Результат интеллектуальной деятельности: ПЕРОРАЛЬНАЯ ОСМОТИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ВИЛДАГЛИПТИНА
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ
В настоящем изобретении предложена новая осмотическая фармацевтическая композиция, содержащая вилдаглиптин или его соль. В настоящем изобретении дополнительно предложен способ получения таких композиций.
УРОВЕНЬ ТЕХНИКИ
Сахарный диабет является распространенным заболеванием, более распространенным в развитых странах. Он характеризуется своими клиническими проявлениями, а именно инсулиннезависимой или приобретенной формой, известной также как диабет 2 типа, и инсулинозависимой или врожденной формой, известной также как диабет 1 типа. Диабет представляет собой метаболическое заболевание, при котором наблюдается высокий уровень сахара в крови в течение длительного периода. Данное расстройство связано с высокой заболеваемостью и смертностью.
Ингибиторы дипептидилпептидазы-4 (ДПП-4) представляют собой класс пероральных лекарственных средств, которые были предложены в 2006 г., указанные ингибиторы представляются простыми в применении и не требуют регулярного контроля глюкозы или регулировок дозы. Ингибиторы ДПП-4 действуют путем ингибирования фермента ДПП-4, многофункционального трансмембранного гликопротеинового фермента, который отщепляет N-терминальные дипептиды от полипептидов с L-пролином или L-аланином в предпоследнем положении. Вилдаглиптин является перорально активным ингибитором ДПП-4. После приема пищи выделяется инсулинотропный гормон глюкагоноподобный пептид-1 (ГПП-1), который в свою очередь индуцирует высвобождение инсулина из поджелудочной железы. Некоторое количество ГПП-1 деактивируется ДПП-4, присутствующей в плазме и эндотелии капилляров кишечника. Поэтому, если ДПП-4 ингибируется, появляется больше доступного ГЛП-1 для стимуляции выделения инсулина из поджелудочной железы. Преимущество этого механизма выделения инсулина заключается в том, что инсулин выделяется только в ответ на прием пищи. Вследствие этого ассоциированные с гипогликемией осложнения, связанные с другими лекарственными средствами при сахарном диабете, менее вероятны при применении ингибитора ДПП-4. В многочисленных клинических исследованиях продемонстрировано, что вилдаглиптин - самостоятельно или в комбинации с другими антидиабетическими агентами - представляет собой эффективное дополнение к диете и физическим упражнениям для улучшения контроля уровня глюкозы в крови у взрослых с сахарным диабетом 2 типа. Вилдаглиптин реализуется на рынке в Европе компанией Novartis в виде таблеток под товарным знаком GALVUS®. Рекомендованная суточная доза вилдаглиптина составляет 100 мг, и указанную суточную дозу вводят в виде одной дозы в количестве 50 мг утром и одной дозы в количестве 50 мг вечером.
Патент США №6,166,063 раскрывает соединение вилдаглиптин, представляющее собой ингибитор ДПП-4, его применение при лечении сахарного диабета 2 типа и фармацевтическую композицию, содержащую указанное соединение. Патент США №6,303,661 раскрывает способы применения перорального ингибитора активности фермента ДПП-4 или ДПП-4-подобного фермента для снижения уровня глюкозы в крови после приема пищи. Патент США №8,143,217 раскрывает способы использования вилдаглиптина в комбинации с инсулином для лечения случаев гипогликемии у больных сахарным диабетом. Заявка на Европейский патент ЕР 1,537,880 А1 относится к составу с замедленным высвобождением, содержащему ингибитор ДПП-4 (вилдаглиптин) и содержащему гидрофильный полимер. Европейский Патент №ЕР 2,191,824 В1 раскрывает состав с замедленным высвобождением, содержащий ингибитор ДПП-4 (вилдаглиптин) и содержащий гидроксипропилметилцеллюлозу. Заявка на Европейский патент ЕР 2,578,208 А1 раскрывает твердую лекарственную форму, полученную путем брикетного прессования либо сжатия между валами и характеризующуюся тем, что она содержит ингибитор ДПП-4. Публикация РСТ №WO 2014/029841 А1 раскрывает композиции с замедленным высвобождением, содержащие амино-С2-С6-алкилнитрат и фиксированные комбинации доз вилдаглиптина.
Существующий продукт вилдаглиптина - это таблетка, полученная с помощью непосредственного прессования с содержанием лекарственного средства 25%. Приверженность пациента к режиму введения лекарственного средства косвенно коррелирует с частотой приема, т.е. большая приверженность наблюдается при введении один раз в сутки по сравнению с двукратным введением. Большинство доступных в настоящее время данных по вилдаглиптину получено при изучении лекарственного средства немедленного высвобождения (GALVUS®) в режиме дозирования два раза в сутки. Существует потребность в разработке состава, содержащего вилдаглиптин для введения один раз в сутки, которая была бы легкой, удобной и обладала минимальными побочными эффектами.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В общем аспекте настоящего изобретения предложена осмотическая фармацевтическая композиция, содержащая: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий вилдаглиптин или его соль и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, которое окружает указанное ядро; (с) покрытие для замедленного высвобождения, содержащее ацетат целлюлозы, которое окружает указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий вилдаглиптин или его соль, который окружает указанное покрытие для замедленного высвобождения.
В другом аспекте настоящего изобретения предложена осмотическая фармацевтическая композиция, содержащая: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, которое окружает указанное ядро; (с) покрытие для замедленного высвобождения, которое содержит от приблизительно 70% до приблизительно 95% ацетатцеллюлозы по массе, которое окружает указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 15 мг до приблизительно 35 мг вилдаглиптина или его соли, который окружает указанное покрытие для замедленного высвобождения.
В другом аспекте настоящего изобретения предложена осмотическая фармацевтическая композиция, содержащая: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, которое окружает указанное ядро; (с) покрытие для замедленного высвобождения, которое содержит от приблизительно 70% до приблизительно 95% ацетатцеллюлозы по массе, которое окружает указанное изолирующее покрытие; (d) слой лекарства для немедленного высвобождения, содержащий от приблизительно 15 мг до приблизительно 35 мг вилдаглиптина или его соли, который окружает указанное покрытие для замедленного высвобождения, и при этом указанная композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 10% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации указанная композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 70% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/ми при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/ми и при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин и при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме крови в диапазоне от приблизительно 5 нг/мл до приблизительно 250 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки. В другом варианте реализации композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме крови в диапазоне от приблизительно 5 нг/мл до приблизительно 30 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки.
В другом варианте реализации композиция способна снижать активность фермента ДДП-4 в плазме крови на величину от приблизительно 60% до приблизительно 90% на протяжении периода, равного 24 часам. В другом варианте реализации композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении периода, равного 24 часам.
В другом аспекте изобретения предложена осмотическая фармацевтическая композиция, содержащая: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий приблизительно 75 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий от приблизительно 80 мг до приблизительно 90 мг полиэтиленоксида, имеющего молекулярную массу от приблизительно 5000000 до приблизительно 8000000; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее от приблизительно 35 мг до приблизительно 45 мг ацетатцеллюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий приблизительно 25 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C, и/или от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом аспекте изобретения предложен набор, содержащий осмотическую фармацевтическую композицию, содержащую: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий вилдаглиптин или его соль и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий вилдаглиптин или его соль, окружающий указанное покрытие для замедленного высвобождения.
В другом аспекте изобретения предложен набор, содержащий осмотическую фармацевтическую композицию, содержащую: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее в массовом отношении от приблизительно 70% до приблизительно 95% ацетатцеллюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 15 мг до приблизительно 35 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 10% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1Н HCl при 37°C ± 0,5°C.
В другом аспекте изобретения предложен способ лечения диабета 2 типа, при этом указанный способ включает введение пациенту, нуждающемуся в этом, осмотической фармацевтической композиции, содержащей: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий вилдаглиптин или его соль и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) ацетатцеллюлозное покрытие для замедленного высвобождения, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий вилдаглиптин или его соль, окружающий указанное покрытие для замедленного высвобождения.
В другом аспекте изобретения предложен способ лечения диабета 2 типа, при этом указанный способ включает введение пациенту, нуждающемуся в этом, осмотической фармацевтической композиции, содержащей: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее в массовом отношении от приблизительно 70% до приблизительно 95% ацетатцеллюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 15 мг до приблизительно 35 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 10% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1Н HCl при 37°C ± 0,5°C.
В другом аспекте изобретения предложен способ получения осмотической фармацевтической композиции, содержащей: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее в массовом отношении от приблизительно 70% до приблизительно 95% ацетатцеллюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 15 мг до приблизительно 35 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro проявляет профиль высвобождения, при котором от приблизительно 10% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1Н HCl при 37°C ± 0,5°C, причем указанный способ включает следующие этапы:
(a) приготовление слоя лекарственного средства в виде гранул, содержащих вилдаглиптин или его соль и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество,
(b) приготовление осмотического слоя в виде гранул, содержащих полиэтиленоксид,
(c) прессование гранул слоя лекарственного средства и осмотического слоя с целью формирования ядра,
(d) нанесение изолирующего покрытия на ядро, полученное на этапе (с),
(e) нанесение на указанное изолирующее покрытие, полученное на этапе (d), покрытия для замедленного высвобождения, содержащего в массовом отношении от приблизительно 70% до приблизительно 95% ацетатцеллюлозы, и
(f) нанесение на покрытие для замедленного высвобождения, полученное на этапе (е), слоя для немедленного высвобождения лекарственного средства, содержащего вилдаглиптин или его соль, с целью получения осмотической фармацевтической композиции.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фигуре 1А и на Фигуре 1В представлен профиль растворения композиции 1А и композиции 1В вилдаглиптина в условиях in vitro, соответственно, в кислотной среде и буферной среде.
На Фигуре 2А представлена степень ингибирования ДПП-4 (в процентах) после введения композиции 1А согласно настоящему изобретению и лекарственного средства GALVUS®.
На Фигуре 2В представлен профиль изменения концентрации лекарственного средства в плазме крови после введения композиции 1А согласно настоящему изобретению и лекарственного средства GALVUS®.
На Фигуре 3А представлена степень ингибирования ДПП-4 (в процентах) после введения композиции 1В согласно настоящему изобретению и лекарственного средства GALVUS®.
На Фигуре 3В представлен профиль изменения концентрации лекарственного средства в плазме крови после введения композиции 1В согласно настоящему изобретению и лекарственного средства GALVUS®.
На Фигуре 4 представлен сравнительный профиль растворения in vitro композиций вилдаглиптина 6 А и 6 В, в кислотной среде и буферной среде.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Основной аспект изобретения раскрывает осмотическую фармацевтическую композицию, содержащую: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий вилдаглиптин или его соль и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее ацетат целлюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий вилдаглиптин или его соль, окружающий указанное покрытие для замедленного высвобождения.
Термин "осмотическая фармацевтическая композиция" относится к усовершенствованной системе доставки твердого лекарственного средства с контролируемым высвобождением с покрытием для замедленного высвобождения и одним или несколькими малыми каналами в нем, просверленными с помощью лазера. Термин "ядро" относится к матричному, или двухслойному, или трехслойному, или многослойному компоненту, содержащему лекарственное средство, осмотический агент и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество.
Термин "покрытие для замедленного высвобождения" относится к полупроницаемой мембране, регулирующей высвобождение лекарственного средства.
В другом аспекте изобретения предложена осмотическая фармацевтическая композиция, содержащая: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее в массовом отношении от приблизительно 70% до приблизительно 95% ацетатцеллюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 15 мг до приблизительно 35 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения.
В другом аспекте изобретения предложена осмотическая фармацевтическая композиция, содержащая: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее в массовом отношении от приблизительно 70% до приблизительно 95% ацетатцеллюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 15 мг до приблизительно 35 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 10% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C.
В одном варианте реализации, композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 15% до приблизительно 25% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений в 0,1 Н HCl при 37°C ± 0,5°C. В другом случае композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 18% до приблизительно 22% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений в 0,1 Н HCl при 37°C ± 0,5°C. В другом случае композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений в 0,1 Н HCl при 37°C ± 0,5°C.
В другом варианте реализации композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 70% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C. В другом случае композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 80% до приблизительно 90% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C. В другом случае композиция в условиях in vitro демонстрирует профиль высвобождения, при котором приблизительно 85% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом случае композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором приблизительно 25% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C.
В другом варианте реализации композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом случае композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором приблизительно 25% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C.
В другом варианте реализации композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом случае композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором приблизительно 90% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом случае композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором приблизительно 90% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме крови в диапазоне от приблизительно 5 нг/мл до приблизительно 250 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки. В другом варианте реализации композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме крови в диапазоне от приблизительно 10 нг/мл до приблизительно 200 нг/мл, или от приблизительно 20 нг/мл до приблизительно 150 нг/мл, или от приблизительно 30 нг/мл до приблизительно 100 нг/мл, или от приблизительно 40 нг/мл до приблизительно 50 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки. В другом варианте реализации композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме крови в диапазоне от приблизительно 5 нг/мл до приблизительно 30 нг/мл через 24 часа после перорального введения один раз в сутки. В другом случае композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме крови в диапазоне от приблизительно 10 нг/мл до приблизительно 20 нг/мл через 24 часа после перорального введения один раз в сутки.
В другом варианте реализации композиция способна снижать активность фермента ДДП-4 в плазме крови на величину от приблизительно 60% до приблизительно 90% на протяжении периода, равного 24 часам. В другом случае композиция способна снижать активность фермента ДДП-4 в плазме крови на величину от приблизительно 70% до приблизительно 80% на протяжении периода, равного 24 часам. В другом случае композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении периода, равного 24 часам. В другом варианте реализации композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении периода, равного 24 часам. В другом варианте реализации композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении 20 часов. В другом варианте реализации композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении 18 часов. В другом варианте реализации композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении 16 часов. В другом варианте реализации композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении 14 часов. В другом варианте реализации композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении 12 часов. В другом варианте реализации композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении 10 часов.
В другом варианте реализации ядро содержит слой лекарственного средства, содержащий от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли. В другом варианте реализации ядро содержит слой лекарственного средства, содержащий от приблизительно 45 мг до приблизительно 90 мг, или от приблизительно 55 мг до приблизительно 85 мг или от приблизительно 65 мг до приблизительно 75 мг вилдаглиптина или его соли. В другом случае ядро содержит слой лекарственного средства, содержащий приблизительно 40 мг, или приблизительно 45 мг, или приблизительно 50 мг, или приблизительно 55 мг, или приблизительно 60 мг, или приблизительно 65 мг, или приблизительно 70 мг, или приблизительно 75 мг, или приблизительно 80 мг, или приблизительно 85 мг или приблизительно 90 мг вилдаглиптина или его соли. В другом варианте реализации слой лекарственного средства содержит приблизительно 75 мг вилдаглиптина или его соли.
В дополнительном варианте реализации слой лекарственного средства содержит фармацевтически приемлемые вспомогательные вещества, такие как набухающие полимеры, такие как полиэтиленоксид с молекулярной массой от приблизительно 100000 до 200000, осмотические агенты, выбранные из группы, состоящей из сахарозы, ксилитола, глюкозы, лактозы, маннитола, хлорида натрия, хлорида калия, эфиров целлюлозы, мальтодекстринов и циклодекстринов. Предпочтительным осмотическим агентом является маннитол и смазывающие (скользящие) вещества, такие как стеарат магния, стеарат цинка, стеарилфумарат натрия. В другом варианте реализации слой лекарственного средства содержит полиэтиленоксид с молекулярной массой от приблизительно 100000 до 200000 в количестве от приблизительно 170 мг до приблизительно 200 мг, или от приблизительно 188 мг до приблизительно 190 мг, или приблизительно 195 мг. В другом варианте реализации слой лекарственного средства содержит осмотический агент маннитол в количестве от приблизительно 30 мг до приблизительно 50 мг, или приблизительно 36 мг.
В другом варианте реализации ядро сдержит осмотический слой, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000. В другом варианте реализации осмотический слой содержит полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000 и присутствует в количестве от приблизительно 70 мг до приблизительно 100 мг, или от приблизительно 80 мг до приблизительно 90 мг. В другом случае осмотический слой содержит полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000 и присутствует в количестве приблизительно 85 мг или 90 мг, или 95 мг.
В другом варианте реализации осмотический слой содержит фармацевтически приемлемые вспомогательные вещества, такие как осмотические агенты, выбранные из группы, состоящей из сахарозы, ксилитола, глюкозы, лактозы, маннитола, хлорида натрия, хлорида калия, эфиров целлюлозы, мальтодекстринов и циклодекстринов. Предпочтительным осмотическим агентом является хлорид натрия, связывающее вещество такое, как гидроксипропилметилцеллюлоза (ГПМЦ), имеющее вязкость в диапазоне от 3сР до 6сР в 2%-ом растворе, и скользящие вещества, такие как стеарат магния, стеарат цинка, стеарилфумарат натрия. В другом варианте реализации осмотический слой содержит осмотический агент, представляющий собой хлорид натрия, в количестве от приблизительно 30 мг до приблизительно 50 мг, или приблизительно 45 мг. В другом варианте реализации осмотический агент, присутствующий в слое лекарственного средства отличается от такового в осмотическом слое. В другом варианте реализации осмотический слой дополнительно содержит гидроксипропилметилцеллюлозу от приблизительно 3% до приблизительно 7% по массе.
В другом варианте реализации массовое отношение вилдаглиптина или его соли и полиэтиленоксида, имеющего молекулярную массу от приблизительно 5000000 до приблизительно 8000000, в ядре варьирует от приблизительно 1:1 до приблизительно 1:10. В другом варианте реализации массовое отношение вилдаглиптина или его соли и полиэтиленоксида, имеющего молекулярную массу от приблизительно 5000000 до приблизительно 8000000, в ядре варьируется от приблизительно 1:1 до приблизительно 1:3. В другом варианте реализации массовое отношение вилдаглиптина или его соли и полиэтиленоксида, имеющего молекулярную массу от приблизительно 5000000 до приблизительно 8000000, в ядре составляет приблизительно 1:1,10, или приблизительно 1:1,15, или приблизительно 1:1,20, или приблизительно 1:1,25.
В другом варианте реализации ядро окружено изолирующим покрытием, выбранным из доступных на рынке систем для покрытия, таких как Opadry®. В другом варианте реализации изолирующее покрытие окружено покрытием для замедленного высвобождения, содержащим в массовом отношении от приблизительно 80% до приблизительно 90% ацетатцеллюлозы. В другом варианте реализации изолирующее покрытие окружено покрытием для замедленного высвобождения, содержащим приблизительно 90% ацетатцеллюлозы по массе.
В другом варианте реализации покрытие для замедленного высвобождения включает, по меньшей мере, один канал, пригодный для осмотического высвобождения вилдаглиптина или его соли из ядра, причем поперечное сечение указанного канала составляет от приблизительно 0,1 мм2 до приблизительно 1 мм2 или приблизительно 0,7 мм2.
В другом варианте реализации покрытие для замедленного высвобождения окружено слоем для немедленного высвобождения лекарственного средства, содержащим от приблизительно 20 мг до приблизительно 30 мг вилдаглиптина или его соли, или приблизительно 25 мг вилдаглиптина или его соли.
В другом варианте реализации суммарное количество вилдаглиптина или его соли составляет в композиции приблизительно 100 мг, или приблизительно 150 мг, или приблизительно 200 мг, или приблизительно 250 мг, или приблизительно 300 мг, при этом приблизительно 90%, или приблизительно 80%, или приблизительно 70%, или приблизительно 60%, или приблизительно 50% вилдаглиптина или его соли находятся в ядре указанной композиции, а приблизительно 10%, или приблизительно 20%, или приблизительно 30%, или приблизительно 40%, или приблизительно 50% вилдаглиптина или его соли находятся в слое для немедленного высвобождения.
В другом варианте реализации композиции согласно настоящему изобретению приготовлены в виде формы для перорального введения, например, таблетка, капсула (в том числе микрокапсула), гранула или порошок; из этих форм более предпочтительной является такая пероральная форма, как таблетка.
В другом аспекте изобретения предложена осмотическая фармацевтическая композиция, содержащая: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий приблизительно 75 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий от приблизительно 80 мг до приблизительно 90 мг полиэтиленоксида, имеющего молекулярную массу от приблизительно 5000000 до приблизительно 8000000; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее от приблизительно 35 мг до приблизительно 45 мг ацетатцеллюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий приблизительно 25 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В одном варианте реализации указанного аспекта от приблизительно 15% до приблизительно 25% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений в 0,1 Н HCl при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 70% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме в диапазоне от приблизительно 5 нг/мл до приблизительно 250 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки. В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме в диапазоне от приблизительно 5 нг/мл до приблизительно 30 нг/мл через 24 часа после однократного суточного перорального введения.
В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину от приблизительно 60% до приблизительно 90% на протяжении периода, равного 24 часам. В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении 24 часов
В другом варианте реализации этого аспекта осмотический слой дополнительно содержит гидроксипропилметилцеллюлозу в массовом отношении от приблизительно 3% до приблизительно 7%. В другом варианте реализации этого аспекта ядро дополнительно содержит, по меньшей мере, один осмотический агент, выбранный из группы, состоящей из сахарозы, ксилитола, глюкозы, лактозы, маннитола, хлорида натрия, хлорида калия, эфиров целлюлозы, мальтодекстринов и циклодекстринов. В другом варианте реализации осмотический агент, присутствующий в слое лекарственного средства, отличается от такового в осмотическом слое. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения содержит приблизительно 40 мг ацетатцеллюлозы. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения включает, по меньшей мере, один канал, пригодный для осмотического высвобождения вилдаглиптина или его соли из ядра, причем поперечное сечение указанного канала составляет от приблизительно 0,1 мм2 до приблизительно 1 мм2 или приблизительно 0,7 мм2.
В другом аспекте изобретения предложена осмотическая фармацевтическая композиция, содержащая: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий приблизительно 75 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий приблизительно 95 мг полиэтиленоксида, имеющего молекулярную массу от приблизительно 5000000 до приблизительно 8000000, (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее от приблизительно 35 мг до приблизительно 45 мг ацетатцеллюлозы, окружающее указанное изолирующее покрытие;, (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 25 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта от приблизительно 15% до приблизительно 25% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений в 0,1 Н HCl при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 70% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли было высвобождено через 24 часа в условиях измерений в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме в диапазоне от приблизительно 5 нг/мл до приблизительно 250 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки. В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме в диапазоне от приблизительно 5 нг/мл до приблизительно 30 нг/мл через 24 часа после перорального введения один раз в сутки.
В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину от приблизительно 60% до приблизительно 90% на протяжении периода, равного 24 часам. В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении 24 часов.
В другом варианте реализации этого аспекта осмотический слой содержит гидроксипропилметилцеллюлозу в массовом отношении от приблизительно 3% до приблизительно 7%. В другом варианте реализации этого аспекта ядро содержит, по меньшей мере, один осмотический агент, выбранный из группы, состоящей из сахарозы, ксилитола, глюкозы, лактозы, маннитола, хлорида натрия, хлорида калия, эфиров целлюлозы, мальтодекстринов и циклодекстринов. В другом варианте реализации этого аспекта осмотический агент, присутствующий в слое лекарственного средства, отличается от такового в осмотическом слое. В варианте реализации этого аспекта покрытие для замедленного высвобождения содержит приблизительно 40 мг ацетатцеллюлозы. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения включает, по меньшей мере, один канал, пригодный для осмотического высвобождения вилдаглиптина или его соли из ядра, причем поперечное сечение указанного канала составляет от приблизительно 0,1 мм2 до приблизительно 1 мм2 или приблизительно 0,7 мм2.
В другом аспекте изобретения предложена осмотическая фармацевтическая композиция, содержащая: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий приблизительно 75 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий приблизительно 85 мг полиэтиленоксида, имеющего молекулярную массу от приблизительно 5000000 до приблизительно 8000000, (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее от приблизительно 35 мг до приблизительно 45 мг ацетатцеллюлозы, окружающее изолирующий слой; (d) слой лекарственного средства для немедленного высвобождения, содержащий приблизительно 25 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта от приблизительно 15% до приблизительно 25% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений в 0,1 Н HCl при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 70% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме в диапазоне от приблизительно 5 нг/мл до приблизительно 250 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки. В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме в диапазоне от приблизительно 5 нг/мл до приблизительно 30 нг/мл через 24 часа после перорального введения один раз в сутки.
В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину от приблизительно 60% до приблизительно 90% на протяжении периода, равного 24 часам. В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении периода, равного 24 часам.
В другом варианте реализации этого аспекта осмотический слой содержит гидроксипропилметилцеллюлозу в массовом отношении от приблизительно 3% до приблизительно 7%. В другом варианте реализации этого аспекта ядро дополнительно содержит, по меньшей мере, один осмотический агент, выбранный из группы, состоящей из сахарозы, ксилитола, глюкозы, лактозы, маннитола, хлорида натрия, хлорида калия, эфиров целлюлозы, мальтодекстринов и циклодекстринов. В другом варианте реализации этого аспекта осмотический агент, присутствующий в слое лекарственного средства, отличается от такового в осмотическом слое. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения содержит приблизительно 40 мг ацетатцеллюлозы. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения включает, по меньшей мере, один канал, пригодный для осмотического высвобождения вилдаглиптина или его соли из ядра, причем поперечное сечение указанного канала составляет от приблизительно 0,1 мм2 до приблизительно 1 мм2 или приблизительно 0,7 мм2.
В другом варианте реализации этого аспекта предложена осмотическая фармацевтическая композиция, содержащая: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий приблизительно 75 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий приблизительно 90 мг полиэтиленоксида, имеющего молекулярную массу от приблизительно 5000000 до приблизительно 8000000, (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее от приблизительно 35 мг до приблизительно 45 мг ацетатцеллюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 25 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта от приблизительно 15% до приблизительно 25% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений в 0,Ш НО при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 70% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме в диапазоне от приблизительно 5 нг/мл до приблизительно 250 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки. В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме в диапазоне от приблизительно 5 нг/мл до приблизительно 30 нг/мл через 24 часа после перорального введения один раз в сутки.
В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину от приблизительно 60% до приблизительно 90% на протяжении периода, равного 24 часам. В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении периода, равного 24 часам.
В другом варианте реализации этого аспекта осмотический слой содержит гидроксипропилметилцеллюлозу в массовом отношении от приблизительно 3% до приблизительно 7%. В другом варианте реализации этого аспекта ядро дополнительно содержит, по меньшей мере, один осмотический агент, выбранный из группы, состоящей из сахарозы, ксилитола, глюкозы, лактозы, маннитола, хлорида натрия, хлорида калия, эфиров целлюлозы, мальтодекстринов и циклодекстринов. В другом варианте реализации этого аспекта осмотический агент, присутствующий в слое лекарственного средства, отличается от такового в осмотическом слое. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения содержит приблизительно 40 мг ацетатцеллюлозы. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения включает, по меньшей мере, один канал, пригодный для осмотического высвобождения вилдаглиптина или его соли из ядра, причем поперечное сечение указанного канала составляет от приблизительно 0,1 мм2 до приблизительно 1 мм2 или приблизительно 0,7 мм2.
В другом варианте реализации этого аспекта предложен набор, содержащий осмотическую фармацевтическую композицию, содержащую: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий вилдаглиптин или его соль и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, окружающее изолирующий слой; (d) слой лекарственного средства для немедленного высвобождения, содержащий вилдаглиптин или его соль, окружающий указанное покрытие для замедленного высвобождения.
В другом аспекте изобретения предложен набор, содержащий осмотическую фармацевтическую композицию, содержащую: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее в массовом отношении от приблизительно 70% до приблизительно 95% ацетатцеллюлозы, окружающее изолирующий слой; (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 15 мг до приблизительно 35 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 10% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C.
В одном варианте реализации этого аспекта от приблизительно 15% до приблизительно 25% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений в 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 70% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме крови в диапазоне от приблизительно 5 нг/мл до приблизительно 250 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки. В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме крови в диапазоне от приблизительно 5 нг/мл до приблизительно 30 нг/мл через 24 часа после перорального введения один раз в сутки.
В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину от приблизительно 60% до приблизительно 90% на протяжении периода, равного 24 часам. В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении периода, равного 24 часам.
В другом варианте реализации этого аспекта ядро содержит приблизительно 75 мг вилдаглиптина или его соли. В варианте реализации этого аспекта массовое отношение вилдаглиптина или его соли и полиэтиленоксида в ядре находится в диапазоне от приблизительно 1:1 до приблизительно 1:3. В другом варианте реализации этого аспекта осмотический слой содержит гидроксипропилметилцеллюлозу в массовом отношении от приблизительно 3% до приблизительно 7%. В другом варианте реализации этого аспекта ядро содержит, по меньшей мере, один осмотический агент, выбранный из группы, состоящей из сахарозы, ксилитола, глюкозы, лактозы, маннитола, хлорида натрия, хлорида калия, эфиров целлюлозы, мальтодекстринов и циклодекстринов. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения содержит по массе приблизительно 90% ацетатцеллюлозы. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения включает, по меньшей мере, один канал, пригодный для осмотического высвобождения вилдаглиптина или его соли из ядра, причем поперечное сечение указанного канала составляет от приблизительно 0,1 мм2 до приблизительно 1 мм2 или приблизительно 0,7 мм2.
В другом аспекте настоящего изобретения предложен способ лечения диабета 2 типа, при этом указанный способ включает введение пациенту, нуждающемуся в этом, осмотической фармацевтической композиции, содержащей: (а) ядро, содержащее: (1) слой лекарственного средства, содержащий вилдаглиптин или его соль и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) ацетатцеллюлозное покрытие для замедленного высвобождения, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий вилдаглиптин или его соль, окружающий указанное покрытие для замедленного высвобождения.
В другом аспекте изобретения предложен способ лечения диабета 2 типа, включающий введение пациенту, нуждающемуся в этом, осмотической фармацевтической композиции, содержащей: (а) ядро, содержащее: (I) слой лекарственного средства, содержащий от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее в массовом отношении от приблизительно 70% до приблизительно 95% ацетатцеллюлозы, окружающее изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 15 мг до приблизительно 35 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 10% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C.
В одном варианте реализации этого аспекта от приблизительно 15% до приблизительно 25% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерения в 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 70% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерения прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерения в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме в диапазоне от приблизительно 5 нг/мл до приблизительно 250 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки. В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме в диапазоне от приблизительно 5 нг/мл до приблизительно 30 нг/мл через 24 часа после перорального введения один раз в сутки.
В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину от приблизительно 60% до приблизительно 90% на протяжении периода, равного 24 часам. В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении периода, равного 24 часам.
В другом варианте реализации этого аспекта ядро содержит приблизительно 75 мг вилдаглиптина или его соли. В другом варианте реализации этого аспекта массовое отношение вилдаглиптина или его соли и полиэтиленоксида в ядре находится в диапазоне от приблизительно 1:1 до приблизительно 1:3. В другом варианте реализации этого аспекта осмотический слой содержит гидроксипропилметилцеллюлозу в массовом отношении от приблизительно 3% до приблизительно 7%. В другом варианте реализации этого аспекта ядро содержит, по меньшей мере, один осмотический агент, выбранный из группы, состоящей из сахарозы, ксилитола, глюкозы, лактозы, маннитола, хлорида натрия, хлорида калия, эфиров целлюлозы, мальтодекстринов и циклодекстринов. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения содержит по массе приблизительно 90% ацетатцеллюлозы. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения включает, по меньшей мере, один канал, пригодный для осмотического высвобождения вилдаглиптина или его соли из ядра, причем поперечное сечение указанного канала составляет от приблизительно 0,1 мм2 до приблизительно 1 мм2 или приблизительно 0,7 мм2.
В другом аспекте изобретения предложен способ получения осмотической фармацевтической композиции, содержащей: (а) ядро, содержащее от приблизительно 35 мг до приблизительно 95 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий полиэтиленоксид, имеющий молекулярную массу от приблизительно 5000000 до приблизительно 8000000, при этом массовое отношение вилдаглиптина или его соли и полиэтиленоксида находится в диапазоне от приблизительно 1:1 до приблизительно 1:5; (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее в массовом отношении от приблизительно 70% до приблизительно 95% ацетатцеллюлозы, окружающее изолирующий слой; (d) слой лекарственного средства для немедленного высвобождения, содержащий от приблизительно 15 мг до приблизительно 35 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 10% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C, причем указанный способ включает следующие этапы:
(a) приготовление слоя лекарственного средства в виде гранул, содержащих вилдаглиптин или его соль, и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество,
(b) приготовление осмотического слоя в виде гранул, содержащих полиэтиленоксид,
(c) прессование гранул слоя лекарственного средства и осмотического слоя с целью получения ядра,
(d) нанесение изолирующего покрытия на ядро, полученное на этапе (с),
(e) нанесение на изолирующее покрытие, полученное на этапе (d), покрытия для замедленного высвобождения, содержащего в массовом отношении от приблизительно 70% до приблизительно 95% ацетатцеллюлозы, и
(f) нанесение на покрытие для замедленного высвобождения, полученное на этапе (е), слоя для немедленного высвобождения лекарственного средства, содержащего вилдаглиптин или его соль, с целью получения осмотической фармацевтической композиции.
В одном варианте реализации этого аспекта от приблизительно 15% до приблизительно 25% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений в 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 70% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений в фосфатном буфере с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при приблизительно 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 30°C/65% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C. В другом варианте реализации этого аспекта композиция сохраняет стабильность при хранении на протяжении периода, составляющего 3 месяца при 40°C/75% относительной влажности, и в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 85% до приблизительно 95% вилдаглиптина или его соли высвобождается через 24 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, фосфатного буфера с рН=6,8 при 37°C ± 0,5°C.
В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме крови в диапазоне от приблизительно 5 нг/мл до приблизительно 250 нг/мл на протяжении, по меньшей мере, 24 часов после перорального введения один раз в сутки. В другом варианте реализации этого аспекта композиция обеспечивает среднюю концентрацию вилдаглиптина или его соли в плазме крови в диапазоне от приблизительно 5 нг/мл до приблизительно 30 нг/мл через 24 часа после перорального введения один раз в сутки.
В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину от приблизительно 60% до приблизительно 90% на протяжении периода, равного 24 часам. В другом варианте реализации этого аспекта композиция способна снижать активность фермента ДДП-4 в плазме крови на величину приблизительно 80% на протяжении периода, равного 24 часам.
В другом варианте реализации этого аспекта ядро содержит приблизительно 75 мг вилдаглиптина или. его соли. В другом варианте реализации этого аспекта массовое отношение вилдаглиптина или его соли и полиэтиленоксида в ядре находится в диапазоне от приблизительно 1:1 до приблизительно 1:3. В другом варианте реализации этого аспекта осмотический слой дополнительно содержит гидроксипропилметилцеллюлозу в массовом отношении от приблизительно 3% до приблизительно 7%. В другом варианте реализации этого аспекта ядро содержит, по меньшей мере, один осмотический агент, выбранный из группы, состоящей из сахарозы, ксилитола, глюкозы, лактозы, маннитола, хлорида натрия, хлорида калия, эфиров целлюлозы, мальтодекстринов и циклодекстринов. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения содержит по массе приблизительно 90% ацетатцеллюлозы. В другом варианте реализации этого аспекта покрытие для замедленного высвобождения включает, по меньшей мере, один канал, пригодный для осмотического высвобождения вилдаглиптина или его соли из ядра, причем поперечное сечение указанного канала составляет от приблизительно 0,1 мм2 до приблизительно 1 мм2 или приблизительно 0,7 мм2.
В другом аспекте изобретения предложен способ получения осмотической фармацевтической композиции, содержащей: (а) ядро, содержащее (I) слой лекарственного средства, содержащий приблизительно 75 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий от приблизительно 80 до приблизительно 90 мг полиэтиленоксида, имеющего молекулярную массу от приблизительно 5000000 до приблизительно 8000000, (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее в массовом отношении от приблизительно 35 мг до приблизительно 45 мг ацетатцеллюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий приблизительно 25 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C, причем указанный способ включает следующие этапы:
(a) приготовление слоя лекарственного в виде гранул, содержащих вилдаглиптин или его соли, и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество,
(b) приготовление осмотического слоя в виде гранул, содержащих полиэтиленоксид,
(c) прессование гранул слоя лекарственного средства и осмотического слоя с целью формирования ядра,
(d) нанесение изолирующего покрытия на ядро, полученное на этапе (с),
(e) нанесение на изолирующее покрытие, полученное на этапе (d), покрытия для замедленного высвобождения, содержащего ацетатцеллюлозу, и
(f) нанесение на покрытие для замедленного высвобождения, полученное на этапе (е), слоя для немедленного высвобождения лекарственного средства, содержащего вилдаглиптин или его соль, с получением осмотической фармацевтической композиции.
В другом аспекте изобретения предложен способ получения осмотической фармацевтической композиции, содержащей: (а) ядро, содержащее (I) слой лекарственного средства, содержащий приблизительно 75 мг вилдаглиптина или его соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество; (II) слой осмотического вещества, содержащий приблизительно 90 мг полиэтиленоксида, имеющего молекулярную массу от приблизительно 5000000 до приблизительно 8000000, (b) изолирующее покрытие, окружающее указанное ядро; (с) покрытие для замедленного высвобождения, содержащее от приблизительно 35 мг до приблизительно 45 мг ацетатцеллюлозы, окружающее указанное изолирующее покрытие; (d) слой лекарственного средства для немедленного высвобождения, содержащий приблизительно 25 мг вилдаглиптина или его соли, окружающий указанное покрытие для замедленного высвобождения, и при этом композиция в условиях in vitro демонстрирует профиль высвобождения, при котором от приблизительно 20% до приблизительно 30% вилдаглиптина или его соли высвобождается через 2 часа в условиях измерений прибором USP Типа 1, при 100 об/мин при использовании 900 мл, 0,1 Н HCl при 37°C ± 0,5°C, причем указанный способ включает следующие этапы:
(a) приготовление слоя лекарственного средства в виде гранул, содержащих вилдаглиптин или его соль и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество,
(b) приготовление осмотического слоя в виде гранул, содержащих полиэтиленоксид,
(c) прессование гранул лекарственного слоя и осмотического слоя с целью получения ядра,
(d) нанесение изолирующего покрытия на ядро, полученное на этапе (с),
(e) нанесение на изолирующее покрытие, полученное на этапе (d), покрытия замедленного высвобождения, содержащего ацетатцеллюлозу, и
(f) нанесение на покрытие для замедленного высвобождения, полученное на этапе (е), слоя для немедленного высвобождения лекарственного средства, содержащего вилдаглиптин или его соль, с целью получения осмотической фармацевтической композиции.
Принимая во внимание, что изобретение было описано в его конкретных вариантах, для специалистов в данной области техники будут понятны некоторые модификации и эквивалентные решения, и они также должны быть включены в объем настоящего изобретения.
ПРИМЕРЫ
ПРИМЕР 1: Осмотическая композиция вилдаглипина

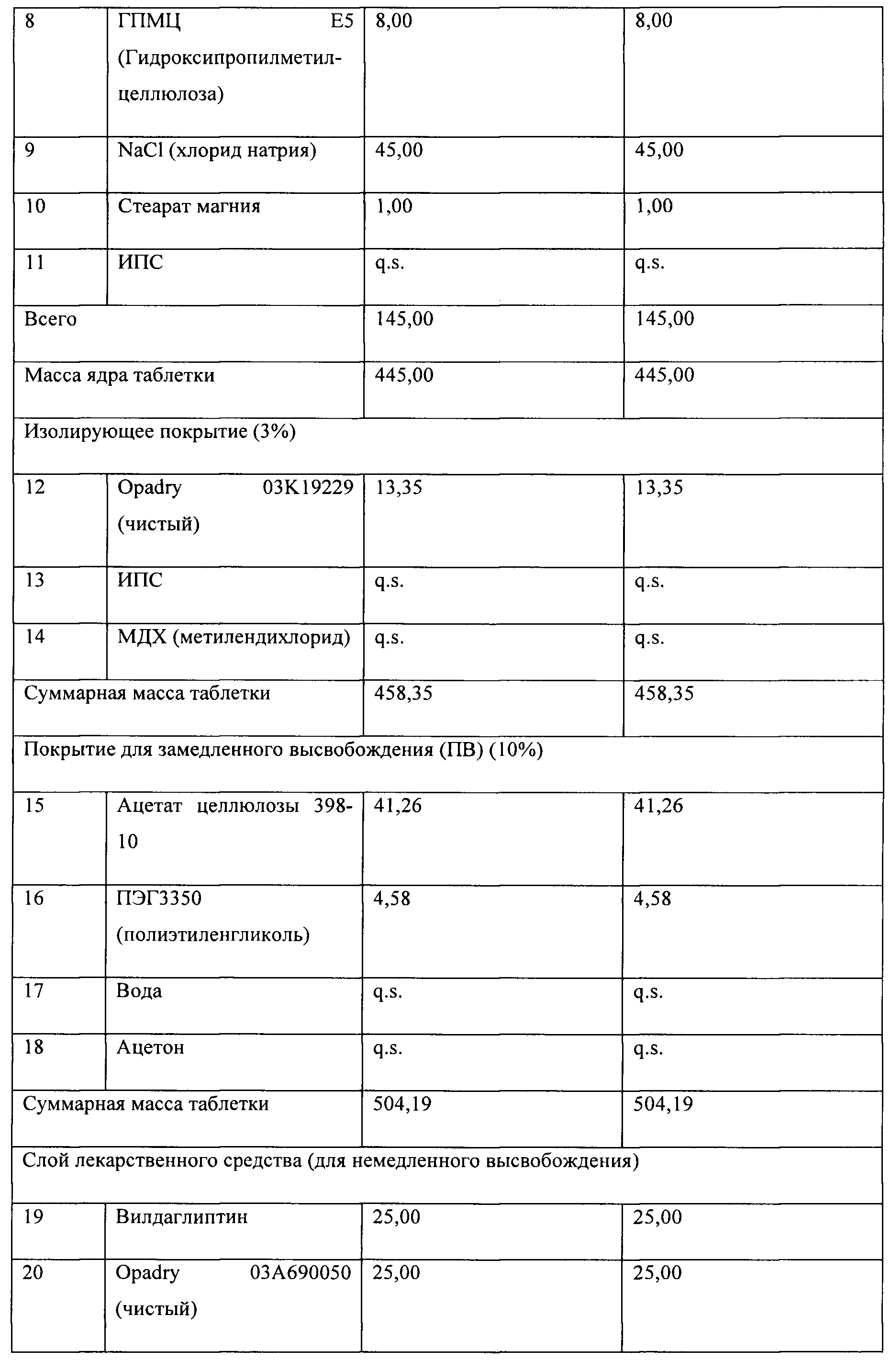

Способ получения:
Слой лекарственного средства:
I. Вилдаглиптин и Polyox N 80 пропускали через сито #40.
II. Отдельно пропускали маннитол через сито #40 и геометрически смешивали со смесью, полученной на этапе 1.
III. Порошковую смесь, полученную на этапе II, гранулировали с ИПС в грануляторе быстрого перемешивания путем распыления ИПС на сухую смесь с целью получения гранул одного размера.
IV. Полученные гранулы высушивали в сушилке с псевдоожиженным слоем при начальной температуре 40°C ± 5°C в течение времени, необходимого для достижения LOD менее 2%.
V. Высушенные гранулы пропускали через сито №40 и дополнительно в течение 5 минут смазывали гранулы предварительно просеянным стеаратом магния.
Осмотический слой:
I. Polyox WSR 303 и ГПМЦ Е-5 пропускали через сито #60.
II. Оксид железа пропускали через сито #60 и смешивали со смесью, полученной на этапе I, в течение 5 минут.
III. Порошковую смесь, полученную на этапе И, гранулировали с ИПС в грануляторе быстрого перемешивания путем распыления ИПС на сухую смесь с целью получения гранул одного размера.
IV. Вышеуказанные гранулы высушивали в сушилке с псевдоожиженным слоем при начальной температуре 40°C ± 5°C в течение времени, необходимого для достижения LOD менее 2%.
V. Высушенные гранулы пропускали через сито #40 и дополнительно в течение 5 минут смазывали гранулы предварительно просеянным стеаратом магния.
Прессование:
I. Полученные два вида смазанных гранул слоя с лекарственным средством и осмотического слоя подвергали прессованию в двуслойную таблетку с использованием круглой неглубокой вогнутой пресс-формы диаметром 10,00 мм в роторной машине двуслойного прессования.
Изолирующее покрытие:
I. Opadry 03K19229 (чистый) диспергировали в ИПС и перемешивали в течение 30 минут. Добавляли к вышеуказанному раствору МДХ и перемешивали в течение 10 минут.
II. Двуслойные таблетки покрывали вышеуказанным раствором до достижения массового отношения, равного 3%.
Покрытие из микропористой полупроницаемой мембраны (ППМ) (замедленного высвобождения)
I. Ацетатцеллюлозу 398-10 диспергировали в ацетоне при перемешивании. PEG 3350 растворяли в воде и смешивали с раствором ацетатцеллюлозы в течение 40 минут.
II. Таблетки, покрытые изолирующим покрытием, покрывали раствором, полученным на этапе I до достижения массового отношения, равного 10%.
Изготовление лекарственного слоя:
I. Вилдаглиптин растворяли в 60% МДЦ при перемешивании.
II. Отдельно Opadry 03А690050 (чистый) диспергировали в ИПС в течение 10 минут и добавляли в раствор оставшиеся 40% МДХ.
III. Раствор Opadry при перемешивании добавляли в лекарственный раствор и смешивали в течение 30 минут.
IV. Далее таблетки, покрытые ППМ, покрывали вышеуказанным раствором лекарственного средства и связывающим веществом до того момента, пока на таблетке не достигался слой с необходимой дозой лекарственного средства (25 мг).
Изготовление каналов
I. Сторону лекарственного слоя вышеуказанной таблетки просверливали иглой диаметром 0,6 мм с использованием лазерной сверлильной машины для изготовления таблеток.
Наружное покрытие:
I. Opadry 03K19229 (чистый) диспергировали в МПа и перемешивали в течение 30 минут. МДХ добавляли к вышеуказанному раствору и смешивали в течение 10 минут.
II. Вышеуказанным раствором покрывали поверхность таблеток (просверленных на стороне лекарственного слоя) до достижения массового отношения, равного 2,5-3%.
ПРИМЕР 2: Тестирование растворимости осмотических фармацевтических композиций Примера 1.
Долю высвобожденного лекарственного средства (в процентах) в условиях in vitro устанавливали с использованием следующей модификации способа изучения растворимости.

После завершения эксперимента по растворению в кислотной стадии кислотную среду тщательно удаляли или переливали, а таблетки высушивали с целью сохранения для экспериментов в буферной стадии.


В экспериментах с композициями из Примеров 1А и 1В в кислотной стадии приблизительно 25% вилдаглиптина или его соли было высвобождено через 2 часа, а в экспериментах в буферной стадии приблизительно 80% вилдаглиптина или его соли было высвобождено через 24 часа, как представлено на Фигуре 1А и Фигуре 1В.
ПРИМЕР 3: тестирование ингибирования ДПП-4 при введении осмотической фармацевтической композиции Примера 1.
Доклинические исследования с целью оценки профиля ФК/ФЭ композиций Примера 1 (1А и 1В) проводили на собаках-самцах породы бигль. Измеряли концентрацию лекарственного средства и активность фермента ДПП-4 в плазме крови и сравнивали с таковыми лекарственного средства GALVUS®. Собаки с массой тела от 10,5 кг до 13,2 кг были получены из питомника при Wockhardt Research Centre, Аурангабад. Собаки содержались в регулируемых условиях питомника со световым циклом 12 ч/12 ч, получали корм один раз в сутки и имели неограниченный доступ к воде. Животные не получали корм в течение 14 часов перед введением лекарственного средства и были распределены на несколько экспериментальных групп (n=4-5) с сопоставимой средней массой тела. После голодания животным вводили однократную пероральную дозу композиций Примера 1 (1А и 1В) (100 мг) и две дозы лекарственного средства GALVUS® (50 мг) в 0 и 10 часов. Пробы крови брали спустя 0 (до введения) и 0,5, 1, 2, 3, 3,25, 3,5, 4, 6, 8, 10, 10,5, 11, 12, 14, 16, 20, 24 и 30 часов после введения дозы лекарственного средства. Отобранную кровь центрифугировали, плазму отделяли и хранили в двух аликвотах при температуре -80°с для измерений активности фермента ДПП-4 и концентрации лекарственного средства. Анализ активности фермента ДПП-4 проводили путем измерения скоростей флуоресценции высвобожденного флуорогенного вещества 7-амино-4-метилкумарина (АМК), отщепленного от субстрата Gly-Pro-АМК в результате активности фермента ДПП-4 при длине волны возбуждения (λex) 360 нм и длине волны излучения (λem) 460 нм. Степень ингибирования ДПП-4 в процентах рассчитывали как 100×(1-At/А0), где А0 - активность фермента, измеренная до поступления дозы лекарственного средства, a At - активность, измеренная после поступления дозы в момент времени t в том же периоде обработки. Концентрации лекарственного средства в плазме крови измеряли с использованием способа ЖХ-МС. Полученные результаты представлены в виде средних величин ± SEM на Фигуре 2А и Фигуре 2В для Примера композиции 1А и на Фигуре 3А и Фигуре 3В для Примера композиции 1В.
Как представлено на Фигуре 2А и Фигуре 2В, лекарственное средство GALVUS® продемонстрировало ингибирование свыше 80% ДПП-4 в период от 0,5 ч до 6 ч после поступления первой дозы и от 10,5 ч до 16 ч после поступления второй дозы. В целом, GALVUS® поддерживал ингибирование свыше 80% ДПП-4 в течение 6 ч после введения каждой дозы. Через 6 ч после введения лекарственное средство GALVUS® продемонстрировало ингибирование ДПП-4 ниже 80%, что коррелирует с концентрацией лекарственного средства в плазме крови. Композиция Примера 1А продемонстрировала ингибирование ДПП-4 свыше 80% в течение периода до 14 часов, что находится в соответствии с концентрацией лекарственного средства в плазме крови. Спад ингибирования ДПП-4 после 20 часов может быть связан с неполным высвобождением лекарственного средства. Композиция Примера 1А продемонстрировала более последовательное и более длительное ингибирование ДПП-4 по сравнению с лекарственным средством GALVUS®.
Как представлено на Фигуре 3А и Фигуре 3В, композиция Примера 1В продемонстрировала ингибирование свыше 80% ДПП-4 на протяжении периода до 8 часов и более 70% на протяжении периода до 14 часов. Активность ДПП-4 коррелирует с концентрацией лекарственного средства в плазме и результатами экспериментов in vitro. В сравнении с лекарственным средством GALVUS® относительная биодоступность достигла 95%. Подобные и сходные показатели ФК/ФД можно предполагать у волонтеров-людей, принимая во внимание корреляции полувыведения и полного выведения лекарственных средств между людьми и собаками.
ПРИМЕР 4: Изучение стабильности композиции вилдаглиптина
Таблетки композиции Примера 1В упаковывали в алюминиевую блистерную упаковку и хранили для определения стабильности в условиях температуры 40°C ± 2°C и 75% относительной влажности (OB) ± 5%ОВ, а также 30°C ± 2°C и 65%ОВ ± 5%ОВ и оценивали на количественное содержание, растворимость и наличие примесей.

Как представлено в вышеприведенной таблице, композиция согласно настоящему изобретению (Пример 1В) сохраняет стабильность при хранении в условиях температуры 40°C ± 2°C и 75%ОВ ± 5%ОВ и 30°C ± 2°C и 65%ОВ ± 5%ОВ. Доказано также, что композиции при этих условиях сохраняют растворимость.
ПРИМЕР 5: Осмотические композиции вилдаглиптина



Способ получения:
Композиции Примера 5 были приготовлены способом, описанным в Примере 1.
ПРИМЕР 6: Сравнительные примеры



Сравнительные примерные композиции Примера 6 были приготовлены с помощью способа, описанного в Примере 1. Сравнительные примерные композиции 6А и 6В тестировали на растворимость in vitro при условиях, приведенных в Примере 2, и сравнивали с композицией 1А согласно настоящему изобретению.
На Фигуре 4 представлены профили растворимости in vitro сравнительных композиций вилдаглиптина 6А и 6В в кислотной стадии и буферной стадии, а в нижеследующей таблице указаны различия между композицией согласно настоящему изобретению и сравнительными примерами:
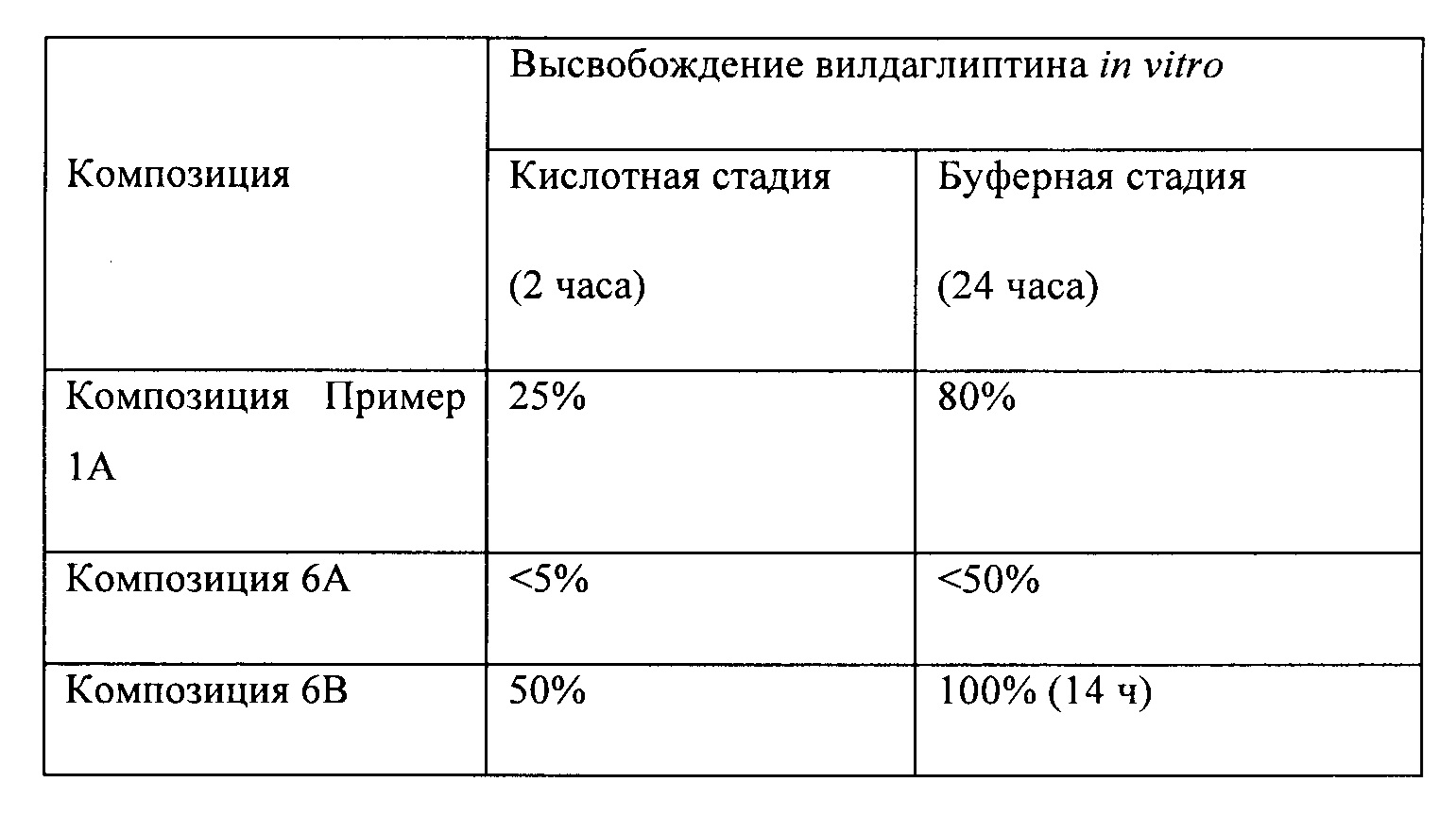
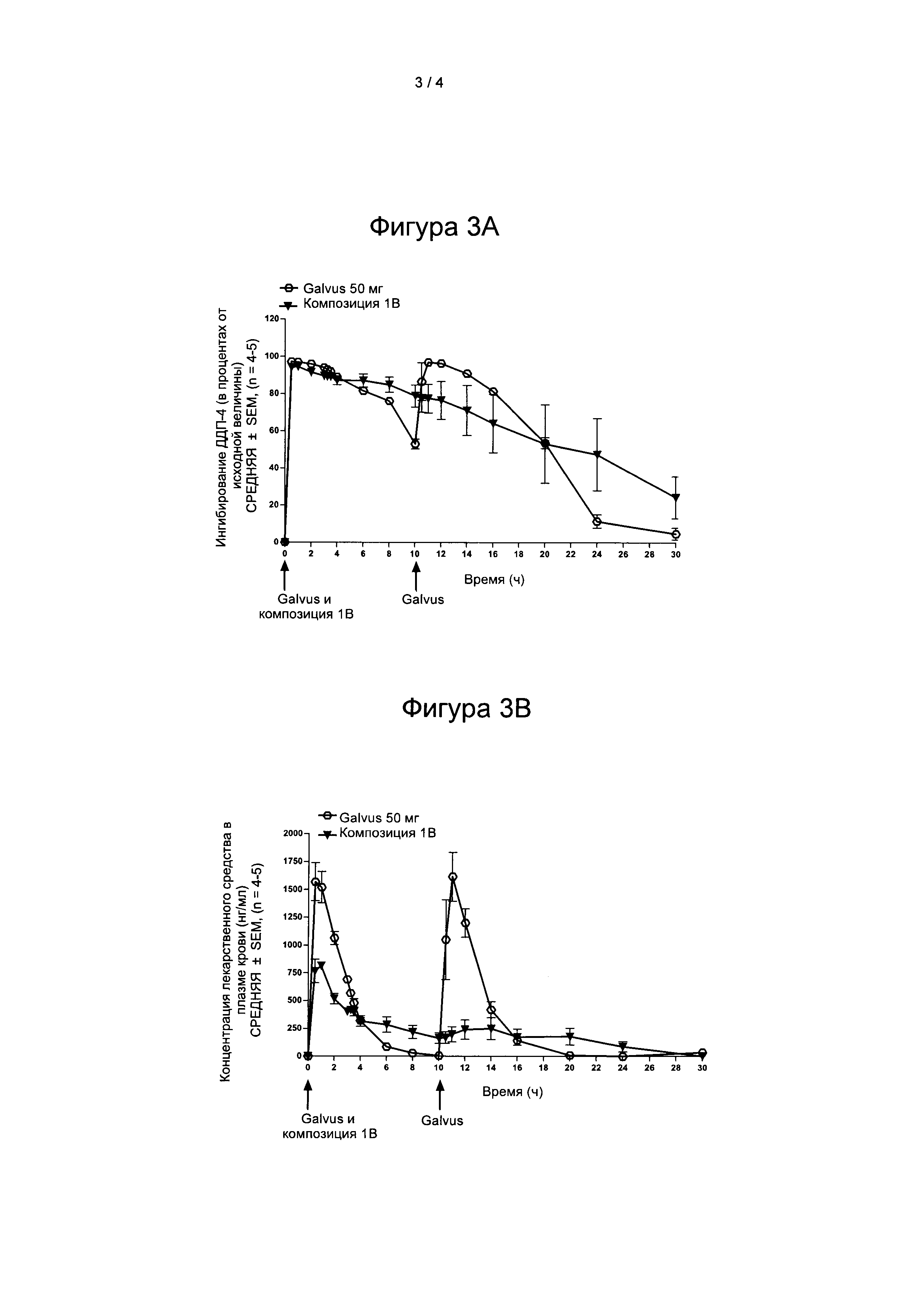
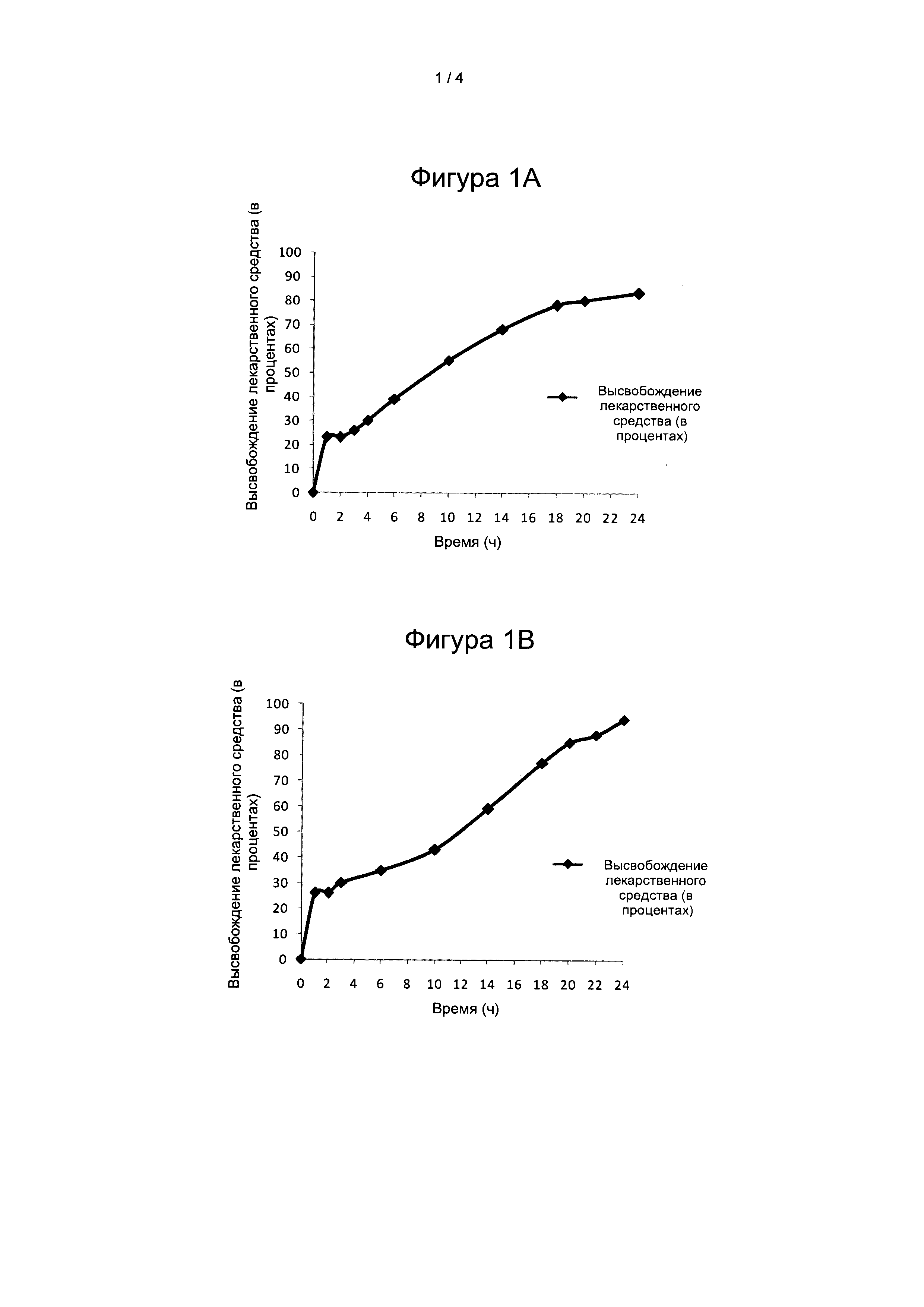
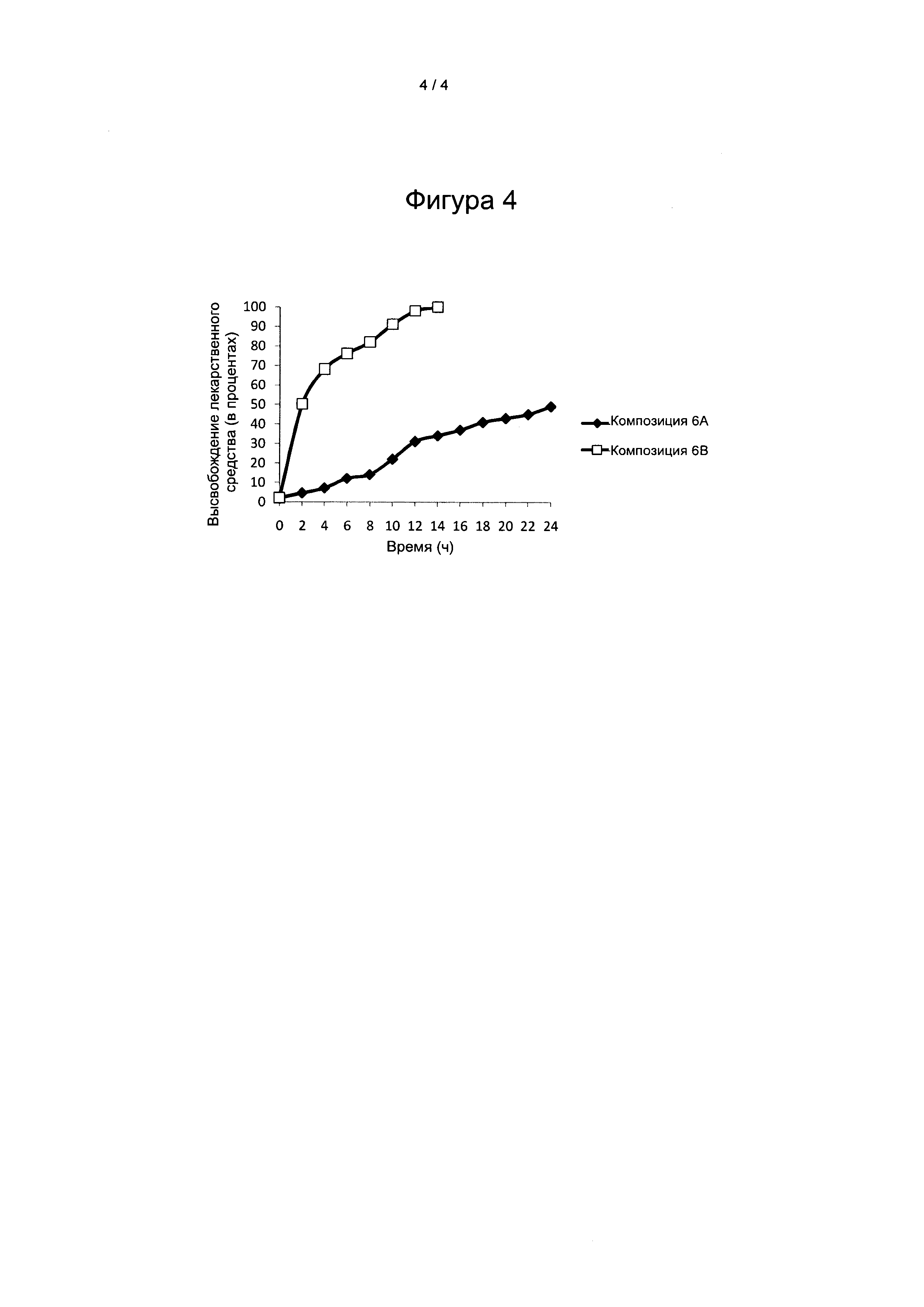
Фармацевтические композиции, содержащие сулбактам и ингибитор бета-лактамазы
Фармацевтические композиции, содержащие бета-лактамный антибиотик, сульбактам и ингибитор бета-лактамаз
Азотсодержащие соединения и их применение
Производные 1,6-диазабицикло[3.2.1]октан-7-она и их применение при лечении бактериальных инфекций
Способы лечения сердечно-сосудистых нарушений
Производные 1,6- диазабицикло [3,2,1] октан-7-она и их применение при лечении бактериальных инфекционных болезней
Чистый тетрагидрат l-аргининовой соли s-(-)-9-фтор-6, 7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1н, -5н-бензо[i,j]хинолизин-2-карбоновой кислоты и способ его получения
Композиции, включающие антибактериальное средство и тазобактам
Кетолидные соединения
Производные 1,6-диазабицикло[3.2.1]октан-7-она и их применение для лечения бактериальных инфекций

