Результат интеллектуальной деятельности: ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АЛПЕЛИСИБ
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым диспергирующимся таблеткам, содержащим соединение 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль.
Уровень техники
Фосфатидилинозит-3-киназы ("PI-3-киназа" или "PI3K") образуют широко экспрессирующееся семейство липидкиназ, который действуют, как ключевые переносчики сигналов в прямом направлении от клеточных рецепторов и как ключевые регуляторы метаболизма и выживания клеток.
Среди двух классов I PI3Ks PI3K класса IA представляют собой гетеродимеры, состоящие из каталитической субъединицы p110 (α, β, δ изоформы), конститутивно ассоциированной с регуляторной субъединицей, которая может представлять собой p85α, p55α, p50α, p85β или p55γ. Эти каталитические субъединицы p110 (α, β, δ изоформы) у людей кодируются тремя генами (PIK3CA, PIK3CB и PIK3CD). Класс 1B PI3K состоит из одного представителя семейства, гетеродимера, который включает каталитическую субъединицу p110γ, ассоциированную с двумя регуляторными субъединицами p101 или p84. Модулярные домены субъединиц p85/55/50 включают домены, гомологичные Src (SH2), которые связывают остатки фосфотирозина в контексте специфической последовательности на активированном рецепторе и цитоплазматических тирозинкиназах, что приводит к активации и локализации класса IA PI3Ks. Класс IB, а также p110β в некоторых случаях активируются непосредственно сшитыми с протеином G рецепторами, с которыми связывается широкое разнообразие пептидных и непептидных лигандов (Stephens et al., Cell 89:105 (1997)); Katso et al., Annu. Rev. Cell Dev. Biol. 17:615-675 (2001)). В результате образовавшиеся фосфолипидные продукты PI3K класса I связывают рецепторы, расположенные выше в пути передачи сигнала с расположенными ниже в пути передачи сигнала клеточными элементами, оказывающими воздействие на пролиферацию, выживание, хемотаксис, направленную миграцию клеток, подвижность, метаболизм, воспалительные и аллергические ответы, транскрипцию и трансляцию (Cantley et al., Cell 64:281 (1991); Escobedo and Williams, Nature 335:85 (1988); Fantl et al., Cell 69:413 (1992)).
Нарушение регуляции PI3K является одним из наиболее распространенных видов нарушения регуляции, ассоциированных с раком и пролиферативными заболеваниями человека (Parsons et al., Nature 436:792 (2005); Hennessey at el., Nature Rev. Drug Disc. 4:988-1004 (2005)). У гена-супрессора опухолей PTEN, который дефосфорилирует фосфоинозитиды в 3'-положении инозитного кольца и поэтому обладает способностью антагонизировать активность PI3K, во многих типах опухолей отсутствует функциональная активность. Гены для изоформы p110α (PIK3CA) амплифицированы и повышенный уровень экспрессии белковых продуктов этих генов был продемонстрирован при некоторых видах рака человека. Для некоторых видов рака человека описаны мутации и транслокации p85α, которая служит для повышающей регуляции комплекса p85-p110. И, наконец, установлено, что соматические миссенс-мутации PIK3CA, которые активируют пути дальнейшей передачи сигнала, достаточно часто встречаются при многих видах рака человека, включая 32% типов колоректального рака, 27% глиобластом, 25% типов рака желудка, 36% типов печеночно-клеточной карциномы и 18-40% раковых заболеваний молочной железы.
2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) является специфическим 2-карбоксамидциклоаминопроизводным мочевины, соединением, которое активно и селективно воздействует на альфа (α)-изоформу класса IA PI3K. Это соединение обладает следующей химической структурой:
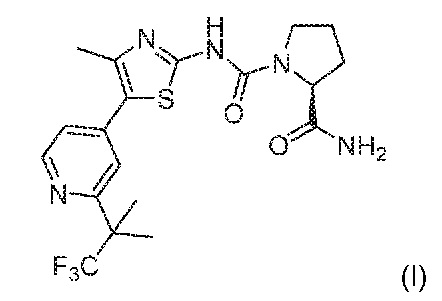
(далее "соединение I"). Соединение I продемонстрировало клиническую эффективность при лечении одним средством пациентов, у которых имелись запущенные сóлидные злокачественные новообразования, в которых происходили измерения в гене PIK3CA в фазе I клинического исследования. К 10 марта 2014 г. из 131 пациента, для которых проводили радиологическую оценку воздействия, у 15 пациентов с сóлидными опухолями с измененным PIK3CA наблюдался частичных ответ на лечение соединением I и у 68 пациентов наблюдалась стабильность заболевания, согласно критериям, приведенным в Response Evaluation Criteria in Solid Tumors (RECIST) guideline version 1.0 (Therasse P. et al., "New Guidelines to Evaluate the Response to Treatment in in Solid Tumors", JNCI National Cancer Inst. (2000), 2(3): 205-216.). Кроме того, соединение I продемонстрировало in vivo зависимую от дозы противоопухолевую зависимость в модели с мутантным PIK3CA или в расширенной модели ксенотрансплантата опухоли, такой как рак молочной железы, головы и шеи и яичников. В настоящее время соединение I исследуется в фазе Ib/II клинического исследования в комбинации с цетуксимабом для лечения пациентов с рецидивирующей или метастатической плоскоклеточной карциномой головы и шеи (NCT01602315).
В зависимости от возраста, состояния пациента, пути введения и типа заболевания, соединение I или его фармацевтически приемлемую соль вводят перорально в эффективной суточной дозе, равной примерно от 1 до 6,5 мг/кг для взрослых или детей. Взрослому пациенту с массой тела, равной 70 кг, соединение I или его фармацевтически приемлемую соль вводят перорально в суточной дозе, равной примерно от 70 мг до 455 мг. Соединение I или его фармацевтически приемлемую соль в настоящее время вводят перорально пациентам один или два раза в сутки в одной или большем количестве твердых таблеток, проглатываемых указанным пациентом. Эти дозированные формы, обеспечивающие эффективное введение указанного средства удобны для введения и являются стабильными. Однако для некоторых групп пациентов пероральное введение лекарственных форм в виде твердой таблетки нежелательно или невыполнимо. В частности, дети, пожилые пациенты и пациенты, страдающие от рака головы и шеи, могут быть неспособны легко проглатывать такие таблетки. Для этих пациентов обычно желательно или необходимо разработать альтернативные дозированные формы или альтернативные способы введения лекарственного средства.
Неожиданно было установлено, что соединение I или его фармацевтически приемлемую соль можно приготовить в виде новой диспергирующейся таблетки, предлагаемой в настоящем изобретении, содержащей большую дозу лекарственного средства (например, примерно от 5% до 50%, предпочтительно примерно от 35% до 45 мас.% в пересчете на общую массу таблетки), и которая диспергируется в водной среде за 5 мин или менее, предпочтительно за 3 мин или менее, и которая образует однородную дисперсию, которая может проходить через сито с номинальным размером отверстий, равным 710 мкм. Эта новая диспергирующаяся таблетка также позволяет удобно и безопасно вводить соединение I или его фармацевтически приемлемую соль пациентам, у которых имеются затруднения с проглатыванием, в фармакологически активной суточной дозе соединения I.
Сущность изобретения
Настоящее изобретение относится к диспергирующейся таблетке, содержащей (a) 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, (b) по меньшей мере один разрыхлитель в общем количестве, равном примерно от 1% до 25 мас.% в пересчете на общую массу таблетки, (c) по меньшей мере одно связующее в общем количестве, равном примерно от 1% до 20 мас.% в пересчете на общую массу таблетки, (d) по меньшей мере одно смазывающее вещество в общем количестве, равном примерно от 1% до 15 мас.% в пересчете на общую массу таблетки, и необязательно (e) один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
Предпочтительно, если диспергирующаяся таблетка, предлагаемая в настоящем изобретении, содержит грануляты, содержащие внутреннюю фазу и наружную фазу, где внутренняя фаза содержит 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль, по меньшей мере один разрыхлитель, по меньшей мере одно связующее и необязательно любые дополнительные эксципиенты, где наружная фаза содержит по меньшей мере один разрыхлитель, по меньшей мере одно смазывающее вещество и необязательно любые дополнительные эксципиенты, при условии, что указанный фармацевтически приемлемый эксципиент во внутренней фазе или наружной фазе не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
В одном варианте осуществления настоящее изобретение относится к диспергирующейся таблетке, содержащей (a) 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, (b) натриевую соль гликолята крахмала в количестве, равном примерно от 2% до 12 мас.% в пересчете на общую массу таблетки, (c) обладающую низкой степенью замещения гидроксипропилцеллюлозу в количестве, равном примерно от 1% до 10 мас.% в пересчете на общую массу таблетки, и (d) стеарилфумарат натрия в количестве, равном примерно от 1% до 7 мас.% в пересчете на общую массу таблетки, и необязательно (e) один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
В одном варианте осуществления настоящее изобретение относится к диспергирующейся таблетке, содержащей (a) 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 35% до 45 мас.% в пересчете на общую массу таблетки, (b) натриевую соль гликолята крахмала в количестве, равном примерно от 5% до 10 мас.% в пересчете на общую массу таблетки, (c) обладающую низкой степенью замещения гидроксипропилцеллюлозу в количестве, равном примерно от 1% до 3 мас.% в пересчете на общую массу таблетки, и (d) стеарилфумарат натрия в количестве, равном примерно от 3% до 5 мас.% в пересчете на общую массу таблетки, и необязательно (e) один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
В одном варианте осуществления диспергирующиеся таблетки, предлагаемые в настоящем изобретении, вводят пациенту, такому как ребенок или взрослый, страдающему от пролиферативного заболевания (например, рака), путем (i) взаимодействия указанной таблетки с проглатываемой жидкостью, (ii) предоставления таблетке возможности диспергироваться в проглатываемой жидкости с образованием диспергированной смеси, например, за 5 мин или менее, предпочтительно за 3 мин или менее, и (iii) проглатывания диспергированной смеси.
В одном варианте осуществления диспергирующиеся таблетки, предлагаемые в настоящем изобретении, можно вводить нуждающемуся в них пациенту путем (i) взаимодействия указанной таблетки с проглатываемой жидкостью (ii), предоставления таблетке возможности диспергироваться в проглатываемой жидкости с образованием диспергированной смеси, например, за 5 мин или менее, предпочтительно за 3 мин или менее, и (iii) введения указанной диспергированной смеси указанному пациенту с использованием или с помощью питательной трубки, предпочтительно гастростомической трубки (G-трубки) или PEG (чрескожной эндоскопической гастростомической ) трубки.
В одном объекте настоящее изобретение относится к способу получения диспергирующейся таблетки, предлагаемой в настоящем изобретении, включающему
(a) формование гранулята, содержащего внутреннюю фазу и наружную фазу, с помощью
(i.) влажного гранулирования внутренней фазы, содержащей 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, по меньшей мере один разрыхлитель в общем количестве, равном примерно от 1% до 15 мас.% в пересчете на общую массу таблетки, по меньшей мере одно связующее в общем количестве, равном примерно от 1% до 20 мас.% в пересчете на общую массу таблетки, и необязательно любые дополнительные фармацевтически приемлемые эксципиенты;
(ii.) добавления по меньшей мере одного разрыхлителя в общем количестве, равном примерно от 1% до 10 мас.% в пересчете на общую массу таблетки, и необязательно любых дополнительных фармацевтически приемлемых эксципиентов к внутренней фазе, сформованной на стадии (a)(i.), и смешивания; и
(iii.) добавления по меньшей мере одного смазывающего вещества в общем количестве, равном примерно от 1% до 15 мас.% в пересчете на общую массу таблетки, к смеси, сформованной на стадии (a)(ii.), и смешивания; и
(b) формование диспергирующейся таблетки путем прессования смеси, полученной на стадии (a)(iii.),
при условии, что любой дополнительный фармацевтически приемлемый эксципиент, использующийся на стадии (a)(i.) или (a)(ii.), не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу. Следует понимать, что стадии (a)(ii) и (a)(iii) приводят к формированию наружной фазы гранулята. В одном варианте осуществления способ дополнительно включает стадию нанесения пленочного покрытия на таблетку.
В одном варианте осуществления настоящее изобретение относится к диспергирующимся таблеткам, предлагаемым в настоящем изобретении, для применения для лечения или предупреждения пролиферативного нарушения, предпочтительно рака.
В одном варианте осуществления настоящее изобретение относится к применению диспергирующейся таблетки, предлагаемой в настоящем изобретении, для лечения или предупреждения пролиферативного заболевания, предпочтительно рака.
В одном варианте осуществления настоящее изобретение относится к применению диспергирующейся таблетки, предлагаемой в настоящем изобретении, для приготовления лекарственного средства, предназначенного для лечения или предупреждения пролиферативного заболевания, предпочтительно рака.
В одном варианте осуществления настоящее изобретение относится к способу лечения или предупреждения пролиферативного заболевания, включающему введение нуждающемуся в нем пациенту одной или большего количества диспергирующихся таблеток, предлагаемых в настоящем изобретении, которые вместе включают терапевтически эффективное количество соединения I или его фармацевтически приемлемой соли.
В одном варианте осуществления настоящее изобретение также относится к упаковке лекарственного средства, содержащей диспергирующиеся таблетки, предлагаемые в настоящем изобретении, и печатные инструкции, относящиеся к одной или большему количеству диспергирующихся таблеток соединения I или его фармацевтически приемлемой соли для перорального введения.
Подробное описание изобретения
Настоящее изобретение относится к диспергирующейся таблетке, содержащей (a) 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, (b) по меньшей мере один разрыхлитель в общем количестве, равном примерно от 1% до 25 мас.% в пересчете на общую массу таблетки, (c) по меньшей мере одно связующее в общем количестве, равном примерно от 1% до 20 мас.% в пересчете на общую массу таблетки, (d) по меньшей мере одно смазывающее вещество в общем количестве, равном примерно от 1% до 15 мас.% в пересчете на общую массу таблетки, и необязательно (e) один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
"Диспергирующаяся таблетка" означает не содержащую покрытие или содержащую пленочное покрытие таблетку, которая диспергируется в водной среде, например, в воде, соде или соке (например овощном соке) до введения.
Предпочтительно, если диспергирующаяся таблетка, предлагаемая в настоящем изобретении, содержит внутреннюю фазу и наружную фазу, где внутренняя фаза содержит 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль, по меньшей мере один разрыхлитель, по меньшей мере одно связующее и необязательно любые дополнительные эксципиенты, где наружная фаза содержит по меньшей мере один разрыхлитель, по меньшей мере одно смазывающее вещество и необязательно любые дополнительные эксципиенты, при условии, что указанный фармацевтически приемлемый эксципиент во внутренней фазе или наружной фазе не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
Настоящее изобретение относится к диспергирующейся таблетке, обладающей большим содержанием лекарственного средства, содержащего соединение I или его фармацевтически приемлемую соль в качестве активного ингредиента. Соединение I или его фармацевтически приемлемая соль содержится в количестве, равном примерно от 5% до 50%, например, примерно от 10% до 50% или от 20% до 50%, или от 30% до 45%, предпочтительно от 35% до 45 мас.% в пересчете на общую массу диспергирующейся таблетки. В частности, количество соединения I или его фармацевтически приемлемой соли может находиться в диапазоне от 35 до 45%, например, примерно от 36% до 41 мас.% в пересчете на общую массу диспергирующейся таблетки.
Некоторые термины, использующиеся в настоящем изобретении, описаны ниже. Если не приведены другие определения, все технические и научные термины, использующиеся в настоящем изобретении, обладают такими же значениями, как обычно понимает специалист в области техники, к которой относится настоящее изобретение.
Приведенные ниже общие определения применимы в настоящем изобретении, если не указано иное:
Термины "содержащий" и "включающий" используются в настоящем изобретении в их допускающем изменения и неограничивающем смысле, если не указано иное.
При использовании в настоящем изобретении, в особенности в контексте приведенной ниже формулы изобретения, термины, применяющиеся в единственном числе, включают множественное число, если в настоящем изобретении не указано иное или это явно не противоречит контексту. Если множественное число используется для соединений, солей и т. п., это также означает одно соединение, соль и т. п.
Термин "фармацевтически приемлемый" в настоящем изобретении определен, как означающий такие соединения, материалы, эксципиенты, композиции и/или дозированные формы, которые с медицинской точки зрения применимы для взаимодействия с тканями субъекта, например, млекопитающего или человека, без проявления чрезмерной токсичности, раздражения, аллергической реакции и других осложнений при разумном соотношении польза/риск.
Термин "суточная доза" означает общее дозированное количество терапевтического средства, вводимого конкретному пациенту в любой суточный или 24-часовой период.
Выражение "эффективное количество" или "терапевтически эффективное количество" при использовании в настоящем изобретении означает количество, достаточное для уменьшения по меньшей мере примерно на 15%, предпочтительно по меньшей мере на 50%, более предпочтительно по меньшей мере на 90%, и наиболее предпочтительно для предупреждения клинически значимого недостатка активности, функции и ответа нуждающегося в нем субъекта. Альтернативно, эффективное количество или терапевтически эффективное количество означает количество, достаточное для обеспечения наблюдаемого улучшения по сравнению с исходными клинически наблюдающимися признаками и симптомами заболевания.
Термин "фармацевтическая композиция" в настоящем изобретении определен, как означающий смесь или раствор, содержащий по меньшей мере один активный ингредиент, вводимый субъекту, например, млекопитающему или человеку, для предупреждения или лечения конкретного заболевания или патологического состояния, которым страдает субъект.
"Внутренняя фаза" означает фазу гранулята или ядра, включающую активный ингредиент - 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль, по меньшей мере один разрыхлитель, по меньшей мере одно связующее и необязательно один или более дополнительных фармацевтически приемлемых эксципиентов.
"Наружная фаза" означает по меньшей мере один разрыхлитель, по меньшей мере одно смазывающее вещество и необязательно один или более дополнительных фармацевтически приемлемых эксципиентов, которые добавляют ко внутренней фазе (гранулятам).
"Общая масса диспергирующейся таблетки" означает массу таблетки, включая внутреннюю и наружную фазу.
Термин "примерно" или "приблизительно" означает отклонение, не превышающее 10%, более предпочтительно 5%, данного значения или диапазона.
Термин "субъект" означает животное. Обычно животным является млекопитающее. Субъект также означает например, приматов (например, людей), коров, овец, коз, лошадей, собак, кошек, кроликов, крыс, мышей, рыб, птиц и т. п. В некоторых вариантах осуществления субъектом является примат. В других вариантах осуществления субъектом является человек.
Настоящее изобретение относится к диспергирующейся таблетке, содержащей (a) 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, (b) по меньшей мере один разрыхлитель в общем количестве, равном примерно от 1% до 25 мас.% в пересчете на общую массу таблетки, (c) по меньшей мере одно связующее в общем количестве, равном примерно от 1% до 20 мас.% в пересчете на общую массу таблетки, (d) по меньшей мере одно смазывающее вещество в общем количестве, равном примерно от 1% до 15 мас.% в пересчете на общую массу таблетки, и необязательно (e) один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
Предпочтительно, если диспергирующаяся таблетка, предлагаемая в настоящем изобретении, содержит внутреннюю фазу и наружную фазу, где внутренняя фаза содержит 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль, по меньшей мере один разрыхлитель, по меньшей мере одно связующее и необязательно любые дополнительные эксципиенты, где наружная фаза содержит по меньшей мере один разрыхлитель и необязательно любые дополнительные эксципиенты.
2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) ("соединение I") является специфическим 2-карбоксамидциклоаминопроизводным мочевины, соединением, которое активно и селективно воздействует на альфа (α)-изоформу класса IA PI3K и обладает следующей химической структурой:
Соединение I и его фармацевтически приемлемые соли описаны в заявке PCT № WO2010/029082, которая во всей своей полноте включена в настоящее изобретение в качестве ссылки, и способы его получения описаны, например, в примере 15 в этой заявке. Предпочтительно, если соединение I находится в форме свободного основания.
Соединение I можно вводить перорально в эффективной суточной дозе, равной примерно от 1 до 6,5 мг/кг взрослым людям или детям. Соединение I можно вводить перорально взрослому человеку с массой тела, равной 70 кг, в суточной дозе, равной примерно от 70 мг до 455 мг, например, примерно от 200 до 400 мг, или примерно от 240 мг до 400 мг, или примерно от 300 мг до 400 мг, или примерно от 350 мг до 400 мг, в виде одной дозы или разделенных доз до четырех раз в сутки. Предпочтительно, если соединение I вводят взрослому человеку с массой тела, равной 70 кг, в суточной дозе, равной от примерно 350 мг до примерно 400 мг.
Настоящее изобретение относится к диспергирующейся таблетке, обладающей большим содержанием лекарственного средства, содержащей соединение I в качестве активного ингредиента. Соединение I содержится в количестве, равном примерно от 5% до 50%, например, примерно от 10% до 50% или примерно от 20% до 50%, или примерно от 30% до 45%, предпочтительно примерно от 35% до 45 мас.% в пересчете на общую массу диспергирующейся таблетки. В частности, количество соединения I может находиться в диапазоне примерно от 35 до 45%, например, примерно от 36% до 41 мас.% в пересчете на общую массу диспергирующейся таблетки.
Подходящие разрыхлители для диспергирующихся таблеток, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, микрокристаллическую целлюлозу, натриевую соль гликолята крахмала, обладающую низкой степенью замещения гидроксипропилцеллюлозу, натриевую соль кроскармеллозы, кальциевую соль кроскармеллозы, сшитый поливинилпирролидон (например, продающийся под торговыми названиями Кросповидон® или Полипласдон® или Kollidone®CL), сшитую альгиновую кислоту, альгинат натрия, альгинат калия, геллановую камедь, кукурузный крахмал, предварительно желатинизированный крахмал, натриевую соль карбоксиметилцеллюлозы, глицин и любую их комбинацию. Предпочтительно используют натриевую соль гликолята крахмала или обладающую низкой степенью замещения гидроксипропилцеллюлозу. Более предпочтительно используют и натриевую соль гликолята крахмала и обладающую низкой степенью замещения гидроксипропилцеллюлозу.
В диспергирующихся таблетках, предлагаемых в настоящем изобретении, разрыхлитель содержится в общем количестве, равном от примерно от 1% до 25%, предпочтительно примерно от 2% до 12%, например, примерно от 5% до 12%, например, примерно от 5 до 10 мас.% в пересчете на общую массу таблетки.
Таким образом, разрыхлителем может быть натриевая соль гликолята крахмала, которая содержится в общем количестве, равном примерно от 2% до 12%, предпочтительно примерно от 5% до 10 мас.% в пересчете на общую массу таблетки. Разрыхлителем также может быть смесь натриевой соли гликолята крахмала, которая содержится в общем количестве, равном примерно от 5% до 10 мас.% в пересчете на общую массу таблетки, и обладающей низкой степенью замещения гидроксипропилцеллюлозы, которая содержится в общем количестве, равном примерно от 2% до 5 мас.% в пересчете на общую массу таблетки.
В одном варианте осуществления разрыхлителем является натриевая соль гликолята крахмала, которая содержится в общем количестве, равном примерно от 2% до 12%, предпочтительно примерно от 5% до 10 мас.% в пересчете на общую массу таблетки.
В одном варианте осуществления разрыхлитель содержит натриевую соль гликолята крахмала, которая содержится в общем количестве, равном примерно от 5% до 10 мас.% в пересчете на общую массу таблетки и обладающую низкой степенью замещения гидроксипропилцеллюлозу, которая содержится в общем количестве, равном примерно от 2% до 5 мас.% в пересчете на общую массу таблетки.
Подходящие связующие для диспергирующихся таблеток, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, микрокристаллическую целлюлозу, обладающую низкой степенью замещения гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, гидроксиметилцеллюлозу, гидроксиэтилцеллюлозу, крахмал 1500, гуаровую камедь, ксантановую камедь, поливинилпирролидон, альгинат натрия и любую их комбинацию. Предпочтительно используют обладающую низкой степенью замещения гидроксипропилцеллюлозу.
В диспергирующихся таблетках, предлагаемых в настоящем изобретении, связующее содержится в общем количестве, равном примерно от 1% до 20%, предпочтительно примерно от 1% до 10%, например, примерно от 2% до 5%, например, примерно от 1% до 3 мас.% в пересчете на общую массу таблетки.
Предпочтительным связующим является обладающая низкой степенью замещения гидроксипропилцеллюлоза. Если она содержится, то содержится в общем количестве, равном примерно от 1 до 10%, например, примерно от 2 до 5%, например, примерно от 1% до 3 мас.% в пересчете на общую массу таблетки.
В одном варианте осуществления связующим является обладающая низкой степенью замещения гидроксипропилцеллюлоза, которая содержится в общем количестве, равном примерно от 1 до 10%, например, примерно от 2 до 5%, например, примерно от 1% до 3 мас.% в пересчете на общую массу таблетки.
Подходящие смазывающие вещества для диспергирующихся таблеток, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, стеарилфумарат натрия, стеарат магния, стеариновую кислоту, гидрированное касторовое масло, стеарат кальция, стеарат алюминия, PEG 4000-8000, тальк, глицерилмоностеарат, глицерилдибегенат (например, продающийся фирмой Gattefossé под торговым названием Compritol® 888 ATO), глицерилпальмитостеариловый эфир (например продающийся фирмой Gattefossé под торговым названием Precerol®), гидрированное хлопковое масло, касторовое масло и любую их комбинацию. Предпочтительно используют стеарилфумарат натрия.
В диспергирующихся таблетках, предлагаемых в настоящем изобретении, смазывающее вещество содержится в общем количестве, равном от примерно 1% до примерно 15%, предпочтительно примерно от 1% до 7%, например, примерно от 3% до 6%, например, примерно от 3% до 5 мас.% в пересчете на общую массу таблетки.
В одном варианте осуществления смазывающим веществом является стеарилфумарат натрия, который содержится в общем количестве, равном примерно от 1% до 7%, например, примерно от 3% до 6%, например, примерно от 3% до 5 мас.% в пересчете на общую массу таблетки.
Диспергирующаяся таблетка, предлагаемая в настоящем изобретении, необязательно может включать один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу. Примеры фармацевтически приемлемых эксципиентов, которые дополнительно можно использовать в настоящем изобретении, включают, но не ограничиваются только ими, один или более разбавители, при условии, что разбавитель не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, или дигидрат дикальцийортофосфата; агенты, способствующие скольжению; поверхностно-активные вещества; агент, маскирующий вкус,, при условии, что агент, маскирующий вкус, не представляет собой сукралозу, или любую их комбинацию.
Предпочтительно, если диспергирующаяся таблетка, предлагаемая в настоящем изобретении, дополнительно содержит дополнительный фармацевтически приемлемый эксципиент, который представляет собой разбавитель, при условии, что этот разбавитель не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, или дигидрат дикальцийортофосфата. Подходящие разбавители для диспергирующихся таблеток, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, маннит (включая альфа, бета и дельта полиморфные формы), сорбит, мальтодекстрин, лактозу (например, моногидрат лактозы), микрокристаллическую целлюлозу, мальтит, ксилит, крахмал (например, кукурузный, маисовый или рисовый) или любую их комбинацию.
Предпочтительным разбавителем является маннит или микрокристаллическая целлюлоза, или смесь маннита и микрокристаллической целлюлозы. Более предпочтительно, разбавителем является дельта полиморфная форма маннита и/или микрокристаллическая целлюлоза.
Общее количество разбавителя может находиться в диапазоне примерно от 20% до 70%, в частности, например, примерно от 30% до 60%, например, примерно от 35% до 55%, например, примерно от 45% до 55%, например, примерно от 45% до 50 мас.% в пересчете на общую массу таблетки.
В одном варианте осуществления диспергирующаяся таблетка, предлагаемая в настоящем изобретении, содержит дополнительный фармацевтически приемлемый эксципиент, который представляет собой разбавитель, при условии, что этот разбавитель не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, или дигидрат дикальцийортофосфата.
В одном варианте осуществления разбавителем является маннит или микрокристаллическая целлюлоза. В одном предпочтительном варианте осуществления разбавителем является маннит и микрокристаллическая целлюлоза. В одном предпочтительном варианте осуществления разбавителем является дельта полиморфная форма маннита и/или микрокристаллическая целлюлоза.
В одном варианте осуществления таблетка содержит дополнительный фармацевтически приемлемый эксципиент, который представляет собой разбавитель, где разбавитель содержится в общем количестве, находящемся в диапазоне от примерно от 30% до 60%, например, примерно от 35% до 55%, например, примерно от 45% до 55%, например, примерно от 50% до 55 мас.% в пересчете на общую массу таблетки, и где указанным разбавителем является маннит и микрокристаллическая целлюлоза.
В одном варианте осуществления таблетка содержит дополнительный фармацевтически приемлемый эксципиент, который представляет собой разбавитель, где разбавитель содержится в общем количестве, находящемся в диапазоне от примерно от 30% до 60%, например, примерно от 35% до 55%, например, примерно от 45% до 55%, например, примерно от 50% до 55 мас.% в пересчете на общую массу таблетки, и где указанным разбавителем является маннит, который содержится в общем количестве, находящемся в диапазоне от примерно от 15% до 20 мас.% в пересчете на общую массу таблетки, и микрокристаллическая целлюлоза, которая содержится в общем количестве, находящемся в диапазоне от примерно от 25% до 35 мас.% в пересчете на общую массу таблетки.
Диспергирующаяся таблетка, предлагаемая в настоящем изобретении, может включать дополнительный фармацевтически приемлемый эксципиент, которым является агент, способствующий скольжению. Подходящие агенты, способствующие скольжению, для диспергирующихся таблеток, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, диоксид кремния, коллоидный диоксид кремния (например, безводный коллоидный диоксид кремния), трисиликат магния, порошкообразную целлюлозу, крахмал, тальк и любую их комбинацию.
Общее количество агента, способствующего скольжению, может находиться в диапазоне примерно от 0,1% до 6%, в частности, примерно от 0,1% до 4%, например, примерно от 0,1% до 2,5%, например, примерно от 0,1 до 1,0 мас.% в пересчете на общую массу таблетки.
В одном варианте осуществления диспергирующаяся таблетка, предлагаемая в настоящем изобретении, содержит дополнительный фармацевтически приемлемый эксципиент, которым является агент, способствующий скольжению.
В одном варианте осуществления таблетка содержит дополнительный фармацевтически приемлемый эксципиент, которым является агент, способствующий скольжению, где общее количество агента, способствующего скольжению, может находиться в диапазоне примерно от 0,1% до 6%, в частности, примерно от 0,1% до 4%, например, примерно от 0,1% до 2,5%, например, примерно от 0,1 до 1,0 мас.% в пересчете на общую массу таблетки.
Диспергирующаяся таблетка, предлагаемая в настоящем изобретении, может включать дополнительный фармацевтически приемлемый эксципиент, которым является поверхностно-активное вещество. Подходящие поверхностно-активные вещества для диспергирующихся таблеток, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, лаурилсульфат натрия, четвертичные аммониевые соли, полисорбаты, эфиры сорбита, полоксамер, витамин E, полиэтиленгликольсукцинат, пальмитат сахарозы, докузат натрия, лецитин и любую их комбинацию.
Общее количество поверхностно-активного вещества может находиться в диапазоне примерно от 0,01% до 4%, в частности, примерно от 0,05% до 2,0%, например, примерно от 0,05% до 1,0 мас.% в пересчете на общую массу таблетки.
В одном варианте осуществления диспергирующаяся таблетка, предлагаемая в настоящем изобретении, содержит дополнительный фармацевтически приемлемый эксципиент, которым является поверхностно-активное вещество.
В одном варианте осуществления диспергирующаяся таблетка, предлагаемая в настоящем изобретении, содержит дополнительный фармацевтически приемлемый эксципиент, которым является поверхностно-активное вещество, где общее количество поверхностно-активного вещества может находиться в диапазоне примерно от 0,01% до 4%, в частности, примерно от 0,05% до 2,0%, например, примерно от 0,05% до 1,0 мас.% в пересчете на общую массу таблетки.
Диспергирующаяся таблетка, предлагаемая в настоящем изобретении, может включать дополнительный фармацевтически приемлемый эксципиент, которым является агент, маскирующий вкус, при условии, что дополнительный фармацевтический агент не представляет собой сукралозу. Агент, маскирующий вкус, может представлять собой подсластитель, вкусовое вещество или их комбинацию.
Подсластителем может быть сахар или заменитель сахара, выбранный из группы, включающей лактозу, маннит, сахарозу, глюкозу, тауматин, неотам, тагатозу, ацесульфам калия, сахаринат натрия, аспартам, трегалозу, сахарин или их комбинацию.
Вкусовое вещество представляет собой вещество, способное усиливать вкус или аромат композиции. Подходящие вкусовые агенты можно выбрать из числа указанных в стандартных справочниках, например, в Fenaroli's Handbook of Flavor Ingredients, 3rd edition (1995). Подходящие вкусовые агенты для диспергирующейся таблетки, предлагаемой в настоящем изобретении, включают, но не ограничиваются только ими, (i.) натуральные вкусовые добавки, которые можно имитировать натуральными или синтетическими агентами, или их комбинации, такие как миндаль, анис, яблоко, абрикос, бергамот, черника, черная смородина, голубика, какао, карамель, вишня, корица, клюква, эвкалипт, инжир, имбирь, виноград, лимон, лакрица, лайм, солод, меласса, мускатный орех, персик, груша, мята перечная, ананас, малина, роза, мята, земляника, мандарин, чай, ваниль, винтергрен и т. п., и (ii) синтетические вкусовые добавки, такие как засахаренные фрукты или аромат жевательной резинки. Вкусовые агенты можно использовать по отдельности или в комбинациях двух или большего количества из них.
Один или более фармацевтически приемлемых эксципиентов специалист в данной области техники может выбрать и использовать в соответствии с настоящим изобретением с учетом конкретных желательных характеристик диспергирующейся таблетки на основании настоящего описания и опыта и подготовки специалиста в данной области техники. Кроме того, абсолютные количества каждого фармацевтически приемлемого эксципиента и количества, относительные по сравнению с другими эксципиентами в диспергирующейся таблетке, предлагаемой в настоящем изобретении, специалист в данной области техники может выбрать с учетом конкретных желательных характеристик диспергирующейся таблетки на основании настоящего описания и опыта и подготовки специалиста в данной области техники.
Следует понимать, что любой данный эксципиент может выполнять более одной функции, например, в качестве разрыхлителя, смазывающего вещества, связующего, разбавителя, агента, способствующего скольжению и/или поверхностно-активного вещества. Например, в предпочтительном варианте осуществления обладающая низкой степенью замещения гидроксипропилцеллюлоза выступает в качестве и разрыхлителя, и связующего.
Диспергирующаяся таблетка, предлагаемая в настоящем изобретении, может не обладать покрытием или обладать пленочным покрытием. Подходящие пленочные покрытия известны и имеются в продаже или из можно приготовить по известным методикам. Обычно материалами пленочного покрытия являются гидрофильные полимеры, такие как полиэтиленгликоль, поливинилпирролидон, поливиниловый спирт, гидроксипропилцеллюлоза, гидроксиметилцеллюлоза и гидроксипропилметилцеллюлоза и т. п. Ингредиенты композиции пленочного покрытия могут включать пластификаторы, использующиеся в обычных количествах, а также замутнители и красители. Обычно материал пленочного покрытия наносят в таком количестве, чтобы сформировать пленочное покрытие, масса которого составляет от примерно 1% до примерно 6 мас.% в пересчете на всю таблетку. Можно использовать сухие смеси, такие как смеси Sepifilm или Opadry, выпускающиеся фирмой Colorcon Corp. Эти продукты представляют собой приготовленные по отдельности сухие премиксы пленкообразующих полимеров, замутнителей, красителей и пластификаторов, которые дополнительно обрабатывают с получением водной суспензии пленочного покрытия.
Пленочное покрытие можно наносить по обычным методикам в подходящем баке для нанесения покрытия или аппарате с псевдоожиженным слоем с использованием воды и/или обычных органических растворителей (например, метилового спирта, этилового спирта, изопропилового спирта), кетонов (ацетон) и т. п.
Предпочтительно, если диспергирующаяся таблетка, предлагаемая в настоящем изобретении, не содержит покрытия.
В предпочтительном варианте осуществления настоящее изобретение относится к диспергирующейся таблетке, содержащей (a) 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, (b) натриевую соль гликолята крахмала в количестве, равном примерно от 2% до 12 мас.% в пересчете на общую массу таблетки, (c) обладающую низкой степенью замещения гидроксипропилцеллюлозу в количестве, равном примерно от 1% до 10 мас.% в пересчете на общую массу таблетки, и (d) стеарилфумарат натрия в количестве, равном примерно от 1% до 7 мас.% в пересчете на общую массу таблетки, и необязательно (e) один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
В предпочтительном варианте осуществления настоящее изобретение относится к диспергирующейся таблетке, содержащей (a) 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 35% до 45 мас.% в пересчете на общую массу таблетки, (b) натриевую соль гликолята крахмала в количестве, равном примерно от 5% до 10 мас.% в пересчете на общую массу таблетки, (c) обладающую низкой степенью замещения гидроксипропилцеллюлозу в количестве, равном примерно от 1% до 3 мас.% в пересчете на общую массу таблетки, и (d) стеарилфумарат натрия в количестве, равном примерно от 3% до 5 мас.% в пересчете на общую массу таблетки, и необязательно (e) один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
В одном варианте осуществления настоящее изобретение относится к диспергирующейся таблетке, содержащей грануляты, содержащие внутреннюю фазу и наружную фазу, где внутренняя фаза содержит 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль, по меньшей мере один разрыхлитель, по меньшей мере одно связующее и необязательно любые дополнительные эксципиенты, где наружная фаза содержит по меньшей мере один разрыхлитель, по меньшей мере одно смазывающее вещество, и необязательно любые дополнительные эксципиенты, и где диспергирующаяся таблетка содержит (a) 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, (b) по меньшей мере один разрыхлитель в общем количестве, равном примерно от 1% до 25 мас.% в пересчете на общую массу таблетки, (c) по меньшей мере одно связующее в общем количестве, равном примерно от 1% до 20 мас.% в пересчете на общую массу таблетки, (d) по меньшей мере одно смазывающее вещество в общем количестве, равном примерно от 1% до 15 мас.% в пересчете на общую массу таблетки, и необязательно (e) один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент во внутренней фазе или наружной фазе не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
В предпочтительном варианте осуществления настоящее изобретение относится к диспергирующейся таблетке, содержащей грануляты, содержащие внутреннюю фазу и наружную фазу, где внутренняя фаза содержит 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль, по меньшей мере один разрыхлитель - натриевую соль гликолята крахмала, по меньшей мере одно связующее - обладающую низкой степенью замещения гидроксипропилцеллюлозу, и необязательно любые дополнительные эксципиенты, где наружная фаза содержит по меньшей мере один разрыхлитель - натриевую соль гликолята крахмала, по меньшей мере одно смазывающее вещество - стеарилфумарат натрия и необязательно любые дополнительные эксципиенты, и где диспергирующаяся таблетка содержит (a) 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, (b) натриевую соль гликолята крахмала в количестве, равном примерно от 2% до 12 мас.% в пересчете на общую массу таблетки, (c) обладающую низкой степенью замещения гидроксипропилцеллюлозу в количестве, равном примерно от 1% до 10 мас.% в пересчете на общую массу таблетки, и (d) стеарилфумарат натрия в количестве, равном примерно от 1% до 7 мас.% в пересчете на общую массу таблетки, и необязательно (e) один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент во внутренней фазе или наружной фазе не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
В предпочтительном варианте осуществления настоящее изобретение относится к диспергирующейся таблетке, содержащей грануляты, содержащие внутреннюю фазу и наружную фазу, где внутренняя фаза содержит 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль, по меньшей мере один разрыхлитель - натриевую соль гликолята крахмала, по меньшей мере одно связующее - обладающую низкой степенью замещения гидроксипропилцеллюлозу, и необязательно любые дополнительные эксципиенты, где наружная фаза содержит по меньшей мере один разрыхлитель - натриевую соль гликолята крахмала, по меньшей мере одно смазывающее вещество - стеарилфумарат натрия, и необязательно любые дополнительные эксципиенты, и где диспергирующаяся таблетка содержит (a) 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 35% до 45 мас.% в пересчете на общую массу таблетки, (b) натриевую соль гликолята крахмала в количестве, равном примерно от 5% до 10 мас.% в пересчете на общую массу таблетки, (c) обладающую низкой степенью замещения гидроксипропилцеллюлозу в количестве, равном примерно от 1% до 3 мас.% в пересчете на общую массу таблетки, и (d) стеарилфумарат натрия в количестве, равном примерно от 3% до 5 мас.% в пересчете на общую массу таблетки, и необязательно (e) один или более дополнительных фармацевтически приемлемых эксципиентов, при условии, что указанный фармацевтически приемлемый эксципиент во внутренней фазе или наружной фазе не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
В соответствии с настоящим изобретением диспергирующиеся таблетки, предлагаемые в настоящем изобретении, которые быстро распадаются в водной среде, такой как вода, сода или сок (например, фруктовый или овощной сок). Быстрый распад можно наблюдать в стандартных тестах, известных специалисту в данной области техники. Время распада предпочтительно измеряют с помощью стандартного теста для диспергирующейся таблетки, описанного в публикации European Pharmacopoeia Eighth edition, Supplement 8.2, page 811 (January 2014) в сочетании с European Pharmacopoeia, Eighth edition, Supplement 8.2, page 285-287, Section 2.9.1 (January 2014), которые обе во всей своей полноте включены в настоящее изобретение в качестве ссылки. "Время распада" означает время, которое необходимо для диспергирующейся таблетки для распада в воде при комнатной температуре в устройстве для определения времени распада. В этом тесте из публикации European Pharmacopoeia (January 2014) определяют время распада таблетки в воде при температуре, равной от 15 до 25°C.
Скорость и полноту распада можно наблюдать визуально. Распад считается завершенным, когда на сите прибора для исследования не остается остаток или когда остаток, остающийся на сите прибора для исследования, состоит из мягкой массы, не содержащей прощупываемую пленку ядра, или на сите находятся только фрагменты нерастворимого покрытия.
Таблетки, предлагаемые в настоящем изобретении, предпочтительно обладают временем распада, равным 5 мин или менее, предпочтительно 3 мин или менее, по данным исследования с помощью описанного выше теста в воде при температуре, равной от 15 до 25°C. Более предпочтительно, если время распада равно 90 с или менее.
Предпочтительно, если диспергирующиеся таблетки, предлагаемые в настоящем изобретении, образуют однородную дисперсию, которая проходит через сито с номинальным размером отверстий, равным 710 мкм, после того, как указанные таблетки полностью диспергированы в водной среде, такой как вода, сода или сок (например, фруктовый или овощной сок). Эту мелкозернистость дисперсии можно измерить с помощью стандартного теста для диспергирующейся таблетки, описанного в публикации European Pharmacopoeia Eighth edition, Supplement 8.2, page 811 (January 2014), которая во всей своей полноте включена в настоящее изобретение в качестве ссылки.
В соответствии с настоящим изобретением диспергирующиеся таблетки, предлагаемые в настоящем изобретении, диспергируют в проглатываемой жидкости, включая, но не ограничиваясь только ими, воду, соду и сок (например, фруктовый сок или овощной сок), до введения субъекту. Предпочтительно, если проглатываемой жидкостью является вода. Полученную диспергированную таблетку можно вводить субъекту или пациенту путем проглатывания или путем введения с помощью питательной трубки, предпочтительно гастростомической трубки (G-трубки) или PEG (чрескожной эндоскопической гастростомической ) трубки.
В одном объекте диспергирующиеся таблетки, предлагаемые в настоящем изобретении, вводят пациенту, такому как ребенок или взрослый, страдающему от пролиферативного заболевания (например, рака), путем (i) взаимодействия указанной таблетки с проглатываемой жидкостью, (ii) предоставления таблетке возможности диспергироваться в проглатываемой жидкости с образованием диспергированной смеси, например, за 5 мин или менее, предпочтительно за 3 мин или менее, и (iii) проглатывания диспергированной смеси.
В одном варианте осуществления настоящее изобретение относится к способу введения диспергирующейся таблетки, предлагаемой в настоящем изобретении, нуждающемуся в ней пациенту, который включает (i) взаимодействие таблетки с проглатываемой жидкостью, (ii) диспергирование таблетки в проглатываемой жидкости с образованием диспергированной смеси, например, за 5 мин или менее, предпочтительно за 3 мин или менее, и (iii) проглатывание диспергированной смеси.
В одном варианте осуществления настоящее изобретение относится к способу введения диспергирующейся таблетки, предлагаемой в настоящем изобретении, нуждающемуся в ней пациенту, который включает инструкции нуждающемуся в ней субъекту по (i) взаимодействию таблетки с проглатываемой жидкостью, (ii) диспергированию таблетки в проглатываемой жидкости с образованием диспергированной смеси, например, за 5 мин или менее, предпочтительно за 3 мин или менее, и (iii) проглатыванию диспергированной смеси.
В другом объекте диспергирующиеся таблетки, предлагаемые в настоящем изобретении, можно вводить нуждающемуся в них пациенту путем (i) взаимодействия указанной таблетки с проглатываемой жидкостью (ii), предоставления таблетке возможности диспергироваться в проглатываемой жидкости с образованием диспергированной смеси, например, за 5 мин или менее, предпочтительно за 3 мин или менее, и (iii) введения указанной диспергированной смеси указанному пациенту с использованием или с помощью питательной трубки, предпочтительно гастростомической трубки (G-трубки) или PEG (чрескожной эндоскопической гастростомической ) трубки.
В одном варианте осуществления настоящее изобретение относится к способу введения диспергирующейся таблетки, предлагаемой в настоящем изобретении, нуждающемуся в ней пациенту, который включает (i) взаимодействие указанной таблетки с проглатываемой жидкостью, (ii) диспергирование таблетки в проглатываемой жидкости с образованием диспергированной смеси, например, за 5 мин или менее, предпочтительно за 3 мин или менее, и (iii) введения указанной диспергированной смеси указанному пациенту с использованием или с помощью питательной трубки, предпочтительно гастростомической трубки (G-трубки) или PEG-трубки.
Диспергирующиеся таблетки, предлагаемые в настоящем изобретении, можно диспергировать, например, в равном от 20 до 50 мл количестве воды при перемешивании.
В соответствии с настоящим изобретением неожиданно было установлено, что эти диспергирующиеся таблетки, содержащие большую дозу лекарственного средства (например, примерно от 5% до 50%, предпочтительно примерно от 35% до 45 мас.% в пересчете на общую массу таблетки) соединения I, способны диспергироваться в водной среде, такой как вода, сода или сок (например, фруктовый или овощной сок), за 5 мин или менее, предпочтительно за 3 мин или менее, можно получить с помощью влажного гранулирования последующим прессованием.
Обычно влажное гранулирование не является предпочтительным для приготовления диспергирующихся таблеток. Влажное гранулирование увеличивает когезию частиц активного ингредиента и увеличивает время распада готовой таблетки, что не согласуется с предпочтениями пациента или положениями European Pharmacopoeia, согласно которой для диспергирующейся таблетки необходимо время распада, равное 3 мин или менее. Согласно изобретению неожиданно было установлено, что конкретное применение соединения I с разрыхлителем в общем количестве, равном примерно от 1% до 25 мас.% в пересчете на общую массу таблетки, связующим в общем количестве, равном примерно от 1% до 20 мас.% в пересчете на общую массу таблетки, и смазывающим веществом в общем количестве, равном примерно от 1% до 15 мас.% в пересчете на общую массу таблетки дает стабильную композицию, обладающую большим содержанием лекарственного средства (например, примерно от 5% до 50%, предпочтительно примерно от 35% до 45 мас.% в пересчете на общую массу таблетки), и которая диспергируется в водной среде за 5 мин или менее, предпочтительно за 3 мин или менее. Кроме того, согласно изобретению неожиданно было установлено, что для обеспечения указанных выше физических и химических характеристик и подходящей стабильности соединение I необходимо приготовить в виде диспергирующейся таблетки, которая не содержит трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
В одном объекте настоящее изобретение относится к способу получения диспергирующейся таблетки, предлагаемой в настоящем изобретении, включающему
(a) формование гранулята, содержащего внутреннюю фазу и наружную фазу, с помощью
(i.) влажного гранулирования внутренней фазы, содержащей 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, по меньшей мере один разрыхлитель в общем количестве, равном примерно от 1% до 15 мас.% в пересчете на общую массу таблетки, по меньшей мере одно связующее в общем количестве, равном примерно от 1% до 20 мас.% в пересчете на общую массу таблетки, и необязательно любые дополнительные фармацевтически приемлемые эксципиенты;
(ii.) добавления по меньшей мере одного разрыхлителя в общем количестве, равном примерно от 1% до 10 мас.% в пересчете на общую массу таблетки, и необязательно любых дополнительных фармацевтически приемлемых эксципиентов к внутренней фазе, сформованной на стадии (a)(i.), и смешивания; и
(iii.) добавления по меньшей мере одного смазывающего вещества в общем количестве, равном примерно от 1% до 15 мас.% в пересчете на общую массу таблетки, к смеси, сформованной на стадии (a)(ii.), и смешивания; и
(b) формование диспергирующейся таблетки путем прессования смеси, полученной на стадии (a)(iii.),
при условии, что любой дополнительный фармацевтически приемлемый эксципиент, использующийся на стадии (a)(i.) или (a)(ii.), не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу. Следует понимать, что стадии (a)(ii) и (a)(iii) приводят к формированию наружной фазы гранулята.
На таблетку необязательно можно нанести пленочное покрытие. В одном варианте осуществления способ дополнительно включает стадию нанесения пленочного покрытия на таблетку.
Процедуры, которые можно использовать могут быть обычными или известными в данной области техники или быть основаны на таких процедурах, например, описанных в публикациях L. Lachman et al. The Theory and Practice of Industrial Pharmacy, 3rd Ed., 1986; H. Sucker et al., Pharmazeutische Technologie, Thieme, 1991, Hagers, Handbuch der Pharmazeutischen Praxis, 4th Ed. (Springer Verlag, 1971) и Remington's pharmaceutical Sciences, 13th Ed. (Mack Publ., Co., 1970) или более поздних изданиях.
В одном варианте осуществления способ, описанный выше, включает один или более дополнительных фармацевтически приемлемых эксципиентов на стадии (a)(i.), которые представляют собой разбавитель, при условии, что разбавитель не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, или дигидрат дикальцийортофосфата. Предпочтительно, если указанным разбавителем является маннит или микрокристаллическая целлюлоза. Более предпочтительно, если разбавителем является маннит и микрокристаллическая целлюлоза.
В одном варианте осуществления способ, описанный выше, включает один или более дополнительных фармацевтически приемлемых эксципиентов на стадии (a)(ii.), которые представляют собой разбавитель, при условии, что разбавитель не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, или дигидрат дикальцийортофосфата. Предпочтительно, если указанным разбавителем является маннит или микрокристаллическая целлюлоза. Более предпочтительно, если разбавителем является маннит и микрокристаллическая целлюлоза.
В одном варианте осуществления способ, описанный выше, включает один или более дополнительных фармацевтически приемлемых эксципиентов на стадии (a)(i.) и стадии (a)(ii.), которые представляют собой разбавитель, при условии, что разбавитель не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, или дигидрат дикальцийортофосфата. Предпочтительно, если указанным разбавителем является маннит или микрокристаллическая целлюлоза. Предпочтительно, если разбавителем является маннит и микрокристаллическая целлюлоза. Более предпочтительно, если разбавителем на стадии (a)(i.) является маннит и микрокристаллическая целлюлоза и разбавителем на стадии (a)(ii.) является микрокристаллическая целлюлоза.
В одном варианте осуществления разбавителем на стадии (a)(i.) является маннит, который содержится в общем количестве, равном примерно от 15% до 20 мас.% в пересчете на общую массу таблетки, и микрокристаллическая целлюлоза, которая содержится в количестве, равном примерно от 15% до 20 мас.% в пересчете на общую массу таблетки, и разбавителем на стадии (a)(ii.) является микрокристаллическая целлюлоза, которая содержится в количестве, равном примерно от 10% до 15 мас.% в пересчете на общую массу таблетки.
В одном варианте осуществления настоящее изобретение относится к способу получения диспергирующейся таблетки, предлагаемой в настоящем изобретении, включающему
(a) формование гранулята, содержащего внутреннюю фазу и наружную фазу, с помощью
(i.) влажного гранулирования внутренней фазы, содержащей 2-амид (S)-пирролидин-1,2-дикарбоновой кислоты 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)-пиридин-4-ил]-тиазол-2-ил}-амид) или его фармацевтически приемлемую соль в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, натриевую соль гликолята крахмала в общем количестве, равном примерно от 1% до 7 мас.% в пересчете на общую массу таблетки, обладающую низкой степенью замещения гидроксипропилцеллюлозу в общем количестве, равном примерно от 1% до 10 мас.% в пересчете на общую массу таблетки, и по меньшей мере один разбавитель;
(ii.) добавления натриевой соли гликолята крахмала в общем количестве, равном примерно от 1% до 5 мас.% в пересчете на общую массу таблетки и по меньшей мере одного разбавителя во внутренней фазе, сформированной на стадии (a)(i.) и смешивания; и
(iii.) добавления стеарилфумарата натрия в общем количестве, равном примерно от 1% до 7 мас.% в пересчете на общую массу таблетки, к смеси, сформованной на стадии (a)(ii.), и смешивания; и
(b) формование диспергирующейся таблетки путем прессования смеси, полученной на стадии (a)(iii.),
при условии, что разбавитель, использующийся на стадии (a)(i.) или (a)(ii.), не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, или дигидрат дикальцийортофосфата.
Предпочтительно, разбавителем на стадии (a)(i.) является маннит и микрокристаллическая целлюлоза и разбавителем на стадии (a)(ii.) является микрокристаллическая целлюлоза.
В одном варианте осуществления разбавителем на стадии (a)(i.) является маннит, который содержится в общем количестве, равном примерно от 15% до 20 мас.% в пересчете на общую массу таблетки, и микрокристаллическая целлюлоза, которая содержится в количестве, равном примерно от 15% до 20 мас.% в пересчете на общую массу таблетки, и разбавителем на стадии (a)(ii.) является микрокристаллическая целлюлоза, которая содержится в количестве, равном примерно от 10% до 15 мас.% в пересчете на общую массу таблетки.
В одном варианте осуществления способ дополнительно включает стадию нанесения пленочного покрытия на таблетку.
В другом варианте осуществления настоящее изобретение относится к способу получения диспергирующейся таблетки, предлагаемой в настоящем изобретении, включающему
(a) формование гранулята, содержащего внутреннюю фазу и наружную фазу, с помощью
(i.) смешивания соединения I или его фармацевтически приемлемой соли в количестве, равном примерно от 5% до 50 мас.% в пересчете на общую массу таблетки, по меньшей мере одного разрыхлителя в общем количестве, равном примерно от 1% до 15 мас.% в пересчете на общую массу таблетки, и необязательно любых дополнительных фармацевтически приемлемых эксципиентов в грануляторе с большим сдвиговым усилием,
(ii.) добавления раствора, содержащего по меньшей мере одно связующее к смеси, полученной на стадии (a)(i.), смачивания и замешивания смеси в грануляторе с большим сдвиговым усилием, где связующее добавляют к смеси, полученной на стадии (a)(i.), в общем количестве, равном примерно от 1% до 20 мас.% в пересчете на общую массу таблетки
(iii.) влажного просеивания указанной смеси, полученной на стадии (a)(ii.), в мельнице с ситами,
(iv.) сушки к смеси, полученной на стадии (a)(iii), в сушилке с псевдоожиженным слоем,
(v.) просеивание смеси, полученной на стадии (a)(iv.), в мельнице с ситами с качающимся колосником,
(vi.) добавления просеянных эксципиентов, содержащих по меньшей мере один разрыхлитель в количестве, равном примерно от 1% до 5 мас.% в пересчете на общую массу таблетки, по меньшей мере одного смазывающего вещества в количестве, равном примерно от 1% до 7 мас.% в пересчете на общую массу таблетки, и необязательно любых дополнительных фармацевтически приемлемых эксципиентов к смеси, полученной на стадии (v.), и смешивания в диффузном смесителе (переворачиванием), и
(b) таблетирование смеси, полученной на стадии (vi.), путем прессования с использованием обычного таблетирующего пресса, предпочтительно ротационного таблетирующего пресса,
при условии, что указанный фармацевтически приемлемый эксципиент, использующийся на стадиях (a)(i.), (a)(vi.), не представляет собой трикальцийфосфат, безводный дикальцийортофосфат, дигидрат дикальцийортофосфата или сукралозу.
В одном варианте осуществления стадия (a)(i.) включает один или более дополнительных фармацевтически приемлемых эксципиентов, которые представляют собой разбавитель, содержащий маннит, который содержится в общем количестве, равном примерно от 15% до 20 мас.% в пересчете на общую массу таблетки, и микрокристаллическую целлюлозу, которая содержится в количестве, равном примерно от 15% до 20 мас.% в пересчете на общую массу таблетки, и стадия (a)(i.) включает один или более дополнительных фармацевтически приемлемых эксципиентов, которые представляют собой разбавитель - микрокристаллическую целлюлозу, которая содержится в количестве, равном примерно от 10% до 15 мас.% в пересчете на общую массу таблетки.
В одном объекте настоящее изобретение относится к диспергирующейся таблетке, приготовленной или полученной способом, описанным выше. Предпочтительно, если диспергирующаяся таблетка не содержит покрытия.
Физическую и химическую стабильность можно исследовать обычным образом, например, диспергирующиеся таблетки можно исследовать путем определения растворения, истираемости, времени распада, изучения продуктов разложения соединения I, внешнего вида и/или с помощью микроскопии, например, после хранения при комнатной температуре, т. е. 25°C, и/или хранения при 40°C.
Диспергирующиеся таблетки могут обладать различной формой и могут обладать круглой, овальной, вытянутой, цилиндрической или любой другой подходящей формой. В предпочтительном варианте осуществления настоящего изобретения диспергирующиеся таблетки, полученные способом, описанным выше, обладают круглой формой. Края диспергирующихся таблеток могут быть скошенными или скругленными. Более предпочтительно, если диспергирующиеся таблетки обладают круглой формой или овальной формой со скошенными краями. Диспергирующиеся таблетки могут обладать насечками, обладать рельефным рисунком или гравировкой.
Диспергирующиеся таблетки, предлагаемые в настоящем изобретении, предпочтительно обладают круглой формой или овальной формой, обладать рельефным рисунком и со скошенными краями.
Диспергирующиеся таблетки, предлагаемые в настоящем изобретении, могут быть окрашены или маркированы для придания им индивидуального внешнего вида, чтобы сделать их легко распознаваемыми. Применение красителей может служить для улучшения внешнего вида, а также идентификации диспергирующихся таблеток. Красители, подходящие для применения для фармацевтические композиции или дозированных форм, обычно включают каротиноиды, оксиды железа или хлорофилл. Диспергирующиеся таблетки, предлагаемые в настоящем изобретении, могут быть маркированы с использованием штампованного кода.
Твердость или стойкость по отношению к истиранию таблеток, предлагаемых в настоящем изобретении, можно определить с помощью стандартных тестов. Можно использовать устройство, такое как устройство для исследования таблеток Kraemer® 3S. В этом тесте определяют стойкость таблеток по отношению к истиранию, измеряемую с помощью силы, необходимой для их разрушения путем истирания.
Твердость таблеток, предлагаемых в настоящем изобретении, меняется в зависимости от массы и диаметра таблеток и прессующей силы. Для таблетки, содержащей примерно 50 мг соединения I в качестве активного ингредиента, обладающей диаметром, равным примерно 7 мм, твердость предпочтительно равна примерно от 25 Н до 75 Н, предпочтительно примерно от 35 Н до 65 Н, наиболее предпочтительно примерно 50 Н и может быть обеспечена путем приложения прессующей силы, равной примерно от 5 до 11 кН. Для таблетки, содержащей примерно 200 мг соединения I в качестве активного ингредиента, обладающей диаметром, равным примерно 6,3 мм, твердость предпочтительно равна от примерно от 120 Н до 180 Н, предпочтительно от примерно от 130 Н до 170 Н, наиболее предпочтительно примерно 150 Н и может быть обеспечена путем приложения прессующей силы, равной примерно от 12 до 20 кН. Для таблеток, обладающих другими массами и диаметрами, предпочтительные значения твердости являются другими.
Таким образом, благоприятные характеристики диспергирующихся таблеток, предлагаемых в настоящем изобретении, можно продемонстрировать с помощью значений твердости и времен распада указанных таблеток. Соответственно, в предпочтительном варианте осуществления настоящее изобретение относится к диспергирующейся таблетке, определенной выше, содержащей 50 мг соединения I или его фармацевтически приемлемой соли после прессования с использованием силы, равной от 5 до 11 кН, с помощью пуансона диаметром 7 мм и стандартных круглых матриц, обладает твердостью, равной от 25 до 75 Н, и временем распада, равным 3 мин или менее. Соответственно, в предпочтительном варианте осуществления настоящее изобретение относится к диспергирующейся таблетке, определенной выше, содержащей 200 мг соединения I или его фармацевтически приемлемой соли после прессования с использованием силы, равной от 12 до 20 кН и пуансона 16,0×6,3 мм и стандартных овалоидных матриц обладает твердостью, равной от 120 до 180 Н, и временем распада, равным 3 мин или менее.
Приведенные выше положения настоящего изобретения определяют диспергирующуюся таблетку с помощью характеристик конкретной таблетки, полученной из описанной фармацевтической композиции. Однако ясно, что настоящее изобретение никоим образом не ограничивается таблетками, обладающими такой массой, диаметром или твердостью, только способом получения, включающим применение такой прессующей силы. Как отмечено выше, определение приведено только для прояснения того, что благоприятные собственные характеристики полученной таблетки включают небольшое время распада в сочетании с высокой степенью твердости.
Диспергирующиеся таблетки, предлагаемые в настоящем изобретении, применимы для лечения или предупреждения пролиферативного нарушения, предпочтительно рака.
Диспергирующиеся таблетки, предлагаемые в настоящем изобретении, являются особенно подходящими для лечения или предупреждения раковых заболеваний, включая, например, саркому, раковые заболевания легких, бронхов, предстательной железы, молочной железы (включая спорадические раковые заболевания молочной железы и страдающих от болезни Коудена), поджелудочной железы, желудочно-кишечного тракта, толстой кишки, прямой кишки, карциному толстой кишки, колоректальную аденому, щитовидной железы, печени, внутрипеченочного желчного протока, печеночно-клеточный, надпочечника, желудка, гастральный, глиому, глиобластому, эндометрия, меланому, почки, почечной лоханки, мочевого пузыря, тела матки, шейки матки, влагалища, яичников, множественную миелому, пищевода, лейкоз, острый миелолейкоз, хронический миелолейкоз, лимфолейкоз, миелолейкоз, головного мозга, полости рта и глотки, гортани, тонкого кишечника, неходжкинскую лимфому, меланому, ворсинчатую аденому толстой кишки, неоплазию, неоплазию эпителиального характера, лимфомы, карциному молочной железы, базально-клеточную карциному, старческий кератоз, головы и шеи, истинную полицитемию, эссенциальную тромбоцитемию, миелофиброз с миелоидной метаплазией и болезнь Вальденстрема. Предпочтительно, если диспергирующиеся таблетки, предлагаемые в настоящем изобретении, применяют для лечения пролиферативного заболевания.
Пролиферативные заболевания опосредуемые альфа-субъединицей PI3K, могут включать такие, которые характеризуются сверхэкспрессией или амплификацией PI3K альфа, соматической мутацией PIK3CA или мутациями зародышевой линии или соматической мутацией PTEN или мутациями и транслокацией p85α, которые обеспечивают повышающую регуляцию комплекса p85-p110. В предпочтительном варианте осуществления раком является опухоль и/или раковая опухоль, опосредуемая альфа-изоформой PI3K.
В одном варианте осуществления пролиферативным заболеванием является рак, выбранный из группы, включающей рак легких, бронхов, предстательной железы, молочной железы (включая спорадические раковые заболевания молочной железы и страдающих от болезни Коудена), толстой кишки, прямой кишки, карциному толстой кишки, колоректальную аденому, поджелудочной железы, желудочно-кишечного тракта, печеночно-клеточный, желудка, гастральный, яичников, плоскоклеточную карциному и головы и шеи.
В предпочтительном варианте осуществления пролиферативным заболеванием является рак головы и шеи.
В предпочтительном варианте осуществления пролиферативным заболеванием является рак молочной железы.
В предпочтительном варианте осуществления диспергирующиеся таблетки, предлагаемые в настоящем изобретении, применимы для лечения пролиферативного нарушения, предпочтительно рака.
В другом варианте осуществления настоящее изобретение относится к применению диспергирующейся таблетки, предлагаемой в настоящем изобретении, для лечения или предупреждения пролиферативного заболевания, предпочтительно рака. Каждый из вариантов осуществления включен в настоящее изобретение в качестве ссылки.
В другом варианте осуществления настоящее изобретение относится к способу лечения или предупреждения пролиферативного заболевания, включающему введение нуждающемуся в нем пациенту одной или большего количества диспергирующихся таблеток, предлагаемых в настоящем изобретении, которые вместе включают терапевтически эффективное количество соединения I или его фармацевтически приемлемой соли. Каждый из вариантов осуществления, приведенных выше, включен в настоящее изобретение в качестве ссылки. Предпочтительно, если субъектом или пациентом, нуждающимся в указанных диспергирующихся таблетках, предлагаемых в настоящем изобретении, являются субъекты или пациенты, страдающие от пролиферативного нарушения, предпочтительно рака.
Активность и характеристики диспергирующихся таблеток, предлагаемых в настоящем изобретении, можно продемонстрировать с помощью стандартных клинических исследований и/или исследований на животных.
В зависимости от возраста, состояния пациента, пути введения и типа заболевания соединение I вводят перорально в эффективной суточной дозе, равной примерно от 1 до 6,5 мг/кг для взрослых или детей. Взрослому пациенту с массой тела, равной 70 кг, соединение I вводят перорально в суточной дозе, равной примерно от 70 мг до 455 мг. Следует понимать, что конкретная доза для любого конкретного пациента зависит от множества факторов, включая возраст, массу тела, общее состояние здоровья, комбинации лекарственного средства с одним или большим количеством активных лекарственных средств, типа и тяжести заболевания. С использованием данных настоящего изобретения специалист в данной области техники должен приобрести опыт и знания для выбора надлежащей дозы для лечения конкретного пациента.
В одном объекте настоящее изобретение также относится к упаковке лекарственного средства, содержащей диспергирующиеся таблетки, предлагаемые в настоящем изобретении, и печатные инструкции, относящиеся к одной или большему количеству диспергирующихся таблеток соединения I или его фармацевтически приемлемой соли для перорального введения.
Приведенный ниже пример иллюстрирует изобретение, описанное выше, но никоим образом не ограничивает объем настоящего изобретения. Другие модели исследования, сами по себе известные специалисту в данной области техники, также могут установить полезные эффекты заявленного изобретения.
Пример 1: Состав диспергирующейся таблетки (50 мг диспергирующихся таблеток, обладающих временем распада, равным менее 3 мин)
|
Таблетку готовят путем формования гранулята, содержащего внутреннюю фазу и наружную фазу, с помощью (i.) влажного гранулирования внутренней фазы, содержащей 50,00 мг соединения I, 23,00 мг микрокристаллической целлюлозы (Avicel PH101), 23,00 мг маннита, 3,0 мг натриевой соли гликолята крахмала и 3,00 мг обладающей низкой степенью замещения гидроксипропилцеллюлозы, и последующего (ii) формования наружной фазы путем смешивания полученной смеси с просеянными 4,06 мг натриевой соли гликолята крахмала и 15,86 мг микрокристаллической целлюлозы (Avicel PH102) и последующего смешивания полученной смеси с просеянными 5,08 мг стеарилфумарата натрия.
Таблетку получают таблетированием смеси, полученной выше на стадии (ii). Композицию, полученную выше на стадии (ii), прессуют с помощью ротационного таблетирующего пресса Fette® 102i с использованием прессующей силы, равной примерно от 5 до 11 кН (предпочтительно примерно 8 кН) с помощью пуансона диаметром 7 мм и стандартных круглых матриц.
Полученная таблетка обладает следующими характеристиками:
|
Пример 2: Состав диспергирующейся таблетки (200 мг диспергирующихся таблеток, обладающих временем распада, равным менее 3 мин)
|
Таблетку готовят путем формования гранулята, содержащего внутреннюю фазу и наружную фазу, с помощью (i.) влажного гранулирования внутренней фазы, содержащей 200,00 мг соединения I, 92,00 мг микрокристаллической целлюлозы (Avicel PH101), 92,00 мг маннита, 12,00 мг натриевой соли гликолята крахмала и 12,00 мг обладающей низкой степенью замещения гидроксипропилцеллюлозы, и последующего (ii) формования наружной фазы путем смешивания полученной смеси с просеянными 16,24 мг натриевой соли гликолята крахмала и 63,44 мг микрокристаллической целлюлозы (Avicel PH102) и последующего смешивания полученной смеси с просеянными 20,32 мг стеарилфумарата натрия.
Таблетку получают таблетированием смеси, полученной выше на стадии (ii). Композицию, полученную выше на стадии (ii), прессуют с помощью ротационного таблетирующего пресса Fette® 102i с использованием прессующей силы, равной примерно от 12 до 20 кН (предпочтительно примерно 16 кН) с пуансоном 16,0×6,3 мм стандартными и овалоидными матрицами.
Полученная таблетка обладает следующими характеристиками:
|
Пример 3: Примеры времен распада
В приведенной ниже таблице представлены примеры времен распада в минутах для таблеток, приготовленных в соответствии с примером 1 или примером 2:
|
В описанных выше тестах указанные таблетки, приготовленные в соответствии с примером 1 или примером 2, исследованы для определения того, распадаются ли таблетки менее, чем за 5 мин при помещении в жидкую среду. Время распада таблетки определяют по методике, описанной в публикации European Pharmacopoeia, Eighth edition, Supplement 8.2, page 285-287, Section 2.9.1 (January 2014), которая во всей своей полноте включена в настоящее изобретение в качестве ссылки. В этих экспериментах диски не использовали.
Полный распад определяется, как состояние, в котором любой остаток таблетки, исключая фрагменты нерастворимого покрытия, оставшийся на сите аппарата для исследования, является мягкой массой, не содержащей прощупываемую пленку ядра.
Для этих исследований используют систему типа багажной сетки. Система типа багажной сетки состоит из 6 прозрачных трубок с открытыми концами (каждая обладает длиной, равной примерно 77,5+2,5 мм, и диаметром, равным примерно 21,85+1,15 мм, и стенкой, толщиной равной примерно 1,9+0,9 мм) удерживаемых 2 пластинами, содержащими 6 отверстий, находящихся на одинаковых расстояниях от центра и на одинаковых расстояниях друг от друга. К нижней части нижней пластины прикреплено проволочное сито из нержавеющей стали с плоским квадратным переплетением с отверстиями размером 2,0+0,2 из проволоки диаметром 0,615+0,045 мм. Части аппарате собраны и жестко скреплены с помощью 3 болтов, проходящих через 2 пластины. Систему типа багажной сетки подвешивают на поднимающем и опускающем устройстве на его оси. Следует понимать, что конструкцию системы типа багажной сетки можно менять при условии, что сохраняются параметры стеклянных трубок и отверстий сетки.
При исследовании распада одну таблетку помещают в каждую из 6 трубок системы типа багажной сетки. Аппарат работает с использованием очищенной воды при комнатной температуре (примерно от 15 до 25°C) в качестве иммерсионной жидкости. Для таблеток определяют степень и скорость распада путем поднимания сетки из жидкости. Результаты, приведенные выше, характеризуют максимальное время распада одной таблетки из шести таблеток в исследовании распада.
Мутантные антигены gas57 и антитела против gas57
[(1н-индол-5-ил)-гетероарилокси]-1-(азабицикло[3.3.1]нонаны, как холинергические лиганды n-achr, предназначенные для лечения психотических и нейродегенеративных нарушений
Лечение туберозного склероза
Органические соединения
Способ получения фармацевтической композиции
Предназначенная для перорального применения фармацевтическая композиция
Менингококковые полипептиды fhbp
Стабилизированные полипептиды инсулиноподобного фактора роста
Органические соединения
Курс лечения с использованием агониста рецептора s1p

