Результат интеллектуальной деятельности: КРИСТАЛЛЫ АЗАБИЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001]
Настоящее изобретение относится к новому кристаллу азабициклического соединения, которое является стабильным и обладает превосходной способностью к всасыванию в полости рта, и является полезным в качестве противоопухолевого агента.
УРОВЕНЬ ТЕХНИКИ
[0002]
В общем, когда соединение используется в качестве эффективного активного ингредиента фармацевтического средства, требуется химическая и физическая стабильность соединения, чтобы сохранить качество стабильным и/или облегчить управление хранением. Следовательно, полученное соединение предпочтительно находится в стабильной кристаллической форме, и, как правило, наиболее стабильная кристаллическая форма часто выбирается в качестве лекарственного вещества для фармацевтического средства.
[0003]
До настоящего времени в качестве противоопухолевого средства сообщалось о множестве ингибиторов HSP90, например, в патентном документе 1 и 2 описаны 3-этил-4-{3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамида (далее также называемого «Соединение 1») в качестве соединения, которое обладает превосходным ингибитором действия HSP90 и проявляет противоопухолевую активность.
[0004]
Между тем, хотя фармацевтическая композиция для перорального введения требует, как правило, не только стабильности эффективного ингредиента, но также отличной абсорбируемости при пероральном введении, в патентном документе 1 и 2 не описываются никакие кристаллы соединения 1, а также стабильность и способность кристаллов к всасыванию в полости рта.
СПИСОК ПРОЦИТИРОВАННОЙ ЛИТЕРАТУРЫ
Патентный документ
[0005]
Патентный документ 1:
Международная публикация № WO2012/093708
Патентный документ 2:
Международная публикация № WO2011/004610
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Проблема, требующая решения с помощью изобретения
[0006]
Целью настоящего изобретения является создание кристалла соединения 1, полезного в качестве противоопухолевого средства, который является стабильным и обладает превосходной способностью к всасыванию полости рта.
Способы решения проблемы
[0007]
Для достижения цели изобретатель настоящего изобретения синтезировал соединение 1 в соответствии со способом изготовления, описанным в патентном документе 1, для получения кристаллической формы I соединения 1. Однако, как описано в примерах ниже, кристаллическая форма I имела проблемы со способностью к всасыванию в полости рта и поэтому они дополнительно исследовали условия кристаллизации. В результате они обнаружили, что кристаллическую форму II можно получить путем добавления и создания суспензии соединения 1 в определенном органическом растворителе и, кроме того, обнаружили, что кристаллическая форма II превосходит по стабильности и способности к всасыванию в полости рта по сравнению с кристаллической формой I, тем самым завершая настоящее изобретение.
[0008]
Таким образом, настоящее изобретение относится к обеспечению следующих [1] - [15].
[0009]
[1] Кристаллическая форма II 3-этил-4-{3-изопропил-4-(4- (1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b]пиридин-1-ил} бензамида, показывающая спектр дифракции рентгеновских лучей на порошке, имеющий, по меньшей мере, три характеристических дифракционных пика при углах (2θ±0,2°), выбранных из группы, состоящей из 7,7°, 8,0°, 11,1°, 12,5°, 12,9 °, 15,2°, 15,8°, 17,2°, 19,0°, 22,5°, 26,1° и 27,4°.
[2] Кристаллическая форма II согласно [1], где кристалл показывает спектр дифракции рентгеновских лучей на порошке, имеющий, по меньшей мере, пять характеристических дифракционных пиков при углах (2θ ±0,2°), выбранных из группы, состоящей из 7,7°, 8,0°, 11,1°, 12,5°, 12,9°, 15,2°, 15,8°, 17,2°, 19,0°, 22,5°, 26,1° и 27,4°
[3] Кристаллическая форма II по [1] или [2], в которой эндотермический пик, определяемый путем дифференциальной сканирующей калориметрии составляет около 270°С.
[4] Фармацевтическая композиция, содержащая кристаллическую форму II по любому из [1] - [3].
[5] Фармацевтическая композиция для перорального введения, включающая кристаллическую форму II в соответствии с любым из [1] - [3].
[6] Противоопухолевое средство, содержащее кристаллическую форму II, согласно любому из [1] - [3].
[7] Применение кристаллической формы II согласно любого с [1] по [3], для производства фармацевтической композиции.
[8] Применение согласно [7], в котором фармацевтическая композиция является фармацевтической композицией для перорального применения.
[9] Применение кристаллической формы II согласно любого из с [1] по [3] для производства противоопухолевого средства.
[10] Кристаллическая форма II согласно любому из с [1] по [3] для использования в качестве лекарственного средства.
[11] Кристаллическая форма II согласно любому из с [1] по [3] для использования при лечении опухоли.
[12] Способ лечения опухоли, включающий введение эффективного количества кристаллической формы II в соответствии с любым из с [1] по [3] субъекту, нуждающемуся в этом.
[13] Способ получения кристаллической формы II по любому из с [1] по [3], включающий стадии:
(1) создание суспензии 3-этил-4-{3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло[3,4-b]пиридин-1-ил} бензамида в органическом растворителе при нагревании с получением суспензии и
(2) получение твердофазного 3-этил-4- {3-изопропил-4- (4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамида из суспензии, полученной на вышеуказанной стадии (1).
[14] Способ получения кристаллической формы II согласно любому из с [1] по [3], в соответствии с [13], где органический растворитель представляет собой 2-пропанол, метилацетат, этилацетат, пропилацетат, бутилацетат, циклопентилметиловый эфир, метилэтилкетон, метилизобутилкетон, ацетон, ацетонитрил или смешанный растворитель из них.
[13] Способ получения кристаллической формы II по любому из с [1] по [3], включающий стадии:
(1) создание суспензии 3-этил-4-{3-изопропил-4- (4- (1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил) -1Н-пиразоло [3,4-b] пиридин-1-ил} бензамида в органическом растворителе, выбранном из группы, состоящей из 2-пропанола, метилацетата, этилацетата, пропилацетата, бутилацетата, циклопентилметилового эфира, метилэтилкетона, метилизобутилкетона, ацетона, ацетонитрила и смешанного растворителя из них для получения суспензии и
(2) получение твердофазного 3-этил-4-{3-изопропил-4- (4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамида из суспензии, полученной на вышеуказанной стадии (1).
ЭФФЕКТ ИЗОБРЕТЕНИЯ
[0010]
В соответствии с настоящим изобретением кристаллическая форма II соединения 1 обладает высокой стабильностью и превосходной способностью к всасыванию в полости рта и полезна в качестве перорального фармацевтического средства.
КРАТКОЕ ОПИСАНИЕ ФИГУР
[0011]
[Фигура 1] Спектр рентгеновской порошковой дифракции кристаллической формы I соединения 1 (вертикальная ось представляет собой интенсивность (cps), а горизонтальная ось представляет собой угол дифракции (2θ±0,2°)).
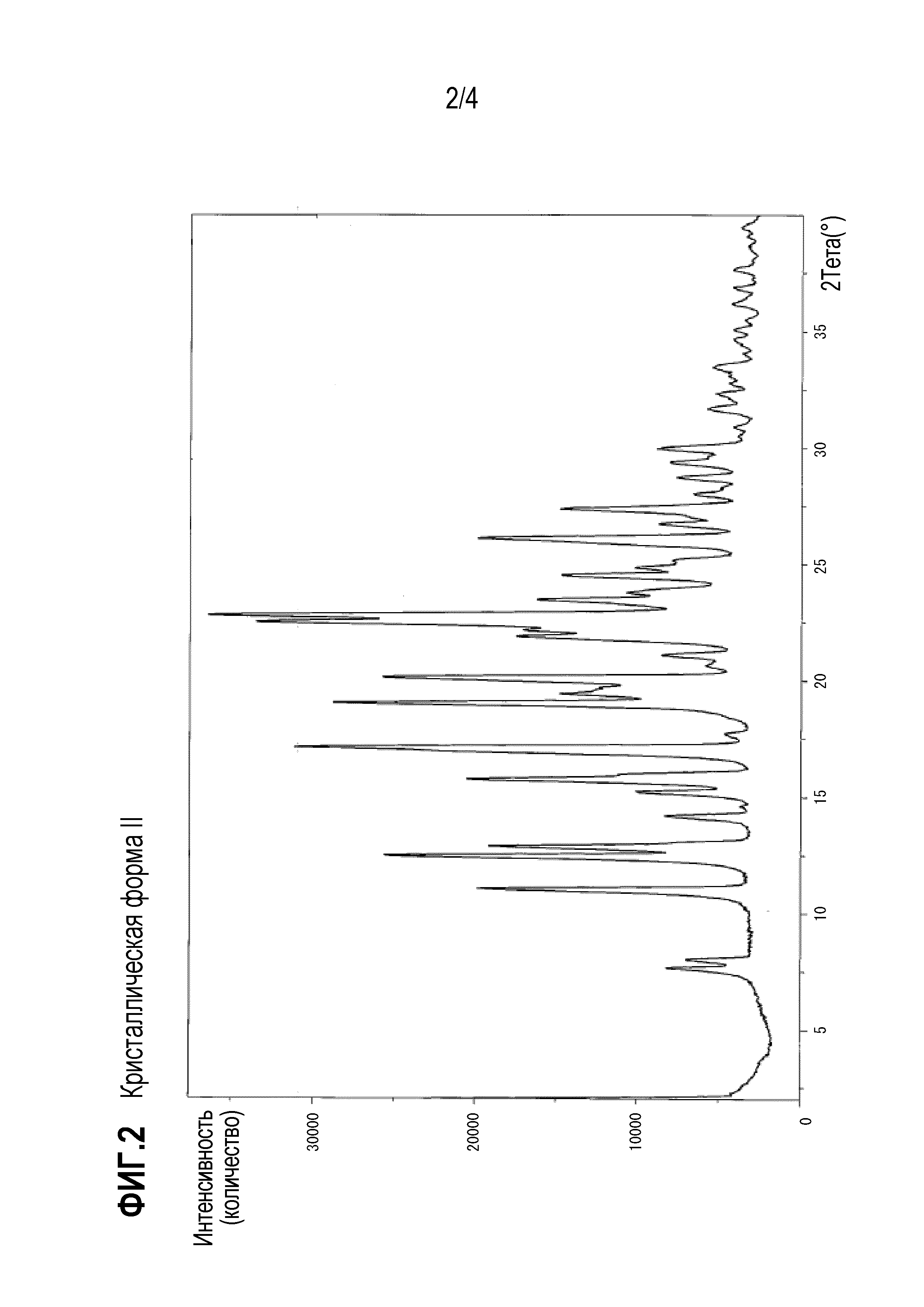
[Фигура 2] Спектр рентгеновской порошковой дифракции кристаллической формы II соединения 1 (вертикальная ось представляет интенсивность (cps), а горизонтальная ось представляет собой угол дифракции (2θ±0,2°)).
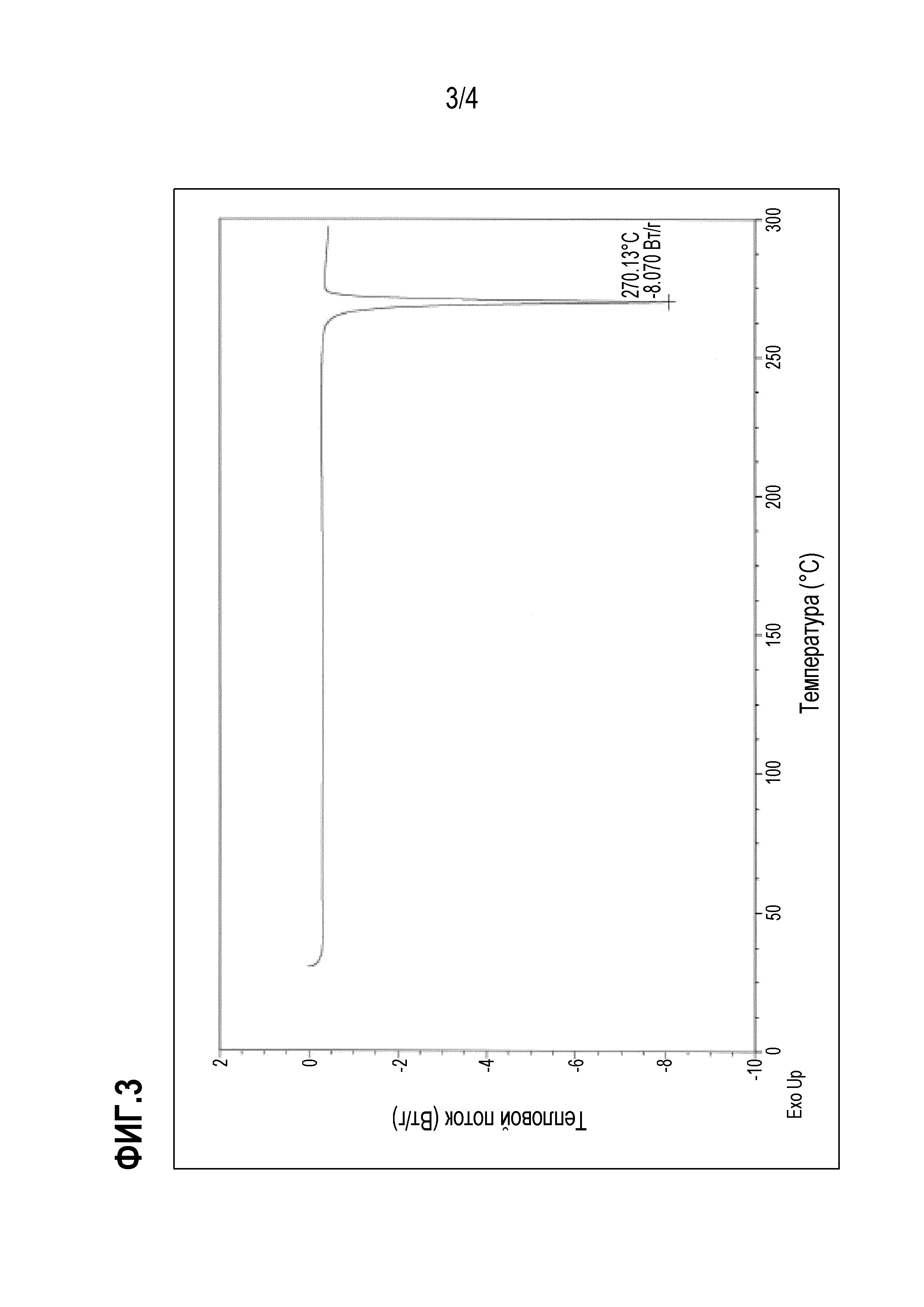
[Фигура 3] Кривая дифференциальной сканирующей калориметрии (DSC) кристаллической формы II соединения 1.
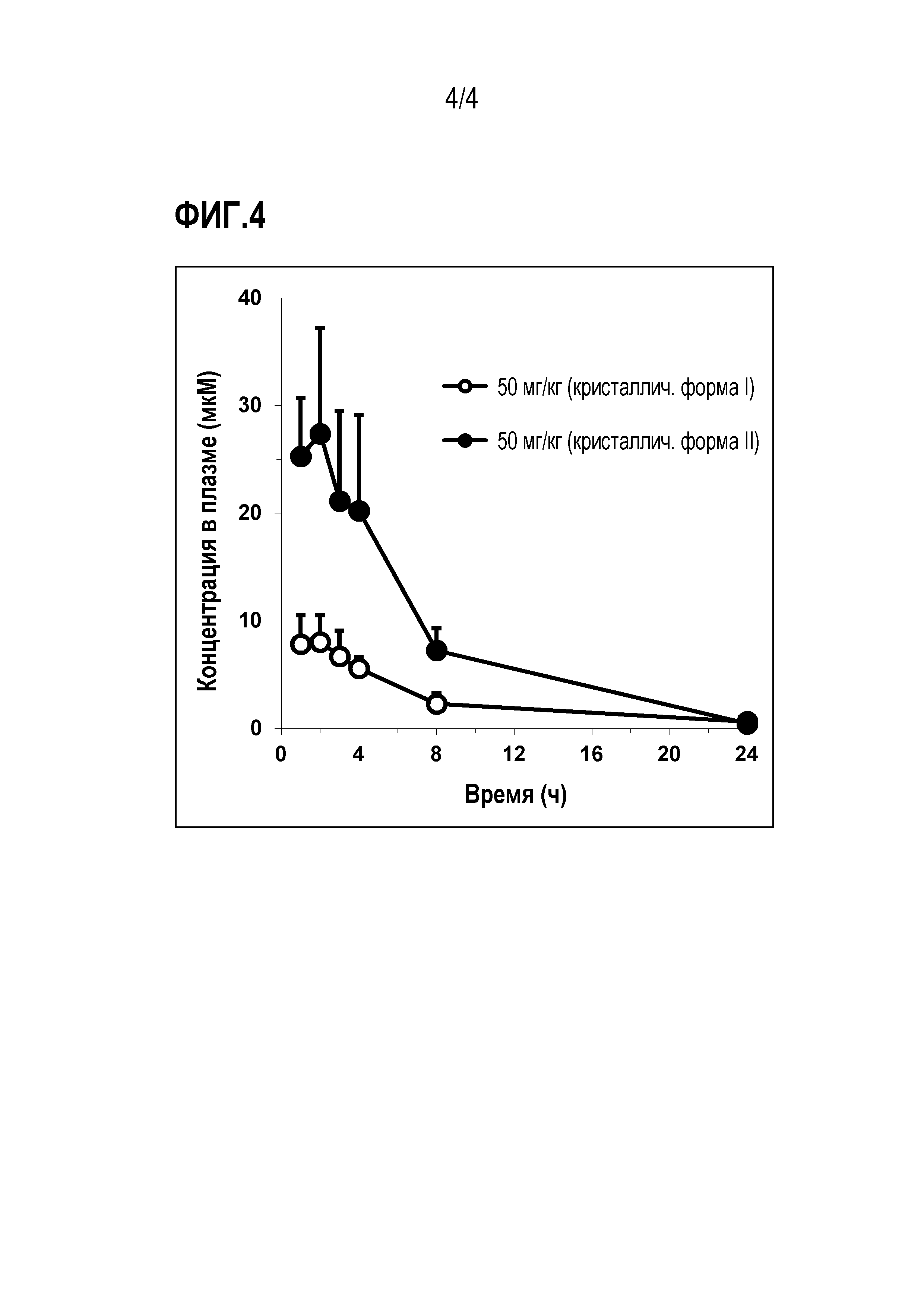
[Фигура 4] Результат измерения концентрации в крови кристаллической формы II соединения 1.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0012]
Соединение 1 по настоящему изобретению представляет собой 3-этил-4-{3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид (представленное формулой ниже). Известно, что соединение 1 демонстрирует ингибирующую активность HSP90 и обладает превосходной противоопухолевой активностью. Соединение 1 может быть синтезировано на основе способа производства, описанного в патентных документах 1 и 2.
[0013]
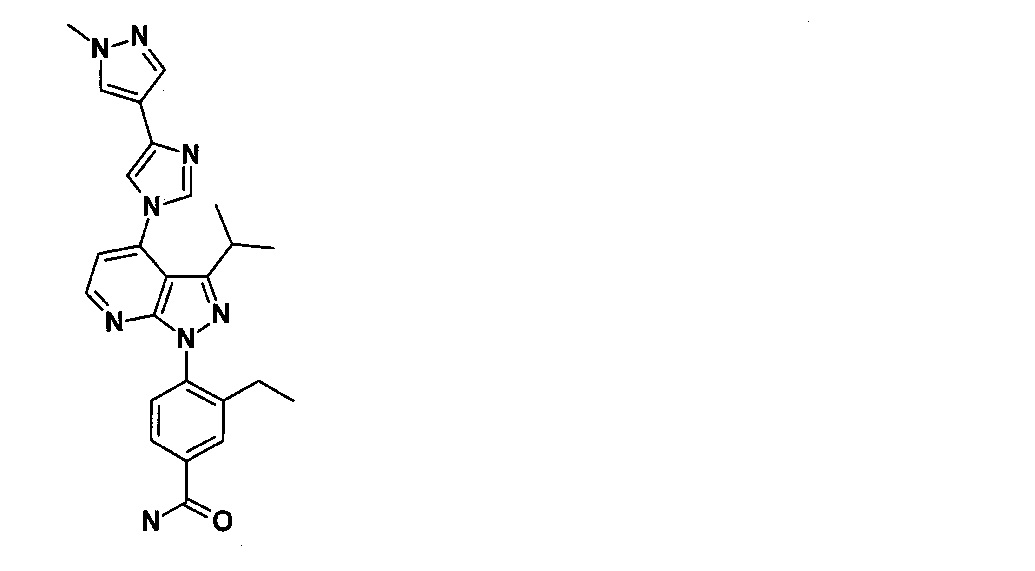
[0014]
Кристалл настоящего изобретения может быть кристаллом, содержащим кристаллическую форму II соединения 1, и может представлять собой монокристалл кристаллической формы II или полиморфную смесь, содержащую кристаллы, отличные от кристаллической формы II. Для кристалла по настоящему изобретению кристаллическая форма II с высокой степенью чистоты является предпочтительной. В частности, химическая чистота кристаллической формы II составляет предпочтительно 90% или более, более предпочтительно 95% или более и особенно предпочтительно 98% или более.
[0015]
Кристаллическая форма II по настоящему изобретению может быть получена путем добавления и создания суспензии соединения 1 в определенном органическом растворителе. В частности, кристаллическая форма II может быть получена способом получения, включающим следующие этапы (1) и (2):
(1) создание суспензии соединения 1 в органическом растворителе для получения суспензии,
(2) получение твердофазного соединения 1 из суспензии, полученной в вышеуказанном этапе (1).
Хотя соединение 1, добавляемое в органический растворитель, может быть кристаллом или нет, предпочтительно использовать кристаллическую форму соединения 1, и, в частности, кристаллическую форму II соединения 1 предпочтительно используют с точки зрения получения кристаллической формы II с высокой степенью чистоты. При кристаллизации по настоящему изобретению может быть использован затравочный кристалл. С точки зрения получения кристаллической формы II с высокой степенью чистоты, кристаллическая форма II является предпочтительной для использования в качестве затравочного кристалла.
[0016]
Примеры органического растворителя, используемого при кристаллизации по настоящему изобретению, включают спирты, такие как метанол, н-пропанол, 2-пропанол и этиленгликоль; сложные алифатические карбоксилатные эфиры, такие как метилацетат, этилацетат, пропилацетат и бутилацетат; простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, циклопентилметиловый эфир, 1,4-диоксан, тетрагидрофуран; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон и циклогексанон; ароматические растворители, такие как толуол, ксилол и хлорбензол; апротонные полярные органические растворители, такие как ацетонитрил, N-метил-2-пирролидон, N,N-диметилформамид, N,N-диметилацетамид, 1,3-диметил-2-имидазолидинон и диметилсульфоксид или смешанные растворители из них. Предпочтительно органический растворитель представляет собой кетон, одноатомный спирт, содержащий 3 или более атомов углерода, двухатомный спирт, сложный алифатический карбоксилат, простой эфир, апротонный полярный органический растворитель или его смешанный растворитель, и более предпочтительно представляет собой 2-пропанол, метилацетат, этилацетат, пропилацетат, бутилацетат, циклопентилметиловый эфир, метилэтилкетон, метилизобутилкетон, ацетон, ацетонитрил и смешанные растворители из них. Особенно предпочтительно с точки зрения чистоты и выхода кристаллической формы II органический растворитель представляет собой метилацетат, метилэтилкетон, ацетон или смешанные растворители из них.
[0017]
С точки зрения чистоты и выхода кристаллической формы II количество (по объему) органического растворителя при кристаллизации по настоящему изобретению предпочтительно составляет 2-30-кратное количество, более предпочтительно 3-20-кратное количество и особенно предпочтительно 4-15 раз по отношению к количеству соединения 1.
[0018]
Предпочтительно при кристаллизации по настоящему изобретению добавляют соединение 1 и создают суспензию в органическом растворителе с нагреванием и кипятят с обратным холодильником в течение длительного времени. Температура нагревания при кристаллизации по настоящему изобретению конкретно не ограничена, если она находится в пределах температуры, подходящей для флегмы, и ее соответствующим образом определяют в зависимости от используемого органического растворителя. Предпочтительно температура нагрева составляет52-126°C.
Продолжительность дефлегмации при кристаллизации по настоящему изобретению составляет предпочтительно 12-60 часов, а более предпочтительно 16-48 часов, в противном случае доза кристаллизации не протекает в достаточной мере, и, следовательно, кристалл высокой чистоты не может быть получен за более короткую продолжительность времени, а с другой стороны, за более длительный период происходит разрушение кристалла, что приводит к низкому выходу.
[0019]
При кристаллизации по настоящему изобретению осадок кристаллической формы II после охлаждения можно собирать после кипячения с обратным холодильником. Температура охлаждения может быть соответствующим образом отрегулирована, она является предпочтительно комнатной температурой.
[0020]
Осадок кристалла может быть выделен и очищен от указанного растворения или смешанного раствора в соответствии с известными способами разделения и очистки, такими как, например, фильтрация, промывка органическим растворителем и сушка при пониженном давлении.
Примеры органического растворителя, используемого для промывки, включают низшие спирты, ацетон и ацетонитрил. Органические растворители, используемые для кристаллизации формы II, могут использоваться для промывки.
[0021]
Кристаллическая форма II по настоящему изобретению, полученная таким образом, представляет собой кристалл, показывающий спектр дифракции рентгеновских лучей на порошке, имеющий 3 или более, предпочтительно 5 или более, более предпочтительно 8 или более и еще более предпочтительно 12 или более характеристических дифракционных пиков при углах ( 2θ± 0,2°), выбранный из группы, состоящей из 7,7°, 8,0 °, 11,1 °, 12,5 °, 12,9 °, 15,2 °, 15,8 °, 17,2 °, 19,0°, 22,5 °, 26,1 ° и 27,4 °, как показано на фигуре 2. Кристаллическая форма II показывает эндотермический пик около 270°C, как показано в результате дифференциальной сканирующей калориметрии (DSC) на фиг.3.
[0022]
Напротив, кристаллическая форма I представляет собой кристалл с рентгеновским спектром порошковой дифракции, имеющим характерные дифракционные пики при углах (2θ±0,2°) 8,1°, 12,1°, 14,0°, 16,2°, 21,5°, 25,4° и 28,3°, как показано на фигуре 1 [0023]
Пиковые значения в спектре рентгеновской порошковой дифракции могут включать некоторые ошибки из-за измерительных приборов или условий измерения, таких как условия считывания пиков. Значения пиков в описании настоящей заявки могут иметь ошибки измерения в диапазоне около ±0,2°.
Эндотермический пик (максимальное значение пика), измеренный в дифференциальной сканирующей калориметрии, может колебаться в зависимости от повышения температуры в минуту, количества образцов и чистоты и т.д. Термин «примерно» в описании к данной заявке, относится к ±5,0°С.
[0024]
Как показано в примерах ниже, кристаллическая форма I и кристаллическая форма II соединения 1 имеют значительно различную абсорбируемость при пероральном введении. Результат, что кристаллическая форма II, стабильная форма, демонстрирует более высокую способность к всасыванию в полости рта по сравнению с кристаллической формой I, метастабильной формой, является неожиданным результатом, потому что метастабильная форма, в целом, имеет более высокую растворимость, чем стабильная форма (Akira Tsuji, ʺNew pharmaceutics", опубликованная NANKODO Co. Ltd.).
[0025]
Кристаллическая форма I соединения 1 переходит в кристаллическую форму II перегонкой с обратным холодильником. Кристаллическая форма II устойчива в условиях высокой температуры и высокой влажности. Поэтому кристаллическая форма II стабильна по сравнению с кристаллической формой I и применима в качестве фармацевтического материала.
Поэтому кристаллическая форма II по настоящему изобретению полезна в качестве эффективного ингредиента фармацевтической композиции и, в частности, полезна в качестве эффективного ингредиента фармацевтической композиции для перорального введения. Кристаллическая форма II по настоящему изобретению полезна в качестве противоопухолевого средства, поскольку соединение 1 демонстрирует превосходную ингибирующую активность HSP90. Целевые раковые заболевания для лечения включают, без ограничения, рак головы и шеи, желудочно-кишечный рак (например, рак пищевода, рак желудка, желудочно-кишечную стромальную опухоль, карциному двенадцатиперстной кишки, гепатокарциному, желчный рак (такой как рак желчного пузыря и желчных протоков), рак поджелудочной железы, рак тонкой кишки и рак толстой кишки (например, колоректальный рак, рак толстой кишки и рак прямой кишки), карциному легкого, карциному молочной железы, рак яичника, карциному матки (например, рак шейки матки и рак тела матки), рак почки, рак мочевого пузыря, рак предстательной железы, уротелиальную карциному, саркому костей и мягких тканей, рак крови (такой как В-клеточная лимфома, хроническая лимфоцитарная лейкемия, периферическая Т-клеточная лимфома, миелодиспластический синдром, острый миелоидный лейкоз и острый лимфоцитарный лейкоз), множественная миелома, рак кожи и мезотелиому.
[0026]
Когда кристаллическая форма II по настоящему изобретению используется в качестве активного ингредиента фармацевтической композиции, его можно смешивать с фармацевтически приемлемым носителем по мере необходимости, и в зависимости от превентивных или терапевтических целей может использоваться множество форм введения. Указанные формы предпочтительно представляют собой пероральный агент, такой как таблетка, капсула, гранула, тонкая гранула и порошкообразное лекарственное средство. Каждая из этих форм введения может быть изготовлена способами для лекарственного препарата, известного специалистам в данной области.
Примеры.
[0027]
Хотя настоящее изобретение конкретно описано ниже со ссылкой на примеры, настоящее изобретение не ограничивается этими вариантами способа осуществления. Хотя настоящее изобретение достаточно описано в примерах, следует понимать, что различные изменения и модификации могут быть сделаны специалистами в данной области техники. Поэтому такие изменения и модификации охватываются настоящим изобретением, если они не отходят от объема настоящего изобретения.
Реактивы, используемые в примерах были коммерчески доступны, если не указано иное. Спектр NMR измеряли, используя тетраметилсилан в качестве внутреннего стандарта, когда тетраэтилсилан был включен в дейтерированный растворитель и с использованием растворителя NMR в качестве внутреннего стандарта, в противном случае, используя AL400 (400 МГц, Japan Electro Optical Laboratory (JEOL)), Mercury 400 (400 МГц, модель Agilent Technologies) или спектрометр модели Inova 400 (400 МГц, Agilent Technologies), оснащенный зондом 400MNMR (Protasis). Все δ значения указаны в частях на миллион.
[0028]
Ниже приведены значения аббревиатуры.
s: Синглет
d: Дуплет
t: Триплет
q: Квадруплет
dd: Двойной дуплет
dt: Двойной триплет
td: Тройной дуплет
tt: Тройной триплет
ddd: Двойной двойной дуплет
ddt: Двойной двойной триплет
dtd: Двойной тройной дуплет
tdd: Тройной двойной дуплет
m: Мультиплет
br: Широкий
brs: Широкий синглет
[0029]
Измерение рентгеновской порошковой дифракции
Дифракцию рентгеновских лучей на порошке измеряли в соответствии со следующими условиями испытаний после легкого измельчения подходящего количества испытываемого материала с использованием агатового раствора по мере необходимости.
Прибор: PANalytical EMPYREAN
Мишень: Cu
Настройка выходного сигнала рентгеновского излучения: 40 мА, 45 кВ
Диапазон сканирования: 2.0-40.0°
Размер шага: 0.026°
Дивергенция щели: автоматическая
Ширина облучения: 10,00 мм
Ширина образца: 10,00 мм
Обращение с приборами, включая обработку данных, осуществлялось в соответствии со способом и процедурой, указанным для каждого прибора.
[0030]
Термический анализ измерений (дифференциальная сканирующая калориметрия (DSC))
DSC была выполнена с учетом следующих условий теста.
Прибор: TA Instruments Q1000
Образец: около 1 мг
Контейнер для образцов: сделан из алюминия
Скорость нагрева: нагревание при 10°С/мин до 300°C
Газ атмосферы: Азот
Скорость потока газа азота: 50 мл/мин
Обращение с приборами, включая обработку данных, осуществлялось в соответствии со способом и процедурой, указанным для каждого прибора.
[0031]
Сравнительный пример 1: Синтез кристаллической формы I 3-этил-4-{3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло[3,4-b] пиридин-1-ил} бензамида
Белое твердое вещество (3,58 г) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации № WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610 добавляли к этанолу (7,84 мл) и перемешивали в течение 2 часов при комнатной температуре. После того, как твердое вещество собирали фильтрованием, твердое вещество промывали этанолом (7,84 мл), с последующей сушкой при пониженном давлении при температуре 70-80°С в течение 20 часов с получением кристаллической формы I (выход: 2,40 г, процентный выход: 61,2%, чистота: 98,21%).
Кристаллическая форма I дает спектр дифракции рентгеновских лучей на порошке с характеристическими пиками при углах дифракции (2θ) 8,1°, 10,9°, 12,1°, 14,0°, 14,9°, 16,2°, 17,7°, 20,2 °, 21,0°, 21,5°, 22,6°, 24,3°, 25,4°, 26,4°, 27,0°, 28,3°, 30,2°, 30,9°, 31,5°, 32,7°, 34,7°, 35,4° и 36,6°, как показано на рисунке 1.
[0032]
1H-NMR (DMSO-d6): δ частей на миллион 9,35 (1H, d, J=4,88 Гц), 8,93 (1H, d, J=1,22 Гц), 8,84 (1H, brs), 8,72 (1H, d, J=1,95 Гц), 8,70 (1H, s), 8,63 (1H, d, J=1,22 Гц), 8,60 (1H, dd, J=8,29, 1,95 Гц), 8,46 (1H, s), 8,25 (1H, d, J=8,29 Гц), 8,22 (1H, brs), 8,12 (1H, d, J=4,88 Гц), 4,59 (3H, s), 3,95 (1H, tt, J=6,83, 6,83 Гц), 3,21 (2H, q, J=7,56 Гц), 1,83 (6H, d, J=6,83 Гц), 1,75 (3H, t, J=7,56 Гц): МСНР (ESI) m/z 455 [M+H].
[0033]
Пример 1: Синтез кристаллической формы II 3-этил-4- {3-изопропил-4- (4- (1-метил-1Н-пиразол-4-ил) -1Н-имидазол-1-ил) -1Н-пиразоло [ 3,4-б] пиридин-1-ил} бензамида
Белое твердое вещество, (4,0 г) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации № WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610 добавляли к ацетону (19,54 мл) и перемешивали в течение 16 часов с нагреванием с обратным холодильником. После предоставления возможности смеси остыть до комнатной температуры, твердое вещество собирали фильтрацией и промывали ацетоном (8,4 мл), с последующей сушкой при пониженном давлении при температуре 70-80°С в течение 16-24 ч с получением кристаллической формы II (выход: 1,59 г, процентный выход: 57,0%, чистота 98,37%).
Кристаллическая форма II дала спектр дифракции рентгеновских лучей на порошке с характерными пиками при углах дифракции (2θ) 7,7°, 8,0°, 11,1°, 12,5°, 12,9°, 14,2°, 15,2°, 15,8 °, 17,2°, 17,7°, 19,0°, 20,2°, 21,1°, 22,5°, 22,8°, 23,5°, 24,5°, 26,1°, 26,7°, 27,4°, 28,0°, 28,7°, 29,4°, 30,0°, 31,7°, 35,1°, 36,2 °, 36,9° и 37,6°, как показано на фигуре 2. Кристаллическая форма II показала эндотермический пик около 270°C, как показано в результате дифференциальной сканирующей калориметрии (DSC) на фиг.3.
[0034]
1H-NMR (DMSO-d6): δ частей на миллион 9,35 (1H, d, J=4,88 Гц), 8,93 (1H, d, J=1,22 Гц), 8,84 (1H, brs), 8,72 (1H, d, J=1,95 Гц), 8,70 (1H, s), 8,63 (1H, d, J=1,22 Гц), 8,60 (1H, dd, J=8,29, 1,95 Гц), 8,46 (1H, s), 8,25 (1H, d, J=8,29 Гц), 8,22 (1H, brs), 8,12 (1H, d, J=4,88 Гц), 4,59 (3H, s), 3,95 (1H, tt, J=6,83, 6,83 Гц), 3,21 (2H, q, J=7,56 Гц), 1,83 (6H, d, J=6,83 Гц), 1,75 (3H, t, J=7,56 Гц): МСНР (ESI) m/z 455 [M+H].
[0035]
Пример 2: Синтез кристаллической формы II 3-этил-4- {3-изопропил-4- (4- (1-метил-1Н-пиразол-4-ил) -1Н-имидазол-1-ил) -1Н-пиразоло [3,4-b] пиридин-1-ил} бензамида
Белое твердое вещество, (400 мг) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации № WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610 добавляли к метилэтилкетону (2,8 мл) и перемешивали в течение 16 часов при нагревании с обратным холодильником. После предоставления возможности смеси остыть до комнатной температуры, твердое вещество собирали фильтрацией и промывали метилэтилкетоном (1,2 мл), с последующей сушкой при пониженном давлении при температуре 70-80°С в течение 16-24 ч с получением кристаллической формы II (выход: 197 мг, процентный выход: 60,9%, чистота 98,83%).
[0036]
Пример 3: Синтез кристаллической формы II 3-этил-4-{3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил) -1Н-имидазол-1-ил) -1Н-пиразоло [ 3,4-б] пиридин-1-ил} бензамида
Белое твердое вещество (400 мг) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации № WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610 добавляли к ацетонитрилу (4,0 мл) и перемешивали в течение 3 часов при нагревании с обратным холодильником. После предоставления возможности смеси остыть до комнатной температуры, твердое вещество собирали фильтрацией и промывали ацетонитрилом (1,2 мл), с последующей сушкой при пониженном давлении при температуре 70-80°С в течение 3 ч с получением кристаллической формы II (выход: 120 мг, процентный выход: 43,0%, чистота 98,25%).
[0037]
Пример 4 Синтез кристаллической формы II 3-этил-4- {3-изопропил-4- (4- (1-метил-1Н-пиразол-4-ил) -1Н-имидазол-1-ил) -1Н-пиразоло [ 3,4-б] пиридин-1-ил} бензамида
Белое твердое вещество (400 мг) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации № WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610 добавляли к метилизобутилкетону (4,0 мл) и перемешивали в течение 3 часов при нагревании с обратным холодильником. После предоставления возможности смеси остыть до комнатной температуры, твердое вещество собирали фильтрацией и промывали метилизобутилкетоном (1,2 мл), с последующей сушкой при пониженном давлении при температуре 70-80°С в течение 3 ч с получением кристаллической формы II (выход: 154 мг, процентный выход: 55,3%, чистота 96,89%).
[0038]
Пример 5: Синтез кристаллической формы II 3-этил-4- {3-изопропил-4- (4- (1-метил-1Н-пиразол-4-ил) -1Н-имидазол-1-ил) -1Н-пиразоло [ 3,4-б] пиридин-1-ил} бензамида
Белое твердое вещество (400 мг) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации №. WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610 добавляли к 2-пропанолу (4,0 мл) и перемешивали в течение 3 часов при нагревании с обратным холодильником. После предоставления возможности смеси остыть до комнатной температуры, твердое вещество собирали фильтрацией и промывали 2-пропанолом (1,2 мл), с последующей сушкой при пониженном давлении при температуре 70-80°С в течение 3 ч с получением кристаллической формы II (выход: 108 мг, процентный выход: 38,8%, чистота 96,83%).
[0039]
Пример 6: Синтез кристаллической формы II 3-этил-4- {3-изопропил-4- (4- (1-метил-1Н-пиразол-4-ил) -1Н-имидазол-1-ил) -1Н-пиразоло [ 3,4-б] пиридин-1-ил} бензамида
Белое твердое вещество (400 мг) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации № WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610 добавляли к этилацетату (4,0 мл) и перемешивали в течение 3 часов при нагревании с обратным холодильником. После предоставления возможности смеси остыть до комнатной температуры, твердое вещество собирали фильтрацией и промывали этилацетатом (1,2 мл), с последующей сушкой при пониженном давлении при температуре 70-80°С в течение 3 ч с получением кристаллической формы II (выход: 156 мг, процентный выход: 56,0%, чистота 96,45%).
[0040]
Пример 7: Синтез кристаллической формы II 3-этил-4- {3-изопропил-4- (4- (1-метил-1Н-пиразол-4-ил) -1Н-имидазол-1-ил) -1Н-пиразоло [ 3,4-б] пиридин-1-ил} бензамида
Белое твердое вещество, (400 мг) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации № WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610, добавляли к бутилацетату (4,0 мл) и перемешивали в течение 3 часов при нагревании с обратным холодильником. После предоставления возможности смеси остыть до комнатной температуры, твердое вещество собирали фильтрацией и промывали бутилацетатом (1,2 мл), с последующей сушкой при пониженном давлении при температуре 70-80°С в течение 3 ч с получением кристаллической формы II (выход: 164 мг, процентный выход: 58.8%, % чистота 96,04%).
[0041]
Пример 8: Синтез кристаллической формы II 3-этил-4- {3-изопропил-4- (4- (1-метил-1Н-пиразол-4-ил) -1Н-имидазол-1-ил) -1Н-пиразоло [ 3,4-б] пиридин-1-ил} бензамида
Белое твердое вещество (400 мг) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации № WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610 добавляли к циклопентилметиловому эфиру (4,0 мл) и перемешивали в течение 3 часов при нагревании с обратным холодильником. После предоставления возможности смеси остыть до комнатной температуры, твердое вещество собирали фильтрацией и промывали циклопентилметиловым эфиром (1,2 мл), с последующей сушкой при пониженном давлении при температуре 70-80°С в течение 3 ч с получением кристаллической формы II (выход: 192 мг, процентный выход: 68,8%, чистота 95,68%).
[0042]
Пример 9: Синтез кристаллической формы II 3-этил-4-{3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил) -1Н-пиразоло [ 3,4-b] пиридин-1-ил} бензамида
Белое твердое вещество, (400 мг) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации № WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610 добавляли к пропилацетату (4,0 мл) и перемешивали в течение 3 часов при нагревании с обратным холодильником. После предоставления возможности смеси остыть до комнатной температуры, твердое вещество собирали фильтрацией и промывали пропилацетатом (1,2 мл), с последующей сушкой при пониженном давлении при температуре 70-80°С в течение 3 ч с получением кристаллической формы II (выход: 172 мг, процентный выход: 61,5%, чистота 96,77%).
[0043]
Пример 10: Синтез кристаллической формы II 3-этил-4- {3-изопропил-4- (4- (1-метил-1Н-пиразол-4-ил) -1Н-имидазол-1-ил) -1Н-пиразоло [ 3,4-б] пиридин-1-ил} бензамида
Белое твердое вещество, (400 мг) 3-этил-4- {3-изопропил-4-(4-(1-метил-1Н-пиразол-4-ил)-1Н-имидазол-1-ил)-1Н-пиразоло [3,4-b] пиридин-1-ил} бензамид, полученный по способу получения, описанному в пояснительной статье к международной публикации № WO2012/093708 и пояснительной статье к международной публикации № WO2011/004610 добавляли к метилацетату (2,2 мл) и перемешивали в течение 16 часов при нагревании с обратным холодильником. После предоставления возможности смеси остыть до комнатной температуры, твердое вещество собирали фильтрованием и промывали метилацетатом (0,94 мл), с последующей сушкой при пониженном давлении при 80°C для получения кристаллической формы II (выход: 215,5 мг, процентный выход: 68,6%, чистота 98,06%).
[0044]
Тестовый пример 1: тестовое измерение концентрации в крови
Были получены соответствующие растворы для введения (50 мг/10 мл/кг) кристаллической формы I и кристаллической формы II. Эти растворы вводили перорально мышам (Balb/cA), выведенным в условиях кормления, в объеме 10 мл/кг массы тела с использованием зонда для перорального введения. После введения мышей возвращали в клетки для мышей, чтобы проверить их условия. Вода и корм были свободно доступны для мышей в клетках. Мышей анестезировали изофлураном 1, 2, 3, 4, 8 и 24 часа после введения и 60 мкл крови собирали из их орбитальных пазух с использованием капиллярных труб для сбора крови. Собранную кровь охлаждали на льду и центрифугировали для отделения плазмы.
AUC0-24 часа рассчитывали методом линейного логарифмического способа трапеции с использованием Phoenix WinNonlin (v6.3.0), программного обеспечения Pharsight, из концентраций соединения 1 в образцах плазмы, измеренных методом множественного контроля реакции с использованием LC-MS/MS.
[0045]
Результаты показаны на рисунке 4 и в таблице 1. Этот тест доказал, что AUC 0-24чса (площадь под кривой концентрации в крови во время 0-24 ч после приема) кристаллической формы II примерно в 3 раза выше, чем у кристаллической формы I. Поэтому кристаллическая форма II в соответствии с настоящим изобретением является превосходно отличной с точки зрения способности к всасыванию в полости рта и полезна в качестве пероральной фармацевтической композиции.
[0046]
Таблица 1
|
[0047]
Тестовый пример 2: Тест на стабильность в твердой фазе (ускоренное испытание)
Прочная стабильность кристалла II формы, полученного в примере 1 после хранения 1, 3 и 6 месяцев при 40°C±2°C/75%RH±5% RH, измерялась при следующих условиях.
Условия хранения: 40°C±2°C/75% RH ±5% RH
Точки измерения: 1, 3 и 6 месяцев
Объем хранения: 6 г
Контейнер для хранения: двойные пластиковые пакеты, бандажные ленты+пластиковые барабаны
Метод подготовки образца раствора: 100 мг кристаллической формы II точно измеряется, к ней добавляют смешанный раствор ацетонитрила-воды (4:1) для растворения кристалла (обрабатывают ультразвуком для растворения, если его трудно растворить), и раствор разбавляют в измерительном цилиндре с точностью до 200 мл. 10 мл этого раствора точно измеряют, и смешанный раствор ацетонитрила-воды (1:1) добавляют к точным 20 мл для использования в качестве образцового раствора.
[0048]
HPLC проводили в следующих условиях.
Колонка: общий фонд Chemicals Evaluation and Research Institute, Япония L- колонка 2 ODS
Размер частиц: 3 мкм, внутренний диаметр: 4,6 мм, длина: 15 см
Длина волны измерения: 220 нм
Подвижная фаза: Подвижная фаза A: 10 ммоль/л фосфатного буфера (рН 6,9), подвижная фаза В: ацетонитрил
Скорость потока: около 1,0 мл/мин
[0049]
Градиент концентрации контролируется путем изменения соотношения смешивания подвижных фаз А и В следующим образом при подаче жидкости.
[0050]
Таблица 2
|
[0051]
Количество сопутствующих примесей в растворах образцов измеряли и оценивали с помощью HPLC, результат которого показан в таблице 3.
Сопутствующие примеси определяются как обнаруженные вещества, отличные от 3-этил-4- {3-изопропил-4- (4- (1-метил-1Н-пиразол-4-ил) -1Н-имидазол-1-ил) -1Н пиразол [3,4-b] пиридин-1-ил} бензамида.
[0052]
Таблица 3
|
[0053]
Как видно из этого результата, кристаллическая форма II соединения 1 производит небольшое количество сопутствующих примесей и демонстрирует отличную стабильность в твердом состоянии.

Противоопухолевое средство для непрерывного внутривенного введения, содержащее цитидиновое производное
Противоопухолевое средство, включающее производное цитидина и карбоплатин
Средство, улучшающее противоопухолевый эффект, содержащее липосомальное средство, содержащее оксалиплатин, и противоопухолевое средство, содержащее липосомальное средство
Новое урациловое соединение или его соль, обладающие ингибирующей активностью относительно дезоксиуридинтрифосфатазы человека
Пиперазиновое соединение, ингибирующее простагландин-d-синтазу
Производное ацилтиомочевины или его соль, и его применение
Противоопухолевый агент, набор и способ лечения рака
Способ детекции дегенеративных мышечных заболеваний и способ определения терапевтической эффективности при заболеваниях
Усилитель действия противоопухолевого средства
Азабициклосоединение и его соль
Азабициклосоединение и его соль
Новое хинолин-замещенное соединение

