Результат интеллектуальной деятельности: ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к области фармацевтического применения соединения гексагидродибензо[a,g]хинолизина и, в частности, к применению соединения гексагидродибензо[a,g]хинолизина в получении лекарственного средства для лечения и/или предупреждения доброкачественной гиперплазии предстательной железы.
Предшествующий уровень техники
Доброкачественная гиперплазия предстательной железы (ВРН) представляет собой физиологическое заболевание, распространенное у мужчин среднего и пожилого возраста. Наряду с неизбежно стареющим населением, частота возникновения ВРН возрастает в более значительной степени, чем раньше, и ВРН стала одним из наиболее распространенных заболеваний мужчин среднего и пожилого возраста в Китае. Статистика показывает, что заболеваемость гиперплазией предстательной железы является очень низкой до возраста 40 лет, при этом от доброкачественной гиперплазии предстательной железы страдает примерно половина мужчин старше возраста 50 лет, и почти 90% 80-летних мужчин страдает от данного заболевания. Доброкачественная гиперплазия предстательной железы представляет собой доброкачественную аденоматозную гиперплазию клеток окружающей зоны простатической уретры, и прогрессивное увеличение железы нарушает структуру простатического отдела уретры и вызывает обструкцию выходного отверстия мочевого пузыря. Исходным клиническим проявлением являются симптомы нижних мочевыводящих путей (LUTS), и в конечном итоге это может перерастать в задержку мочеиспускания, инфекции мочевого пузыря, камни мочевого пузыря и почечную недостаточность и даже угрожает жизни пациента. Вследствие этого, доброкачественная гиперплазия предстательной железы, как одно из распространенных заболеваний у мужчин среднего и пожилого возраста в Китае и заграницей, значительно снижает качество жизни пациентов.
Патогенез доброкачественной гиперплазии предстательной железы является очень сложным и связан с разными ферментами и рецепторами. В настоящее время два лекарственных средства, наиболее широко используемых для лечения ВРН в клинических условиях, представляют собой ингибиторы 5α-редуктазы и антагонисты α1-адренергических рецепторов, чье лечение, соответственно, направлено на объем предстательной железы и гладкомышечное напряжение, которые представляют собой два фактора, вызывающих симптомы доброкачественной гиперплазии предстательной железы. Ингибиторы 5α-редуктазы уменьшают объем предстательной железы и улучшают дизурию посредством ингибирования превращения тестостерона в дигидротестостерон в организме, посредством этого уменьшая содержание дигидротестостерона в предстательной железе. Однако данные лекарственные средства подходят только для лечения пациентов с ВРН с увеличенным объемом предстательной железы и симптомом нижних мочевыводящих путей и часто сопровождаются эректильной дисфункцией, отклоняющейся от нормы эякуляцией, низким либидо и другими побочными эффектами, такими как феминизация молочной железы мужчины, боль в груди и т.д. Таким образом, наиболее широко используемым лекарственным средством в клинических условиях является антагонист α1-адренергических рецепторов.
Адренергические рецепторы (AR) подразделяют на α-рецепторы и β-рецепторы, а α-адренергические рецепторы (α-AR) подразделяют на два типа рецепторов, то есть, α1 и α2. В настоящее время было идентифицировано три вида подтипов си-рецепторов (α1A, α1B и α1D). Исследование показывает, что α1-рецепторы главным образом находятся в матричных компонентах предстательной железы и железистом эпителии, в которых α1A-AR составляют примерно 70% от всех α1-AR в предстательной железе и мочевыделительной системе человека. В половой и мочевыделительной системе α1A-рецепторы главным образом распространены в предстательной железе, уретре и треугольнике мочевого пузыря, и семявыносящем протоке; α1B-рецепторы главным образом распространены в кровеносных сосудах; и α1D-рецепторы главным образом распространены в детрузоре мочевого пузыря и гладких мышцах мочеточника.
В патологических случаях ВРН плотность α1-AR значительно повышена. Кроме того, распространение подтипов α1-AR различно при изменениях возраста. Корреляция между возрастом и распространением является важной для понимания и лечения доброкачественной гиперплазии предстательной железы и симптомов нижних мочевыводящих путей, и для разработки антагонистов α1-адренергических рецепторов. Считается, что α1A-адренергический рецептор является идеальной мишенью для лечения доброкачественной гиперплазии предстательной железы и его блокирование, как было доказано, эффективно снижает частоту сокращения гладких мышц предстательной железы и одновременно улучшает опорожнение мочевого пузыря. Блокирование α1B-адренергического рецептора приводит к релаксации гладких мышц сосудов, артериовенозному расширению, уменьшению периферического сопротивления и другим симптомам, и может вызывать побочные эффекты, такие как головокружение и гипотензия у некоторых пациентов. Активация α1D-адренергического рецептора может приводить к гиперактивности детрузора, а его блокирование может снижать встречаемость симптомов опорожнения, как доказано в экспериментах на животных. Теоретически двойные ингибиторы α1A- и α1D-адренергического рецептора являются очень эффективными лекарственными средствами для контролирования доброкачественной гиперплазии предстательной железы, поскольку они имеют две функции, то есть снижение частоты сокращения гладких мышц предстательной железы и ингибирование дисфункции детрузора и, кроме того, можно избегать сердечнососудистых побочных эффектов, вызванных блокадой α1B-адренергического рецептора.
В качестве первого поколения репрессора α-рецептора разработан феноксибензамин и он используется для эффективного излечения симптомов доброкачественной гиперплазии предстательной железы. Феноксибензамин представляет собой необратимый и неселективный репрессор α1/α2-рецептора и принадлежит к β-галоалкану, и он блокирует α-рецептор в предстательной железе и приводит к расслаблению фиброзной ткани предстательной железы. Феноксибензамин используют в клинических условиях для лечения затрудненного мочеиспускания, вызванного немеханической обструкцией уретры, обусловленной предстательной железой. Феноксибензамин содержит структуру β-хлорэтиламина, которая легко реагирует с другими ферментами в организме, вследствие этого, вызывая токсичность и побочные эффекты. В качестве неселективного репрессора α-рецептора феноксибензамин блокирует α1-рецепторы и пресинаптические α2-рецепторы и вызывает обратную связь в нервных окончаниях с высвобождением норэпинефрина, вызывая посредством этого рефлекторную тахикардию, аритмию и другие побочные эффекты.
Для уменьшения данных побочных эффектов разработано второе поколение антагонистов α1-адренергических рецепторов с высокой селективностью в отношении α1-рецептора (таких как: празозин, теразозин, доксазозин, алфузозин). α1-адренергический рецептор может ослаблять сокращение гладких мышц предстательной железы и уретры, обусловленное симпатической нервной системой, и кинетически уменьшать симптомы уретральной обструкции. Данные лекарственные средства могут эффективно облегчать симптомы нижних мочевыводящих путей и уменьшать побочные эффекты, вызванные вазодилатацией. Лекарственные средства празозин все имеют ядерную структуру хиназолина и их обычно используют для лечения симптомов нижних мочевыводящих путей (LUTS), обусловленных ВРН, в клинических условиях. Однако в связи с широким распространением α1-адренергического рецептора и важными физиологическими функциями применение антагонистов α1-адренергических рецепторов часто приводит к ортостатической гипотензии, головокружению, слабости и другим побочным эффектам.
В последнее время разрешенными лекарственными средствами для лечения доброкачественной гиперплазии предстательной железы являются селективные антагонисты α1A-рецепторов - тамсулозин и силодозин. Тамсулозин считается селективным антагонистом α1A-рецепторов, но обладает плохой селективностью в отношении других α1-рецепторов, и в связи с более ранним появлением на рынке он имеет большую долю на рынке. Но он все еще имеет некоторые побочные эффекты, такие как побочные эффекты на иммунную систему и глазничную область. Кроме того, он может вызывать нарушения эякуляции, более низкое кровяное давление, головные боли и другие побочные эффекты. Лекарственное средство силодозин, которое поступило на рынок недавно, имеет хорошую селективность в отношении рецептора, которая лучше, чем селективность тамсулозина, и в настоящее время его побочные эффекты в клинических условиях меньше, чем побочные эффекты тамсулозина. Вследствие этого, лекарственные средства, имеющие хорошую селективность в отношении α1A-рецептора, несомненно, имеют хорошую перспективу рынка для лечения доброкачественной гиперплазии предстательной железы.
Краткое описание изобретения
Одним объектом настоящего изобретения является обеспечение фармацевтического применения соединения гексагидродибензо[a,g]хинолизина.
Другим объектом настоящего изобретения является обеспечение лекарственного средства с лучшей селективностью в отношении a1A-рецептора для лечения доброкачественной гиперплазии предстательной железы.
В первом аспекте изобретения предложено применение соединения гексагидродибензо[a,g]хинолизина формулы (I) или его фармацевтически приемлемой соли, сольвата, стереоизомера, таутомера для получения лекарственного средства для лечения и/или предупреждения доброкачественной гиперплазии предстательной железы:
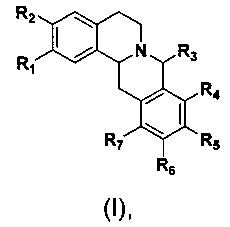
где каждый из R1, R2, R4, R5, R6 и R7 независимо представляет собой водород, гидрокси, галоген, замещенный или незамещенный С1-С6 алкокси, замещенный или незамещенный С1-С6 алкил, замещенный или незамещенный С2-С6 алкенил, замещенный или незамещенный С2-С6 алкинил, замещенный или незамещенный С3-С6 циклоалкил, замещенный или незамещенный бензилокси, при этом заместитель для замещения представляет собой галоген, гидрокси, амино или сульфонил;
R3 представляет собой водород, галоген, незамещенный или галоген-замещенный С1-С6 алкил или незамещенный или галоген-замещенный С1-С6 алкокси;
R1 и R2 могут вместе образовывать замещенный или незамещенный 5-7-членный гетероцикл, при этом заместитель для замещения представляет собой галоген или незамещенный или галоген-замещенный С1-С6 алкил, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
любые два смежных заместителя из R4, R5, R6 и R7 могут вместе образовывать замещенный или незамещенный 5-7-членный гетероцикл, при этом заместитель для замещения представляет собой галоген, незамещенный или галоген-замещенный С1-С6 алкил или незамещенный или галоген-замещенный С1-С6 алкокси, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
конфигурация хирального атома углерода в соединении общей формулы (I) представляет собой R или S.
В одном воплощении каждый из R1, R2, R4, R5, R6 и R7 независимо представляет собой водород, гидрокси, галоген, замещенный или незамещенный С1-С4 алкокси, замещенный или незамещенный С1-С4 алкил, замещенный или незамещенный С2-С4 алкенил, замещенный или незамещенный С2-С4 алкинил, замещенный или незамещенный С3-С6 циклоалкил, замещенный или незамещенный бензилокси, при этом заместитель для замещения представляет собой галоген, гидрокси, амино или сульфонил;
R3 представляет собой водород, галоген, незамещенный или галоген-замещенный С1-С4 алкил, или незамещенный или галоген-замещенный С1-С4 алкокси;
R1 и R2 могут вместе образовывать замещенный или незамещенный 5-7-членный гетероцикл, при этом заместитель для замещения представляет собой галоген или незамещенный или галоген-замещенный С1-С4 алкил, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
любые два смежных заместителя из R4, R5, R6 и R7 могут вместе образовывать замещенный или незамещенный 5-7-членный гетероцикл, при этом заместитель для замещения представляет собой галоген, незамещенный или галоген-замещенный С1-С4 алкил, или незамещенный или галоген-замещенный С1-С4 алкокси, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
конфигурация хирального атома углерода в соединении общей формулы (I) представляет собой либо R или S.
В одном воплощении каждый из R1, R2, R4, R5, R6 и R7 независимо представляет собой водород, гидрокси, галоген, замещенный или незамещенный С1-С4 алкокси, замещенный или незамещенный С1-С4 алкил или замещенный или незамещенный бензилокси, при этом заместитель для замещения может представлять собой галоген, гидрокси или амино;
R3 представляет собой водород, галоген и незамещенный или галоген-замещенный С1-С4 алкил;
R1 и R2 могут вместе образовывать замещенный или незамещенный 5-или 6-членный гетероцикл, при этом заместитель для замещения представляет собой галоген или незамещенный или галоген-замещенный С1-С4 алкил, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
любые два смежных заместителя из R4, R5, R6 и R7 могут вместе образовывать замещенный или незамещенный 5- или 6-членный гетероцикл, при этом заместитель для замещения представляет собой галоген, незамещенный или галоген-замещенный С1-С4 алкил или незамещенный или галоген-замещенный С1-С4 алкокси, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
конфигурация хирального атома углерода в соединении общей формулы (I) представляет собой R или S.
В одном воплощении каждый из R1, R2, R4, R5, R6 и R7 независимо представляет собой водород, гидрокси, галоген, замещенный или незамещенный С1-С4 алкокси, замещенный или незамещенный С1-С4 алкил или замещенный или незамещенный бензилокси, при этом заместитель для замещения представляет собой галоген или гидрокси;
R3 представляет собой водород, галоген или незамещенный или галоген-замещенный С1-С4 алкил;
R1 и R2 могут вместе образовывать 5- или 6-членный гетероцикл, который содержит от 1 до 2 гетероатомов, выбранных из N, О или S;
R5 и R6 могут вместе образовывать 5- или 6-членный гетероцикл, который содержит от 1 до 2 гетероатомов, выбранных из N, О или S;
конфигурация хирального атома углерода в соединении общей формулы (I) представляет собой R или S.
В одном воплощении соединения гексагидродибензо[a,g]хинолизина общей формулы (I) выбраны из следующих соединений:
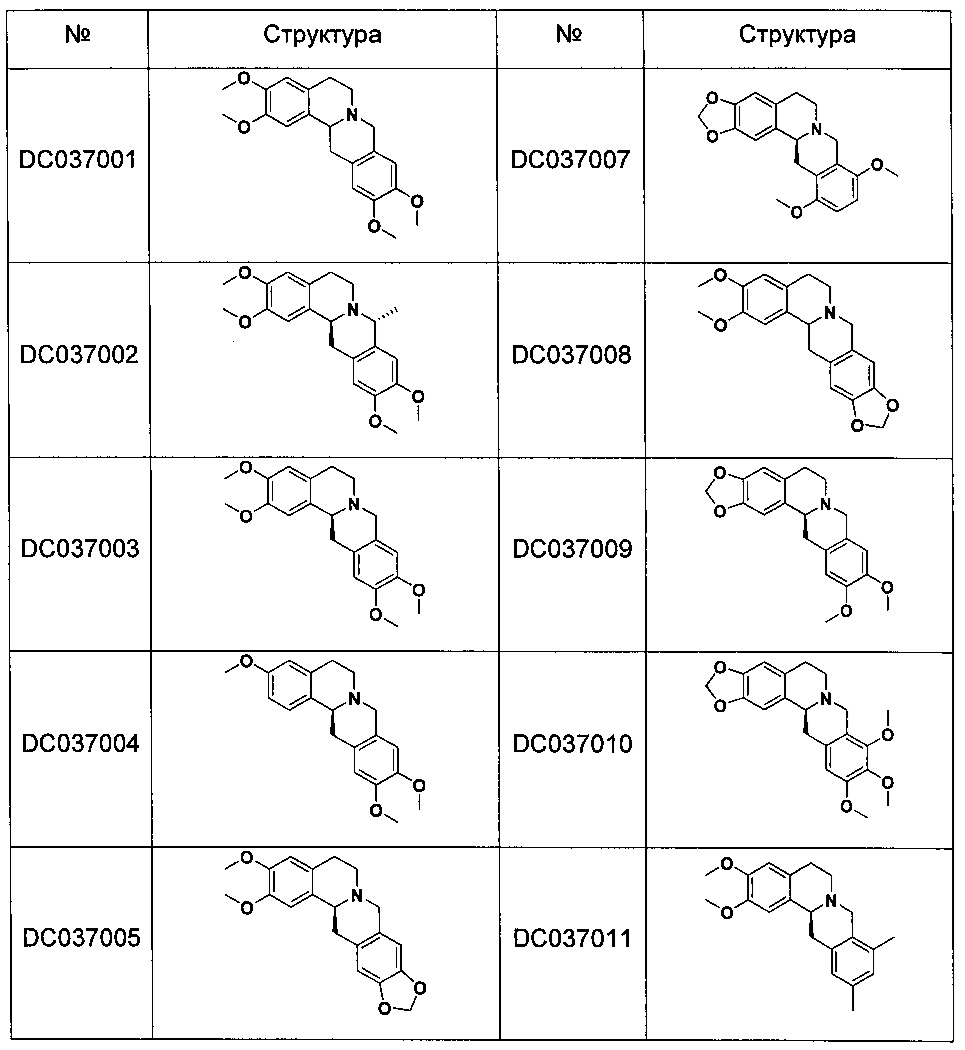
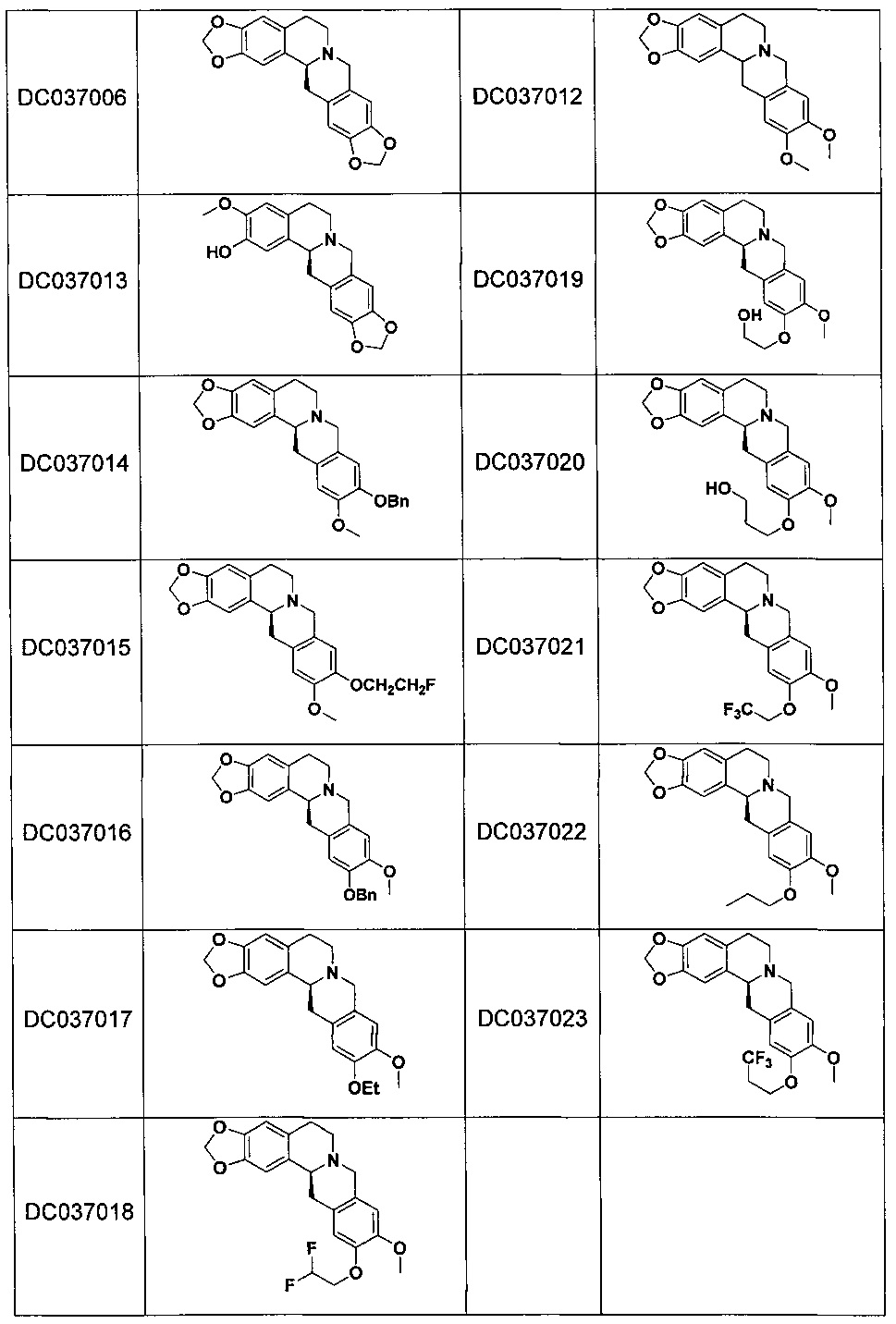
при этом конфигурация неотмеченного хирального атома углерода представляет собой R или S.
В одном воплощении данное лекарственное средство предназначено для селективного связывания с α1A-адренергическими рецепторами.
В одном воплощении IC50 (концентрация полумаксимального ингибирования)(α1B/α1A) составляет больше или равно 2, предпочтительно больше или равно 5, более предпочтительно больше или равно 10, где IC50(α1B/α1A) представляет собой соотношение IC50 указанного лекарственного средства, связывающегося с α1B-адренергическими рецепторами, и IC50 указанного лекарственного средства, связывающегося с α1A-адренергическими рецепторами.
В одном воплощении IC50(α1B/α1A) составляет 2-3000, где IC50(α1B/α1A) представляет собой соотношение IC50 указанного лекарственного средства, связывающегося с α1B-адренергическими рецепторами, и значения IC50 указанного лекарственного средства, связывающегося с α1A-адренергическими рецепторами.
В одном воплощении IC50 указанного лекарственного средства, связывающегося с α1B-адренергическими рецепторами, составляет больше или равно 150 нМ, предпочтительно больше или равно 500 нМ и более предпочтительно больше или равно 1000 нМ.
В одном воплощении указанное лекарственное средство предназначено для селективного ингибирования сокращения гладких мышц мочевыделительной системы.
В одном воплощении указанные гладкие мышцы мочевыделительной системы выбраны из гладких мышц уретры и гладких мышц предстательной железы.
Во втором аспекте изобретения предложен нетерапевтический способ in vitro ингибирования α1A-адренергического рецептора, включающий стадию введения ингибирующего эффективного количества соединения гексагидродибензо[a,g]хинолизина общей формулы (I) или его фармацевтически приемлемой соли, сольвата, стереоизомера, таутомера нуждающемуся в этом субъекту.
В одном воплощении ингибирование является селективным ингибированием α1A-адренергического рецептора.
В одном воплощении объект, подлежащий ингибированию, представляет собой клетку или животную ткань in vitro, экспрессирующую α1A-адренергический рецептор.
В одном воплощении объект, подлежащий ингибированию, представляет собой клетку или животную ткань in vitro, экспрессирующую α1A-адренергический рецептор и α1B-адренергический рецептор, и предпочтительно объект также экспрессирует α1D-адренергический рецептор.
В одном воплощении объект, подлежащий ингибированию, представляет собой гладкую мышцу уретры или гладкую мышцу предстательной железы.
В третьем аспекте изобретения предложен способ получения фармацевтической композиции, содержащей смесь соединения общей формулы (I) или его фармацевтически приемлемой соли, сольвата, стереоизомера или таутомера с фармацевтически приемлемым носителем, с образованием фармацевтической композиции.
В одном воплощении фармацевтически приемлемый носитель выбран из пероральных носителей композиции или инъекционных носителей.
В четвертом аспекте изобретения предложено применение соединения формулы (I) в первом аспекте изобретения для получения ингибитора, селективно связывающегося с α1A-адренергическим рецептором.
В пятом аспекте изобретения предложено применение соединения формулы (I) в первом аспекте изобретения, где соединение применяют в качестве ингибитора для лечения предстательной железы; и/или соединение применяют для антагонистического воздействия на вызванное норэпинефрином сокращение гладких мышц уретры и/или гладких мышц предстательной железы.
В шестом аспекте изобретения предложен ингибитор, селективный в отношении α1A-адренергического рецептора, который содержит ингибирующее эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, стереоизомера, таутомера.
В седьмом аспекте изобретения предложен способ лечения и/или предупреждения доброкачественной гиперплазии предстательной железы, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, стереоизомера, таутомера.
В восьмом аспекте изобретения предложена фармацевтическая композиция для лечения или ингибирования доброкачественной гиперплазии предстательной железы, содержащая (а) фармацевтически приемлемый носитель и (б) соединение формулы (I) или его фармацевтически приемлемую соль, сольват, стереоизомер, таутомер.
В одном воплощении препарат фармацевтической композиции представляет собой пероральный препарат или инъекционный препарат.
Следует понимать, что в настоящем изобретении технические признаки, в особенности, описанные выше и ниже в данном документе (например, раздел Примеры), можно объединять друг с другом, создавая посредством этого новое или предпочтительное техническое решение, которое отдельно не нуждается в точном определении.
Описание графических материалов
Фиг. 1 представляет собой экспериментальную кривую в Примере 4 изобретения, показывающую эффекты положительного лекарственного средства силодозин на уретральное давление крысы (IUP) и давление в периферических артериях (МВР).
Фиг. 2 представляет собой экспериментальную кривую в Примере 4 изобретения, показывающую эффекты DC037009 на уретральное давление крысы (IUP) и давление в периферических артериях (МВР).
На Фиг. 3 показана частота мочеиспускания, средний объем мочеиспускания, общий объем мочеиспускания крыс в группе имитации и группе ВРН в Примере 5 изобретения.
На Фиг. 4 показана частота мочеиспускания, средний объем мочеиспускания, общий объем мочеиспускания крыс в группе имитации, группе солидозина (группа дозировки ВРН-1,3) и DC037009 (группа дозировки ВРН-2,3) в Примере 5 изобретения.
На Фиг. 5 показана кривая концентрация в плазме - время после внутрижелудочного введения и внутривенной инъекции DC037009 крысам в Примере 6 изобретения.
Подробное описание изобретения
Посредством интенсивного и долгосрочного исследования авторы изобретения неожиданно обнаружили, что соединения гексагидродибензо[a,g]хинолизина могут селективно связываться с α1A-адренергическим рецептором и не связываться с α1B-адренергическим рецептором и, таким образом, могут применяться для получения лекарственных средств для контролирования доброкачественной гиперплазии предстательной железы с ослабленными сердечнососудистыми побочными эффектами. На основе данного открытия авторы изобретения осуществили настоящее изобретение.
Определения
Термин "С1-С6 алкокси" относится к прямому или разветвленному алкокси, имеющему 1-6 атомов углерода, такому как метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси и тому подобное.
Термин "С1-С6 алкил" относится к прямому или разветвленному алкилу, имеющему 1-6 атомов углерода, такому как метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил и тому подобное.
Термин "С3-С6 циклоалкил" относится к циклоалкильной группе, имеющей 3-6 атомов углерода, такой как циклопропил, циклобутил, циклопентил, циклогептил и тому подобное.
Термин "С2-С6 алкенил" относится к алкенильной группе, имеющей 1-6 атомов углерода, такой как этенил, пропенил, изопропенил, бутенил, изобутенил, втор-бутенил, трет-бутенил и тому подобное.
Термин "С2-С6 алкинил" относится к алкинильной группе, имеющей 1-6 атомов углерода, такой как этинильная, пропинильная, изо-алкинильная, бутинильная, изо-алкинильная, втор-бутинильная, трет-бутинильная группа и тому подобное.
Термин "сульфонил" относится к группе, имеющей структуру "С1-С6 алкил-SO2-" или "арил-SO2-", такой как метилсульфонил, этилсульфонил, пропилсульфонил, изопропилсульфонил, бутилсульфонил, изо-бутилсульфонил, втор-бутилсульфонил, трет-бутилсульфонил, бензолсульфонил, тозил и тому подобное.
Термин "5-7-членный гетероцикл" относится к циклической структуре, имеющей один или более чем один и предпочтительно 1-3 гетероатома, и указанный гетероцикл является насыщенным или ненасыщенным. В частности, "R1 и R2 могут вместе образовывать гетероцикл" означает, что R1 и R2 образуют гетероциклическое кольцо вместе с углеродной цепью, к которой они присоединены.
Термин "галоген" относится к F, Cl, Br и I.
В том виде, как он используется в данном документе, термин "Rx и Ry (х и y выбраны из 1, 2, 4, 5, 6, 7) могут вместе образовывать замещенный или незамещенный m-членный гетероцикл (m равно 5, 6 или 7)" означает, что Rx, Ry и 1-3 смежных атома углерода вместе образуют замещенный или незамещенный m-членный гетероцикл (m равно 5, 6 или 7).
В том виде, как он используется в данном документе, термин "конфигурация хирального атома углерода представляет собой R или S" означает, что хиральный атом углерода в структурной формуле может представлять собой R конфигурацию, S конфигурацию или их смесь, и предпочтительно представляет собой единственную R конфигурацию или S конфигурацию.
Фармацевтически приемлемая соль, сольват, стереоизомер, таутомер
В том виде, как он используется в данном документе, термин "фармацевтически приемлемая соль" относится к солям, образованным соединением по настоящему изобретению с фармацевтически приемлемой неорганической и органической кислотой. Предпочтительная неорганическая кислота включает (но не ограничивается) следующими кислотами: соляная кислота, бромистоводородная кислота, фосфорная кислота, азотная кислота, серная кислота; и предпочтительная органическая кислота включает (но не ограничивается) следующими кислотами: муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, нафталиндикарбоновая кислота (1,5), азиатиковая кислота, щавелевая кислота, винная кислота, молочная кислота, салициловая кислота, бензойная кислота, пентановая кислота, диэтилуксусная кислота, малоновая кислота, янтарная кислота, фумаровая кислота, пимелиновая кислота, адипиновая кислота, малеиновая кислота, яблочная кислота, сульфаминовая кислота, фенилпропионовая кислота, глюконовая кислота, аскорбиновая кислота, никотиновая кислота, изоникотиновая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота, лимонная кислота и аминокислота.
В том виде, как он используется в данном документе, термин "фармацевтически приемлемый сольват" относится к сольвату, образованному соединением по настоящему изобретению с фармацевтически приемлемым растворителем, где фармацевтически приемлемый растворитель включает (но не ограничивается) следующими растворителями: вода, этанол, метанол, изопропанол, тетрагидрофуран и метиленхлорид.
В том виде, как он используется в данном документе, термин "фармацевтически приемлемый стереоизомер" относится к соединению по настоящему изобретению, в котором хиральный атом углерода представляет собой R конфигурацию или S конфигурацию, или их комбинацию.
Фармацевтическая композиция
Согласно настоящему изобретению также предложена фармацевтическая композиция, которая обладает значительной эффективностью, которая содержит терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли, и один или более чем один фармацевтически приемлемый носитель.
Смесь, образованную самим соединением или его фармацевтически приемлемой солью с фармацевтически приемлемым эксципиентом или разбавителем, можно перорально вводить в форме таблеток, капсул, гранул, порошков или сиропов, или неперорально вводить в форме инъекции. Фармацевтическая композиция предпочтительно содержит от 0,01 до 99 мас. % соединения формулы I или его фармацевтически приемлемой соли в качестве активного ингредиента и более предпочтительно содержит от 0,1 до 90 мас. % активного ингредиента.
Приведенные выше композиции могут быть получены традиционными способами фармации. Примеры доступных фармацевтически приемлемых адъювантов включают эксципиенты (например, производные сахаров, такие как лактоза, сахароза, глюкоза, маннит и сорбит; производные крахмала, такие как кукурузный крахмал, картофельный крахмал, декстрин и карбоксиметилкрахмал; производные целлюлозы, такие как кристаллическая целлюлоза, гидроксипропилцеллюлоза, карбоксиметилцеллюлоза, кальций-карбоксиметилцеллюлоза, натрий-карбоксиметилцеллюлоза; гуммиарабик; декстран; силикатное производное, такое как метасиликат магния-алюминия; фосфатные производные, такие как фосфат кальция; карбонатные производные, такие как карбонат кальция; сульфатные производные, такие как сульфат кальция), связующие вещества (например, желатин, поливинилпирролидон и полиэтиленгликоли), разрыхлители (например, производные целлюлозы, такие как натрий-карбоксиметилцеллюлоза, поливинилпирролидон), смазывающие вещества (например, тальк, стеарат кальция, стеарат магния, спермацет, борная кислота, бензоат натрия, лейцин), стабилизаторы (метилпарабен, пропилпарабен и т.д.), корригенты (традиционные подсластители, кислые агенты и отдушки, и тому подобное), разбавители и инъекционный растворитель (такой как вода, этанол и глицерин, и т.д.).
Соединение по изобретению, его фармацевтически приемлемую соль или его пролекарство или его фармацевтическую композицию можно вводить в разной дозировке, которая меняется в зависимости от возраста, пола, расы и заболеваний.
Новое фармацевтическое применение соединения гексагидродибензо[a,g]хинолизина
Согласно настоящему изобретению предложено применение соединения гексагидродибензо[a,g]хинолизина формулы (I) или его фармацевтически приемлемой соли, сольвата, стереоизомера, таутомера для получения лекарственного средства для лечения и/или предупреждения доброкачественной гиперплазии предстательной железы,
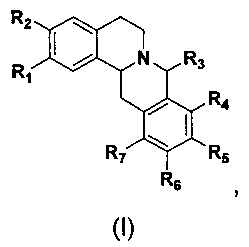
где каждый из R1, R2, R4, R5, R6 и R7 независимо представляет собой водород, гидрокси, галоген, замещенный или незамещенный С1-С6 алкокси, замещенный или незамещенный С1-С6 алкил, замещенный или незамещенный С2-С6 алкенил, замещенный или незамещенный С2-С6 алкинил, замещенный или незамещенный С3-С6 циклоалкил, замещенный или незамещенный бензилокси, при этом заместитель для замещения представляет собой галоген, гидрокси, амино или сульфонил;
R3 представляет собой водород, галоген, незамещенный или галоген-замещенный С1-С6 алкил или незамещенный или галоген-замещенный С1-С6 алкокси;
R1 и R2 могут вместе образовывать замещенный или незамещенный 5-7-членный гетероцикл, при этом заместитель для замещения представляет собой галоген или незамещенный или галоген-замещенный С1-С6 алкил, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
любые два смежных заместителя из R4, R5, R6 и R7 могут вместе образовывать замещенный или незамещенный 5-7-членный гетероцикл, при этом заместитель для замещения представляет собой галоген, незамещенный или галоген-замещенный С1-С6 алкил или незамещенный или галоген-замещенный С1-С6 алкокси, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
конфигурация хирального атома углерода в соединении общей формулы (I) представляет собой R или S.
Предпочтительно, в соединении формулы (I) каждый из R1, R2, R4, R5, R6 и R7 независимо представляет собой водород, гидрокси, галоген, замещенный или незамещенный С1-С4 алкокси, замещенный или незамещенный С1-С4 алкил, замещенный или незамещенный С2-С4 алкенил, замещенный или незамещенный С2-С4 алкинил, замещенный или незамещенный С3-С6 циклоалкил, замещенный или незамещенный бензилокси, при этом заместитель для замещения представляет собой галоген, гидрокси, амино или сульфонил;
R3 представляет собой водород, галоген, незамещенный или галоген-замещенный С1-С4 алкил или незамещенный или галоген-замещенный С1-С4 алкокси;
R1 и R2 могут вместе образовывать замещенный или незамещенный 5-7-членный гетероцикл, при этом заместитель для замещения представляет собой галоген или незамещенный или галоген-замещенный С1-С4 алкил, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
любые два смежных заместителя из R4, R5, R6 и R7 могут вместе образовывать замещенный или незамещенный 5-7-членный гетероцикл, где заместитель для замещения представляет собой галоген, незамещенный или галоген-замещенный С1-С4 алкил или незамещенный или галоген-замещенный С1-С4 алкокси, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
конфигурация хирального атома углерода в соединении общей формулы (I) представляет собой R или S.
Более предпочтительно в соединении формулы (I) каждый из R1, R2, R4, R5, R6 и R7 независимо представляет собой водород, гидрокси, галоген, замещенный или незамещенный С1-С4 алкокси, замещенный или незамещенный С1-С4 алкил или замещенный или незамещенный бензилокси, при этом заместитель для замещения представляет собой галоген, гидрокси или амино;
R3 представляет собой водород, галоген, незамещенный или галоген-замещенный С1-С4 алкил;
R1 и R2 могут вместе образовывать замещенный или незамещенный 5-или 6-членный гетероцикл, при этом заместитель для замещения представляет собой галоген или незамещенный или галоген-замещенный С1-С4 алкил, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
любые два смежных заместителя из R4, R5, R6 и R7 могут вместе образовывать замещенный или незамещенный 5- или 6-членный гетероцикл, при этом заместитель для замещения представляет собой галоген, незамещенный или галоген-замещенный С1-С4 алкил или незамещенный или галоген-замещенный С1-С4 алкокси, и указанный гетероцикл содержит 1-3 гетероатома, выбранных из N, О или S;
конфигурация хирального атома углерода в соединении общей формулы (I) представляет собой R или S.
Еще более предпочтительно в соединении формулы (I) каждый из R1, R2, R4, R5, R6 и R7 независимо представляет собой водород, гидрокси, галоген, замещенный или незамещенный С1-С4 алкокси, замещенный или незамещенный С1-С4 алкил или замещенный или незамещенный бензилокси, где заместитель для замещения представляет собой галоген или гидрокси;
R3 представляет собой водород, галоген, незамещенный или галоген-замещенный С1-С4 алкил;
R1 и R2 могут вместе образовывать 5- или 6-членный гетероцикл, который содержит от 1 до 2 гетероатомов, выбранных из N, О или S;
R5 и R6 могут вместе образовывать 5- или 6-членный гетероцикл, который содержит от 1 до 2 гетероатомов, выбранных из N, О или S; и
конфигурация хирального атома углерода в соединении общей формулы (I) представляет собой R или S.
Галоген представляет собой F, Cl, Br или I.
Наиболее предпочтительные соединения гексагидродибензо[a,g]хинолизина по изобретению, включая их фармацевтически приемлемую соль, сольват, стереоизомер, таутомер, выбраны из следующих соединений:
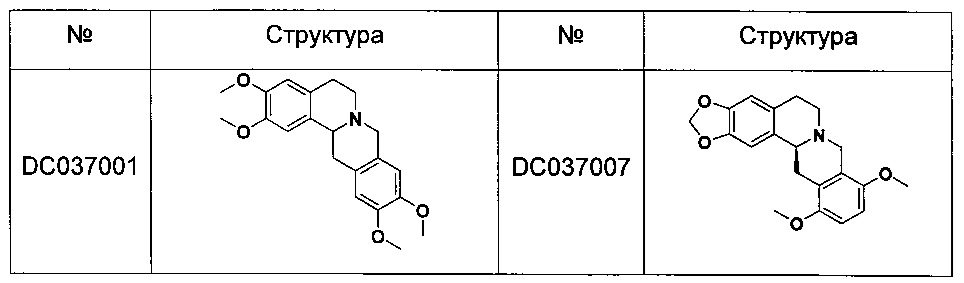
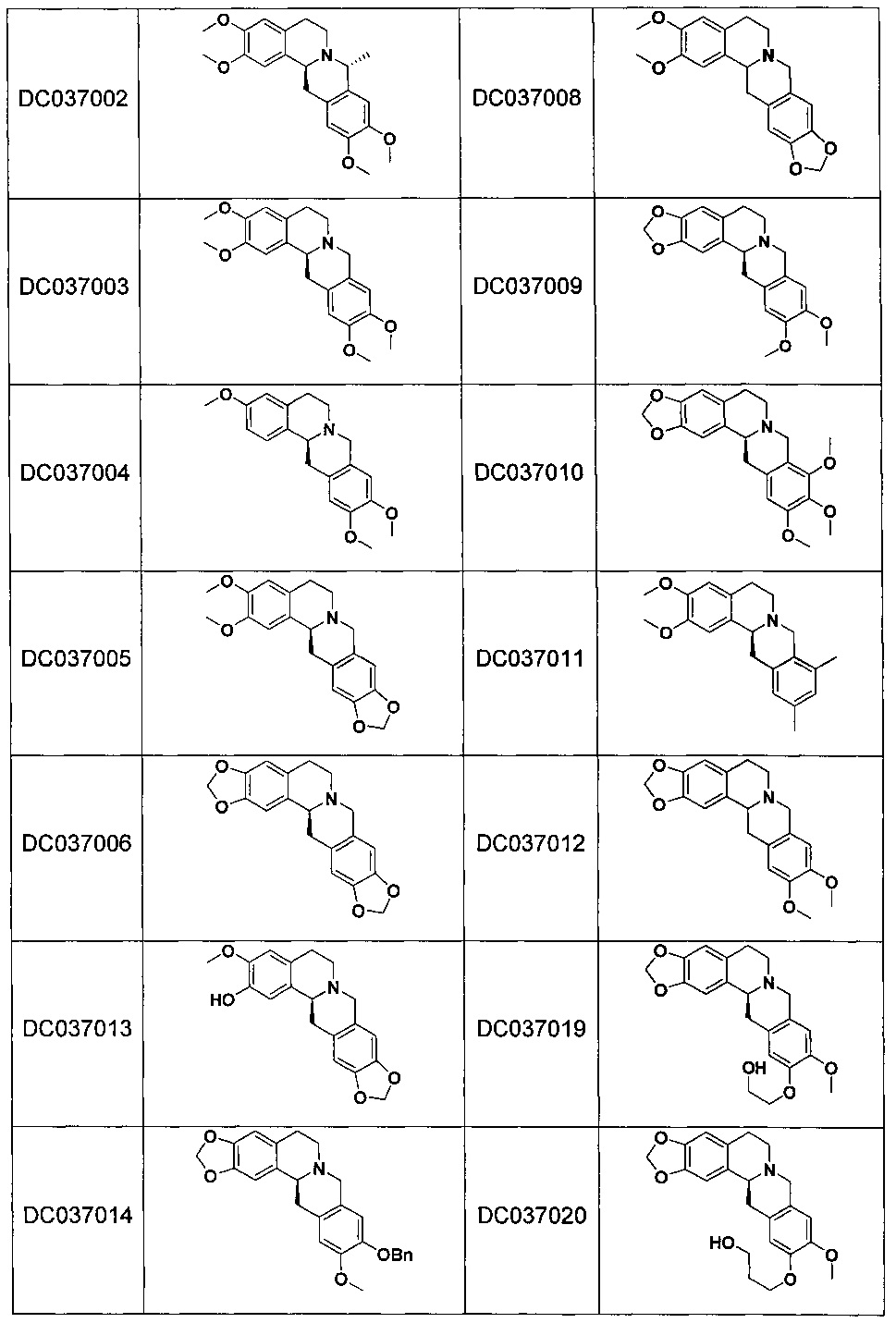
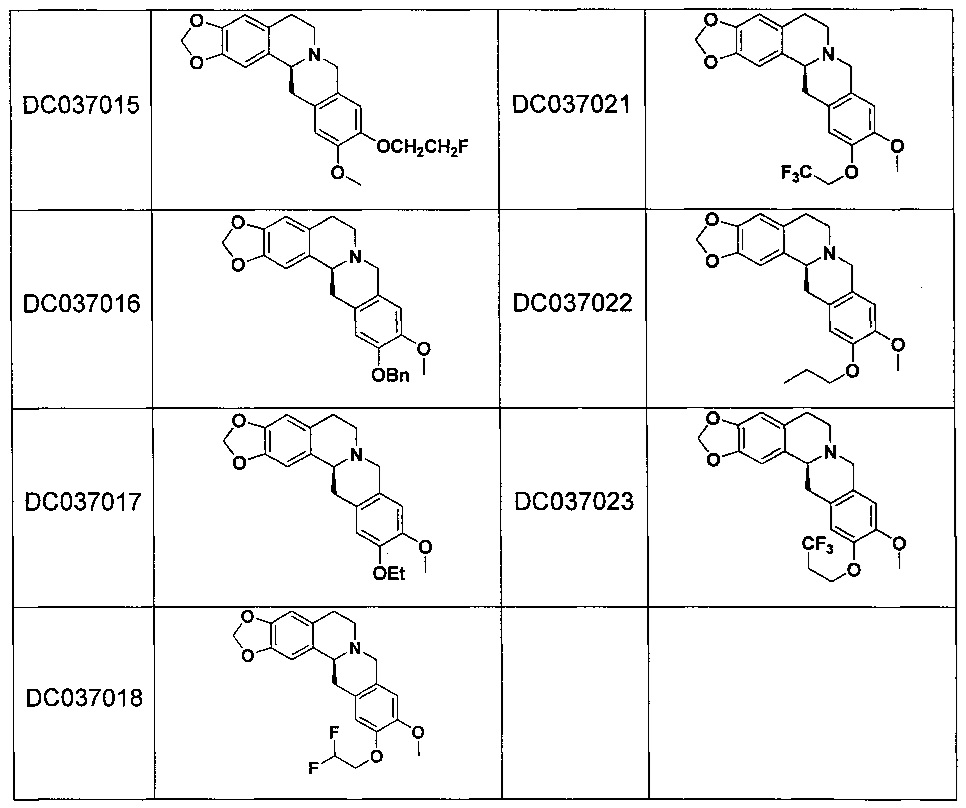
Наиболее предпочтительное соединение по настоящему изобретению представляет собой S-(-)-2,3-метилендиокси-10,11-диметокси-5,8,13,13а-тетрагидро-6H-дибензо[a,g]хинолизин (DC037009), включая его фармацевтически приемлемую соль, сольват, стереоизомер, таутомер.
Фармацевтическая композиция, содержащая соединения гексагидродибензо[a,g]хинолизина
Другим объектом настоящего изобретения является предложение фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, сольват, стереоизомер, таутомер, пролекарство и фармацевтически приемлемый носитель.
Настоящее изобретение относится к лекарственному средству для лечения и/или предупреждения доброкачественной гиперплазии предстательной железы, которое получают, используя соединения гексагидродибензо[a,g]хинолизина, как показано в формуле (I), или их фармацевтически приемлемую соль, сольват, стереоизомер, таутомер, пролекарство или смесь в качестве активного ингредиента.
Лекарственное соединение можно вводить в организм, например, в мышцу, внутрикожно, подкожно, внутривенно, слизистую ткань посредством инъекции, спрея, назальной ингаляции, глазных капель, проникновения, абсорбции, физически или химически опосредованного способа; или лекарственное средство смешивают с другими веществами или покрывают ими перед введением в организм.
При необходимости в упомянутое выше лекарственное средство можно добавлять один или более чем один фармацевтически приемлемый носитель. Носители включают разбавители, эксципиенты, наполнители, связующие вещества, увлажнители, разрыхлители, стимуляторы абсорбции, поверхностно-активное вещество, адсорбционный носитель, смазывающее вещество и тому подобное, которые являются традиционными в фармацевтической области.
Соединения гексагидродибензо[a,g]хинолизина, как показано в формуле (I), или их фармацевтически приемлемую соль, сольват, стереоизомер, таутомер или пролекарство в качестве активного ингредиента применяют отдельно или в комбинации с другими лекарственными соединениями, или смешивают со вспомогательными веществами и готовят в виде различных препаратов, включая таблетки, порошки, пилюли, инъекции, капсулы, пленки, суппозитории, пасту, гранулы и т.д., но не ограничиваясь ими. Приведенные выше разные препараты можно получать согласно традиционным способам в области фармации.
Настоящее изобретение будет дополнительно проиллюстрировано ниже со ссылкой на конкретные примеры. Следует понимать, что данные примеры даны только для иллюстрации изобретения, не для ограничения объема изобретения. Экспериментальные способы без конкретных условий, описанные в следующих примерах, обычно осуществляют в традиционных условиях (например, условия, описанные Sambrook et al., Molecular Cloning: A Laboratory Manual (New York: Cold Spring Harbor Laboratory Press, 1989)) или в соответствии с инструкциями изготовителя. Если не указано иное, доли и процентное содержание рассчитаны по массе. Реагенты и биологические вещества имеются в продаже, если нет подробного описания.
Пример 1: Исследование активности на клетке и селективности лекарственных средств
Клетки HEK293, стабильно экспрессирующие α1A-AR/Gα16, α1B-AR/Gα16, α1D-AR/Gα16, высевали в 96-луночный планшет и культивировали в течение 24 часов. Затем среду удаляли, и 40 мкл сбалансированного солевого раствора Хэнкса (HBSS: содержащего 5,4 мМ KCl, 0,3 мМ Na2HPO4, 0,4 мМ KH2PO4, 4,2 мМ NaHCO3, 1,3 мМ CaCl2, 0,5 мМ MgCl2, 0,6 мМ MgSO4, 137 мМ NaCl, 5,6 мМ D-глюкозу и 250 мкМ сульфинпиразон, рН 7,4), содержащего 2 мкМ Fluo-4 AM, добавляли в каждую лунку и инкубировали при 37°С в течение 45 мин. Краситель удаляли и добавляли 50 мкл HBSS, содержащего либо тестируемое соединение, либо 1% DMSO (диметилсульфоксид) (отрицательный контроль). После инкубации при комнатной температуре в течение 10 мин для считывания значения использовали микропланшет-ридер Flex Station 3. В конкретные моменты времени 25 мкл агониста (фенилэфрин, конечная концентрация 30 нМ) дозировали в лунку автоматически, используя ридер, и изменение интенсивности флуоресценции красителя, обусловленное изменением внутриклеточной концентрации кальция, выявляли при длине волны возбуждения 485 нм и длине волны эмиссии при 525 нм.
После инкубации с разными лекарственными средствами степень реакции клеток на 1-AR агонист фенилэфрин рассчитывали следующим образом:
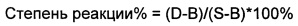 ;
;
где D представляет собой пик сигнала мобилизации кальция, вызванный фенилэфрином после инкубации с тестируемым лекарственным средством, В представляет собой пик сигнала мобилизации кальция, вызванный фенилэфрином после инкубации с 10 мкМ положительным контролем - лекарственным средством тамсулозином, S представляет собой пик сигнала мобилизации кальция, вызванный фенилэфрином после инкубации с отрицательным контролем 1% DMSO.
Степень реакции одного и того же лекарственного средства при разных дозах тестировали с использованием программного обеспечения GraphPad Prism посредством нелинейного регрессионного анализа, и получали кривую доза-ответ, и измеряли значения IC50. Данные получали из трех отдельных экспериментов, и каждый эксперимент включал три дублирующие лунки.
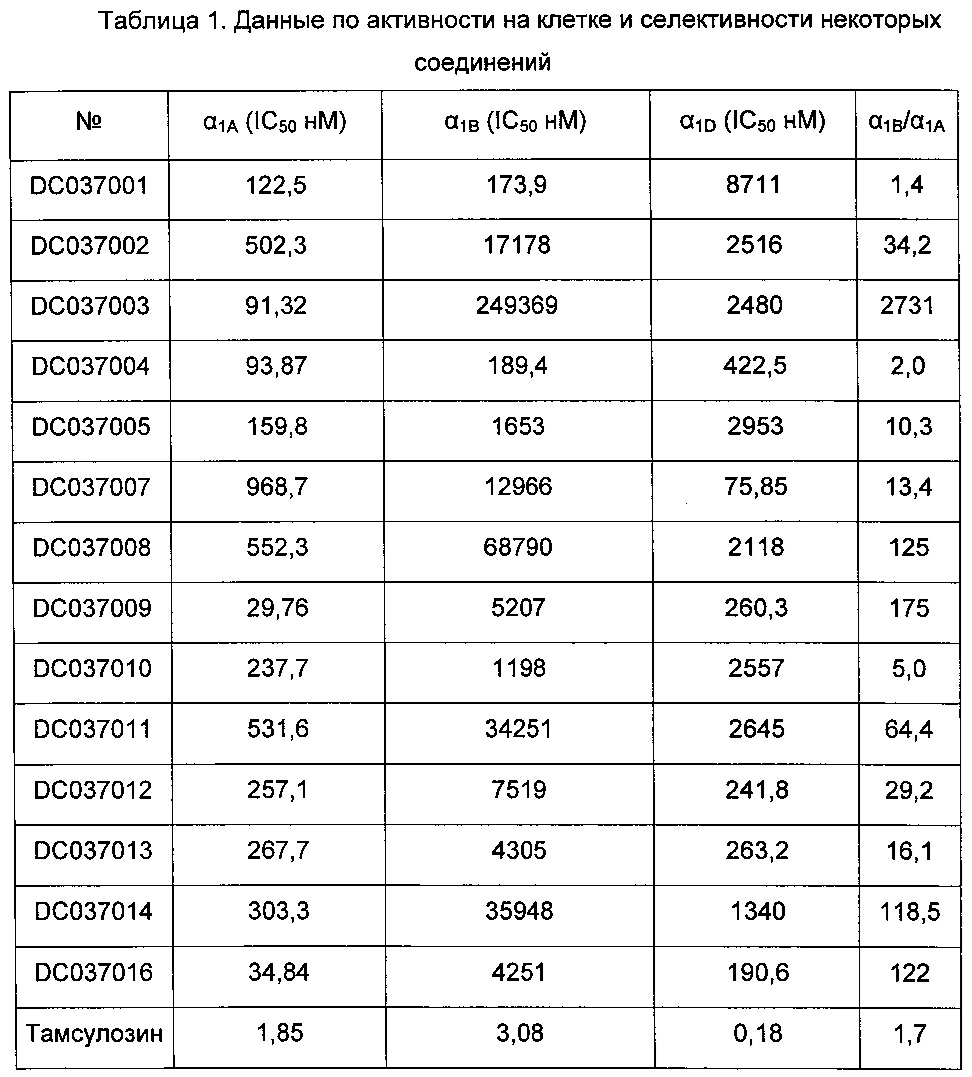
Эксперименты на клетках показали, что, несмотря на то, что активность соединений, включая DC037009, в отношении α1A-рецептора была немного слабее, чем активность положительного лекарственного средства тамсулозин, селективность соединений в отношении α1A-рецептора была значительно лучше, чем селективность тамсулозина, указывая на то, что данные соединения заслуживали дальнейшего исследования.
Пример 2: Исследование активности связывания лекарственных средств с рецептором
Для изучения активности связывания с рецептором использовали анализ Tag-lite. Анализ Tag-lite представлял собой сочетание технологии SNAP-Tag и HTRF (гомогенная флуоресценция с временным разрешением), и его использовали для анализа поверхностных рецепторов на живой клетке. SNAP представляли собой небольшие меченные слитые белки, которые могли необратимо, специфично и ковалентно связываться с субстратами через бензилметил, а субстраты представляли собой бензилметилгуанин и бензилметилцитозин. Субстраты и разные красители образовывали производные, и посредством ковалентной реакции SANP метили красителем. Плазмиду конструировали с использованием pSNAP и α1-AR кодирующего гена. После трансфекции в клетки экспрессировался N-концевой слитый белок SNAP и α1-AR. Субстрат и флуоресцентный краситель HTRF (донорное соединение Lumi4-Tb) образовывали производное, которое вступало во взаимодействие со слитым белком SANP-α1-AR так, что α1-AR метился Tb. После добавления лиганда, меченного акцепторным флуоресцентным красителем, проводили эксперимент по связыванию рецептора и лиганда.
Клетки HEK293, стабильно экспрессирующие SNAP-α1A-AR, SNAP-α1B-AR, SNAP-α1D-AR, высевали в 3 см чашки и культивировали в течение 24 часов. Добавляли 10 нМ Tag-lite SNAP Lumi4Tb и инкубировали в течение 1 ч в инкубаторе. Клетки отделяли и промывали в буфере для мечения 4 раза и затем добавляли в 384-луночный планшет в объеме 10 мкл на лунку. Добавляли 5 мкл тестируемого соединения и 5 мкл известного лиганда, меченного красным флуоресцеином, с постепенно увеличивающейся концентрацией. После инкубации при комнатной температуре в течение 1 часа изменения интенсивности флуоресценции, обусловленные изменениями клеточного рецептора и лиганда, выявляли, используя 665 нм (акцептор) и 620 (донор) одновременно.
После инкубации с разными лекарственными средствами степень реакции клеток на 1-AR известный лиганд, меченный флуоресцеином, рассчитывали следующим образом:
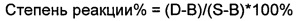 ,
,
где D представляет собой пик сигнала, вызванный известным лигандом, меченным флуоресцеином, после инкубации с тестируемым лекарственным средством, В представляет собой пик сигнала, вызванный 10 мкМ положительным контролем - лекарственным средством тамсулозин, и S представляет собой пик сигнала, вызванный отрицательным контролем DMSO.
Степень реакции одного и того же лекарственного средства при разных дозах тестировали, используя программное обеспечение GraphPad Prism посредством нелинейного регрессионного анализа, и получали кривую доза-ответ, и измеряли значения Ki. Данные получали из трех отдельных экспериментов, и каждый эксперимент включает три дублирующие лунки.
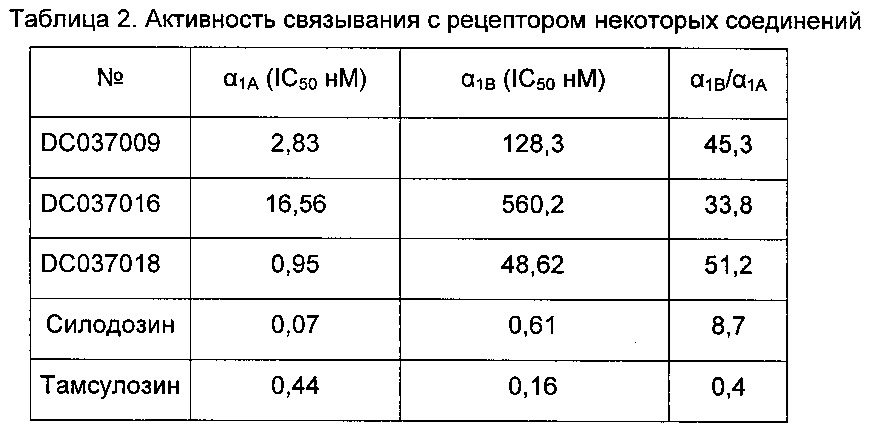
Эксперименты на клетках показали, что, несмотря на то, что активность соединений, включая DC037009, в отношении α1A-рецептора была немного слабее, чем активности положительного лекарственного средства тамсулозин и силодозин, селективность соединений в отношении α1B была значительно лучше, чем селективность лекарственных средств, используемых в качестве положительного контроля, указывая на то, что данные соединения заслуживали дальнейшего исследования.
Пример 3: Исследование активности лекарственных средств в отношении тканей, выделенных из животного
Эксперимент in vitro с гладкой мышцей уретры:
Здоровых крыс-самцов Вистар ударяли головой для того, чтобы вызвать потерю сознания. Брюшную полость и лонное сочленение быстро разрезали так, чтобы быстро удалить простатическую уретру, которою сразу же помещали в чашку, наполненную 4°С раствором Kerbs-Henseleit (раствор K-Н). Окружающую ткань осторожно отделяли, и получали полоски мышцы примерно 3-5 мм, и нижний конец фиксировали на 20 мл термостатической бани, а верхний конец присоединяли к датчику давления. Питательная жидкость представляла собой раствор K-Н, и осуществляли продувку карбогеном (95% кислорода и 5% углекислого газа), и температура бани составляла 37°С, и прикладываемое давление покоя составляло 0,5 г. Питательную жидкость заменяли каждые 6-8 мин. До стабилизации исходного уровня, в трубку бани добавляли норэпинефрин в конечной концентрации 3×10-4 моль/л. Когда кривая сокращения достигала наивысшей точки, сразу же осуществляли промывку для восстановления исходных уровней. При стабилизации исходного уровня, соответственно, добавляли тестируемое лекарственное средство в разных концентрациях и инкубировали 3-5 мин. В трубку бани добавляли 60 мкл 1×10-1 М норэпинефрина в конечной концентрации 3×10-4 моль/л, и записывали кривую сокращения.
Эксперимент с гладкими мышцами сосудов in vitro:
Здоровых крыс-самцов Вистар ударяли головой для того, чтобы вызвать потерю сознания. Брюшную полость быстро разрезали, и вынимали грудную аорту. Прилежащую ткань отделяли и нарезали на 2-3 мм сосудистые кольца. Эксперимент проводили таким же образом, как эксперимент с уретральной гладкой мышцей. Прикладываемое давление покоя составляло 1 г, и при стабилизации исходного уровня в трубку бани добавляли норэпинефрин в конечной концентрации 10-7 моль/л. Когда кривая сокращения достигала наивысшей точки, сразу же осуществляли промывку для восстановления исходных уровней. При стабилизации исходного уровня, соответственно, добавляли тестируемое лекарственное средство в разных концентрациях и инкубировали 3-5 мин. В трубку бани добавляли 20 мкл 1×10-4 М норэпинефрина в конечной концентрации 10-7 моль/л и записывали кривую сокращения.
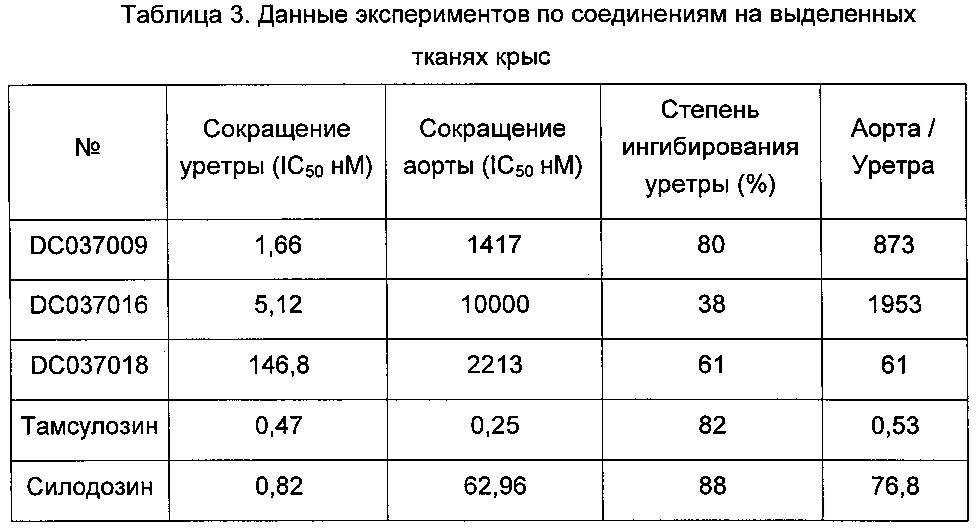
Соединение DC037009 демонстрировало очевидное антагонистическое действие (противодействие) на вызванное норэпинефрином сокращение гладкой мышцы уретры (IC50=1,66 нМ), которое значительно сильнее, чем ингибирование вызванного норэпинефрином сокращения гладких мышц сосудов, и показатель селективности в отношении рецептора достигал 873, и степень подавления уретры была сравнима со степенью подавления положительных лекарственных средств. Селективность DC037009 была лучше, чем селективности тамсулозина и силодозина, поскольку она в 1671 раз превышала селективность тамсулозина и в 11 раз - селективность силодозина. Это указывало на то, что было очень важно уменьшать побочные эффекты существующих лекарственных средств.
Пример 4: Фармакологическое исследование
Крысам-самцам SD (200-450 г) давали наркоз путем интраперитонеального введения уретана (1,25 г/кг). Мочевой пузырь, предстательную железу и уретру выводили на поверхность через среднюю линию живота и разрезание лонного сочленения. Катетер помещали в простатическую уретру через купол мочевого пузыря и закрепляли на шейке мочевого пузыря (везико-уретральное соединение) с помощью шелковой нити. Дистальная уретра под лонной областью была закрыта. Другую часть катетера соединяли с датчиком давления для измерения внутриуретрального давления (IUP). После отделения двенадцатиперстной кишки и вскрытия на уровне примерно 2 см от привратника желудка, интубирования и наложения кисетного шва давали тестируемый образец. Затем осуществляли срединное надрезание сонной артерии, отделяли сонную артерию и измеряли артериальное кровяное давление посредством датчика давления, присоединенного к артериальной канюле. Будьте осторожны с тем, чтобы не повредить нервы и кровеносные сосуды на протяжении процедуры. После операции инъецировали небольшое количество (0,1~0,2 мл) солевого раствора для уравновешивания внутриуретрального давления на уровне примерно 10 см водного столба. Затем осуществляли внутривенную инъекцию с концентрацией 30 мкг/мл гидрохлорида (РНЕ) 1 мл/кг перед введением и через 5 мин, 30 мин, 1 ч, 1,5 ч, 2 ч, 3 ч после введения для того, чтобы вызвать повышение внутриуретрального давления. (Если эффект был стабильным или ослабленным, эксперимент заканчивали). Наблюдали за изменениями внутриуретрального давления и кровяного давления, вызванными фенилэфрином у крыс, и их регистрировали в разные моменты времени перед и после введения.
Экспериментальные результаты показали, что солидозин и DC037009 могли дозозависимо ингибировать вызванное фенилэфрином повышение внутриуретрального давления (IUP) и кровяного давления в периферических артериях (МВР). Ингибирование вызванного фенилэфрином повышения внутриуретрального давления имело следующий вид: DC037009 > Силодозин. Ингибирование вызванного фенилэфрином повышения кровяного давления имело следующий вид: Силодозин > DC037009. Селективность в отношении уретры имела следующий вид: DC037009 > Силодозин. Кроме того, начальная доза DC037009 была значительно ниже, чем доза положительного лекарственного средства силодозин (см. Фиг. 1 и Фиг. 2).
Пример 5: Фармакодинамический эксперимент
Создание модели ВРН на крысах: 6-недельные крысы-самцы SD (n=49) (масса тела примерно 200 г), где 37 крыс составляли модельную группу ВРН и им ежесуточно подкожно инъецировали тестостерона пропионат, а другие 12 крыс составляли группу имитации и им инъецировали оливковое масло в течение 4 недель. Спустя 4 недели 49 крыс получали зондовое кормление в дозировке 30 мл/кг воды, и для каждой крысы отслеживали и регистрировали мочеиспускание (регистрирование каждого диуреза и частоты мочеиспускания за два часа).
Согласно приведенным выше результатам верификации, прежде всего, исключали крыс, имеющих отклоняющееся от нормы мочеиспускание. Затем, нормальных крыс модели ВРН разделяли на 2 группы, по 12 крыс в каждой группе, и 12 крыс группы имитации определяли как контрольную группу. Принципы группирования были следующими: имелись значительные различия массы тела, частоты мочеиспускания в пределах двух часов, единичного диуреза и общего количества мочеиспускания между двумя группами модели ВРН и группой имитации, и отсутствовали значительные различия между двумя группами модели ВНР. Результаты показали, что две группы крыс модели ВРН значительно отличаются от группы имитации по массе тела, частоте мочеиспускания в пределах двух часов и единичному диурезу, в то время как различие в частоте мочеиспускания и общем количестве мочеиспускания отсутствовало (Фиг. 3).
Эксперимент по верификации эффективности. В эксперименте имелось 7 групп: группа имитации, группа силодозина (ВРН-1,3-групповая доза) и группа DC037009 (ВРН-2,3-групповая доза). Крысы получали зондовое кормление в дозировке 5 мл/кг, и каждую дозу соединения растворяли CMCNa (натрий-карбоксиметилцеллюлоза). Спустя 20 мин, крысы получали зондовое кормление в дозировке 30 мл/кг воды. Временной интервал исследования разных доз одного и того же соединения составлял более 24 часов.
И частота мочеиспускания и единичный диурез крыс групп солидозина и DC037009 показали дозозависимую связь. DC037009 было сравнимо с положительным лекарственным соединением, и начальная доза составляла 1 мг/кг. Фармакодинамический эксперимент доказал, что, при определенном общем диурезе, соединение DC037009 могло значительно увеличивать единичный диурез и сокращать частоту мочеиспускания (Фиг. 4).
Пример 6: Фармакокинетическое исследование
Введение крысам лекарственного средства через зондовое кормление. 3 здоровых крыс SD (самец, с массой 200-220 г) держали без пищи в течение 12 часов перед введением, но им позволяли пить воду. Соединение DC037009 получали с 10% DMSO/10% Tween 80/80% солевым раствором и вводили в дозе 20 мг/кг, и объем дозирования составлял 10 мл/кг.
Внутривенное введение крысам. 3 здоровых крыс SD (самец, с массой 200-220 г) держали без пищи в течение 12 часов перед введением, но им позволяли пить воду. Соединение DC037009 получали с 10% DMSO/10% Tween 80/80% солевым раствором и вводили через инъекцию в хвостовую вену крысы в дозе 10 мг/кг, и объем дозирования составлял 5 мл/кг.
Введение через зондовое кормление: 0,25, 0,5, 1,0, 2,0, 3,0, 5,0, 7,0, 9,0 и 24 ч после введения;
Внутривенное введение: 5 мин, 0,25, 0,5, 1,0, 2,0, 3,0, 5,0, 7,0, 9,0 и 24 часа после введения.
В упомянутые выше моменты времени 0,3 мл крови, полученной из постглазного венозного сплетения крысы, помещали в гепаринизированную пробирку, которую центрифугировали при 11000 об./мин. в течение 5 мин, и плазму отделяли и замораживали в холодильнике при -20°С.
После введения крысам лекарственного средства посредством зондового кормления в дозе 20 мг/кг DC037009 время достижения максимальной концентрации в плазме в крысах, Tmax, составляло 0,67±0,29 ч, максимальная концентрация Cmax составляла 453±147 нг/мл, площадь под кривой "концентрация в плазме - время" AUC0-t составляла 2867±798 нг··ч/мл, и период полувыведения t1/2 составлял 3,26±0,82 ч.
После введения 10 мг/кг DC037009 посредством внутривенной инъекции, AUC0-t составляла 4196±141 нг·ч/мл, t1/2 составляло 5,44±0,85 ч, скорость плазменного клиренса CL составляла 2,38±0,08 л/ч/кг и объем распределения в равновесном состоянии Vss составлял 3,49±0,24 л/кг (см. Фиг. 5).
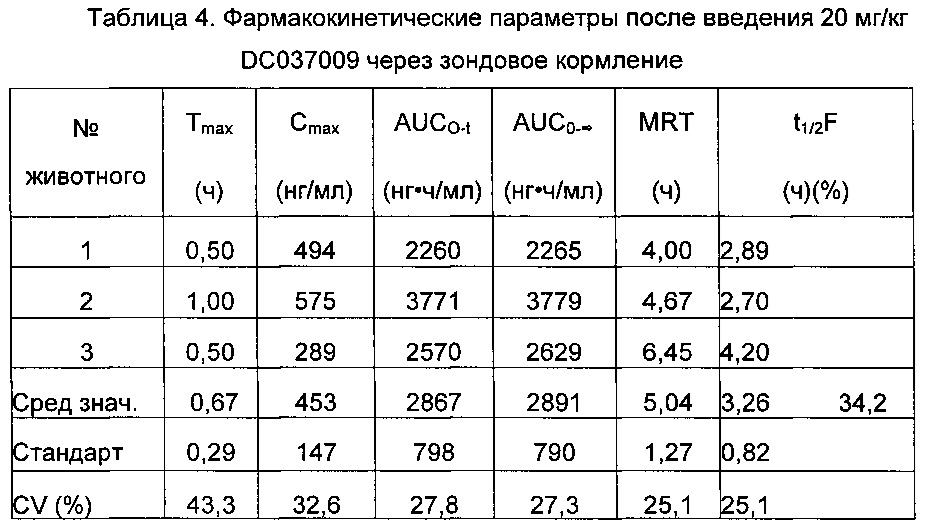
В том, что качается биодоступности и периода полувыведения, период полувыведения и биодоступность соединения DC037009 в крысах составляли 3,26 ч и 34,2%, соответственно, и AUC0-t составляла 2260 нг⋅ч/мл. Предварительные результаты экспериментов по фармакокинетике показали, что данный класс соединений обладал лучшими фармакокинетическими свойствами.
Пример 7: Сравнительное исследование in vitro метаболизма DC037009 у разных видов
Общий объем каждой инкубационной системы составлял 200 мкл, и среда представляла собой 100 мМ фосфатный буферный раствор (PBS, рН 7,4), содержащий конечную концентрацию 3 мкМ DC037009 и 2 мМ NADPH (никотинамидадениндинуклеотидфосфат, восстановленный). Для инкубации использовали 37°С водяную баню. После 3 минут преинкубации, разные белки микросом печени добавляли в смесь буфер-субстрат-кофакторы для начала реакции. Концентрация микросомального белка печени разных видов составляла 1,0 мг/мл. После реагирования в течение 60 мин реакцию останавливали изоволюметрическим ледяным ацетонитрилом. Общий объем холостой пробы составлял 200 мкл, и среда представляла собой 100 мМ фосфатный буферный раствор (PBS, рН 7,4), содержащий конечную концентрацию 3 мкМ DC037009 и термоинактивированные микросомальные белки. Все образцы инкубации делали в двойной повторности.
150 мкл образец из каждого двойного образца инкубации смешивали. Добавляли 300 мкл ацетонитрила, смешивали путем перемешивания на вортексе в течение 1 мин и центрифугировали 5 мин (11000 об./мин.). Супернатант помещали в 10 мл пробирку и сушили при 40°C с потоком азота. Остаток растворяли 100 мкл смеси метанол/вода (1:9, об/об). 10 мкл образец отбирали для анализа UPLC /Q-TOF MS (сверхпроизводительная жидкостная хроматография/квадрупольная времяпролетная масс-спектрометрия).
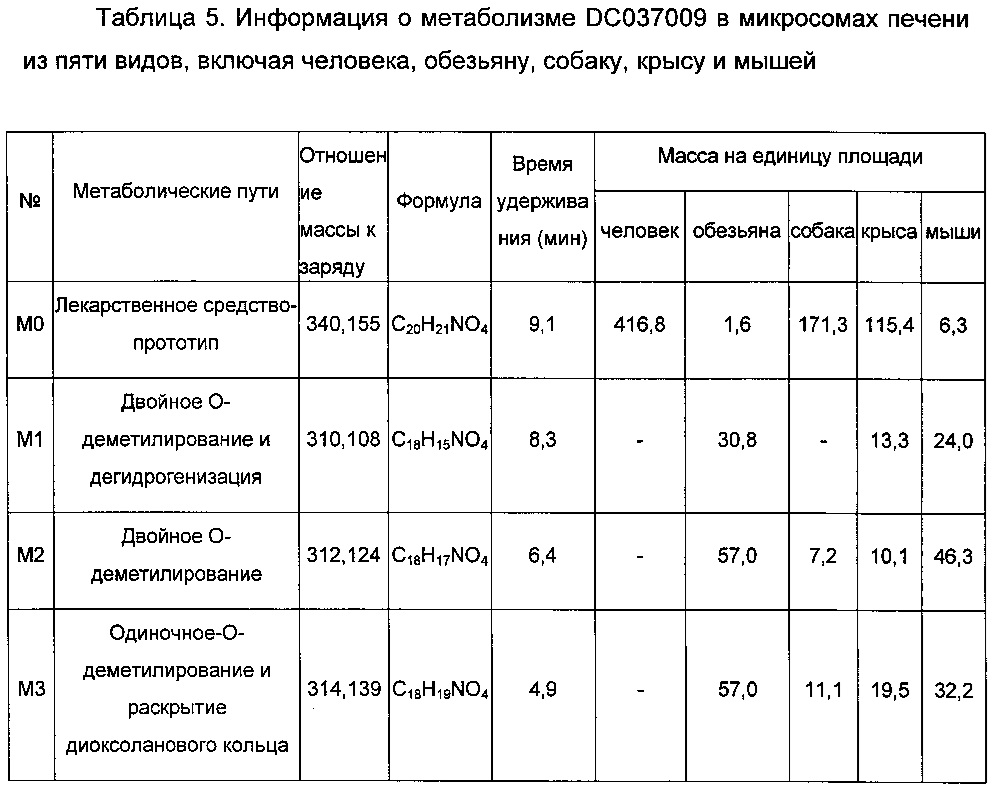
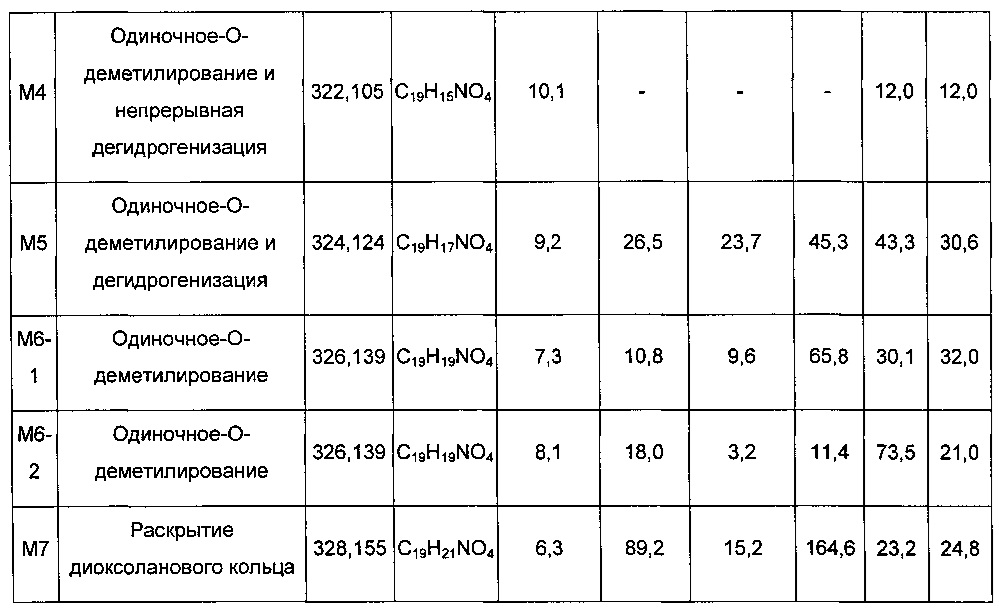
Метаболизм DC037009 в микросоме печени обезьяны и мыши был нестабильным, и выявляли только небольшое количество лекарственного средства-прототипа. Метаболическая устойчивость в микросоме печени собаки и крысы занимала второе место, и метаболическая устойчивость в микросоме печени человека была относительно выше. После 60 мин инкубации приблизительно 26% лекарственного средства-прототипа метаболизировалось. Выявляли четыре типа метаболитов в микросоме печени человека, семь типов метаболитов выявляли в микросоме печени обезьяны, шесть типов метаболитов выявляли в микросоме печени собаки, восемь типов метаболитов выявляли в микросоме печени крысы и восемь типов метаболитов выявляли в микросоме печени мыши. Продукция каждого из метаболитов была NADPH-зависимой. Главный метаболический путь DC037009 в микросоме печени человека представлял собой О-деметилирование, раскрытие диоксоланового кольца и дегидрогенизацию, и относительные доли образованных метаболитов показали, что микросомы печени человека и собаки обладали большим сходством.
Результаты in vitro метаболического различия разных видов показало, что, несмотря на то, что метаболизмы соединения DC037009 были нестабильными в микросомах клеток печени обезьяны и мыши, типы метаболитов в микросомах клеток печени человека явно уменьшались, и стабильность была значительно улучшена, и после 60 мин инкубации только 26% лекарственного средства-прототипа метаболизировалось. Это дополнительно проиллюстрировало то, что DC037009 обладало хорошими метаболическими свойствами лекарственного средства и обладало потенциалом для того, чтобы стать лекарственным средством.
Пример 8: Оценка безопасности

Антагонистическая активность DC037009 в отношении α1А-рецептора в 272 раза превышала его ингибиторную активность в отношении hERG, указывая на то, что соединение имело слабый ингибиторный эффект на сердце и менее вероятно вызывало побочные эффекты на сердце.
Эффект DC037009 на сокращение семенного пузырька, вызванное электростимуляцией подчревного нерва у нормальных крыс
Экспериментальные результаты показали, что DC037009 не имело значительного ингибирующего эффекта на амплитуду сокращения и площадь под кривой сокращения в семенном пузырьке крысы, вызванного электростимуляцией (Р>0,05), указывая на то, что тестируемое соединение не влияло на семенной пузырек крыс с воздействием на его функцию семяизвержения. Это дополнительно доказало, что побочный эффект данного соединения возможно небольшой.
Эксперимент по острой токсичности DC037009 на крысах SD в результате перорального зондового кормления
Крысам SD вводили соединение DC037009 с однократной дозой 500 и 1000 мг/кг посредством перорального зондового кормления. Смерти крыс не наблюдали и после анатомирования не наблюдали явных изменений, связанных с лекарственным средством, и максимальная переносимая доза (MTD) составляла 1000 мг/кг.
Эксперимент по токсичности повторной дозы DC037009 на крысах SD в течение 14 суток
Крысам SD перорально вводили лекарственное средство в дозировке 0, 100 и 200 мг/кг в течение 14 суток, и не наблюдали смерти крыс.
По сравнению с вспомогательной контрольной группой, результат тестирования показателей крови и коагуляции и биохимических показателей сыворотки у животных, которым давали 100 и 200 мг/кг, показал, что изменения показателей крови и коагуляции и изменения биохимических показателей сыворотки не были дозозависимыми и находились в нормальном диапазоне лабораторных показателей. Вследствие этого, не было показано какого-либо токсикологического значения. В макроскопической анатомии все животные не показывали патологических изменений, связанных с вводимым лекарственным средством.
Результаты тестирования острой токсичности и подострой токсичности показали, что токсичность соединения DC037009 у крыс была низкой, и оно было безопасным и надежным. В то же время, соединение DC037009 демонстрировало большое терапевтическое окно, указывая на то, что данное соединение имеет большую ценность для применения.
Пример 9: Фармацевтическое исследование соединения DC037009 Растворимость соединения: 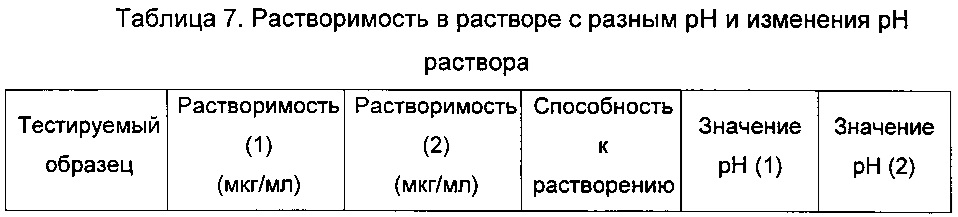
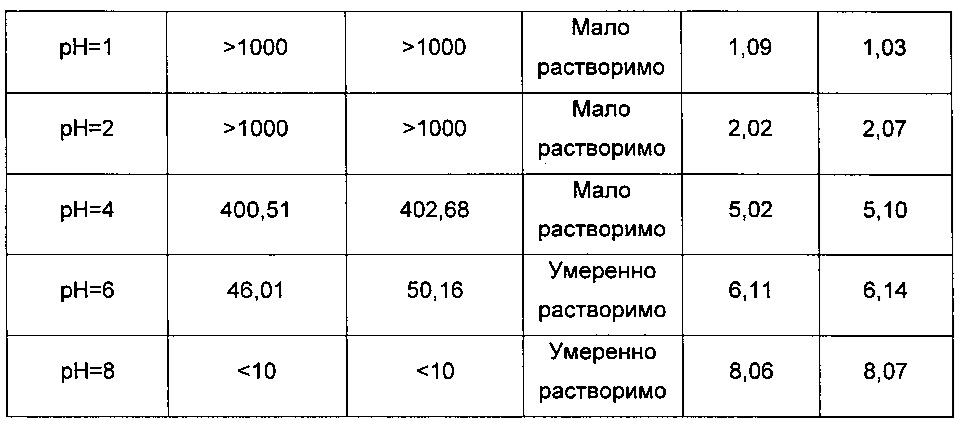
Растворимость в кислом растворе была выше, чем растворимость в щелочном растворе, и рН раствора повышался после добавления соединения DC037009.
Стабильность соединения и определение pKa:
Соединение DC037009 нагревали при 80°С в течение 24 часов, и чистота соединения только варьировала на 0,5%. pKa соединения в разных растворителях составляли pKa (H2O)=7,28, и pKa (СН3ОН)=6,81.
Приведенные выше фармацевтические данные показали, что соединение DC037009 имеет основные свойства для того, чтобы стать лекарственным средством и имеет возможные применения.
Все документы, на которые ссылаются в настоящем изобретении, включены путем ссылки, как если бы каждая ссылка приводилась отдельно в качестве ссылки в настоящей заявке. Кроме того, следует понимать, что после прочтения идей настоящего изобретения, описанных выше, специалист в данной области может вносить разные изменения или модификации изобретения, и данные эквивалентные формы также попадают в объем, как определено прилагаемой формулой изобретения настоящей заявки.
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/bc46ffa77f8e811356aee6268f87ccc9.png)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/5e236127c504567b9962ad890deddfe9.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/ef8d8460ff38e58da883a4e4343754bd.png)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/55514a05c6910841133f0d5442be8601.png)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/16b03748d1155ceba9ede8ea4658aa45.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/541393bded6c01be2a816073000522f1.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/b6fa915d6035947e602cccbc01045fbe.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/f990535297eb8ec0f5537ff06ebacfbf.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/4a3c24ae111c6c6a1cdf597f515925c7.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/2fccc77a51098f7e06fedaeaeffeb306.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/f5a44009e8a2a4bbec5d34c44b5579e1.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/3c7d0c81b6adaa69587d5e9540b95272.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/921f547e1b67aa3f41fe67a815b8d234.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/d0ebd1a8a60854f3f017636f1a201bae.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/fc27e3a2109dbed6f99db6fce79600ff.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/75cc9521ff115be570f83bbf52f733b7.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/44e4460f7f49b0bc7dda13524f57e3d2.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/c08f4637b44a1f1045787111caa844f2.png)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/d6d36b8af76328fe070745330c81218d.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/6fc54441bb51128e8be697b38f54b8ea.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/905cad4ef8c8f115511ee1e8c02c580d.png)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/54e67db87c4de2df139f9fce16e8ce3e.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/0a640202a8889fb986fd5cb91e2fc591.jpg)
![ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ГЕКСАГИДРОДИБЕНЗО[A, G]ХИНОЛИЗИНА](https://fips.edrid.ru/images/rid/f9/07/41/a9ffe3349823b2b8394f1de218a34d43.jpg)
Соединения 2-арилбензофуран-7-формамида, способ их получения и применение
Пирроло[2,1-f][1,2,4]триазиновое соединение, способ его получения и применения
Фторзамещенные циклические аминосоединения и способы их получения, фармацевтические композиции и их применения
Соединения тиенил[3, 2-d]пиримидин-4-он, способ получения, фармацевтические композиции и их применение
Соединение нафтиламида, способ его получения и применение
Соединения 1-(3-аминопропил)-замещенного циклического амина, способ их получения и их фармацевтические композиции и применения
5-ароматическое алкинилзамещенное бензамидное соединение и способ его получения, фармацевтическая композиция и их применение
Кристаллическая форма а соединения и способ ее получения
Бензопроизводные с шестичленным кольцом в качестве ингибитора dpp-4 и их применение
Способ и устройство кодирования сигнала, способ для кодирования объединенного сигнала обратной связи
Способ и устройство кодирования сигнала обратной связи
Устройство и способ управления мощностью
Способ и абонентское оборудование для повторной передачи данных
Система и способ мультиплексирования каналов управления и данных в системе связи с множеством входов и множеством выходов (mimo)
Производные фенилпиримидона, фармацевтические композиции, способы их получения и применения
Способ и система для выявления способности поддержки мобильным терминалом голосовых услуг по адаптивным многопользовательским каналам на одном временном слоте
Система и способ мультиплексирования каналов управления и данных в системе связи с множеством входов и множеством выходов (mimo)
Возбудитель блока генерирования мощности, блок генерирования мощности и оборудование вывода энергии в электрической сети
Способы и устройство для произвольного доступа в системе связи

