Результат интеллектуальной деятельности: ТВЕРДЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ ОНДАНСЕТРОНА С ПРОЛОНГИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ ДЛЯ ЛЕЧЕНИЯ СИМПТОМОВ ТОШНОТЫ, РВОТЫ И ДИАРЕИ
Вид РИД
Изобретение
РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка испрашивает приоритет и преимущество по предварительной заявке США № 61/951074, поданной 11-го марта 2014 года, предварительной заявке США № 61/951092, поданной 11-го марта 2014 года, предварительной заявке США № 61/951112, поданной 11-го марта 2014 года и предварительной заявке США № 62/040136, поданной 21-го августа 2014 года. Содержание каждой из этих заявок полностью включено в данный документ путем ссылки.
УРОВЕНЬ ТЕХНИКИ
Гастроэнтерит, вызываемый вирусами или бактериями, является заболеванием, которое вызывает раздражение и воспаление желудка и кишечника (желудочно-кишечного тракта). Другие причины включают паразитов, пищевые аллергены, реакцию на антибиотики и попадание в пищу ядовитых растений. Рвота, вызванная острым гастроэнтеритом, очень часто встречается у детей и подростков и является весьма распространенной причиной, по которой дети и подростки попадают в отделения неотложной помощи. Раздражение кишечника, вызываемое гастроэнтеритом, по-видимому, является основным стимулом рвоты. По мере того как вирус или бактерии проникают в клетки слизистой оболочки верхних отделов желудочно-кишечного тракта, они приводят к нарушению нормального натриевого и осмотического внутриклеточного баланса, и в результате происходит чрезмерная потеря жидкости внутри клеток, приводя к ее истощению.
Острый гастрит представляет собой раздражение и воспаление слизистой выстилки желудка. Гастрит может быть вызван химическим, термическим или бактериальным стрессом. Например, лекарственные соединения, такие как алкоголь, аспирин и химиотерапевтические средства, могут вызвать приступ гастрита. Аналогичным образом, горячие, острые, необработанные или испорченные продукты могут вызвать приступ. У людей, имеющих гастрит, как правило, наблюдается рвота.
Hyperemesis gravidarum («HG», рвота беременных) представляет собой заболевание, при котором интенсивные и постоянные тошнота и рвота возникают во время беременности. У женщины наблюдается рвота беременных, если она беременна, и ее рвет более три-четырех раз в день так сильно, что это приводит к потере более 3 килограммов веса, ощущениям головокружения и потери ориентации или к обезвоживанию.
Рвота, имеющая любую причину, возникает вследствие стимуляции двух центров, расположенных в головном мозге - хеморецепторной триггерной зоны и рвотного центра. Если человек не может потреблять жидкость, чтобы восполнить ее потерю, то для него может потребоваться внутривенное введение жидкости (регидратация). Известные противорвотные препараты облегчают рвоту путем ингибирования хеморецепторной триггерной зоны (CTZ) в организме или более прямым действием на рвотный центр в мозге.
Синдром раздраженного кишечника (IBS) представляет собой функциональное расстройство желудочно-кишечного тракта (ЖКТ), что означает, что симптомы вызваны изменениями в работе желудочно-кишечного тракта. IBS представляет собой группу симптомов, которые возникают совместно. Основным симптомом IBS является боль и/или дискомфорт в животе. Боль или дискомфорт связаны с изменением частоты или консистенции стула. Изменения в деятельности кишечника может представлять собой хронические или рецидивирующие диарею или запор. У некоторых людей возникают и диарея и запоры, только в разное время. Вздутие или вспучивание в брюшной полости является обычным явлением. Диарея является одним из симптомов, часто связанных с IBS. IBS с диареей иногда называют IBS-D.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем документе описаны твердые лекарственные формы ондансетрона с пролонгированным высвобождением для лечения симптомов тошноты, рвоты или диареи.
В соответствии с аспектами, проиллюстрированными в данном описании, способ лечения пациента включает пероральное введение пациенту твердой пероральной лекарственной формы, включающей: сердцевину, содержащую неионогенный полимерный матрикс, первое количество ондансетрона, диспергированное в матриксе, и соль, диспергированную в матриксе, где первое количество ондансетрона варьирует от примерно 9 мг до примерно 28 мг; первую изолирующую оболочку, окружающую сердцевину, где первая изолирующая оболочка состоит из неионогенного полимерного матрикса; и слой немедленно высвобождающегося лекарственного средства, окружающий первую изолирующую оболочку и содержащий неионогенный полимер и второе количество ондансетрона, диспергированное в нем, где второе количество ондансетрона варьирует от примерно 3 мг до примерно 8 мг; причем высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает лекарственное действие ондансетрона как минимум в течение 16 часов, приводя к снижению частоты любого из рвоты, тошноты или диареи, или их комбинации.
Твердые лекарственные формы с пролонгированным высвобождением раскрыты в данном документе для снижения, лечения или предупреждения любого из тошноты, рвоты или диареи у пациента, симптомов, которые могут быть вызваны различными заболеваниями. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами вирусного гастроэнтерита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами бактериального гастроэнтерита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами гастрита (воспаления стенки желудка) у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами воспалительного заболевания кишечника у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами синдрома раздраженного кишечника у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами холецистита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами диспепсии у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами панкреатита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами аппендицита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами хирургического вмешательства у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами гепатита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами перитонита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами гастроэзофагеальной рефлюксной болезни у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами обструктивного поражения ЖКТ у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами пищевого отравления у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами опухоли у субъекта.
В данном документе раскрыты твердые лекарственные формы с пролонгированным высвобождением. В одном варианте осуществления твердые лекарственные формы с пролонгированным высвобождением раскрыты в данном документе для снижения, лечения или профилактики симптомов гастроэнтерита. В одном варианте осуществления связанным с гастроэнтеритом симптомом является рвота. Для того чтобы оценить снижение вызываемой гастроэнтеритом рвоты после приема твердых пероральных лекарственных форм по настоящему изобретению по сравнению с плацебо, это сравнение может быть проведено между долями пациентов без дальнейшей рвоты на протяжении более 30 минут после первой дозы исследуемого препарата после выписки из отделения неотложной помощи. Вторичной целью может быть сравнение между группами исследуемого препарата и группами плацебо по следующим критериям: частота рвоты на протяжении 4 дней после первой дозы исследуемого препарата; доля пациентов, получающих резервную противорвотную терапию; доля пациентов, получающих жидкость внутривенно; доля пациентов, нуждающихся в госпитализации; доля пациентов, возвращающихся в отделение неотложной помощи/неотложной терапии после первой выписки из него; время для возобновления нормальной деятельности (работа/школа/бытовая деятельность); тяжесть тошноты; и профили побочных эффектов.
В данном документе раскрыты твердые лекарственные формы с пролонгированным высвобождением. Более конкретно, твердые лекарственные формы с пролонгированным высвобождением раскрыты в данном документе для лечения рвоты беременных (HG). Для того, чтобы оценить лечение HG после введения твердых пероральных лекарственных форм по настоящему изобретению может быть измерена тяжесть рвоты. В одном варианте осуществления тяжесть рвоты оценивается, например, по баллам Уникального количественного определения рвоты при беременности (PUQE). Количество баллов варьирует от 3 (наилучшее состояние) до 15 (наихудшее состояние). В одном варианте осуществления тяжесть рвоты оценивается, например, с помощью балльной Визуальной аналоговой шкалы (VAS). Количество баллов в шкале варьирует от 0 (наилучшее состояние) до 50 (наихудшее состояние).
В данном документе раскрыты твердые лекарственные формы с пролонгированным высвобождением. Более конкретно, твердые лекарственные формы с пролонгированным высвобождением раскрыты в данном документе для лечения диареи у субъекта, симптома, который может быть вызван различными заболеваниями. В одном варианте осуществления диарея является побочным эффектом вирусного гастроэнтерита у субъекта. В одном варианте осуществления диарея является побочным эффектом бактериального гастроэнтерита у субъекта. В одном варианте осуществления диарея является побочным эффектом пищевой аллергии у индивидуума. В одном варианте осуществления диарея является побочным эффектом предменструального синдрома (PMS) у субъекта. В одном варианте осуществления диарея является побочным эффектом синдрома раздраженного кишечника у субъекта. В одном варианте осуществления диарея является побочным эффектом непереносимости лактозы у субъекта. В одном варианте осуществления диарея является побочным эффектом паразитов у субъекта. В одном варианте осуществления диарея является побочным эффектом бактериальной инфекции у субъекта. В одном варианте твердые лекарственные формы с пролонгированным высвобождением по настоящему изобретению вводят при лечении синдрома раздраженного кишечника с преобладанием диареи (IBS-D).
В одном варианте осуществления, чтобы оценить уменьшение рвоты после введения твердых пероральных лекарственных форм по настоящему изобретению, могут быть измерены показатели рвоты, такие, как частота, длительность, объем, тяжесть и дистресс. Частота может быть измерена, например, по числу приступов рвоты в течение определенного периода, длительность может быть измерена, например, по количеству часов рвоты, объем может быть измерен, например, в чашках рвоты, тяжесть может быть измерена, например, путем количественной оценки физических симптомов, и дистресс, который пациент испытывает, может быть измерен, например, по полученному стрессу и психологическим симптомам.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта таблетка ондансетрона с пролонгированным высвобождением, которая включает сердцевину, содержащую гидрофильный набухающий матрикс, содержащую ондансетрон или его фармацевтически приемлемую соль и безводный цитрат натрия; первую изолирующую оболочку, содержащую гипромеллозу и plasACRYL™; слой немедленно высвобождающегося лекарственного средства, окружающий первую изолирующую оболочку и содержащий ондансетрон или его фармацевтически приемлемую соль, гипромеллозу и plasACRYL™; и вторую изолирующую оболочку, содержащую гипромеллозу и plasACRYL™ Т20, где слой немедленно высвобождающегося лекарственного средства подобран так, чтобы высвобождать около 1/4 общей дозы ондансетрона в течение примерно 1 часа после перорального введения, и где сердцевина подобрана так, чтобы высвобождать остальную дозу ондансетрона в течение периода до 24 часов с кинетикой нулевого порядка. В одном варианте осуществления сердцевина содержит около 18 мг ондансетрона в виде свободного основания. В одном варианте осуществления сердцевина содержит около 20 мг ондансетрона в виде свободного основания. В одном варианте осуществления сердцевина содержит около 28 мг ондансетрона в виде свободного основания. В одном варианте осуществления безводный цитрат натрия присутствует в концентрации в диапазоне от примерно 50% до примерно 100% по весу гидрофильного набухающего матрикса. В одном варианте осуществления гидрофильным набухающим матриксом сердцевины является METHOCEL™ К4М Premium CR, гипромеллозой первой изолирующей оболочки и второй изолирующей оболочки является METHOCEL™ Е5 Premium LV, а гипромеллозой слоя немедленно высвобождающегося лекарственного средства является METHOCEL™ Е5 Premium LV. В одном варианте осуществления слой немедленно высвобождающегося лекарственного средства содержит около 6 мг ондансетрона.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая лекарственная форма с пролонгированным высвобождением, которая включает внутреннюю часть, где внутренняя часть содержит первую дозу по меньшей мере одного антагониста серотонина; первую оболочку, где первая оболочка непосредственно инкапсулирует внутреннюю часть твердой лекарственной формы; оболочку из слоя лекарственного средства, где оболочка из слоя лекарственного средства непосредственно инкапсулирует первую оболочку, где оболочка из слоя лекарственного средства содержит вторую дозу по меньшей мере одного антагониста серотонина, где оболочка из слоя лекарственного средства составляет, по меньшей мере 4% по весу твердой лекарственной формы, где вторая доза равна по меньшей мере 15% по весу от общей дозы по меньшей мере одного антагониста серотонина в твердой лекарственной форме, и где первая доза равна суммарной дозе минус вторая доза; и вторую оболочку, где вторая оболочка непосредственно инкапсулирует оболочку из слоя лекарственного средства, где внутренняя часть имеет растворимость в воде X, где первая оболочка, оболочка из слоя лекарственного средства и вторая оболочка имеют растворимость в воде по меньшей мере Y, и где Х меньше Y. В одном варианте осуществления по меньшей мере одним антагонистом рецептора серотонина-3 является ондансетрона гидрохлорид. В одном варианте осуществления вторая доза равна по меньшей мере 20% по весу от общей дозы по меньшей мере одного антагониста рецептора серотонина-3 в твердой лекарственной форме. В одном варианте осуществления по меньшей мере одним антагонистом рецептора серотонина-3 является ондансетрона гидрохлорид. В одном варианте осуществления вторая доза равна по меньшей мере 25% по весу от общей дозы по меньшей мере одного антагониста рецептора серотонина-3 в твердой лекарственной форме. В одном варианте осуществления первая оболочка и вторая оболочка включают гидрофильный материал. В одном варианте осуществления слой лекарственного вещества дополнительно содержит гидрофильный материал. В одном варианте осуществления гидрофильным материалом является гипромеллоза. В одном варианте осуществления каждая из первой оболочки и второй оболочки составляют по меньшей мере 1,5% по весу твердой лекарственной формы. В одном варианте осуществления соотношение гипромеллозы и по меньшей мере одного антагониста рецептора серотонина-3 в слое лекарственного вещества составляет примерно 4:6. В варианте осуществления изобретения общее количество гипромеллозы в первой оболочке, слое лекарственного вещества и второй оболочке составляет менее 4% по весу твердой лекарственной формы. В одном варианте осуществления сердцевина дополнительно содержит цитрат натрия в количестве менее 15% по весу сердцевины. В одном из вариантов Х существенно меньше Y, так что вторая доза по существу высвобождается из твердой лекарственной формы менее чем через 12 часов после того, как твердая лекарственная форма подвергается воздействию водной среды, а первая доза по существу высвобождается из твердой лекарственной формы за период от 12 до 24 часов с кинетикой нулевого порядка после того, как твердая лекарственная форма подвергается воздействию водной среды. В одном варианте осуществления водная среда имеет рН в диапазоне от рН 1,5 до рН 7,5. В одном варианте осуществления твердая лекарственная форма спрессована в таблетку. В одном варианте осуществления твердая лекарственная форма сформирована в виде капсулы. В одном варианте осуществления сердцевина дополнительно содержит глицин в количестве менее 20% по весу сердцевины.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта таблетка ондансетрона с пролонгированным высвобождением, изготовленная прессованием сердцевины таблетки с пролонгированным высвобождением и затем покрытием сердцевины таблетки первой изолирующей оболочкой с последующим покрытием оболочкой из лекарственного соединения и, наконец, второй изолирующей оболочкой, где сердцевина таблетки содержит гидрофильный набухающий матрикс, содержащий ондансетрона гидрохлорида и безводный цитрат натрия, где первая изолирующая оболочка содержит гипромеллозу и plasACRYL™, где оболочка с лекарственным веществом содержит ондансетрона гидрохлорид, гипромеллозу и plasACRYL™, и где вторая изолирующая оболочка содержит гипромеллозу и plasACRYL™ T20.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая включает в себя: сердцевину, содержащую неионогенный полимерный матрикс, первое количество первого противорвотного лекарственного соединения или его фармацевтически приемлемой соли, диспергированное в матриксе, и соль, диспергированную в матриксе; первую изолирующую оболочку, окружающую сердцевину, где первая изолирующая оболочка состоит из неионогенного полимерного матрикса; и слой немедленно высвобождающегося лекарственного средства, окружающий первую изолирующую оболочку, где слой немедленно высвобождающегося лекарственного средства содержит неионогенный полимер и второе количество второго противорвотного лекарственного средства или его фармацевтически приемлемой соли, диспергированное в нем, где слой лекарственного вещества подобран так, чтобы высвобождать второе количество противорвотного лекарственного средства в течение периода по меньшей мере 1 ч, где твердая пероральная лекарственная форма подобрана так, чтобы высвобождать первое количество первого противорвотного средства и второе количество второго противорвотного средства за как минимум 16 часов.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая включает в себя: сердцевину, содержащую гипромеллозу, 18 мг ондансетрона или эквивалентное количество соли ондансетрона и безводный цитрат натрия; первую изолирующую оболочку, окружающую сердцевину и содержащую гипромеллозу; и слой немедленно высвобождающегося лекарственного средства, окружающий первую изолирующую оболочку и содержащий гипромеллозу и 6 мг ондансетрона или эквивалентное количество соли ондансетрона, причем слой немедленно высвобождающегося лекарственного средства достаточен для высвобождения ондансетрона за период по меньшей мере 1 час, где общее количество ондансетрона в лекарственной форме высвобождается за 24 часа.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая включает в себя: сердцевину, содержащую неионогенный полимерный матрикс, первое количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное в матриксе, и соль, диспергированную в матриксе; первую изолирующую оболочку, окружающую сердцевину, где первая изолирующая оболочка состоит из неионогенного полимерного матрикса; и слой немедленно высвобождающегося лекарственного средства, окружающий первую изолирующую оболочку, где слой немедленно высвобождающегося лекарственного средства содержит неионогенный полимер и второе количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное в нем, где твердая пероральная лекарственная форма дает в результате in vitro профиль растворения ондансетрона при измерении в лопастном аппарате для растворения 2-го типа при 37°С в водном растворе, содержащем дистиллированную воду, при 50 об./мин, показывающий, что: а) от примерно 15% до 30% от общего количества ондансетрона высвобождается после двух с половиной часов измерения в аппарате; b) от примерно 30% до 50% от общего количества ондансетрона высвобождается через пять часов измерения в аппарате; и с) примерно не менее чем 75% от общего количества ондансетрона высвобождается через пятнадцать часов измерения в аппарате.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыт упакованный фармацевтический препарат, который включает в себя множество твердых пероральных лекарственных форм по настоящему изобретению в запечатанном контейнере и инструкции по пероральному приему лекарственных форм для предупреждения тошноты и рвоты.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыт фармацевтический препарат, который включает в себя множество твердых пероральных лекарственных форм по настоящему изобретению, каждая в отдельном запечатанном корпусе, и инструкции по пероральному приему лекарственных форм для предупреждения тошноты и рвоты.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыт упакованный фармацевтический препарат, который включает в себя множество твердых пероральных лекарственных форм по настоящему изобретению в запечатанном контейнере и инструкции по пероральному приему лекарственных форм для лечения диареи.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыт фармацевтический препарат, который включает в себя множество твердых пероральных лекарственных форм по настоящему изобретению, каждая в отдельном запечатанном корпусе, и инструкции по пероральному приему лекарственных форм для лечения диареи.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая снижает симптомы рвоты у субъекта. В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая снижает необходимость внутривенного введения жидкости у субъектов с гастроэнтеритом или гастритом. В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая снижает частоту госпитализаций у субъектов с гастроэнтеритом или гастритом. В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая снижает длительность пребывания в стационаре у субъектов с гастроэнтеритом или гастритом.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая уменьшает рвоту у пациентов с Hyperemesis gravidarum. Способ уменьшения симптомов рвоты у пациента с Hyperemesis gravidarum включает введение терапевтически эффективного количества твердой пероральной лекарственной формы по настоящему изобретению пациенту один раз в день; и наблюдение за уменьшением симптомов рвоты. В одном варианте осуществления наблюдение за уменьшением симптомов рвоты включает количественную оценку на основе балльной шкалы PUQE или на основе шкалы VAS.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая уменьшает симптомы диареи у субъекта.
Способ уменьшения симптомов, ассоциированных с гастроэнтеритом или гастритом, у пациента включает введение терапевтически эффективного количества твердой пероральной лекарственной формы по настоящему изобретению пациенту один раз в день; и наблюдение за уменьшением симптомов. В одном варианте осуществления наблюдение за уменьшением симптомов включает в себя мониторинг пациента, чтобы количественно оценить по меньшей мере одно из: частоты рвоты, требуется ли пациенту резервная терапия, получает ли пациент жидкость внутривенно, нуждается ли пациент в госпитализации, был ли пациент принят в отделение неотложной помощи/неотложной терапии, время для возобновления нормальной деятельности и тяжесть тошноты.
Способ уменьшения симптомов, ассоциированных с воспалительным заболеванием кишечника, у пациента включает введение пациенту терапевтически эффективного количества твердой пероральной лекарственной формы по настоящему изобретению пациенту один раз в день; и наблюдение за уменьшением симптомов. В одном варианте осуществления наблюдение за уменьшением симптомов включает в себя мониторинг пациента, чтобы количественно оценить по меньшей мере одно из: частоты рвоты, требуется ли пациенту резервная терапия, получает ли пациент жидкость внутривенно, нуждается ли пациент в госпитализации, был ли пациент принят в отделение неотложной помощи/неотложной терапии, время для возобновления нормальной деятельности и тяжесть тошноты.
Способ уменьшения симптомов, ассоциированных с синдромом раздраженного кишечника, у пациента включает введение пациенту терапевтически эффективного количества твердой пероральной лекарственной формы по настоящему изобретению пациенту один раз в день; и наблюдение за уменьшением симптомов. В одном варианте осуществления наблюдение за уменьшением симптомов включает в себя мониторинг пациента, чтобы количественно оценить по меньшей мере одно из: частоты рвоты, требуется ли пациенту резервная терапия, получает ли пациент жидкость внутривенно, нуждается ли пациент в госпитализации, был ли пациент принят в отделение неотложной помощи/неотложной терапии, время для возобновления нормальной деятельности и тяжесть тошноты.
Способ уменьшения симптомов, ассоциированных с диспепсией, у пациента включает введение пациенту терапевтически эффективного количества твердой пероральной лекарственной формы по настоящему изобретению пациенту один раз в день; и наблюдение за уменьшением симптомов. В одном варианте осуществления наблюдение за уменьшением симптомов включает в себя мониторинг пациента, чтобы количественно оценить по меньшей мере одно из: частоты рвоты, требуется ли пациенту резервная терапия, получает ли пациент жидкость внутривенно, нуждается ли пациент в госпитализации, был ли пациент принят в отделение неотложной помощи/неотложной терапии, время для возобновления нормальной деятельности и тяжесть тошноты.
Способ уменьшения симптомов, ассоциированых с Hyperemesis gravidarum, у пациента включает введение пациенту терапевтически эффективного количества твердой пероральной лекарственной формы по настоящему изобретению пациенту один раз в день; и наблюдение за уменьшением симптомов. В одном варианте осуществления наблюдение за уменьшением симптомов включает в себя мониторинг пациента, чтобы количественно оценить по меньшей мере одно из: частоты рвоты, требуется ли пациенту резервная терапия, получает ли пациент жидкость внутривенно, нуждается ли пациент в госпитализации, был ли пациент принят в отделение неотложной помощи/неотложной терапии, время для возобновления нормальной деятельности и тяжесть тошноты.
Способ уменьшения симптомов, ассоциированных с диареей, у пациента включает введение пациенту терапевтически эффективного количества твердой пероральной лекарственной формы по настоящему изобретению один раз в день; и наблюдение за уменьшением симптомов. В одном варианте осуществления наблюдение за уменьшением симптомов включает в себя мониторинг пациента, чтобы количественно оценить по меньшей мере одно из: частоты диареи, тяжести диареи и продолжительности диареи.
Способ уменьшения симптомов диареи, ассоциированных синдромом раздраженного кишечника с преобладающей диареей (IBS-D), у пациента включает введение пациенту терапевтически эффективного количества твердой пероральной лекарственной формы по настоящему изобретению пациенту один раз в день; и наблюдение за уменьшением симптомов. В одном варианте осуществления наблюдение за уменьшением симптомов включает в себя мониторинг пациента, чтобы количественно оценить по меньшей мере одно из: частоты диареи, тяжести диареи, продолжительности диареи и консистенции стула.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Раскрытые в данном описании варианты осуществления будут далее пояснены со ссылкой на прилагаемые чертежи. Приведенные чертежи не обязательно выполнены с соблюдением масштаба, напротив, с акцентом, в общем, направленным на иллюстрацию принципов вариантов осуществления, раскрытых в данном описании.
На фиг.1 показаны профили растворения ондансетрона из двух вариантов осуществления твердых лекарственных форм c пролонгированным высвобождением по настоящему изобретению, которые измеряли с использованием системы растворения 2-го типа (лопастной) в соответствии с USP (Фармакопеей США) при 50 об./мин, при температуре 37±0,5°С в дистиллированной воде в качестве растворяющей среды.
На фиг.2 показан профиль растворения ондансетрона из варианта осуществления твердой лекарственной формы c пролонгированным высвобождением по настоящему изобретению, который измеряли с использованием системы растворения 2-го типа (лопастной) в соответствии с USP при 50 об./мин, при температуре 37±0,5°С в 0,1 N HCl и фосфатном буфере с рН 6,8 в качестве среды растворения.
На фиг.3 показан профиль растворения ондансетрона из варианта осуществления твердой лекарственой формы c пролонгированным высвобождением по настоящему изобретению, который измеряли с использованием системы растворения 2-го типа (лопастной) в соответствии с USP при 50 об./мин, при температуре 37±0,5°С в 0,1 N HCl и фосфатном буфере с рН 6,8 в качестве среды растворения.
На фиг.4 показаны профили растворения ондансетрона из варианта осуществления твердых лекарственных форм c пролонгированным высвобождением по настоящему изобретению, которые измеряли с использованием системы растворения 2-го типа (лопастной) в соответствии с USP при 50 об./мин, при температуре 37±0,5°С в физиологически соответствующей среде при рН в диапазоне рН 1,2 до 7,2, соответствующем значениям рН в желудочно-кишечном тракте.
На фиг.5 показан профиль средней измеренной концентрации ондансетрона в плазме в зависимости от времени, полученный после введения различных вариантов осуществления твердых лекарственных форм с пролонгированным высвобождением по настоящему изобретению и эталонного продукта.
На фиг.6 показан логарифмически преобразованный профиль средней концентрации ондансетрона в зависимости от времени, полученный после введения различных вариантов осуществления твердых лекарственных форм с пролонгированным высвобождением по настоящему изобретению и эталонного продукта.
На фиг.7 показана блок-схема процесса изготовления препаратов ондансетрона гидрохлорида с пролонгированным высвобождением с номерами партий L004-04001, -04003, -04005 и -04007 согласно варианту осуществления настоящего изобретения.
На фиг.8 показана блок-схема процесса изготовления препаратов ондансетрона гидрохлорида с пролонгированным высвобождением для продолжительного введения с номерами партий L004-04002, -04004, -04006 и -04008 согласно варианту осуществления настоящего изобретения.
На фиг.9 показана блок-схема процесса приготовления раствора изолирующей оболочки для лекарственной формы с пролонгированным высвобождением согласно варианту осуществления настоящего изобретения.
На фиг.10 показана блок-схема процесса приготовления суспензии кишечнорастворимой оболочки для лекарственной формы с пролонгированным высвобождением согласно варианту осуществления настоящего изобретения.
На фиг.11 показана блок-схема процесса приготовления суспензии слоя немедленно высвобождающегося лекарственного средства для лекарственной формы с пролонгированным высвобождением согласно варианту осуществления настоящего изобретения.
На фиг.12 показана блок-схема процесса приготовления суспензии для продолжительного высвобождения для номеров партий с L004-04002A по -04002E согласно варианту осуществления настоящего изобретения.
На фиг.13 показана блок-схема процесса приготовления суспензии для продолжительного высвобождения для партий с номерами L004-04002F по -04002J, с -04004A по -04004D, с -04006A по -04006F и с -04008A по -04008B согласно варианту осуществления настоящего изобретения.
На фиг.14 показаны профили растворения для бимодальных таблеток ондансетрона, 28 мг (-04001 и -04001 А), и бимодальных таблеток ондансетрона 36 мг (-04003).
На фиг.15 показан профиль растворения для сердцевин таблеток ондансетрона 28 мг (-04005).
На фиг.16 показаны профили растворения сердцевины таблетки ондансетрона 28 мг (-04007) и бимодальной таблетки ондансетрона 36 мг (-04007 А).
На фиг.17 показаны профили растворения (в мг) для бимодальных таблеток ондансетрона 28 мг (-04001 и -04001A) и бимодальных таблеток ондансетрона 36 мг (-04003, -04007A и -04007B).
На фиг.18 показаны профили растворения (в%) для бимодальных таблеток ондансетрона 28 мг (-04001 И -04001A) и бимодальных таблеток ондансетрона 36 мг (-04003, -04007A и -04007B).
На фиг.19 показаны профили растворения для таблеток с продолжительным высвобождением ондансетрона, 8 мг -04002D, -04002D-042HC, -04002E, -04002F-042HC и -04002J.
На фиг.20 показаны профили растворения для таблеток с продолжительным высвобождением ондансетрона 8 мг (№№ партий с -04004A по -04004D).
На фиг.21 показаны профили растворения для таблеток с продолжительным высвобождением ондансетрона 8 мг (№№ партий с -04006A по -04006D).
На фиг.22 показаны профили растворения для таблеток с продолжительным высвобождением ондансетрона 8 мг (№№ партий с -04008A по -04008B).
На фиг.23 показан профиль линейной средней измеренной концентрации в плазме в зависимости от времени для тестируемого продукта в 1-й день, полученный после введения варианта осуществления твердой лекарственной формы с пролонгированным высвобождением по настоящему изобретению и эталонного продукта.
На фиг.24 показан профиль линейной средней измеренной концентрации в плазме в зависимости от времени для тестируемого продукта во 2-й день, полученный после введения варианта осуществления твердой лекарственной формы с пролонгированным высвобождением по настоящему изобретению и эталонного продукта.
На фиг.25 показан логарифмически преобразованный профиль средней концентрации в зависимости от времени для тестируемого продукта в 1-й день, полученный после введения варианта осуществления твердой лекарственной формы с пролонгированным высвобождением по настоящему изобретению и эталонного продукта.
На фиг.26 показан логарифмически преобразованный профиль средней концентрации в зависимости от времени для тестируемого продукта во 2-й день, полученный после введения варианта осуществления твердой лекарственной формы с пролонгированным высвобождением по настоящему изобретению и эталонного продукта.
На фиг.27 показан линейный общий профиль средней измеренной концентрации в плазме в зависимости от времени для тестируемого продукта и эталонного продукта, полученный после введения варианта осуществления твердой лекарственной формы с пролонгированным высвобождением по настоящему изобретению и эталонного продукта.
На фиг.28 показан логарифмически преобразованный общий профиль средней измеренной концентрации в плазме в зависимости от времени для тестируемого продукта и эталонного продукта, полученный после введения варианта осуществления твердой лекарственной формы с пролонгированным высвобождением по настоящему изобретению и эталонного продукта.
Хотя описанные выше чертежи иллюстрируют раскрытые в настоящее время варианты выполнения, также предусмотрены другие варианты осуществления, что отмечено в обсуждении. В данном описании представлены иллюстративные варианты осуществления путем представления, а не ограничения. Специалисты в данной области могут разработать множество других модификаций и вариантов осуществления, которые входят в объем и сущность принципов вариантов, раскрытых в настоящее время.
ПОДРОБНОЕ ОПИСАНИЕ
Используемые в данном описании следующие термины имеют изложенные ниже определения.
«Гидропатия» относится к шкале характеристик растворимости, сочетающей гидрофобность и гидрофильность аминокислот. Более конкретно, этот термин относится к скользящей шкале, аналогичной шкале рН, которая присваивает относительные величины, представляющие относительный баланс между гидрофобными и гидрофильными компонентами аминокислоты. Типичная шкала описана в Pliska et al., J. Chromatog. 216, 79, 1981, и названа «Относительный гидрофобный характер боковых цепей аминокислот», где глицин имеет значение 0, означающее относительно равный баланс между гидрофобными и гидрофильными компонентами и может называться как относительно «нейтральный», «сбалансированный», «слабо гидрофильный» или «слабо гидрофобный», изо-лейцин имеет положительное значение 1,83 и является сильно гидрофобным, а на противоположном конце шкалы аспарагиновая кислота имеет отрицательное значение -2,15 и может быть охарактеризована как сильно гидрофильная. Такая шкала и характеристики гидропатии, описанные в данном документе, хорошо известны и понятны специалистам в данной области.
«Монолитный» относится к таблеткам, которые не требуют нескольких слоев, специальных форм, осмотических отделов и/или специализированных оболочек, как правило, без стыков или швов, и их можно таблетировать на современном высокоскоростном оборудовании для таблетирования.
Термин «бимодальный», используемый в данном документе, относится к бимодальным профилям высвобождения лекарственного средства (быстрое высвобождение/медленное высвобождение).
«Антагонист серотонина» или «антагонист 5-НТ3-рецепторов» относится к классу лекарственных средств, используемых для профилактики и снятия тошноты и рвоты. Считается, что антагонисты серотонина работают, блокируя эффекты химического соединения серотонина, который вырабатывается в головном мозге и в желудке. Антагонисты 5-НТ3-рецепторов, эффективные для профилактики и снятия тошноты и рвоты, включают, но не ограничиваются ими, доласетрон, гранисетрон, ондансетрон, палоносетрон, трописетрон.
Термин «противорвотное лекарственное средство» включает в себя противорвотное лекарственное средство или его фармацевтически приемлемую соль. Когда используется термин «ондансетрон», он включает в себя фармацевтически приемлемую соль (ондансетрон⋅HCl).
Предложены твердые лекарственные формы с пролонгированным высвобождением. Более конкретно, настоящее изобретение относится к бимодальным твердым лекарственным формам с пролонгированным высвобождением для уменьшения вызываемой гастроэнтеритом рвоты. «Уменьшение вызываемой гастроэнтеритом рвоты» можно измерить путем мониторинга частоты (измеряемой, например, по количеству приступов рвоты в течение определенного периода), продолжительности (измеряемой, например, по количеству часов рвоты), объема (измеряемой, например, в чашках рвоты), тяжести (измеряемой, например, путем количественной оценки физических симптомов), и/или дистресса, который испытывает пациент (измеряемого, например, по полученному стрессу и психологическим симптомам). В одном варианте осуществления твердая лекарственная форма с пролонгированным высвобождением включает в себя внутреннюю часть, где внутренняя часть содержит первую дозу ондансетрона; первую оболочку, где первая оболочка непосредственно инкапсулирует внутреннюю часть твердой лекарственной формы; оболочку из слоя лекарственного средства, где оболочка из слоя лекарственного средства непосредственно инкапсулирует первую оболочку, где оболочка из слоя лекарственного средства содержит вторую дозу ондансетрона, где оболочка из слоя лекарственного средства составляет, по меньшей мере 4% по весу твердой лекарственной формы, где вторая доза равна по меньшей мере 15% по весу от общей дозы ондансетрона в твердой лекарственной форме, и где первая доза равна суммарной дозе минус вторая доза; и вторую оболочку, где вторая оболочка непосредственно инкапсулирует оболочку из слоя лекарственного средства, где внутренняя часть имеет растворимость в воде X, где первая оболочка, оболочка из слоя лекарственного средства и вторая оболочка имеют растворимость в воде по меньшей мере Y, и где Х меньше Y. В одном варианте осуществления твердая лекарственная форма с пролонгированным высвобождением способна высвобождать взрывным образом приблизительно 25% ондансетрона, а затем высвобождать остальной ондансетрон за 16-20 часов с кинетикой нулевого порядка. В одном варианте осуществления твердая лекарственная форма с пролонгированным высвобождением способна высвобождать взрывным образом приблизительно 25% ондансетрона, а затем высвобождать остальной ондансетрон за 20-30 часов с кинетикой нулевого порядка.
Ондансетрон
Ондансетрон является эффективным противорвотным агентом. Ондансетрон действует на центральную и/или периферическую нервную систему, преимущественно блокируя серотониновые 5-НТ3-рецепторы. Гидрохлорид (НСl) ондансетрона является дигидратом, рацемической формой ондансетрона. Ондансетрон имеет эмпирическую формулу С18Н19Ν30⋅HCl-2Η2О, представляющую собой молекулярную массу 365,9. Дигидрат ондансетрона⋅НСl является белым или грязновато белым порошком, который растворим в воде и физиологическом растворе.
Внутренняя часть («сердцевина») твердых лекарственных форм по варианту осуществления настоящего изобретения
Как таблетка проходит через желудочно-кишечный тракт человека, она проходит через изменения значения рН от примерно 1,5 до примерно 7,4. Слюна во рту имеет нейтральный рН, желудок имеет рН примерно 1,5-4,0, а рН кишечника имеет рН примерно между 5,0-7,5. Чтобы лекарственное средство высвобождалось равномерно (с кинетикой нулевого порядка), его растворение не должно зависеть от рН окружающей среды. Внутренняя часть («сердцевина») лекарственной формы по настоящему изобретению может приближаться к доставке лекарственного средства с кинетикой нулевого порядка.
Внутренняя часть - электролитная платформа
В одном варианте осуществления изобретения внутренняя часть («сердцевина») состоит из гидрофильного набухающего матрикса, в которой помещен фармацевтически активный агент («API») и один или несколько электролитов. «Электролитная сердцевина» представляет собой состав с замедленным высвобождением («SR»). Один или несколько электролитов в сочетании либо с API, либо с другой солью, при реакции в водной среде вызывает затвердевание матрикса. Скорость обратной диффузии (наружу) регулируют путем взаимодействия внутренней части с водной средой. Это, в свою очередь, индуцирует протекание реакции затвердевания зависимым от времени образом в направлении от внешней границы вглубь внутренней части; затвердевший продукт реакции, в свою очередь, ограничивает обратную диффузию API, поскольку проникновение водной среды внутрь вызывает усиление затвердевания от внешних границ внутренней части в направлении к центру внутренней части.
Во внутренней части используют явление «высаливания» в коллоидной химии для замедления кинетики набухания и эрозии неионогенного полимерного матрикса, содержащего API и один или несколько электролитов. Присутствие этих электролитических соединений в виде ионизируемых солей позволяет формироваться неразрушающимся диффузионным каналам; используемые в прошлом каналообразующие агенты не были ионизируемыми, поэтому, диффузионные каналы были непредсказуемы, что приводило к плохим профилям высвобождения и потере контроля. Электролиты также способствуют образованию микросреды в таблетке, рН которой опосредуется рКа электролита, тем самым усиливая или подавляя растворимость самого API. По мере гидратации матрикса электролиты и полимер конкурируют за поступающую воду с API, что приводит к программируемой скорости высвобождения. Внутренняя часть, таким образом, способна к равномерному, рН-независимому высвобождению API на протяжении времени до 24 часов, без учета растворимости самого API.
В результате процессов ионного взаимодействия/комплексообразования/молекулярной и/или самоассоциации между лекарственным средством и электролитом, комбинации электролит/лекарственное средство, равномерно диспергированные в набухающем полимере, таком как гидроксипропилметилцеллюлоза (HPMC), изменяют динамику скорости набухания матрицы и эрозии набухающего полимера в соответствии с изменениями рН во внешней среде в диапазоне примерно 1,5-7,0. Эти взаимодействия приводят к контролируемому затвердеванию матрикса. Такое затвердевание отвечает за контроль скорости эрозии полимера/растворения и высвобождения лекарственного средства. По замыслу, растворитель проникает на периферию таблетки, и быстрое первоначальное взаимодействие между лекарственным средством и электролитом, заключенными в полимерный матрикс, вызывает немедленное затвердевание внешней границы таблетки, скорость затвердевания постепенно уменьшается в направлении к центру сердцевины матрикса зависимым от времени образом в течение длительного периода времени (например, в течение 24 часов).
Дифференциальная скорость затвердевания матрикса является движущим принципом во внутренней части, которая зависит от скорости попадания жидкости в сердцевину внутренней части и контролируется ей. С одновременным зависящим от времени снижением целостности слоя геля, снижается и скорость диффузии лекарственного средства. Это явление компенсирует увеличение длины диффузионного пути и уменьшение площади поверхности уменьшающейся сердцевины, которые возникают из-за набухания полимера. Таким образом, достигается лучше контролируемое, предпочтительно, с кинетикой нулевого порядка, высвобождение лекарственного средства. Процесс высвобождения лекарственного средства может быть растянут до 24 часов. Контроль изменений в твердости сердцевины и синхронизации фронта жесткости/набухания и описанных отступающих фазовых границ, а также эрозии границ фронта растворения (т.е. эрозии периферии таблетки), приводит к контролируемому высвобождению лекарственного средства, предпочтительно, включая кинетику нулевого порядка. Необязательно, затвердевание полимерного матрикса легко достижимо путем взаимодействия с двойной солью. Эта комбинация бинарной соли также равномерно диспергирована в полимерном матриксе, который через ионное взаимодействие/комплексообразование/молекулярное и/или самообъединение, увеличивает относительную прочность и жесткость матрикса, приводя к контролируемому высвобождению лекарственного средства с механизмом, аналогичным описанному выше.
Одним гидрофильным матриксным материалом, используемым во внутренней части, является НРМС K4M. Это неионогенный набухающий гидрофильный полимер, производимый «The Dow Chemical Company» под торговым названием «Methocel». НРМС K4M также сокращенно называется НРМС K4MP, где «Р» относится к эфиру целлюлозы высшего качества, разработанному для препаратов с контролируемым высвобождением. «4» в аббревиатуре означает, что полимер имеет номинальную вязкость (при 2%-м содержании в воде) 4000. Процент метоксильных групп и гидроксипропильных групп составляет 19-24 и 7-12 соответственно. Внешне HPMC K4M представляет собой свободно сыпучий снежно-белый порошок, имеющий ограничение по размеру частиц, когда 90% частиц проходят через сито 100 меш. Существуют и другие типы HPMC, такие как K100LVP, K15MP, K100MP, E4MP и E10MP CR с номинальной вязкостью 100, 1500, 100000, 4000 и 10000, соответственно.
Поскольку внутренняя часть состоит из нековалентно связанной матрицы, процесс изготовления представляет собой по сути дела двухэтапный процесс сухого смешивания и прямого прессования.
В одном варианте осуществления соль диспергируют в матриксе с концентрацией в диапазоне от примерно 50% до примерно 100% по весу полимерной матрицы. В одном варианте осуществления соль выбирают из одного или двух членов группы, состоящей из хлорида натрия, бикарбоната натрия, бикарбоната калия, цитрата натрия, бисульфата натрия, сульфита натрия, сульфата магния, хлорида кальция, хлорида калия и карбоната натрия.
Полагают, что в результате взаимодействия между лекарственным средством и солью постепенно образуется комплекс в окружающем набухающем матриксе, поскольку это происходит зависимым от времени образом, по мере того как растворитель, высвобождающий лекарственное средство, проникает внутрь таблетки. Аналогичным образом, поскольку катализатором для инициации высвобождения лекарственного средства является проникновение жидкости внутрь, скорость высвобождения лекарственного средства также контролируется затвердеванием солевого комплекса, распространяющимся внутрь.
Бинарная система солей (например, хлорида кальция и карбоната натрия) также может быть использована, и в этом случае реакция затвердевания может быть функцией взаимодействия солей. Хлорид кальция может быть включен для образования комплекса с карбонатом натрия. В этой комбинации, продуктами реакции являются нерастворимый карбонат кальция и растворимый формирователь каналов, хлорид натрия. Таким образом, карбонат кальция сам внедряется в полимерный матрикс, инициирует затвердевание и медленно растворяется с проникновением жидкости и последующим созданием диффузионных каналов, по мере того как лекарственное средство диффундирует наружу. Аналогичным образом, другие бинарные комбинации солей проявляют зависимые от времени свойства «затвердевания/размягчения».
Необходимое количество соли для можно определить, учитывая растворимость лекарственного вещества, природу полимера и требуемую степень затвердевания матрикса. В случае дилтиазема гидрохлорида в матриксе HPMC, 100 мг бикарбоната натрия обеспечивает затвердевание матрикса, подходящее для контролируемого высвобождения с кинетикой нулевого порядка, тогда как в случае того же количества лекарственного средства в другом полимере, таком как полиэтиленоксид, 50 мг бикарбоната натрия, по-видимому, является идеальным для достижения контролируемого высвобождения с кинетикой нулевого порядка.
Фармацевтически активный ингредиент может быть выбран из группы, состоящей из апрепитанта (Эменда), дексаметазона, доласетрона (Анземета), дронабинола (Маринол), дроперидола (Инсапсина), гранисетрона (Китрила), галоперидола (Галдола), метилпреднизолона (Медрола) Метоклопрамида (Церукала), набилона (Цесамета), ондансетрона (Зофрана), палоносетрона (Алокси), прохлорперазина (Прокомпа), и их фармацевтически приемлемых солей или их комбинаций.
В одном варианте осуществления внутренняя часть твердой лекарственной формы по настоящему изобретению представляет собой гидрофильный набухающий полимерный матрикс, имеющий диспергированное в матриксе фармацевтически эффективное количество по меньшей мере одного антагониста серотонина, чья степень солюбилизации по существу не зависит от рН в диапазоне от 1,5 до рН 7,5, и неорганическую соль, где неорганическая соль присутствует в концентрации в диапазоне от 50% до 100% по весу полимерного матрикса. В одном варианте осуществления неорганическая соль представляет собой цитрат натрия. В одном варианте осуществления гидрофильным набухающим полимерным матриксом является гидроксипропилметилцеллюлоза или полиэтиленоксид.
Внутренняя часть, описанная выше, может быть получена способом, раскрытым в патенте США № 6090411, который включен в настоящее описание путем ссылки из-за идей, раскрытых в нем.
Внутренняя часть - аминокислотная платформа
В одном варианте осуществления изобретения внутренняя часть («сердцевина») состоит из экстрагранулярного (внешнего относительно гранул) гидрофильного полимера, в котором диспергировано множество гранул API, гранулированных по меньшей мере с одной аминокислотой и интрагранулярным (содержащимся в гранулах) полимером. «Аминокислотная сердцевина» или «АА-сердцевина» представляет собой состав с замедленным высвобождением («SR»). Гранулы диспергированы в экстрагранулярном гидрофильном полимере с образованием монолитного матрикса. Экстрагранулярный полимер быстрее гидратируется относительно интрагранулярного полимера. Быстрая гидратация экстрагранулярного полимера способствует получению линейного профиля высвобождения лекарственного средства и примерно 100%-му растворению, хотя увеличение продолжительности высвобождения и снижение эффекта взрывного высвобождения часто присущи лекарственным формам с пролонгированным высвобождением. Линейная скорость высвобождения может быть адаптирована для удовлетворения требований каждого варианта применения путем выбора полимеров для различных скоростей растворения. В одном варианте осуществления достигается время высвобождения от 12 до 24 часов.
Интрагранулярный полимер объединяют с API и по меньшей мере с одной аминокислотой для образования гранул. Интрагранулярный полимер может быть одним или несколькими из следующих полимеров: поливинилацетат, галактоманнановый полисахарид, такой как гидроксипропиловый гуар, гуаровая камедь, камедь плодов рожкового дерева, пектин, гуммиарабик, трагакант, камедь карайи, простые эфиры целлюлозы, такие как гидроксипропилметилцеллюлоза (HPMC), а также другие камеди и простые эфиры целлюлозы, которые будут выбраны специалистом в данной области по свойствам, соответствующим идее настоящего изобретения. В одном варианте осуществления интрагранулярный полимер представляет собой галактоманнановый полисахарид, гуаровую камедь (с диапазоном вязкости 75-6000 сП для 1%-го раствора при 25°С в воде и размером частиц 10-300 мкм).
Интрагранулярный полимер во внутренней части присутствует в количестве от 4% до 45% от общего веса лекарственной формы. Конкретный тип интрагранулярного полимера и количество используемого интрагранулярного полимера выбираются в зависимости от желаемой скорости высвобождения лекарственного средства, вязкости полимера, желаемого количества лекарственного средства и растворимости лекарственного средства. Интрагранулярный полимер гидратируется менее быстро, чем экстрагранулярный полимер. Относительная разница в скорости гидратации между двумя полимерами создает менее вязкий интрагранулярный полимер и более вязкий экстрагранулярный полимер. Со временем разница в вязкости способствует непрерывной эрозии и распаду твердой лекарственной формы.
Аминокислоты могут использоваться в данном варианте осуществления по двум основным причинам. Во-первых, аминокислоты являются фактором, определяющим вязкость полимеров. Как было отмечено выше, с течением времени разница в вязкости между экстрагранулярным и интрагранулярным полимерами способствует непрерывной эрозии и распаду сердцевины, способствуя примерно 100%-му высвобождению лекарственного средства. Другим важным аспектом использования аминокислот в гранулах является то, что гидропатия аминокислоты может использоваться для модулирования растворимости и высвобождения лекарственного средства.
Таким образом, аминокислоту выбирают по гидропатии в зависимости от характеристик растворимости активного вещества. Когда соединение является по меньшей мере умеренно растворимым в воде, то есть, например, умеренно растворимым, растворимым или имеет более высокий уровень растворимости согласно определено в Фармакопее США, используется аминокислота, которая имеет относительно равный баланс между гидрофильной и гидрофобной составляющей, то есть является нейтральной или сбалансированной, или близка к нейтральной, или является относительно более сильно гидрофильной.
Например, растворение и высвобождение растворимых или слаборастворимых способных образовывать ионы лекарственных соединений, таких как верапамил⋅НСl, можно контролировать путем включения одной или нескольких аминокислот в гранулы. Без привязки к какой-либо конкретной теории высвобождения лекарственного средства и растворения, полагают, что природа процесса гранулирования является таковой, что поскольку компоненты состава вступают в тесный молекулярный контакт, гранулирование уменьшает доступную площадь поверхности частиц, тем самым снижая начальную скорость гидратации. В гранулированных составах карбоксильные (COOH-) группы и аминогруппы (NH2/NH3+) аминокислоты имеют возможность взаимодействовать с гидроксильными группами на полимере, таким образом опосредуя набухание, вязкость и гелевые свойства полимера и тем самым контролируя опосредованную набуханием диффузию лекарственного средства. Одновременно карбоксильные группы аминокислот могут также взаимодействовать с соответствующими полярными заместителями в молекуле лекарственного средства, такими как вторичные или третичные амины. Кроме того, гидрофильный и ионогенный характер аминокислот обеспечивает их интенсивную гидратацию в водном растворе. Следовательно, эта аминокислота способствует эрозии, но также конкурирует как с полимером, так и с лекарственным средством за поглощение воды, необходимой для гидратации и растворения.
Однако, когда действующее соединение имеет растворимость ниже умеренной, включая действующие соединения слабо растворимые или нерастворимые, используется комбинация по меньшей мере из двух аминокислот, одна из которых является сильно гидрофобной, а другая является относительно более гидрофильной, чем гидрофобный компонент, т.е. является примерно нейтральной или сбалансированной и до сильно гидрофильной.
Аминокислотный компонент гранул может включать любые фармацевтически приемлемые α-амино или β-аминокислоты, соли α-амино или β-аминокислот, или любую их комбинацию. Примерами подходящих α-аминокислот являются глицин, аланин, валин, лейцин, изолейцин, фенилаланин, пролин, аспарагиновая кислота, глутаминовая кислота, лизин, аргинин, гистидин, серин, треонин, цистеин, аспарагин и глутамин. Примером β-аминокислоты является β-аланин.
Тип аминокислот, используемых в этом варианте осуществления внутренней части, может быть описан как гидрофильные, гидрофобные, соли гидрофильных или гидрофобных аминокислот, или любая их комбинация. Подходящие для использования гидрофобные аминокислоты, включают, но не ограничиваются ими, изолейцин, фенилаланин, лейцин и валин. Кроме того, в гранулах могут быть использованы гидрофильные аминокислоты, такие как глицин, аспартат и глутамат. В конечном счете, любая аминокислота и любая аминокислота в комбинации с другой аминокислотой могут использоваться в настоящем изобретении для повышения растворимости лекарственного средства. Подробный список аминокислот, которые могут быть использованы в настоящем изобретении и гидропатию каждой из них см. в Albert L. Lehninger et al., Principles of Biochemistry 113 (2nd ed. Worth Publishers 1993).
Тип и количество аминокислот могут быть выбраны в зависимости от желаемого количества лекарственного средства, желаемой скорости высвобождения лекарственного средства и растворимости лекарственного средства. Аминокислота в лекарственной форме составляет, как правило, от 4% до 45% от общего веса лекарственной формы. В одном варианте осуществления количество аминокислоты составляет от 11% до 29% по весу от общего веса лекарственной формы.
Гранулы могут быть необязательно смешаны с материалом оболочки, например, стеаратом магния или другими гидрофобными производными стеариновой кислоты. Используемое количество материала оболочки может варьировать от 1% до 3% от общего веса лекарственной формы. Обычно, стеарат магния используется для облегчения производства, например, для улучшения сыпучести, но в настоящем изобретении стеарат магния имеет дополнительное преимущество, замедляя растворение благодаря гидрофобной природе материала оболочки. Поэтому, стеарат магния может использоваться для дополнительной корректировки растворимости лекарственной формы и дополнительного замедления высвобождения лекарственного средства из гранул.
Для дополнительного повышения механических свойств и/или влияния на скорость высвобождения лекарственного средства гранулы могут также содержать небольшие количества инертных фармацевтических наполнителей и связующих/гранулирующих агентов, как обычно принято в данной области. Примеры инертных фармацевтических наполнителей включают: лактозу, сахарозу, мальтозу, мальтодекстрины, декстрины, крахмал, микрокристаллическую целлюлозу, фруктозу, сорбит, гидрофосфат и фосфат кальция. Примеры гранулирующих агентов/ связующих веществ включают крахмал, метилцеллюлозу, гидроксипропилцеллюлозу или гидроксипропилметилцеллюлозу, натриевую соль карбоксиметилцеллюлозы или поливинилпирролидон, гуммиарабик, трагакант и сахарозу. Также могут использоваться другие подходящие наполнители, как очевидно специалисту в данной области. В зависимости от физических и/или химических свойств лекарственного средства, может быть использованы процедура влажного гранулирования (с использованием водной или органической гранулирующей жидкости) или процедура сухого гранулирования (например, комкование или вальцевание).
После гранулирования фармацевтически активного соединения, интрагранулярного полимера, аминокислоты и, необязательно, наполнителей и гидрофобных материалов оболочки, гранулы затем смешивают с экстрагранулярным полимером и диспергируют в нем.
Экстрагранулярный полимер может быть одним или несколькими из следующих полимеров: полиэтиленоксида, галактоманнанового полисахарида, такого как гидроксипропиловый гуар, гуаровая камедь, камедь плодов рожкового дерева, пектин, гуммиарабик, трагакант, камедь карайи, простые эфиры целлюлозы, такие как гидроксипропилметилцеллюлоза (HPMC), а также другие камеди и простые эфиры целлюлозы, которые будут выбраны специалистом в данной области по свойствам, соответствующим идее настоящего изобретения. Экстрагранулярный полимер может представлять собой галактоманнановый полисахарид, такой как гуаровая камедь (с диапазоном вязкости 75-6000 сП для 1%-го раствора при 25°С в воде и размером частиц 10-300 мкм). Как было отмечено выше, экстрагранулярный полимер должен быстро гидратироваться и достигать высокого уровня вязкости за более короткий период времени по сравнению с интрагранулярным полимером.
Разница в скорости гидратации между экстрагранулярным полимером и интрагранулярным полимером достигается тремя основными способами: (1) путем выбора полимеров на основе из различий в размере частиц; (2) путем выбора полимеров на основе различий в молекулярной массе и химическом составе; и (3) путем выбора полимеров на основе комбинации (1) и (2). Хотя данное описание фокусируется в основном на полимерах, выбранных по различиям в размерах частиц, можно достичь результатов по данному изобретению с использованием интрагранулярного полимера с молекулярной массой и/или химическим составом, отличающимся от таковых у экстрагранулярного полимера. Например, полиэтиленоксид может быть использован в качестве интрагранулярного полимера, а гуаровая камедь - в качестве экстрагранулярного полимера.
Размер частиц является еще одной отличительной особенностью коммерческой гуаровой камеди, поскольку более крупные частицы обеспечивают быструю дисперсию, в то время как более мелкие частицы идеально подходят для быстрой гидратации. Поэтому для того, чтобы достичь желаемого результата настоящего изобретения. В одном варианте осуществления, более мелкие частицы используются для экстрагранулярного полимера, а более крупные частицы используются для интрагранулярных полимерных частиц. Брошюра от HERCULES Incorporated с названием «Supercol® Guar Gum, 1997» содержит типичные свойства гуаровой камеди различных марок и размеров частиц. Другие быстро гидратирующиеся экстрагранулярные полимеры, которые могут быть использованы, включают: полиэтиленоксид (РЕО), простые эфиры целлюлозы и полисахариды, такие как гидроксипропиловый гуар, пектин, гумиарабик и трагакант, камедь карайи, смеси вышеуказанных полимеров и любых других полимеров, выбираемых специалистом в данной области по свойствам, соответствующим идее настоящего изобретения. Количество и тип экстрагранулярного полимера выбирают в зависимости от требуемого количества лекарственного средства, скорости высвобождения лекарственного средства и растворимости лекарственного средства. Было найдено, что подходящим является диапазон, составляющий примерно 4-47% (от общего веса таблетки), экстрагранулярного полимера. В одном варианте осуществления диапазон содержания экстрагранулярного полимера составляет от примерно 15% до примерно 47% (от общего веса таблетки).
Во внутреннюю часть может быть включено терапевтическое количество API, например, до примерно 75% от общего веса лекарственной формы. При этом количестве лекарственного средства внутренняя часть приближается к линейному профилю высвобождения с минимальным или отсутствующим эффектом взрывного высвобождения. Однако по желанию квалифицированного специалиста экстрагранулярный полимер может содержать дополнительные количества фармацевтически активного соединения для достижения более быстрого высвобождения лекарственного средства или для индукции эффекта взрывного высвобождения, а также может содержать аминокислоты, чтобы опосредовать растворение фармацевтически активного соединения, описанного выше.
Таблетированная пероральная лекарственная форма с пролонгированным высвобождением, необязательно, может быть покрыта полимерами, пластификаторами, матирующими агентами и красителями, как принято в данной области.
В одном варианте осуществления внутренняя часть твердой лекарственной формы по настоящему изобретению представляет собой: (1) множество гранул, содержащих (а) по меньшей мере, один антагонист серотонина; (b) по меньшей мере одну аминокислоту; и (с) интрагранулярный полимер, причем интрагранулярный полимер содержится в количестве от 4% до 45% по весу от общей лекарственной формы; и (2) гидрофильный экстрагранулярный полимер, в котором диспергированы гранулы; причем экстрагранулярный полимер содержится в количестве от 4% до 47% по весу от общей лекарственной формы и более быстро гидратируется чем интрагранулярный полимер, где аминокислота выбирается по характеристикам гидропатии в зависимости от характеристик растворимости по меньшей мере одного антагониста серотонина и содержится в количестве от 11% до 29% по весу от общей лекарственной формы. В одном варианте осуществления, когда по меньшей мере один антагонист серотонина является по меньшей мере умеренно растворимым в воде, аминокислота имеет относительно равный баланс между гидрофобными и гидрофильными составляющими или является относительно более гидрофильной. В варианте осуществления, когда по меньшей мере один антагонист серотонина меньше чем умеренно растворим в воде, аминокислота представляет собой комбинацию по меньшей мере из двух аминокислот, одна из которых является умеренно или сильно гидрофобной, а другая является относительно более гидрофильной. В одном варианте осуществления интрагранулярный полимер содержит по меньшей мере одно из следующих соединений: поливинилацетат, галактоманнановый полисахарид, выбранный из группы, состоящей из гидроксипропилового гуара, гуаровой камеди, камеди плодов рожкового дерева, пектина, гуммиарабика, трагаканта, камеди карайи, или простые эфиры целлюлозы. В одном варианте осуществления аминокислота выбрана из группы, состоящей из: а) α-аминокислоты b) β-аминокислоты с) комбинации α- и β-аминокислоты. В одном варианте осуществления α-аминокислота является по меньшей мере одним членом, выбранным из группы, состоящей из глицина, аланина, валина, лейцина, изолейцина, фенилаланина, пролина, аспарагиновой кислоты, глутаминовой кислоты, лизина, аргинина, гистидина, серина, треонина, цистеина, аспарагина и глутамина. В одном варианте осуществления комбинация α и β-аминокислот содержит β-аланин, и по меньшей мере одну α-аминокислоту выбирают из группы, состоящей из глицина, аланина, валина, лейцина, изолейцина, фенилаланина, пролина, аспарагиновой кислоты, глутаминовой кислоты, лизина, аргинина, гистидина, серина, треонина, цистеина, аспарагина и глутамина. В одном варианте осуществления аминокислота выбрана из группы, состоящей из: а) сбалансированный аминокислоты, имеющей относительно равный баланс между гидрофобными и гидрофильными составляющими или относительно более гидрофильной аминокислоты, или b) комбинации (i) сбалансированной аминокислоты или относительно более гидрофильной аминокислоты и (ii) гидрофобной аминокислоты. В одном варианте осуществления сбалансированная аминокислота содержит глицин. В одном варианте осуществления изобретения внутренняя часть содержит глицин и гидрофобную аминокислоту, выбранную из изолейцина, валина и фенилаланина. В одном варианте осуществления множество гранул смешивают с гидрофобным материалом оболочки. В одном варианте осуществления изобретения гидрофобный материал оболочки представляет собой стеарат магния. В одном варианте осуществления изобретения гидрофобный материал оболочки составляет от 1% до 3% от общего веса лекарственной формы.
Внутренняя часть, описанная выше, может быть изготовлена способом, описанным в патенте США № 6517868, который включен в настоящее описание путем ссылки из-за идей, раскрытых в нем.
Первая и вторая оболочки
Первая и вторая оболочки бимодальной твердой лекарственной формы с пролонгированным высвобождением по настоящему изобретению являются нефункциональными оболочками, которые действуют в качестве технологических добавок. Первая оболочка и вторая оболочка не оказывают существенного влияния на высвобождение API из лекарственной формы. В одном варианте осуществления первая и вторая оболочка содержат гидрофильный материал. В одном варианте осуществления гидрофильным материалом является гипромеллоза. В одном варианте осуществления гипромеллозой является Methocel E5. В одном варианте осуществления первая и вторая оболочка дополнительно содержат добавку для оболочки plasACRYL™, водную эмульсию глицерилмоностеарата и триэтилцитрата (разработанную Emerson Resources, Inc., Norristown, PA, USA). В одном варианте осуществления plasACRYL™, используемый в первой и второй оболочке относится к классу Т20. В одном варианте осуществления PlasACRYL™ Т20 представляет собой 20%-ю водную суспензию, содержащую противодействующий прилипанию агент, пластификатор и стабилизатор. Гипромеллоза представляет собой не зависящий от рН неионогенный полимер, образованный путем частичного замещения O-метилированными и О-(2-гидроксипропилированными) группами. Категории гипромеллозы варьируют в зависимости от степени замещения, которая влияет на вязкость. HPMC K4M Premium имеет вязкость 3550 мПа⋅с, тогда как НРМС Е5 Premium LV представляет собой полимер с низким классом вязкости, имеющий вязкость 5 мПа⋅с. Гипромеллоза растворима в холодной воде и образует коллоидную вязкую жидкость.
Верхняя оболочка из слоя лекарственного средства
Верхняя оболочка из слоя лекарственного средства твердой лекарственной формы с пролонгированным высвобождением по настоящему изобретению является слоем лекарственного средства с немедленным высвобождением («IR»). В одном варианте осуществления верхняя оболочка из слоя лекарственного средства подобрана так, чтобы обеспечить взрывное высвобождение около 25% API, что при пероральном приеме твердой дозированной формы, привело бы к тому, что около 25% API высвобождалось бы в желудке. В одном варианте осуществления верхняя оболочка из слоя лекарственного средства или слой с немедленным высвобождением лекарственного средства содержит ондансетрона гидрохлорид, гипромеллозу и plasACRYL™. В одном варианте осуществления гипромеллозой, используемой в IR-слое является Methocel E5.
Дополнительные слои - энтеросолюбильная оболочка
В одном варианте осуществления твердая лекарственная форма с пролонгированным высвобождением по настоящему изобретению дополнительно включает в себя энтеросолюбильную оболочку. В одном варианте осуществления слой энтеросолюбильной оболочки расположен между первой оболочкой и верхним слоем лекарственного средства. В одном варианте осуществления, энтеросолюбильной оболочкой является EUDRAGIT® L30D-55. В одном варианте осуществления слоем энтеросолюбильной оболочки является EUDRAGIT® FS 30 D. В одном варианте осуществления энтеросолюбильной оболочкой является SURETERIC®.
Твердые пероральные лекарственные формы по настоящему изобретению
В одном варианте осуществления твердая пероральная лекарственная форма по настоящему изобретению включает в себя в общем 24 мг ондансетрона (или эквивалентное количество ондансетрона-HCl). В одном варианте осуществления, 18 мг ондансетрона присутствует в сердцевине лекарственной формы, и 6 мг ондансетрона присутствует в верхнем слое лекарственного средства.
В одном варианте осуществления твердая пероральная лекарственная форма по настоящему изобретению включает в себя в общем 12 мг ондансетрона (или эквивалентное количество ондансетрона-HCl). В одном варианте осуществления 9 мг ондансетрона присутствует в сердцевине лекарственной формы, и 3 мг ондансетрона присутствует в верхнем слое лекарственного средства.
В одном варианте осуществления твердая пероральная лекарственная форма по настоящему изобретению включает в себя в общем 28 мг ондансетрона (или эквивалентное количество ондансетрона-HCl). В одном варианте осуществления изобретения 20 мг ондансетрона присутствует в сердцевине лекарственной формы, и 8 мг ондансетрона присутствует в верхнем слое лекарственного средства.
В одном варианте осуществления твердая пероральная лекарственная форма по настоящему изобретению включает в себя в общем 36 мг ондансетрона (или эквивалентное количество ондансетрона-HCl). В одном варианте осуществления 28 мг ондансетрона присутствует в сердцевине лекарственной формы, и 8 мг ондансетрона присутствует в верхнем слое лекарственного средства.
Схема введения
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 24 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят взрослому человеку или ребенку (старше 12 лет), страдающим от острого гастроэнтерита, чтобы обеспечить быстрое облегчение симптомов и сохранение эффекта без необходимости повторного введения в течение болезни, которая обычно продолжается приблизительно один день. Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 12 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят ребенку (младше 12 лет), страдающему от острого гастроэнтерита, чтобы обеспечить быстрое облегчение симптомов и сохранение эффекта без необходимости повторного введения в течение болезни, которая обычно продолжается приблизительно один день. Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 24 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят взрослому человеку или ребенку (старше 12 лет), страдающим от продолжительного гастроэнтерита, чтобы обеспечить быстрое облегчение симптомов и сохранение лечебного эффекта. В течение болезни может оказаться необходимым повторное введение (один раз в день). Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 12 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят ребенку (младше 12 лет), страдающему от продолжительного гастроэнтерита, чтобы обеспечить быстрое облегчение симптомов и сохранение лечебного эффекта. В течение болезни может оказаться необходимым повторное введение. Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 28 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят взрослому человеку или ребенку (старше 12 лет), страдающим от продолжительного гастроэнтерита, чтобы обеспечить быстрое облегчение симптомов и сохранение лечебного эффекта. В течение болезни может оказаться необходимым повторное введение. Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 36 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят взрослому человеку или ребенку (старше 12 лет), страдающим от продолжительного гастроэнтерита, чтобы обеспечить быстрое облегчение симптомов и сохранение лечебного эффекта. В течение болезни может оказаться необходимым повторное введение. Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердая пероральная лекарственная форма по настоящему изобретению включает в себя в общем 12 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl). Лекарственную форму разделяют пополам, и дают ребенку в возрасте 4-12 лет, чтобы обеспечить быстрое облегчение симптомов и сохранение лечебного эффекта. В течение болезни может оказаться необходимым повторное введение, например, через каждые 8 часов Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 24 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят беременной пациентке, страдающей от рвоты беременных, чтобы обеспечить быстрое облегчение симптомов и сохранение лечебного эффекта. В течение болезни может оказаться необходимым повторное введение. Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 28 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят беременной пациентке, страдающей от рвоты беременных, чтобы обеспечить быстрое облегчение симптомов и сохранение лечебного эффекта. В течение болезни может оказаться необходимым повторное введение. Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 36 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят беременной пациентке, страдающей от рвоты беременных, чтобы обеспечить быстрое облегчение симптомов и сохранение лечебного эффекта. В течение болезни может оказаться необходимым повторное введение. Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 12 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят пациенту, имеющему синдром раздраженного кишечника с преобладанием диареи (IBS-D), чтобы обеспечить быстрое облегчение симптомов и сохранение лечебного эффекта. В течение болезни может оказаться необходимым повторное введение. Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
В одном варианте осуществления твердую пероральную лекарственную форму по настоящему изобретению, включающую в себя в общем 24 мг ондансетрона (в виде эквивалентного количества ондансетрона-НСl), вводят пациенту, имеющему синдром раздраженного кишечника с преобладанием диареи (IBS-D), чтобы обеспечить быстрое облегчение симптомов и сохранение лечебного эффекта. В течение болезни может оказаться необходимым повторное введение. Высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает действие ондансетрона на протяжении как минимум 16 часов, что приводит к снижению частоты любого из рвоты, тошноты или диареи у пациента.
Следующие примеры приведены, чтобы обеспечить для средних специалистов в этой области полное раскрытие изобретения и описание того, как осуществлять и применять описанное изобретение, и они не предназначены для ограничения объема того, что авторы изобретения считают своим изобретением, а также они не должны создавать представление о том, что представленные ниже эксперименты являются единственно проведенными. Были предприняты усилия для обеспечения точности в отношении используемых чисел (например, количеств, температуры и т.д.), но следует учитывать некоторые экспериментальные ошибки и отклонения. Если не указано иное, части являются весовыми частями, молекулярный вес является средневесовым молекулярным весом, температура приведена в градусах Цельсия, а давление равно или близко к атмосферному.
ПРИМЕРЫ
Пример 1 - изготовление внутренних сердцевин, содержащих 18 мг ондансетрона
Таблица 1. Внутренняя сердцевина, содержащая 18 мг ондансетрона; сердцевина содержит аминокислоту («АА-сердцевина»)
|
Состав с аминокислотой («АА-сердцевина») изготавливали с использованием влажной грануляции с низким усилием сдвига. Микрокристаллическую целлюлозу Avicel® PH-102, ондансетрон-НСl, глицин и НРМС K15M смешивали в V-образном смесителе с объемом 0,028 м3 в течение 10 минут, выгружали и убирали комки, используя Comil, оборудованный ситом 20 меш. Предварительную смесь затем гранулировали в Hobart D300 путем добавления воды к смеси при перемешивании. После добавления воды материал перемешивали в течение еще 2 минут. Материал гранулировали при достаточном, но не слишком сильном увлажнении, поэтому не добавляли дополнительное количество воды. Влажную массу просеивали через сито 8 меш и затем высушивали в сушильном шкафу. Высушенные гранулы перемалывали с использованием Comil с ситом 18 меш, смешивали с экстрагранулярной НРМС K100LV и смазывающим веществом. Прессование конечной смеси проводили на 36-позиционном прессе Kikusui с использованием 8 мм × 15 мм модифицированного овального пресс-инструмента.
Таблица 2. Внутренняя сердцевина, содержащая 18 мг ондансетрона; сердцевина содержит электролит («сердцевина с электролитом»)
|
Состав с электролитом («сердцевину с электролитом») изготавливали путем смешивания и прессования. Все материалы просеивали по отдельности через ручное сито 30 меш, загружали в V-образный смеситель и смешивали в течение 15 минут, а затем добавляли смазывающее вещество. Прессование конечной смеси проводили на 36-позицонном прессе Kikusui с использованием 7 мм × 13 мм модифицированного овального пресс-инструмента.
Пример 2 - первая и вторая изолирующие оболочки; необязательная энтеросолюбильная оболочка
Таблица 3. Состав изолирующей оболочки (внутренней оболочки и верхней оболочки)
|
* размер партии для одной изолирующей оболочки, примерно с 30%-м избытком
Таблица 4. Состав энтеросолюбильной оболочки
|
* Размер партии включает в себя 30%-й избыток
Раствор изолирующей оболочки был изготовлен путем растворения Methocel E5 в воде и последующего добавления PlasACRYL™. Суспензию для энтеросолюбильной оболочки изготавливали путем смешивания воды, триэтилцитрата и PlasACRYL™. Добавляли дисперсию EUDRAGIT®; суспензию перемешивали в течение 30 минут, а затем просеивали через сито 60 меш. Суспензию с действующим веществом изготовляли, сначала растворяя Methocel E5 в воде, и отдельно диспергировали ондансетрон в воде и гомогенизировали. Раствор Methocel затем добавляли к суспензии лекарственного средства, и добавляли PlasACRYL™.
Пример 3 - Верхняя оболочка из слоя лекарственного средства
Таблица 5. Составы оболочки с лекарственным средством
|
* Размер партии включает 18% избытка для учета потерь при изготовлении
Таблетки покрывали требуемыми оболочками, перечисленными в таблицах 6-8. Увеличение веса контролировали путем измерения веса 50 таблеток каждые 10 минут. Благодаря наличию оборудования, первые две партии были покрыты оболочками с использованием лабораторного (R&D) устройства для нанесения оболочек (OʹHara LabMX). Третья партия была изготовлена с использованием cGMP-оборудования, которое будет использоваться для СТМ-производств (веществ для клинических испытаний).
Таблица 6. Параметры нанесения оболочки; продукт 1
|
Таблица 7. Параметры нанесения оболочки; продукт 2
|
Таблица 8. Параметры нанесения оболочки; продукт 3
|
Таблица 9. Общее описание партий
|
Пример 4 - профиль растворения
Таблица 10. Растворение (таблетки ондансетрона с бимодальным высвобождением, 24 мг)
|
В таблице 10 в сочетании с фиг.1 и 2 показан профиль растворения для продуктов 1, 2 и 3. У продукта 1 было первоначальное взрывное высвобождение 25% соединения с последующим замедленным высвобождением в течение 24 часов. У продукта 2 было первоначальное взрывное высвобождение 25% соединения с последующим замедленным высвобождением в течение 24 часов. У продукта 3 было первоначальное взрывное высвобождение 25% соединения с последующей отсрочкой высвобождения в кислой среде.
Пример 5 - изготовление содержащей ондансетрон внутренней сердцевины с электролитом
Сердцевины таблеток ондансетрона-HCl изготавливали сухим смешиванием и прямым прессованием. Детали ингредиентов состава приведены в таблицах 11 и 12. Профиль растворения (с учетом энтеросолюбильной оболочки и оболочки, содержащей 6 мг лекарственного средства с немедленным высвобождением) для этого состава показан на фиг. 3.
Таблица 11. Ондансетрон - электролит 11 - сердцевина таблетки
|
Таблица 12. Состав 11 Ондансетрона-HCl, 22,5 мг
|
Пример 6 - профиль растворения
In vitro растворение проводили в физиологически соответствующей среде в диапазоне рН от 1,2 до 7,2, приближающемся к уровням в желудочно-кишечном тракте. Вследствие различий в растворимости ондансетрона-HCl (API) при разных значениях рН использовали максимум абсорбции для расчета высвобождения растворения, а не калибровочную кривую, построенную с использованием API в воде. Результаты исследования растворения для среды с рH 1,2, 4,5, 6,8, 7,2 и дистилиллированной Н2О приведены на фиг.4.
Пример 7 - in vivo тестирование твердых лекарственных форм
Одноцентровое, рандомизированное, лабораторно-слепое, 4-периодное с 4-последовательностями введения, перекрестное исследование было проведено на здоровых мужчинах и женщинах. Следующие тестируемые продукты вводили субъектам натощак:
|
Продукты введили 28 здоровым мужчинам и женщинам в соответствии с таблицей 13.
|
ВЫБОР ДОЗИРОВОК В ИССЛЕДОВАНИИ
Дозу выбирали, чтобы получить действие, аналогичное действию доступного в продаже состава с немедленным высвобождением (Зофран®, 8 мг) при ежедневном трехкратном введении.
ВЫБОР ДОЗИРОВОК И ВРЕМЯ ВВЕДЕНИЯ ДЛЯ КАЖДОГО СУБЪЕКТА
Субъекты голодали ночью по меньшей мере в течение 10 часов до введения лекарства утром.
Тестируемые продукты 1-3
Одну дозу назначенного тестируемого состава вводили перорально приблизительно с 240 мл воды при температуре окружающей среды, начиная с 07:30, одному субъекту в минуту.
Эталонный продукт
Назначенный эталонный состав вводили перорально (три раза в день, с 8-часовыми интервалами) с приблизительно 240 мл воды при температуре окружающей среды, начиная с 07:30, одному субъекту в минуту. Последующее введение состава проводили во второй половине дня и вечером в 15:30 и 23:30, соответственно.
Голодание продолжали в течение по меньшей мере 4 часов после введения препарата утром, после чего подавали стандартизированный обед. Обед должен был быть завершен не позднее чем через 5 часов после утреннего введения препарата. Все приемы пищи осуществляли в соответствующие моменты времени после этого, но не ранее, чем через 9 часов после утреннего введения препарата. Ужин не подавали ранее 11 часов после утреннего введения препарата, и он должен был быть завершен не позднее чем через 13 часов после утреннего введения препарата. Кроме того, легкие перекусы должны были быть закончены не позднее чем через 13 часов после утреннего введения препарата. Вода была разрешена в неограниченном количестве до 1 часа до введения препарата и начиная с 1 часа после каждого введения препарата.
АНАЛИЗ И ДИАГРАММА ИЗМЕРЕНИЙ ЭФФЕКТИВНОСТИ И БЕЗОПАСНОСТИ
Фармакокинетический анализ
Образцы крови для фармакокинетических измерений собирали до и в течение 32 часов (последовательный отбор образцов) после каждого утреннего введения препарата. Прямым измерением в этом исследовании была концентрация ондансетрона в плазме. Эту концентрацию получали путем анализа плазмы, выделенной из образцов крови, собранных в ходе данного исследования. Общий объем крови, собранный у каждого субъекта (639 мл для мужчин и 653 мл для женщин), считается оказывающим незначительное влияние или вообще не влияющим на фармакокинетические профили препаратов и оценки биоэквивалентности. Кроме того, считается, что он оказывает незначительное влияние на безопасность субъектов.
ИЗМЕРЕНИЯ КОНЦЕНТРАЦИИ ЛЕКАРСТВЕННОГО СОЕДИНЕНИЯ
Тестируемые продукты 1-3 (21 образец крови):
Первый образец крови из каждого периода, то есть пустой образец плазмы, собирали перед введением лекарственного средства, тогда как другие образцы собирали через 0,25, 0,5, 1, 1,5, 2, 2,5, 3, 3,5, 4, 5, 6, 7, 8, 9, 10, 12, 16, 20, 24 и 32 часа после введения лекарственного средства в одну 6-мл пробирку (K2 EDTA Vacutainers).
Эталонный продукт (33 образца крови):
Первый образец крови из каждого периода, то есть пустой образец плазмы, собирали перед введением лекарственного средства, тогда как другие образцы собирали через 0,25, 0,5, 1, 1,5, 2, 2,5, 3, 4, 6, 8, 8,25, 8,5, 9, 9,5, 10, 10,5, 11, 12, 14, 16, 16,25, 16,5, 17, 17,5, 18, 18,5, 19, 20, 22, 24, 28 и 32 часа после утреннего введения лекарства в одну 6-мл пробирку (K2 EDTA Vacutainers). 8-часовой и 16-часовой образцы собирали в течение 5 минут до введения лекарственного средства (дневного и вечернего введения).
Тестируемый продукт ондансетрона 1 (тестируемый продукт 1) относительно эталонного продукта (эталона)
Двадцать шесть (26) субъектов были включены в сравнение между тестируемым препаратом 1 и эталонным препаратом. Обобщенные фармакокинетические параметры и стандарты для сравнительной биодоступности представлены в таблицах 14 и 15. Профиль средней измеренной концентрации в плазме в зависимости от времени, полученный после введения тестируемого продукта 1 и эталонного продукта, изображен на фиг.5, а логарифмически преобразованный профиль средней концентрации относительно времени показан на фиг.6.
Таблица 14. Обобщение основных результатов исследования тестируемого продукта ондансетрона 1 относительно эталонного продукта
|
Для Tmax представлена медиана.
Таблица 15. Сравнение результатов со стандартами по биоэквивалентности - тестируемый продукт ондансетрона 1 относительно эталонного продукта
|
* единицами являются нг/мл для Cmax и нг⋅ч/мл для AUCT и AUC∞
Число субъектов, включенных в статистический анализ этих параметров, составляло n=24 для тестируемого продукта 1 и n=26 для эталонного продукта. Среднее значение Cmax составляло, соответственно, 50,669 нг/мл и 50,731 нг/мл для тестируемого состава 1 и эталонного состава. Соотношение геометрических LS-средних Cmax тестируемого состава 1 и эталонного состава составляло 99,05% (90%-й ДИ: с 92,89 до 105,62%). Таким образом, этот результат показывает, что соотношение и соответствующий 90%-й доверительный интервал соотношения геометрических LS-средних Cmax тестируемого состава 1 и эталонного состава находились в пределах заданного диапазона от 80,00 до 125,00%. Медианное Tmax составляло, соответственно, 3,50 и 17,50 часов для тестируемого состава 1 и эталонного состава. Среднее значение AUCT составляло, соответственно, 659,098 нг⋅ч/мл и 854,517 нг⋅ч/мл для тестируемого состава 1 и эталонного состава. Соотношение геометрических LS-средних AUCT для тестируемого состава 1 и эталонного состава составляло 77,54% (90%-й ДИ: с 73,60 до 81,68%). Таким образом, этот результат показывает, что соотношение и соответствующий 90%-й доверительный интервал соотношения геометрических LS-средних AUCT тестируемого состава 1 и эталонного состава выходят за пределы заданного диапазона от 80,00 до 125,00%. Среднее значение Kel составляло 0,0671 ч-1 для тестируемого состава 1 и 0,1391 часов ч-1 для эталонного состава. Среднее значение T1/2el составляло 11,72 и 5,40 ч для тестируемого состава 1 и эталонного состава, соответственно. Среднее значение AUC∞ составляло, соответственно, 795,397 нг⋅ч/мл и 946,030 нг⋅ч/мл для тестируемого состава 1 и эталонного состава. Соотношение геометрических LS-средних AUC∞ тестируемого состава 1 и эталонного состава составляло 83,95% (90%-й ДИ: с 78,46 до 89,82%). Таким образом, этот результат показывает, что 90%-й доверительный интервал соотношения геометрических LS-средних AUC∞ тестируемого состава 1 и эталонного состава выходит за пределы заданного диапазона от 80,00 до 125,00%. Среднее значение индивидуального соотношения AUCT и AUC∞ (AUCT/∞) составляло, соответственно, 84,61% и 92,07% для тестируемого состава 1 и эталонного состава.
Тестируемый продукт ондансетрона 2 (тестируемый продукт 2) относительно эталонного продукта
Двадцать шесть (26) субъектов были включены в сравнение между тестируемым продуктом 2 и эталонным продуктом. Обобщенные фармакокинетические параметры и стандарты для сравнительной биодоступности представлены в таблицах 16 и 17. Профиль средней измеренной концентрации в плазме в зависимости от времени, полученный после введения тестируемого продукта 2 и эталонного продукта, изображен на фиг.5, а логарифмически преобразованный профиль средней концентрации относительно времени показан на фиг.6.
Таблица 16. Обобщение основных результатов исследования тестируемого продукта ондансетрона 2 относительно эталонного продукта
|
Для Tmax представлена медиана.
Таблица 17. Сравнение результатов со стандартами по биоэквивалентности - тестируемого продукта ондансетрона 2 относительно эталонного продукта
|
* единицами являются нг/мл для Cmax и нг⋅ч/мл для AUCT и AUC∞
Среднее значение Cmax составляло, соответственно, 55,718 нг/мл и 50,731 нг/мл для тестируемого состава 2 и эталонного состава. Соотношение геометрических LS-средних Cmax тестируемого состава 2 и эталонного состава составляло 110,93% (90%-й ДИ: с 104,03 до 118,30%). Таким образом, этот результат показывает, что соотношение и соответствующий 90%-й доверительный интервал соотношения геометрических LS-средних Cmax тестируемого состава 2 и эталонного состава находились в пределах заданного диапазона от 80,00 до 125,00%. Медианное Tmax составляло, соответственно, 4,00 и 17,50 часов для тестируемого состава 2 и эталонного состава. Среднее значение AUCT составляло, соответственно, 730,199 нг⋅ч/мл и 854,517 нг⋅ч/мл для тестируемого состава 2 и эталонного состава. Соотношение геометрических LS-средних AUCT для тестируемого состава 2 и эталонного состава составляло 86,79% (90%-й ДИ: с 82,38 до 91,43%). Таким образом, этот результат показывает, что соотношение и соответствующий 90%-й доверительный интервал соотношения геометрических LS-средних AUCT тестируемого состава 2 и эталонного состава находились в пределах заданного диапазона от 80,00 до 125,00%. Среднее значение Kel составляло 0,0676 ч-1 для тестируемого состава 2 и 0,1391 часов ч-1 для эталонного состава. Среднее значение T1/2el составляло 10,84 и 5,40 ч для тестируемого состава 2 и эталонного состава, соответственно. Среднее значение AUC∞ составляло, соответственно, 847,660 нг⋅ч/мл и 946,030 нг⋅ч/мл для тестируемого состава 2 и эталонного состава. Соотношение геометрических LS-средних AUC∞ тестируемого состава 2 и эталонного состава составляло 91,38% (90%-й ДИ: с 85,57 до 97,58%). Таким образом, этот результат показывает, что соотношение и соответствующий 90%-й доверительный интервал соотношения геометрических LS-средних AUC∞ тестируемого состава 2 и эталонного состава находились в пределах заданного диапазона от 80,00 до 125,00%. Среднее значение индивидуального соотношения AUCT и AUC∞ (AUCT/∞) составляло, соответственно, 87,44% и 92,07% для тестируемого и эталонного составов.
Тестируемый продукт ондансетрона 3 (тестируемый продукт 3) относительно эталонного продукта
Двадцать пять (25) субъектов были включены в группу тестируемого препарата 3 и двадцать шесть (26) субъектов были включены в группу эталонного препарата. Обобщенные фармакокинетические параметры и стандарты для сравнительной биодоступности представлены в таблицах 18 и 19. Профиль средней измеренной концентрации в плазме в зависимости от времени, полученный после введения тестируемого продукта 3 и эталонного продукта, изображен на фиг.5, а логарифмически преобразованный профиль средней концентрации относительно времени показан на фиг.6.
Таблица 18. Обобщение основных результатов исследования тестируемого продукта ондансетрона 3 относительно эталонного продукта
|
Для Tmax представлена медиана.
Таблица 19. Сравнение результатов с стандартами по биоэквивалентности - тестируемого продукта ондансетрона 3 относительно эталонного продукта
|
* единицами являются нг/мл для Cmax и нг⋅ч/мл для AUCT и AUC∞
Количество субъектов, включенных в статистический анализ этих параметров, составляло n=23 для тестируемого состава 3 и n=26 для эталонного состава. Среднее значение Cmax составляло, соответственно, 32,958 нг/мл и 50,731 нг/мл для тестируемого состава 3 и эталонного состава. Соотношение геометрических LS-средних Cmax тестируемого состава 3 и эталонного состава составляло 65,67% (90%-й ДИ: с 61,54 до 70,09%). Этот результат, таким образом, показывает, что соотношение и соответствующий 90%-й доверительный интервал соотношения геометрических LS-средних Cmax тестируемого состава 3 и эталонного состава выходят за пределы заданного диапазона от 80,00 до 125,00%. Медианное Tmax составляло, соответственно, 5,00 и 17,50 часов для тестируемого состава 3 и эталонного состава. Среднее значение AUCT составляло, соответственно, 646,611 нг⋅ч/мл и 854,517 нг⋅ч/мл для тестируемого состава 3 и эталонного состава. Соотношение геометрических LS-средних AUCT для тестируемого состава 3 и эталонного состава составляло 76,47% (90%-й ДИ: с 72,54 до 80,61%). Таким образом, этот результат показывает, что соотношение и соответствующий 90%-й доверительный интервал соотношения геометрических LS-средних AUCT тестируемого состава 3 и эталонного состава выходили за пределы заданного диапазона от 80,00 до 125,00%. Среднее значение Kel составляло 0,0640 ч-1 для тестируемого состава 3 и 0,1391 часов ч-1 для эталонного состава. Среднее значение T1/2el составляло 12,73 и 5,40 ч для тестируемого состава 3 и эталонного состава, соответственно. Среднее значение AUC∞ составляло, соответственно, 830,321 нг⋅ч/мл и 946,030 нг⋅ч/мл для тестируемого состава 3 и эталонного состава. Соотношение геометрических LS-средних AUC∞ тестируемого состава 3 и эталонного состава составляло 88,38% (90%-й ДИ: с 82,53 до 94,65%). Таким образом, этот результат показывает, что соотношение и соответствующий 90%-й доверительный интервал соотношения геометрических LS-средних AUC∞ тестируемого состава 3 и эталонного состава находились в пределах заданного диапазона от 80,00 до 125,00%. Среднее значение индивидуального соотношения AUCT и AUC∞ (AUCT/∞) составляло, соответственно, 80,15% и 92,07% для тестируемого состава 3 и эталонного состава.
Пример 8 - разработка состава пероральной твердой лекарственной формы ондансетрона с пролонгированным высвобождением в течение 36-48 часов
Может быть желательно создание пероральной твердой лекарственной формы ондансетрона с пролонгированным высвобождением в течение 36-48 часов, содержащей 36 мг до 48 мг ондансетрона гидрохлорида.
В таблице 20 представлена композиция сухой смеси для прямого прессования составов сердцевин таблеток с пролонгированным высвобождением, содержащих 20 и 28 мг ондансетрона в виде свободного основания. Материалы, используемые для создания сердцевин таблеток ондансетрона гидрохлорида с пролонгированным высвобождением, были аналогичны перечисленным в таблице 2 из примера 1 для состава бимодальной таблетки 24 мг (18 мг ондансетрона в виде свободного основания в сердцевине таблетки) за исключением того, что использовали гипромеллозу К4М премиального качества для прямого прессования (DC) вместо гипромеллозы K4M премиального качества.
Таблица 20: композиции сухой смеси для прямого прессования составов с пролонгированным высвобождением ондансетрона
|
*: Перед регулировкой силы действия
В таблице 21 представлена композиция сухой смеси прямого прессования для состава сердцевины таблетки с 8 мг свободного основания ондансетрона с оболочкой для продолжительного высвобождения.
Таблица 21: композиция сухой смеси прямого прессования для состава сердцевины таблетки ондансетрона с оболочкой для продолжительного высвобождения
|
Пример 9 - описание процесса сухого смешивания
Сухую смесь получали с использованием лабораторного смесителя PK Blend Master (Patterson-Kelly, East Stroudsburg, PA, USA), оборудованного 1,5-литровой V-образной емкостью для составов с L004-04001 по -04008 в лабораторном масштабе (таблицы 22А и 22В и таблицы 23A и 23B, соответственно). Все материалы по отдельности просеивали через ручное сито 30 меш, загружали в V-образный смеситель и смешивали в течение 15 мин при 25 оборотах в минуту без смазывающего агента, который затем добавляли и перемешивали еще в течение 3 минут. Такой же способ смешивания применяли к партиям -04002, -04004, -04006 и -04004 с оболочкой для продолжительного высвобождения.
Таблица 22А: композиция составов сердцевины таблетки ондансетрона с пролонгированным высвобождением, партии L004-04001 и L003
|
Таблица 22В: композиция составов сердцевины таблетки ондансетрона с пролонгированным высвобождением, партии L005 и L007
|
Таблица 23А: композиция составов сердцевины таблетки ондансетрона с оболочкой для продолжительного введения, партии L004-04002 и L004
|
Таблица 23В: композиция составов сердцевины таблетки ондансетрона с оболочкой для продолжительного введения, партии L006 и L008
|
Схемы процессов изготовления бимодальной таблетки с пролонгированным высвобождением и составов для продолжительного введения представлены на фиг.7 и фиг.8, соответственно.
Пример 10 - описание процесса прессования ядра таблетки
Испытания процесса прессования для партий L004-0401, -04001A, -04005 и -04007 состава с пролонгированным высвобождением были выполнены с использованием гидравлического лабораторного ручного пресса с стандартным сферическим пресс-инструментом диаметром 10,0 мм, тогда как прессование для партии -04003 было проведено с использованием 6-позиционного роторного таблеточного пресса PR6 (SVIAC, Antony, France), оснащенного гравитационным порошковым питателем с 8,0×16,0×2,0 (глубокая овальная сфера) пресс-инструментом D-типа. Сердцевину таблеток - 04007B также прессовали с использованием 6-позиционного роторного таблеточного пресса PR6 с 7,0×14,0 мм пресс-инструментом D-типа с числом «20», нанесенным на верхний пуансон. Сердцевины таблеток из партий L004-04002, -04002C, -04004, -04006 и -04008, покрываемых оболочкой для продолжительного введения, также прессовали с использованием SVIAC со стандартным 6,0 мм сферическим пресс-инструментом D-типа.
Пример 11 - нанесение оболочки на таблетки
Нанесение изолирующей оболочки, энтеросолюбильной оболочки и оболочки для немедленного высвобождения для бимодального лекарственного продукта из составов -04001, -04003 и -04007, а также пленочной оболочки для лекарственных препаратов с продолжительным введением из составов -04002, -04004, -04006 и -04008, выполняли с использованием лабораторного аппарата для создания псевдоожиженного слоя, Aeromatic-Fielder (модель Strea-1, Columbia, MD, USA), снабженного колонкой Wurster. Суспензию для нанесения оболочки распыляли с использованием перистальтического насоса Cole-Parmer (модель 77521-40, Vernon Hills, IL, USA) с шлангами Masterflex # 16.
Водные композиции для изолирующей и энтерсолюбильной оболочки, а также для слоя с немедленным высвобождением, наносимого на покрытые энтеросолюбильной оболочкой таблетки с продолжительным высвобождением, приведены в таблицах 24, 25 и 26, соответственно. Сердцевины таблеток 28 мг из первой попытки прессования состава с пролонгированным высвобождением L004-04005 не покрывали оболочкой. Они предназначались для оценки и сравнения профилей растворения в среде с рН 6,8 относительно партии -04003.
Таблица 24: композиции всех водных суспензий для изолирующих оболочек
|
*: удаляется в процессе нанесения оболочки и сушки
Таблица 25: композиции всех водных суспензий энтеросолюбильных оболочек
|
*: удаляется в процессе нанесения оболочки и сушки
Таблица 26: композиции всех водных суспензий слоев с немедленным высвобождением
|
*: удаляется в процессе нанесения оболочки и сушки
В таблице 27 показаны различные композиции разнообразных вариантов водных суспензий оболочки для продолжительного высвобождения, наносимых на сердцевину таблетки 8 мг. Варианты №1 и №2 суспензии оболочки были изготовлены без талька. Варианты №3, №4, №7 и №9 включали 5,88% талька, тогда как в вариантах №5 и №6 содержание талька было снижено до 1%.
Таблица 27: различные композиции вариантов водных суспензий №№1-8 оболочек для продолжительного высвобождения
|
Пример 12 - изолирующая оболочка таблеток
Водный раствор изолирующей оболочки (6,12 весов.%) изготавливали путем растворения METHOCEL™ E5 в воде и последующего добавления plasACRYL™ с использованием лопастной мешалки (с диаметром приблизительно 50,0 мм), как показано на фиг. 9. В таблице 28 приведены параметры процесса нанесения изолирующей оболочки. На заключительной стадии сушки температура воздуха на входе была установлена на уровне 46°С.
Таблица 28: технологические параметры нанесения изолирующей оболочки с помощью аппарата Aeromatic-Fielder с псевдоожиженным слоем
|
Пример 13 - нанесение на таблетки энтеросолюбильной оболочки
Использовали 24,58%-ю (вес.) водную систему для энтеросолюбильной оболочки, которую получали путем смешивания с водой триэтилцитрата и plasACRYL™ с использованием лопатной мешалки (с диаметром приблизительно 50,0 мм), как показано на фиг.10. Добавляли дисперсию EUDRAGIT®, после чего суспензию перемешивали в течение 30 минут при 400 об./мин, затем просеивали через сито 60 меш. Параметры нанесения энтеросолюбильной оболочки представлены в таблице 29. На заключительной стадии сушки температура воздуха на входе была установлена на уровне 46°С.
Таблица 29: технологические параметры нанесения энтеросолюбильной оболочки с помощью аппарата Aeromatic-Fielder с псевдоожиженным слоем
|
Пример 14 - нанесение на таблетки оболочки с немедленно высвобождающимся лекарственным средством
6,18%-ю (вес.) водную суспензию действующего вещества получали сначала растворением METHOCEL™ E5 в половине заданного количества воды, которая будет использоваться, и отдельно диспергированием ондансетрона в оставшейся воде и перемешиванием при высокой скорости (750-950 об./мин) с использованием лопастной мешалки с диаметром 50,0 мм в течение 90 минут. Раствор METHOCEL™ затем добавляли к суспензии лекарственного средства, и в конце добавляли plasACRYL™, как представлено на фиг. 11. Параметры нанесения энтеросолюбильной оболочки приведены в таблице 30. В течение последней стадии сушки температура воздуха на входе была установлена на уровне 46°C для L001 и поддерживалась на уровне 56-58°С для L001A и L003, а также для последующих составов бимодальных таблеток.
Таблица 30: технологические параметры нанесения оболочки из слоя лекарственного средства с немедленным высвобождением с помощью аппарата Aeromatic-Fielder с псевдоожиженным слоем
|
Пример 15 - таблетки с оболочкой для продолжительного высвобождения
Различные композиции водных суспензий оболочки для продолжительного высвобождения были испытаны при различных степенях увеличения веса таблетки, чтобы оценить, насколько они могут задержать высвобождение лекарственного продукта.
Для композиций водных суспензий оболочки для продолжительного высвобождения (24,12 вес.%), используемых в испытаниях №1 и №2 (фиг.12), EUDRAGIT® RL 30D и за ним plasACRYL™ вносили в подходящий контейнер, а затем смешивали с образованием воронки, используя лопастную мешалку с диаметром 50,0 мм. Затем последовательно добавляли EUDRAGIT® RS 30 и очищенную воду и перемешивали перед просеиванием через сито 250 мкм (60 меш).
Для композиций водных суспензий оболочки для продолжительного высвобождения (20,0 вес.%), используемых в испытаниях №№3-8 (фиг.13), очищенную воду наливали в подходящую емкость и перемешивали, используя лопастную мешалку с диаметром 50,0 мм, с образованием воронки. Затем последовательно добавляли тальк и триэтилцитрат и перемешивали. В конце добавляли EUDRAGIT® RS 30D и RL 30D и перемешивали перед просеиванием через сито 500 мкм (35 меш).
Для лекарственного продукта с продолжительным высвобождением из партий L004-04002D по -04008B была добавлена стадия процесса отверждения путем распыления небольшого количества очищенной воды, равного примерно четверти от общего объема используемой водной суспензии для продолжительного высвобождения или ее половине (при использовании очень небольшого количества водной суспензии для продолжительного высвобождения) при температуре на выходе 50°C, непосредственно перед окончательной фазой сушки в соответствии с рекомендациями поставщика Eudragit Evonik. Однако для 04006E и 04008A стадию отверждения не выполняли, чтобы оценить влияние отверждения на таблетки, покрытые оболочкой.
Пример 16 - дополнительное отверждение таблеток с продолжительным высвобождением
Партии таблеток с продолжительным высвобождением с L004-04002D и -04002F (8 мг ондансетрона в виде свободного основания) дополнительно отверждали без распыления воды в течение 2 часов при температуре воздуха на входе 50°С, получая, соответственно, L004-04002D-2HC (2 часа отверждения) и -04002F- 2HC, для того чтобы оценить влияние дополнительного отверждения на растворимость таблеток с продолжительным высвобождением.
Сухие смеси для прямого прессования составов сердцевин таблеток с пролонгированным высвобождением L004-04001, -04003, -04005 и -04007 были приготовлены с содержанием API 6,64% и 9,30%, соответственно, в то время как составы сердцевин таблеток с продолжительным высвобождением -04002, -04004, -04006 и -04008 были изготовлены с содержанием API 12,44%. Все составы давали высокий выход 99,6% или выше при размере лабораторных партий 0,1 кг.
Обобщенное описание партий
|
Пример 17 - in vitro профили растворения
На фиг.14 представлено сравнение профилей растворения бимодальных круглых выпуклых таблеток ондансетрона 28 мг из партий -04001 и -04001, прессованных с низкой и высокой твердостью, соответственно, овальных выпуклых таблеток 36 мг из партии -04003. Бимодальная таблетка 36 мг из партии -04003 с 41,30% гипромеллозы K4M (агента, замедляющего высвобождение) давала 80% растворение на 36-й час.
Сердцевина таблетки 28 мг из партии -04005 со сниженным содержание агента замедленного высвобождения до 34,30% давала 87% растворения на 36-й час (фиг.15). Партии -04007A и -04007B (фиг.16 и 17) включали 30,0% гипромеллозы (агента замедленного высвобождения). На фиг.16 представлено сравнение профилей растворения сердцевины таблетки ондансетрона 28 мг (-04007) и бимодальной таблетки (-04007A). Оказалось, что оболочка действительно влияла на профили растворения и следует проверить эффект снижения веса энтеросолюбильной оболочки.
На фиг.17 представлено сравнение профилей растворения в мг бимодальных таблеток ондансетрона из составов -04001, -04003 и -04007. Лекарственные продукты -04001, -04001A и -04007A прессовали со стандартным 10,0-мм круглым вогнутым пресс-инструментом, тогда как партию -04003 прессовали с использованием овального вогнутого (8,0×16,0×2,0 мм) пресс-инструмента, а партию -04007B с использованием пресс-инструмента для продолговатой капсулы (7,0×14,0 мм, сверху выдавлено «20»).
Через 36 часов растворения партии -04007A (твердость 24,4 кП) и -04007B (твердость 20,2 кП) показали самый высокие показатели растворимости API, высвобождая более 30 мг из ожидаемых 36 мг. Однако партия -04007B показала более быстрый профиль растворения по сравнению с -04007A.
На фиг.18 представлено сравнение профилей растворения в процентах для бимодальных составов ондансетрона из показанных выше на фиг.17 со скорректированными значениями ожидаемых результатов с учетом фактического среднего веса сердцевины таблетки, составляющего 360,0 мг.
Партию -04009A (твердость 12,6 кП) и 04009B (твердость 16,7 кП) прессовали, используя овальный вогнутый новый пресс-инструмент (7,6×14,0 мм) со средним весом сердцевины таблетки 376,40 мг и 386,87 мг, соответственно. Чтобы получить разницу в твердости всего примерно 4 кП, сила сжатия была увеличена в пять раз с 3923 Н до 21575 Н для партий 04009A и 04009B, соответственно, что указывает на пластическую деформацию сердцевины таблетки с более высокой твердостью, что могло бы объяснить более быстрое высвобождение. На фиг.16 представлены профили растворения в мг относительно времени для бимодальных лекарственных продуктов 04003, 04007A, 04007B, 04009A и 04009B. Более 32 мг из ожидаемых 36 мг высвобождались из бимодальной таблетки 04009B, немного лучше, чем партия 04007B, но немного быстрее. На фиг.17 показаны те же результаты в процентах растворенного вещества.
|
На фиг.19 представлено сравнение профилей растворения круглых выпуклых таблеток с продолжительным введением, 8 мг, из партий -04002D и -04002D-2HC с использованием варианта №1 композиции оболочки (соотношение EUDRAGIT® RS/RL: 3-7); партии -04002E с использованием варианта №2 композиция оболочки (соотношение EUDRAGIT® RS/RL: 7-3), партии -04002F с использованием варианта №3 композиции оболочки (соотношение EUDRAGIT® RS/RL: 9-1) и партии-002J с использованием варианта №4 композиции оболочки (соотношение EUDRAGIT® RS/RL: 8-2). В составе -04002 использовали только МСС-102 в качестве наполнителя, и его время распадения сердцевины составило 15 мин.
Варианты №№ 1 и 2 композиций оболочки для продолжительного введения без талька не были способны задерживать растворение на 2 ч кислотной стадии. Вопреки ожидаемому, дополнительное отверждение в течение двух часов для -04002D - 04002HC не улучшало сопротивление растворению во время кислотной стадии. Однако отвержденные в течение 2 часов -04002F - 04002HC с использованием варианта №3 композиции оболочки, содержащей 5,9% талька (соотношение EUDRAGIT® RS/RL: 9-1), были еще целы через 36 часов, высвободив только около 1%. Партия -04002J, покрытая оболочкой для продолжительного высвобождения с использованием варианта №4 композиции оболочки с 5,9% талька (соотношение EUDRAGIT® RS/RL: 8-2) с 4,9%-м приростом веса, показала очень незначительное высвобождение API в течение 36 часов.
На фиг.20 показано сравнение профилей растворения круглых выпуклых таблеток с продолжительным высвобождением, 8 мг, из состава -04004, изготовленного с МСС-102 и Tablettose 80 с временем распадения сердцевины менее 8 минут. Партии -04004A и -04004B были покрыты оболочкой для продолжительного высвобождения с использованием варианта №5 композиции оболочки (соотношение EUDRAGIT® RS/RL: 8-2 и 1% талька) для увеличения веса 4,9 и 11,0%, соответственно, тогда как партии -04004C и -04004D были покрыты оболочкой с использованием варианта №6 композиции оболочки (соотношение EUDRAGIT® RS/RL: 6-4 с 1% талька) с увеличением веса 4,9 и 10,1%. Все четыре партии показали быстрое профили растворения в течение первых 3 часов, но не смогли высвободить более 75% в течение 36 часов. По неизвестной причине, партия -04004D с двукратным увеличением веса по сравнению с партией -04004C показала более быстрый профиль растворения.
На фиг.21 показано сравнение профилей растворения круглых выпуклых таблеток с продолжительным высвобождением, 8 мг, из партии -04006, полученной с МСС-102, TABLETOSSE® 80 и 4% крахмалгликолята натрия в качестве разрыхлителя, и чье время распадения сердцевины было меньше 2 минут. Партии -04006A и -04006B были покрыты оболочкой для продолжительного высвобождения с использованием варианта №4 композиции оболочки как и -04002J, (соотношение EUDRAGIT® RS/RL: 8-2 и 5,9% талька) для увеличения веса 4,8 и 9,8%, соответственно, тогда как партии -04006C и -04006D были покрыты оболочкой с использованием варианта №7 композиции оболочки (соотношение EUDRAGIT® RS/RL: 7-3 с 5,9% талька) с увеличением веса 4,9 и 9,8%. Максимальное высвобождение API более чем за 36 часов составило 40% для -04006C.
На фиг.22 показано сравнение профилей растворения круглых выпуклых таблеток с продолжительным высвобождением, 8 мг, из состава -04008, полученного с такими же вспомогательными веществами как и L-006, но с увеличением крахмалгликолята натрия до 8%, и для которого время распадения сердцевины было менее 1 мин.
Партии -04008A и -04008B были покрыты оболочкой для продолжительного введения с использованием варианта №8 композиции оболочки (соотношение EUDRAGIT® RS/RL: 6-4 и 5,9% талька) для увеличения веса на 10,1%. Единственное различие между этими двумя партиями в процессе изготовления было то, что для партии L-008A не проводили отверждение после нанесения пленочной оболочки для продолжительного высвобождения. Профили растворения обеих партий были почти одинаковыми. По сравнению с L-006D (с аналогичным увеличением веса, но с оболочкой из варианта №7 композиции (соотношение EUDRAGIT® RS/RL: 7-3 и 5,9% талька), профиль растворения улучшился, но все же не достигал более чем 40% за 36 часов.
Пример 18-3-групповое перекрестное сравнительное исследование биодоступности твердых лекарственных форм
3-групповое перекрестное сравнительное исследование биодоступности после пяти дней введения (один раз в день) твердых лекарственных форм по настоящему изобретению в сравнении с двухдневным введением два раза в день таблеток ондансетрона 8 мг с немедленным высвобождением в сравнении с однократным введением таблеток ондансетрона 24 мг с немедленным высвобождением у здоровых мужчин и женщин-добровольцев/натощак.
Цели:
Основной целью данного исследования было сравнение относительной биодоступности и пиковой и минимальной концентрации между двумя одобренными FDA схемами введения коммерчески доступных таблеток ондансетрона 8 мг с немедленным высвобождением (схемы введения два раза в день Зофрана® 8 мг в течение двух дней и схемы однократного введения Зофрана® 24 мг в виде трех таблеток Зофрана® 8 мг, принимаемых вместе) и испытываемым продуктом, таблетками ондансетрона 24 мг с пролонгированным высвобождением по настоящему изобретению (один раз в день).
Вторичными целями исследования были:
1. Оценка накопления ондансетрона в плазме после введения тестируемого продукта в течение пяти последовательных дней натощак.
2. Оценка безопасности и переносимости состава с пролонгированным высвобождением у здоровых добровольцев.
Методика:
Одноцентровое, рандомизированное, открытое, 3-периодное, с 3 последовательностями введения, перекрестное исследование.
Число субъектов (планируемое и проанализированное):
Планируемое для включения: 18
Включенные: 18
Выбывшие: 0
Проанализированные: 18
Учтенные в фармакокинетическом и статистическом анализе: 18
Учтенные в анализе безопасности: 18
Диагноз и основные критерии включения:
В исследование были включены добровольцы, мужчины и женщины, некурящие или бросившие курить, в возрасте по меньшей мере 18 лет с индексом массы тела выше или равным 18,50 и ниже 30,00 кг/м2. Субъекты имели хорошее состояние здоровья, что было определено по медицинской истории, полному медицинскому осмотру (включая жизненно важные признаки), 12-канальной электрокардиограмме (ЭКГ) и обычным клиническим лабораторным исследованиям (общей биохимии, гематологии, анализу мочи), включая отрицательный анализ на вирус иммунодефицита человека (ВИЧ), гепатит В и С, а также отрицательный результат скрининга мочи на алкоголь, котинин и вызывающие привыкание лекарственные соединения, а также отрицательный качественный тест на беременность по содержанию в сыворотке бета-хорионического гонадотропина человека (ХГЧ) (для женщин).
Тестируемый продукт, дозировка и способ введения:
Название: ондансетрон.
Лекарственная форма/путь введения: бимодальная таблетка по настоящему изобретению (CDT-сердцевина с электролитом)/перорально («Тестируемый продукт»).
Схема лечения-1: однократная доза 24 мг (1×24 мг) один раз в день в течение 5 дней подряд.
Эталонный продукт, дозировка и способ введения:
Название: Зофран®
Лекарственная форма/путь введения: таблетка/пероральная
Схема лечения-2: однократная доза 8 мг (1×8 мг) два раза в день с 8-часовым интервалом в 1-й день 12-часовым интервалом во 2-й день.
Схема лечения-3: однократная доза 24 мг (3×8 мг)
Лечение:
Лечение-1: тестируемый продукт вводят один раз в день в течение 5 дней подряд.
Лечение-2: эталонный продукт вводится дважды в день с 8-часовым интервалом в 1-й день 12-часовым интервалом во 2-й день.
Лечение-3: однократную дозу 24 мг вводят в виде трех эталонных таблеток, принимаемых вместе.
Периоды лечения:
Период 1: 2013/08/08 до 2013/08/12 (лечение-1)
Период 1: 2013/08/08 до 2013/08/09 (лечение-2)
Период 1: 2013/08/08 (лечение-3)
Период 2: 2013/08/17 до 2013/08/21 (лечение-1)
Период 2: 2013/08/17 до 2013/08/18 (лечение-2)
Период 2: 2013/08/17 (лечение-3)
Период 3: 2013/08/26 до 2013/08/30 (лечение-1)
Период 3: 2013/08/26 до 2013/08/27 (лечение-2)
Период 3: 2013/08/26 (лечение -3)
Продолжительность лечения:
Лечение-1: одну дозу ондансетрона 24 мг (1 × бимодальную таблетку 24 мг (CDT-сердцевина с электролитом)) («Тестируемый продукт») вводили перорально один раз в день утром после 10-часового ночного голодания в течение 5 дней подряд.
Лечение-2: одну дозу Зофрана® 8 мг (1 × таблетку 8 мг) вводили перорально два раза в день в течение двух дней подряд с 8-часовым интервалом в 1-й день и с 12-часовым интервалом во 2-й день (первую дозу утром каждый день после 10-часового ночного голодания и вторую дозу во второй половине дня (1 день) или вечером (2-й день)) (всего 4 введения лекарственного препарата).
Лечение-3: одну дозу Зофрана® 24 мг (3 × таблетки 8 мг) перорально вводили после 10-часового ночного голодания.
Период вымывания между первыми введениями лекарственного препарата в каждом периоде исследований должен был составлять 9 календарных дней.
Точки забора образцов крови:
В ходе исследования, в общей сложности 98 образцов крови были собраны следующим образом:
Лечение-1: в 1-й и 2-й день введения, собирали 13 образцов крови в день. Первый образец крови собирали перед введением лекарственного средства (в течение 5 минут), тогда как другие собирали через 1, 2, 3, 4, 5, 6, 8, 10, 12, 14, 16 и 20 ч после введения лекарственного средства.
В 3-й и 4-й день введения, собирали 8 образцов крови в день, причем первый образец крови отбирали перед введением лекарственного средства (в течение 5 минут), тогда как другие собирали через 2, 4, 6, 8, 10, 14 и 18 часов после введения лекарственного средства.
В 5-й день приема лекарственного средства собирали 10 образцов крови, причем первый образец крови отбирали перед введением лекарственного средства (в течение 5 минут), тогда как другие собирали через 2, 4, 6, 8, 10, 14, 18, 24 и 48 часов после введения лекарственного средства.
Всего было собрано по 52 образца для каждого субъекта при этом лечении.
Лечение-2: в 1-й день введения препарата 15 образцов крови были собраны. Первый образец крови собирали до введения препарата утром (в течение 5 минут), в то время как другие были собраны 1, 2, 3, 4, 5, 6, 8, 9, 10, 11, 12, 14, 16 и 20 часов после введения препарата утром. 8-часовой образец крови собирали в течение 5 минут перед введением во второй половине дня.
Во 2-й день введения лекарственного средства, были собраны 17 образцов крови. Первый образец крови собирали до введения лекарственного средства утром (в течение 5 минут), тогда как другие были собраны через 1, 2, 3, 4, 5, 6, 8, 12, 13, 14, 15, 16, 18, 20, 24 и 48 часов после утреннего введения лекарственного средства. 12-часовой образец крови собирали в течение 5 минут до начала вечернего введения лекарственного средства.
Всего было собрано по 32 образца для каждого субъекта при этом лечении.
Лечение-3: в 1-й день введения лекарственного средства были собраны 14 образцов крови. Первый образец крови собирали перед введением лекарственного средства (в течение 5 минут), тогда как другие собирали через 1, 2, 3, 4, 5, 6, 8, 10, 12, 16, 20, 24 и 48 часов после введения лекарственного средства.
Критерии оценки
Аналитический способ:
Анализируемое соединение: ондансетрон в плазме крови человека
Способ: ВЭЖХ с МС/МС-обнаружением
Аналитический диапазон: от 0,500 нг/мл до 300,000 нг/мл
Безопасность:
Безопасность оценивали путем оценки побочных эффектов, стандартных лабораторных тестов, жизненно важных признаков, ЭКГ и физического осмотра.
Математическая модель и статистические методы анализа фармакокинетических параметров
Для основных параметров всасывания и распределения использовали бескамерный подход с допущением логарифмически-линейной терминальной фазы. Для оценки площади под кривой использовали правило трапеции, оценка терминальной фазы основана на максимальном коэффициенте определения. Фармакокинетическими параметрами, представляющие интерес для данного исследования, должны были быть Cmax для каждого дня введения, AUC0-24 для каждого дня введения, Cmin для каждого дня введения и C24 для каждого дня введения. Другие параметры, включая Tmax для каждого дня введения, AUCT, AUC∞, AUCT/∞, Kel и T1/2el, должны были быть вычислены.
Статистический анализ всех фармакокинетических параметров основан на параметрической случайной модели ANOVA. Двусторонний 90%-й доверительный интервал соотношения геометрических LS-средних получен из логарифмически преобразованных фармакокинетических параметров.
Во время лечения с использованием тестируемого продукта, необходимо было провести сравнение Cmax и AUC0-24 в дни 2-5 с Cmax и AUC0-24 в 1-й день для оценки накопления при повторном введении.
Накопление тестируемого состава должно было оцениваться с использованием логарифмически преобразованных Cmax и AUC0-24. Модель дисперсионного анализа (ANOVA) должна была включать день как фиксированный эффект и субъекта как случайный эффект.
ANOVA-модель для сравнения вариантов лечения:
- фиксированные факторы: последовательность, период, лечение
- случайный фактор: субъект (сгруппированный в последовательности)
ANOVA для накопления:
- фиксированные факторы: день
- случайный фактор: субъект
Стандарты для сравнительной биодоступности:
Концентрации ондансетрона в динамике после введения дозы тестируемого состава должны были сравниваться с концентрациями после введения по эталонным схемам. Если было найдено, что концентрация ондансетрона после введения тестируемого состава была аналогичной или выше концентрации после введения по одной или обоим эталонным схемам в большинство моментов времени в течение изучаемого первого 24-часового периода, можно сделать вывод, что тестируемый продукт должен был быть по меньшей мере также эффективен, как существующие схемы, в отношении лечения рвоты и тошноты.
Безопасность:
Описательная статистика.
ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ
Результаты по безопасности:
Девять (9) из 18 субъектов (50,0%), включенных в это исследование, испытали в общей сложности 28 событий побочных эффектов. Все 28 событий побочных эффектов в ходе исследования были легкой степени тяжести. В приведенной ниже таблице представлены число побочных эффектов в результате лечения, классифицированных по степени тяжести и причине:
Таблица 28. Число пациентов с побочными эффектами
|
Шесть (6) субъектов (33,3%) сообщили о 12 побочных эффектах (2 различных класса систем органов и 7 различных предпочтительных терминов) после лечения-1, 5 субъектов (27,8%) сообщили о 7 побочных эффектах (3 различных класса систем органов и 5 различных предпочтительных терминов) после лечения-2, и 6 субъектов (33,3%) сообщили о 9 побочных эффектах (4 различных класса систем органов и 7 различных предпочтительных терминов) после лечения-3. Число субъектов, которые испытывали по меньшей мере один побочный эффект в ходе исследования было одинаковы для всех 3 вариантов лечения.
Побочными эффектами, которые испытывали два или несколько субъектов при любых вариантах лечения, были (лечение-1, лечение-2, лечение-3) аномальные фекалии (2, 0, 0), запор (2, 0, 1), гематома в месте укола в сосуд (2, 3, 2), боль в месте укола в сосуд (0, 1, 1), головная боль (0, 1, 1) и сонливость (0, 1, 1). Кроме того, связанные побочные эффекты, которые испытывали два или несколько субъектов при любых вариантах лечения были (лечение-1, лечение-2, лечение-3) запор (2, 0, 1), головная боль (0, 1, 1) и сонливость (0, 1, 1).
Никаких серьезных побочных эффектов или смертей не было зарегистрировано в ходе этого исследования. Кроме того, в ходе этого исследования не было обнаружено клинически значимых изменений в лабораторных анализах, жизненно важных признаках, ЭКГ или при физическом обследовании.
Никакие побочные эффекты не требовали применения лекарственных препаратов после первого введения исследуемого соединения.
Ни один субъект не был исключен из исследования по соображениям безопасности.
Фармакокинетические результаты:
Сравнения вариантов лечения:
Основные фармакокинетические параметры (Cmin, Cmax, C24 и AUC0-24) каждого варианта лечения были измерены для каждого дня введения препарата. Сравнивали первые 2 дня введения тестируемого продукта с 2 днями введения Зофрана, 8 мг (дважды в день), а также сравнивали первый день введения тестируемого продукта с введением Зофрана, 24 мг. Краткое изложение результатов этих сравнений представлены на фиг.23-28 и в таблицах 29-32.
Таблица 29. Фармакокинетические параметры после введения тестируемого продукта
|
Таблица 30. Фармакокинетические параметры после введения Зофрана 8 мг дважды в день
|
Таблица 31. Фармакокинетические параметры после однократного введения Зофрана 24 мг
|
Таблица 32. Сравнение вариантов лечения (с продолжением)
|
* Единицами являются нг/мл для Cmax, Cmin and C24 и нг*ч/мл для AUC0-24\
** Относится к Зофрану 8 мг два раза в день или Зофрану 24 мг х 1 согласно сравнению
Сравнение концентраций:
Концентрации ондансетрона в выбранные моменты времени после введения тестируемого продукта сравнивали с таковыми после введения Зофрана 8 мг дважды в день и после однократного введения Зофрана 24 мг. Концентрации, измеренные через 10, 12 14 и 16 часов после введения тестируемого продукта в день 1 и через 20 часов после введения во 2-й день сравнивали с соответствующими измеренными концентрациями ондансетрона, достигнутыми после применения других вариантов лечения. Краткое изложение результатов этих сравнений представлено в следующих таблицах.
Таблица 33. Концентрация после введения тестируемого продукта
|
Таблица 34. Концентрация после введения Зофрана 8 мг дважды в день
|
Таблица 35. Концентрация после однократного введения Зофрана 24 мг
|
Н/П: не применимо
Таблица 36. Сравнение концентраций после введения
|
* Относится к Зофрану 8 мг два раза в день или к Зофрану 24 мг х 1 согласно сравнению
Оценка накопления:
Для того чтобы оценить накопление ондансетрона после многократного введения тестируемого продукта, Cmax и AUC0-24 измеряли в течение 5 последовательных дней введения и сравнивали с однократным введением дозы тестируемого продукта в 1-й день. Краткое изложение результатов представлено в следующих таблицах.
Таблица 37. Оценка накопления тестируемого продукта - Cmax
|
* Относится к 2-му, 3-му, 4-му или 5-му дню согласно сравнению
Таблица 38. Оценка накопления тестируемого продукта - AUC0-24
|
* Относится к 2-му, 3-му, 4-му или 5-му дню согласно сравнению
Обсуждение фармакокинетических данных:
Сравнения вариантов лечения:
- Тестируемый продукт относительно Зофрана 8 мг дважды в день:
Результаты, представленные в данном документе, показывают, что Cmin и Cmax за 24 часа, а также AUC0 24, были выше в течение первых двух дней введения тестируемого продукта по сравнению с двумя днями введения Зофрана 8 мг дважды в день.
Было обнаружено, что C24 была выше при введении тестируемого продукта в первый день лечения и сопоставима у тестируемого продукта и Зофрана 8 мг дважды в день во второй день лечения.
- Тестируемый продукт относительно Зофрана 24 мг х 1:
Cmin и C24 были также выше в первый день введения тестируемого продукта по сравнению с однократным (х1) введением Зофрана 24 мг.
Однако Cmax и AUC0-24, достигаемые при введении тестируемого продукта, были примерно на 60% (соотношение 38%) и на 40% (соотношение 59%) ниже Cmax и AUC0-24, достигаемых после введения Зофрана 24 мг х1.
Сравнение концентраций:
- Тестируемый продукт относительно Зофрана 8 мг дважды в день:
Измеренные концентрации на протяжении от 3 до 8 часов после первоначальной дозы были выше после введения тестируемого продукта. Через 10 и 12 часов было обнаружено, что концентрации были ниже после введения тестируемого продукта в первый день лечения; последующие концентрации были аналогичны между двумя группами в первый день.
Из-за более позднего введения второй дозы во 2-й день, форма кривой концентрации для Зофрана 8 мг (дважды в день) была несколько иной, чем в первый день, но в целом результаты были сходными.
- Тестируемый продукт относительно Зофрана 24 мг х 1:
Было обнаружено, что измеренные концентрации в течение 10 часов были ниже после введения тестируемого продукта. Было найдено, что измеренные концентрации через 12 и 16 часов были сопоставимы между двумя группами лечения и выше для тестируемого продукта через 20 и 24 часов.
Оценка накопления
Оценка накопления, проведенная по Cmax, продемонстрировала первое увеличение на 15% между 1-м и 2-м днем, а также продемонстрировала равномерное увеличение оценки в виде соотношения, основанного на обратном преобразовании разницы LS-средних с 3-го по 5-й день (118-125% от Cmax, наблюдаемой в 1-й день), указывая на накопление ондансетрона после многократного введения тестируемого продукта. Аналогичный рост наблюдался для AUC0-24 на 3-й и 4-й день (125-131% от AUC0-24, наблюдаемой в 1-й день) после 22%-го увеличения между 1-м и 2-м днем.
Оценка в виде соотношения, основанного на обратном преобразовании разницы LS-средних для AUC0-24, была аналогичной на 4-й день (131%) и на 5-й день (132%), указывая на то, что стационарное состояние было достигнуто между 4-м и 5-м днями многократного ежедневного введения тестируемого продукта.
Выводы:
Сравнительная биодоступность:
Представленные в данном документе результаты показывают, что биологическая доступность тестируемого продукта не уступает таковой для Зофрана 8 мг, вводимого дважды в день, что представляет собой утвержденную схему предупреждения тошноты и рвоты при химиотерапии, вызывающей умеренную тошноту.
Ключевые моменты в этом сравнении:
- Геометрическое среднее AUC0-24 тестируемого продукта было на 19% выше, чем у Зофрана 8 мг дважды в день (90%-й ДИ 12-28%) в 1-й день введения препарата, на 33% выше (90%-й ДИ 24-43%) на 2-й день.
- Геометрическое среднее Cmax тестируемого продукта было на 18% выше, чем у Зофрана 8 мг дважды в день (90%-й ДИ 9-28%) в 1-й день введения препарата, на 38% выше во 2-й день (90%-й ДИ 30-47%).
- Оба C24 и Cmin тестируемого продукта были выше, чем эти же параметры у обоих Зофрана 8 мг дважды в день и Зофрана 24 мг х 1.
- Уровни ондансетрона были аналогичны или выше после введения тестируемого продукта, чем после введения Зофрана 8 мг дважды в день во все моменты времени, кроме 10 и 12 часов после первоначального введения в 1-й день и 14-20 часов во 2-й день.
-- Через 10 и 12 часов после введения в 1-й день уровни после введения тестируемого продукта были на 107% и 12% выше, чем минимальный уровень ондансетрона через 8 часов после первоначальной дозы Зофрана 8 мг.
-- Через 14-20 часов после первой дозы во 2-й день уровни после введения тестируемого продукта были на 47-159% выше, чем минимальный уровень ондансетрона через 12 часов после исходной дозы Зофрана 8 мг.
- Кроме того, после 12 часов уровни ондансетрона после введения тестируемого продукта были аналогичны или выше уровней после введения Зофрана 24 мг х1, что является утвержденной схемой лечения при химиотерапии, вызывающей умеренную тошноту.
Уровень ондансетрона в плазме после введения тестируемого продукта аналогичен или выше уровня в плазме после введения Зофрана 8 мг два раза в день в большинстве проверенных временных точках, а концентрации в других временных точках значительно превышают минимальные уровни через 8 или 12 часов (1-й и 2-й дни, соответственно) после введения первоначальной дозы для эталонной схемы лечения Зофраном 8 мг два раза в день. Поэтому, справедливо сделать вывод, что эффективность тестируемого продукта по меньшей мере так же хороша, как эффективность Зофрана 8 мг, вводимого два раза в день.
Оценка накопления:
Введение тестируемого продукта один раз в день в течение 5 дней подряд после голодания подтвердило накопление ондансетрона в плазме крови человека. Максимальные концентрации в плазме увеличились с 15% до 25% со 2-го по 5-й день. После первого увеличения на 15%, увеличение примерно на 3% от максимальной концентрации наблюдалось для каждого последующего дня введения. AUC0-24 увеличилась с 22 до 31% со 2-го по 4-й день, а затем стабилизировалась на 32% на 5-й день, что указывает на достижение стационарного состояния.
Безопасность и переносимость испытываемого продукта:
Результаты, представленные в настоящем документе, показывают, что введение тестируемого продукта один раз в день в течение 5 дней подряд было безопасным и хорошо переносилось субъектами, включенными в данное исследование. Кроме того, количество субъектов, которые испытали по меньшей мере один побочный эффект, было сопоставимо между всеми группами лечения, и все 28 событий побочных эффектов в ходе исследования были легкими, демонстрируя, что безопасность и переносимость состава с пролонгированным высвобождением (тестируемого продукта) были похожи на профиль безопасности других методов лечения.
Несмотря на некоторое накопление препарата при многократном введении, не было никаких признаков того, что это приводило к каким-либо проблемам в отношении безопасности. В частности, частота возникновения небольшого увеличения QTc была выше после однократной дозы Зофрана 24 мг с немедленным высвобождением, чем после введения 5 ежедневных доз тестируемого продукта.
Пример 19-2-групповое перекрестное сравнительное исследование биодоступности твердых лекарственных форм
2-группове перекрестное сравнительное исследование биодоступности однократной дозы твердой лекарственной формы по настоящему изобретению в сравнении с однократной дозой ондансетрона 16 мг в суппозиториях у здоровых мужчин и женщин-добровольцев/натощак.
Цели:
Основная цель данного исследования заключалась в изучении относительной биодоступности между тестируемой таблеткой с бимодальным высвобождением 24 мг ондансетрона и вводимым один раз в день суппозиторием ондансетрона, который утвержден во многих странах Европы (суппозитории Зофран® 16 мг). Вторичными целями исследования были:
1. Оценка безопасности и переносимости состава с пролонгированным высвобождением у здоровых добровольцев.
Методология:
Одноцентровое, рандомизированное, однократная доза, лабораторно-слепое, 2-периодное, с 2 последовательностями введения, перекрестное исследование.
Число субъектов (планируемое и проанализированное):
Планируемое для включения: 20
Включенные: 20
Выбывшие: 0
Проанализированные: 18
Учтенные в фармакокинетическом и статистическом анализе: 20
Учтенные в анализе безопасности: 20
Тестируемый продукт, дозировка и способ введения, номер партии:
Название: ондансетрон
Лекарственная форма/путь введения: бимодальная таблетка по настоящему изобретению (CDT-сердцевина с электролитом)/перорально («Тестируемый продукт»).
Схема лечения-1: однократная доза 24 мг (1×24 мг) однократно.
Эталонный продукт, дозировка и способ введения:
Название: суппозитории Зофран® 16 мг («Эталонный продукт»)
Схема лечения-2: однократная доза 16 мг (1×16 мг) однократно.
Последовательности введения соединений в исследовании:
|
Варианты лечения:
Субъекты получали каждый из следующих исследуемых продуктов в одном случае в соответствии со списком рандомизации:
Лечение-1: 1 х тестируемый продукт (бимодальная таблетка ондансетрона 24 мг [CDT-сердцевина с электролитом]); Тест
Лечение-2: 1 х суппозитории Зофрана® 16 мг; Эталон
Выбор и время введения для каждого субъекта:
Субъекты голодали в течение ночи на протяжении по меньшей мере 10 часов до введения препарата.
Лечение-1:
После этого одну дозу тестируемого состава вводили перорально приблизительно с 240 мл воды, имеющей температуру окружающей среды.
Лечение-2:
После этого ректально вводили однократную дозу эталонного состава. Воду не давали при введении суппозиториев.
Голодание продолжали в течение по меньшей мере 4 часов после введения препарата, после чего подавали стандартизированный обед. Ужин и легкие закуски также подавали в соответствующее время после этого, но не ранее, чем через 9 часов после введения препарата. Вода была разрешена в неограниченном количестве до 1 часа до введения препарата и начиная с 1 часа после введения препарата.
График отбора образцов крови
Образцы крови для фармакокинетических измерений собирали перед и до 48 часов после каждого введения лекарственного средства (последовательный сбор образцов). График сбора образцов крови (по часам): 0, 1, 2, 3, 4, 5, 6, 8, 10, 12, 14, 16, 20, 24, и 28.
ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ
Анализ эффективности
Среднее значение Cmin составляло, соответственно, 2,629 нг/мл и 0,793 нг/мл для тестируемого и эталонного составов. Соотношение геометрических LS-средних для Cmin тестируемого и эталонного препаратов составляло 277,21% (90%-й ДИ: с 162,84% до 471,90%).
Среднее значение Cmax составляло, соответственно, 53,055 нг/мл и 24,392 нг/мл для тестируемого и эталонного составов. Соотношение геометрических LS-средних для Cmax тестируемого и эталонного препаратов составляло 219,26% (90%-й ДИ: с 192,74% до 249,43%).
Среднее значение C24 составляло, соответственно, 10,542 нг/мл и 5,570 нг/мл для тестируемого и эталонного составов. Соотношение геометрических LS-средних для C24 тестируемого и эталонного препаратов составляло 203,59% (90%-й ДИ: с 157,81% до 262,65%).
Медианное Tmax составляло 4,00 и 5,00 часов для тестируемого и эталонного составов, соответственно.
Среднее значение AUC0-24 составляло, соответственно, 629,082 нг⋅ч/мл и 321,115 нг ч/мл для тестируемого и эталонного составов. Соотношение геометрических LS-средних для AUC0-24 тестируемого и эталонного препаратов составляло 197,68% (90%-й ДИ: с 173,80% до 224,85%).
Среднее значение AUC0-T составляло, соответственно, 781,788 нг⋅ч/мл и 382,769 нг⋅ч/мл для тестируемого и эталонного составов. Соотношение геометрических LS-средних для AUC0-T тестируемого и эталонного препаратов составляло 212,62% (90%-й ДИ: с 180,62% до 250,30%).
Среднее значение λξ составляло 0,0665 ч-1 для тестируемого состава и 0,0914 ч-1 для эталонного состава. Среднее значение T1/2 составляло 12,69 и 8,16 часов, для тестируемого и эталонного составов, соответственно.
Среднее значение AUC0-∞ составляло, соответственно, 848,005 нг⋅ч/мл и 407,528 нг⋅ч/мл для тестируемого и эталонного составов. Соотношение геометрических LS-средних для AUC0-∞ тестируемого и эталонного препаратов составляло 212,76% (90%-й ДИ: с 183,77% до 246.33%).
Степень воздействия ондансетрона была на 113% выше при введении в виде одной 24 мг бимодальной таблетки, [CDT-сердцевина с электролитом]), чем при введении в виде суппозиториев Зофрана® 16 мг. Это предполагает на 42% более высокую биодоступность для композиции по настоящему изобретению по сравнению с суппозиториями Зофрана®. Кроме того, коэффициент интерсубъектной вариабельности для тестируемого состава был аналогичен или ниже, чем для суппозиториев Зофрана® по всем оцениваемым фармакокинетическим параметрам. Фармакокинетические данные показали, что тестируемый состав обеспечивает более высокие уровни концентрации в плазме, чем суппозитории Зофрана® в течение всего 24-часового периода после введения дозы, таким образом, указывая, что эффективность тестируемого состава была по меньшей мере столь же высокой, как и у суппозиториев Зофрана®.
Вторая цель данного исследования заключалась в том, чтобы оценить безопасность и переносимость состава с бимодальным высвобождением у здоровых добровольцев. Частота и тяжесть побочных эффектов, связанных с составом с бимодальным высвобождением, была сравнима с наблюдаемыми после суппозиториев Зофрана®, хотя воздействие и пиковый уровень ондансетрона были выше после введения состава с бимодальным высвобождением. В целом, протестированные два различных состава были безопасными и хорошо переносились субъектами, включенными в данное исследование.
Пример 20 - (предполагаемое) рандомизированное, плацебо-контролируемое исследование твердых лекарственных форм по настоящему изобретению против рвоты, возникающей из-за предполагаемого острого гастроэнтерита или гастрита
Острый гастроэнтерит представляет собой воспаление желудка, тонкого кишечника или толстой кишки, приводящее к комбинации боли в животе, судорог, тошноты, рвоты и диареи. Острый гастрит является воспалением желудка; больные острым гастритом могут иметь рвоту без диареи. Для целей настоящего протокола, у пациентов должна присутствовать по меньшей мере рвота, как описано ниже, с или без других симптомов.
Острый гастроэнтерит является серьезной медицинской проблемой во всем мире. Хотя смертность является наиболее высокой в развивающихся странах, как среди очень молодых, так и среди пожилых людей, заболеваемость острым гастроэнтеритом является одинаковой для всех возрастов и экономических слоев. В США, большинство случаев острого гастроэнтерита является следствием вирусной инфекции, чаще всего ротавирусной и норовирусой. В США ежегодно насчитывается 15-25 миллионов случаев вирусного гастроэнтерита, что приводит к 3-5 миллионам амбулаторных посещений и к 200000 госпитализациям. Хотя смертность является низкой, из 17000 людей, умирающих от острого гастроэнтерита в США каждый год, 83% являются старше 65 лет.
Большинство случаев являются самоограниченными, проходящими в течение от одного до нескольких дней. Для легких или умеренных случаев, как правило, лечение является симптоматическим. Согласно рекомендациям, детям показана пероральная регидратация. В более тяжелых и длительных случаях могут потребоваться диагностические исследования для выявления возбудителя. В зависимости от тяжести и этиологии, лечение варьирует от изменения питания, включая пероральную регидратацию до внутривенного введения жидкости и приема антибиотиков. Часто используются противорвотные и антидиарейные средства.
Тестируемым продуктом в данном исследовании является пероральный состава ондансетрона с бимодальным высвобождением. Он содержит 24 мг ондансетрона, 6 мг немедленного высвобождения и 18 мг в матриксе с пролонгированным высвобождением. Как описано выше, исследования фармакокинетики на здоровых добровольцах показали быстрое раннее высвобождение с появлением препарата в плазме в течение получаса после введения, аналогично Зофрану с немедленным освобождением, но с удлиненной доступностью лекарственного средства в течение 24 часов. Таким образом, тестируемый продукт должен обеспечивать быстрое избавление от тошноты и рвоты у пациентов, без необходимости немедленного внутривенного введения препарата, а требуя введения только один раз в день для пролонгированного эффекта в течение нескольких дней болезни. Профиль безопасности тестируемого продукта аналогичен профилю для Зофрана 8 мг, как было показано в исследованиях на добровольцах, описанных выше.
|
Оценка исследования
|
Примечания:
аИстория болезни, включая показатели жизненно важных функций (пульс, частота дыхания, артериальное давление, температуру и только вначале высоту и вес), информированное согласие. Для всех пациентов≥50 лет, необязательно для пациентов до 50 лет: общий анализ крови с количеством тромбоцитов, биохимический профиль, анализ мочи. Анализы крови и мочи образцы должны быть проведены ускоренно в начале оценки, но для включения пациента в исследование и проведения лечения не нужно ждать результатов лабораторных исследований (кроме теста на беременность при необходимости), если клинически не показано обратное. После кровопускания может быть установлен солевой замок, чтобы пациенту не нужно было делать дополнительный укол, если ему/ей впоследствии будет необходимо внутривенное введение жидкости. История болезни должна включать в себя анкетирование в отношении провоцирующих/причинных факторов, а также регистрацию числа и характера эпизодов рвоты и диареи. У пациентов с контролируемым диетой сахарным диабетом 2-го типа должно быть измерено содержание сахара крови из пальца.
Для женщин детородного возраста, как определено в критериях исключения: тест на беременность по моче или крови. Отрицательный результат теста на беременность должен быть получен до начала лечения пациента в исследовании.
bПациенты должны наблюдаться в отделении неотложной помощи в течение как минимум 2 часов, с анализом показателей жизненно важных функций по меньшей мере один раз в час и фиксацией вводимого (как перорально и парентерально) и количеством выделяемого. Все эпизоды рвоты и диареи должны быть записаны, и тяжесть тошноты вначале и через каждый час во время нахождения в отделении неотложной помощи должна быть отмечена. После истечения как минимум 2 часов после первоначального введения исследуемого препарата, пациенты могут быть выписаны по показаниям.
cПациенты будут заполнять дневник в Интернете или на бумажном носителе, или с ними будут связываться каждый день после лечения до максимум 4 дней, чтобы определить, были ли у них дальнейшие тошнота и рвота после выписки из отделения неотложной помощи, требуется ли им дальнейшее лечение острого гастроэнтерита, и подробности в отношении продолжающейся, если таковая имеется, болезни. Если симптомы сохраняются в течение 3 или более дней после начала исследования, пациент должен быть проинструктирован, что он должен вернуться для дополнительного осмотра.
dВключая все лекарственные препараты, принятые в течение 7 дней до начала исследования, все виды лечения (включая парентеральное введение жидкости) в день вхождения в исследование, и все лекарственные препараты, включая резервные препараты, с момента начала исследования до момента последнего наблюдения.
еПациенты должны получить первую дозу исследуемого препарата в течение одного часа после подписания информированного согласия.
fПациенты могут принимать исследуемый препарат ежедневно в течение 3 дней после начала исследования (максимум в общей сложности 4 дозы, плюс одна дополнительная, если пациента рвало в течение 15 минут после первой дозы) при сохранении симптомов. Пациенты должны прекратить прием исследуемого препарата, если симптомы исчезли, хотя они могут возобновить прием исследуемого препарата один раз в день, если симптомы повторяются после прекращения приема препарата. Пациенты не должны принимать исследуемый препарат, оставшийся после 4-го дня. Им будут даны специальные конверты, чтобы возвратить любой неиспользованный исследуемый препарат.
Пример 21 - (предполагаемые) способы определения сокращения вызванной гастроэнтеритом продолжительной рвоты после введения твердых лекарственных форм по настоящему изобретению
Для того, чтобы оценить сокращение вызванной гастроэнтеритом рвоты после приема твердых пероральных лекарственных форм по настоящему изобретению, будут измеряться симптомы рвоты, такие как частота, длительность, объем, тяжесть и дистресс. Частота может быть измерена, например, по числу приступов рвоты в течение определенного периода, длительность может быть измерена, например, по количеству часов рвоты, объем может быть измерен, например, в чашках рвоты, тяжесть может быть измерена, например, путем количественной оценки физических симптомов, и дистресс, который пациент испытывает, может быть измерен, например, по полученному стрессу и психологическим симптомам.
Для того, чтобы оценить частоту рвоты и дистресс, связанный с рвотой, можно быть использовать индекс тошноты, рвоты и позывов на рвоту (INVR). В INVR есть вопросы относительно числа эпизодов позывов на рвоту в течение предшествующих 12 часов и дистресса, испытываемого при этих эпизодах.
Пациентам будут вводить твердую пероральную лекарственную форму по настоящему изобретению (бимодальную таблетку 24 мг, описанную выше) или плацебо, и сокращение вызванной гастроэнтеритом рвоты будут оценивать путем наблюдения за одним или несколькими симптомами рвоты, такими как частота, продолжительность, объем, тяжесть и дистресс.
Пример 22 - (предполагаемое) интервенционное исследование для оценки тяжести рвоты после введения твердых лекарственных форм по настоящему изобретению женщинам с рвотой беременных
Желаемый тип исследования: интервенционное
Желаемая схема исследования: Место: рандомизированное
Желаемая классификация конечных точек: исследование безопасности/эффективности
Желаемая интервенционная модель: перекрестное назначение
Желаемая маскировка: двойное слепое (субъект, обслуживающий персонал, исследователь, оценщик результатов)
Желаемая основная цель: лечение
Желаемые основные критерии эффективности:
PUQE-шкала предназначена для оценки степени тяжести при рвоте беременных. PUQE является акронимом Уникального количественного определения рвоты при беременности, единственной утвержденной клинической шкалы для оценки тяжести рвоты. Диапазон варьируется от 3 (наилучшее состояние) до 15 (наихудшее состояние).
За участниками будут наблюдать в течение всего периода пребывания в стационаре, сравнивая изменения от базовой линии в первый период времени со вторым периодом времени.
VAS-шкала предназначена для оценки тяжести рвоты беременных
VAS является визуальный аналоговой шкалой, состоящей из 5 пунктов. Диапазон варьирует от 0 (лучшее состояние) до 50 (худшее состояние).
За участниками будут наблюдать в течение всего периода пребывания в стационаре, сравнивая первый период времени и второй период времени.
Желаемые вторичные критерии эффективности:
Количество доз стандартных противорвотных препаратов (таблеток Зофрана® 8 мг), требующихся в двух разных периодах времени.
Критерий исхода беременности: вес при рождении.
Вес тела при рождении с поправкой на гестационный возраст при родах является критерием исхода беременности после лечения рвоты беременных.
Критерий исхода беременности: оценка по шкале APGAR.
APGAR является наиболее распространенным показателем статуса новорожденных сразу после родов.
Желаемые другие критерии эффективности:
Кетонурия в утренней моче
Кетонурия в утренней моче является простым прямым маркером голодания, связанного с тошнотой и рвотой.
1-я группа: твердая лекарственная форма по настоящему изобретению первая - плацебо второе
В этой группе пациентов будут лечить с помощью твердой лекарственной формой ондансетрона по настоящему изобретению в течение первого периода времени, а затем переключатся на плацебо во втором периоде времени.
2-я группа: плацебо первое - твердая лекарственная форма по настоящему изобретению - вторая
В этой группе пациентов будут лечить с помощью плацебо в течение первого периода времени, а затем с помощью твердой лекарственной формы ондансетрона по настоящему изобретению в течение второго периода времени.
Желаемые критерии
Желаемые критерии включения:
Гестационный возраст 6-12 недель, и тяжелая степень рвоты беременных, определяемая следующим образом: индекс PUQE≥13, связанный с одним или несколькими из следующих условий:
потеря веса > 5% от веса до беременности,
нарушения содержания электролитов,
обезвоживание,
длительность симптомов > 10 дней,
недостаточное потребление продуктов питания и питья.
Пример 23 - (предполагаемое) рандомизированное, плацебо-контролируемое исследование твердых лекарственных форм по настоящему изобретению для лечения синдрома раздраженного кишечника с преобладанием диареи (IBS-D)
Предполагается следующее исследование
|
Пример 24 - (предполагаемое) рандомизированное, двойное слепое, 3-групповое исследование твердых лекарственных форм по настоящему изобретению для лечения синдрома раздраженного кишечника с преобладанием диареи (IBS-D)
Предполагается следующее исследование
|
В настоящем документе описаны твердые лекарственные формы ондансетрона с пролонгированным высвобождением для лечения симптомов тошноты, рвоты или диареи.
В соответствии с аспектами, проиллюстрированными в данном описании, способ лечения пациента включает пероральное введение пациенту твердой пероральной лекарственной формы, включающей: сердцевину, содержащую неионогенный полимерный матрикс, первое количество ондансетрона, диспергированное в матриксе, и соль, диспергированную в матриксе, где первое количество ондансетрона варьирует от примерно 9 мг до примерно 28 мг; первую изолирующую оболочку, окружающую сердцевину, где первая изолирующая оболочка состоит из неионогенного полимерного матрикса; и слой немедленно высвобождающегося лекарственного средства, окружающий первую изолирующую оболочку и содержащий неионогенный полимер и второе количество ондансетрона, диспергированное в нем, где второе количество ондансетрона варьирует от примерно 3 мг до примерно 8 мг; причем высвобождение ондансетрона из твердой пероральной лекарственной формы обеспечивает лекарственное действие ондансетрона как минимум в течение 16 часов, приводя к снижению частоты рвоты, тошноты или диареи, или их комбинации.
Твердые лекарственные формы с пролонгированным высвобождением раскрыты в данном документе для снижения, лечения или предупреждения любого из тошноты, рвоты или диареи у пациента, симптомов, которые могут быть вызваны различными заболеваниями. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами вирусного гастроэнтерита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами бактериального гастроэнтерита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами гастрита (воспаления стенки желудка) у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами воспалительного заболевания кишечника у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами синдрома раздраженного кишечника у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами холецистита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами диспепсии у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами панкреатита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами аппендицита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами хирургического вмешательства у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами гепатита у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами перитонит у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами гастроэзофагеальной рефлюксной болезни у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами обструктивного поражения ЖКТ у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами пищевых отравлений у субъекта. В одном варианте осуществления тошнота, рвота или диарея являются побочными эффектами опухоли у субъекта.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая включает в себя: сердцевину, содержащую неионогенный полимерный матрикс, первое количество первого противорвотного лекарственного соединения или его фармацевтически приемлемой соли, диспергированное в матриксе, и соль, диспергированную в матриксе; первую изолирующую оболочку, окружающую сердцевину, где первая изолирующая оболочка состоит из неионогенного полимерного матрикса; и слой немедленно высвобождающегося лекарственного средства, окружающий первую изолирующую оболочку, где слой немедленно высвобождающегося лекарственного средства содержит неионогенный полимер и второе количество второго противорвотного лекарственного средства или его фармацевтически приемлемой соли, диспергированное в нем, где слой лекарственного вещества подобран так, чтобы высвобождать второе количество противорвотного лекарственного средства в течение периода по меньшей мере 1 ч, где твердая пероральная лекарственная форма подобрана так, чтобы высвобождать первое количество первого противорвотного средства и второе количество второго противорвотного средства за минимальный период 16 часов. В одном варианте осуществления твердая пероральная лекарственная форма дополнительно включает энтеросолюбильную оболочку, окружающую первую изолирующую оболочку. В одном варианте осуществления твердая пероральная лекарственная форма дополнительно содержит вторую изолирующую оболочку, окружающую слой лекарственного средства с немедленным высвобождением, где вторая изолирующая оболочка состоит из неионогенного полимера. В одном варианте осуществления первая изолирующая оболочка дополнительно содержит добавку, такую как plasACRYL™. В одном варианте осуществления соль в сердцевине диспергируют в матриксе с концентрацией в диапазоне от 50% до 100% от веса матрицы. В одном варианте осуществления при воздействии на твердую лекарственную форму водной среды, соль вызывает затвердевание границы по периферии матрикса, причем граница последовательно продвигается внутрь по направлению к его центру, по мере того как водная среда проникает в матрицу, а затвердевшая граница ограничивает скорость, с которой противорвотное лекарственное средство в матриксе высвобождается из таблетки. В одном варианте осуществления твердая пероральная лекарственная форма разработана для высвобождения первого количества противорвотного лекарственного средства и второго количества противорвотного лекарственного средства в течение как минимум 20 часов. В одном варианте осуществления твердая пероральная лекарственная форма разработана для высвобождения первого количества противорвотного лекарственного средства и второго количества противорвотного лекарственного препарата в течение как минимум 24 часов. В одном варианте осуществления первое противорвотное лекарственное средство и второе противорвотное лекарственное средство являются одним и тем же лекарственным средством. В одном варианте осуществления первое противорвотное лекарственное средство и второе противорвотное лекарственное средство являются ондансетроном или эквивалентным количеством соли ондансетрона.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая включает в себя: сердцевину, содержащую гипромеллозу, 18 мг ондансетрона или эквивалентное количество соли ондансетрона и безводный цитрат натрия; первую изолирующую оболочку, окружающую сердцевину и содержащую гипромеллозу; и слой немедленно высвобождающегося лекарственного средства, окружающий первую изолирующую оболочку и содержащий гипромеллозу и 6 мг ондансетрона или эквивалентное количество соли ондансетрона, причем слой немедленно высвобождающегося лекарственного средства достаточен для высвобождения ондансетрона за период по меньшей мере 1 час, где общее количество ондансетрона в лекарственной форме высвобождается за 24 часа. В одном варианте осуществления твердая пероральная лекарственная форма дополнительно включает энтеросолюбильную оболочку, окружающую первую изолирующую оболочку. В одном варианте осуществления твердая пероральная лекарственная форма дополнительно содержит вторую изолирующую оболочку, окружающую слой лекарственного средства с немедленным высвобождением, где вторая изолирующая оболочка состоит из неионогенного полимера. В одном варианте осуществления первая изолирующая оболочка дополнительно содержит добавку, такую как plasACRYL™. В одном варианте осуществления безводный цитрат натрия в сердцевине диспергируют в гипромеллозе с концентрацией в диапазоне от 50% до 100% от веса гипромеллозы. В одном варианте осуществления при воздействии на твердую лекарственную форму водной среды, безводный цитрат натрия вызывает затвердевание границы по периферии гипромеллозы, причем граница последовательно продвигается внутрь по направлению к его центру, по мере того как водная среда проникает в гипромеллозу, а затвердевшая граница ограничивает скорость, с которой ондансетрон в гипромеллозе высвобождается из таблетки. В одном варианте осуществления, когда твердая пероральная лекарственная форма вводится пациенту натощак, достигается Cmax по меньшей мере 50 нг/мл. В одном варианте осуществления, когда твердая пероральная лекарственная форма вводится пациенту натощак, достигается AUC по меньшей мере 700 нг⋅ч/мл.
В соответствии с аспектами, проиллюстрированными в данном описании, в них раскрыта твердая пероральная лекарственная форма, которая включает в себя: сердцевину, содержащую неионогенный полимерный матрикс, первое количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное в матриксе, и соль, диспергированную в матриксе; первую изолирующую оболочку, окружающую сердцевину, где первая изолирующая оболочка состоит из неионогенного полимерного матрикса; и слой немедленно высвобождающегося лекарственного средства, окружающий первую изолирующую оболочку, где слой немедленно высвобождающегося лекарственного средства содержит неионогенный полимер и второе количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное в нем, где твердая пероральная лекарственная форма дает в результате in vitro профиль растворения ондансетрона при измерении в лопастном аппарате для растворения 2-го типа при 37°С в водном растворе, содержащем дистиллированную воду, при 50 об./мин, который показывает, что: а) от примерно 15% до 30% от общего количества ондансетрона высвобождается после двух с половиной часов измерения в аппарате; b) от примерно 30% до 50% от общего количества ондансетрона высвобождается через пять часов измерения в аппарате; и с) примерно не менее чем 75% от общего количества ондансетрона высвобождается через пятнадцать часов измерения в аппарате. В одном варианте осуществления, когда твердая пероральная лекарственная форма вводится пациенту натощак в дозе 24 мг ондансетрона, достигается Cmax по меньшей мере 50 нг/мл. В одном варианте осуществления, когда твердая пероральная лекарственная форма вводится пациенту натощак в дозе 24 мг ондансетрона, достигается AUC по меньшей мере 700 нг⋅ч/мл.
В соответствии с аспектами, проиллюстрированными в данном документе, раскрыт упакованный фармацевтический препарат, который включает в себя множество любых твердых пероральных лекарственных форм по настоящему изобретению в запечатанном контейнере и инструкции по введению лекарственных форм перорально для осуществления профилактики тошноты.
В соответствии с аспектами, проиллюстрированными в данном документе, раскрыт упакованный фармацевтический препарат, который включает в себя множество любых твердых пероральных лекарственных форм по настоящему изобретению в запечатанном контейнере и инструкции по введению лекарственных форм перорально для осуществления профилактики рвоты.
В соответствии с аспектами, проиллюстрированными в данном документе, раскрыт упакованный фармацевтический препарат, который включает в себя множество любых твердых пероральных лекарственных форм по настоящему изобретению в запечатанном контейнере и инструкции по введению лекарственных форм перорально для осуществления профилактики диареи.
В соответствии с аспектами, проиллюстрированными в данном описании, раскрыт фармацевтический препарат, который включает в себя множество любых твердых пероральных лекарственных форм по настоящему изобретению, каждая в отдельном запечатанном корпусе, и инструкции по введению лекарственных форм перорально для осуществления профилактики тошноты.
В соответствии с аспектами, проиллюстрированными в данном описании, раскрыт фармацевтический препарат, который включает в себя множество любых твердых пероральных лекарственных форм по настоящему изобретению, каждая в отдельном запечатанном корпусе, и инструкции по введению лекарственных форм перорально для осуществления профилактики рвоты.
В соответствии с аспектами, проиллюстрированными в данном описании, раскрыт фармацевтический препарат, который включает в себя множество любых твердых пероральных лекарственных форм по настоящему изобретению, каждая в отдельном запечатанном корпусе, и инструкции по введению лекарственных форм перорально для осуществления профилактики диареи.
В соответствии с аспектами, проиллюстрированными в данном описании, раскрыта стандартная лекарственная форма для предупреждения тошноты и/или рвоты для перорального введения пациенту, которая включает в себя комбинацию: ондансетронового компонента с немедленным высвобождением, содержащего стандартную дозу ондансетрона или его фармацевтически приемлемой соли в диапазоне от 4 мг до 8 мг; и ондансетронового компонента с контролируемым высвобождением, содержащего стандартную дозу ондансетрона или его фармацевтически приемлемой соли в диапазоне от 16 мг до 28 мг, причем ондансетроновый компонент с контролируемым высвобождением содержит неионогенный полимерный матрикс, ондансетрон в матриксе и соль, диспергированную в матриксе, и где стандартная лекарственная форма имеет максимальную концентрацию в плазме (Cmax) через примерно 2-5 часа (Tmax) после введения и Cmax, сравнимую с таковой для состава без контролируемого высвобождения ондансетрона, вводимого три раза в день, без снижения общего эффекта лекарственного средства, определяемого по площади под кривой (AUC) зависимости концентрации от времени, что позволяет уменьшить зависящие от концентрации побочные эффекты без снижения эффективности.
В соответствии с аспектами, проиллюстрированными в данном описании, раскрыта стандартная лекарственная форма для предупреждения диареи для перорального введения пациенту, которая включает в себя комбинацию: ондансетронового компонента с немедленным высвобождением, содержащего стандартную дозу ондансетрона или его фармацевтически приемлемой соли в диапазоне от 4 мг до 8 мг; и ондансетронового компонента с контролируемым высвобождением, содержащего стандартную дозу ондансетрона или его фармацевтически приемлемой соли в диапазоне от 16 мг до 28 мг, причем ондансетроновый компонент с контролируемым высвобождением содержит неионогенный полимерный матрикс, ондансетрон в матриксе и соль, диспергированную в матриксе, и где стандартная лекарственная форма имеет максимальную концентрацию в плазме (Cmax) через примерно 2-5 часа (Tmax) после введения и Cmax, сравнимую с таковой для состава без контролируемого высвобождения ондансетрона, вводимого три раза в день, без снижения общего эффекта лекарственного средства, определяемого по площади под кривой (AUC) зависимости концентрации от времени, что позволяет уменьшить зависящие от концентрации побочные эффекты без снижения эффективности.
Упакованный фармацевтический препарат, который включает в себя множество стандартных дозированных форм по настоящему изобретению, может содержаться в запечатанном контейнере и включать в себя инструкции по введению лекарственных форм перорально для осуществления профилактики тошноты.
Упакованный фармацевтический препарат, который включает в себя множество стандартных дозированных форм по настоящему изобретению, может содержаться в запечатанном контейнере и включать в себя инструкции по введению лекарственных форм перорально для осуществления профилактики рвоты.
Упакованный фармацевтический препарат, который включает в себя множество стандартных дозированных форм по настоящему изобретению, может содержаться в запечатанном контейнере и включать в себя инструкции по введению лекарственных форм перорально для осуществления профилактики диареи.
Упакованный фармацевтический препарат, который включает в себя множество стандартных дозированных форм по настоящему изобретению, может содержаться в отдельных запечатанных емкостях и может включать в себя инструкции по введению лекарственных форм перорально для осуществления профилактики тошноты.
Упакованный фармацевтический препарат, который включает в себя множество стандартных дозированных форм по настоящему изобретению, может содержаться в отдельных запечатанных емкостях и может включать в себя инструкции по введению лекарственных форм перорально для осуществления профилактики рвоты.
Упакованный фармацевтический препарат, который включает в себя множество стандартных дозированных форм по настоящему изобретению, может содержаться в отдельных запечатанных емкостях и может включать в себя инструкции по введению лекарственных форм перорально для осуществления профилактики диареи.
Способ профилактики тошноты включает в себя стадию введения пациенту терапевтически эффективного количества твердой пероральной лекарственной формы или стандартной лекарственной формы по настоящему изобретению.
Способ профилактики рвоты включает в себя стадию введения пациенту терапевтически эффективного количества твердой пероральной лекарственной формы или стандартной лекарственной формы по настоящему изобретению.
Способ профилактики диареи включает в себя стадию введения пациенту терапевтически эффективного количества твердой пероральной лекарственной формы или стандартной лекарственной формы по настоящему изобретению.
В соответствии с аспектами, проиллюстрированными в данном описании, раскрыта композиция для приема один раз в день, которая включает: (а) сердцевину, содержащую неионогенный полимерный матрикс, первое количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное в матриксе, и соль, диспергированную в матриксе; (b) первую изолирующую оболочку, окружающую сердцевину, где первая изолирующая оболочка состоит из неионогенного полимерного матрикса; и (с) слой немедленно высвобождающегося лекарственного средства, окружающий энтеросолюбильное покрытие, где слой немедленно высвобождающегося лекарственного средства содержит неионогенный полимер и второе количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное в нем, где слой немедленно высвобождающегося лекарственного средства подобран таким образом, чтобы высвобождать второе количество ондансетрона в течение периода по меньшей мере 1 час, где слой немедленно высвобождающегося лекарственного средства высвобождает второе количество ондансетрона в верхних отделах желудочно-кишечного тракта у человека, где сердцевина высвобождает первое количество ондансетрона в нижней части желудочно-кишечного тракта у человека, где композиция представляет собой таблетку или капсулу, которая содержит от 24 до 40 мг ондансетрона или эквивалентное количество соли ондансетрона и обеспечивает in vivo профиль в плазме, выбранный из: (а) средней Cmax по меньшей мере 50,0 нг/мл; (b) средней AUC0-24 выше 550,0 нг⋅ч/мл; и (с) среднего Tmax в интервале между приблизительно 2,0 часами и 5,0 часами, исходя из однократного введения композиции, содержащей 24 мг ондансетрона. В одном варианте осуществления композиция для введения один раз в день при введении один раз в день человеку натощак биоэквивалентна введению человеку натощак три раза в день стандартной лекарственной формы, содержащей 8 мг ондансетрона. В одном варианте осуществления биоэквивалентность устанавливается 90%-м доверительным интервалом от 0,80 до 1,25 для Cmax и AUC при введении человеку. В одном варианте осуществления характеристики растворимости и растворения являются рН-независимыми. В одном варианте осуществления сердцевина имеет рН-независимый профиль высвобождения при растворении в интервале рН 1,2-6,8. В одном варианте осуществления оба сердцевина и слой немедленного высвобождающегося лекарственного средства имеют рН-независимый профиль высвобождения при растворении в интервале рН 1,2-6,8. В одном варианте осуществления оба сердцевина и слой немедленного высвобождающегося лекарственного средства окружены изолирующей оболочкой, состоящей из неионогенного полимера, который увеличивает гидрофильность композиции, и в результате профиль растворения композиции не зависит от рН.
Все патенты, заявки на патенты и опубликованные ссылки, приведенные в данном документе, полностью включены путем ссылки. Следует принять во внимание, что некоторые из раскрытых выше и другие признаки и функции или их альтернативы могут быть по желанию объединены в множество других различных систем или вариантов применения. Различные непредвиденные или незапланированные в настоящее время альтернативы, модификации, вариации или улучшения в них могут быть впоследствии выполнены специалистами в данной области.
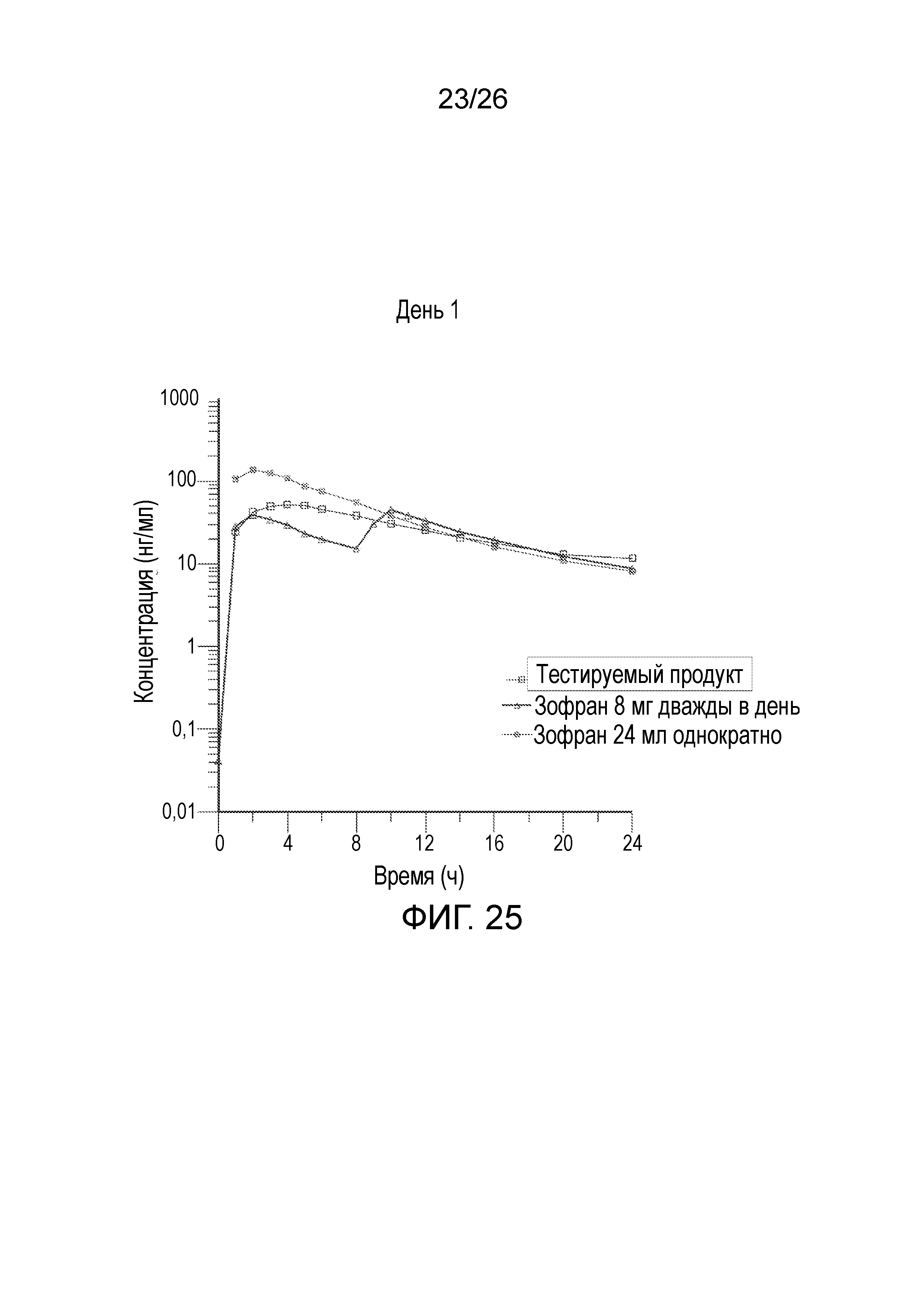
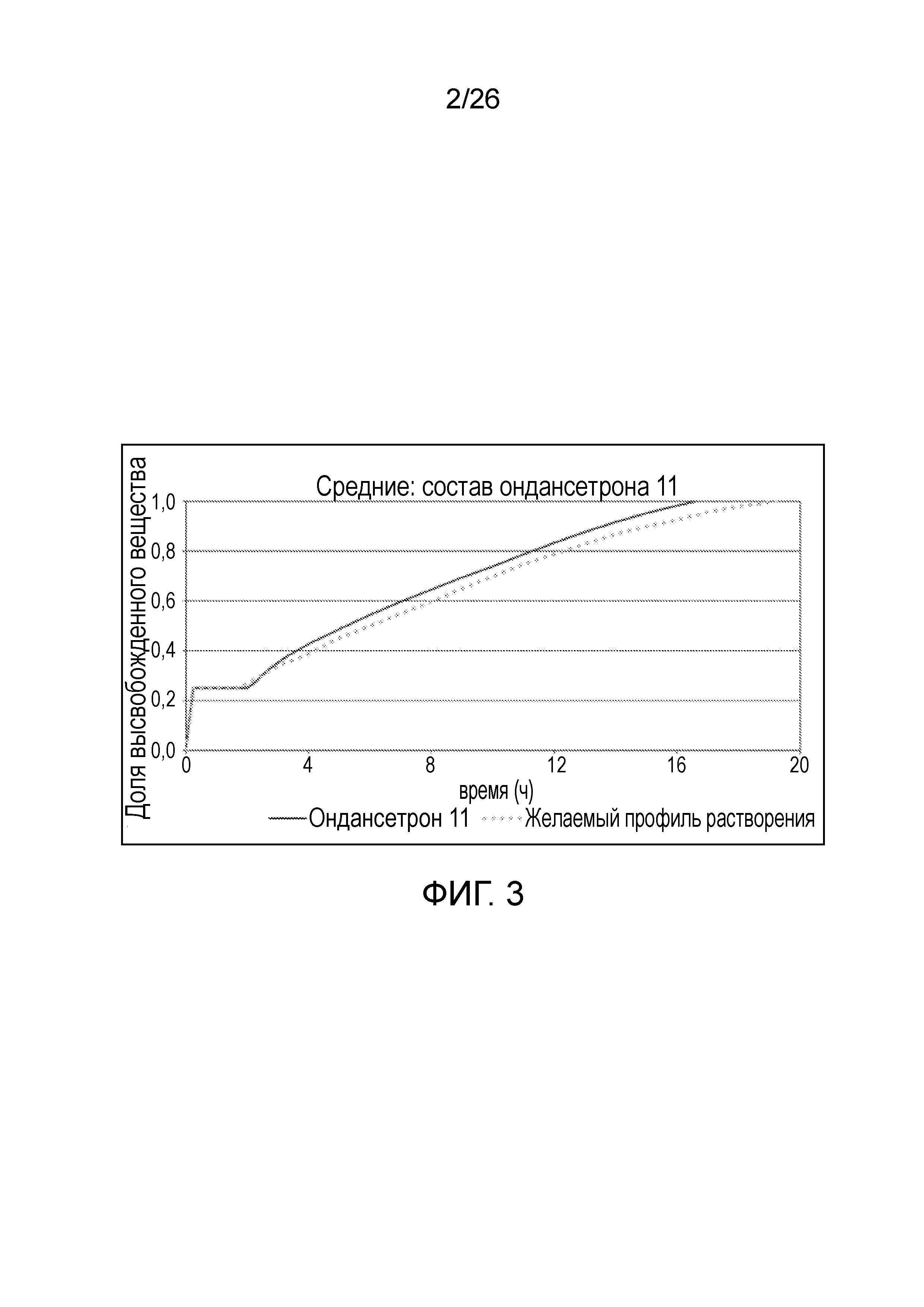

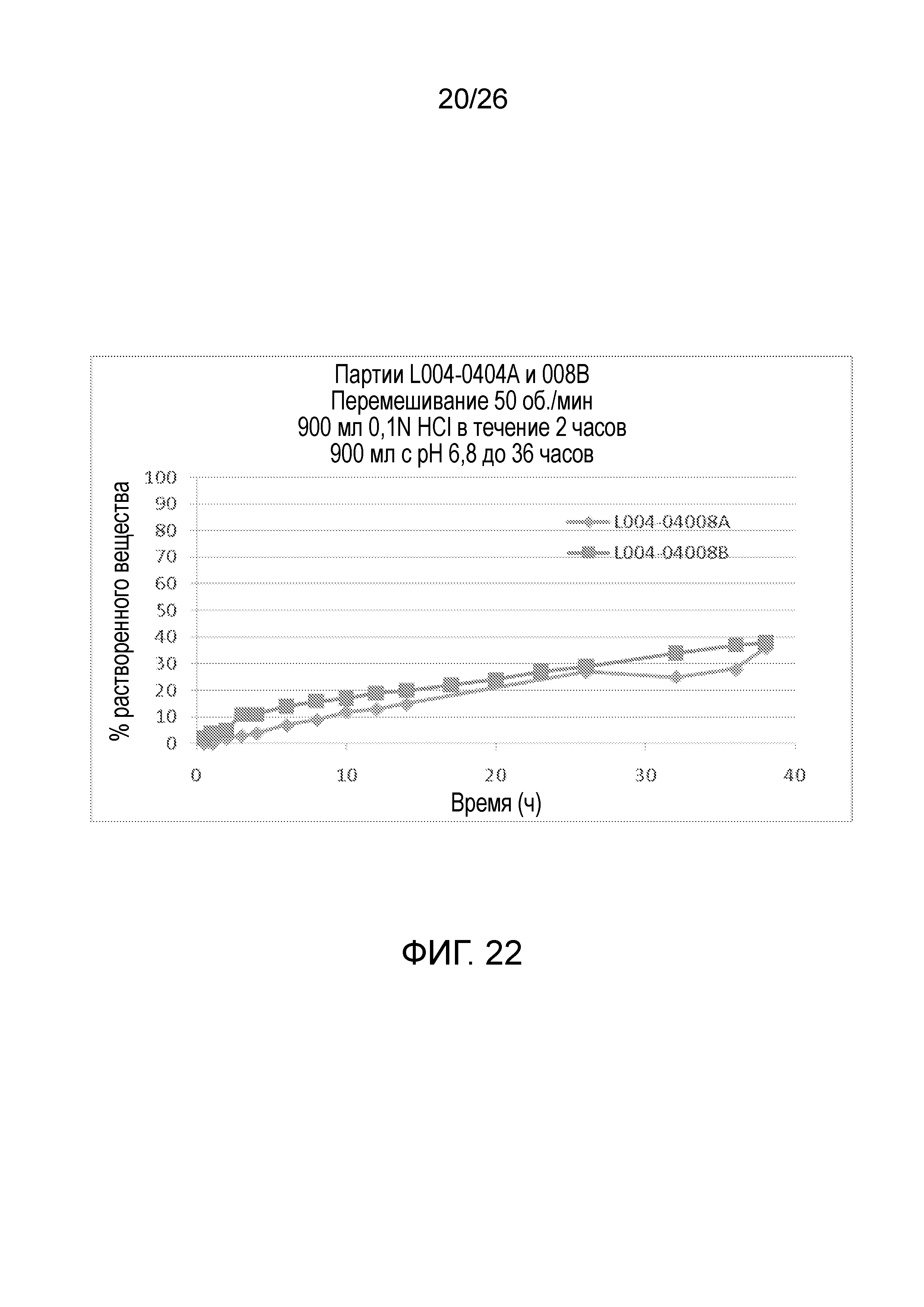
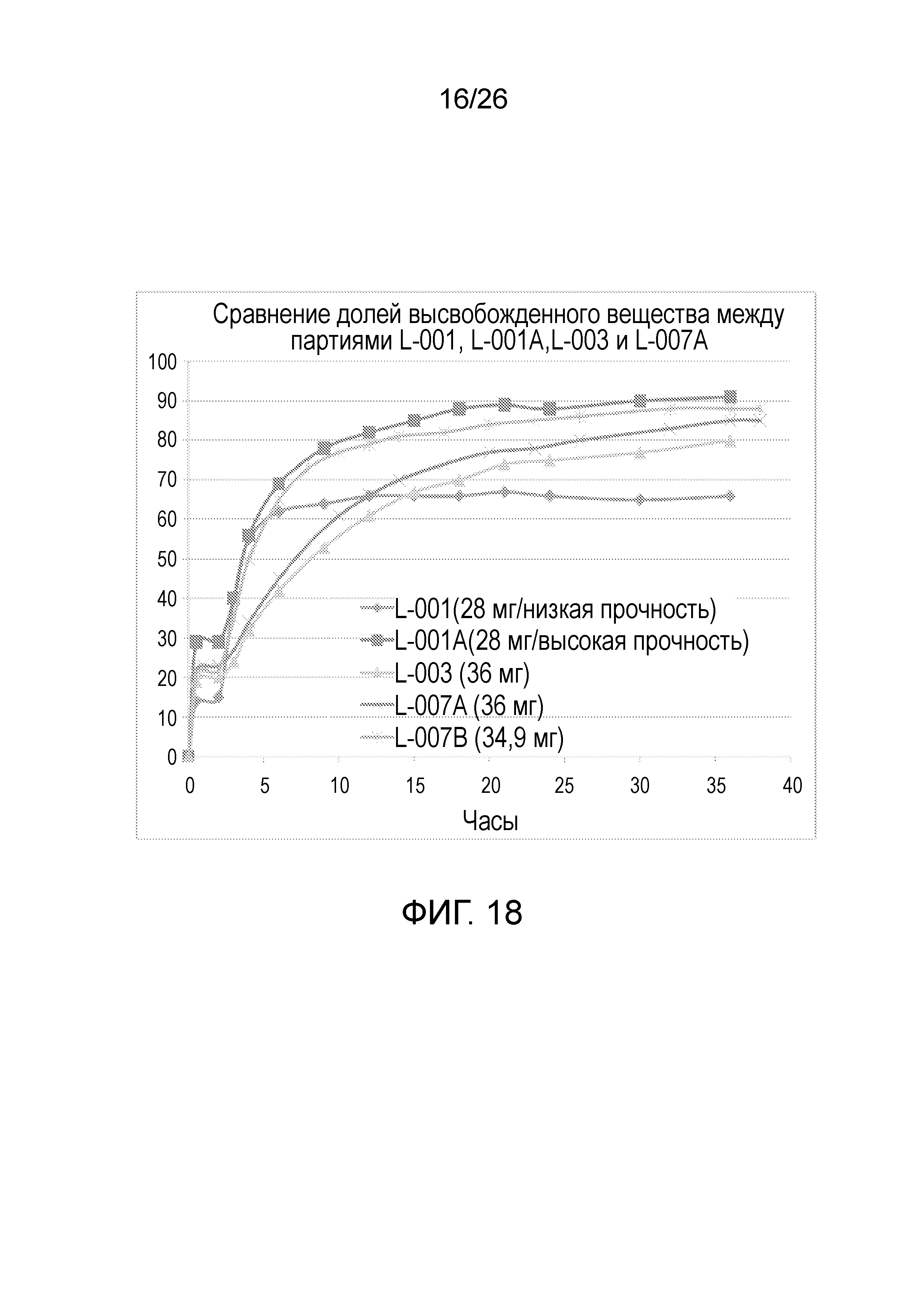
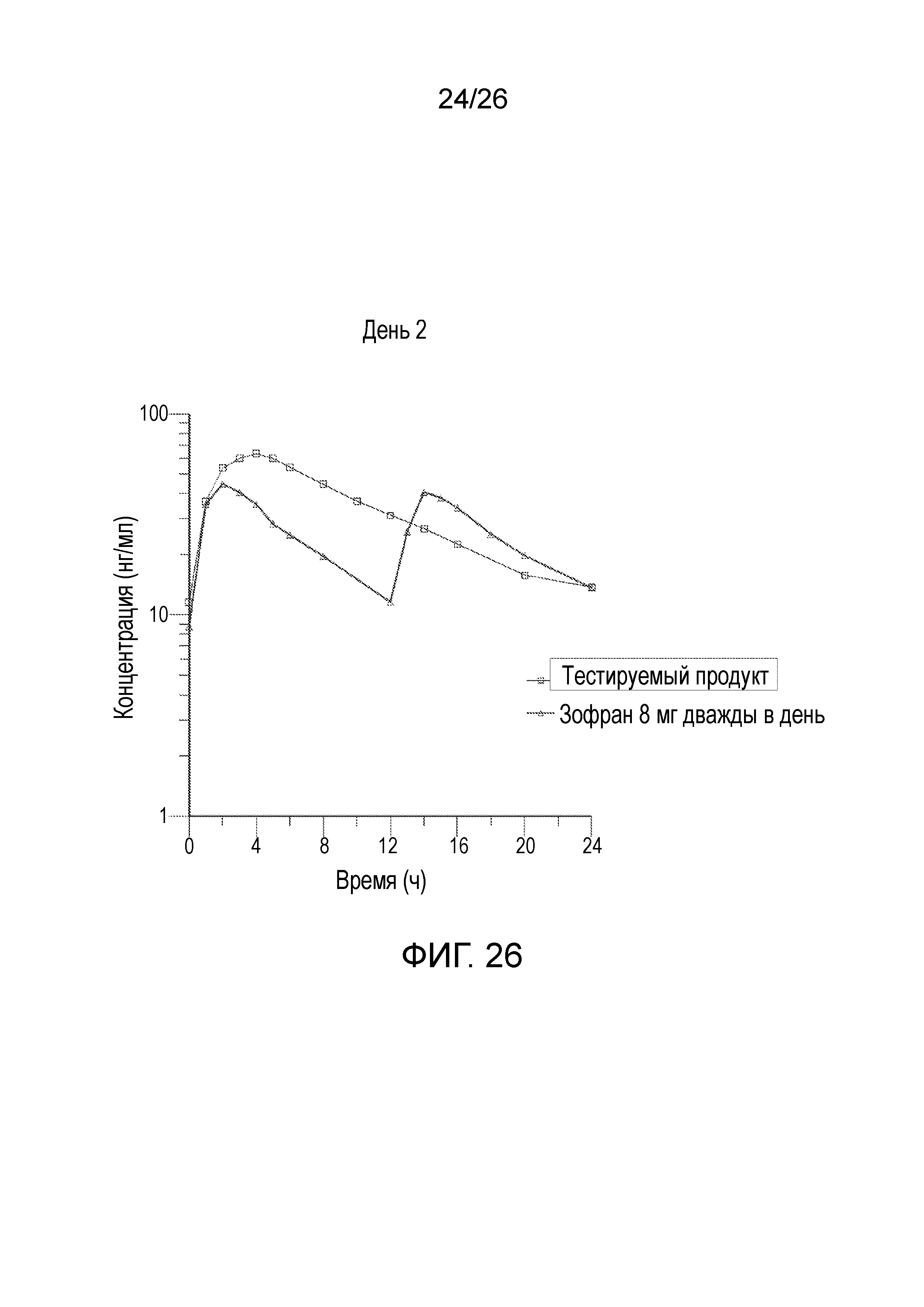
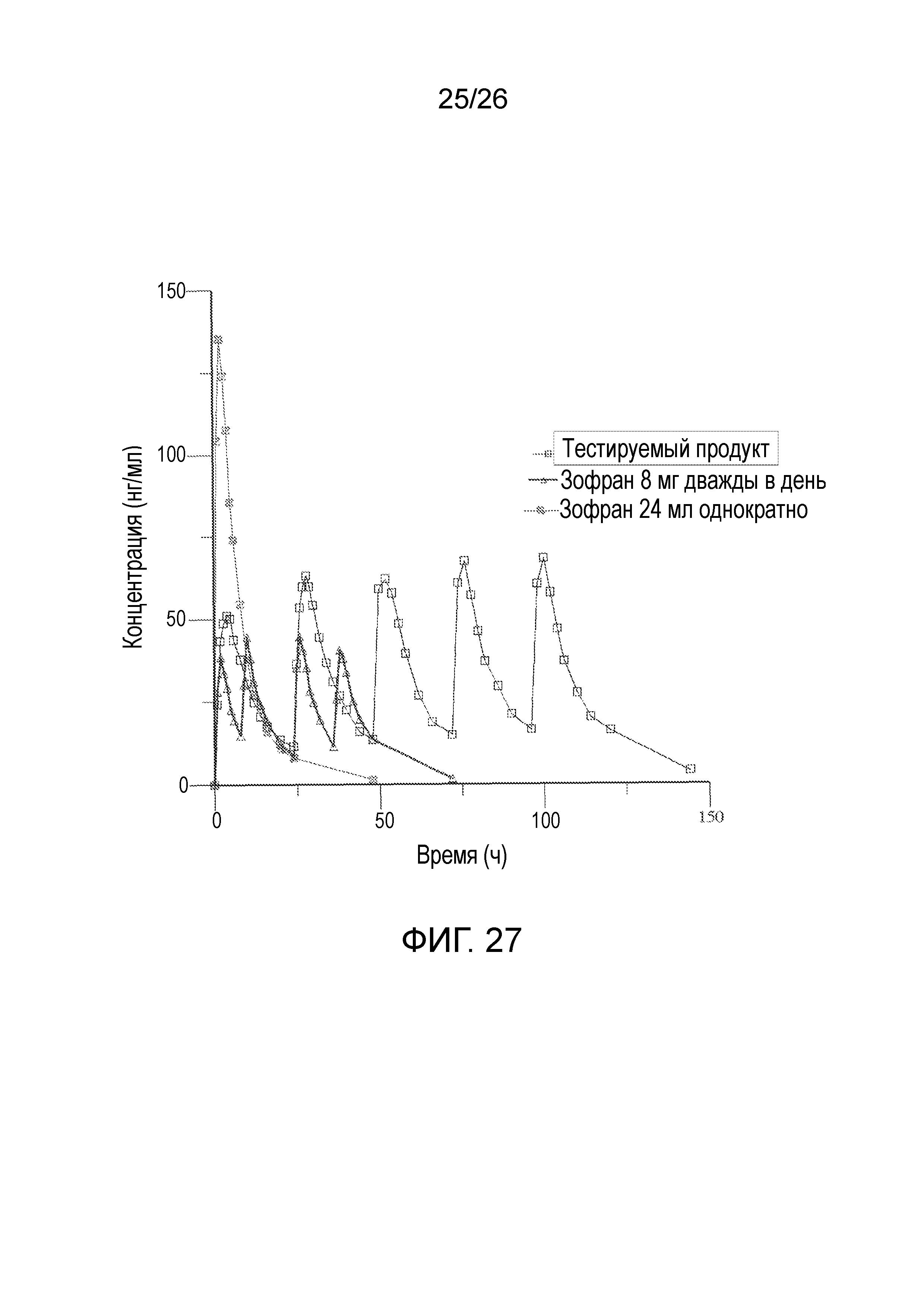
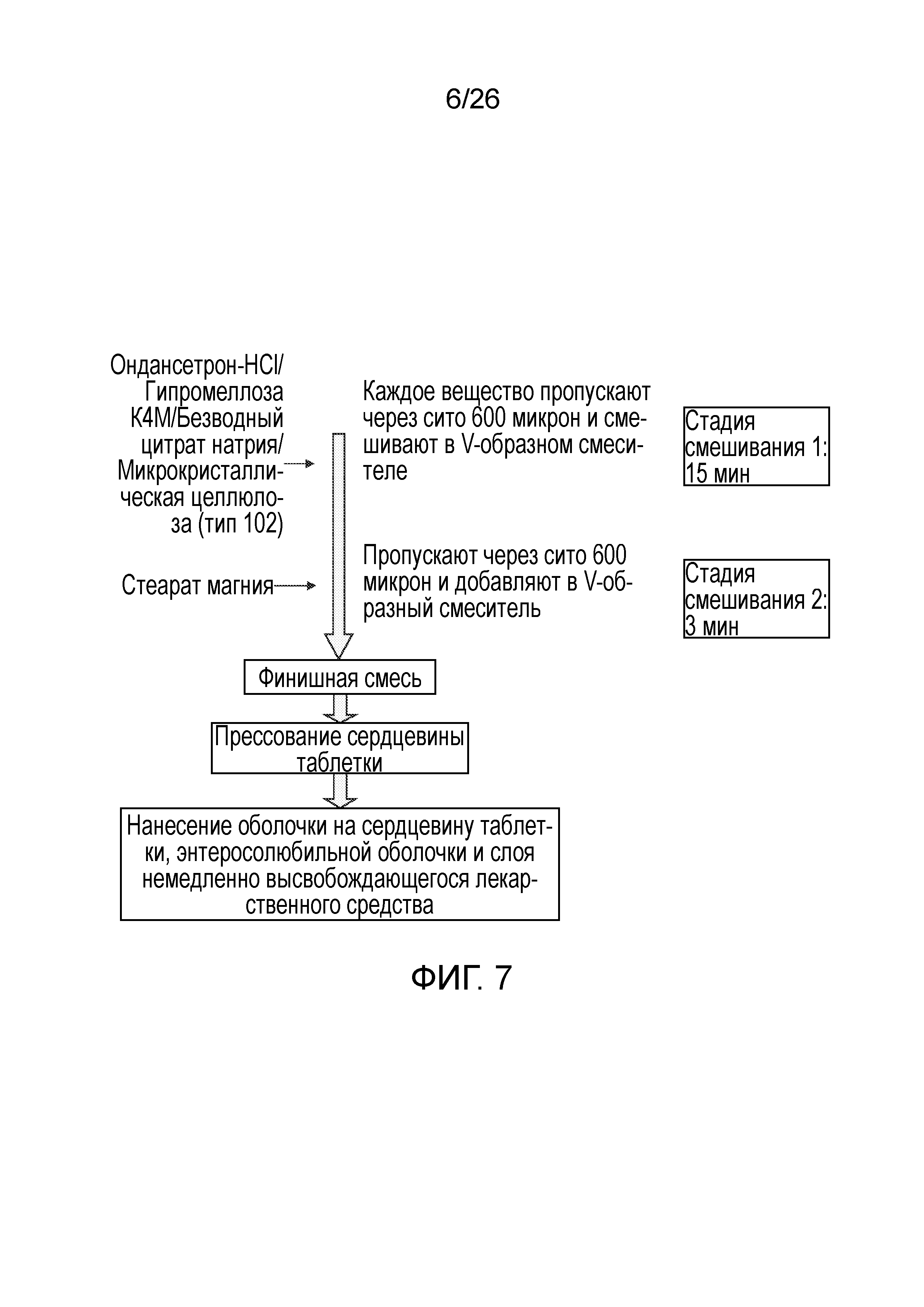
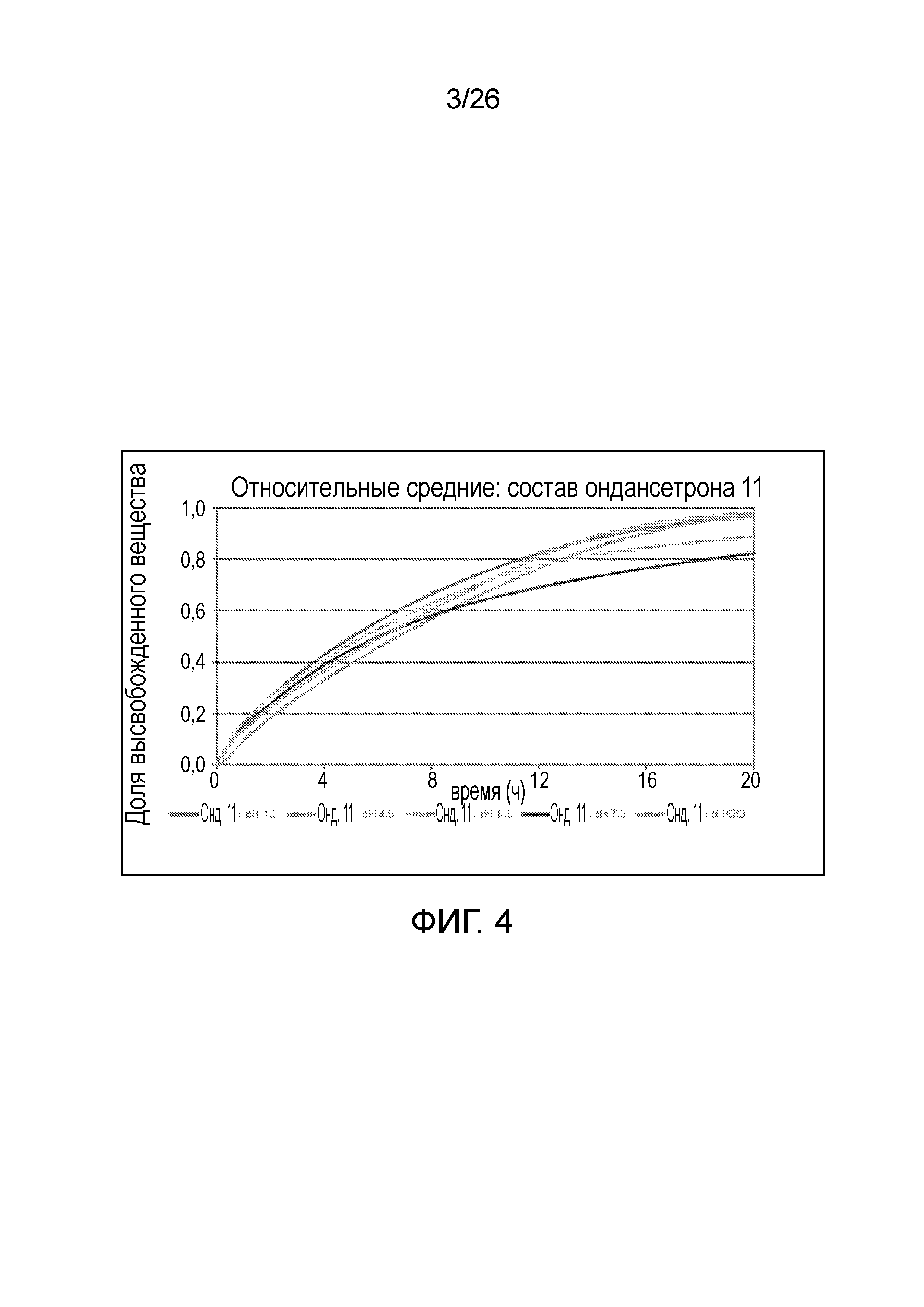
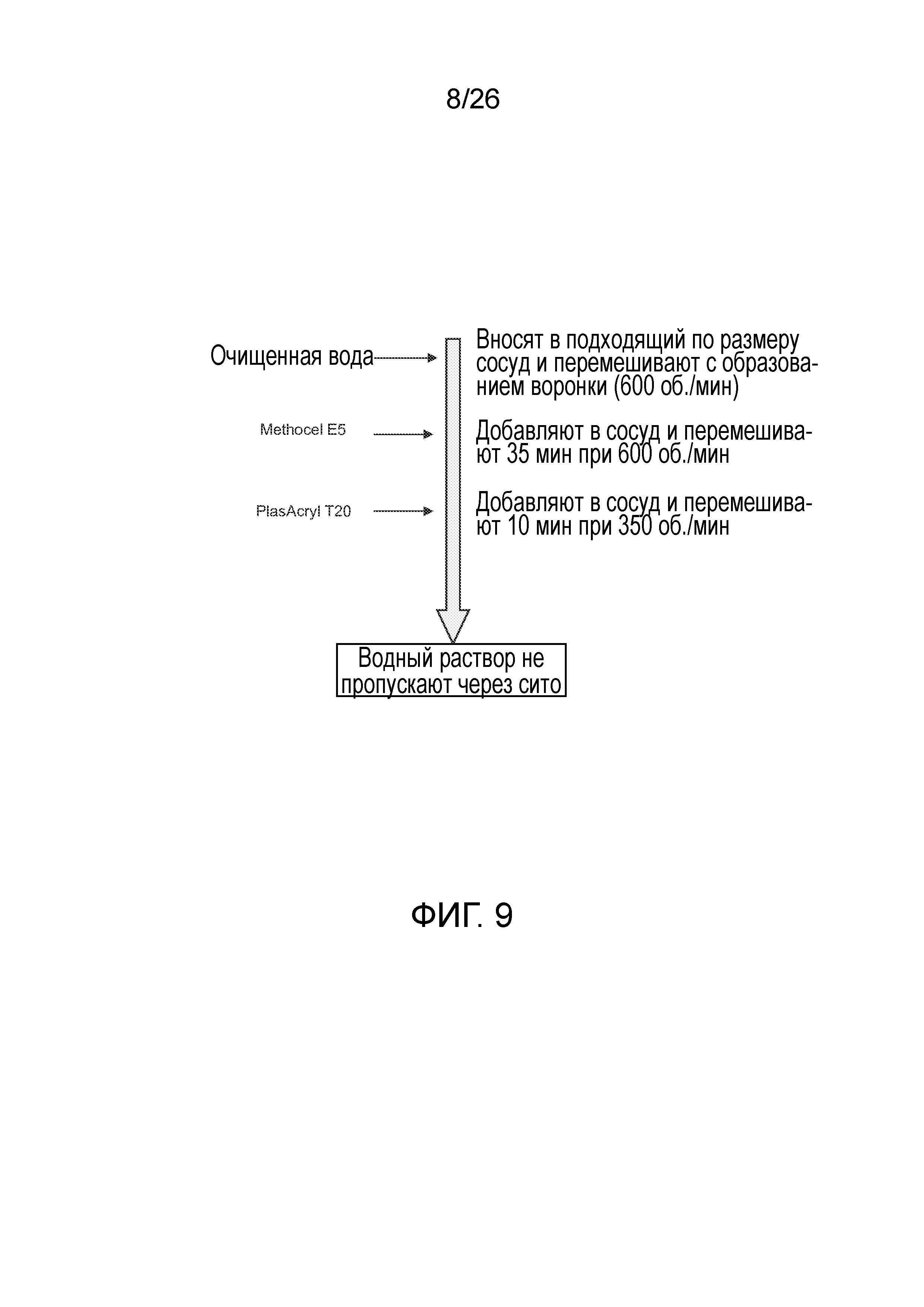
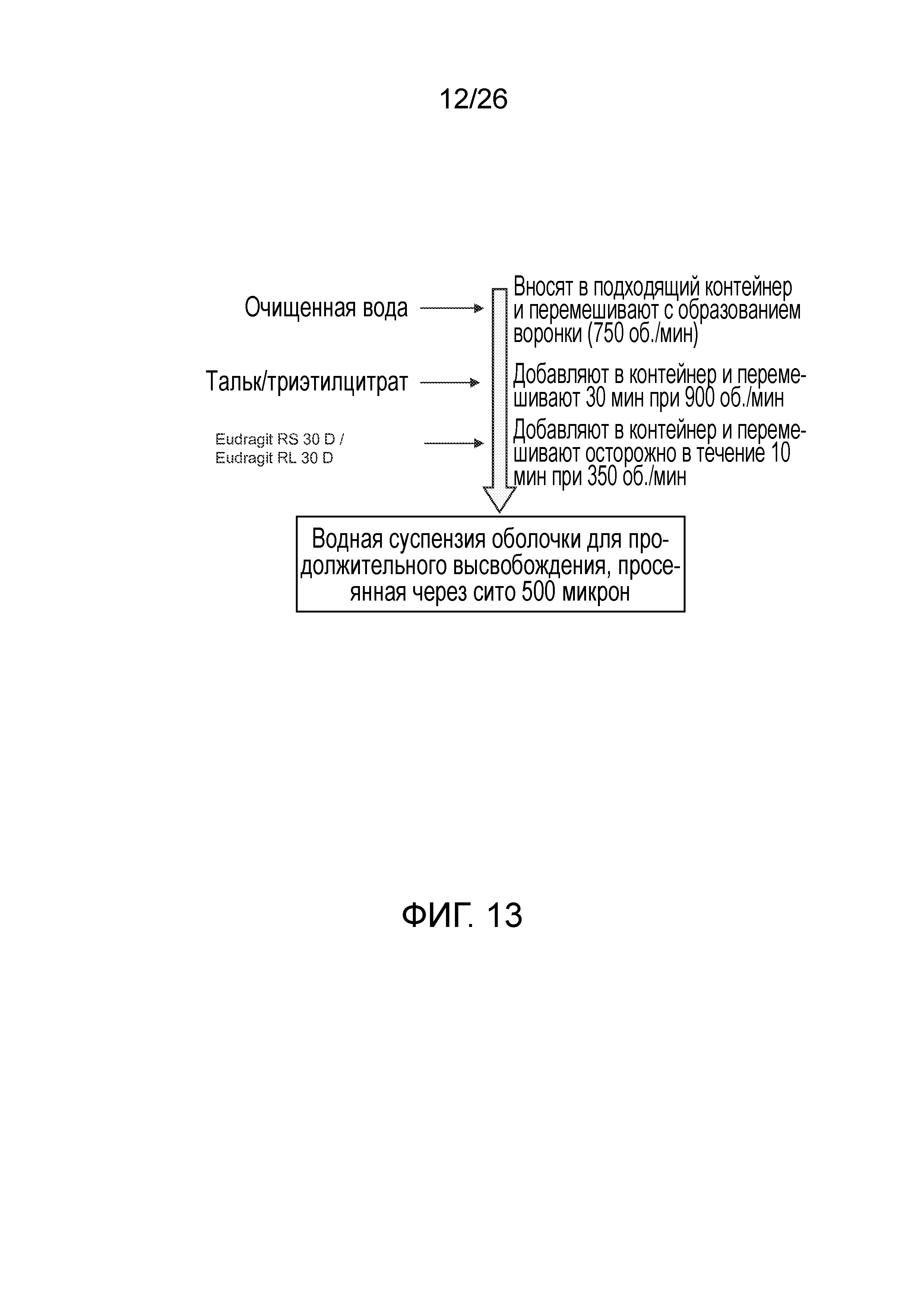
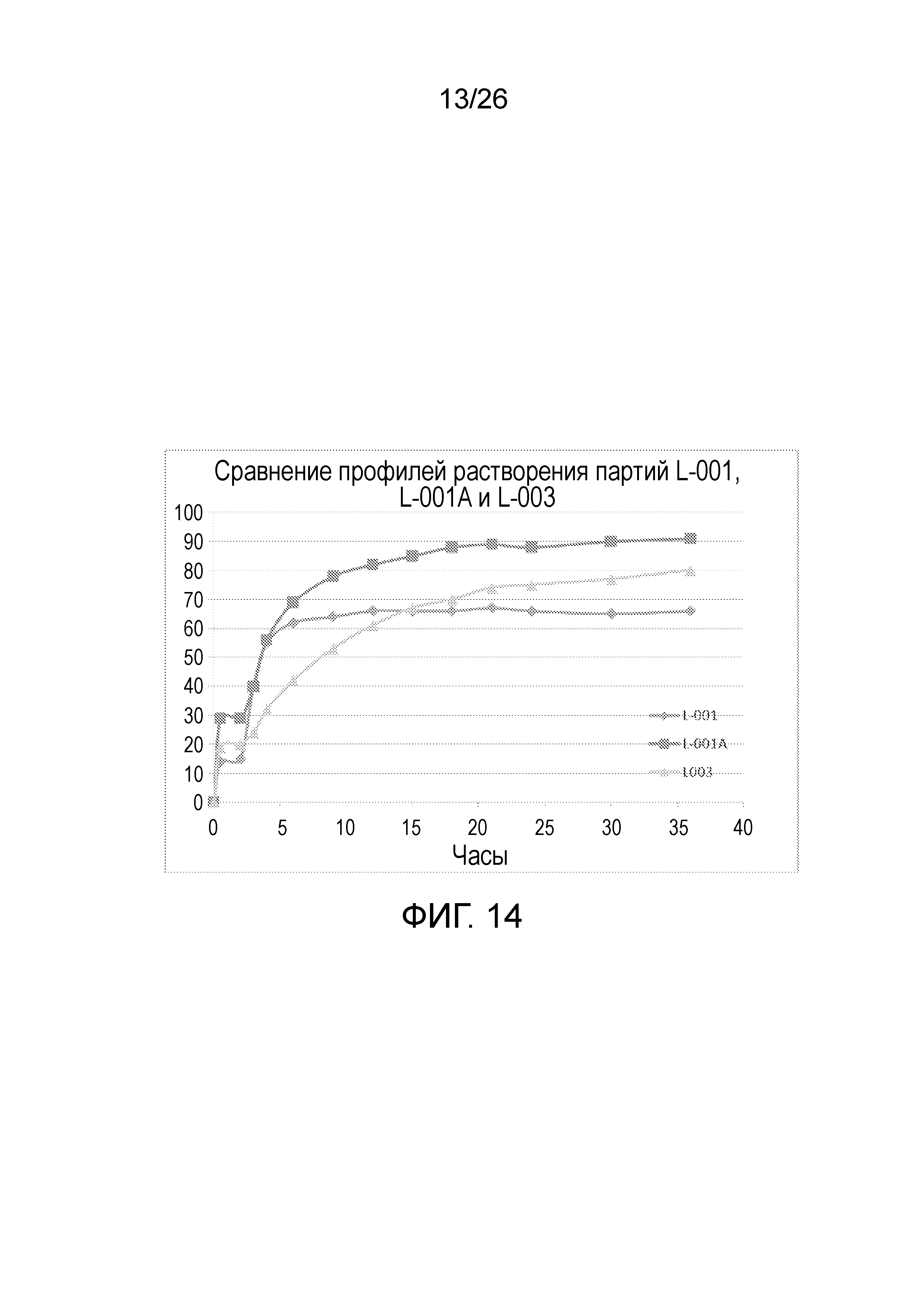
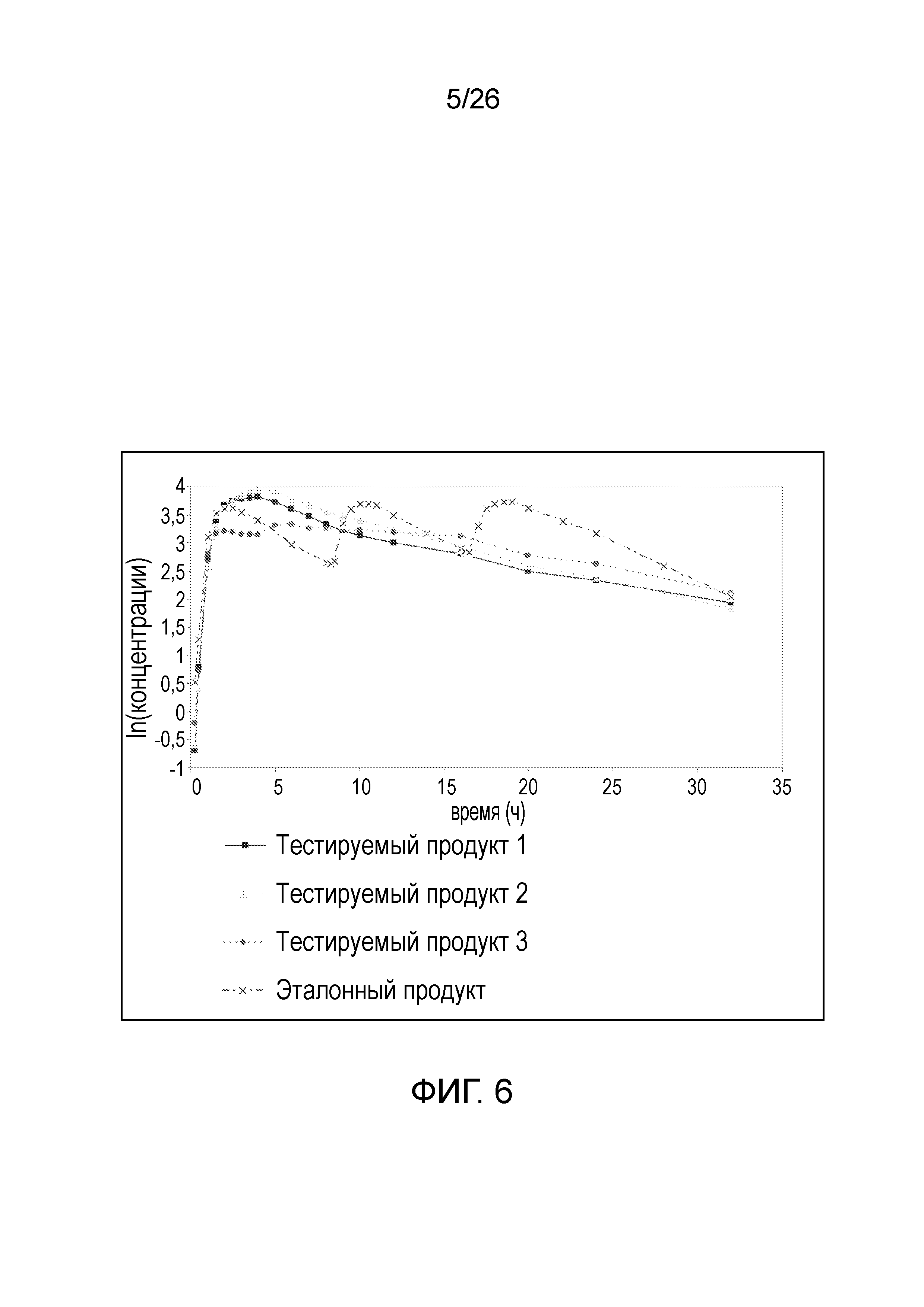
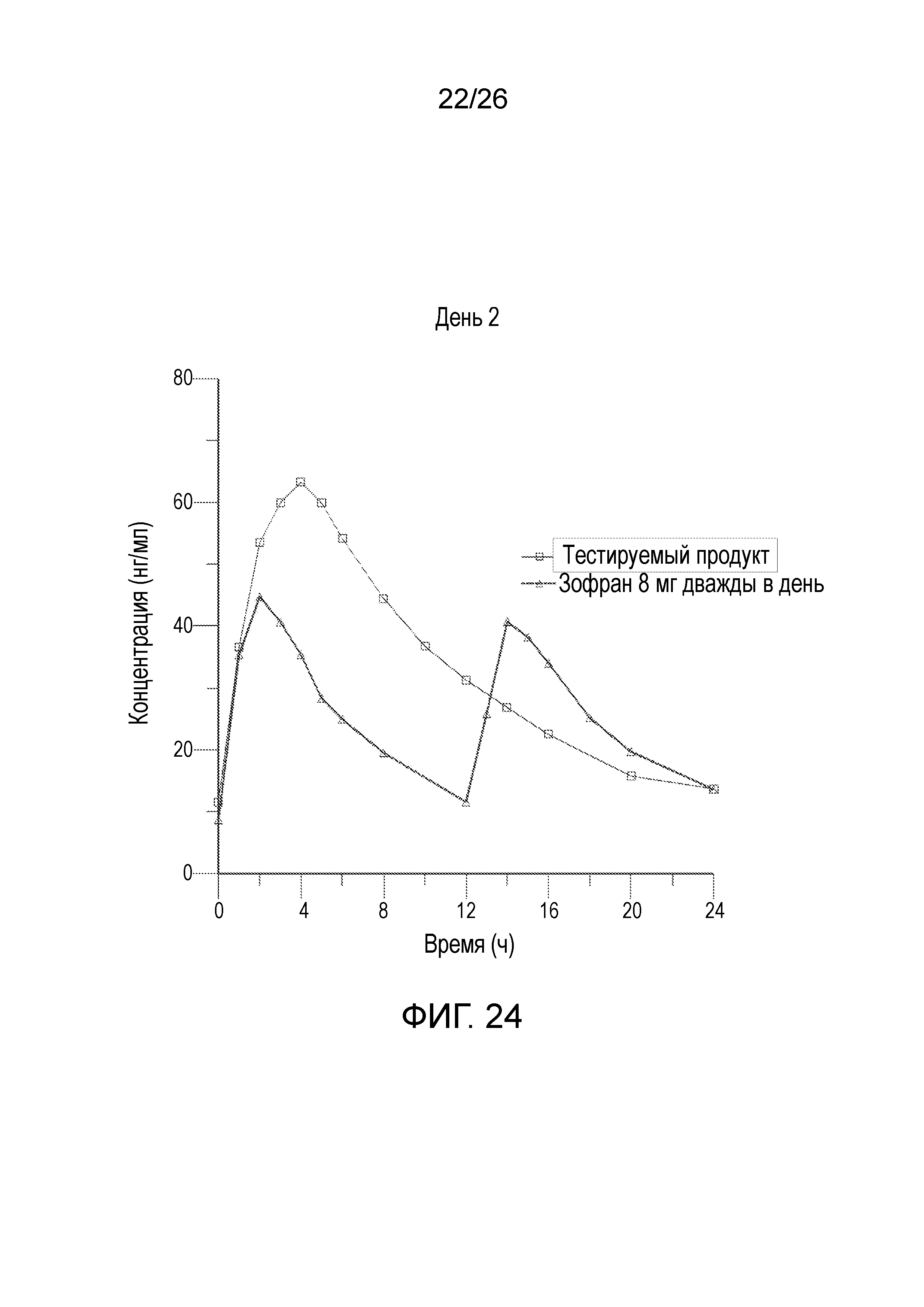
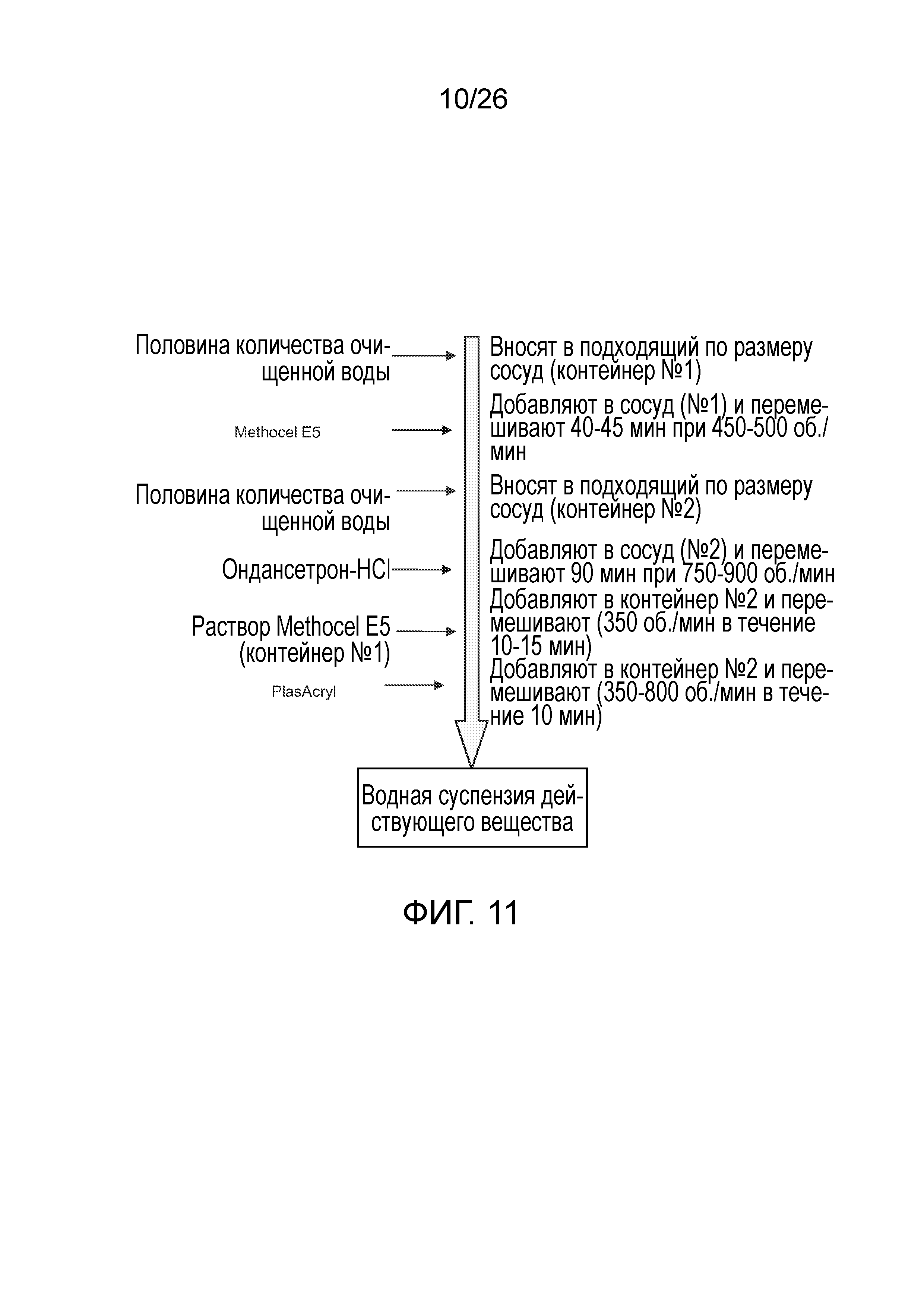
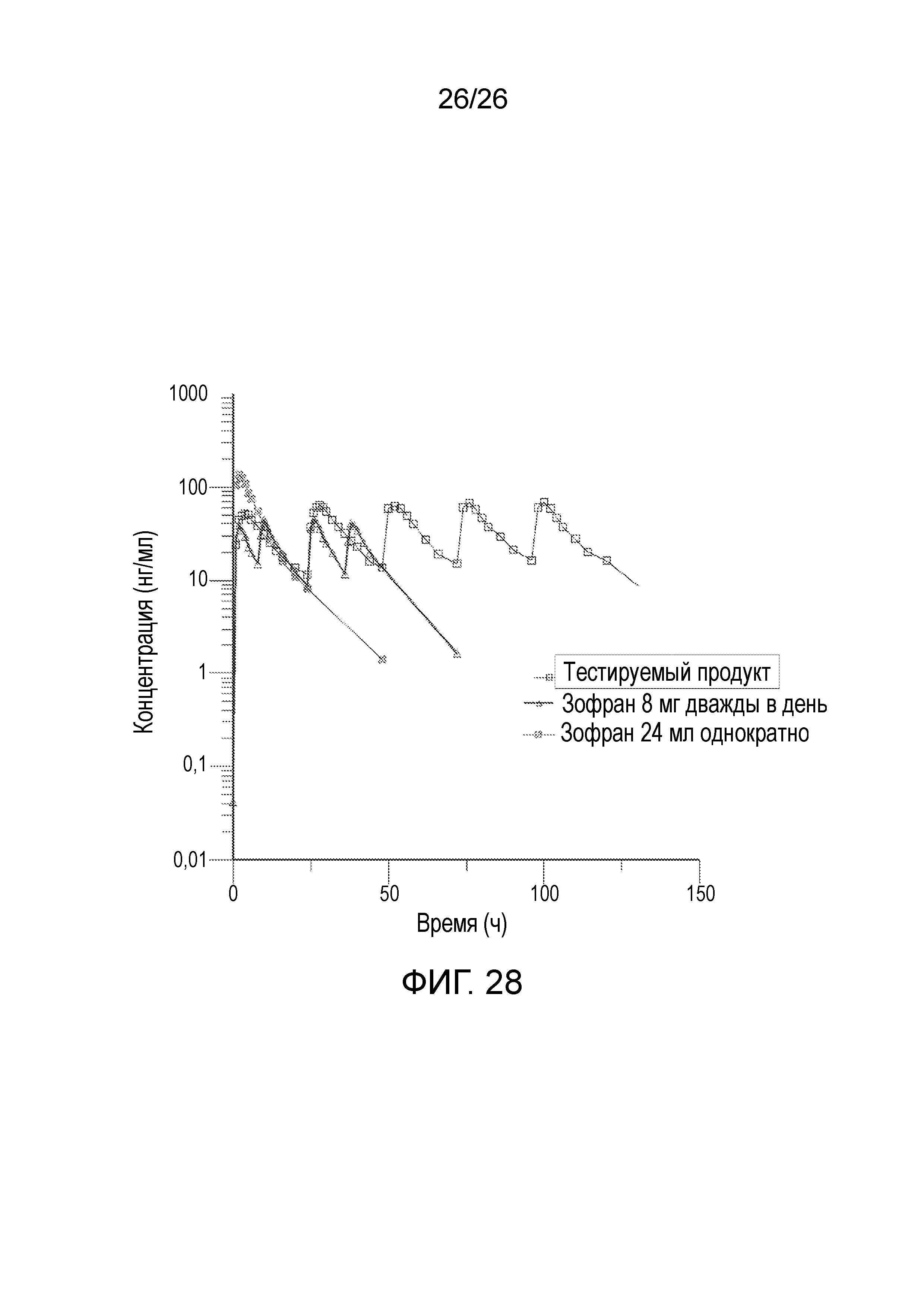
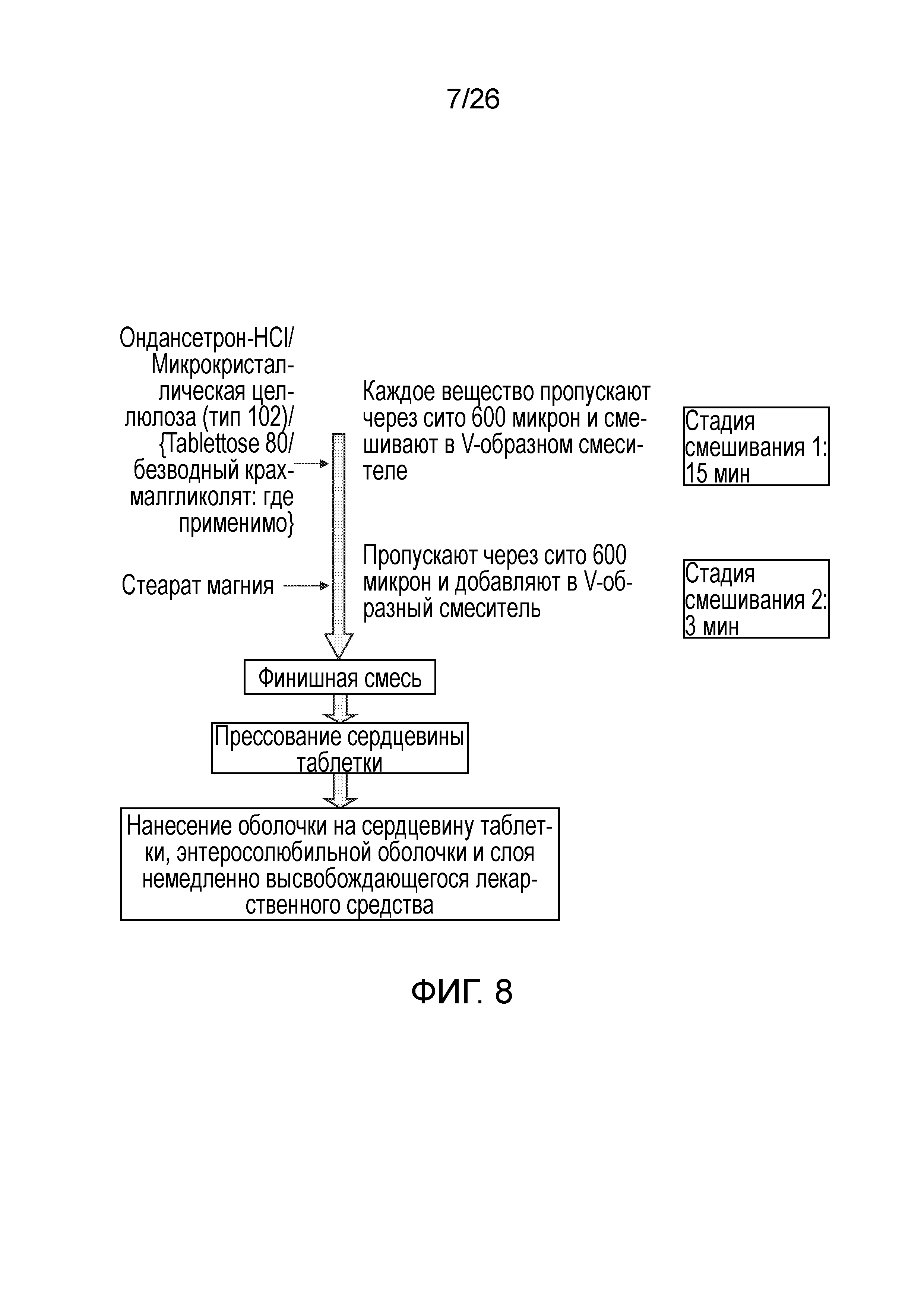
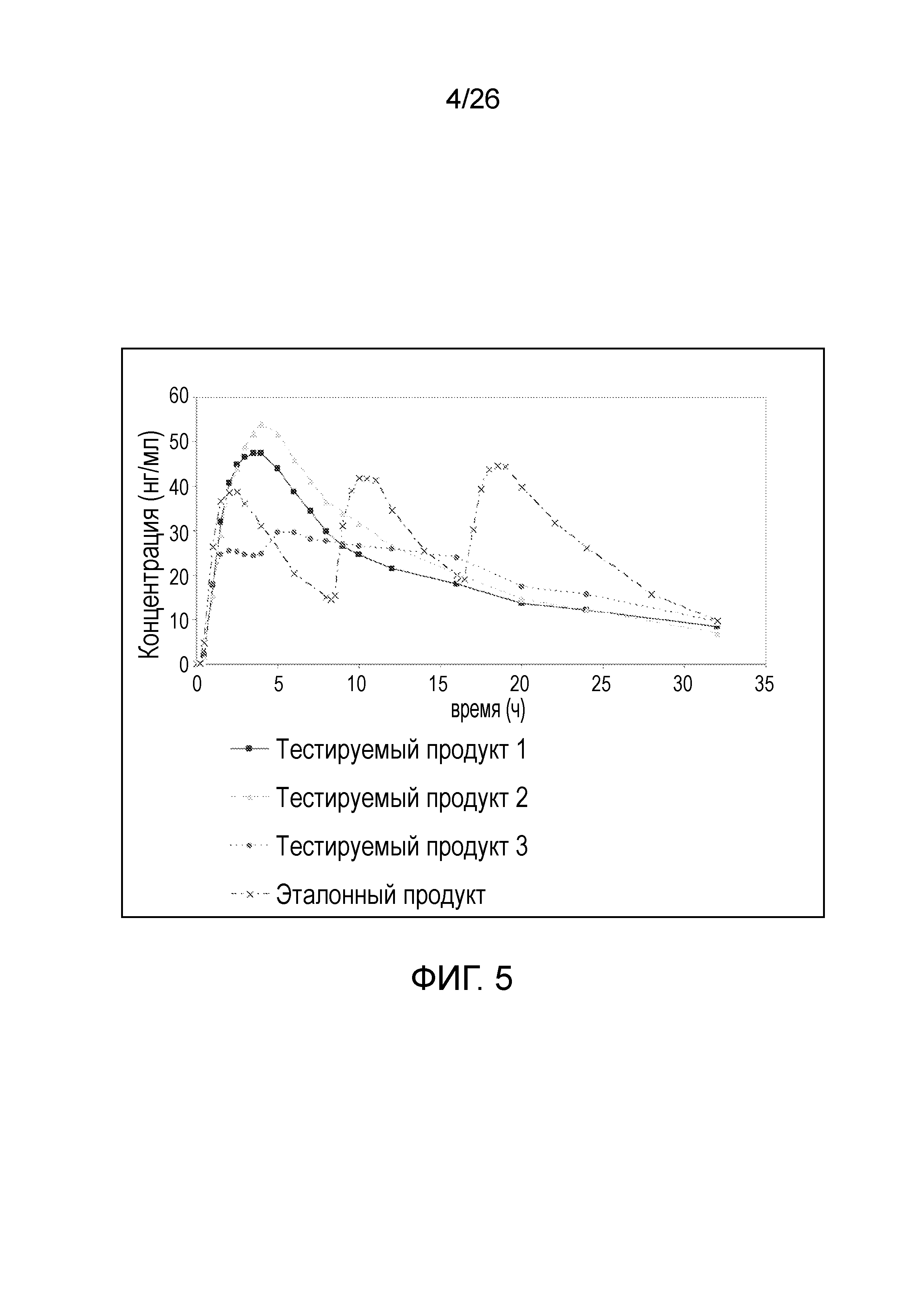
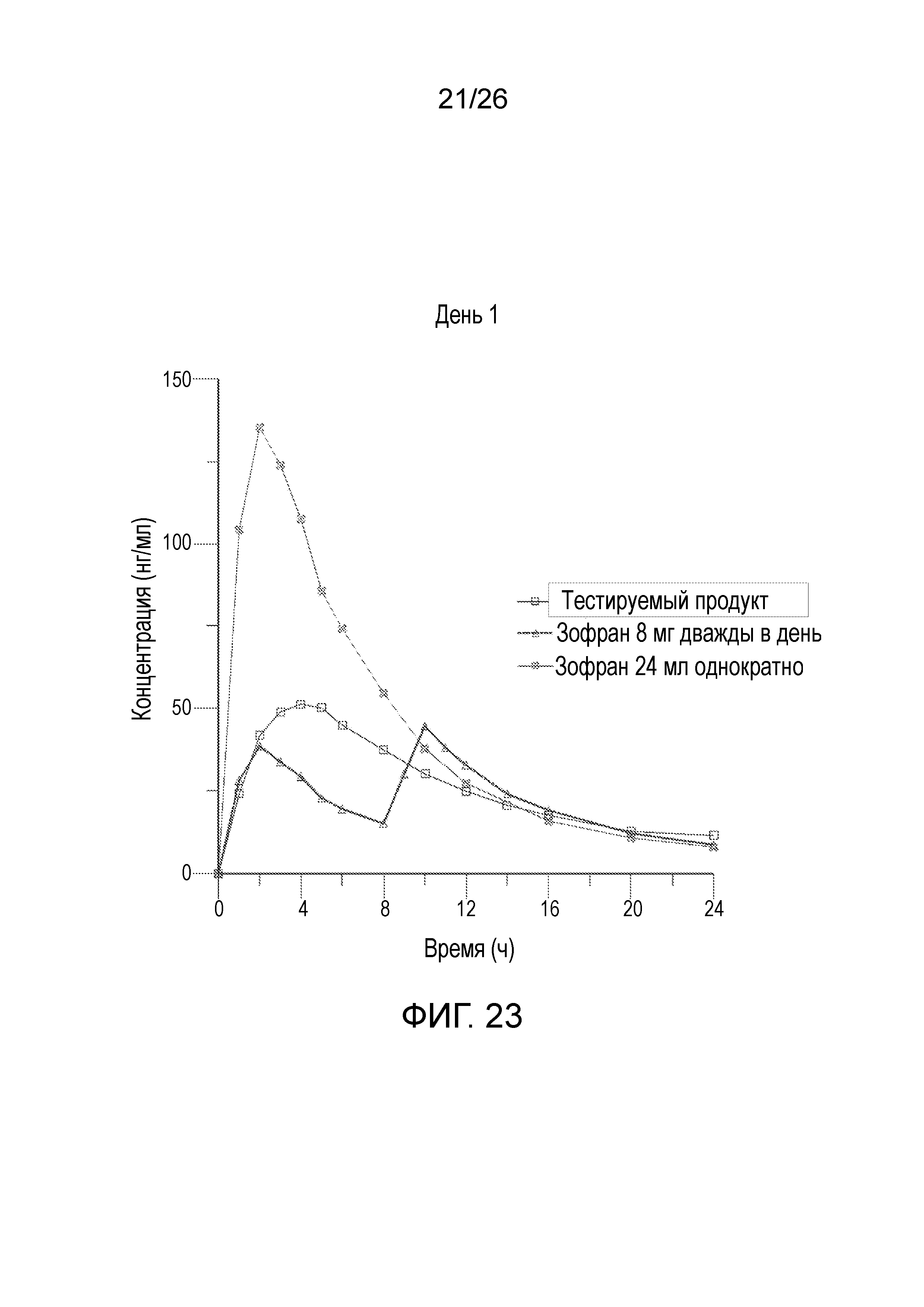
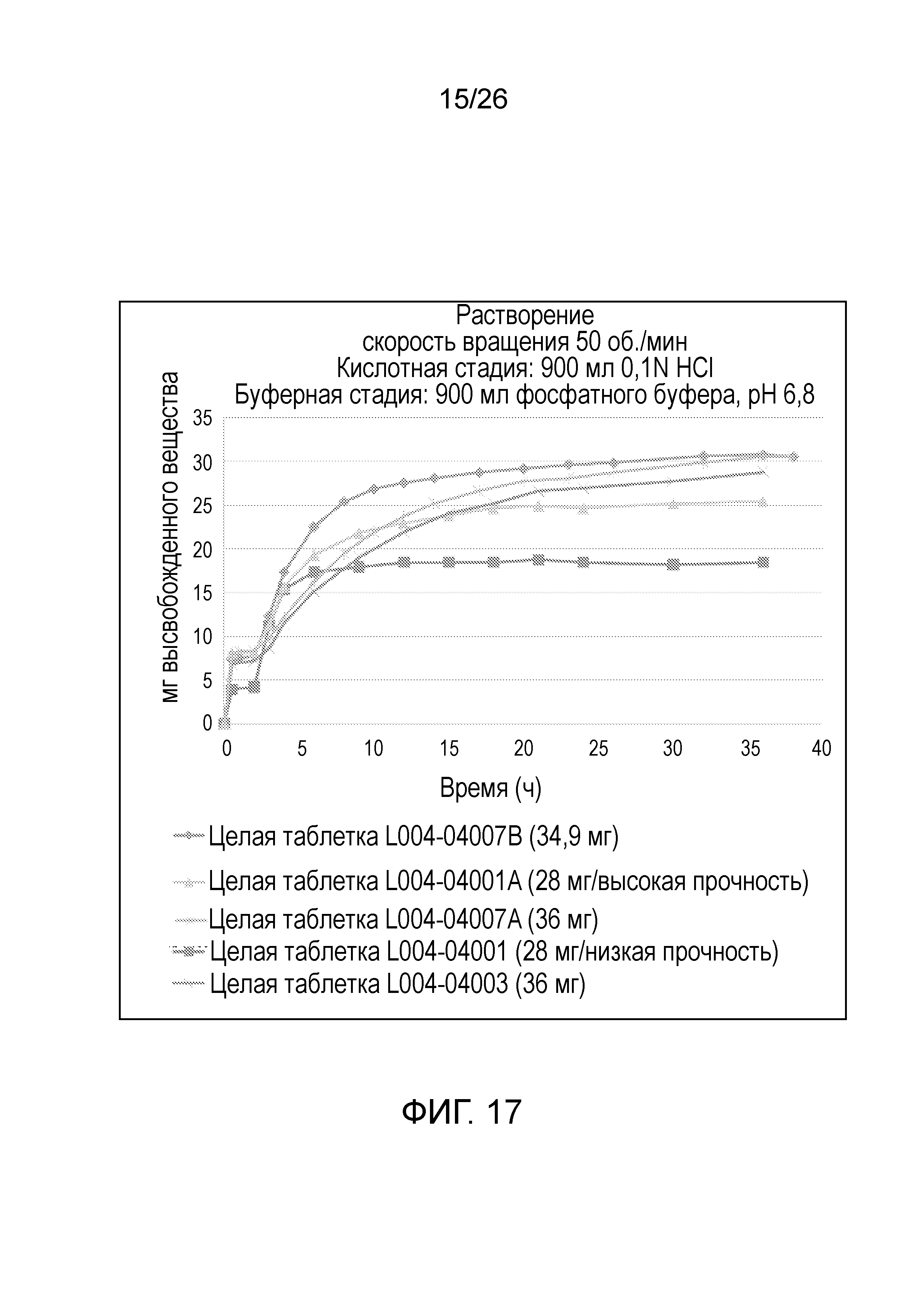
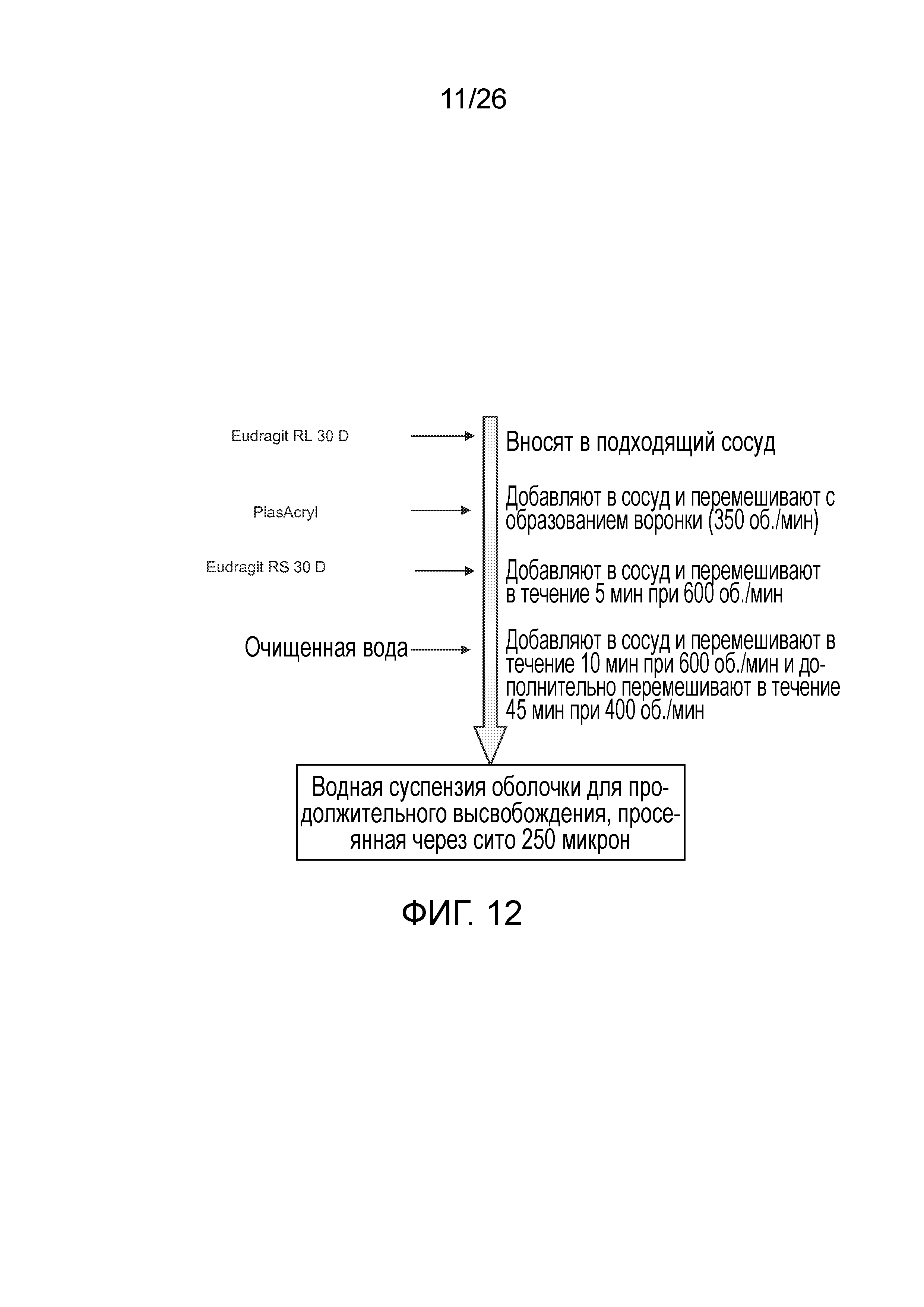
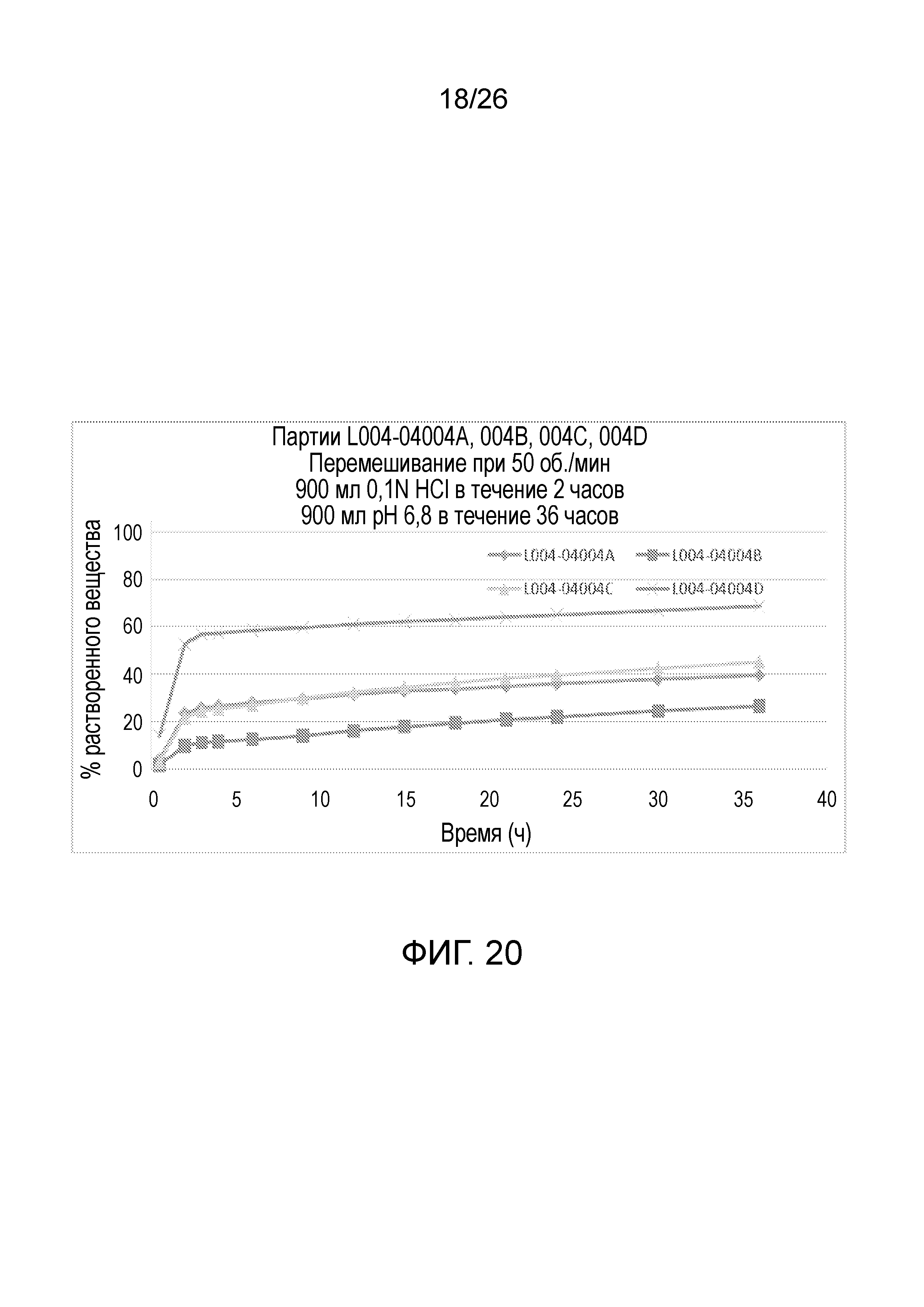
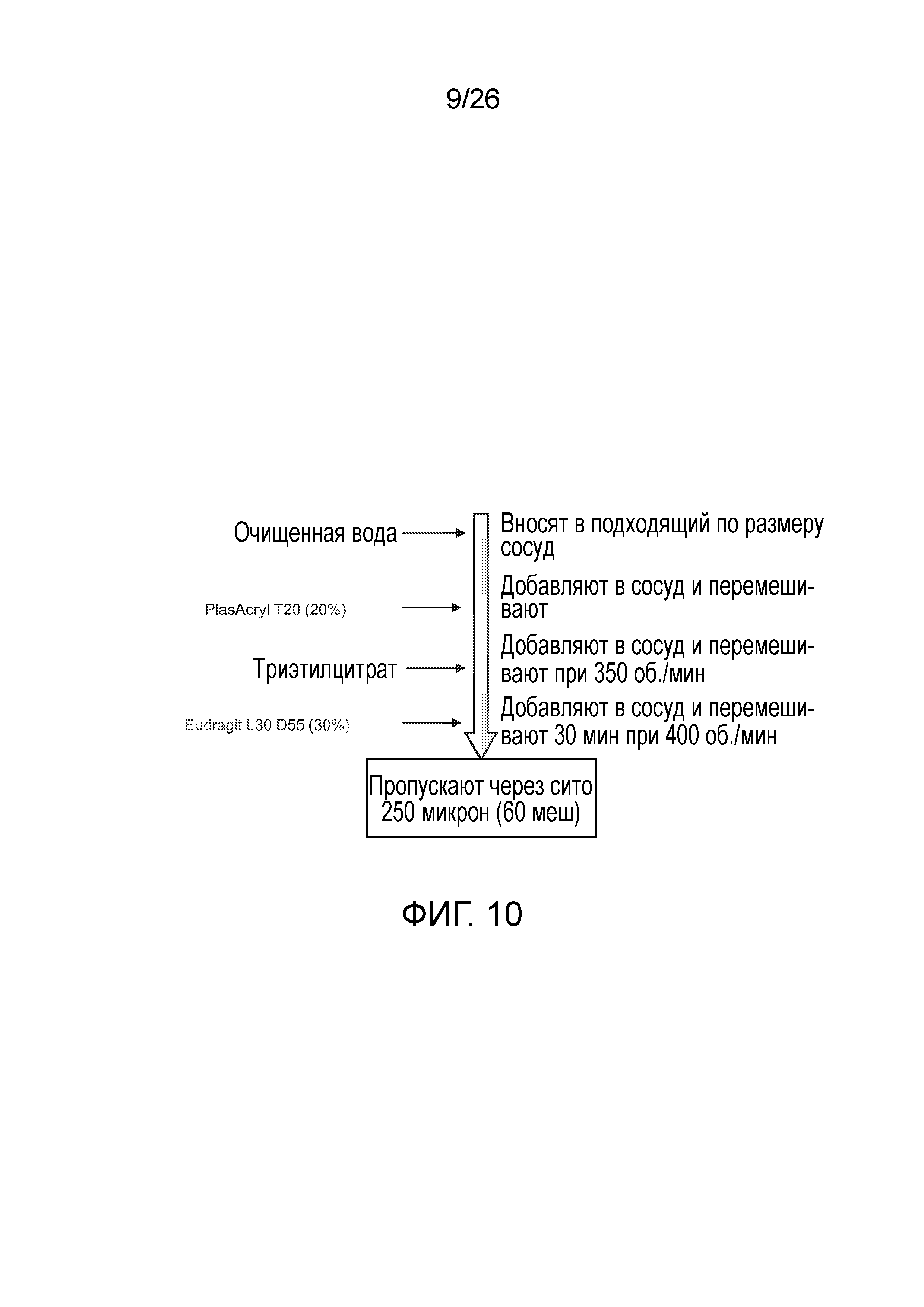
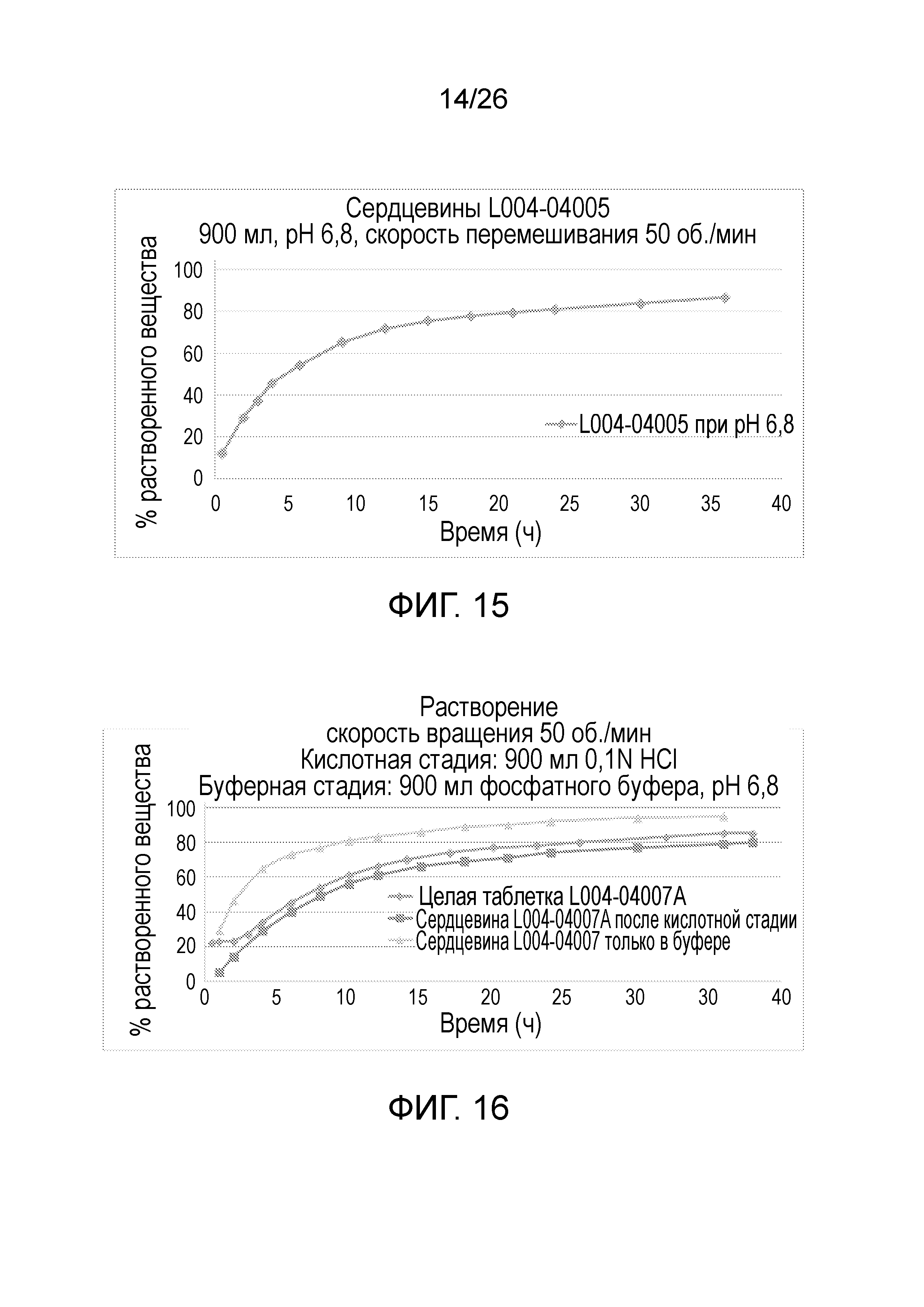
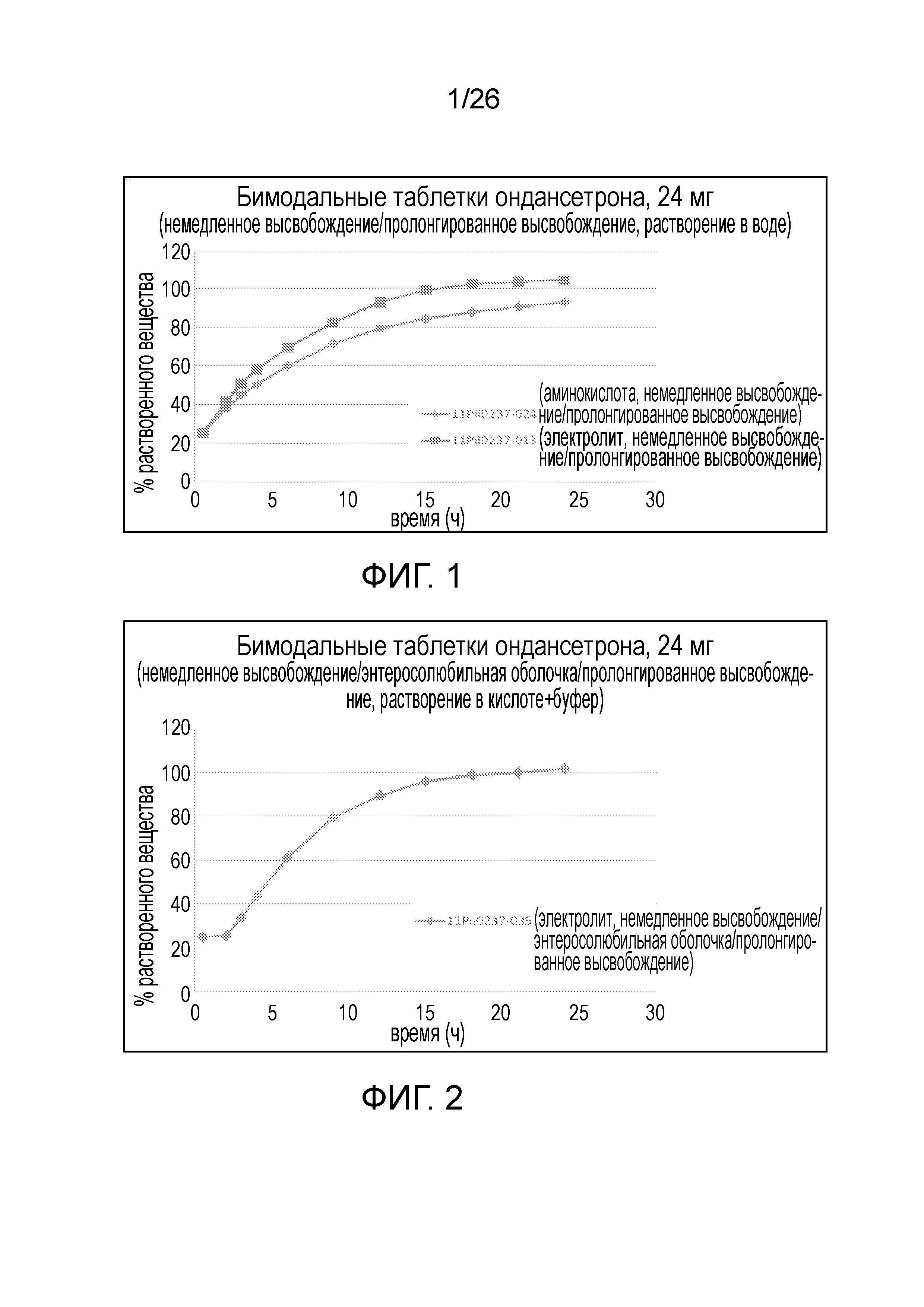
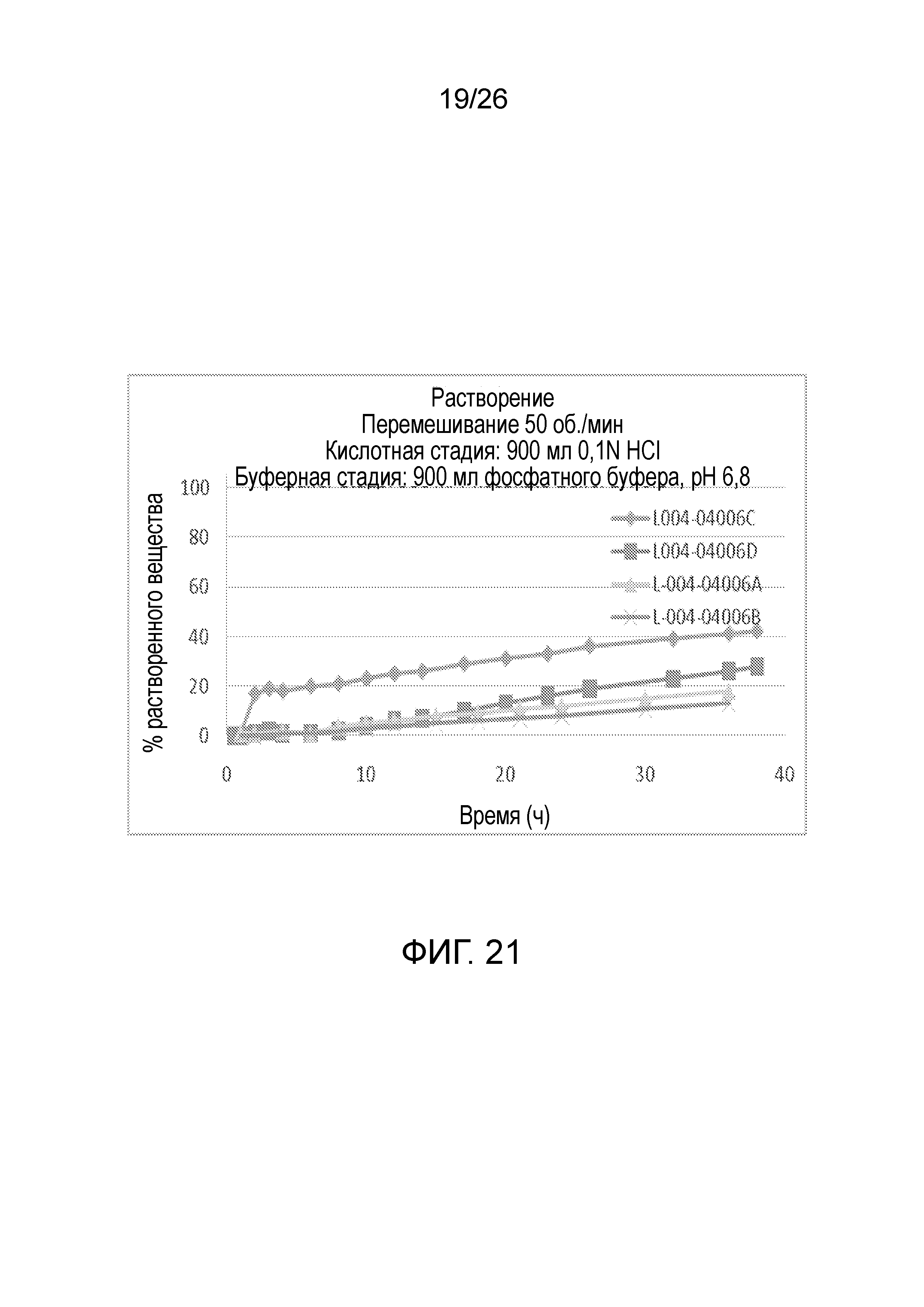
Фармацевтические композиции для лечения от helicobacter pylori
Твердые лекарственные формы, обладающие противорвотным действием, с замедленным высвобождением
Виды комбинированной терапии для лечения рака
Композиция и способ лечения аутоиммунного заболевания
Фармацевтические композиции для лечения от helicobacter pylori
Твердые лекарственные формы, обладающие противорвотным действием, с замедленным высвобождением

