Результат интеллектуальной деятельности: НОВАЯ СОЛЬ КОНДЕНСИРОВАННОГО ПИРИМИДИНОВОГО СОЕДИНЕНИЯ И ЕЕ КРИСТАЛЛ
Вид РИД
Изобретение
Область, к которой относится изобретение
[0001]
Настоящее изобретение относится к новой соли соединения, обладающего ингибиторной активностью в отношении тирозинкиназы Брутона (BTK), и к ее кристаллу.
Предпосылки создания изобретения
[0002]
Известно, что различные протеинкиназы существуют in vivo и вовлечены в регуляцию различных функций. Тирозинкиназа Брутона (BTK) представляет собой протеинкиназу, которая относится к Tec семейству киназ, и представляет собой нерецепторную тирозинкиназу, которая играет важную роль, связанную с контролем, например, пролиферации, выживания, дифференциации и активации B-клеток на пути передачи сигнала от B-клеточного рецептора (BCR) (Непатентная литература 1). Ингибитор, который способен контролировать BTK активность, считается полезным в качестве терапевтического средства для лечения заболеваний, ассоциированных с аномальной гиперактивностью сигнального пути BTK (например, рака).
[0003]
Что касается соединения, обладающего ингибиторной активностью в отношении BTK, известны PCI-32765 (Непатентная литература 2) и соединения, описанные в Патентной литературе 1 и 2.
Соединения, раскрытые в патентной литературе 1 и 2, также известны как демонстрирующие высокую ингибиторную активность, например, в отношении EGFR (Рецептор Эпидермального Фактора Роста) и JAK3 (Janus киназа 3), помимо BTK. Однако, поскольку такой ингибитор нескольких киназ подавляет, например, клеточную пролиферацию путем ингибирования различных сигнальных путей, существуют опасения относительно различных нежелательных побочных эффектов. Например, известно, что EGFR связывается с его лигандом, например, эпидермальным фактором роста (EGF), и участвует в пролиферации и выживании (например, ингибировании апоптоза) различных клеток (Непатентная литература 3). Однако известно, что ингибиторы, направленно действующие на EGFR, вызывают нежелательные побочные эффекты, такие как кожные расстройства и дисфункцию желудочно-кишечного тракта одновременно, и существует множество предположений, что эти нежелательные побочные эффекты могут быть связаны с ингибированием сигнального пути EGFR дикого типа (Непатентная литература 4).
Таким образом, PCI-45292 известен как соединение, которое обладает ингибиторной активностью против BTK со слабой ингибиторной активностью против EGFR (Непатентная литература 5).
Перечень ссылочных документов
Патентная литература
[0004]
Патентная литература 1: WO 2011/090760
Патентная литература 2: WO 2009/158571
Непатентная литература
[0005]
Непатентная литература 1: Curr. Opin. Immunol., 2000 Jun; 12(3): 282-8
Непатентная литература 2: Proc. Natl. Acad. Sci. USA., 2010 Jul 20; 107(29):13075-80
Непатентная литература 3: Nature Rev. Cancer, Vol. 6, pp. 803-811 (2006)
Непатентная литература 4: Nature Rev. Clin. Oncol., Vol. 6, pp. 98-109 (2012)
Непатентная литература 5: American College of Rheumatology Annual Meeting, Atlanta, GA, 6-11 November, 2010)
Сущность изобретения
Техническая задача
[0006]
Целью настоящего изобретения является предоставление соли, которая является ингибитором BTK с высокой селективностью, обладающей высокой ингибиторной активностью в отношении BTK и низкой ингибиторной активностью в отношении других киназ, таких как EGFR, и которая применима в качестве лекарственного ингредиента для фармацевтического продукта.
Решение задачи
[0007]
В результате проведенных серьезных исследований для решения этой проблемы авторами настоящего изобретения было обнаружено, что (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид (Соединение A), представленный следующей формулой (1), обладает высокой ингибиторной активностью в отношении BTK, обладая при этом низкой ингибиторной активностью в отношении других киназ, таких как EGFR, и является применимым в качестве лекарственного средства для лечения рака, аутоиммунных заболеваний или аллергических заболеваний.
[0008]
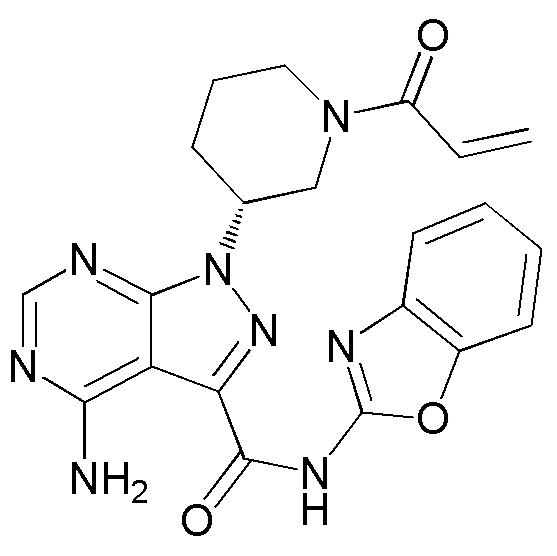 (1)
(1)
[0009]
Затем в ходе исследований физико-химических свойств Соединения A в целях разработки композиции Соединения A, авторами настоящего изобретения было обнаружено, что (1) Соединение A трудно использовать в качестве лекарственного ингредиента для фармацевтического продукта, поскольку оно обладает свойством, как гидрат канального типа, абсорбции влаги в воздухе, когда свободная форма Соединения A находится под воздействием атмосферы высокой влажности, и выделения влаги под воздействием атмосферы низкой влажности, (2) кислотно-аддитивная соль образуется только с винной кислотой, фосфорной кислотой или фумаровой кислотой, что касается кислотно-аддитивной соли Соединения A, и (3), что еше более удивительно, из этих кислотно-аддитивных солей только фумарат не обладает свойством гидрата канального типа, и, таким образом, было создано настоящее изобретение.
[0010]
Более конкретно, для промышленного производства фармацевтического продукта необходимо, чтобы лекарственный ингредиент обладал стабильностью т.д. Однако стабильность т.д. зависят от свойств каждого соединения. Поэтому в комплексном соединении трудно предугадать соль, обладающую подходящими свойствами в качестве лекарственного ингредиента для фармацевтического продукта, и, соответственно, желательно найти для каждого соединения различные соли, которые являются полезными для фармацевтических продуктов. В связи с этим, авторами настоящего изобретения были синтезированы различные соли Соединения A и были исследованы их свойства, стабильность т.д. В конечном итоге, в результате исследования удалось получить соль, такую как фумарат, тартрат, фосфат и магниевая соль. Однако, что касается этих солей и свободной формы, тартрат и свободная форма имели свойство гидрата канального типа и плохую стабильность твердого состояния; фосфат не сохранял свою исходную кристаллическую форму в испытании абсорбции/десорбции влаги и, кроме того, имел плохую стабильность твердого состояния; и магниевая соль имела низкую чистоту кристаллов из-за включения множества аналогичных веществ. Было обнаружено, что только фумарат мог избежать свойства гидрата канального типа и, в то же время, был превосходным в том, что касается получения-эксплуатационной пригодности и воспроизводимости, и стабильным и превосходным в том, что касается абсорбционных свойств, и, таким образом, было совершено настоящее изобретение.
[0011]
Точнее, настоящее изобретение относится к перечисленным ниже 1) - 18).
1) Фумарат (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (Соединение A).
2) гемифумарат (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (Соединение A)⋅.
3) монофумарат (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (Соединение A)⋅.
4) Ингибитор BTK, содержащий фумарат Соединения A в качестве активного ингредиента.
5) Фармацевтическая композиция, содержащая фумарат Соединения A.
6) Противоопухолевое средство или профилактическое и/или терапевтическое средство от аллергических заболеваний, аутоиммунных заболеваний или воспалительных заболеваний, содержащее фумарат Соединения A в качестве активного ингредиента.
7) Противоопухолевое средство против гематологической опухоли или профилактическое и/или терапевтическое средство от аллергического ринита, пыльцевой аллергии, атопического дерматита, ревматоидного артрита или системной красной волчанки, которое содержит фумарат Соединения A в качестве активного ингредиента.
8) Применение фумарата Соединения A для получения ингибитора BTK.
9) Применение фумарата Соединения A для получения фармацевтической композиции.
10) Применение фумарата Соединения A для получения противоопухолевого средства или профилактического и/или терапевтического средства от аллергических заболеваний, аутоиммунных заболеваний или воспалительных заболеваний.
11) Применение фумарата Соединения A для получения противоопухолевого средства против гематологической опухоли или профилактического и/или терапевтического средства от аллергического ринита, пыльцевой аллергии, атопического дерматита, ревматоидного артрита или системной красной волчанки.
12) Фумарат Соединения A для применения в ингибировании BTK.
13) Фумарат Соединения A для применения в качестве лекарственного средства.
14) Фумарат Соединения A для применения в профилактическом и/или терапевтическом средстве от опухоли, аллергического заболевания, аутоиммунного заболевания или воспалительного заболевания.
15) Фумарат Соединения A для применения в профилактике или лечении гематологической опухоли, аллергического ринита, пыльцевой аллергии, атопического дерматита, ревматоидного артрита или системной красной волчанки.
16) Способ ингибирования BTK, включающий введение эффективного количества фумарата Соединения A субъекту, нуждающемуся в этом.
17) Способ профилактики и/или лечения опухоли, аллергического заболевания, аутоиммунного заболевания или воспалительного заболевания, включающий введение эффективного количества фумарата Соединения A субъекту, нуждающемуся в этом.
18) Способ лечения гематологической опухоли, аллергического ринита, пыльцевой аллергии, атопического дерматита, ревматоидного артрита или системной красной волчанки, включающий введение эффективного количества фумарата Соединения A субъекту, нуждающемуся в этом.
Полезные эффекты изобретения
[0012]
Фумарат Соединения A по настоящему изобретению обладает превосходной стабильностью в твердом состоянии в качестве лекарственного ингредиента для фармацевтического продукта и способен избегать свойства гидрата канального типа, по сравнению с Соединением A или солями, отличными от фумарата Соединения A, и является превосходным в том, что касается его получения-эксплуатационной пригодности и воспроизводимости. Кроме того, фумарат Соединения A по настоящему изобретению демонстрирует превосходные характеристики пероральной абсорбции и является чрезвычайно полезным в качестве фармацевтического продукта или в качестве лекарственного ингредиента для фармацевтического продукта.
Краткое описание чертежей
[0013]
Фиг. 1 иллюстрирует спектр порошковой рентгеновской дифракции монофумарата Соединения A (аморфный), синтезированного в Примере 1 (ось ординат представляет интенсивность (имп./сек), и ось абсцисс представляет дифракционный угол (2θ ± 0,1°)).
Фиг. 2 иллюстрирует спектр порошковой рентгеновской дифракции гемифумарата Соединения A (кристалл), синтезированного в Примере 2 (ось ординат представляет интенсивность (имп./сек), и ось абсцисс представляет дифракционный угол (2θ ± 0,1°)).
Фиг. 3 иллюстрирует полученную методом дифференциальной сканирующей калориметрии (ДСК) кривую гемифумарата Соединения A (кристалл), синтезированного в Примере 2.
Фиг. 4 иллюстрирует спектр порошковой рентгеновской дифракции монофумарата Соединения A (кристалл), синтезированного в Примере 3 (ось ординат представляет интенсивность (имп./сек), и ось абсцисс представляет дифракционный угол (2θ ± 0,1°)).
Фиг. 5 иллюстрирует кривую дифференциальной сканирующей калориметрии (ДСК) для монофумарата Соединения A (кристалл), синтезированного в Примере 3.
Фиг. 6 иллюстрирует изотермическую кривую абсорбции/десорбции влаги для Соединения A.
Фиг. 7 иллюстрирует изотермическую кривую абсорбции/десорбции влаги для монофумарата Соединения A (кристалл).
Фиг. 8 иллюстрирует изотермическую кривую абсорбции/десорбции влаги для гемифумарата Соединения A (кристалл).
Фиг. 9 иллюстрирует изотермическую кривую абсорбции/десорбции влаги для гемитартрата Соединения A.
Фиг. 10 иллюстрирует изотермическую кривую абсорбции/десорбции влаги для монофосфата Соединения A.
Фиг. 11 иллюстрирует действие монофумарата Соединения A (кристалл) в мышиной модели коллаген-индуцированного артрита.
Описание вариантов осуществления
[0014]
Далее настоящее изобретение будет описано подробно.
[0015]
"Соединение A", для простоты указываемое так в описании настоящего изобретения, относится к (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамиду в свободной форме.
[0016]
"Фумарат Соединения A", для простоты указываемый так в описании настоящего изобретения, может представлять собой любую из форм солей Соединения A с фумаровой кислотой, включая монофумарат и гемифумарат. Этот термин также используется в значении, охватывающем как кристаллическую форму фумарата Соединения A, так и аморфную форму фумарата Соединения A. Фумарат Соединения A предпочтительно представляет собой монофумарат Соединения A (который может быть сокращенно указан как "Соединение A⋅монофумарат") и гемифумарат Соединения A (который может быть сокращенно указан как "Соединение A⋅гемифумарат"); более предпочтительно монофумарат Соединения A (кристалл), гемифумарат Соединения A (кристалл) и монофумарат Соединения A (аморфный); и особенно предпочтительно монофумарат Соединения A (кристалл) и гемифумарат Соединения A (кристалл).
[0017]
В описании настоящего изобретения термины "кристалл" и "аморфный" используются в их обычных значениях.
[0018]
В некоторых случаях возникают различные кристаллические формы, которые отличаются друг от друга пространственным расположением их атомов и физико-химическими свойствами (полиморфизм). Соль по настоящему изобретению может представлять собой любой из этих полиморфов, и может представлять собой смесь двух или более полиморфов, или смесь кристаллической и аморфной формы.
[0019]
Настоящее изобретение также включает меченную форму фумарата Соединения A, то есть соединение, содержащее один или несколько атомов Соединения A или фумарата, замещенных радиоизотопным элементом или нерадиоизотопным элементом.
[0020]
В этой связи, в порошковом рентгеновском дифракционном спектре дифракционный угол или общая картина являются важными для определения идентичности кристаллов, что касается характера данных. Относительная интенсивность рентгеновского порошкового дифракционного спектра может слегка варьироваться в зависимости от направления роста кристалла, размера частиц или условий измерения, и поэтому не должна строго интерпретироваться.
Числовое значение, полученное из различных дифрактограмм, может иметь незначительное отклонение из-за направления роста кристалла, размера частиц или условий измерения. Поэтому в описании настоящего изобретения термин дифракционный угол (2θ ± 0,1°) в порошковом рентгеновском дифракционном спектре относится к значению, которое может находиться в пределах ±0,1° от указанного значения.
[0021]
Термин "в области", который используют в связи с температурой эндотермического пика на кривой дифференциальной сканирующей калориметрии (ДСК), относится к значению, которое приблизительно представляет собой эту температуру, предпочтительно относится к значению, которое может находиться в пределах ±5°C от указанного значения. Более предпочтительно, относится к значению, которое может находиться в пределах ±2°C от указанного значения.
[0022]
Предпочтительно, монофумарат Соединения A (кристалл) имеет порошковый рентгеновский дифракционный спектр, как показано на Фиг. 4, и/или кривую дифференциальной сканирующей калориметрии (ДСК), как показано на Фиг. 5.
[0023]
В данном случае, характеристические пики монофумарата Соединения A (кристалл) в порошковом рентгеновском дифракционном спектре могут включать 7,2°, 12,4°, 15,6°, 25,9° и 27,6°, более предпочтительно 7,2°, 12,4°, 14,4°, 15,0°, 15,6°, 19,0°, 22,3°, 22,6°, 23,4°, 25,5°, 25,9° и 27,6°, выраженные как дифракционный угол (2θ ± 0,1°).
Монофумарат Соединения A (кристалл) по настоящему изобретению представляет собой кристалл, имеющий по меньшей мере два или более пиков, выбранных из более предпочтительных пиков, описанных выше, предпочтительно кристалл, имеющий по меньшей мере три или более пиков, выбранных из этих пиков, более предпочтительно кристалл, имеющий по меньшей мере пять или более пиков, выбранных из этих пиков, еще более предпочтительно кристалл, имеющий по меньшей мере восемь или более пиков, выбранных из этих пиков, и особенно предпочтительно кристалл, имеющий все из пиков, описанных выше.
[0024]
Эндотермический пик на кривой дифференциальной сканирующей калориметрии (ДСК) монофумарата Соединения A (кристалл) может включать пики в области от 219°C до 224°C, и предпочтительно в области 223°C.
[0025]
Другой предпочтительный вариант осуществления монофумарата Соединения A (кристалл) по настоящему изобретению может представлять собой кристалл, в котором дифракционный угол (2θ ± 0,1°) имеет по меньшей мере два или более, предпочтительно по меньшей мере три или более, и более предпочтительно по меньшей мере пять или более пиков, выбранных из 7,2°, 12,4°, 15,6°, 25,9° и 27,6°, в порошковом рентгеновском дифракционном спектре; и температура пика на кривой дифференциальной сканирующей калориметрии (ДСК) имеет эндотермический пик в области от 219 до 224°C, и предпочтительно в области 223°C. Еще один предпочтительный вариант осуществления может представлять собой кристалл, в котором дифракционный угол (2θ ± 0,1°) в порошковом рентгеновском дифракционном спектре имеет по меньшей мере два или более, предпочтительно по меньшей мере три или более, более предпочтительно по меньшей мере пять или более, еще более предпочтительно по меньшей мере восемь или более, и еще более предпочтительно все из пиков, выбранных из 7,2°, 12,4°, 14,4°, 15,0°, 15,6°, 19,0°, 22,3°, 22,6°, 23,4°, 25,5°, 25,9° и 27,6°; и в то же время, температура пика на кривой дифференциальной сканирующей калориметрии (ДСК) имеет эндотермический пик в области от 219°C до 224°C, и предпочтительно в области 223°C.
[0026]
Предпочтительно, гемифумарат Соединения A (кристалл) имеет порошковый рентгеновский дифракционный спектр, как показано на Фиг. 2, и/или кривую дифференциальной сканирующей калориметрии (ДСК), как показано на Фиг. 3.
[0027]
В данном случае, характеристические пики гемифумарата Соединения A (кристалл) в порошковом рентгеновском дифракционном спектре могут включать 4,5°, 5,8°, 16,6°, 20,2° и 26,4°, и более предпочтительно 4,5°, 5,8°, 11,2°, 12,1°, 12,4°, 13,4°, 16,6°, 17,3°, 18,2°, 20,2°, 26,4° и 27,1°, выраженные как дифракционный угол (2θ ± 0,1°).
Гемифумарат Соединения A (кристалл) по настоящему изобретению представляет собой кристалл, имеющий по меньшей мере два или более пиков, выбранных из более предпочтительных пиков, описанных выше, предпочтительно кристалл, имеющий по меньшей мере три или более пиков, выбранных из этих пиков, более предпочтительно кристалл, имеющий по меньшей мере пять или более пиков, выбранных из этих пиков, еще более предпочтительно кристалл, имеющий по меньшей мере восемь или более пиков, выбранных из этих пиков, и особенно предпочтительно кристалл, имеющий все из этих пиков.
[0028]
Эндотермический пик на кривой дифференциальной сканирующей калориметрии (ДСК) для гемифумарата (кристалл) Соединения A может включать пики в области от 197°C до 199°C, и предпочтительно в области 198°C.
[0029]
Другие предпочтительные варианты осуществления гемифумарата Соединения A (кристалл) по настоящему изобретению могут включать кристалл, в котором дифракционный угол (2θ ± 0,1°) имеет по меньшей мере два или более, предпочтительно по меньшей мере три или более, и более предпочтительно по меньшей мере пять или более пиков, выбранных из 4,5°, 5,8°, 16,6°, 20,2° и 26,4° в порошковом рентгеновском дифракционном спектре; и температура пика на кривой дифференциальной сканирующей калориметрии (ДСК) имеет эндотермический пик в области от 197 до 199°C, и предпочтительно в области 198°C. Некоторые другие варианты осуществления могут включать кристалл, в котором дифракционный угол (2θ ± 0,1°) в порошковом рентгеновском дифракционном спектре имеет по меньшей мере два или более, предпочтительно по меньшей мере три или более, более предпочтительно по меньшей мере пять или более, еще более предпочтительно по меньшей мере восемь или более, и еще более предпочтительно все из пиков, выбранных из 4,5°, 5,8°, 11,2°, 12,1°, 12,4°, 13,4°, 16,6°, 17,3°, 18,2°, 20,2°, 26,4° и 27,1°; и в то же время, температура пика на кривой дифференциальной сканирующей калориметрии (ДСК) имеет эндотермический пик в области от 197°C до 199°C, и предпочтительно в области 198°C.
[0030]
Также возможно получение фумарата Соединения A по настоящему изобретению в виде аморфного вещества. В частности, аморфная форма фумарата Соединения A по настоящему изобретению имеет дифракционную картину, демонстрирующую гало-паттерн, который является широким и неясным, в порошковом рентгеновском дифракционном спектре, и более предпочтительно имеет порошковый рентгеновский дифракционный спектр, как показано на Фиг. 1.
[0031]
Соединение A можно синтезировать, например, в соответствии со Ссылочными Примерами 1 и 2, которые будут описаны ниже. Способ синтеза Соединения A не ограничивается Ссылочными Примерами 1 и 2, которые будут описаны ниже.
Более конкретно, метансульфонилхлорид подвергают взаимодействию с (S)-N-Boc-3-пиперидинолом в присутствии третичного амина, такого как триэтиламин, с получением (S)-трет- бутил-3-(метилсульфонилокси)пиперидин-1-карбоксилата. Затем это соединение подвергают взаимодействию с 3-иод-1H-пиразоло[3,4-d]пиримидин-4-амином в присутствии основания, такого как карбонат калия, с получением (R)-трет-бутил-3-(4-амино-3-иод-1H-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-карбоксилата. Затем это соединение подвергают взаимодействию с палладиевым катализатором и основанием в присутствии бензо[d]оксазол-2-амина, в атмсофере оксида углерода, с получением (R)-трет-бутил-3-(4-амино-3-((бензо[d]оксазол-2-ил)карбамоил)-1H-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-карбоксилата. Затем защитную группу Boc удаляют из этого соединения, которое затем подвергают взаимодействию с акрилоилхлоридом, с получением, таким образом, Соединения A.
[0032]
Монофумарат Соединения A по настоящему изобретению (аморфный) можно получить, например, следующим способом.
К Соединению A добавляют тетрагидрофуран (ТГФ) в количестве 100-300-кратном, и предпочтительно 150-кратном относительно количества соединения, и воду в количестве 0,01-1-кратном, предпочтительно 0,1-кратном относительно количества соединения. К смеси добавляют фумаровую кислоту в молярном количестве, равном количеству Соединения A, и растворяют.
Растворитель отгоняют путем азеотропной перегонки смеси с ТГФ несколько раз, предпочтительно от 2 раз до 5 раз. Таким образом, возможно получение аморфного монофумарата Соединения A в виде белого порошка.
[0033]
Монофумарат Соединения A (кристалл) по настоящему изобретению можно получить в виде белого порошка, например, путем суспендирования монофумарата Соединения A (аморфный) в ацетонитриле в количестве 5-50-кратном, предпочтительно 20-кратном по отношению к нему, и нагревания суспензии в течение 12-72 часов, предпочтительно в течение 24 часов.
[0034]
Гемифумарат Соединения A (кристалл) по настоящему изобретению можно получить в виде белого порошка, например, путем суспендирования монофумарата Соединения A (аморфный) в метилэтилкетоне в количестве 10-100-кратном, предпочтительно 60-кратном по отношению к нему, и нагревания суспензии в течение 12-72 часов, предпочтительно в течение 24 часов.
[0035]
Согласно результатам, полученным для фумарата Соединения A по настоящему изобретению, можно избежать свойств гидрата канального типа Соединения A.
Как правило, в фармацевтическом продукте или лекарственном ингредиенте для фармацевтического продукта, который получают с использованием соединения, для которого удалось избежать свойств гидрата канального типа, известно, что уменьшаются проблемы, связанные с хранением и контролем качества в условиях влажности при хранении; а также, что, когда получают твердый препарат, такой как таблетка или капсула, можно уменьшить проблемы при получении за счет изменения массы активного ингредиента.
Поэтому можно сказать, что, в соответствии с фумаратом Соединения A по настоящему изобретению, можно ожидать стабильного хранения и легкого контроля качества, а также, что касается получения, фумарат Соединения A по настоящему изобретению является превосходным соединением, удобным в обращении.
[0036]
Фумарат Соединения A по настоящему изобретению является превосходным в том, что касается получения-эксплуатационной пригодности и воспроизводимости, по сравнению с другими солями Соединения A. В частности, например, соль Соединения A с хлористоводородной кислотой, серной кислотой, янтарной кислотой, яблочной кислотой, лимонной кислотой или уксусной кислотой не была получена исследовательским способом, описанным в описании настоящего изобретения. Например, в процессе образования натриевой соли Соединения A происходило существенное разложение. В процессе синтеза гемимагниевой соли Соединения A количество аналогичных веществ увеличивалось, и, более того, процедура получения соли была сложной, и было трудно осуществить повторное растворение соли из-за ее низкой растворимости в воде и в органическом растворителе.
Фумарат Соединения A по настоящему изобретению является легким в обращении в качестве лекарственного ингредиента для фармацевтического продукта и способствует промышленному получению фармацевтического продукта, имеющего стабильное качество.
[0037]
Фумарат Соединения A по настоящему изобретению является превосходным в том, что касается стабильности твердого состояния. Для соединения-кандидата, разрабатываемого в качестве фармацевтического продукта, важно, чтобы оно обладало стабильностью твердого состояния для промышленной эксплуатации и поддержания качества. Поэтому фумарат Соединения A по настоящему изобретению обладает превосходными свойствами, необходимыми для фармацевтического продукта или лекарственного ингредиента для фармацевтического продукта.
[0038]
Фумарат Соединения A по настоящему изобретению является превосходным в том, что касается пероральной абсорбции, и способствует получению превосходного фармацевтического продукта с высоким качеством.
[0039]
Из солей Соединения A, фумарат Соединения A по настоящему изобретению является превосходным в том, что касается любой из таких его характеристик, таких как получение-эксплуатационная пригодность, воспроизводимость, стабильность твердого состояния и пероральная абсорбция, при этом избегая свойства гидрата канального типа, которой обладает Соединение A, и поддерживая достаточную растворимость, в качестве лекарственного ингредиента для фармацевтического продукта.
[0040]
Фумарат Соединения A по настоящему изобретению обладает превосходной активностью ингибирования BTK, и является полезным в качестве профилактического и/или терапевтического средства от, например, рака, опухолей и различных иммунных заболеваний (например, аллергических заболеваний, аутоиммунных заболеваний и воспалительных заболеваний). Кроме того, фумарат обладает превосходной селективностью в отношении BTK, а также обладает преимуществом, имея меньше нежелательных побочных эффектов, связанных с ингибированием других киназ (например, EGFR).
[0041]
Фумарат Соединения A по настоящему изобретению обладает превосходной ингибиторной активностью в отношении BTK. "BTK", в соответствии с настоящим описанием, включает BTK человека или млекопитающего, отличного от человека, и BTK предпочтительно представляет собой человеческую BTK. В данном случае, термин "BTK" включает изоформы этого фермента.
Кроме того, благодаря превосходной ингибиторной активности в отношении BTK, фумарат Соединения A по настоящему изобретению является полезным в качестве лекарственного средства для профилактики или лечения заболеваний, ассоциированных с BTK. "Заболевания, ассоциированные с BTK" включают заболевания, частота случаев которых снижается, и имеет место ремиссия, облегчение тяжести и/или полное устранение симптомов в результате делеции, супрессии и/или ингибирования функций BTK. Примеры таких заболеваний включают, без ограничения, раковые заболевания или опухоли, аллергические заболевания, аутоиммунные заболевания, воспалительные заболевания, болезнь трансплантат-против-хозяина, и предпочтительно раковые заболевания, опухоли, аллергические заболевания и аутоиммунные заболевания.
[0042]
Нет никакого конкретного ограничения в том, что касается целевых раковых заболеваний и опухолей, и их примеры включают эпителиальный рак (например, раковые заболевания респираторной системы, раковые заболевания желудочно-кишечной системы, раковые заболевания репродуктивной системы и раковые заболевания системы секреции), саркомы, опухоли гематопоэтической системы, опухоли центральной нервной системы и опухоли периферической нервной системы. Предпочтительными примерами являются опухоли гематопоэтической системы (например, лейкоз, множественная миелома и злокачественная лимфома). Кроме того, нет никакого конкретного ограничения, касающегося типа органов, в которых может развиться опухоль, и примеры включают рак головы и шеи, рак пищевода, рак желудка, рак толстой кишки, ректальный рак, рак печени, рак желчного пузыря/желчных протоков, рак желчных путей, рак поджелудочной железы, рак легких, рак молочной железы, рак яичников, рак шейки матки, рак эндометрия, рак почки, рак мочевого пузыря, рак предстательной железы, тестикулярные опухоли, саркому костей/мягких тканей, гематологические опухоли, множественную миелому, рак кожи, опухоли головного мозга и мезотелиальный рак. Предпочтительные примеры опухолей гематопоэтической системы включают острый лейкоз, острый промиелоцитарный лейкоз, острый лимфобластный лейкоз, хронический миелогенный лейкоз, лимфобластную лимфому, миелопролиферативные новообразования, хронический лимфоцитарный лейкоз, малую лимфоцитарную лимфому, миелодиспластические синдромы, фолликулярную лимфому, MALT лимфому, лимфому маргинальной зоны, лимфоплазмоцитарную лимфому, макроглобулинемию Вальденстрема, лимфому из клеток мантийной зоны, диффузную крупноклеточную B-клеточную лимфому, лимфому Беркитта, экстранодальную NK/T-клеточную лимфому, лимфому Ходжкина и множественную миелому. Особенно предпочтительные примеры включают гематологические опухоли, такие как B-лимфобластный лейкоз/лимфому, фолликулярную лимфому, лимфому из клеток мантийной зоны, нодальную фолликулярную лимфому маргинальной зоны, диффузную крупноклеточную B-клеточную лимфому, лимфому Беркитта, хронический лимфоцитарный лейкоз, малую лимфоцитарную лимфому, макроглобулинемию Вальденстрема, экстранодальную NK/T-клеточную лимфому, лимфому Ходжкина, миелодиспластические синдромы, острый миелогенный лейкоз и острый лимфоцитарный лейкоз.
[0043]
Нет никакого конкретного ограничения в том, что касается целевых аллергических заболеваний, и их примеры включают, например, бронхиальную астму, аллергический ринит, пыльцевую аллергию, атопический дерматит, пищевую аллергию, анафилаксию, лекарственную аллергию, аллергическую сыпь и конъюнктивит. Предпочтительные примеры включают бронхиальную астму, аллергический ринит, пыльцевую аллергию и атопический дерматит; и особенно предпочтительные примеры включают аллергический ринит, пыльцевую аллергию и атопический дерматит.
[0044]
Нет никакого конкретного ограничения в том, что касается целевых аутоиммунных заболеваний, и их примеры включают ревматоидный артрит, системную красную волчанку, дерматосклероз, полимиозит, синдром Шегрена и болезнь Бехчета. Предпочтительные примеры включают ревматоидный артрит и системную красную волчанку, и особенно предпочтительным примером является ревматоидный артрит.
[0045]
Нет никакого конкретного ограничения в том, что касается целевых воспалительных заболеваний, и их примеры включают аппендицит, блефарит, бронхиолит, бронхит, бурсит, цервицит, холангит, холецистит, язвенный колит, болезнь Крона, синдром раздраженной толстой кишки, цистит, дакриоаденит, контактный дерматит, дерматомиозит, энцефалит, эндокардит, эндометрит, эпидидимит, фасциит, мышечный ревматизм, гастроэнтерит, гепатит, абсцесс потовых желез, ларингит, мастит, менингит, миелит, миокардит, нефрит, воспаление яичников, дидимит, панкреатит, паротит, перикардит, перитонит, фарингит, плеврит, флебит, пневмонию, проктит, простатит, пиелонефрит, сальпингит, синусит, стоматит, остеоартрит, синовит, тендинит, тонзилит, увеит, вагинит, васкулит и вульвит. Предпочтительные примеры могут включать язвенный колит, болезнь Крона, синдром раздраженной толстой кишки, контактный дерматит, цистит и остеоартрит. Особенно предпочтительные примеры могут включать контактный дерматит, цистит и остеоартрит.
[0046]
Когда фумарат Соединения A по настоящему изобретению используют в виде фармацевтической композиции, можно использовать различные лекарственные формы, в соответствии с целью профилактики или лечения, путем включения фармацевтических носителей, в соответствии с необходимостью. Лекарственная форма может представлять собой, например, любую форму, такую как пероральный препарат, препарат для инъекций, препарат в форме суппозитория, мазь и пластырь. Любую из этих лекарственных форм можно получить способом формулирования, который широко известен и который традиционно используют специалисты в данной области. В частности, таблетка для перорального введения, таблетка с покрытием, пилюля, гранулированный препарат, препарат в форме порошка и препарат в форме капсул, которые содержат кристаллический фумарат Соединения A в качестве лекарственного ингредиента, являются предпочтительными в качестве стабильного твердого препарата.
[0047]
Что касается фармацевтических носителей, то используют различные органические или неорганические вещества-носители, которые традиционно используются в качестве веществ для формулирования препаратов, и фармацевтические носители включают, например, в качестве эксципиента, связующего, разрыхлителя, смазывающего вещества и агента покрытия в твердые препараты; и в качестве растворителя, добавки, способствующей растворению, суспендирующего вещества, изотонического агента, pH-регулирующего агента, буферного агента и анальгетического средства в жидкие препараты. Кроме того, если необходимо, также можно использовать добавки для формулирования препаратов, такие как антисептик, антиоксидант, краситель, отдушка/корригент и стабилизатор.
Примеры эксципиента включают лактозу, сахарозу, D-маннит, крахмал, кристаллическую целлюлозу и силикат кальция.
Примеры связующего включают гидроксипропилцеллюлозу, метилцеллюлозу, поливинилпирролидон, сахарный порошок и гидроксипропилметилцеллюлозу.
Примеры разрыхлителя включают натрий крахмалгликолят, кармелозу кальция, кроскармелозу натрия, кросповидон, низкозамещенную гидроксипропилцеллюлозу и частично желатинизированный крахмал.
Примеры смазывающего вещества включают тальк, стеарат магния, сложные эфиры жирных кислот сахарозы, стеариновую кислоту и стеарилфумарат натрия.
Примеры агента покрытия включают этилцеллюлозу, аминоалкилметакрилатный сополимер RS, гидроксипропилметилцеллюлозу и сахарозу.
Примеры растворителя включают воду, пропиленгликоль и физиологический солевой раствор.
Примеры добавки, способствующей растворению, включают полиэтиленликоль, этанол, α-циклодекстрин, Macrogol 400 и Полисорбат 80.
Примеры суспендирующего вещества включают каррагенан, кристаллическую целлюлозу, кармелозу натрия, и полиоксиэтилен гидрогенизированное касторовое масло.
Примеры изотонического агента включают хлорид натрия, глицерин и хлорид калия.
Примеры pH-регулирующего агента и буферного агента включают цитрат натрия, хлористоводородную кислоту, молочную кислоту, фосфорную кислоту и дигидрофосфат натрия.
Примеры анальгетического средства включают прокаин гидрохлорид и лидокаин.
Примеры антисептика включают этил пара-оксибензоат, крезол и бензалконийхлорид.
Примеры антиоксиданта включают сульфит натрия, аскорбиновую кислоту и природный витамин E.
Примеры красителя включают оксид титана, сесквиоксид железа, Пищевой Синий № 1 и медно-хлорофилловый комплекс.
Примеры отдушки/корригента включают аспартам, сахарин, трихлоргалактосахарозу, l-ментол и мятную отдушку.
Примеры стабилизатора включают пиросульфит натрия, эдетат натрия, эриторбовую кислоту, оксид магния и дибутилгидрокситолуол.
[0048]
При получении перорального твердого препарата к фумарату Соединения A необязательно добавляют эксципиент, связующее, разрыхлитель, смазывающее вещество, краситель и отдушку/корригент, и затем традиционным способом можно получить, например, таблетку, таблетку с покрытием, гранулированный препарат, препарат в форме порошка или препарат в форме капсул.
При получении препарата для инъекций к фумарату Соединения A добавляют pH регулирующий агент, буферный агент, стабилизатор, изотонический агент и местный анестетик, и, используя традиционные способы, можно получить препараты для подкожных, внутримышечных и внутривенных инъекций.
[0049]
Количество фумарата Соединения A для включения в различные стандартные лекарственные формы может варьироваться в зависимости от симптомов у пациента, которому следует вводить этот фумарат, или в зависимости от формы препарата; однако, как правило, желательно доведение количества до 0,05-1,000 мг в пероральном препарате, до 0,01-500 мг в препарате для инъекций и до 1-1000 мг в препарате в форме суппозитория, в расчете на стандартную лекарственную форму.
[0050]
Кроме того, суточная доза лекарственного средства в лекарственной форме, описанной выше, может варьироваться в зависимости, например, от симптомов, массы тела, возраста и пола пациента, и ее нельзя определять для всех одинаково. Однако дозу обычно доводят до 0,05-5000 мг, и предпочтительно 0,1-1000 мг, в день для взрослого пациента (масса тела: 50 кг), и предпочтительно вводить это количество раз в день или в виде дробных доз примерно два-три раза в день.
Примеры
[0051]
Далее настоящее изобретение будет описано более конкретно при помощи Примеров, но настоящее изобретение не должно ограничиваться ими. Хотя настоящее изобретение в достаточной степени описано примерами, должно быть понятно, что специалисты в данной области смогут осуществить различные изменения или модификации. Поэтому такие изменения или модификации охватываются настоящим изобретением, если только они не являются отступлением от объема настоящего изобретения.
[0052]
Что касается различных реагентов, используемых в примерах, если специально не указано иное, использовали коммерчески доступные продукты. Для колоночной хроматографии на силикагеле использовали PURIF-PACK (зарегистрированная торговая марка) SI, изготовитель Schott Moritex Corp., заполненную диоксидом кремния колонку KP-Sil (зарегистрированная торговая марка), изготовитель Biotage AB, или заполненную диоксидом кремния колонку HP-Sil (зарегистрированная торговая марка), изготовитель Biotage AB. Для колоночной хроматографией на щелочном силикагеле использовали PURIF-PACK (зарегистрированная торговая марка) NH, изготовитель Moritex Corp., или KP-NH (зарегистрированная торговая марка) заполненную колонку, изготовитель Biotage AB. Для тонкослойной хроматографии для фракционирования использовали KIESELGEL TM60F254, Art. 5744, изготовитель Merck KGaA, или пластину с NH2 силикагелем 60F254, изготовитель Wako Pure Chemical Industries, Ltd. Спектр ЯМР измеряли с использованием спектрометра типа AL400 (400 МГц; JEOL, Ltd.), MERCURY400 (400 МГц; Agilent Technologies, Inc.) или спектрометра типа INOVA400 (400 МГц; Agilent Technologies, Inc.), снабженного 400MЯМР зондом (Protasis Corp.), и с использованием тетраметилсилана в качестве внутреннего стандарта в случае, когда дейтерированный растворитель содержал тетраметилсилан, при этом в других случаях использовали ЯМР растворитель в качестве внутреннего стандарта. Все δ значения выражали в м.д.
[0053]
ЖХМС спектр измеряли с использованием ACQUITY SQD (квадрупольного типа), изготовитель Waters Corp., в условиях, описанных ниже.
Колонка: YMC-TRIART C18, изготовитель YMC Co., Ltd., 2,0 × 50 мм, 1,9 мкм
МС детекция: ESI положительный режим
УФ детекция: 254 нм и 210 нм
Колонка, скорость потока: 0,5 мл/мин
Подвижная фаза: вода/ацетонитрил (0,1% муравьиной кислоты)
Объем вводимой пробы: 1 мкл
Градиент (Таблица 1)
[0054]
|
[0055]
Кроме того, очистку обращенно-фазовой препаративной ВЭЖХ осуществляли на препаративной системе, изготовитель Waters Corp., с использованием условий, описанных ниже.
Колонка: использовали YMC-ACTUS TRIART C18, изготовитель YMC Co., Ltd., 20 × 50 мм, 5 мкм, соединенную с YMC-ACTUS TRIART C18, изготовитель YMC Co., Ltd. 20 × 10 мм, 5 мкм.
УФ детекция: 254 нм
МС детекция: ESI положительный режим
Колонка, скорость потока: 25 мл/мин
Подвижная фаза: вода/ацетонитрил (0,1% муравьиной кислоты)
Объем вводимой пробы: 0,1-0,5 мл
[0056]
Аббревиатуры имеют значения, указанные ниже.
с: Синглет
д: Дублет
т: Триплет
кв.: Квартет
дд: Двойной дублет
дт: Двойной триплет
тд: Тройной дублет
тт: Тройной триплет
ддд: Двойной двойной дублет
ддт: Двойной двойной триплет
дтд: Двойной тройной дублет
тдд: Триплет двойной дублет
м: мультиплет
ушир.: Уширенный
ушир.с: Уширенный синглет
CDI: Карбонилдиимидазол
DMSO-d6: Дейтерированный диметилсульфоксид
CDCl3: Дейтерированный хлороформ
CD3OD: Дейтерированный метанол
ТГФ: Тетрагидрофуран
DMF: N,N-диметилформамид
DMA: N,N-диметилацетамид
NMP: 1-Метил-2-пирролидинон
DMSO: Диметилсульфоксид
TFA: Трифторуксусная кислота
WSC: 1-(3-Диметиламинопропил)-3-этилкарбодиимид гидрохлорид
HOBt: 1-Гидроксибензотриазол моногидрат
HATU: (Диметиламино)-N,N-диметил(3H-[1,2,3]триазоло[4,5-b]пиридин-3-илокси)метаниминий гексафторфосфат
DIAD: Диизопропилазодикарбоксилат
TBAF: Тетрабутиламмонийфторид
DIPEA: Диизопропилэтиламин
Boc: Трет-бутоксикарбонил
Boc2O: Ди-трет-бутилдикарбонат
DMAP: Диметиламинопиридин
[0057]
Измерение рентгеновской порошковой дифракции
Рентгеновскую порошковую дифракцию измеряли в соответствии со следующими условиями испытания после растирания в порошок испытываемого вещества, при необходимости, в агатовой ступке.
Устройство: Rigaku MiniFlexII
Мишень: Cu
Выход рентгеновского излучателя: 15 мА, 30 кВ
Область сканирования: 2,0-40,0°
Размер шага: 0,010°
Скорость сканирования: 5,00°/мин.
Щель расходимости: 1,25°
Щель рассеивания: Открытая
Щель для входа светового пучка: Открытая
Обращение с устройствами, включая обработку данных, осуществляли на основании способа и режима, указанного в каждом устройстве.
Числовые значения, получаемые из разных спектров, могут слегка варьироваться в зависимости от направления роста кристалла, размера частиц или условий измерения. Поэтому эти числовые значения не должны строго интерпретироваться.
[0058]
Измерения в термическом анализе (дифференциальная сканирующая калориметрия (ДСК измерения))
ДСК измерения осуществляли в соответствии со следующими условиями испытания.
Устройство: TA Instrument Q1000
Образец: около 1 мг
Контейнер для образца: алюминиевый
Скорость повышения температуры: повышали на 5°C/мин., до 250°C
Атмосферный газ: Азот
Скорость потока газообразного азота: 50 мл/мин.
Обращение с устройствами, включая обработку данных, осуществляли на основании способа и режима, указанного в каждом устройстве.
[0059]
Ссылочный Пример 1 Синтез (R)-трет-бутил-3-(4-амино-3-иод-1H-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-карбоксилата
[0060]
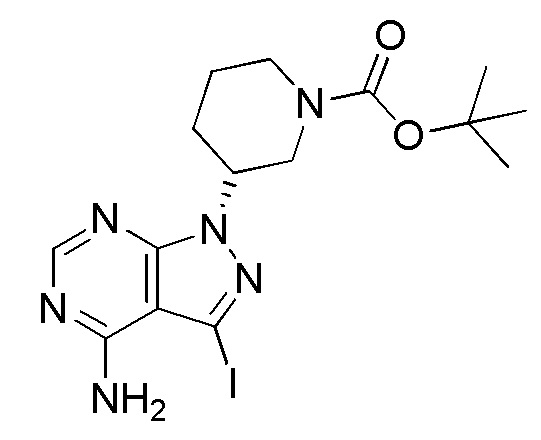
[0061]
(Стадия 1) Синтез (S)-трет-бутил-3-(метилсульфонилокси)пиперидин-1-карбоксилата
20 г (S)-N-Boc-3-пиперидинола растворяли в 100 мл толуола и добавляли 21 мл триэтиламина и 9,2 мл метансульфонилхлорида при 0°C. Смесь перемешивали в течение 1 часа при охлаждении льдом, затем к смеси добавляли этилацетат и воду и органический слой отделяли. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия, насыщенным водным раствором хлорида аммония и водой и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и, таким образом, получали 26,8 г указанного в заголовке соединения в виде бесцветного твердого вещества.
[0062]
(Стадия 2) Синтез (R)-трет-бутил-3-(4-амино-3-иод-1H-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-карбоксилата
Суспензионный раствор 14,6 г 3-иод-1H-пиразоло[3,4-d]пиримидин-4-амина, синтезированного способом, описанным в WO 2007/126841, 25 г (S)-трет-бутил-3-(метилсульфонилокси)пиперидин-1-карбоксилата, полученного на стадии 1, и 69 г карбоната калия в 150 мл DMA нагревали до 100°C и перемешивали в течение 10 часов. Суспензионный раствор охлаждали до комнатной температуры и затем к нему добавляли 300 мл воды. Полученное таким образом твердое вещество собирали фильтрованием и промывали водой и твердое вещество сушили. Таким образом, получали 26,9 г указанного в заголовке соединения в виде желтого твердого вещества.
[0063]
Ссылочный Пример 2 Синтез (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (Соединение A)
[0064]
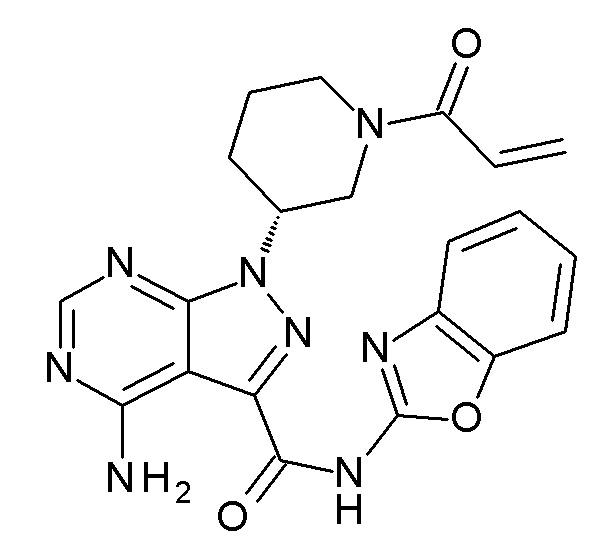
[0065]
(Стадия 1) Синтез (R)-трет-бутил-3-(4-амино-3-((бензо[d]оксазол-2-ил)карбамоил)-1H-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-карбоксилата
300 мг (R)-трет-бутил-3-(4-амино-3-иод-1H-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-карбоксилата, полученного в Ссылочном Примере 1, растворяли в 3 мл NMP. К смеси добавляли 118 мг бензо[d]оксазол-2-амина, 20 мг xantphos и 0,15 мл N-метилморфолина и осуществляли процедуру дегазирования. Затем к смеси добавляли 7,6 мг ацетата палладия и в атмсофере оксида углерода смесь нагревали до 110°C и перемешивали в течение 2 часов. После охлаждения смеси к ней добавляли 4,5 мл метанола и 0,45 мл 5 N водного раствора гидроксида натрия и смесь перемешивали в течение 30 минут при комнатной температуре. Затем pH доводили до 5,3 при помощи 2 N HCl и полученное таким образом твердое вещество собирали фильтрованием. Неочищенный продукт очищали колоночной хроматографией на силикагеле (хлороформ-метанол) и, таким образом, получали 257 мг указанного в заголовке соединения в виде белого твердого вещества.
[0066]
(Стадия 2) Синтез Соединения A
5,0 г (R)-трет-бутил-3-(4-амино-3-((бензо[d]оксазол-2-ил)карбамоил)-1H-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-карбоксилата, полученного на стадии 1, суспендировали в 50 мл ацетонитрила и к смеси добавляли 7,85 г иодида натрия. К смеси добавляли по каплям 6,65 мл триметилсилилхлорида при перемешивании при комнатной температуре и смесь перемешивали в течение 1 часа. К смеси добавляли 87,5 мл воды и 12,5 мл 5 N водного раствора гидроксида натрия и затем систему охлаждали льдом. К смеси добавляли по каплям раствор, полученный путем растворения 0,895 мл акрилоилхлорида в 4,1 мл ацетонитрила, и смесь перемешивали в течение 1 часа при охлаждении льдом. К смеси добавляли 50 мл воды и твердое вещество, полученное таким образом, собирали фильтрованием, промывали водой и сушили при пониженном давлении. Таким образом, получали 4,13 г указанного в заголовке соединения в виде белого твердого вещества (Соединение A).
1H-ЯМР (DMSO-d6): δ м.д. 1,53-1,68 (м, 1H), 1,86-1,98 (м, 1H), 2,08-2,21 (м, 1H), 2,25-2,39 (м, 1H), 2,82-2,95 (м, 0,5H), 3,10-3,22 (м, 0,5H), 3,23-3,37 (м, 0,5H), 3,68-3,78 (м, 0,5H), 4,04-4,14 (м, 0,5H), 4,22-4,38 (м, 1H), 4,52-4,65 (м, 0,5H), 4,67-4,81 (м, 1H), 5,58-5,74 (м, 1H), 6,03-6,19 (м, 1H), 6,68-6,92 (м, 1H), 7,28-7,40 (м, 2H), 7,59-7,71 (м, 2H), 8,22 (ушир.с, 2H), 8,28 (с, 1H), 12,15 (ушир.с, 1H)
[0067]
Ссылочный Пример 3 Исследование (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (Соединение A) и кислотно- или основно-аддитивных солей
[0068]
(1) Исследование кислотно-аддитивных солей Соединения A
Исследование осуществляли на соли, образованной с хлористоводородной кислотой, серной кислотой и фосфорной кислотой из числа неорганических кислот. В частности, Соединение A растворяли в подходящем растворителе и к смеси добавляли каждый тип из этих кислот в подходящем количестве (от 0,5 до 1,5 эквивалентов) и осуществляли перемешивание в течение ночи, для определения таким образом, произошло или нет солеобразование. Как результат, при добавлении хлористоводородной кислоты или серной кислоты к Соединению A происходило заметное разложение, и поэтому исследование останавливали. При добавлении фосфорной кислоты к Соединению A получали монофосфат Соединения A (Ссылочный Пример 4).
Кроме того, исследование осуществляли на соли, образованной с фумаровой кислотой, янтарной кислотой, винной кислотой, яблочной кислотой, лимонной кислотой и уксусной кислотой. В частности, Соединение A растворяли в подходящем растворителе и к смеси добавляли каждый тип из этих кислот в молярном количестве, равном количеству Соединения A, и затем растворитель отгоняли с получением таким образом аморфного соединения каждого типа из этих органических кислот. Затем это аморфное вещество суспендировали при нагревании в органическом растворителе (например, метилэтилкетоне, этаноле, этилацетате и бутилацетате) для определения таким образом, произошло или нет солеобразование. Как результат, не происходило никакого солеобразования с яблочной кислотой, лимонной кислотой или уксусной кислотой. Из аморфного соединения янтарной кислоты, однако, было получено кристаллическое вещество, было невозможно получить сукцинат в количестве, эквивалентом теоретическому количеству. Из фумаровой кислоты был получен монофумарат Соединения A (аморфный), и из винной кислоты был получен гемитартрат Соединения A (Пример 1 и Ссылочный Пример 5).
[0069]
(2) Исследование основно-аддитивных солей Соединения A
Исследование осуществляли на соли, образованной с натрием и магнием из числа неорганических оснований. В частности, Соединение A растворяли в подходящем растворителе и к смеси добавляли каждый тип из этих оснований (от 0,5 до 1,5 эквивалентов) и осуществляли перемешивание в течение ночи для определения таким образом, произошло или нет солеобразование. Как результат, при образовании соли с натрием происходило заметное разложение, и поэтому исследование останавливали. В случае с магнием, получали гемимагниевую соль Соединения A (Ссылочный Пример 6). Однако в гемимагниевой соли Соединения A количество аналогичных веществ увеличивалось, и, кроме того, процедура получения соли была сложной, и было трудно осуществить повторное растворение из-за ее низкой растворимости в органическом растворителе, и поэтому эту соль сочли неподходящей для практического производства.
[0070]
Ссылочный Пример 4 Синтез монофосфата (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида ⋅ (монофосфата Соединения A)
[0071]
К Соединению A, полученному выше (150 мг), добавляли ТГФ (18 мл) и после того, как смесь нагревали до 70°C, к смеси добавляли фосфорную кислоту (27,3 мкл). Затем смесь перемешивали при этой температуре в течение 72 часов и осажденное твердое вещество собирали фильтрованием, затем сушили при пониженном давлении и получали указанное в заголовке соединение в виде белого твердого вещества. Выход (количество): 158 мг, Выход (процент): 85,9%
1H-ЯМР (DMSO-d6): δ м.д. 1,51-1,69 (м, 1H), 1,87-1,97 (м, 1H), 2,09-2,21 (м, 1H), 2,25-2,41 (м, 1H), 2,84-2,95 (м, 0,5H), 3,09-3,22 (м, 0,5H), 3,22-3,38 (м, 0,5H), 3,67-3,83 (м, 0,5H), 4,04-4,17 (м, 0,5H), 4,23-4,40 (м, 1H), 4,54-4,65 (м, 0,5H), 4,66-4,83 (м, 1H), 5,58-5,76 (м, 1H), 6,05-6,22 (м, 1H), 6,70-6,96 (м, 1H), 7,32-7,48 (м, 2H), 7,61-7,75 (м, 2H), 8,15-8,26 (ушир.с, 2H), 8,30 (с, 1H)
[0072]
Ссылочный Пример 5 Синтез геми-L-(+)-тартрата (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида⋅ (гемитартрата Соединения A)
[0073]
Соединение A, полученное выше (600 мг), растворяли в смеси ТГФ (90 мл) и воды (60 мкл) и в полученный раствор вливали L-(+)-тартрат (209 мг) и полностью растворяли. Растворитель отгоняли путем азеотропной перегонки смеси с ТГФ два раза с получением, таким образом, белого твердого вещества. К этому белому твердому веществу (130 мг) добавляли метилэтилкетон (1,5 мл) и осуществляли нагревание суспензии при 70°C в течение 21 часа. Твердое вещество собирали фильтрованием, затем сушили при пониженном давлении и получали продукт в виде белого твердого вещества. Выход (количество): 96 мг, Выход (процент): 85,9%
1H-ЯМР (DMSO-d6): δ м.д. 1,55-1,68 (м, 1H), 1,89-1,97 (м, 1H), 2,10-2,21 (м, 1H), 2,25-2,40 (м, 1H), 2,85-2,95 (м, 0,5H), 3,15-3,65 (м, 1H), 3,68-3,80 (м, 0,5H), 4,05-4,15 (м, 0,5H), 4,24-4,36 (м, 1H), 4,30 (с, 2H), 4,53-4,62 (м, 0,5H), 4,67-4,79 (ушир.с, 1H), 5,67 (дд, J=9,99 Гц, 1H), 6,08-6,18 (м, 1H), 6,71-6,92 (м, 1H), 7,30-7,41 (м, 2H), 7,61-7,73 (м, 2H), 8,22 (ушир.с, 2H), 8,29 (с, 1H)
[0074]
Ссылочный Пример 6 Синтез гемимагниевой соли (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида⋅ (гемимагниевой соли Соединения A)
[0075]
К Соединению A, полученному выше (159,8 мг), добавляли ТГФ (3 мл) и метанол (1 мл) для суспендирования соединения. Затем к смеси добавляли 2M водный раствор гидроксида натрия (185 мкл), и Соединение A полностью растворялось в нем. Затем в смесь вливали хлорид магния (17,6 мг) и смесь перемешивали с получением, таким образом, белого твердого вещества. После добавления воды (2 мл) твердое вещество собирали фильтрованием. Твердое вещество промывали водой (1 мл) два раза и сушили при пониженном давлении с получением, таким образом, белого твердого вещества. К этому белому твердому веществу (50 мг) добавляли метилэтилкетон (1 мл) и нагревание суспензии осуществляли при 70°C в течение 24 часов. Твердое вещество собирали фильтрованием, затем сушили при пониженном давлении и получали продукт в виде белого твердого вещества. Выход (количество): 44,7 мг, Выход (процент): 57,3%
1H-ЯМР (DMSO-d6): δ м.д. 1,52-1,67 (м, 1H), 1,88-2,01 (м, 1H), 2,19-2,41 (м, 2H), 2,75-2,89 (м, 0,5H), 3,07-3,21 (м, 1H), 3,57-3,71 (м, 0,5H), 4,15-4,23 (м, 0,5H), 4,36-4,55 (м, 1H), 4,69-4,88 (м, 2H), 5,60-5,80 (м, 1H), 6,08-6,22 (м, 1H), 6,79-6,96 (м, 1H), 7,08-7,23 (м, 2H), 7,47-7,56 (м, 2H), 8,13-8,22 (ушир.с, 2H), 8,49 (с, 1H), 10,42-10,50 (ушир.с, 1H)
[0076]
Пример 1 Синтез монофумарата (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида⋅ (монофумарата Соединения A (аморфный))
[0077]
Соединение A, полученное в Ссылочном Примере 2 (1,4 г), растворяли в ТГФ (210 мл) и воде (140 мкл) при комнатной температуре и затем в смесь вливали фумаровую кислоту (376 мг) и полностью растворяли в ней. Растворитель отгоняли путем азеотропной перегонки смеси с ТГФ два раза с получением, таким образом, монофумарата Соединения A (аморфная форма) в виде белого порошка. Выход (количество): 1,57 г, Выход (процент): 90,1%
1H-ЯМР (DMSO-d6): δ м.д. 1,53-1,68 (м, 1H), 1,86-1,98 (м, 1H), 2,08-2,21 (м, 1H), 2,25-2,39 (м, 1H), 2,82-2,95 (м, 0,5H), 3,10-3,22 (м, 0,5H), 3,23-3,37 (м, 0,5H), 3,68-3,78 (м, 0,5H), 4,04-4,14 (м, 0,5H), 4,22-4,38 (м, 1H), 4,52-4,65 (м, 0,5H), 4,67-4,81 (м, 1H), 5,58-5,74 (м, 1H), 6,03-6,19 (м, 1H), 6,62 (с, 2H), 6,68-6,92 (м, 1H), 7,28-7,40 (м, 2H), 7,59-7,71 (м, 2H), 8,22 (ушир.с, 2H), 8,28 (с, 1H), 12,15 (ушир.с, 1H)
[0078]
Спектр порошковой рентгеновской дифракции: Показан на Фиг. 1.
[0079]
Пример 2 Синтез гемифумарата (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида⋅ (гемифумарата Соединения A (кристалл))
[0080]
Монофумарат Соединения A, полученный в Примере 1 (аморфный) (450 мг), суспендировали в метилэтилкетоне (27 мл) и осуществляли нагревание суспензии при 80°C в течение 24 часов. Вещество собирали фильтрованием и затем сушили при пониженном давлении с получением гемифумарата Соединения A (кристалл) в виде белого порошка. Выход (количество): 279 мг, Выход (процент): 62,0%
[0081]
1H-ЯМР (DMSO-d6): δ м.д. 1,53-1,68 (м, 1H), 1,86-1,98 (м, 1H), 2,08-2,21 (м, 1H), 2,25-2,39 (м, 1H), 2,82-2,95 (м, 0,5H), 3,10-3,22 (м, 0,5H), 3,23-3,37 (м, 0,5H), 3,68-3,78 (м, 0,5H), 4,04-4,14 (м, 0,5H), 4,22-4,38 (м, 1H), 4,52-4,65 (м, 0,5H), 4,67-4,81 (м, 1H), 5,58-5,74 (м, 1H), 6,03-6,19 (м, 1H), 6,62 (с, 1H), 6,68-6,92 (м, 1H), 7,28-7,40 (м, 2H), 7,59-7,71 (м, 2H), 8,22 (ушир.с, 2H), 8,28 (с, 1H), 12,15 (ушир.с, 1H)
[0082]
Спектр порошковой рентгеновской дифракции: Показан на Фиг. 2.
Характеристический дифракционный угол (2θ ± 0,1°): 4,5°, 5,8°, 11,2°, 12,1°, 12,4°, 13,4°, 16,6°, 17,3°, 18,2°, 20,2°, 26,4°, 27,1°
[0083]
Кривая дифференциальной сканирующей калориметрии (ДСК): Показана на Фиг. 3.
Эндотермический пик на кривой дифференциальной сканирующей калориметрии (ДСК): в области от 197°C до 199°C
[0084]
Пример 3 Синтез монофумарата (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-N-(бензо[d]оксазол-2-ил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида⋅ (монофумарата Соединения A (кристалл))
[0085]
Монофумарат Соединения A, полученный в Примере 1 (аморфный) (500 мг), суспендировали в ацетонитриле (10 мл) и нагревание суспензии осуществляли при 80°C в течение 24 часов. Вещество собирали фильтрованием и затем сушили при пониженном давлении с получением монофумарата Соединения A (кристалл) в виде белого порошка. Выход (количество): 448 мг, Выход (процент): 89,6%
[0086]
1H-ЯМР (DMSO-d6): δ м.д. 1,53-1,68 (м, 1H), 1,86-1,98 (м, 1H), 2,08-2,21 (м, 1H), 2,25-2,39 (м, 1H), 2,82-2,95 (м, 0,5H), 3,10-3,22 (м, 0,5H), 3,23-3,37 (м, 0,5H), 3,68-3,78 (м, 0,5H), 4,04-4,14 (м, 0,5H), 4,22-4,38 (м, 1H), 4,52-4,65 (м, 0,5H), 4,67-4,81 (м, 1H), 5,58-5,74 (м, 1H), 6,03-6,19 (м, 1H), 6,62 (с, 2H), 6,68-6,92 (м, 1H), 7,28-7,40 (м, 2H), 7,59-7,71 (м, 2H), 8,22 (ушир.с, 2H), 8,28 (с, 1H), 12,15 (ушир.с, 1H)
[0087]
Спектр порошковой рентгеновской дифракции: Показан на Фиг. 4.
Характеристический дифракционный угол (2θ ± 0,1°): 7,2°, 12,4°, 14,4°, 15,0°, 15,6°, 19,0°, 22,3°, 22,6°, 23,4°, 25,5°, 25,9° и 27,6°
Кривая дифференциальной сканирующей калориметрии (ДСК): Показана на Фиг. 5.
Эндотермический пик на кривой дифференциальной сканирующей калориметрии (ДСК): в области от 219°C до 224°C
[0088]
Сравнительный Пример 1 Синтез (R)-1-(3-(4-амино-3-(4-феноксифенил)-1H-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-ил)проп-2-ен-1-она (Сравнительное соединение 1)
Синтез осуществляли в соответствии со способом, описанным в WO 2008/121742, с получением указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (DMSO-d6): δ м.д. 1,21-1,28 (м, 1H), 1,42-1,71 (м, 1H), 1,91 (ушир.с, 1H), 2,04-2,36 (м, 2H), 2,91-3,10 (м, 1H), 3,13-3,27 (м, 1H), 3,59-3,76 (м, 1H), 4,04-4,26 (м, 2H), 4,47-4,80 (м, 2H), 5,51-5,78 (м, 1H), 5,96-6,21 (м, 1H), 6,64-6,95 (м, 1H), 7,14 (дд, J=11,46, 8,54 Гц, 6H), 7,40-7,47 (м, 2H), 7,63-7,70 (м, 2H), 8,26 (с, 1H)
[0089]
Эффекты фумарата Соединения A по настоящему изобретению были подтверждены следующими Примерами испытаний. В этой связи, в качестве Соединения A и его соли в представленных Примерах испытаний использовали их кристаллы, если не указано иное.
[0090]
Пример испытания 1 Испытание абсорбции/десорбции влаги
Испытание абсорбции/десорбции влаги осуществляли в отношении Соединения A, монофумарата Соединения A, гемифумарата Соединения A, гемитартрата Соединения A и монофосфата Соединения A для исследования присутствия или отсутствия свойства гидрата канального типа.
[0091]
В испытании абсорбции/десорбции влаги измерения осуществляли в соответствии со следующими условиями.
Специальный кварцевый держатель заполняли примерно 5-10 мг образца, и массу образца при каждой влажности измеряли и регистрировали непрерывно в следующих условиях. Обращения с устройствами, включая обработку данных осуществляли на основании способа и режима, указанного в каждом устройстве.
[0092]
Устройство: VTI SA+ (изготовитель TA Instruments Inc.)
Температура сушки: 60°C
Скорость повышения температуры: 1°C/мин.
Равновесие при сушке: Было подтверждено, что никакого уменьшения 0,01% масс. не происходило через 5 минут, в пределах времени не более 300 минут.
Температура для измерения: 25°C
Равновесие при увлажнении: Было подтверждено, что никакого увеличения 0,01% масс. не происходило через 5 минут, в пределах времени не более 120 минут.
Программа относительной влажности: Увеличение на 5% RH от 5% RH до 95% RH, и уменьшение на 5% RH от 95% RH до 5% RH.
[0093]
Графики, полученные в этих испытаниях, показаны на Фиг. 6 - Фиг. 10. Изменения массы в пределах условий измерений показаны в Таблице 2-1 - Таблице 2-5.
[0094]
|
[0095]
|
[0096]
|
[0097]
|
[0098]
|
[0099]
Как показано в Таблицах 2-1 по 2-5, когда Соединение A увлажняли при относительной влажности от 5 до 95%, которая находится в пределах условий измерений, изменение его массы составляло максимум около 4,4%. Также было подтверждено, что при уменьшении влажности от относительной влажности 95% Соединение A возвращалось почти к исходному состоянию. То есть было обнаружено, что Соединение A обладало свойством гидрата канального типа, который будет абсорбировать/десорбировать влагу в зависимости от влажности.
Подобным образом, гемитартрат Соединения A показал изменение массы максимум около 3,3% в условиях увлажнения при относительной влажности от 5 до 95%. Также было подтверждено, что при понижении влажности от относительной влажности 95% Соединение A возвращалось почти к исходному состоянию. То есть, было обнаружено, что гемитартрат Соединения A также обладал свойством гидрата канального типа, который будет абсорбировать/десорбировать влагу в зависимости от влажности.
Также было обнаружено, что в монофосфате Соединения A кристаллическая форма после испытания абсорбции/десорбции влаги не сохраняла исходную кристаллическую форму.
В отличие от этого, как в монофумарате Соединения A, так и в гемифумарате Соединения A по настоящему изобретению изменение массы поддерживалось на уровне примерно меньше чем 1% увеличения, при относительной влажности 95%, и было обнаружено, что Соединение A возвращалось почти к исходному состоянию при понижении влажности. Таким образом, было подтверждено, что фумарат Соединения A по настоящему изобретению способен избегать свойства гидрата канального типа и обладает превосходными свойствами в качестве фармацевтического продукта или лекарственного ингредиента для фармацевтического продукта.
[0100]
Пример испытания 2 Испытание стабильности твердого состояния (ускоренное испытание)
Стабильность твердого состояния измеряли в указанных ниже условиях в отношении монофумарата Соединения A, гемифумарата Соединения A, гемитартрата Соединения A и монофосфата Соединения A, которые были получены в примерах и Ссылочных Примерах, когда их хранили в течение 2 недель или 4 недель при 40°C/75% RH (закрытые условия и открытые условия).
[0101]
Условия хранения: 40°C/75% RH (закрытые и открытые) (открытые условия относятся к условиям, где крышку стеклянного контейнера снимали и контейнер накрывали KimWipe).
Точки измерения: 2 недели и 4 недели
Используемое для хранения количество: около 30 мг
Контейнер для хранения: коричневый стеклянный контейнер
Способ получения раствора образца: Образец растворяли в 50% ацетонитриле так, чтобы концентрация образца составляла 0,4 мг/мл.
[0102]
Массу аналогичного вещества в растворе образца измеряли при помощи ВЭЖХ анализа. Обращения с устройствами, включая обработку данных осуществляли на основании способа и режима, указанного в каждом устройстве. (Устройство: Shimadzu Corporation SIL-HTc/LC-20AB)
Колонка: InertSustein C18, 4,6 × 150 мм, 3 мкм, изготовитель GL Sciences Inc.
МС детекция: ESI положительный режим
УФ детекция: 220 нм
Температура колонки: 40°C
Скорость потока через колонку: 1,0 мл/мин
Подвижная фаза: A: смешанный раствор 10 ммоль/л фосфатный буфер (pH 6,0):ацетонитрил (17:3), B: ацетонитрил
Объем вводимой пробы: 5 мкл
Градиент: Таблица 3
[0103]
|
[0104]
Таблица 4 представляет результаты оценки измеренной массы аналогичного вещества. В этой Таблице A, B и C относятся к процентам общей массы аналогичного вещества меньше чем 0,1%, 0,1% или больше, и меньше чем 0,5% и 0,5% или больше, соответственно. В данном случае, звездочка (*) означает, что значение было измерено через 2 недели, а остальные измеряли через 4 недели.
[0105]
|
[0106]
В результате было обнаружено, что монофумарат Соединения A и гемифумарат Соединения A образовывали небольшое количество аналогичного вещества и демонстрировали превосходную стабильность твердого вещества по сравнению с гемитартратом Соединения A или монофосфатом Соединения A. Таким образом, было подтверждено, что фумарат Соединения A по настоящему изобретению демонстрирует превосходную стабильность твердого состояния.
[0107]
Пример испытания 3 Испытание стабильности твердого состояния (Испытание в жестких условиях)
Стабильность твердого состояния измеряли в указанных ниже условиях в отношении монофумарата Соединения A, гемитартрата Соединения A и монофосфата Соединения A, которые были получены в Примерах и Ссылочных Примерах, когда их хранили в течение 2 недель или 4 недель при 60°C.
Условия хранения: 60°C (закрытые)
Точка измерения: 2 недели и 4 недели
Используемое для хранения количество: около 30 мг
Контейнер для хранения: коричневый стеклянный контейнер
Способ получения раствора образца: Образец растворяли в 50% ацетонитриле так, чтобы концентрация образца составляла 0,4 мг/мл.
[0108]
Также как в Примере испытания 2, Таблица 5 представляет результаты оценки массы аналогичного вещества в растворе образца, измеренной при помощи ВЭЖХ анализа. В этой Таблице A и B относятся к процентам общей массы аналогичного вещества меньше чем 0,1%, 0,1% или больше и меньше чем 0,5%, соответственно. В данном случае, звездочка (*) означает, что значение было измерено через 2 недели, а остальные измеряли через 4 недели.
[0109]
|
[0110]
В результате было обнаружено, что монофумарат Соединения A образовывал небольшое количество аналогичного вещества и демонстрировал превосходную стабильность твердого вещества по сравнению с гемитартратом Соединения A или монофосфатом Соединения A. Таким образом, было подтверждено, что фумарат Соединения A по настоящему изобретению демонстрирует превосходную стабильность твердого состояния.
[0111]
Пример испытания 4 Измерение концентрации в крови
Для Соединения A, гемифумарата Соединения A и монофумарата Соединения A, которые были получены в Примерах, подготавливали суспензии 50 мг/10 мл/кг, в расчете на молекулярную массу Соединения A, с 0,5% HPMC. Эти растворы для введения перорально вводили мышам (Balb/cA), которых выращивали в условиях откорма, при дозе 10 мл на 1 кг массы тела через зонд для перорального введения. После введения мышей возвращали в клетку и контролировали их состояние. Внутри клетки обеспечивали условия, в которых вода и пища были доступны ad libitum. Через 0,25, 0,5, 1, 2, 4 и 6 часов после введения мышей анестезировали изофлураном, и 60 мкл крови собирали из орбитальной пазухи с использованием пробирки для забора капиллярной крови.
Собранную кровь охлаждали льдом и плазму крови отделяли с использованием процедуры центрифугирования. Мышей после сбора крови возвращали в клетку для содержания животных и состояния после восстановления от анестезии контролировали. После завершения последней процедуры взятия крови мышей умерщвляли путем цервикальной дислокации после проверки уровня анестезии изофлураном.
AUC0-6ч, Cmax и Tmax рассчитывали линейно-логарифмическим методом трапеций с использованием Phoenix WinNonlin (v6.3.0), которая представляет собой программу, разработанную Pharsight Corporation, на основании концентрации Соединения A в каждом образце плазмы крови, измеренной MRM методом с использованием ЖХ-МС/МС.
[0112]
Результаты показаны в Таблице 6. В испытании было обнаружено, касательно Cmax (максимальная концентрация в крови), что гемифумарат Соединения A показал значение, эквивалентное значению Соединения A, а монофумарат Соединения A показал значение примерно в два раза больше, чем Соединение A; и касательно AUC0-6ч (площадь под кривой концентрация в крови-время, 0-6 часов после введения), гемифумарат Соединения A показал значение примерно в два раза больше, чем Соединение A, а монофумарат Соединения A показал значение примерно в 1,3 раза больше, чем Соединение A. Таким образом, было подтверждено, что фумарат Соединения A по настоящему изобретению демонстрирует превосходное свойство абсорбции при пероральном введении.
[0113]
|
[0114]
Пример испытания 5 Измерение ингибиторной активности в отношении BTK (in vitro)
В условиях, установленных для способа измерения ингибиторной активности in vitro соединений в отношении BTK киназной активности, FL-Пептид 2 использовали в качестве субстрата, поскольку он был описан в прайс-листе LabChip (зарегистрированная торговая марка) серии материалов для использования в качестве образцов от PerkinElmer Co., Ltd., и FL-Пептид 2 соответствовал пептидному субстрату в измерении BTK киназной активности. Очищенный рекомбинантный человеческий BTK белок, используемый в испытании, закупали у Carna Biosciences, Inc.
Что касается измерения ингибиторной активности соединений, сначала монофумарат Соединения A последовательно разбавляли диметилсульфоксидом (DMSO). Затем BTK белок, пептидный субстрат (конечная концентрация 1 мкМ), хлорид магния (конечная концентраци 10 мМ), АТФ (конечная концентрация 45 мкМ) и DMSO раствор испытываемых соединений (конечная концентрация DMSO 5%) добавляли к буферному раствору для киназной реакции (20 мМ HEPES (pH 7,5), 2 мМ дитиотреитола, 0,01% Triton X-100) и затем раствор инкубировали в течение 40 минут при 25°C, после чего осуществляли киназную реакцию. Реакцию останавливали путем добавления EDTA для получения конечной концентрации 30 мМ. В завершение, пептидный субстрат, который не был фосфорилирован (S), и фосфорилированный пептид (P) разделяли и осуществляли детекцию методом микроканального капиллярного электрофореза с использованием LabChip EZ Reader II (PerkinElmer, Inc.). Количество фосфорилирования определяли из высоты индивидуальных пиков S и P, и концентрацию соединения, при которой реакция фосфорилирования может подавляться на 50%, определяли как IC50 значение (нМ). Результаты показаны в Таблице 7 ниже.
[0115]
|
[0116]
В результате испытания было обнаружено, что фумарат Соединения A по настоящему изобретению обладал ингибиторной активностью в отношении BTK in vitro.
[0117]
Пример испытания 6 Селективность ингибирования BTK по сравнению с ингибиторной активностью в отношении EGFR киназы (in vitro)
1) Измерение ингибиторной активности в отношении BTK
Ингибиторную активность в отношении BTK измеряли таким же способом, как в Примере испытания 5.
2) Измерение ингибиторной активности в отношении EGFR
Что касается установления условий для способа измерения ингибиторной активности соединения в отношении EGFR киназной активности in vitro, в прайс-листе предлагаемых готовых для применения реагентов для LabChip (зарегистрированная торговая марка) серии от PerkinElmer Co., Ltd. описано, что FL-ПЕПТИД 22 соответствует пептидному субстрату для измерения EGFR киназной активности. Поэтому биотинилированный пептид (биотин- EEPLYWSFPAKKK) был получен путем соотнесения с аминокислотной последовательностью пептида. Очищенный рекомбинантный человеческий EGFR белок, используемый в испытании, закупали у Carna Biosciences, Inc.
Что касается измерения ингибиторной активности соединений, сначала монофумарат Соединения A последовательно разбавляли диметилсульфоксидом (DMSO). Затем EGFR белок, пептидный субстрат (конечная концентрация 250 нМ), хлорид магния (конечная концентрация 10 мМ), хлорид марганца (конечная концентрация 10 мМ), АТФ (конечная концентрация 1,5 мкМ) и DMSO раствор испытываемых соединений (конечная концентрация DMSO 2,5%) добавляли к буферному раствору для киназной реакции (20 мМ HEPES (pH 7,5), 2 мМ дитиотреитола, 0,01% Triton X-100) и затем раствор инкубировали в течение 120 минут при 25°C, после чего осуществляли киназную реакцию. Реакцию останавливали путем добавления EDTA так, чтобы получить конечную концентрацию 24 мМ. Затем добавляли жидкость для детекции, содержащую Eu-меченное антифосфотирозиновое антитело PT66 (PerkinElmer, Inc.) и SURELIGHT APC-SA (PerkinElmer, Inc.), и систему оставляли выстаиваться в течение 2 часов или дольше при комнатной температуре. В завершение, количество флуоресценции после облучения при длине волны возбуждения 337 нм измеряли при двух длинах волн 620 нм и 665 нм при помощи PHERAstar FS (BMG Labtech GmbH). Количество фосфорилирования определяли из отношения количеств флуоресценции при двух длинах волн, и концентрацию соединения, при которой реакция фосфорилирования может подавляться на 50%, определяли как IC50 значение (нМ).
3) Селективность ингибирования BTK
Отношение "ингибиторная активности в отношении EGFR, значение IC50 (нМ)/ингибиторная активность в отношении BTK значение IC50 (нМ)" рассчитывали на основании результатов, полученных в описанных выше разделах 1) и 2), и, таким образом, определяли селективность ингибирования BTK испытываемым соединением.
[0118]
|
[0119]
На основании результатов испытания было обнаружено, что in vitro селективность ингибирования BTK по сравнению с EGFR киназой у монофумарата Соединения A по настоящему изобретению была примерно в 13 раз выше, чем у Сравнительного соединения 1, и монофумарат Соединения A по настоящему изобретению обладал превосходной селективностью ингибирования BTK. На основании этих результатов было показано, что фумарат Соединения A по настоящему изобретению может иметь меньше нежелательных побочных эффектов по сравнению с существующими ингибиторами BTK.
[0120]
Пример испытания 7 Испытание для измерения активности ингибирования пролиферации против клеточных линий, экспрессирующих BTK и EGFR (in vitro), и сравнение селективности
TMD8 клетки, которые представляют собой клеточную линию диффузной крупноклеточной B-клеточной лимфомы, экспрессирующую BTK, суспендировали в RPMI1640 среде (изготовитель Life Technologies Corp.), содержащей 10% фетальную бычью сыворотку. A431 клетки, которые представляют собой EGFR-сверхэкспрессирующую высокоактивированную клеточную линию эпидермоидной карциномы человека, суспендировали в DMEM среде с высоким содержанием глюкозы (изготовитель Life Technologies Corp.), содержащей 10% фетальную бычью сыворотку. Клеточные суспензии инокулировали в каждую лунку 384-луночных плоскодонных микропланшетов и клетки культивировали в течение 1 дня при 37°C в инкубаторе, содержащем 5% газообразного диоксида углерода. Монофумарат Соединения A и Сравнительное соединение 1 растворяли в DMSO, и растворы разбавляли до 500-кратной концентрации относительно конечной концентрации испытываемого соединения с использованием DMSO. DMSO раствор испытываемых соединений разводили при помощи среды, используемой для суспендирования каждого типа клеток, и добавляли в каждую лунку культуральных планшетов так, чтобы конечная концентрация DMSO была 0,2%. Клетки снова культивировали в течение 3 дней при 37°C в инкубаторе, содержащем газообразный 5% диоксид углерода. Подсчет количества клеток до добавления соединений и после культивирования в течение 3 дней в присутствии соединений осуществляли с использованием CELLTITER GLO (изготовитель Promega Corp.) на основании протокола, рекомендованного Promega Corp. Процент ингибирования пролиферации рассчитывали по следующей формуле и определяли концентрацию испытываемого соединения, обеспечивающую ингибирование 50% (GI50 (нМ)).
[0121]
Процент ингибирования пролиферации (%)=(C - T)/(C - C0) × 100
T: Интенсивность люминесценции лунки с испытываемым соединением
C: Интенсивность люминесценции лунки без испытываемого соединения
C0: Интенсивность люминесценции лунки, измеренная до добавления испытываемого соединения
[0122]
Путем сравнения активности ингибирования клеточной пролиферации в отношении A431 клеток, которая зависит от передачи сигнала пролиферации EGFR, и активности ингибирования клеточной пролиферации в отношении TMD8 клеток, которая зависит от передачи сигнала пролиферации BTK, можно оценить влияние соответствующих киназ на клеточном уровне. А именно, при расчете "процент ингибирования клеточной пролиферации A431/процент ингибирования клеточной пролиферации TMD8", предполагается, что чем больше это отношение, тем больше селективность к BTK по сравнению с EGFR в клетках. Значения "процент ингибирования клеточной пролиферации A431/процент ингибирования клеточной пролиферации TMD8" показаны в Таблице 9.
[0123]
|
[0124]
На основании результатов испытания было обнаружено, что монофумарат Соединения A по настоящему изобретению в анализе ингибирования клеточной пролиферации in vitro имел селективность ингибирования BTK по сравнению с EGFR киназой примерно в 65 раз выше, чем селективность Сравнительного соединения 1. Таким образом, было обнаружено, что фумарат Соединения A по настоящему изобретению обладал превосходной селективностью ингибирования BTK, не только в киназе, но также в клетках. На основании полученных результатов было продемонстрировано, что фумарат Соединения A по настоящему изобретению может иметь меньше нежелательных побочных эффектов по сравнению с существующими ингибиторами BTK.
[0125]
Пример испытания 8 Мышиная модель коллаген-индуцированного артрита (профилактический эффект)
Испытание осуществляли в соответствии со способом, описанным в непатентной литературе (Brand DD, et al., Nat Protoc. 2007; 2, 1269-1275, Xu D. et al., JPET, 2012 Apr; 341(1): 90-103). Самцам DBA/1 мышей (CHARLES RIVER LABORATORIES JAPAN, INC.) семинедельного возраста вводили внутрикожно в тыльную часть стопы инъекцию 100 мкл/животное смешанного в равных количествах раствора (эмульсии) 4 мг/мл раствора бычьего коллагена 2 типа (Collagen Research Center) и полного адъюванта Фрейнда (DIFCO Inc.) (первичная иммунизация). Через 21 день после этого мышам внутрикожно в область спинного хребта вводили инъекцию 100 мкл/животное смешанного в равных количествах раствора (эмульсии) 4 мг/мл раствора бычьего коллагена 2 типа (Collagen Research Center) и полного адъюванта Фрейнда (DIFCO) для обеспечения дополнительной иммунизации. Пероральное введение 1 раз в день продолжали в течение 17 дней, при этом устанавливали день начала введения (день 0) как восьмой день после дополнительной иммунизации. Симптомы артрита в день 0, день 3, день 7, день 10, день 14 и день 17 оценивали макроскопически (0: отсутствие изменений, 1: опухание пальца, 2: опухание двух пальцев или более, 3: опухание тыльной части стопы, 4: опухание всех пальцев, распространяющееся на запястье/заплюсну), и получали общую оценку всех четырех конечностей в виде балла для индивидуального животного (максимум 16 баллов). Результаты показаны на Фиг. 11.
[0126]
Фиг. 11 показывает, что группа, которой вводили преднизолон (3 мг/кг), который использовали в качестве положительного контрольного соединения в системе испытания, только в некоторой степени поддерживала повышенную оценку артрита, тогда как группа, которой вводили монофумарат Соединения A по настоящему изобретению (0,381 мг/кг), эффективно снижала оценку артрита. На основании результатов было подтверждено, что фумарат Соединения A по настоящему изобретению имел превосходной терапевтический эффект против ревматоидного артрита, который уже проявился.
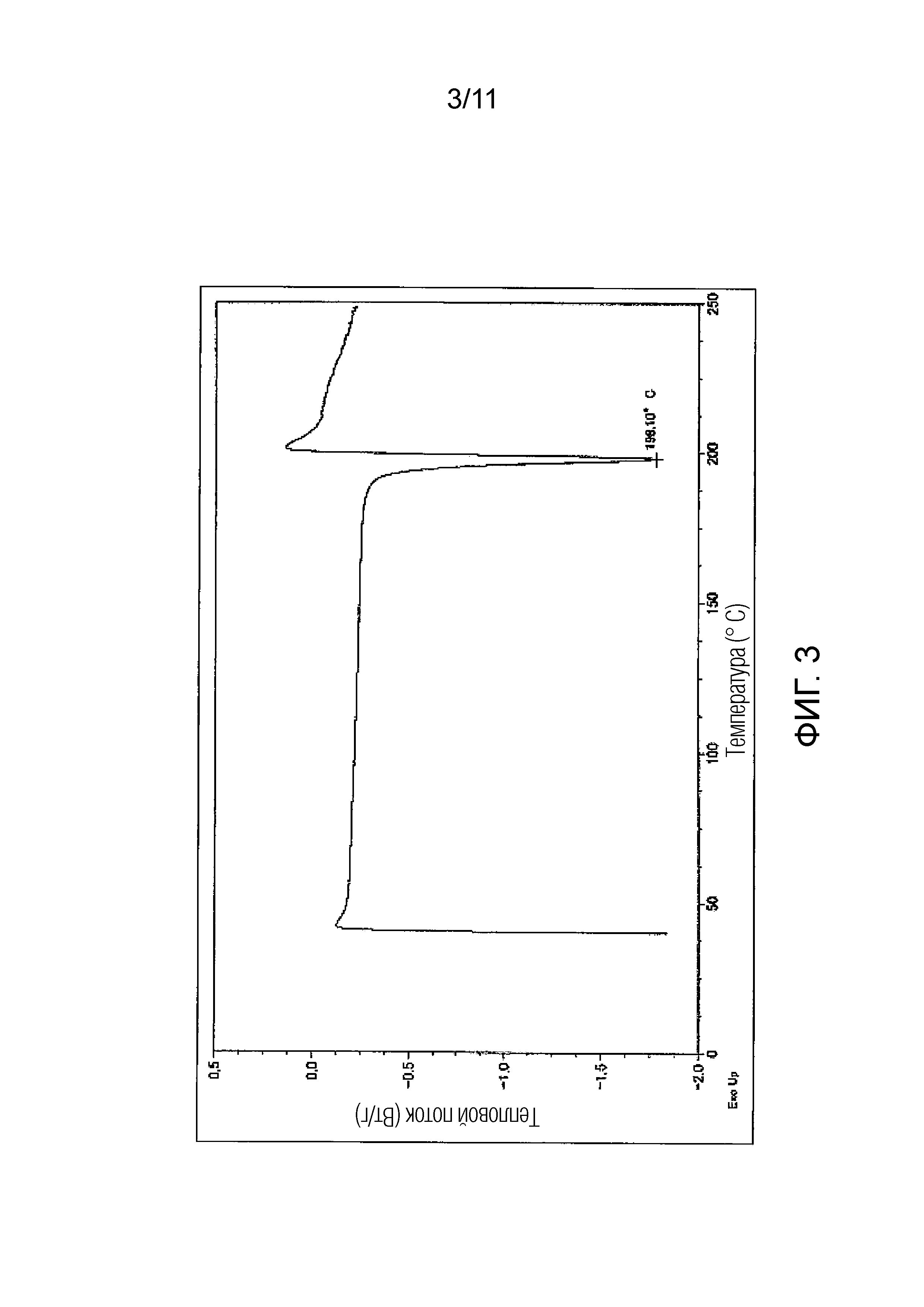
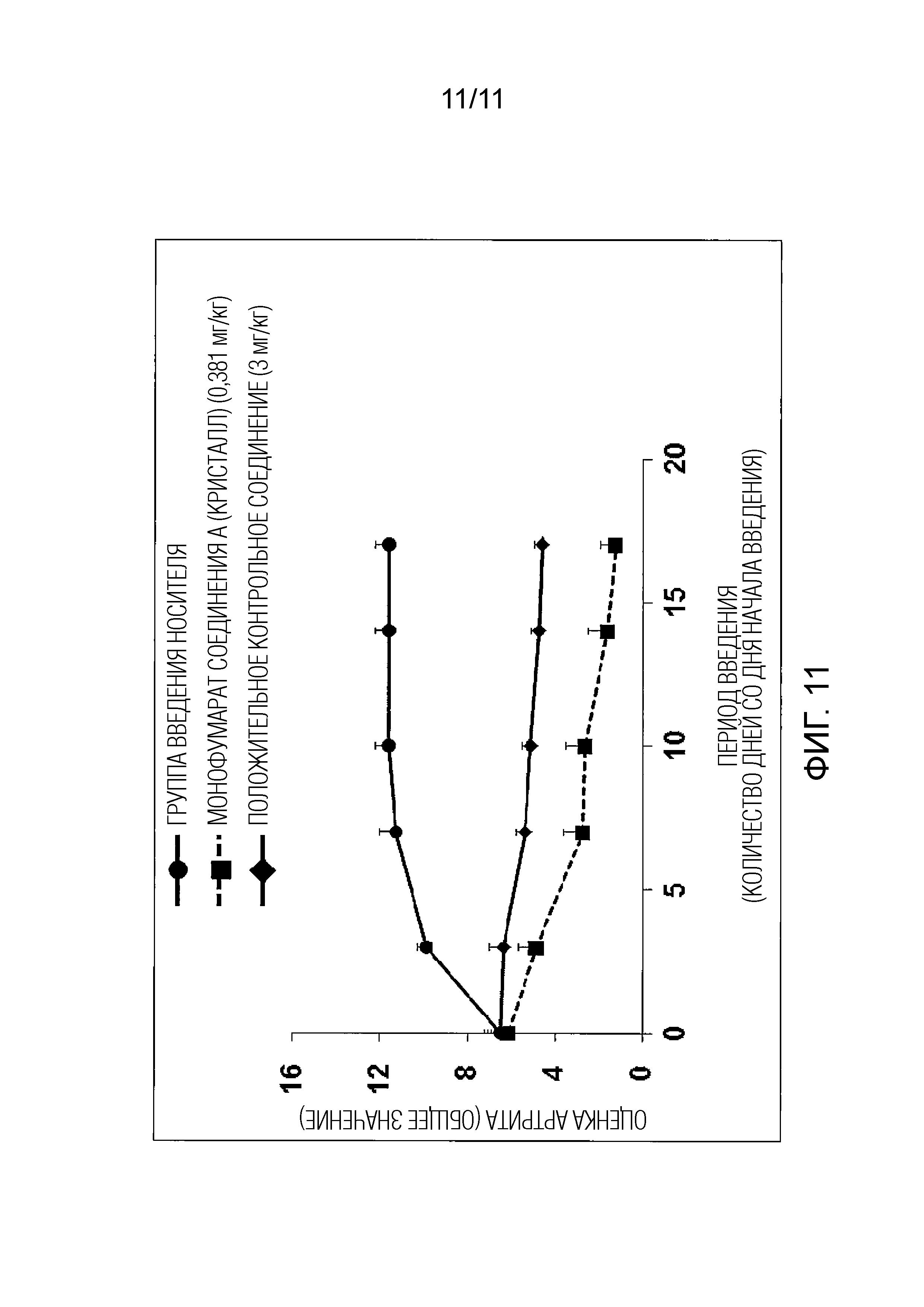
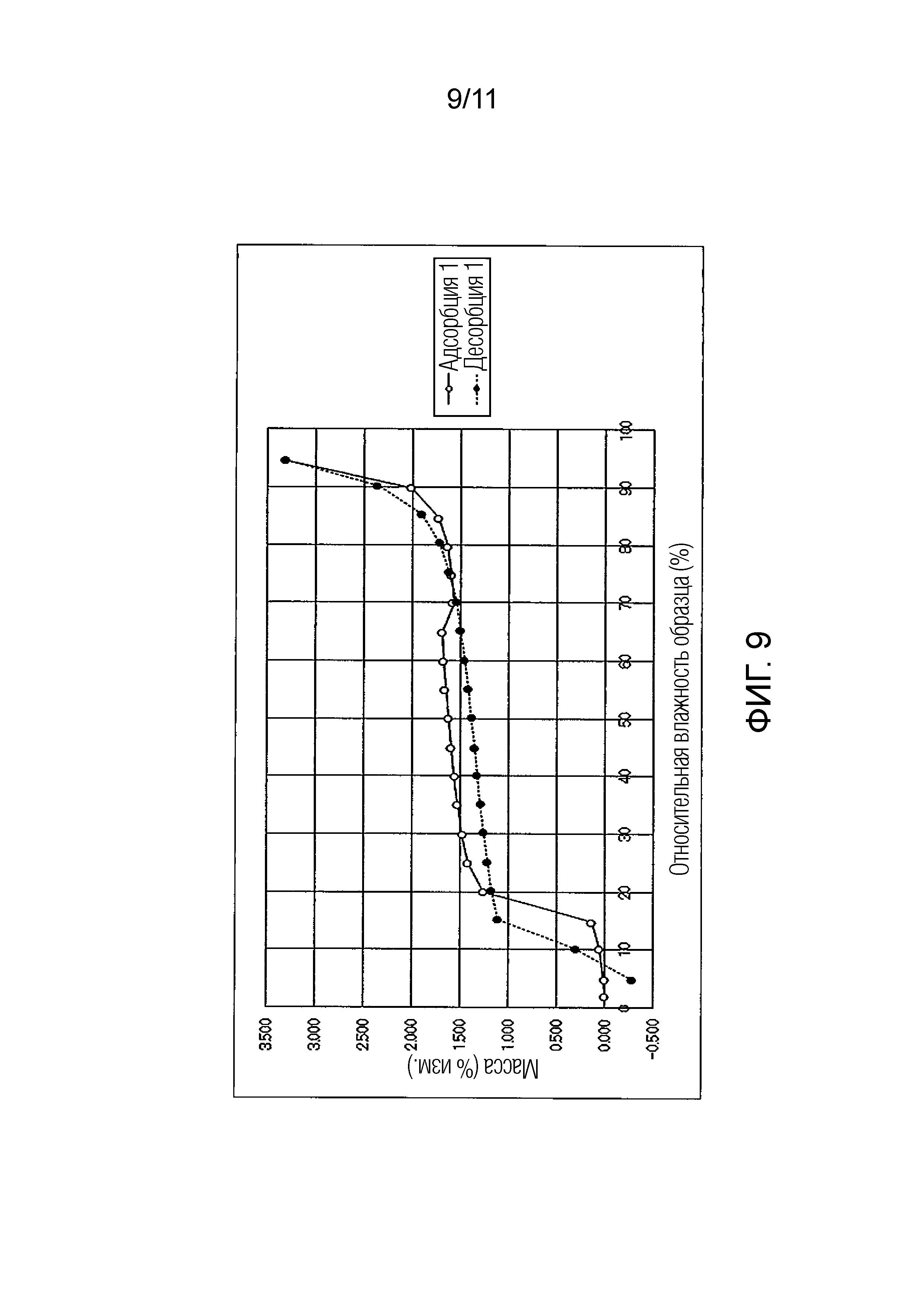
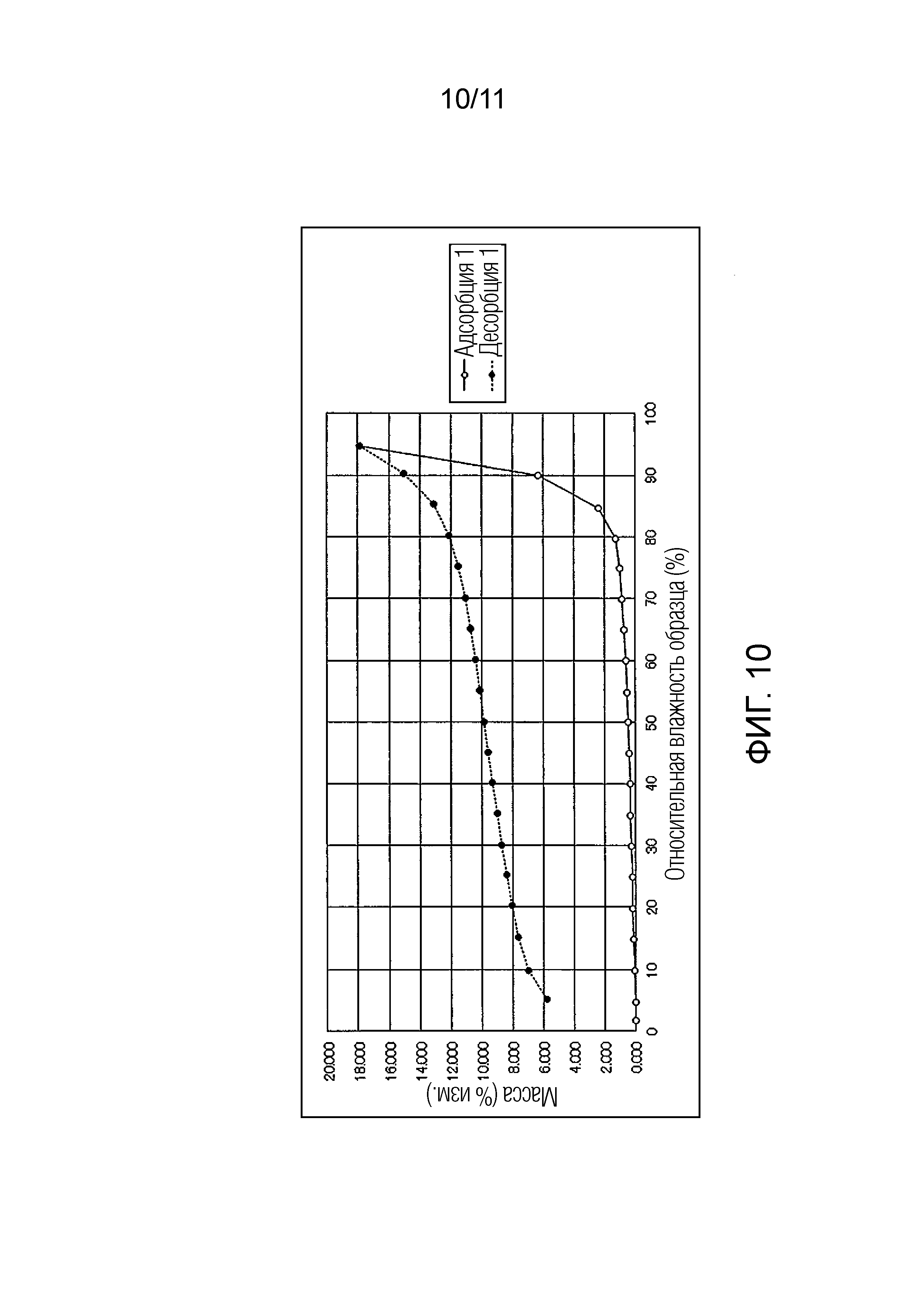
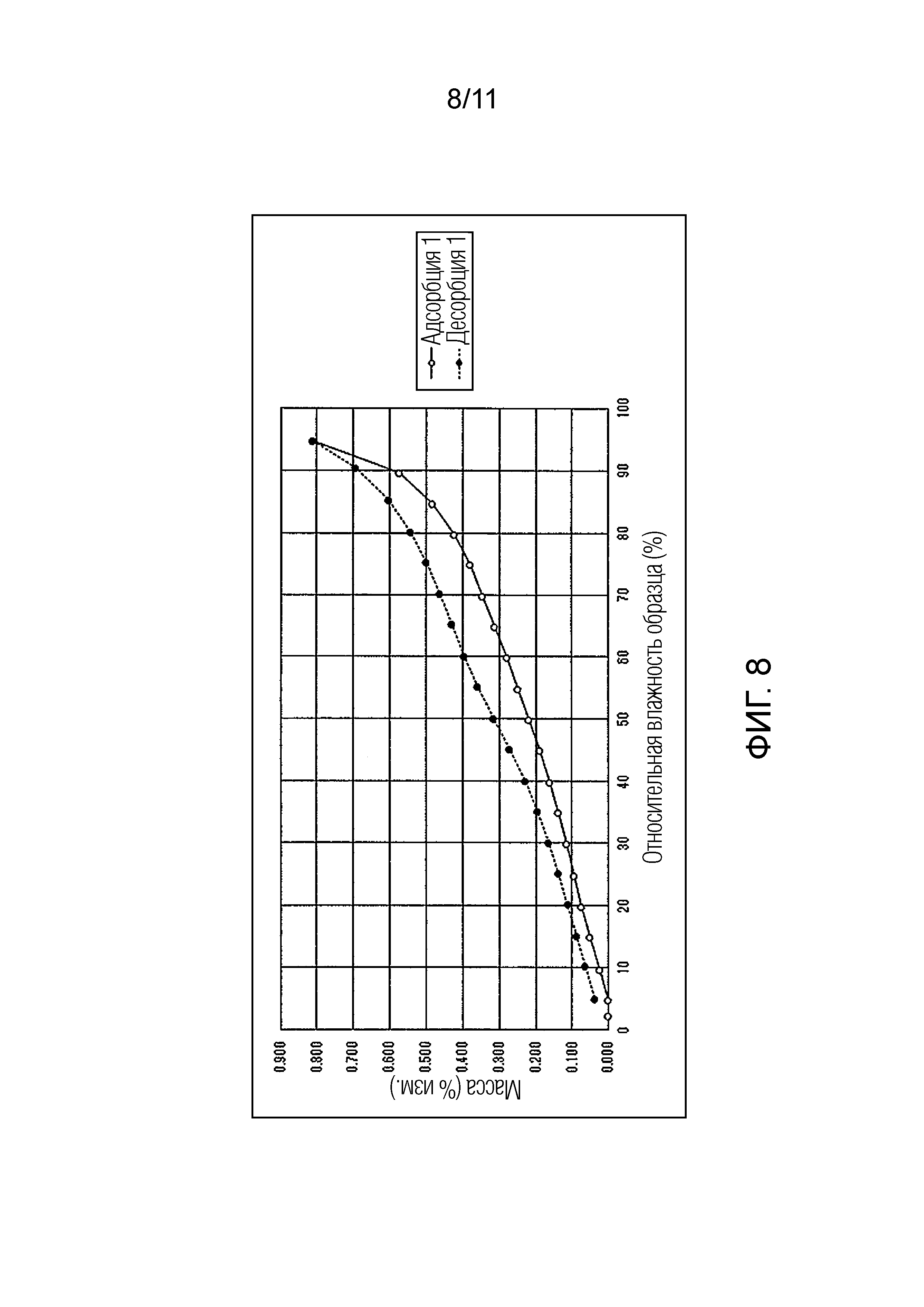
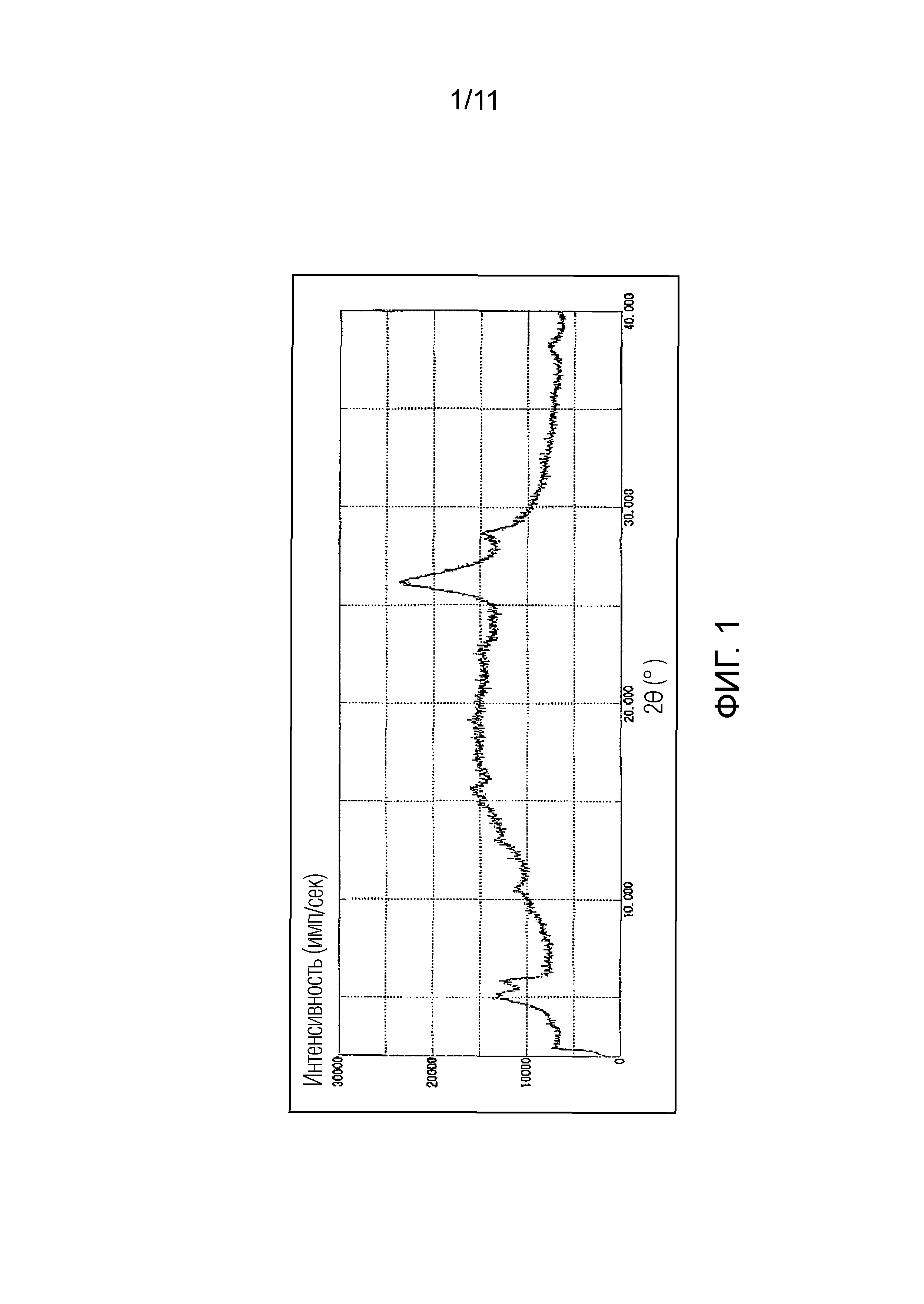
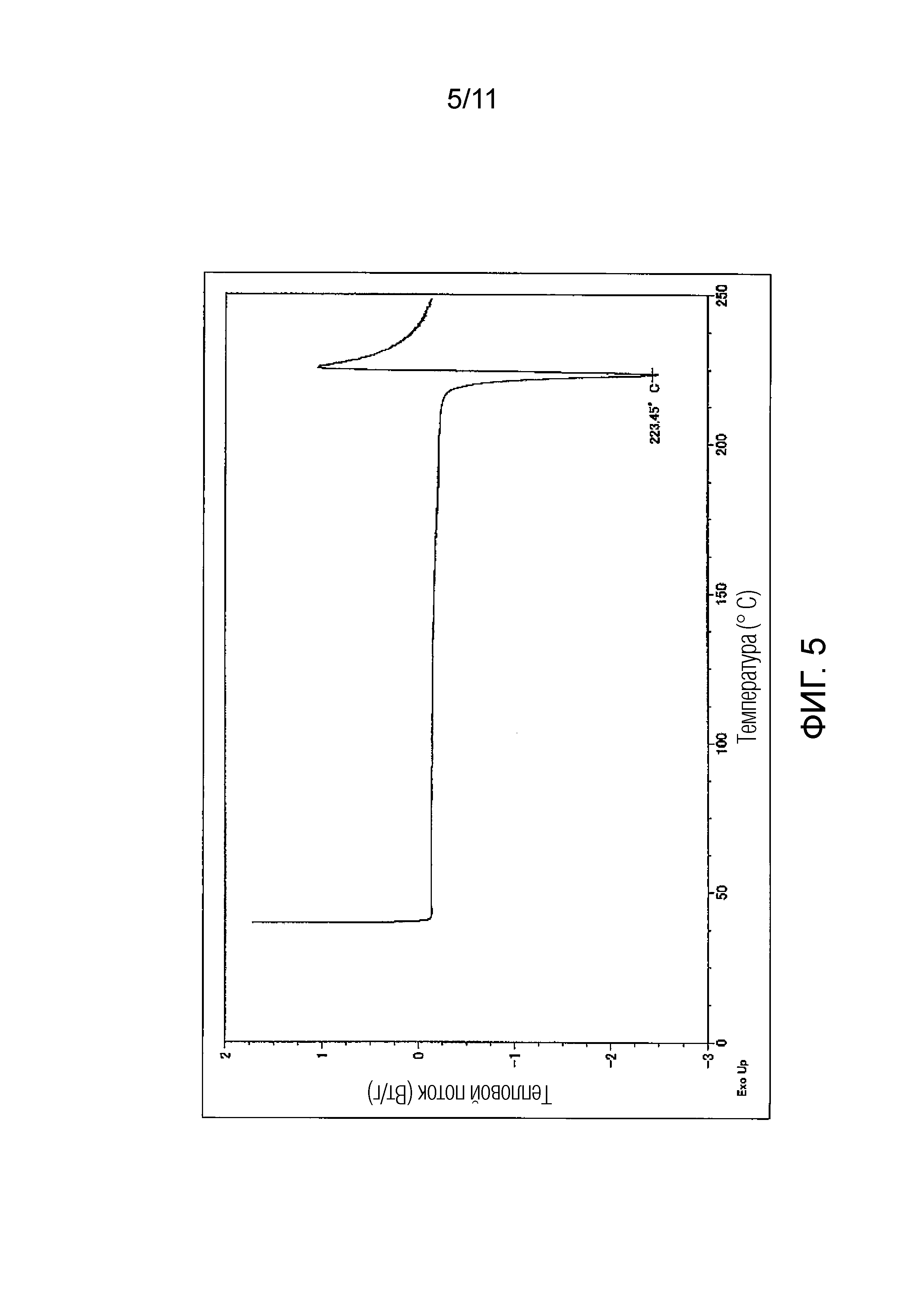
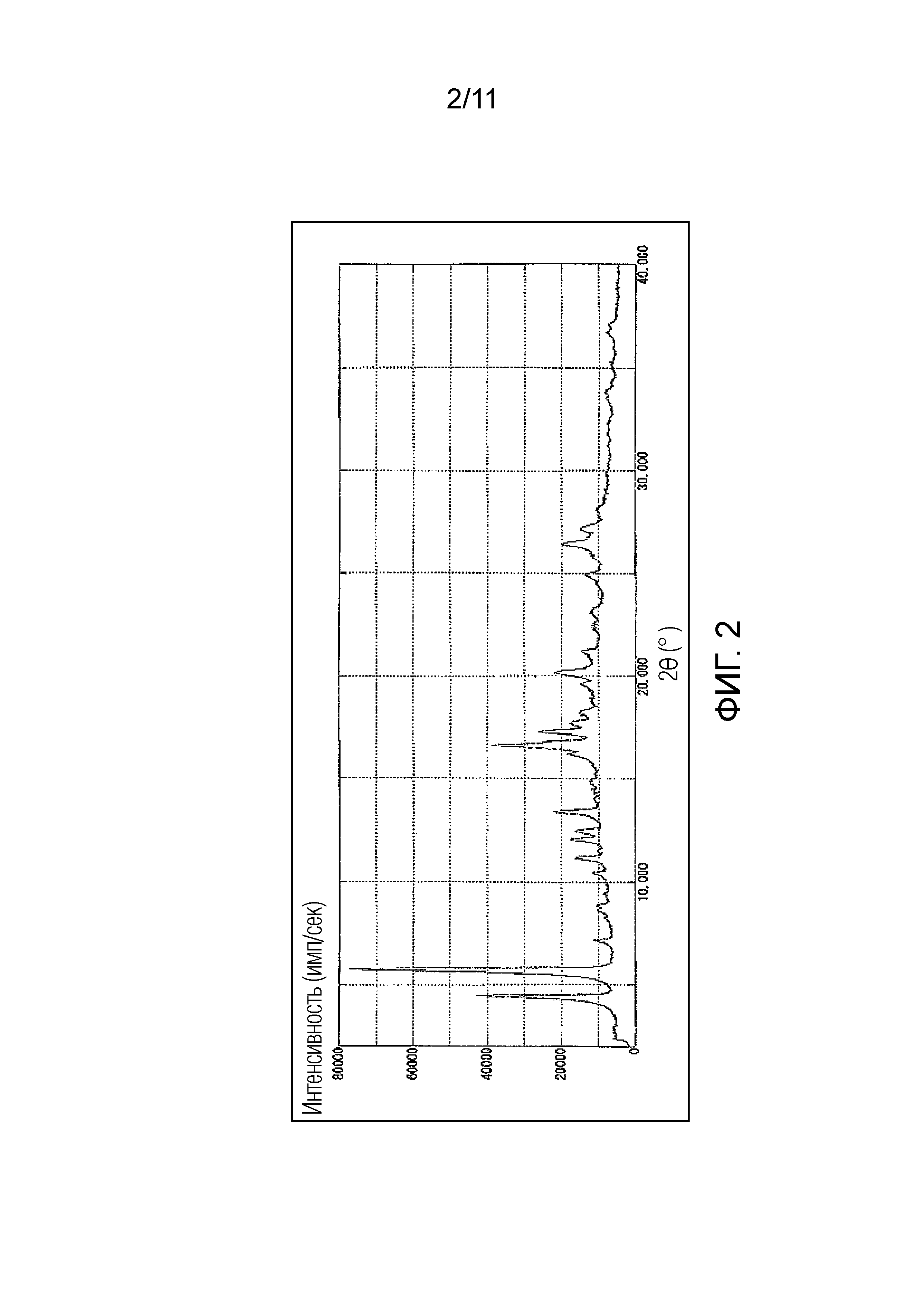
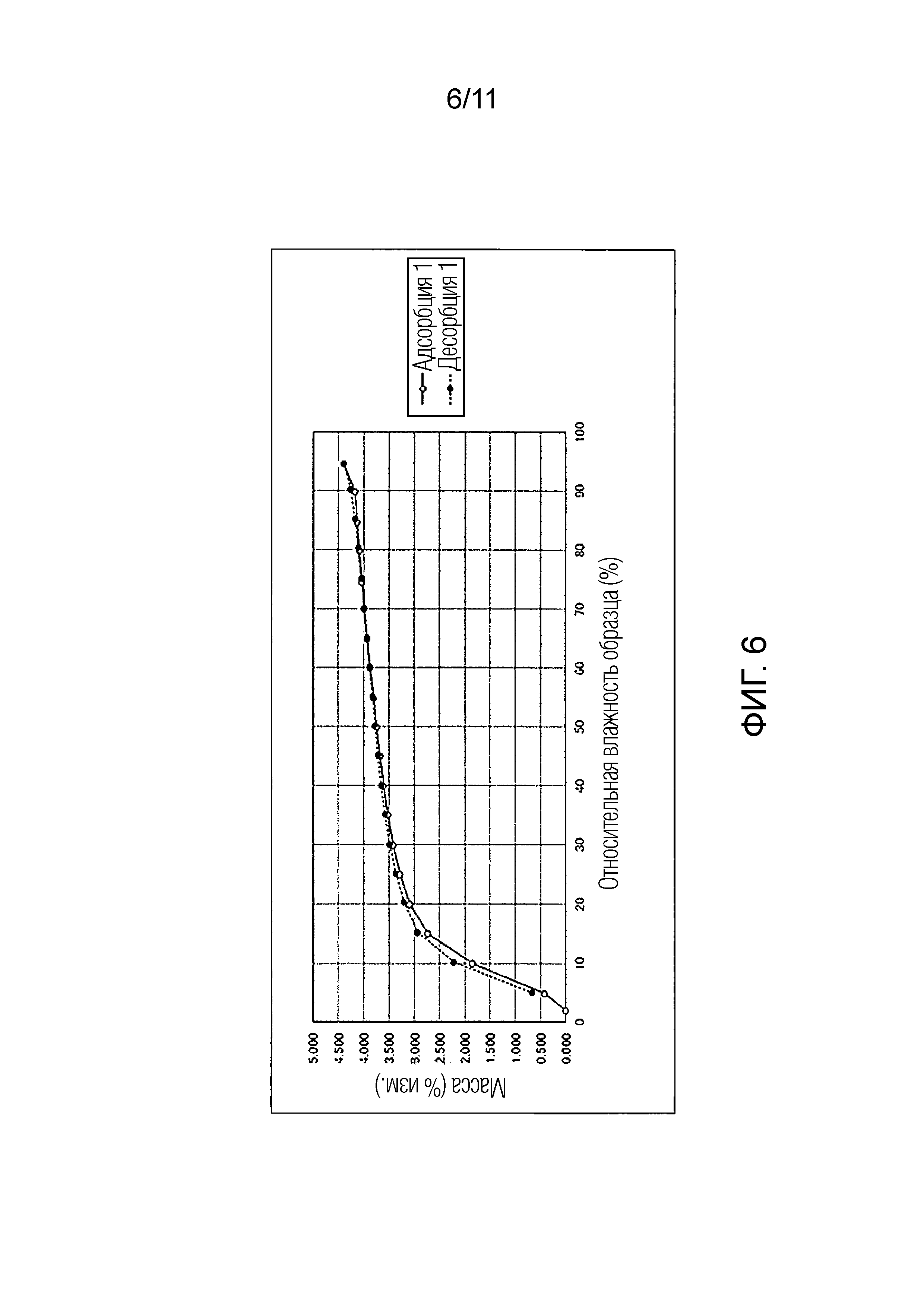

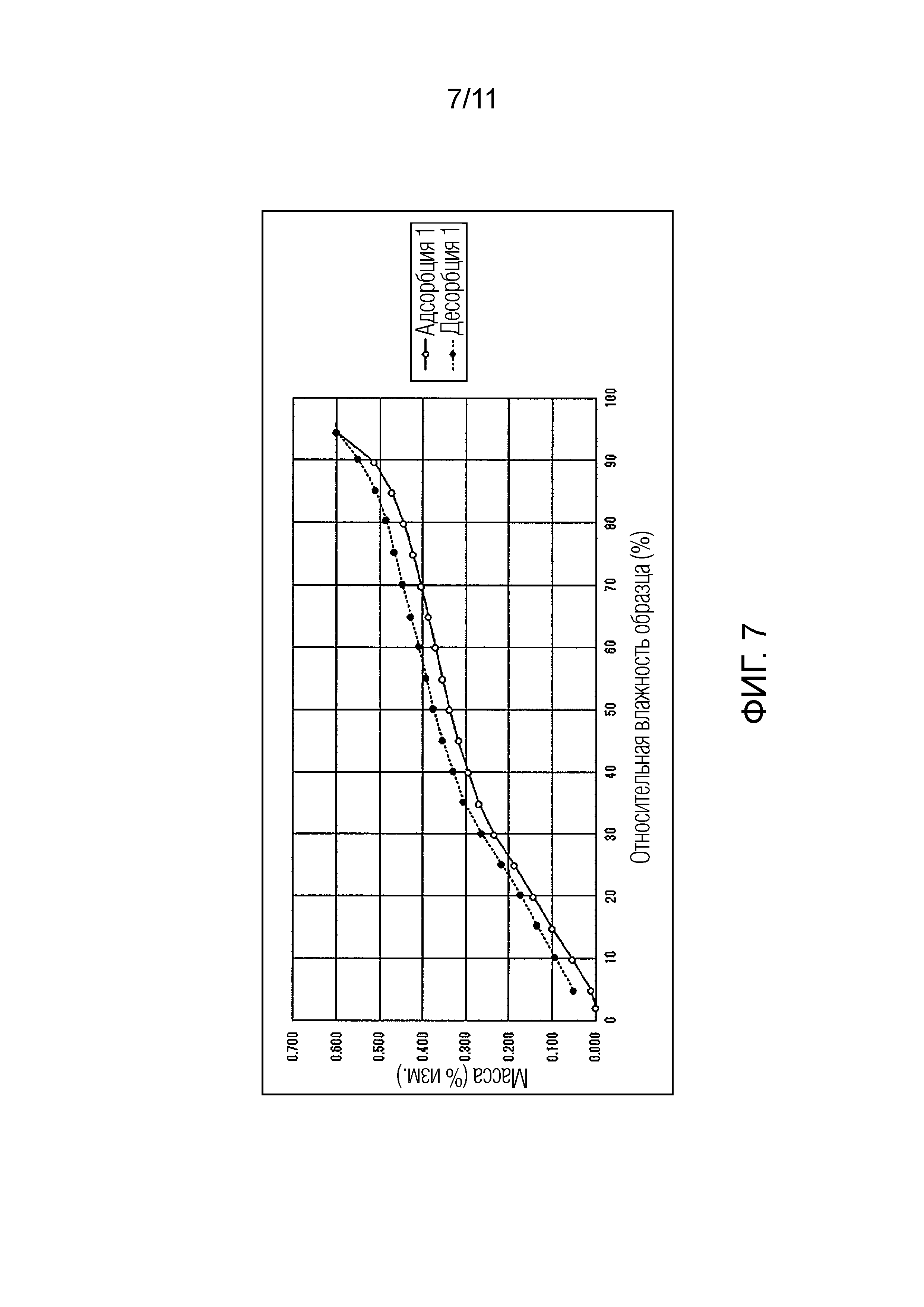
Противоопухолевое средство для непрерывного внутривенного введения, содержащее цитидиновое производное
Противоопухолевое средство, включающее производное цитидина и карбоплатин
Средство, улучшающее противоопухолевый эффект, содержащее липосомальное средство, содержащее оксалиплатин, и противоопухолевое средство, содержащее липосомальное средство
Новое урациловое соединение или его соль, обладающие ингибирующей активностью относительно дезоксиуридинтрифосфатазы человека
Пиперазиновое соединение, ингибирующее простагландин-d-синтазу
Производное ацилтиомочевины или его соль, и его применение
Противоопухолевый агент, набор и способ лечения рака
Способ детекции дегенеративных мышечных заболеваний и способ определения терапевтической эффективности при заболеваниях
Усилитель действия противоопухолевого средства
Азабициклосоединение и его соль
Азабициклосоединение и его соль

