Результат интеллектуальной деятельности: (2R,4R)-5-(5′-ХЛОР-2′-ФТОРБИФЕНИЛ-4-ИЛ)-2-ГИДРОКСИ-4-[(5-МЕТИЛОКСАЗОЛ-2-КАРБОНИЛ)АМИНО]ПЕНТАНОВАЯ КИСЛОТА
Вид РИД
Изобретение
УРОВЕНЬ ТЕХНИКИ
Область техники, к которой относится изобретение
Настоящее изобретение относится к новому соединению и его кристаллической форме, обладающим неприлизин-ингибирующим действием. Изобретение также относится к фармацевтическим композициям, содержащим соединение, способам получения соединения и способам применения соединения для лечения заболеваний, таких как гипертензия, сердечная недостаточность и почечная недостаточность.
Уровень техники
Неприлизин (нейтральная эндопептидаза, EC 3.4.24.11) (НЕП) является связанной эндотелиальной мембраной Zn2+металлопептидазой, найденной во множестве органов и тканей, включая мозг, почки, легкие, желудочно-кишечный тракт, сердце и периферийные сосуды. НЕП разлагается и инактивирует множество эндогенных пептидов, таких как энкефалины, циркулирующий брадикинин, пептиды ангиотензина и натрийуретические пептиды, последние их которых обладают некоторыми действиями, включая, например, вазодилатацию и натрийурез/диурез, а также ингибирование сердечной гипертрофии и фиброз желудочков. Таким образом, НЕП играет важную роль в гомеостазе кровяного давления и здоровье сердечнососудистой системы.
Ингибиторы НЕП, такие как тиорфан, кандоксатрил и кандоксатрилат, изучались в качестве потенциальных терапевтических средств. Соединения, которые ингибируют и НЕП и фермент, превращающий ангиотензин-I (АПФ) также известны, и включают омапатрилат, гемпатрилат и сампатрилат. Что касается ингибиторов вазопептидазы, этот последний класс соединений описан у Robl et al. (1999) Exp. Opin. Ther. Patents 9(12): 1665-1677.
Множество ингибиторов НЕП описано в патенте США № 8,263,629 Coppola et al и патенте США № 8,586,536 Gendron et al. Множество этих соединений обладают одним или более желательными свойствами. Несмотря на эти соединения, однако, все еще остается необходимость в мощном ингибиторе НЕП, который имеет высокую пероральную биодоступность и низкий клиренс по всем протестированным видам. Данное изобретение направлено на эту потребность.
Дополнительно к эффективному применению ингибитора НЕП в качестве терапевтического агента, желательно иметь твердую форму, которую легко производить, и которая имеет приемлемую химическую и физическую стабильность. Например, было бы очень желательно иметь физическую форму, которая является теплостойкой при достаточно высокой температуре, что способствует обработке и хранению продукта. Кристаллические твердые вещества обычно предпочтительнее аморфных форм для улучшения чистоты и стабильности произведенного продукта. Однако получение кристаллических форм органических соединений крайне непредсказуемо. Не существует надежных способов, позволяющих спрогнозировать какая, если она существует, форма органического соединения будет кристаллической. Более того, не существует способов спрогнозировать какая, если она существует, кристаллическая форма будет иметь физические свойства, желательные для применения в качестве фармацевтических агентов. Следовательно, существует необходимость в стабильной кристаллической форме, которая имеет достаточно высокую температуру плавления.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем изобретении представлено новое соединение (1), которое обладает ингибирующим действием на фермент неприлизин (НЕП). Следовательно, ожидается, что это соединение полезно и предпочтительно в качестве терапевтического агента для лечения состояний, таких как гипертензия, легочная гипертензия, сердечная недостаточность и почечная недостаточность.
Один аспект изобретения относится к (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоте (1):
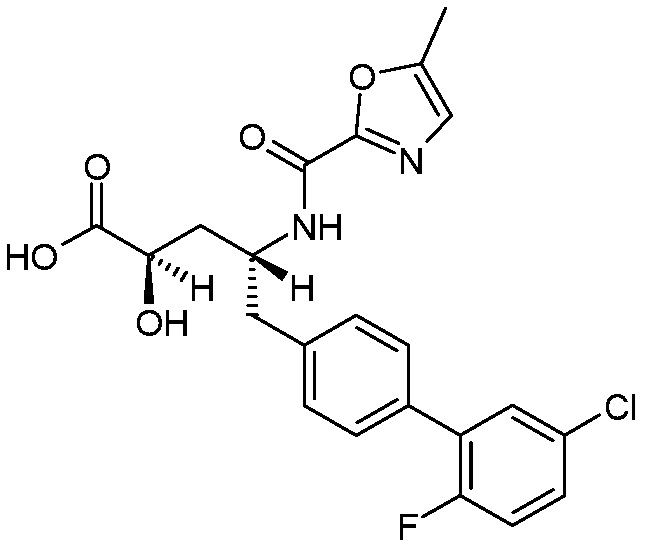 (1),
(1),
или ее фармацевтически приемлемой соли. Другой аспект данного изобретения относится к кристаллической форме соединения 1. В одном варианте, кристаллическая форма (1') не сольватирована.
Другой аспект изобретения относится к фармацевтическим композициям, содержащим один или более фармацевтически приемлемых носителей и соединение 1 или его кристаллическую форму. Такие композиции могут необязательно содержать другие терапевтические агенты, включающие, но не ограниченные ими, антагонист рецептора AT1, ингибитор ангиотензинпревращающего фермента, ингибитор фосфодиэстеразы (ФДЭ), ингибитор ренина, диуретик или их сочетание.
Соединение 1 в соответствии с данным изобретением обладает ингибирующим действием к ферменту НЕП, и поэтому может быть полезным в качестве терапевтического агента для лечения пациентов, страдающих заболеванием или расстройством, которое лечат ингибированием фермента НЕП или повышением уровней его пептидного субстрата. Таким образом, один аспект данного изобретения относится к способу лечения пациентов, страдающих заболеванием или расстройством, которое лечат ингибированием фермента НЕП, включающему введение пациенту терапевтически эффективного количества соединения 1. Другой аспект изобретения относится к способу лечения гипертензии, легочной гипертензии, сердечной недостаточности или почечной недостаточности, включающему введение пациенту терапевтически эффективного количества соединения 1. Другой аспект изобретения относится к способу ингибирования фермента НЕП у пациента, включающему введение пациенту ингибирующего фермент НЕП количества соединения 1.
Так как соединение 1 в соответствии с данным изобретением обладает ингибирующим действием на НЕП, оно также применяется в качестве средства исследования. Следовательно, один аспект данного изобретения относится к способу применения соединения 1 в соответствии с данным изобретением в качестве средства исследования, где способ включает проведение биологического исследования с применением соединения 1. Соединение 1 также может применяться для оценки новых химических соединений. Таким образом, другой аспект данного изобретения относится к способу оценки тестируемого соединения в биологическом исследовании, включающему: (a) проведение биологического исследования с тестируемым соединением с получением первого результата исследования; (b) проведение биологического исследования с соединением 1 с получением второго результата исследования; где стадию (a) проводят до, после или одновременно со стадией (b); и (c) сравнение первого результата исследования со стадии (a) со вторым результатом исследования со стадии (b). Типовые биологические исследования включают исследование ингибирования фермента НЕП. Еще один аспект данного изобретения относится к способу изучения биологической системы или образца, содержащего фермент НЕП, где способ включает: (a) контакт биологической системы или образца с соединением 1; и (b) определение эффектов, вызванных соединением 1 в биологической системе или образце.
Еще один аспект изобретения относится к способам, применяемым для получения соединения 1 или его кристаллической формы.
Еще один аспект изобретения относится к применению соединения 1 или его кристаллической формы для производства лекарственного средства, особенно для производства лекарственного средства, применяемого для лечения гипертензии, сердечной недостаточности или почечной недостаточности. Другой аспект изобретения относится к применению соединения 1 или его кристаллической формы для ингибирования фермента НЕП у пациента. Другие аспекты и варианты изобретения описаны здесь.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Различные аспекты данного изобретения иллюстрированы на прилагаемых чертежах.
На фиг. 1 показана порошковая рентгеновская дифрактограмма (ПРД) кристаллической не сольватированной (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоты (1').
На фиг. 2 показана термограмма дифференциальной сканирующей калориметрии (ДСК) кристаллической не сольватированной формы (1').
На фиг. 3 показан профиль тепловой гравиметрии кристаллической не сольватированной формы (1').
На фиг. 4 показана изотерма динамического поглощения влаги (ДПВ) кристаллической не сольватированной формы (1').
На фиг. 5 представлено изображение оптического поляризационного микроскопа (ОПМ) кристаллической не сольватированной формы (1').
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте изобретение относится к (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоте (1) или ее фармацевтически приемлемой соли.
Соединение 1 в соответствии с данным изобретением содержит два хиральных центра, и поэтому соединение с такой структурой может существовать в различных стереоизомерных формах. Например, атомы углерода могут иметь конкретную (R,R), (S,S), (S,R) или (R,S) конфигурацию, или в них преобладает стереоизомерная форма, имеющая такую конфигурацию. Соединение 1 показано и названо в (R,R) конфигурации. Специалисту должно быть понятно, что незначительные количества других стереоизомеров могут присутствовать в композициях в соответствии с данным изобретением, если не указано иное, при условии, что польза композиции в целом не исключается присутствием таких других изомеров. Отдельные стереоизомеры могут быть получены множеством способов, известных в данной области техники, включая хиральную хроматографию с применением подходящей хиральной неподвижной фазы или подложки, или химическим превращением их в диастереоизомеры, разделением диастереоизомеров обычными средствами, такими как хроматография или перекристаллизация с последующим восстановлением исходного стереоизомера.
Соединение 1 в соответствии с данным изобретением обладает ингибирующим действием на неприлизин (НЕП), то есть соединение способно ингибировать фермент-каталитическую активность. Одним из показателей способности соединения ингибировать активность НЕП является константа ингибирования (pKi). Значение pKi является отрицательным логарифмом к основанию 10 константы диссоциации (Kid), который обычно указывают в молярных единицах. Соединение в соответствии с данным изобретением имеет pKi для НЕП ≥9,0. Другие свойства и применение соединения 1 могут быть продемонстрированы с применением in vitro и in vivo анализов, которые хорошо известны специалистам в данной области техники, включающим, кроме прочего, описанные в патенте США № 8,586,536.
Соединение 1, а также соединения, применяемые в его синтезе, могут также включать изотопно-меченные соединения, то есть в которых один или более атомов обогащены атомами, имеющими атомную массу, отличающуюся от атомной массы, преимущественно имеющейся в природе. Примеры изотопов, которые могут быть введены в соединения, описанные в изобретении, например, включают, но не ограничены ими, 2H, 3H, 13C, 14C, 15N, 18O, 17O, 35S, 36Cl и 18F. Особенный интерес представляет соединение 1, обогащенное тритием или углеродом-14, которое может применяться, например, в исследованиях распределения в тканях; соединение 1, обогащенное дейтерием, особенно то, которое в месте метаболизма превращается в соединение, имеющее большую метаболическую стабильность; и соединение 1, обогащенное позитронно-излучающим изотопом, таким как 11C, 18F, 15O и 13N, которое может применяться, например, в исследованиях позитронно-эмиссионной томографии (ПЭТ).
Химические структуры названы здесь в соответствии с условными обозначениями IUPAC, введенными в программное обеспечение ChemDraw (Perkin Elmer, Inc., Cambridge, MA).
Определения
При описании соединения, композиций, способов и процессов изобретения следующие термины имеют следующие значения, если не указано иначе. Кроме того, в данном описании единственное число включает соответствующие множественные формы, если контекст четко не указывает на иное. Термины "содержащий", "включающий" и "имеющий" являются включающими и означают, что могут быть дополнительные элементы, отличные от перечисленных элементов. Все числа, обозначающие количества ингредиентов, свойства, такие как молекулярная масса, условия реакции и так далее, применяемые здесь, понимаются как модифицированные во всех случаях термином "около", если не указано иначе. Следовательно, представленные здесь цифры являются приблизительными, которые могут варьироваться в зависимости от желаемых свойств, получаемых в соответствии с данным изобретением. По меньшей мере, и не как попытка ограничить применение доктрины эквивалентов формулой изобретения, каждое число, по меньшей мере, должно рассматриваться в свете указанных значимых цифр и с применением обычных методов округления.
Термин ʺоколоʺ или ʺприблизительноʺ, при применении в контексте теплового поведения соединения 1, определен как ±1-3°С. Термин ʺприблизительноʺ, при применении в контексте % дозы соединения 1, выводимого в моче, определяет погрешность, которая обычно составляет почти вдвое от стандартного отклонения или полуширину 95 процентов доверительного интервала. Термин ʺприблизительноʺ в других частях описания может применяться для указания стандартного отклонения или количества отклонения или вариации или разбросанности совокупности данных.
Термин ʺконтролируемое высвобождениеʺ в данном описании является синонимом замедленному высвобождению и продленному высвобождению и относится к количеству лекарственного средства, доставляемому в течение продленного периода времени пациенту. В общем, таблетки и капсулы с контролируемым высвобождением выделяют активное вещество в пациента в течение периодов времени около 8, 12, 16 и 24 часов. С другой стороны, термин ʺнемедленное высвобождениеʺ относится к активному веществу, выделяемому в пациенте в течение короткого периода времени, обычно менее около 30 минут. Термин ʺотсроченное высвобождениеʺ относится к таблеткам и капсулам, которые выделяют фармацевтическую дозу после установленного периода времени. Такие лекарственные формы обычно имеют энтеросолюбильную оболочку для того, чтобы предотвратить выделение в желудке, но позволить выделение в кишечном тракте.
В данном описании, фраза "формулы" или "имеющее формулу" или "имеющее структуру" не является ограничивающей и применяется так же, как и часто применяемый термин "содержащее". Например, если изображена одна структура, понятно, что она охватывает все стереоизомерные и таутомерные формы, если не указано иначе.
В общем, при описании фармацевтических твердых веществ, термин "не сольватированное" означает "без растворителя". Таким образом, если кристаллическая форма в соответствии с данным изобретением описана как "не сольватированная", это означает, что кристаллические частицы содержат по существу только молекулы (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоты; форма не содержит значительных количеств других включенных в решетку молекул растворителя, или другими словами, растворитель по существу не включен в кристаллическую решетку. Термин ʺне сольватированноеʺ также означает не гидрированное или безводное, если вода является растворителем.
Термин ʺтемпература плавленияʺ в данном описании означает температуру, при которой максимальный эндотермический поток тепла наблюдается дифференциальной сканирующей калориметрией, для теплового перехода, который соответствует превращению твердой фазы в жидкую.
Термин "фармацевтически приемлемый" относится к материалу, который не является биологически или по-другому неприемлемым при применении в изобретении. Например, термин "фармацевтически приемлемый носитель" относится к материалу, который может быть введен в композицию и вводиться пациенту, не вызывая неприемлемые биологические эффекты или не взаимодействуя неприемлемым образом с другими компонентами композиции. Такие фармацевтически приемлемые материалы обычно соответствуют требуемым стандартам токсикологического и производственного тестирования, и включают материалы, идентифицированные как подходящие неактивные ингредиенты Управлением США по санитарному надзору за качеством пищевых продуктов и медикаментов.
Термин "фармацевтически приемлемая соль" означает соль, полученную из основания или кислоты, которые приемлемы для введения пациенту, такому как млекопитающее (например, соли, имеющие приемлемую для млекопитающих безопасность для данного режима дозирования). Однако понятно, что соли, включенные в данное изобретение, не обязательно являются фармацевтически приемлемыми солями, например, солями промежуточных соединений, которые не предназначены для введения пациенту. Фармацевтически приемлемые соли могут быть получены из фармацевтически приемлемых неорганических и органических оснований и из фармацевтически приемлемых неорганических и органических кислот. Кроме того, если соединение содержит основную часть, такую как амин, пиридин или имидазол, и кислую часть, такую как карбоновая кислота или тетразол, цвиттерионы могут быть образованы, и они включены в термин "соль", применяемый здесь. Соли, полученные из фармацевтически приемлемых неорганических оснований, включают соли аммония, кальция, меди, окиси железа, железа, лития, магния, марганца, окиси марганца, калия, натрия и цинка, и подобные. Соли, полученные из фармацевтически приемлемых оснований, включают соли первичных, вторичных или третичных аминов, включая замещенные амины, циклические амины, природные амины и подобные, такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и подобные. Соли, полученные из фармацевтически приемлемых неорганических кислот, включают соли борных, карбоновых, галоидных (бромистоводородной, хлористоводородной, фтористоводородной или йодистоводородной), азотной, фосфорной, сульфаминовой и серной кислот. Соли, полученные из фармацевтически приемлемых органических кислот, включают соли алифатических гидроксильных кислот (например, лимонной, глюконовой, гликолевой, молочной, лактобионовой, яблочной и винной кислот), алифатических монокарбоновых кислот (например, уксусной, масляной, муравьиной, пропионовой и трифторуксусной кислот), аминокислот (например, аспарагиновой и глутаминовой кислот), ароматических карбоновых кислот (например, бензойной, п-хлорбензойной, дифенилуксусной, гентизиновой, гиппуровой и трифенилуксусной кислот), ароматических карбоновых кислот (например, о-гидроксибензойной, п-гидроксибензойной, 1-гидроксинафталин-2-карбоновой и 3-гидроксинафталин-2-карбоновой кислот), аскорбиновой, дикарбоновых кислот (например, фумаровой, малеиновой, щавелевой и янтарной кислот), глюкуроновой, миндальной, слизевой, никотиновой, оротовой, памовой, пантотеновой, сульфоновых кислот (например, бензолсульфоновой, камфосульфоновой, эдизиловой, этансульфоновой, изетионовой, метансульфоновой, нафталинсульфоновой, нафталин-1,5-дисульфоновой, нафталин-2,6-дисульфоновой и п-толуолсульфоновой кислоты), ксинафоевой кислоты и подобных.
Термин "терапевтически эффективное количество" означает количество, достаточное для эффективного лечения при введении пациенту, нуждающемуся в таковом, то есть, количество лекарственного средства, необходимое для получения желаемого терапевтического эффекта. Например, терапевтически эффективным количеством для лечения гипертензии является количество соединения, необходимое для, например, снижения, подавления, ликвидации или профилактики симптомов гипертензии, или для лечения первопричин гипертензии. В одном варианте, терапевтически эффективным количеством является количество лекарственного средства, необходимое для снижения кровяного давления, или количество лекарства, необходимое для поддержания нормального кровяного давления. С другой стороны, термин "эффективное количество" означает количество, достаточное для получения желаемого результата, которым не обязательно является терапевтический результат. Например, при изучении системы, содержащей фермент НЕП, "эффективным количеством" может быть количество, необходимое для ингибирования фермента.
Термин "лечить" или "лечение" в данном описании означает обработку или лечение заболевания или медицинского состояния (такого как гипертензия) у пациента, такого как млекопитающее (в частности, человек), которое включает одно или более из следующих: (a) профилактику наступления заболевания или медицинского состояния, т.е. профилактику повторного наступления заболевания или состояния или профилактическое лечение пациента, который предрасположен к заболеванию или медицинскому состоянию; (b) облегчение заболевания или медицинского состояния, т.е. прекращение или снижение заболевания или медицинского состояния у пациента; (c) подавление заболевания или медицинского состояния, т.е. замедление или остановку развития заболевания или медицинского состояния у пациента; или (d) облегчение симптомов заболевания или медицинского состояния у пациента. Например, термин "лечение гипертензии" включает профилактику наступления гипертензии, облегчение гипертензии, подавление гипертензии и облегчение симптомов гипертензии (например, снижение кровяного давления). Термин "субъект" или "пациент" включает млекопитающих, таких как человек, которые нуждаются в лечении или профилактике заболевания, или которых в настоящее время обрабатывают для лечения или профилактики определенного заболевания или медицинского состояния, а также субъекты тестирования, на которых оценивают кристаллическое соединение или которых используют в исследовании, например, в животной модели.
Все другие термины, применяемые здесь, имеют свои обычные значения, понимаемые специалистами в области техники, к которой они принадлежат.
Общие методы синтеза
Соединение 1 в соответствии с данным изобретением и его кристаллическая не сольватированная форма могут быть синтезированы из легко доступных исходных материалов, как описано ниже в примерах. Должно быть понятно, что если указаны типовые или предпочтительные условия процесса (т.е. температуры реакций, время, молярные соотношения реагентов, растворители, давление и т.д.), другие условия процесса также могут применяться, если не указано иначе. Должно быть понятно, что хотя даны конкретные условия процесса (т.е. температуры кристаллизации, время, молярные соотношения реагентов, растворители, давление и т.д.), другие условия процесса также могут применяться, если не указано иначе. В некоторых случаях, реакции или кристаллизацию проводят при комнатной температуре и действительное измерение температуры не производят. Понятно, что комнатная температура означает температуру в пределах, обычно связанных с температурой окружающей среды в лаборатории, и обычно составляет от около 15°C до около 30°C, например, от около 20°C до около 25°C. В других случаях, реакции или кристаллизацию проводят при комнатной температуре, и температуру действительно измеряют и записывают.
Любые молярные соотношения, описанные в способах данного изобретения, могут быть легко определены различными методами, доступными специалистам в данной области техники. Например, такие молярные соотношения могут быть легко определены 1H ЯМР. Альтернативно, элементный анализ и методы ВЭЖХ могут применяться для определения молярного соотношения.
В одном варианте, изобретение относится к (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоте (1) or ее фармацевтически приемлемой соли.
В другом варианте, соединение 1 может быть получено сочетанием этилового эфира (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты с 2-метилоксазол-2-карбоиловой кислотой с получением соединения 1.
В еще одном варианте, соединение 1 может быть получено (a) объединением 2-метилоксазол-2-карбоиловой кислоты и гексафторфосфата N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (ГАТУ) в N,N-диметилформамиде (ДМФ) и перемешиванием при комнатной температуре; (b) добавлением этилового эфира (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты и N,N-диизопропилэтиламина и перемешиванием при комнатной температуре; (c) выделением и последующим растворением твердых веществ в сухом этаноле и сухом тетрагидрофуране; (d) добавлением раствора гидроксида лития в воде; и (e) выделением полученных твердых веществ с получением соединения 1. Полученные на предыдущих стадиях (c) и (e) твердые вещества также могут быть очищены хроматографией.
Получение кристаллической формы обычно осуществляют в подходящем инертном разбавителе, примеры которого включают, но не ограничены ими, ацетон, ацетонитрил, этилацетат, метилэтилкетон, метанол, этанол, изопропанол, изобутанол, дихлорметан, метилтрет-бутиловый эфир, циклопентилметиловый эфир, гексан и подобные, и их смеси, необязательно содержащие воду. Смеси инертных разбавителей (также названные системами растворителей) включают ацетон с водой, ацетонитрил с водой, этанол и этилацетат, этилацетат и гексан, и низшие спирты (C1-6алкил-OH) с водой, например, метанол и воду и изопропанол и воду. Особенно подходящие системы растворителей включают этилацетат и гексан. По завершении кристаллизации, кристаллическое соединение может быть выделено из реакционной смеси любыми обычными методами, такими как осаждение, фильтрация, концентрация, центрифугирование, сушка в вакууме и подобными.
В одном варианте, изобретение относится к кристаллической форме (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоты. В другом варианте, кристаллической формой является не сольватированная кристаллическая форма (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоты (1').
В другом варианте, кристаллическая форма (1') может быть получена (a) растворением (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоты (1) в этилацетате и гексане до полного растворения; и (b) выделением полученных твердых веществ с получением кристаллической формы (1'). Стадию (a) обычно проводят при комнатной температуре.
В еще одном варианте, кристаллическая форма (1') может быть получена (a) сочетанием этилового эфира (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты с 5-метилоксазол-2-карбоилата натрия с получением (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоты; (b) обработкой (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоты этилацетатом и гексаном до полного растворения; и (c) выделением полученного твердого вещества с получением кристаллической формы (1').
Кристаллические свойства
Как хорошо известно в области порошковой рентгеновской дифракции (ПРД), относительные высоты пиков ПРО рентгенограмм зависят от множества факторов, связанных с получением образца и геометрией инструмента, а положения пиков относительно не чувствительны к деталям эксперимента. ПРД, дифференциальную сканирующую калориметрию (ДСК), тепловой гравиметрический анализ (ТГА) и динамическое поглощение влаги (ДПВ) (также называемое анализ поглощения-десорбции влаги) проводят, как описано здесь.
В другом аспекте, изобретение относится к (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановой кислоте в кристаллической форме. В другом варианте, кристаллическая форма не сольватирована (1') и характеризуется Рентгенограммой ПРД, в которой положения пиков, по существу, соответствуют тем, которые показаны на фиг. 1. Вкладка с пунктирными линиями на фиг. 1 показывает у-масштабированное изображение для выделения пиков с более низкой интенсивностью.
Пики с относительными интенсивностями более 1% на площади перечислены в таблице ниже. Эта диаграмма показывает острые дифракционные пики в интервале 5-35° в 2θ. Эти и другие пики в дифракционной диаграмме могут применяться для идентификации этой формы.
|
*Значения 2θ указаны как значение ± 0,20.
Таким образом, в одном варианте, кристаллическая форма 1' характеризуется рентгенограммой ПРД, содержащей дифракционные пики со значениями 2θ 8,48±0,20, 14,19±0,20, 17,03±0,20, 21,15±0,20 и 25,41±0,20.
В другом варианте, кристаллическая форма 1' характеризуется рентгенограммой ПРД, содержащей дифракционные пики со значениями 2θ 7,51±0,20, 8,48±0,20, 14,19±0,20, 17,03±0,20, 17,62±0,20, 17,87±0,20, 20,59±0,20, 21,15±0,20, 21,88±0,20, 24,45±0,20, 24,78±0,20, 25,41±0,20, 25,67±0,20, 27,67±0,20 и 28,22±0,20.
В другом варианте, кристаллическая форма 1' далее характеризуется наличием одного или более дополнительных дифракционных пиков со значениями 2θ, выбранными из 16,09±0,20, 18,70±0,20, 19,21±0,20, 19,40±0,20, 21,64±0,20, 22,25±0,20, 26,43±0,20, 28,55±0,20, 30,73±0,20, 31,10±0,20, 32,64±0,20, 33,14±0,20 и 34,46±0,20; и в еще одном варианте кристаллическое соединение также характеризуется наличием трех или более таких дополнительных дифракционных пиков.
В одном варианте, кристаллическая форма 1' характеризуется ДСК термограммой или следами дифференциальной сканирующей калориметрии по существу в соответствии с той, которая показана на фиг. 2. Кристаллическая форма 1' характеризуется следами дифференциальной сканирующей калориметрии, записанными при скорости нагревания 10°C в минуту, которые показывают максимум в эндотермическом потоке тепла при температуре от около 165°C до около 169°C. ДСК термограмма или следы дифференциальной сканирующей калориметрии иллюстрируют эндотерму плавления с пиком около 167,1°C, наступление при 165,2°C, и с площадью под эндотермой, соответствующую 114 Дж/г. Разложение соединения совпадает с плавлением, и потребление 114 Дж/г для энтальпии плавления в направлении не установлено.
В одном варианте, кристаллическая форма 1' характеризуется профилем ТГА на фиг. 3. Этот профиль показывает отсутствие потери массы до температуры около 150°C; кристаллическое соединение разрушается после плавления, что показывает значительная потеря массы, происходящая в начале приблизительно 159°C.
В одном варианте, кристаллическая форма 1' характеризуется ДПВ изотермой на фиг. 4. Эта форма является не гигроскопическим твердым веществом. Общее полученное приращение влажности меньше, чем 0,025% массовых в условиях 5-70% относительной влажности. Общее приращение влажности меньше, чем 0,235% массовых в условиях 5-90% относительной влажности. Никакого значительного гистерезиса не было найдено между двумя последовательными циклами поглощения-десорбции. Твердое вещество, полученное после циклов поглощения-десорбции, показало такую же рентгенограмму ПРД, как и исходный материал, показывая отсутствие изменений в форме после этого эксперимента.
Кристаллическая форма 1' может быть охарактеризована изображением МПС на фиг. 5, на котором показана эта форма как кристаллическая, двоякопреломляющаяся, с частицами формой от тонких игл до тонких плашек.
Применение
Экстраполяция in vitro-до-in vivo параметров лекарственного средства у пациента продолжает улучшаться (см., например, Chiba et al., AAPS J., 2009 June; 11(2): 262-276). В данном изобретении in vitro ингибирующее действие на человеческий неприлизин оценивают (анализ 1) для определения ингибирующего действия соединения 1 на неприлизин. Порог pKi≥9,0 подтверждается. Однако дополнительные in vivo эксперименты также были проведены для более точного предсказания поведения соединения 1 у пациента.
Что касается поведения in vivo, существует несколько свойств, которые применяют при оценке того, доставляется ли достаточное количество лекарственного средства в плазму для достижения необходимой терапевтической пользы, например, низкого плазменного клиренса в отношении всех тестированных видов, высокой пероральной биодоступности, благоприятного потенцирования реакции циклического гуанозинмонофосфата (цГМФ) и низкого почечного клиренса для пациентов с аномальной функцией почек.
Для данного изобретения, пероральные и внутривенные фармакокинетические исследования проводят на крысах и собаках для определения пероральной биодоступности соединения 1 по сравнению с другими ингибиторами неприлизина (анализ 2). Этот анализ также применяют для определения скорости плазменного клиренса для этих соединений; полагают, что низкая скорость клиренса может предсказать, как долго соединение будет оставаться в кровотоке, т.е., его in vivo стабильность и стойкость без идентификации отдельных вовлеченных процессов элиминирования. Дополнительно, исследуют пероральную биодоступность и скорость плазменного клиренса у обезьян (анализ 4).
Фармакокинетические/фармакодинамические исследования проводят на крысах для определения уровня ингибирования неприлизина соединением 1 по сравнению с другими ингибиторами неприлизина (анализ 3). В этом исследовании измеряют уровень циклического гуанозинмонофосфата (цГМФ). цГМФ является молекулой нижележащего эффектора, связывающей рецептор натрийуретического пептида и, таким образом, служит в качестве эффективного in vivo биомаркера активности натрийуретического пептида. Уровень цГМФ повышается, если животному вводят ингибитор неприлизина по сравнению с плацебо. Один вариант изобретения относится к способу повышения базовых уровней предсердного натрийуретического пептида (ПНП) или цГМФ у пациента с гипертензией, сердечной недостаточностью и почечной недостаточностью, включающему введение пациенту терапевтически эффективного количества соединения 1 или его кристаллической формы. Уровни ПНП и цГМФ измеряют в моче или плазме или обеих жидкостях у пациента. В другом варианте, уровень ПНП или цГМФ повышается, по меньшей мере, ≥1,1-кратно, ≥1,2-кратно, ≥1,3-кратно, ≥1,4-кратно, ≥1,5-кратно, ≥2-кратно, ≥3-кратно, ≥4-кратно или≥5-кратно за 24-часовой период у пациента при введении терапевтически эффективного количества соединения 1 или его кристаллической формы.
Соединение 1 ингибирует фермент НЕП и, следовательно, может быть полезным для лечения и/или профилактики медицинских состояний, чувствительных к ингибированию НЕП. Таким образом ожидается, что пациенты, страдающие заболеванием или расстройством, которое лечат ингибированием фермента НЕП или повышением уровней его пептидных субстратов, могут лечиться введением терапевтически эффективного количества соединения 1. Например ожидается, что через ингибирование НЕП соединение 1 потенцирует биологические эффекты эндогенных пептидов, которые метаболизируются НЕП, таких как натрийуретические пептиды, бомбезин, брадикинины, кальцитоцин, эндотелины, энкефалины, нейротензины, вещество P и вазоактивный пептид кишечника. Таким образом ожидается, что соединение обладает другими физиологическими действиями, например, на почечную, центральную нервную, репродуктивную и желудочно-кишечную системы.
Лекарственные средства выводят из тела пациента различными способами элиминирования, которые, в общем, классифицируются на выведение и биопревращение. Выведение относится к удалению исходного нелетучего лекарственного средства в основном ренальным путем (почки) в мочевой пузырь в моче, а другие пути выведения включают желчь (печень), пот, слюну, молоко (через лактацию) или другие жидкости тела. Летучие лекарственные средства, такие как спиртовые и газообразные анестетики, выводят через легкие в выдыхаемый воздух. С другой стороны, биопревращение, или метаболизм лекарственного средства, относится к химическому превращению лекарственного средства в теле до метаболита, и обычно этот процесс является ферментным. Исключением является тот случай, когда лекарственное средство химически изменено не ферментативно, например, гидролизом сложного эфира. Ферменты, вовлеченные в биопревращение лекарственных средств, расположены в основном в печени. Другие ткани, такие как почки, легкие, тонкий кишечник и кода, также содержат метаболические ферменты.
Фармакокинетические исследования могут применяться для исследования путей выведения у пациента, например, почечного клиренса через выведение введенного лекарственного средства в моче в течение времени. Почечное выведение соединения 1 у крыс, собак и обезьян проводят для оценки почечного выведения в качестве пути выведения (анализ 5). Этот путь выведения важен для пациентов, у которых нарушена почечная функция и они нуждаются в терапиях, которые минимально очищаются через почечное выведение. В одном варианте, почечное выведение соединения 1 или его кристаллической формы у пациента составляет приблизительно≤15%,≤10%,≤5%,≤3%,≤2%,≤1% или≤0,5% введенной дозы в течение более 24 часов.
Как описано в разделе анализов ниже, вместе с соединением 1 в in vitro НЕП ферментный анализ и в in vivo определения плазменного клиренса, пероральной биодоступности и почечного выведения у множества видов животных, используют сравнительные соединения подобной химической структуры. Неожиданно были получены значительные различия в результатах. Хотя отдельные сравнительные соединения демонстрируют свойства, подобные свойствам соединения 1 в одном или более анализах, только соединение 1 демонстрирует, одновременно, высокое ингибирующее действие на человеческий неприлизин, высокую биодоступность, низкий плазменный клиренс, повышенное потенцирование цГМФ и низкое ожидаемое почечное выведение, что делает его особенно полезным при лечении заболевания.
Сердечнососудистые заболевания
Благодаря потенцированию действий вазоактивных пептидов, таких как натрийуретические пептиды и брадикинин, ожидается, что соединение 1 найдет применение в лечении и/или профилактике медицинских состояний, таких как сердечнососудистые заболевания. См., например, Roques et al. (1993) Pharmacol. Rev. 45:87-146 и Dempsey et al. (2009) Amer. J. of Pathology 174(3):782-796. Особенно интересные сердечнососудистые заболевания включают гипертензию и сердечную недостаточность. Гипертензия включает, в качестве иллюстрации, но не ограничения: первичную гипертензию, которая также называется гипертоническая болезнь или идиопатическая гипертензия; вторичную гипертензию; гипертензию с сопутствующей почечной недостаточностью; тяжелую гипертензию с или без сопутствующей почечной недостаточности; легочную гипертензию, включая легочную артериальную гипертензию; и резистентную гипертензию. Сердечная недостаточность включает, в качестве иллюстрации, но не ограничения: хроническую сердечную недостаточность; острую сердечную недостаточность; хроническую сердечную недостаточность, например, с уменьшенной фракцией выброса левого желудочка (также называемую систолической сердечной недостаточностью) или с сохраненной фракцией выброса левого желудочка (также называемую диастолической сердечной недостаточностью); и острую и хроническую декомпенсированную сердечную недостаточность. Таким образом, один вариант изобретения относится к способу лечения гипертензии, особенно, первичной гипертензии или легочной артериальной гипертензии, включающему введение пациенту терапевтически эффективного количества соединения 1.
Для лечения первичной гипертензии, терапевтически эффективным количеством обычно является количество, которое достаточно для снижения кровяного давления пациента. Она включает гипертензию от слабой до умеренной и тяжелую гипертензию. При применении для лечения гипертензии, соединение 1 может вводиться в сочетании с другими терапевтическими агентами, такими как антагонисты альдостерона, ингибиторы альдостеронсинтазы, ингибиторы ангиотензин-превращающего фермента и двойного действия ингибиторы ангиотензин-превращающего фермента/неприлизина, активаторы и стимуляторы ангиотензин-превращающего фермента 2 (АПФ2), вакцины ангиотензина-II, противодиабетические агенты, противолипидные агенты, антитромботические агенты, антагонисты AT1 рецептора и двойного действия антагонист AT1 рецептора/ингибиторы неприлизина, антагонисты β1-адренергического рецептора, двойного действия антагонист β-адренергического рецептора/агонисты α1-рецептора, блокаторы кальциевого канала, диуретики, антагонисты рецептора эндотелина, ингибиторы эндотелин-превращающего фермента, ингибиторы неприлизина, натрийуретические пептиды и их аналоги, антагонисты рецептора клиренса натрийуретического пептида, доноры оксида азота, нестероидные противовоспалительные агенты, ингибиторы фосфодиэстеразы (особенно ингибиторов ФДЭ-V), агонисты рецептора простагландина, ингибиторы ренина, растворимые стимуляторы и активаторы гуанилатциклазы, и их сочетания. В одном особенно предпочтительном варианте изобретения, соединение в соответствии с данным изобретением объединяют с антагонистом рецептора AT1, блокатором кальциевого канала, диуретиком или их сочетанием, и применяют для лечения первичной гипертензии. В другом конкретном варианте изобретения, соединение в соответствии с данным изобретением объединяют с антагонистом рецептора AT1 и применяют для лечения гипертензии с сопутствующей почечной недостаточностью. При применении для лечения резистентной гипертензии, соединение может вводиться в сочетании с другими терапевтическими агентами, такими как ингибиторы альдостеронсинтазы.
Для лечения легочной артериальной гипертензии терапевтически эффективным количеством обычно является количество, которое достаточно для снижения легочного сосудистого сопротивления. Другими целями терапии являются улучшение способности пациента к нагрузке. Например, в клинической ситуации, терапевтически эффективным количеством может быть количество, которое улучшает способность пациента комфортабельно ходить в течение 6 минут (проходя расстояние приблизительно 20-40 метров). При применении для лечения легочной артериальной гипертензии соединение 1 может вводиться в сочетании с другими терапевтическими агентами, такими как антагонисты α-адренергического рецептора, антагонисты β1-адренергического рецептора, агонисты β2-адренергического рецептора, ингибиторы ангиотензин-превращающего фермента, антикоагулянты, блокаторы кальциевого канала, диуретики, антагонисты рецептора эндотелина, ингибиторы ФДЭ-V, аналоги простагландина, селективные ингибиторы обратного захвата серотонина и их сочетания. В одном конкретном варианте, соединение 1 объединяют с ингибитором ФДЭ-V или селективным ингибитором обратного захвата серотонина, и применяют для лечения легочной артериальной гипертензии.
Другой вариант изобретения относится к способу лечения сердечной недостаточности, в частности застойной сердечной недостаточности (включая систолическую и диастолическую сердечную недостаточность), включающему введение пациенту терапевтически эффективного количества соединения 1. Обычно терапевтически эффективным количеством является количество, которое достаточной для снижения кровяного давления и/или улучшения почечных функций. В клинической ситуации, терапевтически эффективным количеством может быть количество, которое достаточно для улучшения сердечной гемодинамики, например, снижения заклиненного давления, давления в правом предсердии, давления заполнения и сосудистого сопротивления. В одном варианте, соединение вводят в виде внутривенной лекарственной формы. При применении для лечения сердечной недостаточности, соединение 1 может вводиться в сочетании с другими терапевтическими агентами, такими как антагонисты рецептора аденозина, разрушители конечного продукта усиленного гликозилирования, антагонисты альдостерона, антагонисты AT1 рецептора, антагонисты β1-адренергического рецептора, двойного действия антагонист β-адренергического рецептора/антагонисты α1-рецептора, ингибиторы химазы, дигоксин, диуретики, ингибиторы эндотелин-превращающего фермента (ЭПФ), антагонисты рецептора эндотелина, натрийуретические пептиды и их аналоги, антагонисты рецептора клиренса натрийуретического пептида, доноры оксида азота, аналоги простагландина, ингибиторы ФДЭ-V, растворимые активаторы и стимуляторы гуанилатциклазы и антагонисты рецептора вазопрессина. В одном конкретном варианте изобретения, соединение 1 объединяют с антагонистом альдостерона, антагонистом β1-адренергического рецептора, антагонистом AT1 рецептора или диуретиком, и применяют для лечения застойной сердечной недостаточности.
Диарея
Ожидается, что в качестве ингибитора НЕП соединение 1 будет ингибировать разложение эндогенных энкефалинов и, следовательно, такие соединения также могут применяться для лечения диареи, включая инфекционную и секреторную/водянистую диарею. См., например, Baumer et al. (1992) Gut 33:753-758; Farthing (2006) Digestive Diseases 24:47-58; и Marçais-Collado (1987) Eur. J. Pharmacol. 144(2):125-132. При применении для лечения диареи соединение 1 может быть объединено с одним или более дополнительными противодиарейными агентами.
Почечная недостаточность
Ожидается, что через потенцирование эффектов вазоактивных пептидов, таких как натрийуретические пептиды и брадикинин, соединение 1 будет улучшать почечную функцию (см. Chen et al. (1999) Circulation 100:2443-2448; Lipkin et al. (1997) Kidney Int. 52:792-801; и Dussaule et al. (1993) Clin. Sci. 84:31-39) и сможет применяться для лечения и/или профилактики почечной недостаточности у пациента с нарушением почечной функции. Заболевания почек, представляющие особенный интерес, включают диабетическую нефропатию, хроническую почечную недостаточность, протеинурию и, в частности, острое повреждение почек (вызванное, например, сердечнососудистой хирургией, химиотерапией или применением контрастных красителей для диагностической визуализации) или острую почечную недостаточность (см. Sharkovska et al. (2011) Clin. Lab. 57:507-515 и Newaz et al. (2010) Renal Failure 32:384-390).
Пациенты с нарушением почечной функции, которые имеют хроническую почечную недостаточность (ХПН) могут быть классифицированы согласно инструкциям National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI). Как только диагностирована хроническая почечная недостаточность, т.е. повреждение почек или скорость клубочковой фильтрации (СКФ) <60 мл/мин/1,73 м2 в течение≥3 месяцев, стадия заболевания может оцениваться согласно классификации ХПН от KDOQI. Она включает 1 стадию (повреждение почек с нормальной или повышенной СКФ): СКФ≥90; 2 стадия (повреждение почек с незначительно пониженной СКФ): СКФ 60-89; 3 стадия (умеренно пониженная СКФ): СКФ 30-59; 4 стадия 4 (сильное пониженная СКФ): СКФ 15-29; и 5 стадия 5 (почечная недостаточность): СКФ <15 (или диализ). СКФ определяется в единицах мл/мин/1,73 м2.
Один вариант включает способ лечения пациента с нарушением почечной функции, включающий введение терапевтически эффективного количества соединения 1 или его кристаллической формы, особенно кристаллической формы 1'. Этот способ также включает лечение пациента с нарушением почечной функции с гипертензией или почечной недостаточностью. При применении для лечения почечной недостаточности, соединение 1 или его кристаллическая форма, особенно кристаллическая форма 1', может вводиться в сочетании с другими терапевтическими агентами, такими как ингибиторы ангиотензин-превращающего фермента, антагонисты AT1 рецептора и диуретики.
Другой вариант включает способ лечения пациента с нарушением почечной функции, имеющего хроническую почечную недостаточность с расчетной скоростью клубочковой фильтрации (рСКФ) от 60 мл/мин/1,73 м2 и 15 мл/мин/1,73 м2, включающий введение пациенту терапевтически эффективного количества соединения 1 или его кристаллической формы, особенно, кристаллической формы 1'. Другой вариант включает способ лечения пациента с нарушением почечной функции, имеющего хроническую почечную недостаточность с расчетной скоростью клубочковой фильтрации (рСКФ) ≥90 мл/мин/1,73 м2 (1 стадия) или рСКФ <15 мл/мин/1,73 м2 (5 стадия), включающий введение пациенту терапевтически эффективного количества соединения 1 или его кристаллической формы, особенно кристаллической формы 1'. Для целей данного изобретения, тяжелая почечная недостаточность может быть классифицирована как рСКФ <30 мл/мин/1,73 м2. В еще одном варианте, включен способ лечения пациента с нарушением почечной функции, имеющего хроническую почечную недостаточность, находящуюся в 1 стадии, 2 стадии, 3 стадии, 4 стадии, 5 стадии или с интервалами рСКФ, соответствующими одной или более из этих стадий, соединением 1 или его кристаллической формой, особенно кристаллической формой 1'.
Профилактическая терапия
Также ожидается, что через потенцирование эффектов натрийуретических пептидов соединение 1 будет полезно в профилактической терапии, благодаря антигипертрофических и антифибротических эффектов натрийуретических пептидов (см. Potter et al. (2009) Handbook of Experimental Pharmacology 191:341-366), например, для профилактики развития сердечной недостаточности после инфаркта миокарда, профилактики артериального рестеноза после ангиопластики, профилактики утолщения стенок кровяных сосудов после операций на сосудах и профилактики диабетической ангиопатии.
Глаукома
Ожидается, что через потенцирование натрийуретических пептидов, соединение 1 будет полезно для лечения глаукомы. См., например, Diestelhorst et al. (1989) International Ophthalmology 12:99-101. При применении для лечения глаукомы соединение 1 может быть объединено с одним или более дополнительными противоглаукомными агентами.
Облегчение боли
Ожидается, что в качестве ингибитора НЕП соединение 1 будет ингибировать разрушение эндогенных энкефалинов и, следовательно, такое соединение может применяться в качестве обезболивающего. См., например, Roques et al. (1980) Nature 288:286-288 и Thanawala et al. (2008) Current Drug Targets 9:887-894. При применении для лечения боли, соединение 1 может быть объединено с одним или более дополнительными антиноцицептивными лекарственными средствами, такими как ингибиторы аминопептидазы N или дипептидилпептидазы III, нестероидные противовоспалительные агенты, ингибиторы обратного захвата моноамина, миорелаксантами, антагонистами рецептора NMDA, агонистами опиоидного рецептора, агонистами рецептора 5-HT1D серотонина и трициклическими антидепрессантами.
Другие области применения
Благодаря свойствам ингибирования НЕП, также ожидается, что соединение 1 будет применяться в качестве противокашлевого средства, а также может применяться для лечения портальной гипертензии, связанной с циррозом печени (см. Sansoe et al. (2005) J. Hepatol. 43:791-798), рака (см. Vesely (2005) J. Investigative Med. 53:360-365), депрессии (см. Noble et al. (2007) Exp. Opin. Ther. Targets 11:145-159), нарушений менструального цикла, преждевременных родов, преэклампсии, эндометриоза, расстройств репродуктивной функции (например, мужского и женского бесплодия, поликистоза яичников, неблагоприятной имплантации) и мужской и женской половой дисфункции, включая мужскую эректильную дисфункцию и расстройство женского сексуального возбуждения. Более конкретно, ожидается, что соединение 1 будет полезно для лечения женской половой дисфункции (см. Pryde et al. (2006) J. Med. Chem. 49:4409-4424), которая часто определяется как трудность или невозможность пациентки получать сексуальное удовлетворение. Она включает множество различных женских половых расстройств, включая, в качестве иллюстрации, но не ограничения, сниженное половое влечение, расстройство сексуального возбуждения, расстройство оргазма и болезненный половой акт. При применении для лечения таких расстройств, особенно женской половой дисфункции, соединение в соответствии с данным изобретением может быть объединено с одним или более другими вторичными агентами: ингибиторы ФДЭ-V, агонисты дофамина, агонисты и/или антагонисты рецептора эстрогена, андрогены и эстрогены. Благодаря свойству ингибирования НЕП, также ожидается, что соединение 1 обладает противовоспалительными свойствами, и может применяться как таковое, особенно при применении в сочетании со статинами.
Недавние исследования подтвердили, что НЕП играет роль в регулировании нервной функции при инсулинозависимых диабетах и алиментарном ожирении. Coppey et al. (2011) Neuropharmacology 60:259-266. Поэтому, благодаря свойству ингибирования НЕП, соединение 1 может применяться для защиты от нервных нарушений, вызванных диабетом или алиментарным ожирением.
Количество соединения 1, вводимое на дозу, или общее количество, вводимое в сутки, может быть заранее определено, или оно может быть определено для конкретного пациента, принимая во внимание множество факторов, включая природу и тяжесть состояния пациента, лечимое состояние, возраст, вес и общее состояние здоровья пациента, переносимость пациентом активного агента, способ введения, фармакологические факторы, такие как активность, эффективность, фармакокинетика и токсикологический профиль соединения и любых вводимых вторичных агентов, и подобные. Лечение пациента, страдающего заболеванием или медицинским состоянием (таким как гипертензия) может начинаться с заранее определенной дозы или дозы, определенной лечащим врачом, и продолжается в течение периода времени, необходимого для профилактики, облегчения, подавления или ослабления симптомов заболевания или медицинского состояния. Пациентов, проходящих такое исследование, обычно контролируют обычными методами для определения эффективности терапии. Например, при лечении гипертензии, измерения кровяного давления может применяться для определения эффективности лечения. Подобные показатели для других заболеваний и состояний, описанных здесь, хорошо известны и легко доступны лечащему врачу. Непрерывный контроль врача обеспечивает введение оптимального количества соединения 1 в любой момент времени, а также способствует определению длительности лечения. Это имеет особое значение, когда также вводятся вторичные агенты, так как их выбор, дозировка и длительность терапии также может потребовать корректировки. Таким образом, режим лечения и схема дозирования может корректироваться в курсе терапии так, чтобы вводить наименьшее количество активного агента, которое демонстрирует желаемую эффективность, а также, чтобы продолжать введение не дольше, чем это необходимо для успешного лечения заболевания или медицинского состояния.
Соединение 1 также может применяться в качестве промежуточного соединения, применяемого для получения кристаллических форм соединения 1, включая, например, кристаллическую форму 1'.
Инструменты исследования
Так как соединение 1 обладает ингибирующим действием на НЕП фермент, его также применяют в качестве инструмента исследования для исследования или изучения биологических систем или образцов, содержащих НЕП фермент, например, для изучения заболеваний, в которых НЕП фермент или его пептидные субстраты играют роль. Любые подходящие биологические системы или образцы, содержащие НЕП фермент, могут применяться в таких исследованиях, которые могут проводиться in vitro или in vivo. Типовые биологические системы или образцы, подходящие для таких исследований, включают, но не ограничены ими, клетки, клеточные экстракты, мембраны плазмы, образцы тканей, выделенные органы, млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи, человек и так далее), и подобные, где млекопитающие представляют особый интерес. В одном конкретном варианте изобретения, активность НЕП фермента у млекопитающих ингибируется введением НЕП-ингибирующего количества соединения 1.
При применении в качестве инструмента исследования, биологическая система или образец, содержащие НЕП фермент, обычно подвергают взаимодействию с ингибирующим НЕП фермент количеством соединения 1. После обработки биологической системы или образца соединением, ингибирующее действие на НЕП фермент определяют с применением обычных методик и оборудования, таких как измерение рецепторного связывания в анализе связывания, или измерение медиированных лигандом изменений в функциональном анализе. Обработка включает контакт клеток или ткани с соединением, введение соединения млекопитающему, например, в.б., п.о., в.в., п.к. или ингаляцией, и так далее. Стадия определения может включать измерение реакции (количественный анализ) или может включать наблюдение (качественный анализ). Измерение реакции включает, например, определение действия соединения на биологическую систему или образец с применением обычных методик и оборудования, таких как испытание активности ферментов и измерение изменений, медиированных ферментным субстратом или продуктом в функциональных анализах. Результаты исследований могут применяться для определения уровня активности, а также количества соединения, необходимого для достижения желаемого результата, то есть, количества, ингибирующего НЕП фермент. Обычно стадия определения включает определение эффекта ингибирования НЕП фермента.
Дополнительно, соединение 1 может применяться в качестве инструмента исследования для оценки других химических соединений, и поэтому также применяется в скрининговых исследованиях для обнаружения, например, новых соединений, обладающих НЕП-ингибирующим действием. Таким образом, соединение 1 применяют в качестве стандарта в анализе для сравнения результатов, полученных с тестируемым соединением и с соединением 1 для идентификации тестируемых соединений, которые обладают примерно равной или превосходящей активностью, если имеется. Например, данные pKi для тестируемого соединения или группы тестируемых соединений сравнивают с данными pKi соединения 1 для идентификации тех тестируемых соединений, которые обладают желаемыми свойствами, например, тестируемых соединений, имеющих значение pKi, равное или превосходящее соединение в соответствии с данным изобретением. Этот аспект изобретения включает, в качестве отдельных вариантов, получение сравнительных данных (с применением подходящих анализов) и анализ данных тестирования для идентификации целевых тестируемых соединений. Таким образом, тестируемое соединение может оцениваться в биологическом анализе, способом, включающим стадии: (a) проведения биологического анализа с тестируемым соединением для получения первого результата анализа; (b) проведения биологического анализа с соединением 1 с получением второго результата анализа; где стадию (a) проводят до, после или одновременно со стадией (b); и (c) сравнение первого результата анализа со стадии (a) со вторым результатом анализа со стадии (b). Типовые биологические анализы включают анализ ингибирования НЕП фермента.
Еще один аспект изобретения относится к способу изучения биологической системы или образца, содержащих НЕП фермент, где способ включает: (a) контакт биологической системы или образца с соединением 1; и (b) определение эффектов, вызванных соединением в биологической системе или образце.
Фармацевтические композиции и составы
Соединение 1 обычно вводят пациенту в форме фармацевтической композиции или состава. Такие фармацевтические композиции могут вводиться пациенту любым приемлемым путем введения, включая, но не ограничиваясь ими, пероральный, ректальный, вагинальный, назальный, ингаляционный, местный (включая чрезкожный), глазной и парентеральный способы введения. Также соединение 1 может вводиться, например, перорально, несколькими дозами в сутки (например, два, три или четыре раза в сутки), однократной суточной дозой или однократной недельной дозой. Должно быть понятно, что любая форма соединения 1 (то есть, свободное основание, свободная кислота, фармацевтически приемлемая соль, сольват и т.д.), которая подходит для конкретного способа введения, может применяться в обсуждаемых здесь фармацевтических композициях.
Следовательно, в одном варианте, изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение 1. Композиция может содержать другие терапевтические и/или препаратообразующие агенты, при желании. При обсуждении композиций, "соединение 1" также может быть названо "активный агент" для отделения его от других компонентов композиции, таких как носитель. Таким образом, понятно, что термин "активный агент" включает соединение 1, а также его фармацевтически приемлемые соли.
Фармацевтические композиции в соответствии с данным изобретением обычно содержат терапевтически эффективное количество соединения 1. Специалисты в данной области техники поймут, однако, что фармацевтическая композиция может содержать больше, чем фармацевтически эффективное количество, например, в сыпучих композициях, или меньше, чем терапевтически эффективное количество, то есть, в отдельных лекарственных формах, разработанных для неоднократного введения для получения терапевтически эффективного количества. Обычно композиция содержит около 0,01-95% масс. активного агента, включая около 0,01-30% масс., например, около 0,01-10% масс., где действительное количество зависит от самой композиции, способа введения, частоты дозирования и так далее. В одном варианте, композиция, подходящая для пероральной лекарственной формы, например, может содержать около 5-70% масс. или около 10-60% масс. активного агента.
Любой обычный носитель или наполнитель может применяться в фармацевтических композициях в соответствии с данным изобретением. Выбор конкретного носителя или наполнителя, или сочетаний носителей или наполнителей, зависит от способа введения, применяемого для лечения конкретного пациента или типа медицинского состояния или болезненного состояния. В связи с этим, получение подходящей композиции для конкретного способа введения находится в компетенции специалиста в области фармацевтики. Дополнительно, носители или наполнители, применяемые в таких композициях, коммерчески доступны. В качестве дополнительной иллюстрации, типовые методы получения составов описаны в Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); и H. C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Типовые примеры материалов, которые могут быть фармацевтически приемлемыми носителями, включают, но не ограничены ими, следующие: сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу, такую как микрокристаллическая целлюлозы, и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; соли жирной кислоты, такие как стеарат магния; порошковый трагакант; солод; желатин; тальк; наполнители, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, конопляное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид; альгиновую кислоту; апирогенную воду; изотонический солевой раствор; раствор Рингера; этиловый спирт; фосфатные буферные растворы; сжатые газы-вытеснители, такие как хлорфторуглероды и гидрофторуглероды; и другие не токсичные совместимые вещества, применяемые в фармацевтических композициях.
В одном варианте данного изобретения, фармацевтически приемлемым носителем является стеарат магния. Например, фармацевтическая композиция может содержать соединение 1 или кристаллическую форму 1' и стеарат магния в соотношении от около 3:1 до около 10:1 соединения 1 кристаллической формы 1' к стеарату магния. Другие соотношения соединения 1 или кристаллической формы 1' к стеарату магния включают, но не ограничены ими, 1:1, 5:1, 15:1, 20:1, 25:1, 30:1 и 50:1.
Фармацевтические композиции обычно получают тщательным и равномерным смешиванием или гомогенизацией активного агента с фармацевтически приемлемым носителем и одним или более необязательными ингредиентами. Полученная однородно смешанная смесь затем может быть сформована или загружена в таблетки, капсулы, пилюли, коробки, картриджи, раздатчики и подобные с применением обычных методов и оборудования.
В одном варианте, фармацевтические композиции подходят для перорального введения. Подходящие композиции для перорального введения могут быть в форме капсул, таблеток, пилюль, лепешек, крахмальных облаток, драже, порошков, гранул; растворов или суспензий в водной или неводной жидкости; жидких эмульсии масло-в-воде или вода-в-масле; эликсиров или сиропов; и подобных; каждая из которых содержит заданное количество активного агента.
Когда она предназначена для перорального введения в твердой лекарственной форме (капсулы, таблетки, пилюли и подобные), композиция обычно содержит активный агент и один или более фармацевтически приемлемых носителей, таких как цитрат натрия, дикальцийфосфат или стеарат магния. Твердые лекарственные формы также могут содержать наполнители или сухие разбавители, такие как крахмалы, микрокристаллическая целлюлоза, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; связующие агенты, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или аравийская камедь; увлажнители, такие как глицерин; разрыхляющие агенты, такие как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, определенные силикаты и/или карбонат натрия; замедлители растворения, такие как парафин; усилители абсорбции, такие как соединения четвертичного аммония; смачивающие агенты, такие как цетиловый спирт и/или глицеролмоностеарат; абсорбенты, такие как каолин и/или бентонитовая глина; смазывающие агенты, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и/или их смеси; красители; и буферы. Для целей данного изобретения термины ʺфармацевтически приемлемые носителиʺ включают все термины, такие как носители, наполнители или твердые разбавители, связующие агенты, увлажнители, замедлители растворения, смачивающие агенты, абсорбенты, смазывающие агенты, красители и буферы, описанные выше.
Разделительные агенты, смачивающие агенты, покрытия, подсластители, вкусовые добавки и отдушки, консерванты и антиоксиданты также могут присутствовать в фармацевтических композициях. Типовые покрытия для таблеток, капсул, пилюль и подобных включают покрытия, применяемые для энтеросолюбильных покрытий, такие как ацетатфталат целлюлозы, фталат полвинилацетата, фталат гидроксипропилметилцеллюлозы, сополимеры метакриловой кислоты-эфира метакриловой кислоты, ацетат-тримеллтат целлюлозы, карбоксиметилэтилцеллюлоза, сукцинат ацетат гидроксипропилметилцеллюлозы, и подобные. Примеры фармацевтически приемлемых антиоксидантов включают: водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфат натрия, сульфит натрия и подобные; маслорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, лецитин, пропилгаллат, альфа-токоферол, и подобные; металл-хелатирующие агенты, такие как лимонная кислота, этилендиаминтетрауксусная кислота, сорбит, винная кислота, фосфорная кислота и подобные.
Композиции также могут быть составлены так, чтобы обеспечить медленное или контролируемое выделение активного агента с применением, например, гидроксипропилметилцеллюлозы в различных долях или других полимерных матриц, липосом и/или микросфер. Кроме того, фармацевтические композиции в соответствии с данным изобретением могут содержать замутнители, и могут быть составлены так, чтобы выделять только или преимущественно активный агент, в определенную часть желудочно-кишечного тракта, необязательно, с задержкой. Примеры заливочных композиций, которые могут применяться, включают полимерные вещества и воски. Активный агент также может быть в микроинкапсулированной форме, необязательно, с одним или более указанных выше наполнителей.
Один вариант изобретения включает пероральную лекарственную форму, содержащую соединение 1 или кристаллическую форму 1' в капсуле, таблетке, жидкости или суспензии. Другой вариант изобретения относится к пероральной лекарственной форме, в которой соединение 1 или кристаллическая форма 1' в теле пациента выделяется сразу же, под контролем или с задержкой. Если в качестве пероральной лекарственной формы применяют капсулу, другой вариант включает капсулу, состоящую из желатина, полисахаридов или синтетических полимеров. В конкретном варианте, капсула содержит гидроксипропилметилцеллюлозу.
Подходящие материалы для капсул в соответствии с данным изобретением выбирают из желатина, производных целлюлозы, крахмала, производных крахмала, хитозана и синтетических полимеров. Если желатин применяют в качестве материала капсулы, он может применяться в смеси с другими добавками, выбранными из полиэтиленгликоля (ПЭГ), глицерина, сорбита, полипропиленгликоля, ПЭО-ППО блок-сополимеров и других полиспиртов и полиэфиров. Если производное целлюлозы применяют в качестве материала капсулы, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, метилцеллюлоза, гидроксиметилцеллюлоза и гидроксиэтилцеллюлоза являются предпочтительными полимерами. Если синтетические пластики применяют в качестве материала капсулы, полиэтилен, поликарбонат, полиэфир, полипропилен и полиэтилентерефталат являются предпочтительными материалами. Особенно предпочтительны полиэтилен, поликарбонат или полиэтилентетрфталат.
Подходящие жидкие лекарственные формы для перорального введения включают, в качестве иллюстрации, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие лекарственные формы обычно включают активный агент и инертный разбавитель, такой как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (например, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и конопляное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и эфиры жирных кислот сорбита, и их смеси. Суспензии могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и эфиры сорбита, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант, и их смеси.
Когда они предназначены для перорального введения, фармацевтические композиции в соответствии с данным изобретением могут быть упакованы в виде единичной лекарственной формы. Термин "единичная лекарственная форма" относится к физически отдельной единице, подходящей для введения пациенту, то есть, каждая единица содержит определенное количество активного агента, рассчитанное для получения желаемого терапевтического эффекта, отдельно или в сочетании с одной или более дополнительными единицами. Например, такими единичными лекарственными формами могут быть капсулы, таблетки, пилюли и подобные.
В другом варианте, композиции в соответствии с данным изобретением подходят для введения ингаляцией, и обычно имеют форму аэрозоля или порошка. Такие композиции обычно вводят с применением хорошо известных устройств, таких как небулайзер, порошковый или дозирующий ингалятор. Небулайзеры выпускают поток воздуха с высокой скоростью, который распыляет композицию в виде водяной пыли, которая переносится в дыхательные пути пациента. Типовые композиции для небулайзера содержат активный агент, растворенный в носителе с получением раствора, или микронизированы и объединены с носителем с получением суспензии или микронизированных частиц вдыхаемого размера. Порошковые ингаляторы вводят активный агент в виде свободнотекучего порошка, который диспергирован в воздухе, вдыхаемом пациентом, во время вдоха. Типовая порошковая композиция включает активный агент, смешанный в сухой смеси с наполнителем, таким как лактоза, крахмал, маннит, декстроза, полимолочная кислота, полилактид-со-гликолид и их сочетания. Дозирующие ингаляторы выделяют отмеренное количество активного агента с помощью сжатого газа-вытеснителя. Типовые дозируемые композиции включают раствор или суспензию активного агента в сжиженном газе-вытеснителе, таком как хлорфторуглерод или гидрофторуглерод. Необязательные компоненты таких композиций включают сорастворители, такие как этанол или пентан, и поверхностно-активные вещества, такие как сорбитантриолеат, олеиновая кислота, лецитин, глицерин и лаурилсульфат натрия. Такие композиции обычно получают добавлением охлажденного или сжатого гидрофторалкана в подходящий контейнер, содержащий активный агент, этанол (если присутствует) и поверхностно-активное вещество (если присутствует). Для получения суспензии активный агент микронизируют и затем объединяют с газом-вытеснителем. Альтернативно, суспензия может быть получена сушкой распылением покрытия поверхностно-активного агента на микронизированные частицы активного агента. Композицию затем загружают в аэрозольный баллон, который составляет часть ингалятора.
Соединение 1 и его композиции также могут вводиться парентерально, например, подкожной, внутривенной, внутримышечной или внутрибрюшинной инъекцией. Для такого введения активный агент представлен в стерильном растворе, суспензии или эмульсии. Типовые растворители для получения таких композиций включают воду, физиологический раствор, электролиты, низкомолекулярные спирты, такие как пропиленгликоль и полиэтиленгликоль, масла, аминокислоты, желатин, сахара, сложные эфиры жирных кислот, такие как этилолеат, и подобные. Парентеральные композиции могут также содержать один или несколько антиоксидантов, солюбилизаторов, стабилизаторов, консервантов, смачивающих агентов, эмульгаторов и диспергирующих агентов. Поверхностно-активные вещества, дополнительные стабилизирующие агенты или агенты, регулирующие рН (кислоты, основания или буферы) и антиоксиданты, особенно полезны для обеспечения стабильности композиции, например, для минимизации или избежания гидролиза сложноэфирных и амидных связей, которые могут присутствовать в соединении. Эти композиции могут оставаться стерильными с помощью стерильной среды для инъекций, стерилизующего агента, фильтрации, облучения или тепла.
Типовые физиологически приемлемые водные носители включают, например, стерильную воду для инъекций, USP; инъекцию декстрозы, USP (например, 2,5, 5,0, 10, 20% декстроза, включая 5% инъекцию декстрозы (D5/W)); инъекцию декстрозы и хлорида натрия, USP (например, декстрозу в количестве от 2,5 до 10% и хлорид натрия от 0,12 (19 мЭкв натрия) до 0,9% (154 мЭкв натрия)); инъекцию маннита, USP, (например, 5, 10, 15, 20 и 25% маннита); инъекцию Рингера, USP (например, 147 мЭкв натрия, 4 мЭкв калия, 4,5 мЭкв кальция и 156 мЭкв хлорида на литр); лактированную инъекцию Рингера, USP (например, 2,7 мЭкв кальция, 4 мЭкв калия, 130 мЭкв натрия и 28 мЭкв лактата на литр); инъекцию хлорида натрия, USP (например, 0,9% хлорида натрия) и подобные.
При введении пациенту соединение 1 обычно разбавляют от около 0,5 мл до около 10 мл водного носителя на мг соединения 1, например, от около 0,6 до около 8 мл на мг.
В одном конкретном варианте, парентеральная композиция содержит водный раствор циклодекстрина в качестве фармацевтически приемлемого носителя. Подходящие циклодекстрины включают циклические молекулы, содержащие шесть или более единиц α-D-глюкопиранозы, связанные в 1,4 положениях такими связями, как в амилазе, β-циклодекстрине или циклогептаамилозе. Типовые циклодекстрины включают производные циклодекстрина, такие как циклодекстрины гидроксипропила и сульфобутилового эфира, такие как гидроксипропил-β-циклодекстрин и β-циклодекстрин сульфобутилового эфира. Типовые буферы для таких композиций включают буферы на основе карбоновой кислоты, такие как цитратные, лактатные и малеатные буферные растворы. В одном варианте изобретения, внутривенная лекарственная форма содержит соединение 1 или кристаллическую форму 1' в буферированном растворе.
В одном варианте, соединением 1 или его фармацевтической композицией является лиофилизированный порошок. Обычно лиофилизированный порошок является стерильным и упакован в герметично закрытые флаконы или ампулы или подобные контейнеры.
Соединение 1 также может вводиться чрезкожно с применением известных систем чрезкожной доставки и наполнителей. Например, соединение 1 может быть смешано с усилителями проницаемости, такими как пропиленгликоль, монолаурат полиэтиленгликоля, азациклоалкан-2-оны и подобные, и введено в пластырь или подобную систему доставки. Дополнительные наполнители, включающие желирующие агенты, эмульгаторы и буферы, могут применяться в таких чрезкожных композициях, при желании.
Вторичные агенты
Соединение 1 может применяться в качестве единственного лечения заболевания или может быть объединено с одним или более дополнительными терапевтическими агентами для получения желаемого терапевтического эффекта. Таким образом, в одном варианте, фармацевтические композиции в соответствии с данным изобретением содержат другие лекарственные средства, которые вводят совместно с соединением 1. Например, композиция может также содержать одно или более лекарственных средства (также называемых "вторичные агенты"). Такие терапевтические агенты хорошо известны в данной области техники и включают антагонисты рецептора аденозина, антагонисты α-адренергического рецептора, антагонисты β1-адренергического рецептора, агонисты β2-адренергического рецептора, двойного действия антагонист β-адренергического рецептора/антагонисты α1-рецептора, разрушители конечного продукта усиленного гликозилирования, антагонисты альдостерона, ингибиторы альдостеронсинтазы, ингибиторы аминопептидазы N, андрогены, ингибиторы антиотензин-превращающего фермента, и двойного действия ингибиторы ангиотензин-превращающего фермента/неприлизина, активаторы и стимуляторы ангиотензин-превращающего фермента 2, вакцины ангиотензина-II, антикоагулянты, противодиабетические агенты, противодиарейные агенты, противоглаукомные агенты, антилипидные агенты, антиноцицептивные агенты, антитромботические агенты, антагонисты рецептора AT1 и двойного действия антагонист рецептора AT1/ингибиторы неприлизина и многофункциональные блокаторы рецептора ангиотензина, антагонисты рецептора брадикинина, блокаторы кальциевого канала, ингибиторы химазы, дигоксин, диуретики, агонисты дофамина, ингибиторы эндотелин-превращающего фермента, антагонисты рецептора эндотелина, ингибиторы HMG-CoA редуктазы, эстрогены, агонисты и/или антагонисты рецептора эстрогена, ингибиторы обратного захвата моноамина, миорелаксанты, натрийуретические пептиды и их аналоги, антагонисты очищающего рецептора натрийуретического пептида, ингибиторы неприлизина, доноры оксида азота, нестероидные противовоспалительные агенты, антагонисты рецептора N-метил d-аспартата, агонисты опиоидного рецептора, ингибиторы фосфодиэстеразы, аналоги простагландина, агонисты рецептора простагландина, ингибиторы ренина, селективные ингибиторы обратного захвата серотонина, блокатор натриевого канала, растворимые стимуляторы и активаторы гуанилатциклазы, трициклические антидепрессанты, антагонисты рецептора вазопрессина, и их сочетания. Конкретные примеры этих агентов представлены ниже.
Конкретный вариант включает фармацевтическую композицию, содержащую соединение 1 или его кристаллическую форму и антагониста рецептора AT1, ингибитор ангиотензин-превращающего фермента, ингибитор фосфодиэстеразы (ФДЭ), ингибитор ренина, диуретик или их сочетание, и необязательно один или более фармацевтически приемлемых носителей.
Следовательно, в еще одном аспекте изобретения, фармацевтическая композиция содержит соединение 1, второй активный агент и фармацевтически приемлемый носитель. Третий, четвертый и так далее активные агенты также могут быть включены в композицию. В комбинированной терапии вводимое количество соединения 1, а также количество вторичных агентов может быть меньше, чем количество, обычно вводимое при монотерапии.
Соединение 1 может быть физически смешано со вторым активным агентом с получением композиции, содержащей оба агента; или каждый агент может присутствовать в отдельных и разных композициях, которые вводят пациенту одновременно или в разное время. Например, соединение 1 может быть объединено со вторым активным агентом с применением обычных методов и оборудования с получением сочетания активных агентов, содержащего соединение 1 и второй активный агент. Дополнительно, активные агенты могут быть объединены с фармацевтически приемлемым носителем с получением фармацевтической композиции, содержащей соединение 1, второй активный агент и фармацевтически приемлемый носитель. В этом варианте, компоненты композиции обычно смешивают или объединяют с получением физической смеси. Физическую смесь затем вводят в терапевтически эффективном количестве с применением любого из описанных путей.
Альтернативно, активные агенты могут оставаться отдельными и разными до введения пациенту. В этом варианте, агенты физические не смешивают друг с другом до введения, но вводят одновременно или в разное время в виде отдельных композиций. Такие композиции могут быть упакованы отдельно или могут быть упакованы вместе в наборе. При введении в разное время, вторичный агент обычно вводят менее чем через 24 часа после введения соединения 1, в интервале от одновременного введения с соединением в соответствии с данным изобретением до около 24 часов после введения. Это также называется последовательным введением. Таким образом, соединение 1 может вводиться перорально одновременного или последовательно с другим активным агентом с применением двух таблеток, по одной для каждого активного агента, где последовательное введение означает введение сразу же после введения соединения 1 или через некоторое определенное время (например, через один час или три часа). Также рассматривается, что вторичный агент может быть введен более чем через 24 часа после введения соединения 1. Альтернативно, сочетание может вводиться различными путями введения, то есть, один перорально и второй ингаляцией.
В одном варианте, набор содержит первую лекарственную форму, содержащую соединение 1, и, по меньшей мере, одну дополнительную лекарственную форму, содержащую один или более вторичных агентов, указанных здесь, в количествах, достаточных для осуществления способов изобретения. Первая лекарственная форма и вторая (или третья и т.д.) лекарственная форма вместе содержат терапевтически эффективное количество активных агентов для лечения или профилактики заболевания или медицинского состояния у пациента.
Вторичный агент, если включен, присутствует в терапевтически эффективном количестве так, что их обычно вводят в количестве, которое дает терапевтически благоприятный эффект при совместном введении с соединением 1 в соответствии с данным изобретением. Вторичный агент может быть в форме фармацевтически приемлемой соли, сольвата, оптически чистого стереоизомера и так далее. Вторичный агент также может быть в форме пролекарства, например, соединения, имеющего группу карбоновой кислоты, которая эстерифицирована. Таким образом, перечисленные вторичные агенты включают все такие формы и коммерчески доступны или могут быть получены с применением обычных методов и реагентов.
В одном варианте, Соединение 1 вводят в сочетании с антагонистом рецептора аденозина, примеры которого включают наксифиллин, ролофиллин, SLV-320, теофиллин и тонафофиллин.
В одном варианте, Соединение 1 вводят в сочетании с антагонистом α-адренергического рецептора, примеры которого включают доксазозин, празозин, тамсулозин и теразозин.
Соединение 1 также может вводиться в сочетании с антагонистом β1-адренергического рецептора ("β1-блокатором"), примеры которого включают ацебутолол, альпренолол, амосулалол, аротинолол, атенолол, бефунолол, бетаксолол, бевантолол, бисопролол, бопиндолол, буциндолол, букумолол, буфетолол, буфуралол, бунитролол, бупранолол, бубридин, бутофилолол, каразолол, картеолол, карведилол, целипролол, цетамолол, клоранолол, дилевалол, эпанолол, эсмолол, инденолол, лабетолол, левобунолол, мепиндолол, метипранолол, метопролол, такой как сукцинат метопролола и тартрат метопролола, мопролол, надолол, надоксолол, небивалол, нипрадилол, окспренолол, пенбутолол, пербутолол, пиндолол, практолол, пронеталол, пропранолол, соталол, суфиналол, талиндол, тертатолол, тилисолол, тимолол, толипролол, ксибенолол и их сочетания. Обычно β1-блокатор вводят в количестве, достаточном для получения около 2-900 мг на дозу.
В одном варианте, соединение 1 вводят в сочетании с агонистом β2-адренергического рецептора, примеры которого включают альбутерол, битолтерол, фенотерол, формотерол, индакатерол, изоэтарин, левалбутерол, метапротеренол, пирбутерол, сальбутамол, сальмефамол, сальметерол, тербуталин, вилантерол и подобные. Обычно, агонист β2-адренергического рецептора вводят в количестве, достаточном для получения около 0,05-500 мкг на дозу.
В одном варианте, соединение 1 вводят в сочетании с разрушителем конечного продукта усиленного гликозилирования (КПУГ), примеры которого включают алагебриум (или ALT-711) и TRC4149.
В другом варианте, соединение 1 вводят в сочетании с антагонистом альдостерона, примеры которого включают эплеренон, спиронолактон и их сочетания. Обычно, антагонист альдостерона вводят в количестве, достаточном для получения около 5-300 мг в сутки.
В одном варианте, Соединение 1 вводят в сочетании с ингибитором аминопептидазы N или дипептидилпептидазы III, примеры которого включают бестатин и PC18 (2-амино-4-метилсульфонилбутантиол, метионинтиол).
Соединение 1 также может вводиться в сочетании с ингибитором ангиотензин-превращающего фермента (АПФ), примеры которого включают аккуприл, алацеприл, беназеприл, беназеприлат, каптоприл, церанаприл, цилазаприл, делаприл, эналаприл, эналаприлат, фозиноприл, фозиноприлат, имидаприл, лизиноприл, моэксиприл, моноприл, мовелтиприл, пентоприл, периндоприл, квинаприл, квинаприлат, рамиприл, рамиприлат, саралазин ацетат, спираприл, темокаприл, трандолаприл, зофеноприл и их сочетания. В конкретном варианте, ингибитор АПФ выбирают из: беназеприла, каптоприла, эналаприла, лизиноприла, рамиприла и их сочетаний. Обычно, ингибитор АПФ вводят в количестве, достаточном для получения около 1-150 мг в сутки.
В другом варианте, Соединение 1 вводят в сочетании с ингибитором двойного действия ангиотензин-превращаюшего фермента/неприлизина (АПФ/НЕП), примеры которого включают: AVE-0848 ((4S,7S,12bR)-7-[3-метил-2(S)-сульфанилбутирамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота); AVE-7688 (илепатрил) и его исходное соединение; BMS-182657 (2-[2-оксо-3(S)-[3-фенил-2(S)-сульфанилпропионамидо]-2,3,4,5-тетрагидро-1H-1-бензазепин-1-ил]уксусная кислота); CGS-35601 (N-[1-[4-метил-2(S)-сульфанилпентанамидо]циклопентилкарбонил]-L-триптофан); фазидотрил; фазидотрилат; эналаприлат; ER-32935 ((3R,6S,9aR)-6-[3(S)-метил-2(S)-сульфанилпентанамидо]-5-оксопергидротиазоло[3,2-a]азепин-3-карбоновая кислота); гемпатрилат; MDL-101264 ((4S,7S,12bR)-7-[2(S)-(2-морфолиноацетилтио)-3-фенилпропионамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота); MDL-101287 ([4S-[4α,7α(R*),12bβ]]-7-[2-(карбоксиметил)-3-фенилпропионамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота); омапатрилат; RB-105 (N-[2(S)-(меркаптометил)-3(R)-фенилбутил]-L-аланин); сампатрилат; SA-898 ((2R,4R)-N-[2-(2-гидроксифенил)-3-(3-меркаптопропионил)тиазолидин-4-илкарбонил]-L-фенилаланин); Sch-50690 (N-[1(S)-карбокси-2-[N2-(метансульфонил)-L-лизиламино]этил]-L-валил-L-тирозин); и их сочетания, также могут быть включены. В одном конкретном варианте, ингибитор АПФ/НЕП выбирают из: AVE-7688, эналаприлата, фазидотрила, фазидотрилата, омапатрилата, сампатрилата и их сочетания.
В одном варианте, Соединение 1 вводят в сочетании с активатором или стимулятором ангиотензин-превращающего фермента 2 (АПФ2).
В одном варианте, Соединение 1 вводят в сочетании вакциной ангиотензина-II, примеры которого включают ATR12181 и CYT006-AngQb.
В одном варианте, Соединение 1 вводят в сочетании с антикоагулянтом, примеры которого включают: кумарины, такие как варфарин; гепарин; и прямые ингибиторы тромбина, такие как аргатробан, бивалирудин, дабигатран и лепирудин.
В еще одном варианте, Соединение 1 вводят в сочетании с противодиабетическим агентом, примеры которого включают инъекционные лекарственные средства и пероральные лекарственные средства, и их сочетания. Примеры инъекционных лекарственных средств включают инсулин и производные инсулина. Примеры пероральных лекарственных средств включают: бигуаниды, такие как метформин; антагонисты глюкагона; ингибиторы α-глюкозидазы, такие как акарбоза и миглитол; ингибиторы дипептидилпептидазы IV (ингибиторы ДПП-IV), такие как алоглиптин, денаглиптин, линаглиптин, саксаглиптин, ситаглиптин и вилдаглиптин; меглитиниды, такие как репаглинид; оксадиазолидиндионы; сульфонилмочевины, такие как хлорпропамид, глимепирид, глипизид, глибурид и толазамид; тиазолидиндионы, такие как пиоглитазон и розиглитазон; и их сочетания.
В другом варианте, Соединение 1 вводят в сочетании с антидиарейными терапиями. Типовые терапевтические опции включают растворы для пероральной регидратации (РПР), лоперамид, дифеноксилат и субсалицилат висмута.
В еще одном варианте, Соединение 1 вводят в сочетании с антиглаукомным агентом, примеры которого включают: α-адренергические агонисты, такие как бримонидин; антагонисты β1-адренергического рецептора; местные β1-блокаторы, такие как бетаксолол, левобунолол и тимолол; ингибиторы карбоангидразы, такие как ацетазоламид, бринзоламид или дорзоламид; холинергические агонисты, такие как цевимелин и DMXB-анабазеин; соединения эпинефрина; миотики, такие как пилокаприн; и аналоги простагландина.
В еще одном варианте, Соединение 1 вводят в сочетании с антилипидным агентом, примеры которого включают: ингибиторы транспортного белка холестериновых эфиров (ТБХЭ), такие как анацетрапиб, далцетрапиб и торцетрапиб; статины, такие как аторвастатин, флувастатин, ловастатин, правастатин, розувастатин и симвастатин; и их сочетания.
В одном варианте, Соединение 1 вводят в сочетании с антитромботическим агентом, примеры которого включают: аспирин; антитромбоцитарные агенты, такие как клопидогрел, празугрел и тиклопидин; гепарин и их сочетания.
В одном варианте, Соединение 1 вводят в сочетании с антагонистом рецептора AT1, также известным как блокаторы рецептора ангиотензина II типа 1 (ARB). Типовые ARB включают абитесартан, азилсартан (например, азилсартан медоксомил), бензиллосартан, кандесартан, кандесартан цилексетил, элизартан, эмбусартан, энолтасосартан, эпросартан, EXP3174, фонсартан, форасартан, глициллосартан, ирбесартан, изотеолин, лозартан, медоксомил, милфасартан, олмесартан (например, олмесартан медоксомил), опомисартан, пратосартан, риписартан, саприсартан, саралазин, сармезин, TAK-591, тазосартан, телмисартан, валсартан, золасартан и их сочетания. В конкретном варианте, ARB выбирают из азилсартана медоксомила, кандесартана цилексетила, эпросартана, ирбесартана, лосартана, олмесартана медоксомила, саприсартана, тасосартана, телмисартана, валсартана и их сочетания. Типовые соли и/или пролекарства включают кандесартан цилексетил, эпросартан мезилат, калиевую соль лосартан и олмесартан медоксомил. Обычно, ARB вводят в количестве, достаточном для получения около 4-600 мг на дозу, где типовая суточная доза составляет 20-320 мг в сутки.
Соединение 1 также могут вводиться в сочетании с агентом двойного действия, таким как антагонист рецептора AT1/ингибитор неприлизина (ARB/НЕП), примеры которого включают соединения, описанные в патентах США №№ 7,879,896 и 8,013,005, оба Allegretti et al., такие как соединение 4'-{2-этокси-4-этил-5-[((S)-2-меркапто-4-метилпентаноиламино)метил]имидазол-1-илметил}-3'-фторбифенил-2-карбоновая кислота.
Соединение 1 также может вводиться в сочетании с многофункциональными блокаторами рецептора ангиотензина, описанными у Kurtz & Klein (2009) Hypertension Research 32:826-834.
В одном варианте, Соединение 1 вводят в сочетании с антагонистом рецептора брадикинина, например, икатибантом (HOE-140). Ожидается, что такая комбинированная терапия может давать преимущество профилактики отека Квинке или других нежелательных последствий повышенных уровней брадикинина.
В одном варианте, Соединение 1 вводят в сочетании с блокатором кальциевого канала, примеры которого включают амлодипин, анипамил, аранипин, барнидипин, бенциклан, бенидипин, бепридил, клентиазем, цилнидипин, циннаризин, дилтиазем, эфонидипин, элгодипин, этафенон, фелодипин, фендилин, флунаризин, галлопамил, исрадипин, лацидипин, лерканидипин, лидофлазин, ломеризин, манидипин, мибефрадил, никардипин, нифедипин, нигулдипин, нилудипин, нилвадипин, нимодипин, низолдипин, нитрендипин, нивалдипин, пергексилин, прениламин, риозидин, семотиадил, теродилин, тиапамил, верапамил и их сочетания. В конкретном варианте, блокатор кальциевого канала выбирают из амлодипина, бепридила, дилтиазема, фелодипина, исрадипина, лацидипина, никардипина, нифедипина, нигулдипина, нилудипина, нимодипина, низолдипина, риозидина, верапамила и их сочетания. Обычно, блокатор кальциевого канала вводят в количестве, достаточном для получения около 2-500 мг на дозу.
В одном варианте, Соединение 1 вводят в сочетании с ингибитором химазы, таким как TPC-806 и 2-(5-формиламино-6-оксо-2-фенил-1,6-дигидропиримидин-1-ил)-N-[{3,4-диоксо-1-фенил-7-(2-пиридилокси)}-2-гептил]ацетамид (NK3201).
В одном варианте, Соединение 1 вводят в сочетании с диуретиком, примеры которого включают: ингибиторы карбоангидразы, такие как ацетазоламид и дихлорфенамид; петлевые диуретики, которые включают производные сульфонамида, такие как ацетазоламид, амбузид, азосемид, буметанид, бутазоламид, хлораминофенамид, клофенамид, клопамид, клорексолон, дисульфамид, этоксзоламид, фуросемид, мефрузид, метазоламид, пиретанид, торсемид, трипамид и ксипамид, а также не сульфонамидные диуретики, такие как этакриновая кислота и другие соединения феноксиуксусной кислоты, такие как тиениловая кислота, индакринон и квинкарбат; осмотические диуретики, такие как маннит; калийсберегающие диуретики, которые включают антагонисты альдостерона, такие как спиронолактон, и ингибиторы Na+ канала, такие как амилорид и триамтерен; тиазид и тиазидоподобные диуретики, такие как альтиазид, бендрофлуметиазид, бензилгидрохлортиазид, бензтиазид, бутиазид, хлорталидон, хлортиазид, циклопентиазид, циклотиазид, эпитиазид, этиазид, фенквизон, флуметиазид, гидрохлортиазид, гидрофлуметиазид, индапамид, метилклотиазид, метикран, метолазон, парафлутизид, политиазид, квинетазон, теклотиазид и трихлорметиазид; и их сочетания. В конкретном варианте, диуретик выбирают из амилорида, буметанида, хлортиазида, хлорталидона, дихлорфенамида, этакриновой кислоты, фуросемида, гидрохлортиазида, гидрофлуметиазида, индапамида, метилклотиазида, метолазона, торсемида, триамтерена и их сочетания. Диуретик вводят в количестве, достаточном для получения около 5-50 мг в сутки, обычно, 6-25 мг в сутки, где обычная доза составляет 6,25 мг, 12,5 мг или 25 мг в сутки.
Соединение 1 также может вводиться в сочетании с ингибитором эндотелин-превращающего фермента (ЭПФ), примеры которого включают фосфорамидон, CGS 26303 и их сочетания.
В конкретном варианте, Соединение 1 вводят в сочетании с антагонистом рецептора эндотелина, примеры которого включают: селективные антагонисты рецептора эндотелина, которые воздействуют на рецептора эндотелина A, такие как авозентан, амбризентан, атразентан, BQ-123, клазозентан, дарузентан, ситаксентан и зиботентан; и двойные антагонисты рецептора эндотелина, которые воздействуют на рецепторы эндотелина A и B, такие как бозентан, мацитентан и тезозентан.
В еще одном варианте, Соединение 1 вводят в сочетании с одним или более ингибиторами HMG-CoA редуктазы, которые также известны как статины. Типовые статины включают аторвастатин, флувастатин, ловастатин, питавастатин, правастатин, розувастатин и симвастатин.
В одном варианте, Соединение 1 вводят в сочетании с ингибитором обратного захвата моноамина, примеры которого включают ингибиторы обратного захвата норэпинефрина, такие как атомоксетин, бупропирон и метаболит бупроприона гидроксибупропион, мапротилин, ребоксетин и вилоксазин; селективные ингибиторы обратного захвата серотонина (СИОЗС), такие как циталопрам и метаболит циталопрама дезметилциталопрам, дапоксетин, эсциталопрам (например, оксалат эсциталопрама), флуоксетин и метаболит флуоксетиндезметила норфлуоксетин, флувоксамин (например, малеат флувоксамина), пароксетин, сертралин и метаболит сертралина деметилсертралин; двойные ингибиторы обратного захвата серотонина-норэпинефрина (ИОЗСН), такие как бицифадин, дулоксетин, милнаципран, нефазодон и венлафаксин; и их сочетания.
В другом варианте, Соединение 1 вводят в сочетании с миорелаксантом, примеры которого включают: каризопродол, хлорхоксазон, циклобензаприн, дифлунизал, метаксалон, метокарбамол и их сочетания.
В одном варианте, Соединение 1 вводят в сочетании с натрийуретическим пептидом или аналогом, примеры которого включают: карперитид, CD-NP (Nile Therapeutics), CU-NP, незиритид, PL-3994 (Palatin Technologies, Inc.), уларитид, цендеритид и соединения, описанные в Ogawa et al (2004) J. Biol. Chem. 279:28625-31. Эти соединения также называются агонистами рецептора-А натрийуретического пептида (НПР-A). В другом варианте, Соединение 1 вводят в сочетании с антагонистом очищающего рецептора натрийуретического пептида (НПР-C), таким как SC-46542, cANF (4-23) и AP-811 (Veale (2000) Bioorg Med Chem Lett 10:1949-52). Например, AP-811 демонстрирует синергию при объединении с ингибитором НЕП, тиорфаном (Wegner (1995) Clin. Exper. Hypert. 17:861-876).
В другом варианте, Соединение 1 вводят в сочетании с ингибитором неприлизина (НЕП), примеры которого включают: AHU-377; кандоксатрил; кандоксатрилат; дексекадотрил (бензиловый эфир (+)-N-[2(R)-(ацетилтиометил)-3-фенилпропионил]глицина); CGS-24128 (3-[3-(бифенил-4-ил)-2-(фосфонометиламино)пропионамидо]пропионовая кислота); CGS-24592 ((S)-3-[3-(бифенил-4-ил)-2-(фосфонометиламино)пропионамидо]пропионовая кислота); CGS-25155 (бензиловый эфир N-[9(R)-(ацетилтиометил)-10-оксо-1-азациклодекан-2(S)илкарбонил]-4(R)гидрокси-L-пролина); производные 3-(1-карбамоилциклогексил)пропионовой кислоты, описанные в WO 2006/027680 Hepworth et al. (Pfizer Inc.); JMV-390-1 (2(R)-бензил-3-(N-гидроксикарбамоил)пропионил-L-изолейцил-L-лейцин); экадотрил; фосфорамидон; ретротиорфан; RU-42827 (2-(меркаптометил)-N-(4-пиридинил)бензолпропионамид); RU-44004 (N-(4-морфолинил)-3-фенил-2-(сульфанилметил)пропионамид); SCH-32615 ((S)-N-[N-(1-карбокси-2-фенилэтил)-L-фенилаланил]-β-аланин) и его пролекарство SCH-34826 ((S)-N-[N-[1-[[(2,2-диметил-1,3-диоксолан-4-ил)метокси]карбонил]-2-фенилэтил]-L-фенилаланил]-β-аланин); сиалорфин; SCH-42495 (этиловый эфир N-[2(S)-(ацетилсульфанилметил)-3-(2-метилфенил)пропионил]-L-метионина); спинорфин; SQ-28132 (N-[2-(меркаптометил)-1-оксо-3-фенилпропил]лейцин); SQ-28603 (N-[2-(меркаптометил)-1-оксо-3-фенилпропил]-β-аланин); SQ-29072 (7-[[2-(меркаптометил)-1-оксо-3-фенилпропил]амино]гептановая кислота); тиорфан и его пролекарство рацекадотрил; UK-69578 (цис-4-[[[1-[2-карбокси-3-(2-метоксиэтокси)пропил]циклопентил]карбонил]амино]циклогексанкарбоновая кислота); UK-447,841 (2-{1-[3-(4-хлорфенил)пропилкарбамоил]циклопентилметил}-4-метоксимасляная кислота); UK-505,749 ((R)-2-метил-3-{1-[3-(2-метилбензотиазол-6-ил)пропилкарбамоил]циклопентил}пропионовая кислота); 5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановая кислота и этиловый эфир 5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановой кислоты (WO 2007/056546); даглутрил [(3S,2'R)-3-{1-[2'-(этоксикарбонил)-4'-фенилбутил]-циклопентан-1-карбониламино}-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусная кислота], описанный в WO 2007/106708 Khder et al. (Novartis AG); и их сочетания. В конкретном варианте, ингибитор НЕП выбирают из AHU-377, кандоксатрила, кандоксатрилата, CGS-24128, фосфорамидона, SCH-32615, SCH-34826, SQ-28603, тиорфана и их сочетаний. В конкретном варианте, ингибитором НЕП является соединение, такое как даглутрил или GS-26303 ([N-[2-(бифенил-4-ил)-1(S)-(1H-тетразол-5-ил)этил]амино]метилфосфоновая кислота), которые являются ингибиторами эндотелин-превращающего фермента (ЭПФ) и НЕП. Другие соединения двойного действия на ЭПФ/НЕП также могут применяться. Ингибитор НЕП вводят в количестве, достаточном для получения около 20-800 мг в сутки, типовые дозы составляют 50-700 мг в сутки, более часто, 100-600 или 100-300 мг в сутки.
В одном варианте, Соединение 1 вводят в сочетании с донором оксида азота, примеры которого включают: никорандил; органические нитраты, такие как тетранитрат пентаэритритола; и сиднонимины, такие как линсидомин и молсидомин.
В еще одном варианте, Соединение 1 вводят в сочетании с нестероидным противовоспалительным агентом (НПВС), примеры которого включают: ацеметацин, ацетилсалициловую кислоту, альклофенак, алминопрофен, амфенак, амиприлозу, алоксиприн, аниролак, апазон, азапропазон, бенорилат, бенокапрофен, безпиперилон, броперамол, буклоксиновую кислоту, карпрофен, клиданак, диклофенак, дифлунисал, дифталон, эноликам, этодолак, эторикоксиб, фенбуфен, фенклофенак, фенклозовую кислоту, фенопрофен, фентиазак, фепразон, флуфенаминовую кислоту, флуфенизал, флупрофен, флурбипрофен, фурофенак, ибуфенак, ибупрофен, индометацин, индопрофен, изоксепак, изоксикам, кетопрофен, кеторолак, лофемизол, лорноксикам, меклофенамат, меклофенамовую кислоту, мефенамовую кислоту, мелоксикам, мезаламин, мипрофен, мофебутазон, набуметон, напроксен, нифлумовую кислоту, оксапрозин, окспинак, оксифенбутазон, фенилбутазон, пироксикам, пирпрофен, пранопрофен, сальсалат, судоксикам, сульфасалазин, сулиндак, супрофен, теноксикам, тиопинак, тиапрофеновую кислоту, тиоксапрофен, толфенамовую кислоту, толметин, трифлумидат, зидометацин, зомепирак и их сочетания. В конкретном варианте НПВС выбирают из этодолака, флурбипрофена, ибупрофена, индометацина, кетопрофена, кеторолака, мелоксикама, напроксена, оксапрозина, пироксикама, и их сочетаний.
В одном варианте, Соединение 1 вводят в сочетании с антагонистом рецептора N-метил d-аспартата (NMDA), примеры которого включают амантадин, декстрометорфоан, декстропропоксифен, кетамин, кетобемидон, мемантин, метадон и так далее.
В еще одном варианте, Соединение 1 вводят в сочетании с агонистом опиоидного рецептора (также называемым опиоидные анальгетики). Типовые агонисты опиоидного рецептора включают: бупренорфин, буторфанол, кодеин, дигидрокодеин, фентанил, гидрокодон, гидроморфон, леваллорфан, леворфанол, меперидин, метадон, морфин, налбуфин, налмефен, налорфин, налоксон, налтрексон, налорфин, оксикодон, оксиморфон, пентазоцин, пропоксифен, трамадол и их сочетания. В определенных вариантах, агонист опиоидного рецептора выбирают из кодеина, дигидрокодеина, гидрокодона, гидроморфона, морфина, оксикодона, оксиморфона, трамадола и их сочетания.
В конкретном варианте, Соединение 1 вводят в сочетании с ингибитором фосфодиэстеразы (ФДЭ), в частности, ингибитором ФДЭ-V. Типовые ингибиторы тФДЭ-V включают аванафил, лоденафил, мироденафил, силденафил (Revatio®), тадалафил (Adcirca®), варденафил (Levitra®) и уденафил.
В другом варианте, Соединение 1 вводят в сочетании с аналогом простагландина (также называемым простаноиды или аналоги простациклина). Типовые аналоги простагландина включают берапрост натрий, биматопрост, эпопростенол, илопрост, латанопрост, тафлупрост и трепростинил, где биматопрост, латанопрост и тафлупрост особенно интересны.
В еще одном варианте, Соединение 1 вводят в сочетании с агонистом рецептора простагландина, примеры которого включают биматопрост, латанопрост, травопрост и так далее.
Соединение 1 также может вводиться в сочетании с ингибитором ренина, примеры которого включают алискирен, эналкирен, ремикирен и их сочетания.
В другом варианте, Соединение 1 вводят в сочетании с селективным ингибитором обратного захвата серотонина (СИОЗС), примеры которого включают: циталопрам и метаболит циталопрама дезметилциталопрам, дапоксетин, эсциталопрам (например, оксалат эсциталопрама), флуоксетин и метаболит флуоксетиндезметила норфлуоксетин, флувоксамин (например, малеат флувоксамина), пароксетин, сертралин и метаболит сертралина деметилсертралин, и их сочетания.
В одном варианте, Соединение 1 вводят в сочетании с агонистом 5-HT1D рецептора серотонина, примеры которого включают триптаны, такие как алмотриптан, авитриптан, элетриптан, фроватриптан, наратриптан, ризатриптан, суматриптан и золмитриптан.
В одном варианте, Соединение 1 вводят в сочетании с блокатором натриевого канала, примеры которого включают карбамазепин, фосфенитоин, ламотригин, лидокаин, мексилетин, окскарбазепин, фенитоин и их сочетания.
В одном варианте, Соединение 1 вводят в сочетании с растворимым стимулятором или активатором гуанилатциклазы, примеры которого включают атацигуат, риоцигуат и их сочетания.
В одном варианте, Соединение 1 вводят в сочетании с трициклическим антидепрессантом (ТЦА), примеры которого включают амитриптилин, амитриптилиноксид, бутриптилин, кломипрамин, демексиптилин, дезипрамин, дибензепин, диметакрин, досулепин, доксепин, имипрамин, имипроминоксид, лофепрамин, мелитрацен, метапрамин, нитроксазепин, нортриптилин, ноксиптилин, пипофезин, пропизепин, протриптилин, квинупрамин и их сочетания.
В одном варианте, Соединение 1 вводят в сочетании с антагонистом рецептора вазопрессина, примеры которого включают кониваптан и толваптан.
Объединенные вторичные терапевтические агенты также могут быть полезны в другой комбинированной терапии с соединением в соответствии с данным изобретением. Например, соединение в соответствии с данным изобретением может быть объединено с диуретиком и ARB, или блокатором кальциевого канала и ARB, или диуретиком и ингибитором АПФ, или блокатором кальциевого канала и статином. Типовые примеры включают сочетание ингибитора АПФ эналаприла (в форме малеата) и диуретика гидрохлортиазида, который подают под торговым наименованием Vaseretic®, или сочетание блокатора кальциевого канала амлодипина (в форме безилата) и ARB олмесартана (в форме пролекарства медоксомила), или сочетанием блокатора кальциевого канала и статина, все могут также применяться с соединением 1. Другие терапевтические агенты, такие как агонисты α2-адренергического рецептора и антагонисты рецептора вазопрессина, могут также применяться в комбинированной терапии. Типовые агонисты α2-адренергического рецептора включают клонидин, дексмедетомидин и гуанфацин.
Представленные ниже композиции иллюстрируют типовые фармацевтические композиции данного изобретения.
Типовые твердые желатиновые капсулы для перорального введения
Соединение в соответствии с данным изобретением (50 г), 440 г высушенной распылением лактозы и 10 г стеарата магния тщательно смешивают. Полученную композицию затем загружают в твердые желатиновые капсулы (500 мг композиции на капсулу). Альтернативно, соединение 1 (20 мг) тщательно смешивают с крахмалом (89 мг), микрокристаллической целлюлозой (89 мг) и стеаратом магния (2 мг). Смесь затем пропускают через сито № 45 меш США и загружают в твердую желатиновую капсулу (200 мг композиции на капсулу).
Альтернативно, соединение 1 (30 г), вторичный агент (20 г), 440 г высушенной распылением лактозы и 10 г стеарата магния тщательно смешивают и обрабатывают, как описано выше.
Типовые желатиновые капсулы для перорального введения
Соединение 1 (100 мг) тщательно смешивают с моноолеатом полиоксиэтиленсорбитана (50 мг) и порошком крахмала (250 мг). Смесь затем загружают в желатиновую капсулу (400 мг композиции на капсулу). Альтернативно, соединение 1 (70 мг) и вторичный агент (30 мг) тщательно смешивают с моноолеатом полиоксиэтиленсорбитана (50 мг) и порошком крахмала (250 мг), и полученную смесь загружают в желатиновую капсулу (400 мг композиции на капсулу).
Альтернативно, соединение 1 (40 мг) тщательно смешивают с микрокристаллической целлюлозой (Avicel PH 103; 259,2 мг) и стеаратом магния (0,8 мг). Смесь затем загружают в желатиновую капсулу (размер №1, белая, непрозрачная) (300 мг композиции на капсулу).
Типовая гидроксипропилметилцеллюлозная (ГПМЦ) капсула для перорального введения
Соединение 1 (50 мг или 100 мг) загружают сразу же в ГПМЦ капсулу.
Типовая таблетка для перорального введения
Соединение 1 (10 мг), крахмал (45 мг) и микрокристаллическую целлюлозу (35 мг) пропускают через сито № 20 меш США и тщательно смешивают. Полученные гранулы сушат при 50-60°C и пропускают через сито № 16 меш США. Раствор поливинилпирролидона (4 мг в виде 10% раствора в стерильной воде) смешивают с карбоксиметилкрахмалом натрия (4,5 мг), стеаратом магния (0,5 мг) и тальком (1 мг), и эту смесь затем пропускают через сито № 16 меш США. Карбоксиметилкрахмал натрия, стеарат магния и тальк затее добавляют к гранулам. После смешивания смесь прессуют в таблеточной машине с получением таблеток массой 100 мг.
Альтернативно, соединение 1 (250 мг) тщательно смешивают с микрокристаллической целлюлозой (400 мг), высокодисперсным диоксидом кремния (10 мг) и стеариновой кислотой (5 мг). Затем смесь прессуют с получением таблеток (665 мг композиции на таблетку).
Альтернативно, соединение 1 (400 мг) тщательно смешивают с кукурузным крахмалом (50 мг), кроскармеллозой натрия (25 мг), лактозой (120 мг) и стеаратом магния (5 мг). Затем смесь прессуют с получением таблетки с одной насечкой (600 мг композиции на таблетку).
Альтернативно, соединение 1 (100 мг) тщательно смешивают с кукурузным крахмалом (100 мг) с водным раствором желатина (20 мг). Смесь сушат и измельчают до тонкоизмельченного порошка. Микрокристаллическую целлюлозу (50 мг) и стеарат магния (5 мг) затем смешивают с желатиновой композицией, гранулируют, и полученную смесь прессуют с получением таблеток (100 мг соединения в соответствии с данным изобретением на таблетку).
Типовая суспензия для перорального введения
Следующие ингредиенты смешивают с получением суспензии, содержащей 100 мг соединения 1 на 10 мл суспензии:
|
Типовая жидкая композиция для перорального введения
Подходящей жидкой композицией является композиция с буфером на основе карбоновой кислоты, таким как цитратный, лактатный и малеатный буферные растворы. Например, соединение 1 (которое может быть предварительно смешано с ДПВO), смешивают со 100 мМ цитратным аммониевым буфером и pH доводят до pH 5, или смешивают со 100 мМ раствора лимонной кислоты и pH доводят до pH 2. Такие растворы также могут включать солюбилизирующий наполнитель, такой как циклодекстрин, например, раствор может включать 10% масс. гидроксипропил-β-циклодекстрин.
Другие подходящие композиции включают 5% раствор NaHCO3 с или без циклодекстрина.
Типовая парентеральная ВВ композиция для введения инъекцией
Соединение 1 (0,2 г) смешивают с 0,4 M буферным раствором ацетата натрия (2,0 мл). pH полученного раствора доводят до pH 4 добавлением 0,5 N водной хлористоводородной кислоты или 0,5 N водного гидроксида натрия, при необходимости, и затем достаточное количество воды для инъекций добавляют до получения общего объема 20 мл. Затем смесь фильтруют через стерильный фильтр (0,22 микрон) с получением стерильного раствора, подходящего для введения инъекцией.
Следующие композиции иллюстрируют типовые фармацевтические композиции в соответствии с данным изобретением.
Пример композиции A
Замороженный раствор, подходящий для получения раствора для инъекций, получают следующим образом:
|
Типовая методика: наполнители, если присутствуют, растворяют в около 80% воды для инъекций и активное соединение 1 или 1' добавляют и растворяют. pH доводят 1 M гидроксидом натрия до 3-4,5, и объем затем доводят до 95% конечного объема водой для инъекций. pH проверяют и доводят, при необходимости, и объем доводят до конечного объема водой для инъекций. Затем композицию стерильно фильтруют через 0,22 микроновый фильтр и помещают в стерильный флакон в асептических условиях. Флакон закрывают крышкой, наклеивают этикетку и хранят в замороженном виде.
Пример композиции B
Лиофилизированный порошок и кристаллическое твердое вещество для получения раствора для инъекций готовят следующим образом:
|
Типовая методика: Наполнители и/или буферные агенты, если присутствуют, растворяют в около 60% воды для инъекций. Активное соединение 1 или 1' добавляют и растворяют, и pH доводят 1 M гидроксидом натрия до 3-4,5, и объем доводят до 95% конечного объема водой для инъекций. pH проверяют и доводят, при необходимости, и объем доводят до конечного объема водой для инъекций. Затем композицию стерильно фильтруют через 0,22 микроновый фильтр и помещают в стерильный флакон в асептических условиях. Затем композицию сушат вымораживанием с применением подходящего цикла лиофилизации. Флакон закрывают крышкой (необязательно под частичным вакуумом или сухим азотом), наклеивают этикетку и хранят в холодильнике.
Пример композиции C
Раствор для инъекций для внутривенного введения пациенту получают из примера композиции В выше следующим образом:
Типовая методика: лиофилизированный порошок из примера композиции В (например, содержащий от 10 до 1000 мг активного соединения 1 или 1') восстанавливают 20 мл стерильной воды, и полученный раствор разбавляют 80 мл стерильного раствора соли в 100 мл инфузионном пакете. Разбавленный раствор затем вводят пациенту внутривенно в течение от 30 до 120 минут.
Типовые композиции для введения ингаляцией
Соединение 1 (0,2 мг) микронизируют и затем смешивают с лактозой (25 мг). Эту смесь затем загружают в желатиновый картридж для ингаляций. Содержимое картриджа вводят с применением порошкового ингалятора, например.
Альтернативно, микронизированное соединение 1 (10 г) диспергируют в растворе, полученном растворением лецитина (0,2 г) в деминерализованной воде (200 мл). Полученную суспензию сушат распылением и затем микронизируют с получением микронизированной композиции, содержащей частицы со средним диаметром менее около 1,5 мкм. Микронизированную композицию затем загружают в картриджи дозирующего ингалятора, содержащие сжатый 1,1,1,2-тетрафторэтан в количестве, достаточном для получения от около 10 мкг до около 500 мкг соединения в соответствии с данным изобретением на дозу при введении ингалятором.
Альтернативно, соединение 1 (25 мг) растворяют в изотоническом солевом растворе с цитратным буфером (pH 5) (125 мл). Смесь перемешивают и обрабатывают ультразвуком до растворения соединения. pH раствора проверяют и доводят, при необходимости, до pH 5 медленным добавлением водного 1 N NaOH. Раствор вводят с применением небулайзера, который обеспечивает от около 10 мкг до около 500 мкг соединения 1 на дозу.
ПРИМЕРЫ
Следующие примеры получения и примеры даны для иллюстрации конкретных вариантов изобретения. Эти конкретные варианты, однако, не ограничивают объем изобретения, если не указано иначе.
Следующие аббревиатуры имеют следующие значения, если не указано иначе, и любые другие аббревиатуры, применяемые здесь и не раскрытые, имеют стандартные общепринятые значения:
|
Если не указано иначе, все материалы, такие как реагенты, исходные материалы и растворители, покупают у коммерческих источников (таких как Sigma-Aldrich, Fluka Riedel-de Haën и подобные) и применяют без дальнейшей очистки.
Реакции проводят в атмосфере азота, если не указано иначе. Развитие реакций отслеживают тонкослойной хроматографией (ТСХ), аналитической высокоэффективной жидкостной хроматографией (анал. ВЭЖХ) и масс-спектрометрией, подробности которых даны в конкретных примерах. В общем, в аналитической ВЭЖХ применяют следующие растворители: растворитель A - 98% H2O/2% MeCN/1,0 мл/л ТФК; растворитель B - 90% MeCN/10% H2O/1,0 мл/л ТФК.
Реакции проводят как описано конкретно в каждом примере получения; обычно реакционные смеси очищают экстрагированием и другими способами очистки, такими как зависимая от температуры и растворителя кристаллизация и осаждение. Кроме того, реакционные смеси обычно очищают препаративной ВЭЖХ, обычно с применением колонок Microsorb C18 и Microsorb BDS и обычных элюентов. Развитие реакций обычно отслеживают жидкостной хроматографией масс спектрометрией (ЖХМС). Характеризацию изомеров проводят спектроскопией ядерного эффекта Оверхаузера (NOE). Характеризацию продуктов реакции обычно проводят массовой и 1H-ЯМР спектрометрией. Для записи ЯМР образцы растворяют в дейтерированном растворителе (CD3OD, CDCl3 или ДМСО-d6) и 1H-ЯМР спектр получают на инструменте Varian Gemini 2000 (400 МГц) в стандартных условиях наблюдения. Масс спектрометрическую идентификацию соединений обычно проводят с применением способа ионизации электрораспылением (ЭРМС) на инструменте Applied Biosystems (Foster City, CA), модель API 150 EX, или на инструменте Agilent (Palo Alto, CA), модель 1200 LC/MSD.
Методы измерения
Порошковая рентгеновская дифракция
Порошковую рентгеновскую дифракцию проводят с применением рентгеновского дифракторметра Bruker D8-Advance. Источником рентгеновского излучения является излучение Cu-Kα при выходном напряжении 40 кВ и токе 40 мА. Инструмент работает в геометрии Брэгга-Брентано, и применяют зеркала Goebel для получения параллельного рентгеновского луча. Любое отклонение луча ограничено 0,2° щелью вертикальной расходимости на источнике и щелями Соллера (2,5°) на источнике и детекторе. Для измерения небольшое количество порошка (5-25 мг) осторожно сжимают в силиконовом держателе образца с нулевым фоном с получением гладкой поверхности и подвергают рентгеновскому облучению. Образцы сканируют в режиме связанного колебания θ-2θ от 2° до 35° в 2θ с размером шага 0,02° и скоростью сканирования 0,3 секунды на шаг. Сбор данных контролируют программным обеспечением Bruker DiffracSuite и анализируют программой Jade (версия 7,5,1). Инструмент калибруют корундовым стандартом в пределах ±0,02° 2θ угла.
Необходимо иметь в виду, что геометрия Брэгга-Брентано, применяемая при сборе данных, склонна к предпочтительной ориентации. В этих условиях возможно, что относительные интенсивности пиков дифракции могут не представлять истинные относительные интенсивности, которые будут получены при идеализированном распределении сферических частиц или из дифрактограммы, смоделированной из монокристаллических данных. Также возможно, что некоторые пики не видны в некоторых дифрактограммах из-за экстенсивной предпочтительной ориентации.
Дифференциальная сканирующая калориметрия
Изменения ДСК проводят с применением модуля TA Instruments Model Q-100 с контроллером Thermal Analyst. Данные собирают и анализируют с применением программы TA Instruments Universal Analysis. Образцы точно взвешивают в закрытом алюминиевом лотке. Через 5 минут периода изотермического уравновешивания при 5°C образец нагревают с применением линейной скорости нагревания 10°C/мин от 0°C до 200°C.
Термогравиметрический анализ
Тепловую гравиметрию проводят с применением модуля TA Instruments Model Q-500, имеющего высокую разрешающую способность. Данные собирают с применением контроллера TA Instruments Thermal Analyst и анализируют с применением программы TA Instruments Universal Analysis. Взвешенный образец помещают в платиновый лоток и сканируют со скоростью нагревания 10°C/мин от температуры окружающей среды до 200°C. Весы и топочные камеры продувают потоком азота во время применения.
Микроскопия в поляризованном свете
Для исследований на оптическом поляризационном микроскопе (ОПМ) образцы исследуют под оптическим микроскопом (Olympus BX51) с кроссполяризационным оптическим фильтром. Изображения собирают камерой PaxCam, контролируемой программой PaxIt Imaging (версия 6,4). Образцы получают на стеклах с легким минеральным маслом в качестве иммерсионной среды. В зависимости от размера частиц, 4×, 10× или 20× объективы применяют для увеличения.
Оценка динамического поглощения влаги
Измерения ДПВ проводят с применением атмосферных микровесов VTI, системы SGA-100 (VTI Corp., Hialeah, FL 33016). Примеряют взвешенный образец, и влажность имеет наименьшее возможное значение (близкое к 0% относительной влажности) в начале анализа. Анализ ДПВ включает скорость сканирования 5% относительной влажности/шаг при полном интервале влажности 5-90%. ДПВ проводят изотермически при 25°C.
Методы синтеза и сравнительные примеры
Следующие соединения синтезируют и оценивают ингибирующее действие на НЕП фермент:
|
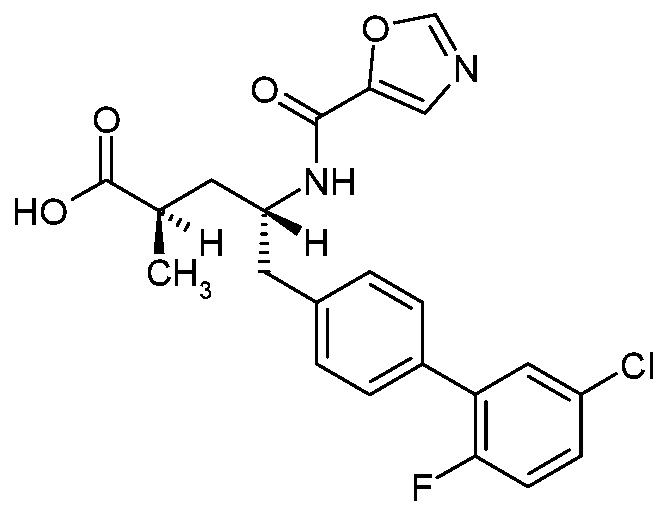
Пример получения 1: этиловый эфир ( 2R,4R )-4-Амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты (соединение 7)
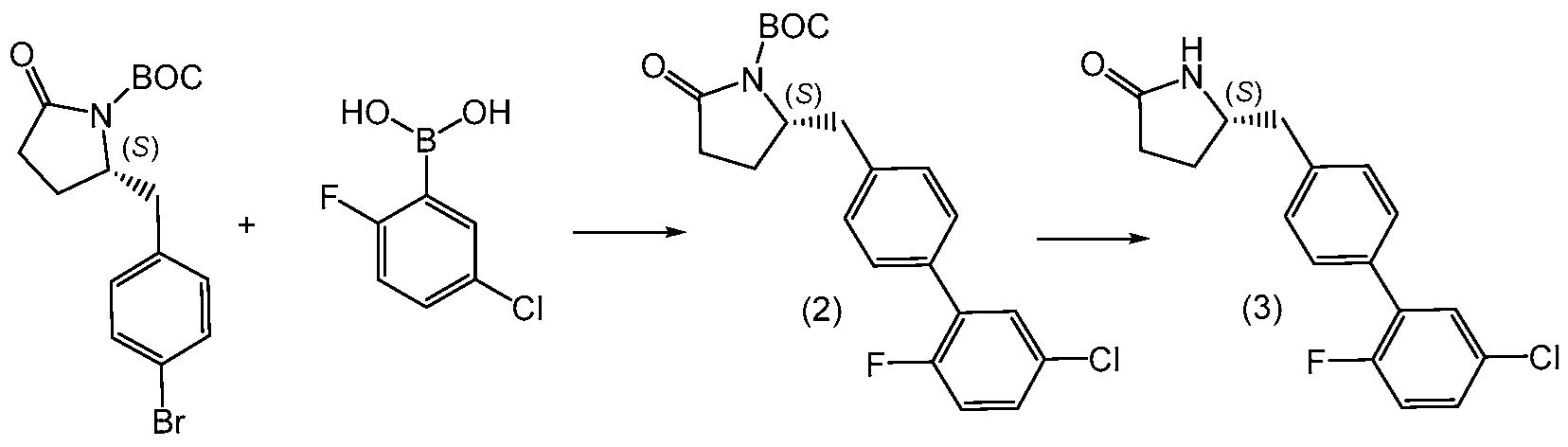
К раствору трет-бутилового эфира (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (25 г, 70,6 ммоль) в 1,4-диоксане (500 мл) добавляют 5-хлор-2-фторфенилбороновую кислоту (24,6 г, 141 ммоль), Pd(PPh3)4 (4,1 г, 3,5 ммоль) и раствор K2CO3 (17,8 г, 141 ммоль) в воде (90 мл), при комнатной температуре под азотом. Смесь нагревают до 60°C и перемешивают в течение ночи. Добавляют воду (500 мл), и растворитель выпаривают. Смесь экстрагируют EtOAc (200 мл×3). Объединенные органические слои промывают насыщенным водным NaCl (300 мл) и фильтруют. Фильтрат концентрируют с получением неочищенного остатка, который очищают хроматографией с получением соединения 2 (22,7 г) в виде светло-желтого твердого вещества. ЖХ-МС: 829,2 [2M+Na+].
К раствору соединения 2 (4,9 г, 12,1 моль) в ДХМ (100 мл) добавляют ТФК (4,5 мл, 60,7 ммоль) при 0°C под азотом, и перемешивают в течение 1 часа. Смесь нагревают до комнатной температуры в течение 1,5 часов. После выпаривания растворителя остаток разбавляют EtOAc (100 мл), затем промывают насыщенным водным NaHCO3 (100 мл×3), водой (100 мл×2), насыщенным водным NaCl (100 мл) и затем сушат над Na2SO4. Смесь фильтруют, и фильтрат концентрируют с получением неочищенного соединения 3. ЖХ-МС: 304 [M+H]+.
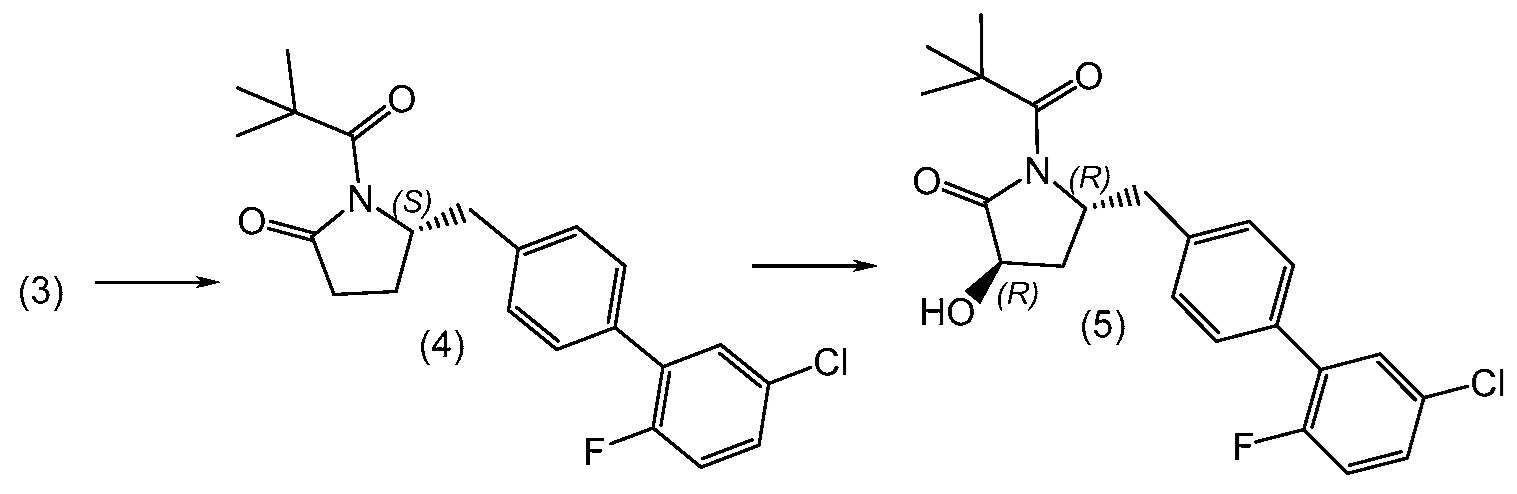
К раствору NaH (2,4 г, 695 ммоль) в ТГФ (200 мл) добавляют по каплям раствор соединения 3 (8,5 г, 278 ммоль) в ТГФ (50 мл) при 0°C под азотом. Смесь нагревают до комнатной температуры и перемешивают в течение 2 часов. После охлаждения до 0°C по каплям добавляют пивалоилхлорид (5 г, 41,7 ммоль) в течение более 30 минут. Смесь нагревают до комнатной температуры и перемешивают в течение 9,5 часов. Реакцию гасят насыщенным водным NH4Cl (250 мл) и экстрагируют EtOAc (400 мл×3). Объединенные органические слои сушат над Na2SO4 и концентрируют с получением неочищенного остатка, который очищают хроматографией с получением соединения 4 (18 г) в виде желтого твердого вещества. ЖХ-МС: 388 [M+H+].
К раствору соединения 4 (9 г, 23,2 ммоль) в ТГФ (200 мл) добавляют по каплям NaHMDS (20,9 мл, 41,8 ммоль) при -78°C под азотом. После перемешивания в течение 1 часа при -78°C раствор (+)-(8,8-дихлоркамфорилсульфонил)оксазиридина (10,4 г, 34,8 ммоль) в ТГФ (50 мл) добавляют по каплям. После перемешивания при -78°C в течение 1 часа, реакцию гасят насыщенным водным NH4Cl (50 мл) и экстрагируют EtOAc (400 мл×3). Объединенные органические слои промывают 1M водной HCl (400 мл), насыщенным водным NaHCO3 (400 мл) и насыщенным водным NaCl (400 мл), сушат над Na2SO4 и концентрируют с получением неочищенного остатка, который очищают хроматографией с получением соединения 5 (8,8 г) в виде белого полутвердого вещества. ЖХ-МС: 426,1 [M+Na+].
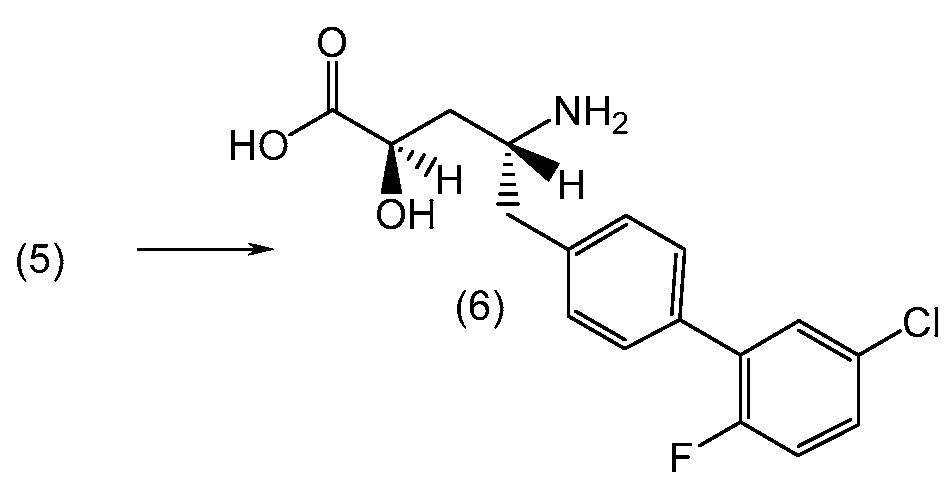
Раствор соединения 5 (8,8 г, 21,8 ммоль) в EtOH (12 мл) добавляют к концентрированной HCl (200 мл) и нагревают при 100°C и перемешивают в течение ночи. Смесь затем концентрируют с получением неочищенного остатка, который очищают промыванием Et2O (100 мл) с получением соединения 6 (7,5 г) в виде твердой соли HCl. ЖХ-МС: 338 [M+H+].
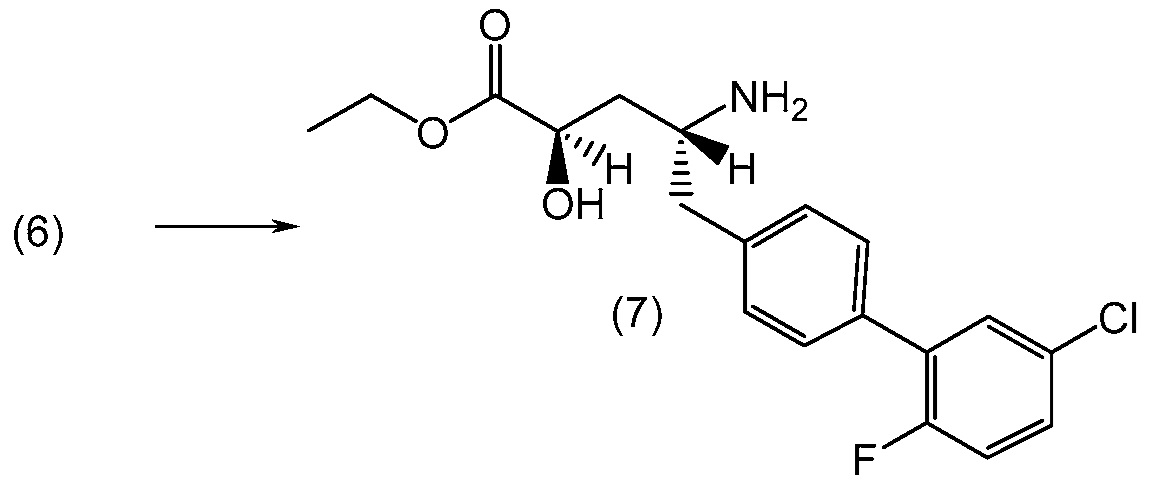
Раствор соединения 6 (7,5 г, 20,1 ммоль) в 1:1 EtOH/HCl (100 мл) нагревают при 50°C в течение ночи. Смесь концентрируют, и неочищенный остаток очищают промыванием Et2O (200 мл) с получением соединения 7 (6,5 г) в виде белой твердой соли HCl. ЖХ-МС: 366,1 [M+H+].
ПРИМЕР 1: ( 2R,4R )-5-(5'-Хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановая кислота (соединение 1)
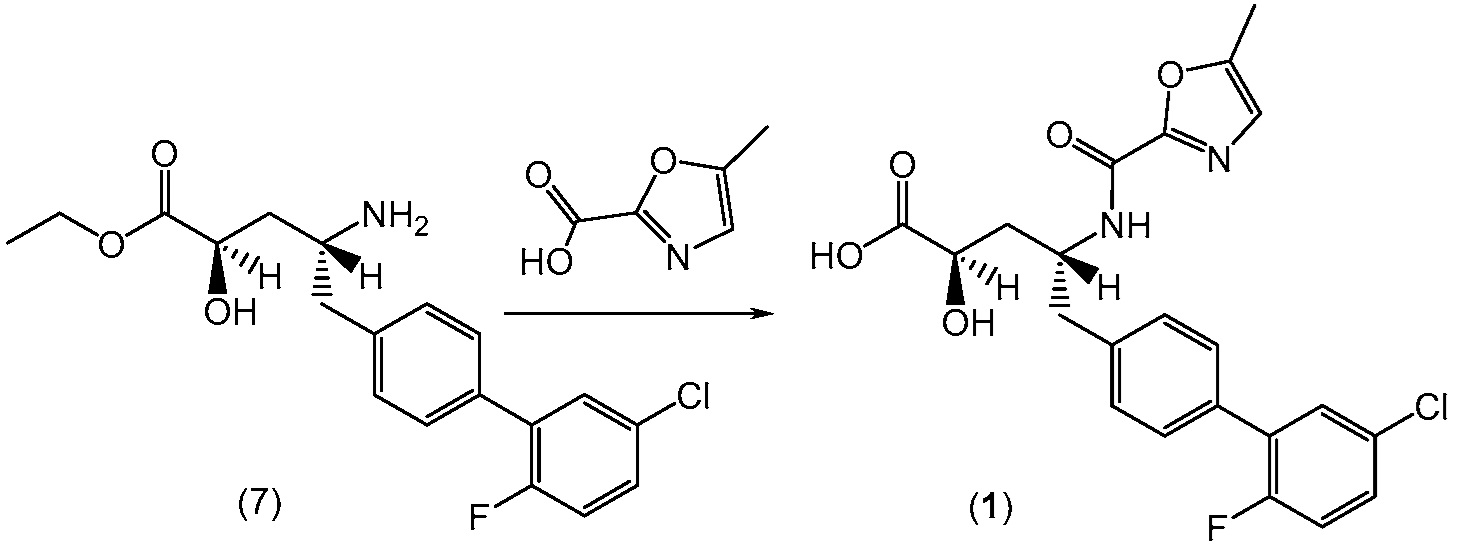
5-Метилоксазол-2-карбоновую кислоту (182 мг, 1,4 ммоль) и ГАТУ (546 мг, 1,4 ммоль) объединяют с ДМФ (3 мл) и перемешивают в течение 15 минут при комнатной температуре. Затем добавляют соединение 7 (500 мг, 1,4 ммоль) и ДИПЭА (716 мкл, 4,1 ммоль). Полученную смесь перемешивают в течение 15 минут при комнатной температуре, затем ЖХ/МС показывает завершение. Растворитель удаляют в вакууме, и неочищенный остаток очищают нормальной флэш-хроматографией (гексан:EtOAC 20-95%) с получением твердого вещества (590 мг, 1,2 ммоль), которое растворяют в сухом EtOH (5 мл) и сухом ТГФ (5 мл). Затем добавляют раствор 1 N LiOH в воде (9,9 мл, 9,9 ммоль). Полученный раствор перемешивают при комнатной температуре в течение 1 часа, затем ЖХ/МС показывает завершение. Растворитель удаляют в вакууме, и неочищенный остаток очищают хроматографией с обращенной фазой с получением соединения 1 (490 мг) в виде белого порошка. МС m/z [M+H]+ рассч. для C22H20ClFN2O5, 447,10; найдено 447,2.
Кристаллическая не сольватированная (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(5-метилоксазол-2-карбонил)амино]пентановая кислота (соединение 1')
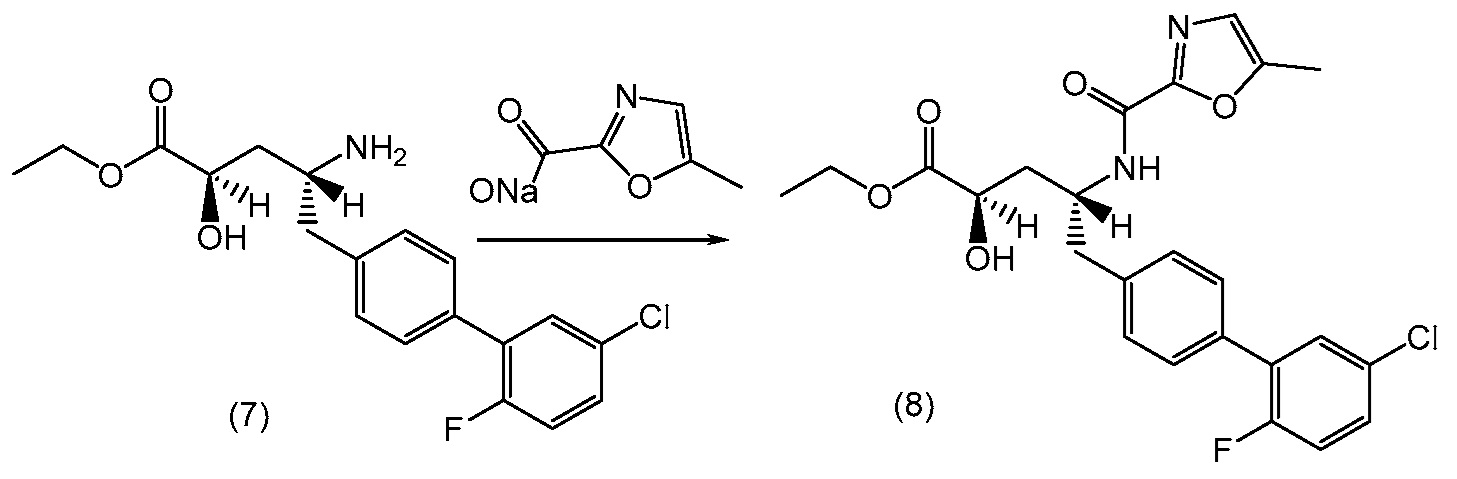
Соединение 7 (HCl; 190,0 г, 472 ммоль) растворяют в ДМФ (2 л) и полученную смесь охлаждают до 0°C. Добавляют 5-метилоксазол-2-карбоксилат натрия (73,9 г, 496 ммоль), затем ДИПЭА (124 мл, 708 ммоль) одной порцией. Добавляют PyBOP (320 г, 614 ммоль) порциями в течение более 20 минут, поддерживая внутреннюю температуру ниже 10°C (6,5°C максимум), и полученную смесь перемешивают при 0°C в течение 1 часа и затем при 20°C в течение 1 часа, отслеживая реакцию. При превращении >99% смесь перемешивают в течение еще 30 минут при комнатной температуре. Добавляют EtOAc (6 л) и воду (5 л), и полученную смесь перемешивают в течение 20 минут. Фазы разделяют, и органический слой промывают 0,5 M HCl (5 л), 5% водным раствором NaHCO3 (5 л) и 5% насыщенным водным раствором NaCl (5 л). Органический слой сушат над Na2SO4 в течение 24 часов при комнатной температуре, затем концентрируют роторным испарением. Добавляют ТГФ (500 мл), и полученную смесь концентрируют с получением соединения 8 в виде густого масла.
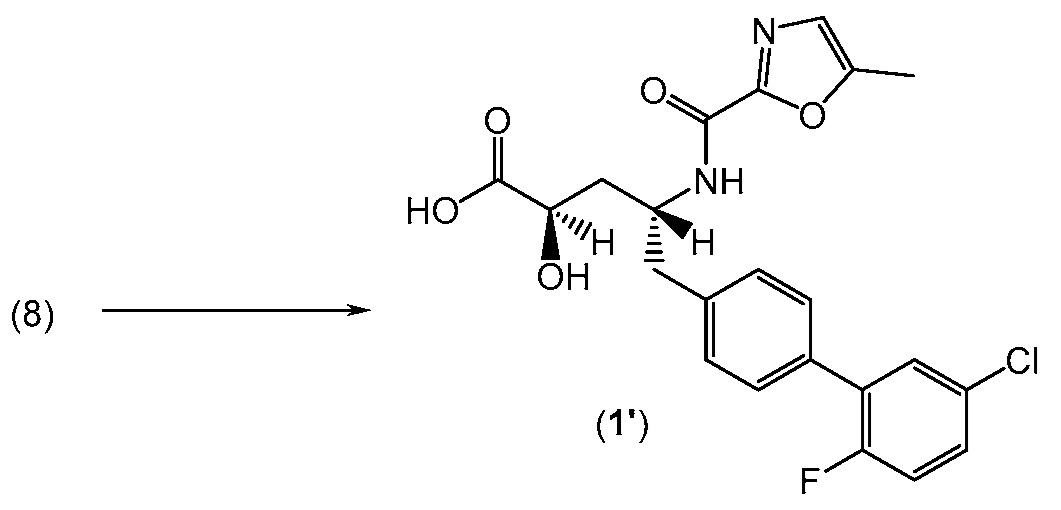
Неочищенное соединение 8 (224 г, 472 ммоль) растворяют в ТГФ (2 л). Добавляют моногидрат LiOH (39,6 г, 944 ммоль), растворенный в воде (400 мл), и полученную смесь перемешивают при комнатной температуре, отслеживая реакцию. Полное превращение наблюдают через 2 часа. Затем реакцию гасят 1M водной HCl (1180 мл, 1180 ммоль). Добавляют EtOAc (2 л) и насыщенный водный NaCl (2 л), и полученную смесь перемешивают в течение 15 минут. Фазы разделяют, и органический слой промывают 10% водным раствором NaCl (3 л) и сушат над Na2SO4 (500 г) в течение ночи, затем растворитель удаляют. К полученному маслу добавляют EtOAc (2 л) и объем снижают до около 500 мл. Полученную суспензию перемешивают при комнатной температуре в течение 30 минут с получением густой трудно перемешиваемой суспензии (образующейся в течение 10 минут). Медленно добавляют гексан (500 мл) (в течение более 10 минут), и полученную свободнотекучую суспензию перемешивают в течение 20 минут при комнатной температуре. Фильтрация и сушка дает соединение 1' (170 г; 99,2% чистый продукт) в виде твердого вещества. Этот продукт анализируют ПРД, ДСК и тепловой гравиметрией, как описано здесь, и определяют как не сольватированный кристаллический продукт. Эти данные представлены на фиг. 1-3.
ПРИМЕР 2: ( 2R,4R )-5-(5'-Хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(3-метоксиихоксазол-5-карбонил)амино]пентановая кислота (сравнительное соединение C2)
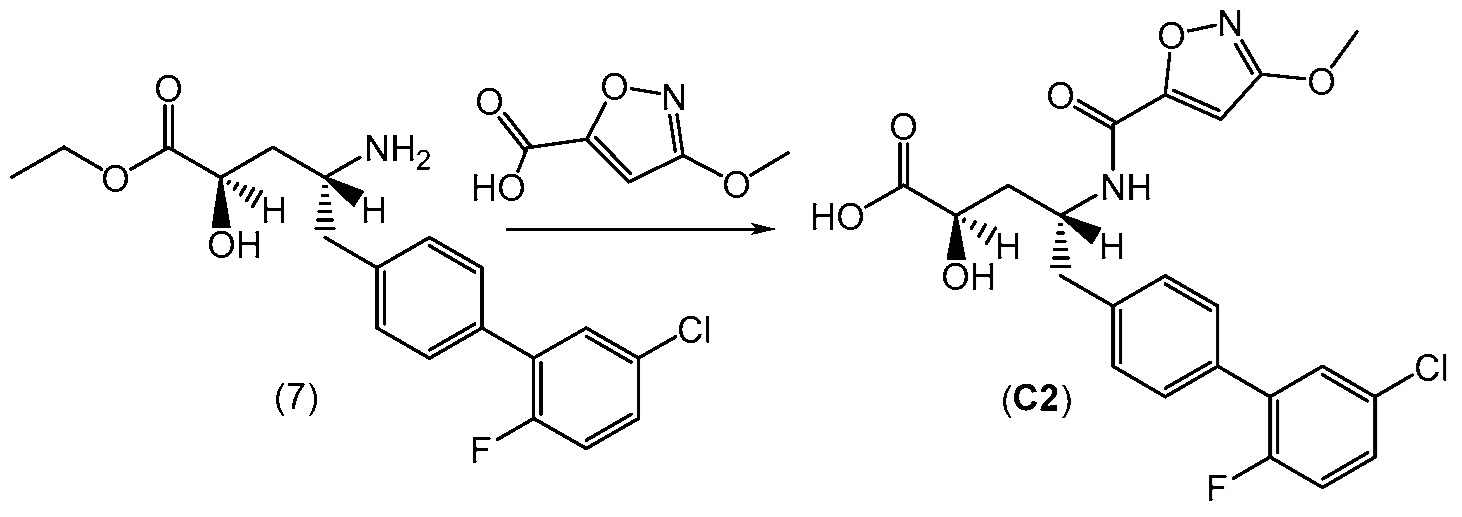
ДИПЭА (227 мкл, 1,3 ммоль) добавляют к раствору 3-метоксиизоксазол-5-карбоновой кислоты (74,7 мг, 522 мкмоль), соединения 7 (175 мг, 435 мкмоль) и ГАТУ (248 мг, 653 мкмоль) в ДМФ (2 мл). Полученную смесь перемешивают в течение 10 минут при комнатной температуре, затем ЖХ/МС показывает завершение. Добавляют 5,0 M водный LiOH (696 мкл, 3,5 ммоль) и смесь перемешивают при комнатной температуре в течение 30 минут. Добавляют концентрированную HCl (~0,4 мл), пока реакционная смесь не станет кислой, и неочищенную смесь очищают хроматографией с обращенной фазой (30-90% MeCN в воде с 0,05% ТФК) с получением сравнительного соединения C2 (132 мг) в виде белого твердого вещества. МС m/z [M+H]+ рассч. для C22H20ClFN2O6, 463,10; найдено 463,2.
Сравнительное соединение C2 описано в примере 19-9 патенте США № 8,586,536 Gendron et al.
Пример получения 2: этиловый эфир ( 2R,4S )-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-метилпентановой кислоты (соединение 11)
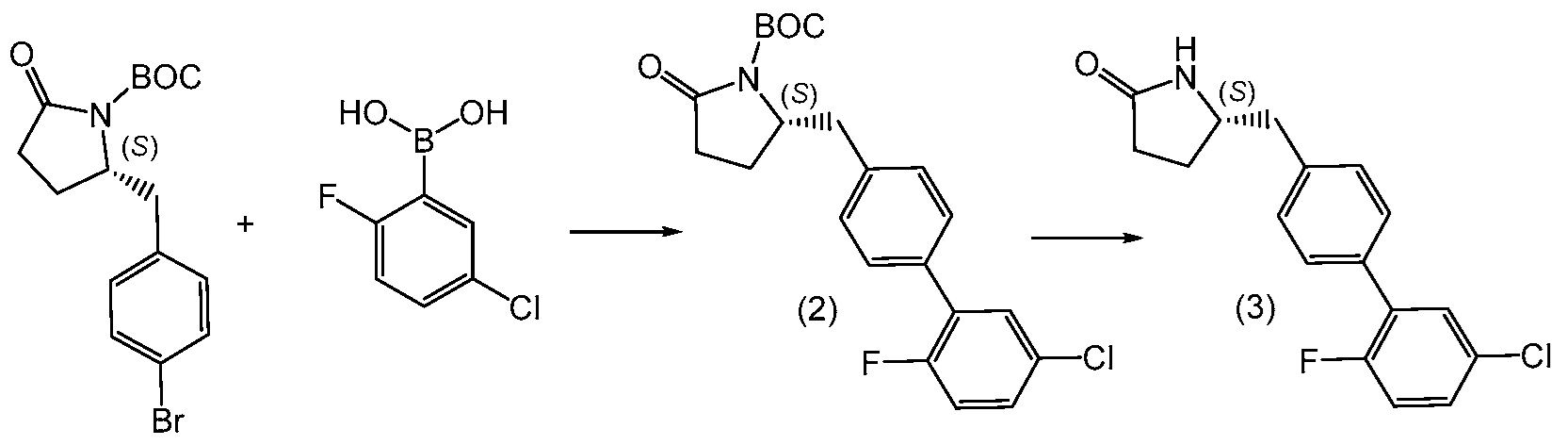
трет-Бутиловый эфир (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (90 г, 254 ммоль), 5-хлор-2-фторфенилбороновую кислоту (48,7 г, 279,4 ммоль), Pd(dppf)2Cl2 (5,6 г, 7,6 ммоль), KF (29,5 г, 508 ммоль) в диоксане (900 мл) и воде (300 мл) объединяют при комнатной температуре под азотом. Полученный раствор нагревают до 85°C и перемешивают в течение 4 часов. Добавляют воду (500 мл), и смесь экстрагируют EtOAc (500 мл×2). Объединенные органические слои промывают насыщенным водным NaCl (500 мл), сушат, концентрируют и очищают хроматографией (ПЭ/EtOAc=6:1 до 3:1) с получением соединения 2 (96 г) в виде желтого твердого вещества. ЖХ-МС: m/z=348[(M-56)++1].
К раствору соединения 2 (20 г, 49,5 ммоль) в безводном ДХМ (200 мл) добавляют ТФК (30 мл) при 0°C под азотом. Смесь нагревают до комнатной температуры и перемешивают при комнатной температуре в течение 2 часов. После выпаривания растворителя остаток разбавляют EtOAc (200 мл), затем промывают насыщенным водным NaHCO3 (200 мл×3) и насыщенным водным NaCl (200 мл×2). Органический слой сушат и концентрируют с получением соединения 3 (14 г, неочищенное) в виде желтого масла. ЖХ-МС: m/z=304[(M++1)], m/z=607[(2M++1)].
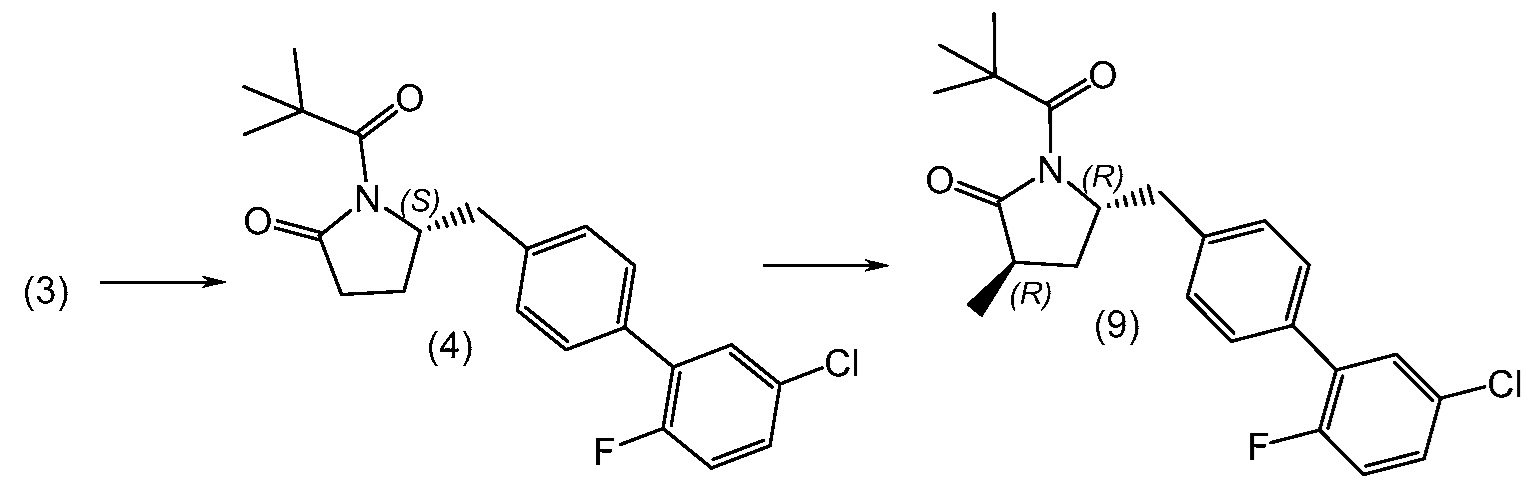
К раствору соединения 3 (14 г, 46 ммоль) в безводном ТГФ (150 мл) добавляют по каплям 2,5 M раствор н-бутиллития в гексане (21 мл, 52,9 ммоль) при 78°C под азотом. Полученную смесь перемешивают при -78°C в течение 1 часа. Пивалоилхлорид (7,2 г, 59,8 ммоль) затем добавляют по каплям при -78°C, и смесь перемешивают при -78°C в течение еще 2 часов. Реакцию гасят водой (200 мл) и экстрагируют EtOAc (200 мл×2). Объединенные органические слои промывают насыщенным водным NaCl (200 мл), сушат над Na2SO4, концентрируют и очищают хроматографией на колонке (ПЭ/EtOAc=50:1) с получением соединения 4 (14,6 г) в виде светло-желтого масла. ЖХ-МС: m/z=388[(M++1)].
К раствору соединения 4 (14,6 г, 37,4 ммоль) в толуоле (200 мл) добавляют по каплям KHMDS (82 мл, 41,1 ммоль) при -78°C под азотом. После перемешивания при -78°C в течение 2 часов, Me2SO4 (4,3 мл, 44,9 ммоль) добавляют по каплям. После перемешивания при -78°C в течение 2 часов реакцию гасят насыщенным водным NH4Cl (100 мл) и экстрагируют EtOAc (100 мл×2). Объединенные органические слои промывают насыщенным водным NaCl (200 мл), сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного продукта, который очищают хроматографией (ПЭ/EtOAc=100:1) с получением соединения 9 (3,4 г) в виде бесцветного масла. ЖХ-МС: m/z=402[(M++1)].
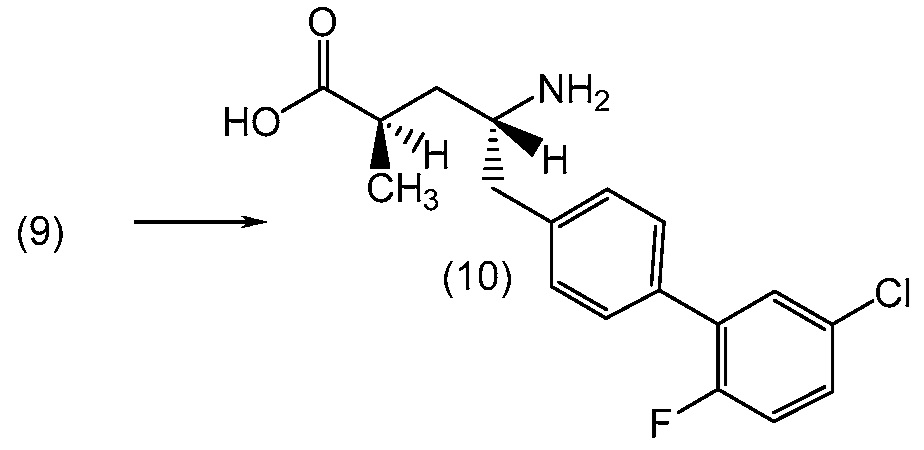
Раствор соединения 9 (3,4 г, 8,5 ммоль) в концентрированной HCl (50 мл) кипятят с обратным холодильником в течение 2 дней. Смесь затем концентрируют при пониженном давлении, и остаток очищают промыванием EtOAc (20 мл) с получением соединения 10 (2,2 г) в виде беловатого твердого вещества соли HCl. ЖХ-МС: m/z=336[(M++1)].
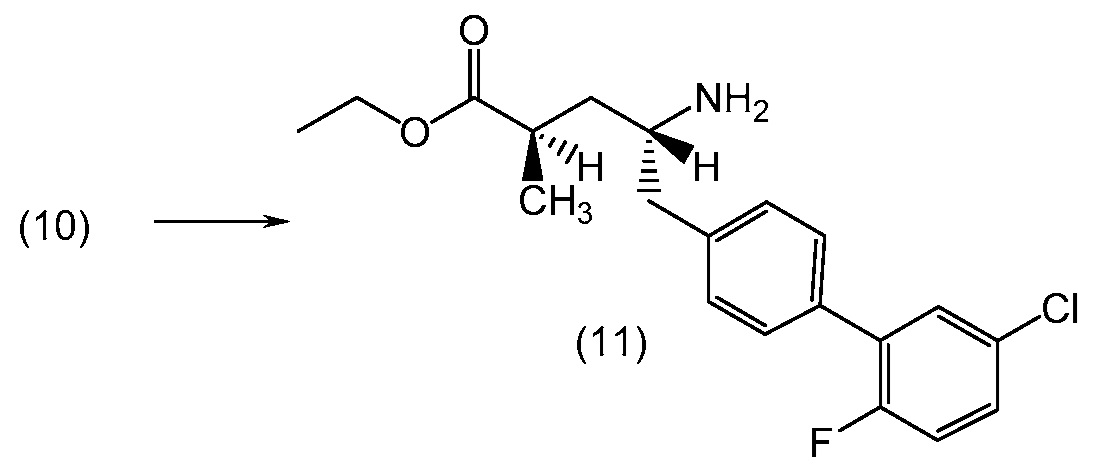
Раствор соединения 10 (2,2 г, 5,9 ммоль) в 4 M HCl в EtOH (15 мл) перемешивают при комнатной температуре в течение 2 часов. Смесь затем концентрируют при пониженном давлении, и остаток очищают промыванием EtOAc (20 мл) с получением соединения 11 (2,3 г) в виде беловатого твердого вещества. ЖХ-МС: m/z=364[(M++1)] 1H ЯМР (300 МГц, ДПВO) δ 7,61-7,52 (м, 3H), 7,47 (ддд, J=8,7, 4,3, 2,7 Гц, 1H), 7,38 (дд, J=13,5, 5,5 Гц, 3H), 3,98 (кв, J=7,1 Гц, 2H), 3,37 (д, J=11,1 Гц, 1H), 3,11 (дд, J=13,7, 5,1 Гц, 1H), 2,90-2,67 (м, 2H), 1,85 (ддд, J=14,0, 9,3, 4,8 Гц, 1H), 1,68-1,54 (м, 1H), 1,13-0,99 (м, 6H).
ПРИМЕР 3: ( 2R,4S )-5-(5'-Хлор-2'-фторбифенил-4-ил)-4-[(2-этилоксазол-5-карбонил)амино]-2-метилпентановая кислота (сравнительное соединение C3)
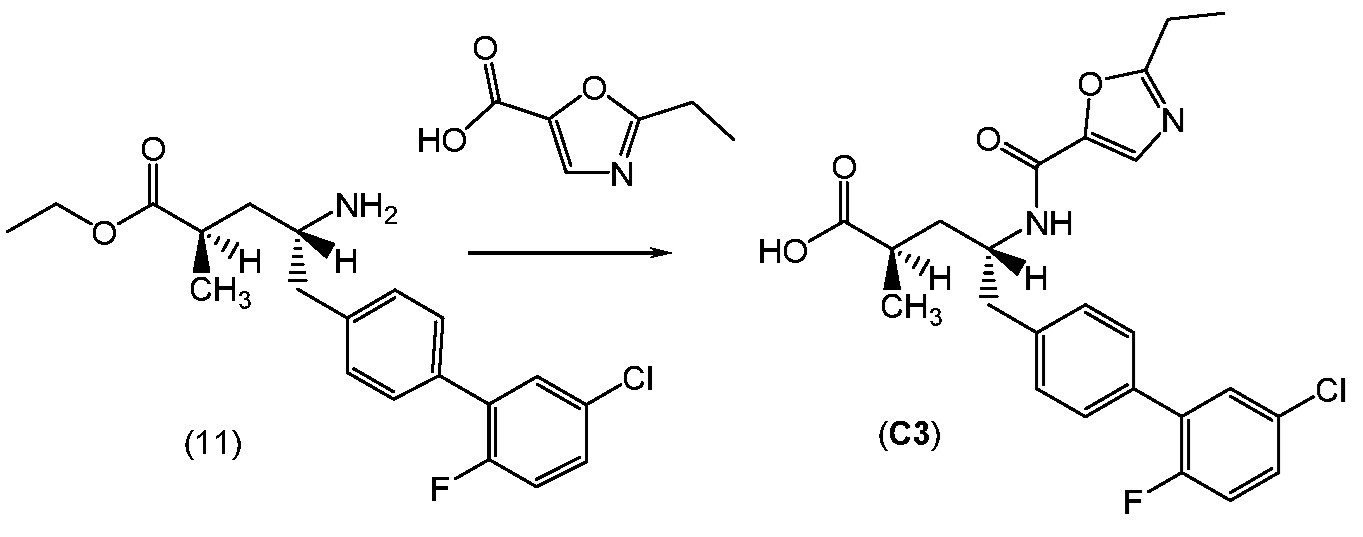
ДИПЭА (228 мкл, 1,3 ммоль) добавляют к раствору 2-этилоксазол-5-карбоновой кислоты (67,9 мг, 481 мкмоль), соединения 11 (175 мг, 437 мкмоль) и ГАТУ (249 мг, 656 мкмоль) в ДМФ (2,0 мл) и перемешивают при комнатной температуре в течение 15 минут. Добавляют 5,0 M водный LiOH (699 мкл, 3,5 ммоль) и смесь перемешивают при комнатной температуре в течение 1,5 часов. Добавляют концентрированную HCl (~0,5 мл), пока смесь не станет кислой, и неочищенную смесь очищают препаративной ВЭЖХ (20-80% MeCN в воде с 0,05% ТФК) с получением сравнительного соединения C3 (170 мг) в виде белого твердого вещества. МС m/z [M+H]+ рассч. для C24H24ClFN2O4, 459,14; найдено 459,05.
Сравнительное соединение C3 описано в примере 71-1 из патента США № 8,263,629 Coppola et al.
ПРИМЕР 4: ( 2R,4S )-5-(5'-Хлор-2'-фторбифенил-4-ил)-2-метил-4-[(оксазол-5-карбонил)амино]пентановая кислота (сравнительное соединение C4)
ДИПЭА (228 мкл, 1,3 ммоль) добавляют к раствору оксазол-5-карбоновой кислоты (49,4 мг, 437 мкмоль), соединения 11 (HCl; 175 мг, 437 мкмоль) и ГАТУ (249 мг, 656 мкмоль) в ДМФ (2 мл). Полученную смесь перемешивают в течение 10 минут при комнатной температуре, затем ЖХ/МС показывает завершение. Добавляют 5,0 M водный LiOH (699 мкл, 3,5 ммоль) и смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют концентрированную HCl (~0,5 мл), пока реакционная смесь не станет кислой, и неочищенную смесь очищают препаративной ВЭЖХ (20-80% MeCN в воде с 0,05% ТФК) с получением сравнительного соединения C4 (103 мг) в виде белого твердого вещества. МС m/z [M+H]+ рассч. для C22H20ClFN2O4, 431,11; найдено 431,1.
Сравнительное соединение C4 описано в примере 68-1 из патента США № 8,263,629 Coppola et al.
АНАЛИЗЫ
Соединение 1 и сравнительные соединения C2, C3 и C4 оценивают в описанных ниже анализах.
Анализ 1: In vitro анализ количественной оценки активности ингибитора на человеческом НЕП
Ингибирующее действие на человеческий неприлизин (EC 3,4,24,11; НЕП) определяют следующим образом.
Рекомбинантный человеческий НЕП получают коммерчески (R&D Systems, Minneapolis, MN, каталожный номер 1182-ZN). Применяют флуорогенный пептидный субстрат Mca-D-Arg-Arg-Leu-Dap-(Dnp)-OH (Medeiros et al. (1997) Braz. J. Med. Biol. Res. 30:1157-62; Anaspec, San Jose, CA).
Анализ проводят в 384-луночных белых непрозрачных планшетах при 37°C с применением флуорогенного пептидного субстрата в концентрации 10 мкМ в аналитическом буфере (50 мМ HEPES, pH 7,5, 100 мМ NaCl, 0,01% сорбитанмонолаурата полиэтиленгликоля (Tween-20), 10 мкМ ZnSO4). Фермент применяют в концентрации, которая дает количественный протеолиз 1 мкМ субстрата через 20 минут при 37°C.
Тестируемые соединения анализируют в интервале концентраций от 10 мкМ до 20 пМ. Тестируемые соединения добавляют к ферменту и инкубируют в течение 30 минут при 37°C перед началом реакции добавлением субстрата. Реакции заканчивают через 20 минут инкубирования при 37°C добавлением ледяной уксусной кислоты до конечной концентрации 3,6% (об./об.).
Планшеты считывают на флуорометре при длине волны возбуждения и испускания 320 нм. Константы ингибирования получают не линейной регрессией данных с применением уравнения (GraphPad Software, Inc., San Diego, CA):
v=v0/[1+(I/K′)]
где v является скоростью реакции, v0 является скоростью ингибированной реакции, I является концентрацией ингибитора и K′ является условной константой ингибирования.
Протестированные в этом анализе соединения имеют следующие значения pKi для человеческого НЕП.
|
Анализ 2: ВВ/ПО фармакокинетические исследование на крысах и собаках
Каждое ФК исследование на крысах или собаках начинают с получением тестируемого соединения. Подходящие массы каждого тестируемого соединения добавляют в объем носителя (например, 5% бикарбонат натрия, 5% декстрозу в H2O) так, чтобы конечная концентрация каждого соединения подходила для введения в количестве 2 мл/кг. Хотя гомогенная суспензия подходит для перорального введения, растворы для внутривенного дозирования стерильно фильтруют (0,2 мкм) до введения, чтобы удостовериться в отсутствии твердых частиц.
В исследовании на крысах, предварительно канюлированных самцов крыс Sprague-Dawley (3 на курс) в возрасте от 8 до 10 недель получают от Harlan Laboratories (Indianapolis, IN). Крысам вводят либо однократную пероральную дозу через зонд, либо однократную внутривенную (через боковую хвостовую вену) дозу тестируемого раствора. Конечная доза составляет обычно 0,5-3 мг/кг. Серийные образцы крови собирают через канюлю, имплантированную в яремную вену через 3 минуты, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов и 24 часа послед введения. Образцы собирают вручную или автоматизированными пробозаборниками. Образцы собирают в пробирки Microtainer, содержащие ЭДТК в качестве антикоагулянта и обрабатывают до плазмы центрифугированием при охлаждении.
В исследовании на собаках, самцы биглей (3 на курс), находящиеся в Agilux Laboratories (Worcester, MA) и весящие 7-12 кг получают однократную пероральную дозу через зонд или однократную внутривенную (через полостной катетер) дозу тестируемого раствора. Конечная доза обычно составляет 0,1-2 мг/кг. Серийные образцы крови собирают через венопункцию через 3 минуты, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 12 часов и 24 часа после введения. Все образцы собирают вручную в пробирки Microtainer, содержащие ЭДТК в качестве антикоагулянта и обрабатывают до плазмы центрифугированием при охлаждении.
Образцы плазмы экстрагируют 3 объемами MeCN, содержащего подходящий внутренний стандарт. Экстракты восстанавливают в 3 объемах воды, содержащей 1% муравьиную кислоту и анализируют ВЭЖХ-объединенным МС/МС. Данные концентрация плазмы-время анализируют с применением программы Phoenix (Pharsight Corp., St. Louis, MO) для расчета фармакокинетических параметров.
Клиренс плазмы определяют из внутривенной части анализа, и он представляет скорость, при которой плазма очищается от лекарственного средства. Она равна дозе, деленной на площадь под кривой концентрация в плазме-время. Кроме клиренса плазмы, для перорально вводимого лекарственного средства также необходимо, чтобы оно достигало эффективных системных уровней после перорального введения. Пероральная биодоступность является показателем воздействия на плазму после перорального введения по отношению к воздействию после внутривенного введения.
Фармакокинетические данные у крыс
|
a ППКпосл является площадью под кривой концентрации в плазме ко времени от времени 0 до времени после введения, в которое определяется последняя измеримая концентрация, оцениваемая линейным методом трапеций
b КЛпосл является дозой, деленной на ППКпосл
c Пероральную биодоступность рассчитывают как ППКпосл после перорального введения, деленная на ППКпосл после внутривенного введения, нормализованная для любых разниц в вводимой дозе, выражают как процент.
Эти данные на крысах показали, что соединение в соответствии с данным изобретением (соединение 1) имеет высокую пероральную биодоступность (67%). Сравнительные соединения C3 и C4 демонстрируют похожую пероральную биодоступность (60% и 60%, соответственно). Однако сравнительное соединение C2 имеет низкую пероральную биодоступность (20%). Эти данные на крысах также показывают, что соединение 1 имеет похожую низкую скорость клиренса по сравнению со сравнительными соединениями C3 и C4 (0,83, 0,60 и 0,67, соответственно). Однако сравнительное соединение C2 очищается намного скорее (средняя КЛпосл 1,7).
Для получения более предсказуемого определения того, как соединение будет вести себя у человека (например, безопасность и эффективность), проводят оценку на втором виде животных.
Фармакокинетические данные на собаках
|
*Больше чем 100% биодоступность может относиться к абсорбции, распределению и/или выведению.
Эти данные на собаках показывают, что соединение в соответствии с данным изобретением (соединение 1) имеет высокую пероральную биодоступность (>100%) и низкий клиренс плазмы. Хотя сравнительное соединение C2 демонстрирует подобную пероральную биодоступность (99%), сравнительные соединения C3 и C4 демонстрируют значительно меньшую пероральную биодоступность (64% и 42%, соответственно). Дополнительно, эти данные на собаках также показывают, что соединение 1 имеет низкую скорость клиренса (0,29) по сравнению со всеми сравнительными соединениями C2, C3 и C4 (0,65, 0,59 и 1,54, соответственно).
В заключение, соединение 1 последовательно демонстрирует высокую пероральную биодоступность и низкий клиренс плазмы у крыс и у собак. Наоборот, сравнительное соединение C2 демонстрирует более низкую биодоступность у крыс, чем соединение 1, и более высокий клиренс плазмы у крыс и собак, чем соединение 1. Сравнительные соединения C3 и C4, хотя и сравнимы по пероральной биодоступности и клиренсу с соединением 1 у крыс, демонстрируют более низкую пероральную биодоступность и высокий клиренс, чем соединение 1, у собак.
Анализ 3: Фармакокинетическое/фармакодинамическое (ФК-ФД) исследование активности к НЕП у пребывающих в сознании нормотензивных крыс Sprague-Dawley
Подходящие массы каждого тестируемого соединения добавляют в объем носителя (например, 5% бикарбонат натрия, 5% декстрозу в H2O) так, чтобы конечная концентрация каждого соединения подходила для введения в дозе 2 мл/кг. Обратите внимание, что гомогенная суспензия считается приемлемой для перорального введения.
Предварительно канюлированных самцов крыс Sprague-Dawley (3 на оцениваемое соединение) в возрасте от 8 до 10 недель получают от Harlan Laboratories (Indianapolis, IN). Крысы получают однократную пероральную дозу вводимого раствора через зонд. Конечная доза обычно составляет 3-30 мг/кг. Серийные образцы крови собирают через канюлю, имплантированную в яремную вену через 3 минуты, 15 минут, 35 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов и 24 часа после введения. Образцы собирают в пробирки Microtainer, содержащие ЭДТК в качестве антикоагулянта и обрабатывают до плазмы центрифугированием с охлаждением. За пятнадцать минут до забора образцов в 45 минут, 2 часа, 4 часа, 6 часов и 24 часа вводят ВВ болюс (30 мг/кг) предсердного натрийуретического пептида (ПНП). Если тестируемое соединение успешно ингибирует неприлизин, один из ключевых медиаторов клиренса ПНП, введенный ПНП будет увеличивать базовые уровни ПНП, тем самым потенцируя подачу нисходящего сигнала. Если тестируемое соединение не ингибирует успешно неприлизин, уровни циркулирования ПНП и нисходящий каскад сигнала останется на базовом уровне в течение 15 минут между введением ПНП болюса и моментом времени, в который берут образец плазмы. Так как связывание ПНП с его рецептором приводит к активации гуанилилциклазы и последующему образованию циклического гуанозинмонофосфата (цГМФ), повышенные уровни цГМФ (>20 нМ) в плазме через 15 минут после ВВ введения ПНП болюса интерпретируется как доказательство происходящего ингибирования неприлизина. Повышенные уровни в плазме цГМФ через 24 часа после введения являются показателем длительность фармакологического действия, аналогично, значения клиренса плазмы являются показателем фармакокинетической стойкости.
Образцы плазмы экстрагируют 3 объемами MeCN, содержащего подходящий внутренний стандарт. Экстракты восстанавливают в 3 объемах воды, содержащей 1% муравьиной кислоты. цГМФ в плазме и концентрации тестируемого соединения количественно оценивают в образцах плазмы ВЭЖХ-объединенным масс-спектрометрическим определением.
|
a Средняя для трех определений
Таким образом, соединение в соответствии с данным изобретением (соединение 1) демонстрирует более чем 2-кратное потенцирование цГМФ через 24 ч после введения по сравнению с соединениями известного уровня техники C2, C3 и C4.
Анализ 4: ВВ/ПО фармакокинетическое исследование соединения 1 на обезьянах
ФК исследование проводят с подходящей массой соединения 1 в объеме носителя (например, 5% бикарбоната натрия, 5% декстрозы в H2O, pH 7,4, для перорального и ПЭГ200:EtOH:вода (40:10:50), pH 7,0, отфильтрован через 0,22 мкМ μM шприц PDVF для ВВ) так, чтобы конечная концентрация составляла 0,5 мл/кг (ВВ) или 2,0 мл/кг (ПО).
Самцы яванских макаков (3 на курс), размещенные в Xenometrics (Stilwell, KS), получают ВВ или ПО дозу соединения 1 в количестве 1 мг/кг. Животные, которым вводят ВВ дозы, имеют доступ к пище без ограничений, и животные, которым вводят ПО дозы, голодают в течение ночи и получают пищу приблизительно через 4 часа после введения дозы. Образцы крови берут у каждого животного в каждый момент времени (перед введением, 0,083 часа, 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 12 часов и 24 часа) через головную, бедренную или клиновидную вену через 48 часов. Все образцы собирают в пробирки K2EDTA и помещают на лед. Образцы обрабатывают до плазмы центрифугированием (3200 об./мин, 10 минут, 5°C) и подкисляют до конечной концентрации 2% уксусной кислоты. Аликвоты плазмы переносят в пробирки 96-луночного планшета и хранят замороженными (-70°C) до биоанализа.
Концентрации соединения 1 в плазме определяют ЖХ/МС/МС. Образцы для исследования плазмы встряхивают и помещают в 96-луночный планшет. Образцы экстрагируют 200 мкл 0,2% муравьиной кислоты в ацетонитриле с внутренним стандартом. Экстракт центрифугируют в течение 10 минут при 3700 об./мин. и надосадочную жидкость смешивают с 0,2% муравьиной кислотой в воде. Образцы (15 мкл) вводят в колонку Thermo (C18 50×2,1 мм) со скоростью потока 0,3 мл/мин. Подвижная фаза A состоит из 0,2% муравьиной кислоты в воде, и подвижная фаза B состоит из 0,2% муравьиной кислоты в ацетонитриле. Градиентное элюирование начинают с от 0% до 95% B от 0,5 до 1,5 мин, хранят при 95% от 1,5 до 1,85 мин, затем градиент от 95% до 0% B от 1,85 до 1,86 мин и останавливают в 2,6 мин. Интервал исследования соединения 1 составляет от 0,0005 до 5 мкг/мл. Фармакокинетические параметры соединения 1 определяют некомпартментным анализом (Model 201 и Model 200 для ВВ и ПО введения, соответственно) с применением Phoenix WinNonlin Version 6,3 (Certara, Sunnyvale, CA) и с применением индивидуальных профилей концентрация в плазме-время для 3 животных.
Клиренс плазмы определяют из внутривенной части исследования, и он представляет скорость, с которой плазма очищается от лекарственного средства. Она равна дозе, деленной на площадь под кривой концентрация в плазме-время. Кроме клиренса плазмы, для перорально вводимого лекарственного средства также необходимо, чтобы оно достигало эффективных системных уровней после перорального введения. Пероральная биодоступность является показателем воздействия на плазму после перорального введения по отношению к воздействию после внутривенного введения.
Фармакокинетические данные на обезьянах
|
Эти данные показывают, что соединение в соответствии с данным изобретением (соединение 1) имеет высокую пероральную биодоступность (приблизительно 60%) и низкий клиренс плазмы (приблизительно 0,36) у самцов яванских макаков. Это согласуется с и похоже на данные клиренса, полученные для крыс и собак.
Анализ 5: Выведение из почек соединения 1 у крыс, собак и обезьян
Важным фактором обеспечивания подходящего долговременного введения лекарственного средства и корректного гомеостаза концентраций лекарственного средства у пациентов является выведение лекарственного средства. В общем, пониженное выведение лекарственного средства приводит к более высоким концентрациям лекарственного средства и большему воздействию лекарственного средства. Для понимания почечного клиренса соединения 1, процент введенной дозы, присутствующий в моче после однократного ВВ введения, оценивают на трех видах животных. Проводят три отдельных исследования на самцах крыс Sprague Dawley, самцах биглей и самцах яванских макаков, соответственно, и методика и результаты эксперимента представлены ниже.
Самца крыс Sprague Dawley rats (N=3), массой тела от 0,348 до 0,362 кг, получают ВВ дозу соединения 1 в количестве 0,5 мг/кг как часть дозирующей кассеты. Соединение 1 растворяют в 5% NaHCO3 в D5W (5% декстроза в воде, pH 7,4) и фильтруют через 0,22 мкМ шприцевой фильтр из поливинилдифторида (ПВДФ) перед введением. Крысы имеют неограниченный доступ к пище до и после введения соединения 1. Образцы мочи собирают в метаболические клетки и выдерживают на замороженном сухом льду во время сбора. Образцы оттаивают, и объем мочи записывают. Аликвоту образца мочи переносят в полипропиленовую пробирку для хранения, замораживают и хранят (-80°C) до биоанализа.
Концентрации соединения 1 в моче крыс определяют ЖХ/МС/МС. Образцы мочи оттаивают и разводят 5-кратно в крысиной K2EDTA плазме. 50 мкл аликвоту разведенной мочи переносят в 96-луночный планшет и экстрагируют 200 мкл объемом 2% муравьиной кислоты в ацетонитриле, содержащей внутренний стандарт. 96-луночный планшет центрифугируют в течение 10 минут при 3700 об./мин., и надосадочную жидкость переносят в новый 96-луночный планшет. Надосадочную жидкость разводят в 0,2% муравьиной кислоте в воде (4-кратное разведение). 10 или 20 мкл объем впрыскивают в колонку Xbridge phenyl (21×50 мм; 5 мк). Подвижная фаза A состоит из 0,2% муравьиной кислоты в воде и подвижная фаза B состоит из 0,2% муравьиной кислоты в ацетонитриле. Градиентное элюирование начинают с от 20% до 95% B от 0,5 до 2,0 мин, выдерживают при 95% от 2,0 до 2,2 мин., затем градиент от 95% до 20% B от 2,2 до 2,3 мин. и останавливают в 3,3 мин. Диапазон результатов количественного анализа соединения 1 составляет от 0,00125 до 25 мкг/мл.
Самцам биглей (N=6, две группы по 3), массой 9,04-10,2 кг и 10,8-11,5 кг, вводят ВВ дозу соединения 1 в количестве 0,1 мг/кг (группа I) и 1,255 мг/кг (группа II) как часть дозирующей кассеты. Соединение 1 растворяют в ПЭГ-200:этаноле:воде (40:10:50) и фильтруют через 0,22 мкМ шприцевой фильтр из поливинилдифторида (ПВДФ) перед введением. Собаки имеют неограниченный доступ к пище до и после введения соединения 1. Образцы мочи собирают в гипотермические пакеты в заранее взвешенные контейнеры, заполненные 0,5 мл ледяной уксусной кислоты. Образцы взвешивают снова и добавляют еще ледяную уксусную кислоту, при необходимости, до конечной концентрации 2%. Образцы замораживают и хранят (-80°C) до биоанализа.
Концентрации соединения 1 в моче собак определяют ЖХ/МС/МС. Исследуемые образцы мочи (разведенные в плазме K2EDTA (1:9)) встряхивают и 25 мкл помещают в 96-луночный планшет. Образцы экстрагируют 100 мкл в ацетонитриле с внутренним стандартом Глибурид. Экстракт центрифугируют в течение 5 минут при 3100 об./мин. и 75 мкл надосадочной жидкости переносят и смешивают с 150 мкл воды. Образцы (12 мкл) впрыскивают в колонку Waters Acquity UPLC BEH C18 (50×2,1 мм, 1,7 мкм) со скоростью потока 0,9 мл/мин. Подвижная фаза A состоит из 95:5:0,1 (об.:об.:об.) воды:ацетонитрила:муравьиной кислоты и подвижная фаза B состоит из 50:50:0,1 (об.:об.:об.) метанола:ацетонитрила:муравьиной кислоты. Градиентное элюирование начинают с от 35% до 90% B от 0,2 до 1,6 мин, выдерживают при 95% от 1,7 до 2,2 мин., затем стадия от 95% до 35% B от 2,20 до 2,30 мин. min. Диапазон результатов количественного анализа соединения 1 составляет от 0,000100 до 1,00 мкг/мл.
Самцам яванских макаков (N=3), массой 4,42-5,81 кг, вводят ВВ дозу соединения 1 в количестве 1 мг/кг. Соединение 1 растворяют в ПЭГ-200:этаноле:воде (40:10:50) и фильтруют через 0,22 мкМ шприцевой фильтр из поливинилдифторида (ПВДФ) перед введением. Обезьяны имеют неограниченный доступ к пище до и после введения соединения 1. Образцы мочи собирают на сухой лед в периоды сбора, объемы образцов записывают, и мочу подкисляют уксусной кислотой до конечной концентрации приблизительно 2%. Аликвоты получают и замораживают (-70°C) до биоанализа.
Концентрации соединения 1 в моче обезьян определяют ЖХ/МС/МС. Образцы мочи оттаивают и разводят в K2EDTA плазме (1:4). Образцы экстрагируют 200 мкл 2% муравьиной кислоты в ацетонитриле с внутренним стандартом. Экстракт центрифугируют в течение 10 минут при 3700 об./мин., и надосадочную жидкость смешивают с 0,2% муравьиной кислотой в воде. Образцы (15 мкл) впрыскивают в колонку Thermo (C18 50×2,1 мм) со скоростью потока 0,3 мл/мин. Подвижная фаза A состоит из 0,2% муравьиной кислоты в воде и подвижная фаза B состоит из 0,2% муравьиной кислоты в ацетонитриле. Градиентное элюирование начинают с от 0% до 95% B от 0,5 до 1,5 мин, выдерживают при 95% от 1,5 до 1,85 мин., затем градиент от 95% до 0% B от 1,85 до 1,86 мин. и останавливают в 2,6 мин. Диапазон результатов количественного анализа соединения 1 составляет от 0,0025 до 25 мкг/мл.
Среднее количество мочи, выведенное в течение периода сбора 24 часа и приблизительной % от введенной дозы, выведенный в моче, представлены в таблице.
|
a Среднее для трех измерений
Выведение почками соединения 1 у крыс составляет приблизительно 0,03% от введенной дозы, у собак приблизительно от 1 до 1,5% от введенной дозы и у обезьян приблизительно 3% от введенной дозы. Эти данные показывают, что соединение 1 плохо выводится почками у трех тестированных видов.
![(2R,4R)-5-(5′-ХЛОР-2′-ФТОРБИФЕНИЛ-4-ИЛ)-2-ГИДРОКСИ-4-[(5-МЕТИЛОКСАЗОЛ-2-КАРБОНИЛ)АМИНО]ПЕНТАНОВАЯ КИСЛОТА](https://fips.edrid.ru/images/rid/6e/31/15/861df34641e0a2e2200146c1d48e4785.jpg)
![(2R,4R)-5-(5′-ХЛОР-2′-ФТОРБИФЕНИЛ-4-ИЛ)-2-ГИДРОКСИ-4-[(5-МЕТИЛОКСАЗОЛ-2-КАРБОНИЛ)АМИНО]ПЕНТАНОВАЯ КИСЛОТА](https://fips.edrid.ru/images/rid/6e/31/15/8e1c918bb4fc4441e8645a84f23358c4.jpg)
![(2R,4R)-5-(5′-ХЛОР-2′-ФТОРБИФЕНИЛ-4-ИЛ)-2-ГИДРОКСИ-4-[(5-МЕТИЛОКСАЗОЛ-2-КАРБОНИЛ)АМИНО]ПЕНТАНОВАЯ КИСЛОТА](https://fips.edrid.ru/images/rid/6e/31/15/6710ac27d1d5df024338290fad60d349.jpg)
![(2R,4R)-5-(5′-ХЛОР-2′-ФТОРБИФЕНИЛ-4-ИЛ)-2-ГИДРОКСИ-4-[(5-МЕТИЛОКСАЗОЛ-2-КАРБОНИЛ)АМИНО]ПЕНТАНОВАЯ КИСЛОТА](https://fips.edrid.ru/images/rid/6e/31/15/c5fe088cc305c7c904f5227c1038eb7c.jpg)
![(2R,4R)-5-(5′-ХЛОР-2′-ФТОРБИФЕНИЛ-4-ИЛ)-2-ГИДРОКСИ-4-[(5-МЕТИЛОКСАЗОЛ-2-КАРБОНИЛ)АМИНО]ПЕНТАНОВАЯ КИСЛОТА](https://fips.edrid.ru/images/rid/6e/31/15/21c6d91332e65e69aaa31f660f70b70d.jpg)
Соединения-агонисты рецептора 5-нт для лечения расстройств познавательной способности
Ингибиторы вируса гепатита с, имеющие жесткую вытянутую цепь и содержащие фрагмент { 2-[4-(бифенил-4-ил)-1н-имидазо-2-ил]пирролидин-1-карбонилметил} амина
Ингибиторы неприлизина
Ингибиторы неприлизина
Соединение для применения в лечении нейрогенной ортостатической гипотензии
(2s,4r)-5-(5'-хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксиметил-2-метилпентановая кислота
Способы лечения с использованием соединения-ингибитора jak
Производные 3-карбоксипропил-аминотетралина и родственные соединения в качестве антагонистов mu-опиоидного рецептора
Кристаллическая форма 4-[2-(2-фторфеноксиметил)фенил]пиперидинового соединения
Кристаллические формы соединения 3-карбоксипропил-аминотетралина
Диамидные соединения, обладающие активностью антагониста мускаринового рецептора и активностью агониста в2 адренергического рецептора
Замещенные аминомасляные производные в качестве ингибиторов неприлизина
Ингибиторы неприлизина
Ингибиторы неприлизина
Ингибиторы неприлизина
Ингибиторы неприлизина
Ингибиторы неприлизина

