Результат интеллектуальной деятельности: Композиция для лечения болезни Паркинсона
Вид РИД
Изобретение
Изобретение относится к медицине, в частности к фармакологии и фармацевтике, и касается назальной формы новой фармацевтической композиции в виде капель, содержащей в качестве действующего вещества 3.4 - дигидрокси - L - фенилаланин (леводопа), распределенной в полимерной матрице на основе биодеградируемого сополимера молочной/гликолевой кислот (полилактидгликолид, PLGA 50/50) для применения при нейродегенеративных заболеваниях, в частности болезни Паркинсона (БП).
БП - это хроническое неуклонно прогрессирующее нейродегенеративное заболевание с широким диапазоном моторных и немоторных нарушений. БП неминуемо инвалидизирует пациентов, значительно снижая качество жизни больных и их близких. Через 10-20 лет 40-75% пациентов умирают, а около 50% выживших требуют постоянного ухода (Hely. М.A. The Sydney multicenter of Parkinson's disease: The inevitability of dementia at 20 years / M.A. Hely, W.G J. Reid, M.A. Adena, G.A. Halliday, J.G.L. Morris // Mov. Disord. 2008. - V. 23. - №6. - P. 837-844). Общая распространенность заболевания в Европе среди людей старше 65 лет составляет 1,6-1,8 случаев на 100 человек. Из-за старения населения ожидается, что значение паркинсонизма как проблемы здравоохранения будет возрастать (Литвиненко И.В. Болезнь Паркинсона. М.: «Миклош», 2010. 216 с.; Handbook of Parkinson's disease / edited by Rajesh Pahwa, Kelly E. Lyons. - 4th ed. - 2007. - P. 501). Прогрессирование заболевания и ухудшение качества жизни пациентов, а также недостаточно эффективное лечение создают серьезную социальную проблему.
До настоящего времени стандартным лекарственным средством при БП остается леводопа. Однако для леводопы характерен короткий период полувыведения из плазмы крови, что даже в лучших условиях современной терапии приводит лишь к пульсирующей стимуляции дофаминэргических нейронов. Следовательно, длительная терапия осложнена двигательными отклонениями и дискинезией, являющихся источником существенных ограничений для некоторых пациентов. Терапевтическая стратегия, которая позволит в конечном итоге доставлять леводопу/дофамин в головной мозг более непрерывным и физиологичным образом, обеспечит преимущества по отношению к стандартной форме, уменьшив двигательные осложнения, что является крайне необходимым для пациентов, страдающих сопутствующими неврологическими или двигательными расстройствами (Olanow CW; Mov. Dis. 2008, 23 (Suppl. 3): S613-S622).
В настоящее время разработаны пероральные формы леводопы с замедленным высвобождением действующего вещества. Использование же липосомальных лекарственных форм обеспечивает пролонгированность действия препаратов и снижение их токсичности. В работе During M.J. и др. было показано, что липосомы с включенным ДОФА, имплантированные в частично денервируемый стриатум крыс, проявляли длительный эффект в течение 40 дней. Малые одноламеллярные везикулы были получены озвучиванием липосомальной дисперсии. Результаты этих исследований свидетельствовали о частичном восстановлении дефицита дофамина у животных с апоморфин-индуцированной моделью БП (During M.J., Freese A., Deutch A.Y., Kibat P.G., Sabel В.A., Langer R., Roth R.H. Biochemical and behavioral recovery in a rodent model of Parkinson's disease following stereotactic implantation of dopamine-contatining liposomes // Exper. Neurol. 1992. V115. P. 193-199). К существенным недостаткам липосомальных препаратов следует отнести высокую стоимость используемых фосфолипидов, а также их низкую стабильность при хранении.
Попытки непрерывного введения леводопы интрадуоденально или с помощью инфузии были осуществлены с помощью амбулаторных насосов и пластырей. Однако такие способы лечения считаются чрезвычайно агрессивными и неудобными, а также могут вызвать дофаминэргические побочные эффекты. При этом даже непрерывный прием леводопы или допа-агонистов связан с периодами отсутствия действия самоограничивающихся веществ. (Nutt JG; Mov. Dis. 2008, 23 (Suppl. 3): S580-4).
Процесс метаболического превращения леводопы в дофамин катализируется ферментом декарбоксилазой ароматических L-аминокислот, убиквитарным ферментом, в наибольших концентрациях присутствующим в слизистой оболочке кишечника, печени, головном мозге и капиллярах головного мозга. Из-за возможного экстрацеребрального метаболизма леводопы необходимо вводить большие дозы данного вещества, чтобы получить высокую концентрацию экстрацеребрального дофамина, которая в свою очередь может вызвать тошноту у некоторых пациентов. Поэтому прием леводопы обычно проводят совместно с пероральным введением ингибитора допа-декарбоксилазы, таким как карбидопа или бенсеразид, что уменьшает на 60-80% дозу леводопы, необходимой для клинического ответа и, таким образом, предотвращает некоторые из ее побочных эффектов путем ингибирования конверсии леводопы в дофамин вне головного мозга. Процесс уменьшения дозы леводопы остается неизученным. Хорошо известны различные составы, содержащие леводопу отдельно или совместно с ингибиторами ферментов, связанных с метаболическим разрушением леводопы, например, ингибиторами декарбоксилазы, такими как карбидопа и бенсеразид, ингибиторами катехол-О-метилтрансферазы, такими как энтакапон и толкапон, и ингибиторами моноаминоксидазы (МАО)-А или МАО-В, такими как моклобемид, разагилин или селегилин, или сафинамид. На сегодняшний день список пероральных препаратов включает таблетки SINEMET® и SINEMET®CR с замедленным высвобождением, содержащие карбидопу или леводопу; таблетки STALEVO®, содержащие карбидопу, энтакапон и леводопу; и таблетки MADOPAR®, содержащие леводопу и бенсеразид.
Таким образом, на сегодняшний день актуальна проблема разработки и составления принципиально новых фармацевтических композиций и способов доставки действующего вещества, которые:
- обеспечат продолжительную стимуляцию L-допы без необходимости расширения состава активных веществ;
- дадут возможность в значительной мере уменьшить дозу леводопы для более эффективного лечения двигательных нарушений;
- сократят вероятность появления побочных эффектов.
Данная задача была решена посредством создания назальной формы в виде капель, содержащей наносомальную форму активного вещества 3.4 - дигидрокси - L - фенилаланин (леводопа).
Ранее была разработана фармацевтическая композиция для лечения БП (Патент РФ 2440100), содержащая пищевую кислоту или комбинацию пищевых кислот, обеспечивающую рН композиции в диапазоне 5,5-6,5, и фармакологически эффективное количество микронизированного L-ДОФА в качестве активного ингредиента, в которой активный ингредиент присутствует в виде частиц на поверхности более крупных частиц носителя. Действие препарата направлено на устранение двигательных флуктуаций у больных, принимающих L-ДОФА для лечения БП. Недостатком композиции является относительно низкая эффективность лечения. Также разработано лекарственное средство (Патент РФ 2545734) для купирования проявлений БП на основе микронизированного L-ДОФА (3-гидрокси-L-тирозина). Однако для достижения оптимальных результатов лечения необходима такая система доставки активного вещества, которая смогла бы в полной мере раскрывать потенциал препарата. Именно к такому техническому результату пришли в результате ряда экспериментов.
Отметим, что разработка систем доставки лекарственных веществ является одним из быстроразвивающихся и перспективных направлений фармакологии. В настоящее время с успехом применяются различные наноразмерные носители, неоспоримым преимуществом которых является защита здоровых клеток от токсического воздействия лекарственного вещества и увеличение биодоступности инновационных препаратов. При этом снижается вероятность деградации включенной субстанции в условиях организма, действие лекарственного вещества пролонгируется, обеспечивается его селективное накопление в области поражения. Лекарственное вещество, форма и технология, используемые для производства лекарственного препарата, сочетаются, поэтому их взаимодействие должно быть изучено. Например, при выборе материалов, используемых в лекарственной форме, важно понимать технологические последствия сочетания отобранных элементов. Аналогичным образом, знание механизма высвобождения лекарственного вещества является ключевым для понимания критических характеристик лекарственного препарата и достижения требуемого профиля высвобождения. Это знание может использоваться в качестве вспомогательного средства при формировании, проектировании и разработке новой технологии.
Сущность данного изобретения заключается в разработке такой формы фармацевтической композиции, которая позволит осуществлять доставку микронизированного 3.4 - дигидрокси - L - фенилаланина без потери эффективности и обеспечит пролонгированность действия. В результате экспериментов было выявлено, что данную задачу можно решить с помощью приготовления фармацевтической композиции в лекарственной форме назальные капли. На финальный результат состава и метода получения полимерной композиции влияют физико-химические свойства самого лекарственного вещества, потому оптимальный состав и процесс получения были сформированы в процессе многочисленных опытов.
I. Фармацевтическая композиция на основе микронизированного 3.4 - дигидрокси - L - фенилаланина
При разработке назального лекарственного средства были рассмотрены положительные и отрицательные стороны уже существующих известных лекарственных форм, используемых как для системного, так и местного применения.
Интраназальные кати. К числу преимуществ данной лекарственной формы относят относительную простоту изготовления и использования, к недостаткам - необходимость использования холодовых условий хранения.
Интраназальные аэрозоли (спреи) - одна из разновидностей фармацевтических аэрозолей. Имеют следующие преимущества: небольшой расход и равномерное распределение действующего вещества по поверхности слизистой; возможность применения в различных условиях; высокая концентрация вещества на месте патологического процесса. Однако имеется и ряд существенных недостатков: газы-вытеснители (пропелленты) могут оказывать раздражающее действие на слизистую носа; требуется обязательная синхронизация введения препарата с моментом вдоха, чего трудно добиться у детей, пожилых пациентов и пациентов с низким уровнем интеллекта; при неумелом использовании существует возможность повреждения слизистой носового хода насадкой аэрозольного баллона; при распылении не исключается возможность попадания веществ в глаза, на кожу лица и т.д.
Назальные гели. Гель - мягкая лекарственная форма для местного применения. Преимущества лекарственной формы: пролонгированное действие; возможность применения на ночь; наличие увлажняющего действия на слизистую носа; благоприятное действие на слизистую оболочку носа при ее сухости, наличии корочек; при попадании на кожу или одежду гель легко смывается водой, не оставляя следов (в отличие от мазей). Недостатки: далеко не все действующие вещества могут вводиться в состав гелей и соответственно использоваться в данной лекарственной форме; нестабильность лекарственной формы; возможность возникновения расслаивания в процессе хранения на ВМС и водную фазу; диффузия действующего вещества в ткани из гелевой лекарственной формы проходит медленнее, чем из раствора; сложность применения при обильном количестве слизистого отделяемого.
Назальные мази. Мази для носа готовятся в асептических условиях. Поскольку они наносятся на влажную слизистую, для повышения их всасываемости добавляют ПАВ. Преимущества, мазевая основа обеспечивает более длительное действие активных веществ на слизистую носа; обладает смягчающим действием на слизистую носа; лекарственная форма мази обладает системным действием и дает возможность совместного введения в один препарат действующих веществ как гидрофобной, так и гидрофильной природы.
Учитывая дисперсность частиц полученной субстанции (леводопа в полилактидгликолидах), необходимость системного воздействия, относительную длительность пребывания на слизистой носа для обеспечения полноты всасывания, регулярность нанесения лекарственного препарата на слизистую носа, а также его применение лицами пожилого возраста, в качестве лекарственных форм выбраны капли, гели, мази. Таким образом, выбор вспомогательных веществ при разработке лекарственной формы средства для назального применения будет обусловлен следующими факторами:
- свойствами вводимого фармакологически активного компонента;
- физико-химическими параметрами слизистой носа и назального секрета;
- физико-химическими и технологическими свойствами самих вспомогательных веществ.
II. Состав фармацевтической композиции на основе микронизированного 3.4 - дигидрокси - L - фенилаланина
В процессе подбора оптимальной формы и состава было разработано несколько вариантов композиций для каждой из выбранных готовых лекарственных форм. В качестве примеров ниже будут приведены более предпочтительные составы. Остальные составы, не указанные в примерах, будут обозначены при описании технологии изготовления.
Пример 1. Вода очищенная как дисперсионная среда
Образец К-3 водн.
Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 - 5,0
Вспомогательные вещества.
бензиловый спирт - 0,10;
сорбитол 70% - 0,02;
гипромеллоза - 0,05-0,50;
вода очищенная - до 10,0
Пример 2. Масла растительные как дисперсионная среда Образец К-3 масл.
Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 - 5,0
Вспомогательные вещества.
твин-80 - 0,01;
масло оливковое - до 10,0
Пример 3. Комбинированные системы как дисперсионная среда
Образец К-1 комб.
Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 - 5,0
Вспомогательные вещества:
масло оливковое - 0,5-2,5;
твин-80 - 0,005-0,02;
пропиленгликоль - 0,05-0,5;
сорбитол 70% - 0,02;
вода очищенная - 2,20-4,43
Пример 4. Комбинированные системы как дисперсионная среда Образец К-2 комб.
Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 - 5,0
Вспомогательные вещества:
масло оливковое - 2,0;
твин-80 - 0,02;
пропиленгликоль - 0,1;
сорбитол 70% - 0,02;
вода очищенная - 2,86
Пример 5. Гель Образец Г-1 гель
Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 - 5,0
Вспомогательные вещества:
натрия гидрофосфат безводный - 0,01;
лимонной кислоты моногидрат - 0,02;
сорбитол 70% - 0,1;
масло оливковое - 2,0;
твин-80 - 0,01;
гипромеллоза - 0,09-0,14;
вода очищенная - 2,72-2,77
Пример 6. Мазь Образец М-3 мазь
Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 - 5,0;
Вспомогательные вещества:
вазелин - 3,0;
вазелиновое масло - 1,5;
софтизан 649 - 0,5
Пример 7. Мазь Образец М-1 мазь
Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 - 5,0
Вспомогательные вещества: вазелин - 5,0
Технология изготовления водных назальных капель
Образец К-1 водн. готовили следующим образом (на 100 мл). Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 в количестве 50,0 отвешивали на весах с точностью 0,01 г, добавляли воду очищенную, до 100 мл. Взбалтывали до получения однородной смеси. Образовывался коллоидный раствор, устойчивый в течение 15-20 мин. Далее происходило частичное разделение системы, и часть частиц оседала на дно сосуда. При оценке стабильности данного образца и пригодности его к использованию, был сделан вывод о необходимости введения загустителей в состав капель, таких как производные целлюлозы, сорбита.
Образцы К-2 водн., К-3 водн. и К-4 водн. готовили также в расчете на 100 мл. Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 в количестве 50,0 отвешивали на весах с точностью 0,01 г. Предварительно готовили растворы гипромеллозы и метилгидроксипропилцеллюлозы различной концентрации от 0,5% до 5%, с шагом концентрации в 0,5%. Сорбитол использовали в виде 70% водного раствора. В части воды очищенной, в зависимости от прописи, растворяли рассчитанное количество динатрия эдетата, натрия цитрата, натрия дигидрофосфата дигидрата, динатрия гидрофосфата дигидрата. К отвешенным в подставку полимерным частицам добавляли оставшуюся воду, взбалтывали, добавляли растворы целлюлозы, в последнюю очередь добавляли растворы солей.
При использовании 2-5% растворов гипромеллозы и метилгидроксипропилцеллюлозы получали устойчивую взвесь (суспезию), не расслаивающуюся при температуре 4°C (температура хранения полимерных частиц с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50) в течение 14 дней (срок наблюдения)).
Технология изготовления назальных капель с растительными маслами
Образец К-1 масл. и образец К-2 масл. готовили следующим образом (на 100 мл). Полимерные частицы с 3,4-дигидрокси-Е-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 в количестве 50,0 отвешивали на весах с точностью 0,01 г, добавляли масло оливковое/подсолнечное в количестве 50,0 г. Взбалтывали до получения однородной смеси. Образовывалась суспензия, устойчивая в течение 60-90 мин. Далее происходило частичное разделение системы, часть частиц оседала на дно сосуда. При оценке стабильности данного образца и пригодности его к использованию был сделан вывод о необходимости введения поверхностно-активных веществ в состав капель, таких как твин-80.
Образцы К-3 масл. и К-4 масл. готовили аналогичным образом с введением в состав твина-80. Это позволило увеличить время седиментации частиц до 10 дней при температуре +4°C (время наблюдения).
При выборе систем испытывались составы с той или иной степенью гидрофильности, что обеспечивало введение пропиленгликоля или цетилового спирта и парафина жидкого.
Для уточнения влияния вспомогательных веществ на седиментационную стабильность полученных комбинированных капель и для выявления характера их функционирования на модельной слизистой приведенный состав был изготовлен в вариантах, указанных в примерах 3 и 4, а также в описании технологии изготовления комбинированных назальных капель.
Технология изготовления комбинированных назальных капель
Образец К-1 комб. готовили следующим образом (на 100 мл). Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 в количестве 50,0 отвешивали на весах с точностью 0,01 г, помещали в подставку, добавляли твин-80, встряхивали, для равномерного распределения твина-80 по полимерным частицам. Добавляли масло оливковое в рассчитанном количестве и опять встряхивали, образовывалась концентрированная суспензия (паста). В ступку помещали пропиленгликоль, сорбитол, рассчитанное количество воды. Растирали до получения однородной смеси. Полученную жидкость добавляли к масляной пасте по частям, при постоянном встряхивании. Образовывалась суспензионно-эмульсионная система, наблюдаемая в том числе и под микроскопом. Следует отметить, что стабильность образовавшейся системы в значительной степени зависит от концентрации вспомогательных веществ.
Наибольшей устойчивости удалось добиться при соотношении ингредиентов, указанном в Примере 4.
Образцы К-2 комб. готовили также в расчете на 100 мл, однако, учитывая значительный объем липофильной фазы, липофильную часть мази готовили отдельно. В подогретую до 50-60°C ступку помещали цетиловый спирт и отвешенные количества масла оливкового и жидкого парафина (вазелина), смесь тщательно растирали до образования однородной массы. В полученную смесь вносили смоченные твином-80 полилактидные частицы, перемешивали и постепенно добавляли воду очищенную. Все полученные образцы отличались седиментационной устойчивостью в течение 10 дней (срок наблюдения).
Технология изготовления назального геля
Образец К-1 гель готовили следующим образом (на 100 мл). Полимерные частицы с 3,4-дигидрокси-L-фенилаланином (L-ДОФА) в полилактидгликолиде 50/50 в количестве 50,0 отвешивали на весах с точностью 0,01 г, помещали в ступку, добавляли твин-80, аккуратно перемешивали скребком, не допуская растирания частиц. Добавляли масло оливковое в рассчитанном количестве и опять перемешивали скребком. Образовывалась концентрированная суспензия (паста). В другую ступку помещали 3-5% раствор гипромеллозы, в который вводили в следующей последовательности: натрия гидрофосфат безводный, сорбитол, лимонной кислоты моногидрат. Растирали до получения однородной смеси. Полученную массу добавляли к масляной пасте по частям при постоянном перемешивании. Образовывалась стабильная система, гетерогенность которой можно было наблюдать под микроскопом.
В настоящее время на фармацевтическом рынке России назальные гели представлены такими лекарственными препаратами как «Галазолин», «Виброцил». Лекарственных средств системного действия при назальном применении выявлено не было. Возможно это связано с точностью дозирования назальных гелей, которые упаковываются в тубы с удлиненным наконечником и дозируются при непосредственном выдавливании из тубы. Для назальных гелей системного применения данный способ не является пригодным, так как требует дозирования на градуированный шпатель или иное приспособление, а только затем введения в носовую полость на слизистую.
В патентной литературе теме назальных форм уделено недостаточное внимание. Все вышеописанные факты были учтены при разработке составов на основе гипромеллозы в концентрации 3-5%.
Технология изготовления назальной мази
В простейшем случае мазевая композиция может быть представлена суспензионной мазью с полимерными частицами на белом парафине (вазелине) в соответствии с составом, указанном в Примере 7.
В подогретую до температуры 50-60°C ступку помещали предварительно отвешенный вазелин, расплавляли его и при постоянном перемешивании постепенно вводили полимерные частицы.
Полученная мазь может быть охарактеризована как достаточно концентрированная масса, поэтому в следующий образец наряду с вазелиновым маслом вводили софтизан 649. В подогретую до температуры 50-60°C ступку помещали предварительно отвешенный вазелин, расплавляли его, добавляли вазелиновое масло, софтизан 649, перемешивали. При постоянном перемешивании постепенно вводили полимерные частицы. Полученная мазь отличалась удовлетворительными характеристиками, пластичностью, не расслаивалась, обладала визуальной однородностью.
Применительно к назальным мазям системного приготовления также целесообразно использование при дозировании градуированного шпателя.
При разработке назальной мази, учитывая длительность ее применения, не применялись макроголы различных марок из-за их осмотической активности и, соответственно, потенциальной возможности негативного влияния на слизистую. Также было признано нецелесообразным использование альгинатов (образование сжимающихся пленок).
III. Выбор оптимального состава фармацевтической композиции на основе микронизированного 3.4 - дигидрокси - L - фенилаланина
Выбор оптимального состава назальных композиций осуществлялся при помощи разработанной модельной системы, имитирующей слизистую оболочку носа.
С целью определения наиболее рациональных составов разработанных назальных композиций с полимерными частицами 3,4-дигидрокси-L-фенилаланина (L-ДОФА) в полилактидгликолиде 50/50 для терапии БП, была проведена оценка их поведения на модельной системе in vitro. Оценка проводилась по показателям площади распределения и степени адгезии на модельной поверхности. Исследование адгезивных свойств разработанных лекарственных средств оценивали по скорости стекания образца по наклонной поверхности модельной подложки, смоченной модельной средой.
Для более точного определения в образцы капель в зависимости от состава был введен 0,001% метиленового синего (капли на водной основе) или капля Судана III.
Исследуемое средство в количестве 5,0 мл, что составляло в пересчете на леводопу дозу вводимого лекарственного вещества, наносили на линию старта предварительно нагретой до 37°C подложки (поверхность в чашке Петри). Подложку поднимали под углами 10° и 70° и по истечении 30 секунд измеряли путь, пройденный передней границей капли жидкости. Скорость стекания v (мм/с) рассчитывали по формуле:
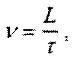
где: L - путь пройденный каплей жидкости, мм;
τ - время стекания, с.
Результаты эксперимента представлены в Таблице 1.
Для измерения площади распределения окрашенное как описано выше исследуемое средство в количестве 5,0 мл, что составляло в пересчете на леводопу дозу вводимого лекарственного вещества, помещали на предварительно нагретую до 37°C поверхность модельной слизистой, расположенную в чашке Петри, накрывали крышкой и термостатировали при данной температуре в течение 15 мин (время возможного нахождения на слизистой), отмечая площадь распределения для каждого образца. Площадь распределения S (см2) определяли при помощи миллиметровой бумаги.
Результаты эксперимента представлены в Таблице 2.
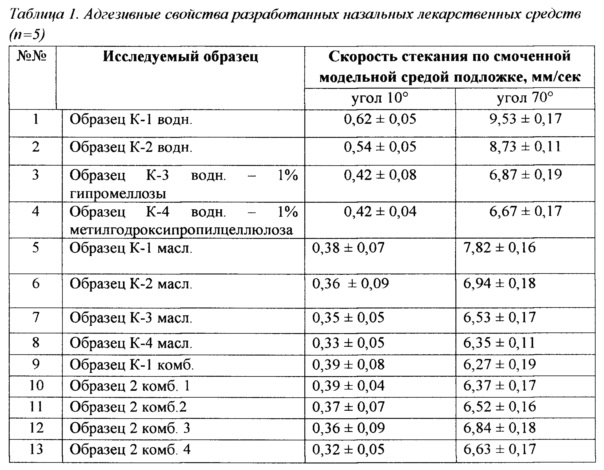
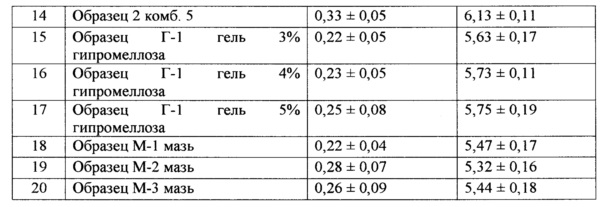

Анализируя результаты проведенного эксперимента можно отметить, что назальные капли на водной основе без включения производных целлюлозы распределяются по модельной поверхности и, обладая низкими адгезионными свойствами, не могут обеспечить точность дозирования за счет вытекания из носовых полостей. Введение гипромеллозы и метилгидроксипропилцеллюлозы позволяет уменьшить площадь распределения на поверхности модельной слизистой. Назальные капли на масляной основе распределялись (Образец К-1 масл) крайне неравномерно.
Разработанные назальные лекарственные средства могут быть расположены по поведению на модельной слизистой (по пригодности к дальнейшим исследованиям) следующим образом: мази → гели → капли на комбинированной основе → капли на масляной основе → капли на основе водных растворов производных целлюлозы → капли на основе водных растворов. Учитывая результаты проведенных экспериментов, для дальнейших исследований были отобраны следующие образцы:
Образец К-3 водн. - 1% гипромеллозы
Образец К-4 водн. - 1% метилгидроксипропилцеллюлоза
Образец К-2 масл.
Образец К-3 масл.
Образец К-4 масл.
Образец К-1 комб.
Образец 2 комб. 1
Образец 2 комб. 2
Образец 2 комб. 3
Образец 2 комб. 4
Образец 2 комб. 5
Образец Г-1 гель 3% гипромеллоза
Образец Г-1 гель 4% гипромеллоза
Образец Г-1 гель 5% гипромеллоза
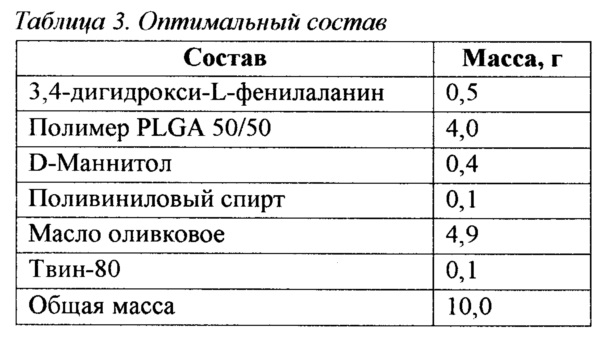
Неожиданно в процессе работы было обнаружено, что уменьшение дозы вспомогательного вещества и добавление его совместно с твин-80 на этапе приготовления готовой композиции дали самый эффективный результат в отношении адгезивных свойств, площади распределения и скорости действия, а также пролонгированности активного вещества, учитывая все химико-физические свойства микронизированной формы 3,4-дигидрокси-L-фенилаланина.
Предпочтительный образец композиции указан в Таблице 3. Способ его получения и эффективность зафиксированы ниже.
IV. Фармакологическая эффективность оптимального состава и сравнительная оценка специфической активности инновационного лекарственного средства на основе 3,4-дигидрокси-1-фенилаланина (леводопа) и полилактидгликолидов на модели БП
Оценка специфической активности инновационного лекарственного средства с оптимальным составом, указанным в Таблице 3, и стандартного препарата Леводопа противопаркинсонического действия проводилась с помощью разработанной модели БП и батареи поведенческих тестов.
Согласно современным рекомендациям, доклиническое изучение активности потенциальных противопаркинсонических препаратов проводят на моделях паркинсонического синдрома. При фармакологическом моделировании БП обычно прибегают либо к различным способам угнетения дофаминергической (ДА) передачи, прежде всего в нигростриатуме, либо к активации холинэргических (ХЭ) нейронов. Два типа этих моделей различаются динамикой развития процесса, а также его проявлениям, и рассматриваются как воспроизводящие, соответственно, акинетико-ригидную или дрожательную формы ПС.
Поскольку разработанное лекарственное средство на основе полимеркасулированного 3,4-дигидрокси-L-фенилаланина (леводопа) в полилактидгликолиды (PLGA) ориентировано на пролонгированное высвобождение действующего вещества, то необходимой динамике постепенного развития и длительности сохранения моделируемой БП в большей степени соответствует модель с угнетением ДА передачи.
Выбор модели
С целью адекватного формирования БП и последующего ее лечения потенциальным антипаркинсоническим средством, была использована фармакологическая модель БП, индуцированная разрушением нигростриарной дофаминэргической системы нейротоксином 6-гидроксидофамином (6-OHDA), введенным в головной мозг крысы.
Модель с использованием 6-OHDA считается моделью ПС у крыс, т.к. они наиболее подвержены действию нейротоксина, однако проявления его эффектов у крыс и обезьян также были изучены. 6-OHDA вызывает характерное изменение позы животных - поворот головы и девиацию хвоста в направлении поврежденной стороны, а также типичные круговые движения и стереотипии (принюхивание, облизывание, покусывание).
Детальное получение фармацевтической композиции
На первом этапе получают раствор ПВС, насыщенного L-ДОФА. В стеклянный сосуд помещают L-ДОФА, добавляют 2% раствор ПВС. Затем сосуд переносят на водяную баню, расположенную на магнитной мешалке, где L-ДОФА растворяют в течение двух часов при постоянном перемешивании со скоростью 400-500 об/мин. Температура в сосуде не должна превышать 37-42°C. Перемешивание смеси в бане продолжается 30-40 мин (без дополнительного подогрева). Затем баня с водой удаляется, перемешивание продолжается еще 30-40 мин до полного растворения твердой фазы.
Затем получают суспензию L-ДОФА в растворе полимера PLGA 50/50 в дихлорметане. В стеклянном бюксе отвешивают L-ДОФА. В стеклянном стакане отвешивают полимер PLGA 50/50 и переносят в реактор. В пластиковом стакане отвешивают дихлорметан. Дихлорметан переносят в реактор порционно и закрывают крышкой из фольги. Растворение полимера проводят при перемешивании лопастной мешалкой перемешивающего устройства со скоростью 300-400 об/мин в течение 20-25 мин.
После полного растворения полимера PLGA 50/50 в дихлорметане в реактор перегружают отвешенную ранее навеску субстанции L-ДОФА, закрывают крышкой, продолжают перемешивание смеси со скоростью 300-400 об/мин в течение 25-30 мин до получения однородной смеси. Полученный раствор-суспензию используют для создания эмульсии.
2% раствор ПВС, насыщенного L-ДОФА, переносят в пластиковый реактор и помещают на водяную баню. Температура реакционной смеси должна поддерживаться в пределах 21-27°C. При интенсивном перемешивании (600-700 об/мин) в центральную часть реактора постепенно добавляют полученную ранее раствор-суспензию L-ДОФА пипеточным дозатором порциями по 50-75 мл. Пластиковый реактор накрывают крышкой из фольги, перемешивание смеси продолжают 25-30 мин. Останавливают нагревание и сливают воду из бани. Пространство емкости бани, свободное от реактора, заполняют льдом.
Далее смесь подвергают второму эмульгированию (гомогенизации). Для этого штатив с гомогенизатором помещают около реактора и погружают до дна насадку Н-27 (шифр S25N-25F). Смесь эмульгируют при 24 тыс. об/мин и комнатной температуре 3 раза по 30-40 с, с двумя перерывами по 0,5 мин. В ходе процесса происходит образование пенистой массы.
Далее полученную эмульсию дополнительно обрабатывают при помощи погружного зонда гомогенизатора (В. Braun, США) ГВ-28, проводят ультразвуковое диспергирование в режиме: 1 мин; 1,5 мин; 1 мин с двумя перерывами по 1 мин (power level 300-320, repeating duty cycle 0.7 s). Из полученного продукта упаривают дихлорметан.
Гомогенизат из реактора переносят в колбу Е-29 вместимостью 10 л. Колбу заполняют примерно на 1/3 часть объема, затем соединяют ее с валами роторного испарителя через стеклянную каплеотбойную ловушку, подключают вакуум от мембранного (или водоструйного) вакуумного насоса. После набора вакуума в системе (примерно 0.4 ед.) включают вращение вала. Смесь упаривают на роторном испарителе (вакуумирование с помощью водоструйного насоса) в режиме: 30-35 мин при комнатной температуре; 15-20 мин при 25°C (в бане); 10 мин при 35°C; 10 мин при 40°C. В начале упаривания дихлорметана (при комнатной температуре и 25°C) имеет место сильное пенообразование и частичное попадание пены в ловушку и приемную колбу, поэтому данный этап процесса следует визуально контролировать и при необходимости впускать воздух в систему через специальный кран для пеногашения. Оставшееся количество смеси постепенно доливают. Процесс заканчивают при достижении вакуума 0,93-0,98 ед. и эффекта запотевания ловушки.
После упаривания дихлорметана суспензию композиции L-ДОФА и полимера PLGA 50/50 в колбе передают на стадию смешения с D-Маннитола. В первую очередь D-Маннитол 41,00±0,01 г растворяют в воде очищенной 287,00±1,00 мл. Добавление воды к навеске D-Маннитола осуществляют не менее чем за 1 ч до начала проведения операции смешивания с полученной ранее суспензией ввиду медленного его растворения.
Далее идет стадия фильтрования суспензии и смешивание с D-Маннитолом. Раствор D-Маннитола переносят в колбу Бунзена. Полученная ранее суспензия в колбе может содержать крупные частицы, для отделения которых проводят процесс фильтрования через стеклянный фильтр с размером пор 40-100 мкм (пор 111). Фильтрование суспензии осуществляют самотеком, при необходимости систему подключают к вакуумной линии. Объединенный фильтрат собирают в приемную колбу. После окончания фильтрования колбу отсоединяют и переносят на магнитную мешалку.
Раствор D-Маннитола, фильтрат суспензии и промывные воды перемешивают в приемной колбе с помощью магнита во фторопластовой оболочке и магнитной мешалки в течение 10-15 мин при 300-400 об/мин. Затем определяют массу смеси в колбе. После этого готовят фильтрат к лиофильному высушиванию.
Смесь фильтрата с D-Маннитолом, полученная на прошлой стадии, равномерно дозируется в пластиковые банки (контейнеры). Процесс производится в помещении с ламинированным потоком воздуха. Наполненные банки прикрывают завинчивающимися крышками и помещают для замораживания в морозильник при температуре минус 70±2°C. Время замораживания составляет не менее 24 ч. Далее идет процесс лиофилизации. Процесс высушивания проводится при комнатной температуре в помещении в течение 45-48 ч. После данного этапа запускается процесс получения капель назальных.
К навеске твина-80 в стакане отливают масло оливковое, перемешивают до однородности смеси. В чашу переносят лиофилизированные полимерные частицы с 3,4-дигидрокси-L-фенилаланином в полилактидгликолиде 50/50. Смесь твина-80 и масла оливкового переносят из стакана, распределяя по всей поверхности лиофилизированных частиц. Включают мешалку со скоростью 50-60 об/мин. Перемешивание ведут 5-10 минут, добиваясь равномерного распределения масляной смеси во всем объеме лиофилизированных частиц. Частями добавляют оставшееся масло оливковое, тщательно перемешивают до образования однородной массы.
Готовые назальные капли представляют собой новую суспензию Леводопа (полимерные частицы с 3,4-дигидрокси-Г-фенилаланином в полилактидгликолиде 50/50 1:9) в растворе твина-80 в масле. Отвешивают на весах с точностью 0,01 г по 10,0 г в стеклянные флаконы и укупоривают крышкой с винтовой резьбой и маслостойкой пипеткой.
Приготовление препаратов. Назальные капли Леводопа-ПК с оптимальным составом, указанным в Таблице 3
Композиционная эмульсия из лиофилизата L-ДОФА (Sigma) в составе полимерных частиц на основе биодеградируемого сополимера молочной и гликолевой кислот (PLGA(Plurac Biochem), 50/50) и вспомогательных веществ (субстанция Леводопа). Размер частиц составлял 250±50 нм, включение действующего вещества - 10±2%. Для введения крысам линии Wistar стандартный препарат растворяли в 1% растворе аскорбиновой кислоты до концентрации 3,2 мг/мл, первоначально доводя рН до 2 посредством добавления раствора 1М HCl. После растворения L-ДОФА рН доводили до 7 с использованием 5M NaOH. Непосредственно перед введением полученный состав Леводопа-ПК доводили до нужной концентрации (по L-ДОФА), чтобы предупредить выход активного вещества до попадания в организм.
Исследование в тесте спонтанной активности передних конечностей
Данный тест предъявлялся через 4 недели после введения 6-OHDA подготовленным животным (n=112) с подтвержденным развитием БП для поиска диапазона эффективных доз с целью их последующего применения при интраназальном введении. Контролем служили крысы (n=7), оперированные по указанной схеме, которым не вводили 6-OHDA, но получавшие эквиобъемное количество стерильного физиологического раствора.
Введение препаратов и дозировки
Все препараты вводили ежедневно, назально, при помощи автоматической пипетки. Объем введения составлял 40 мкл (по 20 мкл в каждый носовой проход), расчет предъявляемых доз производили в пересчете на действующее вещество. Для контроля физического состояния животных проводили еженедельное взвешивание. Эффективность однократного введения испытуемого препарата Леводопа в дозах 0,1-1 мг/кг (в пересчете на действующее вещество) и препарата сравнения L-ДОФА (прототип) в тех же дозах оценивали при тестировании животных в следующем режиме: непосредственно перед введением препаратов, через 30 мин, после курсового введения через 3 суток и через 7 суток ежедневных назальных введений сравниваемых препаратов.
Пример 1. Регистрация спонтанной активности передних конечностей
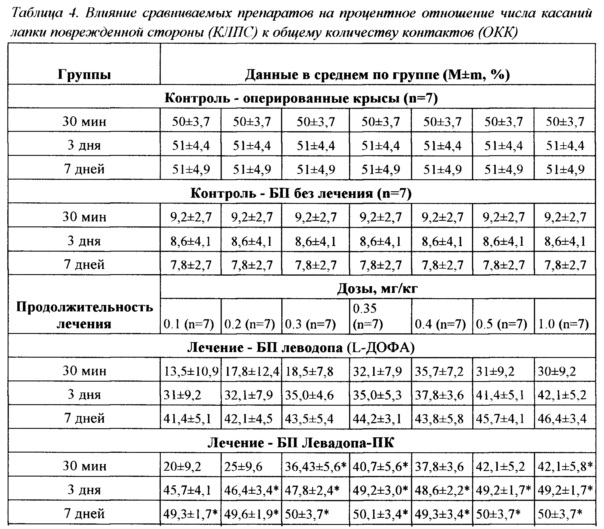
Данный тест предъявлялся через 4 недели после введения 6-OHDA для поиска диапазона эффективных доз с целью их последующего применении при интраназальном введении. Результаты, полученные в тесте спонтанной активности передних конечностей, сведены в Таблицу 4.
Результаты тестирования животных после ежедневного введения препаратов в течение 3 дней показали, что во всем диапазоне испытанных доз показатель приближения к норме после введения Леводопа всегда был больше, чем в контрольной группе без лечения БП. Превышение над результатами группы прототипа наблюдалось при дозах не меньше 0,2 мг/кг (D>0,3 мг/кг). Результаты выполнения теста контрольными животными с БП достоверно отличались от результатов группы оперированного контроля (р<0,05).
Оптимальными для терапии модельной БП при однократном и курсовом трехдневном введении можно считать дозы 0,3-0,5 мг/кг, поскольку повышение доз до 1,0 мг/кг, т.е. в 2-3 раза, не сопровождалось дальнейшим повышением процента правильных решений. Данные, полученные после ежедневного введения препаратов в течение 7 дней, подтверждают такое заключение. Положительные результаты в последействии введения сравниваемых препаратов были более выражены и имели меньший разброс при введении препарата Леводопа-ПК, чем при введении прототипа L-ДОФА, во всем диапазоне испытанных доз 0,1-1 мг/кг.
*Р<0,05
Наряду с этим необходимо отметить, что лечебный эффект семидневного курса лечения сопровождался повышением показателя правильных реакций (касание стенок цилиндра) до уровня «норма», выявленного в группе «оперированный контроль», - до уровня крыс, которым не вводился 6-ОНДА и, в этом смысле, по сохранности дофаминэргической системы - до уровня интактных животных. Такой результат наблюдался практически во всем диапазоне испытанных доз 0,1-1 мг/кг и сохранялся спустя 24 часа от момента последнего введения препаратов Леводопа-ПК, но не прототипа L-ДОФА, что свидетельствует о высокой специфической активности и четком терапевтическом эффекте разработанного средства. Диапазон испытанных доз 0,1-1 мг/кг может рассматриваться как терапевтический или как терапевтическая широта препарата Леводопа-ПК, поскольку сопровождался положительным эффектом при низких дозах (0,1-0,3 мг/кг) и хорошей переносимостью при более высоких дозах (0,35-1 мг/кг). Однако по комплексу показателей: необходимая и достаточная эффективность при однократном (разовом) и многократном курсовом введении, хорошая переносимость, пролонгация лечебного эффекта после отмены курса терапии, а также технических сложностях возникающих при дробном введении доз больших 0,4 мг/кг, для дальнейшего экспериментального исследования была выбрана доза 0,35 мг/кг. Необходимо отметить, что эта доза при переносе ее значения согласно правилам межвидового переноса с крысы на человека будет соответствовать среднему значению разовой терапевтической дозе рекомендуемой для человека.
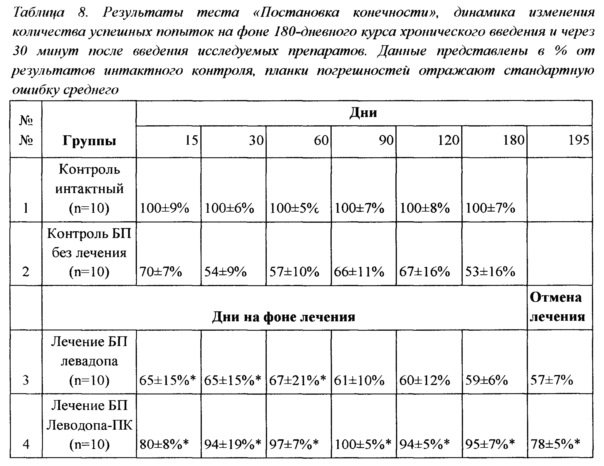
Пример 2. Исследование в тесте «Постановка конечности»
В ходе экспериментов введение препаратов начинали с 5-й недели после операции, после подтверждения развития БП у крыс. Животным контрольных групп вводили не нагруженную L-ДОФА эмульсию. Животные были разделены на 4 группы (n=10) на основе результатов теста «Постановка конечности» таким образом, чтобы средние результаты выполнения теста не различались для всех групп с БП.
Все препараты вводили ежедневно, назально, при помощи автоматической пипетки. Объем введения составлял 40 мкл (по 20 мкл в каждый носовой проход), расчет предъявляемых доз производили в пересчете на действующее вещество. Для контроля физического состояния животных проводили еженедельное взвешивание.
Тест «Постановка конечности» проводили еженедельно в течение всего периода введения препаратов. Животных тестировали дважды - непосредственно до введения препаратов и через 30 минут после их введения. Тестирование животных в таком режиме при хроническом введении позволяло выявить эффективность действия препарата непосредственно после введения и изучить действие препаратов через сутки после введения.
Через 5 недель после операции, до начала введения препаратов, результаты тестирования групп животных с индуцированной БП не превышали 75% от результатов группы интактного контроля, различия были достоверны (р<0,05).
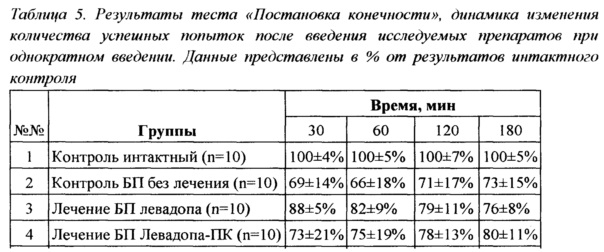
Эффективность однократного введения испытуемого препарата Леводопа-ПК в дозе 0,35 мг/кг (в пересчете на действующее вещество) и препарата сравнения L-ДОФА (прототип) в дозе 0,35 мг/кг исследовали при тестировании животных непосредственно перед введением препаратов, через 30, 60 и 180 минут после введения препаратов. Результаты сведены в Таблицу 5.
Из представленных данных видно, что через 30 минут после введения препаратов среднее количество успешных попыток постановки конечности составляло 100±4% для интактного контроля, 69±14% для контроля БП, 88±5% для группы, получающей L-ДОФА и 73±21% для группы Леводопа-ПК Через час после введения результаты были следующими: 100±5%, 66±180%, 82±90% и 75±19% соответственно.
Р<0.05
Достоверные различия были выявлены между результатами группы контроля БП и группы, получавшей L-ДОФА (р<0,05), через 30 и 60 минут после введения препарата. Через 180 минут после введения препаратов, результаты всех групп животных с БП не отличались между собой, но в группе Леводопа-ПК наметился тренд к повышению количества успешных попыток постановки конечности. Таким образом, эффект однократного введения стандартного препарата продолжался не более часа, действие Леводопа-ПК не привело к значимым изменениям в координации движения крыс. Различия в эффекте исследуемых препаратов можно объяснить замедленным высвобождением лекарственного средства из полимерных частиц Леводопа-ПК, что не позволяет достичь эффективной концентрации при однократном введении. В то же время действие препарата сравнения достоверно увеличивает количество успешных попыток при выполнении теста через 30 минут после введения L-ДОФА с полным исчезновением эффекта к 180 минуте.
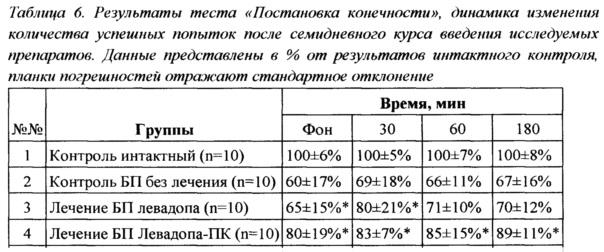
Тестирование животных после ежедневного введения препаратов в течение 7 дней проводили по той же схеме, что и после однократного введения - непосредственно перед введением препаратов, затем через 30, 60 и 180 минут после их введения. В среднем количество успешных попыток постановки конечности до введения препаратов составляло 100±6% для интактного контроля, 60±17% для контроля БП, 65±15% для группы, получающей L-ДОФА, и 80±19% для группы Леводопа-ПК. Результаты выполнения теста контрольными животными с БП достоверно отличались от результатов группы интактного контроля (р<0,05). Достоверные различия были выявлены между группой контроля БП и группой, получавшей Леводопа-ПК (р<0,05). Результаты исследования зафиксированы в Таблице 6.
Важно отметить, что во все исследованные интервалы времени после введения препарата достоверные отличия в результатах теста были обнаружены только после введения Леводопа-ПК. У группы животных, получавших L-ДОФА, достоверных отличий результатов теста от таковых у группы контроля БП зафиксировано не было. Нивелирование эффекта от введения препарата сравнения можно объяснить следующим образом: суточная доза стандартного препарата L-ДОФА в данном эксперименте равна 0,35 мг/кг, что в 60 раз ниже минимальной терапевтической суточной дозы (в пересчете на человека). Можно предположить, что мы наблюдаем характерный для терапии леводопой эффект истощения дозы, который является одной из ключевых проблем использования препарата в клинике и проявился столь быстро из-за использования малых доз препаратов.
Р<0.05
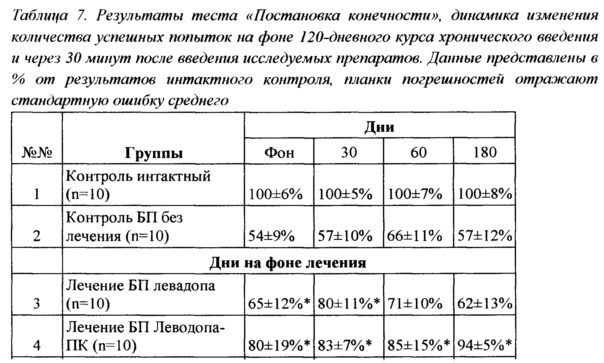
Важно отметить, что введение Леводопа-ПК в течение 7 дней приводило к улучшению координации движений у животных с БП вплоть до очередного введения препарата, то есть эффект сохранялся в течение 24 часов после предыдущего введения. При этом эффект был максимальным в течение 180 мин после введения (89% от результата интактного контроля), но несколько уменьшался через 24 часа после введения (83% от результата интактного контроля при тестировании до очередного введения препарата). Результаты сведены в Таблицу 7.
Р<0.05
Тестирование животных после хронического курса ежедневного введения препаратов в течение 120 дней выявило значительное снижение способности к выполнению теста в контрольной группе животных с БП до 51±12% от интактного контроля. Похожая динамика наблюдалась и для результатов группы животных, получавших L-ДОФА: к 120 дню лечения результаты выполнения теста через 30 минут после введения препарата составили 62±13% от результата группы интактного контроля. В то же время результаты группы, получавшей Леводопа-ПК, составили 94±5% и были достоверно выше по сравнению с животными как группы контроля БП (р<0,05), так группы, получавшей препарат прототипа, что наблюдалось на протяжении всего периода ежедневной терапии.
Как показал анализ данных тестирования и наблюдений, полученных в процессе 180-дневного курса терапии, уже через неделю ежедневного введения Леводопа-ПК проявились преимущества разработанного средства по сравнению со стандартным препаратом L-ДОФА. Возможно, различия в эффекте связаны со стабилизацией концентрации препарата, капсулированного в PLGA, на терапевтическом уровне. Координация движений у животных в группе Леводопа-ПК была достоверно лучше по сравнению с животными в группе контроля БП (р<0,05) начиная с 7 дня терапии и в течение всего остального приема препаратов. С 14 дня терапии и в последующие дни результаты в группе Леводопа-ПК также достоверно отличались от результатов группы L-ДОФА (р<0,05). При этом отличия наблюдали как через 30 минут, так и через 24 часа после введения препаратов. Уже через три недели ежедневного использования Леводопа-ПК через 30 мин после введения препарата способность животных выполнять тест достоверно не отличалась от результатов животных группы интактного контроля. Отсутствие различий сохранялось вплоть до отмены препарата, что свидетельствует о полном восстановлении исследуемых моторных функций животных после введения Леводопа-ПК в составе полимерных частиц, но не прототипа - L-ДОФА. Важно еще раз подчеркнуть, что выраженный терапевтический эффект Леводопа-ПК наблюдался не только непосредственно после введения препарата, но и через 24 часа после приема препарата. Данный факт указывает на компенсацию недостатка дофамина и стабилизацию его уровня в мозге. Кроме того, эффект препарата сохранялся как минимум неделю после его отмены и полностью исчезал только через месяц после прекращения терапии. Результаты эксперимента сведены в Таблицу 8.
Пример 3. Исследование животных в тесте «Открытое поле»
Тест «Открытое поле» проводили дважды - непосредственно перед началом терапии и через 6 недель после начала терапии. Данный тест используют для оценки возможных последствий введения препарата на двигательную и исследовательскую активность экспериментальных животных в условиях свободного поведения в экстремальных для них условиях. Поведение крыс исследовалось в «открытом поле» в соответствии с общепринятыми стандартами. «Открытое поле» - квадратная равномерно освещенная площадка размером 100×100 см, огороженная непрозрачными стенками высотой 40 см. Пол «открытого поля» расчерчен на квадраты.
Методика «Открытое поле» - классическая испытательная система для изучения поведения животных в незнакомой обстановке, основанная на конфликте двух доминирующих мотиваций в незнакомом пространстве - инстинктивной тенденции к исследованию нового окружения и тенденции минимизировать возможную опасность со стороны такового. Тест «Открытое поле» является информативной методикой, позволяющей в частности адекватно оценивать нейротропные, психостимулирующие эффекты действия фармакологических средств. В этом тесте оцениваются двигательная и ориентировочно-исследовательская активность, а также эмоциональная реакция страха, возникающая при изучении незнакомого пространства.
Для испытания крысы по одной помещались на 2 минуты в центр ярко освещенной разграфленной круглой площадки. Учитывались следующие показатели, горизонтальная активность (количество пересеченных секторов), вертикальная активность (число стоек), акты выхода в центр поля, количество актов груминга, заглядываний, количество фекальных болюсов. По соотношению регистрируемых показателей в контрольных и испытуемых группах судили о характере влияния веществ на психоэмоциональное состояние (тревога, страх) и на уровень ориентировочно исследовательской активности. Усиление тревожности отражается в увеличении количества фекальных болюсов и снижении показателей исследовательской активности. Перед испытанием в «открытом поле» за 60 минут до тестирования животных помещали в тихое, слабо освещенное помещение. В этот период исключались любые активные манипуляции.
До начала терапии экспериментальные группы животных не различались ни по одному из регистрируемых параметров. Тестирование через 6 недель после начала терапии выявило достоверное увеличение количества стоек в группе животных получающих Леводопа-ПК по сравнению как с интактным контролем, так и с контрольной группой с БП до 162±75% (р<0,05). Достоверное увеличение вертикальной активности животных свидетельствует о повышении исследовательской активности, снижении тревожности и улучшении общего эмоционального состояния животных. Введение препарата L-ДОФА в той же дозе не привело к достоверному изменению активности животных. Кроме того, выявлено достоверное снижение среднего количества актов груминга у группы животных, получающих Леводопа-ПК, по сравнению с группой интактного контроля (р<0,05). Среднее значение количества актов груминга для группы Леводопа-ПК составил 22±23% от результата интактного контроля. Введение L-ДОФА в той же дозе не приводило к достоверному изменению количество актов груминга у животных. Поскольку в течение эксперимента достоверных различий между контрольными группами выявлено не было, наблюдаемое достоверное изменение количества актов груминга у группы получавшей препарат Леводопа-ПК следует отнести к собственному, не связанному с развитием БП, действию препарата, приводящему к снижению тревожности и ускорению принятия решений в новых для животного условиях.
Пример 4. Исследование животных в тесте «Приподнятый крестообразный лабиринт»
Тест и установка «Приподнятый крестообразный лабиринт» предназначены для изучения поведения грызунов в условиях переменной стрессогенности (при свободном выборе комфортных условий) и позволяет оценить: уровень тревожности животного (по предпочтению темноты/света, боязни высоты, выраженности и динамике поведения «выглядывания»); симптомы неврологического дефицита; привыкание (habituation).
Тест «Приподнятый крестообразный лабиринт» проводили однократно, через 11 недель после начала терапии. Исследование выявило достоверные различия только между группами контроля БП и группы получавшей Леводопа-ПК по снижению количества актов автогруминга до 50% от значения группы интактного контроля (р<0,05), что воспроизводит результаты, полученные в тесте «Открытое поле». Резкое снижение уровня автогруминга в группе получавшей Леводопа-ПК наряду с повышением уровня исследовательской активности в открытом поле и со снижением уровня дефекаций рассматривается как подавление уровня тревожности и страха.
Результаты испытаний Леводопа-ПК в тестах «Открытое поле» и «Приподнятый крестообразный лабиринт» позволяют заключить, что комплексное повышение исследовательских компонентов поведения и снижение показателей психоэмоционального напряжения, уровня страха в стрессогенной среде, указывают на восстановление дофаминэргических функций под влиянием Леводопа-ПК, что говорит о фармакологическом эффекте испытанной дозы. При сравнительной оценке действия препаратов установлено, что выявленный сдвиг в направлении повышения психо-эмоциональной устойчивости и усиления исследовательского поведения более выражен при назальном введении Леводопа-ПК, чем при введении препарата сравнения L-ДОФА.
Других достоверных различий в поведении животных по регистрируемым в тесте параметрам выявлено не было.
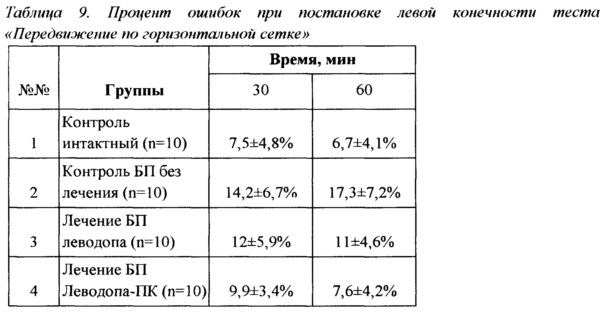
Пример 5. Тест «Передвижение по горизонтальной сетке»
Исследования в тесте «Передвижение по горизонтальной сетке» позволяют дать сравнительную оценку степени сохранности локомоторных и сложнокоординированных двигательных реакций у животных с поврежденной дофаминэргической нигро-стриарной системой относительно поведения интактных животных. Животные по одному помещались в центр горизонтальной квадратной сетки-экрана (1×1 м) на 4 минуты, определялось количество шагов каждой из конечностей по горизонтальной сетке и процент ошибок при постановке каждой из конечностей.
Было проведено однократное исследование животных через 30 и 60 дней терапии с анализом количества шагов каждой из конечностей по горизонтальной сетке, процента ошибок при постановке каждой из конечностей и коэффициента асимметрии через 30 мин после введения препаратов.
Количество шагов как левой, так и правой ногой достоверно не отличалось у животных всех групп. Процент ошибок при постановке левой конечности составлял 7,5±4,8% для интактного контроля, 14,2±6,7% для контроля БП, 12±5,9% для группы, получающей леводопу, и 9,9±3,4% для группы Леводопа-ПК. Процент ошибок группы контроля БП достоверно отличался от такового у интактного контроля в два раза (р<0.05), однако результаты группы, получавшей Леводопа-ПК, а также группы, получавшей препарат сравнения, от интактного контроля достоверно не отличались. При исследовании процента ошибок при постановке правой конечности достоверных различий между группами обнаружено не было.
Коэффициент асимметрии через 30 минут после введения препаратов составлял 0,9±4,6% для интактного контроля, 5,8±4,9% для контроля БП, 6,7±3,3% для группы, получающей леводопу, и 1,3±4,3% для группы Леводопа-ПК (представлены абсолютные значения параметра). Коэффициенты асимметрии групп контроля БП и L-ДОФА достоверно отличались от такового группы интактного контроля (р<0.05) и группы Леводопа-ПК. Коэффициент асимметрии группы животных, получавшей Леводопа-ПК, был наиболее близок к значению группы интактного контроля и достоверно отличался от контроля БП и группы L-ДОФА.
Полученные результаты теста «Передвижение по горизонтальной сетке» полностью согласуются с результатами, полученными в тесте «Постановка конечности» и подтверждают как наличие моторных дисфункций у животных с индуцированной БП, так и значимо более высокую эффективность назальной терапии Леводопа-ПК по сравнению со стандартным препаратом в той же дозе.
Результаты эксперимента сведены в Таблицу 9.
Во всех проведенных тестах отсутствие достоверных отличий при манипуляции правой конечностью объясняется односторонним характером повреждений s. nigra в используемой модели БП, влияющим в основном на возможности противоположной поврежденной части мозга стороны тела.
Техническим результатом были установлены пролонгированность действия и биодоступность, а именно после получения и экспериментального обоснования модели БП, вызванная внутримозговым введением 6-гидроксидофамина крысам. Новая фармацевтическая композиция показала следующее.
- Интраназальное введение Леводопа-ПК в дозах 0,1-1,0 мг/кг по L-ДОФА привело к достоверному улучшению координации движений животных с БП в тесте «Цилиндр-касания передних конечностей» через трех- и семидневные курсы ежедневного назального введения. Установлены оптимальные терапевтические дозы лечения для данного теста.
- Курсовое интраназальное введение Леводопа-ПК в дозе 0,35 мг/кг по L-ДОФА привело к достоверному улучшению координации движений животных с БП через неделю ежедневного назального введения. Терапевтический эффект сохранялся в течение всего периода введения, начиная с 7 дня, и через неделю после отмены препарата.
- Обнаружено пролонгированное действие Леводопа-ПК, которое выражалось в сохранении эффективности препарата через 24 часа после однократного введения и на протяжении всего периода лечения, в отличие от стандартного препарата, эффективность действия которого снижалась по мере курсового введения.
- Обнаруженное влияние Леводопа-ПК на психоэмоциональное состояние животных, приводящее к уменьшению тревожности и увеличению исследовательского интереса, является собственным эффектом препарата и может быть полезным для улучшения самочувствия пациентов при лечении БП.
Фармацевтическая субстанция для лечения инфицированных ран различного генеза
Комбинированная композиция для лечения инфицированных ран различного генеза
Фармацевтическая субстанция для лечения инфицированных ран различного генеза
Способ получения рекомбинантного антиангиогенного полипептида
Рекомбинантный гибридный ингибитор ангиогенеза и способ его получения
Сбор гепатопротекторного действия
Способ повышения напряжения кислорода в крови пациентов с хронической сердечной недостаточностью
N-карбоксиметилимидазо[4,5-е]бензо[1,2-с;3,4-с']дифуроксан
Лекарственный препарат для лечения болезни паркинсона
N-карб(аргинил)оксиметилимидазо[4,5-е]бензо[1,2-с;3,4-с']дифуроксан, ингибирующий агрегацию тромбоцитов
Гетеромерные пептиды на основе имидазо[4,5-е]бензо[1,2-с;3,4-с']дифуроксана, ингибирующие агрегацию тромбоцитов
Средство, обладающее хронокорректирующей и адаптогенной активностью
Средство хронобиологической коррекции

