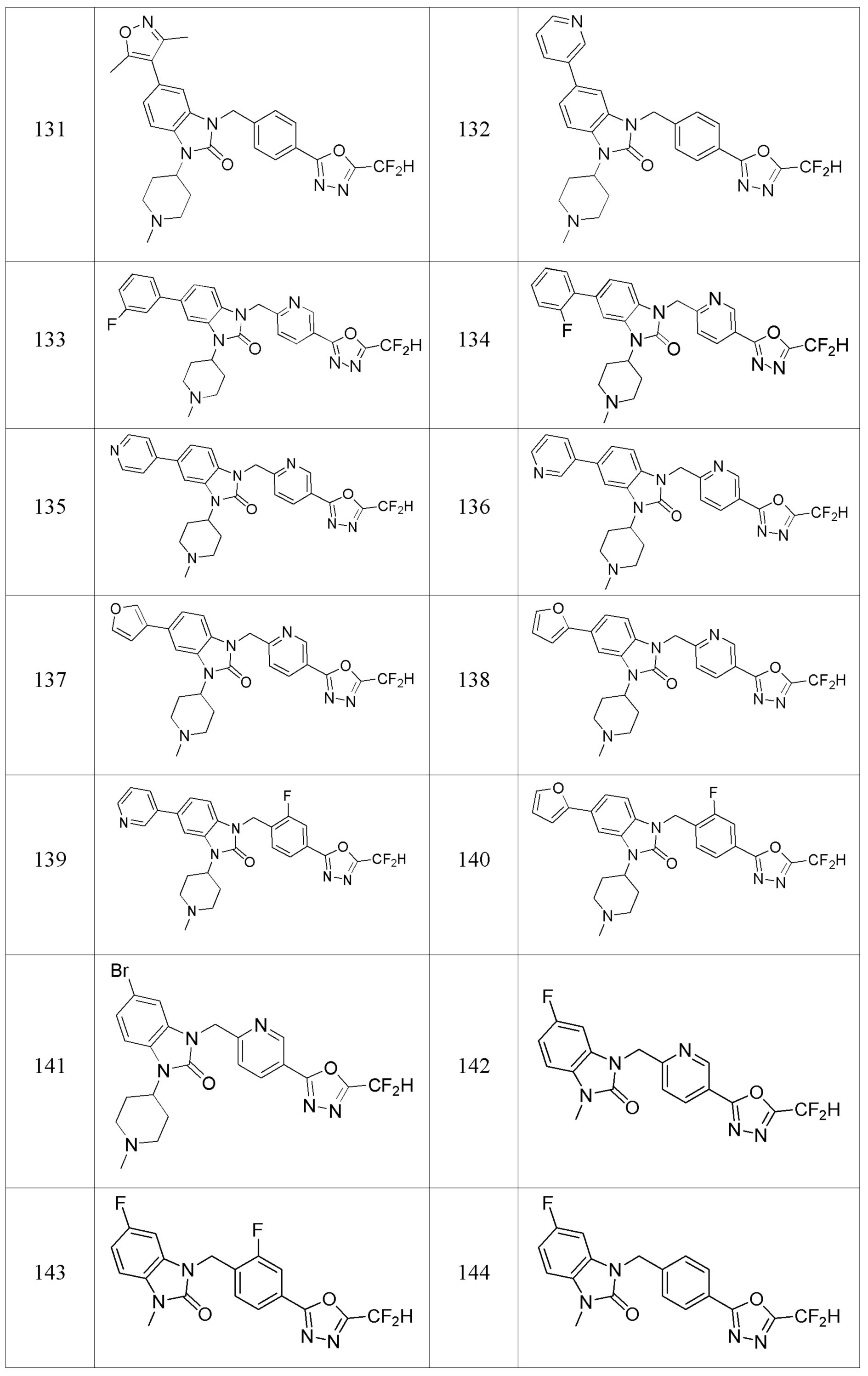
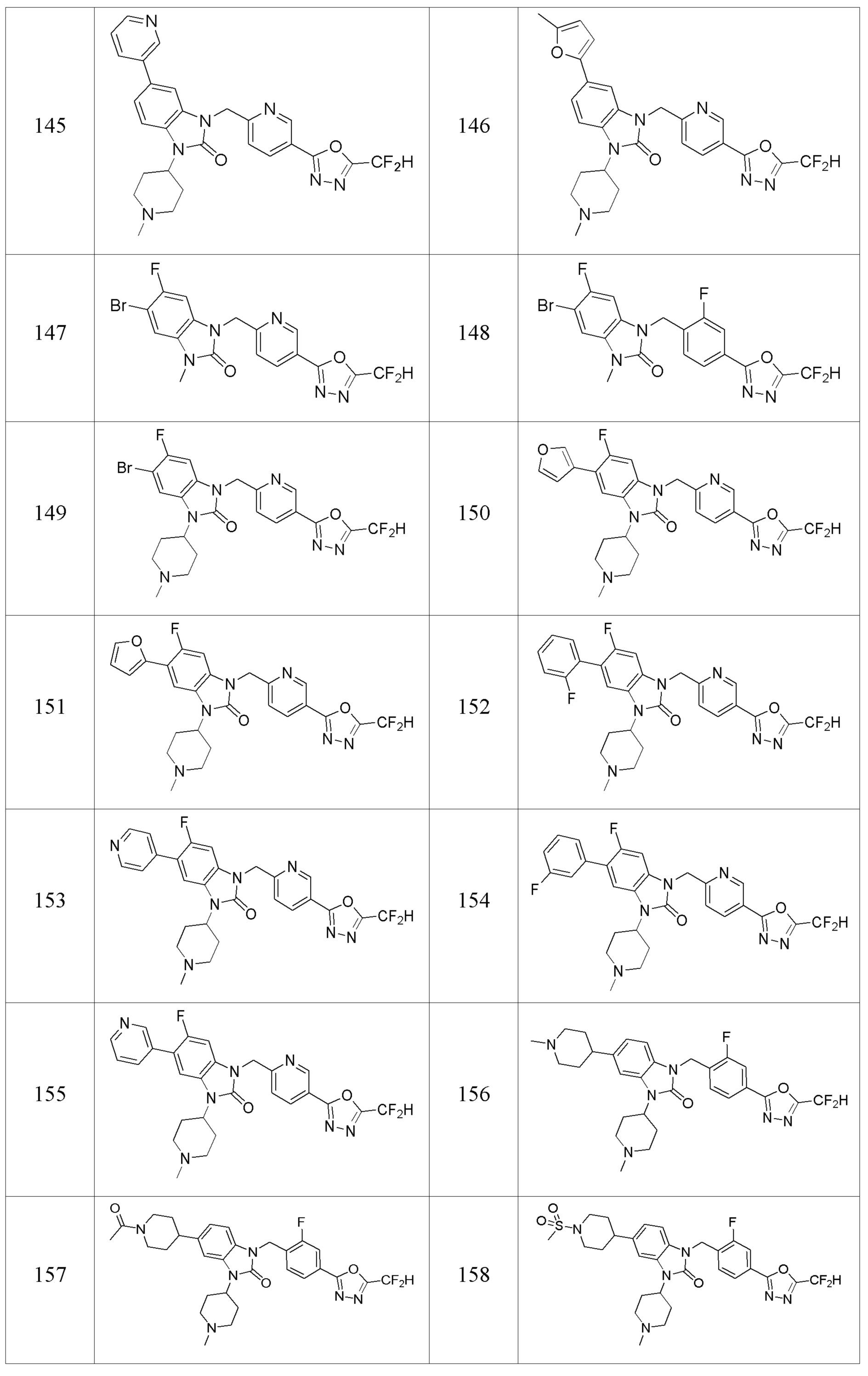
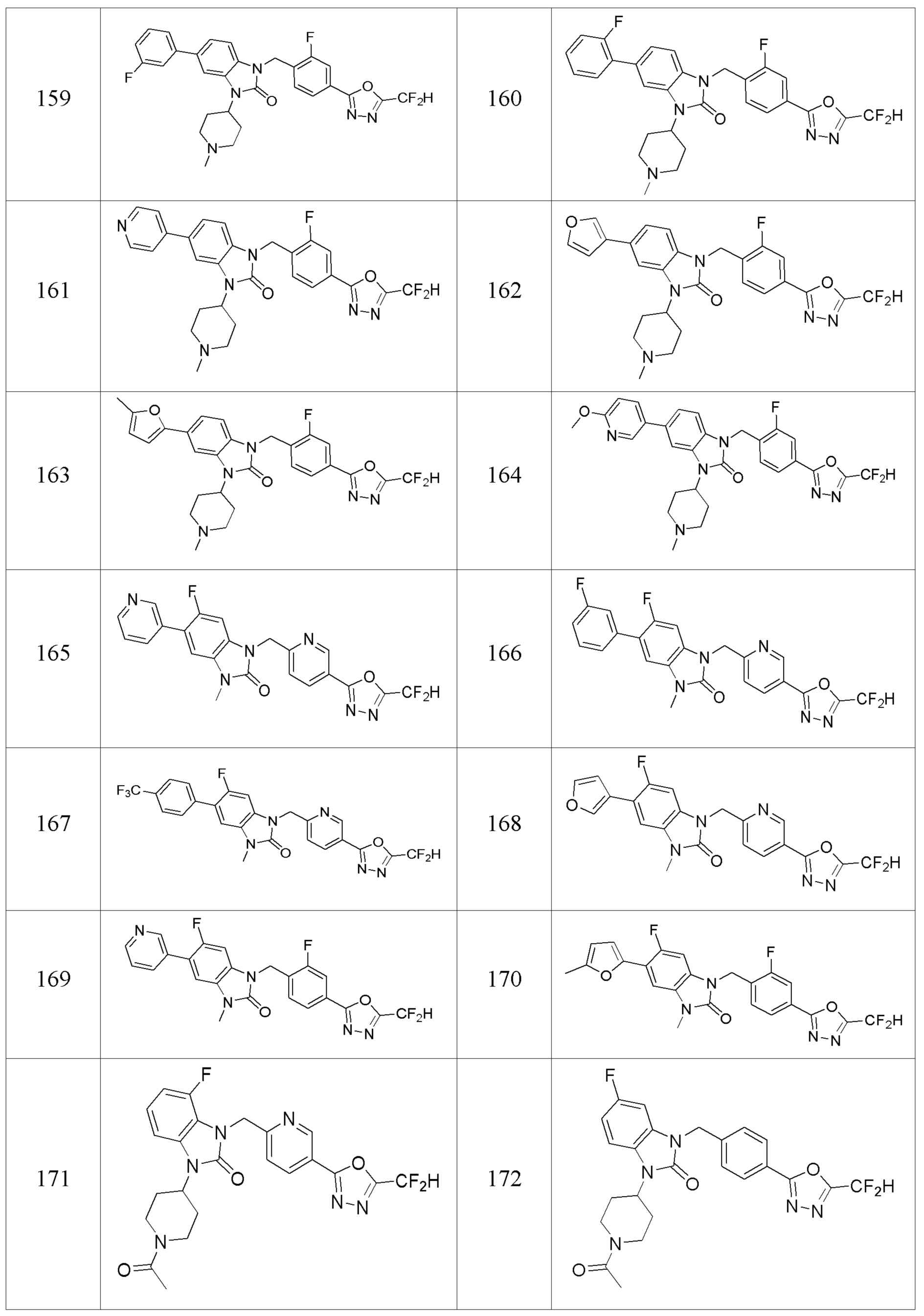
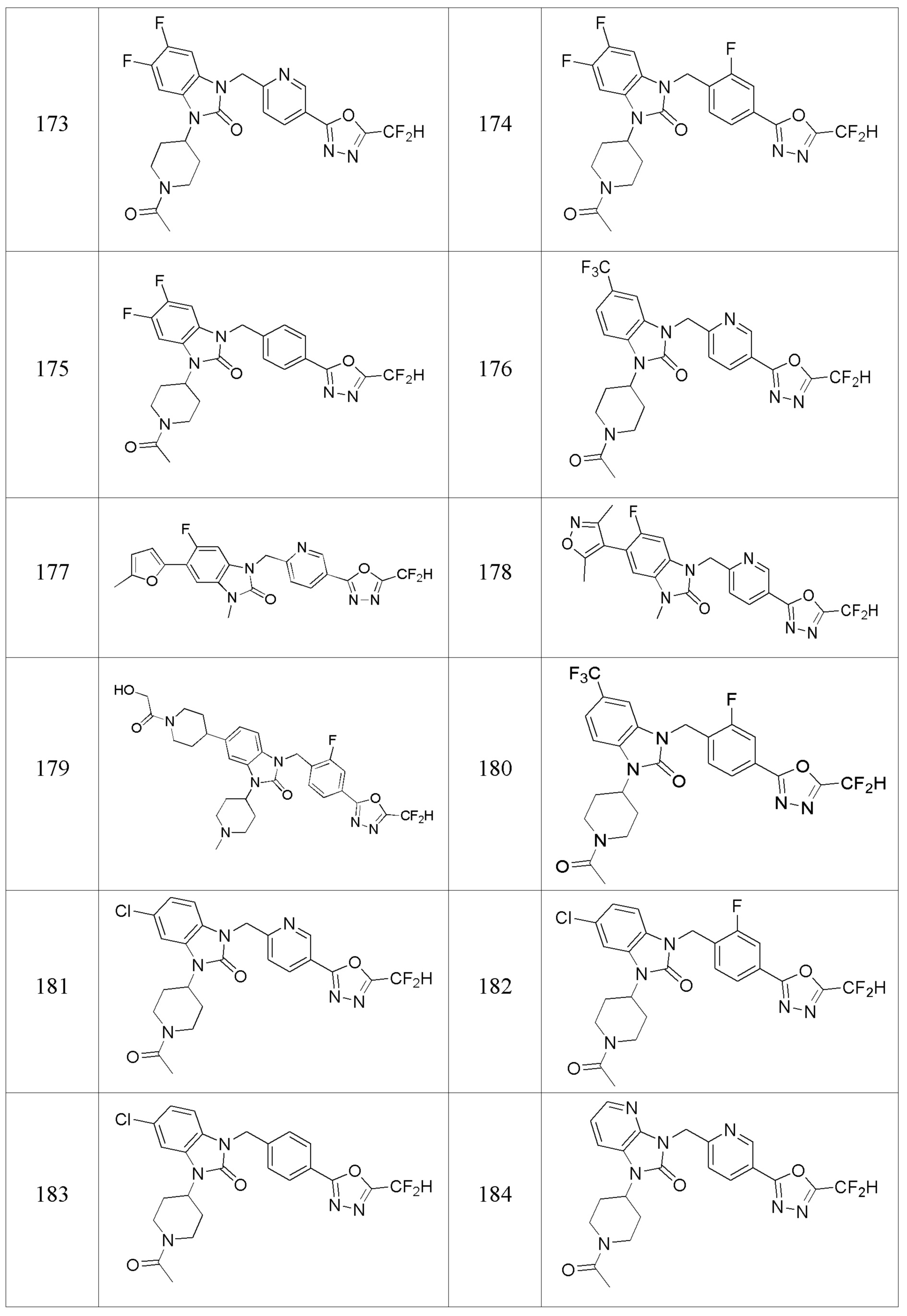
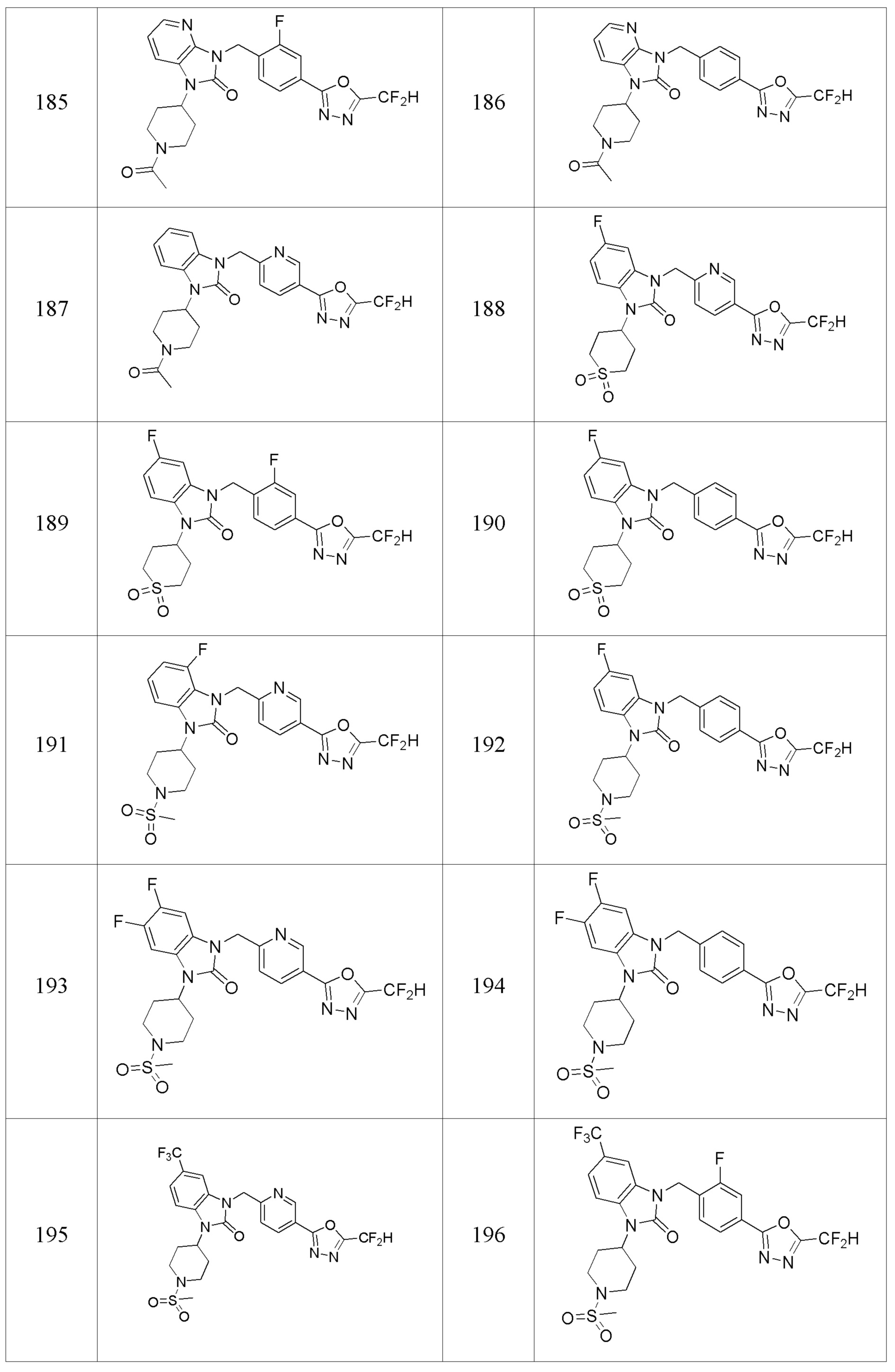
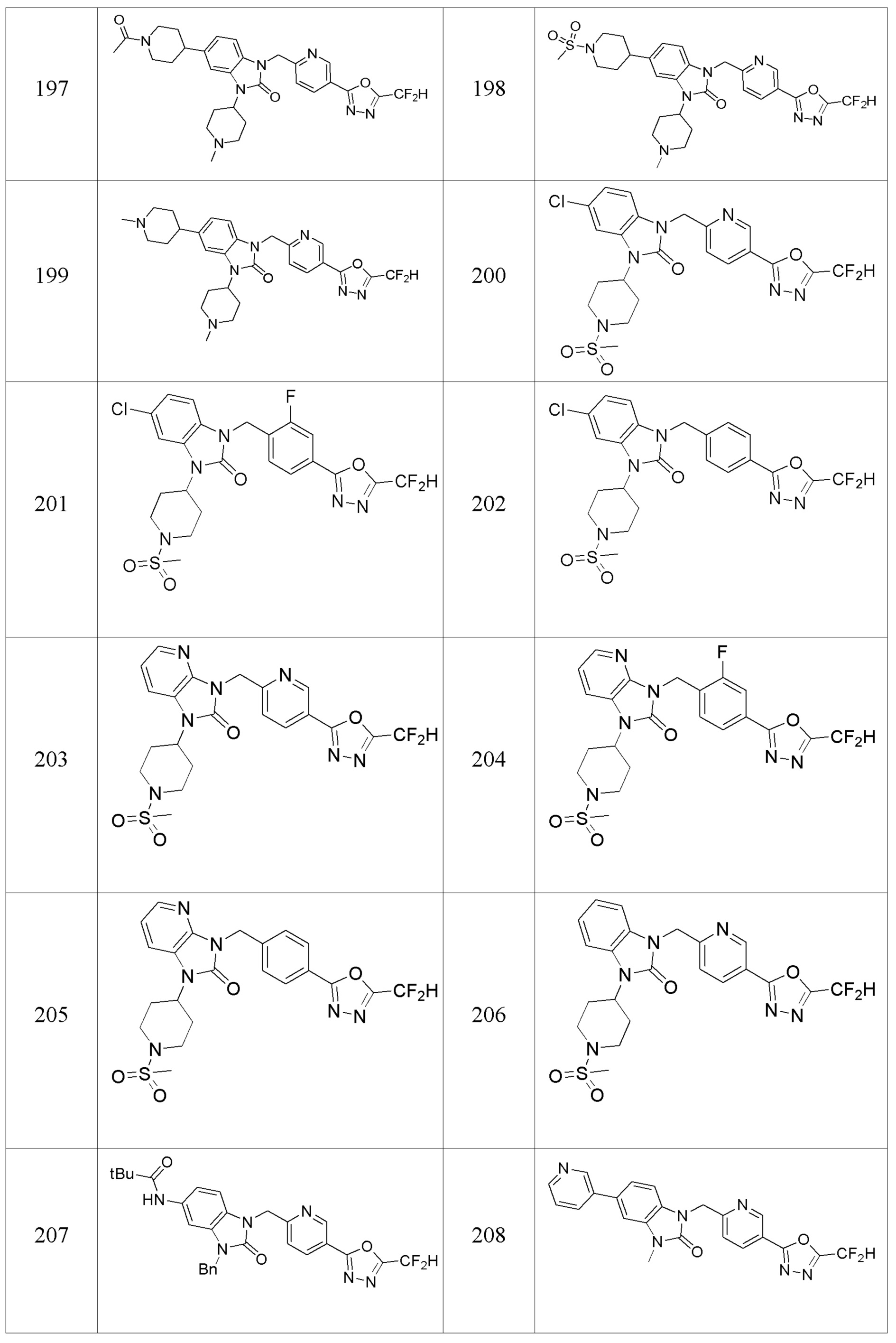
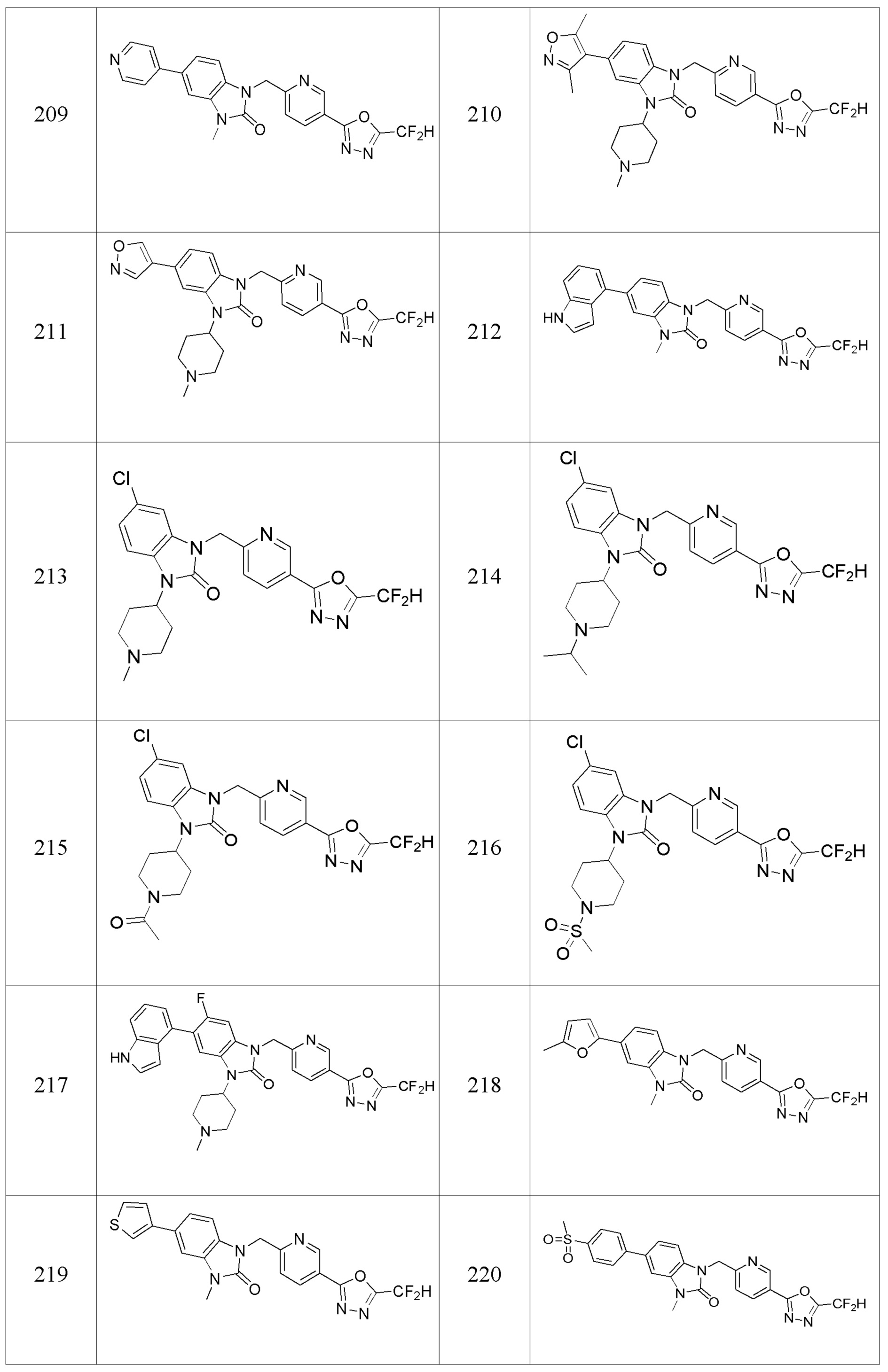
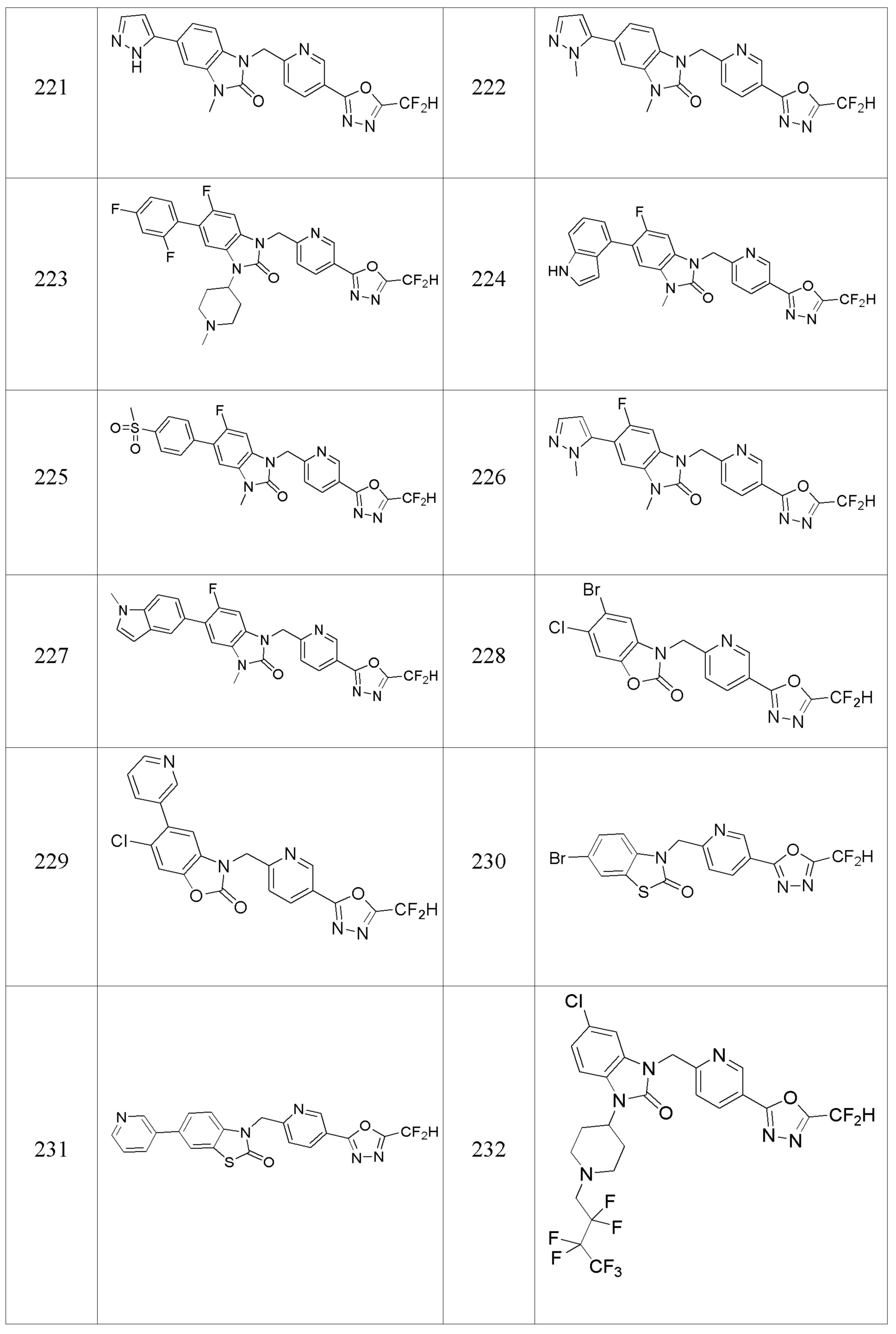
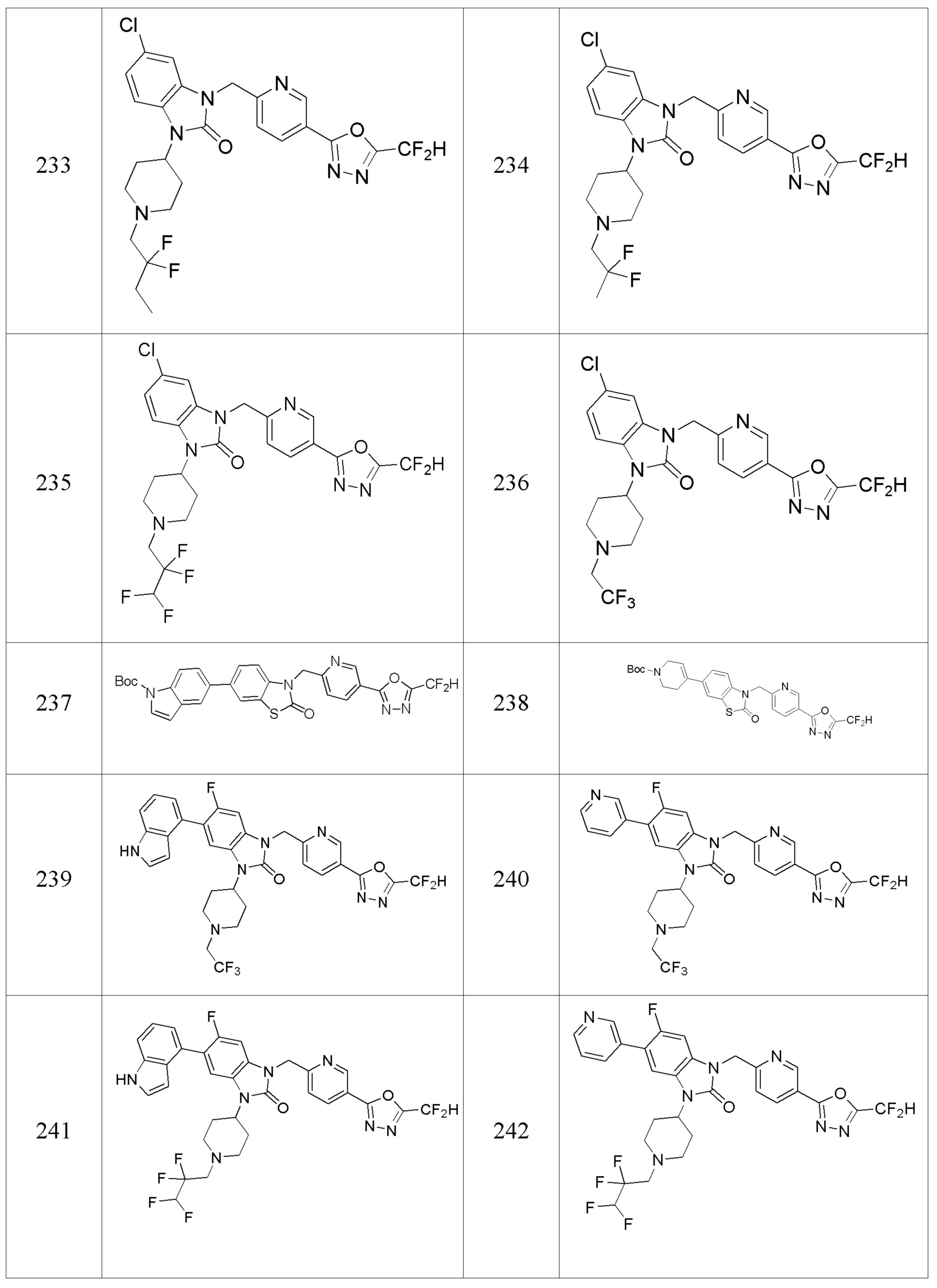
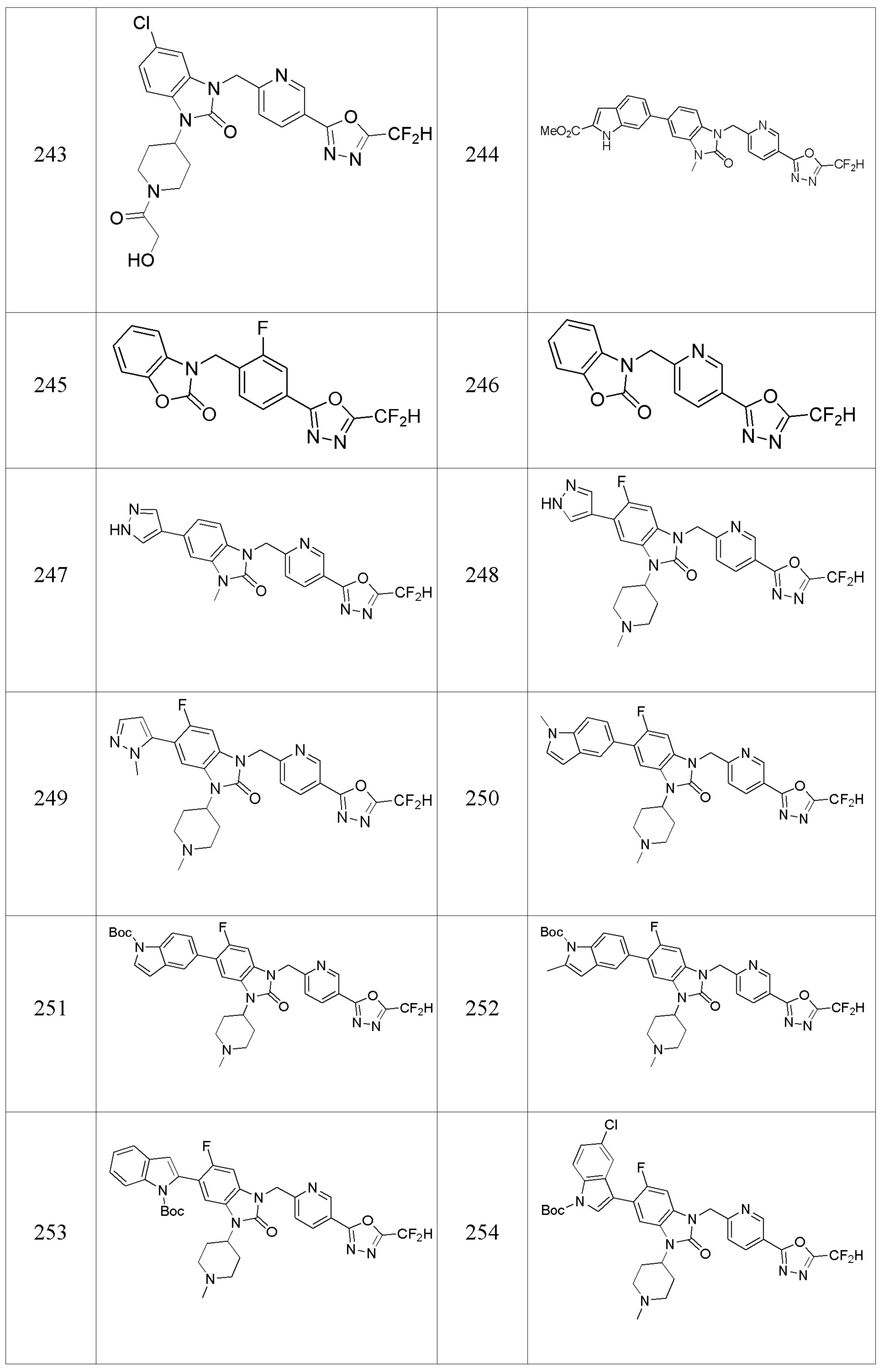
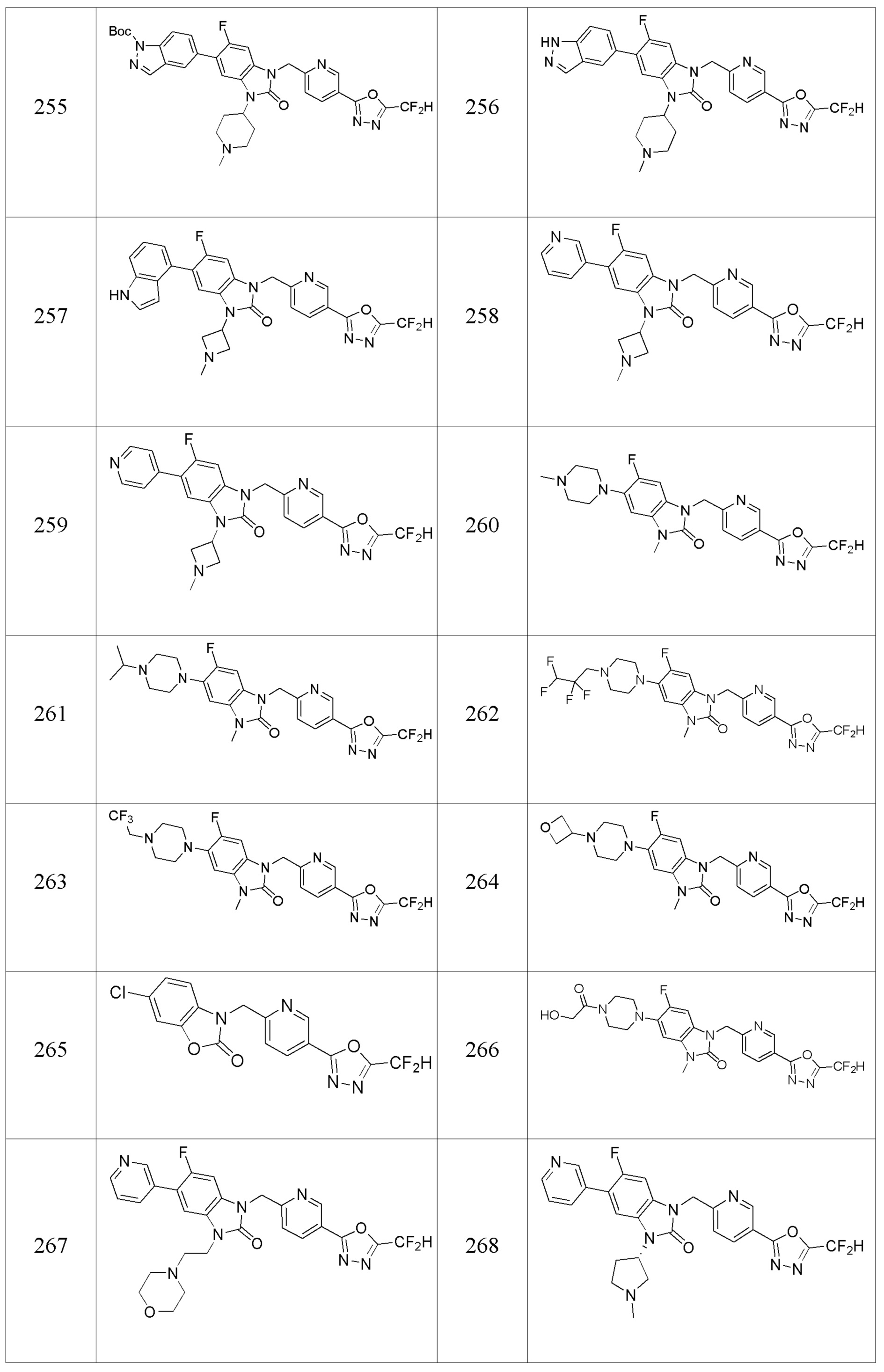
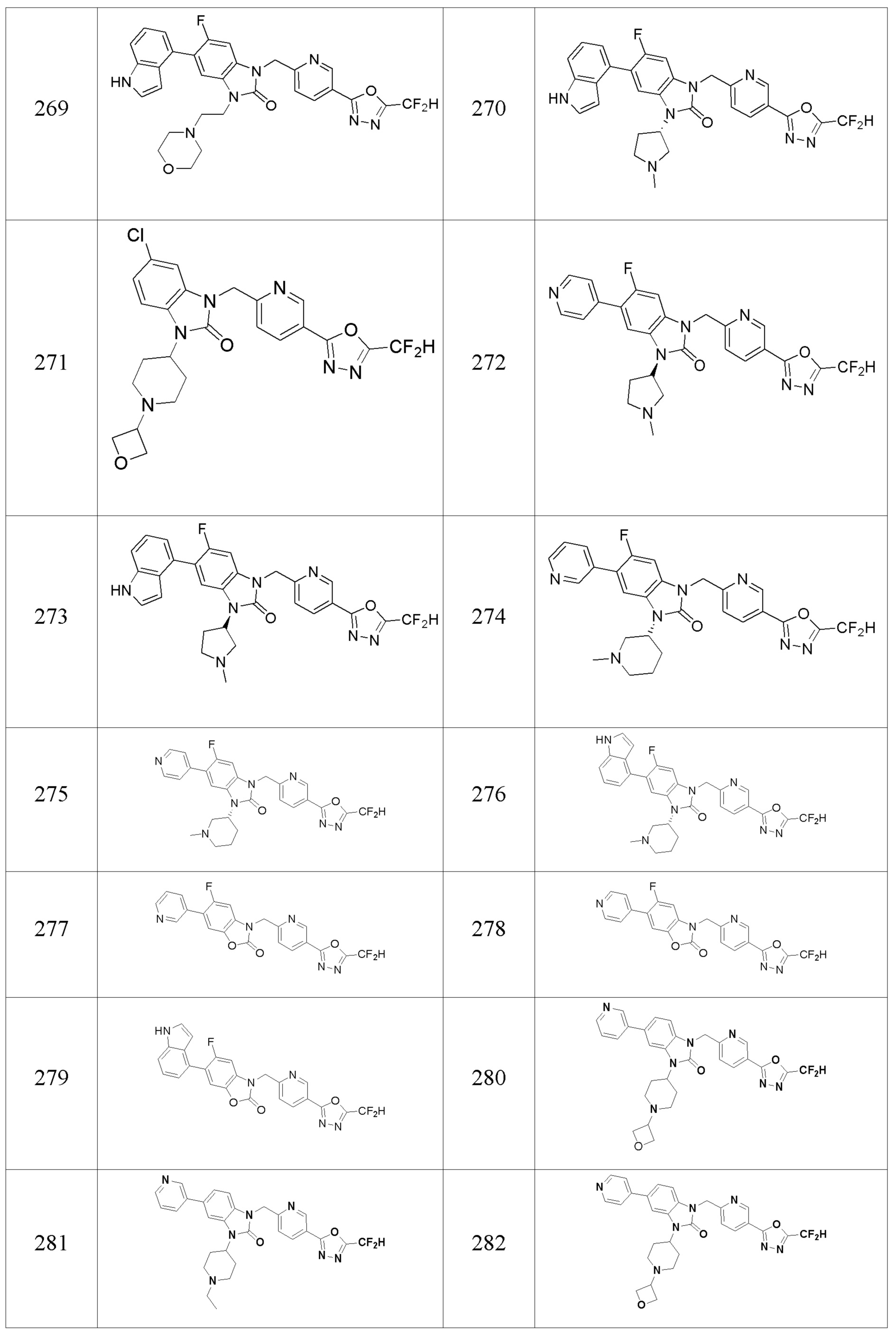
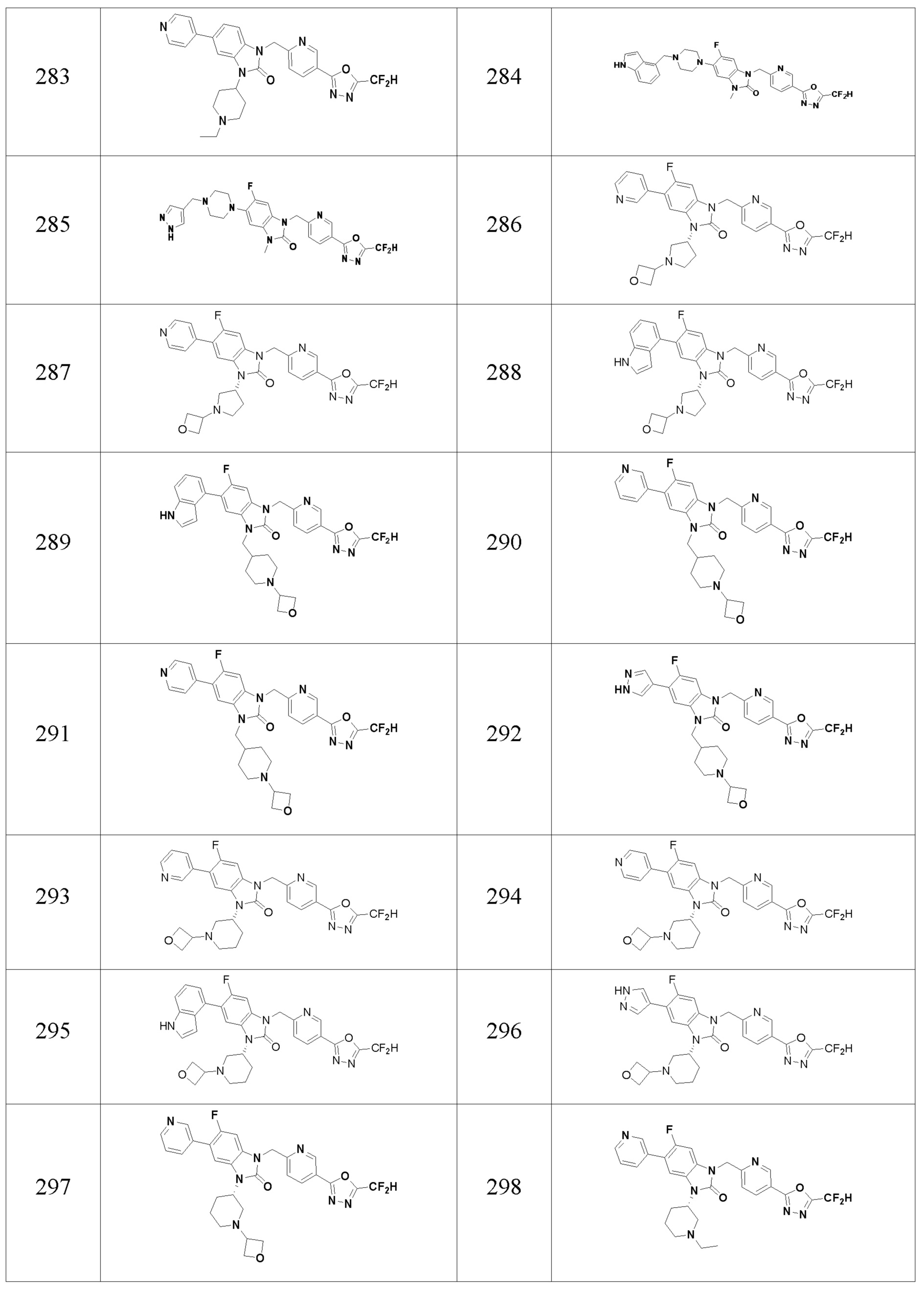
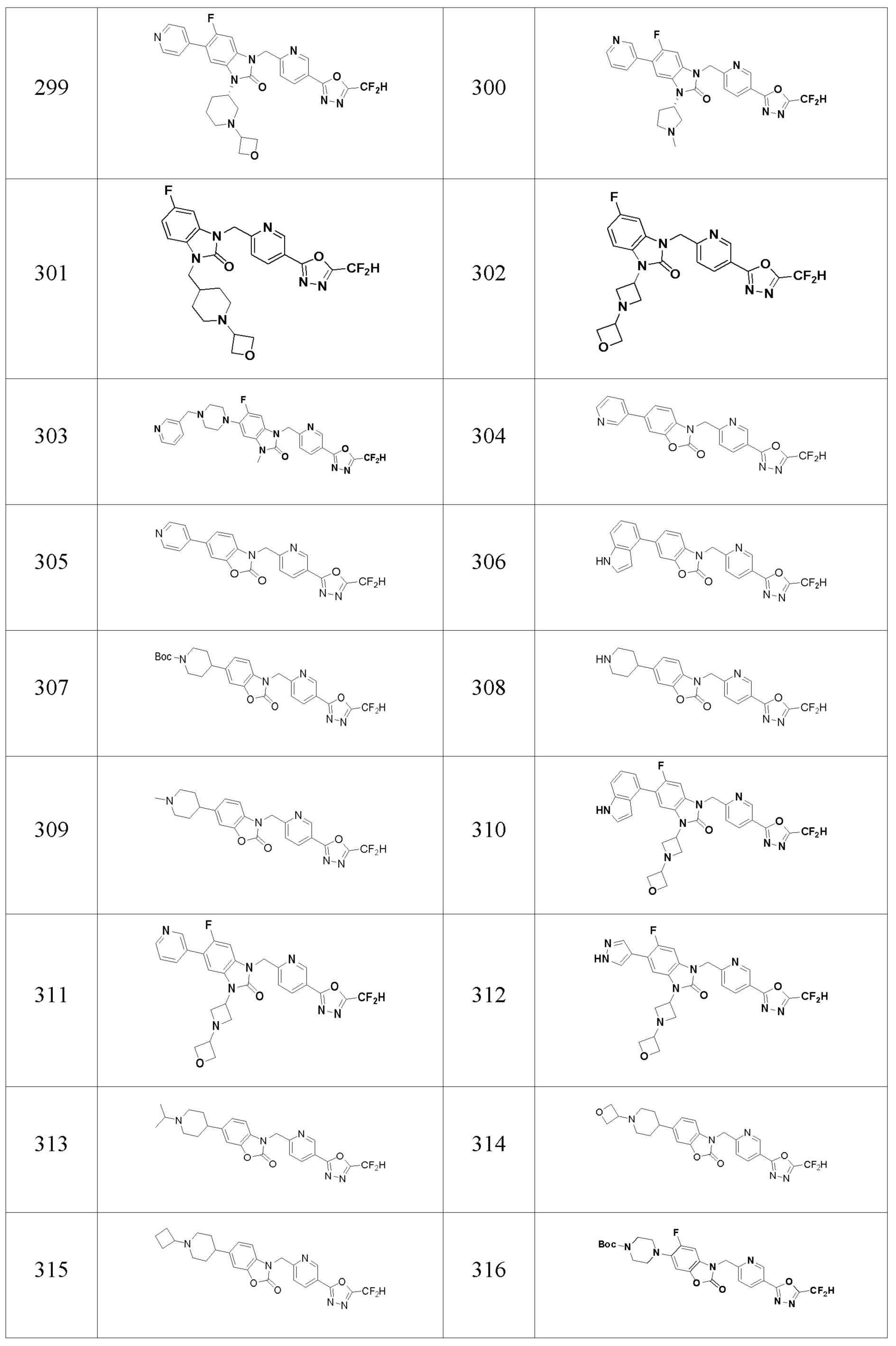
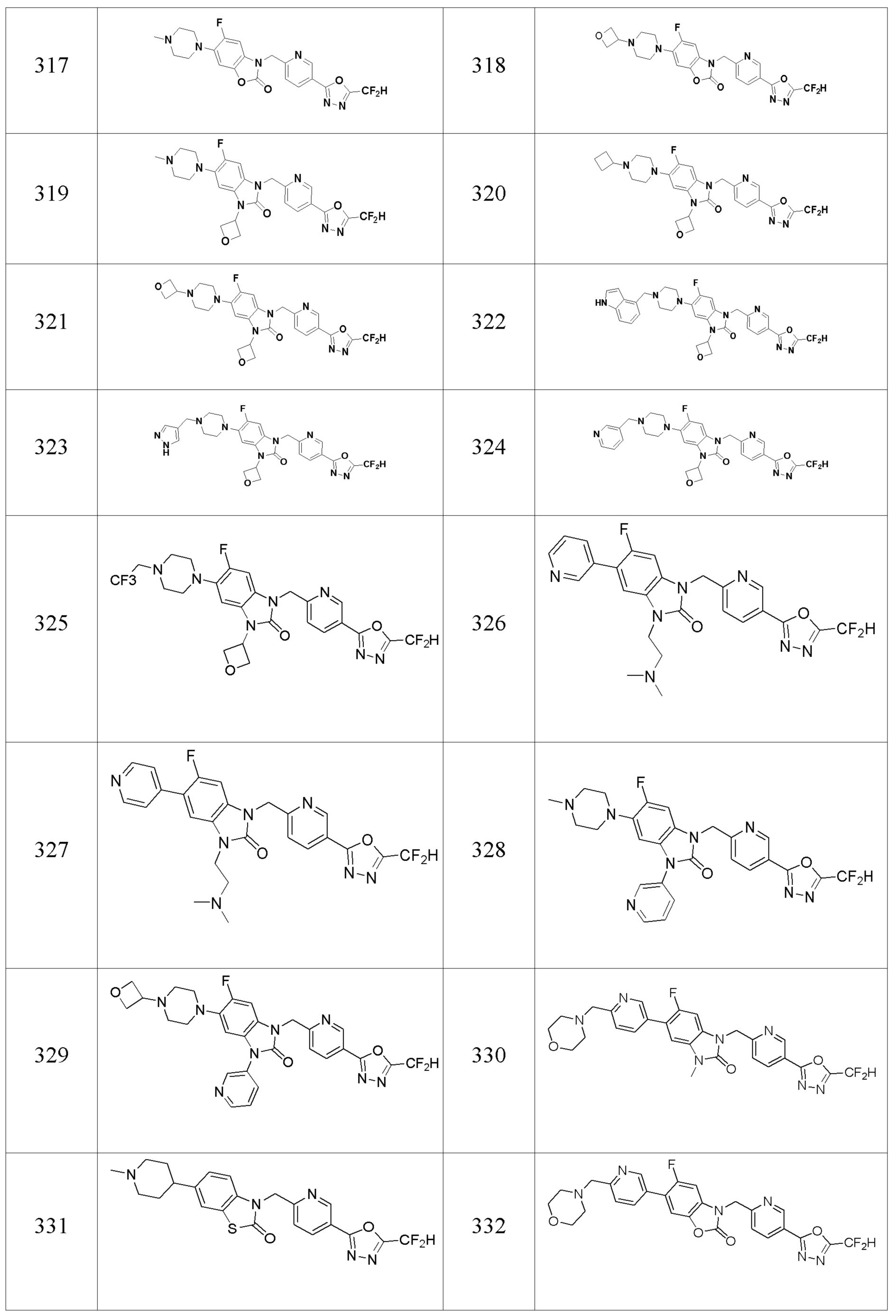
Способ получения производных соединений 1,3,4-оксадиазола
Настоящее изобретение представляет способ получения производных соединений 1,3,4-оксадиазола, представленных химической формулой I, их стереоизомеров или их фармацевтически приемлемых солей.
Предпочтительный способ получения производных соединений 1,3,4-оксадиазола, представленных химической формулой I, их стереоизомеров или их фармацевтически приемлемых солей является таким же, как показан на следующих формулах реакции 1-17, и в настоящий документ также включен способ получения, модифицированный на уровне, очевидном для специалиста в данной области техники.
В каждой из [Формула реакции 1] - [Формула реакции 17], «A» может быть фениленом, таким как арилен, но особенно не ограничен им. «A» может быть гетероариленом. Также в [Формула реакции 1] - [Формула реакции 17], R1 - R5, Z1 - Z4, a, b и X каждый является по существу таким же, как определен в химической формуле I, и L3 является C1-4 алкиленом. В [Формула реакции 1] - [Формула реакции 17], «галоген» означает атом галогена F, Cl, Br или I. Также в [Формула реакции 1] - [Формула реакции 17], «PG» означает «защитную группу» и может включать трет-бутилоксикарбонил (Boc),бензилоксикарбонил (Cbz) или подобные. Кроме того, в [Формула реакции 1] - [Формула реакции 17], «X1» означает O или S.
[Формула реакции 1]
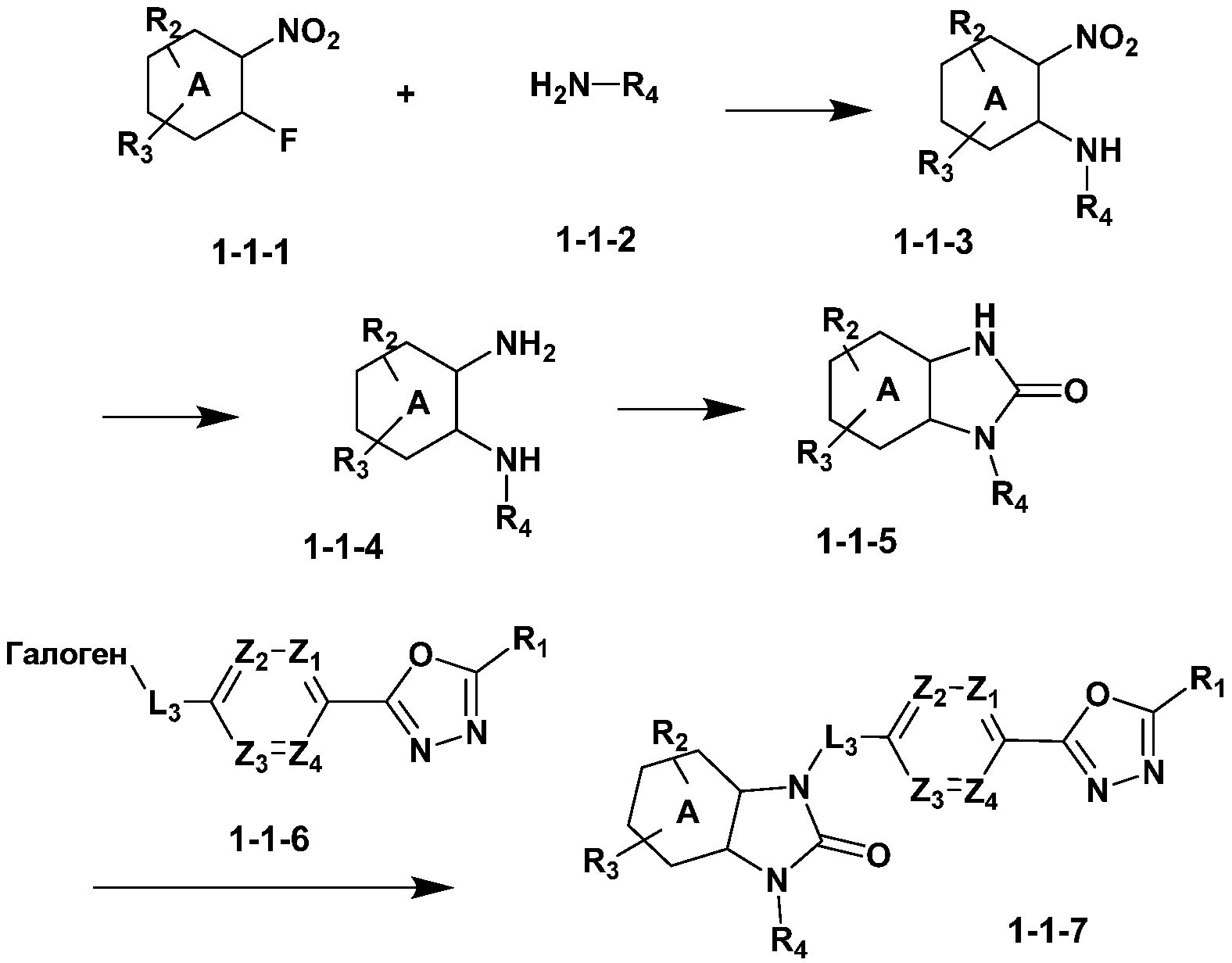
Вышеуказанная [Формула реакции 1] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензимидазолоновую структуру, и соединение химической формулы 1-1-1 взаимодействует с соединением химической формулы 1-1-2 с получением соединения химической формулы 1-1-3. Затем, полученное соединение применяют для получения соединения химической формулы 1-1-4. После этого, соединение химической формулы 1-1-4 подвергают реакции циклизации с получением соединения химической формулы 1-1-5. После этого, полученное соединение подвергают реакции замещения с соединением химической формулы 1-1-6 с получением соединения химической формулы 1-1-7.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 1, включают соединения 2, 3, 11-13, 24-26, 34-37, 142-144, 147, 148 и подобные.
[Формула реакции 2]
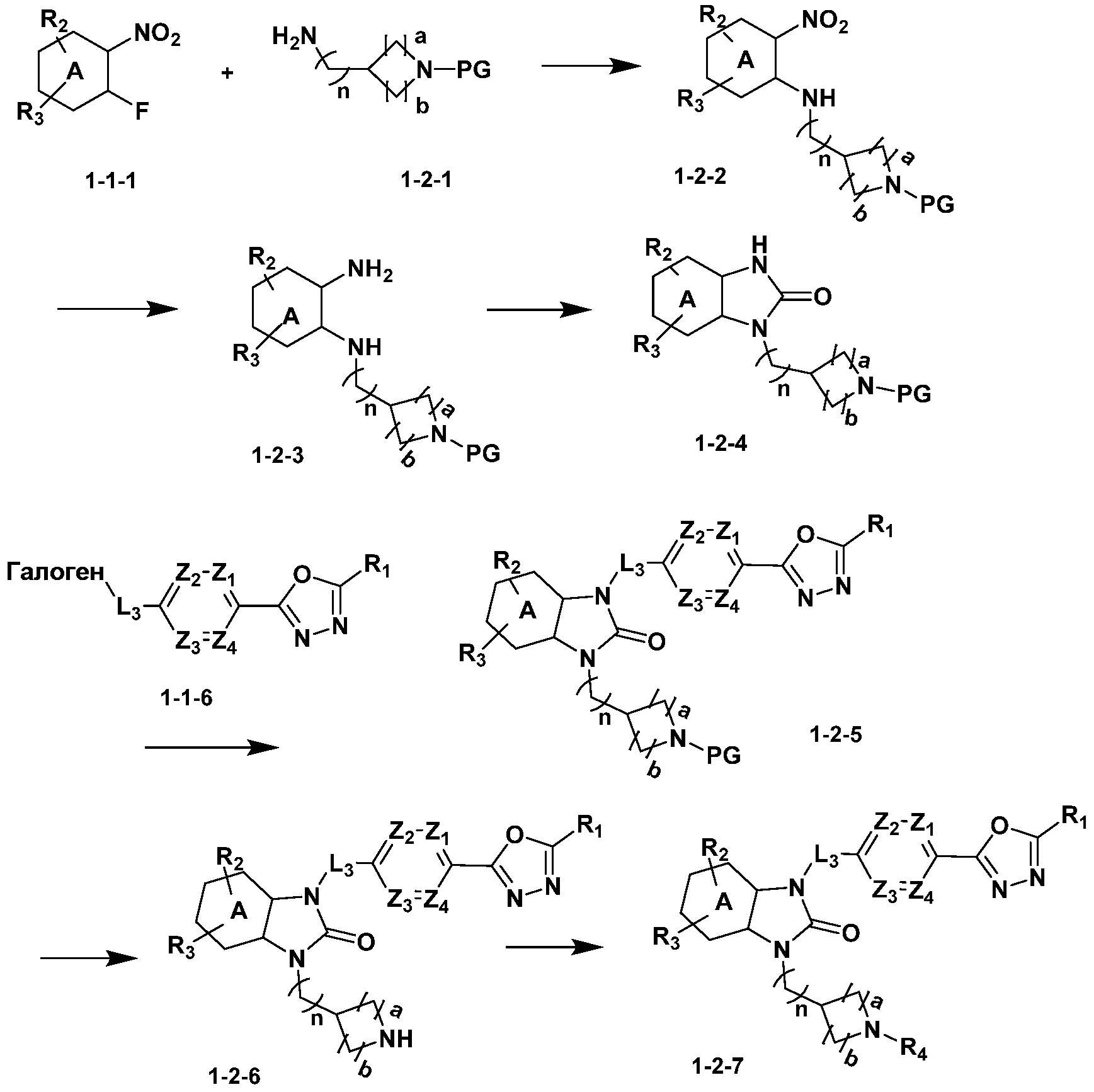
Вышеуказанная [Формула реакции 2] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензимидазолоновую структуру, и соединение химической формулы 1-1-1 подвергают реакции замещения с соединением химической формулы 1-2-1 с получением соединения химической формулы 1-2-2, и затем подвергают реакции восстановления с получением соединения химической формулы 1-2-3. После этого, соединение химической формулы 1-2-3 подвергают реакции циклизации с получением соединения химической формулы 1-2-4, и затем подвергают реакции замещения с соединением химической формулы 1-1-6 с получением соединения химической формулы 1-2-5. Защитную группу удаляют из соединения химической формулы 1-2-5 с получением соединения химической формулы 1-2-6, и затем проводят реакцию замещения, реакцию восстановительного аминирования и реакцию ацилирования проводят с получением соединения химической формулы 1-2-7.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 2 включают соединения 40-77, 79-108, 113, 125, 132, 141, 149, 172-176, 180-186, 191-196, 200-206, 213-216, 232-236, 243, 271, 301, 302 и подобные.
[Формула реакции 3]
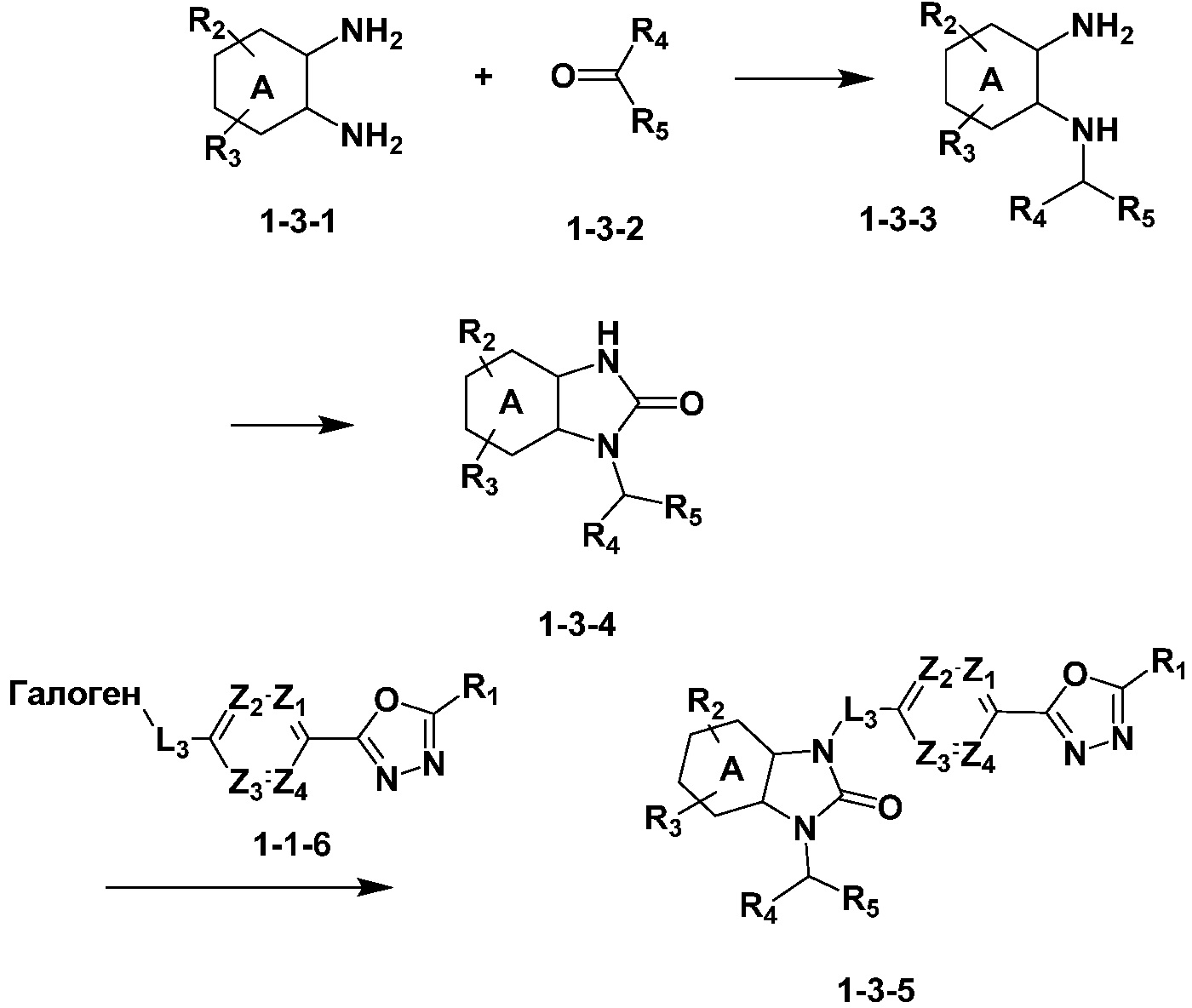
Вышеуказанная [Формула реакции 3] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензимидазолоновую структуру, и соединение химической формулы 1-3-1 подвергают реакцию восстановительного аминирования с соединением химической формулы 1-3-2 с получением соединения химической формулы 1-3-3, и затем подвергают реакции циклизации с получением соединения химической формулы 1-3-4. После этого, соединение химической формулы 1-3-4 подвергают реакции замещения с соединением химической формулы 1-1-6 с получением соединения химической формулы 1-3-5.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 3 включают соединения 27, 28, 29, 109, 188, 189, 190 и подобные.
[Формула реакции 4]
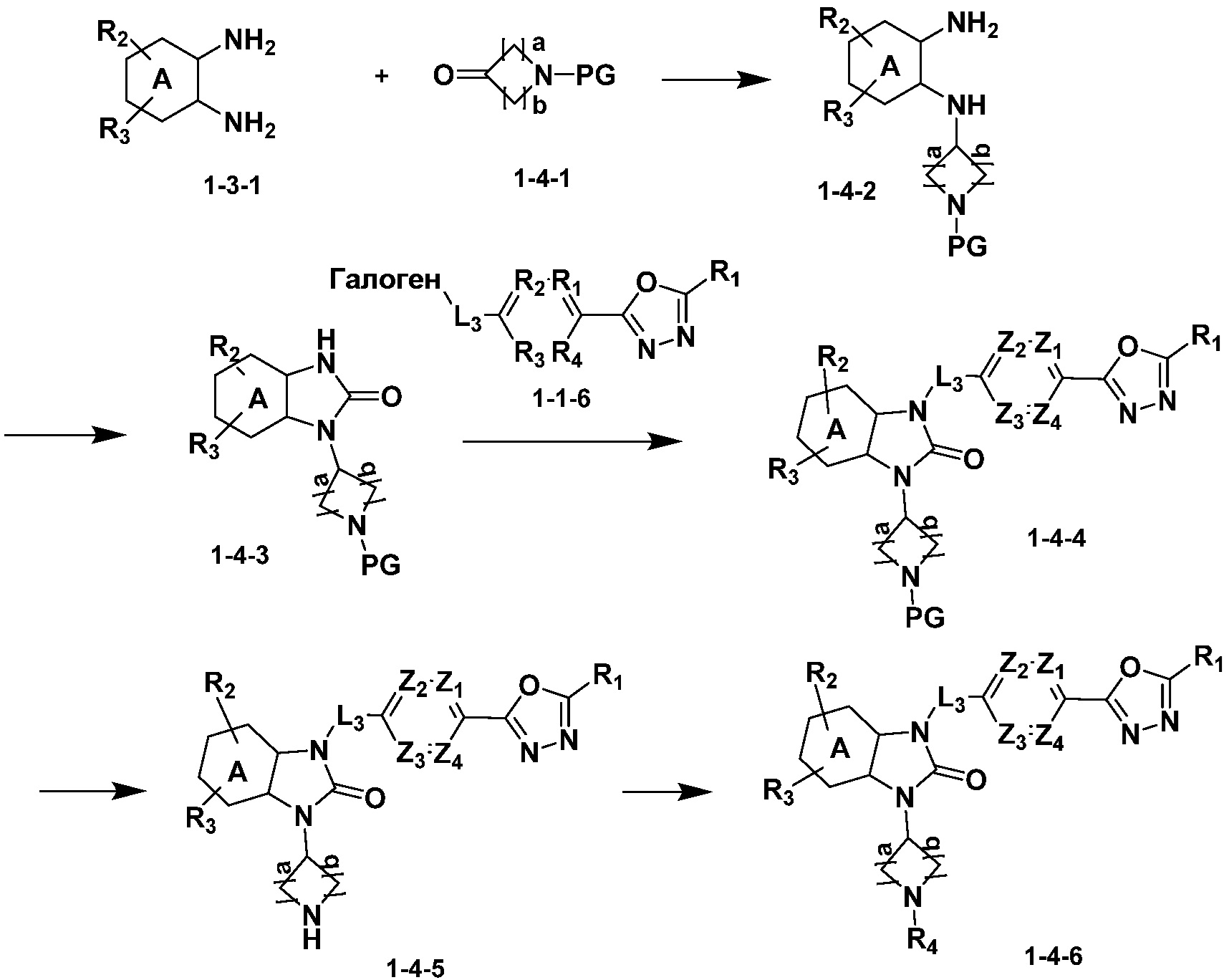
Вышеуказанная [Формула реакции 4] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензимидазолоновую структуру, и соединение химической формулы 1-3-1 подвергают реакцию восстановительного аминирования с соединением химической формулы 1-4-1 с получением соединения химической формулы 1-4-2, и затем подвергают реакции циклизации с получением соединения химической формулы 1-4-3. После этого, соединение химической формулы 1-4-3 подвергают реакции замещения с соединением химической формулы 1-1-6 с получением соединения химической формулы 1-4-4. Защитную группу удаляют из соединения химической формулы 1-4-4 с получением соединения химической формулы 1-4-5, и затем реакцию замещения, реакцию восстановительного аминирования и реакцию ацилирования проводят с получением соединения химической формулы 1-4-6.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 4 включают соединения 1, 4-7, 14-23, 78, 187 и подобные.
[Формула реакции 5]
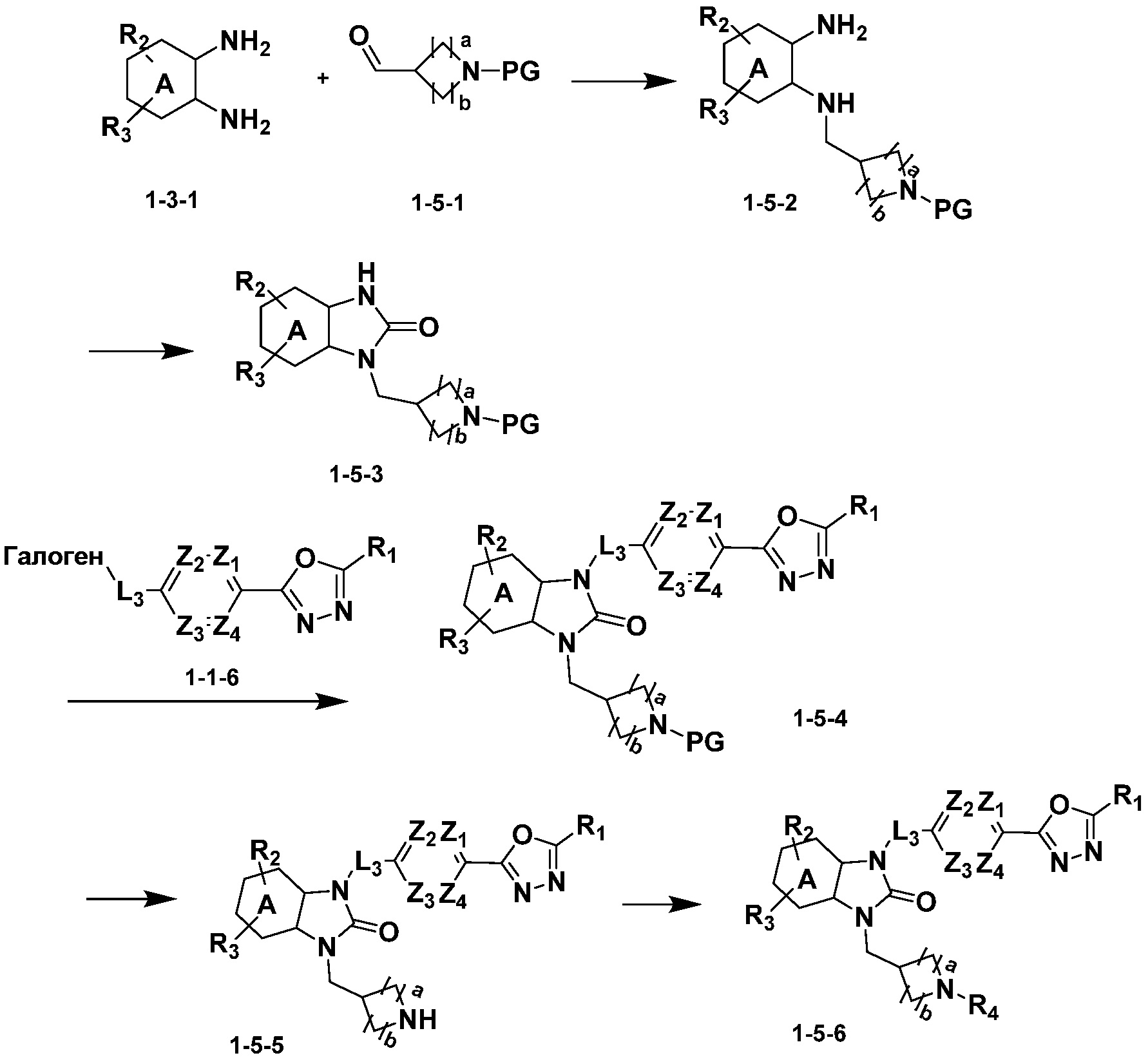
Вышеуказанная [Формула реакции 5] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензимидазолоновую структуру, и соединение химической формулы 1-3-1 подвергают реакцию восстановительного аминирования с соединением химической формулы 1-5-1 с получением соединения химической формулы 1-5-2, и затем подвергают реакции циклизации с получением соединения химической формулы 1-5-3. После этого, соединение химической формулы 1-5-3 подвергают реакции замещения с соединение химической формулы 1-1-6 с получением соединения химической формулы 1-5-4. Защитную группу удаляют из соединения химической формулы 1-5-4 с получением соединения химической формулы 1-5-5, и затем реакцию замещения, реакцию восстановительного аминирования и реакцию ацилирования проводят с получением соединения химической формулы 1-5-6.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 5 включают соединения 10, 38, 39 и подобные.
[Формула реакции 6]
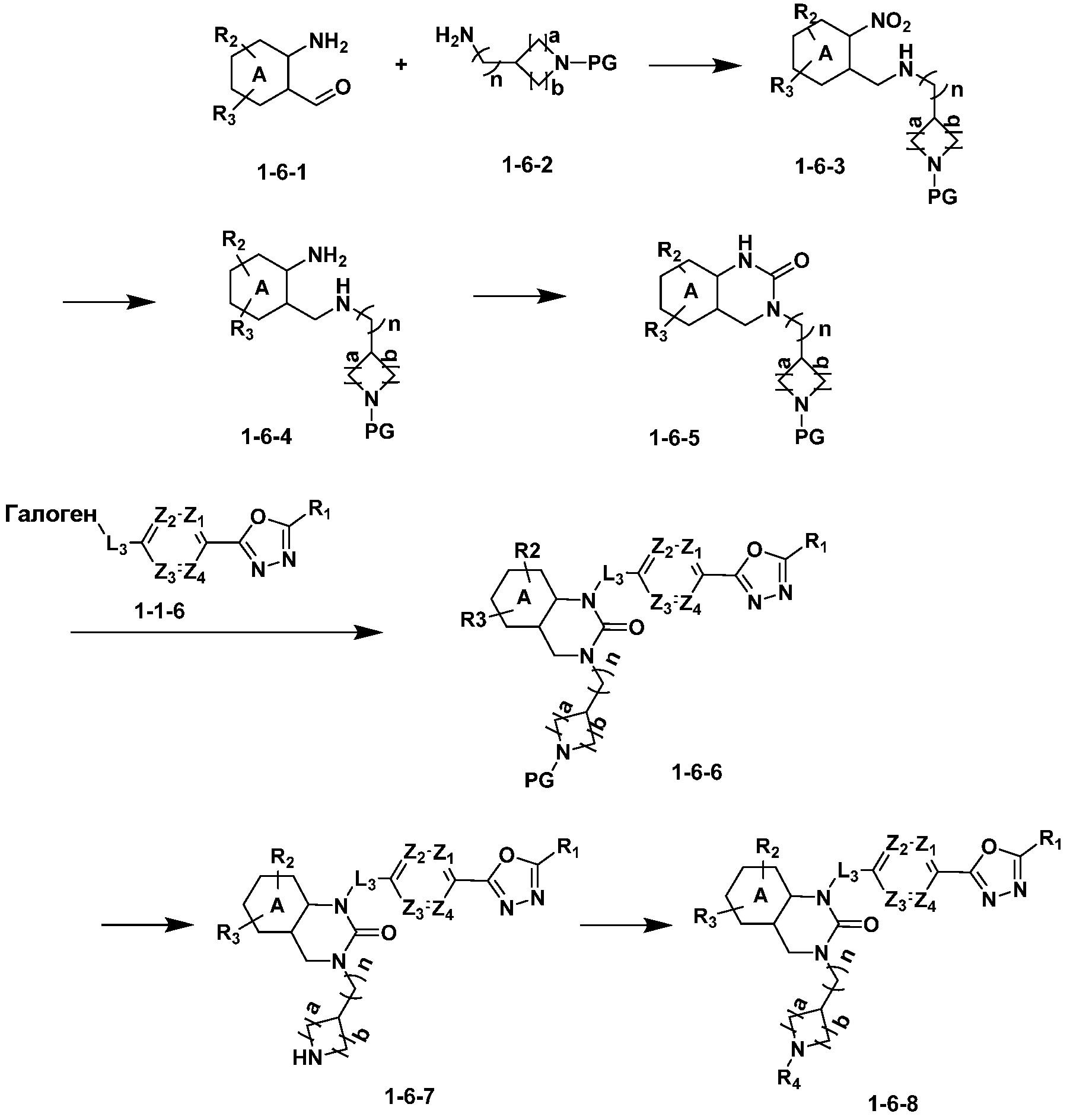
Вышеуказанная [Формула реакции 6] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих структуру 3,4-дигидрохиназолина-2(1H)-она, и соединение химической формулы 1-6-1 подвергают реакцию восстановительного аминирования с соединением химической формулы 1-6-2 с получением соединения химической формулы 1-6-3, и затем подвергают реакции восстановления с получением соединения химической формулы 1-6-4. После этого, реакции циклизации проводят с получением соединения химической формулы 1-6-5, после чего соединение химической формулы 1-6-5 подвергают реакции замещения с соединением химической формулы 1-1-6 с получением соединения химической формулы 1-6-6. Защитную группу удаляют из соединения химической формулы 1-6-6 с получением соединения химической формулы 1-6-7, и затем реакцию замещения, реакцию восстановительного аминирования и реакцию ацилирования проводят с получением соединения химической формулы 1-6-8.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 6 включают соединения 8, 9, 32, 33 и подобные.
[Формула реакции 7]
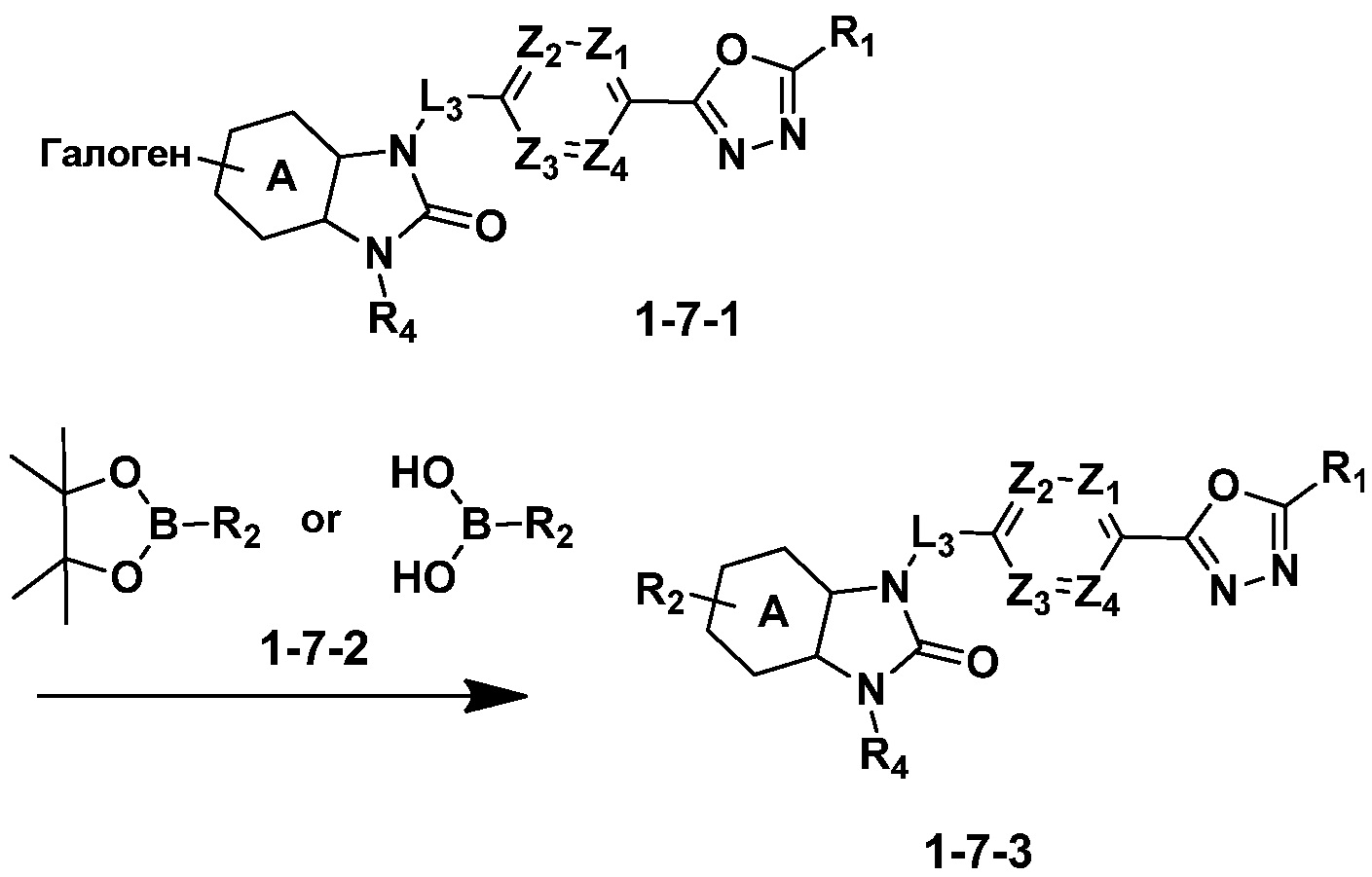
Вышеуказанная [Формула реакции 7] показывает способ синтеза соединений 1,3,4-оксадиазола имеющих бензимидазолоновую структуру, и соединение химической формулы 1-7-1 подвергают C-C сочетанию (реакция Сузуки) с соединением химической формулы 1-7-2 с получением соединения химической формулы 1-7-3.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 7, включают соединения 110-124, 126-131, 133-140, 145, 146, 150-155, 159-170, 177, 178, 208-212, 217-227, 239-244, 247-259, 267-270, 272-283, 286-300, 304-306, 310-312, 326, 327, 332, 333, 340, 341 и подобные.
[Формула реакции 8]
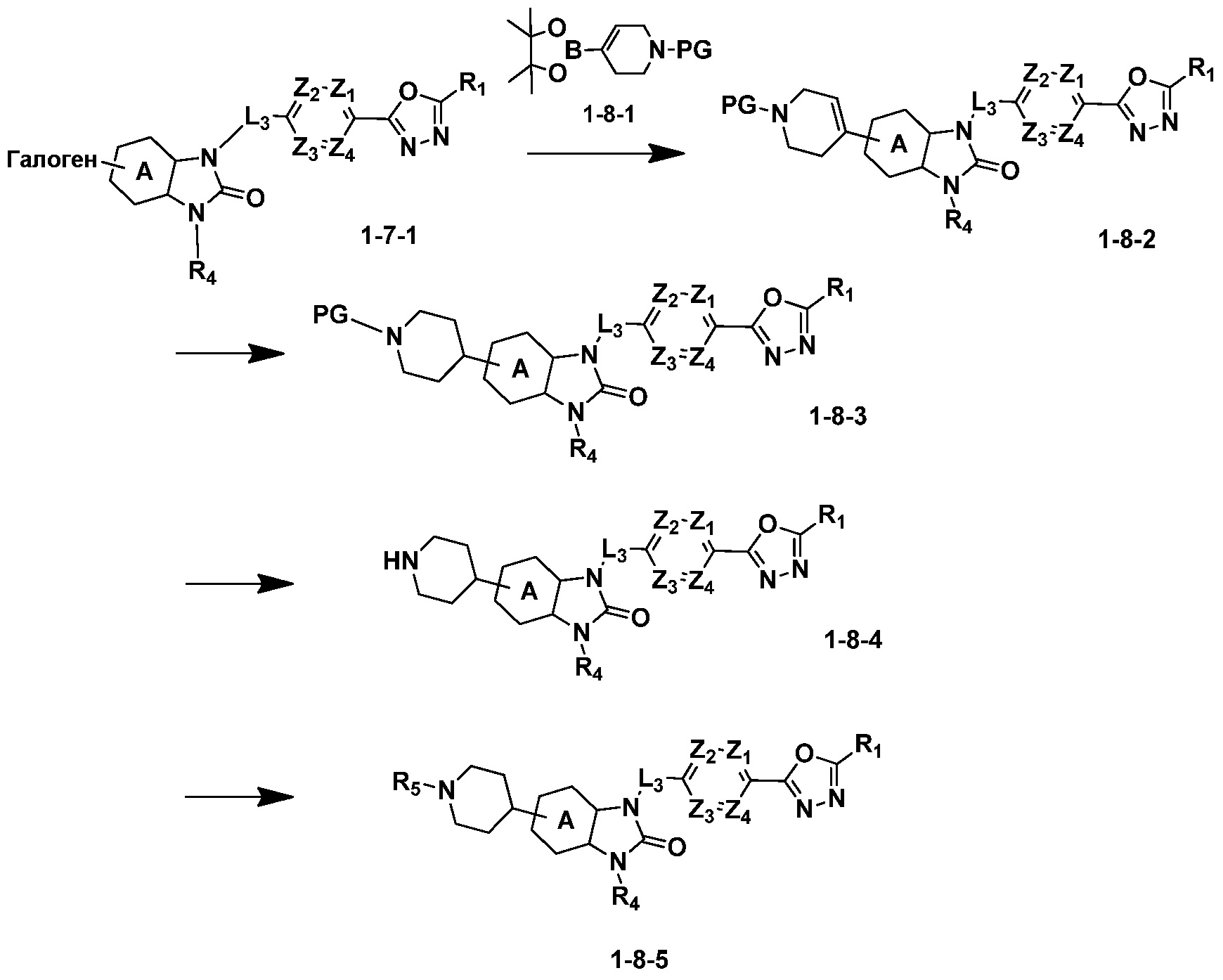
Вышеуказанная [Формула реакции 8] показывает способ синтеза соединений 1,3,4-оксадиазола имеющих бензимидазолоновую структуру, и соединение химической формулы 1-7-1 подвергают C-C сочетанию (Реакция Сузуки) с соединением химической формулы 1-8-1 с получением соединения химической формулы 1-8-2, после чего реакцию восстановления проводят с получением соединения химической формулы 1-8-3. После этого, защитную группу удаляют из соединения химической формулы 1-8-3 с получением соединения химической формулы 1-8-4, и затем реакцию замещения, реакцию восстановительного аминирования и реакцию ацилирования проводят с получением соединения химической формулы 1-8-5.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 8 включают соединения 156, 157, 158, 179, 197, 198, 199, 307-309, 313-315, 331, 334-336 и подобные.
[Формула реакции 9]
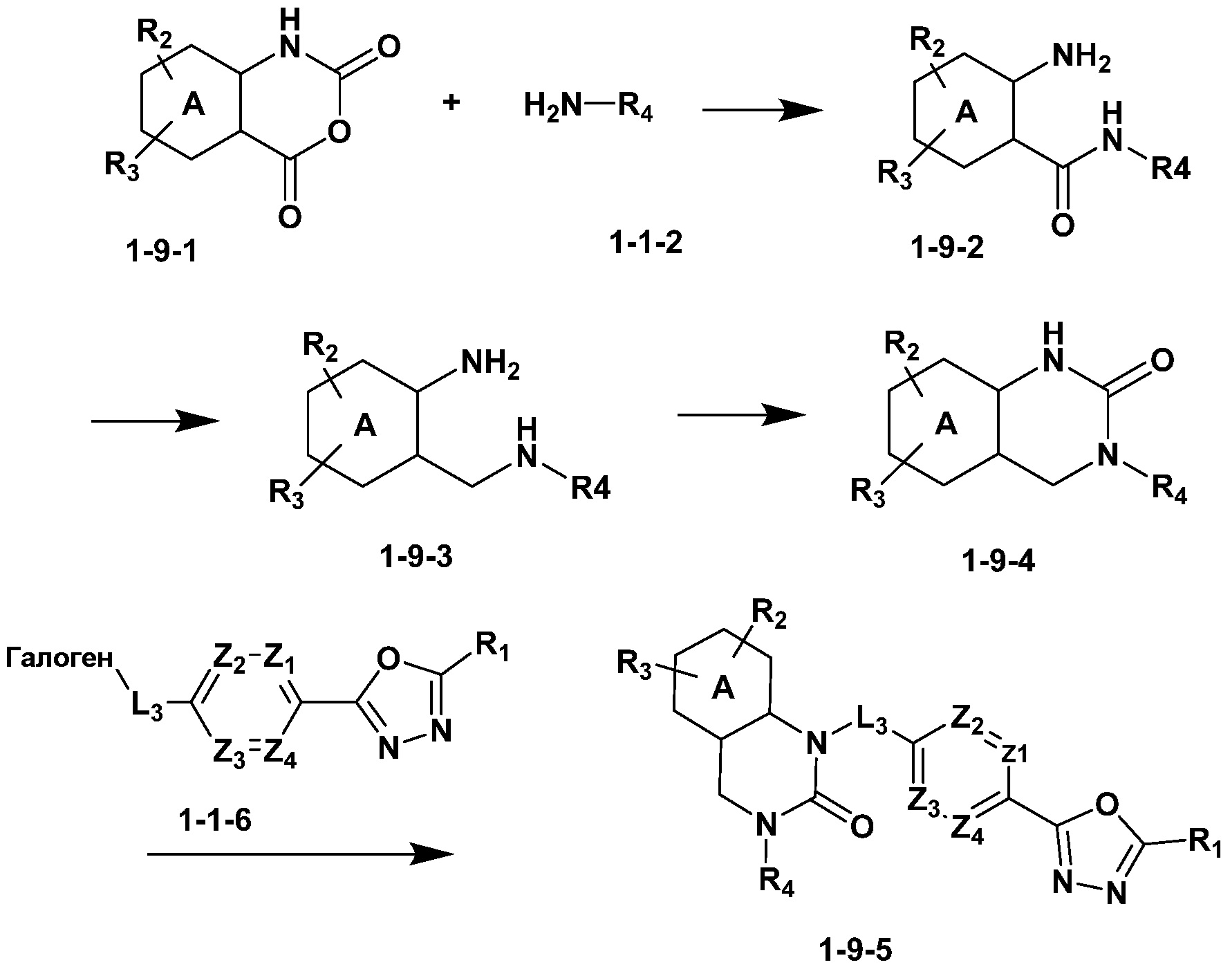
Вышеуказанная [Формула реакции 9] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих структуру 3,4-дигидрохиназолин-2(1H)-она, и соединение химической формулы 1-9-1 взаимодействует с соединением химической формулы 1-1-2 с получением соединения химической формулы 1-9-2, и затем реакцию восстановления проводят с получением соединения химической формулы 1-9-3. После этого, соединение химической формулы 1-9-3 подвергают реакции циклизации с получением соединения химической формулы 1-9-4, и затем подвергают реакции замещения с соединением химической формулы 1-1-6 с получением соединения химической формулы 1-9-5.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 9 включают соединения 30, 31 и подобные.
[Формула реакции 10]
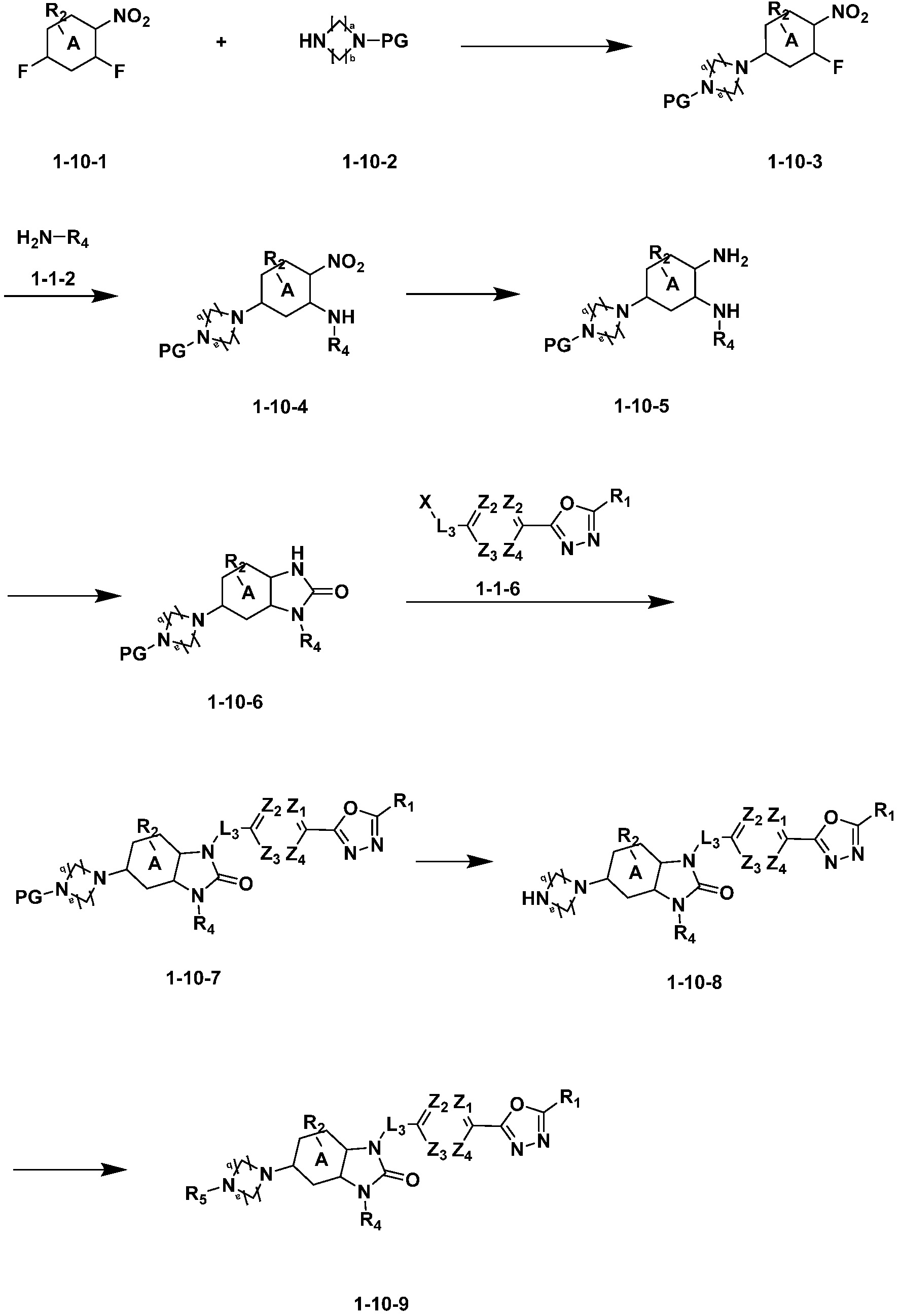
Вышеуказанная [Формула реакции 10] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензимидазолоновую структуру, и соединение химической формулы 1-10-1 взаимодействует с соединением химической формулы 1-10-2 с получением соединения химической формулы 1-10-3, и затем подвергают реакции замещения с соединением химической формулы 1-1-2 с получением соединения химической формулы 1-10-4. Затем, соединение химической формулы 1-10-4 подвергают реакции восстановления с получением соединения химической формулы 1-10-5, и затем реакцию циклизации проводят с получением соединения химической формулы 1-10-6. После этого, соединение химической формулы 1-10-6 подвергают реакции замещения с соединение химической формулы 1-1-6 с получением соединения химической формулы 1-10-7. Защитную группу удаляют из соединение химической формулы 1-10-7 с получением соединения химической формулы 1-10-8, и затем реакцию восстановительного аминирования, реакцию алкилирования и реакцию ацилирования проводят с получением соединения химической формулы 1-10-9.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 10 включают соединения 260-264, 266, 284, 285, 303, 319-325, 328, 329, 330 и подобные.
[Формула реакции 11]
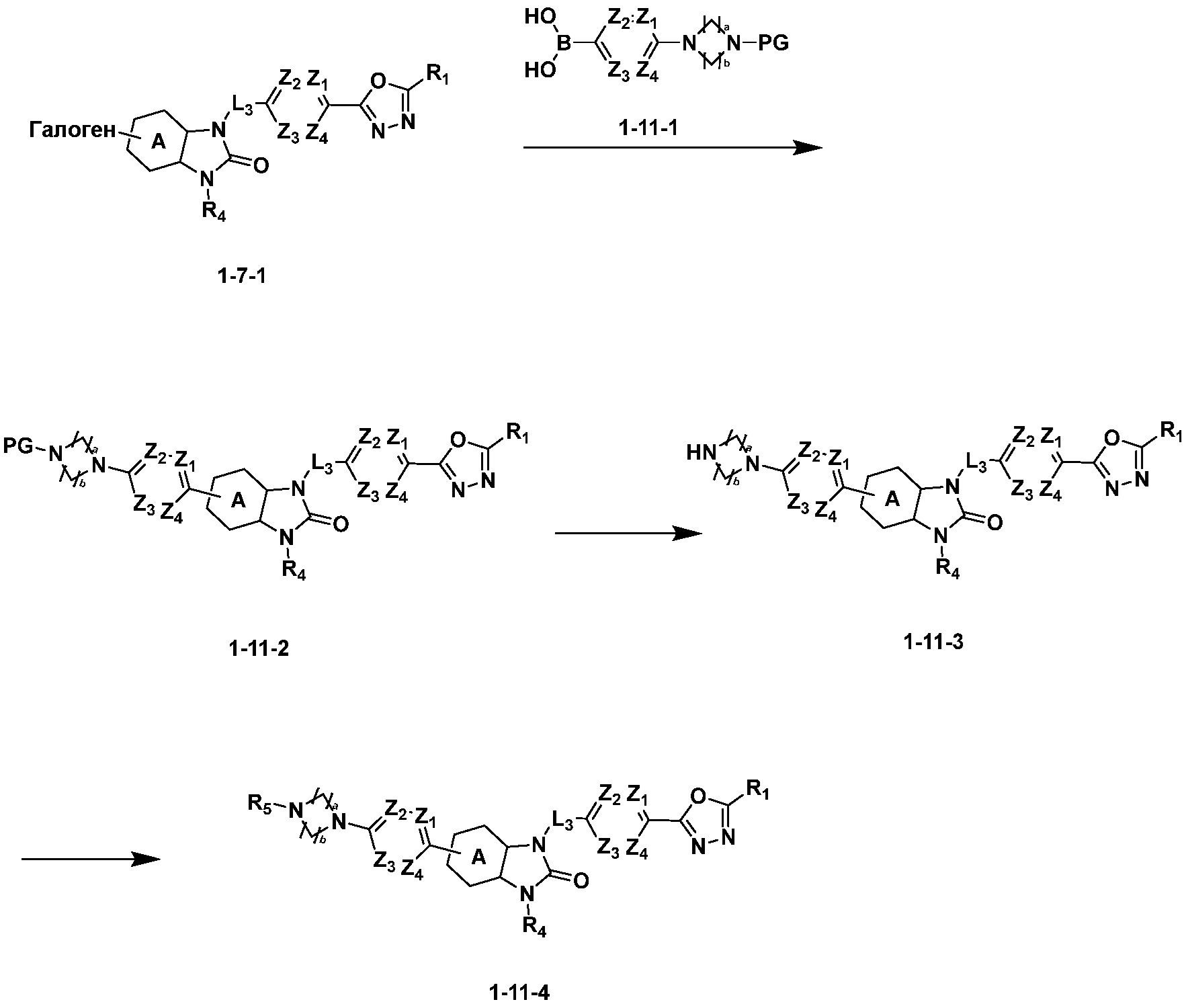
Вышеуказанная [Формула реакции 11] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензимидазолоновую структуру, и соединение химической формулы 1-7-1 подвергают C-C сочетанию (Реакция Сузуки) с соединением химической формулы 1-11-1 с получением соединения химической формулы 1-11-2. Защитную группу удаляют из соединения химической формулы 1-11-2 с получением соединения химической формулы 1-11-3, и затем реакцию восстановительного аминирования, реакцию алкилирования и реакцию ацилирования проводят с получением соединения химической формулы 1-11-4.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 11 включают соединения 337-339, 342-344, 358-368, и т.д.
[Формула реакции 12]
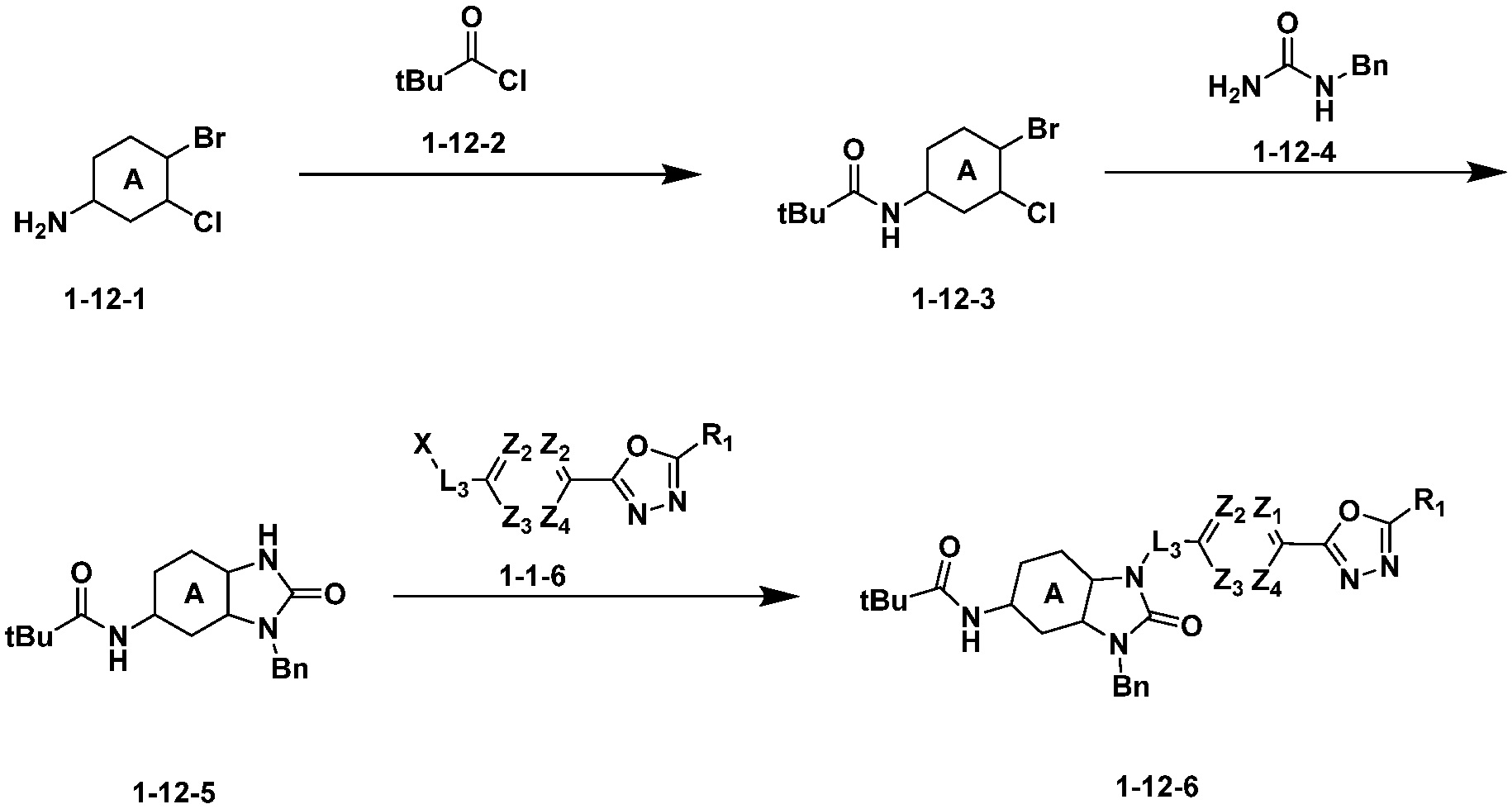
Вышеуказанная [Формула реакции 12] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензимидазолоновую структуру, и соединение химической формулы 1-12-1 взаимодействует с соединением химической формулы 1-12-2 с получением соединения химической формулы 1-12-3. Затем полученное соединение подвергают реакции с соединением химической формулы 1-12-4 с получением циклизованного соединения химической формулы 1-12-5. После этого, соединение химической формулы 1-12-5 подвергают реакции замещения с соединением химической формулы 1-1-6 с получением соединения химической формулы 1-12-6.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 12, включают соединение 207 и подобные.
[Формула реакции 13]
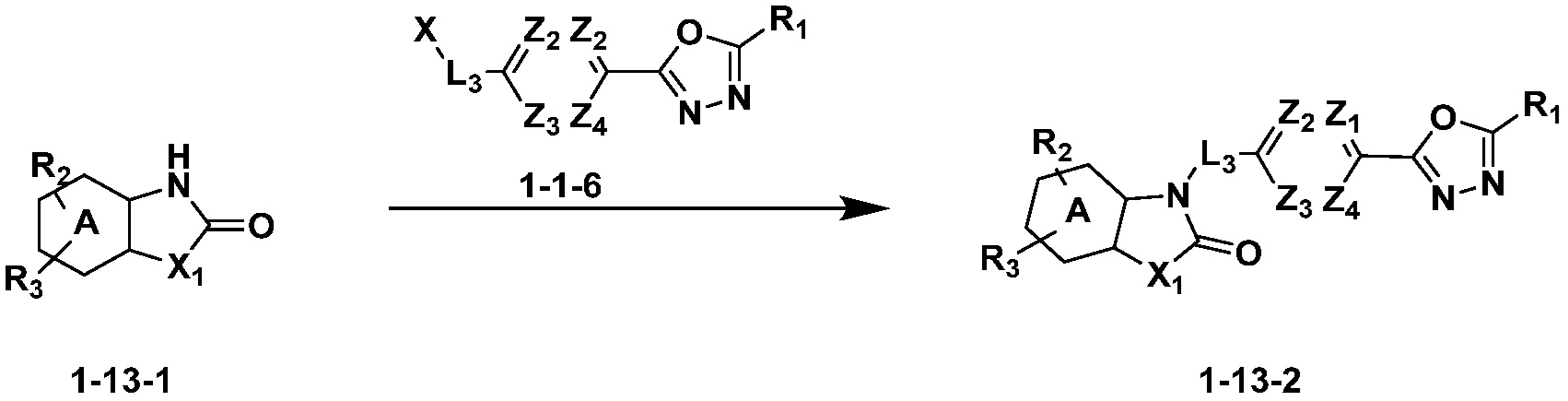
Вышеуказанная [Формула реакции 13] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензоксазолоновую или бензотиазолоновую структуру, и соединение химической формулы 1-13-1 подвергают реакции замещения с соединением химической формулы 1-1-6 с получением соединения химической формулы 1-13-2.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 13, включают соединения 228, 230, 245, 246, 265 и подобные.
[Формула реакции 14]
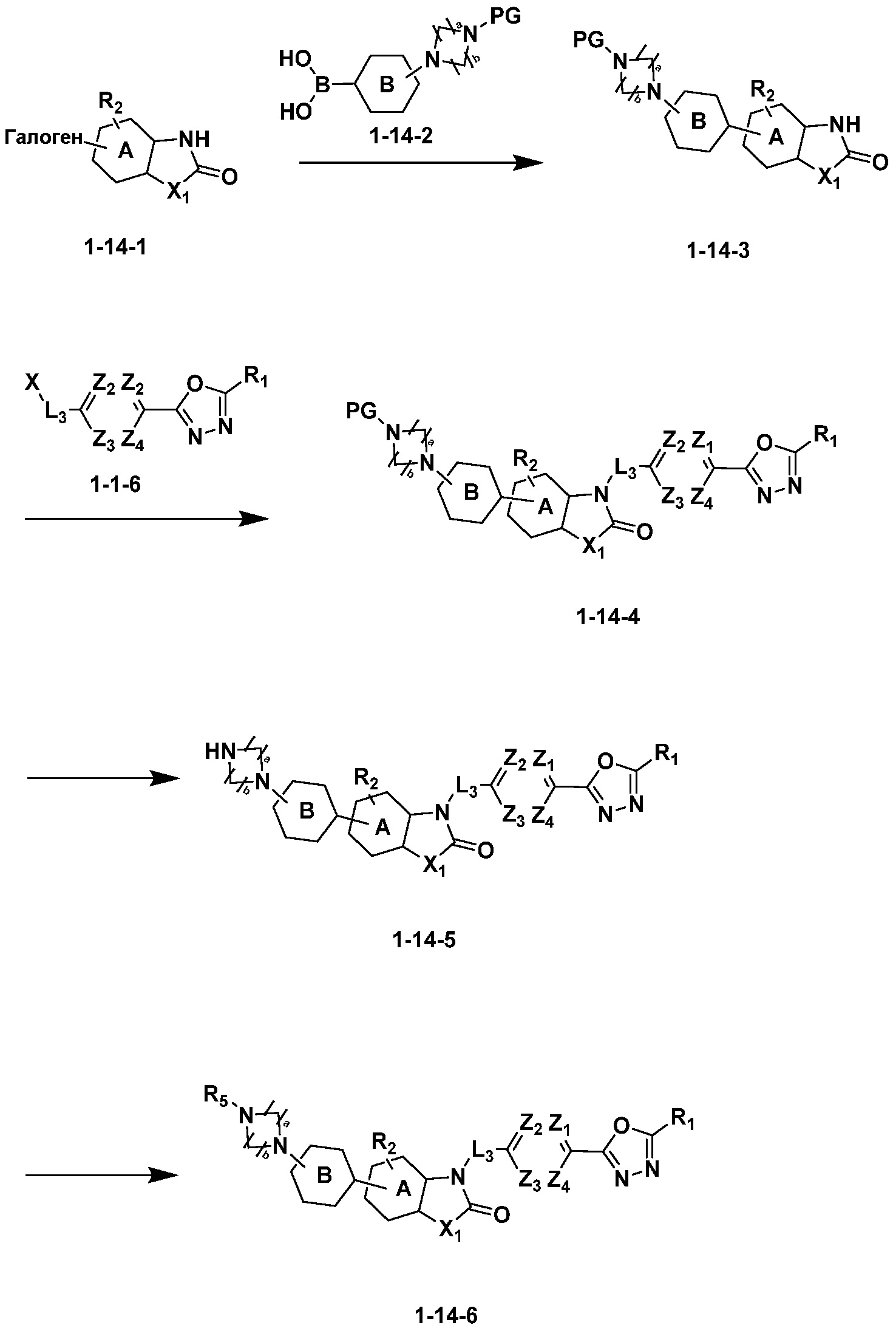
Вышеуказанная [Формула реакции 14] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензоксазолоновую или бензотиазолоновую структуру, и соединение химической формулы 1-14-1 подвергают C-C сочетанию (Реакция Сузуки) с соединением химической формулы 1-14-2 с получением соединения химической формулы 1-14-3. Соединение химической формулы 1-14-3 подвергают реакции замещения с соединением химической формулы 1-1-6 с получением соединения химической формулы 1-14-4. Защитную группу удаляют из соединения химической формулы 1-14-4 с получением соединения химической формулы 1-14-5, и затем реакцию восстановительного аминирования, реакцию алкилирования и реакцию ацилирования проводят с получением соединения химической формулы 1-14-6.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 14 включают соединения 345-351 и подобные.
[Формула реакции 15]
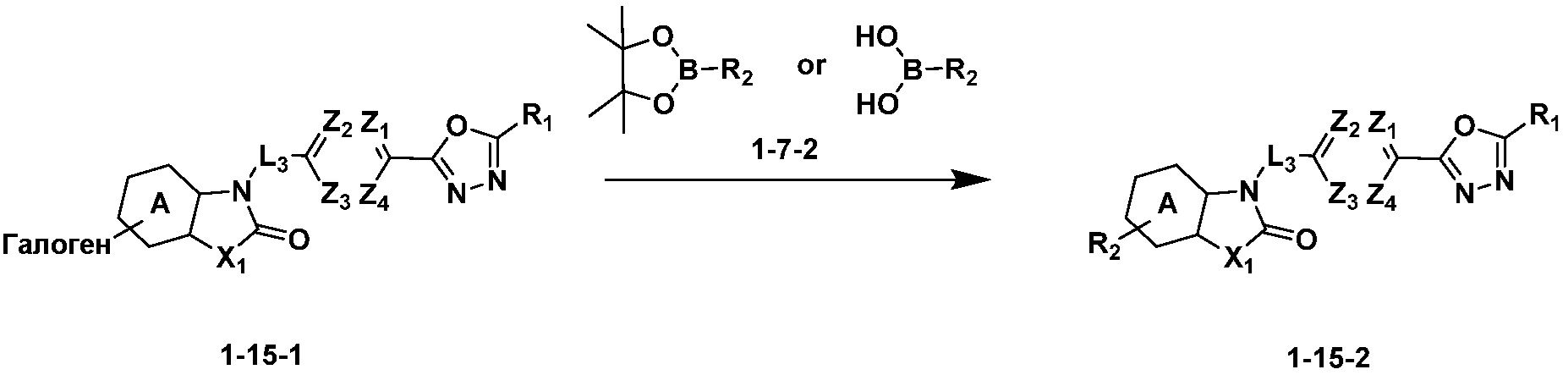
Вышеуказанная [Формула реакции 15] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензоксазолоновую или бензотиазолоновую структуру, и соединение химической формулы 1-15-1 подвергают C-C сочетанию (Реакция Сузуки) с соединением химической формулы 1-7-2 с получением соединения химической формулы 1-15-2.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 15 включают соединения 229, 231, 237, 238 и подобные.
[Формула реакции 16]
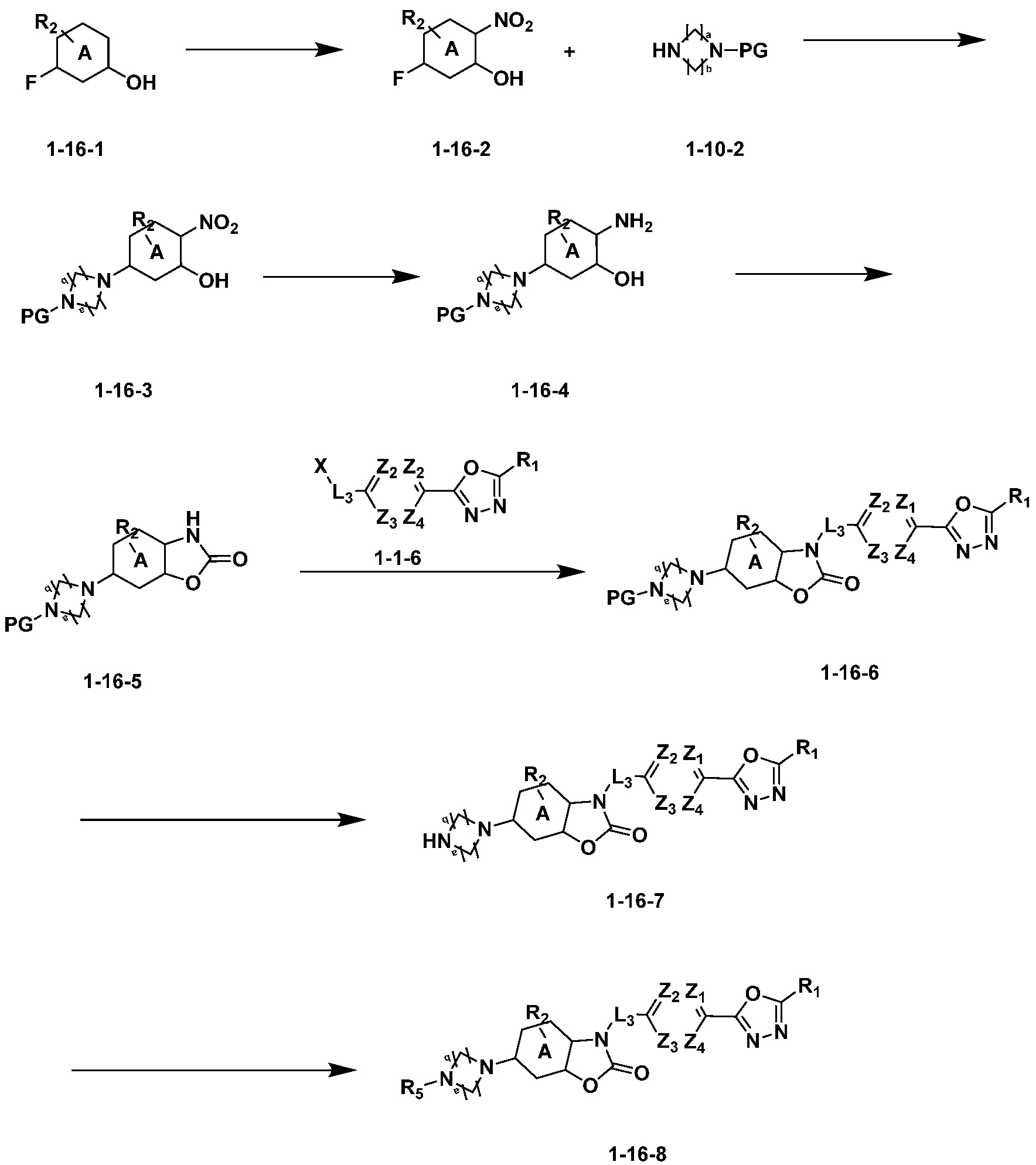
Вышеуказанная [Формула реакции 16] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющего бензоксазолоновую структуру, и соединение химической формулы 1-16-1 подвергают реакции нитрования с получением соединения химической формулы 1-16-2, и затем подвергают реакции с соединением химической формулы 1-10-2 с получением соединения химической формулы 1-16-3. После этого, полученное соединение подвергают реакции восстановления с соединением химической формулы 1-16-3 с получением соединения химической формулы 1-16-4, и затем реакцию циклизации проводят с получением соединения химической формулы 1-16-5. Затем, соединение химической формулы 1-16-5 подвергают реакции замещения с соединение химической формулы 1-1-6 с получением соединения химической формулы 1-16-6. Защитную группу удаляют из соединения химической формулы 1-16-6 с получением соединения химической формулы 1-16-7, и затем реакцию восстановительного аминирования, реакцию алкилирования и реакцию ацилирования проводят с получением соединения химической формулы 1-16-8.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 16 включают соединения 316-318 и подобные.
[Формула реакции 17]
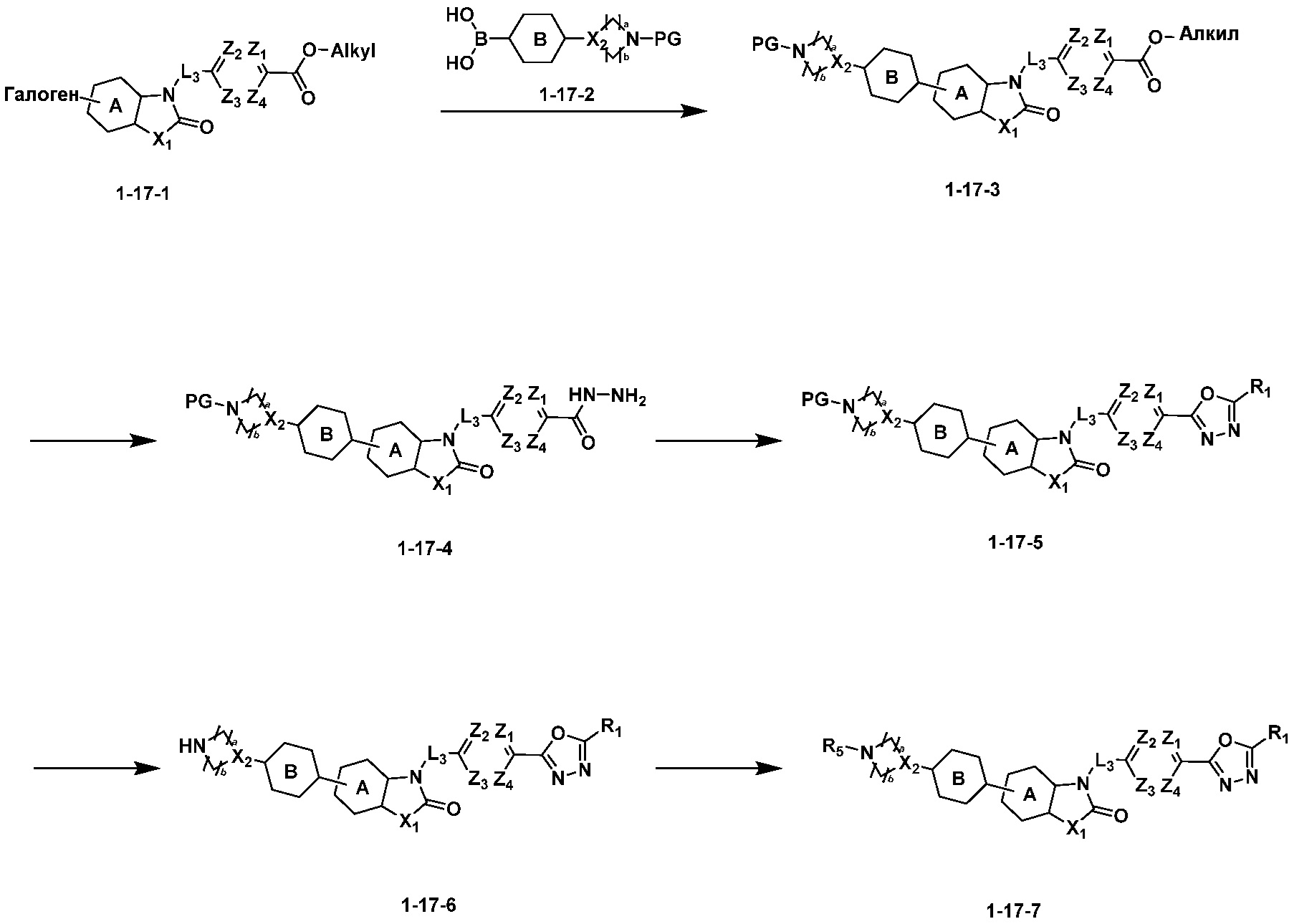
Вышеуказанная [Формула реакции 17] показывает способ синтеза соединений 1,3,4-оксадиазола, имеющих бензоксазолоновую или бензотиазолоновую структуру, и соединение химической формулы 1-17-1 подвергают C-C сочетанию (Реакция Сузуки) с соединением химической формулы 1-17-2, к которому добавляют защитную группу, с получением соединения химической формулы 1-17-3, затем подвергают реакции с гидразином с получением соединения химической формулы 1-17-4, и затем подвергают реакции с дифторуксусным ангидридом с получением соединения химической формулы 1-17-5. После этого, защитную группу удаляют из соединения химической формулы 1-17-5 с получением соединения химической формулы 1-17-6, и затем реакцию восстановительного аминирования, реакцию алкилирования и реакцию ацилирования проводят с получением соединения химической формулы 1-17-7.
В настоящем изобретении, примеры соединений, полученных способом, показанным в вышеуказанной формуле реакции 17 включают соединения 352-357 и подобные.
В дальнейшем настоящее изобретение будет описано более подробно с помощью следующих примеров и экспериментальных примеров. Однако следующие примеры и подобные приведены только с целью иллюстрации настоящего изобретения, и, таким образом, объем настоящего изобретения ими не ограничивается.
Получение производных соединений 1,3,4-оксадиазола
Конкретный способ получения соединений, представленных химической формулой I, такой, как описан ниже.
Пример 1: Синтез соединения 1, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
[Стадия 1] Синтез трет-бутил 4-((2-аминофенил)амино)пиперидин-1-карбоксилата
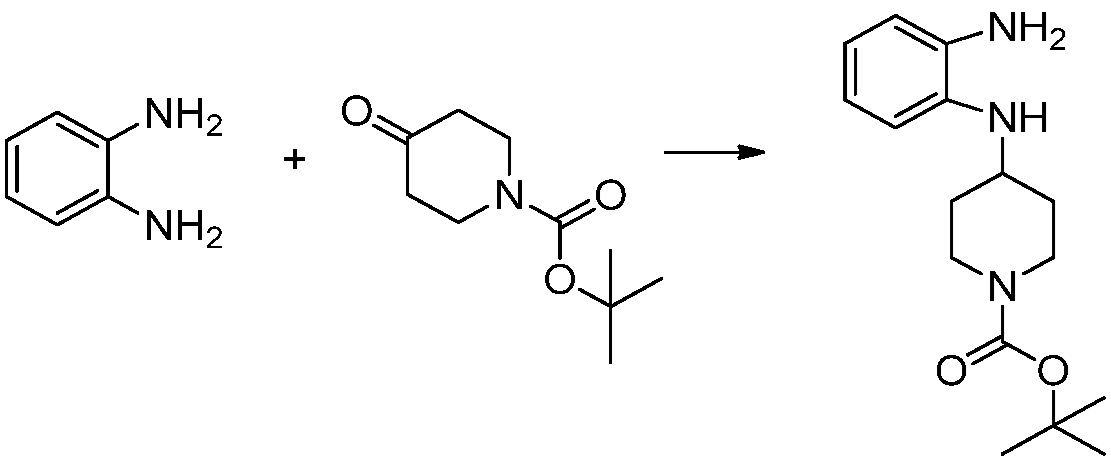
Трет-бутил 4-оксопиперидин-1-карбоксилат (3,000 г, 15,056 ммоль), бензол-1,2-диамин (4,885 г, 45,169 ммоль), триацетоксиборгидрид натрия (6,382 г, 30,113 ммоль) и уксусную кислоту (1,724 мл, 30,113 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = 0-50%) и концентрируют с получением указанного в заголовке соединения (3,500 г, 79,8%) в виде бесцветного масла.
[Стадия 2] Синтез трет-бутил 4-(2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
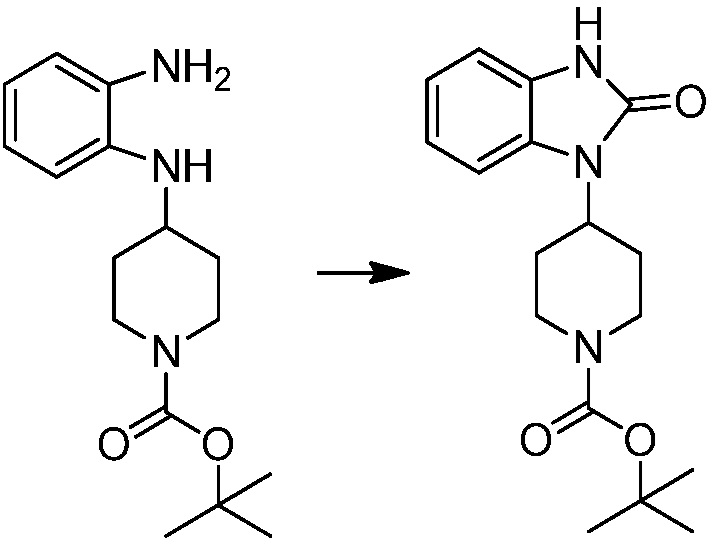
Трет-бутил 4-((2-аминофенил)амино)пиперидин-1-карбоксилат (3,500 г, 12,011 ммоль) полученный на стадии 1 растворяют в дихлорметане (5 мл), после чего триметиламин (1,674 мл, 12,011 ммоль) и трифосген (14,257 г, 48,044 ммоль) добавляют туда при 0°C, затем перемешивают при той же температуре в течение 30 минут, и затем дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = 0-50%) и концентрируют с получением указанного в заголовке соединения (1,000 г, 26,2%) в виде белого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(3-(2-фтор-4-(метоксикарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
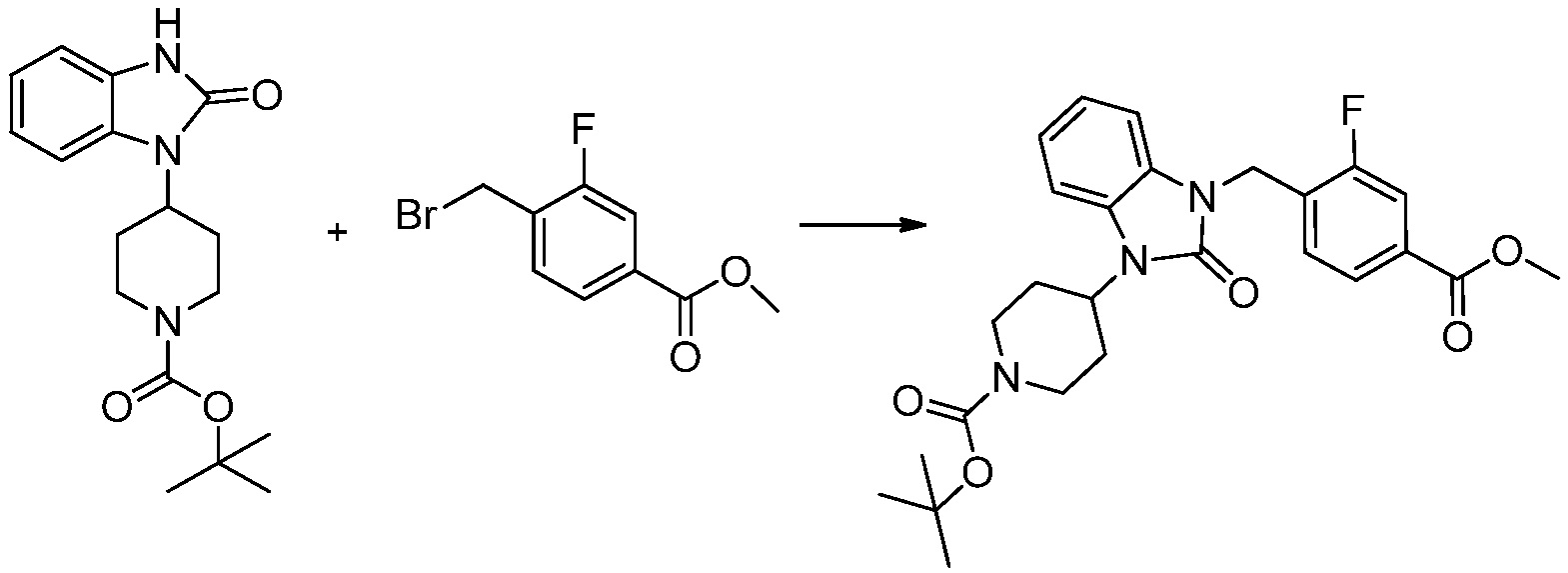
Трет-бутил 4-(2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,000 г, 3,151 ммоль) полученный на стадии 2 растворяют в N,N-диметилформамиде (30 мл) при 0°C, после чего гидрид натрия (60,00%, 0,189 г, 4,726 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. Метил 4-(бромметил)-3-фторбензоат (0,778 г, 3,151 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,670 г, 44,0%) в виде бесцветного масла.
[Стадия 4] Синтез трет-бутил 4-(3-(2-фтор-4-(гидразинкарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
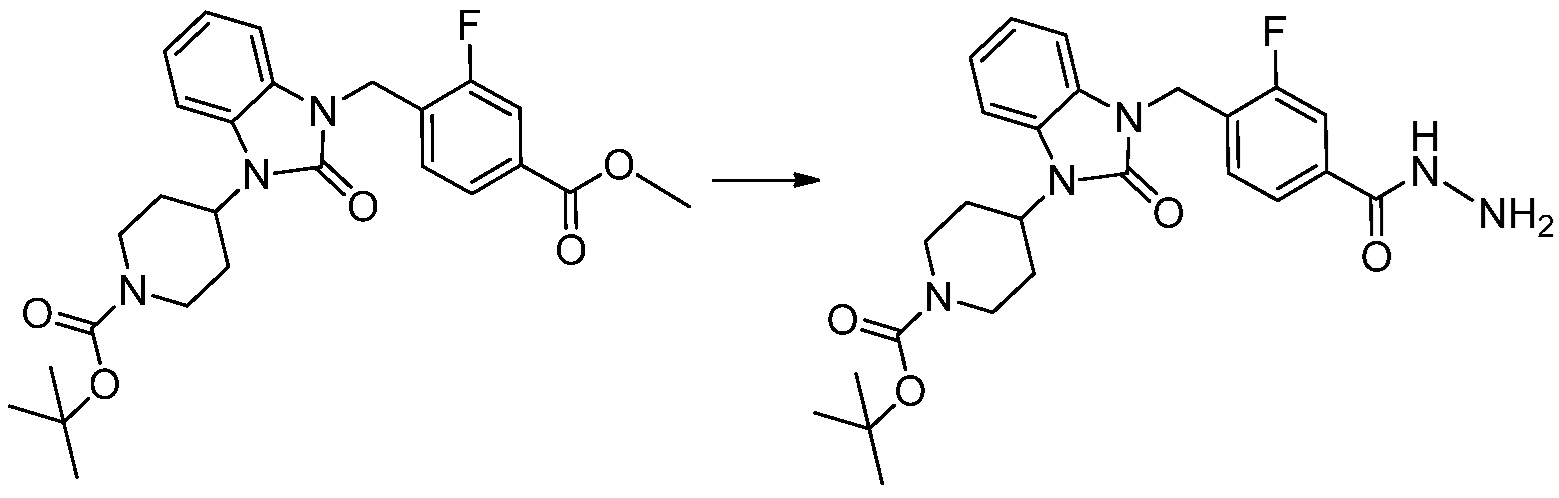
Трет-бутил 4-(3-(2-фтор-4-(метоксикарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,670 г, 1,386 ммоль) полученный на стадии 3 и моногидрат гидразина (1,347 мл, 27,712 ммоль) растворяют в этаноле (10 мл) at 90°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,670 г, 100,0%, бесцветное масло).
[Стадия 5] Синтез трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
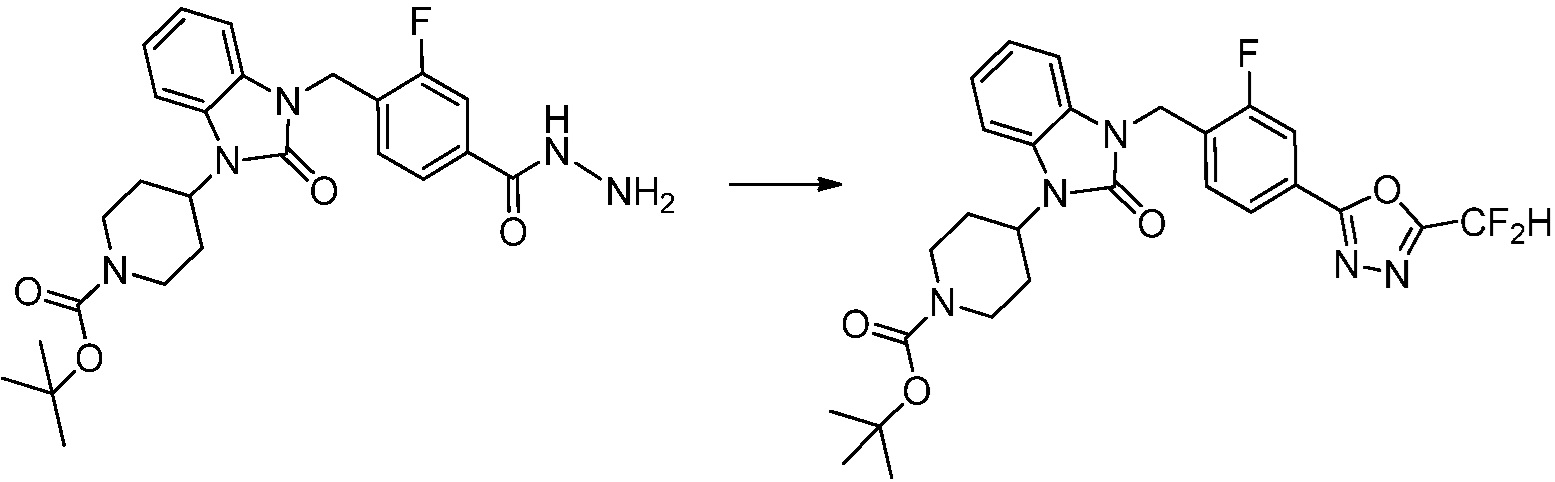
Трет-бутил 4-(3-(2-фтор-4-(гидразинкарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,670 г, 1,386 ммоль) полученный на стадии 4, 2,2-дифторуксусный ангидрид (0,517 мл, 4,157 ммоль) и имидазол (0,283 г, 4,157 ммоль) растворяют в дихлорметане (10 мл) при 45°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,600 г, 79,7%) в виде белого пенистого твердого вещества.
[Стадия 6] Синтез 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она 2,2,2-трифторацетата
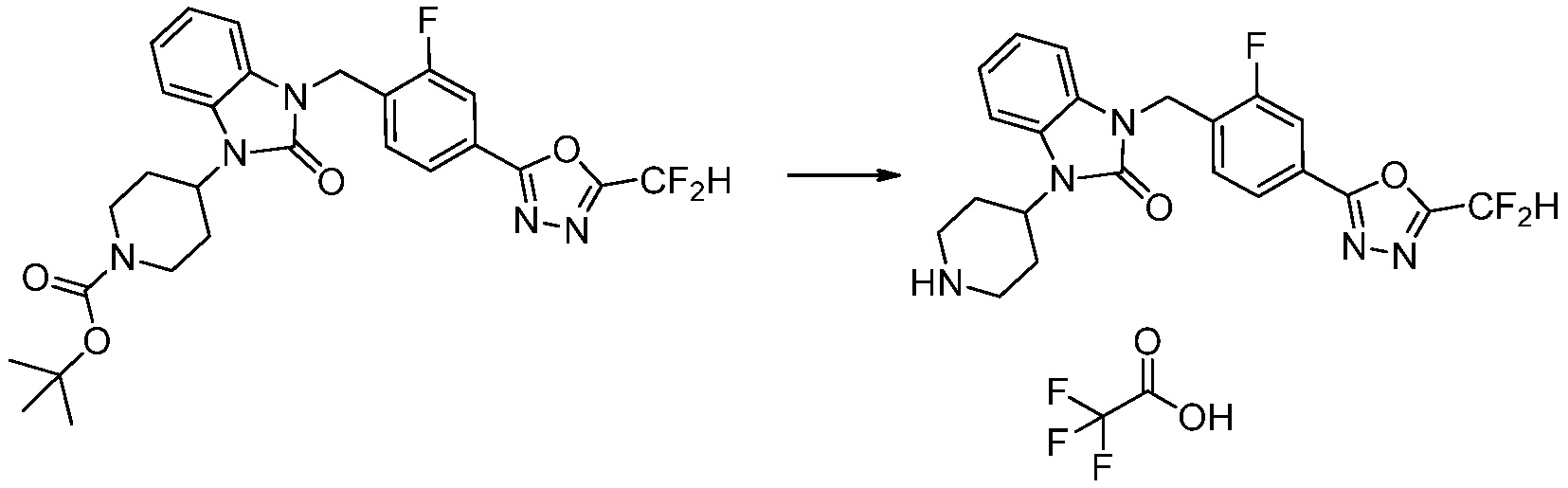
Трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,300 г, 0,552 ммоль) полученный на стадии 5 и трифторуксусную кислоту (0,845 мл, 11,039 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,290 г, 94,3%, коричневое масло).
[Стадия 7] Синтез соединения 1
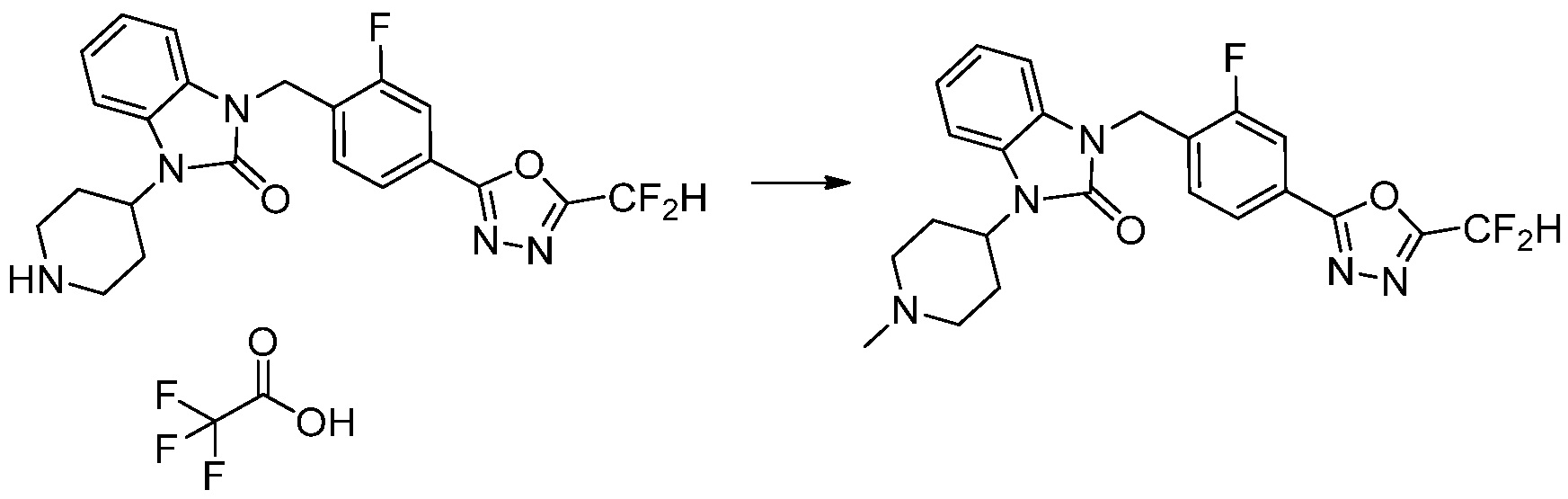
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,289 г, 0,518 ммоль) полученный на стадии 6, формальдегид (0,031 г, 1,037 ммоль), N,N-диизопропилэтиламин (0,090 мл, 0,518 ммоль) и триацетоксиборгидрид натрия (0,220 г, 1,037 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-12%) и концентрируют с получением указанного в заголовке соединения (0,220 г, 92,8%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,82 (м, 2H), 7,47-7,39 (м, 2H), 7,12-7,04 (м, 2H), 7,06 (с, 0,25H), 6,98-6,95 (м, 1H), 6,93 (с, 0,5H), 6,80 (с, 0,25H), 5,21 (с, 2H), 4,55-4,51 (м, 1H), 3,22 (д, J=11,8 Гц, 2H), 2,66-2,60 (м, 2H), 2,40-2,34 (м, 2H), 1,91-1,87 (м, 2H); МСНР (ЭР) m/z 458,0 (M+ + 1).
Пример 2: Синтез соединения 2, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез N-(2-морфолиноэтил)-2-нитроанилина
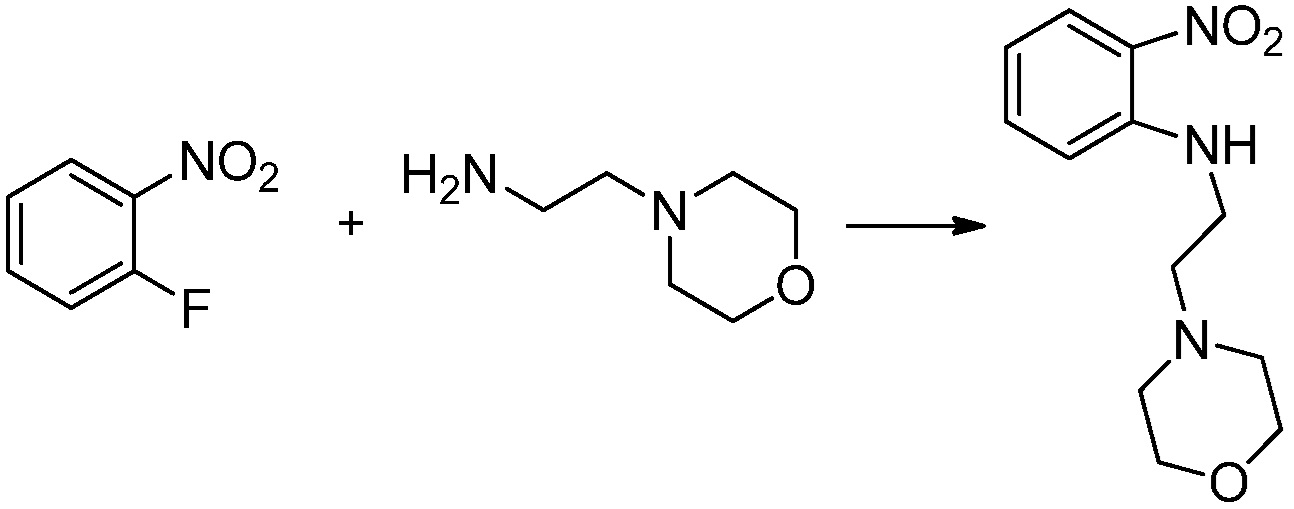
1-фтор-2-нитробензол (6,000 г, 42,523 ммоль), 2-морфолиноэтан-1-амин (6,090 г, 46,775 ммоль) и карбонат калия (11,754 г, 85,046 ммоль) растворяют в тетрагидрофуране (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (5,500 г, 51,5%) в виде бесцветного масла.
[Стадия 2] Синтез N1-(2-морфолиноэтил)бензол-1,2-диамина
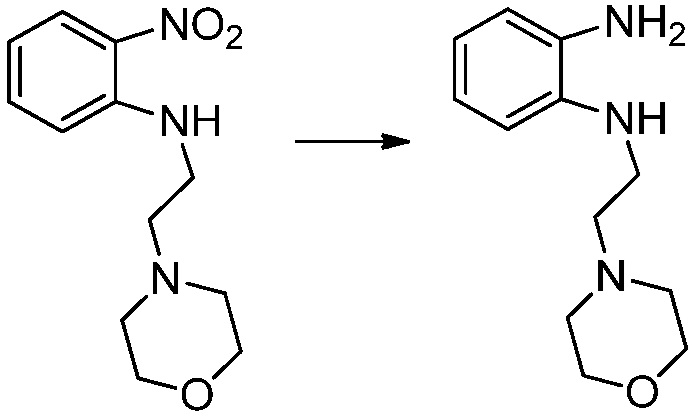
N-(2-морфолиноэтил)-2-нитроанилин (4,500 г, 17,908 ммоль) полученный на стадии 1, растворяют в метаноле (30 мл) при комнатной температуре, после чего 10%-Pd/C (450 мг) медленно добавляют туда и перемешивают при той же температуре в течение 12 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (3,500 г, 88,3%, коричневое масло).
[Стадия 3] Синтез 1-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
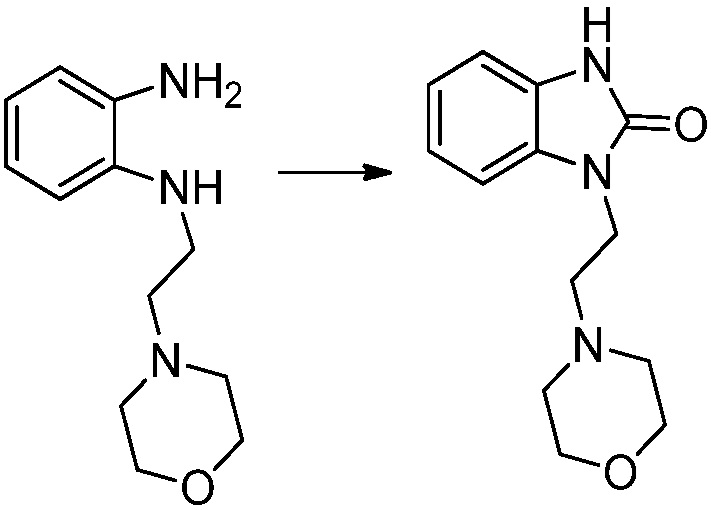
N1-(2-морфолиноэтил)бензол-1,2-диамин (2,770 г, 12,517 ммоль), полученный на стадии 2 и 1,1'-карбонилдиимидазол (КДИ, 2,030 г, 12,517 ммоль) растворяют в тетрагидрофуране (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (2,240 г, 72,4%) в виде бесцветного масла.
[Стадия 4] Синтез метил 3-фтор-4-((3-(2-морфолиноэтил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)бензоата
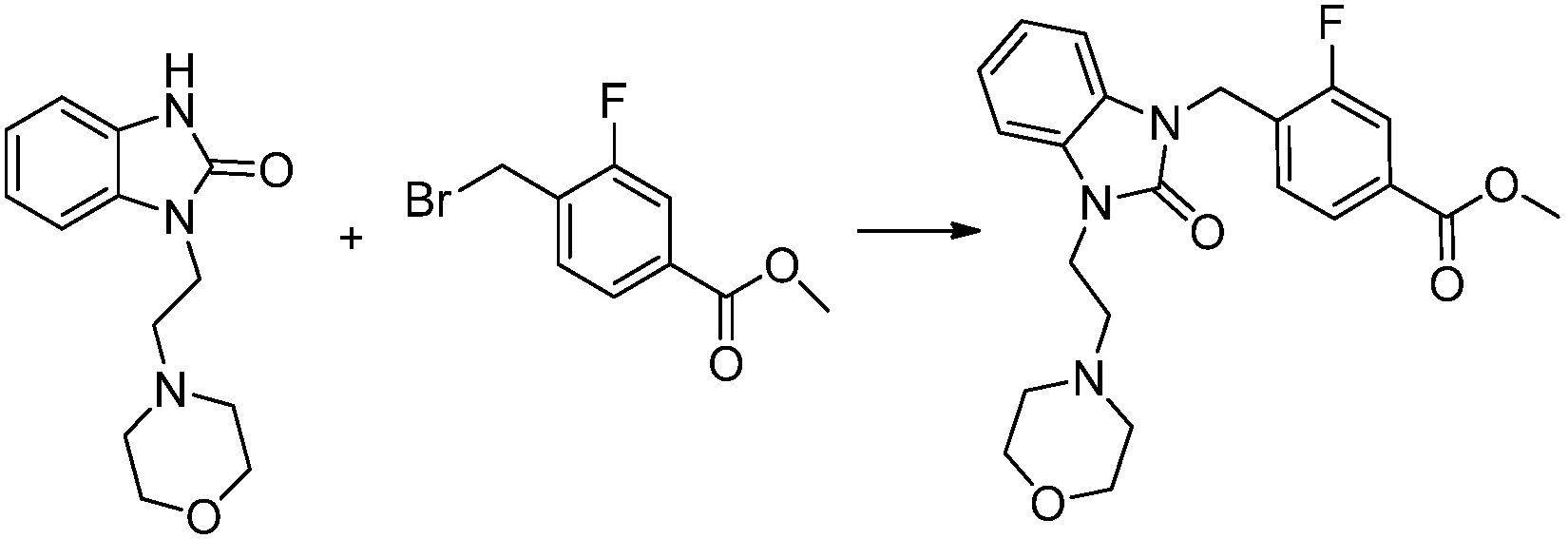
1-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (3,000 г, 12,131 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (30 мл) при 0°C, после чего гидрид натрия (60,00%, 0,728 г, 18,197 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. Метил 4-(бромметил)-3-фторбензоат (2,997 г, 12,131 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 18 часов. выпавшее в осадок твердое вещество фильтруют, затем промывают гексан, и затем сушат с получением указанного в заголовке соединения (2,200 г, 43,9%) в виде бесцветного масла.
[Стадия 5] Синтез 3-фтор-4-((3-(2-морфолиноэтил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)бензогидразид
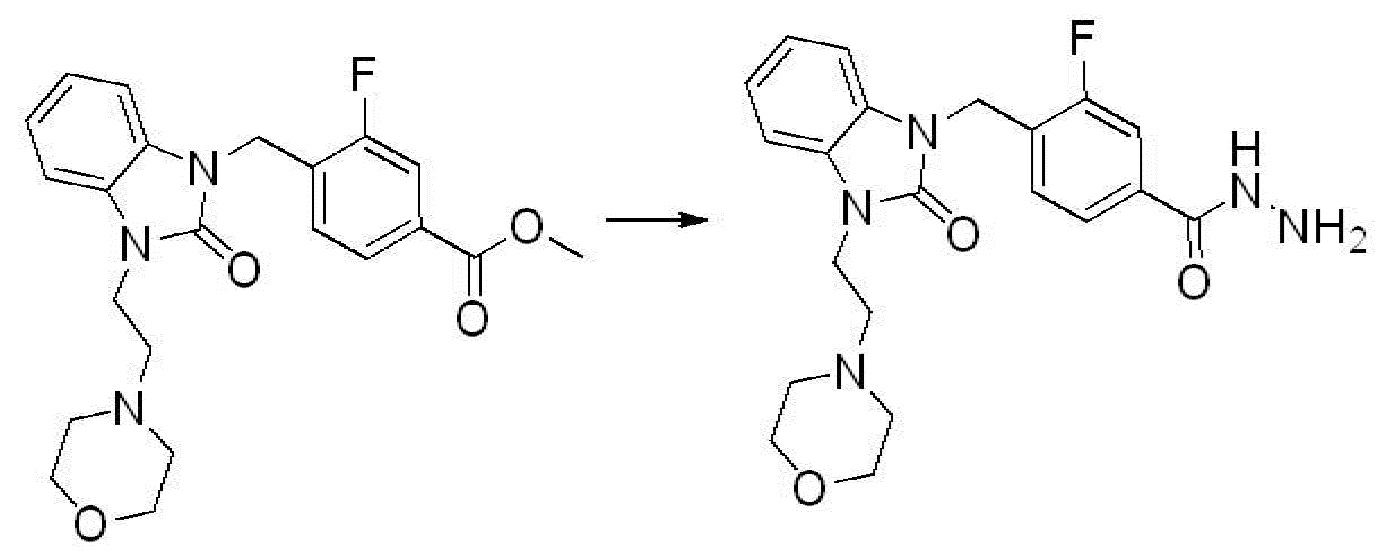
Метил 3-фтор-4-((3-(2-морфолиноэтил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)бензоат (2,200 г, 5,321 ммоль), полученный на стадии 4, и моногидрат гидразина (5,172 мл, 106,422 ммоль) растворяют в этаноле (20 мл) at 90°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (2,200 г, 100,0%, бесцветное масло).
[Стадия 6] Синтез соединения 2
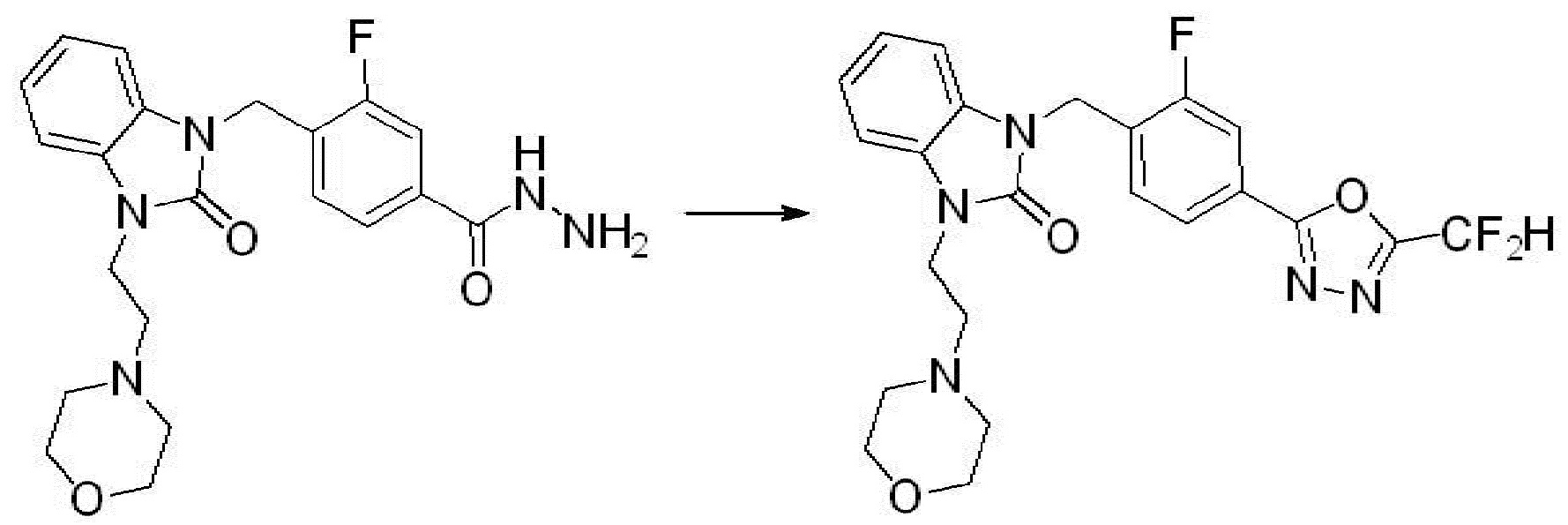
3-фтор-4-((3-(2-морфолиноэтил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)бензогидразид (2,150 г, 5,200 ммоль), полученный на стадии 5, 2,2-дифторуксусный ангидрид (1,939 мл, 15,600 ммоль) и триэтиламин (2,174 мл, 15,600 ммоль) растворяют в дихлорметане (20 мл) при 45°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (1,550 г, 63,0%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,90-7,85 (м, 2H), 7,52 (дд, J=8,0, 7,5 Гц, 1H), 7,19-7,13 (м, 2H), 7,12-7,09 (м, 1H), 7,06 (с, 0,25H), 6,98 (д, J=7,6 Гц, 1H), 6,93 (с, 0,5H), 6,80 (с, 0,25H), 5,21 (с, 2H), 4,36 (т, J=6,9 Гц, 2H), 3,96 (т, J=4,8 Гц, 4H), 3,51-3,24 (м, 6H); МСНР (ЭР) m/z 474,3 (M+ + 1).
Пример 3: Синтез соединения 3, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(2-(диметиламино)этил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез N1,N1-диметил-N2-(2-нитрофенил)этан-1,2-диамина
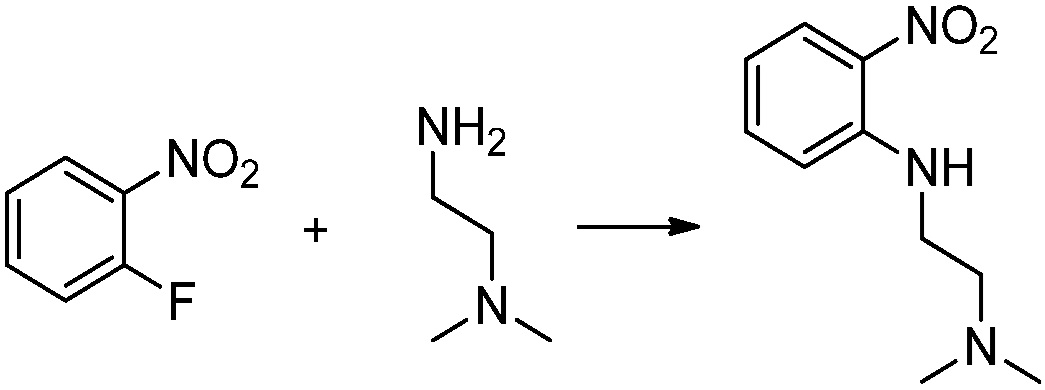
1-фтор-2-нитробензол (2,000 г, 14,174 ммоль), N1,N1-диметилэтан-1,2-диамин (1,548 мл, 14,174 ммоль) и карбонат калия (3,918 г, 28,349 ммоль) растворяют в тетрагидрофуране (30 мл) при 70°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (2,500 г, 84,3%) в виде красного масла.
[Стадия 2] Синтез N1-(2-(диметиламино)этил)бензол-1,2-диамина
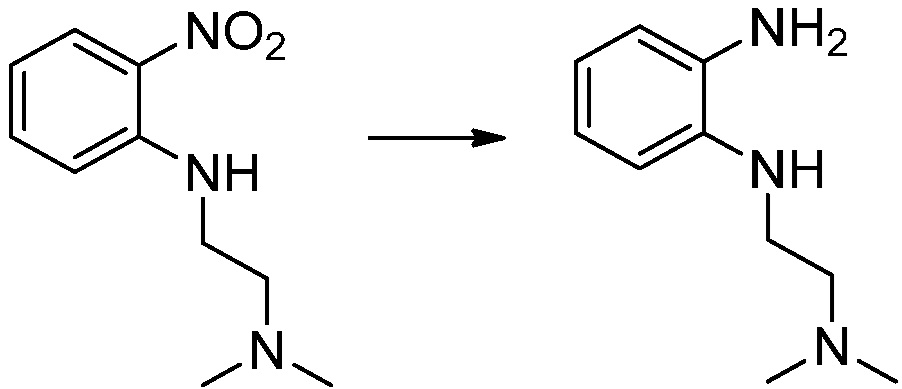
N1,N1-диметил-N2-(2-нитрофенил)этан-1,2-диамин (2,500 г, 11,947 ммоль), полученный на стадии 1, растворяют в метаноле (30 мл) при комнатной температуре, после чего 10%-Pd/C (250 мг) медленно добавляют туда и перемешивают при той же температуре в течение 12 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (2,100 г, 98,0%, черное масло).
[Стадия 3] Синтез 1-(2-(диметиламино)этил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
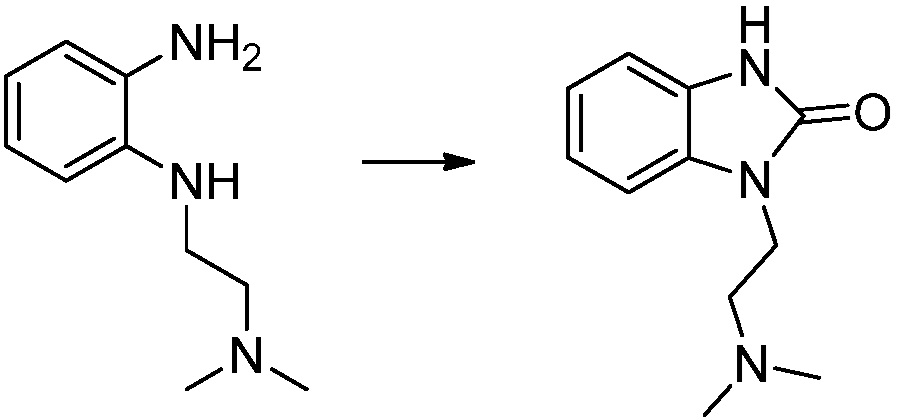
N1-(2-(диметиламино)этил)бензол-1,2-диамин (2,100 г, 11,714 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 2,089 г, 12,886 ммоль) растворяют в тетрагидрофуране (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (1,700 г, 70,7%) в виде черного масла.
[Стадия 4] Синтез соединения 3
1-(2-(диметиламино)этил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,300 г, 1,462 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,088 г, 2,192 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,466 г, 1,608 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,300 г, 49,5%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (дд, J=2,2, 0,7 Гц, 1H), 8,30 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (дд, J=8,0, 0,4 Гц, 1H), 7,11-7,02 (м, 3H), 7,06 (с, 0,25H), 6,95-6,93 (м, 1H), 6,93 (с, 0,5H), 6,80 (с, 0,25H), 5,29 (с, 2H), 4,07 (т, J=7,1 Гц, 2H), 2,70 (т, J=7,1 Гц, 2H), 2,34 (с, 6H); МСНР (ЭР) m/z 415,4 (M+ + 1).
Пример 4: Синтез соединения 4, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
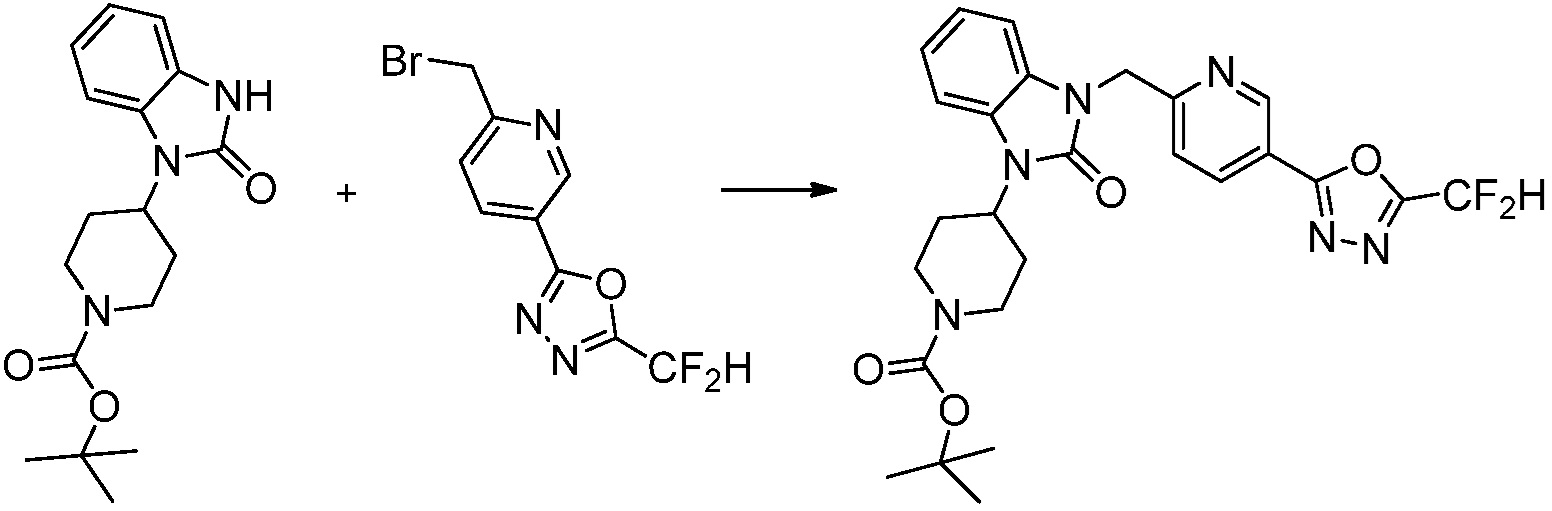
Трет-бутил 4-(2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,200 г, 0,630 ммоль) растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,038 г, 0,945 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,201 г, 0,693 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,210 г, 63,3%) в виде бесцветного масла.
[Стадия 2] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетата
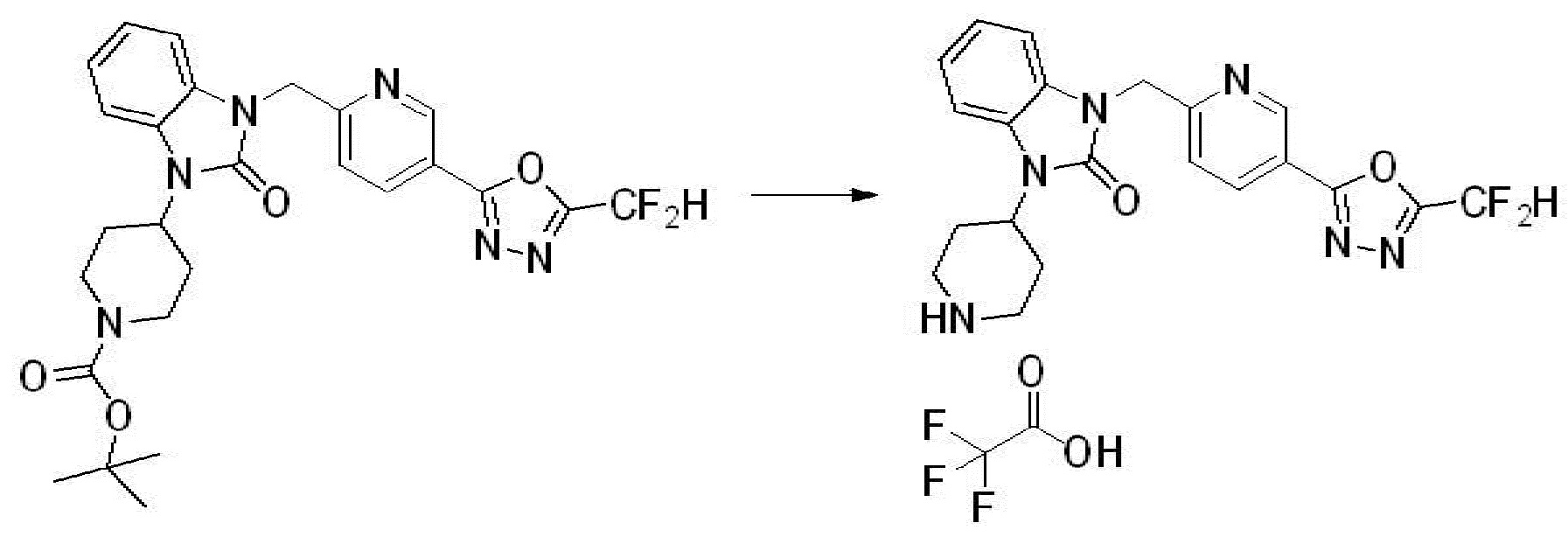
Трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,250 г, 0,475 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,727 мл, 9,496 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Указанное в заголовке соединение применяют без дополнительной очистки (0,250 г, 97,4%, желтое масло).
[Стадия 3] Синтез соединения 4
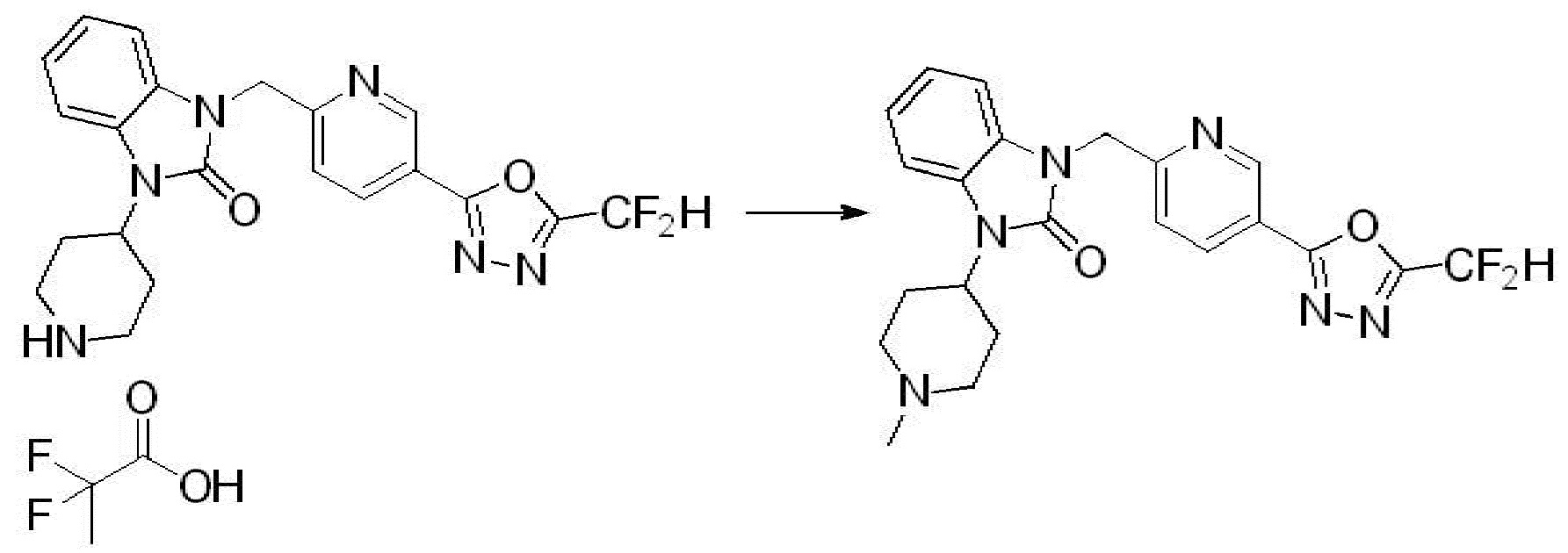
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,141 г, 0,261 ммоль), полученный на стадии 2, и N,N-диизопропилэтиламин (0,045 мл, 0,261 ммоль) растворяют в дихлорметане (10 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем формальдегид (37,00% раствор, 0,039 мл, 0,522 ммоль) и триацетоксиборгидрид натрия (0,111 г, 0,522 ммоль) добавляют туда и дополнительно перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,080 г, 69,6%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=2,2 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,44-7,39 (м, 2H), 7,13-7,03 (м, 2H), 7,07 (с, 0,25H), 6,97 (д, J=1,3 Гц, 1H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 5,30 (с, 2H), 4,68-4,62 (м, 1H), 3,48-3,43 (м, 2H), 2,85-2,79 (м, 2H), 2,67 (с, 3H), 2,00-1,96 (м, 2H), 1,45-1,41 (м, 2H); МСНР (ЭР) m/z 441,5 (M+ + 1).
Пример 5: Синтез соединения 5, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
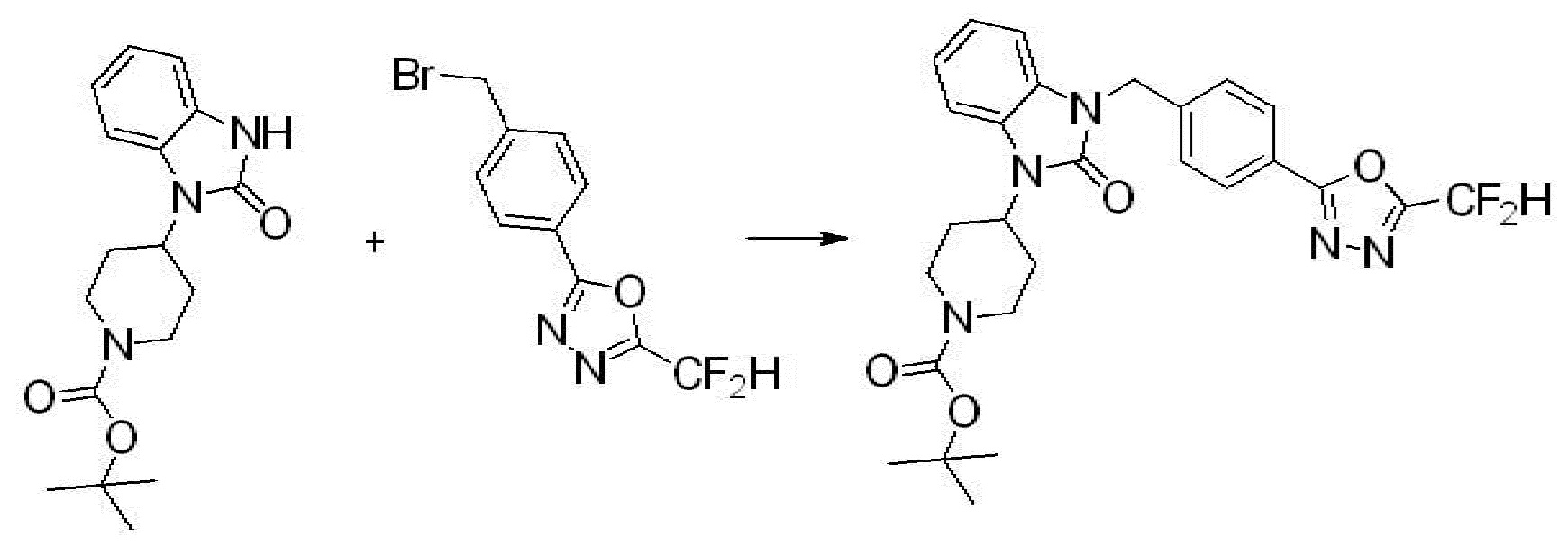
Трет-бутил 4-(2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,200 г, 0,630 ммоль) растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,038 г, 0,945 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,200 г, 0,693 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,150 г, 45,3%) в виде бесцветного масла.
[Стадия 2] Синтез 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетата
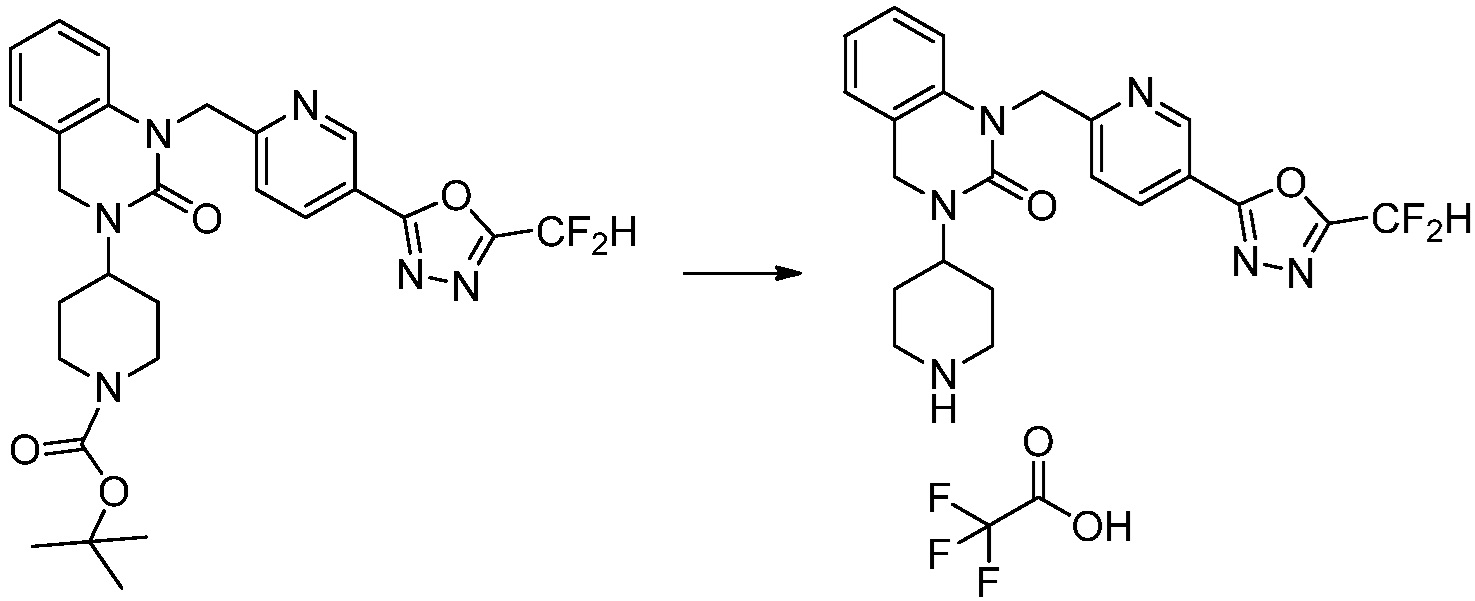
Трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,150 г, 0,285 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,437 мл, 5,708 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Указанное в заголовке соединение применяют без дополнительной очистки (0,150 г, 97,4%, желтое масло).
[Стадия 3] Синтез соединения 5
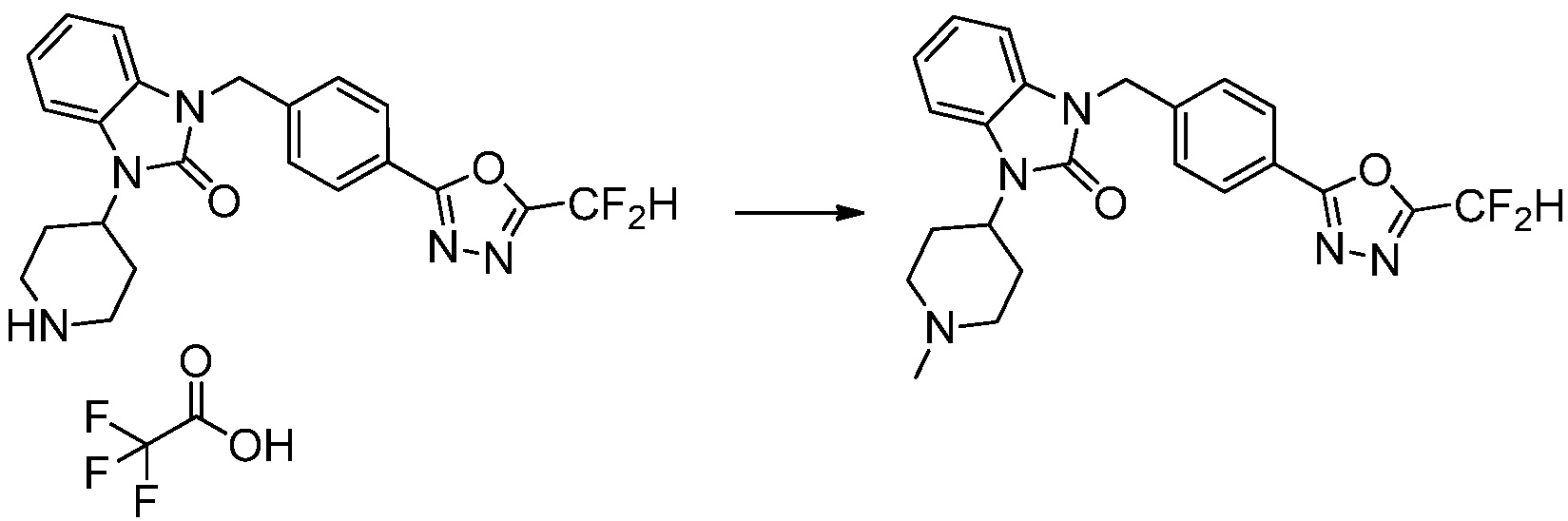
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,150 г, 0,278 ммоль), полученный на стадии 2, и N,N-диизопропилэтиламин (0,048 мл, 0,278 ммоль) растворяют в дихлорметане (10 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют формальдегид (37,00% раствор, 0,041 мл, 0,556 ммоль) и триацетоксиборгидрид натрия (0,118 г, 0,556 ммоль) и дополнительно перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,090 г, 73,7%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 8,05 (дд, J=6,7, 1,7 Гц, 2H), 7,47-7,40 (м, 3H), 7,10-6,99 (м, 2H), 7,06 (с, 0,25H), 6,91 (с, 0,5H), 6,86 (дд, J=7,8, 0,9 Гц, 1H), 6,78 (с, 0,25H), 5,13 (с, 2H), 4,68-4,62 (м, 1H), 3,48-3,44 (м, 2H), 2,83-2,68 (м, 5H), 1,97 (дд, J=12,6, 2,5 Гц, 2H), 1,44-1,39 (м, 2H); МСНР (ЭР) m/z 440,3 (M+ + 1).
Пример 6: Синтез соединения 6, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез бензил 3-((2-аминофенил)амино)пирролидин-1-карбоксилата
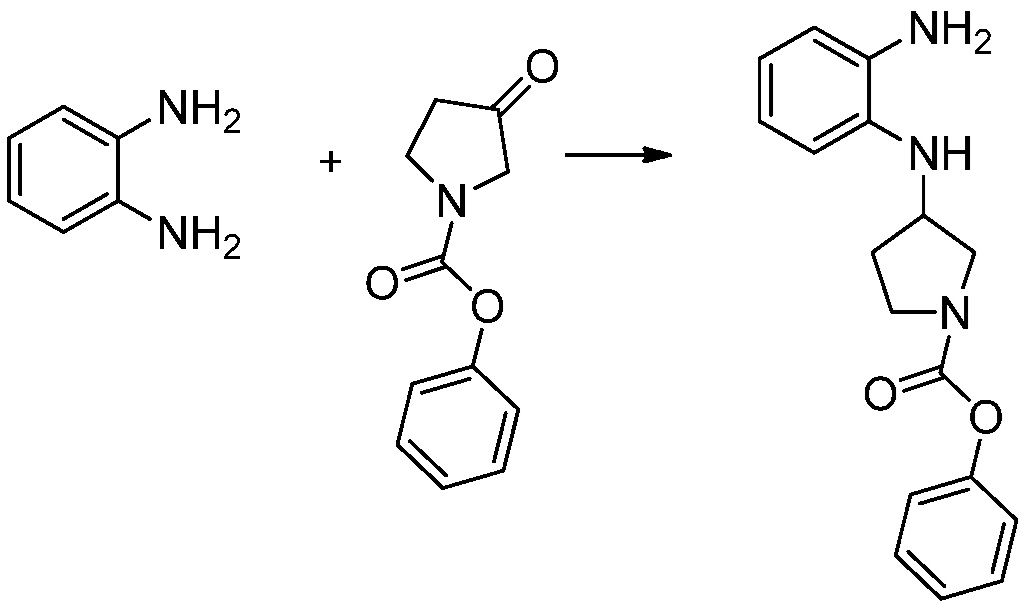
Бензил 3-оксопирролидин-1-карбоксилат (2,000 г, 9,122 ммоль), бензол-1,2-диамин (2,959 г, 27,367 ммоль), триацетоксиборгидрид натрия (3,867 г, 18,245 ммоль) и уксусную кислоту (1,044 мл, 18,245 ммоль) растворяют в 1,2-дихлорэтане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (1,620 г, 57,0%) в виде желтого твердого вещества.
[Стадия 2] Синтез бензил 3-(2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилата
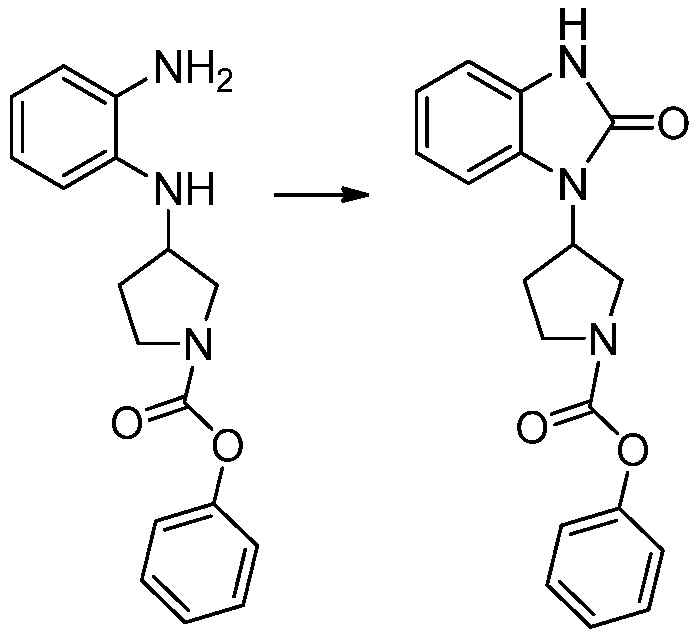
Бензил 3-оксопирролидин-1-карбоксилат (1,620 г, 7,389 ммоль), полученный на стадии 1, и 1,1'-карбонилдиимидазол (КДИ, 1,198 г, 7,389 ммоль) растворяют в тетрагидрофуране (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,900 г, 36,1%) в виде коричневого пенистого твердого вещества.
[Стадия 3] Синтез бензил 3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилата
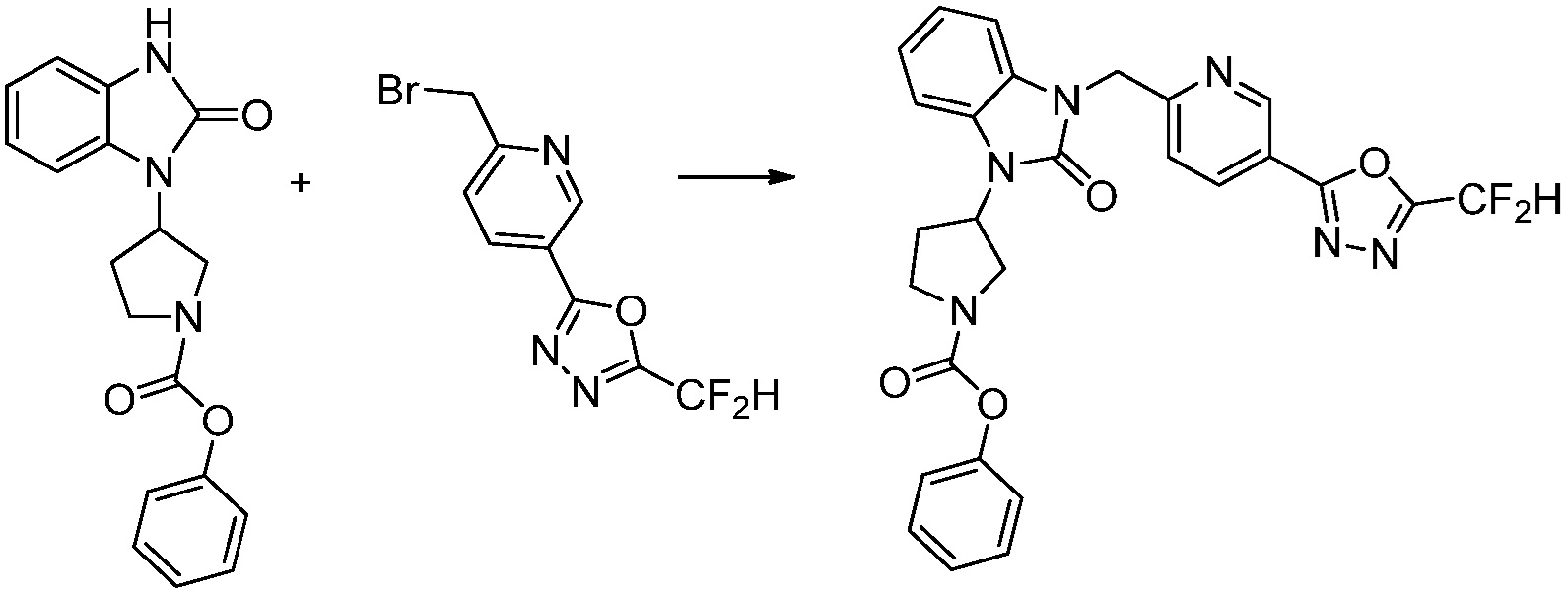
Бензил 3-(2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (0,200 г, 0,593 ммоль), полученный на стадии 2, растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,036 г, 0,889 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,189 г, 0,652 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,180 г, 55,6%) в виде бесцветного масла.
[Стадия 4] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
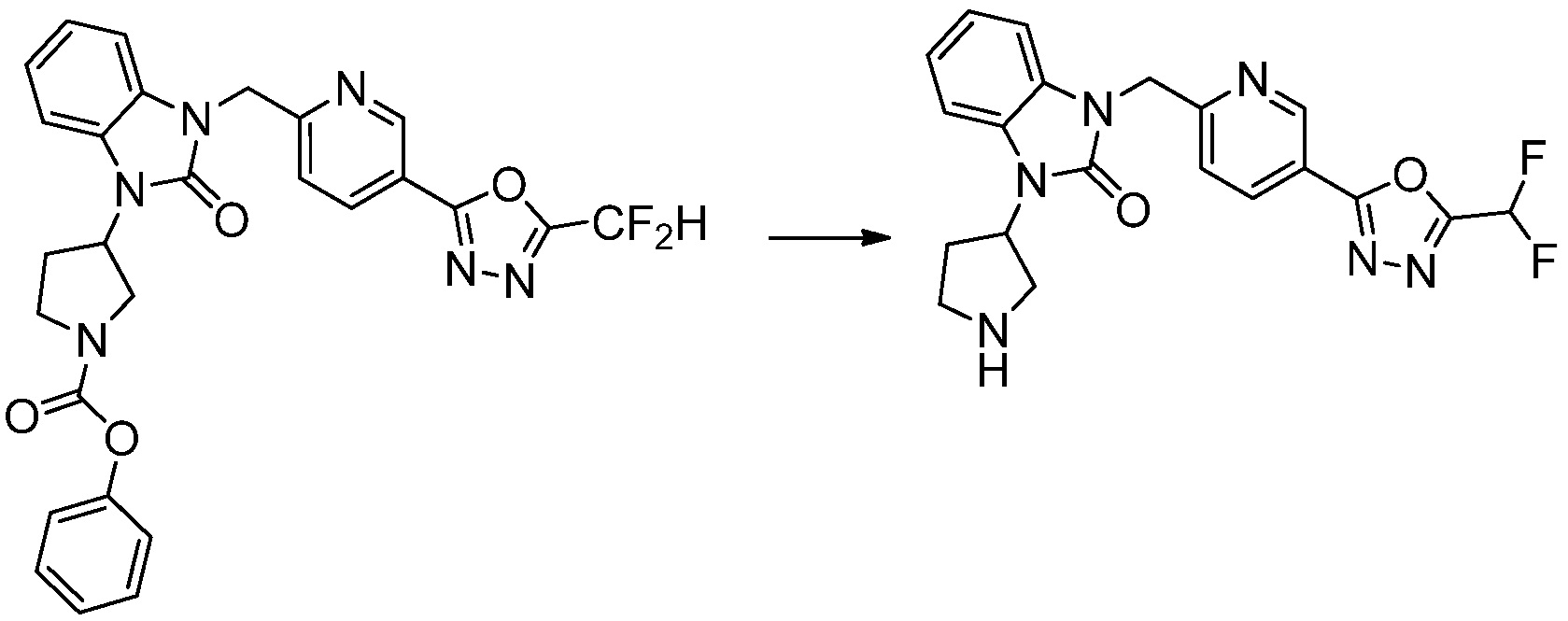
Бензил 3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (0,180 г, 0,329 ммоль), полученный на стадии 3, растворяют в метаноле (10 мл) при комнатной температуре, после чего 10%-Pd/C (18 мг) медленно добавляют туда и перемешивают при той же температуре в течение 12 часов в присутствии присоединенного баллона с водородом. Указанное в заголовке соединение применяют без дополнительной очистки (0,070 г, 51,5%, белое твердое вещество).
[Стадия 5] Синтез соединения 6
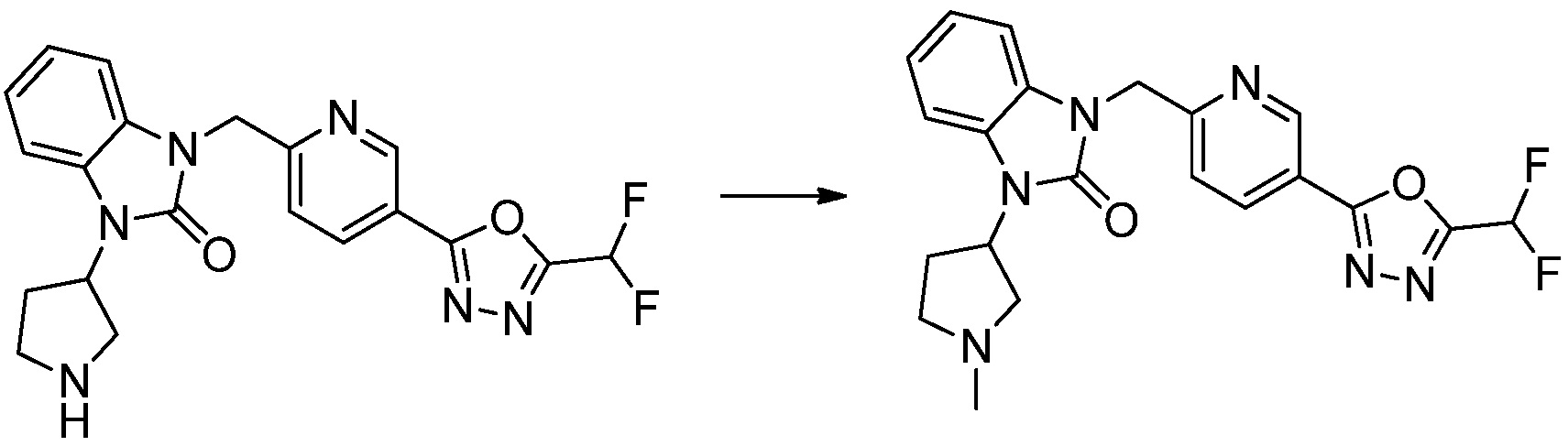
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,070 г, 0,170 ммоль) полученный на стадии 4, триацетоксиборгидрид натрия (0,072 г, 0,339 ммоль) и формальдегид (37,00% раствор, 0,025 мл, 0,339 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 30 минут. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,040 г, 55,3%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (дд, J=2,2, 0,9 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,55-7,53 (м, 1H), 7,43-7,41 (м, 1H), 7,16-7,10 (м, 1H), 7,08-7,02 (м, 1H), 7,06 (с, 0,25H), 6,97-6,95 (м, 1H), 6,93 (с, 0,5H), 6,81 (с, 0,25H), 5,31-5,23 (м, 3H), 3,23-3,21 (м, 2H), 3,12-3,07 (м, 1H), 2,86-2,84 (м, 1H), 2,57 (с, 3H), 2,45-2,34 (м, 2H); МСНР (ЭР) m/z 427,4 (M+ + 1).
Пример 7: Синтез соединения 7, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилазетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 3-((2-аминофенил)амино)азетидин-1-карбоксилата
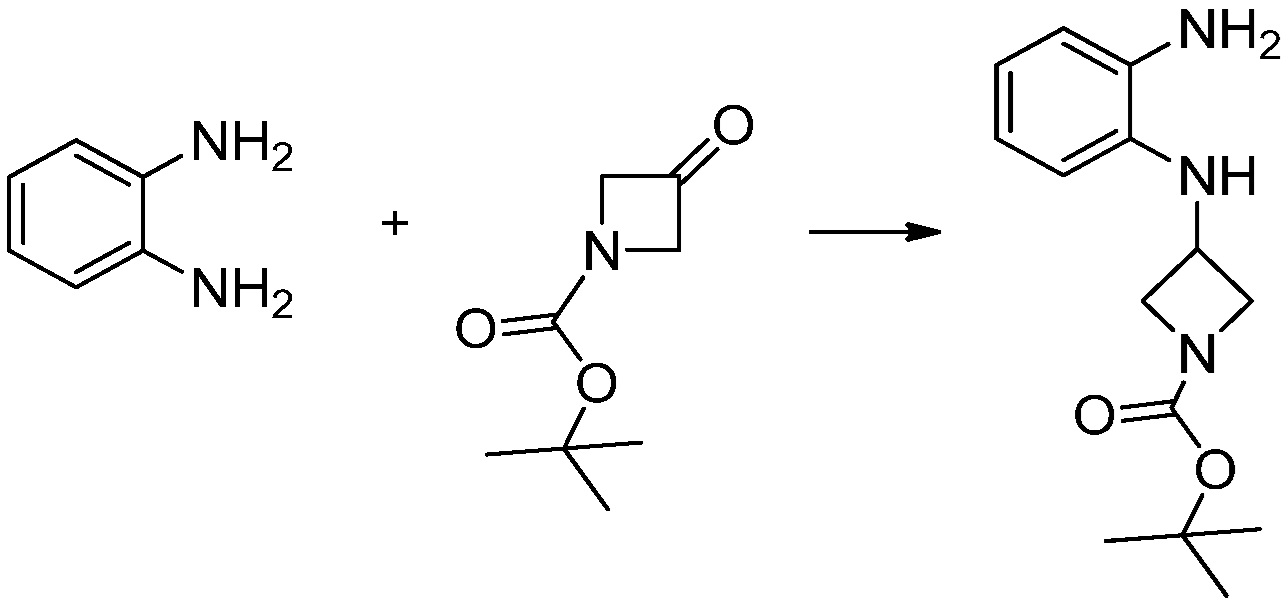
Трет-бутил 3-оксоазетидин-1-карбоксилат (2,000 г, 11,682 ммоль), бензол-1,2-диамин (3,790 г, 35,047 ммоль), триацетоксиборгидрид натрия (4,952 г, 23,364 ммоль) и уксусную кислоту (1,337 мл, 23,364 ммоль) растворяют в 1,2-дихлорэтане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (1,900 г, 61,8%) в виде желтого твердого вещества.
[Стадия 2] Синтез трет-бутил 3-(2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилата
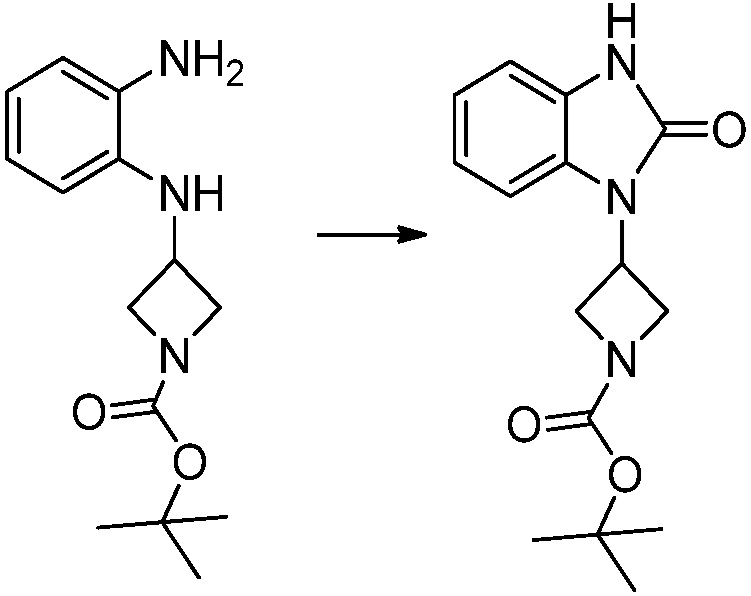
Трет-бутил 3-((2-аминофенил)амино)азетидин-1-карбоксилат (1,900 г, 7,215 ммоль), полученный на стадии 1, и 1,1'-карбонилдиимидазол (КДИ, 1,170 г, 7,215 ммоль) растворяют в тетрагидрофуране (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,900 г, 43,1%) в виде коричневого пенистого твердого вещества.
[Стадия 3] Синтез трет-бутил 3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилата
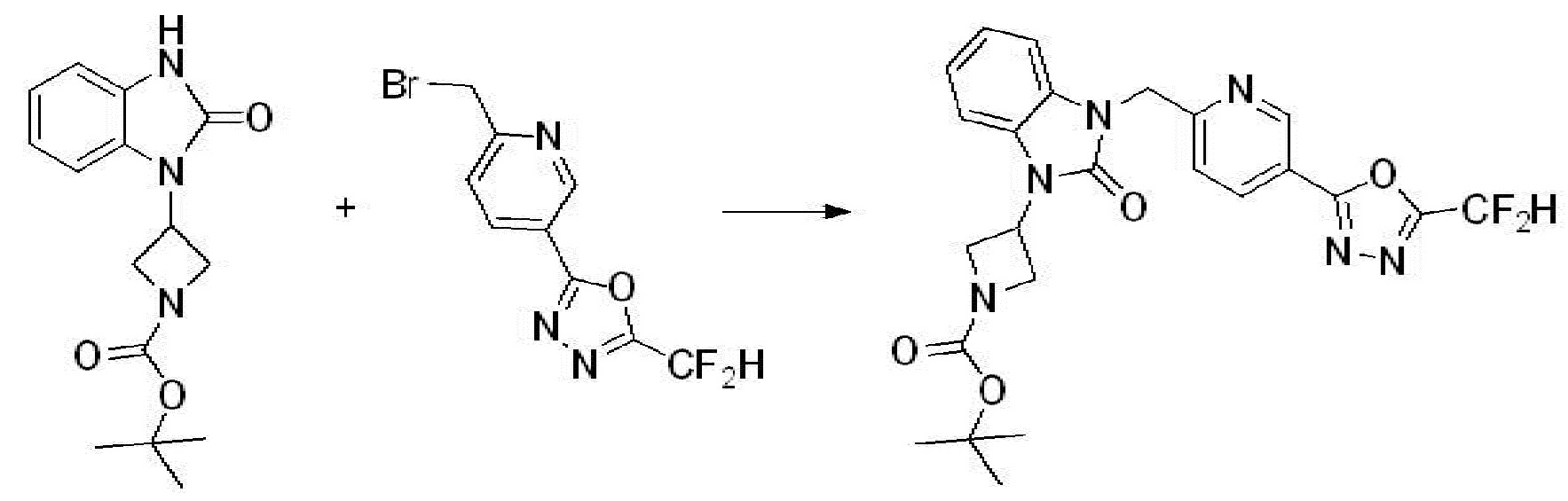
Трет-бутил 3-(2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилат (0,200 г, 0,691 ммоль), полученный на стадии 2, растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,041 г, 1,037 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,221 г, 0,760 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 29,0%) в виде бесцветного масла.
[Стадия 4] Синтез 1-(азетидин-3-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетата
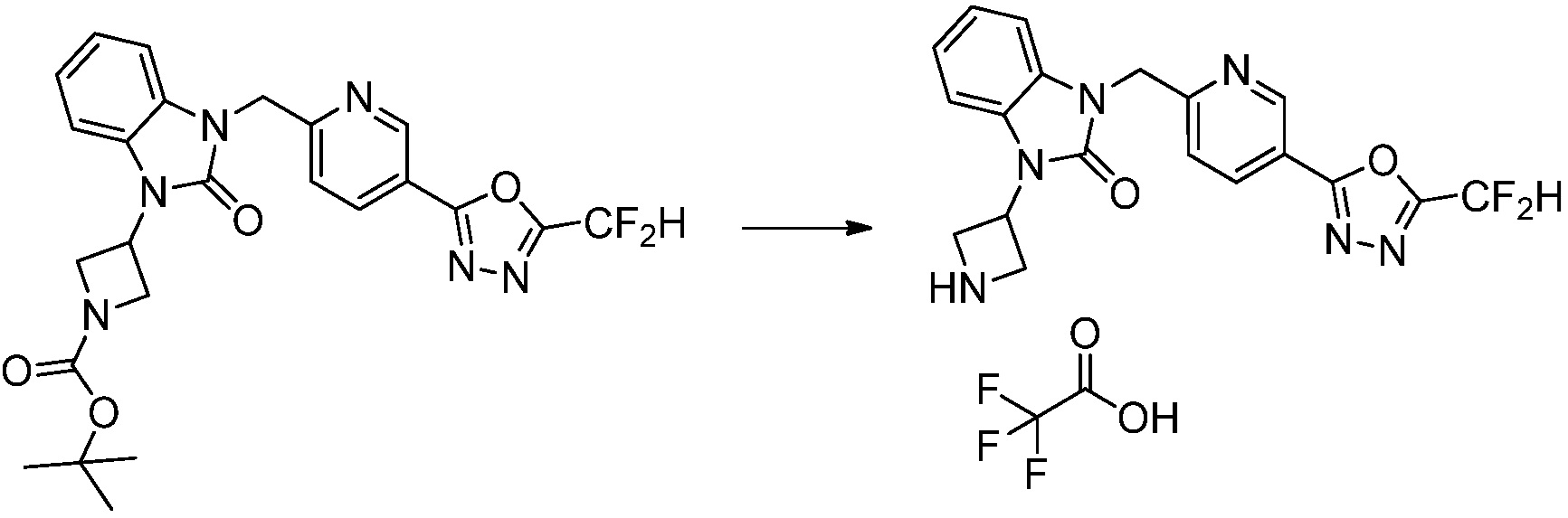
Трет-бутил 3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилат (0,100 г, 0,201 ммоль), полученный на стадии 3, и трифторуксусную кислоту (0,307 мл, 4,012 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Указанное в заголовке соединение применяют без дополнительной очистки (0,100 г, 97,3%, желтое масло).
[Стадия 5] Синтез соединения 7
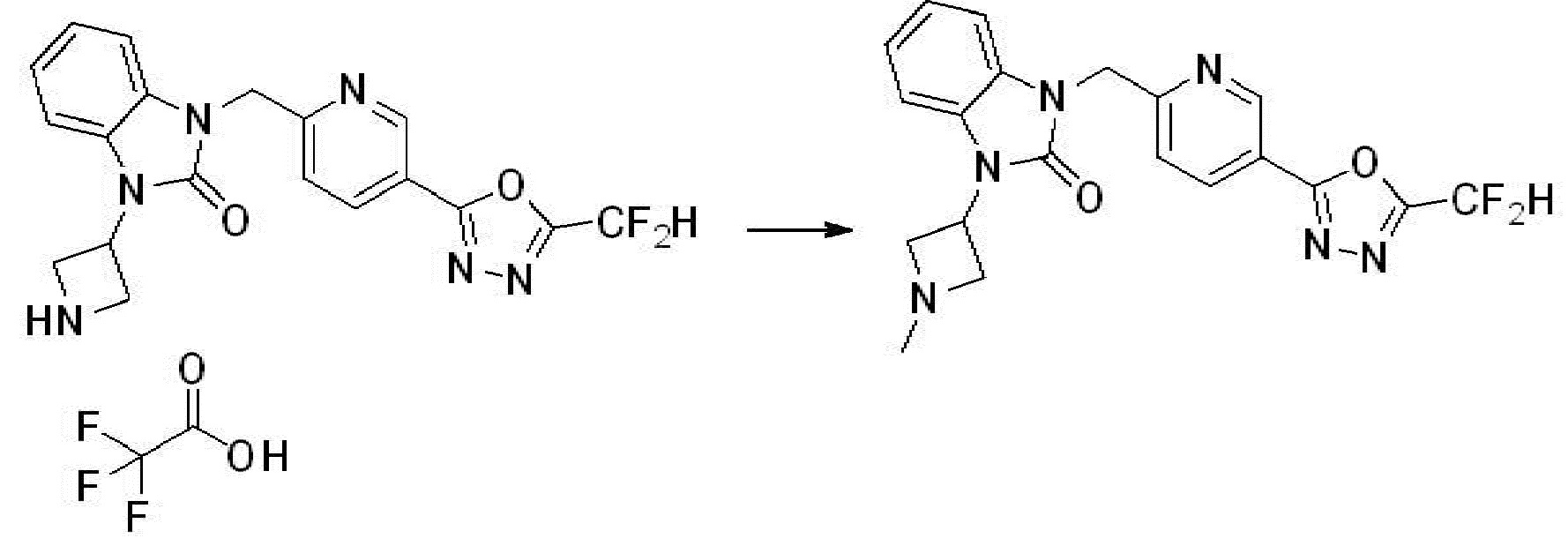
1-(азетидин-3-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,100 г, 0,195 ммоль), полученный на стадии 4, и N,N-диизопропилэтиламин (0,034 мл, 0,195 ммоль) растворяют в дихлорметане (10 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем формальдегид (37,00% раствор, 0,029 мл, 0,390 ммоль) и триацетоксиборгидрид натрия (0,083 г, 0,390 ммоль) добавляют туда и дополнительно перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,050 г, 62,1%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (дд, J=2,2, 0,7 Гц, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 7,47 (дд, J=8,2, 0,5 Гц, 1H), 7,21-7,01 (м, 3H), 7,07 (с, 0,25H), 6,97 (д, J=19,6 Гц, 1H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 5,40-5,36 (м, 1H), 5,30 (с, 2H), 4,79-4,65 (м, 4H), 3,10 (с, 3H); МСНР (ЭР) m/z 413,4 (M+ + 1).
Пример 8: Синтез соединения 8, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-3,4-дигидрохиназолин-2(1H)-он
[Стадия 1] Синтез трет-бутил 4-((2-нитробензил)амино)пиперидин-1-карбоксилата
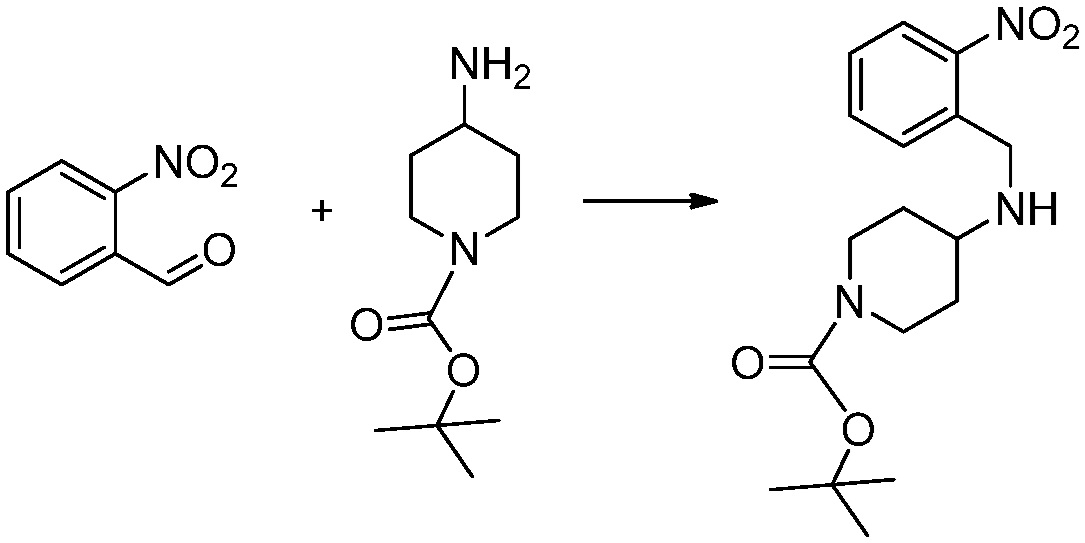
2-нитробензальдегид (3,000 г, 19,852 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (3,976 г, 19,852 ммоль), триацетоксиборгидрид натрия (8,415 г, 39,704 ммоль) и уксусную кислоту (2,273 мл, 39,704 ммоль) растворяют в 1,2-дихлорэтане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (2,500 г, 37,5%) в виде бесцветного масла.
[Стадия 2] Синтез трет-бутил 4-((2-аминобензил)амино)пиперидин-1-карбоксилата
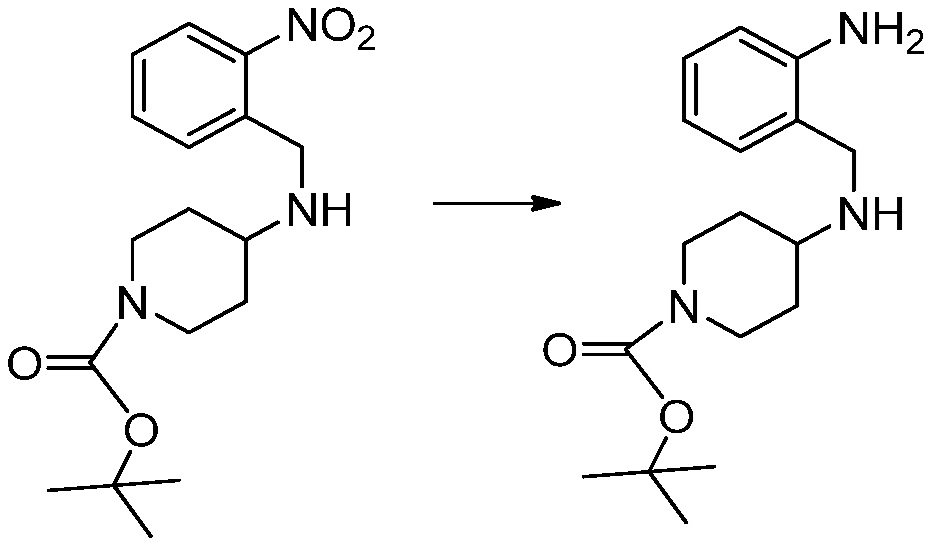
Трет-бутил 4-((2-нитробензил)амино)пиперидин-1-карбоксилат (2,500 г, 7,454 ммоль), полученный на стадии 1, растворяют в метанол (30 мл) при комнатной температуре, после чего 10%-Pd/C (250 мг) медленно добавляют туда и перемешивают при той же температуре в течение 12 часов в присутствии присоединенного баллона с водородом. Реакционную смесь was фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (2,000 г, 87,9%, бесцветное масло).
[Стадия 3] Синтез трет-бутил 4-(2-оксо-1,4-дигидрохиназолин-3(2H)-ил)пиперидин-1-карбоксилата
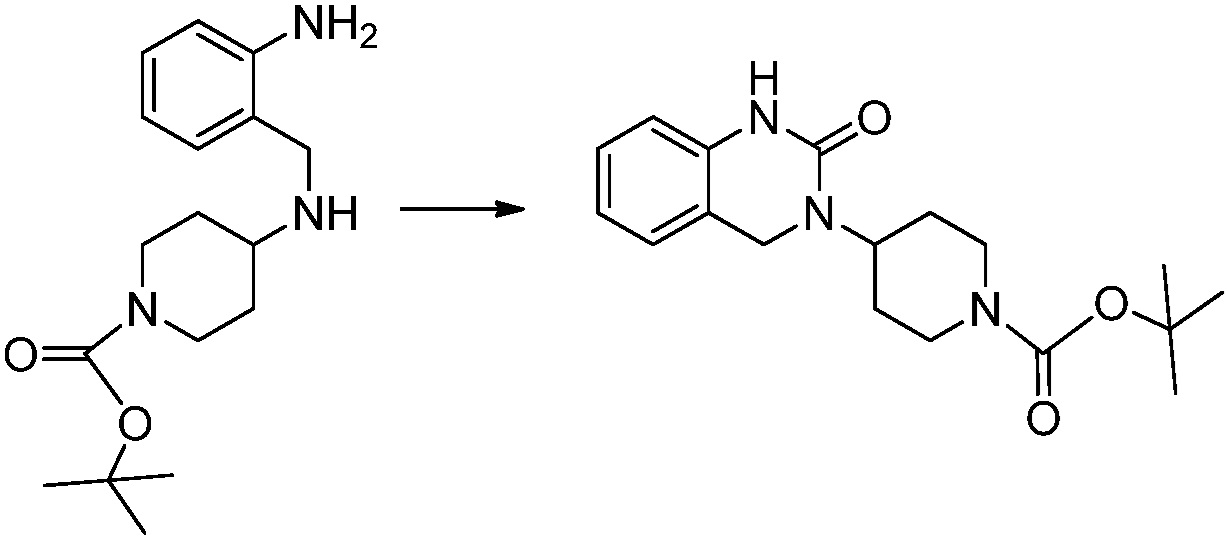
Трет-бутил 4-((2-аминобензил)амино)пиперидин-1-карбоксилат (2,000 г, 6,548 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 1,168 г, 7,203 ммоль) растворяют в тетрагидрофуране (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,900 г, 41,5%) в виде белого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-1,4-дигидрохиназолин-3(2H)-ил)пиперидин-1-карбоксилата
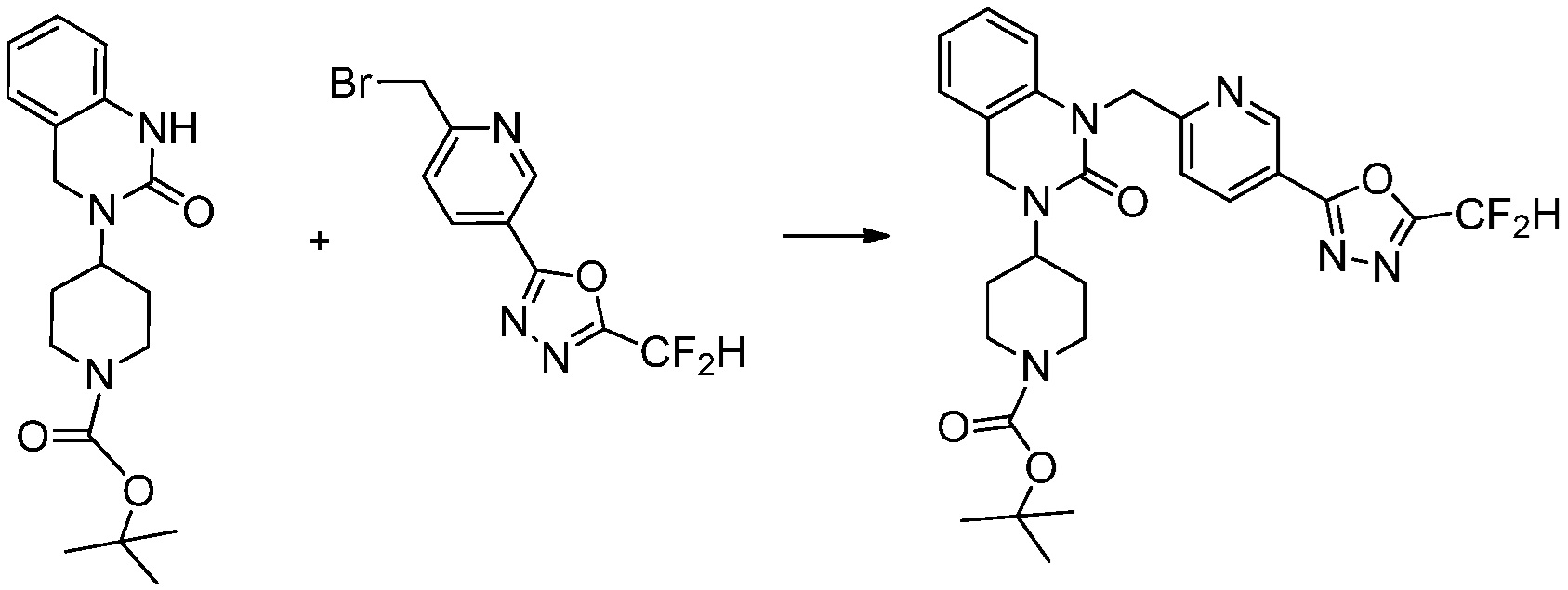
Трет-бутил 4-(2-оксо-1,4-дигидрохиназолин-3(2H)-ил)пиперидин-1-карбоксилат (0,200 г, 0,603 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,036 г, 0,905 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,193 г, 0,664 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,160 г, 49,0%) в виде бесцветного масла.
[Стадия 5] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-3,4-дигидрохиназолин-2(1H)-он 2,2,2-трифторацетата
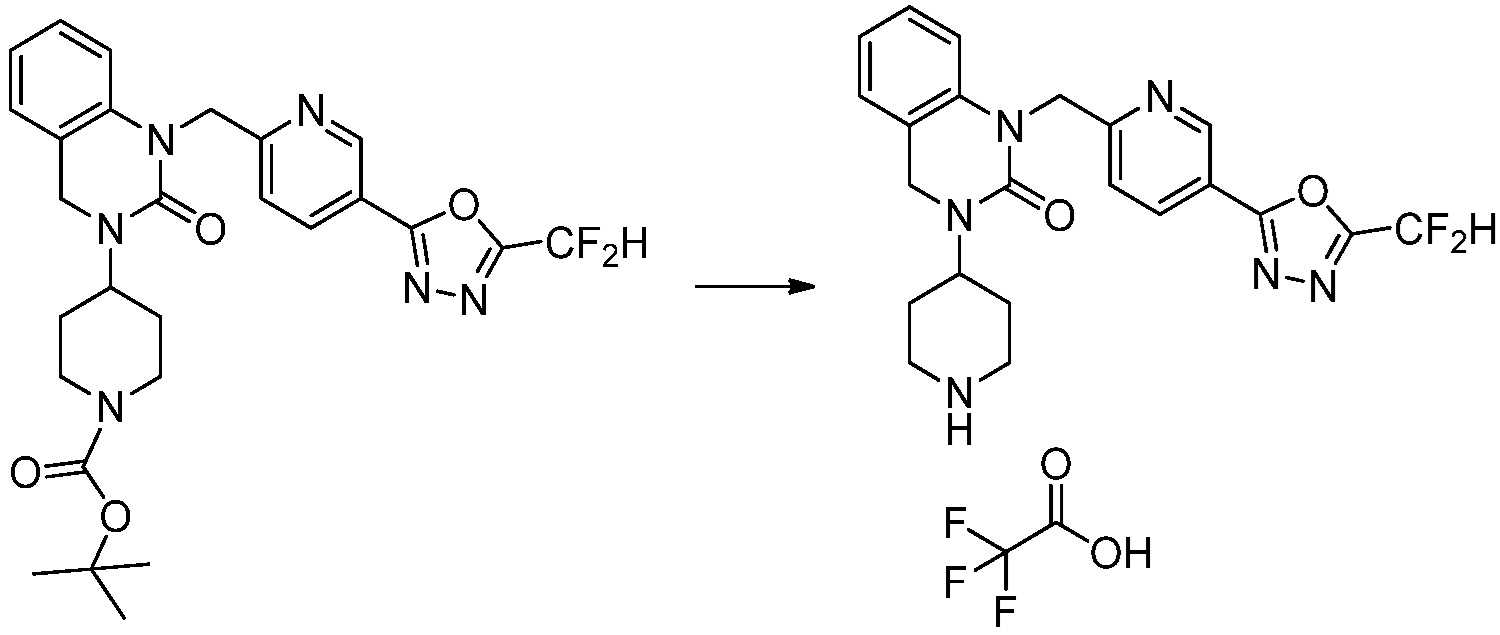
Трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-1,4-дигидрохиназолин-3(2H)-ил)пиперидин-1-карбоксилат (0,160 г, 0,296 ммоль), полученный на стадии 4, и трифторуксусную кислоту (0,453 мл, 5,920 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Указанное в заголовке соединение применяют без дополнительной очистки (0,160 г, 97,5%, желтое масло).
[Стадия 6] Синтез соединения 8
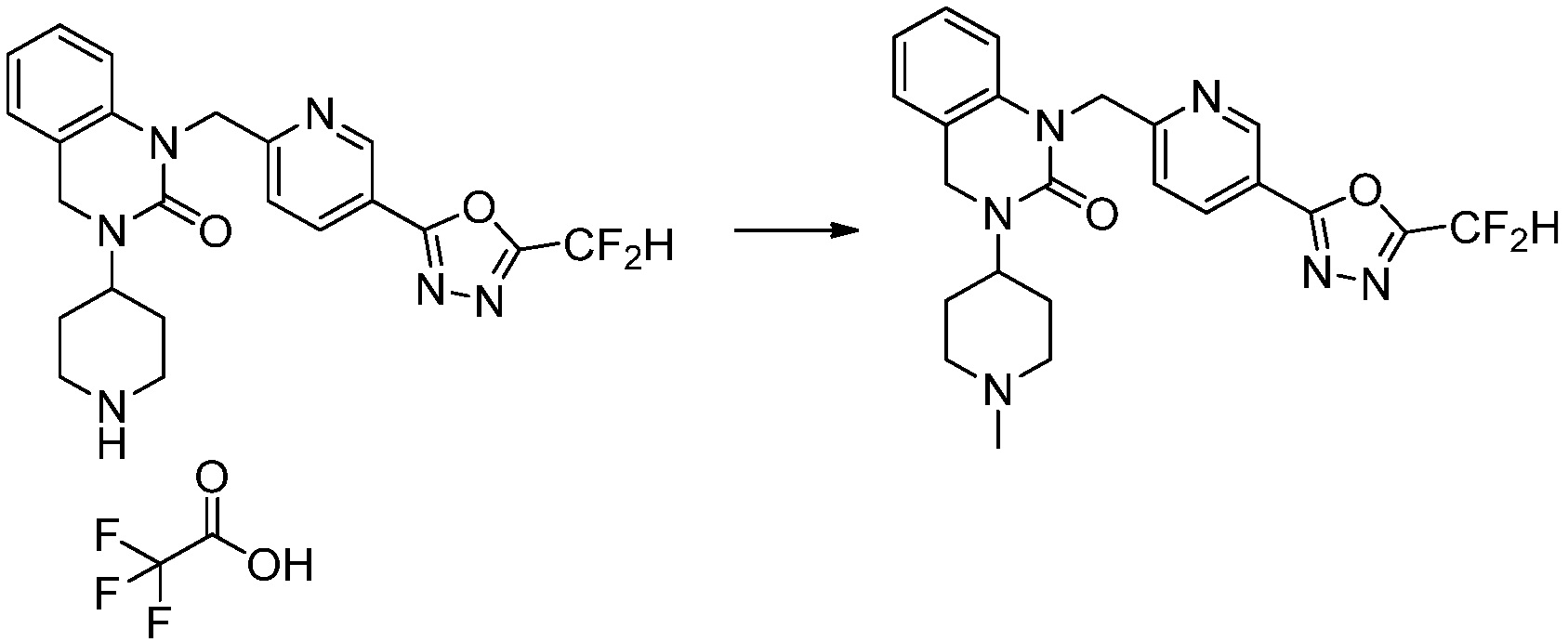
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-3,4-дигидрохиназолин-2(1H)-он 2,2,2-трифторацетат (0,160 г, 0,289 ммоль), полученный на стадии 5, и N,N-диизопропилэтиламин (0,050 мл, 0,289 ммоль) растворяют в дихлорметане (10 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем формальдегид (37,00% раствор, 0,043 мл, 0,577 ммоль) и триацетоксиборгидрид натрия (0,122 г, 0,577 ммоль) добавляют туда и дополнительно перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 83,9%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,31-9,30 (м, 1H), 8,30 (дд, J=8,3, 2,2 Гц, 1H), 7,40 (д, J=8,3 Гц, 1H), 7,16-7,12 (м, 2H), 7,07 (с, 0,25H), 7,02-7,01 (м, 1H), 6,94 (с, 0,5H), 6,81 (с, 0,25H), 6,72 (д, J=8,1 Гц, 1H), 5,31 (с, 2H), 4,64-4,58 (м, 1H), 4,41 (с, 2H), 3,49-3,39 (м, 2H), 2,64-2,58 (м, 5H), 2,35-2,26 (м, 2H), 1,91-1,88 (м, 2H), 1,47-1,44 (м, 2H); МСНР (ЭР) m/z 455,4 (M+ + 1).
Пример 9: Синтез соединения 9, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилазетидин-3-ил)-3,4-дигидрохиназолин-2(1H)-он
[Стадия 1] Синтез трет-бутил 3-((2-нитробензил)амино)азетидин-1-карбоксилата
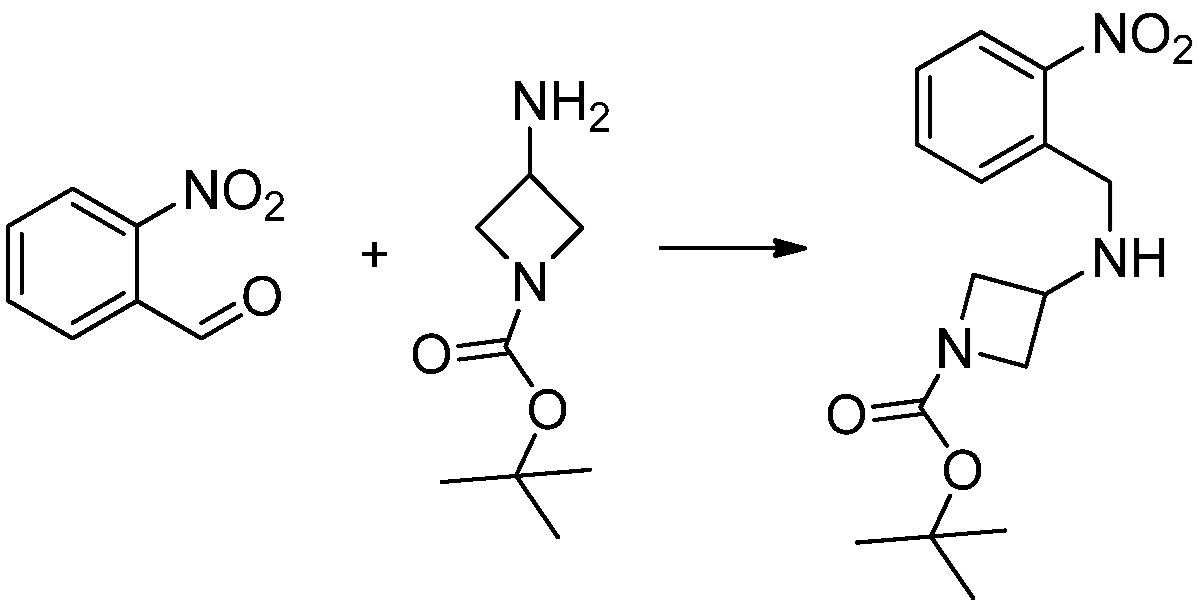
2-нитробензальдегид (3,000 г, 19,852 ммоль), трет-бутил 3-аминоазетидин-1-карбоксилат (3,419 г, 19,852 ммоль), триацетоксиборгидрид натрия (8,415 г, 39,704 ммоль) и уксусную кислоту (2,273 мл, 39,704 ммоль) растворяют в 1,2-дихлорэтане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = 0-50%) и концентрируют с получением указанного в заголовке соединения (3,600 г, 59,0%) в виде бесцветного масла.
[Стадия 2] Синтез трет-бутил 3-((2-аминобензил)амино)азетидин-1-карбоксилата
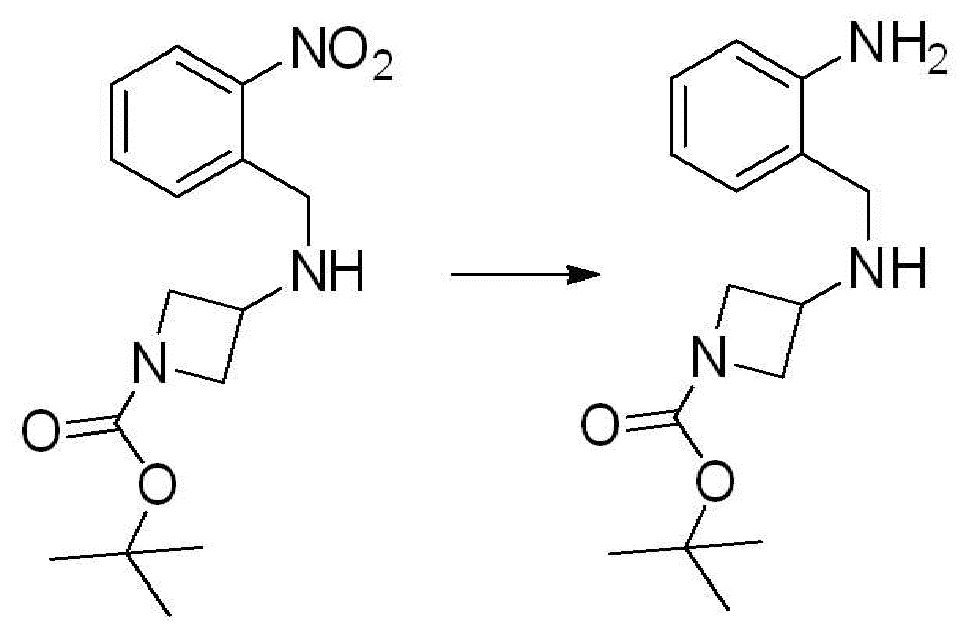
Трет-бутил 3-((2-нитробензил)амино)азетидин-1-карбоксилат (3,600 г, 11,713 ммоль), полученный на стадии 1, растворяют в метаноле (30 мл) при комнатной температуре, после чего туда медленно добавляют 10%-Pd/C (360 мг) и перемешивают при той же температуре в течение 12 часов в присутствии присоединенного баллона с водородом. Реакционную смесь was фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (3,000 г, 92,3%, бесцветное масло).
[Стадия 3] Синтез трет-бутил 3-(2-оксо-1,4-дигидрохиназолин-3(2H)-ил)азетидин-1-карбоксилата
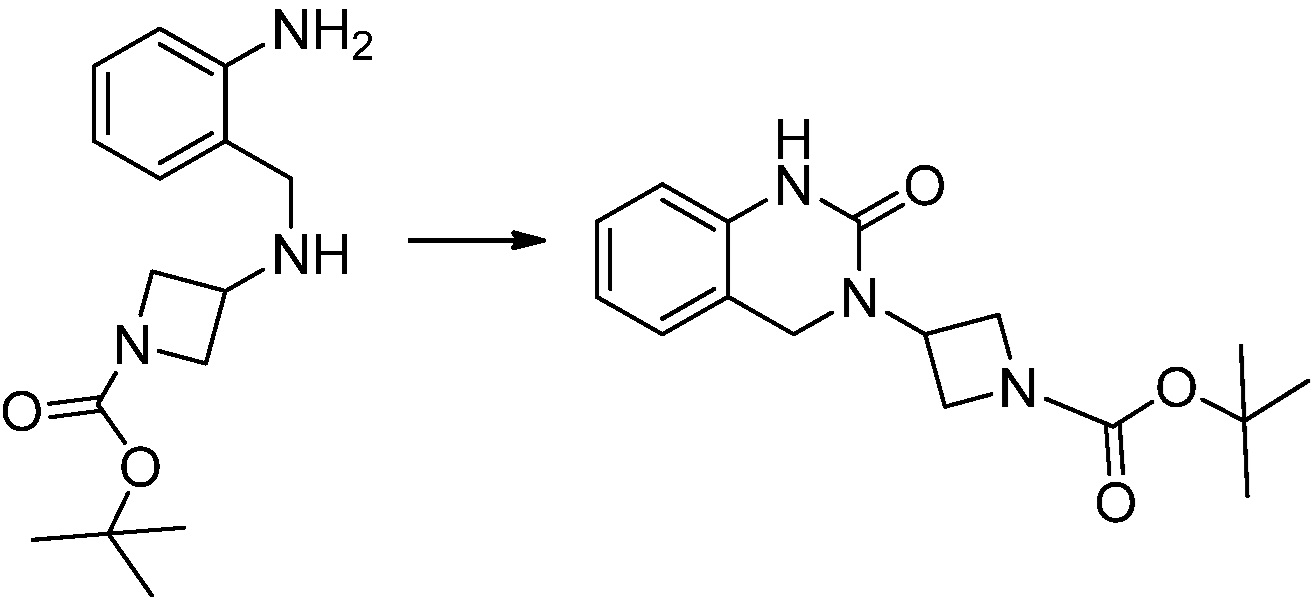
Трет-бутил 3-((2-аминобензил)амино)азетидин-1-карбоксилат (3,000 г, 10,816 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 1,929 г, 11,897 ммоль) растворяют в тетрагидрофуране (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (1,300 г, 39,6%) в виде белого твердого вещества.
[Стадия 4] Синтез трет-бутил 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-1,4-дигидрохиназолин-3(2H)-ил)азетидин-1-карбоксилат
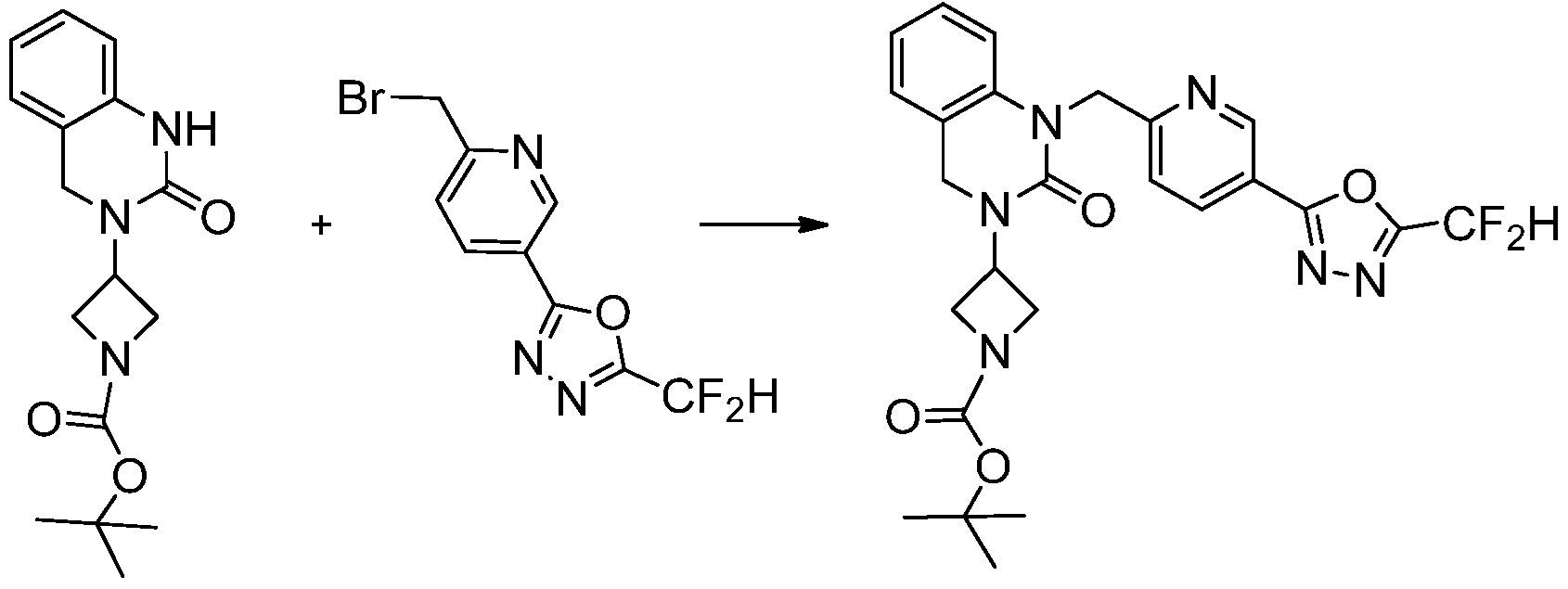
Трет-бутил 3-(2-оксо-1,4-дигидрохиназолин-3(2H)-ил)азетидин-1-карбоксилат (0,200 г, 0,659 ммоль) полученный на стадии 3 растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,040 г, 0,989 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,210 г, 0,725 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,150 г, 44,4%) в виде бесцветного масла.
[Стадия 5] Синтез 3-(азетидин-3-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,4-дигидрохиназолин-2(1H)-он 2,2,2-трифторацетата
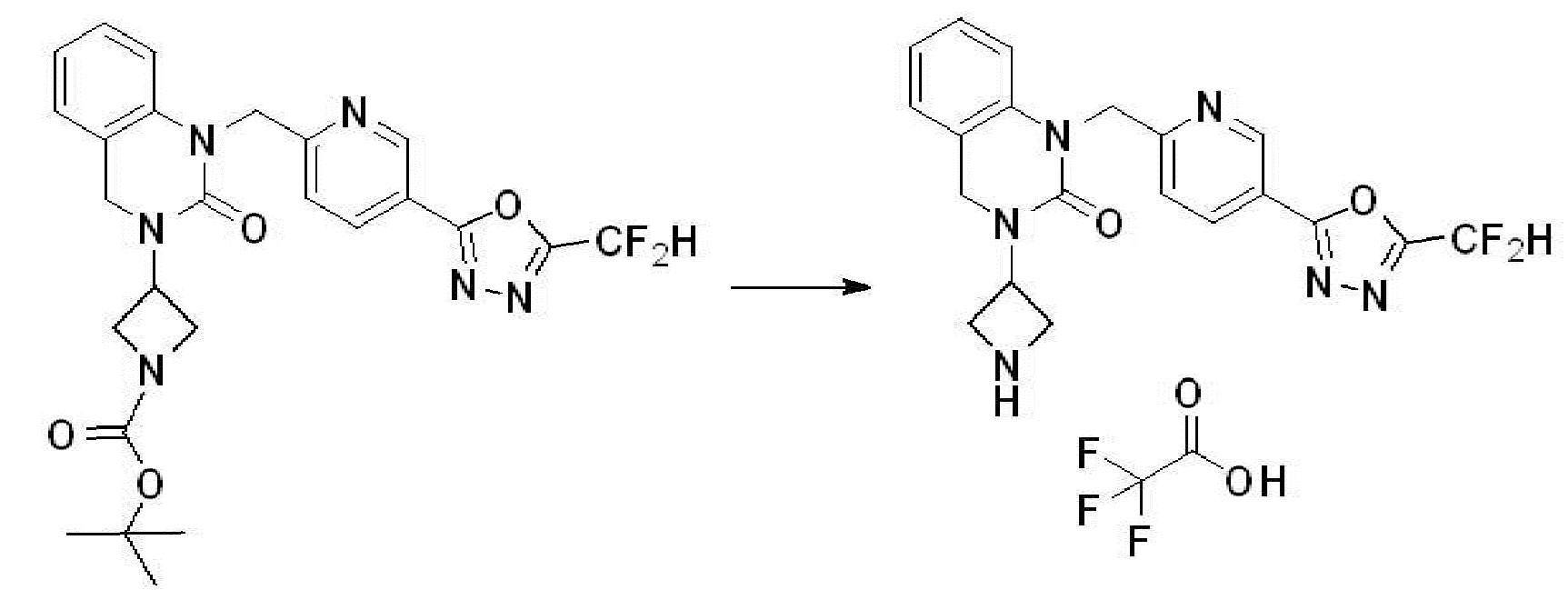
Трет-бутил 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-1,4-дигидрохиназолин-3(2H)-ил)азетидин-1-карбоксилат (0,150 г, 0,293 ммоль), полученный на стадии 4, и трифторуксусную кислоту (0,448 мл, 5,853 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Указанное в заголовке соединение применяют без дополнительной очистки (0,150 г, 97,4%, желтое масло).
[Стадия 6] Синтез соединения 9
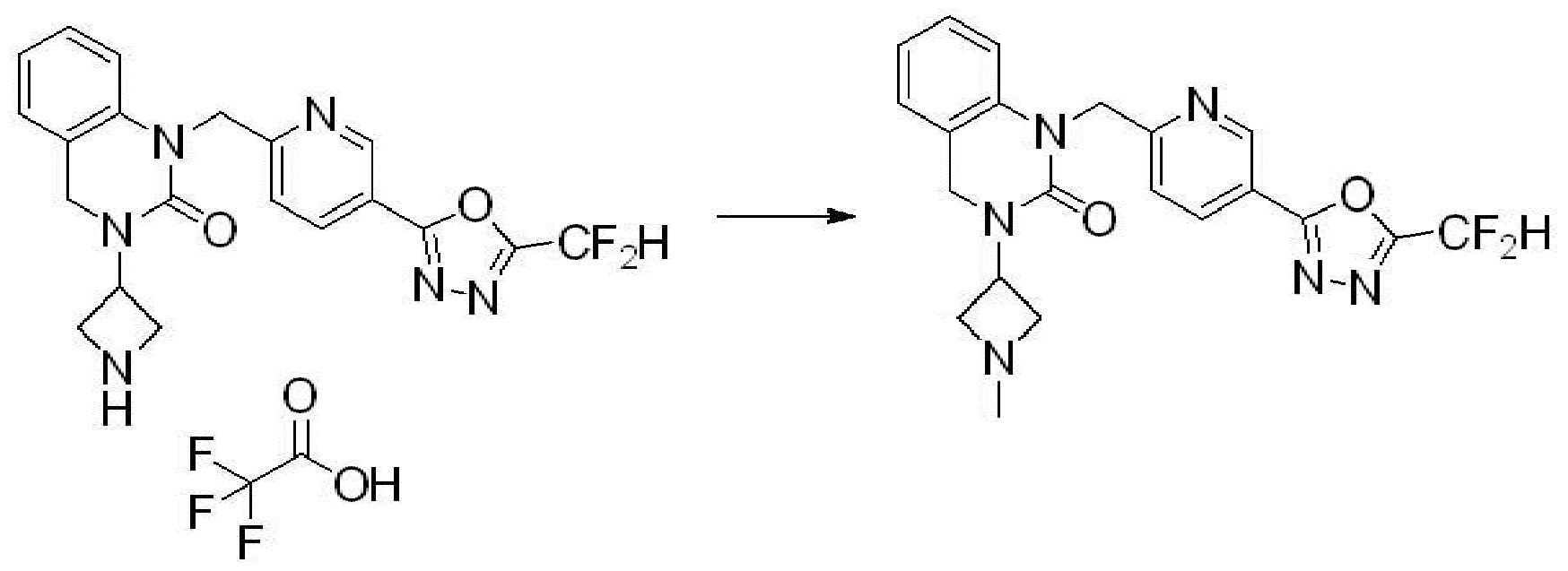
3-(азетидин-3-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,4-дигидрохиназолин-2(1H)-он 2,2,2-трифторацетат (0,150 г, 0,285 ммоль), полученный на стадии 5, и N,N-диизопропилэтиламин (0,050 мл, 0,285 ммоль) растворяют в дихлорметане (10 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют формальдегид (37,00% раствор, 0,042 мл, 0,570 ммоль) и триацетоксиборгидрид натрия (0,121 г, 0,570 ммоль) и дополнительно перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 82,3%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (дд, J=2,2, 0,8 Гц, 1H), 8,34 (дд, J=8,3, 2,2 Гц, 1H), 7,42 (дд, J=8,3, 0,8 Гц, 1H), 7,19-7,12 (м, 2H), 7,08 (с, 0,25H), 7,08-7,03 (м, 1H), 7,01 (с, 0,5H), 6,95 (с, 0,25H), 6,74 (д, J=8,0 Гц, 1H), 5,31 (с, 2H), 4,71-4,67 (м, 1H), 4,49 (с, 2H), 4,32-4,28 (м, 2H), 4,19-4,15 (м, 2H), 2,83 (с, 3H); МСНР (ЭР) m/z 427,3 (M+ + 1).
Пример 10: Синтез соединения 10, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-((1-метилпиперидин-4-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(((2-аминофенил)амино)метил)пиперидин-1-карбоксилата
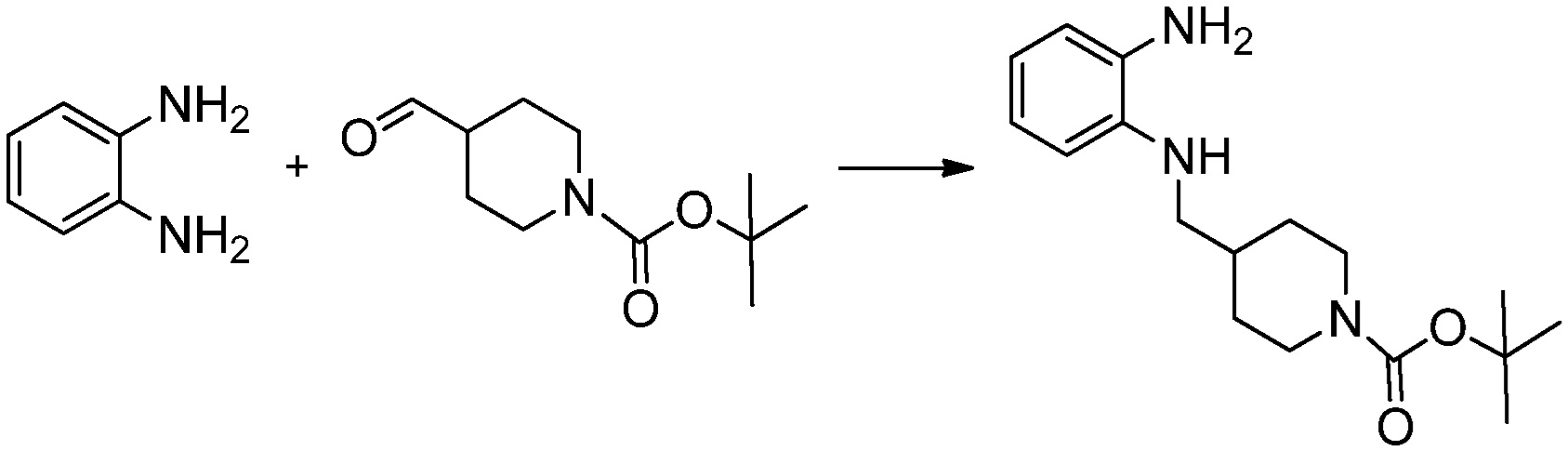
Бензол-1,2-диамин (1,700 г, 15,720 ммоль), трет-бутил 4-формилпиперидин-1-карбоксилат (10,059 г, 47,161 ммоль), триацетоксиборгидрид натрия (6,664 г, 31,441 ммоль) и уксусную кислоту (1,800 мл, 31,441 ммоль) растворяют в 1,2-дихлорэтане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (1,350 г, 28,1%) в виде белого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-((2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилата
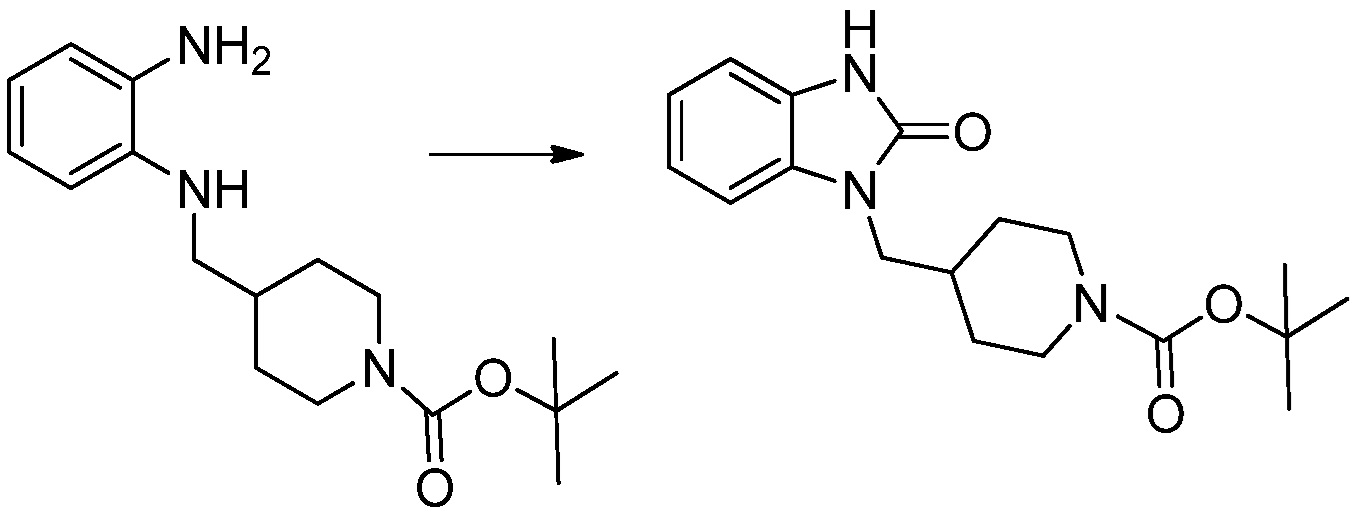
Трет-бутил 4-(((2-аминофенил)амино)метил)пиперидин-1-карбоксилат (1,350 г, 4,420 ммоль), полученный на стадии 1, и 1,1'-карбонилдиимидазол (КДИ, 0,788 г, 4,862 ммоль) растворяют в тетрагидрофуране (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (1 г, 68,3%) в виде белого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-((3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилата
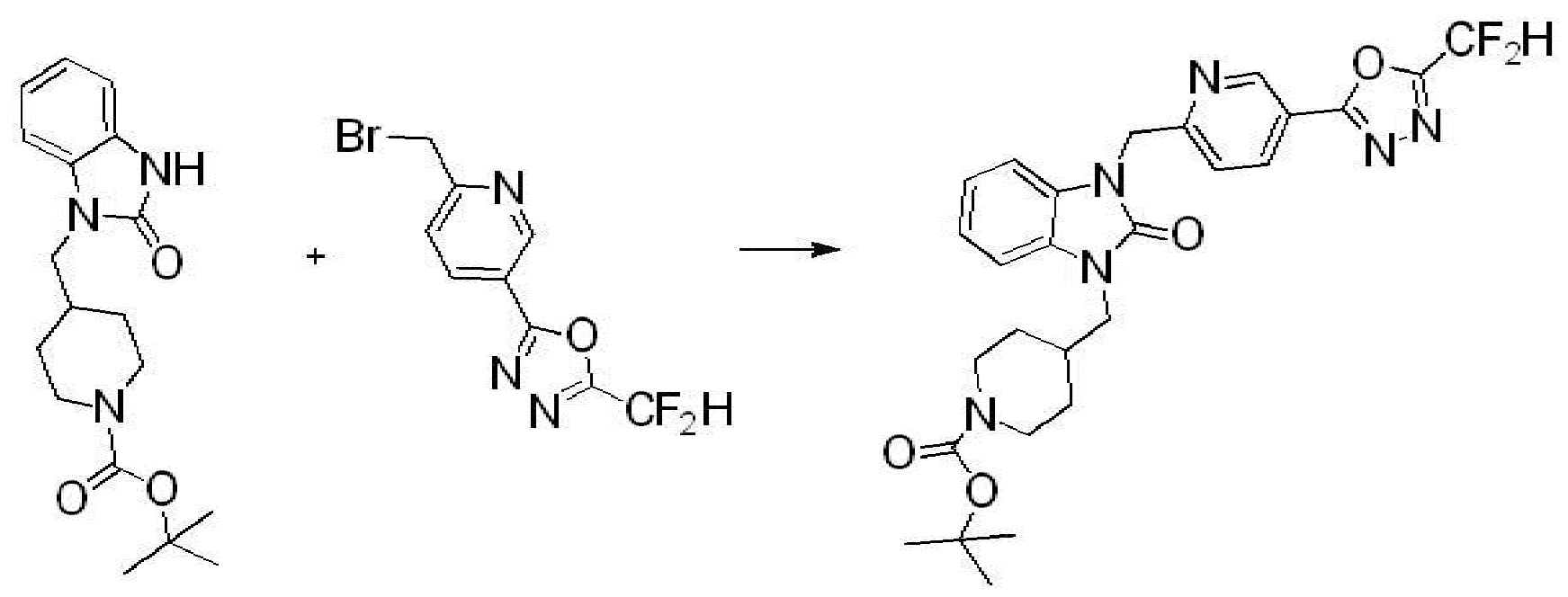
Трет-бутил 4-((2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилат (0,200 г, 0,603 ммоль), полученный на стадии 2, растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,036 г, 0,905 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,193 г, 0,664 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,150 г, 46,0%) в виде бесцветного масла.
[Стадия 4] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-илметил)-1,3-дигидро-2H-бензо[d]имидазол-2-она 2,2,2-трифторацетата
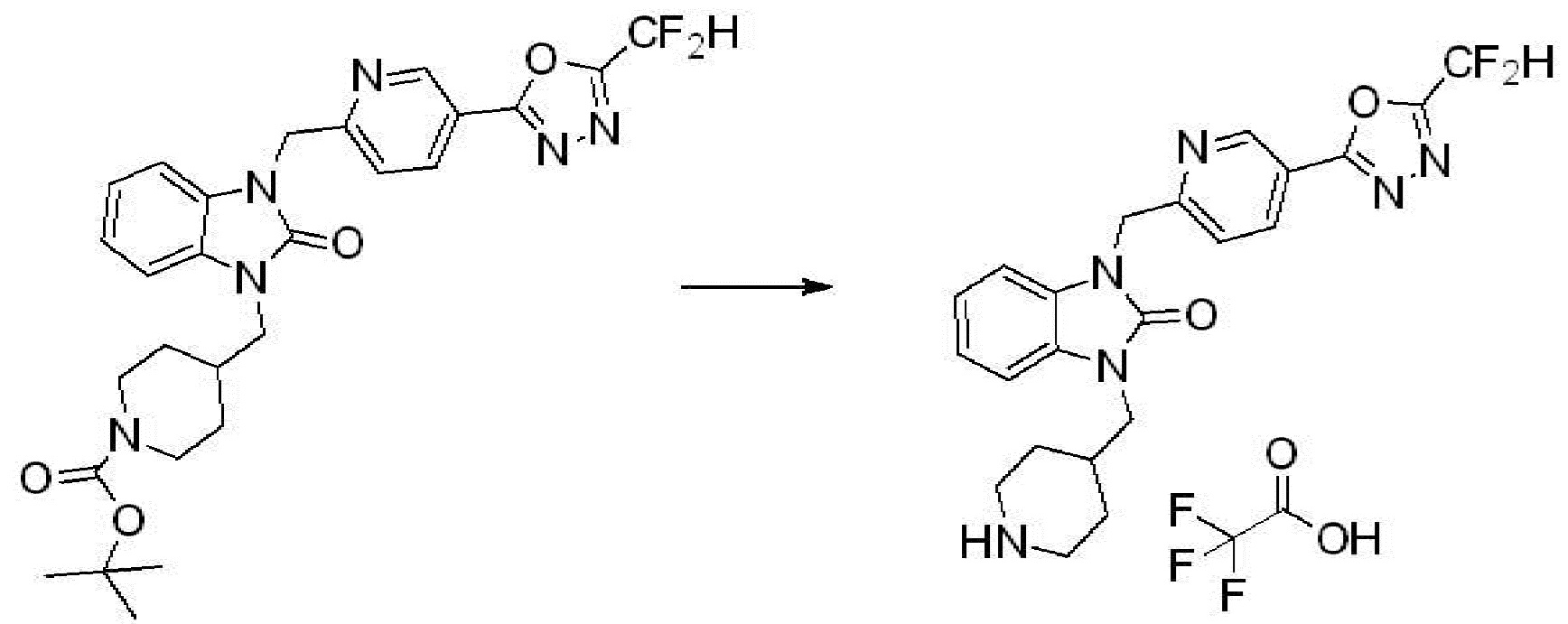
Трет-бутил 4-((3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилат (0,150 г, 0,277 ммоль), полученный на стадии 3, и трифторуксусную кислоту (0,425 мл, 5,550 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Указанное в заголовке соединение применяют без дополнительной очистки (0,150 г, 97,5%, желтое масло).
[Стадия 5] Синтез соединения 10
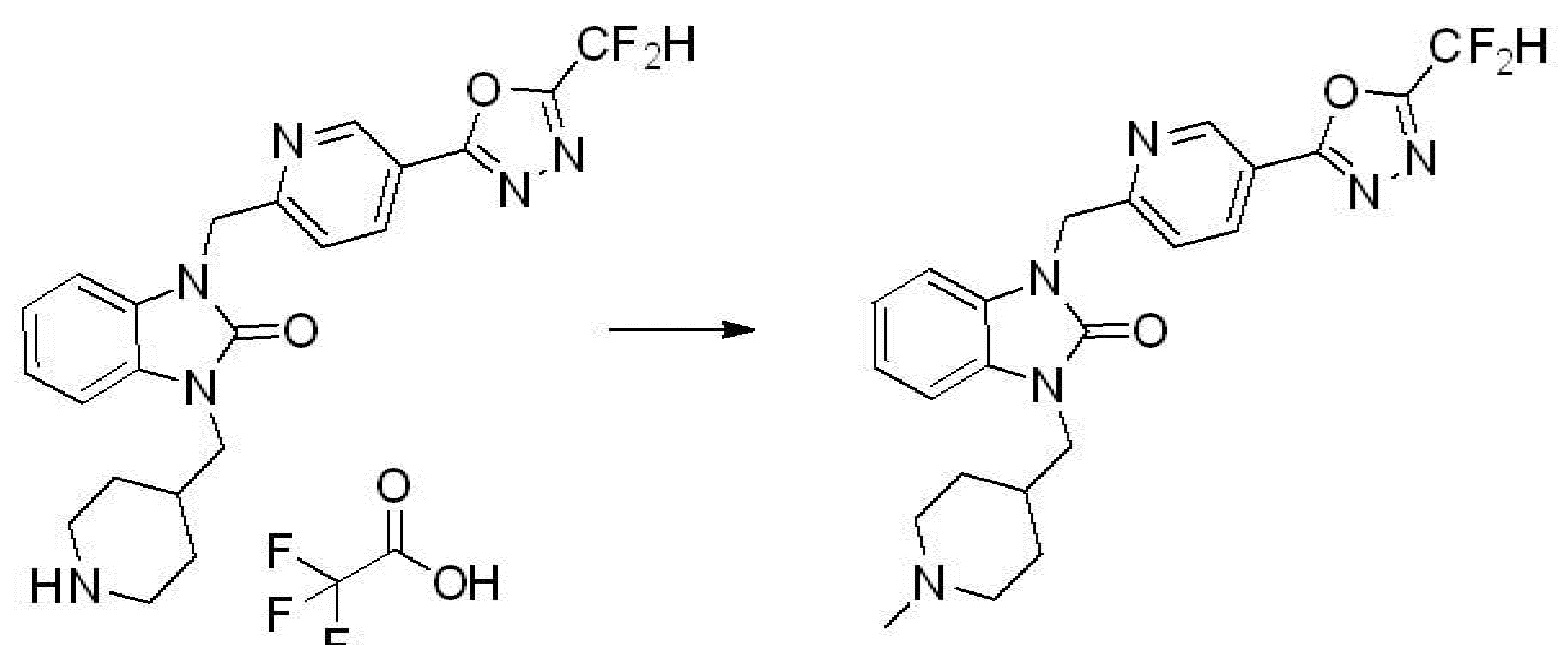
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(пиперидин-4-илметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,150 г, 0,271 ммоль), полученный на стадии 4, и N,N-диизопропилэтиламин (0,047 мл, 0,271 ммоль) растворяют в дихлорметане (10 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем формальдегид (37,00% раствор, 0,040 мл, 0,542 ммоль) и туда добавляют триацетоксиборгидрид натрия (0,115 г, 0,542 ммоль) и дополнительно перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,080 г, 65,1%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,6 Гц, 1H), 8,34 (дд, J=8,2, 2,1 Гц, 1H), 7,42 (д, J=8,0 Гц, 1H), 7,15-6,69 (м, 4H), 7,07 (с, 0,25H), 6,96 (с, 0,5H), 6,81 (с, 0,25H), 5,31 (с, 2H), 3,88 (д, J=7,2 Гц, 2H), 3,42-3,39 (м, 2H), 2,67 (с, 3H), 2,55-2,50 (м, 2H), 2,05-2,04 (м, 1H), 1,93-1,84 (м, 2H), 1,47-1,42 (м, 2H); МСНР (ЭР) m/z 455,4 (M+ + 1).
Пример 11: Синтез соединения 11, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(оксетан-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез N-(2-нитрофенил)оксетан-3-амина
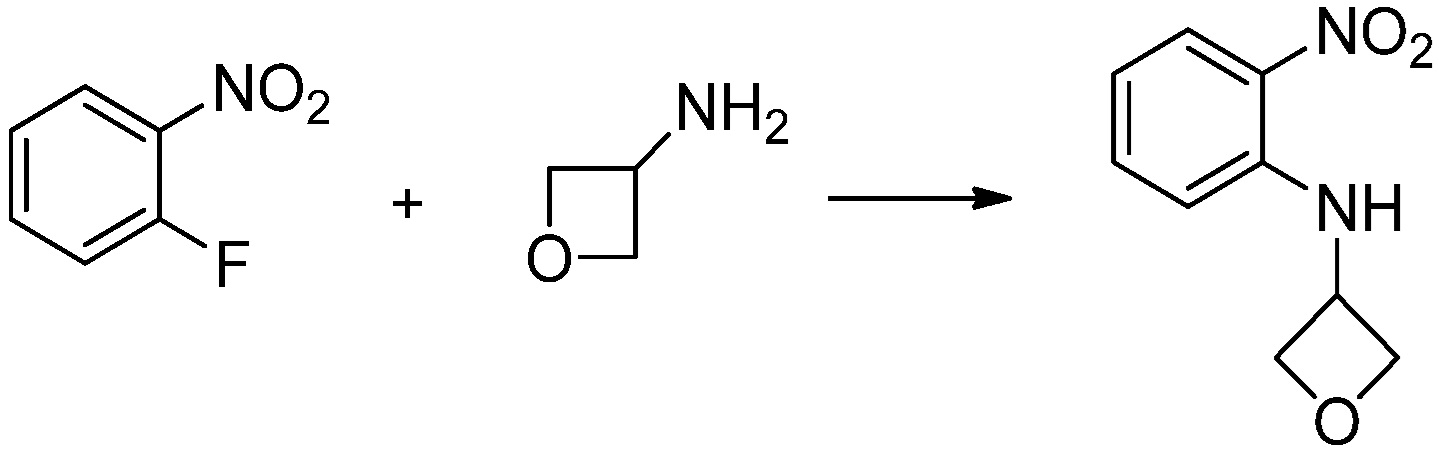
1-фтор-2-нитробензол (2,000 г, 14,174 ммоль), оксетан-3-амин (1,140 г, 15,592 ммоль) и карбонат калия (3,918 г, 28,349 ммоль) растворяют в тетрагидрофуране (30 мл) при 70°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (1,750 г, 63,6%) в виде бесцветного масла.
[Стадия 2] Синтез N1-(оксетан-3-ил)бензол-1,2-диамина
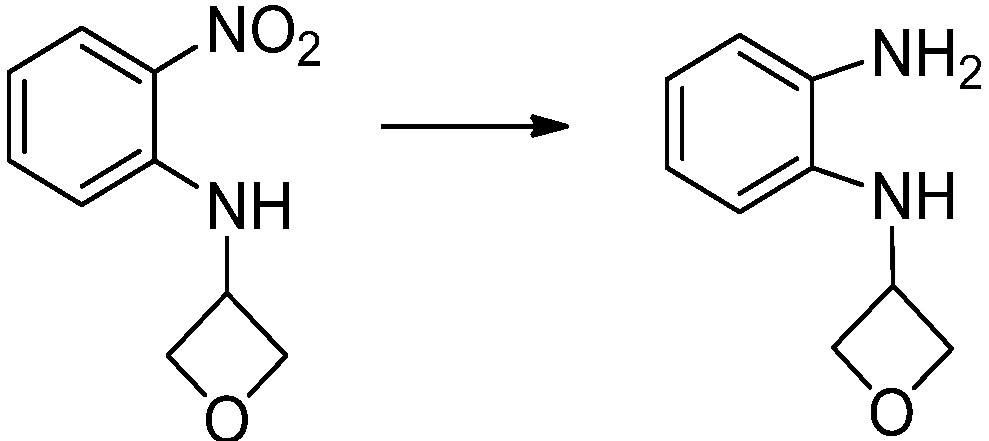
N-(2-нитрофенил)оксетан-3-амин (1,750 г, 9,012 ммоль), полученный на стадии 1, растворяют в метаноле (30 мл) при комнатной температуре, после чего туда медленно добавляют 10%-Pd/C (170 мг) и перемешивают при той же температуре в течение 12 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (1,000 г, 67,6%, желтое масло).
[Стадия 3] Синтез 1-(оксетан-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
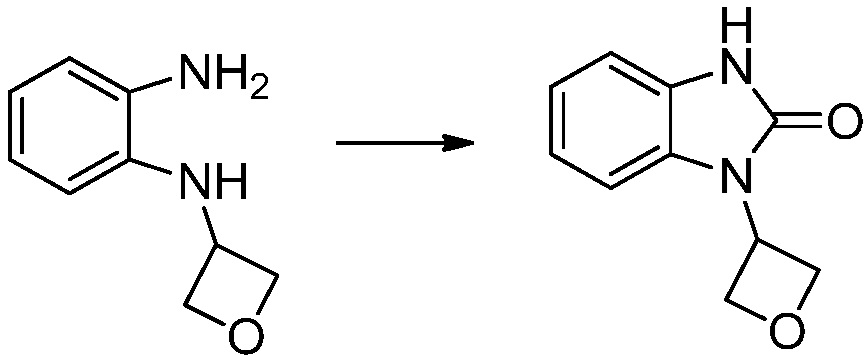
N1-(оксетан-3-ил)бензол-1,2-диамин (1,000 г, 6,090 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 1,086 г, 6,699 ммоль) растворяют в тетрагидрофуране (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,400 г, 34,5%) в виде коричневого твердого вещества.
[Стадия 4] Синтез соединения 11
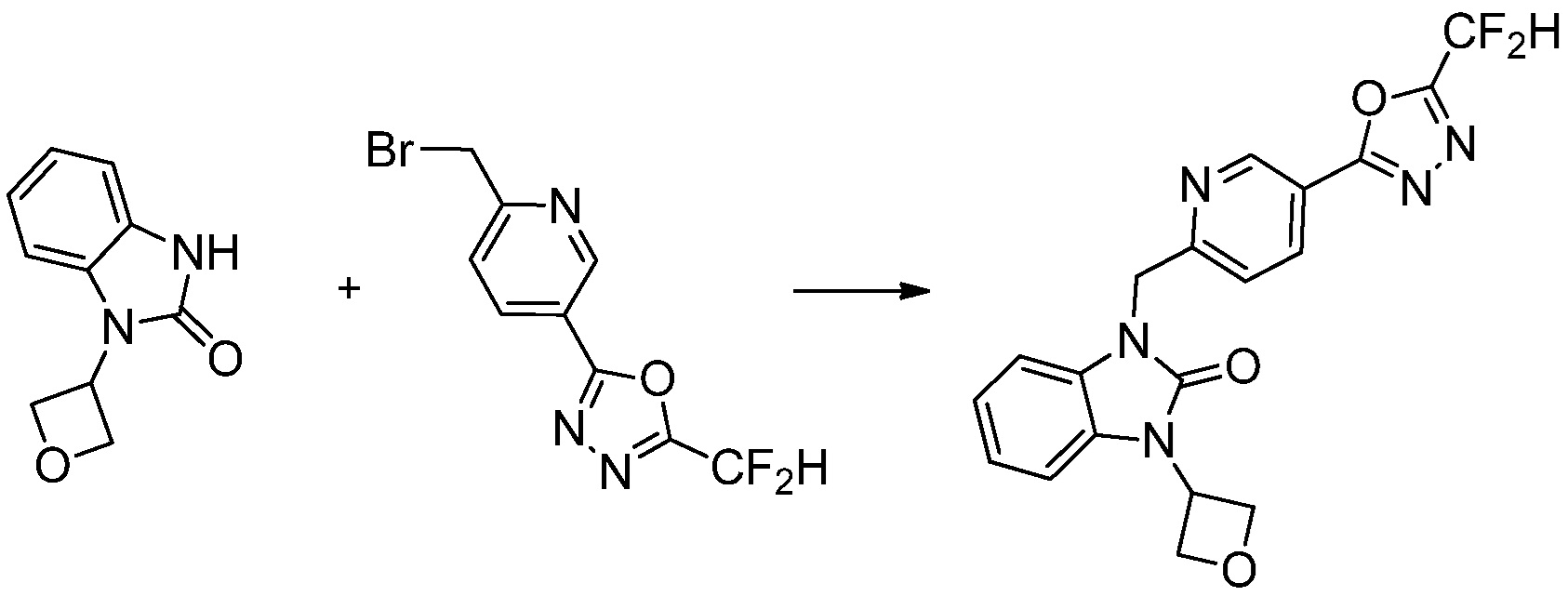
1-(оксетан-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,526 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,032 г, 0,789 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,168 г, 0,578 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 52,4%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,31-9,30 (м, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,75-7,73 (м, 1H), 7,45 (дд, J=8,2, 0,8 Гц, 1H), 7,22 (тд, J=7,7, 1,3 Гц, 1H), 7,14 (тд, J=7,7, 1,1 Гц, 1H), 7,06-7,04 (м, 1H), 7,07 (с, 0,25H), 6,94 (с, 0,5H), 6,81 (с, 0,25H), 5,77-5,70 (м, 1H), 5,30 (с, 2H), 5,26-5,23 (м, 2H), 5,18-5,15 (м, 2H); МСНР (ЭР) m/z 400,3 (M+ + 1).
Пример 12: Синтез соединения 12, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(тетрагидро-2H-пиран-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез N-(2-нитрофенил)тетрагидро-2H-пиран-4-амина
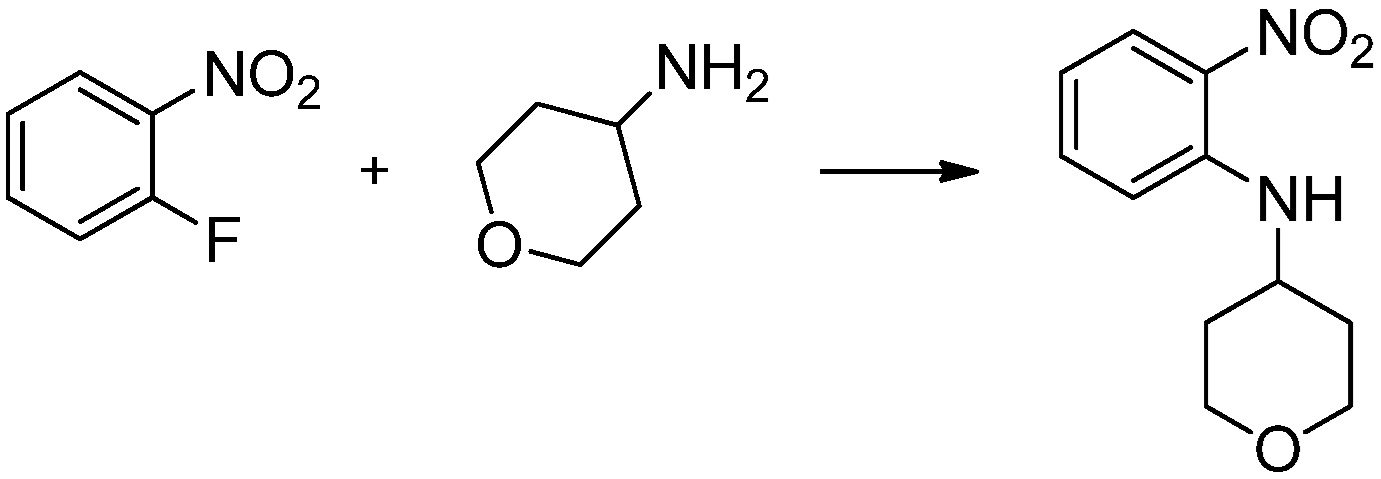
1-фтор-2-нитробензол (2,000 г, 14,174 ммоль), тетрагидро-2H-пиран-4-амин (1,577 г, 15,592 ммоль) и карбонат калия (3,918 г, 28,349 ммоль) растворяют в тетрагидрофуране (30 мл) при 70°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (1,800 г, 57,1%) в виде желтого масла.
[Стадия 2] Синтез N1-(тетрагидро-2H-пиран-4-ил)бензол-1,2-диамина
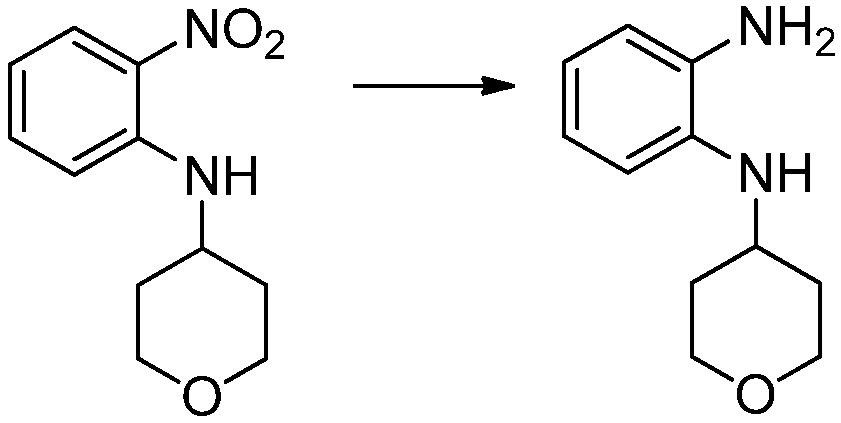
N-(2-нитрофенил)тетрагидро-2H-пиран-4-амин (1,800 г, 8,099 ммоль), полученный на стадии 1, растворяют в метаноле (30 мл) при комнатной температуре, после чего 0,1%-Pd/C (180 мг) медленно добавляют туда и перемешивают при той же температуре в течение 12 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (1,710 г, 109,8%, желтое масло).
[Стадия 3] Синтез 1-(тетрагидро-2H-пиран-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
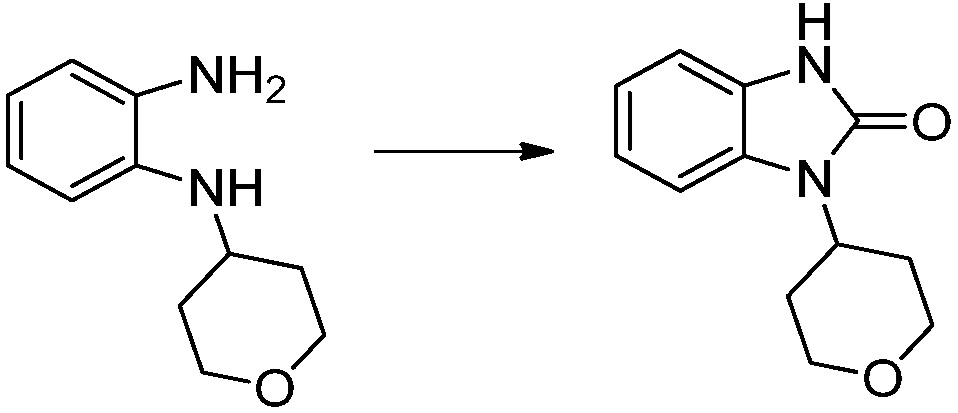
N1-(тетрагидро-2H-пиран-4-ил)бензол-1,2-диамин (1,710 г, 8,894 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 1,586 г, 9,784 ммоль) растворяют в тетрагидрофуране (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,900 г, 46,4%) в виде черного твердого вещества.
[Стадия 4] Синтез соединения 12
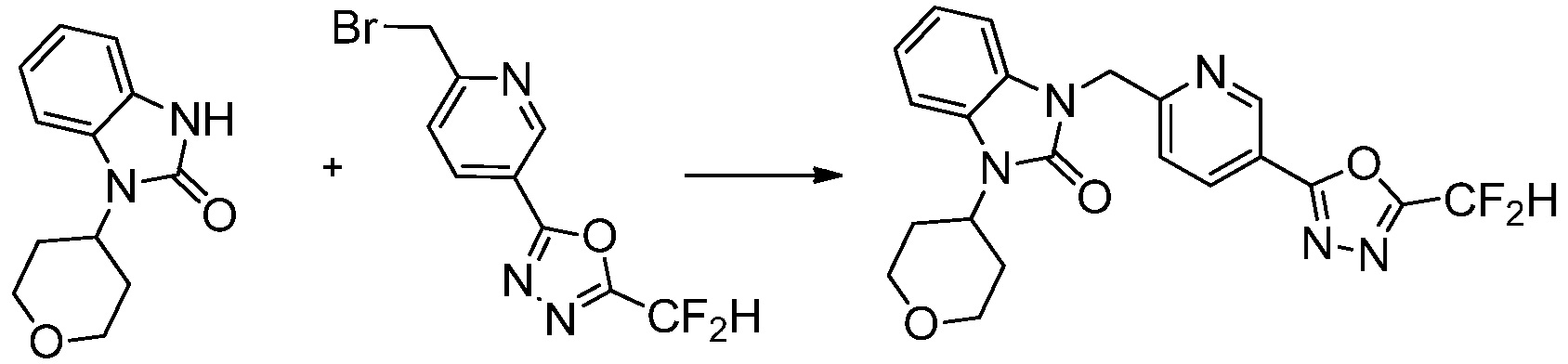
1-(тетрагидро-2H-пиран-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,458 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,027 г, 0,687 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,146 г, 0,504 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = 0-70%) и концентрируют с получением указанного в заголовке соединения (0,080 г, 40,9%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (дд, J=2,2, 0,8 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,41-7,39 (м, 1H), 7,28-7,26 (м, 1H), 7,11-7,00 (м, 3H), 7,07 (с, 0,25H), 6,94 (с, 0,5H), 6,81 (с, 0,25H), 5,30 (с, 2H), 4,68-4,61 (м, 1H), 4,17-4,13 (м, 2H), 3,61-3,54 (м, 2H), 2,59-2,48 (м, 2H), 1,84-1,80 (м, 2H); МСНР (ЭР) m/z 428,3 (M+ + 1).
Пример 13: Синтез соединения 13, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез 4-((2-нитрофенил)амино)тетрагидро-2H-тиопиран 1,1-диоксида
1-фтор-2-нитробензол (1,710 г, 12,119 ммоль), 4-аминотетрагидро-2H-тиопиран 1,1-диоксид (1,808 г, 12,119 ммоль) и триэтиламин (3,378 мл, 24,238 ммоль) растворяют в N,N-диметилформамиде (30 мл) at 90°C, после чего полученный раствор перемешивают при той же температуре в течение 24 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Этилацетат (20 мл) и гексан (10 мл) добавляют в полученный концентрат и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают гексаном, и затем сушат с получением указанного в заголовке соединения (1,730 г, 52,8%) в виде желтого твердого вещества.
[Стадия 2] Синтез 4-((2-аминофенил)амино)тетрагидро-2H-тиопиран 1,1-диоксида
4-((2-нитрофенил)амино)тетрагидро-2H-тиопиран 1,1-диоксид (1,730 г, 8,268 ммоль), полученный на стадии 1, растворяют в метаноле (30 мл) при комнатной температуре, после чего туда медленно добавляют 10%-Pd/C (170 мг) и перемешивают при той же температуре в течение 12 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (0,310 г, 15,6%, белое твердое вещество).
[Стадия 3] Синтез 1-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
4-((2-аминофенил)амино)тетрагидро-2H-тиопиран 1,1-диоксид (0,310 г, 1,290 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 0,230 г, 1,419 ммоль) растворяют в тетрагидрофуране (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (0,220 г, 64,0%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 13
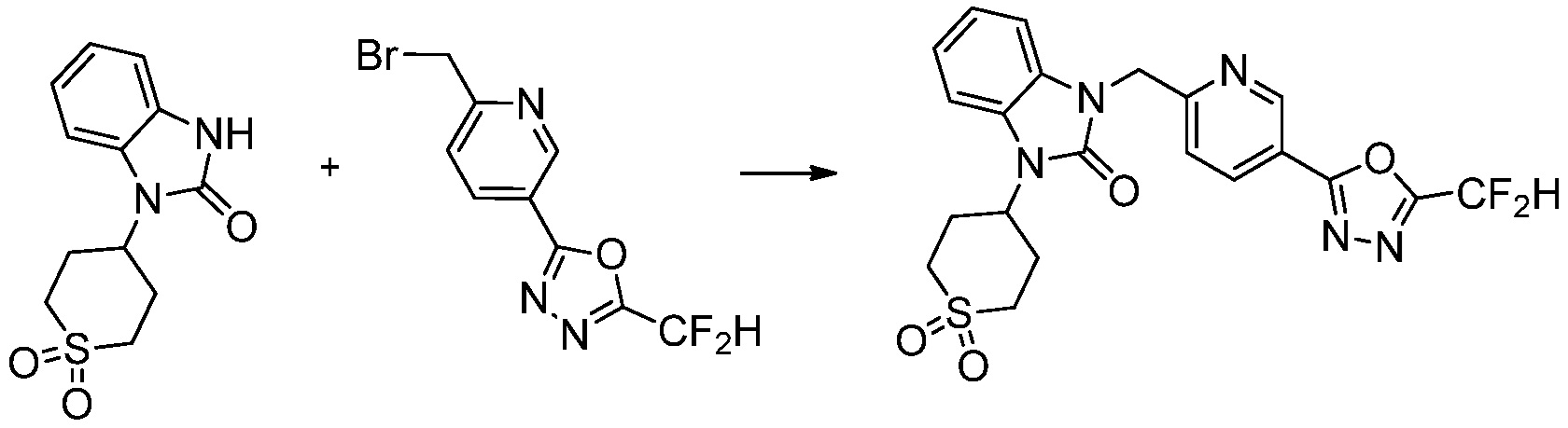
1-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,375 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,023 г, 0,563 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,120 г, 0,413 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0 to 70%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 56,0%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,2, 0,8 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (дд, J=8,2, 0,5 Гц, 1H), 7,29-7,27 (м, 1H), 7,12-7,01 (м, 3H), 7,07 (с, 1H), 6,94 (с, 1H), 6,81 (с, 1H), 5,28 (с, 2H), 3,32-3,08 (м, 6H), 2,28-2,24 (м, 2H); МСНР (ЭР) m/z 476,4 (M+ + 1).
Пример 14: Синтез соединения 14, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-этилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 14
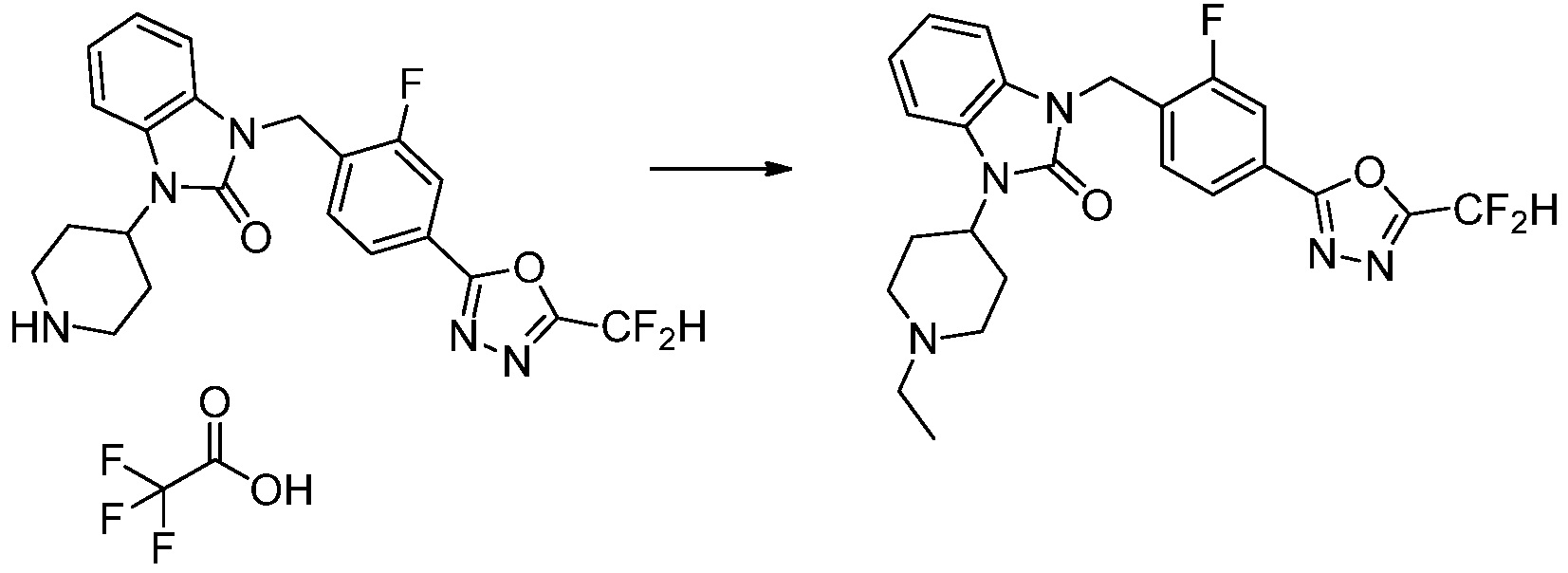
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,323 г, 0,579 ммоль), ацетальдегид (0,051 г, 1,159 ммоль), N,N-диизопропилэтиламин (0,101 мл, 0,579 ммоль) и триацетоксиборгидрид натрия (0,246 г, 1,159 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 8 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,180 г, 65,9%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,85-7,82 (м, 2H), 7,48-7,42 (м, 1H), 7,36-7,32 (м, 1H), 7,10-7,07 (м, 2H), 7,05-7,02 (м, 1H), 7,07 (с, 1H), 6,94 (с, 1H), 6,79 (с, 1H), 5,21 (с, 2H), 4,51-4,46 (м, 1H), 3,17-3,14 (м, 2H), 2,52-2,49 (м, 4H), 2,19-2,13 (м, 2H), 1,90-1,87 (м, 2H), 1,17-1,14 (м, 3H).
Пример 15: Синтез соединения 15, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-изопропилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 15
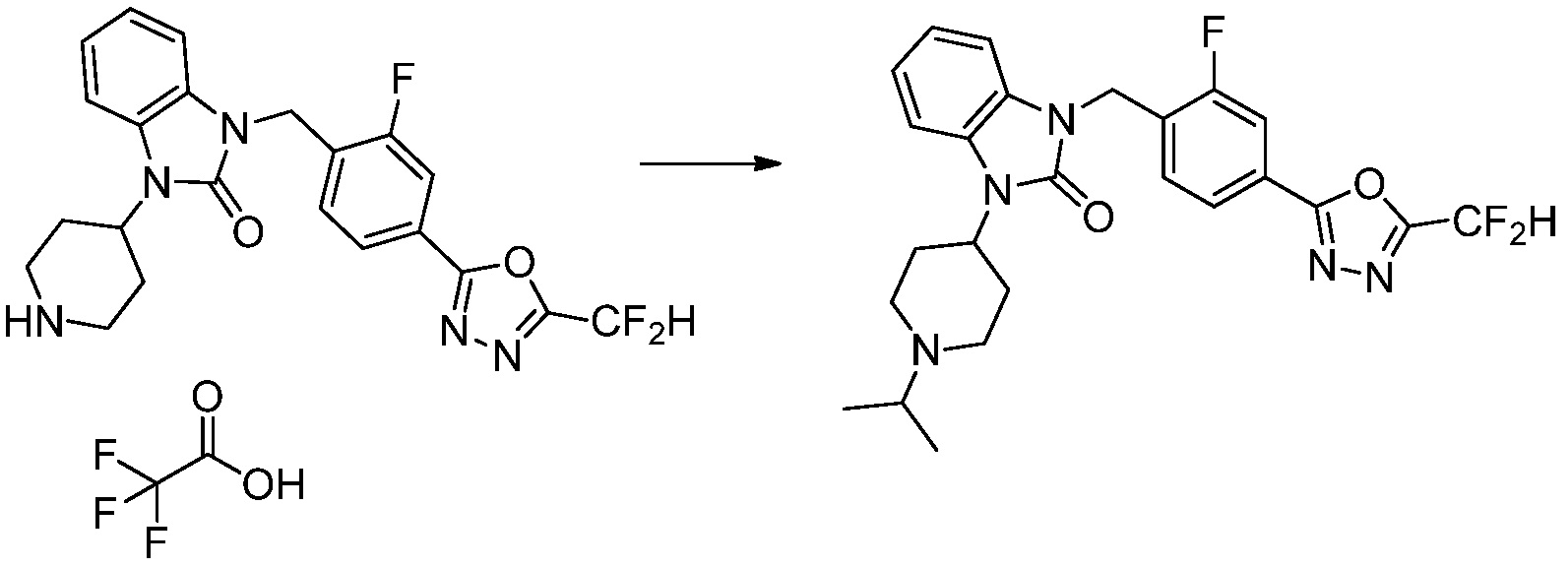
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,300 г, 0,538 ммоль), ацетон (0,080 мл, 1,076 ммоль), N,N-диизопропилэтиламин (0,094 мл, 0,538 ммоль) и триацетоксиборгидрид натрия (0,228 г, 1,076 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 8 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,200 г, 76,5%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,88-7,83 (м, 2H), 7,50-7,43 (м, 2H), 7,13-7,05 (м, 2H), 6,98-6,95 (м, 1H), 7,07 (с, 0,25H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,21 (с, 2H), 4,60-4,58 (м, 1H), 3,32-3,29 (м, 3H), 2,70-2,62 (м, 4H), 1,96-1,93 (м, 2H), 1,29-1,23 (м, 6H); МСНР (ЭР) m/z 486,3 (M+ + 1).
Пример 16: Синтез соединения 16, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 16
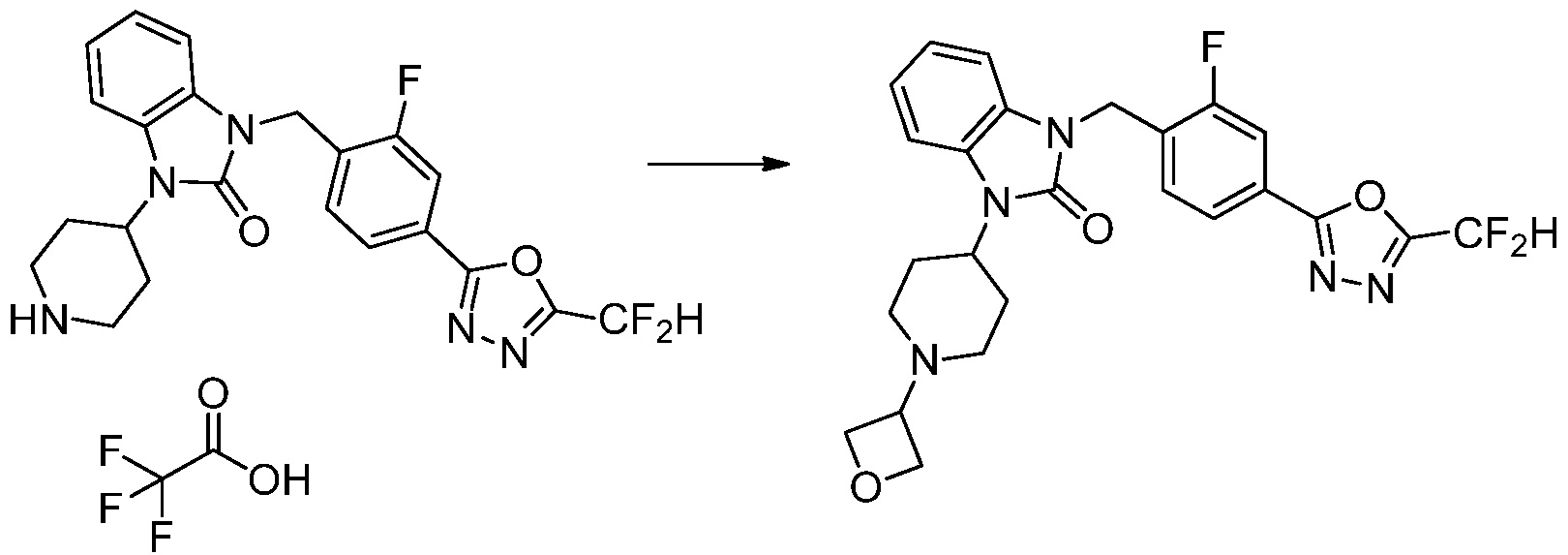
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,300 г, 0,538 ммоль), оксетан-3-он (0,078 г, 1,076 ммоль), N,N-диизопропилэтиламин (0,094 мл, 0,538 ммоль) и триацетоксиборгидрид натрия (0,228 г, 1,076 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 8 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,180 г, 67,0%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,85-7,81 (м, 2H), 7,46-7,42 (м, 1H), 7,36-7,31 (м, 1H), 7,20-7,05 (м, 2H), 6,98-6,90 (м, 1H), 7,07 (с, 0,25H), 6,91 (с, 0,5H), 6,78 (с, 0,25H), 5,20 (с, 2H), 4,71-4,64 (м, 4H), 4,50-4,46 (м, 1H), 3,57-3,54 (м, 1H), 2,95-2,92 (м, 2H), 2,54-2,49 (м, 2H), 2,05-2,02 (м, 2H), 1,89-1,86 (м, 2H); МСНР (ЭР) m/z 500,4 (M+ + 1).
Пример 17: Синтез соединения 17, 1-(1-ацетилпиперидин-4-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 17
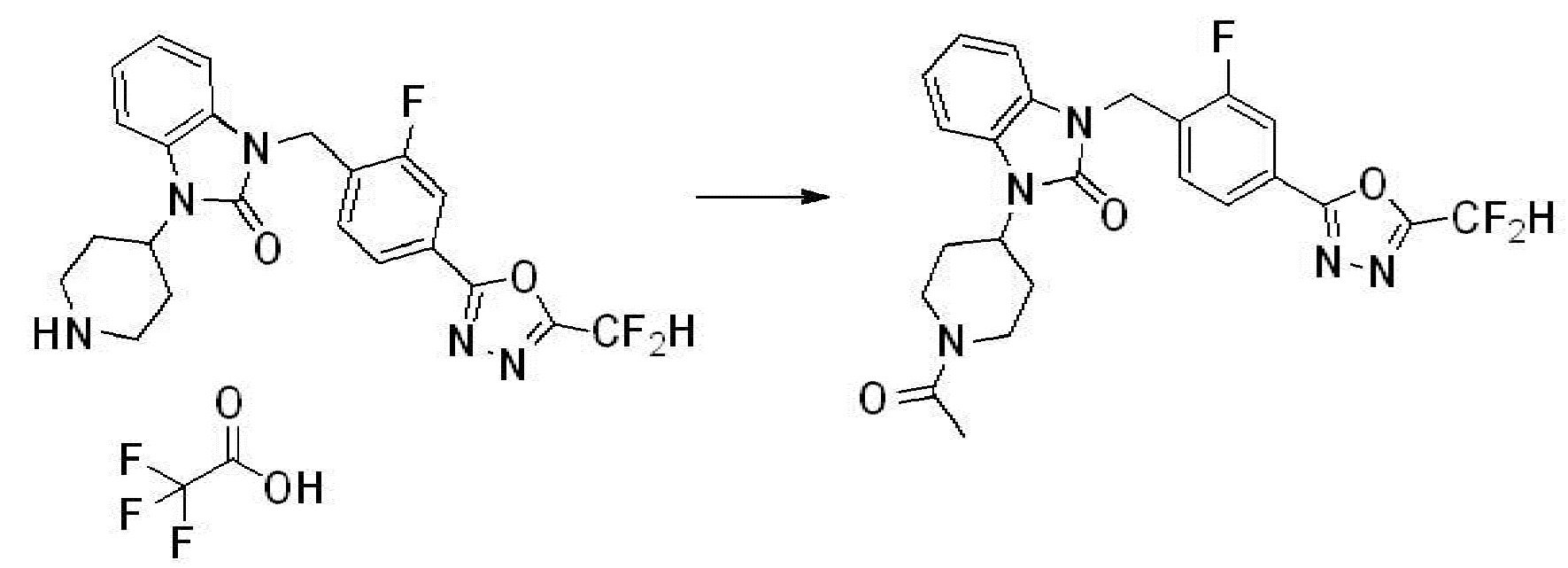
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,300 г, 0,538 ммоль), ацетилхлорид (0,077 мл, 1,076 ммоль), и N,N-диизопропилэтиламин (0,187 мл, 1,076 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 8 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 42,1%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,90-7,83 (м, 2H), 7,48-7,44 (м, 1H), 7,16-7,11 (м, 3H), 7,09-7,04 (м, 1H), 7,07 (с, 1H), 6,92 (с, 1H), 6,79 (с, 1H), 5,20 (с, 2H), 4,91-4,87 (м, 1H), 4,65-4,60 (м, 1H), 4,15-4,11 (м, 1H), 3,30-3,26 (м, 1H), 2,73-2,69 (м, 1H), 2,40-2,32 (м, 2H), 2,18 (с, 3H), 1,98-1,94 (м, 2H); МСНР (ЭР) m/z 486,3 (M+ + 1).
Пример 18: Синтез соединения 18, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 18
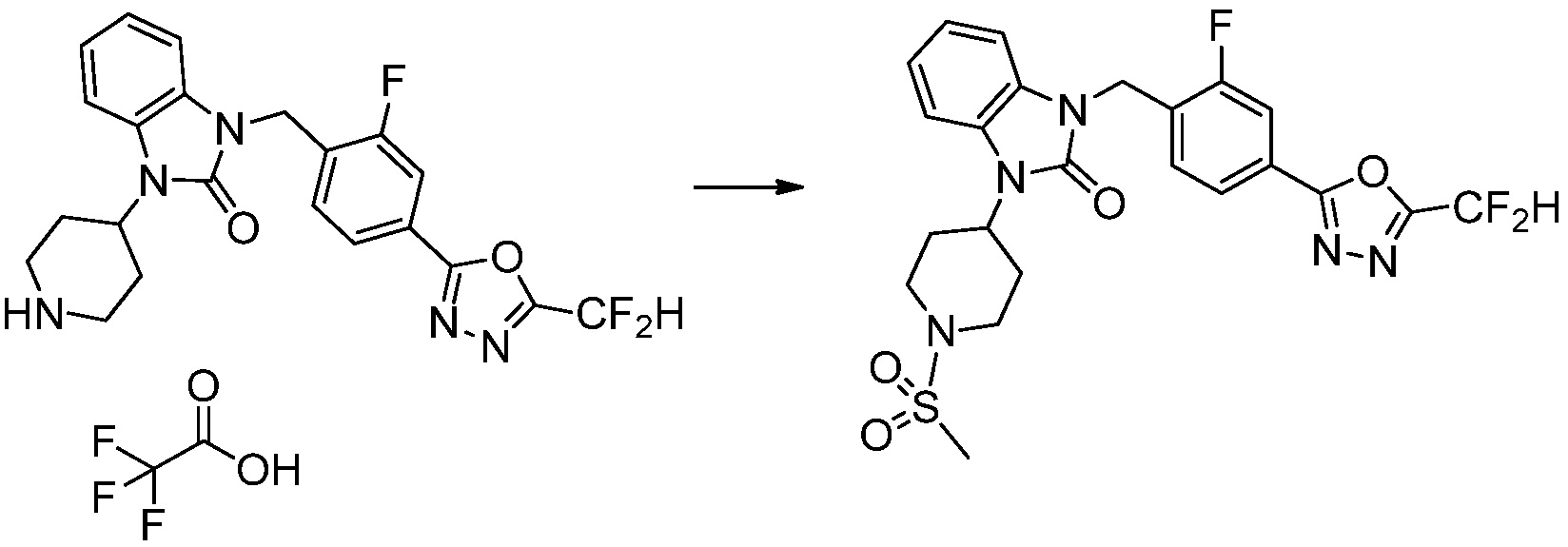
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,300 г, 0,538 ммоль), метансульфонилхлорид (0,083 мл, 1,076 ммоль) и N,N-диизопропилэтиламин (0,187 мл, 1,076 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 8 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,150 г, 53,4%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,90-7,84 (м, 2H), 7,48-7,44 (м, 1H), 7,26-7,24 (м, 1H), 7,15-7,06 (м, 2H), 7,07 (с, 0,25H), 7,06-7,01 (м, 1H), 6,91 (с, 0,5H), 6,79 (с, 0,25H), 5,22 (с, 2H), 4,60-4,56 (м, 1H), 4,07-4,04 (м, 2H), 2,94-2,87 (м, 5H), 2,61-2,56 (м, 2H), 2,06-1,98 (м, 2H); МСНР (ЭР) m/z 507,2 (M+ + 1).
Пример 19: Синтез соединения 19, 1-(1-((1H-индол-7-ил)метил)пиперидин-4-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 19
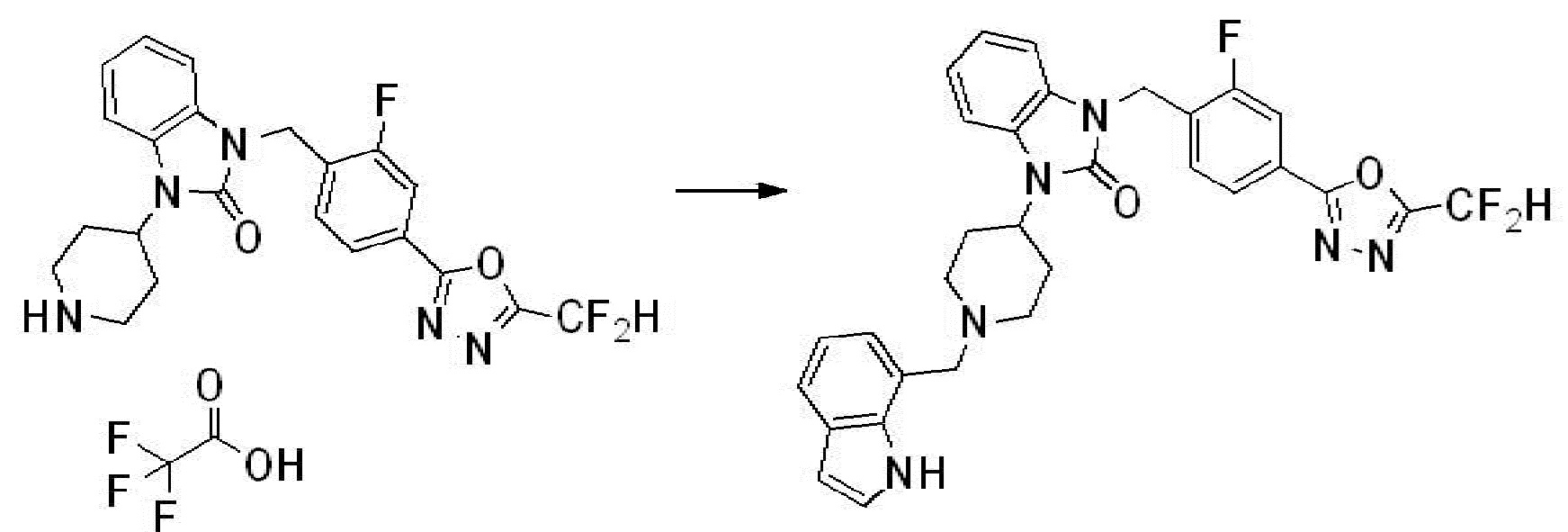
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,200 г, 0,359 ммоль), 1H-индол-7-карбальдегид (0,078 г, 0,538 ммоль), N,N-диизопропилэтиламин (0,062 мл, 0,359 ммоль) и триацетоксиборгидрид натрия (0,152 г, 0,718 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 8 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 53,5%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,80 (шс, 1H), 7,88-7,83 (м, 2H), 7,62 (д, J=7,6 Гц, 1H), 7,49-7,46 (м, 1H), 7,33-7,32 (м, 1H), 7,26-7,23 (м, 1H), 7,16-6,98 (м, 5H), 7,07 (с, 0,25H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 6,59-6,57 (м, 1H), 5,21 (с, 2H), 4,39-4,37 (м, 1H), 3,96 (с, 2H), 3,19-3,17 (м, 2H), 2,64-2,60 (м, 2H), 2,07-2,02 (м, 2H), 1,91-1,88 (м, 2H); МСНР (ЭР) m/z 574,4 (M+ + 1).
Пример 20: Синтез соединения 20, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез бензил 3-(3-(2-фтор-4-(метоксикарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилата
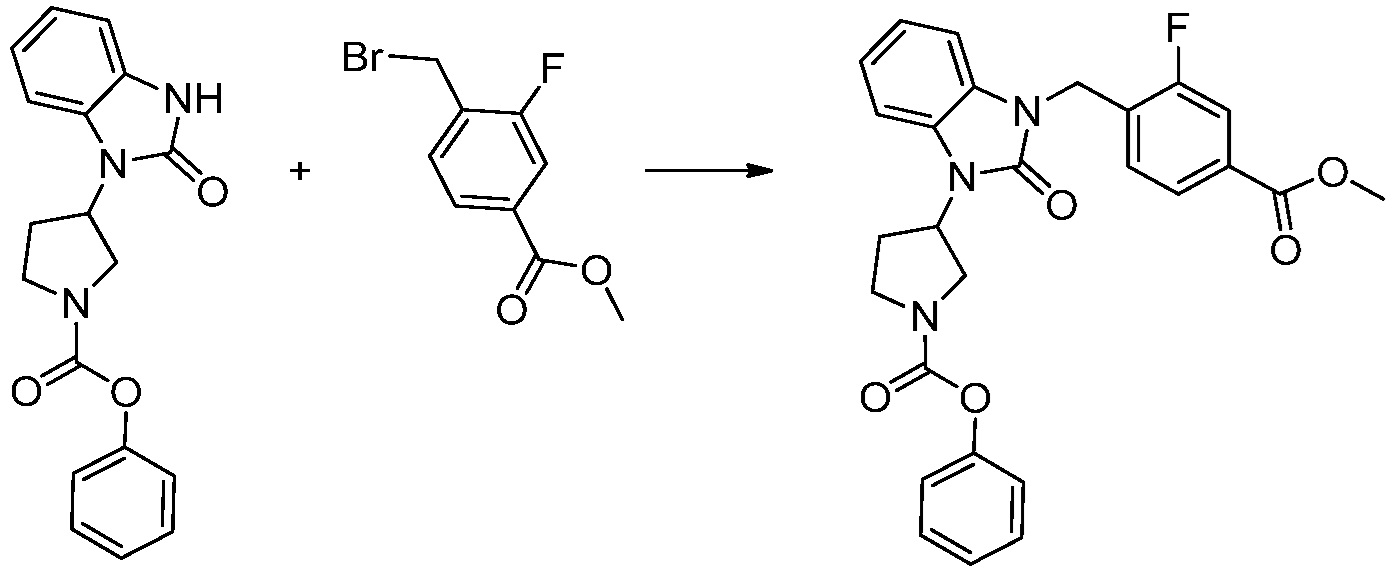
Бензил 3-(2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (0,580 г, 1,719 ммоль) растворяют в N,N-диметилформамиде (30 мл) при 0°C, после чего гидрид натрия (60,00%, 0,103 г, 2,579 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. Метил 4-(бромметил)-3-фторбензоат (0,425 г, 1,719 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,780 г, 90,1%) в виде бесцветного масла.
[Стадия 2] Синтез бензил 3-(3-(2-фтор-4-(гидразинкарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилата
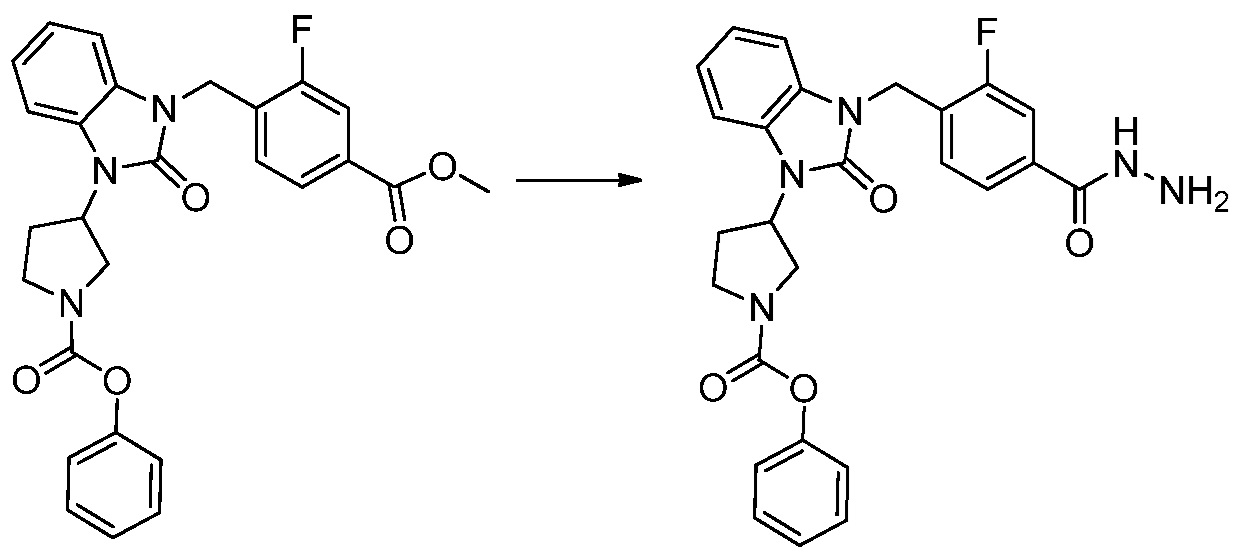
Бензил 3-(3-(2-фтор-4-(метоксикарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (0,780 г, 1,549 ммоль), полученный на стадии 1, и моногидрат гидразина (1,506 мл, 30,981 ммоль) растворяют в этаноле (10 мл) при 80°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,780 г, 100,0%, бесцветное масло).
[Стадия 3] Синтез бензил 3-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилата
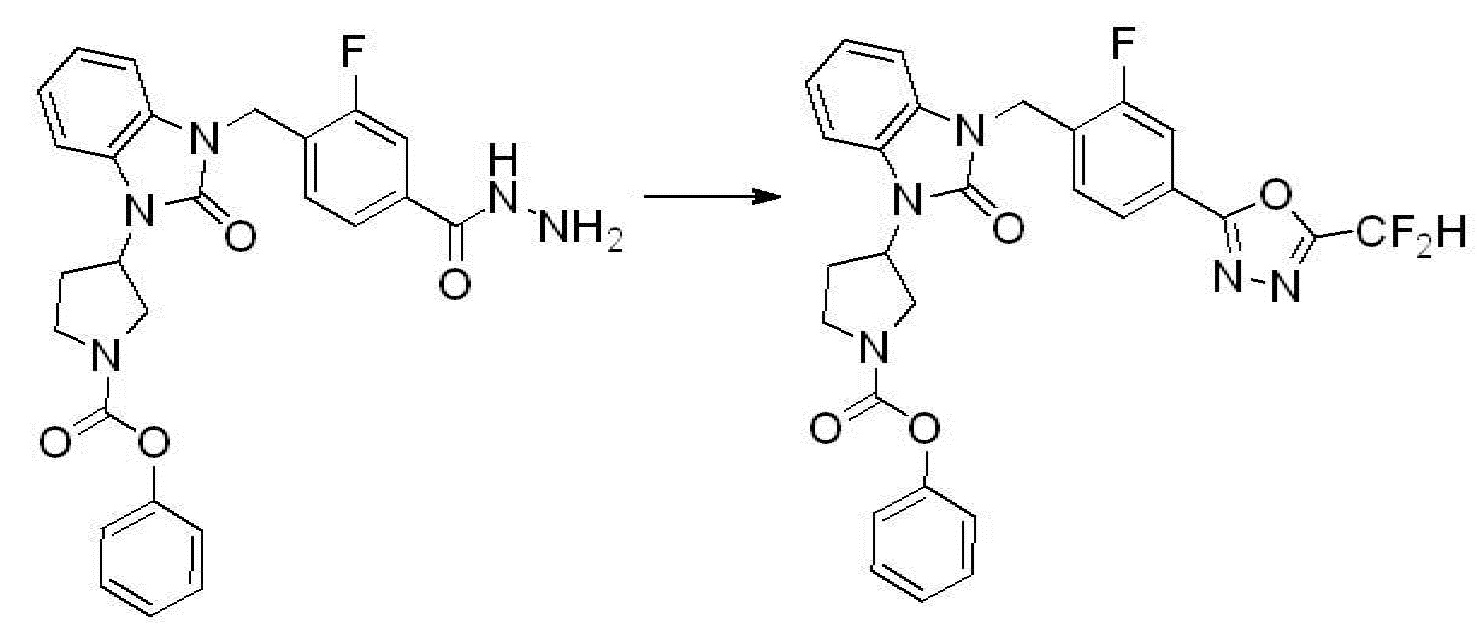
Бензил 3-(3-(2-фтор-4-(гидразинкарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (0,780 г, 1,549 ммоль), полученный на стадии 2, 2,2-дифторуксусный ангидрид (0,578 мл, 4,647 ммоль) и имидазол (0,316 г, 4,647 ммоль) растворяют в дихлорметане (30 мл) при 45°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,700 г, 80,2%) в виде бесцветного масла.
[Стадия 4] Синтез 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
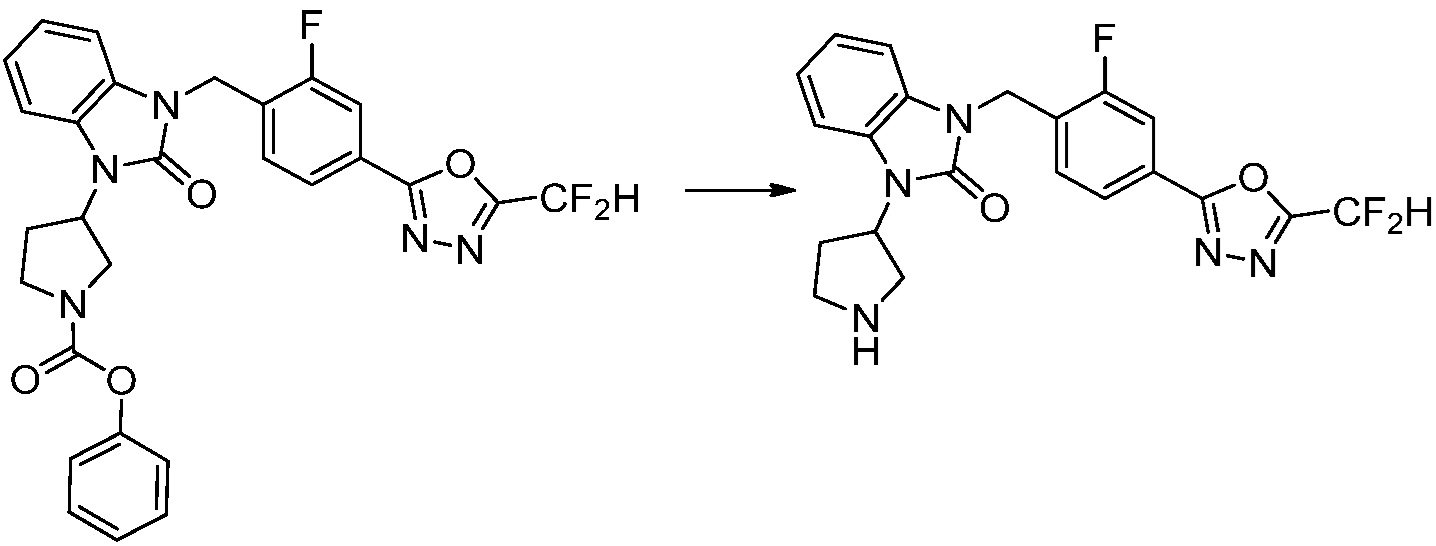
Бензил 3-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (0,700 г, 1,242 ммоль), полученный на стадии 3, растворяют в метаноле (20 мл) при комнатной температуре, после чего туда медленно добавляют 10%-Pd/C (70 мг) и перемешивают при той же температуре в течение 12 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (0,500 г, 93,7%, белое твердое вещество).
[Стадия 5] Синтез соединения 20
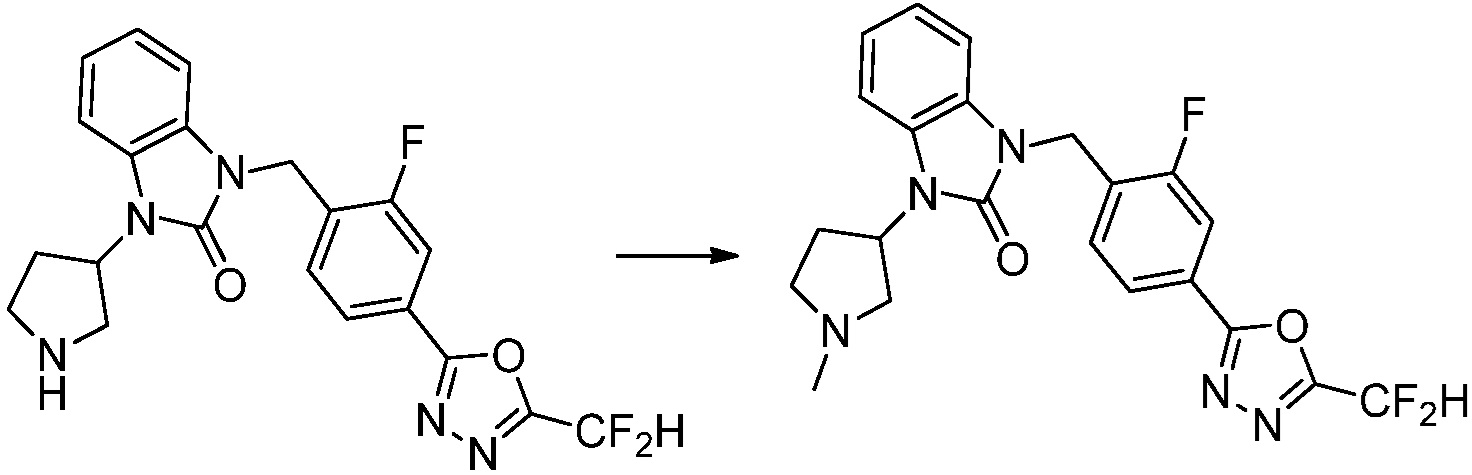
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,190 г, 0,442 ммоль), полученный на стадии 4, формальдегид (0,027 г, 0,885 ммоль) и триацетоксиборгидрид натрия (0,188 г, 0,885 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 56,1%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,83 (м, 2H), 7,60-7,58 (м, 1H), 7,46 (т, J=7,8 Гц, 1H), 7,14-7,05 (м, 2H), 6,97-6,92 (м, 1H), 7,07 (с, 0,25H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,26-5,23 (м, 1H), 5,20 (с, 2H), 3,17-3,13 (м, 2H), 2,98-2,93 (м, 1H), 2,73-2,68 (м, 1H), 2,51 (с, 3H), 2,38-2,31 (м, 2H); МСНР (ЭР) m/z 444,4 (M+ + 1).
Пример 21: Синтез соединения 21, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-(оксетан-3-ил)пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 21
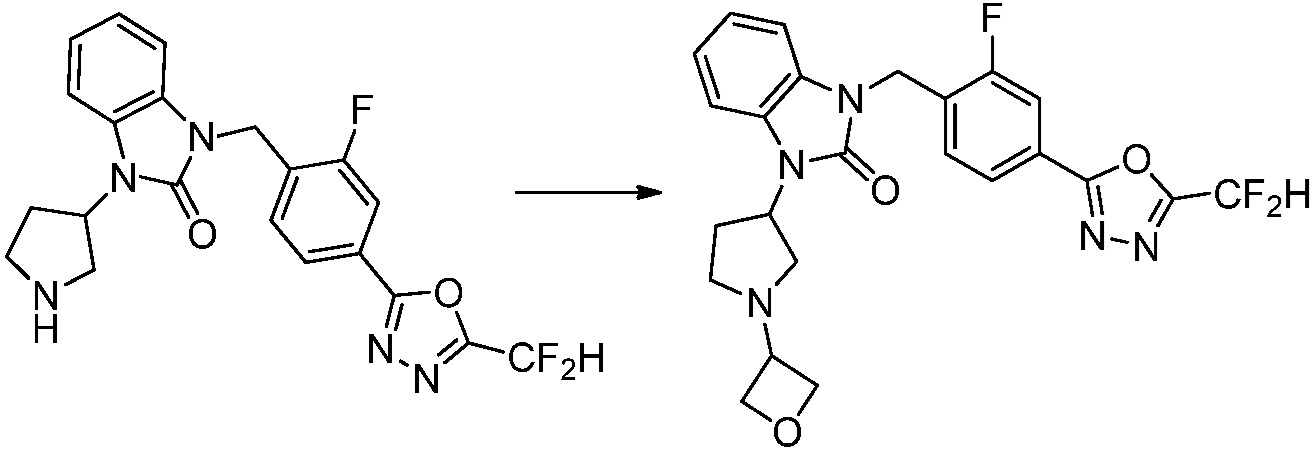
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,210 г, 0,489 ммоль), оксетан-3-он (0,070 г, 0,978 ммоль) и триацетоксиборгидрид натрия (0,207 г, 0,978 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 42,1%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,88-7,77 (м, 3H), 7,49-7,45 (м, 1H), 7,18-7,05 (м, 2H), 6,99-6,97 (м, 1H), 7,07 (с, 0,25H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,30-5,28 (м, 1H), 5,21 (с, 2H), 4,78-4,65 (м, 4H), 3,73-3,70 (м, 1H), 3,12-3,08 (м, 2H), 2,71-2,66 (м, 1H), 2,42-2,28 (м, 3H); МСНР (ЭР) m/z 486,4 (M+ + 1).
Пример 22: Синтез соединения 22, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилазетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 3-(3-(2-фтор-4-(метоксикарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилата
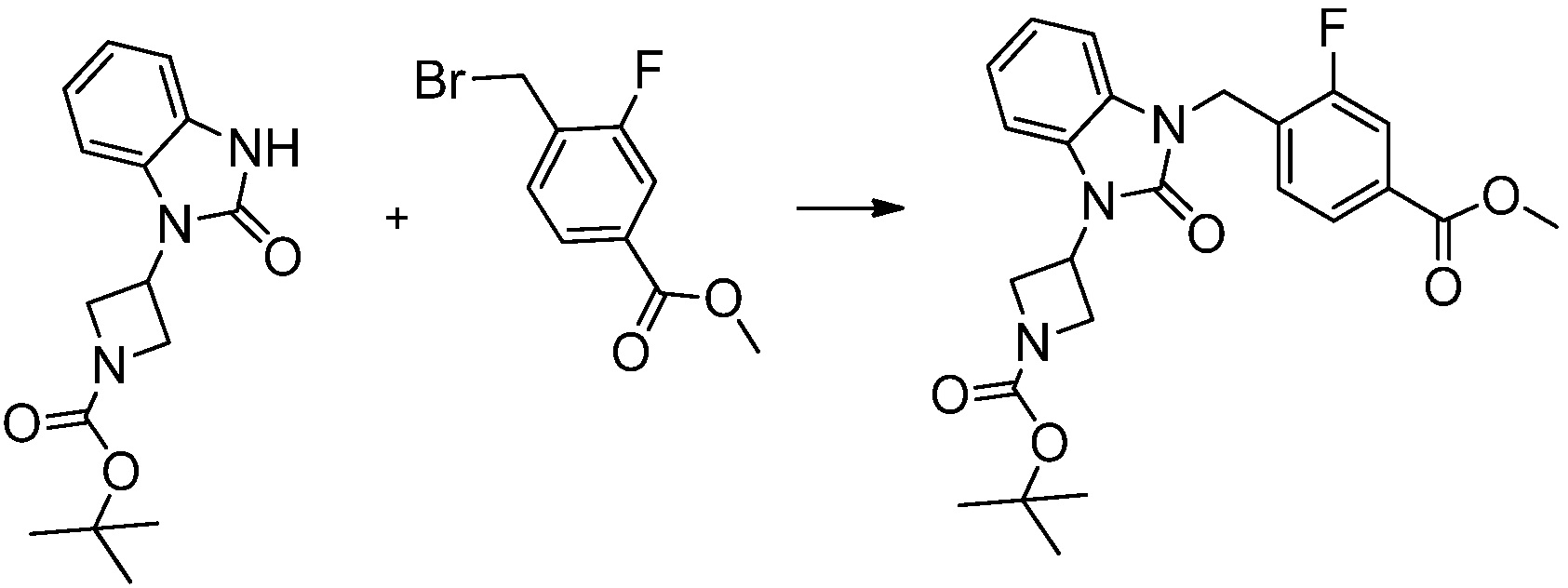
Трет-бутил 3-(2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилат (0,570 г, 1,970 ммоль) растворяют в N,N-диметилформамиде (30 мл) при 0°C, после чего гидрид натрия (60,00%, 0,118 г, 2,955 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. Метил 4-(бромметил)-3-фторбензоат (0,487 г, 1,970 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,610 г, 68,0%) в виде бесцветного масла.
[Стадия 2] Синтез трет-бутил 3-(3-(2-фтор-4-(гидразинкарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилата
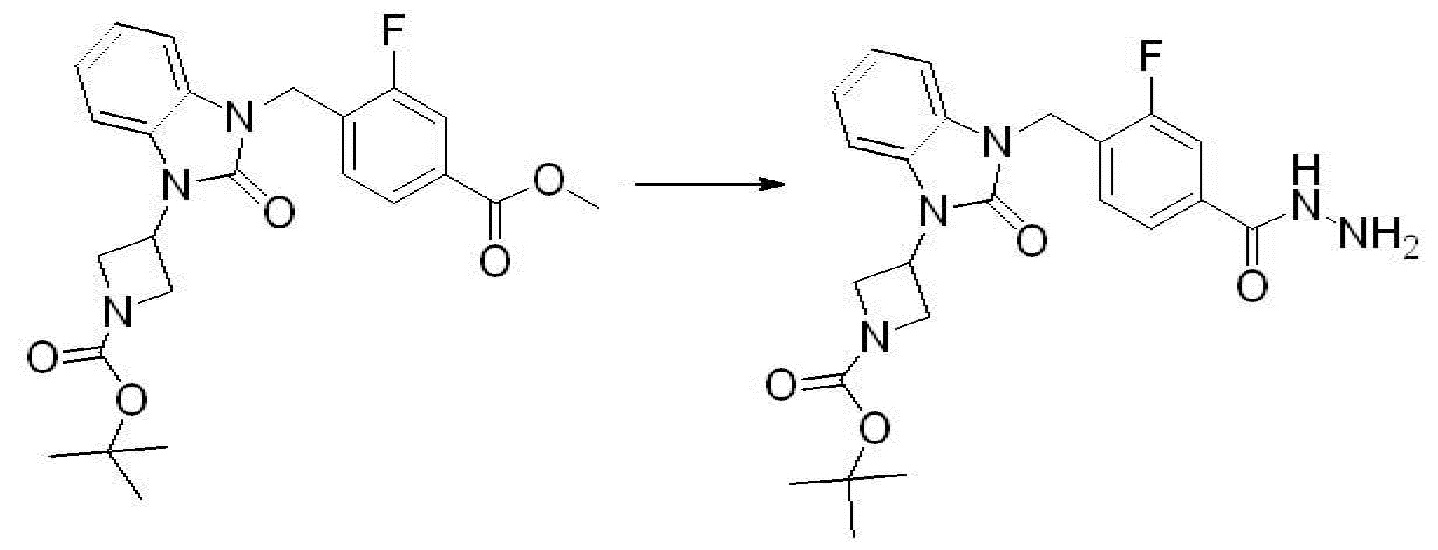
Трет-бутил 3-(3-(2-фтор-4-(метоксикарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилат (0,610 г, 1,339 ммоль), полученный на стадии 1, и моногидрат гидразина (1,302 мл, 26,784 ммоль) растворяют в этаноле (10 мл) при 80°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,610 г, 100,0%, бесцветное масло).
[Стадия 3] Синтез трет-бутил 3-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилата
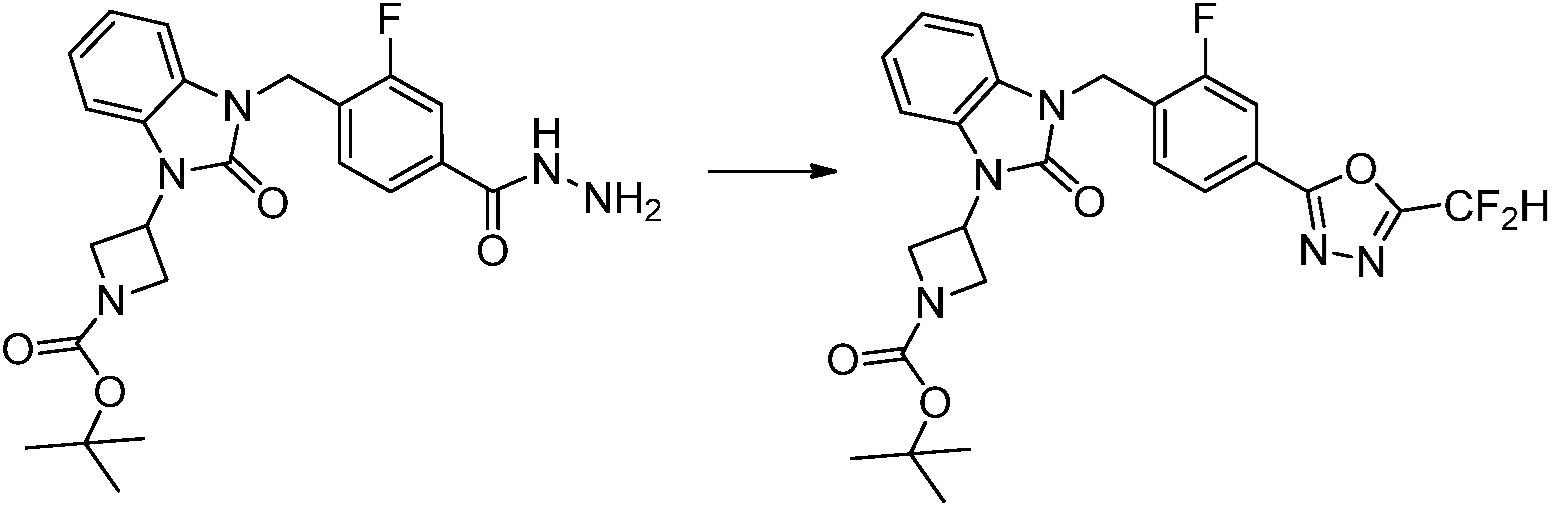
Трет-бутил 3-(3-(2-фтор-4-(гидразинкарбонил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилат (0,610 г, 1,339 ммоль), полученный на стадии 2, 2,2-дифторуксусный ангидрид (0,499 мл, 4,018 ммоль) и имидазол (0,274 г, 4,018 ммоль) растворяют в дихлорметане (30 мл) при 45°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,600 г, 86,9%) в виде бесцветного масла.
[Стадия 4] Синтез 1-(азетидин-3-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетата
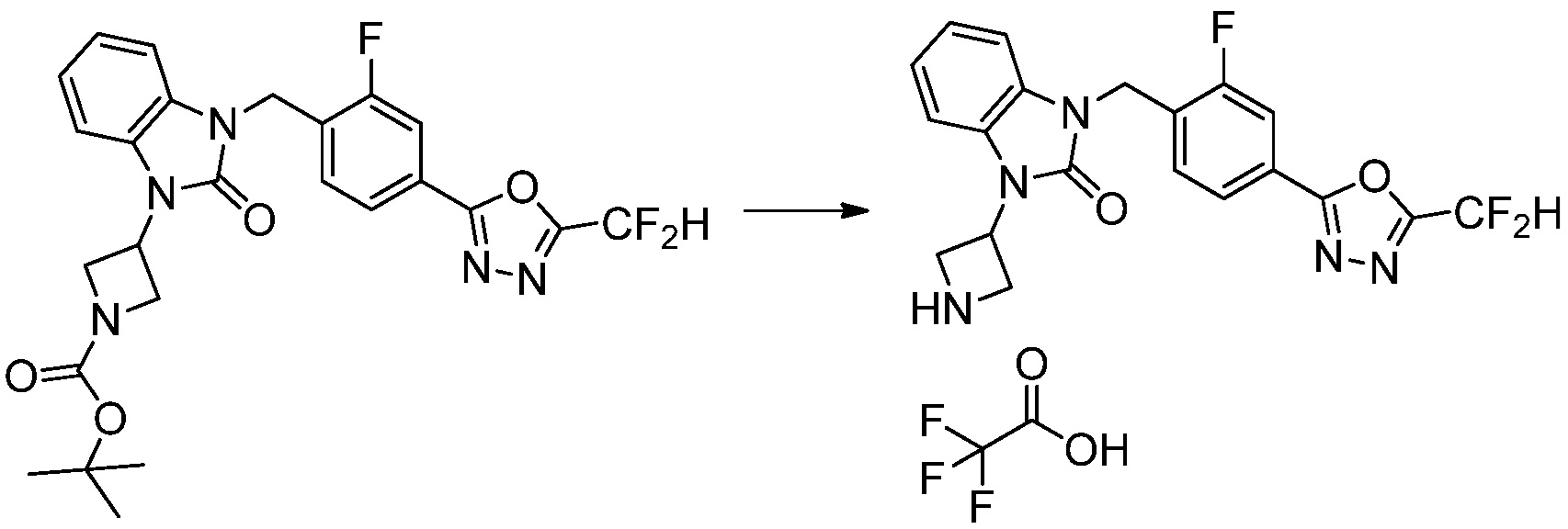
Трет-бутил 3-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилат (0,600 г, 1,164 ммоль), полученный на стадии 3, и трифторуксусную кислоту (1,783 мл, 23,279 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,600 г, 97,4%, желтое масло).
[Стадия 5] Синтез соединения 22
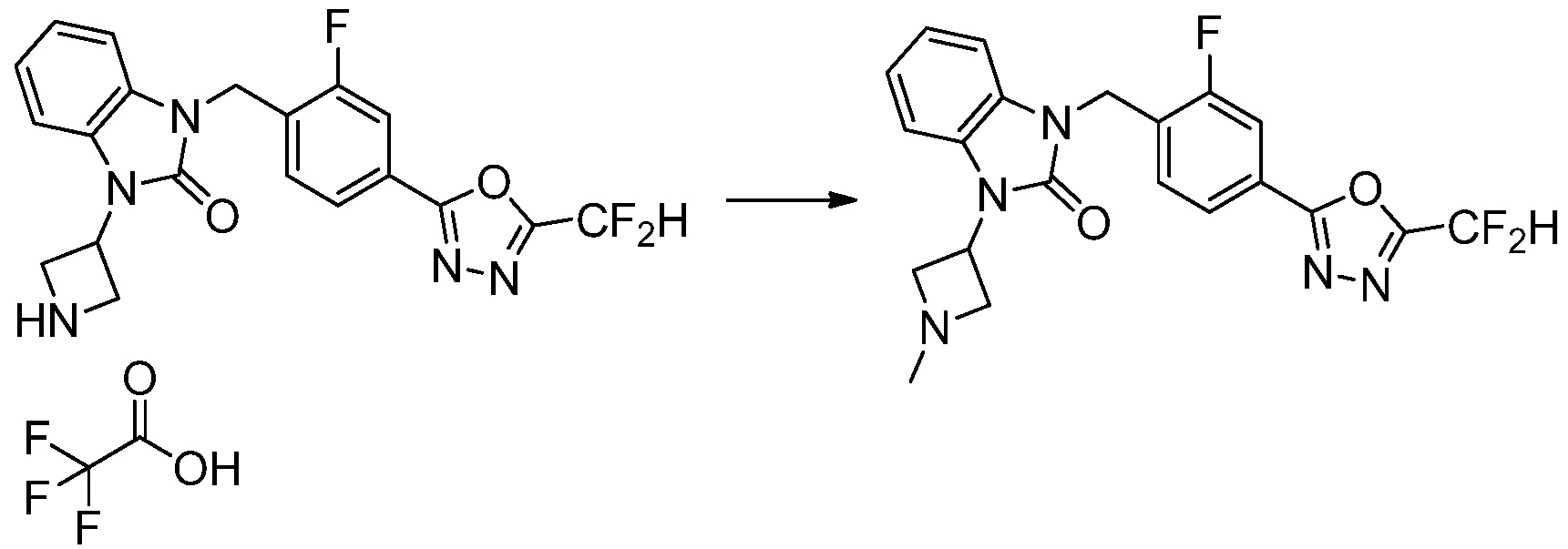
1-(азетидин-3-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,300 г, 0,567 ммоль), полученный на стадии 4, и N,N-диизопропилэтиламин (0,099 мл, 0,567 ммоль) растворяют в дихлорметане (10 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем формальдегид (0,034 г, 1,133 ммоль) и триацетоксиборгидрид натрия (0,240 г, 1,133 ммоль) добавляют туда и дополнительно перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,160 г, 65,8%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,84 (м, 2H), 7,50-7,42 (м, 2H), 7,17-7,05 (м, 2H), 7,00-6,98 (м, 1H), 7,05 (с, 0,25H), 6,92 (с, 0,5H), 6,79 (с, 0,5H), 5,21-5,15 (м, 3H), 4,24-4,16 (м, 4H), 2,75 (с, 3H); МСНР (ЭР) m/z 430,4 (M+ + 1).
Пример 23: Синтез соединения 23, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-(оксетан-3-ил)азетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 23
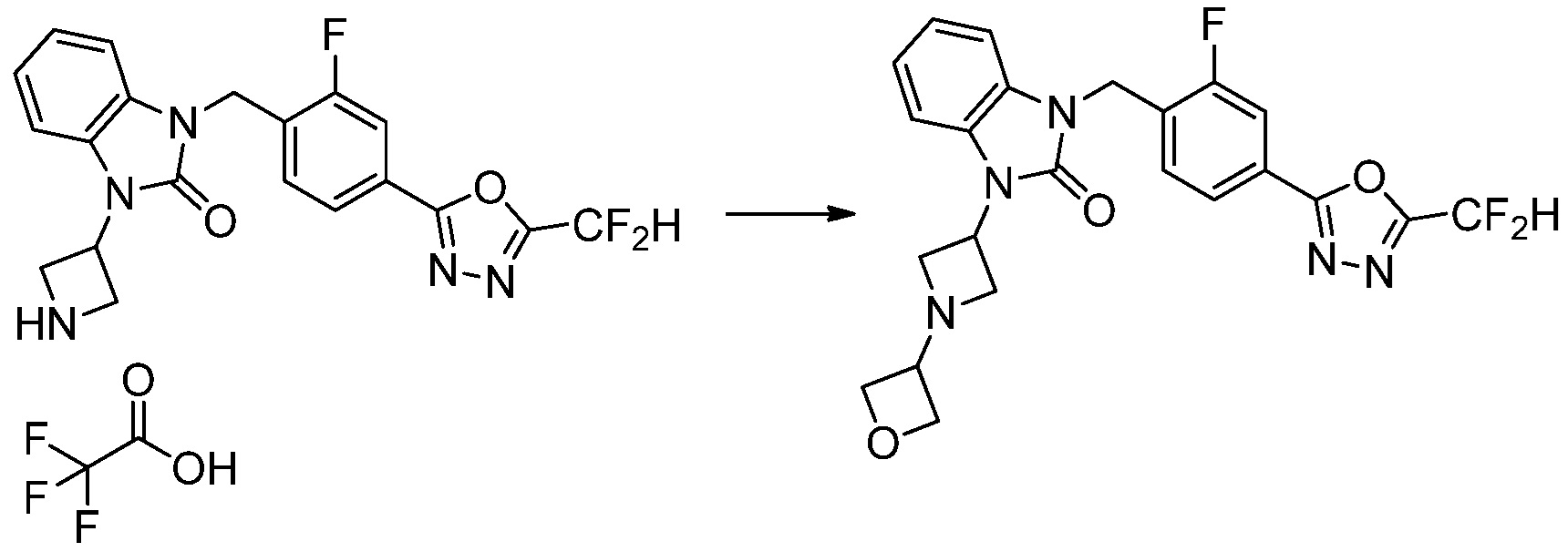
1-(азетидин-3-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,300 г, 0,567 ммоль) и N,N-диизопропилэтиламин (0,099 мл, 0,567 ммоль) растворяют в дихлорметане (10 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют оксетан-3-он (0,082 г, 1,133 ммоль) и триацетоксиборгидрид натрия (0,240 г, 1,133 ммоль) и дополнительно перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 37,4%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,83 (м, 2H), 7,63-7,61 (м, 1H), 7,48-7,45 (м, 1H), 7,18-7,09 (м, 2H), 7,05 (с, 0,25H), 7,00-6,98 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,19 (с, 2H), 5,15-5,10 (м, 1H), 4,80-4,77 (м, 2H), 4,65-4,62 (м, 2H), 3,99-3,96 (м, 3H), 3,87-3,83 (м, 2H); МСНР (ЭР) m/z 472,3 (M+ + 1).
Пример 24: Синтез соединения 24, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(2-оксаспиро[3,3]гептан-6-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез N-(2-(2-окса-6-азаспиро[3,3]гептан-6-ил)этил)-2-нитроанилина
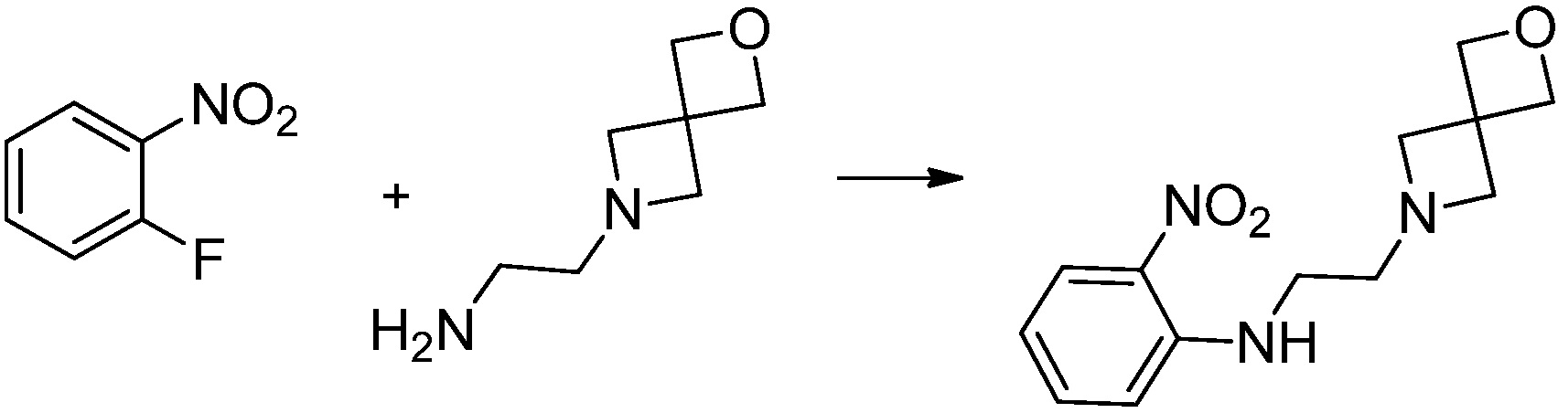
1-фтор-2-нитробензол (0,400 г, 2,835 ммоль), 2-(2-окса-6-азаспиро[3,3]гептан-6-ил)этан-1-амин (0,403 г, 2,835 ммоль) и карбонат калия (0,784 г, 5,670 ммоль) растворяют в тетрагидрофуране (20 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 2 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,388 г, 52,0%) в виде желтого масла.
[Стадия 2] Синтез N1-(2-(2-окса-6-азаспиро[3,3]гептан-6-ил)этил)бензол-1,2-диамина
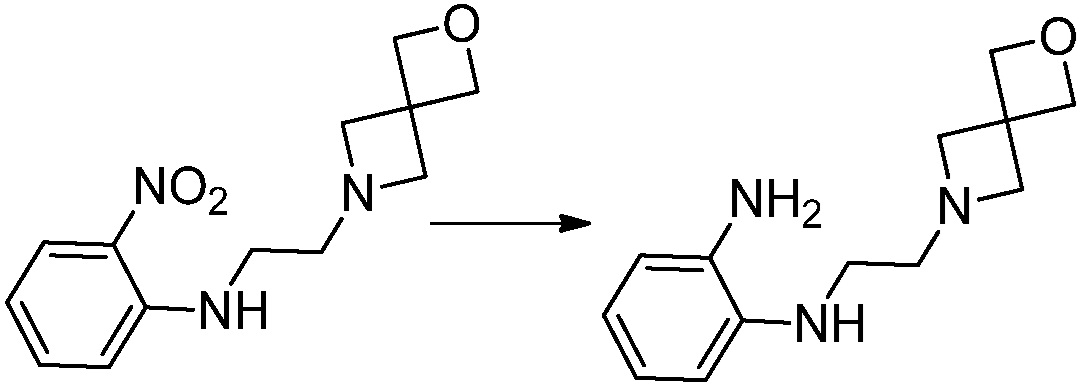
N-(2-(2-окса-6-азаспиро[3,3]гептан-6-ил)этил)-2-нитроанилин (0,388 г, 1,474 ммоль), полученный на стадии 1 растворяют в этаноле (15 мл) и перемешивают при комнатной температуре, после чего туда медленно добавляют 10%-Pd/C (40 мг) при той же температуре и перемешивают при той же температуре в течение 3 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (0,366 г, 106,5%, коричневое масло).
[Стадия 3] Синтез 1-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
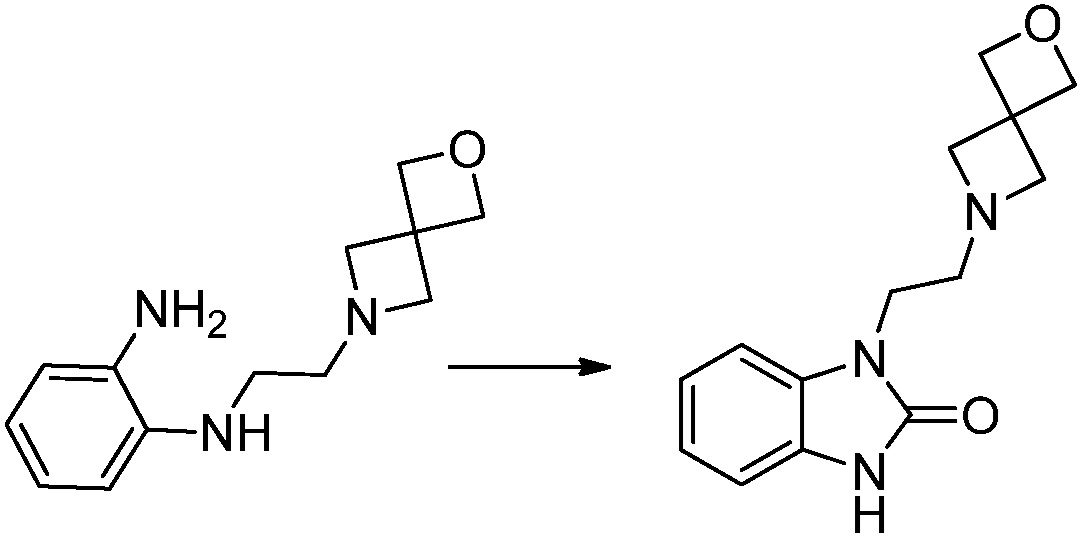
N1-(2-(2-окса-6-азаспиро[3,3]гептан-6-ил)этил)бензол-1,2-диамин (0,366 г, 1,569 ммоль), полученный на стадии 2 и 1,1'-карбонилдиимидазол (0,254 г, 1,569 ммоль) растворяют в тетрагидрофуране (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,330 г, 81,1%) в виде коричневого масла.
[Стадия 4] Синтез соединения 24
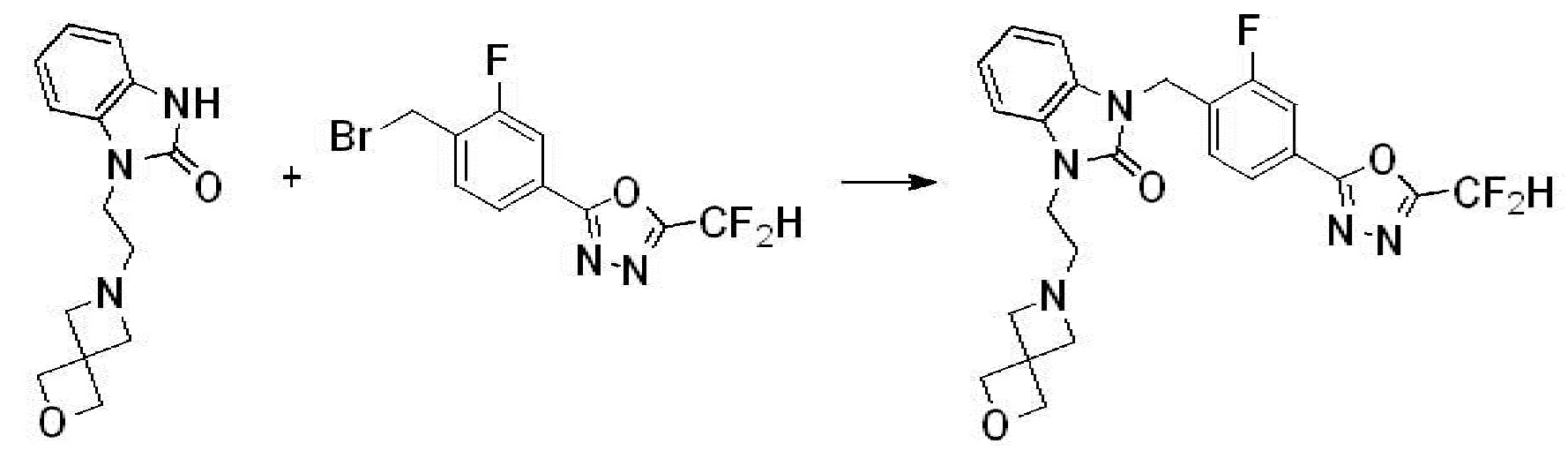
1-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,386 ммоль), полученный на стадии 3, и гидрид натрия (60,00%, 0,017 г, 0,424 ммоль) растворяют в N,N-диметилформамиде (4 мл) при 0°C, после чего 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,130 г, 0,424 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,027 г, 14,4%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,88 MГц- 7,82 (м, 2H), 7,46 (т, J=7,6 Гц, 1H), 7,15-6,79 (м, 5H), 5,21 (с, 2H), 4,72 (с, 4H), 3,91 (т, J=6,8 Гц, 2H), 3,42 (с, 4H), 2,77 (т, J=6,8 Гц, 2H); МСНР (ЭР) m/z 486,4 (M+ + H).
Пример 25: Синтез соединения 25, 1-(2-(2-окса-6-азаспиро[3,3]гептан-6-ил)этил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 25
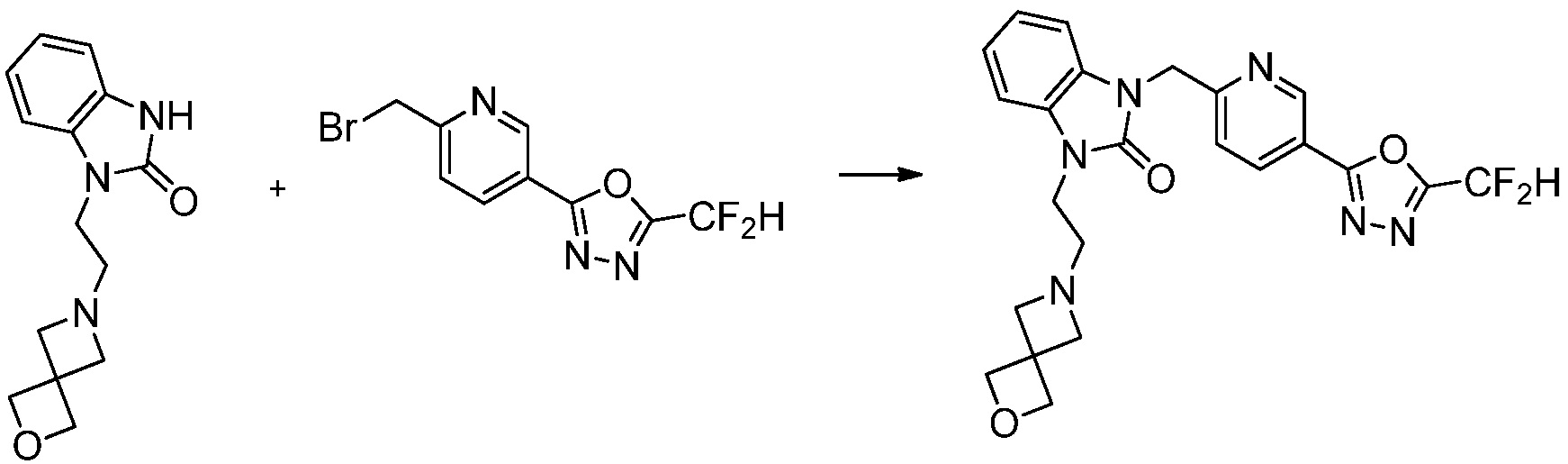
1-(2-(2-окса-6-азаспиро[3,3]гептан-6-ил)этил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,115 г, 0,443 ммоль), полученный на стадии 3 соединения 24, и гидрид натрия (60,00%, 0,020 г, 0,488 ммоль) растворяют в N,N-диметилформамиде (4 мл) при 0°C, после чего 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,142 г, 0,488 ммоль) добавляют в полученный раствор, и затем перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,083 г, 40,0%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,2, 0,8 Гц, 1H), 8,30 (дд, J=8,2, 2,2 Гц, 1H), 7,38 (дд, J=8,3, 0,7 Гц, 1H), 7,10-7,09 (м, 1H), 7,08-6,79 (м, 4H), 5,28 (с, 2H), 4,70 (с, 4H), 3,89 (т, J=6,8 Гц, 2H), 3,40 (с, 4H), 2,76 (т, J=6,8 Гц, 2H); МСНР (ЭР) m/z 469,4 (M+ + H).
Пример 26: Синтез соединения 26, 1-(2-(2-окса-6-азаспиро[3,3]гептан-6-ил)этил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 26
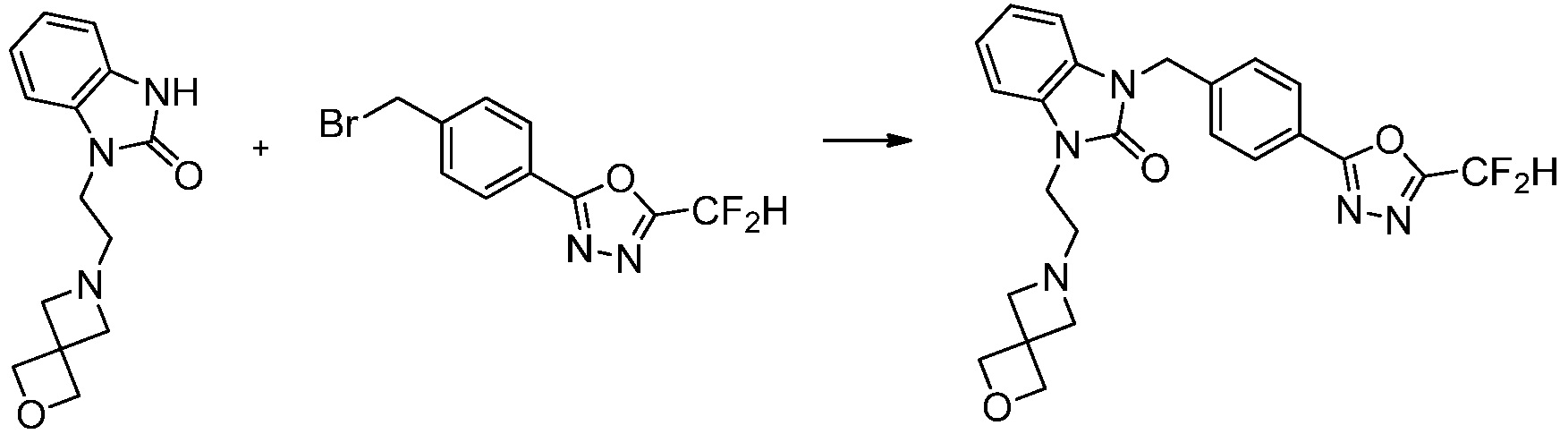
1-(2-(2-окса-6-азаспиро[3,3]гептан-6-ил)этил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,115 г, 0,443 ммоль), полученный на стадии 3 соединения 24, и гидрид натрия (60,00%, 0,020 г, 0,488 ммоль) растворяют в N,N-диметилформамиде (4 мл) при 0°C, после чего 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,142 г, 0,488 ммоль) добавляют в полученный раствор, и затем перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,083 г, 40,0%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,2, 0,8 Гц, 1H), 8,30 (дд, J=8,2, 2,2 Гц, 1H), 7,38 (дд, J=8,3, 0,7 Гц, 1H), 7,10-7,09 (м, 1H), 7,08-6,79 (м, 4H), 5,28 (с, 2H), 4,70 (с, 4H), 3,89 (т, J=6,8 Гц, 2H), 3,40 (с, 4H), 2,76 (т, J=6,8 Гц, 2H); МСНР (ЭР) m/z 469,4 (M+ + H).
Пример 27: Синтез соединения 27, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(2-оксаспиро[3,3]гептан-6-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез N1-(2-оксаспиро[3,3]гептан-6-ил)бензол-1,2-диамина
2-оксаспиро[3,3]гептан-6-он (1,000 г, 9,247 ммоль) и 1,2-фенилендиамин (2,074 г, 18,495 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (2,352 г, 11,097 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением указанного в заголовке соединения (1,120 г, 59,3%) в виде оранжевого твердого вещества.
[Стадия 2] Синтез 1-(2-оксаспиро[3,3]гептан-6-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
N1-(2-оксаспиро[3,3]гептан-6-ил)бензол-1,2-диамин (1,120 г, 5,483 ммоль), полученный на стадии 1, и 1,1'-карбонилдиимидазол (0,889 г, 5,483 ммоль) растворяют в тетрагидрофуране (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Этилацетат добавляют в полученный концентрат и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют, затем промывают этилацетатом, и затем сушат с получением указанного в заголовке соединения (0,556 г, 44,0%) в виде розового твердого вещества.
[Стадия 3] Синтез соединения 27
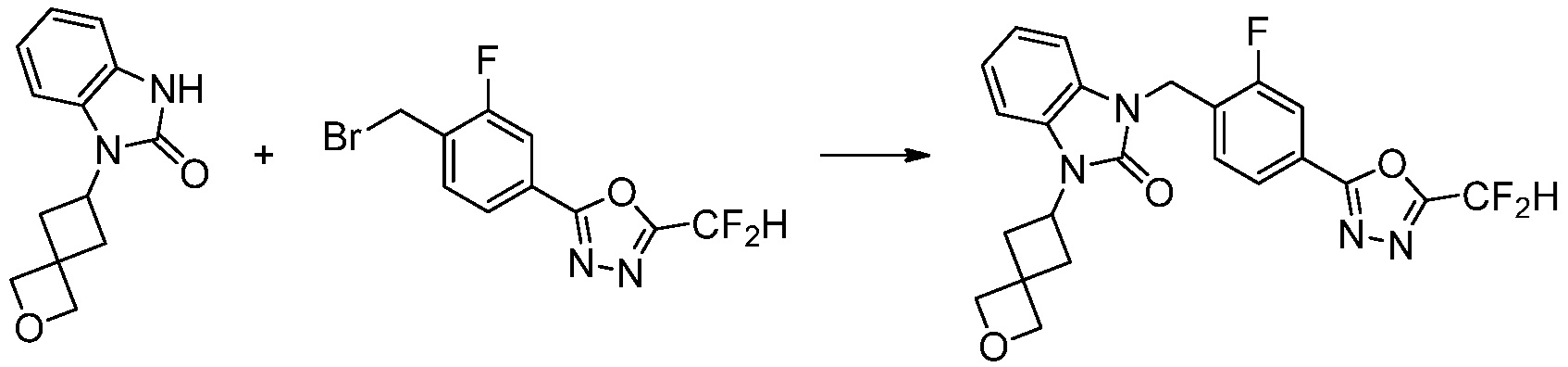
1-(2-оксаспиро[3,3]гептан-6-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,434 ммоль), полученный на стадии 2, и гидрид натрия (60,00%, 0,019 г, 0,478 ммоль) растворяют в N,N-диметилформамиде (3 мл) при 0°C, после чего 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,147 г, 0,478 ммоль) добавляют в полученный раствор, и затем перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением указанного в заголовке соединения (0,136 г, 68,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,88-7,84 (м, 2H), 7,47 (т, J=7,8 Гц, 1H), 7,14-6,79 (м, 5H), 5,19 (с, 2H), 4,87 (с, 2H), 4,82 (с, 2H), 4,74-4,68 (м, 1H), 3,19-3,14 (м, 2H), 2,85-2,79 (м, 2H); МСНР (ЭР) m/z 457,4 (M+ + H).
Пример 28: Синтез соединения 28, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(2-оксаспиро[3,3]гептан-6-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 28
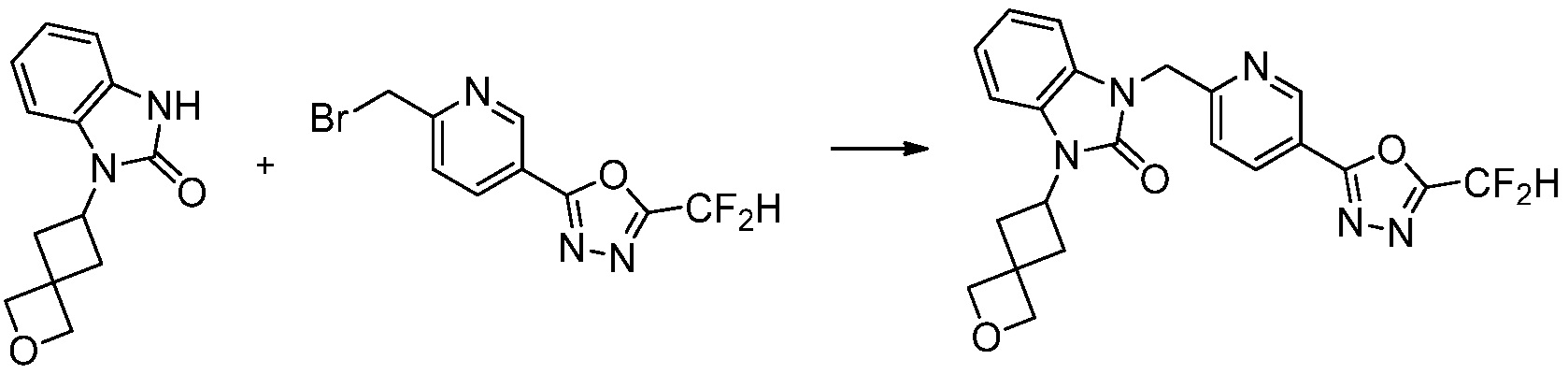
1-(2-оксаспиро[3,3]гептан-6-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,434 ммоль), полученный на стадии 2 соединения 27, и гидрид натрия (60,00%, 0,019 г, 0,478 ммоль) растворяют в N,N-диметилформамиде (3 мл) при 0°C, после чего 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,139 г, 0,478 ммоль) добавляют в полученный раствор, и затем перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением указанного в заголовке соединения (0,118 г, 61,8%) в виде светло-оранжевого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (дд, J=2,2, 0,7 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,10-6,81 (м, 5H), 5,27 (с, 2H), 4,86 (с, 2H), 4,80 (с, 2H), 4,74-4,70 (м, 1H), 3,19-3,16 (м, 2H), 2,84-2,78 (м, 2H); МСНР (ЭР) m/z 440,4 (M+ + H).
Пример 29: Синтез соединения 29, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(2-оксаспиро[3,3]гептан-6-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 29
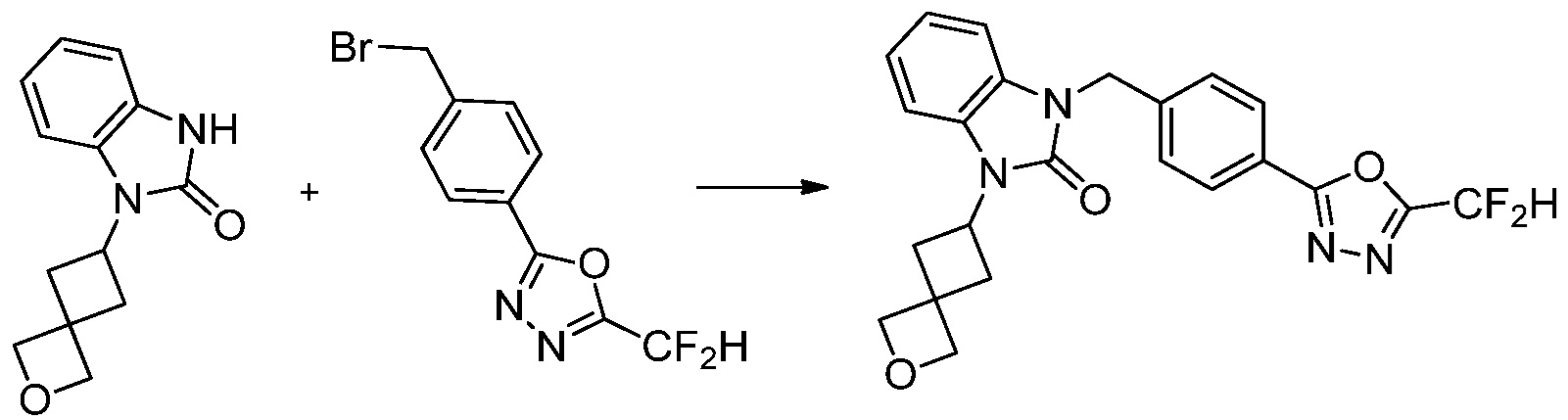
1-(2-оксаспиро[3,3]гептан-6-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,434 ммоль), полученный на стадии 2 соединения 27, и гидрид натрия (60,00%, 0,019 г, 0,478 ммоль) растворяют в N,N-диметилформамиде (3 мл) при 0°C, после чего 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,138 г, 0,478 ммоль) добавляют в полученный раствор и затем перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением указанного в заголовке соединения (0,137 г, 72,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,09 (д, J=7,7 Гц, 2H), 7,49 (д, J=7,8 Гц, 2H), 7,12-6,79 (м, 5H), 5,14 (с, 2H), 4,87 (с, 2H), 4,82 (с, 2H), 4,75-4,71 (м, 1H), 3,17 (т, J=10,3 Гц, 2H), 2,82 (т, J=9,9 Гц, 2H); МСНР (ЭР) m/z 439,4 (M+ + H).
Пример 30: Синтез соединения 30, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(2-метоксиэтил)-3,4-дигидрохиназолин-2(1H)-он
[Стадия 1] Синтез 2-амино-N-(2-метоксиэтил)бензамид
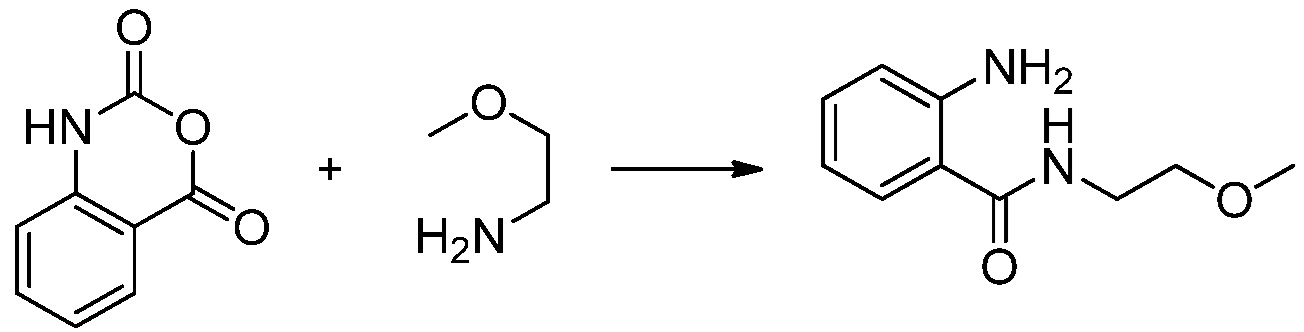
2H-бензо[d][1,3]оксазин-2,4(1H)дион (10,000 г, 61,301 ммоль), 2-метоксиэтан-1-амин (4,604 г, 61,301 ммоль) и триэтиламин (8,544 мл, 61,301 ммоль) растворяют в этаноле (50 мл) при 80°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (9,800 г, 82,3%) в виде бесцветного масла.
[Стадия 2] Синтез 2-(((2-метоксиэтил)амино)метил)анилин
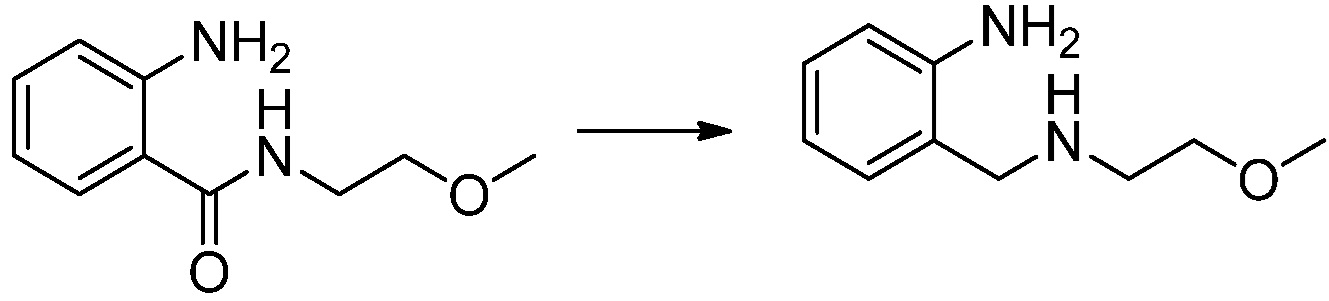
2-амино-N-(2-метоксиэтил)бензамид (3,670 г, 18,895 ммоль), полученный на стадии 1, растворяют в дихлорметане (5 мл), после чего туда добавляют алюмогидрид лития (2,00 M раствор в ТГФ, 25,508 мл, 51,017 ммоль) при 0°C, затем перемешивают при той же температуре в течение 30 часов, затем дополнительно перемешивают при 60° в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (3,300 г, 96,9%, бесцветное масло).
[Стадия 3] Синтез 3-(2-метоксиэтил)-3,4-дигидрохиназолин-2(1H)-она
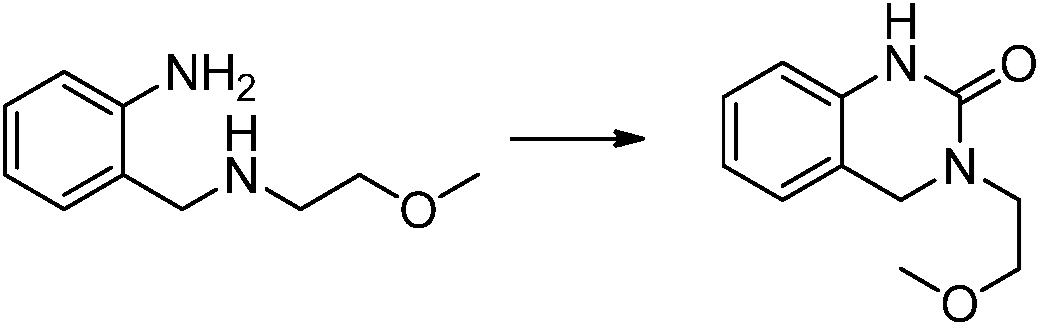
2-(((2-метоксиэтил)амино)метил)анилин (2,700 г, 14,979 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 2,429 г, 14,979 ммоль) растворяют в тетрагидрофуране (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (2,000 г, 64,7%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 30
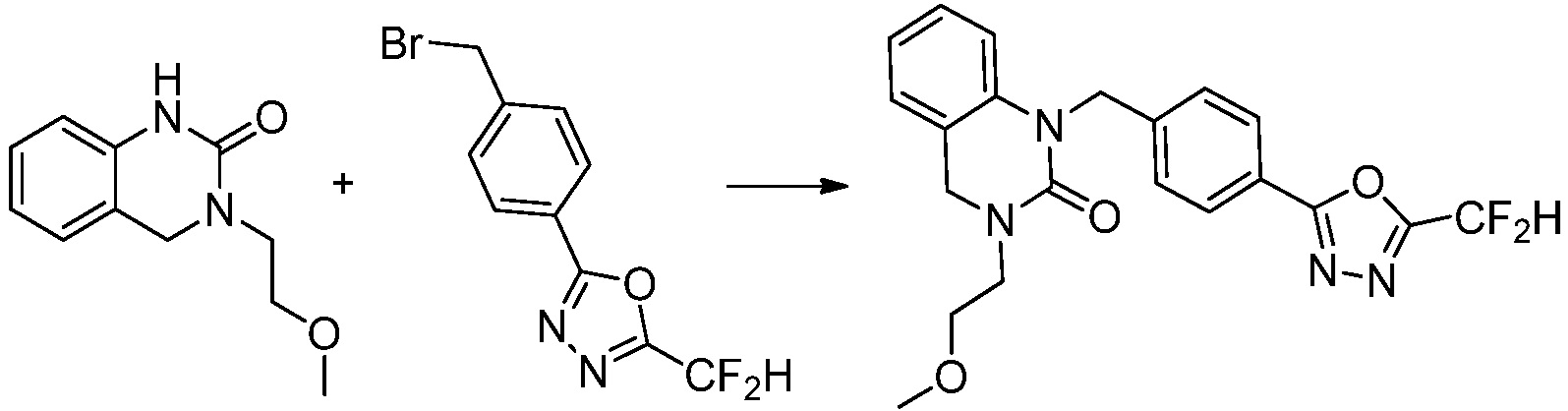
3-(2-метоксиэтил)-3,4-дигидрохиназолин-2(1H)-он (0,100 г, 0,485 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,029 г, 0,727 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,140 г, 0,485 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,060 г, 29,9%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 8,07-8,05 (м, 2H), 7,46-7,44 (м, 2H), 7,10-7,08 (м, 2H), 7,04 (с, 0,25H), 6,98-6,96 (м, 1H), 6,91 (с, 0,5H), 6,78 (с, 0,25H), 6,62-6,59 (м, 1H), 5,21 (с, 2H), 4,65 (с, 2H), 3,71-3,70 (м, 4H), 3,41 (с, 3H); МСНР (ЭР) m/z 415,3 (M+ + 1).
Пример 31: Синтез соединения 31, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(2-метоксиэтил)-3,4-дигидрохиназолин-2(1H)-он
[Стадия 1] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(2-метоксиэтил)-3,4-дигидрохиназолин-2(1H)-она
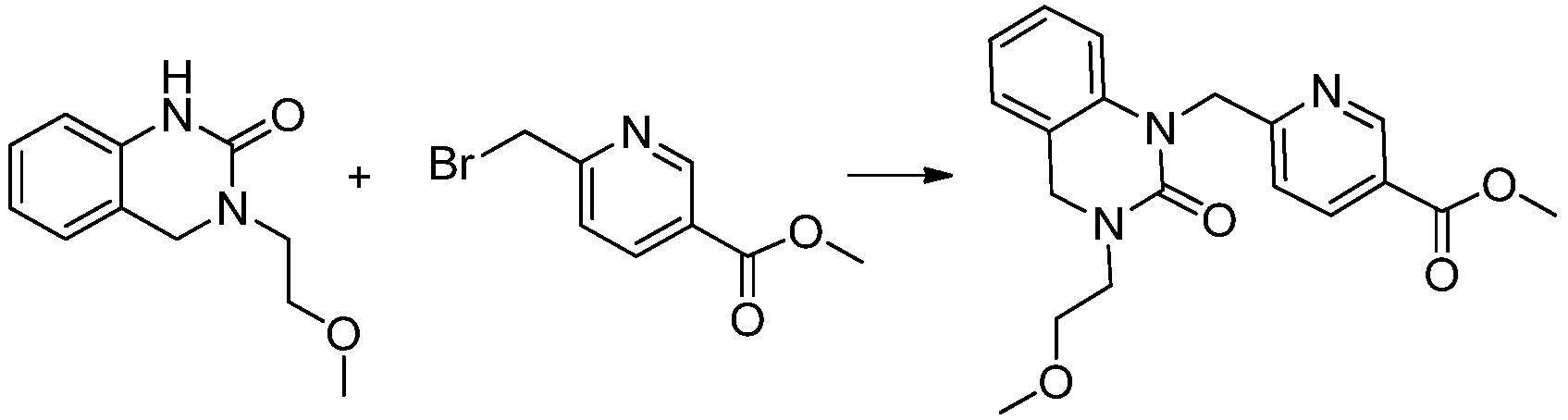
3-(2-метоксиэтил)-3,4-дигидрохиназолин-2(1H)-он (0,100 г, 0,485 ммоль) растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,029 г, 0,727 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,141 г, 0,485 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,080 г, 39,7%) в виде бесцветного масла.
[Стадия 2] Синтез 6-((3-(2-метоксиэтил)-2-оксо-3,4-дигидрохиназолин-1(2H)-ил)метил)никотиногидразида
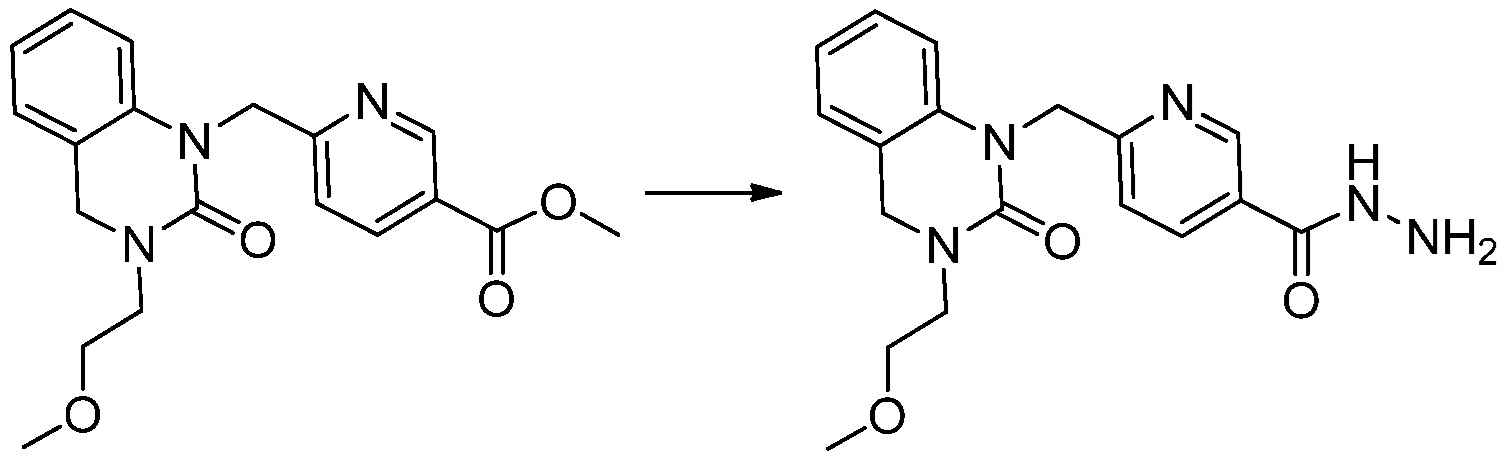
Метил 6-((3-(2-метоксиэтил)-2-оксо-3,4-дигидрохиназолин-1(2H)-ил)метил)никотинат (0,300 г, 0,844 ммоль), полученный на стадии 1, и моногидрат гидразина (0,821 мл, 16,883 ммоль) растворяют в этаноле (20 мл) при 80°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,300 г, 100,0%, белое твердое вещество).
[Стадия 3] Синтез соединения 31
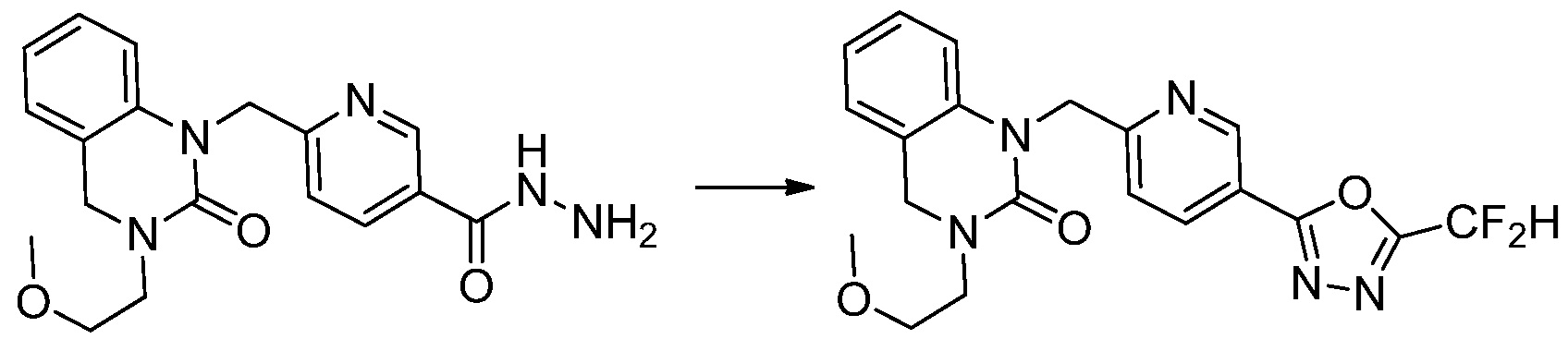
6-((3-(2-метоксиэтил)-2-оксо-3,4-дигидрохиназолин-1(2H)-ил)метил)никотиногидразид (0,240 г, 0,675 ммоль), полученный на стадии 2, 2,2-дифторуксусный ангидрид (0,252 мл, 2,026 ммоль) и имидазол (0,138 г, 2,026 ммоль) растворяют в дихлорметане (10 мл) при 45°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,150 г, 53,5%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (дд, J=2,2, 0,8 Гц, 1H), 8,29 (дд, J=8,3, 2,2 Гц, 1H), 7,44-7,42 (м, 1H), 7,10-7,07 (м, 2H), 7,07 (с, 0,25H), 6,99-6,96 (м, 1H), 6,94 (с, 0,5H), 6,81 (с, 0,25H), 6,69-6,67 (м, 1H), 5,31 (с, 2H), 4,65 (с, 2H), 3,72-3,69 (м, 4H), 3,40 (с, 3H); МСНР (ЭР) m/z 416,4 (M+ + 1).
Пример 32: Синтез соединения 32, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилазетидин-3-ил)-3,4-дигидрохиназолин-2(1H)-он
[Стадия 1] Синтез трет-бутил 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-1,4-дигидрохиназолин-3(2H)-ил)азетидин-1-карбоксилата
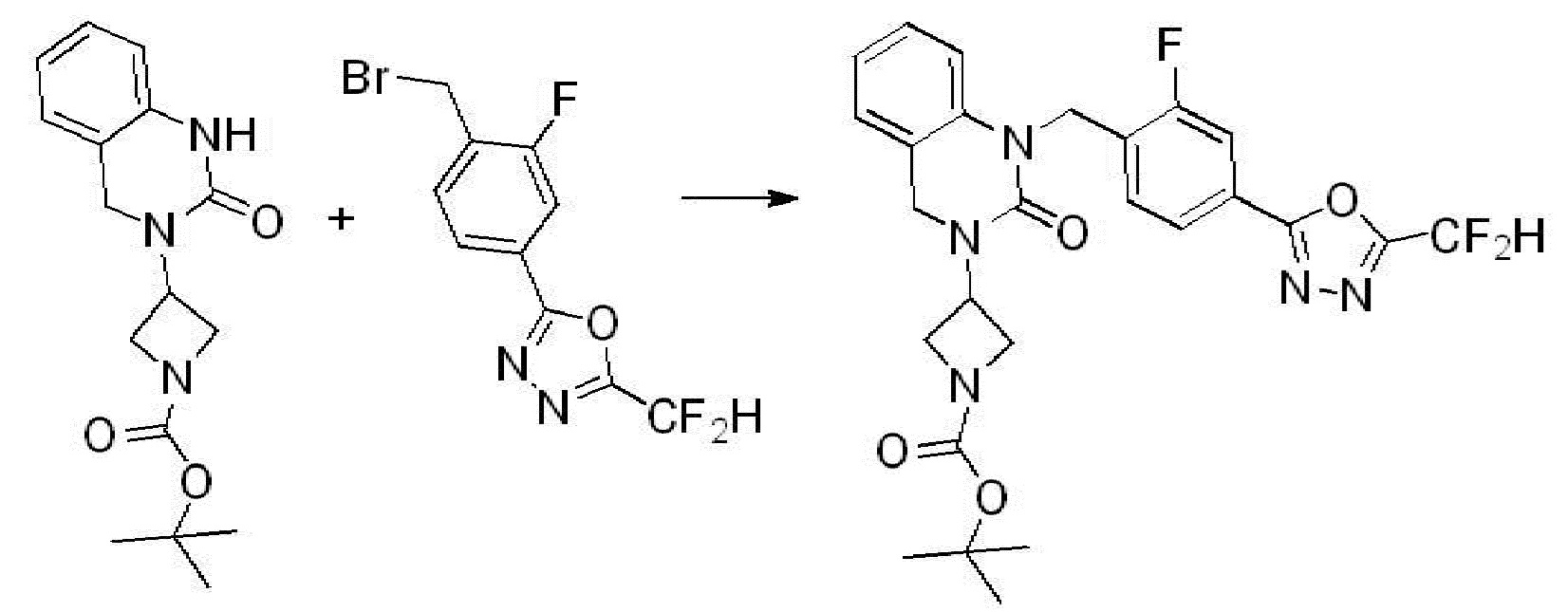
Трет-бутил 3-(2-оксо-1,4-дигидрохиназолин-3(2H)-ил)азетидин-1-карбоксилат (0,469 г, 1,546 ммоль) растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,093 г, 2,319 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,475 г, 1,546 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,410 г, 50,1%) в виде бесцветного масла.
[Стадия 2] Синтез 3-(азетидин-3-ил)-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,4-дигидрохиназолин-2(1H)-он 2,2,2-трифторацетата
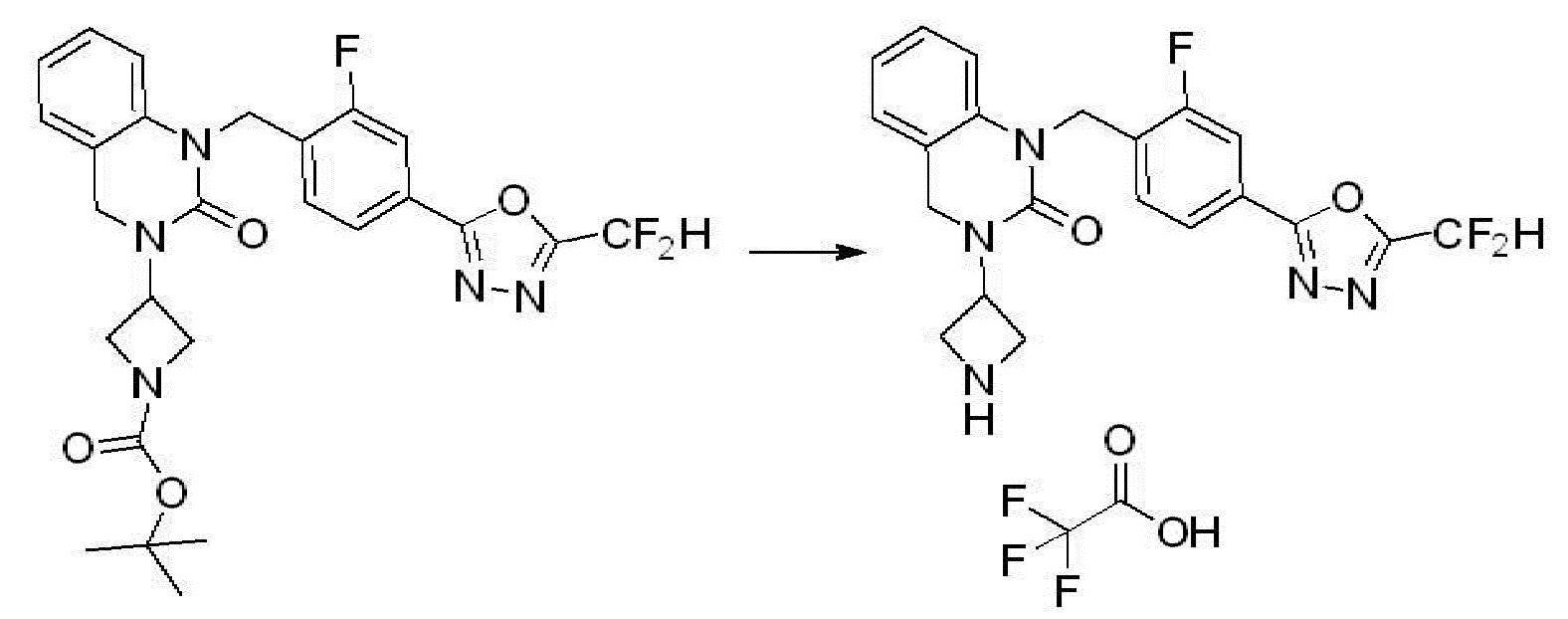
Трет-бутил 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-1,4-дигидрохиназолин-3(2H)-ил)азетидин-1-карбоксилат (0,420 г, 0,793 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,607 мл, 7,932 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение трех часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,420 г, 97,4%, коричневое масло).
[Стадия 3] Синтез соединения 32
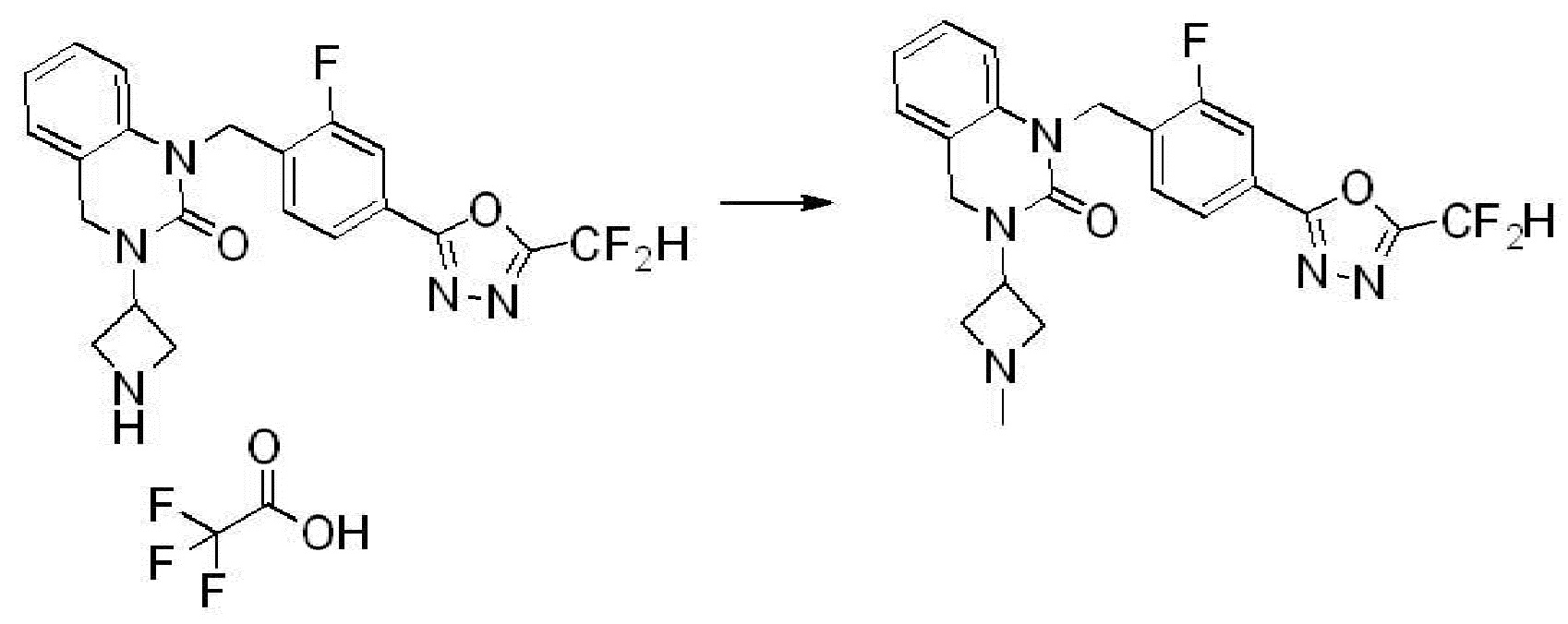
3-(азетидин-3-ил)-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,4-дигидрохиназолин-2(1H)-он 2,2,2-трифторацетат (0,200 г, 0,368 ммоль), полученный на стадии 2, формальдегид (0,022 г, 0,736 ммоль), N,N-диизопропилэтиламин (0,064 мл, 0,368 ммоль) и триацетоксиборгидрид натрия (0,156 г, 0,736 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 67,4%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,80 (м, 2H), 7,30-7,28 (м, 1H), 7,15-7,12 (м, 2H), 7,05 (с, 0,25H), 7,05-7,00 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 6,62 (д, J=8,2 Гц, 1H), 5,24 (д, J=3,8 Гц, 2H), 4,70-4,60 (м, 1H), 4,52 (с, 2H), 4,00-3,93 (м, 2H), 2,87-2,85 (м, 1H), 2,78-2,76 (м, 1H); МСНР (ЭР) m/z 476,4 (M+ + 1).
Пример 33: Синтез соединения 33, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-3,4-дигидрохиназолин-2(1H)-он
[Стадия 1] Синтез трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-1,4-дигидрохиназолин-3(2H)-ил)пиперидин-1-карбоксилата
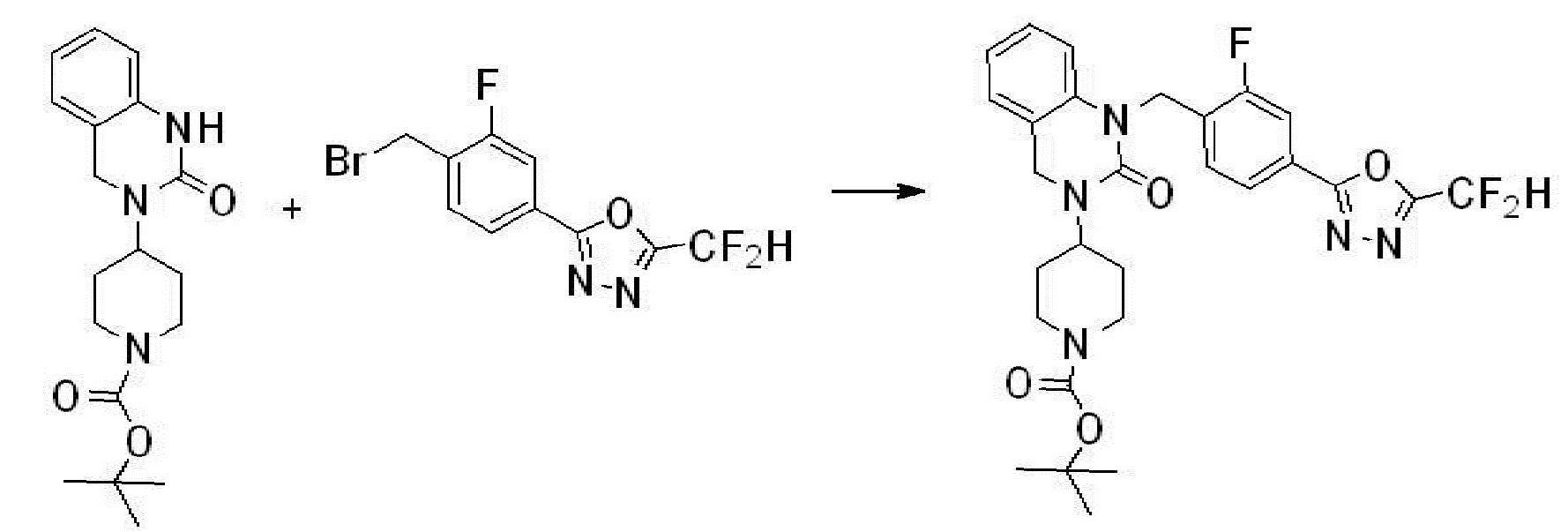
Трет-бутил 4-(2-оксо-1,4-дигидрохиназолин-3(2H)-ил)пиперидин-1-карбоксилат (0,685 г, 2,067 ммоль) растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,124 г, 3,100 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,635 г, 2,067 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,530 г, 46,0%) в виде бесцветного масла.
[Стадия 2] Синтез 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-3,4-дигидрохиназолин-2(1H)-она 2,2,2-трифторацетата
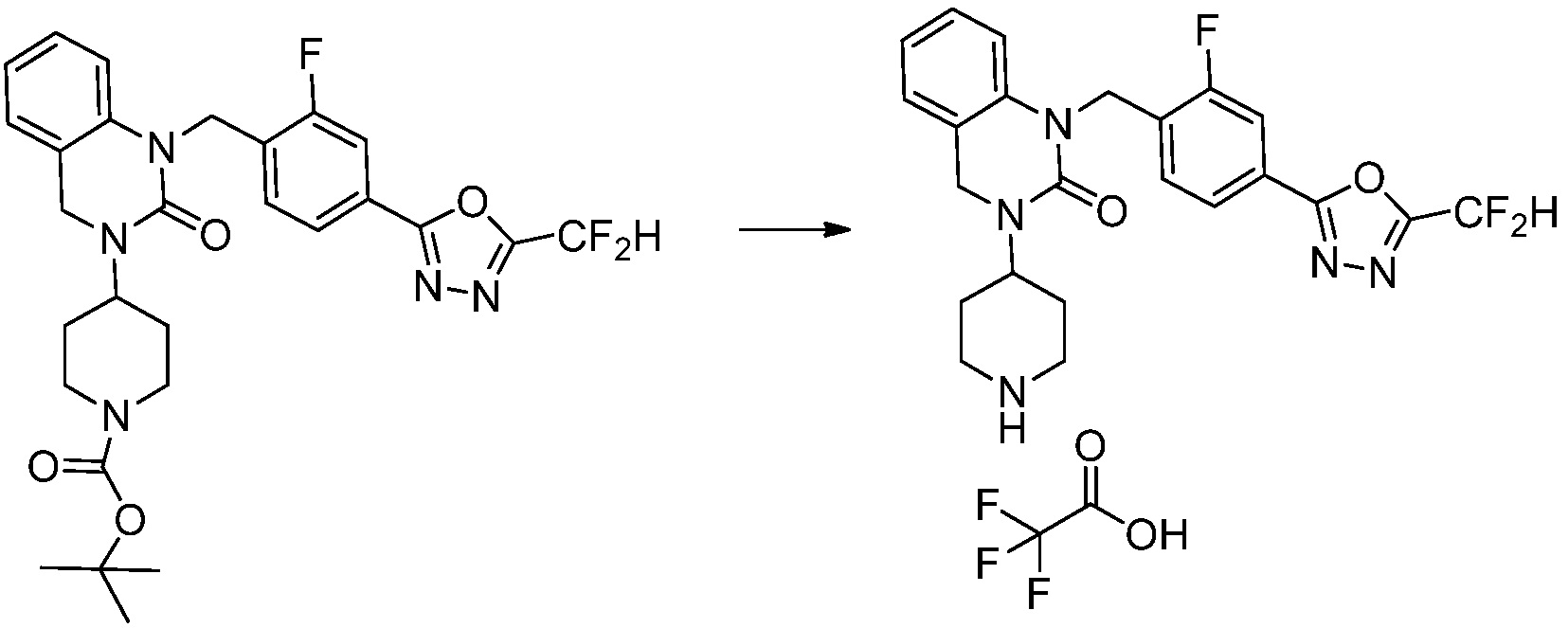
Трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-1,4-дигидрохиназолин-3(2H)-ил)пиперидин-1-карбоксилат (0,530 г, 0,951 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,728 мл, 9,506 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,530 г, 97,6%, коричневое масло).
[Стадия 3] Синтез соединения 33
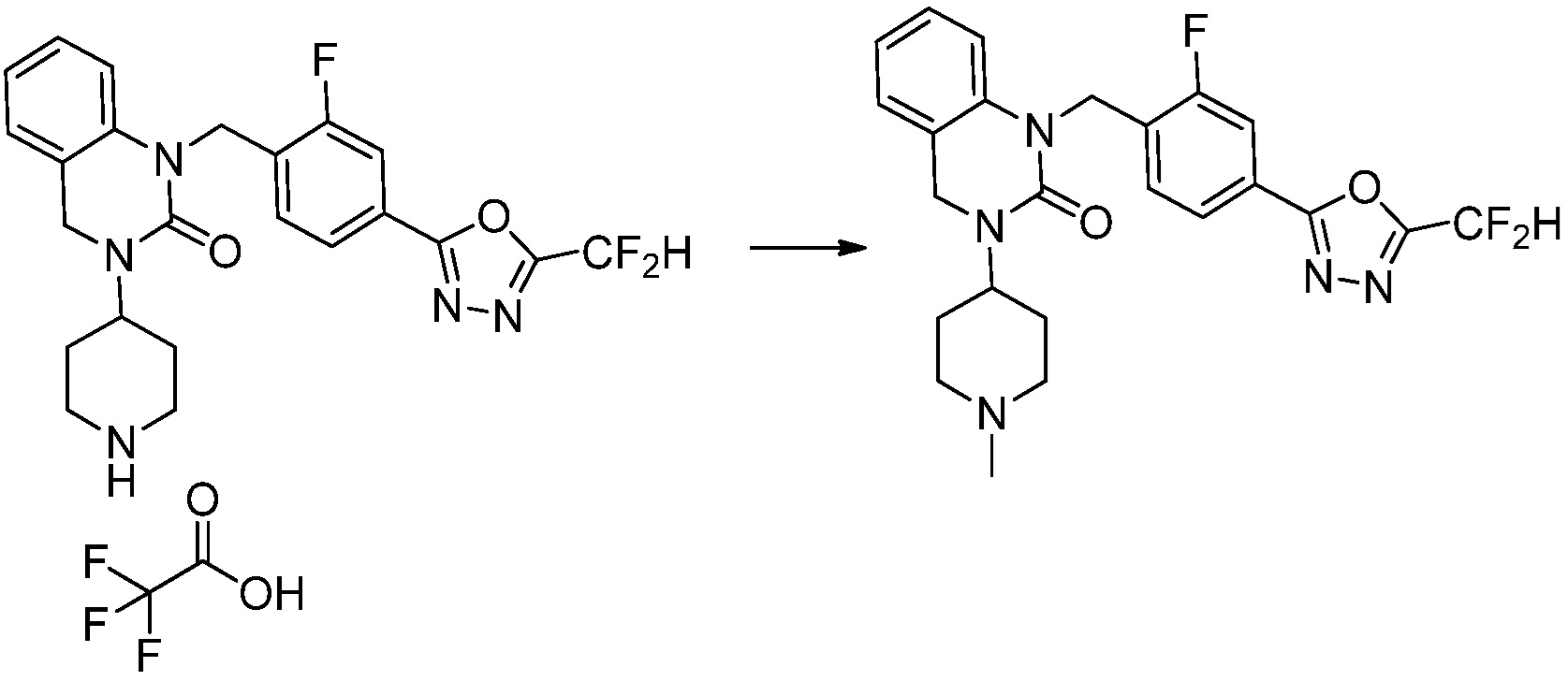
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-3,4-дигидрохиназолин-2(1H)-он 2,2,2-трифторацетат (0,200 г, 0,350 ммоль), полученный на стадии 2, формальдегид (0,021 г, 0,700 ммоль), N,N-диизопропилэтиламин (0,061 мл, 0,350 ммоль) и триацетоксиборгидрид натрия (0,148 г, 0,700 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 60,6%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,86-7,78 (м, 2H), 7,28-7,24 (м, 1H), 7,18-7,14 (м, 2H), 7,05 (с, 0,25H), 7,05-7,00 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 6,63 (д, J=7,9 Гц, 1H), 5,22 (с, 2H), 4,68-4,61 (м, 1H), 4,39 (с, 2H), 3,56-3,53 (м, 2H), 2,81-2,74 (м, 5H), 2,43-2,34 (м, 2H), 1,95-1,91 (м, 2H); МСНР (ЭР) m/z 472,4 (M+ + 1).
Пример 34: Синтез соединения 34, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(тетрагидро-2H-пиран-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 34
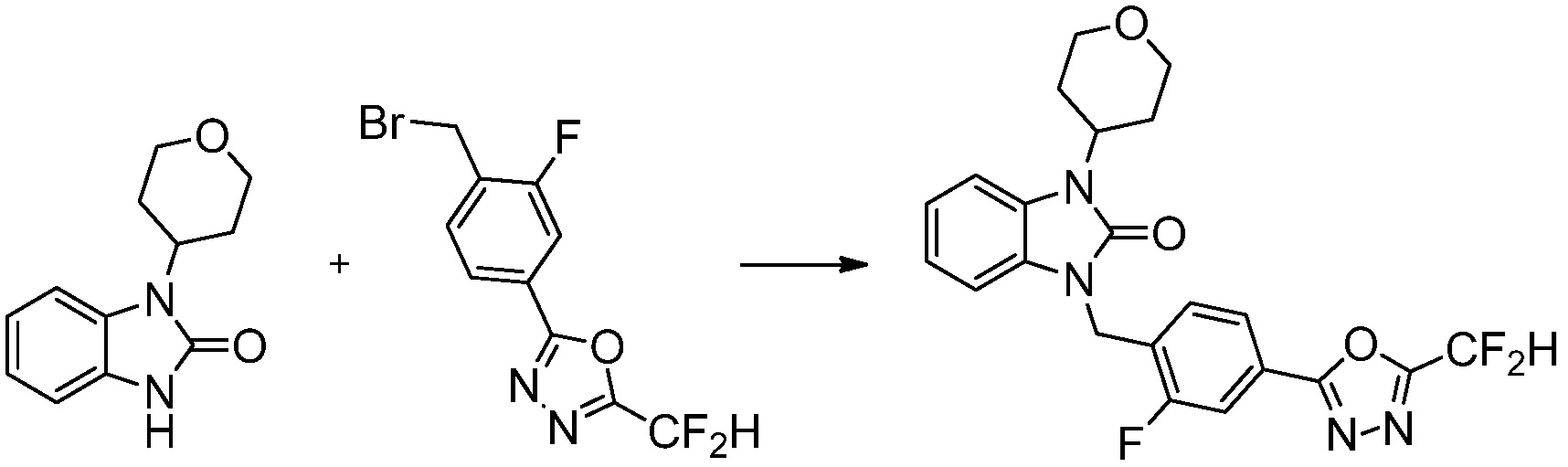
1-(тетрагидро-2H-пиран-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,458 ммоль) растворяют в N,N-диметилформамиде (5 мл) при 0°C, после чего гидрид натрия (60,00%, 0,027 г, 0,687 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,141 г, 0,458 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,070 г, 34,4%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,89-7,84 (м, 2H), 7,48 (т, J=7,8 Гц, 1H), 7,30-7,25 (м, 1H), 7,14-7,05 (м, 2H), 7,05 (с, 0,25H), 7,02-6,98 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,22 (с, 2H), 4,69-4,63 (м, 1H), 4,19-4,13 (м, 2H), 3,63-3,57 (м, 2H), 2,60-2,50 (м, 2H), 1,85-1,81 (м, 2H); МСНР (ЭР) m/z 445,3 (M+ + 1).
Пример 35: Синтез соединения 35, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 35
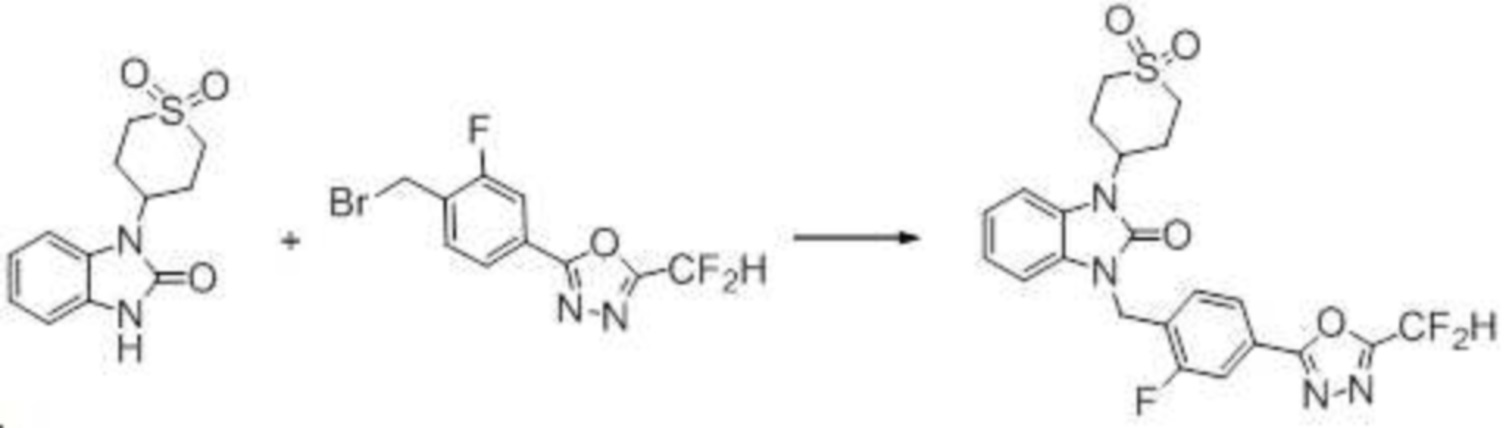
1-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,563 ммоль) растворяют в N,N-диметилформамиде (5 мл) при 0°C, после чего гидрид натрия (60,00%, 0,034 г, 0,845 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,173 г, 0,563 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,080 г, 28,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,89-7,85 (м, 2H), 7,50-7,46 (м, 1H), 7,31-7,28 (м, 1H), 7,17-7,10 (м, 2H), 7,05 (с, 0,25H), 7,03-7,01 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,21 (с, 2H), 4,80-4,73 (м, 1H), 3,30-2,97 (м, 6H), 2,29-2,24 (м, 2H); МСНР (ЭР) m/z 493,3 (M+ + 1).
Пример 36: Синтез соединения 36, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(2-(диметиламино)этил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 36
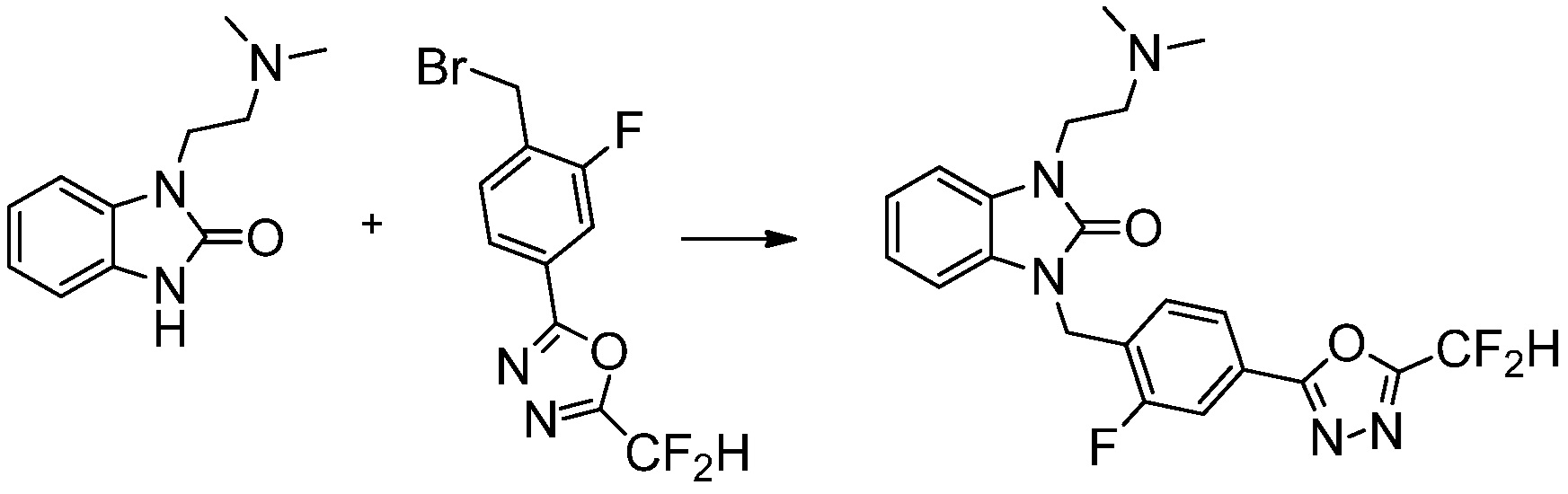
1-(2-(диметиламино)этил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,731 ммоль) растворяют в N,N-диметилформамиде (5 мл) при 0°C, после чего гидрид натрия (60,00%, 0,044 г, 1,096 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,224 г, 0,731 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 34,9%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,87 ~ 7,82 (м, 2H), 7,50 ~ 7,45 (м, 1H), 7,13 ~ 7,06 (м, 3H), 7,05 (с, 0,25H), 6,96 ~ 6,94 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,21 (с, 2H), 4,08 (т, J=7,1 Гц, 2H), 2,71 (т, J=7,1 Гц, 2H), 2,36 (с, 6H).
Пример 37: Синтез соединения 37, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(оксетан-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 37
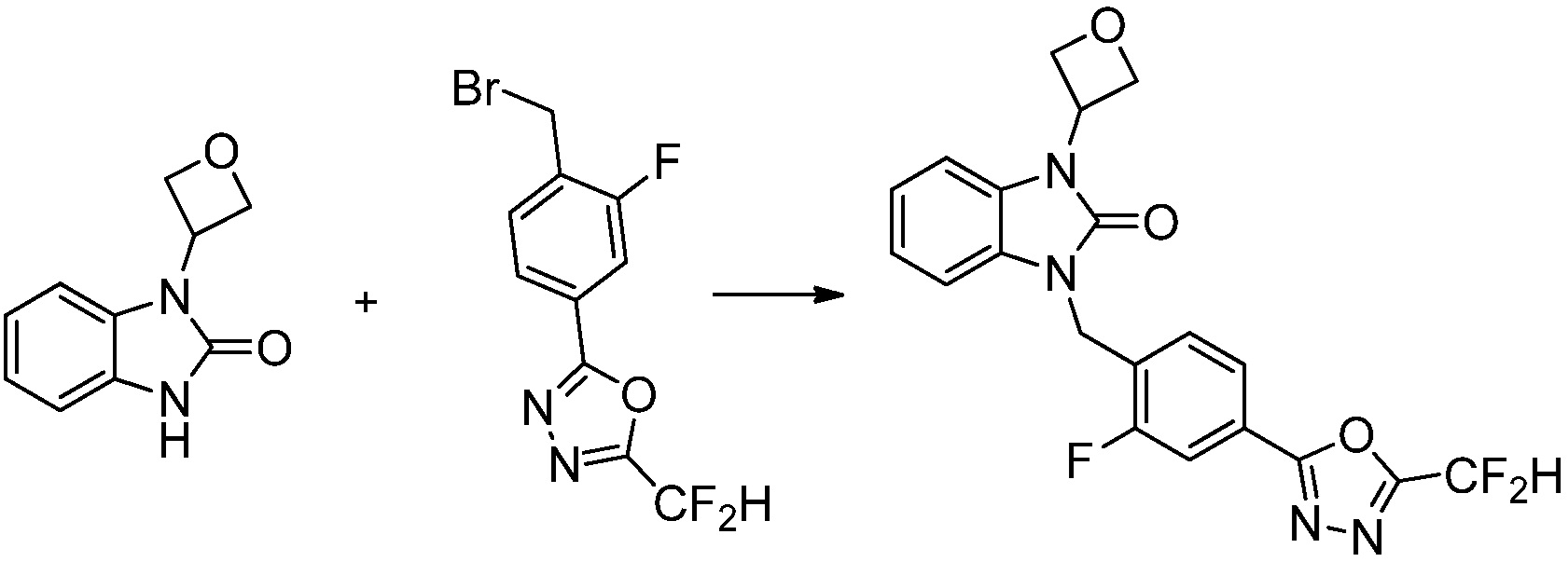
1-(оксетан-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 1,052 ммоль) растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,063 г, 1,577 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,646 г, 2,103 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,160 г, 36,5%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,85-7,81 (м, 2H), 7,72-7,70 (м, 1H), 7,48-7,45 (м, 1H), 7,21-7,11 (м, 2H), 7,04 (с, 0,25H), 7,04-7,01 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,72-5,66 (м, 1H), 5,21-5,11 (м, 6H).
Пример 38: Синтез соединения 38, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилата
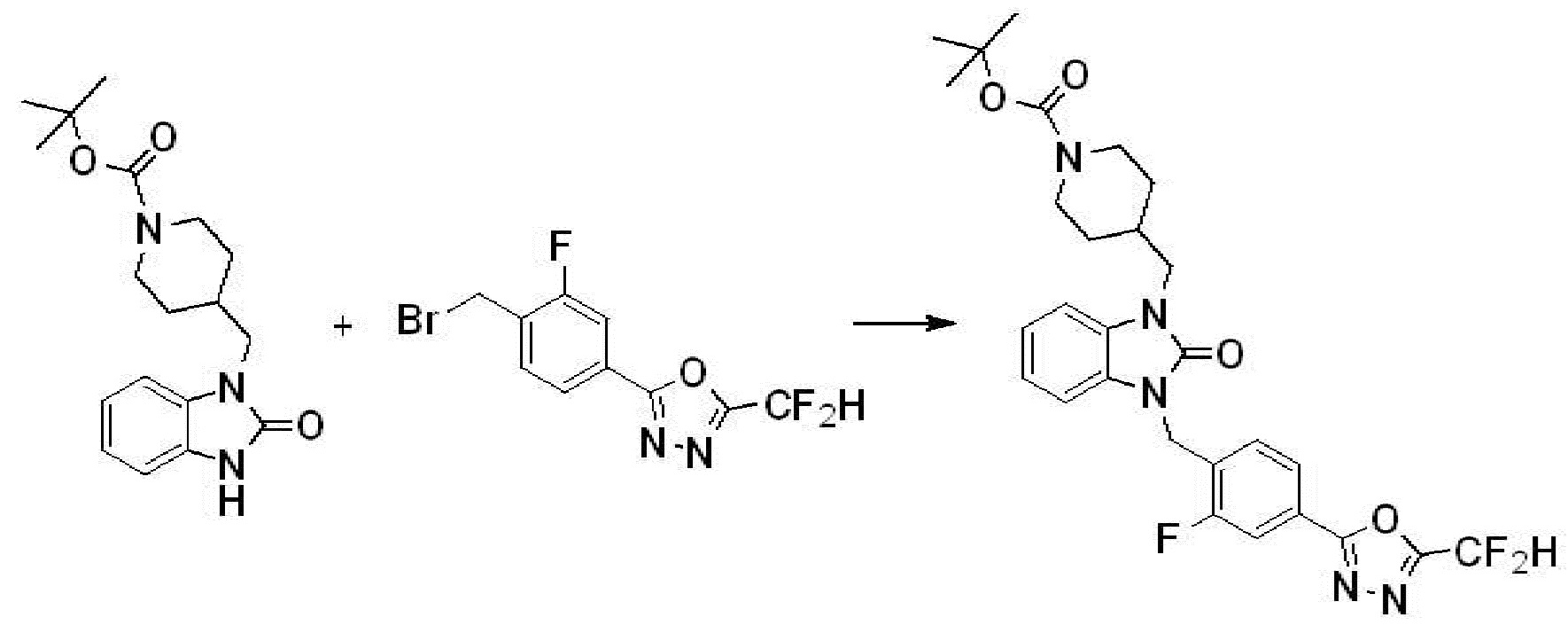
Трет-бутил 4-((2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилат (0,437 г, 1,319 ммоль) растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,079 г, 1,978 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,810 г, 2,637 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,550 г, 74,8%) в виде бесцветного масла.
[Стадия 2] Синтез 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-илметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетата
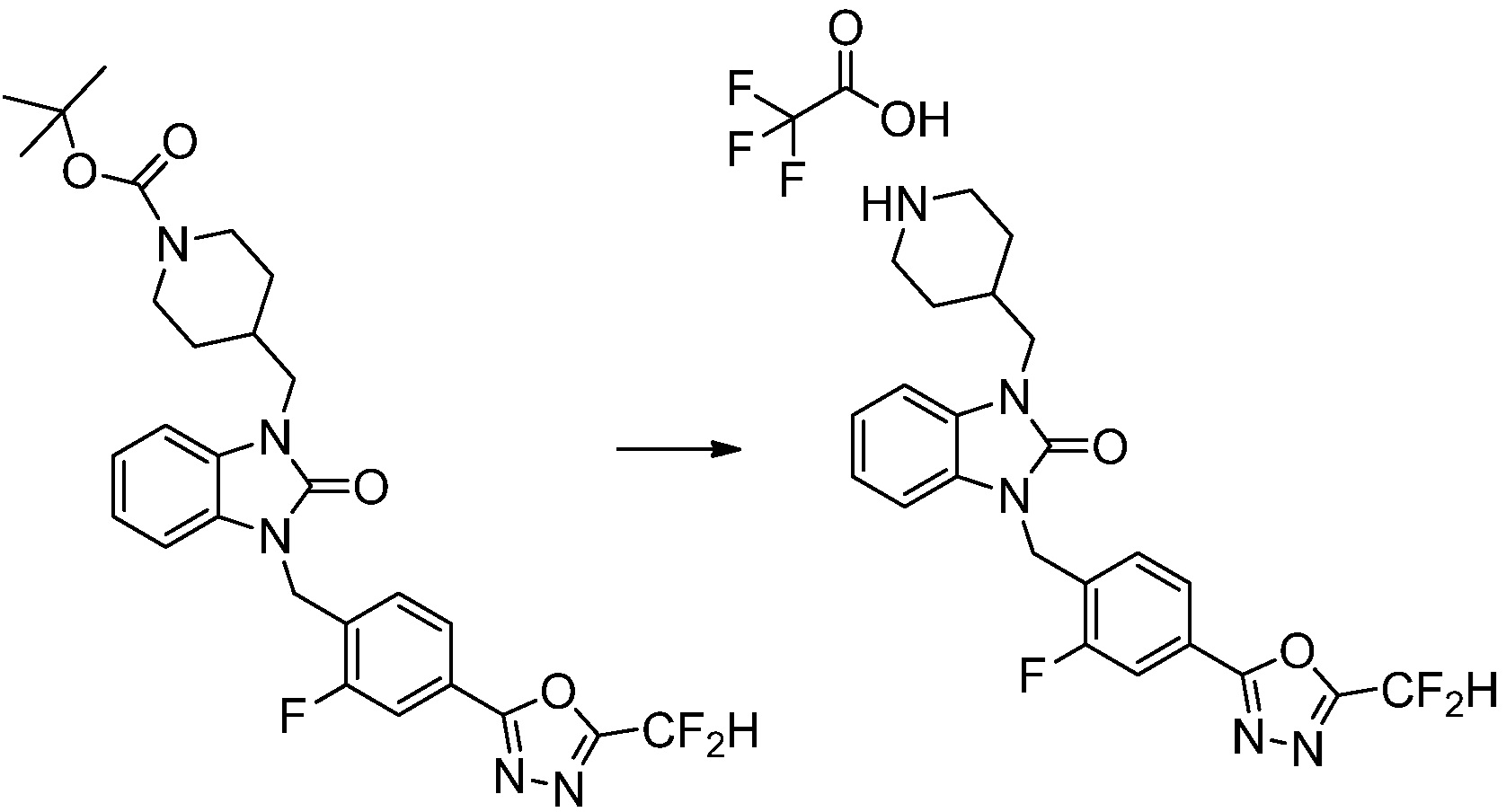
Трет-бутил 4-((3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилат (0,550 г, 0,986 ммоль), полученный на стадии 1 и трифторуксусную кислоту (1,511 мл, 19,728 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,570 г, 101,1%, коричневое масло).
[Стадия 3] Синтез соединения 38
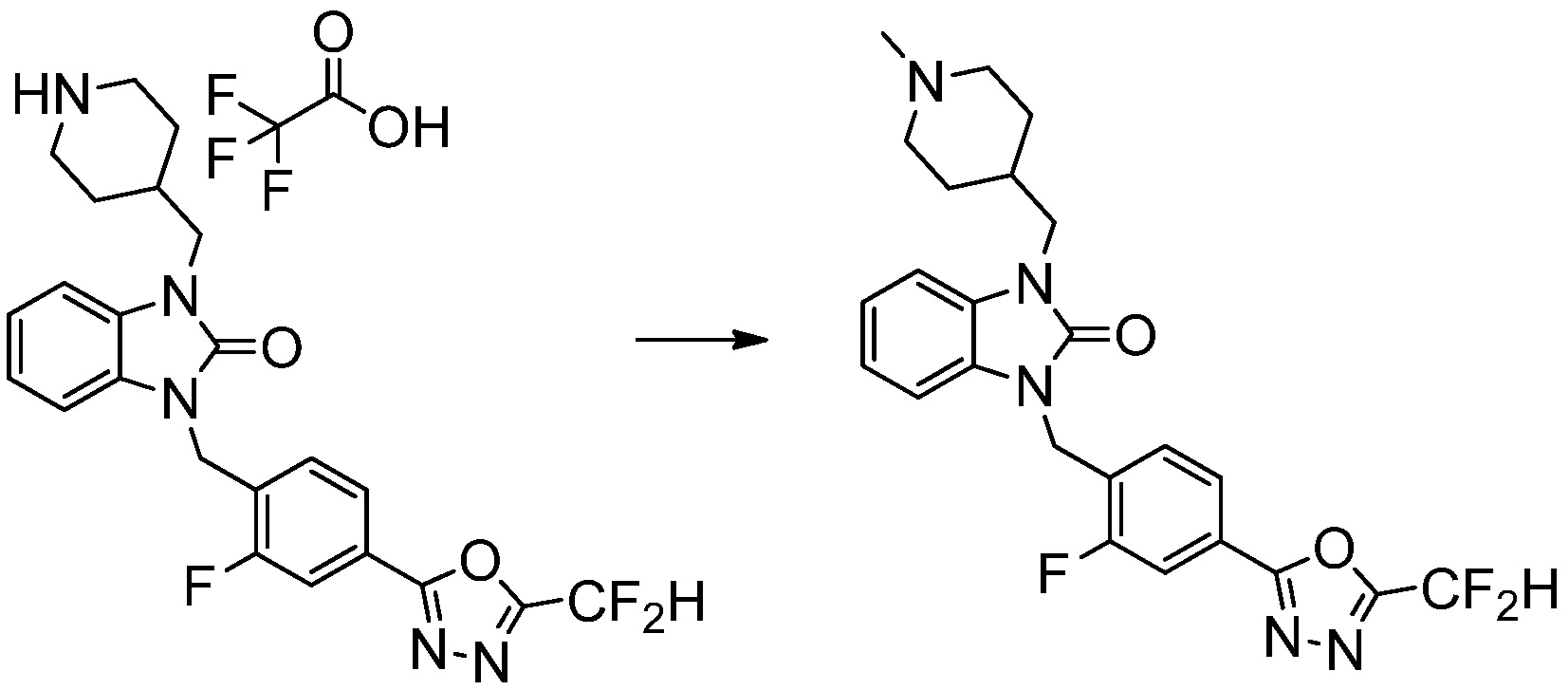
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-илметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,200 г, 0,350 ммоль), полученный на стадии 2, и N,N-диизопропилэтиламин (0,061 мл, 0,350 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем формальдегид (0,021 г, 0,700 ммоль) и триацетоксиборгидрид натрия (1,558 г, 7,349 ммоль) добавляют туда и дополнительно перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,150 г, 90,9%) в виде бесцветного масла.
МСНР (ЭР) m/z 558,4 (M+ + 1).
Пример 39: Синтез соединения 39, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 39
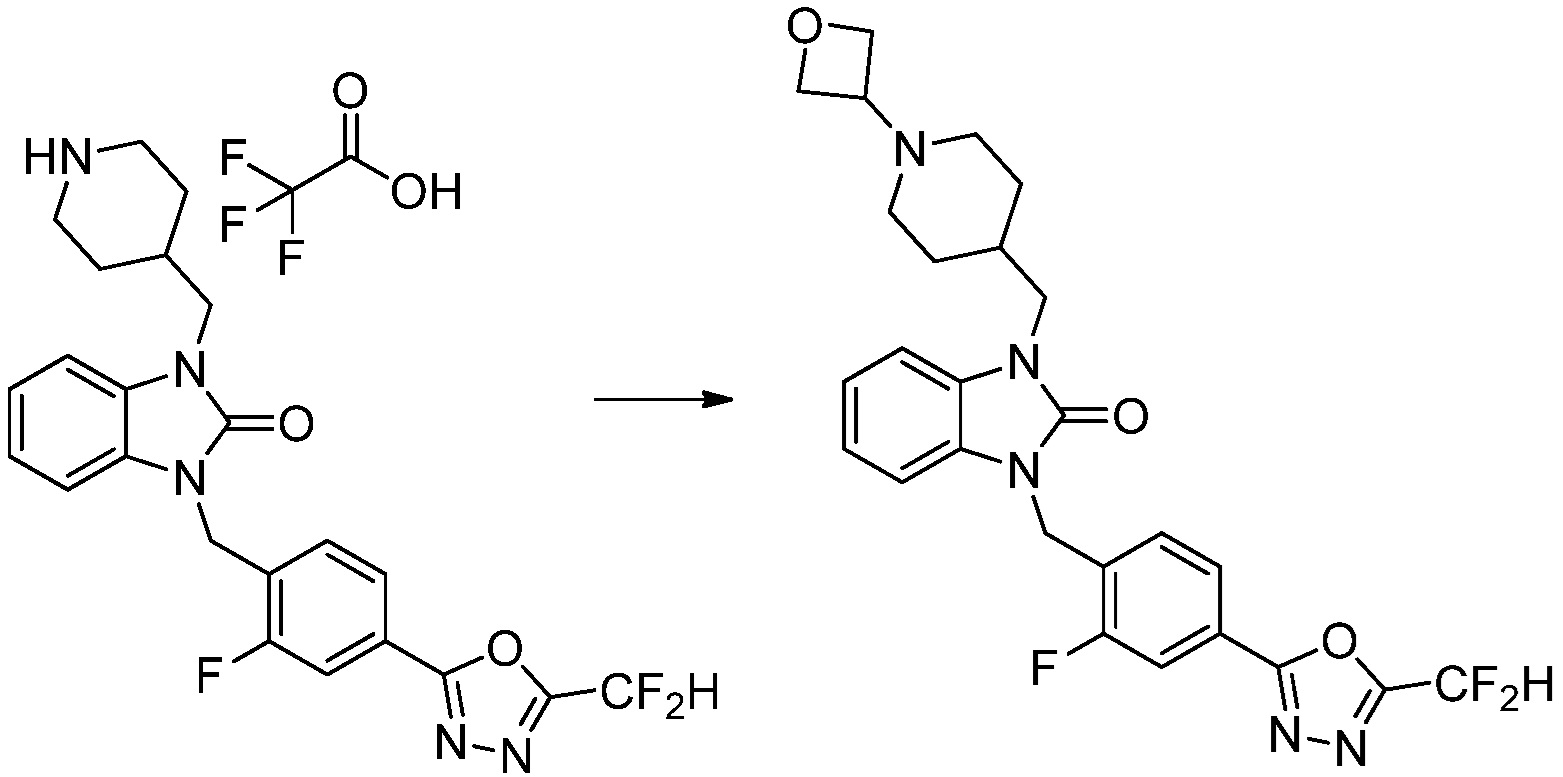
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-илметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (0,200 г, 0,350 ммоль) и N,N-диизопропилэтиламин (0,061 мл, 0,350 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем оксетан-3-он (0,050 г, 0,700 ммоль) и триацетоксиборгидрид натрия (1,558 г, 7,349 ммоль) добавляют туда и дополнительно перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 55,6%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 7,86 ~ 7,81 (м, 2H), 7,45 (дд, J=8,2, 7,5 Гц, 1H), 7,13 ~ 7,01 (м, 3H), 7,02 (с, 1H), 6,96 ~ 6,94 (м, 1H), 6,91 (с, 1H), 6,78 (с, 1H), 5,00 (с, 2H), 4,65 ~ 4,58 (м, 4H), 3,83 (д, J=7,1 Гц, 2H), 3,47 ~ 3,43 (м, 1H), 2,77 ~ 2,74 (м, 2H), 2,00 ~ 1,90 (м, 1H), 1,84 ~ 1,72 (м, 4H), 1,51 ~ 1,47 (м, 2H).
Пример 40: Синтез соединения 40, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-этилпиперидин-4-ил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((5-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилата
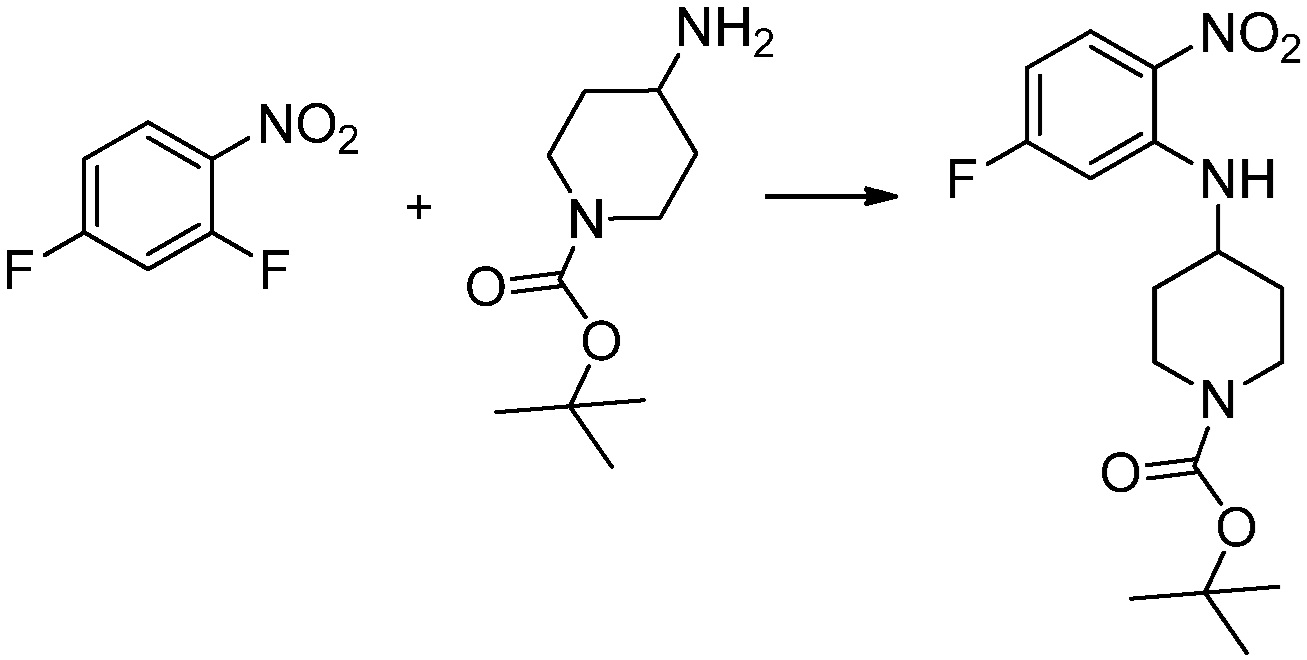
2,4-дифтор-1-нитробензол (2,000 г, 12,572 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (2,518 г, 12,572 ммоль), карбонат калия (2,085 г, 15,086 ммоль) и йодид калия (0,021 г, 0,126 ммоль) растворяют в N,N-диметилформамиде (30 мл) при 80°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (3,550 г, 83,2%) в виде желтой жидкости.
[Стадия 2] Синтез трет-бутил 4-((2-амино-5-фторфенил)амино)пиперидин-1-карбоксилата
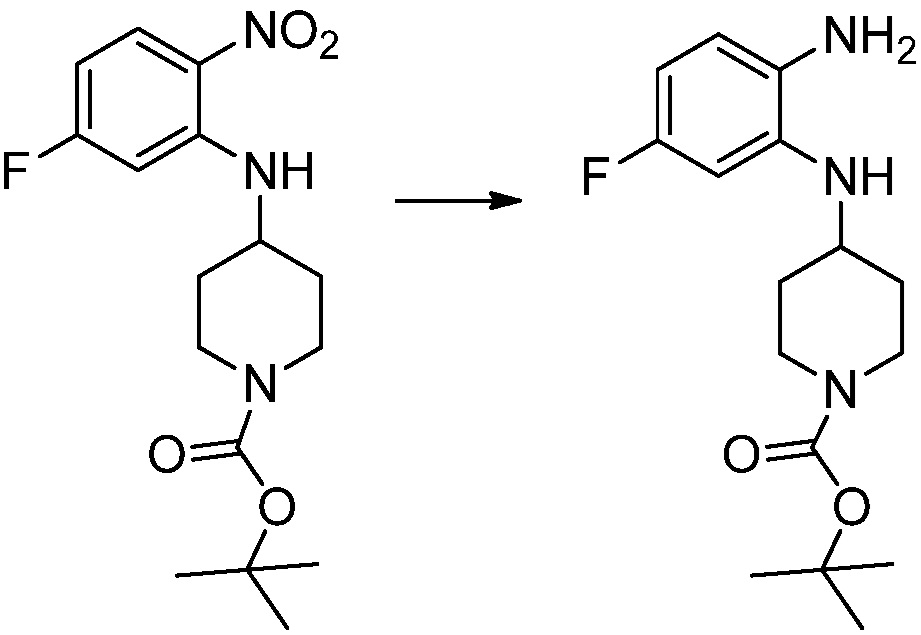
Трет-бутил 4-((5-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилат (3,550 г, 10,461 ммоль), полученный на стадии 1, и Pd/C (45,00%, 0,247 г, 1,046 ммоль) растворяют в метаноле (45 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (2,460 г, 76,0%) в виде красного твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(6-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
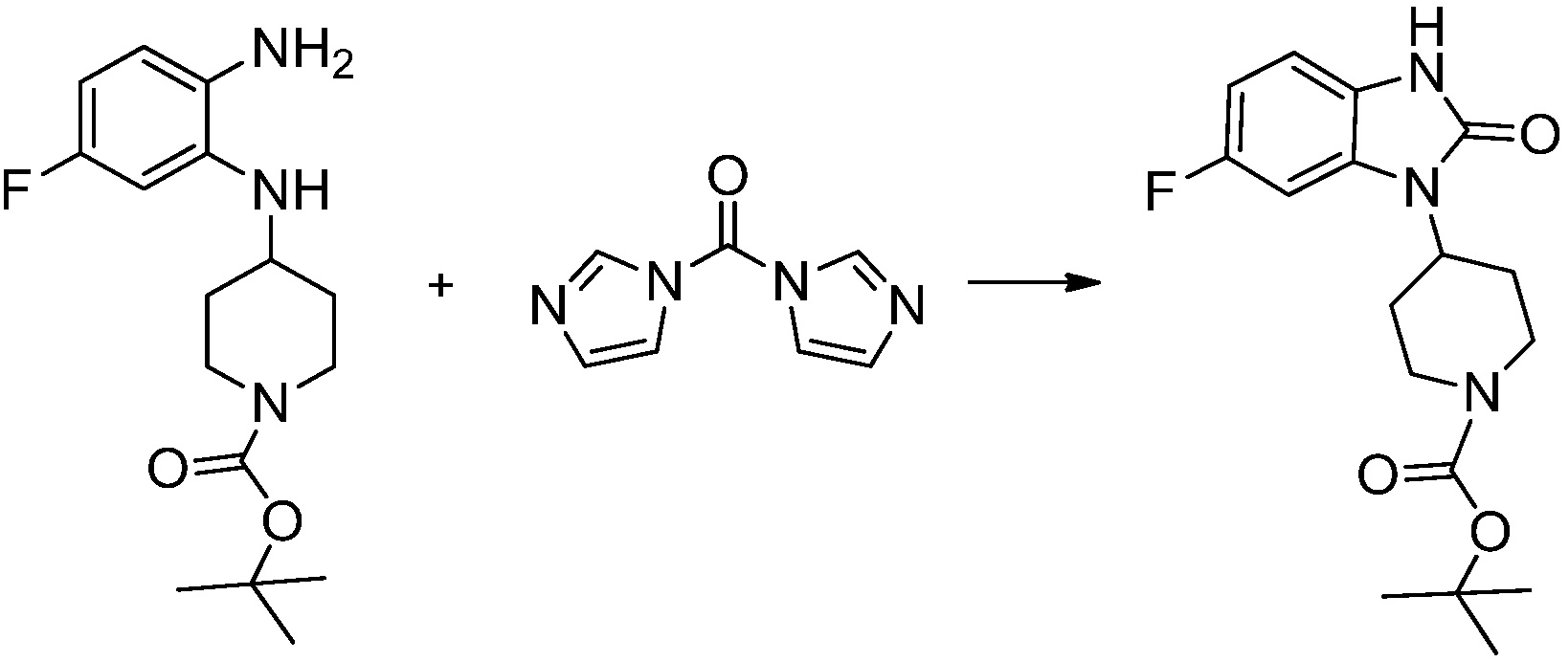
Трет-бутил 4-((2-амино-5-фторфенил)амино)пиперидин-1-карбоксилат (6,520 г, 21,074 ммоль), полученный на стадии 2, растворяют в N,N-диметилформамиде (210 мл) при 0°C, после чего ди(1H-имидазол-1-ил)метанон (4,101 г, 25,288 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0 to 80%) и концентрируют с получением указанного в заголовке соединения в виде фиолетового твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
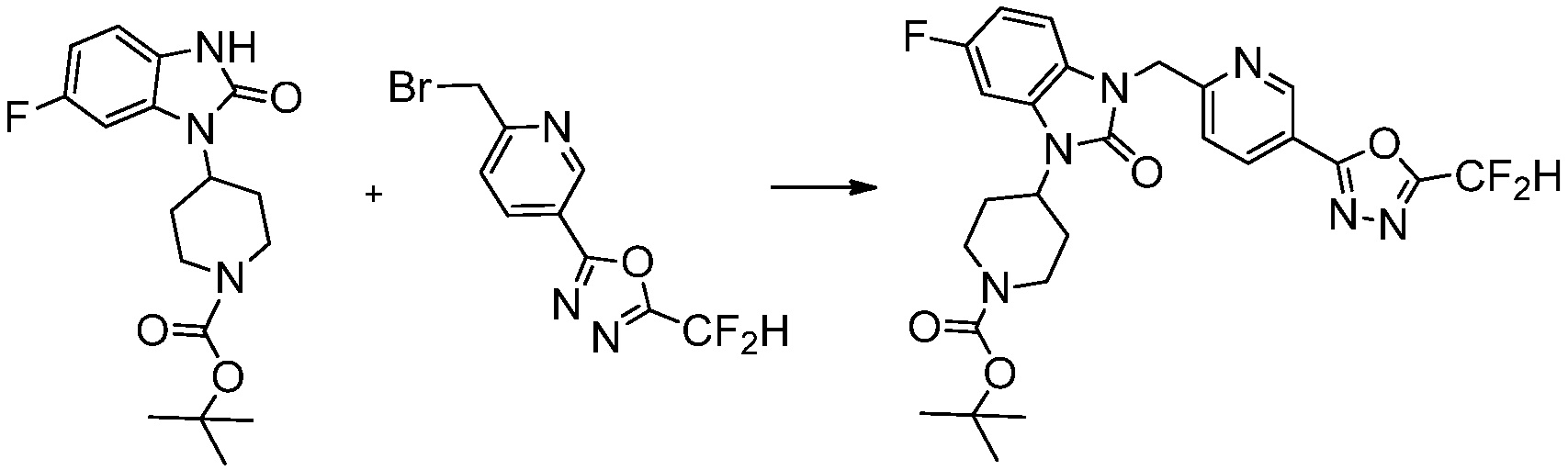
Трет-бутил 4-(6-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,200 г, 3,578 ммоль), полученный на стадии 3 растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,215 г, 5,367 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 20 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,557 г, 5,367 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-60%) и концентрируют с получением указанного в заголовке соединения (1,400 г, 71,9%) в виде коричневого твердого вещества.
[Стадия 5] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
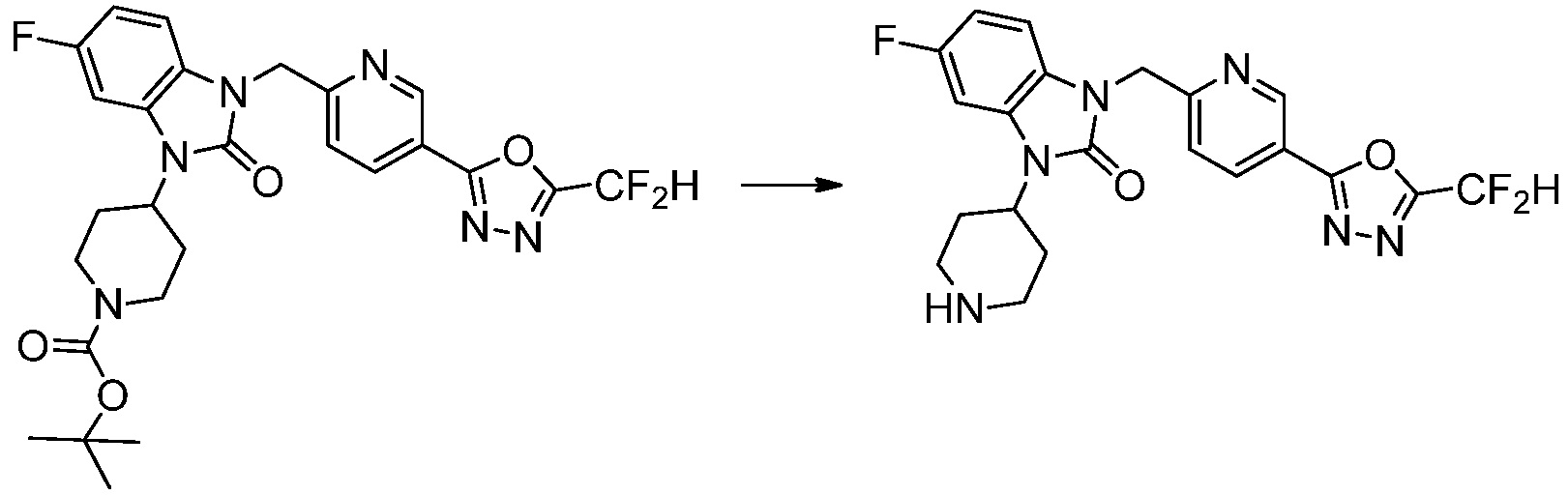
Трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,400 г, 2,571 ммоль), полученный на стадии 4, растворяют в дихлорметане (5 мл), после чего трифторуксусную кислоту (3,937 мл, 51,420 ммоль) добавляют в полученный раствор при 0° и перемешивают при комнатной температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (1,100 г, 96,3%, коричневое твердое вещество).
[Стадия 6] Синтез соединения 40
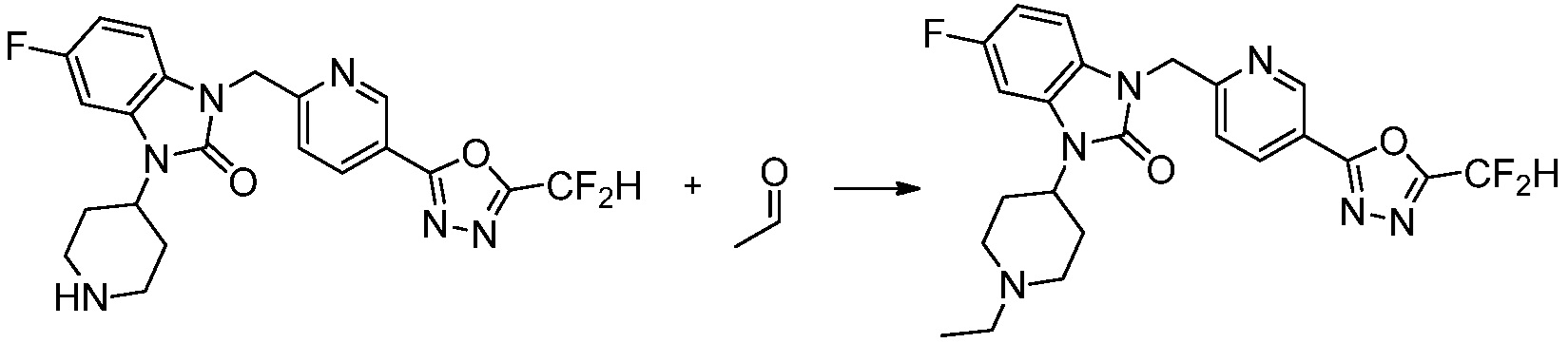
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5, ацетальдегид (0,020 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (1,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,071 г, 66,4%) в виде коричневой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (дд, J=2,1, 0,6 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,38 (д, J=8,2 Гц, 1H), 7,18 (дд, J=9,2, 2,3 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,86 (дд, J=8,6, 4,5 Гц, 1H), 6,71 (тд, J=9,0, 1,7 Гц, 1H), 5,24 (с, 2H), 4,51-4,42 (м, 1H), 3,21 (д, J=11,8 Гц, 2H), 2,60-2,46 (м, 4H), 2,21 (тд, J=12,1, 2,0 Гц, 2H), 1,86 (дд, J=12,1, 2,3 Гц, 2H), 1,15 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 473,3 (M+ + 1).
Пример 41: Синтез соединения 41, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 41
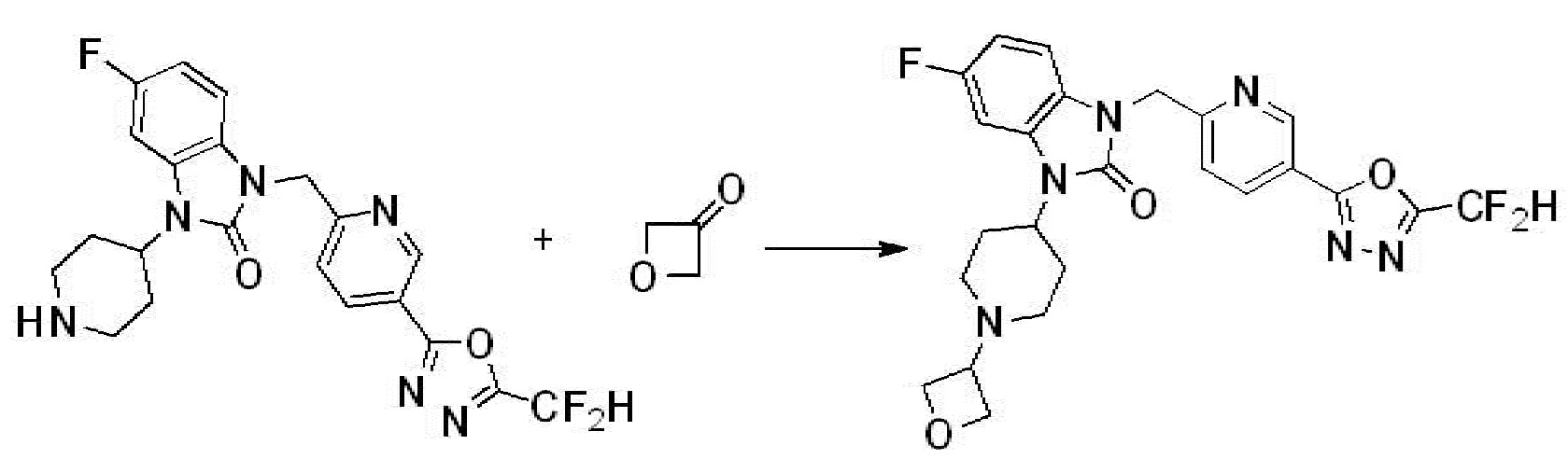
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 40, оксетан-3-он (0,032 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (1,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,084 г, 74,1%) в виде коричневой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (дд, J=2,2, 0,7 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,39 (дд, J=8,2, 0,6 Гц, 1H), 7,11 (дд, J=9,1, 2,3 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,87 (дд, J=8,6, 4,5 Гц, 1H), 6,72 (тд, J=9,0, 2,3 Гц, 1H), 5,24 (с, 2H), 4,66 (дт, J=10,6, 5,2 Гц, 4H), 4,46-4,38 (м, 1H), 3,58-3,52 (м, 1H), 2,94-2,92 (м, 2H), 2,50-2,40 (м, 2H), 2,07-2,04 (м, 2H), 1,86 (дд, J=12,1, 2,4 Гц, 2H); МСНР (ЭР) m/z 501,4 (M+ + 1).
Пример 42: Синтез соединения 42, 3-(1-ацетилпиперидин-4-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 42
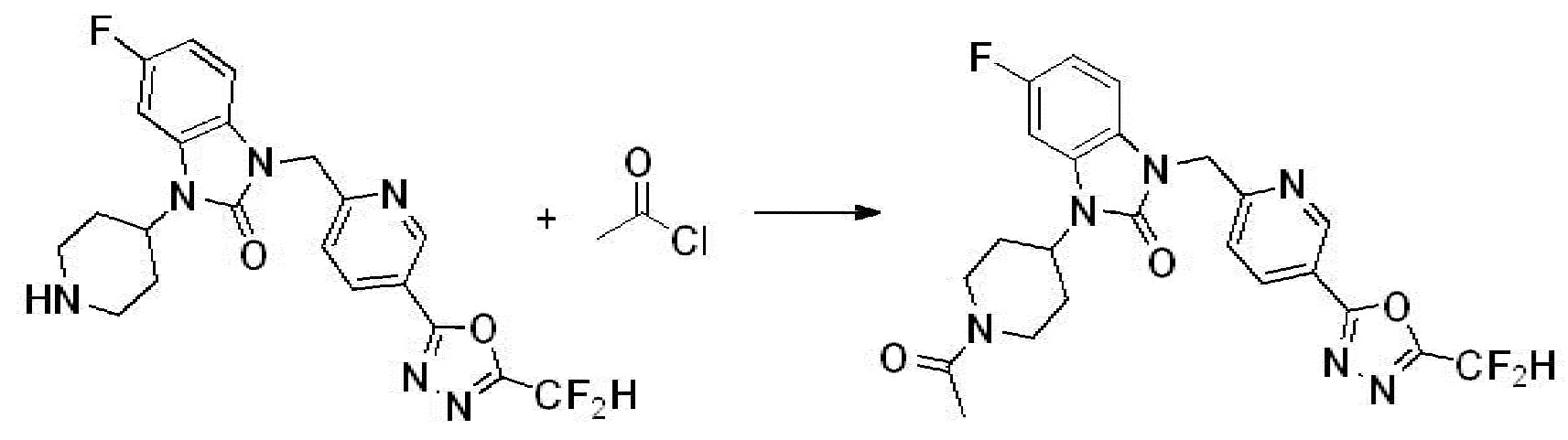
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 40, ацетилхлорид (0,032 мл, 0,450 ммоль), и триэтиламин (0,063 мл, 0,450 ммоль) растворяют в дихлорметане (1,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (0,030 г, 27,4%) в виде коричневой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (с, 1H), 8,33 (д, J=8,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,06-6,73 (м, 4H), 5,24 (с, 2H), 4,88 (д, J=13,0 Гц, 1H), 4,53 (т, J=12,3 Гц, 1H), 4,02 (д, J=12,4 Гц, 1H), 3,24 (т, J=12,7 Гц, 1H), 2,67 (т, J=12,8 Гц, 1H), 2,36-2,25 (м, 2H), 2,17 (с, 3H), 1,93 (т, J=13,3 Гц, 2H); МСНР (ЭР) m/z 487,4 (M+ + 1).
Пример 43: Синтез соединения 43, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 43
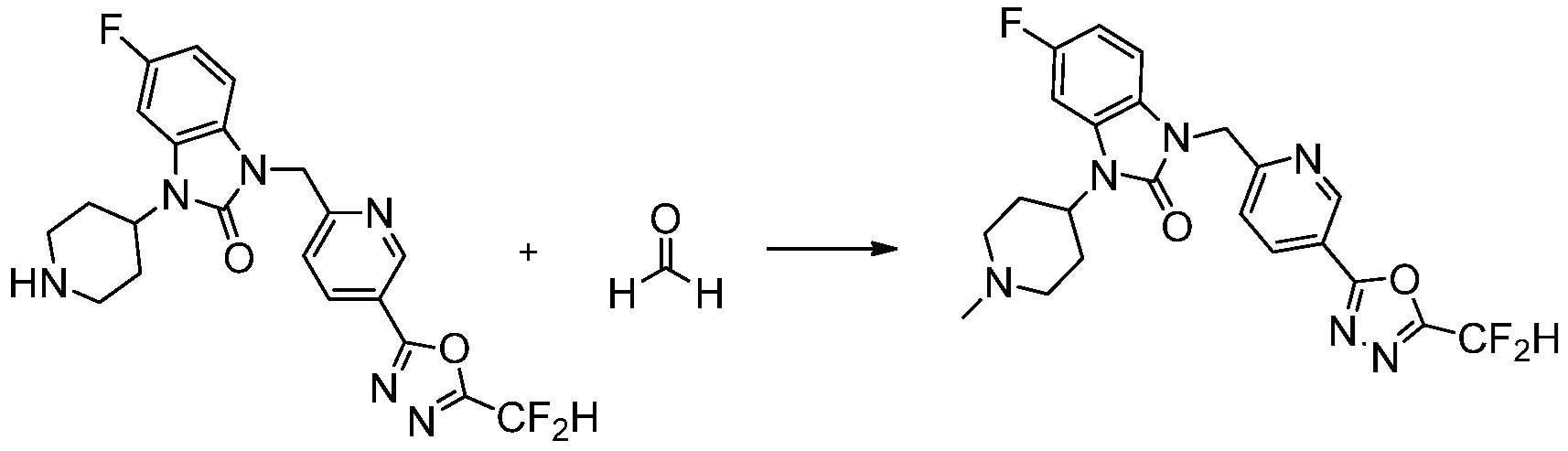
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 40, уксусную кислоту (0,014 мл, 0,248 ммоль), триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) и формальдегид (0,014 г, 0,450 ммоль) растворяют в дихлорметане (1,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-15%) и концентрируют с получением указанного в заголовке соединения (0,051 г, 49,8%) в виде коричневой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=2,0 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,38 (д, J=8,2 Гц, 1H), 7,16 (дд, J=9,2, 2,3 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,86 (дд, J=8,6, 4,5 Гц, 1H), 6,72 (тд, J=9,0, 2,2 Гц, 1H), 5,24 (с, 2H), 4,51-4,42 (м, 1H), 3,13 (д, J=11,8 Гц, 2H), 2,58-2,48 (м, 2H), 2,40 (с, 3H), 2,28 (тд, J=12,1, 1,9 Гц, 2H), 1,84 (дд, J=12,3, 2,3 Гц, 2H); МСНР (ЭР) m/z 459,3 (M+ + 1).
Пример 44: Синтез соединения 44, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(1-(тетрагидро-2H-пиран-4-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 44
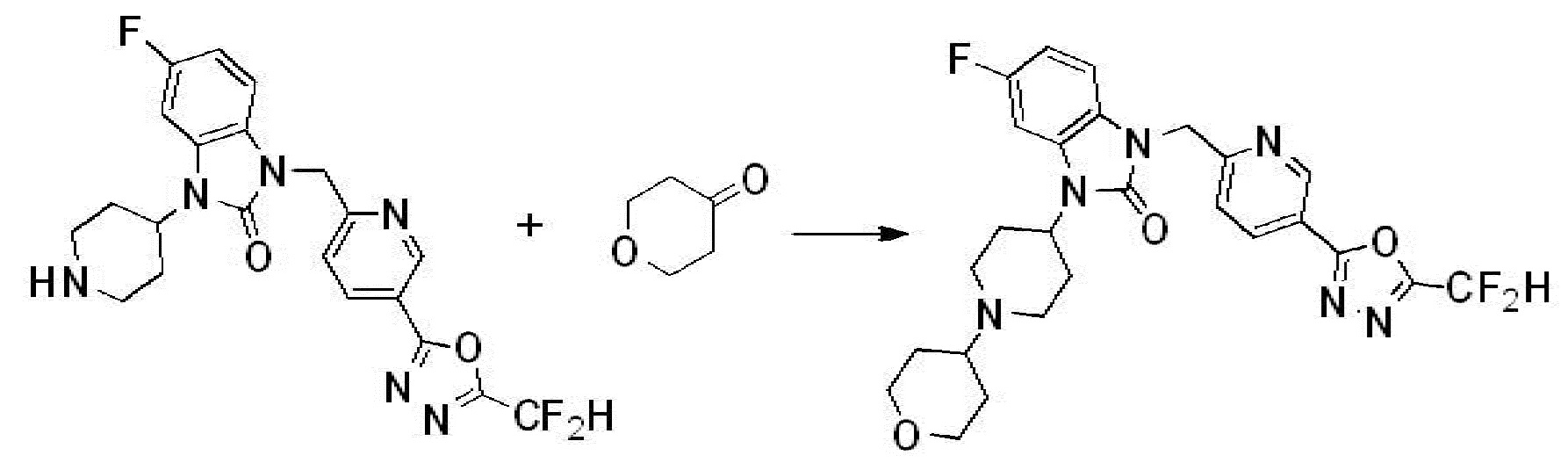
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 40, тетрагидро-4H-пиран-4-он (0,045 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,076 г, 63,5%) в виде желтой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,2, 0,8 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,2 Гц, 1H), 7,17 (дд, J=9,2, 1,7 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,87 (дд, J=8,6, 4,5 Гц, 1H), 6,73 (тд, J=9,0, 2,3 Гц, 1H), 5,25 (с, 2H), 4,46-4,39 (м, 1H), 4,05 (дд, J=11,3, 3,9 Гц, 2H), 3,42-3,37 (м, 2H), 3,19 (д, J=8,8 Гц, 2H), 2,70-2,65 (м, 1H), 2,50-2,39 (м, 4H), 1,90-1,80 (м, 4H), 1,72-1,61 (м, 2H); МСНР (ЭР) m/z 539,6 (M+ + 1).
Пример 45: Синтез соединения 45, 3-(1-циклобутилпиперидин-4-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 45
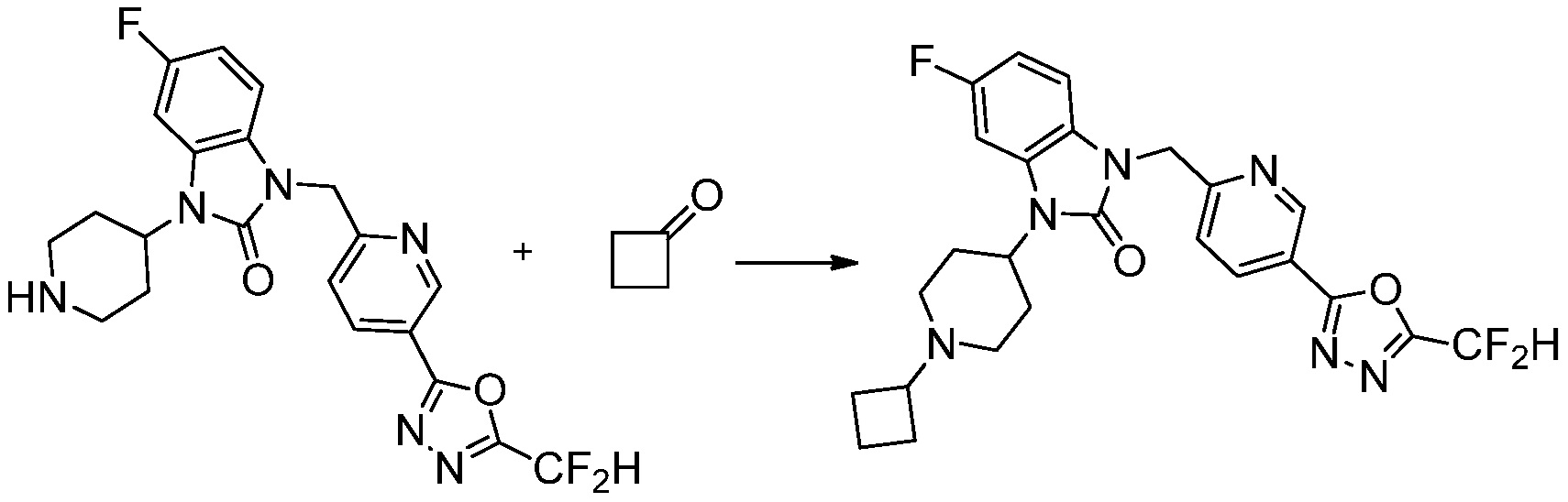
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный синтезом со стадии 5 соединения 40, циклобутанон (0,032 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,097 г, 86,5%) в виде желтой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (дд, J=2,2, 0,8 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (дд, J=8,2, 0,7 Гц, 1H), 7,22-7,19 (м, 1H), 6,93 (т, J=51,6 Гц, 1H), 6,87 (дд, J=8,6, 4,5 Гц, 1H), 6,74 (тд, J=9,0, 2,1 Гц, 1H), 5,26 (с, 2H), 4,51-4,43 (м, 1H), 3,17 (д, J=11,7 Гц, 2H), 2,92-2,84 (м, 1H), 2,55-2,46 (м, 2H), 2,11-2,01 (м, 6H), 1,86 (дд, J=12,2, 2,3 Гц, 2H), 1,79-1,66 (м, 2H); МСНР (ЭР) m/z 499,4 (M+ + 1).
Пример 46: Синтез соединения 46, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(1-изопропилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 46
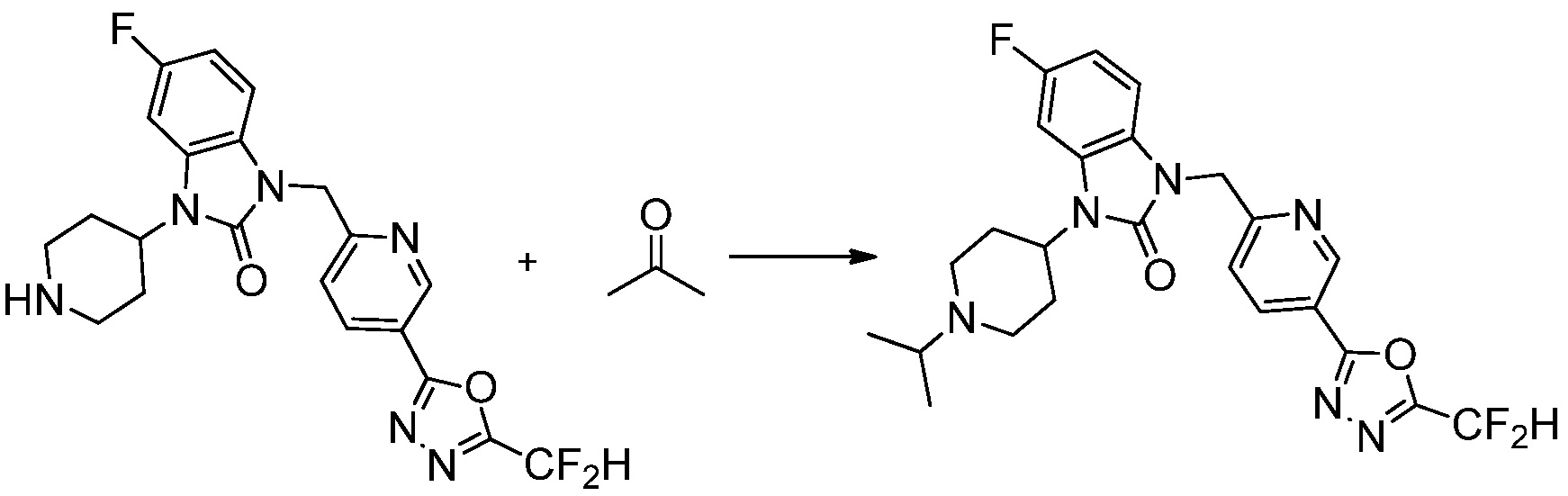
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 40, пропан-2-он (0,026 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,076 г, 69,6%) в виде желтой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (дд, J=2,2, 0,7 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,38 (д, J=8,2 Гц, 1H), 7,30 (дд, J=9,2, 2,3 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,86 (дд, J=8,6, 4,5 Гц, 1H), 6,72 (тд, J=9,0, 2,2 Гц, 1H), 5,24 (с, 2H), 4,56-4,48 (м, 1H), 3,26 (д, J=11,1 Гц, 2H), 3,18-3,11 (м, 1H), 2,70-2,52 (м, 4H), 1,90 (дд, J=12,2, 2,8 Гц, 2H), 1,19 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 487,5 (M+ + 1).
Пример 47: Синтез соединения 47, 3-(1-циклогексилпиперидин-4-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 47
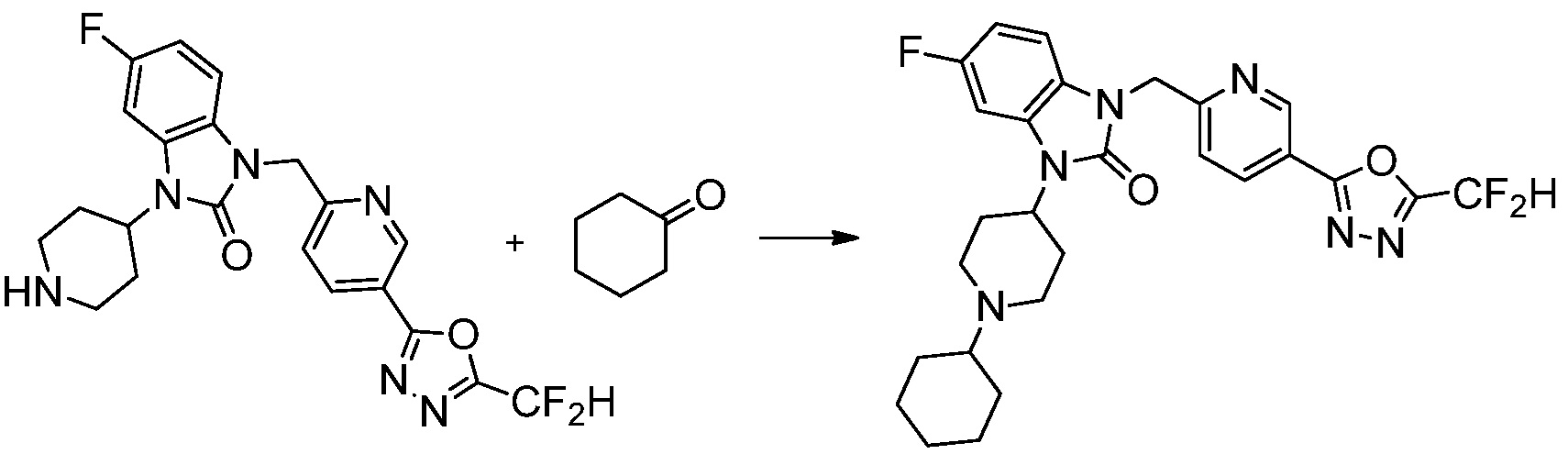
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 40, циклогексанон (0,044 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,094 г, 79,4%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (дд, J=2,2, 0,8 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,39-7,35 (м, 2H), 6,92 (т, J=51,6 Гц, 1H), 6,86 (дд, J=8,6, 4,5 Гц, 1H), 6,73 (тд, J=9,0, 2,3 Гц, 1H), 5,24 (с, 2H), 4,61-4,54 (м, 1H), 3,40 (д, J=9,5 Гц, 2H), 2,90-2,68 (м, 5H), 2,06-2,04 (м, 2H), 1,94-1,86 (м, 4H), 1,70-1,67 (м, 1H), 1,41-1,26 (м, 4H), 1,18-1,08 (м, 1H); МСНР (ЭР) m/z 527,5 (M+ + 1).
Пример 48: Синтез соединения 48, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((4-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилата
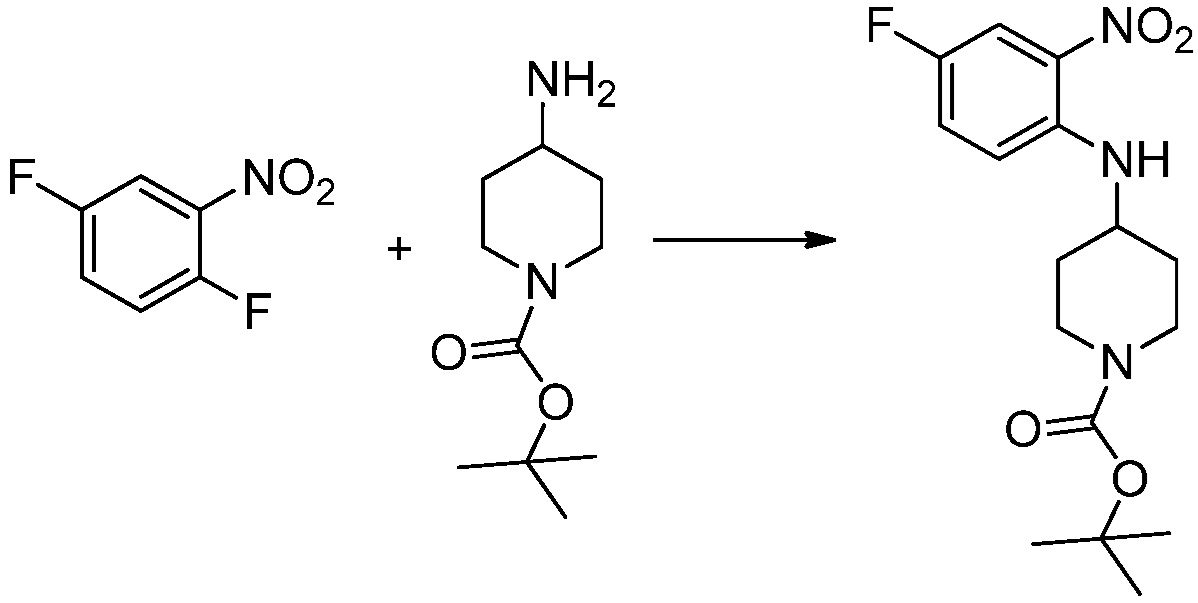
1,4-дифтор-2-нитробензол (2,000 г, 12,572 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (2,518 г, 12,572 ммоль), карбонат калия (2,085 г, 15,086 ммоль) и йодид калия (0,021 г, 0,126 ммоль) растворяют в N,N-диметилформамиде (30 мл) при 80°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (4,230 г, 99,1%) в виде красного твердого вещества.
[Стадия 2] Синтез трет-бутил 4-((2-амино-4-фторфенил)амино)пиперидин-1-карбоксилата
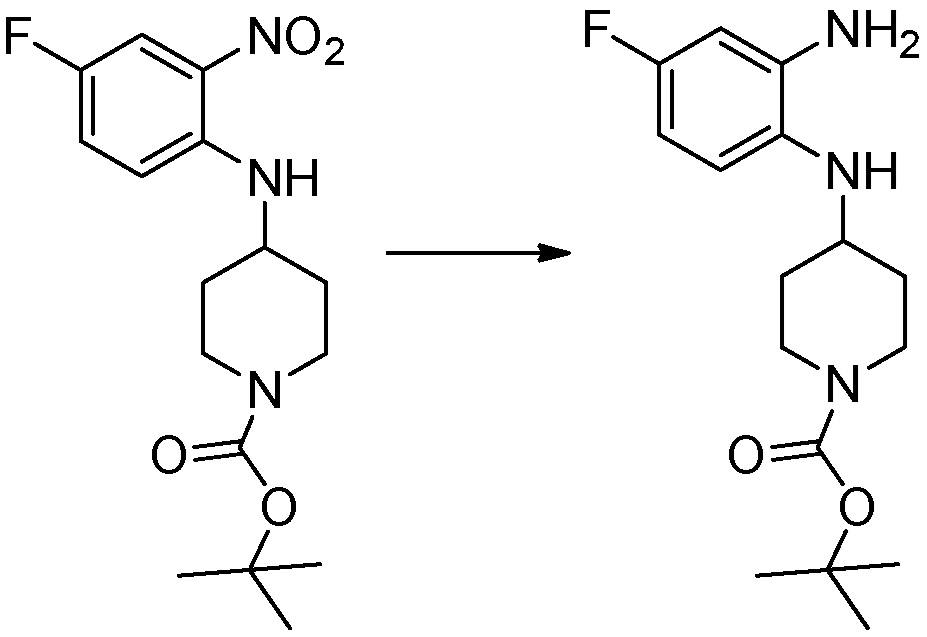
Трет-бутил 4-((4-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилат (4,230 г, 12,464 ммоль), полученный на стадии 1, и Pd/C (10%, 0,295 г, 1,246 ммоль) смешивают в метаноле (50 мл) при комнатной температуре, после чего полученную суспензию перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (3,260 г, 84,5%) в виде красного твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат
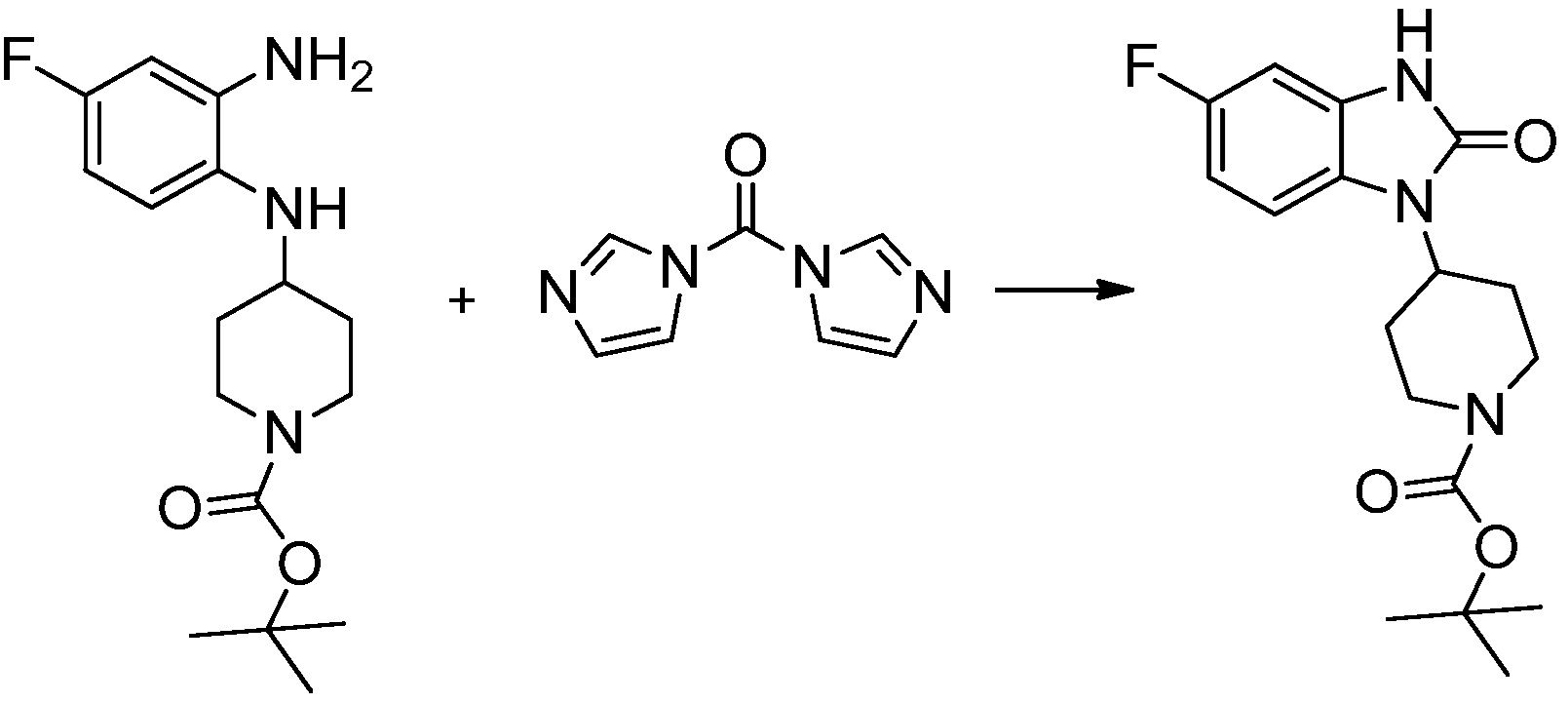
Трет-бутил 4-((2-амино-4-фторфенил)амино)пиперидин-1-карбоксилат (7,300 г, 23,595 ммоль), полученный на стадии 2 и ди(1H-имидазол-1-ил)метанон (4,017 г, 24,775 ммоль) растворяют в N,N-диметилформамиде (150 мл) при 0°C, после чего полученный раствор перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (6,950 г, 87,8%) в виде коричневого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
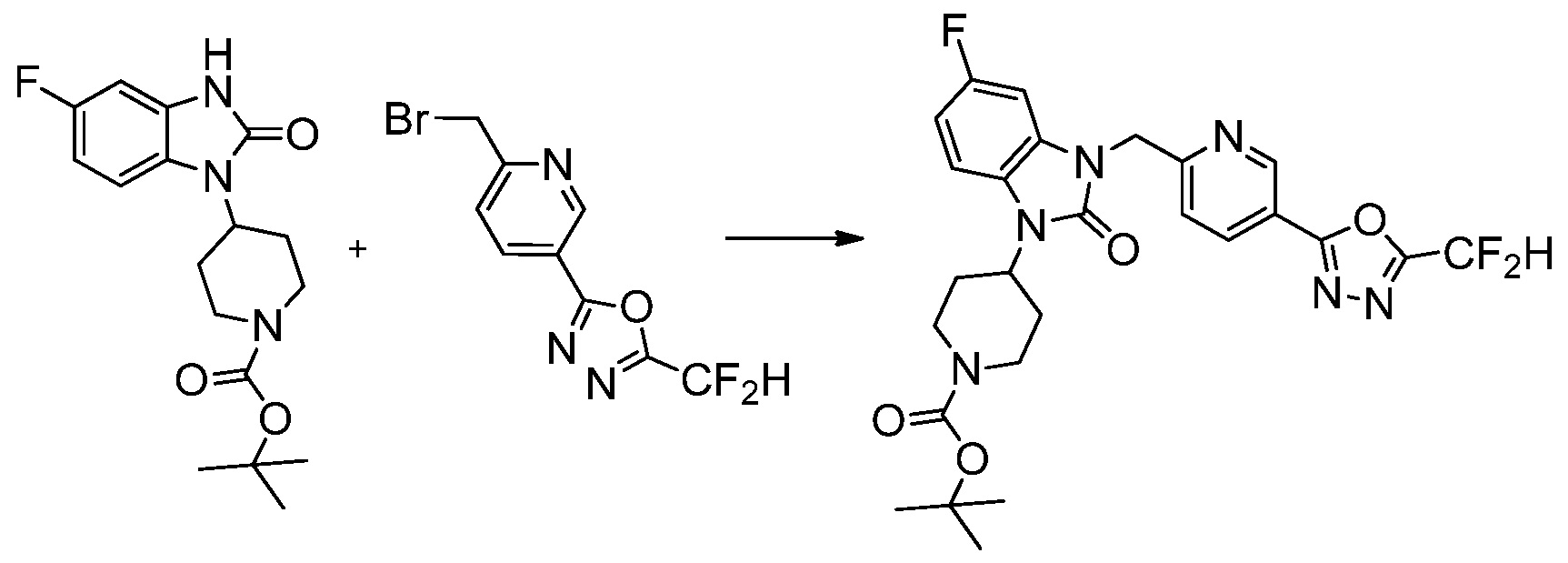
2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (4,000 г, 13,790 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (140 мл) при 0°C, после чего гидрид натрия (60,00%, 0,717 г, 17,927 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. Трет-бутил 4-(5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (6,012 г, 17,927 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-40%) и концентрируют с получением указанного в заголовке соединения (4,680 г, 62,3%) в виде оранжевого твердого вещества.
[Стадия 5] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
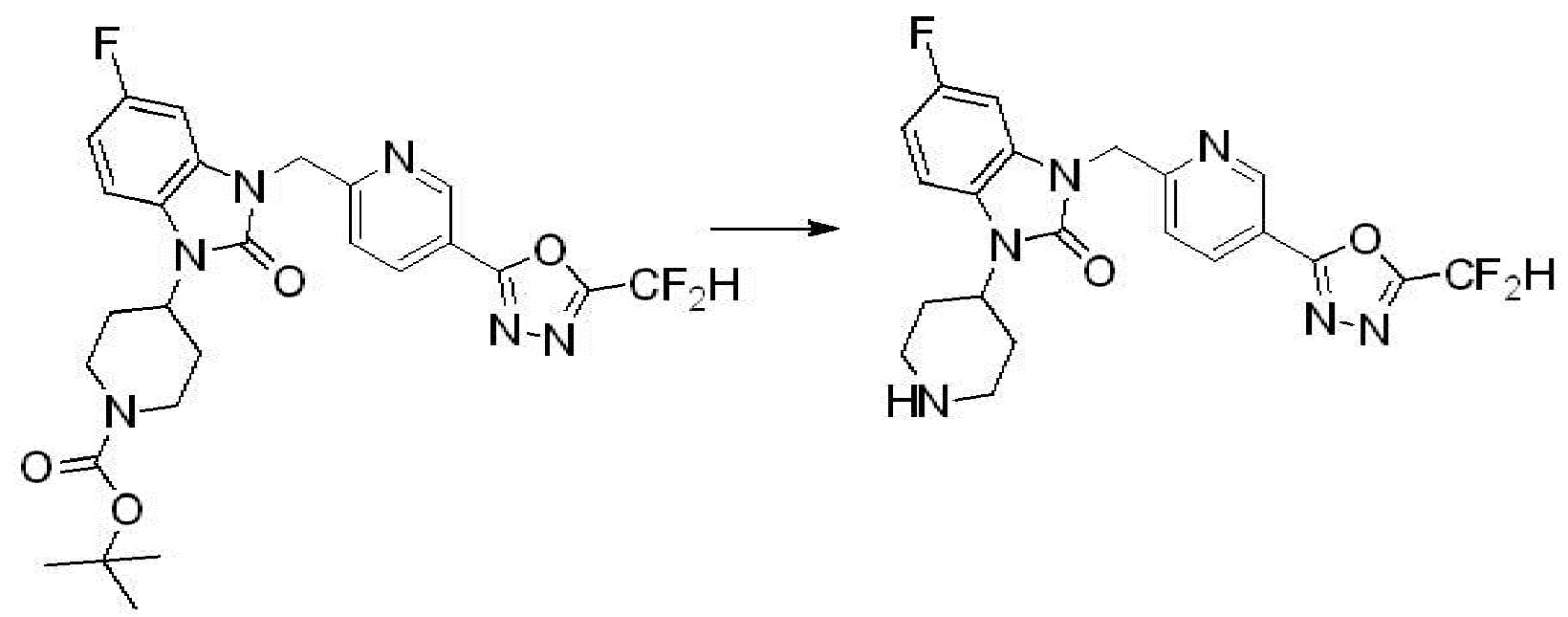
Трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (4,680 г, 8,594 ммоль), полученный на стадии 4 растворяют в дихлорметане (80 мл) при 0°C, после чего трифторуксусную кислоту (13,162 мл, 171,888 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 4,5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (3,710 г, 97,1%) в виде светло-коричневого твердого вещества.
[Стадия 6] Синтез соединения 48
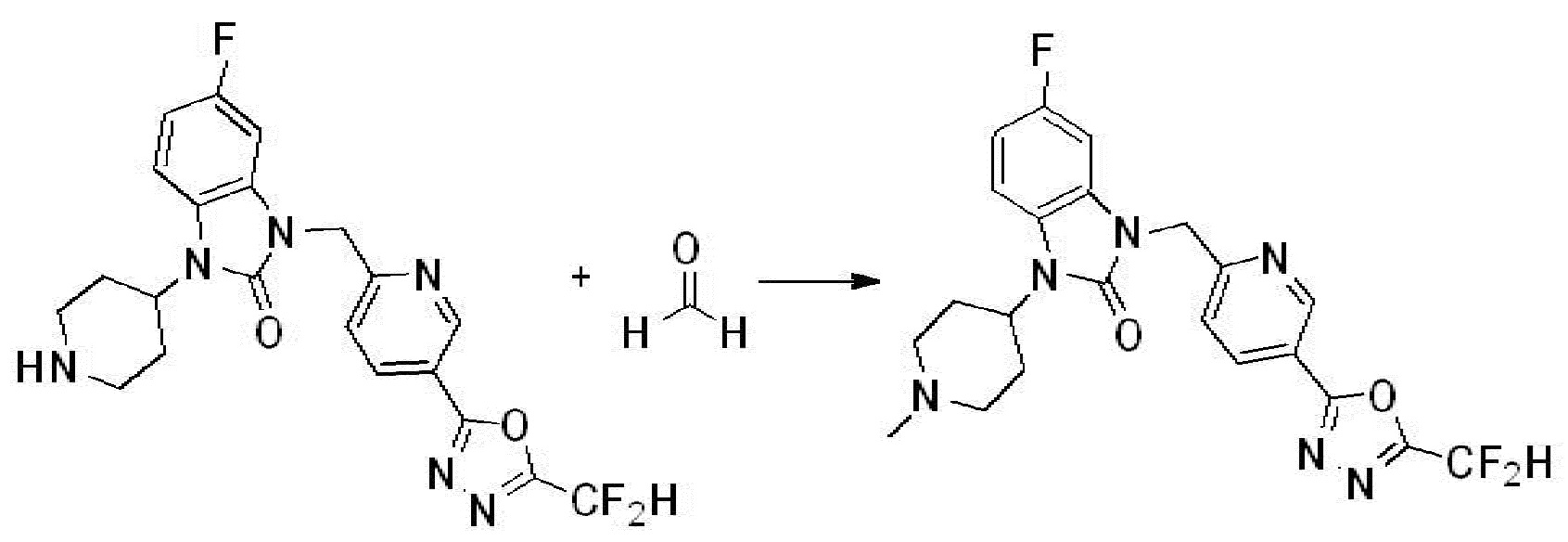
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5, формальдегид (0,014 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,065 г, 63,0%) в виде желтой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (с, 1H), 8,33 (д, J=8,0 Гц, 1H), 7,41 (д, J=7,8 Гц, 1H), 7,26-7,24 (м, 1H), 7,05-6,74 (м, 3H), 5,23 (с, 2H), 4,45-4,39 (м, 1H), 3,04 (д, J=10,6 Гц, 2H), 2,50-2,44 (м, 2H), 2,36 (с, 3H), 2,19 (т, J=11,3 Гц, 2H), 1,84 (д, J=10,6 Гц, 2H); МСНР (ЭР) m/z 459,3 (M+ + 1).
Пример 49: Синтез соединения 49, 1-(1-(2-оксаспиро[3,3]гептан-6-ил)пиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 49
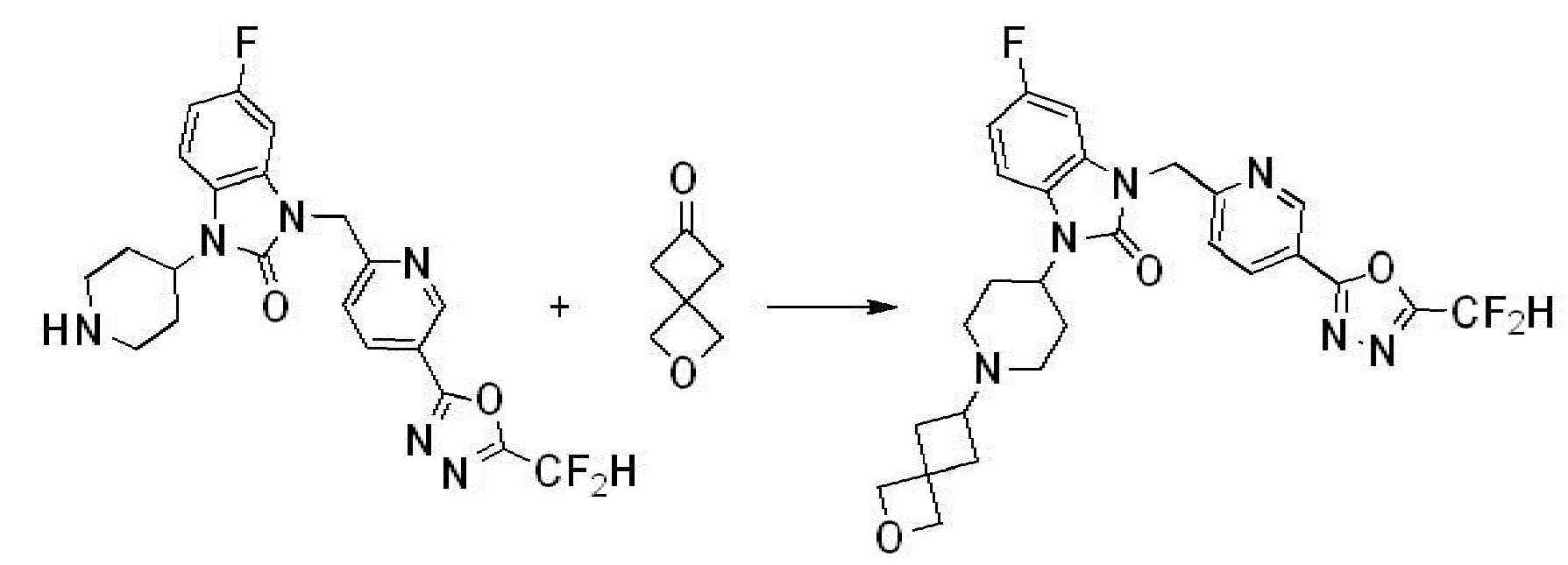
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, 2-оксаспиро[3,3]гептан-6-он (0,050 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,070 г, 57,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,7 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=7,2 Гц, 1H), 7,22 (с, 1H), 7,05-6,74 (м, 3H), 5,23 (с, 2H), 4,72 (с, 2H), 4,61 (с, 2H), 4,45-4,30 (м, 1H), 3,15-2,95 (м, 2H), 2,90-2,50 (м, 1H), 2,50-2,25 (м, 4H), 2,20-1,85 (м, 6H); МСНР (ЭР) m/z 541,6 (M+ + 1).
Пример 50: Синтез соединения 50, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-этилпиперидин-4-ил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 50
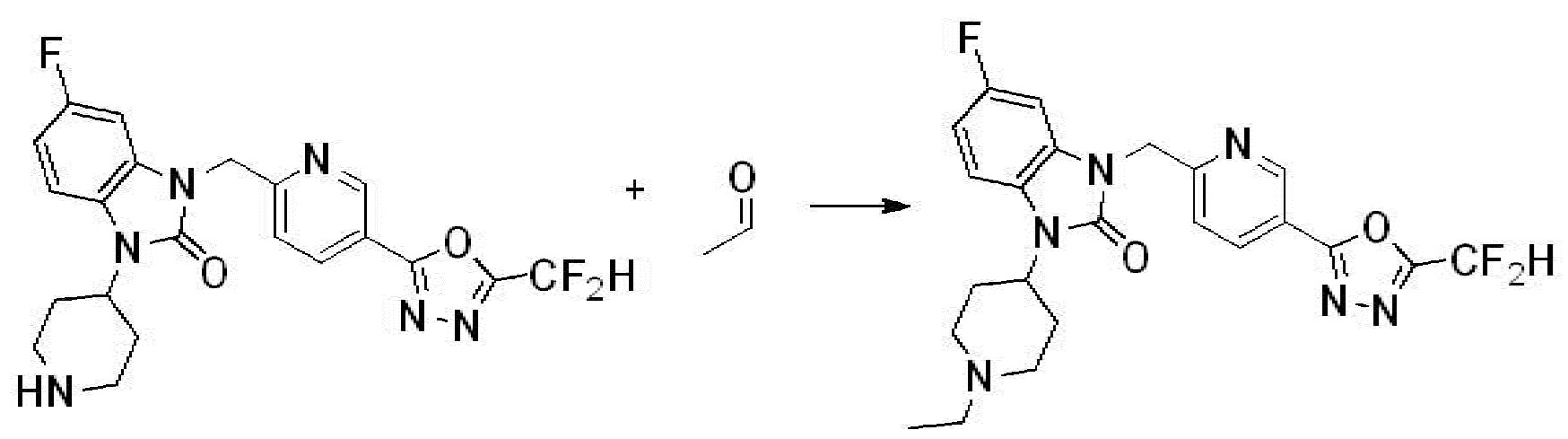
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, ацетальдегид (0,020 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,063 г, 59,4%) в виде желтой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (с, 1H), 8,32 (д, J=8,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,28-7,23 (м, 1H), 6,93 (т, J=51,6 Гц, 1H), 6,77-6,73 (м, 2H), 5,23 (с, 2H), 4,45-4,39 (м, 1H), 3,12 (д, J=11,5 Гц, 2H), 2,50-2,40 (м, 4H), 2,12 (т, J=11,8 Гц, 2H), 1,85 (д, J=11,7 Гц, 2H), 1,12 (т, J=7,1 Гц, 3H); МСНР (ЭР) m/z 473,6 (M+ + 1).
Пример 51: Синтез соединения 51, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-изопропилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 51
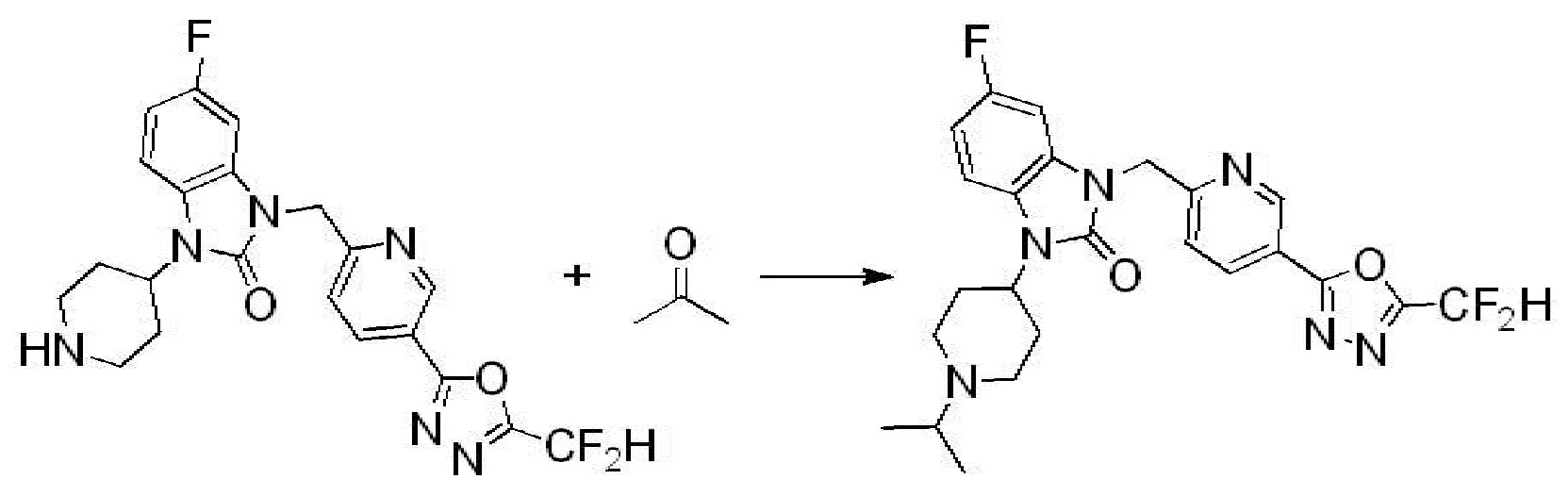
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, пропан-2-он (0,026 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,068 г, 62,5%) в виде желтой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (т, J=1,1 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=8,3 Гц, 1H), 7,31-7,28 (м, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,78-6,72 (м, 2H), 5,23 (с, 2H), 4,42-4,35 (м, 1H), 3,04 (д, J=9,9 Гц, 2H), 2,87-2,80 (м, 1H), 2,49-2,34 (м, 4H), 1,87-1,84 (м, 2H), 1,09 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 487,6 (M+ + 1).
Пример 52: Синтез соединения 52, 1-(1-циклобутилпиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 52
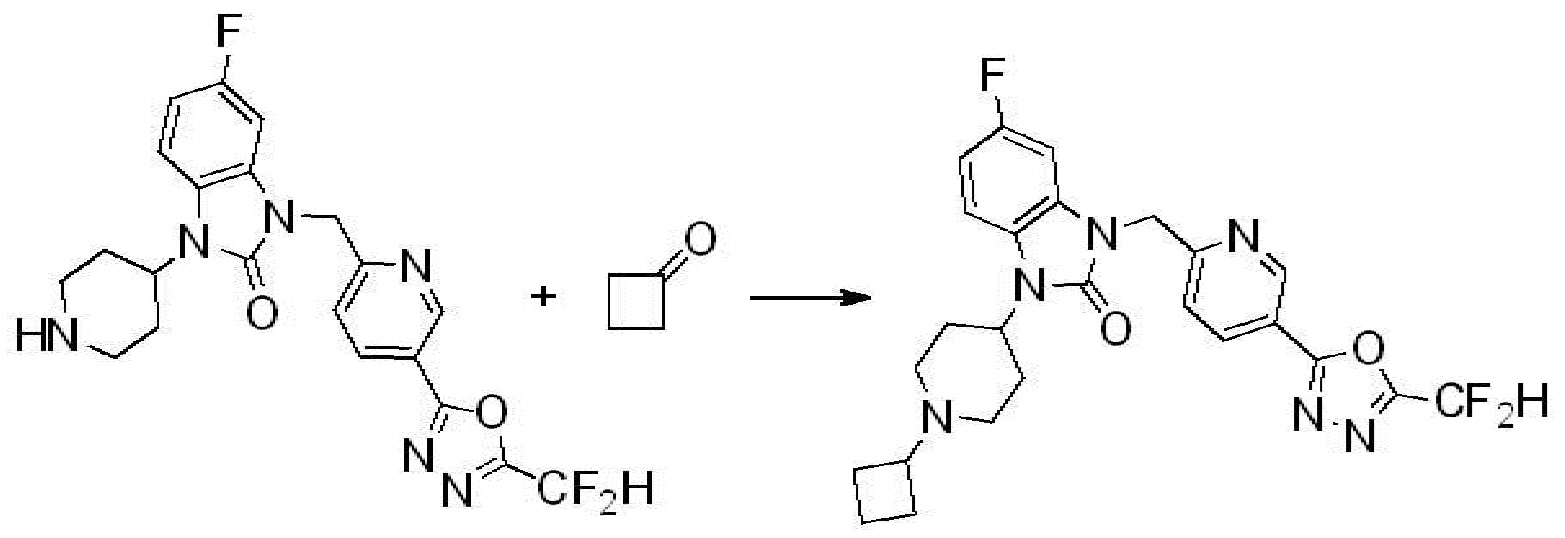
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 45, циклобутанон (0,032 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,096 г, 85,3%) в виде прозрачной жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (т, J=1,1 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,2 Гц, 1H), 7,29-7,26 (м, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,79-6,73 (м, 2H), 5,23 (с, 2H), 4,45-4,37 (м, 1H), 3,06 (д, J=11,4 Гц, 2H), 2,82-2,74 (м, 1H), 2,46-2,37 (м, 2H), 2,09-2,03 (м, 2H), 1,96-1,85 (м, 6H), 1,76-1,64 (м, 2H); МСНР (ЭР) m/z 499,6 (M+ + 1).
Пример 53: Синтез соединения 53, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 53
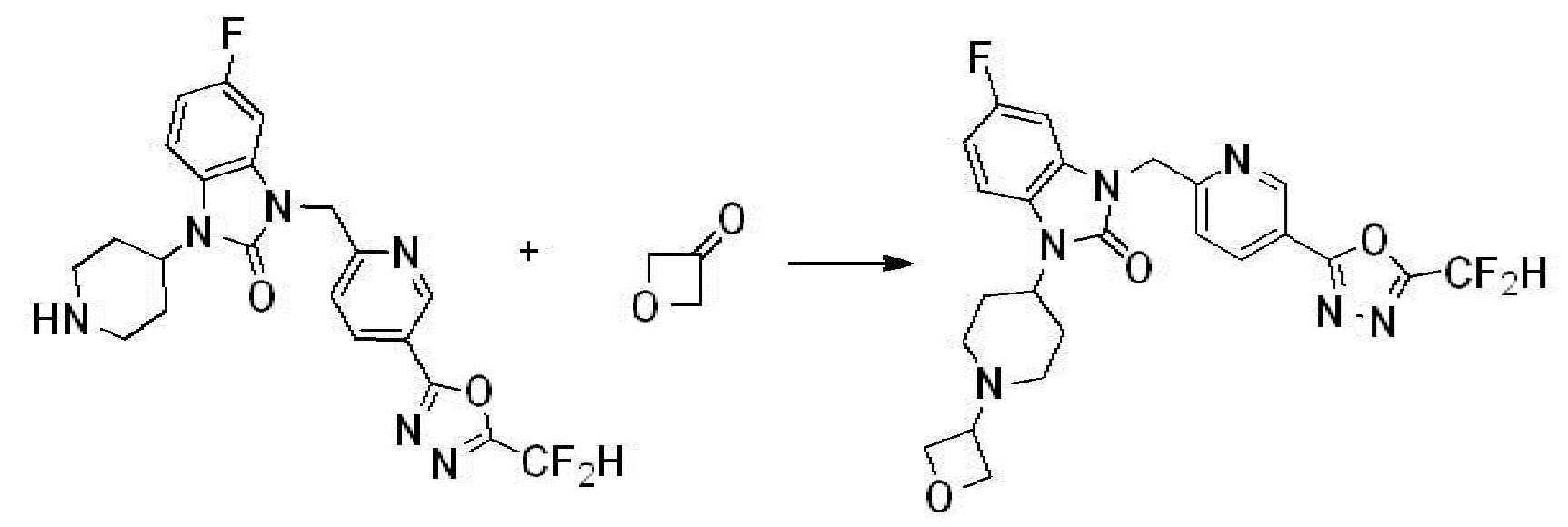
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, оксетан-3-он (0,032 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия(0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 97,6%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (т, J=1,1 Гц, 1H), 8,32 (дд, J=8,2, 2,1 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,23 (дд, J=8,4, 4,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,79-6,74 (м, 2H), 5,22 (с, 2H), 4,68-4,60 (м, 4H), 4,46-4,37 (м, 1H), 3,56-3,49 (м, 1H), 2,90 (д, J=11,5 Гц, 2H), 2,50-2,40 (м, 2H), 2,05-1,99 (м, 2H), 1,87-1,85 (м, 2H); МСНР (ЭР) m/z 501,5 (M+ + 1).
Пример 54: Синтез соединения 54, 1-(1-циклогексилпиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 54
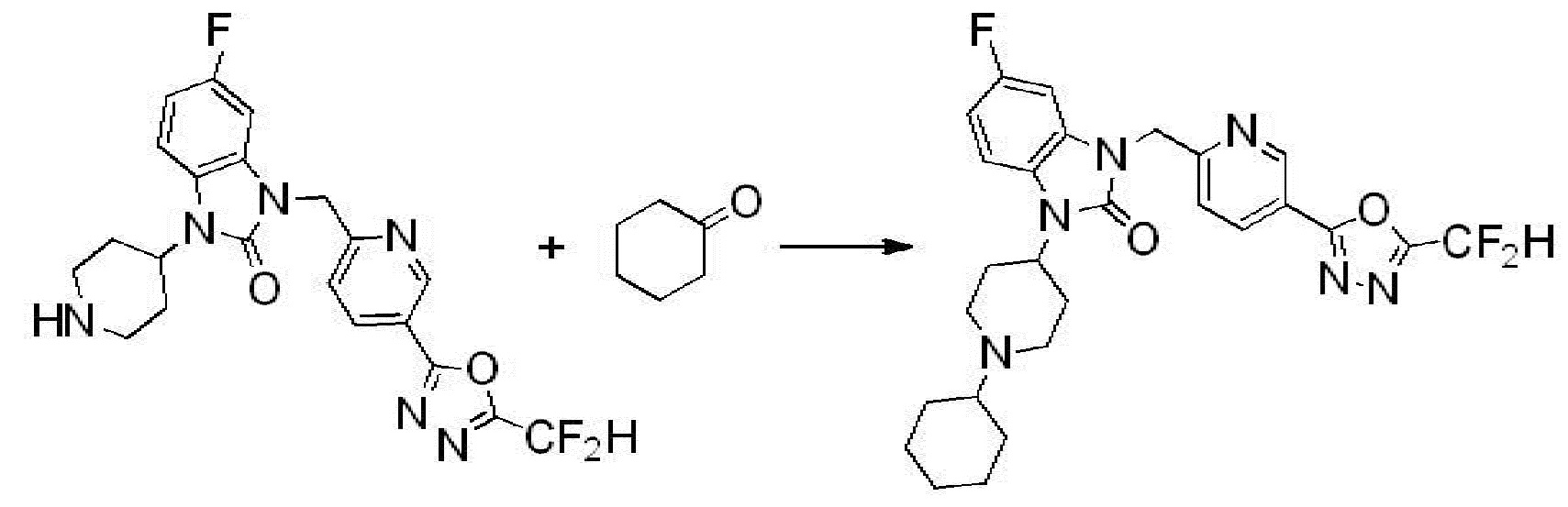
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, циклогексанон (0,044 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,077 г, 64,9%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=1,7 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,0 Гц, 1H), 7,29-7,26 (м, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,78-6,72 (м, 2H), 5,23 (с, 2H), 4,40-4,35 (м, 1H), 3,06-3,05 (м, 2H), 2,46-2,37 (м, 5H), 1,88-1,79 (м, 6H), 1,63 (д, J=12,2 Гц, 1H), 1,30-1,19 (м, 4H), 1,14-1,07 (м, 1H); МСНР (ЭР) m/z 527,6 (M+ + 1).
Пример 55: Синтез соединения 55, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(тетрагидро-2H-пиран-4-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 55
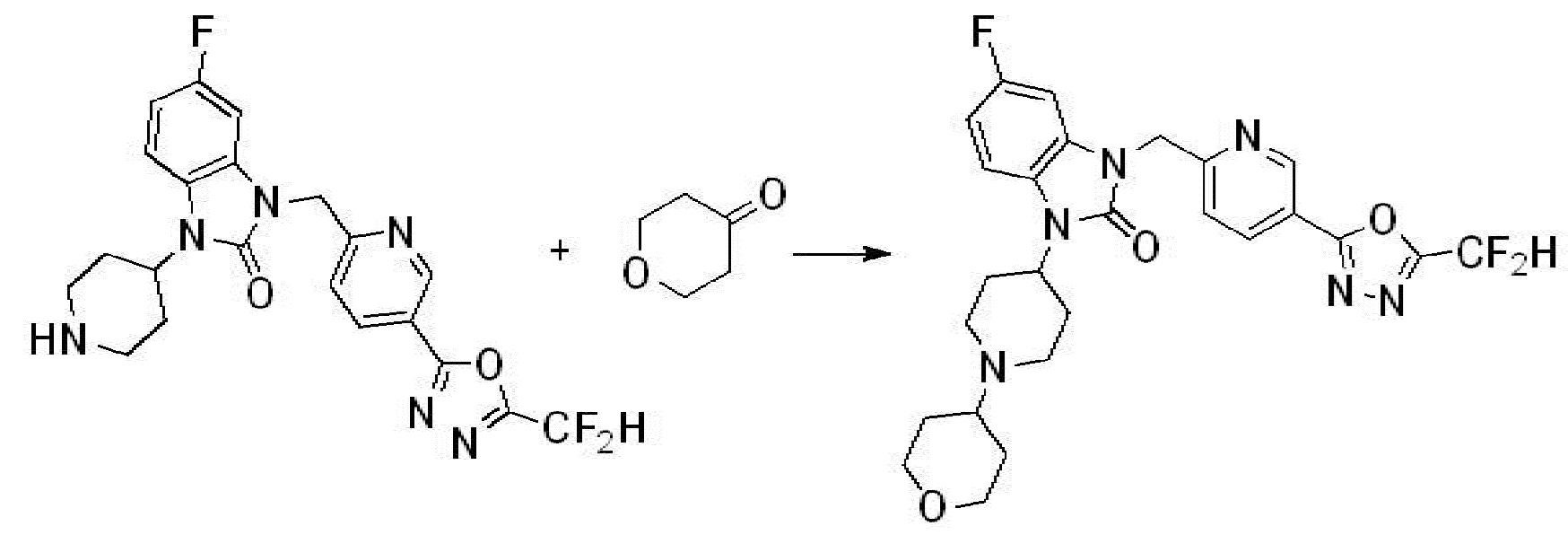
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, тетрагидро-4H-пиран-4-он (0,045 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,070 г, 58,9%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,6 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,30-7,20 (м, 1H), 6,92 (т, J=51,7 Гц, 1H), 6,79-6,73 (м, 2H), 5,23 (с, 2H), 4,42-4,35 (м, 1H), 4,03 (дд, J=11,2, 3,8 Гц, 2H), 3,41-3,35 (м, 2H), 3,13 (д, J=8,2 Гц, 2H), 2,65-2,50 (м, 1H), 2,44-2,34 (м, 4H), 1,89-1,85 (м, 2H), 1,80-1,77 (м, 2H), 1,68-1,67 (м, 2H); МСНР (ЭР) m/z 529,4 (M+ + 1).
Пример 56: Синтез соединения 56, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(1-метоксипропан-2-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 56
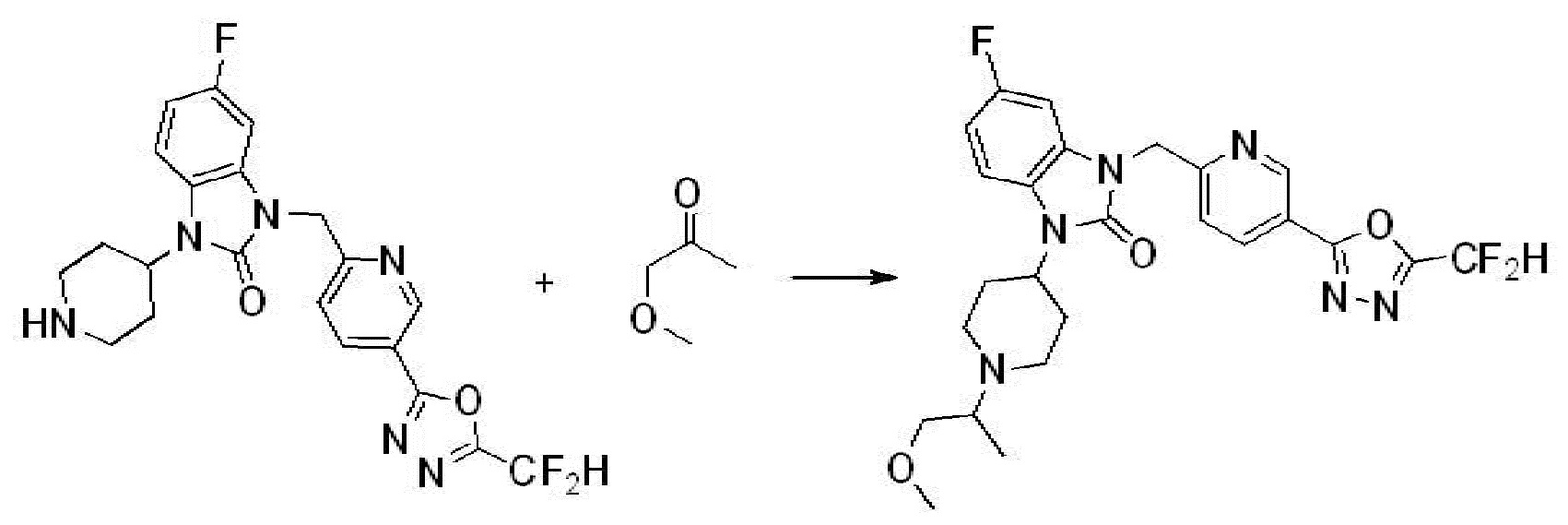
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, 1-метоксипропан-2-он (0,040 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,039 г, 33,6%) в виде желтой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=2,1 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,5 Гц, 1H), 7,36-7,20 (м, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,79-6,72 (м, 2H), 5,23 (с, 2H), 4,46-4,35 (м, 1H), 3,49 (дд, J=9,7, 6,0 Гц, 1H), 3,35-3,28 (м, 4H), 3,10-2,95 (м, 2H), 2,94-2,86 (м, 1H), 2,50-2,40 (м, 4H), 1,84-1,82 (м, 2H), 1,07 (д, J=6,6 Гц, 3H); МСНР (ЭР) m/z 517,4 (M+ + 1).
Пример 57: Синтез соединения 57, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(тетрагидро-2H-тиопиран-4-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 57
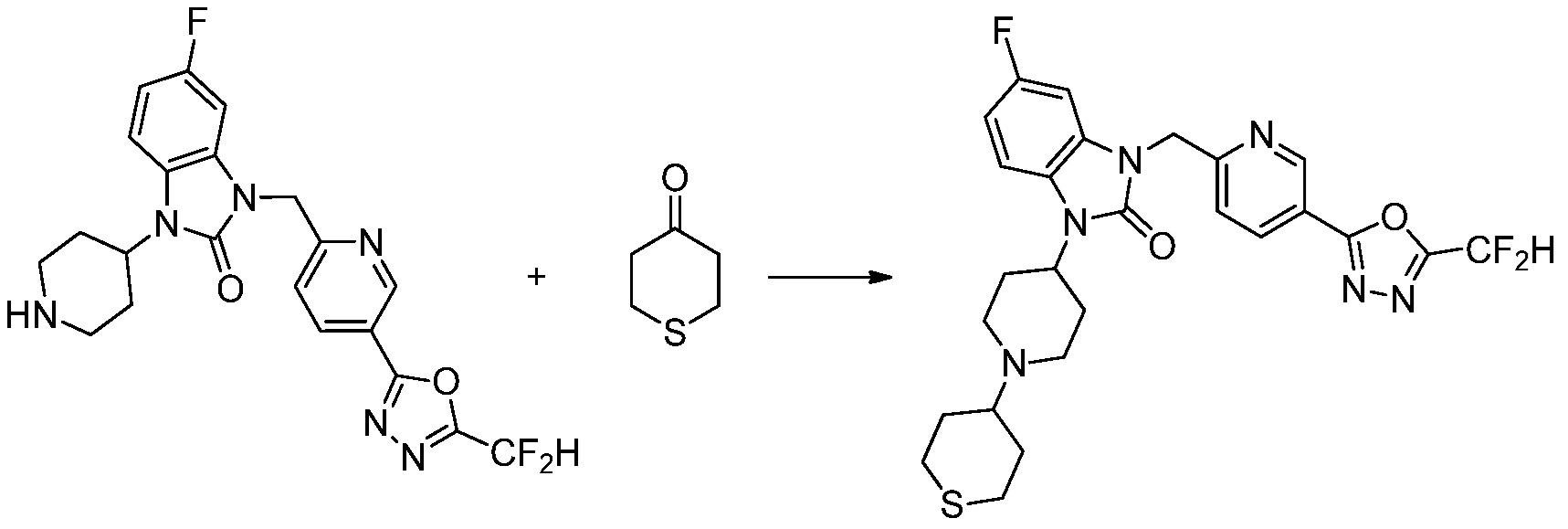
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, тетрагидро-4H-тиопиран-4-он (0,052 г, 0,450 ммоль), уксусную кислоту (0,014 мл, 0,248 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,025 г, 20,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=2,0 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,35-7,15 (м, 1H), 7,05-6,73 (м, 3H), 5,23 (с, 2H), 4,50-4,30 (м, 1H), 3,20-2,85 (м, 2H), 2,73-2,70 (м, 4H), 2,70-2,35 (м, 4H), 2,25-2,15 (м, 2H), 1,19-1,85 (м, 3H), 1,78-1,69 (м, 2H); МСНР (ЭР) m/z 545,6 (M+ + 1).
Пример 58: Синтез соединения 58, 1-(1-(1,4-диоксаспиро[4,5]декан-8-ил)пиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 58
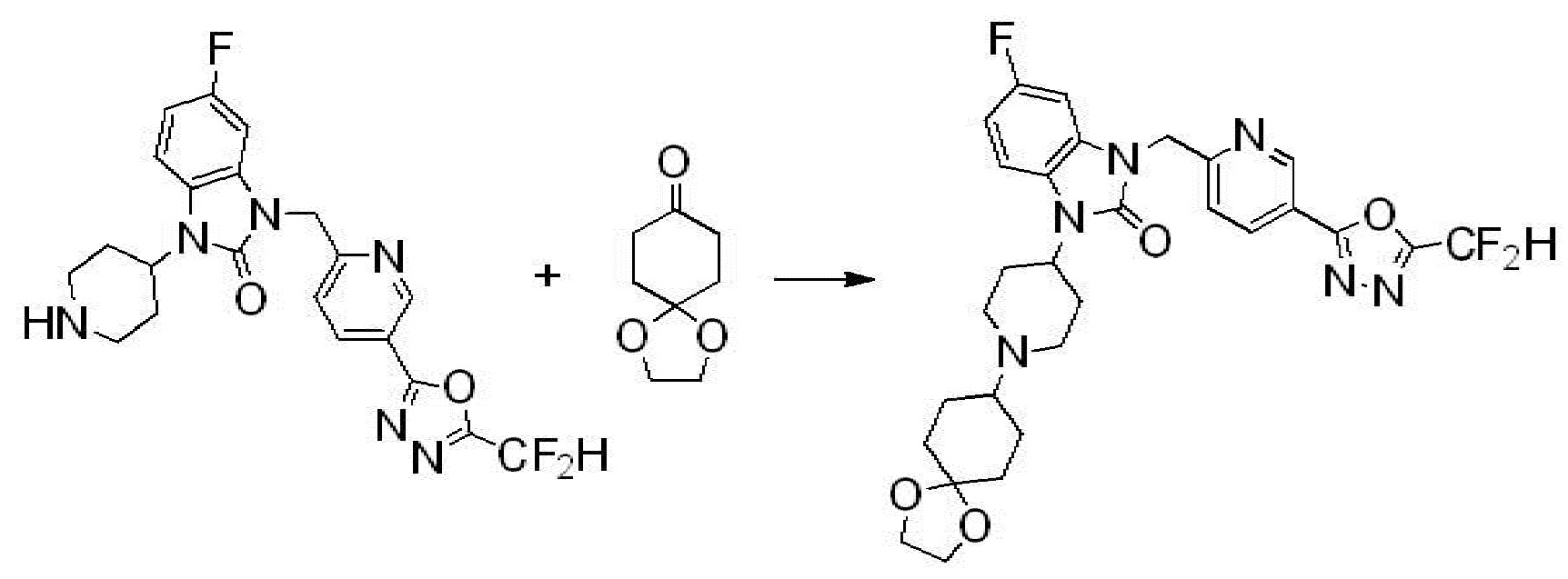
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, 1,4-диоксаспиро[4,5]декан-8-он (0,070 г, 0,450 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,050 г, 38,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (дд, J=2,2, 0,8 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (дд, J=8,2, 0,7 Гц, 1H), 7,27-7,24 (м, 1H), 7,05-6,72 (м, 3H), 5,22 (с, 2H), 4,40-4,35 (м, 1H), 3,93 (с, 4H), 3,06-3,04 (м, 2H), 2,51-2,42 (м, 4H), 1,85-1,81 (м, 6H), 1,67-1,52 (м, 4H); МСНР (ЭР) m/z 585,6 (M+ + 1).
Пример 59: Синтез соединения 59, 1-(1'-ацетил-[1,4'-бипиперидин]-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 59
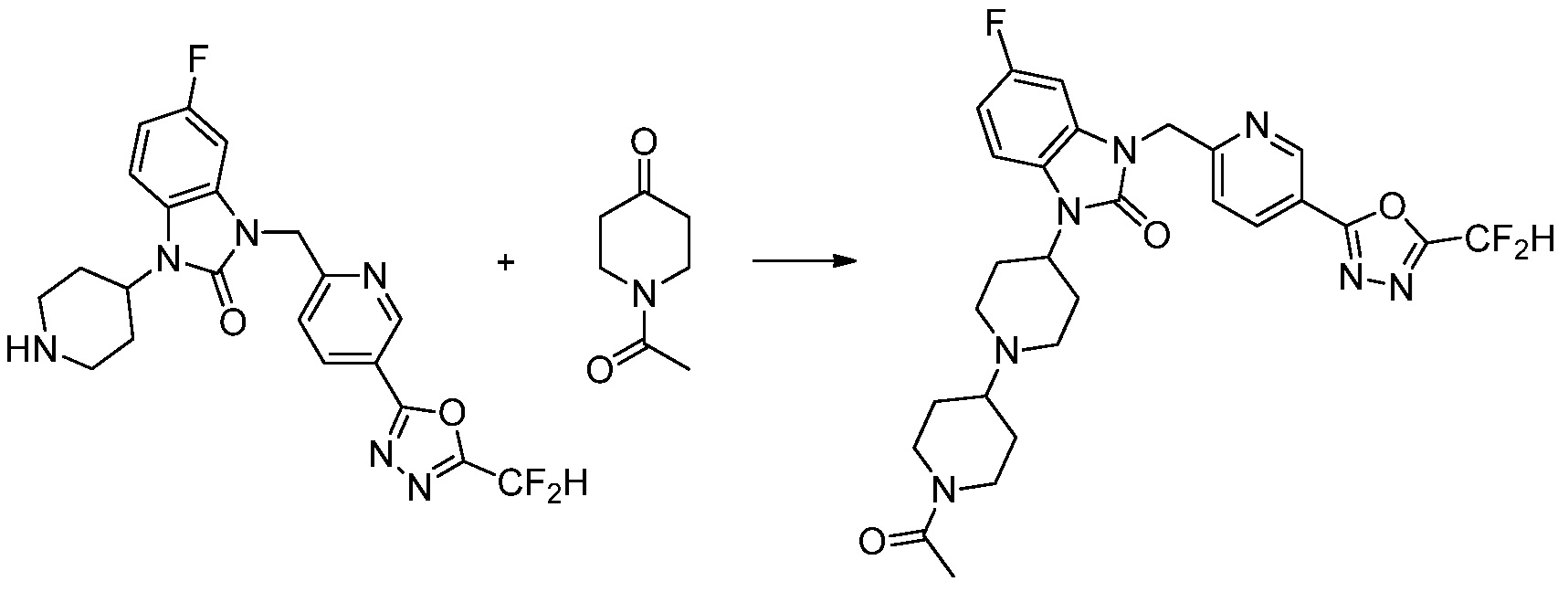
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, 1-ацетилпиперидин-4-он (0,064 г, 0,450 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,063 г, 48,9%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (дд, J=2,1, 0,6 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,39 (д, J=8,2 Гц, 1H), 7,23-7,19 (м, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,77-6,72 (м, 2H), 5,21 (с, 2H), 4,67-4,63 (м, 1H), 4,36-4,35 (м, 1H), 3,88-3,85 (м, 1H), 3,07-3,06 (м, 3H), 2,63-2,56 (м, 2H), 2,56-2,39 (м, 4H), 2,07 (с, 3H), 1,91-1,84 (м, 4H), 1,52-1,38 (м, 2H); МСНР (ЭР) m/z 570,6 (M+ + 1).
Пример 60: Синтез соединения 60, 1-(1-ацетилпиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 60
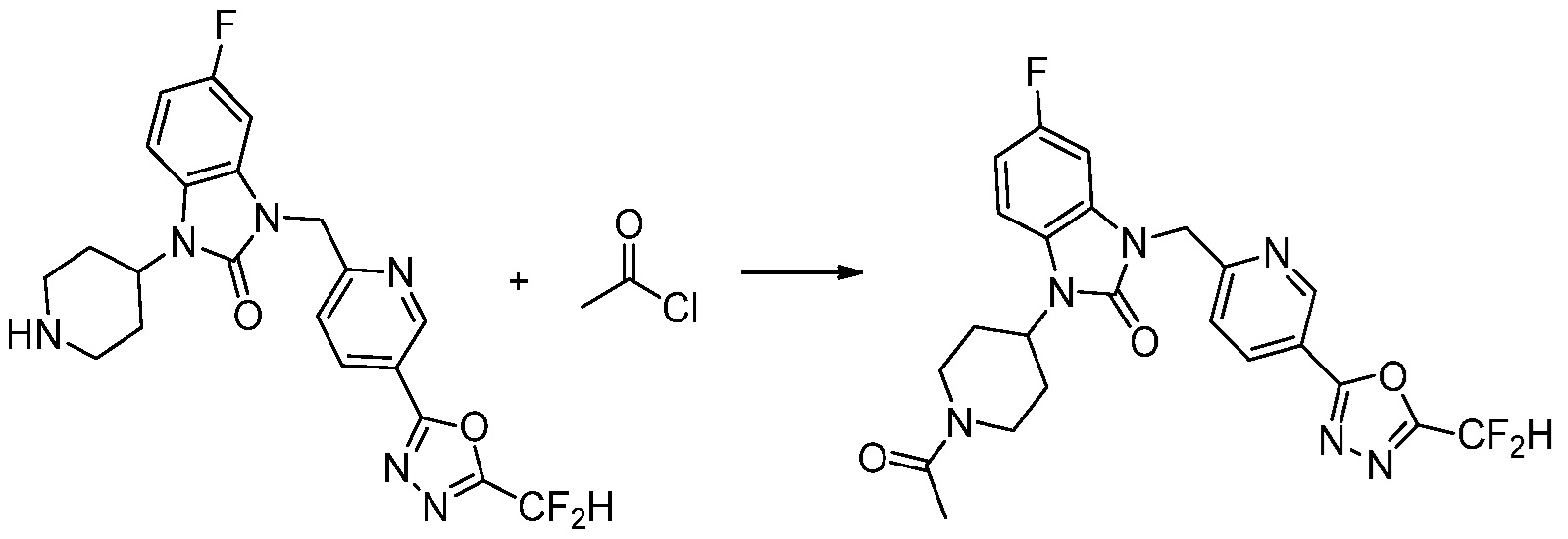
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, триэтиламин (0,063 мл, 0,450 ммоль) и ацетилхлорид (0,032 мл, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,085 г, 77,7%) в виде желтой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (дд, J=10,9, 9,5 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (дд, J=8,2, 0,7 Гц, 1H), 7,05-6,73 (м, 4H), 5,22 (д, J=1,9 Гц, 2H), 4,84 (дт, J=13,6, 2,0 Гц, 1H), 4,59-4,50 (м, 1H), 4,01-3,98 (м, 1H), 3,23 (тд, J=13,3, 2,2 Гц, 1H), 2,67 (тд, J=13,1, 2,3 Гц, 1H), 2,37-2,21 (м, 2H), 2,15 (с, 3H), 1,96-1,87 (м, 2H); МСНР (ЭР) m/z 487,6 (M+ + 1).
Пример 61: Синтез соединения 61, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(morpholine-4-carbonyl)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 61
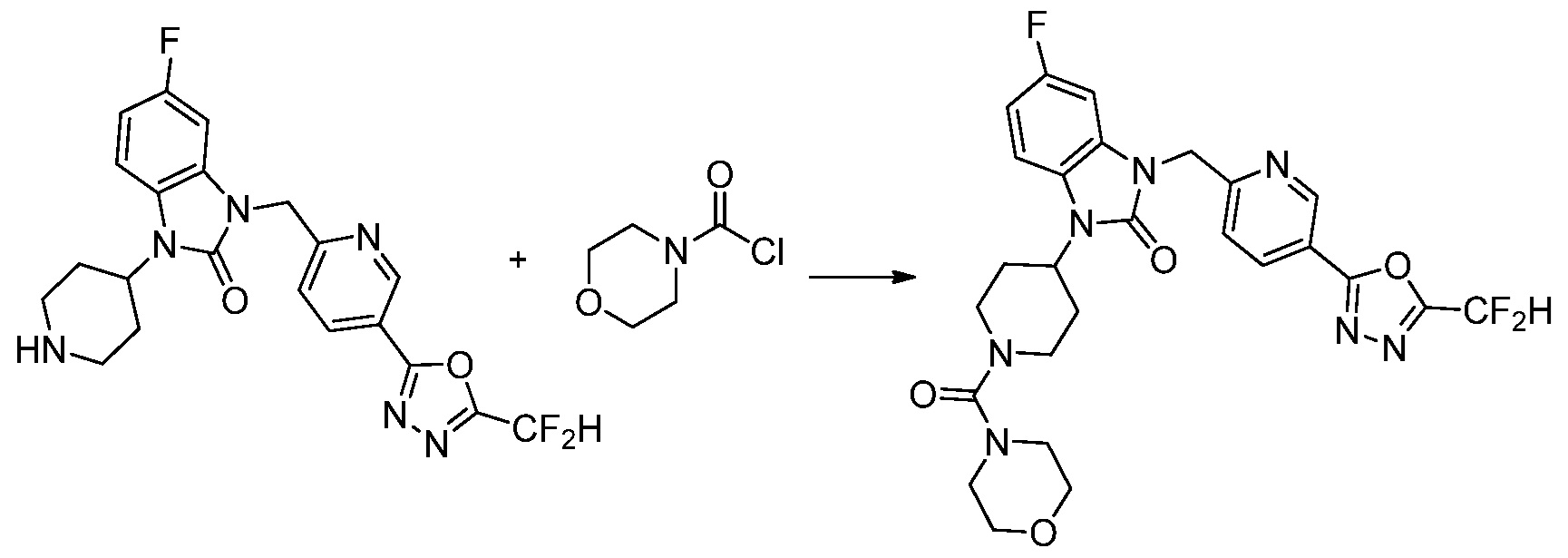
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, растворяют в дихлорметане (2 мл) при комнатной температуре, после чего триэтиламин (0,063 мл, 0,450 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 10 минут. Морфолин-4-карбонилхлорид (0,067 г, 0,450 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,057 г, 45,1%) в виде прозрачной жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=2,1 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,43 (д, J=8,2 Гц, 1H), 7,06-6,75 (м, 4H), 5,23 (с, 2H), 4,51-4,43 (м, 1H), 3,88 (д, J=13,6 Гц, 2H), 3,70 (т, J=4,7 Гц, 4H), 3,31 (т, J=4,7 Гц, 4H), 2,95 (т, J=12,0 Гц, 2H), 2,44-2,33 (м, 2H), 1,87 (дд, J=12,2, 2,3 Гц, 2H); МСНР (ЭР) m/z 558,6 (M+ + 1).
Пример 62: Синтез соединения 62, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(4-(трифторметил)бензоил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 62
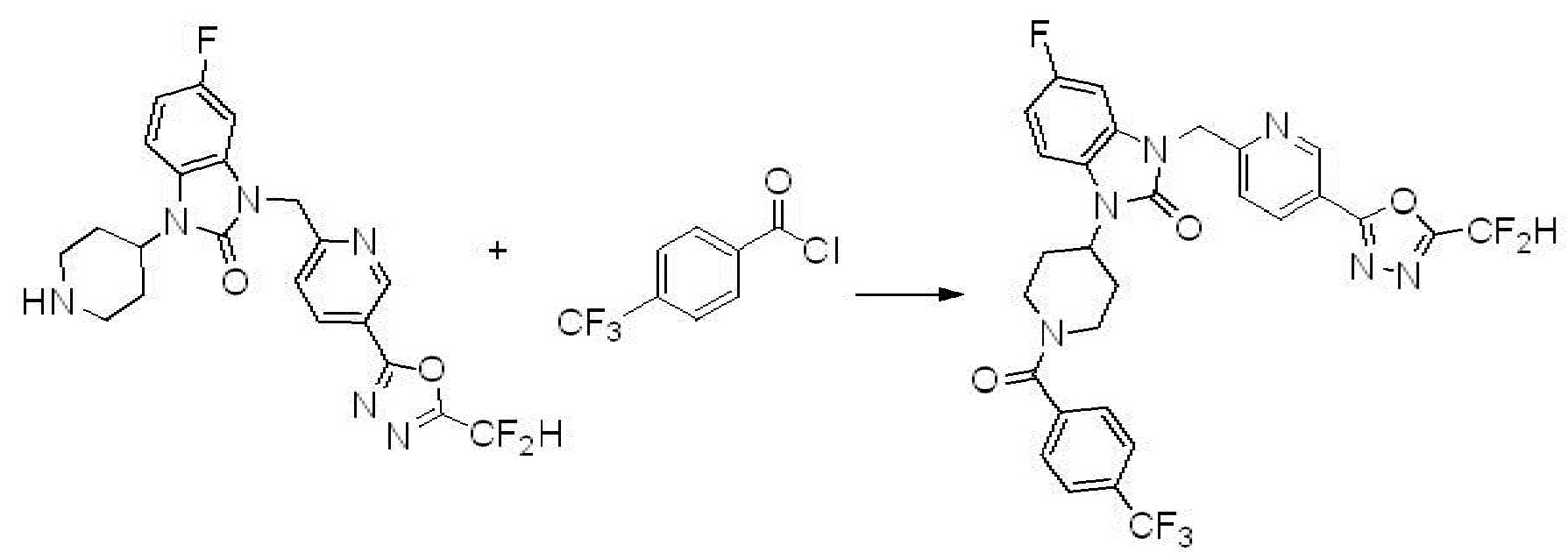
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, растворяют в дихлорметане (2 мл) при комнатной температуре, после чего триэтиламин (0,063 мл, 0,450 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 10 минут. 4-(трифторметил)бензоилхлорид (0,094 г, 0,450 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол = 0-5%) и концентрируют с получением указанного в заголовке соединения (0,106 г, 76,2%) в виде желтой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=14,6 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,68, 7,57 (ABкв, J=42,1, 8,3 Гц, 4H), 7,43 (д, J=8,2 Гц, 1H), 7,05-6,76 (м, 4H), 5,22 (с, 2H), 5,10-4,80 (м, 1H), 4,58-4,50 (м, 1H), 4,00-3,68 (м, 1H), 3,35-3,05 (м, 1H), 3,05-2,75 (м, 1H), 2,60-2,25 (м, 2H), 2,15-1,65 (м, 2H); МСНР (ЭР) m/z 617,6 (M+ + 1).
Пример 63: Синтез соединения 63, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-изоникотиноилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 63
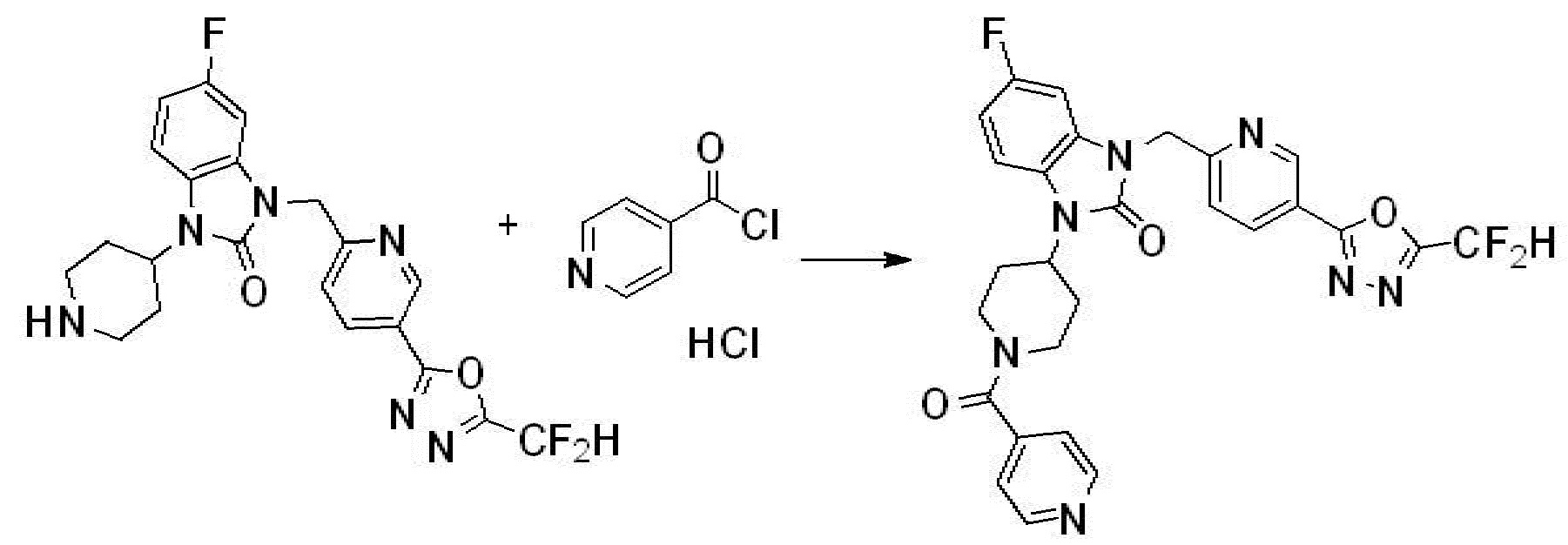
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, триэтиламин (0,094 мл, 0,675 ммоль) и гидрохлорид изоникотиноилхлорида (0,080 г, 0,450 ммоль) растворяют в дихлорметане (2 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 10 минут и дополнительно перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,074 г, 59,8%) в виде прозрачной жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (д, J=2,2 Гц, 1H), 8,69 (д, J=5,8 Гц, 2H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (д, J=8,2 Гц, 1H), 7,34-7,32 (м, 2H), 7,05-6,76 (м, 4H), 5,20 (д, J=1,0 Гц, 2H), 4,93 (д, J=11,2 Гц, 1H), 4,56-4,48 (м, 1H), 3,80 (д, J=12,4 Гц, 1H), 3,21 (т, J=12,3 Гц, 1H), 2,90 (т, J=11,9 Гц, 1H), 2,49-2,34 (м, 2H), 2,01 (д, J=9,7 Гц, 1H), 1,86 (д, J=10,9 Гц, 1H); МСНР (ЭР) m/z 551,6 (M+ + 2).
Пример 64: Синтез соединения 64, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(пиримидин-5-карбонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 64
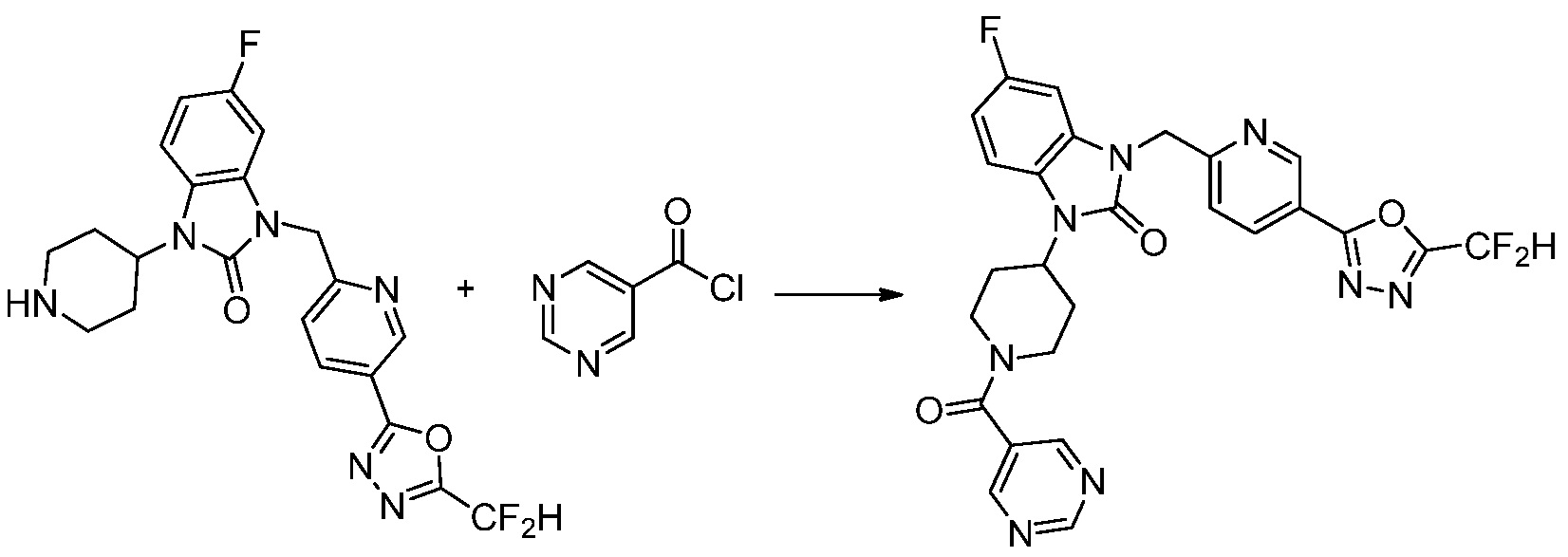
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, растворяют в дихлорметане (2 мл) при комнатной температуре, после чего триэтиламин (0,063 мл, 0,450 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 10 минут. Пиримидин-5-карбонилхлорид (0,064 г, 0,450 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,065 г, 52,6%) в виде желтой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (с, 1H), 9,24 (дд, J=2,1, 0,6 Гц, 1H), 8,85 (с, 2H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (дд, J=8,2, 0,5 Гц, 1H), 7,04-6,75 (м, 4H), 5,21 (с, 2H), 5,08-4,75 (м, 1H), 4,59-4,51 (м, 1H), 4,00-3,75 (м, 1H), 3,50-3,15 (м, 1H), 3,10-2,85 (м, 1H), 2,60-2,33 (м, 2H), 2,20-1,80 (м, 2H); МСНР (ЭР) m/z 551,6 (M+ + 1).
Пример 65: Синтез соединения 65, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(5-метилизооксазол-4-карбонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 65
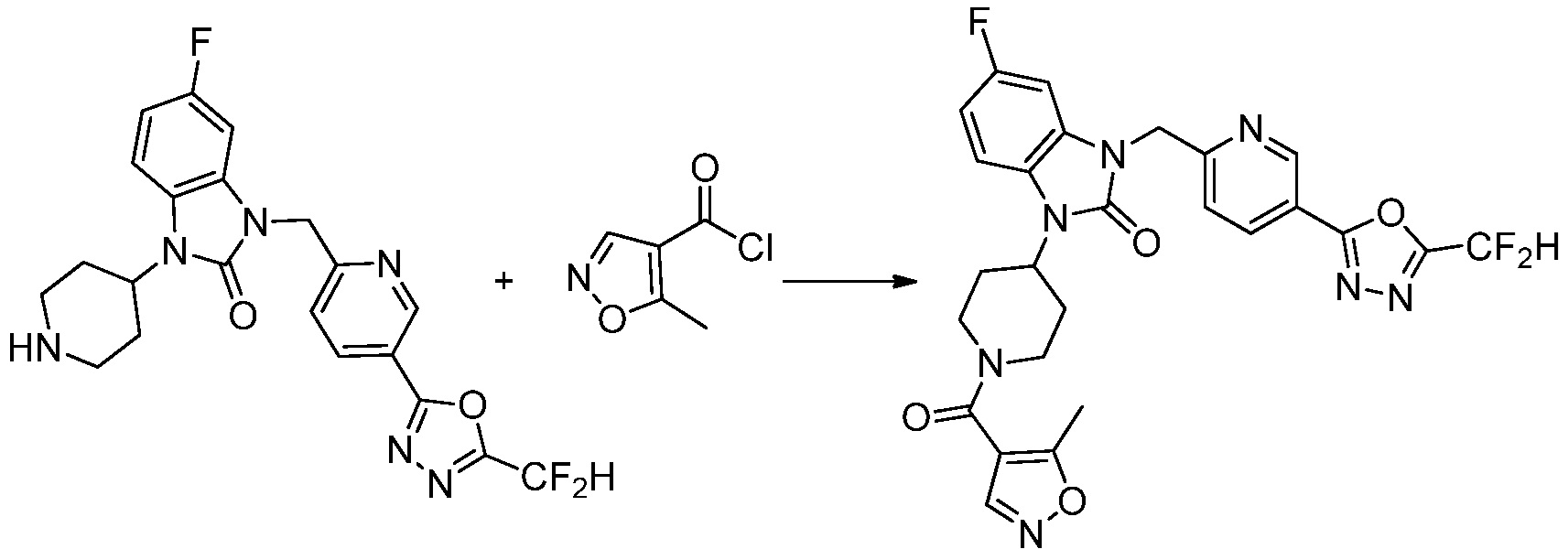
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, растворяют в дихлорметане (2 мл) при комнатной температуре, после чего триэтиламин (0,063 мл, 0,450 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 10 минут. 5-метилизооксазол-4-карбонилхлорид (0,065 г, 0,450 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,036 г, 28,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,6 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,44 (д, J=8,1 Гц, 1H), 7,05-6,76 (м, 4H), 5,23 (с, 2H), 4,82 (д, J=14,0 Гц, 2H), 4,64-4,56 (м, 1H), 3,06 (т, J=13,0 Гц, 2H), 2,48-2,36 (м, 5H), 2,01 (дд, J=12,8, 2,5 Гц, 2H); МСНР (ЭР) m/z 554,6 (M+ + 1).
Пример 66: Синтез соединения 66, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(5-метилфуран-2-карбонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 66
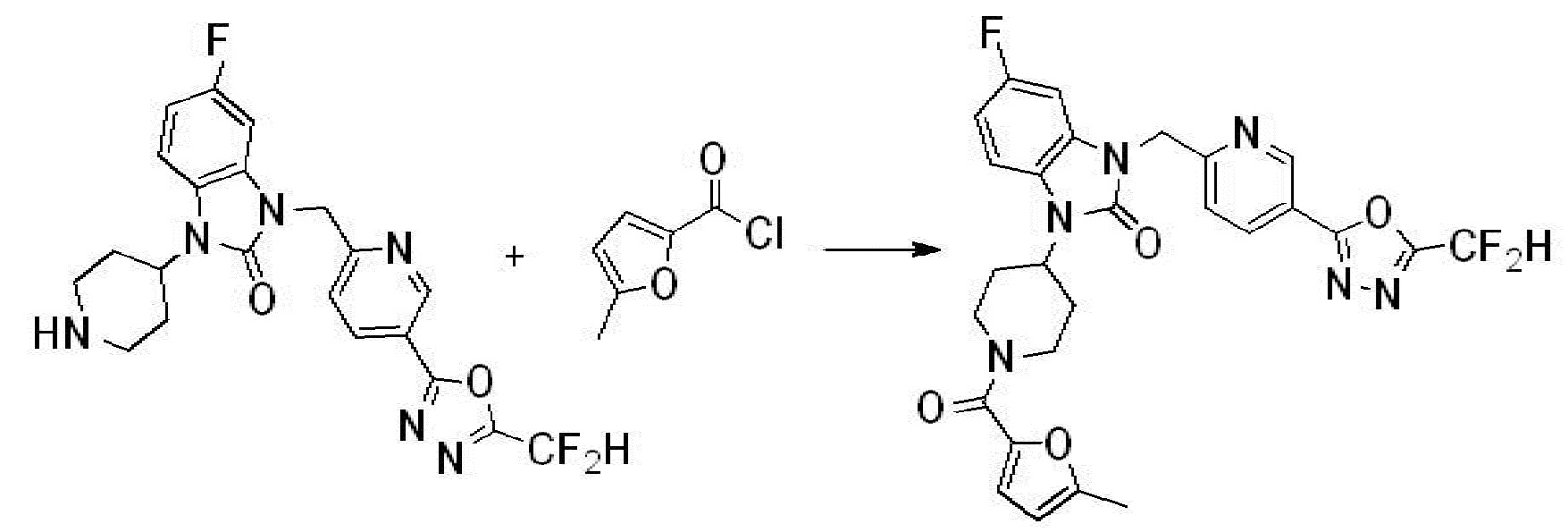
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, растворяют в дихлорметане (2 мл) при комнатной температуре, после чего триэтиламин (0,063 мл, 0,450 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 10 минут. 5-метилфуран-2-карбонилхлорид (0,065 г, 0,450 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,089 г, 71,8%) в виде коричневой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (дд, J=2,2, 0,8 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (дд, J=8,2, 0,7 Гц, 1H), 7,05-6,72 (м, 5H), 6,08 (дд, J=3,4, 1,0 Гц, 1H), 5,22 (с, 2H), 4,79 (д, J=13,6 Гц, 2H), 4,67-4,58 (м, 1H), 3,20-2,95 (м, 2H), 2,43-2,34 (м, 5H), 1,95 (дд, J=12,1, 2,2 Гц, 2H); МСНР (ЭР) m/z 553,6 (M+ + 1).
Пример 67: Синтез соединения 67, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(3-метоксипропаноил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 67
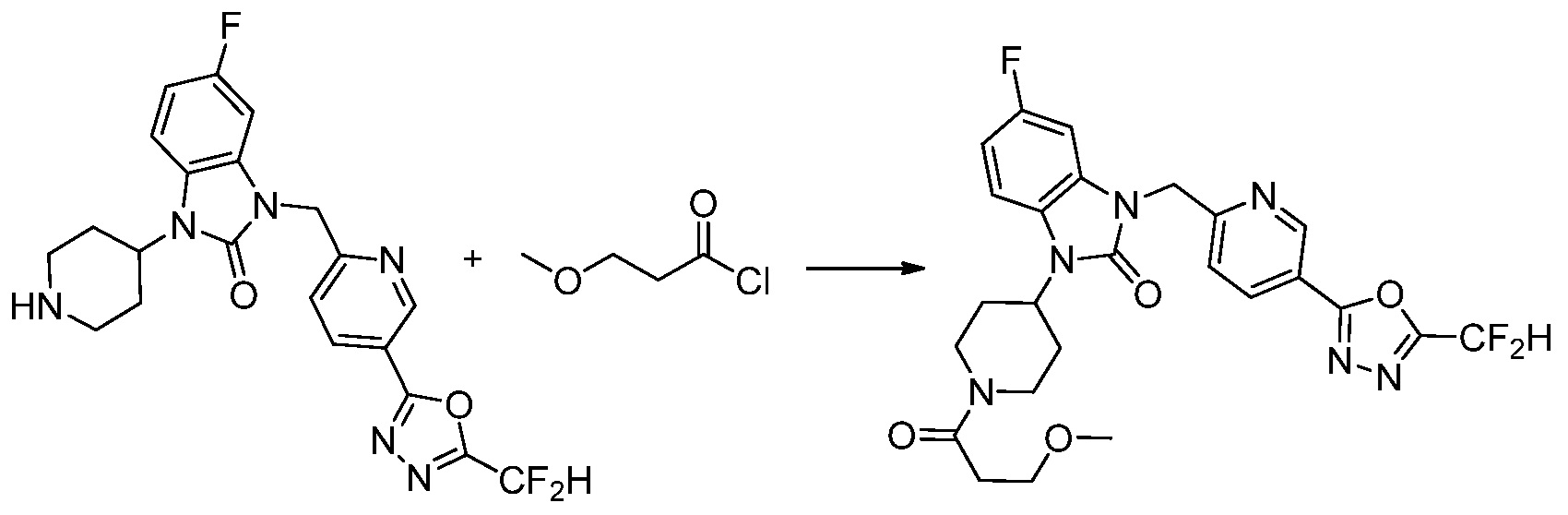
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, растворяют в дихлорметане (2 мл) при комнатной температуре, после чего триэтиламин (0,063 мл, 0,450 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 10 минут. 3-метоксипропаноилхлорид (0,055 г, 0,450 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,065 г, 54,5%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,23 (т, J=1,1 Гц, 1H), 8,30 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,2 Гц, 1H), 7,04-6,69 (м, 4H), 5,20 (с, 2H), 4,84 (д, J=13,3 Гц, 2H), 4,58-4,51 (м, 1H), 4,08 (д, J=13,5 Гц, 2H), 3,76-3,65 (м, 2H), 3,34 (с, 3H), 3,18 (т, J=8,3 Гц, 2H), 2,74-2,54 (м, 3H), 2,35-2,19 (м, 2H), 1,88 (т, J=11,5 Гц, 2H); МСНР (ЭР) m/z 531,6 (M+ + 1).
Пример 68: Синтез соединения 68, Метил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат
[Стадия 1] Синтез соединения 68
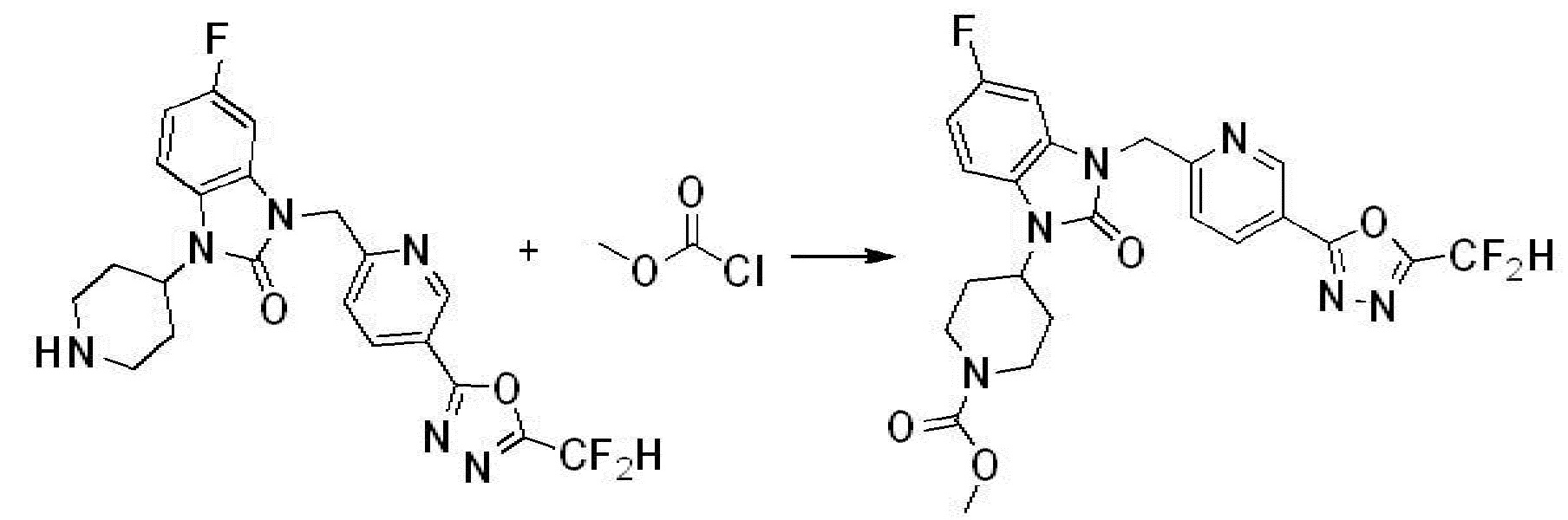
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 48, растворяют в дихлорметане (2 мл) при комнатной температуре, после чего триэтиламин (0,063 мл, 0,450 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 10 минут. Метил карбонохлоридат (0,043 г, 0,450 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,034 г, 30,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=2,0 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (д, J=8,2 Гц, 1H), 7,05-6,74 (м, 4H), 5,23 (с, 2H), 4,54-4,20 (м, 3H), 3,73 (с, 3H), 3,05-2,80 (м, 2H), 2,35-2,25 (м, 2H), 1,87 (д, J=11,4 Гц, 2H); МСНР (ЭР) m/z 503,6 (M+ + 1).
Пример 69: Синтез соединения 69, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
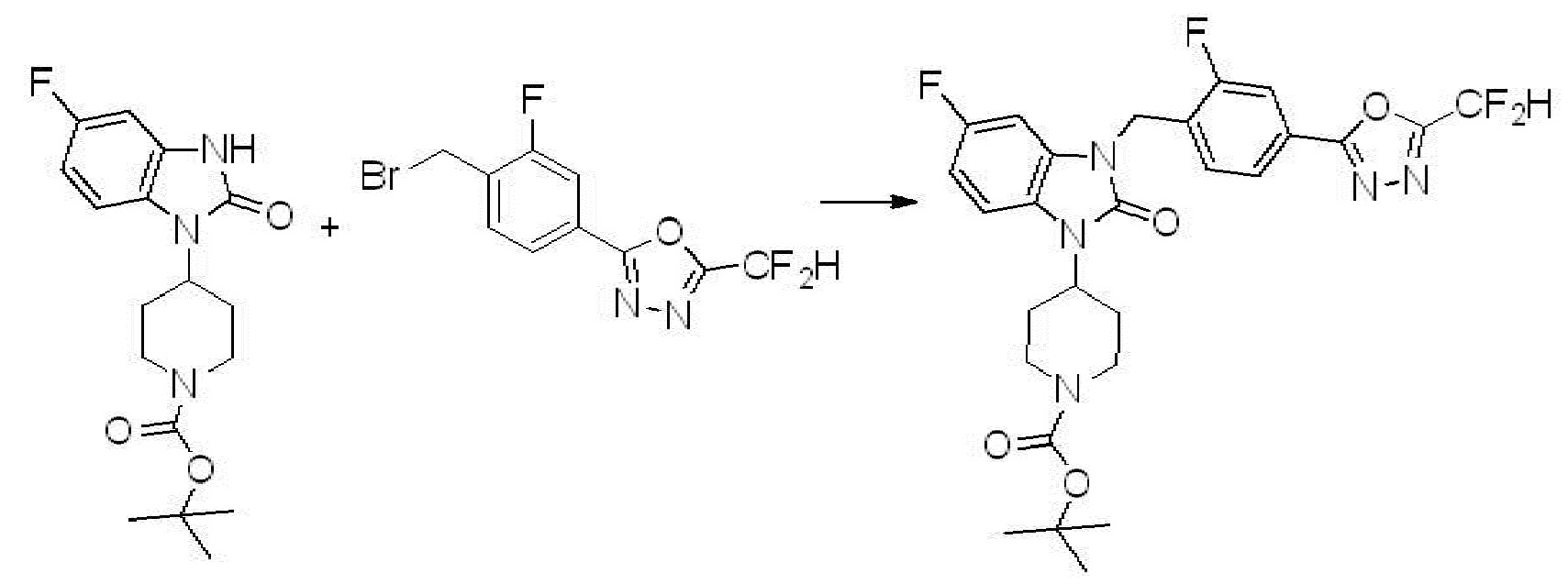
Трет-бутил 4-(5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,300 г, 3,876 ммоль), полученный на стадии 3 соединения 48, растворяют в N,N-диметилформамиде (40 мл) при 0°C, после чего гидрид натрия (60,00%, 0,202 г, 5,039 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (1,547 г, 5,039 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0 to 60%) и концентрируют с получением указанного в заголовке соединения (1,420 г, 65,2%) в виде светло-коричневого твердого вещества.
[Стадия 2] Синтез 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
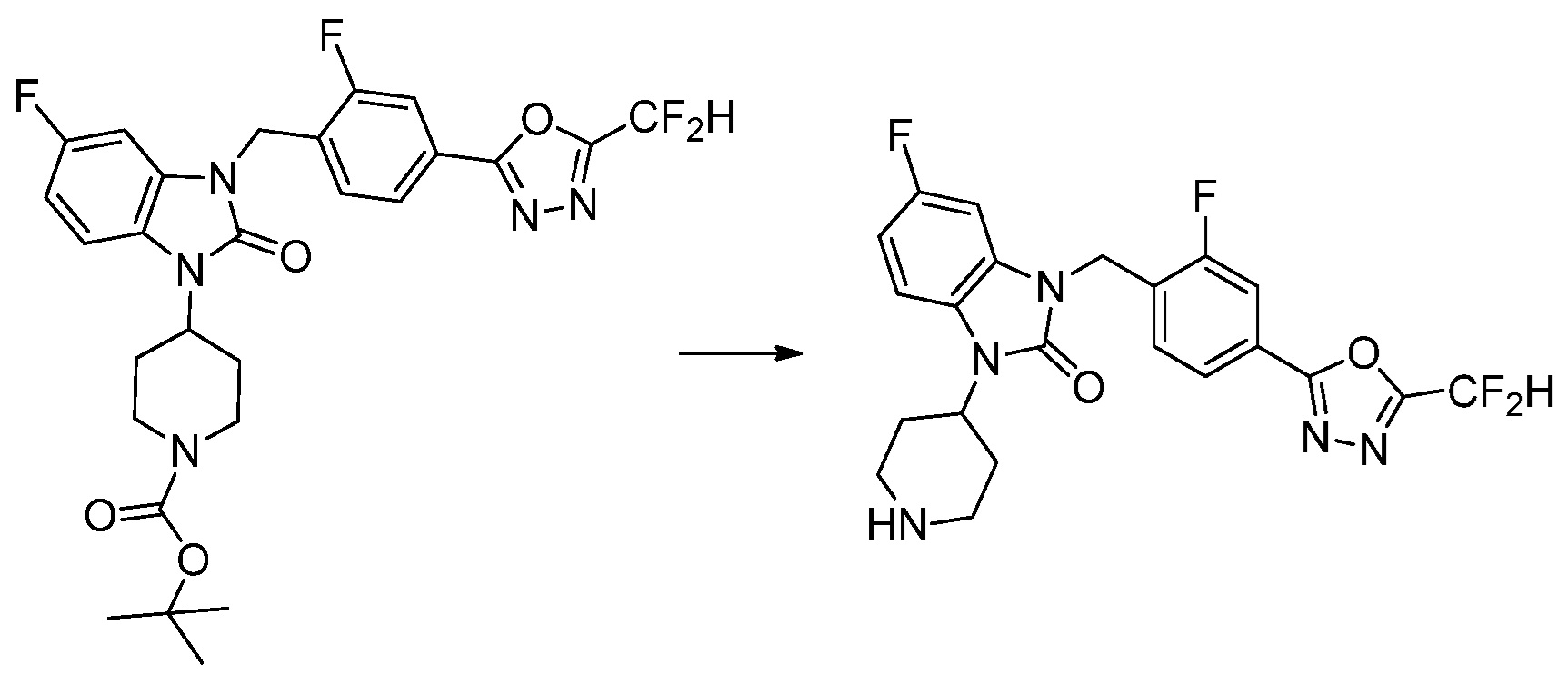
Трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,420 г, 2,529 ммоль), полученный на стадии 1, растворяют в дихлорметане (15 мл) при 0°C, после чего трифторуксусную кислоту (1,936 мл, 25,288 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=50-100%) и концентрируют с получением продукта, после чего полученный продукт снова очищают хроматографией (SiO2, 40 г картридж; дихлорметан/метанол=0-30%) и концентрируют с получением указанного в заголовке соединения (0,940 г, 80,6%) в виде светло-желтого твердого вещества.
[Стадия 3] Синтез соединения 69
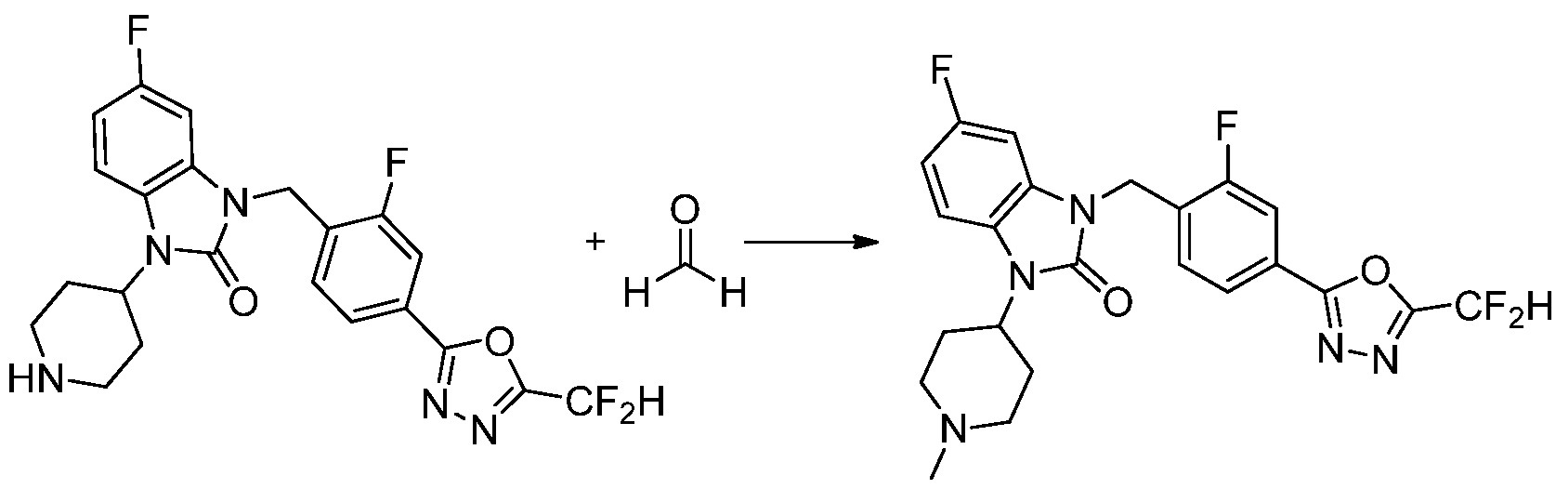
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,092 г, 0,433 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и формальдегид (0,013 г, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,037 г, 35,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,85-7,82 (м, 2H), 7,44 (т, J=7,8 Гц, 1H), 7,23 (дд, J=8,7, 4,3 Гц, 1H), 7,03-6,68 (м, 3H), 5,14 (с, 2H), 4,44-4,36 (м, 1H), 3,02 (д, J=11,7 Гц, 2H), 2,51-2,40 (м, 2H), 2,34 (с, 3H), 2,16 (тд, J=12,0, 2,1 Гц, 2H), 1,84-1,80 (м, 2H); МСНР (ЭР) m/z 476,4 (M+ + 1).
Пример 70: Синтез соединения 70, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(1-изопропилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 70
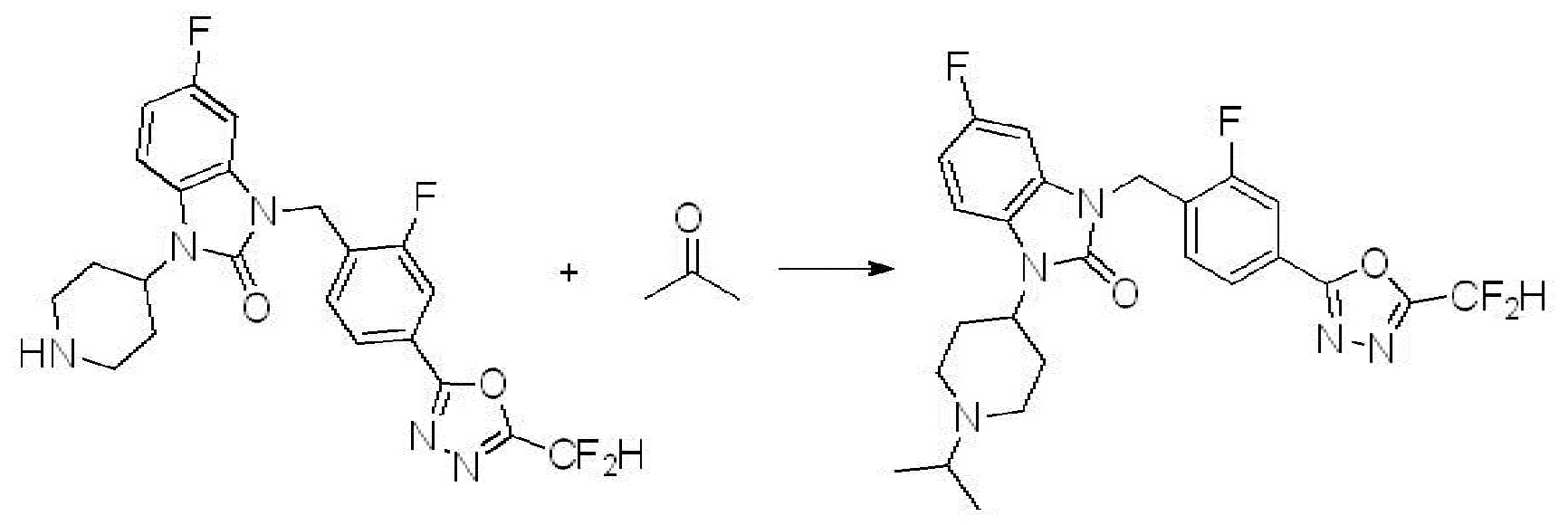
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 69, триацетоксиборгидрид натрия (0,092 г, 0,433 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и пропан-2-он (0,025 г, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,070 г, 63,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,85-7,82 (м, 2H), 7,44 (т, J=7,8 Гц, 1H), 7,28 (дд, J=9,0, 4,6 Гц, 1H), 6,90-6,68 (м, 3H), 5,28 (с, 2H), 4,40-4,33 (м, 1H), 3,02 (д, J=9,5 Гц, 2H), 2,84-2,78 (м, 1H), 2,46-2,31 (м, 4H), 1,84 (д, J=11,4 Гц, 2H), 1,07 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 504,6 (M+ + 1).
Пример 71: Синтез соединения 71, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 71
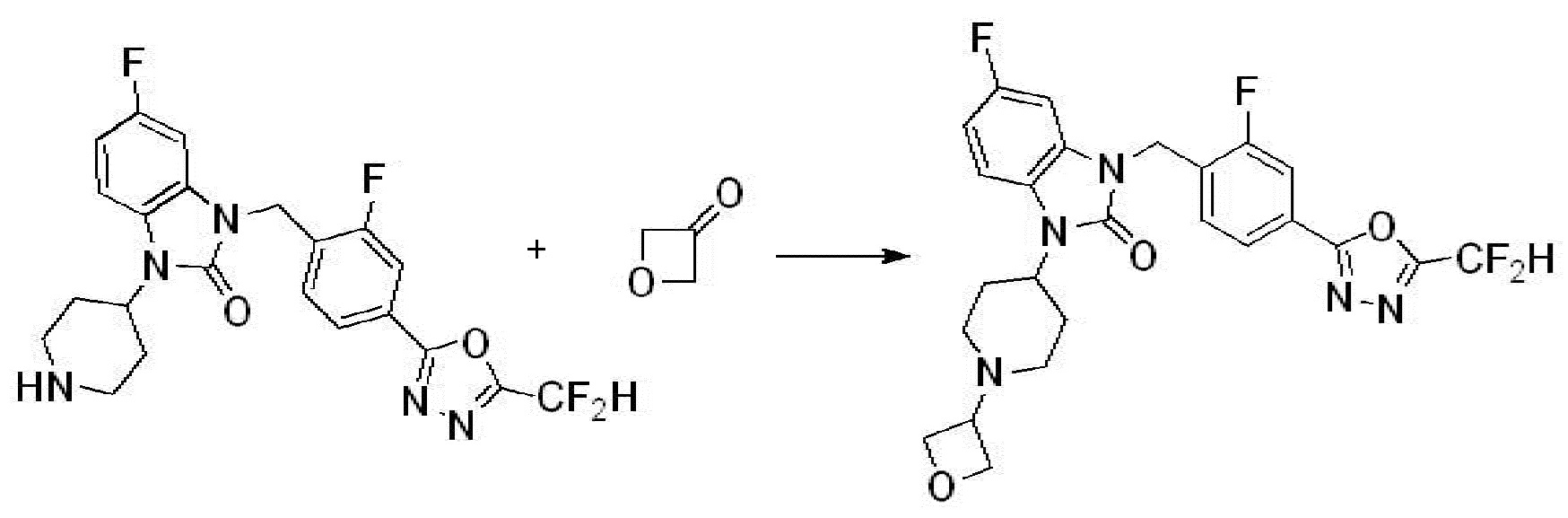
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 69, триацетоксиборгидрид натрия (0,092 г, 0,433 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и оксетан-3-он (0,031 г, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,077 г, 68,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,84-7,82 (м, 2H), 7,44 (т, J=7,8 Гц, 1H), 7,23 (дд, J=8,7, 4,3 Гц, 1H), 7,02-6,69 (м, 3H), 5,13 (с, 2H), 4,64 (дт, J=20,7, 6,4 Гц, 4H), 4,45-4,36 (м, 1H), 3,56-3,49 (м, 1H), 2,90 (д, J=11,5 Гц, 2H), 2,50-2,39 (м, 2H), 2,02 (т, J=10,9 Гц, 2H), 1,85 (дд, J=12,1, 2,3 Гц, 2H); МСНР (ЭР) m/z 518,6 (M+ + 1).
Пример 72: Синтез соединения 72, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(1-(тетрагидро-2H-пиран-4-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 72
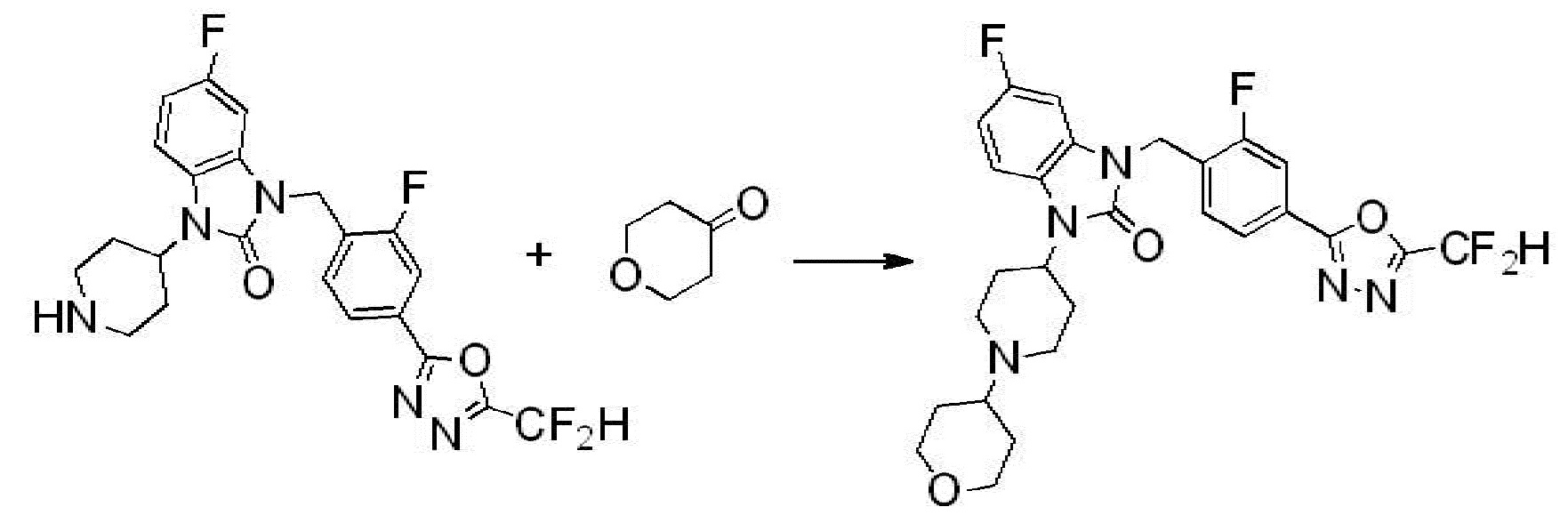
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 69, триацетоксиборгидрид натрия (0,092 г, 0,433 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и тетрагидро-4H-пиран-4-он (0,043 г, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,048 г, 40,4%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,84-7,82 (м, 2H), 7,44 (т, J=7,8 Гц, 1H), 7,23 (дд, J=8,7, 4,3 Гц, 1H), 7,02-6,69 (м, 3H), 5,13 (с, 2H), 4,64 (дт, J=20,7, 6,4 Гц, 4H), 4,45-4,36 (м, 1H), 3,56-3,49 (м, 1H), 2,90 (д, J=11,5 Гц, 2H), 2,50-2,39 (м, 2H), 2,02 (т, J=10,9 Гц, 2H), 1,85 (дд, J=12,1, 2,3 Гц, 2H); МСНР (ЭР) m/z 546,6 (M+ + 1).
Пример 73: Синтез соединения 73, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(1-(тетрагидро-2H-тиопиран-4-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 73
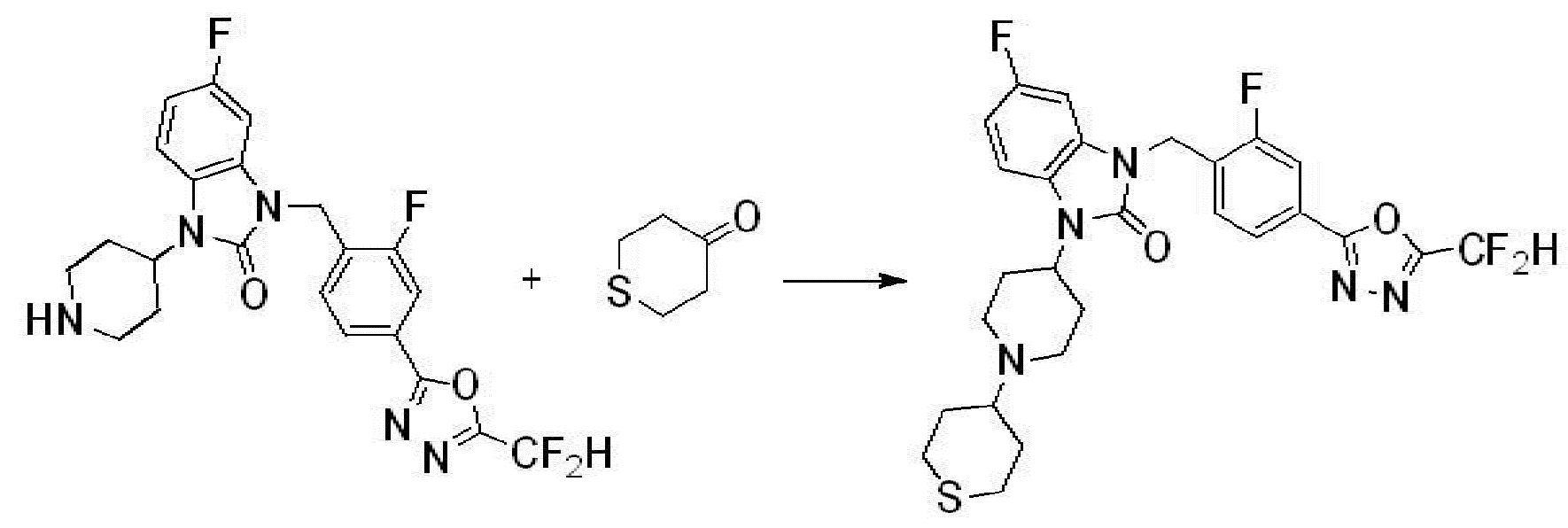
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 69, триацетоксиборгидрид натрия (0,092 г, 0,433 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и тетрагидро-4H-тиопиран-4-он (0,050 г, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,054 г, 44,4%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,85-7,82 (м, 2H), 7,44 (т, J=7,8 Гц, 1H), 7,22 (дд, J=8,6, 4,2 Гц, 1H), 7,03-6,68 (м, 3H), 5,14 (с, 2H), 4,38-4,30 (м, 1H), 2,97 (д, J=11,0 Гц, 2H), 2,71-2,69 (м, 4H), 2,53-2,33 (м, 5H), 2,16-2,13 (м, 2H), 1,83 (дд, J=11,6, 2,3 Гц, 2H), 1,77-1,67 (м, 2H); МСНР (ЭР) m/z 562,6 (M+ + 1).
Пример 74: Синтез соединения 74, 1-(1-циклобутилпиперидин-4-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 74
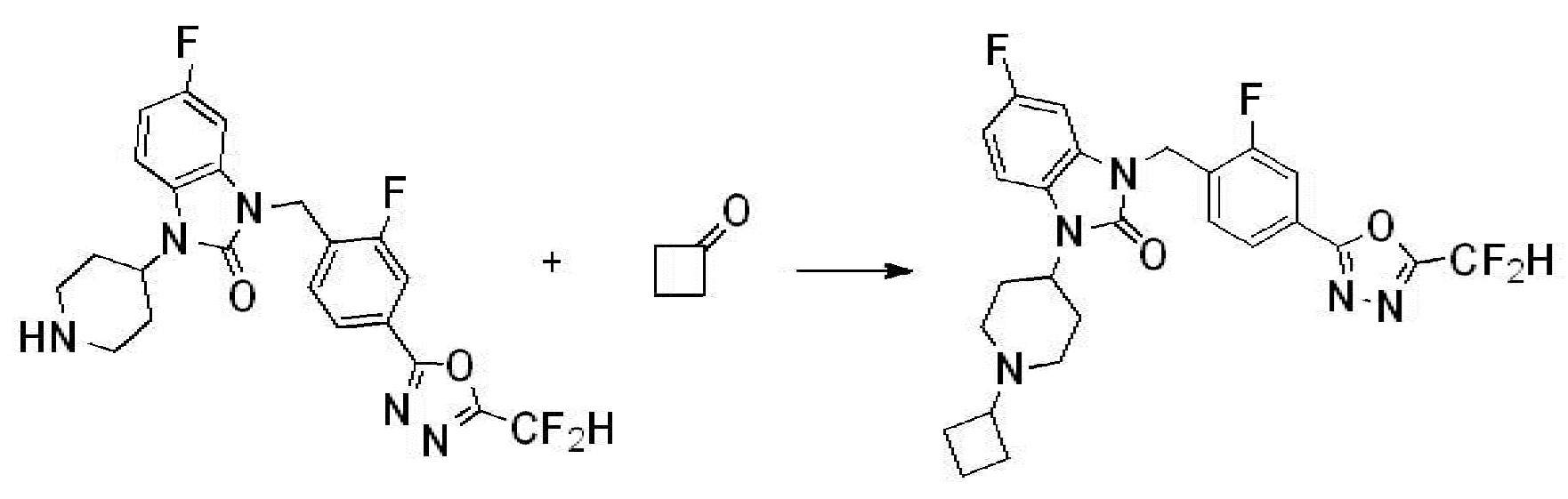
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 69, триацетоксиборгидрид натрия (0,092 г, 0,433 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и циклобутанон (0,030 г, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,079 г, 70,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,81 (д, J=8,6 Гц, 2H), 7,47-7,40 (м, 2H), 7,03-6,77 (м, 2H), 6,68 (дд, J=8,3, 2,5 Гц, 1H), 5,12 (с, 2H), 4,56-4,48 (м, 1H), 3,29 (д, J=11,7 Гц, 2H), 3,06-2,98 (м, 1H), 2,76-2,67 (м, 2H), 2,28-2,20 (м, 4H), 2,15-2,08 (м, 2H), 1,89-1,63 (м, 4H); МСНР (ЭР) m/z 516,5 (M+ + 1).
Пример 75: Синтез соединения 75, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(1-(тетрагидро-2H-тиопиран-4-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 75
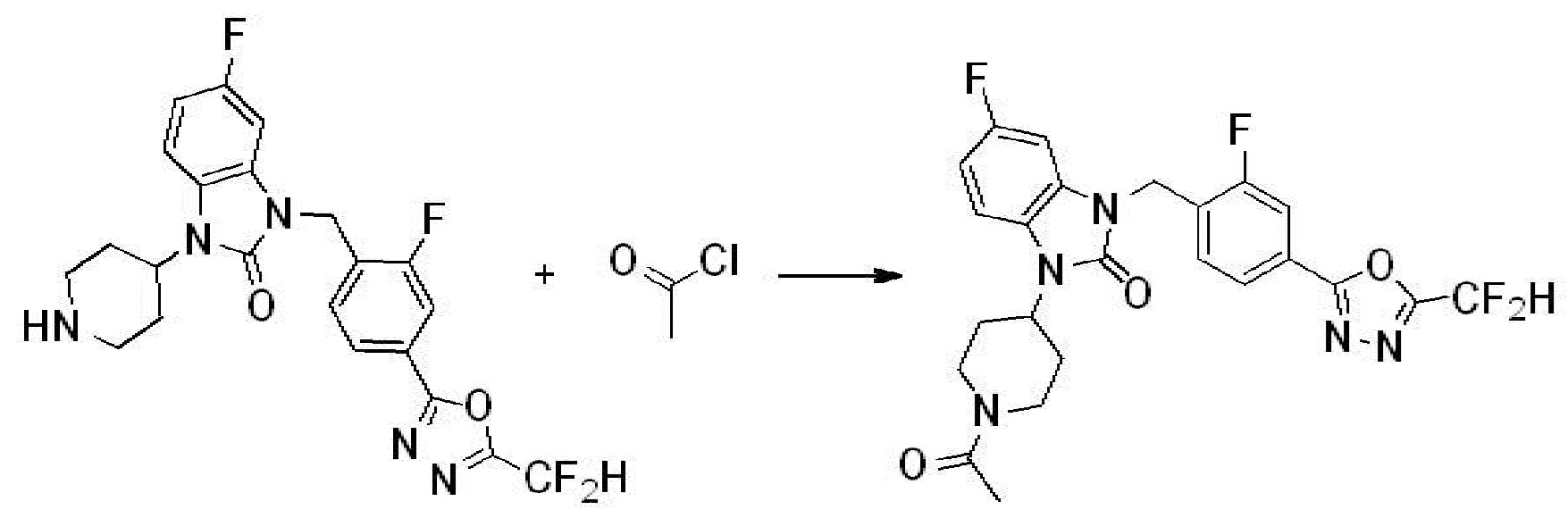
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 69, триэтиламин (0,060 мл, 0,433 ммоль) и ацетилхлорид (0,023 мл, 0,325 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,101 г, 92,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,82-7,79 (м, 2H), 7,44 (т, J=7,7 Гц, 1H), 7,01 (дд, J=8,2, 3,9 Гц, 1H), 6,99-6,69 (м, 3H), 5,12 (с, 2H), 4,83 (д, J=13,6 Гц, 1H), 4,55-4,47 (м, 1H), 3,99 (д, J=13,8 Гц, 1H), 3,24-3,18 (м, 1H), 2,67-2,61 (м, 1H), 2,36-2,20 (м, 2H), 2,13 (с, 3H), 1,89 (т, J=11,4 Гц, 2H); МСНР (ЭР) m/z 504,6 (M+ + 1).
Пример 76: Синтез соединения 76, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фтор-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((3-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилат
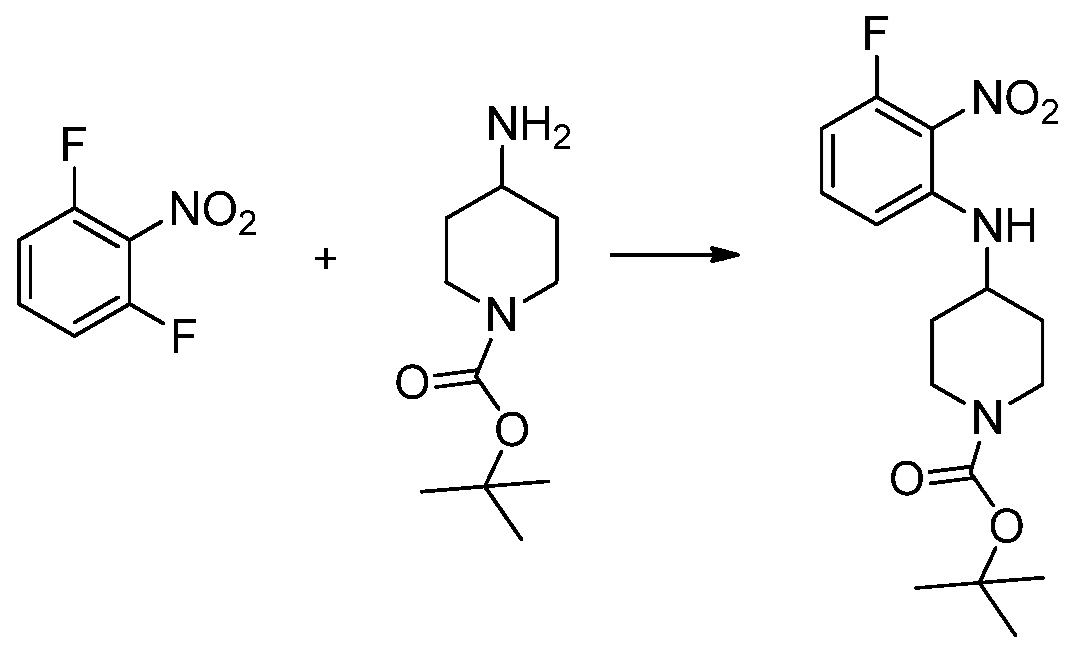
1,3-дифтор-2-нитробензол (3,000 г, 18,857 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (3,777 г, 18,857 ммоль), карбонат калия (3,127 г, 22,629 ммоль) и йодид калия (0,031 г, 0,189 ммоль) растворяют в N,N-диметилформамиде (150 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-40%) и концентрируют с получением указанного в заголовке соединения (4,300 г, 67,2%) в виде красного твердого вещества.
[Стадия 2] Синтез трет-бутил 4-((2-амино-3-фторфенил)амино)пиперидин-1-карбоксилата
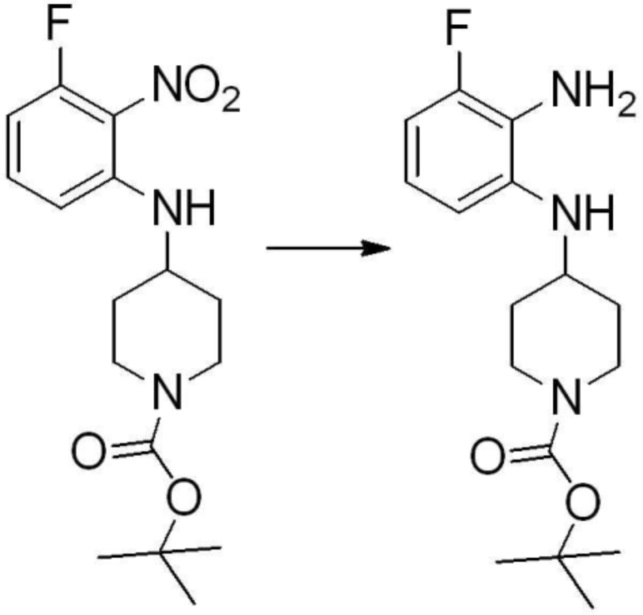
Трет-бутил 4-((3-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилат (4,300 г, 12,671 ммоль), полученный на стадии 1, растворяют в метанол (120 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через целит для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (3,260 г, 83,2%) в виде желтого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(4-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
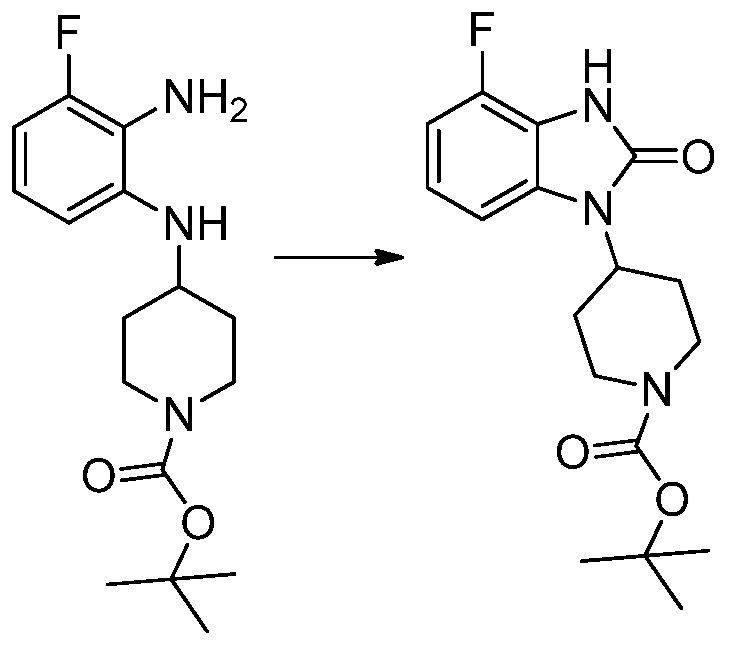
Трет-бутил 4-((2-амино-3-фторфенил)амино)пиперидин-1-карбоксилат (3,260 г, 10,537 ммоль), полученный на стадии 2 растворяют в N,N-диметилформамиде (100 мл) при 0°C, после чего 1,1'-карбонилдиимидазол (КДИ, 1,879 г, 11,591 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0 to 70%) и концентрируют с получением указанного в заголовке соединения (2,320 г, 65,7%) в виде фиолетового твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(4-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
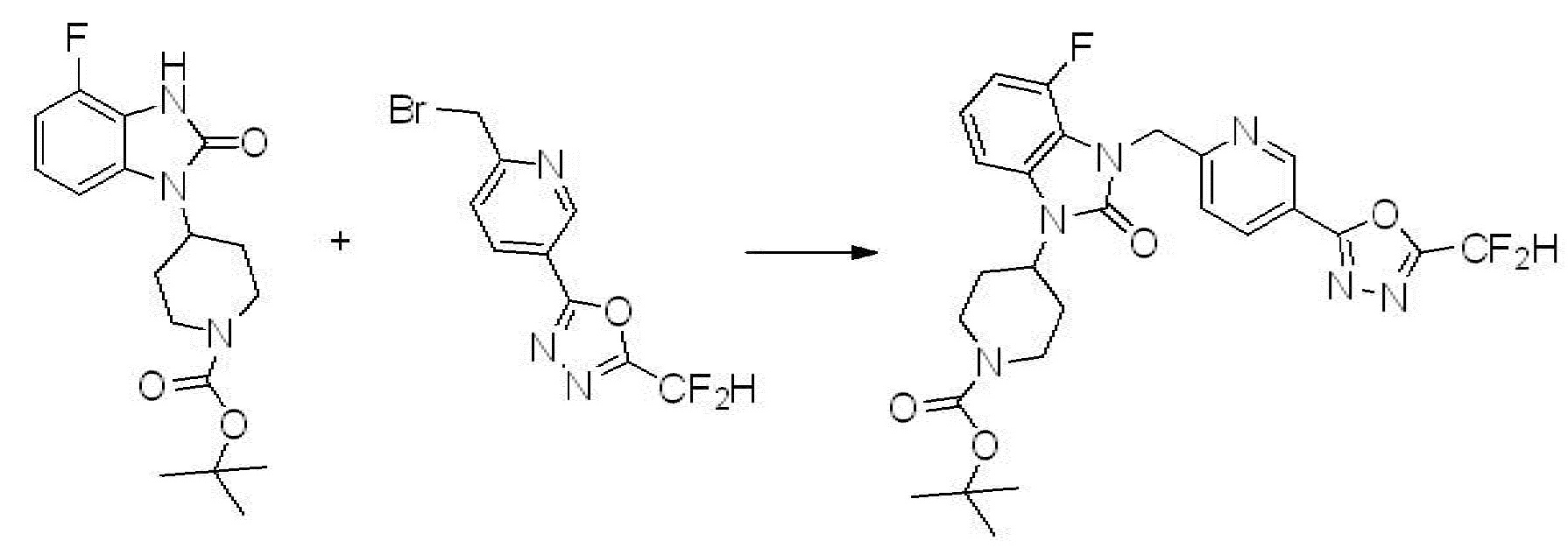
Трет-бутил 4-((2-амино-3-фторфенил)амино)пиперидин-1-карбоксилат (3,260 г, 10,537 ммоль), полученный на стадии 3 растворяют в N,N-диметилформамиде (100 мл) при 0°C, после чего 1,1'-карбонилдиимидазол (КДИ, 1,879 г, 11,591 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (2,320 г, 65,7%) в виде фиолетового твердого вещества.
[Стадия 5] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
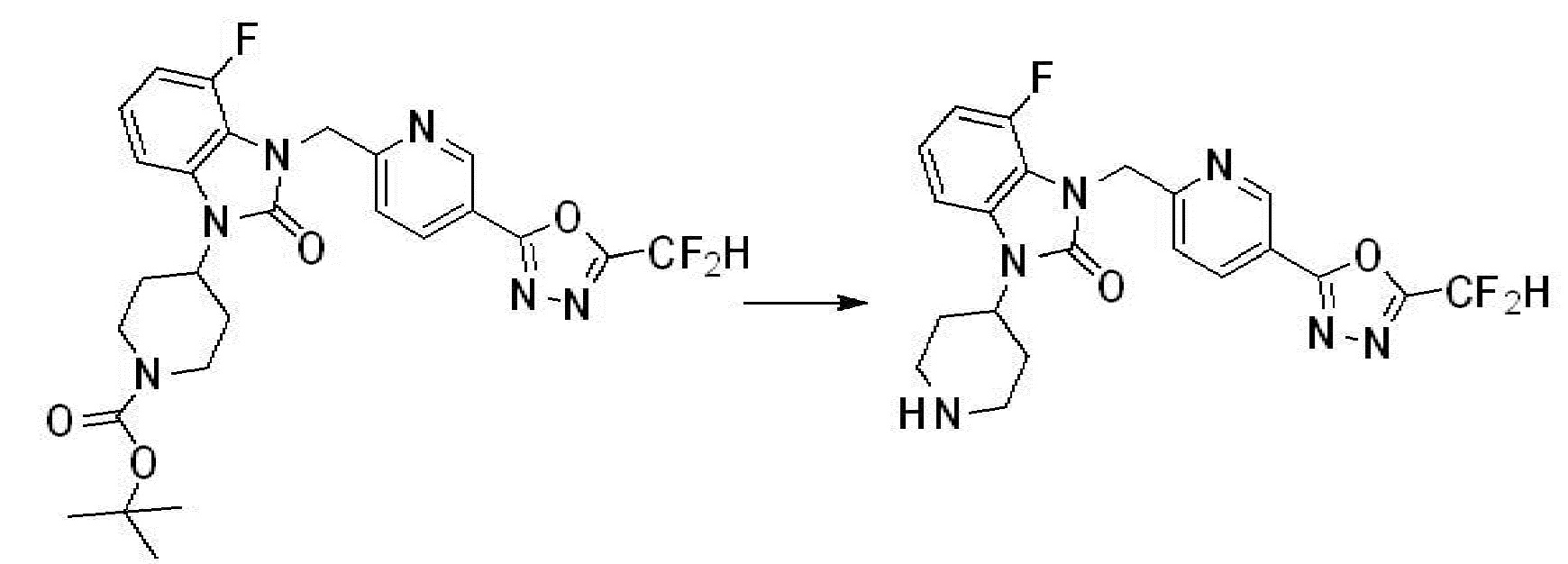
Трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,650 г, 4,866 ммоль), полученный на стадии 4, растворяют в дихлорметане (50 мл) при 0°C, после чего трифторуксусную кислоту (7,453 мл, 97,330 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-50%) и концентрируют с получением указанного в заголовке соединения (1,800 г, 83,2%) в виде коричневого твердого вещества.
[Стадия 6] Синтез соединения 76
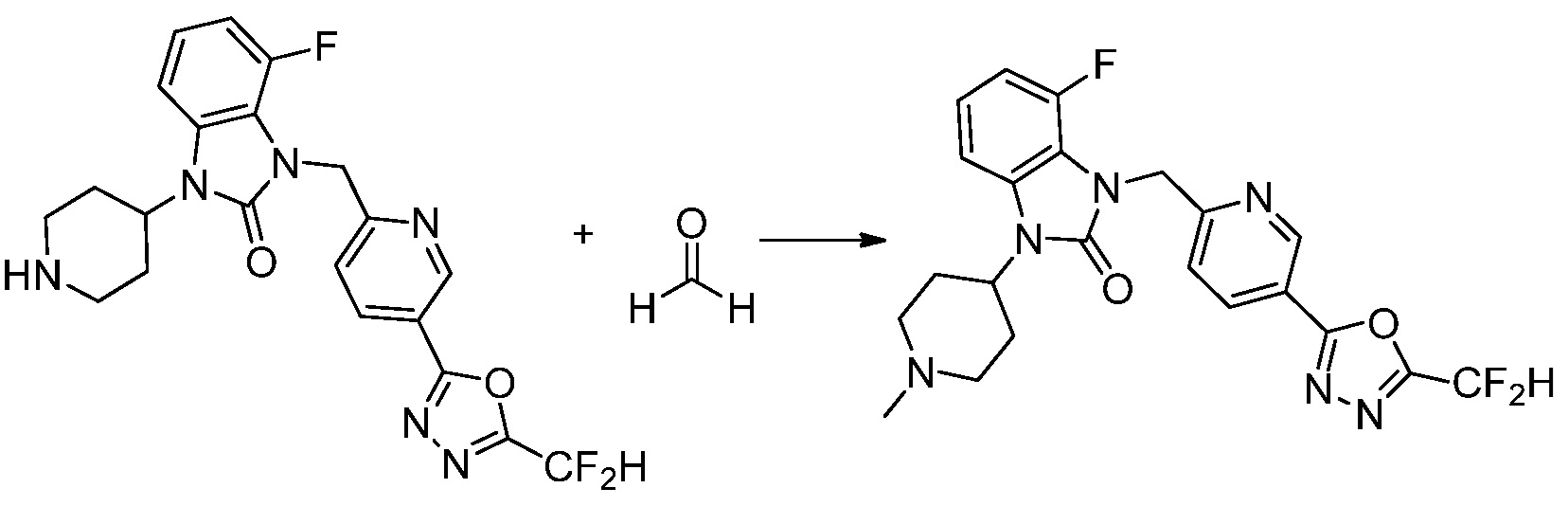
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5, триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль), уксусную кислоту (0,013 мл, 0,225 ммоль) и формальдегид (0,014 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,040 г, 39,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (д, J=2,1 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,32 (д, J=8,2 Гц, 1H), 7,14 (д, J=8,0 Гц, 1H), 7,04-6,72 (м, 3H), 5,42 (с, 2H), 4,47-4,38 (м, 1H), 3,02 (д, J=11,6 Гц, 2H), 2,54-2,44 (м, 2H), 2,34 (с, 3H), 2,19-2,13 (м, 2H), 1,87-1,83 (м, 2H); МСНР (ЭР) m/z 459,3 (M+ + 1).
Пример 77: Синтез соединения 77, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фтор-1-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 77
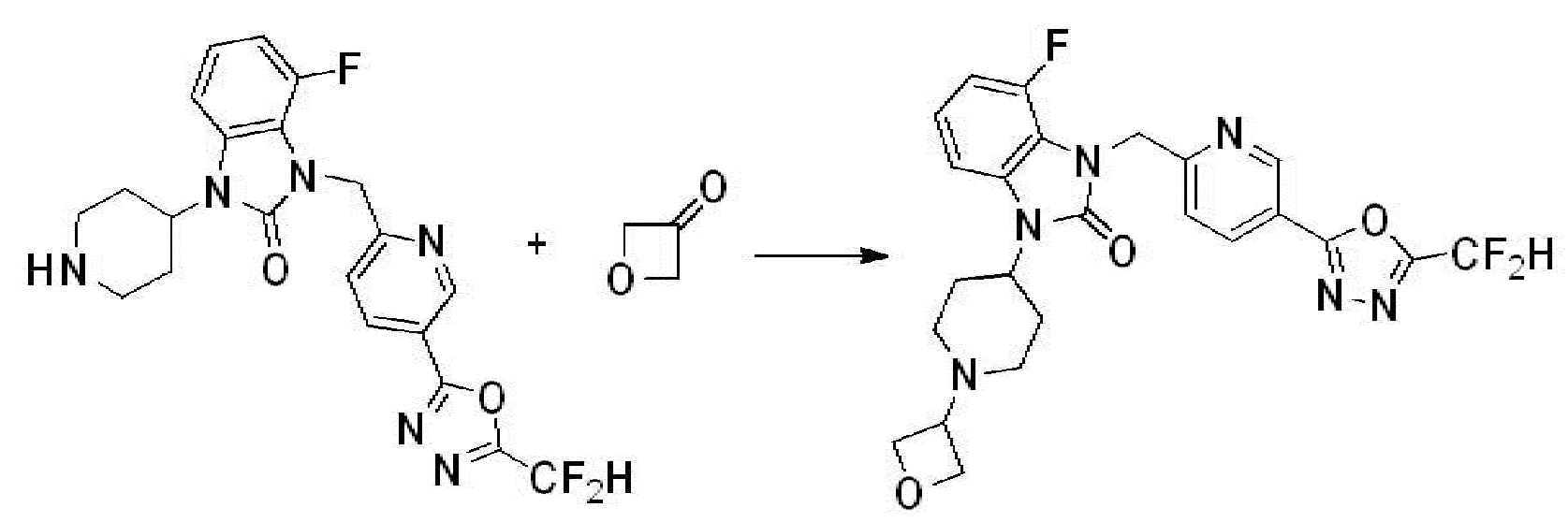
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 76, триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль), уксусную кислоту (0,013 мл, 0,225 ммоль) и оксетан-3-он (0,032 г, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,047 г, 41,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,22 (д, J=2,0 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,32 (д, J=8,2 Гц, 1H), 7,15 (д, J=7,9 Гц, 1H), 7,04-6,72 (м, 3H), 5,41 (с, 2H), 4,64 (дт, J=22,3, 6,4 Гц, 4H), 4,47-4,39 (м, 1H), 3,55-3,49 (м, 1H), 2,90 (д, J=11,5 Гц, 2H), 2,53-2,43 (м, 2H), 2,01 (тд, J=11,8, 1,9 Гц, 2H), 1,89-1,85 (м, 2H); МСНР (ЭР) m/z 501,6 (M+ + 1).
Пример 78: Синтез соединения 78, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 78
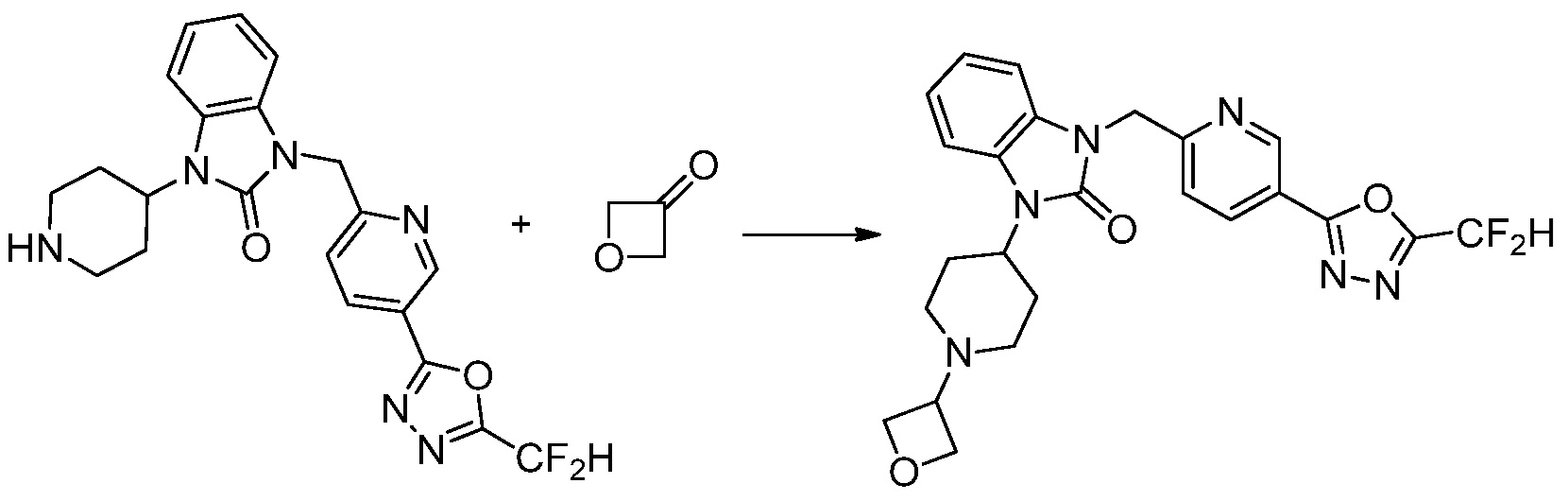
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,235 ммоль), триацетоксиборгидрид натрия (0,099 г, 0,469 ммоль), уксусную кислоту (0,013 мл, 0,235 ммоль) и оксетан-3-он (0,034 г, 0,469 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,053 г, 46,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (дд, J=2,2, 0,7 Гц, 1H), 8,28 (дд, J=8,2, 2,2 Гц, 1H), 7,34 (дд, J=12,3, 4,0 Гц, 2H), 7,09-6,78 (м, 4H), 5,26 (д, J=2,4 Гц, 2H), 4,64 (дт, J=19,7, 6,4 Гц, 4H), 4,47-4,39 (м, 1H), 3,55-3,49 (м, 1H), 2,89 (д, J=11,5 Гц, 2H), 2,54-2,44 (м, 2H), 2,01 (тд, J=11,8, 1,9 Гц, 2H), 1,88-1,84 (м, 2H); МСНР (ЭР) m/z 483,0 (M+ + 1).
Пример 79: Синтез соединения 79, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
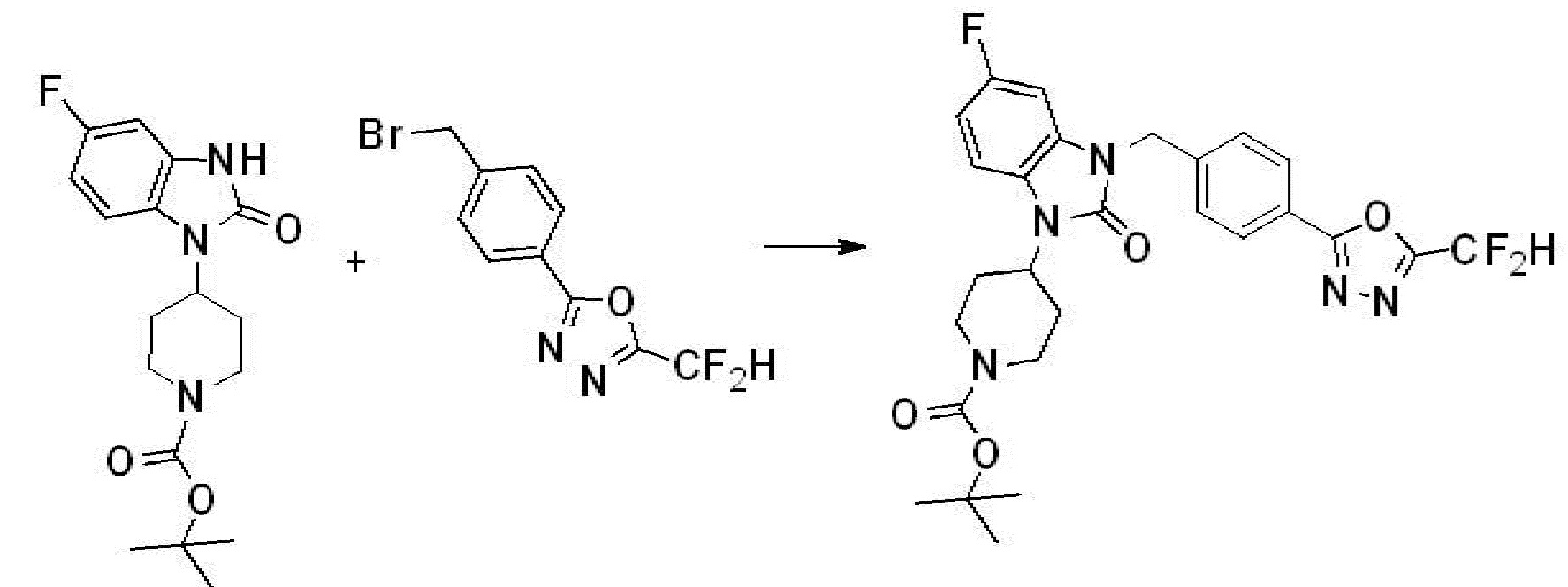
Трет-бутил 4-(5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,300 г, 3,876 ммоль) растворяют в N,N-диметилформамиде (40 мл) при 0°C, после чего гидрид натрия (60,00%, 0,202 г, 5,039 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (1,457 г, 5,039 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (1,880 г, 89,2%) в виде светло-коричневого твердого вещества.
[Стадия 2] Синтез 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
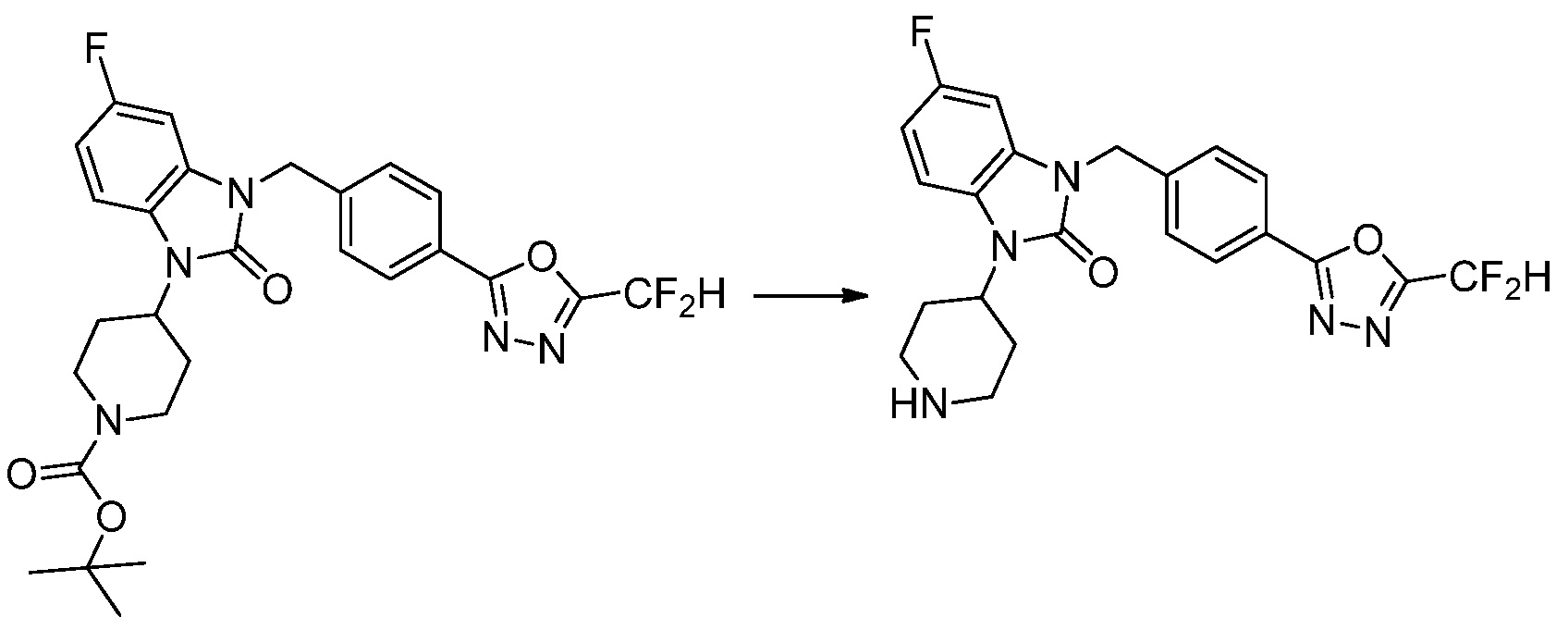
Трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,880 г, 3,459 ммоль), полученный на стадии 1, растворяют в дихлорметане (20 мл) при 0°C, после чего трифторуксусную кислоту (2,649 мл, 34,587 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=50-100%) и концентрируют с получением продукта, после чего полученный продукт снова очищают хроматографией (SiO2, 40 г картридж; дихлорметан/метанол=0-40%) и концентрируют с получением указанного в заголовке соединения (1,230 г, 80,2%) в виде светло-желтого твердого вещества.
[Стадия 3] Синтез соединения 79
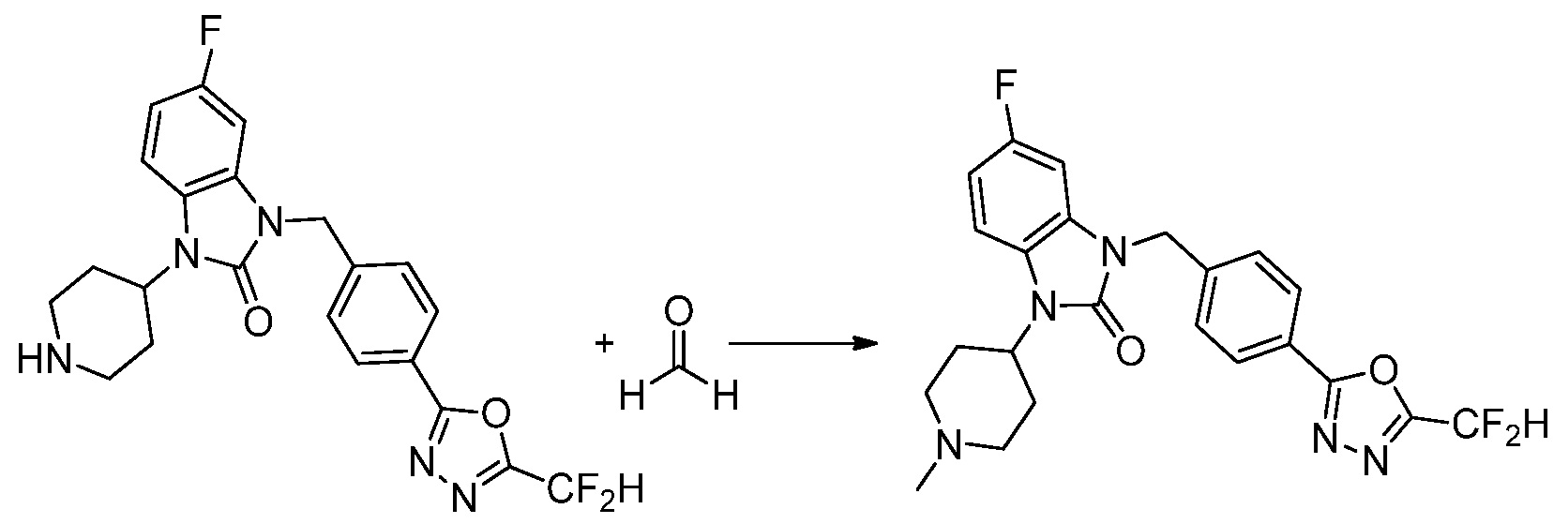
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,226 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,096 г, 0,451 ммоль), уксусную кислоту (0,013 мл, 0,226 ммоль) и формальдегид (0,014 г, 0,451 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,067 г, 65,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,05 (д, J=8,3 Гц, 2H), 7,44 (д, J=8,3 Гц, 2H), 7,21 (дд, J=8,7, 4,3 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 6,73 (тд, J=9,4, 2,8 Гц, 1H), 6,56 (дд, J=8,4, 2,5 Гц, 1H), 5,09 (с, 2H), 4,44-4,36 (м, 1H), 3,02-2,99 (м, 2H), 2,50-2,39 (м, 2H), 2,33 (с, 3H), 2,18-2,12 (м, 2H), 1,84-1,80 (м, 2H); МСНР (ЭР) m/z 458,3 (M+ + 1).
Пример 80: Синтез соединения 80, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-1-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 80
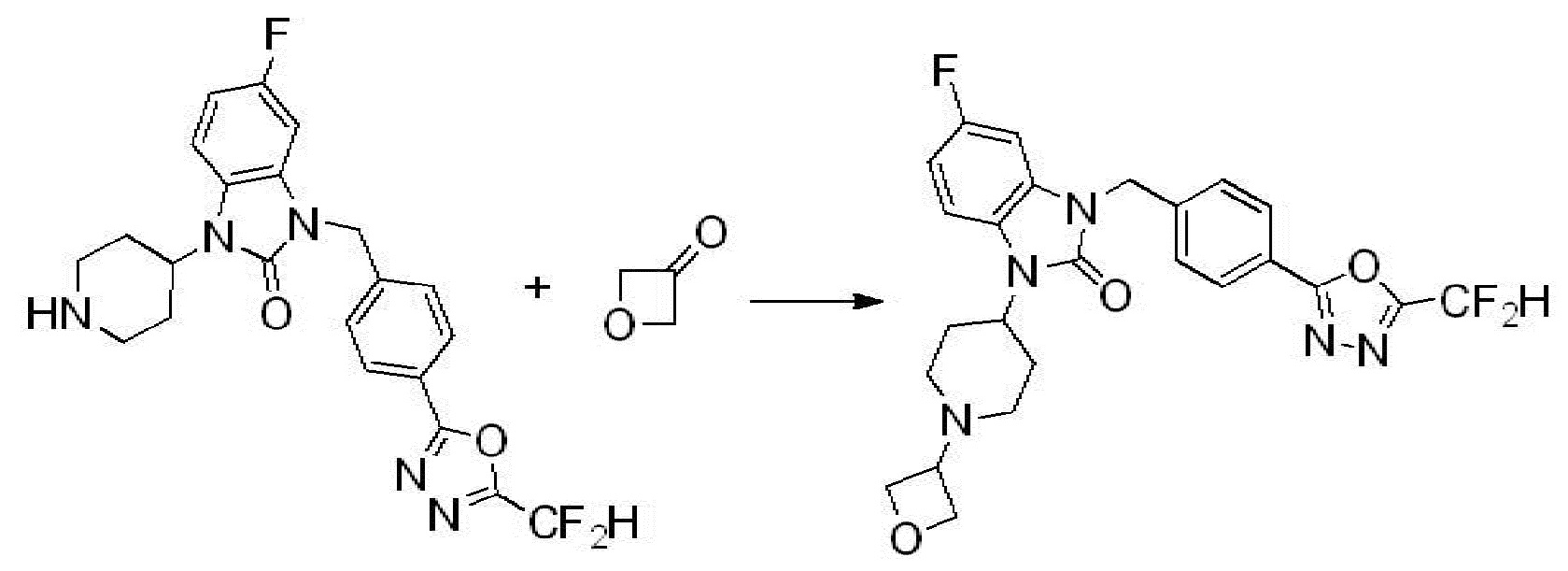
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,226 ммоль), полученный на стадии 2 соединения 79, триацетоксиборгидрид натрия (0,096 г, 0,451 ммоль), уксусную кислоту (0,013 мл, 0,226 ммоль) и оксетан-3-он (0,033 г, 0,451 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,086 г, 76,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,05 (д, J=8,4 Гц, 2H), 7,44 (д, J=8,4 Гц, 2H), 7,23 (дд, J=8,7, 4,3 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 6,76 (тд, J=9,1, 2,5 Гц, 1H), 6,58 (дд, J=8,4, 2,5 Гц, 1H), 5,09 (с, 2H), 4,63 (дт, J=22,9, 6,4 Гц, 4H), 4,45-4,37 (м, 1H), 3,55-3,48 (м, 1H), 2,89 (д, J=11,5 Гц, 2H), 2,49-2,39 (м, 2H), 2,01 (тд, J=11,8, 1,9 Гц, 2H), 1,87-1,83 (м, 2H); МСНР (ЭР) m/z 500,6 (M+ + 1).
Пример 81: Синтез соединения 81, 5,6-дихлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((2-амино-4,5-дихлорфенил)амино)пиперидин-1-карбоксилата
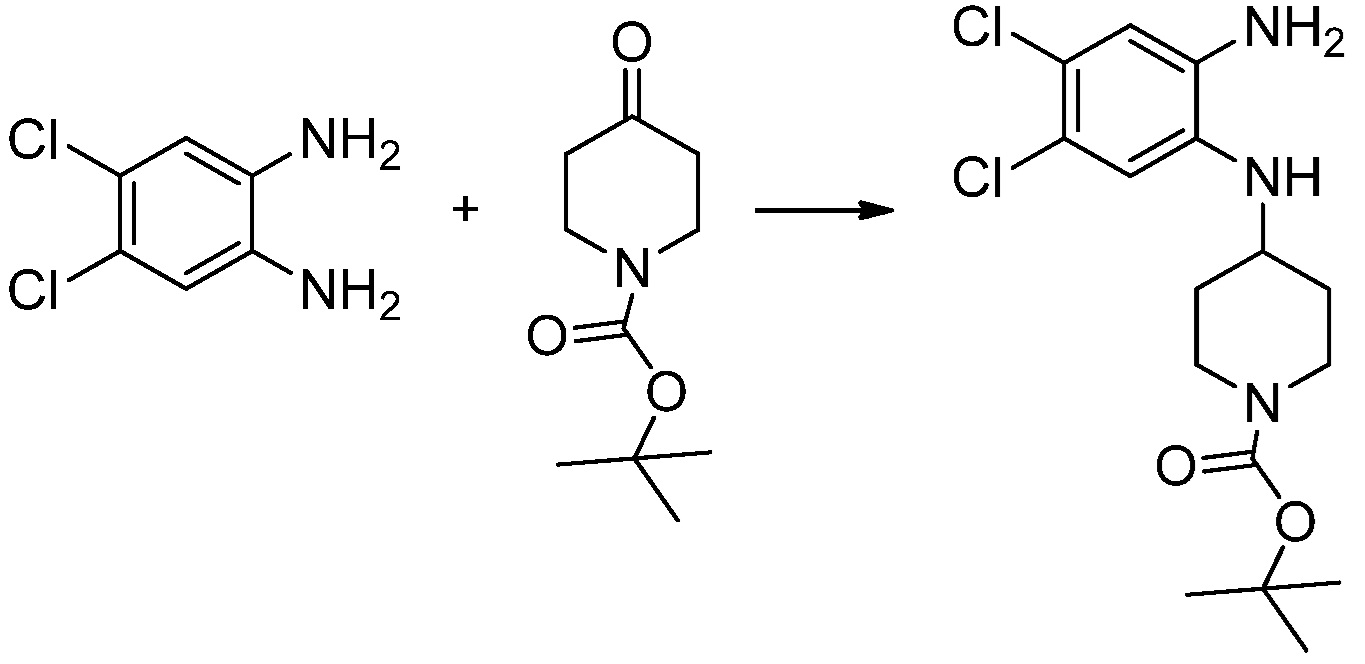
4,5-дихлорбензол-1,2-диамин (5,000 г, 28,244 ммоль), трет-бутил 4-оксопиперидин-1-карбоксилат (6,753 г, 33,893 ммоль), триацетоксиборгидрид натрия (11,972 г, 56,488 ммоль) и уксусную кислоту (1,617 мл, 28,244 ммоль) растворяют в дихлорметане (280 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (4,380 г, 43,0%) в виде коричневого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(5,6-дихлор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
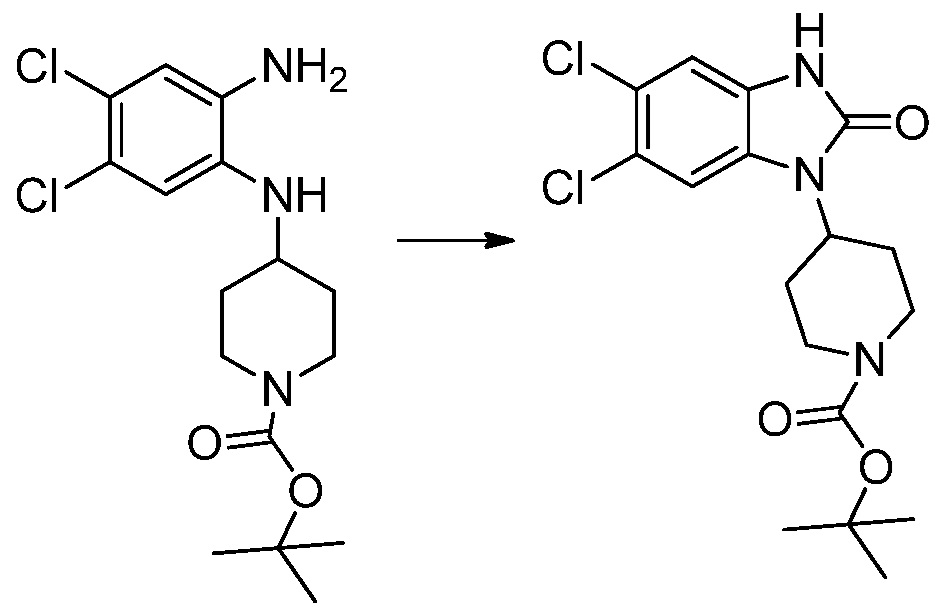
Трет-бутил 4-((2-амино-4,5-дихлорфенил)амино)пиперидин-1-карбоксилат (4,380 г, 12,157 ммоль), полученный на стадии 1 растворяют в тетрагидрофуране (120 мл), после чего 1,1'-карбонилдиимидазол (КДИ, 2,760 г, 17,020 ммоль) добавляют в полученный раствор при 0° и перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают водой, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (4,110 г, 87,5%) в виде серого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(5,6-дихлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
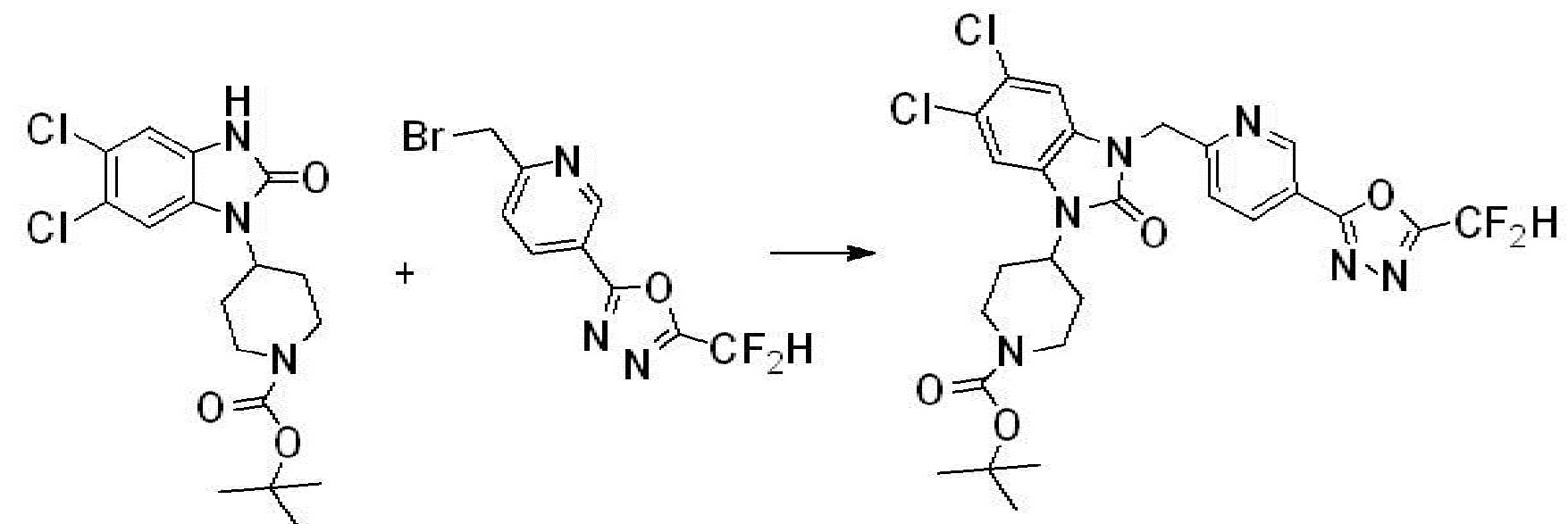
Трет-бутил 4-(5,6-дихлор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,800 г, 7,249 ммоль), полученный на стадии 2, гидрид натрия (60,00%, 0,377 г, 9,423 ммоль) и 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,733 г, 9,423 ммоль) растворяют в N,N-диметилформамиде (70 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (3,240 г, 75,1%) в виде желтого твердого вещества.
[Стадия 4] Синтез 5,6-дихлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
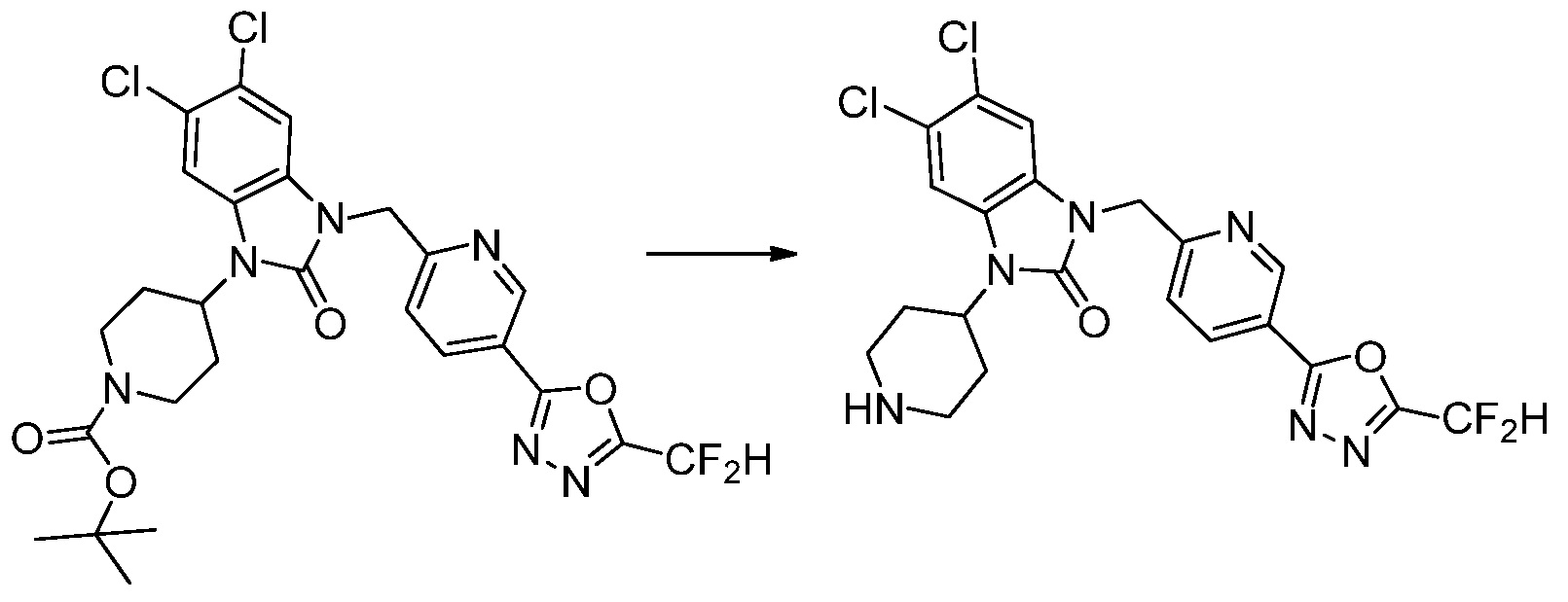
Трет-бутил 4-(5,6-дихлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (3,240 г, 5,441 ммоль), полученный на стадии 3 растворяют в дихлорметане (50 мл) при 0°C, после чего трифторуксусную кислоту (8,334 мл, 108,829 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; дихлорметан/метанол=0-30%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 80 г картридж; дихлорметан/метанол=0-30%) и концентрируют с получением указанного в заголовке соединения (2,316 г, 85,9%) в виде коричневого твердого вещества.
[Стадия 5] Синтез соединения 81
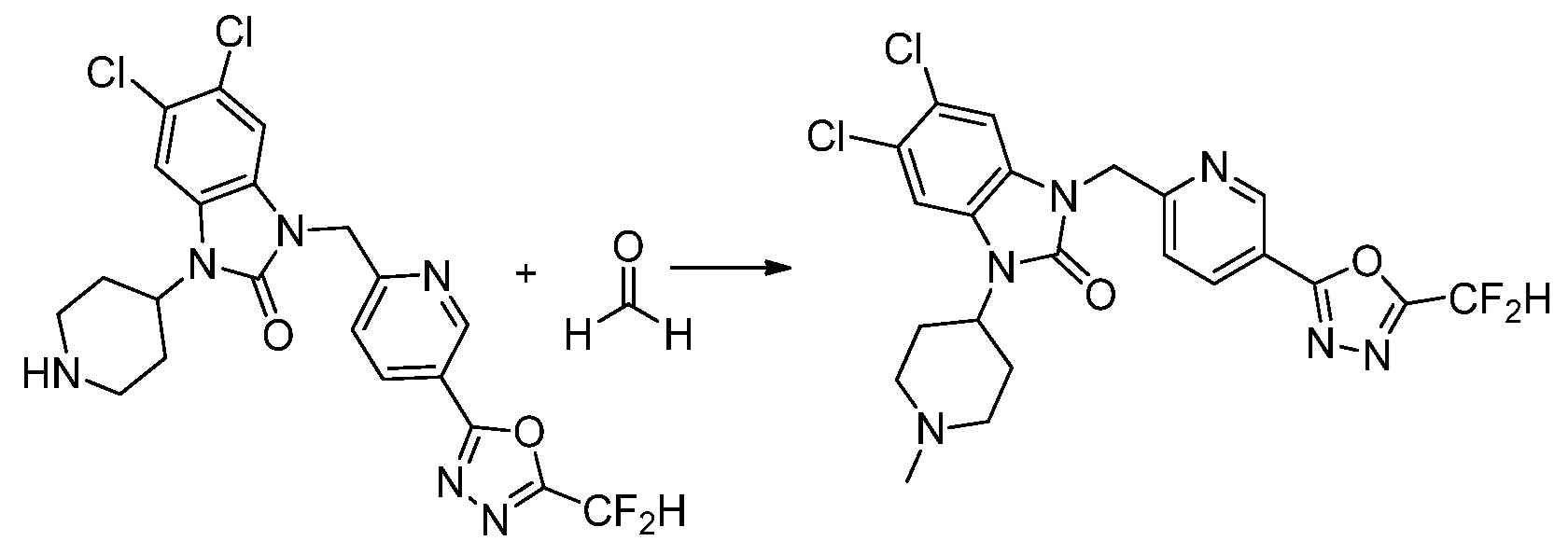
5,6-дихлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,202 ммоль), полученный на стадии 4, триацетоксиборгидрид натрия (0,086 г, 0,404 ммоль), уксусную кислоту (0,012 мл, 0,202 ммоль) и формальдегид (0,012 г, 0,404 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,051 г, 49,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=1,6 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,74-7,40 (м, 2H), 7,07 (с, 1H), 6,92 (т, J=51,6 Гц, 1H), 5,21 (с, 2H), 4,41-4,32 (м, 1H), 3,02 (д, J=11,6 Гц, 2H), 2,45-2,34 (м, 5H), 2,18-2,11 (м, 2H), 1,84-1,80 (м, 2H); МСНР (ЭР) m/z 511,5 (M+ + 2).
Пример 82: Синтез соединения 82, 5,6-дихлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 82
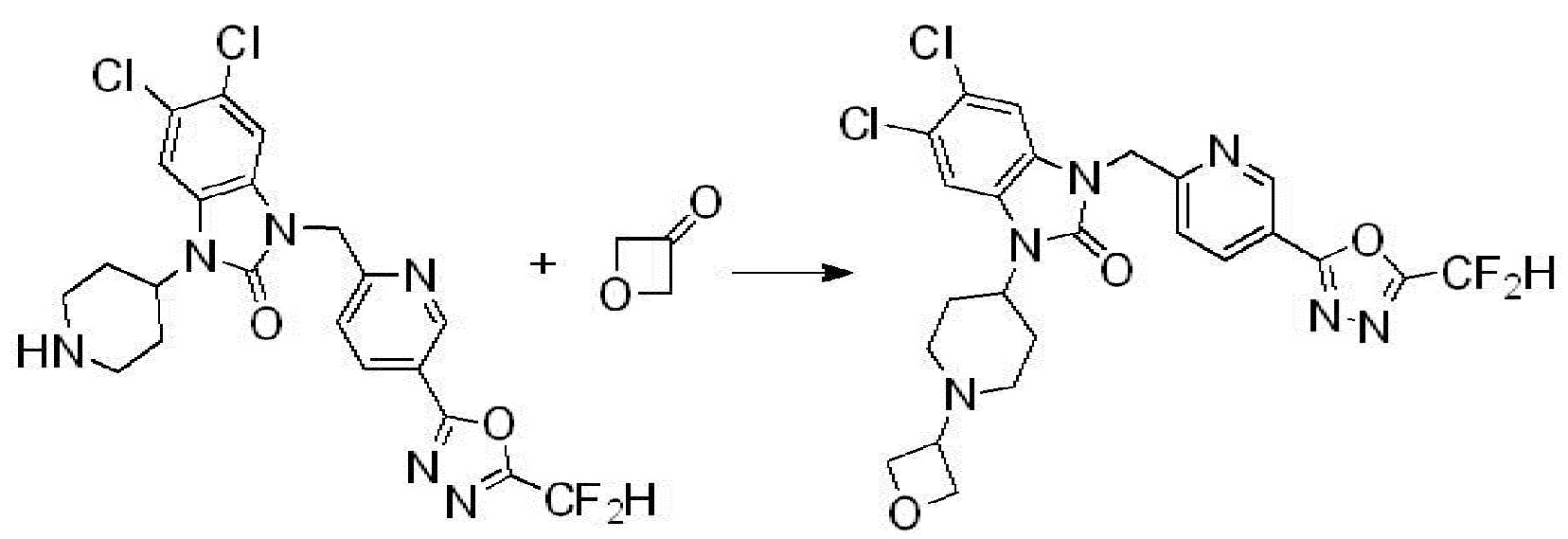
5,6-дихлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,202 ммоль), полученный на стадии 4 соединения 81, триацетоксиборгидрид натрия (0,086 г, 0,404 ммоль), уксусную кислоту (0,012 мл, 0,202 ммоль) и оксетан-3-он (0,029 г, 0,404 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,091 г, 82,0%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (дд, J=2,2, 0,6 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (дд, J=8,2, 0,4 Гц, 1H), 7,38 (с, 1H), 7,08 (с, 1H), 6,92 (т, J=51,6 Гц, 1H), 5,20 (с, 2H), 4,65 (дт, J=19,9, 6,4 Гц, 4H), 4,41-4,33 (м, 1H), 3,56-3,50 (м, 1H), 2,93-2,90 (м, 2H), 2,45-2,35 (м, 2H), 2,04-1,98 (м, 2H), 1,87-1,83 (м, 2H); МСНР (ЭР) m/z 553,5 (M+ + 2).
Пример 83: Синтез соединения 83, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((2-амино-4,5-дифторфенил)амино)пиперидин-1-карбоксилата
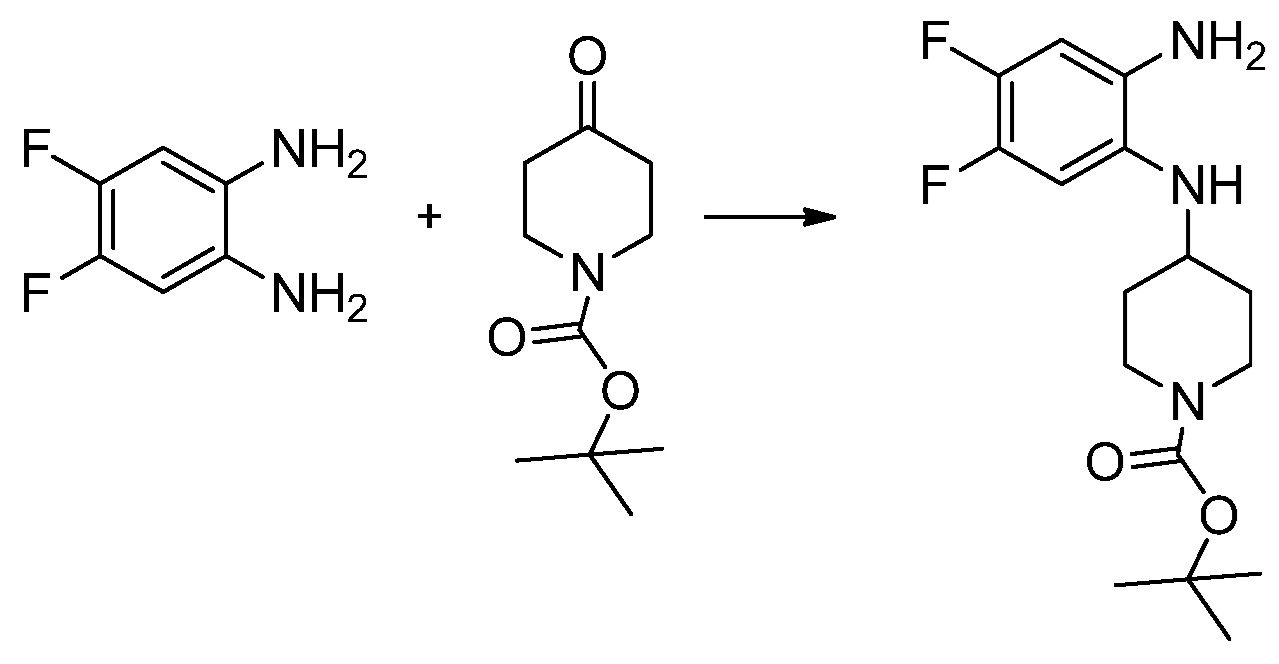
4,5-дифторбензол-1,2-диамин (4,000 г, 27,755 ммоль), трет-бутил 4-оксопиперидин-1-карбоксилат (5,807 г, 29,142 ммоль) и триацетоксиборгидрид натрия (11,765 г, 55,509 ммоль) растворяют в дихлорметане (270 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-40%) и концентрируют с получением указанного в заголовке соединения (5,970 г, 65,7%) в виде коричневой жидкости.
[Стадия 2] Синтез трет-бутил 4-(5,6-дифтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
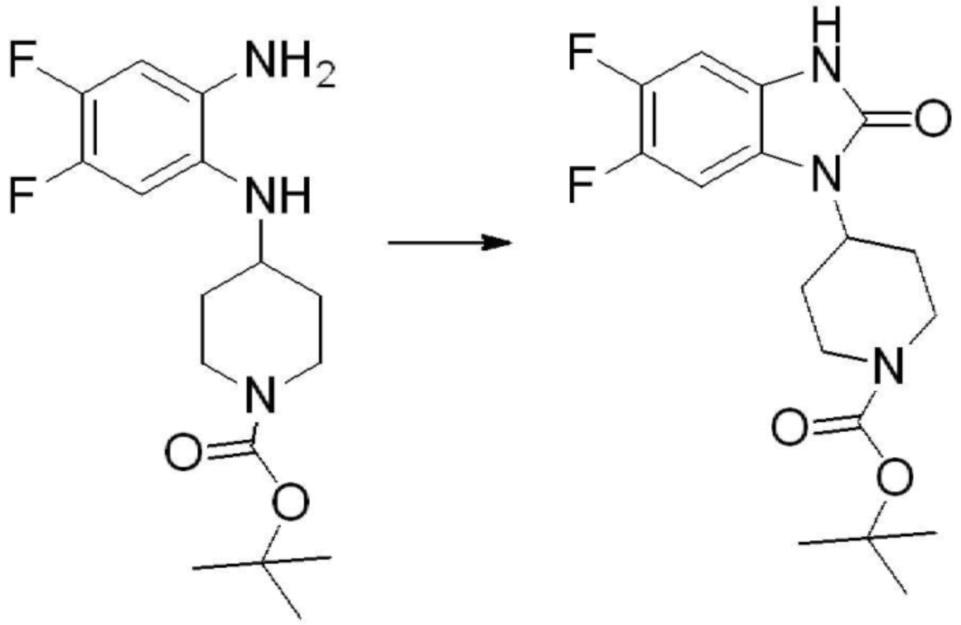
Трет-бутил 4-((2-амино-4,5-дифторфенил)амино)пиперидин-1-карбоксилат (5,970 г, 18,236 ммоль), полученный на стадии 1 растворяют в тетрагидрофуране (180 мл) при 0°C, после чего 1,1'-карбонилдиимидазол (КДИ, 3,548 г, 21,883 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-80%) и концентрируют с получением указанного в заголовке соединения (5,060 г, 78,5%) в виде коричневого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
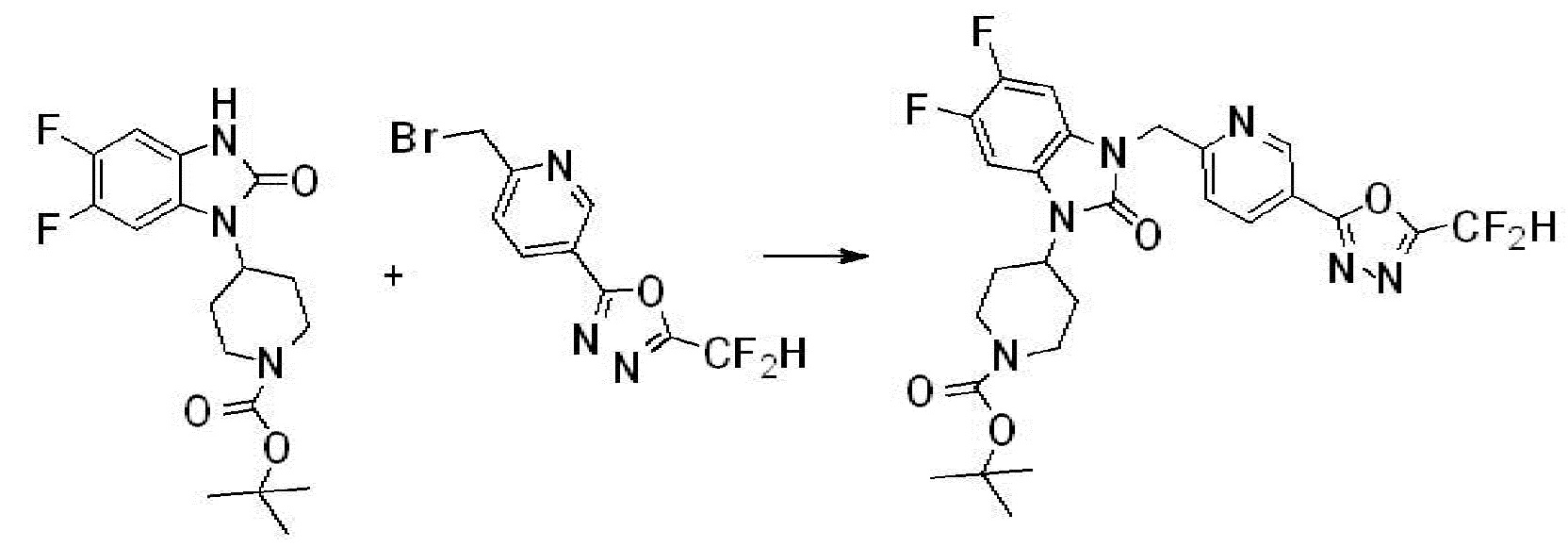
Трет-бутил 4-(5,6-дифтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,600 г, 4,528 ммоль), полученный на стадии 2 растворяют в N,N-диметилформамиде (50 мл) при 0°C, после чего гидрид натрия (60,00%, 0,272 г, 6,792 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,707 г, 5,886 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (1,580 г, 62,0%) в виде коричневого твердого вещества.
[Стадия 4] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
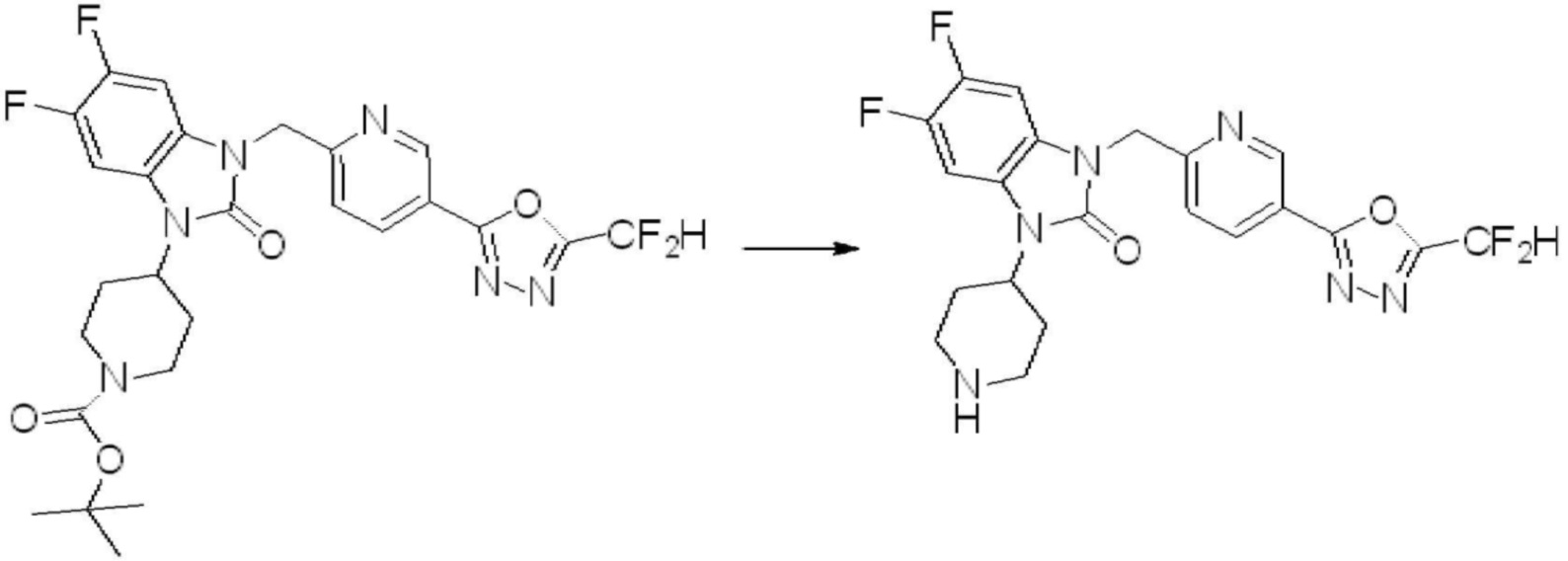
Трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,580 г, 2,809 ммоль), полученный на стадии 3, растворяют в дихлорметане (19 мл) при 0°C, после чего трифторуксусную кислоту (2,151 мл, 28,087 ммоль) добавляют в полученный раствор и перемешивают при комнатной температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=50-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 40 г картридж; дихлорметан/метанол=0-30%) и концентрируют с получением указанного в заголовке соединения (0,513 г, 39,5%) в виде коричневого твердого вещества.
[Стадия 5] Синтез соединения 83
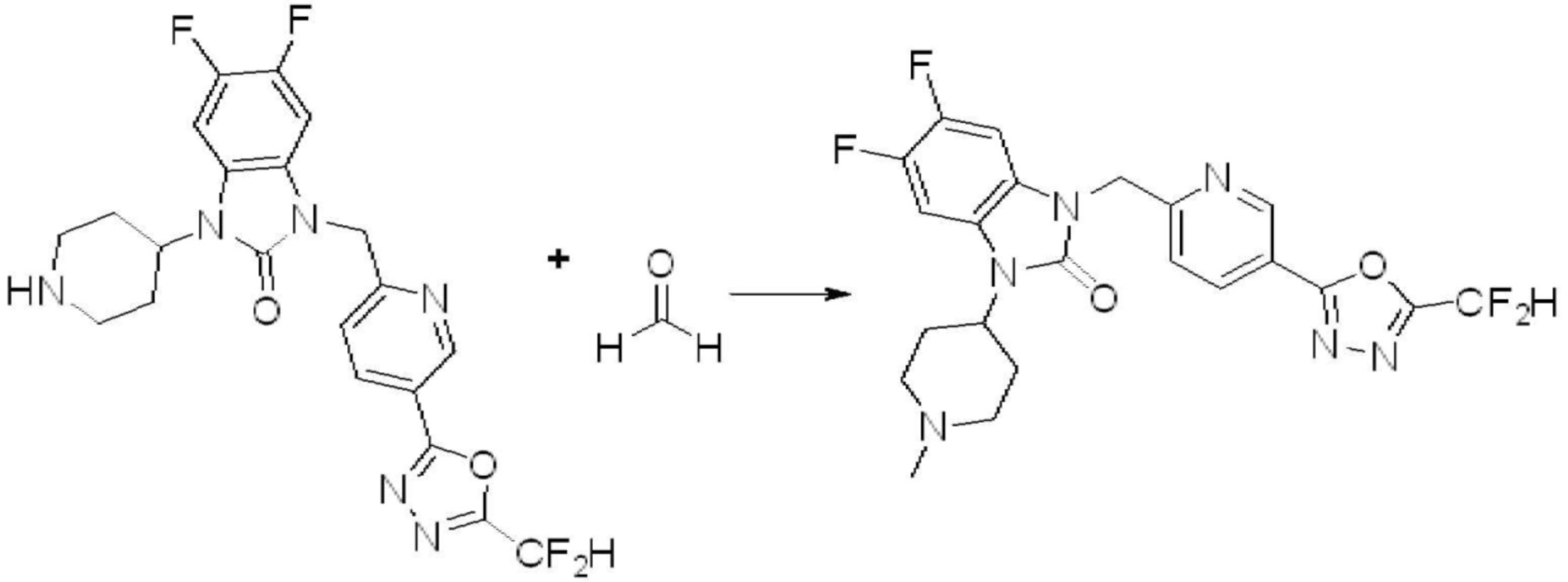
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,216 ммоль), полученный на стадии 4, триацетоксиборгидрид натрия (0,092 г, 0,433 ммоль), уксусную кислоту (0,012 мл, 0,216 ммоль) и формальдегид (0,013 г, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,070 г, 67,5%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (дд, J=2,2, 0,7 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (дд, J=8,2, 0,6 Гц, 1H), 7,16 (дд, J=10,4, 6,7 Гц, 1H), 6,91 (т, J=51,6 Гц, 1H), 6,86 (дд, J=9,7, 6,8 Гц, 1H), 5,20 (с, 2H), 4,41-4,32 (м, 1H), 3,01 (дд, J=9,8, 1,9 Гц, 2H), 2,44-2,33 (м, 5H), 2,14 (тд, J=12,0, 2,1 Гц, 2H), 1,84-1,80 (м, 2H); МСНР (ЭР) m/z 477,6 (M+ + 1).
Пример 84: Синтез соединения 84, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 84
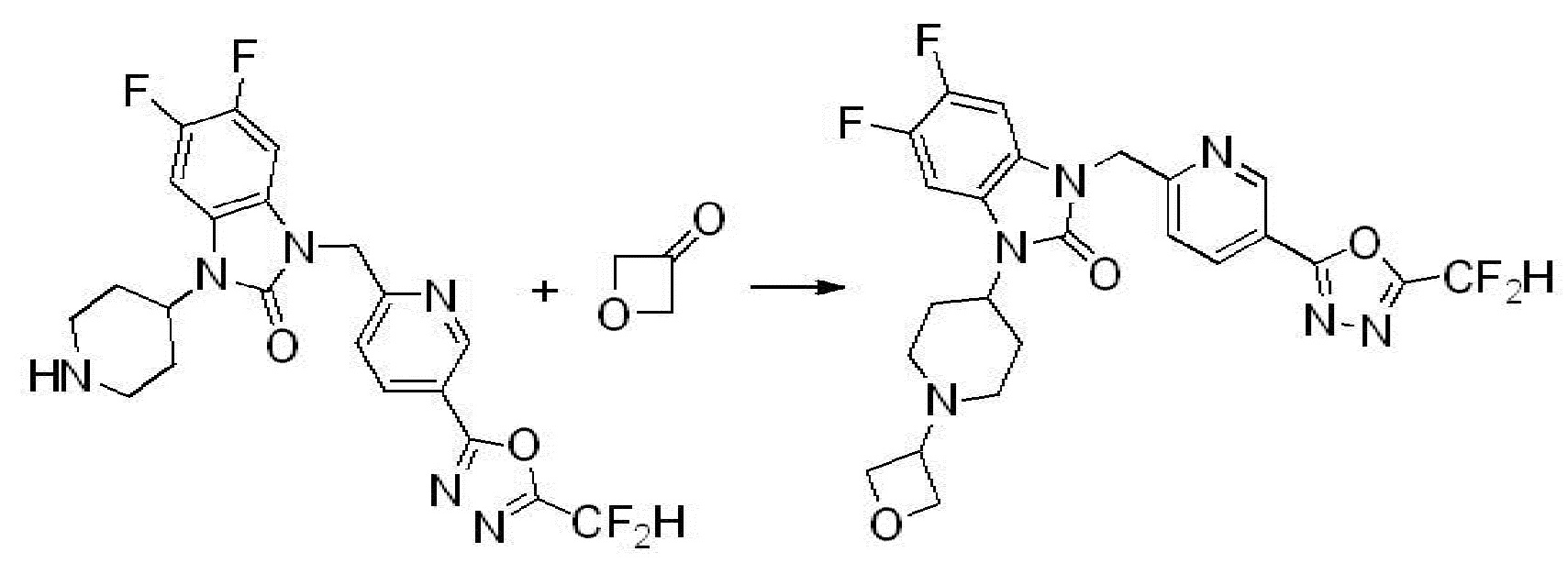
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,216 ммоль), полученный на стадии 4 соединения 83, триацетоксиборгидрид натрия (0,092 г, 0,433 ммоль), уксусную кислоту (0,012 мл, 0,216 ммоль) и оксетан-3-он (0,031 г, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,071 г, 63,1%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (дд, J=2,2, 0,8 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (дд, J=8,2, 0,7 Гц, 1H), 7,16 (дд, J=10,3, 6,7 Гц, 1H), 6,91 (т, J=51,6 Гц, 1H), 6,87 (дд, J=9,7, 6,8 Гц, 1H), 5,19 (с, 2H), 4,97 (дт, J=21,5, 200,2 Гц, 4H), 4,41-4,33 (м, 1H), 3,55-3,49 (м, 1H), 2,90 (д, J=11,6 Гц, 2H), 2,43-2,33 (м, 2H), 2,01 (тд, J=11,8, 2,0 Гц, 2H), 1,86-1,83 (м, 2H); МСНР (ЭР) m/z 519,6 (M+ + 1).
Пример 85: Синтез соединения 85, 5,6-дихлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(5,6-дихлор-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
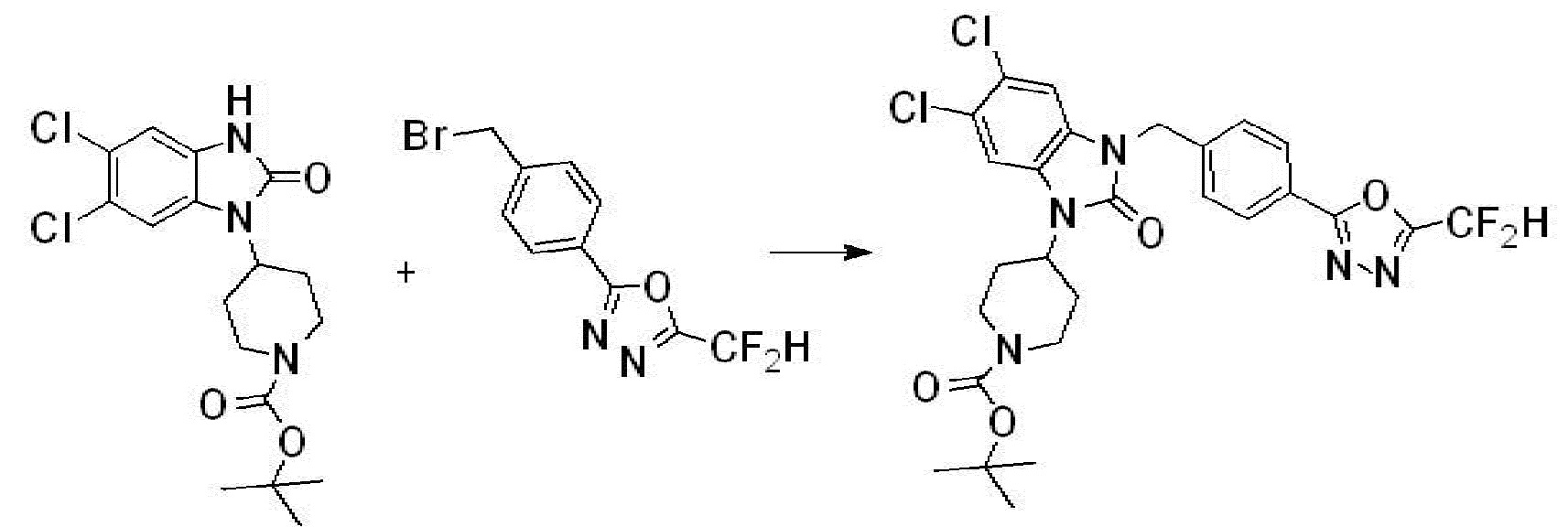
Трет-бутил 4-(5,6-дихлор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,050 г, 5,307 ммоль), полученный на стадии 2 соединения 81 растворяют в N,N-диметилформамиде (50 мл) при 0°C, после чего гидрид натрия (60,00%, 0,318 г, 7,961 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (1,994 г, 6,899 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (2,120 г, 67,2%) в виде коричневого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(5,6-дихлор-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
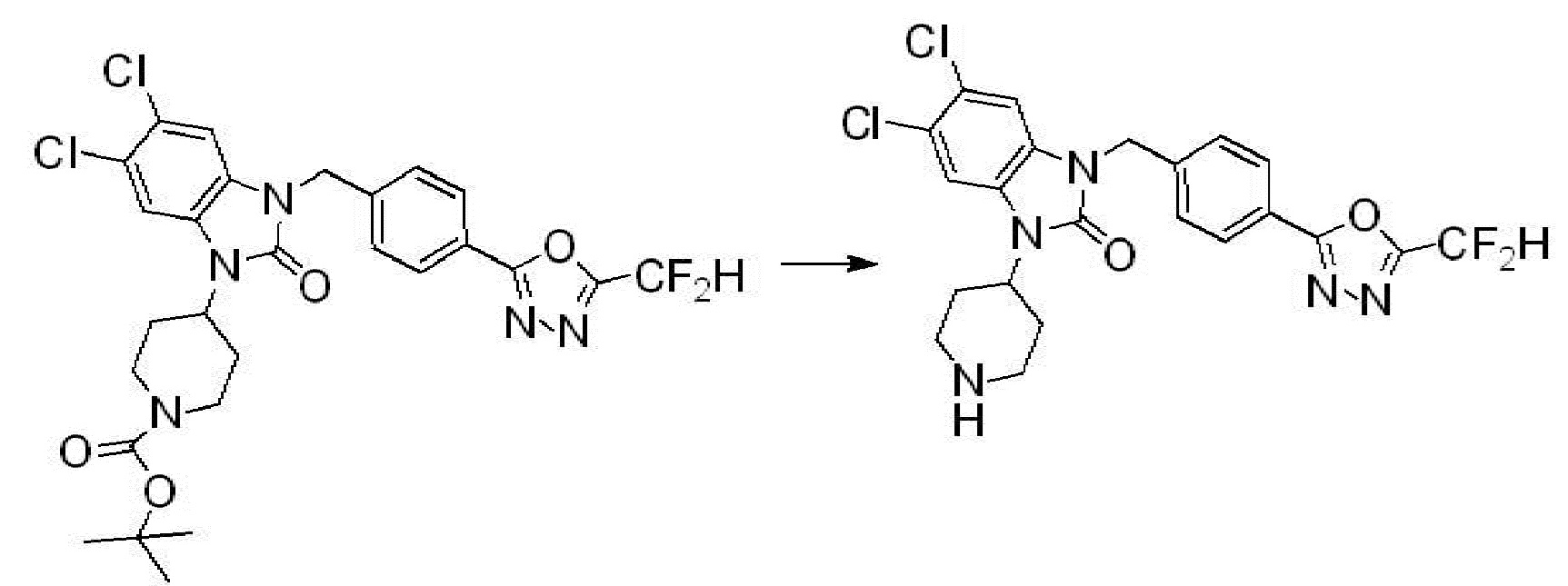
Трет-бутил 4-(5,6-дихлор-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,120 г, 3,566 ммоль), полученный на стадии 1 и трифторуксусную кислоту (2,731 мл, 35,664 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; дихлорметан/метанол=0-30%) и концентрируют с получением указанного в заголовке соединения (0,195 г, 9,2%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 85
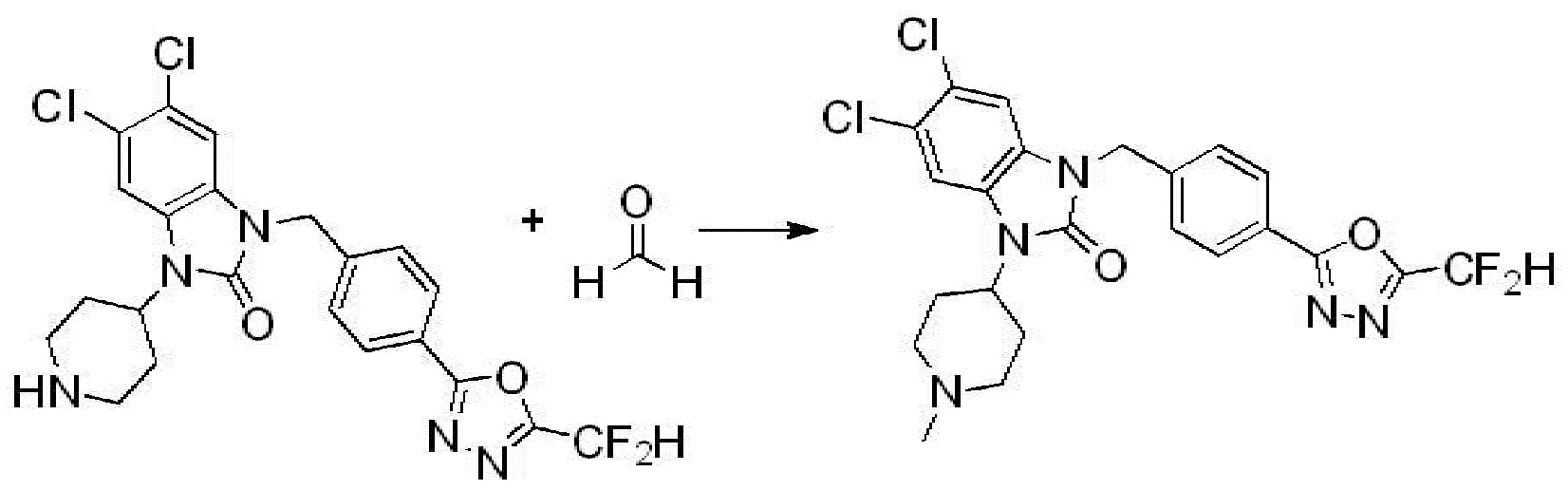
5,6-дихлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,202 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,086 г, 0,405 ммоль), уксусную кислоту (0,012 мл, 0,202 ммоль) и формальдегид (0,012 г, 0,405 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,065 г, 63,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,07 (д, J=8,4 Гц, 2H), 7,44-7,41 (м, 3H), 6,90 (т, J=51,7 Гц, 1H), 6,87 (с, 1H), 5,09 (с, 2H), 4,43-4,35 (м, 1H), 3,05-3,02 (м, 2H), 2,46-2,35 (м, 5H), 2,16 (тд, J=12,0, 2,1 Гц, 2H), 1,85-1,81 (м, 2H); МСНР (ЭР) m/z 510,5 (M+ + 2).
Пример 86: Синтез соединения 86, 5,6-дихлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 86
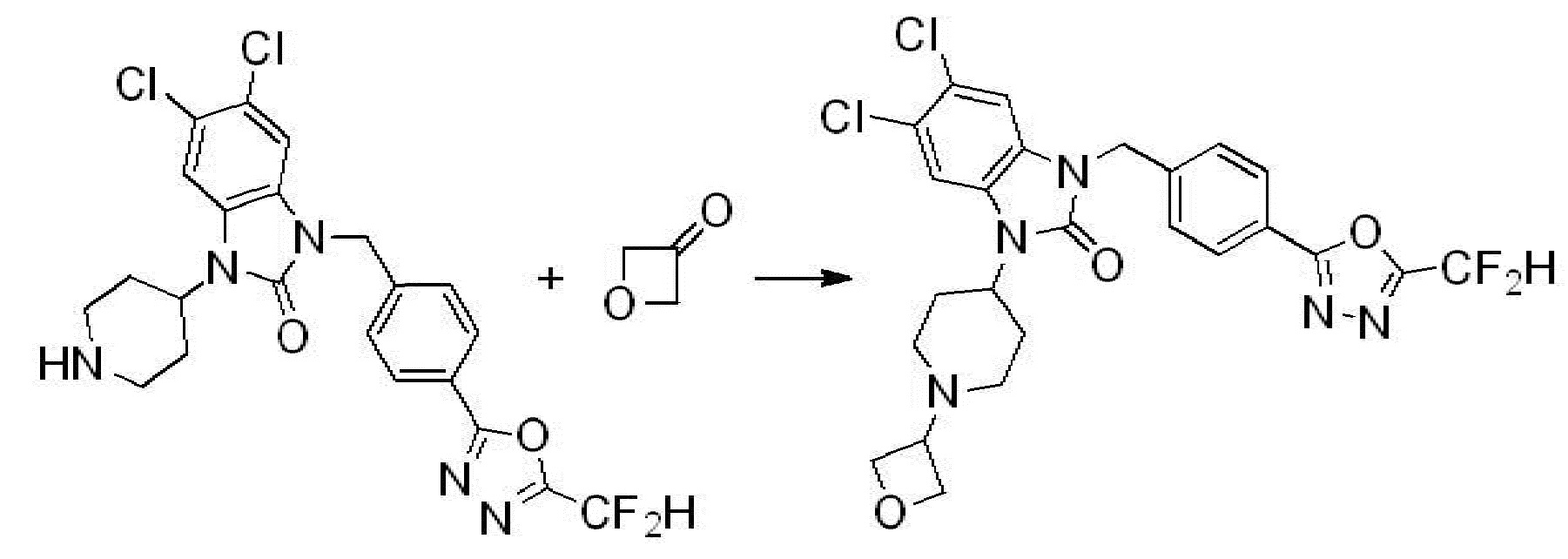
5,6-дихлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,202 ммоль), полученный на стадии 2 соединения 85, триацетоксиборгидрид натрия (0,086 г, 0,405 ммоль), уксусную кислоту (0,012 мл, 0,202 ммоль) и оксетан-3-он (0,029 г, 0,405 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,056 г, 50,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,08-8,06 (м, 2H), 7,43 (д, J=8,5 Гц, 2H), 7,39 (с, 1H), 6,90 (т, J=51,7 Гц, 1H), 6,89 (с, 1H), 5,09 (с, 2H), 4,66 (дт, J=19,4, 6,4 Гц, 4H), 4,43-4,35 (м, 1H), 3,57-3,51 (м, 1H), 2,92 (д, J=11,6 Гц, 2H), 2,45-2,35 (м, 2H), 2,05-1,99 (м, 2H), 1,88-1,84 (м, 2H); МСНР (ЭР) m/z 552,5 (M+ + 2).
Пример 87: Синтез соединения 87, 5,6-дихлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(5,6-дихлор-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
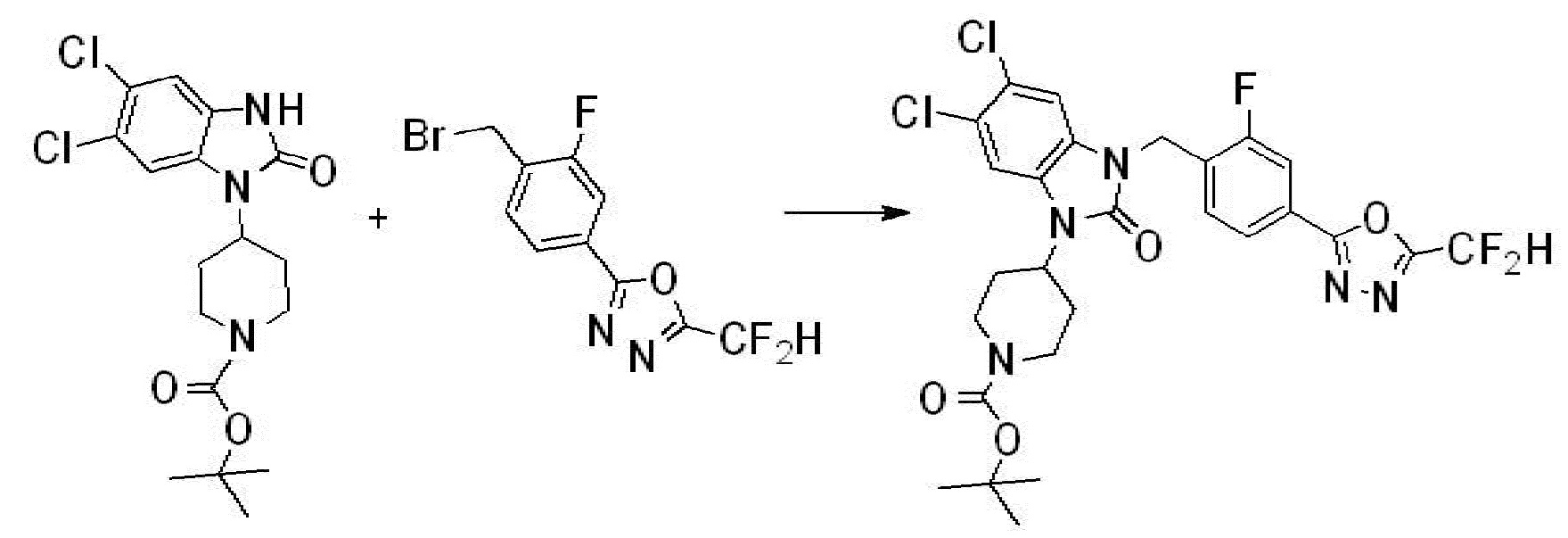
Трет-бутил 4-(5,6-дихлор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,050 г, 5,307 ммоль), полученный на стадии 2 соединения 81 растворяют в N,N-диметилформамиде (50 мл) при 0°C, после чего гидрид натрия (60,00%, 0,318 г, 7,961 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (2,119 г, 6,899 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (2,157 г, 66,4%) в виде светло-коричневого твердого вещества.
[Стадия 2] Синтез 5,6-дихлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
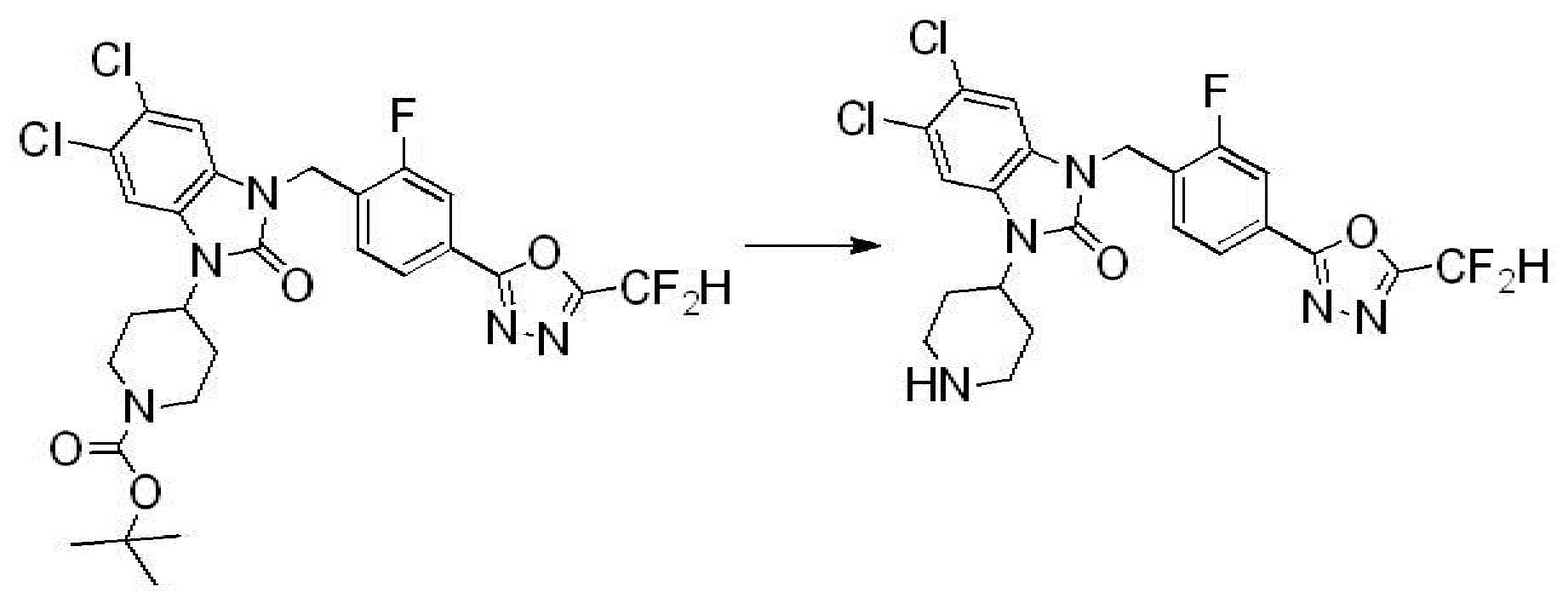
Трет-бутил 4-(5,6-дихлор-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,157 г, 3,522 ммоль), полученный на стадии 1 и трифторуксусную кислоту (2,697 мл, 35,219 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; дихлорметан/метанол=0-50%) и концентрируют с получением указанного в заголовке соединения (1,268 г, 70,3%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 87
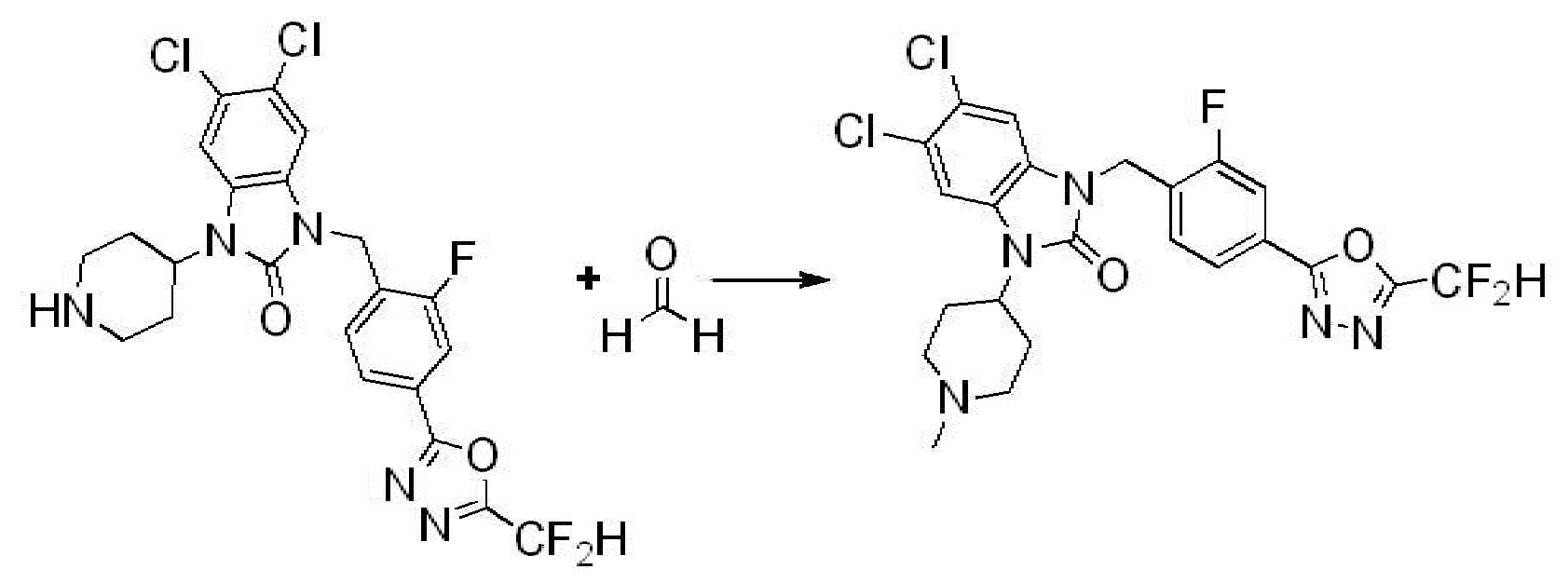
5,6-дихлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,195 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,083 г, 0,390 ммоль), уксусную кислоту (0,011 мл, 0,195 ммоль) и формальдегид (0,012 г, 0,390 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,061 г, 58,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,84-7,82 (м, 2H), 7,43-7,39 (м, 2H), 6,99 (с, 1H), 6,90 (т, J=51,6 Гц, 1H), 5,12 (с, 2H), 4,40-4,32 (м, 1H), 3,02 (д, J=11,7 Гц, 2H), 2,44-2,31 (м, 5H), 2,17-2,11 (м, 2H), 1,81 (дд, J=12,0, 2,2 Гц, 2H); МСНР (ЭР) m/z 526,5 (M+).
Пример 88: Синтез соединения 88, 5,6-дихлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 88
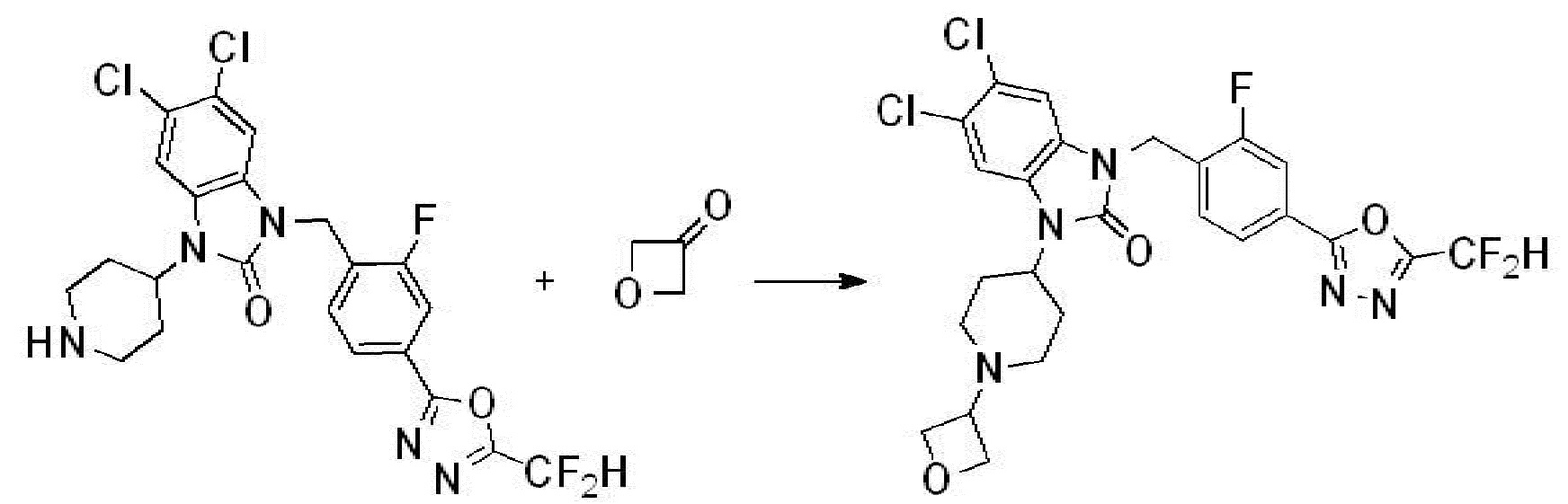
5,6-дихлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,195 ммоль), полученный на стадии 2 соединения 87, триацетоксиборгидрид натрия (0,083 г, 0,390 ммоль), уксусную кислоту (0,011 мл, 0,195 ммоль) и оксетан-3-он (0,028 г, 0,390 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,067 г, 60,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,86-7,83 (м, 2H), 7,44 (т, J=6,5 Гц, 1H), 7,38 (с, 1H), 7,01 (с, 1H), 6,90 (т, J=51,7 Гц, 1H), 5,12 (с, 2H), 4,65 (дт, J=19,6, 6,4 Гц, 4H), 4,41-4,33 (м, 1H), 3,56-3,50 (м, 1H), 2,92 (д, J=11,6 Гц, 2H), 2,45-2,34 (м, 2H), 2,04-1,99 (м, 2H), 1,85 (дд, J=12,0, 2,2 Гц, 2H); МСНР (ЭР) m/z 568,5 (M+ ).
Пример 89: Синтез соединения 89, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-метилпиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((2-нитро-4-(трифторметил)фенил)амино)пиперидин-1-карбоксилата
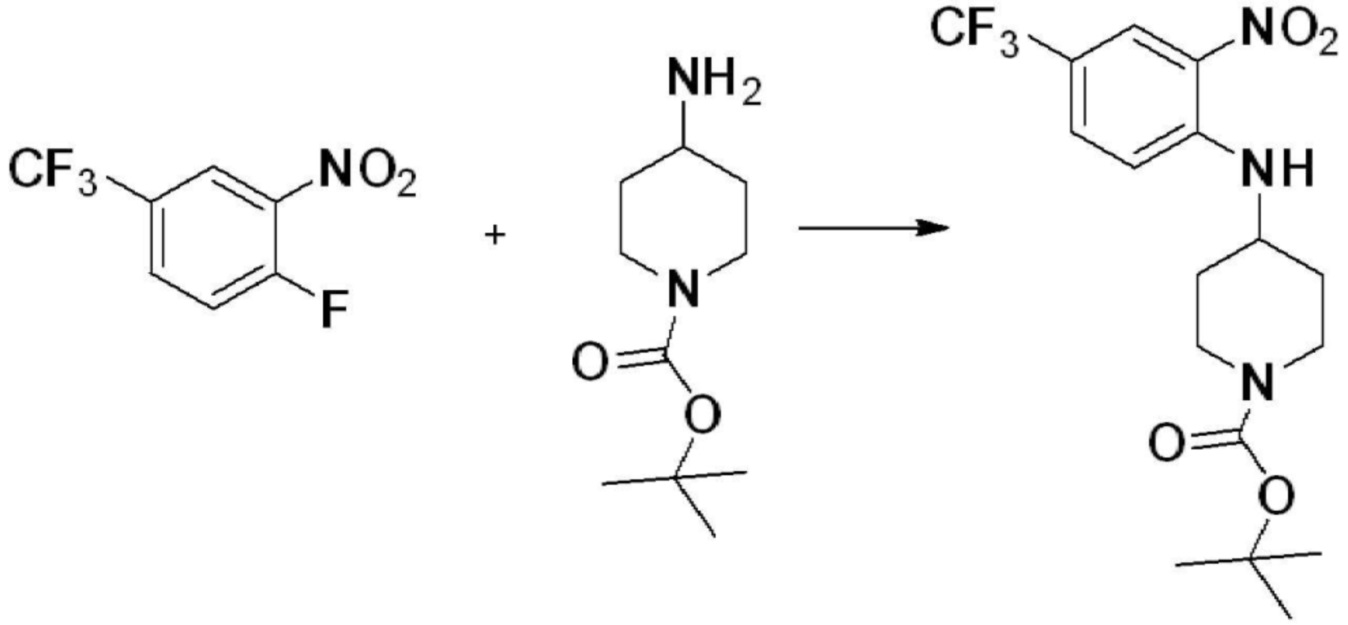
1-фтор-2-нитро-4-(трифторметил)бензол (5,000 г, 23,912 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (5,029 г, 25,108 ммоль), карбонат калия (6,609 г, 47,824 ммоль) и йодид калия (0,397 г, 2,391 ммоль) растворяют в N,N-диметилформамиде (240 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (7,920 г, 85,1%) в виде желтого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-((2-амино-4-(трифторметил)фенил)амино)пиперидин-1-карбоксилата
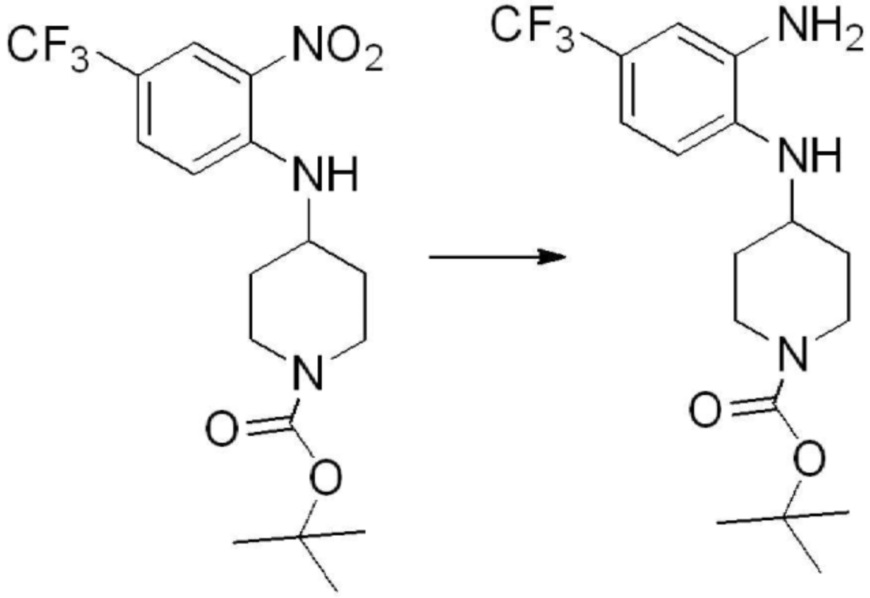
Трет-бутил 4-((2-нитро-4-(трифторметил)фенил)амино)пиперидин-1-карбоксилат (7,920 г, 20,340 ммоль), полученный на стадии 1, и Pd/C (10%, 0,481 г, 2,034 ммоль) растворяют в метаноле (200 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (7,130 г, 97,5%, фиолетовое твердое вещество).
[Стадия 3] Синтез трет-бутил 4-(2-оксо-5-(трифторметил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
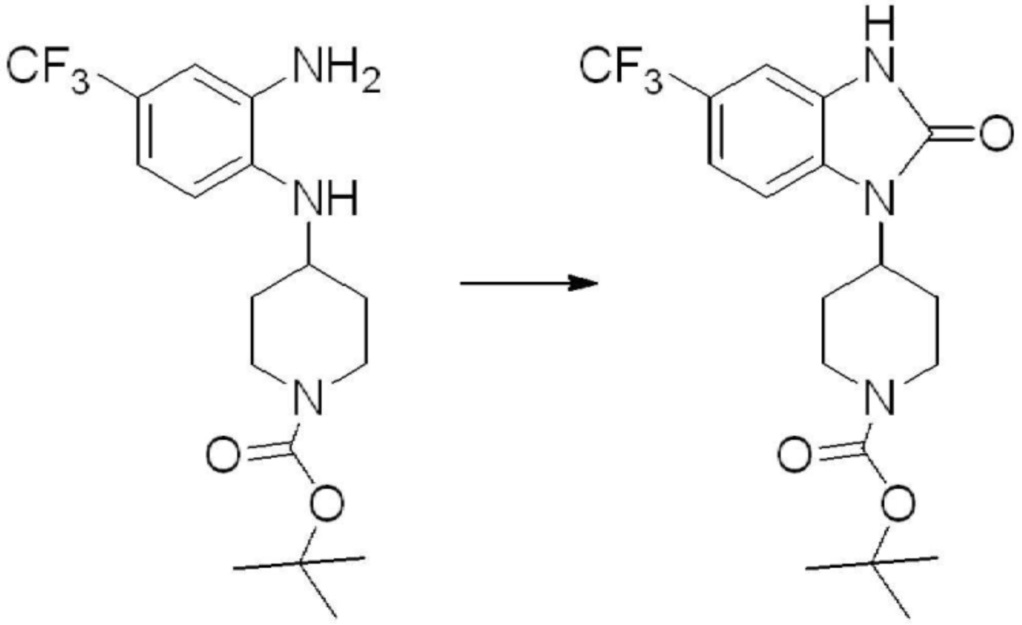
Трет-бутил 4-((2-амино-4-(трифторметил)фенил)амино)пиперидин-1-карбоксилат (7,130 г, 19,839 ммоль), полученный на стадии 2, растворяют в N,N-диметилформамиде (200 мл), после чего 1,1'-карбонилдиимидазол (КДИ, 4,504 г, 27,775 ммоль) добавляют в полученный раствор при 0° и перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (7,610 г, 99,5%) в виде серого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-5-(трифторметил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
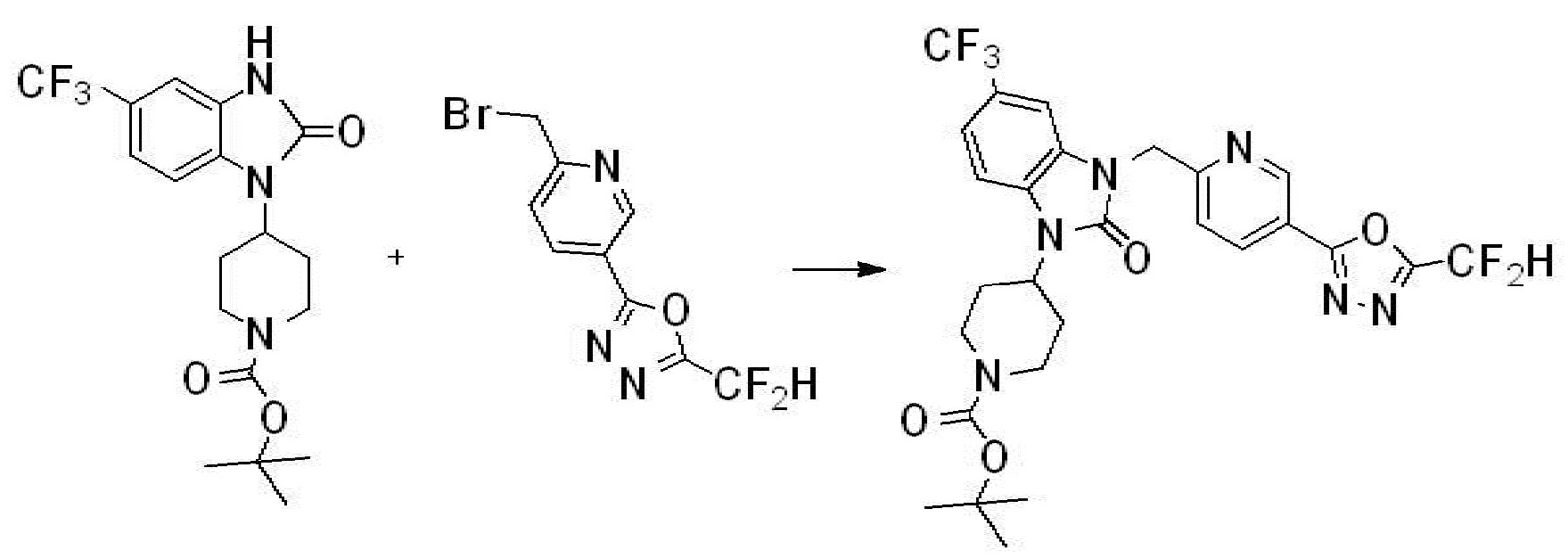
Трет-бутил 4-(2-оксо-5-(трифторметил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,500 г, 6,487 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (65 мл) при 0°C, после чего гидрид натрия (60,00%, 0,389 г, 9,730 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,446 г, 8,433 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (2,516 г, 65,2%) в виде коричневого твердого вещества.
[Стадия 5] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
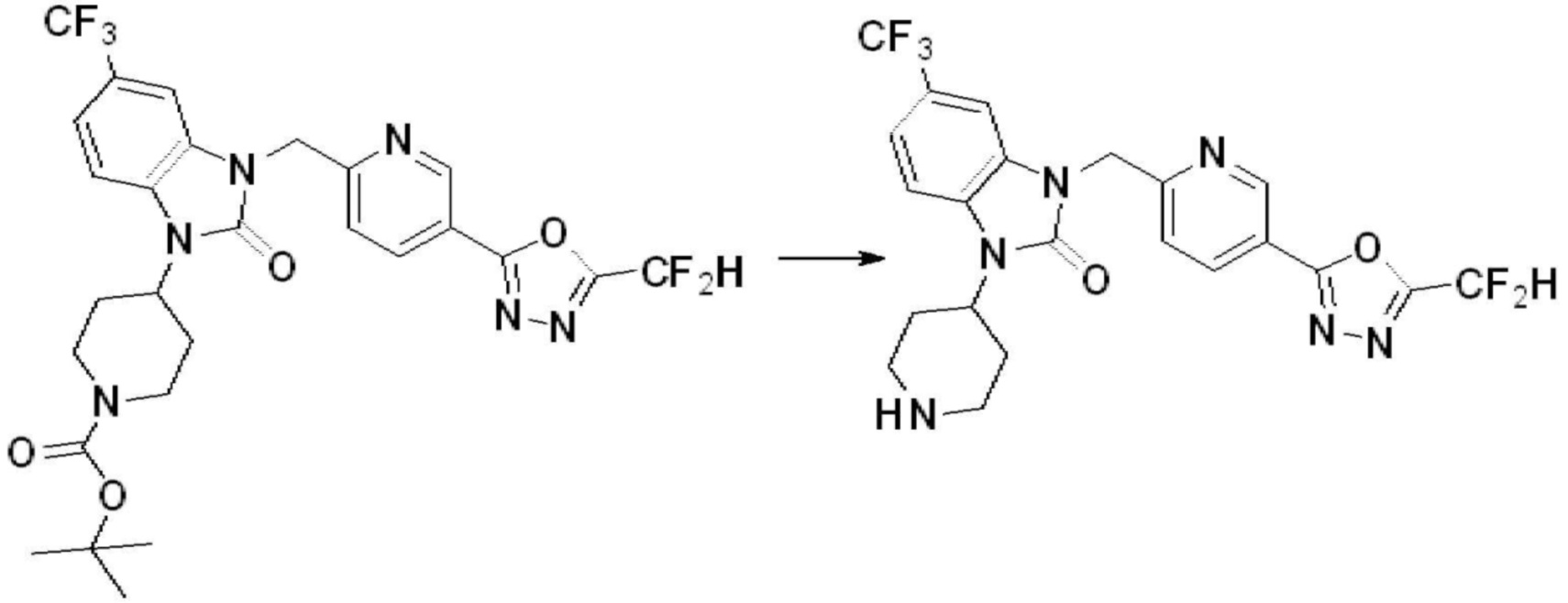
Трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-5-(трифторметил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,510 г, 4,222 ммоль), полученный на стадии 4, и трифторуксусную кислоту (3,233 мл, 42,218 ммоль) растворяют в дихлорметане (40 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=50-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 40 г картридж; дихлорметан/метанол=0-50%) и концентрируют с получением указанного в заголовке соединения (1,573 г, 75,4%) в виде коричневого твердого вещества.
[Стадия 6] Синтез соединения 89
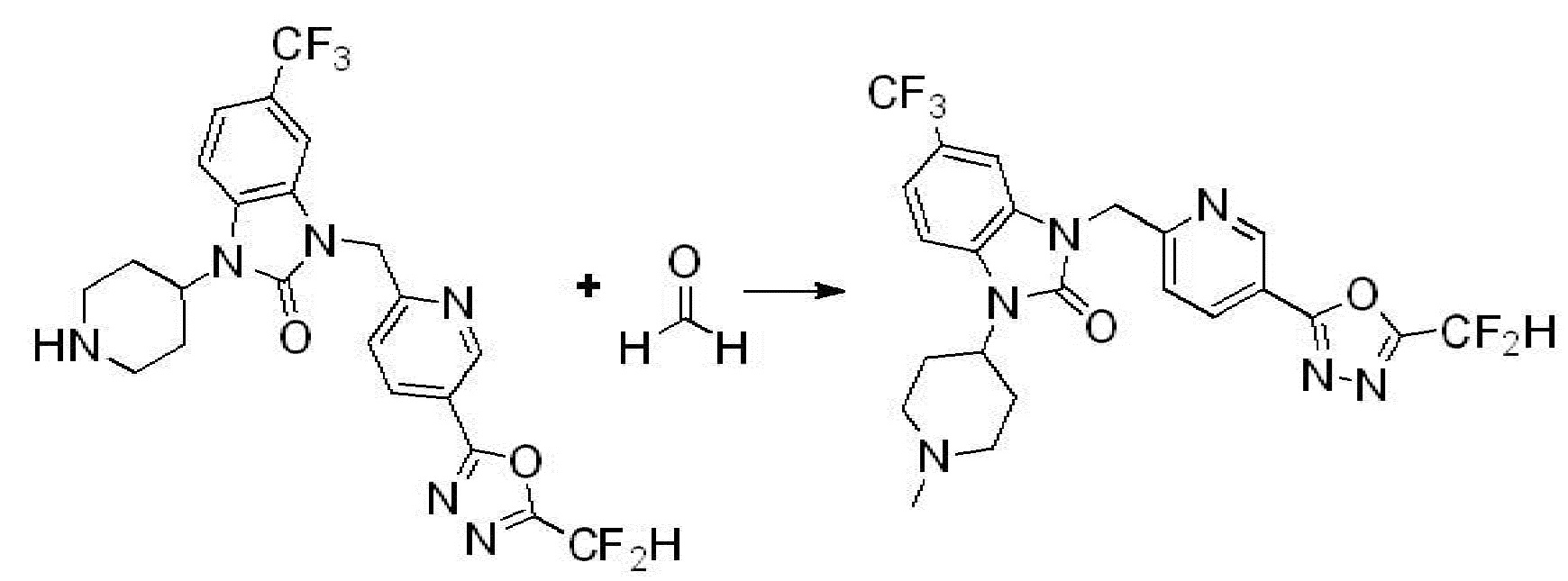
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,202 ммоль), полученный на стадии 5, триацетоксиборгидрид натрия (0,086 г, 0,405 ммоль), уксусную кислоту (0,012 мл, 0,202 ммоль) и формальдегид (0,012 г, 0,405 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,061 г, 59,3%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=2,1 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,44-7,25 (м, 4H), 6,91 (т, J=51,6 Гц, 1H), 5,29 (с, 2H), 4,48-4,39 (м, 1H), 3,03 (д, J=11,7 Гц, 2H), 2,55-2,43 (м, 2H), 2,34 (с, 3H), 2,17 (тд, J=12,0, 2,0 Гц, 2H), 1,86-1,82 (м, 2H); МСНР (ЭР) m/z 509,3 (M+ + 1).
Пример 90: Синтез соединения 90, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-(оксетан-3-ил)пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 90
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,202 ммоль), полученный на стадии 5 соединения 89, триацетоксиборгидрид натрия (0,086 г, 0,405 ммоль), уксусную кислоту (0,012 мл, 0,202 ммоль) и оксетан-3-он (0,029 г, 0,405 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,083 г, 74,9%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26-9,25 (м, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,44-7,34 (м, 3H), 7,26 (с, 1H), 6,92 (т, J=51,6 Гц, 1H), 5,28 (с, 2H), 4,64 (дт, J=22,9, 6,4 Гц, 4H), 4,49-4,40 (м, 1H), 3,56-3,50 (м, 1H), 2,91 (д, J=11,5 Гц, 2H), 2,52-2,42 (м, 2H), 2,05-2,00 (м, 2H), 1,89-1,85 (м, 2H); МСНР (ЭР) m/z 551,6 (M+ + 1).
Пример 91: Синтез соединения 91, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-5-(трифторметил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
Трет-бутил 4-(2-оксо-5-(трифторметил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,500 г, 6,487 ммоль), полученный на стадии 3 соединения 89, растворяют в N,N-диметилформамиде (65 мл) при 0°C, после чего гидрид натрия (60,00%, 0,389 г, 9,730 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (2,590 г, 8,433 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (2,821 г, 71,1%) в виде белого твердого вещества.
[Стадия 2] Синтез 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-она 2,2,2-трифторацетата
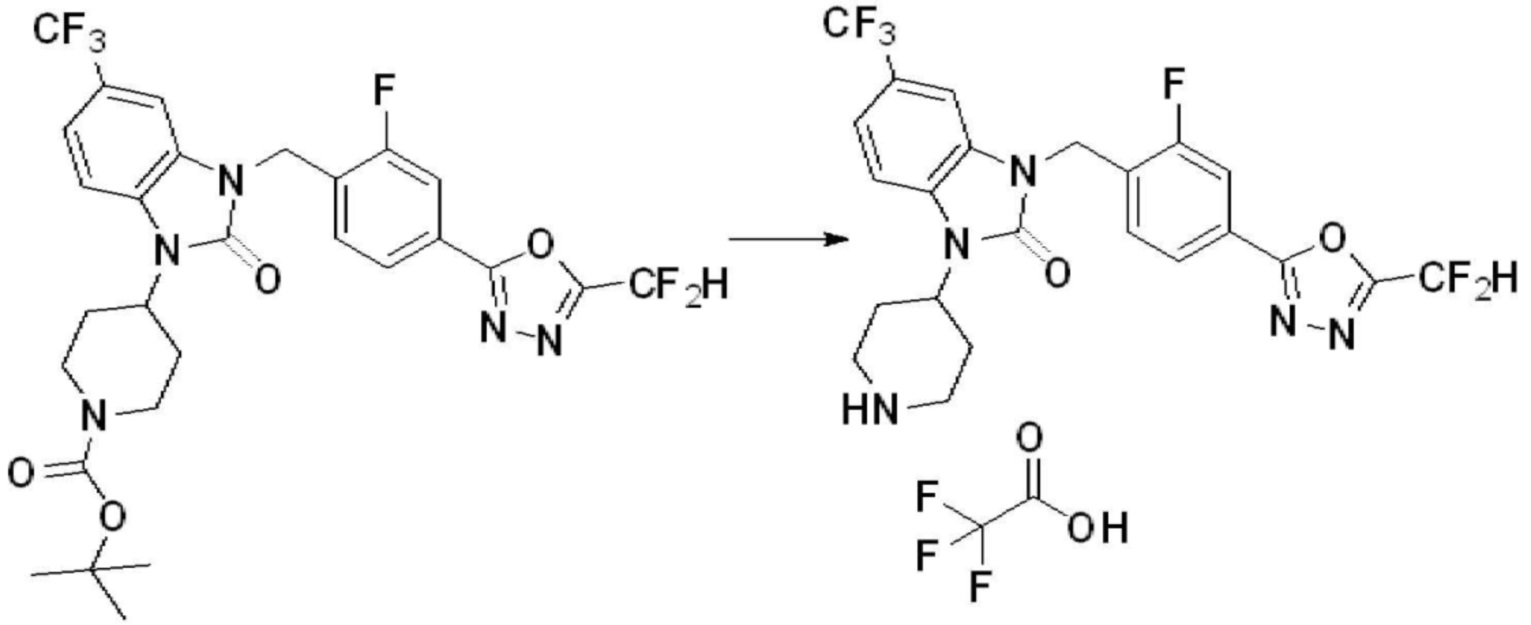
Трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-5-(трифторметил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (3,500 г, 5,723 ммоль), полученный на стадии 1 и трифторуксусную кислоту (4,383 мл, 57,232 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (4,770 г, 133,3%, светло-желтое твердое вещество).
[Стадия 3] Синтез соединения 91
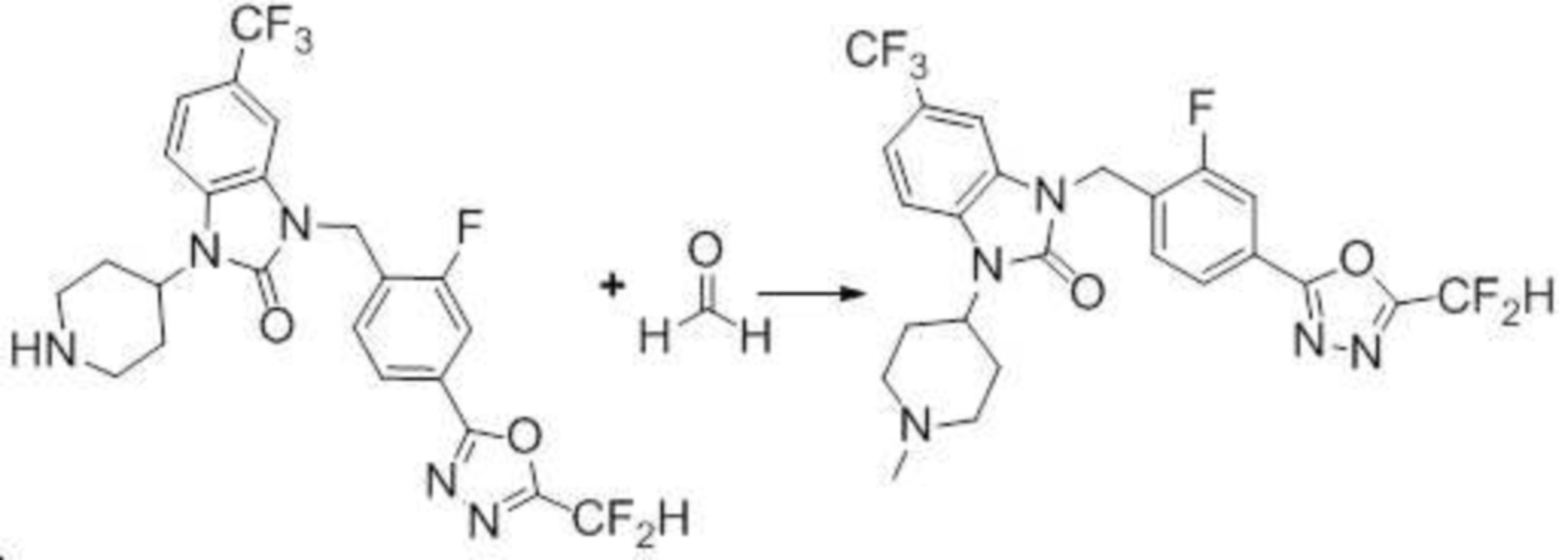
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,196 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,083 г, 0,391 ммоль), уксусную кислоту (0,011 мл, 0,196 ммоль) и формальдегид (0,012 г, 0,391 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,079 г, 77,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,85-7,83 (м, 2H), 7,46-7,32 (м, 3H), 7,19 (с, 1H), 6,89 (т, J=51,7 Гц, 1H), 5,20 (с, 2H), 4,47-4,39 (м, 1H), 3,02 (д, J=11,6 Гц, 2H), 2,51-2,41 (м, 2H), 2,34 (с, 3H), 2,19-2,12 (м, 2H), 1,85-1,81 (м, 2H); МСНР (ЭР) m/z 526,1 (M+ + 1).
Пример 92: Синтез соединения 92, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-(оксетан-3-ил)пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 92
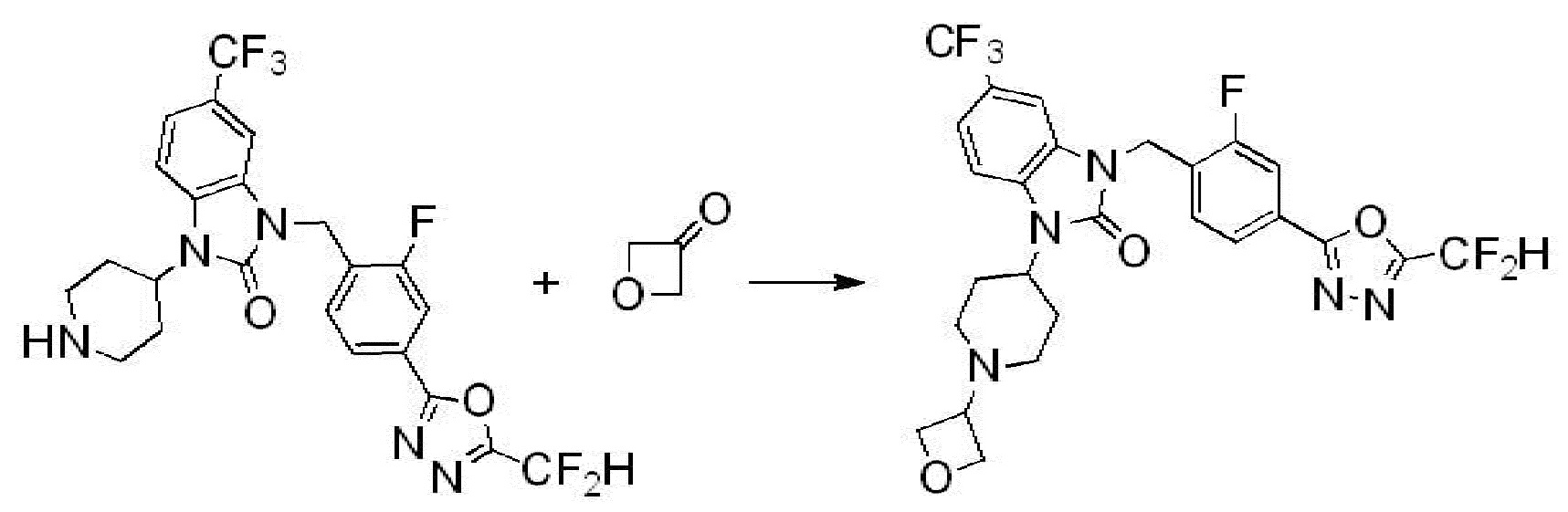
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,196 ммоль), полученный на стадии 2 соединения 91, триацетоксиборгидрид натрия (0,083 г, 0,391 ммоль), уксусную кислоту (0,011 мл, 0,196 ммоль) и оксетан-3-он (0,028 г, 0,391 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,093 г, 83,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,84 (д, J=9,1 Гц, 1H), 7,83 (с, 1H), 7,47-7,34 (м, 3H), 7,21 (с, 1H), 6,90 (т, J=51,7 Гц, 1H), 5,20 (с, 2H), 4,64 (дт, J=23,1, 6,4 Гц, 4H), 4,48-4,40 (м, 1H), 3,56-3,50 (м, 1H), 2,91 (д, J=11,5 Гц, 2H), 2,51-2,41 (м, 2H), 2,05-2,00 (м, 2H), 1,88-1,85 (м, 2H); МСНР (ЭР) m/z 568,2 (M+ + 1).
Пример 93: Синтез соединения 93, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5,6-дифтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5,6-дифтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
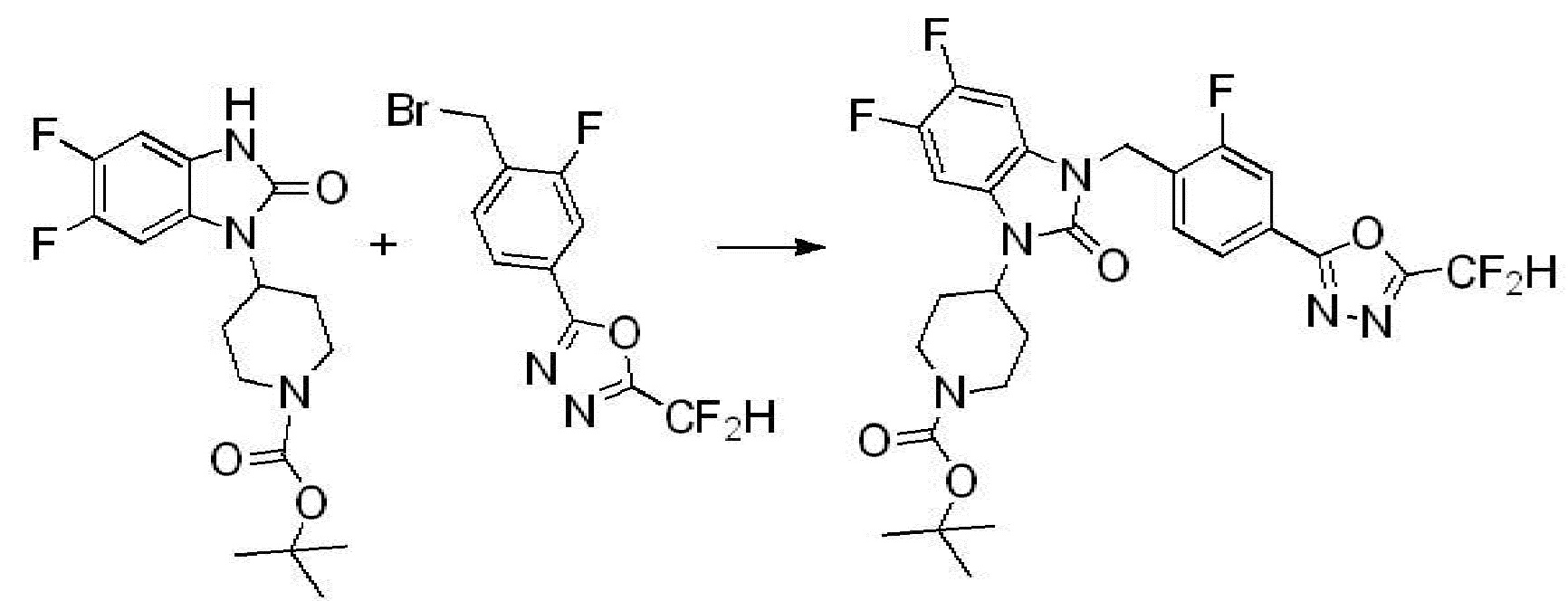
Трет-бутил 4-(5,6-дифтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,600 г, 4,528 ммоль), полученный на стадии 2 соединения 83, растворяют в N,N-диметилформамиде (50 мл) при 0°C, после чего гидрид натрия (60,00%, 0,272 г, 6,792 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (1,807 г, 5,886 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (1,270 г, 48,4%) в виде светло-желтого твердого вещества.
[Стадия 2] Синтез 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
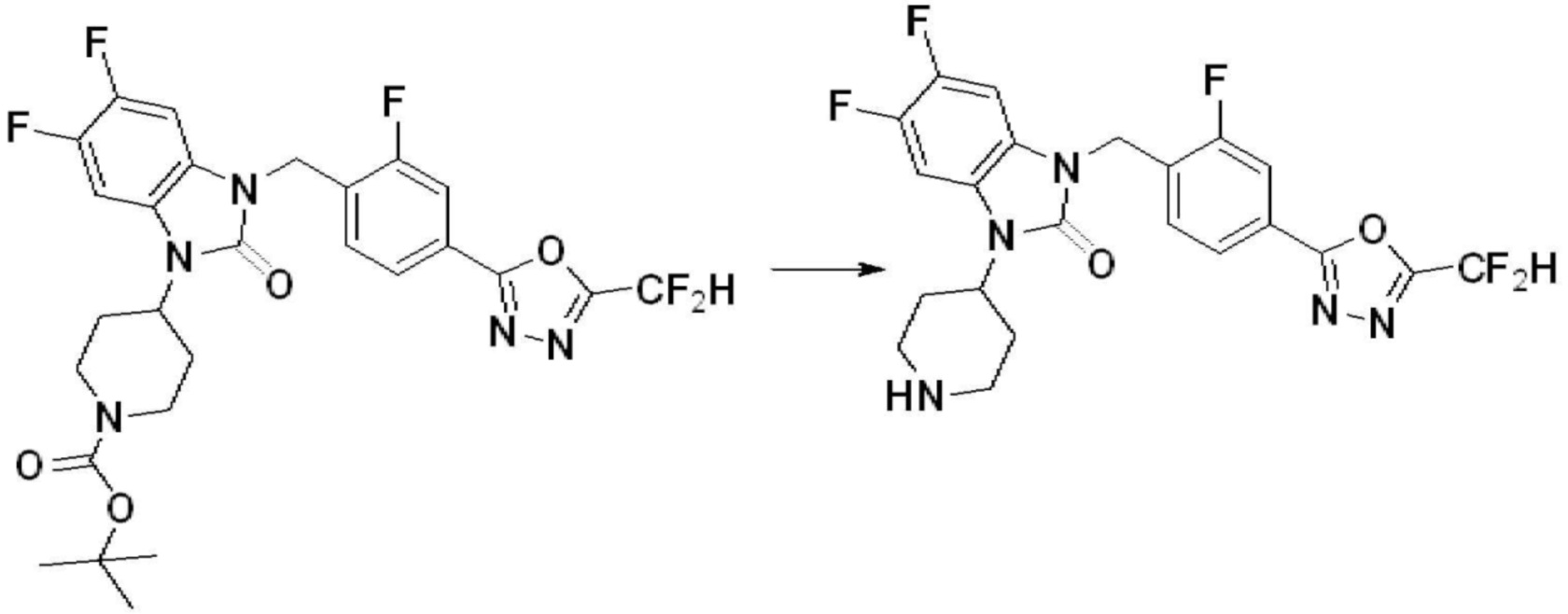
Трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5,6-дифтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,270 г, 2,191 ммоль), полученный на стадии 1, и трифторуксусную кислоту (1,678 мл, 21,914 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; дихлорметан/метанол=0-50%) и концентрируют с получением указанного в заголовке соединения (0,401 г, 38,1%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 93
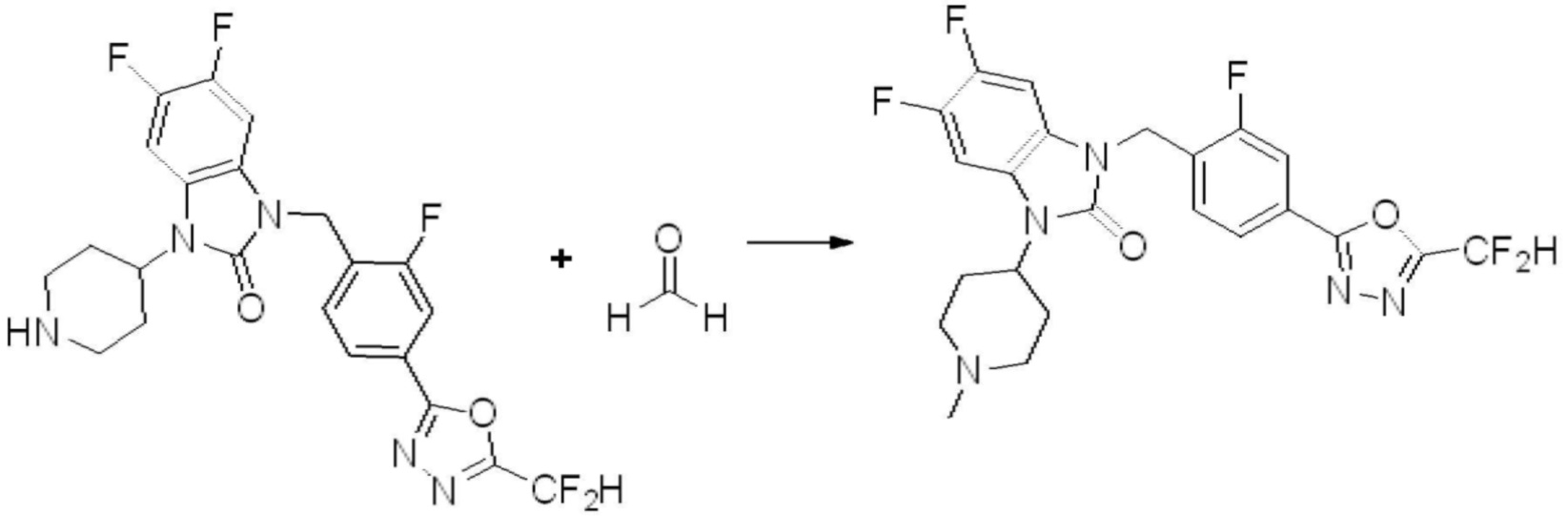
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,209 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,088 г, 0,417 ммоль), уксусную кислоту (0,012 мл, 0,209 ммоль) и формальдегид (0,013 г, 0,417 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,051 г, 49,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,84 (д, J=8,7 Гц, 2H), 7,44 (т, J=7,6 Гц, 1H), 7,17 (дд, J=10,3, 6,7 Гц, 1H), 7,03-6,76 (м, 2H), 5,12 (с, 2H), 4,41-4,32 (м, 1H), 3,01 (д, J=11,7 Гц, 2H), 2,43-2,33 (м, 5H), 2,17-2,11 (м, 2H), 1,83-1,79 (м, 2H); МСНР (ЭР) m/z 494,4 (M+ + 1).
Пример 94: Синтез соединения 94, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5,6-дифтор-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 94
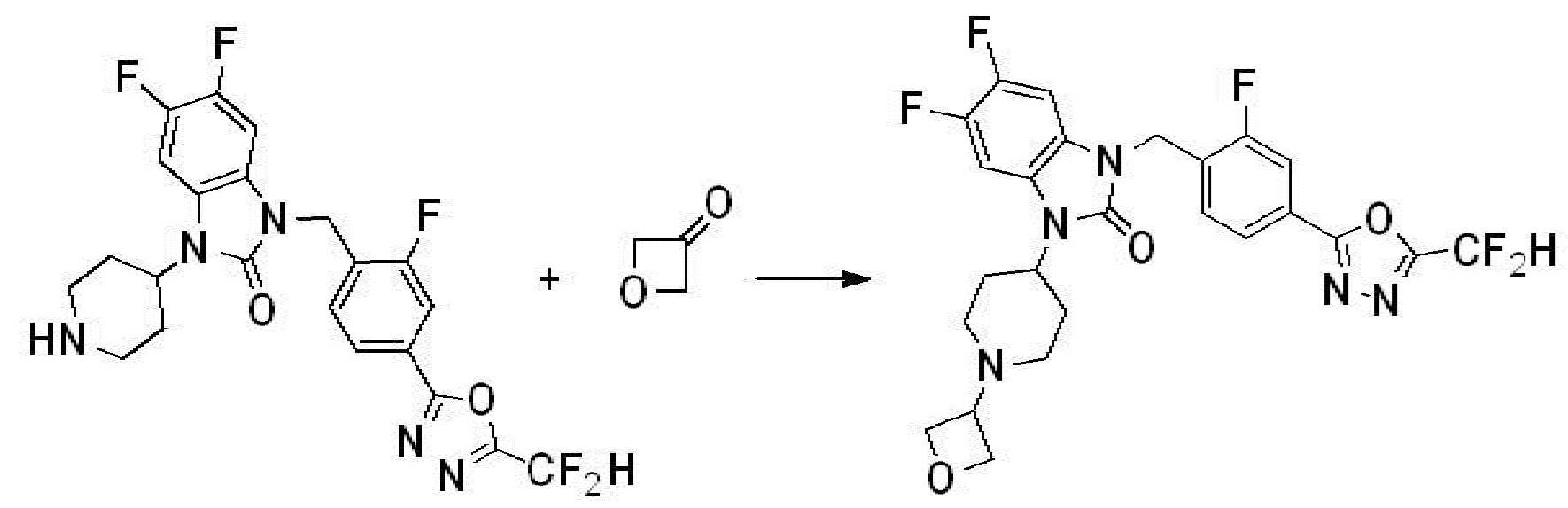
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,209 ммоль), полученный на стадии 2 соединения 93, триацетоксиборгидрид натрия (0,088 г, 0,417 ммоль), уксусную кислоту (0,012 мл, 0,209 ммоль) и оксетан-3-он (0,030 г, 0,417 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,083 г, 74,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,84-7,81 (м, 2H), 7,44 (т, J=7,6 Гц, 1H), 7,16 (дд, J=10,3, 6,7 Гц, 1H), 7,03-6,77 (м, 2H), 5,11 (с, 2H), 4,69 (дт, J=21,2, 36,2 Гц, 4H), 4,41-4,32 (м, 1H), 3,55-3,49 (м, 1H), 2,90 (д, J=11,5 Гц, 2H), 2,43-2,33 (м, 2H), 2,03-1,98 (м, 2H), 1,85-1,82 (м, 2H); МСНР (ЭР) m/z 536,2 (M+ + 1).
Пример 95: Синтез соединения 95, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
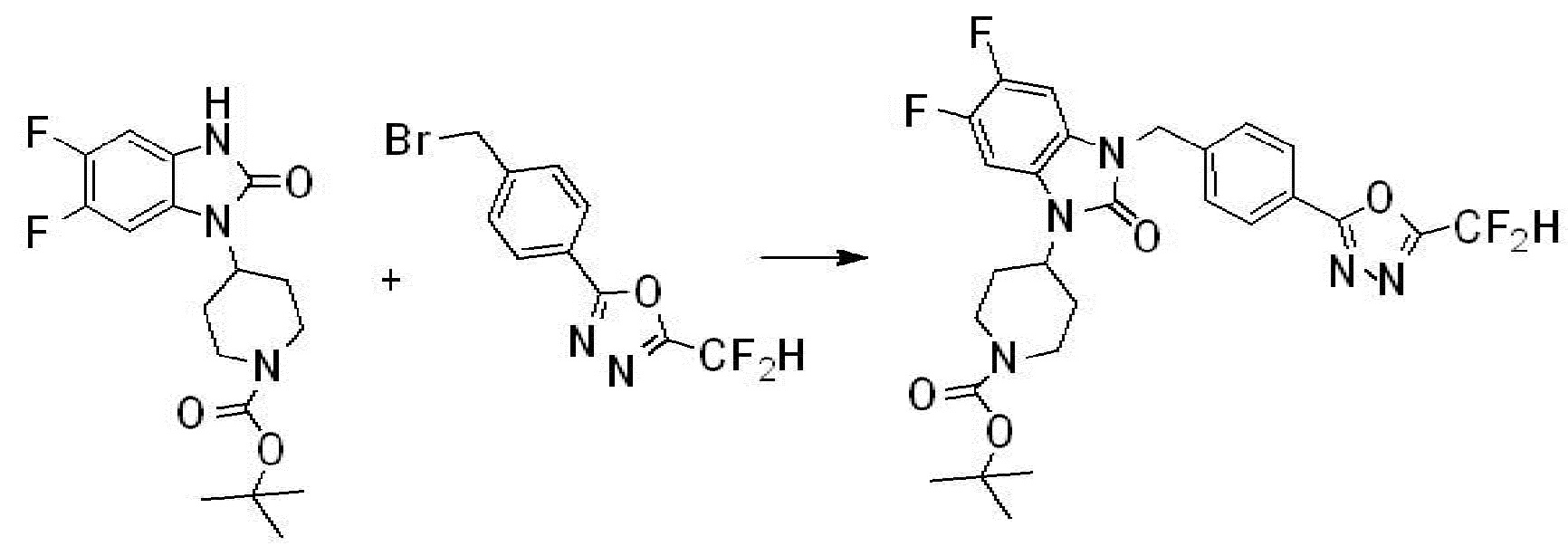
Трет-бутил 4-(5,6-дифтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,600 г, 4,528 ммоль), полученный на стадии 2 соединения 83 растворяют в N,N-диметилформамиде (45 мл) при 0°C, после чего гидрид натрия (60,00%, 0,272 г, 6,792 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (1,702 г, 5,886 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (1,869 г, 73,5%) в виде светло-желтого твердого вещества.
[Стадия 2] Синтез 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
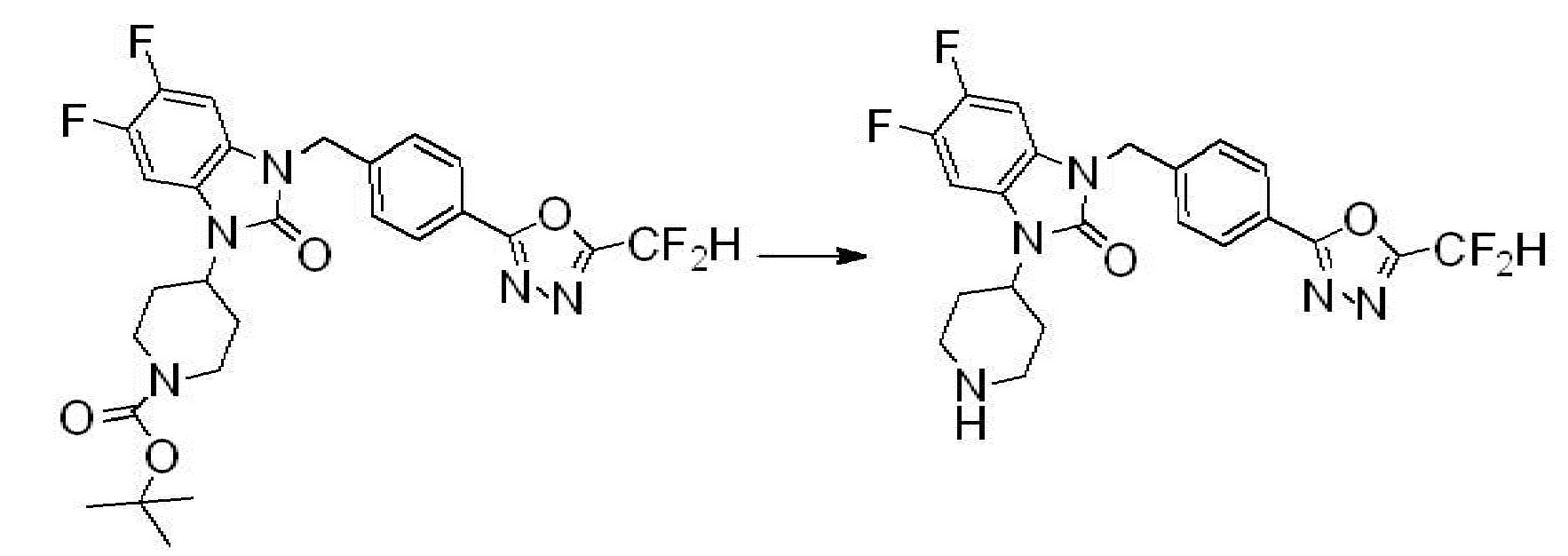
Трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,869 г, 3,328 ммоль), полученный на стадии 1, и трифторуксусную кислоту (2,549 мл, 33,283 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=50-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 40 г картридж; дихлорметан/1% водный раствор метанола=0-50%) и концентрируют с получением указанного в заголовке соединения (1,152 г, 75,0%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 95
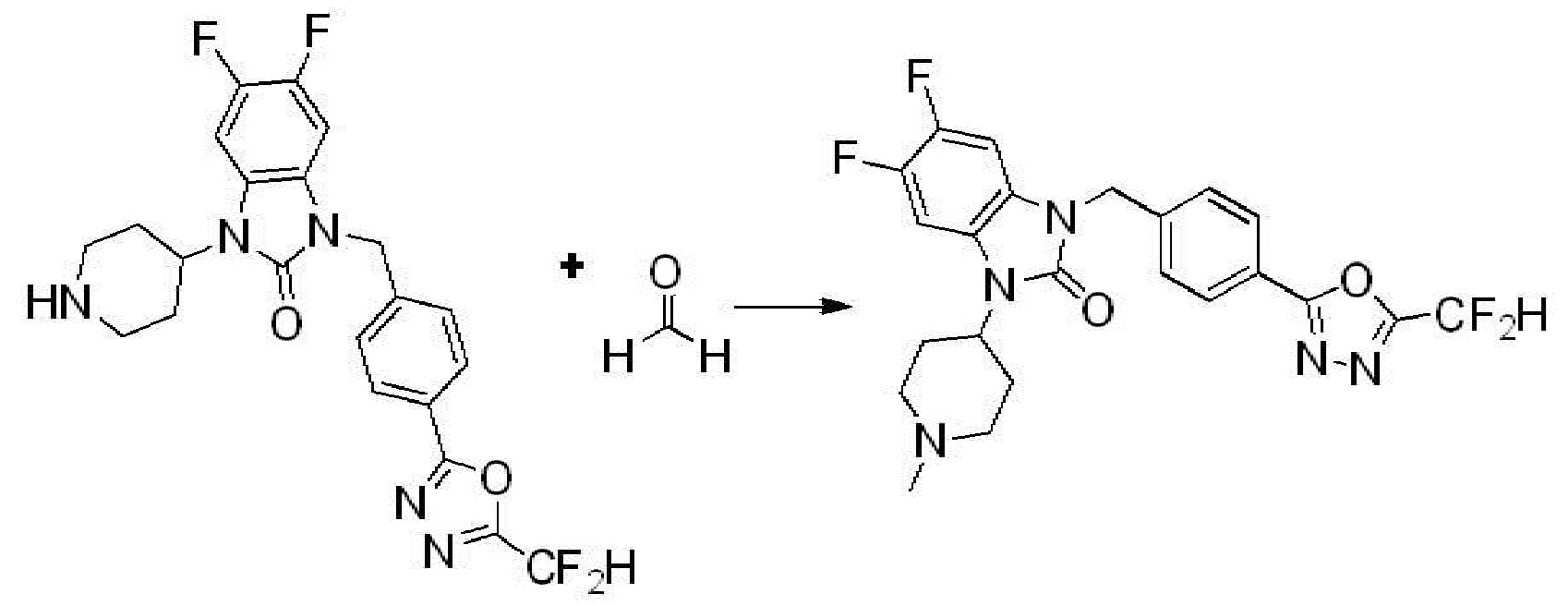
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,092 г, 0,433 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и формальдегид (0,013 г, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,061 г, 59,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,05 (д, CDCJ=8,4 Гц, 2H), 7,42 (д, J=8,4 Гц, 2H), 7,18 (дд, J=10,5, 6,6 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 6,63 (дд, J=9,6, 6,8 Гц, 1H), 5,07 (с, 2H), 4,42-4,34 (м, 1H), 3,02 (д, J=11,7 Гц, 2H), 2,45-2,33 (м, 5H), 2,18-2,13 (м, 2H), 1,84-1,80 (м, 2H); МСНР (ЭР) m/z 476,3 (M+ + 1).
Пример 96: Синтез соединения 96, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 96
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 95, триацетоксиборгидрид натрия (0,092 г, 0,433 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и оксетан-3-он (0,031 г, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,099 г, 88,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,09 (дд, J=7,5, 2,7 Гц, 2H), 7,45 (д, J=8,4 Гц, 2H), 7,18 (дд, J=10,3, 6,7 Гц, 1H), 6,90 (т, J=51,7 Гц, 1H), 6,65 (дд, J=9,6, 6,8 Гц, 1H), 5,10 (с, 2H), 4,67 (дт, J=20,8, 6,4 Гц, 4H), 4,45-4,37 (м, 1H), 3,58-3,53 (м, 1H), 2,92 (д, J=11,4 Гц, 2H), 2,45-2,35 (м, 2H), 2,04 (тд, J=11,8, 1,9 Гц, 2H), 1,89-1,86 (м, 2H); МСНР (ЭР) m/z 518,5 (M+ + 1).
Пример 97: Синтез соединения 97, 5-хлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((5-хлор-2-нитрофенил)амино)пиперидин-1-карбоксилата
4-хлор-2-фтор-1-нитробензол (5,000 г, 28,484 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (5,990 г, 29,908 ммоль), карбонат калия (7,873 г, 56,967 ммоль) и йодид калия(0,473 г, 2,848 ммоль) растворяют в N,N-диметилформамиде (300 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Реакционную смесь фильтруют через бумажный фильтр для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (9,450 г, 93,2%) в виде оранжевой жидкости.
[Стадия 2] Синтез трет-бутил 4-((2-амино-5-хлорфенил)амино)пиперидин-1-карбоксилата
Трет-бутил 4-((5-хлор-2-нитрофенил)амино)пиперидин-1-карбоксилат (9,450 г, 26,558 ммоль), полученный на стадии 1 и Ni Ренея 10% масс. растворяют в метаноле (200 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении. Затем, воду выливают в полученный концентрат и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (8,270 г, 95,6%, коричневое твердое вещество).
[Стадия 3] Синтез трет-бутил 4-(6-хлор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
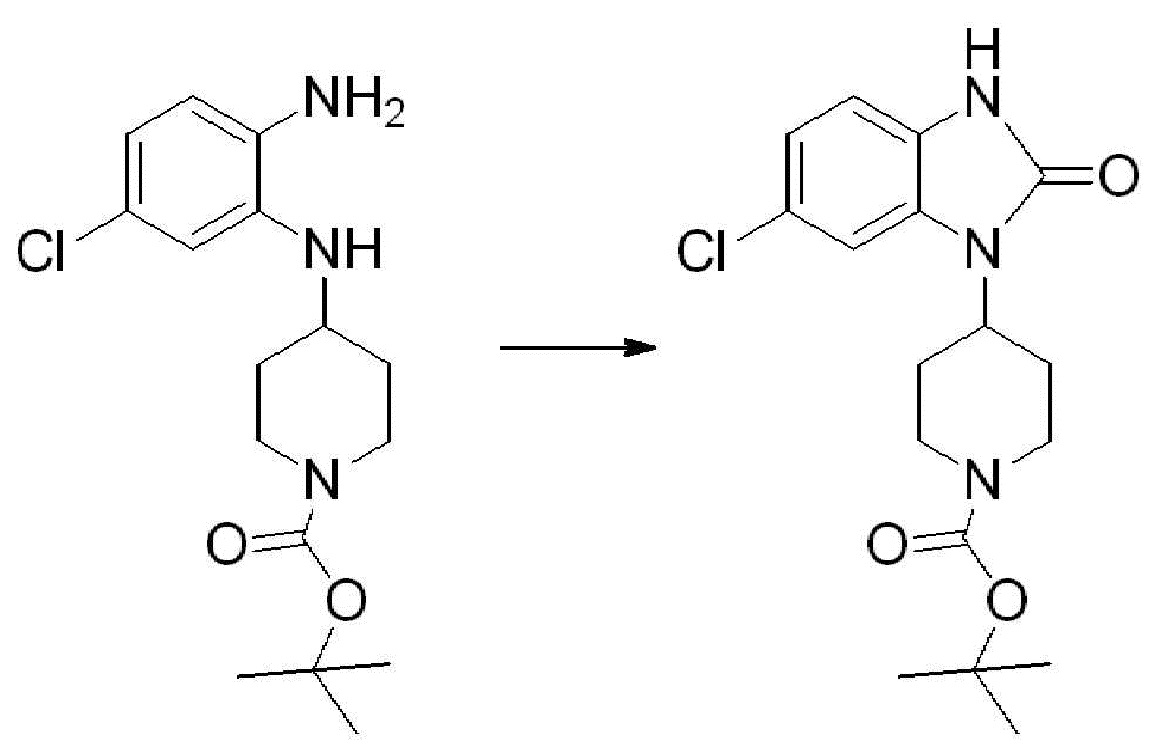
Трет-бутил 4-((2-амино-5-хлорфенил)амино)пиперидин-1-карбоксилат (8,270 г, 25,381 ммоль), полученный на стадии 2 и 1,1'-карбонилдиимидазол (КДИ, 5,762 г, 35,533 ммоль) растворяют в N,N-диметилформамиде (250 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (6,720 г, 75,3%) в виде коричневого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(6-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
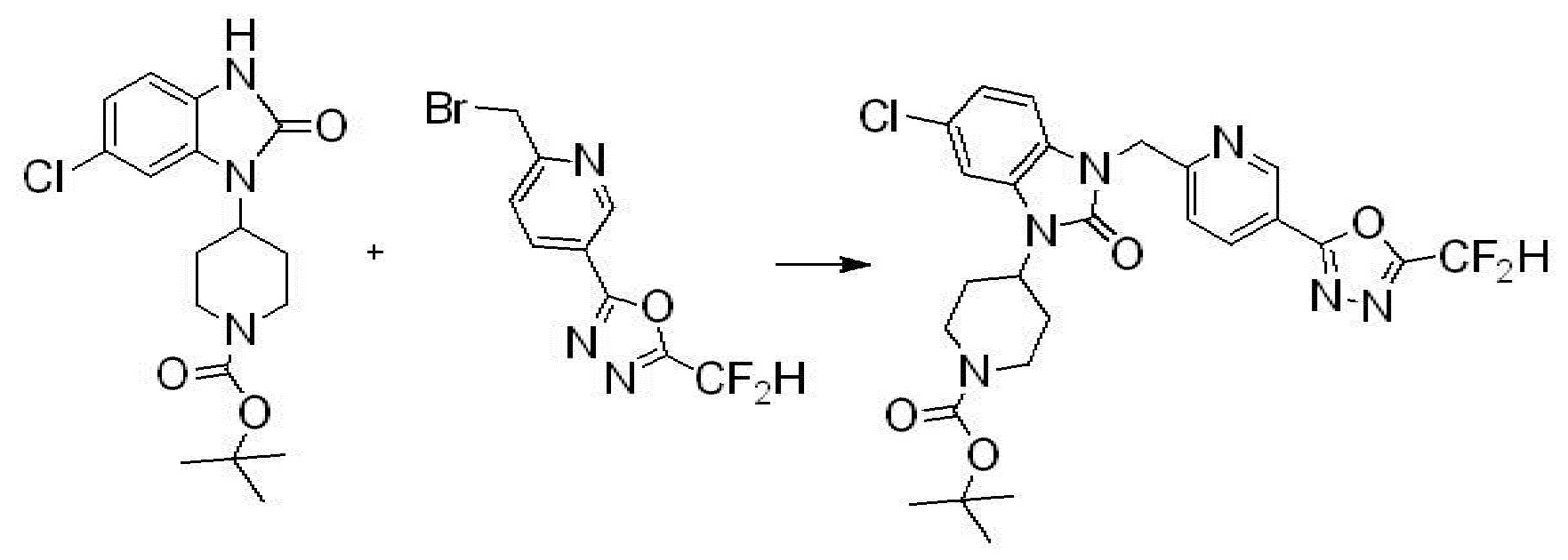
Трет-бутил 4-(6-хлор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,200 г, 6,253 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (50 мл) при 0°C, после чего гидрид натрия (60,00%, 0,375 г, 9,380 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,358 г, 8,129 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2,5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (2,124 г, 60,5%) в виде коричневого твердого вещества.
[Стадия 5] Синтез 5-хлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
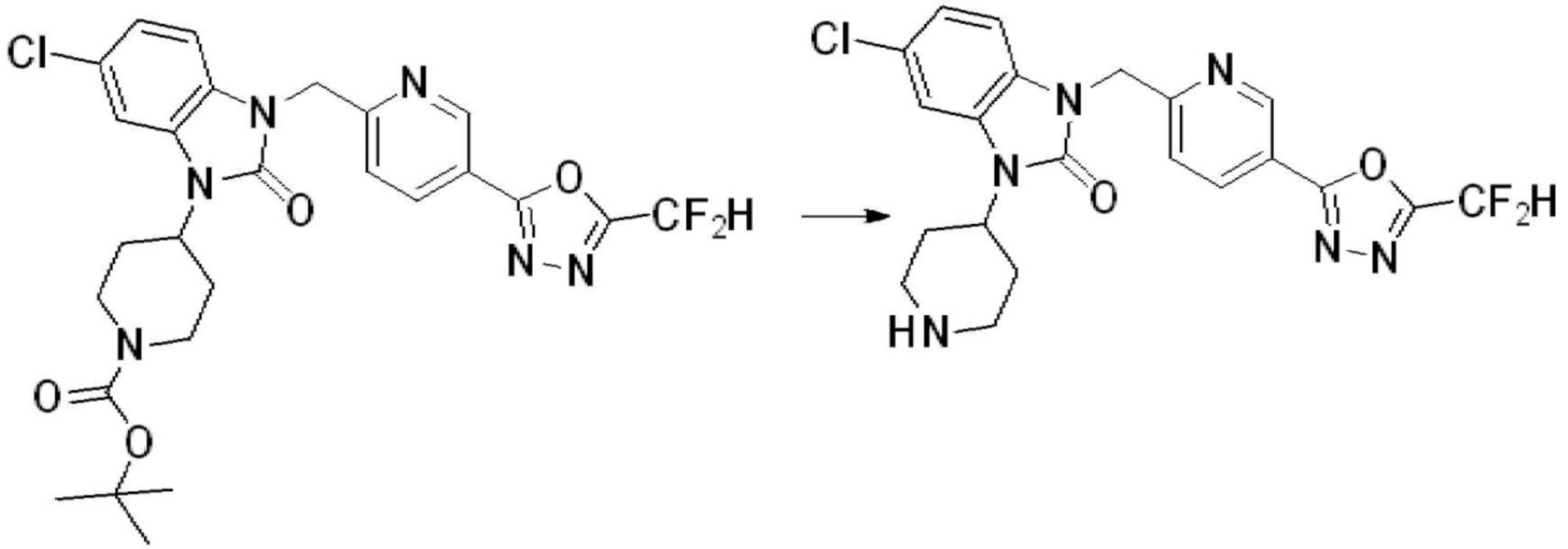
Трет-бутил 4-(6-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,124 г, 3,786 ммоль), полученный на стадии 4, и трифторуксусную кислоту (2,899 мл, 37,862 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=80-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-70%) и концентрируют с получением указанного в заголовке соединения (1,210 г, 69,3%) в виде коричневого твердого вещества.
[Стадия 6] Синтез соединения 97
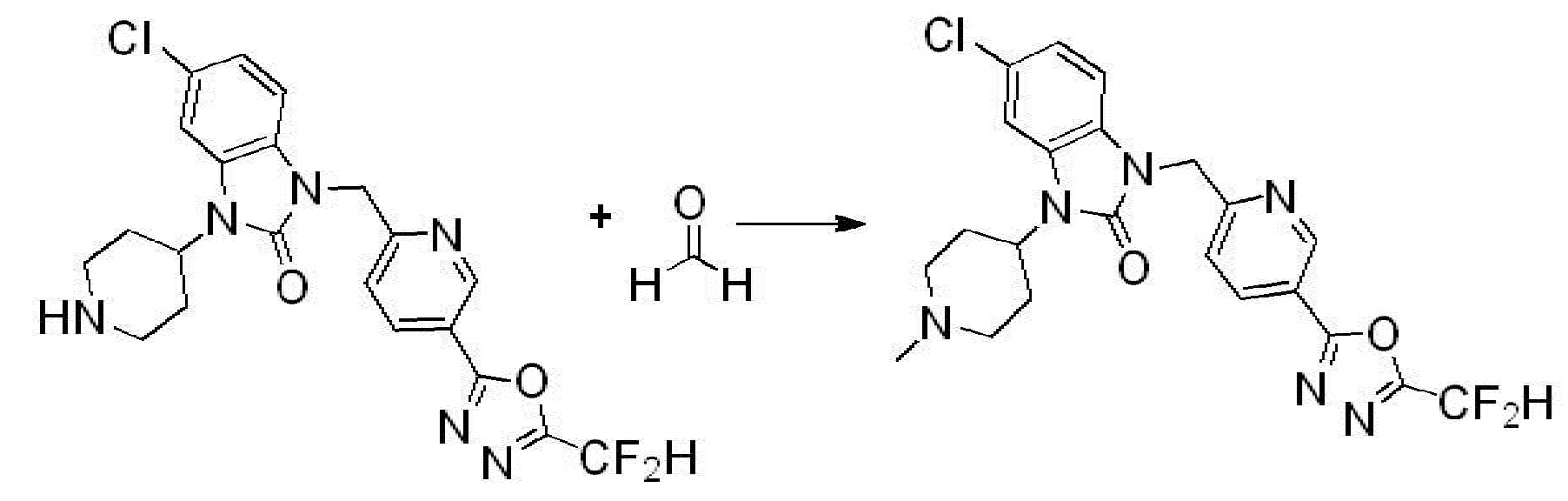
5-хлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5, триацетоксиборгидрид натрия (0,092 г, 0,434 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и формальдегид (0,020 г, 0,651 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,065 г, 62,8%) в виде красного твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (дд, J=2,3, 0,4 Гц, 1H), 8,29 (дд, J=8,2, 2,2 Гц, 1H), 7,37 (д, J=7,9 Гц, 1H), 7,32 (д, J=1,8 Гц, 1H), 7,04-6,78 (м, 3H), 5,23 (с, 2H), 4,42-4,34 (м, 1H), 3,01 (д, J=11,6 Гц, 2H), 2,48-2,38 (м, 2H), 2,33 (с, 3H), 2,14 (тд, J=12,0, 2,1 Гц, 2H), 1,83-1,80 (м, 2H); МСНР (ЭР) m/z 475,5 (M+ + 1).
Пример 98: Синтез соединения 98, 5-хлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 98
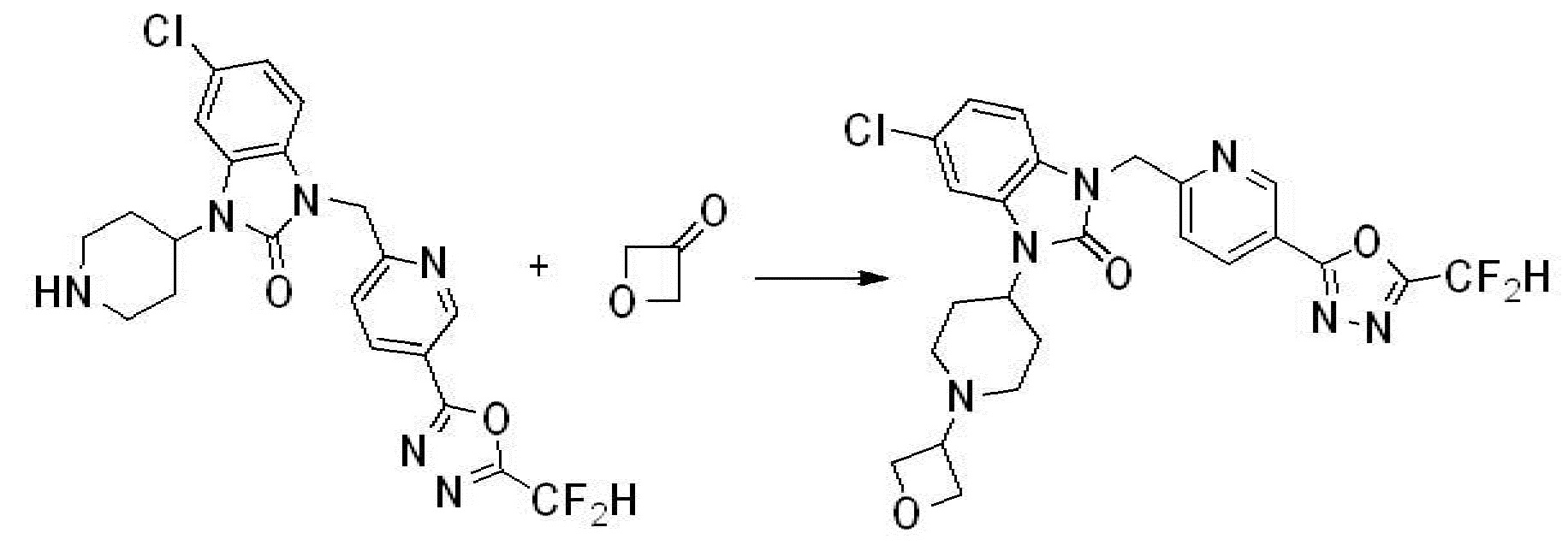
5-хлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 97, триацетоксиборгидрид натрия (0,092 г, 0,434 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и оксетан-3-он (0,047 г, 0,651 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,072 г, 64,1%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,23 (дд, J=2,2, 0,7 Гц, 1H), 8,29 (дд, J=8,2, 2,2 Гц, 1H), 7,38 (д, J=8,2 Гц, 1H), 7,30 (д, J=1,8 Гц, 1H), 7,04-6,78 (м, 3H), 5,22 (с, 2H), 4,64 (дт, J=16,8, 6,4 Гц, 4H), 4,43-4,34 (м, 1H), 3,56-3,49 (м, 1H), 2,91 (д, J=11,5 Гц, 2H), 2,48-2,37 (м, 2H), 2,04-1,98 (м, 2H), 1,86-1,83 (м, 2H); МСНР (ЭР) m/z 517,4 (M+ + 1).
Пример 99: Синтез соединения 99, 5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(6-хлор-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
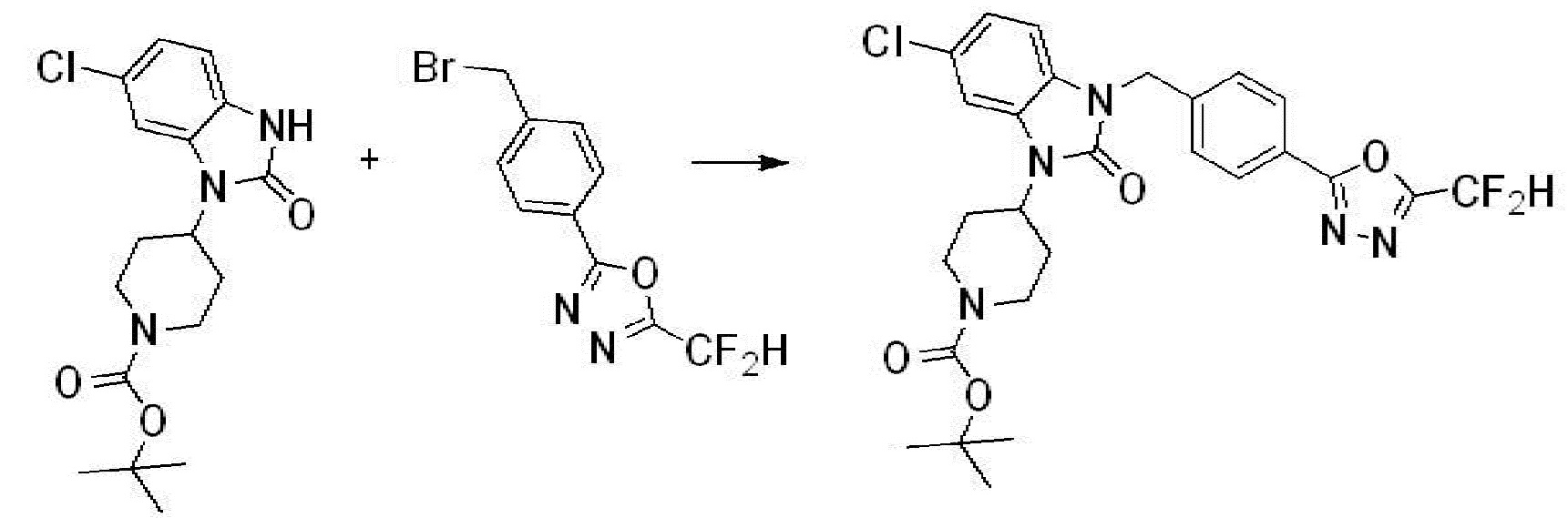
Трет-бутил 4-(6-хлор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,200 г, 6,253 ммоль), полученный на стадии 3 соединения 97 растворяют в N,N-диметилформамиде (60 мл) при 0°C, после чего гидрид натрия (60,00%, 0,375 г, 9,380 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (2,350 г, 8,129 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (2,251 г, 64,3%) в виде коричневого твердого вещества.
[Стадия 2] Синтез 5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
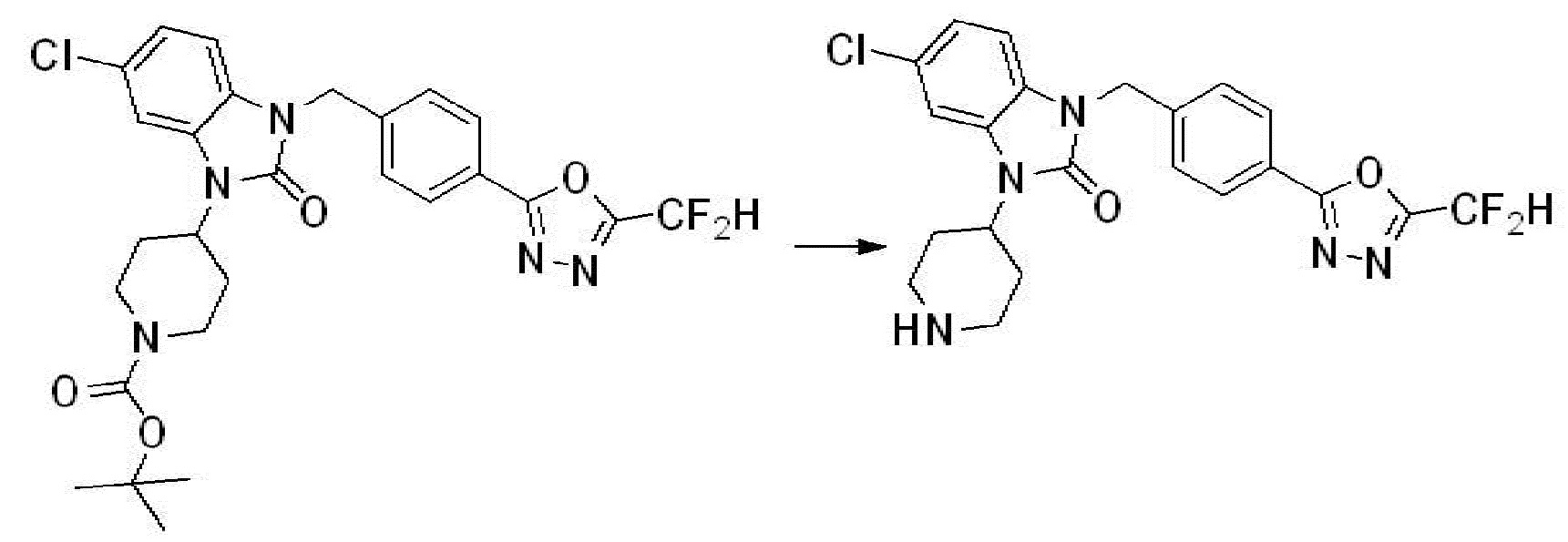
Трет-бутил 4-(6-хлор-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,251 г, 4,020 ммоль), полученный на стадии 1, и трифторуксусную кислоту (3,078 мл, 40,196 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=80-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-60%) и концентрируют с получением указанного в заголовке соединения (1,746 г, 94,5%) в виде желтого твердого вещества.
[Стадия 3] Синтез соединения 99
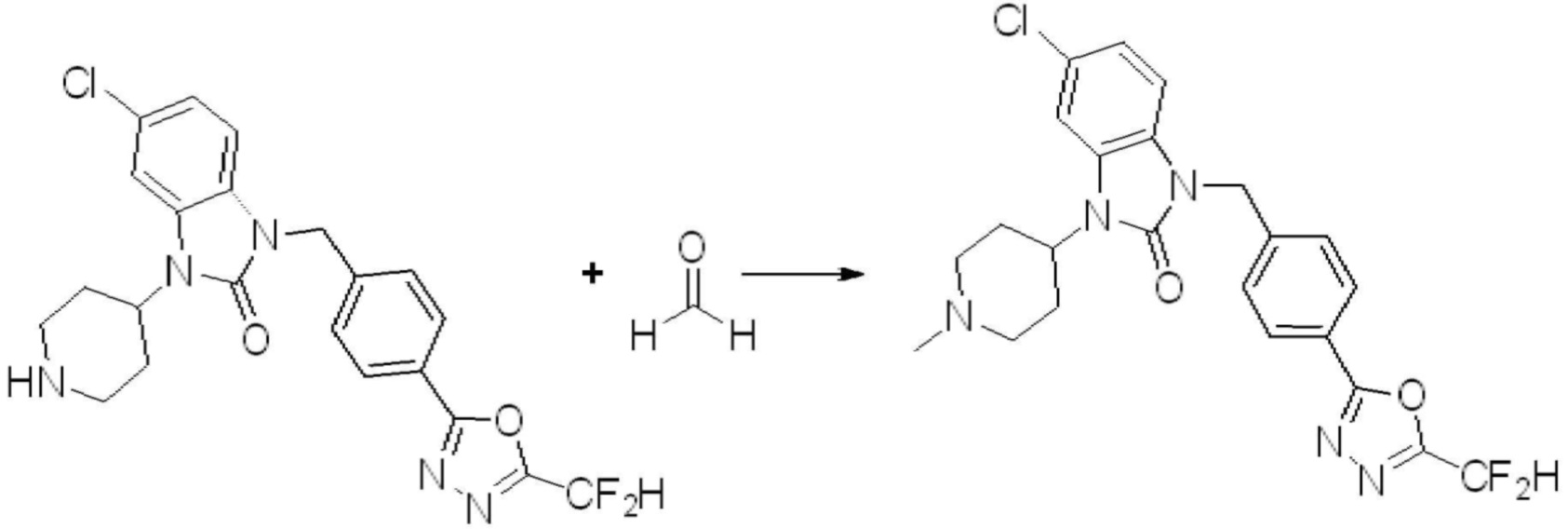
5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и формальдегид (0,020 г, 0,652 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,051 г, 49,9%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,04 (д, J=8,4 Гц, 2H), 7,42 (д, J=8,5 Гц, 2H), 7,33 (д, J=1,8 Гц, 1H), 6,94 (дд, J=8,3, 1,9 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 6,71 (д, J=8,4 Гц, 1H), 5,11 (с, 2H), 4,44-4,36 (м, 1H), 3,03-3,01 (м, 2H), 2,49-2,38 (м, 2H), 2,34 (с, 3H), 2,15 (тд, J=12,0, 2,1 Гц, 2H), 1,89-1,79 (м, 2H); МСНР (ЭР) m/z 474,4 (M+ + 1).
Пример 100: Синтез соединения 100, 5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 100
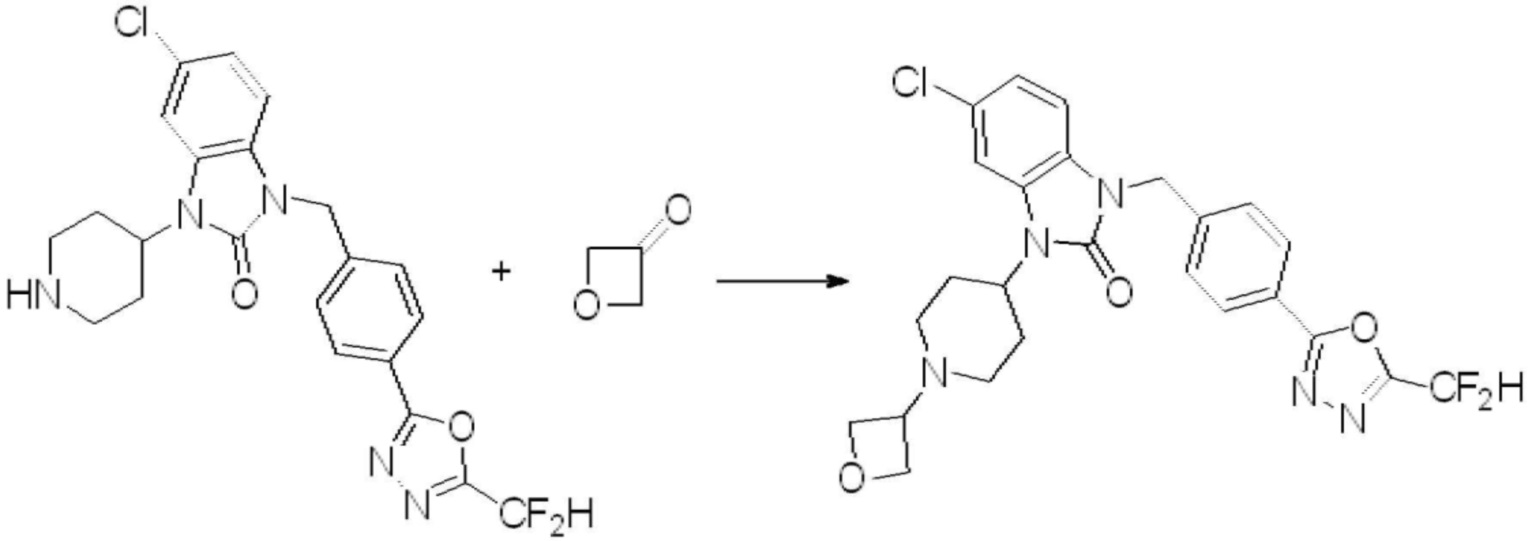
5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 99, триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль), уксусную кислоту (0,012 мл, 0,217 ммоль) и оксетан-3-он (0,047 г, 0,652 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,052 г, 46,6%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,05 (дд, J=6,6, 1,7 Гц, 2H), 7,43 (д, J=8,2 Гц, 2H), 7,32 (д, J=1,8 Гц, 1H), 6,97 (дд, J=8,4, 1,9 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 6,73 (д, J=8,4 Гц, 1H), 5,11 (с, 2H), 4,66 (дт, J=16,8, 6,4 Гц, 4H), 4,45-4,36 (м, 1H), 3,57-3,51 (м, 1H), 2,92 (д, J=11,5 Гц, 2H), 2,49-2,39 (м, 2H), 2,06-2,00 (м, 2H), 1,88-1,85 (м, 2H); МСНР (ЭР) m/z 516,5 (M+ + 1).
Пример 101: Синтез соединения 101, 5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(6-хлор-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
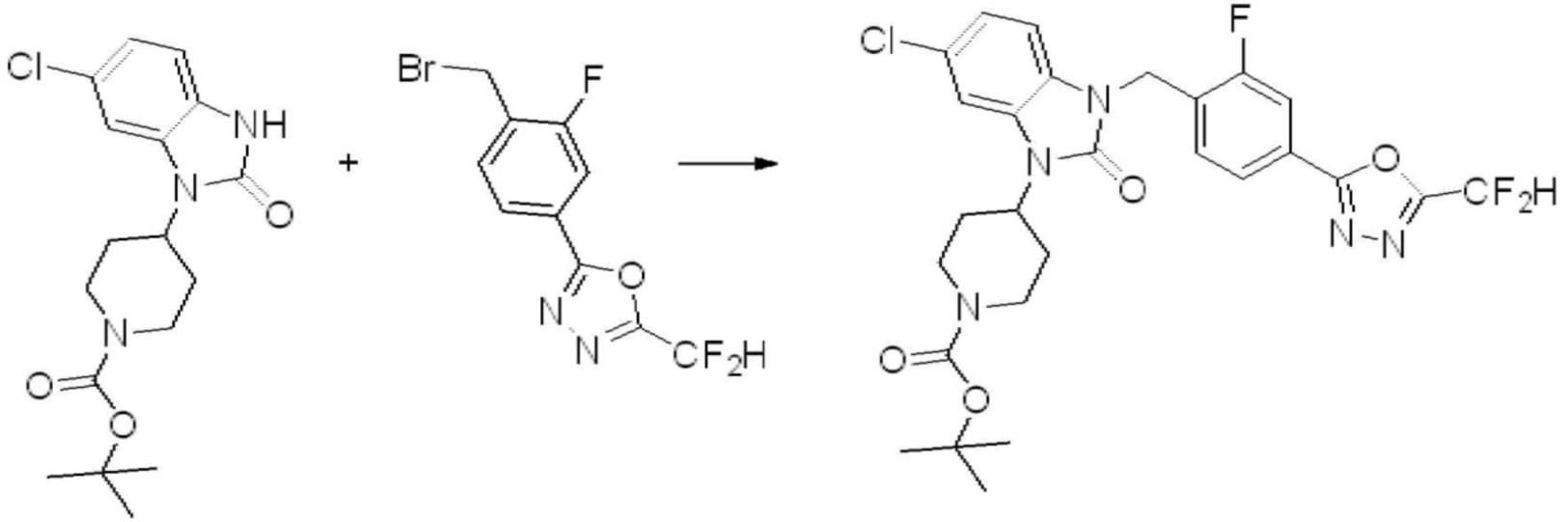
Трет-бутил 4-(6-хлор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,200 г, 6,253 ммоль), полученный на стадии 3 соединения 97, растворяют в N,N-диметилформамиде (60 мл) при 0°C, после чего гидрид натрия (60,00%, 0,375 г, 9,380 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (2,496 г, 8,129 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (2,693 г, 74,5%) в виде коричневого твердого вещества.
[Стадия 2] Синтез 5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
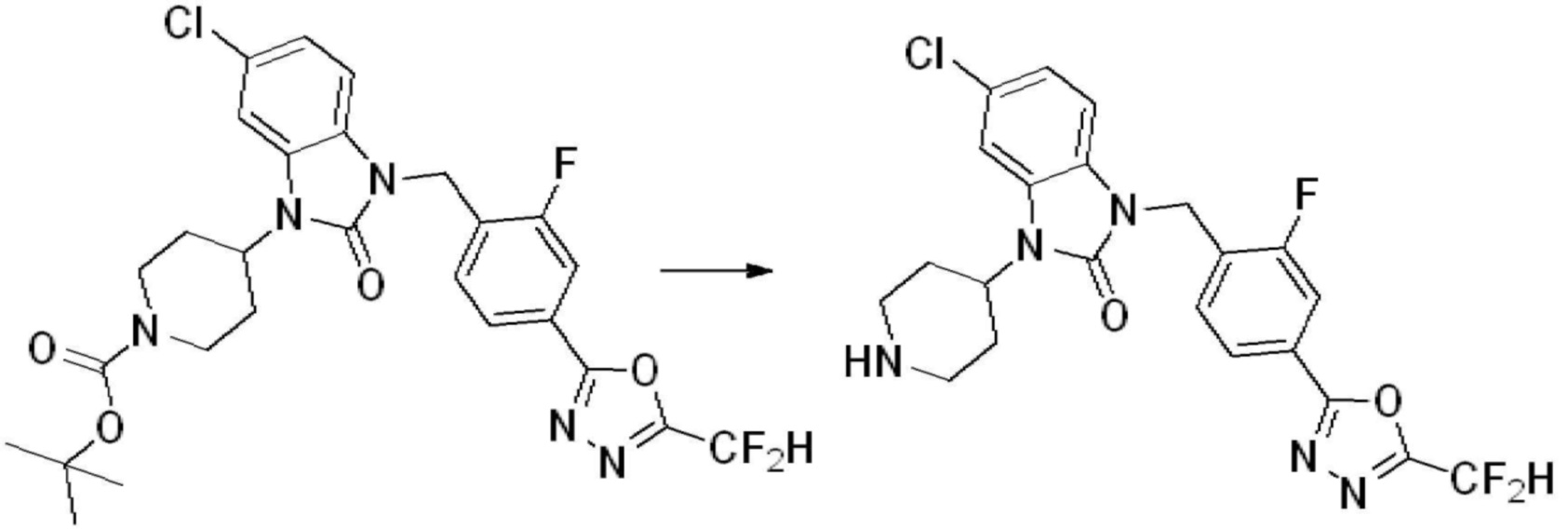
Трет-бутил 4-(6-хлор-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,693 г, 4,659 ммоль), полученный на стадии 1, и трифторуксусную кислоту (3,568 мл, 46,593 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=80-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-60%) и концентрируют с получением указанного в заголовке соединения (2,155 г, 96,8%) в виде желтого твердого вещества.
[Стадия 3] Синтез соединения 101
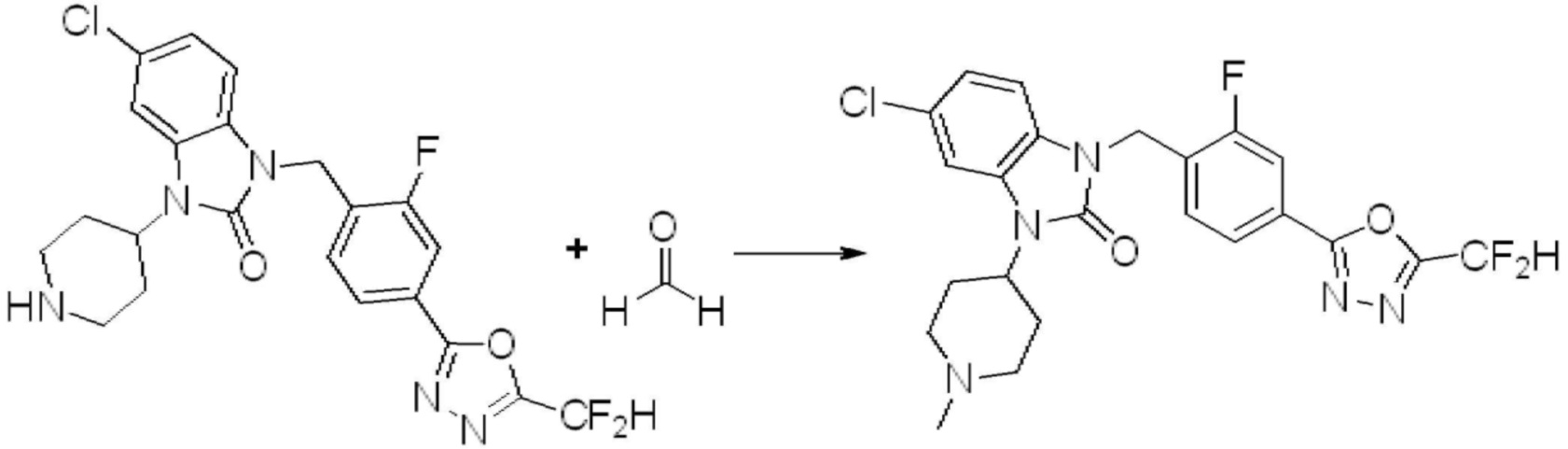
5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,209 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль), уксусную кислоту (0,012 мл, 0,209 ммоль) и формальдегид (0,019 г, 0,628 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,058 г, 56,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,84-7,80 (м, 2H), 7,41 (т, J=7,8 Гц, 1H), 7,33 (д, J=1,8 Гц, 1H), 6,99 (дд, J=8,4, 1,8 Гц, 1H), 6,89 (т, J=51,6 Гц, 1H), 6,83 (д, J=8,4 Гц, 1H), 5,15 (с, 2H), 4,43-4,35 (м, 1H), 3,02 (д, J=11,6 Гц, 2H), 2,48-2,38 (м, 2H), 2,34 (с, 3H), 2,15 (тд, J=11,9, 1,9 Гц, 2H), 1,82 (дд, J=12,0, 2,2 Гц, 2H); МСНР (ЭР) m/z 492,3 (M+ + 1).
Пример 102: Синтез соединения 102, 5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 102
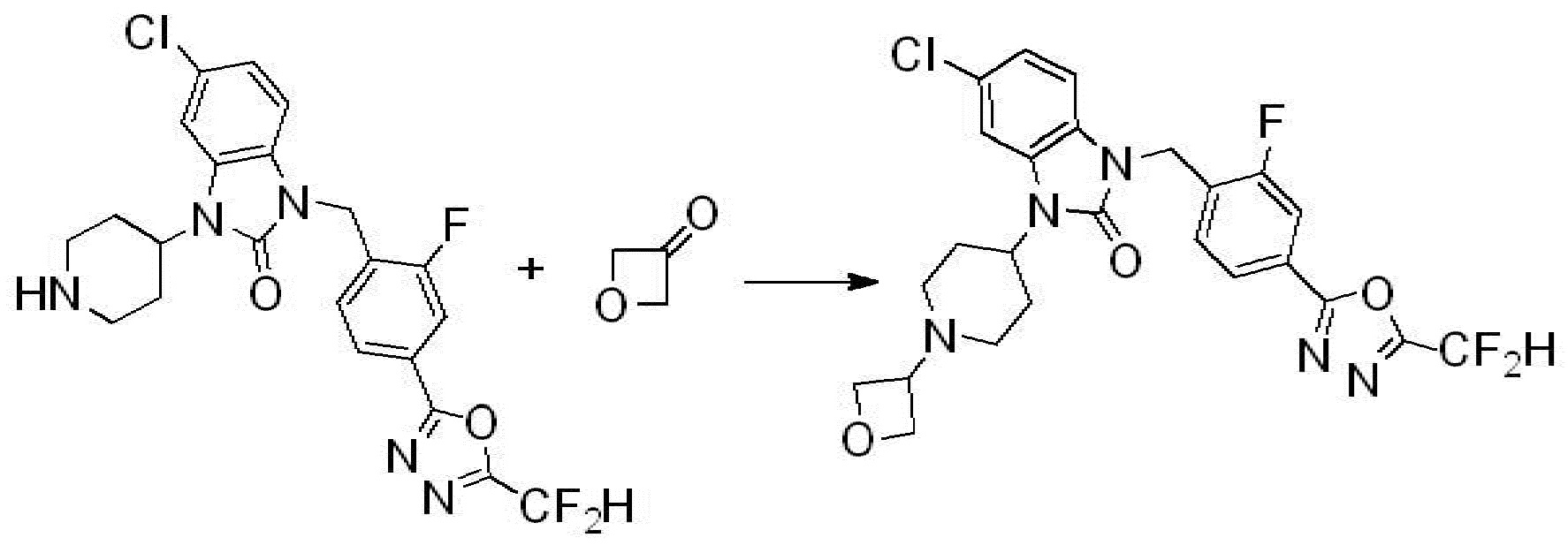
5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,209 ммоль), полученный на стадии 2 соединения 101, триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль), уксусную кислоту (0,012 мл, 0,209 ммоль) и оксетан-3-он (0,045 г, 0,628 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,083 г, 74,2%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,83-7,80 (м, 2H), 7,41 (т, J=7,8 Гц, 1H), 7,31 (д, J=1,8 Гц, 1H), 7,02-6,77 (м, 3H), 5,14 (с, 2H), 4,65 (дт, J=17,0, 5,9 Гц, 4H), 4,43-4,34 (м, 1H), 3,56-3,50 (м, 1H), 2,91 (д, J=11,4 Гц, 2H), 2,47-2,38 (м, 2H), 2,04-1,99 (м, 2H), 1,85 (дд, J=12,0, 2,2 Гц, 2H); МСНР (ЭР) m/z 536,3 (M+ + 1).
Пример 103: Синтез соединения 103, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез трет-бутил 4-((2-нитропиридин-3-ил)амино)пиперидин-1-карбоксилата
3-фтор-2-нитропиридин (5,000 г, 35,189 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (7,400 г, 36,948 ммоль), карбонат калия (9,727 г, 70,378 ммоль) и йодид калия (0,584 г, 3,519 ммоль) растворяют в N,N-диметилформамиде (300 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Реакционную смесь фильтруют через бумажный фильтр для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении. Затем, воду выливают в полученный концентрат и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (11,450 г, 100,9%, оранжевая жидкость).
[Стадия 2] Синтез трет-бутил 4-((2-аминопиридин-3-ил)амино)пиперидин-1-карбоксилата
Трет-бутил 4-((2-нитропиридин-3-ил)амино)пиперидин-1-карбоксилат (11,450 г, 35,518 ммоль), полученный на стадии 1, и Pd/C (10%, 0,840 г, 3,552 ммоль) растворяют в метаноле (250 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (9,820 г, 94,6%, черное твердое вещество).
[Стадия 3] Синтез трет-бутил 4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилата
Трет-бутил 4-((2-аминопиридин-3-ил)амино)пиперидин-1-карбоксилат (9,820 г, 33,586 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 8,169 г, 50,380 ммоль) растворяют в N,N-диметилформамиде (150 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (5,770 г, 54,0%) в виде коричневого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилата
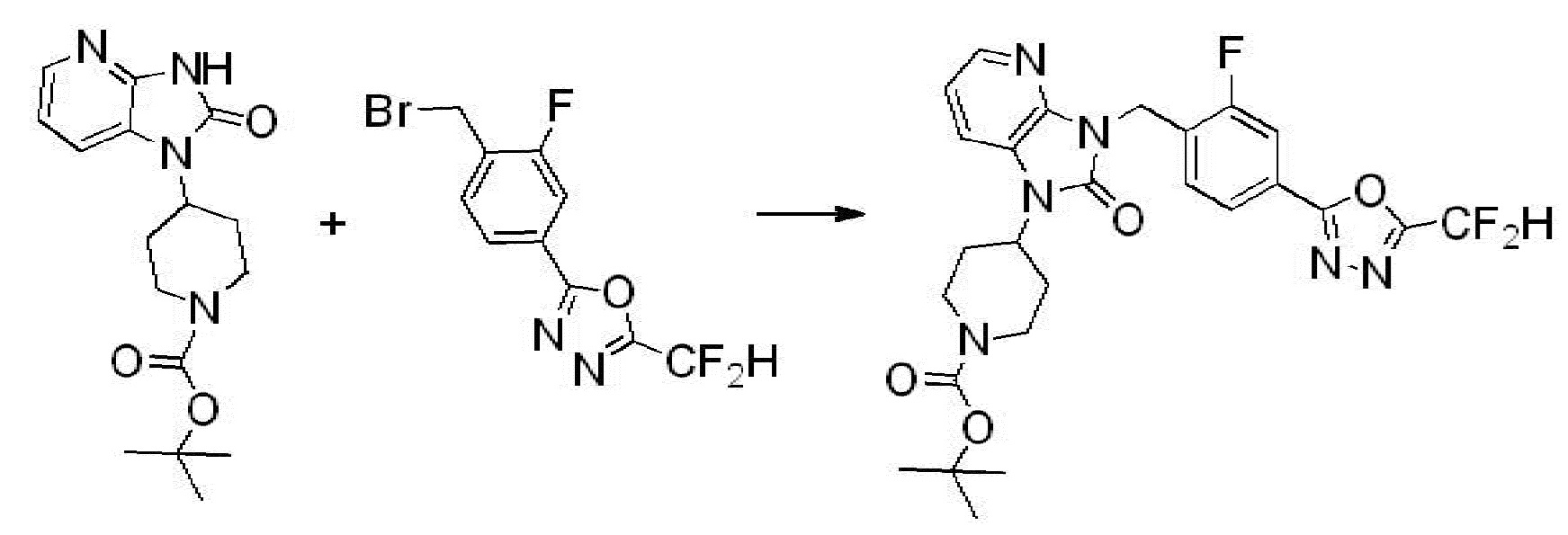
Трет-бутил 4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат (1,900 г, 5,968 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (60 мл) при 0°C, после чего гидрид натрия (60,00%, 0,358 г, 8,952 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (2,382 г, 7,758 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (0,940 г, 28,9%) в виде коричневого твердого вещества.
[Стадия 5] Синтез 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-она
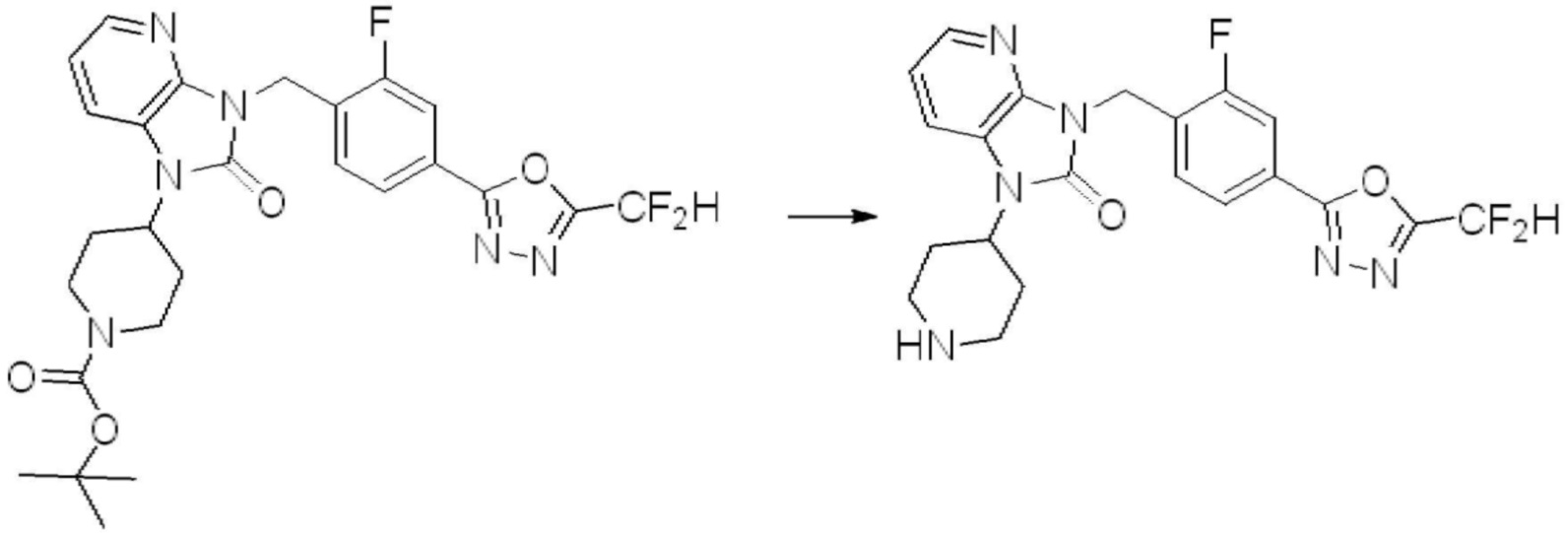
Трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат (0,940 г, 1,727 ммоль), полученный на стадии 4 и трифторуксусную кислоту (1,322 мл, 17,266 ммоль) растворяют в дихлорметане (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=80-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-80%) и концентрируют с получением указанного в заголовке соединения (0,587 г, 76,4%) в виде желтого твердого вещества.
[Стадия 6] Синтез соединения 103
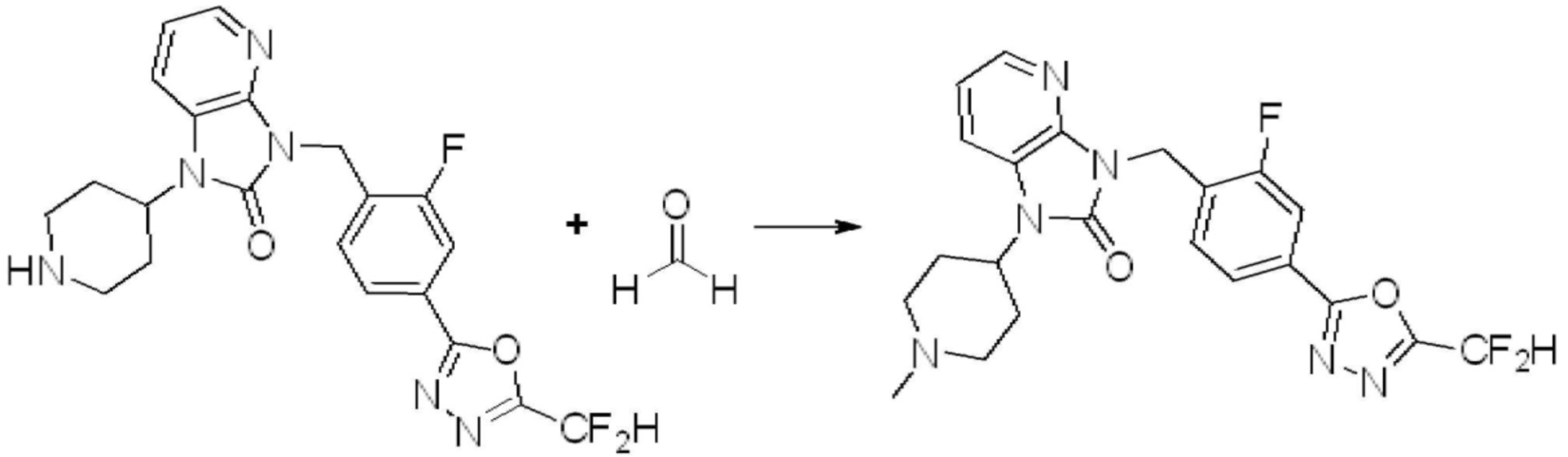
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5, триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) и формальдегид (0,020 г, 0,675 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,048 г, 46,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,00 (дд, J=5,2, 1,2 Гц, 1H), 7,79 (д, J=8,9 Гц, 2H), 7,50 (дд, J=7,9, 1,2 Гц, 1H), 7,41 (т, J=7,6 Гц, 1H), 6,97 (дд, J=7,9, 5,2 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 5,29 (с, 2H), 4,46-4,40 (м, 1H), 3,00 (д, J=11,7 Гц, 2H), 2,41-2,30 (м, 5H), 2,14 (тд, J=12,0, 2,0 Гц, 2H), 1,86-1,83 (м, 2H); МСНР (ЭР) m/z 459,4 (M+ + 1).
Пример 104: Синтез соединения 104, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез соединения 104
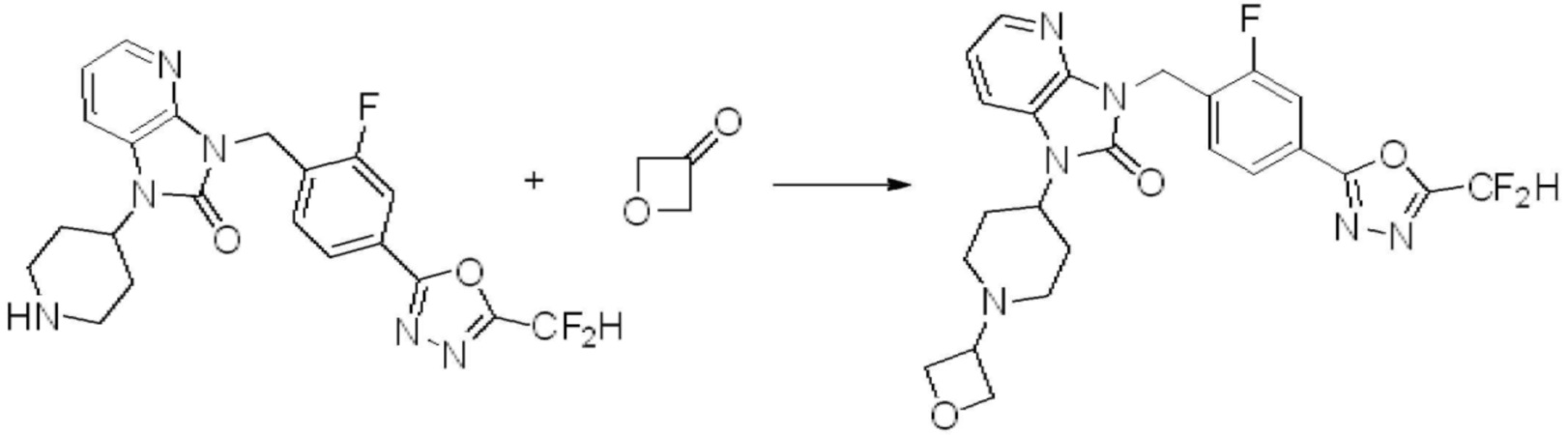
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 103, триацетоксиборгидрид натрия (0,095 г, 0,450 ммоль) и оксетан-3-он (0,049 г, 0,675 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,066 г, 58,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,01 (дд, J=5,2, 1,0 Гц, 1H), 7,79-7,77 (м, 2H), 7,52 (дд, J=7,8, 1,0 Гц, 1H), 7,42 (т, J=7,7 Гц, 1H), 7,00 (дд, J=7,9, 5,3 Гц, 1H), 6,89 (т, J=51,3 Гц, 1H), 5,28 (с, 2H), 4,62 (дт, J=26,4, 6,4 Гц, 4H), 4,48-4,40 (м, 1H), 3,54-3,48 (м, 1H), 2,88 (д, J=11,6 Гц, 2H), 2,41-2,30 (м, 2H), 2,02-1,97 (м, 2H), 1,87 (дд, J=11,8, 2,0 Гц, 2H); МСНР (ЭР) m/z 501,4 (M+ + 1).
Пример 105: Синтез соединения 105, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилата
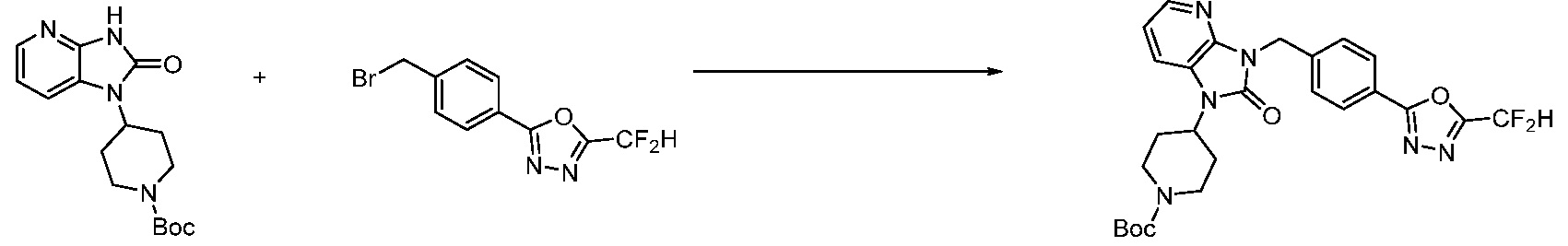
Трет-бутил 4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат (1,900 г, 5,968 ммоль), полученный на стадии 3 соединения 103 растворяют в N,N-диметилформамиде (60 мл) при 0°C, после чего гидрид натрия (60,00%, 0,358 г, 8,952 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (2,243 г, 7,758 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (1,125 г, 35,8%) в виде коричневого твердого вещества.
[Стадия 2] Синтез 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-она
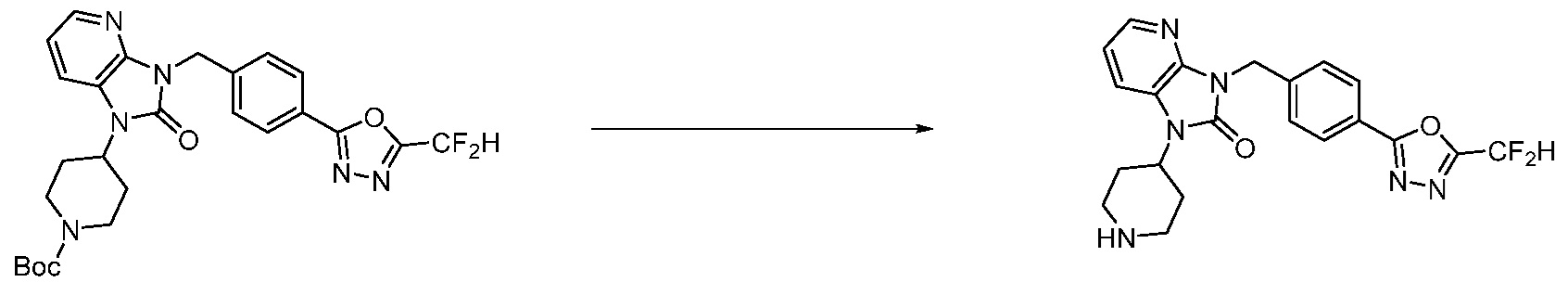
Трет-бутил 4-(3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат (1,125 г, 2,136 ммоль), полученный на стадии 1, и трифторуксусную кислоту (1,636 мл, 21,362 ммоль) растворяют в дихлорметане (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=80-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-80%) и концентрируют с получением указанного в заголовке соединения (0,830 г, 91,1%) в виде желтого твердого вещества.
[Стадия 3] Синтез соединения 105
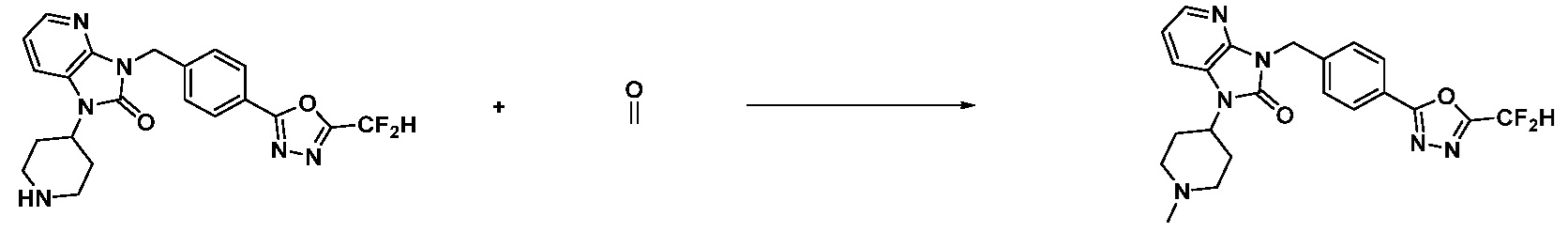
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,235 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,099 г, 0,469 ммоль) и формальдегид (0,021 г, 0,704 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,024 г, 23,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,05-8,01 (м, 3H), 7,62 (д, J=8,2 Гц, 2H), 7,48 (дд, J=7,9, 1,0 Гц, 1H), 6,96 (дд, J=7,8, 5,2 Гц, 1H), 6,88 (т, J=51,8 Гц, 1H), 5,22 (с, 2H), 4,47-4,39 (м, 1H), 3,00 (д, J=11,8 Гц, 2H), 2,40-2,30 (м, 5H), 2,17-2,11 (м, 2H), 1,83 (дд, J=11,8, 2,0 Гц, 2H); МСНР (ЭР) m/z 441,5 (M+ + 1).
Пример 106: Синтез соединения 106, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез соединения 106
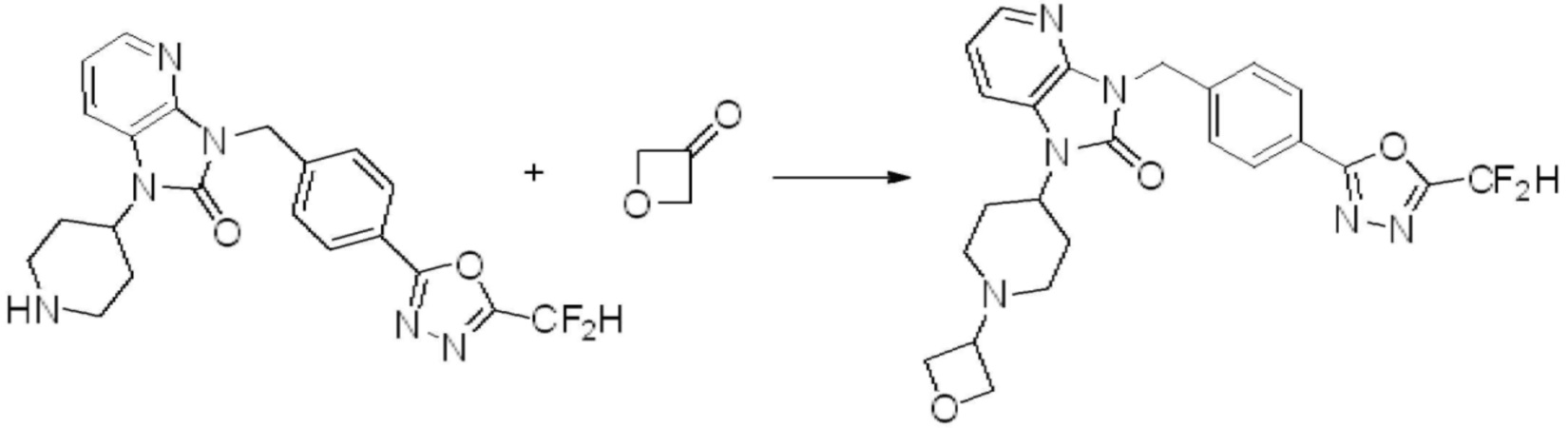
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,235 ммоль), полученный на стадии 2 соединения 105, триацетоксиборгидрид натрия (0,099 г, 0,469 ммоль) и оксетан-3-он (0,051 г, 0,704 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,080 г, 70,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,02-8,00 (м, 3H), 7,61 (д, J=8,3 Гц, 2H), 7,49 (дд, J=7,8, 1,1 Гц, 1H), 6,98 (дд, J=7,8, 5,2 Гц, 1H), 6,88 (т, J=51,7 Гц, 1H), 5,19 (с, 2H), 4,61 (дт, J=26,4, 6,4 Гц, 4H), 4,46-4,38 (м, 1H), 3,53-3,46 (м, 1H), 2,87 (д, J=11,6 Гц, 2H), 2,38-2,28 (м, 2H), 2,01-1,95 (м, 2H), 1,85 (дд, J=11,9, 2,1 Гц, 2H); МСНР (ЭР) m/z 483,3 (M+ + 1).
Пример 107: Синтез соединения 107, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилата
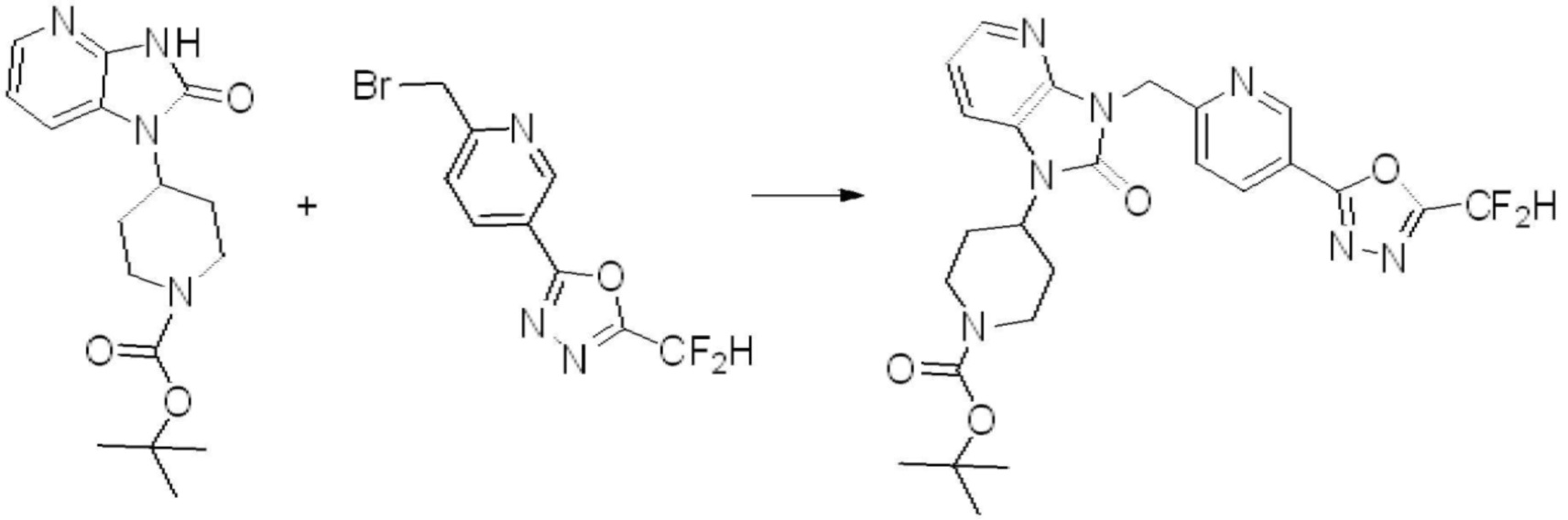
2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,900 г, 6,550 ммоль) растворяют в N,N-диметилформамиде (60 мл) при 0°C, после чего гидрид натрия (60,00%, 0,393 г, 9,825 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. Трет-бутил 4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат (2,711 г, 8,515 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-80%) и концентрируют с получением указанного в заголовке соединения (1,814 г, 52,5%) в виде коричневого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилата
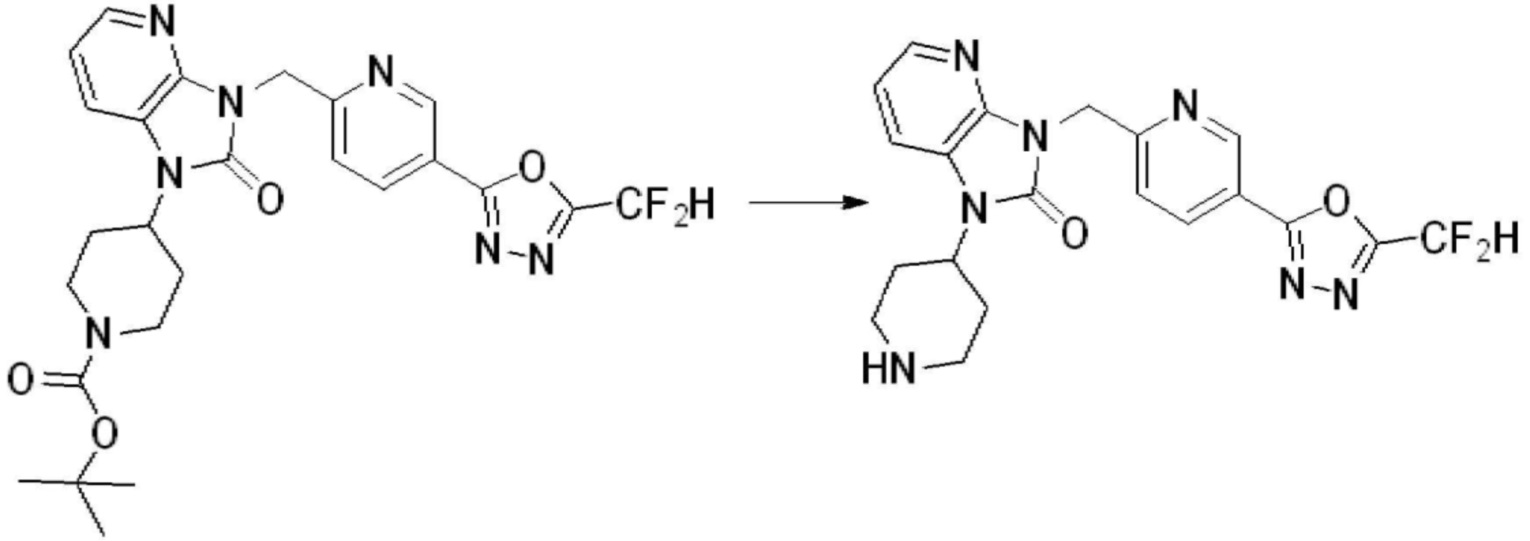
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (1,814 г, 4,244 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,325 мл, 4,244 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=80-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-80%) и концентрируют с получением указанного в заголовке соединения (1,029 г, 46,0%) в виде коричневого твердого вещества.
[Стадия 3] Синтез соединения 107
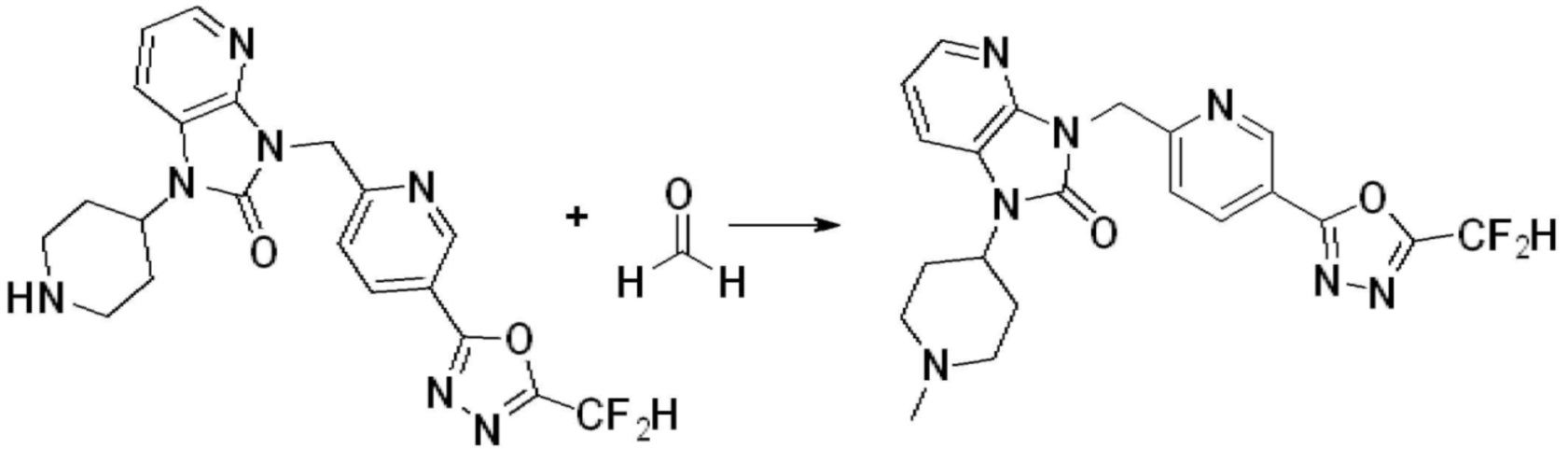
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,234 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (0,099 г, 0,468 ммоль) и формальдегид (0,021 г, 0,702 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,031 г, 30,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,22 (д, J=1,7 Гц, 1H), 8,30 (дд, J=8,2, 2,2 Гц, 1H), 7,99 (дд, J=5,2, 1,1 Гц, 1H), 7,56 (дд, J=7,9, 1,1 Гц, 1H), 7,43 (д, J=8,2 Гц, 1H), 6,98 (дд, J=7,9, 5,2 Гц, 1H), 6,91 (т, J=51,7 Гц, 1H), 5,39 (с, 2H), 4,54-4,45 (м, 1H), 3,10 (д, J=11,8 Гц, 2H), 2,51-2,41 (м, 2H), 2,38 (с, 3H), 2,27-2,21 (м, 2H), 1,88 (дд, J=12,3, 2,2 Гц, 2H); МСНР (ЭР) m/z 442,5 (M+ + 1).
Пример 108: Синтез соединения 108, 3-(1-ацетилпиперидин-4-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 108
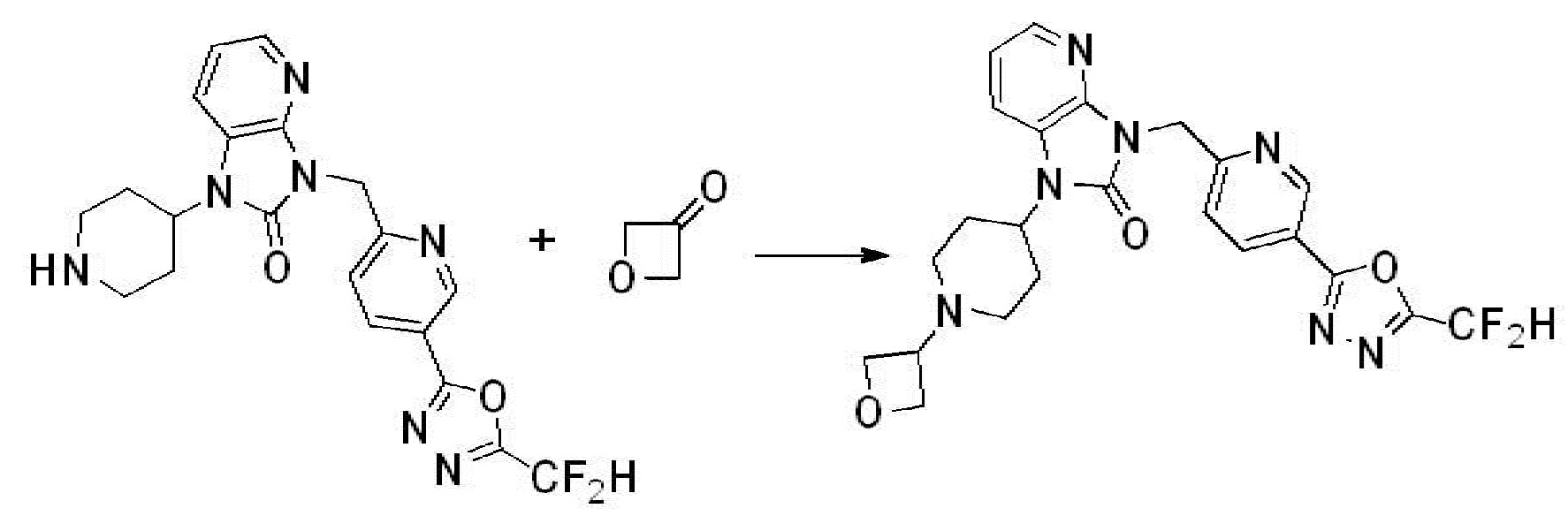
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 2 соединения 107, ацетилхлорид (0,032 мл, 0,450 ммоль) и триэтиламин (0,063 мл, 0,450 ммоль) растворяют в дихлорметане (1,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (0,030 г, 27,4%) в виде коричневой жидкости.
1H ЯМР (400 MГц, CDCl3) δ 9,19 (д, J=1,6 Гц, 1H), 8,28 (дд, J=8,2, 2,2 Гц, 1H), 7,98 (дд, J=5,2, 0,9 Гц, 1H), 7,51 (дд, J=7,8, 0,9 Гц, 1H), 7,42 (д, J=8,2 Гц, 1H), 6,98 (дд, J=7,8, 5,2 Гц, 1H), 6,90 (т, J=51,6 Гц, 1H), 5,36 (с, 2H), 4,62 (дт, J=23,8, 6,3 Гц, 4H), 4,48-4,40 (м, 1H), 3,53-3,42 (м, 1H), 2,88 (д, J=11,5 Гц, 2H), 2,41-2,31 (м, 2H), 2,02-1,97 (м, 2H), 1,89-1,86 (м, 2H); МСНР (ЭР) m/z 484,4 (M+ + 1).
Пример 109: Синтез соединения 109, 1-(2-оксаспиро[3,3]гептан-6-ил)-3-((5-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 109
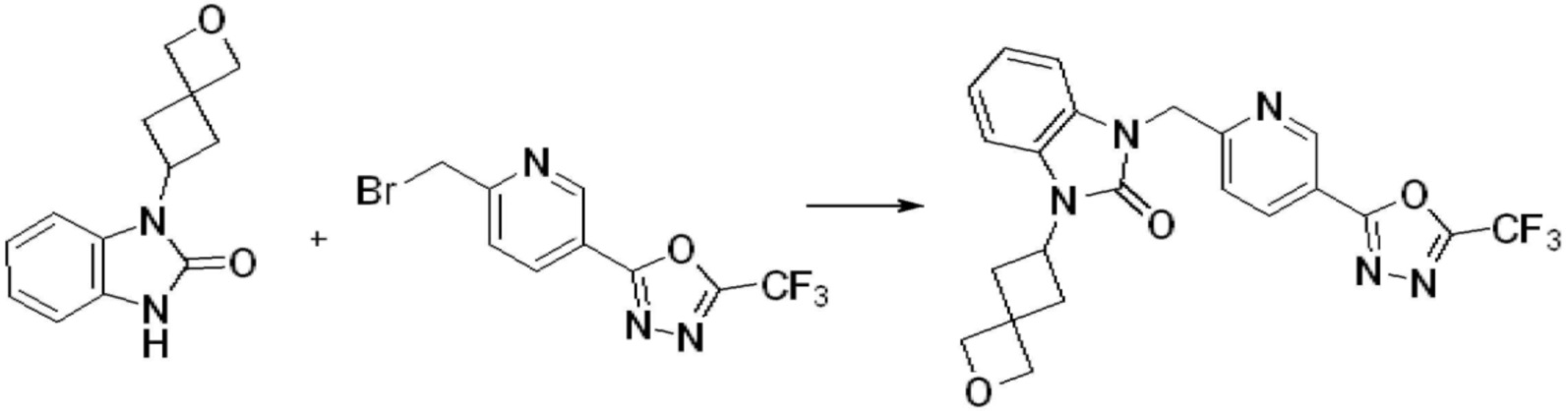
1-(2-оксаспиро[3,3]гептан-6-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,050 г, 0,217 ммоль), полученный на стадии 2 соединения 27, и гидрид натрия (60,00%, 0,010 г, 0,239 ммоль) растворяют в N,N-диметилформамиде (3 мл) при 0°C, после чего 2-(6-(бромметил)пиридин-3-ил)-5-(трифторметил)-1,3,4-оксадиазол (0,074 г, 0,239 ммоль) добавляют в полученный раствор и затем перемешивают при той же температуре в течение 2 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=60-100%) и концентрируют с получением указанного в заголовке соединения (0,011 г, 11,1%) в виде желтого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (дд, J=2,2, 0,8 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,44 (д, J=8,2 Гц, 2H), 7,14-7,04 (м, 4H), 6,98-6,97 (м, 1H), 5,29 (с, 2H), 4,87 (с, 2H), 4,82 (с, 2H), 4,75-4,69 (м, 1H), 3,20-3,15 (м, 2H), 2,85-2,80 (м, 2H); МСНР (ЭР) m/z 458,3 (M+ + H).
Пример 110: Синтез соединения 110, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-метил-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез 4-бром-N-метил-2-нитроанилина
1-фтор-2-нитро-4-(трифторметил)бензол (5,000 г, 22,727 ммоль), метанамин (2,00 M раствор, 13,636 мл, 27,273 ммоль), карбонат калия (6,282 г, 45,455 ммоль) и йодид калия (0,377 г, 2,273 ммоль) растворяют в N,N-диметилформамиде (230 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (4,750 г, 90,5%) в виде оранжевого твердого вещества.
[Стадия 2] 4-бром-N1-метилбензол-1,2-диамина
4-бром-N-метил-2-нитроанилин (4,750 г, 20,558 ммоль), полученный на стадии 1, и Ni Ренея (0,48 г, 10% масс.) растворяют в метаноле (200 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении. Затем, воду выливают в полученный концентрат и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (3,980 г, 96,3%, черная жидкость).
[Стадия 3] Синтез 5-бром-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-она
4-бром-N1-метилбензол-1,2-диамин (3,980 г, 19,794 ммоль), полученный на стадии 2 и 1,1'-карбонилдиимидазол (КДИ, 4,814 г, 29,691 ммоль) растворяют в N,N-диметилформамиде (150 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-80%) и концентрируют с получением указанного в заголовке соединения (0,530 г, 11,8%) в виде фиолетового твердого вещества.
[Стадия 4] Синтез 5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-она
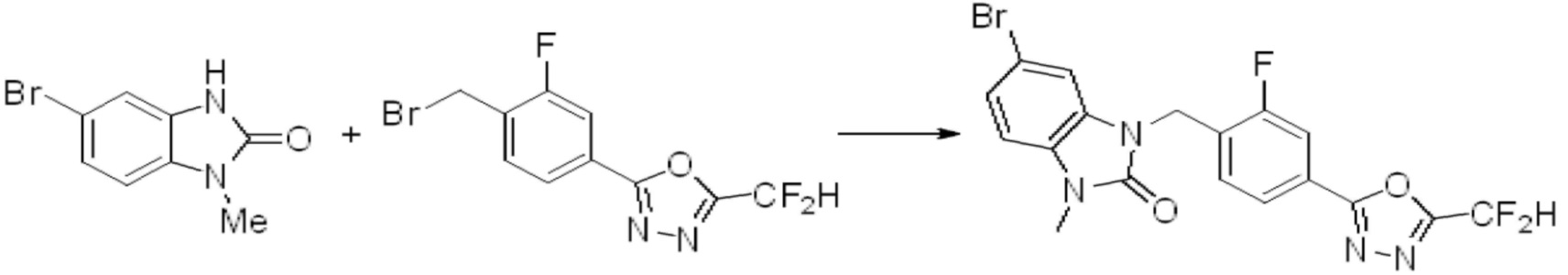
5-бром-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,530 г, 2,334 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,140 г, 3,501 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,932 г, 3,034 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (0,485 г, 45,8%) в виде коричневого твердого вещества.
[Стадия 5] Синтез соединения 110
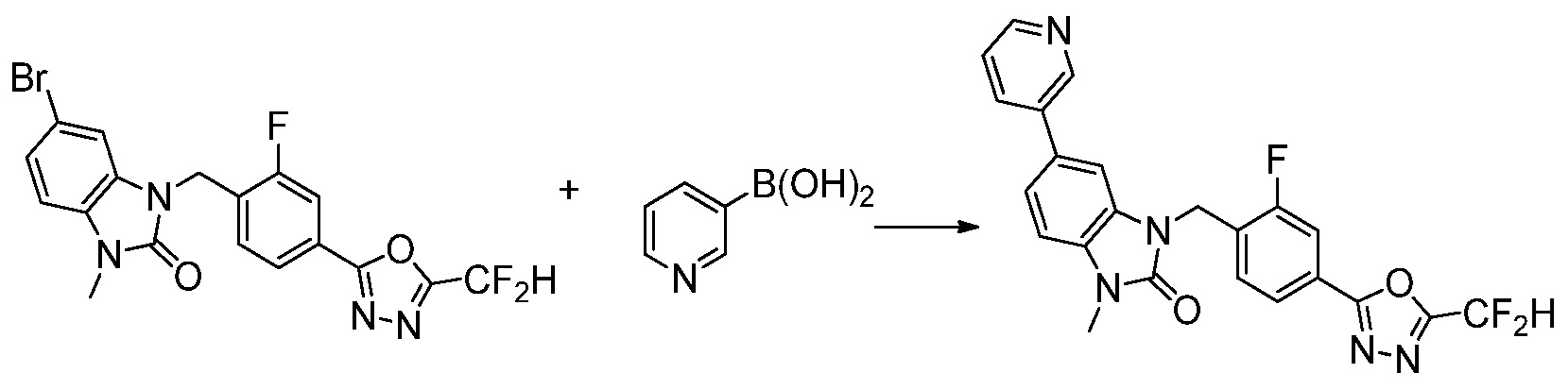
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 0,441 ммоль), полученный на стадии 4, пиридин-3-илбороновую кислоту (0,108 г, 0,883 ммоль), карбонат натрия (0,234 г, 2,206 ммоль) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия с дихлорметаном (0,018 г, 0,022 ммоль) смешивают в 1,2-диметоксиэтане (2,3 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-3%) и концентрируют с получением указанного в заголовке соединения (0,067 г, 33,8%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,75 (д, J=1,8 Гц, 1H), 8,53 (дд, J=4,8, 1,6 Гц, 1H), 7,84-7,76 (м, 3H), 7,49 (т, J=7,8 Гц, 1H), 7,33-7,30 (м, 2H), 7,13 (д, J=1,4 Гц, 1H), 7,09 (д, J=8,1 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 5,24 (с, 2H), 3,49 (с, 3H); МСНР (ЭР) m/z 451,9 (M+ + 1).
Пример 111: Синтез соединения 111, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-метил-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 111
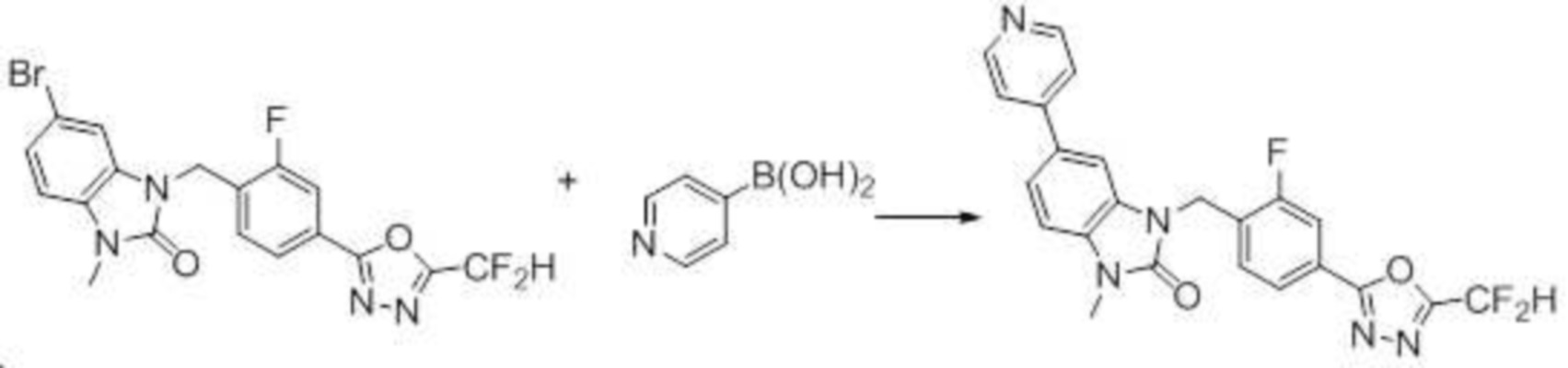
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 0,441 ммоль), полученный на стадии 4 соединения 110, пиридин-4-илбороновую кислоту (0,108 г, 0,883 ммоль), карбонат натрия (0,234 г, 2,206 ммоль) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия с дихлорметаном (0,018 г, 0,022 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-3%) и концентрируют с получением указанного в заголовке соединения (0,019 г, 9,3%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,63 (с, 2H), 7,88-7,83 (м, 2H), 7,52 (т, J=7,8 Гц, 1H), 7,45-7,41 (м, 3H), 7,22 (д, J=1,3 Гц, 1H), 7,11 (д, J=8,2 Гц, 1H), 6,89 (т, J=51,6 Гц, 1H), 5,26 (с, 2H), 3,51 (с, 3H); МСНР (ЭР) m/z 452,0 (M+ + 1).
Пример 112: Синтез соединения 112, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(3-фторфенил)-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 112
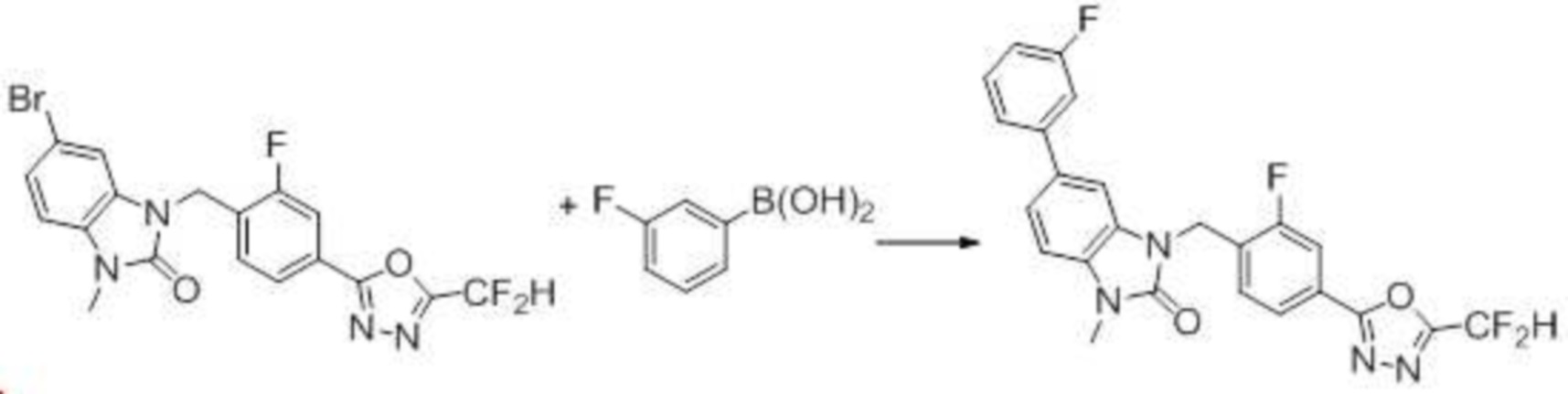
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 0,441 ммоль), полученный на стадии 4 соединения 110, (3-фторфенил)бороновую кислоту (0,074 г, 0,530 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,014 г, 0,022 ммоль) и карбонат цезия (0,288 г, 0,883 ммоль) смешивают в 1,4-диоксане (2,25 мл)/воде (0,75 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=20-50%) и концентрируют с получением указанного в заголовке соединения (0,035 г, 16,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,63 (с, 2H), 7,88-7,83 (м, 2H), 7,52 (т, J=7,8 Гц, 1H), 7,45-7,41 (м, 3H), 7,22 (д, J=1,3 Гц, 1H), 7,11 (д, J=8,2 Гц, 1H), 6,89 (т, J=51,6 Гц, 1H), 5,26 (с, 2H), 3,51 (с, 3H); МСНР (ЭР) m/z 469,2 (M+ + 1).
Пример 113: Синтез соединения 113, 5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((5-бром-4-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилата
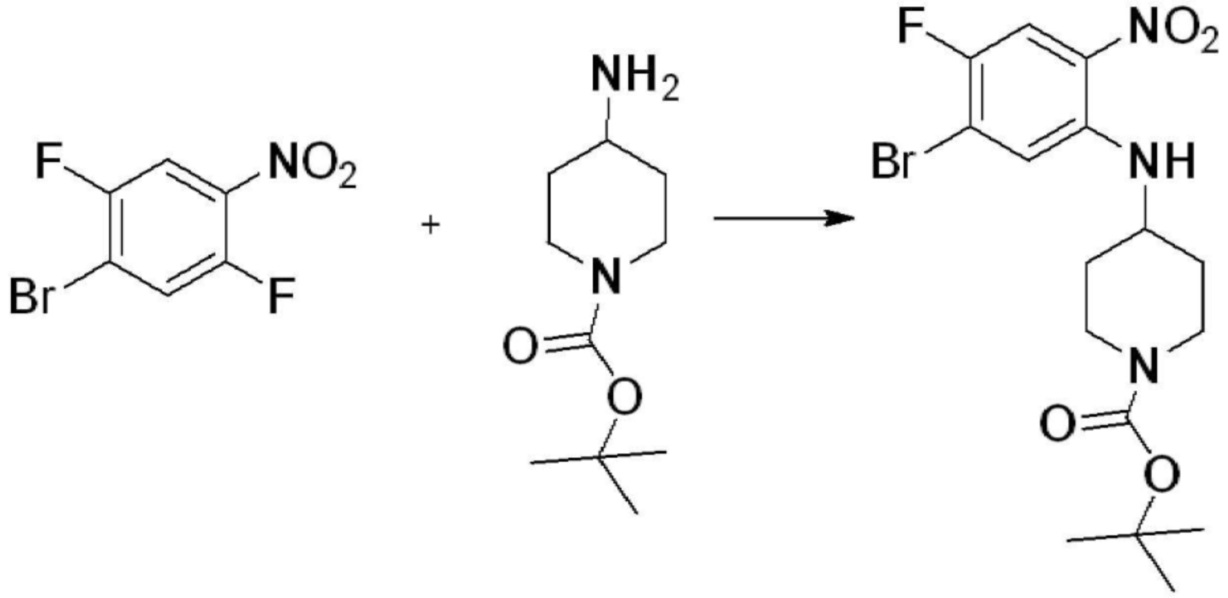
1-бром-2,5-дифтор-4-нитробензол (5,000 г, 21,009 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (5,049 г, 25,211 ммоль) и карбонат калия (5,807 г, 42,019 ммоль) растворяют в N,N-диметилформамиде (30 мл) при 80°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (6,000 г, 68,3%) в виде красного твердого вещества.
[Стадия 2] Синтез трет-бутил 4-((2-амино-5-бром-4-фторфенил)амино)пиперидин-1-карбоксилата
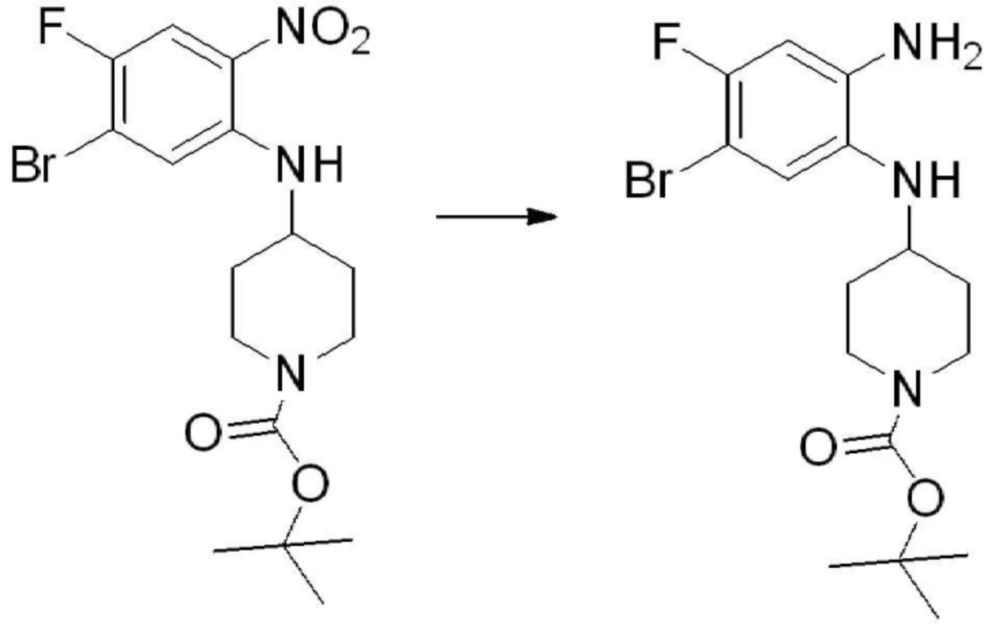
Трет-бутил 4-((5-бром-4-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилат (6,000 г, 14,345 ммоль), полученный на стадии 1, растворяют в метаноле (50 мл) при комнатной температуре, после чего Ni Ренея (30 мг) медленно добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (5,560 г, 99,8%, черное масло).
[Стадия 3] Синтез трет-бутил 4-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
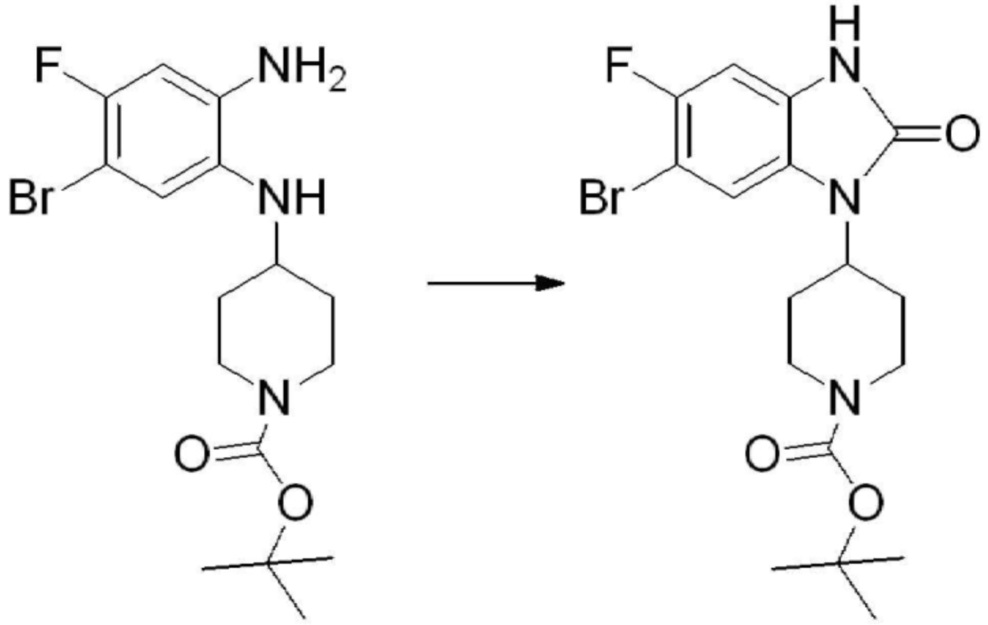
Трет-бутил 4-((2-амино-5-бром-4-фторфенил)амино)пиперидин-1-карбоксилат (5,560 г, 14,320 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 3,483 г, 21,479 ммоль) растворяют в N,N-диметилформамиде (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (3,800 г, 64,1%) в виде белого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(6-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
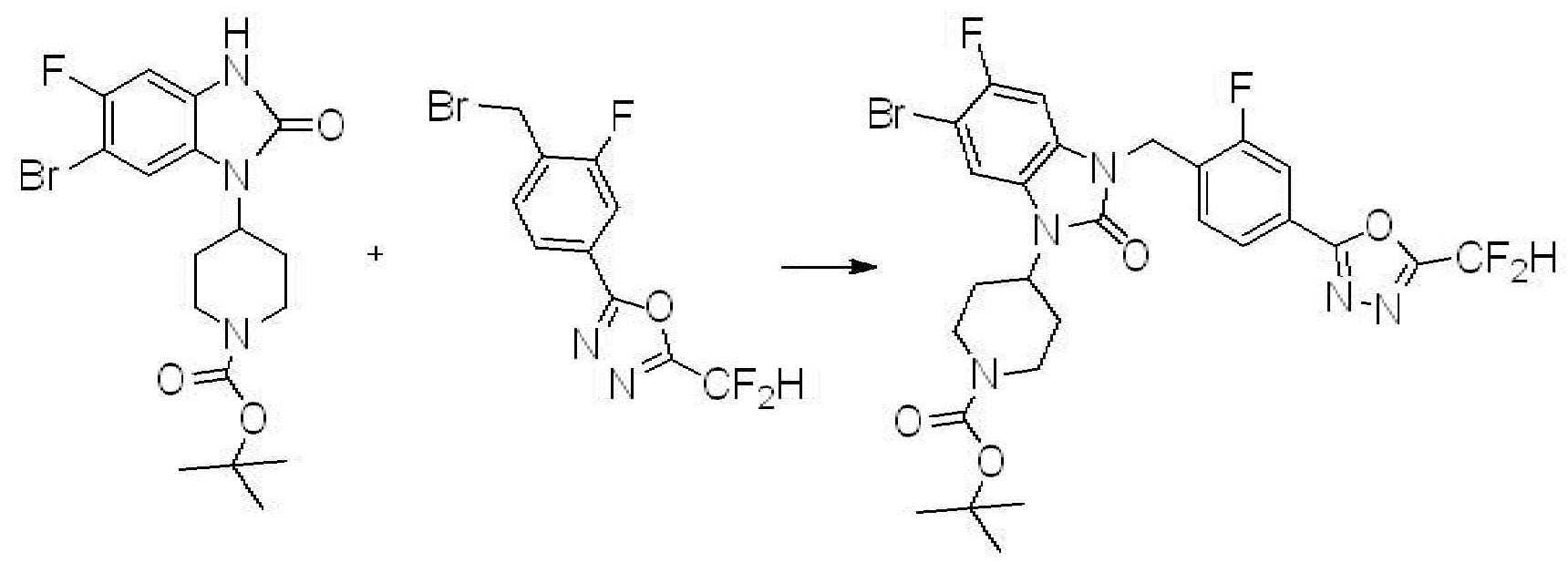
Трет-бутил 4-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,000 г, 4,828 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (30 мл) при 0°C, после чего гидрид натрия (60,00%, 0,290 г, 7,241 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (1,482 г, 4,828 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (1,800 г, 58,2%) в виде коричневого масла.
[Стадия 5] Синтез 5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
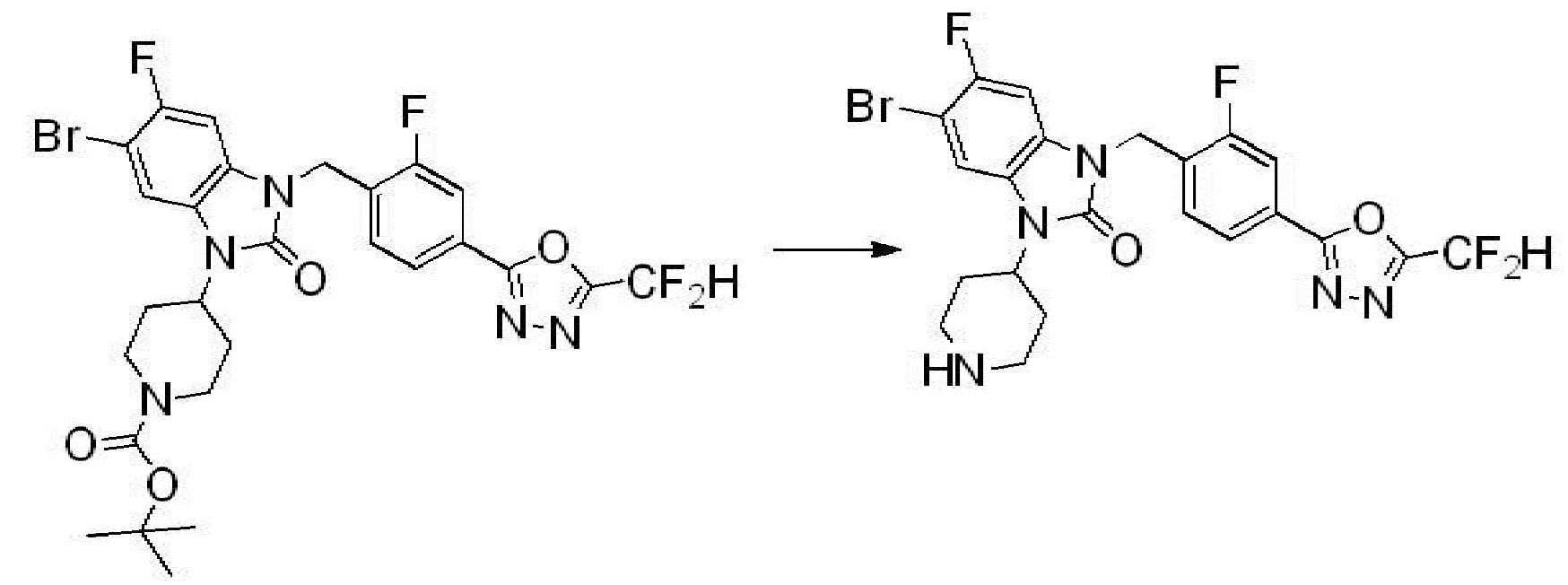
Трет-бутил 4-(6-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,700 г, 2,654 ммоль), полученный на стадии 4, и трифторуксусную кислоту (2,033 мл, 26,545 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (1,000 г, 69,7%, коричневое масло).
[Стадия 6] Синтез соединения 113
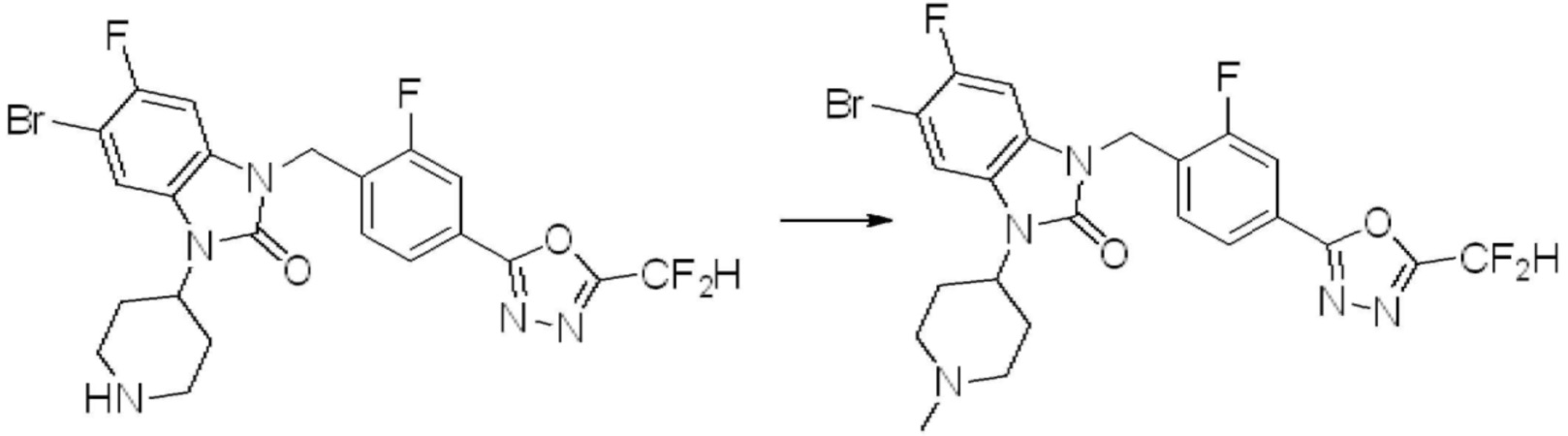
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (1,000 г, 1,851 ммоль), полученный на стадии 5, формальдегид (0,111 г, 3,702 ммоль), уксусную кислоту (0,212 мл, 3,702 ммоль) и триацетоксиборгидрид натрия (0,784 г, 3,702 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Этилацетат (10 мл) и гексан (30 мл) добавляют в полученный концентрат и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают гексан, и затем сушат с получением указанного в заголовке соединения (0,830 г, 80,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 7,99 (д, J=5,8 Гц, 1H), 7,91-7,84 (м, 2H), 7,69 (с, 0,25H), 7,56 (с, 0,5H), 7,45-7,40 (м, 2,25H), 5,22 (с, 2H), 4,59-4,58 (м, 1H), 3,52-3,49 (м, 2H), 3,18-3,12 (м, 2H), 2,80-2,73 (м, 5H), 1,95-1,91 (м, 2H).
Пример 114: Синтез соединения 114, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-5-(фуран-2-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 114
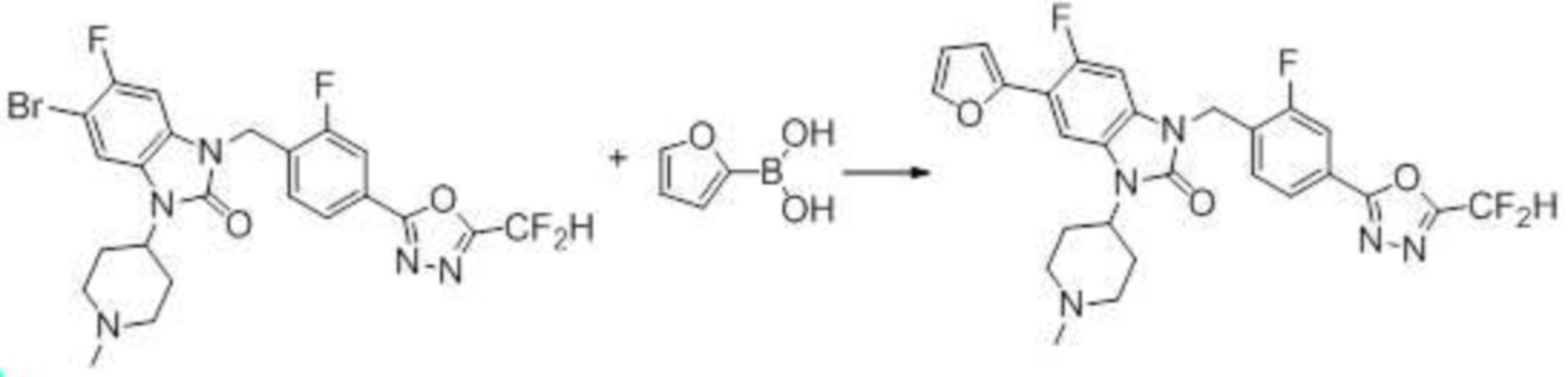
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,271 ммоль), фуран-2-илбороновую кислоту (0,061 г, 0,541 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (0,018 г, 0,027 ммоль) и карбонат цезия (0,132 г, 0,406 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,120 г, 81,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,89-7,85 (м, 2H), 7,68 (д, J=6,1 Гц, 1H), 7,50-7,46 (м, 2H), 7,05 (с, 0,25H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 6,78-6,73 (м, 2H), 6,41-6,50 (м, 1H), 5,17 (с, 2H), 4,46-4,40 (м, 1H), 3,09-3,06 (м, 2H), 2,56-2,49 (м, 2H), 2,39 (с, 3H), 2,23-2,17 (м, 2H), 1,89-1,85 (м, 2H); МСНР (ЭР) m/z 542,3 (M+ + 1).
Пример 115: Синтез соединения 115, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-5-(фуран-3-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 115
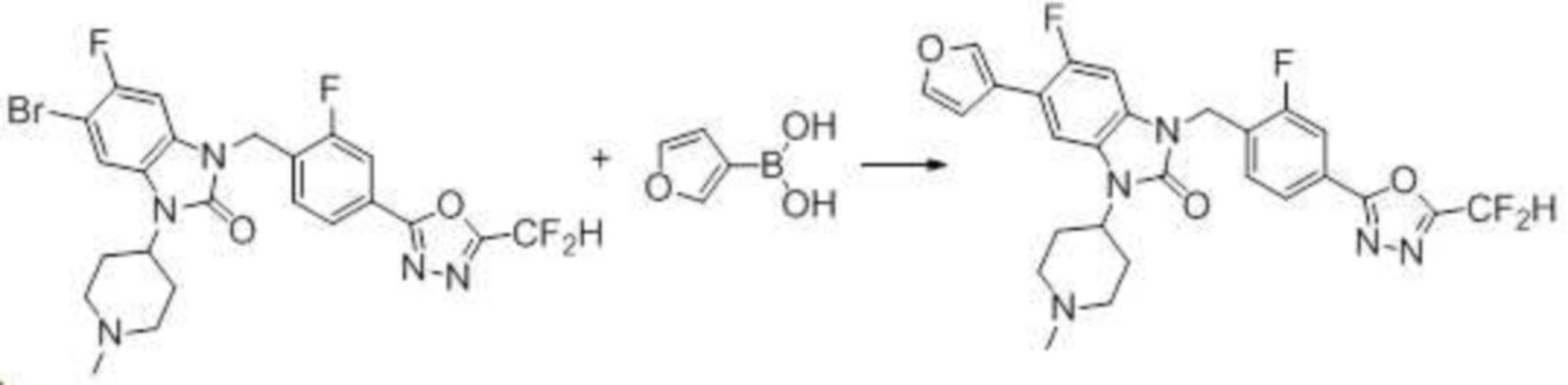
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,271 ммоль), фуран-3-илбороновую кислоту (0,061 г, 0,541 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (0,018 г, 0,027 ммоль) и карбонат цезия (0,132 г, 0,406 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,130 г, 88,7%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,89-7,85 (м, 2H), 7,82-7,81 (м, 1H), 7,50-7,45 (м, 2H), 7,34 (д, J=6,0 Гц, 1H), 7,05 (с, 0,25H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 6,79-6,77 (м, 2H), 5,31 (с, 2H), 4,47-4,39 (м, 1H), 3,07-3,04 (м, 2H), 2,55-2,45 (м, 2H), 2,37 (с, 3H), 2,23-2,18 (м, 2H), 1,89-1,86 (м, 2H); МСНР (ЭР) m/z 542,3 (M+ + 1).
Пример 116: Синтез соединения 116, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-5-(2-фторфенил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 116
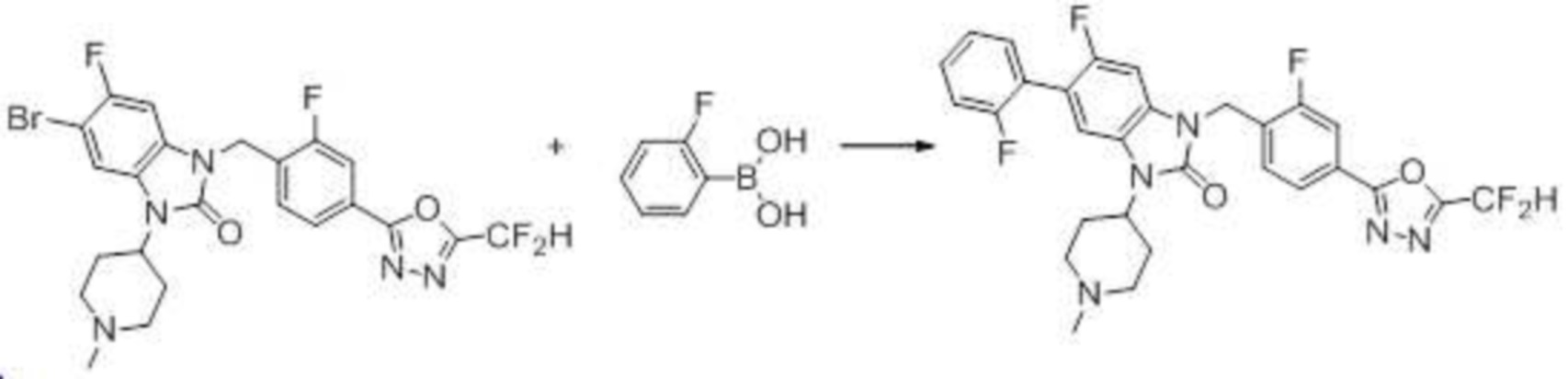
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,271 ммоль), (2-фторфенил)бороновую кислоту (0,076 г, 0,541 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (0,018 г, 0,027 ммоль) и карбонат цезия (0,132 г, 0,406 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 71,4%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 7,89 (д, J=9,1 Гц, 1H), 7,53 (т, J=7,7 Гц, 1H), 7,40-7,33 (м, 2H), 7,28-7,27 (м, 1H), 7,23-7,13 (м, 1H), 7,05 (с, 0,25H), 6,92 (с, 0,5H), 6,83 (д, J=9,2 Гц, 1H), 6,79 (с, 0,25H), 5,20 (с, 2H), 4,48-4,42 (м, 1H), 3,04 (д, J=11,7 Гц, 1H), 2,51-2,41 (м, 2H), 2,34 (с, 3H), 2,21-2,15 (м, 2H), 1,89-1,86 (м, 2H); МСНР (ЭР) m/z 570,3 (M+ + 1).
Пример 117: Синтез соединения 117, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-(1-метилпиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 117
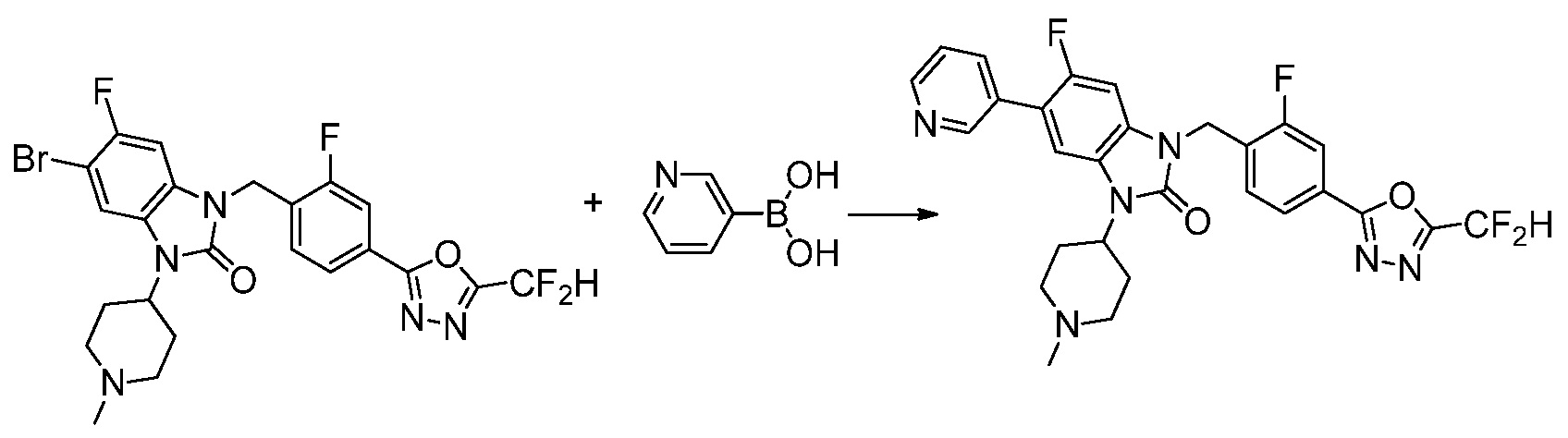
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,271 ммоль), пиридин-3-илбороновую кислоту (0,067 г, 0,541 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (0,018 г, 0,027 ммоль) и карбонат цезия (0,132 г, 0,406 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,090 г, 60,2%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 8,74 (с, 1H), 8,60 (дд, J=4,8, 1,5 Гц, 1H), 7,88 (д, J=8,9 Гц, 2H), 7,82 (дд, J=8,0, 1,7 Гц, 1H), 7,52 (т, J=7,6 Гц, 1H), 7,37 (дд, J=7,9, 4,9 Гц, 1H), 7,32 (д, J=6,2 Гц, 1H), 7,05 (с, 0,25H), 6,93 (с, 0,5H), 6,85 (д, J=9,7 Гц, 1H), 6,79 (с, 0,25H), 5,20 (с, 2H), 4,49-4,41 (м, 1H), 3,05-3,02 (м, 2H), 2,54-2,43 (м, 2H), 2,35 (с, 3H), 2,22-2,16 (м, 2H), 1,89-1,86 (м, 2H); МСНР (ЭР) m/z 553,4 (M+ + 1).
Пример 118: Синтез соединения 118, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-(1-метилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 118
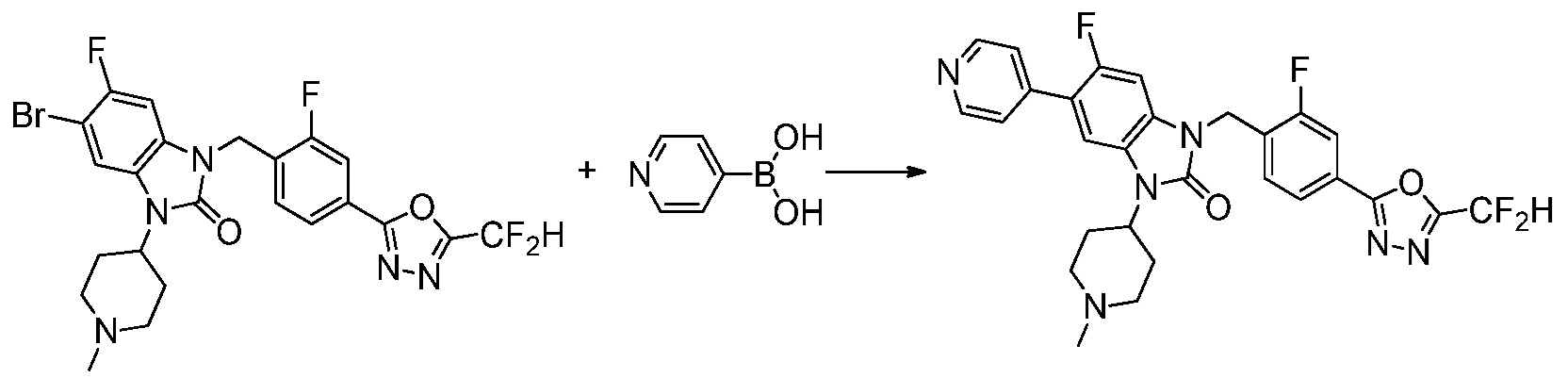
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,271 ммоль), пиридин-4-илбороновую кислоту (0,067 г, 0,541 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (0,018 г, 0,027 ммоль) и карбонат цезия (0,132 г, 0,406 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 66,9%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 8,66 (д, J=5,6 Гц, 1H), 7,88 (д, J=8,9 Гц, 2H), 7,52 (т, J=7,7 Гц, 1H), 7,44 (д, J=4,6 Гц, 2H), 7,34 (д, J=6,2 Гц, 1H), 7,05 (с, 0,25H), 6,92 (с, 0,5H), 6,85 (д, J=10,0 Гц, 1H), 6,79 (с, 0,25H), 5,20 (с, 2H), 4,49-4,41 (м, 1H), 3,06-3,04 (м, 2H), 2,53-2,43 (м, 2H), 2,36 (с, 3H), 2,23-2,17 (м, 2H), 1,89-1,86 (м, 2H); МСНР (ЭР) m/z 553,3 (M+ + 1).
Пример 119: Синтез соединения 119, 5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((4-бром-2-нитрофенил)амино)пиперидин-1-карбоксилата
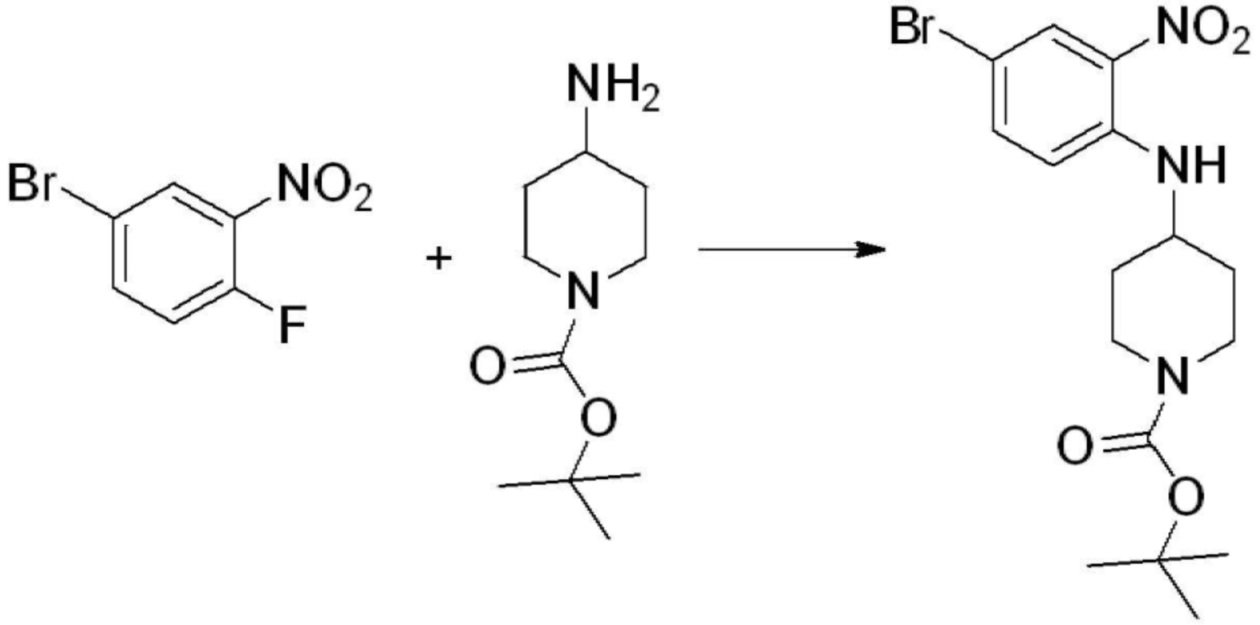
4-бром-1-фтор-2-нитробензол (5,000 г, 22,727 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (5,007 г, 25,000 ммоль), карбонат калия (6,282 г, 45,455 ммоль) и йодид калия (0,377 г, 2,273 ммоль) растворяют в N,N-диметилформамиде (300 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Реакционную смесь фильтруют через стеклянный фильтр для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (9,000 г, 98,9%, оранжевое твердое вещество).
[Стадия 2] Синтез трет-бутил 4-((2-амино-4-бромфенил)амино)пиперидин-1-карбоксилата
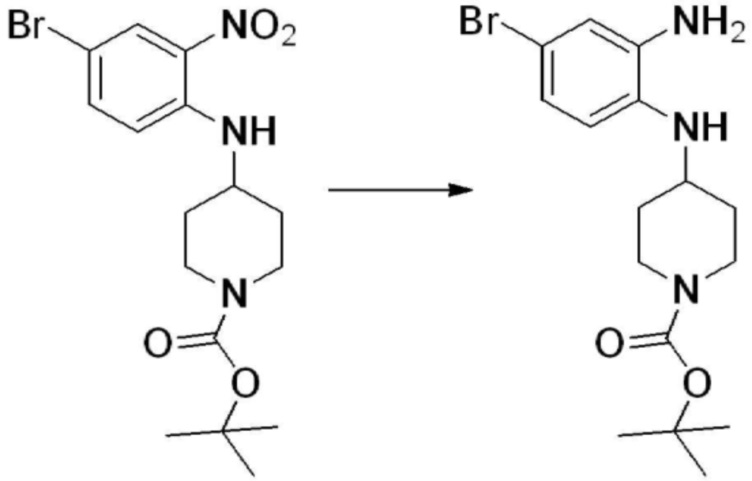
Трет-бутил 4-((4-бром-2-нитрофенил)амино)пиперидин-1-карбоксилат (11,000 г, 27,481 ммоль), полученный на стадии 1, и никель Ренея (1,1 г, 10% масс.) растворяют в метаноле (250 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (9,580 г, 94,1%, красное твердое вещество).
[Стадия 3] Синтез трет-бутил 4-(5-бром-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
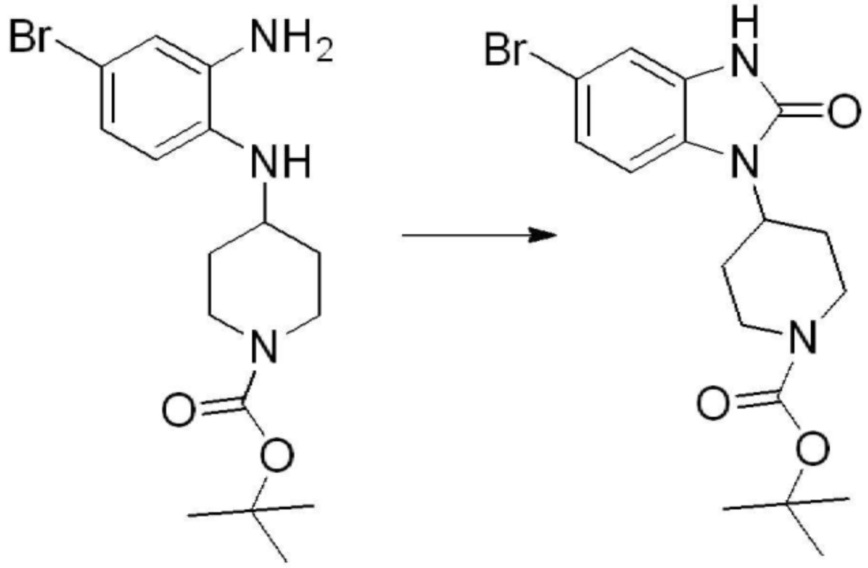
Трет-бутил 4-((2-амино-4-бромфенил)амино)пиперидин-1-карбоксилат (9,580 г, 25,872 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 5,873 г, 36,220 ммоль) растворяют в дихлорметане (200 мл) при комнатной температуре, после чего полученный раствор перемешивают при 65° в течение 3 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-80%) и концентрируют с получением указанного в заголовке соединения (7,090 г, 69,2%) в виде розового твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
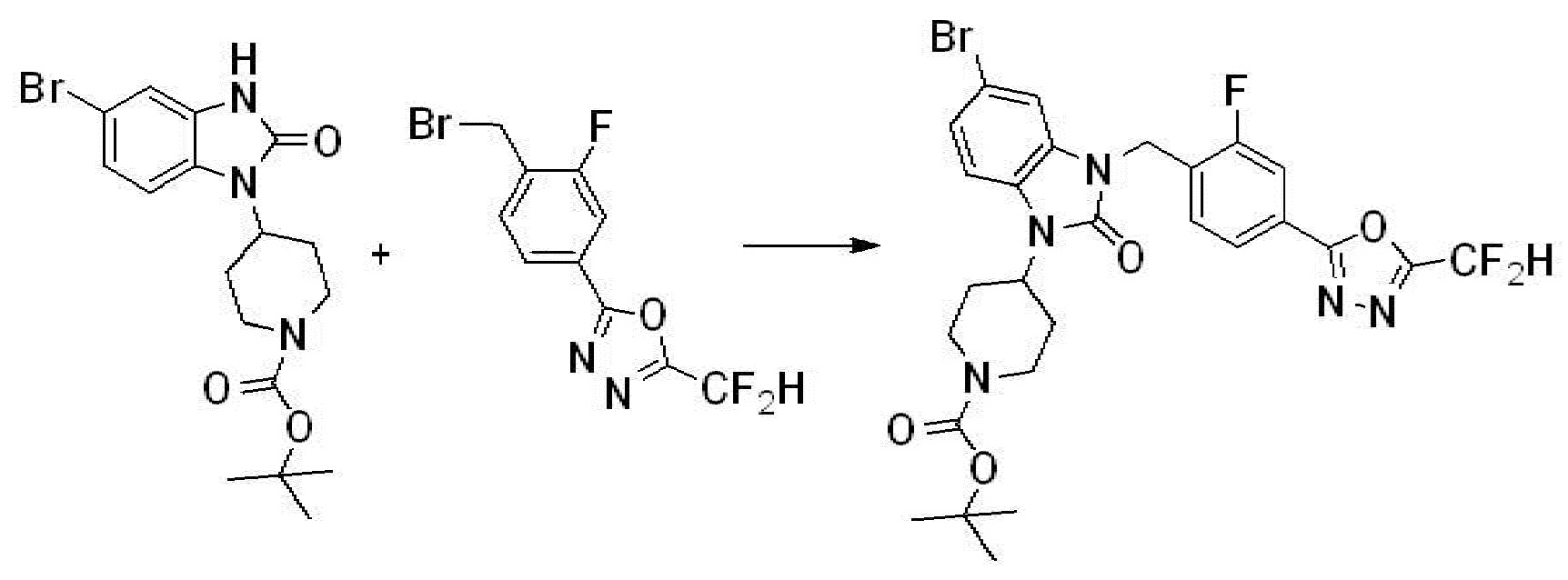
Трет-бутил 4-(5-бром-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,300 г, 5,804 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (50 мл) при 0°C, после чего гидрид натрия (60,00%, 0,348 г, 8,706 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (2,317 г, 7,545 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (3,415 г, 94,5%) в виде коричневого твердого вещества.
[Стадия 5] Синтез соединения 119
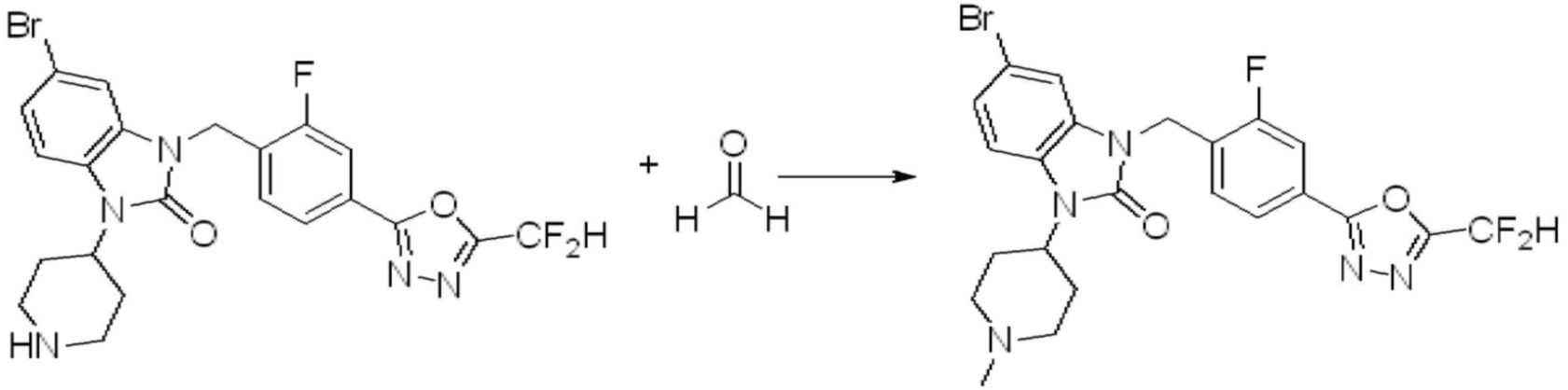
Трет-бутил 4-(5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат, полученный на стадии 4 применяют для проведения реакции снятия защиты по существу таким же образом, как показано на стадии 5 Примера 113 с получением 5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она.
Затем, 5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (2,072 г, 3,966 ммоль), триацетоксиборгидрид натрия (1,681 г, 7,932 ммоль) и формальдегид (0,357 г, 11,898 ммоль) растворяют в дихлорметане (40 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,362 г, 17,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,83 (м, 2H), 7,42 (т, J=7,7 Гц, 1H), 7,18 (с, 2H), 7,07 (с, 1H), 6,90 (т, J=51,6 Гц, 1H), 5,15 (с, 2H), 4,45-4,36 (м, 1H), 3,02 (д, J=11,8 Гц, 2H), 2,48-2,38 (м, 2H), 2,34 (с, 3H), 2,15 (тд, J=11,9, 2,0 Гц, 2H), 1,90-1,80 (м, 2H); МСНР (ЭР) m/z 538,1 (M+ + 1).
Пример 120: Синтез соединения 120, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(3-фторфенил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 120
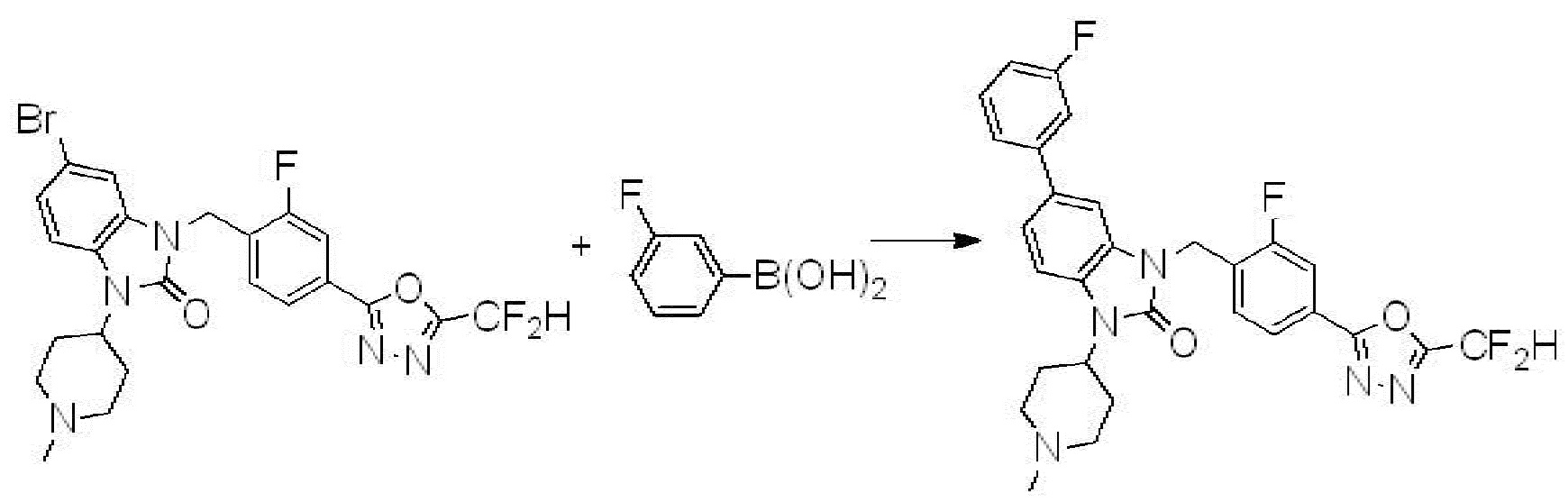
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,280 ммоль), полученный на стадии 5 соединения 119, (3-фторфенил)бороновую кислоту (0,047 г, 0,336 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) и карбонат цезия (0,182 г, 0,559 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,074 г, 48,2%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,82 (м, 2H), 7,48 (т, J=7,8 Гц, 1H), 7,40-7,33 (м, 2H), 7,28-7,26 (м, 2H), 7,19 (дт, J=10,2, 2,1 Гц, 1H), 7,15 (д, J=1,6 Гц, 1H), 7,01-6,97 (м, 1H), 6,90 (т, J=51,7 Гц, 1H), 5,24 (с, 2H), 4,50-4,42 (м, 1H), 3,06 (д, J=11,7 Гц, 2H), 2,59-2,48 (м, 2H), 2,39 (с, 3H), 2,23 (т, J=11,5 Гц, 2H), 1,87 (дд, J=11,9, 2,5 Гц, 2H); МСНР (ЭР) m/z 551,9 (M+ + 1).
Пример 121: Синтез соединения 121, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(2,5-дифторфенил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 121
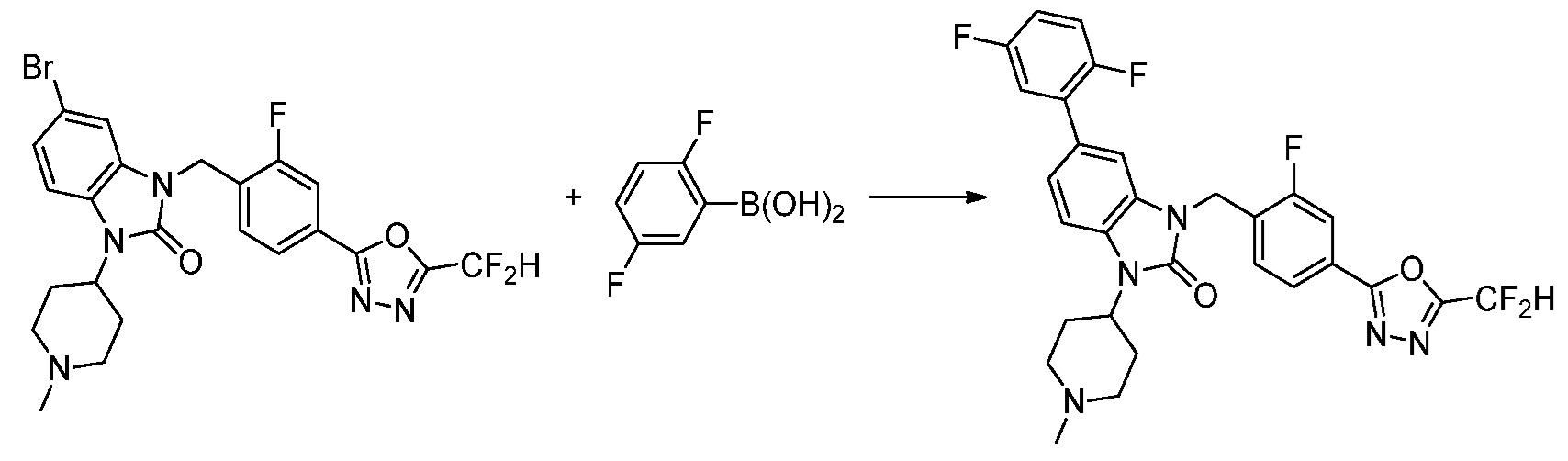
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,224 ммоль), полученный на стадии 5 соединения 119, (2,5-дифторфенил)бороновую кислоту (0,042 г, 0,268 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,007 г, 0,011 ммоль) и карбонат цезия (0,146 г, 0,447 ммоль) смешивают в 1,4-диоксане (1,7 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,032 г, 24,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,86-7,82 (м, 2H), 7,47 (т, J=7,8 Гц, 1H), 7,40 (д, J=8,3 Гц, 1H), 7,22 (д, J=8,3 Гц, 1H), 7,14 (с, 1H), 7,10-7,05 (м, 2H), 6,98-6,93 (м, 1H), 6,89 (т, J=51,7 Гц, 1H), 5,23 (с, 2H), 4,51-4,42 (м, 1H), 3,05 (д, J=11,6 Гц, 2H), 2,56-2,46 (м, 2H), 2,37 (с, 3H), 2,19 (т, J=11,2 Гц, 2H), 1,87 (дд, J=12,0, 2,2 Гц, 2H); МСНР (ЭР) m/z 570,0 (M+ + 1).
Пример 122: Синтез соединения 122, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-метилпиперидин-4-ил)-5-(4-(трифторметил)фенил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 122
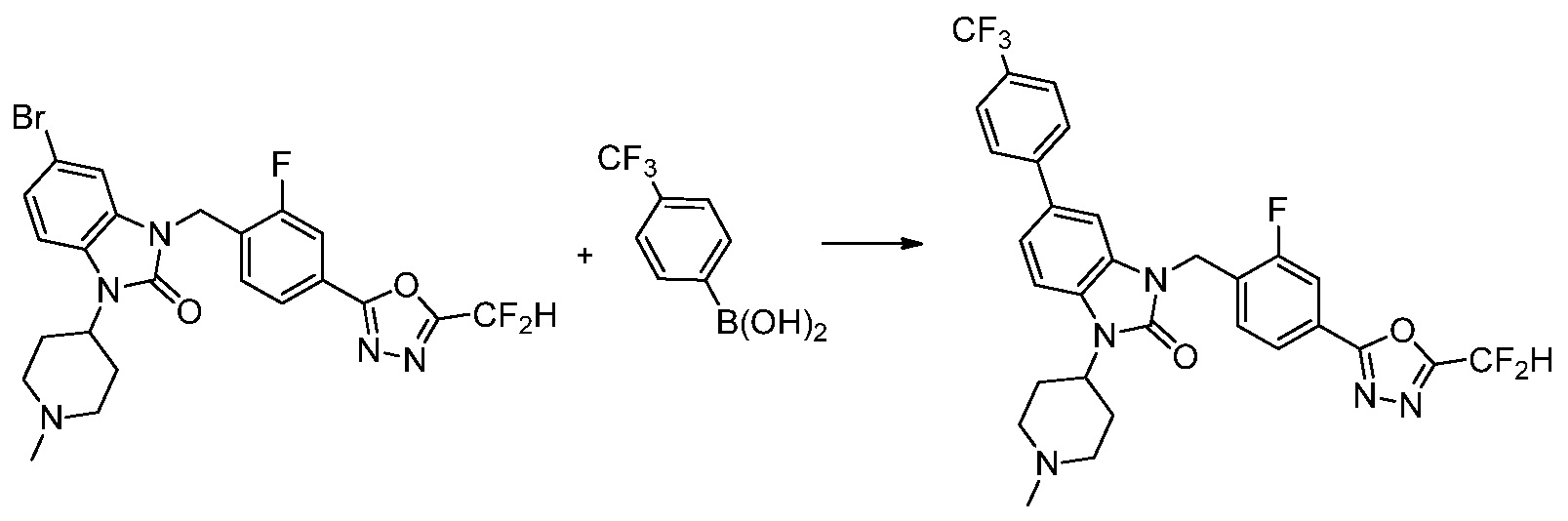
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,224 ммоль), полученный на стадии 5 соединения 119, (4-(трифторметил)фенил)бороновую кислоту (0,051 г, 0,268 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,007 г, 0,011 ммоль) и карбонат цезия (0,146 г, 0,447 ммоль) смешивают в 1,4-диоксане (1,7 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,033 г, 24,6%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,14 (д, J=7,6 Гц, 1H), 7,87-7,84 (м, 2H), 7,67-7,58 (м, 4H), 7,50 (т, J=7,8 Гц, 1H), 7,30 (дд, J=8,3, 1,3 Гц, 1H), 7,18 (с, 1H), 6,89 (т, J=51,7 Гц, 1H), 5,24 (с, 2H), 4,48-4,41 (м, 1H), 3,12 (д, J=11,3 Гц, 2H), 2,64-2,61 (м, 2H), 2,61-2,20 (м, 5H), 1,89 (д, J=10,2 Гц, 2H); МСНР (ЭР) m/z 602,1 (M+ + 1).
Пример 123: Синтез соединения 123, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(2,5-дифторфенил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 123
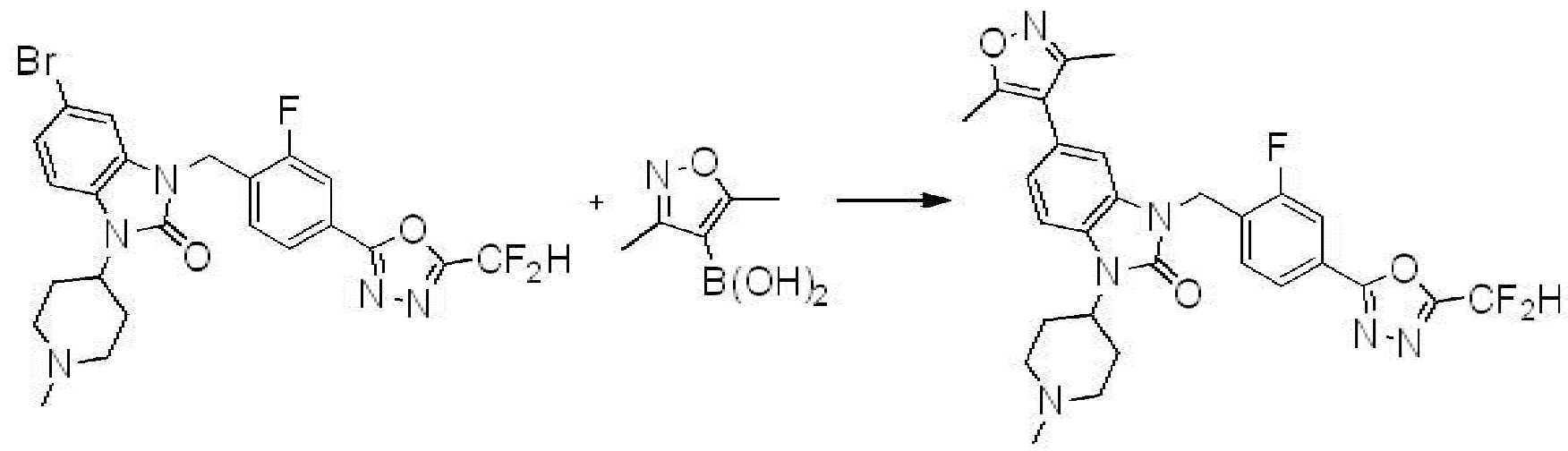
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,224 ммоль), полученный на стадии 5 соединения 119, (2,5-дифторфенил)бороновую кислоту (0,042 г, 0,268 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,007 г, 0,011 ммоль) и карбонат цезия (0,146 г, 0,447 ммоль) смешивают в 1,4-диоксане (1,7 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,032 г, 24,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,86-7,82 (м, 2H), 7,55 (т, J=7,7 Гц, 1H), 7,37 (д, J=8,2 Гц, 1H), 6,92 (дд, J=8,2, 1,6 Гц, 1H), 6,90 (т, J=51,6 Гц, 1H), 6,85 (д, J=1,3 Гц, 1H), 5,19 (с, 2H), 4,50-4,41 (м, 1H), 3,05 (д, J=11,6 Гц, 2H), 2,54-2,46 (м, 2H), 2,36 (с, 3H), 2,33 (с, 3H), 2,22-2,16 (м, 5H), 1,87 (дд, J=12,0, 2,2 Гц, 2H); МСНР (ЭР) m/z 554,2 (M+ + 2).
Пример 124: Синтез соединения 124, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(5-метилфуран-2-ил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 124
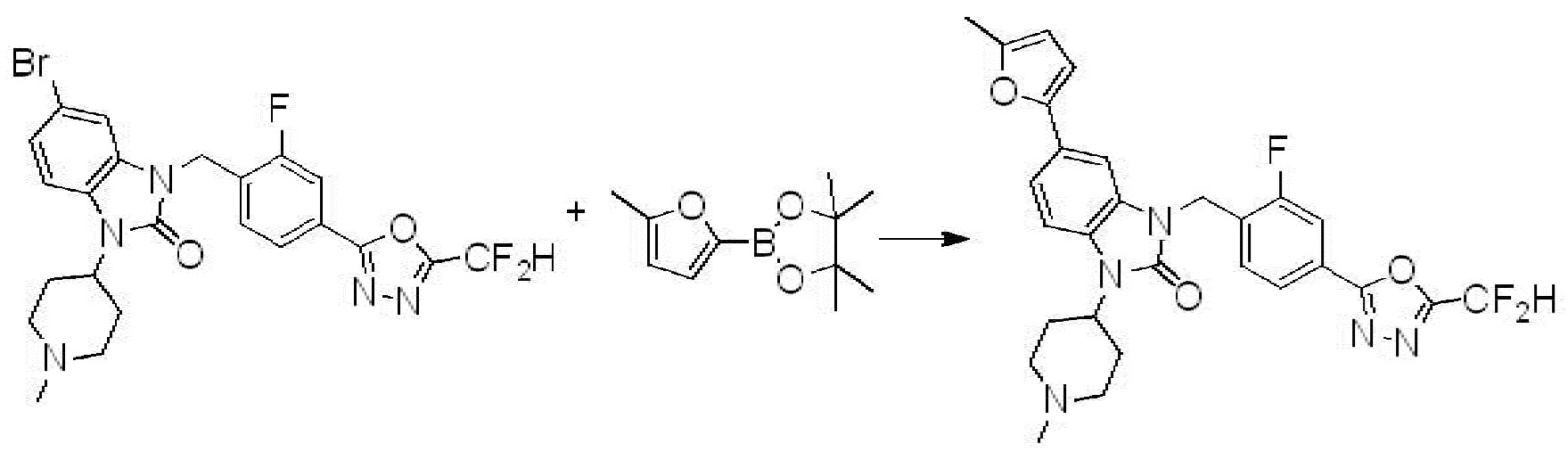
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,224 ммоль), полученный на стадии 5 соединения 119, 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан (0,056 г, 0,268 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,007 г, 0,011 ммоль) и карбонат цезия (0,146 г, 0,447 ммоль) смешивают в 1,4-диоксане (1,7 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,030 г, 25,2%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,80 (м, 2H), 7,41 (т, J=7,6 Гц, 1H), 7,34-7,28 (м, 2H), 7,19 (д, J=1,0 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 6,41 (д, J=3,2 Гц, 1H), 6,01-6,00 (м, 1H), 5,28 (с, 2H), 4,47-4,39 (м, 1H), 3,04 (д, J=11,7 Гц, 2H), 2,54-2,43 (м, 2H), 2,36 (с, 3H), 2,34 (с, 3H), 2,20-2,15 (м, 2H), 1,86 (дд, J=12,1, 2,2 Гц, 2H); МСНР (ЭР) m/z 538,0 (M+ + 1).
Пример 125: Синтез соединения 125, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-метилпиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 125
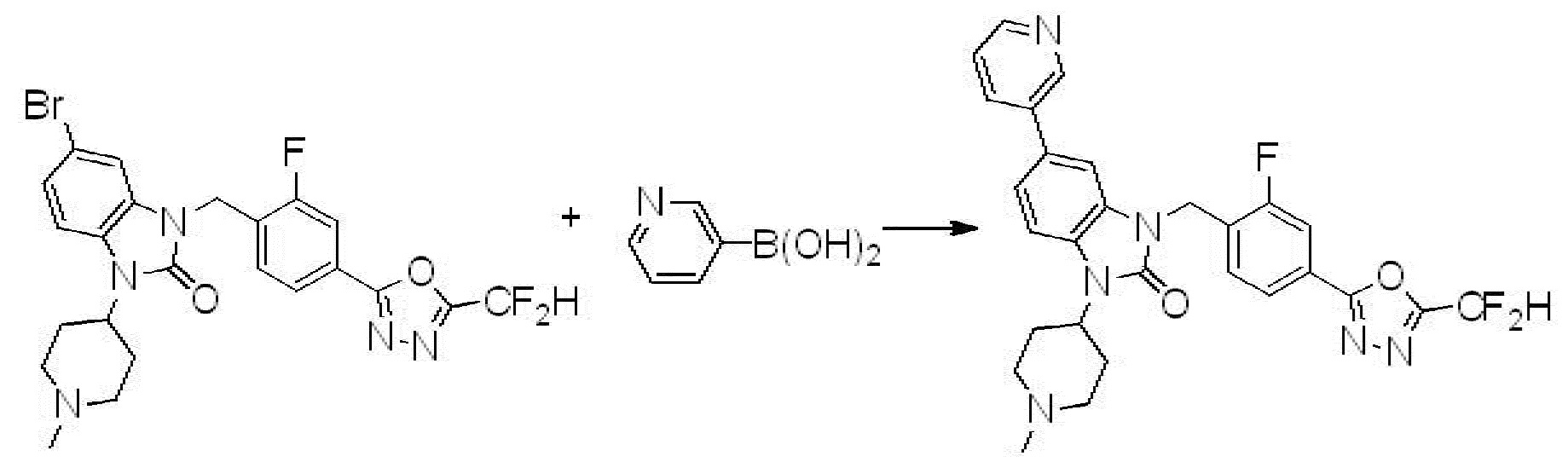
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,280 ммоль), полученный на стадии 5 соединения 119, пиридин-3-илбороновую кислоту (0,041 г, 0,336 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) и карбонат цезия (0,182 г, 0,559 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,077 г, 51,6%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,75-8,74 (м, 1H), 8,53 (дд, J=4,8, 1,6 Гц, 1H), 7,85-7,81 (м, 2H), 7,77 (дт, J=8,0, 1,6 Гц, 1H), 7,48 (т, J=7,8 Гц, 1H), 7,42 (д, J=8,3 Гц, 1H), 7,32 (ддд, J=7,9, 4,8, 0,6 Гц, 1H), 7,27-7,14 (м, 1H), 7,14 (д, J=1,6 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 5,23 (с, 2H), 4,49-4,41 (м, 1H), 3,03 (д, J=11,6 Гц, 2H), 2,55-2,45 (м, 2H), 2,35 (с, 3H), 2,20-2,15 (м, 2H), 1,86 (дд, J=12,0, 2,3 Гц, 2H); МСНР (ЭР) m/z 535,3 (M+ + 1).
Пример 126: Синтез соединения 126, 5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
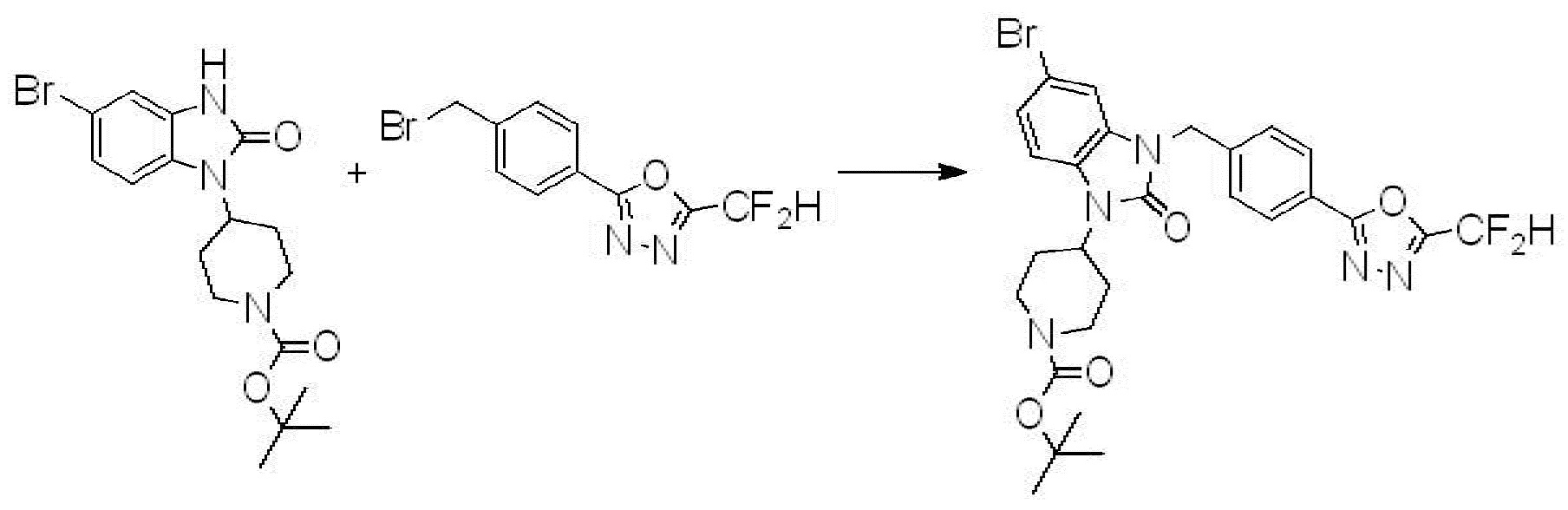
Трет-бутил 4-(5-бром-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,300 г, 5,804 ммоль), полученный на стадии 3 соединения 119 растворяют в N,N-диметилформамиде (50 мл) при 0°C, после чего гидрид натрия (60,00%, 0,348 г, 8,706 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (2,181 г, 7,545 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (3,390 г, 96,6%) в виде коричневого твердого вещества.
[Стадия 2] Синтез 5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
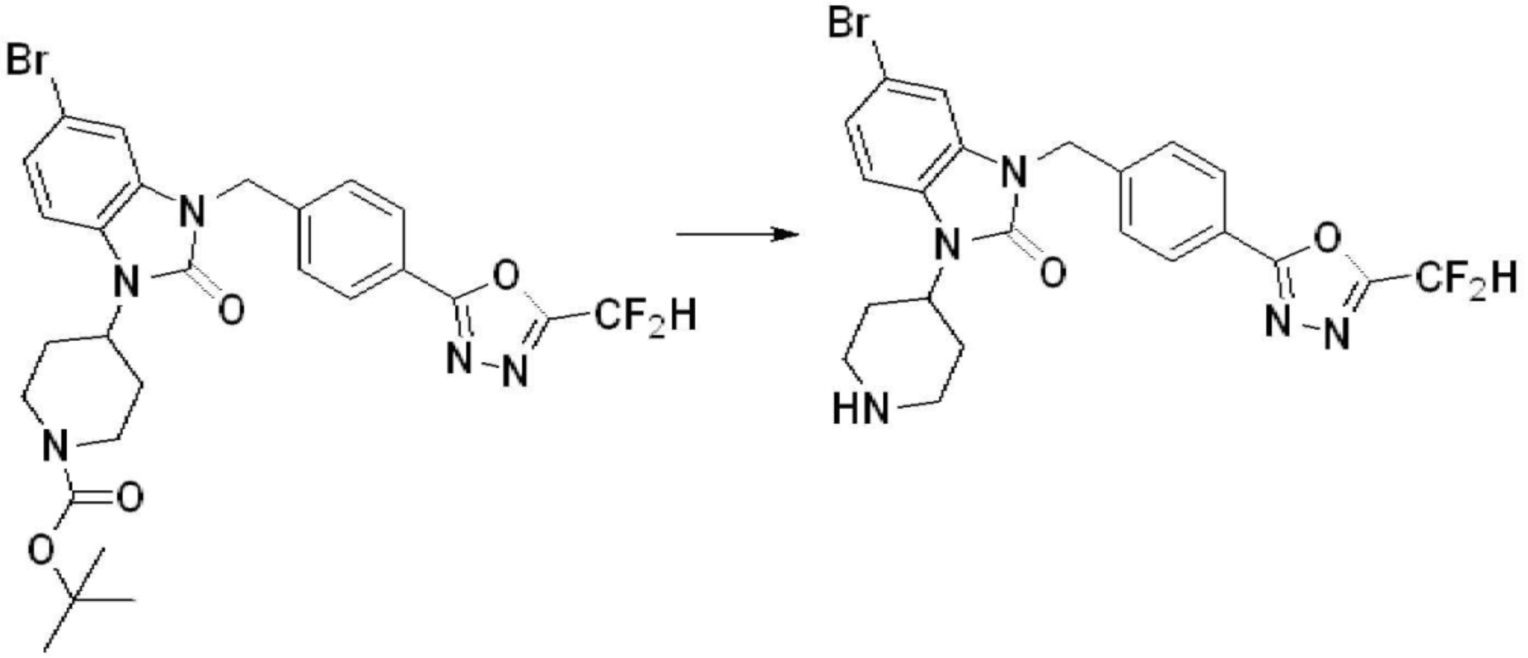
Трет-бутил 4-(5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (3,390 г, 5,608 ммоль), полученный на стадии 1 и трифторуксусную кислоту (4,295 мл, 56,084 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=80-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0 to 80%) и концентрируют с получением указанного в заголовке соединения (2,154 г, 76,2%) в виде коричневого твердого вещества.
[Стадия 3] Синтез соединения 126
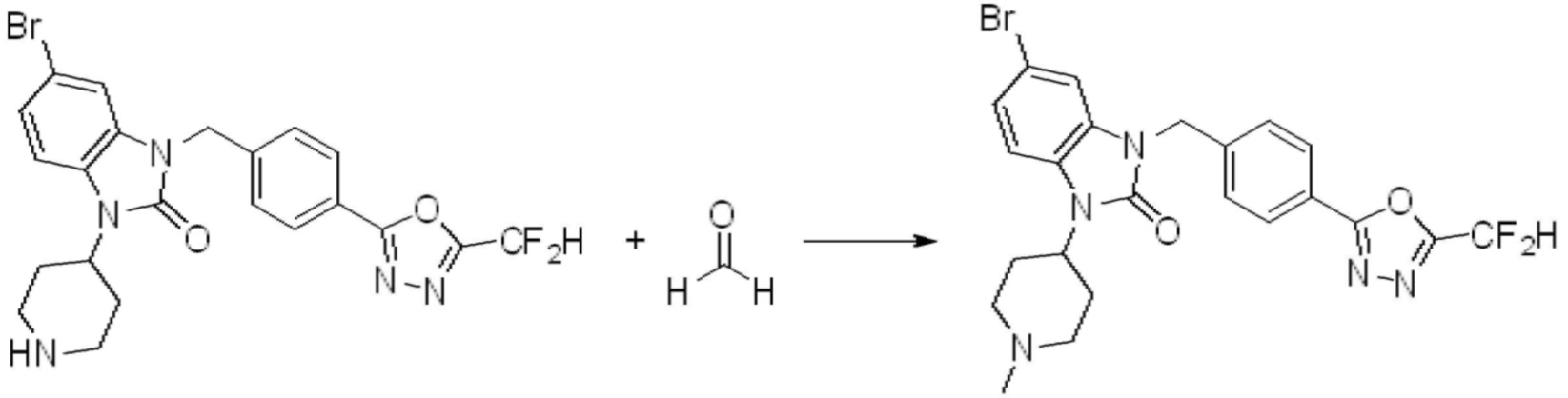
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (1,320 г, 2,616 ммоль), полученный на стадии 2, триацетоксиборгидрид натрия (1,109 г, 5,233 ммоль) и формальдегид (0,236 г, 7,849 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,760 г, 56,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,09 (д, J=8,4 Гц, 2H), 7,45 (д, J=8,4 Гц, 2H), 7,02-7,15 (м, 2H), 6,96 (д, J=1,4 Гц, 1H), 6,90 (т, J=51,8 Гц, 1H), 5,11 (с, 2H), 4,46-4,38 (м, 1H), 3,02 (д, J=11,8 Гц, 2H), 2,49-2,39 (м, 2H), 2,35 (с, 3H), 2,19-2,13 (м, 2H), 1,84 (дд, J=11,9, 2,4 Гц, 2H); МСНР (ЭР) m/z 518,2 (M+ + 1).
Пример 127: Синтез соединения 127, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 127
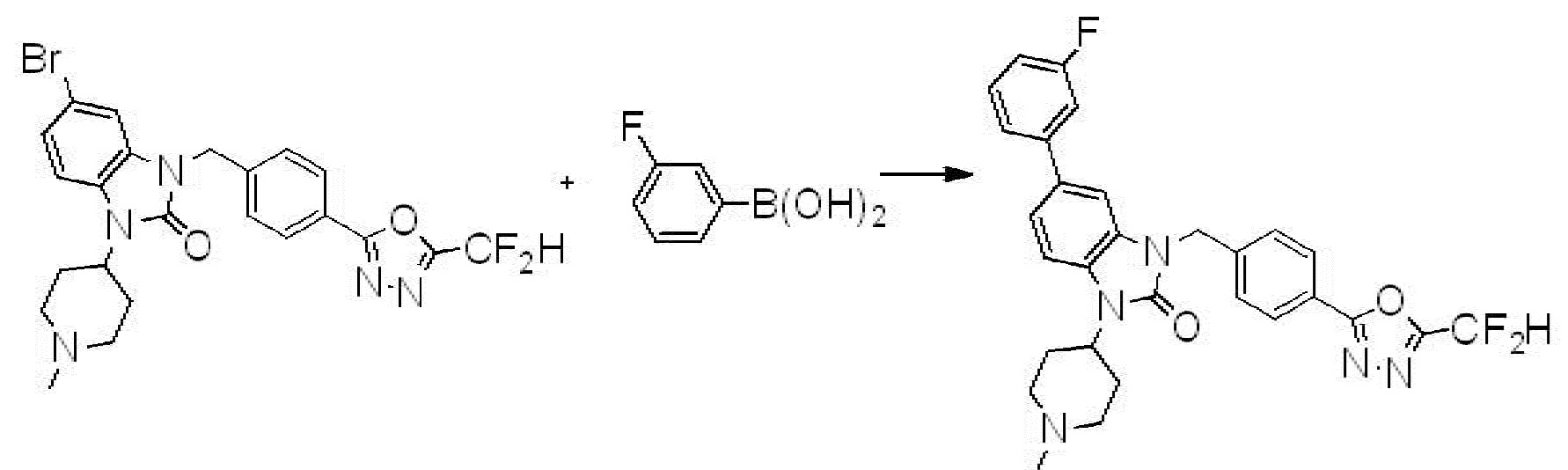
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,231 ммоль), полученный на стадии 3 соединения 126, пиридин-3-илбороновую кислоту (0,034 г, 0,278 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) и карбонат цезия (0,151 г, 0,463 ммоль) смешивают в 1,4-диоксане (1,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,060 г, 50,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,06 (дд, J=6,7, 1,8 Гц, 2H), 7,49 (д, J=8,5 Гц, 2H), 7,40-7,12 (м, 5H), 7,01 (д, J=1,7 Гц, 1H), 7,00-6,95 (м, 1H), 6,88 (т, J=51,5 Гц, 1H), 5,19 (с, 2H), 4,51-4,43 (м, 1H), 3,05 (д, J=11,6 Гц, 2H), 2,58-2,48 (м, 2H), 2,38 (с, 3H), 2,22 (т, J=11,4 Гц, 2H), 1,88 (дд, J=11,9, 2,2 Гц, 2H); МСНР (ЭР) m/z 535,3 (M+ + 1).
Пример 128: Синтез соединения 128, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-(3-фторфенил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 128
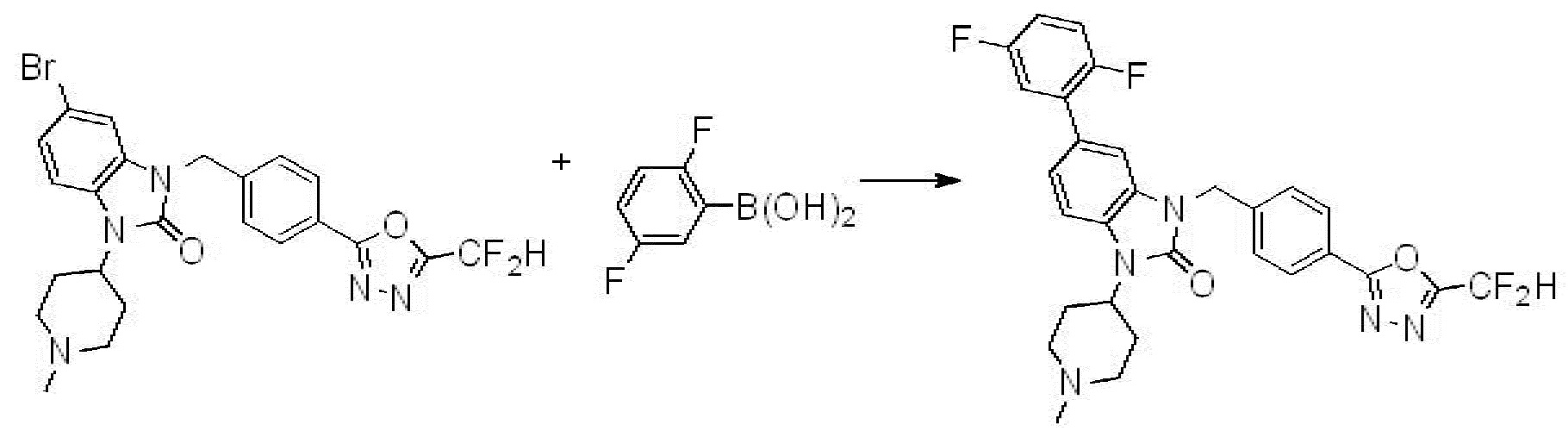
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,231 ммоль), полученный на стадии 3 соединения 126, (3-фторфенил)бороновую кислоту (0,039 г, 0,278 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) и карбонат цезия (0,151 г, 0,463 ммоль) смешивают в 1,4-диоксане (1,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,081 г, 65,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,05 (дд, J=6,7, 1,8 Гц, 2H), 7,48 (д, J=8,5 Гц, 2H), 7,39 (д, J=8,3 Гц, 1H), 7,19 (дт, J=8,3, 1,3 Гц, 1H), 7,07-6,75 (м, 5H), 5,17 (с, 2H), 4,50-4,42 (м, 1H), 3,03 (д, J=11,6 Гц, 2H), 2,56-2,45 (м, 2H), 2,35 (с, 3H), 2,20-2,14 (м, 2H), 1,87 (дд, J=11,9, 2,3 Гц, 2H); МСНР (ЭР) m/z 552,1 (M+ + 1).
Пример 129: Синтез соединения 129, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-(2,5-дифторфенил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 129
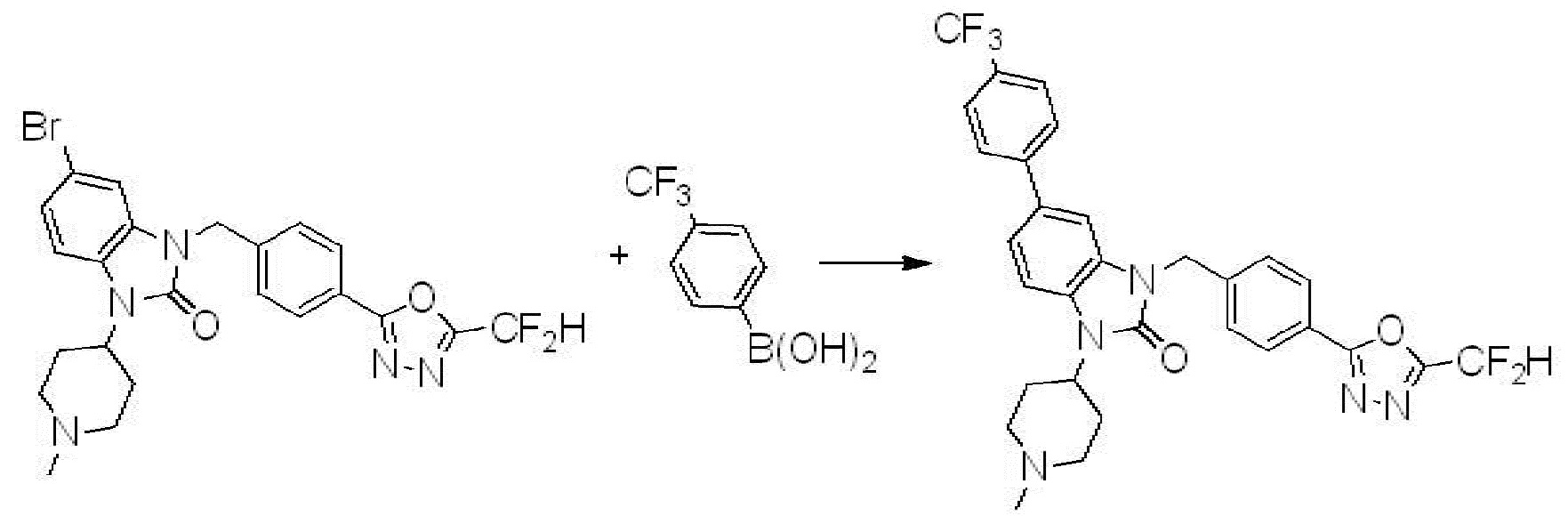
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,231 ммоль), полученный на стадии 3 соединения 126, (2,5-дифторфенил)бороновую кислоту (0,044 г, 0,278 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) и карбонат цезия (0,151 г, 0,463 ммоль) смешивают в 1,4-диоксане (1,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,096 г, 75,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,06 (д, J=8,4 Гц, 2H), 7,63 (д, J=8,2 Гц, 2H), 7,54 (д, J=8,1 Гц, 2H), 7,49 (д, J=8,4 Гц, 2H), 7,42 (д, J=8,3 Гц, 1H), 7,27 (дд, J=8,6, 2,0 Гц, 1H), 7,03 (д, J=1,6 Гц, 1H), 6,88 (т, J=51,7 Гц, 1H), 5,20 (с, 2H), 4,52-4,44 (м, 1H), 3,07 (д, J=11,7 Гц, 2H), 2,61-2,51 (м, 2H), 2,40 (с, 3H), 2,26 (т, J=11,7 Гц, 2H), 1,90-1,87 (м, 2H); МСНР (ЭР) m/z 584,0 (M+ + 1).
Пример 130: Синтез соединения 130, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-5-(4-(трифторметил)фенил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 130
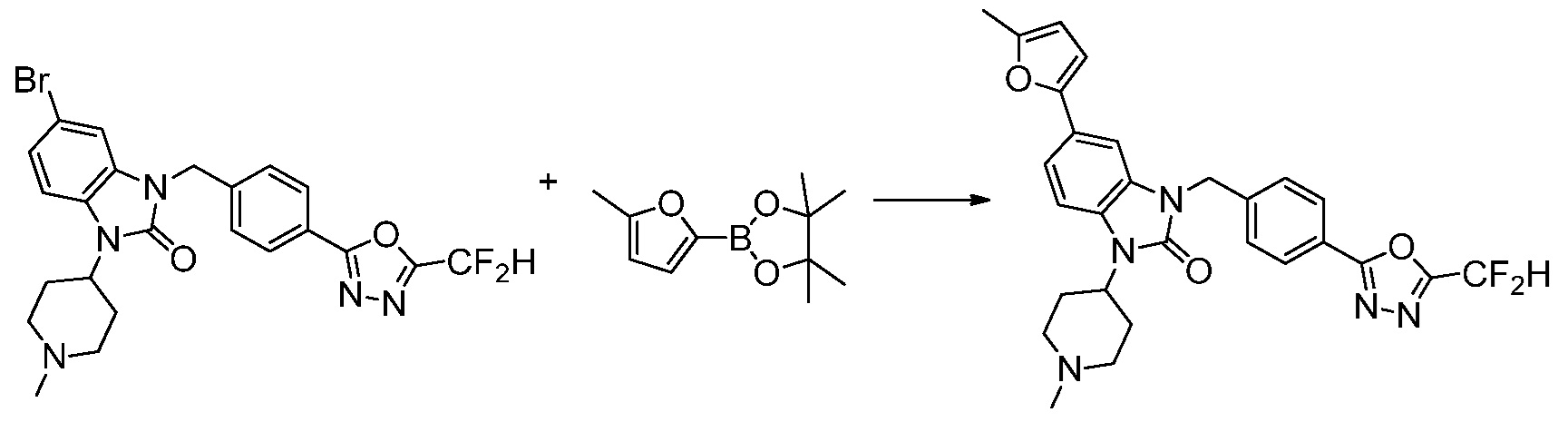
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,231 ммоль), полученный на стадии 3 соединения 126, (4-(трифторметил)фенил)бороновую кислоту (0,053 г, 0,278 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) и карбонат цезия (0,151 г, 0,463 ммоль) смешивают в 1,4-диоксане (1,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,081 г, 60,0%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,05 (д, J=8,4 Гц, 2H), 7,47 (д, J=8,4 Гц, 2H), 7,32-7,26 (м, 2H), 7,09 (с, 1H), 6,88 (т, J=51,7 Гц, 1H), 6,37 (д, J=3,2 Гц, 1H), 5,98 (дд, J=3,2, 1,0 Гц, 1H), 5,16 (с, 2H), 4,46-4,38 (м, 1H), 3,02 (д, J=11,6 Гц, 2H), 2,53-2,43 (м, 2H), 2,34 (с, 3H), 2,31 (с, 3H), 2,16-2,13 (м, 2H), 1,85 (дд, J=12,0, 2,2 Гц, 2H); МСНР (ЭР) m/z 519,9 (M+ + 1).
Пример 131: Синтез соединения 131, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-(3,5-диметилизооксазол-4-ил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 131
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,231 ммоль), полученный на стадии 3 соединения 126, (3,5-диметилизооксазол-4-ил)бороновую кислоту (0,039 г, 0,278 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) и карбонат цезия (0,151 г, 0,463 ммоль) смешивают в 1,4-диоксане (1,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,067 г, 54,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,05 (д, J=8,4 Гц, 2H), 7,47 (д, J=8,4 Гц, 2H), 7,36 (д, J=8,2 Гц, 1H), 7,02-6,76 (м, 2H), 6,66 (д, J=8,7 Гц, 1H), 5,15 (с, 2H), 4,49-4,41 (м, 1H), 3,03 (д, J=11,5 Гц, 2H), 2,54-2,44 (м, 2H), 2,35 (с, 3H), 2,25 (с, 3H), 2,20-2,14 (м, 2H), 2,11 (с, 3H), 1,87 (дд, J=11,9, 2,2 Гц, 2H); МСНР (ЭР) m/z 536,3 (M+ + 1).
Пример 132: Синтез соединения 132, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 132
5-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,231 ммоль), полученный на стадии 3 соединения 126, пиридин-3-илбороновую кислоту (0,034 г, 0,278 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) и карбонат цезия (0,151 г, 0,463 ммоль) смешивают в 1,4-диоксане (1,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,060 г, 50,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,70 (д, J=1,8 Гц, 1H), 8,52 (дд, J=4,8, 1,6 Гц, 1H), 8,08-8,05 (м, 2H), 7,74-7,71 (м, 1H), 7,48 (д, J=8,6 Гц, 2H), 7,42 (д, J=8,2 Гц, 1H), 7,31-7,26 (м, 1H), 7,26-7,23 (м, 1H), 7,00 (д, J=1,6 Гц, 1H), 6,88 (т, J=51,7 Гц, 1H), 5,20 (с, 2H), 4,51-4,43 (м, 1H), 3,04 (д, J=11,6 Гц, 2H), 2,56-2,46 (м, 2H), 2,36 (с, 3H), 2,21-2,16 (м, 2H), 1,88 (дд, J=11,9, 2,3 Гц, 2H); МСНР (ЭР) m/z 517,3(M+ + 1).
Пример 133: Синтез соединения 133, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(3-фторфенил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((5-бром-2-нитрофенил)амино)пиперидин-1-карбоксилата
4-бром-2-фтор-1-нитробензол (5,000 г, 22,727 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (4,552 г, 22,727 ммоль) и карбонат калия (6,282 г, 45,455 ммоль) растворяют в N,N-диметилформамиде (150 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 4 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением указанного в заголовке соединения (8,800 г, 96,7%) в виде желтого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-((2-амино-5-бромфенил)амино)пиперидин-1-карбоксилата
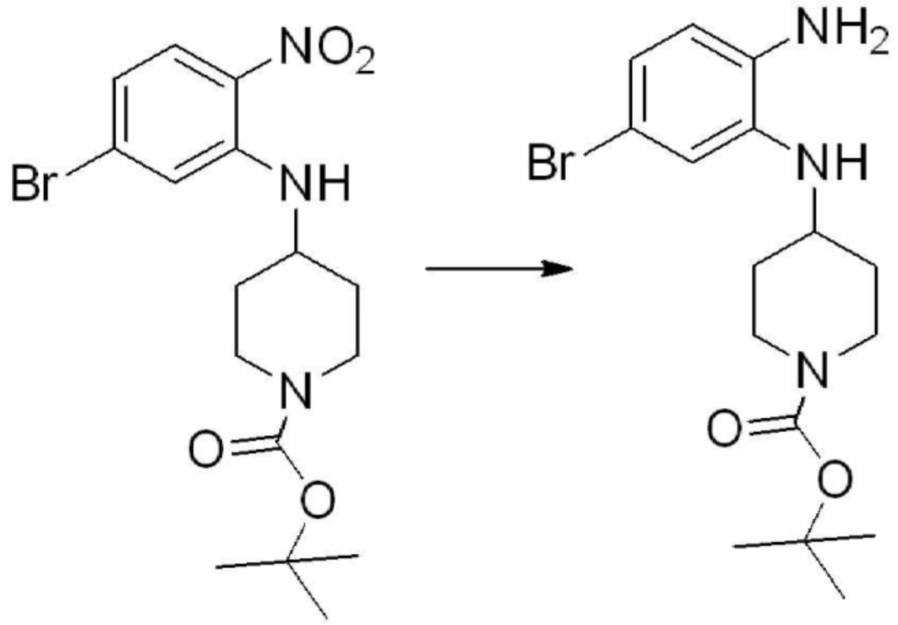
Трет-бутил 4-((5-бром-2-нитрофенил)амино)пиперидин-1-карбоксилат (8,800 г, 21,985 ммоль), полученный на стадии 1 растворяют в этанол (200 мл) и перемешивают при комнатной температуре, после чего никель Ренея (800 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (7,440 г, 91,4%, коричневое твердое вещество).
[Стадия 3] Синтез трет-бутил 4-(6-бром-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
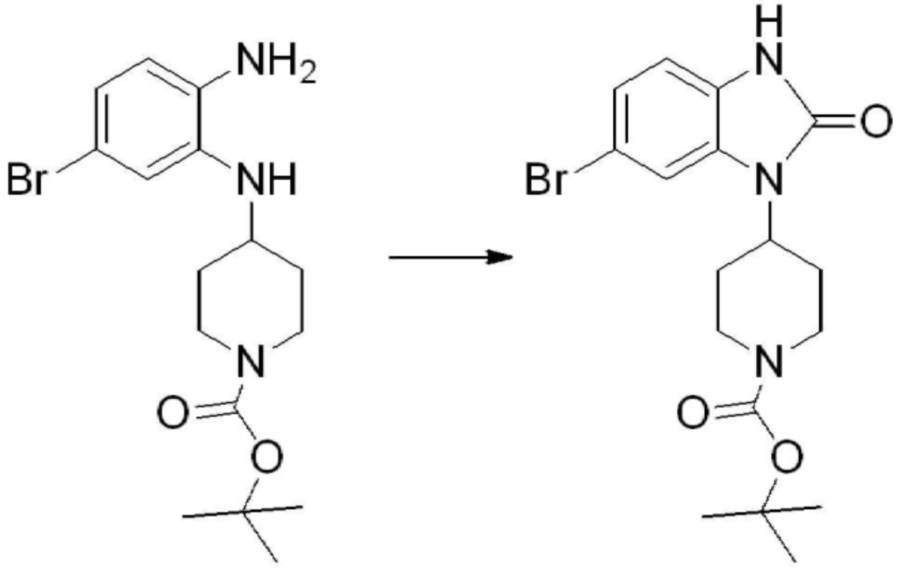
Трет-бутил 4-((2-амино-5-бромфенил)амино)пиперидин-1-карбоксилат (7,440 г, 20,092 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 3,584 г, 22,102 ммоль) смешивают в дихлорметан (200 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-40%) и концентрируют с получением указанного в заголовке соединения (5,460 г, 68,6%) в виде светло-коричневого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
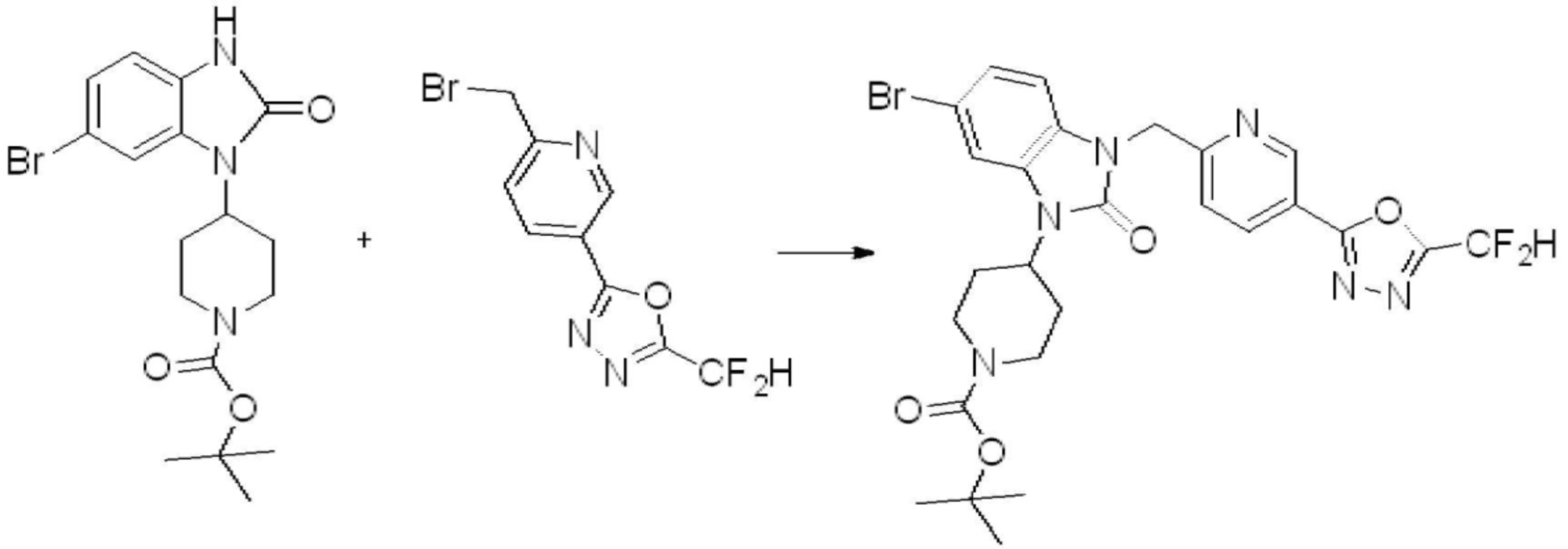
Трет-бутил 4-(6-бром-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (3,000 г, 7,570 ммоль), полученный на стадии 3, и гидрид натрия (60,00%, 0,333 г, 8,327 ммоль) растворяют в N,N-диметилформамиде (100 мл) при 0°C, после чего 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,415 г, 8,327 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 2 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (3,450 г, 75,3%) в виде белого твердого вещества.
[Стадия 5] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
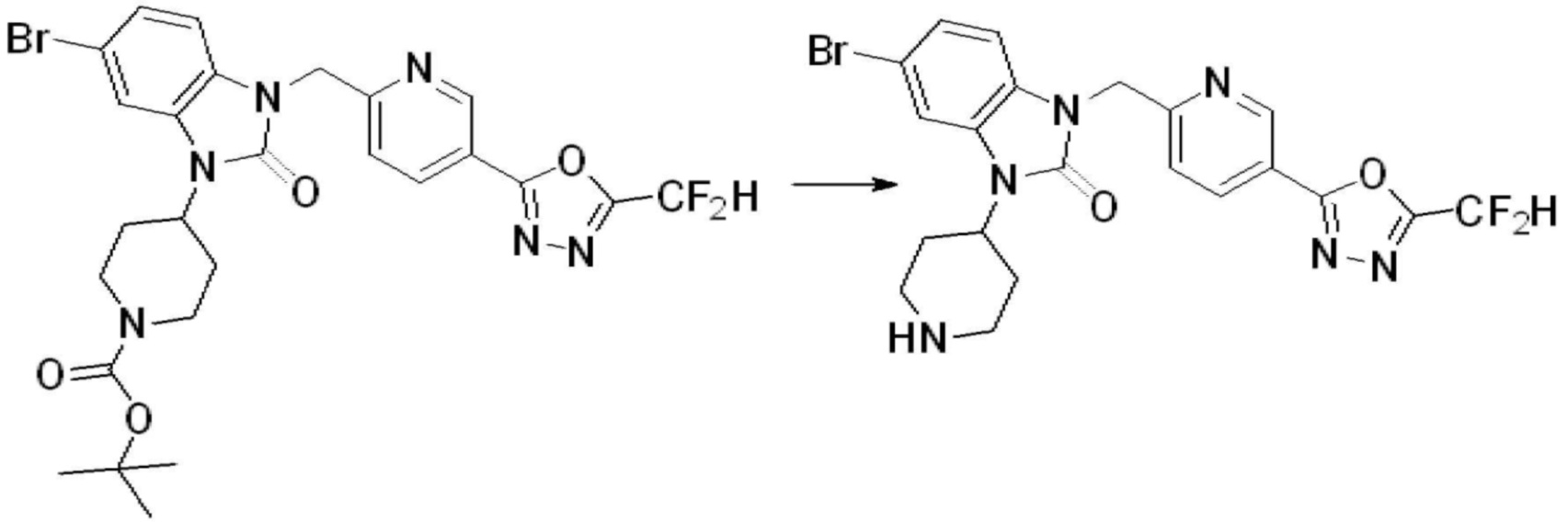
Трет-бутил 4-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (3,450 г, 5,698 ммоль), полученный на стадии 4, и трифторуксусную кислоту (2,618 мл, 34,190 ммоль) растворяют в дихлорметане (40 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 7 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (2,300 г, 79,9%, светло-желтое твердое вещество).
[Стадия 6] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (2,300 г, 4,552 ммоль), полученный на стадии 5, и триацетоксиборгидрид натрия (1,929 г, 9,103 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего формальдегид (37,00% раствор, 0,513 мл, 6,827 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (2,000 г, 84,6%) в виде светло-желтого твердого вещества.
[Стадия 7] Синтез соединения 133
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,289 ммоль), полученный на стадии 6, (3-фторфенил)бороновую кислоту (0,048 г, 0,347 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,009 г, 0,014 ммоль) и карбонат цезия (0,282 г, 0,866 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,018 г, 11,7%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=2,1 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,51 (с, 1H), 7,45 (д, J=8,3 Гц, 1H), 7,43-7,27 (м, 1H), 7,34 (д, J=7,6 Гц, 1H), 7,25-7,23 (м, 2H), 7,08-6,82 (м, 3H), 5,34 (с, 2H), 4,54-4,48 (м, 1H), 3,10 (д, J=11,1 Гц, 2H), 2,61-2,58 (м, 2H), 2,40 (с, 3H), 2,25 (т, J=10,6 Гц, 2H), 1,92 (д, J=10,3 Гц, 2H); МСНР (ЭР) m/z 535,3 (M+ + H).
Пример 134: Синтез соединения 134, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(2-фторфенил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 134
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,289 ммоль), полученный на стадии 6 соединения 133, (2-фторфенил)бороновую кислоту (0,048 г, 0,347 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,009 г, 0,014 ммоль) и карбонат цезия (0,282 г, 0,866 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,026 г, 16,8%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=2,1 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,50 (с, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,40 (тд, J=7,7, 1,7 Гц, 1H), 7,33-7,29 (м, 1H), 7,23-7,20 (м, 2H), 7,17-7,13 (м, 1H), 7,08-6,82 (м, 2H), 5,34 (с, 2H), 4,53-4,46 (м, 1H), 3,07 (д, J=11,7 Гц, 2H), 2,57-2,50 (м, 2H), 2,37 (с, 3H), 2,24-2,18 (м, 2H), 1,91 (д, J=10,1 Гц, 2H); МСНР (ЭР) m/z 535,4 (M+ + H).
Пример 135: Синтез соединения 135, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 135
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,289 ммоль), полученный на стадии 6 соединения 133, пиридин-4-илбороновую кислоту (0,043 г, 0,347 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,009 г, 0,014 ммоль) и карбонат цезия (0,282 г, 0,866 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,011 г, 7,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=2,2 Гц, 1H), 8,66 (д, J=6,0 Гц, 2H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,60 (шс, 1H), 7,51-7,46 (м, 3H), 7,34 (дд, J=8,2, 1,4 Гц, 1H), 7,13-6,82 (м, 2H), 5,34 (с, 2H), 4,54-4,53 (м, 1H), 3,13 (д, J=9,4 Гц, 2H), 2,62 (шс, 2H), 2,43 (с, 3H), 2,30-2,29 (м, 2H), 1,94 (д, J=10,6 Гц, 2H); МСНР (ЭР) m/z 518,3 (M+ + H).
Пример 136: Синтез соединения 136, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 136
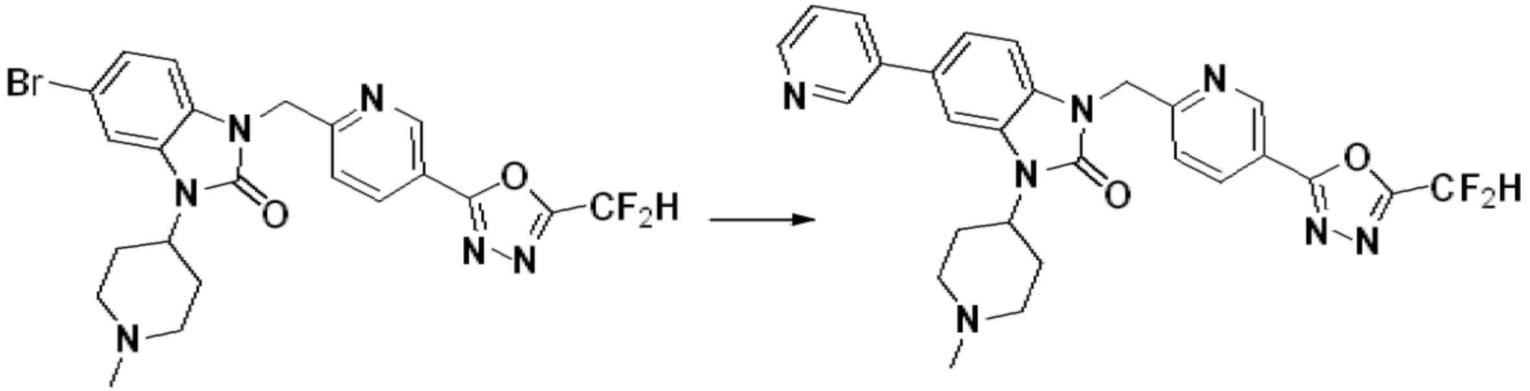
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,289 ммоль), полученный на стадии 6 соединения 133, пиридин-3-илбороновую кислоту (0,043 г, 0,347 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,009 г, 0,014 ммоль) и карбонат цезия (0,282 г, 0,866 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,010 г, 6,7%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,5 Гц, 1H), 8,82 (д, J=1,9 Гц, 1H), 8,60 (дд, J=4,8, 1,4 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,89 (шс, 1H), 7,54 (шс, 1H), 7,46 (д, J=8,2 Гц, 1H), 7,39-7,36 (м, 1H), 7,26 (д, J=8,1 Гц, 1H), 7,11-6,82 (м, 2H), 5,35 (с, 2H), 4,54-4,53 (м, 1H), 3,12-3,11 (м, 2H), 2,62 (шс, 2H), 2,42 (с, 3H), 2,29-2,28 (м, 2H), 1,94 (д, J=10,5 Гц, 2H); МСНР (ЭР) m/z 518,4 (M+ + H).
Пример 137: Синтез соединения 137, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(фуран-3-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 137
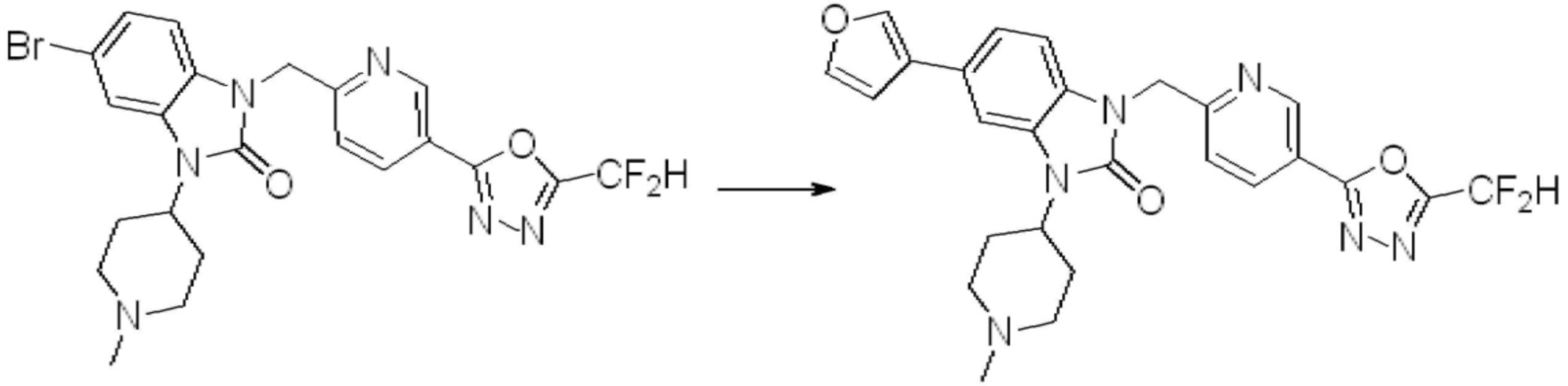
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,289 ммоль), полученный на стадии 6 соединения 133, фуран-3-илбороновую кислоту (0,039 г, 0,347 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,009 г, 0,014 ммоль) и карбонат цезия (0,282 г, 0,866 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,016 г, 10,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=2,1 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,72 (с, 1H), 7,49 (т, J=1,7 Гц, 1H), 7,43-7,41 (м, 2H), 7,17 (дд, J=8,1, 1,4 Гц, 1H), 7,08-6,82 (м, 2H), 6,71 (с, 1H), 5,32 (с, 2H), 4,49-4,46 (м, 1H), 3,10 (д, J=10,6 Гц, 2H), 2,60-2,58 (м, 2H), 2,41 (с, 3H), 2,25 (т, J=10,6 Гц, 2H), 1,91 (д, J=10,2 Гц, 2H); МСНР (ЭР) m/z 507,3 (M+ + H).
Пример 138: Синтез соединения 138, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(фуран-2-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 138
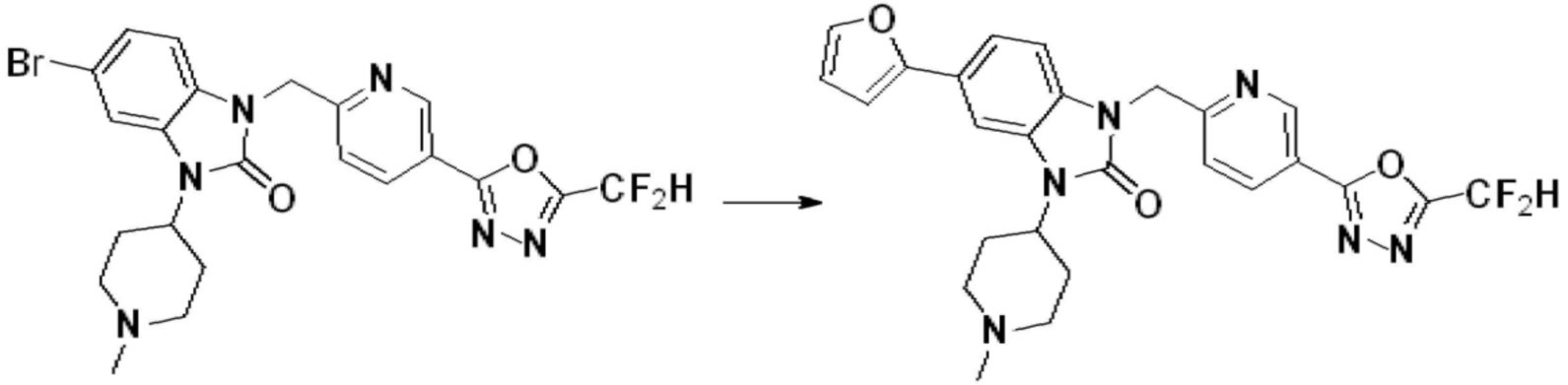
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,289 ммоль), полученный на стадии 6 соединения 133, фуран-2-илбороновую кислоту (0,039 г, 0,347 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,009 г, 0,014 ммоль) и карбонат цезия (0,282 г, 0,866 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,008 г, 5,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 79,32 (д, J=1,5 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,66 (шс, 1H), 7,46 (д, J=1,2 Гц, 1H), 7,42 (д, J=8,2 Гц, 1H), 7,37 (д, J=8,2 Гц, 1H), 7,08-6,82 (м, 2H), 6,64 (шс, 1H), 6,49-6,48 (м, 1H), 5,32 (с, 2H), 4,50-4,49 (м, 1H), 3,12-3,11 (м, 2H), 2,61 (шс, 2H), 2,43 (с, 3H), 2,26-2,25 (м, 2H), 1,92 (д, J=11,2 Гц, 2H); МСНР (ЭР) m/z 507,2 (M+ + H).
Пример 139: Синтез соединения 139, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(6-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
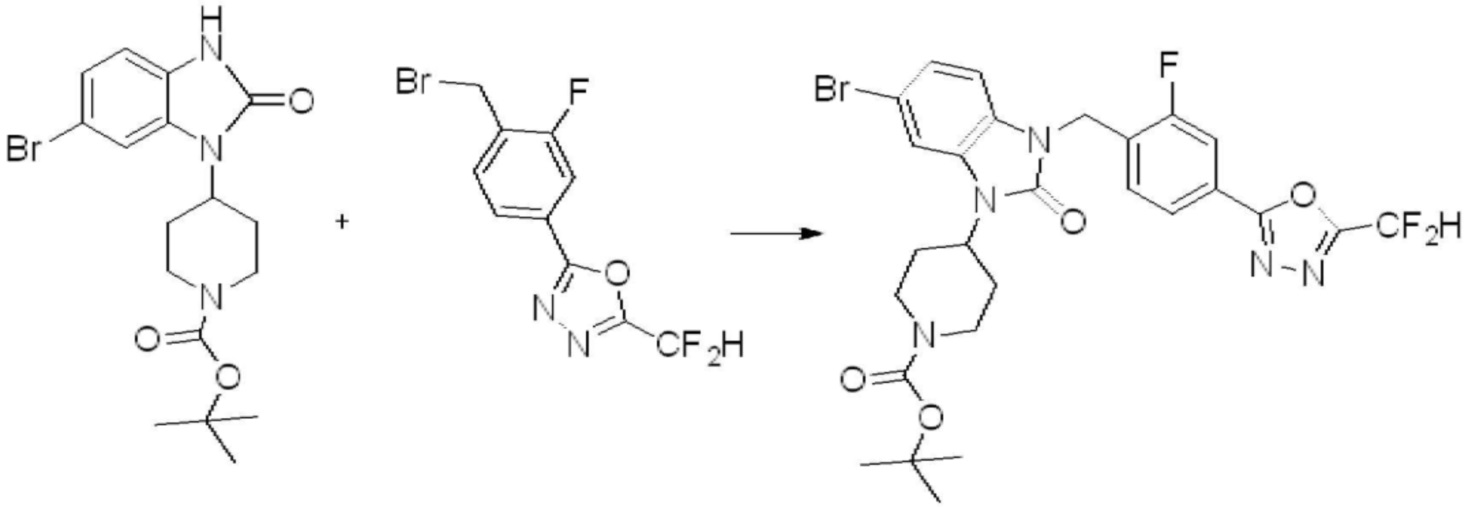
Трет-бутил 4-(6-бром-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,460 г, 6,208 ммоль), полученный на стадии 3 соединения 133, и гидрид натрия (60,00%, 0,273 г, 6,828 ммоль) растворяют в N,N-диметилформамиде (100 мл) при 0°C, после чего 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (2,097 г, 6,828 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 2 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (3,900 г, 100,9%) в виде белого твердого вещества.
[Стадия 2] Синтез 5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
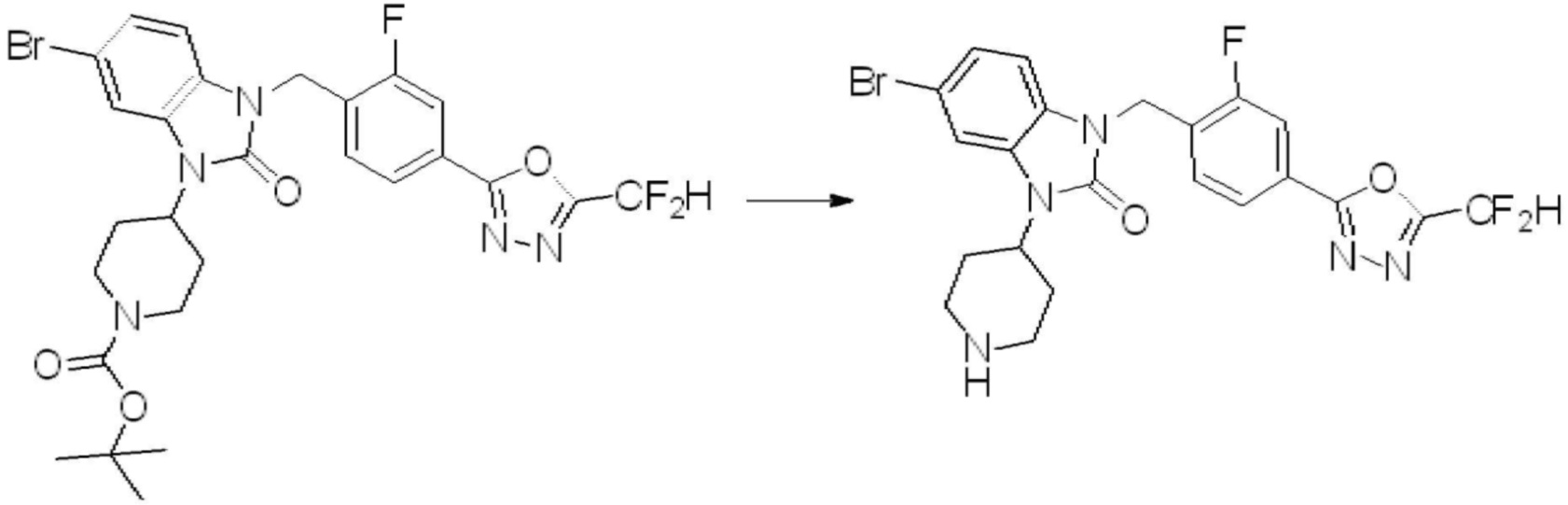
Трет-бутил 4-(6-бром-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (3,900 г, 6,266 ммоль), полученный на стадии 1, и трифторуксусную кислоту (2,399 мл, 31,328 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 6 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (3,080 г, 94,1%, светло-желтое твердое вещество).
[Стадия 3] Синтез 5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
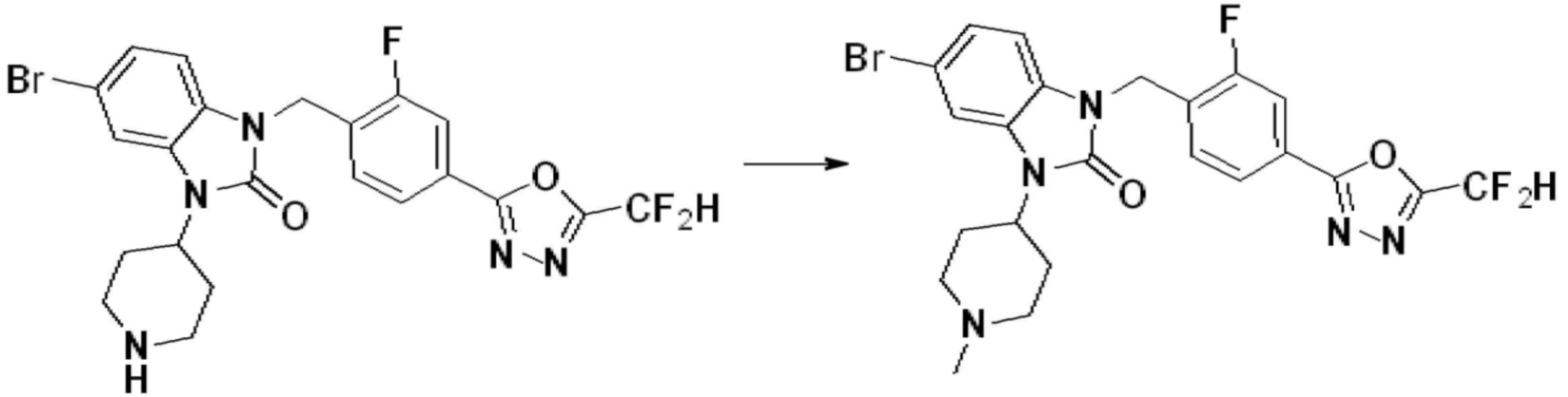
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (3,080 г, 5,897 ммоль), полученный на стадии 2 и триацетоксиборгидрид натрия (2,499 г, 11,793 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего формальдегид (37,00% раствор, 0,665 мл, 8,845 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (2,840 г, 89,8%) в виде светло-желтого твердого вещества.
[Стадия 4] Синтез соединения 139
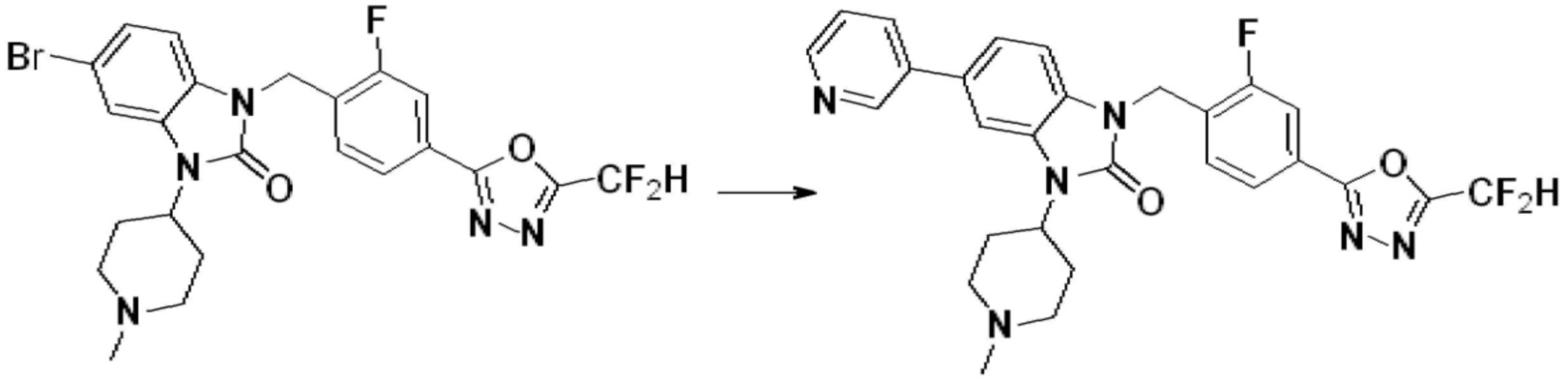
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,280 ммоль), полученный на стадии 3, пиридин-3-илбороновую кислоту (0,047 г, 0,336 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,009 г, 0,014 ммоль) и карбонат цезия (0,273 г, 0,839 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,091 г, 60,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,81 (д, J=1,8 Гц, 1H), 8,60 (дд, J=4,7, 1,3 Гц, 1H), 7,90-7,86 (м, 3H), 7,52-7,48 (м, 2H), 7,39-7,36 (м, 1H), 7,28-7,26 (м, 1H), 7,08-6,80 (м, 2H), 5,26 (с, 2H), 4,54-4,48 (м, 1H), 3,09 (д, J=9,8 Гц, 2H), 2,59-2,57 (м, 2H), 2,40 (с, 3H), 2,26-2,24 (м, 2H), 1,91 (д, J=12,0 Гц, 2H); МСНР (ЭР) m/z 535,3 (M+ + H).
Пример 140: Синтез соединения 140, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(фуран-2-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 140
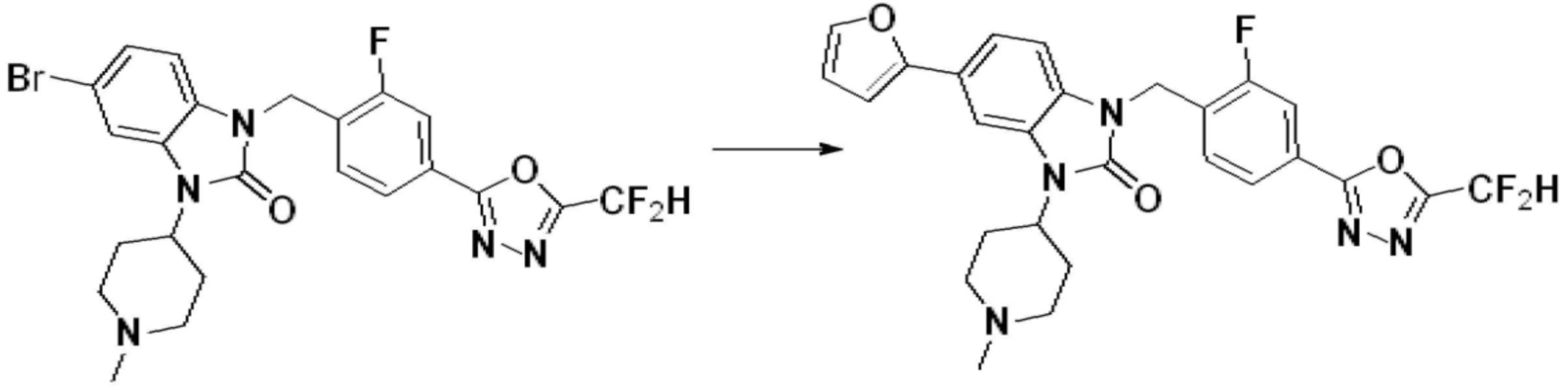
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,280 ммоль), полученный на стадии 3 соединения 139, фуран-2-илбороновую кислоту (0,038 г, 0,336 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,009 г, 0,014 ммоль) и карбонат цезия (0,273 г, 0,839 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,106 г, 72,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,88-7,84 (м, 2H), 7,71 (с, 1H), 7,49-7,45 (м, 2H), 7,40 (с, 1H), 7,18 (дд, J=8,1, 1,5 Гц, 1H), 7,05-6,79 (м, 2H), 6,69 (с, 1H), 5,22 (с, 2H), 4,51-4,43 (м, 1H), 3,08 (д, J=11,4 Гц, 2H), 2,57-2,51 (м, 2H), 2,39 (с, 3H), 2,22 (т, J=11,5 Гц, 2H), 1,89 (д, J=10,0 Гц, 2H); МСНР (ЭР) m/z 524,2 (M+ + H).
Пример 141: Синтез соединения 141, 5-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез метил 6-((6-бром-3-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)никотината
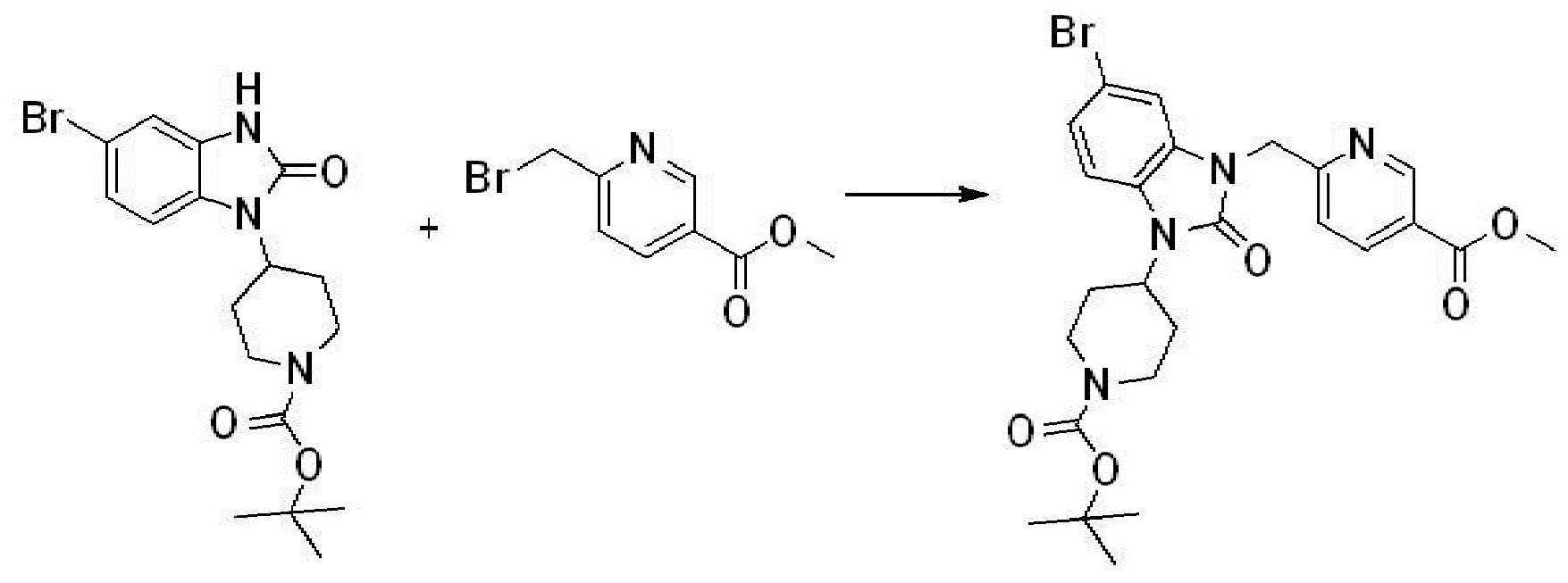
Трет-бутил 4-(5-бром-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,300 г, 5,804 ммоль), полученный на стадии 3 соединения 119, растворяют в N,N-диметилформамиде (60 мл) при 0°C, после чего гидрид натрия (60,00%, 0,348 г, 8,706 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. Метил 6-(бромметил)никотинат (1,736 г, 7,545 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = 0-80%) и концентрируют с получением указанного в заголовке соединения (2,568 г, 81,1%) в виде коричневого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(5-бром-3-((5-(гидразинкарбонил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
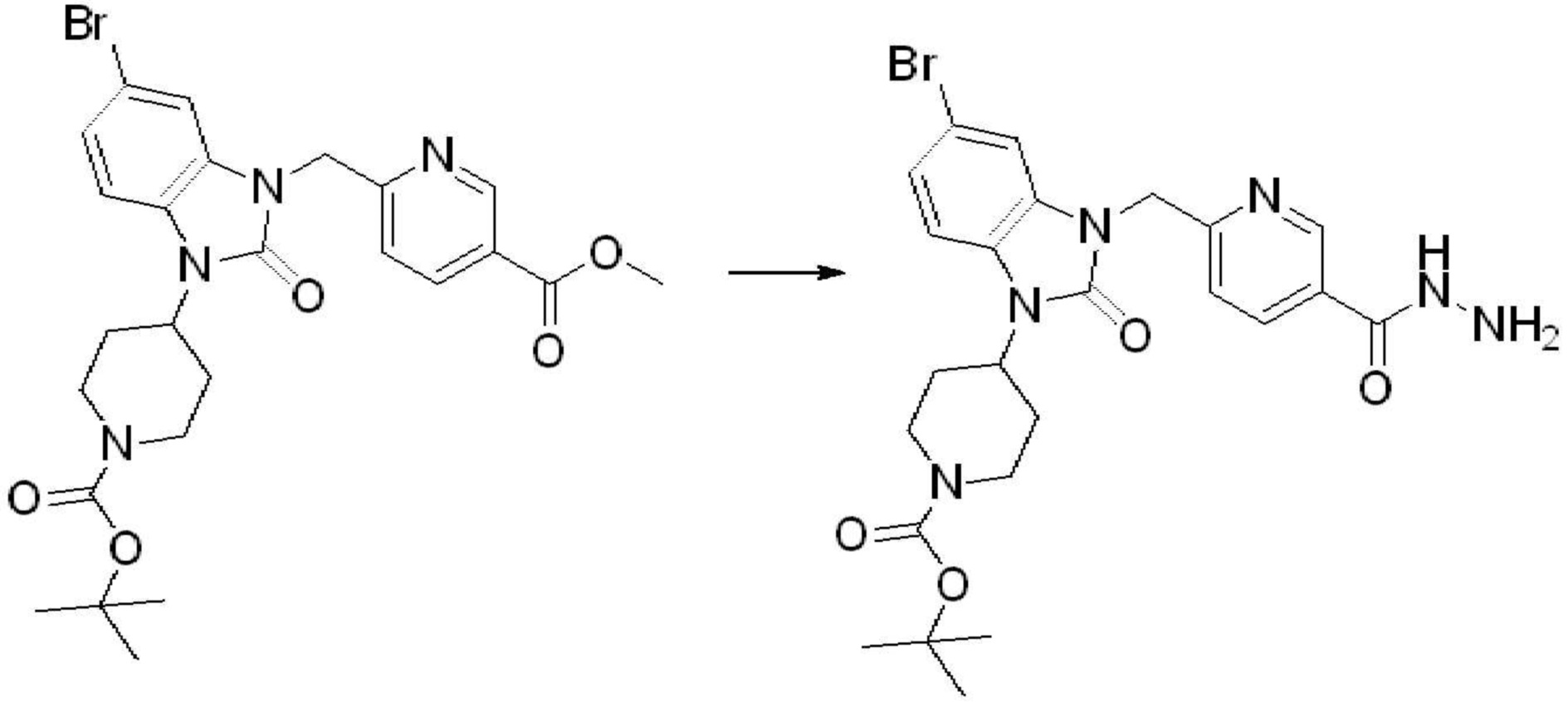
Метил 6-((6-бром-3-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)никотинат (2,568 г, 4,708 ммоль), полученный на стадии 1, и моногидрат гидразина (6,364 г, 127,122 ммоль) смешивают в этаноле (25 мл) при комнатной температуре, после чего полученную суспензию перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. выпавшее в осадок твердое вещество фильтруют, затем промывают этанол, и затем сушат с получением указанного в заголовке соединения (1,680 г, 65,4%) в виде белого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(5-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
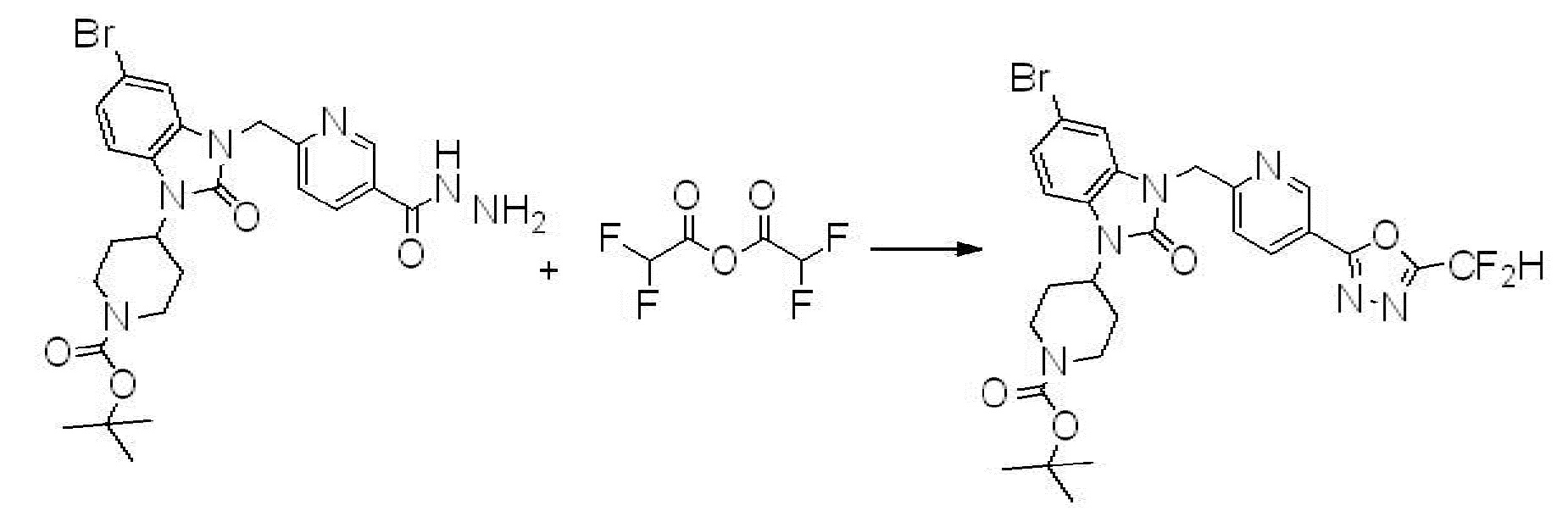
Трет-бутил 4-(5-бром-3-((5-(гидразинкарбонил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,680 г, 3,080 ммоль), полученный на стадии 2, 2,2-дифторуксусный ангидрид (2,489 мл, 20,021 ммоль) и имидазол (0,629 г, 9,240 ммоль) растворяют в дихлорметане (17 мл) при комнатной температуре, после чего полученный раствор перемешивают при 60° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (1,600 г, 85,8%) в виде желтого твердого вещества.
[Стадия 4] Синтез 5-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
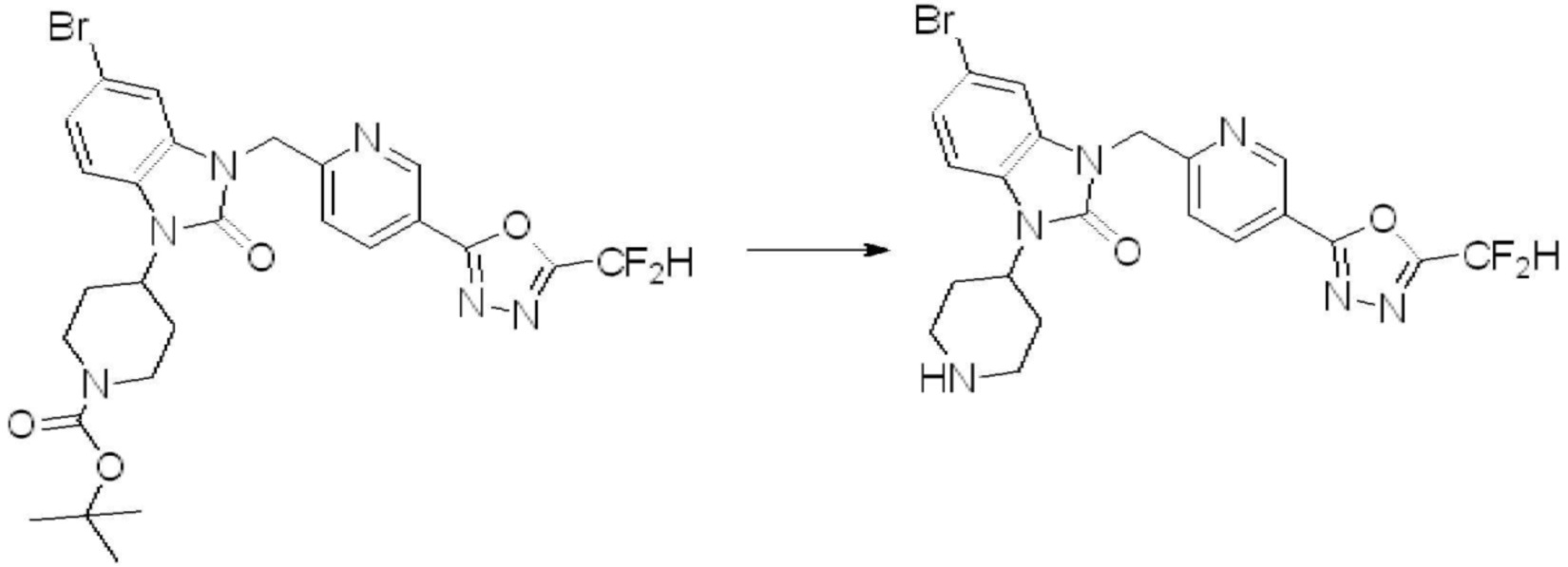
Трет-бутил 4-(5-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,600 г, 2,643 ммоль), полученный на стадии 3, и трифторуксусную кислоту (2,024 мл, 26,427 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=80-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-80%) и концентрируют с получением указанного в заголовке соединения (1,166 г, 87,3%) в виде желтого твердого вещества.
[Стадия 5] Синтез соединения 141
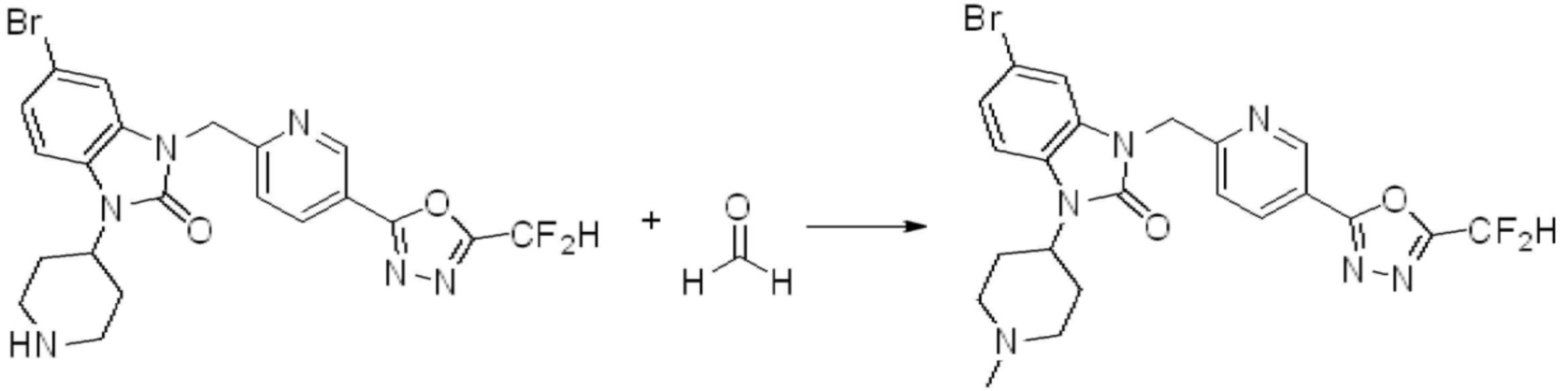
5-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (1,166 г, 2,307 ммоль), полученный на стадии 4, формальдегид (37,00%, 0,562 г, 6,922 ммоль) и триацетоксиборгидрид натрия (0,978 г, 4,615 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (1,018 г, 85,0%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,7 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,3 Гц, 1H), 7,18 (с, 2H), 7,13 (с, 1H), 6,93 (т, J=51,7 Гц, 1H), 5,24 (с, 2H), 4,44-4,38 (м, 1H), 3,02 (д, J=11,8 Гц, 2H), 2,55-2,35 (м, 2H), 2,35 (с, 3H), 2,15 (т, J=11,0 Гц, 2H), 1,84 (дд, J=11,8, 2,2 Гц, 2H); МСНР (ЭР) m/z 519,2 (M+ + 1).
Пример 142: Синтез соединения 142, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез 4-фтор-N-метил-2-нитроанилина
1,4-дифтор-2-нитробензол (3,000 г, 18,857 ммоль), метанамин (33,00% раствор, 3,915 мл, 56,572 ммоль), карбонат калия (5,212 г, 37,715 ммоль) и йодид калия (0,313 г, 1,886 ммоль) растворяют в N,N-диметилформамиде (90 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 6 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-10%) и концентрируют с получением указанного в заголовке соединения (2,690 г, 83,8%) в виде красного твердого вещества.
[Стадия 2] Синтез 4-фтор-N1-метилбензол-1,2-диамина
4-фтор-N-метил-2-нитроанилин (2,690 г, 15,811 ммоль), полученный на стадии 1 и Pd/C (45,00%, 0,374 г, 1,581 ммоль) растворяют в метанол (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (2,070 г, 93,4%, коричневая жидкость).
[Стадия 3] Синтез 5-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-она
4-фтор-N1-метилбензол-1,2-диамин (2,070 г, 14,769 ммоль), полученный на стадии 2 и 1,1'-карбонилдиимидазол (КДИ, 2,874 г, 17,723 ммоль) растворяют в тетрагидрофуране (70 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. выпавшее в осадок твердое вещество фильтруют, затем промывают с дихлорметаном, и затем сушат с получением указанного в заголовке соединения (0,600 г, 24,4%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 142
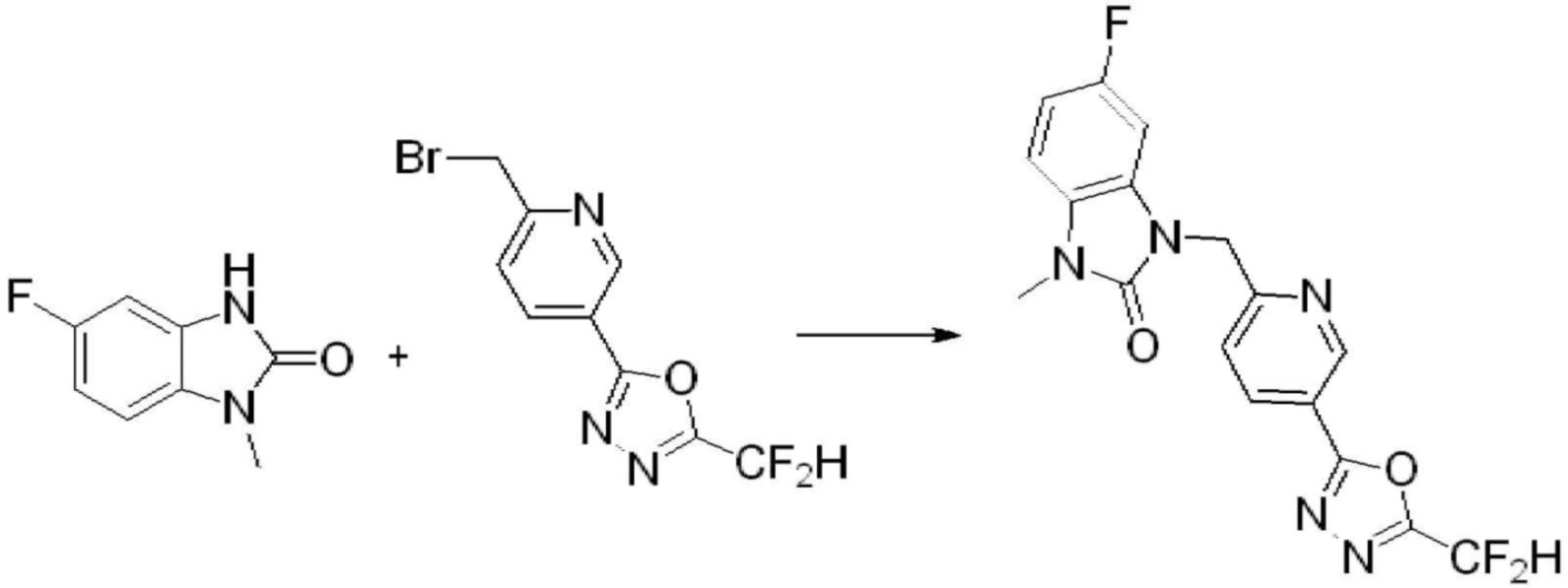
5-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 1,204 ммоль), полученный на стадии 3 растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,072 г, 1,805 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 20 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,454 г, 1,565 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,195 г, 43,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=2,1 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (д, J=8,3 Гц, 1H), 7,05-6,72 (м, 4H), 5,24 (с, 2H), 3,45 (с, 3H); МСНР (ЭР) m/z 376,0 (M+ + 1).
Пример 143: Синтез соединения 143, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 143
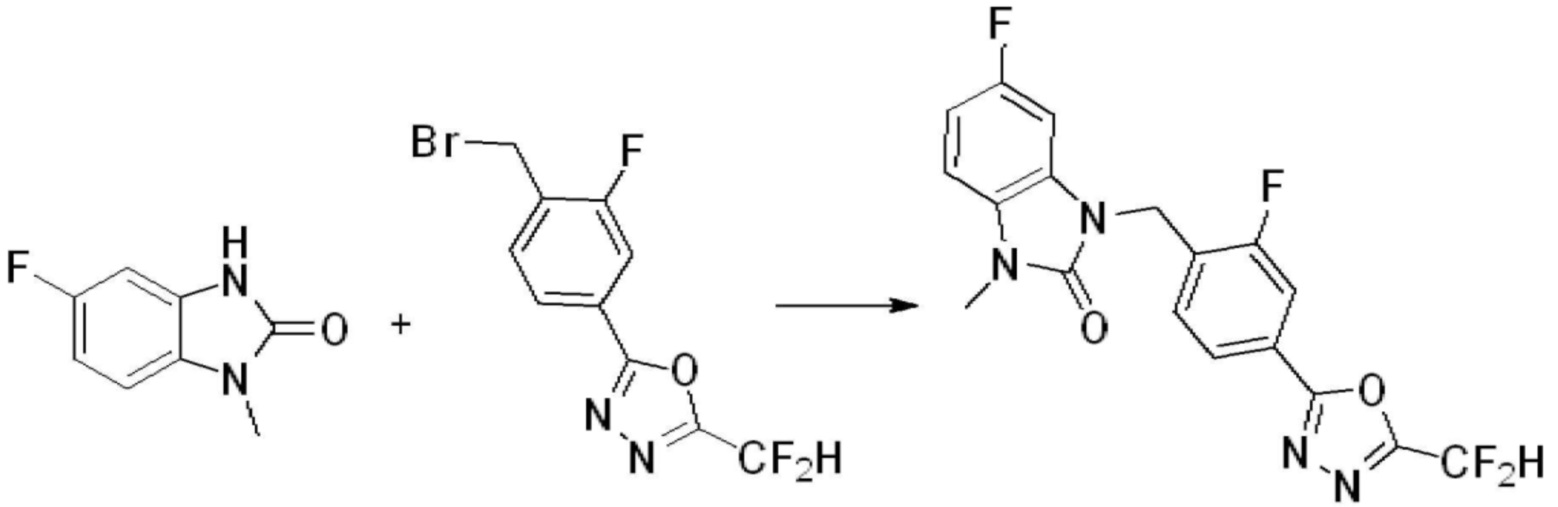
5-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 1,204 ммоль), полученный на стадии 3 соединения 142 растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,072 г, 1,805 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 20 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,480 г, 1,565 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,302 г, 63,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,86-7,82 (м, 2H), 7,47 (т, J=7,8 Гц, 1H), 7,03-6,70 (м, 4H), 5,16 (с, 2H), 3,45 (с, 3H); МСНР (ЭР) m/z 393,2 (M+ + 1).
Пример 144: Синтез соединения 144, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 144
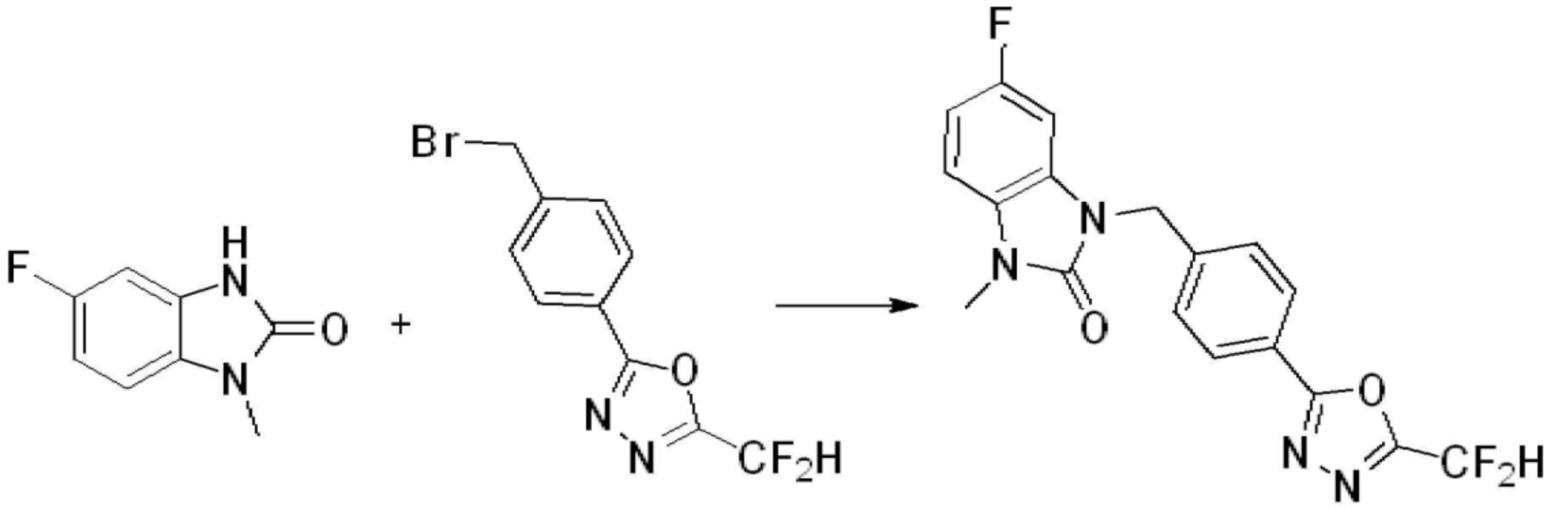
5-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 1,204 ммоль), полученный на стадии 3 соединения 142 растворяют в N,N-диметилформамиде (12 мл) при 0°C, после чего гидрид натрия (60,00%, 0,072 г, 1,805 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 20 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,452 г, 1,565 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,362 г, 80,4%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,02-7,96 (м, 2H), 7,42 (д, J=8,6 Гц, 2H), 7,02-6,72 (м, 3H), 6,55 (дд, J=8,4, 2,4 Гц, 1H), 5,07 (с, 2H), 3,41 (с, 3H); МСНР (ЭР) m/z 374,9 (M+ + 1).
Пример 145: Синтез соединения 145, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-метилпиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 145
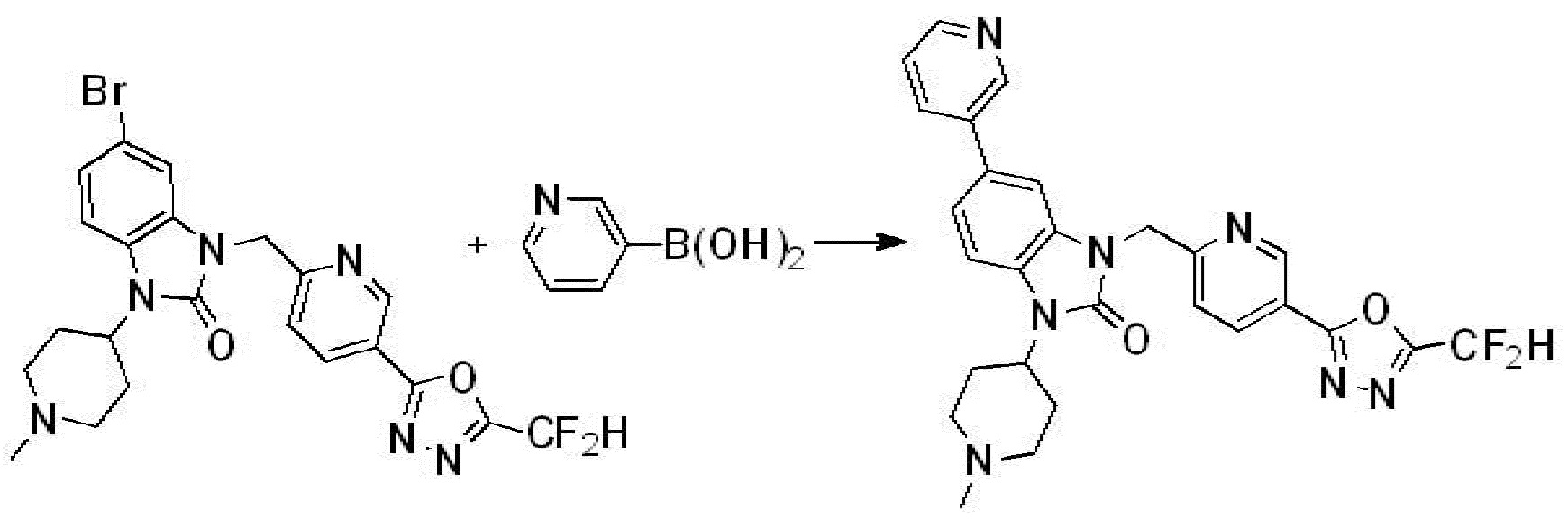
5-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,231 ммоль), полученный на стадии 5 соединения 141, пиридин-3-илбороновую кислоту (0,034 г, 0,277 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) и карбонат цезия (0,151 г, 0,462 ммоль) смешивают в 1,4-диоксане (1,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,018 г, 15,3%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,2, 0,6 Гц, 1H), 8,74 (с, 1H), 8,54 (с, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,78 (д, J=7,7 Гц, 1H), 7,45-7,42 (м, 2H), 7,32-7,18 (м, 3H), 6,91 (т, J=51,6 Гц, 1H), 5,33 (с, 2H), 4,52-4,44 (м, 1H), 3,06 (д, J=11,8 Гц, 2H), 2,57-2,48 (м, 2H), 2,37 (с, 3H), 2,23-2,16 (м, 2H), 1,89 (дд, J=12,1, 2,2 Гц, 2H); МСНР (ЭР) m/z 518,3 (M+ + 1).
Пример 146: Синтез соединения 146, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(5-метилфуран-2-ил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 146
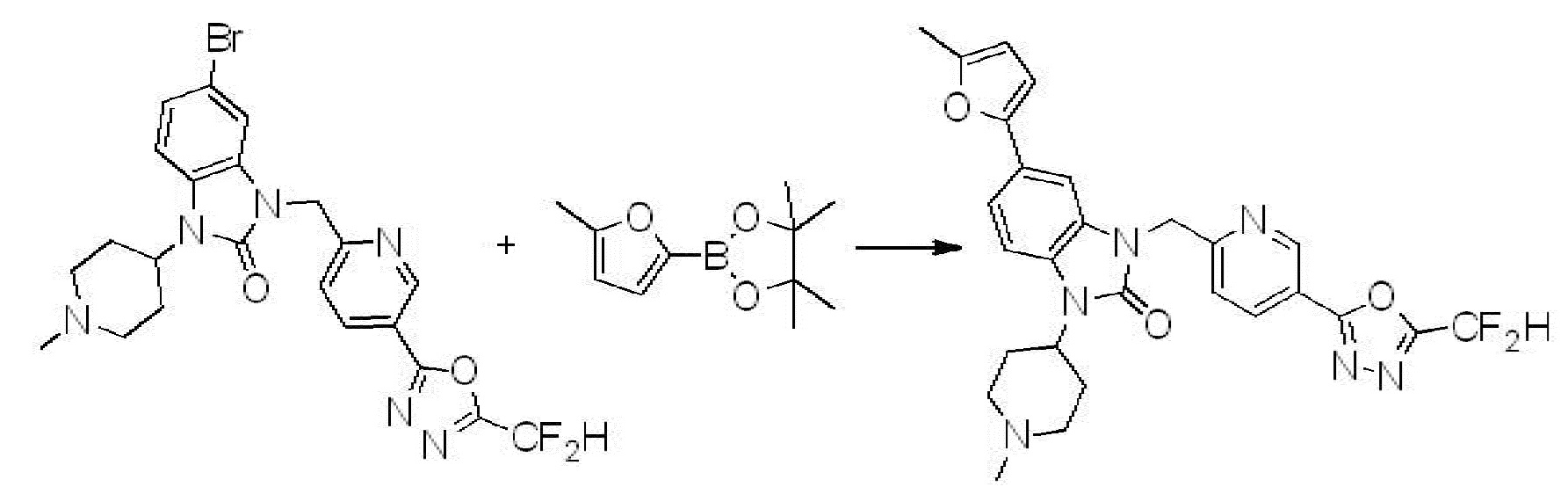
5-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,231 ммоль), полученный на стадии 5 соединения 141, 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан (0,058 г, 0,277 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) и карбонат цезия (0,151 г, 0,462 ммоль) смешивают в 1,4-диоксане (1,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,022 г, 18,2%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30-9,29 (м, 1H), 8,30 (дд, J=8,2, 2,2 Гц, 1H), 7,37 (д, J=8,2 Гц, 1H), 7,32-7,26 (м, 2H), 7,19 (с, 1H), 6,91 (т, J=51,6 Гц, 1H), 6,40 (д, J=3,2 Гц, 1H), 5,99 (дд, J=3,1, 1,0 Гц, 1H), 5,31 (с, 2H), 4,49-4,40 (м, 1H), 3,04 (д, J=11,6 Гц, 2H), 2,53-2,44 (м, 2H), 2,36 (с, 3H), 2,32 (с, 3H), 2,16 (т, J=5,4 Гц, 2H), 1,87 (дд, J=12,1, 2,1 Гц, 2H); МСНР (ЭР) m/z 520,9 (M+ + 1).
Пример 147: Синтез соединения 147, 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез 5-бром-4-фтор-N-метил-2-нитроанилина
1-бром-2,5-дифтор-4-нитробензол (10,000 г, 42,019 ммоль), метанамин (33,00% раствор, 5,754 мл, 46,220 ммоль), карбонат калия (11,614 г, 84,037 ммоль) и йодид калия (0,698 г, 4,202 ммоль) растворяют в N,N-диметилформамиде (250 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 3 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Воду (250 мл) добавляют в реакционную смесь и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют, затем промывают воде, и затем сушат с получением указанного в заголовке соединения (10,130 г, 96,8%) в виде красного твердого вещества.
[Стадия 2] Синтез 5-бром-4-фтор-N1-метилбензол-1,2-диамина
5-бром-4-фтор-N-метил-2-нитроанилин (12,480 г, 50,112 ммоль), полученный на стадии 1 и никель Ренея (1,2 г, 10% масс.) растворяют в метаноле (250 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (10,110 г, 92,1%, коричневая жидкость).
[Стадия 3] Синтез 6-бром-5-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-она
5-бром-4-фтор-N1-метилбензол-1,2-диамин (10,110 г, 46,152 ммоль), полученный на стадии 2, 1,1'-карбонилдиимидазол (КДИ, 9,729 г, 59,997 ммоль) и триэтиламин (7,076 мл, 50,767 ммоль) растворяют в тетрагидрофуране (200 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. выпавшее в осадок твердое вещество фильтруют и сушат с получением указанного в заголовке соединения (8,486 г, 75,0%) в виде фиолетового твердого вещества.
[Стадия 4] Синтез соединения 147
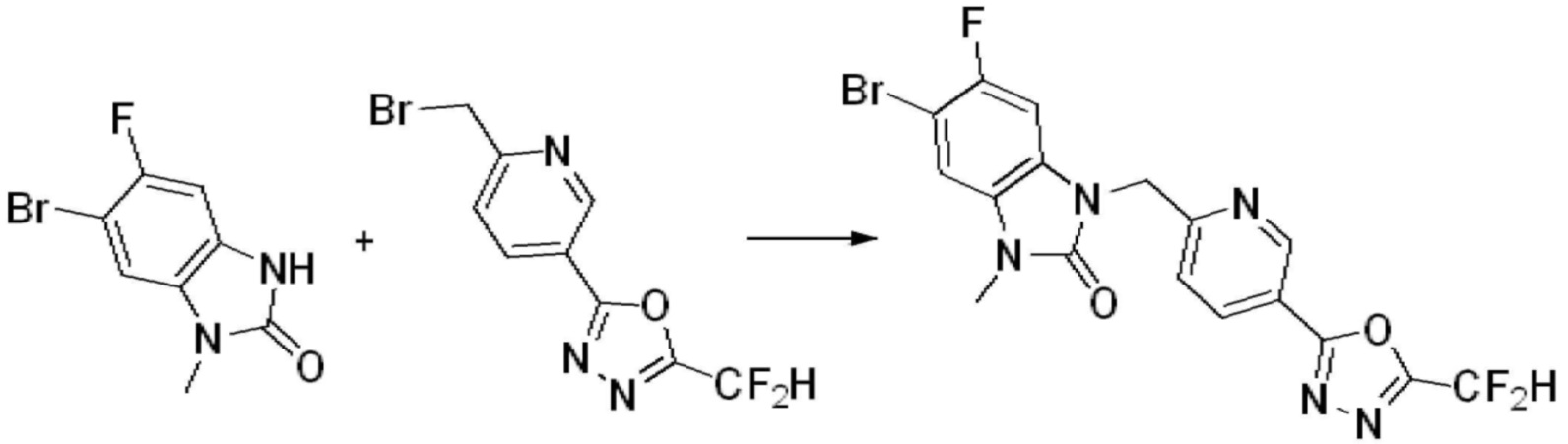
6-бром-5-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (1,489 г, 6,076 ммоль), полученный на стадии 3 растворяют в N,N-диметилформамиде (60 мл) при 0°C, после чего гидрид натрия (60,00%, 0,365 г, 9,114 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,939 г, 6,684 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=30-70%) и концентрируют с получением указанного в заголовке соединения (1,145 г, 41,5%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,2, 0,7 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,46 (д, J=8,2 Гц, 1H), 7,13 (д, J=5,6 Гц, 1H), 6,93 (т, J=51,6 Гц, 1H), 6,86 (д, J=8,1 Гц, 1H), 5,24 (с, 2H), 3,45 (с, 3H); МСНР (ЭР) m/z 454,1 (M+ + 1).
Пример 148: Синтез соединения 148, 5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 148
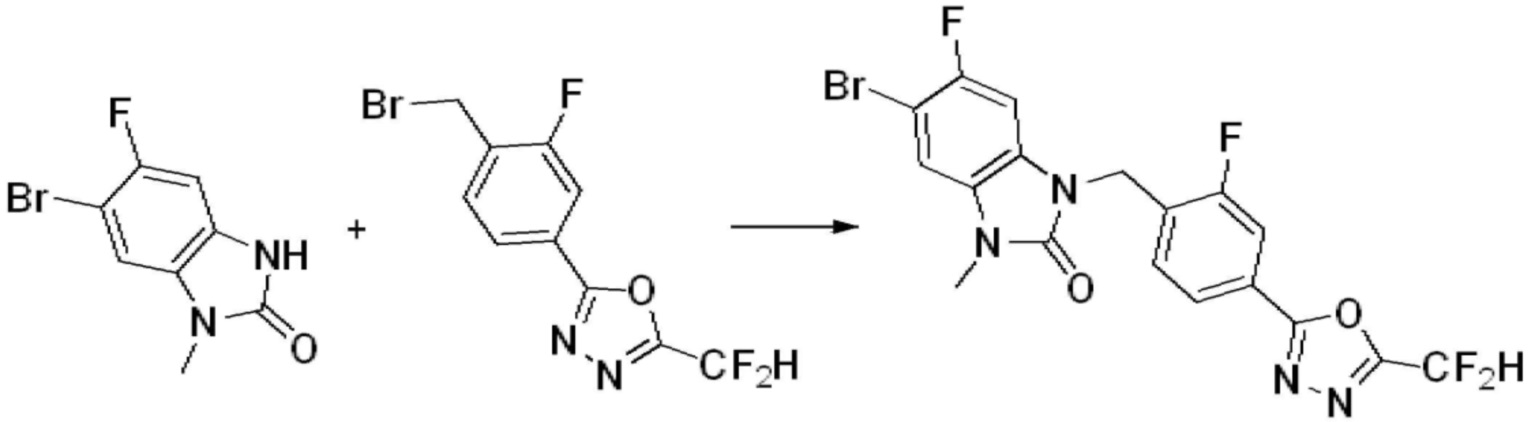
6-бром-5-фтор-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,300 г, 1,224 ммоль), полученный на стадии 3 соединения 147 растворяют в N,N-диметилформамиде (12 мл) при 0°C, после чего гидрид натрия (60,00%, 0,073 г, 1,836 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,414 г, 1,347 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 5 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-60%) и концентрируют с получением указанного в заголовке соединения (0,384 г, 66,6%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 7,89 (дд, J=10,3, 1,6 Гц, 1H), 7,84 (дд, J=8,0, 1,7 Гц, 1H), 7,59 (д, J=6,0 Гц, 1H), 7,54 (т, J=51,3 Гц, 1H), 7,41 (т, J=7,8 Гц, 1H), 7,36 (д, J=9,0 Гц, 1H), 5,21 (с, 2H), 3,35 (с, 3H); МСНР (ЭР) m/z 473,2 (M+ + 1).
Пример 149: Синтез соединения 149, 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
[Стадия 1] Синтез трет-бутил 4-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
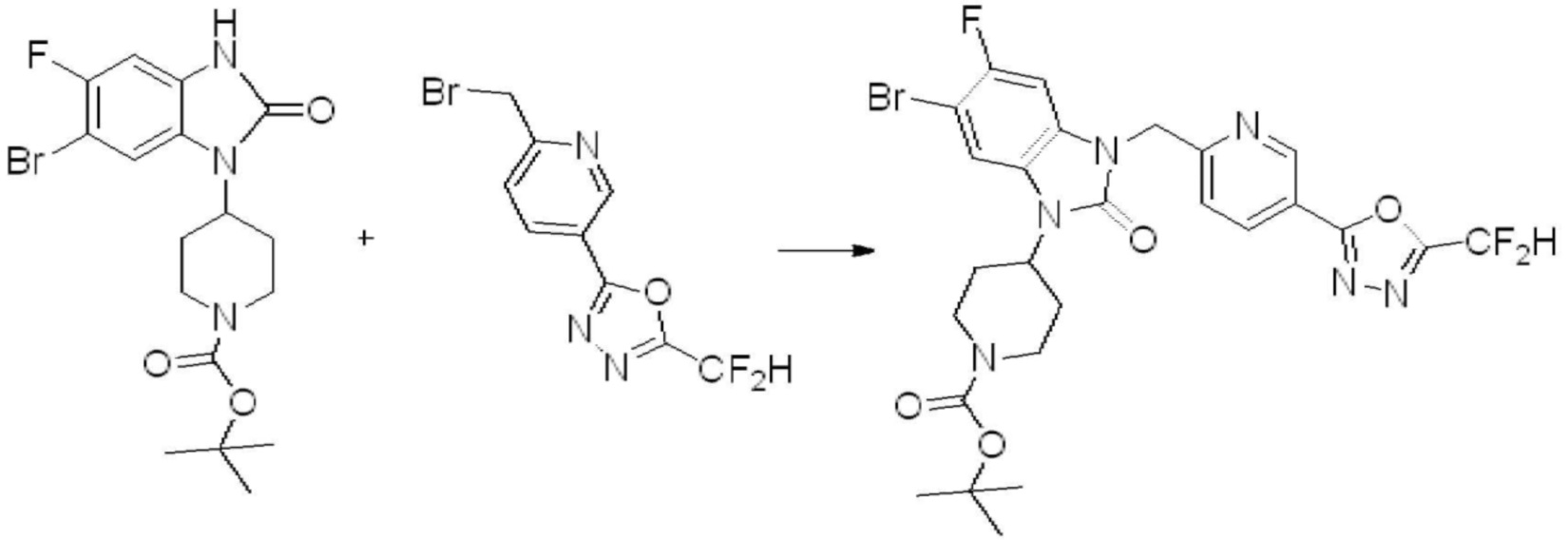
Трет-бутил 4-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил) пиперидин-1-карбоксилат (1,500 г, 3,621 ммоль) растворяют в N,N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,174 г, 4,345 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,050 г, 3,621 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (1,500 г, 66,5%) в виде бесцветного масла.
[Стадия 2] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она 2,2,2-трифторацетата
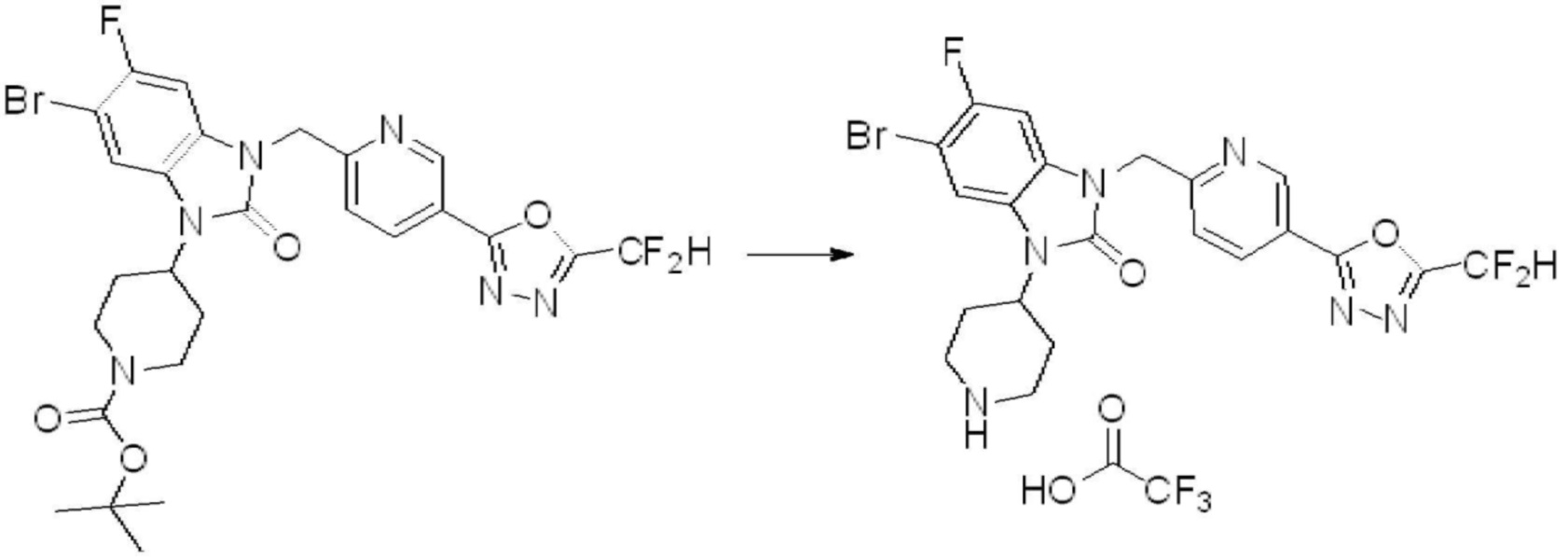
Трет-бутил 4-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,500 г, 2,406 ммоль), полученный на стадии 1, и трифторуксусную кислоту (1,842 мл, 24,060 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (1,400 г, 91,3%, желтое твердое вещество).
[Стадия 3] Синтез соединения 149
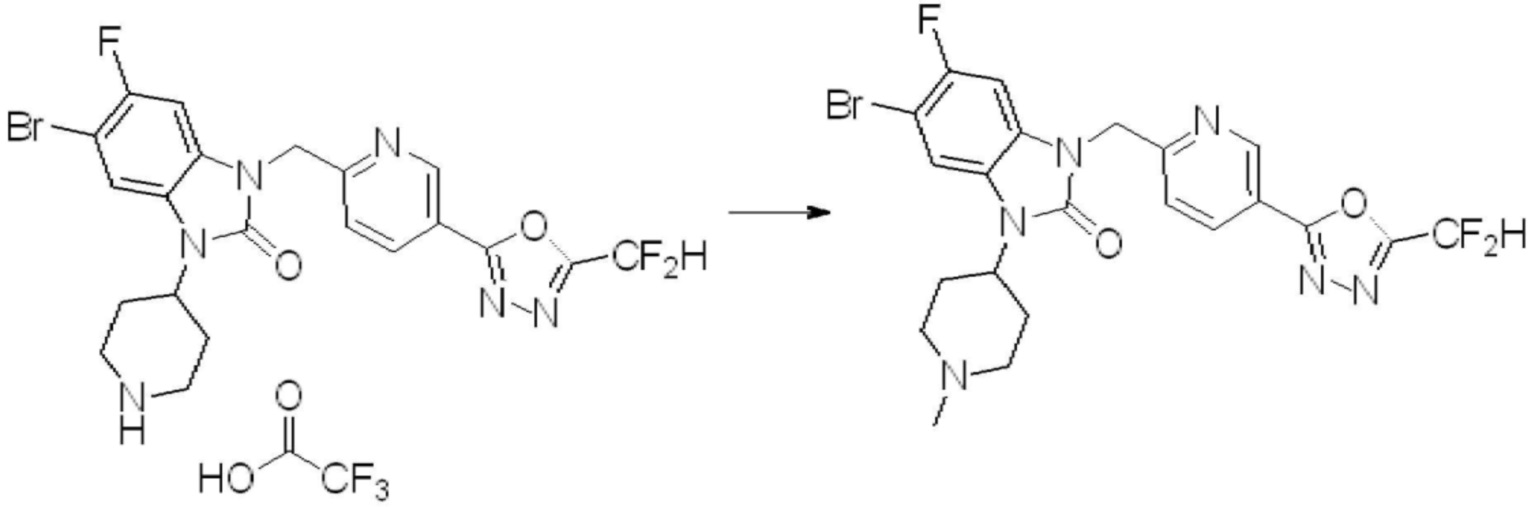
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он 2,2,2-трифторацетат (1,400 г, 2,197 ммоль), полученный на стадии 2, формальдегид (0,132 г, 4,393 ммоль), N,N-диизопропилэтиламин (0,383 мл, 2,197 ммоль) и триацетоксиборгидрид натрия (0,931 г, 4,393 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (1,300 г, 110,1%) в виде белого пенистого твердого вещества.
МСНР (ЭР) m/z 538,4 (M+ + 1).
Пример 150: Синтез соединения 150, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(фуран-3-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 150
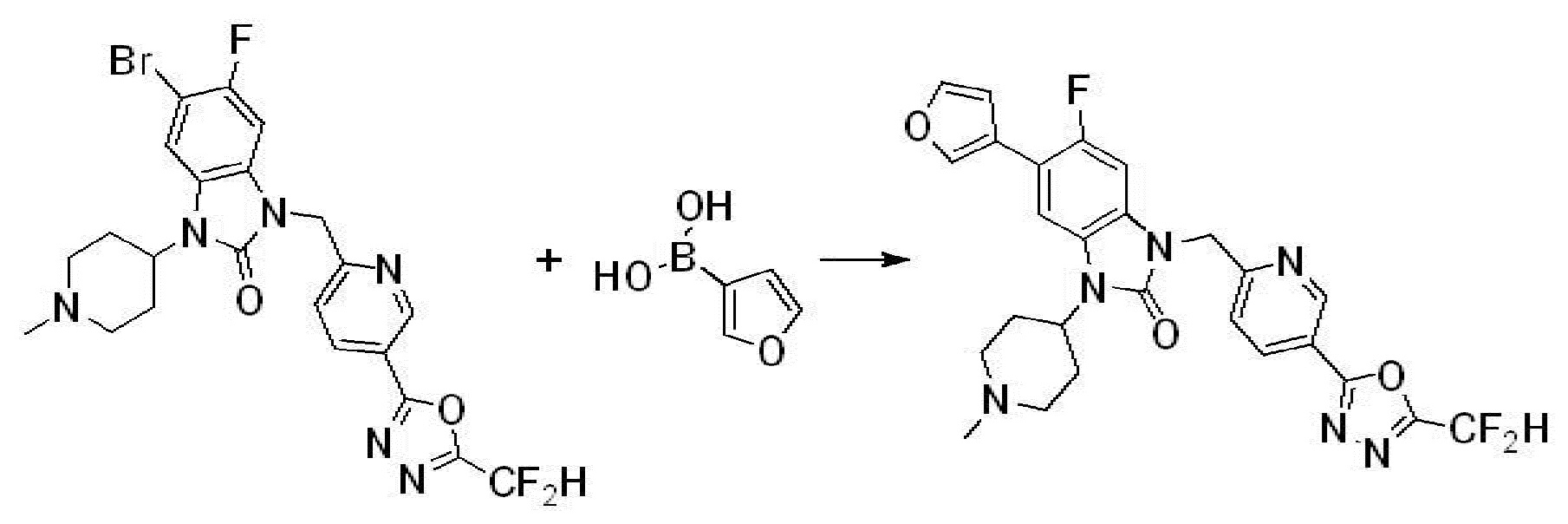
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), фуран-3-илбороновую кислоту (0,047 г, 0,419 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,018 г, 0,028 ммоль) и карбонат цезия (0,136 г, 0,419 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 75,1%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,6 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,83 (с, 1H), 7,51 (т, J=1,7 Гц, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,38 (д, J=5,9 Гц, 1H), 7,07 (с, 0,25H), 6,94 (с, 0,5H), 6,83 (с, 0,25H), 6,85-6,82 (м, 2H), 5,27 (с, 2H), 4,52-4,51 (м, 1H), 3,50 (с, 3H), 3,10-3,08 (м, 2H), 2,60-2,10 (м, 4H), 1,91-1,88 (м, 2H).
Пример 151: Синтез соединения 151, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(фуран-2-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 151
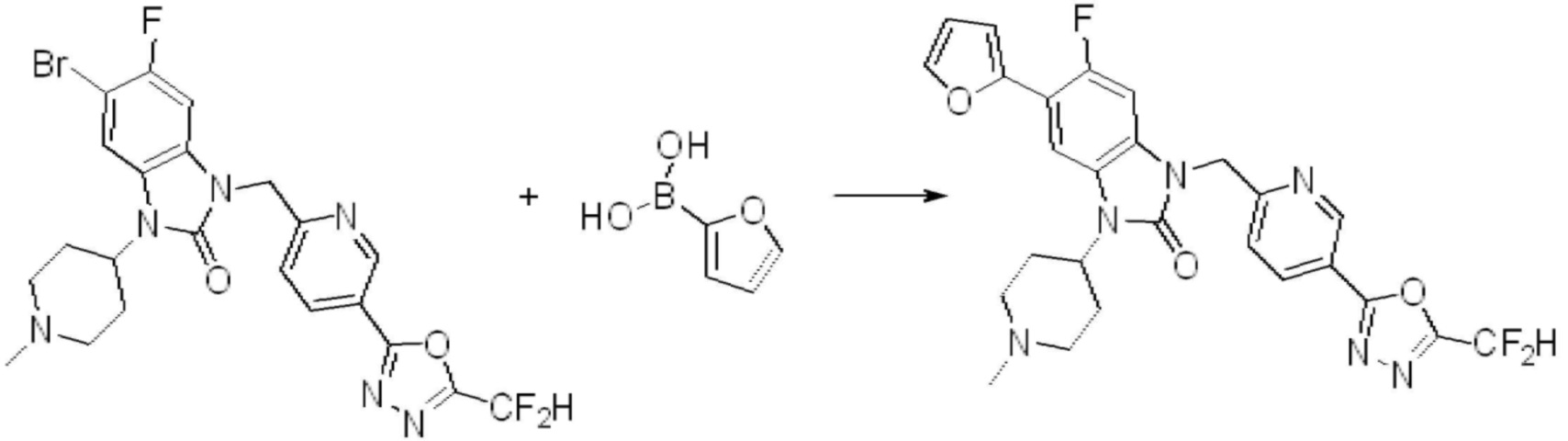
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), фуран-2-илбороновую кислоту (0,047 г, 0,419 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,018 г, 0,028 ммоль) и карбонат цезия (0,136 г, 0,419 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,080 г, 54,6%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=2,1 Гц, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 7,70 (д, J=6,0 Гц, 1H), 7,50 (д, J=1,6 Гц, 1H), 4,23 (с, 1H), 7,07 (с, 0,25H), 6,94 (с, 0,0,5H), 6,84 (д, J=10,4 Гц, 1H), 6,81 (с, 0,25H), 6,76 (т, J=6,2 Гц, 1H), 6,52-6,51 (м, 1H), 5,27 (с, 2H), 4,50-4,40 (м, 1H), 3,10-3,07 (м, 2H), 2,58-2,54 (м, 2H), 2,40 (с, 3H), 2,22-2,18 (м, 2H), 1,90-1,87 (м, 2H).
Пример 152: Синтез соединения 152, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(2-фторфенил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 152
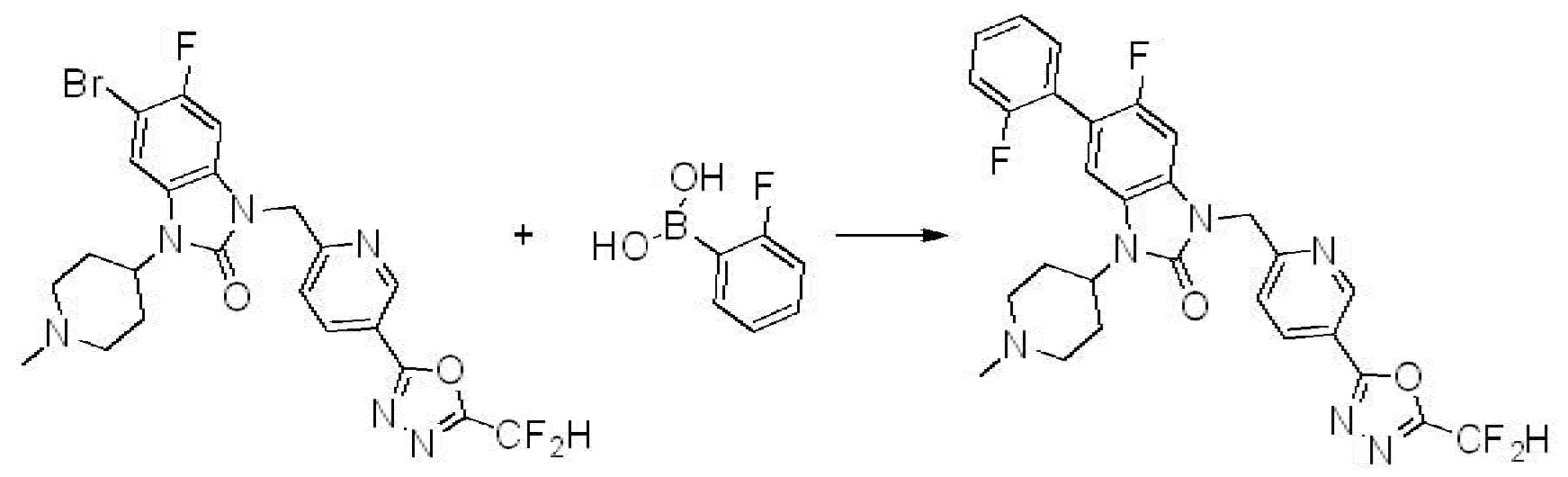
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), (2-фторфенил)бороновую кислоту (0,059 г, 0,419 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,018 г, 0,028 ммоль) и карбонат цезия (0,136 г, 0,419 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,030 г, 19,5%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) 400 MГц, CDJ=1,7 Гц, 1H), 8,39 (дд, J=8,3, 2,1 Гц, 1H), 7,49 (д, J=8,3 Гц, 1H), 7,41-7,34 (м, 2H), 7,28-7,27 (м, 1H), 7,24-7,14 (м, 2H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,90 (д, J=9,2 Гц, 1H), 6,82 (с, 0,25H), 5,30 (с, 2H), 4,50-4,44 (м, 1H), 3,05-3,02 (м, 2H), 2,51-2,42 (м, 2H), 2,35 (с, 3H), 2,23-2,19 (м, 2H), 1,91-1,88 (м, 2H).
Пример 153: Синтез соединения 153, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 153
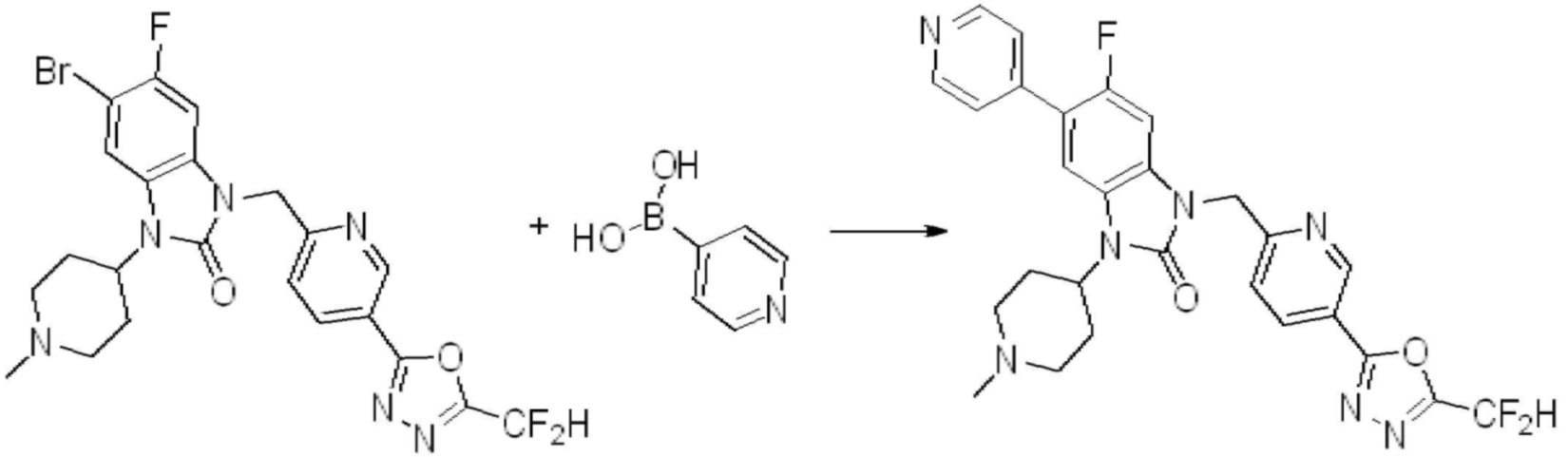
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), пиридин-4-илбороновую кислоту (0,051 г, 0,419 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,018 г, 0,028 ммоль) и карбонат цезия (0,136 г, 0,419 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,060 г, 40,1%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (дд, J=2,2, 0,7 Гц, 1H), 8,68-8,67 (м, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,50 (дд, J=8,3, 0,6 Гц, 1H), 7,45 (д, J=4,6 Гц, 1H), 7,33 (д, J=5,9 Гц, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,94-6,92 (м, 1H), 6,82 (с, 0,25H), 5,30 (с, 2H), 4,50-4,44 (м, 1H), 3,08-3,05 (м, 2H), 2,54-2,44 (м, 2H), 2,38 (с, 3H), 2,24-2,19 (м, 2H), 1,92-1,89 (м, 2H).
Пример 154: Синтез соединения 154, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(3-фторфенил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 154
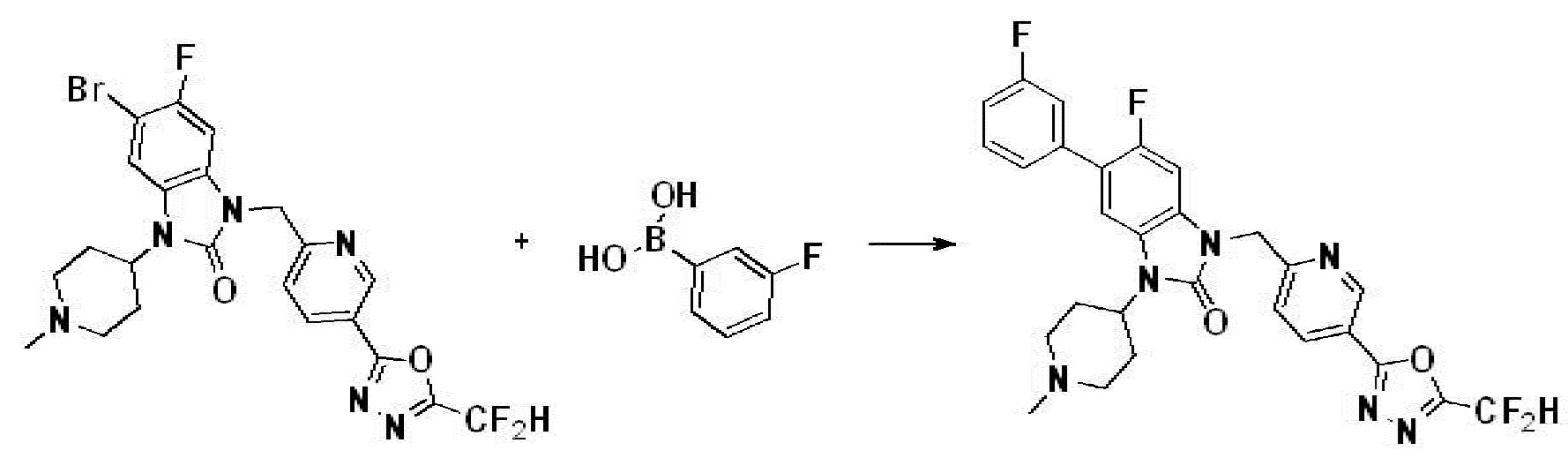
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), (3-фторфенил)бороновую кислоту (0,059 г, 0,419 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,018 г, 0,028 ммоль) и карбонат цезия (0,136 г, 0,419 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,040 г, 25,9%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (дд, J=2,2, 0,8 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,49-7,47 (м, 1H), 7,43-7,38 (м, 2H), 7,28-7,27 (м, 1H), 7,22-7,20 (м, 1H), 7,10-7,05 (м, 1H), 7,07 (с, 0,25H), 6,95 (с, 0,5H), 6,90 (д, J=9,8 Гц, 1H), 6,82 (с, 0,25H), 5,29 (с, 2H), 4,53-4,41 (м, 1H), 3,09-3,07 (м, 2H), 2,42-2,40 (м, 2H), 2,40 (с, 3H), 2,39-0,20 (м, 2H), 1,91-1,88 (м, 2H).
Пример 155: Синтез соединения 155, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 155
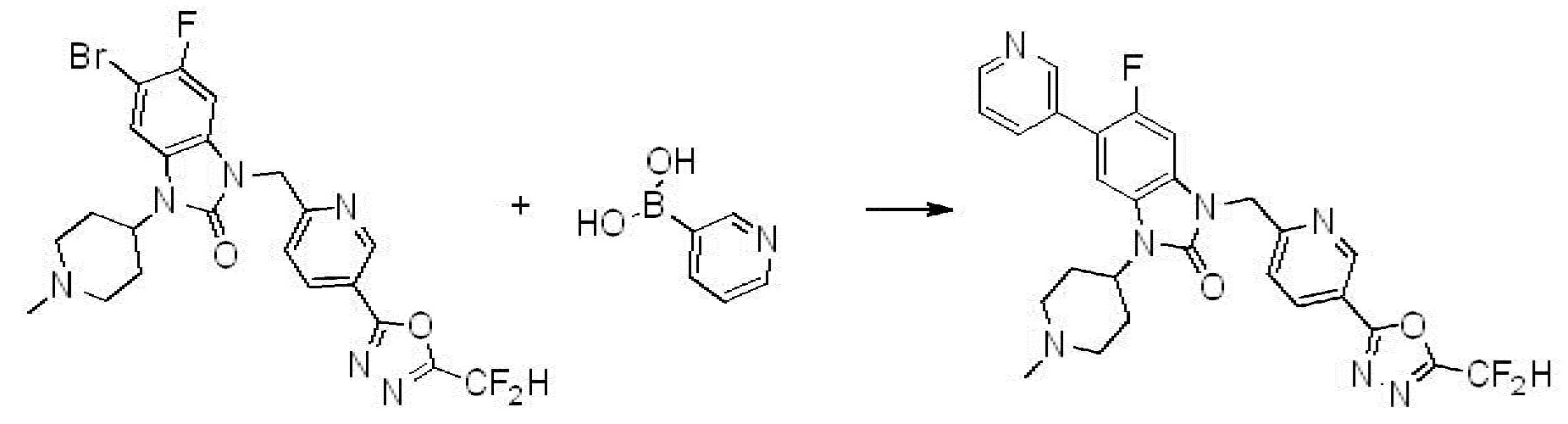
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), пиридин-3-илбороновую кислоту (0,051 г, 0,419 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,018 г, 0,028 ммоль) и карбонат цезия (0,136 г, 0,419 ммоль) смешивают в 1,4-диоксане (6 мл)/воде (2 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,030 г, 20,1%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,33-9,32 (м, 1H), 8,75 (с, 1H), 8,62 (д, J=3,7 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,82 (д, J=4,0 Гц, 1H), 7,49 (д, J=8,0 Гц, 1H), 7,38 (дд, J=7,9, 4,8 Гц, 1H), 7,30-7,28 (м, 1H), 7,08 (с, 1H), 6,95 (с, 1H), 6,93 (д, J=9,7 Гц, 1H), 6,82 (с, 1H), 5,31 (с, 2H), 4,49-4,43 (м, 1H), 3,07-3,04 (м, 2H), 2,53-2,36 (м, 2H), 2,23 (с, 3H), 2,20-2,17 (м, 2H), 1,91-1,88 (м, 2H).
Пример 156: Синтез соединения 156, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,5-бис(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-3,6-дигидропиридин-1(2H)-карбоксилата
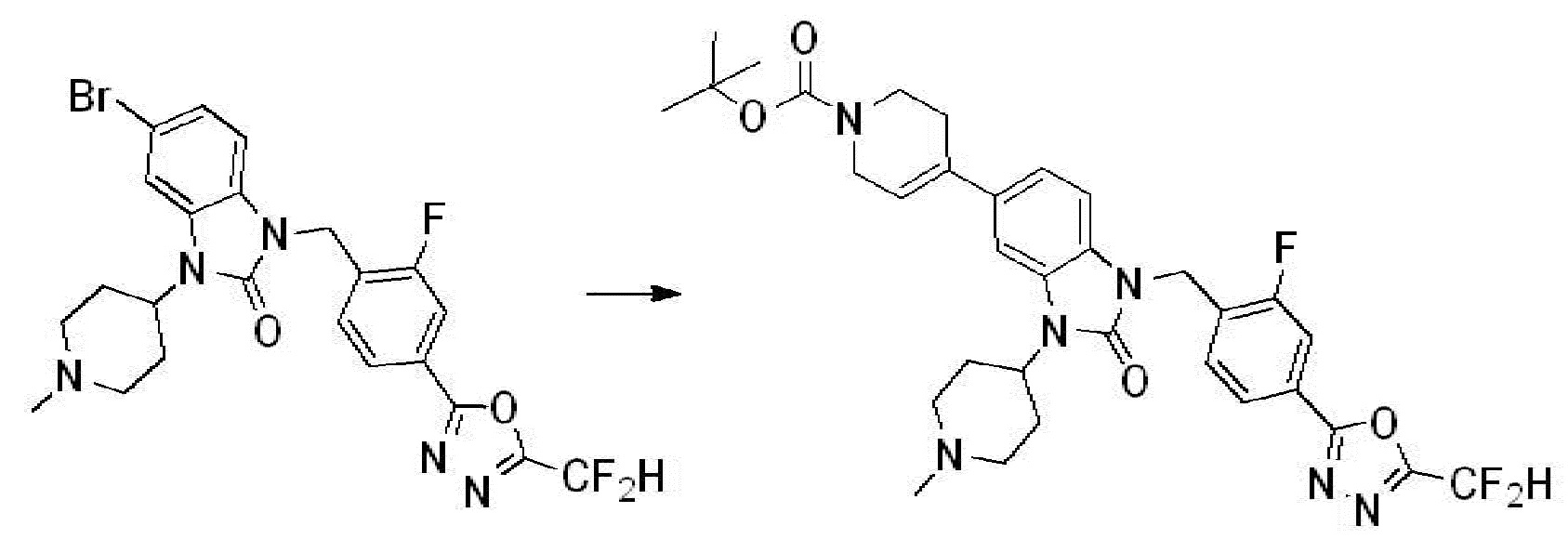
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (1,000 г, 1,864 ммоль), полученный на стадии 3 соединения 139, трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,692 г, 2,237 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,061 г, 0,093 ммоль) и карбонат цезия (1,822 г, 5,593 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (1,120 г, 94,1%) в виде коричневого масла.
[Стадия 2] Синтез трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперидин-1-карбоксилата
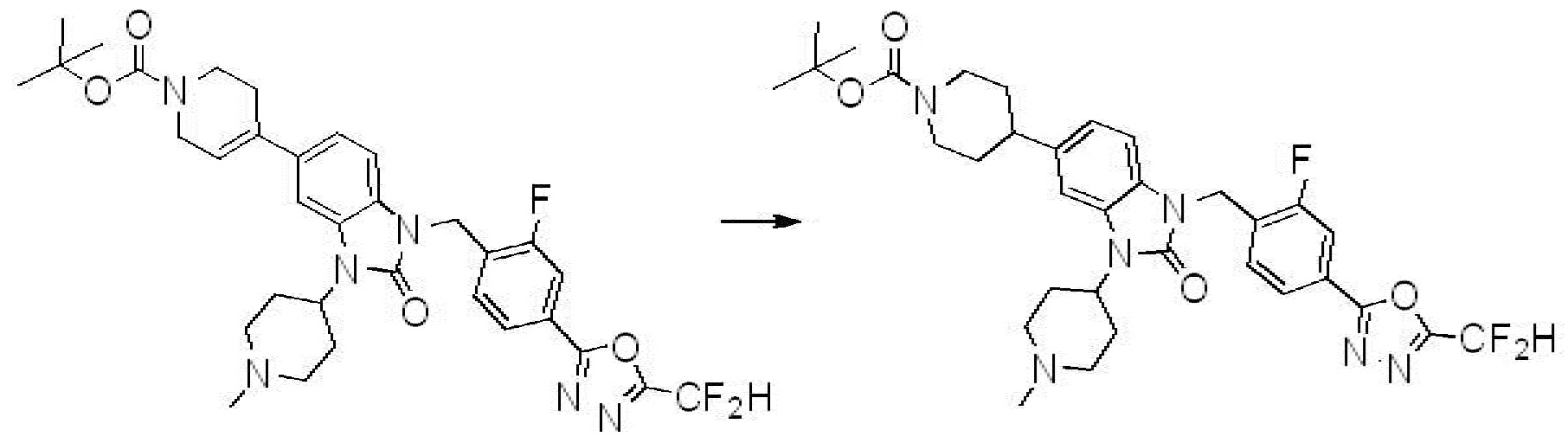
Трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (1,120 г, 1,754 ммоль), полученный на стадии 1 растворяют в метанол (30 мл, 0,058 M) и подвергают взаимодействию с применением Н-куба (10%-Pd/C, 40°C, 2 бар H2, 0,5 мл/мин скорость потока). Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,886 г, 78,9%, коричневое масло).
[Стадия 3] Синтез 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-5-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
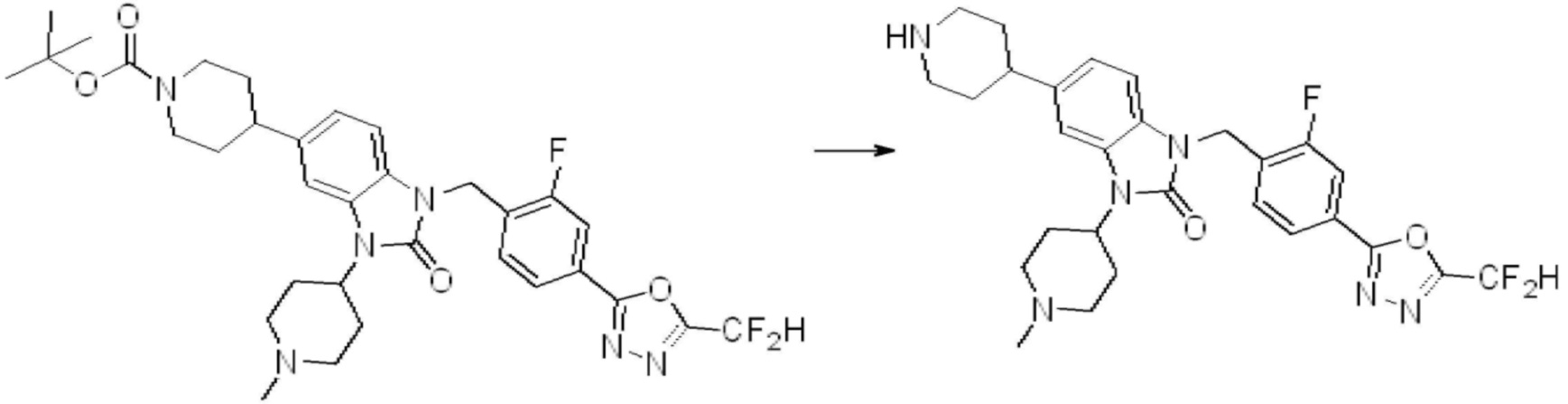
Трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперидин-1-карбоксилат (0,886 г, 1,383 ммоль), полученный на стадии 2, и трифторуксусную кислоту (0,794 мл, 10,371 ммоль) растворяют в дихлорметане (7 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,668 г, 89,4%, коричневое масло).
[Стадия 4] Синтез соединения 156
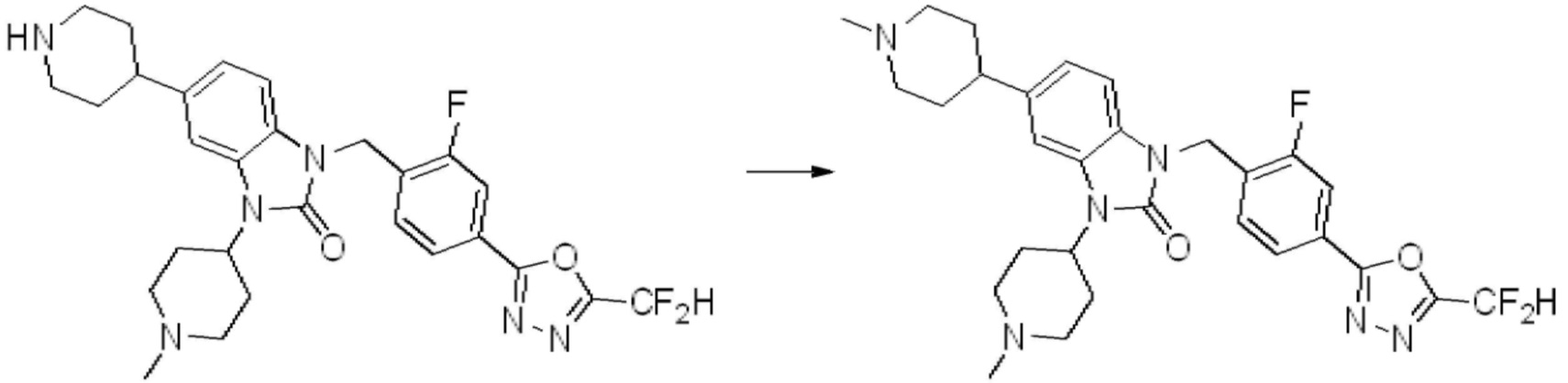
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-5-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,185 ммоль), полученный на стадии 3, формальдегид (38,00%, 0,022 г, 0,277 ммоль) и триацетоксиборгидрид натрия (0,078 г, 0,370 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-15%) и концентрируют с получением указанного в заголовке соединения (0,042 г, 40,9%) в виде желтого масла.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,82 (м, 2H), 7,47 (т, J=7,8 Гц, 1H), 7,24 (с, 1H), 7,05-6,79 (м, 3H), 5,19 (с, 2H), 4,48-4,42 (м, 1H), 3,05 (д, J=11,2 Гц, 4H), 2,57-2,48 (м, 3H), 2,40-2,39 (м, 6H), 2,23-2,14 (м, 4H), 1,92-1,85 (м, 6H); МСНР (ЭР) m/z 555,3 (M+ + H).
Пример 157: Синтез соединения 157, 5-(1-ацетилпиперидин-4-ил)-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 157
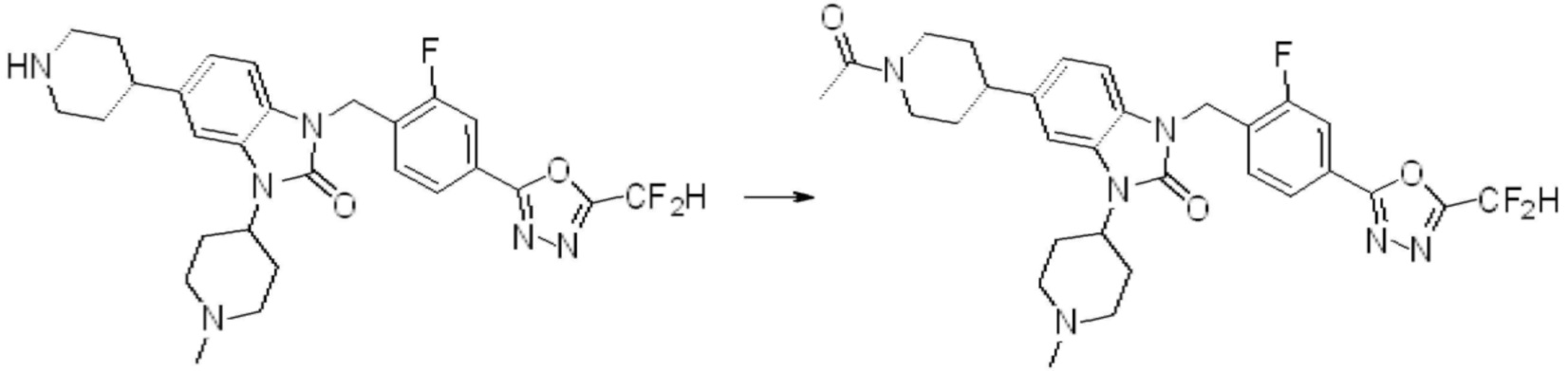
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-5-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,185 ммоль), полученный на стадии 3 соединения 156 и триэтиламин (0,052 мл, 0,370 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего ацетилхлорид (0,020 мл, 0,277 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,053 г, 49,2%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,83 (м, 2H), 7,47 (т, J=7,8 Гц, 1H), 7,19 (шс, 1H), 7,05-6,79 (м, 3H), 5,19 (с, 2H), 4,83-4,78 (м, 1H), 4,47-4,46 (м, 1H), 3,97-3,93 (м, 1H), 3,21-3,07 (м, 3H), 2,80-2,74 (м, 1H), 2,65-2,52 (м, 3H), 2,41 (с, 3H), 2,25-2,20 (м, 2H), 2,16 (с, 3H), 1,92-1,86 (м, 4H), 1,70-1,59 (м, 2H); МСНР (ЭР) m/z 583,3 (M+ + H).
Пример 158: Синтез соединения 158, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-5-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 158
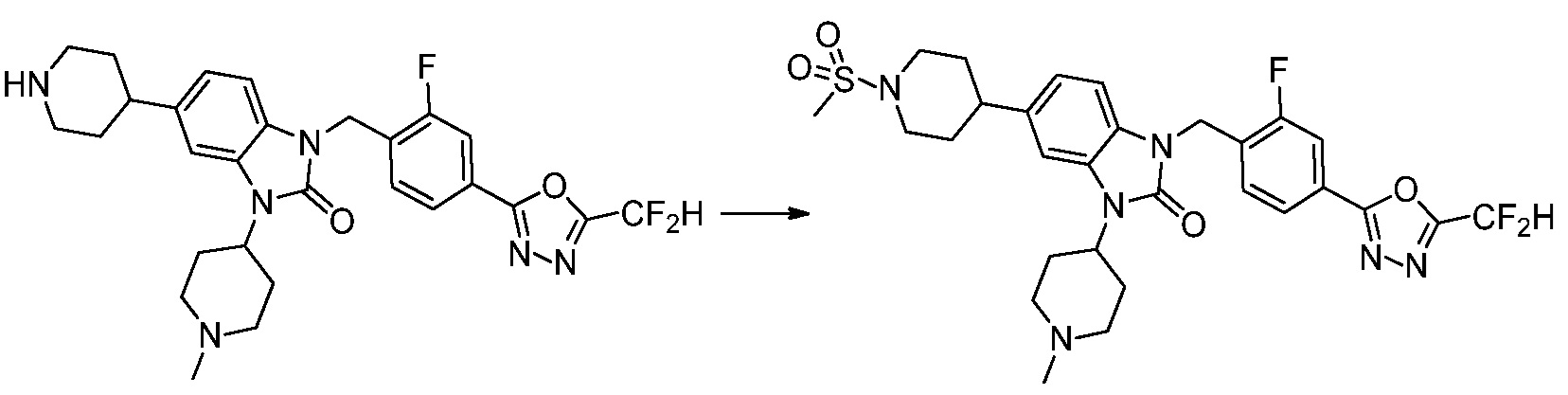
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-5-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,185 ммоль), полученный на стадии 3 соединения 156 и триэтиламин (0,052 мл, 0,370 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего метансульфонилхлорид (0,021 мл, 0,277 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,039 г, 34,1%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,83 (м, 2H), 7,47 (т, J=7,8 Гц, 1H), 7,21 (шс, 1H), 7,05-6,79 (м, 3H), 5,19 (с, 2H), 4,50-4,44 (м, 1H), 3,96 (д, J=11,8 Гц, 2H), 3,08 (д, J=9,4 Гц, 2H), 3,02 (шс, 1H), 2,85 (с, 3H), 2,80-2,74 (м, 2H), 2,70-2,66 (м, 1H), 2,65-2,61 (м, 2H), 2,54 (с, 3H), 2,23 (т, J=10,4 Гц, 2H), 1,96 (д, J=10,8 Гц, 2H), 1,90-1,84 (м, 3H); МСНР (ЭР) m/z 619,3 (M+ + H).
Пример 159: Синтез соединения 159, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(3-фторфенил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 159
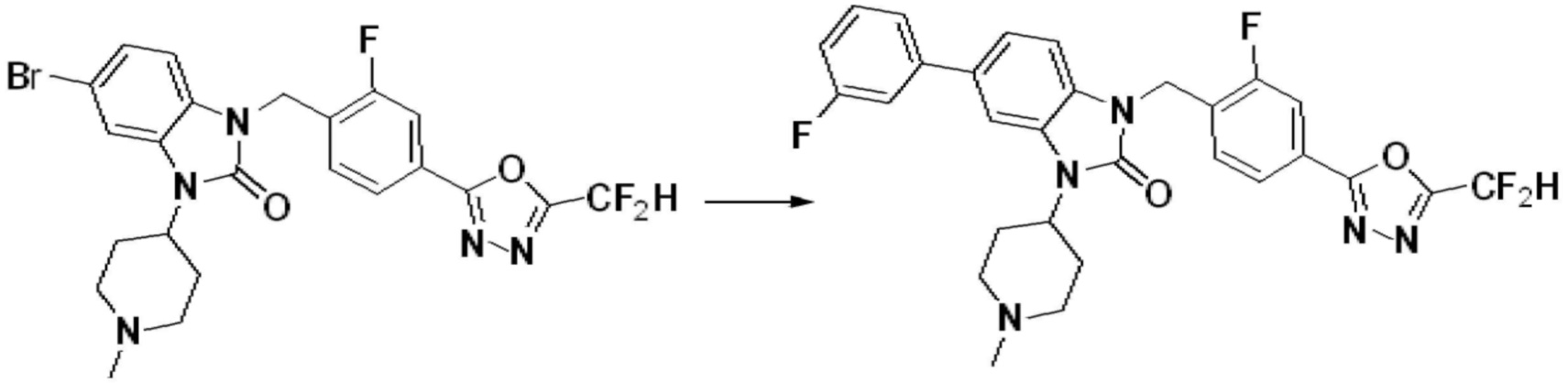
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,186 ммоль), полученный на стадии 3 соединения 139, (3-фторфенил)бороновую кислоту (0,031 г, 0,224 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,182 г, 0,559 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,047 г, 4,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,89-7,85 (м, 2H), 7,51-7,48 (м, 2H), 7,42-7,37 (м, 1H), 7,33 (д, J=7,8 Гц, 1H), 7,27-7,22 (м, 2H), 7,06-6,80 (м, 3H), 5,24 (с, 2H), 4,53-4,47 (м, 1H), 3,08 (д, J=11,5 Гц, 2H), 2,57 (q, J=11,1 Гц, 2H), 2,39 (с, 3H), 2,23 (т, J=11,6 Гц, 2H), 1,91 (д, J=10,1 Гц, 2H); МСНР (ЭР) m/z 552,3 (M+ + H).
Пример 160: Синтез соединения 160, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(2-фторфенил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 160
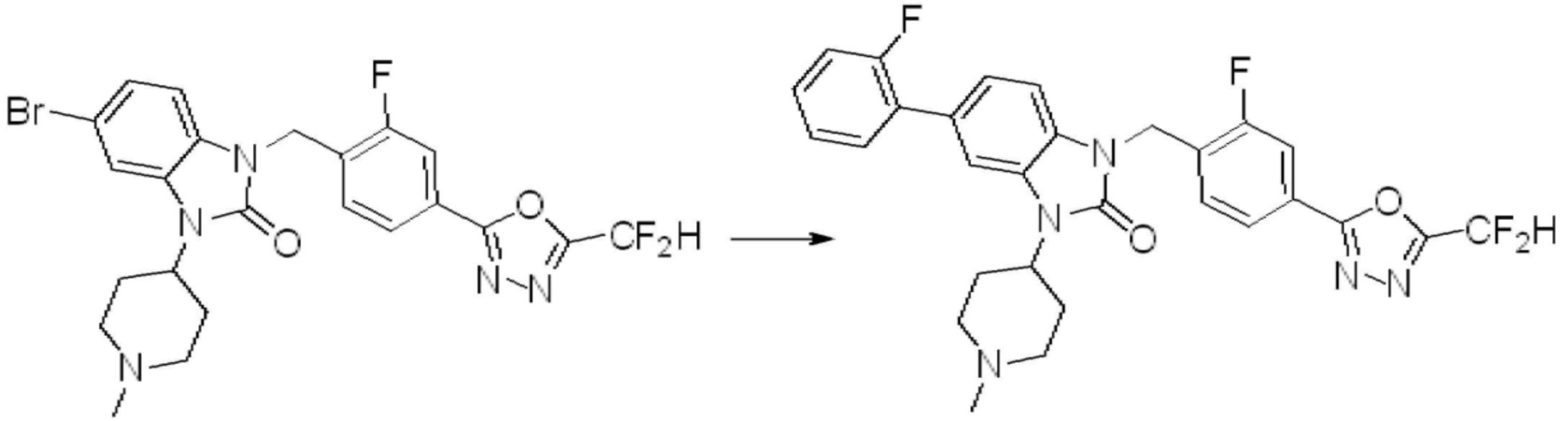
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,186 ммоль), полученный на стадии 3 соединения 139, (2-фторфенил)бороновую кислоту (0,031 г, 0,224 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,182 г, 0,559 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,054 г, 52,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,89-7,85 (м, 2H), 7,52-7,49 (м, 2H), 7,41 (тд, J=7,7, 1,7 Гц, 1H), 7,35-7,30 (м, 1H), 7,24-7,12 (м, 3H), 7,05-6,80 (м, 2H), 5,24 (с, 2H), 4,52-4,45 (м, 1H), 3,06 (д, J=11,5 Гц, 2H), 2,59-2,51 (м, 2H), 2,37 (с, 3H), 2,21 (т, J=11,4 Гц, 2H), 1,90 (д, J=10,2 Гц, 2H); МСНР (ЭР) m/z 552,2 (M+ + H).
Пример 161: Синтез соединения 161, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 161
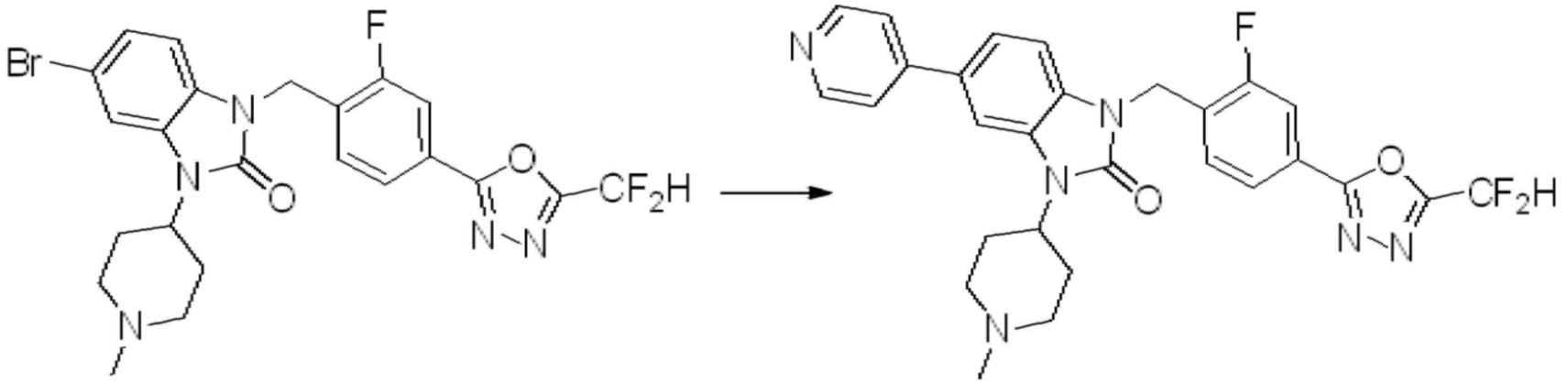
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,280 ммоль), полученный на стадии 3 соединения 139, пиридин-4-илбороновую кислоту (0,041 г, 0,336 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,009 г, 0,014 ммоль) и карбонат цезия (0,273 г, 0,839 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,034 г, 22,7%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,64 (д, J=3,0 Гц, 2H), 7,88-7,84 (м, 2H), 7,56 (с, 1H), 7,51-7,47 (м, 3H), 7,33 (дд, J=8,2, 1,4 Гц, 1H), 7,08-6,79 (м, 2H), 5,30 (с, 2H), 4,51-4,47 (м, 1H), 3,08 (д, J=11,1 Гц, 2H), 2,58-2,56 (м, 2H), 2,39 (с, 3H), 2,23 (т, J=11,4 Гц, 2H), 1,91 (д, J=10,1 Гц, 2H); МСНР (ЭР) m/z 535,3 (M+ + H).
Пример 162: Синтез соединения 162, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(фуран-3-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 162
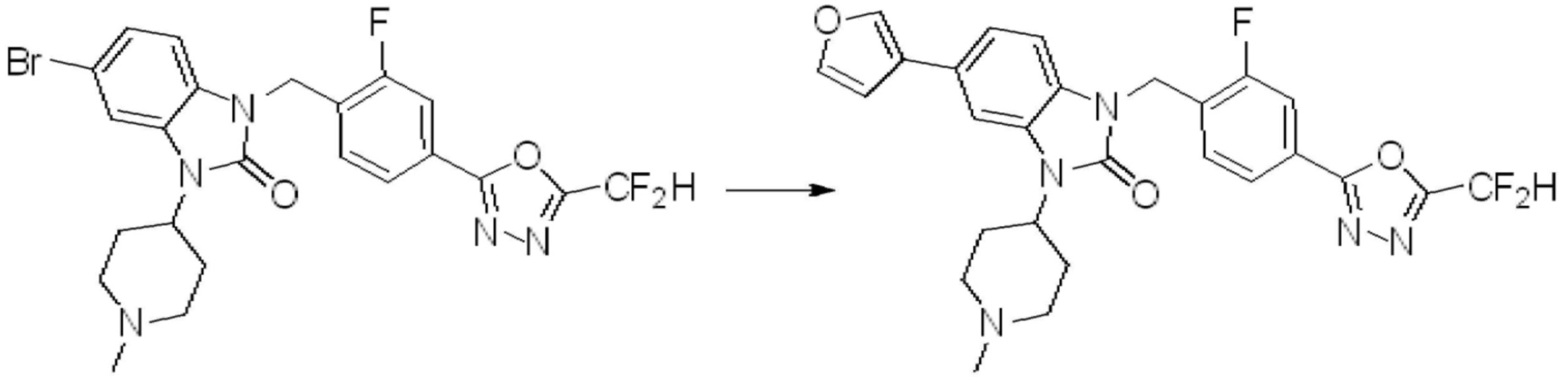
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,186 ммоль), полученный на стадии 3 соединения 139, фуран-3-илбороновую кислоту (0,025 г, 0,224 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,182 г, 0,559 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,042 г, 43,0%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,95-7,88 (м, 3H), 7,69 (д, J=1,3 Гц, 1H), 7,56 (т, J=1,7 Гц, 1H), 7,46 (т, J=7,8 Гц, 1H), 7,35-7,09 (м, 3H), 6,90 (д, J=1,0 Гц, 1H), 5,28 (с, 2H), 4,50-4,44 (м, 1H), 3,10 (д, J=11,8 Гц, 2H), 2,69-2,59 (м, 2H), 2,42 (с, 3H), 2,30 (тд, J=12,2, 2,1 Гц, 2H), 1,86 (дд, J=12,5, 2,5 Гц, 2H); МСНР (ЭР) m/z 524,2 (M+ + H).
Пример 163: Синтез соединения 163, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(5-метилфуран-2-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 163
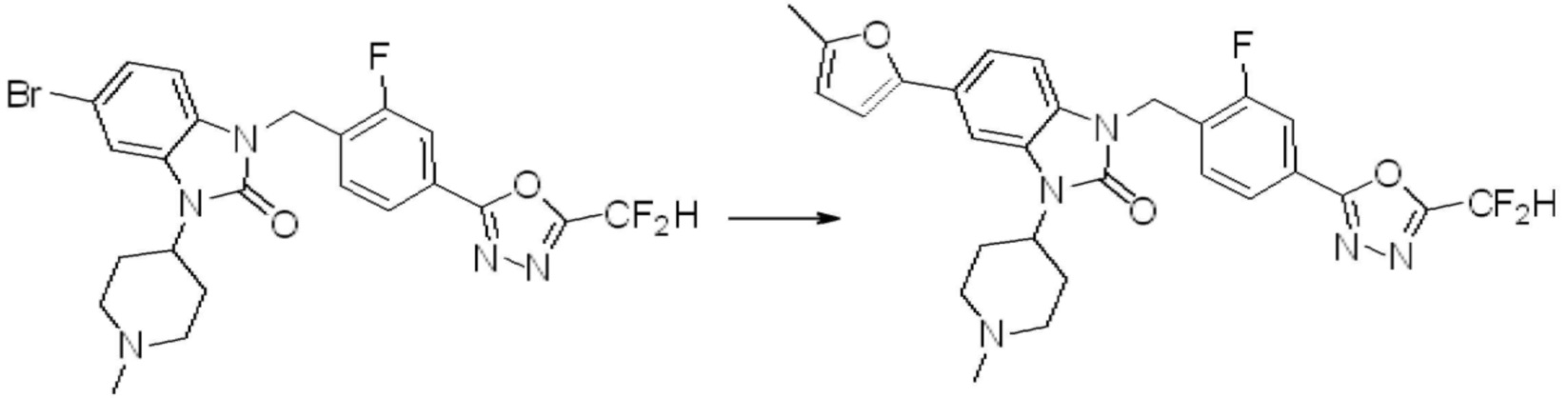
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,186 ммоль), полученный на стадии 3 соединения 139, 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан (0,047 г, 0,224 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,182 г, 0,559 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,043 г, 42,9%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,87-7,82 (м, 2H), 7,56 (с, 1H), 7,45 (т, J=7,8 Гц, 1H), 7,32 (дд, J=8,2, 1,4 Гц, 1H), 7,05-6,79 (м, 2H), 6,51-6,50 (м, 1H), 6,05-6,04 (м, 1H), 5,20 (с, 2H), 4,49-4,43 (м, 1H), 3,10 (д, J=11,2 Гц, 2H), 2,61-2,56 (м, 2H), 2,41-2,38 (м, 6H), 2,25 (т, J=11,4 Гц, 2H), 1,89 (д, J=10,4 Гц, 2H); МСНР (ЭР) m/z 538,2 (M+ + H).
Пример 164: Синтез соединения 164, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(6-метоксипиридин-3-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 164
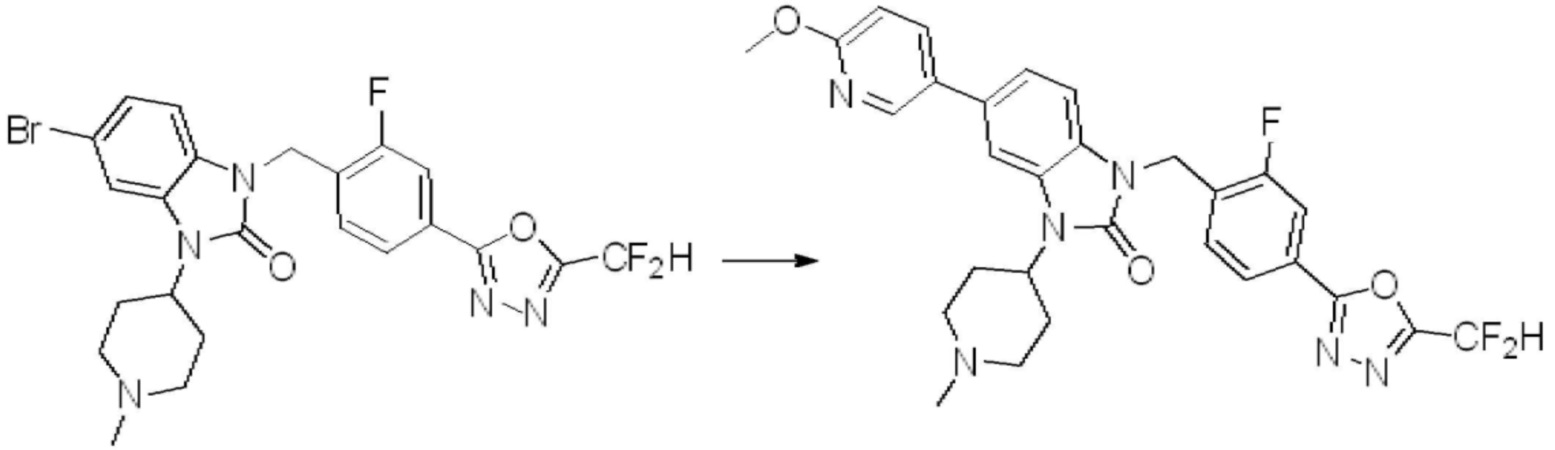
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,186 ммоль), полученный на стадии 3 соединения 139, (6-метоксипиридин-3-ил)бороновую кислоту (0,034 г, 0,224 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,182 г, 0,559 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,013 г, 12,4%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,32 (д, J=2,5 Гц, 1H), 7,87-7,83 (м, 2H), 7,75 (дд, J=8,6, 2,5 Гц, 1H), 7,48 (т, J=7,8 Гц, 1H), 7,43 (с, 1H), 7,18 (дд, J=8,1, 1,5 Гц, 1H), 7,05-6,79 (м, 3H), 5,23 (с, 2H), 4,51-4,44 (м, 1H), 3,96 (с, 3H), 3,05 (д, J=11,5 Гц, 2H), 2,59-2,51 (м, 2H), 2,37 (с, 3H), 2,20 (т, J=11,4 Гц, 2H), 1,89 (д, J=11,9 Гц, 2H); МСНР (ЭР) m/z 565,2 (M+ + H).
Пример 165: Синтез соединения 165, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 165
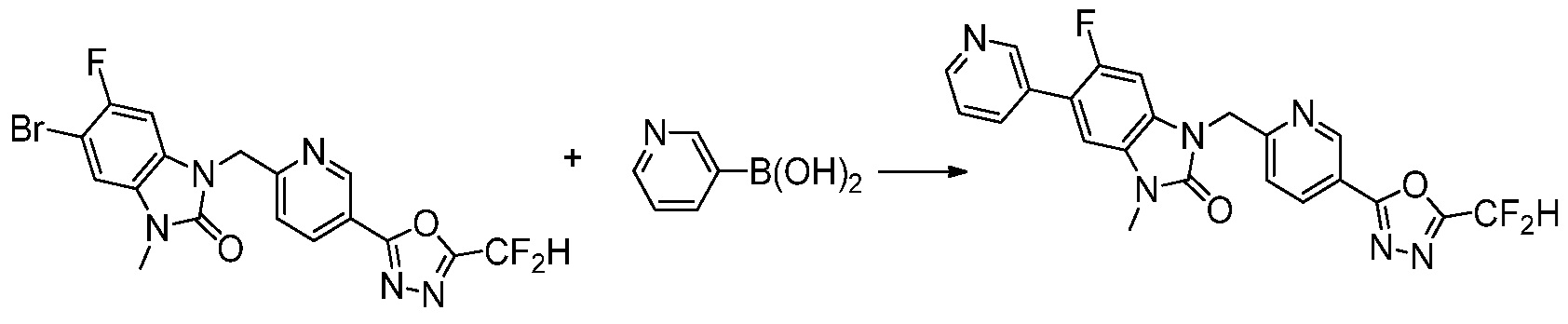
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,220 ммоль), полученный на стадии 4 соединения 147, пиридин-3-илбороновую кислоту (0,032 г, 0,264 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,007 г, 0,011 ммоль) и карбонат цезия (0,143 г, 0,440 ммоль) смешивают в 1,4-диоксане (1,7 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,007 г, 7,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (д, J=1,5 Гц, 1H), 8,77 (с, 1H), 8,60 (д, J=3,5 Гц, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 7,85 (д, J=8,3 Гц, 1H), 7,49 (д, J=8,4 Гц, 1H), 7,37 (дд, J=7,7, 4,7 Гц, 1H), 7,06-6,80 (м, 3H), 5,30 (с, 2H), 3,51 (с, 3H); МСНР (ЭР) m/z 453,3 (M+ + 1).
Пример 166: Синтез соединения 166, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(3-фторфенил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 166
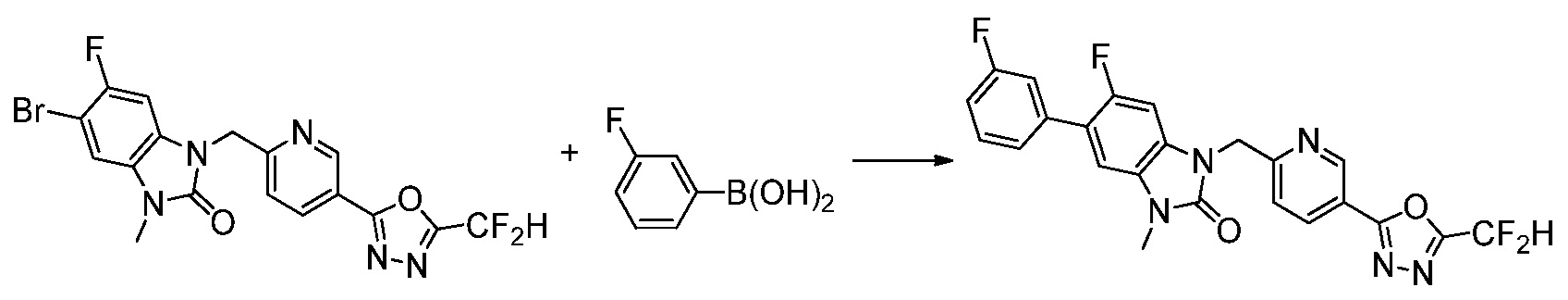
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,220 ммоль), полученный на стадии 4 соединения 147, (3-фторфенил)бороновую кислоту (0,037 г, 0,264 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,007 г, 0,011 ммоль) и карбонат цезия (0,143 г, 0,440 ммоль) смешивают в 1,4-диоксане (1,7 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=50-100%) и концентрируют с получением указанного в заголовке соединения (0,018 г, 17,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (д, J=2,2 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,48 (д, J=8,4 Гц, 1H), 7,42-7,36 (м, 1H), 7,30-7,20 (м, 2H), 7,07-6,80 (м, 4H), 5,28 (с, 2H), 3,49 (с, 3H); МСНР (ЭР) m/z 470,1 (M+ + 1).
Пример 167: Синтез соединения 167, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(4-(трифторметил)фенил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 167
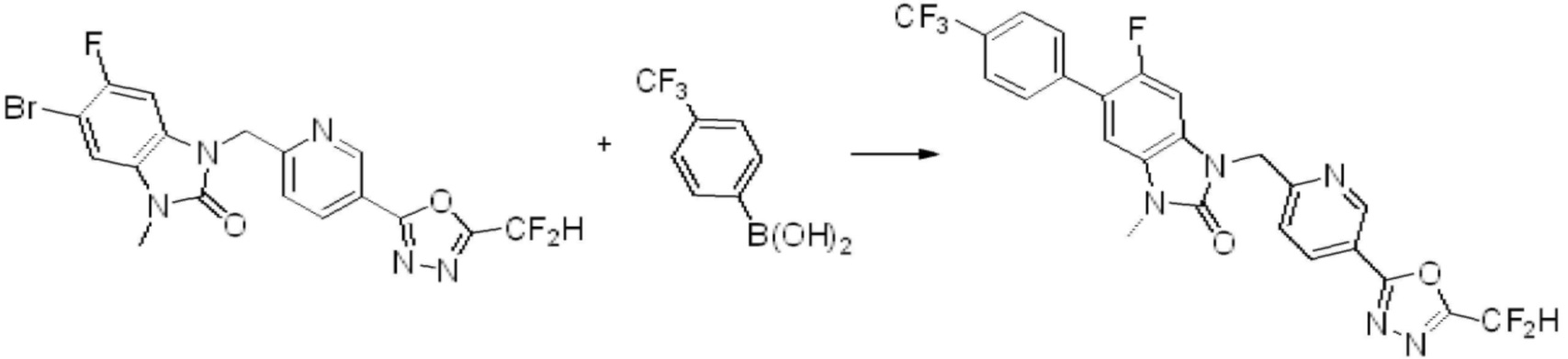
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,220 ммоль), полученный на стадии 4 соединения 147, (4-(трифторметил)фенил)бороновую кислоту (0,050 г, 0,264 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,007 г, 0,011 ммоль) и карбонат цезия (0,143 г, 0,440 ммоль) смешивают в 1,4-диоксане (1,7 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=50-100%) и концентрируют с получением указанного в заголовке соединения (0,019 г, 16,6%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (дд, J=2,2, 0,7 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,69 (д, J=8,3 Гц, 2H), 7,62 (д, J=8,0 Гц, 2H), 7,49 (дд, J=8,2, 0,6 Гц, 1H), 7,05-6,80 (м, 3H), 5,29 (с, 2H), 3,50 (с, 3H); МСНР (ЭР) m/z 520,1 (M+ + 1).
Пример 168: Синтез соединения 168, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(фуран-3-ил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 168
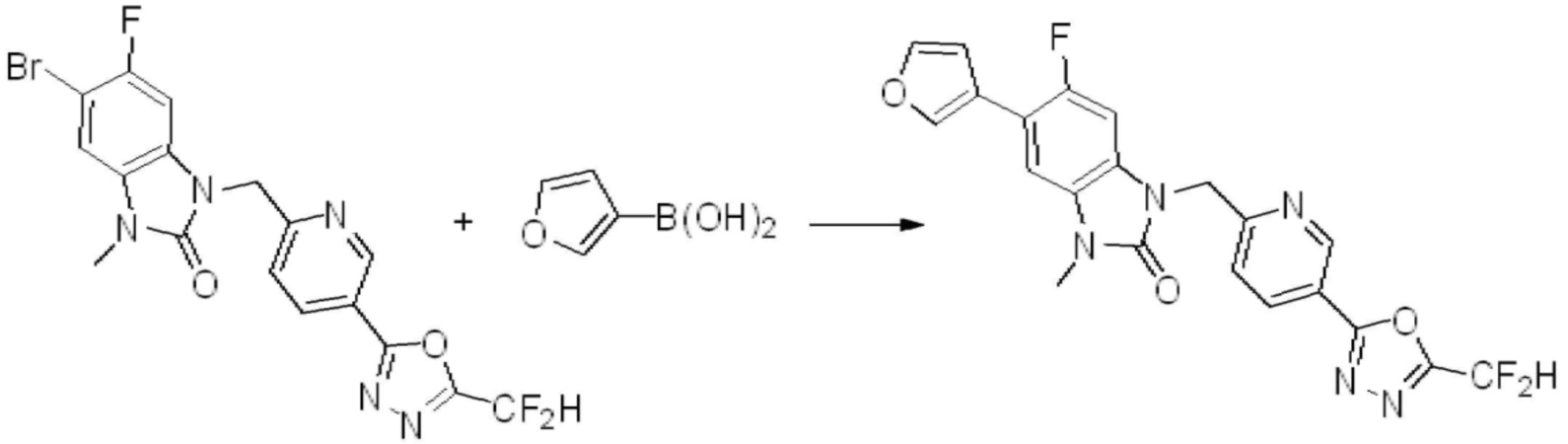
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,220 ммоль), полученный на стадии 4 соединения 147, фуран-3-илбороновую кислоту (0,030 г, 0,264 ммоль), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия дихлорид (0,007 г, 0,011 ммоль) и карбонат цезия (0,143 г, 0,440 ммоль) смешивают в 1,4-диоксане (1,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; гексан/этилацетат=50-100%) и концентрируют с получением указанного в заголовке соединения (0,033 г, 33,7%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (дд, J=2,2, 0,7 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,83-7,82 (м, 1H), 7,50-7,49 (м, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,05-6,74 (м, 4H), 5,26 (с, 2H), 3,50 (с, 3H); МСНР (ЭР) m/z 442,0 (M+ + 1).
Пример 169: Синтез соединения 169, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-метил-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 169
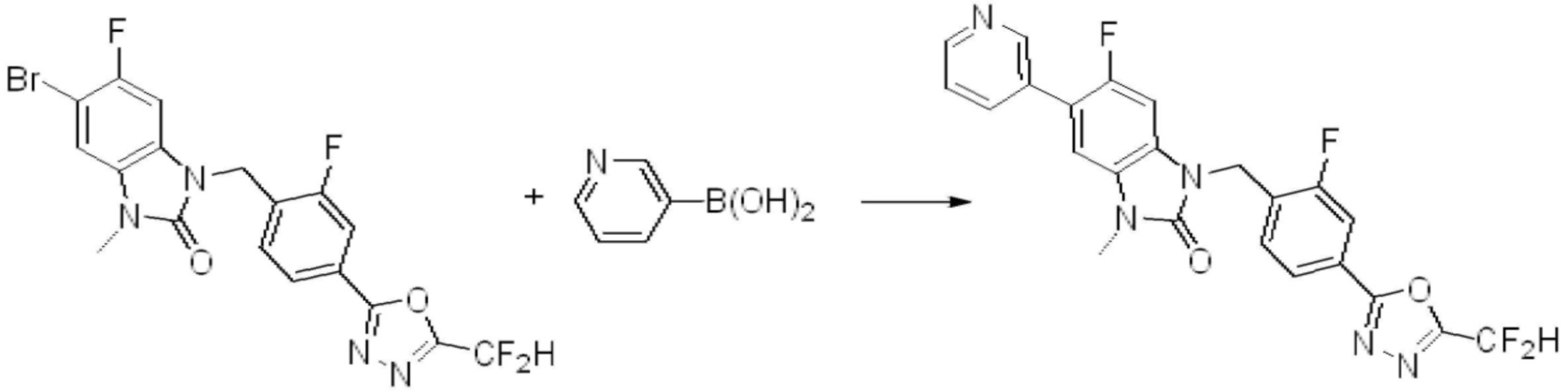
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,318 ммоль), полученный на стадии 1 соединения 148, пиридин-3-илбороновую кислоту (0,047 г, 0,382 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,010 г, 0,016 ммоль) и карбонат цезия (0,207 г, 0,637 ммоль) смешивают в 1,4-диоксане (1,7 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,107 г, 71,3%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,75 (с, 1H), 8,58 (дд, J=4,8, 1,5 Гц, 1H), 7,87-7,81 (м, 3H), 7,52 (т, J=7,5 Гц, 1H), 7,37-7,34 (м, 1H), 6,99 (д, J=6,2 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 6,84 (д, J=9,8 Гц, 1H), 5,20 (с, 2H), 3,49 (с, 3H); МСНР (ЭР) m/z 469,8 (M+ + 1).
Пример 170: Синтез соединения 170, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-метил-5-(5-метилфуран-2-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 170
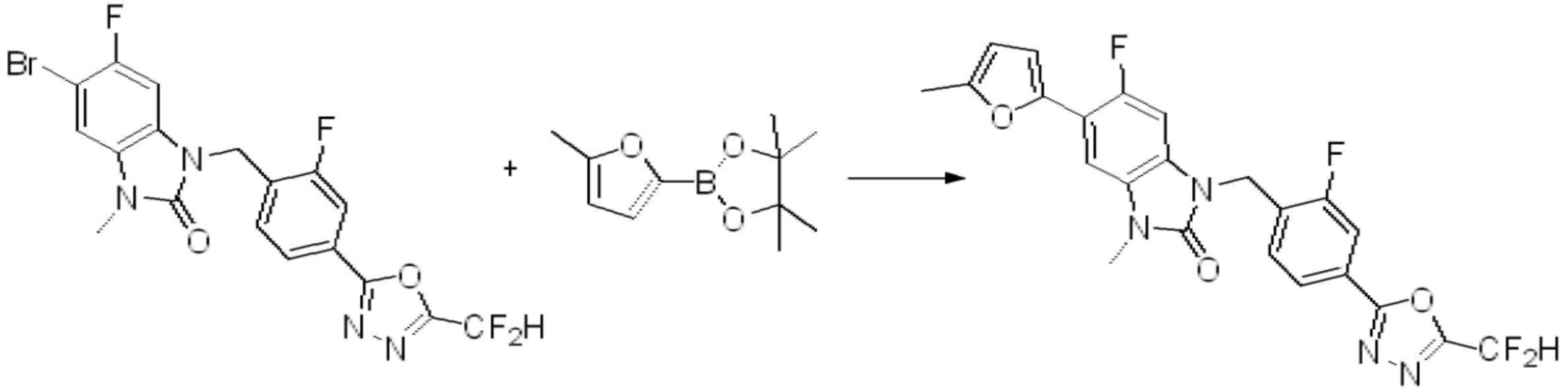
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,318 ммоль), полученный на стадии 1 соединения 148, 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан (0,079 г, 0,382 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,010 г, 0,016 ммоль) и карбонат цезия (0,207 г, 0,637 ммоль) смешивают в 1,4-диоксане (1,7 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=30-100%) и концентрируют с получением указанного в заголовке соединения (0,098 г, 65,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,88-7,84 (м, 2H), 7,51-7,47 (м, 1H), 7,36 (д, J=6,1 Гц, 1H), 7,03-6,73 (м, 2H), 6,64 (т, J=3,6 Гц, 1H), 6,10-6,09 (м, 1H), 5,16 (с, 2H), 3,51 (с, 3H), 2,39 (д, J=0,5 Гц, 3H); МСНР (ЭР) m/z 473,2 (M+ + 1).
Пример 171: Синтез соединения 171, 1-(1-ацетилпиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 171
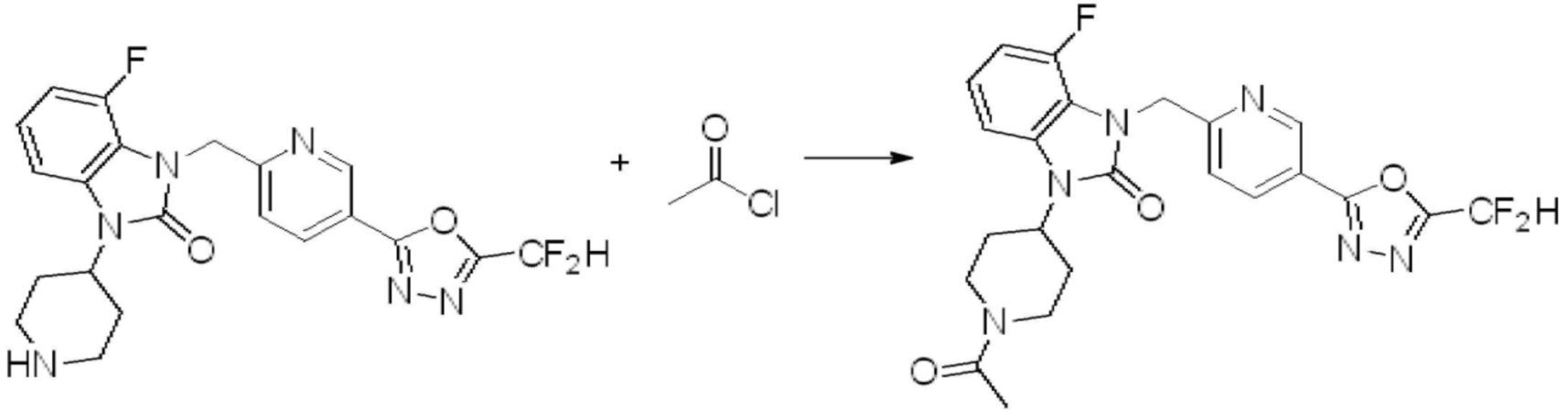
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 76, триэтиламин (0,063 мл, 0,450 ммоль) и ацетилхлорид (0,032 мл, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,060 г, 54,8%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,22 (д, J=1,6 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,33 (д, J=8,3 Гц, 1H), 7,04-6,73 (м, 4H), 5,41 (с, 2H), 4,88-4,85 (м, 1H), 4,60-4,52 (м, 1H), 4,02-3,99 (м, 1H), 3,26-3,20 (м, 1H), 2,70-2,63 (м, 1H), 2,41-2,26 (м, 2H), 2,16 (с, 3H), 1,97-1,90 (м, 2H); МСНР (ЭР) m/z 487,0 (M+ + 1).
Пример 172: Синтез соединения 172, 1-(1-ацетилпиперидин-4-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 172
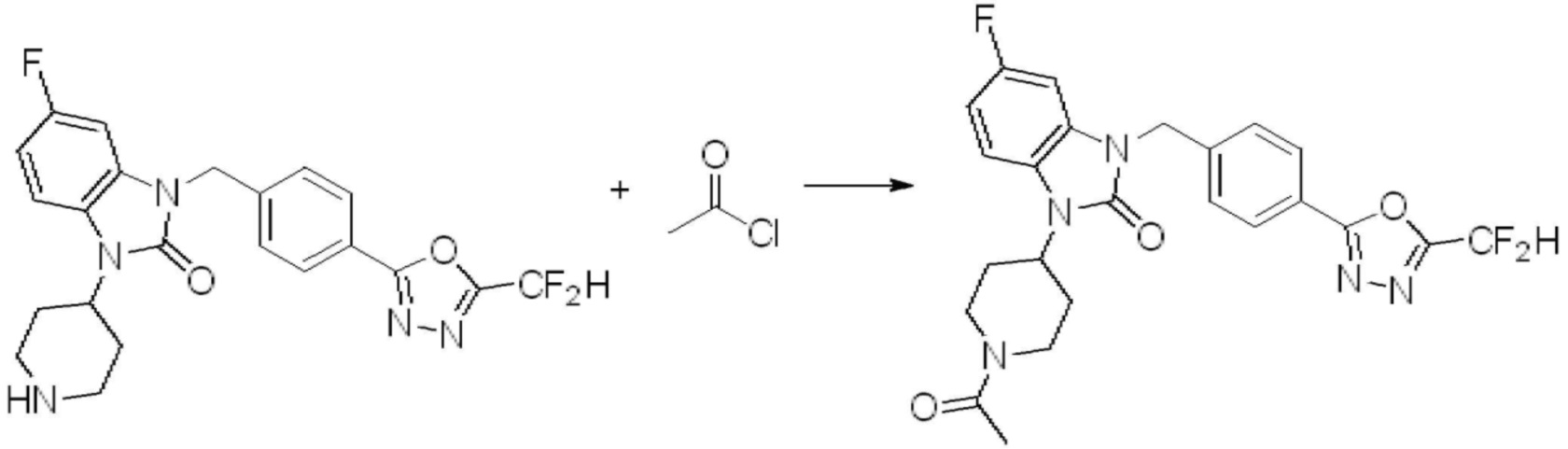
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,226 ммоль), полученный на стадии 2 соединения 79, триэтиламин (0,063 мл, 0,451 ммоль) и ацетилхлорид (0,032 мл, 0,451 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,097 г, 88,5%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,05 (дд, J=6,7, 1,7 Гц, 2H), 7,45 (д, J=8,4 Гц, 2H), 7,02-6,72 (м, 3H), 6,59 (дд, J=8,3, 2,4 Гц, 1H), 5,27 (с, 2H), 4,87-4,84 (м, 1H), 4,58-4,50 (м, 1H), 4,00 (д, J=13,6 Гц, 1H), 3,26-3,19 (м, 1H), 2,69-2,63 (м, 1H), 2,37-2,22 (м, 2H), 2,15 (с, 3H), 1,95-1,88 (м, 2H); МСНР (ЭР) m/z 486,1 (M+ + 1).
Пример 173: Синтез соединения 173, 1-(1-ацетилпиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 173
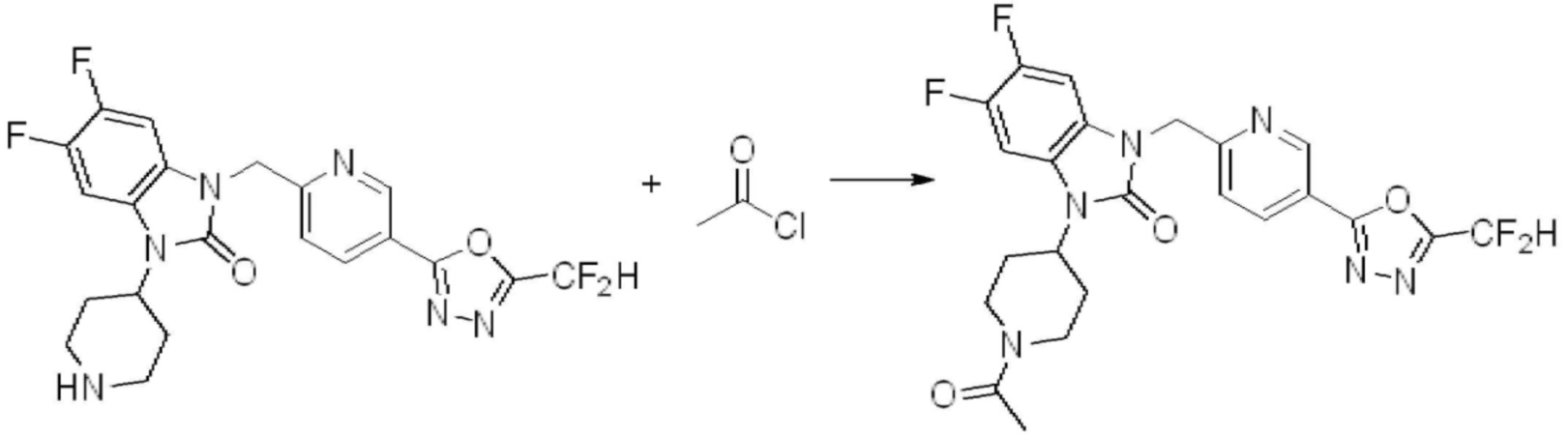
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,216 ммоль), полученный на стадии 4 соединения 83, триэтиламин (0,060 мл, 0,433 ммоль) и ацетилхлорид (0,031 мл, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,052 г, 48,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (дд, J=2,2, 0,7 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,44 (д, J=8,2 Гц, 1H), 7,05-6,79 (м, 3H), 5,19 (д, J=1,9 Гц, 2H), 4,88-4,84 (м, 1H), 4,51-4,44 (м, 1H), 4,01 (дд, J=13,8, 2,1 Гц, 1H), 3,26-3,19 (м, 1H), 2,69-2,62 (м, 1H), 2,34-2,18 (м, 2H), 2,15 (с, 3H), 1,95-1,87 (м, 2H); МСНР (ЭР) m/z 505,0 (M+ + 1).
Пример 174: Синтез соединения 174, 1-(1-ацетилпиперидин-4-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5,6-дифтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 174
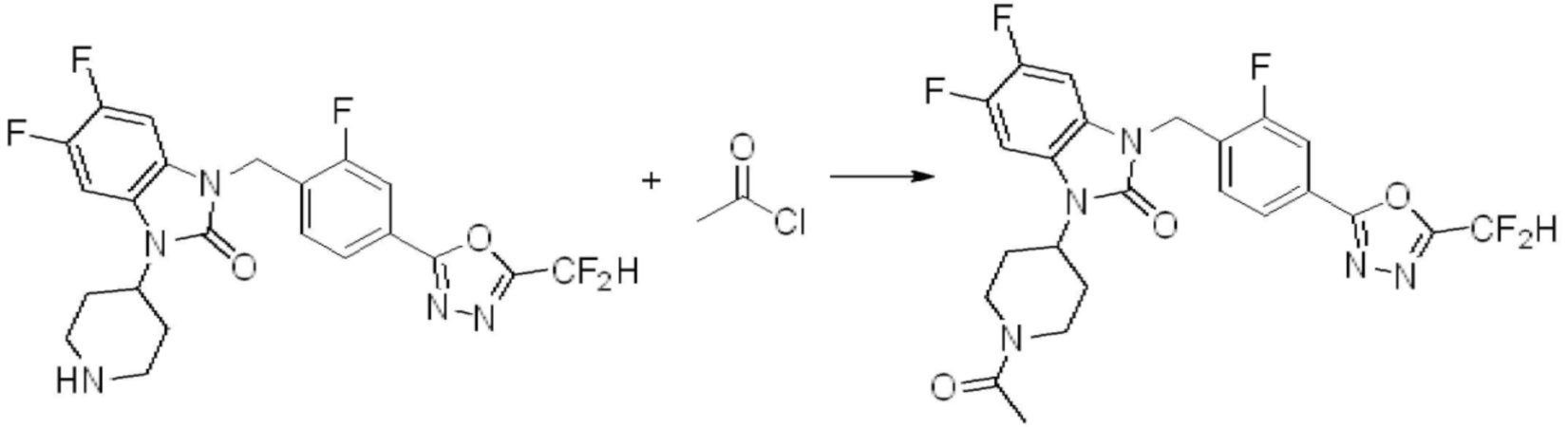
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,209 ммоль), полученный на стадии 2 соединения 93, триэтиламин (0,058 мл, 0,417 ммоль) и ацетилхлорид (0,030 мл, 0,417 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,099 г, 91,1%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,85-7,81 (м, 2H), 7,45 (т, J=7,7 Гц, 1H), 7,03-6,77 (м, 3H), 5,10 (с, 2H), 4,87-4,84 (м, 1H), 4,51-4,43 (м, 1H), 4,02-3,99 (м, 1H), 3,24-3,18 (м, 1H), 2,67-2,61 (м, 1H), 2,35-2,17 (м, 2H), 2,14 (с, 3H), 1,93-1,86 (м, 2H); МСНР (ЭР) m/z 521,9 (M+ + 1).
Пример 175: Синтез соединения 175, 1-(1-ацетилпиперидин-4-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 175
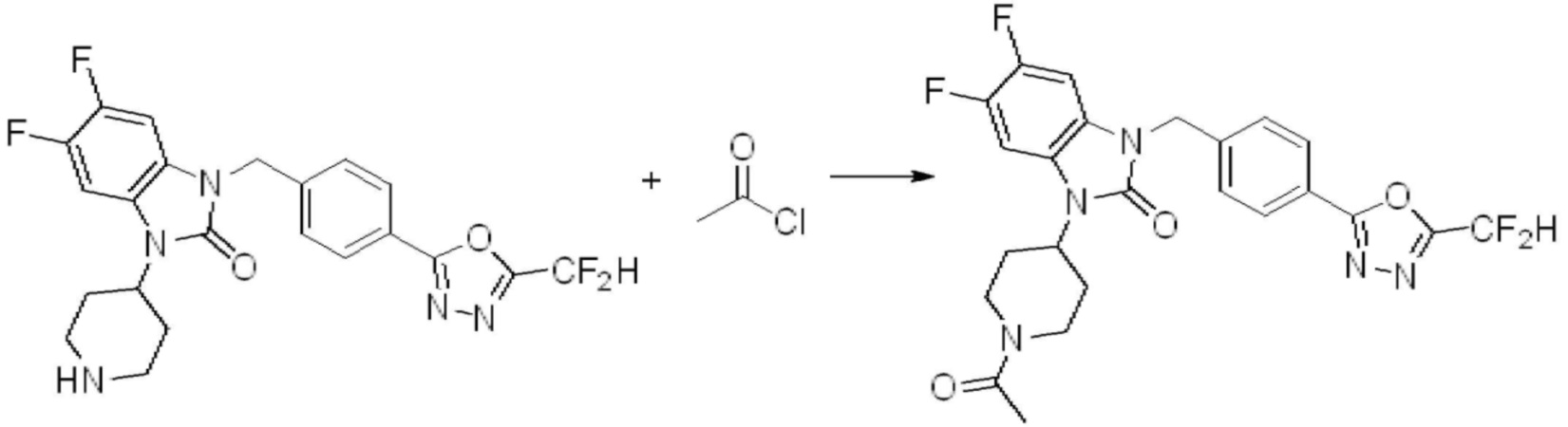
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 95, триэтиламин (0,060 мл, 0,433 ммоль) и ацетилхлорид (0,031 мл, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,079 г, 72,4%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,07 (д, J=8,3 Гц, 2H), 7,43 (д, J=8,3 Гц, 2H), 6,96 (дд, J=10,1, 6,6 Гц, 1H), 6,90 (т, J=51,7 Гц, 1H), 6,66 (дд, J=9,5, 6,8 Гц, 1H), 5,05 (с, 2H), 4,87 (д, J=13,6 Гц, 1H), 4,54-4,45 (м, 1H), 4,01 (д, J=13,8 Гц, 1H), 3,26-3,19 (м, 1H), 2,69-2,63 (м, 1H), 2,35-2,19 (м, 2H), 2,16 (с, 3H), 1,95-1,88 (м, 2H); МСНР (ЭР) m/z 504,1 (M+ + 1).
Пример 176: Синтез соединения 176, 1-(1-ацетилпиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 176
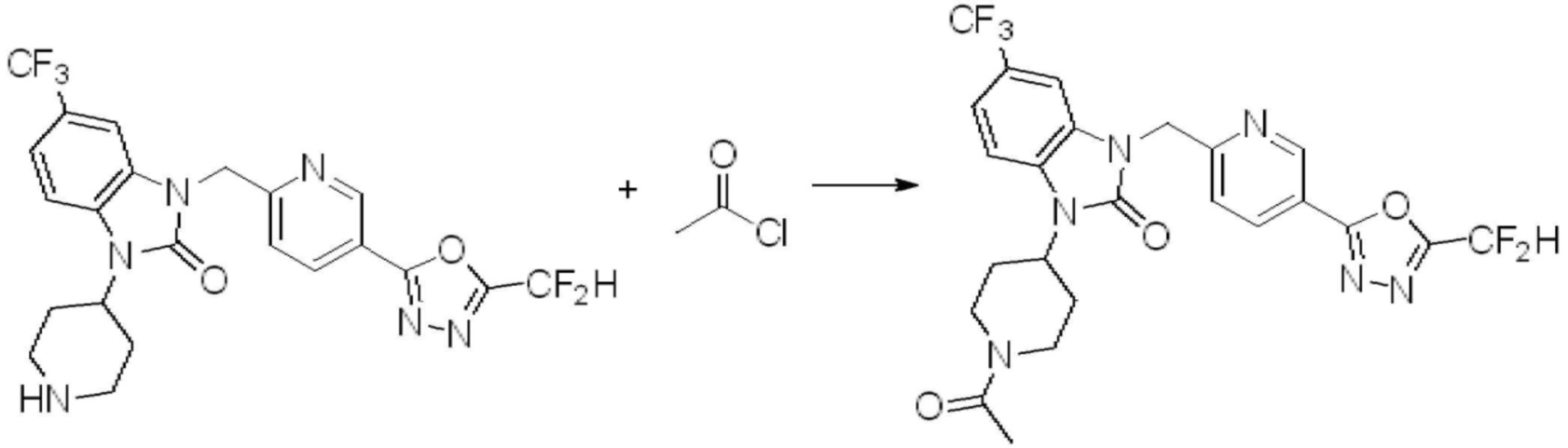
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,202 ммоль), полученный на стадии 5 соединения 89, триэтиламин (0,056 мл, 0,405 ммоль) и ацетилхлорид (0,029 мл, 0,405 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,083 г, 76,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,2, 0,7 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,45 (дд, J=6,0, 6,5 Гц, 1H), 7,36 (дд, J=8,3, 0,8 Гц, 1H), 7,29 (д, J=1,3 Гц, 1H), 7,19 (д, J=8,3 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 5,29 (с, 2H), 4,89 (дд, J=11,5, 2,0 Гц, 1H), 4,62-4,54 (м, 1H), 4,03 (дд, J=12,0, 2,0 Гц, 1H), 3,28-3,21 (м, 1H), 2,68 (тд, J=13,1, 2,0 Гц, 1H), 2,41-2,26 (м, 2H), 2,17 (с, 3H), 1,97-1,91 (м, 2H); МСНР (ЭР) m/z 537,1 (M+ + 1).
Пример 177: Синтез соединения 177, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(5-метилфуран-2-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 177
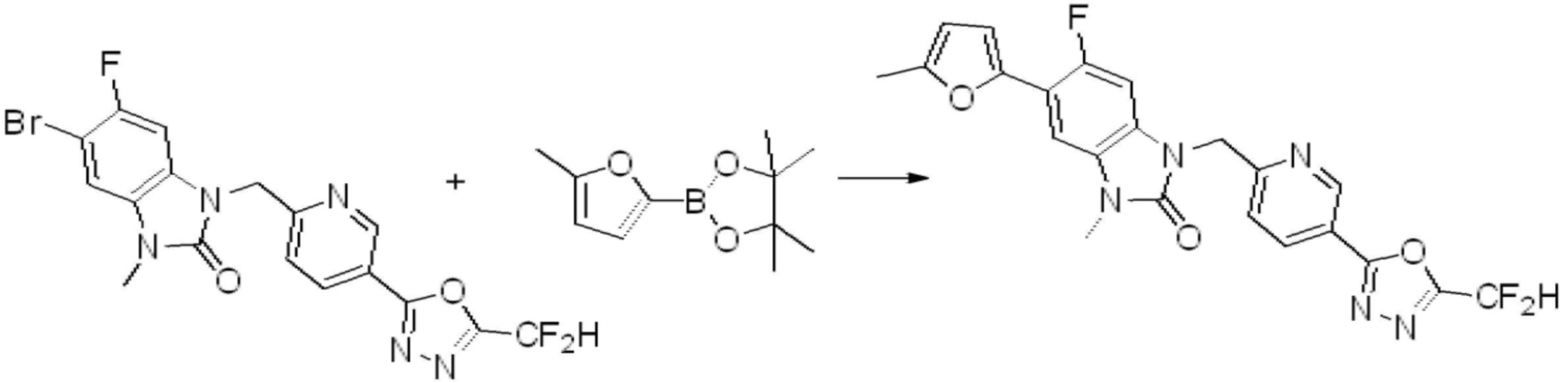
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,264 ммоль), полученный на стадии 4 соединения 147, 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан (0,066 г, 0,317 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,013 ммоль) и карбонат цезия (0,172 г, 0,528 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=30-100%) и концентрируют с получением указанного в заголовке соединения (0,055 г, 45,8%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,6 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,44 (д, J=8,2 Гц, 1H), 7,34 (д, J=6,1 Гц, 1H), 7,05-6,76 (м, 2H), 6,61 (т, J=3,6 Гц, 1H), 6,08-6,07 (м, 1H), 5,23 (с, 2H), 3,50 (с, 3H), 2,38 (с, 3H); МСНР (ЭР) m/z 457,1 (M+ + 1).
Пример 178: Синтез соединения 178, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(3,5-диметилизоксазол-4-ил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 178
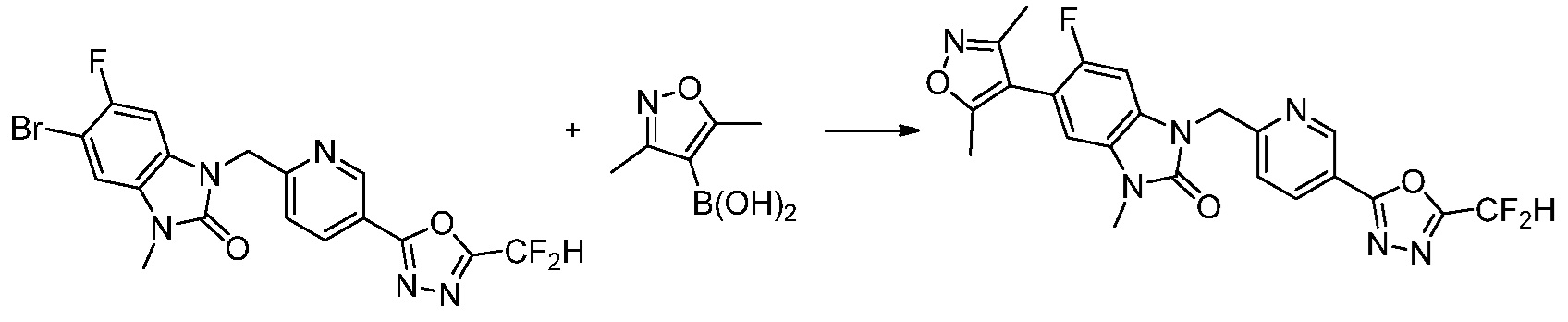
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,264 ммоль), полученный на стадии 4 соединения 147, (3,5-диметилизооксазол-4-ил)бороновую кислоту (0,045 г, 0,317 ммоль), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия дихлорид (0,009 г, 0,013 ммоль) и карбонат цезия (0,172 г, 0,528 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=30-100%) и концентрируют с получением указанного в заголовке соединения (0,044 г, 35,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,7 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,51 (д, J=8,2 Гц, 1H), 7,05-6,79 (м, 2H), 6,74 (д, J=5,8 Гц, 1H), 5,27 (с, 2H), 3,47 (с, 3H), 2,31 (с, 3H), 2,17 (с, 3H); МСНР (ЭР) m/z 471,2 (M+ + 1).
Пример 179: Синтез соединения 179, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(1-(2-гидроксиацетил)пиперидин-4-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 179
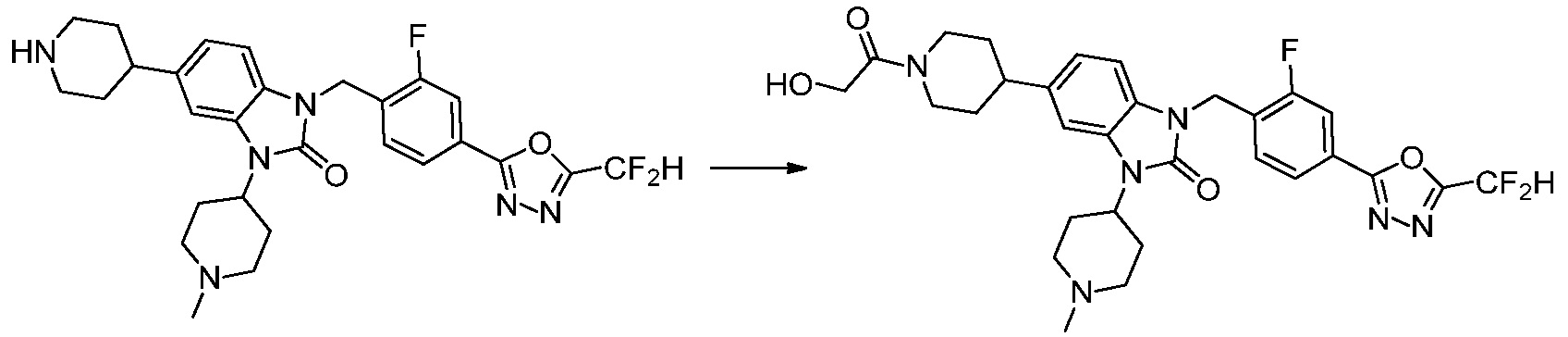
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-5-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 0,370 ммоль), полученный на стадии 3 соединения 156, 2-гидроксиуксусную кислоту (0,034 г, 0,444 ммоль), 3-оксид гексафторфосфат 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (ГАТУ, 0,281 г, 0,740 ммоль) и N,N-диизопропилэтиламин (0,322 мл, 1,850 ммоль) растворяют в N,N-диметилформамиде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением продукта. Затем, полученный продукт снова очищают хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,034 г, 15,4%) в виде желтого масла.
1H ЯМР (400 MГц, CDCl3) δ 7,86-7,82 (м, 2H), 7,46 (т, J=7,8 Гц, 1H), 7,28 (шс, 1H), 7,05-6,79 (м, 3H), 5,18 (с, 2H), 4,76 (д, J=13,3 Гц, 1H), 4,50-4,44 (м, 1H), 4,22 (кв, J=14,2 Гц, 2H), 3,63 (д, J=13,5 Гц, 1H), 3,14-3,07 (м, 3H), 2,83-2,68 (м, 2H), 2,54-2,51 (м, 2H), 2,40 (с, 3H), 2,23 (т, J=10,5 Гц, 2H), 1,93-1,85 (м, 4H), 1,72-1,61 (м, 2H); МСНР (ЭР) m/z 599,3 (M+ + H).
Пример 180: Синтез соединения 180, 1-(1-ацетилпиперидин-4-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 180
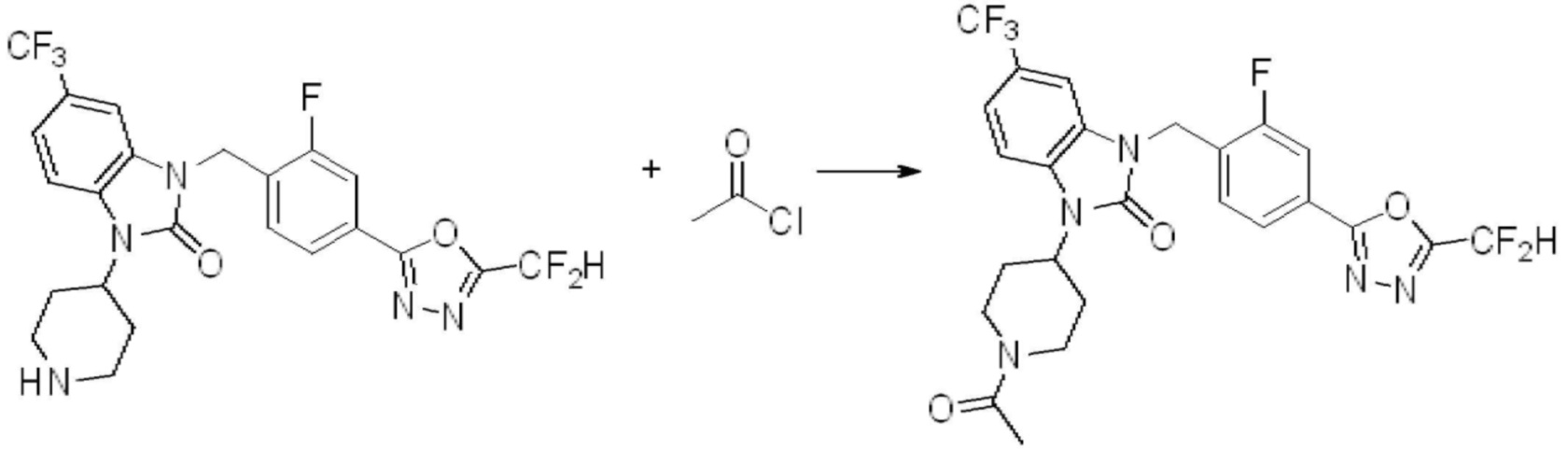
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,196 ммоль), полученный на стадии 2 соединения 91, триэтиламин (0,055 мл, 0,391 ммоль) и ацетилхлорид (0,028 мл, 0,391 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенным водным раствором выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,093 г, 85,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,85-7,82 (м, 2H), 7,46 (т, J=7,7 Гц, 1H), 7,35 (дд, J=8,3, 0,8 Гц, 1H), 7,22-7,18 (м, 2H), 6,90 (т, J=51,7 Гц, 1H), 5,19 (с, 2H), 4,89-4,85 (м, 1H), 4,60-4,52 (м, 1H), 4,03-4,00 (м, 1H), 3,27-3,20 (м, 1H), 2,67 (тд, J=13,9, 3,3 Гц, 1H), 2,40-2,24 (м, 2H), 2,15 (с, 3H), 1,92 (т, J=14,5 Гц, 2H); МСНР (ЭР) m/z 554,2 (M+ + 1).
Пример 181: Синтез соединения 181, 3-(1-ацетилпиперидин-4-ил)-5-хлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 181
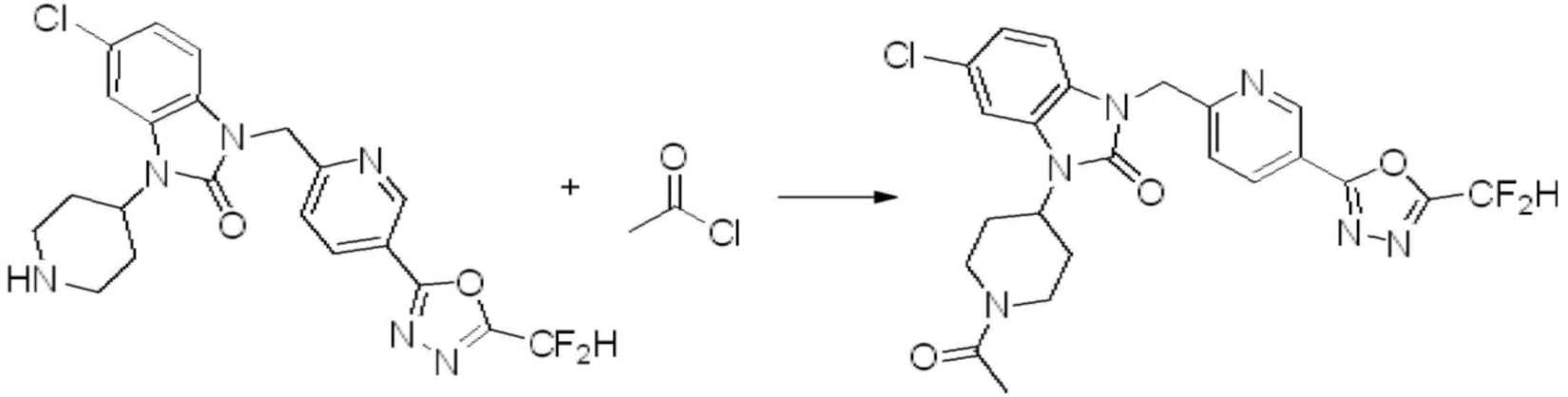
5-хлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 97, триэтиламин (0,060 мл, 0,434 ммоль) и ацетилхлорид (0,031 мл, 0,434 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,081 г, 73,8%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,23 (дд, J=2,2, 0,7 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,39 (д, J=8,2 Гц, 1H), 7,09 (д, J=1,8 Гц, 1H), 7,05-6,79 (м, 3H), 5,22 (д, J=2,0 Гц, 2H), 4,86 (дд, J=11,5, 2,1 Гц, 1H), 4,54-4,46 (м, 1H), 4,02-3,99 (м, 1H), 3,26-3,18 (м, 1H), 2,69-2,62 (м, 1H), 2,39-2,23 (м, 2H), 2,15 (с, 3H), 1,94-1,87 (м, 2H); МСНР (ЭР) m/z 503,1 (M+ + 1).
Пример 182: Синтез соединения 182, 3-(1-ацетилпиперидин-4-ил)-5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 182
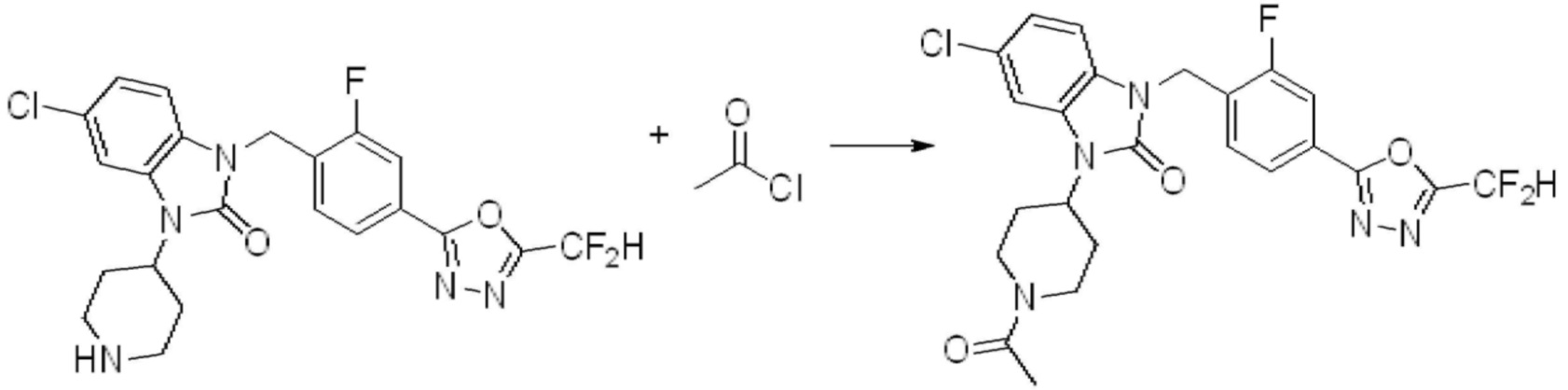
5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,209 ммоль), полученный на стадии 2 соединения 101, триэтиламин (0,058 мл, 0,419 ммоль) и ацетилхлорид (0,030 мл, 0,419 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,049 г, 44,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,82 (д, J=8,6 Гц, 2H), 7,43 (т, J=7,5 Гц, 1H), 7,09 (д, J=1,8 Гц, 1H), 7,03-6,77 (м, 3H), 5,14 (с, 2H), 4,90-4,85 (м, 1H), 4,54-4,46 (м, 1H), 4,03-4,00 (м, 1H), 3,23 (тд, J=13,2, 2,3 Гц, 1H), 2,70-2,63 (м, 1H), 2,39-2,22 (м, 2H), 2,17 (с, 3H), 1,94-1,87 (м, 2H); МСНР (ЭР) m/z 520,1 (M+ + 1).
Пример 183: Синтез соединения 183, 3-(1-ацетилпиперидин-4-ил)-5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 183
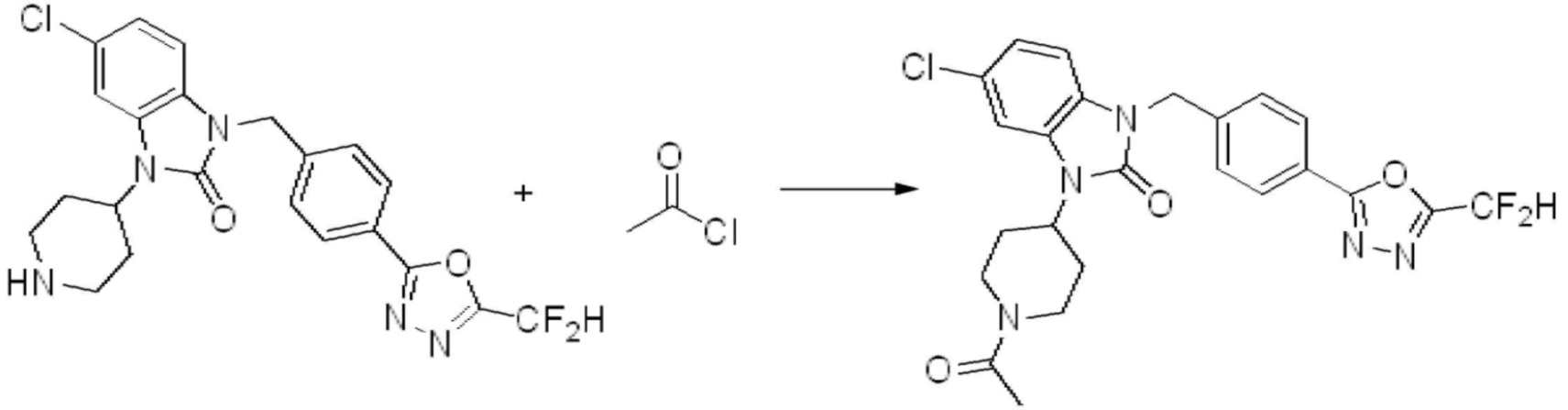
5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 99, триэтиламин (0,061 мл, 0,435 ммоль) и ацетилхлорид (0,031 мл, 0,435 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,084 г, 76,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,07-8,04 (м, 2H), 7,43 (д, J=8,5 Гц, 2H), 7,10 (д, J=1,8 Гц, 1H), 6,97 (дд, J=8,4, 1,8 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 6,74 (д, J=8,4 Гц, 1H), 5,10 (с, 2H), 4,90-4,86 (м, 1H), 4,55-4,48 (м, 1H), 4,04-4,00 (м, 1H), 3,23 (тд, J=13,2, 2,1 Гц, 1H), 2,70-2,64 (м, 1H), 2,40-2,23 (м, 2H), 2,17 (с, 3H), 1,96-1,88 (м, 2H); МСНР (ЭР) m/z 502,2 (M+ + 1).
Пример 184: Синтез соединения 184, 1-(1-ацетилпиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез соединения 184
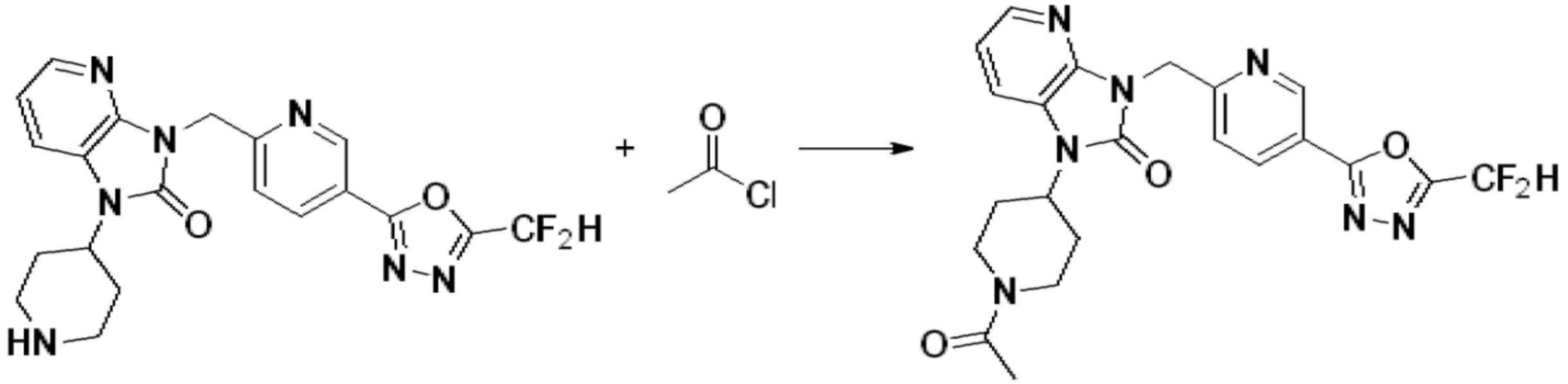
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,234 ммоль), полученный на стадии 2 соединения 107, триэтиламин (0,065 мл, 0,468 ммоль) и ацетилхлорид (0,033 мл, 0,468 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = 0-5%) и концентрируют с получением указанного в заголовке соединения (0,089 г, 81,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,19 (д, J=2,2 Гц, 1H), 8,29 (дд, J=8,2, 2,2 Гц, 1H), 7,98 (дд, J=5,2, 1,2 Гц, 1H), 7,43 (д, J=8,2 Гц, 1H), 7,31 (дд, J=7,9, 1,3 Гц, 1H), 6,97 (дд, J=7,8, 5,2 Гц, 1H), 6,91 (т, J=51,6 Гц, 1H), 5,36 (с, 2H), 4,86-4,82 (м, 1H), 4,61-4,55 (м, 1H), 4,00-3,97 (м, 1H), 3,25-3,18 (м, 1H), 2,64 (тд, J=13,0, 2,2 Гц, 1H), 2,25-2,18 (м, 2H), 2,13 (с, 3H), 1,99-1,90 (м, 2H); МСНР (ЭР) m/z 471,4 (M+ + 2).
Пример 185: Синтез соединения 185, 1-(1-ацетилпиперидин-4-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез соединения 185
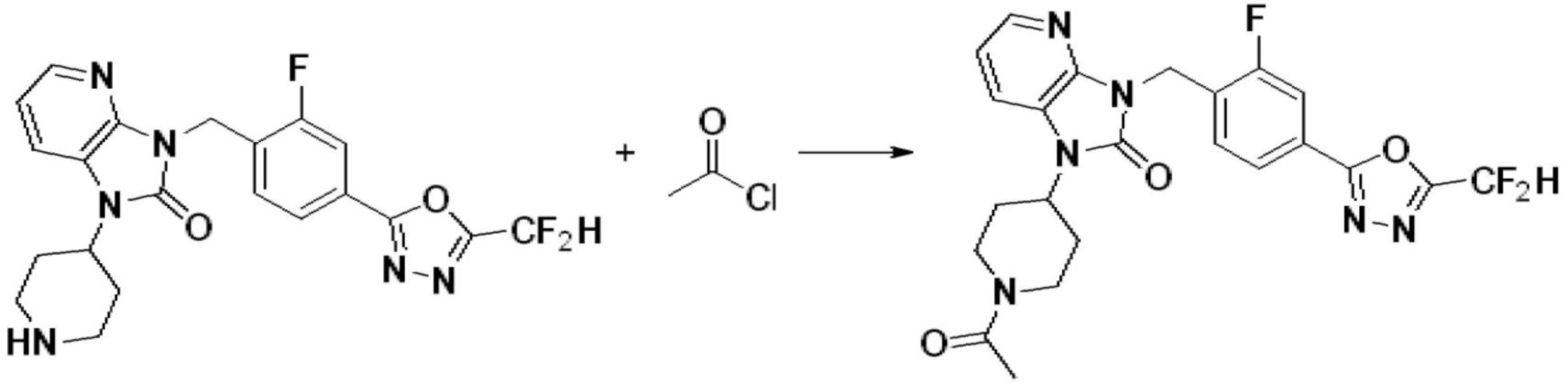
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 101, триэтиламин (0,063 мл, 0,450 ммоль) и ацетилхлорид (0,032 мл, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,082 г, 75,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,01 (дд, J=5,2, 1,3 Гц, 1H), 7,80-7,76 (м, 2H), 7,43 (т, J=7,7 Гц, 1H), 7,31 (дд, J=7,9, 1,3 Гц, 1H), 6,99 (дд, J=7,8, 5,2 Гц, 1H), 6,89 (т, J=51,7 Гц, 1H), 5,27 (д, J=3,9 Гц, 2H), 4,86-4,82 (м, 1H), 4,61-4,53 (м, 1H), 3,99 (дд, J=12,0, 1,9 Гц, 1H), 3,25-3,18 (м, 1H), 2,64 (тд, J=13,0, 2,1 Гц, 1H), 2,27-2,17 (м, 2H), 2,14 (с, 3H), 1,97-1,89 (м, 2H); МСНР (ЭР) m/z 487,0 (M+ + 1).
Пример 186: Синтез соединения 186, 1-(1-ацетилпиперидин-4-ил)-3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез соединения 186
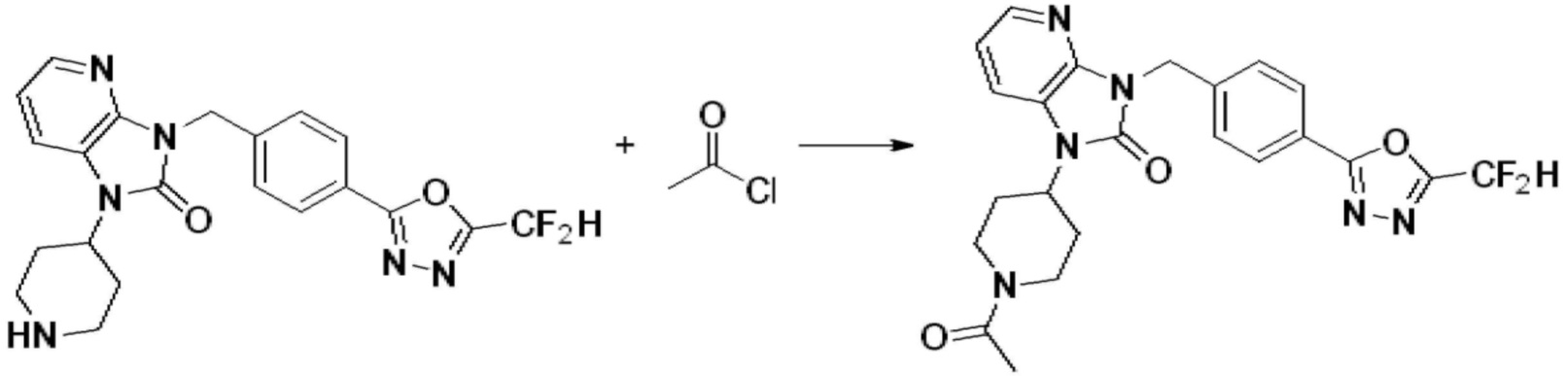
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,235 ммоль), полученный на стадии 2 соединения 105, триэтиламин (0,065 мл, 0,469 ммоль) и ацетилхлорид (0,033 мл, 0,469 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,093 г, 85,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,01-7,98 (м, 3H), 7,59 (д, J=8,4 Гц, 2H), 7,28 (дд, J=7,8, 1,3 Гц, 1H), 6,95 (дд, J=7,8, 5,2 Гц, 1H), 6,87 (т, J=51,7 Гц, 1H), 5,17 (с, 2H), 4,81 (дд, J=11,5, 2,1 Гц, 1H), 4,58-4,50 (м, 1H), 3,98-3,95 (м, 1H), 3,23-3,16 (м, 1H), 2,62 (тд, J=13,0, 2,1 Гц, 1H), 2,25-2,11 (м, 5H), 1,93-1,85 (м, 2H); МСНР (ЭР) m/z 469,2 (M+ + 1).
Пример 187: Синтез соединения 187, 1-(1-ацетилпиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 187
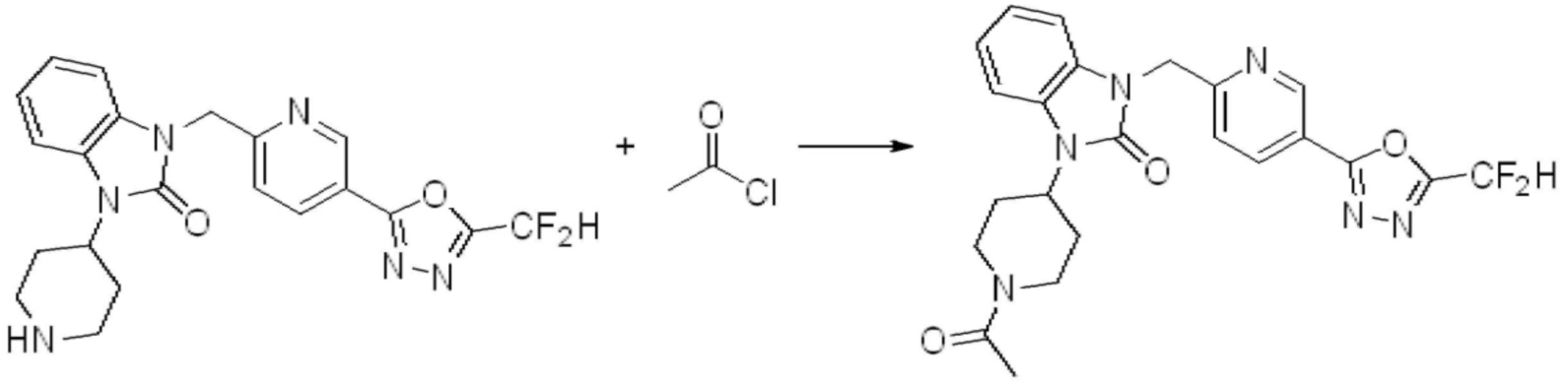
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,235 ммоль), триэтиламин (0,065 мл, 0,469 ммоль) и ацетилхлорид (0,033 мл, 0,469 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,085 г, 77,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,23 (д, J=2,1 Гц, 1H), 8,27 (дд, J=8,2, 2,2 Гц, 1H), 7,37 (д, J=8,2 Гц, 1H), 7,11-6,78 (м, 5H), 5,24 (д, J=1,9 Гц, 2H), 4,83 (дд, J=11,5, 2,0 Гц, 1H), 4,60-4,51 (м, 1H), 3,99 (дд, J=11,9, 1,9 Гц, 1H), 3,22 (тд, J=13,2, 2,1 Гц, 1H), 2,65 (тд, J=13,0, 2,2 Гц, 1H), 2,41-2,25 (м, 2H), 2,13 (с, 3H), 1,95-1,87 (м, 2H); МСНР (ЭР) m/z 468,9 (M+ + 1).
Пример 188: Синтез соединения 188, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез 4-((4-фтор-2-нитрофенил)амино)тетрагидро-2H-тиопиран 1,1-диоксид
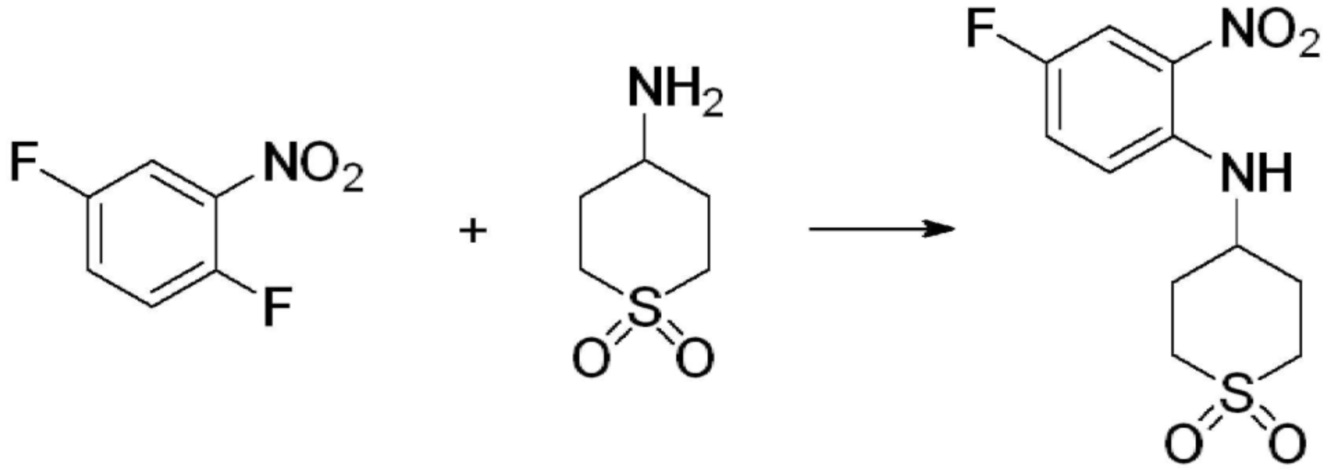
1,4-дифтор-2-нитробензол (0,500 г, 3,143 ммоль), 1,1-диоксид 4-аминотетрагидро-2H-тиопирана (0,563 г, 3,771 ммоль), карбонат калия (0,869 г, 6,286 ммоль) и йодид калия (0,052 г, 0,314 ммоль) растворяют в N,N-диметилформамиде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=30-100%) и концентрируют с получением указанного в заголовке соединения (0,536 г, 59,2%) в виде желтого твердого вещества.
[Стадия 2] Синтез 4-((2-амино-4-фторфенил)амино)тетрагидро-2H-тиопиран 1,1-диоксид
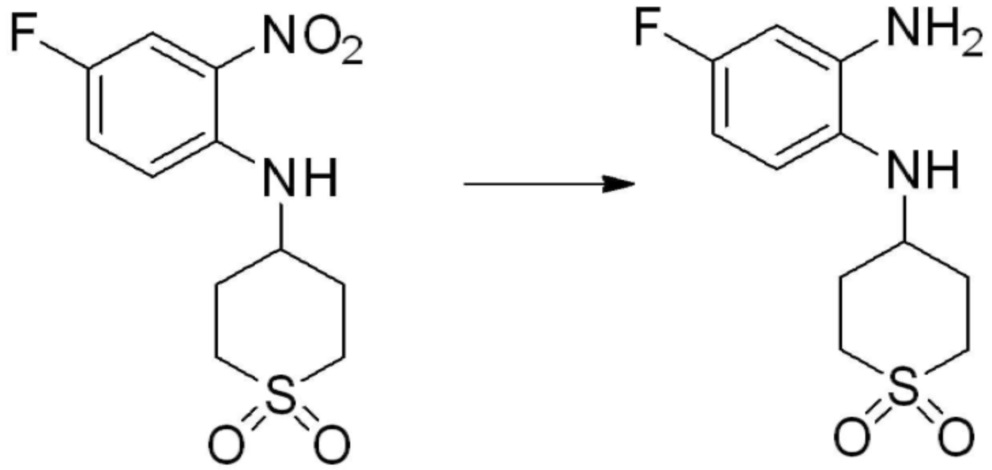
1,1-диоксид 4-((4-фтор-2-нитрофенил)амино)тетрагидро-2H-тиопиран (0,536 г, 1,859 ммоль), полученный на стадии 1 и карбид палладия (45,00%, 0,044 г, 0,186 ммоль) растворяют в этаноле (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (0,477 г, 99,3%, коричневое твердое вещество).
[Стадия 3] Синтез 1-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
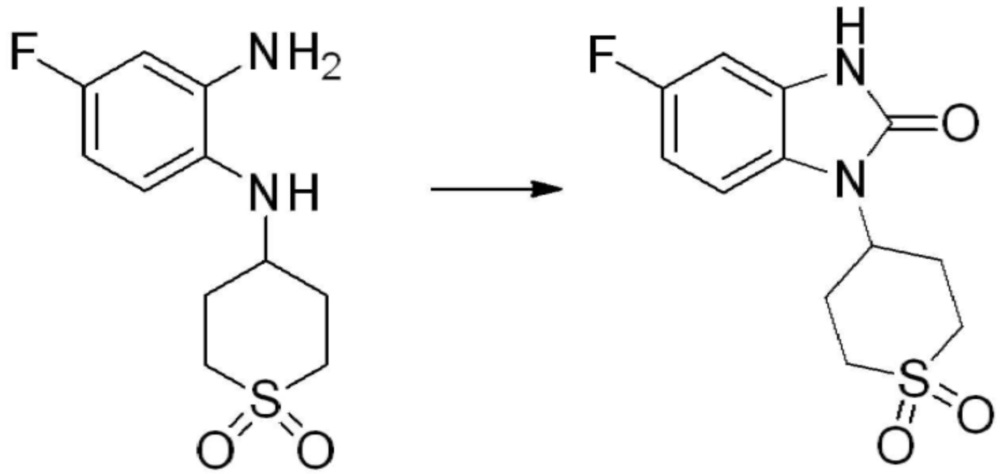
1,1-диоксид 4-((2-амино-4-фторфенил)амино)тетрагидро-2H-тиопиран (0,500 г, 1,936 ммоль), полученный на стадии 2, 1,1'-карбонилдиимидазол (КДИ, 0,408 г, 2,516 ммоль) и триэтиламин (0,270 мл, 1,936 ммоль) растворяют в тетрагидрофуране (12 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Выпавшее в осадок твердое вещество фильтруют и сушат с получением указанного в заголовке соединения (0,383 г, 69,6%) в виде серого твердого вещества.
[Стадия 4] Синтез соединения 188
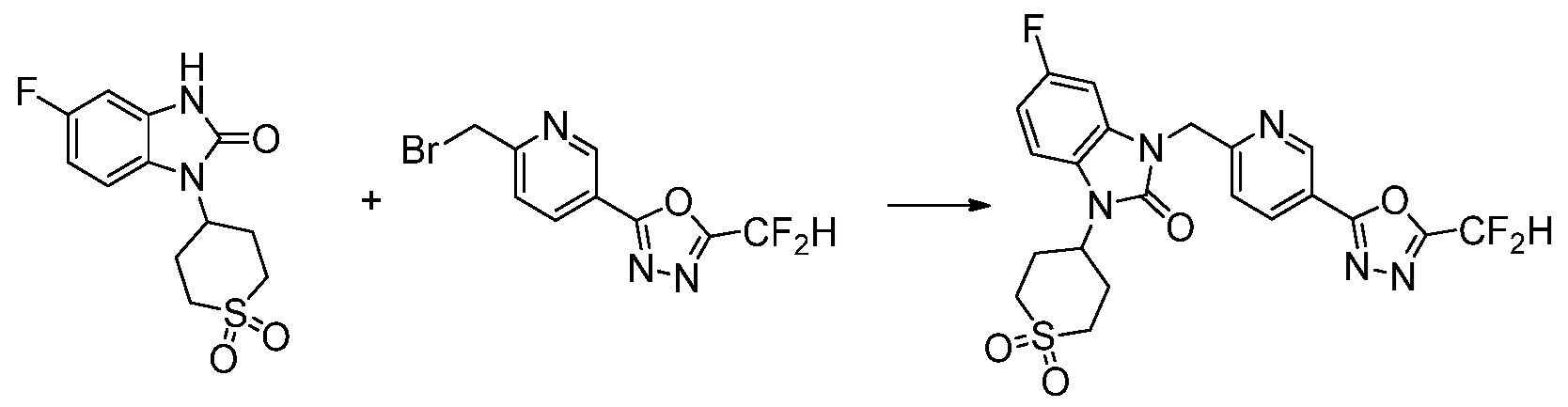
1-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,126 г, 0,443 ммоль), полученный на стадии 3 растворяют в N,N-диметилформамиде (2 мл) при 0°C, после чего гидрид натрия (60,00%, 0,023 г, 0,576 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,141 г, 0,487 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-3%) и концентрируют с получением указанного в заголовке соединения (0,120 г, 54,9%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (дд, J=2,2, 0,8 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,45 (дд, J=8,2, 0,7 Гц, 1H), 7,17 (дд, J=9,4, 4,2 Гц, 1H), 7,06-6,80 (м, 3H), 5,24 (с, 2H), 4,76-4,68 (м, 1H), 3,31-3,18 (м, 4H), 3,09-2,98 (м, 2H), 2,27-2,23 (м, 2H); МСНР (ЭР) m/z 493,2 (M+ + 1).
Пример 189: Синтез соединения 189, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 189
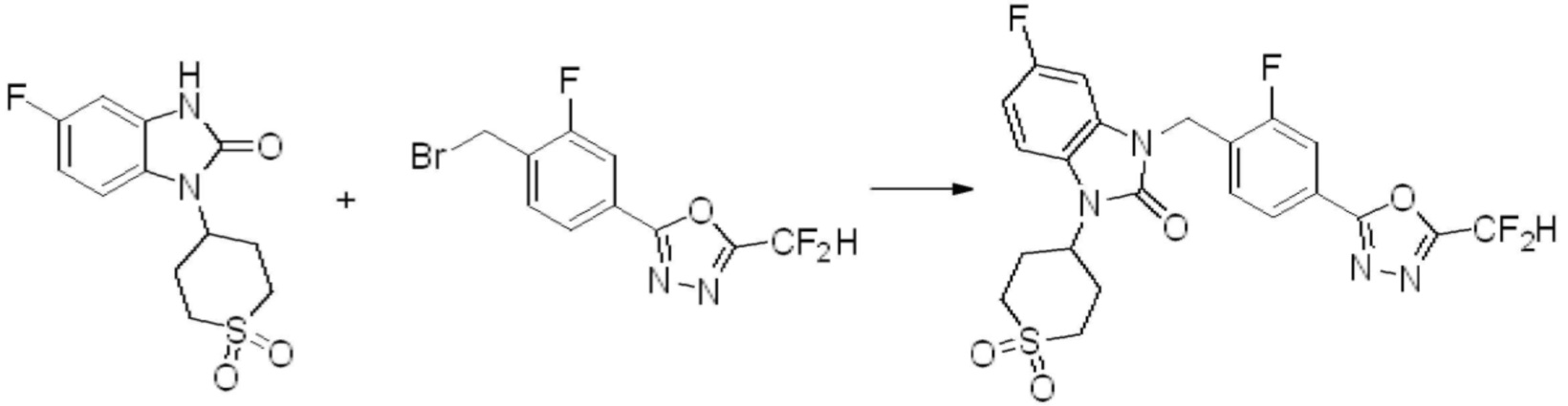
1-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,126 г, 0,443 ммоль), полученный на стадии 3 соединения 188, растворяют в N,N-диметилформамиде (2 мл) при 0°C, после чего гидрид натрия (60,00%, 0,023 г, 0,576 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,150 г, 0,487 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-3%) и концентрируют с получением указанного в заголовке соединения (0,132 г, 58,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,85-7,81 (м, 2H), 7,45 (т, J=7,7 Гц, 1H), 7,14 (дд, J=8,7, 4,2 Гц, 1H), 7,03-6,73 (м, 3H), 5,12 (с, 2H), 4,73-4,65 (м, 1H), 3,31-3,15 (м, 4H), 3,06-2,94 (м, 2H), 2,23-2,19 (м, 2H); МСНР (ЭР) m/z 511,3 (M+ + 1).
Пример 190: Синтез соединения 190, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 190
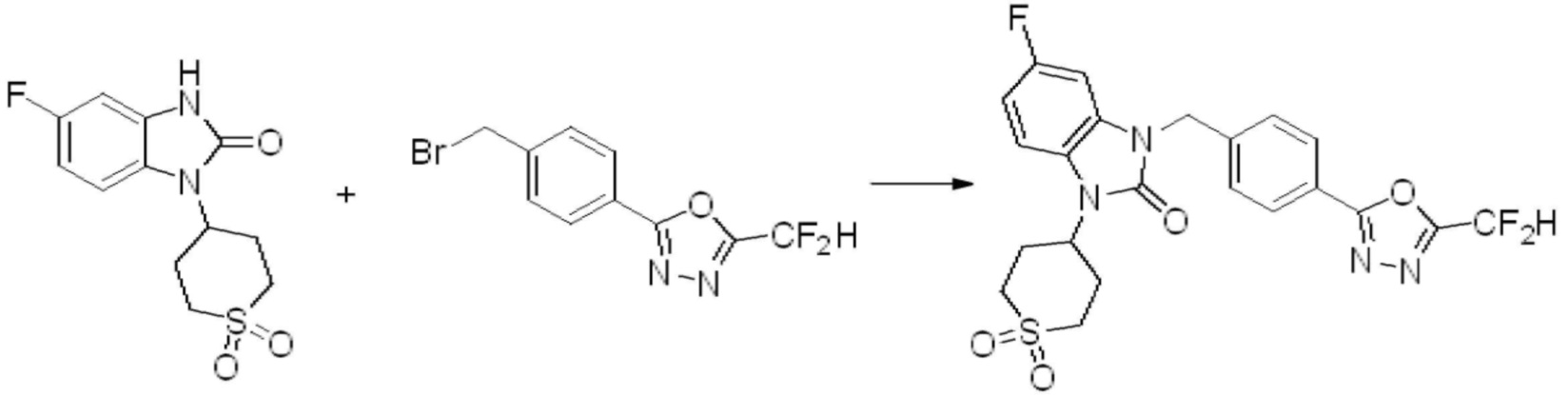
1-(1,1-диоксидотетрагидро-2H-тиопиран-4-ил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,126 г, 0,443 ммоль), полученный на стадии 3 соединения 188, растворяют в N,N-диметилформамиде (2 мл) при 0°C, после чего гидрид натрия (60,00%, 0,023 г, 0,576 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,141 г, 0,487 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-3%) и концентрируют с получением указанного в заголовке соединения (0,130 г, 59,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,06 (дд, J=8,4, 1,8 Гц, 2H), 7,44 (д, J=8,6 Гц, 2H), 7,15 (дд, J=8,7, 4,2 Гц, 1H), 6,90 (т, J=51,7 Гц, 1H), 6,82-6,77 (м, 1H), 6,62 (дд, J=8,2, 2,4 Гц, 1H), 5,09 (с, 2H), 4,76-4,68 (м, 1H), 3,32-3,16 (м, 4H), 3,07-2,96 (м, 2H), 2,25-2,21 (м, 2H); МСНР (ЭР) m/z 494,1 (M+ + 1).
Пример 191: Синтез соединения 191, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фтор-1-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 191
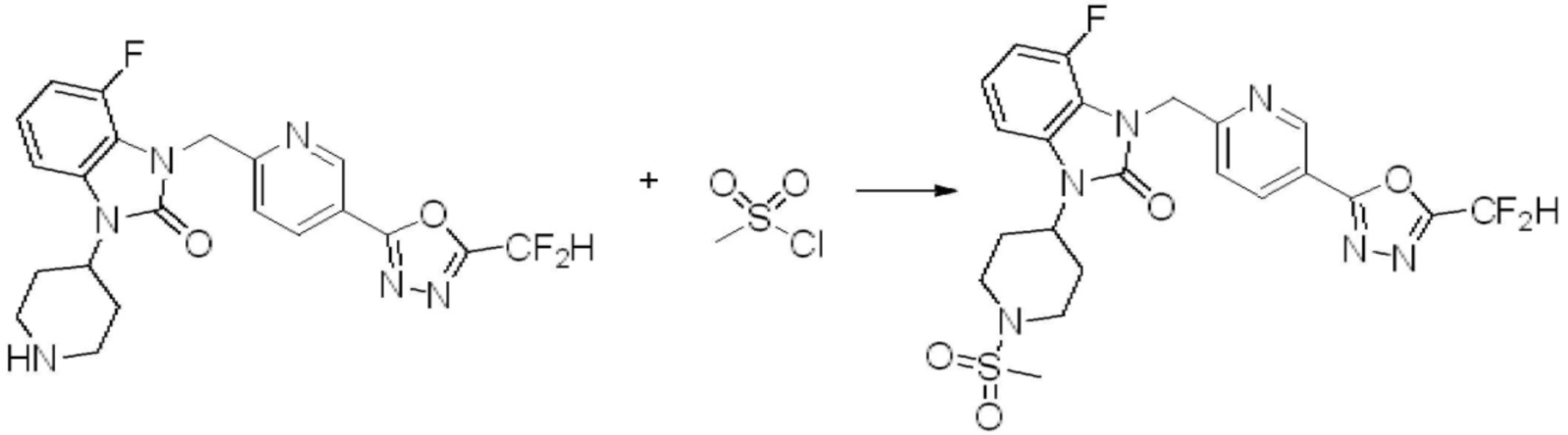
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 76, триэтиламин (0,063 мл, 0,450 ммоль) и метансульфонилхлорид (0,035 мл, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,079 г, 67,5%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,23 (д, J=1,8 Гц, 1H), 8,33 (дд, J=8,2, 2,0 Гц, 1H), 7,34 (д, J=8,2 Гц, 1H), 7,05-6,76 (м, 4H), 5,42 (с, 2H), 4,58-4,50 (м, 1H), 4,03 (д, J=12,3 Гц, 2H), 2,92-2,86 (м, 5H), 2,60-2,50 (м, 2H), 1,99 (дд, J=12,3, 2,2 Гц, 2H); МСНР (ЭР) m/z 523,0 (M+ + 1).
Пример 192: Синтез соединения 192, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-1-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 192
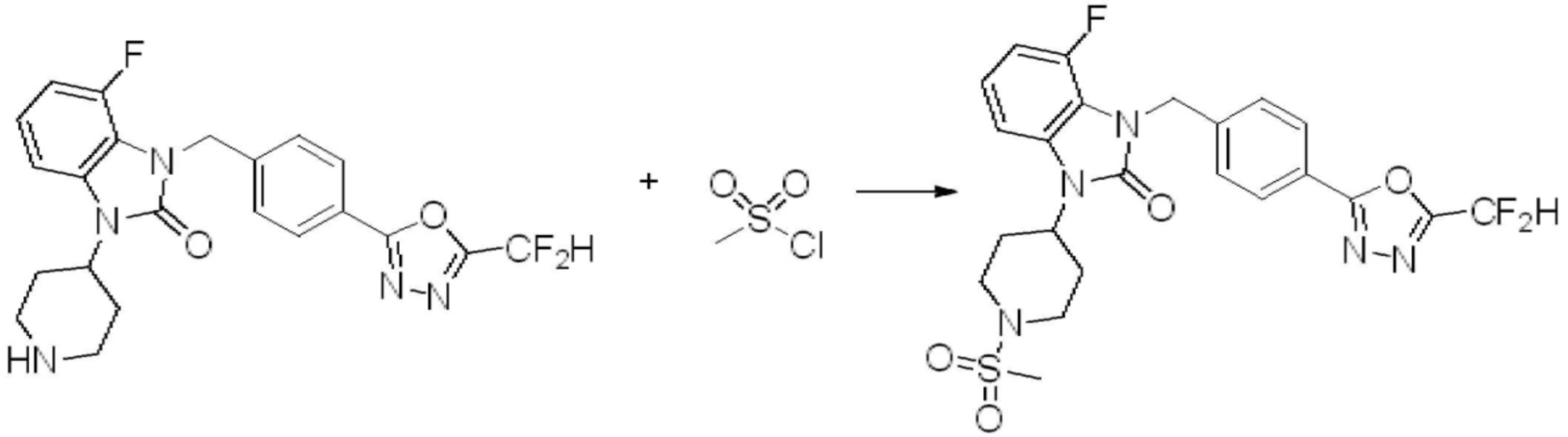
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5-фтор-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,226 ммоль), полученный на стадии 2 соединения 79, триэтиламин (0,063 мл, 0,451 ммоль) и метансульфонилхлорид (0,035 мл, 0,451 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,087 г, 74,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,04 (д, J=8,4 Гц, 2H), 7,66-7,05 (м, 3H), 7,34 (дд, J=8,7, 4,5 Гц, 1H), 7,16 (дд, J=9,0, 2,5 Гц, 1H), 6,91-6,86 (м, 1H), 5,16 (с, 2H), 4,44-4,37 (м, 1H), 3,73 (д, J=11,8 Гц, 2H), 2,97-2,91 (м, 5H), 2,45-2,33 (м, 2H), 1,86 (д, J=10,0 Гц, 2H); МСНР (ЭР) m/z 522,2 (M+ + 1).
Пример 193: Синтез соединения 193, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-3-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 193
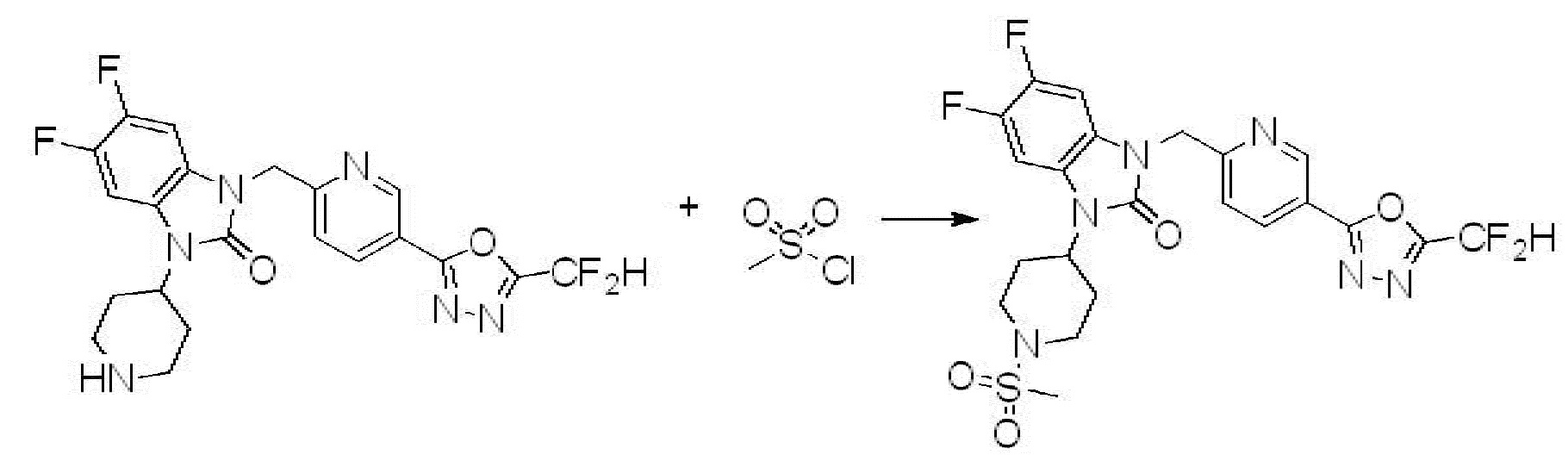
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,216 ммоль), полученный на стадии 4 соединения 83, триэтиламин (0,060 мл, 0,433 ммоль) и метансульфонилхлорид (0,033 мл, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,073 г, 62,6%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=2,2 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,07-6,80 (м, 3H), 5,21 (с, 2H), 4,50-4,44 (м, 1H), 4,04 (дд, J=10,2, 2,2 Гц, 2H), 2,91-2,85 (м, 5H), 2,53-2,42 (м, 2H), 1,97 (дд, J=12,2, 2,4 Гц, 2H); МСНР (ЭР) m/z 541,2 (M+ + 1).
Пример 194: Синтез соединения 194, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-3-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 194
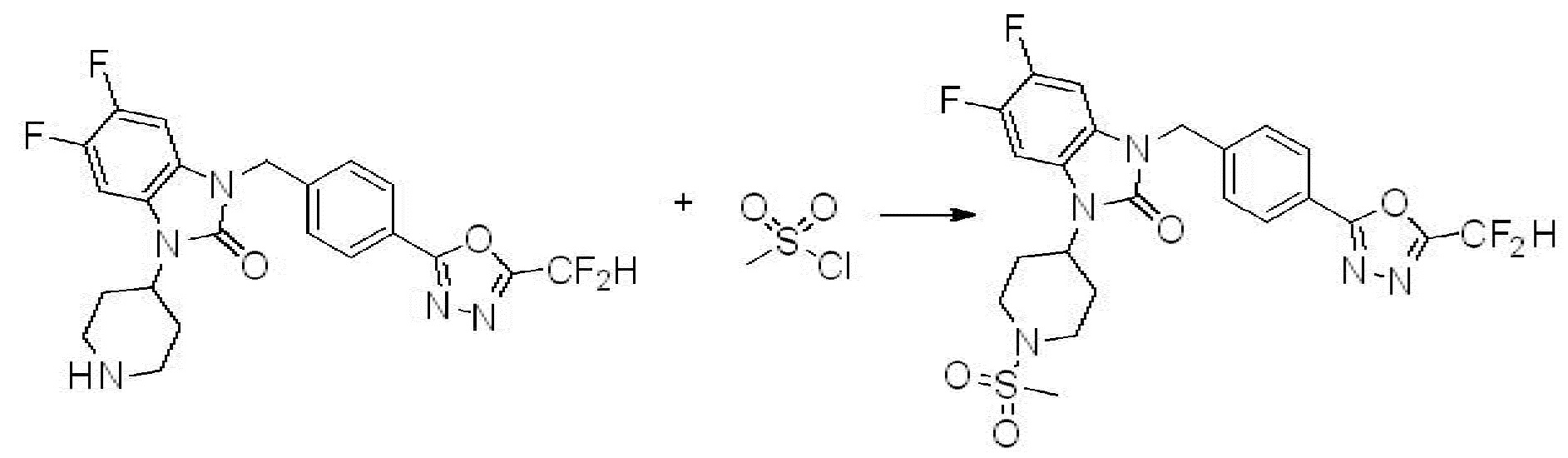
1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,6-дифтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 95, триэтиламин (0,060 мл, 0,433 ммоль) и метансульфонилхлорид (0,034 мл, 0,433 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,087 г, 74,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,10 (д, J=8,4 Гц, 2H), 7,45 (д, J=8,5 Гц, 2H), 7,08-7,03 (м, 2H), 6,67 (дд, J=9,5, 6,7 Гц, 1H), 5,09 (с, 2H), 4,53-4,45 (м, 1H), 4,05 (дд, J=10,3, 2,1 Гц, 2H), 2,92-2,85 (м, 5H), 2,53-2,43 (м, 2H), 1,98 (дд, J=12,2, 2,5 Гц, 2H); МСНР (ЭР) m/z 540,3 (M+ + 1).
Пример 195: Синтез соединения 195, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-(метилсульфонил)пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 195
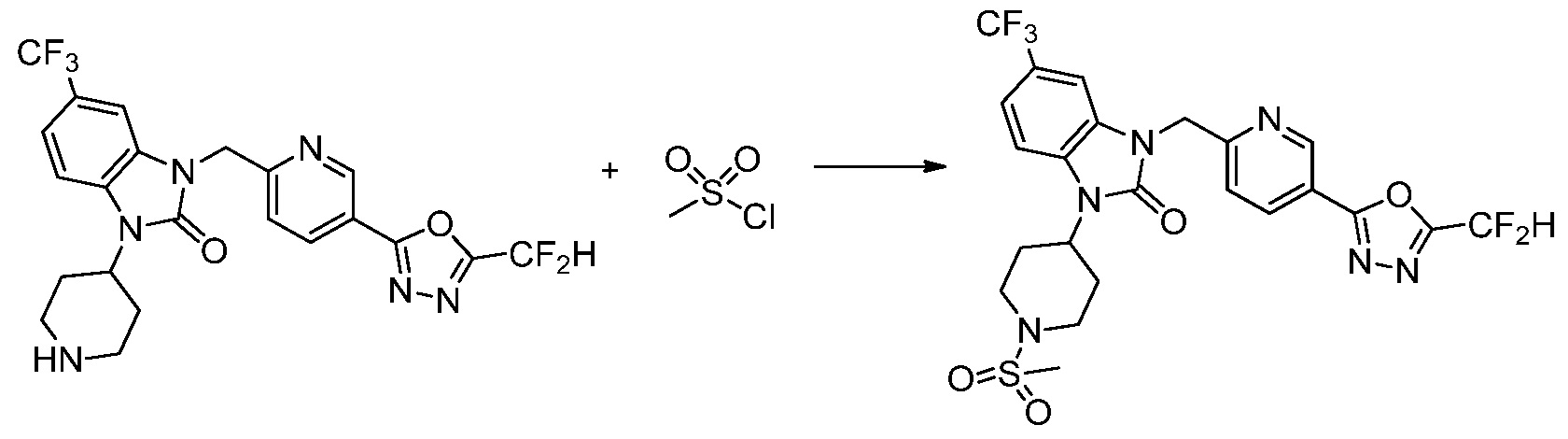
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,202 ммоль), полученный на стадии 5 соединения 89, триэтиламин (0,056 мл, 0,405 ммоль) и метансульфонилхлорид (0,031 мл, 0,405 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,074 г, 63,5%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (дд, J=2,2, 0,7 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,44 (дд, J=8,2, 0,7 Гц, 1H), 7,37 (дд, J=8,4, 0,8 Гц, 1H), 7,29-7,26 (м, 2H), 6,92 (т, J=51,6 Гц, 1H), 5,29 (с, 2H), 4,58-4,50 (м, 1H), 4,03 (дд, J=10,3, 2,0 Гц, 2H), 2,92-2,86 (м, 5H), 2,59-2,48 (м, 2H), 1,98 (дд, J=12,3, 2,4 Гц, 2H); МСНР (ЭР) m/z 573,3 (M+ + 1).
Пример 196: Синтез соединения 196, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-(метилсульфонил)пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 196
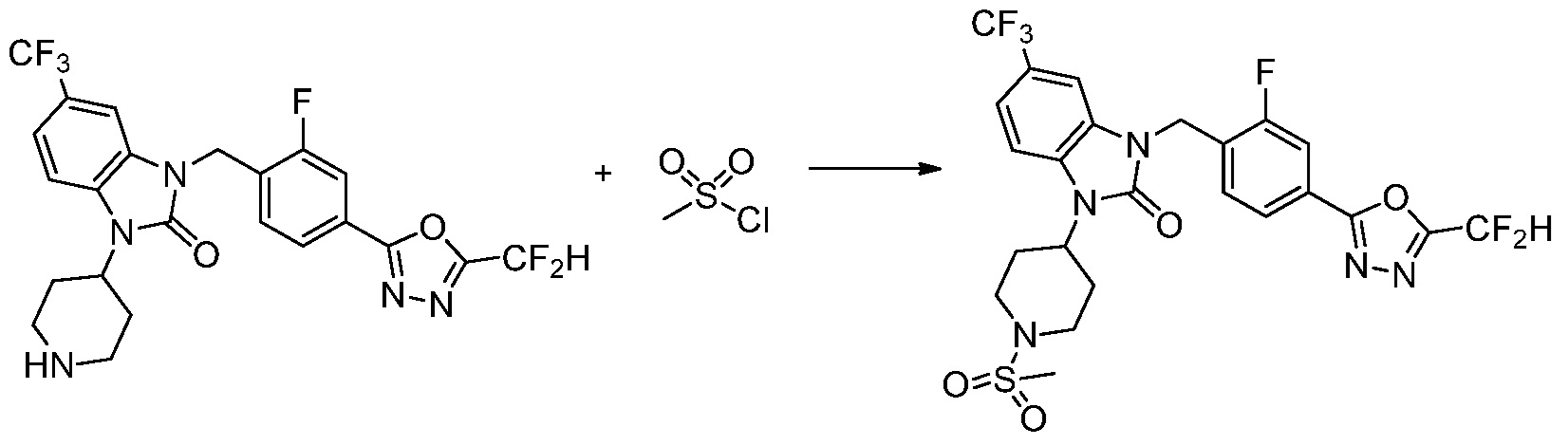
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-5-(трифторметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,196 ммоль), полученный на стадии 2 соединения 91, триэтиламин (0,055 мл, 0,391 ммоль) и метансульфонилхлорид (0,030 мл, 0,391 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,091 г, 78,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,85 (д, J=8,8 Гц, 2H), 7,46 (т, J=7,6 Гц, 1H), 7,39 (д, J=8,3 Гц, 1H), 7,29 (д, J=8,4 Гц, 1H), 7,23 (с, 1H), 6,90 (т, J=51,7 Гц, 1H), 5,21 (с, 2H), 4,58-4,50 (м, 1H), 4,03 (д, J=12,2 Гц, 2H), 2,92-2,87 (м, 5H), 2,59-2,48 (м, 2H), 1,99-1,96 (м, 2H); МСНР (ЭР) m/z 554,2 (M+ + 1).
Пример 197: Синтез соединения 197, 5-(1-ацетилпиперидин-4-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-3,6-дигидропиридин-1(2H)-карбоксилата
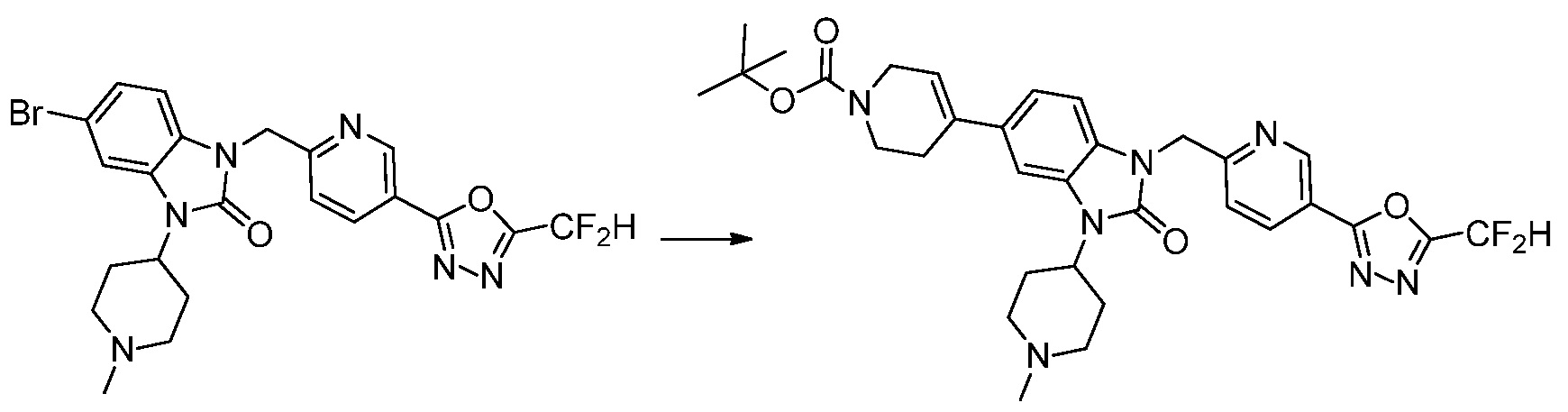
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (1,000 г, 1,925 ммоль), полученный на стадии 6 соединения 133, трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,714 г, 2,311 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,063 г, 0,096 ммоль) и карбонат цезия (1,882 г, 5,776 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,552 г, 46,1%) в виде оранжевого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперидин-1-карбоксилата
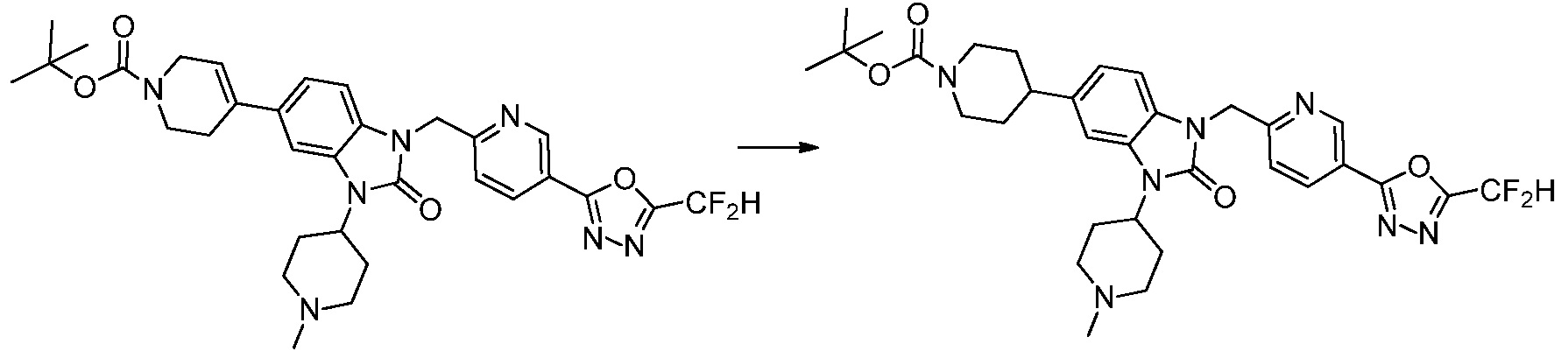
Трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,621 г, 0,972 ммоль), полученный на стадии 1 растворяют в метанол (20 мл) и подвергают взаимодействию с применением Н-куба (10%-Pd/C, 2 атм, 40°C, 0,5 мл/мин). Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,476 г, 76,4%, желтое масло).
[Стадия 3] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-5-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
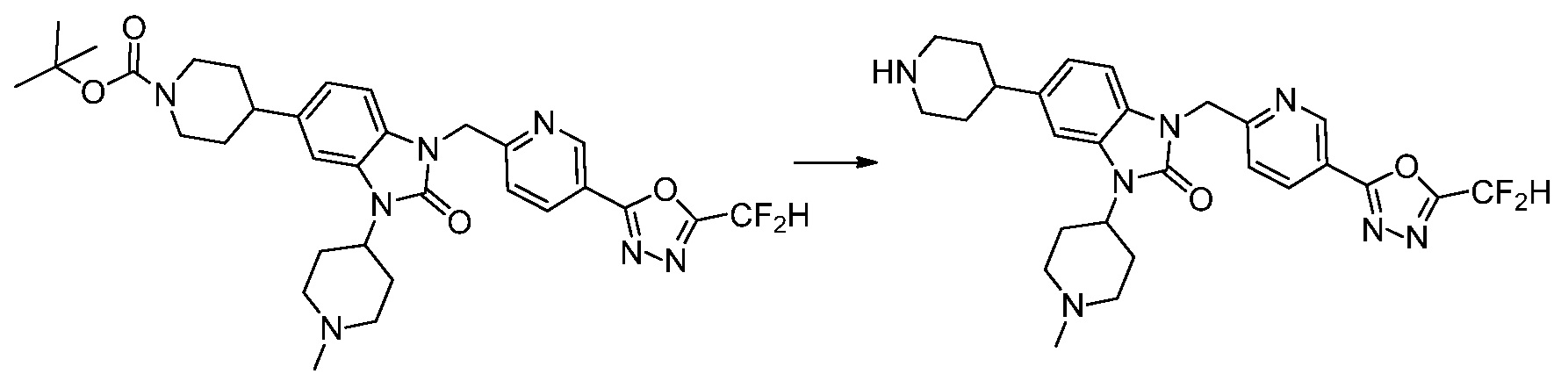
Трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперидин-1-карбоксилат (0,476 г, 0,743 ммоль), полученный на стадии 2, и трифторуксусную кислоту (0,284 мл, 3,715 ммоль) растворяют в дихлорметане (6 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 6 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,374 г, 93,1%, оранжевое масло).
[Стадия 4] Синтез соединения 197
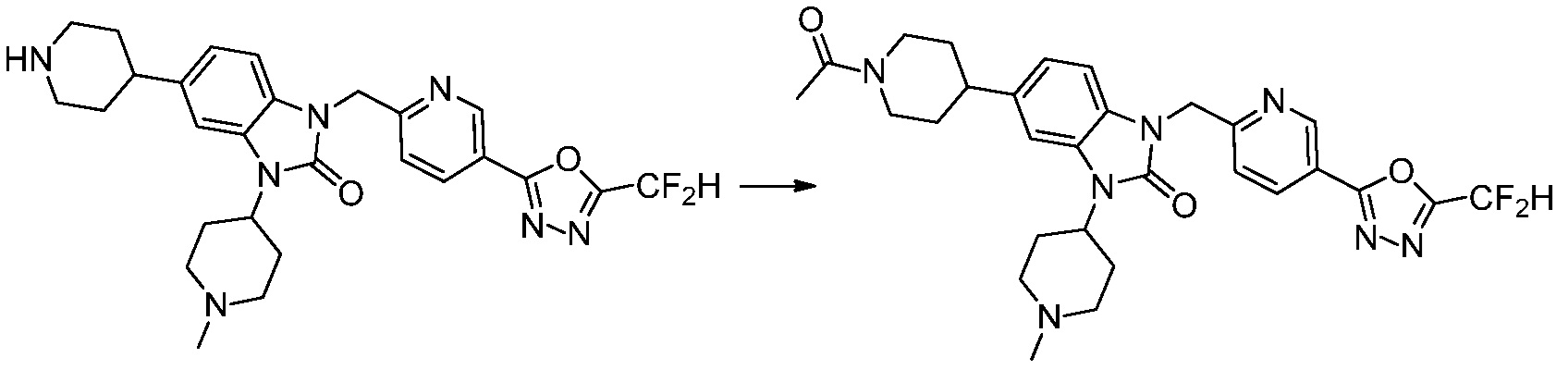
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-5-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,191 ммоль), полученный на стадии 3 и триэтиламин (0,053 мл, 0,382 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего ацетилхлорид (0,022 г, 0,286 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,056 г, 51,8%) в виде желтого масла.
1H ЯМР (400 MГц, CDCl3) δ 7,87 ~ 7,83 (м, 2H), 7,47 (т, J=7,8 Гц, 1H), 7,19 (шс, 1H), 7,05 ~ 6,79 (м, 3H), 5,19 (с, 2H), 4,83 ~ 4,78 (м, 1H), 4,47 ~ 4,46 (м, 1H), 3,97 ~ 3,93 (м, 1H), 3,21 ~ 3,07 (м, 3H), 2,80 ~ 2,74 (м, 1H), 2,65 ~ 2,52 (м, 3H), 2,41 (с, 3H), 2,25 ~ 2,20 (м, 2H), 2,16 (с, 3H), 1,92 ~ 1,86 (м, 4H), 1,70 ~ 1,59 (м, 2H); МСНР (ЭР) m/z 583,3 (M+ + H).
Пример 198: Синтез соединения 198, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-5-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 198
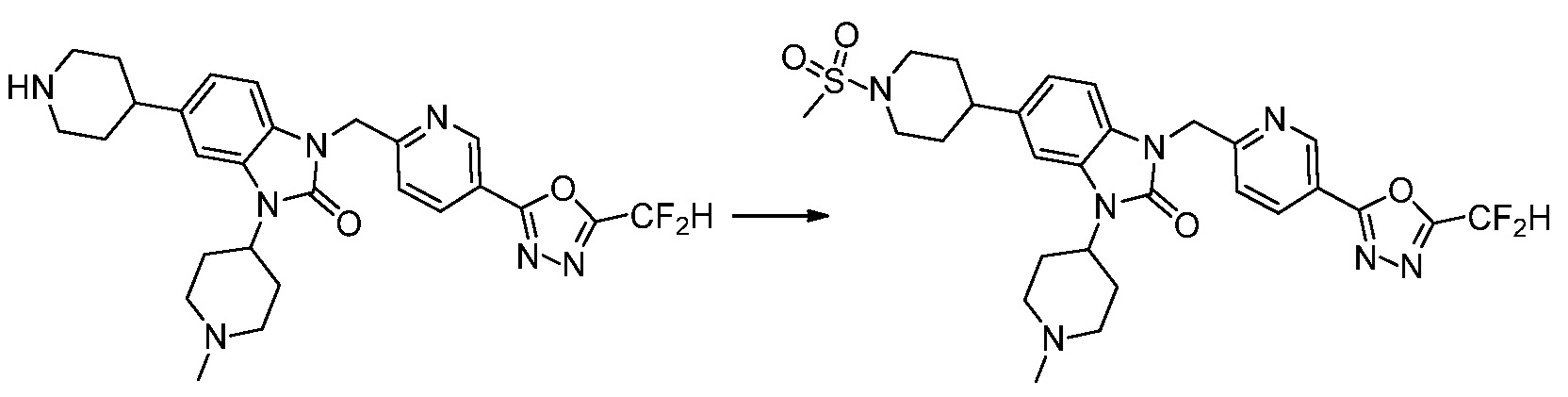
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-5-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,191 ммоль), полученный на стадии 3 соединения 197 и триэтиламин (0,053 мл, 0,382 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего метансульфонилхлорид (0,022 мл, 0,286 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,039 г, 33,9%) в виде желтого масла.
1H ЯМР (400 MГц, CDCl3) δ 7,87 ~ 7,83 (м, 2H), 7,47 (т, J=7,8 Гц, 1H), 7,21 (шс, 1H), 7,05 ~ 6,79 (м, 3H), 5,19 (с, 1H), 4,50 ~ 4,44 (м, 1H), 3,96 (д, J=11,8 Гц, 2H), 3,08 (д, J=9,4 Гц, 2H), 3,02 (шс, 1H), 2,85 (с, 3H), 2,80 ~ 2,74 (м, 2H), 2,70 ~ 2,66 (м, 1H), 2,65 ~ 2,61 (м, 2H), 2,54 (с, 3H), 2,23 (т, J=10,4 Гц, 2H), 1,96 (д, J=10,8 Гц, 2H), 1,90 ~ 1,84 (м, 3H); МСНР (ЭР) m/z 619,3 (M+ + H).
Пример 199: Синтез соединения 199, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,5-бис(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 199
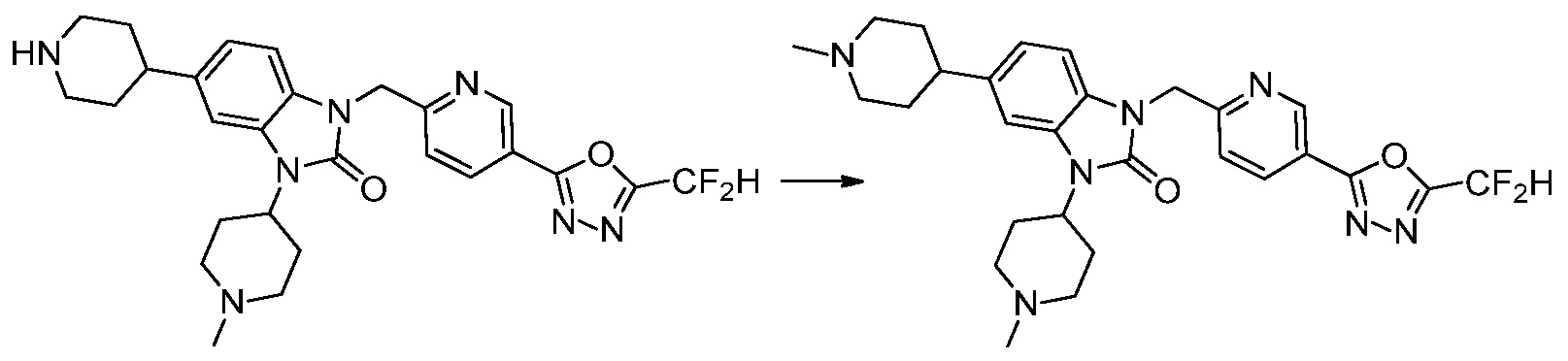
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-5-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,191 ммоль), полученный на стадии 3 соединения 197 и формальдегид (37,00%, 0,023 г, 0,286 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,081 г, 0,382 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,025 г, 24,3%) в виде желтого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,30-9,29 (м, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,23 (с, 1H), 7,07-6,81 (м, 3H), 5,28 (с, 2H), 4,48-4,41 (м, 1H), 3,03 (т, J=11,3 Гц, 4H), 2,57-2,47 (м, 3H), 2,38 (с, 3H), 2,36 (с, 3H), 2,19 (т, J=11,2 Гц, 2H), 2,11-2,06 (м, 2H), 1,87-1,83 (м, 6H); МСНР (ЭР) m/z 538,4 (M+ + H).
Пример 200: Синтез соединения 200, 5-хлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 200
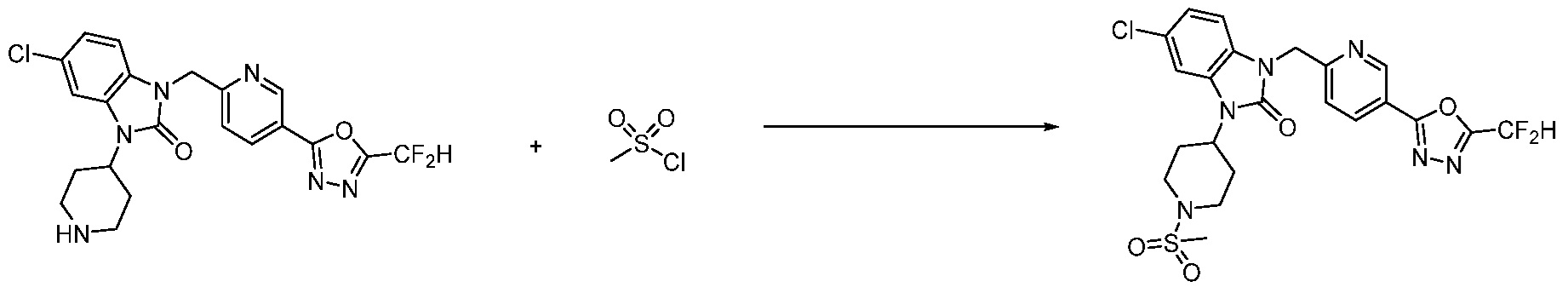
5-хлор-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 97, триэтиламин (0,060 мл, 0,434 ммоль) и метансульфонилхлорид (0,034 мл, 0,434 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,086 г, 73,8%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=2,0 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,2 Гц, 1H), 7,18 (д, J=1,8 Гц, 1H), 7,06-6,80 (м, 3H), 5,24 (с, 2H), 4,52-4,43 (м, 1H), 4,03 (дд, J=10,3, 2,0 Гц, 2H), 2,91-2,85 (м, 5H), 2,58-2,47 (м, 2H), 1,98 (дд, J=12,3, 2,2 Гц, 2H); МСНР (ЭР) m/z 539,2 (M+ + 1).
Пример 201: Синтез соединения 201, 5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 201
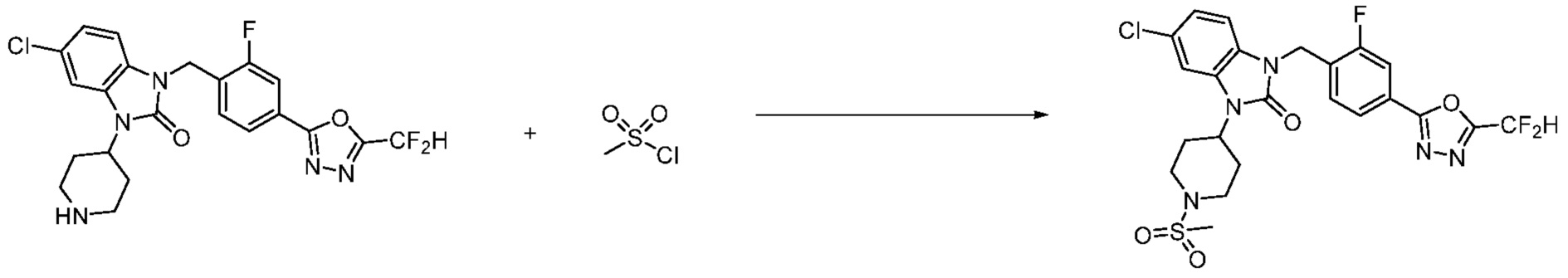
5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,209 ммоль), полученный на стадии 2 соединения 101, триэтиламин (0,058 мл, 0,419 ммоль) и метансульфонилхлорид (0,032 мл, 0,419 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,086 г, 73,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,82 (д, J=8,8 Гц, 2H), 7,42 (т, J=7,5 Гц, 1H), 7,18 (д, J=1,8 Гц, 1H), 7,03-6,78 (м, 3H), 5,15 (с, 2H), 4,50-4,42 (м, 1H), 4,02 (дд, J=10,2, 2,0 Гц, 2H), 2,90-2,84 (м, 5H), 2,56-2,46 (м, 2H), 1,96 (дд, J=12,3, 2,2 Гц, 2H); МСНР (ЭР) m/z 556,2 (M+ + 1).
Пример 202: Синтез соединения 202, 5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 202
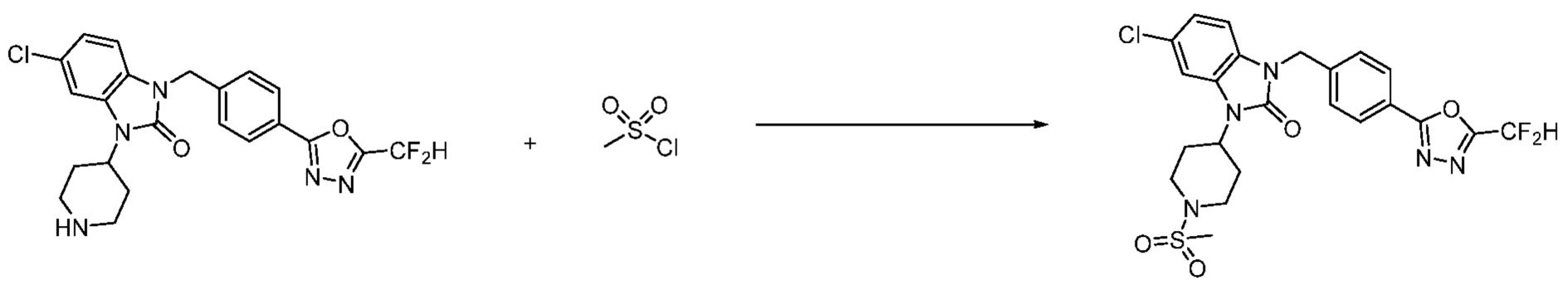
5-хлор-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 2 соединения 99, триэтиламин (0,061 мл, 0,435 ммоль) и метансульфонилхлорид (0,034 мл, 0,435 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = 0-5%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 85,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,04 (дд, J=6,7, 1,8 Гц, 2H), 7,42 (д, J=8,5 Гц, 2H), 7,17 (д, J=1,8 Гц, 1H), 7,03-6,74 (м, 3H), 5,10 (с, 2H), 4,51-4,43 (м, 1H), 4,02 (дд, J=10,2, 2,0 Гц, 2H), 2,88-2,85 (м, 5H), 2,57-2,46 (м, 2H), 1,96 (дд, J=12,3, 2,3 Гц, 2H); МСНР (ЭР) m/z 538,1 (M+ + 1).
Пример 203: Синтез соединения 203, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез соединения 203
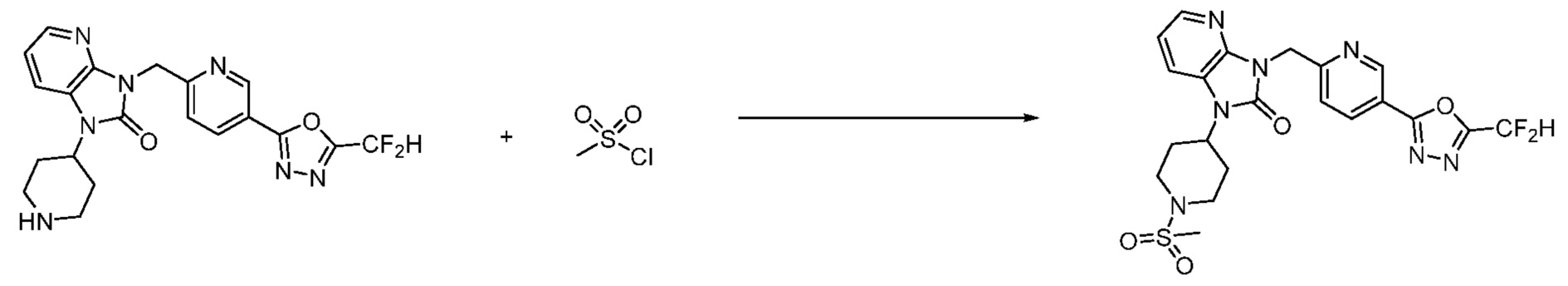
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,234 ммоль), полученный на стадии 2 соединения 107, триэтиламин (0,065 мл, 0,468 ммоль) и метансульфонилхлорид (0,036 мл, 0,468 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,041 г, 34,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,23 (д, J=1,7 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 8,04 (дд, J=5,2, 1,0 Гц, 1H), 7,48-7,42 (м, 2H), 7,05-6,78 (м, 2H), 5,40 (с, 2H), 4,63-4,54 (м, 1H), 4,04 (дд, J=10,3, 2,0 Гц, 2H), 2,91-2,85 (м, 5H), 2,50-2,40 (м, 2H), 2,02 (дд, J=12,4, 2,4 Гц, 2H); МСНР (ЭР) m/z 506,2 (M+ + 1).
Пример 204: Синтез соединения 204, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез соединения 204
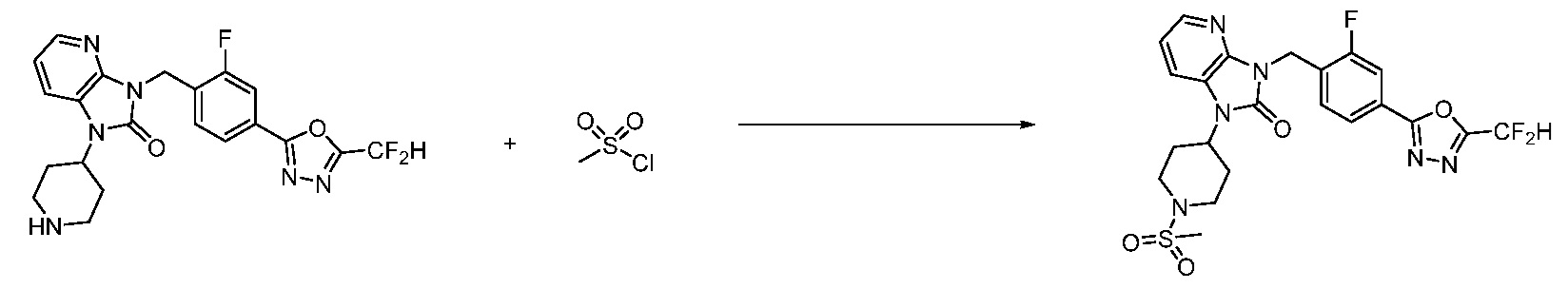
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,225 ммоль), полученный на стадии 5 соединения 103, триэтиламин (0,063 мл, 0,450 ммоль) и метансульфонилхлорид (0,035 мл, 0,450 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,103 г, 87,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,03 (дд, J=5,2, 1,0 Гц, 1H), 7,80-7,77 (м, 2H), 7,45-7,41 (м, 2H), 7,03-6,77 (м, 2H), 5,28 (с, 2H), 4,59-4,50 (м, 1H), 4,00 (дд, J=10,4, 1,9 Гц, 2H), 2,89-2,83 (м, 5H), 2,47-2,37 (м, 2H), 1,98 (дд, J=12,3, 2,3 Гц, 2H); МСНР (ЭР) m/z 523,1 (M+ + 1).
Пример 205: Синтез соединения 205, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он
[Стадия 1] Синтез соединения 205
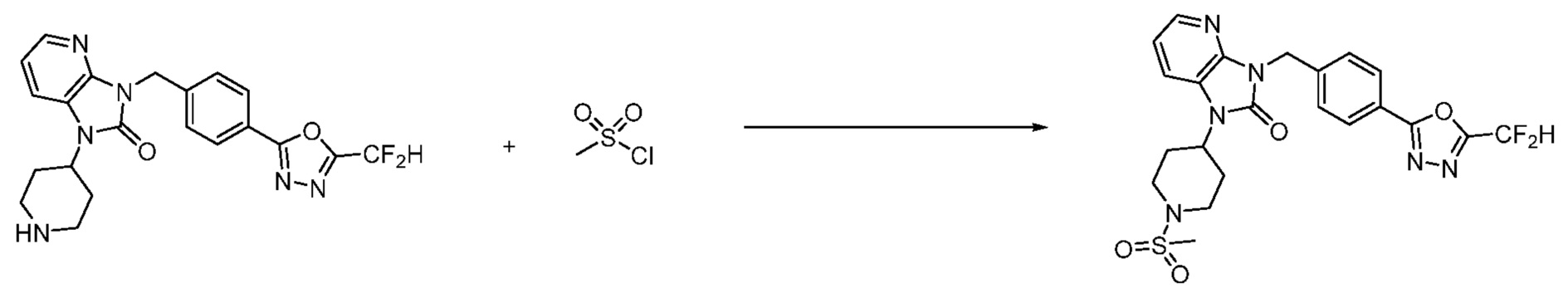
3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-имидазо[4,5-b]пиридин-2-он (0,100 г, 0,235 ммоль), полученный на стадии 2 соединения 105, триэтиламин (0,065 мл, 0,469 ммоль) и метансульфонилхлорид (0,036 мл, 0,469 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = 0-5%) и концентрируют с получением указанного в заголовке соединения (0,066 г, 55,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,06-8,03 (м, 3H), 7,63 (д, J=8,3 Гц, 2H), 7,40 (дд, J=7,9, 1,2 Гц, 1H), 7,03-6,76 (м, 2H), 5,22 (с, 2H), 4,59-4,51 (м, 1H), 4,01 (дд, J=10,3, 2,1 Гц, 2H), 2,89-2,83 (м, 5H), 2,46-2,36 (м, 2H), 1,97 (дд, J=12,3, 2,3 Гц, 2H); МСНР (ЭР) m/z 505,1 (M+ + 1).
Пример 206: Синтез соединения 206, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 206
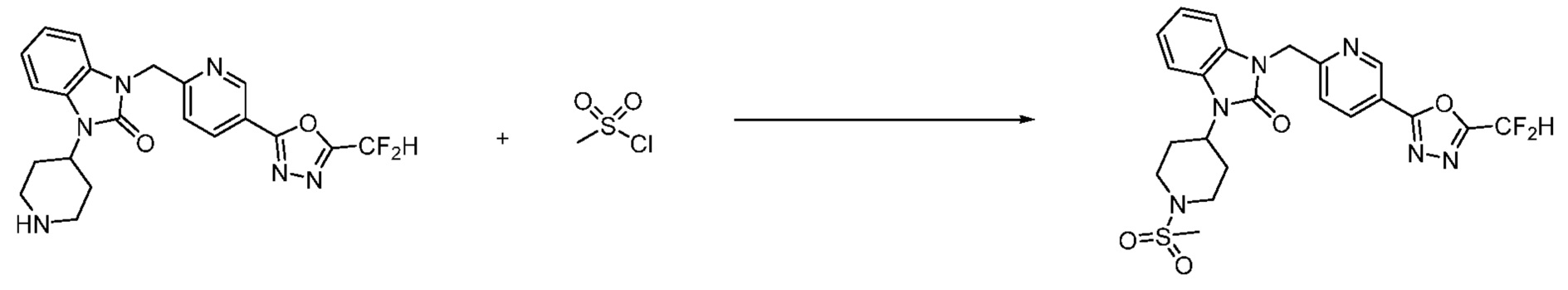
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,235 ммоль), триэтиламин (0,065 мл, 0,469 ммоль) и метансульфонилхлорид (0,036 мл, 0,469 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,048 г, 40,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=1,8 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=23,8 Гц, 1H), 7,22 (д, J=7,5 Гц, 1H), 7,12-6,79 (м, 4H), 5,28 (с, 2H), 4,59-4,51 (м, 1H), 4,03 (дд, J=10,1, 2,1 Гц, 2H), 2,92-2,85 (м, 5H), 2,62-2,52 (м, 2H), 1,98 (дд, J=12,3, 2,3 Гц, 2H); МСНР (ЭР) m/z 505,0 (M+ + 1).
Пример 207: Синтез соединения 207, N-(3-бензил-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиваламид
[Стадия 1] Синтез N-(4-бром-3-хлорфенил)пиваламида
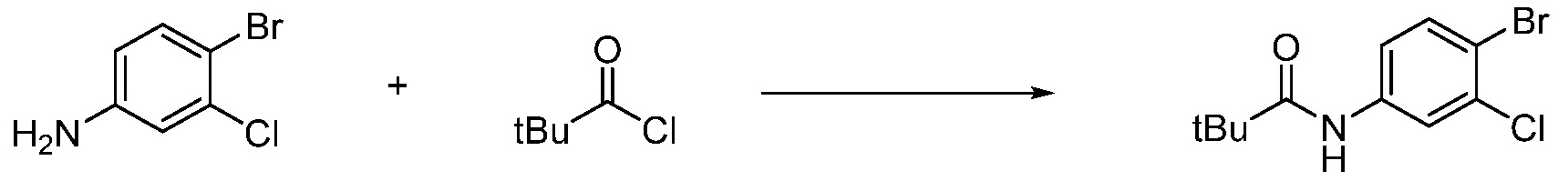
4-бром-3-хлоранилин (0,500 г, 2,422 ммоль), триэтиламин (0,405 мл, 2,906 ммоль) и триметилацетилхлорид (0,328 мл, 2,664 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,684 г, 97,2%) в виде белого твердого вещества.
[Стадия 2] Синтез N-(3-бензил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиваламида
N-(4-бром-3-хлорфенил)пиваламид (0,150 г, 0,516 ммоль), полученный на стадии 1, 1-бензилмочевину (0,310 г, 2,065 ммоль), BrettPhos палладий G3 (0,023 г, 0,026 ммоль) и фосфат калия (0,263 г, 1,239 ммоль) растворяют в трет-бутаноле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при 110° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Реакционную смесь was фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 65,9%) в виде желтого твердого вещества.
[Стадия 3] Синтез соединения 207
N-(3-бензил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиваламид (0,110 г, 0,340 ммоль), полученный на стадии 2 растворяют в N,N-диметилформамиде (3 мл) при 0°C, после чего гидрид натрия (60,00%, 0,020 г, 0,510 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,109 г, 0,374 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2%) и концентрируют с получением указанного в заголовке соединения (0,065 г, 35,9%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,23 (д, J=1,7 Гц, 1H), 8,26 (дд, J=8,2, 2,2 Гц, 1H), 7,55 (д, J=1,8 Гц, 1H), 7,44 (с, 1H), 7,34-7,20 (м, 6H), 6,91-6,78 (м, 3H), 5,26 (с, 2H), 5,05 (с, 2H), 1,25 (с, 9H); МСНР (ЭР) m/z 533,1 (M+ + 1).
Пример 208: Синтез соединения 208, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез 5-бром-N-метил-2-нитроанилина
4-бром-2-фтор-1-нитробензол (5,000 г, 22,727 ммоль), метанамин (0,777 г, 25,000 ммоль), карбонат калия (6,282 г, 45,455 ммоль) и йодид калия (0,377 г, 2,273 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Гексан добавляют в реакционную смесь и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют и сушат с получением указанного в заголовке соединения (4,760 г, 90,6%) в виде желтого твердого вещества.
[Стадия 2] Синтез 5-бром-N1-метилбензол-1,2-диамина
5-бром-N-метил-2-нитроанилин (4,760 г, 20,602 ммоль), полученный на стадии 1, и никель Ренея (400 мг, 10% масс.) растворяют в метаноле (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (4,000 г, 96,6%, черная жидкость).
[Стадия 3] Синтез 6-бром-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-она

5-бром-N1-метилбензол-1,2-диамин (4,000 г, 19,894 ммоль), полученный на стадии 2, триэтиламин (2,773 мл, 19,894 ммоль) и 1,1'-карбонилдиимидазол (КДИ, 3,871 г, 23,872 ммоль) растворяют в дихлорметане (60 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. выпавшее в осадок твердое вещество фильтруют, затем промывают гексан, и затем сушат с получением указанного в заголовке соединения (3,935 г, 87,1%) в виде коричневого твердого вещества.
[Стадия 4] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-она
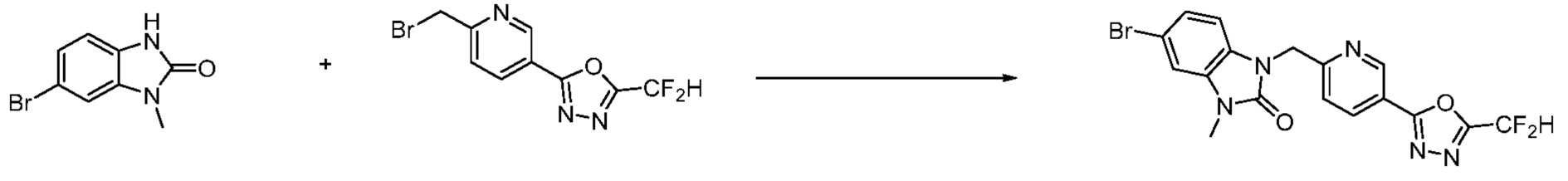
6-бром-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (1,930 г, 8,500 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (40 мл) при 0°C, после чего гидрид натрия (60,00%, 0,510 г, 12,750 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,712 г, 9,350 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=20-50%) и концентрируют с получением указанного в заголовке соединения (2,060 г, 55,6%) в виде светло-коричневого твердого вещества.
[Стадия 5] Синтез соединения 208
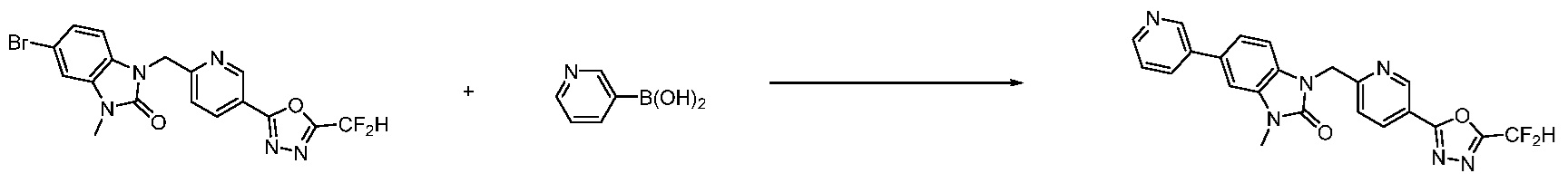
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,344 ммоль), полученный на стадии 4, пиридин-3-илбороновую кислоту (0,051 г, 0,413 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,224 г, 0,688 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,081 г, 54,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (д, J=2,1 Гц, 1H), 8,83 (с, 1H), 8,57 (с, 1H), 8,29 (дд, J=8,2, 2,2 Гц, 1H), 7,81 (д, J=7,9 Гц, 1H), 7,42 (д, J=8,2 Гц, 1H), 7,33 (дд, J=7,4, 4,7 Гц, 1H), 7,21 (дд, J=8,1, 1,6 Гц, 1H), 7,16 (д, J=1,4 Гц, 1H), 7,04-6,78 (м, 2H), 5,28 (с, 2H), 3,50 (с, 3H); МСНР (ЭР) m/z 533,1 (M+ + 1).
Пример 209: Синтез соединения 209, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 209
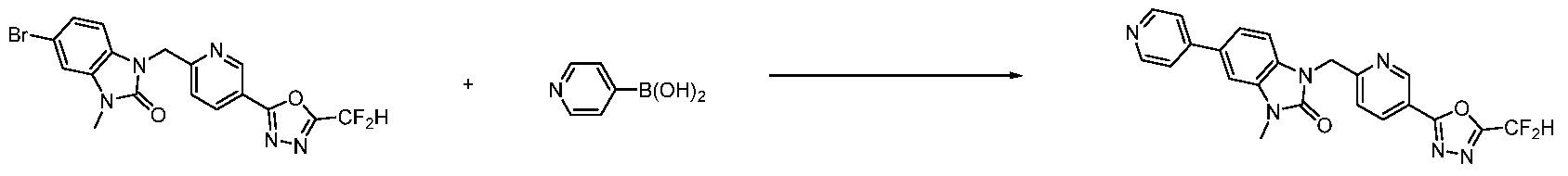
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,344 ммоль), полученный на стадии 4 соединения 208, пиридин-4-илбороновую кислоту (0,051 г, 0,413 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,224 г, 0,688 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,034 г, 22,8%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,1, 0,6 Гц, 1H), 8,64 (с, 2H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,49-7,44 (м, 3H), 7,33 (дд, J=8,1, 1,6 Гц, 1H), 7,25 (д, J=1,5 Гц, 1H), 7,07-6,79 (м, 2H), 5,31 (с, 2H), 3,53 (с, 3H); МСНР (ЭР) m/z 436,1 (M+ + 1).
Пример 210: Синтез соединения 210, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(3,5-диметилизоксазол-4-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 210
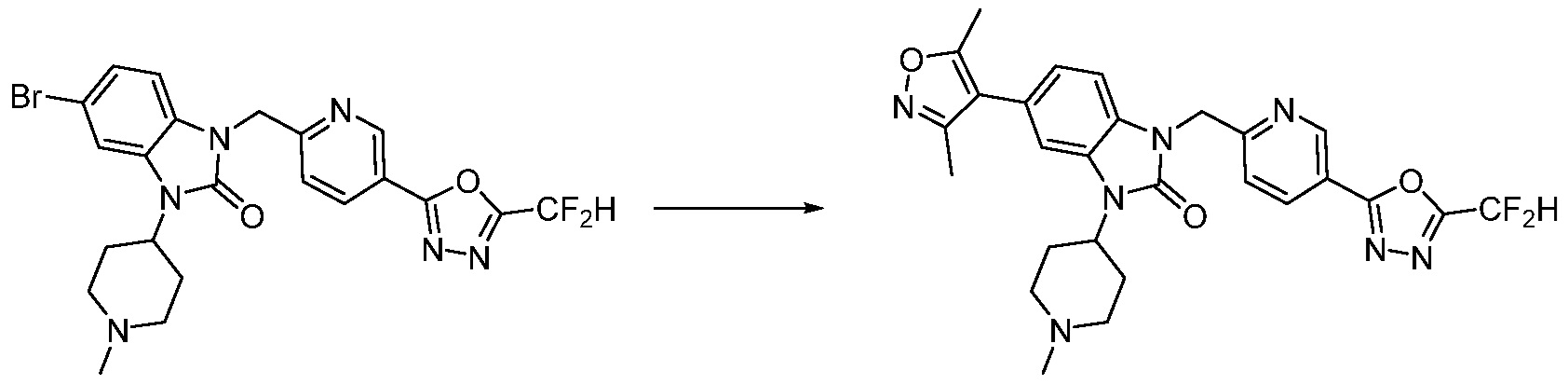
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 0,385 ммоль), (3,5-диметилизооксазол-4-ил)бороновую кислоту (0,065 г, 0,462 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,013 г, 0,019 ммоль) и карбонат цезия (0,376 г, 1,155 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 48,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (с, 1H), 8,37 (д, J=8,2 Гц, 1H), 7,50 (д, J=8,2 Гц, 1H), 7,28 (с, 1H), 7,25 (шс, 1H), 7,08-6,82 (м, 3H), 5,33 (с, 2H), 4,50-4,51 (м, 1H), 3,08-3,09 (м, 2H), 2,55-2,56 (м, 2H), 2,40 (с, 6H), 2,25-2,26 (м, 5H), 1,92 (д, J=11,2 Гц, 2H); МСНР (ЭР) m/z 536,3 (M+ + 1).
Пример 211: Синтез соединения 211, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(изоксазол-4-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 211
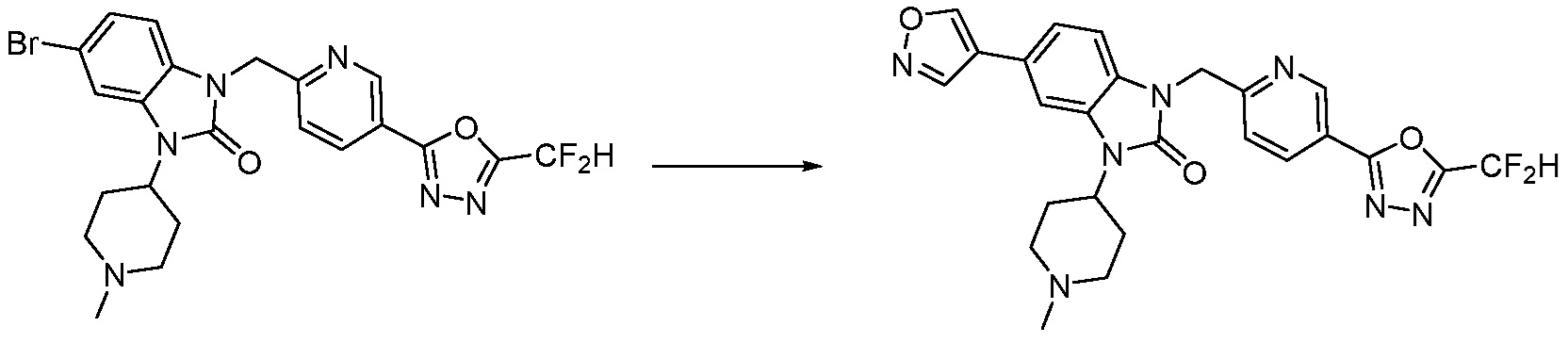
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 0,385 ммоль), бензофуран-2-илбороновую кислоту (0,057 г, 0,462 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,013 г, 0,019 ммоль) и карбонат цезия (0,376 г, 1,155 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,008 г, 4,1%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=2,0 Гц, 1H), 8,80 (шс, 1H), 8,61 (с, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,54 (шс, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,17 (д, J=8,1 Гц, 1H), 7,08-6,82 (м, 2H), 5,32 (с, 2H), 4,54-4,55 (м, 1H), 3,16-3,17 (м, 2H), 2,69 (шс, 2H), 2,48 (с, 3H), 2,39-2,35 (м, 2H), 1,94 (д, J=10,6 Гц, 2H); МСНР (ЭР) m/z 508,4 (M+ + 1).
Пример 212: Синтез соединения 212, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(1H-индол-4-ил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 212
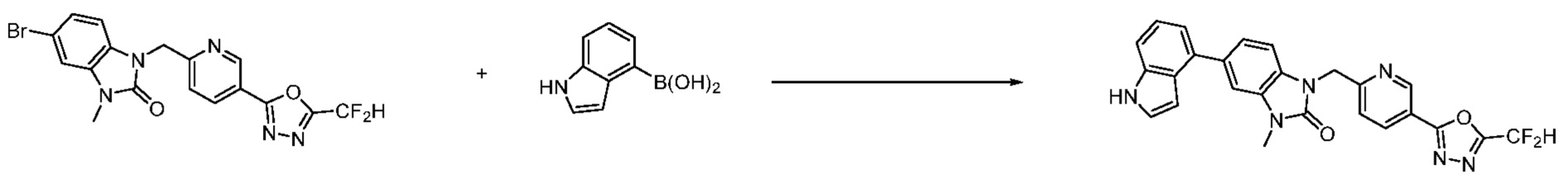
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,344 ммоль), полученный на стадии 4 соединения 208, (1H-индол-4-ил)бороновую кислоту (0,066 г, 0,413 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,224 г, 0,688 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,049 г, 30,2%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=1,6 Гц, 1H), 8,65 (с, 1H), 8,28 (дд, J=8,2, 2,2 Гц, 1H), 7,44-7,34 (м, 4H), 7,24-7,22 (м, 2H), 7,15 (дд, J=7,3, 0,7 Гц, 1H), 7,03 (д, J=7,9 Гц, 1H), 6,91 (т, J=52,0 Гц, 1H), 6,68 (т, J=2,1 Гц, 1H), 5,34 (с, 2H), 3,52 (с, 3H); МСНР (ЭР) m/z 473,3 (M+ + 1).
Пример 213: Синтез соединения 213, 5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-((4-хлор-2-нитрофенил)амино)пиперидин-1-карбоксилата
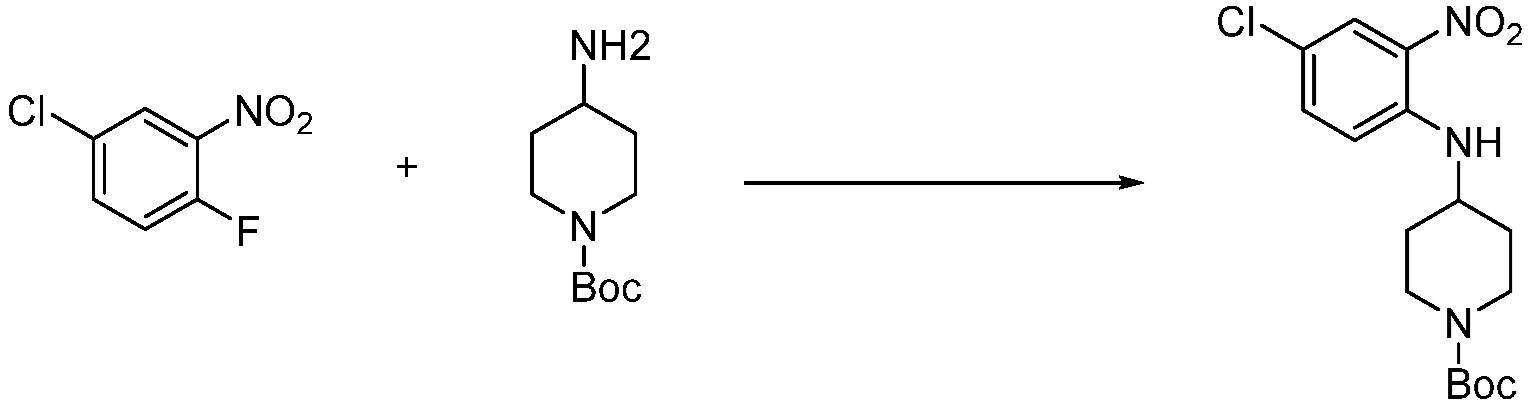
4-хлор-1-фтор-2-нитробензол (5,000 г, 28,484 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилат (6,275 г, 31,332 ммоль), карбонат калия (7,873 г, 56,967 ммоль) и йодид калия (0,473 г, 2,848 ммоль) растворяют в N,N-диметилформамиде (140 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воде (200 мл) добавляют в реакционную смесь и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют, затем промывают гексан, и затем сушат с получением указанного в заголовке соединения (10,100 г, 99,7%) в виде оранжевого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-((2-амино-4-хлорфенил)амино)пиперидин-1-карбоксилата

Трет-бутил 4-((4-хлор-2-нитрофенил)амино)пиперидин-1-карбоксилат (10,100 г, 28,385 ммоль), полученный на стадии 1 и никель Ренея (1 г, 10% масс.) растворяют в метанол (140 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов в присутствии баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (9,190 г, 99,4%, черная жидкость).
[Стадия 3] Синтез трет-бутил 4-(5-хлор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
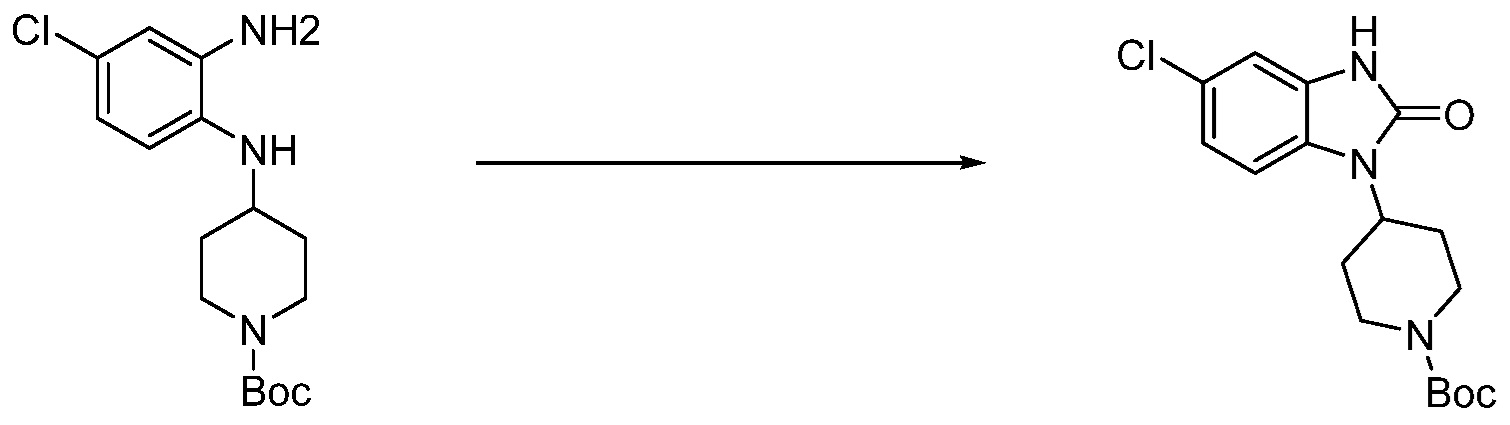
Трет-бутил 4-((2-амино-4-хлорфенил)амино)пиперидин-1-карбоксилат (9,190 г, 28,204 ммоль), полученный на стадии 2, триэтиламин (3,931 мл, 28,204 ммоль) и 1,1'-карбонилдиимидазол (КДИ, 5,488 г, 33,845 ммоль) растворяют в дихлорметане (80 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=30-70%) и концентрируют с получением указанного в заголовке соединения (5,660 г, 57,0%) в виде красного твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
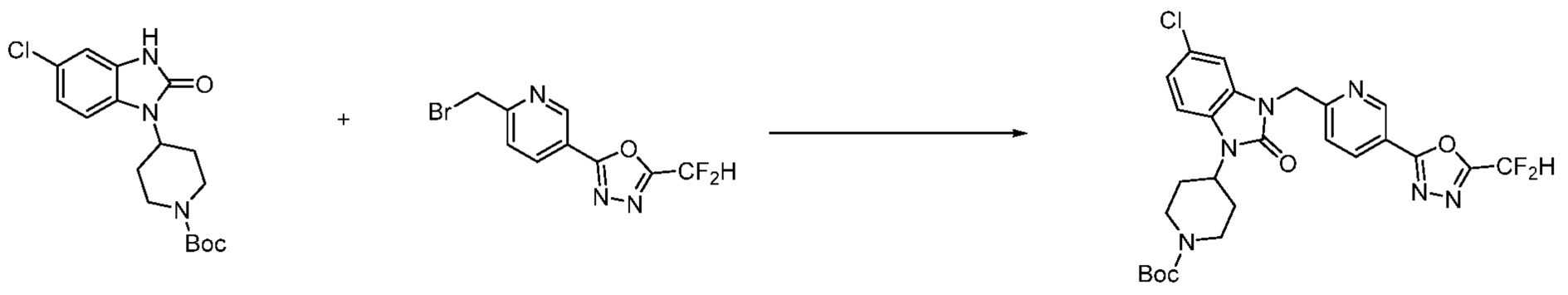
Трет-бутил 4-(5-хлор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (3,000 г, 8,527 ммоль), полученный на стадии 3 растворяют в N,N-диметилформамиде (45 мл) при 0°C, после чего гидрид натрия (60,00%, 0,512 г, 12,790 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,721 г, 9,380 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=30 to 60%) и концентрируют с получением указанного в заголовке соединения (3,270 г, 68,4%) в виде светло-желтого твердого вещества.
[Стадия 5] Синтез 5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
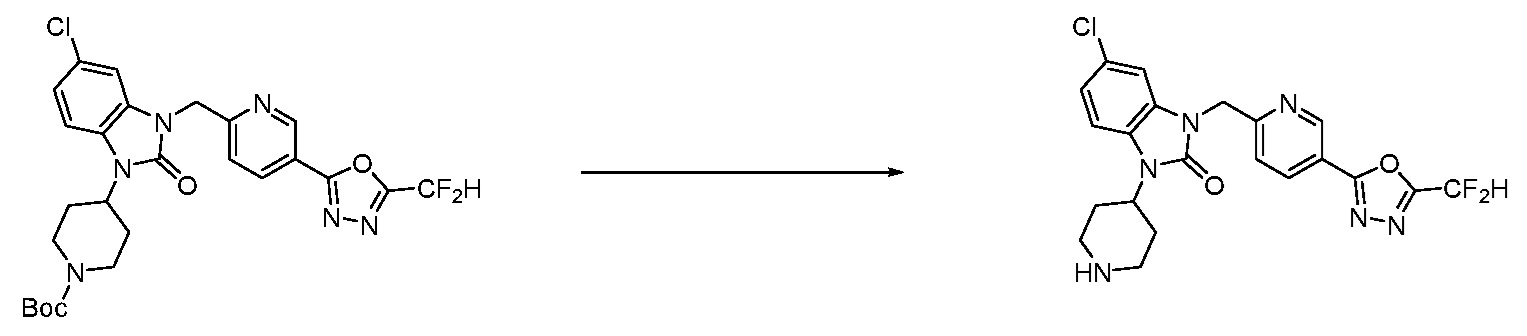
Трет-бутил 4-(5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (3,270 г, 5,829 ммоль), полученный на стадии 4 и трифторуксусную кислоту (4,464 мл, 58,290 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=80-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-70%) и концентрируют с получением указанного в заголовке соединения (2,100 г, 78,2%) в виде светло-желтого твердого вещества.
[Стадия 6] Синтез соединения 213
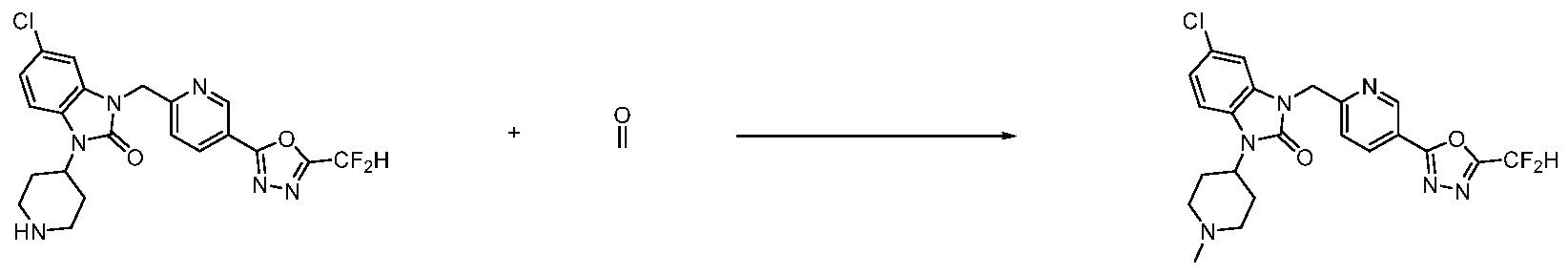
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5, формальдегид (0,020 г, 0,651 ммоль) и триацетоксиборгидрид натрия (0,092 г, 0,434 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,079 г, 77,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (с, 1H), 8,31 (дд, J=8,2, 1,9 Гц, 1H), 7,38 (д, J=8,2 Гц, 1H), 7,20 (д, J=8,4 Гц, 1H), 7,04-6,79 (м, 3H), 5,22 (с, 2H), 4,42-4,35 (м, 1H), 3,00 (д, J=11,5 Гц, 2H), 2,47-2,37 (м, 2H), 2,32 (с, 3H), 2,14 (т, J=11,8 Гц, 2H), 1,82 (д, J=11,8 Гц, 2H); МСНР (ЭР) m/z 477,2 (M+ + 1).
Пример 214: Синтез соединения 214, 5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-изопропилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 214
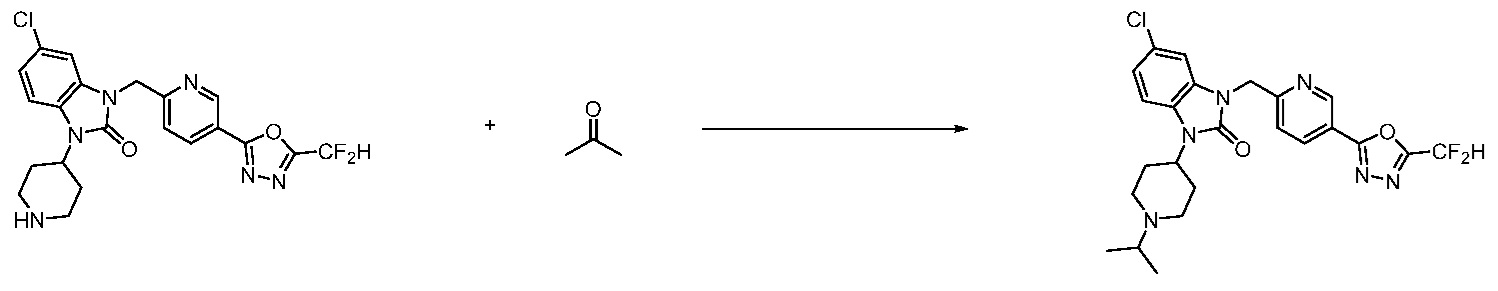
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 213, пропан-2-он (0,038 г, 0,651 ммоль) и триацетоксиборгидрид натрия (0,092 г, 0,434 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,075 г, 68,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=2,1 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,2 Гц, 1H), 7,26 (т, J=4,2 Гц, 1H), 7,05-6,79 (м, 3H), 5,24 (с, 2H), 4,41-4,35 (м, 1H), 3,03 (д, J=7,9 Гц, 2H), 2,85-2,79 (м, 1H), 2,43-2,31 (м, 4H), 1,86 (д, J=9,0 Гц, 2H), 1,07 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 503,1 (M+ + 1).
Пример 215: Синтез соединения 215, 1-(1-ацетилпиперидин-4-ил)-5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 215
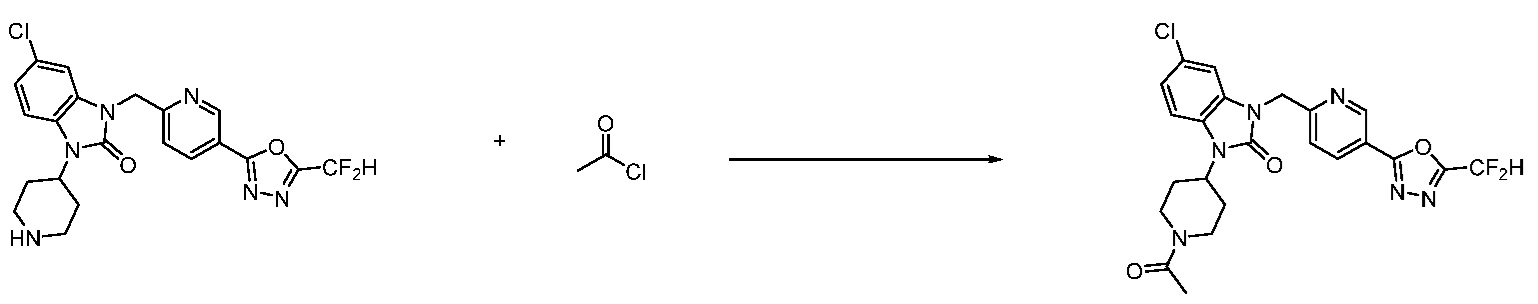
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 213, ацетилхлорид (0,031 мл, 0,434 ммоль) и триэтиламин (0,060 мл, 0,434 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,096 г, 88,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,1, 0,6 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (д, J=8,2 Гц, 1H), 7,05-7,00 (м, 3H), 6,93 (т, J=51,6 Гц, 1H), 5,23 (д, J=1,9 Гц, 2H), 4,87 (дд, J=11,6, 1,9 Гц, 1H), 4,58-4,50 (м, 1H), 4,01 (д, J=13,8 Гц, 1H), 3,26-3,20 (м, 1H), 2,70-2,63 (м, 1H), 2,40-2,22 (м, 2H), 2,16 (с, 3H), 1,92 (т, J=14,0 Гц, 2H); МСНР (ЭР) m/z 503,1 (M+ + 1).
Пример 216: Синтез соединения 216, 5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-(метилсульфонил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 216
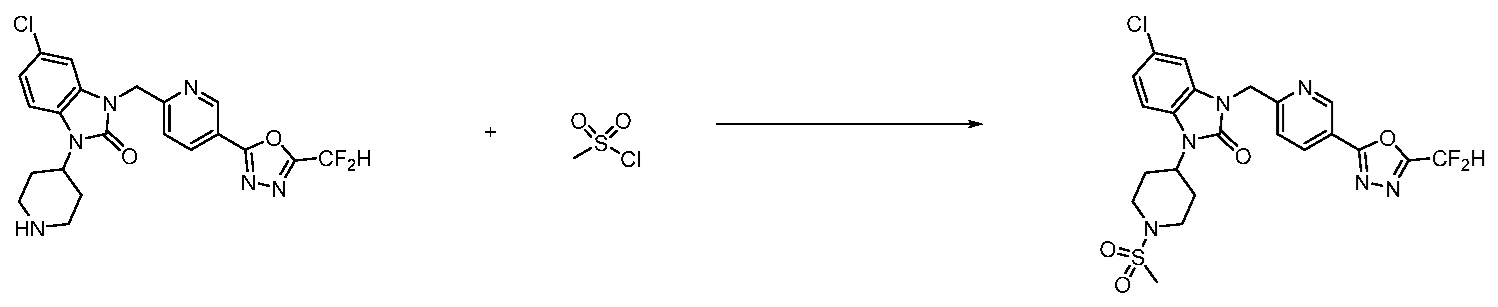
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 213, метансульфонилхлорид (0,034 мл, 0,434 ммоль) и триэтиламин (0,060 мл, 0,434 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,079 г, 67,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=1,4 Гц, 1H), 8,35 (дд, J=8,2, 2,0 Гц, 1H), 7,42 (д, J=8,2 Гц, 1H), 7,13-6,79 (м, 4H), 5,24 (с, 2H), 4,56-4,47 (м, 1H), 4,03 (д, J=12,3 Гц, 2H), 2,90-2,85 (м, 5H), 2,56-2,45 (м, 2H), 1,97 (д, J=10,7 Гц, 2H); МСНР (ЭР) m/z 539,3 (M+ + 1).
Пример 217: Синтез соединения 217, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 217
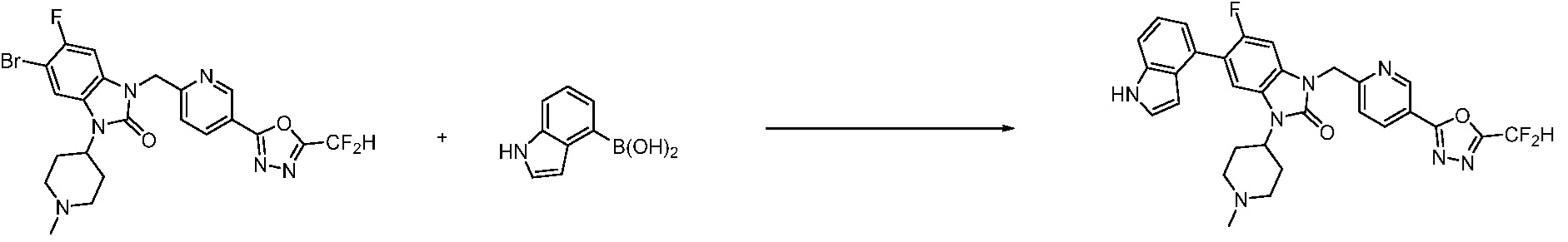
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, (1H-индол-4-ил)бороновую кислоту (0,054 г, 0,335 ммоль), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия дихлорид (0,009 г, 0,014 ммоль) и карбонат цезия (0,182 г, 0,558 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,026 г, 16,2%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,8 Гц, 1H), 8,62 (с, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,42-7,39 (м, 2H), 7,26-7,20 (м, 2H), 7,10 (д, J=7,2 Гц, 1H), 7,08 (т, J=146,7 Гц, 1H), 6,89 (д, J=9,3 Гц, 1H), 6,40 (д, J=1,6 Гц, 1H), 5,30 (с, 2H), 4,47-4,39 (м, 1H), 2,99 (д, J=11,6 Гц, 2H), 2,52-2,42 (м, 2H), 2,30 (с, 3H), 2,14 (т, J=11,1 Гц, 2H), 1,87 (д, J=10,1 Гц, 2H); МСНР (ЭР) m/z 574,2 (M+ + 1).
Пример 218: Синтез соединения 218, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-5-(5-метилфуран-2-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 218
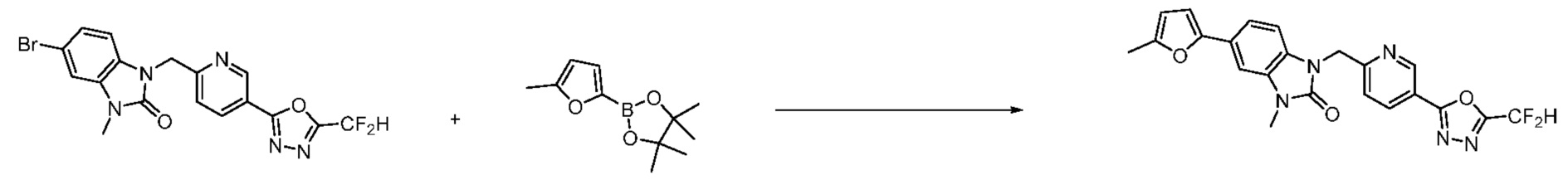
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,344 ммоль), полученный на стадии 4 соединения 208, 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан (0,086 г, 0,413 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,224 г, 0,688 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2%) и концентрируют с получением указанного в заголовке соединения (0,073 г, 48,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=1,6 Гц, 1H), 8,29 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,2 Гц, 1H), 7,29 (дд, J=8,2, 1,5 Гц, 1H), 7,26 (д, J=1,3 Гц, 1H), 7,05-6,79 (м, 2H), 6,44 (д, J=3,2 Гц, 1H), 6,03-6,02 (м, 1H), 5,27 (с, 2H), 3,50 (с, 3H), 2,36 (д, J=0,2 Гц, 3H); МСНР (ЭР) m/z 437,9 (M+ + 1).
Пример 219: Синтез соединения 219, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-5-(тиофен-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 219

5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,344 ммоль), полученный на стадии 4 соединения 208, тиофен-3-илбороновую кислоту (0,053 г, 0,413 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,224 г, 0,688 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2%) и концентрируют с получением указанного в заголовке соединения (0,065 г, 43,0%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,17 (д, J=1,8 Гц, 1H), 8,20 (дд, J=8,2, 2,2 Гц, 1H), 7,31 (д, J=8,2 Гц, 1H), 7,28-7,22 (м, 3H), 7,16 (дд, J=8,1, 1,6 Гц, 1H), 7,09 (д, J=1,4 Гц, 1H), 6,85 (д, J=8,1 Гц, 1H), 6,81 (т, J=51,6 Гц, 1H), 5,19 (с, 2H), 3,41 (с, 3H); МСНР (ЭР) m/z 440,1 (M+ + 1).
Пример 220: Синтез соединения 220, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-5-(4-(метилсульфонил)фенил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 220
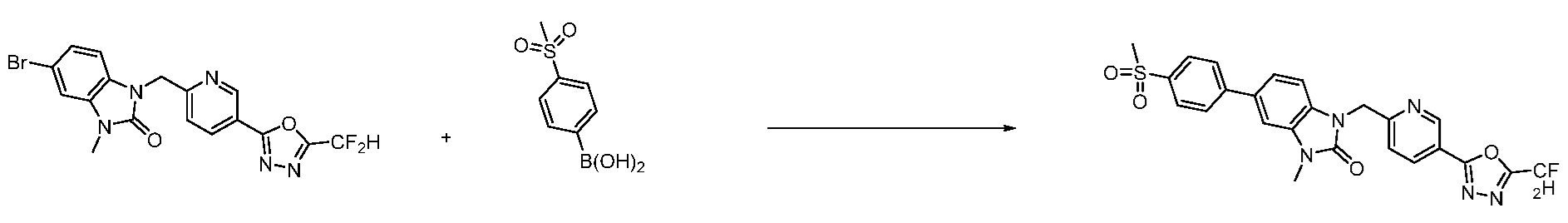
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,344 ммоль), полученный на стадии 4 соединения 208, (4-(метилсульфонил)фенил)бороновую кислоту (0,083 г, 0,413 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,224 г, 0,688 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2%) и концентрируют с получением указанного в заголовке соединения (0,096 г, 54,4%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (д, J=2,0 Гц, 1H), 8,30 (дд, J=8,2, 2,2 Гц, 1H), 7,95 (д, J=8,4 Гц, 2H), 7,72 (дд, J=6,8, 1,7 Гц, 2H), 7,44 (д, J=8,2 Гц, 1H), 7,27 (дд, J=8,2, 1,5 Гц, 1H), 7,22 (д, J=1,5 Гц, 1H), 7,05 (д, J=8,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 5,30 (с, 2H), 3,52 (с, 3H), 3,06 (с, 3H); МСНР (ЭР) m/z 512,3 (M+ + 1).
Пример 221: Синтез соединения 221, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-5-(1H-pyrazole-5-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 221
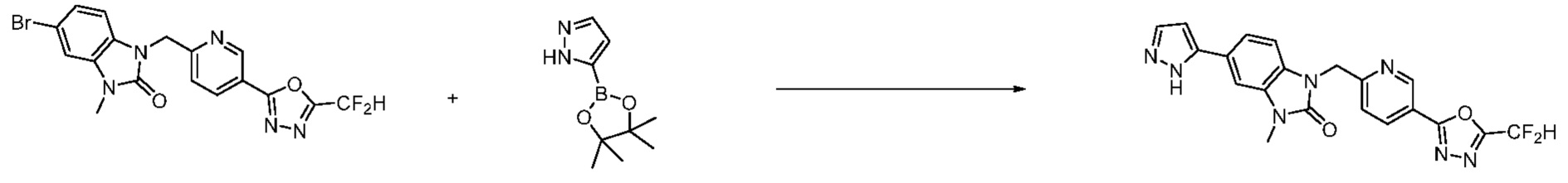
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,344 ммоль), полученный на стадии 4 соединения 208, 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (0,080 г, 0,413 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,224 г, 0,688 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,022 г, 15,1%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=1,7 Гц, 1H), 8,29 (дд, J=8,2, 2,2 Гц, 1H), 7,59 (д, J=2,2 Гц, 1H), 7,46-7,39 (м, 3H), 6,95 (д, J=8,1 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,57 (д, J=1,0 Гц, 1H), 5,29 (с, 2H), 3,47 (с, 3H); МСНР (ЭР) m/z 424,3 (M+ + 1).
Пример 222: Синтез соединения 222, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-5-(1-метил-1H-пиразол-5-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 222
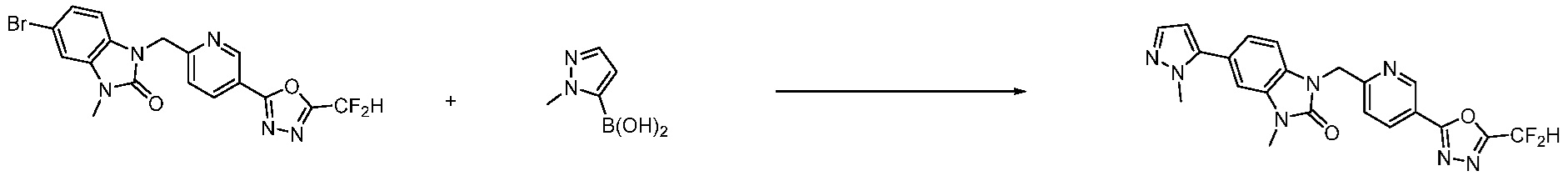
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,344 ммоль), полученный на стадии 4 соединения 208, (1-метил-1H-пиразол-5-ил)бороновую кислоту (0,052 г, 0,413 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,224 г, 0,688 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2%) и концентрируют с получением указанного в заголовке соединения (0,018 г, 11,8%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,8 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,50 (д, J=1,9 Гц, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,09-6,79 (м, 4H), 6,27 (д, J=1,9 Гц, 1H), 5,32 (с, 2H), 3,86 (с, 3H), 3,51 (с, 3H); МСНР (ЭР) m/z 438,0 (M+ + 1).
Пример 223: Синтез соединения 223, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(2,4-дифторфенил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 223

5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, (2,4-дифторфенил)бороновую кислоту (0,053 г, 0,335 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) и карбонат цезия (0,182 г, 0,558 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,042 г, 26,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,6 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,46 (д, J=8,2 Гц, 1H), 7,32-7,26 (м, 1H), 7,21 (д, J=5,8 Гц, 1H), 7,05-6,79 (м, 4H), 5,26 (с, 2H), 4,47-4,39 (м, 1H), 3,01 (д, J=11,6 Гц, 2H), 2,49-2,38 (м, 2H), 2,33 (с, 3H), 2,16 (т, J=11,1 Гц, 2H), 1,86 (дд, J=11,9, 2,0 Гц, 2H); МСНР (ЭР) m/z 571,1 (M+ + 1).
Пример 224: Синтез соединения 224, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 224
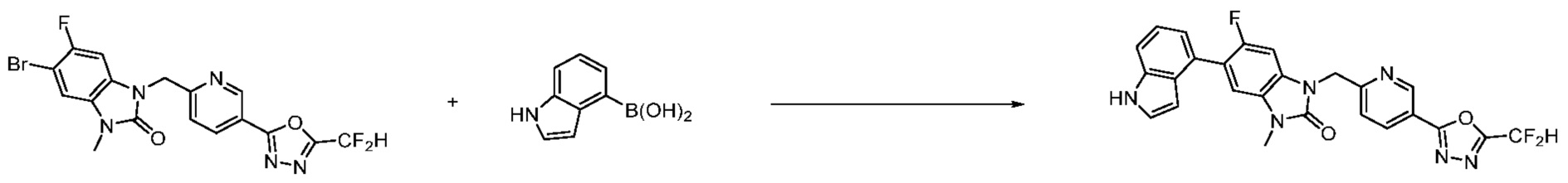
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,330 ммоль), полученный на стадии 4 соединения 147, (1H-индол-4-ил)бороновую кислоту (0,064 г, 0,396 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,215 г, 0,660 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,054 г, 33,6%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,6 Гц, 1H), 8,59 (с, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,38 (д, J=8,1 Гц, 1H), 7,26-7,20 (м, 2H), 7,13 (д, J=6,1 Гц, 2H), 6,92 (т, J=51,7 Гц, 1H), 6,87 (д, J=9,4 Гц, 1H), 6,44 (д, J=2,0 Гц, 1H), 5,30 (с, 2H), 3,46 (с, 3H); МСНР (ЭР) m/z 491,3 (M+ + 1).
Пример 225: Синтез соединения 225, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(4-(метилсульфонил)фенил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 225
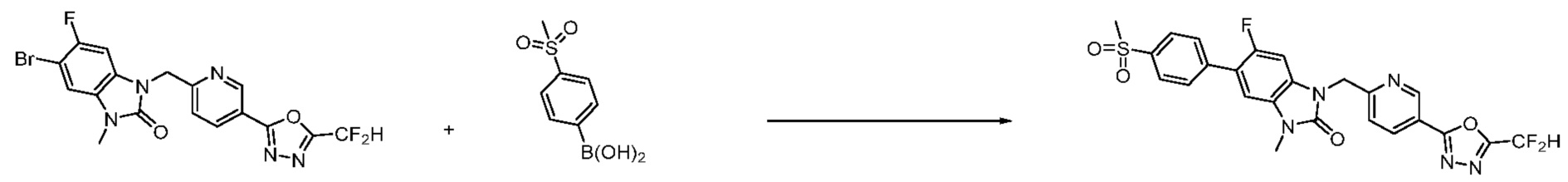
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,330 ммоль), полученный на стадии 4 соединения 147, (4-(метилсульфонил)фенил)бороновую кислоту (0,079 г, 0,396 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,215 г, 0,660 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2%) и концентрируют с получением указанного в заголовке соединения (0,073 г, 41,5%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=1,8 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,96 (д, J=8,4 Гц, 2H), 7,69 (дд, J=8,3, 1,3 Гц, 2H), 7,61 (д, J=110,4 Гц, 1H), 7,00 (д, J=6,2 Гц, 1H), 6,93 (т, J=51,6 Гц, 1H), 6,89 (д, J=10,0 Гц, 1H), 5,27 (с, 2H), 3,49 (с, 3H), 3,07 (с, 3H); МСНР (ЭР) m/z 530,3 (M+ + 1).
Пример 226: Синтез соединения 226, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(1-метил-1H-пиразол-5-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 226
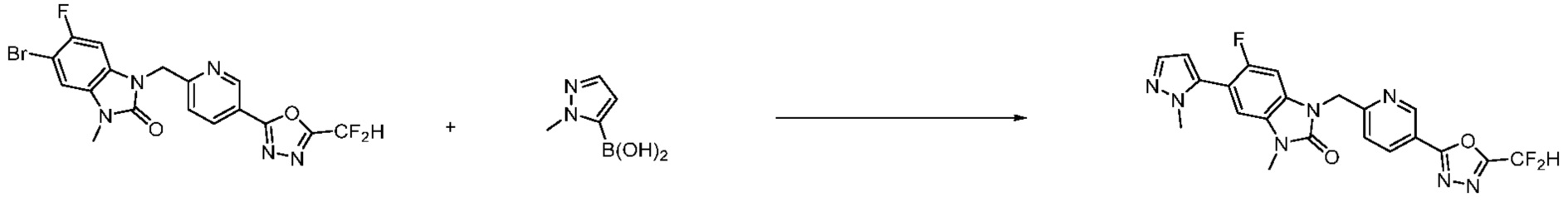
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,330 ммоль), полученный на стадии 4 соединения 147, (1-метил-1H-пиразол-5-ил)бороновую кислоту (0,050 г, 0,396 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,215 г, 0,660 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2%) и концентрируют с получением указанного в заголовке соединения (0,014 г, 9,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (д, J=1,6 Гц, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 7,54 (д, J=1,9 Гц, 1H), 7,51 (д, J=8,2 Гц, 1H), 7,06-6,80 (м, 3H), 6,29 (д, J=1,9 Гц, 1H), 5,29 (с, 2H), 3,78 (д, J=1,5 Гц, 3H), 3,48 (с, 3H); МСНР (ЭР) m/z 456,22 (M+ + 1).
Пример 227: Синтез соединения 227, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(1-метил-1H-индол-5-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 227
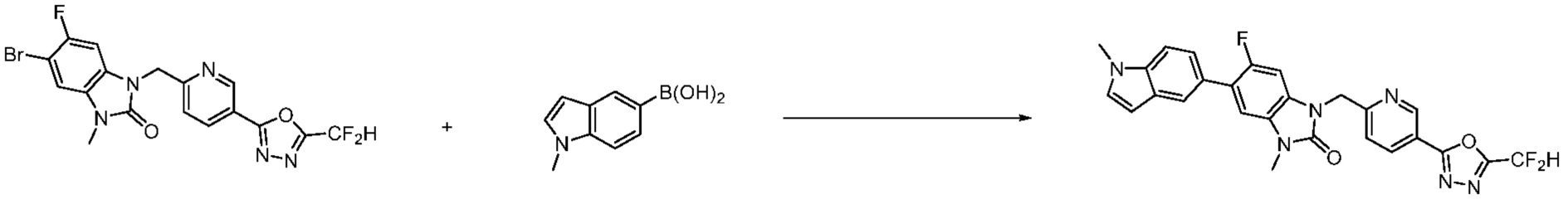
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,330 ммоль), полученный на стадии 4 соединения 147, (1-метил-1H-индол-5-ил)бороновую кислоту (0,069 г, 0,396 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,011 г, 0,017 ммоль) и карбонат цезия (0,215 г, 0,660 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2%) и концентрируют с получением указанного в заголовке соединения (0,047 г, 28,3%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (д, J=1,6 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,74 (д, J=1,1 Гц, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,37 (с, 2H), 7,09-6,80 (м, 4H), 6,52 (д, J=3,1 Гц, 1H), 5,29 (с, 2H), 3,82 (с, 3H), 3,50 (с, 3H); МСНР (ЭР) m/z 505,4 (M+ + 1).
Пример 228: Синтез соединения 228, 5-бром-6-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез 5-бром-6-хлорбензо[d]оксазол-2(3H)-она
2-амино-4-бром-5-хлорфенол (0,200 г, 0,899 ммоль) и 1,1'-карбонилдиимидазол (КДИ, 0,219 г, 1,348 ммоль) растворяют в тетрагидрофуране (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,190 г, 85,1%) в виде белого твердого вещества.
[Стадия 2] Синтез соединения 228
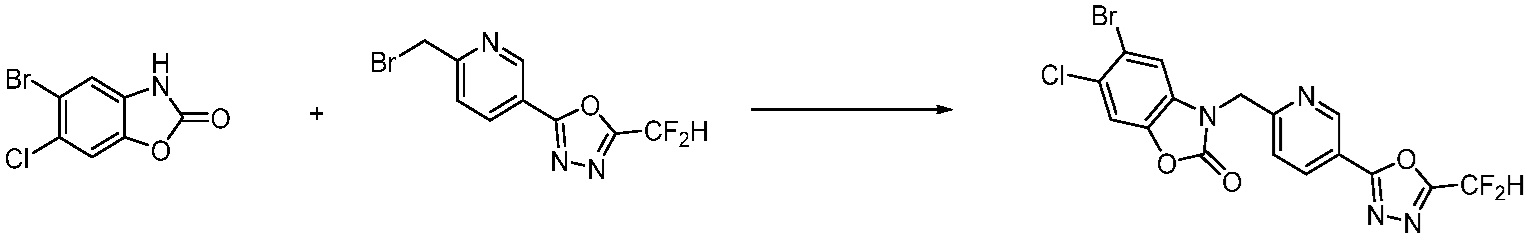
5-бром-6-хлорбензо[d]оксазол-2(3H)-он (0,190 г, 0,765 ммоль), полученный на стадии 1, растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,046 г, 1,147 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,244 г, 0,841 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,253 г, 72,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (дд, J=2,2, 0,8 Гц, 1H), 8,45 (дд, J=8,2, 2,2 Гц, 1H), 7,57 (д, J=8,2 Гц, 1H), 7,39 (с, 1H), 7,33 (с, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,83 (с, 0,25H), 5,20 (с, 2H); МСНР (ЭР) m/z 459,1 (M+ + 1).
Пример 229: Синтез соединения 229, 6-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-(пиридин-3-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 229
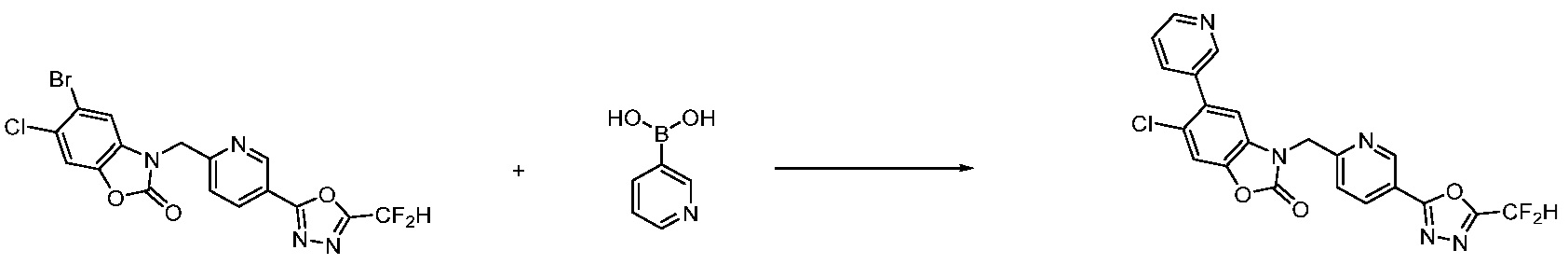
5-бром-6-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]оксазол-2(3H)-он (0,200 г, 0,437 ммоль), пиридин-3-илбороновую кислоту (0,054 г, 0,437 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,028 г, 0,044 ммоль) и карбонат цезия (0,214 г, 0,656 ммоль) смешивают в 1,4-диоксане (10 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 50,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,2, 0,8 Гц, 1H), 8,65 (дд, J=4,9, 1,6 Гц, 1H), 8,62 (д, J=1,6 Гц, 1H), 8,43 (дд, J=8,2, 2,2 Гц, 1H), 7,77 ~ 7,74 (м, 1H), 7,59 (дд, J=8,2, 0,7 Гц, 1H), 7,44 (с, 1H), 7,41 ~ 7,36 (м, 1H), 7,07 (с, 0,25H), 7,03 (с, 1H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 5,23 (с, 2H); МСНР (ЭР) m/z 456,3 (M+ + 1).
Пример 230: Синтез соединения 230, 6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]тиазол-2(3H)-он
[Стадия 1] Синтез соединения 230
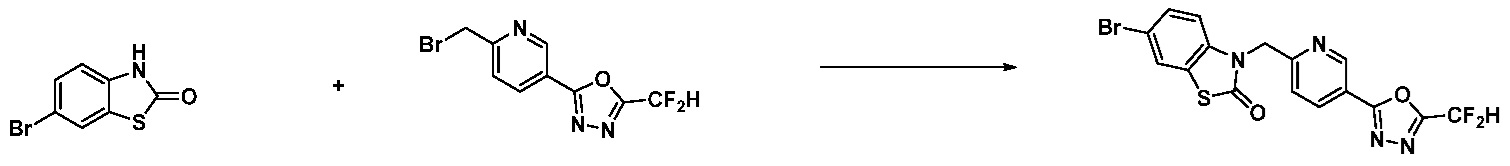
6-бромбензо[d]тиазол-2(3H)-он (0,300 г, 1,304 ммоль) растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,078 г, 1,956 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,378 г, 1,304 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 3 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,309 г, 54,0%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (дд, J=2,2, 0,8 Гц, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 7,59 (д, J=1,9 Гц, 1H), 7,46 (дд, J=8,2, 0,8 Гц, 1H), 7,37 (дд, J=8,6, 2,0 Гц, 1H), 7,07 (с, 0,25H), 7,02 (д, J=8,6 Гц, 1H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 5,34 (с, 2H).
Пример 231: Синтез соединения 231, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(пиридин-3-ил)бензо[d]тиазол-2(3H)-он
[Стадия 1] Синтез соединения 231
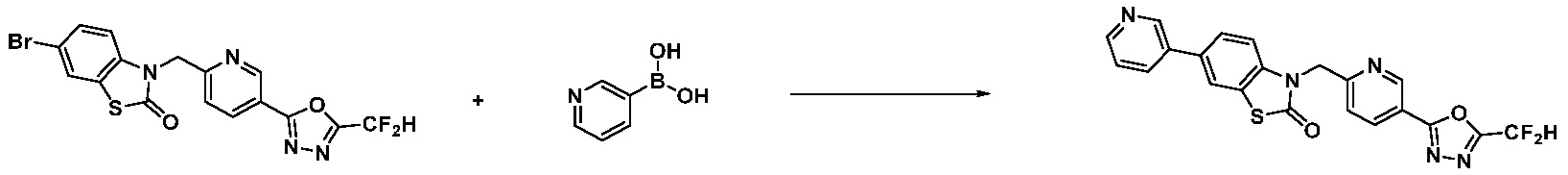
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]тиазол-2(3H)-он (0,170 г, 0,387 ммоль), пиридин-3-илбороновую кислоту (0,062 г, 0,503 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,025 г, 0,039 ммоль) и карбонат цезия (0,189 г, 0,581 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 25 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 65,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ d 9,33 (д, J=1,6 Гц, 1H), 8,82 (с, 1H), 8,65 ~ 8,60 (м, 1H), 8,39 (дд, J=9,0, 1,4 Гц, 1H), 7,85 ~ 7,80 (м, 1H), 7,69 (д, J=1,6 Гц, 1H), 7,51 ~ 7,46 (м, 2H), 7,45 ~ 7,40 (м, 1H), 7,24 (д, J=8,4 Гц, 1H), 7,07 (с, 0,25H), 6,94 (с, 0,5H), 6,81 (с, 0,25H), 5,42 (с, 2H); МСНР (ЭР) m/z 438,2 (M+ + 1).
Пример 232: Синтез соединения 232, 5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-(2,2,3,3,4,4,4-гептафторбутил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 232
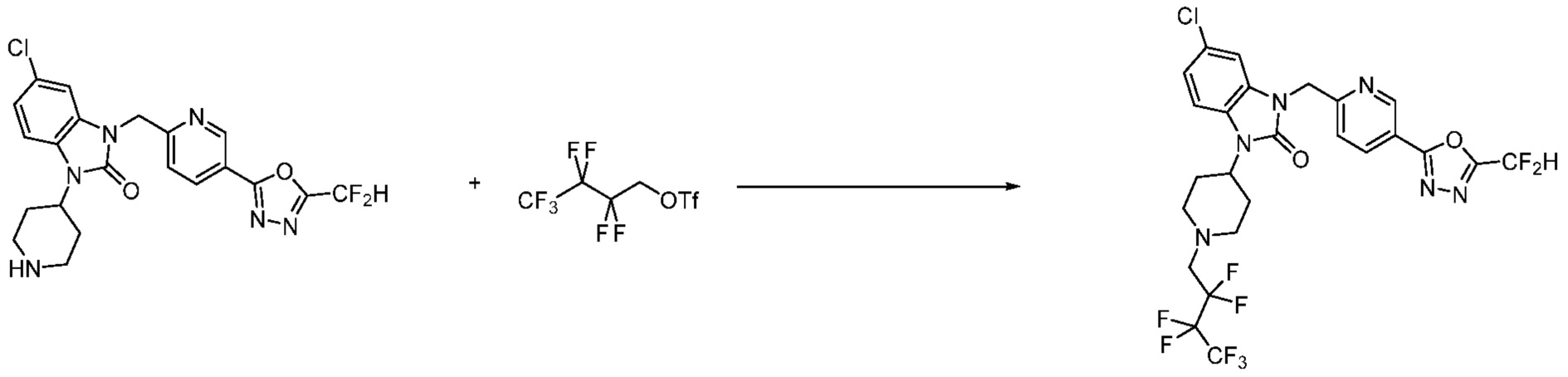
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 213, карбонат калия (0,060 г, 0,434 ммоль) и 2,2,3,3,4,4,4-гептафторбутил трифторметансульфонат (0,079 г, 0,239 ммоль) растворяют в ацетонитриле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,051 г, 36,5%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=1,5 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,16-6,79 (м, 4H), 5,23 (с, 2H), 4,42-4,34 (м, 1H), 3,15-3,07 (м, 4H), 2,61 (т, J=11,2 Гц, 2H), 2,50-2,40 (м, 2H), 1,82 (дд, J=11,9, 2,2 Гц, 2H); МСНР (ЭР) m/z 644,9 (M+ + 1).
Пример 233: Синтез соединения 233, 5-хлор-1-(1-(2,2-дифторбутил)пиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 233
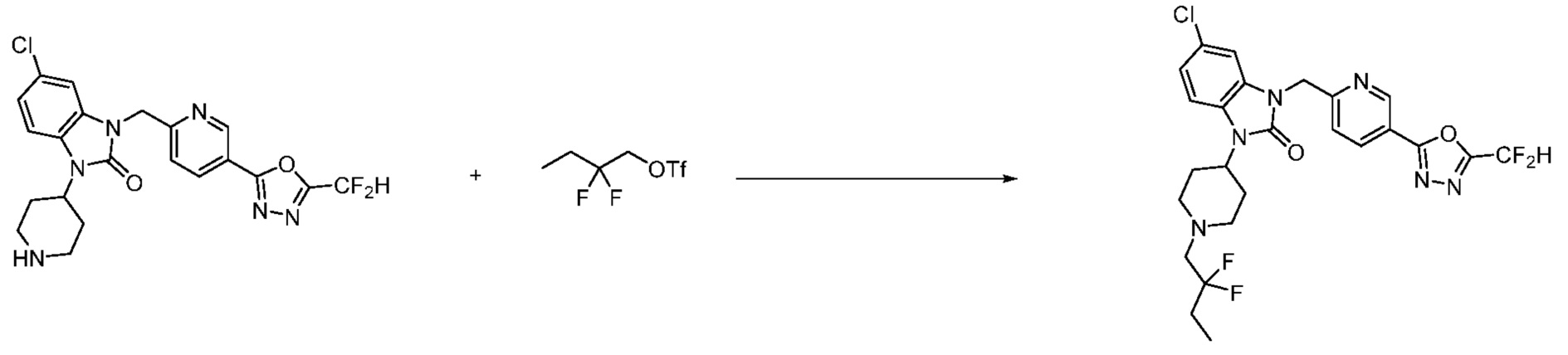
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 213, карбонат калия (0,060 г, 0,434 ммоль) и 2,2-дифторбутил трифторметансульфонат (0,058 г, 0,239 ммоль) растворяют в ацетонитриле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,079 г, 65,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,5 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,15-6,79 (м, 4H), 5,23 (с, 2H), 4,35-4,29 (м, 1H), 3,14-3,09 (м, 2H), 2,73 (т, J=13,8 Гц, 2H), 2,49-2,39 (м, 4H), 2,05-1,91 (м, 2H), 1,81-1,80 (м, 2H), 1,03 (т, J=7,5 Гц, 3H); МСНР (ЭР) m/z 552,9 (M+ + 1).
Пример 234: Синтез соединения 234, 5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-(2,2-дифторпропил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 234
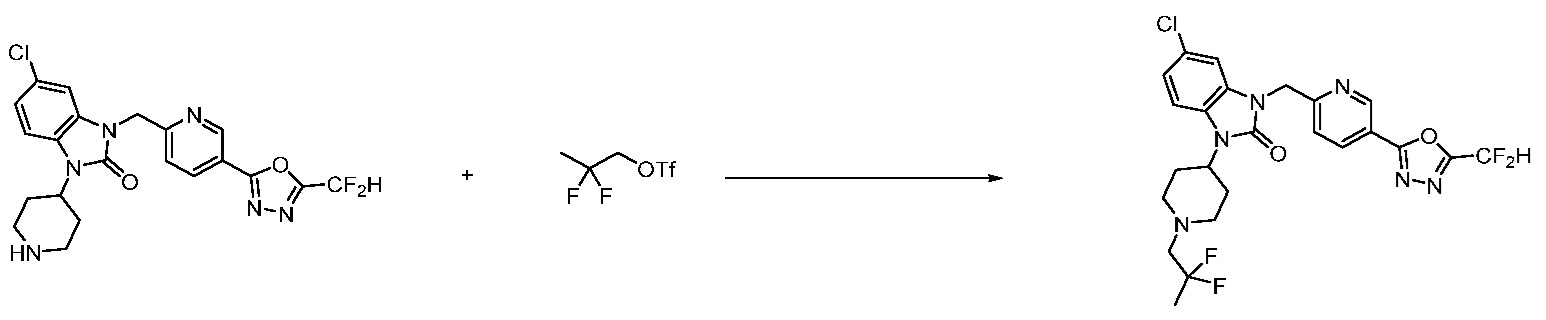
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 213, карбонат калия (0,060 г, 0,434 ммоль) и 2,2-дифторпропил трифторметансульфонат (0,054 г, 0,239 ммоль) растворяют в ацетонитриле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,082 г, 70,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,2, 0,7 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (дд, J=8,2, 0,6 Гц, 1H), 7,15 (д, J=8,4 Гц, 1H), 7,05-6,79 (м, 3H), 5,22 (с, 2H), 4,37-4,30 (м, 1H), 3,13-3,07 (м, 2H), 2,73 (т, J=13,5 Гц, 2H), 2,49-2,38 (м, 4H), 1,82-1,78 (м, 2H), 1,67 (т, J=18,7 Гц, 3H); МСНР (ЭР) m/z 540,9 (M+ + 1).
Пример 235: Синтез соединения 235, 5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-(2,2,3,3-тетрафторпропил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 235
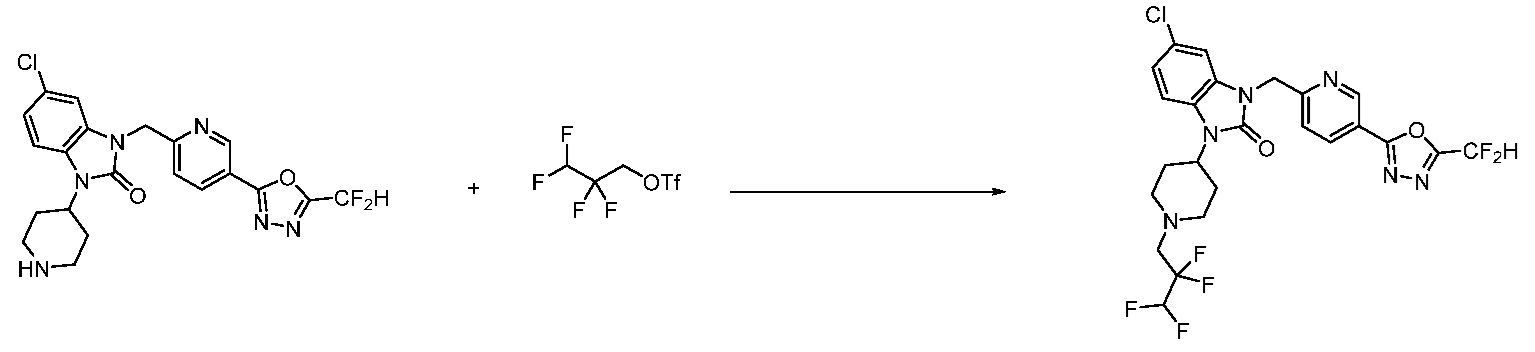
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 213, карбонат калия (0,060 г, 0,434 ммоль) и 2,2,3,3-тетрафторпропил трифторметансульфонат (0,063 г, 0,239 ммоль) растворяют в ацетонитриле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,101 г, 80,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,1, 0,7 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (дд, J=8,2, 0,5 Гц, 1H), 7,12-6,92 (м, 4H), 6,20-5,90 (м, 1H), 5,22 (с, 2H), 4,37-4,29 (м, 1H), 3,11 (д, J=10,8 Гц, 2H), 2,96 (т, J=14,1 Гц, 2H), 2,55-2,39 (м, 4H), 1,82 (дд, J=11,1, 3,1 Гц, 2H); МСНР (ЭР) m/z 576,9 (M+ + 1).
Пример 236: Синтез соединения 236, 5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 236
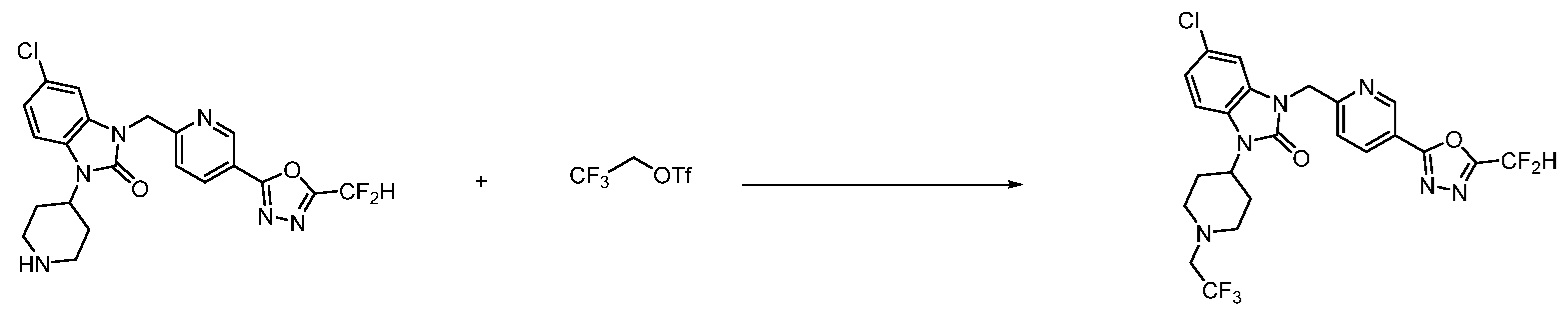
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 213, карбонат калия (0,060 г, 0,434 ммоль) и 2,2,2-трифторэтил трифторметансульфонат (0,055 г, 0,239 ммоль) растворяют в ацетонитриле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,087 г, 73,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=2,1 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,18 (д, J=8,4 Гц, 1H), 7,05-6,79 (м, 3H), 5,23 (с, 2H), 4,42-4,34 (м, 1H), 3,12 (д, J=11,5 Гц, 2H), 3,05 (кв, J=9,6 Гц, 2H), 2,59 (т, J=11,3 Гц, 2H), 2,50-2,40 (м, 2H), 1,82 (дд, J=11,9, 2,2 Гц, 2H); МСНР (ЭР) m/z 545,2 (M+ + 1).
Пример 237: Синтез соединения 237, трет-бутил 5-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)-1H-индол-1-карбоксилат
[Стадия 1] Синтез соединения 237
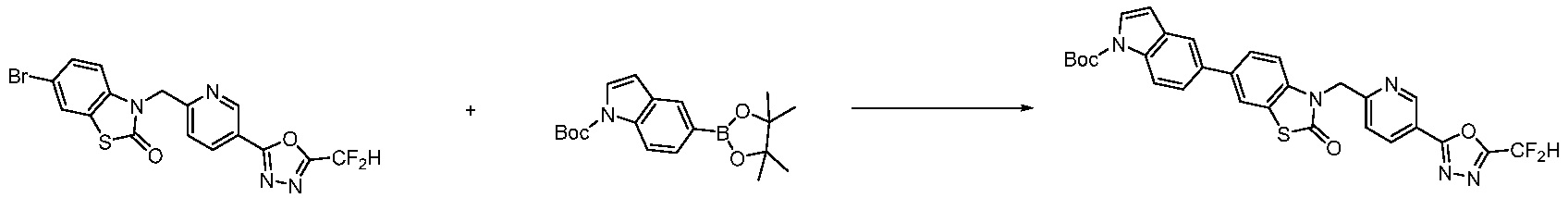
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]тиазол-2(3H)-он (0,150 г, 0,342 ммоль), трет-бутил 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилат (0,141 г, 0,410 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,022 г, 0,034 ммоль) и карбонат цезия (0,167 г, 0,512 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,013 г, 6,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,34 (дд, J=2,2, 0,7 Гц, 1H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 8,21 (д, J=8,4 Гц, 1H), 7,74 ~ 7,71 (м, 2H), 7,65 (д, J=3,6 Гц, 1H), 7,54 ~ 7,48 (м, 3H), 7,17 (д, J=8,4 Гц, 1H), 7,07 (с, 0,25H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 6,62 (дд, J=3,7, 0,5 Гц, 1H), 5,42 (с, 2H), 1,70 (с, 9H).
Пример 238: Синтез соединения 238, трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)-3,6-дигидропиридин-1(2H)-карбоксилат
[Стадия 1] Синтез соединения 238
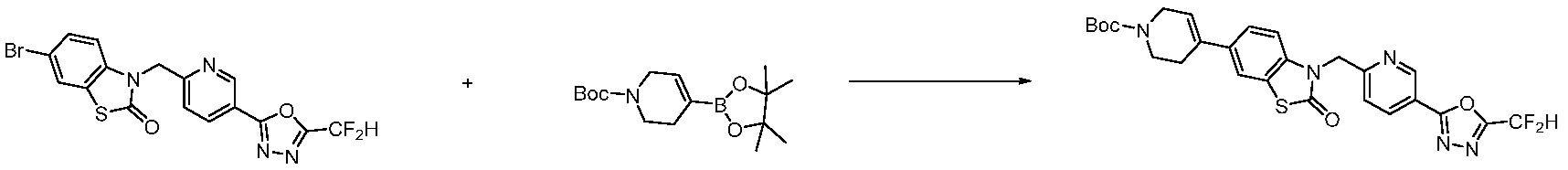
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]тиазол-2(3H)-он (0,700 г, 1,594 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,591 г, 1,912 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,104 г, 0,159 ммоль) и карбонат цезия (0,779 г, 2,391 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,442 г, 51,2%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 ~ 9,30 (м, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,48 ~ 7,44 (м, 2H), 7,28 ~ 7,25 (м, 1H), 7,07 (с, 0,25H), 7,06 (д, J=8,6 Гц, 1H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 6,00 (с, 1H), 5,37 (с, 2H), 4,05 ~ 4,00 (м, 2H), 3,64 (т, J=5,6 Гц, 2H), 2,50 ~ 2,49 (м, 2H), 1,52 (с, 9H).
Пример 239: Синтез соединения 239, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
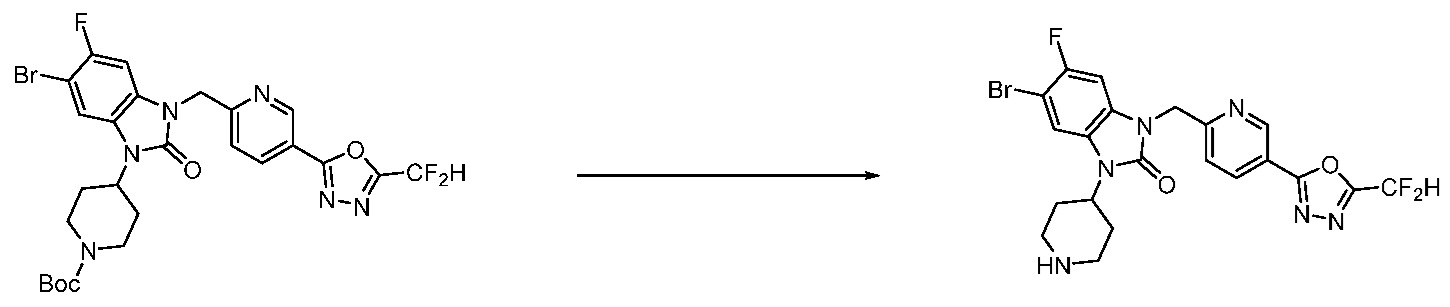
Трет-бутил 4-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,750 г, 4,411 ммоль), полученный на стадии 1 соединения 149, и трифторуксусную кислоту (3,378 мл, 44,111 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=50-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-50%) и концентрируют с получением указанного в заголовке соединения (2,100 г, 91,0%) в виде коричневого твердого вещества.
[Стадия 2] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
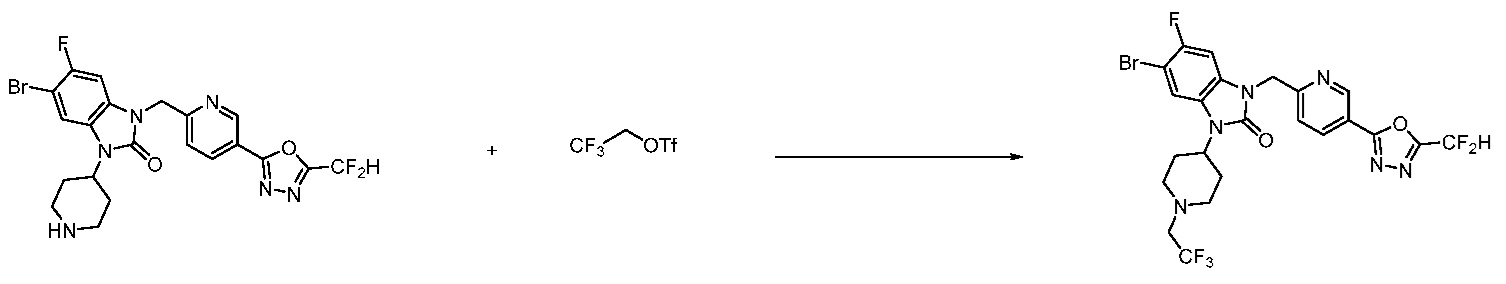
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,400 г, 0,764 ммоль), полученный на стадии 1, карбонат калия (0,211 г, 1,529 ммоль) и 2,2,2-трифторэтил трифторметансульфонат (0,177 г, 0,764 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2%) и концентрируют с получением указанного в заголовке соединения (0,380 г, 82,1%) в виде желтого твердого вещества.
[Стадия 3] Синтез соединения 239
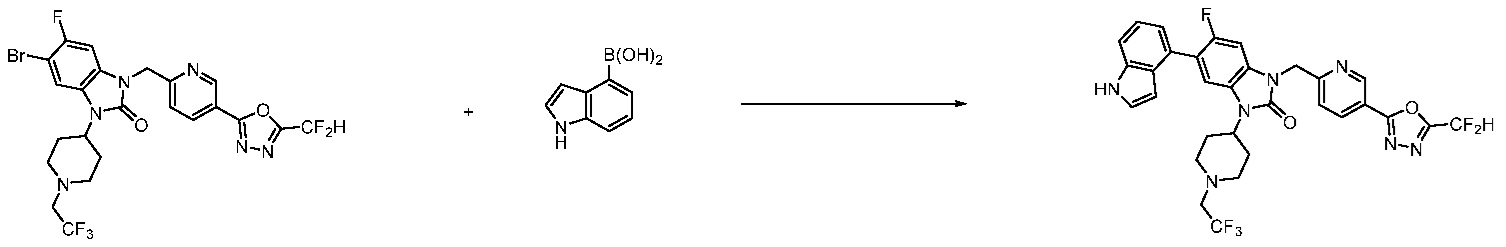
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,248 ммоль), полученный на стадии 2, (1H-индол-4-ил)бороновую кислоту (0,048 г, 0,297 ммоль), карбонат цезия (0,161 г, 0,496 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,045 г, 28,3%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (дд, J=2,2, 0,7 Гц, 1H), 8,51 (с, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,48 (дд, J=8,2, 0,5 Гц, 1H), 7,42 (д, J=8,1 Гц, 1H), 7,39 (д, J=5,9 Гц, 1H), 7,30-7,23 (м, 2H), 7,16 (д, J=7,2 Гц, 1H), 6,93 (т, J=51,6 Гц, 1H), 6,92 (д, J=10,0 Гц, 1H), 6,44 (д, J=2,2 Гц, 1H), 5,29 (с, 2H), 4,44-4,36 (м, 1H), 3,09 (д, J=11,3 Гц, 2H), 3,01 (кв, J=9,6 Гц, 2H), 2,61-2,43 (м, 4H), 1,86 (дд, J=11,7, 2,1 Гц, 2H); МСНР (ЭР) m/z 642,0 (M+ + 1).
Пример 240: Синтез соединения 240, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(пиридин-3-ил)-3-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 240
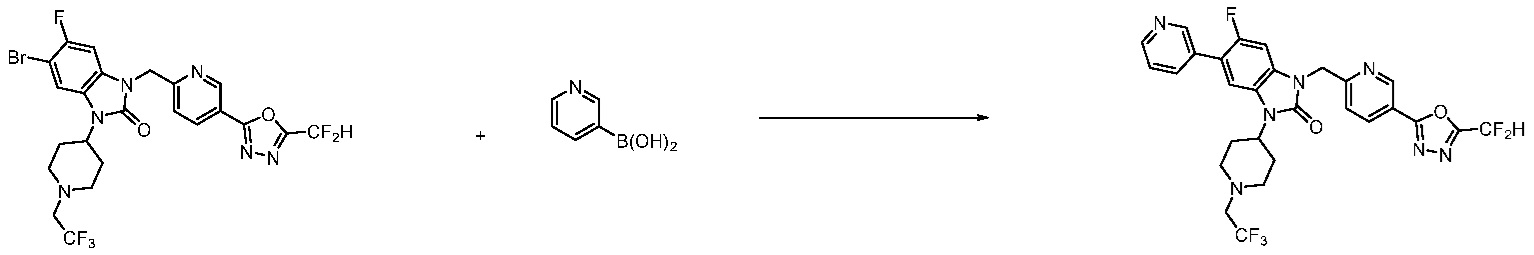
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,248 ммоль), полученный на стадии 2 соединения 239, пиридин-3-илбороновую кислоту (0,037 г, 0,297 ммоль), карбонат цезия (0,161 г, 0,496 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,123 г, 82,0%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (дд, J=2,1, 0,6 Гц, 1H), 8,74 (с, 1H), 8,59 (д, J=4,0 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,83 (дд, J=7,9, 1,4 Гц, 1H), 7,48 (д, J=8,2 Гц, 1H), 7,38 (дд, J=7,8, 4,8 Гц, 1H), 7,21 (д, J=6,1 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,91 (д, J=9,0 Гц, 1H), 5,26 (с, 2H), 4,43-4,35 (м, 1H), 3,13 (д, J=11,5 Гц, 2H), 3,05 (кв, J=9,6 Гц, 2H), 2,62 (т, J=11,4 Гц, 2H), 2,52-2,24 (м, 2H), 1,86 (дд, J=11,8, 2,0 Гц, 2H); МСНР (ЭР) m/z 604,1 (M+ + 1).
Пример 241: Синтез соединения 241, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(1-(2,2,3,3-тетрафторпропил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(2,2,3,3-тетрафторпропил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
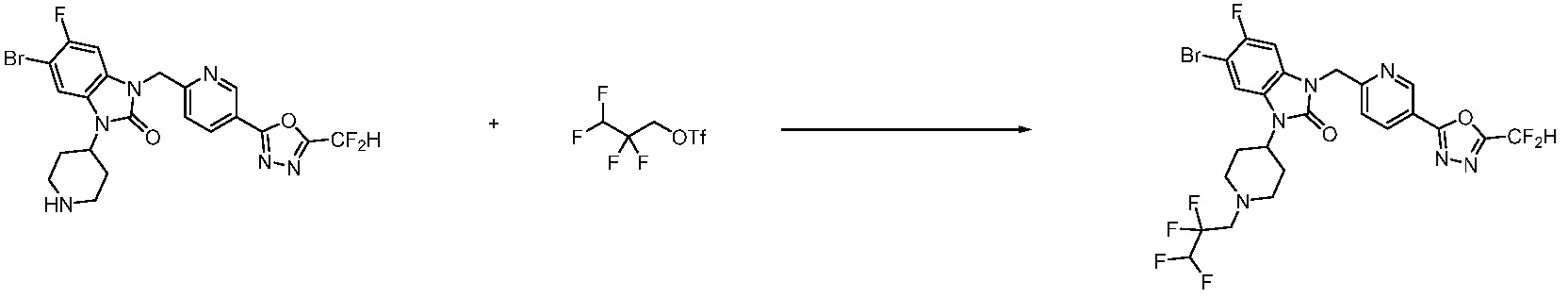
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,400 г, 0,764 ммоль), полученный на стадии 1 соединения 239, карбонат калия (0,211 г, 1,529 ммоль) и 2,2,3,3-тетрафторпропил трифторметансульфонат (0,202 г, 0,764 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2%) и концентрируют с получением указанного в заголовке соединения (0,350 г, 71,8%) в виде желтого твердого вещества.
[Стадия 2] Синтез соединения 241
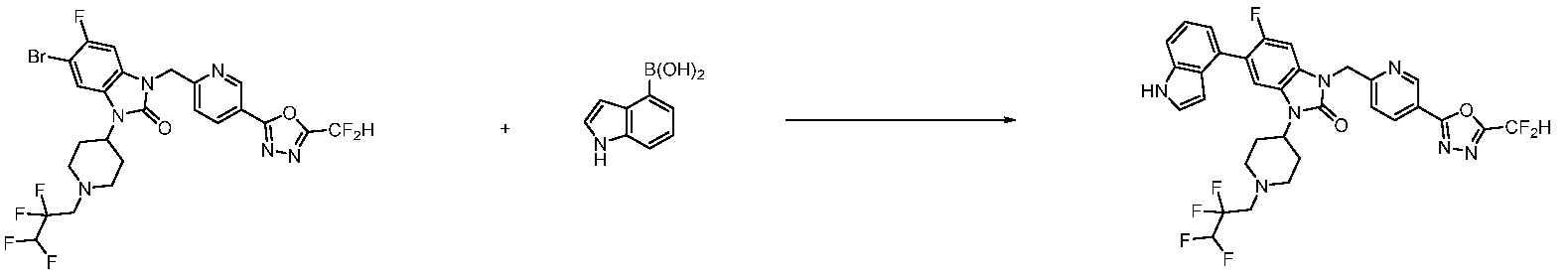
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(2,2,3,3-тетрафторпропил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,235 ммоль), полученный на стадии 1, (1H-индол-4-ил)бороновую кислоту (0,045 г, 0,282 ммоль), карбонат цезия (0,153 г, 0,471 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,112 г, 70,3%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,7 Гц, 1H), 8,54 (с, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,48 (д, J=8,2 Гц, 1H), 7,43 (д, J=13,6 Гц, 1H), 7,37 (д, J=5,9 Гц, 1H), 7,29 (д, J=7,4 Гц, 1H), 7,26-7,24 (м, 1H), 7,17 (д, J=12,0 Гц, 1H), 7,05-6,80 (м, 2H), 6,47 (д, J=1,8 Гц, 1H), 6,13-5,84 (м, 1H), 5,29 (с, 2H), 4,40-4,36 (м, 1H), 3,09-3,07 (м, 2H), 2,92 (т, J=13,9 Гц, 2H), 2,52-2,41 (м, 4H), 1,86-1,85 (м, 2H); МСНР (ЭР) m/z 673,9 (M+ + 1).
Пример 242: Синтез соединения 242, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(пиридин-3-ил)-3-(1-(2,2,3,3-тетрафторпропил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 242
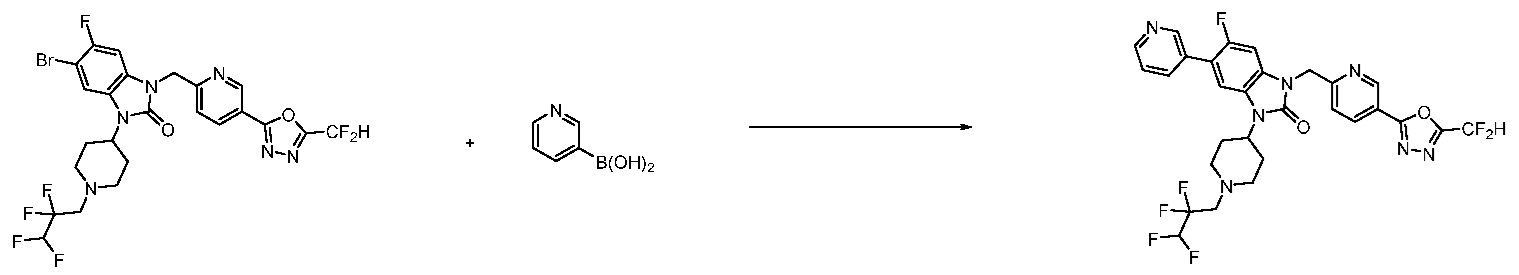
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(2,2,3,3-тетрафторпропил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,235 ммоль), полученный на стадии 1 соединения 241, пиридин-3-илбороновую кислоту (0,035 г, 0,282 ммоль), карбонат цезия (0,153 г, 0,471 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,012 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,095 г, 63,3%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (дд, J=2,1, 0,6 Гц, 1H), 8,71 (с, 1H), 8,56 (д, J=3,7 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,80 (дд, J=8,0, 1,8 Гц, 1H), 7,46 (д, J=8,2 Гц, 1H), 7,35 (дд, J=7,8, 4,9 Гц, 1H), 7,13 (д, J=6,1 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,91 (д, J=9,7 Гц, 1H), 6,16-5,86 (м, 1H), 5,24 (с, 2H), 4,34-4,27 (м, 1H), 3,10 (д, J=8,8 Гц, 2H), 2,94 (т, J=14,1 Гц, 2H), 2,54-2,41 (м, 4H), 1,84 (д, J=9,8 Гц, 2H); МСНР (ЭР) m/z 636,2 (M+ + 1).
Пример 243: Синтез соединения 243, 5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-(2-гидроксиацетил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 243
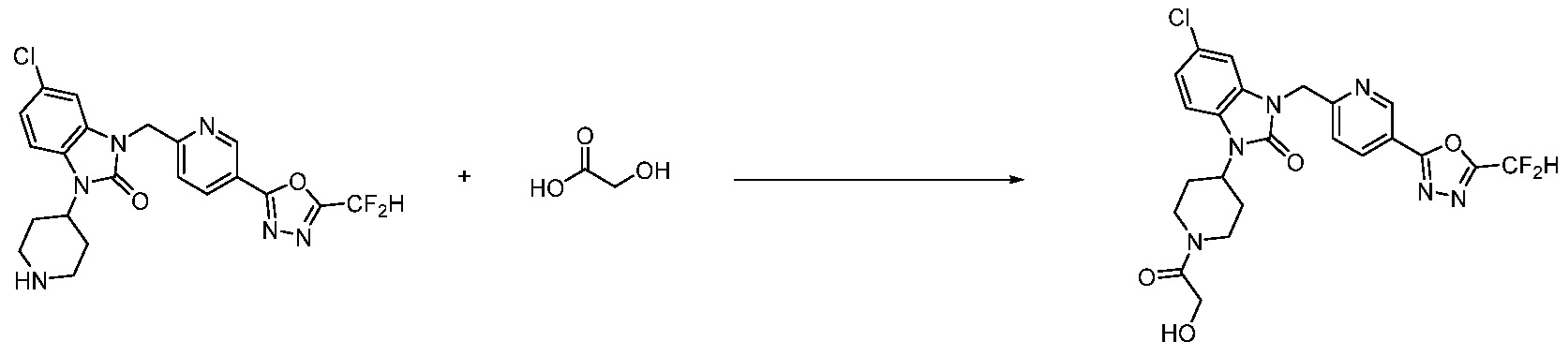
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 213, 2-гидроксиуксусную кислоту (0,018 г, 0,239 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид (ЭДК, 0,067 г, 0,434 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt, 0,059 г, 0,434 ммоль) и триэтиламин (0,091 мл, 0,651 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,084 г, 74,4%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (д, J=1,6 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,03-6,98 (м, 3H), 6,92 (т, J=51,6 Гц, 1H), 5,21 (с, 2H), 4,83-4,82 (м, 1H), 4,58-4,50 (м, 1H), 4,21 (кв, J=13,9 Гц, 2H), 3,68 (д, J=13,7 Гц, 1H), 3,19-3,12 (м, 1H), 2,81 (т, J=12,1 Гц, 1H), 2,37-2,26 (м, 2H), 1,95 (д, J=12,1 Гц, 2H); МСНР (ЭР) m/z 519,4 (M+ + 1).
Пример 244: Синтез соединения 244, метил 6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-1H-индол-2-карбоксилат
[Стадия 1] Синтез 1-(трет-бутил) 2-метил 6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-1H-индол-1,2-дикарбоксилата
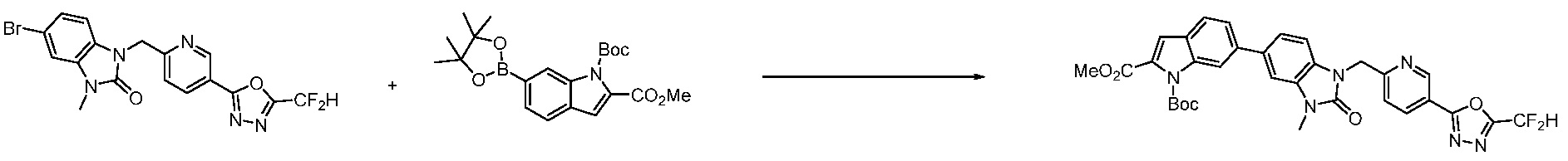
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 0,458 ммоль), полученный на стадии 4 соединения 208, 1-(трет-бутил) 2-метил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1,2-дикарбоксилат (0,221 г, 0,550 ммоль), карбонат цезия (0,299 г, 0,917 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,015 г, 0,023 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,150 г, 50,4%) в виде коричневого твердого вещества.
[Стадия 2] Синтез соединения 244

1-(трет-бутил) 2-метил 6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-1H-индол-1,2-дикарбоксилат (0,150 г, 0,238 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,182 мл, 2,379 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Дихлорметан (10 мл) добавляют в полученный концентрат и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют, затем промывают с дихлорметаном, и затем сушат с получением указанного в заголовке соединения (0,075 г, 59,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,6 Гц, 1H), 9,04 (с, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,73 (д, J=8,4 Гц, 1H), 7,56 (с, 1H), 7,44 (д, J=8,2 Гц, 1H), 7,38 (дд, J=8,4, 1,5 Гц, 1H), 7,31 (дд, J=8,1, 1,6 Гц, 1H), 7,26-7,22 (м, 2H), 7,01 (д, J=3,3 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 5,33 (с, 2H), 3,95 (с, 3H), 3,54 (с, 3H); МСНР (ЭР) m/z 531,4 (M+ + 1).
Пример 245: Синтез соединения 245, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 245
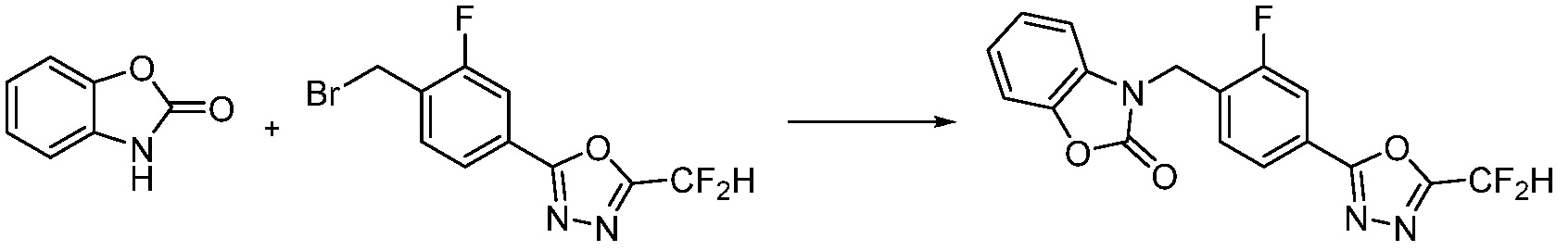
Бензо[d]оксазол-2(3H)-он (0,100 г, 0,740 ммоль), 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,250 г, 0,814 ммоль), карбонат калия (0,153 г, 1,110 ммоль) и йодид калия (0,012 г, 0,074 ммоль) растворяют в N,N-диметилформамиде (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Этилацетат добавляют в полученный концентрат и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют, затем промывают этилацетат, и затем сушат с получением указанного в заголовке соединения (0,191 г, 71,4%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 7,93-7,89 (м, 2H), 7,67 (д, J=7,4 Гц, 1H), 7,64-7,43 (м, 1H), 7,41-7,39 (м, 1H), 7,23-7,14 (м, 3H), 5,23 (с, 2H); МСНР (ЭР) m/z 362,3 (M+ + 1).
Пример 246: Синтез соединения 246, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 246
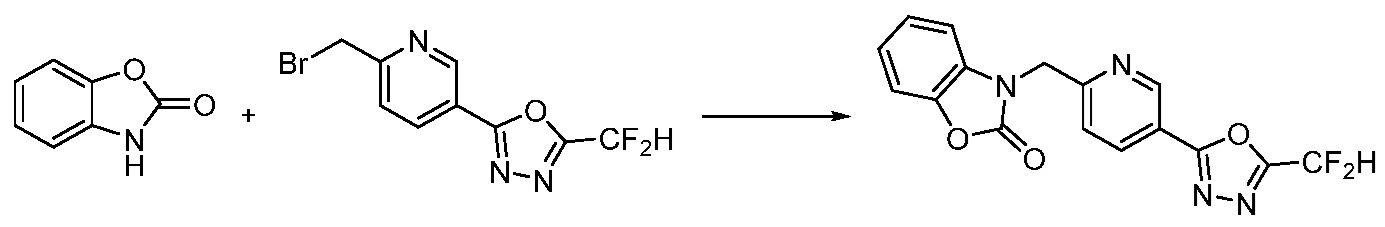
Бензо[d]оксазол-2(3H)-он (0,115 г, 0,851 ммоль), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,247 г, 0,851 ммоль), карбонат калия (0,176 г, 1,277 ммоль) и йодид калия (0,014 г, 0,085 ммоль) растворяют в N,N-диметилформамиде (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Этилацетат добавляют в полученный концентрат и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют, затем промывают этилацетат, и затем сушат с получением указанного в заголовке соединения (0,160 г, 54,6%) в виде светло-оранжевого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,14 (д, J=1,6 Гц, 1H), 8,46 (дд, J=8,2, 2,2 Гц, 1H), 7,72 (д, J=8,3 Гц, 1H), 7,70-7,45 (м, 1H), 7,41-7,39 (м, 1H), 7,19-7,14 (м, 3H), 5,32 (с, 2H); МСНР (ЭР) m/z 345,3 (M+ + 1).
Пример 247: Синтез соединения 247, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-5-(1H-пиразол-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 247
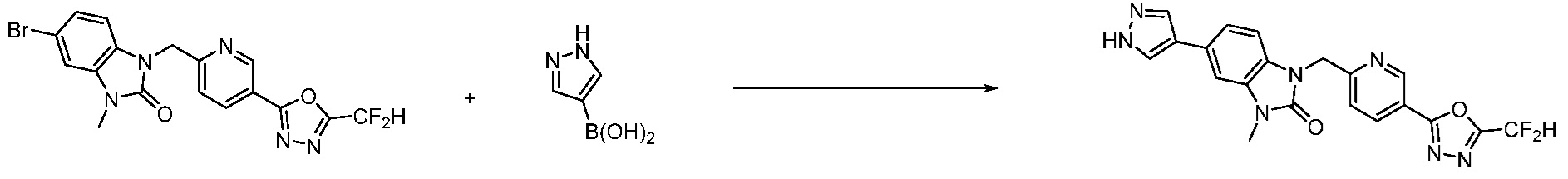
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,344 ммоль), полученный на стадии 4 соединения 208, (1H-пиразол-4-ил)бороновую кислоту (0,050 г, 0,447 ммоль), карбонат цезия (0,224 г, 0,688 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,022 г, 0,034 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,064 г, 43,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=1,6 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,90-7,75 (м, 2H), 7,42 (д, J=8,3 Гц, 1H), 7,17-7,10 (м, 2H), 6,94 (д, J=8,1 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 5,29 (с, 2H), 3,51 (с, 3H); МСНР (ЭР) m/z 424,0 (M+ + 1).
Пример 248: Синтез соединения 248, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-5-(1H-пиразол-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 248
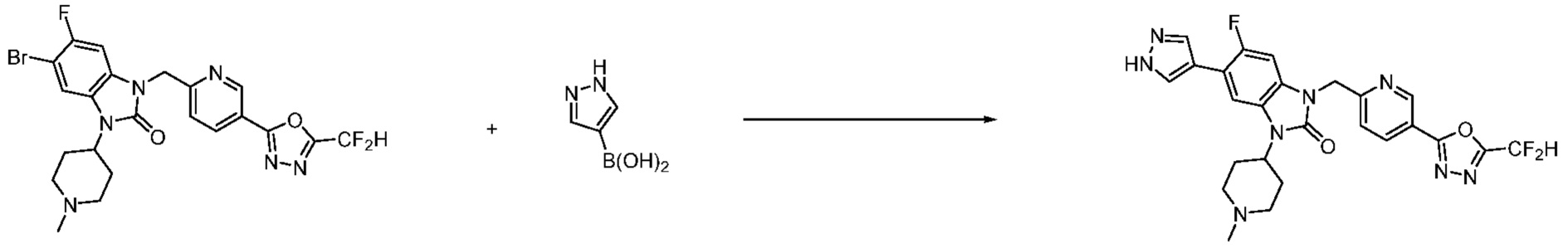
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, (1H-пиразол-4-ил)бороновую кислоту (0,037 г, 0,335 ммоль), карбонат цезия (0,182 г, 0,558 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,041 г, 28,0%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=2,0 Гц, 1H), 8,32 (дд, J=8,2, 2,1 Гц, 1H), 7,80 (д, J=1,1 Гц, 2H), 7,75 (д, J=6,1 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,78 (д, J=9,9 Гц, 1H), 5,24 (с, 2H), 4,60-4,52 (м, 1H), 3,18 (д, J=11,4 Гц, 2H), 2,82-2,73 (м, 2H), 2,46 (с, 3H), 2,35 (т, J=11,4 Гц, 2H), 1,90 (д, J=10,7 Гц, 2H); МСНР (ЭР) m/z 525,6 (M+ + 1).
Пример 249: Синтез соединения 249, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1-метил-1H-пиразол-5-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 249
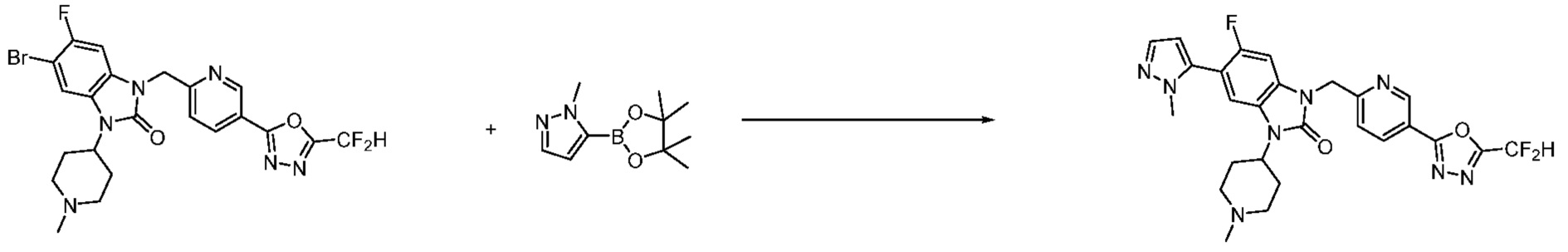
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (0,070 г, 0,335 ммоль), карбонат цезия (0,182 г, 0,558 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,041 г, 27,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,8 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,50-7,47 (м, 2H), 7,19 (д, J=5,8 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,90 (д, J=9,2 Гц, 1H), 6,24 (д, J=1,8 Гц, 1H), 5,25 (с, 2H), 4,44-4,35 (м, 1H), 3,74 (д, J=1,2 Гц, 3H), 3,00 (д, J=11,6 Гц, 2H), 2,47-2,37 (м, 2H), 2,31 (с, 3H), 2,14 (т, J=11,1 Гц, 2H), 1,84 (дд, J=11,8, 2,0 Гц, 2H); МСНР (ЭР) m/z 539,5 (M+ + 1).
Пример 250: Синтез соединения 250, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1-метил-1H-индол-5-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 250
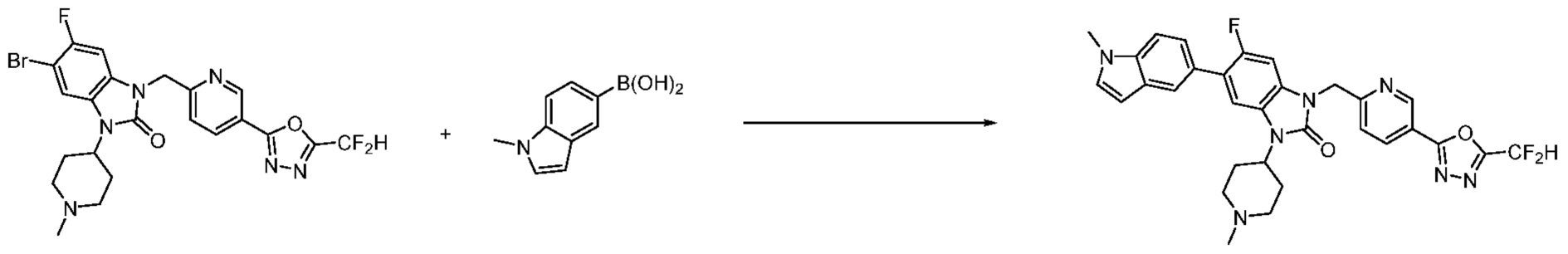
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, (1-метил-1H-индол-5-ил)бороновую кислоту (0,059 г, 0,335 ммоль), карбонат цезия (0,182 г, 0,558 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,3 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,101 г, 61,6%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,6 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,70 (д, J=1,0 Гц, 1H), 7,42 (д, J=8,2 Гц, 1H), 7,37 (д, J=6,3 Гц, 1H), 7,33 (с, 2H), 7,06-6,79 (м, 3H), 6,50 (д, J=3,1 Гц, 1H), 5,26 (с, 2H), 4,47-4,39 (м, 1H), 3,78 (с, 3H), 3,01 (д, J=11,6 Гц, 2H), 2,57-2,47 (м, 2H), 2,32 (с, 3H), 2,16 (т, J=11,1 Гц, 2H), 1,87 (д, J=10,1 Гц, 2H); МСНР (ЭР) m/z 588,5 (M+ + 1).
Пример 251: Синтез соединения 251, трет-бутил 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-1H-индол-1-карбоксилат
[Стадия 1] Синтез соединения 251
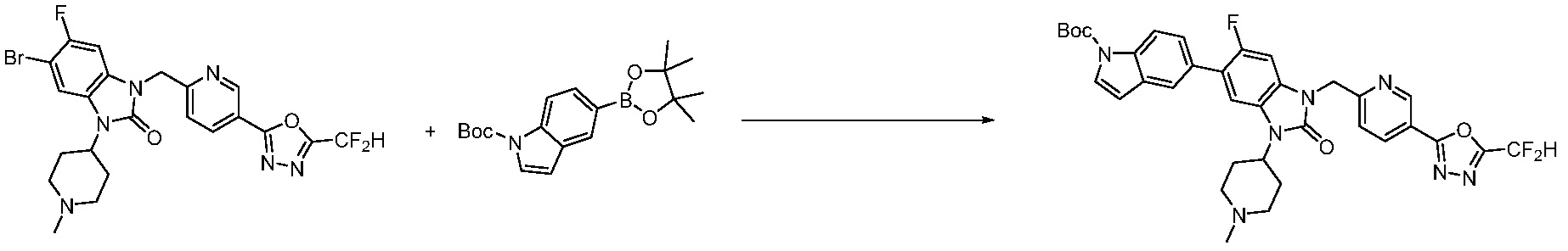
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, трет-бутил 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилат (0,115 г, 0,335 ммоль), карбонат цезия (0,182 г, 0,558 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,124 г, 65,9%) в виде красного твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,6 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 8,15 (д, J=8,4 Гц, 1H), 7,64-7,62 (м, 2H), 7,45-7,40 (м, 2H), 7,34 (д, J=6,3 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,85 (д, J=9,8 Гц, 1H), 6,59 (д, J=3,6 Гц, 1H), 5,27 (с, 2H), 4,48-4,40 (м, 1H), 3,02 (д, J=11,6 Гц, 2H), 2,55-2,45 (м, 2H), 2,33 (с, 3H), 2,20-2,15 (м, 2H), 1,89-1,86 (м, 2H), 1,68 (с, 9H); МСНР (ЭР) m/z 674,19 (M+ + 1).
Пример 252: Синтез соединения 252, трет-бутил 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-2-метил-1H-индол-1-карбоксилат
[Стадия 1] Синтез соединения 252
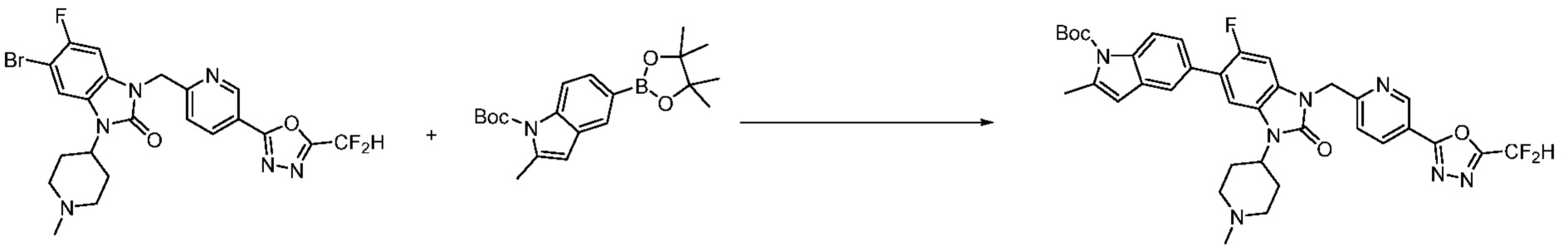
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, трет-бутил 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилат (0,120 г, 0,335 ммоль), карбонат цезия (0,182 г, 0,558 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,112 г, 58,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=1,8 Гц, 1H), 8,34 (дд, J=17,7, 11,7 Гц, 1H), 8,10 (д, J=8,7 Гц, 1H), 7,51 (с, 1H), 7,43 (д, J=8,2 Гц, 1H), 7,33 (т, J=7,9 Гц, 2H), 6,92 (т, J=51,7 Гц, 1H), 6,83 (д, J=9,7 Гц, 1H), 6,33 (с, 1H), 5,26 (с, 2H), 4,47-4,39 (м, 1H), 3,03-3,01 (м, 2H), 2,60 (с, 3H), 2,55-2,43 (м, 2H), 2,20 (с, 3H), 2,20-2,14 (м, 2H), 1,88-1,86 (м, 2H), 1,68 (с, 9H); МСНР (ЭР) m/z 688,48 (M+ + 1).
Пример 253: Синтез соединения 253, трет-бутил 2-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-1H-индол-1-карбоксилат
[Стадия 1] Синтез соединения 253
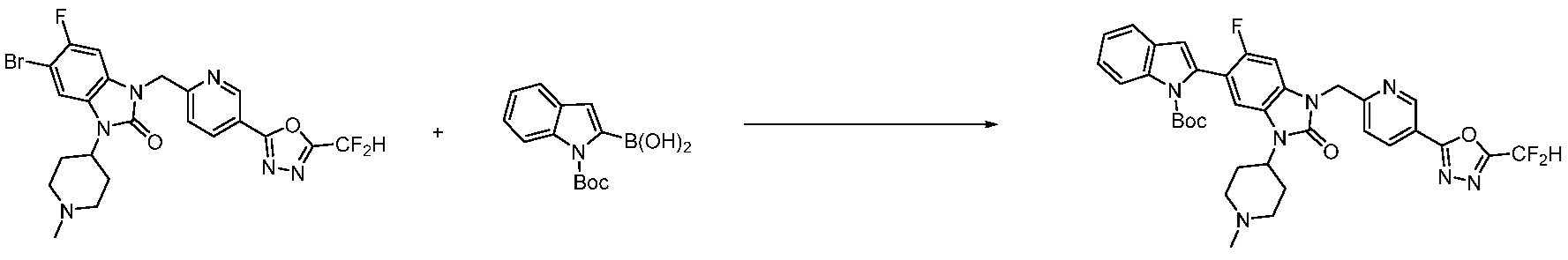
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, (1-(трет-бутоксикарбонил)-1H-индол-2-ил)бороновую кислоту (0,087 г, 0,335 ммоль), карбонат цезия (0,182 г, 0,558 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,139 г, 73,8%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (д, J=2,0 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 8,13 (д, J=8,3 Гц, 1H), 7,52 (д, J=7,6 Гц, 1H), 7,43 (д, J=8,2 Гц, 1H), 7,34 (д, J=5,8 Гц, 1H), 7,30 (тд, J=7,8, 1,1 Гц, 1H), 7,21 (т, J=7,5 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,82 (д, J=9,2 Гц, 1H), 6,56 (с, 1H), 5,25 (с, 2H), 4,44-4,36 (м, 1H), 3,00-2,98 (м, 2H), 2,53-2,43 (м, 2H), 2,30 (с, 3H), 2,17-2,11 (м, 2H), 1,87-1,84 (м, 2H), 1,37 (с, 9H); МСНР (ЭР) m/z 674,19 (M+ + 1).
Пример 254: Синтез соединения 254, трет-бутил 2-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-1H-индол-1-карбоксилат
[Стадия 1] Синтез соединения 254
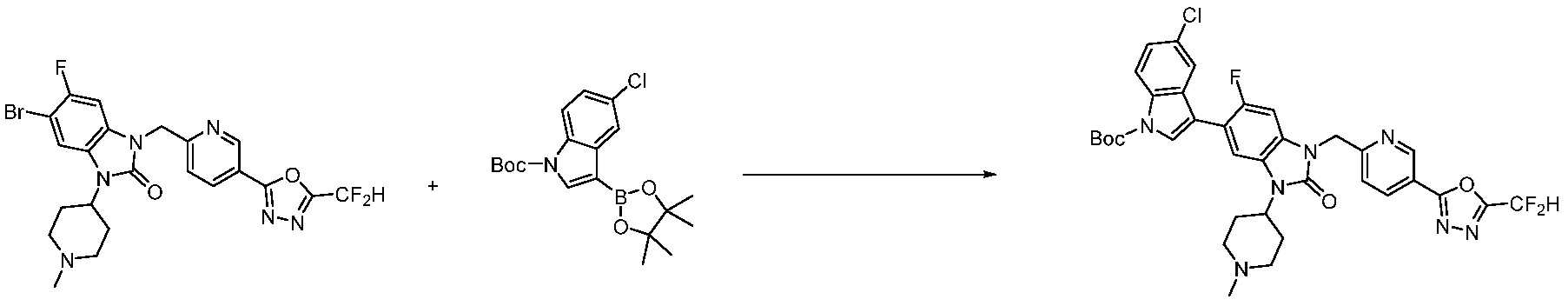
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, трет-бутил 5-хлор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилат (0,127 г, 0,335 ммоль), карбонат цезия (0,182 г, 0,558 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,134 г, 68,0%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=1,6 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 8,10 (д, J=8,8 Гц, 1H), 7,66 (с, 1H), 7,46-7,42 (м, 2H), 7,33 (д, J=5,8 Гц, 1H), 7,26 (дд, J=8,8, 2,1 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,90 (д, J=9,3 Гц, 1H), 5,27 (с, 2H), 4,47-4,38 (м, 1H), 3,02-2,99 (м, 2H), 2,52-2,43 (м, 2H), 2,31 (с, 3H), 2,19-2,13 (м, 2H), 1,88-1,86 (м, 2H), 1,67 (с, 9H); МСНР (ЭР) m/z 708,40 (M+ + 1).
Пример 255: Синтез соединения 255, трет-бутил 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)-1H-индазол-1-карбоксилат
[Стадия 1] Синтез соединения 255
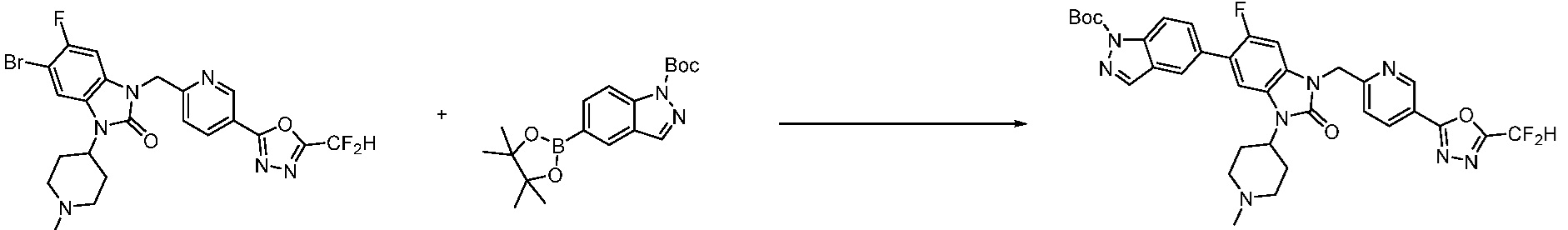
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, трет-бутил 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индазол-1-карбоксилат (0,115 г, 0,335 ммоль), карбонат цезия (0,182 г, 0,558 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,067 г, 35,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (с, 1H), 8,34 (дд, J=8,2, 1,7 Гц, 1H), 8,20-8,18 (м, 2H), 7,81 (с, 1H), 7,65 (д, J=8,7 Гц, 1H), 7,46 (д, J=8,2 Гц, 1H), 7,34 (д, J=6,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,88 (д, J=9,8 Гц, 1H), 5,27 (с, 2H), 4,47-4,41 (м, 1H), 3,02 (д, J=11,2 Гц, 2H), 2,53-2,45 (м, 2H), 2,32 (с, 3H), 2,17 (т, J=11,4 Гц, 2H), 1,87 (д, J=10,3 Гц, 2H), 1,72 (с, 9H); МСНР (ЭР) m/z 675,04 (M+ + 1).
Пример 256: Синтез соединения 256, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индазол-5-ил)-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 256
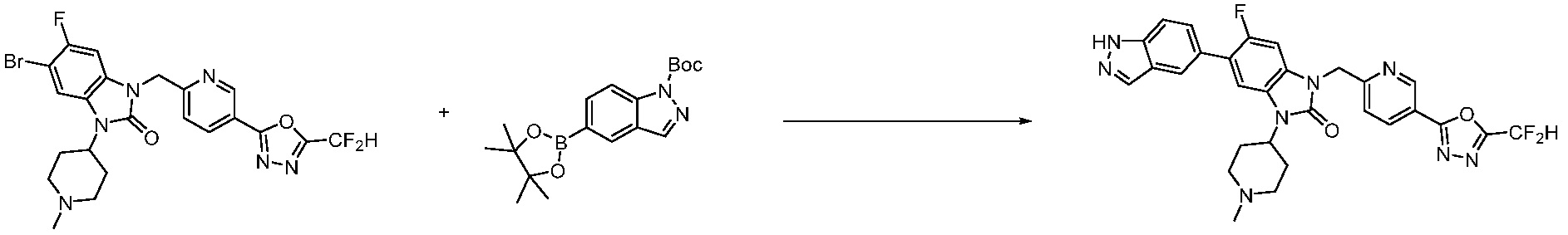
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 3 соединения 149, трет-бутил 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индазол-1-карбоксилат (0,115 г, 0,335 ммоль), карбонат цезия (0,182 г, 0,558 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,036 г, 22,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 12,37 (с, 1H), 9,29 (д, J=1,4 Гц, 1H), 8,35 (дд, J=8,2, 2,0 Гц, 1H), 7,77 (с, 1H), 7,49-7,41 (м, 5H), 6,93 (т, J=51,6 Гц, 1H), 6,84 (д, J=9,8 Гц, 1H), 5,28 (с, 2H), 4,61-4,55 (м, 1H), 3,17 (д, J=10,9 Гц, 2H), 2,81-2,72 (м, 2H), 2,42 (с, 3H), 2,31 (т, J=12,0 Гц, 2H), 1,95 (д, J=10,8 Гц, 2H); МСНР (ЭР) m/z 575,02 (M+ + 1).
Пример 257: Синтез соединения 257, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(1-метилазетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 3-((5-бром-4-фтор-2-нитрофенил)амино)азетидин-1-карбоксилата
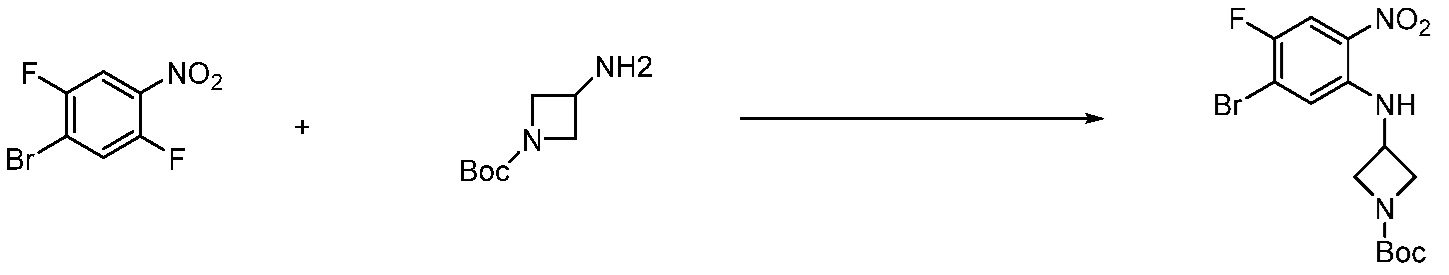
1-бром-2,5-дифтор-4-нитробензол (5,000 г, 21,009 ммоль), трет-бутил 3-аминоазетидин-1-карбоксилат (4,704 г, 27,312 ммоль), карбонат калия (5,807 г, 42,019 ммоль) и йодид калия (0,349 г, 2,101 ммоль) растворяют в N,N-диметилформамиде (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 5 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (7,450 г, 90,9%) в виде желтого твердого вещества.
[Стадия 2] Синтез трет-бутил 3-((2-амино-5-бром-4-фторфенил)амино)азетидин-1-карбоксилата
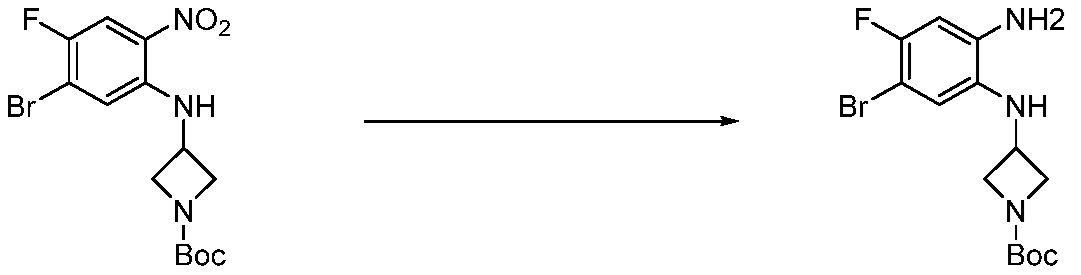
Трет-бутил 3-((5-бром-4-фтор-2-нитрофенил)амино)азетидин-1-карбоксилат (7,450 г, 19,092 ммоль), полученный на стадии 1 растворяют в метанол (100 мл) и перемешивают при комнатной температуре, после чего никель Ренея (750 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при 60° в течение 18 часов в присутствии присоединенного баллона с водородом, и затем реакцию останавливают понижением температуры до комнатной температуры. Реакционную смесь was фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (6,830 г, 99,3%, черное твердое вещество).
[Стадия 3] Синтез трет-бутил 3-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилата
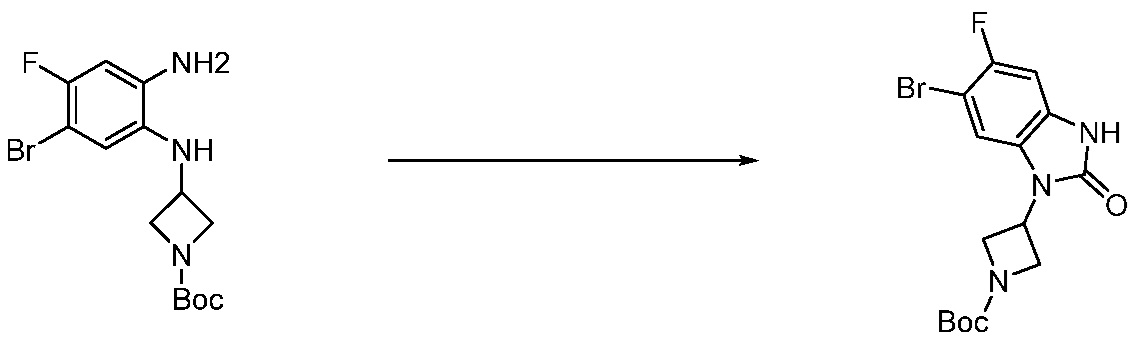
Трет-бутил 3-((2-амино-5-бром-4-фторфенил)амино)азетидин-1-карбоксилат (7,450 г, 20,681 ммоль), полученный на стадии 2, триэтиламин (2,883 мл, 20,681 ммоль) и 1,1'-карбонилдиимидазол (КДИ, 4,360 г, 26,886 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; метанол/дихлорметан=0-3%) и концентрируют с получением указанного в заголовке соединения (2,820 г, 35,3%) в виде коричневого твердого вещества.
[Стадия 4] Синтез трет-бутил 3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилата
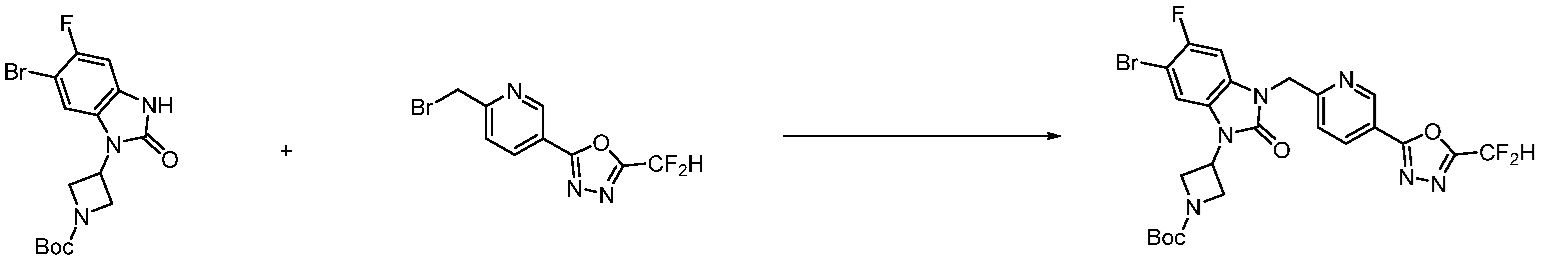
Трет-бутил 3-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилат (2,810 г, 7,276 ммоль), полученный на стадии 3 растворяют в N,N-диметилформамиде (40 мл) при 0°C, после чего гидрид натрия (60,00%, 0,436 г, 10,913 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,321 г, 8,003 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0 to 60%) и концентрируют с получением указанного в заголовке соединения (2,950 г, 68,1%) в виде коричневого твердого вещества.
[Стадия 5] Синтез 1-(азетидин-3-ил)-6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-она
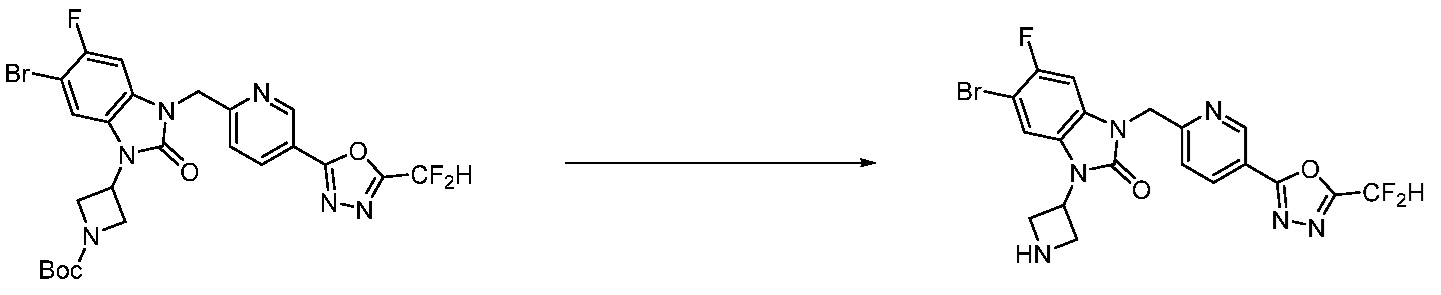
Трет-бутил 3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилат (2,900 г, 4,871 ммоль), полученный на стадии 4, и трифторуксусную кислоту (3,730 мл, 48,708 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=80-100%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-60%) и концентрируют с получением указанного в заголовке соединения (2,290 г, 94,9%) в виде коричневого твердого вещества.
[Стадия 6] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилазетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
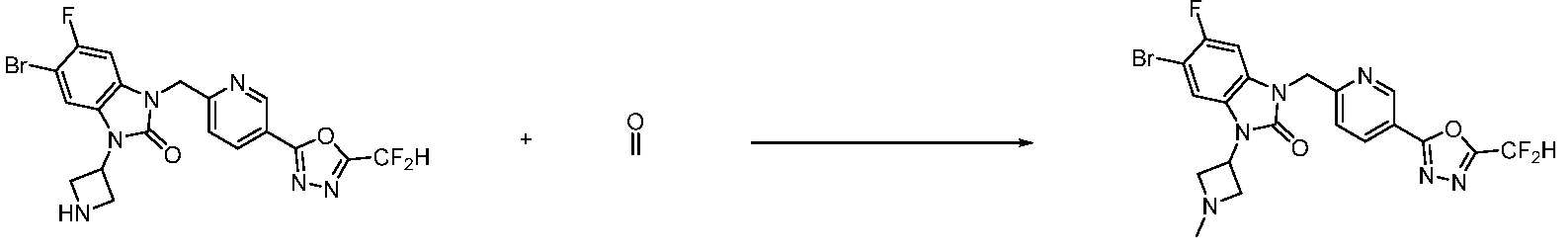
1-(азетидин-3-ил)-6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он (1,540 г, 3,109 ммоль), полученный на стадии 5, триацетоксиборгидрид натрия (1,318 г, 6,219 ммоль) и формальдегид (37,00% раствор в воде, 0,701 мл, 9,328 ммоль) растворяют в дихлорметане (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,570 г, 36,0%) в виде коричневого твердого вещества.
[Стадия 7] Синтез соединения 257
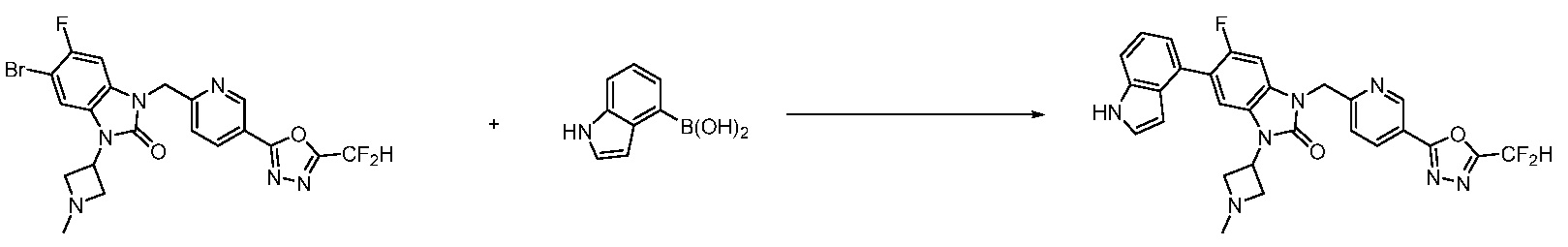
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилазетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,140 г, 0,275 ммоль), полученный на стадии 6, (1H-индол-4-ил)бороновую кислоту (0,053 г, 0,330 ммоль), карбонат цезия (0,179 г, 0,550 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,036 г, 23,7%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,7 Гц, 1H), 8,51 (с, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 7,77 (д, J=6,1 Гц, 1H), 7,50 (д, J=8,2 Гц, 1H), 7,42 (д, J=8,1 Гц, 1H), 7,29-7,24 (м, 2H), 7,18 (д, J=7,3 Гц, 1H), 6,93 (т, J=51,6 Гц, 1H), 6,91 (д, J=9,4 Гц, 1H), 6,49 (д, J=1,9 Гц, 1H), 5,28 (с, 2H), 5,07-5,00 (м, 1H), 3,84-3,78 (м, 4H), 2,43 (с, 3H); МСНР (ЭР) m/z 545,98 (M+ + 1).
Пример 258: Синтез соединения 258, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилазетидин-3-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 258
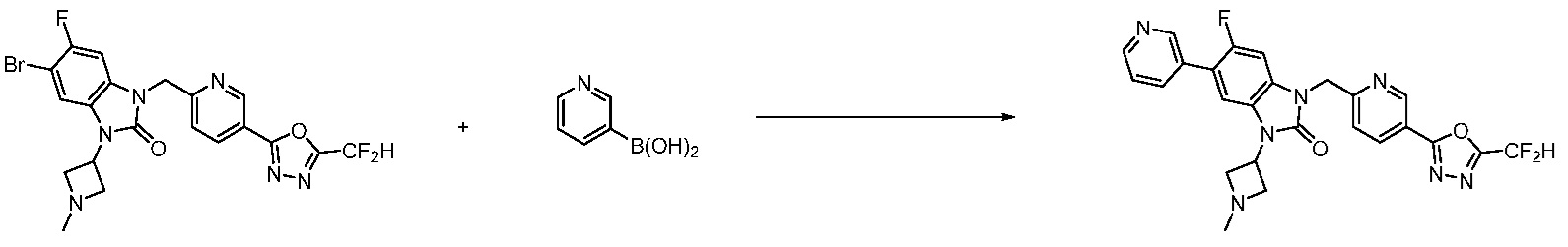
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилазетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,140 г, 0,275 ммоль), полученный на стадии 6 соединения 257, пиридин-3-илбороновую кислоту (0,041 г, 0,330 ммоль), карбонат цезия (0,179 г, 0,550 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,051 г, 36,6%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,8 Гц, 1H), 8,75 (с, 1H), 8,56 (д, J=3,8 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,83 (дд, J=7,9, 1,5 Гц, 1H), 7,75 (д, J=6,3 Гц, 1H), 7,48 (д, J=8,2 Гц, 1H), 7,35 (дд, J=7,8, 4,8 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,90 (д, J=9,8 Гц, 1H), 5,24 (с, 2H), 5,12-5,05 (м, 1H), 3,91-5,05 (м, 4H), 2,51 (с, 3H); МСНР (ЭР) m/z 508,20 (M+ + 1).
Пример 259: Синтез соединения 259, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилазетидин-3-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 259
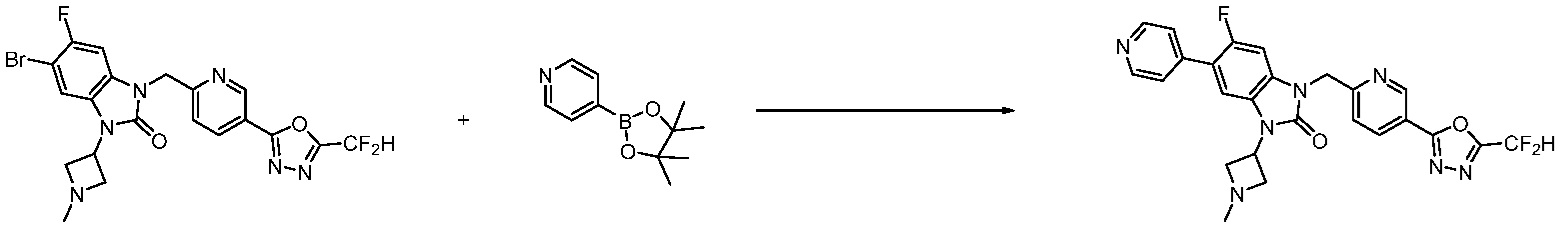
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилазетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,140 г, 0,275 ммоль), полученный на стадии 6 соединения 257, 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (0,068 г, 0,330 ммоль), карбонат цезия (0,179 г, 0,550 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,021 г, 14,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=1,7 Гц, 1H), 8,65 (д, J=5,8 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,81 (д, J=6,3 Гц, 1H), 7,49 (д, J=8,2 Гц, 1H), 7,45 (д, J=4,6 Гц, 1H), 7,05-6,79 (м, 2H), 5,25 (с, 2H), 5,14-5,07 (м, 1H), 3,94-3,87 (м, 4H), 2,55 (с, 3H); МСНР (ЭР) m/z 508,6 (M+ + 1).
Пример 260: Синтез соединения 260, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(4-метилпиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(2,5-дифтор-4-нитрофенил)пиперазин-1-карбоксилата
1,2,4-трифтор-5-нитробензол (3,660 г, 20,669 ммоль), карбонат калия (2,999 г, 21,702 ммоль) и трет-бутил пиперазин-1-карбоксилат (4,042 г, 21,702 ммоль) растворяют в N,N-диметилформамиде (80 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением указанного в заголовке соединения (5,660 г, 79,8%) в виде желтого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(2-фтор-5-(метиламино)-4-нитрофенил)пиперазин-1-карбоксилата
Трет-бутил 4-(2,5-дифтор-4-нитрофенил)пиперазин-1-карбоксилат (5,660 г, 16,486 ммоль), полученный на стадии 1, метиламин (33,00% раствор in EtOH, 1,368 мл, 19,783 ммоль), карбонат калия (4,557 г, 32,971 ммоль) и йодид калия (0,274 г, 1,649 ммоль) растворяют в N,N-диметилформамиде (80 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (3,470 г, 59,4%) в виде оранжевого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(4-амино-2-фтор-5-(метиламино)фенил)пиперазин-1-карбоксилата

Трет-бутил 4-(2-фтор-5-(метиламино)-4-нитрофенил)пиперазин-1-карбоксилат (3,460 г, 9,764 ммоль), полученный на стадии 2 растворяют в метаноле (40 мл) и перемешивают при комнатной температуре, после чего 10%-Pd/C (350 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь was фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (3,020 г, 95,3%) в виде фиолетового твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(6-фтор-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилата
Трет-бутил 4-(4-амино-2-фтор-5-(метиламино)фенил)пиперазин-1-карбоксилат (3,020 г, 9,309 ммоль), полученный на стадии 3, триэтиламин (1,298 мл, 9,309 ммоль) и 1,1'-карбонилдиимидазол (КДИ, 1,811 г, 11,171 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (3,000 г, 92,0%) в виде коричневого твердого вещества.
[Стадия 5] Синтез трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилата
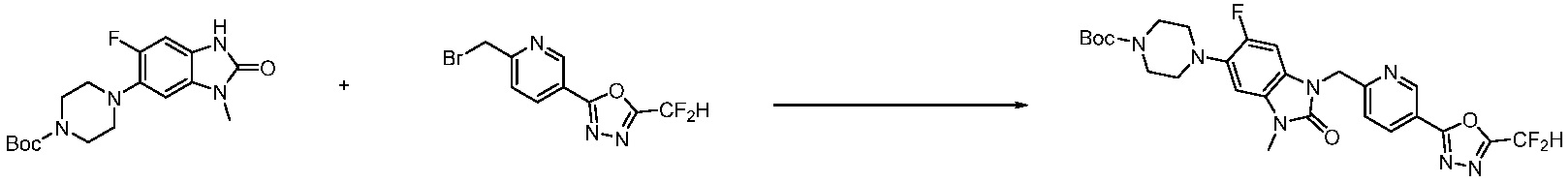
Трет-бутил 4-(6-фтор-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилат (3,000 г, 8,562 ммоль), полученный на стадии 4 растворяют в N,N-диметилформамиде (40 мл) при 0°C, после чего гидрид натрия (60,00%, 0,514 г, 12,843 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,732 г, 9,418 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=40-80%) и концентрируют с получением указанного в заголовке соединения (2,950 г, 61,6%) в виде коричневого твердого вещества.
[Стадия 6] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она

Трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилат (3,000 г, 5,361 ммоль), полученный на стадии 5, и трифторуксусную кислоту (4,106 мл, 53,615 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=50-100%) и концентрируют с получением указанного в заголовке соединения (2,150 г, 87,3%) в виде красного твердого вещества.
[Стадия 7] Синтез соединения 260
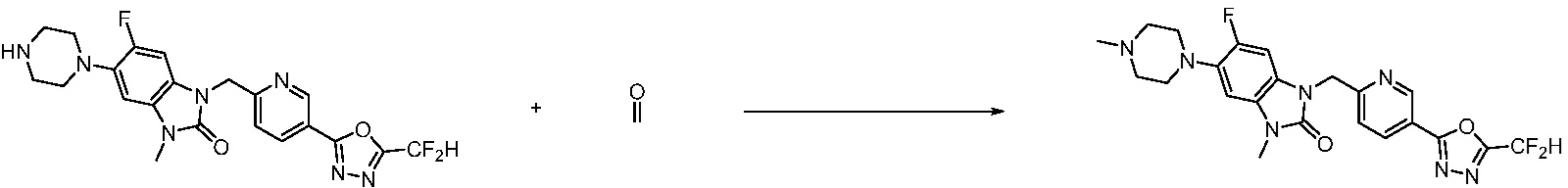
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,218 ммоль), полученный на стадии 6, триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) и формальдегид (0,020 г, 0,653 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,071 г, 68,5%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,22 (д, J=1,6 Гц, 1H), 8,27 (дд, J=8,2, 2,2 Гц, 1H), 7,37 (д, J=8,2 Гц, 1H), 6,91 (т, J=51,6 Гц, 1H), 6,70 (д, J=11,2 Гц, 1H), 6,61 (д, J=7,0 Гц, 1H), 5,18 (с, 2H), 3,41 (с, 3H), 3,03 (т, J=4,2 Гц, 4H), 2,26-2,26 (м, 4H), 2,31 (с, 3H); МСНР (ЭР) m/z 474,27 (M+ + 1).
Пример 261: Синтез соединения 261, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(4-изопропилпиперазин-1-ил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 261
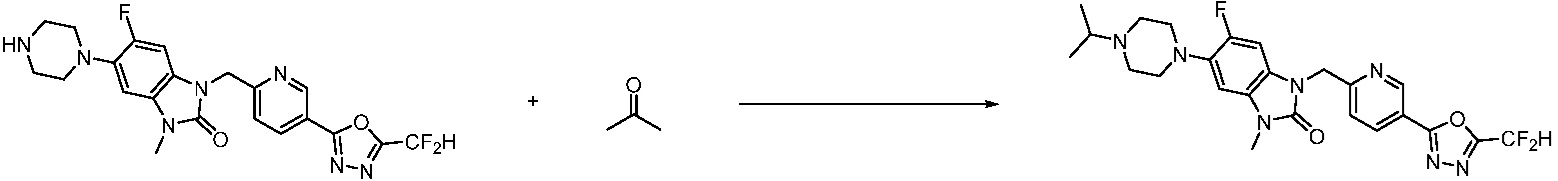
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,218 ммоль), полученный на стадии 6 соединения 260, триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) и пропан-2-он (0,038 г, 0,653 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,107 г, 97,9%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,23 (д, J=1,6 Гц, 1H), 8,28 (дд, J=8,2, 2,2 Гц, 1H), 7,38 (д, J=8,2 Гц, 1H), 6,91 (т, J=51,6 Гц, 1H), 6,71 (д, J=11,2 Гц, 1H), 6,62 (д, J=7,0 Гц, 1H), 5,18 (с, 2H), 3,41 (с, 3H), 3,05-3,03 (м, 4H), 2,74-2,67 (м, 5H), 1,06 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 502,05 (M+ + 1).
Пример 262: Синтез соединения 262, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(4-(2,2,3,3-тетрафторпропил)пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 262
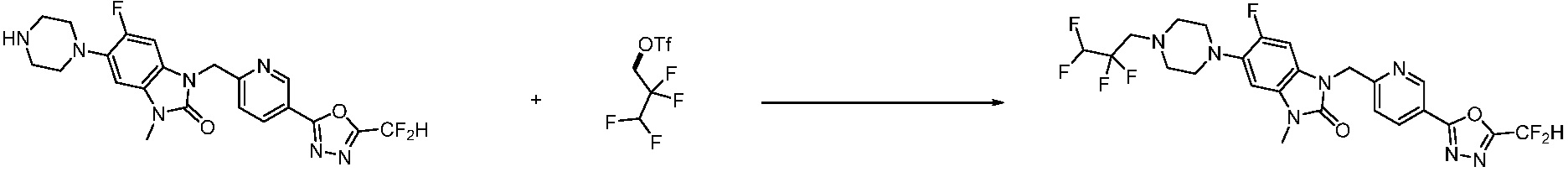
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,218 ммоль), полученный на стадии 6 соединения 260, карбонат калия (0,060 г, 0,435 ммоль) и 2,2,3,3-тетрафторпропил трифторметансульфонат (0,063 г, 0,239 ммоль) растворяют в ацетонитриле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-3%) и концентрируют с получением указанного в заголовке соединения (0,095 г, 75,8%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (д, J=1,7 Гц, 1H), 8,30 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,74 (д, J=11,2 Гц, 1H), 6,63 (д, J=6,9 Гц, 1H), 6,16-5,86 (м, 1H), 5,20 (с, 2H), 3,42 (с, 3H), 3,03 (т, J=4,6 Гц, 4H), 2,95 (т, J=14,1 Гц, 2H), 2,80 (т, J=4,6 Гц, 4H); МСНР (ЭР) m/z 573,96 (M+ + 1).
Пример 263: Синтез соединения 263, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(4-(2,2,2-трифторэтил)пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 263
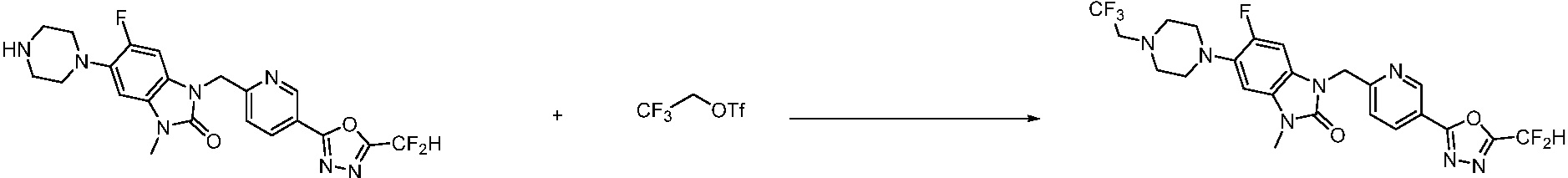
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,218 ммоль), полученный на стадии 6 соединения 260, карбонат калия (0,060 г, 0,435 ммоль) и 2,2,2-трифторэтил трифторметансульфонат (0,056 г, 0,239 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,092 г, 78,2%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (д, J=1,7 Гц, 1H), 8,29 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,73 (д, J=11,2 Гц, 1H), 6,63 (д, J=6,9 Гц, 1H), 5,19 (с, 2H), 3,42 (с, 3H), 3,06-2,98 (м, 6H), 2,84 (т, J=4,7 Гц, 4H); МСНР (ЭР) m/z 541,94 (M+ + 1).
Пример 264: Синтез соединения 264, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(4-(оксетан-3-ил)пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 264
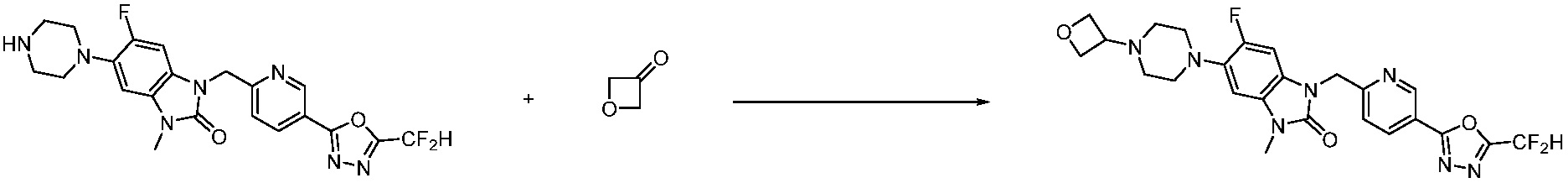
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,218 ммоль), полученный на стадии 6 соединения 260, триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) и оксетан-3-он (0,047 г, 0,653 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,055 г, 48,7%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (с, 1H), 8,29 (дд, J=8,2, 2,0 Гц, 1H), 7,39 (д, J=8,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,73 (д, J=11,2 Гц, 1H), 6,63 (д, J=6,9 Гц, 1H), 5,19 (с, 2H), 4,64 (тд, J=12,4, 6,1 Гц, 4H), 3,59-3,52 (м, 1H), 3,42 (с, 3H), 3,10-3,00 (м, 4H), 2,60-2,45 (м, 4H); МСНР (ЭР) m/z 516,08 (M+ + 1).
Пример 265: Синтез соединения 265, 6-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 265
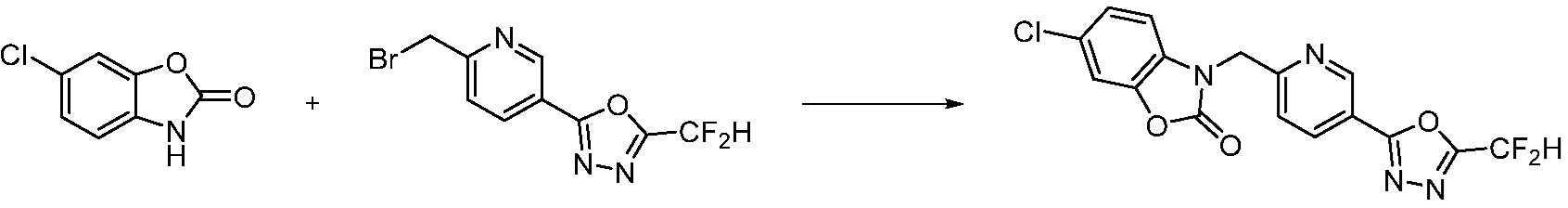
6-хлорбензо[d]оксазол-2(3H)-он (0,113 г, 0,666 ммоль), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,213 г, 0,733 ммоль), карбонат калия (0,138 г, 1,000 ммоль) и йодид калия (0,011 г, 0,067 ммоль) растворяют в N,N-диметилформамиде (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего дихлорметан добавляют в полученный концентрат и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают с дихлорметаном, и затем сушат с получением указанного в заголовке соединения (0,124 г, 49,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,13 (д, J=2,1 Гц, 1H), 8,46 (дд, J=8,2, 2,2 Гц, 1H), 7,73 (д, J=8,2 Гц, 1H), 7,70-7,44 (м, 2H), 7,27 (дд, J=8,3, 1,8 Гц, 1H), 7,21 (д, J=8,4 Гц, 1H), 5,33 (с, 2H); МСНР (ЭР) m/z 379,3 (M+ + 1).
Пример 266: Синтез соединения 266, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(4-(2-гидроксиацетил)пиперазин-1-ил)-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 266
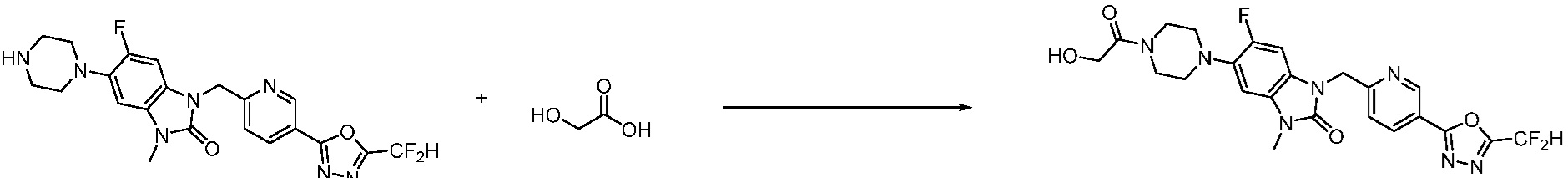
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,218 ммоль), полученный на стадии 6 соединения 260, 2-гидроксиуксусную кислоту (0,018 г, 0,239 ммоль), 1-этил-3-(3-диметиламинопропил) карбодиимид (ЭДК, 0,068 г, 0,435 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt, 0,059 г, 0,435 ммоль) и триэтиламин (0,061 мл, 0,435 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,061 г, 54,5%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (д, J=1,6 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,77 (д, J=11,0 Гц, 1H), 6,61 (д, J=6,9 Гц, 1H), 5,20 (с, 2H), 4,18 (с, 2H), 3,81 (т, J=4,9 Гц, 2H), 3,62 (с, 1H), 3,45-3,35 (м, 5H), 3,05-2,95 (м, 4H); МСНР (ЭР) m/z 518,00 (M+ + 1).
Пример 267: Синтез соединения 267, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(2-морфолиноэтил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез 5-бром-4-фтор-N-(2-морфолиноэтил)-2-нитроанилина
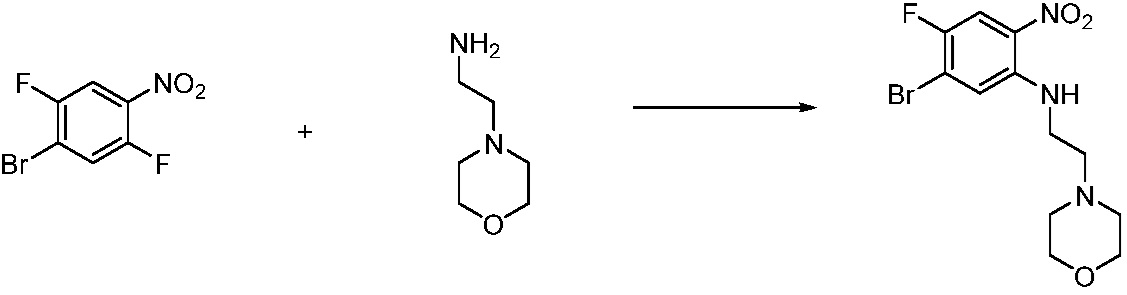
1-бром-2,5-дифтор-4-нитробензол (2,000 г, 8,404 ммоль), 2-морфолиноэтан-1-амин (1,313 г, 10,084 ммоль) и N,N-диизопропилэтиламин (2,196 мл, 12,606 ммоль) растворяют в тетрагидрофуране (5 мл) при 65°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего этилацетат (20 мл) и гексан (10 мл) добавляют в полученный концентрат и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают гексан, и затем сушат с получением указанного в заголовке соединения (2,500 г, 85,4%) в виде желтого твердого вещества.
[Стадия 2] Синтез 5-бром-4-фтор-N1-(2-морфолиноэтил)бензол-1,2-диамина
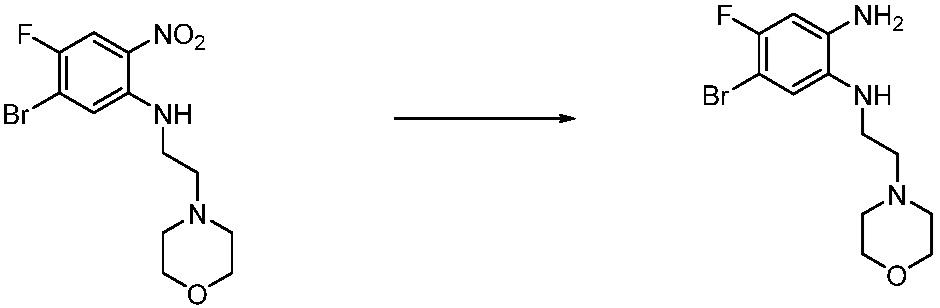
5-бром-4-фтор-N-(2-морфолиноэтил)-2-нитроанилин (2,500 г, 7,180 ммоль), полученный на стадии 1 растворяют в этанол (50 мл) при комнатной температуре, после чего никель Ренея медленно добавляют туда и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (1,800 г, 78,8%, черное твердое вещество).
[Стадия 3] Синтез 6-бром-5-фтор-1-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
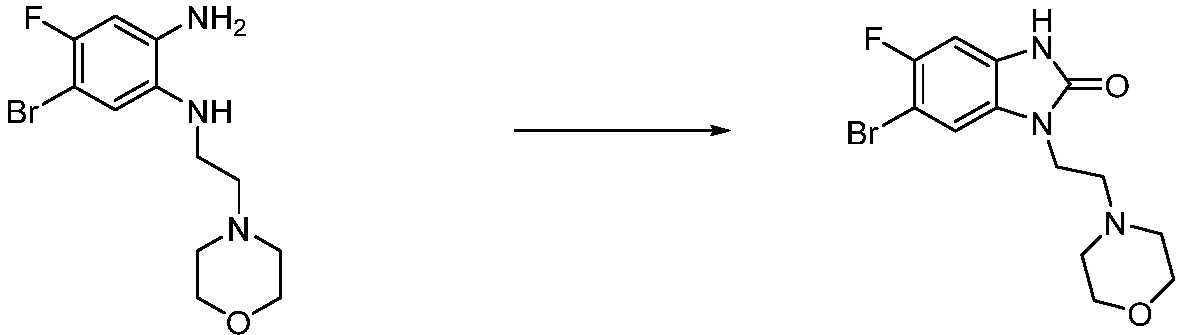
5-бром-4-фтор-N1-(2-морфолиноэтил)бензол-1,2-диамин (1,800 г, 5,657 ммоль), полученный на стадии 2 и 1,1'-карбонилдиимидазол (КДИ, 1,376 г, 8,485 ммоль) растворяют в тетрагидрофуране (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (1,230 г, 63,2%) в виде красного твердого вещества.
[Стадия 4] Синтез метил 6-((5-бром-6-фтор-3-(2-морфолиноэтил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)никотинат
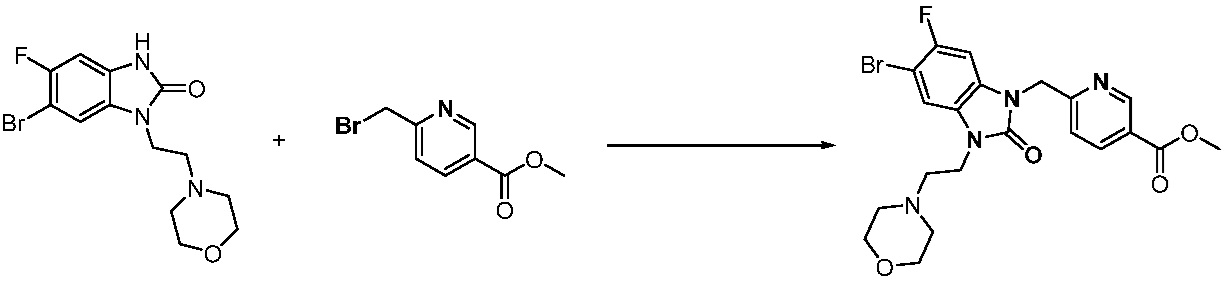
6-бром-5-фтор-1-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (2,000 г, 5,811 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (30 мл) при 0°C, после чего гидрид натрия (60,00%, 0,349 г, 8,716 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. Метил 6-(бромметил)никотинат (1,337 г, 5,811 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (1,160 г, 40,5%) в виде бесцветного масла.
[Стадия 5] Синтез 6-((5-бром-6-фтор-3-(2-морфолиноэтил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)никотиногидразида
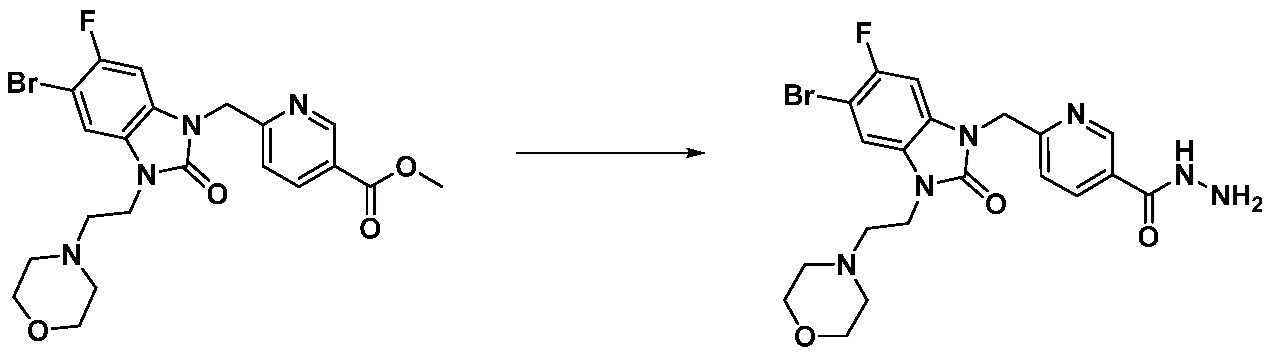
метил 6-((5-бром-6-фтор-3-(2-морфолиноэтил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)никотинат (1,160 г, 2,351 ммоль), полученный на стадии 4 и моногидрат гидразина (2,286 мл, 47,027 ммоль) растворяют в этаноле (10 мл) при 80°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,652 г, 56,2%) в виде белого твердого вещества.
[Стадия 6] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
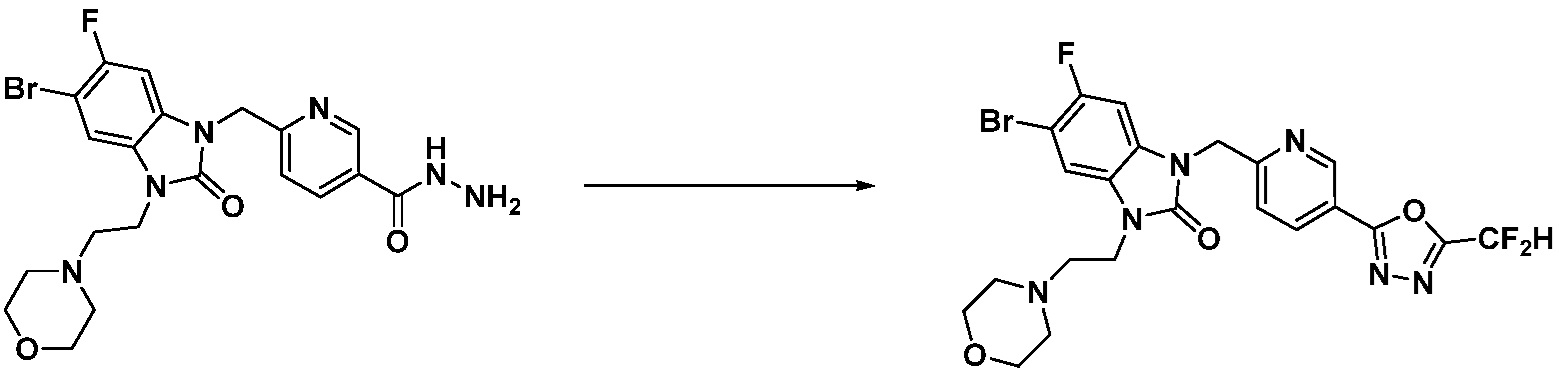
6-((5-бром-6-фтор-3-(2-морфолиноэтил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)никотиногидразид (0,652 г, 1,322 ммоль), полученный на стадии 5, 2,2-дифторуксусный ангидрид (0,493 мл, 3,965 ммоль) и имидазол (0,270 г, 3,965 ммоль) растворяют в дихлорметане (30 мл) при 45°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (0,600 г, 82,0%) в виде белого пенистого твердого вещества.
[Стадия 7] Синтез соединения 267
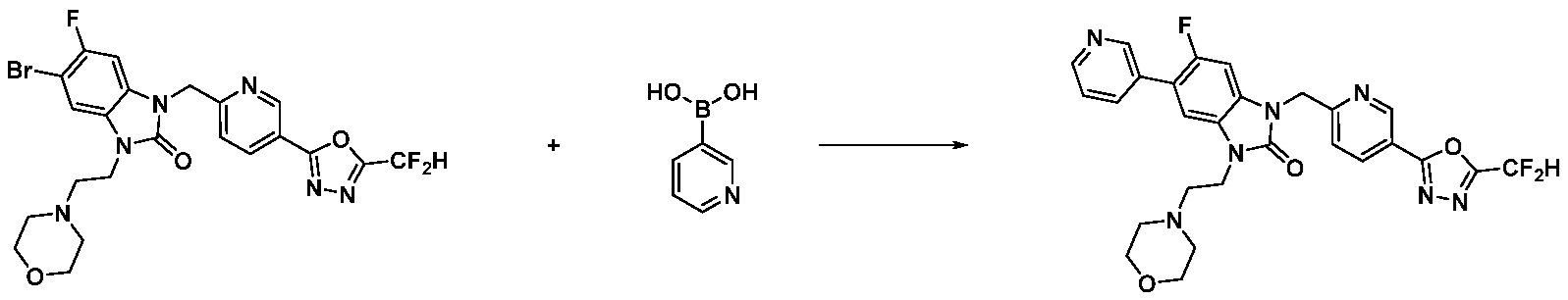
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,181 ммоль), полученный на стадии 6, пиридин-3-илбороновую кислоту (0,033 г, 0,271 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,012 г, 0,018 ммоль) и карбонат цезия (0,118 г, 0,361 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,060 г, 60,2%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,5 Гц, 1H), 8,76 (с, 1H), 8,62 ~ 8,61 (м, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,87 (дд, J=7,9, 1,9 Гц, 1H), 7,51 (д, J=8,4 Гц, 1H), 7,40 (дд, J=7,9, 4,6 Гц, 1H), 7,08 (с, 0,25H), 7,07 (д, J=6,2 Гц, 1H), 6,95 (с, 0,5H), 6,92 (д, J=9,8 Гц, 1H), 6,80 (с, 0,25H), 4,70 (с, 2H), 4,16 ~ 4,09 (м, 2H), 3,69 (т, J=4,6 Гц, 4H), 2,76 (т, J=6,7 Гц, 2H), 2,58 (т, J=4,5 Гц, 4H).
Пример 268: Синтез соединения 268, (S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпирролидин-3-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил (S)-3-((5-бром-4-фтор-2-нитрофенил)амино)пирролидин-1-карбоксилата
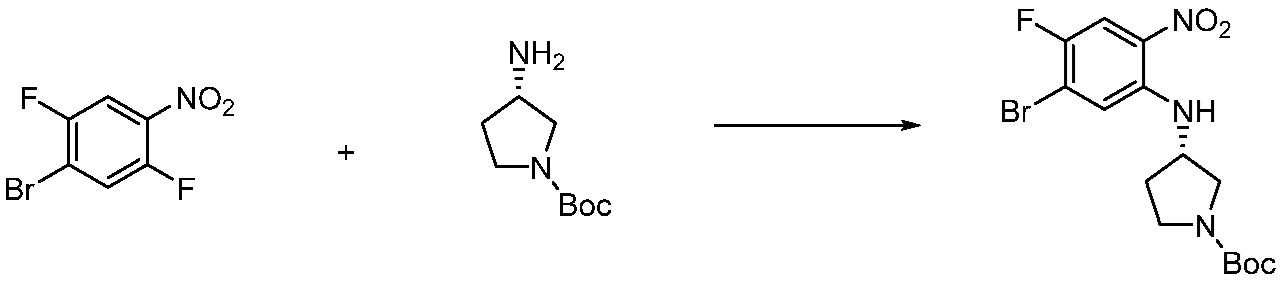
1-бром-2,5-дифтор-4-нитробензол (2,000 г, 8,404 ммоль), трет-бутил (S)-3-аминопирролидин-1-карбоксилат (1,878 г, 10,084 ммоль) и N,N-диизопропилэтиламин (2,196 мл, 12,606 ммоль) растворяют в тетрагидрофуране (5 мл) при 65°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего этилацетат (20 мл) и гексан (10 мл) добавляют в полученный концентрат и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают гексан, и затем сушат с получением указанного в заголовке соединения (1,450 г, 42,7%) в виде желтого твердого вещества.
[Стадия 2] Синтез трет-бутил (S)-3-((2-амино-5-бром-4-фторфенил)амино)пирролидин-1-карбоксилата
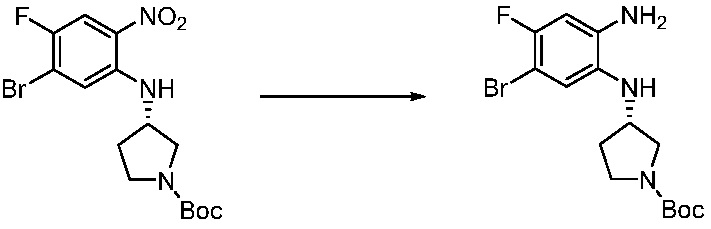
Трет-бутил (S)-3-((5-бром-4-фтор-2-нитрофенил)амино)пирролидин-1-карбоксилат (1,45 г, 3,587 ммоль), полученный на стадии 1 растворяют в этаноле (50 мл) при комнатной температуре, после чего никель Ренея медленно добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (1,23 г, 91,6%, черное твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-3-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилата
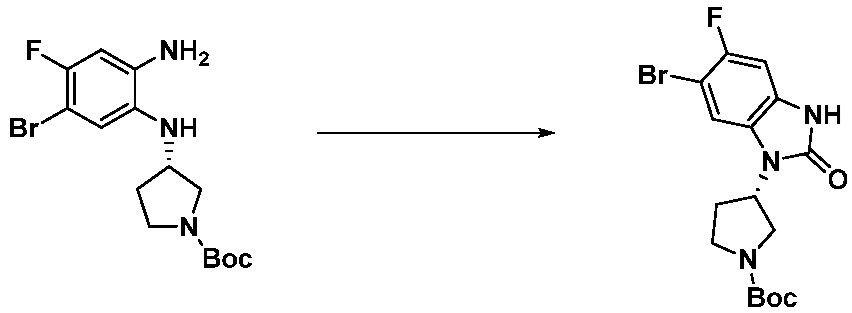
Трет-бутил (S)-3-((2-амино-5-бром-4-фторфенил)амино)пирролидин-1-карбоксилат (1,230 г, 3,287 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 0,799 г, 4,930 ммоль) растворяют в тетрагидрофуране (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,830 г, 63,1%) в виде белого твердого вещества.
[Стадия 4] Синтез метил (S)-6-((5-бром-3-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-6-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)никотината
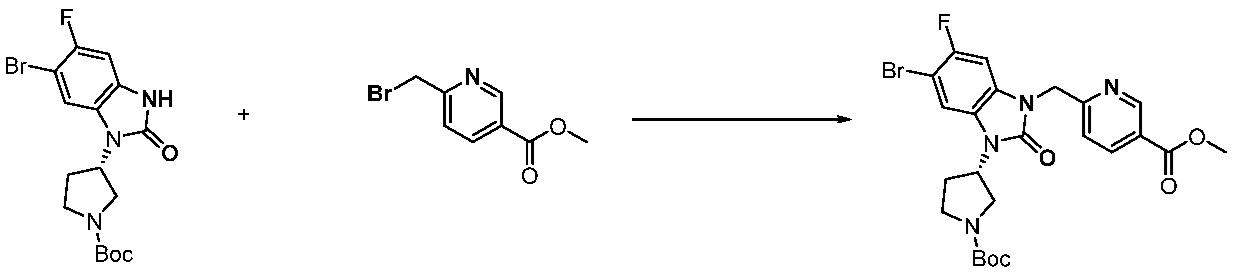
Трет-бутил (S)-3-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (0,830 г, 2,074 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (30 мл) при 0°C, после чего гидрид натрия (60,00%, 0,124 г, 3,111 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. Метил 6-(бромметил)никотинат (0,525 г, 2,281 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,634 г, 55,6%) в виде белого пенистого твердого вещества.
[Стадия 5] Синтез трет-бутил (S)-3-(6-бром-5-фтор-3-((5-(гидразинкарбонил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилата
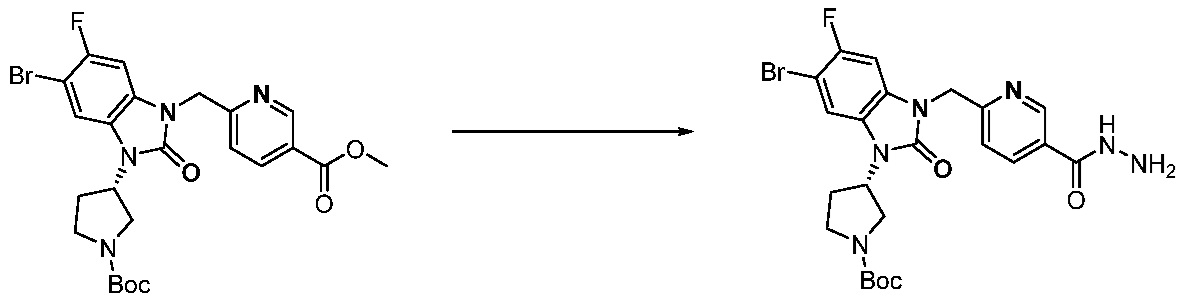
Метил (S)-6-((5-бром-3-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-6-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)никотинат (0,634 г, 1,154 ммоль), полученный на стадии 4 и моногидрат гидразина (1,122 мл, 23,080 ммоль) растворяют в этаноле (10 мл) при 80°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (0,634 г, 100,0%, белое твердое вещество).
[Стадия 6] Синтез трет-бутил (S)-3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилата

Трет-бутил (S)-3-(6-бром-5-фтор-3-((5-(гидразинкарбонил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (0,634 г, 1,154 ммоль), полученный на стадии 5, 2,2-дифторуксусный ангидрид (0,430 мл, 3,462 ммоль) и имидазол (0,236 г, 3,462 ммоль) растворяют в дихлорметане (30 мл) при 45°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением указанного в заголовке соединения (0,500 г, 71,1%) в виде бесцветного масла.
[Стадия 7] Синтез (S)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
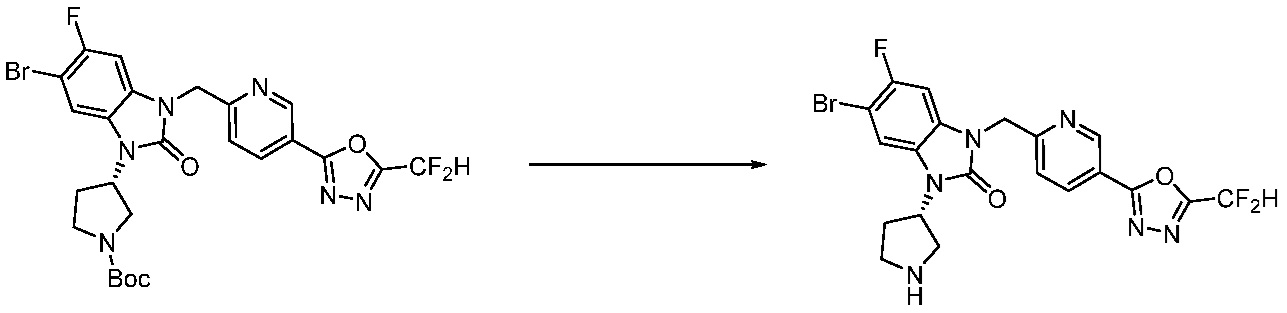
Трет-бутил (S)-3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (0,500 г, 0,820 ммоль), полученный на стадии 6, и трифторуксусную кислоту (0,628 мл, 8,205 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,349 г, 83,5%, желтое масло).
[Стадия 8] Синтез (S)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
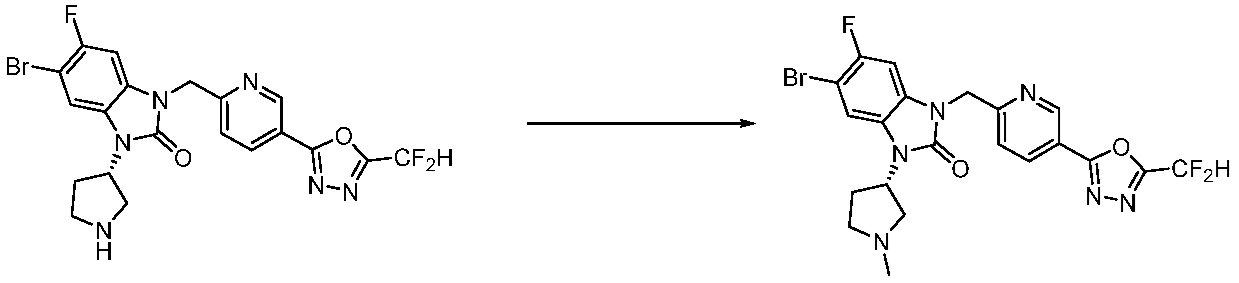
(S)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,349 г, 0,685 ммоль), полученный на стадии 7, формальдегид (0,041 г, 1,371 ммоль) и триацетоксиборгидрид натрия (0,290 г, 1,371 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,300 г, 83,7%) в виде белого пенистого твердого вещества.
[Стадия 9] Синтез соединения 268
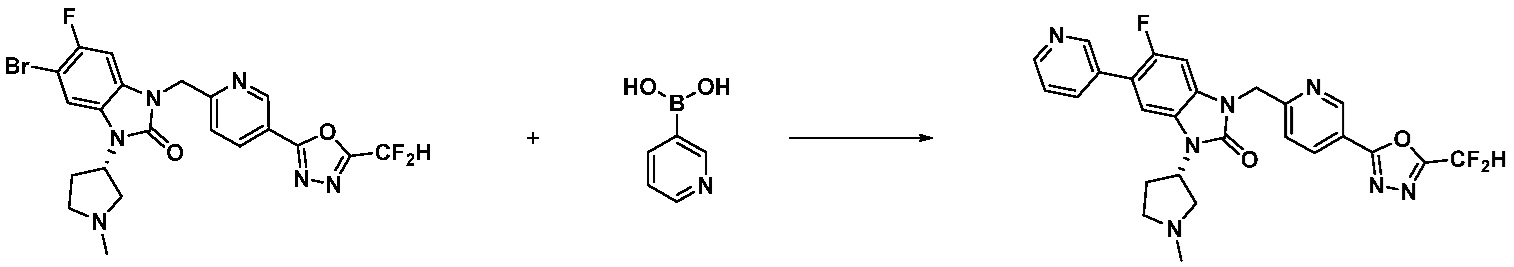
(S)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,191 ммоль), пиридин-3-илбороновую кислоту (0,035 г, 0,287 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,012 г, 0,019 ммоль) и карбонат цезия (0,125 г, 0,382 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,010 г, 10,0%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,6 Гц, 1H), 8,78 (с, 1H), 8,61 (д, J=3,5 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,97 (д, J=6,3 Гц, 1H), 7,88 (дд, J=7,8, 1,6 Гц, 1H), 7,50 (д, J=8,2 Гц, 1H), 7,41 ~ 7,38 (м, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,91 (д, J=10,0 Гц, 1H), 6,82 (с, 0,25H), 5,25 (с, 2H), 3,17 ~ 3,15 (м, 2H), 1,70 ~ 1,60 (м, 1H), 2,45 ~ 2,38 (м, 5H), 2,35 ~ 2,20 (м, 2H).
Пример 269: Синтез соединения 269, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 269
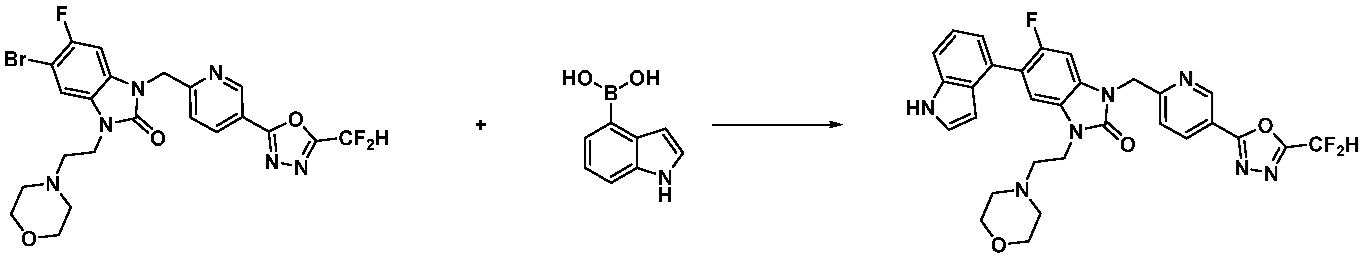
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(2-морфолиноэтил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 0,361 ммоль), (1H-индол-4-ил)бороновую кислоту (0,087 г, 0,542 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,024 г, 0,036 ммоль) и карбонат цезия (0,236 г, 0,723 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 46,9%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,35 (д, J=1,6 Гц, 1H), 8,41 ~ 8,38 (м, 2H), 7,52 (д, J=8,2 Гц, 1H), 7,45 (д, J=8,1 Гц, 1H), 7,31 ~ 7,26 (м, 3H), 7,22 (д, J=5,9 Гц, 1H), 7,17 (д, J=7,3 Гц, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,91 (д, J=9,4 Гц, 1H), 6,82 (с, 0,25H), 6,48 (д, J=2,1 Гц, 1H), 5,32 (с, 2H), 4,17 ~ 4,08 (м, 2H), 3,70 (т, J=4,5 Гц, 4H), 2,76 (т, J=6,7 Гц, 2H), 2,60 ~ 2,58 (м, 4H).
Пример 270: Синтез соединения 270, (S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(1-метилпирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 270
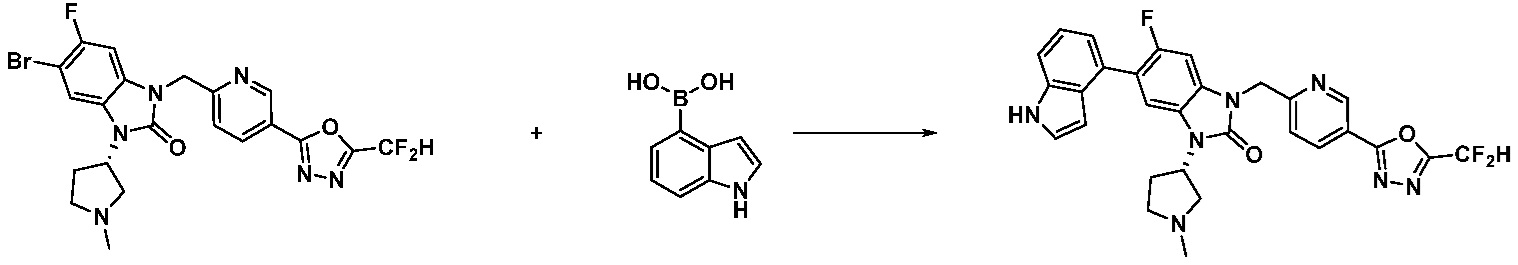
(S)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,191 ммоль), (1H-индол-4-ил)бороновую кислоту (0,046 г, 0,287 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,012 г, 0,019 ммоль) и карбонат цезия (0,125 г, 0,382 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,030 г, 28,1%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,34 ~ 9,34 (м, 1H), 8,67 (с, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,99 (д, J=6,2 Гц, 1H), 7,52 (д, J=8,0 Гц, 1H), 7,46 (д, J=8,1 Гц, 1H), 7,30 ~ 7,26 (м, 2H), 7,21 (д, J=7,3 Гц, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,91 (д, J=9,6 Гц, 1H), 6,82 (с, 0,25H), 6,53 ~ 6,52 (м, 1H), 5,31 (с, 2H), 3,10 ~ 2,95 (м, 2H), 1,85 ~ 1,70 (м, 1H), 2,50 ~ 2,40 (м, 6H), 2,15 ~ 2,10 (м, 1H); МСНР (ЭР) m/z 560,6 (M+ + 1).
Пример 271: Синтез соединения 271, 5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1-(оксетан-3-ил)пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 271
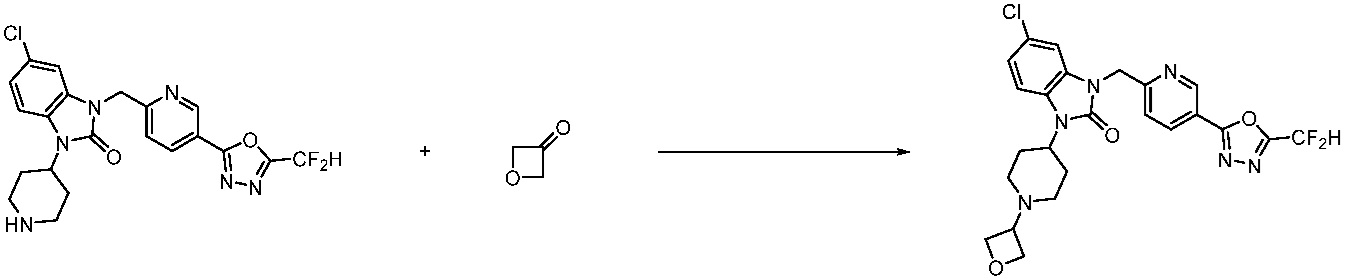
5-хлор-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,217 ммоль), полученный на стадии 5 соединения 213, оксетан-3-он (0,047 г, 0,651 ммоль) и триацетоксиборгидрид натрия (0,092 г, 0,434 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,071 г, 63,3%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=1,6 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,22 (д, J=135,8 Гц, 1H), 7,23 (д, J=8,5 Гц, 1H), 7,05-6,79 (м, 3H), 5,22 (с, 2H), 4,64 (тд, J=14,4, 7,5 Гц, 4H), 4,45-4,36 (м, 1H), 3,55-3,49 (м, 1H), 2,89 (д, J=11,5 Гц, 2H), 2,49-2,38 (м, 2H), 2,04-1,98 (м, 2H), 1,85 (дд, J=12,0, 2,2 Гц, 2H); МСНР (ЭР) m/z 519,2 (M+ + 1).
Пример 272: Синтез соединения 272, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпирролидин-3-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил (R)-3-((5-бром-4-фтор-2-нитрофенил)амино)пирролидин-1-карбоксилата
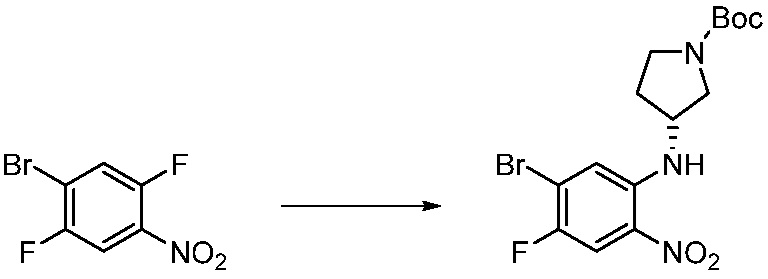
1-бром-2,5-дифтор-4-нитробензол (2,000 г, 8,404 ммоль), трет-бутил (R)-3-аминопирролидин-1-карбоксилат (1,722 г, 9,244 ммоль) и N,N-диизопропилэтиламин (2,196 мл, 12,606 ммоль) растворяют в тетрагидрофуране (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при 60° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (3,370 г, 99,2%, оранжевое твердое вещество).
[Стадия 2] Синтез трет-бутил (R)-3-((2-амино-5-бром-4-фторфенил)амино)пирролидин-1-карбоксилата
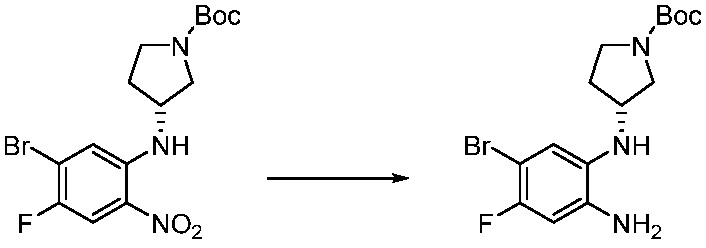
Трет-бутил (R)-3-((5-бром-4-фтор-2-нитрофенил)амино)пирролидин-1-карбоксилат (3,370 г, 8,337 ммоль), полученный на стадии 1 растворяют в метаноле (100 мл) и перемешивают при комнатной температуре, после чего никель Ренея (350 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (3,110 г, 99,7%, коричневое масло).
[Стадия 3] Синтез трет-бутил (R)-3-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилата
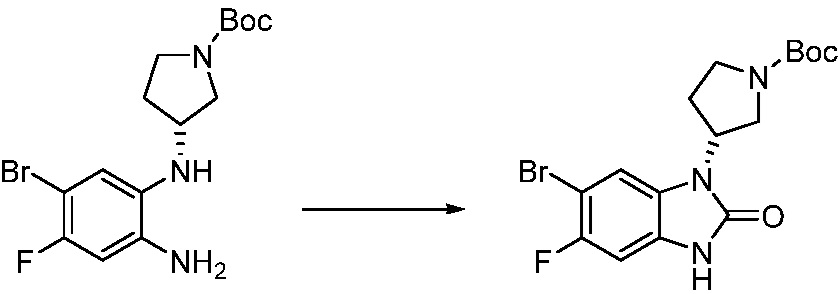
Трет-бутил (R)-3-((2-амино-5-бром-4-фторфенил)амино)пирролидин-1-карбоксилат (3,110 г, 8,310 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 1,347 г, 8,310 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. выпавшее в осадок твердое вещество фильтруют, затем промывают с дихлорметаном, и затем сушат с получением указанного в заголовке соединения (2,050 г, 61,6%) в виде фиолетового твердого вещества.
[Стадия 4] Синтез трет-бутил (R)-3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилата
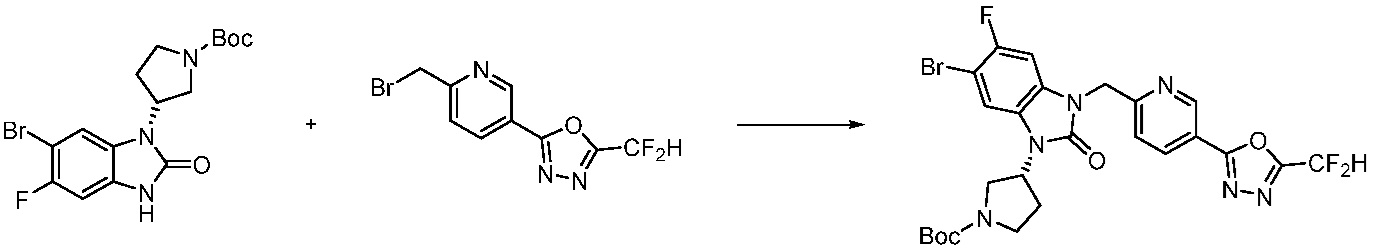
Трет-бутил (R)-3-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (2,050 г, 5,122 ммоль), полученный на стадии 3, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,634 г, 5,634 ммоль), карбонат калия (1,062 г, 7,683 ммоль) и йодид калия (0,085 г, 0,512 ммоль) растворяют в N,N-диметилформамиде (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением указанного в заголовке соединения (1,400 г, 44,9%) в виде светло-желтого твердого вещества.
[Стадия 5] Синтез (R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
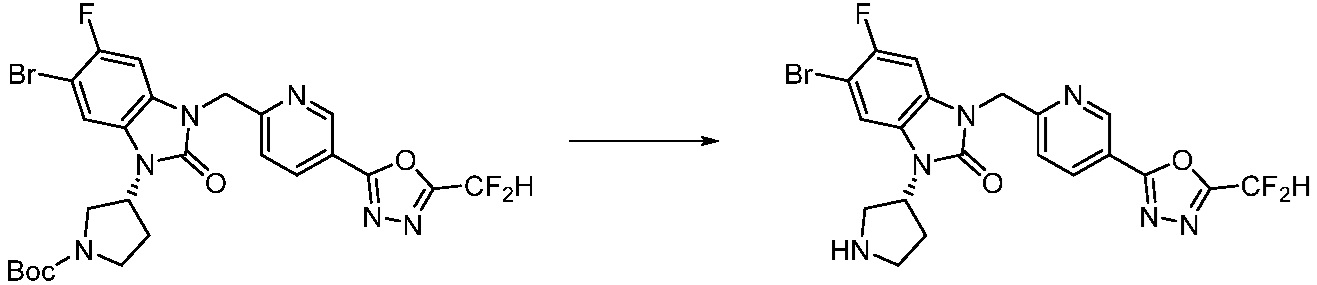
Трет-бутил (R)-3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пирролидин-1-карбоксилат (1,400 г, 2,297 ммоль), полученный на стадии 4 и трифторуксусную кислоту (1,056 мл, 13,784 ммоль) растворяют в дихлорметане (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (1,030 г, 88,0%, оранжевое твердое вещество).
[Стадия 6] Синтез (R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
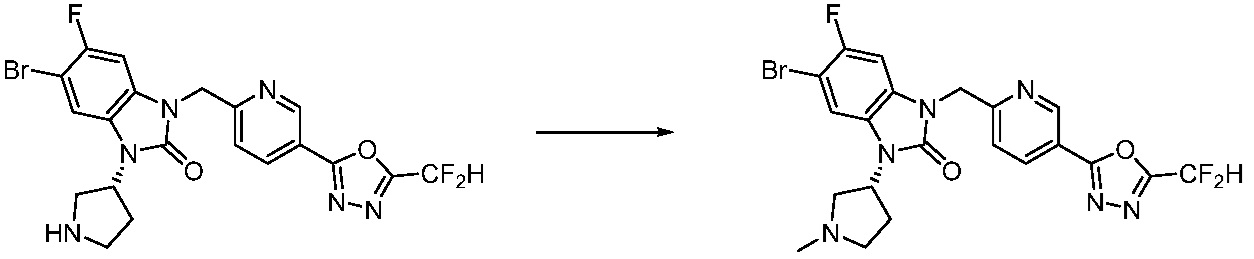
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,500 г, 0,982 ммоль), полученный на стадии 5, и формальдегид (38,00% раствор, 0,108 мл, 1,473 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,416 г, 1,964 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,440 г, 85,6%) в виде белого твердого вещества.
[Стадия 7] Синтез соединения 272
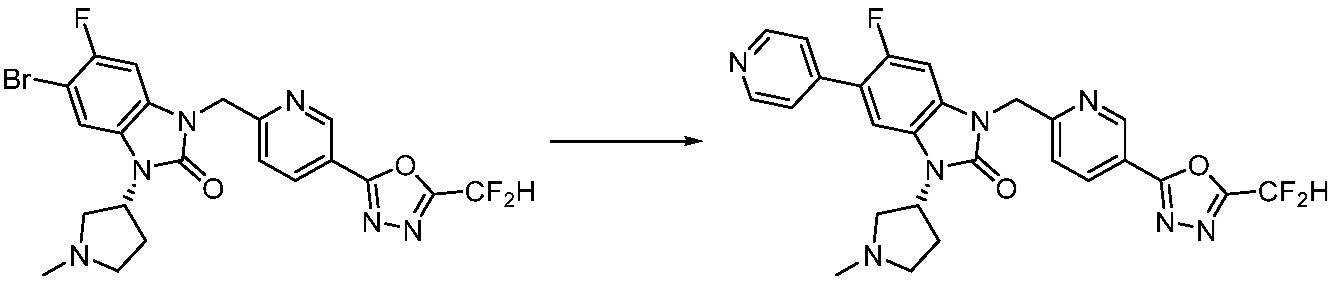
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,191 ммоль), полученный на стадии 6, пиридин-4-илбороновую кислоту (0,028 г, 0,229 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,010 ммоль) и карбонат цезия (0,187 г, 0,573 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,005 г, 5,0%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,6 Гц, 1H), 8,68 (д, J=5,8 Гц, 2H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 8,09 (д, J=6,6 Гц, 1H), 7,52-7,48 (м, 3H), 7,08-6,82 (м, 2H), 5,33-5,24 (м, 3H), 3,19-3,13 (м, 2H), 2,65 (т, J=9,6 Гц, 1H), 2,46 (с, 3H), 2,43-2,35 (м, 2H), 2,19-2,16 (м, 1H); МСНР (ЭР) m/z 522,5 (M+ + 1).
Пример 273: Синтез соединения 273, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(1-метилпирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 273
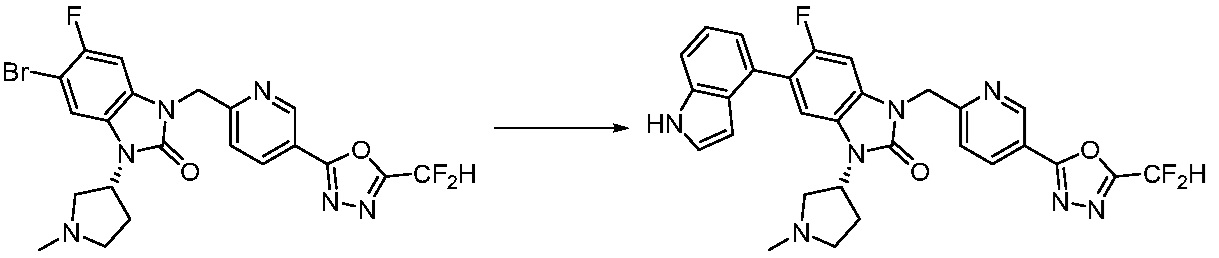
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,191 ммоль), (1H-индол-4-ил)бороновую кислоту (0,037 г, 0,229 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,010 ммоль) и карбонат цезия (0,187 г, 0,573 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,015 г, 14,0%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,34 (д, J=1,6 Гц, 1H), 8,51 (с, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 8,03 (д, J=6,3 Гц, 1H), 7,51 (д, J=8,2 Гц, 1H), 7,45 (д, J=8,0 Гц, 1H), 7,31-7,27 (м, 3H), 7,22 (д, J=7,3 Гц, 1H), 7,08-6,82 (м, 2H), 6,55 (с, 1H), 5,31-5,26 (м, 3H), 3,11 (дд, J=10,2, 4,0 Гц, 1H), 3,00-2,98 (м, 1H), 2,71 (т, J=9,5 Гц, 1H), 2,40-2,33 (м, 5H), 2,25-2,24 (м, 1H); МСНР (ЭР) m/z 560,5 (M+ + 1).
Пример 274: Синтез соединения 274, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-3-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил (R)-3-((5-бром-4-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилата
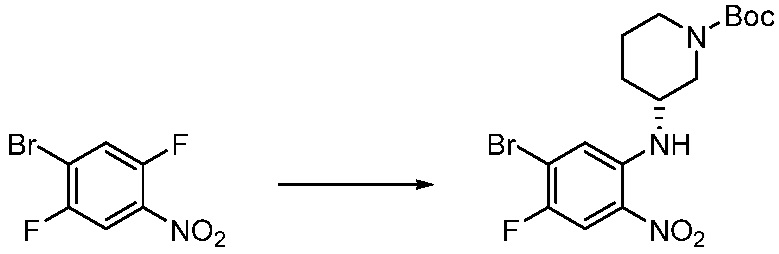
1-бром-2,5-дифтор-4-нитробензол (2,000 г, 8,404 ммоль), трет-бутил (R)-3-аминопиперидин-1-карбоксилат (1,851 г, 9,244 ммоль) и N,N-диизопропилэтиламин (2,196 мл, 12,606 ммоль) растворяют в тетрагидрофуране (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при 60° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (3,290 г, 93,6%, оранжевое твердое вещество).
[Стадия 2] Синтез трет-бутил (R)-3-((2-амино-5-бром-4-фторфенил)амино)пиперидин-1-карбоксилата
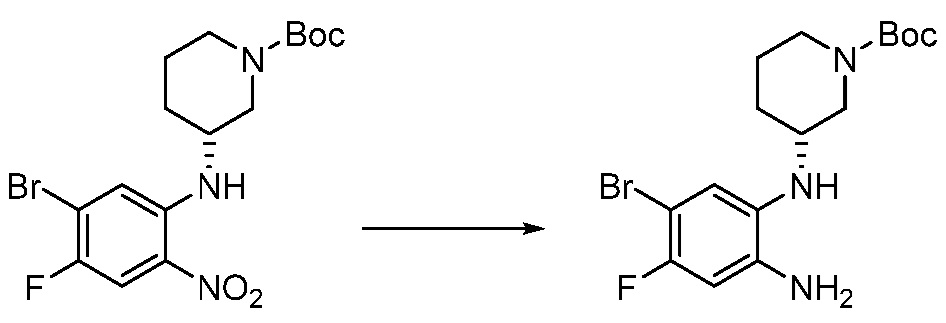
Трет-бутил (R)-3-((5-бром-4-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилат (3,290 г, 7,866 ммоль), полученный на стадии 1, растворяют в метаноле (100 мл) и перемешивают при комнатной температуре, после чего никель Ренея (350 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (2,990 г, 97,9%, коричневое масло).
[Стадия 3] Синтез трет-бутил (R)-3-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
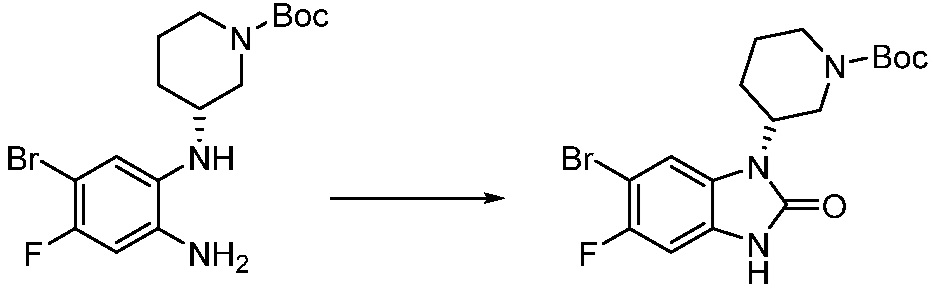
Трет-бутил (R)-3-((2-амино-5-бром-4-фторфенил)амино)пиперидин-1-карбоксилат (2,990 г, 7,701 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (1,249 г, 7,701 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=5-40%) и концентрируют с получением продукта. Затем, диэтиловый эфир добавляют в полученный продукт и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают диэтиловым эфиром, и затем сушат с получением указанного в заголовке соединения (1,440 г, 45,1%) в виде светло-коричневого твердого вещества.
[Стадия 4] Синтез трет-бутил (R)-3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
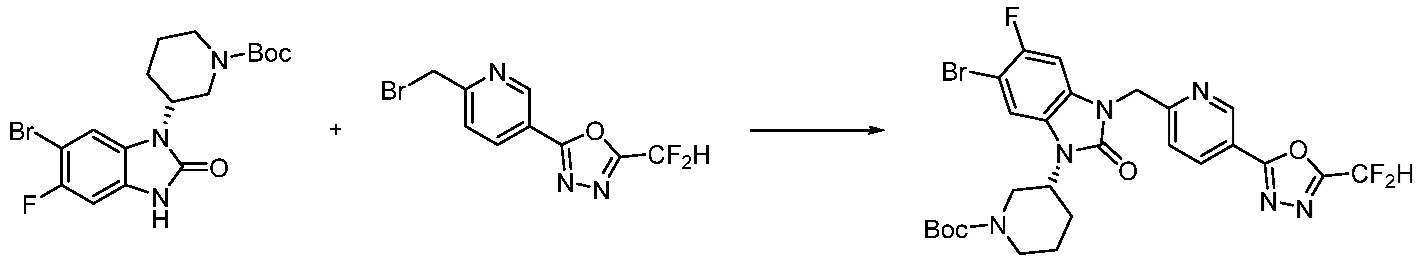
Трет-бутил (R)-3-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,440 г, 3,476 ммоль), полученный на стадии 3, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,109 г, 3,824 ммоль), карбонат калия (0,721 г, 5,214 ммоль) и йодид калия (0,058 г, 0,348 ммоль) растворяют в N,N-диметилформамиде (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением указанного в заголовке соединения (1,680 г, 77,5%) в виде светло-желтого твердого вещества.
[Стадия 5] Синтез (R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
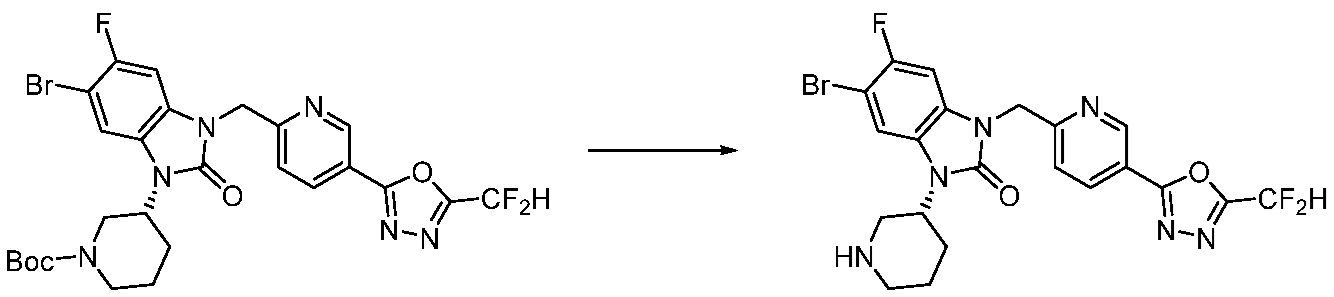
Трет-бутил (R)-3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (1,680 г, 2,695 ммоль), полученный на стадии 4, и трифторуксусную кислоту (1,238 мл, 16,169 ммоль) растворяют в дихлорметане (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (1,290 г, 91,5%, оранжевое твердое вещество).
[Стадия 6] Синтез (R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
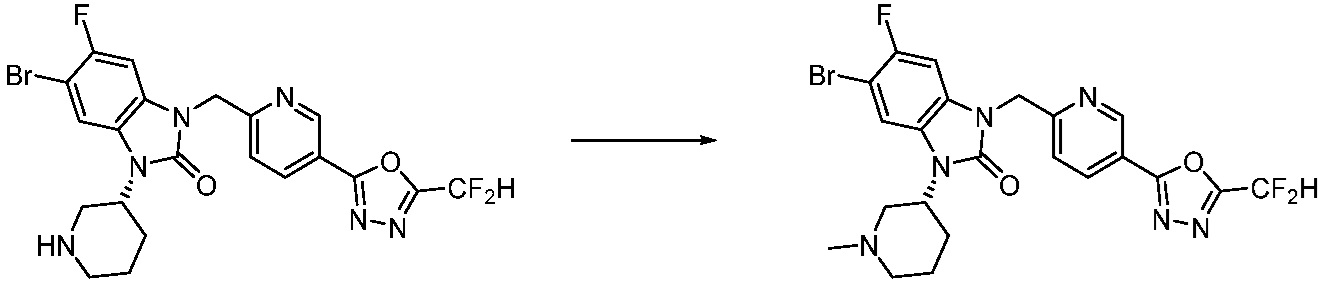
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,600 г, 1,147 ммоль), полученный на стадии 5, и триацетоксиборгидрид натрия (0,486 г, 2,293 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего формальдегид (38,00% раствор, 0,126 мл, 1,720 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,360 г, 58,4%) в виде светло-желтого твердого вещества.
[Стадия 7] Синтез соединения 274
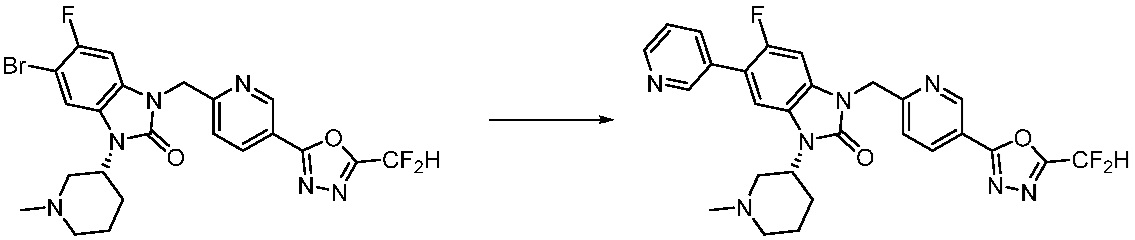
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,186 ммоль), полученный на стадии 6, пиридин-3-илбороновую кислоту (0,027 г, 0,223 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,182 г, 0,558 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,054 г, 54,2%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,7 Гц, 1H), 8,76 (с, 1H), 8,61 (дд, J=4,8, 1,4 Гц, 1H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 7,84 (дд, J=7,9, 1,8 Гц, 1H), 7,49 (д, J=8,2 Гц, 1H), 7,41-7,37 (м, 1H), 7,18 (д, J=6,2 Гц, 1H), 7,08-6,82 (м, 2H), 5,27 (с, 2H), 4,50-4,43 (м, 1H), 3,00 (дд, J=10,5, 3,9 Гц, 1H), 2,89 (д, J=11,2 Гц, 1H), 2,73 (т, J=10,9 Гц, 1H), 2,37 (с, 3H), 2,30-2,26 (м, 1H), 2,08-2,05 (м, 1H), 1,99-1,96 (м, 1H), 1,92-1,88 (м, 1H), 1,83-1,80 (м, 1H); МСНР (ЭР) m/z 536,5 (M+ + 1).
Пример 275: Синтез соединения 275, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-3-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 275
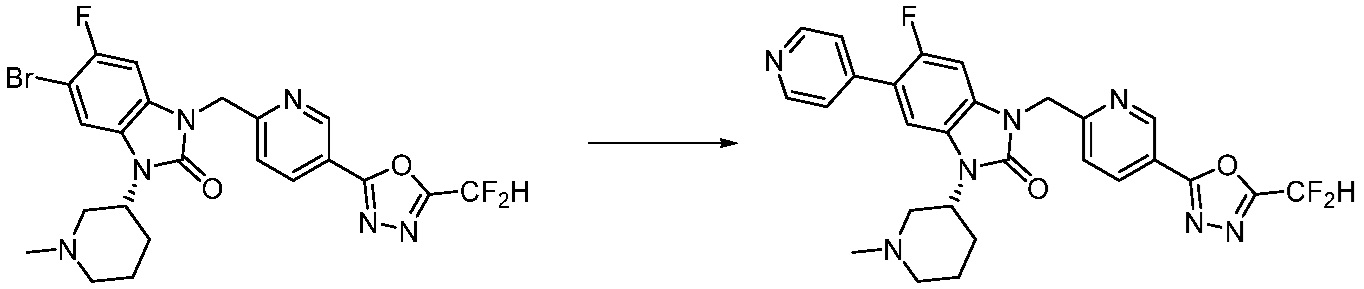
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,186 ммоль), пиридин-4-илбороновую кислоту (0,027 г, 0,223 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,182 г, 0,558 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,054 г, 54,2%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (д, J=1,8 Гц, 1H), 8,67 (д, J=6,0 Гц, 2H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 7,50-7,45 (м, 3H), 7,22 (д, J=6,1 Гц, 1H), 7,08-6,82 (м, 2H), 5,26 (с, 2H), 4,49-4,43 (м, 1H), 3,72-3,70 (м, 1H), 3,04 (д, J=10,5 Гц, 1H), 2,79 (т, J=10,9 Гц, 1H), 2,40-2,31 (м, 4H), 2,14-2,08 (м, 1H), 1,85-1,79 (м, 2H), 1,28-1,27 (м, 1H); МСНР (ЭР) m/z 536,5 (M+ + 1).
Пример 276: Синтез соединения 276, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(1-метилпиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 276
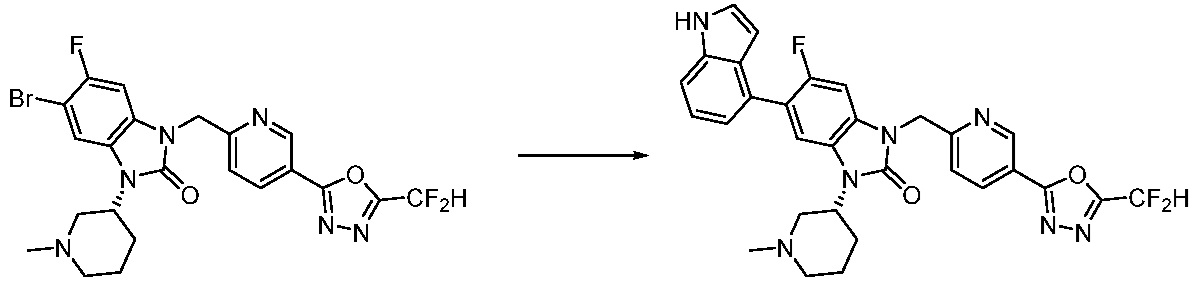
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-метилпиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,186 ммоль), (1H-индол-4-ил)бороновую кислоту (0,036 г, 0,223 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,182 г, 0,558 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,080 г, 74,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,34 (д, J=1,6 Гц, 1H), 8,51 (шс, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,51 (д, J=8,2 Гц, 1H), 7,45 (д, J=8,1 Гц, 1H), 7,36 (д, J=5,9 Гц, 1H), 7,32-7,27 (м, 2H), 7,18 (д, J=7,3 Гц, 1H), 7,08-6,82 (м, 2H), 6,49-6,48 (м, 1H), 5,32-5,26 (м, 2H), 4,55-4,48 (м, 1H), 3,03 (дд, J=10,5, 3,8 Гц, 1H), 2,88 (д, J=11,2 Гц, 1H), 2,75 (т, J=11,0 Гц, 1H), 2,36 (с, 3H), 2,29-2,20 (м, 1H), 2,07-1,97 (м, 1H), 1,89-1,81 (м, 3H); МСНР (ЭР) m/z 574,6 (M+ + 1).
Пример 277: Синтез соединения 277, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(пиридин-3-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез 6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фторбензо[d]оксазол-2(3H)-она
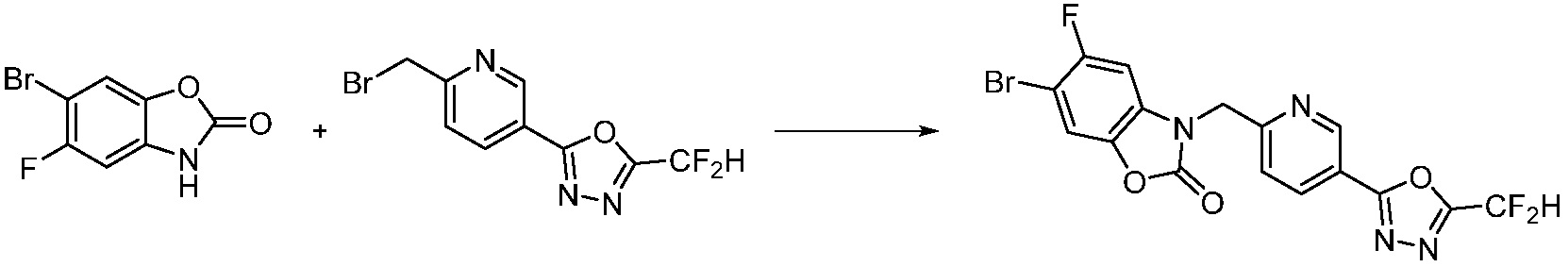
6-бром-5-фторбензо[d]оксазол-2(3H)-он (3,000 г, 12,930 ммоль), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (3,938 г, 13,577 ммоль), карбонат калия (2,681 г, 19,396 ммоль) и йодид калия (0,215 г, 1,293 ммоль) растворяют в N,N-диметилформамиде (150 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Этилацетат добавляют в полученный концентрат и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют, затем промывают этилацетат, и затем сушат с получением указанного в заголовке соединения. Полученный фильтрат дополнительно концентрируют при пониженном давлении, после чего метанол добавляют в полученный концентрат и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают метанол, и затем сушат с получением указанного в заголовке соединения (4,650 г, 81,5%) в виде оранжевого твердого вещества.
[Стадия 2] Синтез соединения 277
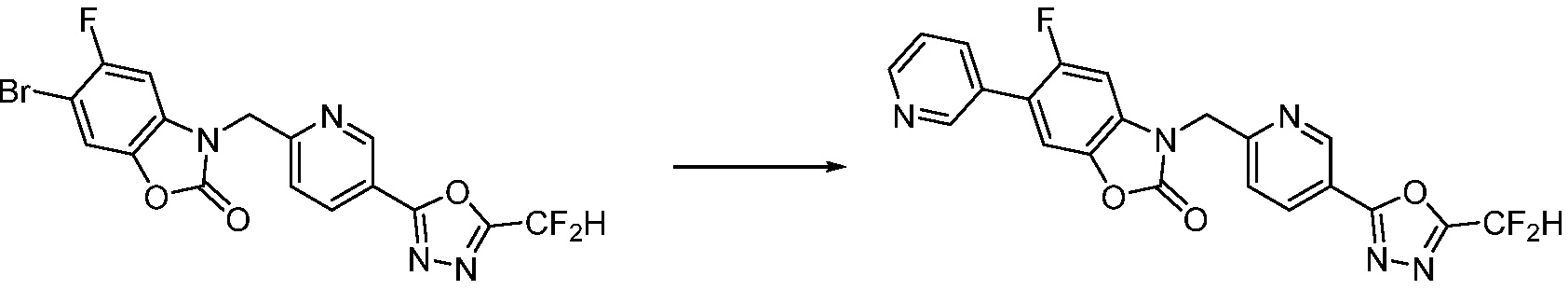
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фторбензо[d]оксазол-2(3H)-он (0,100 г, 0,227 ммоль), полученный на стадии 1, пиридин-3-илбороновую кислоту (0,033 г, 0,272 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,007 г, 0,011 ммоль) и карбонат цезия (0,222 г, 0,680 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=20-60%) и концентрируют с получением указанного в заголовке соединения (0,061 г, 61,3%) в виде оранжевого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,34 (д, J=2,1 Гц, 1H), 8,76 (с, 1H), 8,64 (дд, J=5,2, 1,2 Гц, 1H), 8,46 (дд, J=8,2, 2,2 Гц, 1H), 7,85-7,82 (м, 1H), 7,62 (д, J=7,7 Гц, 1H), 7,42-7,39 (м, 1H), 7,32 (д, J=6,0 Гц, 1H), 7,09-6,83 (м, 2H), 5,25 (с, 2H); МСНР (ЭР) m/z 440,4 (M+ + 1).
Пример 278: Синтез соединения 278, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(пиридин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 278
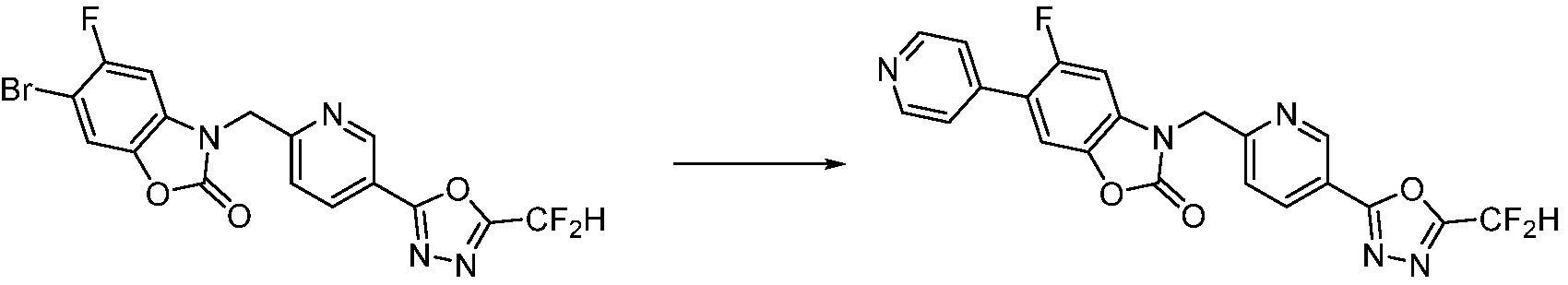
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фторбензо[d]оксазол-2(3H)-он (0,100 г, 0,227 ммоль), пиридин-4-илбороновую кислоту (0,033 г, 0,272 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,007 г, 0,011 ммоль) и карбонат цезия (0,222 г, 0,680 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением указанного в заголовке соединения (0,067 г, 67,3%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,6 Гц, 1H), 8,71-8,69 (м, 2H), 8,46 (дд, J=8,2, 2,2 Гц, 1H), 7,62 (д, J=8,2 Гц, 1H), 7,46-7,44 (м, 2H), 7,35 (д, J=5,9 Гц, 1H), 7,09-6,83 (м, 2H), 5,25 (с, 2H); МСНР (ЭР) m/z 440,4 (M+ + 1).
Пример 279: Синтез соединения 279, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1H-индол-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 279
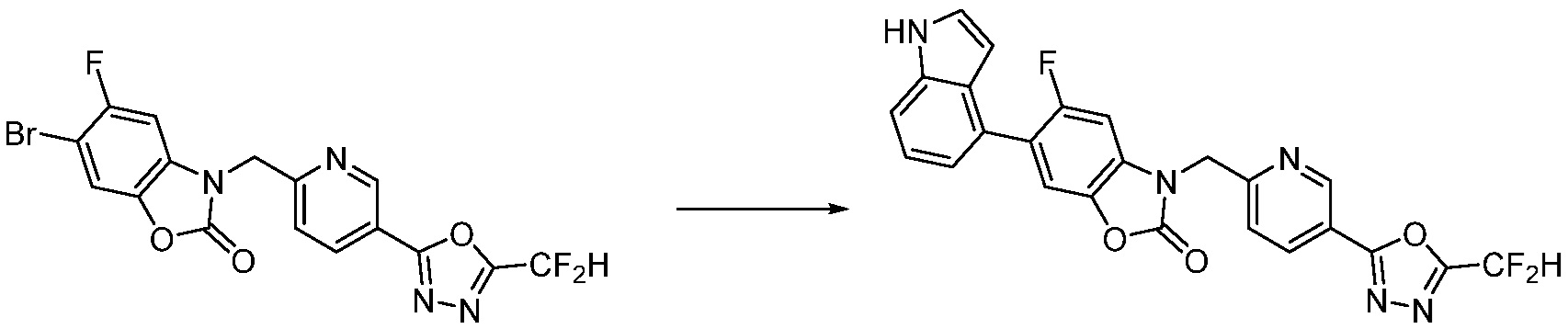
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фторбензо[d]оксазол-2(3H)-он (0,100 г, 0,227 ммоль), (1H-индол-4-ил)бороновую кислоту (0,044 г, 0,272 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,007 г, 0,011 ммоль) и карбонат цезия (0,222 г, 0,680 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением продукта. Затем, диэтиловый эфир и гексан добавляют в полученный продукт и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают гексан, и затем сушат с получением указанного в заголовке соединения (0,021 г, 19,4%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,36 (д, J=2,1 Гц, 1H), 8,46 (дд, J=8,2, 2,2 Гц, 1H), 8,32 шс, 1H), 7,62 (д, J=8,2 Гц, 1H), 7,48-7,44 (м, 2H), 7,31-7,27 (м, 2H), 7,16 (д, J=6,3 Гц, 1H), 7,09-6,83 (м, 2H), 6,48-6,47 (м, 1H), 5,27 (с, 2H); МСНР (ЭР) m/z 478,4 (M+ + 1).
Пример 280: Синтез соединения 280, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-(оксетан-3-ил)пиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 280
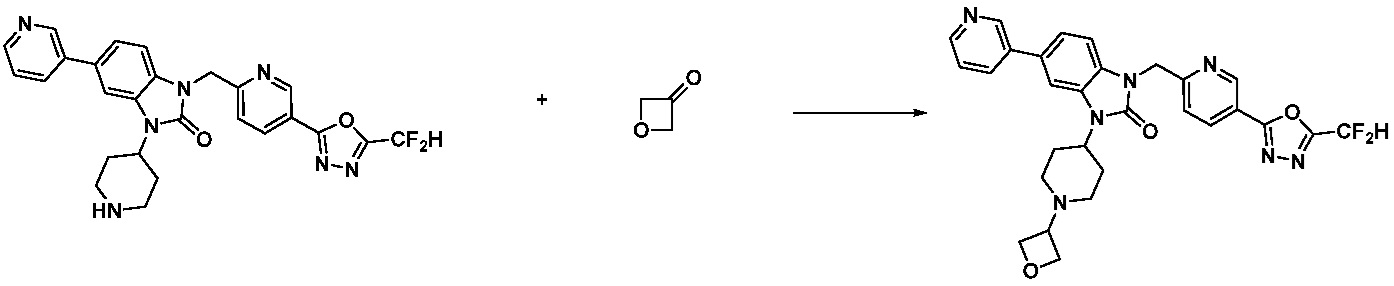
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,162 г, 0,322 ммоль), оксетан-3-он (0,046 г, 0,643 ммоль) и триацетоксиборгидрид натрия (0,136 г, 0,643 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 55,5%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 ~ 9,30 (м, 1H), 8,84 (д, J=1,9 Гц, 1H), 8,60 (дд, J=4,8, 1,4 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,91 ~ 7,87 (м, 1H), 7,53 (д, J=1,4 Гц, 1H), 7,47 ~ 7,44 (м, 1H), 7,41 ~ 7,38 (м, 1H), 7,25 (дд, J=8,1, 1,5 Гц, 1H), 7,11 (д, J=8,2 Гц, 1H), 7,07 (с, 0,25H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 5,33 (с, 2H), 4,72 ~ 4,66 (м, 4H), 4,53 ~ 4,47 (м, 1H), 3,59 ~ 3,56 (м, 1H), 2,98 ~ 2,95 (м, 2H), 2,61 ~ 2,52 (м, 2H), 2,17 ~ 2,06 (м, 2H), 1,95 ~ 1,92 (м, 2H).
Пример 281: Синтез соединения 281, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-этилпиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 281
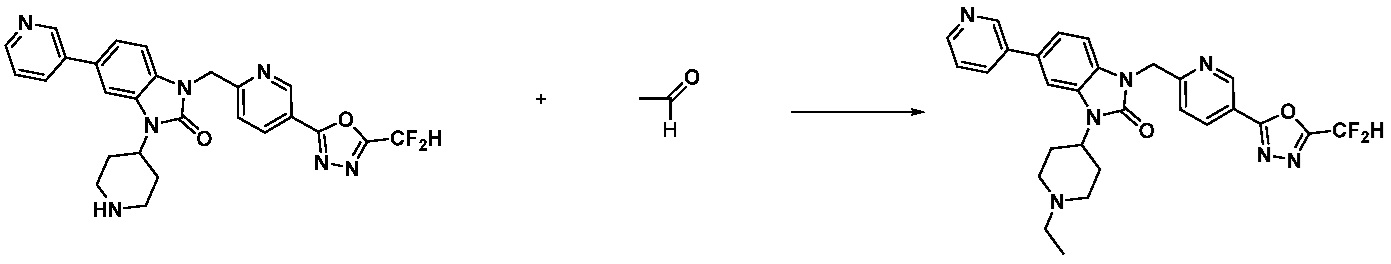
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,143 г, 0,284 ммоль), ацетальдегид (0,025 г, 0,568 ммоль) и триацетоксиборгидрид натрия (0,120 г, 0,568 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,080 г, 53,0%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (д, J=1,6 Гц, 1H), 8,82 (д, J=1,3 Гц, 1H), 8,57 (д, J=3,9 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,92 (дт, J=8,0, 1,9 Гц, 1H), 7,61 (д, J=1,2 Гц, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,37 (дд, J=7,8, 4,8 Гц, 1H), 7,26 (дд, J=8,2, 1,4 Гц, 1H), 7,09 (д, J=8,2 Гц, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,81 (с, 0,25H), 5,33 (с, 2H), 4,63 ~ 4,54 (м, 1H), 3,33 ~ 3,31 (м, 2H), 2,76 ~ 2,64 (м, 4H), 2,37 ~ 2,32 (м, 2H), 1,97 ~ 1,94 (м, 2H), 1,25 ~ 1,19 (м, 3H).
Пример 282: Синтез соединения 282, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-(оксетан-3-ил)пиперидин-4-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 282
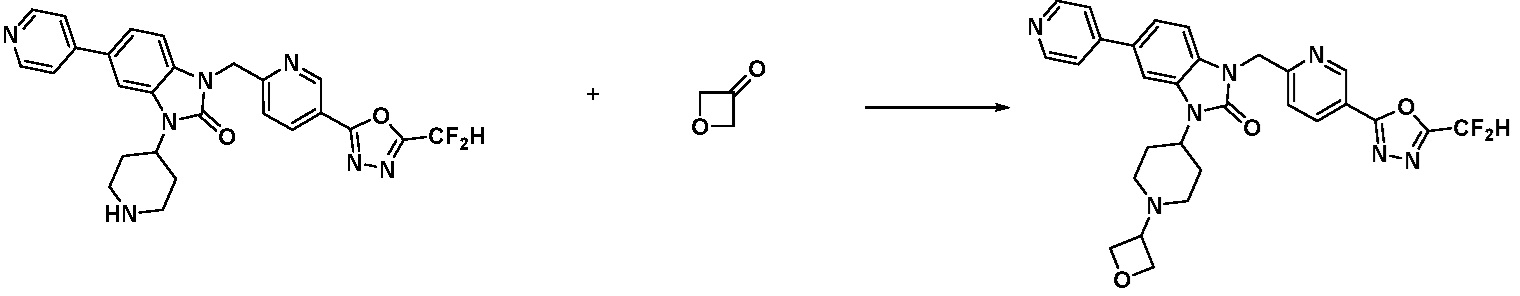
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,199 ммоль), оксетан-3-он (0,029 г, 0,397 ммоль) и триацетоксиборгидрид натрия (0,084 г, 0,397 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,065 г, 58,5%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (дд, J=2,2, 0,7 Гц, 1H), 8,66 (дд, J=4,6, 1,5 Гц, 2H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,58 (д, J=1,4 Гц, 1H), 7,54 (дд, J=4,6, 1,6 Гц, 2H), 7,46 (дд, J=8,2, 0,5 Гц, 1H), 7,34 (дд, J=8,2, 1,5 Гц, 1H), 7,12 (д, J=8,2 Гц, 1H), 7,07 (с, 0,25H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 5,32 (с, 2H), 4,73 ~ 4,66 (м, 4H), 4,60 ~ 4,40 (м, 1H), 3,60 ~ 3,56 (м, 1H), 3,30 ~ 2,99 (м, 2H), 2,60 ~ 2,56 (м, 2H), 2,12 ~ 2,05 (м, 2H), 1,95 ~ 1,92 (м, 2H).
Пример 283: Синтез соединения 283, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-этилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 283
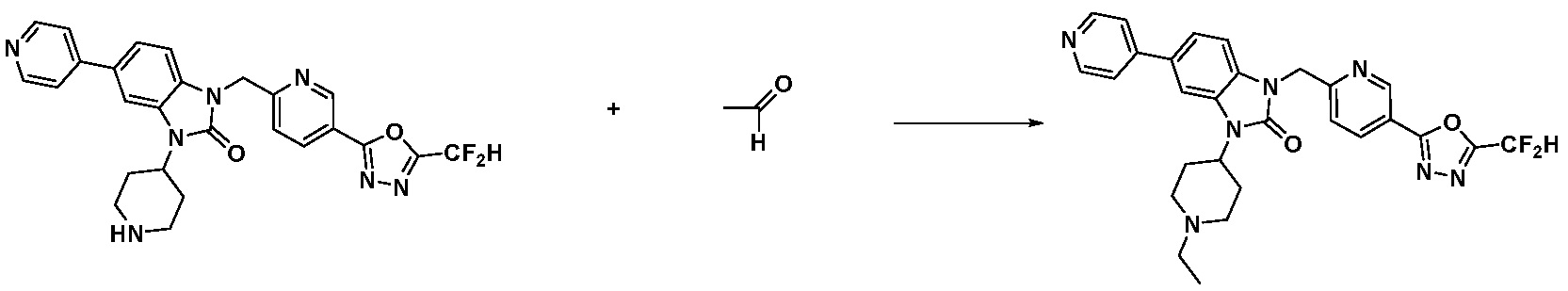
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(пиперидин-4-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,199 ммоль), ацетальдегид (0,017 г, 0,397 ммоль) и триацетоксиборгидрид натрия (0,084 г, 0,397 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,050 г, 47,4%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (дд, J=2,2, 0,7 Гц, 1H), 8,65 (дд, J=4,6, 1,6 Гц, 2H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,70 (д, J=1,2 Гц, 1H), 7,56 (дд, J=4,6, 1,6 Гц, 2H), 7,47 (дд, J=8,3, 0,6 Гц, 1H), 7,35 (дд, J=8,2, 1,6 Гц, 1H), 7,12 (д, J=8,2 Гц, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 5,34 (с, 2H), 4,65 ~ 4,60 (м, 1H), 3,37 ~ 3,34 (м, 2H), 2,76 ~ 2,66 (м, 4H), 2,39 ~ 2,33 (м, 2H), 1,99 ~ 1,94 (м, 2H), 1,22 (т, J=7,2 Гц, 3H).
Пример 284: Синтез соединения 284, 5-(4-((1H-индол-4-ил)метил)пиперазин-1-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 284
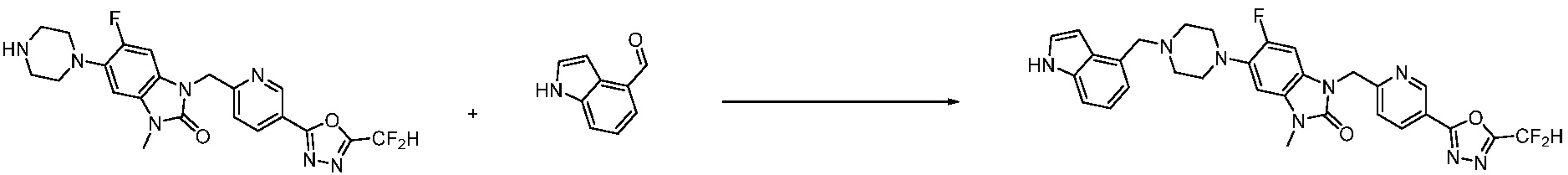
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,218 ммоль), полученный на стадии 6 соединения 260, 1H-индол-4-карбальдегид (0,063 г, 0,435 ммоль) и триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,099 г, 77,0%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (д, J=2,0 Гц, 1H), 8,71 (с, 1H), 8,26 (дд, J=8,2, 2,2 Гц, 1H), 7,36 (д, J=8,2 Гц, 1H), 7,27 (д, J=7,6 Гц, 1H), 7,17 (т, J=2,8 Гц, 1H), 7,14-7,07 (м, 2H), 6,92 (т, J=51,6 Гц, 1H), 6,74-6,72 (м, 2H), 6,64 (д, J=7,0 Гц, 1H), 5,20 (с, 2H), 3,86 (с, 2H), 3,42 (с, 3H), 3,06-3,05 (м, 4H), 2,80-2,65 (м, 4H); МСНР (ЭР) m/z 589,2 (M+ + 1).
Пример 285: Синтез соединения 285, 5-(4-((1H-пиразол-4-ил)метил)пиперазин-1-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 285
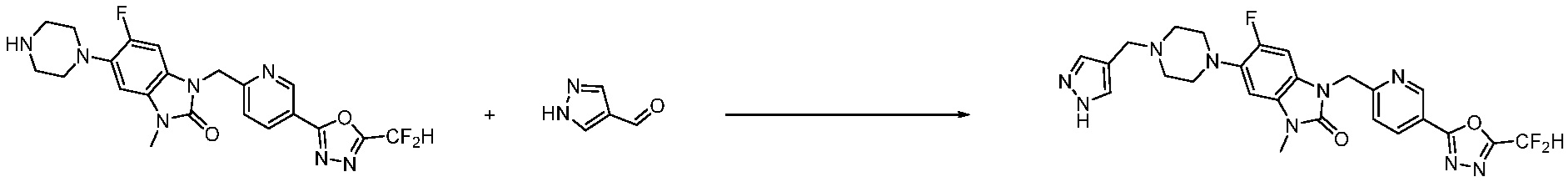
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,218 ммоль), полученный на стадии 6 соединения 260, 1H-пиразол-4-карбальдегид (0,042 г, 0,435 ммоль) и триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,079 г, 67,1%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,24 (дд, J=2,2, 0,7 Гц, 1H), 8,29 (дд, J=8,2, 2,2 Гц, 1H), 7,52 (с, 2H), 7,39 (дд, J=8,2, 0,5 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,72 (д, J=11,2 Гц, 1H), 6,62 (д, J=7,0 Гц, 1H), 5,20 (с, 2H), 3,53 (с, 2H), 3,41 (с, 3H), 3,10-2,95 (м, 4H), 2,70-2,55 (м, 4H); МСНР (ЭР) m/z 540,2 (M+ + 1).
Пример 286: Синтез соединения 286, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пирролидин-3-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез (R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
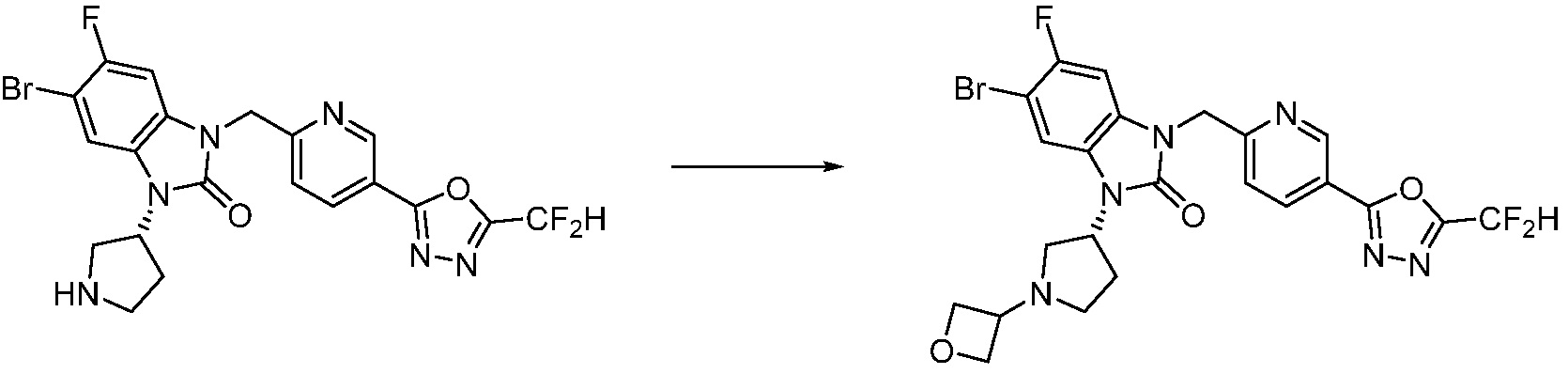
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,500 г, 0,982 ммоль) и оксетан-3-он (0,094 мл, 1,473 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,416 г, 1,964 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-3%) и концентрируют с получением указанного в заголовке соединения (0,300 г, 54,1%) в виде белого твердого вещества.
[Стадия 2] Синтез соединения 286
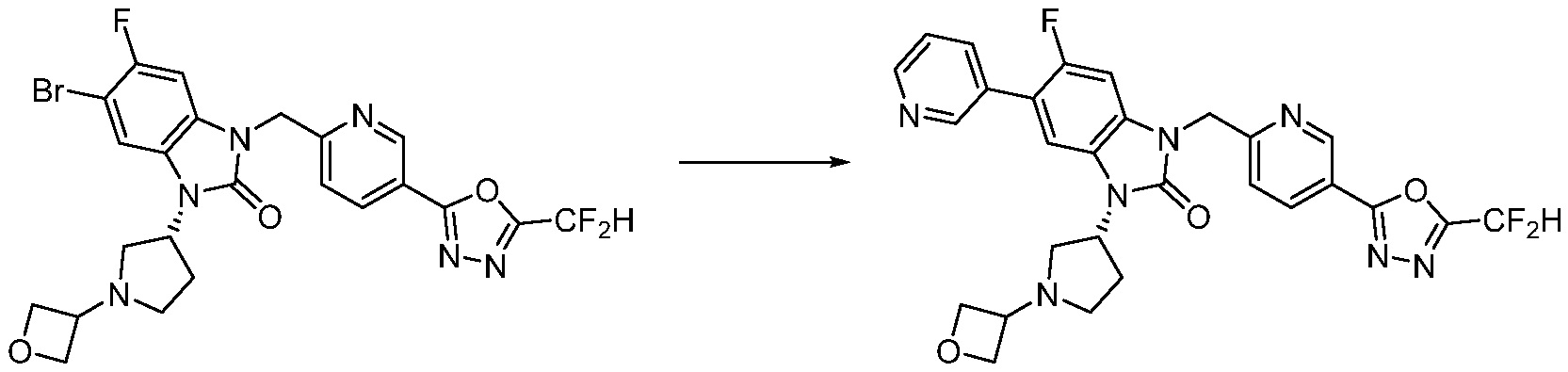
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,177 ммоль), полученный на стадии 1, пиридин-3-илбороновую кислоту (0,026 г, 0,212 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,173 г, 0,531 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,024 г, 24,1%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,8 Гц, 1H), 8,84 (с, 1H), 8,59 (д, J=4,0 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 8,15 (д, J=6,6 Гц, 1H), 7,93 (дд, J=8,0, 1,7 Гц, 1H), 7,51 (д, J=8,2 Гц, 1H), 7,41-7,38 (м, 1H), 7,08-6,82 (м, 2H), 5,33-5,24 (м, 3H), 4,79-4,73 (м, 2H), 4,68 (т, J=5,9 Гц, 1H), 4,61 (т, J=6,0 Гц, 1H), 3,70-3,66 (м, 1H), 3,17-3,12 (м, 2H), 2,64 (т, J=9,6 Гц, 1H), 2,66-2,64 (м, 1H), 2,32-2,20 (м, 2H); МСНР (ЭР) m/z 564,5 (M+ + 1).
Пример 287: Синтез соединения 287, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пирролидин-3-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 287
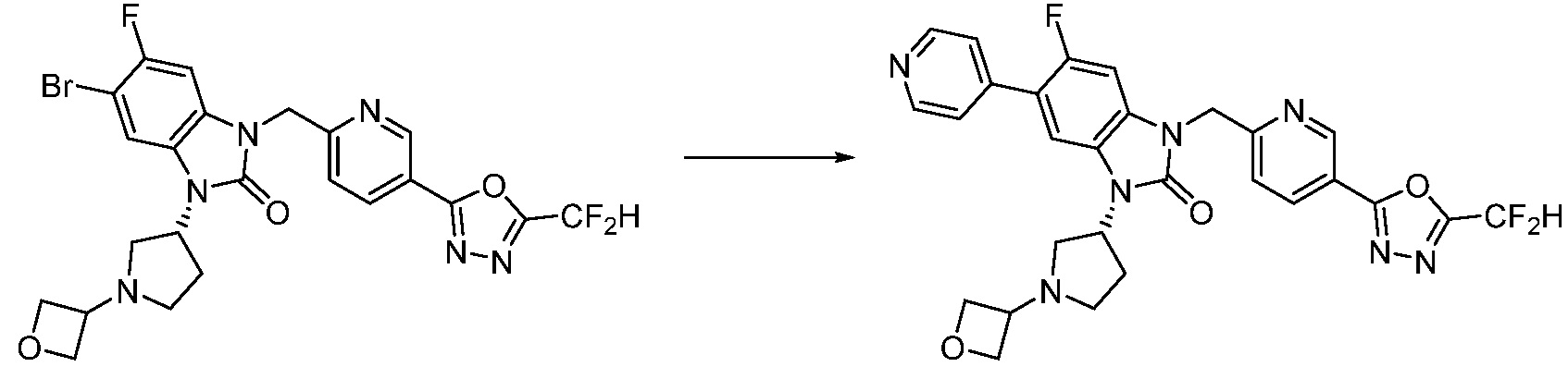
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,177 ммоль), пиридин-4-илбороновую кислоту (0,026 г, 0,212 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,173 г, 0,531 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,049 г, 49,2%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,6 Гц, 1H), 8,68 (д, J=5,6 Гц, 2H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 8,33 (д, J=6,6 Гц, 1H), 7,57 (д, J=4,7 Гц, 2H), 7,51 (д, J=8,2 Гц, 2H), 7,08-6,82 (м, 2H), 5,33-5,24 (м, 3H), 4,83-4,76 (м, 2H), 4,70 (т, J=5,9 Гц, 1H), 4,63 (т, J=5,9 Гц, 1H), 3,68-3,65 (м, 1H), 3,19-3,15 (м, 2H), 2,60 (т, J=9,6 Гц, 1H), 2,43-2,40 (м, 1H), 2,30-2,20 (м, 2H); МСНР (ЭР) m/z 564,5 (M+ + 1).
Пример 288: Синтез соединения 288, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(1-(оксетан-3-ил)пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 288
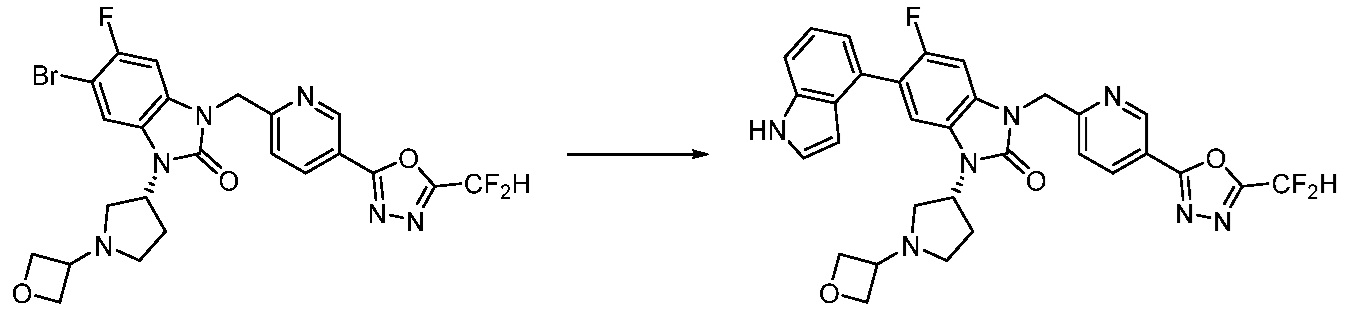
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пирролидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,177 ммоль), (1H-индол-4-ил)бороновую кислоту (0,006 г, 0,035 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,173 г, 0,531 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,032 г, 30,1%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,35 (д, J=1,7 Гц, 1H), 8,41-8,39 (м, 2H), 8,03 (д, J=6,2 Гц, 1H), 7,53 (д, J=8,2 Гц, 1H), 7,45 (д, J=8,0 Гц, 1H), 7,33-7,24 (м, 4H), 7,08-6,83 (м, 2H), 6,58 (с, 1H), 5,32-5,27 (м, 3H), 4,68-4,62 (м, 4H), 3,74-3,71 (м, 1H), 3,13 (дд, J=10,0, 3,9 Гц, 1H), 3,02 (т, J=6,6 Гц, 1H), 2,74 (т, J=9,5 Гц, 1H), 2,39-2,26 (м, 3H); МСНР (ЭР) m/z 602,5 (M+ + 1).
Пример 289: Синтез соединения 289, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(((5-бром-4-фтор-2-нитрофенил)амино)метил)пиперидин-1-карбоксилата
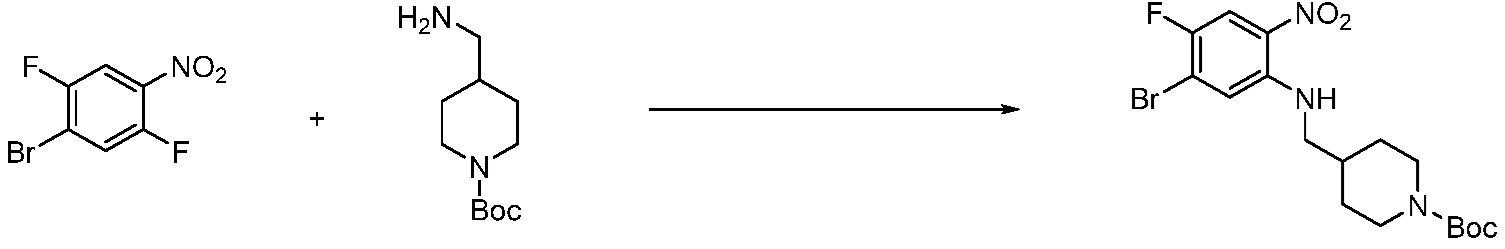
1-бром-2,5-дифтор-4-нитробензол (5,000 г, 21,009 ммоль), трет-бутил 4-(аминометил)пиперидин-1-карбоксилат (5,853 г, 27,312 ммоль), карбонат калия (5,807 г, 42,019 ммоль) и йодид калия (0,349 г, 2,101 ммоль) растворяют в N,N-диметилформамиде (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 5 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (7,910 г, 87,1%) в виде оранжевого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(((2-амино-5-бром-4-фторфенил)амино)метил)пиперидин-1-карбоксилата

Трет-бутил 4-(((5-бром-4-фтор-2-нитрофенил)амино)метил)пиперидин-1-карбоксилат (7,910 г, 18,298 ммоль), полученный на стадии 1, растворяют в метаноле (100 мл) и перемешивают при комнатной температуре, после чего никель Ренея (800 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (7,340 г, 99,7%, коричневое твердое вещество).
[Стадия 3] Синтез трет-бутил 4-((6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилата
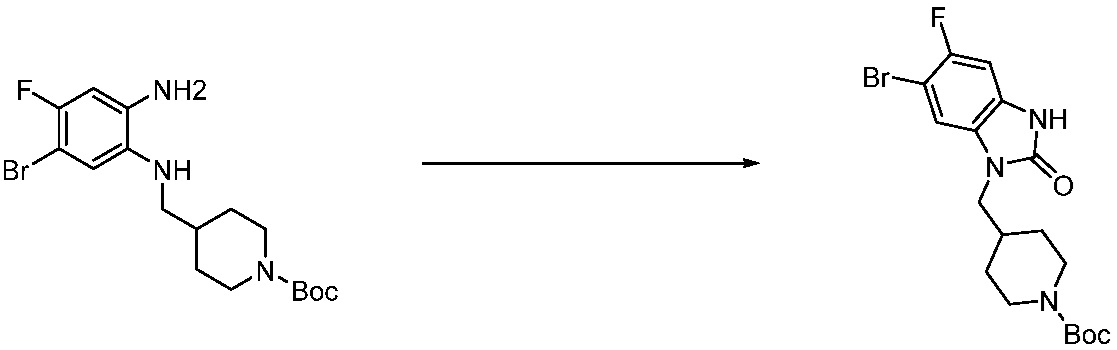
Трет-бутил 4-(((2-амино-5-бром-4-фторфенил)амино)метил)пиперидин-1-карбоксилат (7,340 г, 18,245 ммоль), полученный на стадии 2, триэтиламин (2,543 мл, 18,245 ммоль) и 1,1'-карбонилдиимидазол (КДИ, 3,550 г, 21,894 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=20-80%) и концентрируют с получением указанного в заголовке соединения (6,670 г, 85,4%) в виде фиолетового твердого вещества.
[Стадия 4] Синтез трет-бутил 4-((6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилата
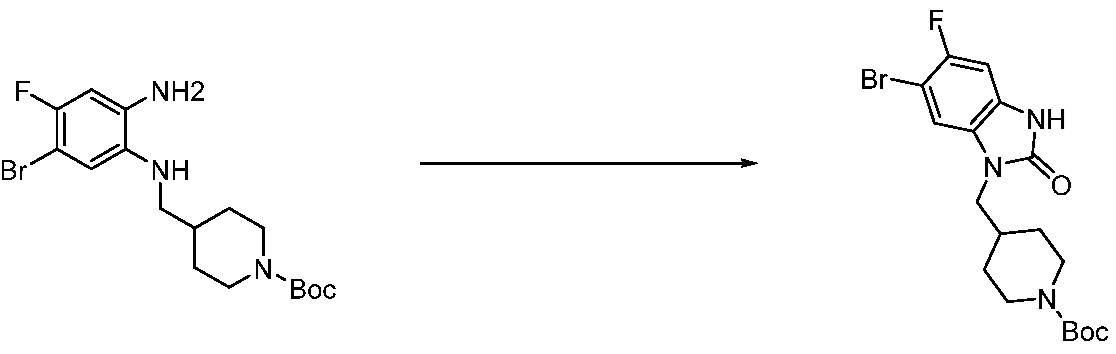
Трет-бутил 4-((6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилат (4,270 г, 9,970 ммоль), полученный на стадии 3, гидрид натрия (60,00%, 0,598 г, 14,954 ммоль) и 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (3,181 г, 10,967 ммоль) растворяют в N,N-диметилформамиде (50 мл), после чего полученный раствор перемешивают при 0° в течение 30 минут и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=25-60%) и концентрируют с получением указанного в заголовке соединения (4,100 г, 64,5%) в виде желтого твердого вещества.
[Стадия 5] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-4-илметил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
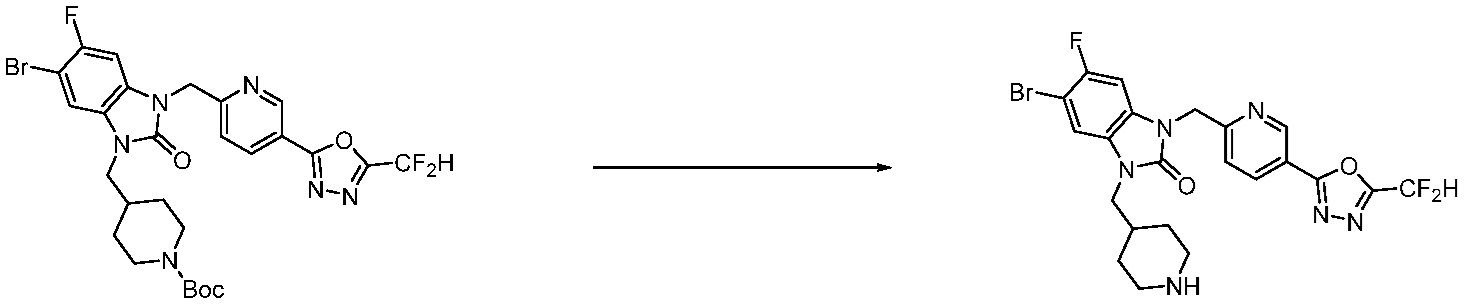
Трет-бутил 4-((6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилат (4,100 г, 6,432 ммоль), полученный на стадии 4, и трифторуксусную кислоту (4,925 мл, 64,318 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (3,640 г, 105,3%, коричневое твердое вещество).
[Стадия 6] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
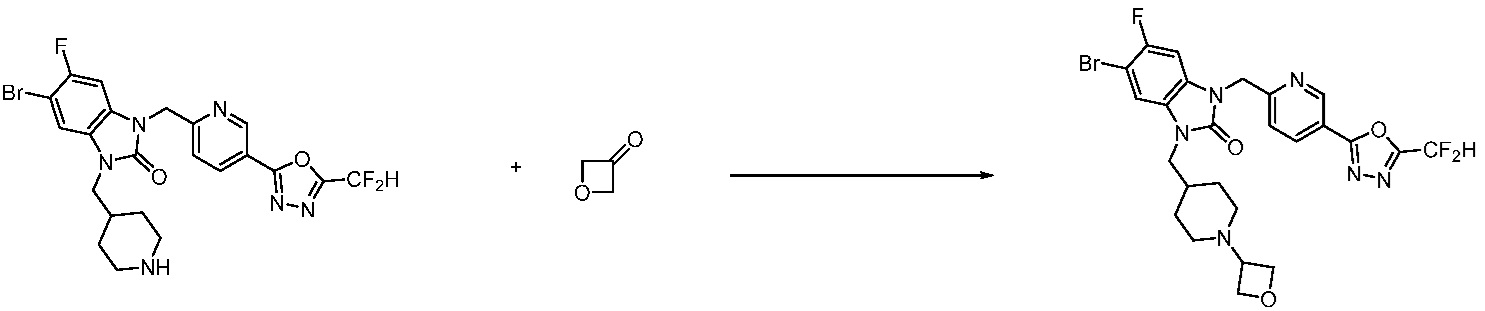
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-4-илметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (3,450 г, 6,421 ммоль), полученный на стадии 5, триацетоксиборгидрид натрия (2,722 г, 12,841 ммоль) и оксетан-3-он (1,388 г, 19,262 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (3,210 г, 84,3%) в виде коричневого твердого вещества.
[Стадия 7] Синтез соединения 289
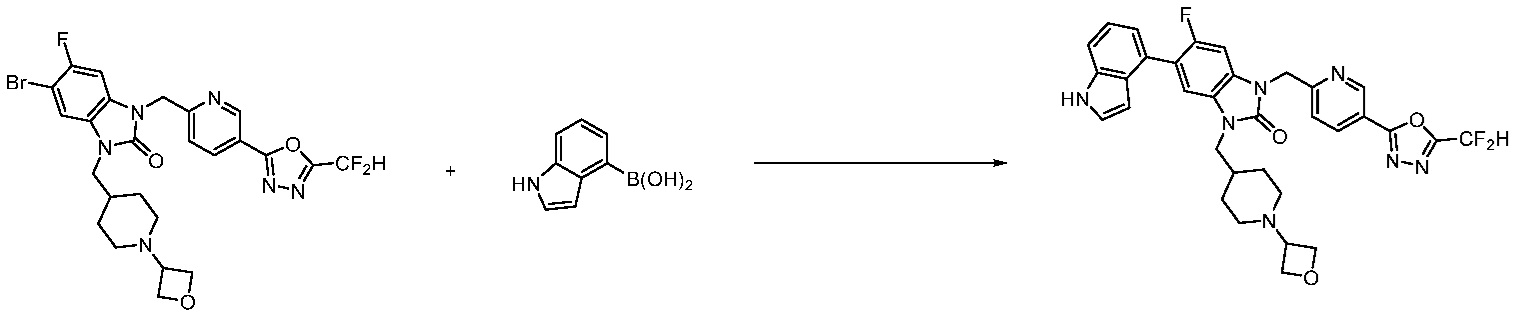
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,253 ммоль), полученный на стадии 6, (1H-индол-4-ил)бороновую кислоту (0,049 г, 0,303 ммоль), карбонат цезия (0,165 г, 0,506 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,013 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,081 г, 50,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,9 Гц, 1H), 8,87 (с, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,44 (д, J=8,2 Гц, 1H), 7,38 (д, J=8,1 Гц, 1H), 7,26-7,20 (м, 2H), 7,14-7,10 (м, 2H), 6,91 (т, J=51,7 Гц, 1H), 6,88 (д, J=9,4 Гц, 1H), 6,41 (д, J=1,9 Гц, 1H), 5,29 (с, 2H), 4,59 (тд, J=12,1, 5,9 Гц, 4H), 3,82 (д, J=7,0 Гц, 2H), 3,44-3,37 (м, 1H), 2,72 (д, J=11,4 Гц, 2H), 1,94-1,90 (м, 1H), 1,78-1,70 (м, 4H), 1,51-1,41 (м, 2H); МСНР (ЭР) m/z 631,5 (M+ + 1).
Пример 290: Синтез соединения 290, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 290
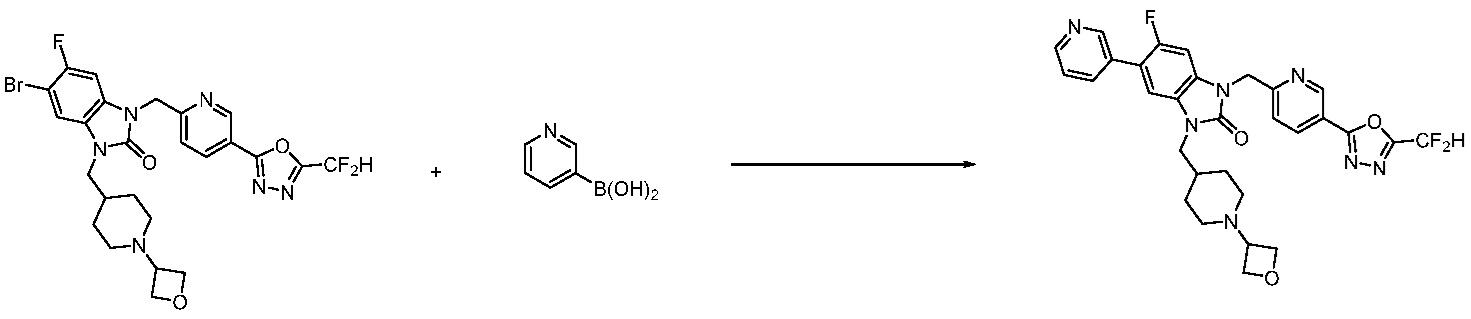
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,253 ммоль), полученный на стадии 6 соединения 289, пиридин-3-илбороновую кислоту (0,037 г, 0,303 ммоль), карбонат цезия (0,165 г, 0,506 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,013 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,035 г, 23,7%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,6 Гц, 1H), 8,72 (с, 1H), 8,58 (с, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,82 (дд, J=7,9, 1,6 Гц, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,36 (дд, J=7,8, 4,8 Гц, 1H), 6,96 (д, J=6,1 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,89 (д, J=9,8 Гц, 1H), 5,27 (с, 2H), 4,59 (тд, J=12,2, 6,0 Гц, 4H), 3,84 (д, J=7,1 Гц, 2H), 3,46-3,39 (м, 1H), 2,74 (д, J=11,3 Гц, 2H), 1,98-1,90 (м, 1H), 1,82-1,71 (м, 4H), 1,52-1,42 (м, 2H); МСНР (ЭР) m/z 592,5 (M+ + 1).
Пример 291: Синтез соединения 291, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 291
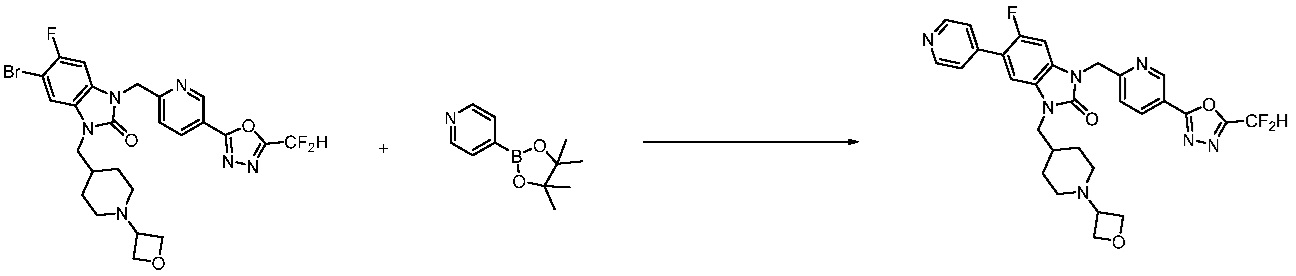
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,253 ммоль), полученный на стадии 6 соединения 289, 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (0,062 г, 0,303 ммоль), карбонат цезия (0,165 г, 0,506 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,013 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 66,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (д, J=1,6 Гц, 1H), 8,63 (д, J=4,9 Гц, 2H), 8,34 (дд, J=8,2, 2,1 Гц, 1H), 7,46 (д, J=8,2 Гц, 1H), 7,41 (д, J=4,6 Гц, 2H), 6,98 (д, J=6,0 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,88 (д, J=10,1 Гц, 1H), 5,26 (с, 2H), 4,58 (тд, J=13,1, 6,6 Гц, 4H), 3,83 (д, J=7,1 Гц, 2H), 3,44-3,38 (м, 1H), 2,73 (д, J=11,2 Гц, 2H), 1,96-1,90 (м, 1H), 1,81-1,70 (м, 4H), 1,48-1,41 (м, 2H); МСНР (ЭР) m/z 592,6 (M+ + 1).
Пример 292: Синтез соединения 292, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-5-(1H-пиразол-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 292
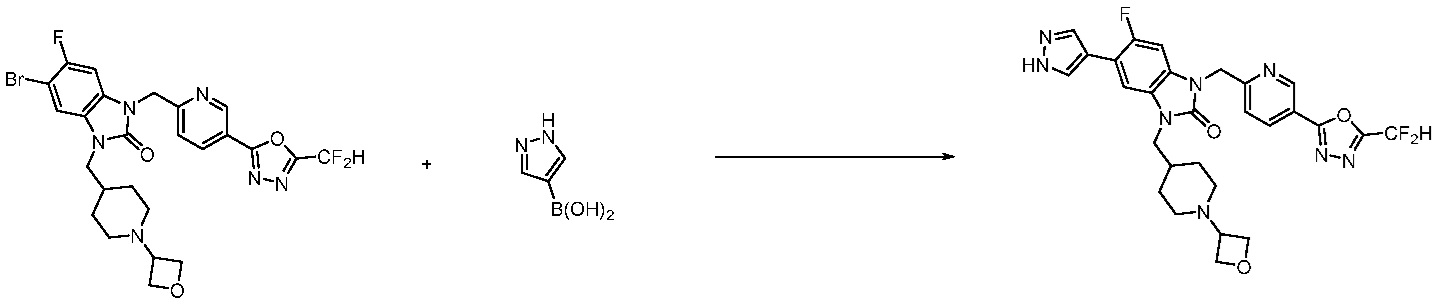
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,253 ммоль), полученный на стадии 6 соединения 289, (1H-пиразол-4-ил)бороновую кислоту (0,034 г, 0,303 ммоль), карбонат цезия (0,165 г, 0,506 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,008 г, 0,013 ммоль) смешивают в 1,4-диоксане (2,1 мл)/воде (0,7 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,072 г, 48,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,6 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,94 (с, 2H), 7,43 (д, J=8,2 Гц, 1H), 7,14 (д, J=6,0 Гц, 1H), 7,05-6,79 (м, 2H), 5,25 (с, 2H), 4,63 (тд, J=14,8, 7,7 Гц, 4H), 3,86 (д, J=7,2 Гц, 2H), 3,48-3,42 (м, 1H), 2,78 (д, J=11,1 Гц, 2H), 2,01-1,97 (м, 1H), 1,85-1,73 (м, 4H), 1,61-1,52 (м, 2H); МСНР (ЭР) m/z 581,3 (M+ + 1).
Пример 293: Синтез соединения 293, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пиперидин-3-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 293
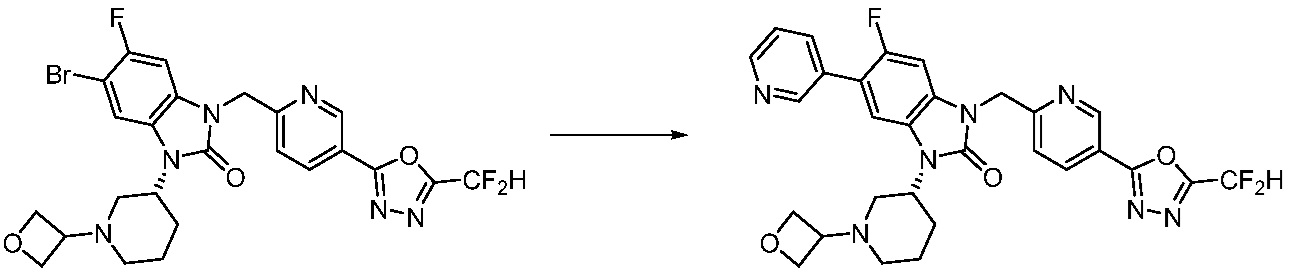
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,173 ммоль), пиридин-3-илбороновую кислоту (0,025 г, 0,207 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,169 г, 0,518 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,030 г, 30,1%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,7 Гц, 1H), 8,77 (с, 1H), 8,62 (д, J=4,8 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,85 (дд, J=7,9, 1,6 Гц, 1H), 7,50 (д, J=8,2 Гц, 1H), 7,42-7,39 (м, 1H), 7,16 (д, J=6,1 Гц, 1H), 7,08-6,82 (м, 2H), 5,27-5,26 (м, 2H), 4,70 (т, J=6,5 Гц, 1H), 4,68-4,63 (м, 3H), 4,50-4,44 (м, 1H), 3,63-3,60 (м, 1H), 2,91 (д, J=10,3 Гц, 1H), 2,80 (д, J=10,6 Гц, 1H), 2,70 (т, J=10,8 Гц, 1H), 2,33-2,29 (м, 1H), 2,06-1,99 (м, 2H), 1,94-1,92 (м, 2H); МСНР (ЭР) m/z 578,6 (M+ + 1).
Пример 294: Синтез соединения 294, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пиперидин-3-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 294
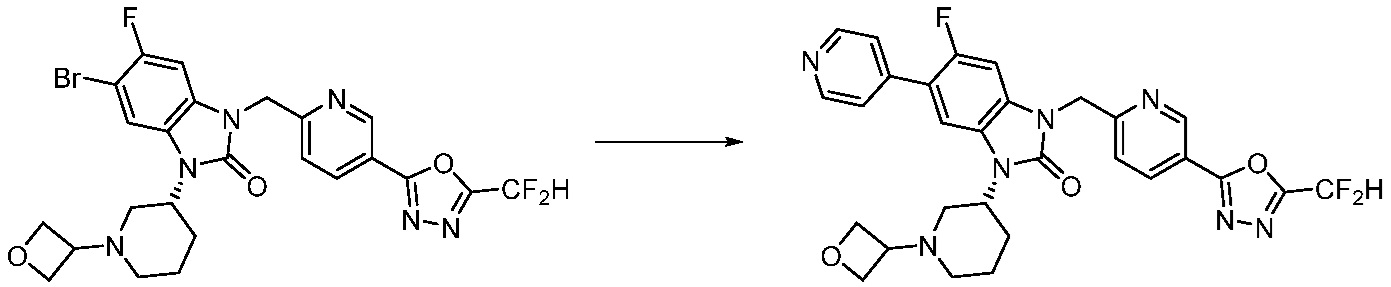
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,173 ммоль), пиридин-4-илбороновую кислоту (0,025 г, 0,207 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,169 г, 0,518 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,008 г, 8,0%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=2,0 Гц, 1H), 8,70 (д, J=5,7 Гц, 2H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,51 (д, J=8,2 Гц, 1H), 7,47 (д, J=4,7 Гц, 2H), 7,20 (д, J=6,0 Гц, 1H), 7,08-6,82 (м, 2H), 5,31-5,22 (м, 2H), 4,73-4,69 (м, 1H), 4,66-4,64 (м, 3H), 4,46-4,42 (м, 1H), 3,64-3,61 (м, 1H), 2,91-2,89 (м, 1H), 2,81 (д, J=11,6 Гц, 1H), 2,72 (т, J=10,8 Гц, 1H), 2,36-2,70 (м, 1H), 2,07-2,04 (м, 1H), 1,97-1,93 (м, 2H), 1,84-1,80 (м, 1H); МСНР (ЭР) m/z 578,6 (M+ + 1).
Пример 295: Синтез соединения 295, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(1-(оксетан-3-ил)пиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 295
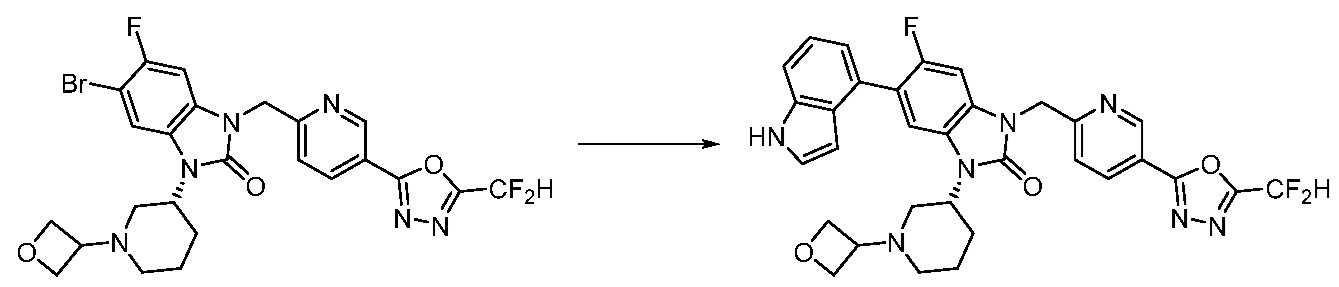
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,173 ммоль), (1H-индол-4-ил)бороновую кислоту (0,033 г, 0,207 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,169 г, 0,518 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,013 г, 12,2%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,34 (дд, J=2,1, 0,7 Гц, 1H), 8,43-8,39 (м, 2H), 7,52 (д, J=8,2 Гц, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,34-7,29 (м, 2H), 7,18 (д, J=7,3 Гц, 1H), 7,08-6,83 (м, 2H), 6,48-6,47 (м, 1H), 5,34-5,25 (м, 2H), 4,70-4,63 (м, 4H), 4,54-4,48 (м, 1H), 3,61-3,57 (м, 1H), 2,91 (дд, J=10,2, 3,9 Гц, 1H), 2,77 (д, J=9,9 Гц, 1H), 2,68 (т, J=10,8 Гц, 1H), 2,31-2,27 (м, 1H), 2,04 (д, J=10,9 Гц, 1H), 1,90-1,87 (м, 2H), 1,80-1,76 (м, 1H); МСНР (ЭР) m/z 616,2 (M+ + 1).
Пример 296: Синтез соединения 296, (R)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пиперидин-3-ил)-5-(1H-пиразол-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 296
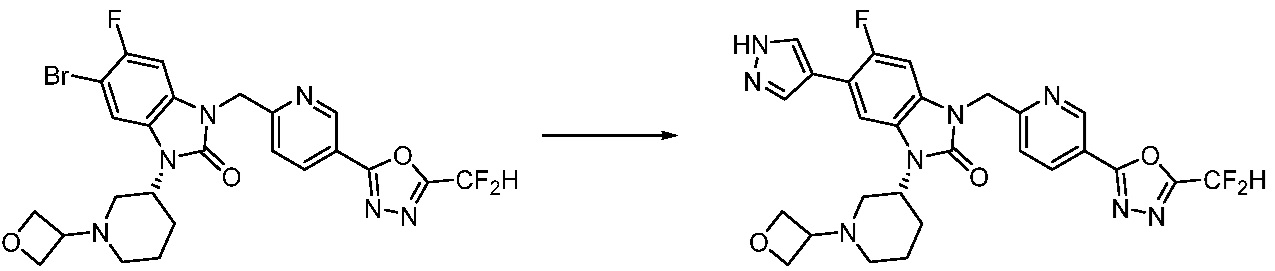
(R)-5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пиперидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,173 ммоль), (1H-пиразол-4-ил)бороновую кислоту (0,023 г, 0,207 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,006 г, 0,009 ммоль) и карбонат цезия (0,169 г, 0,518 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,008 г, 8,2%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,5 Гц, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 7,98 (с, 2H), 7,46 (д, J=8,2 Гц, 1H), 7,32 (д, J=6,0 Гц, 1H), 7,08-6,82 (м, 2H), 5,24-5,24 (м, 2H), 4,75 (т, J=6,5 Гц, 1H), 4,70-4,63 (м, 3H), 4,55-4,50 (м, 1H), 3,64-3,61 (м, 1H), 2,92-2,85 (м, 2H), 2,76 (т, J=10,8 Гц, 1H), 2,45-2,40 (м, 1H), 2,07-2,00 (м, 3H), 1,90-1,87 (м, 1H); МСНР (ЭР) m/z 567,5 (M+ + 1).
Пример 297: Синтез соединения 297, (S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пиперидин-3-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил (S)-3-((5-бром-4-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилата
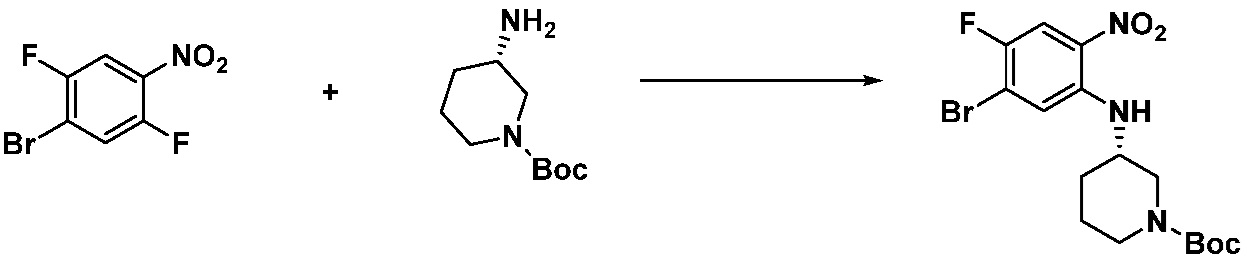
1-бром-2,5-дифтор-4-нитробензол (4,000 г, 16,807 ммоль), трет-бутил (S)-3-аминопирролидин-1-карбоксилат (3,757 г, 20,169 ммоль) и N,N-диизопропилэтиламин (3,513 мл, 20,169 ммоль) растворяют в тетрагидрофуране (30 мл) при 65°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (5,140 г, 73,1%) в виде желтого твердого вещества.
[Стадия 2] Синтез трет-бутил (S)-3-((2-амино-5-бром-4-фторфенил)амино)пиперидин-1-карбоксилата
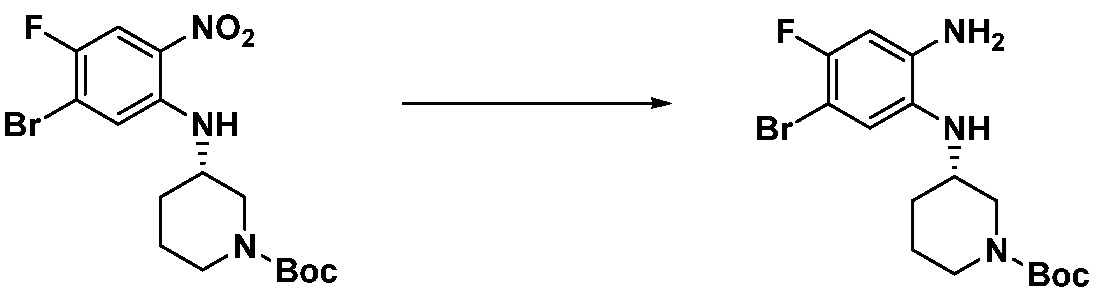
Трет-бутил (S)-3-((5-бром-4-фтор-2-нитрофенил)амино)пиперидин-1-карбоксилат (5,140 г, 12,289 ммоль), полученный на стадии 1, растворяют в этаноле (50 мл) при комнатной температуре, после чего никель Ренея медленно добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (4,500 г, 94,3%, желтое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-3-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
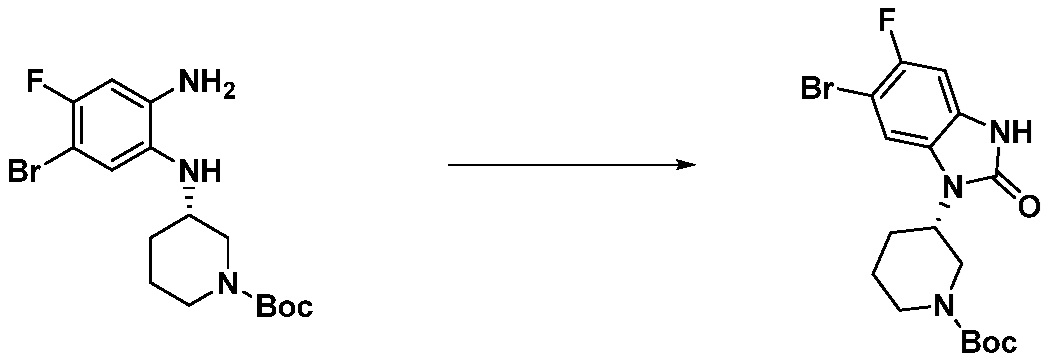
Трет-бутил (S)-3-((2-амино-5-бром-4-фторфенил)амино)пиперидин-1-карбоксилат (5,140 г, 13,238 ммоль), полученный на стадии 2, и 1,1'-карбонилдиимидазол (КДИ, 3,220 г, 19,857 ммоль) растворяют в тетрагидрофуране (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (3,190 г, 58,2%) в виде белого твердого вещества.
[Стадия 4] Синтез метил (S)-6-((5-бром-3-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-6-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)никотината
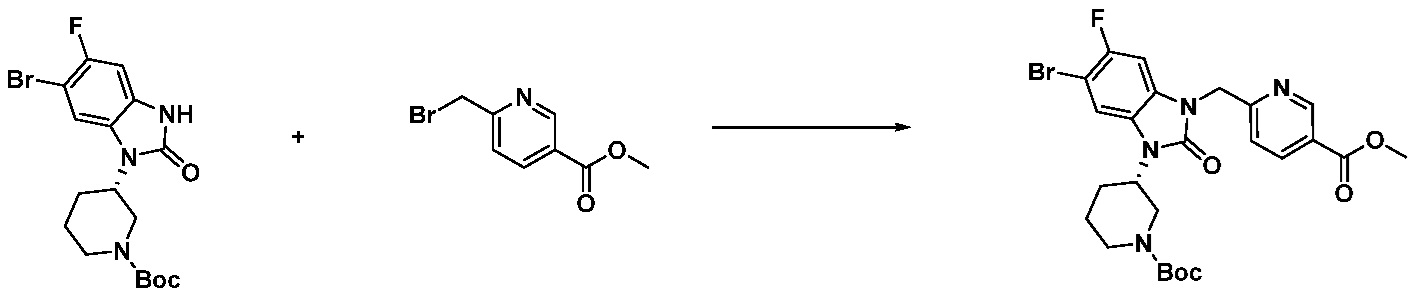
Трет-бутил (S)-3-(6-бром-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (3,190 г, 7,700 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (30 мл) при 0°C, после чего гидрид натрия (60,00%, 0,462 г, 11,550 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. Метил 6-(бромметил)никотинат (2,126 г, 9,240 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (2,570 г, 59,2%) в виде белого пенистого твердого вещества.
[Стадия 5] Синтез трет-бутил (S)-3-(6-бром-5-фтор-3-((5-(гидразинкарбонил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
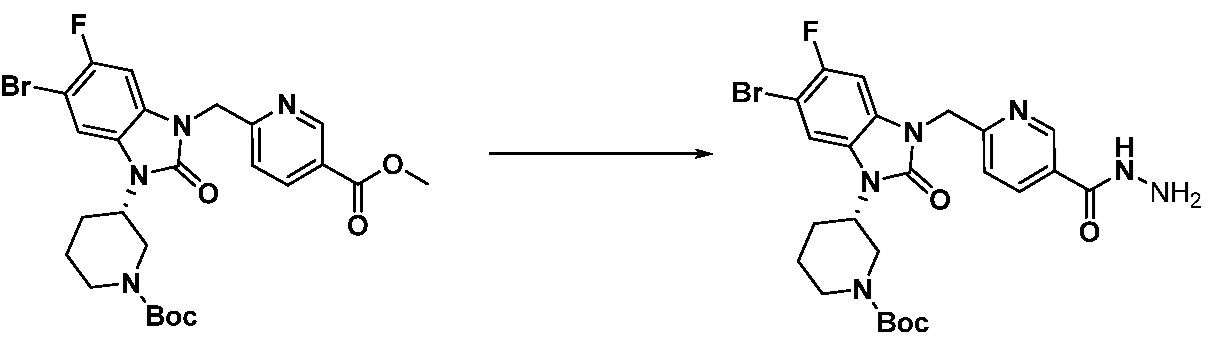
Метил (S)-6-((5-бром-3-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-6-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)никотинат (2,570 г, 4,561 ммоль), полученный на стадии 4, и моногидрат гидразина (4,434 мл, 91,229 ммоль) растворяют в этаноле (20 мл) при 80°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего указанное в заголовке соединение применяют без дополнительной очистки (2,570 г, 100,0%, белое твердое вещество).
[Стадия 6] Синтез трет-бутил (S)-3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
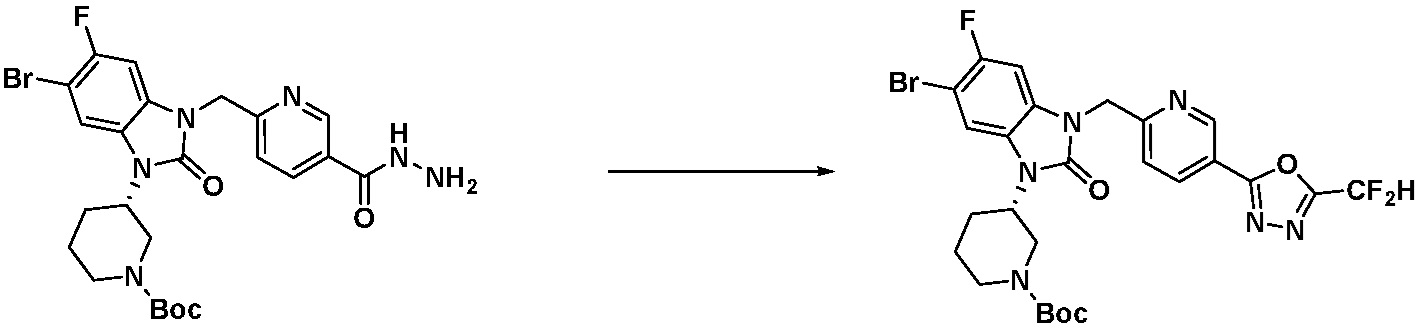
Трет-бутил (S)-3-(6-бром-5-фтор-3-((5-(гидразинкарбонил)пиридин-2-ил)метил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (2,700 г, 4,792 ммоль), полученный на стадии 5, 2,2-дифторуксусный ангидрид (1,787 мл, 14,376 ммоль) и имидазол (0,979 г, 14,376 ммоль) растворяют в дихлорметане (30 мл) при 45°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (2,400 г, 80,3%) в виде белого пенистого твердого вещества.
[Стадия 7] Синтез трет-бутил (S)-3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-6-(пиридин-3-ил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
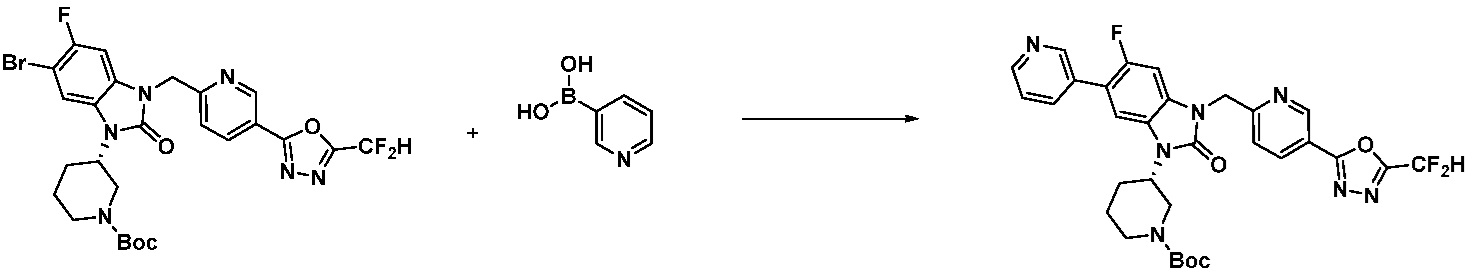
Трет-бутил (S)-3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,600 г, 0,962 ммоль), полученный на стадии 6, пиридин-3-илбороновую кислоту (0,177 г, 1,444 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2, 0,070 г, 0,096 ммоль) и карбонат цезия (0,627 г, 1,925 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (0,410 г, 68,5%) в виде коричневого масла.
[Стадия 8] Синтез (S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-3-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
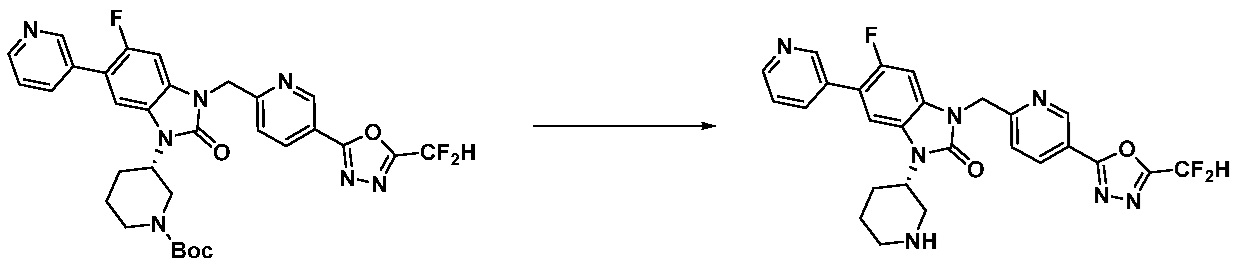
Трет-бутил (S)-3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-6-(пиридин-3-ил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,410 г, 0,660 ммоль), полученный на стадии 7, и трифторуксусную кислоту (0,505 мл, 6,596 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,360 г, 104,7%, желтое масло).
[Стадия 9] Синтез соединения 297
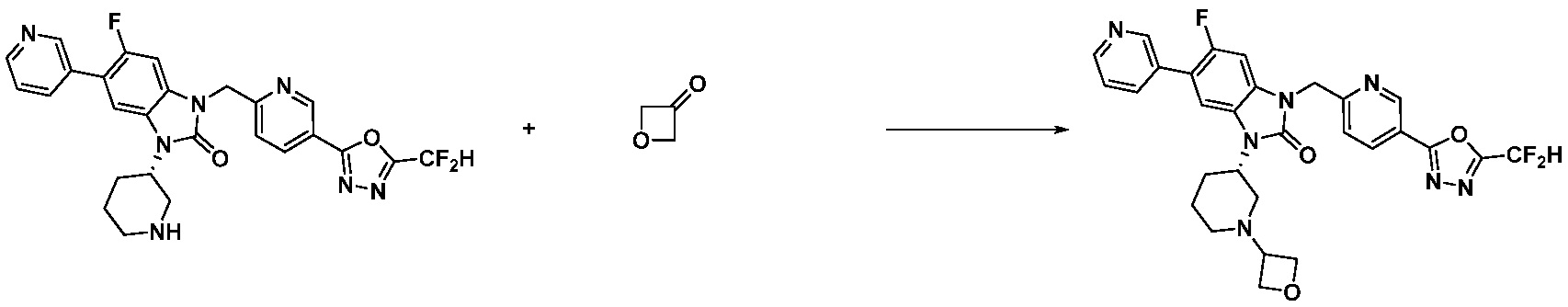
(S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-3-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,144 г, 0,276 ммоль), полученный на стадии 8, оксетан-3-он (0,040 г, 0,552 ммоль) и триацетоксиборгидрид натрия (0,117 г, 0,552 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 62,7%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (дд, J=2,1, 0,6 Гц, 1H), 8,76 (с, 1H), 8,60 (д, J=3,6 Гц, 1H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 7,87 ~ 7,85 (м, 1H), 7,49 (д, J=7,8 Гц, 1H), 7,40 (дд, J=7,7, 5,2 Гц, 1H), 7,18 (д, J=6,2 Гц, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,92 (д, J=9,8 Гц, 1H), 6,82 (с, 0,25H), 5,30 (с, 2H), 4,70 ~ 4,62 (м, 4H), 4,50 ~ 4,47 (м, 1H), 3,62 ~ 3,59 (м, 1H), 2,92 ~ 2,78 (м, 2H), 2,72 ~ 2,67 (м, 1H), 2,33 ~ 2,29 (м, 1H), 2,04 ~ 1,81 (м, 4H).
Пример 298: Синтез соединения 298, (S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-этилпиперидин-3-ил)-6-фтор-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 298
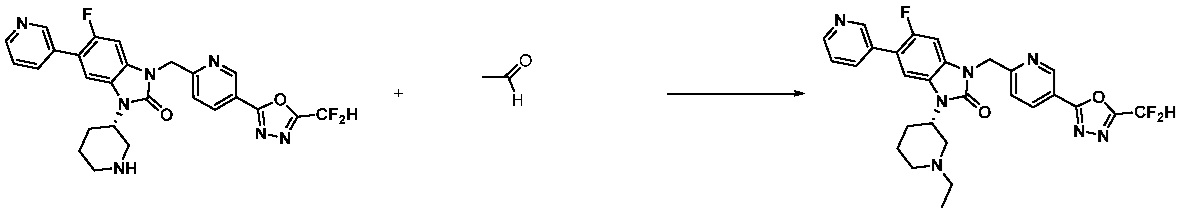
(S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-3-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,139 г, 0,267 ммоль), ацетальдегид (0,655 мл, 11,741 ммоль) и триацетоксиборгидрид натрия (0,113 г, 0,533 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 68,3%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,6 Гц, 1H), 8,78 (с, 1H), 8,59 (дд, J=4,8, 1,6 Гц, 1H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 7,90 ~ 7,87 (м, 1H), 7,49 (д, J=8,2 Гц, 1H), 7,40 (дд, J=4,9, 0,4 Гц, 1H), 7,28 ~ 7,25 (м, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,90 (д, J=9,8 Гц, 1H), 6,82 (с, 0,25H), 5,30 (с, 2H), 4,61 ~ 4,54 (м, 1H), 3,22 ~ 3,07 (м, 2H), 2,88 ~ 2,82 (м, 1H), 2,64 ~ 2,57 (м, 2H), 2,41 ~ 2,37 (м, 1H), 2,14 ~ 2,03 (м, 1H), 1,98 ~ 1,86 (м, 3H), 1,15 (т, J=7,2 Гц, 3H).
Пример 299: Синтез соединения 299, (S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)пиперидин-3-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил (S)-3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-6-(пиридин-4-ил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилата
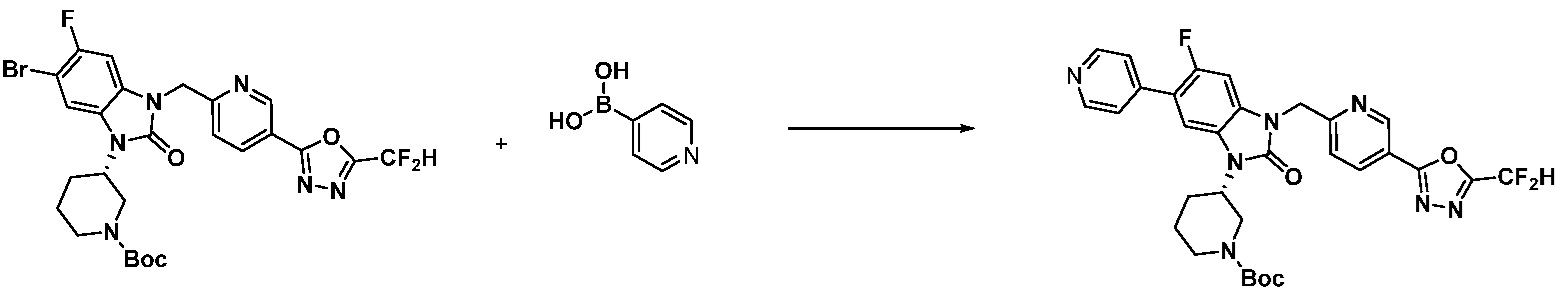
Трет-бутил (S)-3-(6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,800 г, 1,283 ммоль), пиридин-4-илбороновую кислоту (0,237 г, 1,925 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2, 0,094 г, 0,128 ммоль) и карбонат цезия (0,836 г, 2,566 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (0,514 г, 64,4%) в виде коричневого масла.
[Стадия 2] Синтез (S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-3-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
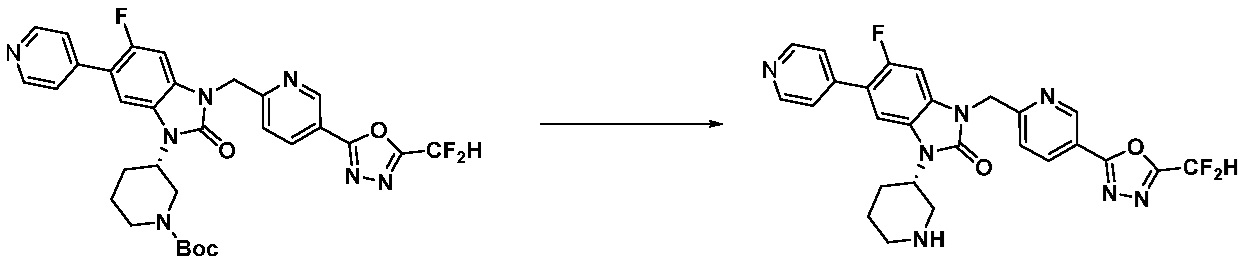
Трет-бутил (S)-3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-6-(пиридин-4-ил)-2,3-дигидро-1H-бензо[d]имидазол-1-ил)пиперидин-1-карбоксилат (0,514 г, 0,827 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,633 мл, 8,269 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,450 г, 104,4%, желтое масло).
[Стадия 3] Синтез соединения 299
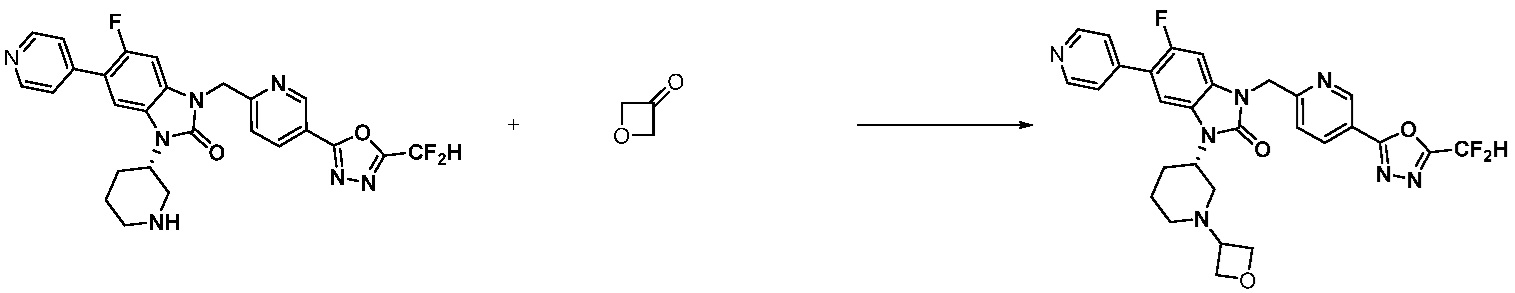
(S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-3-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,130 г, 0,249 ммоль), полученный на стадии 2, оксетан-3-он (0,036 г, 0,499 ммоль) и триацетоксиборгидрид натрия (0,106 г, 0,499 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,080 г, 55,6%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (дд, J=2,1, 0,7 Гц, 1H), 8,68 (д, J=5,6 Гц, 2H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,51 ~ 7,47 (м, 3H), 7,21 (д, J=6,1 Гц, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,93 (д, J=10,1 Гц, 1H), 6,82 (с, 0,25H), 5,31 (с, 2H), 4,68 ~ 4,58 (м, 4H), 4,48 ~ 4,42 (м, 1H), 3,63 ~ 3,60 (м, 1H), 2,92 ~ 2,88 (м, 1H), 2,82 ~ 2,79 (м, 1H), 2,79 ~ 2,69 (м, 1H), 2,35 ~ 2,31 (м, 1H), 2,05 ~ 1,92 (м, 3H), 1,81 ~ 1,79 (м, 1H).
Пример 300: Синтез соединения 300, (S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(1-этилпиперидин-3-ил)-6-фтор-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 300
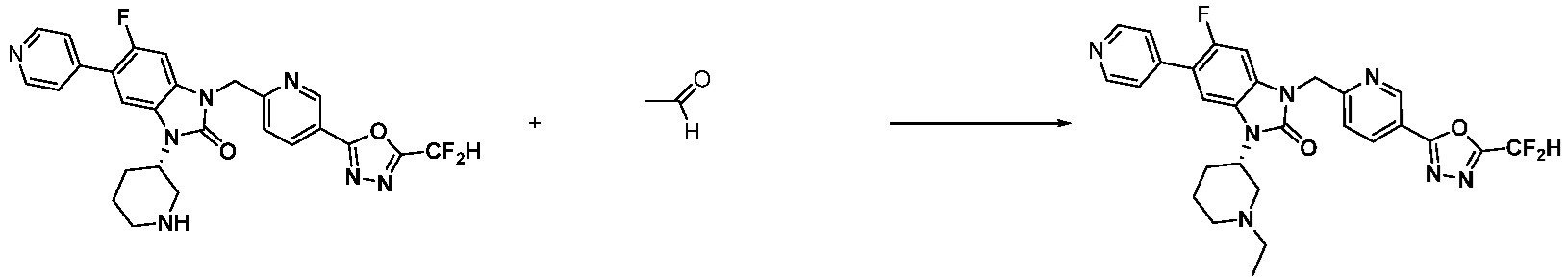
(S)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(пиперидин-3-ил)-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,168 г, 0,322 ммоль), ацетальдегид (0,791 мл, 14,191 ммоль) и триацетоксиборгидрид натрия (0,137 г, 0,644 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,110 г, 62,1%) в виде белого пенистого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (дд, J=2,2, 0,7 Гц, 1H), 8,65 ~ 8,64 (м, 2H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 7,52 ~ 7,47 (м, 3H), 7,34 (д, J=6,2 Гц, 1H), 7,80 (с, 0,25H), 6,95 (с, 0,5H), 6,90 (д, J=10,2 Гц, 1H), 6,82 (с, 0,25H), 5,25 (с, 2H), 4,61 ~ 4,55 (м, 1H), 3,25 ~ 3,21 (м, 1H), 3,12 ~ 3,09 (м, 1H), 2,89 (т, J=11,0 Гц, 1H), 2,67 ~ 2,60 (м, 2H), 2,45 ~ 2,40 (м, 1H), 2,21 ~ 2,15 (м, 1H), 2,18 ~ 1,87 (м, 3H), 1,20 ~ 1,15 (м, 3H).
Пример 301: Синтез соединения 301, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-((1-(оксетан-3-ил)пиперидин-4-ил)метил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(((4-фтор-2-нитрофенил)амино)метил)пиперидин-1-карбоксилата
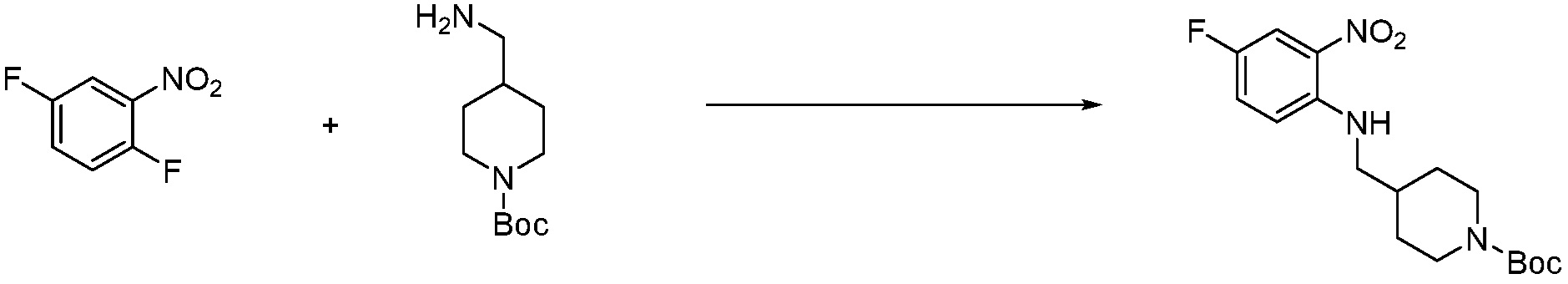
1,4-дифтор-2-нитробензол (0,300 г, 1,886 ммоль), трет-бутил 4-(аминометил)пиперидин-1-карбоксилат (0,525 г, 2,451 ммоль), карбонат калия (0,521 г, 3,771 ммоль) и йодид калия (0,031 г, 0,189 ммоль) растворяют в N,N-диметилформамиде (7 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,484 г, 72,6%) в виде оранжевого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(((2-амино-4-фторфенил)амино)метил)пиперидин-1-карбоксилата
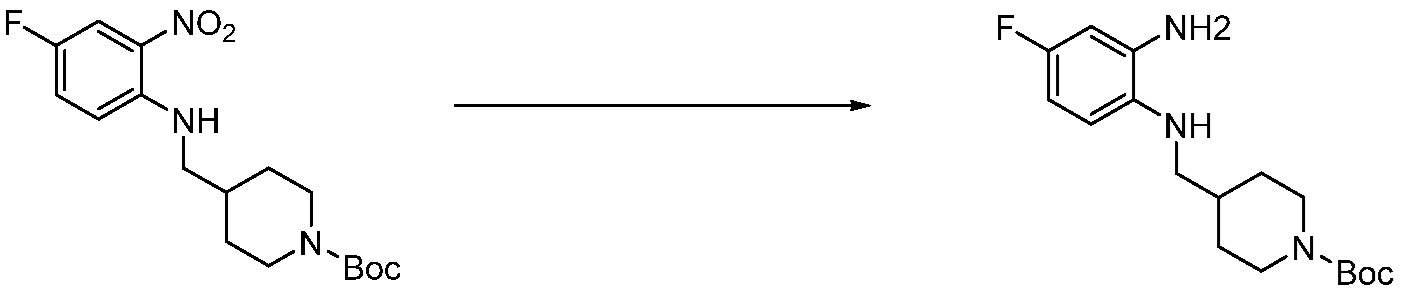
Трет-бутил 4-(((4-фтор-2-нитрофенил)амино)метил)пиперидин-1-карбоксилат (0,484 г, 1,370 ммоль), полученный на стадии 1 растворяют в метанол (13 мл) и перемешивают при комнатной температуре, после чего 10%-Pd/C (50 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (0,426 г, 96,2%, коричневое твердое вещество).
[Стадия 3] Синтез трет-бутил 4-((5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилата
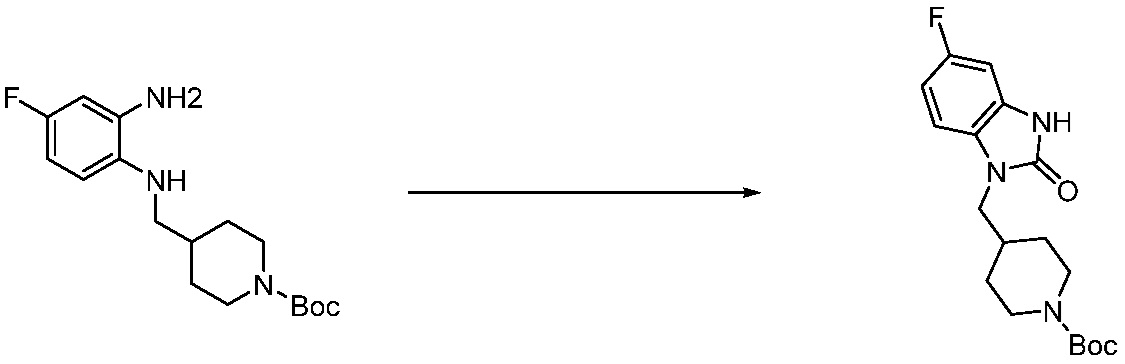
Трет-бутил 4-(((2-амино-4-фторфенил)амино)метил)пиперидин-1-карбоксилат (0,426 г, 1,317 ммоль), полученный на стадии 2, триэтиламин (0,184 мл, 1,317 ммоль) и 1,1'-карбонилдиимидазол (КДИ, 0,256 г, 1,581 ммоль) растворяют в дихлорметане (7 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,154 г, 33,5%) в виде коричневого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-((3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилата
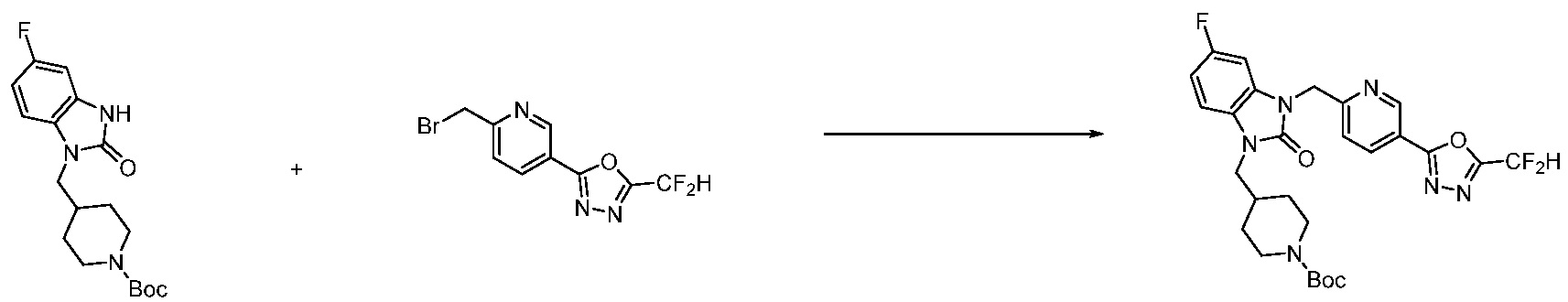
Трет-бутил 4-((5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилат (0,154 г, 0,441 ммоль), полученный на стадии 3, гидрид натрия (60,00%, 0,026 г, 0,661 ммоль) и 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,141 г, 0,485 ммоль) растворяют в N,N-диметилформамиде (3 мл), после чего полученный раствор перемешивают при 0° в течение 10 минут и дополнительно перемешивают при комнатной температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,150 г, 60,9%) в виде белого твердого вещества.
[Стадия 5] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-илметил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
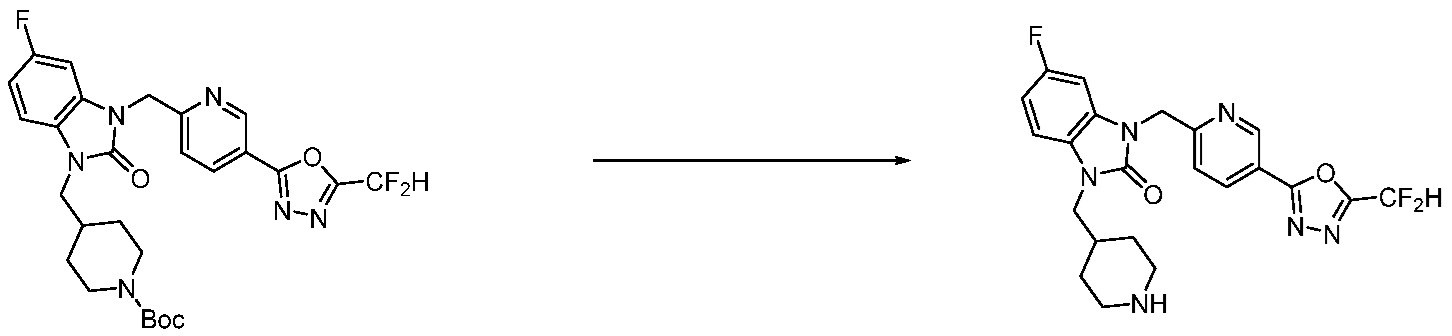
Трет-бутил 4-((3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)метил)пиперидин-1-карбоксилат (0,150 г, 0,269 ммоль), полученный на стадии 4, и трифторуксусную кислоту (0,206 мл, 2,685 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,120 г, 97,5%, коричневое твердое вещество).
[Стадия 6] Синтез соединения 301
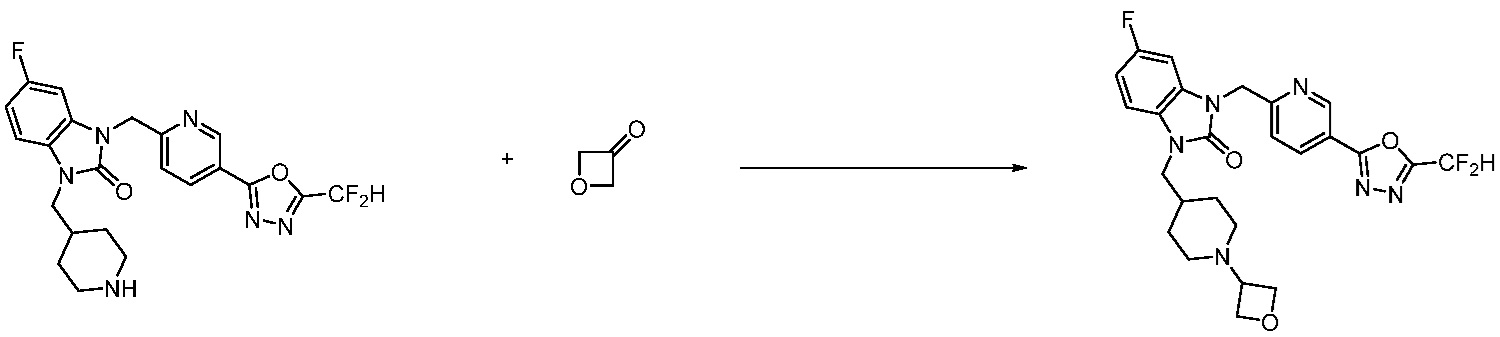
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(пиперидин-4-илметил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,120 г, 0,262 ммоль), полученный на стадии 5, оксетан-3-он (0,057 г, 0,785 ммоль) и триацетоксиборгидрид натрия (0,111 г, 0,524 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,087 г, 64,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,23 (дд, J=2,1, 0,6 Гц, 1H), 8,31 (дд, J=8,2, 2,2 Гц, 1H), 7,40 (д, J=8,2 Гц, 1H), 7,04-6,71 (м, 4H), 5,22 (с, 2H), 4,58 (тд, J=12,7, 6,3 Гц, 4H), 3,44-3,37 (м, 1H), 2,72 (д, J=11,4 Гц, 2H), 1,93-1,86 (м, 1H), 1,79-1,68 (м, 4H), 1,49-1,39 (м, 2H); МСНР (ЭР) m/z 515,5 (M+ + 1).
Пример 302: Синтез соединения 302, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1-(1-(оксетан-3-ил)азетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 3-((4-фтор-2-нитрофенил)амино)азетидин-1-карбоксилата
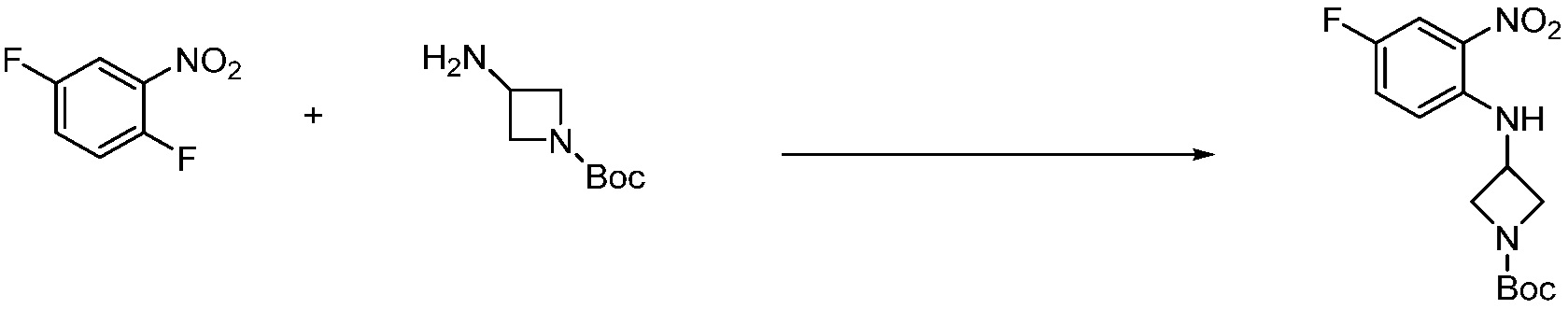
1,4-дифтор-2-нитробензол (0,300 г, 1,886 ммоль), трет-бутил 3-аминоазетидин-1-карбоксилат (0,422 г, 2,451 ммоль), карбонат калия (0,521 г, 3,771 ммоль) и йодид калия (0,031 г, 0,189 ммоль) растворяют в N,N-диметилформамиде (7 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,571 г, 97,3%) в виде оранжевого твердого вещества.
[Стадия 2] Синтез трет-бутил 3-((2-амино-4-фторфенил)амино)азетидин-1-карбоксилата

Трет-бутил 3-((4-фтор-2-нитрофенил)амино)азетидин-1-карбоксилат (0,571 г, 1,834 ммоль), полученный на стадии 1, растворяют в метаноле (16 мл) и перемешивают при комнатной температуре, после чего 10%-Pd/C (50 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (0,532 г, 103,2%, коричневое твердое вещество).
[Стадия 3] Синтез трет-бутил 3-(5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилата

Трет-бутил 3-((2-амино-4-фторфенил)амино)азетидин-1-карбоксилат (0,532 г, 1,891 ммоль), полученный на стадии 2, триэтиламин (0,264 мл, 1,891 ммоль) и 1,1'-карбонилдиимидазол (КДИ, 0,368 г, 2,269 ммоль) растворяют в дихлорметане (8 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,222 г, 38,3%) в виде коричневого твердого вещества.
[Стадия 4] Synthesis 1-(азетидин-3-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-она
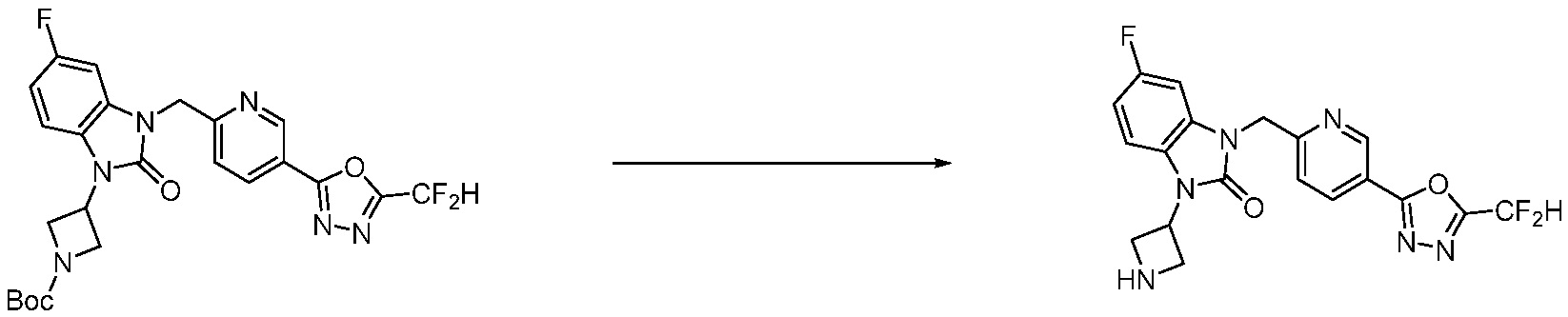
Трет-бутил 3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилат (0,320 г, 0,620 ммоль), полученный на стадии 3, и трифторуксусную кислоту (0,474 мл, 6,196 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,250 г, 96,9%, коричневое твердое вещество).
[Стадия 5] Синтез 1-(азетидин-3-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-она
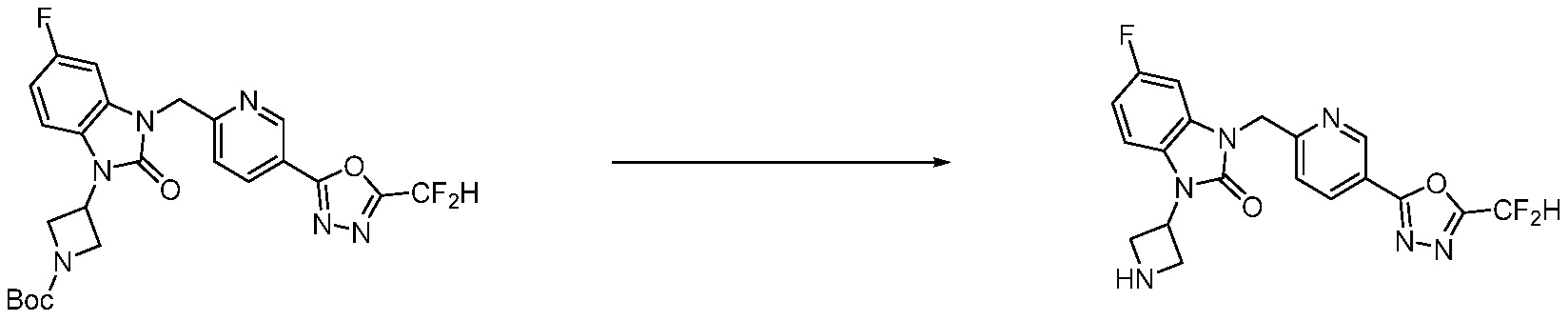
Трет-бутил 3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-1-ил)азетидин-1-карбоксилат (0,320 г, 0,620 ммоль), полученный на стадии 4 и трифторуксусную кислоту (0,474 мл, 6,196 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,250 г, 96,9%, коричневое твердое вещество).
[Стадия 6] Синтез соединения 302

1-(азетидин-3-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,250 г, 0,600 ммоль), полученный на стадии 5, оксетан-3-он (0,130 г, 1,801 ммоль) и триацетоксиборгидрид натрия (0,255 г, 1,201 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,038 г, 13,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=1,6 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,60 (дд, J=8,7, 4,4 Гц, 1H), 7,43 (д, J=8,2 Гц, 1H), 7,05-6,76 (м, 3H), 5,22 (с, 2H), 5,14-5,07 (м, 1H), 4,76 (т, J=6,7 Гц, 2H), 4,60 (дд, J=6,6, 5,3 Гц, 2H), 3,98-3,90 (м, 3H), 3,82 (т, J=8,3 Гц, 2H); МСНР (ЭР) m/z 473,5 (M+ + 1).
Пример 303: Синтез соединения 303, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(4-(пиридин-3-илметил)пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 303

1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,218 ммоль), полученный на стадии 4 соединения 147, никотинальдегид (0,070 г, 0,653 ммоль) и триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,062 г, 51,5%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,23 (дд, J=2,1, 0,7 Гц, 1H), 8,54 (д, J=1,6 Гц, 1H), 8,47 (дд, J=4,8, 1,6 Гц, 1H), 8,28 (дд, J=8,2, 2,2 Гц, 1H), 7,66 (тд, J=4,8, 2,6 Гц, 1H), 7,38 (дд, J=8,2, 0,4 Гц, 1H), 7,26-7,22 (м, 1H), 6,91 (т, J=51,6 Гц, 1H), 6,72 (д, J=11,2 Гц, 1H), 6,62 (д, J=7,0 Гц, 1H), 5,18 (с, 2H), 3,56 (с, 2H), 3,41 (с, 3H), 3,03 (т, J=4,4 Гц, 4H), 2,70-2,50 (м, 4H); МСНР (ЭР) m/z 551,4 (M+ + 1).
Пример 304: Синтез соединения 304, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(пиридин-3-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез 6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]оксазол-2(3H)-она
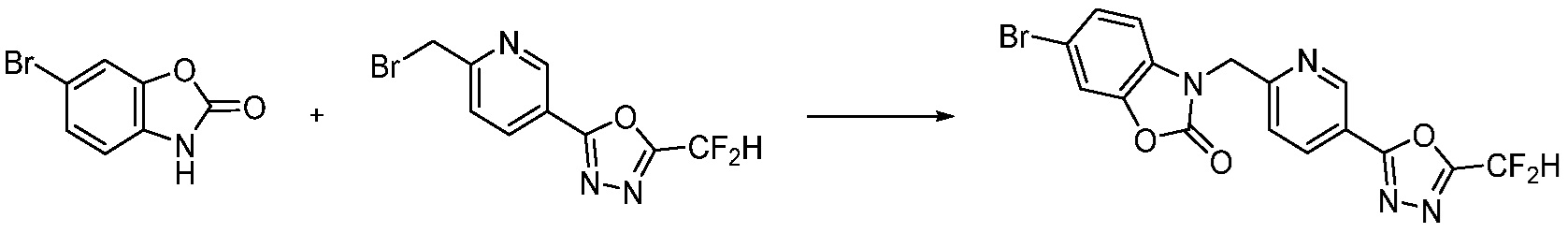
6-бромбензо[d]оксазол-2(3H)-он (1,000 г, 4,672 ммоль), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,491 г, 5,140 ммоль), карбонат калия (0,969 г, 7,009 ммоль) и йодид калия (0,078 г, 0,467 ммоль) растворяют в N,N-диметилформамиде (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Метанол добавляют в полученный концентрат и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют, затем промывают метанол, и затем сушат с получением указанного в заголовке соединения (1,250 г, 63,2%) в виде светло-желтого твердого вещества.
[Стадия 2] Синтез соединения 304
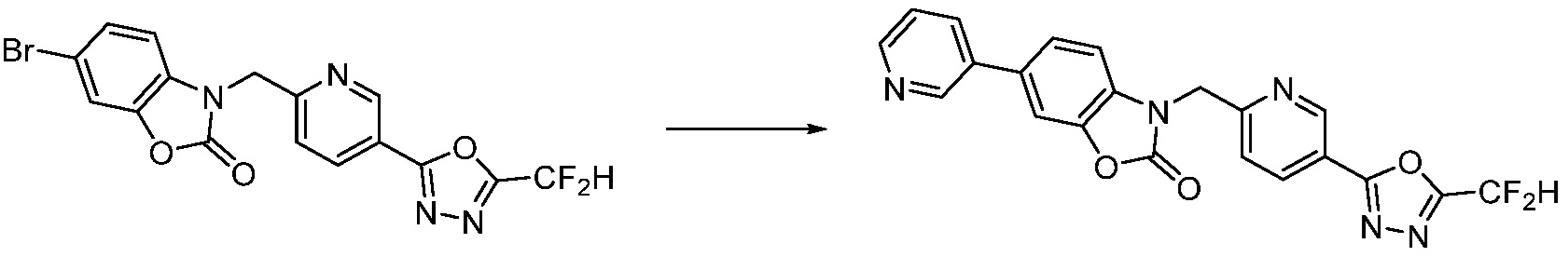
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,236 ммоль), полученный на стадии 1, пиридин-3-илбороновую кислоту (0,035 г, 0,284 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,008 г, 0,012 ммоль) и карбонат цезия (0,231 г, 0,709 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-40%) и концентрируют с получением указанного в заголовке соединения (0,008 г, 8,0%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,4 Гц, 1H), 8,82 (д, J=1,6 Гц, 1H), 8,72 (дд, J=75,5, 1,1 Гц, 1H), 8,43 (дд, J=8,2, 2,2 Гц, 1H), 7,86-7,83 (м, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,49 (д, J=1,4 Гц, 1H), 7,41-7,38 (м, 2H), 7,14 (д, J=8,1 Гц, 1H), 7,08-6,83 (м, 1H), 5,19 (с, 2H); МСНР (ЭР) m/z 422,4 (M+ + 1).
Пример 305: Синтез соединения 305, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(пиридин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 305
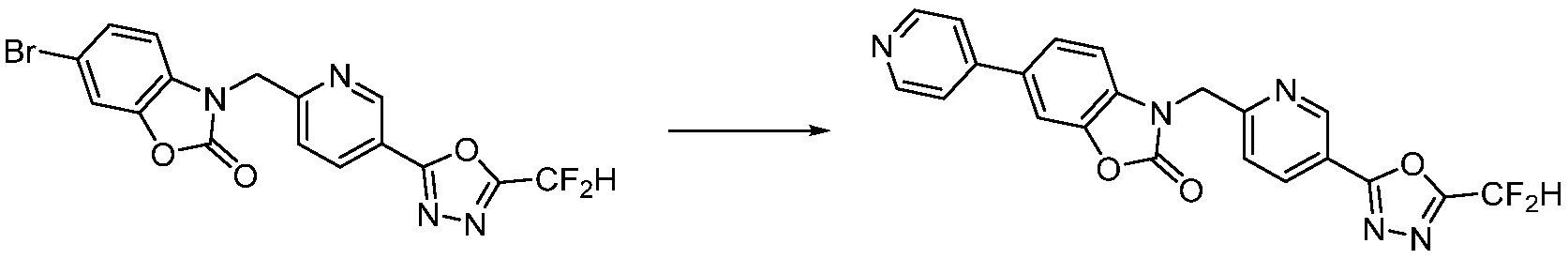
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,236 ммоль), пиридин-4-илбороновую кислоту (0,035 г, 0,284 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,008 г, 0,012 ммоль) и карбонат цезия (0,231 г, 0,709 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-40%) и концентрируют с получением указанного в заголовке соединения (0,012 г, 12,1%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=2,1 Гц, 1H), 8,68 (д, J=6,2 Гц, 2H), 8,43 (дд, J=8,2, 2,2 Гц, 1H), 7,60 (д, J=8,2 Гц, 1H), 7,55 (д, J=1,4 Гц, 1H), 7,49-7,46 (м, 3H), 7,16 (д, J=8,2 Гц, 1H), 7,08-6,83 (м, 1H), 5,28 (с, 2H); МСНР (ЭР) m/z 422,4 (M+ + 1).
Пример 306: Синтез соединения 306, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1H-индол-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 306
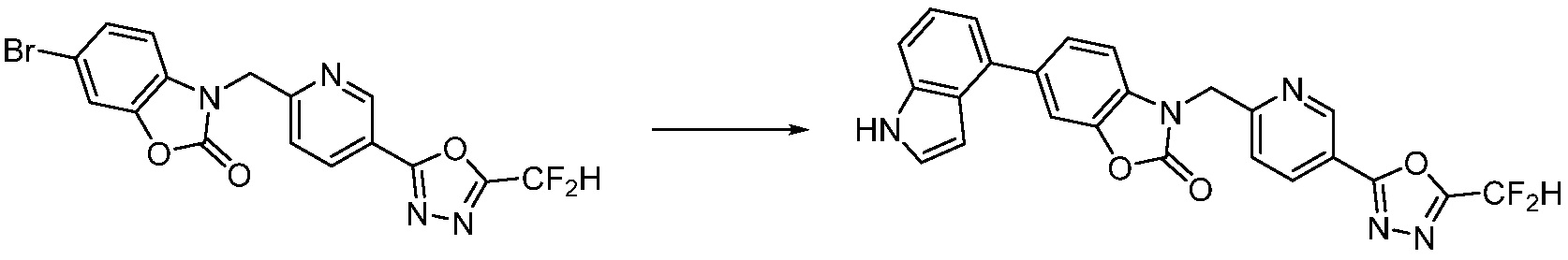
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,236 ммоль), (1H-индол-4-ил)бороновую кислоту (0,046 г, 0,284 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,008 г, 0,012 ммоль) и карбонат цезия (0,231 г, 0,709 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=5-20%) и концентрируют с получением указанного в заголовке соединения (0,007 г, 6,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,35 (д, J=1,6 Гц, 1H), 8,43 (дд, J=8,2, 2,2 Гц, 1H), 4,38 (шс, 1H), 7,62 (д, J=1,4 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,51 (дд, J=8,1, 1,5 Гц, 1H), 7,43 (д, J=8,1 Гц, 1H), 7,31-7,27 (м, 3H), 7,16 (д, J=7,3 Гц, 1H), 7,11-6,83 (м, 2H), 6,70-6,68 (м, 1H), 5,30 (с, 2H); МСНР (ЭР) m/z 460,5 (M+ + 1).
Пример 307: Синтез соединения 307, трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиперидин-1-карбоксилат
[Стадия 1] Синтез трет-бутил 4-(2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиперидин-1-карбоксилата
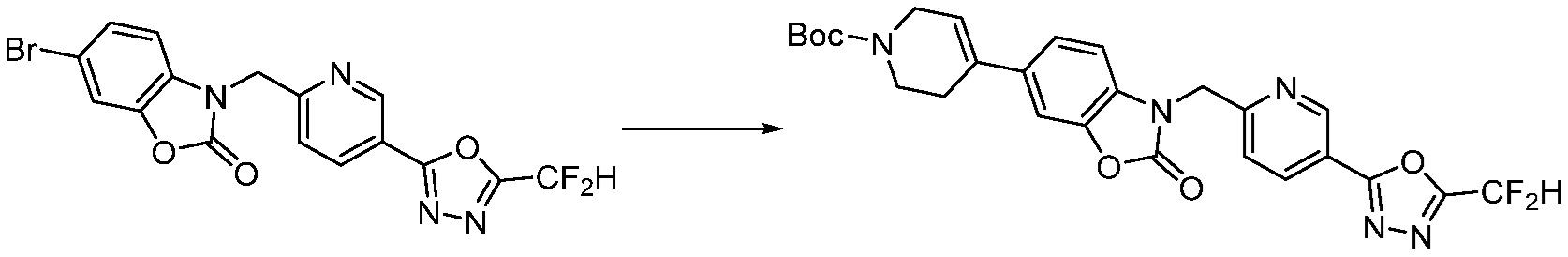
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]оксазол-2(3H)-он (0,800 г, 1,890 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,701 г, 2,269 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,062 г, 0,095 ммоль) и карбонат цезия (1,848 г, 5,671 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-30%) и концентрируют с получением указанного в заголовке соединения (0,286 г, 28,8%) в виде желтого твердого вещества.
[Стадия 2] Синтез соединения 307
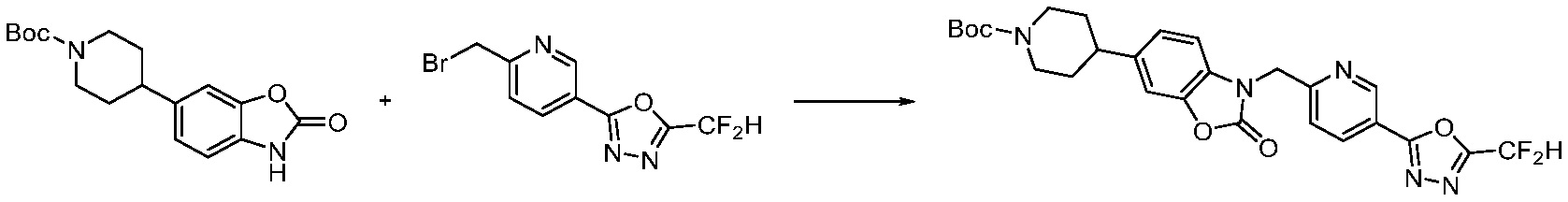
Трет-бутил 4-(2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиперидин-1-карбоксилат (0,390 г, 1,225 ммоль), полученный на стадии 1, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,391 г, 1,347 ммоль), карбонат калия (0,254 г, 1,837 ммоль) и йодид калия (0,020 г, 0,122 ммоль) растворяют в N,N-диметилформамиде (8 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-40%) и концентрируют с получением указанного в заголовке соединения (0,116 г, 18,0%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=2,2 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,54 (д, J=8,2 Гц, 1H), 7,12 (д, J=1,4 Гц, 1H), 7,08-6,82 (м, 3H), 5,22 (с, 2H), 4,26-4,25 (м, 2H), 2,81 (т, J=12,0 Гц, 2H), 2,72-2,65 (м, 1H), 1,82 (д, J=12,9 Гц, 2H), 1,65-1,58 (м, 2H), 1,50 (с, 9H); МСНР (ЭР) m/z 428,4 (M+ + 1).
Пример 308: Синтез соединения 308, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(пиперидин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 308
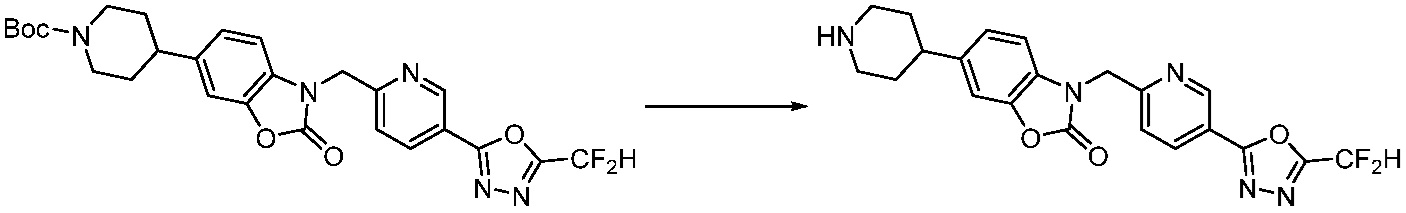
Трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиперидин-1-карбоксилат (0,116 г, 0,220 ммоль) и трифторуксусную кислоту (0,118 мл, 1,539 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение получают без дополнительного процесса очистки (0,093 г, 99,0%, желтое твердое вещество).
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=2,3 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,53 (д, J=8,2 Гц, 1H), 7,15 (д, J=1,4 Гц, 1H), 7,08-6,82 (м, 3H), 5,22 (с, 2H), 3,24 (д, J=12,0 Гц, 2H), 2,78 (тд, J=12,2, 2,3 Гц, 2H), 2,70-2,6388 (м, 1H), 1,84-1,83 (м, 2H), 1,72-1,64 (м, 2H); МСНР (ЭР) m/z 428,5 (M+ + 1).
Пример 309: Синтез соединения 309, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-метилпиперидин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 309
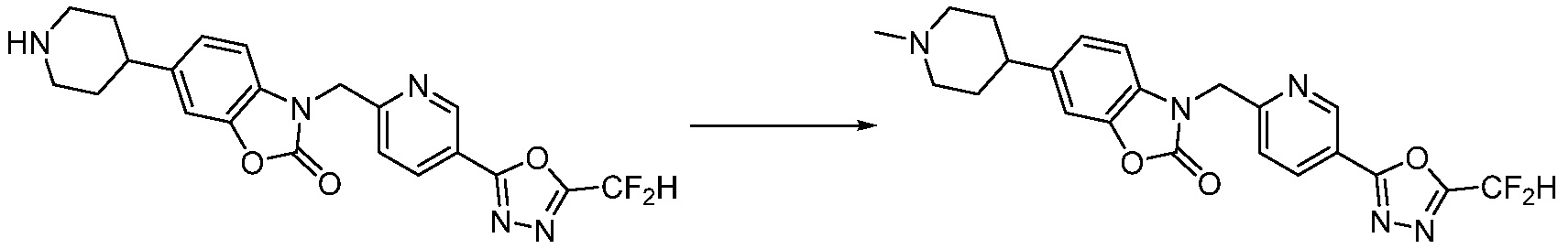
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(пиперидин-4-ил)бензо[d]оксазол-2(3H)-он (0,090 г, 0,211 ммоль) и формальдегид (37,00% раствор, 0,024 мл, 0,316 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,089 г, 0,421 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,026 г, 28,0%) в виде светло-желтого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (д, J=1,4 Гц, 1H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 7,52 (д, J=8,2 Гц, 1H), 7,14 (д, J=1,4 Гц, 1H), 7,08-6,82 (м, 3H), 5,21 (с, 2H), 2,98 (д, J=11,6 Гц, 2H), 2,53-2,48 (м, 1H), 2,32 (с, 3H), 1,84-1,75 (м, 2H), 1,47-1,25 (м, 4H); МСНР (ЭР) m/z 442,3 (M+ + 1).
Пример 310: Синтез соединения 310, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(1H-индол-4-ил)-3-(1-(оксетан-3-ил)азетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез 5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)азетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
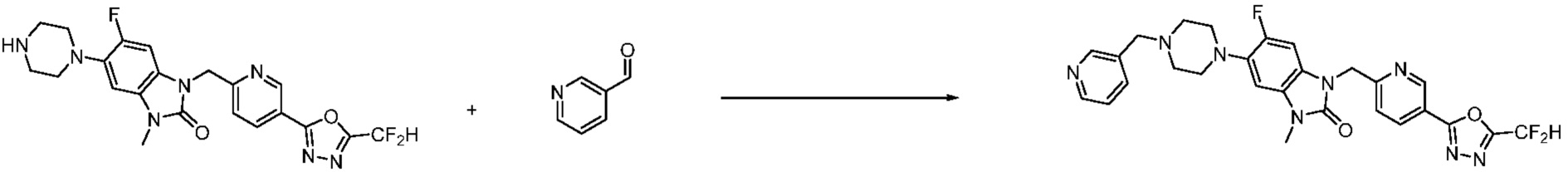
1-(азетидин-3-ил)-6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он (2,390 г, 4,826 ммоль), полученный на стадии 5 соединения 257, оксетан-3-он (0,695 г, 9,651 ммоль) и триацетоксиборгидрид натрия (2,046 г, 9,651 ммоль) растворяют в дихлорметане (45 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,530 г, 19,9%) в виде белого твердого вещества.
[Стадия 2] Синтез соединения 310
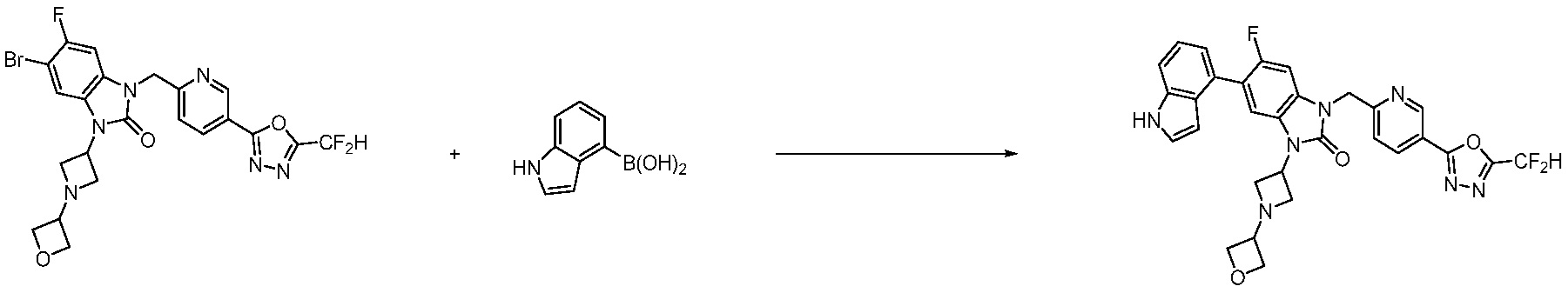
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)азетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,272 ммоль), полученный на стадии 1, (1H-индол-4-ил)бороновую кислоту (0,053 г, 0,326 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) и карбонат цезия (0,177 г, 0,544 ммоль) смешивают в 1,4-диоксане (2 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,115 г, 71,7%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,7 Гц, 1H), 8,78 (с, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,79 (д, J=6,0 Гц, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,38 (д, J=8,0 Гц, 1H), 7,26-7,16 (м, 3H), 6,92 (т, J=51,6 Гц, 1H), 6,91 (д, J=8,8 Гц, 1H), 6,46 (с, 1H), 5,27 (с, 2H), 5,14-5,07 (м, 1H), 4,67 (т, J=6,7 Гц, 2H), 4,56 (т, J=6,0 Гц, 2H), 3,93-3,87 (м, 3H), 3,82 (т, J=8,0 Гц, 2H); МСНР (ЭР) m/z 588,7 (M+ + 1).
Пример 311: Синтез соединения 311, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)азетидин-3-ил)-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 311
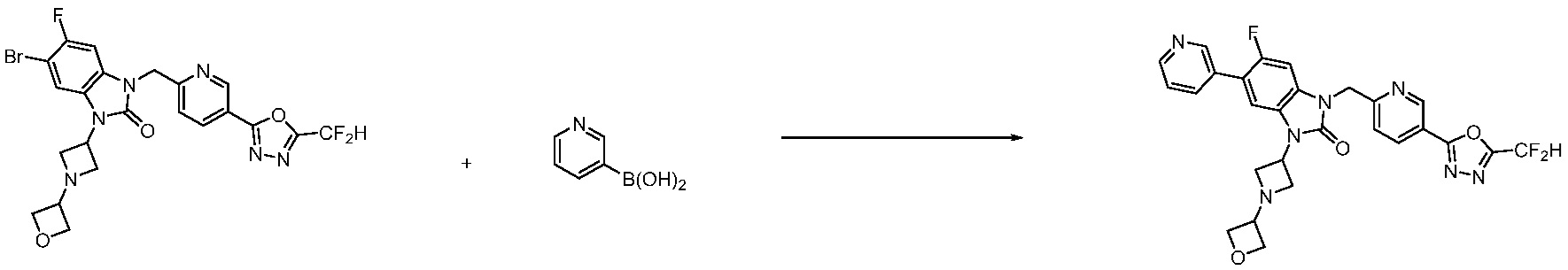
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)азетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,272 ммоль), полученный на стадии 1 соединения 310, пиридин-3-илбороновую кислоту (0,040 г, 0,326 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,007 г, 0,014 ммоль) и карбонат цезия (0,177 г, 0,544 ммоль) смешивают в 1,4-диоксане (2 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,087 г, 58,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (д, J=1,6 Гц, 1H), 8,73 (с, 1H), 8,54 (дд, J=4,7, 1,2 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,82 (дд, J=7,9, 1,6 Гц, 1H), 7,71 (д, J=6,3 Гц, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,34 (дд, J=7,8, 4,9 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,90 (д, J=10,3 Гц, 1H), 5,23 (с, 2H), 5,12-5,05 (м, 1H), 4,71 (т, J=6,7 Гц, 2H), 4,56 (т, J=6,0 Гц, 2H), 3,95-3,89 (м, 3H), 3,82 (т, J=8,1 Гц, 2H); МСНР (ЭР) m/z 550,6 (M+ + 1).
Пример 312: Синтез соединения 312, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)азетидин-3-ил)-5-(1H-пиразол-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 312

5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(1-(оксетан-3-ил)азетидин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,272 ммоль), полученный на стадии 1 соединения 310, (1H-пиразол-4-ил)бороновую кислоту (0,037 г, 0,326 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (0,009 г, 0,014 ммоль) и карбонат цезия (0,177 г, 0,544 ммоль) смешивают в 1,4-диоксане (2 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,074 г, 50,2%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CH3OD) δ 9,19 (д, J=1,6 Гц, 1H), 8,46 (дд, J=8,2, 2,2 Гц, 1H), 8,01 (с, 2H), 7,88 (д, J=6,2 Гц, 1H), 7,60 (д, J=8,1 Гц, 1H), 7,22 (т, J=51,6 Гц, 1H), 6,99 (д, J=10,6 Гц, 1H), 5,31 (с, 2H), 5,23-5,15 (м, 1H), 4,90-4,79 (м, 2H), 4,65 (дд, J=6,9, 4,9 Гц, 2H), 4,18 (т, J=7,7 Гц, 2H), 4,13-4,08 (м, 1H), 3,96 (т, J=8,3 Гц, 2H); МСНР (ЭР) m/z 539,5 (M+ + 1).
Пример 313: Синтез соединения 313, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-изопропилпиперидин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 313
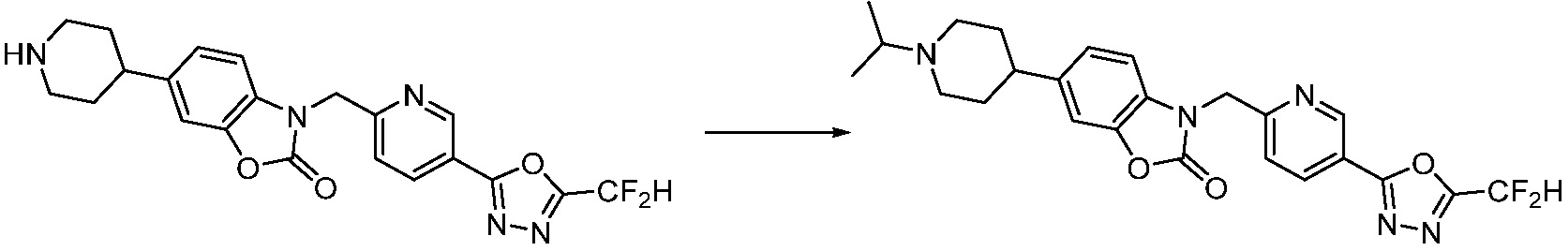
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(пиперидин-4-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,234 ммоль) и ацетон (0,020 г, 0,351 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего в полученный раствор добавляют триацетоксиборгидрид натрия (0,099 г, 0,468 ммоль) и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,036 г, 32,8%) в виде оранжевого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (д, J=1,4 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,52 (д, J=8,2 Гц, 1H), 7,15 (д, J=1,4 Гц, 1H), 7,08-6,82 (м, 3H), 5,21 (с, 2H), 3,16 (д, J=11,5 Гц, 2H), 3,03-2,99 (м, 1H), 2,60-2,54 (м, 1H), 2,40 (тд, J=11,5, 3,4 Гц, 2H), 2,03-1,87 (м, 4H), 1,16 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 470,5 (M+ + 1).
Пример 314: Синтез соединения 314, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(оксетан-3-ил)пиперидин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 314
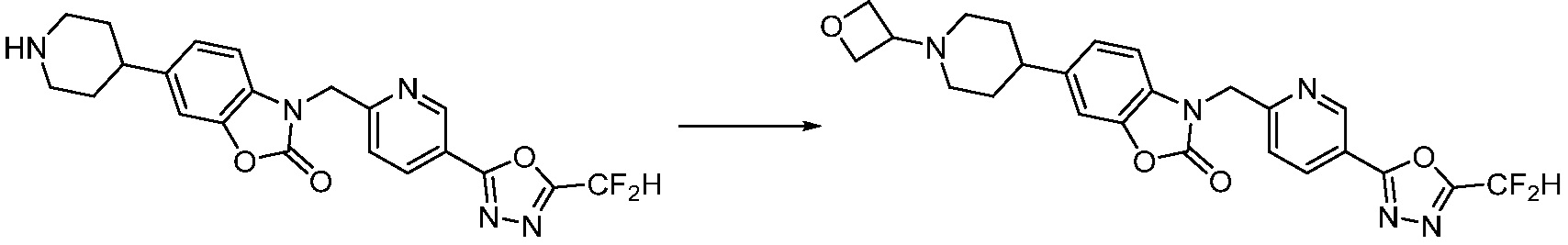
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(пиперидин-4-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,234 ммоль) и оксетан-3-он (0,025 г, 0,351 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,099 г, 0,468 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 4 г картридж; этилацетат/1%-гексан aqueous раствор=70-100%) и концентрируют с получением указанного в заголовке соединения (0,062 г, 54,8%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (д, J=1,5 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,53 (д, J=8,2 Гц, 1H), 7,15 (д, J=1,4 Гц, 1H), 7,08-6,82 (м, 3H), 5,21 (с, 2H), 4,70-4,64 (м, 4H), 3,53-3,50 (м, 1H), 2,88 (д, J=11,3 Гц, 2H), 2,58-2,52 (м, 1H), 1,97-1,77 (м, 6H); МСНР (ЭР) m/z 484,5 (M+ + 1).
Пример 315: Синтез соединения 315, 6-(1-циклобутилпиперидин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 315
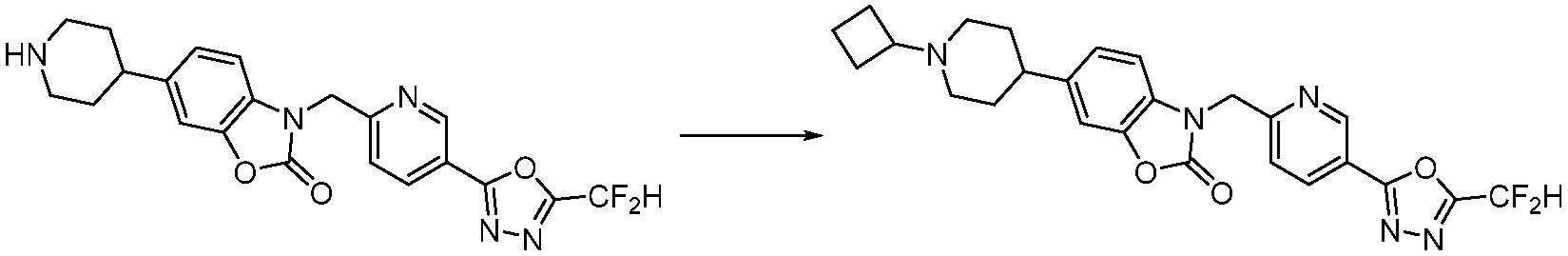
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(пиперидин-4-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,234 ммоль) и циклобутанон (0,025 г, 0,351 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,099 г, 0,468 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,025 г, 22,2%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,4 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,53 (д, J=8,2 Гц, 1H), 7,14 (д, J=1,4 Гц, 1H), 7,08-6,82 (м, 3H), 5,22 (с, 2H), 3,27 (д, J=10,9 Гц, 2H), 3,01-2,97 (м, 1H), 2,65-2,57 (м, 2H), 2,26-2,22 (м, 2H), 2,18-2,10 (м, 3H), 2,02-1,98 (м, 2H), 1,91-1,88 (м, 2H), 1,79-1,77 (м, 1H), 1,76-1,71 (м, 1H); МСНР (ЭР) m/z 482,6 (M+ + 1).
Пример 316: Синтез соединения 316, трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиперазин-1-карбоксилат
[Стадия 1] Синтез 4,5-дифтор-2-нитрофенола
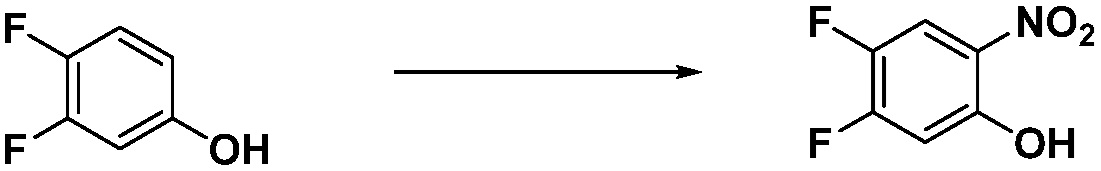
3,4-дифторфенол (7,160 г, 55,039 ммоль) и нитрат железа (III)-9H2O (22,236 г, 55,039 ммоль) смешивают в этаноле (50 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 16 часов и охлаждают до комнатной температуры. Затем, растворитель удаляют из реакционной смеси при пониженном давлении, после чего этилацетат (20 мл) и гексан (10 мл) добавляют в полученный концентрат и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают гексан, и затем сушат с получением указанного в заголовке соединения (7,850 г, 81,5%) в виде желтого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(2-фтор-5-гидрокси-4-нитрофенил)пиперазин-1-карбоксилата
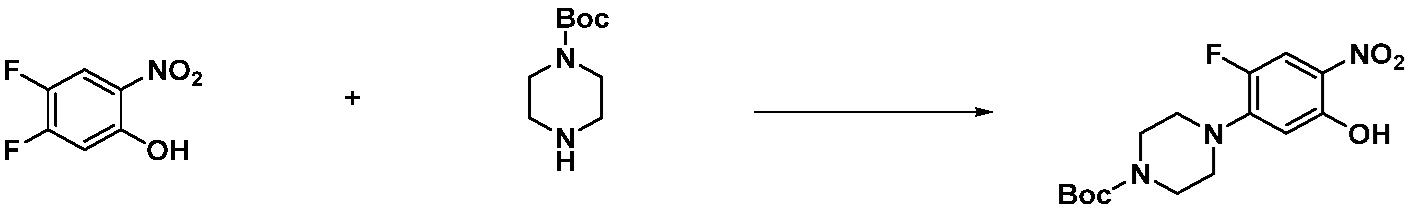
4,5-дифтор-2-нитрофенол (7,850 г, 44,834 ммоль), полученный на стадии 1, трет-бутил пиперазин-1-карбоксилат (16,702 г, 89,668 ммоль) и карбонат калия (12,393 г, 89,668 ммоль) смешивают в толуоле (50 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 18 часов и охлаждают до комнатной температуры. Затем, в реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Этилацетат (20 мл) и гексан (10 мл) добавляют в полученный концентрат и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают гексан, и затем сушат с получением указанного в заголовке соединения (8,300 г, 54,2%) в виде желтого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(4-амино-2-фтор-5-гидроксифенил)пиперазин-1-карбоксилата
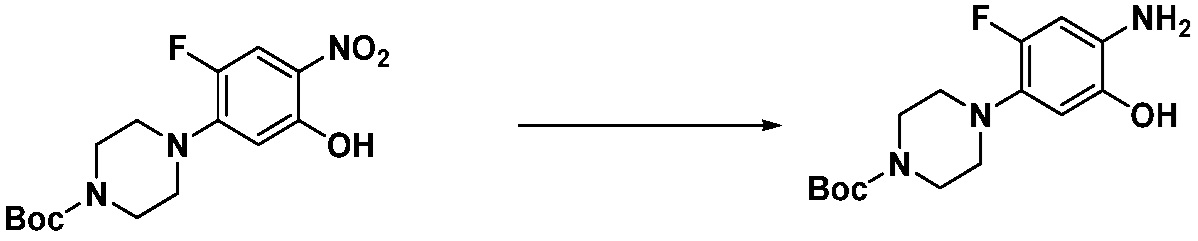
Трет-бутил 4-(2-фтор-5-гидрокси-4-нитрофенил)пиперазин-1-карбоксилат (0,780 г, 2,285 ммоль), полученный на стадии 2 растворяют в этанол (50 мл) при комнатной температуре, после чего никель Ренея медленно добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (0,500 г, 70,3%, белое твердое вещество).
[Стадия 4] Синтез трет-бутил 4-(5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиперазин-1-карбоксилата
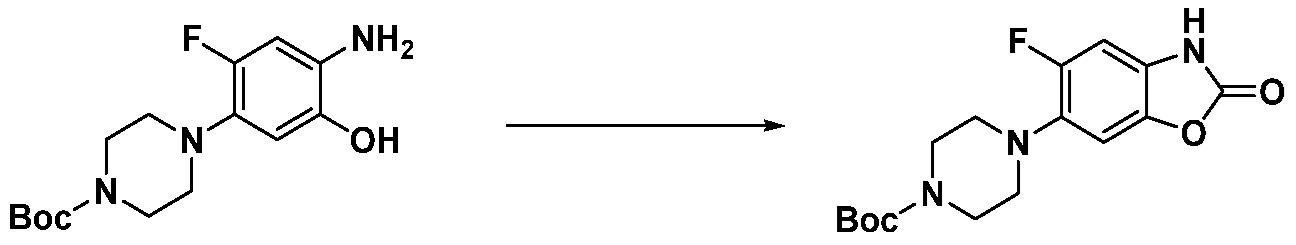
Трет-бутил 4-(4-амино-2-фтор-5-гидроксифенил)пиперазин-1-карбоксилат (0,500 г, 1,606 ммоль), полученный на стадии 3, и 1,1'-карбонилдиимидазол (КДИ, 0,391 г, 2,409 ммоль) растворяют в тетрагидрофуране (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,205 г, 37,8%) в виде белого твердого вещества.
[Стадия 5] Синтез соединения 316
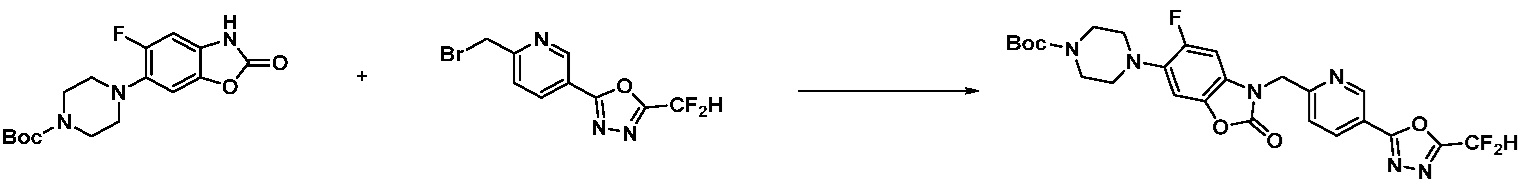
Трет-бутил 4-(5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиперазин-1-карбоксилат (0,205 г, 0,608 ммоль), полученный на стадии 4 растворяют в N,N-диметилформамиде (10 мл) при 0°C, после чего гидрид натрия (60,00%, 0,028 г, 0,699 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,212 г, 0,729 ммоль) добавляют в реакционную смесь и дополнительно перемешивают при комнатной температуре в течение 2 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,200 г, 60,2%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (дд, J=2,2, 0,7 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,54 (дд, J=8,2, 0,7 Гц, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,92 (д, J=6,8 Гц, 1H), 6,82 (д, J=10,6 Гц, 1H), 6,82 (с, 0,25H), 5,16 (с, 2H), 3,59 (т, J=5,0 Гц, 4H), 2,96 (т, J=4,8 Гц, 4H), 1,48 (с, 9H).
Пример 317: Синтез соединения 317, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(4-метилпиперазин-1-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(пиперазин-1-ил)бензо[d]оксазол-2(3H)-она
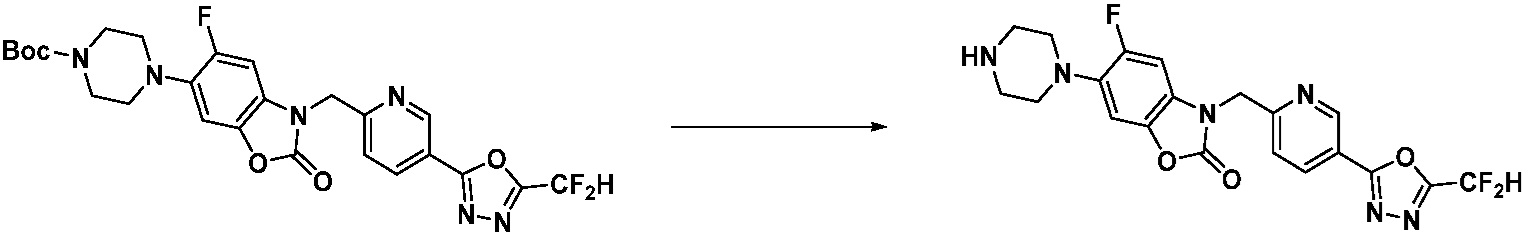
Трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиперазин-1-карбоксилат (0,300 г, 0,549 ммоль) и трифторуксусную кислоту (0,420 мл, 5,489 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,210 г, 85,7%, желтое твердое вещество).
[Стадия 2] Синтез соединения 317
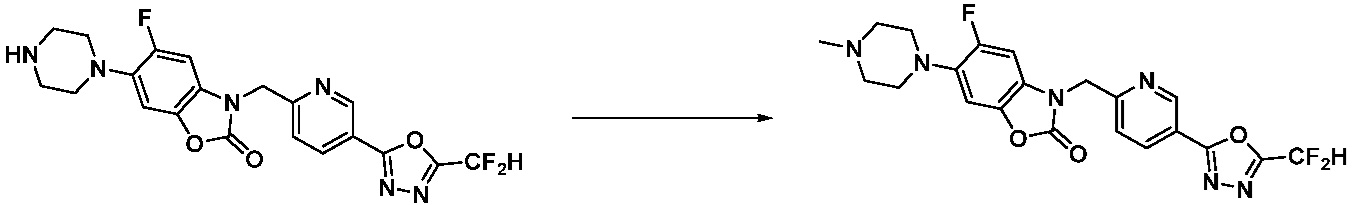
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(пиперазин-1-ил)бензо[d]оксазол-2(3H)-он (0,110 г, 0,246 ммоль), полученный на стадии 1, формальдегид (37,00% раствор, 0,037 мл, 0,493 ммоль) и триацетоксиборгидрид натрия (0,104 г, 0,493 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,061 г, 53,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (дд, J=2,2, 0,7 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,54 (дд, J=8,2, 0,6 Гц, 1H), 7,08 (с, 0,25H), 6,95 (д, J=6,9 Гц, 1H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 6,80 (д, J=10,7 Гц, 1H), 5,16 (с, 2H), 3,07 (т, J=4,7 Гц, 4H), 2,67 ~ 2,63 (м, 4H), 2,37 (с, 3H).
Пример 318: Синтез соединения 318, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(4-(оксетан-3-ил)пиперазин-1-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 318
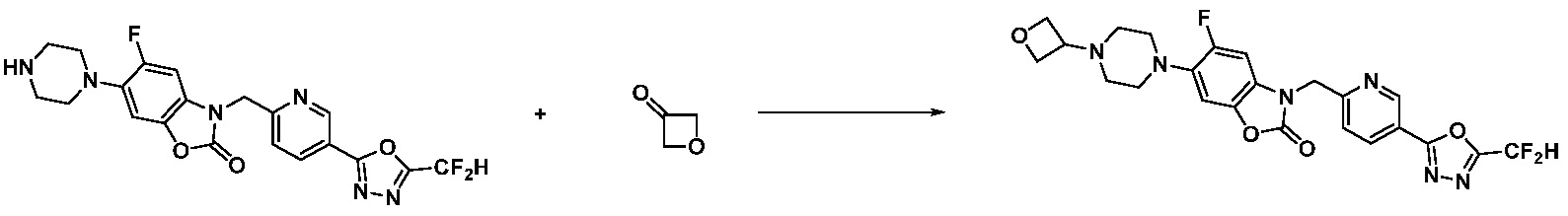
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(пиперазин-1-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,224 ммоль), 3-оксетан (0,026 мл, 0,448 ммоль) и триацетоксиборгидрид натрия (0,095 г, 0,448 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. В реакционную смесь выливают в воду и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,040 г, 35,5%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (дд, J=2,2, 0,7 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,55 (дд, J=8,2, 0,6 Гц, 1H), 7,08 (с, 0,25H), 6,96 ~ 6,95 (м, 1H), 6,95 (с, 0,5H), 6,82 (с, 0,25H), 6,82 ~ 6,80 (м, 1H), 5,17 (с, 2H), 4,73 ~ 4,64 (м, 4H), 3,62 ~ 3,56 (м, 1H), 3,10 ~ 3,08 (м, 4H), 2,55 ~ 2,52 (м, 4H).
Пример 319: Синтез соединения 319, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(4-метилпиперазин-1-ил)-3-(оксетан-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(2-фтор-4-нитро-5-(оксетан-3-иламино)фенил)пиперазин-1-карбоксилата
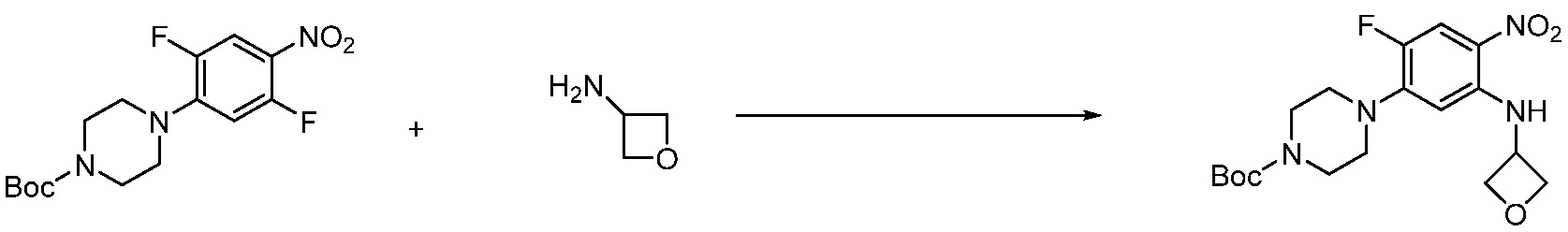
Трет-бутил 4-(2,5-дифтор-4-нитрофенил)пиперазин-1-карбоксилат (4,640 г, 13,515 ммоль), оксетан-3-амин (1,087 г, 14,866 ммоль), карбонат калия (3,736 г, 27,029 ммоль) и йодид калия (0,224 г, 1,351 ммоль) растворяют в N,N-диметилформамиде (60 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (5,520 г, 103,0%, желтое твердое вещество).
[Стадия 2] Синтез трет-бутил 4-(4-амино-2-фтор-5-(оксетан-3-иламино)фенил)пиперазин-1-карбоксилата
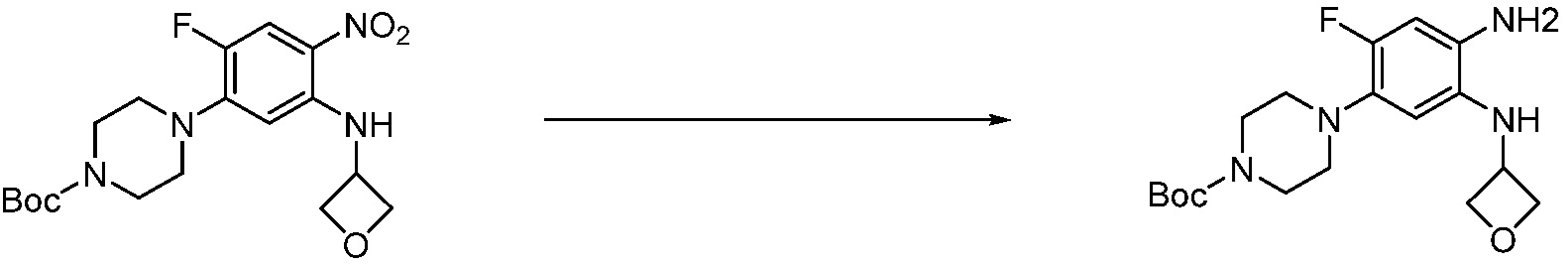
Трет-бутил 4-(2-фтор-4-нитро-5-(оксетан-3-иламино)фенил)пиперазин-1-карбоксилат (5,520 г, 13,925 ммоль), полученный на стадии 1, растворяют в метаноле (60 мл) и перемешивают при комнатной температуре, после чего 10%-Pd/C (550 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (4,870 г, 95,4%, фиолетовое твердое вещество).
[Стадия 3] Синтез трет-бутил 4-(6-фтор-3-(оксетан-3-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилата

Трет-бутил 4-(4-амино-2-фтор-5-(оксетан-3-иламино)фенил)пиперазин-1-карбоксилат (4,870 г, 13,290 ммоль), полученный на стадии 2, триэтиламин (1,852 мл, 13,290 ммоль) и 1,1'-карбонилдиимидазол (КДИ, 2,801 г, 17,277 ммоль) растворяют в дихлорметане (70 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (4,710 г, 90,3%) в виде фиолетового твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилата
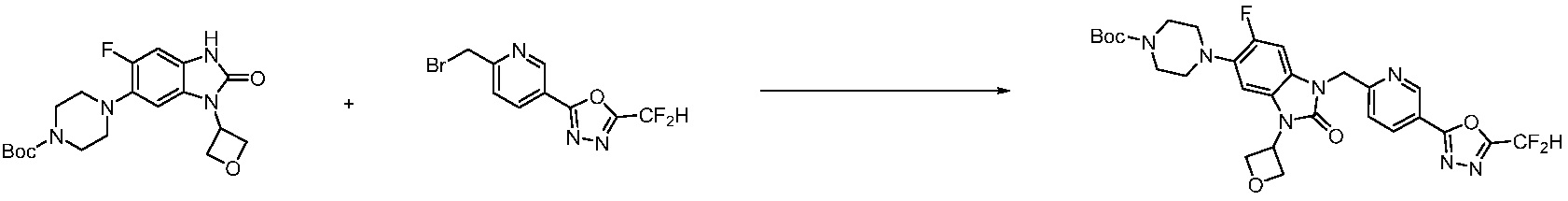
Трет-бутил 4-(6-фтор-3-(оксетан-3-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилат (2,000 г, 5,096 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (25 мл), после чего гидрид натрия (60,00%, 0,306 г, 7,645 ммоль) добавляют в полученный раствор при 0°C, затем перемешивают при той же температуре в течение 30 минут, и затем дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=30-70%) и концентрируют с получением указанного в заголовке соединения (1,960 г, 63,9%) в виде коричневого твердого вещества.
[Стадия 5] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она

Трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилат (1,960 г, 3,258 ммоль), полученный на стадии 4, и трифторуксусную кислоту (2,495 мл, 32,580 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-30%) и концентрируют с получением указанного в заголовке соединения (1,000 г, 61,2%) в виде коричневого твердого вещества.
[Стадия 6] Синтез соединения 319
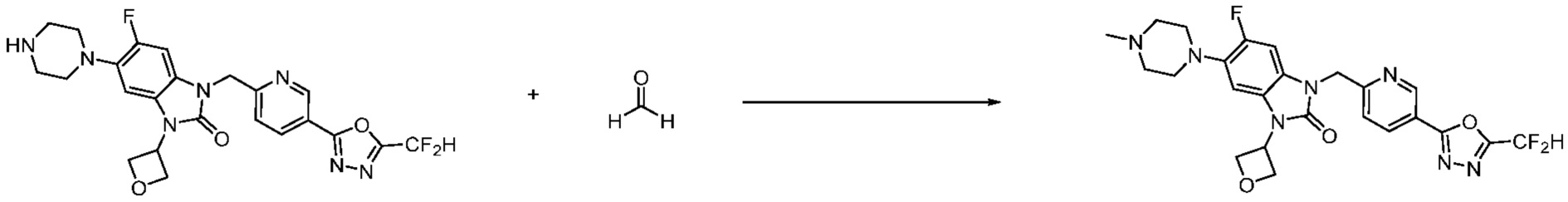
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,199 ммоль), полученный на стадии 5, триацетоксиборгидрид натрия (0,085 г, 0,399 ммоль) и формальдегид (0,018 г, 0,598 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,077 г, 75,0%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,6 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,41 (дд, J=12,0, 7,7 Гц, 2H), 6,92 (т, J=51,6 Гц, 1H), 6,81 (д, J=11,2 Гц, 1H), 5,66-5,60 (м, 1H), 5,19 (с, 2H), 5,14 (д, J=6,7 Гц, 4H), 3,11 (т, J=4,4 Гц, 4H), 2,70-2,50 (м, 4H), 2,36 (с, 3H); МСНР (ЭР) m/z 516,3 (M+ + 1).
Пример 320: Синтез соединения 320, 5-(4-циклобутилпиперазин-1-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 320
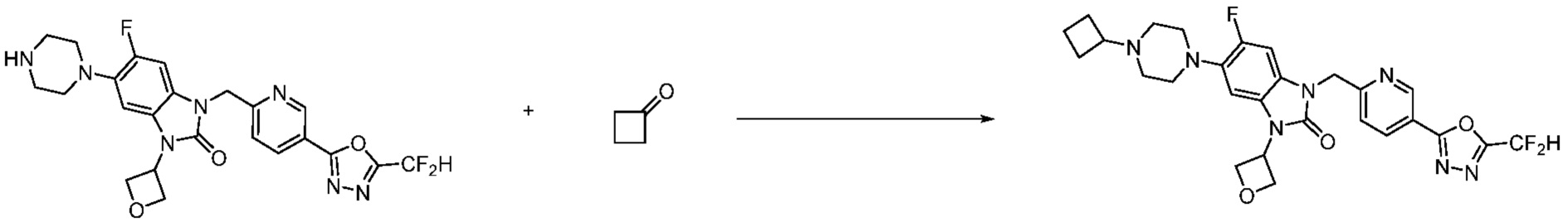
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,199 ммоль), полученный на стадии 5 соединения 319, триацетоксиборгидрид натрия (0,085 г, 0,399 ммоль) и циклобутанон (0,042 г, 0,598 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,073 г, 66,3%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (дд, J=2,1, 0,6 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,42 (д, J=7,5 Гц, 2H), 6,92 (т, J=51,6 Гц, 1H), 6,81 (д, J=11,2 Гц, 1H), 5,66-5,59 (м, 1H), 5,19 (с, 2H), 5,19-5,10 (м, 4H), 3,11 (т, J=4,5 Гц, 4H), 2,87-2,79 (м, 1H), 2,70-2,45 (м, 4H), 2,16-2,02 (м, 2H), 1,98-1,89 (м, 2H), 1,77-1,65 (м, 2H); МСНР (ЭР) m/z 556,3 (M+ + 1).
Пример 321: Синтез соединения 321, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(4-(оксетан-3-ил)пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 321
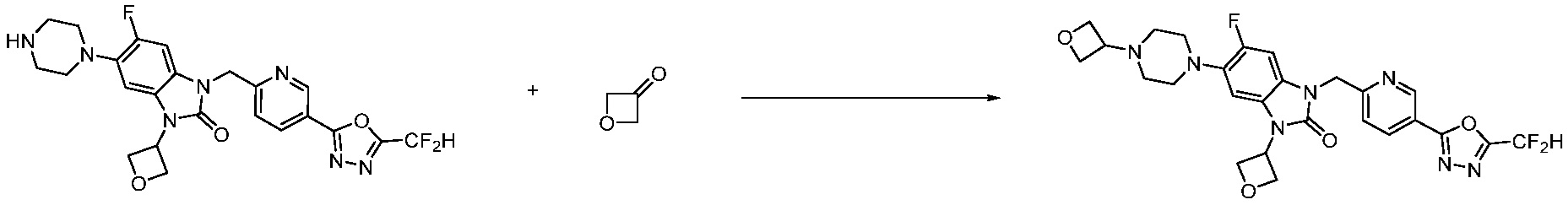
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,199 ммоль), полученный на стадии 5 соединения 319, триацетоксиборгидрид натрия (0,085 г, 0,399 ммоль) и оксетан-3-он (0,043 г, 0,598 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,081 г, 72,5%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=2,1 Гц, 1H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,43 (д, J=7,4 Гц, 2H), 6,92 (т, J=51,6 Гц, 1H), 6,83 (д, J=11,2 Гц, 1H), 5,65-5,60 (м, 1H), 5,19 (с, 2H), 5,14 (тд, J=9,8, 5,3 Гц, 4H), 4,66 (тд, J=12,1, 5,9 Гц, 4H), 3,61-3,55 (м, 1H), 3,13 (т, J=4,6 Гц, 4H), 2,70-2,50 (м, 4H); МСНР (ЭР) m/z 558,3 (M+ + 1).
Пример 322: Синтез соединения 322, 5-(4-((1H-индол-4-ил)метил)пиперазин-1-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 322
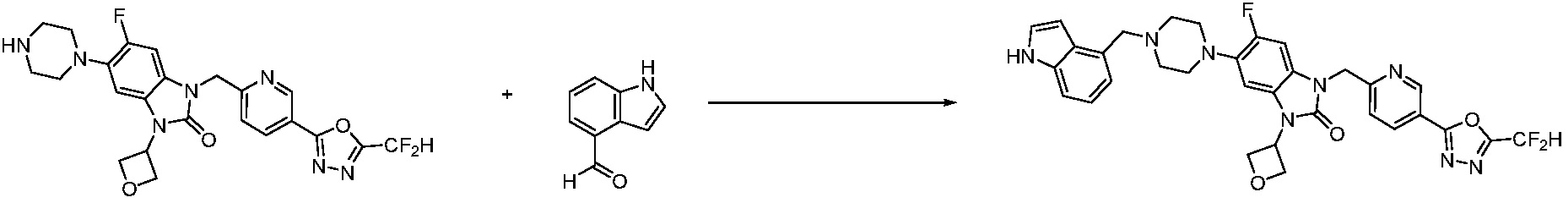
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,199 ммоль), полученный на стадии 5 соединения 319, триацетоксиборгидрид натрия (0,085 г, 0,399 ммоль) и 1H-индол-4-карбальдегид (0,087 г, 0,598 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,078 г, 61,9%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,6 Гц, 1H), 8,32 (дд, J=8,1, 2,2 Гц, 2H), 7,40 (т, J=8,3 Гц, 2H), 7,31 (д, J=7,8 Гц, 1H), 7,21 (т, J=2,8 Гц, 1H), 7,17-7,10 (м, 2H), 6,92 (т, J=51,6 Гц, 1H), 6,80 (д, J=11,2 Гц, 1H), 6,74-6,73 (м, 1H), 5,66-5,59 (м, 1H), 5,19 (с, 2H), 5,12 (д, J=6,8 Гц, 4H), 3,88 (с, 2H), 3,10 (т, J=4,3 Гц, 4H), 2,80-2,60 (м, 4H); МСНР (ЭР) m/z 632,4 (M+ + 1).
Пример 323: Синтез соединения 323, 5-(4-((1H-пиразол-4-ил)метил)пиперазин-1-ил)-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 323
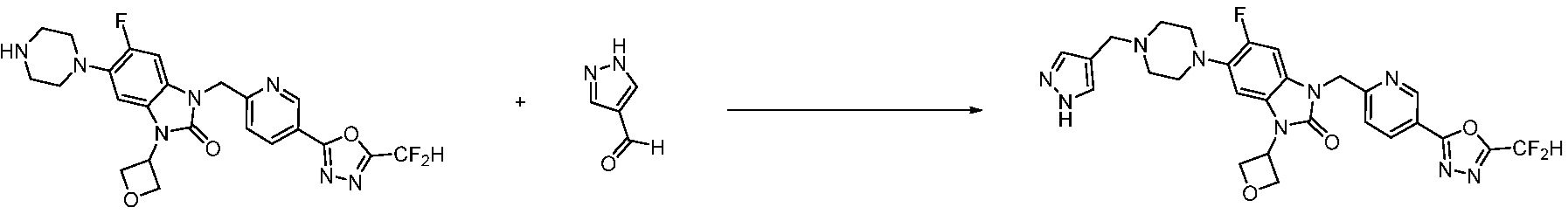
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,199 ммоль), полученный на стадии 5 соединения 319, триацетоксиборгидрид натрия (0,085 г, 0,399 ммоль) и 1H-пиразол-4-карбальдегид (0,057 г, 0,598 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,094 г, 80,7%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (с, 1H), 8,32 (дд, J=8,2, 1,8 Гц, 1H) 7,54 (с, 2H), 7,43-7,39 (м, 2H), 6,92 (т, J=50,8 Гц, 1H), 6,81 (д, J=11,3 Гц, 1H), 5,66-5,59 (м, 1H), 5,19-5,12 (м, 6H), 3,56 (с, 2H), 3,20-3,00 (м, 4H), 2,80-2,50 (м, 4H); МСНР (ЭР) m/z 582,3 (M+ + 1).
Пример 324: Синтез соединения 324, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(4-(пиридин-3-илметил)пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 324
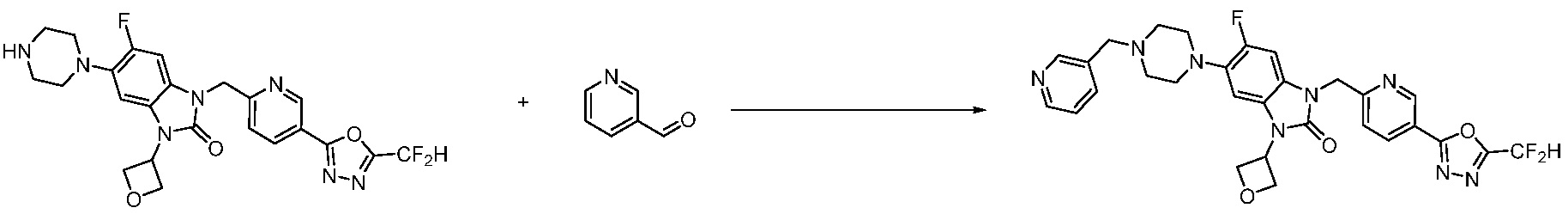
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,199 ммоль), полученный на стадии 5 соединения 319, триацетоксиборгидрид натрия (0,085 г, 0,399 ммоль) и никотинальдегид (0,064 г, 0,598 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,085 г, 72,0%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,26 (д, J=1,6 Гц, 1H), 8,55 (д, J=1,5 Гц, 1H), 8,50 (дд, J=4,7, 1,4 Гц, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,72 (д, J=7,8 Гц, 1H), 7,42 (т, J=7,2 Гц, 2H), 7,26 (дд, J=8,1, 4,5 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,82 (д, J=11,2 Гц, 1H), 5,66-5,59 (м, 1H), 5,19 (с, 2H), 5,16-5,10 (м, 4H), 3,60 (с, 2H), 3,10 (т, J=4,4 Гц, 4H), 2,80-2,60 (м, 4H); МСНР (ЭР) m/z 593,6 (M+ + 1).
Пример 325: Синтез соединения 325, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(4-(пиридин-3-илметил)пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 325
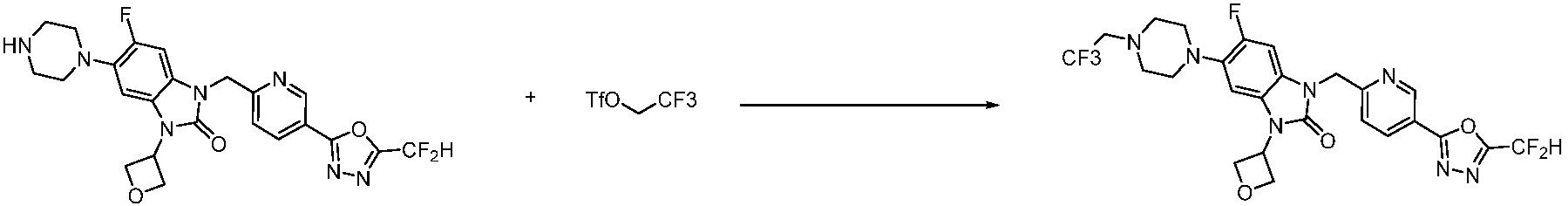
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-(оксетан-3-ил)-5-(пиперазин-1-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,100 г, 0,199 ммоль), полученный на стадии 5 соединения 319, триэтиламин (0,056 мл, 0,399 ммоль) и 2,2,2-трифторэтил трифторметансульфонат (0,093 г, 0,399 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,102 г, 87,3%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=1,6 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,44 (д, J=8,2 Гц, 2H), 6,92 (т, J=51,6 Гц, 1H), 6,83 (д, J=11,2 Гц, 1H), 5,67-5,60 (м, 1H), 5,20 (с, 2H), 5,18-5,11 (м, 4H), 3,12 (т, J=4,6 Гц, 4H), 3,04 (кв, J=9,5 Гц, 2H), 2,88 (т, J=4,5 Гц, 4H); МСНР (ЭР) m/z 584,2 (M+ + 1).
Пример 326: Синтез соединения 326, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(2-(диметиламино)этил)-6-фтор-5-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 326
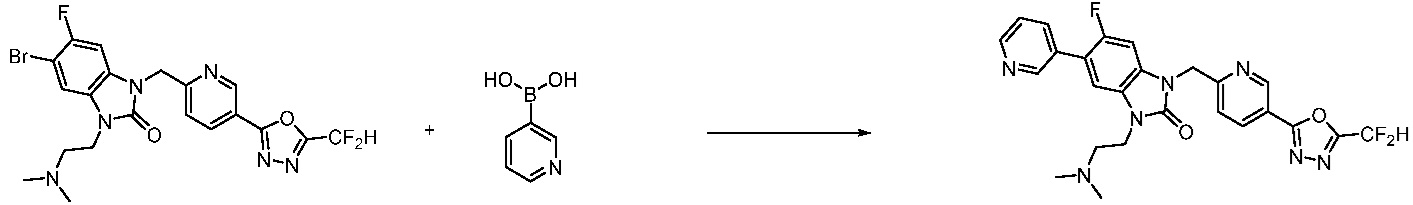
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(2-(диметиламино)этил)-6-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,293 ммоль), пиридин-3-илбороновую кислоту (0,054 г, 0,440 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,019 г, 0,029 ммоль) и карбонат цезия (0,239 г, 0,733 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,060 г, 40,1%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,6 Гц, 1H), 8,78 (с, 1H), 8,62 ~ 8,61 (м, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,88 ~ 7,86 (м, 1H), 7,50 (д, J=8,2 Гц, 1H), 7,40 (дд, J=7,9, 4,9 Гц, 1H), 7,13 ~ 7,11 (м, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,91 (д, J=9,8 Гц, 1H), 6,82 (с, 0,25H), 5,31 (с, 2H), 4,15 ~ 4,11 (м, 2H), 2,79 ~ 2,75 (м, 2H), 2,40 (с, 6H).
Пример 327: Синтез соединения 327, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(2-(диметиламино)этил)-6-фтор-5-(пиридин-4-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 327
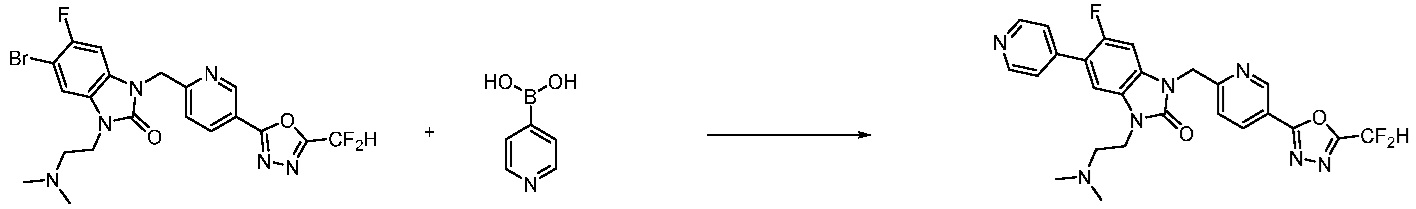
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(2-(диметиламино)этил)-6-фтор-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,293 ммоль), пиридин-4-илбороновую кислоту (0,054 г, 0,440 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,019 г, 0,029 ммоль) и карбонат цезия (0,239 г, 0,733 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл), после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом натрия, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,050 г, 33,5%) в виде коричневого масла.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (с, 1H), 8,69 (д, J=4,9 Гц, 2H), 8,39 (дд, J=8,2, 1,8 Гц, 1H), 7,51 ~ 7,46 (м, 3H), 7,14 (д, J=6,0 Гц, 1H), 7,08 (с, 0,25H), 6,95 (с, 0,5H), 6,91 (д, J=10,1 Гц, 1H), 6,82 (с, 0,25H), 5,30 (с, 2H), 4,11 (т, J=6,6 Гц, 2H), 2,74 (т, J=6,5 Гц, 2H), 2,37 (с, 6H).
Пример 328: Синтез соединения 328, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(4-метилпиперазин-1-ил)-3-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез трет-бутил 4-(2-фтор-4-нитро-5-(пиридин-3-иламино)фенил)пиперазин-1-карбоксилата
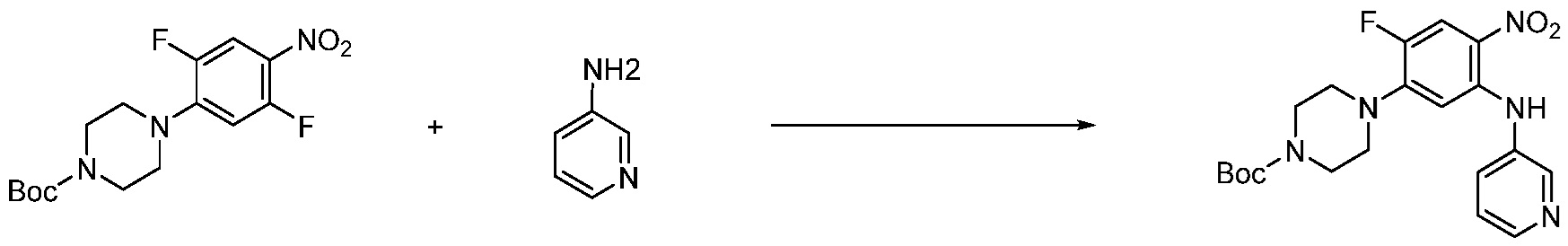
Пиридин-3-амин (0,663 г, 7,049 ммоль) растворяют в тетрагидрофуране (40 мл), после чего бис(триметилсилил)амид лития (26,00%, 4,536 г, 7,049 ммоль) медленно добавляют в полученный раствор и перемешивают, сохраняя температуру 0° и затем туда добавляют трет-бутил 4-(2,5-дифтор-4-нитрофенил)пиперазин-1-карбоксилат (2,200 г, 6,408 ммоль) и перемешивают при комнатной температуре в течение 18 часов. В реакционную смесь выливают в воду, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (0,504 г, 18,8%) в виде красного твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(4-амино-2-фтор-5-(пиридин-3-иламино)фенил)пиперазин-1-карбоксилата

Трет-бутил 4-(2-фтор-4-нитро-5-(пиридин-3-иламино)фенил)пиперазин-1-карбоксилат (0,504 г, 1,207 ммоль), полученный на стадии 1, растворяют в метаноле (5 мл)/тетрагидрофуран (2 мл) и перемешивают при комнатной температуре, после чего 10%-Pd/C (50 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при той же температуре в течение 18 часов в присутствии присоединенного баллона с водородом. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (0,460 г, 98,3%, коричневое твердое вещество).
[Стадия 3] Синтез трет-бутил 4-(6-фтор-2-оксо-3-(пиридин-3-ил)-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилат

Трет-бутил 4-(4-амино-2-фтор-5-(пиридин-3-иламино)фенил)пиперазин-1-карбоксилат (0,430 г, 1,110 ммоль), полученный на стадии 2, триэтиламин (0,155 мл, 1,110 ммоль) и 1,1'-карбонилдиимидазол (КДИ, 0,216 г, 1,332 ммоль) растворяют в дихлорметане (6 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением указанного в заголовке соединения (0,449 г, 97,9%) в виде коричневого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-2-оксо-3-(пиридин-3-ил)-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилата
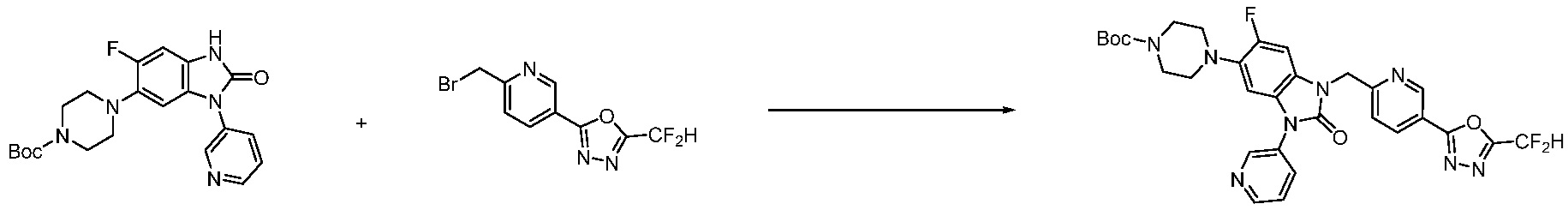
Трет-бутил 4-(6-фтор-2-оксо-3-(пиридин-3-ил)-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилат (0,440 г, 1,064 ммоль), полученный на стадии 3, растворяют в N,N-диметилформамиде (6 мл), после чего гидрид натрия (60,00%, 0,064 г, 1,596 ммоль) медленно добавляют в полученный раствор и перемешивают в течение 30 минут сохраняя температуру 0°. Затем, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,340 г, 1,171 ммоль) добавляют в полученную смесь и дополнительно перемешивают при комнатной температуре в течение 4 часов. В реакционную смесь выливают в воду и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (0,490 г, 74,0%) в виде коричневого твердого вещества.
[Стадия 5] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(пиперазин-1-ил)-3-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-она
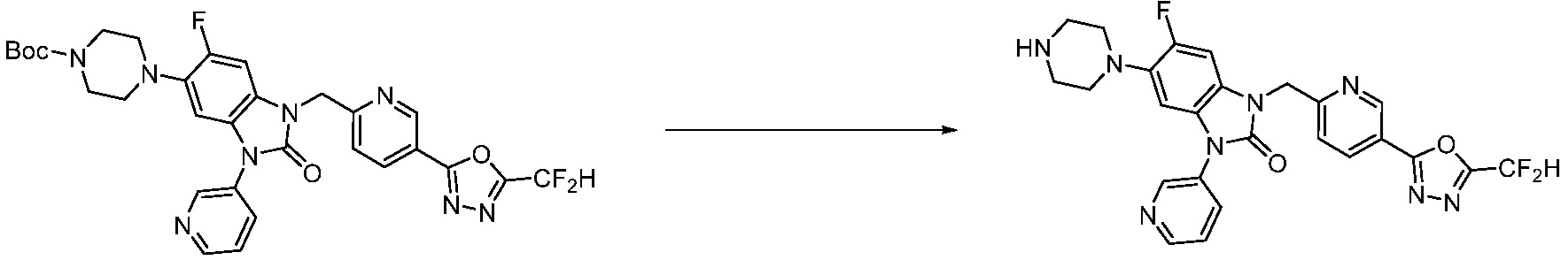
Трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-2-оксо-3-(пиридин-3-ил)-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиперазин-1-карбоксилат (0,490 г, 0,787 ммоль), полученный на стадии 4, и трифторуксусную кислоту (0,603 мл, 7,870 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-60%) и концентрируют с получением указанного в заголовке соединения (0,267 г, 64,9%) в виде коричневого твердого вещества.
[Стадия 6] Синтез соединения 328
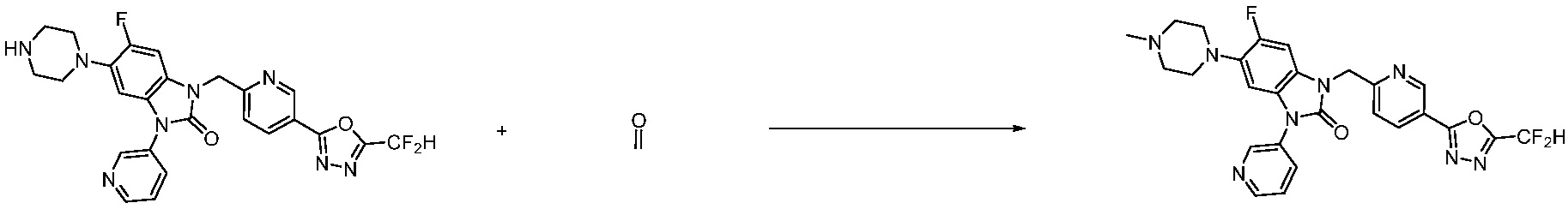
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(пиперазин-1-ил)-3-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,130 г, 0,249 ммоль), полученный на стадии 5, триацетоксиборгидрид натрия (0,105 г, 0,498 ммоль) и формальдегид (37,00%, 0,061 г, 0,746 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,086 г, 64,5%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (д, J=1,6 Гц, 1H), 8,84 (д, J=2,3 Гц, 1H), 8,65 (дд, J=4,7, 1,3 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,93 (тд, J=5,0, 2,7 Гц, 1H), 7,52-7,49 (м, 2H), 6,92 (т, J=51,6 Гц, 1H), 6,87 (д, J=11,1 Гц, 1H), 6,73 (д, J=7,0 Гц, 1H), 5,26 (с, 2H), 3,10-2,90 (м, 4H), 2,70-2,40 (м, 4H), 2,32 (с, 3H); МСНР (ЭР) m/z 537,5 (M+ + 1).
Пример 329: Синтез соединения 329, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(4-(оксетан-3-ил)пиперазин-1-ил)-3-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 329
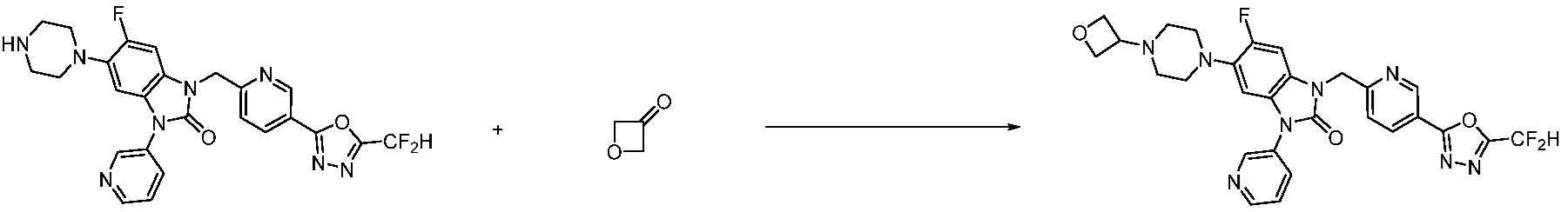
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-5-(пиперазин-1-ил)-3-(пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,130 г, 0,249 ммоль), полученный на стадии 5 соединения 328, триацетоксиборгидрид натрия (0,105 г, 0,498 ммоль) и оксетан-3-он (0,054 г, 0,746 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,113 г, 78,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,5 Гц, 1H), 8,84 (д, J=2,3 Гц, 1H), 8,67 (дд, J=4,8, 1,5 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,95-7,92 (м, 1H), 7,54-7,50 (м, 2H), 6,92 (т, J=51,6 Гц, 1H), 6,89 (д, J=11,1 Гц, 1H), 6,75 (д, J=7,0 Гц, 1H), 5,27 (с, 2H), 4,64 (тд, J=11,6, 5,5 Гц, 4H), 3,60-3,53 (м, 1H), 3,03 (т, J=4,5 Гц, 4H), 2,60-2,45 (м, 4H); МСНР (ЭР) m/z 579,3 (M+ + 1).
Пример 330: Синтез соединения 330, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(6-(морфолинометил)пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 330
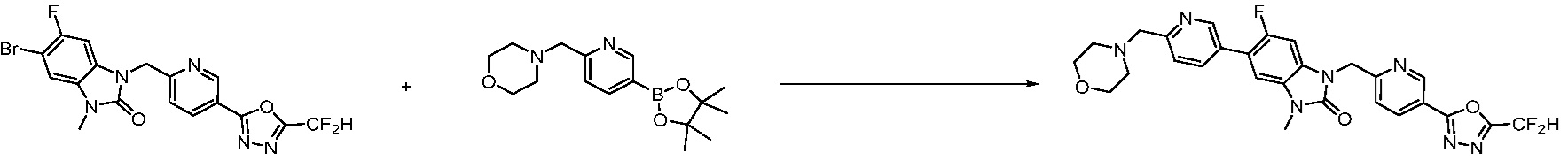
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 0,440 ммоль), полученный на стадии 4 соединения 147, 4-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)метил)морфолин (0,161 г, 0,528 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,014 г, 0,022 ммоль) и карбонат цезия (0,170 г, 0,881 ммоль) смешивают в 1,4-диоксане (2,5 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,188 г, 77,5%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,25 (д, J=1,6 Гц, 1H), 8,66 (с, 1H), 8,32 (дд, J=8,2, 2,2 Гц, 1H), 7,78 (тд, J=5,0, 2,7 Гц, 1H), 7,45 (д, J=8,2 Гц, 2H), 6,95 (д, J=6,2 Гц, 1H), 6,91 (т, J=51,6 Гц, 1H), 6,86 (д, J=9,8 Гц, 1H), 5,25 (с, 2H), 3,71 (т, J=4,6 Гц, 4H), 3,67 (с, 2H), 3,46 (с, 3H), 2,51 (т, J=4,4 Гц, 4H); МСНР (ЭР) m/z 553,4 (M+ + 1).
Пример 331: Синтез соединения 331, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-метилпиперидин-4-ил)бензо[d]тиазол-2(3H)-он
[Стадия 1] Синтез трет-бутил 4-(2-оксо-2,3-дигидробензо[d]тиазол-6-ил)-3,6-дигидропиридин-1(2H)-карбоксилата
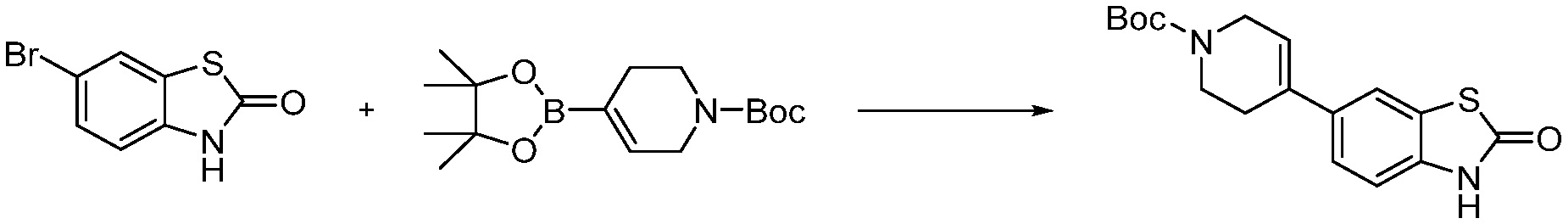
6-бромбензо[d]тиазол-2(3H)-он (1,500 г, 6,519 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (2,419 г, 7,823 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2, 0,239 г, 0,326 ммоль) и карбонат цезия (6,372 г, 19,558 ммоль) растворяют в 1,4-диоксане (30 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=5-30%) и концентрируют с получением указанного в заголовке соединения (0,700 г, 14,7%) в виде белого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(2-оксо-2,3-дигидробензо[d]тиазол-6-ил)пиперидин-1-карбоксилата
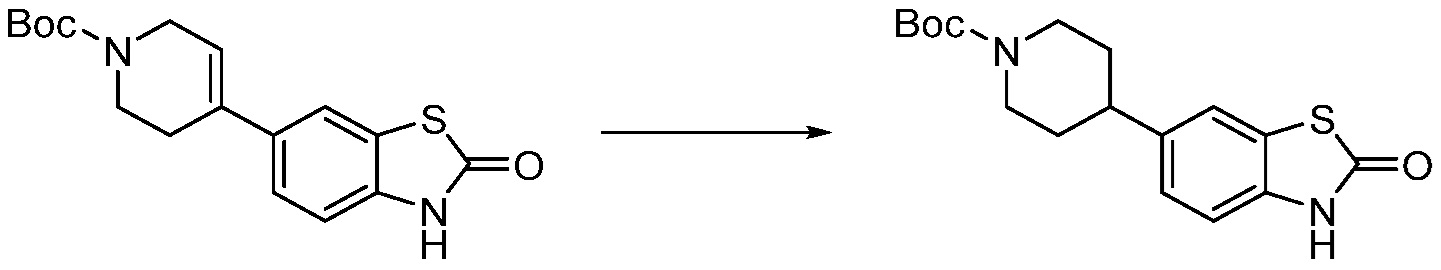
Трет-бутил 4-(2-оксо-2,3-дигидробензо[d]тиазол-6-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,700 г, 2,106 ммоль), полученный на стадии 1, растворяют в метаноле (50 мл) и перемешивают при комнатной температуре, после чего 10%-Pd/C (70 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают при 50° в течение 18 часов в присутствии присоединенного баллона с водородом, и затем реакцию останавливают понижением температуры до комнатной температуры. Реакционную смесь фильтруют через слой целита для удаления твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем указанное в заголовке соединение применяют без дополнительной очистки (0,217 г, 30,8%, светло-желтое твердое вещество).
[Стадия 3] Синтез трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)пиперидин-1-карбоксилата
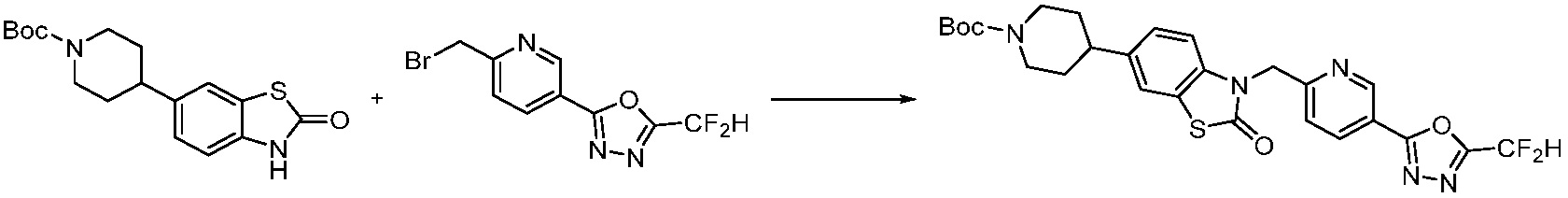
Трет-бутил 4-(2-оксо-2,3-дигидробензо[d]тиазол-6-ил)пиперидин-1-карбоксилат (0,217 г, 0,649 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,207 г, 0,714 ммоль), карбонат калия (0,135 г, 0,973 ммоль) и йодид калия (0,011 г, 0,065 ммоль) растворяют в N,N-диметилформамиде (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-20%) и концентрируют с получением указанного в заголовке соединения (0,100 г, 28,4%) в виде желтого твердого вещества.
[Стадия 4] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(пиперидин-4-ил)бензо[d]тиазол-2(3H)-она
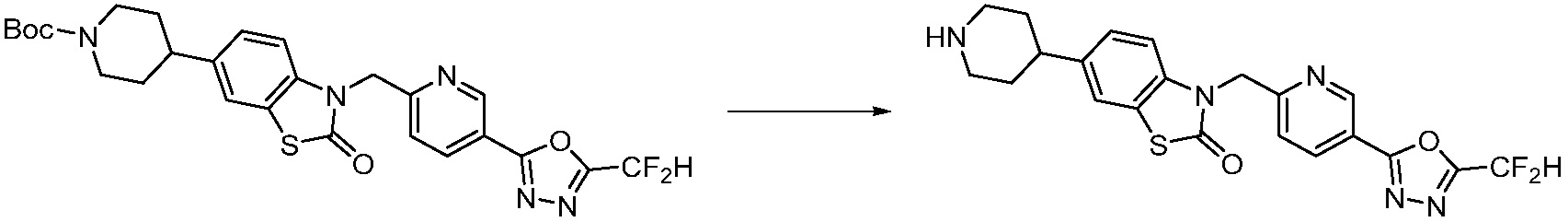
Трет-бутил 4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)пиперидин-1-карбоксилат (0,100 г, 0,184 ммоль), полученный на стадии 3, и трифторуксусную кислоту (0,014 мл, 0,184 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,064 г, 78,4%, желтое масло).
[Стадия 5] Синтез соединения 331
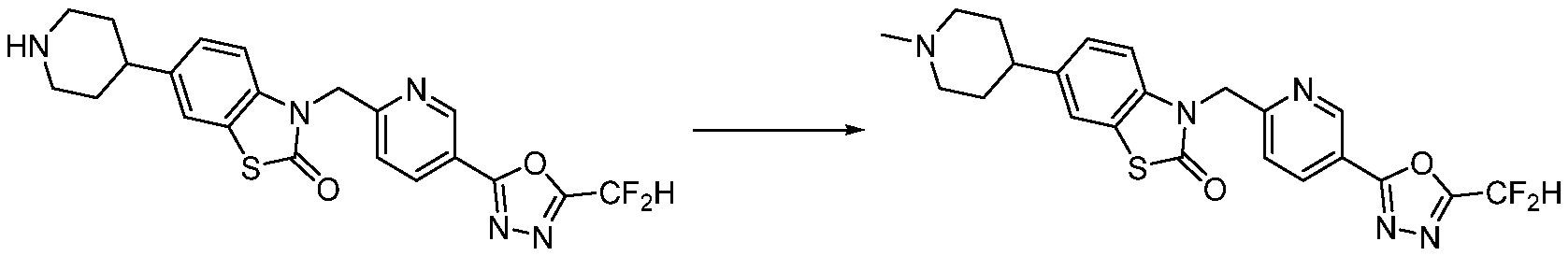
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(пиперидин-4-ил)бензо[d]тиазол-2(3H)-он (0,064 г, 0,144 ммоль), полученный на стадии 4, формальдегид (37,00% раствор, 0,016 мл, 0,216 ммоль) и триацетоксиборгидрид натрия (0,061 г, 0,289 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенным водным раствором хлорида натрия was выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,055 г, 83,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (с, 1H), 8,36 (д, J=8,0 Гц, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,35 (с, 1H), 7,14 (д, J=8,2 Гц, 1H), 7,08-6,82 (м, 2H), 5,36 (с, 2H), 3,20 (д, J=8,9 Гц, 1H), 2,61-2,58 (м, 1H), 2,51 (с, 3H), 2,33-2,32 (м, 2H), 2,06-2,04 (м, 2H), 1,91-1,88 (м, 2H); МСНР (ЭР) m/z 458,4 (M+ + 1).
Пример 332: Синтез соединения 332, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(морфолинометил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 332
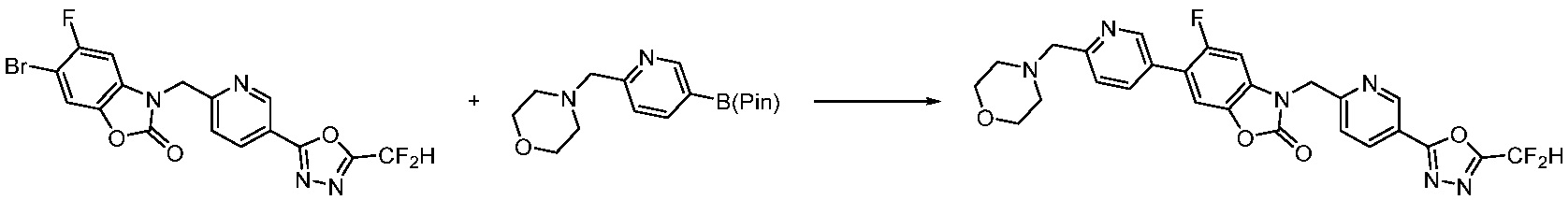
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фторбензо[d]оксазол-2(3H)-он (0,100 г, 0,227 ммоль), 4-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)метил)морфолин (0,083 г, 0,272 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,007 г, 0,011 ммоль) и карбонат цезия (0,148 г, 0,453 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрируют с получением указанного в заголовке соединения (0,009 г, 7,4%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,8 Гц, 1H), 8,71 (с, 1H), 8,46 (дд, J=8,2, 2,2 Гц, 1H), 7,83 (д, J=7,1 Гц, 1H), 7,62-7,60 (м, 2H), 7,30 (д, J=6,0 Гц, 1H), 7,09-6,83 (м, 2H), 5,24 (с, 2H), 3,82 (шс, 6H), 2,64 (шс, 4H); МСНР (ЭР) m/z 539,4 (M+ + 1).
Пример 333: Синтез соединения 333, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(6-(морфолинометил)пиридин-3-ил)бензо[d]тиазол-2(3H)-он
[Стадия 1] Синтез соединения 333
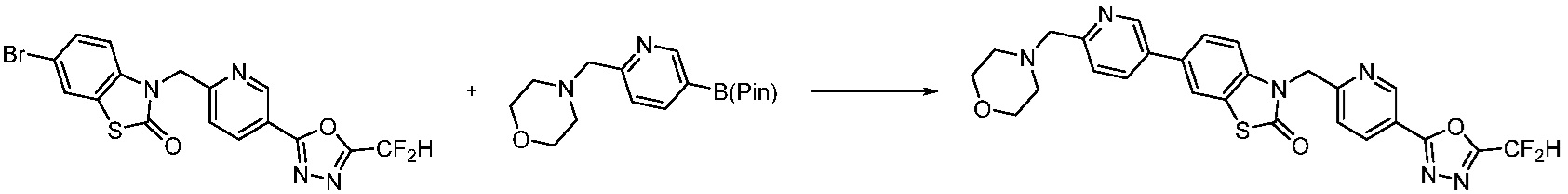
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)бензо[d]тиазол-2(3H)-он (0,100 г, 0,228 ммоль), 4-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)метил)морфолин (0,083 г, 0,273 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,007 г, 0,011 ммоль) и карбонат цезия (0,148 г, 0,455 ммоль) смешивают в 1,4-диоксане (1,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрируют с получением указанного в заголовке соединения (0,034 г, 27,8%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,6 Гц, 1H), 8,75 (д, J=2,1 Гц, 1H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 7,82 (дд, J=8,1, 2,3 Гц, 1H), 7,67 (д, J=1,7 Гц, 1H), 7,51-7,45 (м, 3H), 7,23 (д, J=8,4 Гц, 1H), 7,08-6,82 (м, 1H), 5,41 (с, 2H), 3,79-3,75 (м, 6H), 2,59 (шс, 4H); МСНР (ЭР) m/z 537,4 (M+ + 1).
Пример 334: Синтез соединения 334, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-метилпиперидин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 334
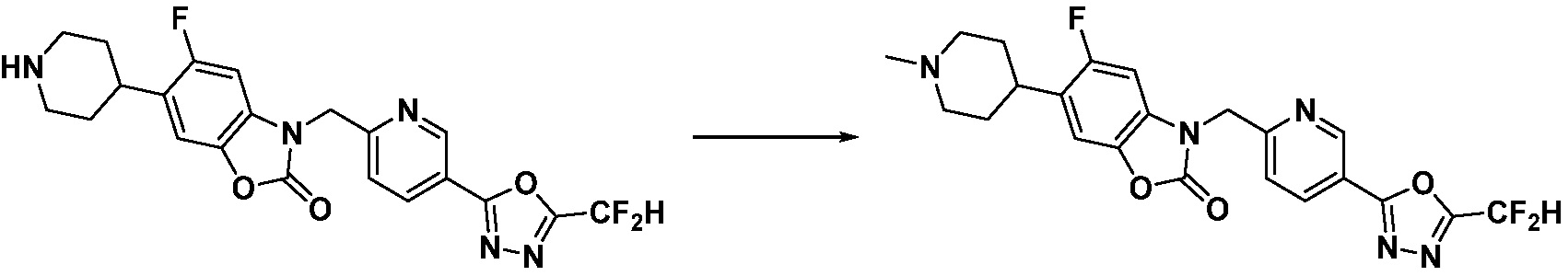
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(пиперидин-4-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,225 ммоль) и раствор формальдегида (37,00%, 0,025 мл, 0,337 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,095 г, 0,449 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,060 г, 58,2%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (с, 1H), 8,42 (д, J=7,9 Гц, 1H), 7,56 (д, J=8,2 Гц, 1H), 7,15 (д, J=5,6 Гц, 1H), 6,95 (т, J=51,6 Гц, 1H), 6,78 (д, J=9,0 Гц, 1H), 5,18 (с, 2H), 3,12-3,09 (м, 2H), 2,94-2,88 (м, 1H), 2,43 (с, 3H), 2,06-2,05 (м, 2H), 1,94-1,84 (м, 4H); МСНР (ЭР) m/z 460,4 (M+ + 1).
Пример 335: Синтез соединения 335, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-изопропилпиперидин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 335
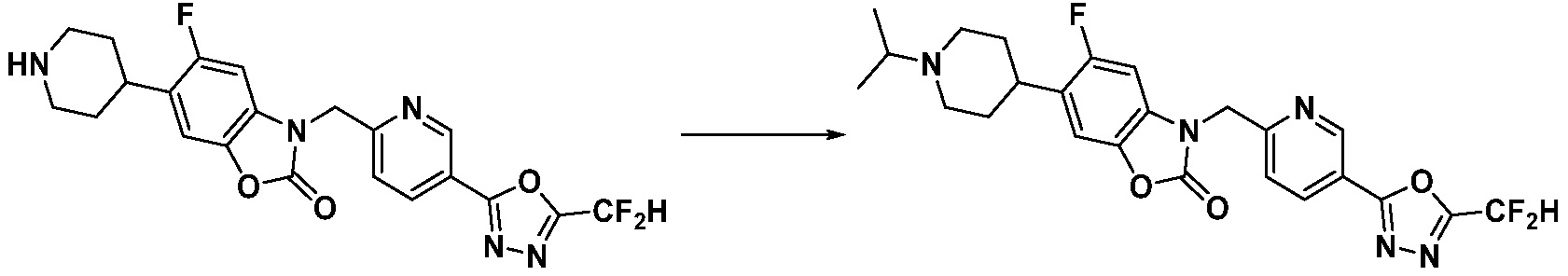
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(пиперидин-4-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,225 ммоль) и ацетон (0,025 мл, 0,337 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,095 г, 0,449 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,056 г, 51,2%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,7 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,56 (д, J=16,2 Гц, 1H), 7,17 (д, J=5,7 Гц, 1H), 7,08-6,82 (м, 1H), 6,76 (д, J=9,0 Гц, 1H), 5,18 (с, 2H), 3,12-3,09 (м, 2H), 2,92-2,91 (м, 2H), 2,40-2,35 (м, 2H), 1,88-1,87 (м, 4H), 1,16 (д, J=6,2 Гц, 6H); МСНР (ЭР) m/z 488,5 (M+ + 1).
Пример 336: Синтез соединения 336, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-(оксетан-3-ил)пиперидин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 336
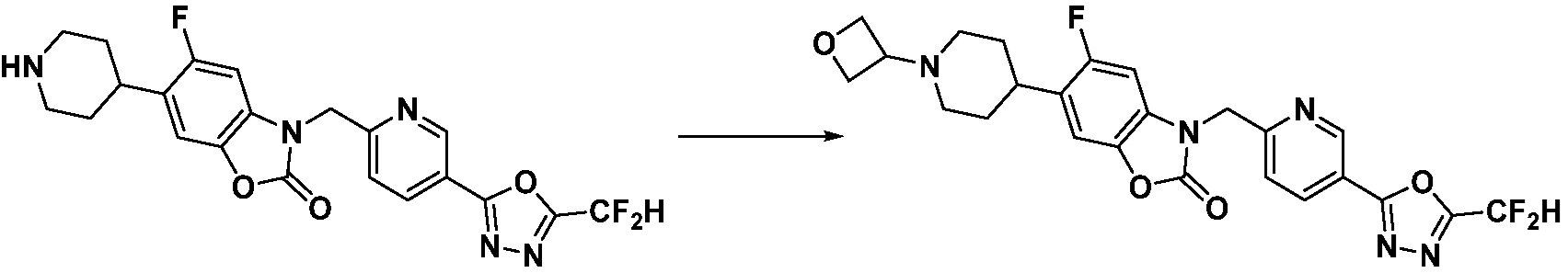
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(пиперидин-4-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,635 ммоль) и оксетан-3-он (0,061 мл, 0,953 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,269 г, 1,271 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,042 г, 13,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,6 Гц, 1H), 8,42 (дд, J=8,2, 2,2 Гц, 1H), 7,56 (д, J=8,2 Гц, 1H), 7,16 (д, J=5,6 Гц, 1H), 7,08-6,82 (м, 1H), 6,79 (д, J=9,0 Гц, 1H), 5,32 (с, 2H), 4,74-4,70 (м, 4H), 3,58-3,57 (м, 1H), 2,93-2,92 (м, 3H), 2,03-2,02 (м, 2H), 1,65-1,64 (м, 4H); МСНР (ЭР) m/z 502,4 (M+ + 1).
Пример 337: Синтез соединения 337, трет-бутил 4-(5-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиримидин-2-ил)пиперазин-1-карбоксилат
[Стадия 1] Синтез соединения 337
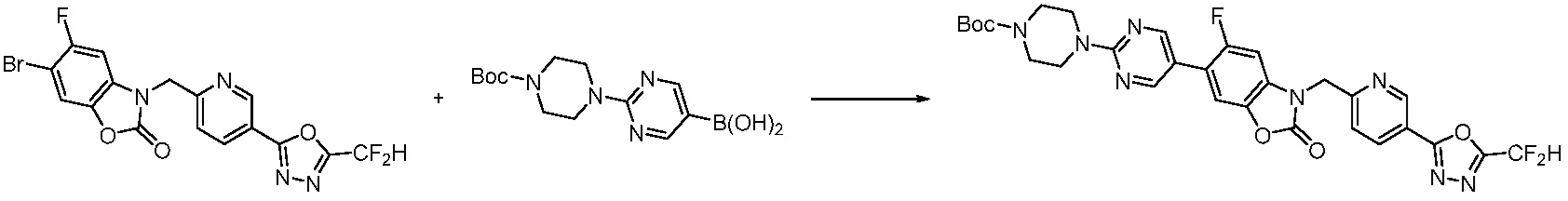
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фторбензо[d]оксазол-2(3H)-он (0,500 г, 1,133 ммоль), 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)пиримидин-5-ил)бороновую кислоту (0,418 г, 1,360 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,037 г, 0,057 ммоль) и карбонат цезия (0,739 г, 2,267 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-70%) и концентрируют с получением указанного в заголовке соединения (0,200 г, 28,3%) в виде светло-коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,9 Гц, 1H), 8,51 (с, 2H), 8,45 (дд, J=8,2, 2,1 Гц, 1H), 7,60 (д, J=8,2 Гц, 1H), 7,23 (д, J=5,9 Гц, 1H), 7,09-6,83 (м, 2H), 5,23 (с, 2H), 3,93-3,91 (м, 4H), 3,57-3,55 (м, 4H), 1,52 (с, 9H); МСНР (ЭР) m/z 625,2 (M+ + 1).
Пример 338: Синтез соединения 338, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(2-(4-метилпиперазин-1-ил)пиримидин-5-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(2-(пиперазин-1-ил)пиримидин-5-ил)бензо[d]оксазол-2(3H)-она
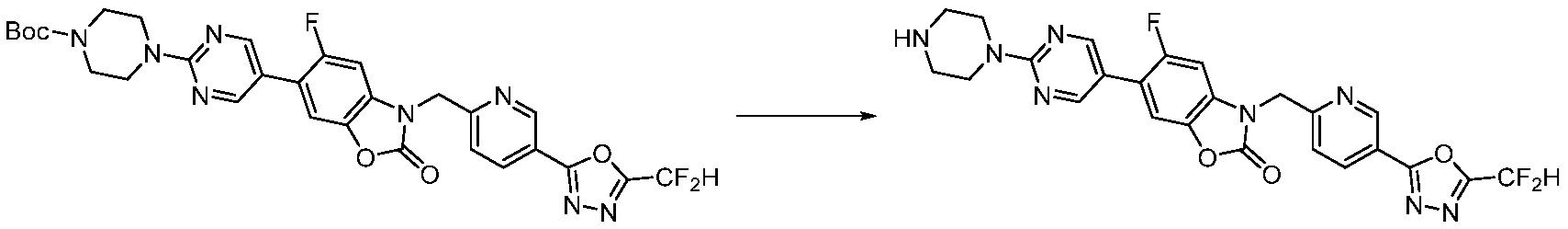
Трет-бутил 4-(5-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиримидин-2-ил)пиперазин-1-карбоксилат (0,241 г, 0,386 ммоль) и трифторуксусную кислоту (0,295 мл, 3,859 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,132 г, 65,2%, коричневое твердое вещество).
[Стадия 2] Синтез соединения 338
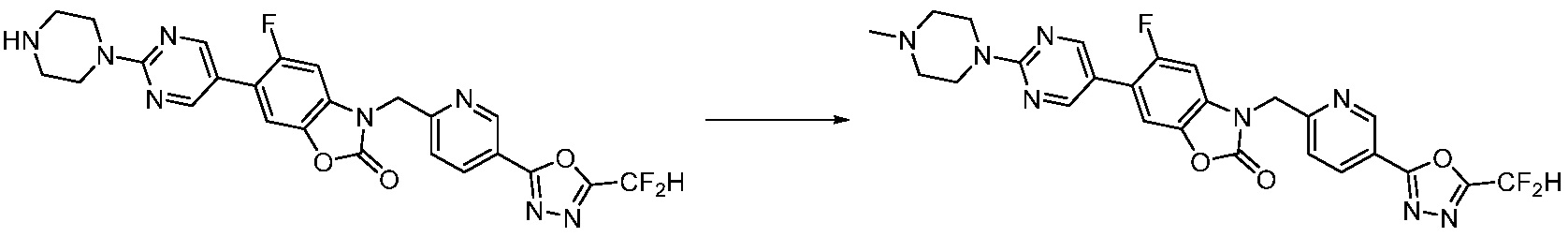
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(2-(пиперазин-1-ил)пиримидин-5-ил)бензо[d]оксазол-2(3H)-он (0,070 г, 0,133 ммоль), полученный на стадии 1, и формальдегид (37,00% раствор, 0,015 мл, 0,200 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,057 г, 0,267 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 47 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,046 г, 64,0%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 9,14 (д, J=1,8 Гц, 1H), 8,55 (д, J=1,4 Гц, 2H), 8,47 (дд, J=8,2, 2,2 Гц, 1H), 7,75 (д, J=11,9 Гц, 1H), 7,69-7,43 (м, 2H), 7,36 (д, J=10,0 Гц, 1H), 5,35 (с, 2H), 3,79-3,77 (м, 4H), 2,38-2,36 (м, 4H), 2,22 (с, 3H); МСНР (ЭР) m/z 539,1 (M+ + 1).
Пример 339: Синтез соединения 339, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(2-(4-(оксетан-3-ил)пиперазин-1-ил)пиримидин-5-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 339
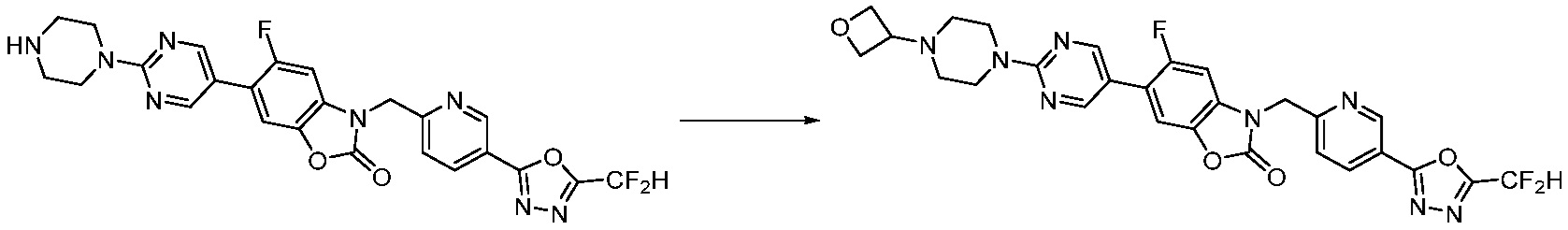
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(2-(пиперазин-1-ил)пиримидин-5-ил)бензо[d]оксазол-2(3H)-он (0,060 г, 0,114 ммоль) и оксетан-3-он (0,012 г, 0,172 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,048 г, 0,229 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрируют с получением указанного в заголовке соединения (0,026 г, 39,1%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 9,13 (д, J=2,0 Гц, 1H), 8,56 (д, J=1,2 Гц, 2H), 8,47 (дд, J=8,2, 2,2 Гц, 1H), 7,75 (д, J=8,3 Гц, 1H), 7,69-7,47 (м, 2H), 7,37 (д, J=10,0 Гц, 1H), 5,35 (с, 2H), 4,57 (т, J=6,5 Гц, 2H), 4,49 (т, J=6,1 Гц, 2H), 3,81-3,80 (м, 4H), 3,50-3,49 (м, 1H), 2,34-2,33 (м, 4H); МСНР (ЭР) m/z 581,5 (M+ + 1)
Пример 340: Синтез соединения 340, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-метил-5-(6-(морфолинометил)пиридин-3-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 340
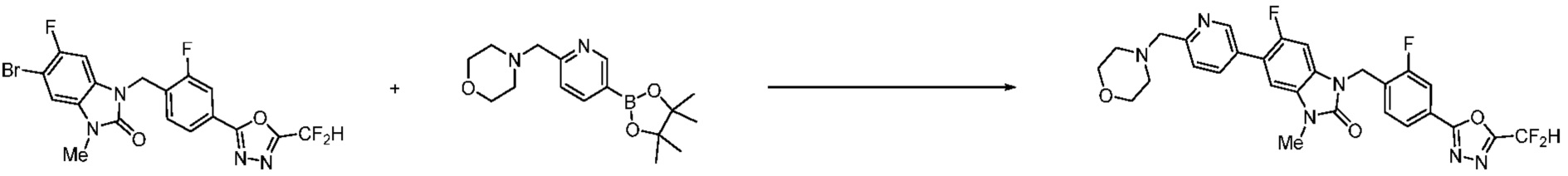
5-бром-1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,200 г, 0,424 ммоль), полученный на стадии 1 соединения 148, 4-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)метил)морфолин (0,155 г, 0,509 ммоль), карбонат цезия (0,277 г, 0,849 ммоль) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,014 г, 0,021 ммоль) смешивают в 1,4-диоксане (2 мл)/воде (0,6 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,135 г, 55,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,67 (с, 1H), 7,83 (д, J=8,7 Гц, 2H), 7,79 (д, J=8,1 Гц, 1H), 7,51-7,45 (м, 2H), 6,96 (д, J=6,2 Гц, 1H), 6,90 (т, J=51,6 Гц, 1H), 6,81 (д, J=9,8 Гц, 1H), 5,17 (с, 2H), 3,72 (т, J=4,5 Гц, 4H), 3,67 (с, 2H), 3,46 (с, 3H), 2,60-2,45 (м, 4H); МСНР (ЭР) m/z 569,35 (M+ + 1).
Пример 341: Синтез соединения 341, трет-бутил 4-(5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пиперазин-1-карбоксилат
[Стадия 1] Синтез соединения 341
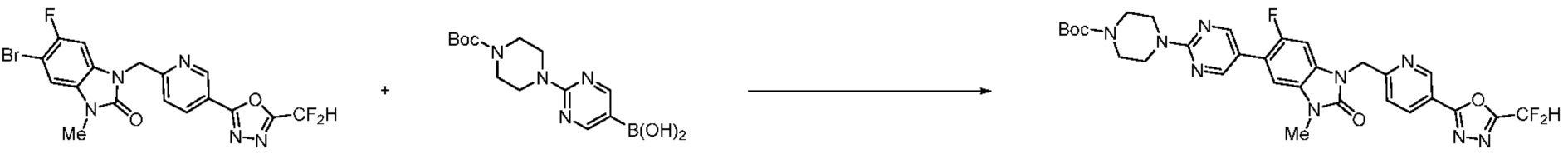
5-бром-1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (1,500 г, 3,302 ммоль), полученный на стадии 4 соединения 147, (2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)пиримидин-5-ил)бороновую кислоту (1,221 г, 3,963 ммоль), карбонат цезия (2,152 г, 6,605 ммоль) и [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) дихлорид (Pd(dtbpf)Cl2, 0,108 г, 0,165 ммоль) смешивают в 1,4-диоксане (9 мл)/воде (3 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Реакционную смесь фильтруют через стеклянный фильтр для удаления твердого вещества, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный фильтрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=50-70%) и концентрируют с получением указанного в заголовке соединения (0,590 г, 28,0%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,29 (д, J=1,9 Гц, 1H), 8,48 (с, 2H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,47 (д, J=8,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,89 (д, J=6,2 Гц, 1H), 6,86 (д, J=9,8 Гц, 1H), 5,27 (с, 2H), 3,86 (т, J=5,1 Гц, 4H), 3,52 (т, J=5,0 Гц, 4H), 3,49 (с, 3H), 1,49 (с, 9H); МСНР (ЭР) m/z 638,38 (M+ + 1).
Пример 342: Синтез соединения 342, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(2-(4-метилпиперазин-1-ил)пиримидин-5-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 342
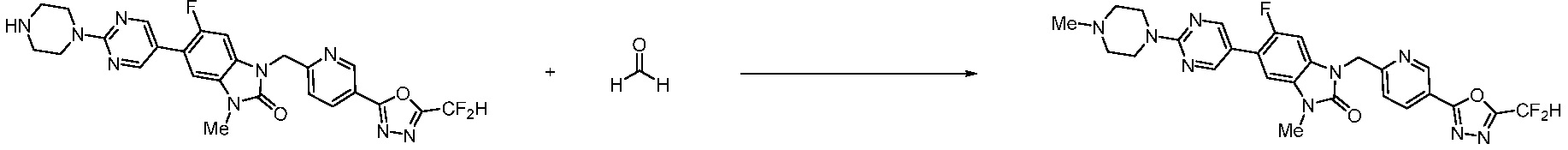
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(2-(пиперазин-1-ил)пиримидин-5-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 1 соединения 341, триацетоксиборгидрид натрия (0,118 г, 0,558 ммоль) и формальдегид (37,00%, 0,068 г, 0,837 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,141 г, 91,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,9 Гц, 1H), 8,44 (д, J=1,2 Гц, 2H), 8,33 (дд, J=8,2, 2,2 Гц, 1H), 7,45 (д, J=8,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,88 (д, J=6,2 Гц, 1H), 6,84 (д, J=9,8 Гц, 1H), 5,25 (с, 2H), 3,88 (т, J=4,9 Гц, 4H), 3,47 (с, 3H), 2,48 (т, J=5,0 Гц, 4H), 2,34 (с, 3H); МСНР (ЭР) m/z 552,35 (M+ + 1).
Пример 343: Синтез соединения 343, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(2-(4-(оксетан-3-ил)пиперазин-1-ил)пиримидин-5-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он
[Стадия 1] Синтез соединения 343
1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-3-метил-5-(2-(пиперазин-1-ил)пиримидин-5-ил)-1,3-дигидро-2H-бензо[d]имидазол-2-он (0,150 г, 0,279 ммоль), полученный на стадии 1 соединения 341, триацетоксиборгидрид натрия (0,118 г, 0,558 ммоль) и оксетан-3-он (0,060 г, 0,837 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,115 г, 69,1%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,27 (д, J=1,6 Гц, 1H), 8,45 (д, J=1,4 Гц, 2H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,46 (д, J=8,2 Гц, 1H), 6,92 (т, J=51,6 Гц, 1H), 6,88 (д, J=6,2 Гц, 1H), 6,85 (д, J=9,8 Гц, 1H), 5,26 (с, 2H), 4,67 (тд, J=7,5, 5,6 Гц, 4H), 3,91 (т, J=4,9 Гц, 4H), 3,55-3,47 (м, 1H), 3,47 (с, 3H), 2,40 (т, J=4,9 Гц, 4H); МСНР (ЭР) m/z 594,24 (M+ + 1).
Пример 344: Синтез соединения 344, трет-бутил 4-(5-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиридин-2-ил)пиперазин-1-карбоксилат
[Стадия 1] Синтез трет-бутил 4-(5-(5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиридин-2-ил)пиперазин-1-карбоксилата
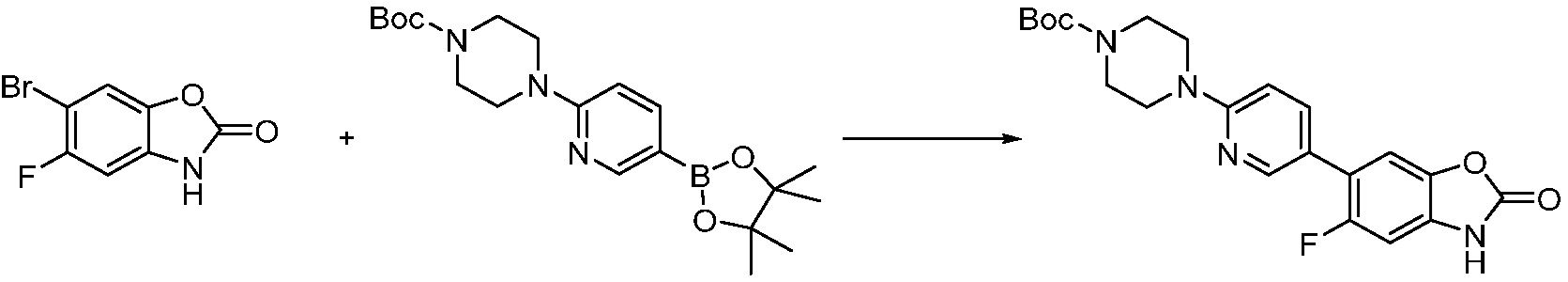
6-бром-5-фторбензо[d]оксазол-2(3H)-он (0,500 г, 2,155 ммоль), трет-бутил 4-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)пиперазин-1-карбоксилат (1,007 г, 2,586 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,070 г, 0,108 ммоль) и карбонат цезия (1,404 г, 4,310 ммоль) смешивают в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 30 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Диэтиловый эфир (20 мл) добавляют в полученный концентрат и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют, затем промывают диэтиловым эфиром, и затем сушат с получением указанного в заголовке соединения (0,783 г, 87,7%) в виде коричневого твердого вещества.
[Стадия 2] Синтез соединения 344
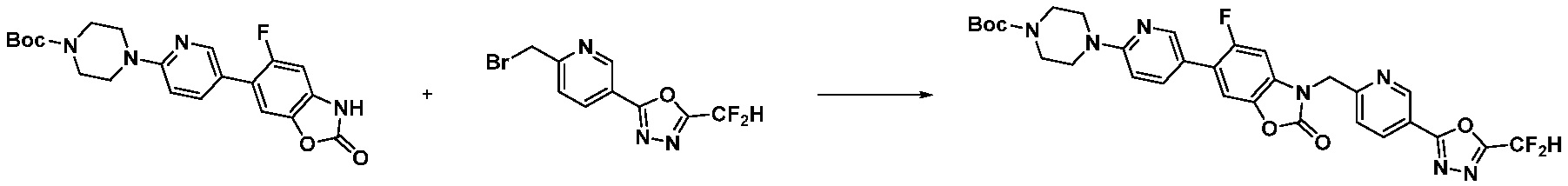
Трет-бутил 4-(5-(5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиридин-2-ил)пиперазин-1-карбоксилат (0,783 г, 1,889 ммоль), полученный на стадии 1, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,603 г, 2,078 ммоль), карбонат калия (0,392 г, 2,834 ммоль) и йодид калия (0,031 г, 0,189 ммоль) растворяют в N,N-диметилформамиде (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстрагирование с этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Диэтиловый эфир добавляют в полученный концентрат и перемешивают, после чего выпавшее в осадок твердое вещество фильтруют, затем промывают диэтиловым эфиром, и затем сушат с получением указанного в заголовке соединения (0,794 г, 67,4%) в виде серого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,9 Гц, 1H), 8,44 (дд, J=8,2, 2,1 Гц, 1H), 8,34 (с, 1H), 7,67 (д, J=7,4 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,26 (д, J=6,0 Гц, 1H), 7,09-6,83 (м, 2H), 6,74 (д, J=8,2 Гц, 1H), 5,23 (с, 2H), 3,62-3,60 (м, 8H), 1,51 (с, 9H); МСНР (ЭР) m/z 624,4 (M+ + 1).
Пример 345: Синтез соединения 345, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(пиперазин-1-ил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 345
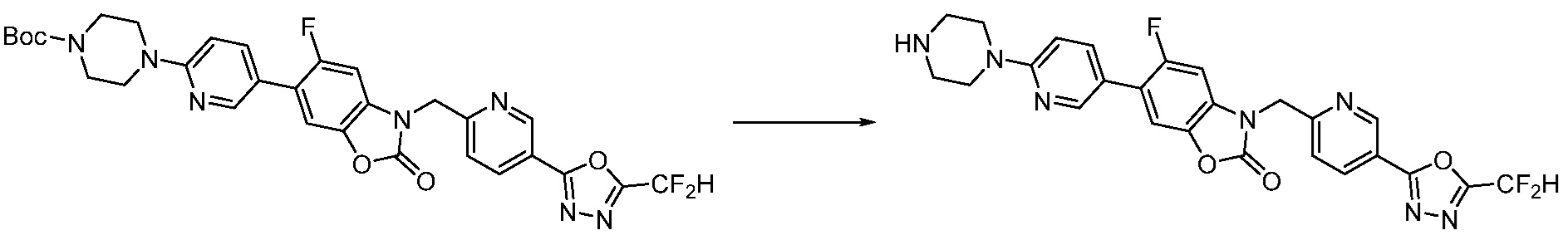
Трет-бутил 4-(5-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиридин-2-ил)пиперазин-1-карбоксилат (0,045 г, 0,072 ммоль) и трифторуксусную кислоту (0,028 мл, 0,361 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия и дихлорметан добавляют в полученный концентрат и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают гексан, и затем сушат с получением указанного в заголовке соединения (0,023 г, 60,9%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 9,15 (д, J=1,9 Гц, 1H), 8,47 (дд, J=8,2, 2,2 Гц, 1H), 8,28 (с, 1H), 7,75 (д, J=8,3 Гц, 1H), 7,71-7,45 (м, 3H), 7,33 (д, J=10,0 Гц, 1H), 6,89 (д, J=8,6 Гц, 1H), 5,35 (с, 2H), 3,46-3,36 (м, 6H), 2,86 (шс, 2H); МСНР (ЭР) m/z 524,1 (M+ + 1).
Пример 346: Синтез соединения 346, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 346
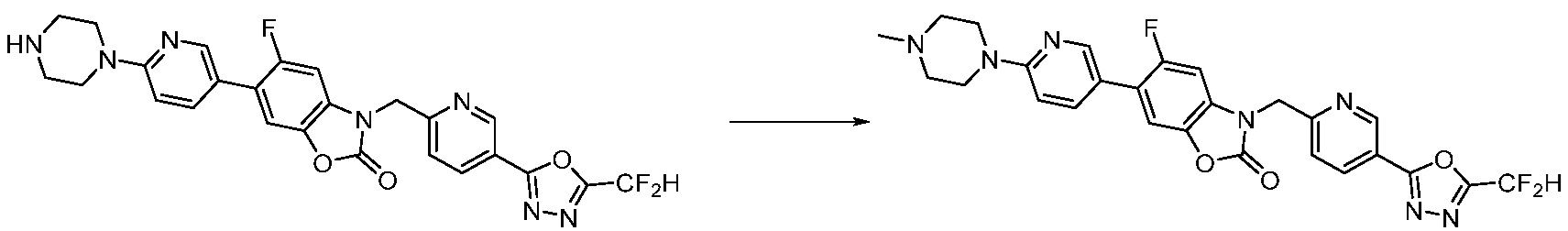
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(пиперазин-1-ил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он (0,200 г, 0,382 ммоль) и формальдегид (37,00% раствор, 0,043 мл, 0,573 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,162 г, 0,764 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением продукта. Затем, диэтиловый эфир добавляют в полученный продукт и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают диэтиловым эфиром, и затем сушат с получением указанного в заголовке соединения (0,011 г, 5,4%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,9 Гц, 1H), 8,44 (дд, J=7,5, 2,8 Гц, 1H), 8,33 (с, 1H), 7,66-7,64 (м, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,25 (д, J=4,5 Гц, 1H), 6,96-6,83 (м, 2H), 6,73 (д, J=8,9 Гц, 1H), 5,23 (с, 2H), 3,72 (шс, 4H), 2,68 (шс, 4H), 2,45 (с, 3H); МСНР (ЭР) m/z 538,4 (M+ + 1).
Пример 347: Синтез соединения 347, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(6-(4-этилпиперазин-1-ил)пиридин-3-ил)-5-фторбензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 347
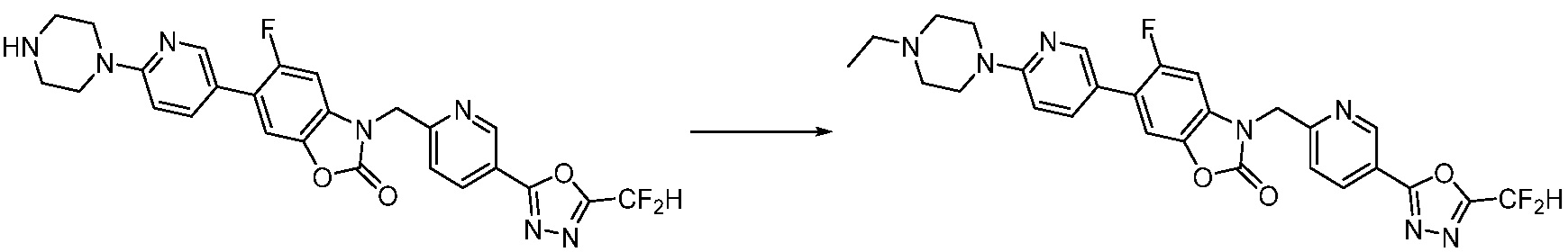
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(пиперазин-1-ил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он (0,200 г, 0,382 ммоль), ацетальдегид (0,032 мл, 0,573 ммоль) и триацетоксиборгидрид натрия (0,162 г, 0,764 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,011 г, 5,2%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=2,1 Гц, 1H), 8,44 (дд, J=8,2, 2,2 Гц, 1H), 8,32 (шс, 1H), 7,65-7,63 (м, 1H), 7,58 (д, J=8,2 Гц, 1H), 7,25 (д, J=6,0 Гц, 1H), 6,95-6,82 (м, 2H), 6,72 (д, J=8,9 Гц, 1H), 3,70-3,67 (м, 4H), 2,64-2,63 (м, 4H), 2,54 (кв, J=7,2 Гц, 2H), 1,19 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 552,4 (M+ + 1).
Пример 348: Синтез соединения 348, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(5-морфолинопиридин-3-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез 5-фтор-6-(5-морфолинопиридин-3-ил)бензо[d]оксазол-2(3H)-она
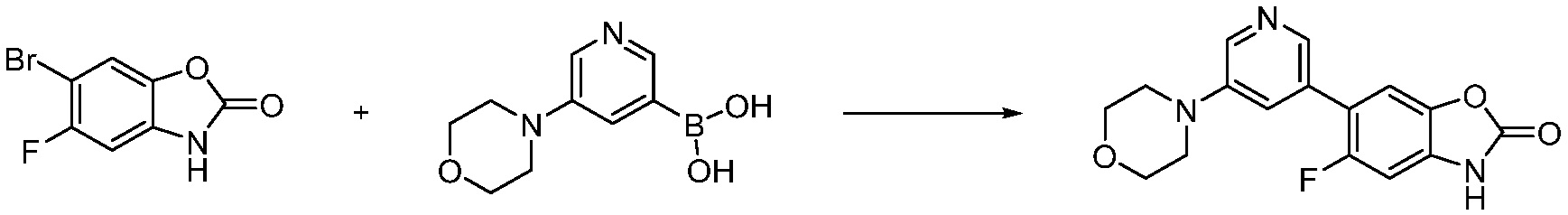
6-бром-5-фторбензо[d]оксазол-2(3H)-он (0,200 г, 0,862 ммоль), (2-морфолинопиридин-4-ил)бороновую кислоту (0,215 г, 1,034 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,028 г, 0,043 ммоль) и карбонат цезия (0,562 г, 1,724 ммоль) растворяют в 1,4-диоксане (15 мл)/воде (0,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением указанного в заголовке соединения (0,064 г, 23,5%) в виде желтого твердого вещества.
[Стадия 2] Синтез соединения 348
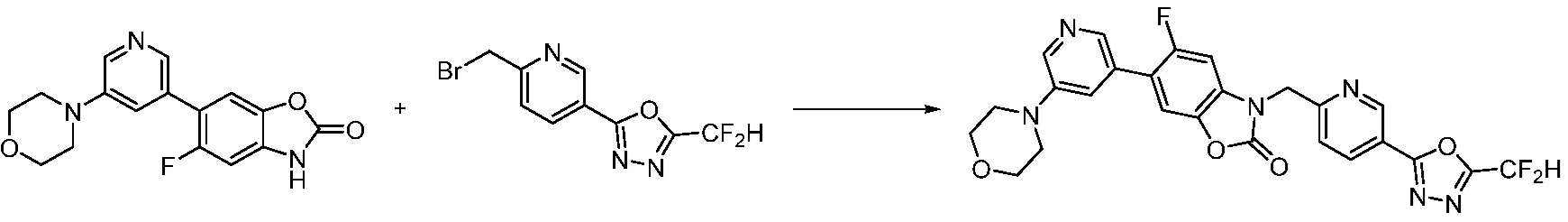
5-фтор-6-(5-морфолинопиридин-3-ил)бензо[d]оксазол-2(3H)-он (0,064 г, 0,203 ммоль), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,071 г, 0,244 ммоль), полученный на стадии 1, карбонат калия (0,042 г, 0,304 ммоль) и йодид калия (0,003 г, 0,020 ммоль) растворяют в N,N-диметилформамиде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением указанного в заголовке соединения (0,016 г, 15,0%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,8 Гц, 1H), 8,46 (дд, J=8,2, 2,1 Гц, 1H), 8,30 (д, J=2,5 Гц, 1H), 8,24 (с, 1H), 7,62 (д, J=8,2 Гц, 1H), 7,37 (с, 1H), 7,31 (д, J=5,9 Гц, 1H), 7,09-6,83 (м, 2H), 5,25 (с, 2H), 3,92 (т, J=4,8 Гц, 4H), 3,29 (т, J=4,8 Гц, 4H); МСНР (ЭР) m/z 525,1 (M+ + 1).
Пример 349: Синтез соединения 349, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(4-изопропилпиперазин-1-ил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 349
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(пиперазин-1-ил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он (0,200 г, 0,382 ммоль), ацетон (0,042 мл, 0,573 ммоль) и триацетоксиборгидрид натрия (0,162 г, 0,764 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,056 г, 25,9%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,8 Гц, 1H), 8,43 (дд, J=8,2, 2,0 Гц, 1H), 8,32 (с, 1H), 7,63 (д, J=8,9 Гц, 1H), 7,58 (д, J=8,2 Гц, 1H), 7,24 (д, J=6,0 Гц, 1H), 7,08-6,82 (м, 2H), 6,71 (д, J=8,9 Гц, 1H), 5,22 (с, 2H), 3,70 (т, J=4,8 Гц, 4H), 2,92-2,89 (м, 1H), 2,77 (т, J=4,9 Гц, 4H), 1,16 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 566,2 (M+ + 1).
Пример 350: Синтез соединения 350, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 350
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(пиперазин-1-ил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он (0,200 г, 0,382 ммоль), оксетан-3-он (0,037 мл, 0,573 ммоль) и триацетоксиборгидрид натрия (0,162 г, 0,764 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенным водным раствором хлорида натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрируют с получением указанного в заголовке соединения (0,011 г, 5,0%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,9 Гц, 1H), 8,44 (дд, J=8,2, 2,2 Гц, 1H), 8,33 (с, 1H), 7,66 (д, J=8,9 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,25 (д, J=6,0 Гц, 1H), 7,09-6,83 (м, 2H), 6,74 (д, J=8,9 Гц, 1H), 5,23 (с, 2H), 4,73 (шс, 4H), 3,71-3,61 (м, 6H), 2,53 (шс, 4H); МСНР (ЭР) m/z 580,3 (M+ + 1).
Пример 351: Синтез соединения 351, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 351
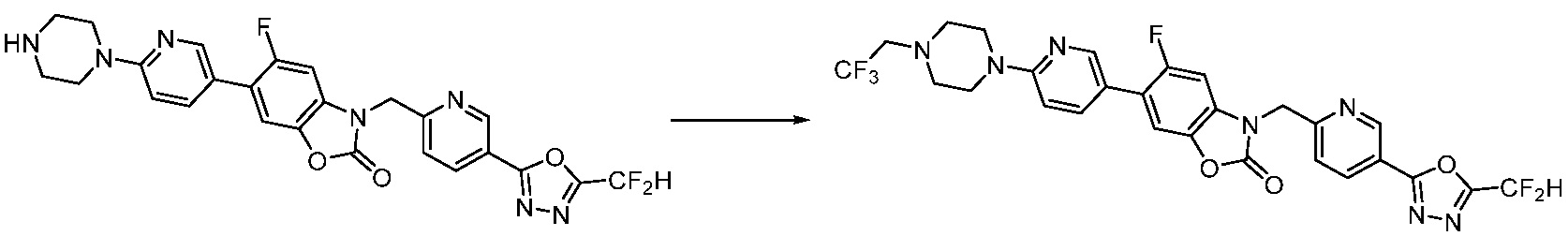
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(6-(пиперазин-1-ил)пиридин-3-ил)бензо[d]оксазол-2(3H)-он (0,200 г, 0,382 ммоль), 2,2,2-трифторэтил трифторметансульфонат (0,133 г, 0,573 ммоль) и карбонат калия (0,106 г, 0,764 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением указанного в заголовке соединения (0,028 г, 12,1%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,8 Гц, 1H), 8,44 (дд, J=8,2, 2,0 Гц, 1H), 8,33 (с, 1H), 7,65 (д, J=8,5 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,25 (д, J=6,0 Гц, 1H), 7,09-6,83 (м, 2H), 6,73 (д, J=8,9 Гц, 1H), 5,23 (с, 2H), 3,66-3,65 (м, 4H), 3,06 (кв, J=9,5 Гц, 2H), 2,82 (т, J=4,9 Гц, 4H); МСНР (ЭР) m/z 606,2 (M+ + 1).
Пример 352: Синтез соединения 352, трет-бутил 4-(4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилат
[Стадия 1] Синтез метил 6-((6-бром-2-оксобензо[d]тиазол-3(2H)-ил)метил)никотината
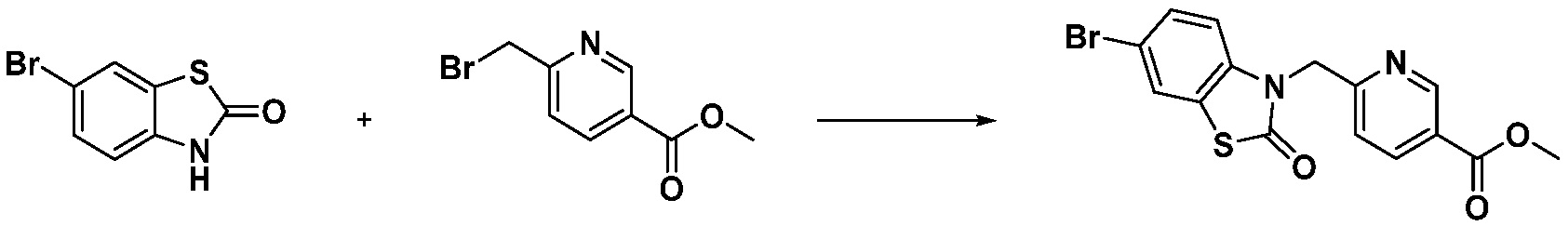
6-бромбензо[d]тиазол-2(3H)-он (1,000 г, 4,346 ммоль), метил 6-(бромметил)никотинат (2,000 г, 8,693 ммоль), карбонат калия (1,201 г, 8,693 ммоль) и йодид калия (0,072 г, 0,435 ммоль) растворяют в N,N-диметилформамиде (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=5-15%) и концентрируют с получением продукта. Затем, диэтиловый эфир добавляют в полученный продукт и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают диэтиловым эфиром, и затем сушат с получением указанного в заголовке соединения (0,700 г, 42,5%) в виде светло-желтого твердого вещества.
[Стадия 2] Синтез метил 6-((6-(1-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1H-пиразол-4-ил)-2-оксобензо[d]тиазол-3(2H)-ил)метил)никотинат
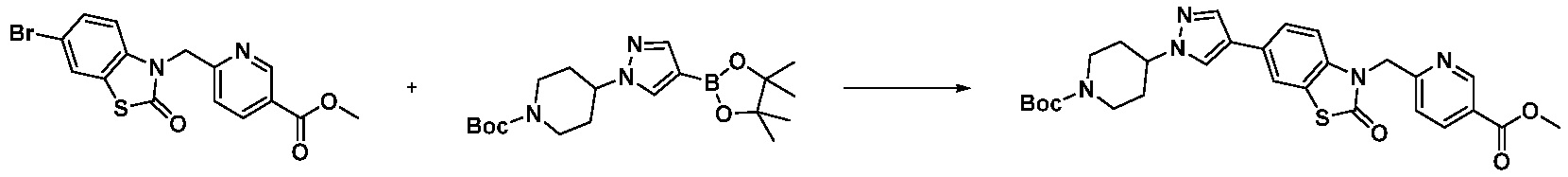
The метил 6-((6-бром-2-оксобензо[d]тиазол-3(2H)-ил)метил)никотинат (0,700 г, 1,846 ммоль), полученный на стадии 1, трет-бутил 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилат (0,836 г, 2,215 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,060 г, 0,092 ммоль) и карбонат цезия (1,203 г, 3,692 ммоль) растворяют в 1,4-диоксане (12 мл)/воде (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=20-60%) и концентрируют с получением указанного в заголовке соединения (0,920 г, 90,7%) в виде белого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(4-(3-((5-(гидразинкарбонил)пиридин-2-ил)метил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилата
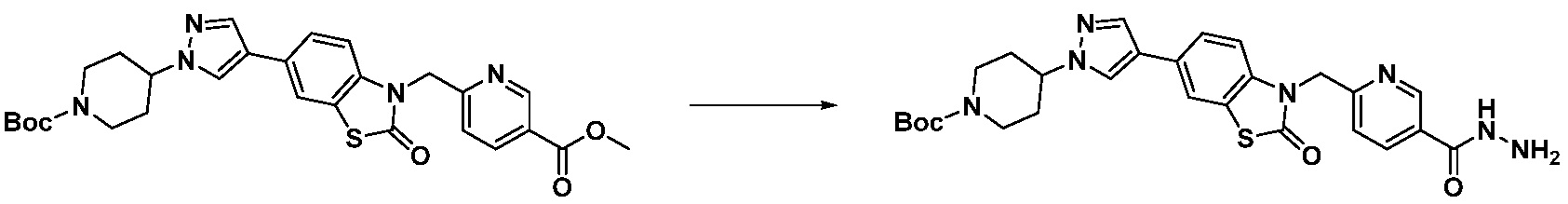
Метил 6-((6-(1-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1H-пиразол-4-ил)-2-оксобензо[d]тиазол-3(2H)-ил)метил)никотинат (0,920 г, 1,674 ммоль), полученный на стадии 2 и моногидрат гидразина (0,813 мл, 16,738 ммоль) растворяют в этаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80° в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. выпавшее в осадок твердое вещество фильтруют, затем промывают этанол, и затем сушат с получением указанного в заголовке соединения (0,717 г, 77,9%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 352
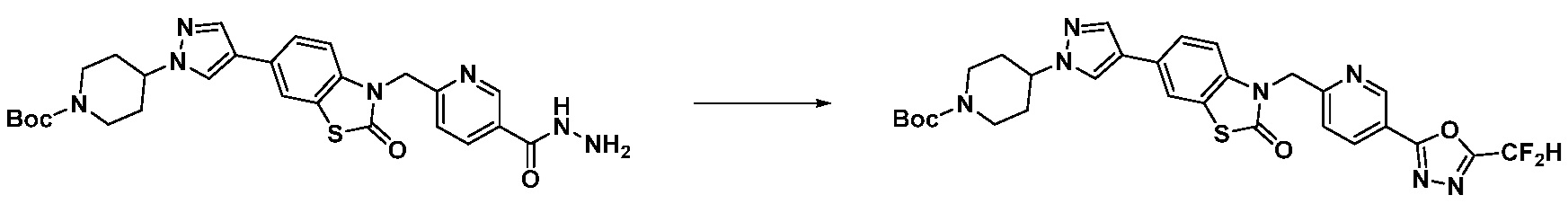
Трет-бутил 4-(4-(3-((5-(гидразинкарбонил)пиридин-2-ил)метил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилат (0,717 г, 1,304 ммоль), полученный на стадии 3, и имидазол (0,266 г, 3,913 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего 2,2-дифторуксусный ангидрид (0,487 мл, 3,913 ммоль) добавляют в полученный раствор, затем нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстрагирование с дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, затем дегидратируют безводным сульфатом магния, затем фильтруют, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=5-40%) и концентрируют с получением указанного в заголовке соединения (0,856 г, 107,6%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,9 Гц, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 7,74 (с, 1H), 7,64 (с, 1H), 7,57 (д, J=1,6 Гц, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,36 (дд, J=8,4, 1,7 Гц, 1H), 7,10 (д, J=8,4 Гц, 1H), 7,08-6,82 (м, 1H), 5,38 (с, 2H), 4,33-4,27 (м, 1H), 2,98-2,90 (м, 2H), 2,18 (д, J=10,8 Гц, 2H), 1,97-1,93 (м, 2H), 1,50 (с, 9H); МСНР (ЭР) m/z 610,3 (M+ + 1).
Пример 353: Синтез соединения 353, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(1-метилпиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]тиазол-2(3H)-он
[Стадия 1] Синтез соединения 353
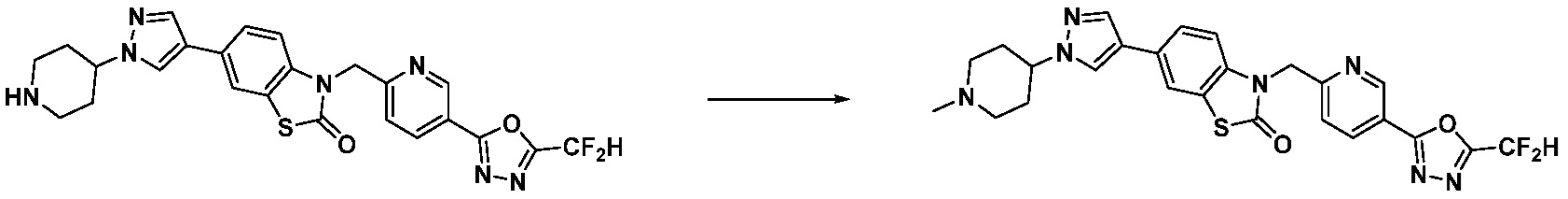
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]тиазол-2(3H)-он (0,100 г, 0,196 ммоль) и формальдегид (38,00% раствор, 0,022 мл, 0,294 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,083 г, 0,393 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,068 г, 66,2%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,28 (с, 1H), 8,33 (дд, J=8,2, 1,6 Гц, 1H), 7,70 (с, 1H), 7,64 (с, 1H), 7,54 (с, 1H), 7,44 (д, J=8,2 Гц, 1H), 7,33 (д, J=8,4 Гц, 1H), 7,07 (с, 1H), 7,05-6,81 (м, 1H), 5,35 (с, 2H), 4,20-4,16 (м, 1H), 3,04 (д, J=11,6 Гц, 1H), 2,36 (с, 3H), 2,26-2,15 (м, 6H); МСНР (ЭР) m/z 524,3 (M+ + 1).
Пример 354: Синтез соединения 354, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(1-изопропилпиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]тиазол-2(3H)-он
[Стадия 1] Синтез соединения 354

3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]тиазол-2(3H)-он (0,100 г, 0,196 ммоль) и триацетоксиборгидрид натрия (0,083 г, 0,393 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего ацетон (0,022 мл, 0,294 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,025 г, 23,1%) в виде светло-оранжевого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,9 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,72 (с, 1H), 7,66 (с, 1H), 7,56 (д, J=1,6 Гц, 1H), 7,46 (д, J=8,2 Гц, 1H), 7,35 (дд, J=8,4, 1,6 Гц, 1H), 7,10-6,82 (м, 2H), 5,38 (с, 2H), 4,19-4,15 (м, 1H), 3,09-3,08 (м, 2H), 2,89-2,88 (м, 2H), 2,41-2,40 (м, 2H), 2,26-2,24 (м, 2H), 2,08-2,06 (м, 2H), 1,14 (д, J=6,0 Гц, 6H); МСНР (ЭР) m/z 552,4 (M+ + 1).
Пример 355: Синтез соединения 355, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(1-(оксетан-3-ил)пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]тиазол-2(3H)-он
[Стадия 1] Синтез соединения 355
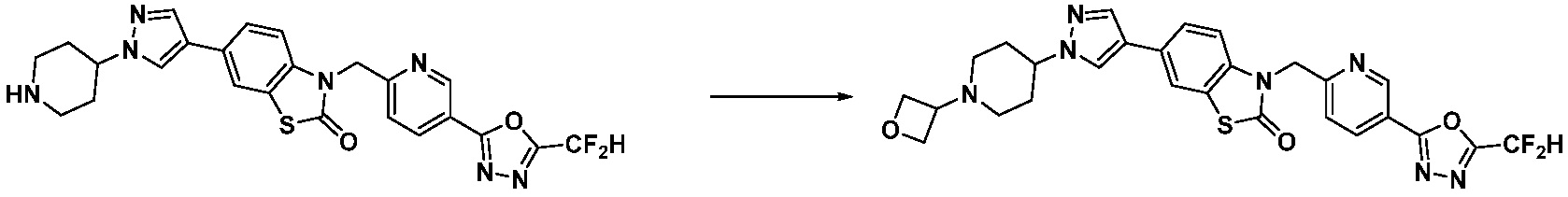
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]тиазол-2(3H)-он (0,100 г, 0,196 ммоль) и триацетоксиборгидрид натрия (0,083 г, 0,393 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего 3-оксетанoн (0,019 мл, 0,294 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=50-90%) и концентрируют с получением указанного в заголовке соединения (0,045 г, 40,5%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,9 Гц, 1H), 8,36 (дд, J=8,2, 2,2 Гц, 1H), 7,73 (с, 1H), 7,65 (с, 1H), 7,56 (д, J=1,4 Гц, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,35 (дд, J=8,4, 1,6 Гц, 1H), 7,10-6,82 (м, 2H), 5,38 (с, 2H), 4,72-4,64 (м, 4H), 4,21-4,17 (м, 1H), 3,58-3,55 (м, 1H), 2,93-2,91 (м, 2H), 2,26-2,23 (м, 2H), 2,16-2,06 (м, 4H); МСНР (ЭР) m/z 566,3 (M+ + 1).
Пример 356: Синтез соединения 356, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]тиазол-2(3H)-он
[Стадия 1] Синтез соединения 356
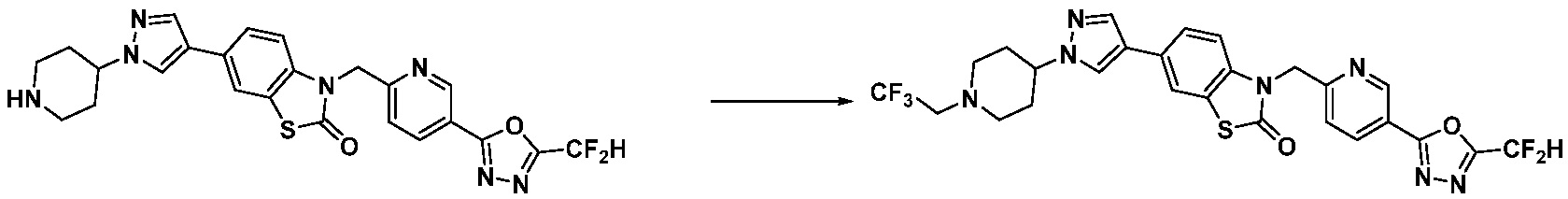
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]тиазол-2(3H)-он (0,100 г, 0,196 ммоль), 2,2,2-трифторэтил трифторметансульфонат (0,042 мл, 0,294 ммоль) и карбонат калия (0,054 г, 0,393 ммоль) растворяют в ацетонитриле (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80°C в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=20-60%) и концентрируют с получением указанного в заголовке соединения (0,075 г, 64,6%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=2,0 Гц, 1H), 8,37 (дд, J=8,2, 2,1 Гц, 1H), 7,73 (с, 1H), 7,65 (с, 1H), 7,57 (с, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,36 (дд, J=8,4, 1,3 Гц, 1H), 7,11-6,82 (м, 2H), 5,39 (с, 2H), 4,21-4,15 (м, 1H), 3,15-3,04 (м, 4H), 2,62 (т, J=10,9 Гц, 2H), 2,20-2,07 (м, 4H); МСНР (ЭР) m/z 592,2 (M+ + 1).
Пример 357: Синтез соединения 357, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(1-((1-фторциклопропил)метил)пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]тиазол-2(3H)-он
[Стадия 1] Синтез соединения 357
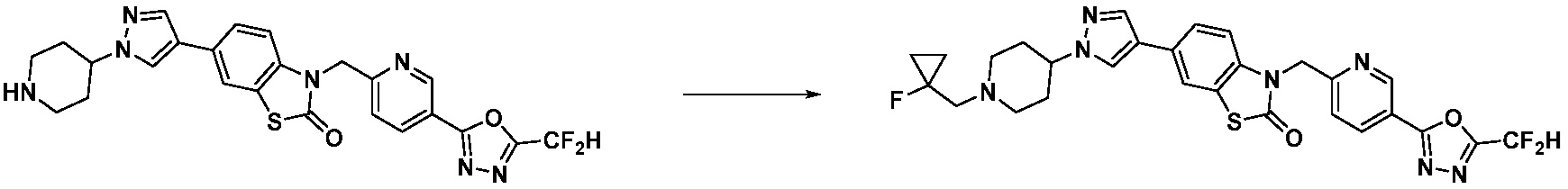
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]тиазол-2(3H)-он (0,100 г, 0,196 ммоль) и 1-фторциклопропан-1-карбальдегид (0,026 г, 0,294 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,083 г, 0,393 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=50-90%) и концентрируют с получением указанного в заголовке соединения (0,067 г, 58,7%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,30 (с, 1H), 8,35 (д, J=8,2 Гц, 2H), 7,72 (с, 1H), 7,65 (с, 1H), 7,55 (с, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,35 (д, J=8,4 Гц, 1H), 7,09-6,82 (м, 2H), 5,37 (с, 2H), 4,21-4,16 (м, 1H), 3,23 (д, J=11,6 Гц, 2H), 2,82 (д, J=21,4 Гц, 2H), 2,36 (т, J=11,3 Гц, 2H), 2,22-2,16 (м, 2H), 2,13-2,05 (м, 2H), 1,15-1,07 (м, 2H), 0,68-0,62 (м, 2H); МСНР (ЭР) m/z 582,3 (M+ + 1).
Пример 358: Синтез соединения 358, трет-бутил 4-(4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилат
[Стадия 1] Синтез соединения 358
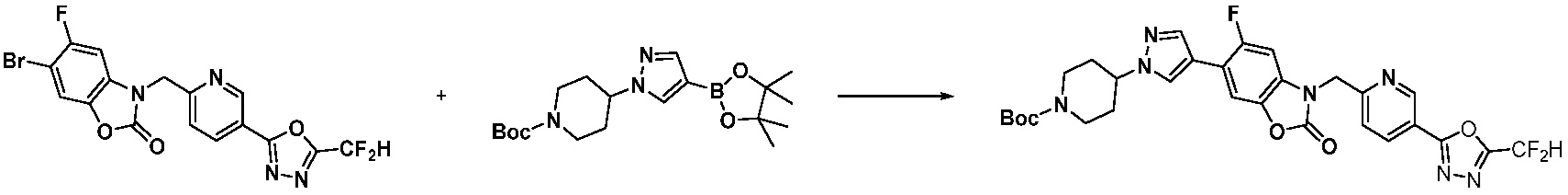
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фторбензо[d]оксазол-2(3H)-он (2,000 г, 4,534 ммоль), трет-бутил 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилат (2,053 г, 5,440 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,148 г, 0,227 ммоль) и карбонат цезия (2,954 г, 9,067 ммоль) смешивают в 1,4-диоксане (20 мл)/воде (5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=20-70%) и концентрируют с получением указанного в заголовке соединения (0,491 г, 17,7%) в виде светло-оранжевого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,8 Гц, 1H), 8,44 (дд, J=8,2, 2,2 Гц, 1H), 7,83 (с, 1H), 7,78 (д, J=2,2 Гц, 1H), 7,58 (д, J=8,2 Гц, 1H), 7,39 (д, J=5,9 Гц, 1H), 7,08-6,83 (м, 2H), 5,21 (с, 2H), 4,36-4,30 (м, 1H), 2,96-2,89 (м, 2H), 2,19 (д, J=10,3 Гц, 2H), 2,00-1,96 (м, 2H), 1,51 (с, 9H); МСНР (ЭР) m/z 612,2 (M+ + 1).
Пример 359: Синтез соединения 359, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 359
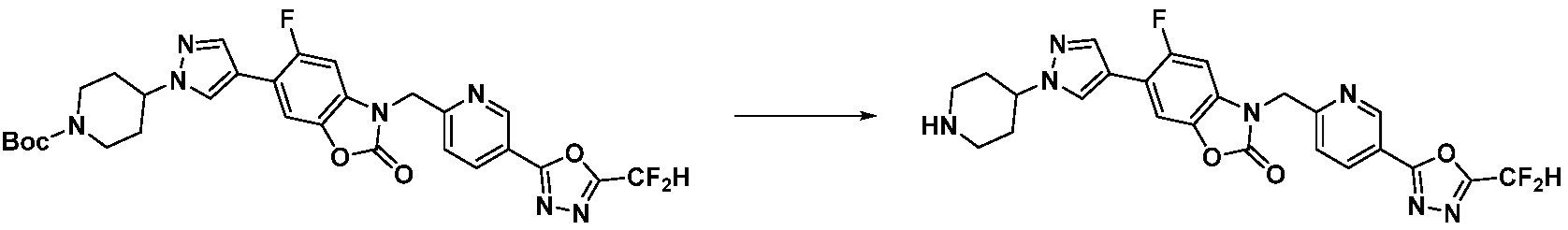
Трет-бутил 4-(4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилат (0,591 г, 0,966 ммоль) и трифторуксусную кислоту (0,370 мл, 4,832 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 6 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Указанное в заголовке соединение применяют без дополнительной очистки (0,515 г, 104,2%, светло-коричневое твердое вещество).
1H ЯМР (400 MГц, ДМСО-d6) δ 9,14 (д, J=1,9 Гц, 1H), 8,47 (дд, J=8,2, 2,1 Гц, 1H), 8,18 (с, 1H), 7,94 (с, 1H), 7,81 (д, J=6,1 Гц, 1H), 7,74 (д, J=8,2 Гц, 1H), 7,70-7,45 (м, 1H), 7,31 (д, J=10,4 Гц, 1H), 5,32 (с, 2H), 4,43-4,40 (м, 1H), 3,36-3,27 (м, 2H), 2,90-2,88 (м, 2H), 2,12 (д, J=11,9 Гц, 1H), 2,02-1,99 (м, 2H); МСНР (ЭР) m/z 512,0 (M+ + 1).
Пример 360: Синтез соединения 360, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-(1-метилпиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 360
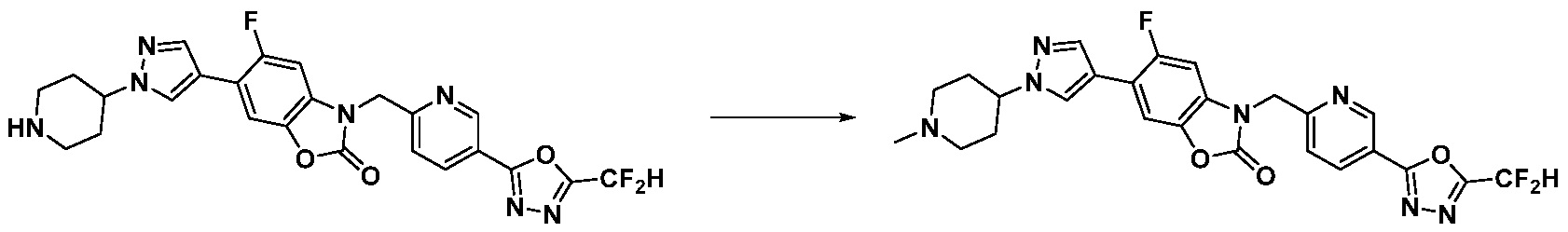
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,196 ммоль) и формальдегид (38,00% раствор, 0,021 мл, 0,293 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,083 г, 0,391 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,023 г, 22,4%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (с, 1H), 8,43 (дд, J=8,1, 1,6 Гц, 1H), 7,81-7,80 (м, 2H), 7,58 (д, J=8,2 Гц, 1H), 7,38 (д, J=5,8 Гц, 1H), 7,08-6,83 (м, 2H), 5,21 (с, 2H), 4,26-4,25 (м, 1H), 3,15-3,14 (м, 2H), 2,46 (с, 3H), 2,36-2,19 (м, 6H); МСНР (ЭР) m/z 526,2 (M+ + 1).
Пример 361: Синтез соединения 361, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-(1-изопропилпиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 361
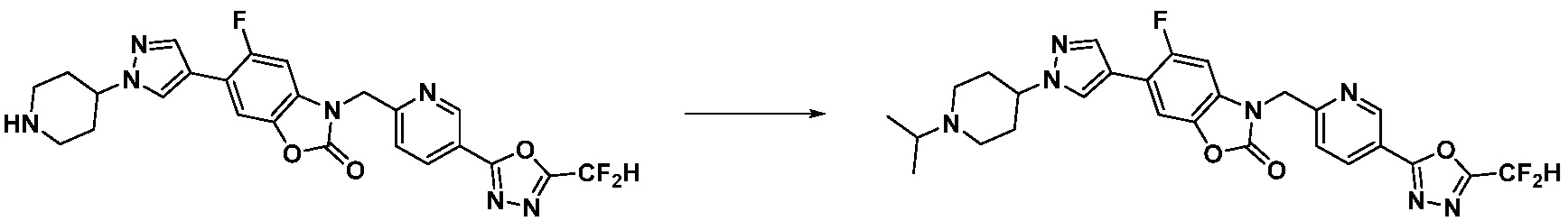
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,196 ммоль) и ацетон (0,022 мл, 0,293 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,083 г, 0,391 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,018 г, 16,6%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,9 Гц, 1H), 8,43 (дд, J=8,2, 2,1 Гц, 1H), 7,81-7,80 (м, 2H), 7,57 (д, J=8,2 Гц, 1H), 7,38 (д, J=5,8 Гц, 1H), 7,08-6,82 (м, 2H), 5,21 (с, 2H), 4,20-4,15 (м, 1H), 3,51-3,49 (м, 2H), 3,11-3,08 (м, 1H), 2,37-2,36 (м, 2H), 2,25-2,19 (м, 2H), 2,13-2,11 (м, 2H), 1,32-1,13 (м, 6H); МСНР (ЭР) m/z 554,5 (M+ + 1).
Пример 362: Синтез соединения 362, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-(1-(оксетан-3-ил)пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 362
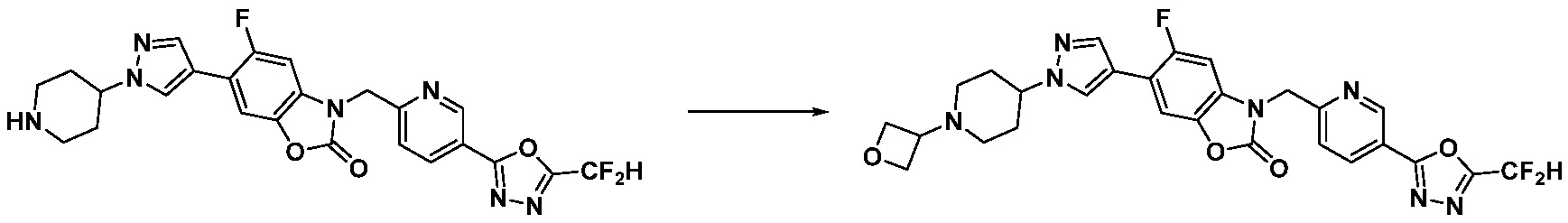
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,196 ммоль) и 3-оксетанон (0,019 мл, 0,293 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,083 г, 0,391 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрируют с получением указанного в заголовке соединения (0,056 г, 5,1%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=1,8 Гц, 1H), 8,43 (дд, J=8,2, 2,2 Гц, 1H), 7,81-7,80 (м, 2H), 7,58 (д, J=8,2 Гц, 1H), 7,38 (д, J=5,9 Гц, 1H), 7,08-6,83 (м, 2H), 5,32 (с, 2H), 4,72-4,64 (м, 4H), 4,24-4,19 (м, 1H), 3,58-3,49 (м, 1H), 2,91 (д, J=9,6 Гц, 1H), 2,23-2,22 (м, 2H), 2,18-2,03 (м, 4H); МСНР (ЭР) m/z 568,4 (M+ + 1).
Пример 363: Синтез соединения 363, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 363
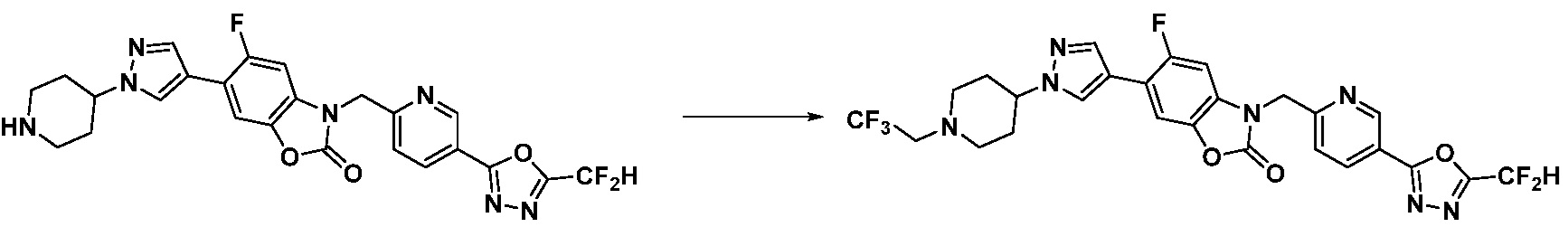
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)бензо[d]оксазол-2(3H)-он (0,100 г, 0,196 ммоль), 2,2,2-трифторэтил трифторметансульфонат (0,042 мл, 0,293 ммоль) и карбонат калия (0,054 г, 0,391 ммоль) растворяют в ацетонитриле (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80°C в течение 18 часов, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=10-40%) и концентрируют с получением указанного в заголовке соединения (0,006 г, 5,2%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,33 (д, J=1,8 Гц, 1H), 8,43 (дд, J=8,2, 2,1 Гц, 1H), 7,82 (с, 1H), 7,79 (д, J=2,2 Гц, 1H), 7,58 (д, J=8,2 Гц, 1H), 7,39 (д, J=5,9 Гц, 1H), 7,08-6,83 (м, 2H), 5,21 (с, 2H), 4,21-4,20 (м, 1H), 3,16-3,05 (м, 4H), 2,63 (т, J=10,5 Гц, 1H), 2,19-2,12 (м, 4H); МСНР (ЭР) m/z 594,3 (M+ + 1).
Пример 364: Синтез соединения 364, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(2-(4-метилпиперазин-1-ил)пиридин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 364
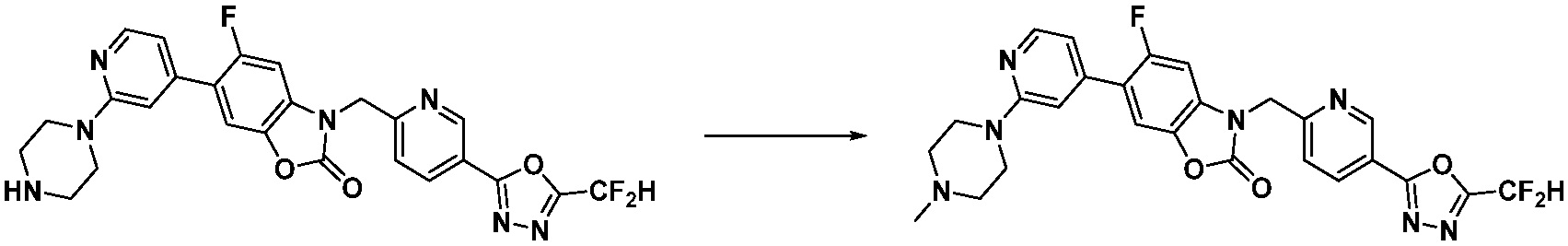
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(2-(пиперазин-1-ил)пиридин-4-ил)бензо[d]тиазол-2(3H)-он (0,100 г, 0,191 ммоль) и формальдегид (38,00% раствор, 0,021 мл, 0,287 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,081 г, 0,382 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,072 г, 70,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (с, 1H), 8,44 (дд, J=8,2, 2,1 Гц, 1H), 8,24 (д, J=5,6 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,31 (д, J=5,8 Гц, 1H), 7,08-6,82 (м, 2H), 6,74 (шс, 2H), 5,23 (с, 2H), 3,62 (т, J=4,9 Гц, 4H), 2,55 (т, J=4,9 Гц, 4H), 2,37 (с, 3H); МСНР (ЭР) m/z 538,3 (M+ + 1).
Пример 365: Синтез соединения 365, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(2-(4-изопропилпиперазин-1-ил)пиридин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 365
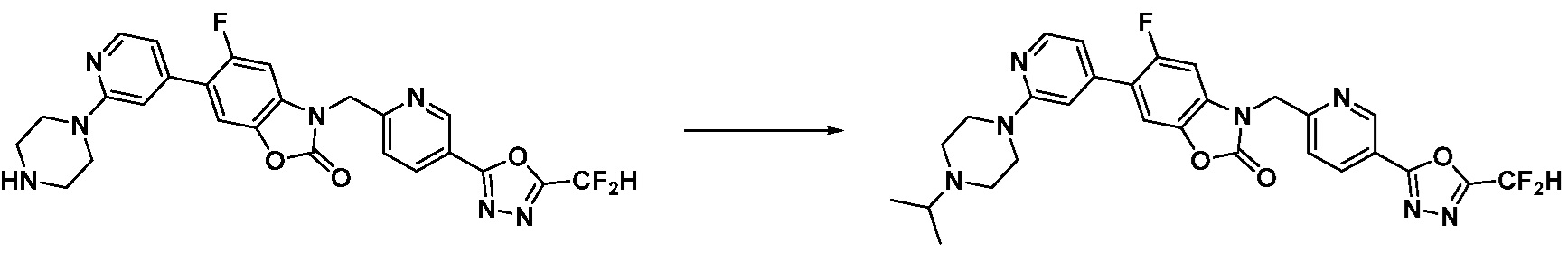
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(2-(пиперазин-1-ил)пиридин-4-ил)бензо[d]тиазол-2(3H)-он (0,100 г, 0,191 ммоль) и ацетон (0,021 мл, 0,287 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,081 г, 0,382 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,072 г, 66,6%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,5 Гц, 1H), 8,43 (дд, J=8,2, 2,1 Гц, 1H), 8,23 (д, J=5,7 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,31 (д, J=5,8 Гц, 1H), 7,08-7,82 (м, 2H), 6,73 (шс, 2H), 5,22 (с, 2H), 3,60 (т, J=4,9 Гц, 4H), 2,76-2,72 (м, 1H), 2,65 (т, J=4,9 Гц, 4H), 1,10 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 566,1 (M+ + 1).
Пример 366: Синтез соединения 366, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(2-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 366
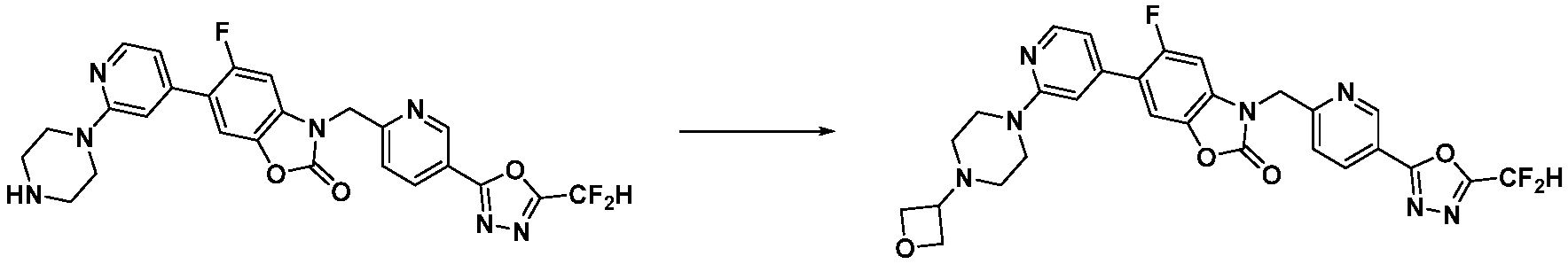
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(2-(пиперазин-1-ил)пиридин-4-ил)бензо[d]тиазол-2(3H)-он (0,100 г, 0,191 ммоль) и 3-оксетанoн (0,018 мл, 0,287 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,081 г, 0,382 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрируют с получением указанного в заголовке соединения (0,030 г, 27,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,32 (д, J=2,0 Гц, 1H), 8,44 (дд, J=8,2, 2,1 Гц, 1H), 8,25 (д, J=5,2 Гц, 1H), 7,60 (д, J=8,2 Гц, 1H), 7,32 (д, J=5,9 Гц, 1H), 7,09-6,83 (м, 2H), 6,77-6,74 (м, 2H), 5,23 (с, 2H), 4,73-4,67 (м, 4H), 4,15 (т, J=7,2 Гц, 4H), 3,57-3,54 (м, 1H), 2,47 (т, J=5,0 Гц, 4H); МСНР (ЭР) m/z 580,4 (M+ + 1).
Пример 367: Синтез соединения 367, 6-(2-(4-циклобутилпиперазин-1-ил)пиридин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фторбензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез соединения 367
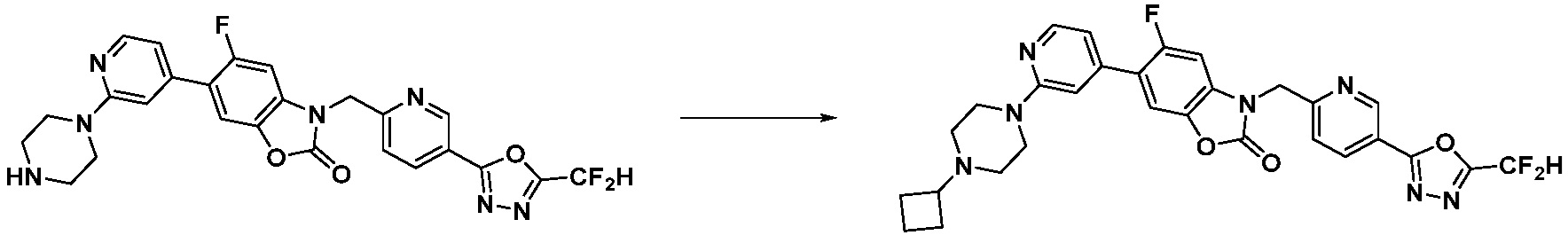
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(2-(пиперазин-1-ил)пиридин-4-ил)бензо[d]тиазол-2(3H)-он (0,100 г, 0,191 ммоль) и циклобутанон (0,021 г, 0,287 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,081 г, 0,382 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстрагирование с дихлорметаном, затем фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрируют с получением указанного в заголовке соединения (0,052 г, 47,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 9,31 (д, J=1,8 Гц, 1H), 8,44 (дд, J=8,2, 2,1 Гц, 1H), 8,24 (д, J=5,3 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,31 (д, J=5,8 Гц, 1H), 7,08-6,82 (м, 2H), 6,74-6,43 (м, 2H), 5,23 (с, 2H), 3,61 (т, J=4,9 Гц, 4H), 2,80-2,76 (м, 1H), 2,11-2,07 (м, 2H), 1,97-1,92 (м, 2H), 1,77-1,73 (м, 2H); МСНР (ЭР) m/z 578,4 (M+ + 1).
Пример 368: Синтез соединения 368, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-6-(2-(пиперазин-1-ил)пиридин-4-ил)бензо[d]оксазол-2(3H)-он
[Стадия 1] Синтез трет-бутил 4-(4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиридин-2-ил)пиперазин-1-карбоксилата
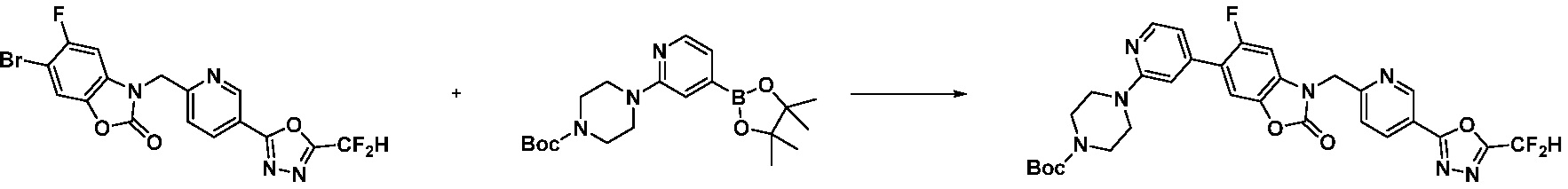
6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фторбензо[d]оксазол-2(3H)-он (2,000 г, 4,534 ммоль), трет-бутил 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)пиперазин-1-карбоксилат (2,118 г, 5,440 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,148 г, 0,227 ммоль) и карбонат цезия (2,954 г, 9,067 ммоль) смешивают в 1,4-диоксане (10 мл)/воде (2,5 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100° в течение 20 минут, и затем реакцию останавливают понижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=5 to 20%) и концентрируют с получением указанного в заголовке соединения (1,260 г, 44,6%) в виде серого твердого вещества.
[Стадия 2] Синтез соединения 368
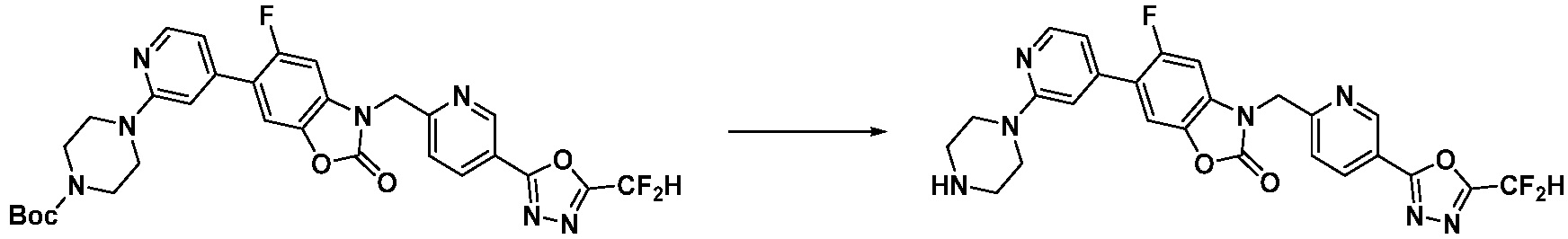
Трет-бутил 4-(4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фтор-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)пиридин-2-ил)пиперазин-1-карбоксилат (1,000 г, 1,604 ммоль), полученный на стадии 1 и трифторуксусную кислоту (0,982 мл, 12,829 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 5 часов. Насыщенный водный раствор гидрокарбоната натрия и дихлорметан добавляют в реакционную смесь и перемешивают для отфильтровывания выпавшего в осадок твердого вещества, затем промывают с дихлорметаном, и затем сушат с получением указанного в заголовке соединения (0,830 г, 98,9%) в виде розового твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 9,14 (д, J=1,8 Гц, 1H), 8,49 (дд, J=8,2, 2,2 Гц, 1H), 8,22 (д, J=5,2 Гц, 1H), 7,77 (д, J=8,3 Гц, 1H), 7,73-7,45 (м, 2H), 7,41 (д, J=10,2 Гц, 1H), 7,04 (с, 1H), 6,92 (д, J=4,8 Гц, 1H), 5,38 (с, 2H), 3,75 (шс, 4H), 3,17 (шс, 4H); МСНР (ЭР) m/z 524,1 (M+ + 1).
Протокол измерения и анализа активности соединения по изобретению
<Экспериментальный Пример 1> Идентификация ингибирования активности HDAC фермента (in vitro)
Селективный ингибитор HDAC6 является важным для селективности ингибирования HDAC1, что является причиной побочных эффектов, и, таким образом, идентифицируют селективность к ферменту HDAC1/6 и клеточную селективность (HDAC1: ацетилирование гистона/HDAC6: ацетилирование тубулина).
1. Способ эксперимента
Ингибирующую способность к HDAC ферменту тестируемого продукта измеряют с применением HDAC1 Fluorimetric Drug Discovery Assay Kit (Enzolifesciences: BML-AK511) и рекомбинантного HDAC6 человека (Calbiochem: 382180). Для анализа HDAC1, образцы обрабатывают в концентрации 100, 1000 и 10000 нМ. Для анализа HDAC6, образцы обрабатывают в концентрации 0,1, 1, 10, 100 и 1000 нМ. После вышеуказанной обработки образца, реакцию продолжают при 37° в течение 60 минут, затем обрабатывают проявителем, и затем подвергают взаимодействию при 37° в течение 30 минут, после чего измеряют интенсивность флуоресценции (Ex 390, Em 460) с применением FlexStatin3 (Molecular device).
2. Результаты эксперимента
Результаты измерения ингибирующей активность к ферменту HDAC, полученные согласно вышеуказанному способу эксперимента, показаны в следующей таблице 2.
|
Согласно таблице 2, можно определить, что производные соединения 1,3,4-оксадиазола согласно настоящему изобретению показывают превосходную селективность к ферменту HDAC1/6.
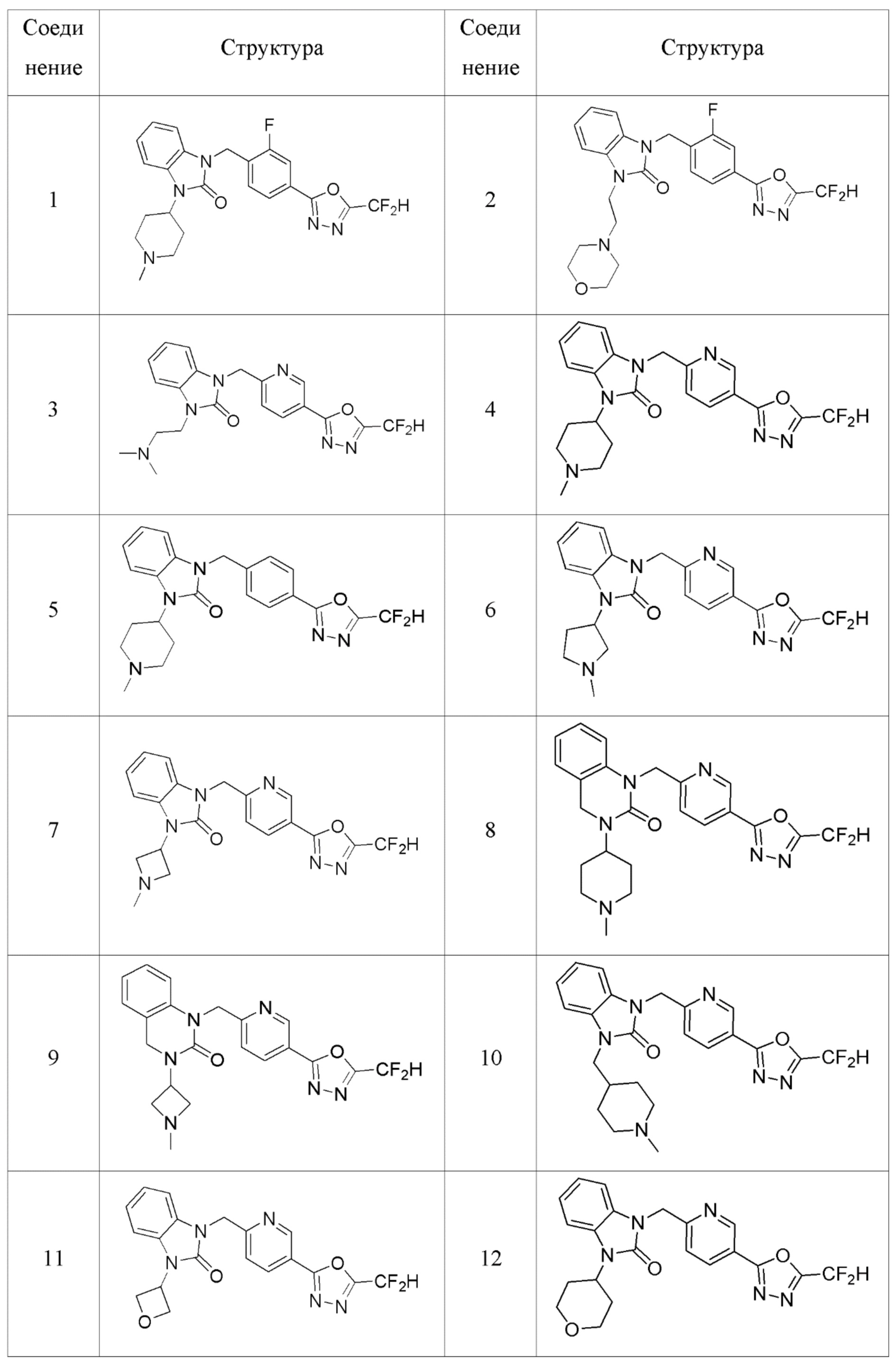
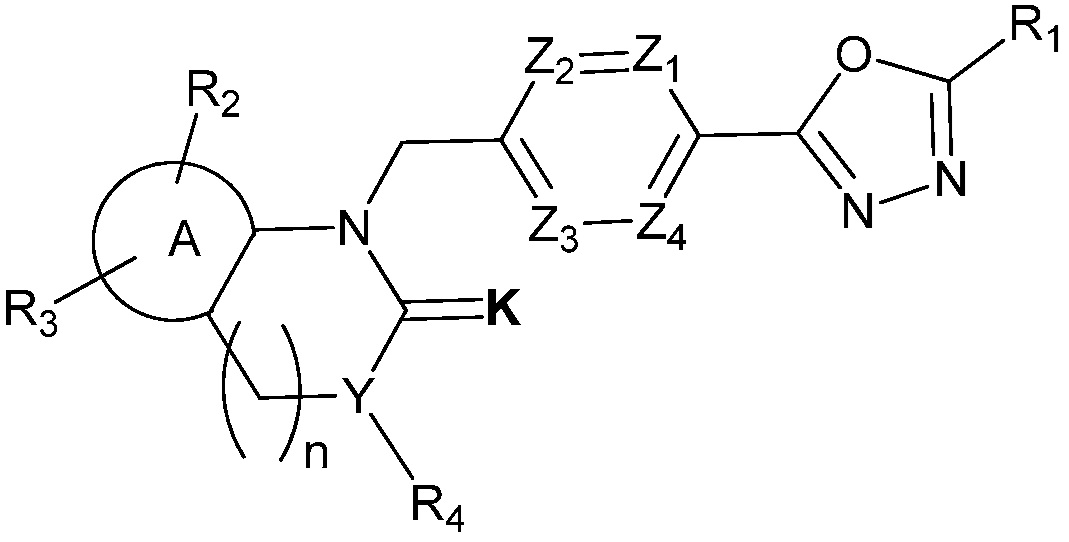
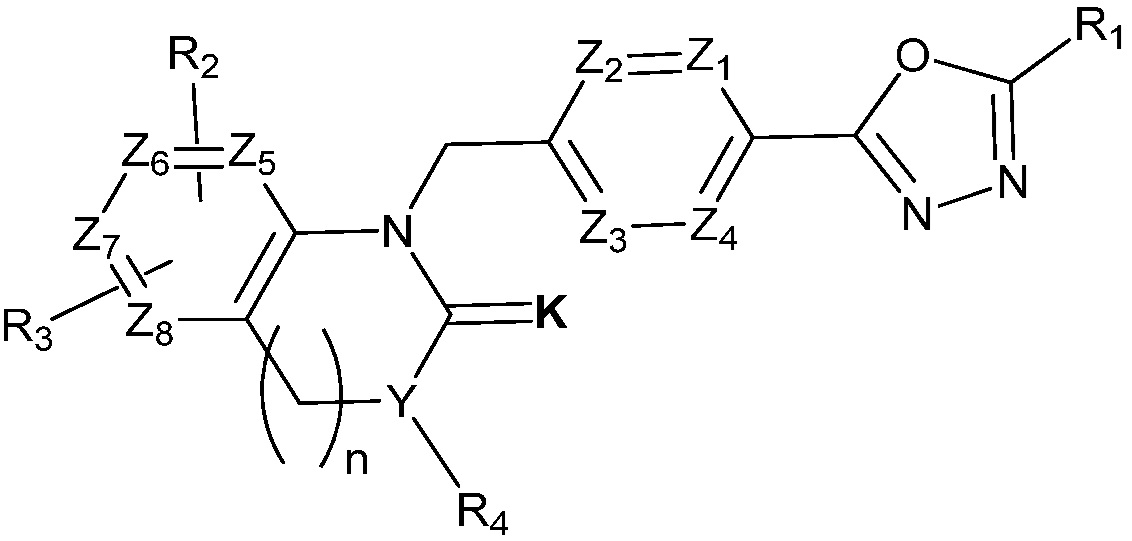
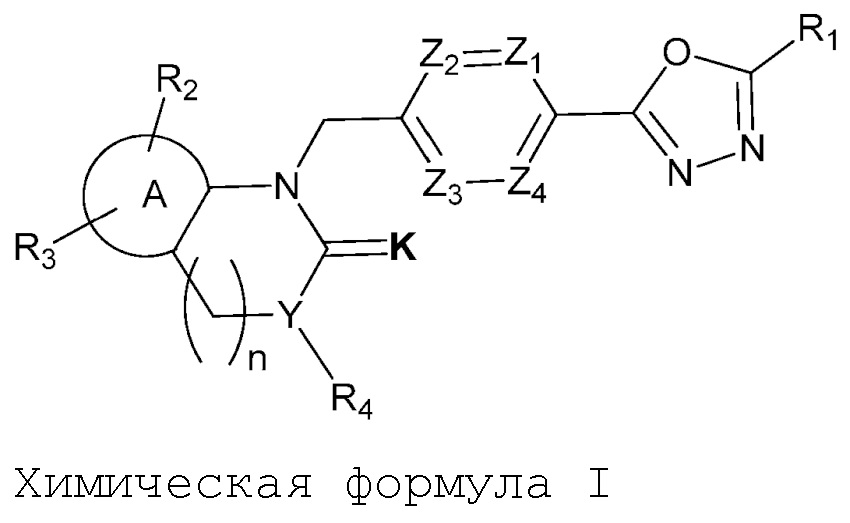
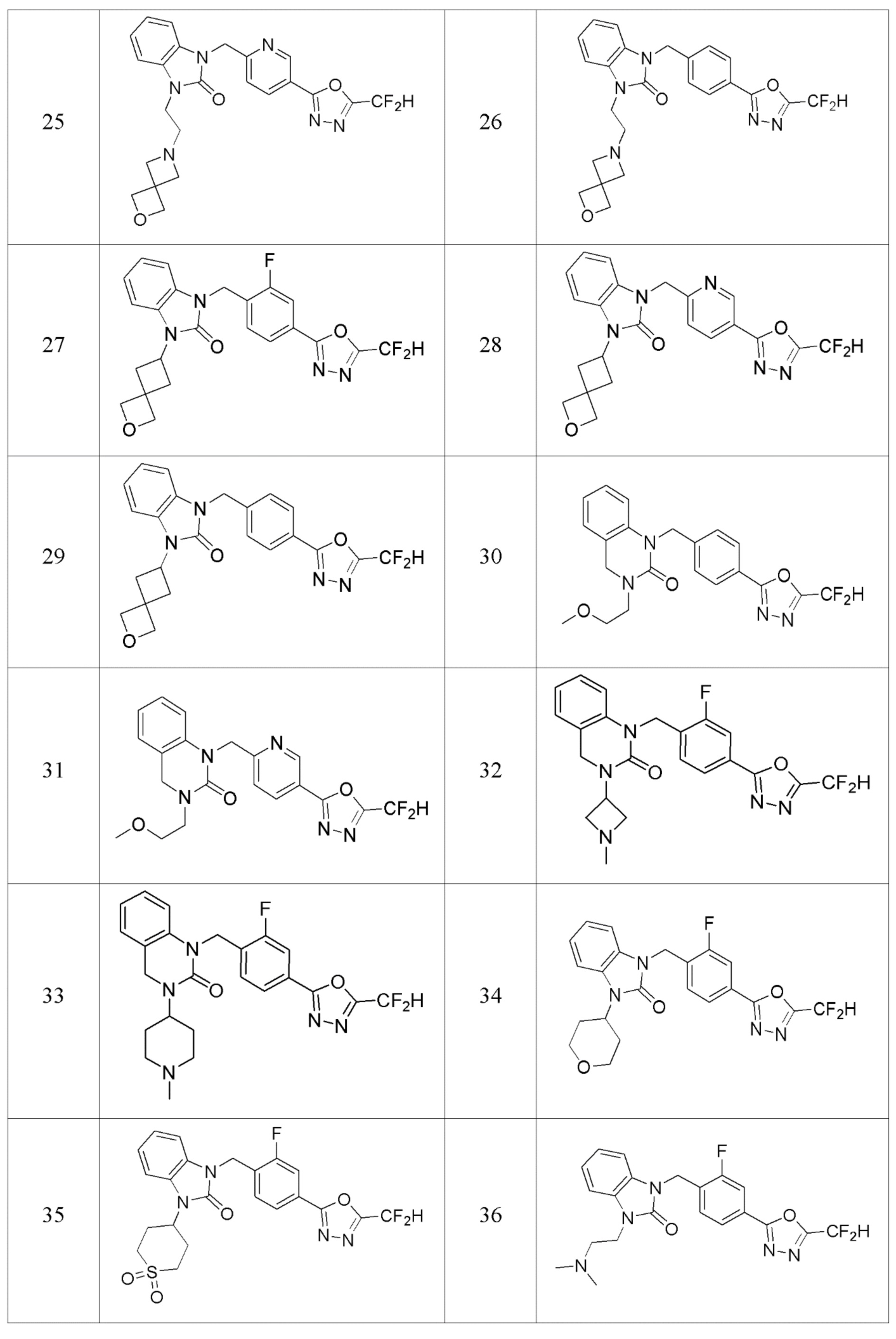
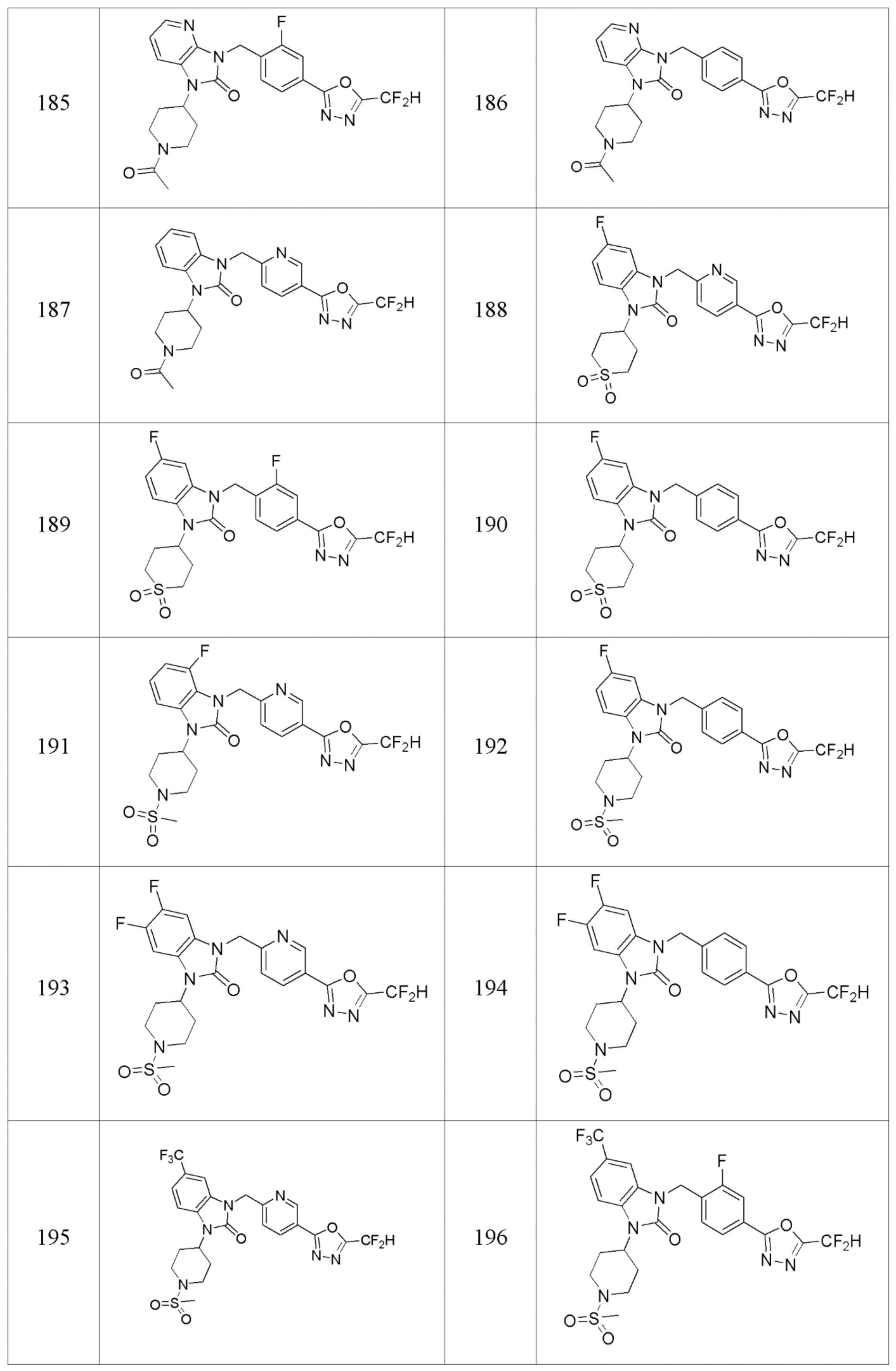
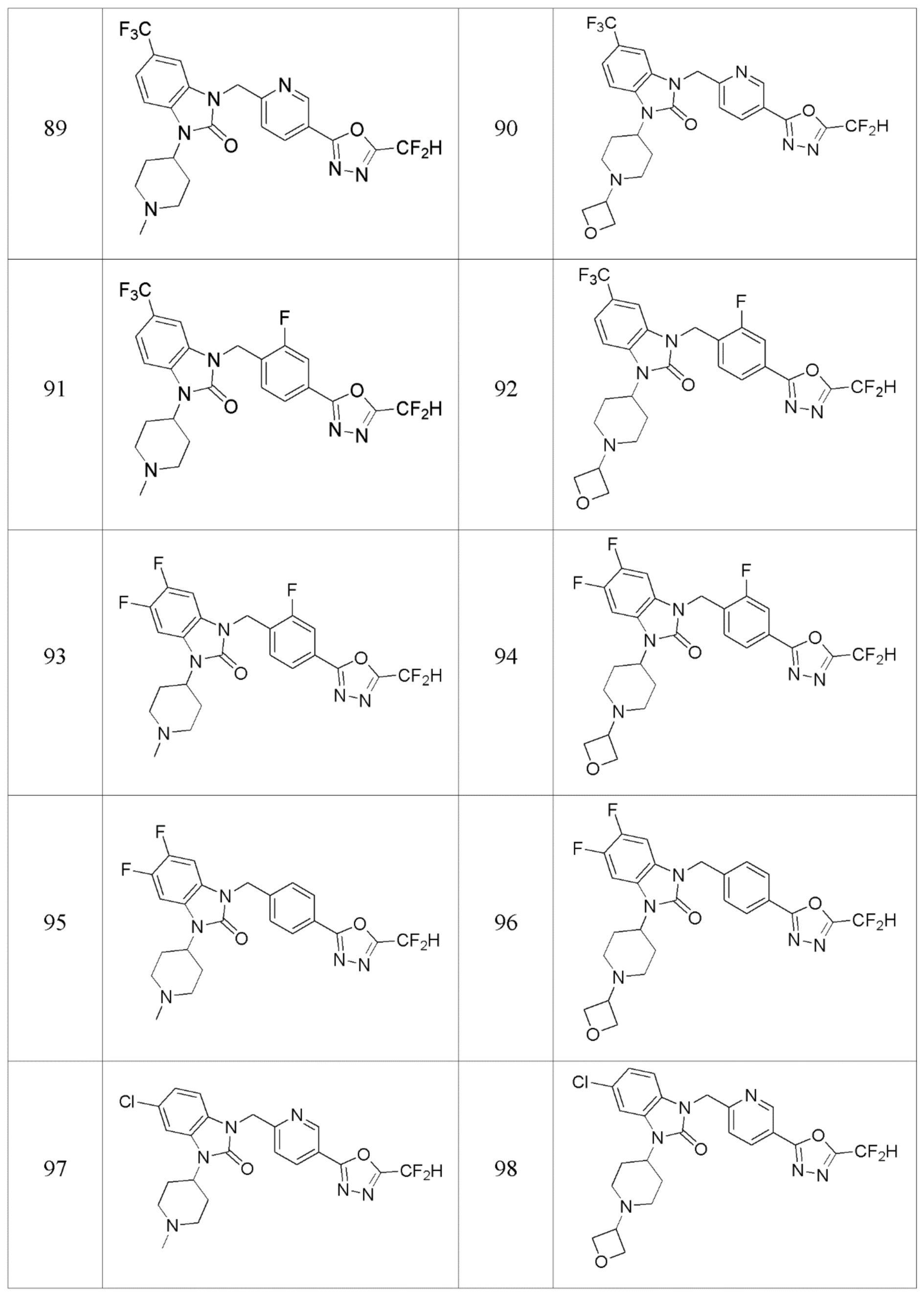
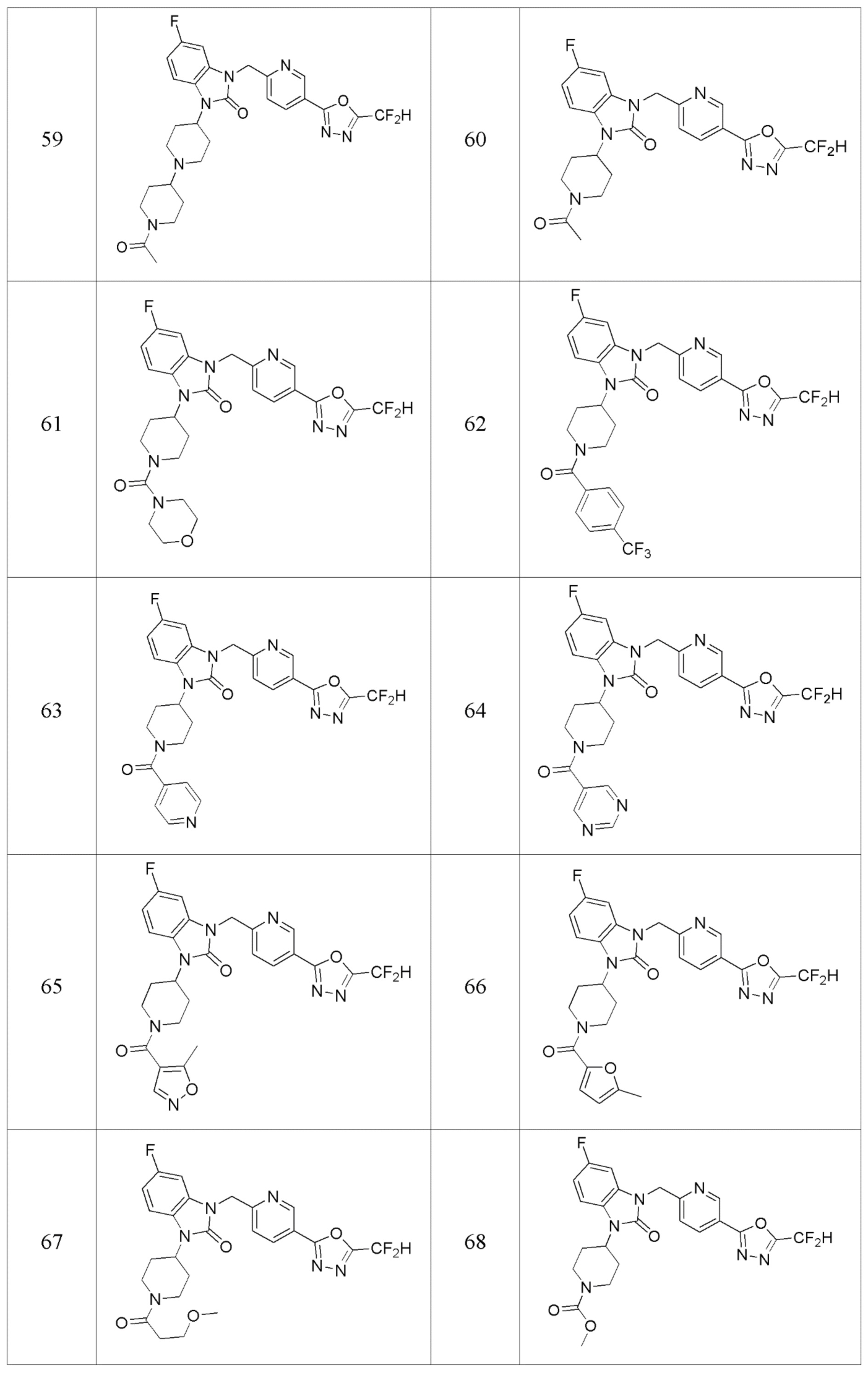
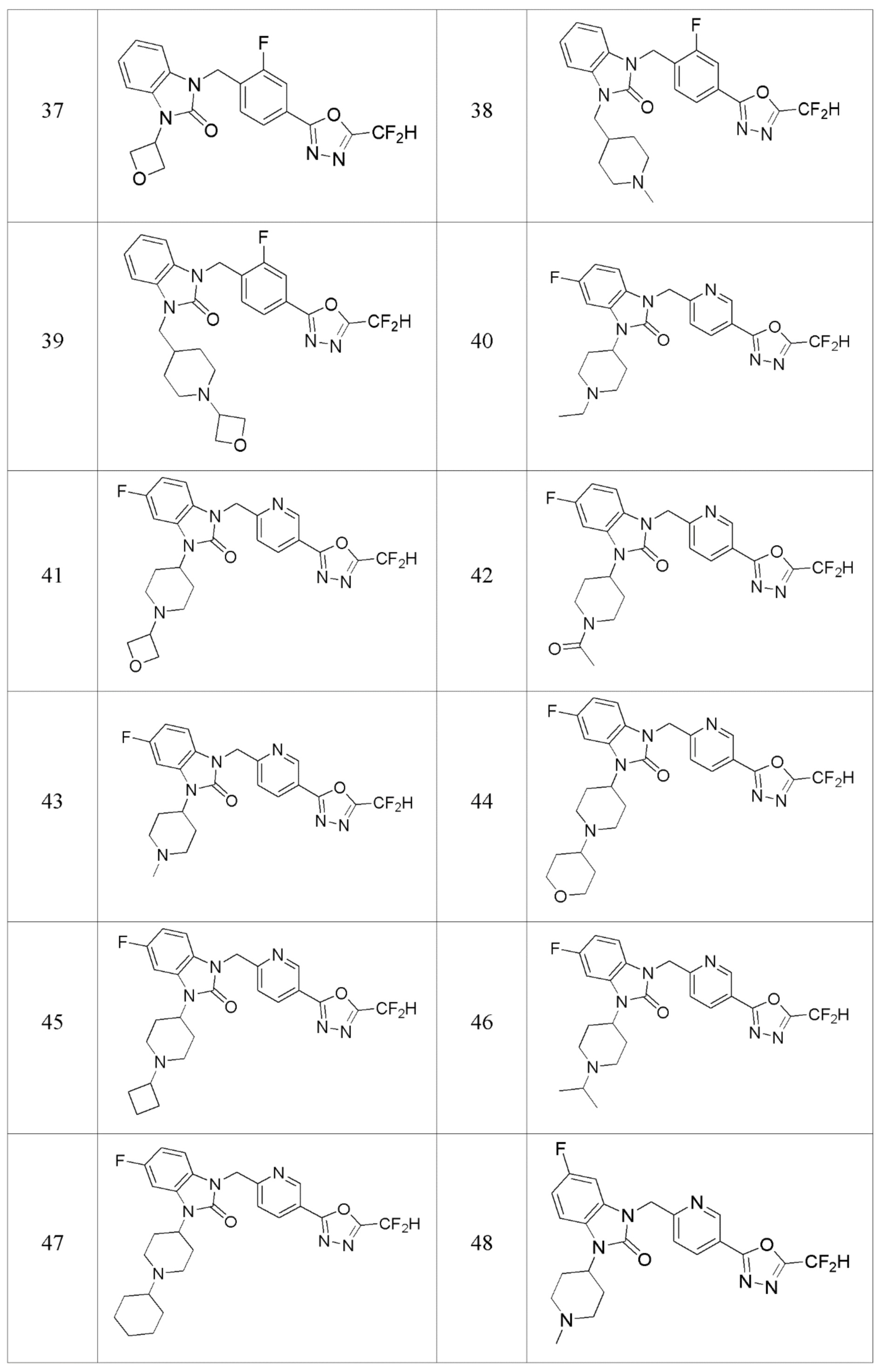
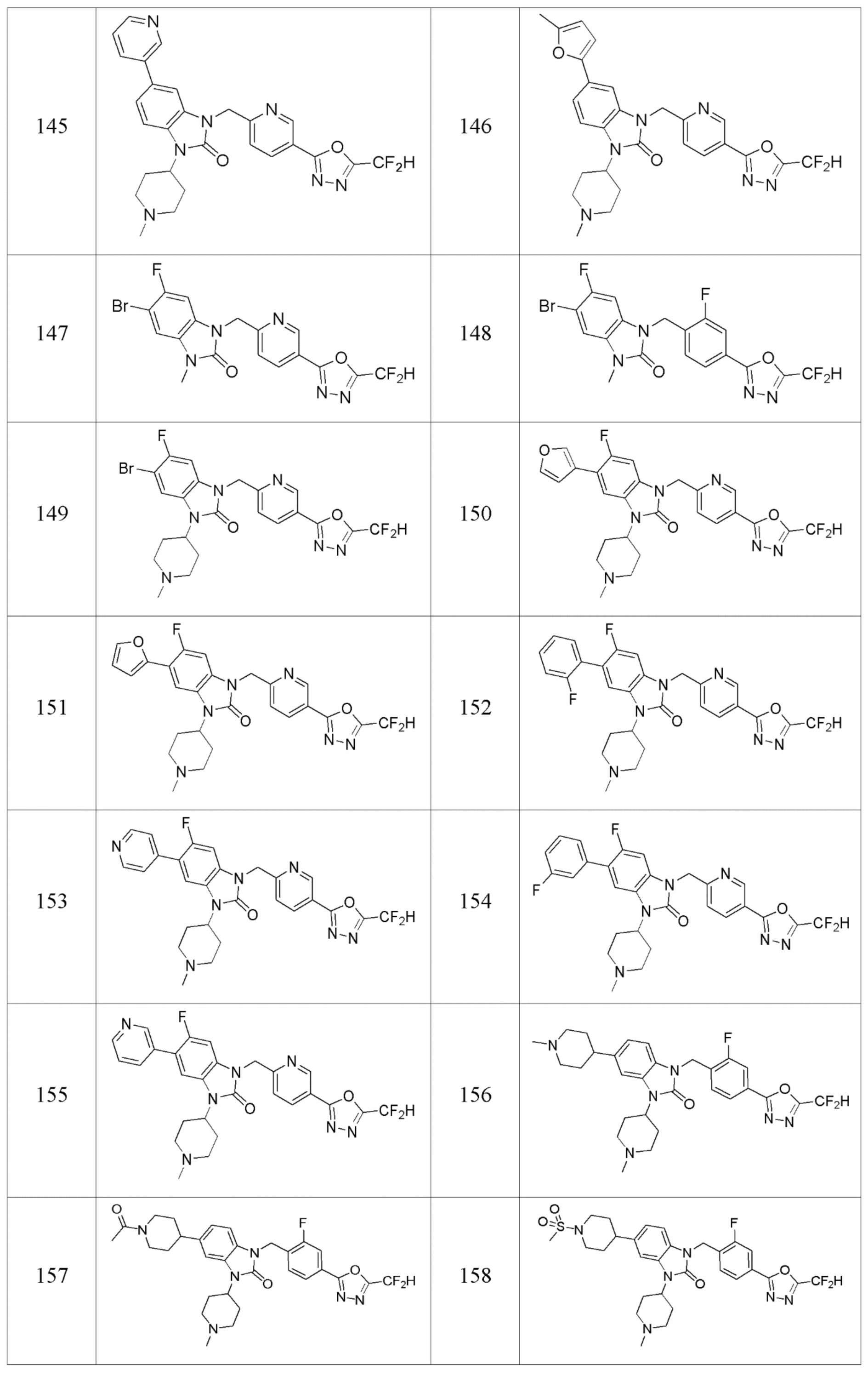
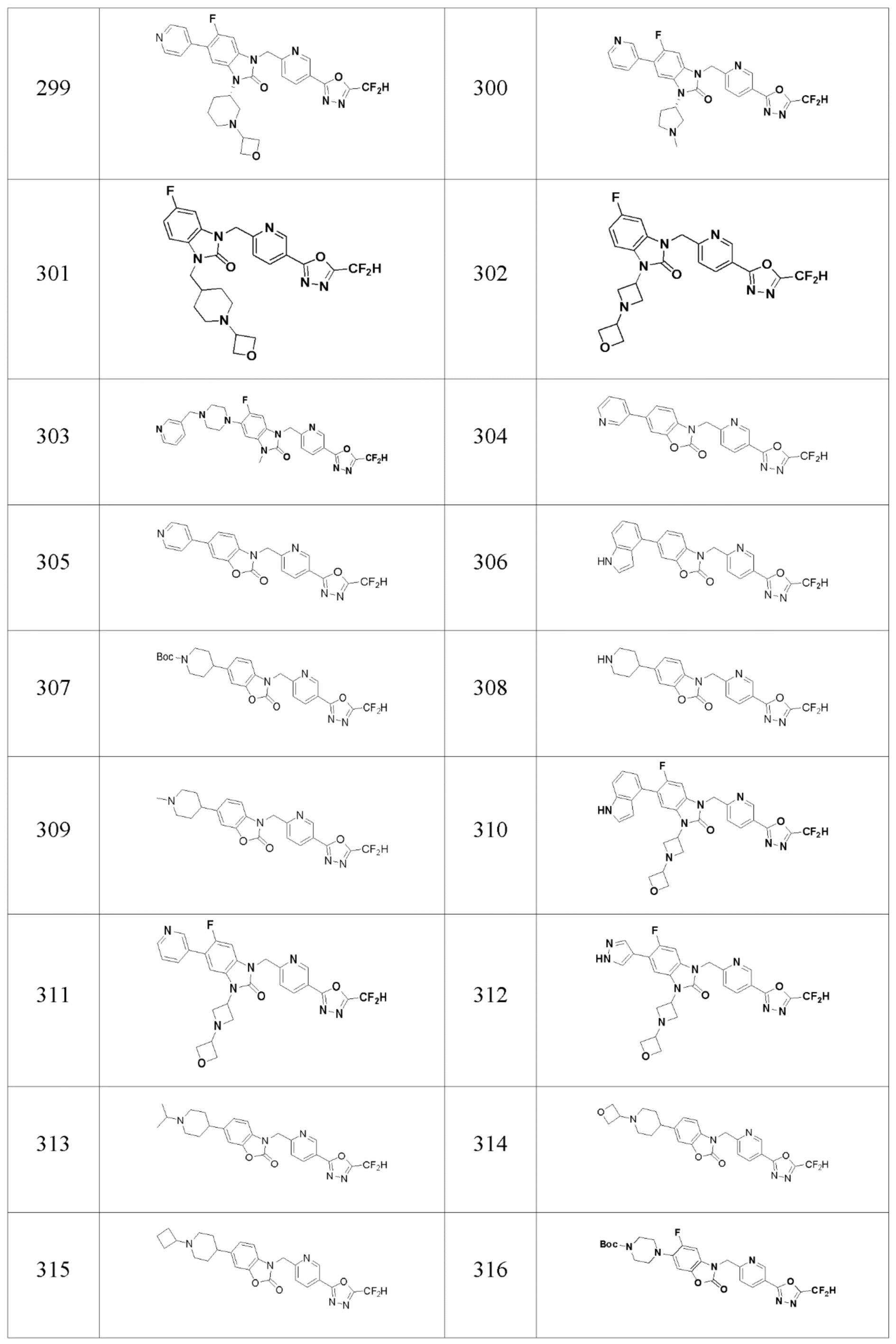
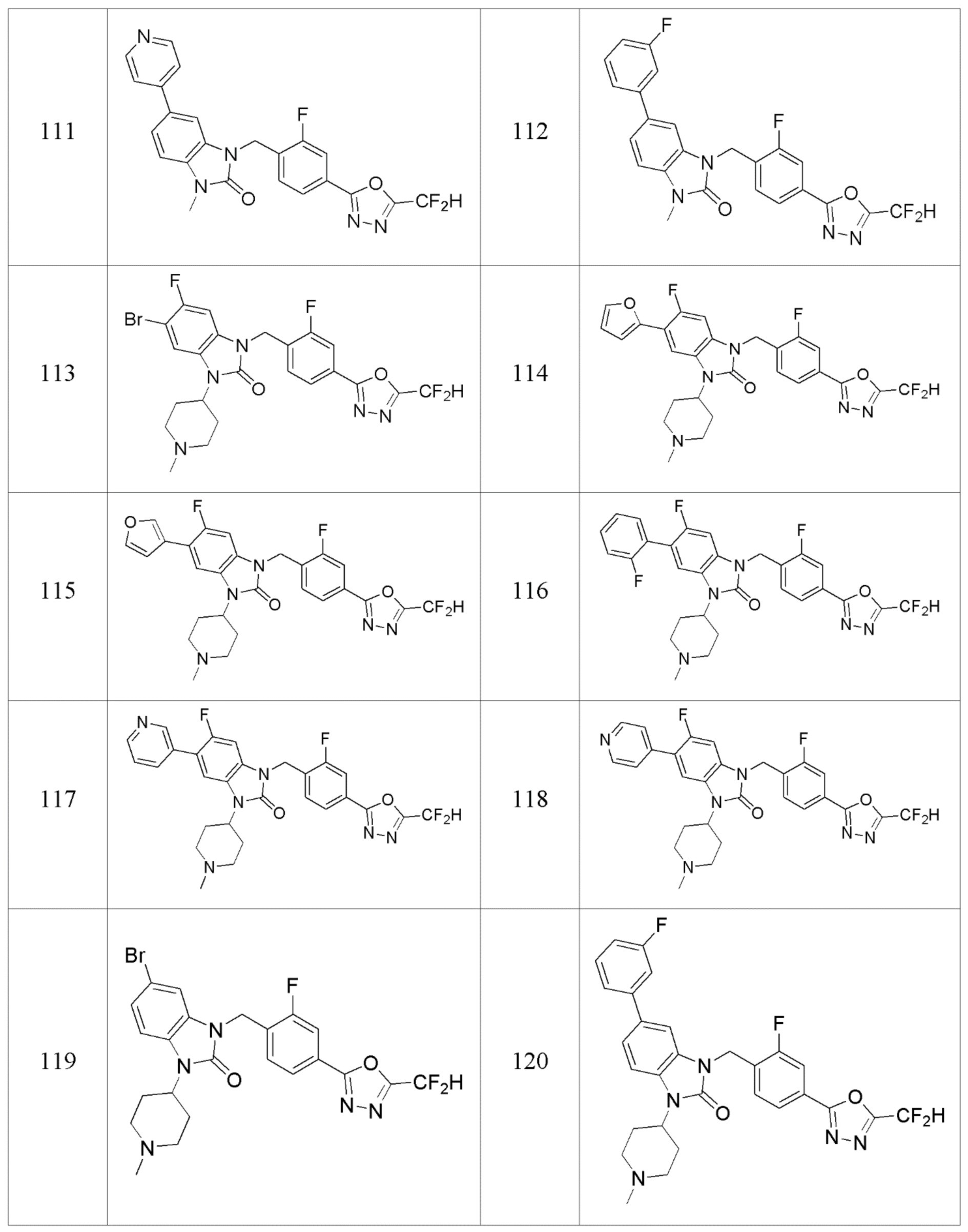
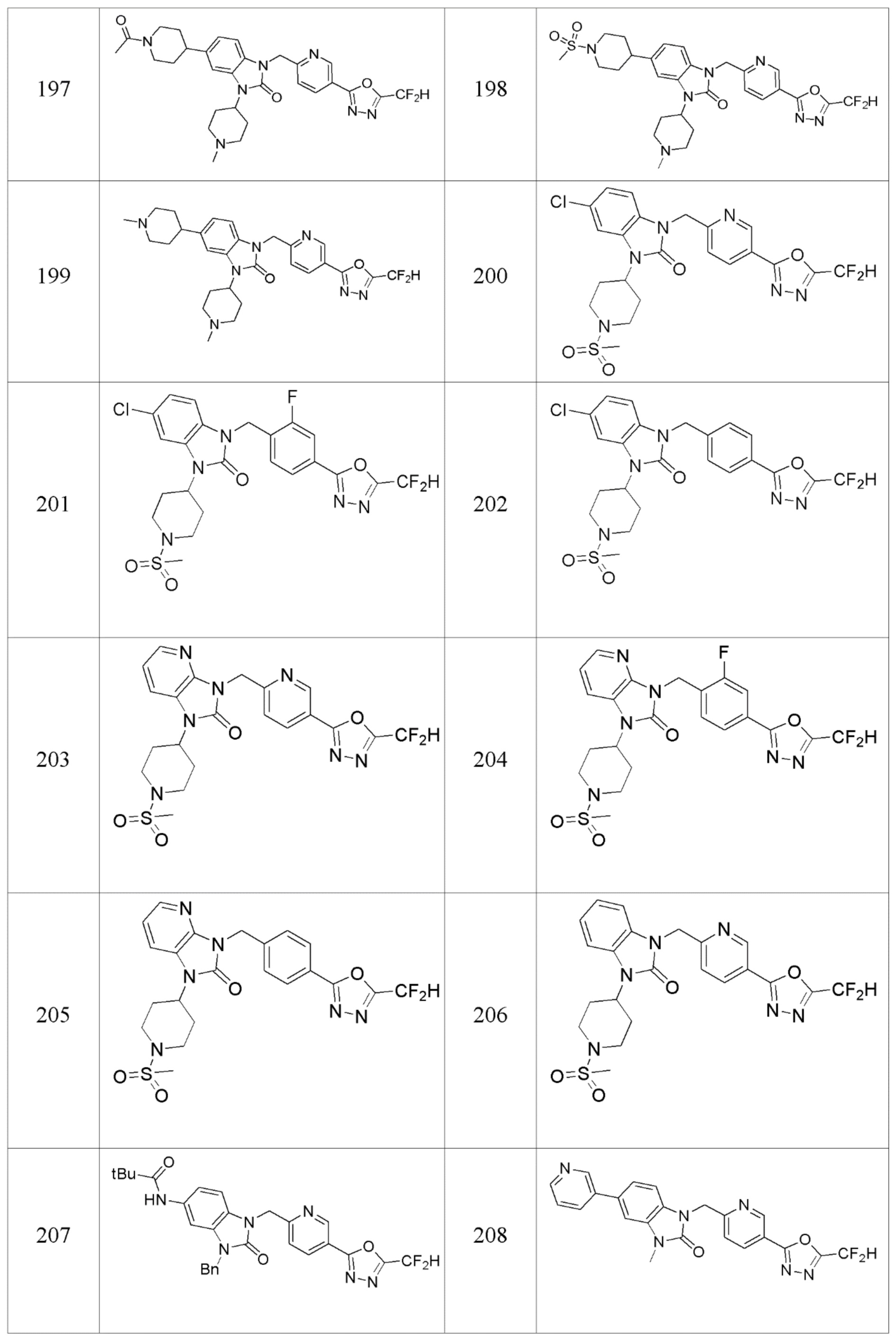
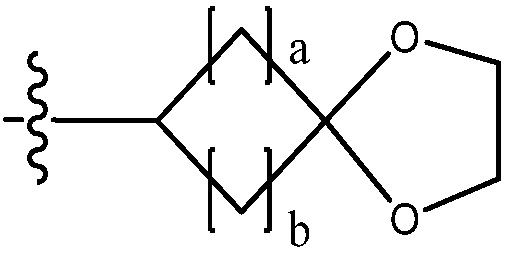
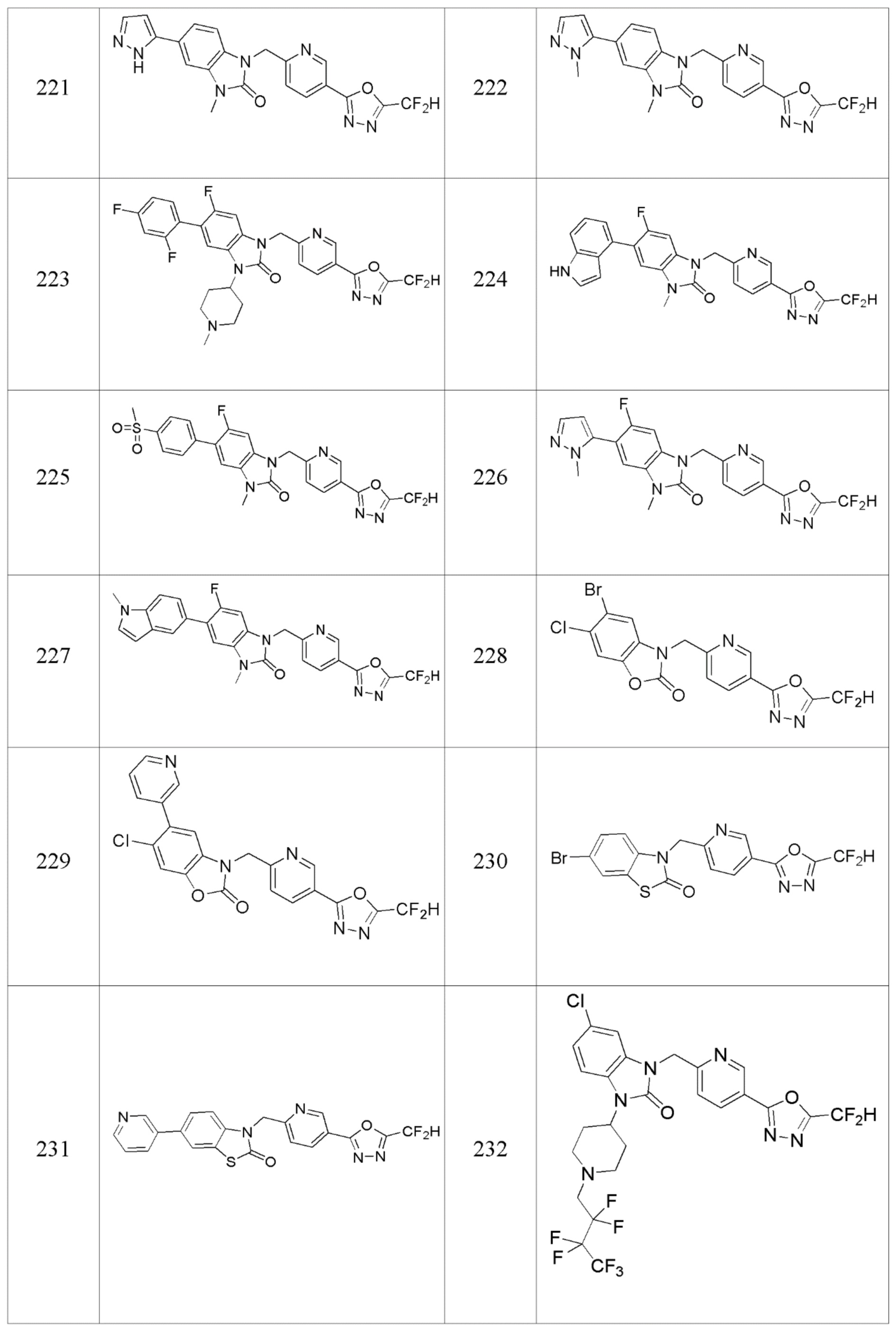
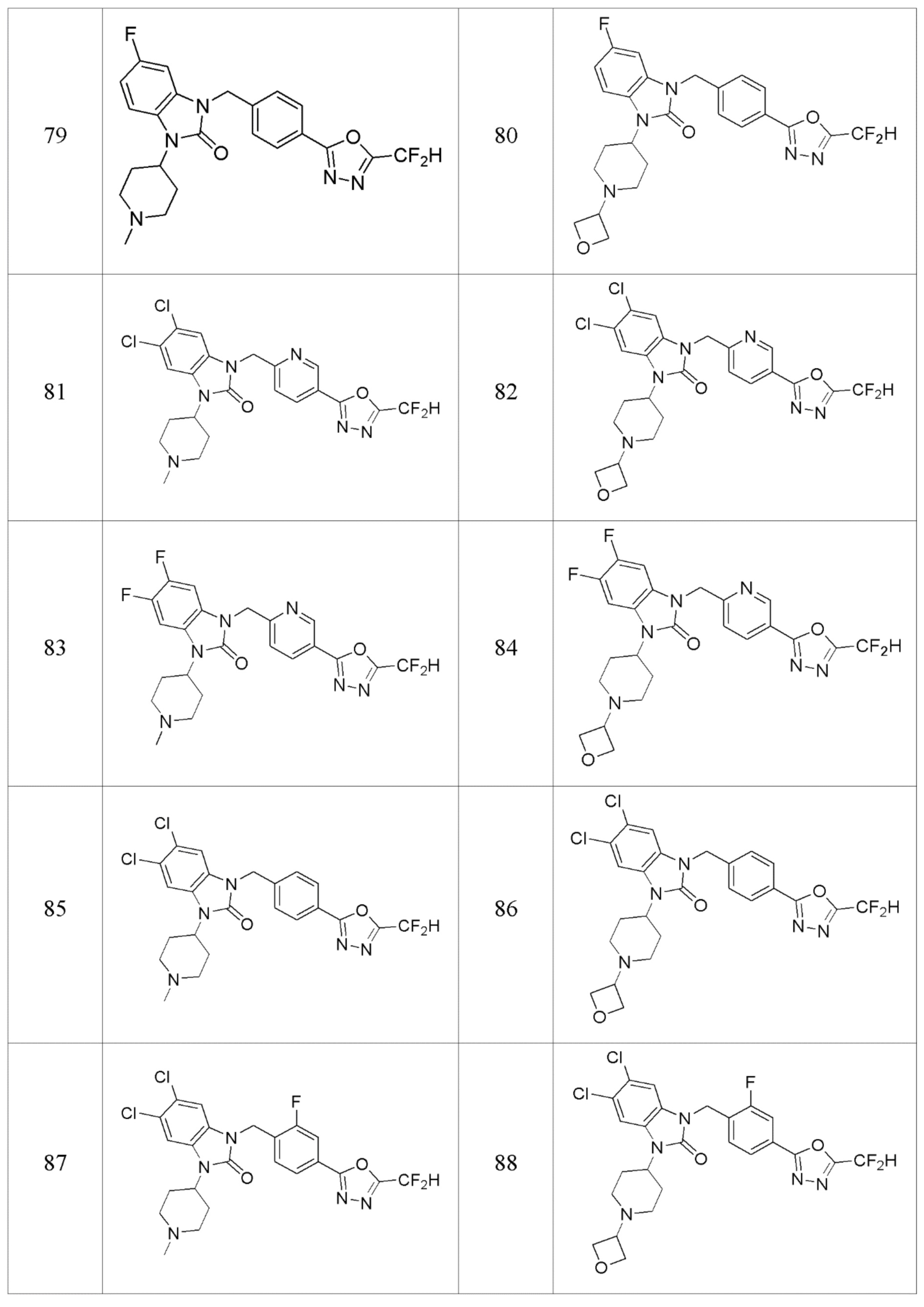
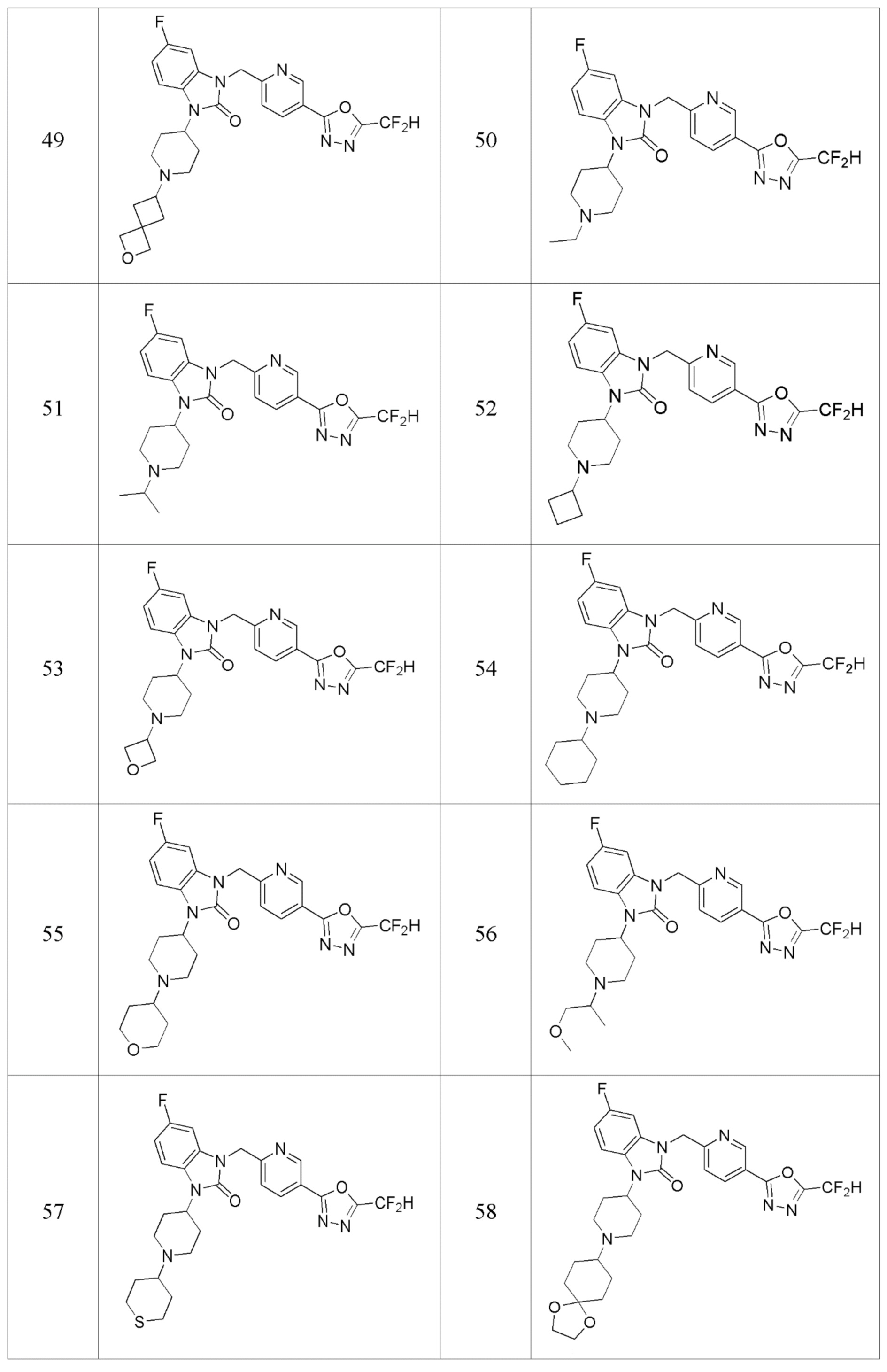
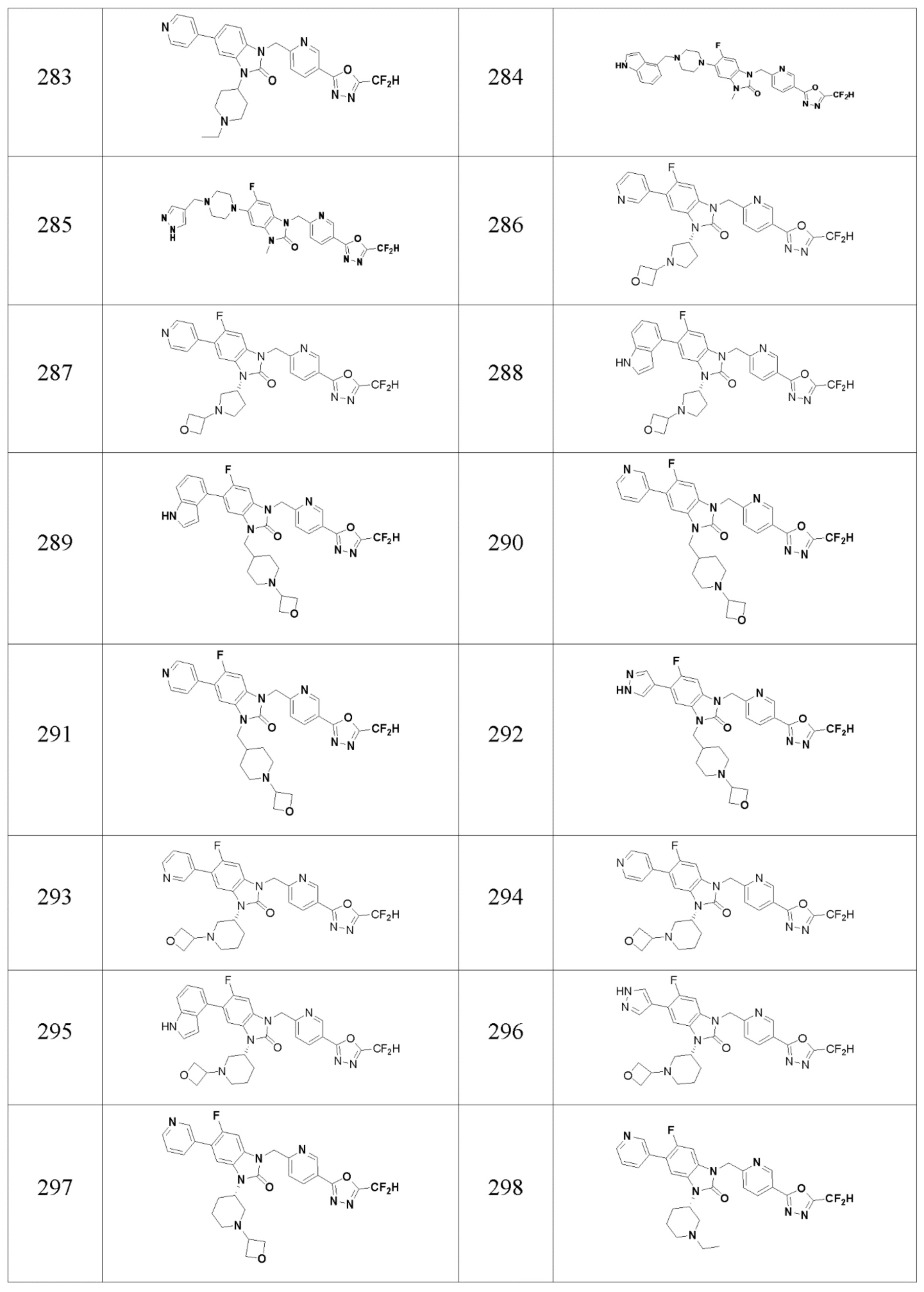
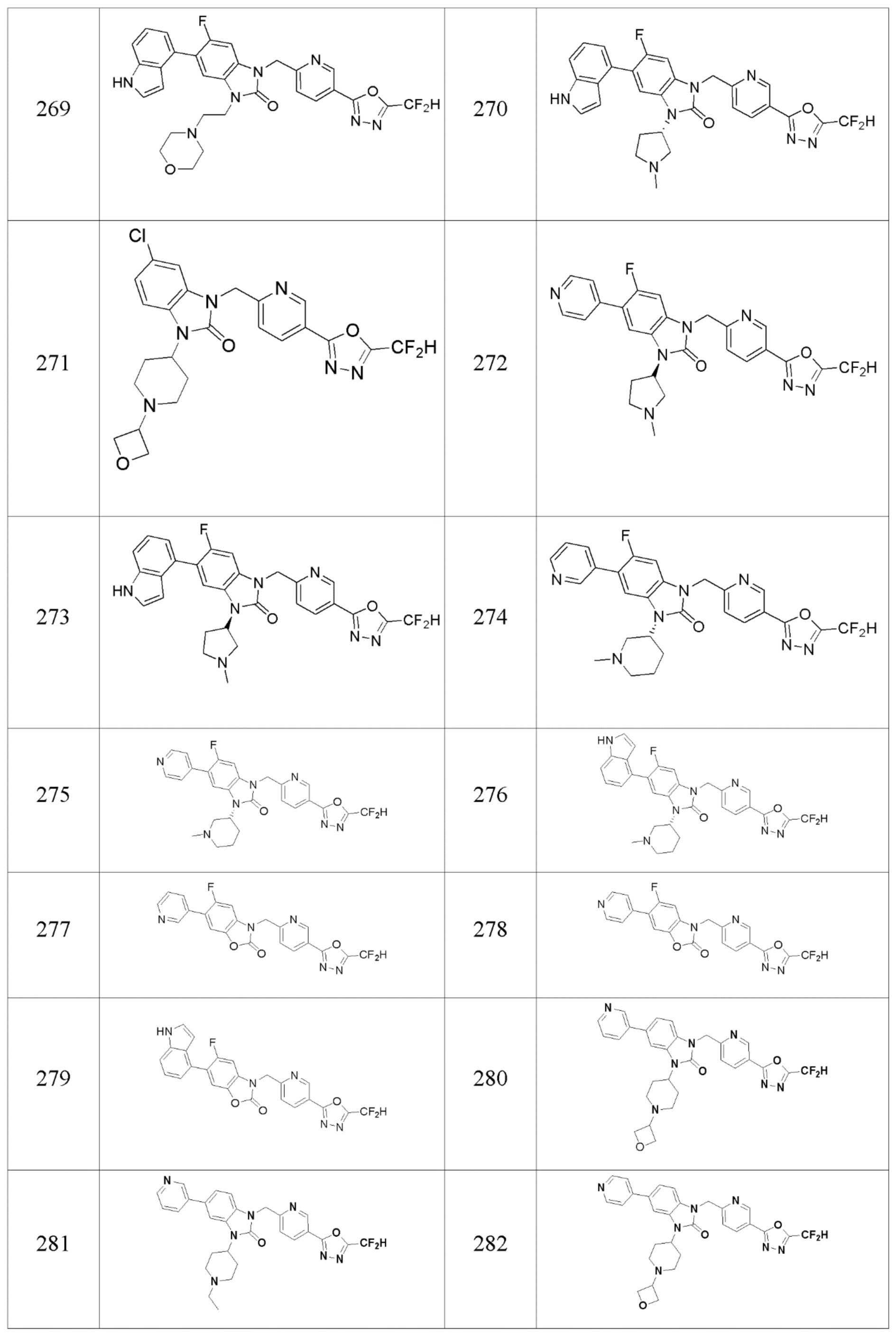
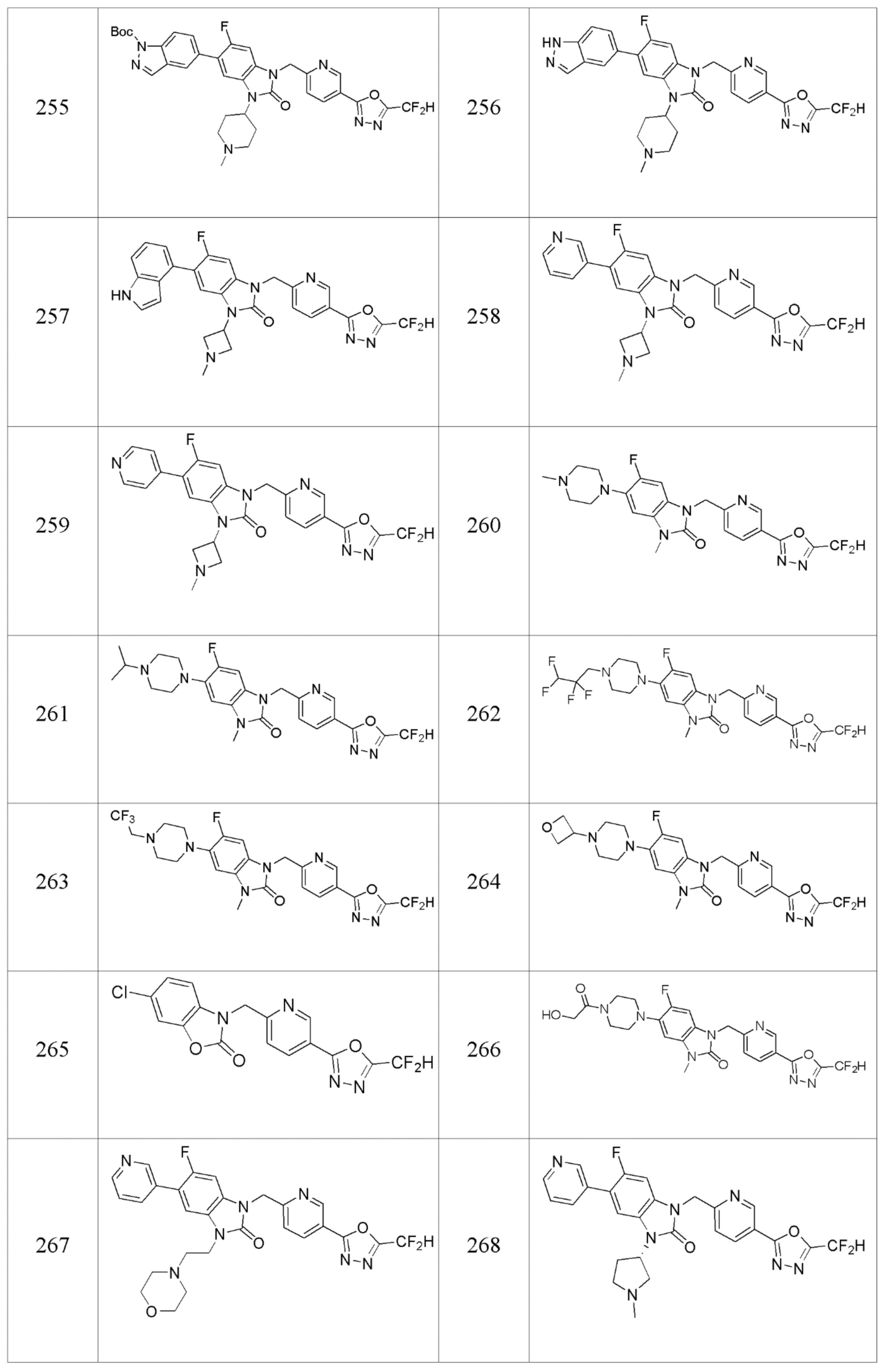
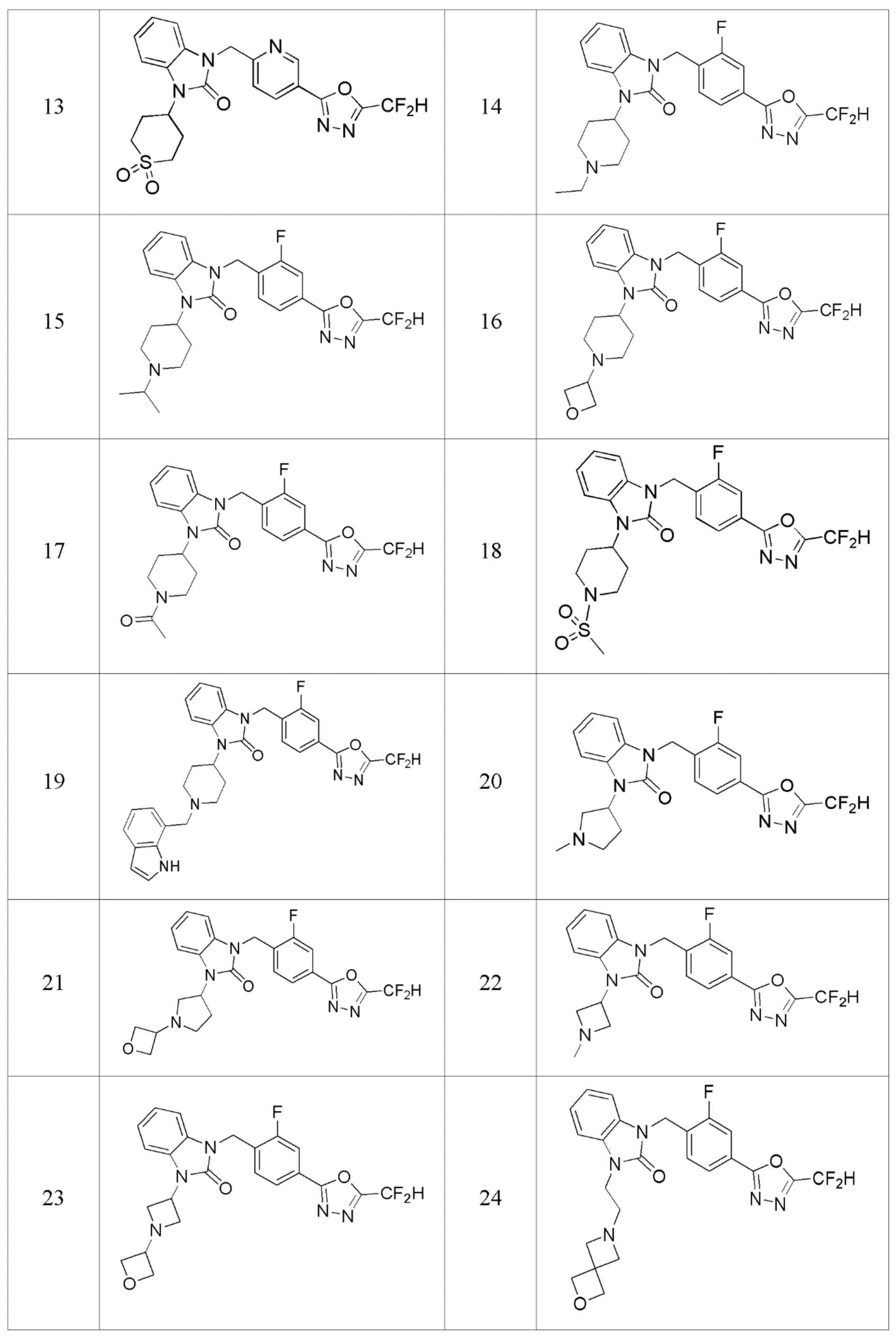
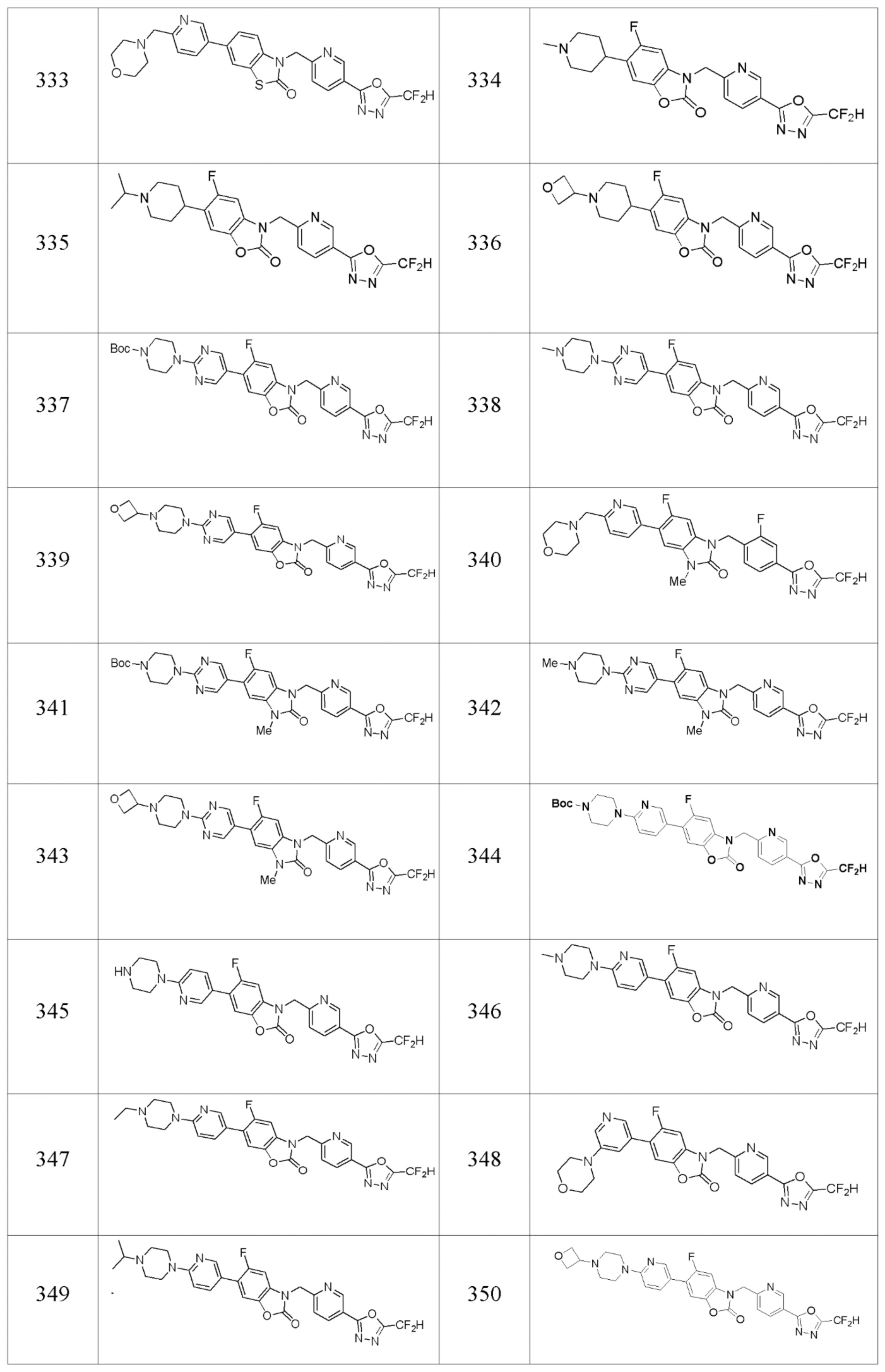
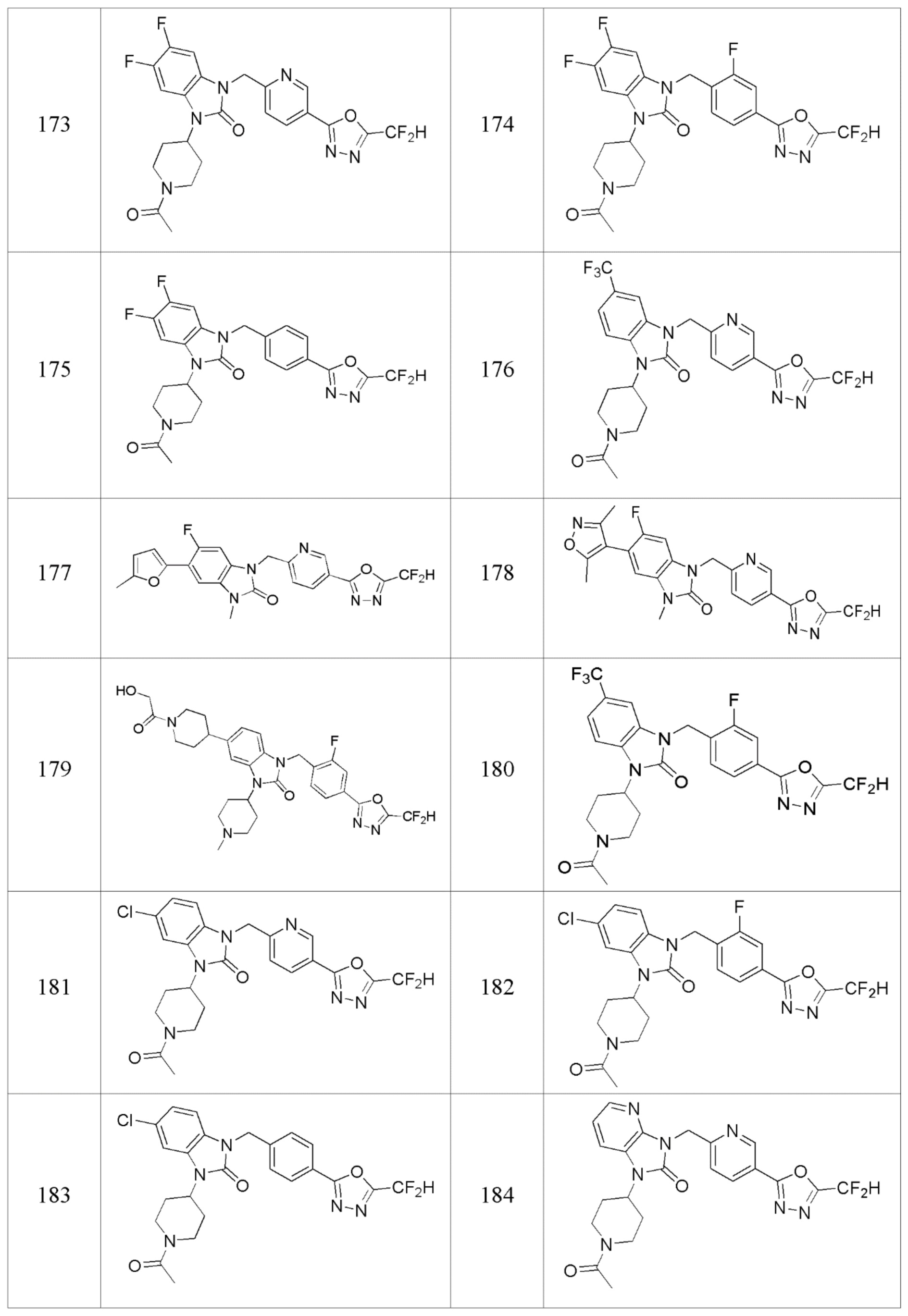
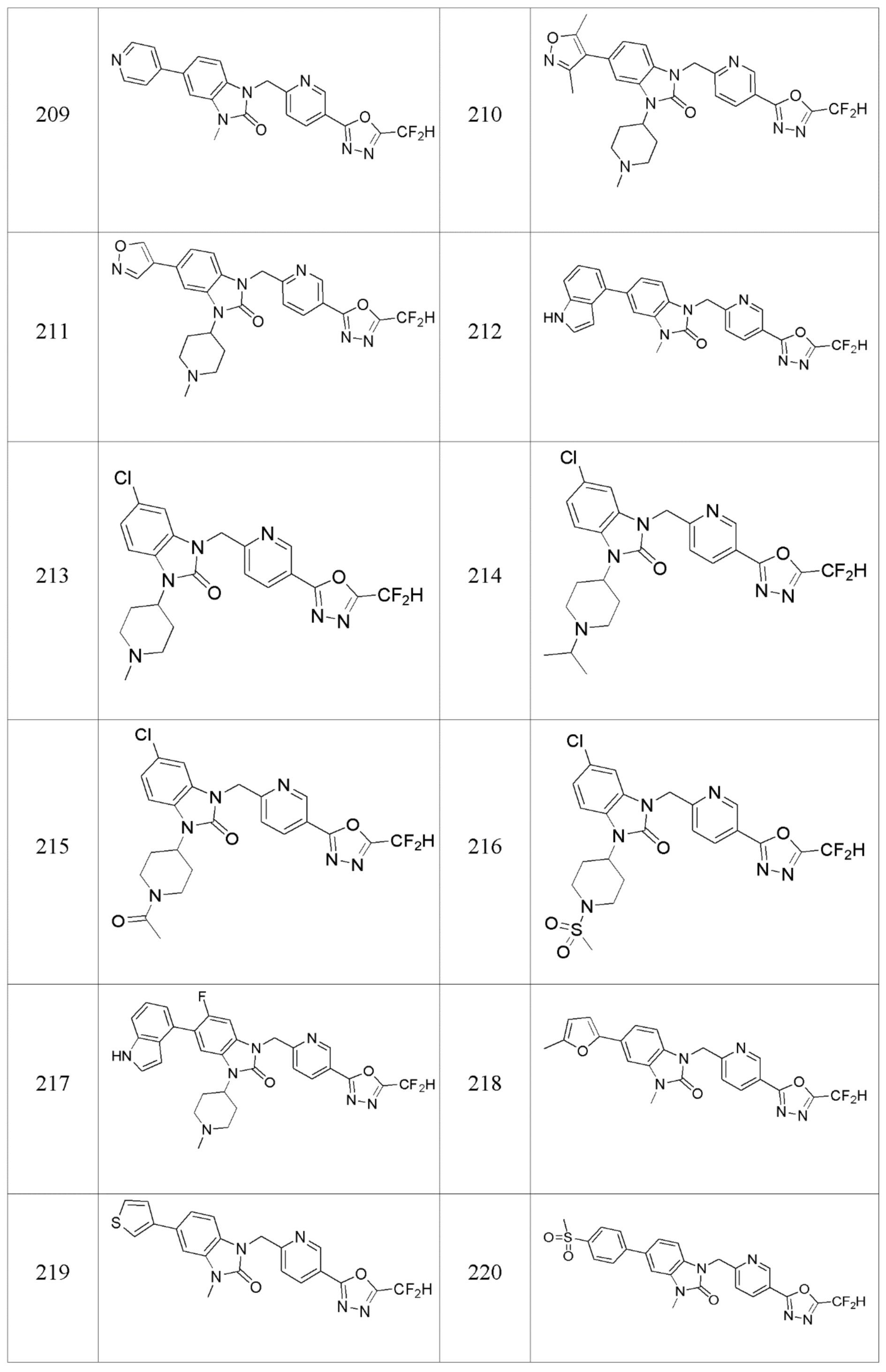
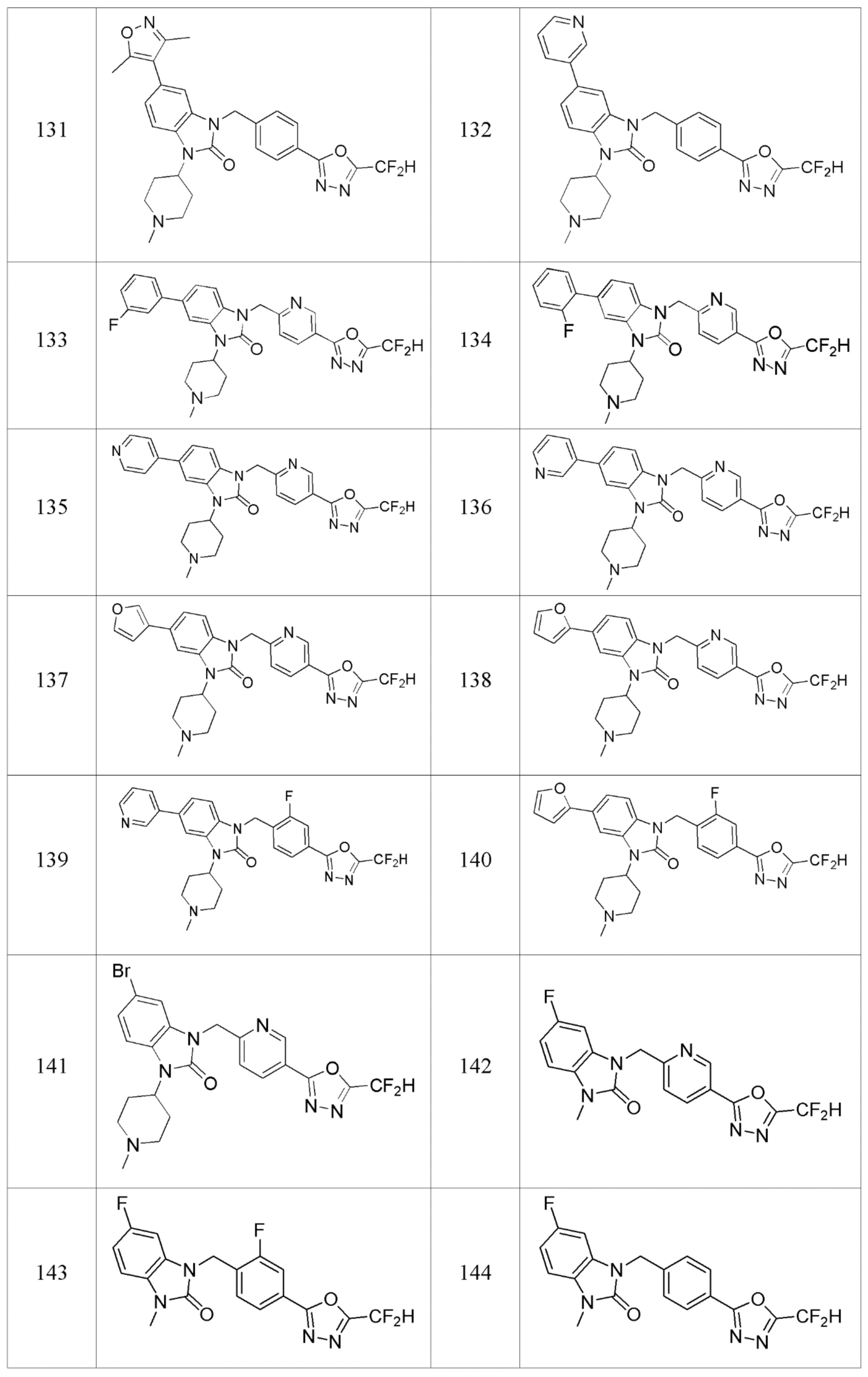
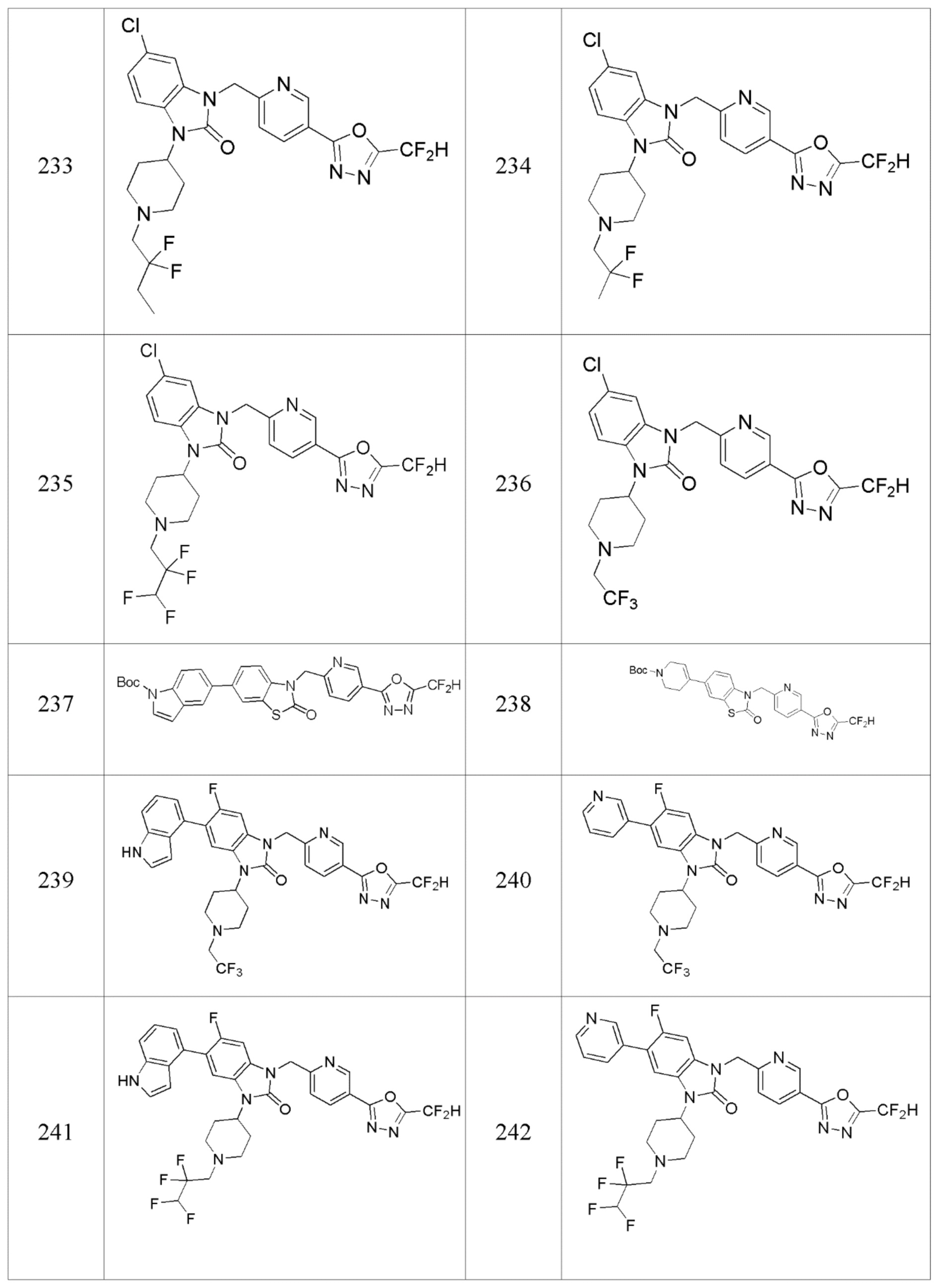
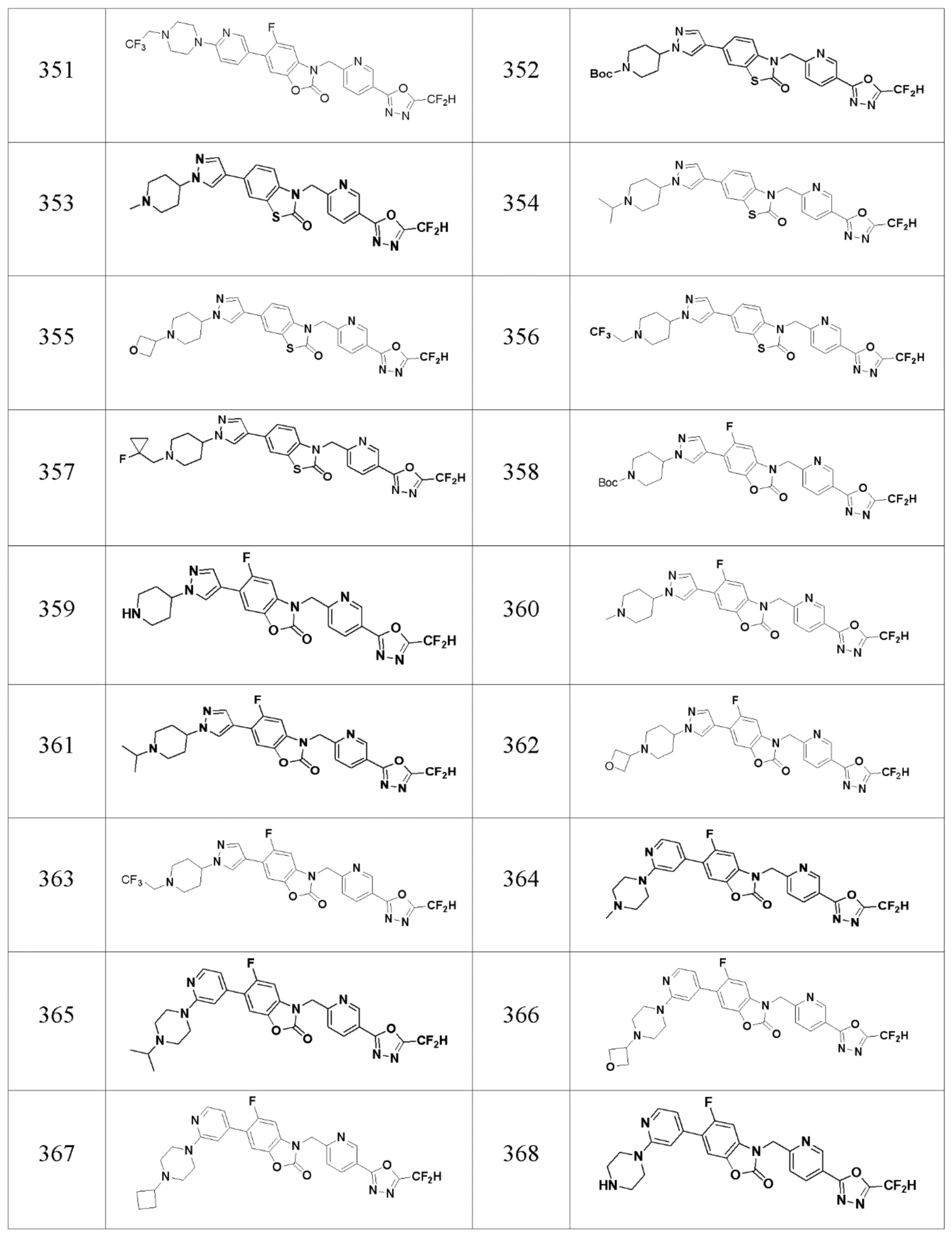
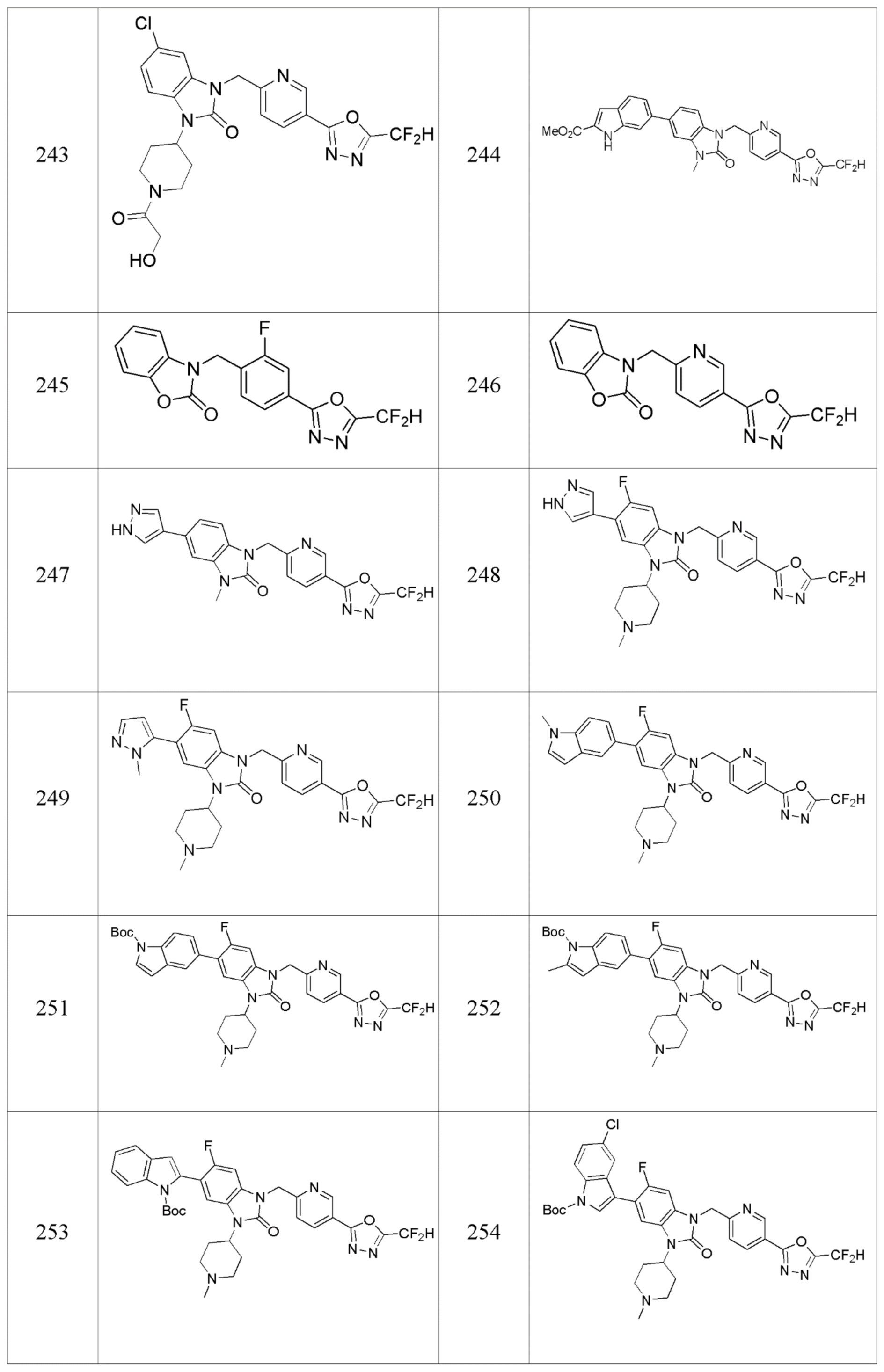
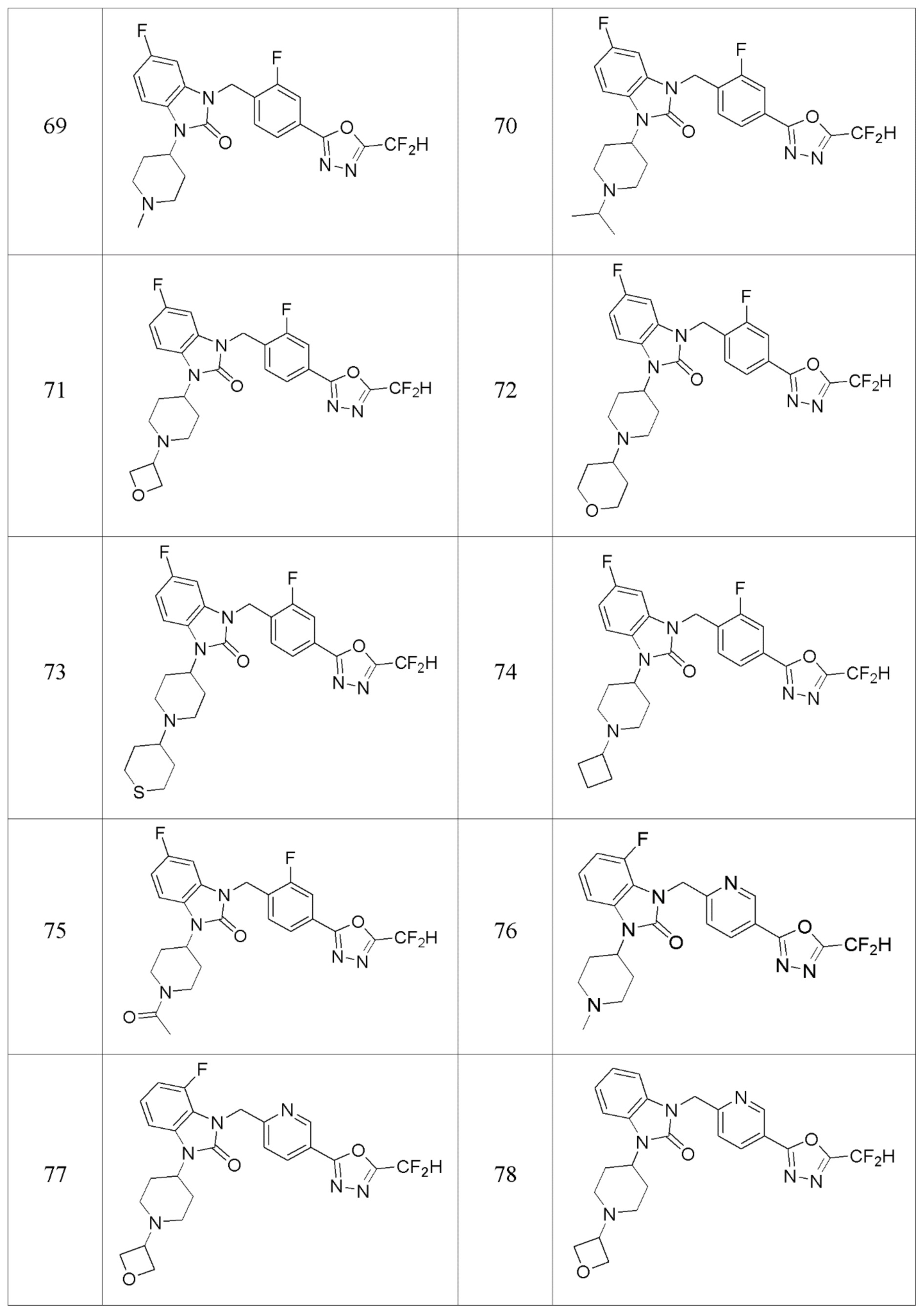
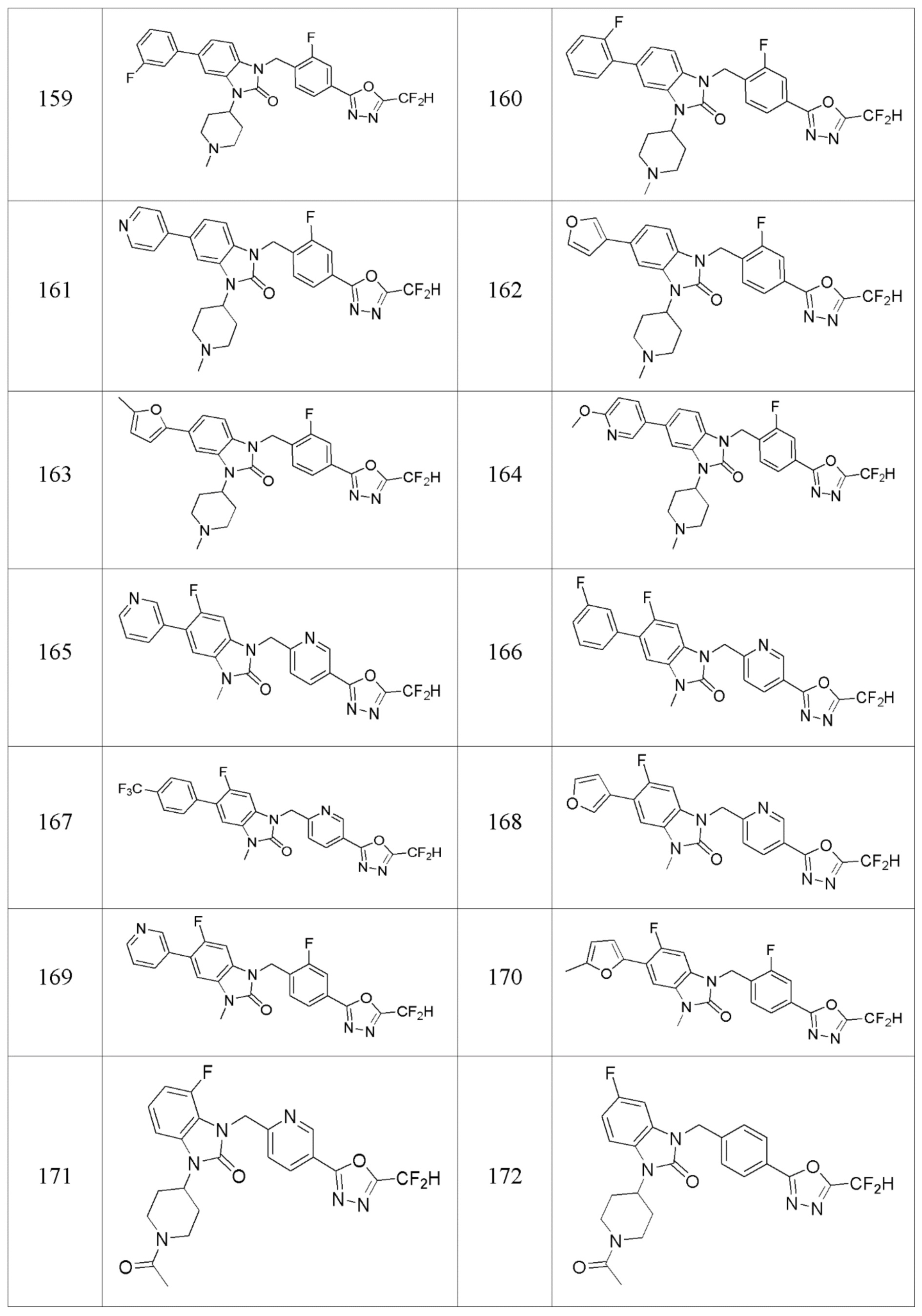
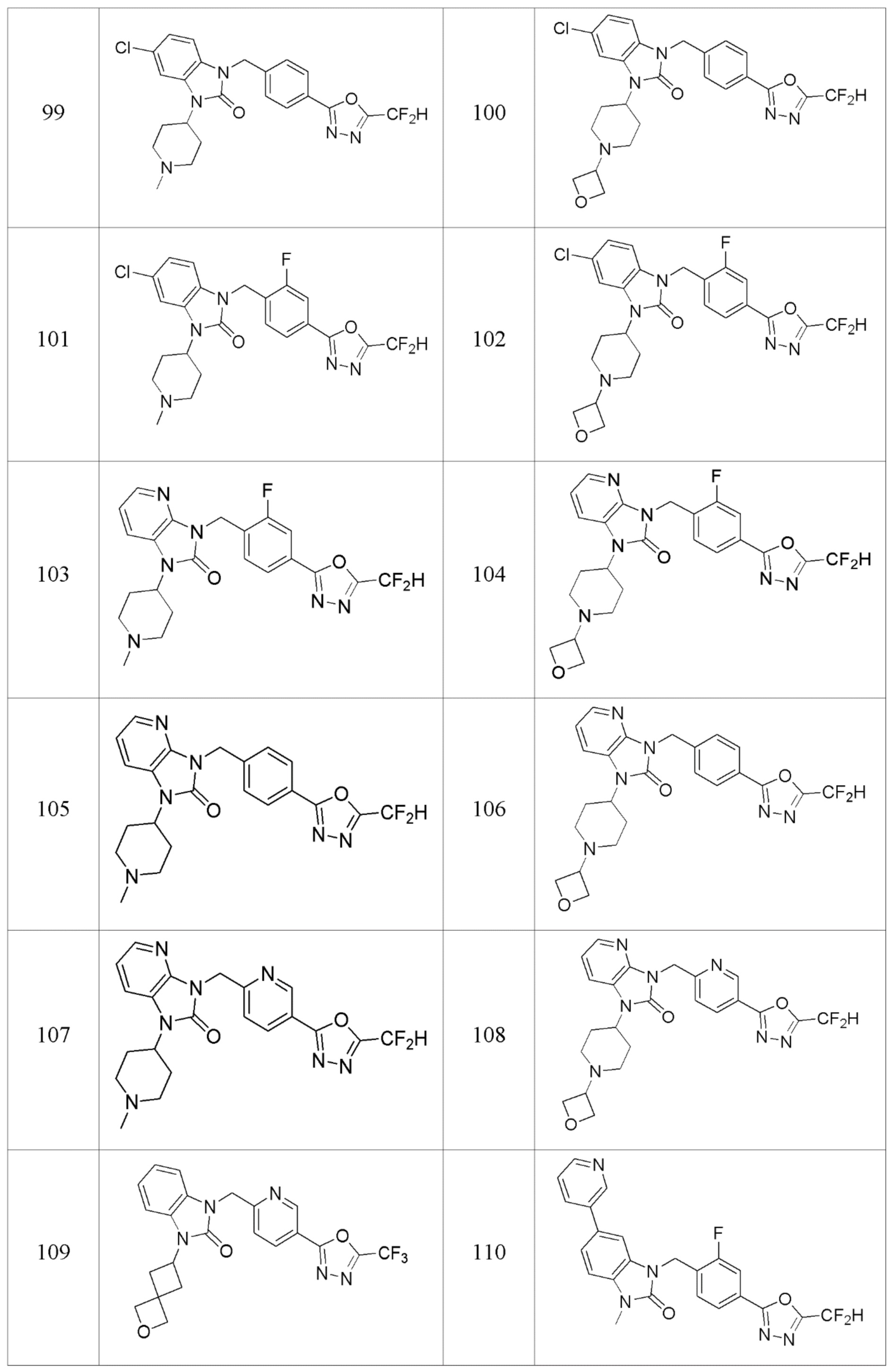
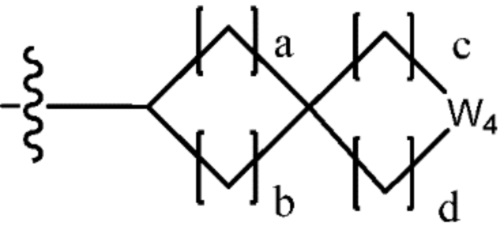
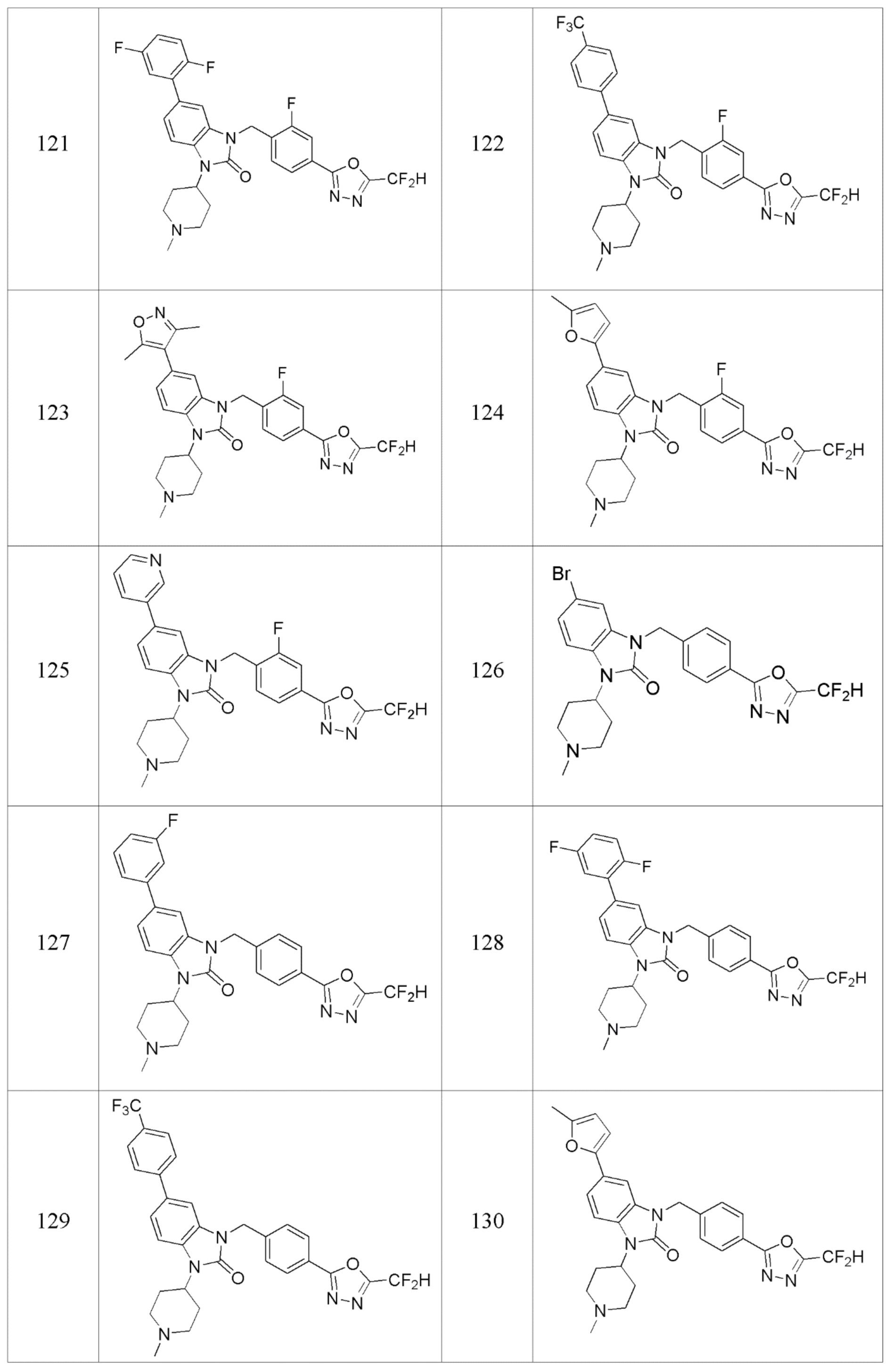
Биарил- или гетероциклические биарилзамещенные производные циклогексена в качестве ингибиторов сетр
Липидный преконцентрат с замедленным высвобождением фармакологически активного вещества и фармацевтическая композиция, содержащая его
Липидный преконцентрат анионных фармакологически активных веществ с замедленным высвобождением и содержащая его фармацевтическая композиция
Новые соединения для селективных ингибиторов гистондеацетилазы и фармацевтическая композиция, включающая такие соединения
Липидный преконцентрат аналогов gnrh с замедленным высвобождением и содержащая его фармацевтическая композиция
Липидный предконцентрат катионного фармакологически активного вещества с длительным высвобождением и содержащая его фармацевтическая композиция
Новые соединения в качестве ингибиторов гистондеацетилазы 6 и содержащие их фармацевтические композиции
Производные гетероциклические алкильные соединения в качестве селективных ингибиторов гистондеацетилазы и содержащие их фармацевтические композиции
Производные оксадиазоламина в качестве ингибитора гистондеацетилазы 6 и содержащая их фармацевтическая композиция
Производные 1,3,4-оксадиазолсульфамида в качестве ингибитора гистондеацетилазы 6 и содержащая их фармацевтическая композиция
Кондиционер
Производные оксадиазоламина в качестве ингибитора гистондеацетилазы 6 и содержащая их фармацевтическая композиция
1,3,4-оксадиазольные производные соединения в качестве ингибитора гистондеацетилазы 6 и фармацевтическая композиция, содержащая их
Гомофталимидные производные 1,3,4-оксадиазола как ингибитор гистондезацетилазы 6 и содержащая их фармацевтическая композиция

