Результат интеллектуальной деятельности: ТРИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ГИРАЗЫ
Вид РИД
Изобретение
Область, к которой относится изобретение
[0001] Настоящее раскрытие относится к области медицинской химии и, в частности, к соединениям и их фармацевтическим композициям, которые являются полезными в качестве антибиотиков. В частности, трициклические соединения-ингибиторы гиразы ингибируют ферменты ДНК Гиразу B (GyrB) и Топоизомеразу IV (ParE). Также предусматриваются соответствующие способы лечения бактериальных инфекций и способы получения соединений с использованием новых промежуточных соединений.
Описание предшествующего уровня техники
[0002] Бактериальные инфекции являются постоянной медицинской проблемой, поскольку антибактериальные лекарственные средства в конечном счете вызывают резистентность в бактериях, против которых их используют. Соответственно, существует потребность в новых лекарственных средствах с эффективностью против патогенных бактерий для применения в лечении и профилактике бактериальных инфекций.
[0003] Одним объектом, на который направлена разработка антибактериальных лекарственных средств, являются ферменты ДНК Гираза B (GyrB) и Топоизомераза IV (ParE), необходимые для репликации ДНК. Ингибиторы гиразы были раскрыты в RE40245, который включен в настоящую заявку посредством ссылки во всей его полноте.
[0004] Некоторые ингибиторы гиразы имеют тенденцию ингибировать ген специфических калиевых каналов сердца человека (hERG), который кодирует белки ионных каналов, называемые hERG каналами, и могут привести в качестве побочного эффекта к повышенному риску потенциально фатальных сердечных аритмий из-за нарушений реполяризации потенциала действия сердечной мышцы.
[0005] GyrB ферментный карман подробно охарактеризован в Wigley, D.B. et al., Nature, 351(6328), 624-629, 1991. См. также Tsai F.T., et al., The high-resolution crystal structure of a 24-kDa gyrase B fragment from E. coli complexed with one of the most potent coumarin inhibitors, clorobiocin, Proteins. 1997 May; 28(1):41-52.
[0006] ParE ферментный карман подробно охарактеризован в Bellon, S., et al. Crystal structures of Escherichia coli topoisomerase IV ParE subunit (24 and 43 kilodaltons): a single residue dictates differences in novobiocin potency against topoisomerase IV and DNA gyrase, Antimicrob. Agents Chemother. 48: 1856-1864 (2004). Эти ссылочные документы включены в настоящую заявку посредством ссылки во всей их полноте.
[0007] В отличие от этого, патентные публикации, указывающие в качестве авторов изобретения Hurley et al., направлены на ингибиторы протеинкиназы, которые являются полезными для протеинкиназа-опосредованных заболеваний и состояний, таких как рак. См., например, US 2008/0051414, US 2009/0143399 и US 2009/0099165.
[0008] PCT/US2012/029104, Предварительная патентная заявка США №61/700159 и Заявка PCT, которая испрашивает приоритет по предварительной патентной заявке США №61/700159 (WO 2014/043272), права пользования на которые переданы или переуступлены тем же правопреемникам, как в настоящей заявке, раскрывают трициклические ингибиторы гиразы и включены в настоящую заявку посредством ссылки во всей их полноте.
Сущность изобретения
[0009] Трициклические соединения-ингибиторы гиразы формулы I ингибируют ферменты ДНК Гиразу B (GyrB) и Топоизомеразу IV (ParE).
[0010] Также предусматриваются способы применения соединения для лечения бактериальных инфекций и способы получения соединений.
[0011] Эти и другие соответствующие аспекты описаны более подробно ниже.
Подробное Описание изобретения
[0012] Соединения, раскрытые в PCT/US2012/029104, предварительной патентной заявке США №61/700159 и заявке PCT, которая испрашивает приоритет по указанной заявке (WO 2014/043272), сильно ингибируют GyrB и ParE рецепторы и обладают сильной грамотрицательной активностью. В некоторых аспектах, любые виды трициклических соединений, являющихся ингибиторами гиразы, ранее раскрытые в PCT/US2012/029104 или в предварительной патентной заявке США №61/700159 или в заявке PCT, которая испрашивает приоритет по предварительной патентной заявке США №61/700159 (WO 2014/043272), в настоящем изобретении исключаются. Один аспект грамотрицательной антибактериальной активности представляет собой способность соединения проникать через клеточную стенку грамотрицательных бактерий и затем избегать быстрого оттока из клетки. В PCT/US2012/029104, US 61/700159 и заявке PCT, которая испрашивает приоритет по этой заявке (WO 2014/043272), наиболее сильнодействующие и активные соединения содержат основный амин, такой как заряженный диамин в R4 положении формулы I, и, таким образом, считаются особенно полезными для грамотрицательной активности. Эти содержащие R4 диамин соединения содержат основный амин, который типично заряжен в физиологических pH условиях. Эта заряженная молекула является малой и полярной, и, не связывая это с теорией, считалось, что она способна проникать в клетки грамотрицательных бактерий путем проникновения через порины. Эта молекула также существует в равновесии, с небольшой (2-5%) частью нейтрального вещества, которое, не связывая это с теорией, как считалось, проникает через внутреннюю мембрану. Попадая в цитозоль, соединение существует в основном как полярная заряженная молекула, и менее вероятно, что оно будет подвергаться воздействию эффлюксных насосов, в отличие от нейтральных молекул, таких как соединения с не-оснóвным спиртом в R4 положении. Например, виды соединений, раскрытые в заявке PCT, которая испрашивает приоритет по предварительной патентной заявке США №61/700159 (WO 2014/043272), такие как

 или
или 
[0013] содержат не-основный спирт в R4 положении, но имеют относительно низкую грамотрицательную активность по сравнению с наиболее активными оснóвными аминовыми соединениями. Ec (E. coli) MIC значения для трех соединений, указанных выше, составляют >64 мкг/мл; 8 мкг/мл; и >32 мкг/мл, соответственно. Не связывая это с теорией, считается, что a) серный L линкер и b) 10-членный гетероарил, содержащий два конденсированных кольца R2 группы, могут затруднять проникновение соединения через порины в клеточную стенку грамотрицательных бактерий.
[0014] Было бы желательно, чтобы сильнодействующие соединения, обладающие грамотрицательной активностью, также избегали серьезных побочных эффектов, являющихся результатом нецелевого связывания. Сердечно-сосудистые побочные эффекты являются основной причиной для отказа от лекарственного средства. Ингибирование hERG (гена специфических калиевых каналов сердца человека) используют в качестве предиктивного in vitro ферментативного скрининга для исключения соединений с сердечно-сосудистыми побочными эффектами, особенно пролонгированием QTc интервала (Valentin, J. British Journal of Pharmacology 2010, 159, 5-11). Было бы желательным в данной области, чтобы соединения не имели нежелательных побочных эффектов ингибирования hERG канала. Заявка PCT, которая испрашивает приоритет по предварительной патентной заявке США №61/700159 (WO 2014/043272), раскрывает, что, как было обнаружено, специфические соединения, содержащие диамин в R4 положении, являются неожиданно и значительно более селективными в hERG анализе, чем многие другие испытанные соединения, содержащие диамин в R4 положении, с сохранением при этом грамотрицательной активности. Эти виды соединений, а именно,  и
и  содержат CF группы в обоих Y и Z положениях и содержат гидроксиэтильный заместитель на R2. Для этих видов с положительными hERG результатами, однако, было обнаружено, что замена оснóвного амина нейтральным спиртом в R4 положении в довольно схожих соединениях (CF в обоих Y и Z положениях и гидроксиэтильный заместитель на R2) приводила к аналогично положительным hERG результатам, но грам-отрицательная активность снижалась. Также было бы желательным, чтобы соединения могли иметь положительные hERG значения и сохранять при этом грамотрицательную активность.
содержат CF группы в обоих Y и Z положениях и содержат гидроксиэтильный заместитель на R2. Для этих видов с положительными hERG результатами, однако, было обнаружено, что замена оснóвного амина нейтральным спиртом в R4 положении в довольно схожих соединениях (CF в обоих Y и Z положениях и гидроксиэтильный заместитель на R2) приводила к аналогично положительным hERG результатам, но грам-отрицательная активность снижалась. Также было бы желательным, чтобы соединения могли иметь положительные hERG значения и сохранять при этом грамотрицательную активность.
[0015] Кроме того, в данной области также могли бы быть полезными соединения, которые, помимо того, что они обладают грамотрицательной активностью и отсутствием описанного выше нежелательного побочного эффекта ингибирования hERG канала, можно было бы вводить перорально. Однако многие антибиотики ограничены внутривенным введением из-за отсутствия способности прохождения через слизистую оболочку тонкого кишечника, что способствует пероральной абсорбции. Многие факторы влияют на пероральную абсорбцию и, следовательно, на пероральную биодоступность. Некоторые факторы относятся к молекулярным свойствам, таким как растворимость, скорость растворения и всасываемость средства, которые хорошо известны. Однако другие молекулярные свойства, такие как молекулярная гибкость, количество вращаемых связей или площадь полярной поверхности, менее изучены. Не связывая это с теорией, считается, что комбинация этих молекулярных свойств делает соединение перорально биодоступным. Трудно предсказать, какая комбинация молекулярных свойств приведет к соединению, обладающему пероральной биодоступностью, поэтому эмпирическое испытание является полезным для определения пероральной биодоступности.
[0016] Для достижения этих целей использовали химическую вариативность по различным R группам на ABC кольце. Доступные для растворителя R группы, т.е. R2 и R4, обеспечивали наилучшую возможность для химической вариативности, оставляя при этом остальные группы расположенными во внутренней части белка, при нахождении в связанной конформации, для взаимодействия с карманами активных центров GyrB и PyrE.
[0017] Совершенно неожиданным оказалось, что, в некоторых аспектах, соединения, раскрытые в настоящей заявке, могли одновременно: 1) сильно ингибировать GyrB и ParE и сохранять грамотрицательную активность; и 2) избегать нежелательного побочного эффекта ингибирования hERG канала; и кроме того, в некоторых аспектах, обеспечивать отличную пероральную биодоступность.
[0018] Пероральную биодоступность можно оценить различными способами, такими как измерение клеточной проницаемости в модели Caco-2 и измерение фармакокинетических свойств путем расчета отношения AUC после перорального введения к AUC после внутривенного введения. Оба способа используют для оценки соединений в настоящем изобретении, как проиллюстрировано в Примерах 8-9. В некоторых аспектах, соединения, представленные в настоящей заявке, показывают самую высокую мембранную (Caco-2 клеточного монослоя) проницаемость среди всех испытанных соединений. Caco-2 клеточная линия представляет собой стабильную клеточную линию из клеток гетерогенной эпителиальной колоректальной аденокарциномы человека, которые при культивировании в специфических условиях становятся дифференцированными и поляризованными таким образом, что их фенотип, морфологически и функционально, напоминает энтероциты, выстилающие тонкий кишечник. Таким образом, Caco-2 монослой широко используют в фармацевтической промышленности в качестве in vitro модели слизистой оболочки тонкого кишечника человека для прогнозирования абсорбции перорально-вводимых лекарственных средств.
[0019] Некоторые аспекты соединений формулы I подробно описаны ниже. В Формуле I выше, аспекты переменных на ABC кольце, т.е. L, R2, R4, R8, X, Y и Z, подробно обсуждаются в PCT/US2012/029104 и предварительной патентной заявке США №61/700159 или заявке PCT, которая испрашивает приоритет по предварительной патентной заявке США №61/700159 (WO 2014/043272), и соответствующие аспекты, такие как взаимодействия соединений с GryB и ParE, включены в настоящую заявку посредством ссылки.
[0020] В конктексте настоящего изобретения, термин “арил” относится к необязательно-замещенной моноциклической и конденсированной бициклической углеводородной группе. Любая моноциклическая или конденсированная кольцевая бициклическая система, которая обладает характеристиками ароматичности, что касается электронного распределения в кольцевой системе, включена в это определение. Типично, кольцевые системы содержат 5-12 атомов в качестве кольцевых членов. “Гетероарил” относится к необязательно замещенным ароматическим моноциклическим и конденсированным бициклическим гетероциклам, содержащим один или несколько гетероатомов, выбранных из N, O и S. Включение гетероатома делает возможным включение 5-членных колец, а также 6-членных колец.
[0021] В конктексте настоящего изобретения, термин “алкил” включает линейные и разветвленные и циклические одновалентные заместители. Примеры включают метил, этил, пропил, изопропил и циклопропил. Где это указано, алкильные заместители могут содержать 1-1°C (от 1 до 10 атомов углерода), например 1-3C, 1-6C, или 1-8C.
[0022] В конктексте настоящего изобретения, “углеводородный остаток” относится к остатку, который содержит только углерод и водород. Углеводородный остаток может быть насыщенным или ненасыщенным, алифатическим или ароматическим, с линейной цепью, с разветвленной цепью или циклическим, включая одно кольцо, конденсированную кольцевую систему, связанную мостиковой связью кольцевую систему или спиро кольцевую систему, или комбинацию углеводородных групп. Углеводородный остаток, однако, когда это специально указано, может содержать гетероатомы в дополнение к углеродным и водородным членам остатка как заместителя. Таким образом, когда он специально указывается как содержащий такие гетероатомы, углеводородный остаток также может содержать гетероатомы, такие как O, S или N в “скелете” углеводородного остатка. Углеводородная группа может включать комбинацию углеводород-содержащих групп, например, гетероциклическую группу, связанную с гетероалкилом, содержащим комбинацию линейной алкильной и циклоалкильной группы.
[0023] В конктексте настоящего изобретения, “циклический остаток” относится к циклическому углеводородному остатку, который содержит только углерод и водород. Циклический остаток, однако, когда это специально указано, может содержать гетероатомы в дополнение к углеродным и водородным членам остатка как заместителя. Таким образом, когда он специально указывается как содержащий такие гетероатомы, углеводородный остаток также может содержать гетероатомы, такие как O, S или N в “скелете” циклического остатка. В некоторых аспектах, когда это специально указано, циклический остаток представляет собой циклоалифатический или циклогетероалифатический остаток. Насыщенный циклоалифатический или насыщенный циклогетероалифатический остаток относится к кольцу, содержащему насыщенные связи между кольцевыми членами.
[0024] В конктексте настоящего изобретения, “ненасыщенный циклический остаток” относится к по меньшей мере частично ненасыщенному или ароматическому циклическому углеводородному остатку, который содержит только углерод и водород. Ненасыщенный циклический остаток, однако, когда это специально указано, может содержать гетероатомы в дополнение к углеродным и водородным членам остатка как заместителя. Таким образом, когда он специально указывается как содержащий такие гетероатомы, ненасыщенный гетероциклический остаток также может содержать гетероатомы, такие как O, S или N в “скелете” ненасыщенного циклического остатка.
[0025] Термин “члены” или “членный”, в контексте гетероциклических и гетероарильных групп, относится ко всем атомам, углероду и гетероатомам N, O и/или S, которые образуют кольцо. Таким образом, примером 6-членного насыщенного циклогетероалифатического кольца является пиперидин, и примером 6-членного гетероарильного кольца является пиридин. Конечно, термин “члены” или “членный” также относится к количеству атомов углерода в не содержащих гетероаты или не содержащих циклические части группах.
[0026] Связанная конформация относится к конформации (т.е. пространственному расположению атомов), которую могло бы принимать трициклическое соединение-ингибиторы гиразы в случае связывания с карманом активного центра GyrB/ParE во внутренней части фермента. При использовании, соединение может взаимодействовать с карманом активного центра и ингибировать активность АТФазы. Когда соединение связывается с карманом активного центра GyrB/ParE, некоторые заместители взаимодействуют с некоторыми аминокислотами, и, таким образом, способность таких заместителей свободно вращаться вокруг связи ограничивается. Таким образом, можно произвести более полезные измерения для определения расстояний, имеющих значение для определения размеров подходящих заместителей. Если указано, измерения основаны на относительных положениях заместителей на соединении, в то время как оно гипотетически связано с карманом активного центра сGyrB/ParE. Ссылки на связанную конформацию, относящиеся к соединению, не следует интерпретировать как в буквальном смысле охватывающие карман активного центра GyrB/ParE в комбинации с соединением. Связанную конформацию характеризуют через измерения, полученные от трехмерных структур из данных рентгеноструктурного анализа на ингибиторе, образующем комплекс с белковым конструктом, который типично охватывает 24 или 46 кДа АТФ-связывающих домена одного или нескольких репрезентативных бактериальных GyrB или ParE ортологов. Учитывая высокую степень идентичности последовательностей между GyrB и ParE ферментами в наиболее патогенных организмах, представляющих интерес, структурная информация, полученная от ортолога белка из любого клинически релевантного патогена, должна быть достаточной для описания связанной конформации. Вкратце, кристаллографические структуры получают с использованием следующих способов: Белки, представляющие интерес (например, E. faecalis GyrB, E. coli GyrB, F. tularensis ParE или E. coli ParE), получают в стандартной E. coli системе экспрессии. Открытые рамки считывания клонируют в экспрессирующую плазмиду (например, pET28a) и экспрессируют в подходящем E. coli экспрессирующем штамме (например, BL21 (DE3)). Для кристаллографии 24 кДа и 46 кДа АТФ связывающие домены клонируют с C(His)6 tag для облегчения очистки методом аффинной хроматографии на иммобилизованных ионах металла. Эта стадия грубой хроматографической очистки типично дает больше чем 80% чистого белка. Стадии тонкой очистки, включая ионообменную и эксклюзионную хроматографию, осуществляют, если это необходимо, до достижения удовлетворительной (>95%) чистоты. После получения очищенного белка получают комплексы GyrB или ParE и молекулы ингибитора, представляющего интерес, путем смешивания стехиометрического избытка ингибитора, представляющего интерес, с рекомбинантным белком-мишенью в растворе и кристаллизации комплекса с использованием общепринятых способов кристаллизации (типично диффузии паров, как описано Drenth J. (1999) в Principles of protein x-ray crystallography. 2nd ed. Springer, New York). После кристаллизации получают данные рентгеноструктурного анализа на монокристаллах комплексов белок-ингибитор с использованием монохроматического рентгеновского излучения, генерируемого вращающимся анодом или источником синхротронного излучения. Обработку данных рентгеноструктурного анализа, анализ и последующее разрешение и уточнение структуры осуществляют с использованием общепринятых способов расчета (см. обзор Drenth J. (1999) в Principles of protein x-ray crystallography. 2nd ed. Springer, New York).
[0027] Взаимодействующие заместители на соединении, которые взаимодействуют с карманом активного центра GyrB/ParE, включают такие заместители, которые будут расположены во внутренней части белка, когда соединение находится в связанной конформации. Взаимодействия взаимодействующих заместителей, как правило, включают гидрофобные взаимодействия (которые способствуют контакту липофильных поверхностей на ингибиторе и кармана активного центра) и электростатические взаимодействия, такие как вандерваальсовские, диполь-дипольные, кулоновские взаимодействия, или образование водородных связей между атомами на соединении и атомами в кармане активного центра GyrB/ParE. Например, R8, RX, RY и RZ взаимодействуют с различными частями внутренней части белка. Когда R8, RX, RY или RZ представляет собой NH2 или NHR (где R представляет собой, например, низшую алкильную группу), H атом(атомы) на азоте могут взаимодействовать с электроотрицательными атомами, такими как азот или кислород, проксимально расположенными в кармане активного центра GyrB/ParE, с которым может связываться соединение. Когда R8, RX, RY и RZ являются неполярными (например, метильная группа), взаимодействующий заместитель также может электростатически взаимодействовать с атомом во внутренней части белка через вандерваальсовские взаимодействия и десольватировать смежные липофильные поверхности в кармане активного центра с образованием благоприятных гидрофобных взаимодействий. Кроме того, в некоторых аспектах, форма и размер активного центра могут налагать ограничения на размеры заместителей в соединении, которые должны быть стерически совместимы с карманом активного центра.
[0028] Где это указано, могут быть приведены размеры заместителя, и они должны соотноситься с размерами кармана, в котором соединение будет расположено, находясь в связанной конформации. Например, может быть приведена длина заместителя на основании расстояния от атома на трициклическом каркасе до атома заместителя, который находится дальше всего от трициклического каркаса, т.е. концевого атома. Расстояние измеряют на основании расстояния от центра первого атома, такого как C на трициклическом каркасе, до центра концевого атома. Расстояние измеряют от одной точки до другой по прямой линии, независимо от того, что связи в заместителе не располагаются в одну линию, как, например, этильный или OH заместитель.
[0029] Соединения, представленные в настоящей заявке, имеют структуру формулы I

Формула I
или их фармацевтически приемлемые соли, сложные эфиры и пролекарства.
L может представлять собой O, S, NH или CH2;
[0030] X может представлять собой CH, и R8 может представлять собой NHCH3.
[0031] В некоторых аспектах, R8 может представлять собой пролекарство-содержащий заместитель, где пролекарство расщепляется с образованием соединения, которое имеет размеры, соответствующие длине от углерода трициклического каркаса до кармана активного центра, на основании кристаллографических данных, как описано выше. Эти пролекарства, такие как Формула II, среди прочего, описаны более подробно ниже.
[0032] Когда Z не объединен с R4 с образованием конденсированного кольца, каждый из Y и Z независимо может представлять собой CRY или CRZ, соответственно, или N, где каждый из RY и RZ независимо представляет собой H, галоген, такой как F, Cl, или Br, CH3, CF3, CHF2, CH2F или CN.
[0033] В некоторых аспектах Z может представлять собой C, связанный с R4. Хотя это не связано с теорией, активность и/или селективность можно повысить, поскольку конформационная энтропия уменьшается, когда Z объединяется с R4 с образованием конденсированного кольца. Когда Z объединяется с R4 с образованием конденсированного кольца, Y может представлять собой N или CRY, где RY представляет собой H, галоген, например, F, Cl или Br, CH3, CF3, CHF2, CH2F или CN.
[0034] Когда Z объединен или не объединен с R4 с образованием конденсированного кольца, CRY может представлять собой CH или CF, например, CF. Когда Z не объединен с R4 с образованием конденсированного кольца, CRZ может представлять собой CF или CH, например, CH.
[0035] В некоторых аспектах, Z может представлять собой C, связанный с R4, где соединение имеет структуру формулы VI

Формула VI
R4l может представлять собой CR10, CR10CR11, NR12, O или S. R4m может представлять собой CR10, CR10CR11 или NR12. R4n может представлять собой CR10, CR10CR11, NR12, O или S. Каждый из R10, R11 и R12 независимо представляет собой H или не-интерферирующий заместитель, где по меньшей мере один из R10, R11 и R12 представляет собой OH-содержащий заместитель. OH-содержащий заместитель может представлять собой OH или C1-C5 углеводородный остаток, замещенный по меньшей мере одним -OH, например, одним или 2 -OH, где C1-C5 углеводородный остаток может содержать 0-2 гетероатома, например, 0-1 гетероатом, выбранных из O или S. R10, R11 и R12 может представлять собой не-интерферирующий заместитель, такой как H, C1-C5 алкил, необязательно замещенный группой CN или OH. Когда R4l, R4m, R4n, R4o представляет собой N и содержит OH-содержащий заместитель, в некоторых аспектах C1-C5 алкил, такой как C2 алкил, разделяет N и -OH на OH-содержащем заместителе. Два смежных R4l, R4m и R4n вместе могут образовывать конденсированное кольцо, например, C или N в D кольце могут образовывать кольцо с двумя из R10, R11 и R12. В некоторых аспектах, R10, R11 и/или R12 не содержат N, такой как первичный или вторичный амин. В некоторых аспектах, ни один из R4l, R4m, R4n и R4o не содержит основный амин, который обсуждается в настоящей заявке.
R4o может представлять собой a) связь, где образуется 7-членное D кольцо, где R4n может представлять собой CH, CH2, S, NH, O, CHF или CF2; или b) состоящую из 1 члена связь в скелете D кольца, где образуется 8-членное D кольцо, где состоящая из 1 члена связь может представлять собой CH, CH2, S, NH, O, CHF или CF2. Как обсуждается выше, член представляет собой C, O, S или N кольцевой член, такой как CH, CH2, CHF или CF2, NH, O или S.
В некоторых аспектах, R4l представляет собой O, CH2 или CHOH. В некоторых аспектах, R4m представляет собой CHOH, CH(CH2)2OH или CHCH(OH)CH2OH. В некоторых аспектах, R4n представляет собой CH2. В некоторых аспектах, R4o представляет собой связь.
[0036] Два смежных не-интерферирующих заместителя на R4l и R4m могут образовывать одно или несколько конденсированных колец. Пунктирные линии указывают необязательную двойную связь, когда два смежных R4l, R4m и R4n представляют собой CR10, и R4o представляет собой CH или N. Таким образом, в некоторых аспектах Формула VI может иметь структуру формулы VIa:
 ,
,
Формула VIa
[0037] Пунктирные линии указывают заместители на R4l и R4m, которые образуют конденсированное кольцо E, которое, необязательно, может быть замещено не-интерферирующим заместителем.
[0038] В некоторых аспектах, часть E кольца, связывающая R4l и R4m, представляет собой C1-C10 углеводородный остаток, содержащий 0-5 O или S, присоединенный к D кольцу по R4l и R4m, который необязательно замещен OH, CN, =O, =NOH, =NOCH3, Br, F, Cl, SO3H, или NO2. CN, =NOH, =NOCH3 и NO2 не являются аминами и, таким образом, не являются оснóвными аминами.
[0039] Например, R4l и R4m вместе с двумя не-интерферирующими заместителями могут образовывать конденсированное 3, 4, 5 или 6-членное E кольцо, содержащее 0 или 1 атом O, S или N, необязательно замещенное галогеном, таким как хлор. E кольцо может быть конденсированным, спиро или связанным мостиковой связью.
[0040] Кроме того, -R4o-R4n-R4m-R4l-, т.е.  может быть выбран из следующих групп:
может быть выбран из следующих групп:
-CH2CH(CH2OH)O-, -CH2CH(CH2CH2OH)O-, -CH2CH((CHOH)CH2OH)CH(OH)-, -CH2CH(OH)CH2-, -CH2CH(OH)CH(OH)-, 
 ,
,  .
.
[0041] В некоторых аспектах, D кольцо может содержать O, S или N в своей структуре.
[0042] В некоторых аспектах, два смежных не-интерферирующих заместителя на R4m и R4n могут образовывать одно или несколько конденсированных колец. Пунктирные линии указывают необязательную двойную связь, когда два из R4l, R4m и R4n представляют собой CR10 и R4o представляет собой CH или N. Таким образом, в некоторых аспектах Формула VI может иметь структуру формулы VIb:

Формула VIb
[0043] В некоторых аспектах, часть F кольца, связывающая R4m и R4n, представляет собой C1-C15 углеводородный остаток, содержащий 0-5 O, S или N гетероатомов, присоединенный к D кольцу по R4m и R4n, необязательно замещенный OH, CN, =O, =NOH, =NOCH3, Br, F, Cl, SO3H или NO2 заместителем.
[0044] Хотя это не связано с теорией, целесообразно избегать пространственной затрудненности F кольца и избегать его вмешательства в связывание соединения с активным центром фермента. Таким образом, в некоторых аспектах, если F кольцо присутствует, R4o может иметь значение, отличное от связи. Когда R4o представляет собой состоящую из 1 члена связь, часть F кольца, связывающая R4m и R4n, в случае присутствия, может представлять собой незамещенный C1 остаток или C1, замещенный представляющим собой небольшую группу заместителем, таким как F или OH заместитель, с образованием незамещенного циклопропильного остатка (или замещенного небольшим заместителем) с R4m и R4n. Когда R4o представляет собой состоящую из 1 члена связь, часть F кольца, связывающая R4m и R4n, может представлять собой C2-C10 углеводородный остаток, содержащий 0-5 O, S или N гетероатомов, однако, положение на F кольце непосредственно примыкающее к R4n, может быть незамещенным или замещено представляющим собой небольшую группу заместителем, таким как F или OH.
[0045] Например, R4m и R4n вместе с двумя не-интерферирующими заместителями могут образовывать конденсированное 3, 4, 5 или 6-членное F кольцо, содержащее 0 или 1 атом O, S или N, необязательно замещенное галогеном, таким как фтор, или OH. F кольцо может быть конденсированным, спиро или связанным мостиковой связью.
[0046] Хотя это не связано с теорией, целесообразно избегать пространственной затрудненности D кольца и избегать его вмешательства в связывание соединения с активным центром фермента. В некоторых аспектах, D кольцо не выступает больше чем примерно на 3 Å ниже плоскости A, B и C колец в направлении нижней стороны связывающего кармана GyrB/ParE в связанной конформации; и D кольцо стерически не влияет на R2, когда соединение находится в связанной конформации.
[0047] В некоторых аспектах, соединение формулы VI может представлять собой одно из следующих соединений:

[0048] Представленные выше соединения также могут быть получены с использованием R2 заместителей, которые описаны в настоящей заявке.
[0049] Не связывая это с теорией, R2 может быть полезным для придания селективности и активности против эукариотных АТФ-связывающих белков, таких как киназы и HSP90. Таким образом, одно из преимуществ соединений включает исключение токсичности в результате нецелевого связывания, например, с киназой частично из-за селективности R2 как части соединения. Как правило, в некоторых аспектах, соединения не являются сильными ингибиторами для эукариотных киназ.
[0050] В некоторых аспектах, R2 представляет собой фенил, тиадиазолил, такой как 1,3,4-тиадиазолил, пиридинил или пиримидинил, необязательно замещенный не-интерферирующим заместителем, где 2 необязательных не-интерферирующих заместителя могут объединяться с образованием конденсированного кольца; или R2 представляет собой пролекарство-содержащий заместитель, обсуждаемый более подробно ниже.
[0051] Доступные для растворителя стороны карманов активного центра GyrB/ParE позволяют частям соединения подвергаться воздействию окружающего растворителя, в процессе использования. В некоторых аспектах, не-интерферирующие заместители могут быть водорастворимыми для обеспечения совместимости с водной растворяющей средой. Доли заместителей в направлении потенциальной растворяющей среды не являются критическими, но специалистам в данной области должно быть понятно, что пространственно незатрудненные заместители являются полезными. Таким образом, доли доступных для растворителя заместителей могут быть разными.
[0052] В отличие от “взаимодействующего заместителя” некоторые положения молекулы могут быть описаны как допускающие “не-интерферирующие заместители”. Эту терминологию используют, поскольку заместители в этих положениях, вообще говоря, имеют меньшее отношение к активности молекулы в целом. Можно использовать широкий ряд различных заместителей в этих положениях, и средний специалист сможет определить, является или нет конкретный произвольный заместитель “не-интерферирующим”.
[0053] В конктексте настоящего изобретения, “не-интерферирующий заместитель” представляет собой заместитель, который оставляет способность соединения, раскрытого в настоящей заявке, такого как соединения формулы I, ингибировать бактериальный рост по меньшей мере одного типа бактерий качественно неизмененной, например, грамотрицательных бактерий. Например, не-интерферирующий заместитель должен оставлять способность соединения обеспечивать антибактериальную эффективность на основании минимальной ингибирующей концентрации (MIC) меньше чем 32 мкг/мл или на основании ингибировани АТФазной активности ДНК Гиразы B (GyrB) или Топоизомеразы IV (ParE) меньше чем 10 нМ. Таким образом, заместитель может изменять степень ингибирования на основании MIC или АТФазной активности. Однако, до тех пор, пока соединение, раскрытое в настоящей заявке, такое как соединения формулы I, сохраняет способность ингибировать бактериальную/АТФазную активность, заместитель будет классифицироваться как “не-интерферирующий”. Существует ряд анализов для определения MIC или способности любого соединения ингибировать АТФазную активность ДНК Гиразы B (GyrB) или Топоизомеразы IV (ParE), известных в данной области, и некоторые проиллюстрированы в Примерах ниже. Например, объединенный спектрофотометрический анализ, в котором измеряют фермент-зависимое высвобождение неорганического фосфата в результате гидролиза АТФ, определяет ингибиторную активность произвольно выбранного соединения в процессе инкубации с GyrB или ParE при добавлении АТФ.
[0054] В некоторых аспектах, в дополнение к способности ингибировать бактериальную активность, не-интерферирующий заместитель оставляет способность соединения быть более селективным в hERG анализе, чем его амин-содержащий аналог. Кроме того, в некоторых аспектах, не-интерферирующий заместитель будет оставлять способность соединения к достижению большей пероральной биодоступности (измеренной на основании Caco и/или %F), чем достигается его амин-содержащим аналогом.
[0055] Характерные признаки, относящиеся к активности молекулы, четко определены. Положения, занимаемые “не-интерферирующими заместителями”, могут быть замещены традиционными группами, как это известно из предшествующего уровня техники. Нецелесообразно исследовать внешние пределы таких замещений. Релевантные характерные признаки соединений являются такими, которые подробно описаны в настоящей заявке.
[0056] Не-интерферирующие заместители на R2 кольце, которые могут быть доступны для растворителя в связанной конформации, могут включать крупные заместители, такие как пролекарства.
[0057] Не-интерферирующие заместители R2 включают C1-C10 углеводородный остаток, содержащий 0-5 O, S или N атомов в своем скелете, необязательно замещенный одним или несколькими заместителями OH, =O или NH2, где два заместителя на R2 могут образовывать конденсированное кольцо. Не-интерферирующие заместители включают COOH, NH2, OH, CH3, CH2CH3, NH2, CH2NH2, NHCH3, CH2CH2NH2, CH2CH2OH, CH(CH3)OH, CH(CH3)OCH3, COOH, CONHOCH2CH2N(CH3), CONHOCH3, CH(CH3)OCH2OCH3, CH2COOH, CH2COOCH3, 

 или
или  .
.
В некоторых аспектах R2 может представлять собой один из следующих заместителей:






 или
или  .
.
В некоторых аспектах, R2 не может представлять собой  .
.
Как обсуждается в PCT/US2012/029104, предварительной патентной заявке США №61/700159 или заявке PCT, которая испрашивает приоритет по предварительной патентной заявке США №61/700159 (WO 2014/043272), соединение доступно для растворителя в связанной конформации по R4 оси связи и в повороте на 0-90°° против часовой стрелки от R4 оси связи. Выборы пролекарств и заместителей на R4 поэтому могут варьировать. При выборе R4 заместителя, в некоторых аспектах R4 группы пространственно не влияют на R2 или Z группы в связанной конформации. Опытному специалисту должно быть понятно, что для того, чтобы избежать пространственного влияния, атомы на R4 не должны приближаться к атомам на R2 или Rz (в связанной конформации) так, чтобы межатомные расстояния между ближайшими атомами были меньше, чем суммы их ванн-дер-ваальсовых радиусов.
[0058] Кроме того, в некоторых аспектах, R4 заместитель не выступает больше чем примерно на 3 Å ниже плоскости A, B и C колец в направлении связывающего кармана GyrB/ParE в связанной конформации. “В направлении нижней стороны связывающего кармана GyrB/ParE” относится к части, не выступающей больше чем на около 3 Å ниже плоскости, в пределах примерно 5-6 связей от точки присоединения R4 к каркасу. Таким образом, части R4, которые простираются на больше чем около 5-6 связей от точки присоединения R4 к C кольцу, могут выступать больше чем на около 3 Å ниже плоскости A, B и C колец, поскольку эти части не подвергаются сдерживанию нижней частью GyrB/ParE связывающего кармана.
[0059] Расстояние определяют как расстояние равное длине перпендикуляра от плоскости, проходящей через центры атомов трициклического каркаса, до центра наиболее удаленного атома (от плоскости) на R4 заместителе в связанной конформации.
[0060] Когда R4 не объединяется с Z с образованием конденсированного кольца, R4 может представлять собой C3-C20 алифатический углеводородный остаток, содержащий 1-6 гетероатомов, выбранных из O, S и N, где один гетероатом из 1-6 гетероатомов представляет собой N в структуре углеводородного остатка, и где N присоединен к C кольцу. В некоторых аспектах, C3-C20 алифатический углеводородный остаток замещен по меньшей мере одним гидроксильным заместителем и 0-3 не-интерферирующими заместителями. В некоторых аспектах, R4 может содержать необязательные заместители на C3-C20 алифатическом углеводородном остатке, такие как OH, =O, CN, галоген, такой как F, NOCH3, CF3, OCH3, OCH2CH3. R4 не содержит основный амин. В некоторых аспектах, R4 не содержит N, такой как первичный или вторичный амин (за исключением N, связанного с C кольцом).
[0061] Заместители, что касается R4, такие как CN, =NOH, =NOCH3 и NO2, не являются аминами и, таким образом, не считаются оснóвными аминами. Кроме того, когда, например, морфолинил присоединен к C кольцу через N в морфолиниле, морфолинил не считается основным амином в этом случае. Аминовый заместитель на замещенном R4, например, замещенный морфолинил R4, однако может считаться оснóвным амином, таким как первичный (например, NH2) или вторичный амин (NH-алкил). Оснóвные амины, в некоторых аспектах, не включены в R4 положение в соединениях формулы I, содержащих R4 с гидроксил-содержащим заместителем.
[0062] R4 может представлять собой необязательно замещенный C3-C20 алифатический углеводородный остаток, например, один из следующих:





Например, R4 может представлять собой один из следующих:







 и
и  .
.
В некоторых аспектах R4 не является:
 .
.
[0063] Соединение может представлять собой одно из соединений, проиллюстрированных в Примерах.
[0064] Пролекарства также можно получить из соединений формулы I. Термин “пролекарство”, в конктексте настоящего изобретения, означает соединения, которые могут преобразовываться in vivo в активные исходные соединения, определенные в настоящей заявке.
[0065] Кроме того, пролекарства могут обладать повышенной пероральной биодоступностью по сравнению с исходным лекарственным средством. Хотя преимущества пролекарств широко признаны, часто такие преимущества пролекарств не достигаются. Таким образом, необходимы существенные усилия и исследования для разработки эффективных пролекарств.
[0066] Пролекарства, раскрытые в настоящей заявке, могут обладать меньшей антибактериальной активностью по сравнению с исходным антибактериальным средством и, соответственно, являются менее разрушительными для пищеварительного тракта. Поскольку эти пролекарства преобразуются в крови в активное антибактериальное средство, они являются системно активными. Таким образом, пролекарство может сохранять полезные эффекты лечения бактериальной инфекции, избегая при этом существенных побочных эффектов исходного антибактериального средства на желудочно-кишечный тракт.
[0067] Кроме того, пролекарство может иметь повышенную водорастворимость по сравнению с исходным антибактериальным средством, позволяя, таким образом, разработать более лучшую композицию для внутривенного введения.
[0068] В некоторых аспектах, пролекарство может иметь структуру формулы II:

Формула II
R2, R4 и R9 имеют значения, описанные выше.
[0069] Лекарственное средство формулы II может расщепляться эстеразой в крови и преобразовываться в активное антибактериальное средство, имеющее формулу I.
[0070] R8b или R8c, каждый независимо, могут представлять собой H или C1-C6 алкил, например, C1-C4 алкил, такой как метил, этил или третичный бутил. Например, R8b может представлять собой метил, R8c может представлять собой H; или R8c может представлять собой третичный бутил, и R8c может представлять собой H. В некоторых случаях, один из R8b или R8c представляет собой H или оба представляют собой H.
[0071] В некоторых аспектах, R8d представляет собой  ,
,  или его фармацевтически приемлемую соль. Фармацевтически приемлемые соли известны из уровня техники и включают катионы металлов, например, натриевую, магниевую, кальциевую или калиевую соль, а также включают аминовые катионы, такие как NH4+ или алкилированные амины.
или его фармацевтически приемлемую соль. Фармацевтически приемлемые соли известны из уровня техники и включают катионы металлов, например, натриевую, магниевую, кальциевую или калиевую соль, а также включают аминовые катионы, такие как NH4+ или алкилированные амины.
[0072] Q может представлять собой CH или N, например, CH.
[0073] R8e может представлять собой (CR8g2)n-основный амин, где n имеет значение 0-2, например 1, и где каждый R8g независимо может представлять собой H или C1-C3 алкил, например, H2, HCH2 или CH2CH2. Как правило, основный амин представляет собой солюбилизирующую группу, которая повышает растворимость лекарственного средства или пролекарства в водных средах, таких как кровь, при введении субъекту.
[0074] Основный амин может представлять собой NR8hR8i, где R8h и R8i независимо выбраны из группы, состоящей из H, необязательно замещенного C1-C4 алкила, где необязательные заместители могут представлять собой OH, NH2, или NHCH3, где R8h и R8i могут объединяться с образованием конденсированного кольца, содержащего 1-3 N или 0-3 O или S гетероатома. Например, основные амины, в контексте пролекарств, могут включать пиперазинил, морфолинил, C1-C2 алкиламин, такой как метиламин, C1-C2 диалкиламин, такой как диметиламин, или NH2.
[0075] Например, R8d может представлять собой  .
.
[0076] В некоторых аспектах, R8f представляет собой водород или C1-C6 алкил, такой как метил, этил, пропил или изопропил, или C1-C6 алкил, такой как метил, этил, пропил или изопропил, необязательно замещенный OH или NH2 заместителем. Например, R8f может представлять собой CH2OH, CHOHCH3 или (CH2)4NH2. R8f также может представлять собой метил.
[0077] Кроме того, R8e и R8f могут объединяться с образованием кольца; например, R8d может представлять собой
 или
или  .
.
[0078] R8c может представлять собой  , такой как
, такой как  или его фармацевтически приемлемую соль, как описано в настоящей заявке. R8j и R8k независимо могут представлять собой H, C1-C8 углеводородный остаток, такой как C1-C8 алкил, например, третичный бутил, или бензил.
или его фармацевтически приемлемую соль, как описано в настоящей заявке. R8j и R8k независимо могут представлять собой H, C1-C8 углеводородный остаток, такой как C1-C8 алкил, например, третичный бутил, или бензил.
[0079] Например, в некоторых аспектах, R8c может представлять собой  или
или  .
.
[0080] В некоторых аспектах, пролекарство может иметь структуру формулы II’:

[0081] Формула II’
[0082] где R группы имеют значение, определенное выше.
[0083] Как правило, более чем один пролекарственный заместитель может присутствовать на соединении.
[0084] Пролекарство также может иметь структуру формулы IV или V:
 или
или 
или фармацевтически приемлемая соль такого соединения, описанная в настоящей заявке.
[0085] Любая подходящая группа R2, описанная в настоящей заявке, включающая OH группу или замещенная группой OH, может обеспечить возможность фосфорилирования, чтобы выйти на Формулу IV. Таким образом, R2a содержит кислородный остаток, происходящий из R2, где R2 содержит OH группу, где OH из R2 замещается на кислородный остаток в R2a при фосфорилировании, и где кислородный остаток связан с P в фосфатной группе.
[0086] В числе прочих, примеры подходящих R2 групп включают следующие, которые показаны ниже, как присоединенные к O линкеру, хотя можно использовать и другие линкеры:

[0087] Любая подходящая группа R4, описанная в настоящей заявке, включающая OH группу или замещенная группой OH, может обеспечить возможность фосфорилирования, чтобы выйти на Формулу V. Таким образом, R4a содержит кислородный остаток, происходящий из не- пролекарственной группы R4. Таким образом, если не-пролекарственная группа R4 содержит OH группу, OH группа из R4 замещается на кислородный остаток в R4a при фосфорилировании, где кислородный остаток связан с P в фосфатной группе.
[0088] R2 и R4 заместители, которые раскрыты в PCT/US2012/029104, в предварительной патентной заявке США №61/700159 или в заявке PCT, которая испрашивает приоритет по предварительной патентной заявке США №61/700159, могут быть дополнительно замещены OH, как известно из уровня техники.
[0089] Пролекарство Формулы IV или Формулы V может расщепляться фосфатазой в крови и преобразовываться в активное антибактериальное средство, содержащее R2 или R4 группу, соответственно, содержащую гидрокси группу. R2a или R4a могут происходить из активного антибактериального соединения, содержащего гидрокси-замещенную R2 или R4 группу, соответственно, где при образовании пролекарства гидрокси становится точкой присоединения к фосфату.
[0090] Когда формула пролекарства, например, Формула II-V, включает R2, R4 или R8 группу, можно использовать любую подходящую R2, R4 или R8 группу, описанную в настоящей заявке.
[0091] Также предусматривается фармацевтически приемлемая соль, сложный эфир или пролекарство соединений, описанных в настоящей заявке. Специалистам в данной области должно быть понятно, что различные пролекарства, соли, гидраты, сольваты и полиморфы могут быть получены из соединений, раскрытых в настоящей заявке, и что различные изотопно-замещенные варианты (через, например, замещение водорода дейтерием, углерода изотопом 13C, азота изотопом 15N или фосфора изотопом 32P), известные как “изотопомеры”, также могут быть легко получены. Все такие производные предусматриваются как охватываемые объемом настоящего изобретения.
[0092] Многие соединения могут быть в форме соли. Специалистам в области медицинской химии должно быть понятно, что выбор соли не является критичным, и фармацевтически приемлемые соли можно получить хорошо известными способами. Такие соли подробно обсуждаются в Handbook of Pharmaceutical Salts: Properties, Selection and Use. (P. Heinrich Stahl and Camille G. Wermuth, eds.) International Union of Pure and Applied Chemistry, Wiley-VCH 2002 and L.D. Bighley, S.M. Berge, D.C. Monkhouse, in “Encyclopedia of Pharmaceutical Technology’. Eds. J. Swarbrick and J.C. Boylan, Vol. 13, Marcel Dekker, Inc., New York, Basel, Hong Kong 1995, pp. 453-499.
[0093] Соединения, раскрываемые в настоящей заявке, включают такие структуры, которые описаны в различных примерах, и их фармацевтически приемлемые соли, сложные эфиры и пролекарства. В некоторых вариантах осуществления, соединение присутствует в фармацевтической композиции или в лекарственной форме, при этом фармацевтическая композиция или лекарственная форма обеспечивает эффективное антибиотическое количество соединения для лечения или профилактики инфекции.
[0094] В другом аспекте, настоящее раскрытие относится к фармацевтической композиции, включающей одно или несколько физиологически приемлемых поверхностно-активных веществ, дополнительных носителей, разбавителей, эксципиентов, мягчителей, суспендирующих веществ, пленкообразующих веществ и образующих покрытие веществ или комбинацию таких веществ; и композицию, раскрытую в настоящей заявке. Приемлемые дополнительные носители или разбавители для терапевтического применения хорошо известны в фармацевтике и описаны, например, в Remington’s Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), который включен в настоящую заявку посредством ссылки во всей его полноте. Консерванты, стабилизаторы, красители, подсластители, ароматизаторы, отдушки и т.п. могут присутствовать в фармацевтической композиции. Например, бензоат натрия, аскорбиновая кислота и сложные эфиры п-гидроксибензойной кислоты могут быть добавлены в качестве консервантов. Кроме того, можно использовать антиоксиданты и суспендирующие вещества. В различных вариантах осуществления спирты, сложные эфиры, сульфатированные алифатические спирты и т.п. можно использовать в качестве поверхностно-активных веществ; сахарозу, глюкозу, лактозу, крахмал, микрокристаллическую целлюлозу, кристаллическую целлюлозу, маннит, светлый безводный силикат, алюминат магния, алюмометасиликат магния, синтетический алюмосиликат, карбонат кальция, кислый карбонат натрия, гидрофосфат кальция, кальций карбоксиметилцеллюлозу и т.п. можно использовать в качестве эксципиентов; стеарат магния, тальк, отвержденное масло и т.п. можно использовать в качестве мягчителей; кокосовое масло, оливковое масло, кунжутное масло, арахисовое масло, сою можно использовать в качестве суспендирующих веществ или смазывающих веществ; ацетатфталат целлюлозы в качестве производного углевода, такого как целлюлоза или сахар, или метилацетат-метакрилатный сополимер как производное поливинила можно использовать в качестве суспендирующих веществ; и пластификаторы, такие как эфирфталаты и т.п., можно использовать в качестве суспендирующих веществ.
[0095] Термин “фармацевтическая композиция” относится к смеси соединения, раскрытого в настоящей заявке, с другими химическими компонентами, такими как разбавители или дополнительные носители. Фармацевтическая композиция способствует введению соединения в организм. Существует множество способов введения фармацевтической композиции, известных из уровня техники, включая, но не ограничиваясь этим, пероральное введение, введение с использованием инъекции, аэрозоля, парентеральное и местное введение. В некоторых вариантах осуществления обеспечиваются фармацевтически приемлемые соли соединений, раскрытых в настоящей заявке.
[0096] Термин “носитель” относится к химическому соединению, которое способствует инкорпорированию соединения в клетки или ткани.
[0097] Термин “разбавитель” относится к химическим соединениям, разбавленным в воде, которые будут растворять композицию, представляющую интерес, а также стабилизировать биологически активную форму соединения. Соли, растворенные в буферных растворах, в данной области техники используют в качестве разбавителей. Одним из широко используемых буферных растворов является фосфатно-буферный солевой раствор, поскольку он имитирует солевые условия крови человека. Поскольку буферные соли могут контролировать pH раствора при низких концентрациях, буферный разбавитель редко модифицирует биологическую активность соединения. В конктексте настоящего изобретения, “эксципиент” относится к инертному веществу, которое добавляют к композиции для обеспечения, без ограничения, объема, консистентности, стабильности, связывающей способности, смазывающей способности, дезинтегрирующей способности и т.д. композиции. “Разбавитель” представляет собой тип эксципиента.
[0098] Термин “физиологически приемлемый” относится к носителю или разбавителю, который не оказывает негативного влияния на биологическую активность и свойства соединения.
[0099] Фармацевтические соединения, описанные в настоящей заявке, можно вводить пациенту, такому как человек, per se или в фармацевтических композициях, где они смешаны с другим активным ингредиент(ингредиентами), как в комбинированной терапии, или с подходящими носителями или эксципиентом(эксципиентами). В некоторых вариантах осуществления лекарственная форма включает такие формы, в которых соединение вводят per se. Кроме того, лекарственная форма может включать фармацевтическую композицию. В любом случае, лекарственная форма может включать достаточное количество соединения для лечения бактериальной инфекции как часть конкретного протокола введения, как должно быть понятно специалистам в данной области. Способы формулирования и введения соединений, раскрытых в настоящей заявке, можно найти в “Remington’s Pharmaceutical Sciences”, Mack Publishing Co., Easton, PA, 18th edition, 1990.
[0100] Подходящие пути введения могут включать, например, пероральное, ректальное, трансмукозальное, местное или интестинальное введение; парентеральную доставку, включая внутримышечные, подкожные, внутривенные, интрамедуллярные инъекции, а также интратекальные, прямые интравентрикулярные, интраперитонеальные, интраназальные или интраокулярные инъекции. Соединение также можно вводить в лекарственных формах замедленного или контролируемого высвобождения, включая депо инъекции, осмотические насосы, пилюли, чрескожные (включая электроперенос) пластыри и т.п., для пролонгированного и/или рассчитанного по времени, прерывистого введения с предварительно определенной скоростью.
[0101] Фармацевтические композиции можно получить способом, который как таковой является известным, например, с использованием традиционных способов смешивания, растворения, гранулирования, дражирования, измельчения, эмульгирования, инкапсулирования, захватывания или таблетирования.
[0102] Фармацевтические композиции можно формулировать любым традиционным способом с использованием одного или нескольких физиологически приемлемых носителей, включающих эксципиенты и вспомогательные добавки, которые способствуют переработке активных соединений в препараты, которые можно использовать фармацевтически. Подходящая композиция зависит от выбранного пути введения. Любой из хорошо известных методов, разбавителей, носителей и эксципиентов можно использовать как подходящий и как известный из уровня техники; например, см. Remington’s Pharmaceutical Sciences, указанный выше.
[0103] Препараты для инъекций можно получить в традиционных формах, либо в виде жидких растворов или суспензий, либо в виде твердых форм, подходящих для растворения или суспендирования в жидкости перед введением инъекции, либо в виде эмульсий. Подходящие эксципиенты представляют собой, например, воду, солевой раствор, декстрозу, маннит, лактозу, лецитин, альбумин, глутамат натрия, цистеин гидрохлорид и т.п. Кроме того, если желательно, фармацевтические композиции для инъекций могут содержать минорные количества нетоксичных вспомогательных веществ, таких как смачивающие вещества, регулирующие pH буферные вещества и т.п. Физиологически совместимые буферы включают, но не ограничиваются этим, раствор Хэнкса, раствор Рингера или физиологический солевой буфер. Если желательно, можно использовать средства, повышающие абсорбцию.
[0104] Для трансмукозального введения в композиции можно использовать пенетранты, подходящие для барьера, через который должно проникать средство.
[0105] Фармацевтические композиции для парентерального введения, например, путем болюсной инъекции или непрерывной инфузии, включают водные растворы активных соединений в водорастворимой форме. Кроме того, суспензии активных соединений можно получить в виде подходящих масляных суспензий для инъекций. Водные суспензии для инъекций могут содержать вещества, которые повышают вязкость суспензии, такие как натрий карбоксиметилцеллюлоза, сорбит или декстран. Необязательно, суспензия также может содержать подходящие стабилизаторы или вещества, которые повышают растворимость соединений, что делает возможным получение высококонцентрированных растворов. Композиции для инъекций могут быть представлены в стандартной лекарственной форме, например, в ампулах или в многодозовых контейнерах, с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях и могут содержать вспомогательные вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие вещества. Альтернативно, активный ингредиент может быть в порошкообразной форме для реструктурирования с использованием подходящего носителя, например, стерильной апирогенной воды, перед использованием.
[0106] Для перорального введения, композицию можно легко сформулировать путем объединения представляющих интерес композиций с фармацевтически приемлемыми носителями, хорошо известными в данной области техники. Такие носители, которые можно использовать в дополнение к катионному полимерному носителю, позволяют формулировать композиции в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п., для перорального приема пациентом, принимающим лечение. Фармацевтические препараты для перорального применения можно получить путем объединения активного соединения с твердым эксципиентом, необязательно измельчения полученной смеси и переработки смеси в гранулы после добавления подходящих вспомогательных веществ, если желательно, с получением таблеток или сердцевин драже. Подходящие эксципиенты представляют собой, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; препараты на основе целлюлозы, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий карбоксиметилцеллюлоза и/или поливинилпирролидон (PVP), например, Повидон. Если желательно, могут быть добавлены разрыхлители, такие как сшитый поливинилпирролидон (например, Кросповидон), агар или альгиновая кислота или ее соль, такая как альгинат натрия. Для сердцевин драже обеспечивают подходящие покрытия. Для этих целей можно использовать концентрированные растворы сахаров, которые необязательно содержат аравийскую камедь, тальк, поливинилпирролидон, гелеобразный карбопол, полиэтиленгликоль и/или диоксид титана, растворы для получения лаковых покрытий и подходящие органические растворители или смеси растворителей. Красители или пигмент могут быть добавлены к покрытиям для таблеток или драже для идентификации или в качестве отличительного признака для определения различных комбинаций доз активных соединений.
[0107] Фармацевтические препараты, которые можно использовать перорально, включают твердые капсулы из желатина, а также мягкие герметичные капсулы, полученные из желатина и пластификатора, такого как глицерин или сорбит. Твердые капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими, такими как крахмалы, и/или смазывающими веществами, такими как тальк или стеарат магния, и, необязательно, стабилизаторами. В мягких капсулах, активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы. Все композиции для перорального введения должны быть представлены в дозах, подходящих для такого введения.
[0108] Для буккального введения, композиции могут принимать форму таблеток или пастилок для рассасывания, сформулированных обычным способом. Предусматривается введение в слизистую оболочку щеки и сублигвальное введение.
[0109] Для введения путем ингаляции можно осуществить удобную доставку композиции в форме распыляемого аэрозоля, подаваемого из находящихся под давлением упаковок или небулайзера, с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае подаваемого под давлением аэрозоля, единицу дозирования можно определить путем обеспечения клапана для доставки отмеренного количества. Капсулы и картриджи, например, из желатина для применения в ингаляторе или инсуффляторе могут представлять собой такие, которые содержат порошкообразную смесь соединения и подходящей основы для порошка, такой как лактоза или крахмал.
[0110] Кроме того, в настоящей заявке раскрываются различные фармацевтические композиции, хорошо известные в фармацевтике, для применений, которые включают интраокулярную, интраназальную и интрааурикулярную доставку. Подходящие пенетранты для этих применений широко известны в данной области техники. Такие подходящие фармацевтические композиции наиболее часто и предпочтительно формулируют таким образом, чтобы они были стерильными, изотоничными и забуференными для стабильности и комфорта. Фармацевтические композиции для интраназальной доставки могут также включать капли и спреи, часто получаемые таким образом, чтобы воспроизводить во многих отношениях носовые секреты для обеспечения поддержания нормального биения ресничек. Как раскрыто в Remington’s Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), который включен в настоящую заявку посредством ссылки во всей его полноте и хорошо известен специалистам в данной области, подходящие композиции наиболее часто и предпочтительно являются изотоничными, слегка забуференными для поддержания pH в пределах от 5,5 до 6,5 и наиболее часто и предпочтительно включают антимикробные консерванты и подходящие стабилизаторы для лекарственного средства. Фармацевтические композиции для интрааурикулярной доставки включают суспензии и мази для местного нанесения в ухо. Традиционные растворители для таких композиций для ушного введения включают глицерин и воду.
[0111] Композиции также могут быть сформулированы в ректальные композиции, такие как суппозитории или микроклизмы с удержанием, например, содержащие традиционные основы для суппозиториев, такие как масло какао или другие глицериды.
[0112] Помимо композиций, описанных выше, композиции также могут быть сформулированы в виде депо препарата. Такие долгодействующие композиции можно вводить путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. Таким образом, например, соединения могут быть сформулированы с подходящими полимерными или гидрофобными веществами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами, или в виде умеренно растворимых производных, например, в виде умеренно растворимой соли.
[0113] Для гидрофобных соединений подходящий фармацевтический носитель может представлять собой систему со-растворителей, включающую бензиловый спирт, неполярное поверхностно-активное вещество, смешиваемый с водой органический полимер и водную фазу. Обычно используемая система со-растворителей представляет собой VPD систему со-растворителей, которая представляет собой раствор 3% масс./об. бензилового спирта, 8% масс./об. неполярного поверхностно-активного вещества Полисорбата 80™ и 65% масс./об. полиэтиленгликоля 300, доведенную до нужного объема в абсолютном этаноле. Естественно, пропорции системы со-растворителей могут существенно варьироваться, не нарушая ее характеристики растворимости и токсичности. Кроме того, компоненты системы со-растворителецй могут варьироваться: например, можно использовать другие низкотоксичные неполярные поверхностно-активные вещества вместо ПОЛИСОРБАТА 80™; размер фракции полиэтиленгликоля может варьировать; можно использовать другие биосовместимые полимеры вместо полиэтиленгликоля, например, поливинилпирролидон; и можно использовать другие сахара или полисахариды вместо декстрозы.
[0114] Способы лечения бактериальных инфекций могут включать введение терапевтически эффективного количества терапевтических соединений, описанных в настоящей заявке. Лечение бактериальной инфекции также может включать профилактическое введение терапевтических соединений для профилактики инфекции или предотвращения распространения инфекции у субъекта при угрозе заражения, такого как субъект, которого оперируют или у которого должна быть хирургическая операция, субъект с ослабленным иммунитетом или субъект, который, если ему не вводить соединение, иначе будет иметь риск заражения. Соединения показывают ингибиторную активность против широкого спектра бактерий, включая H. influenzae, E. coli, S. aureus, E. faecalis, E. facium, K. pneumonia, A. baumannii, S. pneumoniae и P. aeruginosa. Соединения показывают активность против наиболее резистентных штаммов, например метициллин-резистентного Staphylococcus aureus (MRSA). Кроме того, соединения показывают широкий спектр активности против всех Категорий A, B и C бактериальных патогенов биологической защиты, включая B. anthracis, B. pseudomallei, B. mallei, F. tularensis и Y. psetis. См. Примеры. Соединения обладают отличной относительной антибиотической активностью при относительно низкой концентрации. Кроме того, соединения могут проявлять сильную антибактериальную активность против различных патогенов человека и животных, включая грамположительные и грамотрицательные бактерии. В одном варианте осуществления бактериальная инфекция, которую можно лечить или облегчить, представляет собой MRSA.
[0115] Способы лечения бактериальных инфекций также включают интраабдоминальную инфекцию, инфекцию мочевых путей или пневмоэнтерит. Интраабдоминальные инфекции включают различные инфекции, такие как перитонит, апендицит, абсцессы, сепсис и холецистит, которые могут быть осложненными или неосложненными. Соединение, раскрытое в настоящей заявке, также можно использовать для лечения инфекций мочевых путей, которые могут быть вызваны E. coli. Кроме того, соединения, раскрытые в настоящей заявке, являются полезными для лечения пневмоэнтерита, который может быть вызван B. pseudomallei.
[0116] Композиции или фармацевтические композиции, описанные в настоящей заявке, можно вводить субъекту любым подходящим способом. Неограничивающие примеры способов введения включают, среди прочих, (a) введение пероральным путем, которое включает введение в форме капсулы, таблетки, гранулы, спрея, сиропа или других таких формах; (b) введение не пероральным путем, например, таким как ректальный, вагинальный, интрауретральный, интраокулярный, интраназальный или интрааурикулярный, которое включает введение в виде водной суспензии, масляного препарата или т.п. или в виде капель, спрея, суппозитория, бальзама, мази или т.п.; (c) введение через инъекцию, подкожно, интраперитонеально, внутривенно, внутримышечно, внутрикожно, интраорбитально, интракапсулярно, интраспинально, интрастернально или т.п., включая доставку при помощи инфузионного насоса; а также (d) введение местным путем; как сочтет целесообразным специалист в данной области, для приведения активного соединения в контакт с живой тканью.
[0117] Фармацевтические композиции, подходящие для введения, включают композиции, где активные ингредиенты содержатся в количестве, эффективном для достижения предполагаемого назначения. В некоторых вариантах осуществления, терапевтически эффективное количество соединения представляет собой количество, эффективное для лечения бактериальной инфекции, например, у млекопитающего субъекта (например, человека). Терапевтически эффективное количество соединений, раскрытых в настоящей заявке, необходимое в качестве дозы, будет зависеть от пути введения, типа животного, включая человека, подлежащего лечению, и физических характеристик данного конкретного животного. Дозу можно регулировать для достижения желаемого эффекта, но она будет зависеть от таких факторов, как масса тела, режим питания, сопутствующая лекарственная терапия и другие факторы, которые должны быть известны специалистам в области медицины. Более конкретно, терапевтически эффективное количество означает количество соединения, эффективное для предотвращения, облегчения или ослабления тяжести симптомов заболевания или пролонгирования выживания субъекта, подлежащего лечению. Определение терапевтически эффективного количества вполне сможет осуществить специалист в данной области, особенно в свете подробного раскрытия, представленного в настоящей заявке.
[0118] Как будет очевидно специалистам в данной области, полезная in vivo доза, которую можно вводить, и конкретный путь введения будут варьировать в зависимости от возраста, массы тела и вида млекопитающего, подлежащего лечению, конкретных используемых соединений и специфического назначения, для которого используют эти соединения. Определение эффективных уровней доз, которые представляют собой уровни доз, необходимые для достижения желаемого результата, сможет осуществить специалист в данной области с использованием рутинных фармакологических способов. Типично, для человека клинические применения продуктов начинают при более низких уровнях доз, повышая уровень доз вплоть до достижения желаемого эффекта. Альтернативно, можно использовать приемлемые in vitro испытания для установления полезных доз и путей введения композиций, указанных в способах по настоящему изобретению, с использованием хорошо известных фармакологических способов.
[0119] В испытаниях на животных, отличных от человека, применение потенциальных продуктов начинают при более высоких уровнях доз, снижая дозу до тех пор, пока желаемый эффект больше не достигается и неблагоприятные побочные эффекты исчезают. Доза может варьировать в широких пределах, в зависимости от желаемых эффектов и терапевтического показания. Типично, дозы могут составлять от около 10 микрограмм/кг до около 100 мг/кг массы тела, предпочтительно от около 100 микрограмм/кг до около 10 мг/кг массы тела. Альтернативно, дозы могут быть основаны и рассчитываться на основании площади поверхности тела пациента, как это известно специалистам в данной области.
[0120] Точную композицию, путь введения и дозу для фармацевтических композиций может выбрать лечащий врач в свете состояния пациента. (См., например, Fingl et al., 1975, в “The Pharmacological Basis of Therapeutics”, который включен в настоящую заявку посредством ссылки во всей его полноте, особенно главу 1 на стр. 1). В некоторых вариантах осуществления, доза композиции, которую вводят пациенту, может находиться в диапазоне от около 0,5 до около 1000 мг/кг массы тела пациента. Доза может представлять собой разовую дозу или последовательно вводимые две или более доз в течение одного или нескольких дней, как это необходимо для пациента. В случаях, когда для введения человеку дозы соединений были установлены для по меньшей мере некоторых состояний, можно использовать те же самые дозы или дозы, которые составляют от около 0,1% до около 500%, более предпочтительно от около 25% до около 250% от установленной для человека дозы. Когда доза для введения человеку вообще не установлена, как в случае новых открытых фармацевтических композиций, подходящую дозу для введения человеку можно вывести из ED50 или ID50 значений или других соответствующих значений, полученных из in vitro или in vivo исследований, подтвержденных испытаниями токсичности и испытаниями эффективности на животных.
[0121] Следует отметить, что лечащий врач должен знать, как и когда нужно закончить, прервать или отрегулировать введение из-за токсичности или дисфункции органов. И наоборот, лечащий врач должен также знать, как адаптировать лечение к более высоким уровням доз, если клинический ответ не был адекватным (предотвращая при этом токсичность). Уровень вводимой дозы при лечении расстройства, представляющего интерес, будет варьировать в зависимости от тяжести состояния, подлежащего лечению, и от пути введения. Тяжесть состояния можно, например, оценить, частью, при помощи стандартных прогностических способов оценки. Кроме того, доза и, возможно, частота введения также могут варьировать в соответствии с возрастом, массой тела и ответом индивидуального пациента. Программу, аналогичную обсуждаемой выше, можно использовать в ветеринарной медицине.
[0122] Хотя точную дозу можно определить в зависимости от конкретного лекарственного средства, в большинстве случаев можно сделать некоторые обобщения, касающиеся дозы. Ежедневный режим приема лекарственного средства для взрослого человека может включать, например, пероральную дозу от около 0,1 мг до 2000 мг активного ингредиента, предпочтительно от около 1 мг до около 500 мг, например, от 5 до 200 мг. В других вариантах осуществления используют внутривенную, подкожную или внутримышечную дозу активного ингредиента от около 0,01 мг до около 100 мг, предпочтительно от около 0,1 мг до около 60 мг, например, от около 1 до около 40 мг. В случаях введения фармацевтически приемлемой соли, дозы можно определить в расчете на свободную кислоту. В некоторых вариантах осуществления, композицию вводят от 1 до 4 раз в день. Альтернативно, композиции можно вводить путем непрерывной внутривенной инфузии, предпочтительно при дозе до около 1000 мг в день. Как будет понятно специалистам в данной области, в некоторых ситуациях, возможно, будет необходимо вводить соединения, раскрытые в настоящей заявке, в количествах, которые превышают, или даже намного превышают указанный выше предпочтительный диапазон доз, для эффективного и агрессивного лечения особенно агрессивных заболеваний или инфекций. В некоторых вариантах осуществления, соединения можно вводить в течение определенного периода непрерывного лечения, например, в течение недели или больше, или в течение нескольких месяцев или лет.
[0123] Размер дозировки и интервал можно регулировать индивидуально для обеспечения уровней в плазме активного вещества, которые являются достаточными для поддержания антибиотических эффектов или минимальной эффективной концентрации (MEC). MEC будет варьироваться для каждого соединения, но может быть определена из in vitro данных. Дозы, необходимые для достижения MEC, будут зависеть от индивидуальных характеристик и пути введения. Однако можно использовать ВЭЖХ анализы или биоанализы для определения концентраций в плазме.
[0124] Интервалы между введениями лекарственного средства также можно определить с использованием значения MEC. Композиции следует вводить с использованием режима введения, который поддерживает уровни в плазме выше MEC в течение 10-90% времени, предпочтительно в течение 30-90%, и наиболее предпочтительно в течение 50-90% времени.
[0125] В случаях местного применения или селективного приема, эффективная локальная концентрация лекарственного средства может не быть связана с концентрацией в плазме.
[0126] Количество вводимой композиции может зависеть от субъекта, подлежащего лечению, от массы тела субъекта, тяжести инфекции, способа введения и мнения лечащего врача, прописывающего лечение.
[0127] Композиции, раскрытые в настоящей заявке, можно оценить на эффективность и токсичность с использованием известных способов. Например, токсикологию соединения можно установить путем определения in vitro токсичности в отношении клеточной линии, такой как клеточная линия млекопитающего и, предпочтительно, человека. Результаты таких испытаний часто предсказывают токсичность у животных, таких как млекопитающие или, более конкретно, человек. Альтернативно, токсичность конкретных соединений в животной модели, такой как мыши, крысы, кролики или обезьяны, можно определить с использованием известных способов. Эффективность конкретного соединения можно установить с использованием некоторых общеизвестных методов, таких как in vitro методы, животные модели, или клинические испытания на людях. Существуют общепризнанные in vitro модели для почти каждого класса состояний. Подобным образом, приемлемые животные модели можно использовать для установления эффективности химических веществ для лечения таких состояний. При выборе модели для определения эффективности специалист должен руководствоваться сведениями, известными из уровня техники, чтобы выбрать подходящую модель, дозу, и путь, и схему введения. Конечно, клинические испытания на людях также можно использовать для определения эффективности соединения для человека.
[0128] Композиции, если желательно, могут быть представлены в упаковке или дозирующем устройстве, которые могут содержать одну или несколько стандартных лекарственных форм, содержащих активный ингредиент. Упаковка, например, может включать металлическую или пластиковую фольгу, такую как блистерная упаковка. Упаковка или дозирующее устройство могут содержать прилагаемые инструкции по применению препарата. Упаковка или дозирующее устройство также может содержать прилагаемое к контейнеру уведомление в форме, установленной правительственным органом, регулирующим производство, применение или продажу фармацевтических препаратов, которое информирует об одобрении этим органом данной формы лекарственного средства для введения человеку или применению в ветеринарии. Такое уведомление, например, может быть в форме этикетки, одобренной Управлением США по санитарному надзору за качеством пищевых продуктов и медикаментов, или одобренного листка-вкладыша. Композиции, включающие соединение, сформулированное в совместимом фармацевтическом носителе, также могут быть получены, помещены в соответствующий контейнер и помечены как предназначенные для лечения указанного состояния.
[0129] В некоторых вариантах осуществления, в фармацевтической промышленности является стандартной практикой обеспечивать по существу чистое вещество при формулировании фармацевтических композиций. Поэтому в некоторых вариантах осуществления “по существу чистый” относится к степени чистоты, необходимой для формулирования фармацевтических препаратов, которые могут включать, например, небольшое количество другого вещества, которое не будет влиять на соответствие требованиям для фармацевтического применения. В некоторых вариантах осуществления по существу чистое соединение содержит по меньшей мере около 96% масс. соединения, например, по меньшей мере около 97%, 98%, 99%, или 100% соединения.
[0130] Термины “приблизительно”, “около” и “по существу”, в конктексте настоящего изобретения, означают количество близкое к указанному количеству, которое тем не менее осуществляет желаемую функцию или обеспечивает достижение желаемого результата. Например, термины “приблизительно”, “около” и “по существу” могут относиться к количеству в пределах меньше чем 10%, в пределах меньше чем 5%, в пределах меньше чем 1%, в пределах меньше чем 0,1% и в пределах меньше чем 0,01% от указанного количества.
[0131] ПРИМЕРЫ
[0132] Способы синтеза исходных веществ, таких как биссульфон, можно найти в PCT/US2012/029104, предварительной патентной заявке США №61/700159 или заявке PCT, которая испрашивает приоритет по предварительной патентной заявке США №61/700159 (WO 2014/043272). Общая процедура для синтеза представляет собой следующую:

Биссульфон сначала обрабатывали при помощи R2 и K2CO3, с последующим добавлением R4 в одном сосуде. Конечный продукт получали путем удаления защитной группы Boc при помощи TFA.
Общие экспериментальные способы включают следующие:
[0133] 1H ЯМР спектры регистрировали на Bruker Avance III 400 МГц и Bruker Fourier 300 МГц, и TMS использовали в качестве внутреннего стандарта.
[0134] ЖХМС данные получали на квадрупольном масс-спектрометре на системе Agilent LC/MSD 1200 Series (Колонка: ODS 2000 (50×4,6 мм, 5 мкм), работающей в ES (+) или (-) режиме ионизации; T=30°C; скорость потока=1,5 мл/мин; длина волны детектора: 214 нм.
[0135] Препаративную ВЭЖХ осуществляли в следующих условиях: (Колонка: Welchrom C18, 150×20 мм); Длина волны 220 нм; Подвижная фаза: A MeCN (0,1% TFA); B вода (0,1% TFA); Скорость потока: 25 мл/мин; Объем вводимой пробы: 2 мл; Время анализа: 30 мин; Уравновешивание: 5 мин.
Пример 1: Синтез R4 компонентов

Синтез 3-{[(4-метилфенил)сульфонил]амино}пропановой кислоты (2)

[0136] К раствору соединения (1) (45,0 г, 0,51 моль) и NaOH (40,4 г, 1,02 моль) в воде (500 мл) добавляли по каплям хлор(4-метилфенил)сульфон (96 г, 0,51 моль) при 70°C, затем перемешивали при 70°C в течение 1 часа, затем охлаждали. Подкисление реакционной смеси до pH=1 концентрированной HCl давало неочищенный продукт в виде густого осадка. Продукт выделяли фильтрованием и перекристаллизовывали из этанола с получением 90 г соединения 2 в виде белого твердого вещества (выход=73%). 1H ЯМР (400 МГц, DMSO-d6): δ 2,33-2,36 (м, 2H), 2,50-2,51 (м, 3H), 2,89-2,91 (д, J=6 Гц, 2H), 7,39-7,41 (д, J=8 Гц, 2H), 7,59 (с, 1H), 7,67-7,69 (д, J=8 Гц, 2H).
Синтез 3-{[(4-метилфенил)сульфонил]амино}-1-пирролидинилпропан-1-она (3)

[0137] К раствору соединения (2) (90,0 г, 0,37 моль) в SOCl2 (900 мл), который перемешивали при температуре кипения с обратным холодильником в течение 2,5 часов, затем концентрировали и остаток растворяли в DCM, добавляли пирролидин (71,1 г, 0,99 моль) в DCM, затем перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли водой и экстрагировали при помощи DCM (100 мл ×3), промывали насыщенным солевым раствором, сушили над Na2SO4 и фильтровали, фильтрат упаривали для удаления растворителя. Остаток очищали на силикагеле (DCM/MeOH=80/1) с получением соединения 3 (97 г, 88%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6): d 1,83-1,88 (м, 2H), 1,91-1,96 (м, 2H), 2,42 (с, 3H), 2,43-2,49 (т, 2H), 3,17-3,2 (м, 2H), 3,28-3,31 (т, 2H), 3,40-3,43 (т, 2H), 5,67-5,70 (т, 2H), 7,29-7,31 (д, J=8 Гц, 2H), 7,74-7,62 (д, J=8 Гц, 2H); МС рассчитано: 296; МС найдено: 297 ([M+H]+).
Синтез 3-{[(4-метилфенил)сульфонил]проп-2-ениламино}-1- пирролидинилпропан-1-она (4)

[0138] К раствору соединения (3) (97,0 г, 0,33 моль) и соединения (5) (59,4 г, 0,50 моль) в DMF (500 мл) добавляли гидрид натрия (60% масс. в минеральном масле, 19,8 мг, 0,50 моль) при 0°C, затем перемешивали при 95°C в течение 3,5 часов. Реакционную смесь разбавляли водой и экстрагировали при помощи DCM (100 мл ´3), промывали насыщенным солевым раствором, сушили над Na2SO4 и фильтровали, фильтрат упаривали для удаления растворителя. Остаток очищали на силикагеле (DCM/MeOH=120/1) с получением соединения (6) (35 г, 31%) в виде светло-желтого масла. 1H ЯМР (400 МГц, CDCl3): d 1,82-1,88 (м, 2H), 1,92-1,99 (м, 2H), 2,43 (с, 3H), 2,63-2,67 (т, 2H), 3,39-3,44 (м, 6H), 3,82-3,83 (д, J=6,4 Гц, 2H), 5,12-5,20 (м, 2H), 5,60-5,70 (м, 1H), 7,29-7,32 (д, J=8 Гц, 2H), 7,69-7,71 (д, J=8,4 Гц, 2H); МС рассчитано: 336; МС найдено: 337 ([M+H]+).
Синтез 3-[(4-метилфенил)сульфонил]-3-азабицикло[3.2.0]гептан-6-она (5)

[0139] Раствор соединения 6 (35 г, 0,10 моль) в 1,2-дихлорэтане (250 мл) добавляли в течение 20 минут к раствору трифторметансульфонового ангидрида (58 г, 0,20 моль) в 1,2-дихлорэтане (100 мл). Затем медленно добавляли раствор коллидина (19 г, 0,15 моль) в 1,2-дихлорэтане (100 мл) в течение 30 минут. Затем реакционную смесь нагревали при 90°C в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали. Остаток гидролизовали в двухфазной системе H2O-CCl4. Органические слои сушили над Na2SO4 и фильтровали, фильтрат упаривали для удаления растворителя. Остаток очищали на силикагеле (PE/EA=10/1) с получением соединения 7 (15 г, 54%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): d 2,45 (с, 3H), 2,67-2,72 (м, 1H), 2,85-2,92 (м, 1H), 2,94-3,00 (м, 2H), 3,27-3,35 (м, 1H), 3,59-3,66 (м, 2H), 3,17-3,12 (м, 2H), 3,83-3,86 (д, J=10 Гц, 1H), 7,34-7,36 (д, J=8 Гц, 2H), 7,69-7,71 (д, J=8,4 Гц, 2H); МС рассчитано: 265; МС найдено: 266([M+H]+).
Синтез 3-[(4-метилфенил)сульфонил]-3-азабицикло[3.2.0]гептан-6-ола(6)

[0140] К раствору соединения (7) (15 г, 0,1 моль) в MeOH (150 мл) добавляли по порциям NaBH4 (4,29 г, 0,2 моль) при охлаждении на ледяной бане, затем реакционную смесь перемешивали в течение 45 минут при комнатной температуре. Реакционную смесь гасили водой, экстрагировали при помощи EA (50 мл ´3), объединенные органические слои сушили над Na2SO4, концентрировали и очищали колоночной хроматографией на силикагеле (PE:EA=8:1-4:1) с получением соединения 8 (14 г, 92%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): d 1,73-1,79 (м, 1H), 2,12-2,14 (м, 1H), 2,44 (с, 3H), 2,52-2,63 (м, 3H), 2,67-2,75 (м, 1H), 2,96-3,01 (м, 1H), 3,42-3,43 (д, J=4 Гц, 1H), 3,86-3,88 (д, J=10,8 Гц, 1H), 4,23-4,25 (м, 1H), 7,34-7,36 (д, J=8,4 Гц, 2H), 7,72-7,74 (д, J=8,4 Гц, 1H);
МС рассчитано: 267; МС найдено: 268 ([M+H]+)
Синтез трет-бутил 6-гидрокси-3-азабицикло[3.2.0]гептан-3-карбоксилата (7)

[0141] К раствору нафталина (26,8 г, 0,2 моль) в диметоксиэтане (100 мл) добавляли натрий (4,2 мг, 0,2 ммоль) и полученную темно-зеленую смесь перемешивали в течение 3 часов. Соединение 8 (14 г, 0,05 моль) в диметоксиэтане (50 мл) при -60°C медленно обрабатывали раствором нафталенида натрия до тех пор, пока сохранялся светло-зеленый цвет. Реакционную смесь затем гасили при -78°C путем добавления воды. Смеси давали нагреться до 0°C и добавляли ди-трет-бутилдикарбонат. Через 3 часа смесь разбавляли при помощи EA, экстрагировали при помощи EA (50 мл ´3), промывали насыщенным солевым раствором, объединенные органические слои сушили над Na2SO4, концентрировали и очищали колоночной хроматографией на силикагеле (PE:EA=8:1-4:1) с получением соединения 9 (7 г, 63%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3): d 1,48 (с, 9H), 1,59-1,61 (м, 1H), 2,46-2,53 (м, 1H), 2,56-2,64 (м, 1H), 3,02-3,23 (м, 3H), 3,45-3,48 (м, 1H), 4,00-4,03 (м, 1H), 4,25-4,28 (т, 1H); МС рассчитано: 213; МС найдено: 113[M++1-Boc]
Синтез 3-азабицикло[3.2.0]гептан-6-ола (8)

[0142] К раствору соединения (7) (10 г, 0,1 моль) в DCM (150 мл) добавляли TFA (4,29 г, 0,2 моль) по порциям при охлаждении на ледяной бане, затем реакционную смесь перемешивали в течение 45 минут при комнатной температуре. Реакционную смесь концентрировали и использовали на следующей стадии реакции.
Пример 2: Синтез соединений

[0143] Бис-сульфон 9 (11,80 г, 17,23 ммоль) растворяли в NMP (60 мл), затем добавляли 2-метилпиримидин-5-ол 1 (7,59 г, 68,93 ммоль). Получали гомогенный раствор. Добавляли K2CO3 (9,53 г, 68,93 ммоль) и полученную суспензию нагревали до 100°C в течение 1 часа, затем добавляли аминоспирт (7,32 г, 34,46 ммоль) и полученную смесь нагревали до 100°C еще в течение одного часа, охлаждали до комнатной температуры и в смесь вливали воду (450 мл) при перемешивании. Смесь охлаждали до 0°C, фильтровали, промывали осажденные вещества водой (2´25 мл) и сушили с получением около 12 г неочищенного продукта в виде белого твердого вещества. Неочищенное твердое вещество растворяли в дихлорметане и добавляли силикагель. Растворители удаляли. Остаток очищали флэш-хроматографией на силикагеле (EtOAc/гексан: 20% до 50% до 90%) с получением чистого соединения 10 в виде белого твердого вещества (7,76 г, 75%). LC-MS: M+1: 536,30.
[0144] Соединение 10 растворяли в 50 мл TFA и перемешивали в течение 1 минуты при комнатной температуре. После удаления растворителя добавляли воду (50 мл) и EtOH (25 мл). Гомогенный раствор нейтрализовали при помощи 1н раствора NaOH (около 150 мл, PH>10). Образовалось смолистое твердое вещество, и его выделяли. Смолистое твердое вещество суспендировали в воде (50 мл), и это смолистое твердое вещество разбивали на мелкие кусочки шпателем. Осажденные вещества фильтровали, промывали водой два раза и сушили на воздухе с получением 4,40 грамм очищенного соединения 11 в виде белого твердого вещества (85%, в целом 63% исходя из 1). ЖХ-МС:M+1:436,24. 1H ЯМР (300 МГц, DMSO) d (м.д.): 11,66 (с, 1H), 8,72 (с, 2H), 7,15 (д, J=11,2, 1H), 6,29 (д, J=9,7, 1H), 5,55 (шир.с, 1H), 5,11 (с, 1H), 4,64 (д, J=9,9, 1H), 4,20 (д, J=7,6, 1H), 4,03 (д, J=12,3, 1H), 3,60 (м, 2H), 3,12 (шир. м, 1H), 2,85 (с, 3H), 2,67 (с, 3H), 1,48 (м, 1H).
Схема:

[0145] Смесь 3-азабицикло[3,1,0]гексан-6-ола (10 мг, 0,1 ммоль), биссульфона (50мг, 0,1 ммоль) и K2CO3 (40 мг, 0,3 ммоль) в NMP (2 мл) перемешивали в течение ночи при комнатной температуре, затем добавляли 2-метилпиримидин-5-ол (330 мг, 3 ммоль) и полученную смесь нагревали до 80°C в течение ночи. Неочищенный продукт очищали при помощи ВЭЖХ с получением соединения в виде белого твердого вещества (15 г, 35%). ЖХ-МС:M+1:540.
[0146] Полученное на предыдущей стадии соединение (10 мг, 0,02 ммоль) растворяли в 1 мл TFA и перемешивали в течение 1 минуты при комнатной температуре. После удаления растворителей остаток очищали при помощи ВЭЖХ с получением конечного соединения (6 мг, 68%). ЖХ-МС:M+1:440.

[0147] SM III (198 мг, 0,419 ммоль) и II (95 мг, 1,0 экв.) растворяли в 1 мл NMP. К этой смеси добавляли Na2CO3 (133 мг, 3 экв.). Полученную смесь перемешивали при комнатной температуре в течение 5 часов. К реакционной смеси добавляли воду и экстрагировали этилацетатом три раза. Органический слой собирали, объединяли и концентрировали с получением масла. Используя хроматографию с нормальными фазами на системе ISCO, выделяли желаемый продукт в виде желтого полутвердого вещества (71%). ЖХ-МС:M+1:506.

[0148] SM III (50 мг, 0,1 ммоль) и I (58 мг, 5,0 экв.) растворяли в 0,5 мл DMF. К этой смеси добавляли KOtBu (55 мг, 5 экв.). Полученную смесь нагревали при 150°C в течение 5 часов. К реакционной смеси добавляли воду и экстрагировали этилацетатом три раза. Органический слой собирали, объединяли и концентрировали с получением масла. Используя обращенно-фазовую хроматографию на системе ISCO, выделяли желаемый продукт в виде не совсем белого твердого вещества (40%). Выделяли около 25 мг SM III. К высушенному продукту из этой реакции добавляли 4 н раствор HCl в диоксане (0,5 мл) и перемешивали в течение 30 минут. После концентрирования остаток очищали обращенно-фазовой хроматографией на системе ISCO с получением желаемого продукта в виде белых твердых частиц (5 мг, 11%). ЖХ-МС:M+1:444.

[0149] Представленные выше два соединения синтезировали с использованием таких же процедур, которые описаны выше.
Пример 3: Синтез соединений с N-линкером

трет-бутил(6-фтор-4-(6-гидрокси-3-азабицикло[3.2.0]гептан-3-ил)-2-(метилсульфонил)-9H-пиримидо[4,5-b]индол-8-ил)(метил)карбамат.
Смесь бис-сульфона (1,0 г, 2,12 ммоль), амина (448,7 мг, 2,12 ммоль) и K2CO3 (292,3 мг, 2,12 ммоль) в NMP (7 мл) перемешивали в течение 24 часов при комнатной температуре. ЖХ/МС анализ показал завершение реакции. К смеси добавляли воду (200 мл) и образовавшийся осадок фильтровали, промывали водой (2´15 мл) и сушили. Получали 0,8 г порошкообразного продукта (выход: 80%). MS (ESI) m/z 506 (M+H)+.

3-(6-фтор-8-(метиламино)-2-((2-метилпиримидин-5-ил)амино)-9H-пиримидо[4,5-b]индол-4-ил)-3-азабицикло[3.2.0]гептан-6-ол.
Смесь моно-сульфона (50,6 мг, 0,1 ммоль), 2-метилпиримидин-5-амина (109 мг, 1,0 ммоль) и K2CO3 (138,1 мг, 1,0 ммоль) в NMP (0,5 мл) нагревали до 140°C в течение 17 часов, охлаждали до комнатной температуры, добавляли TFA (15 мл) и перемешивали в течение 5 минут. После удаления растворителя остаток очищали препаративной ВЭЖХ с получением желаемого продукта. MS (ESI) m/z 435 (M+H)+.
Пример 4: Синтез соединения, где Z и R4 объединены
Общая схема:

Промежуточное соединение бис-метилсульфонил 1 использовали в качестве исходного вещества для получения соединений формулы I. R4 метилсульфонил соединения 1 селективно замещали 2-метилпиримидин-5-олом в присутствии карбоната калия при комнатной температуре с получением продукта 3. Затем осуществляли региоселективное бромирование соединения 3 при помощи NBS при 40°C в DMF с получением соединения 4, которое затем преобразовывали в виниловое соединение 5 путем сочетания по методу Стилле. Дигидроксилирование in situ с последующим окислительным расщеплением соединения 5 непосредственно давало альдегид 6. Различные R2 фрагменты затем могут быть введены на этой стадии синтеза. Типично, OR2 может замещать R2 метилсульфонил соединения 6 путем нагревания реакционной смеси при 90°C в присутствии основания. Когда R2 был отличным от R4 2-метилпиримидин-5-ола, использовали более 3 эквивалентов R2OH для минимизирования бис-метилпиримидинового продукта. Продукт замещения 7 затем обрабатывали амином 8 для преобразования в соединение 9. Температура реакции варьировала от комнатной температуры до 80°C в зависимости от реактивности амина 8. Циклическое соединение 12 получали из соединения 9 через введение C-H карбена через in situ трехстадийную последовательность иминирования соединения 9, образования диазония 11 и введения 11 с образованием соединения 12. Защитную Boc группу соединения 12 затем удаляли при помощи TFA при комнатной температуре с получением соединения Формулы I.
Пример 4a: Синтез соединения 

Соединение 3. К смеси соединения 1 (14,18 г, 30 ммоль) и 2-метилпиримидин-5-ола (4,29 г, 39 ммоль) в безводном DMF (30 мл) добавляли K2CO3 (10,8 г, 78 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 7 часов. Смесь затем разбавляли EtOAc (100 мл) и водой (100 мл). Водный слой экстрагировали и обратно экстрагировали при помощи EtOAc (100 мл ´2). Объединенные органические слои сушили над Na2SO4 и концентрировали на роторном испарителе при 40°C. Возможные следовые количества EtOAc затем удаляли путем совместного испарения с дихлорметаном в вакууме. Неочищенный липкий продукт (17,2 г) использовали на следующей стадии без дополнительной очистки. ЖХ-МС:M+1:503,5.
[0150] Соединение 4. К раствору неочищенного соединения 3 в безводном DMF (30 мл) при 40°C добавляли NBS (5,34 г, 30 ммоль). Смесь нагревали при 40°C в течение 2 часов и добавляли дополнительное количество NBS (5,34 г, 30 ммоль). Реакцию продолжали при 40°C в течение ночи. Смесь затем очищали колоночной хроматографией на силикагеле (40-60% EtOAc в гексане). Около половины общего объема объединенных фракций удаляли на роторном испарителе. Оставшийся раствор затем промывали водой (3´100 мл), сушили над Na2SO4 и концентрировали на роторном испарителе при 45°C с получением указанного в заголовке соединения в виде желтого твердого вещества (13,95 г, 80%). ЖХ-МС:M+1:582,3.

[0151] Соединение 16: Соединение 4 (1,16 г, 2 ммоль) в безводном DMF (6 мл) нагревали при 90°C и атмосферу заменяли азотом. Добавляли аллилтрибутилстаннан (993 мг, 3 ммоль) с последующим добавлением каталитического количества Pd(PPh3)2Cl2 (140 мг, 0,2 ммоль). Полученную смесь нагревали в атмосфере азота в течение 2 часов. Смесь охлаждали и очищали колоночной хроматографией на силикагеле (60-80% EtOAc в гексане). Около половины объема объединенных фракций продукта частично концентрировали на роторном испарителе. Смесь затем промывали водой (3´70 мл), сушили над Na2SO4 и концентрировали на роторном испарителе при 45°C с получением указанного в заголовке соединения в виде желтого твердого вещества (998,4 мг, 92%). ЖХ-МС:M+1:543,6.
[0152] Соединение 20: Смесь соединения 16 (813,9 мг, 1,5 ммоль), 2-метилпиримидин-5-ола (330,3 мг, 3 ммоль) и K2CO3 (967,4 мг, 7 ммоль) в безводном DMF (2 мл) нагревали при 100°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и очищали хроматографией на колонке C18 с получением указанного в заголовке продукта в виде желтого твердого вещества (773 мг, 90%). ЖХ-МС:M+1:573,6.
[0153] Соединение 22: Смесь соединения 20 (57 мг, 0,1 ммоль) и N-метилморфолин-N-оксида (35 мг, 0,3 ммоль) растворяли в 1:1 смеси диоксан:tBuOH (1 мл). Затем добавляли каталитическое количество 4% водного раствора OsO4 (2 капли) и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли воду (~0,5 мл) и сульфит натрия (50 мг) и перемешивали при комнатной температуре для гашения избытка окисляющих реагентов, реакционную смесь кипятили с обратным холодильником при 100°C в течение 2 часов в присутствии 10% раствора NaOH (1 мл). Продукт очищали при помощи ВЭЖХ и обрабатывали при помощи TFA (~0,3 мл) с получением указанного в заголовке соединения в виде розового твердого вещества (15,8 мг, 40%). ЖХ-МС: M+1: 397,1.
[0154] Пример 4b: Синтез:

и аналогов с другим R2.

[0155] Промежуточное соединение 4 использовали для получения соединений Примера 4b. Соединение сначала преобразовывали в соответствующий аллил 16 путем сочетания по методу Стилле. R2 метилсульфонил соединения 16 затем замещали группой OR2 в присутствии карбоната калия при 100°C с получением продукта 17.
[0156] Пример 4c: Синтез:


[0157] Промежуточное соединение 17, показанное выше, можно использовать для получения остова соединений Примера 4c: R4 группа соединения 17 может быть селективно гидролизована с образованием гидрокси 23, который при обработке при помощи POCl3 преобразуется в Cl 24. Реакция сочетания по методу Сузуки соединения 24 с винилбороновой кислотой или сочетание по методу Стилле соединения 24 с трибутил(винил)оловом обеспечивает винил 25. Это соединение затем подвергают реакции обмена с замыканием кольца с образованием предварительного промежуточного соединения 26. Соединения, содержащие OH-содержащий заместитель в D кольце, могут быть образованы из соединения 26 через различные реакции алкена, такие как дигидроксилирование, реакция Дильса-Альдера, циклопропанирование и т.д.
Пример 4d

[0158] Соединение 27: Смесь соединения 20 (1,145 г, 2 ммоль) и гидроксида натрия (800 мг, 20 ммоль) в диоксане (10 мл) и воде (10 мл) кипятили с обратным холодильником при 100°C в течение 1 дня. Смесь затем очищали хроматографией на колонке C18. Собранные фракции экстрагировали при помощи DCM (100 мл) и водный слой обратно экстрагировали при помощи DCM (50 мл ´2). Объединенные органические слои концентрировали на роторном испарителе с получением указанного в заголовке соединения в виде желтого твердого вещества (800 мг, 83,2%). ЖХ-МС:M+1:481,6.
[0159] Соединение 28: К раствору соединения 27 (800 мг, 1,665 ммоль) в POCl3 (6 мл) добавляли диэтилизопропиламин (430 мг, 3,33 ммоль). Полученный раствор нагревали при 70°C в течение 1 часа, затем при 80°C в течение 1 дня. После охлаждения реакционной смеси до комнатной температуры смесь выливали в химический стакан, содержащий около 100 г льда. Осадок растворяли при помощи DCM (80 мл) и раствор затем экстрагировали. Водный слой обратно экстрагировали при помощи DCM (50 мл ´2). Объединенные органические слои концентрировали на роторном испарителе и очищали хроматографией на колонке C18. Собранные фракции экстрагировали при помощи DCM и концентрировали на роторном испарителе с получением указанного в заголовке продукта в виде твердого вещества персикового цвета (525 мг, 79%). ЖХ-МС:M+1:399,1.
[0160] Соединение 29: К раствору соединения 28 (525 мг, 1,32 ммоль) в безводном DMF (2 мл) добавляли трибутил(винил)олово (835 мг, 2,63 ммоль). Атмосферу содержащего смесь раствора продували азотом, затем добавляли Pd(PPh3)2Cl2 (92,1 ммоль, 0,132 ммоль). Реакционную смесь нагревали при 90°C в течение 2 часов и затем добавляли дополнительное количество Pd(PPh3)2Cl2 (92,1 ммоль, 0,132 ммоль). Реакцию продолжали при 90°C в течение 1,5 часов. Смесь затем охлаждали до комнатной температуры и очищали хроматографией на колонке C18. Собранные фракции экстрагировали при помощи DCM и органический слой концентрировали на роторном испарителе с получением указанного в заголовке продукта в виде красновато-коричневого твердого вещества (421,3 мг, 82%). ЖХ-МС:M+1:391,4.
[0161] Соединение 30: К раствору соединения 29 (42,1 мг, 0,11 ммоль) в DCM (2 мл) добавляли катализатор Граббса (первого поколения, 16,5 мг, 0,02 ммоль). Смесь перемешивали при комнатной температуре в течение 1 дня. Смесь затем концентрировали на роторном испарителе и очищали при помощи ВЭЖХ с получением указанного в заголовке соединения в виде желтого твердого вещества (21,9 мг, 55%).
Пример 5: Синтез пролекарств
Пример 5a: Синтез соединения

ОБЩАЯ СХЕМА

Пример 5b: Синтез соединения

Хлорметил (6-фтор-4-(6-гидрокси-3-азабицикло[3.2.0]гептан-3-ил)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-8-ил)(метил)карбамат (2)

[0162] Смесь соединения 1 (0,180 г, 0,413 ммоль) и ди-изопропилэтиламина (0,267 г, 2,07 ммоль) в CH2Cl2 (10 мл) охлаждали до 0°C в атмосфере азота. Хлорметилхлорформиат (0,061 мл, 0,475 ммоль), растворенный в CH2Cl2 (0,5 мл), по каплям добавляли в реакционную смесь через шприц. Полученный желтый раствор перемешивали в течение 1 часа и затем концентрировали при пониженном давлении. Неочищенный продукт очищали при помощи ВЭЖХ с получением соединения 2 (0,080 г, 0,152 ммоль, 37%) в виде желтого твердого вещества. ЖХ/МС (ESI, M+H+)=528.
((Ди-трет-бутоксифосфорил)окси)метил (6-фтор-4-(6-гидрокси-3-азабицикло[3.2.0]гептан-3-ил)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-8-ил)(метил)карбамат (3)

[0163] Смесь соединения 2 (0,080 г, 0,152 ммоль), иодида натрия (0,037 г, 0,246 ммоль) и тетра-н-бутиламмоний ди-трет-бутилфосфата (0,222 г, 0,491 ммоль) в безводном ТГФ (10,0 мл) перемешивали в течение 22 часов при 23°C. После перемешивания в течение 22 часов полученную гетерогенную смесь фильтровали и очищали при помощи ВЭЖХ с получением соединения 3 (0,060 г, 0,085 ммоль, 56%) в виде белого твердого вещества. ЖХ/МС (ESI, M+H+)=702.
(Фосфоноокси)метил (6-фтор-4-(6-гидрокси-3-азабицикло[3.2.0]гептан-3-ил)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-8-ил)(метил)карбамат (4)

[0164] Смесь соединения 3 (0,060 г, 0,085 ммоль) в трифторуксусной кислоте (3,0 мл) перемешивали в течение 15 минут при 23°C. Трифторуксусную кислоту выпаривали при пониженном давлении, и неочищенный продукт очищали при помощи ВЭЖХ с получением соединения 4 (0,043 г, 0,072 ммоль, 85%) в виде белого твердого вещества. ЖХ/МС (ESI, M+H+)=590.
Пример 5c: Синтез пролекарств
ОБЩАЯ СХЕМА

Синтез Пролекарств:


трет-бутил 6-гидрокси-3-азабицикло[3.2.0]гептанe-3-карбоксилат (2)
[0165] К раствору соединения 1 (0,525 г, 2,48 ммоль) в безводном MeOH (5,0 мл) добавляли NaBH4 (0,093 г, 2,48 ммоль) в атмосфере азота при 23°C. Реакционную смесь перемешивали в течение 15 минут и реакцию контролировали при помощи ЖХ/МС. После завершения реакции смесь обрабатывали ледяной водой (25 мл) и экстрагировали при помощи EtOAc (50 мл ´3). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт 2 в виде светло-желтого масла использовали для следующей реакции без дополнительной очистки. ЖХ/МС (ESI, M+H+)=214.
трет-бутил 6-((диизопропоксифосфорил)окси)-3-азабицикло[3.2.0]гептан-3-карбоксилат (3)
[0166] К раствору соединения 2 (0,525 г, 2,46 ммоль) в безводном ТГФ (5,0 мл) добавляли t-BuOK (4,96 мл, 4,96 ммоль, 1,0 M раствор в ТГФ) в атмосфере азота при 23°C. Полученную смесь перемешивали в течение 15 минут и затем диизопропилфосфорохлоридат (1,00 г, 4,96 ммоль), растворенный в ТГФ (0,5 мл), добавляли по каплям через шприц. После перемешивания в течение 2 часов смесь обрабатывали ледяной водой (50 мл) и экстрагировали при помощи EtOAc (100 мл ´3). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт 3 использовали для следующей реакции без дополнительной очистки. ЖХ/МС (ESI, M+H+)=378.
3-азабицикло[3.2.0]гептан-6-илдиизопропилфосфат (4)
[0167] Смесь соединения 3 в трифторуксусной кислоте (1,5 мл) перемешивали в течение 15 минут при 23°C. Трифторуксусную кислоту выпаривали при пониженном давлении и неочищенный продукт 4 в виде оранжевого масла использовали для следующей реакции без дополнительной очистки. ЖХ/МС (ESI, M+H+)=278.
трет-бутил (4-(6-((диизопропоксифосфорил)окси)-3-азабицикло[3.2.0]гептан-3-ил)-6-фтор-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-8-ил)(метил)карбамат (6)

[0168] Смесь соединения 5 (0,426 г, 0,800 ммоль), 3-азабицикло[3.2.0]гептан-6-илдиизопропилфосфата 4 (0,670 г, 2,40 ммоль) и K2CO3 (0,553 г, 4,00 ммоль) в NMP (3,5 мл) перемешивали в течение 2 часов при 100°C. После перемешивания в течение 2 часов реакцию контролировали при помощи ЖХ/МС. Полученную гетерогенную смесь охлаждали до 23°C и очищали при помощи ВЭЖХ с получением соединения 6 (0,265 г, 0,379 ммоль, 47%) в виде белого твердого вещества. ЖХ/МС (ESI, M+H+)=700.
3-(6-фтор-8-(метиламино)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-4-ил)-3-азабицикло[3.2.0]гептан-6-ил дигидрофосфат (7),
3-(6-фтор-8-(метиламино)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-4-ил)-3-азабицикло[3.2.0]гептан-6-ил изопропил гидрофосфат (8) и
3-(6-фтор-8-(метиламино)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-4-ил)-3-азабицикло[3.2.0]гептан-6-ил диизопропилфосфат (9)

[0169] Смесь соединения 6 (0,265 г, 0,379 ммоль) в трифторуксусной кислоте (3,5 мл) перемешивали в течение 24 часов при 80°C в атмосфере азота. Трифторуксусную кислоту выпаривали при пониженном давлении и неочищенный продукт очищали при помощи ВЭЖХ с получением 3-(6-фтор-8-(метиламино)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-4-ил)-3-азабицикло[3.2.0]гептан-6-ил дигидрофосфата 7 (0,055 г, 0,107 ммоль, 28%) в виде желтого твердого вещества (ЖХ/МС (ESI, M+H+)=516), 3-(6-фтор-8-(метиламино)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-4-ил)-3-азабицикло[3.2.0]гептан-6-ил изопропил гидрофосфата 8 (0,055 г, 0,099 ммоль, 26%) в виде светло-желтого твердого вещества (ЖХ/МС (ESI, M+H+)=558) и 3-(6-фтор-8-(метиламино)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-4-ил)-3-азабицикло[3.2.0]гептан-6-ил диизопропилфосфата 9 (0,040 г, 0,067 ммоль, 18%) в виде белого твердого вещества (ЖХ/МС (ESI, M+H+)=600).
Пример 5d: Синтез соединения

[0170] Пролекарство, указанное выше (ЖХ/МС (ESI, M+H+)=476), получали с использованием процедур, аналогичных описанным в Примере 5c выше.
Пример 5e: Синтез соединения

ОБЩАЯ СХЕМА


N-(6-фтор-4-(6-гидрокси-3-азабицикло[3.2.0]гептан-3-ил)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-8-ил)-N-метилформамид (2)

[0171] Смесь соединения 1 (0,170 г, 0,390 ммоль) в муравьиной кислоте (1,5 мл) перемешивали в течение 45 минут при 60°C в атмосфере азота. Избыток муравьиной кислоты выпаривали при пониженном давлении, и неочищенный продукт очищали при помощи ВЭЖХ с получением ди-формильного аддукта. Этот ди-формильный аддукт медленно гидролизовали с получением моно-формильного продукта в MeCN и H2O с 5% TFA. Растворитель удаляли при пониженном давлении и неочищенный продукт очищали при помощи ВЭЖХ с получением N-(6-фтор-4-(6-гидрокси-3-азабицикло[3.2.0]гептан-3-ил)-2-((2-метилпиримидин-5-ил)окси)-9H-пиримидо[4,5-b]индол-8-ил)-N-метилформамида 2 (0,075 г, 0,162 ммоль, 42%) в виде белого твердого вещества (ЖХ/МС (ESI, M+H+)=464).
Пример 6: Определение антибактериальной эффективности
[0172] Колонии H. influenzae, E. coli, S. aureus, A.baumannii, S. pneumoniae, P. aeruginosa и B. thailandensis извлекали из чашек Петри после культивирования в течение ночи и ресуспендировали в 3 мл DPBS раствора. Поглощение считывали при 600 нм и суспензии разбавляли до OD=0,1.
[0173] Культуры бактерий добавляли к подходящей питательной среде и 98 мкл смеси высевали в колонки 1-11 96-луночного плоскодонного культурального планшета. В колонку 12 высевали только среду.
[0174]
|
[0175] 2 мкл серии разведений соединения в 100% DMSO добавляли в колонки 1-10. Планшеты встряхивали в аппарате для встряхивания планшетов в течение 1 минуты.
[0176] Смеси клеток и среды разбавляли 1000x в DPBS и 100 мкл высевали на соответствующую среду и инкубировали в течение ночи для подсчета КОЕ.
[0177] Планшеты инкубировали в течение ночи при 35°С. Планшеты с Н. influenzae и S. pneumoniae инкубировали с 5% СО2.
[0178] 10 мкл аламарового синего (Invitrogen) добавляли в планшеты и планшеты встряхивали в течение 1 минуты в аппарате для встряхивания планшетов. Планшеты инкубировали при 35°С в течение 1 часа. Планшеты считывали визуально, при этом любое изменение цвета от синего считывали как выживание.
Пример 7: Определение ингибирования hERG
[0179] Cerep автоматизированный пэтч-кламп анализ с использованием клеток К1 яичников китайского хомячка использовали для измерения hERG IC50 значений. Степень ингибирования (%) определяли путем измерения амплитуды следового тока, который индуцировали при помощи 1 сек. пробного импульса до - 40 мВ после 2 сек импульса до +20 мВ, до и после инкубации с лекарственным средством (разницу в токе нормализовали к контролю и умножали на 100, с получением процента ингибирования).
[0180] Кривые концентрация (log) - ответ подгоняли к логистическому уравнению (три параметра, предполагающие полное блокирование тока при очень высоких концентрациях испытываемого соединения) для определения значений 50% ингибирующей концентрации (IC50). Кривые зависимости концентрация-ответ для каждого соединения строили на основании процентного уменьшения амплитуды тока при использовании последовательных концентраций. В Таблицах 2 и 3 ниже представлены MIC и hERG IC50 значения для испытанных соединений.
[0181] Сравнение показывает лучшие hERG значения для -ОН содержащих аналогов, с сохранением при этом грамотрицательной активности, например, в Е. coli.











Пример 8: Проницаемость Сасо-2 клеточного монослоя
[0182] Клеточную линию Сасо-2 карциномы толстой кишки человека (Сасо-2) использовали в качестве модельной системы для кишечной эпителиальной проницаемости. Существует сходство в свойствах поглощения и проницаемости барьера между этой клеточной системой и эпителиальным слоем тонкого кишечника. См. Hidalgo I.J., et al., Gastroenterology. 1989 Mar; 96(3):736-49. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Профиль потенциальных пероральных/в.в. кандидатов показан в Таблице 4.
Пример 9: in vivo РК мыши (параллельное взятие проб)
[0183] Типичный протокол представляет собой введение одной внутривенной и пероральной (через зонд) дозы со сбором крови у каждого животного путем сердечной пункции в выбранных (6-8) точках времени (n=3 мышей на точку времени) в пробирку, содержащую соответствующий антикоагулянт. Кровь центрифугируют и полученную плазму извлекают для последующего анализа. После соответствующей подготовки образца определяют концентрации испытываемого соединения в плазме методом ЖХ-МС/МС и фармакокинетические данные (такие как площадь под кривой или AUC) профиля концентрация-время после перорального введения сравнивают с соответствующими фармакокинетическими данными после внутривенного введения. Отношение AUC после перорального введения к AUC после внутривенного введения дает пероральную биодоступность (%F), как показано для выбранных соединений в Таблице 4 ниже. Кроме того, показаны Сасо значения.
[0184] Первые два амин-содержащих аналога в Таблице 4 были указаны в заявке РСТ, которая испрашивает приоритет по предварительной патентной заявке США №61/700159, как два амин-содержащих соединения, имеющих неожиданно благоприятные hERG значения, но значения пероральной биодоступности существенно ниже по сравнению с -ОН содержащими соединениями.


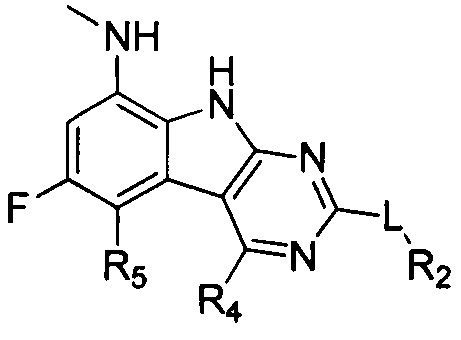
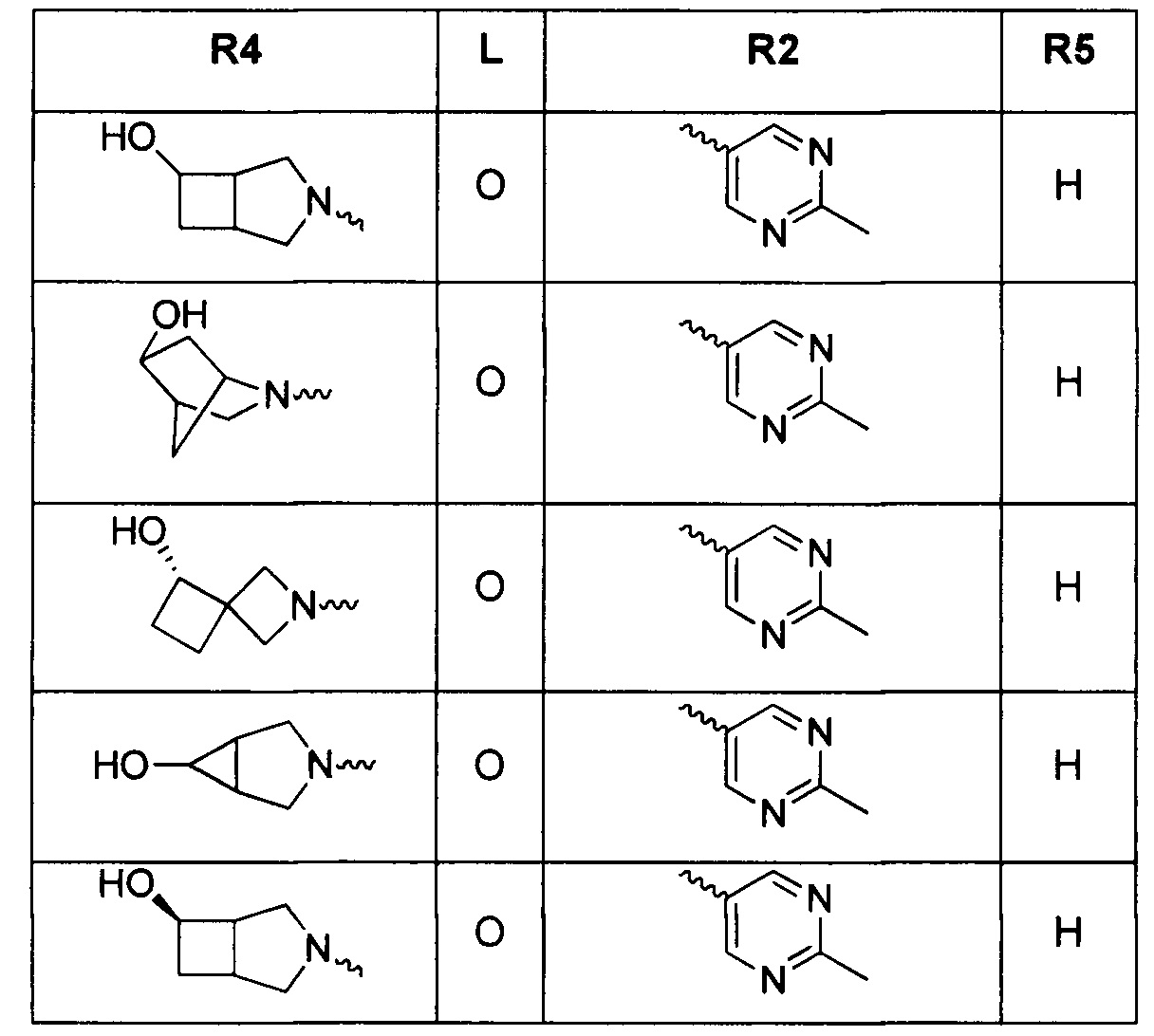
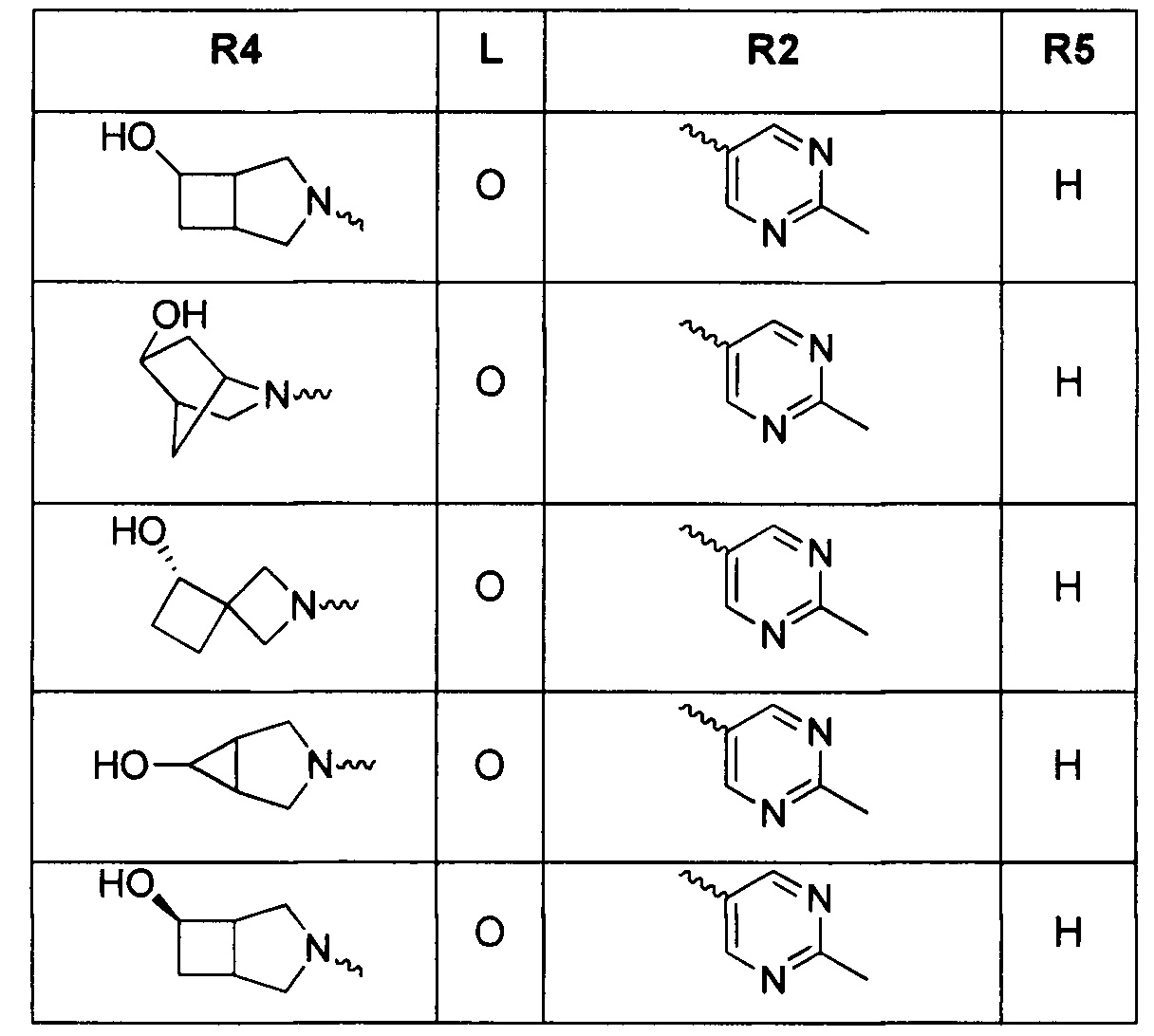
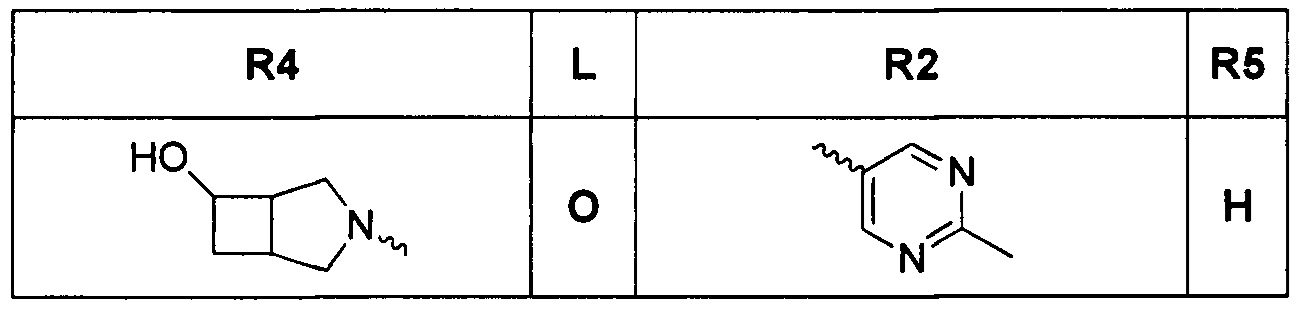
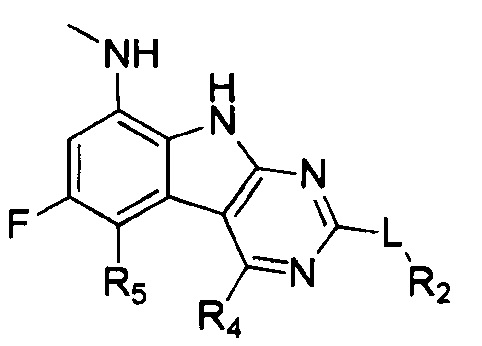
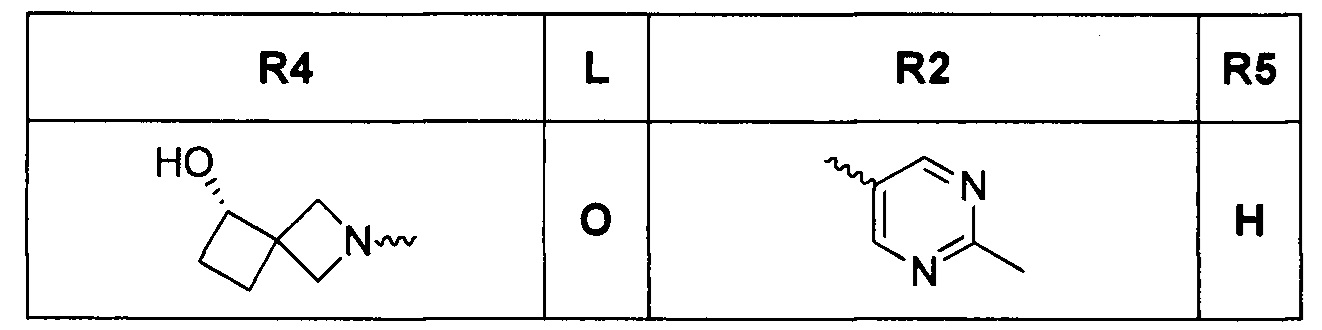
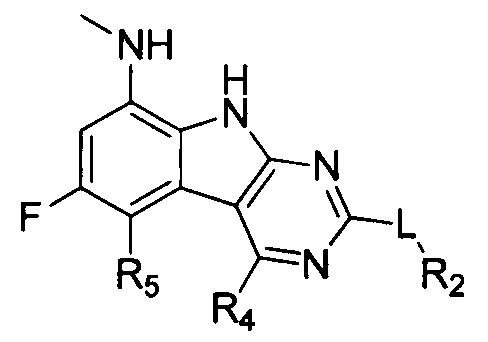
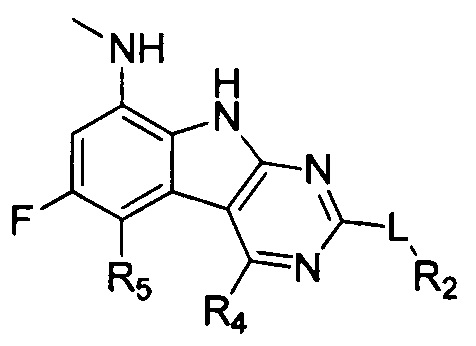
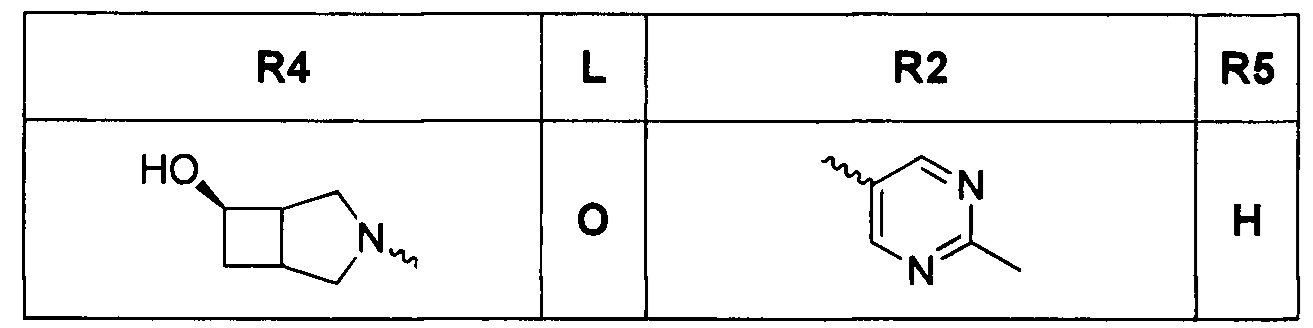
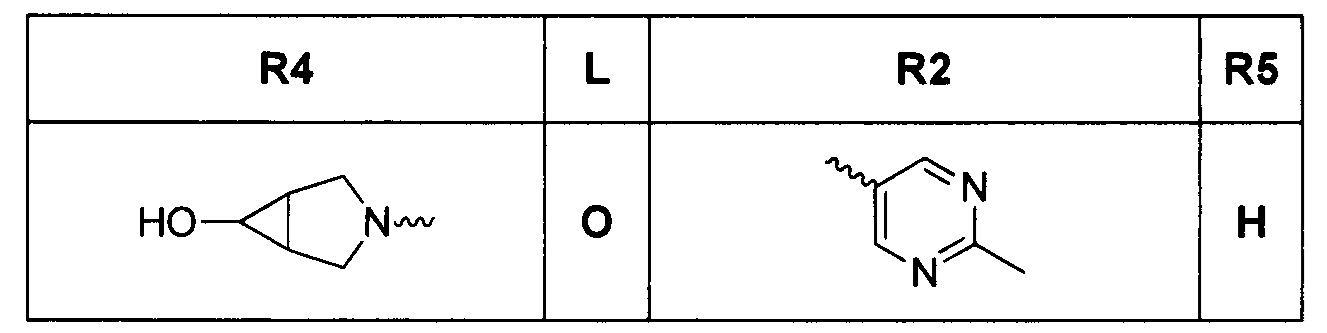
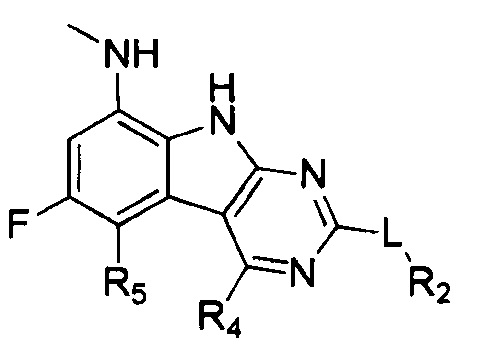
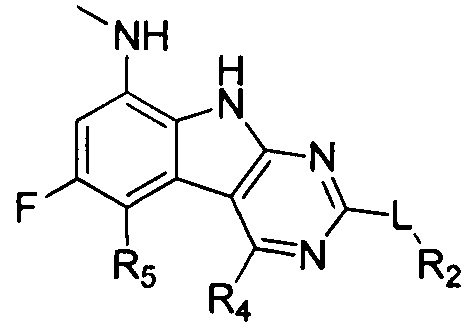
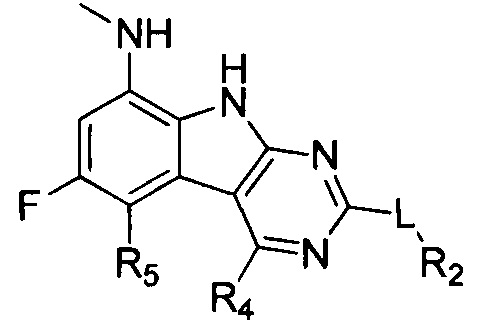
Способ общей обработки кварцевой оптики для уменьшения оптического повреждения
Способ и система для обработки оптических элементов с использованием магнитореологической чистовой обработки
Архитектура многопроходного усилителя для лазерных систем большой мощности
Способ и система конвергентного полирования
Оценка видоизменения ткани с использованием оптоволоконного устройства
Способы получения оксазолидинонов и содержащих их композиций
Оксазолидинонсодержащие димерные соединения, композиции и способы их получения и использования
Трициклические ингибиторы гиразы
Кристаллические частицы для приготовления твердых лекарственных форм для лечения бактериальных инфекций, реакционная смесь, содержащая такие частицы, и фармацевтическая композиция для лечения бактериальных инфекций
Оксазолидиноны и способ их очистки

