Результат интеллектуальной деятельности: ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ
Вид РИД
Изобретение
[001] Ангиогенез представляет собой процесс, в ходе которого из существующей сосудистой сети могут вырастать новые кровеносные сосуды. Этот процесс может происходить при заживлении ран, например, при восстановлении кровотока при повреждении тканей, например, при повреждении руки. Однако избыточный ангиогенез может быть инициирован при специфических патологических состояниях, например, при опухолях, ВМД (возрастной макулярной дегенерации), ревматоидном артрите, псориазе и т.д. В таких обстоятельствах может существовать тенденция к нежелательному образованию новых кровеносных сосудов с возникновением патологических тканей, которые питаются и повреждают нормальные ткани. Например, раковые клетки могут попадать в кровоток посредством новых кровеносных сосудов и проникать в нормальные ткани.
[002] VEGF (фактор роста эндотелия сосудов) и его рецептор VEGFR-2 (также называемый KDR, рецептор, содержащий домен вставки киназы) могут образовывать основной путь для образования новых кровеносных сосудов. Было показано, что ингибирование KDR может вызвать апоптоз эндотелиальных клеток, что в последующем блокирует процесс ангиогенеза (Rubin М. Tuder, Chest, 2000; 117: 281). Таким образом, ингибиторы KDR можно применять для лечения заболеваний, связанных с ангиогенезом.
[003] FGF (фактор роста фибробластов) представляет собой проангиогенетическую молекулу, также как и VEGF. В ходе ангиогенеза, считается, что VEGF является критическим для процесса неоваскуляризации. Ось FGF (фактор роста фибробластов)/FGFR (рецептор фактора роста фибробластов) играет роль в функциональном созревании новообразованных сосудов. Кроме того, аберрантная активация членов семейства FGF и их родственных рецепторов была обнаружена при нескольких видах рака, таких как рак молочной железы, рак мочевого пузыря и рак предстательной железы. Также повышен FGFR1 и его связывающие партнеры FGF1, FGF2, FGF8b и FGF 17. В других типах опухолей FGFR1 участвует в качестве онкогена, экспрессия которого увеличивается по сравнению с нормальной тканью. Следовательно, блокада сигналов FGF/FGFR может быть полезной для лечения рака, связанного с активацией FGF/FGFR.
[004] Нейроэндокринные опухоли (НЭО) представляют собой редкий рак гормональной системы, обычно медленнорастущий. НЭО относятся к нейроэндокринным новообразованиям (НЭН), которые представляют собой редкие опухоли, возникающие из эмбриологических нейроэндокринных клеток, которые имеют нейроэндокринные биомаркеры и могут продуцировать полипептидные гормоны. Поскольку НЭН имеют длительный естественный курс развития заболевания с относительно высоким показателем пятилетней выживаемости по сравнению с другими опухолями, существует большое количество выживших пациентов с НЭН. НЭН возникает в различных органах и тканях по всему телу.
[005] По классификации Всемирной организации здравоохранения (ВОЗ) выделают три основных типа НЭН: малой степени (G1), средней степени (G2) и высокой степени (G3) (G3 также известна как нейроэндокринная карцинома, НЭК). Первые два типа (G1 и G2) в совокупности называются НЭО. НЭН показывают скорость митоза (митотический индекс ≤20/10 НРБ) и/или индекс пролиферации Ki67 (≤20%) опухолевых клеток. В целом, чем выше степень (т.е. чем выше показатель митоза и индекс пролиферации Ki-67), тем более агрессивной является опухоль и тем хуже прогноз для пациента. Хотя НЭН малой и средней степеней показывают медленную прогрессию опухоли, а ранние НЭН можно успешно лечить хирургическим вмешательством, пациенты с продвинутыми НЭН не чувствительны к химиотерапии и варианты лечения для них очень ограничены. Таким образом, НЭН высоких степеней представляют собой тип опухоли, трудно поддающийся лечению.
[006] НЭН, возникающие в поджелудочной железе, можно отличить от тех, которые возникают в других местах желудочно-кишечного тракта. Хотя первоначально предполагалось, что панкреатические нейроэндокринные опухоли (ПНЭО) возникают из островковых клеток поджелудочной железы, недавняя работа подтверждает мнение о том, что ПНЭО возникают из стволоподобной не островковой протоковой прогениторной клетки, что также подтверждает переход в номенклатуре от рака островковых клеток к панкреатической нейроэндокринной опухоли. В настоящее время лекарственные средства для терапии ПНЭО включают соматостатин, интерферон, химиотерапевтические препараты и молекулярные лекарственные средства направленного действия (такие как сунитиниб (Sunitinib) и эверолимус (Everolimus)). Пациенты с ПНЭО не чувствительны к традиционной химиотерапии, и было доказано, что только две целевые терапии продлевают выживаемость без прогрессирования (PFS) для пациентов с ПНЭО высоких степеней.
[007] Экстрапанкреатические нейроэндокринные новообразования (экстрапанкреатические НЭО) зарождаются в органах или тканях, отличных от поджелудочной железы, из которых наиболее распространенными являются желудочно-кишечные нейроэндокринные опухоли (ЖК-НЭО), обнаруживаемые в желудке, двенадцатиперстной кишке, тонком кишечнике, аппендиксе, слепой кишке, толстой кишке и прямой кишке. В настоящее время только аналоги соматостатина длительного действия (SSA, включая октреотид и ланреотид) получили одобрение Управления по контролю качества пищевых продуктов и лекарственных средств США (FDA) для применения при лечении экстрапанкреатических НЭО. Однако из-за своей низкой противоопухолевой активности SSA в основном применяют в клинических исследованиях, в которых участвуют пациенты с опухолями класса G1, поскольку такие опухоли имеют лучший прогноз и медленный темп роста.
[008] Соединение формулы А (термины «соединение А» и «соединение формулы А» используются в настоящем документе взаимозаменяемо), т.е. N-(2-(диметиламино)этил)-1-(3-((4-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)-метансульфонамид и/или его фармацевтически приемлемая соль описаны в заявке на патент США №13/510249 (заявка '249), которая является национальным этапом для заявки PCT/CN 2010/078997, поданной 23 ноября 2010 года, по которой в настоящее время выдан патент США №8658658 (патент '658). Патент '658 включен в настоящее описание посредством ссылки во всей его полноте.

N-(2-(диметиламино)этил)-1-(3-((4-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)метансульфонамид
[009] Твердотельные кристаллические формы I и II соединения формулы А, то есть форма I N-(2-(диметиламино)этил)-1-(3-((4-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)-метансульфонамида и форма II N-(2-(диметиламино)этил)-1-(3-((4-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)-метансульфонамида, и способы их получения были описаны в патенте '658.
[010] В настоящем документе описан способ лечения субъекта, для которого признана необходимость лечения НЭО, включающий введение указанному субъекту, нуждающемуся в этом, эффективного количества соединения формулы А и/или его фармацевтически приемлемой соли. В некоторых вариантах реализации указанное соединение формулы А представляет собой форму I. В некоторых вариантах реализации указанное соединение формулы А представляет собой по существу чистую форму I. В некоторых вариантах реализации указанное соединение формулы А представляет собой форму II. В некоторых вариантах реализации указанное соединение формулы А представляет собой по существу чистую форму II. В некоторых вариантах реализации указанное соединение формулы А и/или его фармацевтически приемлемую соль микронизируют с величиной D90, составляющей менее или равной 20,0 мкм, например, от 1,0 до 20,0 мкм, или в диапазоне от 2,0 до 12,0 мкм. В некоторых вариантах реализации форму I, по существу чистую форму I, форму II или по существу чистую форму II микронизируют с величиной D90, составляющей менее или равной 20,0 мкм, например, в диапазоне от 1,0 до 20,0 мкм или в диапазоне от 2,0 до 12,0 мкм. В некоторых вариантах реализации значение D90 составляет от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации значение D90 составляет от 2,0 до 5,0 мкм, например, значение D90 составляет 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации значение D90 составляет от 9,0 до 12,0 мкм, например, значение D90 составляет 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 составляет менее или равно 11,0 мкм или составляет менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 составляет менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 составляет менее или равно 4,0 мкм.
[011] В некоторых вариантах реализации указанные нейроэндокринные опухоли представляют собой панкреатические нейроэндокринные опухоли. В некоторых вариантах реализации указанные нейроэндокринные опухоли представляют собой экстрапанкреатические нейроэндокринные опухоли. В некоторых вариантах реализации указанные нейроэндокринные опухоли представляют собой желудочно-кишечные нейроэндокринные опухоли.
[012] Также в настоящем документе раскрыт способ лечения субъекта, для которого признана необходимость лечения НЭО, включающий введение указанному субъекту, нуждающемуся в этом, эффективного количества фармацевтической композиции, содержащей
по меньшей мере одно фармацевтически приемлемое вспомогательное вещество и
по меньшей мере один активный ингредиент, выбранный из соединения А и его фармацевтически приемлемых солей. В некоторых вариантах реализации по меньшей мере один активный ингредиент представляет собой соединение А. В некоторых вариантах реализации соединение А представляет собой форму I. В некоторых вариантах реализации соединение А представляет собой по существу чистую форму I. В некоторых вариантах реализации соединение А представляет собой форму II. В некоторых вариантах реализации соединение формулы А представляет собой по существу чистую форму П. В некоторых вариантах реализации соединение А, форму I, по существу чистую форму I, форму II или по существу чистую форму II микронизируют с величиной D90, составляющей менее или равной 20,0 мкм, например, в диапазоне от 1,0 до 20,0 мкм или в диапазоне от 2,0 до 12,0 мкм. В некоторых вариантах реализации значение D90 составляет от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации значение D90 составляет от 2,0 до 5,0 мкм, например, значение D90 составляет 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации значение D90 составляет от 9,0 до 12,0 мкм, например, значение D90 составляет 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 составляет менее или равно 11,0 мкм или составляет менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 составляет менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 составляет менее или равно 4,0 мкм.
[013] Также в настоящем документе описана первая фармацевтическая композиция, содержащая
микронизированное соединение А и/или
микронизированную по меньшей мере одну фармацевтически приемлемую соль соединения А, и
по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
В некоторых вариантах реализации указанное микронизированное соединение А и/или указанная микронизированная по меньшей мере одна фармацевтически приемлемая соль соединения А имеет величину D90, составляющую менее или равную 20,0 мкм, например, в диапазоне от 1,0 до 20,0 мкм, или в диапазоне от 2,0 до 12,0 мкм. В некоторых вариантах реализации значение D90 составляет от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации значение D90 составляет от 2,0 до 5,0 мкм, например, значение D90 составляет 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации значение D90 составляет от 9,0 до 12,0 мкм, например, значение D90 составляет 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 составляет менее или равно 11,0 мкм или составляет менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 составляет менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 составляет менее или равно 4,0 мкм.
[014] В некоторых вариантах реализации указанной первой фармацевтической композиции указанное микронизированное соединение А представляет собой микронизированную форму I. В некоторых вариантах реализации указанной первой фармацевтической композиции указанное микронизированное соединение А представляет собой микронизированную по существу чистую форму I.
[015] В некоторых вариантах реализации указанной первой фармацевтической композиции указанное микронизированное соединение А представляет собой микронизированную форму II. В некоторых вариантах реализации указанной первой фармацевтической композиции указанное микронизированное соединение А представляет собой микронизированную по существу чистую форму II.
[016] В некоторых вариантах реализации указанной первой фармацевтической композиции указанная микронизированная форма I или микронизированная по существу чистая форма I имеет значение D90, составляющее менее или равное 20,0 мкм, например, в диапазоне от 1,0 до 20,0 мкм или в диапазоне от 2,0 до 12,0 мкм. В некоторых вариантах реализации указанной первой фармацевтической композиции указанная микронизированная форма I или микронизированная по существу чистая форма I имеет значение D90 в диапазоне от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации первой фармацевтической композиции указанная микронизированная форма I или микронизированная по существу чистая форма I имеет значение D90 в диапазоне от 2,0 до 5,0 мкм, например, имеет значение D90 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации первой фармацевтической композиции указанная микронизированная форма I или микронизированная по существу чистая форма I имеет значение D90 в диапазоне от 9,0 до 12,0 мкм, например, имеет значение D90 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 менее или равно 11,0 мкм или менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 4,0 мкм.
[017] В некоторых вариантах реализации указанной первой фармацевтической композиции указанная микронизированная форма II или микронизированная по существу чистая форма II имеет значение D90 менее или равное 20,0 мкм, например, в диапазоне от 1,0 до 20,0 мкм или в диапазоне от 2,0 до 12,0 мкм. В некоторых вариантах реализации указанной первой фармацевтической композиции указанная микронизированная форма II или микронизированная по существу чистая форма II имеет значение D90 в диапазоне от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации первой фармацевтической композиции указанная микронизированная форма II или микронизированная по существу чистая форма II имеет значение D90 в диапазоне от 2,0 до 5,0 мкм, например, имеет значение D90 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации первой фармацевтической композиции указанная микронизированная форма II или микронизированная по существу чистая форма II имеет значение D90 в диапазоне от 9,0 до 12,0 мкм, например, имеет значение D90 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 менее или равно 11,0 мкм или менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 4,0 мкм.
[018] В некоторых вариантах реализации указанной первой фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из разбавителей (таких как маннит, микрокристаллическая целлюлоза, крахмал, лактоза, декстрин, сорбит), разрыхлителей (таких как крахмал гликолят натрия), грануляционных связующих (таких как поливинилпирролидон (ПВП)) и скользящих веществ (например, диоксида кремния и стеарата магния). В некоторых вариантах реализации указанный разбавитель может присутствовать в количестве от примерно 35% до примерно 90% по массе композиции. В некоторых вариантах реализации указанный разбавитель может присутствовать в количестве от примерно 0,1% до примерно 5% по массе композиции. В некоторых вариантах реализации указанный разбавитель может присутствовать в количестве от примерно 0,5% до примерно 10% по массе композиции. В некоторых вариантах реализации указанные связующие для грануляции могут присутствовать в количестве от примерно 0,5% до примерно 5% по массе композиции. В некоторых вариантах реализации первой фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из маннита, микрокристаллической целлюлозы, крахмал гликолята натрия, поливинилпирролидона (ПВП) и стеарата магния. В некоторых вариантах реализации первой фармацевтической композиции по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из микрокристаллической целлюлозы, крахмал гликолята натрия, диоксида кремния и стеарата магния. В некоторых вариантах реализации первой фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из микрокристаллической целлюлозы, крахмал гликолята натрия и стеарата магния. В некоторых вариантах реализации первой фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из микрокристаллической целлюлозы и стеарата магния.
[019] В некоторых вариантах реализации первой фармацевтической композиции указанная фармацевтическая композиция находится в форме таблетки или капсулы, при этом указанное микронизированное соединение А, такое как микронизированная форма I/по существу чистая форма I или микронизированная форма II/по существу чистая форма II и/или микронизированная по меньшей мере одна фармацевтически приемлемая соль указанного соединения может присутствовать в количестве 1, 5, 10, 15, 20, 25, 50, 75, 80, 85, 90, 95, 100, 125, 150, 200, 250, 300, 400 и 500 мг в таблетке или капсуле, например, в капсуле.
[020] Также в настоящем документе описана вторая фармацевтическая композиция, содержащая
микронизированное соединение А и/или
по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
В некоторых вариантах реализации распределение частиц по размерам (PSD) микронизированного соединения A D90 меньше или равно 20,0 мкм, например, от 1,0 до 20,0 мкм или от 2,0 до 12,0 мкм. В некоторых вариантах реализации значение D90 составляет от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации значение D90 составляет от 2,0 до 5,0 мкм, например, значение D90 составляет 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации значение D90 составляет от 9,0 до 12,0 мкм, например, значение D90 составляет 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 менее или равно 11,0 мкм или менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 4,0 мкм.
[021] В некоторых вариантах реализации указанной второй фармацевтической композиции указанное микронизированное соединение А представляет собой микронизированную форму I. В некоторых вариантах реализации указанной второй фармацевтической композиции указанное микронизированное соединение А представляет собой микронизированную по существу чистую форму I.
[022] В некоторых вариантах реализации указанной второй фармацевтической композиции указанное микронизированное соединение А представляет собой микронизированную форму И. В некоторых вариантах реализации указанной второй фармацевтической композиции указанное микронизированное соединение А представляет собой микронизированную по существу чистую форму II.
[023] В некоторых вариантах реализации указанной второй фармацевтической композиции указанная микронизированная форма I или микронизированная по существу чистая форма I имеет значение D90, составляющее менее или равное 20,0 мкм, например, в диапазоне от 1,0 до 20,0 мкм или в диапазоне от 2,0 до 12,0 мкм. В некоторых вариантах реализации указанной второй фармацевтической композиции указанная микронизированная форма II или микронизированная по существу чистая форма II имеет значение D90 в диапазоне от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации второй фармацевтической композиции указанная микронизированная форма I или микронизированная по существу чистая форма I имеет значение D90 в диапазоне от 2,0 до 5,0 мкм, например, имеет значение D90 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации второй фармацевтической композиции указанная микронизированная форма I или микронизированная по существу чистая форма I имеет значение D90 в диапазоне от 9,0 до 12,0 мкм, например, имеет значение D90 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 менее или равно 11,0 мкм или менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 4,0 мкм.
[024] В некоторых вариантах реализации указанной второй фармацевтической композиции указанная микронизированная форма II или микронизированная по существу чистая форма I имеет значение D90, составляющее менее или равное 20,0 мкм, например, в диапазоне от 1,0 до 20,0 мкм или в диапазоне от 2,0 до 12,0 мкм. В некоторых вариантах реализации указанной второй фармацевтической композиции указанная микронизированная форма II или микронизированная по существу чистая форма II имеет значение D90 в диапазоне от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации второй фармацевтической композиции указанная микронизированная форма II или микронизированная по существу чистая форма II имеет значение D90 в диапазоне от 2,0 до 5,0 мкм, например, имеет значение D90 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации второй фармацевтической композиции указанная микронизированная форма II или микронизированная по существу чистая форма II имеет значение D90 в диапазоне от 9,0 до 12,0 мкм, например, имеет значение D90 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 менее или равно 11,0 мкм или менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 4,0 мкм.
[025] В некоторых вариантах реализации указанной второй фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из разбавителей (таких как маннит, микрокристаллическая целлюлоза, крахмал, лактоза, декстрин, сорбит), разрыхлителей (таких как крахмал гликолят натрия), связующих для гранулирования (таких как поливинилпирролидон (ПВП)) и скользящих веществ (например, диоксида кремния и стеарата магния). В некоторых вариантах реализации указанный разбавитель может присутствовать в количестве от примерно 35% до примерно 90% по массе композиции. В некоторых вариантах реализации указанный разбавитель может присутствовать в количестве от примерно 0,1% до примерно 5% по массе композиции. В некоторых вариантах реализации указанный разбавитель может присутствовать в количестве от примерно 0,5% до примерно 10% по массе композиции. В некоторых вариантах реализации указанные связующие для грануляции могут присутствовать в количестве от примерно 0,5% до примерно 5% по массе композиции. В некоторых вариантах реализации второй фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из маннита, микрокристаллической целлюлозы, крахмал гликолята натрия, поливинилпирролидона (ПВП) и стеарата магния. В некоторых вариантах реализации второй фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из микрокристаллической целлюлозы, крахмал гликолята натрия, диоксида кремния и стеарата магния. В некоторых вариантах реализации второй фармацевтической композиции по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из микрокристаллической целлюлозы, крахмал гликолята натрия и стеарата магния. В некоторых вариантах реализации второй фармацевтической композиции по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из микрокристаллической целлюлозы и стеарата магния.
[026] В некоторых вариантах реализации второй фармацевтической композиции указанная фармацевтическая композиция находится в форме таблетки или капсулы, при этом указанное микронизированное соединение А может присутствовать в количестве 1, 5, 10, 15, 20, 25, 50, 75, 80, 85, 90, 95, 100, 125, 150, 200, 250, 300, 400 и 500 мг в таблетке или капсуле, например, в капсуле.
[027] Также в настоящем документе описана третья фармацевтическая композиция, содержащая
микронизированное соединение А, которое представляет собой форму I или по существу чистую форму I, и
по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
[028] В некоторых вариантах реализации указанной третьей фармацевтической композиции указанная микронизированная форма 1 или микронизированная по существу чистая форма I имеет значение D90, составляющее менее или равное 20,0 мкм, например, в диапазоне от 1,0 до 20,0 мкм или в диапазоне от 2,0 до 12,0 мкм. В некоторых вариантах реализации указанной третьей фармацевтической композиции указанная микронизированная форма I или микронизированная по существу чистая форма I имеет значение D90 в диапазоне от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации третьей фармацевтической композиции указанная микронизированная форма I или микронизированная по существу чистая форма I имеет значение D90 в диапазоне от 2,0 до 5,0 мкм, например, имеет значение D90 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации третьей фармацевтической композиции указанная микронизированная форма I или микронизированная по существу чистая форма I имеет значение D90 в диапазоне от 9,0 до 12,0 мкм, например, имеет значение D90 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 менее или равно 11,0 мкм или менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 4,0 мкм.
[029] В некоторых вариантах реализации указанной третьей фармацевтической композиции по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из разбавителей (таких как маннит, микрокристаллическая целлюлоза, крахмал, лактоза, декстрин, сорбит), разрыхлителей (таких как крахмал гликолят натрия), связующих для гранулирования (таких как поливинилпирролидон (ПВП)) и скользящих веществ (например, диоксида кремния и стеарата магния). В некоторых вариантах реализации указанный разбавитель может присутствовать в количестве от примерно 35% до примерно 90% по массе композиции. В некоторых вариантах реализации указанный разбавитель может присутствовать в количестве от примерно 0,1% до примерно 5% по массе композиции. В некоторых вариантах реализации указанный разбавитель может присутствовать в количестве от примерно 0,5% до примерно 10% по массе композиции. В некоторых вариантах реализации указанные связующие для грануляции могут присутствовать в количестве от примерно 0,5% до примерно 5% по массе композиции. В некоторых вариантах реализации третьей фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из маннита, микрокристаллической целлюлозы, крахмал гликолята натрия, поливинилпирролидона (ПВП) и стеарата магния. В некоторых вариантах реализации третьей фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из микрокристаллической целлюлозы, крахмал гликолята натрия, диоксида кремния и стеарата магния. В некоторых вариантах реализации третьей фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из микрокристаллической целлюлозы, крахмал гликолята натрия и стеарата магния. В некоторых вариантах реализации третьей фармацевтической композиции указанное по меньшей мере одно фармацевтически приемлемое вспомогательное вещество выбрано из микрокристаллической целлюлозы и стеарата магния.
[030] В некоторых вариантах реализации третьей фармацевтической композиции указанная фармацевтическая композиция находится в форме таблетки или капсулы, при этом указанное микронизированное соединение А, которое представляет собой форму I или по существу чистую форму I, может присутствовать в количестве 1, 5, 10, 15, 20, 25, 50, 75, 80, 85, 90, 95, 100, 125, 150, 200, 250, 300, 400 и 500 мг в таблетке или капсуле, например, в капсуле.
[031] Также в настоящем документе описан способ лечения субъекта с признанной необходимостью лечения нейроэндокринных опухолей (НЭО), включающий введение указанному субъекту, нуждающемуся в этом, эффективного количества первой фармацевтической композиции, включая каждый из ее вариантов реализации, описанных выше.
[032] Также в настоящем документе описан способ лечения субъекта с признанной необходимостью лечения нейроэндокринных опухолей (НЭО), включающий введение указанному субъекту, нуждающемуся в этом, эффективного количества второй фармацевтической композиции, включая каждый из ее вариантов реализации, описанных выше.
[033] Также в настоящем документе описан способ лечения субъекта с признанной необходимостью лечения нейроэндокринных опухолей (НЭО), включающий введение указанному субъекту, нуждающемуся в этом, эффективного количества третьей фармацевтической композиции, включая каждый из ее вариантов реализации, описанных выше.
[034] В некоторых вариантах реализации указанные нейроэндокринные опухоли представляют собой панкреатические нейроэндокринные опухоли. В некоторых вариантах реализации указанные нейроэндокринные опухоли представляют собой экстрапанкреатические нейроэндокринные опухоли. В некоторых вариантах реализации указанные нейроэндокринные опухоли представляют собой желудочно-кишечные нейроэндокринные опухоли.
[035] Также в настоящем документе описан способ лечения субъекта, для которого признана необходимость лечения по меньшей мере одного заболевания, реагирующего на ингибирование FGFR1, такого как рак, и/или по меньшей мере одного заболевания, реагирующего на ингибирование KDR, такого как нарушения, связанные с ангиогенезом, включающий введение указанному субъекту, нуждающемуся в этом, эффективного количества фармацевтической композиции, выбранной из первой, второй и третьей фармацевтической композиции, включая каждый из вариантов их реализации, описанный выше. В некоторых вариантах реализации связанные с ангиогенезом расстройства, описанные в настоящем документе, включают, но не ограничиваются ими, рак и возрастную макулярную дегенерацию. В некоторых вариантах реализации указанный рак, как описано в настоящем документе, включают, но не ограничиваются ими, рак легких, рак головы и шеи, колоректальный рак, рак поджелудочной железы, рак толстой кишки, рак молочной железы, рак яичников, рак предстательной железы, рак желудка, рак почки, рак печени, рак мозга, рак кости, рак щитовидной железы, нейроэндокринные опухоли, саркому, такие как саркома мягких тканей и лейкоз.
[036] Также в настоящем документе описан способ получения таблетки или капсул, включающий:
смешивание по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения А, его фармацевтически приемлемых солей и формы 1, по существу чистой формы I, формы II или по существу чистой формы II соединения А, с по меньшей мере одним фармацевтически приемлемым вспомогательным веществом, где соединение А, фармацевтически приемлемые соли и форму I, по существу чистую форму I, форму II или по существу чистую форму II соединения А микронизируют с распределением частиц по размерам (PSD) D90 менее или равным 20 мкм,
сухое смешивание, влажное гранулирование или уплотнения прокаткой полученной смеси и
наполнение в капсулы сухой смешанной, влажной гранулированной или уплотненной прокаткой смеси или прессование в таблетки сухой смешанной, мокрой гранулированной или уплотненной прокаткой смеси.
[037] В некоторых вариантах реализации способа получения таблетки или капсул соединение А, его фармацевтически приемлемые соли и форму I, по существу чистую форму I, форму II или по существу чистую форму II соединения А микронизируют с величиной PSD D90 в диапазоне от 1,0 до 20,0 мкм или от 2,0 до 12,0 мкм. В некоторых вариантах реализации способа получения таблетки или капсул соединение А, его фармацевтически приемлемые соли и форму I, по существу чистую форму I, форму II или по существу чистую форму II соединения А микронизируют с величиной PSD D90 в диапазоне от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации способа получения таблетки или капсул соединение А, его фармацевтически приемлемые соли и форму 1, по существу чистую форму I, форму II или по существу чистую форму II соединения А микронизируют с величиной PSD D90 в диапазоне от 2,0 до 5,0 мкм, например, с величиной D90 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации способа получения таблетки или капсул соединение А, его фармацевтически приемлемые соли и форму I, по существу чистую форму I, форму II или по существу чистую форму II соединения А микронизируют с величиной PSD D90 в диапазоне от 9,0 до 12,0 мкм, например, с величиной D90 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 менее или равно 11,0 мкм или менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 4,0 мкм.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[038] На фиг. 1 показаны противоопухолевые эффекты формы I соединения А в модели опухоли NCI-H716.
[039] На фиг. 2 показаны ФК-профили соединения А у группы 1 собак породы бигль после однократного перорального дозирования капсулы А в дозе 30 мг формы I соединения А/кг массы тела в период 1.
[040] На фиг. 3 показаны ФК-профили соединения А у группы 1 собак породы бигль после однократного перорального дозирования капсулы А в дозе 30 мг формы I соединения А/кг массы тела в период 2.
[041] На фиг. 4 показаны ФК-профили соединения А у группы 2 собак породы бигль после однократного перорального дозирования капсулы А в дозе 30 мг формы I соединения А/кг массы тела в период 1.
[042] На фиг. 5 показаны ФК-профили соединения А у группы 2 собак породы бигль после однократного перорального дозирования капсулы А в дозе 30 мг формы I соединения А/кг массы тела в период 2.
[043] На фиг. 6 показаны профили средняя концентрации от времени соединения А в плазме собак породы бигль после однократного перорального дозирования капсул А и В, содержащих различные составы (n=6, расчет средней концентрации только по животным без рвоты).
[044] На фиг. 7 приведена рентгеновская порошковая дифрактограмма формы 1 соединения А, совпадающая с фиг. 1 патента '658.
[045] На фиг. 8 приведена рентгеновская порошковая дифрактограмма II соединения А, совпадающая с фиг. 5 патента '658.
[046] Следующие сокращения и термины имеют указанные значения на протяжении всего документа:
[047] Если явно не указано иное, термин в единственном числе означает «один или более».
[048] Термин «значение D90» означает, что 90% (по объему) частиц имеют размер менее или равный приведенному значению. Например, значение D90 20,0 мкм означает, что 90% (по объему) частиц имеют размер менее или равный 20,0 мкм; значение D90 10,0 мкм означает, что 90% (по объему) частиц имеют размер менее или равный 10,0 мкм.
[049] Используемый в настоящем документе термин «фармацевтически приемлемый» относится к веществу, которое в рамках здравого медицинского суждения подходит для применения в контакте с телом человека и других животных без чрезмерной токсичности, раздражения, аллергического ответа или других проблем, соразмерно с разумным соотношением выгода/риск.
[050] «Фармацевтически приемлемые соли» включают, но не ограничиваются ими, соли с неорганическими кислотами, такими как гидрохлорат, гидробромат, фосфат, бифосфат, сульфат, сульфинат, нитрат и тому подобное; а также соли с органическими кислотами, такие как малат, малеат, манделат, фумарат, тартрат, сукцинат, цитрат, аспартат, глутамат, атролактат, глюконат, пропионат, лактат, камфорсульфонат, метансульфонат, этансульфонат, нафталинсульфонат, п-толуолсульфонат, 2-гидроксиэтилсульфонат, гидроксибутират, бензоат, салицилат, стеарат и алканоат, такие как ацетат, соль HOOC-(CH2)n-COOH, где n равно 0-4, и тому подобное. При необходимости «фармацевтически приемлемые соли» также могут быть солями присоединения оснований и включают, но не ограничиваются ими, соль натрия, соль калия, соль кальция, соль алюминия, соль лития и соль аммония.
[051] Кроме того, если описанное в настоящем документе соединение получают в виде соли присоединения кислоты, свободное основание можно получить путем подщелачивания раствора кислой соли. И наоборот, если продукт представляет собой свободное основание, соль присоединения кислоты, в частности, фармацевтически приемлемую соль присоединения кислоты можно получить при помощи растворения свободного основания в подходящем органическом растворителе и обработки указанного раствора кислотой, в соответствии с обычными способами получения соли присоединения кислоты из основных соединений. Специалистам в данной области техники будут понятны различные способы синтеза, которые можно использовать в рамках обычных экспериментов для получения нетоксичных фармацевтически приемлемых солей присоединения.
[052] Используемый в настоящем документе термин «эффективное количество» относится к количеству соединения А или его фармацевтически приемлемой соли, формы I, по существу чистой формы I, формы II или по существу чистой формы II, которое при введении субъекту будет вызывать биологическое или медицинский ответ субъекта, например, улучшать или устранять один или более симптомов заболевания, облегчать или вылечивать заболевание, замедлять или отсрочивать прогрессирование заболевания и т.д.
[053] Используемый здесь термин «субъект» относится к животному. Обычно животное представляет собой человека или другое животное, особенно человека и других млекопитающих, например, приматов, коров, овец, коз, лошадей, собак, кошек, кроликов и тому подобное. В некоторых вариантах реализации настоящего описания субъект представляет собой человека.
[054] Термин "Форма I "относится к форме I N-(2-(диметиламино)этил)-1-(3-((4-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)метансульфонамида, имеющей пики (2θ), выбранные из следующих: 7,0, 8,0 и 8,6, для каждого из углов дифракции±0,2 градуса (2θ). В некоторых вариантах реализации рентгеновская порошковая дифрактограмма формы I, описанной в настоящем документе, может иметь пики (2θ), выбранные из следующих: 7,0, 8,0, 8,6, 11,0, 11,8, для каждого из углов дифракции ±0,2 градуса (2θ). В некоторых вариантах осуществления форма I, описанная в настоящем документе, имеет рентгеновскую порошковую дифрактограмму такую, как показано на фиг. 7.
[055] Термин «Форма II» относится к форме II N-(2-(диметиламино)этил)-1-(3-((4-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)метансульфонамида, имеющей пики (2θ), выбранные из следующих: 6,8, 9,8, 10,5 и 10,7, для каждого из углов дифракции ±0,2 градуса (2θ). В некоторых вариантах реализации рентгеновская порошковая дифрактограмма формы II описанной в настоящем документе, может иметь пики (2θ), выбранные из следующих: 6,8, 9,8, 10,5, 10,7, 13,6, 15,0, для каждого из углов дифракции ±0,2 градуса (2θ). В некоторых вариантах осуществления форма II, описанная в настоящем документе, может иметь рентгеновскую порошковую дифрактограмму такую, как показано на фиг. 8.
[056] Термин «по существу чистая форма I» относится к соединению А, в котором по меньшей мере 75 мас. % соединения А представляет собой форму I. Например, «по существу чистая форма I» относится к соединению А, в котором по меньшей мере 75%, 80%, 85%, 90%, 95% или 100% по массе соединения А представляет собой форму I.
[057] Термин «по существу чистая форма II» относится к соединению А, в котором по меньшей мере 75 мас. % соединения А представляет собой форму II. Например, «по существу чистая форма II» относится к соединению А, в котором по меньшей мере 75%, 80%, 85%, 90%, 95% или 100% по массе соединения А представляет собой форму II.
[058] Слово «примерно» в сочетании со значением расширяет его до диапазона ±20% от указанного значения. Например, «примерно 5%» означает диапазон от 4% до 6%.
[059] В некоторых вариантах реализации указанного изобретения по меньшей мере один активный фармацевтический ингредиент, выбранный из соединения формулы А (соединение А) и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А может быть полезным для лечения нейроэндокринных опухолей.
[060] В некоторых вариантах реализации способ лечения субъекта, имеющего нейроэндокринные опухоли, и для которого признана необходимость их лечения, включает введение указанному субъекту эффективного количества по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А для лечения указанных нейроэндокринных опухолей.
[061] В некоторых вариантах реализации способ лечения субъекта, имеющего нейроэндокринные опухоли, и для которого признана необходимость их лечения, включает введение указанному субъекту эффективного количества формы I N-(2-(диметиламино)этил)-1-(3-((4)-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)метансульфонамида для лечения указанных нейроэндокринных опухолей.
[062] В некоторых вариантах реализации способ лечения субъекта, имеющего нейроэндокринные опухоли, и для которого признана необходимость их лечения, включает введение указанному субъекту эффективного количества формы II N-(2-(диметиламино)этил)-1-(3-((4)-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)метансульфонамида для лечения указанных нейроэндокринных опухолей.
[063] В некоторых вариантах реализации способ лечения субъекта, имеющего нейроэндокринные опухоли, и для которого признана необходимость их лечения, включает введение указанному субъекту, для которого признана потребность в лечении, эффективного количества фармацевтической композиции, содержащей: по меньшей мере одно фармацевтически приемлемое вспомогательное вещество и соединение формулы А и/или их фармацевтически приемлемых солей для обеспечения указанного лечения.
[064] В некоторых вариантах реализации способ лечения субъекта, имеющего нейроэндокринные опухоли, и для которого признана необходимость их лечения, включает введение указанному субъекту эффективного количества фармацевтической композиции, содержащей: по меньшей мере одно фармацевтически приемлемое вспомогательное вещество и форму I N-(2-(диметиламино)этил)-1-(3-((4)-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)метансульфонамида для обеспечения указанного лечения.
[065] В некоторых вариантах реализации способ лечения субъекта, имеющего нейроэндокринные опухоли, и для которого признана необходимость их лечения, включает введение указанному субъекту эффективного количества фармацевтической композиции, содержащей: по меньшей мере одно фармацевтически приемлемое вспомогательное вещество и форму II N-(2-(диметиламино)этил)-1-(3-((4)-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)метансульфонамида для обеспечения указанного лечения.
[066] Во всех вариантах реализации, описанных в настоящем документе выше и далее, соединение А, по меньшей мере одна его фармацевтически приемлемая соль, форма I и форма II N-(2-(диметиламино)этил)-1-(3-((4-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)метансульфонамида и по существу чистая форма I и форма II N-(2-(диметиламино)этил)-1-(3-((4-((2-метил-1H-индол-5-ил)окси)пиримидин-2-ил)амино)фенил)метансульфонамида могут быть микронизированы, например, с величиной D90 менее или равной 20,0 мкм, например, в диапазоне от 1,0 до 20,0 мкм или в диапазоне от 2,0 до 12,0 мкм, например, от 1,0 до 2,0 мкм, от более 2,0 до 3,0 мкм, от более 3,0 до 4,0 мкм, от более 4,0 до 6,0 мкм, от более 6,0 до 8,0 мкм, от более 8,0 до 10,0 мкм или от более 10,0 до 12,0 мкм. В некоторых вариантах реализации значение D90 составляет от 2,0 до 5,0 мкм, например, значение D90 составляет 3,0, 3,5 или 4,0 мкм. В некоторых вариантах реализации значение D90 составляет от 9,0 до 12,0 мкм, например, значение D90 составляет 9,5 или 10,0 мкм. В предпочтительном варианте реализации значение D90 менее или равно 11,0 мкм или менее или равно 10,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 6,0 мкм. В более предпочтительном варианте реализации значение D90 менее или равно 4,0 мкм.
[067] Количество по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, эффективного для достижения желательный биологического эффекта, может зависеть от ряда факторов, например, предполагаемого применения, способа введения и клинического состояния пациента. Суточная доза может составлять, например, от 0,1 мг до 3 г/сутки (например, от 0,5 мг до 2 г/сутки, например, от 50 мг до 1 г/сутки). Однодозовые препараты, которые можно вводить перорально, включают, например, таблетки или капсулы. Кроме того, например, указанный по меньшей мере один активный фармацевтический ингредиент, выбранный из соединения формулы А и/или его фармацевтически приемлемых солей и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А может присутствовать в количестве 1,5, 10, 15, 20, 25, 50, 75, 80, 85, 90, 95, 100, 125, 150, 200, 250, 300, 400 и 500 мг в капсуле или таблетке.
[068] Для терапии приведенных выше состояний указанный по меньшей мере один активный фармацевтический ингредиент, выбранный из соединения формулы А и/или его фармацевтически приемлемых солей и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, можно применять в качестве самого соединения, но обычно каждый из них применяют в виде фармацевтической композиции с одним или более фармацевтически приемлемыми вспомогательными веществами. Типичные вспомогательные вещества должны быть совместимы с другими ингредиентами композиции и не вредны для здоровья пациента. Вспомогательное вещество может быть твердым или жидким или и тем, и другим, и может быть приготовлен с соединением формулы А, таким как форма I и/или форма II, описанные в настоящем документе, в виде разовой дозы, например, в виде таблетки или капсулы, которую можно получать с содержанием от 0,05 до 95 мас. % соединения формулы А, описанного в настоящем документе. Фармацевтические композиции, описанные в настоящем документе, можно получать известными фармацевтическими способами, такими способы, включающими смешивание указанного по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей, и форм I и II соединения формулы А, с фармацевтически приемлемыми вспомогательными веществами.
[069] В некоторых вариантах репрезентативные вспомогательные вещества включают, но не ограничиваются ими: разбавители (такие как целлюлоза, крахмал, лактоза, маннит, декстрин, сорбит и т.д.), поверхностно-активные вещества (такие как лаурилсульфат натрия, полоксамер и т.д.), солюбилизаторы (такие как поливинилпирролидон, полиэтиленгликоль и т.д.), разрыхлители (такие как крахмал гликолят натрия, PVPP, кроскармеллоза и т.д.) и скользящее вещество и смазывающее вещество (такие как SiO2, стеарат магния и т.д.). Другие примерные вспомогательные вещества включают микрокристаллическую целлюлозу, цитрат натрия, карбонат кальция, дикальцийфосфат, глицин, разрыхлители, такие как крахмал, натриевая сшитая карбоксиметилцеллюлоза, сложные силикаты и полиэтиленгликоль с высокой молекулярной массой, связующие для грануляции (такие как поливинилпирролидон, сахароза, желатин и гуммиарабик) и смазывающие вещества (такие как стеариновая кислота, стеарилфумарат натрия и тальк). Другие примерные вспомогательные вещества включают разбавители (такие как маннит и микрокристаллическая целлюлоза) и скользящие вещества (такие как стеарат магния или диоксид кремния). Другие примерные вспомогательные вещества включают разбавители (такие как маннит и микрокристаллическая целлюлоза) и разрыхлители (такие как натрий крахмал гликолят). Другие примерные вспомогательные вещества включают разбавители (такие как маннит и микрокристаллическая целлюлоза), разрыхлители (такие как крахмал гликолят натрия) и связующие для грануляции (такие как поливинилпирролидон).
[070] В некоторых вариантах реализации указанный по меньшей мере один активный фармацевтический ингредиент, выбранный из соединения формулы А и/или его фармацевтически приемлемых солей и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, могут быть объединены с по меньшей мере одним компонентом, таким как вспомогательное вещество, выбранное из подсластителей, деликатных ароматизаторов, окрашивающих веществ, красителей и эмульгаторов.
[071] В некоторых вариантах реализации форма I или форма II, описанные в настоящем документе, не могут быть преобразованы в рецептуру с одним или несколькими фармацевтически приемлемыми разбавителями. В других вариантах реализации форма I или форма II, описанные в настоящем документе, могут быть полностью или частично превращены в одну или несколько других форм, включая нетвердою форму, при приготовлении с одним или более фармацевтически приемлемыми разбавителями. Типичные разбавители включают, но не ограничиваются ими, воду, этанол, пропиленгликоль, глицерин и их смеси. В некоторых вариантах реализации форма I или форма II, описанные в настоящем документе, могут быть растворены при приготовлении в виде фармацевтической композиции. Соответственно, в таких случаях «растворения» указанные форма I или форма II больше не существуют в соответствующей кристаллической форме в фармацевтической композиции и эквивалентны любой растворенной форме соединения А.
[072] В некоторых вариантах реализации указанный по меньшей мере один активный фармацевтический ингредиент, выбранный из соединения формулы А и/или его фармацевтически приемлемых солей и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, может быть приготовлен в подходяще виде.
[073] Фармацевтические композиции, описанные в настоящем документе, могут быть такими, которые подходят для орального и перорального (например, подъязычного) введения, хотя подходящий способ введения может зависеть в каждом отдельном случае от природы и тяжести состояния, подлежащего лечению, и от природы указанного по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, используемого в каждом случае для приготовления фармацевтической композиции. Также настоящее изобретение относится к покрытым композициям и композициям с покрытием с замедленным высвобождением. Возможны составы, устойчивые к кислоте и желудочному соку. Подходящие покрытия, устойчивые к желудочному соку, включают ацетатфталат целлюлозы, фталат поливинилацетата, фталат гидроксипропилметилцеллюлозы, анионные полимеры метакриловой кислоты и метилметакрилат.
[074] Подходящие фармацевтические композиции для перорального введения, полученные из указанного по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей, и формы 1, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А может быть в виде отдельных единиц, таких как, например, капсулы, саше и таблетки, включая таблетки для рассасывания, каждая из которых может быть приготовлена с определенным количеством по меньшей мере одного активного фармацевтического ингредиента, описанного в настоящем документе; а также в формах, выбранных из порошков, гранул, растворов, суспензий в водной или неводной жидкости, эмульсий типа масло-в-воде и вода-в-масле. Указанные композиции можно, как уже упоминалось, получать любым подходящим способом, таким как способы, которые включают стадию, на которой указанный по меньшей мере один активный фармацевтический ингредиент, выбранный из соединения формулы А и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, и вспомогательное вещество (которое может состоять из одного или нескольких дополнительных ингредиентов, включая разбавители) приводят в контакт. Указанные композиции обычно могут быть получены путем однородного и гомогенного смешивания по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, с жидким и/или мелкодисперсным твердым вспомогательным веществом, после чего можно формировать продукт. Так, например, таблетку можно получать прессованием или формованием порошка или гранул указанного по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, где это необходимо, с одним или несколькими дополнительными ингредиентами. Прессованные таблетки можно получать путем таблетирования указанного по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, в свободнотекущей форме, такой как, например, порошок или гранулы, смешанного, где это целесообразно, со связующим, скользящим веществом, инертным разбавителем и/или одним (или более) поверхностно-активным/диспергирующим агентом (агентами) в подходящем аппарате. Формованные таблетки можно получать путем формования указанного по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, в виде порошка, а затем увлажнения инертным жидким разбавителем в подходящем аппарате. Композиции также можно получать методом влажной грануляции. Так, например, композицию можно получить влажным гранулированием путем смешивания указанного по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, формы II, по существу чистой Форма II соединения формулы А, с одним или более необязательными дополнительными ингредиентами, подходящим растворителем и связующим с получением влажного гранулята, сушкой указанного влажного гранулята и измельчением высушенного гранулята. Указанный способ может дополнительно включать добавление по меньшей мере одного смазывающего вещества к указанному высушенному измельченному грануляту и прессование указанного высушенного измельченного гранулята с получением таблеток. Указанные необязательные дополнительные ингредиенты могут включать, например, по меньшей мере один разбавитель и/или по меньшей мере один дезинтегрирующий агент. Подходящий растворитель может представлять собой воду. В некоторых вариантах реализации указанный разбавитель выбран из карбоната кальция, фосфата кальция (двухосновного и/или трехосного), сульфата кальция, порошкообразной целлюлозы, декстратов, декстрина, фруктозы, каолина, лактитола, безводной лактозы, моногидрата лактозы, мальтозы, маннита, микрокристаллической целлюлозы, сорбита, сахарозы и крахмала. В некоторых вариантах реализации связующее может присутствовать в количестве от примерно 35% до примерно 90% по массе таблетки. В некоторых вариантах реализации указанное связующее вещество может быть выбрано из аравийской камеди, альгиновой кислоты, карбомера, карбоксиметилцеллюлозы натрия, декстрина, этилцеллюлозы, желатина, глюкозы, гуаровой смолы, гидроксипропилцеллюлозы, мальтозы, метилцеллюлозы, полиэтиленоксида и повидона. В некоторых примерных вариантах реализации указанное связующее присутствует в количестве от примерно 0,5% до примерно 5% по массе таблетки. В других примерных вариантах реализации вышеупомянутые составы содержат примерно 0,05-5 г указанного по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, на миллилитр или на грамм указанного состава.
[075] Описанные в настоящем документе композиции можно вводить местно или системно.
[076] Фармацевтические композиции, подходящие для перорального (сублингвального) введения, могут содержать таблетки для рассасывания, которые можно получать из указанного по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, Форма II, по существу чистая форма II соединения формулы А, с ароматизатором, обычно выбранным из сахарозы, гуммиарабика, трагаканта, и пастилки.
[077] Фармацевтические композиции, описанные в настоящем документе, также могут быть такими, которые подходят для парентерального введения, для введения с помощью ингаляционного аэрозоля или через имплантированный резервуар. Твердые вспомогательные вещества, например, крахмал, лактоза, микрокристаллическая целлюлоза, силикат алюминия, жидкие вспомогательные вещества, например, вода для инъекций, поливиниловый спирт, неионогенные поверхностно-активные вещества и кукурузное масло, а также любые ингредиенты, подходящие для введения внутрь. Другие вспомогательные вещества, обычно применяемые в фармацевтической композиции, включают красители, консерванты, агенты, улучшающие вкус, и антиоксиданты, такие как витамин Е, витамин А, ВНТ и ВНА.
[078] Соединение формулы А, такое как форма I или форма II, описанные в настоящем документе, также можно вводить внутрибрюшинно. Как раствор, так и суспензию указанных соединений можно получать путем растворения или суспендирования указанного соединения в воде, содержащей подходящие поверхностно-активные вещества. Диспергированные суспензии можно получать с применением глицерина, полиэтиленгликоля (ПЭГ) или их смеси с подходящими маслами. К указанным составам можно добавлять консерванты для предотвращения роста микроорганизмов во время применения.
[079] Инъекционные составы препараты включают раствор или суспензию в стерилизованной воде и стерилизованный порошок. Во всех случаях указанные составы должны быть стерилизованы, их должно быть легко удалять из шприца, они должны быть стабильны в условиях производства и хранения и как можно более чистыми в отношении загрязнений и воздействия микроорганизмов. Наполнители могут представлять собой растворители или диспергирующие агенты и включают воду, спирт и некоторые подходящие масла.
[080] Указанный по меньшей мере один активный фармацевтический ингредиент, выбранный из соединения формулы А и/или его фармацевтически приемлемых солей, и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, также можно вводить в комбинации с одним или более активными ингредиентами. При введении в виде комбинации указанные активные ингредиенты могут быть составлены в виде отдельных композиций, которые вводят в одно и то же время или последовательно в разное время, или указанные активные ингредиенты можно вводить в единичной лекарственной форме, то есть в отдельной композиции, при условии, что активные ингредиенты не являются, в указанной единичной лекарственной форме, несовместимыми с другими активными ингредиентами или с составом, или иным образом нежелательно объединены в одну композицию.
[081] В некоторых вариантах реализации можно вводить указанный по меньшей мере один активный фармацевтический ингредиент, выбранный из соединения формулы А и/или его фармацевтически приемлемых солей и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, с одним или более других агентов, известных для лечения по меньшей мере одного заболевания, реагирующего на ингибирование FGFR1, такого как рак, и/или по меньшей мере одного заболевания, реагирующего на ингибирование KDR, такого как расстройства, связанные с ангиогенезом.
[082] Выражение «совместная терапия» (или «комбинированная терапия») или «в сочетании с», используемое в настоящем документе, определяет применение указанного по меньшей мере одного активного фармацевтического ингредиента, выбранного из соединения формулы А и/или фармацевтически приемлемых солей и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, описанных в настоящем документе, и одного или более других активных ингредиентов, таких как, например, противоопухолевые агенты. Используемый в настоящем документе термин «противоопухолевый агент» относится к любому агенту или лечению, которое вводится субъекту с раком с целью лечения рака, включая: лучевую терапию; иммунотерапию; ДНК-разрушающие химиотерапевтические агенты; и химиотерапевтические агенты, которые нарушают репликацию клеток.
[083] Неограничивающие примеры ДНК-разрушающих химиотерапевтических агентов включают ингибиторы топоизомеразы I (например, иринотекан, топотекан, камптотецин и их аналоги или метаболиты, и доксорубицин); ингибиторы топоизомеразы II (например, этопозид, тенипозид и даунорубицин); алкилирующие агенты (например, мелфалан, хлорамбуцил, бусульфан, тиотепа, ифосфамид, кармустин, ломустин, семустин, стрептозоцин, дакарбазин, метотрексат, митомицин С и циклофосфамид); ДНК-интеркаляторы (например, цисплатин, оксалиплатин и карбоплатин); ДНК-интеркаляторы и генераторы свободных радикалов, такие как блеомицин; и нуклеозидные миметики (например, 5-фторурацил, капецитабин, гемцитабин, флударабин, цитарабин, меркаптопурин, тиогуанин, пентостатин и гидроксимочевина).
[084] Химиотерапевтические агенты, которые нарушают репликацию клеток, включают: паклитаксел, доцетаксел и родственные аналоги; винкристин, винбластин и родственные аналоги; талидомид и родственные аналоги (например, СС-5013 и СС-4047); ингибиторы белковой тирозинкиназы (например, иматиниба мезилат и гефитиниб); ингибиторы протеасомы (например, бортезомиб); ингибиторы NF-каппа В, включая ингибиторы I каппа В-киназы; антитела, которые связываются с белками, сверхэкспрессируемыми в раках, и тем самым снижают регуляцию клеточной репликации (например, трастузумаб, ритуксимаб, цетуксимаб и бевацизумаб); и другие ингибиторы белков или ферментов, про которые известно, что они чрезмерно экспрессируются или активируются в раках, ингибирование которых снижает регуляцию клеточной репликации.
[085] При совместной терапии введение каждого активного ингредиента может происходить последовательно в режиме, обеспечивающем благоприятные эффекты от комбинации лекарственных средств; и/или совместное введение вышеуказанных компонентов может происходить по существу одновременно (например, как в виде единичной дозированной формы, такой как капсула, имеющей фиксированное отношение активных ингредиентов, или в нескольких отдельных капсулах для каждого активного ингредиента, и т.д.).
[086] Таким образом, для способов, описанных в настоящем документе, не ограничена последовательность введения; указанный по меньшей мере один активный фармацевтический ингредиент, выбранный из соединения формулы А и/или его фармацевтически приемлемых солей и формы I, по существу чистой формы I, формы II, по существу чистой формы II соединения формулы А, описанных в настоящем документе, можно вводить до введения одного или более других активных ингредиентов, или в то же самое время, или после.
[087] Приведены следующие неограничивающие примеры.
Пример 1: Составы
[088] Если не указано иное, значения D90 были получены методом лазерного анализа распределения частиц по размерам с применением Malvern 3000 и HYDRO MV или эквивалента с показателем преломления частиц при 1,59, показателем поглощения 0,01, скоростью мешалки 2500 об/мин и без ультразвука. Подготовка проб для измерения включала: размещение примерно 30 мг образца в стеклянной бутылке емкостью 20 мл; добавление 20 капель 0,25% Triton Х-100 (мас./об.)-вода для увлажнения образца, а затем добавление 10 мл очищенной воды, диспергировали с помощью ультразвука в течение 2 минут.Образец анализировали в течение 2 минут.
[089] Соединение А, используемое в следующих примерах, представляет собой форму I.
[090] Капсулы и таблетки, описанные в примерах, анализировали на скорость высвобождения соединения А в 900 мл водной среды (USP pH 4,5 ацетатный буфер) при вращении 75 об/мин в соответствии с первым методом (корзиной), описанным в China Pharmacopoeia (издание 2010 г.).
Способ приготовления 1 - влажное гранулирование А. Капсулы 50 мг капсулы, капсулы 200 мг
[091] Состав капсулы 200 мг (200 мг капсулы №1), компоненты которого представлены в таблице 1, получали из 500 г микронизированного соединения А (PSD, D90=9,6 мкм), 277,5 г маннита, 150 грамм микрокристаллической целлюлозы, 50 г крахмал гликолята натрия, 12,5 г поливинилпирролидона (ПВП) К30 и 10 г стеарата магния. Смешивали микронизированное соединение А (500 г, D90=9,6 мкм), маннит (277,5 г), микрокристаллическую целлюлозу (150 г) и крахмал гликолят натрия (50 г). Получали 5% (мас./об.) водный раствор ПВП-К30 (12,5 г) и добавляли в качестве связующего для получения гранул способом влажной грануляции. Добавляли стеарат магния (10 г) и перемешивали в течение 3 минут. Конечной смесью, содержащей 200 мг соединения А, заполняли каждую капсулу размера №0 с получением продукта соединения А в виде капсул 200 мг (200 мг капсулы №1). Накопительное выделение соединения А из капсулы 200 мг составляло 86,7% за 30 мин. Испытания на стабильность показали, что указанный образец капсулы был стабилен при 25°C/60% отн. вл. в течение 12 месяцев и при 40°С/75% отн. вл. в течение 6 месяцев без какого-либо изменения содержимого по анализу и без обнаружения деградации. Тест на стабильность был продолжительным.
[092] Аналогичным образом готовили капсулы других доз. Например, 50 мг капсулы микронизированного соединения A (D90=9,6 мкм) (50 мг капсула №1) получали путем заполнения конечной смесью, содержащей 50 мг соединения А, полученной аналогично тому, как указано выше, каждой капсулы размера №3, при этом компоненты приведены в таблице 1. Кумулятивное выделение соединения А из капсулы 50 мг составляло 90,8% за 30 мин. Испытания на стабильность показали, что указанный образец капсулы был стабилен при 25°С/60% отн. вл. в течение 12 месяцев и при 40°C/75% отн. вл. в течение 6 месяцев без какого-либо изменения содержимого по анализу и без обнаружения деградации.

[093] Другой состав капсулы 50 мг (50 мг капсулы №2), компоненты которого представлены в таблице 2, получали из 15 г микронизированного соединения A (PSD, D90=3,8 мкм), 19,9875 г маннита, 6,75 грамм микрокристаллической целлюлозы, 2,25 г крахмал гликолята натрия, 0,5625 г поливинилпирролидона (ПВП) К30 и 0,45 г стеарата магния. Смешивали микронизированное соединение А (форма 1,15 г, D90=3,8 мкм), маннит (19,9875 г), микрокристаллическую целлюлозу (6,75 г) и крахмал гликолят натрия (50 г). Получали водный раствор ПВП-К30 (12,5 г) 3% (мас./об.) и добавляли в качестве связующего для получения гранул способом влажной грануляции. Полученные влажные гранулы высушивали, затем к высушенным гранулам добавляли стеарат магния (0,45 г) и перемешивали в течение 3 минут.Конечной смесью, содержащей 50 мг соединения А, заполняли каждую капсулу размера №3 с получением продукта соединения А в виде капсул 50 мг (50 мг капсулы №2). Кумулятивное выделение соединения А из капсулы 50 мг составляло 89,2% за 30 мин.
[094] Другой состав капсулы 50 мг (50 мг капсулы №3), компоненты которого представлены в таблице 2, получали из 5 г микронизированного соединения A (PSD, D90=3,8 мкм), 15,37 г маннита, 5,6 грамм микрокристаллической целлюлозы, 1,4 г крахмал гликолята натрия, 0,35 г поливинилпирролидона (ПВП) К30 и 0,28 г стеарата магния. Смешивали микронизированное соединение А (5 г, D90=3,8 мкм), маннит (15,37 г), микрокристаллическую целлюлозу (5,6 г) и крахмал гликолят натрия (1,4 г). Получали водный раствор ПВП-К30 (0,35 г) 3% (мас./об.) и добавляли в качестве связующего для получения гранул способом влажной грануляции. Полученные влажные гранулы высушивали, затем к высушенным гранулам добавляли стеарат магния (0,28 г) и перемешивали в течение 3 минут. Конечной смесью, содержащей 50 мг соединения А, заполняли каждую капсулу размера №1 с получением продукта соединения А в виде капсул 50 мг (50 мг капсулы №3). Кумулятивное выделение соединения А из капсулы 50 мг составляло 86,5% за 30 мин.
[095] Другой состав капсулы 50 мг (50 мг капсулы №4), компоненты которого представлены в таблице 2, получали из 120 г микронизированного соединения А (PSD, D90=5,7 мкм), 73,92 г маннита, 135,48 грамм микрокристаллической целлюлозы, 18 г крахмал гликолята натрия, 9 г поливинилпирролидона (ПВП) К30 и 3,6 г стеарата магния. Смешивали микронизированное соединение А (120 г), маннит (73,92 г), микрокристаллическую целлюлозу (135,48 г) и крахмал гликолят натрия (18 г). Получали водный раствор ПВП-К30 (9 г) 10% (мас./об.) и добавляли в качестве связующего для получения гранул способом влажной грануляции. Полученные влажные гранулы высушивали, затем к высушенным гранулам добавляли стеарат магния и перемешивали в течение 3 минут. Конечной смесью, содержащей 50 мг соединения А, заполняли каждую капсулу размера №3 с получением продукта соединения А в виде капсул 50 мг (50 мг капсулы №4). Накопительное выделение соединения А из капсулы 50 мг составляло 89,2% за 30 мин.

В. таблетка 300 мг, таблетка 250 мг, таблетка 200 мг
[096] Состав таблетки 300 мг, компоненты которого представлены в таблице 3, получали из 4200 г микронизированного соединения A (PSD, D90=4,8 мкм), 3360 г маннита, 1512 г микрокристаллической целлюлозы, 504 г крахмал гликолята натрия, 403,2 г повидона (ПВП) К30 и 100,8 г стеарата магния. Смешивали микронизированное соединение А, маннит, микрокристаллическую целлюлозу и крахмал гликолят натрия. Получали водный раствор ПВП-К30 12% (мас./об.) и добавляли в качестве связующего для получения гранул способом влажной грануляции. Полученные влажные гранулы высушивали, затем к высушенным гранулам добавляли стеарат магния и перемешивали в течение 3 минут. Конечную смесь прессовали в продолговатую таблетку с силой сжатия 184 кг для таблетки 300 мг, 164 кг для таблетки 250 мг и 116 кг для таблетки 200 мг, которая содержит 300 мг соединения А на таблетку. Таблетки покрывали пленкой путем распыления Опадрай II (Opadry® II). Кумулятивное выделение соединения А из таблетки 300 мг составляло 90,3% за 30 мин.
[097] Таблетки с другими дозами (таблетка 250 мг и таблетка 200 мг, компоненты которых приведены в таблице 3), получали аналогичным образом.

Способ приготовления 2 - Прямое смешивание
А. 50 мг капсулы
[098] Микронизированную форму I соединения А (150 г, D90=10,0 мкм) и микрокристаллическую целлюлозу (238,05 г) просеивали и перемешивали до гомогенности. Затем добавляли стеарат магния (1,95 г) и перемешивали. Конечной смесью, содержащей 50 мг соединения А, заполняли каждую капсулу размера №3 с получением продукта соединения А в виде капсул 50 мг (50 мг капсулы №5), компоненты которых приведены в таблице 4. Кумулятивное выделение соединения А из капсулы 50 мг составляло 96,5% за 30 мин.
[099] Альтернативно, микронизированную форму I соединения А (25 г, D90=3,5 мкм) и микрокристаллическую целлюлозу (39,675 г) просеивали и перемешивали до гомогенности. Затем добавляли стеарат магния (0,325 г) и перемешивали. Конечной смесью, содержащей 50 мг соединения А, заполняли каждую капсулу размера №3 с получением продукта соединения А в виде капсул 50 мг (50 мг капсулы №6), компоненты которых приведены в таблице 4. Кумулятивное выделение соединения А из капсулы 50 мг составляло 95,3% за 30 мин. Испытания на стабильность показали, что указанные образцы капсул были стабильны при 25°C/60% отн. вл. в течение 12 месяцев и при 40°C/75% отн. вл. в течение 6 месяцев без какого-либо изменения содержимого по анализу и без обнаружения деградации.
[0100] Альтернативно, микронизированную форму I соединения А (25 г, D90=5,2 мкм) и микрокристаллическую целлюлозу (39,675 г) просеивали и перемешивали до гомогенности. Затем добавляли стеарат магния (0,325 г) и перемешивали. Конечной смесью, содержащей 50 мг соединения А, заполняли каждую капсулу размера №3 с получением продукта соединения А в виде капсул 50 мг (50 мг капсулы №7), компоненты которых приведены в таблице 4. Кумулятивное выделение соединения А из капсулы 50 мг составляло 83,8% за 30 мин.
[0101] Кроме того, микронизированную форму I соединения А (10 г, D90=8,1 мкм) и микрокристаллическую целлюлозу (15,87 г) просеивали и перемешивали до гомогенности. Затем добавляли стеарат магния (0,13 г) и перемешивали. Конечной смесью, содержащей 50 мг соединения А, заполняли каждую капсулу размера №3 с получением продукта соединения А в виде капсул 50 мг (50 мг капсулы №8), компоненты которых приведены в таблице 4. Кумулятивное выделение соединения А из капсулы 50 мг составляло 98,1% за 30 мин.
[0102] Кроме того, микронизированную форму I соединения А (5,0 г, D90=2,1 мкм) и микрокристаллическую целлюлозу (7,935 г) просеивали и перемешивали до гомогенности. Затем добавляли стеарат магния (0,065 г) и перемешивали. Конечной смесью, содержащей 50 мг соединения А, заполняли каждую капсулу размера №3 с получением продукта соединения А в виде капсул 50 мг (50 мг капсулы №9), компоненты которых приведены в таблице 4. Кумулятивное выделение соединения А из капсулы 50 мг составляло 92,8% за 30 мин.
[0103] Кроме того, микронизированную форму I соединения А (10 г, D90=3,4 мкм), микрокристаллическую целлюлозу (5,1 г), маннит (16,69 г), крахмал гликолят натрия (1,7 г) и диоксид кремния (0,17 г) просеивали и перемешивали до гомогенности. Затем добавляли стеарат магния (0,34 г) и перемешивали в течение 3 минут. 50 мг капсулы с микронизированным соединением А (50 мг капсулы №10), компоненты которых приведены в таблице 4, получали путем заполнения конечной смесью, содержащей 50 мг соединения А, каждой капсулы размера №3. Кумулятивное выделение соединения А из капсулы 50 мг составляло 90,7% за 30 мин.

В. Капсула 200 мг
[0104] Микронизированную форму I соединения А (200 г, D90=10,0 мкм), микрокристаллическую целлюлозу (178 г), крахмал гликолят натрия (9 г) и диоксид кремния (9 г) просеивали и перемешивали до гомогенности. Затем добавляли стеарат магния (4 г) и перемешивали в течение 3 минут.200 мг капсулы с микронизированным соединением А (200 мг капсулы №2) получали путем заполнения конечной смесью, содержащей 200 мг соединения А, каждой капсулы размера №0. Кумулятивное выделение соединения А из капсулы 200 мг составляло 75,1% за 30 мин. Испытания на стабильность показали, что указанный образец капсулы был стабилен при 25°C/60% отн. вл. в течение 12 месяцев и при 40°C/75% отн. вл. в течение 6 месяцев без какого-либо изменения содержимого по анализу и без обнаружения деградации.
[0105] В качестве альтернативы микронизированную форму I соединения А (1050 г, D90=10,0 мкм), микрокристаллическую целлюлозу (726,6 г), крахмал гликолят натрия (75,6 г) просеивали и перемешивали до гомогенности. Затем добавляли стеарат магния (37,8 г) и перемешивали в течение 3 минут.200 мг капсулы с микронизированным соединением А (200 мг капсулы №3) получали путем заполнения конечной смесью, содержащей 200 мг соединения А, каждой капсулы размера №0. Кумулятивное выделение соединения А из капсулы 200 мг составляло 84,6% за 30 мин. Испытания на стабильность показали, что указанный образец капсулы был стабилен при 25°C/60% отн. вл. в течение 3 месяцев и при 40°C/75% отн. вл. в течение 3 месяцев без какого-либо изменения содержимого по анализу и без обнаружения деградации.

С. Капсула 25 мг
[0106] Микронизированную форму I соединения А (27,8 г, D90=3,3 мкм) и микрокристаллическую целлюлозу (205,1 г) просеивали и перемешивали до гомогенности. Затем добавляли стеарат магния (0,67 г) и перемешивали в течение 5 минут. Конечной смесью, содержащей 25 мг соединения А, заполняли каждую капсулу размера №1 с получением продукта соединения А в виде капсул 25 мг. Кумулятивное выделение соединения А из капсулы 25 мг составляло 100% за 30 мин.


Пример 2: Противоопухолевое действие соединения А в ксенографтной модели Н716
[0107] Сообщалось, что клеточная линия NCI-H716 с колоректальной аденокарциномой человека имеет характеристики эндокринных клеток, таких как активность допа-декарбоксилазы и цитоплазматические плотные ядра. Для подтверждения нейроэндокринных характеристик указанной клеточной линии были получены подкожные опухоли NCI-H716 у голых мышей, и на опухолевой ткани подкожной ксенографтной модели проводили патологическую диагностику с окрашиванием гематоксилином и эозином (НЕ) и иммуногистохимическим (IHC) окрашиванием. Клетки NCI-H716 морфологически показали диффузный характер роста. Неопластические клетки были однородными и имели крупные ядра часто с видными ядрышками. Окрашивание IHC показало, что опухолевая ткань была положительной для CgA (хромогранин A), Syn (синаптофизин) и CD56 (молекула адгезии нейронных клеток 1), что является важным стандартом для диагностики нейроэндокринных опухолей. Индекс Ki-67 составлял примерно 80%. Основываясь на результатах морфологических и иммуногистохимических исследований, ксенографтную NCI-H716 можно считать нейроэндокринной карциномой. Материалы и способы
[0108] Линия опухолевых клеток человека: клеточную линию NCI-H716 приобрели у АТСС (CCL-251™) и инкубировали в среде RPMI1640 (Rosewell Park Memorial Institute 1640) с добавлением 10% фетальной бычьей сыворотки (FBS) при температуре 37°C в инкубаторе с 5% CO2.
[0109] Животные: мышей BALB/c, бестимусных самцов в возрасте 6-8 недель, приобретали в Shanghai SLAC Laboratory Animal Co. Ltd. Мышей размещали в особых условиях без патогенов. Животных содержали в следующих условиях: 12-часовые циклы день-ночь, температура 20~25°C и влажность 40% ~ 70%, животные имели свободный доступ к пище, стерильной в отношении излучения Со60, и к воде, стерилизованной автоклавированием. Мышей содержали по четыре штуки в клетке.
[0110] Испытуемое изделие и состав: соединение А растворяли в 0,5% CMC-Na, перемешивали вихревым способом в течение 3 мин, обрабатывали ультразвуком в течение 15 мин и хранили при температуре 4°C. Соединение А приготовили в концентрации 2,0, 4,0 и 8,0 мг/мл один раз в неделю.
[0111] Изучение инокуляции опухолевых клеток и противоопухолевых функций: когда клетки NCI-H716 достигали конфлюэнтности примерно 80-90%, их отделяли трипсином-ЭДТА и собирали после центрифугирования, затем суспендировали в бессывороточной среде. Каждой мыши подкожно в правый бок вводили 0,2 мл суспензии клеток, содержащей 5×106 опухолевых клеток с Martrigel (1:1).
[0112] Когда опухоли NCI-H716 достигли в среднем примерно 300 мм3, мышей с опухолью рандомизировали на группы и обрабатывали носителем и соединением А. Соединение А в 0,5% CMC-Na, как описано выше, вводили мышам в дозе 20, 40 и 80 мг/кг дважды в сутки в течение трех недель, а группа носителя получала 0,5% CMC-Na перорально дважды в сутки. Объем дозирования составлял 10 мл/кг массы тела. Дважды в неделю штангенциркулем измеряли длину и ширину опухоли и по следующей формуле рассчитывали объем опухоли (ОО): ОО=ширина2 × длина/2.
[0113] В конце исследования опухоли удаляли и рассчитывали ингибирование роста опухоли (TGI) и ингибирование массы опухоли (IRTW) по следующим уравнениям, где V0 - данные в день 0 (D0) (начальный день) и Vt - данные в день измерения t (Dt):
TGI=[1-(Vt-V0)лек.ср-во/(Vt-V0)носитель]×100%
IRTW(%)=[(TWноситель-TWлечение)/TWноситель]×100%
[0114] Статистический анализ: Для сравнения с контролем носителем применяли t-критерий Стьюдента. Различия считали статистически значимыми при Р<0,05.
Результаты
[0115] Как показано на фиг. 1, в модели NCI-H716 соединение А демонстрирует дозозависимый противоопухолевый эффект.В конце исследования рост опухоли Н716 был подавлен на 57,1%, 73,9% и 80,0% при введении соединения А в дозе 20, 40 и 80 мг/кг дважды в сутки, соответственно. Кроме того, соединение А уменьшало массу опухоли более чем на 60% при введении в средней и высокой дозах. Когда группу, обработанную соединением А, сравнивали с группой, обработанной носителем (таблица 7), то наблюдали статистически значимое различие. У мышей, обработанных соединением А, не наблюдалось каких-либо клинических признаков токсичности.

*, P<0,05; **, P<0,01 по сравнению с группой, обработанной носителем
[0116] Ксенографтную NCI-H716 можно рассматривать как нейроэндокринную карциному. Соединение А ингибировало рост NCI-H716 дозозависимым образом, что указывает на то, что соединение А будет иметь терапевтический потенциал для лечения нейроэндокринных карцином или нейроэндокринных опухолей в клинике.
Пример 3: Анализ жизнеспособности клеток на NCI-H716
Клеточная линия
NCI-H716: клеточная линия атерокарциномы человека, приобретенная у АТСС (Американская коллекция типовых культур).
Культуральная среда: RPMI 1640, содержащая 10% FBS.
Материалы
Соединение: Соединение А (партия №100223, чистота соединения 96,7%) было синтезирована в Hutchison Medipharma Limited;
Комплект ССК-8 (набор для подсчета клеток Cell Counting Kit-8): Марка: Dojindo, номер по каталогу СК04-01;
Считыватель микропланшетов: Модель: Labsystems Multiskan КЗ, марка: Thermo.
Обработка клеток
[0117] Клетки NCI-H716 разбавляли средой RPMI-1640, содержащей 10% FBS. 90 мкл разбавленных клеток добавляли в каждую лунку 96-луночного планшета, так что каждая лунка содержала 1×104 клеток. Затем клетки инкубировали при температуре 37°C 5% CO2 в течение ночи.
[0118] Испытуемое соединение, растворенное в ДМСО, разбавляли до 30, 10, 3,33, 1,11, 0,37, 0,12, 0,04, 0,01 мкМ бессывороточной средой RPMI-1640, содержащей 5% ДМСО. Затем 10 мкл разбавленного соединения добавляли к 90 мкл вышеуказанных клеточных культур (конечная концентрация ДМСО в реакционной смеси должна составлять 0,5%) и инкубировали при температуре 37°C и 5% CO2 в течение 72 часов. Анализ ССК-8
[0119] 10 мкл раствора ССК-8 добавляли к клеткам и инкубировали при температуре 37°C и 5% СО2 в течение 1 часа. Оптическую плотность каждой лунки измеряли при 450 нм и 630 нм, соответственно, на Labsystems Multiskan K.
Анализ данных

[0D соед.]450-630: оптическая плотность лунки, содержащей клетки, обработанные соединением;
[OD клетки]450-630: оптическая плотность лунок, содержащих не обработанные соединением клетки;
[OD фона] 450-630: оптическая плотность лунок, содержащих только культуральную среду.
[0120] IC50: IC50 рассчитывали на основе кривой ингибирования, установленной программным обеспечением XL-Fit 2.0.
[0121] Результат: IC50 соединения А при ингибировании жизнеспособности клеток NCI-H716 определили как 0,159 мкМ.
Пример 4: Фармакокинетика соединения А у собак породы бигль после однократного перорального введения двух различных составов в капсулах, содержащих соединение А
[0122] Данное исследование ФК у собак включало два периода перекрестных исследований с отмывочным периодом, составляющим 1 неделю. Двенадцать собак породы бигль разделяли на две группы, по 3 самца и 3 самки в каждой группе. Дозировка составляла 30 мг/кг, по одной капсуле на каждую собаку. В период 1 группе 1 вводили капсулу А (композицию микронизированного соединения А, 50 мг капсулы №8, приготовленную в примере 1), а группе 2 вводили капсулу В (композиция не микронизированного соединения А). Капсулу В получали способом, описанным выше для 50 мг капсулы №8. Каждая капсула А и капсула В содержат 50 мг соединения А. Разница между капсулой А и капсулой В заключается в том, что соединение А в капсуле А представляет собой микронизированную форму I соединения А с D90=8,1 мкм, тогда как соединение А в капсуле В представляет собой не микронизированную форму I соединения А с D90=39,2 мкм. Кумулятивное выделение соединения А составляло 98,1% из 50 мг капсулы №8 и 75,1% из капсулы В за 30 мин.
[0123] После отмывочного периода, составлявшего одну неделю, группе 1 вводили капсулу В, а группе 2 вводили капсулу А.
[0124] Образцы крови собирали в разные моменты времени после дозирования в каждом периоде. Подробная информация о группах и временных точках для сбора крови приведена в таблице 8.

[0125] Для предварительной обработки плазмы собаки применяли метод осаждения белка с ацетонитрилом, содержащим фенацетин (внутренний стандарт, IS). Концентрацию соединения А в плазме определяли с помощью жидкостной хроматографии-тандемной масс-спектрометрии (ЖХ-МС/МС) в условиях градиентного элюирования мобильной фазой А (деионизированная вода, содержащая 0,1% муравьиной кислоты) и мобильной фазой В (ацетонитрил, содержащий 0,1% муравьиной кислоты). Использованные хроматографические условия приведены в таблице 9.


Анализ образцов
[0126] Для анализа образцов использовали колонку Symmetry С18 (2,1×50 мм, 3,5 мкм). Применяли мониторинг множественных реакций (MRM) и положительный режим с ионизацией электрораспылением (ESI), и соответствующие детектирующие ионы (Q1/Q3) соединения А составляли 481,2/329,3. ФК-параметры рассчитывали с использованием некомпартментного анализа при помощи программного обеспечения Kinetica. Биоэквивалентность для двух капсул оценивали при помощи статистических тестов по содержанию соединения А в плазме с помощью метода доверительных интервалов и по Tmax соединения А в плазме с помощью рангового критерия Фридмана.
А. Подготовка исходного раствора и рабочих растворов
[0127] Соединение А в виде порошка (23,68 мг) взвешивали и растворяли в 942 мкл ДМСО. Применяли вихревую мешалку и ультразвук до полного растворения порошка. Основной исходный раствор соединения А был получен при 25 мг/мл. Исходный раствор соединения А (40 мкл) добавляли в 960 мкл ацетонитрила. После перемешивания вихревым способом получали вторичный исходный раствор соединения А при 1,0 мг/мл.
[0128] Подготовка рабочих растворов для калибровочного стандарта (С) и контроля качества (QC) показана в таблице 10 и таблице 11, соответственно.


В. Подготовка калибровочной кривой и образцов для контроля качества
[0129] Рабочий раствор калибровочного стандарта (С) или контрольного образца (QC) (10 мкл) добавляли в 190 мкл плазмы собак, не получавших ранее лечения. После перемешивания вихревым способом в течение 1 мин плазму с добавкой (50 мкл) переносили в пустую пробирку объемом 1,5 мл. Для осаждения белка добавляли 150 мкл ацетонитрила, содержащего 500 нг/мл фенацетина (внутренний стандарт, IS). Пробирку встряхивали в течение 2 мин, а затем центрифугировали при 14000 об/мин при температуре 4°C в течение 10 мин. Супернатант (150 мкл) переносили в пробирку объемом 1,5 мл и затем добавляли 150 мкл деионизированной воды. После перемешивания вихревым способом в течение 1 мин для анализа использовали одну аликвоту конечного раствора (10 мкл).
[0130] Пустой образец был таким же, как и образцы для калибровочной кривой, за исключением ацетонитрила вместо рабочих растворов соединений.
[0131] Двойной пустой образец готовили в том же виде, что и пустой образец, за исключением того, что добавленный раствор ацетонитрила не содержал фенацетина.
С. Получение образцов плазмы
[0132] Образец плазмы (по 50 мкл каждый), полученный в ходе эксперимента на животных, переносили в пробирку объемом 1,5 мл и для осаждения белка добавляли 150 мкл ацетонитрила, содержащего 500 нг/мл фленацетина. После перемешивания вихревым способом в течение в течение 2 мин смесь центрифугировали (14000 об/мин при 4°C в течение 10 мин). Супернатант (150 мкл) переносили в пустую пробирку объемом 1,5 мл и затем добавляли 150 мкл деионизированной воды. После перемешивания вихревым способом в течение 1 мин одну аликвоту конечного раствора (10 мкл) вводили в систему ЖХ-МС/МС для анализа.
Анализ данных
[0133] Пиковые области соединения А и IS были интегрированы при помощи программного обеспечения Analyst 1.4.1. Стандартные кривые были получены по стандартным образцам при построении площадей пиков соотношений соединения А к IS против теоретических концентраций соединения А. Кривая регрессии соединения А была линейной с весовым коэффициентом 1/х2. Теоретические концентрации соединения А в калибровочных стандартах составляли 2,50, 5,0, 10, 20, 50, 200, 500 и 1000 нг/мл, соответственно. Концентрации соединения А в образцах плазмы собак определяли с использованием стандартных кривых. Фармакокинетические параметры рассчитывали на основе данных концентрация-время в плазме с использованием программного обеспечения Thermo Kinetica® (версия 4.4.1, Thermo Electron Corporation). Использовали некомпартментный анализ. Биоэквивалентность двух указанных капсул оценивали в ходе статистических тестов по содержанию соединения А в плазме собак методом доверительных интервалов и по Tmax соединения А в плазме собак с помощью рангового критерия Фридмана.
Результаты
[0134] Профиль ФК соединения А у каждой собаки показан на фиг. 2 - фиг. 5. Средние концентрации соединения А в плазме, нанесенные на график по временным точкам, показаны на фиг. 6.
[0135] Фактические дозы для собак 3, 4, 6, 7, 8 и 10 были бы ниже, чем в предусмотрено дизайном эксперимента, на основании времени появления и вида рвоты, поэтому ФК данные этих собак не использовали для вычисления среднего значения. Средние значения основных ФК-параметров у собак приведены в таблице 12.

Примечание: ФК-параметры у собак под номерами 3, 4, 6, 7, 8 и 10 не использовались для расчета среднего из-за рвоты после дозирования.
[0136] Основываясь на ФК-профилях соединения А и наблюдениях у клетки, капсула А обладает меньшей вариабельностью между пациентами и вызвала меньше случаев рвоты, чем капсула В. По сравнению с капсулой В капсула А имела более высокое значение AUC после однократного перорального введения у собак. Не было существенной статистической разницы в среднем значении Tmax для капсулы А и капсулы В у собак породы бигль.
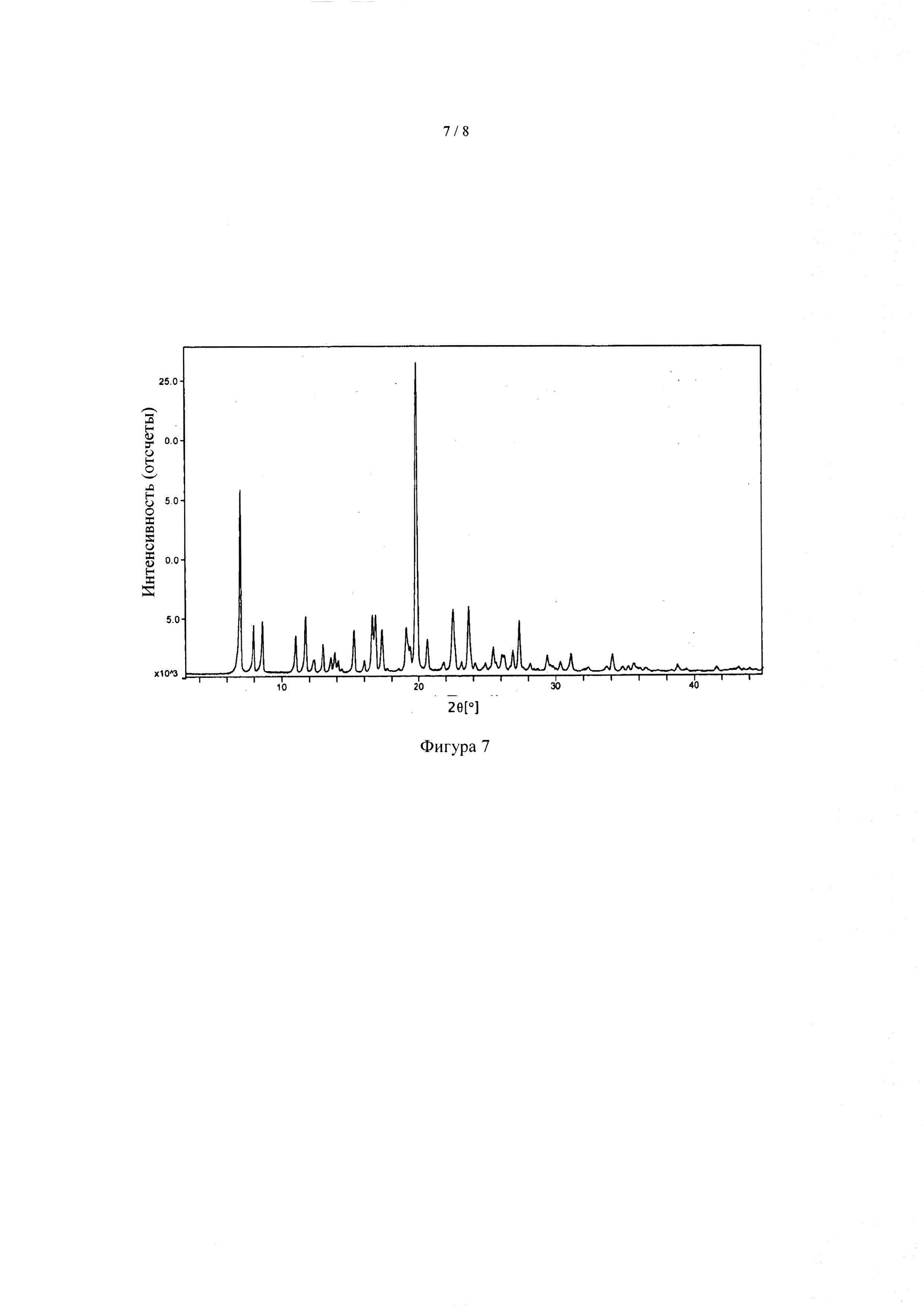
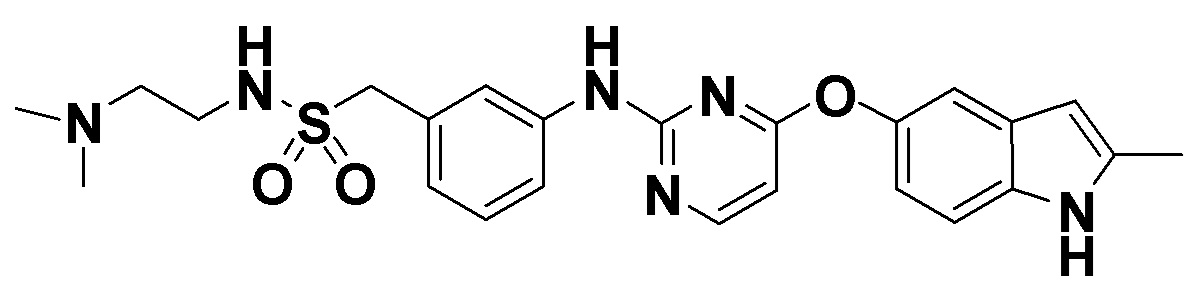
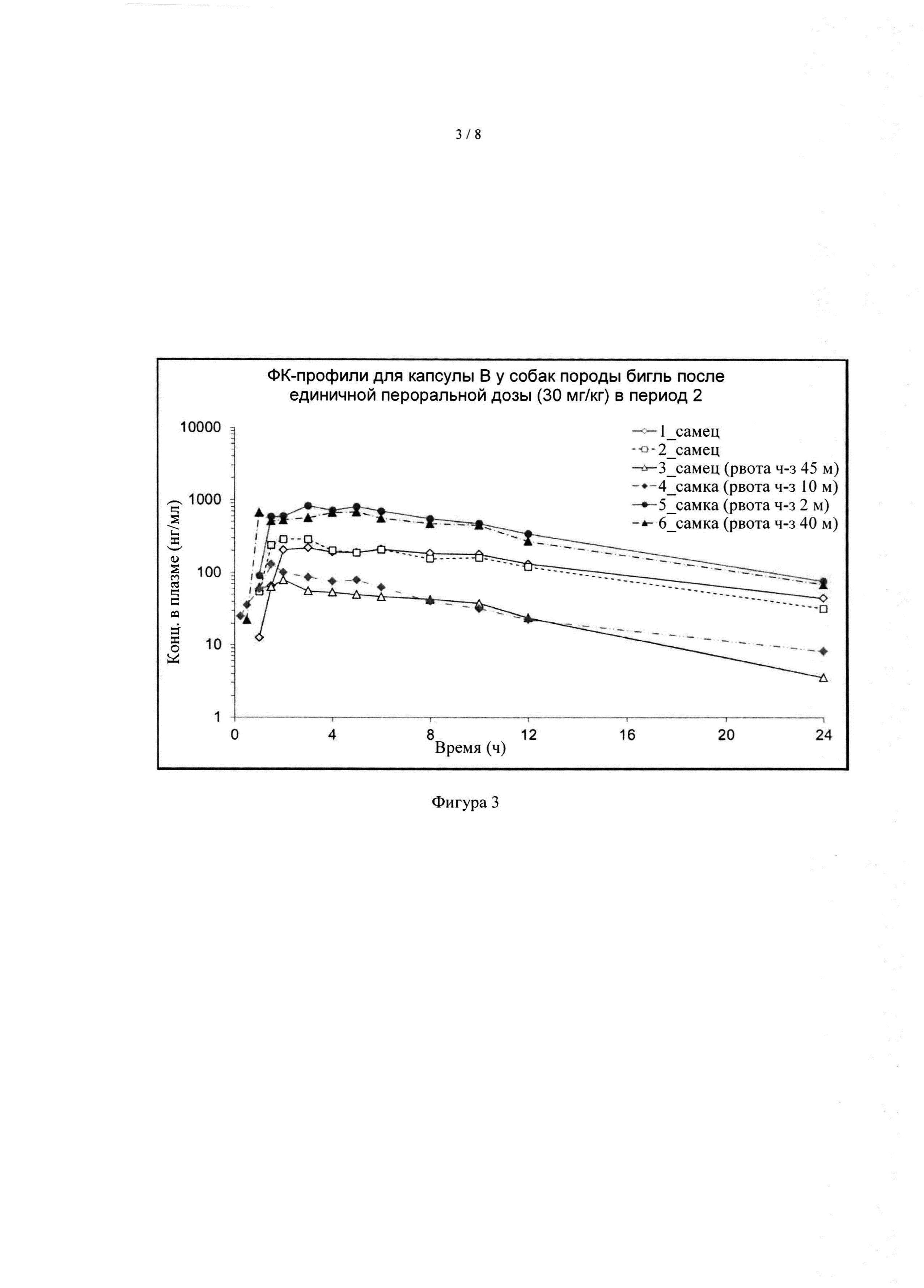
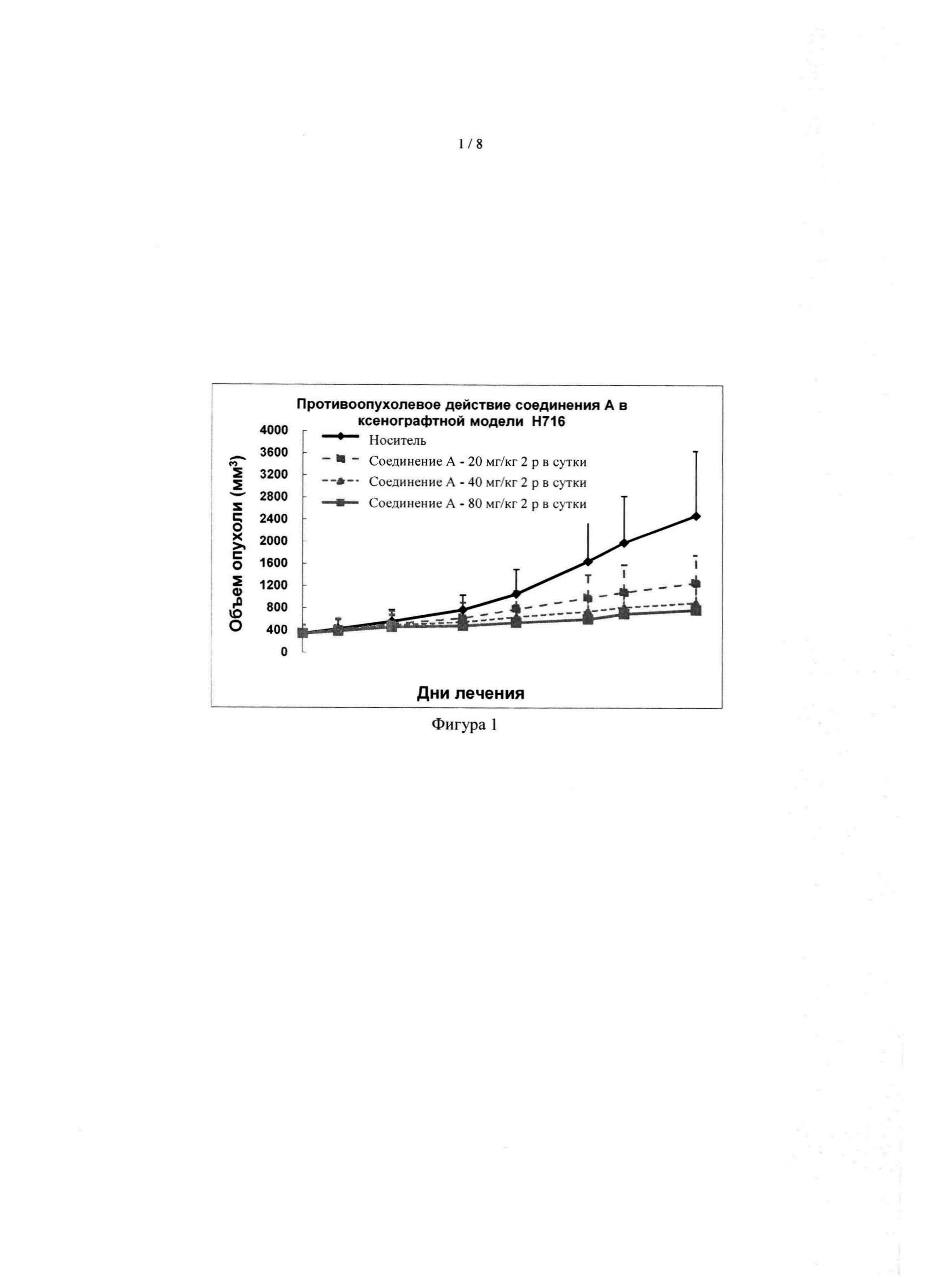
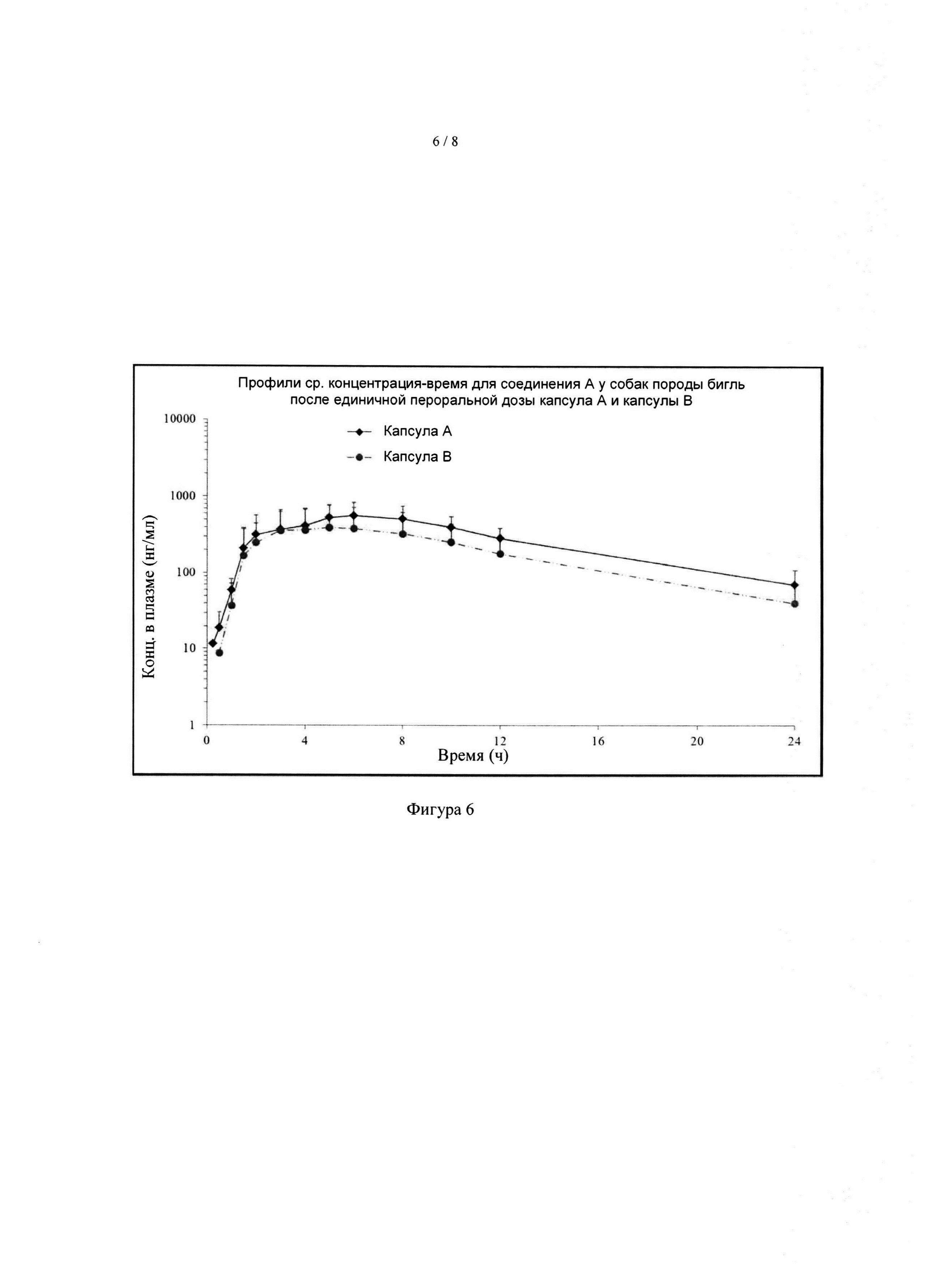
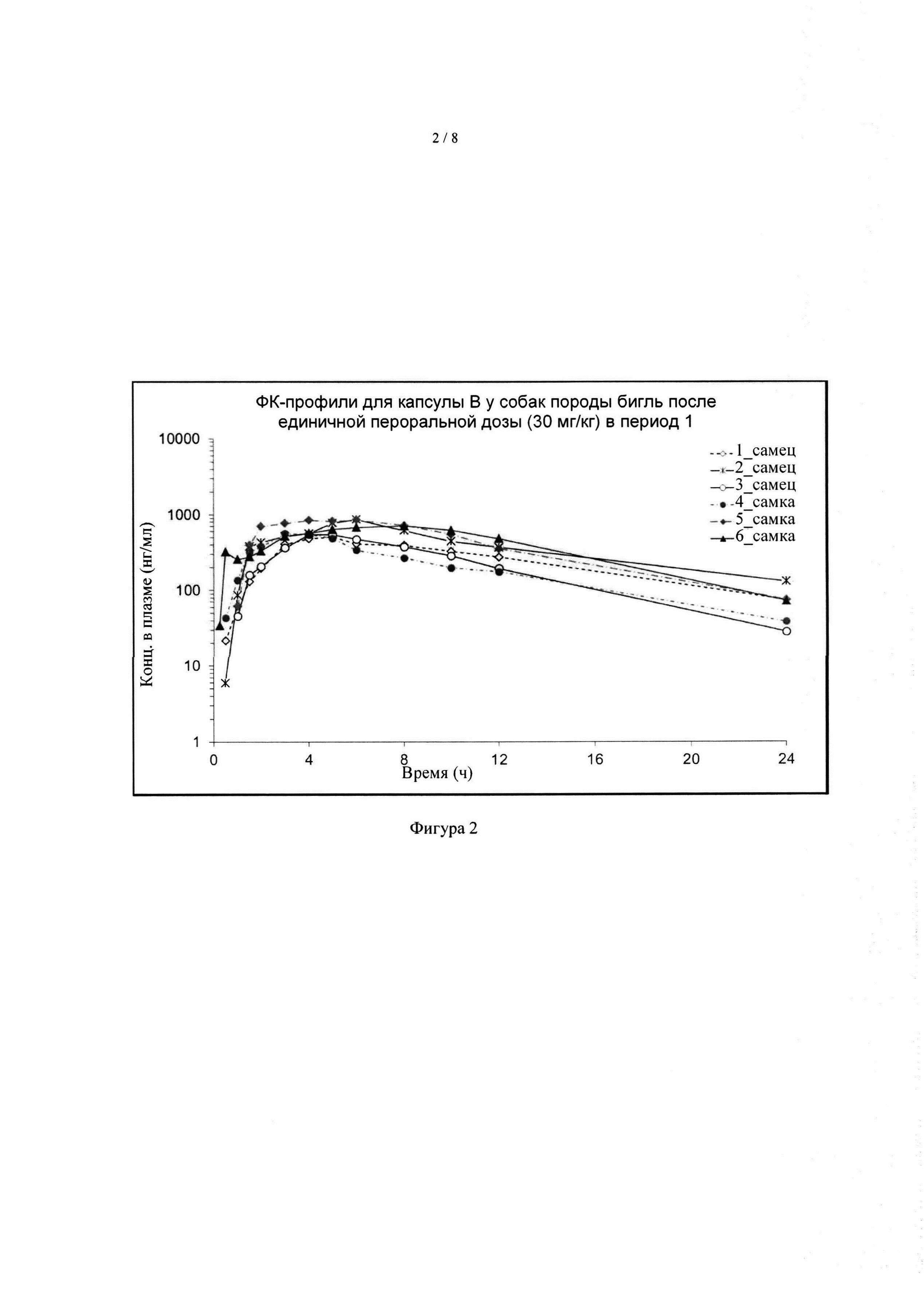
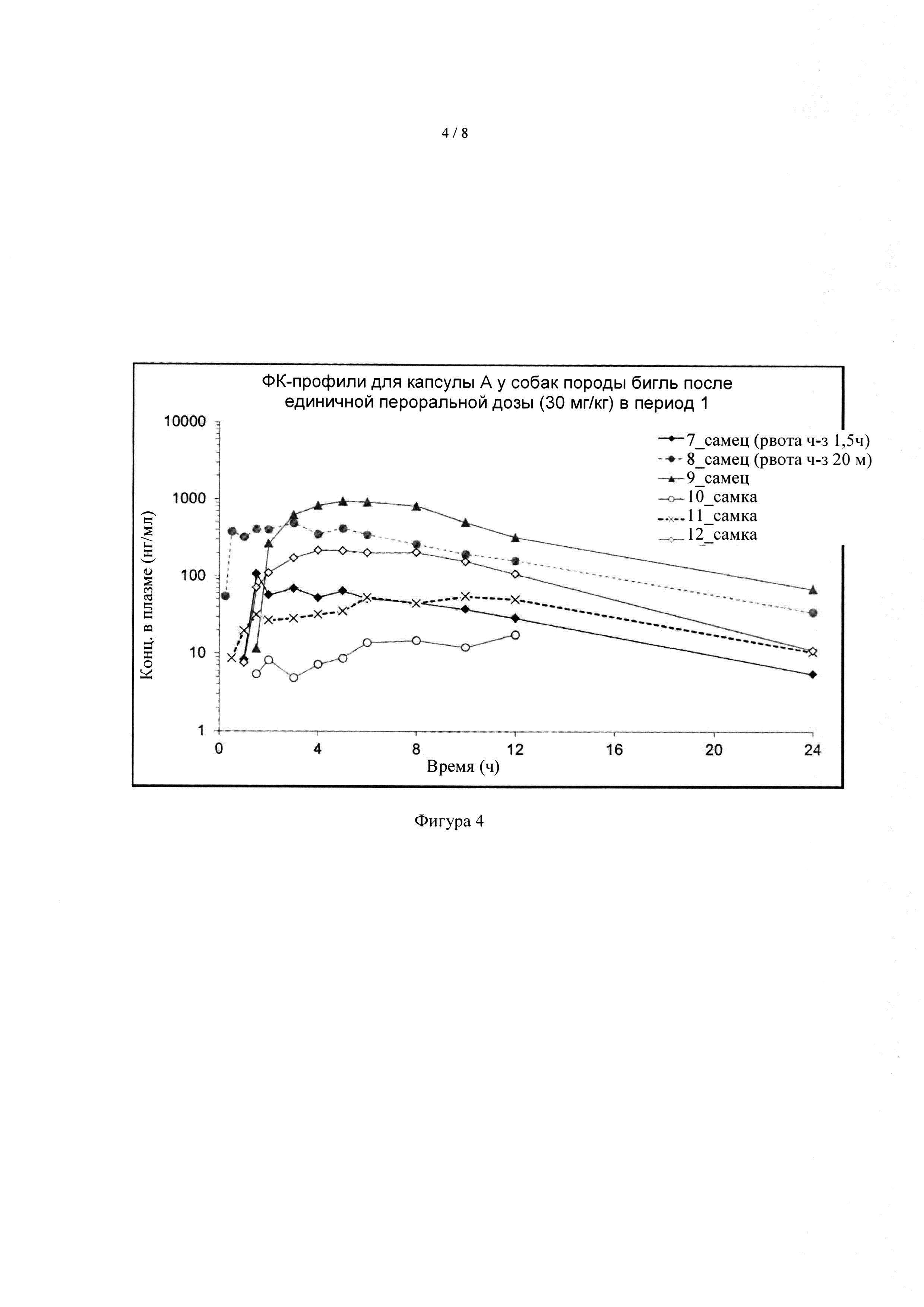
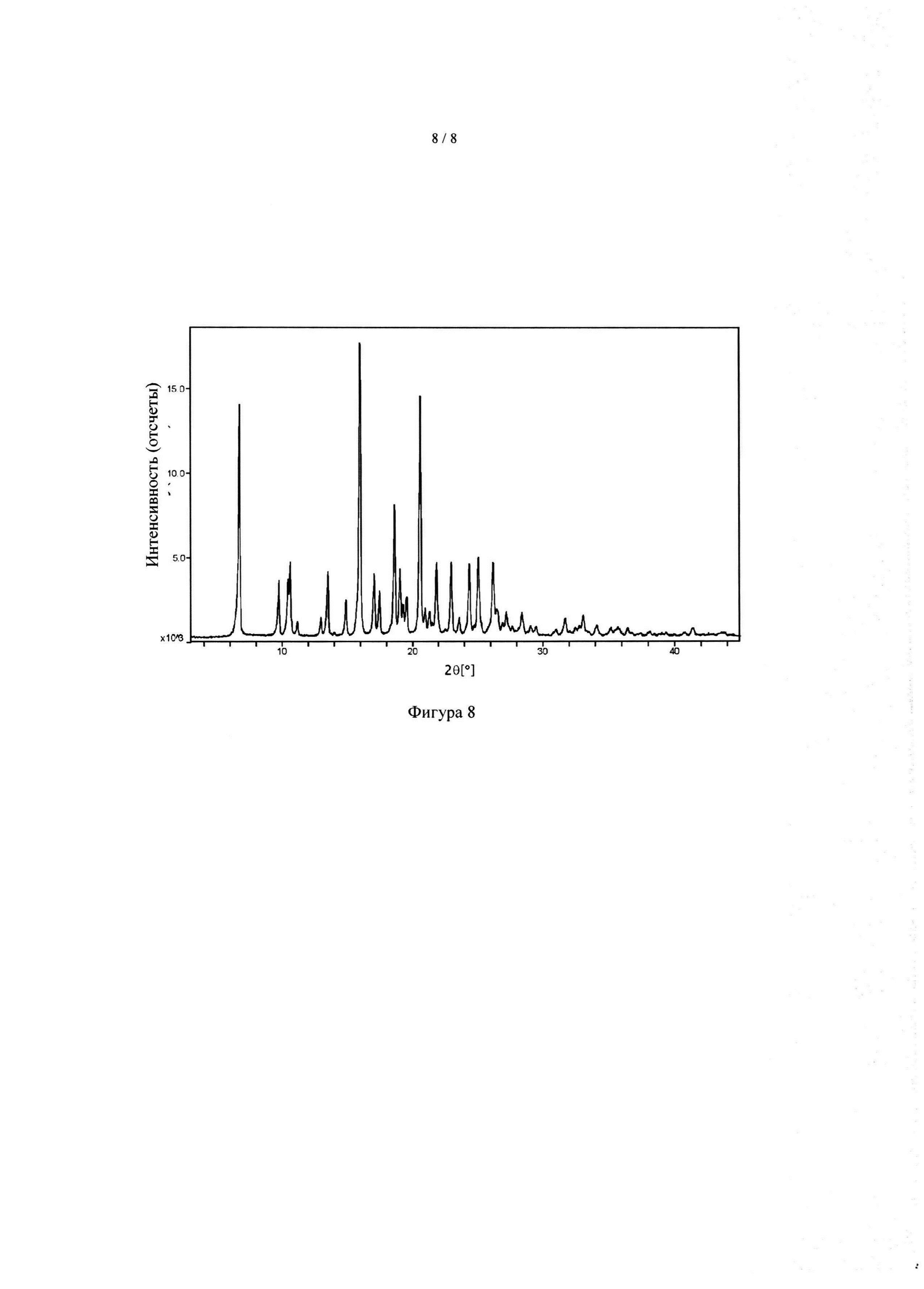
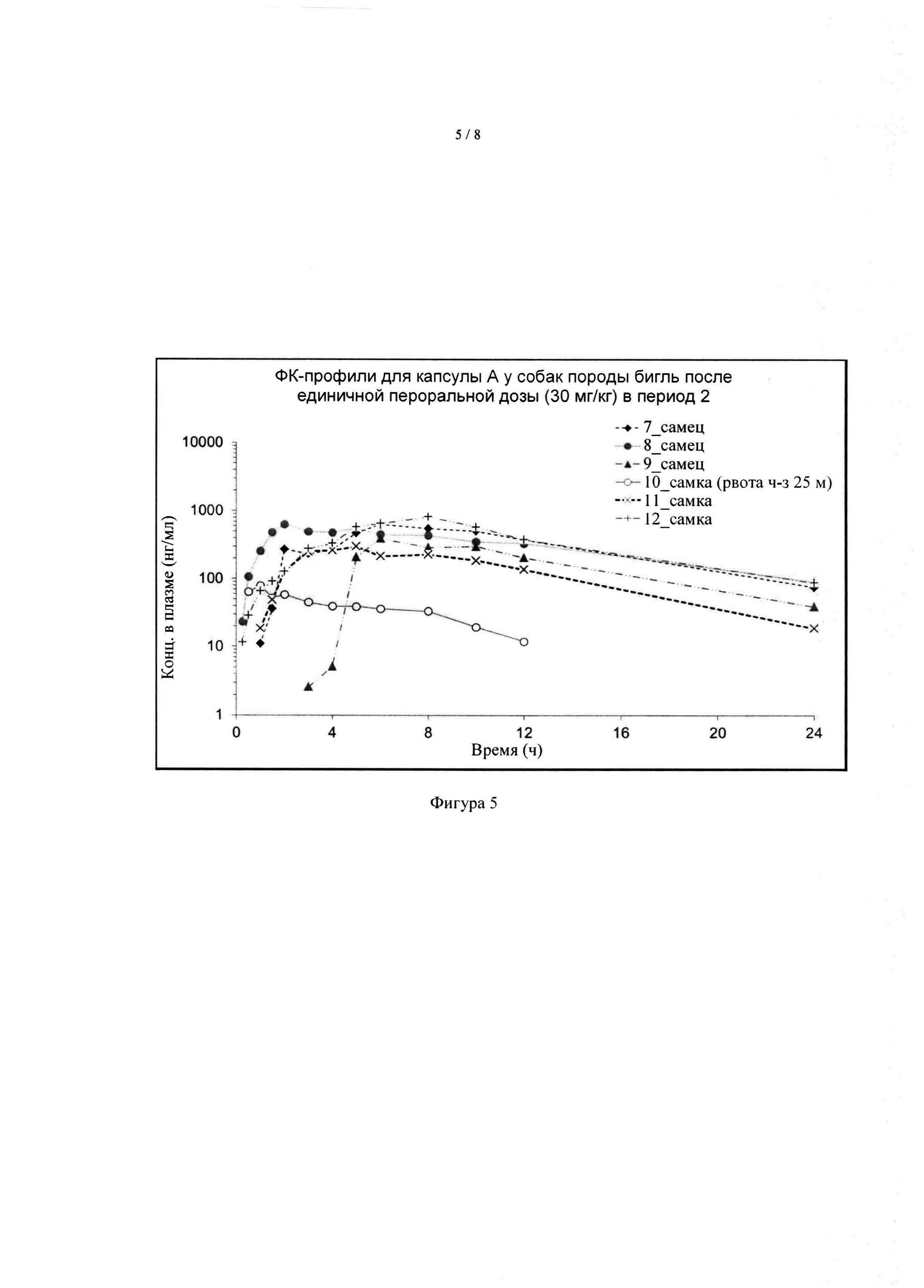
Соединение, некоторые его новые формы, фармацевтические композиции на его основе и способы получения и применения
Соединения хиназолина
Конденсированные гетероарилы и их применение
Соединения пиримидинилиндола
Пирролопиримидиновые соединения и их применения
Замещенные пиридопиразины как новые ингибиторы syk

