Результат интеллектуальной деятельности: НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям, обладающим ингибирующей гистондеацетилазу 6 (HDAC6) активностью, их изомерам, их фармацевтически приемлемым солям, к их применению при получении терапевтических лекарственных средств, к содержащим их фармацевтическим композициям, к способу лечения заболевания с использованием указанной композиции и к способам получения указанных новых соединений.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Пост-трансляционные модификации, такие как ацетилирование, являются очень важными регулирующими модулями, лежащими в основе биологических процессов в клетках, и жестко регулируются множеством ферментов. Гистоны представляют собой главные белковые компоненты хроматина и выступают в качестве бобин, вокруг которых накручены нити ДНК. Кроме того, баланс ацетилирования и деацетилирования гистонов играет важнейшую роль в регуляции экспрессии генов.
Гистондезацетилазы (HDAC) представляют собой ферменты, которые удаляют ацетильные группы из остатков лизина на гистоновых белках хроматина, и, как известно, связаны с молчанием генов и индуцируют остановку клеточного цикла, ангиогенное ингибирование, иммунную регуляцию, гибель клеток и т.д. (Hassig et al., Curr. Opin. Chem. Biol. 1997, 1, 300-308). Кроме того, сообщалось, что ингибирование ферментативной функции HDAC индуцирует апоптоз раковых клеток in vivo за счет снижения активности факторов, ассоциированных с выживанием раковых клеток, и активации факторов, ассоциированных с апоптозом раковых клеток (Warrell et al., J. Natl. Cancer Inst. 1998, 90, 1621-1625).
У человека были идентифицированы 18 HDAC, и их подразделяют на четыре класса на основе их гомологии с дрожжевыми HDAC. Из них 11 HDAC используют цинк в качестве кофактора, и они могут быть разделены на три группы: класс I (HDAC 1, 2, 3 и 8), класс II (IIа: HDAC4, 5, 7 и 9; IIb: HDAC6 и 10), класс IV (HDAC, 11). Кроме того, для 7 HDAC из класса III (SIRT 1-7) в качестве кофактора вместо цинка требуется NAD+ (Bolden et al., Nat. Rev. Drug Discov. 2006, 5(9), 769-784).
На стадии доклинической или клинической разработки находятся различные ингибиторы HDAC, но на сегодняшний день в качестве противораковых агентов были идентифицированы только неселективные ингибиторы HDAC, а для лечения кожной Т-клеточной лимфомы были утверждены только вориностат (SAHA) и ромидепсин (FK228). Однако, как известно, неселективные ингибиторы HDAC вызывают побочные эффекты, такие как усталость и тошнота, обычно при больших дозах (Piekarz et al., Pharmaceuticals 2010, 3, 2751-2767). Такие побочные эффекты, как сообщается, вызваны ингибированием HDAC класса I. Из-за таких побочных эффектов использование неселективных ингибиторов HDAC при разработке лекарственных средств, отличных от противоопухолевых препаратов, было ограничено (Witt et al., Cancer Letters, 2009, 277, 8-21).
В то же время сообщалось, что селективное ингибирование HDAC класса II не проявляет токсичности, показываемой при ингибировании HDAC класса I. Кроме того, при разработке селективных ингибиторов HDAC могут быть преодолены побочные эффекты, такие как токсичность, которая вызывается неселективным ингибированием HDAC. Таким образом, существует возможность разработки селективных ингибиторов HDAC в качестве терапевтических средств, эффективных для лечения различных заболеваний (Matthias et al., Mol. Cell. Biol. 2008, 28, 1688-1701).
Известно, что HDAC6, член класса IIb HDAC, присутствует в основном в цитоплазме и участвует в деацетилировании ряда негистоновых субстратов (HSP90, кортактин и т.д.), включая тубулин (Yao et al., Mol. Cell 2005, 18, 601-607). HDAC6 содержит два каталитических домена, и домен цинкового пальца на C-конце может связываться с убиквитинированными белками. Известно, что HDAC6 имеет ряд негистоновых белков в качестве субстратов, и, таким образом, играет важную роль при различных заболеваниях, включая злокачественное новообразование, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания и нейродегенеративные заболевания (Santo et al., Blood 2012 119: 2579-258; Vishwakarma et al., International Immunopharmacology 2013, 16, 72-78; Hu et al., J. Neurol. Sci. 2011, 304, 1-8).
Таким образом, существует потребность в разработке селективных ингибиторов HDAC6 для лечения злокачественного новообразования, воспалительных заболеваний, аутоиммунных заболеваний, неврологических заболеваний и нейродегенеративных заболеваний, которые не вызывают никаких побочных эффектов в отличие от неселективных ингибиторов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
Объектом настоящего изобретения являются новые соединения, обладающие селективной ингибирующей HDAC6 активностью, их изомеры или их фармацевтически приемлемые соли.
Другим объектом настоящего изобретения являются фармацевтические композиции, содержащие новые соединения, обладающие селективной ингибирующей HDAC6 активностью, их изомеры или их фармацевтически приемлемые соли.
Еще одним объектом настоящего изобретения являются способы получения новых соединений.
Еще одним объектом настоящего изобретения являются фармацевтические композиции, которые содержат вышеуказанное соединение, для предупреждения или лечения ассоциированных с активностью HDAC6 заболеваний, включая злокачественное новообразование, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания и нейродегенеративные заболевания.
Еще одним объектом настоящего изобретения является применение соединений при получении терапевтических лекарственных средств против ассоциированных с активностью HDAC6 заболеваний.
И еще одним объектом настоящего изобретения являются способы лечения ассоциированных с активностью HDAC6 заболеваний, которые включают введение терапевтически эффективного количества фармацевтических композиций, содержащих указанные соединения.
ТЕХНИЧЕСКОЕ РЕШЕНИЕ ЗАДАЧИ
Авторы настоящего изобретения обнаружили новые соединения производные диметилпиперазина, в частности, соединения производные диметилпиперазингидроксамовой кислоты, которые обладают ингибирующей гистондеацетилазу 6 (HDAC6) активностью, и нашли, что эти соединения могут быть использованы для ингибирования или лечения заболеваний, ассоциированных с активностью гистондеацетилазы 6 (HDAC6), таким образом создав настоящее изобретение.
Новые ингибиторы HDAC6
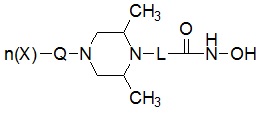
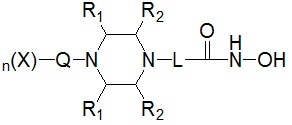
Для достижения вышеуказанных целей в настоящем изобретении предложены соединения, представленные следующей формулой I, их изомеры или их фармацевтически приемлемые соли:
Формула I
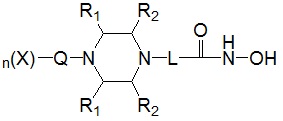
где
* R1 представляет собой водород или -CH3,
* R2 представляет собой водород или -CH3, при условии, что R2 представляет собой -CH3, когда R1 представляет собой водород, и R2 представляет собой водород, когда R1 представляет собой -CH3,
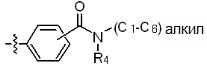
* L представляет собой -(C4-C5 алкил)-; -(C1-C3 алкил)-L1-; -C(=O)-L1- или -S(=O)2-L1-,
где -(C4-C5 алкил)- и -(C1-C3 алкил)- могут быть незамещенными или замещены -CH3,
L1 представляет собой -(C3-C6)циклоалкил-; 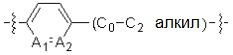 ;
;  ;
;  или
или 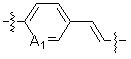 ,
,
A1 и A2, каждый, независимо, представляют собой -N- или -CR3-, при условии, что оба, A1 и A2, не могут представлять собой -N-,
R3 представляет собой водород, -F, -Cl, -Br, -I или -OH, и
A3 представляет собой -NH- или -O-,
* Q выбран из группы, включающей -(C1-C6)алкил-; -(C2-C6)алкенил-; -C(=O)-; -C(=S)-; -S(=O)2- или 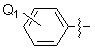 ,
,
где -(C1-C6)алкил- и -(C2-C6)алкенил- могут быть незамещенными, или каждый, независимо друг от друга, может быть замещен от 1 до 3 группами -CH3 или атомами галогена,
Q1 представляет собой водород; -F, -Cl, -Br или -I,
* n обозначает целое число 0, 1 или 2, при условии, что n обозначает 0, когда Q представляет собой , n обозначает 1, когда Q представляет собой -C(=O)-, -C(=S)- или -S(=O)2-, и n обозначает 1 или 2, когда Q представляет собой -(C1-C6)алкил- или -(C2-C6)алкенил-, и
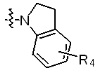
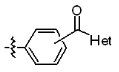
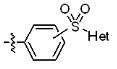
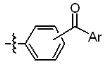
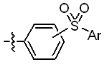
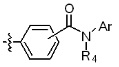
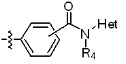
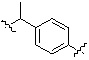
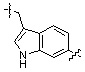
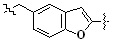
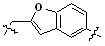
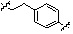
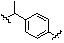
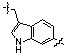
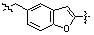
* X может быть выбран из группы, включающей -C1-C6 алкил; -C3-C6 циклоалкил; -C2-C6 алкенил; -C3-C6 циклоалкенил; -(C0-C2 алкил)Ar; -OAr; -(C0-C2 алкил)Het; нафтил и следующие группы:
 ,
,  ,
,  ,
, 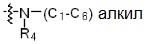 ,
, 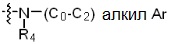 ,
, 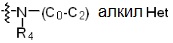 ,
, 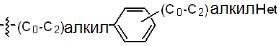 ,
, 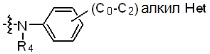 ,
, 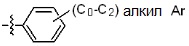 ,
, 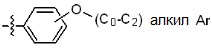 ,
, 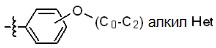 ,
, 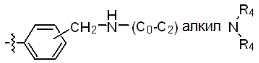 ,
, 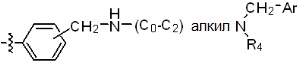 ,
,  ,
, 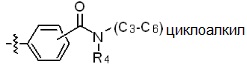 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,
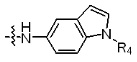
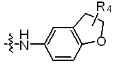
где R4 представляет собой H или -C1-C4 алкил,
-C0-C2 алкил, -C2-C6 алкенил и -C1-C6 алкил могут быть незамещенными или замещены от 1 до 2 -CH3 группами; от 1 до 3 группами -F или их комбинацией,
Ar представляет собой C6 моноциклическую ароматическую группу, которая может быть незамещенной или замещена одним или несколькими атомами галогена; группами -OH; -NH2; -C1-C6 алкил; -O(C1-C6)алкил; -C3-C6 циклоалкенил; -NH(C1-C6 алкил); -N(C1-C3 алкил)2; -CH2N(C1-C3 алкил)2; -S(=O)2-(C1-C3 алкил) или фенил, где -C1-C3 алкил; -C1-C6 алкил и -C3-C6 циклоалкенил, каждый, могут быть независимо замещены от 1 до 5 группами -F или -CH3, и
Het представляет собой 4-6-членное гетероароматическую или неароматическую кольцевую группу, содержащую от 1 до 3 элементов, выбранных из группы, включающей N, O и S, содержащую от 0 до 3 двойных связей, и может быть незамещенной или замещена одним или несколькими атомами галогена; группами -C1-C6 алкил; -C(=O)(C1-C3 алкил); -S(=O)2(C1-C3 алкил) или бензил, где -C1-C3 алкил и -C1-C6 алкил, каждый, могут быть необязательно замещены -OH; от 1 до 5 группами -F или -CH3.
Как используется в настоящем документе, термин «C0 алкил» означает, что углерод отсутствует, поэтому представляет собой связь. Например, «-(C0 алкил)Ar» означает -Ar.
Соединения, представленные формулой I в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой II или формулой III:
Формула II
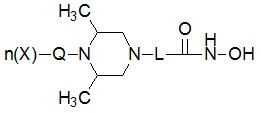
Формула III
где
* L представляет собой -(C5 алкил)-; -(C1-C2 алкил)-L1-; -C(=O)-L1- или -S(=O)2-L1-,
где -(C5 алкил)- и -(C1-C2 алкил)- имеют прямую цепь и могут быть незамещенными или замещены -CH3,
L1 представляет собой -(C3-C6) циклоалкил-; 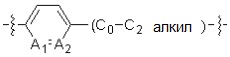 ; ; или ,
; ; или ,
A1 и A2, каждый, независимо, представляют собой -N- или -CR3-, при условии, что оба, A1 и A2, не могут представлять собой -N-,
R3 представляет собой водород, -F или -OH, и
A3 представляет собой -NH- или -O-,
* Q выбран из группы, включающей -(C1-C3)алкил-; -C(=O)-; -C(=S)-; -S(=O)2- или ,
где -(C1-C3)алкил- может быть незамещенным или замещен от 1 до 3 группами -CH3 или атомами галогена,
Q1 представляет собой водород; -F или -Cl,
* n обозначает целое число 0 или 1, при условии, что n обозначает 0, когда Q представляет собой , и n обозначает 1, когда Q представляет собой -C(=O)-, -C(=S)-, -S(=O)2- или -(C1-C3)алкил-, и
* X может быть выбран из группы, включающей -C1-C6 алкил; -C3-C6 циклоалкил; -C2-C6 алкенил; -C3-C6 циклоалкенил; -(C0-C2 алкил)Ar; -OAr; -(C0-C2 алкил)Het; нафтил; и следующие группы:
, , , 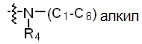 , , , , , , , , , , , , , , , , , ,
, , , , , , , , , , , , , , , , , ,
где R4 представляет собой H или -C1-C4 алкил,
-C0-C2 алкил; -C2-C6 алкенил и -C1-C6 алкил могут быть незамещенными или замещены 1 или 2 группами -CH3 или от 1 до 3 группами -F,
Ar представляет собой C6 моноциклическую ароматическую группу, которая может быть незамещенной или замещена одним или несколькими атомами галогена; группами -OH; -NH2; -C1-C6 алкил; -O(C1-C6)алкил; -C3-C6 циклоалкенил; -NH(C1-C6 алкил); -N(C1-C3 алкил)2; -CH2N(C1-C3 алкил)2; -S(=O)2-(C1-C3 алкил) или фенил, где -C1-C3 алкил; -C1-C6 алкил и -C3-C6 циклоалкенил, каждый, могут быть независимо замещены от 1 до 5 группами -F или -CH3, и
Het представляет собой 4-6-членное гетероароматическую или неароматическую кольцевую группу, содержащую от 1 до 3 элементов, выбранных из группы, включающей N; O и S, содержащую от 0 до 3 двойных связей, и может быть незамещенной или замещена одним или несколькими атомами галогена; группами -C1-C6 алкил; -C(=O)(C1-C3 алкил); -S(=O)2(C1-C3 алкил) или бензил, где -C1-C3 алкил и -C1-C6 алкил, каждый, могут быть необязательно замещены -OH; или от 1 до 5 группами -F или -CH3.
В предпочтительном варианте осуществления настоящего изобретения L, Q и X в формулах I, II и III могут быть определены следующим образом:
* L представляет собой -CH2-L1-,
где
L1 представляет собой  ; ;
; ;  ; или ,
; или ,
A1 и A2, каждый, независимо, представляют собой -N- или -CR3-, при условии, что оба, A1 и A2, не могут представлять собой -N-,
R3 представляет собой водород, -F или -OH, и
A3 представляет собой -NH- или -O-,
* Q представляет собой -CH2-, -C(=O)- или -S(=O)2-, и
* X может быть выбран из группы, включающей -C1-C6 алкил; -(C0-C2 алкил)Ar; -(C0-C2 алкил)Het; -OAr или следующие группы:
, , , , ,
где R4 представляет собой H или -C1-C4 алкил,
-C0-C2 алкил и -C1-C6 алкил могут быть незамещенными или замещены 1 или 2 группами -CH3 и/или от 1 до 3 группами -F, и
Ar и Het, каждый, независимо, имеют значения, определенные в формулах от I до III.
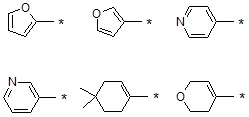
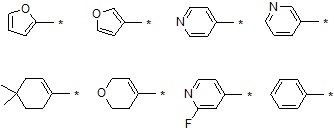
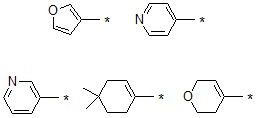
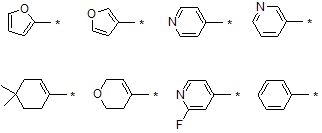
В варианте осуществления настоящего изобретения гетероциклическая группа (Het), указанная как X или заместитель в формулах от I до III выше, может иметь структуру, выбранную из следующей группы:
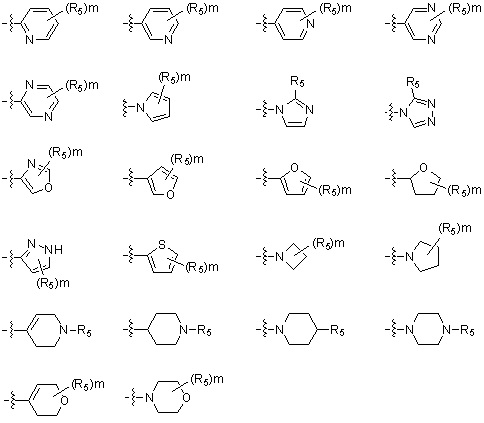
где
R5, каждый, независимо, представляют собой водород; -F; -Cl; -C1-C6 алкил; -C(=O)(C1-C3 алкил); -S(=O)2(C1-C3 алкил) или бензил, где -C1-C3 алкил и -C1-C6 алкил, каждый, могут быть необязательно замещены -OH; от 1 до 5 группами -F или -CH3,
m обозначает целое число 0, 1, 2 или 3, и
Het является незамещенной, когда m обозначает 0, и Het может быть независимо замещена R5, когда m обозначает 1, 2 или 3.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-1:
Формула I-1
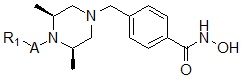
где
A представляет собой -C(=O)-; -CH2-; -NH(C=O)-; -C(=S)- или -S(=O)2-, и
R1 представляет собой C1-C4 алкил с прямой или разветвленной цепью; C1-C4 алкенил с прямой или разветвленной цепью; C3-C5 циклоалкил; -OC6C5; -(C0-C2 алкил)CF3; фенил (который может быть незамещенным или замещен C1-C3 алкилом; пирролом; -OCH3; -CF3; -F; -Cl или -OH); пиридил; фуранил; тиофенил; бензил (который может быть незамещенным или замещен одной или несколькими группами C1-C3 алкил; пиррол; -OCH3; -CF3; -F; -Cl или -OH); фенетил; нафтил; оксазолил; пиримидинил; пиразолил или пиразинил.
Соединения, представленные формулой I-1, предпочтительно, являются соединениями 82, 83, 84, 98, 99, 100, 120, 121, 122, 123, 125, 126, 127, 128, 145, 146, 147, 148, 149, 159, 160, 161, 177, 184, 188, 204, 211, 212, 213, 214, 222, 223, 224, 225, 232, 255, 265, 266, 267, 270, 272, 275, 290, 291, 292, 293, 294, 295, 296, 297, 354, 355, 403, 404 и 405, как описано в настоящем документе.
Соединения, представленные формулой I, могут являться соединениями, изображенными следующей формулой I-2:
Формула I-2
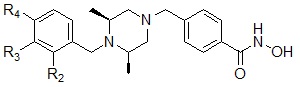 ,
,
где R2, R3 или R4, каждый, независимо, могут представлять собой водород или могут быть выбраны из следующей группы:
 .
.
Соединения, представленные формулой I-2, предпочтительно, являются соединениями 309, 327, 328, 329, 330, 331, 332, 342, 343, 344, 345, 346 и 347, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-3:
Формула I-3
 ,
,
где R5 может быть выбран из группы, включающей C1-C4 алкил с прямой или разветвленной цепью; -COCH3; -CH2CF3 и -SO2CH3.
Соединения, представленные формулой I-3, предпочтительно, являются соединениями 481, 482, 483, 484 и 485, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, представленными формулой I-4:
Формула I-4
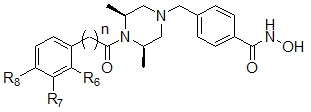 ,
,
где n обозначает целое число 0, 1 или 2, и R6, R7 или R8, каждый, независимо, могут представлять собой водород или могут быть выбраны из следующей группы:
 .
.
Соединения, представленные формулой I-4, предпочтительно, являются соединениями 234, 242, 243, 244, 245, 246, 247, 283, 284, 285, 286, 288, 326 и 340, как описано в настоящем документе.
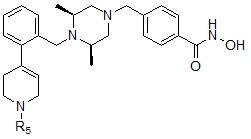
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, представленными формулой I-5:
Формула I-5
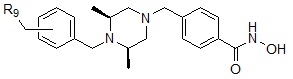 ,
,
где R9 может быть выбран из морфолина; пиперидина; пирролидина; азетидина; пиперазина; первичного или вторичного -C1-C2 алкиламина; 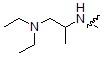 или
или 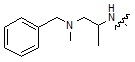 , где пирролидин может быть незамещенным или замещен одной или несколькими группами -F или -Cl, азетидин может быть незамещенным или замещен одной или несколькими группами -F или -Cl, и пиперазин может быть незамещенным или замещен одной или несколькими группами C1-C5 алкил с прямой или разветвленной цепью; бензила; -COCH3; -CH2CF3 или -SO2CH3.
, где пирролидин может быть незамещенным или замещен одной или несколькими группами -F или -Cl, азетидин может быть незамещенным или замещен одной или несколькими группами -F или -Cl, и пиперазин может быть незамещенным или замещен одной или несколькими группами C1-C5 алкил с прямой или разветвленной цепью; бензила; -COCH3; -CH2CF3 или -SO2CH3.
Соединения, представленные формулой I-5, предпочтительно, являются соединениями 356, 376, 382, 383, 384, 385, 411, 412, 413, 426, 427, 428, 429, 430, 431, 452, 453, 454, 455, 456, 457, 466, 467, 468 и 486, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-6A или формулой I-6B:
Формула I-6A
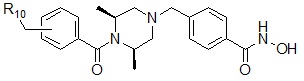 ,
,
Формула I-6B
 ,
,
где R10 может быть выбран из пирролидина; пиперидина; первичного или вторичного C1-C2 алкиламина; пиперазина; морфолина; или  , где пирролидин может быть незамещенным или замещен одной или несколькими группами -F или -Cl, и пиперазин может быть незамещенным или замещен одной или несколькими группами с прямой или разветвленной цепью C1-C5 алкил; -COCH3; -CH2CF3 или -SO2CH3.
, где пирролидин может быть незамещенным или замещен одной или несколькими группами -F или -Cl, и пиперазин может быть незамещенным или замещен одной или несколькими группами с прямой или разветвленной цепью C1-C5 алкил; -COCH3; -CH2CF3 или -SO2CH3.
Соединения, представленные формулой I-6A, предпочтительно, являются соединениями 423, 424, 425, 432, 433, 434, 435, 439, 440, 441, 442, 443, 444, 458, 459, 460, 461, 462 и 463, как описано в настоящем документе. Более того, соединения, представленные формулой I-6B, предпочтительно, представляют собой соединение 446, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-7:
Формула I-7
 ,
,
где X может быть выбран из -OH; -F; -Cl или -Br.
Соединения, представленные формулой I-7, предпочтительно, являются соединениями 386 и 387, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-8:
Формула I-8
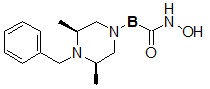 ,
,
где B может быть выбран из
 ;
;  ;
;  ;
;  ;
; 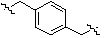 ;
; 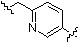 ;
; 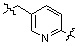 ;
; 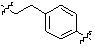 ;
;  ;
;  ;
;  ;
;  ;
; 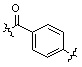 ;
; 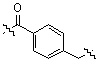 ;
; 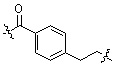 ;
; 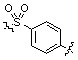 ;
;  ;
;  ,
,
где w представляет собой водород, или может быть замещен -F; -Cl или -OH.
Соединения, представленные формулой I-8, предпочтительно, являются соединениями 154, 171, 172, 173, 194, 218, 219, 520, 571, 574, 652, 812, 813, 814, 818, 820, 822, 823 и 824, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-9:
Формула I-9
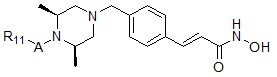 ,
,
где A представляет собой -C(=O)-; -CH2-; -NH(C=O)-; -C(=S)- или -S(=O)2-, и R11 может быть выбран из фуранила; бензила; замещенного пирролом фенила или замещенного пирролом анилинила.
Соединения, представленные формулой I-9, предпочтительно, являются соединениями 321, 322 и 323, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-10:
Формула I-10
 .
.
Соединения, представленные формулой I-10, предпочтительно, являются соединением 472, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-11:
Формула I-11
 .
.
Соединения, представленные формулой I-11, предпочтительно, являются соединением 402, как описано в настоящем документе.
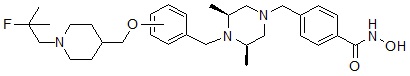
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-12:
Формула I-12
 .
.
Соединения, представленные формулой I-12, предпочтительно, являются соединениями 380 и 388, как описано в настоящем документе.
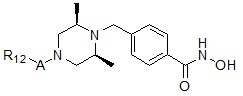
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-13:
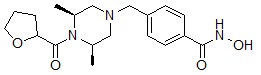
Формула I-13
 ,
,
где A представляет собой -C(=O)-; -CH2-; -CH(CH3)-; -NH(C=O)-; -NCH3(C=O)-; -C(=S)- или -S(=O)2-, и R12 может быть выбран из C1-C4 алкила с прямой или разветвленной цепью; фенил (который может быть незамещенным или замещен одним или несколькими группами C1-C3 алкил с прямой или разветвленной цепью; -F; -Cl; CF3; -OCH3; - первичный или вторичный C1-C2 алкиламин; -SO2CH3; тиофенил; пиррол; пиразол; фуранил; триазолил или имидазолил); пиридинил; тиофенил; фуранил; бензил (который может быть незамещенным или замещен -F, -Cl или -OCH3); индол (который может быть незамещенным или замещен одной или несколькими группами C1-C3 алкил с прямой или разветвленной цепью), дигидробензофуранил; фениламин (который может быть незамещенным или замещен C1-C3 алкилом с прямой или разветвленной цепью); индолин или нафтил.
Соединения, представленные формулой I-13, предпочтительно, являются соединениями 80, 81, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 118, 162, 163, 164, 165, 166, 167, 168, 183, 185, 186, 187, 189, 196, 197, 215, 220, 230, 231, 233, 256, 268, 271, 273, 274, 298, 299, 300, 301, 302, 303, 304 и 305, как описано в настоящем документе.
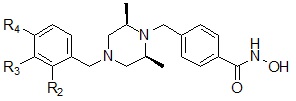
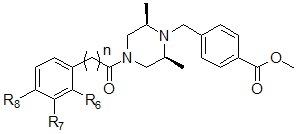
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-14:
Формула I-14
 ,
,
где R2, R3 и R4, каждый, независимо, могут представлять собой водород или могут быть выбраны из следующей группы:
 .
.
Соединения, представленные формулой I-14, предпочтительно, являются соединениями 348, 349, 350, 351, 352, 396, 400 и 401, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-15:
Формула I-15
 ,
,
где n обозначает целое число 0, 1 или 2, и R6, R7 и R8, каждый, независимо, могут представлять собой водород или могут быть выбраны из следующей группы:
 .
.
Соединения, представленные формулой I-15, предпочтительно, являются соединениями 250, 251, 252, 253, 257, 258, 259, 260, 261, 262, 263, 276, 277, 278, 279 и 280, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-16:
Формула I-16
 ,
,
где B может быть выбран из
 ; ;
; ;  или
или  , где w может быть замещен -F или -Cl.
, где w может быть замещен -F или -Cl.
Соединения, представленные формулой I-16, предпочтительно, являются соединениями 174, 175, 176 и 195, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-17:
Формула I-17
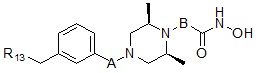 ,
,
где A представляет собой -C(=O)-; -CH2-; -NH(C=O)-; -C(=S)- или -S(=O)2-, и B может быть выбран из  или
или  . Также, R13 может быть выбран из пирролидина и -C1-C2 первичного или вторичного алкиламина.
. Также, R13 может быть выбран из пирролидина и -C1-C2 первичного или вторичного алкиламина.
Соединения, представленные формулой I-17, предпочтительно, являются соединениями 475, 476, 478, 479, 480 и 487, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-18:
Формула I-18
 .
.
Соединения, представленные формулой I-18 предпочтительно, представляют собой соединение 477, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-19:
Формула I-19
 ,
,
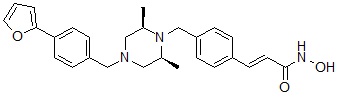
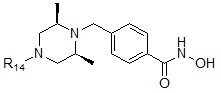
где R14 может быть выбран из 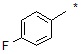 или
или 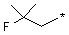 .
.
Соединения, представленные формулой I-19, предпочтительно, являются соединениями 119 и 193, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-20:
Формула I-20
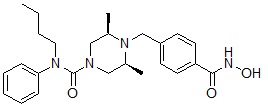 .
.
Соединения, представленные формулой I-20 предпочтительно, представляют собой соединение 198, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-21:
Формула I-21
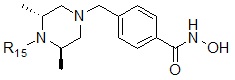 ,
,
где R15 может быть выбран из  или
или  .
.
Соединения, представленные формулой I-21, предпочтительно, являются соединениями 248 и 249, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-22:
Формула I-22
 .
.
Соединения, представленные формулой I-22, предпочтительно, являются соединениями 569 и 573, как описано в настоящем документе.
Соединения, представленные формулой I, в соответствии с настоящим изобретением, могут являться соединениями, изображенными следующей формулой I-23:
Формула I-23
 ,
,
где B может быть выбран из
; ; или , где w представляет собой водород; -F; -Cl или -OH.
Соединения, представленные формулой I-23, предпочтительно, являются соединениями 609, 653 и 696, как описано в настоящем документе.
Соединения, представленные формулой I, формулой II, формулой III и формулами от I-1 до I-23, показаны в таблице 1 далее.
Таблица 1: Названия соединений производных диметилпиперазина
|
Предпочтительно, соединения, представленные формулой I, или их фармацевтически приемлемые соли в соответствии с настоящим изобретением могут быть выбраны из группы, включающей соединения 081, 082, 083, 084, 098, 099, 100, 106, 107, 108, 109, 110, 112, 120, 121, 122, 123, 125, 126, 127, 128, 145, 146, 147, 148, 149, 159, 160, 161, 171, 173, 174, 175, 177, 186, 188, 193, 194, 195, 196, 198, 211, 214, 219, 248, 249, 250, 251, 252, 255, 265, 266, 267, 272, 283, 284, 285, 286, 292, 295, 297, 305, 326, 328, 329, 330, 332, 342, 343, 344, 345, 346, 349, 354, 356, 376, 380, 382, 383, 384, 385, 386, 387, 388, 403, 411, 413, 430, 431, 432, 433, 434, 435, 439, 440, 441, 442, 443, 444, 452, 453, 454, 455, 456, 467, 468, 481, 482, 483, 484, 485, 486, 569, 696, 813 и 823. Более предпочтительно, соединения, представленные формулой I, или их фармацевтически приемлемые соли в соответствии с настоящим изобретением могут быть выбраны из группы, включающей соединения 082, 083, 084, 098, 100, 120, 121, 122, 123, 125, 126, 127, 128, 145, 146, 148, 149, 159, 160, 161, 171, 174, 177, 194, 211, 249, 255, 283, 305, 326, 328, 329, 330, 332, 342, 343, 344, 345, 346, 349, 354, 356, 376, 382, 383, 387, 388, 411, 413, 431, 439, 441, 444, 452, 453, 454, 467, 468, 481, 482, 483, 484, 485, 696 и 813.
Как используется в настоящем документе, термин «фармацевтически приемлемая соль» означает любую соль, которая обычно используется в фармацевтической области. Примеры фармацевтически приемлемых солей включают, но ими не ограничиваются, соли с неорганическими ионами, такими как ионы кальция, калия, натрия или магния, соли с неорганическими кислотами, такими как хлористоводородная кислота, азотная кислота, фосфорная кислота, бромноватая кислота, йодноватая кислота, перхлорная кислота, винная кислота или серная кислота, соли с органическими кислотами, такими как уксусная кислота, трифторуксусная кислота, лимонная кислота, малеиновая кислота, янтарная кислота, щавелевая кислота, бензойная кислота, винная кислота, фумаровая кислота, миндальная кислота, пропионовая кислота, лимонная кислота, молочная кислота, гликолевая кислота, глюконовая кислота, галактуроновая кислота, глутаминовая кислота, глутаровая кислота, глюкуроновая кислота, аспарагиновая кислота, аскорбиновая кислота, угольная кислота, ванилиновая кислота, йодистоводородная кислота или тому подобное, соли с сульфоновыми кислотами, такими как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота или нафталинсульфоновая кислота, соли с аминокислотами, такими как глицин, аргинин или лизин, и соли с аминами, такими как триметиламин, триэтиламин, аммиак, пиридин или пиколин.
В настоящем изобретении предпочтительные соли включают соли с хлористоводородной кислотой, трифторуксусной кислотой, лимонной кислотой, бромистой кислотой, малеиновой кислотой, фосфорной кислотой, серной кислотой, винной кислотой и т.п., и предпочтительные примеры таких соединений включают соединения 230, 245, 250, 251, 253, 266, 270, 271, 273, 274, 275, 290, 291, 292, 293, 294, 295, 296, 297, 478 и 486, как описано в настоящем документе.
Соединения, представленные формулой I, могут содержать один или несколько асимметрических атомов углерода, и, таким образом, могут существовать в виде рацематов, рацемических смесей, отдельных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Соединения формулы I могут быть разделены на такие изомеры способами, известными в данной области, например, хроматографией на колонке или ВЭЖХ. В качестве альтернативы, стереоизомеры соединений формулы I могут быть синтезированы путем стереоспецифического синтеза с использованием оптически чистых исходных материалов и/или реагентов известной конфигурации.
Способы получения новых ингибиторов HDAC6
В настоящем изобретении предложены способы получения новых соединений формулы I, их изомеров или их фармацевтически приемлемых солей.
Предпочтительные способы получения новых соединений формулы I, их изомеров или их фармацевтически приемлемых солей показаны на реакционных схемах от 1 до 23 далее, а также включают модификации, очевидные для специалистов в данной области техники.
Реакционная схема 1
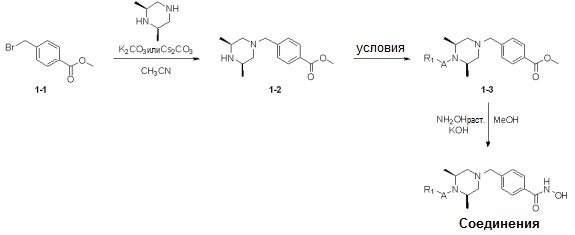
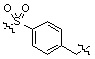
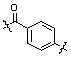
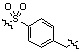
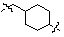
Условия: a) R1-CH2-Br (или -I, -Cl, -OTf, -OMs), K2CO3 (или Cs2CO3)/CH3CN (или ДМФ); b) R1-CHO, Na(CN)BH3, AcOH/ТГФ; c) R1-COCl, TEA/CH2Cl2; d) R1CO2H, HOBt, EDCI, DIPEA/CH2Cl2; e) R1-NCO, TEA/CH2Cl2; f) 4-нитрофенил 3-фторбензилкарбамат, TEA/ДМФ; g) Ac2O, TEA/CH2Cl2; h) R1-SO2Cl, TEA/CH2Cl2; i) R1CO2H, HATU, DIPEA/ДМФ.
Таблица 2
|
Как показано на реакционной схеме 1, выше, амин (2S,6R)-2,6-диметилпиперазин подвергают реакции алкилирования с метил (4-бромметил)бензоатом для получения соединения 1-2, которое затем подвергают реакции алкилирования, реакции ацилирования или реакции восстановительного аминирования в условиях от a) до i), получая таким образом соединения 1-3. В заключение, полученные соединения 1-3 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия с получением соединений 82, 83, 84, 98, 99, 100, 120, 121, 122, 123, 125, 126, 127, 128, 145, 146, 147, 148, 149, 159, 160, 161, 177, 184, 188, 204, 211, 212, 213, 214, 222, 223, 224, 225, 232, 255, 265, 266, 267, 270, 272, 275, 290, 291, 292, 293, 294, 295, 296, 297, 354, 355, 403, 404 и 405.
Реакционная схема 2
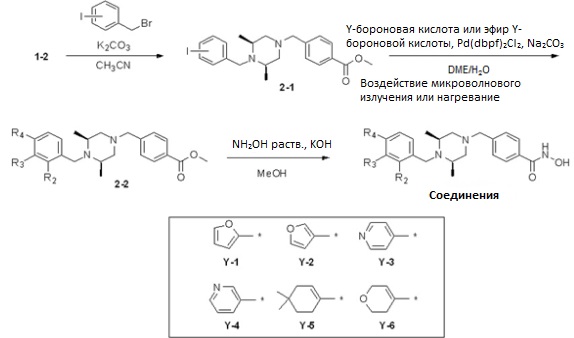
Таблица 3
|
Как показано на реакционной схеме 2, выше, соединение 1-2 алкилируют для получения соединения 2-1, которое затем подвергают реакции Сузуки с бороновой кислотой или сложным эфиром бороновой кислоты, каждое из которых содержит группы от Y-1 до Y-6, получая таким образом соединения 2-2. В заключение, полученные соединения 2-2 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия с получением соединений 309, 327, 328, 329, 330, 331, 332, 342, 343, 344, 345, 346 и 347.
Реакционная схема 3
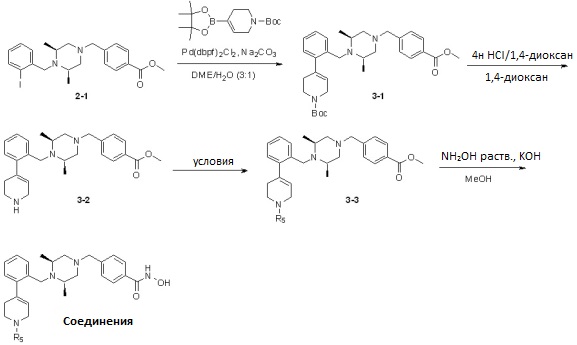
Условия: a) MsCl, TEA/CH2Cl2; b) Ac2O, TEA/CH2Cl2; c) R5-OTf, K2CO3/CH3CN; d) R5-X, TEA/CH2Cl2.
Таблица 4
|
Как показано на реакционной схеме 3, выше, соединение 2-1 подвергают реакции Сузуки с трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилатом для получения соединения 3-1, которое затем подвергают удалению защитных групп в кислотных условиях, получая таким образом соединение 3-2. Полученное соединение 3-2 подвергают реакциям замещения в условиях от a) до d), выше, с получением соединений 3-3, которые затем подвергают реакции взаимодействия с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 481, 482, 483, 484 и 485.
Реакционная схема 4
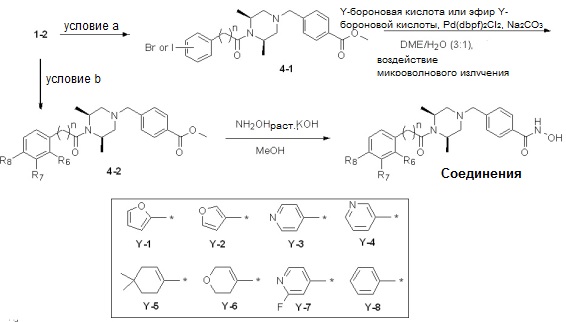
Условия: a) (йод, хлор или бром)бензоилхлорид, TEA/CH2Cl2; b) 2-([1,1’-бифенил]-3-ил)уксусная кислота, HOBt, EDCI, DIPEA/CH2Cl2.
Таблица 5
|
Как показано на реакционной схеме 4, выше, соединение 1-2 подвергают взаимодействию с бензоилхлоридом или бензоилбромидом для получения соединения 4-1, которое затем подвергают реакции Сузуки с бороновой кислотой или сложным эфиром бороновой кислоты, содержащими каждую из групп от Y-1 до Y-8, получая таким образом соединения 4-2. Альтернативно, соединение 1-2 подвергают реакции конденсации амида с 2-([1,1’-бифенил]-3-ил)уксусной кислотой, получая таким образом соединения 4-2. Полученные соединения 4-2 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 234, 242, 243, 244, 245, 246, 247, 283, 284, 285, 286, 288, 326 и 340.
Реакционная схема 5
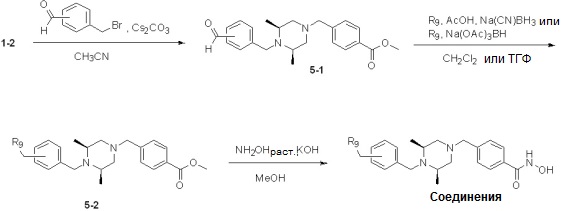
Таблица 6
|
Как показано на реакционной схеме 5, выше, соединение 1-2 подвергают реакции замещения с орто-, мета- или пара-(бромметил)бензальдегидом с получением соединений 5-1, которые затем подвергают реакции восстановительного аминирования с аминосоединениями, получая таким образом соединения 5-2. В заключение, полученные аминосоединения 5-2 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 356, 376, 382, 383, 384, 385, 411, 412, 413, 426, 427, 428, 429, 430, 431, 452, 453, 454, 455, 456, 457, 466, 467, 468 и 486.
Реакционная схема 6
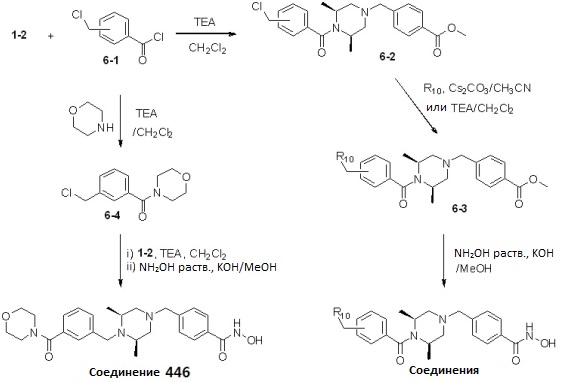
Таблица 7
|
Как показано на реакционной схеме 6, выше, получают соединение 6-1 и подвергают его реакции ацилирования с соединением 1-2 для получения соединения 6-2. Полученное соединение 6-2 подвергают реакции замещения с аминосоединениями с получением соединений 6-3, которые затем подвергают реакции взаимодействия с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 423, 424, 425, 432, 433, 434, 435, 439, 440, 441, 442, 443, 444, 458, 459, 460, 461, 462 и 463.
Альтернативно, соединение 6-1 подвергают взаимодействию с морфолином для получения соединения 6-4. Полученное соединение 6-4 и соединение 1-2 подвергают реакции замещения и затем подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединение 446.
Реакционная схема 7
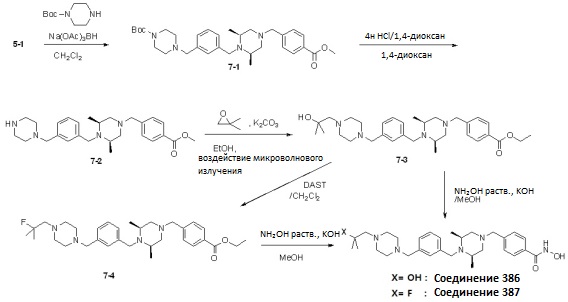
Как показано на реакционной схеме 7, соединение 5-1 подвергают реакции восстановительного аминирования с 4-трет-бутил пиперазин-1-карбоксилатом и затем подвергают удалению защитных групп в кислотных условиях для получения соединения 7-2. Полученное соединение 7-2 подвергают взаимодействию с 2,2-диметилоксираном для получения соединения 7-3, которое затем подвергают реакции взаимодействия с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединение 386. Далее, соединение 7-3 подвергают взаимодействию с DAST для замещения гидроксильной группы на фтор, получая таким образом соединение 7-4. Полученное соединение 7-4 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединение 387.
Реакционная схема 8
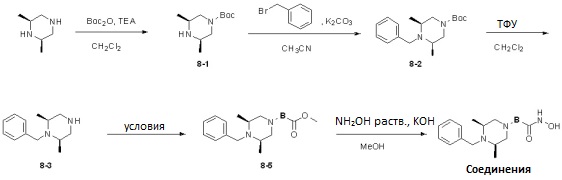
Условия: a) 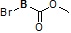 (8-4), основание/CH3CN или DCM; b)
(8-4), основание/CH3CN или DCM; b) 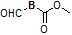 (8-4), восстанавливающий агент/CH2Cl2; c)
(8-4), восстанавливающий агент/CH2Cl2; c) 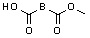 (8-4), EDC, HOBt, DIPEA/CH2Cl2
(8-4), EDC, HOBt, DIPEA/CH2Cl2
Таблица 8
|
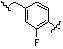
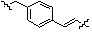
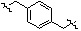
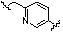
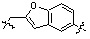
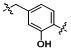
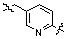
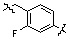
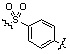
Как показано на реакционной схеме 8, в (2S,6R)-2,6-диметилпиперазин вводят защитную группу Boc и подвергают взаимодействию с бензилбромидом, после чего его подвергают удалению защитных групп в кислотных условиях, получая таким образом соединение (8-3). Полученное соединение 8-3 подвергают реакции алкилирования или реакции восстановительного аминирования с соединением 8-4, как показано в условиях от a) до c), получая таким образом соединения 8-5. Полученные соединения 8-5 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 154, 171, 172, 173, 194, 218, 219, 520, 571, 574, 652, 812, 813, 814, 818, 820, 822, 823 и 824.
Реакционная схема 9
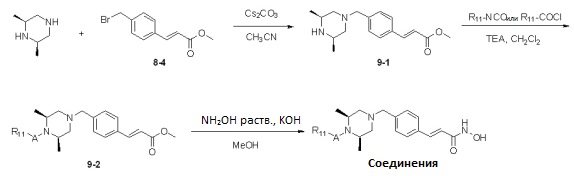
Таблица 9
|
Как показано на реакционной схеме 9, выше, соединение 8-4 подвергают взаимодействию с (2S,6R)-2,6-диметилпиперазином для получения соединения 9-1, которое затем подвергают реакции замещения с изоцианатом или карбонилхлоридом, содержащими заместители, представленные R11, получая таким образом соединение 9-2. Полученные соединения 9-2 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 321, 322 и 323.
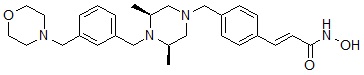
Реакционная схема 10
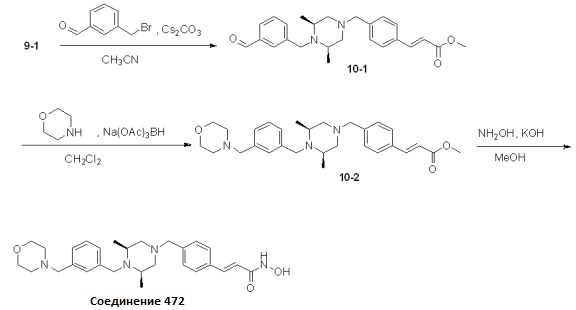
Как показано на реакционной схеме 10, выше, соединение 9-1 подвергают реакции алкилирования с 3-(бромметил)бензальдегидом для получения соединения 10-1. Полученное соединение 10-1 подвергают реакции восстановительного аминирования с морфолином для получения соединения 10-2, которое затем подвергают реакции взаимодействия с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединение 472.
Реакционная схема 11
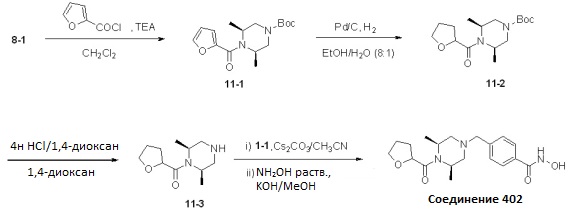
Как показано на реакционной схеме 11 выше, соединение 8-1 подвергают взаимодействию с фуран-2-карбонилхлоридом для получения соединения 11-1. Полученное соединение 11-1 гидрируют для получения соединения 11-2, которое затем подвергают удалению защитных групп Boc в кислотных условиях для получения соединения 11-3. Полученное соединение 11-3 подвергают взаимодействию с соединением 1-1 и затем подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединение 402.
Реакционная схема 12
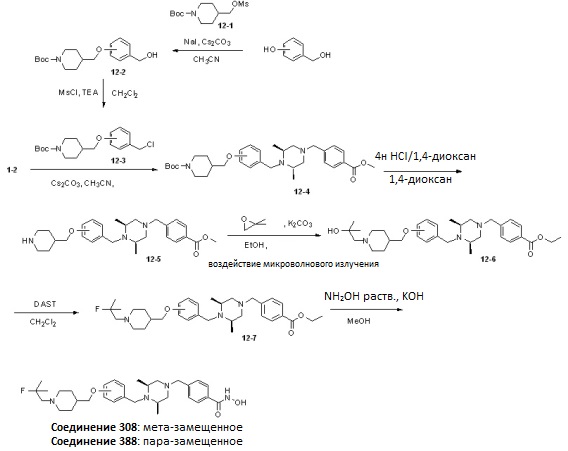
Как показано на реакционной схеме 12, выше, 3-гидроксибензиловый спирт и 4-гидроксибензиловый спирт подвергают реакции замещения с соединением 12-1 с получением соединений 12-2, которые затем подвергают реакции взаимодействия с MsCl с получением соединений 12-3. Соединения 12-3 подвергают взаимодействию с соединением 1-2 с получением соединений 12-4, которые подвергают удалению защитных групп в кислотных условиях и подвергают взаимодействию с 2,2-диметилоксираном с получением соединений 12-6. Полученные соединения 12-6 подвергают взаимодействию с DAST для замещения гидроксильной группы на фтор, получая таким образом соединения 12-7. В заключение, полученные соединения 12-7 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 380 и 388.
Реакционная схема 13
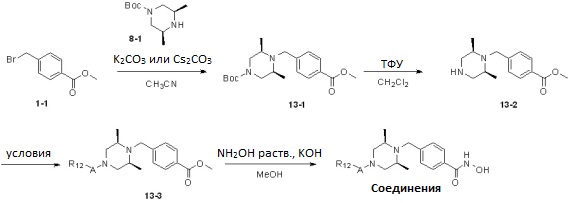
Условия: a) R12-CH2-Br (I или -Cl), K2CO3 (или Cs2CO3)/CH3CN (или ДМФ); b) R12-CHO, NaCNBH3, AcOH/ТГФ или AcOH/CH2ClCH2Cl; c) R12-COX, TEA/CH2Cl2; d) R12CO2H, HOBt, EDCI, DIPEA/CH2Cl2; e) R12-NCO, TEA/CH2Cl2; f) R12-NCS, TEA/CH2Cl2; g) 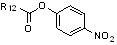 , ДМФ; h) R12-SO2Cl, TEA/CH2Cl2.
, ДМФ; h) R12-SO2Cl, TEA/CH2Cl2.
Таблица 10
|
Как показано на реакционной схеме 13, выше, соединение 8-1 и соединение 1-1 подвергают реакции алкилирования для получения соединения 13-1, которое затем подвергают удалению защитных групп в кислотных условиях для получения соединения 13-2. Полученное соединение 13-2 подвергают реакции алкилирования, реакции ацилирования или реакции восстановительного аминирования в условиях от a) до h), получая таким образом соединения 13-3. В заключение, полученные соединения 13-3 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 80, 81, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 118, 162, 163, 164, 165, 166, 167, 168, 183, 185, 186, 187, 189, 196, 197, 215, 220, 230, 231, 233, 256, 268, 271, 273, 274, 298, 299, 300, 301, 302, 303, 304 и 305.
Реакционная схема 14
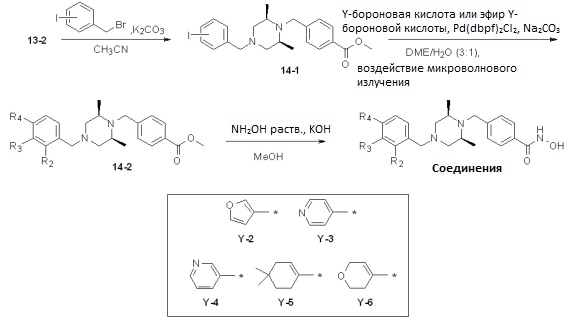
Таблица 11
|
Как показано на реакционной схеме 14, выше, соединение 13-2 подвергают реакции алкилирования для получения соединения 14-1, которое затем подвергают реакции Сузуки с бороновой кислотой или сложным эфиром бороновой кислоты, каждый из которых содержит от Y-2 до Y-6, получая таким образом соединения 14-2. В заключение, полученные соединения 14-2 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 348, 349, 350, 351, 352, 396, 400 и 401.
Реакционная схема 15
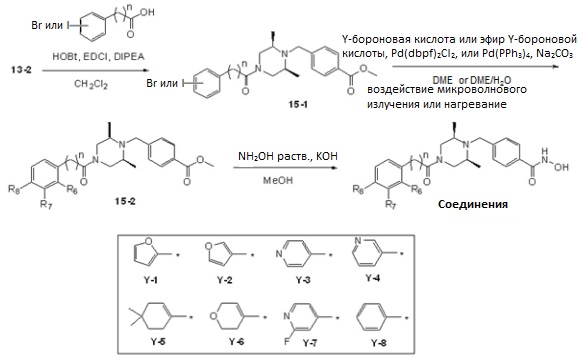
Таблица 12
|
Как показано на реакционной схеме 15, выше, соединение 13-2 подвергают реакции конденсации амида с 2-, 3-, 4-йодбензойной кислотой или 2-(3-бромфенил)уксусной кислотой для получения соединения 15-1, которое затем подвергают реакции Сузуки с бороновой кислотой или сложным эфиром бороновой кислоты, каждое из которых содержит группы от Y-1 до Y-8, получая таким образом соединения 15-2. В заключение, полученные соединения 15-2 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 250, 251, 252, 253, 257, 258, 259, 260, 261, 262, 263, 276, 277, 278, 279 и 280.
Реакционная схема 16
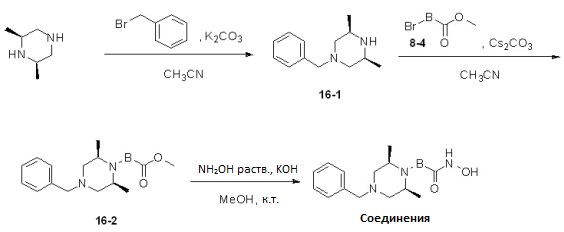
Таблица 13
|
Как показано на реакционной схеме 16, выше, (2S,6R)-2,6-диметилпиперазин подвергают взаимодействию с бензилбромидом для введения в него бензильной группы, получая таким образом соединение 16-1, которое затем подвергают реакции алкилирования с соединением 8-4, получая таким образом соединения 16-2. В заключение, полученные соединения 16-2 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 174, 175, 176 и 195.
Реакционная схема 17
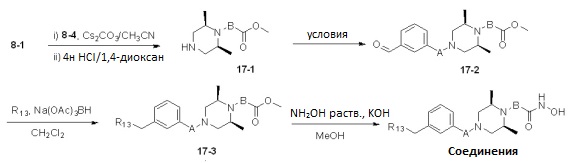
Условия: a) (3-бромметил)бензальдегид, Cs2CO3/CH3CN; b) 3-формилбензойная кислота, HOBt, EDCI, DIPEA/CH2Cl2.
Таблица 14
|
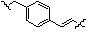
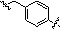
Как показано на реакционной схеме 17, выше, соединение 8-1 подвергают реакции алкилирования с соединением 8-4 и подвергают удалению защитных групп в кислотных условиях, получая таким образом соединение 17-1. Полученное соединение 17-1 подвергают реакции алкилирования или реакции конденсации амида в условиях a) или b) с получением соединений 17-2, которые затем подвергают реакции восстановительного аминирования с пирролидином или диэтиламином, получая таким образом соединения 17-3. В заключение, полученные соединения 17-3 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 475, 476, 478, 479, 480 и 487.
Реакционная схема 18
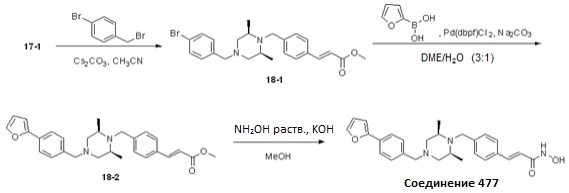
Как показано на реакционной схеме 18, выше, соединение 17-1 подвергают реакции алкилирования с 4-бромбензилбромидом для получения соединения 18-1, которое затем подвергают реакции Сузуки с фуран-2-илборной кислотой, получая таким образом соединение 18-2. В заключение, полученное соединение 18-2 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединение 477.
Реакционная схема 19
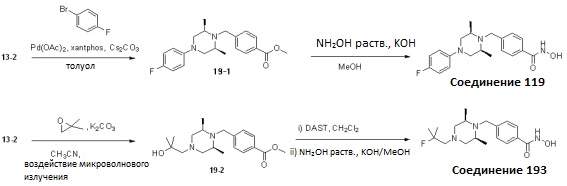
Как показано на реакционной схеме 19, выше, соединение 13-2 подвергают реакции Бухвальда с 1-бром-4-фторбензолом для получения соединения 19-1, которое затем подвергают реакции взаимодействия с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединение 119.
Альтернативно, соединение 13-2 подвергают взаимодействию с 2,2-диметилоксираном для получения соединения 19-2, которое затем подвергают взаимодействию с DAST для замещения гидроксильной группы на фтор и затем подвергают реакции взаимодействия с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединение 193.
Реакционная схема 20
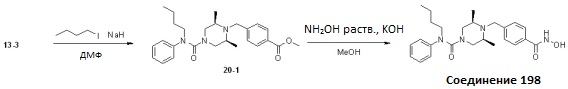
Как показано на реакционной схеме 20, выше, соединение 13-3 подвергают реакции алкилирования с бутилйодидом для получения соединения 20-1, которое затем подвергают реакции взаимодействия с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединение 198.
Реакционная схема 21
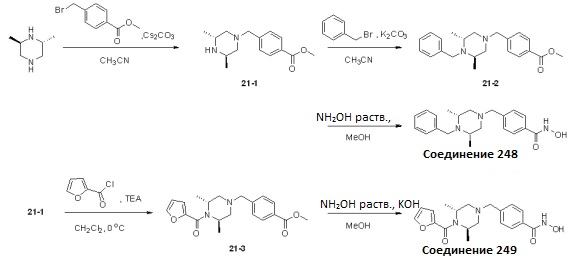
Как показано на реакционной схеме 21, выше, (2R,6R)-2,6-диметилпиперазин подвергают реакции алкилирования с соединением 1-1 для получения соединения 21-1, которое затем подвергают реакции алкилирования или реакции ацилирования, получая таким образом соединение 21-2 или соединение 21-3. В заключение, полученное соединение 21-2 или соединение 21-3 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 248 и 249.
Реакционная схема 22
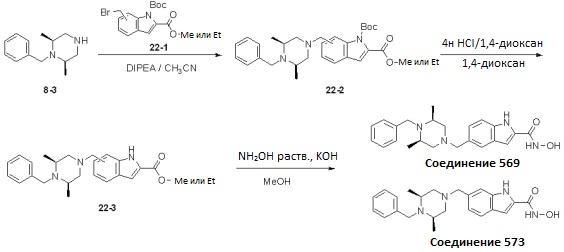
Как показано на реакционной схеме 22, выше, (2S,6R)-1-бензил-2,6-диметилпиперазин подвергают реакции алкилирования с соединением 8-3 и соединением 22-1 с получением соединений 22-2, которое затем подвергают удалению защитных групп для удаления трет-бутилоксикарбонильной группы, получая таким образом соединения 22-3. В заключение, соединения 22-3 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 569 и 573.
Реакционная схема 23
Условия: a) 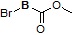 (8-4), TEA/CH3CN; b)
(8-4), TEA/CH3CN; b) 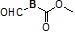 (8-4), восстанавливающий агент/CH2Cl2.
(8-4), восстанавливающий агент/CH2Cl2.
Таблица 15
|
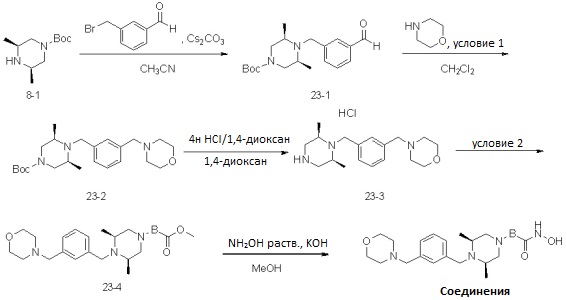
Как показано на реакционной схеме 23, выше, соединение 8-1 подвергают реакции алкилирования для получения соединения 23-1, которое затем подвергают реакции восстановительного аминирования с морфолином для получения соединения 23-2, которое затем подвергают удалению защитных групп для удаления трет-бутилоксикарбонильной группы, получая таким образом соединение 23-3. Соединение 23-3 подвергают реакции алкилирования с соединением 8-4 с получением соединений 23-4. В заключение, полученные соединения 23-4 подвергают взаимодействию с водным раствором гидроксиламина и гидроксидом калия, получая таким образом соединения 609, 653 и 696.
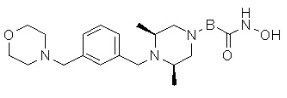
Композиции, содержащие новые соединения, обладающие ингибирующей HDAC6 активностью, их применение и способ лечения заболеваний
В настоящем изобретении предложена фармацевтическая композиция для предупреждения или лечения ассоциированных с активностью гистондеацетилазы 6 (HDAC6) заболеваний, которая содержит в качестве активного ингредиента соединение, представленное следующей формулой I, его изомер или его фармацевтически приемлемая соль:
Формула I
где L, R1, R2, Q, X и n имеют значения, определенные выше.
Фармацевтическая композиция в соответствии с настоящим изобретением демонстрирует замечательный эффект по лечению или предупреждению заболеваний, ассоциированных с активностью гистондеацетилазы 6 (HDAC6) путем селективного ингибирования гистондеацетилаза 6 (HDAC6).
Ассоциированные с активностью гистондеацетилазы 6 (HDAC6) заболевания включают злокачественное новообразование, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания и нейродегенеративные заболевания. В частности, эти ассоциированные с активностью гистондеацетилазы 6 (HDAC6) заболевания включают рак легких, рак толстой кишки, рак молочной железы, рак простаты, рак печени, рак мозга, рак яичников, рак желудка, рак кожи, рак поджелудочной железы, глиому, глиобластому, лейкемию, лимфома, множественную миелому, солидный рак, болезнь Вильсона, спиноцеребеллярную атаксию, прионное заболевание, болезнь Паркинсона, болезнь Хантингтона, боковой амиотрофический склероз, амилоидоз, болезнь Альцгеймера, алкогольную болезнь печени, спинальную мышечную атрофию, ревматоидный артрит и остеоартрит, а также расстройства или заболевания, связанные с аномальной функцией гистондеацетилазы.
В варианте осуществления изобретения соединение, представленное формулой I в соответствии с настоящим изобретением, может являться соединением, представленным формулой II или формулой III. В другом варианте осуществления изобретения соединение, представленное формулой I в соответствии с настоящим изобретением, может являться одним из соединений, представленных формулами от I-1 до I-23. В еще одном варианте осуществления изобретения соединение, представленное формулой I в соответствии с настоящим изобретением, может являться одним из соединений от 080 до 824.
Фармацевтически приемлемой солью является такая, как описано выше в отношении фармацевтически приемлемой соли соединения, представленного формулой I в соответствии с настоящим изобретением.
Предназначенная для введения фармацевтическая композиция по настоящему изобретению может дополнительно содержать, в дополнение к соединению формулы I, его изомеру или его фармацевтически приемлемой соли, по меньшей мере, один фармацевтически приемлемый носитель. Фармацевтически приемлемый носитель, используемый в настоящем изобретении, может представлять собой, по меньшей мере, один из следующих: физиологический солевой раствор, стерильная вода, раствор Рингера, забуференный солевой раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и смеси двух или более из них. При необходимости, композиция может содержать другие обычные добавки, такие как антиоксидант, буфер или бактериостатический агент. Кроме того, композиция может быть изготовлена в виде инъекционных лекарственных препаратов, таких как растворы, суспензии, эмульсии и т.д., пилюль, капсул, гранул и таблеток, используя разбавитель, диспергирующий агент, поверхностно-активное вещество, связующее вещество и смазывающее вещество. Таким образом, композиция по настоящему изобретению может быть представлена в виде пластырей, жидкостей, пилюль, капсул, гранул, таблеток, суппозиториев и т.д. Эти препараты могут быть получены либо с помощью обычных методов, которые используются для изготовления лекарственной формы в данной области, или по способу, раскрытому в Remington’s Pharmaceutical Science (самое последнее издание), Mack Publishing Company, Easton, PA. Кроме того, композиция по настоящему изобретению могут быть изготовлена в виде различных препаратов, в зависимости от заболеваний или компонентов.
Композиция по настоящему изобретению могут быть введена перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно), в зависимости от предполагаемого использования. Доза фармацевтической композиции варьирует в зависимости от веса пациента, возраста, пола, состояния здоровья, диеты, времени введения, способа введения, скорости экскреции, тяжести заболевания и тому подобное.
Фармацевтическая композиция по настоящему изобретению может дополнительно содержать, помимо соединения, представленного формулой I, его изомера или его фармацевтически приемлемой соли, один или несколько активных ингредиентов, которые демонстрируют идентичную или сходную с ним медицинскую эффективность.
В настоящем изобретении предложены способ предупреждения или лечения ассоциированных с активностью гистондеацетилазы 6 заболеваний, который включает введение терапевтически эффективного количества соединения, представленного формулой I, его изомера или его фармацевтически приемлемой соли.
Как используется в настоящем документе, термин «терапевтически эффективное количество» относится к количеству соединения, представленного формулой I, которое эффективно для предупреждения или лечения ассоциированных с активностью гистондеацетилазы 6 заболеваний.
В настоящем изобретении предложен также способ селективного ингибирования HDAC6, который включает введение соединения формулы I, его изомера или его фармацевтически приемлемой соли млекопитающим, включая людей.
Способ предупреждения или лечения ассоциированного с активностью гистондеацетилазы 6 заболевания в соответствии с настоящим изобретением включает ингибирование или предупреждение заболевания, а также устранение самого заболевания, до появления симптомов, путем введения соединения, представленного формулой I. При лечении заболеваний величина профилактической или терапевтической дозы конкретного активного ингредиента будет изменяться в зависимости от характера и тяжести заболевания или состояния, а также может изменяться в зависимости от способа, с помощью которого вводят активный ингредиент. Доза и частота дозирования будут также варьироваться в зависимости от возраста, веса тела и реакции отдельного пациента. Подходящие режимы дозирования могут быть легко выбраны специалистами в данной области с учетом этих факторов. Кроме того, способ предупреждения или лечения ассоциированного с активностью гистондеацетилазы 6 заболевания в соответствии с настоящим изобретением может дополнительно включать введение вместе с соединением, представленным формулой I, терапевтически эффективного количества дополнительного активного агента, полезного для лечения заболевания, где дополнительный активный агент может проявлять синергетический эффект с соединением формулы I или вспомогательный эффект.
Настоящее изобретение предусматривает также применение соединения, представленного формулой I, его изомера или его фармацевтически приемлемой соли при получении лекарственного средства, предназначенного для лечения ассоциированного с активностью гистондеацетилазы 6 заболевания. При получении лекарственного средства соединение, представленное формулой I, может быть смешано с фармацевтически приемлемым адъювантом, разбавителем, носителем или тому подобное и объединено с другими активными агентами, так что активные ингредиенты могут проявлять синергетические эффекты.
Характерные признаки, указанные в отношении применения, композиции и способа лечения по настоящему изобретению, могут быть соответствующим образом объединены, если они не противоречат друг другу.
ПОЛОЖИТЕЛЬНЫЙ ЭФФЕКТ ИЗОБРЕТЕНИЯ
Соединения, представленные формулой I, их изомеры или их фармацевтически приемлемые соли могут селективно ингибировать HDAC6, и, таким образом, демонстрируют превосходное действие по предотвращению или лечению ассоциированных с активностью гистондеацетилазы 6 заболеваний.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение описано более подробно со ссылкой на примеры, примеры получения и экспериментальные примеры. Следует понимать, однако, что эти примеры приведены только в иллюстративных целях и не предназначены для ограничения объема настоящего изобретения.
Получение новых соединений производных диметилпиперазина
Конкретные способы получения соединений формулы I приведены ниже.
Пример 1: Синтез соединения 80
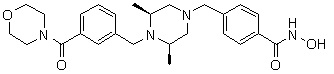
(4-(((2S,6R)-4-бензоил-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-бензоил-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли бензоилхлорид (0,049 мл, 0,420 ммоль) и TEA (0,106 мл, 0,763 ммоль). Смесь перемешивали при температуре 0°C в течение 1 часа, и в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, затем экстрагировали метиленхлоридом. Органический слой отделяли и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,124 г, 88,7%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 80
Метил 4-(((2S,6R)-4-бензоил-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,060 г, 0,164 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,2 мл, 3,275 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,091 г, 1,637 ммоль). Смесь перемешивали при комнатной температуре в течение 20 минут, и в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, затем экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 80 (0,045 г, 74,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, 2 H, J=8,2 Гц), 7,46-7,39 (м, 5 H), 7,34 (д, 2 H, J=8,8 Гц), 3,67 (с, 2 H), 3,17 (с, 2 H), 2,69-2,61 (м, 2 H), 2,60-2,57 (м, 2 H), 1,82-1,76 (м, 2 H), 0,83 (д, 6 H, J=6,12 Гц); ЖХМС (ES) m/z 368,0 (M++1).
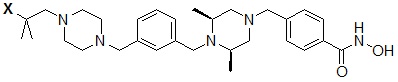
Пример 2: Синтез соединения 81
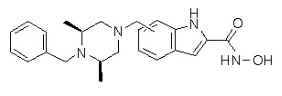
(4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,050 г, 0,191 ммоль) растворяли в ацетонитриле (2 мл) и затем добавляли бензилбромид (0,023 мл, 0,210 ммоль) и K2CO3 (0,053 г, 0,381 ммоль). Смесь нагревали и перемешивали при температуре 80°C в течение 1 часа, и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,022 г, 32,8%) в виде бесцветного масла.
Стадия 2: Синтез соединения 81
Метил 4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,022 г, 0,062 ммоль) растворяли в метаноле (2 мл) и затем добавляли гидроксиламин (0,023 мл, 0,210 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,053 г, 0,381 ммоль). Смесь перемешивали при комнатной температуре в течение 20 минут, и в реакционную смесь добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 81 (0,003 г, 13,6%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,65 (д, 2 H, J=8,1 Гц), 7,34-7,23 (м, 7 H), 3,73 (с, 2 H), 3,39 (с, 2 H), 2,64 (д, 2 H, J=10,4 Гц), 2,56-2,54 (м, 2 H), 1,79 (т, 2 H, J=10,6 Гц), 0,88 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 354,2 (M++1).
Пример 3: Синтез соединения 82
(4-(((3R,5S)-4-ацетил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-ацетил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (2 мл) и затем добавляли ацетангидрид (0,072 мл, 0,763 ммоль) и TEA (0,266 мл, 1,907 ммоль). Смесь перемешивали при температуре 0°C в течение 1 часа и добавляли насыщенный водный раствор гидрокарбоната натрия, затем экстрагировали метиленхлоридом. Органический слой сушили безводным сульфатом магния, и затем концентрировали при пониженном давлении с получением желаемого соединения (0,115 г, 99,1%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 82
Метил 4-(((3R,5S)-4-ацетил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,052 г, 0,171 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,209 мл, 3,417 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,096 г, 1,708 ммоль). Смесь перемешивали при комнатной температуре в течение 20 минут, и затем концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Концентрат очищали с помощью PTLC (100%-ный этилацетат) с получением желаемого соединения 82 (0,016 г, 30,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,73 (д, 2 H, J=8,3 Гц), 7,50 (д, 2 H, J=8,3 Гц), 4,51-4,50 (м, 1 H), 4,11-4,10 (м, 1 H), 3,57 (с, 2 H), 2,71 (д, 2 H, J=11,5 Гц), 2,21-2,20 (м, 2 H), 2,11 (с, 3 H), 1,40-1,38 (м, 6 H); ЖХМС (ES) m/z 306,2 (M++1).
Пример 4: Синтез соединения 83
(4-(((3R,5S)-4-бензоил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-бензоил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли бензоил хлорид (0,049 мл, 0,420 ммоль) и TEA (0,106 мл, 0,763 ммоль). Смесь перемешивали при температуре 0°C в течение 1 часа, и в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, затем метиленхлорид. Органический слой отделяли и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,124 г, 88,7%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 83
Метил 4-(((3R,5S)-4-бензоил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,060 г, 0,164 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,2 мл, 3,275 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,091 г, 1,637 ммоль). Смесь перемешивали при комнатной температуре в течение 20 минут, и в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, затем экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 83 (0,045 г,74,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,72 (д, 2 H, J=8,3 Гц), 7,49 (д, 2 H, J=8,2 Гц), 7,46-7,44 (м, 3 H), 7,36-7,34 (м, 2 H), 3,59 (с, 2 H), 2,71 (д, 2 H, J=10,7 Гц), 2,26 (дд, 2 H, J=10,2, 3,6 Гц), 1,39 (д, 6 H, J=6,7 Гц).
Пример 5: Синтез соединения 84
(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоата
(2S,6R)-2,6-Диметилпиперазин (50,000 г, 437,867 ммоль) и Cs2CO3 (171,199 г, 525,440 ммоль) растворяли в ацетонитриле (200 мл) при температуре 0°C, и в раствор добавляли метил 4-(бромметил)бензоат (формула 1-1, 80,242 г, 350,293 ммоль), затем перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь фильтровали через стеклянный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. К концентрату добавляли гексан (100 мл) и перемешивали, и выпавший твердый осадок фильтровали, промывали гексаном и сушили с получением желаемого соединения (85,200 г, 74,2%) в виде твердого вещества белого цвета.
Стадия 2: Синтез метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 30,000 г, 114,351 ммоль), бензилбромид (14,961 мл, 125,786 ммоль) и K2CO3 (23,707 г, 171,527 ммоль) растворяли в ацетонитриле (150 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Реакционную смесь фильтровали через стеклянный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 120 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением желаемого соединения (22,400 г, 55,6%) в виде твердого вещества белого цвета.
Стадия 3: Синтез соединения 84
Метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 15,000 г, 42,557 ммоль), гидроксиламин (52,061 мл, 851,136 ммоль, 50,00%-ный водный раствор) и гидроксид калия (23,879 г, 425,568 ммоль) растворяли в метаноле (300 мл) при температуре 0°C, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 120 г; метанол/метиленхлорид=от 0% до 20%) и концентрировали, и затем полученное вещество кристаллизовали из диэтилового эфира (200 мл) и метиленхлорида (50 мл) при температуре 25°C и фильтровали. Полученный твердый продукт промывали диэтиловым эфиром и сушили с получением соединения 84 (12,580 г, 83,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,18 (ушир. с, 1 H), 9,03 (ушир. с, 1 H), 7,70 (д, 2 H, J=8,2 Гц), 7,36-7,33 (м, 4 H), 7,28 (дд, 2 H, J=7,5, 7,5 Гц), 7,18 (дд, 1 H, J=7,2, 7,2 Гц), 3,73 (с, 2 H), 3,44 (с, 2 H), 2,64 (д, 2 H, J=10,6 Гц), 2,61-2,53 (м, 2 H), 1,81 (т, 2 H, J=10,5 Гц), 0,90 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 354,2 (M++1).
Пример 6: Синтез соединения 98
((2S,6R)-4-(4-(гидроксикарбамоил)бензил)-N-изопропил-2,6-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-изопропилкарбамоил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли 2-изоцианатопропан (0,041 мл, 0,419 ммоль) и TEA (0,080 мл, 0,572 ммоль). Смесь перемешивали при температуре 0°C в течение 2 часов, и в реакционную смесь добавляли воду, затем экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении с получением желаемого соединения (0,120 г, 90,6%) в виде твердого вещества желтого цвета.
Стадия 2: Синтез соединения 98
Метил 4-(((3R,5S)-4-изопропилкарбамоил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,060 г, 0,173 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,211 мл, 3,454 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,097 г, 1,727 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и добавляли насыщенный водный раствор гидрокарбоната натрия, затем экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 98 (0,040 г, 66,5%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,72 (д, 2 H, J=8,3 Гц), 7,46 (д, 2 H, J=8,2 Гц), 4,06-4,03 (м, 2 H), 3,96-3,93 (м, 1 H), 3,54 (с, 2 H), 2,68 (д, 2 H, J=11,1 Гц), 2,15 (дд, 2 H, J=10,2, 3,6 Гц), 1,29 (д, 6 H, J=6,8 Гц), 1,14 (д, 6 H, J=6,6 Гц); ЖХМС (ES) m/z 349,1 (M++1).
Пример 7: Синтез соединения 99
((2S,6R)-N-(3-хлорфенил)-4-(4-(гидроксикарбамоил)бензил)-2,6-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-((4-(3-хлорфенилкарбамоил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли 3-хлорфенилизоцианат (0,051 мл, 0,419 ммоль) и TEA (0,080 мл, 0,572 ммоль). Смесь перемешивали при температуре 0°C в течение 2 часов, и в реакционную смесь добавляли воду, затем экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении с получением желаемого соединения (0,018 г, 68,1%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 99
Метил 4-((4-(3-хлорфенилкарбамоил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,120 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,147 мл, 2,404 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,067 г, 1,202 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 99 (0,040 г, 66,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,74 (д, 2 H, J=8,0 Гц), 7,53-7,52 (м, 1 H), 7,45 (д, 2 H, J=8,1 Гц), 7,29-7,28 (м, 1 H), 7,24-7,22 (м, 1 H), 7,01 (д, 1 H, J=7,8 Гц), 4,21-4,20 (м, 2 H), 3,56 (с, 2 H), 2,74 (д, 2 H, J=11,4 Гц), 2,22 (дд, 2 H, J=10,2, 3,6 Гц), 1,25 (д, 6 H, J=6,5 Гц).
Пример 8: Синтез соединения 100
(4-(((3R,5S)-3,5-диметил-4-(фенилсульфонил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметилпиперазин-4-(фенилсульфонил-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли бензолсульфонилхлорид (0,054 мл, 0,419 ммоль) и TEA (0,080 мл, 0,572 ммоль). Смесь перемешивали при температуре 0°C в течение 2 часов и затем перемешивали при комнатной температуре в течение 3 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,048 г, 31,3%) в виде твердого вещества бледно-желтого цвета.
Стадия 2: Синтез соединения 100
Метил 4-(((3R,5S)-3,5-диметилпиперазин-4-(фенилсульфонил-1-ил)метил)бензоат (формула 1-3, 0,048 г, 0,119 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,146 мл, 2,385 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,067 г, 1,193 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 100 (0,034 г, 70,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,84 (д, 2 H, J=8,1 Гц), 7,68 (д, 2 H, J=8,3 Гц), 7,63-7,61 (м, 1 H), 7,58-7,54 (м, 2 H), 7,41 (д, 2 H, J=8,3 Гц), 4,06-4,03 (м, 2 H), 3,41 (с, 2 H), 2,53 (д, 2 H, J=11,3 Гц), 1,85-1,81 (м, 2 H), 1,43 (д, 6 H, J=6,9 Гц); ЖХМС (ES) m/z 404,1 (M++1).
Пример 9: Синтез соединения 103
((3R,5S)-4-(4-(гидроксикарбамоил)бензил)-N-изопропил-3,5-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(изопропилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли 2-изоцианатопропан (0,041 мл, 0,419 ммоль) и TEA (0,079 мл, 0,572 ммоль). Смесь перемешивали в течение 2 часов, повышая при этом температуру от 0°C до комнатной температуры, и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,094 г, 70,1%).
Стадия 2: Синтез соединения 103
Метил 4-(((2S,6R)-4-(изопропилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,045 г, 0,130 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,158 мл, 2,590 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,073 г, 1,295 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 103 (0,020 г, 44,3%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,69 (д, 2 H, J=8,2 Гц), 7,50 (д, 2 H, J=8,2 Гц), 3,91-3,82 (м, 5 H), 2,67-2,61 (м, 2 H), 2,55-2,50 (м, 2 H), 1,13 (д, 6 H, J=6,6 Гц), 1,03 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 349,2 (M++1).
Пример 10: Синтез соединения 104
((3R,5S)-N-(3-хлорфенил)-4-(4-(гидроксикарбамоил)бензил)-3,5-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(3-хлорфенилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли 3-хлорфенилизоцианат (0,051 мл, 0,419 ммоль) и TEA (0,079 мл, 0,572 ммоль). Смесь перемешивали в течение 2 часов, повышая при этом температуру от 0°C до комнатной температуры, и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,094 г, 70,1%).
Стадия 2: Синтез соединения 104
Метил 4-(((2S,6R)-4-(3-хлорфенилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,065 г, 0,156 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,191 мл, 3,126 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,088 г, 1,563 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, который затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 104 (0,035 г, 53,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,67 (с, 1 H), 7,67 (д, 2 H, J=8,2 Гц), 7,63-7,62 (м, 1 H), 7,41-7,38 (м, 3 H), 7,24 (дд, 1 H, J=10,2, 3,6 Гц), 7,46 (дд, 1 H, J=10,2, 3,6 Гц), 3,95 (д, 2 H, J=12,8 Гц), 3,77 (с, 2 H), 2,68-2,65 (м, 2 H), 2,53-2,52 (м, 2 H), 0,97 (д, 6 H, J=6,1 Гц).
Пример 11: Синтез соединения 105
(4-(((2S,6R)-2,6-диметил-4-(фенилсульфонил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(фенилсульфонил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли фенилсульфонилхлорид (0,065 г, 0,306 ммоль) и TEA (0,116 мл, 0,835 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,108 г, 88,8%) в виде бесцветного масла.
Стадия 2: Синтез соединения 105
Метил 4-(((2S,6R)-2,6-диметил-4-(фенилсульфонил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,045 г, 0,115 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,137 мл, 2,236 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,063 г, 1,118 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 105 (0,027 г, 59,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,76-7,74 (м, 3 H), 7,69-7,68 (м, 2 H), 7,61 (д, 2 H, J=8,3 Гц), 7,28 (д, 2 H, J=8,1 Гц), 3,71 (с, 2 H), 3,44 (д, 2 H, J=10,5 Гц), 2,65-2,64 (м, 2 H), 2,02 (т, 2 H, J=10,8 Гц), 0,91 (д, 6 H, J=6,2 Гц); ЖХМС (ES) m/z 404,1 (M++1).
Пример 12: Синтез соединения 106
(4-(((2S,6R)-4-(4-хлорбензоил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-((4-(4-хлорбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли 4-хлорбензоилхлорид (0,054 мл, 0,419 ммоль) и TEA (0,159 мл, 1,144 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,142 г, 92,9%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 106
Метил 4-((4-(4-хлорбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,070 г, 0,175 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,214 мл, 3,492 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,098 г, 1,746 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 106 (0,028 г, 39,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,3 Гц), 7,51 (д, 2 H, J=8,4 Гц), 7,48 (д, 2 H, J=8,3 Гц), 7,41 (д, 2 H, J=8,4 Гц), 4,38-4,37 (м, 1 H), 3,88 (с, 2 H), 3,51-3,50 (м, 1 H), 3,06-3,05 (м, 1 H), 2,82-2,80 (м, 1 H), 2,67-2,58 (м, 2 H), 1,10 (с, 3 H), 0,92-0,88 (м, 3 H); ЖХМС (ES) m/z 402,1 (M++1).
Пример 13: Синтез соединения 107
(4-(((2S,6R)-4-(3-хлорбензоил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-((4-(3-хлорбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,278 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли 3-хлорбензоилхлорид (0,039 мл, 0,306 ммоль) и TEA (0,116 мл, 0,835 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,100 г, 89,6%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 107
Метил 4-((4-(3-хлорбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,050 г, 0,125 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,153 мл, 2,494 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,070 г, 1,247 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 107 (0,025 г, 49,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, 2 H, J=8,2 Гц), 7,54 (д, 1 H, J=7,9 Гц), 7,52-7,46 (м, 2 H), 7,42 (д, 2 H, J=8,0 Гц), 7,36 (д, 1 H, J=7,5 Гц), 4,25 (д, 1 H, J=9,4 Гц), 3,78 (с, 2 H), 2,98-2,96 (м, 2 H), 2,71-2,68 (м, 3 H), 1,02 (с, 3 H), 0,84 (с, 3 H); ЖХМС (ES) m/z 402,1 (M++1).
Пример 14: Синтез соединения 108
(4-(((2S,6R)-4-(2-хлорбензоил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-((4-(2-хлорбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,278 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли 2-хлорбензоилхлорид (0,039 мл, 0,306 ммоль) и TEA (0,116 мл, 0,835 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении с получением желаемого соединения (0,110 г, 98,6%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 108
Метил 4-((4-(2-хлорбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,055 г, 0,137 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,168 мл, 2,744 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,077 г, 1,372 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 108 (0,025 г, 45,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 11,13 (с, 1 H), 8,97 (с, 1 H), 7,68 (д, 2 H, J=8,0 Гц), 7,53 (д, 1 H, J=7,2 Гц), 7,44-7,35 (м, 5 H), 4,28 (д, 2 H, J=11,9 Гц), 3,78 (с, 2 H), 3,06 (д, 1 H, J=12,8 Гц), 2,92-2,89 (м, 1 H), 2,74-2,61 (м, 3 H), 1,02 (д, 3 H, J=5,9 Гц), 0,81-0,78 (м, 3 H); ЖХМС (ES) m/z 402,1 (M++1).
Пример 15: Синтез соединения 109
(4-(((2S,6R)-4-((4-хлорфенил)сульфонил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-((4-хлорфенил)сульфонил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли 4-хлорбензол-1-сульфонилхлорид (0,065 г, 0,306 ммоль) и TEA (0,116 мл, 0,835 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,108 г, 88,8%) в виде бесцветного масла.
Стадия 2: Синтез соединения 109
Метил 4-(((2S,6R)-4-((4-хлорфенил)сульфонил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,05 г, 0,114 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,140 мл, 2,289 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,064 г, 1,144 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 109 (0,035 г, 69,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,78 (д, 2 H, J=8,4 Гц), 7,66 (д, 4 H, J=7,2 Гц), 7,43 (д, 2 H, J=7,9 Гц), 3,83 (с, 2 H), 3,52 (д, 2 H, J=11,3 Гц), 2,21-2,20 (м, 2 H), 3,52 (д, 2 H, J=11,3 Гц), 1,01 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 438,0 (M++1).
Пример 16: Синтез соединения 110
(4-(((2S,6R)-4-((2-хлорфенил)сульфонил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-((2-хлорфенил)сульфонил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли 2-хлорбензол-1-сульфонилхлорид (0,042 мл, 0,306 ммоль) и TEA (0,116 мл, 0,835 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,105 г, 68,3%) в виде бесцветного масла.
Стадия 2: Синтез соединения 110
Метил 4-(((2S,6R)-4-((2-хлорфенил)сульфонил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,060 г, 0,137 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,168 мл, 2,746 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,077 г, 1,373 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 110 (0,037 г, 61,5%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,10 (с, 1 H), 9,00 (с, 1 H), 7,98 (д, 1 H, J=7,3 Гц), 7,71-7,64 (м, 4 H), 7,60-7,52 (м, 1 H), 7,37 (д, 2 H, J=8,1 Гц), 3,75 (с, 2 H), 3,52 (д, 2 H, J=11,3 Гц), 2,60-2,56 (м, 4 H), 0,93 (д, 6 H, J=5,8 Гц); ЖХМС (ES) m/z 438,1 (M++1).
Пример 17: Синтез соединения 111
(4-(((2S,6R)-2,6-диметил-4-пиколиноилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-пиколиноилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли пиколиноил хлорид (0,075 г, 0,419 ммоль) и TEA (0,159 мл, 1,144 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат=100%) и концентрировали с получением желаемого соединения (0,046 г, 32,8%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 111
Метил 4-(((2S,6R)-2,6-диметил-4-пиколиноилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,02 г, 0,054 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,067 мл, 1,089 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,03 г, 0,544 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 111 (0,001 г, 5,0%) в виде бесцветного масла.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,65 (дд, 1 H, J=10,2, 3,6 Гц), 8,62 (д, 1 H, J=3,0 Гц), 7,90 (дд, 1 H, J=10,2, 3,6 Гц), 7,70 (д, 2 H, J=8,4 Гц), 7,55-7,51 (м, 3 H), 4,42 (д, 1 H, J=12,3 Гц), 3,89 (с, 2 H), 3,49 (д, 1 H, J=12,7 Гц), 3,12-3,11 (м, 1 H), 2,85-2,84 (м, 1 H), 2,72-2,63 (м, 2 H), 1,12 (д, 3 H, J=3,6 Гц), 0,92 (д, 3 H, J=5,2 Гц).
Пример 18: Синтез соединения 112
(4-(((2S,6R)-2,6-диметил-4-никотиноилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил (((2S,6R)-2,6-диметил-4-никотиноилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли никотиноил хлорид (0,075 г, 0,419 ммоль) и TEA (0,159 мл, 1,144 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,119 г, 85,0%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 112
Метил (((2S,6R)-2,6-диметил-4-никотиноилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,060 г, 0,163 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,200 мл, 3,266 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,092 г, 1,633 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении и экстрагировали этилацетатом и водой. Органический слой концентрировали при пониженном давлении с получением соединения 112 (0,001 г, 1,7%) в виде бесцветного масла.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,65 (дд, 1 H, J=10,2, 3,6 Гц), 8,62 (д, 1 H, J=3,0 Гц), 7,90 (дд, 1 H, J=10,2, 3,6 Гц), 7,70 (д, 2 H, J=8,4 Гц), 7,55-7,51 (м, 3 H), 4,42 (д, 1 H, J=12,3 Гц), 3,89 (с, 2 H), 3,49 (д, 1 H, J=12,7 Гц), 3,12-3,11 (м, 1 H), 2,85-2,84 (м, 1 H), 2,72-2,63 (м, 2 H), 1,12 (д, 3 H, J=3,6 Гц), 0,92 (д, 3 H, J=5,2 Гц); ЖХМС (ES) m/z 369,2 (M++1).
Пример 19: Синтез соединения 113
(4-(((2S,6R)-2,6-диметил-4-(пиридин-3-илсульфонил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(пиридин-3-илсульфонил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли пиридин-3-сульфонилхлорид (0,090 г, 0,419 ммоль) и TEA (0,159 мл, 1,144 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,105 г, 68,3%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 113
Метил 4-(((2S,6R)-2,6-диметил-4-(пиридин-3-илсульфонил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,600 г, 0,149 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,182 мл, 2,974 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,830 г, 1,487 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 113 (0,033 г, 54,9%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 8,93 (д, 1 H, J=2,3 Гц), 8,84 (дд, 1 H, J=10,2, 3,6 Гц), 8,22-8,20 (м, 1 H), 7,66-7,65 (м, 1 H), 7,65 (д, 2 H, J=8,3 Гц), 7,42 (д, 2 H, J=8,2 Гц), 3,83 (с, 2 H), 3,58 (д, 2 H, J=11,5 Гц), 2,74-2,72 (м, 2 H), 2,24 (т, 2 H, J=10,8 Гц), 1,01 (д, 6 H, J=6,2 Гц); ЖХМС (ES) m/z 405,1 (M++1).
Пример 20: Синтез соединения 114
(4-(((2S,6R)-2,6-диметил-4-(тиофен-2-карбонил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(тиофен-2-карбонил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли тиофен-2-карбонилхлорид (0,046 мл, 0,419 ммоль) и TEA (0,159 мл, 1,144 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,127 г, 89,5%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 114
Метил 4-(((2S,6R)-2,6-диметил-4-(тиофен-2-карбонил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,070 г, 0,188 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,230 мл, 3,759 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,105 г, 1,879 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 114 (0,013 г, 18,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,70 (д, 2 H, J=8,4 Гц), 7,65 (дд, 1 H, J=10,2, 3,6 Гц), 7,52 (д, 2 H, J=8,4 Гц), 7,40 (дд, 1 H, J=10,2, 3,6 Гц), 7,12 (дд, 1 H, J=10,2, 3,6 Гц), 4,20-4,19 (м, 2 H), 3,89 (с, 2 H), 3,03-2,97 (м, 2 H), 2,70-2,62 (м, 2 H), 1,05 (д, 6 H, J=5,6 Гц); ЖХМС (ES) m/z 374,1 (M++1).
Пример 21: Синтез соединения 115
(4-(((2S,6R)-4-(фуран-2-карбонил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(фуран-2-карбонил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (3 мл) и добавляли фуран-2-карбонилхлорид (0,040 мл, 0,419 ммоль) и TEA (0,159 мл, 1,144 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,115 г, 84,7%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 115
Метил 4-(((2S,6R)-4-(фуран-2-карбонил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,060 г, 0,168 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,206 мл, 3,367 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,095 г, 1,683 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 115 (0,007 г, 11,6%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,71-7,69 (м, 3 H), 7,53 (д, 2 H, J=8,4 Гц), 7,04 (дд, 1 H, J=10,2, 3,6 Гц), 6,59 (дд, 1 H, J=10,2, 3,6 Гц), 4,31 (д, 2 H, J=13,1 Гц), 3,90 (с, 2 H), 2,85-2,82 (м, 2 H), 2,69-2,65 (м, 2 H), 1,08 (д, 6 H, J=5,4 Гц); ЖХМС (ES) m/z 358,1 (M++1).
Пример 22: Синтез соединения 118
(4-(((2S,6R)-4-(2-хлорбензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-хлорбензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,278 ммоль) растворяли в ацетонитриле (2 мл) и затем добавляли 2-хлорбензилбромид (0,038 мл, 0,292 ммоль) и K2CO3 (0,096 г, 0,696 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, и затем реакционную смесь концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,080 г, 74,3%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 118
Метил 4-(((2S,6R)-4-(2-хлорбензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,040 г, 0,103 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,126 мл, 2,068 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,058 г, 1,034 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 118 (0,018 г, 43,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,65 (д, 2 H, J=8,2 Гц), 7,37-7,33 (м, 4 H), 7,33-7,31 (м, 2 H), 3,74 (с, 2 H), 3,41 (с, 2 H), 2,65-2,62 (м, 2 H), 2,55-2,51 (м, 2 H), 1,83-1,82 (м, 2 H), 0,88 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 388,1 (M++1).
Пример 23: Синтез соединения 119
(4-(((2S,6R)-4-(4-фторфенил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(4-фторфенил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Pd(OAc)2 (0,006 г, 0,028 ммоль) и xantphos (0,003 г, 0,006 ммоль) растворяли в толуоле и затем добавляли 1-бром-4-фторбензол (0,049 г, 0,278 ммоль), метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,278 ммоль) и Cs2CO3 (0,227 г, 0,696 ммоль). Смесь нагревали и перемешивали при температуре 100°C в течение 17 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=15%) и концентрировали с получением желаемого соединения (0,007 г, 7,1%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 119
Метил 4-(((2S,6R)4-(4-фторфенил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 19-1, 0,007 г, 0,020 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,024 мл, 0,393 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,011 г, 0,196 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 119 (0,003 г, 58,0%) в виде твердого вещества бледно-коричневого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,71 (д, 2 H, J=8,2 Гц), 7,53 (д, 2 H, J=8,2 Гц), 6,98-6,95 (м, 4 H), 4,12 (с, 2 H), 3,41 (д, 2 H, J=11,4 Гц), 2,81-2,78 (м, 2 H), 2,51 (т, 2 H, J=11,1 Гц), 1,12 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 358,1 (M++1).
Пример 24: Синтез соединения 120
(4-(((3R,5S)-4-(2-хлорбензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-хлорбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (1 мл) и затем добавляли 2-хлорбензоилхлорид (0,058 мл, 0,801 ммоль) и TEA (0,106 мл, 0,762 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой отделяли и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,076 г, 49,7%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 120
Метил 4-(((3R,5S)-4-(2-хлорбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,070 г, 0,175 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,214 мл, 3,492 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,098 г, 1,746 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 120 (0,029 г, 41,3%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,1 Гц), 7,41-7,40 (м, 3 H), 7,33 (д, 2 H, J=8,0 Гц), 3,51 (с, 2 H), 2,79-2,61 (м, 4 H), 2,14-2,13 (м, 2 H), 1,36 (д, 3 H, J=6,8 Гц), 1,24-1,18 (м, 3 H).
Пример 25: Синтез соединения 121
(4-(((3R,5S)-4-(3-хлорбензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-хлорбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (1 мл) и затем добавляли 3-хлорбензоилхлорид (0,080 г, 0,457 ммоль) и TEA (0,106 мл, 0,762 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой отделяли и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,062 г, 40,6%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 121
Метил 4-(((3R,5S)-4-(3-хлорбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,125 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,153 мл, 2,494 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,070 г, 1,247 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 121 (0,021 г, 41,9%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, 2 H, J=8,0 Гц), 7,47-7,41 (м, 3 H), 7,32 (д, 2 H, J=8,0 Гц), 7,29 (с, 1 H), 3,49 (с, 2 H), 2,61-2,50 (м, 4 H), 2,16-2,12 (м, 2 H), 1,28 (д, 6 H, J=5,6 Гц).
Пример 26: Синтез соединения 122
(4-(((3R,5S)-4-(4-хлорбензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-хлорбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (1 мл) и затем добавляли 4-хлорбензоилхлорид (0,080 г, 0,457 ммоль) и TEA (0,106 мл, 0,762 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой отделяли и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,059 г, 38,6%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 122
Метил 4-(((3R,5S)-4-(4-хлорбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,125 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,153 мл, 2,494 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,070 г, 1,247 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 122 (0,014 г, 27,9%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,10 (ушир. с, 1 H), 9,05 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,1 Гц), 7,50 (д, 2 H, J=8,3 Гц), 7,43 (д, 2 H, J=8,1 Гц), 7,38 (д, 2 H, J=8,4 Гц), 4,10 (ушир. с, 2 H), 3,54 (с, 2 H), 2,63 (д, 2 H, J=11,1 Гц), 2,15 (дд, 2 H, J=11,3, 4,1 Гц), 1,29 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 402,8 (M++1).
Пример 27: Синтез соединения 123
(4-(((3R,5S)-3,5-диметил-4-пиколиноилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-пиколиноилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (1 мл) и затем добавляли пиколиноил хлорид (0,065 г, 0,457 ммоль) и TEA (0,106 мл, 0,762 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой отделяли и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,042 г, 30,0%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 123
Метил 4-(((3R,5S)-3,5-диметил-4-пиколиноилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,035 г, 0,095 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,117 мл, 1,905 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,053 г, 0,953 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 123 (0,011 г, 31,3%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,56-8,55 (м, 1 H), 7,82 (д, 2 H, J=8,0 Гц), 3,71 (с, 2 H), 2,96 (ушир. с, 2 H), 2,56 (ушир. с, 2 H), 1,02 (ушир. с, 3 H), 0,88 (ушир. с, 3 H); ЖХМС (ES) m/z 369,4 (M++1).
Пример 28: Синтез соединения 125
(4-(((3R,5S)-4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 6,000 г, 22,870 ммоль) и TEA (4,781 мл, 34,305 ммоль) растворяли в метиленхлориде (120 мл) при температуре 0°C и в раствор добавляли фуран-2-карбонилхлорид (2,488 мл, 25,157 ммоль), который затем перемешивали при той же температуре в течение 1 часа. В реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 80 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением желаемого соединения (7,440 г, 91,3%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 125
Метил 4-(((3R,5S)-4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 7,440 г, 20,874 ммоль), гидроксиламин (25,536 мл, 417,485 ммоль, 50,00%-ный водный раствор) и гидроксид калия (11,713 г, 208,742 ммоль) растворяли в метаноле (150 мл) при температуре 0°C, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали, и затем концентрировали при пониженном давлении с получением соединения 125 (5,330 г, 71,4%) в виде твердого вещества абрикосового цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,23 (ушир. с, 1 H), 9,06 (ушир. с, 1 H), 7,81 (с, 1 H), 7,74 (д, 2 H, J=7,6 Гц), 7,42 (д, 2 H, J=5,8 Гц), 6,95 (д, 1 H, J=3,3 Гц), 6,61-6,60 (м, 1 H), 4,49 (ушир. с, 2 H), 3,54 (с, 2 H), 2,68 (д, 2 H, J=11,0 Гц), 2,14 (д, 2 H, J=8,2 Гц), 1,25 (д, 6 H, J=5,6 Гц); ЖХМС (ES) m/z 358,2 (M++1).
Пример 29: Синтез соединения 126
(4-(((3R,5S)-3,5-диметил-4-(тиофен-2-карбонил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(тиофен-2-карбонил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (1 мл) и затем добавляли тиофен-2-карбонилхлорид (0,067 г, 0,457 ммоль) и TEA (0,106 мл, 0,762 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой отделяли и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,061 г, 43,0%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 126
Метил 4-(((3R,5S)-3,5-диметил-4-(тиофен-2-карбонил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,134 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,164 мл, 2,685 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,075 г, 1,342 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 126 (0,019 г, 37,9%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,72-7,71 (м, 3 H), 7,41-7,36 (м, 3 H), 7,10 (ушир. с, 1 H), 4,43 (ушир. с, 2 H), 3,52 (с, 2 H), 2,67-2,64 (м, 2 H), 2,14-2,12 (м, 2 H), 1,36 (д, 6 H, J=5,5 Гц).
Пример 30: Синтез соединения 127
(4-(((3R,5S)-4-(2-хлорбензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-хлорбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в ацетонитриле (2 мл) и затем добавляли 1-(бромметил)-2-хлорбензол (0,094 г, 0,457 ммоль) и K2CO3 (0,105 г, 0,762 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,035 г, 23,7%) в виде масла белого цвета.
Стадия 2: Синтез соединения 127
Метил 4-(((3R,5S)-4-(2-хлорбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,030 г, 0,078 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,095 мл, 1,551 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,044 г, 0,775 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Соединение 127 (0,011 г, 36,6%) получали в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,78-7,68 (м, 3 H), 7,32-7,19 (м, 6 H), 3,68 (с, 2 H), 3,44 (с, 2 H), 2,69-2,66 (м, 4 H), 1,86-1,83 (м, 2 H), 0,78 (ушир. с, 6 H).
Пример 31: Синтез соединения 128
(4-(((3R,5S)-4-(3-хлорбензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-хлорбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в ацетонитриле (2 мл) и затем добавляли 1-(бромметил)-3-хлорбензол (0,094 г, 0,457 ммоль) и K2CO3 (0,105 г,0,762 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,041 г, 27,8%) в виде масла белого цвета.
Стадия 2: Синтез соединения 128
Метил 4-(((3R,5S)-4-(3-хлорбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,035 г, 0,090 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,111 мл, 1,809 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,051 г, 0,905 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Соединение 128 (0,015 г, 42,7%) получали в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, 2 H, J=9,6 Гц), 7,38 (с, 1 H), 7,30 (д, 2 H, J=4,0 Гц), 7,27-7,20 (м, 3 H), 3,69 (с, 2 H), 3,40 (с, 2 H), 2,64-2,54 (м, 4 H), 1,82-1,75 (м, 2 H), 0,84 (д, 6 H, J=8,0 Гц).
Пример 32: Синтез соединения 145
(4-(((3R,5S)-4-(фуран-2-илметил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(фуран-2-илметил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,300 г, 1,144 ммоль) и 2-фуральдегид (0,104 мл, 1,258 ммоль) растворяли в метиленхлориде (5 мл), и при температуре 40°C добавляли уксусную кислоту (0,069 мл, 1,144 ммоль), затем перемешивали в течение 1 часа. В реакционную смесь добавляли Na(CN)BH3 (0,072 г, 1,144 ммоль), затем перемешивали при той же температуре в течение 3 дней. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,028 г, 7,2%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 145
Метил 4-(((3R,5S)-4-(фуран-2-илметил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,028 г, 0,082 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,100 мл, 1,635 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,046 г, 0,818 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 145 (0,013 г, 46,3%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,10 (ушир. с, 1 H), 9,00 (ушир. с, 1 H), 7,67 (д, 2 H, J=8,1 Гц), 7,59 (д, 1 H, J=2,4 Гц), 7,30 (д, 2 H, J=8,1 Гц), 6,41-6,40 (м, 1 H), 6,28-6,27 (м, 1 H), 3,86 (с, 2 H), 3,38 (с, 2 H), 2,60 (д, 2 H, J=10,0 Гц), 2,44-2,40 (м, 2 H), 1,74 (т, 2 H, J=10,6 Гц), 0,87 (д, 6 H, J=6,4 Гц).
Пример 33: Синтез соединения 146
(4-(((3R,5S)-3,5-диметил-4-(2-фенилацетил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2-фенилацетил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) растворяли в метиленхлориде (1 мл) и затем добавляли 2-фенилацетилхлорид (0,065 г, 0,419 ммоль) и TEA (0,106 мл, 0,762 ммоль). Смесь перемешивали при комнатной температуре в течение 15 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой отделяли и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,042 г, 29,0%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 146
Метил 4-(((3R,5S)-3,5-диметил-4-(2-фенилацетил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,030 г, 0,079 ммоль) растворяли в метаноле (0,5 мл) и добавляли гидроксиламин (0,096 мл, 1,577 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,044 г, 0,788 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением соединения 146 (0,014 г, 46,5%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,0 Гц), 7,34 (д, 2 H, J=8,0 Гц), 7,31-7,27 (м, 2 H), 7,22-7,20 (м, 3 H), 4,41 (ушир. с, 1 H), 4,12 (ушир. с, 1 H), 3,47-3,33 (м, 2 H), 3,31 (с, 2 H), 2,62-2,59 (м, 2 H), 2,02-2,01 (м, 2 H), 1,22 (ушир. с, 6 H).
Пример 34: Синтез соединения 147
(4-(((3R,5S)-4-этил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-этил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), йодэтан (0,037 мл, 0,457 ммоль) и K2CO3 (0,105 г, 0,762 ммоль) растворяли в ацетонитриле (1 мл) и перемешивали при комнатной температуре в течение 7 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,059 г, 53,3%) в виде твердого вещества бледно-желтого цвета.
Стадия 2: Синтез соединения 147
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,040 г, 0,174 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,213 мл, 3,487 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,098 г, 1,744 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 147 (0,010 г, 19,7%) получали в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,65 (д, 2 H, J=7,8 Гц), 7,20 (д, 2 H, J=7,9 Гц), 2,73 (кв, 2 H, J=7,6 Гц), 2,61-2,55 (м, 4 H), 1,70-1,66 (м, 2 H), 0,90 (д, 6 H, J=5,9 Гц), 0,81 (т, 3 H, J=7,0 Гц).
Соединение 35: Синтез соединения 148
(4-(((3R,5S)-3,5-диметил-4-пропилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-пропилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), йодпропан (0,045 мл, 0,457 ммоль) и K2CO3 (0,105 г, 0,762 ммоль) растворяли в ацетонитриле (1 мл) и перемешивали при комнатной температуре в течение 7 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,054 г, 46,5%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 148
Метил 4-(((3R,5S)-3,5-диметил-4-пропилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,040 г, 0,131 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,161 мл, 3,487 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,074 г, 1,314 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 148 (0,017 г, 42,4%) получали в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,66 (д, 2 H, J=8,0 Гц), 7,21 (д, 2 H, J=8,0 Гц), 3,35 (с, 2 H), 2,60-2,52 (м, 6 H), 1,68 (т, 2 H, J=10,4 Гц), 1,32-1,30 (м, 2 H), 0,90 (д, 6 H, J=6,0 Гц), 0,76 (т, 3 H, J=7,4 Гц).
Пример 36: Синтез соединения 149
(4-(((3R,5S)-3,5-диметил-4-(2,2,2-трифторэтил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2,2,2-трифторэтил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 2,2,2-трифторэтил трифторметан сульфонат (0,066 мл, 0,457 ммоль) и K2CO3 (0,105 г, 0,762 ммоль) растворяли в ацетонитриле (1 мл) и перемешивали при комнатной температуре в течение 12 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,062 г, 47,2%) в виде масла белого цвета.
Стадия 2: Синтез соединения 149
Метил 4-(((3R,5S)-3,5-диметил-4-(2,2,2-трифторэтил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,030 г, 0,087 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,107 мл, 1,742 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,049 г, 1,744 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 149 (0,011 г, 36,6%) получали в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, 2 H, J=7,9 Гц), 7,31 (д, 2 H, J=8,0 Гц), 3,41 (с, 2 H), 3,28 (с, 2 H), 2,70-2,63 (м, 2 H), 2,60-2,49 (м, 2 H), 1,73 (т, 2 H, J=10,5 Гц), 0,96 (д, 6 H, J=6,2 Гц).
Пример 37: Синтез соединения 154
(6-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)-N-гидроксигексанамид)
Стадия 1: Синтез этил 6-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)гексаноата
(2S,6R)-1-Бензил-2,6-диметилпиперазин (формула 8-3, 0,100 г, 0,489 ммоль) растворяли в ацетонитриле (1 мл) и затем добавляли этил 6-бромгексаноат (формула 8-4, 0,131 г, 0,587 ммоль) и Cs2CO3 (0,319 г, 0,979 ммоль). Смесь перемешивали при комнатной температуре в течение 20 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=20%) и концентрировали с получением желаемого соединения (0,090 г, 53,1%) в виде масла белого цвета.
Стадия 2: Синтез соединения 154
Этил 6-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)гексаноат (формула 8-5, 0,040 г, 0,115 ммоль) растворяли в метаноле (0,5 мл) и добавляли гидроксиламин (0,141 мл, 2,309 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,065 г, 1,154 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 154 (0,021 г, 54,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,33-7,24 (м, 4 H), 7,17-7,15 (м, 1 H), 3,69 (с, 2 H), 2,68-2,53 (м, 4 H), 2,13 (т, 2 H, J=7,2 Гц), 1,86 (т, 2 H, J=7,4 Гц), 1,66 (т, 2 H, J=10,6 Гц), 1,45-1,34 (м, 4 H), 1,20-1,18 (м, 2 H), 0,84 (д, 6 H, J=8,0 Гц).
Пример 38: Синтез соединения 159
((2S,6R)-4-(4-(гидроксикарбамоил)бензил)-2,6-диметил-N-фенилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(фенилкарбамоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (соединение 1-2, 0,150 г, 0,572 ммоль) растворяли в метиленхлориде (2 мл) и затем добавляли фенил изоцианат (0,069 мл, 0,629 ммоль) и TEA (0,119 мл, 0,858 ммоль). Смесь перемешивали при температуре 0°C в течение 2 часов и затем перемешивали при комнатной температуре в течение 2 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,171 г, 78,4%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 159
Метил 4-(((3R,5S)-3,5-диметил-4-(фенилкарбамоил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,100 г, 0,262 ммоль) растворяли в метаноле (2 мл) и затем добавляли гидроксиламин (0,321 мл, 5,243 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,147 г, 2,621 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 159 (0,030 г, 29,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 8,22 (с, 1 H), 7,69 (д, 2 H, J=8,1 Гц), 7,44 (д, 2 H, J=7,4 Гц), 7,34-7,32 (м, 2 H), 6,92-6,89 (м, 1 H), 4,20-4,17 (м, 2 H), 7,50 (с, 2 H), 2,64 (д, 2 H, J=11,1 Гц), 2,12-2,08 (м, 2 H), 1,26 (д, 6 H, J=6,7 Гц); ЖХМС (ES) m/z 383,2 (M++1).
Пример 39: Синтез соединения 160
((2S,6R)-4-(4-(гидроксикарбамоил)бензил)-2,6-диметил-N-о-толилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(о-толилкарбамоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,200 г, 0,762 ммоль) растворяли в метиленхлориде (3 мл) и затем добавляли о-толилизоцианат (0,103 мл, 0,839 ммоль) и TEA (0,159 мл, 1,144 ммоль). Смесь перемешивали при температуре 0°C в течение 2 часов и затем перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,030 г, 10,0%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 160
Метил 4-(((3R,5S)-3,5-диметил-4-(о-толилкарбамоил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,030 г, 0,076 ммоль) растворяли в метаноле (2 мл) и добавляли гидроксиламин (0,093 мл, 1,517 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,043 г, 0,759 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 160 (0,010 г, 33,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,10 (с, 1 H), 9,00 (с, 1 H), 7,85 (с, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,43 (д, 2 H, J=8,0 Гц), 7,17-7,10 (м, 3 H), 7,03 (дд, 1 H, J=10,2, 3,6 Гц), 3,54 (с, 2 H), 2,65 (д, 2 H, J=11,2 Гц), 2,16 (с, 3 H), 2,12-2,13 (м, 1 H),1,30 (д, 6 H, J=6,6 Гц); ЖХМС (ES) m/z 397,2 (M++1).
Пример 40: Синтез соединения 161
((2S,6R)-N-бензил-4-(4-(гидроксикарбамоил)бензил)-2,6-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(бензилкарбамоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,150 г, 0,572 ммоль) растворяли в метиленхлориде (2 мл) и затем добавляли бензил изоцианат (0,078 мл, 0,629 ммоль) и TEA (0,119 мл, 0,858 ммоль) при температуре 0°C. Смесь перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,517 г, 69,4%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 161
Метил 4-(((3R,5S)-4-(бензилкарбамоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,100 г, 0,253 ммоль) растворяли в метаноле (2 мл) и добавляли гидроксиламин (0,309 мл, 5,057 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,114 г, 2,529 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 161 (0,062 г, 61,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,10 (с, 1 H), 9,10 (с, 1 H), 7,71 (д, 2 H, J=8,0 Гц), 7,41 (д, 2 H, J=7,9 Гц), 7,31-7,27 (м, 2 H), 7,24-7,18 (м, 3 H), 6,91-6,90 (м, 1 H), 4,25 (д, 2 H, J=5,5 Гц), 4,04-4,03 (м, 2 H), 3,51 (с, 2 H), 2,60 (д, 2 H, J=11,0 Гц), 2,08-2,05 (м, 2 H), 1,22 (д, 6 H, J=6,6 Гц).
Пример 41: Синтез соединения 162
((3R,5S)-4-(4-(гидроксикарбамоил)бензил)-3,5-диметил-N-фенилпиперазин-1-карбоксамид)
Стадия 1: Синтез 4-(((2S,6R)-2,6-диметил-4-(фенилкарбамоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,572 ммоль) растворяли в метиленхлориде (4 мл) и затем добавляли фенил изоцианат (0,069 мл, 0,629 ммоль) и TEA (0,119 мл, 0,858 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,217 г, 99,5%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 162
Метил 4-(((2S,6R)-2,6-диметил-4-(фенилкарбонил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,107 г, 0,280 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,343 мл, 5,610 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,157 г, 2,805 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 162 (0,041 г, 38,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,20 (с, 1 H), 9,00 (с, 1 H), 8,48 (с, 1 H), 7,68 (д, 2 H, J=8,0 Гц), 7,45-7,42 (м, 4 H), 7,22 (дд, 2 H, J=10,2, 3,6 Гц), 6,93-6,91 (м, 1 H), 3,97 (д, 2 H, J=13,3 Гц), 3,78 (с, 2 H), 2,63-2,60 (м, 1 H), 0,97 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 383,2 (M++1).
Пример 42: Синтез соединения 163
((3R,5S)-4-(4-(гидроксикарбамоил)бензил)-N-(2-метоксифенил)-3,5-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-метоксифенилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,572 ммоль) растворяли в метиленхлориде (4 мл) и затем добавляли 2-метоксифенил изоцианат (0,094 г, 0,629 ммоль) и TEA (0,119 мл, 0,858 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,169 г, 71,8%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 163
Метил 4-(((2S,6R)-4-(2-метоксифенилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,100 г, 0,243 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,297 мл, 4,860 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,136 г, 2,430 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 163 (0,060 г, 59,9%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68-7,65 (м, 3 H), 7,56 (д, 1 H, J=8,0 Гц), 7,39 (д, 2 H, J=8,2 Гц), 7,00-6,98 (м, 2 H), 6,87-6,85 (м, 1 H), 3,88 (д, 2 H, J=11,6 Гц), 3,78 (с, 3 H), 3,76 (с, 2 H), 2,69-2,63 (м, 2 H), 2,55-2,52 (м, 2 H), 0,96 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 413,2 (M++1).
Пример 43: Синтез соединения 164
((3R,5S)-4-(4-(гидроксикарбамоил)бензил)-N-(3-метоксифенил)-3,5-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(3-метоксифенилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,572 ммоль) растворяли в метиленхлориде (4 мл) и затем добавляли 3-метоксифенил изоцианат (0,094 г, 0,629 ммоль) и TEA (0,119 мл, 0,858 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,210 г, 89,3%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 164
Метил 4-(((2S,6R)-4-(3-метоксифенилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,100 г, 0,243 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,297 мл, 4,860 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,136 г, 2,430 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 164 (0,025 г, 24,9%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,10 (с, 1 H), 9,00 (с, 1 H), 8,46 (с, 1 H), 7,67 (д, 2 H, J=8,2 Гц), 7,43 (д, 2 H, J=8,2 Гц), 7,14-7,11 (м, 1 H), 7,09-7,02 (м, 1 H), 6,57 (дд, 1 H, J=10,2, 3,6 Гц), 3,95 (д, 2 H, J=12,8 Гц), 3,77 (с, 2 H), 3,69 (с, 3 H), 2,35-2,54 (м, 2 H), 2,50-2,48 (м, 2 H), 0,96 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 413,2 (M++1).
Пример 44: Синтез соединения 165
((3R,5S)-4-(4-(гидроксикарбамоил)бензил)-N-(4-метоксифенил)-3,5-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(4-метоксифенилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,572 ммоль) растворяли в метиленхлориде (4 мл) и затем добавляли 4-метоксифенил изоцианат (0,094 г, 0,629 ммоль) и TEA (0,119 мл, 0,858 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,022 г, 9,4%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 165
Метил 4-(((2S,6R)-4-(4-метоксифенилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,022 г, 0,053 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,065 мл, 1,069 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,03 г, 0,535 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 165 (0,008 г, 36,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,31 (с, 1 H), 7,67 (д, 2 H, J=8,2 Гц), 7,40 (д, 2 H, J=8,2 Гц), 7,31 (д, 2 H, J=9,1 Гц), 6,80 (д, 2 H, J=9,1 Гц), 6,80 (д, 2 H, J=9,1 Гц), 3,94 (д, 2 H, J=12,3 Гц), 3,76 (с, 2 H), 3,69 (с, 3 H), 2,66-2,25 (м, 4 H), 0,96 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 413,2 (M++1).
Пример 45: Синтез соединения 166
((3R,5S)-4-(4-(гидроксикарбамоил)бензил)-3,5-диметил-N-о-толилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(о-толилкарбамоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,572 ммоль) растворяли в метиленхлориде (4 мл) и затем добавляли о-толилизоцианат (0,078 мл, 0,629 ммоль) и TEA (0,119 мл, 0,858 ммоль) при температуре 0°C. Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,182 г, 80,5%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 166
Метил 4-(((2S,6R)-2,6-диметил-4-(о-толилкарбамоил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,082 г, 0,207 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,254 мл, 4,147 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,116 г, 2,073 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 166 (0,010 г, 12,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,04 (с, 1 H), 7,66 (д, 2 H, J=8,1 Гц), 7,34 (д, 2 H, J=8,1 Гц), 7,16-7,10 (м, 3 H), 7,04-7,02 (м, 1 H), 3,90 (д, 2 H, J=13,6 Гц), 3,76 (с, 2 H), 2,69-2,63 (м, 2 H), 2,54-2,50 (м, 2 H), 2,12 (с, 3 H), 0,97 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 397,2 (M++1).
Пример 46: Синтез соединения 167
((3R,5S)-4-(4-(гидроксикарбамоил)бензил)-3,5-диметил-N-м-толилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(м-толилкарбамоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,572 ммоль) растворяли в метиленхлориде (4 мл) и затем добавляли м-толилизоцианат (0,084 мл, 0,629 ммоль) и TEA (0,119 мл, 0,858 ммоль) при температуре 0°C. Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,170 г, 75,2%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 167
Метил 4-(((2S,6R)-2,6-диметил-4-(м-толилкарбамоил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,070 г, 0,177 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,217 мл, 3,540 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,099 г, 1,770 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 167 (0,008 г, 36,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,40 (с, 1 H), 7,68 (д, 2 H, J=8,2 Гц), 7,41 (д, 2 H, J=8,2 Гц), 7,26-7,24 (м, 2 H), 7,11 (дд, 1 H, J=10,2, 3,6 Гц), 6,74 (д, 2 H, J=7,2 Гц), 3,96 (д, 2 H, J=13,0 Гц), 3,77 (с, 2 H), 2,65-2,59 (м, 4 H), 2,24 (с, 3 H), 0,96 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 397,2 (M++1).
Пример 47: Синтез соединения 168
((3R,5S)-N-бензил-4-(4-(гидроксикарбамоил)бензил)-3,5-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(бензилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,572 ммоль) растворяли в метиленхлориде (4 мл) и затем добавляли бензил изоцианат (0,078 мл, 0,629 ммоль) и TEA (0,119 мл, 0,858 ммоль) при температуре 0°C. Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,115 г, 50,9%) в виде твердого вещества бледно-желтого цвета.
Стадия 2: Синтез соединения 168
Метил 4-(((2S,6R)-4-(бензилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,060 г, 0,152 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,186 мл, 3,034 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,085 г, 1,517 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 168 (0,045 г, 74,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,05 (с, 1 H), 9,00 (с, 1 H), 7,67 (д, 2 H, J=8,2 Гц), 7,42 (д, 2 H, J=8,2 Гц), 7,30-7,19 (м, 5 H), 7,04-7,06 (м, 1 H), 4,22 (д, 2 H, J=5,8 Гц), 3,83 (д, 2 H, J=12,1 Гц), 3,75 (с, 2 H), 2,56-2,54 (м, 2 H), 2,45-2,43 (м, 2 H), 0,92 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 397,2 (M++1).
Пример 48: Синтез соединения 171
(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-2-фтор-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-2-фторбензоата
(2S,6R)-1-бензил-2,6-диметилпиперазин (формула 8-3, 0,200 г, 0,979 ммоль) и метил 4-формил-2-фторбензоат (формула 8-4, 0,178 г, 0,979 ммоль) растворяли в тетрагидрофуране (5 мл), и при комнатной температуре добавляли уксусную кислоту (0,028 мл, 0,979 ммоль), затем перемешивали в течение 1 часа. Далее в реакционную смесь добавляли Na(CN)BH3 (0,062 г, 0,979 ммоль), затем перемешивали при той же температуре в течение 2 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,028 г, 7,2%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 171
Метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-2-фторбензоат (формула 8-5, 0,100 г, 0,264 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,323 мл, 5,284 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,148 г, 2,642 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и 40 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 171 (0,034 г, 33,9%) получали в виде твердого вещества бледно-оранжевого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,00 (ушир. с, 1 H), 9,10 (ушир. с, 1 H), 7,47 (дд, 1 H, J=7,5, 7,5 Гц), 7,32 (д, 2 H, J=7,6 Гц), 7,26 (дд, 2 H, J=7,5, 7,5 Гц), 7,17-7,13 (м, 2 H), 3,71 (с, 2 H), 3,42 (с, 2 H), 2,63-2,53 (м, 4 H), 1,81 (т, 2 H, J=10,4 Гц), 0,88 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 372,2 (M++1).
Пример 49: Синтез соединения 172
((E)-3-(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)фенил)-N-гидроксиакриламид)
Стадия 1: Синтез (E)-метил 3-(4-(диметоксиметил)фенил)акрилата
(E)-3-(4-Формилфенил)акриловую кислоту (1,000 г, 5,676 ммоль) растворяли в метаноле (10 мл) и при температуре 0°C добавляли SOCl2 (0,824 мл, 11,353 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,870 г, 64,9%) в виде твердого вещества бледно-желтого цвета.
Стадия 2: Синтез (E)-метил 3-(4-формилфенил)акрилата
(E)-Метил 3-(4-(диметоксиметил)фенил)акрилат (0,870 г, 3,682 ммоль) растворяли в метаноле (15 мл) при комнатной температуре и добавляли HCl (15 мл, 1н раствор в метаноле) при той же температуре, затем перемешивали в течение 2 часов и 30 минут. Реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия. Выпавший твердый осадок фильтровали и сушили с получением желаемого соединения (0,695 г, 99,2%) в виде твердого вещества бледно-желтого цвета.
Стадия 3: Синтез (E)-метил 3-(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)фенил)акрилата
(2S,6R)-1-Бензил-2,6-диметилпиперазин (формула 8-3, 0,050 г, 0,245 ммоль) и (E)-метил 3-(4-формилфенил)акрилат (0,047 г, 0,245 ммоль) растворяли в тетрагидрофуране (1 мл) и добавляли уксусную кислоту (0,007 мл, 0,245 ммоль) при комнатной температуре, затем перемешивали в течение 1 часа. В реакционную смесь добавляли Na(CN)BH3 (0,015 г, 0,245 ммоль), затем перемешивали при той же температуре в течение 2 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,012 г, 13,0%) в виде твердого вещества белого цвета.
Стадия 4: Синтез соединения 172
(E)-Метил 3-(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 8-5, 0,050 г, 0,135 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,165 мл, 2,699 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,076 г, 1,350 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 172 (0,024 г, 47,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,60 (ушир. с, 1 H), 7,44 (д, 2 H, J=8,0 Гц), 7,32-7,24 (м, 7 H), 7,15 (дд, 1 H, J=7,1, 7,1 Гц), 6,39 (д, 1 H, J=15,8 Гц), 3,70 (с, 2 H), 3,37 (с, 2 H), 2,62 (д, 2 H, J=10,7 Гц), 2,56-2,52 (м, 2 H), 1,77 (т, 2 H, J=10,5 Гц), 0,87 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 380,5 (M++1).
Пример 50: Синтез соединения 173
(2-(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)фенил)-N-гидроксиацетамид)
Стадия 1: Синтез метил 2-(4-(бромметил)фенил)ацетата
2-(4-(Бромметил)фенил)уксусную кислоту (3,000 г, 13,096 ммоль) растворяли в метаноле (30 мл) и при температуре 0°C добавляли SOCl2 (1,900 мл, 26,193 ммоль), затем перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (3,140 г, 98,6%) в виде бесцветного масла.
Стадия 2: Синтез метил 2-(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)фенил)ацетата
(2S,6R)-1-Бензил-2,6-диметилпиперазин (формула 8-3, 0,200 г, 0,979 ммоль), метил 2-(4-(бромметил)фенил)ацетат (формула 8-4, 0,238 г, 0,979 ммоль) и Cs2CO3 (0,478 г, 1,468 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре. Реакционный раствор перемешивали при той же температуре в течение 2 часов, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,273 г, 76,1%) в виде масла бледно-желтого цвета.
Стадия 3: Синтез соединения 173
Метил 2-(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)фенил)ацетат (формула 8-5, 0,173 г, 0,472 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,577 мл, 9,441 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,265 г, 4,720 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 173 (0,140 г, 80,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 10,60 (ушир. с, 1 H), 8,80 (brs,1 H), 7,31 (д, 2 H, J=7,3 Гц), 7,27-7,24 (м, 2 H), 7,17-7,15 (м, 5 H), 3,69 (с, 2 H), 3,33 (с, 2 H), 3,22 (с, 2 H), 2,61 (д, 2 H, J=10,1 Гц), 2,53-2,49 (м, 2 H), 1,75 (т, 2 H, J=10,5 Гц), 0,87 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 368,4 (M++1).
Пример 51: Синтез соединения 174
(4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)-2-фтор-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)-2-фторбензоата
(3R,5S)-1-бензил-3,5-диметилпиперазин (формула 16-1, 0,200 г, 0,979 ммоль) растворяли в ацетонитриле (3 мл) и затем добавляли метил 4-(бромметил)-2-фторбензоат (формула 8-4, 0,254 г, 1,028 ммоль) и Cs2CO3 (0,478 г, 1,468 ммоль). Смесь перемешивали при температуре 80°C в течение 2 часов и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=15%) и концентрировали с получением желаемого соединения (0,211 г, 58,2%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 174
Метил 4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)-2-фторбензоат (формула 16-2, 0,100 г, 0,270 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,330 мл, 5,339 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,151 г, 2,699 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 174 (0,058 г, 57,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,46-7,44 (м, 1 H), 7,31-7,23 (м, 5 H), 7,20-7,16 (м, 2 H), 3,72 (с, 2 H), 3,40 (с, 2 H), 2,65 (д, 2 H, J=10,4 Гц), 2,56-2,54 (м, 2 H), 1,82-1,80 (м, 2 H), 0,85 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 372,4 (M++1).
Пример 52: Синтез соединения 175
((E)-3-(4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)фенил)-N-гидроксиакриламид)
Стадия 1: Синтез (3R,5S)-1-бензил-3,5-диметилпиперазина
(2S,6R)-2,6-Диметилпиперазин (1,000 г, 8,757 ммоль) и K2CO3 (1,724 г, 13,136 ммоль) растворяли в ацетонитриле (50 мл), и при температуре 0°C добавляли бензилбромид (1,092 мл, 9,195 ммоль), и смесь перемешивали при той же температуре в течение 1 часа и 30 минут. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (1,170 г, 65,4%) в виде масла желтого цвета.
Стадия 2: Синтез (E)-метил 3-(4-(гидроксиметил)фенил)акрилата
(E)-3-(4-(Гидроксиметил)фенил)акриловую кислоту (0,300 г, 1,577 ммоль) растворяли в метаноле (5 мл), и при комнатной температуре добавляли NaBH4 (0,063 г,1,656 ммоль), затем перемешивали при той же температуре в течение 50 минут. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,284 г, 93,7%) в виде твердого вещества белого цвета.
Стадия 3: Синтез (E)-метил 3-(4-(бромметил))фенил)акрилата
(E)-Метил 3-(4-(гидроксиметил)фенил)акрилат (0,284 г, 1,478 ммоль) растворяли в толуоле (10 мл), и при температуре 0°C добавляли PBr3 (0,042 мл,0,443 ммоль), затем перемешивали при комнатной температуре в течение 30 минут. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,260 г, 69,0%) в виде твердого вещества белого цвета.
Стадия 4: Синтез (E)-метил 3-(4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)фенил)акрилата
(3R,5S)-1-Бензил-3,5-диметилпиперазин (формула 16-1, 0,200 г, 0,979 ммоль), (E)-метил 3-(4-(бромметил))фенил)акрилат (формула 8-4, 0,250 г, 0,979 ммоль) и Cs2CO3 (0,478 г, 1,468 ммоль) растворяли в ацетонитриле (5 мл) при температуре 80°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа и 40 минут. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=15%) и концентрировали с получением желаемого соединения (0,078 г, 21,1%) в виде масла желтого цвета.
Стадия 5: Синтез соединения 175
(E)-метил 3-(4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 16-2, 0,078 г, 0,206 ммоль) растворяли в метаноле (1,5 мл) и затем добавляли гидроксиламин (0,252 мл, 4,121 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,116 г, 2,061 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 175 (0,040 г, 51,1%) в виде твердого вещества оранжевого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 10,00 (с, 2 H), 7,45 (д, 2 H, J=8,0 Гц), 7,36-7,21 (м, 8 H), 6,40 (д, 1 H, J=15,7 Гц), 3,71 (с, 2 H), 3,39 (с, 2 H), 2,64 (д, 2 H, J=10,6 Гц), 2,59-2,54 (м, 2 H), 1,81-1,76 (м, 2 H), 0,87 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 380,4 (M++1).
Пример 53: Синтез соединения 176
(2-(4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)фенил)-N-гидроксиацетамид)
Стадия 1: Синтез метил 2-(4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)фенил)ацетата
(3R,5S)-1-Бензил-3,5-диметилпиперазин (формула 16-1, 0,200 г, 0,979 ммоль), метил 2-(4-(бромметил)фенил)ацетат (формула 8-4, 0,238 г, 0,979 ммоль) и Cs2CO3 (0,478 г, 1,468 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре. Реакционный раствор перемешивали при той же температуре в течение 5 часов, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,274 г, 76,4%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 176
Метил 2-(4-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)фенил)ацетат (формула 16-2, 0,143 г, 0,390 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,477 мл, 7,804 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,219 г, 3,9902 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (5 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 176 (0,105 г, 73,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 10,50 (с, 1 H), 9,00 (с, 1 H), 7,32-7,21 (м, 1 H), 7,16 (д, 2 H, J=7,9 Гц), 3,69 (с, 2 H), 3,37 (с, 2 H), 3,22 (с, 2 H), 2,62 (д, 2 H, J=10,8 Гц), 2,56-2,52 (м, 1 H), 1,80-1,75 (м, 1 H), 0,88 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 368,5 (M++1).
Пример 54: Синтез соединения 177
(фенил (2S,6R)-4-(4-(гидроксикарбамоил)бензил)-2,6-диметилпиперазин-1-карбоксилат)
Стадия 1: Синтез фенил (2S,6R)-4-(4-(метоксикарбонил)бензил)-2,6-диметилпиперазин-1-карбоксилата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,200 г, 0,762 ммоль) растворяли в метиленхлориде (5 мл) и затем добавляли фенилхлорформиат (0,105 мл, 0,839 ммоль) и TEA (0,211 мл, 1,525 ммоль). Смесь перемешивали при температуре 0°C в течение 2 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,066 г, 22,6%) в виде бесцветного масла.
Стадия 2: Синтез соединения 177
Фенил (2S,6R)-4-(4-(метоксикарбонил)бензил)-2,6-диметилпиперазин-1-карбоксилат (формула 1-3, 0,066 г, 0,173 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,211 мл, 3,451 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,097 г, 1,726 ммоль). Смесь перемешивали при комнатной температуре в течение 10 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 177 (0,062 г, 61,8%) в виде твердого вещества бледно-оранжевого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,00 (с, 1 H), 9,00 (с, 1 H), 7,72 (д, 2 H, J=8,0 Гц), 7,43 (д, 2 H, J=8,0 Гц), 7,39-7,36 (м, 1 H), 7,22 (дд, 1 H, J=10,2, 3,6 Гц), 7,10 (д, 2 H, J=7,6 Гц), 4,15-4,13 (м, 1 H), 3,56 (с, 2 H), 2,66 (д, 2 H, J=11,4 Гц), 2,21-2,17 (м, 1 H), 1,35 (д, 6 H, J=6,7 Гц); ЖХМС (ES) m/z 384,2 (M++1).
Пример 55: Синтез соединения 183
(4-(((2S,6R)-2,6-диметил-4-фенетилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-фенетилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) и фенилацетальдегид (0,049 мл, 0,419 ммоль) растворяли в тетрагидрофуране (2 мл), и при комнатной температуре добавляли уксусную кислоту (0,021 мл, 0,381 ммоль), затем перемешивали в течение 1 часа. В реакционную смесь добавляли Na(CN)BH3 (0,024 г,0,381 ммоль), затем перемешивали при той же температуре в течение 17 часов. В реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 4 г, этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,040 г, 28,6%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 183
Метил 4-(((2S,6R)-2,6-диметил-4-фенетилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,045 г, 0,123 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,150 мл, 2,456 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,069 г, 1,228 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 183 (0,040 г, 88,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,66 (д, 2 H, J=8,0 Гц), 7,38 (д, 2 H, J=8,0 Гц), 7,26-7,17 (м, 5 H), 3,73 (с, 2 H), 2,80 (д, 2 H, J=10,1 Гц), 2,71 (т, 2 H, J=7,8 Гц), 2,55-2,53 (м, 2 H), 2,42 (т, 2 H, J=7,8 Гц), 1,83-1,80 (м, 2 H), 0,91 (д, 6 H, J=6,1 Гц).
Пример 56: Синтез соединения 184
(4-(((3R,5S)-3,5-диметил-4-фенетилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-фенетилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) и фенилацетальдегид (0,049 мл, 0,419 ммоль) растворяли в тетрагидрофуране (2 мл), и при комнатной температуре добавляли уксусную кислоту (0,021 мл, 0,381 ммоль), затем перемешивали в течение 1 часа. В смесь добавляли Na(CN)BH3 (0,024 г,0,381 ммоль), затем перемешивали при той же температуре в течение 17 часов. Реакционную смесь промывали водным раствором хлорида натрия и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=30%) и концентрировали с получением желаемого соединения (0,010 г, 7,2%) в виде бесцветного масла.
Стадия 2: Синтез соединения 184
Метил 4-(((3R,5S)-3,5-диметил-4-фенетилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,010 г, 0,027 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,033 мл, 0,546 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,015 г, 0,273 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 184 (0,007 г, 69,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,50 (с, 1 H), 9,00 (с, 1 H), 7,70 (д, 2 H, J=8,2 Гц), 7,35 (д, 2 H, J=8,0 Гц), 7,28 (д, 2 H, J=8,0 Гц), 7,18 (д, 2 H, J=8,2 Гц), 3,43 (с, 1 H), 2,83-2,79 (м, 2 H), 2,72-2,59 (м, 6 H), 1,77-1,72 (м, 2 H), 1,00 (д, 6 H, J=6,1 Гц).
Пример 57: Синтез соединения 185
((3R,5S)-4-(4-(гидроксикарбамоил)бензил)-3,5-диметил-N-(1-метил-1H-индол-5-ил)пиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(1-метил-1H-индол-5-илкарбамоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 5-изоцианато-1-метил-1H-индол (0,079 г, 0,457 ммоль) и TEA (0,106 мл, 0,762 ммоль) растворяли в метиленхлориде (1 мл) и затем перемешивали при комнатной температуре в течение 3 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,061 г, 36,8%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 185
Метил 4-(((2S,6R)-2,6-диметил-4-(1-метил-1H-индол-5-илкарбамоил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,040 г, 0,092 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,113 мл, 1,841 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,052 г, 0,921 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 185 (0,018 г, 44,9%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,30 (с, 1 H), 7,67 (д, 2 H, J=8,2 Гц), 7,57 (д, 1 H, J=1,7 Гц), 7,38 (д, 2 H, J=8,2 Гц), 7,27 (д, 1 H, J=8,8 Гц), 7,23-7,22 (м, 1 H), 7,17 (дд, 1 H, J=10,2, 3,6 Гц), 6,29 (дд, 1 H, J=10,2, 3,6 Гц), 3,99-3,96 (м, 2 H), 3,77 (с, 2 H), 3,73 (с, 3 H), 2,62-2,52 (м, 4 H), 0,97 (д, 6 H, J=6,0 Гц).
Пример 58: Синтез соединения 186
((3R,5S)-N-(3-(1H-пиррол-1-ил)фенил)-4-(4-(гидроксикарбамоил)бензил)-3,5-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(3-(1H-пиррол -1-ил)фенилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 1-(3-изоцианатофенил)-1H-пиррол (0,084 г, 0,457 ммоль) и TEA (0,106 мл, 0,762 ммоль) растворяли в метиленхлориде (1 мл) и затем перемешивали при комнатной температуре в течение 3 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,059 г, 34,7%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 186
Метил 4-(((2S,6R)-4-(3-(1H-пиррол-1-ил)фенилкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,040 г, 0,090 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,110 мл, 1,792 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,050 г, 0,896 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 186 (0,013 г, 32,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,67 (ушир. с, 1 H), 7,78-7,76 (м, 3 H), 7,36-7,22 (м, 6 H), 7,09 (д, 2 H, J=8,0 Гц), 6,23 (с, 2 H), 3,97 (д, 2 H, J=12,0 Гц), 3,75 (с, 2 H), 2,67-2,61 (м, 4 H), 0,84 (д, 6 H, J=4,8 Гц).
Пример 59: Синтез соединения 187
((3R,5S)-N-(2,3-дигидробензофуран-5-ил)-4-(4-(гидроксикарбамоил)бензил)-3,5-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2,3-дигидробензофуран-5-илкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 5-изоцианато-2,3-дигидробензофуран (0,074 г, 0,457 ммоль) и TEA (0,106 мл, 0,762 ммоль) растворяли в метиленхлориде (1 мл) и затем перемешивали при комнатной температуре в течение 3 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,070 г, 43,4%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 187
Метил 4-(((2S,6R)-4-(2,3-дигидробензофуран-5-илкарбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,040 г, 0,094 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,116 мл, 1,889 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,053 г, 0,945 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 187 (0,020 г, 49,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,27 (с, 1 H), 7,66 (д, 2 H, J=8,1 Гц), 7,37 (д, 2 H, J=20,0 Гц), 7,29 (д, 1 H, J=1,2 Гц), 7,04-7,02 (м, 1 H), 6,60 (д, 1 H, J=8,0 Гц), 4,45 (т, 2 H, J=8,7 Гц), 3,92 (д, 2 H, J=12,0 Гц), 3,74 (с, 2 H), 3,11 (т, 2 H, J=8,6 Гц), 2,61-2,56 (м, 4 H), 0,96 (д, 6 H, J=6,0 Гц).
Пример 60: Синтез соединения 188
(4-(((3R,5S)-3,5-диметил-4-(3-фенилпропаноил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-фенилпропаноил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,070 г, 0,267 ммоль) растворяли в метиленхлориде (1 мл) и затем добавляли 3-фенилпропаноилхлорид (0,054 г, 0,320 ммоль) и TEA (0,074 мл, 0,534 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой отделяли и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,097 г, 92,2%) в виде масла белого цвета.
Стадия 2: Синтез соединения 188
Метил 4-(((3R,5S)-3,5-диметил-4-(3-фенилпропаноил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,040 г, 0,101 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,124 мл, 2,028 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,057 г, 1,014 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой концентрировали при пониженном давлении с получением соединения 188 (0,019 г, 47,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,70 (д, 2 H, J=8,4 Гц), 7,38 (д, 2 H, J=8,0 Гц), 7,27-7,14 (м, 5 H), 4,39 (ушир. с, 1 H), 4,00 (ушир. с, 1 H), 3,48 (с, 2 H), 2,80-2,78 (м, 2 H), 2,69-2,66 (м, 2 H), 2,58-2,57 (м, 2 H), 2,01-1,98 (м, 2 H), 1,19 (ушир. с, 6 H).
Пример 61: Синтез соединения 189
(4-(((2S,6R)-2,6-диметил-4-(м-толилкарбамотиоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(м-толилкарбамотиоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (соединение 13-2, 0,070 г, 0,267 ммоль), 1-изотиоцианат-3-метилбензол (0,048 г, 0,320 ммоль) и TEA (0,106 мл, 0,762 ммоль) растворяли в метиленхлориде (1 мл) и затем перемешивали при комнатной температуре в течение 6 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,092 г, 83,8%) в виде твердого вещества желтого цвета.
Стадия 2: Синтез соединения 189
Метил 4-(((2S,6R)-2,6-диметил-4-(м-толилкарбамотиоил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,040 г, 0,097 ммоль) растворяли в метаноле (0,5 мл) и затем добавляли гидроксиламин (0,119 мл, 1,944 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,055 г, 0,972 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 189 (0,021 г, 52,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,65 (д, 2 H, J=8,0 Гц), 7,30 (д, 2 H, J=8,0 Гц), 7,14 (т, 1 H, J=8,0 Гц), 7,05-7,04 (м, 1 H), 6,88 (д, 1 H, J=8,0 Гц), 4,55 (д, 2 H, J=12,0 Гц), 3,75 (с, 2 H), 2,91-2,85 (м, 2 H), 2,55-2,51 (м, 2 H), 2,48 (с, 3 H), 0,98 (д, 6 H, J=6,1 Гц).
Пример 62: Синтез соединения 193
(4-(((2S,6R)-4-(2-фтор-2-метилпропил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-гидрокси-2-метилпропил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль) растворяли в ацетонитриле (2 мл), и добавляли 2,2-диметилоксиран (0,041 г, 0,572 ммоль) и K2CO3 (0,105 г, 0,762 ммоль). Смесь перемешивали при температуре 80°C в течение 48 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=50%) и концентрировали с получением желаемого соединения (0,040 г, 31,4%) в виде масла коричневого цвета.
Стадия 2: Синтез метил 4-(((2S,6R)-4-(2-фтор-2-метилпропил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-4-(2-гидрокси-2-метилпропил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 19-2, 0,080 г, 0,239 ммоль) растворяли в метиленхлориде (4 мл) и при температуре 0°C добавляли DAST (0,042 г, 0,263 ммоль), затем перемешивали при комнатной температуре в течение 40 минут. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=50%) и концентрировали с получением желаемого соединения (0,065 г, 80,8%) в виде масла желтого цвета.
Стадия 3: Синтез соединения 193
Метил 4-(((2S,6R)-4-(2-фтор-2-метилпропил)-2,6-диметилпиперазин-1-ил)метил)бензоат (0,065 г, 0,193 ммоль) растворяли в метаноле (2 мл) и затем добавляли гидроксиламин (0,236 мл, 3,864 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,108 г, 1,932 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 193 (0,030 г, 46,0%) в виде твердого вещества бледно-оранжевого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,65 (д, 2 H, J=8,2 Гц), 7,35 (д, 2 H, J=8,2 Гц), 3,72 (с, 2 H), 2,74 (д, 2 H, J=10,0 Гц), 2,57-2,53 (м, 2 H), 2,35 (д, 2 H, J=22,1 Гц), 1,95-1,93 (м, 2 H), 1,29 (д, 6 H, J=22,1 Гц), 0,89 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 338,2 (M++1).
Пример 63: Синтез соединения 194
(6-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксиникотинамид)
Стадия 1: Синтез метил 6-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)никотината
(2S,6R)-1-Бензил-2,6-диметилпиперазин (формула 8-3, 0,200 г, 0,831 ммоль) растворяли в ацетонитриле (4 мл) и затем добавляли метил 6-(бромметил)никотинат (формула 8-4, 0,191 г, 0,831 ммоль) и Cs2CO3 (0,541 г, 1,661 ммоль). Смесь перемешивали при комнатной температуре в течение 17 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,183 г, 62,3%) в виде твердого вещества бледно-желтого цвета.
Стадия 2: Синтез соединения 194
Метил 6-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)никотинат (формула 8-5, 0,100 г, 0,283 ммоль) растворяли в метаноле (2 мл) и затем добавляли гидроксиламин (0,346 мл, 5,658 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,159 г, 2,829 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 194 (0,032 г, 31,9%) получали в виде твердого вещества бледно-оранжевого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,75 (д, 1 H, J=1,4 Гц), 7,94 (дд, 1 H, J=10,2, 3,6 Гц), 7,35-7,33 (м, 2 H), 7,30-7,24 (м, 3 H), 7,17-7,15 (м, 1 H), 3,73 (с, 2 H), 3,48 (с, 2 H), 2,67 (д, 2 H, J=9,9 Гц), 2,58-2,54 (м, 2 H), 1,88-1,83 (м, 2 H), 0,90 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 355,2 (M++1).
Пример 64: Синтез соединения 195
(6-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)-N-гидроксиникотинамид)
Стадия 1: Синтез метил 6-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)никотината
(3R,5S)-1-Бензил-3,5-диметилпиперазин (формула 16-1, 0,150 г, 0,734 ммоль) растворяли в ацетонитриле (4 мл) и затем добавляли метил 4-(бромметил)никотинат (формула 8-4, 0,169 г, 0,734 ммоль) и Cs2CO3 (0,718 г, 2,203 ммоль). Смесь перемешивали при комнатной температуре в течение 17 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,108 г, 41,6%) в виде твердого вещества желтого цвета.
Стадия 2: Синтез соединения 195
Метил 6-(((2S,6R)-4-бензил-2,6-диметилпиперазин-1-ил)метил)никотинат (формула 16-2, 0,050 г, 0,141 ммоль) растворяли в метаноле (2 мл) и затем добавляли гидроксиламин (0,173 мл, 2,829 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,079 г, 1,415 ммоль). Смесь перемешивали при комнатной температуре в течение 20 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 195 (0,022 г, 43,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,75 (с, 1 H), 7,99 (д, 1 H, J=8,2 Гц), 7,50 (д, 1 H, J=8,2 Гц), 7,35-7,21 (м, 5 H), 3,82 (с, 2 H), 3,44 (с, 2 H), 2,73-2,60 (м, 4 H), 1,78 (т, 2 H, J=9,9 Гц), 0,88 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 355,2 (M++1).
Пример 65: Синтез соединения 196
((3R,5S)-4-(4-(гидроксикарбамоил)бензил)-N,3,5-триметил-N-фенилпиперазин-1-карбоксамид)
Стадия 1: Синтез 4-нитрофенил метил(фенил)карбамата
N-Метиланилин (0,300 г, 2,800 ммоль) и TEA (0,776 мл, 5,600 ммоль) растворяли в метиленхлориде (5 мл) и при температуре 0°C добавляли 4-нитрофенил хлорформиат (0,621 г, 3,080 ммоль), затем перемешивали при той же температуре в течение 2 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,709 г, 93,0%) в виде масла желтого цвета.
Стадия 2: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(метил(фенил)карбамоил)пиперазин-1-ил)метил)бензоата
4-Нитрофенил метил(фенил)карбамат (0,500 г, 1,836 ммоль), метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,549 г, 1,836 ммоль) и TEA (0,382 мл, 2,755 ммоль) растворяли в N,N-диметилформамиде (20 мл). Реакционный раствор перемешивали при температуре 100°C в течение 17 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=50%) и концентрировали с получением желаемого соединения (0,373 г, 51,4%) в виде масла коричневого цвета.
Стадия 3: Синтез соединения 196
Метил 4-(((2S,6R)-2,6-диметил-4-(метил(фенил)карбамоил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,110 г, 0,278 ммоль) растворяли в метаноле (2 мл) и затем добавляли гидроксиламин (0,340 мл, 5,563 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,156 г, 2,781 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 196 (0,092 г, 83,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 10,90 (с, 1 H), 9,10 (с, 1 H), 7,64 (д, 2 H, J=8,3 Гц), 7,35-7,30 (м, 4 H), 7,12-7,08 (м, 3 H), 3,65 (с, 2 H), 3,47 (д, 2 H, J=12,8 Гц), 3,07 (с, 3 H), 2,41 (т, 1 H, J=11,5 Гц), 2,33-2,28 (м, 2 H), 0,86 (д, 6 H, J=6,4 Гц); ЖХМС (ES) m/z 397,2 (M++1).
Пример 66: Синтез соединения 197
(N-гидрокси-4-(((2S,6R)-4-(индолин-1-карбонил)-2,6-диметилпиперазин-1-ил)метил)бензамид)
Стадия 1: Синтез 4-нитрофенил индолин-1-карбоксилата
Индолин (0,300 г, 2,518 ммоль) и TEA (0,698 мл, 5,035 ммоль) растворяли в метиленхлориде (5 мл), и при температуре 0°C добавляли 4-нитрофенил хлорформиат (0,558 г, 2,769 ммоль), затем перемешивали при той же температуре в течение 30 минут. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,668 г, 93,3%) в виде твердого вещества бледно-коричневого цвета.
Стадия 2: Синтез метил 4-(((2S,6R)-4-(индолин-1-карбонил)-2,6-диметилпиперазин-1-ил)метил)бензоата
4-Нитрофенил индолин-1-карбоксилат (0,300 г, 1,055 ммоль), метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,315 г, 1,055 ммоль) и TEA (0,439 мл, 3,166 ммоль) растворяли в N,N-диметилформамиде (6 мл) при температуре 100°C, и затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=50%) и концентрировали с получением желаемого соединения (0,282 г, 65,6%) в виде твердого вещества коричневого цвета.
Стадия 3: Синтез соединения 197
Метил 4-(((2S,6R)-4-(индолин-1-карбонил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,100 г, 0,245 ммоль) растворяли в метаноле (2 мл) и затем добавляли гидроксиламин (0,300 мл, 4,908 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,138 г, 2,454 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 197 (0,068 г, 67,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,67 (д, 2 H, J=8,3 Гц), 7,41 (д, 2 H, J=8,1 Гц), 7,18 (д, 1 H, J=7,3 Гц), 7,10-7,08 (м, 1 H), 7,00 (д, 1 H, J=7,8 Гц), 6,87-6,85 (м, 1 H), 3,83-3,78 (м, 4 H), 3,54 (д, 2 H, J=12,6 Гц), 2,98 (т, 2 H, J=8,2 Гц), 2,73 (т, 2 H, J=11,7 Гц), 2,61-2,59 (м, 2 H), 0,92 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 409,2 (M++1).
Пример 67: Синтез соединения 198
((3R,5S)-N-бутил-4-(4-(гидроксикарбамоил)бензил)-3,5-диметил-N-фенилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(бутилфенилкарбамоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметил-4-(фенилкарбамоил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,500 г, 1,311 ммоль) и NaH (0,033 г, 1,376 ммоль) растворяли в N,N-диметилформамиде (10 мл) и перемешивали при комнатной температуре в течение 1 часа. Затем в реакционную смесь добавляли 1-йодбутан (0,164 мл, 1,442 ммоль), после чего перемешивали при той же температуре в течение 3 часов. Далее в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=50%) и концентрировали с получением желаемого соединения (0,115 г, 20,1%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 198
Метил 4-(((3R,5S)-4-(бутилфенилкарбамоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 20-1, 0,060 г, 0,137 ммоль) растворяли в метаноле (2 мл) и затем добавляли гидроксиламин (0,168 мл, 2,742 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,077 г, 1,371 ммоль). Смесь перемешивали при комнатной температуре в течение 20 минут, и в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 198 (0,038 г, 63,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,95 (с, 1 H), 7,62 (д, 2 H, J=8,2 Гц), 7,35-7,30 (м, 4 H), 7,12-7,06 (м, 3 H), 3,63 (с, 2 H), 3,52-3,48 (м, 2 H), 3,43 (д, 2 H, J=13,1 Гц), 2,39-2,37 (м, 2 H), 2,33-2,26 (м, 2 H), 1,43-1,37 (м, 2 H), 1,28-1,20 (м, 2 H), 0,92-0,90 (м, 3 H), 0,89-0,87 (м, 6 H); ЖХМС (ES) m/z 439,3 (M++1).
Пример 68: Синтез соединения 204
(4-(((3R,5S)-3,5-диметил-4-(4,4,4-трифторбутил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4,4,4-трифторбутил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,300 г, 1,004 ммоль) растворяли в ацетонитриле (5 мл) и затем добавляли 1,1,1-трифтор-4-йодбутан (0,129 мл, 1,004 ммоль) и Cs2CO3 (0,318 г, 2,510 ммоль). Смесь нагревали при кипячении с обратным холодильником в течение 17 часов, и затем охлаждали до комнатной температуры. Реакционную смесь фильтровали через стеклянный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=50%) и концентрировали с получением желаемого соединения (0,115 г, 30,8%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 204
Метил 4-(((3R,5S)-3,5-диметил-4-(4,4,4-трифторбутил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,065 г, 0,175 ммоль) растворяли в метаноле (2 мл) и затем добавляли гидроксиламин (0,214 мл, 3,491 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,098 г, 1,745 ммоль). Смесь перемешивали при комнатной температуре в течение 40 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Соединение 204 (0,023 г, 35,3%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,20 (с, 1 H), 9,00 (с, 1 H), 7,70 (д, 2 H, J=7,2 Гц), 7,34 (д, 2 H, J=7,6 Гц), 3,42 (с, 2 H), 2,62-2,59 (м, 6 H), 2,34-2,33 (м, 2 H), 2,72-1,66 (м, 2 H), 1,25-1,24 (м, 2 H), 0,94 (д, 6 H, J=5,1 Гц); ЖХМС (ES) m/z 374,2 (M++1).
Пример 69: Синтез соединения 211
(4-(((3R,5S)-4-(2-фторбензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-фторбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 1-(бромметил)-2-фторбензол (0,055 мл, 0,457 ммоль) и K2CO3 (0,105 г, 0,762 ммоль) растворяли в ацетонитриле (1 мл) и перемешивали при комнатной температуре в течение 7 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,067 г, 47,4%) в виде масла белого цвета.
Стадия 2: Синтез соединения 211
Метил 4-(((3R,5S)-4-(2-фторбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,135 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,165 мл, 2,699 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,076 г, 1,350 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 211 (0,031 г, 61,8%) получали в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=7,3 Гц), 7,63-7,60 (м, 1 H), 7,30 (д, 2 H, J=7,8 Гц), 3,79 (с, 2 H), 3,43 (с, 2 H), 2,68-2,56 (м, 4 H), 1,81 (т, 2 H, J=10,4 Гц), 0,87 (д, 6 H, J=6,0 Гц).
Пример 70: Синтез соединения 212
(4-(((3R,5S)-4-(2,5-дифторбензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2,5-дифторбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 2-(бромметил)-1,4-дифторбензол (0,059 мл, 0,457 ммоль) и K2CO3 (0,105 г, 0,762 ммоль) растворяли в ацетонитриле (1 мл) и перемешивали при комнатной температуре в течение 7 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,057 г, 38,5%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 212
Метил 4-(((3R,5S)-4-(2,5-дифторбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,129 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,157 мл, 2,574 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,072 г, 1,287 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 212 (0,025 г, 49,9%) получали в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=7,8 Гц), 7,39-7,35 (м, 1 H), 7,30 (д, 2 H, J=7,9 Гц), 7,18-7,15 (м, 1 H), 3,70 (с, 2 H), 3,44 (с, 2 H), 2,69-2,60 (м, 4 H), 1,83 (т, 2 H, J=10,4 Гц), 0,84 (д, 6 H, J=6,0 Гц).
Пример 71: Синтез соединения 213
(4-(((3R,5S)-3,5-диметил-4-(2,4,5-трифторбензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2,4,5-трифторбензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 1-(бромметил)-2,4,5-трифторбензол (0,103 г, 0,457 ммоль) и K2CO3 (0,105 г, 0,762 ммоль) растворяли в ацетонитриле (1 мл) и перемешивали при комнатной температуре в течение 7 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,071 г, 45,8%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 213
Метил 4-(((3R,5S)-3,5-диметил-4-(2,4,5-трифторбензил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,123 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,150 мл, 2,460 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,069 г, 1,230 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 213 (0,024 г, 47,9%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,64 (д, 2 H, J=8,0 Гц), 7,60-7,58 (м, 1 H), 7,48-7,46 (м, 1 H), 7,11 (д, 2 H, J=8,0 Гц), 3,37 (с, 2 H), 3,22 (с, 2 H), 2,69-2,55 (м, 4 H), 1,79 (т, 2 H, J=10,2 Гц), 0,84 (д, 6 H, J=6,1 Гц).
Пример 72: Синтез соединения 214
(4-(((3R,5S)-4-(3,5-бис(трифторметил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3,5-бис(трифторметил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 1-(бромметил)-3,5-бис(трифторметил)бензол (0,084 мл, 0,457 ммоль) и K2CO3 (0,105 г, 0,762 ммоль) растворяли в ацетонитриле (1 мл) и перемешивали при комнатной температуре в течение 7 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=5%) и концентрировали с получением желаемого соединения (0,075 г, 40,3%) в виде масла белого цвета.
Стадия 2: Синтез соединения 214
Метил 4-(((3R,5S)-4-(3,5-бис(трифторметил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,102 ммоль) растворяли в метаноле (1 мл) и затем добавляли гидроксиламин (0,125 мл, 2,047 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,057 г, 1,024 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Соединение 214 (0,021 г, 41,9%) получали в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,06 (с, 2 H), 7,91 (с, 2 H), 7,67 (д, 2 H, J=6,7 Гц), 7,22 (д, 2 H, J=7,3 Гц), 3,89 (с, 2 H), 3,41 (с, 2 H), 2,70-2,55 (м, 4 H), 1,83 (т, 2 H, J=10,1 Гц), 0,82 (д, 6 H, J=5,9 Гц).
Пример 73: Синтез соединения 215
(4-(((2S,6R)-2,6-диметил-4-(2,4,5-трифторбензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(2,4,5-трифторбензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 1-(бромметил)-2,4,5-трифторбензол (0,103 г, 0,457 ммоль) и K2CO3 (0,105 г, 0,762 ммоль) растворяли в ацетонитриле (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 7 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,071 г, 45,8%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 215
Метил 4-(((2S,6R)-2,6-диметил-4-(2,4,5-трифторбензил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,050 г, 0,123 ммоль), гидроксиламин (0,150 мл, 2,460 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,069 г, 1,230 ммоль) растворяли в метаноле (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 215 (0,024 г, 47,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,64 (д, 2 H, J=8,04 Гц), 7,60-7,46 (м, 3 H), 7,11 (д, 2 H, J=8,04 Гц), 3,67 (с, 2 H), 3,17 (с, 2 H), 2,69-2,61 (м, 2 H), 2,60-2,57 (м, 2 H), 1,82-1,76 (м, 2 H), 0,83 (д, 6 H, J=6,12 Гц).
Пример 74: Синтез соединения 218
(4-(2-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)этил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(2-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)этил)бензоата
(2S,6R)-1-Бензил-2,6-диметилпиперазин (формула 8-3, 0,200 г, 0,831 ммоль) и DIPEA (0,363 мл, 2,077 ммоль) растворяли в ацетонитриле (5 мл) и при комнатной температуре добавляли метил 4-(2-бромэтил)бензоат (формула 8-4, 0,404 г, 1,661 ммоль). Смесь перемешивали при температуре 60°C в течение 17 часов, и затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат концентрировали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,075 г, 24,6%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 218
Метил 4-(2-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)этил)бензоат (формула 8-5, 0,075 г, 0,205 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,125 мл, 0,2046 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,266 г, 4,093 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 218 (0,058 г, 77,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,64 (д, 2 H, J=8,2 Гц), 7,35 (д, 2 H, J=7,4 Гц), 7,31-7,27 (м, 2 H), 7,17-7,16 (м, 1 H), 3,72 (с, 2 H), 2,80 (д, 2 H, J=10,0 Гц), 2,74-2,71 (м, 2 H), 2,46-2,42 (м, 2 H), 1,82-1,80 (м, 2 H), 0,93 (д, 6 H, J=6,1 Гц).
Пример 75: Синтез соединения 219
(4-(1-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)этил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(1-бромэтил)бензоата
Метил 4-(1-гидроксиэтил)бензоат (0,500 г, 2,775 ммоль) растворяли в толуоле (10 мл), и при температуре 0°C добавляли PBr3 (0,079 мл, 0,832 ммоль). Смесь перемешивали при той же температуре в течение 1 часа и 20 минут, и реакционную смесь концентрировали при пониженном давлении с получением желаемого соединения (0,410 г, 60,8%) в виде масла пурпурного цвета.
Стадия 2: Синтез метил 4-(1-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)этил)бензоата
(3R,5S)-1-Бензил-3,5-диметилпиперазин (формула 8-3, 0,200 г, 0,831 ммоль), метил 4-(1-бромэтил)бензоат (формула 8-4, 0,222 г, 0,914 ммоль) и Cs2CO3 (0,812 г, 2,492 ммоль) растворяли в N,N-диметилформамиде (4 мл) при комнатной температуре, и реакционный раствор перемешивали при температуре 50°C в течение 17 часов. Реакционную смесь фильтровали через стеклянный фильтр для удаления твердых продуктов, и к фильтрату добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,168 г, 55,2%) в виде масла бледно-желтого цвета.
Стадия 3: Синтез соединения 219
Метил 4-(1-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)этил)бензоат (формула 8-5, 0,080 г, 0,218 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,134 мл, 2,183 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,284 г, 4,366 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, и реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 219 (0,051 г, 63,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,10 (с, 1 H), 9,10 (с, 1 H), 7,67 (д, 2 H, J=6,9 Гц), 7,31-7,23 (м, 6 H), 7,16-7,14 (м, 1 H), 3,68 (с, 2 H), 2,82 (д, 2 H, J=11,0 Гц), 2,53-2,82 (м, 3 H), 1,74 (т, 1 H, J=10,2 Гц), 1,64 (т, 1 H, J=10,5 Гц), 1,24 (д, 3 H, J=6,7 Гц), 0,90 (д, 3 H, J=6,0 Гц), 0,86-0,84 (м, 1 H), 0,81 (д, 3 H, J=6,7 Гц).
Пример 76: Синтез соединения 220
(4-(((2S,6R)-2,6-диметил-4-(1-фенилэтил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез (1-бромэтил)бензола
(1-Гидроксиэтил)бензол (1,000 г, 8,186 ммоль) и PBr3 (0,233 мл, 2,456 ммоль) растворяли в толуоле (10 мл) при температуре 0°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. В реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением желаемого соединения (0,900 г, 59,4%) в виде масла бледно-коричневого цвета.
Стадия 2: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(1-фенилэтил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,200 г, 0,669 ммоль), (1-бромэтил)бензол (0,136 г, 0,736 ммоль) и Cs2CO3 (0,654 г, 2,008 ммоль) растворяли в N,N-диметилформамиде (4 мл) при комнатной температуре, и реакционный раствор перемешивали при температуре 50°C в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=25%) и концентрировали с получением желаемого соединения (0,22 г, 89,7%) в виде бесцветного масла.
Стадия 3: Синтез соединения 220
Метил 4-(((2S,6R)-2,6-диметил-4-(1-фенилэтил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,110 г, 0,300 ммоль) растворяли в метаноле (3 мл) и затем добавляли гидроксиламин (0,184 мл, 3,001 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,391 г, 6,003 ммоль). Смесь перемешивали при комнатной температуре в течение 20 минут, и затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 220 (0,063 г, 57,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,63 (д, 2 H, J=8,3 Гц), 7,35-7,33 (м, 2 H), 7,30-7,26 (м, 4 H), 7,24-7,21 (м, 1 H), 3,69 (с, 2 H), 2,85-2,83 (м, 1 H), 2,55-2,50 (м, 3 H), 1,75-1,74 (м, 1 H), 1,63-1,62 (м, 1 H), 1,25 (д, 3 H, J=6,8 Гц), 0,91-0,90 (м, 1 H), 0,88 (д, 3 H, J=4,9 Гц), 0,78 (д, 3 H, J=6,1 Гц).
Пример 77: Синтез соединения 222
(4-(((3R,5S)-4-(2-(3-фторфенил)ацетил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-(3-фторфенил)ацетил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 2-(3-фторфенил)уксусную кислоту (0,088 г, 0,572 ммоль), HOBt (0,077 г, 0,572 ммоль), EDCI (0,110 г, 0,572 ммоль) и DIPEA (0,133 мл, 0,762 ммоль) растворяли в метиленхлориде (2 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 20 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,047 г, 30,9%) в виде твердого вещества бледно-желтого цвета.
Стадия 2: Синтез соединения 222
Метил 4-(((3R,5S)-4-(2-(3-фторфенил)ацетил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,040 г, 0,100 ммоль), гидроксиламин (0,123 мл, 2,008 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,056 г, 1,004 ммоль) растворяли в метаноле (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 222 (0,019 г, 47,4%) в виде твердого вещества оранжевого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,64 (д, 2 H, J=8,0 Гц), 7,32-7,27 (м, 1 H), 7,24 (д, 2 H, J=8,0 Гц), 7,04-7,01 (м, 3 H), 4,35 (ушир. с, 1 H), 4,07 (ушир. с, 1 H), 3,75-3,62 (м, 2 H), 3,46 (с, 2 H), 2,63-2,52 (м, 2 H), 1,99-1,73 (м, 2 H), 1,20 (д, 6 H, J=17,2 Гц).
Пример 78: Синтез соединения 223
(4-(((3R,5S)-3,5-диметил-4-(2-(3-(трифторметил)фенил)ацетил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2-(3-(трифторметил)фенил)ацетил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 2-(3-(трифторметил)фенил)уксусная кислота (0,117 г, 0,572 ммоль), HOBt (0,077 г, 0,572 ммоль), EDCI (0,110 г, 0,572 ммоль) и DIPEA (0,133 мл, 0,762 ммоль) растворяли в метиленхлориде (2 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 20 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 12 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,037 г, 21,6%) в виде бесцветного масла.
Стадия 2: Синтез соединения 223
Метил 4-(((3R,5S)-3,5-диметил-4-(2-(3-(трифторметил)фенил)ацетил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,030 г, 0,067 ммоль), гидроксиламин (0,082 мл, 1,338 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,038 г, 0,669 ммоль) растворяли в метаноле (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 223 (0,013 г, 43,2%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,64 (д, 2 H, J=8,0 Гц), 7,56-7,48 (м, 4 H), 7,20 (д, 2 H, J=8,4 Гц), 4,36 (ушир. с, 1 H), 4,12 (ушир. с, 1 H), 3,92-3,67 (ушир. с, 1 H), 3,48 (с, 2 H), 2,61 (д, 2 H, J=11,2 Гц), 2,05-2,00 (м, 2 H), 1,23 (д, 6 H, J=31,6 Гц).
Пример 79: Синтез соединения 224
(4-(((3R,5S)-4-(2-(3-хлорфенил)ацетил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-(3-хлорфенил)ацетил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 2-(3-хлорфенил)уксусную кислоту (0,098 г, 0,572 ммоль), HOBt (0,077 г, 0,572 ммоль), EDCI (0,110 г, 0,572 ммоль) и DIPEA (0,133 мл, 0,762 ммоль) растворяли в метиленхлориде (2 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 20 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,042 г, 26,6%) в виде бесцветного масла.
Стадия 2: Синтез соединения 224
Метил 4-(((3R,5S)-4-(2-(3-хлорфенил)ацетил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,035 г, 0,084 ммоль), гидроксиламин (0,103 мл, 1,687 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,047 г, 0,844 ммоль) растворяли в метаноле (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 224 (0,020 г, 57,0%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,62 (д, 2 H, J=7,6 Гц), 7,31-7,23 (м, 4 H), 7,15 (д, 2 H, J=6,4 Гц), 4,35 (ушир. с, 1 H), 4,07 (ушир. с, 1 H), 3,76-3,47 (м, 2 H), 3,30 (с, 2 H), 2,62 (д, 2 H, J=10,8 Гц), 1,98 (ушир. с, 2 H), 1,23 (д, 6 H, J=25,2 Гц).
Пример 80: Синтез соединения 225
(4-(((3R,5S)-4-(3-фторбензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-фторбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (соединение 1-2, 0,150 г, 0,572 ммоль), 3-фторбензилбромид (0,077 мл, 0,629 ммоль) и K2CO3 (0,119 г, 0,858 ммоль) растворяли в ацетонитриле (4 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 4 г; этилацетат/гексан=от 0% до 40%) и концентрировали с получением желаемого соединения (0,110 г, 51,9%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 225
Метил 4-(((3R,5S)-4-(3-фторбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (соединение 1-3, 0,060 г, 0,162 ммоль), гидроксиламин (0,099 мл, 1,620 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,211 г, 3,239 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа, и затем концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 225 (0,044 г, 73,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,65 (д, 2 H, J=8,0 Гц), 7,31-7,27 (м, 1 H), 7,22 (д, 2 H, J=8,0 Гц), 7,17-7,13 (м, 2 H), 6,97-6,96 (м, 1 H), 3,70 (с, 2 H), 3,43 (с, 2 H), 2,64 (д, 2 H, J=11,2 Гц), 2,57-2,56 (м, 2 H), 1,78 (т, 2 H, J=10,6 Гц), 0,84 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 372,2 (M++1).
Пример 81: Синтез соединения 230
((3R,5S)-4-(4-(гидроксикарбамоил)бензил)-N-(3-метоксибензил)-3,5-диметилпиперазин-1-карбоксамид (соль трифторуксусной кислоты))
Стадия 1: Синтез 4-нитрофенил 3-метоксибензил карбамата
3-Метоксибензил амин (0,100 г, 0,729 ммоль) и TEA (0,152 мл, 1,093 ммоль) растворяли в метиленхлориде (2 мл) и при температуре 0°C добавляли 4-нитрофенилхлорформиат (0,162 г, 0,802 ммоль), затем перемешивали при той же температуре в течение 2 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Полученный продукт использовали без дополнительной очистки (0,200 г, 90,8%, масло желтого цвета).
Стадия 2: Синтез метил 4-(((2S,6R)-4-((3-метоксибензил)карбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
4-Нитрофенил 3-метоксибензил карбамат (0,130 г, 0,430 ммоль), метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,129 г, 0,430 ммоль) и TEA (0,179 мл, 1,290 ммоль) растворяли в N,N-диметилформамиде (4 мл) при комнатной температуре, и реакционный раствор перемешивали при температуре 100°C в течение 3 часов. Затем реакционную смесь концентрировали при пониженном давлении. Полученный продукт использовали без дополнительной очистки (0,180 г, 98,4%, масло коричневого цвета).
Стадия 3: Синтез соединения 230
Метил 4-(((2S,6R)-4-((3-метоксибензил)карбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,180 г, 0,423 ммоль), гидроксиламин (0,517 мл, 8,460 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,237 г, 4,230 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали с получением соединения 230 (0,033 г, 18,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,11 (ушир. с, 1 H), 8,95 (ушир. с, 1 H), 7,64 (д, 2 H, J=8,4 Гц), 7,39 (д, 2 H, J=8,0 Гц), 7,17 (дд, 1 H, J=7,0, 7,0 Гц),7,03 (дд, 1 H, J=6,0, 6,0 Гц), 6,75-6,72 (м, 2 H), 4,16 (д, 2 H, J=5,6 Гц), 3,80 (д, 2 H, J=12,4 Гц), 3,72 (с, 2 H), 3,68 (с, 3 H), 2,53 (д, 2 H, J=10,4 Гц), 2,42-2,38 (м, 2 H), 0,89 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 427,3 (M++1).
Пример 82: Синтез соединения 231
((3R,5S)-N-(3-фторбензил)-4-(4-(гидроксикарбамоил)бензил)-3,5-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез 4-нитрофенил 3-фторбензилкарбамата
(3-Фторфенил)метиламин (0,200 г, 1,598 ммоль) и TEA (0,243 г, 2,397 ммоль) растворяли в метиленхлориде (2 мл), и при температуре 0°C добавляли 4-нитрофенил хлорформиат (0,354 г, 1,758 ммоль), затем перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Полученный продукт использовали без дополнительной очистки (0,410 г, 88,4%, твердое вещество желтого цвета).
Стадия 2: Синтез метил 4-(((2S,6R)-4-((3-фторбензил)карбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
4-Нитрофенил 3-фторбензилкарбамат (0,200 г, 0,689 ммоль), метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,216 г, 0,724 ммоль) и TEA (0,287 мл, 2,067 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и реакционный раствор перемешивали при температуре 100°C в течение 2 часов и 30 минут. Затем реакционную смесь концентрировали при пониженном давлении с использованием V10. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением желаемого соединения (0,178 г, 62,5%) в виде масла бледно-коричневого цвета.
Стадия 3: Синтез соединения 231
Метил 4-(((2S,6R)-4-((3-фторбензил)карбамоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,100 г, 0,242 ммоль), гидроксиламин (0,296 мл, 4,837 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,136 г, 2,418 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (10 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 231 (0,055 г, 54,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,64 (д, 2 H, J=8,0 Гц), 7,38 (д, 2 H, J=8,0 Гц), 7,33-7,27 (м, 1 H), 7,10 (т, 1 H, J=5,8 Гц), 7,06-6,98 (м, 3 H), 4,20 (д, 2 H, J=5,6 Гц), 3,79 (д, 2 H, J=12,8 Гц), 3,72 (с, 2 H), 2,52 (т, 2 H, J=11,6 Гц), 2,42-2,38 (м, 2 H), 0,89 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 415,2 (M++1).
Пример 83: Синтез соединения 232
((2S,6R)-N-(3-фторбензил)-4-(4-(гидроксикарбамоил)бензил)-2,6-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-((3-фторбензил)карбамоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
4-Нитрофенил 3-фторбензилкарбамат (0,220 г, 0,758 ммоль), метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,209 г, 0,796 ммоль) и TEA (0,315 мл, 2,274 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и реакционный раствор перемешивали при температуре 100°C в течение 2 часов и 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением желаемого соединения (0,185 г, 59,0%) в виде бесцветного масла.
Стадия 2: Синтез соединения 232
Метил 4-(((3R,5S)-4-((3-фторбензил)карбамоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,100 г, 0,242 ммоль), гидроксиламин (0,296 мл, 4,837 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,136 г, 2,418 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 232 (0,026 г, 25,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,70 (д, 2 H, J=8,0 Гц), 7,48 (д, 2 H, J=8,4 Гц), 7,31-7,25 (м, 1 H), 7,06 (д, 1 H, J=7,6 Гц), 6,98 (д, 1 H, J=10,0 Гц), 6,93-6,88 (м, 1 H), 4,35 (с, 2 H), 4,07-4,04 (м, 2 H), 3,54 (с, 2 H), 2,67 (д, 2 H, J=11,2 Гц), 2,17 (дд, 2 H, J=10,2, 3,6 Гц), 1,31 (д, 6 H, J=6,8 Гц).
Пример 84: Синтез соединения 233
(4-(((2S,6R)-4-(2-(3-хлорфенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-(3-хлорфенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоата
2-(3-Хлорфенил)уксусную кислоту (0,200 г, 0,669 ммоль), метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,126 г, 0,736 ммоль), EDCI (0,257 г, 1,339 ммоль), HOBt (0,205 г, 1,339 ммоль) и DIPEA (0,584 мл, 3,347 ммоль) растворяли в метиленхлориде (5 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 10% до 70%) и концентрировали с получением желаемого соединения (0,187 г, 67,3%) в виде бесцветного масла.
Стадия 2: Синтез соединения 233
Метил 4-(((2S,6R)-4-(2-(3-хлорфенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,100 г, 0,241 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,295 мл, 4,820 ммоль) и гидроксид калия (0,135 г, 2,410 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. К концентрату добавляли диэтиловый эфир (3 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 233 (0,053 г, 52,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,63 (д, 2 H, J=8,4 Гц), 7,33 (д, 2 H, J=8,4 Гц), 7,27-7,24 (м, 3 H), 7,16-7,14 (м, 1 H), 4,10 (д, 1 H, J=12,4 Гц), 3,81 (д, 1 H, J=13,2 Гц), 3,76-3,65 (м, 4 H), 2,86-2,52 (м, 1 H), 2,43 (д, 1 H, J=12,4 Гц), 2,38-2,28 (м, 2 H), 0,90 (т, 6 H, J=5,4 Гц); ЖХМС (ES) m/z 416,1 (M++1).
Пример 85: Синтез соединения 234
(4-(((3R,5S)-4-(2-([1,1’-бифенил]-3-ил)ацетил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-([1,1’-бифенил]-3-ил)ацетил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 2-([1,1’-бифенил]-3-ил)уксусную кислоту (0,097 г, 0,457 ммоль), HOBt (0,077 г, 0,572 ммоль), EDCI (0,110 г, 0,572 ммоль) и DIPEA (0,099 г, 0,762 ммоль) растворяли в метиленхлориде (2 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 7 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,114 г, 65,5%) в виде твердого вещества желтого цвета.
Стадия 2: Синтез соединения 234
Метил 4-(((3R,5S)-4-(2-([1,1’-бифенил]-3-ил)ацетил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 4-2, 0,050 г, 0,110 ммоль), гидроксиламин (0,134 мл, 2,190 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,061 г, 1,095 ммоль) растворяли в метаноле (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. В реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением соединения 234 (0,027 г, 53,9%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,70 (д, 2 H, J=7,6 Гц), 7,68 (д, 2 H, J=8,0 Гц), 7,58-7,30 (м, 8 H), 7,21 (м, 1 H), 4,39 (ушир. с, 1 H), 4,14 (ушир. с, 1 H) 3,83-3,64 (м, 2 H), 3,48 (с, 2 H), 2,62 (д, 2 H, J=10,8 Гц), 2,29 (ушир. с, 2 H), 1,22 (д, 6 H, J=20 Гц).
Пример 86: Синтез соединения 242
(4-(((3R,5S)-4-(3-(фуран-2-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез 3-йодбензоилхлорида
3-Йодбензойную кислоту (0,200 г, 0,806 ммоль) растворяли в SOCl2 (1,170 мл, 16,128 ммоль) и перемешивали при температуре 100°C в течение 2 часов, и реакционную смесь концентрировали при пониженном давлении. Полученный продукт использовали без дополнительной очистки (0,215 г, 100,0%, масло коричневого цвета).
Стадия 2: Метил 4-(((3R,5S)-4-(3-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,215 г, 0,807 ммоль), 3-йодбензоилхлорид (0,233 мл, 0,887 ммоль) и TEA (0,224 мл, 1,613 ммоль) растворяли в метиленхлориде (5 мл) при температуре 0°C, и реакционный раствор перемешивали при той же температуре в течение 2 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 30% до 50%) и концентрировали с получением желаемого соединения (0,365 г, 91,8%) в виде бесцветного масла.
Стадия 3: Метил 4-(((3R,5S)-4-(3-(фуран-2-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (1 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,346 г, 0,703 ммоль), фуран-2-илборной кислоты (0,118 г, 1,054 ммоль), Pd(dppf)Cl2 (0,029 г, 0,035 ммоль) и Na2CO3 (0,223 г, 2,108 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли 1н. водный раствор соляной кислоты, после чего экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 25%) и концентрировали с получением желаемого соединения (0,301 г, 99,0%) в виде масла коричневого цвета.
Стадия 4: Синтез соединения 242
Метил 4-(((3R,5S)-4-(3-(фуран-2-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 4-2, 0,050 г, 0,116 ммоль), гидроксиламин (0,141 мл, 2,312 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,065 г, 1,156 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 242 (0,034 г, 67,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,79 (с, 1 H), 7,78-7,70 (м, 3 H), 7,64 (с, 1 H), 7,48 (т, 1 H, J=7,7 Гц), 7,37 (д, 2 H, J=8,0 Гц), 7,24 (д, 1 H, J=7,6 Гц), 7,08 (д, 1 H, J=3,4 Гц), 6,63-6,61 (м, 1 H), 3,53 (с, 2 H), 2,63 (ушир. с, 2 H), 2,18-2,14 (м, 2 H), 1,31 (ушир. с, 6 H).
Пример 87: Синтез соединения 243
(4-(((3R,5S)-4-(3-(фуран-3-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(фуран-3-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (1 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,120 г, 0,244 ммоль), фуран-3-илборной кислоты (0,041 г, 0,366 ммоль), Pd(dbpf)Cl2 (0,008 г, 0,012 ммоль) и Na2CO3 (0,077 г, 0,731 ммоль), и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли 1н. водный раствор соляной кислоты, после чего экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 30%) и концентрировали с получением желаемого соединения (0,050 г, 47,0%) в виде твердого вещества коричневого цвета.
Стадия 2: Синтез соединения 243
4-(((3R,5S)-4-(3-(Фуран-3-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 4-2, 0,050 г, 0,114 ммоль), гидроксиламин (0,140 мл, 2,289 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,064 г, 1,144 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 243 (0,029 г, 57,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,29 (с, 1 H), 7,76-7,70 (м, 3 H), 7,66 (д, 1 H, J=8,0 Гц), 7,46-7,38 (м, 3 H), 7,20 (д, 1 H, J=7,6 Гц), 7,05-7,04 (м, 1 H), 3,54 (с, 2 H), 2,68-2,64 (м, 2 H), 2,19-2,15 (м, 2 H), 1,32 (ушир. с, 6 H).
Пример 88: Синтез соединения 244
(4-(((3R,5S)-4-(3-(3,6-дигидро-2H-пиран-4-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(3,6-дигидро-2H-пиран-4-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (1 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,120 г, 0,244 ммоль), 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,077 г, 0,366 ммоль), Pd(dbpf)Cl2 (0,008 г, 0,012 ммоль) и Na2CO3( 0,077 г, 0,731 ммоль), и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли 1н. водный раствор соляной кислоты, после чего экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 50%) и концентрировали с получением желаемого соединения (0,049 г, 44,8%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 244
Метил 4-(((3R,5S)-4-(3-(3,6-дигидро-2H-пиран-4-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 4-2, 0,049 г, 0,113 ммоль), гидроксиламин (0,139 мл, 2,266 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,064 г, 1,133 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 244 (0,013 г, 24,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,72 (д, 2 H, J=8,0 Гц), 7,50 (д, 1 H, J=7,7 Гц), 7,45-7,37 (м, 4 H), 7,22 (д, 1 H, J=7,4 Гц), 6,33 (с, 1 H), 4,23-4,22 (м, 2 H), 3,82 (т, 2 H, J=5,3 Гц), 3,53 (с, 2 H), 2,68-2,63 (м, 2 H), 2,18-2,14 (м, 2 H), 1,30 (ушир. с, 6 H).
Пример 89: Синтез соединения 245
(4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-4-ил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-4-ил)бензоил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (1 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,200 г, 0,406 ммоль), гидрата пиридин-4-бороновой кислоты (0,086 г, 0,609 ммоль), Pd(dbpf)Cl2 (0,013 г, 0,020 ммоль) и Na2CO3 (0,129 г, 1,219 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,167 г, 92,8%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 245
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-4-ил)бензоил)пиперазин-1-ил)метил)бензоат (формула 4-2, 0,105 г, 0,236 ммоль), гидроксиламин (0,288 мл, 4,712 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,132 г, 2,356 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Затем концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, 0,1%-ный водный раствор трифторуксусной кислоты/ацетонитрил=от 5% до 80%) и концентрировали с получением соединения 245 (0,072 г, 54,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,91 (д, 2 H, J=6,6 Гц), 8,24 (д, 2 H, J=6,0 Гц), 8,05 (д, 1 H, J=7,9 Гц), 7,98 (с, 1 H), 7,81 (д, 2 H, J=7,9 Гц), 7,68 (т, 1 H, J=7,7 Гц), 7,61-7,56 (м, 3 H), 4,78 (ушир. с, 1 H), 4,24-4,00 (м, 3 H), 3,23-3,10 (м, 2 H), 1,36 (ушир. с, 6 H).
Пример 90: Синтез соединения 246
(4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез 4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (1,2 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,100 г, 0,203 ммоль), пиридин-3-илборной кислоты (0,050 г, 0,406 ммоль), Na2CO3 (0,065 г, 0,609 ммоль) и Pd(dbpf)Cl2 (0,007 г, 0,010 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 80%) и концентрировали с получением желаемого соединения (0,080 г, 88,8%) в виде твердого вещества желтого цвета.
Стадия 2: Синтез соединения 246
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)бензоат (формула 4-2, 0,065 г, 0,147 ммоль), гидроксиламин (0,090 мл, 1,465 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,191 г, 2,931 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 2 часов, и затем концентрировали. Концентрат промывали насыщенным водным раствором гидрокарбоната натрия и дистиллированной водой, получая таким образом соединение 246 (0,035 г, 53,7%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3): 8,93 (д, 1 H, J=2,3 Гц), 8,60 (м, 1 H), 7,06 (дд, 1 H, J=8,0, 1,6 Гц), 7,82-7,77 (м, 1 H), 7,72-7,69 (м, 3 H), 7,58-7,48 (м, 2 H), 7,39-7,35 (м, 3 H), 3,37-3,28 (м, 2 H), 2,63 (м, 2 H), 2,19 (дд, 2 H, J=11,4, 4,0 Гц), 1,32 (с, 6 H).
Пример 91: Синтез соединения 247
((4-(((3R,5S)-4-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-3-карбонил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(4,4-диметилциклогекс-1-енил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан (0,9 мл)/вода (0,3 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,100 г, 0,203 ммоль), 2-(4,4-диметилциклогекс-1-енил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,096 г, 0,406 ммоль), Na2CO3 (0,065 г, 0,609 ммоль) и Pd(dbpf)Cl2 (0,007 г, 0,010 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 40%) и концентрировали с получением желаемого соединения (0,030 г, 31,1%) в виде твердого вещества желтого цвета.
Стадия 2: Синтез соединения 247
Метил 4-(((3R,5S)-4-(3-(4,4-диметилциклогекс-1-енил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 4-2, 0,030 г, 0,063 ммоль), гидроксиламин (0,039 мл, 0,632 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,082 г, 1,264 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 2 часов. Затем реакционную смесь концентрировали. Концентрат промывали насыщенным водным раствором гидрокарбоната натрия и дистиллированной водой, получая таким образом желаемое соединение 247 (0,021 г, 69,9%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3): 7,71 (д, 1 H, J=8,0 Гц), 7,47-7,45 (м, 1 H), 7,38-7,32 (м, 4 H), 7,18 (д, 1 H, J=7,6 Гц), 6,16 (с, 1 H), 3,51 (м, 2 H), 2,62 (м, 2 H), 2,38 (м, 2 H), 2,15 (м, 2 H), 1,98 (м, 2 H), 1,50-1,46 (м, 2 H), 2,29 (м, 6 H), 0,94 (с, 6 H).
Пример 92: Синтез соединения 248 (4-(((3R,5R)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5R)-4-бензил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5R)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 21-1, 0,050 г, 0,191 ммоль), бензилбромид (0,023 мл, 0,191 ммоль) и K2CO3 (0,040 г, 0,286 ммоль) растворяли в ацетонитриле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь фильтровали через пластиковый фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением желаемого соединения (0,038 г, 56,6%) в виде бесцветного масла.
Стадия 2: Синтез соединения 248
Метил 4-(((3R,5R)-4-бензил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 21-2, 0,038 г, 0,108 ммоль), гидроксиламин (0,132 мл, 2,156 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,060 г, 1,078 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 248 (0,018 г, 47,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,71 (д, 2 H, J=8,2 Гц), 7,44 (д, 2 H, J=8,2 Гц), 7,38 (д, 2 H, J=7,3 Гц), 7,29 (дд, 1 H, J=7,4, 7,4 Гц), 7,23-7,22 (м, 1 H), 4,02 (д, 1 H, J=13,4 Гц), 3,58 (д, 1 H, J=13,5 Гц), 3,43-3,42 (м, 2 H), 2,92-2,88 (м, 2 H), 2,49-2,47 (м, 2 H), 2,25-2,25 (м, 2 H), 1,09 (д, 6 H, J=6,4 Гц); ЖХМС (ES) m/z 354,2 (M++1).
Пример 93: Синтез соединения 249
(4-(((3R,5R)-4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5R)-4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5R)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 21-1, 0,050 г, 0,191 ммоль) и TEA (0,053 мл, 0,381 ммоль) растворяли в метиленхлориде (2 мл), и при температуре 0°C добавляли при температуре 0°C 2-фуроилхлорид (0,019 мл, 0,191 ммоль), затем перемешивали при той же температуре в течение 3 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 40%) и концентрировали с получением желаемого соединения (0,033 г, 48,6%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 249
Метил 4-(((3R,5R)-4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 21-3, 0,033 г, 0,093 ммоль), гидроксиламин (0,113 мл, 1,852 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,052 г, 0,926 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 249 (0,019 г, 57,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,73 (д, 2 H, J=7,9 Гц), 7,66 (с, 1 H), 7,48 (д, 2 H, J=7,9 Гц), 7,01 (д, 1 H, J=3,3 Гц), 6,58 (с, 1 H), 4,17-4,13 (м, 2 H), 3,67 (д, 1 H, J=13,5 Гц), 3,53 (д, 1 H, J=13,8 Гц), 2,68-2,65 (м, 2 H), 2,42-2,38 (м, 2 H), 1,35 (д, 6 H, J=6,4 Гц); ЖХМС (ES) m/z 358,1 (M++1).
Пример 94: Синтез соединения 250
(4-(((2S,6R)-4-(2-([1,1’-бифенил]-3-ил)ацетил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-(3-бромфенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 2,000 г, 6,693 ммоль), 2-(3-бромфенил)уксусную кислоту (1,583 г, 7,363 ммоль), EDCI (2,566 г, 13,386 ммоль), HOBt (2,050 г, 13,386 ммоль) и DIPEA (5,845 мл, 33,466 ммоль) растворяли в метиленхлориде (5 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 40% до 70%) и концентрировали с получением желаемого соединения (2,230 г, 72,5%) в виде бесцветного масла.
Стадия 2: Синтез метил 4-(((2S,6R)-4-(2-(3-бромфенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоата
1,2-Диметоксиэтан (2 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-(3-бромфенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,100 г, 0,218 ммоль), фенилборной кислоты (0,029 г, 0,239 ммоль), Pd(PPh3)4 (0,013 г, 0,011 ммоль) и Na2CO3 (0,069 г,0,653 ммоль) при комнатной температуре, и реакционный раствор перемешивали при температуре 100°C в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 40%) и концентрировали с получением желаемого соединения (0,043 г, 43,3%) в виде бесцветного масла.
Стадия 3: Синтез соединения 250
Метил 4-(((2S,6R)-4-(2-([1,1’-бифенил]-3-ил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,043 г, 0,094 ммоль), гидроксиламин (0,115 мл, 1,884 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,053 г, 0,942 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали с получением соединения 250 (0,015 г, 34,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,55-7,37 (м, 9 H), 7,30 (д, 2 H, J=7,7 Гц), 7,22 (дд, 1 H, J=7,2, 7,2 Гц), 7,12 (д, 2 H, J=7,0 Гц), 4,55-4,48 (м, 2 H), 4,38 (д, 1 H, J=14,7 Гц), 4,15 (д, 1 H, J=15,0 Гц), 3,91 (д, 1 H, J=15,0 Гц), 3,68 (д, 1 H, J=6,5 Гц), 3,67 (д, 1 H, J=14,5 Гц), 3,35-3,27 (м, 1 H), 2,97-2,91 (м, 3 H), 1,54 (д, 3 H, J=5,7 Гц), 1,31 (д, 3 H, J=6,4 Гц).
Пример 95: Синтез соединения 251
(4-(((2S,6R)-2,6-диметил-4-(2-(3-(пиридин-4-ил)фенил)ацетил)пиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(2-(3-(пиридин-4-ил)фенил)ацетил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-(3-бромфенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,200 г, 0,435 ммоль), пиридин-4-илбороновой кислоты (0,059 г, 0,479 ммоль), Pd(PPh3)4 (0,025 г, 0,022 ммоль) и Na2CO3 (0,138 г, 1,306 ммоль) при комнатной температуре, и реакционную смесь нагревали при кипячении с обратным холодильником в течение 17 часов и затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,123 г, 61,7%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 251
Метил 4-(((2S,6R)-2,6-диметил-4-(2-(3-(пиридин-4-ил)фенил)ацетил)пиперазин-1-ил)метил)бензоат (формула 15-2, 0,100 г, 0,219 ммоль), гидроксиламин (0,267 мл, 4,371 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,123 г, 2,186 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали с получением соединения 251 (0,006 г, 6,0%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,65 (д, 2 H, J=5,9 Гц), 7,69-7,64 (м, 5 H), 7,45 (дд, 1 H, J=7,7, 7,7 Гц), 7,34 (д, 3 H, J=8,3 Гц), 4,16 (д, 1 H, J=12,8 Гц), 3,91 (д, 1 H, J=13,1 Гц), 3,82 (д, 2 H, J=8,0 Гц), 3,71 (с, 2 H), 2,90 (дд, 1 H, J=10,4, 13,3 Гц), 2,41-2,32 (м, 2 H), 0,96-0,85 (м, 6 H).
Пример 96: Синтез соединения 252
(4-(((2S,6R)-4-(2-(3-(3,6-дигидро-2H-пиран-4-ил)фенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-(3-(3,6-дигидро-2H-пиран-4-ил)фенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-(3-бромфенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,200 г, 0,435 ммоль), 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,101 г, 0,479 ммоль), Pd(PPh3)4 (0,025 г, 0,022 ммоль) и Na2CO3 (0,138 г, 1,306 ммоль) при комнатной температуре, и реакционную смесь нагревали при кипячении с обратным холодильником в течение 17 часов, и затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 30% до 100%) и концентрировали с получением желаемого соединения (0,084 г, 41,7%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 252
Метил 4-(((2S,6R)-4-(2-(3-(3,6-дигидро-2H-пиран-4-ил)фенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,050 г, 0,108 ммоль), гидроксиламин (0,132 мл, 2,162 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,061 г, 1,081 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 252 (0,016 г, 31,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CH3OD) δ 7,70-7,69 (м, 2 H), 7,43 (д, 2 H, J=8,3 Гц), 7,34 (д, 2 H, J=5,8 Гц), 7,30-7,28 (м, 1 H), 7,16-7,16 (м, 1 H), 6,20-6,19 (м, 1 H), 4,32-4,30 (м, 3 H), 3,94 (т, 2 H, J=5,5 Гц), 3,86-3,77 (м, 5 H), 2,85 (дд, 1 H, J=11,3, 11,3 Гц), 2,59-2,56 (м, 1 H), 2,52-2,50 (м, 3 H), 2,19-2,18 (м, 1 H), 1,06 (д, 3 H, J=6,1 Гц), 0,95 (д, 3 H, J=6,2 Гц); ЖХМС (ES) m/z 464,2 (M++1).
Пример 97: Синтез соединения 253
(4-(((2S,6R)-4-(2-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-3-ил)ацетил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-3-ил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-(3-бромфенил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,150 г, 0,327 ммоль), 2-(4,4-диметилцикло-1-гексенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,085 г, 0,359 ммоль), Pd(PPh3)4 (0,019 г, 0,016 ммоль) и Na2CO3 (0,104 г, 0,980 ммоль) при комнатной температуре, и реакционную смесь нагревали при кипячении с обратным холодильником в течение 17 часов, и затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,089 г, 55,8%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 253
Метил 4-(((2S,6R)-4-(2-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-3-ил)ацетил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,050 г, 0,102 ммоль), гидроксиламин (0,125 мл, 2,046 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,057 г, 1,023 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали с получением соединения 253 (0,015 г, 29,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,65 (д, 2 H, J=8,2 Гц), 7,33-7,26 (м, 4 H), 7,23 (дд, 1 H, J=7,6, 7,6 Гц), 7,07 (д, 1 H, J=6,9 Гц), 6,07 (с, 1 H), 4,15 (д, 1 H, J=11,0 Гц), 3,85 (д, 1 H, J=13,2 Гц), 3,76-3,65 (м, 4 H), 2,85 (дд, 1 H, J=11,3, 11,3 Гц), 2,47-2,44 (м, 1 H), 2,37 (с, 3 H), 2,25 (с, 1 H), 1,98 (с, 1 H), 1,49 (т, 2 H, J=6,3 Гц), 0,94 (с, 9 H), 0,89 (д, 3 H, J=5,8 Гц); ЖХМС (ES) m/z 490,3 (M++1).
Пример 98: Синтез соединения 255
(4-(((3R,5S)-4-(3-(1H-пиррол-1-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(1H-пиррол-1-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 1-2, 0,200 г, 0,762 ммоль), 1-(3-(бромметил)фенил)-1H-пиррола (0,198 г, 0,839 ммоль) и K2CO3 (0,211 г, 1,525 ммоль) растворяли в ацетонитриле (2 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 8 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,164 г, 51,5%) в виде бесцветного масла.
Стадия 2: Синтез соединения 255
Метил 4-(((3R,5S)-4-(3-(1H-пиррол-1-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,120 ммоль), гидроксиламин (0,146 мл, 2,395 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,067 г, 1,197 ммоль) растворяли в метаноле (1 мл) при температуре 25°C, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 255 (0,024 г, 47,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,61 (д, 2 H, J=8,4 Гц), 7,45-7,42 (м, 1 H), 7,32 (д, 2 H, J=5,2 Гц), 7,26 (д, 2 H, J=2,4 Гц), 7,22-7,21 (м, 1 H), 7,13-7,11 (м, 2 H) 6,22-6,21 (м, 2 H), 3,80 (с, 2 H), 3,33 (с, 2H), 2,63-2,57 (м, 4 H), 1,79-1,72 (м, 2 H), 0,85 (д, 6 H, J=16,8 Гц).
Пример 99: Синтез соединения 256
(4-(((2S,6R)-4-(3-(1H-пиррол-1-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(3-(1H-пиррол-1-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,200 г, 0,762 ммоль), 1-(3-(бромметил)фенил)-1H-пиррол (0,198 г, 0,839 ммоль) и K2CO3 (0,211 г, 1,525 ммоль) растворяли в ацетонитриле (2 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 8 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,177 г, 55,6%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 256
Метил 4-(((2S,6R)-4-(3-(1H-пиррол-1-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,050 г, 0,120 ммоль), гидроксиламин (0,146 мл, 2,395 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,067 г, 1,197 ммоль) растворяли в метаноле (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, и затем концентрировали при пониженном давлении с получением соединения 256 (0,031 г, 61,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,61 (д, 2 H, J=8,4 Гц), 7,41-7,30 (м, 7 H), 7,14 (д, 2 H, J=7,2 Гц), 6,22-6,21 (м, 2 H, J=2,4 Гц), 3,70 (с, 2 H), 3,42 (с, 2H), 2,66 (д, 2 H, J=10 Гц), 2,57-2,52 (м, 2 H), 1,83-1,77 (м, 2 H), 0,85 (д, 6 H, J=10,4 Гц).
Пример 100: Синтез соединения 257
(4-(((2S,6R)-4-(3-(фуран-2-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(3-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 7,000 г, 28,224 ммоль), 3-йодбензойную кислоту (8,145 г, 31,046 ммоль), EDCI (10,821 г, 56,447 ммоль), HOBt (7,628 г, 56,447 ммоль) и N,N-диизопропилэтиламин (18,238 г, 141,118 ммоль) растворяли в метиленхлориде (150 мл) при комнатной температуре, и реакционный раствор перемешивали в течение ночи при той же температуре. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 30% до 50%) и концентрировали с получением желаемого соединения (10,737 г, 77,3%) в виде твердого вещества белого цвета.
Стадия 2: Синтез метил 4-(((2S,6R)-4-(3-(фуран-2-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((2S,6R)-4-(3-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,400 г, 0,812 ммоль), фуран-2-илборной кислоты (0,136 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли 1н водный раствор соляной кислоты, после чего экстрагировали метиленхлоридом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 50%) и концентрировали с получением желаемого соединения (0,030 г, 8,5%) в виде масла желтого цвета.
Стадия 3: Синтез соединения 257
Метил 4-(((2S,6R)-4-(3-(фуран-2-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,030 г, 0,069 ммоль), гидроксиламин (0,084 мл, 1,378 ммоль, 50%-ный водный раствор) и гидроксид калия (0,039 г, 0,689 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 257 (0,012 г, 41,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,79-7,79 (м, 2 H), 7,77-7,68 (м, 3 H), 7,50 (т, 1 H, J=7,7 Гц), 7,41 (д, 2 H, J=7,9 Гц), 7,29 (д, 1 H, J=7,6 Гц), 7,07 (д, 1 H, J=3,3 Гц), 6,63-6,62 (м, 1 H), 4,30-4,27 (м, 1 H), 3,78 (с, 2 H), 3,01-2,99 (м, 1 H), 2,73-2,71 (м, 1 H), 2,56-2,51 (м, 3 H), 1,03 (с, 3 H), 0,82 (с, 3 H).
Пример 101: Синтез соединения 258
(4-(((2S,6R)-4-(3-(фуран-3-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(3-(фуран-3-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((2S,6R)-4-(3-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,400 г, 0,812 ммоль), фуран-3-илборной кислоты (0,136 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли 1н водный раствор соляной кислоты, после чего экстрагировали метиленхлоридом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 50%) и концентрировали с получением желаемого соединения (0,061 г, 17,4%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 258
Метил 4-(((2S,6R)-4-(3-(фуран-3-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,061 г, 0,141 ммоль), гидроксиламин (0,173 мл, 2,830 ммоль, 50%-ный водный раствор) и гидроксид калия (0,079 г, 1,415 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 258 (0,016 г, 25,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,29 (с, 1 H), 7,77 (с, 1 H), 7,72-7,68 (м, 3 H), 7,64 (с, 1 H), 7,47-7,41 (м, 3 H), 7,25 (д, 1 H, J=7,6 Гц), 7,03 (с, 1 H), 4,31-4,28 (м, 1 H), 3,79 (с, 2 H), 3,00-2,98 (м, 1 H), 2,71-2,68 (м, 1 H), 2,56-2,51 (м, 3 H), 1,03 (с, 3 H), 0,82 (с, 3 H).
Пример 102: Синтез соединения 259
(4-(((2S,6R)-4-(3-(3,6-дигидро-2H-пиран-4-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(3-(3,6-дигидро-2H-пиран-4-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((2S,6R)-4-(3-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,400 г, 0,812 ммоль), 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,256 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 20% до 65%) и концентрировали с получением желаемого соединения (0,290 г, 79,6%) в виде твердого вещества коричневого цвета.
Стадия 2: Синтез соединения 259
Метил 4-(((2S,6R)-4-(3-(3,6-дигидро-2H-пиран-4-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,060 г, 0,134 ммоль), гидроксиламин (0,164 мл, 2,675 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,075 г, 1,338 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 259 (0,010 г, 16,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,0 Гц), 7,54 (д, 1 H, J=7,9 Гц), 7,44-7,41 (м, 4 H), 7,28 (д, 1 H, J=7,7 Гц), 6,33 (с, 1 H), 4,24-4,23 (м, 3 H), 3,83 (т, 2 H, J=5,4 Гц), 3,78 (с, 2 H), 2,98-2,96 (м, 1 H), 2,70-2,68 (м, 1 H), 2,55-2,51 (м, 3 H), 2,46 (с, 2 H), 1,02 (с, 3 H), 0,82 (с, 3 H).
Пример 103: Синтез соединения 260
(4-(((2S,6R)-2,6-диметил-4-(3-(пиридин-4-ил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(3-(пиридин-4-ил)бензоил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((2S,6R)-4-(3-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,400 г, 0,812 ммоль), гидрата пиридин-4-бороновой кислоты (0,172 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 50% до 65%) и концентрировали с получением желаемого соединения (0,354 г, 98,3%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 260
Метил 4-(((2S,6R)-2,6-диметил-4-(3-(пиридин-4-ил)бензоил)пиперазин-1-ил)метил)бензоат (формула 15-2, 0,060 г, 0,135 ммоль), гидроксиламин (0,165 мл, 2,706 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,076 г, 1,353 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 260 (0,003 г, 5,5%) в виде твердого вещества бледно-коричневого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,67-8,66 (м, 2 H), 7,91 (д, 1 H, J=7,9 Гц), 7,81 (с, 1 H), 7,77-7,75 (м, 2 H), 7,68 (д, 2 H, J=8,2 Гц), 7,61 (т, 1 H, J=7,7 Гц), 7,49 (д, 1 H, J=7,6 Гц), 7,43-7,41 (м, 2H), 4,31-4,29 (м, 1 H), 3,79 (с, 2 H), 3,02-3,02 (м, 1 H), 2,73-2,68 (м, 1 H), 2,67-2,55 (м, 3 H), 1,04 (с, 3 H), 0,86 (с, 3 H).
Пример 104: Синтез соединения 261
(4-(((2S,6R)-2,6-диметил-4-(3-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(3-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((2S,6R)-4-(3-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,328 г, 0,665 ммоль), пиридин-3-илборной кислоты (0,090 г, 0,732 ммоль), Pd(dbpf)Cl2 (0,022 г, 0,033 ммоль) и Na2CO3 (0,155 г, 1,464 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 50% до 65%) и концентрировали с получением желаемого соединения (0,244 г, 82,8%) в виде твердого вещества коричневого цвета.
Стадия 2: Синтез соединения 261
Метил 4-(((2S,6R)-2,6-диметил-4-(3-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)бензоат (формула 15-2, 0,060 г, 0,135 ммоль), гидроксиламин (0,165 мл, 2,706 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,076 г, 1,353 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 261 (0,014 г, 23,3%)в виде твердого вещества бледно-коричневого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,91-8,90 (м, 1 H), 8,59-8,58 (м, 1 H), 8,12-8,09 (м, 1 H), 7,81 (д, 1 H, J=7,8 Гц), 7,72 (с, 1 H), 7,67 (д, 2 H, J=8,1 Гц), 7,56 (т, 1 H, J=7,7 Гц), 7,51-7,47 (м, 1 H), 7,43-7,39 (м, 3 H), 4,29-4,26 (м, 1 H), 3,77 (с, 2 H), 3,44-3,42 (м, 1 H), 3,01-2,99 (м, 1 H), 2,71-2,66 (м, 1 H), 2,58 (ушир. с, 2 H), 1,01 (с, 3 H), 0,81 (с, 3 H).
Пример 105: Синтез соединения 262
(4-(((2S,6R)-4-(3-(2-фторпиридин-4-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(3-(2-фторпиридин-4-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((2S,6R)-4-(3-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,400 г, 0,812 ммоль), (2-фторпиридин-4-ил)бороновой кислоты (0,172 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 50%) и концентрировали с получением желаемого соединения (0,366 г, 97,5%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 262
Метил 4-(((2S,6R)-4-(3-(2-фторпиридин-4-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,060 г, 0,130 ммоль), гидроксиламин (0,159 мл, 2,600 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,073 г, 1,300 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 262 (0,009 г, 14,1%) в виде твердого вещества бледно-коричневого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,77 (д, 1 H, J=8,0 Гц), 7,67-7,65 (м, 3 H), 7,54 (т, 1 H, J=7,7 Гц), 7,47-7,44 (м, 2 H), 7,38 (д, 2 H, J=8,1 Гц), 6,60-6,59 (м, 1 H), 6,51 (дд, 1 H, J=6,8, 1,6 Гц), 4,26-4,24 (м, 1 H), 3,77 (с, 2 H), 2,99-2,97 (м, 1 H), 2,72-2,70 (м, 1 H), 2,66-2,54 (м, 3 H), 1,01 (с, 3 H), 0,81 (с, 3 H).
Пример 106: Синтез соединения 263
(4-(((2S,6R)-4-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-3-карбонил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-3-карбонил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((2S,6R)-4-(3-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,400 г, 0,812 ммоль), 2-(4,4-диметилциклогекс-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,288 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль), и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 20%) и концентрировали с получением желаемого соединения (0,065 г, 16,8%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 263
Метил 4-(((2S,6R)-4-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-3-карбонил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,065 г, 0,137 ммоль), гидроксиламин (0,167 мл, 2,735 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,077 г, 1,367 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 263 (0,044 г, 68,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,66 (д, 2 H, J=8,1 Гц), 7,49 (д, 1 H, J=8,0 Гц), 7,38-7,34 (м, 4 H), 7,21 (д, 1 H, J=7,6 Гц), 6,15-6,13 (м, 1 H), 4,24-4,22 (м, 1 H), 3,76 (с, 2 H), 2,95-2,93 (м, 1 H), 2,67-2,50 (м, 4 H), 2,37 (с, 2 H), 1,96 (с, 2 H), 1,47 (т, 2 H, J=6,3 Гц), 1,05 (с, 3 H), 0,92 (с, 6 H), 0,80 (с, 3 H).
Пример 107: Синтез соединения 265
(4-(((3R,5S)-3,5-диметил-4-(нафталин-2-илметил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(нафталин-2-илметил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,150 г, 0,572 ммоль), Cs2CO3 (0,279 г, 0,858 ммоль) и 2-(бромметил)нафталин (0,126 г, 0,572 ммоль) растворяли в ацетонитриле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 48 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Полученный продукт использовали без дополнительной очистки (0,230 г, 99,9%).
Стадия 2: Синтез соединения 265
Метил 4-(((3R,5S)-3,5-диметил-4-(нафталин-2-илметил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,230 г, 0,571 ммоль), гидроксиламин (0,699 мл, 11,428 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,321 г, 5,714 ммоль) растворяли в смеси метанол (3 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат кристаллизовали из метанола (5 мл) с получением соединения 265 (0,042 г, 18,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,87-7,82 (м, 4 H), 7,66 (д, 2 H, J=8,0 Гц), 7,50 (д, 1 H, J=9,0 Гц), 7,47-7,44 (м, 2 H), 7,23 (д, 2 H, J=8,0 Гц), 3,88 (с, 2 H), 3,41 (с, 2 H), 2,67-2,61 (м, 4 H), 1,84 (т, 2 H, J=10,1 Гц), 0,93 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 404,2 (M++1).
Пример 108: Синтез соединения 266
(4-(((3R,5S)-3,5-диметил-4-(пиридин-4-илметил)пиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(пиридин-4-илметил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,150 г, 0,572 ммоль), Cs2CO3 (0,279 г, 0,858 ммоль) и 4-(бромметил)пиридин (0,098 г, 0,572 ммоль) растворяли в ацетонитриле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,200 г, 99,0%).
Стадия 2: Синтез соединения 266
Метил 4-(((3R,5S)-3,5-диметил-4-(пиридин-4-илметил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,200 г, 0,566 ммоль), гидроксиламин (0,692 мл, 11,317 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,317 г, 5,658 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали с получением желаемого соединения 266 (0,019 г, 9,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CH3OD) δ 8,77 (д, 2 H, J=6,7 Гц), 8,17 (д, 2 H, J=6,7 Гц), 7,90 (д, 2 H, J=8,3 Гц), 7,67 (д, 2 H, J=8,3 Гц), 4,45 (с, 2 H), 4,18 (с, 2 H), 3,41 (д, 2 H, J=11,4 Гц), 3,03-3,00 (м, 4 H), 1,04 (д, 6 H, J=6,0 Гц).
Пример 109: Синтез соединения 267
(4-(((3R,5S)-4-(3-хлор-2-(трифторметил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-хлор-2-(трифторметил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,150 г, 0,572 ммоль), Cs2CO3 (0,279 г,0,858 ммоль) и 1-(бромметил)-3-хлор-2-(трифторметил)бензол (0,156 г, 0,572 ммоль) растворяли в ацетонитриле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением желаемого соединения (0,203 г, 78,0%).
Стадия 2: Синтез соединения 267
Метил 4-(((3R,5S)-4-(3-хлор-2-(трифторметил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,100 г, 0,220 ммоль), гидроксиламин (0,269 мл, 4,396 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,123 г, 2,198 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 267 (0,044 г, 43,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 9,80 (ушир. с, 2 H), 7,82 (с, 1 H), 7,71-7,65 (м, 4 H), 7,33 (д, 2 H, J=3,9 Гц), 4,58 (с, 2 H), 3,44-3,35 (м, 3 H), 2,67 (д, 2 H, J=10,3 Гц), 2,60 (с, 2 H), 1,82 (т, 2 H, J=9,4 Гц), 0,83 (с, 6 H); ЖХМС (ES) m/z 456,1 (M++1).
Пример 110: Синтез соединения 268
(4-(((2S,6R)-2,6-диметил-4-(нафталин-2-илметил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(нафталин-2-илметил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,502 ммоль), Cs2CO3 (0,245 г, 0,753 ммоль) и 2-(бромметил)нафталин (0,111 г, 0,502 ммоль) растворяли в ацетонитриле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,200 г, 99,0%).
Стадия 2: Синтез соединения 268
Метил 4-(((2S,6R)-2,6-диметил-4-(нафталин-2-илметил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,200 г, 0,497 ммоль), гидроксиламин (0,608 мл, 9,937 ммоль) и гидроксид калия (0,279 г, 4,969 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (3 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили, и полученный продукт очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением соединения 268 (0,075 г, 37,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,13 (с, 1 H), 8,99 (с, 1 H), 7,90-7,86 (м, 3 H), 7,78 (с, 1 H), 7,67 (д, 2 H, J=7,2 Гц), 7,50-7,47 (м, 3 H), 7,42 (д, 2 H, J=7,8 Гц), 3,76 (с, 2 H), 3,57 (с, 2 H), 2,71 (д, 2 H, J=10,6 Гц), 2,60-2,56 (м, 2 H), 1,86 (т, 2 H, J=10,5 Гц), 0,88 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 404,2 (M++1).
Пример 111: Синтез соединения 270
(N-гидрокси-4-(((3R,5S)-4-изопентил-3,5-диметилпиперазин-1-ил)метил)бензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-4-изопентил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,150 г, 0,572 ммоль), Cs2CO3 (0,279 г, 0,858 ммоль) и 1-бром-3-метилбутан (0,086 мл, 0,720 ммоль) растворяли в N,N-диметилформамиде (100 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,190 г, 99,9%).
Стадия 2: Синтез соединения 270
Метил 4-(((3R,5S)-4-изопентил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,190 г, 0,571 ммоль), гидроксиламин (0,699 мл, 11,429 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,321 г, 5,715 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали с получением соединения 270 (0,025 г, 13,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,23 (ушир. с, 1 H), 9,10 (ушир. с, 1 H), 7,75 (д, 2 H, J=8,2 Гц), 7,42 (д, 2 H, J=8,1 Гц), 3,71 (с, 2 H), 3,45-3,45 (м, 2 H), 3,18-3,18 (м, 2 H), 3,01 (д, 2 H, J=11,3 Гц), 2,28 (т, 2 H, J=11,3 Гц), 1,67-1,60 (м, 1 H), 1,52-1,48 (м, 2 H), 1,22 (д, 6 H, J=6,3 Гц), 0,93 (д, 6 H, J=6,6 Гц); ЖХМС (ES) m/z 334,2 (M++1).
Пример 112: Синтез соединения 271
(N-гидрокси-4-(((2S,6R)-4-изопентил-2,6-диметилпиперазин-1-ил)метил)бензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((2S,6R)-4-изопентил-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,502 ммоль), Cs2CO3 (0,245 г, 0,753 ммоль) и 1-бром-3-метилбутан (0,060 мл, 0,502 ммоль) растворяли в ацетонитриле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,170 г, 101,9%).
Стадия 2: Синтез соединения 271
Метил 4-(((2S,6R)-4-изопентил-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,170 г, 0,511 ммоль), гидроксиламин (0,626 мл, 10,226 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,287 г, 5,113 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали с получением соединения 271 (0,097 г, 56,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,18 (ушир. с, 1 H), 9,00 (ушир. с, 1 H), 7,67 (д, 2 H, J=8,3 Гц), 7,41 (д, 2 H, J=8,2 Гц), 3,74 (с, 2 H), 2,70 (д, 2 H, J=10,8 Гц), 2,56-2,53 (м, 2 H), 2,19 (т, 2 H, J=7,6 Гц), 1,70 (т, 2 H, J=10,6 Гц), 1,57-1,54 (м, 1 H), 1,32-1,26 (м, 2 H), 0,89 (д, 6 H, J=6,2 Гц), 0,86 (д, 6 H, J=6,6 Гц); ЖХМС (ES) m/z 334,2 (M++1).
Пример 113: Синтез соединения 272
(4-(((3R,5S)-3,5-диметил-4-(3-метилбут-2-ен-1-ил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-метилбут-2-ен-1-ил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,150 г, 0,572 ммоль), Cs2CO3 (0,279 г, 0,858 ммоль) и 1-бром-3-метил-2-бутен (0,067 мл, 0,572 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и реакционный раствор перемешивали при температуре 100°C в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,123 г, 65,1%) в сыром виде.
Стадия 2: Синтез соединения 272
Метил 4-(((3R,5S)-3,5-диметил-4-(3-метилбут-2-ен-1-ил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,070 г, 0,212 ммоль), гидроксиламин (0,259 мл, 4,237 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,119 г, 2,118 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 272 (0,030 г, 42,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,10 (ушир. с, 1 H), 9,00 (ушир. с, 1 H), 7,69 (д, 2 H, J=8,0 Гц), 7,33 (д, 2 H, J=8,0 Гц), 5,30 (с, 1 H), 3,40 (с, 2 H), 3,35 (с, 2 H), 3,29 (д, 2 H, J=6,1 Гц), 2,62-2,60 (м, 4 H), 1,74-1,72 (м, 2 H), 1,69 (с, 3 H), 1,60 (с, 3 H), 0,92 (д, 6 H, J=7,0 Гц); ЖХМС (ES) m/z 332,2 (M++1).
Пример 114: Синтез соединения 273
(N-гидрокси-4-(((2S,6R)-4-изопропил-2,6-диметилпиперазин-1-ил)метил)бензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((2S,6R)-4-изопропил-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,502 ммоль), Cs2CO3 (0,245 г, 0,753 ммоль) и 2-йодпропан (0,050 мл, 0,502 ммоль) растворяли в ацетонитриле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,150 г, 98,2%).
Стадия 2: Синтез соединения 273
Метил 4-(((2S,6R)-4-изопропил-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,150 г, 0,493 ммоль), гидроксиламин (0,603 мл, 10,226 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,276 г, 4,927 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали с получением соединения 273 (0,097 г, 64,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,20 (с, 1 H), 9,36 (с, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,46 (д, 2 H, J=8,2 Гц), 3,93 (с, 2 H), 3,44-3,41 (м, 1 H), 3,35 (д, 2 H, J=11,6 Гц), 2,92-2,90 (м, 2 H), 2,77-2,74 (м, 2 H), 1,25 (д, 6 H, J=6,6 Гц), 1,08 (д, 6 H, J=4,8 Гц); ЖХМС (ES) m/z 306,2 (M++1).
Пример 115: Синтез соединения 274
(4-(((2S,6R)-4-бутил-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((2S,6R)-4-бутил-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,150 г, 0,502 ммоль), Cs2CO3 (0,245 г, 0,753 ммоль) и 1-йодбутан (0,057 мл, 0,502 ммоль) растворяли в ацетонитриле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,160 г, 100,1%).
Стадия 2: Синтез соединения 274
Метил 4-(((2S,6R)-4-бутил-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,160 г, 0,502 ммоль), гидроксиламин (0,615 мл, 10,049 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,282 г, 5,024 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали с получением соединения 274 (0,087 г, 54,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, D2O) δ 7,66 (д, 2 H, J=8,3 Гц), 7,51 (д, 2 H, J=8,4 Гц), 4,40 (с, 2 H), 3,58 (д, 2 H, J=12,4 Гц), 3,37-3,36 (м, 2 H), 3,07-2,97 (м, 4 H), 1,57-1,54 (м, 2 H), 1,39 (д, 6 H, J=6,3 Гц), 1,26-1,20 (м, 2 H), 0,79 (т, 3 H, J=7,4 Гц); ЖХМС (ES) m/z 320,2 (M++1).
Пример 116: Синтез соединения 275
(4-(((3R,5S)-4-бутил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-4-бутил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,150 г, 0,502 ммоль), Cs2CO3 (0,245 г, 0,753 ммоль) и 1-йодбутан (0,057 мл, 0,502 ммоль) растворяли в ацетонитриле (3 мл), и реакционную смесь нагревали при кипячении с обратным холодильником в течение 17 часов, и затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,120 г, 98,9%).
Стадия 2: Синтез соединения 275
Метил 4-(((3R,5S)-4-бутил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,120 г, 0,377 ммоль), гидроксиламин (0,461 мл, 7,537 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,211 г, 3,768 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали с получением желаемого соединения 275 (0,088 г, 73,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, D2O) δ 7,66 (д, 2 H, J=8,3 Гц), 7,47 (д, 2 H, J=8,2 Гц), 4,15-4,15 (м, 2 H), 3,57-3,56 (м, 2 H), 3,44 (д, 2 H, J=13,2 Гц), 3,22 (т, 2 H, J=8,5 Гц), 2,91-2,90 (м, 2 H), 1,58-1,54 (м, 2 H), 1,28 (д, 6 H, J=6,4 Гц), 0,82 (т, 3 H, J=7,4 Гц); ЖХМС (ES) m/z 320,2 (M++1).
Пример 117: Синтез соединения 276
(4-(((2S,6R)-4-(2-(фуран-2-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 4,000 г, 16,128 ммоль), 2-йодбензойная кислота (4,654 г, 17,741 ммоль), EDCI (6,183 г, 32,255 ммоль), HOBt (4,359 г, 32,255 ммоль) и N,N-диизопропилэтиламин (10,422 г, 80,639 ммоль) растворяли в метиленхлориде (100 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 30% до 50%) и концентрировали с получением желаемого соединения (7,630 г, 96,1%) в виде твердого вещества белого цвета.
Стадия 2: Синтез метил 4-(((2S,6R)-4-(2-(фуран-2-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,400 г, 0,812 ммоль), фуран-2-илборной кислоты (0,136 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г,0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 30%) и концентрировали с получением желаемого соединения (0,211 г, 60,1%) в виде твердого вещества белого цвета.
Стадия 3: Синтез соединения 276
Метил 4-(((2S,6R)-4-(2-(фуран-2-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,060 г, 0,139 ммоль), гидроксиламин (0,170 мл, 2,775 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,078 г, 1,387 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 276 (0,017 г, 27,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,80-7,65 (м, 4 H), 7,50-7,47 (м, 1 H), 7,40-7,30 (м, 3 H), 7,26 (т, 1 H, J=6,8 Гц), 6,69 (дд, 1 H, J=27,4, 9,3 Гц), 6,61-6,51 (м, 1 H), 4,36-4,33 (м, 1 H), 3,76-3,61 (м, 2 H), 3,09-2,99 (м, 1 H), 2,83-2,77 (м, 1 H), 2,69-2,55 (м, 3 H), 1,03 (д, 3 H, J=5,9 Гц), 0,70 (м, 3 H).
Пример 118: Синтез соединения 277
(4-(((2S,6R)-4-(2-(фуран-3-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-(фуран-3-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 15-1, 0,400 г, 0,812 ммоль), фуран-3-илборной кислоты (0,136 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 30%) и концентрировали с получением желаемого соединения (0,186 г, 52,8%) в виде твердого вещества коричневого цвета.
Стадия 2: Синтез соединения 277
Метил 4-(((2S,6R)-4-(2-(фуран-3-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,060 г, 0,139 ммоль), гидроксиламин (0,170 мл, 2,775 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,078 г, 1,387 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 277 (0,023 г, 37,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,82 (с, 1 H), 7,77-7,64 (м, 3 H), 7,58-7,52 (м, 1 H), 7,46 (т, 1 H, J=7,5 Гц), 7,36 (кв, 2 H, J=8,1 Гц), 7,29-7,25 (м, 2 H), 6,67-6,66 (м, 1 H), 4,31 (д, 1 H, J=10,5 Гц), 3,74 (с, 1 H), 3,62 (кв, 2 H, J=18,9 Гц), 2,90 (м, 1 H), 2,76-2,68 (м, 1 H), 2,62-2,56 (м, 2 H), 1,00-0,98 (м, 3 H), 0,66 (м, 3 H).
Пример 119: Синтез соединения 278
(4-(((2S,6R)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-4-(2-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-1, 0,400 г, 0,812 ммоль), 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,256 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г,0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) растворяли в смеси 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) при комнатной температуре, и реакционный раствор перемешивали в течение ночи при температуре 100°C. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 50%) и концентрировали с получением желаемого соединения (0,272 г, 74,6%) в виде твердого вещества коричневого цвета.
Стадия 2: Синтез соединения 278
Метил 4-(((2S,6R)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,060 г, 0,134 ммоль), гидроксиламин (0,164 мл, 2,675 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,075 г, 1,338 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 278 (0,049 г, 81,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, 2 H, J=8,0 Гц), 7,45-7,39 (м, 3 H), 7,31 (т, 2 H, J=7,5 Гц), 7,23-7,18 (м, 1 H), 5,83-5,76 (м, 1 H), 4,32-4,29 (м, 1 H), 4,15-4,10 (м, 2 H), 3,78-3,73 (м, 4 H), 3,06 (д, 1 H, J=12,6 Гц), 2,78 (кв, 1 H, J=10,9 Гц), 2,67-2,56 (м, 2 H), 2,24-2,10 (м, 1 H), 1,03-1,02 (м, 3 H), 0,80-0,78 (м, 3 H).
Пример 120: Синтез соединения 279
(4-(((2S,6R)-2,6-диметил-4-(2-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(2-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-4-(2-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-1, 0,400 г, 0,812 ммоль), пиридин-3-илбороновую кислоту (0,150 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) растворяли в смеси 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) при комнатной температуре, и реакционный раствор перемешивали в течение ночи при температуре 100°C. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 50% до 60%) и концентрировали с получением желаемого соединения (0,348 г, 96,7%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 279
Метил 4-(((2S,6R)-2,6-диметил-4-(2-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)бензоат (формула 15-2, 0,070 г, 0,158 ммоль), гидроксиламин (0,193 мл, 3,156 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,089 г, 1,578 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 279 (0,062 г, 88,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,62-8,53 (м, 2 H), 7,82-7,76 (м, 1 H), 7,67 (д, 2 H, J=8,0 Гц), 7,60-7,43 (м, 4 H), 7,34 (дд, 3 H, J=36,4, 7,5 Гц), 4,20 (д, 1 H, J=12,5 Гц), 3,72-3,60 (м, 1 H), 3,55-3,41 (м, 1 H), 3,06-2,86 (м, 1 H), 2,66-2,63 (м, 1 H), 2,28-2,22 (м, 1 H), 1,99 (ушир. с, 1 H), 1,06 (ушир. с, 1 H), 0,92-0,91 (м, 3 H), 0,68-0,62 (м, 3 H).
Пример 121: Синтез соединения 280
(4-(((2S,6R)-4-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-карбонил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-карбонил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-4-(2-йодбензоил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-1, 0,400 г, 0,812 ммоль), 2-(4,4-диметилциклогекс-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,288 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) растворяли в смеси 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) при комнатной температуре, и реакционный раствор перемешивали в течение ночи при температуре 100°C. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 10% до 20%) и концентрировали с получением желаемого соединения (0,274 г, 71,1%) в виде твердого вещества коричневого цвета.
Стадия 2: Синтез соединения 280
Метил 4-(((2S,6R)-4-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-карбонил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 15-2, 0,060 г, 0,135 ммоль), гидроксиламин (0,165 мл, 2,706 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,076 г, 1,353 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 280 (0,049 г, 75,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,69-7,67 (м, 2 H), 7,38-7,34 (м, 3 H), 7,31-7,24 (м, 2 H), 7,16 (т, 1 H, J=8,7 Гц), 5,56 (с, 1 H), 4,29 (дд, 1 H, J=31,1, 13,2 Гц), 3,81-3,71 (м, 2 H), 3,09 (т, 1 H, J=14,5 Гц), 2,84-2,62 (м, 2 H), 2,46-2,33 (м, 3 H), 2,10 (ушир. с, 1 H), 1,88-1,73 (м, 2 H), 1,44-1,38 (м, 2 H), 1,05-1,00 (м, 3 H), 0,95-0,89 (м, 6 H), 0,81-0,67 (м, 3 H).
Пример 122: Синтез соединения 283
(4-(((3R,5S)-4-(2-(фуран-2-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез 2-йодбензоилхлорида
2-Йодбензойную кислоту (4,000 г, 16,128 ммоль) и SOCl2 (23,399 мл, 322,555 ммоль) перемешивали в течение ночи при температуре 100°C, и затем реакционную смесь концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (4,295 г, 99,9%, масло коричневого цвета).
Стадия 2: Синтез метил 4-(((3R,5S)-4-(2-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 4,295 г, 16,119 ммоль), 2-йодбензоилхлорид (4,652 г, 17,731 ммоль) и TEA (4,469 мл, 32,237 ммоль) растворяли в метиленхлориде (70 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 30% до 50%) и концентрировали с получением желаемого соединения (7,120 г, 89,7%) в виде твердого вещества белого цвета.
Стадия 3: Синтез метил 4-(((3R,5S)-4-(2-(фуран-2-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,400 г, 0,812 ммоль), фуран-2-илборной кислоты (0,136 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 0% до 15%) и концентрировали с получением желаемого соединения (0,040 г, 11,4%) в виде бесцветного масла.
Стадия 4: Синтез соединения 283
Метил 4-(((3R,5S)-4-(2-(фуран-2-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 4-2, 0,040 г, 0,092 ммоль), гидроксиламин (0,113 мл, 1,850 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,052 г, 0,925 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 283 (0,016 г, 39,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,78-7,68 (м, 4 H), 7,46 (т, 1 H, J=7,6 Гц), 7,41-7,21 (м, 4 H), 6,73 (дд, 1 H, J=33,6, 3,4 Гц), 6,61-6,59 (м, 1 H), 4,70-4,65 (м, 1 H), 3,56-3,41 (м, 3 H), 2,72-2,66 (м, 1 H), 2,41-2,33 (м, 1 H), 2,18-2,03 (м, 1 H), 1,63-1,60 (м, 1 H), 1,33 (т, 3 H, J=9,8 Гц), 1,16 (д, 2 H, J=6,8 Гц), 0,86 (д, 1 H, J=6,8 Гц).
Пример 123: Синтез соединения 284
(4-(((3R,5S)-4-(2-(фуран-3-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-(фуран-3-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,400 г, 0,812 ммоль), фуран-3-илборной кислоты (0,136 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль), и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 0% до 10%) и концентрировали с получением желаемого соединения (0,128 г, 36,5%) в виде бесцветного масла.
Стадия 2: Синтез соединения 284
Метил 4-(((3R,5S)-4-(2-(фуран-3-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 4-2, 0,128 г, 0,296 ммоль), гидроксиламин (0,362 мл, 5,919 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,166 г, 2,959 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 284 (0,038 г, 29,5%)в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,85-7,75 (м, 2 H), 7,68 (д, 2 H, J=8,2 Гц), 7,60-7,51 (м, 1 H), 7,46-7,35 (м, 2 H), 7,33-7,21 (м, 3 H), 6,82-6,66 (м, 1 H), 4,69-4,54 (м, 1 H), 3,52-3,49 (м, 1 H), 3,27-3,25 (м, 1 H), 2,70-2,56 (м, 1 H), 2,37-2,34 (м, 1 H), 2,16-2,04 (м, 1 H), 1,86-1,82 (м, 1 H), 1,39-1,21 (м, 4 H), 1,12 (д, 2 H, J=6,8 Гц), 0,82 (д, 1 H, J=6,8 Гц).
Пример 124: Синтез соединения 285
(4-(((3R,5S)-3,5-диметил-4-(2-(пиридин-4-ил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2-(пиридин-4-ил)бензоил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,400 г, 0,812 ммоль), гидрата пиридин-4-бороновой кислоты (0,172 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль), и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 50% до 60%) и концентрировали с получением желаемого соединения (0,129 г, 35,9%) в виде твердого вещества бледно-желтого цвета.
Стадия 2: Синтез соединения 285
Метил 4-(((3R,5S)-3,5-диметил-4-(2-(пиридин-4-ил)бензоил)пиперазин-1-ил)метил)бензоат (формула 4-2, 0,060 г, 0,135 ммоль), гидроксиламин (0,165 мл, 2,706 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,076 г, 1,353 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 285 (0,016 г, 26,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,64 (д, 2 H, J=5,8 Гц), 7,67 (д, 2 H, J=8,0 Гц), 7,61-7,41 (м, 6 H), 7,30 (д, 1 H, J=8,0 Гц), 4,34 (ушир. с, 1 H), 3,24-3,17 (м, 3 H), 2,24 (д, 1 H, J=11,0 Гц), 1,44-1,40 (м, 1 H), 1,29 (д, 3 H, J=6,7 Гц), 1,07 (д, 3 H, J=6,6 Гц).
Пример 125: Синтез соединения 286
(4-(((3R,5S)-3,5-диметил-4-(2-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил ((3R,5S)-3,5-диметил-4-(2-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,400 г, 0,812 ммоль), пиридин-3-илборной кислоты (0,150 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль), и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=50%) и концентрировали с получением желаемого соединения (0,292 г, 81,1%) в виде твердого вещества желтого цвета.
Стадия 2: Синтез соединения 286
Метил ((3R,5S)-3,5-диметил-4-(2-(пиридин-3-ил)бензоил)пиперазин-1-ил)метил)бензоат (формула 4-2, 0,060 г, 0,135 ммоль), гидроксиламин (0,165 мл, 2,706 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,076 г, 1,353 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 286 (0,050 г, 83,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,58-8,57 (м, 2 H), 7,78 (д, 1 H, J=7,9 Гц), 7,68 (д, 2 H, J=8,1 Гц), 7,57-7,43 (м, 5 H), 7,30 (д, 2 H, J=7,9 Гц), 4,31-4,29 (м, 1 H), 3,26-3,18 (м, 3 H), 2,23 (д, 1 H, J=11,2 Гц), 1,37-1,35 (м, 1 H), 1,28 (д, 3 H, J=6,7 Гц), 1,07 (д, 3 H, J=6,7 Гц).
Пример 126: Синтез соединения 288
(4-(((3R,5S)-4-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,400 г, 0,812 ммоль), 2-(4,4-диметилциклогекс-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,288 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г,0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 0% до 10%) и концентрировали с получением желаемого соединения (0,143 г, 37,0%) в виде твердого вещества коричневого цвета.
Стадия 2: Синтез соединения 288
Метил 4-(((3R,5S)-4-(4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 4-3, 0,060 г, 0,126 ммоль), гидроксиламин (0,155 мл, 2,528 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,071 г, 1,264 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 288 (0,047 г, 78,0%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,71 (д, 2 H, J=8,0 Гц), 7,36-7,26 (м, 4 H), 7,21-7,19 (м, 2 H), 5,55 (с, 1 H), 4,48 (ушир. с, 1 H), 3,50-3,47 (м, 2 H), 2,69 (д, 1 H, J=11,0 Гц), 2,40-2,37 (м, 1 H), 2,12-1,98 (м, 3 H), 1,86-1,81 (м, 2 H), 1,41-1,38 (м, 2 H), 1,34 (д, 3 H, J=6,6 Гц), 1,17 (д, 3 H, J=6,6 Гц), 0,93-0,91 (м, 6 H).
Пример 127: Синтез соединения 290
(4-(((3R,5S)-3,5-диметил-4-(4-метилбензоил)пиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-метилбензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 4-метилбензоилхлорид (0,060 мл, 0,457 ммоль) и TEA (0,077 г, 0,762 ммоль) растворяли в метиленхлориде (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 3 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,078 г, 53,8%) в виде твердого вещества бледно-желтого цвета.
Стадия 2: Синтез соединения 290
Метил 4-(((3R,5S)-3,5-диметил-4-(4-метилбензоил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,131 ммоль), гидроксиламин (0,161 мл, 2,628 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,074 г, 1,314 ммоль) растворяли в метаноле (0,5 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционный раствор добавляли насыщенный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%) и концентрировали с получением соединения 290 (0,019 г, 37,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, D2O) δ 7,67 (д, 2 H, J=8,4 Гц), 7,53 (д, 2 H, J=8,3 Гц), 7,21 (д, 2 H, J=7,8 Гц), 7,17 (д, 2 H, J=8,2 Гц), 4,38 (с, 2 H), 3,20 (м, 4 H), 2,24 (с, 3 H), 1,29 (ушир. с, 6 H).
Пример 128: Синтез соединения 291
(N-гидрокси-4-(((3R,5S)-4-(4-метоксибензоил)-3,5-диметилпиперазин-1-ил)метил)бензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-метоксибензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 4-метоксибензоилхлорид (0,062 мл, 0,457 ммоль) и TEA (0,077 г, 0,762 ммоль) растворяли в метиленхлориде (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 3 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,088 г, 58,2%) в виде твердого вещества бледно-желтого цвета.
Стадия 2: Синтез соединения 291
Метил 4-(((3R,5S)-4-(4-метоксибензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,126 ммоль), гидроксиламин (0,154 мл, 2,522 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,071 г, 1,261 ммоль) растворяли в метаноле (0,5 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%) и концентрировали с получением соединения 291 (0,023 г, 45,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, D2O) δ 7,67 (д, 2 H, J=8,4 Гц), 7,52 (д, 2 H, J=8,3 Гц), 7,24 (д, 2 H, J=8,8 Гц), 6,93 (д, 2 H, J=8,8 Гц), 4,38 (с, 2 H), 3,73 (с, 3 H), 3,36-3,20 (м, 4 H), 1,29 (ушир. с, 6 H).
Пример 129: Синтез соединения 292
(4-(((3R,5S)-3,5-диметил-4-пивалоилпиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4- пивалоилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), пивалоилхлорид (0,055 г, 0,457 ммоль) и TEA (0,077 г, 0,762 ммоль) растворяли в метиленхлориде (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 3 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,105 г, 79,5%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 292
Метил 4-(((3R,5S)-3,5-диметил-4-пивалоилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,144 ммоль), гидроксиламин (0,177 мл, 2,886 ммоль) и гидроксид калия (0,081 г, 1,443 ммоль) растворяли в метаноле (0,5 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%) и концентрировали с получением желаемого соединения 292 (0,018 г, 35,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, D2O) δ 7,66 (д, 2 H, J=8,3 Гц), 7,52 (д, 2 H, J=8,2 Гц), 4,75-4,73 (м, 2 H), 4,35 (с, 2 H), 3,34 (м, 2 H), 3,13 (м, 2 H), 1,31 (ушир. с, 6 H), 1,12 (с, 9 H).
Пример 130: Синтез соединения 293
(4-(((3R,5S)-3,5-диметил-4-пропионилпиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-пропионилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), пропионилхлорид (0,042 г, 0,457 ммоль) и TEA (0,077 г, 0,762 ммоль) растворяли в метиленхлориде (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 3 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,113 г, 93,1%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 293
Метил 4-(((3R,5S)-3,5-диметил-4-пропионилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,157 ммоль), гидроксиламин (0,192 мл, 3,141 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,088 г, 1,570 ммоль) растворяли в метаноле (0,5 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%) и концентрировали с получением соединения 293 (0,027 г, 53,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, D2O) δ 7,66 (д, 2 H, J=7,9 Гц), 7,52 (д, 2 H, J=8,0 Гц), 4,75-4,73 (м, 2 H), 4,37 (с, 2 H), 3,40-3,37 (м, 2 H), 3,12-3,08 (м, 2 H), 2,42-2,27 (м, 2 H), 1,31 (ушир. с, 6 H), 0,95 (т, 3 H, J=11,6 Гц).
Пример 131: Синтез соединения 294
(4-(((3R,5S)-4-бутирил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-4-бутирил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), бутирил хлорид (0,049 г, 0,457 ммоль) и TEA (0,077 г, 0,762 ммоль) растворяли в метиленхлориде (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 3 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,077 г, 60,8%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 294
Метил 4-(((3R,5S)-4-бутирил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,150 ммоль), гидроксиламин (0,184 мл, 3,008 ммоль) и гидроксид калия (0,084 г, 1,504 ммоль) растворяли в метаноле (0,5 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%) и концентрировали с получением соединения 294 (0,024 г, 47,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, D2O) δ 7,67 (д, 2 H, J=6,5 Гц), 7,52 (д, 2 H, J=6,1 Гц), 4,73-4,71 (м, 2 H), 4,35 (с, 2 H), 3,40-3,37 (м, 2 H), 3,11-3,09 (м, 2 H), 2,35-2,23 (м, 2 H), 1,44 (кв, 6 H, J=14,8, 7,4 Гц), 1,33 (ушир. с, 3 H), 1,22 (ушир. с, 3 H), 0,77 (т, 3 H, J=7,4 Гц).
Пример 132: Синтез соединения 295
(4-(((3R,5S)-4-(циклопропанкарбонил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-4-(циклопропанкарбонил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), циклопропанкарбонилхлорид (0,042 мл, 0,457 ммоль) и TEA (0,077 г, 0,762 ммоль) растворяли в метиленхлориде (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 3 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,105 г, 83,4%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 295
Метил 4-(((3R,5S)-4-(циклопропанкарбонил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,151 ммоль), гидроксиламин (0,185 мл, 3,026 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,085 г, 1,513 ммоль) растворяли в метаноле (0,5 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%) и концентрировали с получением соединения 295 (0,021 г, 41,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, D2O) δ 7,66 (д, 2 H, J=8,2 Гц), 7,53 (д, 2 H, J=8,2 Гц), 4,73-4,71 (м, 2 H), 4,38 (с, 2 H), 3,43-3,40 (м, 2 H), 3,14-3,12 (м, 2 H), 1,83-1,25 (м, 1 H), 1,28 (ушир. с, 6 H), 0,79-0,75 (м, 4 H).
Пример 133: Синтез соединения 296
(N-гидрокси-4-(((3R,5S)-4-изобутирил-3,5-диметилпиперазин-1-ил)метил)бензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-4-изобутирил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), изобутирил хлорид (0,048 мл, 0,457 ммоль) и TEA (0,077 г, 0,762 ммоль) растворяли в метиленхлориде (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 3 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,097 г, 76,5%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 296
Метил 4-(((3R,5S)-4-изобутирил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,150 ммоль), гидроксиламин (0,184 мл, 3,008 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,084 г, 1,504 ммоль) растворяли в метаноле (0,5 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%) и концентрировали с получением соединения 296 (0,025 г, 49,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, D2O) δ 7,67 (д, 2 H, J=8,2 Гц), 7,52 (д, 2 H, J=8,2 Гц), 4,73 (м, 1 H), 4,49 (м, 1 H), 4,37 (с, 2 H), 3,41-3,38 (м, 2 H), 3,13-3,09 (м, 2 H), 2,83-2,79 (м, 1 H), 1,33 (ушир. с, 3 H), 1,22 (ушир. с, 3 H), 0,95-0,93 (м, 6 H).
Пример 134: Синтез соединения 297
(4-(((3R,5S)-3,5-диметил-4-(3-метилбутаноил)пиперазин-1-ил)метил)-N-гидроксибензамид (соль трифторуксусной кислоты))
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-метилбутаноил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 3-метилбутаноилхлорид (55 мг, 0,457 ммоль) и TEA (0,077 г, 0,762 ммоль) растворяли в метиленхлориде (1 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 3 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением желаемого соединения (0,078 г, 59,1%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 297
Метил 4-(((3R,5S)-3,5-диметил-4-(3-метилбутаноил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,050 г, 0,144 ммоль), гидроксиламин (0,177 мл, 2,886 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,081 г, 1,443 ммоль) растворяли в метаноле (0,5 мл) при температуре 25°C, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%) и концентрировали с получением желаемого соединения 297 (0,021 г, 41,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, D2O) δ 7,66 (д, 2 H, J=8,3 Гц), 7,52 (д, 2 H, J=8,3 Гц), 4,78-4,75 (м, 1 H), 4,44-4,43 (м, 1 H), 4,37 (с, 2 H), 3,40-3,38 (м, 2 H), 3,10-3,09 (м, 2 H), 2,32-2,08 (м, 2 H), 1,91-1,84 (м, 1 H), 1,32 (ушир. с, 3 H), 1,25 (ушир. с, 1 H), 0,82-0,72 (м, 6 H).
Пример 135: Синтез соединения 298
(4-(((2S,6R)-2,6-диметил-4-(3-(трифторметил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(3-(трифторметил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,200 г, 0,762 ммоль), 3-(трифторметил)бензальдегид (0,133 г, 0,762 ммоль) и уксусную кислоту (0,022 мл, 0,762 ммоль) растворяли в 1,2-этиленхлориде (4 мл) и перемешивали при комнатной температуре в течение 2 часов, и затем добавляли Na(CN)BH3 (0,048 г, 0,762 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,246 г, 76,7%) в виде бесцветного масла.
Стадия 2: Синтез соединения 298
Метил 4-(((2S,6R)-2,6-диметил-4-(3-(трифторметил)бензил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,100 г, 0,238 ммоль), гидроксиламин (0,291 мл, 4,757 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,133 г, 2,378 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 298 (0,061 г, 60,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,67-7,56 (м, 6 H), 7,28 (д, 2 H, J=8,2 Гц), 3,73 (с, 2 H), 3,50 (с, 2 H), 2,65-2,26 (м, 4 H), 1,85 (т, 2 H, J=10,4 Гц), 0,91 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 422,1 (M++1).
Пример 136: Синтез соединения 299
(4-(((2S,6R)-4-(4-(диметиламино)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(4-(диметиламино)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 4-(диметиламино)бензальдегид (0,057 г, 0,381 ммоль) и уксусную кислоту (0,011 мл, 0,381 ммоль) растворяли в 1,2-этиленхлориде (4 мл) и перемешивали при комнатной температуре в течение 2 часов, и затем добавляли Na(CN)BH3 (0,024 г, 0,381 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,062 г, 41,1%) в виде бесцветного масла.
Стадия 2: Синтез соединения 299
Метил 4-(((2S,6R)-4-(4-(диметиламино)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,062 г, 0,157 ммоль), гидроксиламин (0,192 мл, 3,135 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,088 г, 1,567 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (5 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 299 (0,041 г, 66,0%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,65 (д, 2 H, J=8,2 Гц), 7,36 (д, 2 H, J=8,1 Гц), 7,07 (д, 2 H, J=8,6 Гц), 6,67 (д, 2 H, J=8,6 Гц), 3,72 (с, 2 H), 3,27 (с, 2 H), 2,63 (д, 2 H, J=10,5 Гц), 2,55-2,51 (м, 2 H), 1,73 (т, 2 H, J=10,6 Гц), 0,87 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 397,2 (M++1).
Пример 137: Синтез соединения 300
(4-(((2S,6R)-2,6-диметил-4-(4-(метилсульфонил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(4-(метилсульфонил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 4-(метилсульфонил)бензальдегид (0,070 г, 0,381 ммоль) и уксусную кислоту (0,011 мл, 0,381 ммоль) растворяли в 1,2-этиленхлориде (4 мл) и перемешивали при комнатной температуре в течение 2 часов, и затем добавляли Na(CN)BH3 (0,024 г, 0,381 ммоль), потом перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 10% до 70%) и концентрировали с получением желаемого соединения (0,058 г, 35,3%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 300
Метил 4-(((2S,6R)-2,6-диметил-4-(4-(метилсульфонил)бензил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,058 г, 0,135 ммоль), гидроксиламин (0,165 мл, 2,694 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,076 г, 1,347 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 300 (0,015 г, 25,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,87 (д, 2 H, J=8,2 Гц), 7,65 (д, 2 H, J=8,1 Гц), 7,57 (д, 2 H, J=8,1 Гц), 7,30 (д, 2 H, J=8,0 Гц), 3,74 (с, 2 H), 3,52 (с, 2 H), 3,21 (с, 3 H), 2,65 (д, 2 H, J=11,8 Гц), 2,60-2,56 (м, 2 H), 1,85 (т, 2 H, J=10,4 Гц), 0,93 (д, 6 H, J=6,8 Гц); ЖХМС (ES) m/z 432,2 (M++1).
Пример 138: Синтез соединения 301
(4-(((2S,6R)-4-(4-(1H-имидазол-1-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(4-(1H-имидазол-1-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 4-(1H-имидазол-1-ил)бензальдегид (0,066 г, 0,381 ммоль) и уксусную кислоту (0,011 мл, 0,381 ммоль) растворяли в 1,2-этиленхлориде (4 мл), и перемешивали при комнатной температуре в течение 2 часов и затем добавляли Na(CN)BH3 (0,024 г, 0,381 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,065 г, 40,7%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 301
Метил 4-(((2S,6R)-4-(4-(1H-имидазол-1-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,065 г, 0,155 ммоль), гидроксиламин (0,190 мл, 3,106 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,087 г, 1,553 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 301 (0,037 г, 56,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,18 (ушир. с, 1 H), 9,00 (ушир. с, 1 H), 8,24 (с, 1 H), 7,74 (с, 1 H), 7,67 (д, 2 H, J=8,0 Гц), 7,60 (д, 2 H, J=8,3 Гц), 7,42 (д, 4 H, J=8,0 Гц), 7,10 (с, 1 H), 3,86 (с, 2 H), 3,46 (с, 2 H), 2,68 (д, 2 H, J=10,6 Гц), 2,60-2,58 (м, 2 H), 1,83 (т, 2 H, J=10,6 Гц), 0,89 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 420,2 (M++1).
Пример 139: Синтез соединения 302
(4-(((2S,6R)-2,6-диметил-4-(4-(тиофен-2-ил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(4-(тиофен-2-ил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 4-(тиофен-2-ил)бензальдегид (0,072 г, 0,381 ммоль) и уксусную кислоту (0,011 мл, 0,381 ммоль) растворяли в 1,2-этиленхлориде (4 мл) и перемешивали при комнатной температуре в течение 2 часов, и затем добавляли Na(CN)BH3 (0,024 г, 0,381 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,022 г, 13,3%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 302
Метил 4-(((2S,6R)-2,6-диметил-4-(4-(тиофен-2-ил)бензил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,022 г, 0,051 ммоль), гидроксиламин (0,062 мл, 1,012 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,028 г, 0,506 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 302 (0,019 г, 86,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,65 (д, 2 H, J=8,1 Гц), 7,61 (д, 2 H, J=8,0 Гц), 7,53 (д, 1 H, J=5,0 Гц), 7,49 (д, 1 H, J=3,2 Гц), 7,33-7,31 (м, 4 H), 7,13 (дд, 1 H, J=4,4, 4,4 Гц), 3,74 (с, 2 H), 3,41 (с, 2 H), 2,67 (д, 2 H, J=9,5 Гц), 2,56-2,51 (м, 2 H), 1,82 (т, 2 H, J=10,6 Гц), 0,90 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 436,2 (M++1).
Пример 140: Синтез соединения 303
(4-(((2S,6R)-4-(4-(фуран-2-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(4-(фуран-2-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 4-(фуран-2-ил)бензальдегид (0,066 г, 0,381 ммоль) и уксусную кислоту (0,011 мл, 0,381 ммоль) растворяли в 1,2-этиленхлориде (4 мл), и перемешивали при комнатной температуре в течение 2 часов, и затем добавляли Na(CN)BH3 (0,024 г, 0,381 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,053 г, 33,2%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 303
Метил 4-(((2S,6R)-4-(4-(фуран-2-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,053 г, 0,127 ммоль), гидроксиламин (0,155 мл, 2,533 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,071 г, 1,266 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (5 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 303 (0,045 г, 84,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,74 (с, 1 H), 7,73-7,63 (м, 4 H), 7,33 (д, 2 H, J=8,2 Гц), 7,26 (д, 2 H, J=8,1 Гц), 6,91 (д, 1 H, J=3,1 Гц), 6,59 (дд, 1 H, J=3,4, 1,8 Гц), 3,72 (с, 2 H), 3,41 (с, 2 H), 2,65 (д, 2 H, J=10,8 Гц), 2,56-2,55 (м, 2 H), 1,81-1,79 (м, 2 H), 0,91 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 420,2 (M++1).
Пример 141: Синтез соединения 304
(4-(((2S,6R)-4-(4-(4H-1,2,4-триазол-4-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(4-(4H-1,2,4-триазол-4-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 4-(4H-1,2,4-триазол-4-ил)бензальдегид (0,066 г, 0,381 ммоль) и уксусную кислоту (0,011 мл, 0,381 ммоль) растворяли в 1,2-этиленхлориде (4 мл), и перемешивали при комнатной температуре в течение 2 часов, и затем добавляли Na(CN)BH3 (0,024 г, 0,381 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,064 г, 40,0%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 304
Метил 4-(((2S,6R)-4-(4-(4H-1,2,4-триазол-4-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-3, 0,064 г, 0,153 ммоль), гидроксиламин (0,187 мл, 3,051 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,086 г, 1,526 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 304 (0,016 г, 24,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,18 (ушир. с, 1 H), 9,28 (с, 1 H), 9,00 (ушир. с, 1 H), 8,23 (с, 1 H), 7,81 (д, 2 H, J=8,5 Гц), 7,67 (д, 2 H, J=8,2 Гц), 7,47 (д, 2 H, J=8,5 Гц), 7,41 (д, 2 H, J=8,4 Гц), 3,75 (с, 2 H), 3,47 (с, 2 H), 2,68 (д, 2 H, J=10,2 Гц), 2,61-2,26 (м, 2 H), 1,84 (т, 2 H, J=10,6 Гц), 0,89 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 421,2 (M++1).
Пример 142: Синтез соединения 305
(4-(((2S,6R)-2,6-диметил-4-(2-(2-метил-1H-имидазол-1-ил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(2-(2-метил-1H-имидазол-1-ил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 13-2, 0,100 г, 0,381 ммоль), 2-(2-метил-1H-имидазол-1-ил)бензальдегид (0,071 г, 0,381 ммоль) и уксусную кислоту (0,011 мл, 0,381 ммоль) растворяли в 1,2-этиленхлориде (4 мл) и перемешивали при комнатной температуре в течение 2 часов, и затем добавляли Na(CN)BH3 (0,024 г, 0,381 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,066 г, 40,0%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 305
Метил 4-(((2S,6R)-2,6-диметил-4-(2-(2-метил-1H-имидазол-1-ил)бензил)пиперазин-1-ил)метил)бензоат (формула 13-3, 0,066 г, 0,153 ммоль), гидроксиламин (0,187 мл, 3,052 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,086 г, 1,526 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (5 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 305 (0,036 г, 54,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,66 (д, 2 H, J=8,2 Гц), 7,56 (дд, 1 H, J=7,6, 1,5 Гц), 7,49 (ддд, 1 H, J=7,5, 7,5, 1,4 Гц), 7,43 (dt, 1 H, J=7,5, 7,5, 1,7 Гц), 7,37 (д, 2 H, J=8,2 Гц), 7,31 (дд, 1 H, J=7,7, 1,2 Гц), 7,15 (д, 1 H, J=1,3 Гц), 6,91 (д, 1 H, J=1,2 Гц), 3,75 (с, 2 H), 3,12-3,04 (м, 2 H), 2,48-2,46 (м, 4 H), 2,05 (с, 3 H), 1,68 (т, 2 H, J=10,7 Гц), 0,85 (д, 6 H, J=7,0 Гц); ЖХМС (ES) m/z 434,2 (M++1).
Пример 143: Синтез соединения 309
(4-(((3R,5S)-4-(4-(фуран-3-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-(фуран-3-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
смесь 1,2-диметоксиэтан (1 мл)/вода (0,5 мл) добавляли к смеси метил 4-(((3R,5S)-4-(4-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,100 г, 0,209 ммоль), фуран-3-илборной кислоты (0,028 г, 0,251 ммоль), Pd(dbpf)Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,066 г, 0,627 ммоль) при комнатной температуре, и нагревали путем воздействия микроволнового излучения при температуре 120°C в течение 20 минут. Реакционную смесь фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,052 г, 59,4%) в виде твердого вещества бледно-коричневого цвета.
Стадия 2: Синтез соединения 309
Метил 4-(((3R,5S)-4-(4-(фуран-3-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 2-2, 0,030 г, 0,072 ммоль), гидроксиламин (0,088 мл, 1,434 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,040 г, 0,717 ммоль) растворяли в метаноле (0,5 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 309 (0,021 г, 69,8%) в виде твердого вещества бледно-оранжевого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,15 (с, 1 H), 7,73 (с, 1 H), 7,65 (д, 2 H, J=8,1 Гц), 7,55 (д, 2 H, J=8,1 Гц), 7,31-7,27 (м, 4 H), 3,72 (с, 1 H), 3,39 (с, 2 H), 2,67-2,64 (м, 2 H), 2,58-2,56 (м, 2 H), 1,83-1,73 (м, 2 H), 0,90 (с, 3 H), 0,88 (с, 3 H).
Пример 144: Синтез соединения 321
((2S,6R)-N-(3-(1H-пиррол-1-ил)фенил)-4-(4-((E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)бензил)-2,6-диметилпиперазин-1-карбоксамид)
Стадия 1: Синтез (E)-метил 3-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилата
(2S,6R)-2,6-Диметилпиперазин (0,500 г, 4,379 ммоль), (E)-метил 3-(4-(бромметил)фенил)акрилат (формула 8-4, 1,173 г, 4,598 ммоль) и Cs2CO3 (2,140 г, 6,568 ммоль) при комнатной температуре растворяли в ацетонитриле (10 мл), и реакционный раствор перемешивали при той же температуре в течение 5 часов. Затем реакционную смесь фильтровали через стеклянный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 15%) и концентрировали с получением желаемого соединения (1,090 г, 86,3%) в виде твердого вещества оранжевого цвета.
Стадия 2: Синтез (E)-метил 3-(4-(((3R,5S)-4-((3-(1H-пиррол-1-ил)фенил)карбамоил)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 9-1, 0,050 г, 0,173 ммоль) и TEA (0,036 мл, 0,260 ммоль) растворяли в метиленхлориде (2 мл), и при температуре 0°C добавляли 1-(3-изоцианатофенил)-1H-пиррол (0,034 г, 0,182 ммоль), затем перемешивали при той же температуре в течение 1 часа и 40 минут. Затем реакционную смесь фильтровали через слой из целита для удаления твердых продуктов, и к фильтрату добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 40%) и концентрировали с получением желаемого соединения (0,050 г, 61,0%) в виде твердого вещества белого цвета.
Стадия 3: Синтез соединения 321
(E)-Метил 3-(4-(((3R,5S)-4-((3-(1H-пиррол-1-ил)фенил)карбамоил)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 9-2, 0,050 г, 0,106 ммоль), гидроксиламин (0,129 мл, 2,116 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,059 г, 1,058 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 321 (0,048 г, 95,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,90 (ушир. с, 1 H), 8,41 (с, 1 H), 7,73-7,72 (м, 1 H), 7,52 (д, 1 H, J=7,7 Гц), 7,42-7,28 (м, 6 H), 7,24 (дд, 2 H, J=2,1, 2,1 Гц), 7,12 (дд, 1 H, J=7,9, 1,3 Гц), 6,45 (д, 1 H, J=15,9 Гц), 6,26 (дд, 1 H, J=2,1, 2,1 Гц), 4,24 (т, 2 H, J=5,0 Гц), 3,53 (с, 2 H), 2,69 (д, 2 H, J=11,1 Гц), 2,14 (д, 2 H, J=7,4 Гц), 1,31 (д, 6 H, J=6,6 Гц); ЖХМС (ES) m/z 474,2 (M++1).
Пример 145: Синтез соединения 322
((E)-3-(4-(((3R,5S)-4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)фенил)-N-гидроксиакриламид)
Стадия 1: Синтез (E)-метил 3-(4-(((3R,5S)-4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 9-1, 0,050 г, 0,173 ммоль) и TEA (0,036 мл, 0,260 ммоль) растворяли в метиленхлориде (2 мл) и при температуре 0°C добавляли фуран-2-карбонилхлорид (0,018 мл, 0,182 ммоль), затем перемешивали при той же температуре в течение 1 часа и 30 минут. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,053 г, 79,9%) в виде бесцветного масла.
Стадия 2: Синтез соединения 322
(E)-Метил 3-(4-(((3R,5S)-4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 9-2, 0,053 г, 0,139 ммоль), гидроксиламин (0,170 мл, 2,772 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,078 г, 1,386 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 322 (0,027 г, 50,8%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,50 (ушир. с, 1 H), 7,82 (с, 1 H), 7,46 (д, 2 H, J=7,8 Гц), 7,34 (д, 2 H, J=7,8 Гц), 7,10 (д, 1 H, J=15,7 Гц), 6,95 (с, 1 H), 6,61 (д, 1 H, J=1,5 Гц), 6,39 (д, 1 H, J=15,8 Гц), 4,49-4,48 (м, 2 H), 3,50 (с, 2 H), 2,70 (д, 2 H, J=11,0 Гц), 2,12 (д, 2 H, J=7,2 Гц), 1,37 (д, 6 H, J=6,2 Гц); ЖХМС (ES) m/z 384,1 (M++1).
Пример 146: Синтез соединения 323
((E)-3-(4-(((3R,5S)-3,5-диметил-4-(2-фенилацетил)пиперазин-1-ил)метил)фенил)-N-гидроксиакриламид)
Стадия 1: Синтез ((E)-метил 3-(4-(((3R,5S)-3,5-диметил-4-(2-фенилацетил)пиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 9-1, 0,050 г, 0,173 ммоль) и TEA (0,036 мл, 0,260 ммоль) растворяли в метиленхлориде (2 мл) и при температуре 0°C добавляли 2-фенилацетил хлорид (0,024 мл, 0,182 ммоль), затем перемешивали при той же температуре в течение 2 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением желаемого соединения (0,037 г, 52,5%) в виде бесцветного масла.
Стадия 2: Синтез соединения 323
(E)-Метил 3-(4-(((3R,5S)-3,5-диметил-4-(2-фенилацетил)пиперазин-1-ил)метил)фенил)акрилат (формула 9-2, 0,037 г, 0,091 ммоль), гидроксиламин (0,111 мл, 1,820 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,051 г, 0,910 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 323 (0,021 г, 56,6%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, D2O) δ 9,90 (ушир. с, 1 H), 7,48 (д, 2 H, J=8,0 Гц), 7,35 (д, 2 H, J=8,1 Гц), 7,32-7,28 (м, 2 H), 7,23-7,17 (м, 4 H), 6,41 (д, 1 H, J=15,8 Гц), 4,42 (с, 1 H), 4,11 (с, 1 H), 3,77 (д, 1 H, J=13,8 Гц), 3,62 (д, 1 H, J=15,9 Гц), 3,48 (с, 2 H), 2,68-2,62 (м, 2 H), 2,01 (д, 2 H, J=8,0 Гц), 0,88-0,85 (м, 6 H); ЖХМС (ES) m/z 408,2 (M++1).
Пример 147: Синтез соединения 326
(4-(((3R,5S)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (3 мл) добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,400 г, 0,812 ммоль), 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,256 г, 1,219 ммоль), Pd(dbpf)Cl2 (0,026 г, 0,041 ммоль) и Na2CO3 (0,258 г, 2,437 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,098 г, 26,8%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 326
Метил 4-(((3R,5S)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 4-2, 0,098 г, 0,218 ммоль), гидроксиламин (0,267 мл, 4,370 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,123 г, 2,185 ммоль) при комнатной температуре растворяли в метаноле (3 мл), и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 326 (0,010 г, 9,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,1 Гц), 7,41-7,23 (м, 6 H), 5,81 (с, 1 H), 4,49 (br, 1 H), 4,15-4,14 (м, 2 H), 3,78 (т, 2 H, J=5,4 Гц), 3,49 (с, 2 H), 2,71-2,68 (м, 2 H), 2,15-2,02 (м, 3 H), 1,34 (д, 3 H, J=6,7 Гц), 1,13 (д, 3 H, J=6,7 Гц).
Пример 148: Синтез соединения 327
(4-(((3R,5S)-4-(2-(фуран-2-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-(фуран-2-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (2 мл) добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,250 г, 0,523 ммоль), фуран-2-илборной кислоты (0,088 г, 0,784 ммоль), Pd(dbpf)Cl2 (0,017 г, 0,026 ммоль) и Na2CO3 (0,166 г, 1,568 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 0% до 10%) и концентрировали с получением желаемого соединения (0,066 г, 30,0%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 327
Метил 4-(((3R,5S)-4-(2-(фуран-2-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 2-2, 0,066 г, 0,157 ммоль), гидроксиламин (0,192 мл, 3,140 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,088 г, 1,570 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 327 (0,044 г, 66,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,95 (д, 1 H, J=7,7 Гц), 7,80 (с, 1 H), 7,70 (д, 2 H, J=8,1 Гц), 7,56-7,54 (м, 1 H), 7,33-7,23 (м, 4 H), 6,71 (д, 1 H, J=3,3 Гц), 6,64-6,62 (м, 1 H), 3,79 (с, 2 H), 2,69-2,60 (м, 4 H), 1,85 (т, 2 H, J=10,4 Гц), 0,76 (д, 6 H, J=6,0 Гц).
Пример 149: Синтез соединения 328
(4-(((3R,5S)-4-(2-(фуран-3-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-(фуран-3-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (2 мл) добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,250 г, 0,523 ммоль), фуран-3-илборной кислоты (0,088 г, 0,784 ммоль), Pd(dbpf)Cl2 (0,017 г, 0,026 ммоль) и Na2CO3 (0,166 г, 1,568 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 0% до 5%) и концентрировали с получением желаемого соединения (0,020 г, 9,3%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 328
Метил 4-(((3R,5S)-4-(2-(фуран-3-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 2-2, 0,020 г, 0,049 ммоль), гидроксиламин (0,060 мл, 0,975 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,027 г, 0,487 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 328 (0,010 г, 50,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,85-7,84 (м, 2 H), 7,76-7,76 (м, 1 H), 7,69 (д, 2 H, J=8,1 Гц), 7,28 (т, 4 H, J=8,0 Гц), 7,19 (т, 1 H, J=7,5 Гц), 6,76-6,75 (м, 1 H), 3,69 (с, 2 H), 2,65 (д, 2 H, J=10,3 Гц), 2,58-2,55 (м, 2 H), 1,84 (т, 2 H, J=10,5 Гц), 0,76 (д, 1 H, J=6,1 Гц).
Пример 150: Синтез соединения 329
(4-(((3R,5S)-3,5-диметил-4-(2-(пиридин-4-ил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2-(пиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (2 мл) при комнатной температуре добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,250 г, 0,523 ммоль), гидрата пиридин-4-бороновой кислоты (0,110 г, 0,784 ммоль), Pd(dbpf)Cl2 (0,017 г, 0,026 ммоль) и Na2CO3 (0,166 г, 1,568 ммоль), и реакционный раствор перемешивали в течение ночи при температуре 100°C. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 20% до 60%) и концентрировали с получением желаемого соединения (0,139 г, 62,1%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 329
Метил 4-(((3R,5S)-3,5-диметил-4-(2-(пиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоат (формула 2-2, 0,088 г, 0,204 ммоль), гидроксиламин (0,250 мл, 4,083 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,115 г, 2,042 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 329 (0,076 г, 86,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,65-8,63 (м, 2 H), 7,85 (д, 1 H, J=7,7 Гц), 7,69 (д, 2 H, J=8,2 Гц), 7,43-7,38 (м, 3 H), 7,32-7,27 (м, 3 H), 7,18-7,16 (м, 1 H), 3,58 (с, 2 H), 2,58 (д, 2 H, J=10,2 Гц), 2,44-2,43 (м, 2 H), 1,82 (т, 2 H, J=10,4 Гц), 0,75 (д, 6 H, J=6,2 Гц).
Пример 151: Синтез соединения 330
(4-(((3R,5S)-3,5-диметил-4-(2-(пиридин-3-ил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2-(пиридин-3-ил)бензил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (2 мл) при комнатной температуре добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,250 г, 0,523 ммоль), пиридин-3-илборной кислоты (0,096 г, 0,784 ммоль), Pd(dbpf)Cl2 (0,017 г, 0,026 ммоль) и Na2CO3 (0,166 г, 1,568 ммоль), и реакционный раствор перемешивали в течение ночи при температуре 100°C. Затем насыщенный водный раствор гидрокарбоната натрия в реакционный раствор добавляли, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 20% до 60%) и концентрировали с получением желаемого соединения (0,083 г, 36,8%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 330
Метил 4-(((3R,5S)-3,5-диметил-4-(2-(пиридин-3-ил)бензил)пиперазин-1-ил)метил)бензоат (формула 2-2, 0,083 г, 0,192 ммоль), гидроксиламин (0,235 мл, 3,841 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,108 г, 1,921 ммоль) при комнатной температуре растворяли в метаноле (3 мл), и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционный раствор концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили. Полученный продукт очищали с помощью колоночной хроматографии (Waters, C18, колонка C19*100, 0,1%-ный водный раствор трифторуксусной кислоты/ацетонитрил=от 5% до 80%), после чего соединение пропускали через картридж (смола PL-HCO3 MP, метанол) и затем концентрировали с получением соединения 330 (0,012 г, 14,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,61-8,59 (м, 1 H), 8,55 (д, 1 H, J=2,1 Гц), 7,82 (дд, 2 H, J=13,4, 4,8 Гц), 7,68 (д, 2 H, J=8,1 Гц), 7,50-7,47 (м, 1 H), 7,40 (т, 1 H, J=7,0 Гц), 7,38-7,26 (м, 3 H), 7,18-7,16 (м, 1 H), 3,56 (с, 2 H), 2,58 (д, 2 H, J=10,0 Гц), 2,46-2,43 (м, 2 H), 1,82 (т, 2 H, J=10,4 Гц), 0,75 (д, 6 H, J=6,2 Гц).
Пример 152: Синтез соединения 331
(4-(((3R,5S)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-ил)метил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-ил)метил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (2 мл) при комнатной температуре добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,250 г, 0,523 ммоль), 2-(4,4-диметилциклогекс-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,185 г, 0,784 ммоль), Pd(dbpf)Cl2 (0,017 г, 0,026 ммоль) и Na2CO3 (0,166 г, 1,568 ммоль), и реакционный раствор нагревали путем воздействия микроволнового излучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционный раствор добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 0% до 5%) и концентрировали с получением желаемого соединения (0,044 г, 18,4%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 331
Метил 4-(((3R,5S)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-ил)метил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 2-2, 0,044 г, 0,096 ммоль), гидроксиламин (0,118 мл, 1,928 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,054 г, 0,964 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 331 (0,038 г, 85,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,67 (д, 3 H, J=8,1 Гц), 7,22-7,20 (м, 3 H), 7,10 (т, 1 H, J=7,2 Гц), 5,40 (с, 1 H), 3,60 (с, 2 H), 2,64 (д, 2 H, J=10,4 Гц), 2,16 (с, 2 H), 1,94 (с, 2 H), 1,82 (т, 2 H, J=10,5 Гц), 1,47 (т, 2 H, J=6,2 Гц), 1,00 (с, 6 H), 0,78 (д, 6 H, J=6,1 Гц).
Пример 153: Синтез соединения 332
(4-(((3R,5S)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (2 мл) при комнатной температуре добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,250 г, 0,523 ммоль), 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,165 г, 0,784 ммоль), Pd(dbpf)Cl2 (0,017 г, 0,026 ммоль) и Na2CO3 (0,166 г, 1,568 ммоль), и реакционный раствор перемешивали в течение ночи при температуре 100°C. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,108 г, 47,6%) в виде твердого вещества бледно-коричневого цвета.
Стадия 2: Синтез соединения 332
Метил 4-(((3R,5S)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 2-2, 0,108 г, 0,249 ммоль), гидроксиламин (0,304 мл, 4,970 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,139 г, 2,485 ммоль) при комнатной температуре растворяли в метаноле (2 мл), и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили. Полученный продукт очищали с помощью колоночной хроматографии (Waters, C18, колонка C19*100, 0,1%-ный водный раствор трифторуксусной кислоты/ацетонитрил=от 5% до 80%), после чего соединение пропускали через картридж (смола PL-HCO3 MP, метанол), и затем концентрировали с получением соединения 332 (0,017 г, 15,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,73-7,68 (м, 3 H), 7,29-7,20 (м, 3 H), 7,14 (т, 1 H, J=6,9 Гц), 7,04-7,02 (м, 1 H), 5,60 (с, 1 H), 4,19-4,19 (м, 2 H), 3,82 (т, 2 H, J=5,3 Гц), 3,64 (с, 2 H), 2,66-2,57 (м, 4 H), 2,26 (с, 2 H), 1,84 (т, 2 H, J=10,3 Гц), 0,79-0,78 (м, 6 H).
Пример 154: Синтез соединения 340
(4-(((3R,5S)-4-(4-(фуран-2-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-бромбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
4-Бромбензоилхлорид (1,000 г, 4,557 ммоль), метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 1,315 г, 5,012 ммоль) и TEA (1,263 мл, 9,113 ммоль) растворяли в метиленхлориде (20 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 2 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 30% до 50%) и концентрировали с получением желаемого соединения (2,007 г, 98,9%) в виде твердого вещества белого цвета.
Стадия 2: Синтез метил 4-(((3R,5S)-4-(4-(фуран-2-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан/вода (об./об.=3/1) (4 мл) добавляли к смеси метил 4-(((3R,5S)-4-(4-бромбензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 4-1, 0,500 г, 1,123 ммоль), фуран-2-илборной кислоты (0,138 г, 1,235 ммоль), Pd(dbpf)Cl2 (0,037 г, 0,056 ммоль) и Na2CO3 (0,262 г, 2,470 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 30 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,163 г, 33,5%) в виде твердого вещества белого цвета.
Стадия 3: Синтез соединения 340
Метил 4-(((3R,5S)-4-(4-(фуран-2-ил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 4-2, 0,100 г, 0,231 ммоль), гидроксиламин (0,283 мл, 4,624 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,130 г, 2,312 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, и выпавший твердый осадок фильтровали и сушили с получением соединения 340 (0,049 г, 48,7%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,85 (д, 1 H, J=7,7 Гц), 7,76-7,71 (м, 4 H), 7,40-7,36 (м, 3 H), 7,24 (д, 1 H, J=7,9 Гц), 7,01 (д, 1 H, J=3,0 Гц), 6,60-6,59 (м, 1 H), 3,51-3,47 (м, 2 H), 2,62-2,60 (м, 2 H), 2,15-2,09 (м, 2 H), 1,27 (ушир. с, 6 H).
Пример 155: Синтез соединения 342
(4-(((3R,5S)-4-(3-(фуран-2-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(фуран-2-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,100 г, 0,209 ммоль), фуран-2-илборной кислоты (0,047 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали. Полученный продукт растворяли в растворителе, и раствор соединения пропускали через картридж (смола PL-HCO3 MP, только метанол) и затем концентрировали с получением желаемого соединения (0,064 г, 73,2%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 342
Метил 4-(((3R,5S)-4-(3-(фуран-2-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (соединение 2-2, 0,064 г, 0,153 ммоль), гидроксиламин (0,094 мл, 1,529 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,172 г, 3,058 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении с получением соединения 342 (0,031 г, 48,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,10 (ушир. с, 1 H), 9,00 (ушир. с, 1 H), 7,75 (д, 1 H, J=1,3 Гц), 7,69 (д, 2 H, J=7,8 Гц), 7,66 (с, 1 H), 7,52 (д, 1 H, J=7,5 Гц), 7,35-7,31 (м, 3 H), 7,28 (д, 1 H, J=7,5 Гц), 6,90 (д, 1 H, J=3,0 Гц), 6,58 (дд, 1 H, J=3,4, 1,8 Гц), 3,75 (с, 2 H), 3,44 (с, 2 H), 2,66-2,57 (м, 4 H), 1,83 (т, 2 H, J=10,5 Гц), 0,91 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 420,2 (M++1).
Пример 156: Синтез соединения 343
(4-(((3R,5S)-4-(3-(фуран-3-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(фуран-2-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 2-1, 0,100 г, 0,209 ммоль), фуран-3-илборной кислоты (0,047 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль) и нагревали при температуре 120°C в течение 20 минут с помощью микроволнового облучения, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали. Полученный продукт растворяли в растворителе, и раствор соединения пропускали через картридж (смола PL-HCO3 MP, только метанол), и затем концентрировали с получением желаемого соединения (0,061 г, 69,7%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 343
Метил 4-(((3R,5S)-4-(3-(фуран-3-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 2-2, 0,061 г, 0,146 ммоль), гидроксиламин (0,089 мл, 1,457 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,164 г, 2,915 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 100%) и концентрировали с получением соединения 343 (0,008 г, 13,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CH3OD) δ 7,89-7,89 (м, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,57 (дд, 1 H, J=1,6, 1,6 Гц), 7,54 (с, 1 H), 7,44 (д, 3 H, J=7,9 Гц), 7,33-7,30 (м, 2 H), 6,80-6,79 (м, 1 H), 3,95 (с, 2 H), 3,54 (с, 2 H), 2,77-2,17 (м, 4 H), 1,97 (т, 2 H, J=10,8 Гц), 1,13 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 420,2 (M++1).
Пример 157: Синтез соединения 344
(4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-3-ил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-3-ил)бензил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,100 г, 0,209 ммоль), пиридин-3-илборной кислоты (0,051 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали. Полученный продукт растворяли в растворителе, и раствор соединения пропускали через картридж (смола PL-HCO3 MP, только метанол), и затем концентрировали с получением желаемого соединения (0,085 г, 94,7%) в виде бесцветного масла.
Стадия 2: Синтез соединения 344
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-3-ил)бензил)пиперазин-1-ил)метил)бензоат (формула 2-2, 0,085 г, 0,198 ммоль), гидроксиламин (0,121 мл, 1,979 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,222 г, 3,958 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и реакционный раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр, снабженный картриджем с безводным сульфатом натрия, для удаления твердого остатка и водного слоя, и фильтрат концентрировали при пониженном давлении с получением соединения 344 (0,055 г, 64,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,86 (д, 1 H, J=1,9 Гц), 8,58 (д, 1 H, J=3,4 Гц), 8,04 (д, 1 H, J=7,8 Гц), 7,68 (д, 3 H, J=8,1 Гц), 7,54-7,48 (м, 2 H), 7,43 (д, 2 H, J=4,8 Гц), 7,26 (д, 2 H, J=8,0 Гц), 3,81 (с, 2 H), 3,41 (с, 2 H), 2,66 (д, 2 H, J=11,0 Гц), 2,63-2,59 (м, 2 H), 1,82 (т, 2 H, J=10,2 Гц), 0,93 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 431,2 (M++1).
Пример 158: Синтез соединения 345
(4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-4-ил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 2-1, 0,100 г, 0,209 ммоль), пиридин-4-илборной кислоты (0,051 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали. Полученный продукт растворяли в растворителе, и раствор соединения пропускали через картридж (смола PL-HCO3 MP, только метанол) и затем концентрировали с получением желаемого соединения (0,088 г, 98,0%) в виде бесцветного масла.
Стадия 2: Синтез соединения 345
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоат (формула 4-2, 0,040 г, 0,093 ммоль), гидроксиламин (0,031 г, 0,931 ммоль) и гидроксид калия (0,105 г, 1,862 ммоль) смешивали в метаноле (3 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 30 минут. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением соединения 345 (0,005 г, 12,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,64 (д, 2 H, J=5,2 Гц), 7,74 (с, 1 H), 7,62-7,61 (м, 5 H), 7,47-7,45 (м, 2 H), 7,25 (д, 2 H, J=7,8 Гц), 3,82 (с, 2 H), 3,41 (с, 2 H), 2,68-2,61 (м, 4 H), 1,83 (т, 2 H, J=9,9 Гц), 0,92 (д, 6 H, J=5,8 Гц); ЖХМС (ES) m/z 431,2 (M++1).
Пример 159: Синтез соединения 346
(4-(((3R,5S)-4-(3-(3,6-дигидро-2H-пиран-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(3,6-дигидро-2H-пиран-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,100 г, 0,209 ммоль), 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,088 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали. Полученный продукт растворяли в растворителе, и раствор соединения пропускали через картридж (смола PL-HCO3 MP, только метанол), и затем концентрировали с получением желаемого соединения (0,068 г, 74,9%) в виде масла бледно-коричневого цвета.
Стадия 2: Синтез соединения 346
Метил 4-(((3R,5S)-4-(3-(3,6-дигидро-2H-пиран-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 2-2, 0,068 г, 0,156 ммоль), гидроксиламин (0,096 мл, 1,565 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,176 г, 3,130 ммоль) смешивали в метаноле (3 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 30 минут. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали. Полученный продукт растворяли в растворителе, и раствор соединения пропускали через картридж (смола PL-HCO3 MP, 100% метанол), и затем концентрировали с получением соединения 346 (0,005 г, 7,3%) в виде твердого вещества абрикосового цвета.
1H ЯМР (400 МГц, CH3OD) δ 7,72 (д, 2 H, J=8,2 Гц), 7,42 (д, 3 H, J=8,4 Гц), 7,31-7,28 (м, 3 H), 6,17 (т, 1 H, J=1,5 Гц), 4,32-4,30 (м, 2 H), 3,95-3,92 (м, 4 H), 3,52 (с, 2 H), 2,75-2,69 (м, 4 H), 2,39-2,52 (м, 2 H), 1,94 (т, 2 H, J=11,2 Гц), 1,11 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 436,2 (M++1).
Пример 160: Синтез соединения 347
(4-(((3R,5S)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-3-ил)метил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-3-ил)метил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((3R,5S)-4-(3-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 0,100 г, 0,209 ммоль), 2-(4,4-диметилцикло-1-гексенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,099 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и органический слой концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18, колонка 19*100 мм, смесь 0,1%-ная трифторуксусная кислота/ацетонитрил=от 5% до 80%) и концентрировали. Полученный продукт растворяли в растворителе, и раствор соединения пропускали через картридж (смола PL-HCO3 MP, только метанол), и затем концентрировали с получением желаемого соединения (0,064 г, 66,5%) в виде масла коричневого цвета.
Стадия 2: Синтез соединения 347
Метил 4-(((3R,5S)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-3-ил)метил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 2-2, 0,064 г, 0,139 ммоль), гидроксиламин (0,085 мл, 1,389 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,156 г, 2,779 ммоль) смешивали в метаноле (3 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 347 (0,020 г, 31,2%) в виде твердого вещества оранжевого цвета.
1H ЯМР (400 МГц, CH3OD) δ 7,72 (д, 2 H, J=8,1 Гц), 7,43 (д, 2 H, J=8,0 Гц), 7,40 (с, 1 H), 7,28-7,23 (м, 3 H), 6,06 (с, 1 H), 3,93 (с, 2 H), 3,52 (с, 2 H), 2,76-2,69 (м, 4 H), 2,45-2,45 (м, 2 H), 2,03-2,02 (м, 2 H), 1,95 (т, 2 H, J=10,6 Гц), 1,57 (т, 2 H, J=6,4 Гц), 1,13 (д, 6 H, J=6,0 Гц), 1,00 (с, 6 H); ЖХМС (ES) m/z 462,3 (M++1).
Пример 161: Синтез соединения 348
(4-(((2S,6R)-4-(2-(фуран-3-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-(фуран-3-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-йодбензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 14-1, 0,100 г, 0,209 ммоль), фуран-3-илборной кислоты (0,047 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,090 г, 102,9%, сырой).
Стадия 2: Синтез соединения 348
Метил 4-(((2S,6R)-4-(2-(фуран-3-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 14-2, 0,090 г, 0,215 ммоль), гидроксиламин (0,132 мл, 2,150 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,241 г, 4,301 ммоль) смешивали в метаноле (3 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 30 минут. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 15%) и концентрировали с получением соединения 348 (0,023 г, 25,5%) в виде твердого вещества бледно-оранжевого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,13 (с, 1 H), 8,98 (с, 1 H), 8,99 (д, 1 H, J=1,6 Гц), 7,98-7,98 (м, 1 H), 7,76 (дд, 1 H, J=1,7, 1,7 Гц), 7,67 (д, 2 H, J=8,6 Гц), 7,44-7,39 (м, 4 H), 7,33-7,29 (м, 2 H), 6,84 (дд, 1 H, J=1,8, 0,7 Гц), 3,75 (с, 2 H), 3,37 (с, 2 H), 2,68 (д, 2 H, J=10,7 Гц), 1,85-1,84 (м, 2 H), 0,89 (д, 6 H, J=6,1 Гц).
Пример 162: Синтез соединения 349
(4-(((2S,6R)-2,6-диметил-4-(2-(пиридин-3-ил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(2-(пиридин-3-ил)бензил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтана (4 мл)/воды (1 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-йодбензил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 14-1, 0,100 г, 0,209 ммоль), пиридин-3-илборной кислоты (0,051 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,090 г, 100,2%).
Стадия 2: Синтез соединения 349
Метил 4-(((2S,6R)-2,6-диметил-4-(2-(пиридин-3-ил)бензил)пиперазин-1-ил)метил)бензоат (формула 14-2, 0,090 г, 0,210 ммоль), гидроксиламин (0,128 мл, 2,095 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,235 г, 4,190 ммоль) смешивали в метаноле (3 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), и выпавший твердый осадок фильтровали и сушили с получением соединения 349 (0,041 г, 45,5%) в виде твердого вещества абрикосового цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,60-8,56 (м, 2 H), 7,86 (д, 1 H, J=7,6 Гц), 7,65 (д, 2 H, J=7,6 Гц), 7,49-7,38 (м, 4 H), 7,35 (д, 2 H, J=7,9 Гц), 7,28 (д, 1 H, J=6,5 Гц), 3,70 (с, 2 H), 3,30 (с, 2 H), 2,50-2,48 (м, 4 H), 1,72 (т, 2 H, J=10,2 Гц), 0,83 (д, 6 H, J=5,9 Гц); ЖХМС (ES) m/z 431,2 (M++1).
Пример 163: Синтез соединения 350
(4-(((2S,6R)-2,6-диметил-4-(2-(пиридин-4-ил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(2-(пиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан (3 мл)/вода (1 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-йодбензил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 14-1, 0,100 г, 0,209 ммоль), пиридин-4-илборной кислоты (0,051 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль), и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт пропускали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,090 г, 100,2%).
Стадия 2: Синтез соединения 350
Метил 4-(((2S,6R)-2,6-диметил-4-(2-(пиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоат (формула 14-2, 0,090 г, 0,210 ммоль), гидроксиламин (0,128 мл, 2,095 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,235 г, 4,190 ммоль) при комнатной температуре смешивали в метаноле (3 мл), и смесь перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 15%) и концентрировали с получением соединения 350 (0,026 г, 28,8%) в виде твердого вещества бледно-оранжевого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,52 (д, 2 H, J=6,1 Гц), 7,70 (д, 2 H, J=8,2 Гц), 7,49 (д, 2 H, J=6,4 Гц), 7,50-7,47 (м, 5 H), 7,42-7,40 (м, 2 H), 7,29-7,27 (м, 1 H), 3,85 (с, 2 H), 3,42 (с, 2 H), 2,56 (д, 2 H, J=10,6 Гц), 2,47-2,46 (м, 2 H), 1,79 (т, 2 H, J=10,7 Гц), 0,97 (д, 6 H, J=6,2 Гц).
Пример 164: Синтез соединения 351
(4-(((2S,6R)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан (3 мл)/вода (1 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-йодбензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 14-1, 0,100 г, 0,209 ммоль), 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,088 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г,0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 60%) и концентрировали с получением желаемого соединения (0,017 г, 18,7%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 351
Метил 4-(((2S,6R)-4-(2-(3,6-дигидро-2H-пиран-4-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 14-2, 0,017 г, 0,039 ммоль), гидроксиламин (0,024 мл, 0,391 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,044 г, 0,782 ммоль) смешивали в метаноле (1 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 30 минут. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр, снабженный картриджем с безводным сульфатом натрия, для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 351 (0,012 г, 70,4%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,71 (д, 2 H, J=8,3 Гц), 7,49 (д, 2 H, J=8,3 Гц), 7,38-7,37 (м, 1 H), 7,24-7,22 (м, 2 H), 7,12-7,11 (м, 1 H), 5,61 (с, 1 H), 4,26-4,24 (м, 2 H), 3,90-3,87 (м, 4 H), 3,48 (с, 2 H), 2,70 (д, 2 H, J=11,0 Гц), 2,69-2,64 (м, 2 H), 2,36-2,35 (м, 2 H), 1,90 (т, 2 H, J=10,9 Гц), 1,03 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 436,2 (M++1).
Пример 165: Синтез соединения 352
(4-(((2S,6R)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-ил)метил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-ил)метил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан (3 мл)/вода (1 мл) добавляли к смеси метил 4-(((2S,6R)-4-(2-йодбензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 14-1, 0,100 г, 0,209 ммоль), 2-(4,4-диметилцикло-1-гексенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,099 г, 0,418 ммоль), Pd(dbpf)2Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,044 г, 0,418 ммоль) и нагревали с помощью микроволнового облучения при температуре 120°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 10%) и концентрировали с получением желаемого соединения (0,013 г, 13,5%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 352
Метил 4-(((2S,6R)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-2-ил)метил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 14-2, 0,013 г, 0,028 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,017 мл, 0,282 ммоль) и гидроксид калия (0,032 г, 0,564 ммоль) смешивали в метаноле (1 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 30 минут. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью PTLC (диоксид кремния; 100%-ный этилацетат) и концентрировали с получением соединения 352 (0,007 г, 53,7%) в виде твердого вещества оранжевого цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,71 (д, 2 H, J=7,1 Гц), 7,49 (д, 2 H, J=6,9 Гц), 7,37-7,36 (м, 1 H), 7,20-7,18 (м, 2 H), 7,07-7,04 (м, 1 H), 5,43 (с, 1 H), 3,91 (с, 2 H), 3,48 (с, 2 H), 2,72-2,66 (м, 4 H), 2,24 (с, 2 H), 1,96 (с, 2 H), 1,88 (т, 2 H, J=10,4 Гц), 1,50 (т, 2 H, J=6,2 Гц), 1,03 (д, 12 H, J=3,9 Гц); ЖХМС (ES) m/z 462,2 (M++1).
Пример 166: Синтез соединения 354
(4-(((3R,5S)-3,5-диметил-4-(оксазол-4-илметил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез оксазол-4-илметил метансульфоната
Оксазол-4-илметанол (0,300 г, 3,028 ммоль) и TEA (0,633 мл, 4,541 ммоль) смешивали в метиленхлориде (5 мл) при температуре 0°C, и в смесь добавляли MsCl (0,258 мл, 3,330 ммоль), затем перемешивали при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением желаемого соединения (0,119 г, 22,2%) в виде масла оранжевого цвета.
Стадия 2: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(оксазол-4-илметил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,150 г, 0,572 ммоль), оксазол-4-илметил метансульфонат (0,111 г, 0,629 ммоль) и Cs2CO3 (0,279 г, 0,858 ммоль) добавляли к N,N-диметилформамиду (3 мл) при температуре 80°C, и смесь перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 15%) и концентрировали с получением желаемого соединения (0,054 г, 27,5%) в виде масла бледно-желтого цвета.
Стадия 3: Синтез соединения 354
Метил 4-(((3R,5S)-3,5-диметил-4-(оксазол-4-илметил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,054 г, 0,157 ммоль), гидроксиламин (0,096 мл, 1,572 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,176 г, 3,145 ммоль) смешивали в метаноле (1 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр, снабженный картриджем с безводным сульфатом натрия, для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 354 (0,003 г, 5,5%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,16 (с, 1 H), 7,92 (с, 1 H), 7,72 (д, 2 H, J=8,1 Гц), 7,41 (д, 2 H, J=8,0 Гц), 3,98 (с, 2 H), 3,51 (с, 2 H), 2,73 (д, 2 H, J=11,8 Гц), 2,70-2,66 (м, 2 H), 1,93 (т, 2 H, J=10,8 Гц), 1,19 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 345,2 (M++1).
Пример 167: Синтез соединения 355
(4-(((3R,5S)-3,5-диметил-4-(пиримидин-5-илметил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(пиримидин-5-илметил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,200 г, 0,762 ммоль), 5-(хлорметил)пиримидин (0,108 г, 0,839 ммоль) и Cs2CO3 (0,373 г, 1,144 ммоль) добавляли к N,N-диметилформамиду (3 мл) при комнатной температуре, и смесь перемешивали при температуре 120°C в течение 48 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 15%) и концентрировали с получением желаемого соединения (0,027 г, 10,0%) в виде твердого вещества желтого цвета.
Стадия 2: Синтез соединения 355
Метил 4-(((3R,5S)-3,5-диметил-4-(пиримидин-5-илметил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,027 г, 0,076 ммоль), гидроксиламин (0,047 мл, 0,762 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,085 г, 1,523 ммоль) смешивали в метаноле (1 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр, снабженный картриджем с безводным сульфатом натрия, для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 355 (0,003 г, 11,1%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CH3OD) δ 9,03 (с, 1 H), 8,83 (с, 1 H), 7,74 (д, 2 H, J=8,3 Гц), 7,46 (д, 2 H, J=8,3 Гц), 3,92 (с, 2 H), 3,56 (с, 2 H), 2,78 (д, 2 H, J=10,7 Гц), 2,72-2,68 (м, 2 H), 1,97 (т, 2 H, J=10,8 Гц), 0,93 (д, 6 H, J=6,6 Гц); ЖХМС (ES) m/z 356,1 (M++1).
Пример 168: Синтез соединения 356
(4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,500 г, 1,906 ммоль), 3-(бромметил)бензальдегид (0,417 г, 2,096 ммоль) и Cs2CO3 (0,931 г, 2,859 ммоль) смешивали в ацетонитриле (20 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли воду, после чего экстрагировали этилацетатом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 12 г; этилацетат/гексан=от 0% до 60%) и концентрировали с получением желаемого соединения (0,530 г, 73,1%) в виде масла бледно-коричневого цвета.
Стадия 2: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль), морфолин (0,050 мл, 0,578 ммоль) и уксусную кислоту (0,015 мл, 0,526 ммоль) смешивали в тетрагидрофуране (5 мл) при комнатной температуре и смесь перемешивали. Потом в смесь добавляли Na(CN)BH3 (0,033 г, 0,526 ммоль), затем перемешивали при той же температуре в течение 17 часов. Далее в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 4 г; этилацетат/гексан=от 20% до 100%) и концентрировали с получением желаемого соединения (0,109 г, 45,9%) в виде бесцветного масла.
Стадия 3: Синтез соединения 356
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,050 г, 0,111 ммоль), гидроксиламин (0,135 мл, 2,214 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,072 г, 1,107 ммоль) смешивали в метаноле (2 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр, снабженный картриджем с безводным сульфатом натрия, для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 356 (0,034 г, 67,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,66 (д, 2 H, J=8,0 Гц), 7,27-7,22 (м, 5 H), 7,10 (с, 1 H), 3,71 (с, 2 H), 3,55 (с, 4 H), 3,44 (с, 2 H), 3,39 (с, 2 H), 2,63 (д, 2 H, J=10,5 Гц), 2,57-2,55 (м, 2 H), 2,32 (с, 4 H), 1,78 (т, 2 H, J=10,5 Гц), 0,89 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 453,2 (M++1).
Пример 169: Синтез соединения 376
(4-(((3R,5S)-3,5-диметил-4-(4-(морфолинометил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,200 г, 0,762 ммоль), 4-(бромметил)бензальдегид (0,167 г, 0,839 ммоль) и Cs2CO3 (0,373 г, 1,144 ммоль) смешивали в ацетонитриле (5 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 5% до 70%) и концентрировали с получением желаемого соединения (0,131 г, 45,2%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-(морфолинометил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,131 г, 0,344 ммоль) и морфолин (0,033 г, 0,379 ммоль) смешивали в метиленхлориде (4 мл) при комнатной температуре, и смесь перемешивали в течение 30 минут. Потом добавляли Na(OAc)3BH (0,109 г, 0,516 ммоль), затем перемешивали при той же температуре в течение 17 часов. Далее в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,134 г, 86,2%) в виде масла бледно-желтого цвета.
Стадия 3: Синтез соединения 376
Метил 4-(((3R,5S)-3,5-диметил-4-(4-(морфолинометил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,134 г, 0,297 ммоль), гидроксиламин (0,181 мл, 2,967 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,386 г, 5,934 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 376 (0,030 г, 22,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,18 (ушир. с, 1 H),9,04 (ушир. с, 1 H), 7,70 (д, 2 H, J=8,2 Гц), 7,35 (д, 2 H, J=8,1 Гц), 7,28 (д, 2 H, J=8,0 Гц), 7,21 (д, 2 H, J=8,0 Гц), 3,71 (с, 2 H), 3,56 (т, 4 H, J=4,4 Гц), 3,43 (с, 2 H), 3,41 (с, 2 H), 2,63 (д, 2 H, J=10,4 Гц), 2,58-2,53 (м, 2 H), 2,32 (с, 4 H), 1,80 (т, 2 H, J=10,4 Гц), 0,90 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 453,2 (M++1).
Пример 170: Синтез соединения 380
(4-(((3R,5S)-4-(3-((1-(2-фтор-2-метилпропил)пиперидин-4-ил)метокси)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез трет-бутил 4-((3-(гидроксиметил)фенокси)метил)пиперидин-1-карбоксилата
3-(Гидроксиметил)фенол (0,500 г, 4,028 ммоль), трет-бутил 4-((метилсульфонилoxy)метил)пиперидин-1-карбоксилат (формула 12-1, 1,300 г, 4,430 ммоль), Cs2CO3 (1,968 г, 6,042 ммоль) и NaI (0,030 г, 0,201 ммоль) смешивали в ацетонитриле (50 мл) при комнатной температуре, и смесь нагревали при кипячении с обратным холодильником в течение 5 часов, и затем охлаждали до комнатной температуры. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором NaCl, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 10% до 60%) и концентрировали с получением желаемого соединения (0,790 г, 61,0%) в виде бесцветного масла.
Стадия 2: Синтез трет-бутил 4-((3-(гидроксиметил)фенокси)метил)пиперидин-1-карбоксилата
Трет-бутил 4-((3-(гидроксиметил)фенокси)метил)пиперидин-1-карбоксилат (формула 12-2, 0,500 г, 1,556 ммоль) и TEA (0,434 мл, 3,111 ммоль) смешивали в метиленхлориде (50 мл) при температуре 0°C и в смесь добавляли MsCl (0,132 мл, 1,711 ммоль). После этого смесь перемешивали при той же температуре в течение 1 часа и затем перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 40%) и концентрировали с получением желаемого соединения (0,204 г, 38,6%) в виде бесцветного масла.
Стадия 3: Синтез трет-бутил 4-((3-(((2S,6R)-4-(4-(метоксикарбонил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенокси)метил)пиперидин-1-карбоксилата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,150 г, 0,572 ммоль), трет-бутил 4-((3-(хлорметил)фенокси)метил)пиперидин-1-карбоксилат (формула 12-3, 0,194 г, 0,572 ммоль) и Cs2CO3 (0,279 г, 0,858 ммоль) смешивали в ацетонитриле (10 мл) при комнатной температуре, и смесь нагревали при кипячении с обратным холодильником в течение 5 часов и затем охлаждали до комнатной температуры. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 10% до 70%) и концентрировали с получением желаемого соединения (0,181 г, 56,0%) в виде масла бледно-желтого цвета.
Стадия 4: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиперидин-4-илметокси)бензил)пиперазин-1-ил)метил)бензоата
Трет-бутил 4-((3-(((2S,6R)-4-(4-(метоксикарбонил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенокси)метил)пиперидин-1-карбоксилат (формула 12-4, 0,180 г, 0,318 ммоль) добавляли к 1,4-диоксану (5 мл) при комнатной температуре, и в смесь добавляли HCl (4,00 M раствор в 1,4-диоксане, 0,795 мл, 3,182 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,141 г, 95,2%, масло бледно-желтого цвета).
Стадия 5: Синтез этил 4-(((3R,5S)-4-(3-((1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)метокси)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
К смеси метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиперидин-4-илметокси)бензил)пиперазин-1-ил)метил)бензоата (формула 12-5, 0,150 г, 0,322 ммоль), 2,2-диметилоксирана (0,044 мл, 0,483 ммоль) и K2CO3 (0,089 г, 0,644 ммоль) добавляли этанол (4 мл) и нагревали с помощью микроволнового облучения при температуре 110°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,091 г, 52,5%) в виде бесцветного масла.
Стадия 6: Синтез этил 4-(((3R,5S)-4-(3-((1-(2-фтор-2-метилпропил)пиперидин-4-ил)метокси)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Этил 4-(((3R,5S)-4-(3-((1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)метокси)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 12-6, 0,090 г, 0,163 ммоль) растворяли в метиленхлориде (5 мл) и при температуре 0°C добавляли DAST (0,029 г, 0,179 ммоль). Смесь перемешивали при той же температуре в течение 30 минут и затем перемешивали при комнатной температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 8 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением желаемого соединения (0,088 г, 97,4%) в виде масла бледно-коричневого цвета.
Стадия 7: Синтез соединения 380
Этил 4-(((3R,5S)-4-(3-((1-(2-фтор-2-метилпропил)пиперидин-4-ил)метокси)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 12-7, 0,088 г, 0,159 ммоль), гидроксиламин (0,097 мл, 1,589 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,207 г, 3,178 ммоль) растворяли в метаноле (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 380 (0,027 г, 31,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,70 (д, 2 H, J=8,1 Гц), 7,34 (д, 2 H, J=8,1 Гц), 7,18 (дд, 1 H, J=8,0, 8,0 Гц), 6,91-6,89 (м, 2 H), 6,73 (д, 1 H, J=8,5 Гц), 3,78 (д, 2 H, J=5,4 Гц), 3,68 (с, 2 H), 3,51 (с, 2 H), 2,91 (д, 2 H, J=11,3 Гц), 2,64 (д, 2 H, J=10,8 Гц), 2,58-2,56 (м, 2 H), 2,41 (д, 2 H, J=22,9 Гц), 2,07 (т, 2 H, J=10,8 Гц), 1,81 (т, 2 H, J=10,6 Гц), 1,71 (д, 3 H, J=11,5 Гц), 1,33 (с, 3 H), 1,28-1,24 (м, 5 H), 0,89 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 541,3 (M++1).
Пример 171: Синтез соединения 382
(4-(((3R,5S)-3,5-диметил-4-(3-(пиперидин-1-илметил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиперидин-1-илметил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,150 г, 0,394 ммоль) и пиперидин (0,038 мл, 0,434 ммоль) растворяли в метиленхлориде (10 мл), и раствор перемешивали при комнатной температуре в течение 30 минут. В реакционный раствор добавляли Na(OAc)3BH (0,125 г, 0,591 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,095 г, 53,6%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 382
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиперидин-1-илметил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,044 г, 0,098 ммоль), гидроксиламин (0,060 мл, 0,979 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,127 г, 1,957 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 382 (0,037 г, 83,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, 2 H, J=8,2 Гц), 7,30-7,28 (м, 3 H), 7,22-7,20 (м, 2 H), 7,08 (д, 1 H, J=6,2 Гц), 3,72 (с, 2 H), 3,41 (с, 2 H), 3,40 (с, 2 H), 2,65-2,62 (м, 2 H), 2,58-2,53 (м, 2 H), 2,29 (с, 4 H), 1,80 (т, 2 H, J=10,7 Гц), 1,49-1,46 (м, 4 H), 1,40-1,38 (м, 2 H), 0,90 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 451,2 (M++1).
Пример 172: Синтез соединения 383
(4-(((3R,5S)-4-(3-(((R)-3-фторпирролидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(((R)-3-фторпирролидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,150 г, 0,394 ммоль) и гидрохлорид (R)-3-фторпирролидина (0,054 г, 0,434 ммоль) растворяли в метиленхлориде (10 мл), и раствор перемешивали при комнатной температуре в течение 30 минут. В реакционный раствор добавляли Na(OAc)3BH (0,125 г, 0,591 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,120 г, 67,1%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 383
Метил 4-(((3R,5S)-4-(3-(((R)-3-фторпирролидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,060 г, 0,132 ммоль), гидроксиламин (0,081 мл, 1,323 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,172 г, 2,646 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 383 (0,048 г, 79,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,58 (ушир. с, 1 H), 7,67 (д, 2 H, J=8,0 Гц), 7,28-7,17 (м, 5 H), 7,11 (с, 1 H), 5,18 (д, 1 H, J=55,7 Гц), 3,72 (с, 2 H), 3,39 (с, 2 H), 2,80-2,71 (м, 2 H), 2,64 (д, 2 H, J=10,5 Гц), 2,60-2,55 (м, 2 H), 2,31-2,27 (м, 1 H), 2,17-1,89 (м, 2 H), 1,79 (т, 2 H, J=10,4 Гц), 0,89 (д, 6 H, J=5,9 Гц).
Пример 173: Синтез соединения 384
(4-(((3R,5S)-4-(3-((3,3-дифторазетидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((3,3-дифторазетидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,150 г, 0,394 ммоль) и гидрохлорид 3,3-дифторазетидина (0,056 г, 0,434 ммоль) растворяли в метиленхлориде (4 мл), и раствор перемешивали при комнатной температуре в течение 30 минут. В реакционный раствор добавляли Na(OAc)3BH (0,125 г, 0,591 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,131 г, 72,6%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 384
Метил 4-(((3R,5S)-4-(3-((3,3-дифторазетидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,058 г, 0,127 ммоль), гидроксиламин (0,078 мл, 1,268 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,165 г, 2,535 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 384 (0,037 г, 63,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,2 Гц), 7,32 (д, 2 H, J=8,2 Гц), 7,27-7,24 (м, 3 H), 7,11-7,10 (м, 1 H), 3,72 (с, 2 H), 3,70 (с, 2 H), 3,58 (т, 4 H, J=12,5 Гц), 3,43 (с, 2 H), 2,65-2,63 (м, 2 H), 2,59-2,53 (м, 2 H), 1,81 (т, 2 H, J=10,6 Гц), 0,89 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 459,2 (M++1).
Пример 174: Синтез соединения 385
(4-(((3R,5S)-4-(3-((4-бензилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((4-бензилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,150 г, 0,394 ммоль) и 1-бензилпиперазин (0,076 г, 0,434 ммоль) растворяли в метиленхлориде (4 мл), и раствор перемешивали при комнатной температуре в течение 30 минут. В реакционный раствор добавляли Na(OAc)3BH (0,125 г, 0,591 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,158 г, 74,1%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 385
Метил 4-(((3R,5S)-4-(3-((4-бензилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,095 г, 0,176 ммоль), гидроксиламин (0,107 мл, 1,757 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,229 г, 3,514 ммоль) при комнатной температуре растворяли в метаноле (3 мл), и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 385 (0,084 г, 88,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,67 (д, 2 H, J=8,2 Гц), 7,33-7,22 (м, 10 H), 7,09-7,07 (м, 1 H), 3,71 (с, 2 H), 3,45 (с, 2 H), 3,44 (с, 2 H), 3,40 (с, 2 H), 2,65-2,62 (м, 2 H), 2,56-2,52 (м, 2 H), 2,37-2,34 (м, 8 H), 1,79 (т, 2 H, J=10,5 Гц), 0,89 (д, 6 H, J=5,9 Гц); ЖХМС (ES) m/z 542,3 (M++1).
Пример 175: Синтез соединения 386
(N-гидрокси-4-(((3R,5S)-4-(3-((4-(2-гидрокси-2-метилпропил)пиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензамид)
Стадия 1: Синтез трет-бутил 4-(3-(((2S,6R)-4-(4-(метоксикарбонил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензил)пиперазин-1-карбоксилата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,500 г, 1,314 ммоль) и трет-бутил пиперазин-1-карбоксилат (0,269 г, 1,446 ммоль) растворяли в метиленхлориде (10 мл), и раствор перемешивали при комнатной температуре в течение 30 минут. В реакционный раствор добавляли Na(OAc)3BH (0,418 г, 1,971 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,303 г, 41,9%) в виде масла желтого цвета.
Стадия 2: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиперазин-1-илметил)бензил)пиперазин-1-ил)метил)бензоата
Трет-бутил 4-(3-(((2S,6R)-4-(4-(метоксикарбонил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензил)пиперазин-1-карбоксилат (формула 7-1, 0,303 г, 0,550 ммоль) растворяли в 1,4-диоксане (5 мл) при комнатной температуре и в раствор добавляли HCl (4,00 M раствор в 1,4-диоксане, 1,375 мл, 5,502 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,235 г, 94,8%, масло желтого цвета).
Стадия 3: Синтез этил 4-(((3R,5S)-4-(3-((4-(2-гидрокси-2-метилпропил)пиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиперазин-1-илметил)бензил)пиперазин-1-ил)метил)бензоат (формула 7-2, 0,190 г, 0,422 ммоль), оксид изобутилена (0,380 мл, 4,216 ммоль) и K2CO3 (0,583 г, 4,216 ммоль) добавляли к этанолу (5 мл) и нагревали с помощью микроволнового облучения при температуре 110°C в течение 20 минут, затем охлаждали до комнатной температуры для прекращения реакции. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали и концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,196 г, 84,2%, масло желтого цвета).
Стадия 4: Синтез соединения 386
Этил 4-(((3R,5S)-4-(3-((4-(2-гидрокси-2-метилпропил)пиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 7-3, 0,050 г, 0,093 ммоль), гидроксиламин (0,057 мл, 0,932 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,121 г, 1,863 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 386 (0,032 г, 65,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,17 (ушир. с, 1 H)9,01 (ушир. с, 1 H)7,70 (д, 2 H, J=8,0 Гц), 7,34 (д, 2 H, J=7,8 Гц), 7,27 (с, 1 H), 7,23-7,21 (м, 1 H), 7,09 (д, 1 H, J=6,6 Гц), 4,06 (ушир. с, 1 H)3,72 (с, 2 H), 3,43 (с, 2 H), 3,42 (с, 2 H), 2,65-2,62 (м, 2 H), 2,55-2,54 (м, 2 H).
Пример 176: Синтез соединения 387
(4-(((3R,5S)-4-(3-((4-(2-фтор-2-метилпропил)пиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез этил 4-(((3R,5S)-4-(3-((4-(2-гидрокси-2-метилпропил)пиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Этил 4-(((3R,5S)-4-(3-((4-(2-гидрокси-2-метилпропил)пиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 7-3, 0,150 г, 0,287 ммоль) растворяли в метиленхлориде (5 мл), и при температуре 0°C добавляли DAST (0,042 мл, 0,316 ммоль). Смесь перемешивали при той же температуре в течение 30 минут, и затем перемешивали при комнатной температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,098 г, 63,4%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 387
Этил 4-(((3R,5S)-4-(3-((4-(2-гидрокси-2-метилпропил)пиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 7-4, 0,098 г, 0,177 ммоль), гидроксиламин (0,108 мл, 1,770 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,230 г, 3,540 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр, снабженный картриджем с безводным сульфатом натрия, для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 387 (0,089 г, 93,0%) в виде твердого вещества абрикосового цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, 2 H, J=7,9 Гц), 7,29 (д, 3 H, J=7,7 Гц), 7,23-7,21 (м, 2 H), 7,09-7,08 (м, 1 H), 3,72 (с, 2 H), 3,43 (с, 2 H), 3,41 (с, 2 H), 2,65-2,62 (м, 2 H), 2,56-2,54 (м, 2 H), 2,47-2,43 (м, 4 H), 2,40 (д, 2 H, J=23,3 Гц), 2,35-2,33 (м, 4 H), 1,80 (т, 2 H, J=10,4 Гц), 1,32 (с, 3 H), 1,26 (с, 3 H), 0,90 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 526,3 (M++1).
Пример 177: Синтез соединения 388
(4-(((3R,5S)-4-(4-((1-(2-фтор-2-метилпропил)пиперидин-4-ил)метокси)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид
Стадия 1: Синтез трет-бутил 4-((4-(гидроксиметил)фенокси)метил)пиперидин-1-карбоксилата
4-(Гидроксиметил)фенол (0,500 г, 4,028 ммоль), трет-бутил 4-((метилсульфонилокси)метил)пиперидин-1-карбоксилат (формула 12-1, 1,300 г, 4,430 ммоль), Cs2CO3 (1,968 г, 6,042 ммоль) и NaI (0,030 г, 0,201 ммоль) смешивали в ацетонитриле (20 мл) при комнатной температуре, и смесь нагревали при кипячении с обратным холодильником в течение 17 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением желаемого соединения (0,552 г, 42,6%) в виде бесцветного масла.
Стадия 2: Синтез трет-бутил 4-((4-(хлорметил)фенокси)метил)пиперидин-1-карбоксилата
Трет-бутил 4-((4-(гидроксиметил)фенокси)метил)пиперидин-1-карбоксилат (формула 12-2, 0,500 г, 1,556 ммоль) и TEA (0,434 мл, 3,111 ммоль) растворяли в метиленхлориде (30 мл) при температуре 0°C, и в раствор добавляли метансульфонилхлорид (0,132 мл, 1,711 ммоль), затем перемешивали при той же температуре в течение 2 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 40%) и концентрировали с получением желаемого соединения (0,136 г, 25,7%) в виде твердого вещества белого цвета.
Стадия 3: Синтез трет-бутил 4-((4-(((2S,6R)-4-(4-(метоксикарбонил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенокси)метил)пиперидин-1-карбоксилата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), трет-бутил 4-((4-(хлорметил)фенокси)метил)пиперидин-1-карбоксилат (формула 12-3, 0,130 г, 0,381 ммоль), Cs2CO3 (0,186 г, 0,572 ммоль) и NaI (0,003 г, 0,019 ммоль) смешивали в ацетонитриле (5 мл) при комнатной температуре, и смесь нагревали при кипячении с обратным холодильником в течение 3 часов и затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 10% до 60%) и концентрировали с получением желаемого соединения (0,130 г, 60,3%) в виде масла желтого цвета.
Стадия 4: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-(пиперидин-4-илметокси)бензил)пиперазин-1-ил)метил)бензоата (гидрохлоридная соль)
Трет-бутил 4-((4-(((2S,6R)-4-(4-(метоксикарбонил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенокси)метил)пиперидин-1-карбоксилат (формула 12-4, 0,150 г, 0,265 ммоль) растворяли в 1,4-диоксане (5 мл) при комнатной температуре, и в раствор добавляли HCl (4,00 M раствор в 1,4-диоксане, 1,326 мл, 5,303 ммоль), затем перемешивали при той же температуре в течение 17 часов. Выпавший твердый продукт промывали диэтиловым эфиром и сушили с получением желаемого соединения (0,126 г, 94,6%) в виде твердого вещества белого цвета.
Стадия 5: Синтез этил 4-(((3R,5S)-4-(4-((1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)метокси)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Гидрохлорид метил 4-(((3R,5S)-3,5-диметил-4-(4-(пиперидин-4-илметокси)бензил)пиперазин-1-ил)метил)бензоата (формула 12-5, 0,102 г, 0,203 ммоль), 1,1-диметилоксиран (0,073 г, 1,016 ммоль) и K2CO3 (0,281 г, 2,032 ммоль) добавляли к этанолу (3 мл) и нагревали с помощью микроволнового облучения при температуре 110°C в течение 20 минут, затем охлаждали до комнатной температуры. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,102 г, 93,4%, масло бледно-желтого цвета).
Стадия 6: Синтез этил 4-(((3R,5S)-4-(4-((1-(2-фтор-2-метилпропил)пиперидин-4-ил)метокси)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Этил 4-(((3R,5S)-4-(4-((1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)метокси)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 12-6, 0,102 г, 0,185 ммоль) растворяли в метиленхлориде (5 мл), и при температуре 0°C добавляли DAST (0,027 мл, 0,203 ммоль). Смесь перемешивали при той же температуре в течение 30 минут и затем перемешивали при комнатной температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,083 г, 81,1%, масло бледно-коричневого цвета).
Стадия 7: Синтез соединения 388
Этил 4-(((3R,5S)-4-(4-((1-(2-фтор-2-метилпропил)пиперидин-4-ил)метокси)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 12-7, 0,083 г, 0,150 ммоль), гидроксиламин (0,092 мл, 1,499 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,195 г, 2,998 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 388 (0,073 г, 90,1%) в виде твердого вещества абрикосового цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, 2 H, J=8,2 Гц), 7,29 (д, 2 H, J=8,1 Гц), 7,21 (д, 2 H, J=8,6 Гц), 6,84 (д, 2 H, J=8,6 Гц), 3,77 (д, 2 H, J=5,9 Гц), 3,68 (с, 2 H), 3,40 (с, 2 H), 2,91 (д, 2 H, J=11,5 Гц), 2,62 (д, 2 H, J=9,8 Гц), 2,55-2,53 (м, 2 H), 2,41 (д, 2 H, J=22,9 Гц), 2,08 (т, 2 H, J=10,8 Гц), 1,81-1,69 (м, 6 H), 1,33 (с, 3 H), 1,29 (с, 4 H), 0,92 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 541,3 (M++1).
Пример 178: Синтез соединения 396
(4-(((2S,6R)-4-(3-(фуран-3-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(3-(фуран-3-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан (1 мл)/вода (0,5 мл) добавляли к смеси метил 4-(((2S,6R)-4-(3-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 14-1, 0,100 г, 0,209 ммоль), фуран-3-илборной кислоты (0,028 г, 0,251 ммоль), Pd(dbpf)Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,066 г, 0,627 ммоль), и раствор нагревали путем воздействия микроволнового излучения при температуре 120°C в течение 20 минут, и затем фильтровали через целит и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,051 г, 58,3%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 396
Метил 4-(((2S,6R)-4-(3-(фуран-3-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 14-2, 0,030 г, 0,072 ммоль), гидроксиламин (0,088 мл, 1,434 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,040 г, 0,717 ммоль) смешивали в метаноле (0,5 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 396 (0,020 г, 66,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,18 (с, 1 H), 7,73 (с, 1 H), 7,64 (д, 2 H, J=8,0 Гц), 7,49-7,47 (м, 2 H), 7,34-7,18 (м, 4 H), 6,94 (д, 1 H, J=0,96 Гц), 3,72 (с, 2 H), 3,41 (с, 2 H), 2,68-2,65 (м, 2 H), 2,55-2,51 (м, 2 H), 1,84-1,79 (м, 2 H), 0,91 (с, 3H), 0,89 (с, 3 H).
Пример 179: Синтез соединения 400
(4-(((2S,6R)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-4-ил)метил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-4-ил)метил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан (1 мл)/вода (0,5 мл) добавляли к смеси метил 4-(((2S,6R)-4-(4-йодбензил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 14-1, 0,100 г, 0,209 ммоль), (4,4-диметилциклогекс-1-ен-1-ил)бороновой кислоты (0,038 г, 0,251 ммоль), Pd(dbpf)Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,066 г, 0,627 ммоль), и раствор нагревали путем воздействия микроволнового излучения при температуре 120°C в течение 20 минут, после чего фильтровали через целит и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,05 г, 51,9%) в виде твердого вещества бледно-коричневого цвета.
Стадия 2: Синтез соединения 400
Метил 4-(((2S,6R)-4-((4’,4’-диметил-2’,3’,4’,5’-тетрагидро-[1,1’-бифенил]-4-ил)метил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 14-3, 0,030 г, 0,065 ммоль), гидроксиламин (0,080 мл, 1,303 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,037 г, 0,651 ммоль) смешивали в метаноле (0,5 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 400 (0,023 г, 76,5%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,64 (д, 2 H, J=7,9 Гц), 7,36 (д, 2 H, J=8,1 Гц), 7,24-7,20 (м, 4 H), 6,09 (ушир. с, 1 H), 3,71 (с, 2 H), 2,64-2,51 (м, 4 H), 2,37 (ушир. с, 2 H), 1,97 (ушир. с, 2 H), 1,80-1,75 (м, 2 H), 1,49-1,46 (м, 2 H), 0,93-0,89 (м, 12 H).
Пример 180: Синтез соединения 401
(4-(((2S,6R)-4-(4-(3,6-дигидро-2H-пиран-4-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-4-(4-(3,6-дигидро-2H-пиран-4-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Смесь 1,2-диметоксиэтан (1 мл)/вода (0,5 мл) добавляли к смеси метил 4-(((2S,6R)-4-(4-йодбензил)-2,6-диметилпиперазин-1-ил)метил)бензоата (формула 14-1, 0,100 г, 0,209 ммоль), (3,6-дигидро-2H-пиран-4-ил)бороновой кислоты (0,032 г, 0,251 ммоль), Pd(dbpf)Cl2 (0,007 г, 0,010 ммоль) и Na2CO3 (0,066 г, 0,627 ммоль) и затем нагревали путем воздействия микроволнового излучения при температуре 120°C в течение 20 минут. Реакционный раствор фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,029 г, 31,9%) в виде твердого вещества бледно-коричневого цвета.
Стадия 2: Синтез соединения 401
Метил 4-(((2S,6R)-4-(4-(3,6-дигидро-2H-пиран-4-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 14-2, 0,025 г, 0,058 ммоль), гидроксиламин (0,070 мл, 1,151 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,032 г, 0,575 ммоль) смешивали в метаноле (0,5 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением соединения 401 (0,011 г, 43,9%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,66 (д, 2 H, J=8,12 Гц), 7,40-7,36 (м, 4 H), 7,25 (д, 2 H, J=8,12 Гц), 6,23 (ушир. с, 1 H), 4,21 (с, 2 H), 3,81 (т, 2 H, J=5,3 Гц), 3,73 (с, 2 H), 3,39 (с, 2 H), 2,66-2,54 (м, 4 H), 2,47-2,43 (м, 2 H), 1,82-1,76 (м, 2 H), 0,88 (с, 3H), 0,87 (с, 3 H).
Пример 181: Синтез соединения 402
(4-(((3R,5S)-3,5-диметил-4-(тетрагидрофуран-2-карбонил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез (3R,5S)-трет-бутил 4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-карбоксилата
(3R,5S)-трет-Бутил 3,5-диметилпиперазин-1-карбоновую кислоту (формула 8-1, 0,500 г, 2,333 ммоль), фуран-2-карбонилхлорид (0,276 мл, 2,800 ммоль) и TEA (0,976 мл, 7,000 ммоль) растворяли в метиленхлориде (10 мл) при температуре 0°C, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционный раствор добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением желаемого соединения (0,620 г, 86,2%) в виде бесцветного масла.
Стадия 2: Синтез (3R,5S)-трет-бутил 3,5-диметил-4-(тетрагидрофуран-2-карбонил)пиперазин-1-карбоксилат
(3R,5S)-трет-Бутил 4-(фуран-2-карбонил)-3,5-диметилпиперазин-1-карбоксилат (формула 11-1, 0,100 г, 0,324 ммоль) растворяли в смеси этанол/вода (8:1) (2 мл) при комнатной температуре, и медленно добавляли Pd/C (0,010 г). После подсоединения баллона с водородом к сосуду с раствором перемешивали при той же температуре в течение 2 часов. Реакционную смесь фильтровали через слой из целита для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении для удаления растворителя. Продукт использовали без дополнительной очистки (0,100 г, 98,7%, бесцветное масло).
Стадия 3: Синтез ((2S,6R)-2,6-диметилпиперазин-1-ил)(тетрагидрофуран-2-ил)метанона (гидрохлоридная соль)
(3R,5S)-трет-Бутил 3,5-диметил-4-(тетрагидрофуран-2-карбонил)пиперазин-1-карбоксилат (формула 11-2, 0,540 г, 1,729 ммоль) и HCl (4,00 M раствор в 1,4-диоксане, 8,643 мл, 34,571 ммоль) растворяли в 1,4-диоксане (20 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и продукт использовали без дополнительной очистки (0,400 г, 109,0%, бесцветное масло).
Стадия 4: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(тетрагидрофуран-2-карбонил)пиперазин-1-ил)метил)бензоата
Гидрохлорид ((2S,6R)-2,6-диметилпиперазин-1-ил)(тетрагидрофуран-2-ил)метанона (формула 11-3, 0,400 г, 1,608 ммоль), метил 4-(бромметил)бензоат (формула 1-1, 0,368 г, 1,608 ммоль) и Cs2CO3 (1,572 г, 4,824 ммоль) растворяли в ацетонитриле (15 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Реакционную смесь фильтровали через стеклянный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 40%) и концентрировали с получением желаемого соединения (0,196 г, 33,8%) в виде бесцветного масла.
Стадия 5: Синтез соединения 402
Метил 4-(((3R,5S)-3,5-диметил-4-(тетрагидрофуран-2-карбонил)пиперазин-1-ил)метил)бензоат (0,140 г, 0,388 ммоль), гидроксиламин (0,238 мл, 3,884 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,436 г, 7,768 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 402 (0,067 г, 47,7%) в виде твердого вещества абрикосового цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,10 (ушир. с, 1 H) 9,00 (ушир. с, 1 H) 7,73 (д, 2 H, J=8,1 Гц), 7,43 (д, 2 H, J=8,0 Гц), 4,63-4,56 (м, 1 H), 4,37-4,35 (м, 2 H), 3,75-3,72 (м, 2 H), 3,35 (с, 2 H), 2,65-2,62 (м, 2 H), 2,11-2,09 (м, 3 H), 1,88-1,80 (м, 3 H), 1,35-1,21 (м, 6 H), 1,35-1,21 (м, 6 H); ЖХМС (ES) m/z 362,1 (M++1).
Пример 182: Синтез соединения 403
(4-(((3R,5S)-3,5-диметил-4-(1H-пиразол-3-карбонил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(1H-пиразол-3-карбонил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), 1H-пиразол-3-карбоновую кислоту (0,047 г, 0,419 ммоль), HATU (0,290 г, 0,762 ммоль) и DIPEA (0,333 мл, 1,906 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,150 г, 110,4%, масло желтого цвета).
Стадия 2: Синтез соединения 403
Метил 4-(((3R,5S)-3,5-диметил-4-(1H-пиразол-3-карбонил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,150 г, 0,421 ммоль), гидроксиламин (0,257 мл, 4,209 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,472 г, 8,417 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и насыщенный водный раствор гидрокарбоната натрия (20 мл) и к концентрату добавляли метиленхлорид (10 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали водой и сушили с получением соединения 403 (0,046 г, 30,6%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 13,12 (ушир. с, 1 H),11,19 (ушир. с, 1 H),3,02 (brs 1 H),7,79 (с, 1 H), 7,73 (д, 2 H, J=8,2 Гц), 7,45 (д, 2 H, J=8,3 Гц), 6,54 (д, 1 H, J=2,0 Гц), 4,69 (ушир. с, 1 H),3,55 (с, 2 H), 2,68-2,65 (м, 2 H), 2,16-2,12 (м, 2 H), 1,35 (д, 6 H, J=6,7 Гц); ЖХМС (ES) m/z 358,2 (M++1).
Пример 183: Синтез соединения 404
(4-(((3R,5S)-3,5-диметил-4-(пиразин-2-карбонил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(пиразин-2-карбонил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль), пиразин-2-карбоновую кислоту (0,052 г, 0,419 ммоль), HATU (0,290 г, 0,762 ммоль) и DIPEA (0,333 мл, 1,906 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,150 г, 106,8%, масло коричневого цвета).
Стадия 2: Синтез соединения 404
Метил 4-(((3R,5S)-3,5-диметил-4-(пиразин-2-карбонил)пиперазин-1-ил)метил)бензоат (формула 1-3, 0,150 г, 0,408 ммоль), гидроксиламин (0,250 мл, 4,082 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,458 г, 8,165 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18; смесь 0,1%-ный водный раствор трифторуксусной кислоты/ацетонитрил=от 5% до 80%), после чего его пропускали через картридж SPE (PL-HCO3 MP SPE) и концентрировали с получением соединения 404 (0,043 г, 28,6%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CD3OD) δ 8,85 (д, 1 H, J=1,5 Гц), 8,75 (д, 1 H, J=2,5 Гц), 8,65 (дд, 1 H, J=2,6, 1,6 Гц), 7,82 (д, 2 H, J=8,2 Гц), 7,60 (д, 2 H, J=8,2 Гц), 4,10 (ушир. с, 2 H), 3,18 (ушир. с, 2 H), 2,85 (ушир. с, 2 H), 1,52 (д, 6 H, J=7,1 Гц); ЖХМС (ES) m/z 370,1 (M++1).
Пример 184: Синтез соединения 405
(N-гидрокси-4-(((3R,5S)-4-изоникотиноил-3,5-диметилпиперазин-1-ил)метил)бензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-изоникотиноил-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,100 г, 0,381 ммоль) и TEA (0,159 мл, 1,144 ммоль) растворяли в метиленхлориде (4 мл) при комнатной температуре и в раствор добавляли изоникотиноилхлорид (0,081 г, 0,572 ммоль), затем перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,150 г, 107,1%, масло бледно-желтого цвета).
Стадия 2: Синтез соединения 405
Метил 4-(((3R,5S)-4-изоникотиноил-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-3, 0,150 г, 0,408 ммоль), гидроксиламин (0,250 мл, 4,082 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,458 г, 8,165 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18; смесь 0,1%-ный водный раствор трифторуксусной кислоты/ацетонитрил=от 5% до 80%), после чего его пропускали через картридж SPE (PL-HCO3 MP SPE) и концентрировали с получением соединения 405 (0,043 г, 28,6%)в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 8,73 (д, 2 H, J=6,2 Гц), 7,79 (д, 2 H, J=8,2 Гц), 7,60-7,55 (м, 4 H), 3,89 (ушир. с, 2 H), 2,96 (ушир. с, 2 H), 2,61 (ушир. с, 2 H), 1,45 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 369,1 (M++1).
Пример 185: Синтез соединения 411
(4-(((3R,5S)-4-(4-(((R)-3-фторпирролидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-(((R)-3-фторпирролидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,100 г, 0,263 ммоль) растворяли в метиленхлориде (1 мл) при комнатной температуре и в смесь добавляли (R)-3-фторпирролидин (0,036 г, 0,289 ммоль), затем перемешивали в течение 30 минут. В смесь добавляли Na(OAc)3BH (0,061 г, 0,289 ммоль), затем перемешивали в течение ночи при той же температуре. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,059 г, 49,7%) в виде твердого вещества бледно-коричневого цвета.
Стадия 2: Синтез соединения 411
Метил 4-(((3R,5S)-4-(4-(((R)-3-фторпирролидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,059 г, 0,130 ммоль), гидроксиламин (0,159 мл, 2,601 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,073 г, 1,301 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 2 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (10 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали водой и сушили с получением соединения 411 (0,011 г, 17,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,67 (д, 1 H, J=11,2 Гц), 7,43-7,20 (м, 6 H), 5,25-5,11 (м, 1 H), 3,72 (с, 2 H), 3,55 (с, 2 H), 3,43 (с, 2 H), 2,81-2,53 (м, 7 H), 2,35-2,08 (м, 2 H), 1,90-1,78 (м, 3 H), 0,91-0,89 (м, 6 H).
Пример 186: Синтез соединения 412
(4-(((3R,5S)-4-(4-((3,3-дифторазетидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-((3,3-дифторазетидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,100 г, 0,263 ммоль) растворяли в метиленхлориде (1 мл) при комнатной температуре и в смесь добавляли 3,3-дифторазетидин (0,037 г, 0,289 ммоль), затем перемешивали в течение 30 минут. В реакционную смесь добавляли Na(OAc)3BH (0,061 г, 0,289 ммоль), перемешивали в течение ночи при той же температуре. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,073 г, 60,6%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 412
Метил 4-(((3R,5S)-4-(4-((3,3-дифторазетидин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,073 г, 0,160 ммоль), гидроксиламин (0,195 мл, 3,191 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,090 г, 1,595 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 2 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (10 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали водой и сушили с получением соединения 412 (0,014 г, 18,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,2 Гц), 7,30-7,19 (м, 6 H), 3,71-3,67 (м, 4 H), 3,59-3,52 (м, 5 H), 3,41 (м, 1 H), 2,64-2,62 (м, 4 H), 1,80 (т, 2 H, J=10,4 Гц), 0,90-0,88 (м, 6 H).
Пример 187: Синтез соединения 413
(4-(((3R,5S)-4-(4-((4-бензилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-((4-бензилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,100 г, 0,263 ммоль) растворяли в метиленхлориде (1 мл) и в смесь добавляли 1-бензилпиперазин (0,049 мл, 0,289 ммоль), затем перемешивали в течение 30 минут. В реакционную смесь добавляли Na(OAc)3BH (0,061 г, 0,289 ммоль), затем перемешивали в течение ночи при той же температуре. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,074 г, 51,8%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 413
Метил 4-(((3R,5S)-4-(4-((4-бензилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,074 г, 0,137 ммоль), гидроксиламин (0,168 мл, 2,742 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,077 г, 1,371 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 2 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (10 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали водой и сушили с получением соединения 413 (0,053 г, 71,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,67 (д, 2 H, J=8,0 Гц), 7,32-7,14 (м, 11 H), 3,70 (с, 2 H), 3,44 (м, 6 H), 3,22 (м, 2 H), 2,68-2,64 (м, 4 H), 2,35 (м, 6 H), 1,78 (т, 2 H, J=10,4 Гц), 0,90-0,88 (м, 6 H).
Пример 188: Синтез соединения 423
(4-(((3R,5S)-3,5-диметил-4-(3-(пирролидин-1-илметил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез 3-(хлорметил)бензоилхлорида
3-(Бромметил)бензойную кислоту (4,301 г, 20,000 ммоль) и SOCl2 (43,793 мл, 600,000 ммоль) смешивали при температуре 50°C, и смесь перемешивали в течение ночи при той же температуре, затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и продукт использовали без дополнительной очистки (3,778 г, 99,9%, жидкость коричневого цвета).
Стадия 2: Синтез метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 2,622 г, 9,993 ммоль), 3-(хлорметил)бензоилхлорид (формула 6-1, 3,778 г, 19,986 ммоль) и TEA (2,770 мл, 19,986 ммоль) растворяли в метиленхлориде (40 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 3 часов. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; 100% метиленхлорид) и концентрировали с получением желаемого соединения (2,047 г, 49,4%) в виде жидкости коричневого цвета.
Стадия 3: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(пирролидин-1-илметил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,178 г, 0,388 ммоль), пирролидин (0,032 мл, 0,388 ммоль) и TEA (0,108 мл, 0,777 ммоль) растворяли в метиленхлориде (2 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,155 г, 88,8%) в виде жидкости бледно-желтого цвета.
Стадия 4: Синтез соединения 423
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(пирролидин-1-илметил)бензоил)пиперазин-1-ил)метил)бензоат (формула 6-3, 0,155 г, 0,345 ммоль), гидроксиламин (0,422 мл, 6,895 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,193 г, 3,448 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением соединения 423 (0,070 г, 44,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,2 (ушир. с, 1 H), 9,03 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,6 Гц), 7,43 (д, 2 H, J=8,2 Гц), 7,38-7,32 (м, 2 H), 7,24 (с, 1 H), 7,20-7,17 (м, 2 H), 4,06 (ушир. с, 2 H), 3,58-3,54 (м, 4 H), 2,68-2,61 (м, 2 H), 2,40 (с, 4 H), 2,16-2,12 (м, 2 H), 1,69-1,67 (м, 4 H), 1,21-1,28 (м, 6 H).
Пример 189: Синтез соединения 424
(4-(((3R,5S)-3,5-диметил-4-(3-(пиперидин-1-илметил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиперидин-1-илметил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,165 г, 0,359 ммоль), пиперидин (0,036 мл, 0,359 ммоль) и Cs2CO3 (0,142 г, 0,431 ммоль) растворяли в ацетонитриле (1,5 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,127 г, 76,0%) в виде бесцветной жидкости.
Стадия 2: Синтез соединения 424
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(пиперидин-1-илметил)бензоил)пиперазин-1-ил)метил)бензоат (формула 6-3, 0,127 г, 0,273 ммоль), гидроксиламин (0,334 мл, 5,461 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,153 г, 2,731 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 424 (0,101 г, 79,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,72 (д, 2 H, J=8,2 Гц), 7,43 (д, 2 H, J=8,1 Гц), 7,38-7,30 (м, 2 H), 7,23 (с, 1 H), 7,20-7,18 (м, 1 H), 3,54 (с, 2 H), 3,44 (с, 2 H), 2,63-2,61 (м, 2 H), 2,30 (м, 4 H), 2,16-2,12 (м, 2 H), 1,49-1,46 (м, 4 H), 1,38-1,37 (м, 2 H), 1,29 (с, 6 H).
Пример 190: Синтез соединения 425
(4-(((3R,5S)-4-(3-((диэтиламино)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((диэтиламино)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,165 г, 0,359 ммоль), диэтиламин гидрохлорид (0,039 г, 0,359 ммоль) и Cs2CO3 (0,142 г, 0,431 ммоль) растворяли в ацетонитриле (1,5 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. Затем в реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,115 г, 71,1%) в виде бесцветной жидкости.
Стадия 2: Синтез соединения 425
Метил 4-(((3R,5S)-4-(3-((диэтиламино)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-3, 0,115 г, 0,255 ммоль), гидроксиламин (0,312 мл, 5,106 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,143 г, 2,553 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением соединения 425 (0,061 г, 53,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,2 (ушир. с, 1 H), 9,03 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,43 (д, 2 H, J=8,2 Гц), 7,36-7,34 (м, 2 H), 7,25 (с, 1 H), 7,18-7,16 (м, 1 H), 3,54 (с, 4 H), 2,63-2,61 (м, 2 H), 2,46-2,41 (м, 4 H), 2,16-2,12 (м, 2 H), 1,29 (м, 6 H), 0,98-0,94 (м, 6 H).
Пример 191: Синтез соединения 426
(4-(((3R,5S)-4-(3-((диэтиламино)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((диэтиламино)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль) и диэтиламин гидрохлорид (0,086 г, 0,788 ммоль) растворяли в метиленхлориде (10 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В раствор добавляли Na(OAc)3BH (0,223 г, 1,051 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,026 г, 11,3%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 426
Метил 4-(((3R,5S)-4-(3-((диэтиламино)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,026 г, 0,059 ммоль), гидроксиламин (0,073 мл, 1,188 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,033 г, 0,594 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционный раствор концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 426 (0,019 г, 72,9%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 1,10 (ушир. с, 1 H), 9,00 (ушир. с, 1 H), 7,69 (д, 2 H, J=8,3 Гц), 7,33 (д, 2 H, J=8,1 Гц), 7,29 (с, 1 H), 7,24-7,18 (м, 2 H), 7,12-7,10 (м, 1 H), 3,72 (с, 2 H), 3,50 (с, 2 H), 3,43 (с, 2 H), 2,68-2,54 (м, 4 H), 2,43 (кв, 4 H, J=7,1 Гц), 1,80 (т, 2 H, J=10,5 Гц), 0,96 (т, 6 H, J=7,1 Гц), 0,90 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 439,3 (M++1).
Пример 192: Синтез соединения 427
(4-(((3R,5S)-3,5-диметил-4-(3-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль) и пирролидин (0,066 мл, 0,788 ммоль) растворяли в метиленхлориде (4 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В раствор добавляли Na(OAc)3BH (0,223 г, 1,051 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,161 г, 70,3%) в виде бесцветного масла.
Стадия 2: Синтез соединения 427
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,086 г, 0,197 ммоль), гидроксиламин (0,242 мл, 3,949 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,111 г, 1,974 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 427 (0,033 г, 38,3%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,10 (ушир. с, 1 H), 9,00 (ушир. с, 1 H), 7,70 (д, 2 H, J=8,2 Гц), 7,35 (д, 2 H, J=8,1 Гц), 7,26 (с, 1 H), 7,22-7,21 (м, 2 H), 7,11-7,09 (м, 1 H), 3,72 (с, 2 H), 3,54 (с, 2 H), 3,44 (с, 2 H), 2,65-2,55 (м, 4 H), 2,40 (с, 4 H), 1,81 (т, 2 H, J=10,5 Гц), 1,69 (с, 4 H), 0,90 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 437,3 (M++1).
Пример 193: Синтез соединения 428
(4-(((3R,5S)-3,5-диметил-4-(3-((4-метилпиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-метилпиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль) и 1-метилпиперазин (0,088 мл, 0,788 ммоль) растворяли в метиленхлориде (4 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В раствор добавляли Na(OAc)3BH (0,223 г, 1,051 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,152 г, 62,2%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 428
Метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-метилпиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,075 г, 0,161 ммоль), гидроксиламин (0,197 мл, 3,228 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,091 г, 1,614 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 428 (0,031 г, 41,2%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,2 Гц), 7,30 (д, 2 H, J=8,1 Гц), 7,27 (с, 1 H), 7,23-7,21 (м, 2 H), 7,09-7,08 (м, 1 H), 3,72 (с, 2 H), 3,43 (с, 2 H), 3,42 (с, 2 H), 2,63 (д, 2 H, J=10,2 Гц), 2,58-2,51 (м, 2 H), 2,36-2,33 (м, 8 H), 1,80 (т, 2 H, J=10,5 Гц), 0,90 (д, 6 H, J=6,1 Гц).
Пример 194: Синтез соединения 429
(4-(((3R,5S)-4-(3-((4-этилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((4-этилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль) и 1-этилпиперазин (0,100 мл, 0,788 ммоль) растворяли в метиленхлориде (4 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В раствор добавляли Na(OAc)3BH (0,223 г, 1,051 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,099 г, 39,3%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 429
Метил 4-(((3R,5S)-4-(3-((4-этилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,055 г, 0,115 ммоль), гидроксиламин (0,141 мл, 2,298 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,064 г, 1,149 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 429 (0,031 г, 56,2%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,10 (ушир. с, 1 H), 9,00 (ушир. с, 1 H), 7,70 (д, 2 H, J=8,3 Гц), 7,34 (д, 2 H, J=8,2 Гц), 7,27 (с, 1 H), 7,23-7,21 (м, 2 H), 7,09-7,08 (м, 1 H), 3,72 (с, 2 H), 3,43 (с, 4 H), 2,63 (д, 2 H, J=10,2 Гц), 2,59-2,51 (м, 2 H), 2,36-2,33 (м, 8 H), 2,28 (кв, 2 H, J=7,2 Гц), 1,81 (т, 2 H, J=10,5 Гц), 0,97 (т, 3 H, J=7,2 Гц), 0,90 (д, 6 H, J=6,1 Гц).
Пример 195: Синтез соединения 430
(N-гидрокси-4-(((3R,5S)-4-(3-((4-изопропилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((4-изопропилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль) и 1-изопропилпиперазин (0,113 мл, 0,788 ммоль) растворяли в метиленхлориде (4 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В раствор добавляли Na(OAc)3BH (0,223 г, 1,051 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,225 г, 86,9%) в виде масла бледно-коричневого цвета.
Стадия 2: Синтез соединения 430
Метил 4-(((3R,5S)-4-(3-((4-изопропилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,100 г, 0,203 ммоль), гидроксиламин (0,248 мл, 4,059 ммоль, 50,00%-ный водный раствор) и хлорид калия (0,114 г, 2,030 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 430 (0,055 г, 54,9%)в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,10 (ушир. с, 1 H), 9,00 (ушир. с, 1 H), 7,70 (д, 2 H, J=8,0 Гц), 7,35 (д, 2 H, J=8,0 Гц), 7,09-7,08 (м, 2 H), 3,72 (с, 2 H), 3,43 (с, 2 H), 3,42 (с, 2 H), 2,65-2,62 (м, 2 H), 2,59-2,51 (м, 3 H), 2,41-2,34 (м, 8 H), 1,81 (т, 2 H, J=10,3 Гц), 0,94 (д, 6 H, J=6,5 Гц), 0,90 (д, 6 H, J=6,0 Гц).
Пример 196: Синтез соединения 431
(4-(((3R,5S)-4-(3-((4-ацетилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((4-ацетилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль) и 1-ацетилпиперазин (0,101 г, 0,788 ммоль) растворяли в метиленхлориде (4 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,223 г, 1,051 ммоль), затем перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,253 г, 97,7%) в виде масла бледно-коричневого цвета.
Стадия 2: Синтез соединения 431
Метил 4-(((3R,5S)-4-(3-((4-ацетилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,070 г, 0,142 ммоль), гидроксиламин (0,174 мл, 2,842 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,080 г, 1,421 ммоль) растворяли в метаноле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 431 (0,067 г, 95,5%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,1 Гц), 7,32 (д, 2 H, J=8,1 Гц), 7,28 (с, 1 H), 7,25-7,23 (м, 2 H), 7,12-7,11 (м, 1 H), 3,73 (с, 2 H), 3,43-3,39 (м, 6 H), 2,64 (д, 2 H, J=10,1 Гц), 2,59-2,53 (м, 2 H), 2,34 (т, 2 H, J=4,8 Гц), 2,28 (т, 2 H, J=5,0 Гц), 0,90 (д, 6 H, J=6,1 Гц).
Пример 197: Синтез соединения 432
(4-(((3R,5S)-3,5-диметил-4-(3-((4-метилпиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-метилпиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,200 г, 0,482 ммоль), 1-метилпиперазин (0,059 мл, 0,530 ммоль) и Cs2CO3 (0,188 г, 0,578 ммоль) растворяли в ацетонитриле (2 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,163 г, 70,5%) в виде жидкости бледно-желтого цвета.
Стадия 2: Синтез соединения 432
Метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-метилпиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)бензоат (формула 6-3, 0,163 г, 0,340 ммоль), гидроксиламин (0,416 мл, 6,794 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,191 г, 3,397 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 432 (0,095 г, 58,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,72 (д, 2 H, J=8,2 Гц), 7,43 (д, 2 H, J=8,2 Гц), 7,38-7,30 (м, 2 H), 7,23-7,18 (м, 2 H), 3,54-3,47 (м, 6 H), 2,67-2,61 (м, 2 H), 2,33 (м, 8 H), 2,16-2,13 (м, 5 H), 1,28-1,23 (м, 6 H).
Пример 198: Синтез соединения 433
(4-(((3R,5S)-4-(3-((4-этилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((4-этилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,200 г, 0,482 ммоль), 1-этилпиперазин (0,067 мл, 0,530 ммоль) и Cs2CO3 (0,188 г, 0,578 ммоль) растворяли в ацетонитриле (2 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,165 г, 69,4%) в виде жидкости бледно-желтого цвета.
Стадия 2: Синтез соединения 433
Метил 4-(((3R,5S)-4-(3-((4-этилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-3, 0,165 г, 0,334 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,409 мл, 6,686 ммоль) и гидроксид калия (0,188 г, 3,343 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 433 (0,116 г, 70,0%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,2 (ушир. с, 1 H), 9,03 (ушир. с, 1 H), 7,89 (д, 2 H, J=7,8 Гц), 7,72 (д, 2 H, J=8,2 Гц), 7,44-7,30 (м, 2 H), 7,23 (с, 1 H), 7,20-7,19 (м, 1 H), 4,51 (с, 4 H), 2,67-2,61 (м, 2 H), 2,34-2,25 (м, 8 H), 2,14 (дд, 2 H, J=11,3, 4,1 Гц), 1,28-1,23 (м, 6 H), 0,98-0,85 (м, 3 H).
Пример 199: Синтез соединения 434
(N-гидрокси-4-(((3R,5S)-4-(3-((4-изопропилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((4-изопропилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,200 г, 0,482 ммоль), 1-изопропилпиперазин (0,076 мл, 0,530 ммоль) и Cs2CO3 (0,188 г, 0,578 ммоль) растворяли в ацетонитриле (2 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,170 г, 69,5%) в виде жидкости бледно-желтого цвета.
Стадия 2: Синтез соединения 434
Метил 4-(((3R,5S)-4-(3-((4-изопропилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-3, 0,170 г, 0,335 ммоль), гидроксиламин (0,410 мл, 6,699 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,188 г, 3,349 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 434 (0,128 г, 75,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,2 (ушир. с, 1 H), 9,02 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,43 (д, 2 H, J=8,2 Гц), 7,39-7,30 (м, 2 H), 7,23-7,19 (с, 2 H), 3,54 (с, 2 H), 3,47 (с, 2 H), 2,67-2,60 (м, 3 H), 2,43-2,33 (м, 9 H), 2,15 (дд, 1 H, J=11,3, 4,0 Гц), 1,29-1,18 (м, 6 H), 0,96-0,94 (м, 6 H).
Пример 200: Синтез соединения 435
(4-(((3R,5S)-4-(3-((4-ацетилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((4-ацетилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,200 г, 0,482 ммоль), 1-ацетилпиперазин (0,068 г, 0,530 ммоль) и Cs2CO3 (0,188 г, 0,578 ммоль) растворяли в ацетонитриле (2 мл), и раствор перемешивали в течение ночи при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,186 г, 76,2%) в виде жидкости бледно-желтого цвета.
Стадия 2: Синтез соединения 435
Метил 4-(((3R,5S)-4-(3-((4-ацетилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-3, 0,186 г, 0,367 ммоль), гидроксиламин (0,449 мл, 7,346 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,206 г, 3,673 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 435 (0,121 г, 64,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,2 (ушир. с, 1 H), 9,04 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,1 Гц), 7,42-7,33 (м, 4 H), 7,25-7,20 (с, 2 H), 4,52 (ушир. с, 2 H), 3,53 -3,52 (м, 4 H), 3,41-3,40 (м, 4 H), 2,67-2,62 (м, 2 H), 2,36-2,34 (м, 2 H), 2,29-2,27 (м, 2 H), 2,16-2,13 (м, 2 H), 1,97 (с, 3 H), 1,29 (с, 6 H).
Пример 201: Синтез соединения 439
(4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,200 г, 0,482 ммоль), морфолин (0,069 г, 0,530 ммоль) и Cs2CO3 (0,188 г, 0,578 ммоль) растворяли в ацетонитриле (2 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 10%) и концентрировали с получением желаемого соединения (0,163 г, 66,6%) в виде жидкости бледно-желтого цвета.
Стадия 2: Синтез соединения 439
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензоил)пиперазин-1-ил)метил)бензоат (формула 6-3, 0,163 г, 0,321 ммоль), гидроксиламин (0,392 мл, 6,416 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,180 г, 3,208 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 439 (0,105 г, 64,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,71 (д, 2 H, J=8,2 Гц), 7,39-7,33 (м, 4 H), 7,22 (с, 1 H), 7,22-7,20 (м, 1 H), 4,41 (ушир. с, 1 H) 3,57-3,55 (м, 4 H), 3,48 (с, 4 H), 2,65-2,61 (м, 2 H), 2,34 (с, 4 H), 2,15-2,11 (м, 2 H), 1,29-1,23 (м, 6 H).
Пример 202: Синтез соединения 440
(4-(((3R,5S)-4-(3-(((1-(диэтиламино)пропан-2-ил)амино)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(((1-(диэтиламино)пропан-2-ил)амино)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорфенил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,200 г, 0,482 ммоль), N1,N1-диэтилпропан-1,2-диамин (0,069 г, 0,530 ммоль) и Cs2CO3 (0,188 г, 0,578 ммоль) растворяли в ацетонитриле (2 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 10%) и концентрировали с получением желаемого соединения (0,163 г, 66,6%) в виде жидкости бледно-желтого цвета.
Стадия 2: Синтез соединения 440
Метил 4-(((3R,5S)-4-(3-(((1-(диэтиламино)пропан-2-ил)амино)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-3, 0,163 г, 0,321 ммоль), гидроксиламин (0,392 мл, 6,416 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,180 г, 3,208 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 440 (0,105 г, 64,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,71 (д, 2 H, J=8,2 Гц), 7,38-7,32 (м, 4 H), 7,25 (с, 1 H), 7,18-7,16 (м, 1 H), 4,37 (ушир. с, 2 H), 3,83-3,80 (м, 1 H), 3,67-3,64 (м, 1 H), 3,52 (с, 2 H), 2,67-2,57 (м, 3 H), 2,45-2,37 (м, 2 H), 2,35-2,26 (м, 2 H), 2,25-2,11 (м, 5 H), 1,29 (с, 6 H), 0,93-0,86 (м, 9 H).
Пример 203: Синтез соединения 441
(4-(((3R,5S)-4-(3-(((1-(бензил(метил)амино)пропан-2-ил)амино)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(((1-(бензил(метил)амино)пропан-2-ил)амино)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,200 г, 0,482 ммоль), N1-бензил-N1-метилпропан-1,2-диамин (0,095 г, 0,530 ммоль) и Cs2CO3 (0,188 г, 0,578 ммоль) растворяли в ацетонитриле (2 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 5%) и концентрировали с получением желаемого соединения (0,167 г, 62,2%) в виде жидкости бледно-желтого цвета.
Стадия 2: Синтез соединения 441
Метил 4-(((3R,5S)-4-(3-(((1-(бензил(метил)амино)пропан-2-ил)амино)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-3, 0,167 г, 0,300 ммоль), гидроксиламин (0,367 мл, 5,999 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,168 г, 3,000 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 441 (0,134 г, 80,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,71 (д, 2 H, J=8,2 Гц), 7,37-7,30 (м, 4 H), 7,28-7,19 (м, 6 H), 7,18-7,16 (м, 1 H), 4,42 (ушир. с, 2 H), 3,83-3,79 (м, 1 H), 3,67-3,64 (м, 1 H), 3,50 (с, 2 H), 3,46-3,43 (м, 1 H), 3,31 (с, 1 H), 2,74-2,69 (м, 1 H), 2,60 (м, 1 H), 2,33-2,28 (м, 2 H), 2,13-2,08 (м, 3 H), 1,27 (с, 6 H), 0,93-0,91 (м, 3 H); ЖХМС (ES) m/z 558,4 (M++1).
Пример 204: Синтез соединения 442
(N-гидрокси-4-(((3R,5S)-4-(3-((4-изопентилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-((4-изопентилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,200 г, 0,482 ммоль), 1-изобутилпиперазин трифторацетат (0,136 г, 0,530 ммоль) и Cs2CO3 (0,188 г, 0,578 ммоль) растворяли в ацетонитриле (2 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,162 г, 64,6%) в виде жидкости бледно-желтого цвета.
Стадия 2: Синтез соединения 442
Метил 4-(((3R,5S)-4-(3-((4-изопентилпиперазин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-3, 0,162 г, 0,311 ммоль), гидроксиламин (0,381 мл, 6,226 ммоль, 50,00%-ный водный раствор) и гидроксид калия растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 442 (0,121 г, 74,6%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,2 (ушир. с, 1 H), 9,03 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,43 (д, 2 H, J=8,1 Гц), 7,38-7,30 (м, 2 H), 7,23-7,19 (м, 2 H), 4,50 (ушир. с, 2 H), 3,54-3,47 (м, 4 H), 2,62 (м, 2 H), 2,34 (м, 6 H), 2,26-2,22 (м, 3 H), 2,16-2,12 (м, 3 H), 1,56-1,53 (м, 1 H), 1,31-1,25 (м, 8 H), 0,86-0,84 (м, 6 H).
Пример 205: Синтез соединения 443
(4-(((3R,5S)-3,5-диметил-4-(3-((4-(2,2,2-трифторэтил)пиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-(2,2,2-трифторэтил)пиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,200 г, 0,482 ммоль), 1-(2,2,2-трифторэтил)пиперазин трифторацетат (0,141 г, 0,530 ммоль) и Cs2CO3 (0,188 г, 0,578 ммоль) растворяли в ацетонитриле (2 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,164 г, 62,3%) в виде жидкости бледно-желтого цвета.
Стадия 2: Синтез соединения 443
Метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-(2,2,2-трифторэтил)пиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)бензоат (0,148 г, 0,270 ммоль), гидроксиламин (0,331 мл, 5,408 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,152 г, 2,704 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 443 (0,119 г, 80,5%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,2 (ушир. с, 1 H), 9,08 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,39-7,19 (м, 6 H), 4,47 (ушир. с, 2 H), 3,52-3,44 (м, 4 H), 3,17-3,10 (м, 2 H), 2,61 (с, 6 H), 2,36 (с, 4 H), 2,14-2,12 (м, 2 H), 1,29 (м, 6 H).
Пример 206: Синтез соединения 444
(4-(((3R,5S)-3,5-диметил-4-(3-((4-(метилсульфонил)пиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-(метилсульфонил)пиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,200 г, 0,482 ммоль), 1-(метилсульфонил)пиперазин (0,087 г, 0,530 ммоль) и Cs2CO3 (0,188 г, 0,578 ммоль) растворяли в ацетонитриле (2 мл) при комнатной температуре, и раствор перемешивали в течение ночи при той же температуре. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,164 г, 62,8%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 444
Метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-(метилсульфонил)пиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)бензоат (формула 6-3, 0,059 г, 0,109 ммоль), гидроксиламин (0,133 мл, 2,178 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,061 г, 1,089 ммоль) растворяли в метаноле (1,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 444 (0,042 г, 71,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,2 (ушир. с, 1 H), 9,01 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,42-7,33 (м, 4 H), 7,25-7,21 (м, 2 H), 4,47 (ушир. с, 2 H), 3,55-3,53 (м, 4 H), 3,10 (м, 4 H), 2,87 (м, 3 H), 2,69-2,56 (м, 2 H), 2,45 (с, 4 H), 2,16-2,12 (м, 2 H), 1,29 (м, 6 H).
Пример 207: Синтез соединения 446
(4-(((3R,5S)-3,5-диметил-4-(3-(морфолин-4-карбонил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез 3-(хлорметил)бензоилхлорида
Смесь 3-(бромметил)бензойной кислоты (1,000 г, 4,650 ммоль) и SOCl2 (10,120 мл, 139,509 ммоль) перемешивали при комнатной температуре и перемешивали при температуре 70°C в течение 16 часов, затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении, и продукт использовали без дополнительной очистки (0,879 г, 100,0%, масло коричневого цвета).
Стадия 2: Синтез (3-(хлорметил)фенил)(морфолино)метанона
3-(Хлорметил)бензоилхлорид (формула 6-1, 0,879 г, 4,650 ммоль), морфолин (0,409 мл, 4,650 ммоль) и TEA (1,289 мл, 9,300 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 15% до 40%) и концентрировали с получением желаемого соединения (0,415 г, 37,2%) в виде бесцветного масла.
Стадия 3: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(морфолин-4-карбонил)бензил)пиперазин-1-ил)метил)бензоата
(3-(Хлорметил)фенил)(морфолино)метанон (формула 6-4, 0,415 г, 1,730 ммоль), метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,454 г, 1,730 ммоль) и Cs2CO3 (1,127 г, 3,459 ммоль) растворяли в ацетонитриле (2 мл), и раствор перемешивали при комнатной температуре в течение 16 часов и затем перемешивали при температуре 100°C в течение 6 часов, затем охлаждали до комнатной температуры для прекращения реакции. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,381 г, 47,3%) в виде масла желтого цвета.
Стадия 4: Синтез соединения 446
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(морфолин-4-карбонил)бензил)пиперазин-1-ил)метил)бензоат (0,095 г, 0,204 ммоль), гидроксиламин (0,250 мл, 4,085 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,115 г, 2,043 ммоль) растворяли в метаноле (2 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. К полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Соединение 446 использовали без дополнительной очистки (0,064 г, 67,4%, твердое вещество белого цвета).
1H-ЯМР (400 МГц, CDCl3) δ 11,2 (ушир. с, 1 H), 9,01 (ушир. с, 1 H), 7,69 (д, 2 H, J=8,0 Гц), 7,48-7,34 (м, 5 H), 7,21 (д, 1 H, J=7,1 Гц), 3,75 (с, 2 H), 3,61 (ушир. с, 7 H), 3,44 (с, 3 H), 2,66-2,63 (м, 4 H), 1,84-1,78 (м, 2 H), 0,87 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 467,2 (M++1).
Пример 208: Синтез соединения 452
(4-(((3R,5S)-3,5-диметил-4-(3-((4-(метилсульфонил)пиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез 1-бензил-4-(метилсульфонил)пиперазина
1-Бензилпиперазин (2,000 г, 11,347 ммоль), MsCl (0,922 мл, 11,914 ммоль) и TEA (2,372 мл, 17,020 ммоль) растворяли в метиленхлориде (100 мл), и раствор перемешивали при той же температуре в течение 3 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат кристаллизовали из диэтилового эфира (20 мл) и гексана (5 мл) при комнатной температуре и фильтровали, и полученный твердый продукт промывали гексаном и сушили с получением желаемого соединения (0,857 г, 29,7%) в виде твердого вещества белого цвета.
Стадия 2: Синтез 1-(метилсульфонил)пиперазина
1-Бензил-4-(метилсульфонил)пиперазин (0,700 г, 2,752 ммоль) растворяли в метаноле (30 мл) при комнатной температуре и к раствору медленно добавляли Pd/C (0,075 г), и затем раствор помещали в атмосферу водорода из баллона, затем перемешивали при той же температуре в течение 3 часов. Реакционную смесь фильтровали через слой из целита для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении для удаления растворителя. Продукт использовали без дополнительной очистки (0,335 г, 74,1%, твердое вещество белого цвета).
Стадия 3: Метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-(метилсульфонил)пиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль) и 1-(метилсульфонил)пиперазин (0,129 г, 0,788 ммоль) растворяли в метиленхлориде (5 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,223 г, 1,051 ммоль), затем еще перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,254 г, 91,4%) в виде масла бледно-желтого цвета.
Стадия 4: Синтез соединения 452
Метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-(метилсульфонил)пиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,254 г, 0,480 ммоль), гидроксиламин (0,588 мл, 9,608 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,270 г, 4,804 ммоль) растворяли в метаноле (7 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления твердых продуктов и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр, снабженный картриджем с безводным сульфатом натрия, для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 452 (0,115 г, 45,2%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=7,7 Гц), 7,31 (д, 2 H, J=7,8 Гц), 7,26-7,24 (м, 3 H), 7,12 (с, 1 H), 3,72 (с, 2 H), 3,51 (с, 2 H), 3,42 (с, 2 H), 3,10 (с, 4 H), 2,87 (с, 3 H), 2,64 (д, 2 H, J=10,7 Гц), 2,57-2,55 (м, 2 H), 2,44 (с, 4 H), 1,81 (т, 2 H, J=10,3 Гц), 0,90 (д, 6 H, J=5,8 Гц).
Пример 209: Синтез соединения 453
(4-(((3R,5S)-3,5-диметил-4-(3-((4-(2,2,2-трифторэтил)пиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез трет-бутил 4-(2,2,2-трифторэтил)пиперазин-1-карбоксилата
трет-Бутил пиперазин-1-карбоксилат (2,000 г, 10,738 ммоль), 2,2,2-трифторэтил трифторметансульфонат (1,625 мл, 11,275 ммоль) и Cs2CO3 (4,198 г, 12,886 ммоль) растворяли в ацетонитриле (100 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Реакционную смесь фильтровали через стеклянный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении для удаления растворителя. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (2,150 г, 74,7%) в виде бесцветного масла.
Стадия 2: Синтез 1-(2,2,2-трифторэтил)пиперазина
трет-Бутил 4-(2,2,2-трифторэтил)пиперазин-1-карбоксилат (2,000 г, 7,455 ммоль) растворяли в смеси метиленхлорид (10 мл)/трифторуксусная кислота (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и концентрат кристаллизовали из этилацетата (20 мл) при комнатной температуре и фильтровали, и полученный твердый продукт промывали этилацетатом и сушили с получением желаемого соединения (0,457 г, 23,1%) в виде твердого вещества белого цвета.
Стадия 3: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-(2,2,2-трифторэтил)пиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль) и 1-(2,2,2-трифторэтил)пиперазин (0,209 г, 0,788 ммоль) растворяли в метиленхлориде (5 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,223 г, 1,051 ммоль), затем дополнительно перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,115 г, 41,1%) в виде масла бледно-желтого цвета.
Стадия 4: Синтез соединения 453
Метил 4-(((3R,5S)-3,5-диметил-4-(3-((4-(2,2,2-трифторэтил)пиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,115 г, 0,216 ммоль), гидроксиламин (0,264 мл, 4,318 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,121 г, 2,159 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр, снабженный картриджем с безводным сульфатом натрия, для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18; смесь 0,1%-ный водный раствор трифторуксусной кислоты/ацетонитрил=от 0% до 30%), после чего его пропускали через картридж SPE (PL-HCO3 MP SPE) и концентрировали с получением соединения 453 (0,012 г, 10,4%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,17 (ушир. с, 1 H), 9,01 (ушир. с, 1 H), 7,70 (д, 2 H, J=8,3 Гц), 7,35 (д, 2 H, J=8,2 Гц), 7,27 (с, 1 H), 7,23-7,22 (м, 2 H), 7,10-7,09 (м, 1 H), 3,72 (с, 2 H), 3,44 (с, 4 H), 3,14 (кв, 2 H, J=10,4 Гц), 2,68-2,65 (м, 2 H), 2,61-2,53 (м, 6 H), 2,37-2,33 (м, 4 H), 1,81 (т, 2 H, J=10,3 Гц), 0,90 (д, 6 H, J=6,0 Гц).
Пример 210: Синтез соединения 454
(N-гидрокси-4-(((3R,5S)-4-(3-((4-изопентилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензамид)
Стадия 1: Синтез трет-бутил 4-изопентилпиперазин-1-карбоксилата
трет-Бутил пиперазин-1-карбоксилат (2,000 г, 10,738 ммоль), 1-бром-3-метилбутан (1,352 мл, 11,275 ммоль) и Cs2CO3 (4,198 г, 12,886 ммоль) растворяли в ацетонитриле (150 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Реакционную смесь фильтровали через стеклянный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении для удаления растворителя. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 10%) и концентрировали с получением желаемого соединения (1,220 г, 44,3%) в виде бесцветного масла.
Стадия 2: Синтез 1-изопентилпиперазин трифторацетата
трет-Бутил 4-изопентилпиперазин-1-карбоксилат (1,000 г, 3,900 ммоль) растворяли в смеси метиленхлорид (10 мл)/трифторуксусная кислота (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и концентрат кристаллизовали из этилацетата (20 мл) при комнатной температуре и фильтровали, и полученный твердый продукт промывали этилацетатом и сушили с получением желаемого соединения (0,929 г, 94,0%) в виде твердого вещества белого цвета.
Стадия 3: Синтез метил 4-(((3R,5S)-4-(3-((4-изопентилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,220 г, 0,578 ммоль) и трифторацетат 1-изопентилпиперазина (0,220 г, 0,867 ммоль) растворяли в метиленхлориде (5 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,245 г, 1,156 ммоль), затем дополнительно перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,232 г, 77,0%) в виде масла бледно-желтого цвета.
Стадия 4: Синтез соединения 454
Метил 4-(((3R,5S)-4-(3-((4-изопентилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,232 г, 0,446 ммоль), гидроксиламин (0,545 мл, 8,910 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,250 г, 4,455 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 454 (0,067 г, 29,6%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,2 Гц), 7,30 (д, 2 H, J=8,1 Гц), 7,27 (с, 1 H), 7,23-7,21 (м, 2 H), 7,09-7,08 (м, 1 H), 3,72 (с, 2 H), 3,42 (с, 2 H), 3,41 (с, 2 H), 2,63 (д, 2 H, J=10,2 Гц), 2,58-2,53 (м, 2 H), 2,34-2,27 (м, 8 H), 2,24 (т, 2 H, J=7,6 Гц), 1,80 (т, 2 H, J=10,5 Гц), 1,57-1,53 (м, 1 H), 1,31-1,25 (м, 2 H), 0,89 (д, 6 H, J=6,1 Гц), 0,85 (д, 6 H, J=6,6 Гц).
Пример 211: Синтез соединения 455
(4-(((3R,5S)-4-(3-(((1-(диэтиламино)пропан-2-ил)амино)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(((1-(диэтиламино)пропан-2-ил)амино)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль) и N1,N1-диэтилпропан-1,2-диамин (0,103 г, 0,788 ммоль) растворяли в метиленхлориде (5 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,223 г, 1,051 ммоль), затем дополнительно перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,149 г, 57,3%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 455
Метил 4-(((3R,5S)-4-(3-(((1-(диэтиламино)пропан-2-ил)амино)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,149 г, 0,301 ммоль), гидроксиламин (0,368 мл, 6,024 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,169 г, 3,012 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 455 (0,064 г, 42,9%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,70 (д, 2 H, J=8,2 Гц), 7,34 (д, 2 H, J=8,0 Гц), 7,26 (с, 1 H), 7,24-7,18 (м, 2 H), 3,79-3,55 (м, 4 H), 3,43 (с, 2 H), 2,63 (д, 2 H, J=10,1 Гц), 2,59-2,54 (м, 2 H), 2,46-2,37 (м, 2 H), 2,34-2,25 (м, 2 H), 2,23-2,15 (м, 2 H), 1,81 (т, 2 H, J=10,4 Гц), 0,92-0,86 (м, 15 H).
Пример 212: Синтез соединения 456
(4-(((3R,5S)-4-(3-(((1-(бензил(метил)амино)пропан-2-ил)амино)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(3-(((1-(бензил(метил)амино)пропан-2-ил)амино)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,200 г, 0,526 ммоль) и N1-бензил-N1-метилпропан-1,2-диамин (0,141 г, 0,788 ммоль) растворяли в метиленхлориде (5 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,223 г, 1,051 ммоль), затем дополнительно перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,252 г, 88,3%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 456
Метил 4-(((3R,5S)-4-(3-(((1-(бензил(метил)амино)пропан-2-ил)амино)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,252 г, 0,464 ммоль), гидроксиламин (0,568 мл, 9,286 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,261 г, 4,643 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 456 (0,196 г, 77,6%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,69 (д, 2 H, J=8,2 Гц), 7,33 (д, 2 H, J=8,2 Гц), 7,31-7,20 (м, 8 H), 7,12 (д, 1 H, J=6,5 Гц), 3,80-3,57 (м, 4 H), 3,39 (с, 2 H), 2,70-2,69 (м, 1 H), 2,61 (д, 2 H, J=10,8 Гц), 2,55-2,53 (м, 2 H), 2,34-2,29 (м, 1 H), 2,12-2,09 (м, 1 H), 1,79 (т, 2 H, J=10,4 Гц), 0,92 (д, 3 H, J=6,1 Гц), 0,88 (д, 6 H, J=6,0 Гц).
Пример 213: Синтез соединения 457
(4-(((3R,5S)-3,5-диметил-4-(2-(морфолинометил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез 2-(бромметил)бензальдегида
2-(Бромметил)бензонитрил (2,500 г, 12,752 ммоль) растворяли в метиленхлориде (30 мл) при температуре 0°C и в раствор добавляли DIBAL-H (1,00 M раствор, 13,390 мл, 13,390 ммоль), затем перемешивали при той же температуре в течение 2 часов. Затем при температуре 0°C в реакционную смесь добавляли водный раствор соляной кислоты, затем перемешивали в течение 30 минут. После завершения реакции в реакционную смесь добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 40 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением желаемого соединения (0,400 г, 15,8%) в виде масла коричневого цвета.
Стадия 2: Синтез метил 4-(((3R,5S)-4-(2-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 0,500 г, 1,906 ммоль), 2-(бромметил)бензальдегид (0,379 г, 1,906 ммоль) и Cs2CO3 (0,931 г, 2,859 ммоль) растворяли в ацетонитриле (10 мл), и раствор перемешивали при той же температуре в течение 17 часов. В реакционную смесь добавляли воду, затем экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 40%) и концентрировали с получением желаемого соединения (0,116 г, 16,0%) в виде масла бледно-коричневого цвета.
Стадия 3: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2-(морфолинометил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(2-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,116 г, 0,305 ммоль) и морфолин (0,040 мл, 0,457 ммоль) растворяли в метиленхлориде (10 мл), и раствор перемешивали при комнатной температуре в течение 30 минут. В реакционный раствор добавляли Na(OAc)3BH (0,129 г, 0,610 ммоль), затем дополнительно перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением желаемого соединения (0,076 г, 55,2%) в виде масла бледно-желтого цвета.
Стадия 4: Синтез соединения 457
Метил 4-(((3R,5S)-3,5-диметил-4-(2-(морфолинометил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,076 г, 0,168 ммоль), гидроксиламин (0,018 мл, 3,366 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,094 г, 1,683 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр, снабженный картриджем с безводным сульфатом натрия, для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 457 (0,060 г, 78,8%) в виде твердого вещества бледно-желтого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,77 (д, 1 H, J=7,8 Гц), 7,71 (д, 2 H, J=8,1 Гц), 7,33 (д, 2 H, J=8,1 Гц), 7,22-7,21 (м, 1 H), 7,11-7,08 (м, 2 H), 3,86 (с, 2 H), 3,52 (с, 4 H), 3,46 (с, 2 H), 3,43 (с, 2 H), 2,68 (д, 2 H, J=10,8 Гц), 2,65-2,61 (м, 2 H), 2,33 (с, 4 H), 1,87 (т, 2 H, J=10,3 Гц), 0,80 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 453,2 (M++1).
Пример 214: Синтез соединения 458
(4-(((3R,5S)-3,5-диметил-4-(4-(морфолинометил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез 4-(хлорметил)бензоилхлорида
Смесь 4-(бромметил)бензойной кислоты (5,000 г, 23,251 ммоль) и SOCl2 (8,444 мл, 116,257 ммоль) перемешивали при комнатной температуре и перемешивали при температуре 50°C в течение 17 часов, затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и продукт использовали без дополнительной очистки (4,400 г, 100,1%, твердое вещество бледно-фиолетового цвета).
Стадия 2: Синтез метил 4-(((3R,5S)-4-(4-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 4,400 г, 23,275 ммоль), 4-(хлорметил)бензоилхлорид (формула 6-1, 3,053 г, 11,638 ммоль) и TEA (6,488 мл, 46,551 ммоль) растворяли в метиленхлориде (150 мл) при температуре 0°C, и раствор перемешивали при комнатной температуре в течение 3 часов. В реакционную смесь добавляли воду, затем экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 40 г; этилацетат/гексан=от 5% до 70%) и концентрировали и затем добавляли полученный продукт очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 5% до 20%) и концентрировали с получением желаемого соединения (1,720 г, 17,8%) в виде твердого вещества оранжевого цвета.
Стадия 3: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-(морфолинометил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,100 г, 0,241 ммоль), морфолин (0,031 мл, 0,362 ммоль) и Cs2CO3 (0,157 г, 0,482 ммоль) растворяли в ацетонитриле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,107 г, 95,4%) в виде бесцветного масла.
Стадия 4: Синтез соединения 458
Метил 4-(((3R,5S)-3,5-диметил-4-(4-(морфолинометил)бензоил)пиперазин-1-ил)метил)бензоат (формула 6-3, 0,107 г, 0,230 ммоль), гидроксиламин (0,281 мл, 4,596 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,129 г, 2,298 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр, снабженный картриджем с безводным сульфатом натрия, для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 458 (0,034 г, 31,6%) в виде твердого вещества бледно-оранжевого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,70 (д, 2 H, J=8,0 Гц), 7,36-7,34 (м, 4 H), 7,28 (д, 2 H, J=8,0 Гц), 4,12 (ушир. с, 2 H),3,57 (т, 4 H, J=4,4 Гц), 3,52 (с, 2 H), 3,48 (с, 2 H), 2,62-2,61 (м, 2 H), 2,35 (с, 4 H), 2,13 (дд, 2 H, J=11,2, 4,2 Гц), 1,28-1,25 (м, 6 H); ЖХМС (ES) m/z 467,2 (M++1).
Пример 215: Синтез соединения 459
(4-(((3R,5S)-3,5-диметил-4-(4-(пиперидин-1-илметил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-пиперидин-1-илметил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,100 г, 0,241 ммоль), пиперидин (0,031 мл, 0,362 ммоль) и Cs2CO3 (0,157 г, 0,482 ммоль) растворяли в ацетонитриле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,101 г, 90,4%) в виде бесцветного масла.
Стадия 2: Синтез соединения 459
Метил 4-(((3R,5S)-3,5-диметил-4-(4-пиперидин-1-илметил)бензоил)пиперазин-1-ил)метил)бензоат (формула 6-3, 0,101 г, 0,218 ммоль), гидроксиламин (0,267 мл, 4,357 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,122 г, 2,179 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением соединения 459 (0,073 г, 72,1%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,72 (д, 2 H, J=8,2 Гц), 7,42 (д, 2 H, J=8,2 Гц), 7,33 (д, 2 H, J=8,1 Гц), 7,27 (д, 2 H, J=8,1 Гц), 4,12 (ушир. с, 1 H), 3,54 (с, 2 H), 3,43 (с, 2 H), 2,64-2,62 (м, 2 H), 2,31 (с, 4 H), 2,15 (дд, 2 H, J=11,4, 4,2 Гц), 1,52-1,46 (м, 4 H), 1,39-1,38 (м, 2 H), 1,29 (д, 6 H, J=6,1 Гц); ЖХМС (ES) m/z 465,2 (M++1).
Пример 216: Синтез соединения 460
(4-(((3R,5S)-4-(4-(((R)-3-фторпирролидин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-(((R)-3-фторпирролидин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,100 г, 0,241 ммоль), (R)-3-фторпирролидин (0,045 г, 0,362 ммоль) и Cs2CO3 (0,157 г, 0,482 ммоль) растворяли в ацетонитриле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,103 г, 91,4%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 460
Метил 4-(((3R,5S)-4-(4-(((R)-3-фторпирролидин-1-ил)метил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-3, 0,103 г, 0,220 ммоль), гидроксиламин (0,269 мл, 4,406 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,124 г, 2,203 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением соединения 460 (0,082 г, 79,4%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,19 (ушир. с, 1 H), 9,04 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,43 (д, 2 H, J=8,1 Гц), 7,36 (д, 2 H, J=8,1 Гц), 7,29 (д, 2 H, J=8,1 Гц), 5,20 (дт, 1 H, J=55,5, 5,8 Гц), 4,15 (ушир. с, 2 H), 3,62 (с, 2 H), 3,55 (с, 2 H), 2,82-2,73 (м, 2 H), 2,68-2,61 (м, 3 H), 2,33-2,28 (м, 1 H), 2,17-2,13 (м, 3 H), 1,94-1,89 (м, 1 H), 1,29 (д, 6 H, J=5,3 Гц).
Пример 217: Синтез соединения 461
(4-(((3R,5S)-3,5-диметил-4-(4-(пирролидин-1-илметил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-(пирролидин-1-илметил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,100 г, 0,241 ммоль), пирролидин (0,030 мл, 0,362 ммоль) и Cs2CO3 (0,157 г, 0,482 ммоль) растворяли в ацетонитриле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,102 г, 94,1%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 461
Метил 4-(((3R,5S)-3,5-диметил-4-(4-(пирролидин-1-илметил)бензоил)пиперазин-1-ил)метил)бензоат (формула 6-3, 0,102 г, 0,227 ммоль), гидроксиламин (0,278 мл, 4,537 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,127 г, 2,269 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением соединения 461 (0,071 г, 69,5%) в виде твердого вещества бледно-желтого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,18 (ушир. с, 1 H), 9,04 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,43 (д, 2 H, J=8,1 Гц), 7,35 (д, 2 H, J=8,0 Гц), 7,27 (д, 2 H, J=8,0 Гц), 4,19 (ушир. с, 2 H), 3,58 (с, 2 H), 3,55 (с, 2 H), 2,63 (д, 2 H, J=10,0 Гц), 2,41 (с, 4 H), 2,15 (дд, 2 H, J=11,4, 4,2 Гц), 1,70-1,68 (м, 4 H), 1,29 (д, 6 H, J=6,1 Гц).
Пример 218: Синтез соединения 462
(4-(((3R,5S)-3,5-диметил-4-(4-((4-метилпиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-((4-метилпиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,100 г, 0,241 ммоль), 1-метилпиперазин (0,040 мл, 0,362 ммоль) и Cs2CO3 (0,157 г, 0,482 ммоль) растворяли в ацетонитриле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,102 г, 88,4%) в виде бесцветного масла.
Стадия 2: Синтез соединения 462
Метил 4-(((3R,5S)-3,5-диметил-4-(4-((4-метилпиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)бензоат (формула 6-3, 0,102 г, 0,213 ммоль), гидроксиламин (0,261 мл, 4,262 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,120 г, 2,131 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали водой, и сушили с получением соединения 462 (0,057 г, 55,8%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,20 (ушир. с, 1 H), 9,04 (ушир. с, 1 H), 7,72 (д, 2 H, J=8,2 Гц), 7,43 (д, 2 H, J=8,1 Гц), 7,33 (д, 2 H, J=8,1 Гц), 7,27 (д, 2 H, J=7,8 Гц), 4,21 (ушир. с, 2 H), 3,54 (с, 2 H), 3,47 (с, 2 H), 2,63 (д, 2 H, J=11,4 Гц), 2,33-2,32 (м, 8 H), 2,17-2,14 (м, 5 H), 1,30 (с, 6 H); ЖХМС (ES) m/z 480,2 (M++1).
Пример 219: Синтез соединения 463
(4-(((3R,5S)-3,5-диметил-4-(4-((4-(метилсульфонил)пиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-((4-(метилсульфонил)пиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-(хлорметил)бензоил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 6-2, 0,100 г, 0,241 ммоль), 1-(метилсульфонил)пиперазин (0,059 г, 0,362 ммоль) и Cs2CO3 (0,157 г, 0,482 ммоль) растворяли в ацетонитриле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,126 г, 96,3%) в виде бесцветного масла.
Стадия 2: Синтез соединения 463
Метил 4-(((3R,5S)-3,5-диметил-4-(4-((4-(метилсульфонил)пиперазин-1-ил)метил)бензоил)пиперазин-1-ил)метил)бензоат (формула 6-3, 0,126 г, 0,297 ммоль), гидроксиламин (0,364 мл, 5,950 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,167 г, 2,975 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением соединения 463 (0,065 г, 53,2%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,72 (д, 2 H, J=8,3 Гц), 7,42 (д, 2 H, J=8,1 Гц), 7,36 (д, 2 H, J=8,1 Гц), 7,30 (д, 2 H, J=8,2 Гц), 4,25 (ушир. с, 2 H), 3,55 (с, 4 H), 3,11 (т, 4 H, J=4,5 Гц), 2,87 (с, 3 H), 2,63 (д, 2 H, J=10,3 Гц), 2,47 (с, 4 H), 2,15 (дд, 2 H, J=11,4, 4,1 Гц), 1,29-1,28 (м, 6 H).
Пример 220: Синтез соединения 466
(4-(((3R,5S)-3,5-диметил-4-(4-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 1-2, 1,000 г, 3,812 ммоль), 4-(бромметил)бензальдегид (0,759 г, 3,812 ммоль) и Cs2CO3 (2,484 г, 7,623 ммоль) растворяли в ацетонитриле (25 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. Реакционную смесь фильтровали через бумажный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении для удаления растворителя. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 40 г; этилацетат/гексан=от 0% до 25%) и концентрировали с получением желаемого соединения (0,934 г, 64,4%) в виде твердого вещества белого цвета.
Стадия 2: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,180 г, 0,473 ммоль) и пирролидин (0,044 мл, 0,520 ммоль) растворяли в метиленхлориде (2 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,150 г, 0,710 ммоль), затем дополнительно перемешивали при той же температуре в течение 16 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 30% до 100%) и концентрировали с получением желаемого соединения (0,098 г, 47,5%) в виде твердого вещества желтого цвета.
Стадия 3: Синтез соединения 466
Метил 4-(((3R,5S)-3,5-диметил-4-(4-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,098 г, 0,225 ммоль) и гидроксиламин (0,275 мл, 4,490 ммоль, 50,00%-ный водный раствор) растворяли в метаноле (3 мл) при комнатной температуре и в раствор добавляли гидроксид калия (0,126 г, 2,245 ммоль), h затем перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 466 (0,055 г, 56,0%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 10,9 (ушир. с, 1 H), 9,07 (ушир. с, 1 H), 7,69 (д, 2 H, J=7,9 Гц), 7,34 (д, 2 H, J=7,8 Гц), 7,26 (д, 2 H, J=7,8 Гц), 7,20 (д, 2 H, J=7,8 Гц), 3,70 (с, 2 H), 3,51 (с, 2 H), 3,42 (с, 2 H), 2,66-2,64 (м, 2 H), 2,60-2,55 (м, 2 H), 2,39 (с, 4 H), 1,79 (т, 2 H, J=10,4 Гц), 1,66 (с, 4 H), 0,90 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 437,3 (M++1).
Пример 221: Синтез соединения 467
(4-(((3R,5S)-3,5-диметил-4-(4-(пиперидин-1-илметил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-(пиперидин-1-илметил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,180 г, 0,473 ммоль) и пиперидин (0,052 мл, 0,520 ммоль) растворяли в метиленхлориде (2 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,150 г, 0,710 ммоль), затем дополнительно перемешивали при той же температуре в течение 16 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 30% до 100%) и концентрировали с получением желаемого соединения (0,149 г, 70,1%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 467
Метил 4-(((3R,5S)-3,5-диметил-4-(4-(пиперидин-1-илметил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,149 г, 0,332 ммоль) и гидроксиламин (0,406 мл, 6,637 ммоль, 50,00%-ный водный раствор) растворяли в метаноле (3 мл) при комнатной температуре и в раствор добавляли гидроксид калия (0,186 г, 3,318 ммоль), затем перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 467 (0,118 г, 78,8%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,2 (ушир. с, 1 H), 9,04 (ушир. с, 1 H), 7,69 (д, 2 H, J=7,7 Гц), 7,33 (д, 2 H, J=7,7 Гц), 7,26 (д, 2 H, J=7,7 Гц), 7,18 (д, 2 H, J=7,8 Гц), 3,70 (с, 2 H), 3,42 (с, 2 H), 2,62 (д, 2 H, J=10,4 Гц), 2,55 (м, 2 H), 2,27 (с, 4 H), 1,79 (т, 2 H, J=10,4 Гц), 1,48-1,45 (м, 4 H), 1,37-1,36 (м, 2 H), 0,90 (д, 6 H, J=5,9 Гц); ЖХМС (ES) m/z 451,2 (M++1).
Пример 222: Синтез соединения 468
(4-(((3R,5S)-3,5-диметил-4-(4-((4-метилпиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(4-((4-метилпиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,180 г, 0,473 ммоль) и 1-метилпиперазин (0,058 мл, 0,520 ммоль) растворяли в метиленхлориде (2 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,150 г, 0,710 ммоль), затем дополнительно перемешивали при той же температуре в течение 16 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 30% до 100%) и концентрировали с получением желаемого соединения (0,193 г, 87,7%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 468
Метил 4-(((3R,5S)-3,5-диметил-4-(4-((4-метилпиперазин-1-ил)метил)бензил)пиперазин-1-ил)метил)бензоат (формула 5-2, 0,192 г, 0,413 ммоль) и гидроксиламин (0,506 мл, 8,264 ммоль, 50,00%-ный водный раствор) растворяли в метаноле (3 мл) при комнатной температуре и в раствор добавляли гидроксид калия (0,232 г, 4,132 ммоль), затем перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 468 (0,041 г, 21,5%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,1 (ушир. с, 1 H), 9,06 (ушир. с, 1 H), 7,68 (д, 2 H, J=8,1 Гц), 7,31 (д, 2 H, J=7,9 Гц), 7,27 (д, 2 H, J=8,0 Гц), 7,18 (д, 2 H, J=7,9 Гц), 3,70 (с, 2 H), 3,42 (с, 2 H), 3,39 (с, 2 H), 2,67-2,61 (м, 2 H), 2,59-2,54 (м, 2 H), 2,43-2,17 (м, 8 H), 2,13 (с, 3 H), 1,79 (т, 2 H, J=11,1 Гц), 0,89 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 466,3 (M++1).
Пример 223: Синтез соединения 472
((E)-3-(4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)фенил)-N-гидроксиакриламид)
Стадия 1: Синтез (E)-метил 3-(4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 9-1, 0,300 г, 1,040 ммоль), 3-(бромметил)бензальдегид (0,207 г, 1,040 ммоль) и Cs2CO3 (0,678 г, 2,081 ммоль) растворяли в ацетонитриле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом и экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,250 г, 59,2%) в виде масла желтого цвета.
Стадия 2: Синтез (E)-метил 3-(4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 10-1, 0,250 г, 0,616 ммоль) и морфолин (0,060 мл, 0,677 ммоль) растворяли в метиленхлориде (3 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,196 г, 0,924 ммоль), затем дополнительно перемешивали при той же температуре в течение 16 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 30% до 100%) и концентрировали с получением желаемого соединения (0,186 г, 63,1%) в виде масла желтого цвета.
Стадия 3: Синтез соединения 472
(E)-Метил 3-(4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)фенил)акрилат (формула 10-2, 0,186 г, 0,389 ммоль) и гидроксиламин (0,476 мл, 7,788 ммоль, 50,00%-ный водный раствор) растворяли в метаноле (4 мл) при комнатной температуре и в раствор добавляли гидроксид калия (0,219 г, 3,894 ммоль), затем перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18; смесь ацетонитрил/0,1% трифторуксусная кислота=от 5% до 70%), после чего его пропускали через картридж SPE (PL-HCO3 MP SPE) и концентрировали с получением желаемого соединения 472 (0,142 г, 63,4%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 10,8 (ушир. с, 1 H), 9,06 (ушир. с, 1 H), 7,51-7,49 (м, 2 H), 7,45-7,41 (м, 1 H), 7,38-7,19 (м, 4 H), 7,17-7,11 (м, 2 H), 6,44-6,41 (м, 1 H), 3,71 (с, 2 H), 3,56 (с, 4 H), 3,43-3,41 (м, 4 H), 2,65-2,62 (м, 4 H), 2,33 (ушир. с, 4 H), 1,82-1,72 (м, 2 H), 0,89 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 479,3 (M++1).
Пример 224: Синтез соединения 475
((E)-3-(4-(((2S,6R)-2,6-диметил-4-(3-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)фенил)-N-гидроксиакриламид)
Стадия 1: Синтез (3R,5S)-трет-бутил 3,5-диметилпиперазин-1-карбоксилат
(2S,6R)-2,6-Диметилпиперазин (10,000 г, 87,573 ммоль) и TEA (24,278 мл, 175,147 ммоль) растворяли в метиленхлориде (150 мл) при комнатной температуре и в раствор добавляли Boc2O (20,119 мл, 87,573 ммоль), затем перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 80 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (15,393 г, 82,0%) в виде твердого вещества желтого цвета.
Стадия 2: Синтез (3R,5S)-трет-бутил 4-(4-((E)-3-метокси-3-оксопроп-1-ен-1-ил)бензил)-3,5-диметилпиперазин-1-карбоксилат
(3R,5S)-трет-Бутил 3,5-диметилпиперазин-1-карбоксилат (формула 8-1, 5,000 г, 23,332 ммоль), метил 3-(4-бромметил)циннамат (формула 8-4, 5,952 г, 23,332 ммоль) и Cs2CO3 (15,204 г, 46,664 ммоль) смешивали в ацетонитриле (150 мл) при комнатной температуре, и смесь нагревали при кипячении с обратным холодильником и затем охлаждали до комнатной температуры. Реакционную смесь фильтровали через бумажный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении для удаления растворителя. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 80 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (6,800 г, 75,0%) в виде масла желтого цвета.
Стадия 3: Синтез (E)-метил 3-(4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилата
(3R,5S)-трет-Бутил 4-(4-((E)-3-метокси-3-оксопроп-1-ен-1-ил)бензил)-3,5-диметилпиперазин-1-карбоксилат (6,800 г, 17,503 ммоль) и HCl (4,00 M диоксан раствор, 21,879 мл, 87,516 ммоль) растворяли в 1,4-диоксане (100 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Выпавший твердый осадок фильтровали, промывали гексаном и сушили с получением желаемого соединения (2,970 г, 58,8%) в виде твердого вещества коричневого цвета.
Стадия 4: Синтез (E)-метил 3-(4-(((2S,6R)-4-(3-формилбензил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 17-1, 0,300 г, 1,040 ммоль), 3-(бромметил)бензальдегид (0,207 г, 1,040 ммоль) и Cs2CO3 (0,678 г, 2,081 ммоль) растворяли в ацетонитриле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. Реакционную смесь фильтровали через бумажный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении для удаления растворителя. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,293 г, 69,3%) в виде твердого вещества белого цвета.
Стадия 5: Синтез (E)-метил 3-(4-(((2S,6R)-2,6-диметил-4-(3-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((2S,6R)-4-(3-формилбензил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 17-2, 0,130 г, 0,320 ммоль) и пирролидин (0,029 мл, 0,352 ммоль) растворяли в метиленхлориде (3 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,102 г, 0,480 ммоль), затем дополнительно перемешивали при той же температуре в течение 16 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,062 г, 42,0%) в виде масла желтого цвета.
Стадия 6: Синтез соединения 475
(E)-Метил 3-(4-(((2S,6R)-2,6-диметил-4-(3-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)фенил)акрилат (формула 17-3, 0,062 г, 0,133 ммоль) и гидроксиламин (0,163 мл, 2,664 ммоль, 50,00%-ный водный раствор) растворяли в метаноле (2 мл) при комнатной температуре и в раствор добавляли гидроксид калия (0,075 г, 1,332 ммоль), затем перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (10 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали гексаном и сушили с получением соединения 475 (0,039 г, 63,8%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,58 (ушир. с, 1 H), 7,45-7,40 (м, 2 H), 7,33-7,31 (м, 2 H), 7,27-7,21 (м, 3 H), 7,18-7,12 (м, 2 H), 6,39-6,35 (м, 1 H), 3,71 (с, 2 H), 3,53 (с, 2 H), 2,65-2,63 (м, 3 H), 2,55-2,50 (м, 3 H), 2,39-2,33 (м, 4 H), 1,78 (т, 2 H, J=10,4 Гц), 1,67 (с, 4 H), 0,89 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 463,2 (M++1).
Пример 225: Синтез соединения 476
((E)-3-(4-(((2S,6R)-4-(3-((диэтиламино)метил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенил)-N-гидроксиакриламид)
Стадия 1: Синтез (E)-метил 3-(4-(((2S,6R)-4-(3-((диэтиламино)метил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((2S,6R)-4-(3-формилбензил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 17-2, 0,130 г, 0,320 ммоль) и диэтиламин (0,039 г, 0,352 ммоль) растворяли в метиленхлориде (3 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,102 г, 0,480 ммоль), затем дополнительно перемешивали при той же температуре в течение 16 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,061 г, 41,1%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 476
(E)-Метил 3-(4-(((2S,6R)-4-(3-((диэтиламино)метил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 17-3, 0,061 г, 0,134 ммоль) и гидроксиламин (0,164 мл, 2,683 ммоль, 50,00%-ный водный раствор) растворяли в метаноле (2 мл) при комнатной температуре и в раствор добавляли гидроксид калия (0,075 г, 1,342 ммоль), затем перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 476 (0,026 г, 41,2%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 10,7 (ушир. с, 1 H), 9,04 (ушир. с, 1 H), 7,47-7,45 (м, 3 H), 7,37-7,36 (м, 2 H), 7,25-7,21 (м, 2 H), 7,16-7,11 (м, 2 H), 6,42-6,38 (м, 1 H), 3,72 (с, 2 H), 3,50 (с, 2 H), 3,37 (с, 2 H), 2,65-2,63 (м, 2 H), 2,62-2,50 (м, 2 H), 2,42 (кв, 4 H, J=7,1 Гц ), 1,78 (т, 2 H, J=10,4 Гц), 0,95 (т, 6 H, J=7,1 Гц), 0,87 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 465,3 (M++1).
Пример 226: Синтез соединения 477
((E)-3-(4-(((2S,6R)-4-(4-(фуран-2-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенил)-N-гидроксиакриламид)
Стадия 1: Синтез (E)-метил 3-(4-(((2S,6R)-4-(4-бромбензил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 17-1, 0,200 г, 0,694 ммоль), 1-(бромметил)-4-бромбензол (0,173 г, 0,694 ммоль) и Cs2CO3 (0,452 г, 1,387 ммоль) растворяли в ацетонитриле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. Реакционную смесь фильтровали через бумажный фильтр для удаления твердых продуктов, и фильтрат концентрировали при пониженном давлении для удаления растворителя. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,209 г, 65,9%) в виде твердого вещества белого цвета.
Стадия 2: Синтез (E)-метил 3-(4-(((2S,6R)-4-(4-(фуран-2-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилата
Смесь 1,2-диметоксиэтан (0,9 мл)/вода (0,1 мл) добавляли к смеси (E)-метил 3-(4-(((2S,6R)-4-(4-бромбензил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилата (формула 18-1, 0,100 г, 0,219 ммоль), фуран-2-бороновой кислоты (0,027 г, 0,240 ммоль), Pd(dbpf)Cl2 (0,007 г, 0,011 ммоль) и Na2CO3 (0,070 г, 0,656 ммоль) при комнатной температуре, и раствор перемешивали при температуре 50°C в течение 16 часов, и затем охлаждали до комнатной температуры для прекращения реакции. В реакционную смесь добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 4 г; метанол/метиленхлорид=от 0% до 3%) и концентрировали с получением желаемого соединения (0,080 г, 82,4%) в виде твердого вещества белого цвета.
Стадия 3: Синтез соединения 477
(E)-Метил 3-(4-(((2S,6R)-4-(4-(фуран-2-ил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 18-2, 0,077 г, 0,174 ммоль), гидроксиламин (0,213 мл, 3,478 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,098 г, 1,739 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали гексаном и сушили с получением соединения 477 (0,075 г, 96,8%) в виде твердого вещества коричневого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,23 (ушир. с, 1 H), 7,73-7,72 (м, 1 H), 7,65-7,63 (м, 1 H), 7,50-7,48 (м, 1 H), 7,38-7,36 (м, 2 H), 7,33-7,27 (м, 3 H), 7,25-7,23 (м, 1 H), 7,06-7,02 (м, 1 H), 6,90 (д, 1 H, J=3,3 Гц), 6,59-6,57 (м, 1 H), 6,37-6,32 (м, 1 H), 3,70 (с, 2 H), 3,39 (с, 2 H), 2,66-2,53 (м, 4 H), 1,82-1,76 (м, 2 H), 0,90 (д, 6 H, J=5,9 Гц); ЖХМС (ES) m/z 446,2 (M++1).
Пример 227: Синтез соединения 478
(4-(((2S,6R)-4-(3-((диэтиламино)метил)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид (гидрохлоридная соль))
Стадия 1: Синтез метил 4-(((2S,6R)-4-(3-((диэтиламино)метил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-4-(3-формилбензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 17-2, 0,150 г, 0,394 ммоль) и диэтиламин гидрохлорид (0,065 г, 0,591 ммоль) растворяли в метиленхлориде (4 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,167 г, 0,788 ммоль), затем дополнительно перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 5% до 10%) и концентрировали с получением желаемого соединения (0,054 г, 31,3%) в виде масла оранжевого цвета.
Стадия 2: Синтез соединения 478
Метил 4-(((2S,6R)-4-(3-((диэтиламино)метил)бензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 17-3, 0,054 г, 0,123 ммоль), гидроксиламин (0,151 мл, 2,468 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,069 г, 1,234 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,045 г, 83,1%, масло бледно-коричневого цвета).
Стадия 3: Синтез гидрохлоридной соли соединения 478
4-(((2S,6R)-4-(3-((Диэтиламино)метил)бензил)-2,6-диметилпиперазин-1-ил)метил)-N-гидроксибензамид (0,048 г, 0,110 ммоль) и HCl (4,00 M раствор в диоксане, 0,137 мл, 0,548 ммоль) растворяли в 1,4-диоксане (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли этилацетат (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали этилацетатом и сушили с получением соединения 478 (0,039 г, 75,0%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ 7,65 (д, 2 H, J=7,6 Гц), 7,51 (д, 2 H, J=8,4 Гц), 7,46-7,42 (м, 4 H), 4,52 (с, 2 H), 2,22 (с, 2 H), 2,20 (с, 2 H), 3,47-3,44 (м, 4 H), 3,08-3,05 (м, 6 H), 1,42 (д, 6 H, J=5,7 Гц), 1,17 (т, 6 H, J=7,0 Гц).
Пример 228: Синтез соединения 479
(4-(((2S,6R)-2,6-диметил-4-(3-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((2S,6R)-2,6-диметил-4-(3-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((2S,6R)-4-(3-формилбензил)-2,6-диметилпиперазин-1-ил)метил)бензоат (формула 17-2, 0,150 г, 0,394 ммоль) и пирролидин (0,049 мл, 0,591 ммоль) растворяли в метиленхлориде (4 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,167 г, 0,788 ммоль), затем дополнительно перемешивали при той же температуре в течение 17 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 5% до 10%) и концентрировали с получением желаемого соединения (0,122 г, 71,0%) в виде масла оранжевого цвета.
Стадия 2: Синтез соединения 479
Метил 4-(((2S,6R)-2,6-диметил-4-(3-(пирролидин-1-илметил)бензил)пиперазин-1-ил)метил)бензоат (формула 17-3, 0,122 г, 0,280 ммоль), гидроксиламин (0,343 мл, 5,601 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,157 г, 2,801 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 479 (0,069 г, 56,4%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,11 (ушир. с, 1 H), 9,05 (ушир. с, 1 H), 7,66 (д, 2 H, J=8,1 Гц), 7,41 (д, 2 H, J=8,1 Гц), 7,26-7,22 (м, 2 H), 7,15 (дд, 2 H, J=7,3, 7,3 Гц), 3,74 (с, 2 H), 3,54 (с, 2 H), 3,39 (с, 2 H), 2,65 (д, 2 H, J=10,8 Гц), 2,56-2,55 (м, 2 H), 2,40 (с, 4 H), 1,79 (т, 2 H, J=10,6 Гц), 1,67 (с, 4 H), 0,87 (д, 6 H, J=6,1 Гц).
Пример 229: Синтез соединения 480
((E)-3-(4-(((2S,6R)-2,6-диметил-4-(3-(пирролидин-1-илметил)бензоил)пиперазин-1-ил)метил)фенил)-N-гидроксиакриламид)
Стадия 1: Синтез (E)-метил 3-(4-(((2S,6R)-4-(3-формилбензоил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 17-1, 0,300 г, 1,040 ммоль), 3-формилбензойную кислоту (0,172 г, 1,144 ммоль), EDCI (0,399 г, 2,081 ммоль), HOBt (0,319 г, 2,081 ммоль) и DIPEA (0,921 мл, 5,201 ммоль) растворяли в метиленхлориде (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 30% до 40%) и концентрировали с получением желаемого соединения (0,386 г, 88,2%) в виде твердого вещества белого цвета.
Стадия 2: Синтез (E)-метил 3-(4-(((2S,6R)-2,6-диметил-4-(3-(пирролидин-1-илметил)бензоил)пиперазин-1-ил)метил)фенил)акрилат
(E)-Метил 3-(4-(((2S,6R)-4-(3-формилбензоил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 17-2, 0,190 г, 0,452 ммоль) и пирролидин (0,035 г, 0,497 ммоль) растворяли в метиленхлориде (3 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,144 г, 0,678 ммоль), затем дополнительно перемешивали при той же температуре в течение 12 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,105 г, 49,0%) в виде твердого вещества белого цвета.
Стадия 3: Синтез соединения 480
(E)-Метил 3-(4-(((2S,6R)-2,6-диметил-4-(3-(пирролидин-1-илметил)бензоил)пиперазин-1-ил)метил)фенил)акрилат (формула 17-3, 0,100 г, 0,210 ммоль) и гидроксиламин (0,257 мл, 4,205 ммоль, 50,00%-ный водный раствор) растворяли в метаноле (3 мл) при комнатной температуре и в раствор добавляли гидроксид калия (0,118 г, 2,103 ммоль), затем перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 480 (0,049 г, 48,8%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,45-7,30 (м, 9 H), 6,51-6,46 (м, 1 H), 4,41-4,38 (м, 1 H), 3,85 (с, 2 H), 3,67 (с, 2 H), 3,54-3,48 (м, 1 H), 3,04-3,00 (м, 1 H), 2,79-2,76 (м, 1 H), 2,70-3,67 (м, 1 H), 2,54 (с, 6 H), 1,80 (с, 4 H), 1,14-1,08 (м, 3 H), 0,93-0,92 (м, 3 H); ЖХМС (ES) m/z 477,2 (M++1).
Пример 230: Синтез соединения 481
(4-(((3R,5S)-3,5-диметил-4-(2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез трет-бутил 4-(2-(((2S,6R)-4-(4-(метоксикарбонил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата
Смесь 1,2-диметоксиэтан (15 мл)/вода (5 мл) добавляли к смеси метил 4-(((3R,5S)-4-(2-йодбензил)-3,5-диметилпиперазин-1-ил)метил)бензоата (формула 2-1, 1,600 г, 3,345 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат (2,068 г, 6,689 ммоль), Pd(dbpf)2Cl2 (0,109 г, 0,167 ммоль) и Na2CO3 (0,709 г, 6,689 ммоль) при комнатной температуре, и раствор перемешивали при температуре 50°C в течение 17 часов и затем охлаждали до комнатной температуры для прекращения реакции. В реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 5% до 50%) и концентрировали с получением желаемого соединения (1,090 г, 61,1%) в виде масла коричневого цвета.
Стадия 2: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2-(1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоата
трет-Бутил 4-(2-(((2S,6R)-4-(4-(метоксикарбонил)бензил)-2,6-диметилпиперазин-1-ил)метил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилат (формула 3-1, 1,080 г, 2,024 ммоль) растворяли в 1,4-диоксане (15 мл) при комнатной температуре и в раствор добавляли HCl (4,00 M раствор в 1,4-диоксане, 5,059 мл, 20,236 ммоль), затем перемешивали при той же температуре в течение 17 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли этилацетат (200 мл) и метанол (10 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали этилацетатом и сушили с получением желаемого соединения (0,851 г, 89,5%) в виде твердого вещества бледно-желтого цвета.
Стадия 3: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоата
Гидрохлорид метил 4-(((3R,5S)-3,5-диметил-4-(2-(1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоата (формула 3-2, 0,060 г, 0,128 ммоль) и TEA (0,053 мл, 0,383 ммоль) растворяли в метиленхлориде (4 мл) при комнатной температуре и в раствор добавляли MsCl (0,015 мл, 0,191 ммоль), затем перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,055 г, 84,2%) в виде масла бледно-желтого цвета.
Стадия 4: Синтез соединения 481
Метил 4-(((3R,5S)-3,5-диметил-4-(2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоат (формула 3-3, 0,055 г, 0,107 ммоль), гидроксиламин (0,131 мл, 2,150 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,060 г, 1,075 ммоль) растворяли в смеси метанол (2 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 481 (0,048 г, 87,1%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,72 (д, 1 H, J=8,2 Гц), 7,70 (д, 2 H, J=8,2 Гц), 7,32 (д, 2 H, J=8,1 Гц), 7,24 (дд, 1 H, J=7,5, 7,5 Гц), 7,15 (дд, 1 H, J=7,4, 7,4 Гц), 7,04 (д, 1 H, J=7,4 Гц), 5,59 (с, 1 H), 3,83 (д, 2 H, J=2,1 Гц), 3,62 (с, 2 H), 3,45 (с, 2 H), 3,40-3,38 (м, 2 H), 2,65 (д, 2 H, J=10,7 Гц), 2,61-2,57 (м, 2 H), 2,40 (с, 2 H), 1,85 (т, 2 H, J=10,3 Гц), 0,78 (д, 6 H, J=6,0 Гц).
Пример 231: Синтез соединения 482
(4-(((3R,5S)-4-(2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Гидрохлорид метил 4-(((3R,5S)-3,5-диметил-4-(2-(1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоата (формула 3-2, 0,060 г, 0,128 ммоль) и TEA (0,053 мл, 0,383 ммоль) растворяли в метиленхлориде (4 мл) при комнатной температуре и в раствор добавляли ацетангидрид (0,020 г, 0,191 ммоль), затем перемешивали при той же температуре в течение 1 часа. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 5%) и концентрировали с получением желаемого соединения (0,046 г, 75,8%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 482
Метил 4-(((3R,5S)-4-(2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 3-3, 0,046 г, 0,097 ммоль), гидроксиламин (0,118 мл, 1,934 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,054 г, 0,967 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 482 (0,036 г, 78,1%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,05 (ушир. с, 1 H), 9,12 (ушир. с, 1 H), 7,71 (д, 3 H, J=7,9 Гц), 7,36 (д, 2 H, J=7,9 Гц), 7,23 (дд, 1 H, J=7,7, 7,7 Гц), 7,14 (дд, 1 H, J=7,4, 7,4 Гц), 7,03 (д, 1 H, J=7,5 Гц), 5,55 (с, 1 H), 4,08 (д, 2 H, J=21,4 Гц), 3,68-3,62 (м, 4 H), 3,46 (с, 2 H), 2,68-2,57 (м, 4 H), 2,35 (с, 1 H), 2,25 (с, 1 H), 2,07 (д, 3 H, J=8,5 Гц), 1,85 (т, 2 H, J=10,0 Гц), 0,78 (д, 6 H, J=5,9 Гц).
Пример 232: Синтез соединения 483
(4-(((3R,5S)-3,5-диметил-4-(2-(1-(2,2,2-трифторэтил)-1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(2-(1-(2,2,2-трифторэтил)-1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-3,5-диметил-4-(2-(1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоат (0,050 г, 0,115 ммоль), 2,2,2-трифторэтил трифторметансульфонат (формула 3-2, 0,025 мл, 0,173 ммоль) и K2CO3 (0,032 г, 0,231 ммоль) растворяли в ацетонитриле (4 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 4 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением желаемого соединения (0,032 г, 53,9%) в виде масла желтого цвета.
Стадия 2: Синтез соединения 483
Метил 4-(((3R,5S)-3,5-диметил-4-(2-(1-(2,2,2-трифторэтил)-1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоат (формула 3-3, 0,032 г, 0,062 ммоль), гидроксиламин (0,076 мл, 1,241 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,035 г, 0,621 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18; смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%), после чего его пропускали через картридж SPE (PL-HCO3 MPS PE) и концентрировали с получением соединения 483 (0,007 г, 21,8%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,03 (с, 1 H), 7,71-7,69 (м, 3 H), 7,39-7,38 (м, 2 H), 7,22 (дд, 1 H, J=7,3, 7,3 Гц), 7,13 (дд, 1 H, J=7,2, 7,2 Гц), 7,03 (д, 1 H, J=7,4 Гц), 5,49 (с, 1 H), 3,62 (с, 2 H), 3,47 (с, 2 H), 3,31-3,28 (м, 4 H), 2,88 (т, 2 H, J=5,0 Гц), 2,68-2,61 (м, 4 H), 2,33-2,30 (м, 2 H), 1,85-1,84 (м, 2 H), 0,79 (д, 6 H, J=5,7 Гц).
Пример 233: Синтез соединения 484
(N-гидрокси-4-(((3R,5S)-4-(2-(1-изопропил-1,2,3,6-тетрагидропиридин-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-(1-изопропил-1,2,3,6-тетрагидропиридин-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Гидрохлорид метил 4-(((3R,5S)-3,5-диметил-4-(2-(1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоата (формула 3-2, 0,150 г, 0,319 ммоль) и TEA (0,222 мл, 1,596 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и в раствор добавляли 2-йодпропан (0,075 г, 0,479 ммоль), затем нагревали при кипячении с обратным холодильником в течение 5 часов, затем охлаждали до комнатной температуры для прекращения реакции. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 20%) и концентрировали, и затем полученный продукт очищали с помощью хроматографии (Waters, C18; смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%), после чего его пропускали через картридж SPE (PL-HCO3 MP SPE) и концентрировали с получением желаемого соединения (0,032 г, 21,1%) в виде масла бледно-желтого цвета.
Стадия 2: Синтез соединения 484
Метил 4-(((3R,5S)-4-(2-(1-изопропил-1,2,3,6-тетрагидропиридин-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 3-3, 0,032 г, 0,067 ммоль), гидроксиламин (0,082 мл, 1,345 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,038 г, 0,673 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали водой и сушили с получением соединения 484 (0,006 г, 18,7%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,78 (д, 1 H, J=6,7 Гц), 7,74 (д, 2 H, J=8,3 Гц), 7,47 (д, 2 H, J=8,1 Гц), 7,20 (дд, 1 H, J=6,5, 6,5 Гц), 7,13 (дд, 1 H, J=7,2, 7,2 Гц), 7,03 (д, 1 H, J=7,4 Гц), 5,55 (с, 1 H), 3,75 (с, 2 H), 3,57 (с, 2 H), 3,30 (с, 2 H), 2,87-2,81 (м, 3 H), 2,76-2,69 (м, 4 H), 2,42 (с, 2 H), 1,97 (т, 2 H, J=10,4 Гц), 1,19 (д, 6 H, J=6,5 Гц), 0,87 (д, 6 H, J=6,0 Гц).
Пример 234: Синтез соединения 485
(4-(((3R,5S)-4-(2-(1-этил-1,2,3,6-тетрагидропиридин-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-(2-(1-этил-1,2,3,6-тетрагидропиридин-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Гидрохлорид метил 4-(((3R,5S)-3,5-диметил-4-(2-(1,2,3,6-тетрагидропиридин-4-ил)бензил)пиперазин-1-ил)метил)бензоата (формула 3-2, 0,150 г, 0,319 ммоль) и TEA (0,222 мл, 1,596 ммоль) растворяли в метиленхлориде (4 мл) при комнатной температуре и в раствор добавляли 2-йодпропан (0,048 мл, 0,479 ммоль), затем перемешивали при той же температуре в течение 3 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 20%) и концентрировали с получением желаемого соединения (0,048 г, 32,6%) в виде жидкости желтого цвета.
Стадия 2: Синтез соединения 485
Метил 4-(((3R,5S)-4-(2-(1-этил-1,2,3,6-тетрагидропиридин-4-ил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 3-3, 0,048 г, 0,104 ммоль), гидроксиламин (0,127 мл, 2,080 ммоль, 50,00%-ный водный раствор) и гидроксид калия (0,058 г, 1,040 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении с получением соединения 485 (0,033 г, 68,6%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,70 (д, 3 H, J=8,2 Гц), 7,35 (д, 2 H, J=8,1 Гц), 7,20 (дд, 1 H, J=7,5, 7,5 Гц), 7,12 (дд, 1 H, J=7,5, 7,5 Гц), 7,00 (дд, 1 H, J=7,5, 1,3 Гц), 5,49 (с, 1 H), 3,62 (с, 2 H), 3,46 (с, 2 H), 3,03 (с, 2 H), 2,66-2,56 (м, 6 H), 2,45 (кв, 2 H, J=7,2 Гц), 2,28 (ушир. с, 2 H), 1,84 (т, 2 H, J=10,4 Гц), 1,07 (т, 3 H, J=7,2 Гц), 0,78 (д, 6 H, J=6,1 Гц).
Пример 235: Синтез соединения 486
(4-(((3R,5S)-4-(4-((4-ацетилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензамид (гидрохлоридная соль))
Стадия 1: Синтез метил 4-(((3R,5S)-4-(4-((4-ацетилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоата
Метил 4-(((3R,5S)-4-(4-формилбензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-1, 0,180 г, 0,473 ммоль) и 1-(пиперазин-1-ил)этанон (0,067 г, 0,520 ммоль) растворяли в метиленхлориде (2 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,150 г, 0,710 ммоль), затем дополнительно перемешивали при той же температуре в течение 16 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Экстракт фильтровали через пластиковый фильтр с удалением твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния; картридж 4 г; этилацетат/гексан=от 30% до 100%) и концентрировали с получением желаемого соединения (0,112 г, 48,2%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 486
Метил 4-(((3R,5S)-4-(4-((4-ацетилпиперазин-1-ил)метил)бензил)-3,5-диметилпиперазин-1-ил)метил)бензоат (формула 5-2, 0,112 г, 0,228 ммоль) и гидроксиламин (0,279 мл, 4,563 ммоль, 50,00%-ный водный раствор) растворяли в метаноле (3 мл) при комнатной температуре и в раствор добавляли гидроксид калия (0,128 г, 2,282 ммоль), затем перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (Waters, C18; смесь ацетонитрил/0,1%-ный водный раствор трифторуксусной кислоты=от 5% до 70%), после чего его пропускали через картридж SPE (PL-HCO3 MP SPE) и концентрировали с получением соединения 486 (0,049 г, 43,5%) в виде твердого вещества белого цвета.
Стадия 3: Синтез гидрохлоридной соли соединения 486
Соединение 486 (0,049 г, 0,099 ммоль) и HCl (4,00 M раствор в диоксане, 0,124 мл, 0,496 ммоль) растворяли в 1,4-диоксане (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Выпавший твердый осадок фильтровали, промывали этилацетатом и сушили с получением желаемого соединения (0,034 г, 63,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,72 (д, 2 H, J=8,2 Гц), 7,61-7,55 (м, 4 H), 7,52 (д, 2 H, J=8,0 Гц), 4,59 (с, 2 H), 4,39 (с, 2 H), 4,23 (с, 2 H), 3,71-3,69 (м, 8 H), 3,56-3,46 (м, 6 H), 3,06 (м, 3 H), 2,11 (с, 3 H), 1,51-1,50 (м, 6 H); ЖХМС (ES) m/z 494,2 (M++1).
Пример 236: Синтез соединения 487
((E)-3-(4-(((2S,6R)-4-(3-((диэтиламино)метил)бензоил)-2,6-диметилпиперазин-1-ил)метил)фенил)-N-гидроксиакриламид)
Стадия 1: Синтез (E)-метил 3-(4-(((2S,6R)-4-(3-((диэтиламино)метил)бензоил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилата
(E)-Метил 3-(4-(((2S,6R)-4-(3-формилбензоил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 17-2, 0,190 г, 0,452 ммоль) и диэтиламин (0,054 г, 0,497 ммоль) растворяли в метиленхлориде (3 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли Na(OAc)3BH (0,144 г, 0,678 ммоль), затем дополнительно перемешивали при той же температуре в течение 12 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (0,060 г, 27,8%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 487
(E)-Метил 3-(4-(((2S,6R)-4-(3-((диэтиламино)метил)бензоил)-2,6-диметилпиперазин-1-ил)метил)фенил)акрилат (формула 17-3, 0,060 г, 0,126 ммоль) и гидроксиламин (0,154 мл, 2,512 ммоль, 50,00%-ный водный раствор) растворяли в метаноле (2 мл) при комнатной температуре и в раствор добавляли гидроксид калия (0,070 г, 1,256 ммоль), затем перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением соединения 487 (0,006 г, 9,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,57-7,53 (м, 1 H), 7,51-7,46 (м, 3 H), 7,45-7,39 (м, 4 H), 7,36-7,34 (м, 1 H), 6,45 (д, 1 H, J=15,9 Гц), 4,42-4,39 (м, 1 H), 3,87-3,82 (м, 2 H), 3,53-3,50 (м, 1 H), 3,07-3,01 (м, 1 H), 2,83-2,77 (м, 2 H), 2,73-2,68 (м, 4 H), 2,57 (с, 1 H), 1,29 (м, 2 H), 1,15-1,11 (м, 9 H), 0,93-0,88 (м, 3 H); ЖХМС (ES) m/z 479,2 (M++1).
Пример 237: Синтез соединения 520
(3-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N-гидрокси-1H-индол-6-карбоксамид)
Стадия 1: Синтез метил 3-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-1H-индол-6-карбоксилата
Метил 3-формил-1H-индол-6-карбоксилат (формула 8-4, 0,500 г, 2,461 ммоль), (2S,6R)-1-бензил-2,6-диметилпиперазин (0,553 г, 2,707 ммоль) и STAB (0,782 г, 3,691 ммоль) растворяли в метиленхлориде (20 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 5 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 30%) и концентрировали с получением желаемого соединения (0,551 г, 57,2%) в виде твердого вещества цвета слоновой кости.
Стадия 2: Синтез соединения 520
Метил 3-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-1H-индол-6-карбоксилат (формула 8-5, 0,150 г, 0,383 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,234 мл, 3,831 ммоль) и гидроксид калия (0,215 г, 3,831 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 520 (0,149 г, 99,4%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,80 (с, 1 H), 7,56 (д, 2 H, J=8,3 Гц), 7,42 (д, 1 H, J=8,3 Гц), 7,34-7,25 (м, 5 H), 7,16 (т, 1 H, J=7,2 Гц), 3,70 (с, 2 H), 3,55 (с, 2 H), 2,72 (д, 2 H, J=10,0 Гц), 2,55-2,53 (м, 2 H), 1,78 (т, 2 H, J=10,6 Гц), 0,89 (с, 3 H), 0,87 (с, 3 H); ЖХМС (ES) m/z 393,0 (M++1).
Пример 238: Синтез соединения 569
(5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N-гидрокси-1H-индол-2-карбоксамид)
Стадия 1: Синтез 1-(трет-бутил) 2-этил 5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-1H-индол-1,2-дикарбоксилата
1-(трет-Бутил) 2-этил 5-(бромметил)-1H-индол-1,2-дикарбоксилат (формула 22-1, 2,000 г, 5,232 ммоль), DIPEA (2,779 мл, 15,697 ммоль) и (2S,6R)-1-бензил-2,6-диметилпиперазин (1,069 г, 5,232 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Выпавший твердый осадок фильтровали, промывали ацетонитрилом и сушили с получением желаемого соединения (1,100 г, 41,6%) в виде твердого вещества белого цвета.
Стадия 2: Синтез этил 5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-1H-индол-2-карбоксилата
1-(трет-Бутил) 2-этил 5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-1H-индол-1,2-дикарбоксилата (формула 22-2, 1,100 г, 2,175 ммоль) и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 2,719 мл, 10,877 ммоль) смешивали в 1,4-диоксане (2 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 16 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли диэтиловый эфир (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали диэтиловым эфиром и сушили с получением желаемого соединения (0,832 г, 94,3%) в виде твердого вещества бледно-розового цвета.
Стадия 3: Синтез соединения 569
Этил 5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-1H-индол-2-карбоксилат (формула 22-3, 0,100 г, 0,247 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,151 мл, 2,466 ммоль) и гидроксид калия (0,138 г, 2,466 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 569 (0,079 г, 81,6%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,46 (с, 1 H), 7,38-7,28 (м, 5 H), 7,24-7,20 (м, 1 H), 7,11 (дд, 1 H, J=8,4, 1,3 Гц), 6,79 (с, 1 H), 3,91 (с, 2 H), 3,53 (с, 2 H), 2,79 (д, 2 H, J=11,0 Гц), 2,72-2,68 (м, 2 H), 1,93 (т, 2 H, J=11,1 Гц), 1,11 (с, 3 H), 1,10 (с, 3 H); ЖХМС (ES) m/z 393,0 (M++1).
Пример 239: Синтез соединения 571
(5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензофуран-2-карбоксамид)
Стадия 1: Синтез метил 5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)бензофуран-2-карбоксилата
Метил 5-(бромметил)бензофуран-2-карбоксилат (формула 8-4, 0,500 г, 1,858 ммоль), DIPEA (0,720 г, 5,574 ммоль) и (2S,6R)-1-бензил-2,6-диметилпиперазин (0,380 г, 1,858 ммоль) растворяли в ацетонитриле (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и выпавший твердый осадок фильтровали, промывали ацетонитрилом и сушили с получением желаемого соединения (0,200 г, 27,4%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 571
Метил 5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)бензофуран-2-карбоксилат (формула 8-5, 0,050 г, 0,127 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,078 мл, 1,274 ммоль) и гидроксид калия (0,071 г, 1,274 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 571 (0,010 г, 19,4%) в виде твердого вещества бледно-розового цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,60 (с, 1 H), 7,49 (д, 1 H, J=8,4 Гц), 7,38-7,34 (м, 3 H), 7,31 (т, 1 H, J=7,5 Гц), 7,25-7,21 (м, 2 H), 3,92 (с, 2 H), 3,56 (с, 2 H), 2,77 (д, 2 H, J=10,8 Гц), 2,73-2,66 (м, 2 H), 1,94 (т, 2 H, J=10,9 Гц), 1,12 (с, 3 H), 1,10 (с, 3 H); ЖХМС (ES) m/z 392,2 (M+-1).
Пример 240: Синтез соединения 573
(6-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N-гидрокси-1H-индол-2-карбоксамид)
Стадия 1: Синтез 1-(трет-бутил) 2-метил 6-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-1H-индол-1,2-дикарбоксилата
1-(трет-Бутил) 2-метил 6-(бромметил)-1H-индол-1,2-дикарбоксилат (формула 22-1, 0,410 г, 1,113 ммоль), DIPEA (0,432 г, 3,340 ммоль) и (2S,6R)-1-бензил-2,6-диметилпиперазин (0,227 г, 1,113 ммоль) растворяли в ацетонитриле (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 40 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,224 г, 40,9%) в виде жидкости желтого цвета.
Стадия 2: Синтез метил 6-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-1H-индол-2-карбоксилата (гидрохлоридная соль)
1-(трет-Бутил) 2-метил 6-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-1H-индол-1,2-дикарбоксилат (формула 22-2, 0,224 г, 0,456 ммоль) и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 0,570 мл, 2,278 ммоль) растворяли в 1,4-диоксане (2 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли диэтиловый эфир (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали, промывали диэтиловым эфиром и сушили с получением желаемого соединения (0,167 г, 85,6%) в виде твердого вещества цвета охры.
Стадия 3: Синтез соединения 573
Метил 6-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-1H-индол-2-карбоксилат (формула 22-3, 0,050 г, 0,117 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,071 мл, 1,168 ммоль) и гидроксид калия (0,066 г, 1,168 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 573 (0,025 г, 53,4%) в виде твердого вещества желтого цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,49 (д, 1 H, J=8,3 Гц), 7,37 (д, 2 H, J=7,4 Гц), 7,31 (т, 3 H, J=7,5 Гц), 7,23 (т, 1 H, J=7,4 Гц), 7,00 (д, 1 H, J=8,2 Гц), 6,81 (с, 1 H), 3,91 (с, 2 H), 3,55 (с, 2 H), 2,79 (д, 2 H, J=10,9 Гц), 2,71-2,68 (м, 2 H), 1,93 (т, 2 H, J=10,7 Гц), 1,11 (с, 3 H), 1,10 (с, 3 H); ЖХМС (ES) m/z 391,2 (M+-1)
Пример 241: Синтез соединения 574
(2-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксибензофуран-5-карбоксамид)
Стадия 1: Синтез метил 2-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)бензофуран-5-карбоксилата
Метил 2-(бромметил)бензофуран-5-карбоксилат (формула 8-4, 0,691 г, 2,568 ммоль), DIPEA (1,364 мл, 7,704 ммоль) и (2S,6R)-1-бензил-2,6-диметилпиперазин (0,525 г, 2,568 ммоль) растворяли в ацетонитриле (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 40 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,460 г, 45,6%) в виде твердого вещества цвета слоновой кости.
Стадия 2: Синтез соединения 574
Метил 2-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)бензофуран-5-карбоксилат (формула 8-5, 0,100 г, 0,254 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,155 мл, 2,541 ммоль) и гидроксид калия (0,143 г, 2,541 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 574 (0,013 г, 13,0%) в виде твердого вещества бледно-розового цвета.
1H-ЯМР (400 МГц, CD3OD) δ 8,00 (д, 1 H, J=1,4 Гц), 7,72 (дд, 1 H, J=8,6, 1,8 Гц), 7,50 (д, 1 H, J=8,7 Гц), 7,38-7,20 (м, 5 H), 6,80 (с, 1 H), 3,90 (с, 2 H), 3,70 (с, 2 H), 2,85 (д, 1 H, J=10,7 Гц), 2,76-2,71 (м, 2 H), 2,07 (т, 2 H, J=11,0 Гц), 1,12 (с, 3 H), 1,10 (с, 3 H); ЖХМС (ES) m/z 394,0 (M++1).
Пример 242: Синтез соединения 609
(5-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)-N-гидроксипиколинамид)
Стадия 1: Синтез трет-бутил (3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-карбоксилата
(3R,5S)-трет-Бутил 3,5-диметилпиперазин-1-карбоксилат (формула 8-1, 2,730 г, 12,739 ммоль), 3-(бромметил)бензальдегид (2,789 г, 14,013 ммоль) и Cs2CO3 (8,301 г, 25,478 ммоль) смешивали в ацетонитриле (50 мл) при комнатной температуре, и смесь перемешивали при той же температуре в течение 18 часов. Реакционную смесь фильтровали через бумажный фильтр для удаления твердых продуктов, и фильтрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 120 г; этилацетат/гексан=от 5% до 30%) и концентрировали с получением желаемого соединения (2,730 г, 64,5%) в виде масла желтого цвета.
Стадия 2: Синтез трет-бутил (3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-карбоксилат
трет-Бутил (3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-карбоксилат (формула 23-1, 1,246 г, 3,749 ммоль), морфолин (0,656 мл, 7,497 ммоль) и уксусную кислоту (0,429 мл, 7,497 ммоль) смешивали в метиленхлориде (15 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли Na(CN)BH3 (0,942 г, 14,995 ммоль), затем дополнительно перемешивали при той же температуре в течение 4 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 24 г; метанол/метиленхлорид=от 0% до 10%) и концентрировали с получением желаемого соединения (1,272 г, 84,0%) в виде масла желтого цвета.
Стадия 3: Синтез 4-(3-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензил)морфолин (гидрохлоридная соль)
трет-Бутил (3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-карбоксилат (формула 23-2, 1,270 г, 3,147 ммоль) и HCl (4,00 M раствор в 1,4-диоксане, 3,934 мл, 15,735 ммоль) растворяли в метиленхлориде (15 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 18 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Продукт использовали без дополнительной очистки (0,855 г, 89,5%, масло желтого цвета).
Стадия 4: Синтез метил 5-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)пиколинат
Метил 5-формилпиколинат (0,200 г, 1,211 ммоль), 4-(3-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензил)морфолин (0,735 г, 2,422 ммоль) и уксусную кислоту (0,139 мл, 2,422 ммоль) смешивали в метиленхлориде (8 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли Na(CN)BH3 (0,304 г, 4,844 ммоль), затем дополнительно перемешивали при той же температуре в течение 15 часов. Затем в реакционную смесь добавляли насыщенный водный раствор гидрокарбоната натрия, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 60% до 100%) и концентрировали с получением желаемого соединения (0,317 г, 57,8%) в виде масла желтого цвета.
Стадия 5: Синтез соединения 609
Метил 5-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)пиколинат (формула 23-4, 0,059 г, 0,130 ммоль) и гидроксиламин (0,333 мл, 2,594 ммоль, 50,00%-ный водный раствор) смешивали в метаноле (1 мл) и при температуре 0°C добавляли гидроксид калия (0,130 мл, 1,297 ммоль, 10,00 M водный раствор). После этого смесь перемешивали при той же температуре в течение 5 минут и затем перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь концентрировали при пониженном давлении и концентрат очищали с помощью колоночной хроматографии (Waters, C18; смесь 0,1%-ный водный раствор муравьиной кислоты (метановая кислота)/ацетонитрил=от 5% до 30%), после чего его пропускали через картридж SPE (PL-HCO3 MP SPE) и концентрировали с получением соединения 609 (0,006 г, 10,2%) в виде масла коричневого цвета.
1H ЯМР (400 МГц, CD3OD) δ 8,52 (с, 1 H), 7,99-7,87 (м, 2 H), 7,36 (с, 1 H), 7,28-7,27 (м, 2 H), 7,21-7,20 (м, 1 H), 3,90 (с, 2 H), 3,69-3,67 (м, 4 H), 3,54-3,52 (м, 4 H), 2,73-2,65 (м, 4 H), 2,45 (с, 4 H), 1,96 (т, 2 H, J=10,6 Гц), 1,09 (д, 6 H, J=6,0 Гц); ЖХМС (ES) m/z 454,5 (M++1).
Пример 243: Синтез соединения 652
(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N,2-дигидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-2-гидроксибензоата
Метил 4-(бромметил)-2-гидроксибензоат (формула 8-4, 1,144 г, 4,668 ммоль), TEA (0,776 мл, 5,602 ммоль) и (2S,6R)-1-бензил-2,6-диметилпиперазин (0,954 г, 4,668 ммоль) растворяли в ацетонитриле (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 40 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,583 г, 33,9%) в виде жидкости желтого цвета.
Стадия 2: Синтез соединения 652
Метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-2-гидроксибензоат (формула 8-5, 0,050 г, 0,136 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,083 мл, 1,357 ммоль) и гидроксид калия (0,076 г, 1,357 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 652 (0,035 г, 70,0%) в виде твердого вещества розового цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,61 (д, 1 H, J=8,1 Гц), 7,39 (д, 2 H, J=7,1 Гц), 7,33 (т, 2 H, J=7,5 Гц), 7,25 (т, 1 H, J=7,2 Гц), 6,90 (с, 1 H), 6,86 (д, 1 H, J=8,1 Гц), 3,96 (с, 2 H), 3,45 (с, 2 H), 2,78-2,72 (м, 4 H), 1,99-1,94 (м, 4 H), 1,15 (с, 3 H), 1,32 (с, 3 H); ЖХМС (ES) m/z 370,2 (M++1).
Пример 244: Синтез соединения 653
(4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)-N,2-дигидроксибензамид)
Стадия 1: Синтез трет-бутил (3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-карбоксилата
трет-Бутил (3R,5S)-4-(3-формилбензил)-3,5-диметилпиперазин-1-карбоксилат (формула 23-1, 10,439 г, 31,401 ммоль) растворяли в метиленхлориде (150 мл) при комнатной температуре и в раствор добавляли морфолин (2,747 мл, 31,401 ммоль), затем перемешивали при той же температуре в течение 1 часа. В реакционную смесь добавляли STAB (13,310 г, 62,802 ммоль), затем дополнительно перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 80 г; этилацетат/гексан=от 0% до 80%) и концентрировали с получением желаемого соединения (11,080 г, 87,4%) в виде бесцветной жидкости.
Стадия 2: Синтез 4-(3-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензил)морфолина (гидрохлоридная соль)
трет-Бутил (3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-карбоксилат (формула 23-2, 11,080 г, 27,456 ммоль) и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 34,320 мл, 137,278 ммоль) растворяли в 1,4-диоксане (2 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли диэтиловый эфир (150 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением желаемого соединения (7,800 г, 83,6%) в виде твердого вещества белого цвета.
Стадия 3: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)-2-гидроксибензоата
Гидрохлорид 4-(3-(((2S,6R)-2,6-диметилпиперазин-1-ил)метил)бензил)морфолина (формула 23-3, 0,832 г, 2,448 ммоль) и метил 4-(бромметил)-2-гидроксибензоат (0,600 г, 2,448 ммоль), TEA (0,407 мл, 2,938 ммоль) растворяли в ацетонитриле (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционный раствор добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 40 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением желаемого соединения (0,441 г, 38,5%) в виде жидкости желтого цвета.
Стадия 4: Синтез соединения 653
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)-2-гидроксибензоат (формула 23-4, 0,050 г, 0,107 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,065 мл, 1,069 ммоль) и гидроксид калия (0,060 г, 1,069 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 653 (0,033 г, 83,7%) в виде твердого вещества красновато-коричневого цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,65 (д, 1 H, J=8,0 Гц), 7,38 (с, 1 H), 7,30 (д, 2 H, J=5,2 Гц), 7,23-7,22 (м, 1 H), 6,81 (с, 1 H), 6,74 (д, 2 H, J=7,8 Гц), 3,92 (с, 2 H), 3,71-3,69 (м, 4 H), 3,54 (с, 2 H), 3,41 (с, 2 H), 2,77-2,67 (м, 4 H), 2,47 (м, 4 H), 1,93-1,90 (м, 2 H), 1,11 (с, 3 H), 1,10 (с, 3 H); ЖХМС (ES) m/z 469,3 (M++1).
Пример 245: Синтез соединения 696
(4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)-3-фтор-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)-3-фторбензоата
4-(3-(((2S,6R)-2,6-Диметилпиперазин-1-ил)метил)бензил)морфолин (формула 23-3, 1,376 г, 4,048 ммоль), TEA (0,673 мл, 4,857 ммоль) и метил 4-(бромметил)-3-фторбензоат (1,000 г, 4,048 ммоль) растворяли в ацетонитриле (150 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 80 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,438 г, 35,5%) в виде жидкости желтого цвета.
Стадия 2: Синтез соединения 696
Метил 4-(((3R,5S)-3,5-диметил-4-(3-(морфолинометил)бензил)пиперазин-1-ил)метил)-3-фторбензоат (формула 23-4, 0,100 г, 0,213 ммоль), гидроксиламин (0,070 г, 2,130 ммоль) и гидроксид калия (0,119 г, 2,130 ммоль) растворяли в метаноле (2 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 3 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 696 (0,025 г, 25,0%) в виде твердого вещества желтого цвета.
1H-ЯМР (400 МГц, CD3OD) δ 7,58-7,20 (м, 7 H), 3,90 (с, 2 H), 3,71-3,68 (м, 4 H), 3,57 (с, 2 H), 3,53 (с, 2 H), 2,77 (д, 2 H, J=10,2 Гц), 2,71-2,67 (м, 2 H), 2,46-2,45 (м, 4 H), 1,99 (т, 2 H, J=10,8 Гц), 1,10 (с, 3 H), 1,09 (с, 3 H); ЖХМС (ES) m/z 471,2 (M++1).
Пример 246: Синтез соединения 812
(5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксипиколинамид)
Стадия 1: Синтез метил 5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)пиколината
Метил 5-формилпиколинат (формула 8-4, 0,200 г, 0,831 ммоль), (2S,6R)-1-бензил-2,6-диметилпиперазин (0,151 г, 0,914 ммоль) и STAB (0,264 г, 1,246 ммоль) растворяли в метиленхлориде (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,116 г, 39,5%) в виде жидкости желтого цвета.
Стадия 2: Синтез соединения 812
Метил 5-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)пиколинат (формула 8-5, 0,116 г, 0,328 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,100 мл, 1,641 ммоль) и гидроксид калия (0,092 г, 1,641 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 812 (0,088 г, 75,7%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ 8,47 (с, 1 H), 8,07 (д, 1 H, J=7,9 Гц), 7,83 (д, 1 H, J=7,9 Гц), 7,38-7,24 (м, 5 H), 3,91 (с, 2 H), 3,53 (с, 2 H), 2,75-2,68 (м, 4 H), 2,09-2,03 (м, 2 H), 1,11 (с, 3 H), 1,10 (с, 3 H); ЖХМС (ES) m/z 353,1 (M++1).
Пример 247: Синтез соединения 813
(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-3-фтор-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-3-фторбензоата
Метил 4-(бромметил)-3-фторбензоат (формула 8-4, 0,200 г, 0,831 ммоль), (2S,6R)-1-бензил-2,6-диметилпиперазин (0,226 г, 0,914 ммоль) и карбонат натрия (0,230 г, 1,661 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,135 г, 43,9%) в виде твердого вещества цвета слоновой кости.
Стадия 2: Синтез соединения 813
Метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-3-фторбензоат (формула 8-5, 0,135 г, 0,364 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,111 мл, 1,822 ммоль) и гидроксид калия (0,102 г, 1,822 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 813 (0,087 г, 64,3%) в виде твердого вещества цвета слоновой кости.
1H-ЯМР (400 МГц, CDCl3) δ 8,47 (с, 1 H), 8,07 (д, 1 H, J=7,9 Гц), 7,83 (д, 2 H, J=8,2 Гц), 7,38-7,24 (м, 5 H), 3,91 (с, 2 H), 3,53 (с, 2 H), 2,75-2,68 (м, 4 H), 2,10-2,04 (м, 2 H), 1,11 (с, 3 H), 1,10 (с, 3 H); ЖХМС (ES) m/z 372,1 (M++1).
Пример 248: Синтез соединения 814
(4-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-карбонил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-карбонил)бензоата
Метил 4-(хлоркарбонил)бензоат (формула 8-4, 0,200 г, 0,831 ммоль), (2S,6R)-1-бензил-2,6-диметилпиперазин (0,181 г, 0,914 ммоль) и TEA (0,232 мл, 1,661 ммоль) растворяли в метиленхлориде (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 12 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,152 г, 49,9%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 814
Метил 4-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-карбонил)бензоат (формула 8-4, 0,152 г, 0,415 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,127 мл, 2,074 ммоль) и гидроксид калия (0,116 г, 2,074 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 814 (0,118 г, 77,4%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ 7,66 (д, 2 H, J=6,7 Гц), 7,20-6,99 (м, 7 H), 3,61 (с, 2 H), 2,74-2,68 (м, 2 H), 2,49 (м, 2 H), 2,37-2,33 (м, 2 H), 0,92 (с, 3 H), 0,71 (с, 3 H); ЖХМС (ES) m/z 368,1 (M++1).
Пример 249: Синтез соединения 818
(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)сульфонил)-N-гидроксибензамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)сульфонил)бензоата
Метил 4-(хлорсульфонил)бензоат (формула 8-4, 0,200 г, 0,852 ммоль), (2S,6R)-1-бензил-2,6-диметилпиперазин (0,186 г, 0,938 ммоль) и TEA (0,236 мл, 1,705 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением желаемого соединения (0,181 г, 52,8%) в виде твердого вещества белого цвета.
Стадия 2: Синтез соединения 818
Метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)сульфонил)бензоат (формула 8-5, 0,100 г, 0,248 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,076 мл, 1,242 ммоль) и гидроксид калия (0,070 г, 1,242 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 818 (0,042 г, 41,9%) в виде твердого вещества цвета слоновой кости.
1H-ЯМР (400 МГц, CD3OD)) δ 8,00 (д, 2 H, J=8,3 Гц), 7,81 (д, 2 H, J=8,4 Гц), 7,32-7,25 (м, 4 H), 7,19 (т, 1 H, J=7,0 Гц), 3,82 (с, 2 H), 3,52 (д, 2 H, J=11,2 Гц), 2,73-2,68 (м, 2 H), 2,19 (т, 2 H, J=10,6 Гц), 1,07 (с, 3 H), 1,05 (с, 3 H); ЖХМС (ES) m/z 404,1 (M++1).
Пример 250: Синтез соединения 820
(2-(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)сульфонил)фенил)-N-гидроксиацетамид)
Стадия 1: Синтез метил 2-(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)сульфонил)фенил)ацетата
Метил 2-(4-(хлорсульфонил)фенил)ацетат (формула 8-4, 0,200 г, 0,804 ммоль), (2S,6R)-1-бензил-2,6-диметилпиперазин (0,176 г, 0,885 ммоль) и TEA (0,223 мл, 1,608 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. В реакционную смесь добавляли воду, после чего экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 20%) и концентрировали с получением желаемого соединения (0,198 г, 59,2%) в виде бесцветной жидкости.
Стадия 2: Синтез соединения 820
Метил 2-(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)сульфонил)фенил)ацетат (формула 8-5, 0,198 г, 0,475 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,145 мл, 2,377 ммоль) и гидроксид калия (0,133 г, 2,377 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 820 (0,124 г, 62,5%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ 7,67 (д, 2 H, J=7,6 Гц), 7,37-7,15 (м, 7 H), 3,74 (с, 2 H), 3,49 (д, 2 H, J=7,8 Гц), 3,21 (с, 2 H), 2,74 (м, 2 H), 2,23-2,18 (м, 2 H), 0,99 (с, 3 H), 0,98 (с, 3 H); ЖХМС (ES) m/z 418,1 (M++1).
Пример 251: Синтез соединения 822
(4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)-N-гидроксициклогексан-1-карбоксамид)
Стадия 1: Синтез метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)циклогексан-1-карбоксилата
Метил 4-формилциклогексан-1-карбоксилат (формула 8-4, 0,150 г, 0,881 ммоль), (2S,6R)-1-бензил-2,6-диметилпиперазин (0,212 г, 0,881 ммоль) и STAB (0,280 г, 1,322 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением желаемого соединения (0,175 г, 55,4%) в виде бесцветной жидкости.
Стадия 2: Синтез соединения 822
Метил 4-(((3R,5S)-4-бензил-3,5-диметилпиперазин-1-ил)метил)циклогексан-1-карбоксилат (формула 8-5, 0,175 г, 0,488 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,149 мл, 2,441 ммоль) и гидроксид калия (0,137 г, 2,441 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 822 (0,050 г, 28,5%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ 7,38 (д, 2 H, J=7,4 Гц), 7,32-7,28 (м, 2 H), 7,21 (т, 1 H, J=7,3 Гц), 3,82 (с, 2 H), 2,72-2,67 (м, 4 H), 2,19-1,96 (м, 3 H), 1,83-1,67 (м, 6 H), 1,48-1,45 (м, 5 H), 1,03 (с, 6 H); ЖХМС (ES) m/z 360,2 (M++1).
Пример 252: Синтез соединения 823
(2-(4-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-карбонил)фенил)-N-гидроксиацетамид)
Стадия 1: Синтез метил 2-(4-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-карбонил)фенил)ацетата
4-(2-Метокси-2-оксоэтил)бензойную кислоту (формула 8-4, 0,150 г, 0,772 ммоль), (2S,6R)-1-бензил-2,6-диметилпиперазин (0,186 г, 0,772 ммоль), EDC (0,296 г, 1,545 ммоль), HOBt (0,237 г, 1,545 ммоль) и DIPEA (0,684 мл, 3,862 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением желаемого соединения (0,133 г, 45,3%) в виде бесцветной жидкости.
Стадия 2: Синтез соединения 823
Метил 2-(4-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-карбонил)фенил)ацетат (формула 8-5, 0,133 г, 0,350 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,107 мл, 1,748 ммоль) и гидроксид калия (0,098 г, 1,748 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 823 (0,015 г, 11,2%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ 7,36 (д, 2 H, J=7,5 Гц), 7,30 (т, 3 H, J=7,5 Гц), 7,25-7,21 (м, 5 H), 4,44 (д, 1 H, J=9,6 Гц), 3,84-3,75 (м, 2 H), 3,51 (д, 1 H, J=11,2 Гц), 3,23 (м, 2 H), 2,95-2,88 (м, 1 H), 2,69 (м, 1 H), 2,53 (м, 1 H), 1,11 (с, 3 H), 0,88 (с, 3 H); ЖХМС (ES) m/z 382,2 (M++1).
Пример 253: Синтез соединения 824
(3-(4-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-карбонил)фенил)-N-гидроксипропанамид)
Стадия 1: Синтез метил 3-(4-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-карбонил)фенил)пропаноата
4-(3-Метокси-3-оксопропил)бензойную кислоту (формула 8-4, 0,150 г, 0,724 ммоль), (2S,6R)-1-бензил-2,6-диметилпиперазин (0,174 г, 0,724 ммоль), EDC (0,278 г, 1,448 ммоль), HOBt (0,222 г, 1,448 ммоль) и DIPEA (0,641 мл, 3,620 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 16 часов. В реакционную смесь добавляли воду, после чего экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (диоксид кремния, картридж 4 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением желаемого соединения (0,089 г, 31,2%) в виде прозрачной жидкости.
Стадия 2: Синтез соединения 824
Метил 3-(4-((3R,5S)-4-бензил-3,5-диметилпиперазин-1-карбонил)фенил)пропаноат (формула 8-5, 0,089 г, 0,226 ммоль), гидроксиламин (50,00%-ный водный раствор, 0,069 мл, 1,128 ммоль) и гидроксид калия (0,063 г, 1,128 ммоль) растворяли в смеси метанол (1 мл)/тетрагидрофуран (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл), затем перемешивали. Выпавший твердый осадок фильтровали и сушили с получением соединения 824 (0,052 г, 58,3%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ 7,36 (д, 2 H, J=7,4 Гц), 7,30 (т, 2 H, J=7,5 Гц), 7,24 (т, 3 H, J=8,6 Гц), 7,26-7,22 (м, 3 H), 7,18 (д, 2 H, J=7,8 Гц), 4,44 (д, 1 H, J=8,0 Гц), 3,85-3,76 (м, 2 H), 3,53 (д, 1 H, J=9,0 Гц), 2,92-2,87 (м, 3 H), 2,69 (м, 2 H), 2,53 (м, 1 H), 2,30 (м, 2 H), 1,11 (с, 3 H), 0,90 (с, 3 H); ЖХМС (ES) m/z 396,2 (M++1).
Структурные формулы соединений 080-824, полученных, как описано выше, представлены в таблице 16 далее.
[Таблица 16] Структурные формулы соединений производных диметилпиперазина
|
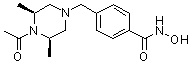
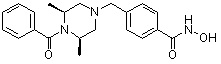
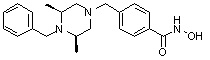
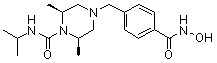
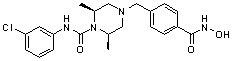
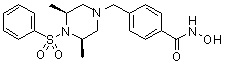
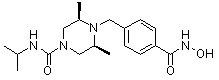
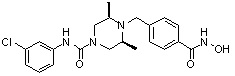
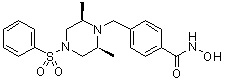
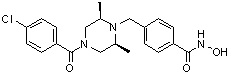
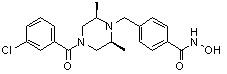
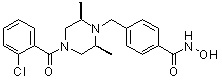
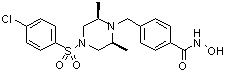
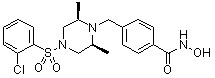
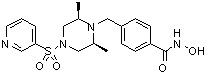
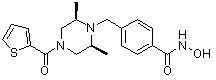
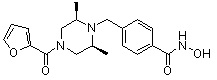
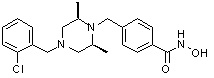
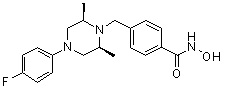
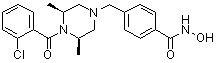
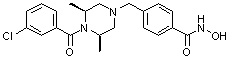
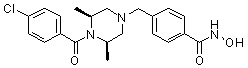
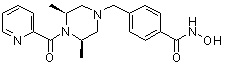
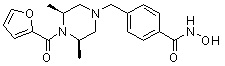
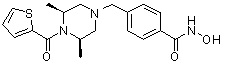
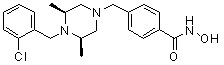
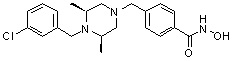
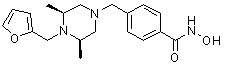
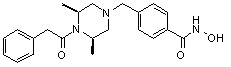
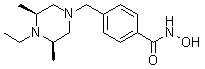
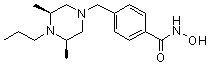
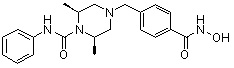
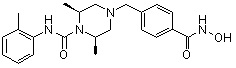
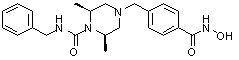
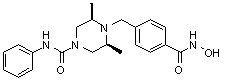
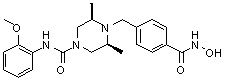
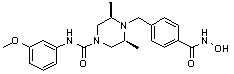
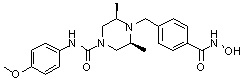
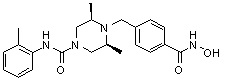
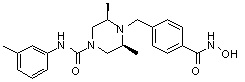
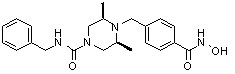
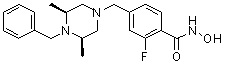
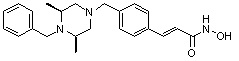
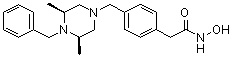
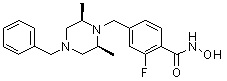
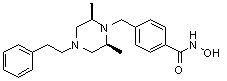
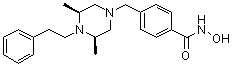
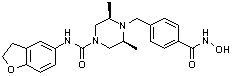
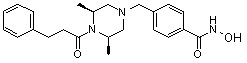
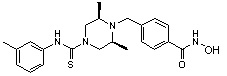
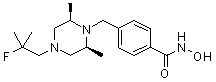
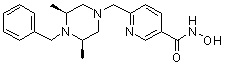
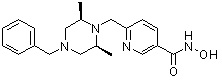
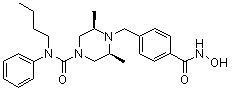
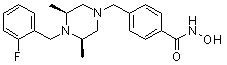
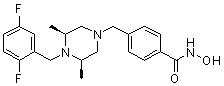
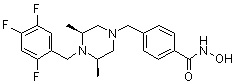
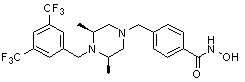
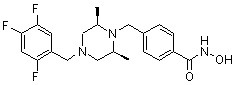
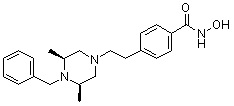
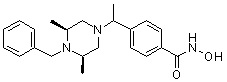
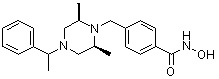
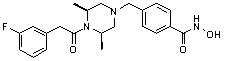
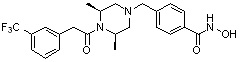
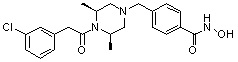
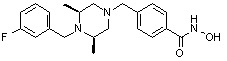
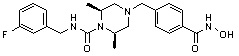
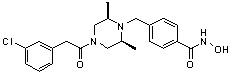
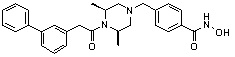
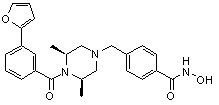
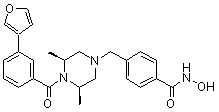
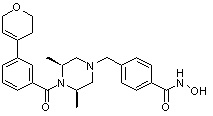
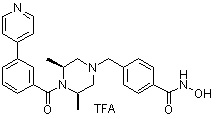
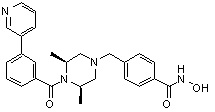
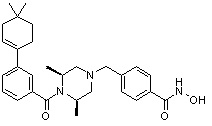
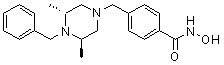
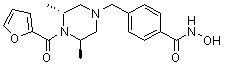
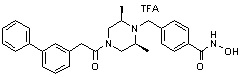
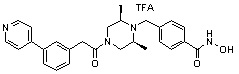
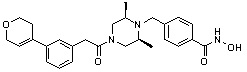
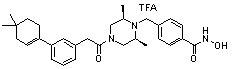
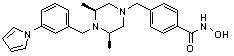
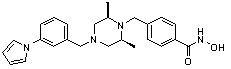
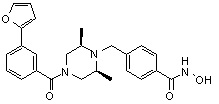
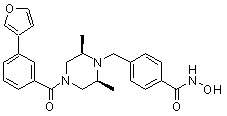
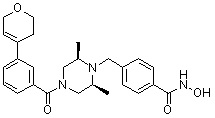
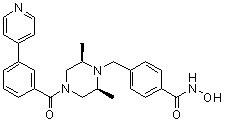
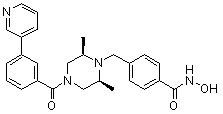
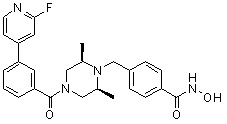
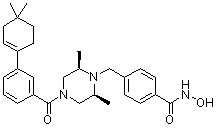
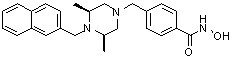
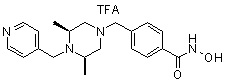
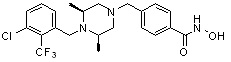
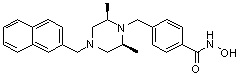
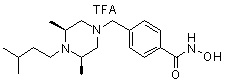
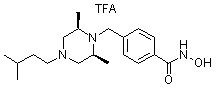
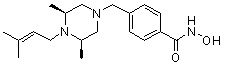
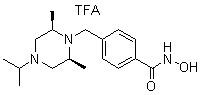
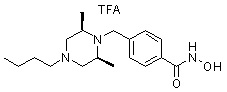
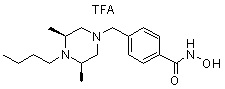
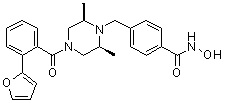
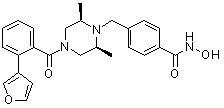
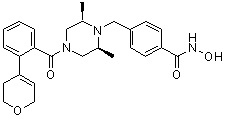
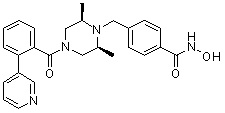
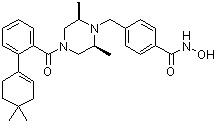
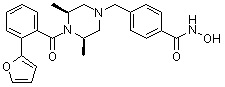
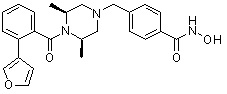
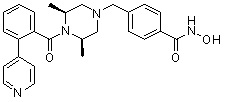
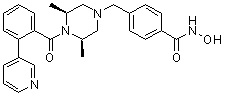
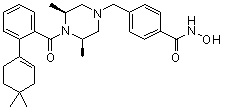
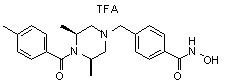
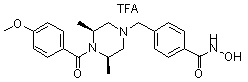
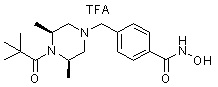
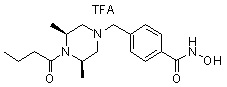
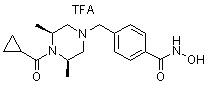
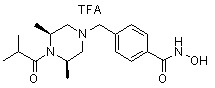
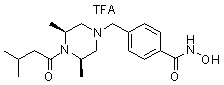
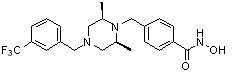
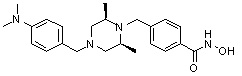
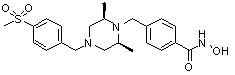
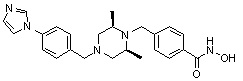
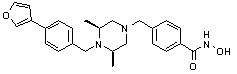
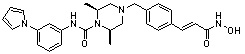
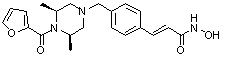
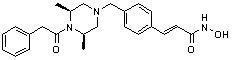
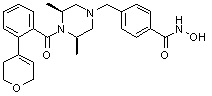
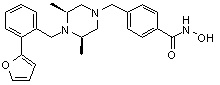
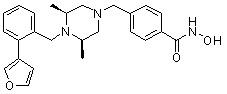
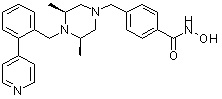
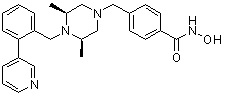
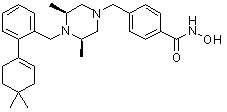
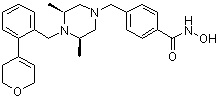
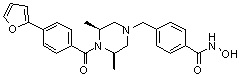
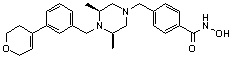
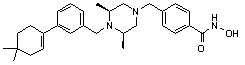
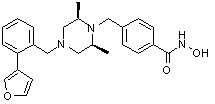
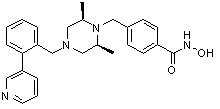
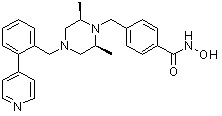
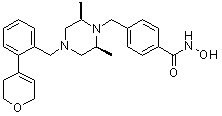
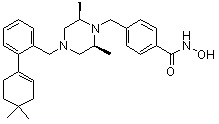
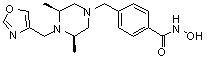
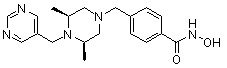
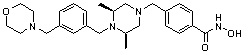
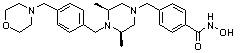
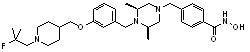
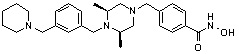
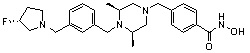
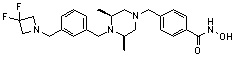
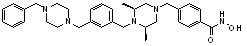
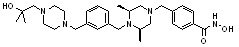
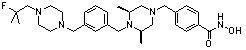
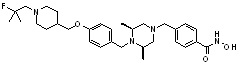
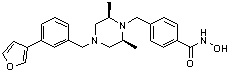
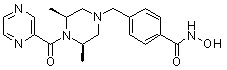
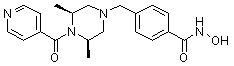
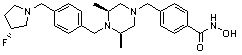
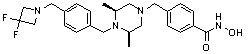
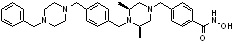
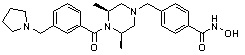
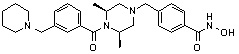
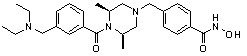
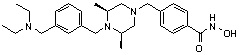
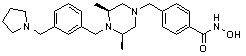
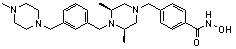
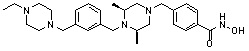
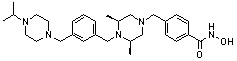
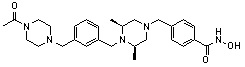
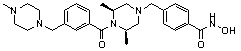
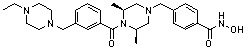
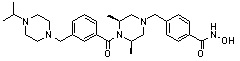
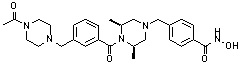
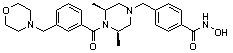
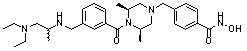
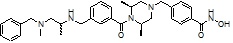
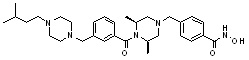
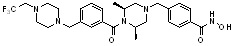
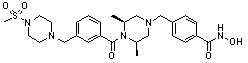
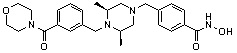
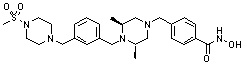
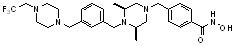
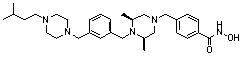
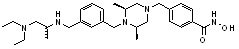
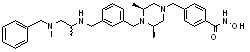
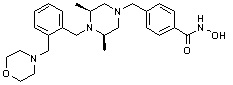
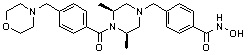
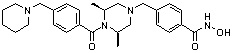
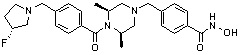
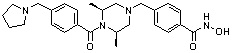
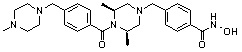
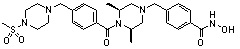
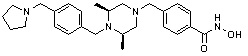
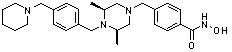
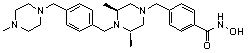
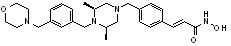
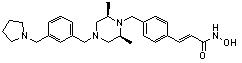
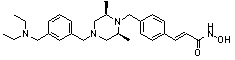
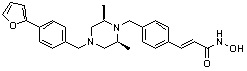
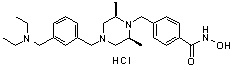
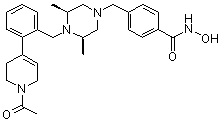
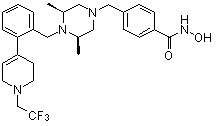
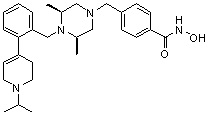
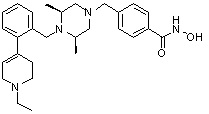
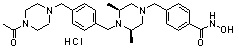
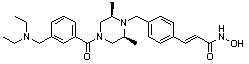
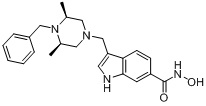
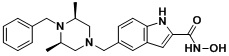
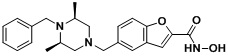
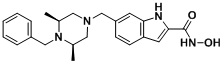
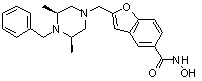
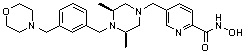
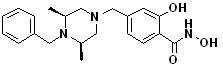
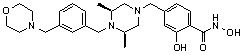
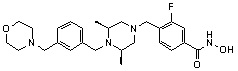
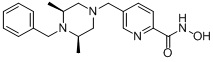
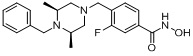
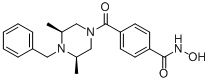
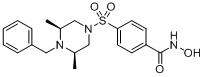
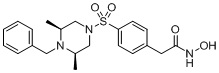
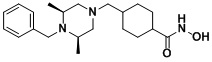
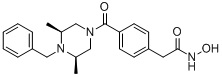
Экспериментальный пример 1: анализы ингибирования ферментативной активности HDAC ( in vitro )
Для исследования HDAC6 селективности соединений формулы I по настоящему изобретению с помощью анализов ингибирования ферментативной активности HDAC1 и HDAC6, эксперимент проводили с использованием в качестве контроля обычного вещества.
Ферментативную активность HDAC определяли с использованием набора HDAC Fluorimetric Drug Discovery Kit (BML-AK511, 516, Enzo Life Science). Для исследования ферментативной активности HDAC1 в качестве источника фермента использовали человеческий рекомбинантный HDAC1 (BML-SE456), в качестве субстрата использовали Fluor de Lys®-"SIRT1 (BNL-KI177). 5-кратно разведенные соединения высевали в 96-луночный планшет, и затем в каждую лунку планшета добавляли 0,3 мкг фермента и 10 мкМ субстрата, и давали возможность взаимодействовать при температуре 30°C в течение 60 минут. Затем добавляли Fluor de Lys® Developer II (BML-KI176) и давали возможность взаимодействовать в течение 30 минут, после чего измеряли значение флуоресценции (Ex 360, Em 460) с помощью мультипланшетного ридера (Flexstation 3, Molecular Device). Фермент HDAC6 исследовали с использованием рекомбинантного человеческого HDAC6 (382180) (Calbiochem) в соответствии с тем же протоколом, что и метод исследования ферментативной активности HDAC1. На основании полученных значений рассчитывали каждое значение IC50 с использованием программы GraphPad Prism4.0.
[Таблица 17] Результаты анализов ингибирования ферментативной активности HDAC
|
Как можно видеть из таблицы 16, выше, соединения производные диметилпиперазин гидроксамовой кислоты по настоящему изобретению в анализах ингибирования активности HDAC1 и HDAC6 показали изобретение приблизительно от 2 до 5500 раз выше селективное ингибирующую активность в отношении HDAC6.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Соединения, представленные формулой I, их изомеры или их фармацевтически приемлемые соли могут быть использованы для предупреждения или лечения ассоциированных с активностью гистондеацетилазы 6 заболеваний.
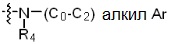
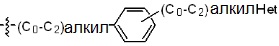
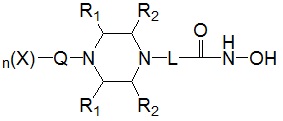
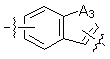
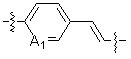
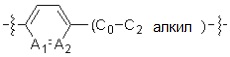
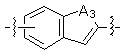
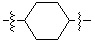
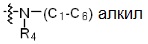
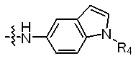
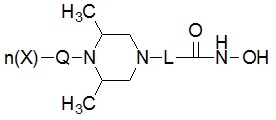
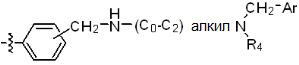
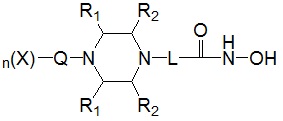
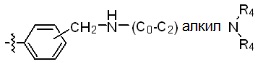
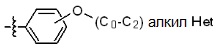
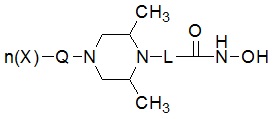
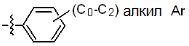
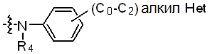
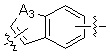
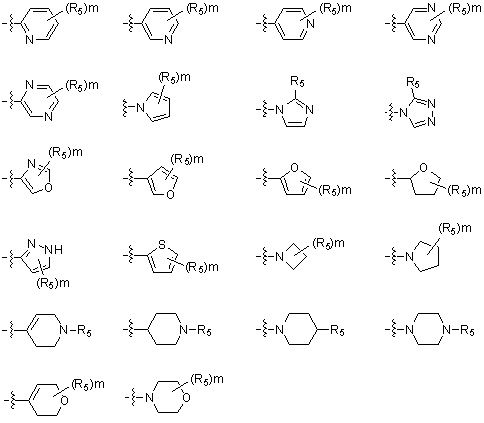
Биарил- или гетероциклические биарилзамещенные производные циклогексена в качестве ингибиторов сетр
Липидный преконцентрат с замедленным высвобождением фармакологически активного вещества и фармацевтическая композиция, содержащая его
Липидный преконцентрат анионных фармакологически активных веществ с замедленным высвобождением и содержащая его фармацевтическая композиция
Новые соединения для селективных ингибиторов гистондеацетилазы и фармацевтическая композиция, включающая такие соединения
Липидный преконцентрат аналогов gnrh с замедленным высвобождением и содержащая его фармацевтическая композиция
Липидный предконцентрат катионного фармакологически активного вещества с длительным высвобождением и содержащая его фармацевтическая композиция
Производные гетероциклические алкильные соединения в качестве селективных ингибиторов гистондеацетилазы и содержащие их фармацевтические композиции
Производные оксадиазоламина в качестве ингибитора гистондеацетилазы 6 и содержащая их фармацевтическая композиция
Производные 1,3,4-оксадиазолсульфамида в качестве ингибитора гистондеацетилазы 6 и содержащая их фармацевтическая композиция
Производные 1,3,4-оксадиазолсульфонамида в качестве ингибиторов деацетилазы гистонов 6 и фармацевтическая композиция, содержащая их
Циклоалкениларильные производные для ингибирования транспортного белка холестериновых эфиров
Новые соединения для селективных ингибиторов гистондеацетилазы и фармацевтическая композиция, включающая такие соединения
Производные гетероциклические алкильные соединения в качестве селективных ингибиторов гистондеацетилазы и содержащие их фармацевтические композиции
Производные 1,3,4-оксадиазолсульфонамида в качестве ингибиторов деацетилазы гистонов 6 и фармацевтическая композиция, содержащая их
1,3,4,-оксадизоламидное производное соединение в качестве ингибитора гистондеацетилазы 6 и содержащая его фармацевтическая композиция

