Результат интеллектуальной деятельности: СПОСОБЫ ПОЛУЧЕНИЯ ОКСАЗОЛИДИНОНОВ И СОДЕРЖАЩИХ ИХ КОМПОЗИЦИЙ
Вид РИД
Изобретение
ОПИСАНИЕ УРОВНЯ ТЕХНИКИ
[0001] Оксазолидиноны находят широкое применение в качестве фармацевтических агентов для лечения и профилактики таких медицинских заболеваний как, бактериальные инфекции и атеросклероз. Ценность этих соединений стимулирует поиск эффективных способов их синтеза, таких как описанные в US 20070049759 реакции перекрестного сочетания на медь содержащих катализаторах.
US 20070155798, который приводится здесь для ссылки во всей его целостности, раскрывает сильнодействующие антибактериальные оксазолидиноны, содержащие замещенные пиридил фенильные фрагменты. Первоначально эти фрагменты были введены синтетическим путем по реакции сочетания в присутствии соединений олова, однако, из-за токсичности последних их применение в фармацевтическом синтезе является нежелательными. Таким образом, существует необходимость в синтетических способах получения замещенных (пиридинил)фенил оксазолидинонов без применения олово содержащих реагентов.
Область применения изобретения
[0002] Новые способы являются полезными для получения оксазолидинон содержащих соединений.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
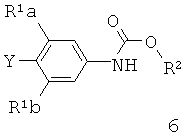
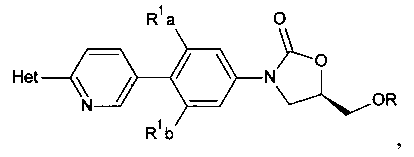
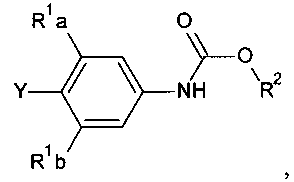
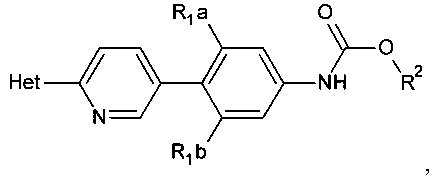
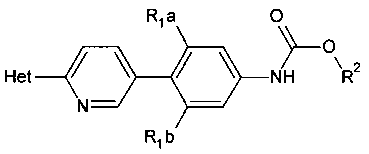
[0003] Способ синтеза соединений структурной формулы
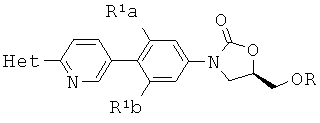
где
R представляет собой Н,
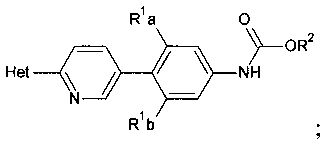
R1a и R1b независимо выбраны из группы, состоящей из Н и F, при условии, что, по меньшей мере, один из R1a и R1b представляет собой F,
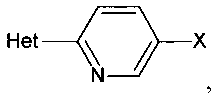
Het представляет собой необязательно замещенный пяти- или шестичленный гетероцикл, содержащий, по меньшей мере, один из атомов N, О или S,
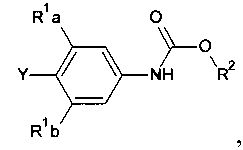
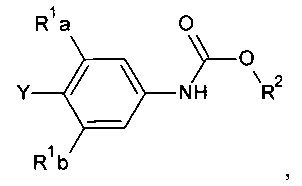
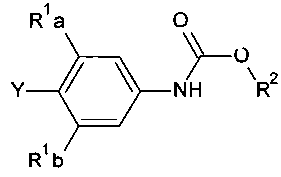
способ включает обработку соединения, имеющего структурную формулу
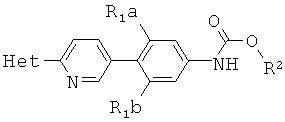
где R2 выбирают из группы, состоящей из необязательно замещенного бензила и необязательно замещенного C1-С6 алкила, сильным основанием или литий органической солью с последующим добавлением глицидил бутирата к полученному аниону в условиях для получения
.
[0004] В некоторых аспектах стадия обработки осуществляется в присутствии соединений, облегчающих протекание реакции, например, 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинона.
[0005] В некоторых вариантах осуществления изобретения, способ включает дополнительную стадию взаимодействия
с POCl3, POCl(OBn)2, или P(N-iPr2)(O-tBu)2 в условиях для получения
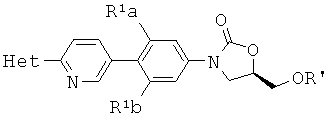
где R' представляет собой PO(OH)2.
[0006] Способ может также включать обработку соединения структурной формулы
где R' представляет собой РО(ОН)2 основанием в условиях для получения соединения структурной формулы
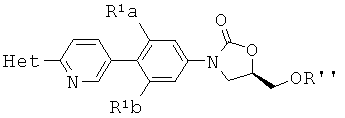
где R'' представляет собой фармацевтически приемлемую соль РО(ОН)2. В некоторых аспектах основание представляет собой натрий содержащее основание. В некоторых аспектах R'' представляет собой PO3Na2.
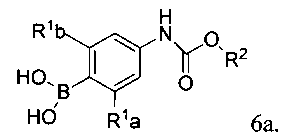
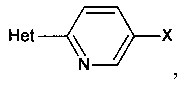
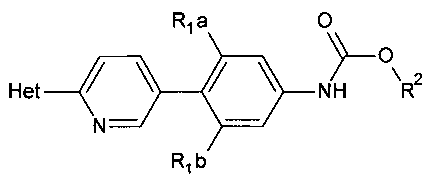
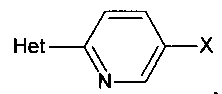
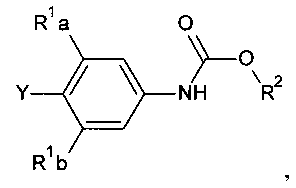
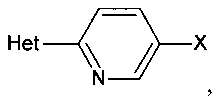
[0007] Отдельный способ получения интермедиата или дополнительная стадия, осуществляемая до стадий, указанных выше, включает реакцию сочетания первого интермедиата структурной формулы
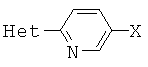
где Х представляет собой уходящую группу, выбранную из Cl, Br, I или трифторметансульфоната, со вторым интермедиатом структурной формулы
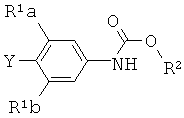
где Y выбирают из группы, состоящей из ZnCl, BF3 и BR3R4, где R3 и R4 независимо выбирают из группы, состоящей из ОН и необязательно замещенных C1-С6 моно и двухатомных спиртов, и где R3 и R4 вместе могут образовывать кольцо в условиях для получения соединения структурной формулы
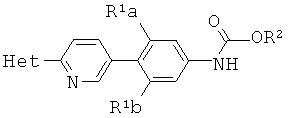 .
.
[0008] В некоторых аспектах реакция сочетания осуществляется в присутствии палладиевого комплекса, такого как фосфиновый лиганд, связанный с палладием, например, дихлорбис(трифенилфосфин)палладий(II), тетракис(трифенилфосфин)палладий(0) или Pd2(dba)3 (трис(дибензилиденацетон)дипалладий(0).
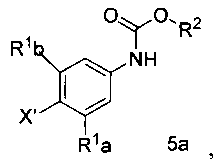
[0009] Отдельный способ получения интермедиата или дополнительная стадия, осуществляемая до стадии сочетания, указанной выше, включает
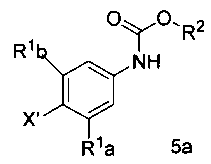
а) обработку арилгалида структурной формулы 5а
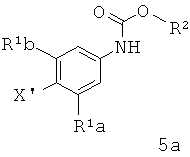
где X1 представляет собой уходящую группу, сильным основанием, таким как н-бутил литий и затем взаимодействие полученного аниона с эфиром триалкилборной кислоты в условиях для получения
 ; или
; или
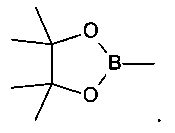
б) обработку арилгалида структурной формулы 5a палладиевым катализатором, таким как PdCl2(dppf)2 бис((дифенилфосфино)ферроцен-палладий дихлорид и дипинаколатным эфиром дибороновой кислоты в условиях для получения
.
[0010] В некоторых вариантах осуществления изобретения Y выбирают из группы, состоящей из В(ОН)2, BF3 и
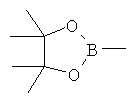 .
.
[0011] В некоторых вариантах осуществления изобретения Het выбирают из группы, состоящей из необязательно замещенного пиррола, фурана, пиперазина, пиперидина, имидазола, 1,2,4-триазола, 1,2,3-триазола, тетразола, пиразола, пирролидина, оксазола, изоксазола, оксадиазола, пиридина, пиримидина, тиазола или пиразина, такого как необязательно замещенная тетразольная группа, например, 2-метил-тетразол-5-ил.
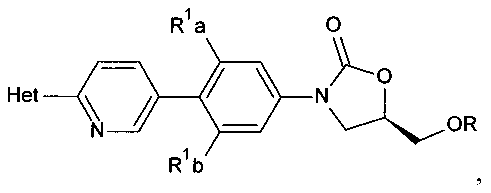
[0012] В некоторых вариантах осуществления изобретения способ также включает обработку соединения структурной формулы
глицидиловым эфиром, таким как глицидил бутират.В некоторых аспектах глицидиловый эфир имеет R стереохимию, такую как R-(-)-глицидил бутират. Эта стадия обработки может быть осуществлена в присутствии гексаметилдисилазида.
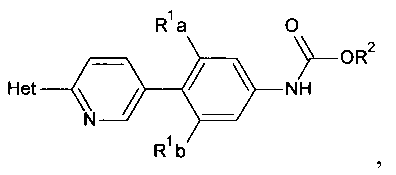
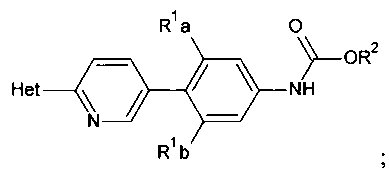
[0013] Соединения, полученные способом, описанным здесь, включают
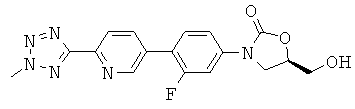 ,
,
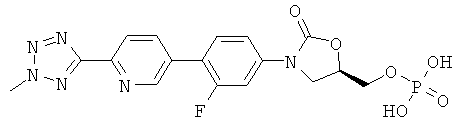 ,
,
и
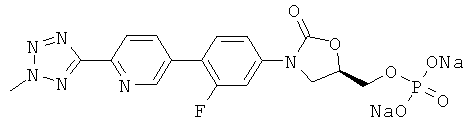
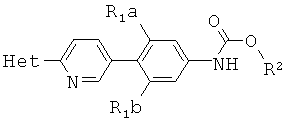
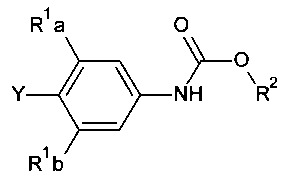
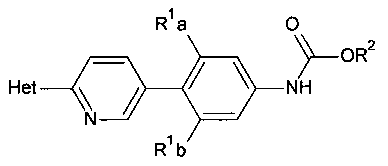
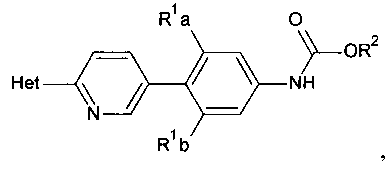
[0014] В некоторых вариантах осуществления изобретения соединение имеет структурную формулу:
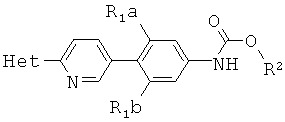
где;
R1a и R1b независимо выбраны из группы, состоящей из Н и F, при условии, что, по меньшей мере, один из R1a и R1b представляет собой F,
R2 выбирают из группы, состоящей из необязательно замещенного бензила и необязательно замещенного C1-С6 алкила, и
Het представляет собой необязательно замещенный пяти- или шестичленный гетероцикл, содержащий, по меньшей мере, один из атомов N, О или S.
[0015] В некоторых вариантах осуществления изобретения соединение имеет следующую структурную формулу:
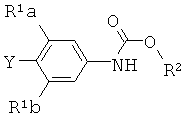
где
R1a и R1b независимо выбраны из группы, состоящей из Н и F, при условии, что, по меньшей мере, один из R1a и R1b представляет собой F,
R2 выбирают из группы, состоящей из необязательно замещенного бензила и необязательно замещенного C1-С6 алкила, и
Y выбирают из группы, состоящей из ZnCl, BF3 и BR3R4, где R3 и R4 независимо выбирают из группы, состоящей из ОН и необязательно замещенных C1-С6 моно и двухатомных спиртов, и где R3 и R4 вместе могут образовывать кольцо.
[0016] В некоторых аспектах композиция содержит соединение, описанное здесь и полученное в соответствии со способом, описанным здесь, и димер, имеющий следующую структурную формулу или фармацевтически приемлемую соль этого димера.
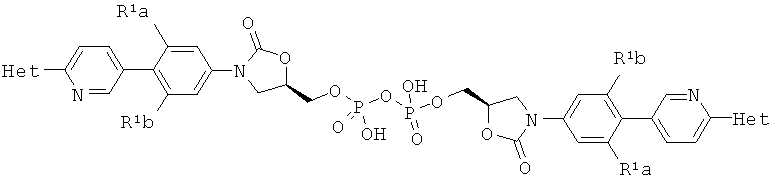
где R1a и R1b являются независимо выбранными из Н и F, при условии, что, по меньшей мере, один из R1a и R1b представляет собой F,
Het представляет собой необязательно замещенный пяти- или шестичленный гетероцикл, содержащий, по меньшей мере, один из атомов N, О или S.
[0017] В некоторых аспектах R1a представляет собой F и R1b представляет собой Н и Het представляет собой 2-метил-тетразол-5-ил.
[0018] Также в вариантах осуществления изобретения композиция содержит соединение, которое описано здесь и получено в соответствии с процессом, представленным выше, причем в композиции отсутствуют примеси олова.
ДЕТАЛЬНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ.
[0019] Представлены способы синтеза замещенных (пиридил)фенилоксазолидинонов
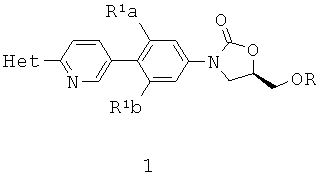
где Het представляет собой необязательно замещенный пяти- или шестичленный гетероцикл, содержащий, по меньшей мере, один из атомов N, О или S, например, необязательно замещеные фрагменты тетразолила, оксазолила, триазолила, оксадиазолила, тиазолила и изоксазолила. В некоторых аспектах Het представляет собой необязательно замещенный тетразолил, например, 2-метил-тетразол-5-ил.
R1a и R1b независимо выбирают из Н и F, при условии, что, по меньшей мере, один из R1a и R1b представляет собой F,
R выбирают из Н, PO(OH)2 и фармацевтически приемлемые соли РО(ОН)2.
[0020] По меньшей мере, если не указано иное, технические термины принимают их обычное значение, в частности представленное в Словаре Научных и технических Терминов McGraw-Hill, 6 издание (McGraw-Hill Dictionary of Scientific and Technical Terms, 6th edition).
[0021] В некоторых вариантах осуществления изобретения способы включают получение замещенных N-(пиридинил)арилоксазолидинонов по следующей схеме:
[0022] Схема 1
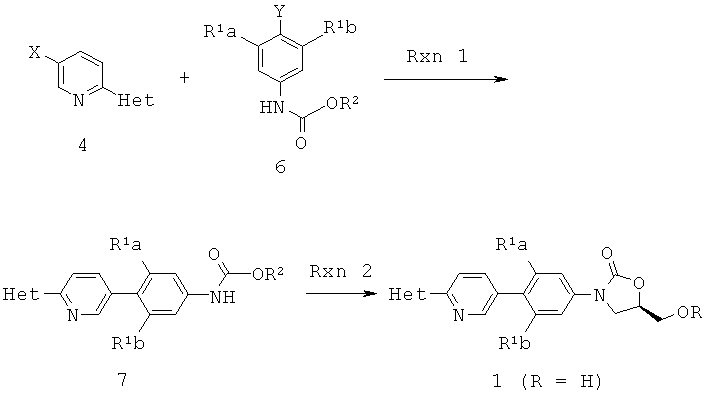
[0023] В Схеме 1, первый интермедиат (4) взаимодействует в Rxn 1 со вторым интермедиатом (6) с получением продукта соединения (7), который затем обрабатывают в Rxn 2 глицидиловым эфиром с получением соединения (1).
Схема 2
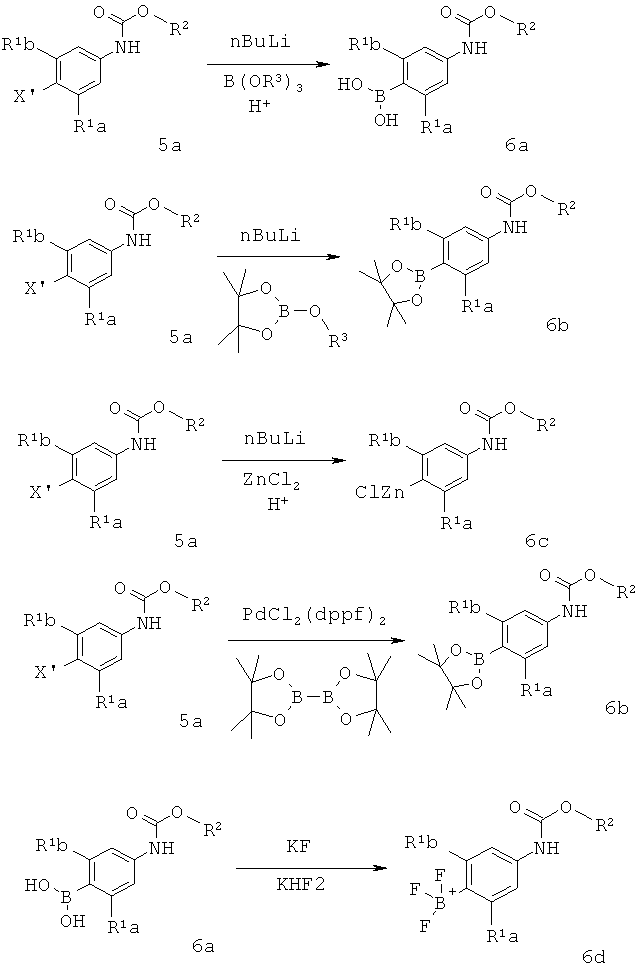
[0024] В Схеме 2, интемедиат 6 может быть получен обработкой интермедиата 5a 2 эквивалентами сильного основания, например, C1-С6 алкил лития, в частности, н-бутил лития или т-бутил лития, затем следует добавление соответствующего электрофила, такого как ZnCl2 или В(OR)3, например, C1-С6 триалкоксибороната, в частности триизопропил бороната. Обработка водой полученной реакционной смеси, где электрофил представляет собой триалкоксиборонатный эфир, дает бороновую кислоту 6a. Если дианион 5а обработать циклическим боронатным эфиром, то эфир циклической бороновой кислоты 6b может быть выделен. Если электрофил представляет собой ZnCl2, то цинковый реагент 6 с может быть выделен. Альтернативно бороновые кислоты могут получены по процедуре борирования Мияуры (Miyaura Top. Curr. Chem. 2002, 219, 11-59). По этой реакции диэфир дибороновой кислоты, такой как дипинаколат эфир дибороновой кислоты присоединяется к арилгалиду (5а), используя палладиевый катализатор. Полученный эфир бороновой кислоты 6b может быть гидролизован водным раствором кислоты с получением бороновой кислоты 6а. Также производное трифторбората 6d может быть получено из бороновой кислоты 6а обработкой KF и/или KHF2.
[0025] В приведенных выше схемах Х представляет собой уходящую группу. В некоторых вариантах осуществления изобретения Х выбирают из Cl, Br, I и трифторметан сульфоната.
[0026] X1 представляет собой уходящую группу. В некоторых вариантах осуществления изобретения, Х представляет собой галоген, такой как Cl, Br или I.
[0027] Het представляет собой необязательно замещенный пяти- или шестичленный гетероцикл, содержащий, по меньшей мере, один из атомов N, О или S, включая необязательно замещенный пиррол, фуран, пиперазин, пиперидин, имидазол, 1,2,4-триазол, тетразол, пиразол, пирролидин, оксазол, изоксазол, оксадиазол, пиридин, пиримидин, тиазол или пиразин. В некоторых аспектах Het представляет собой необязательно замещенный тетразолил или 2-метил-тетразол-5-ил. В некоторых вариантах осуществления изобретения Het является незамещенным или имеет 1 или 2 заместителя.
[0028] R1a и R1b независимо выбирают из Н и F, при условии, что, по меньшей мере, один из R1a и R1b представляет собой F;
Y выбирают из ZnCl, BF3 и BR3R4, где R3 и R4 независимо выбирают из ОН и необязательно замещенных C1-С6 моно и двухатомных спиртов и где R3 и R4 вместе могут образовать кольцо. В некоторых вариантах осуществления изобретения, Y представляет собой В(ОН)2 или пинаколатоборат, а именно, ,
такой как В(ОН)2. C1-С6 моно и двухатомные спирты могут быть необязательно замещены C1-C4 алкилом. Реакция Негиши (Negishi) может быть осуществлена для получения соединений, где Y представляет собой ZnCl (Negishi: Chem. Ind. 1988, 33, 381-407).
[0029] В некоторых вариантах осуществления Het может быть незамещенным или необязательно замещенными одним или более заместителями, например, независимо выбранными из группы, состоящей из галогена, гидрокси, амино, С1-4 алкиламино, ди(С1-4 алкил)амино, циано, нитро, C1-4 алкила, С1-4 алкокси, C1-4 ацила, C1-4 тиоалкила, C1-4 тиооксоалкила, замещенного галогеном С1-4 алкила и замещенного галогеном С1-4 алкокси.
[0030] Также в Схеме 1, R2 представляет собой необязательно замещенный бензил или необязательно замещенный C1-С6 алкил. В некоторых вариантах осуществления изобретения, бензил и C1-С6 алкил являются незамещенными или независимо друг от друга необязательно замещенными галогеном или алкокси, таким как C1-C4 алкилокси. В некоторых вариантах осуществления изобретения R2 представляет собой бензил и R представляет собой Н.
[0031] Подходящие катализаторы для реакции кросс-сочетания Rxn 1 представляют собой палладиевые комплексы, например, палладиево фосфиновые комплексы или дихлорбис(трифенилфосфин)палладий(II), тетракис(трифенилфосфин)палладий(0) и полученные in situ из Pd2(dba)3 (dba = бензилиденацетон) в присутствии РСу3 (трициклогексилфосфина). Соотношение Pd комплекса к субстрату не является критическим для осуществления реакции, но приблизительно 1 молярный % комплекса (по отношению или к 4, или к 6) является приемлемым.
[0032] Циклизация с получением оксазолидинонового кольца осуществляется в Rxn 2 путем обработки 7 сильным основанием, например, гексаметилдисилазидом лития или литий органической солью, например, н-бутил литием в присутствии 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинона (DMPU), затем следует добавление глицидилового эфира, такого как, R-(-)-глицидиловый эфир, в частности бутирата с получением соединения 1 (R = Н). В одном варианте осуществления изобретения используется гексаметилдисилазид лития в качестве основания и тетрагидрофуран (ТГФ) в качестве растворителя с DMPU, присутствующим для облегчения реакции, при температуре приблизительно между 0°С и 30°С, и стехиометрии 7 к глициловому эфиру приблизительно 1:1 на молярной основе.
[0033] Если желательно, соединение 1 (R = Н) может в дальнейшем быть превращено в дигидрофосфат, например, обработкой с POCl3, в соответствии с хорошо известными методами. Например, соединение 1 (R = Н) может быть обработано POCl3, затем следует гашение водой или двустадийный процесс, использующий защищенную форму оксихлорида фосфора, такого как: POCl(OBn)2, где на первой стадии получается фосфатный триэфир, а на второй стадии снимается защитная группа (например, H2/Pd-C для снятия групп бензилового эфира). Альтернативно, 5-гидроксиметил-оксазолидинон может быть обработан P(N-iPr2)(O-tBu)2, затем следует окисление окисляющим реагентом, таким как mCPBA (м-хлорпероксибензойная кислота) с последующей обработкой основанием или водным раствором кислоты для снятия групп трет-бутилового эфира.
[0034] Полученный дигидрофосфат 1 (R = PO(OH)2) может быть в дальнейшем превращен в фармацевтически приемлемую соль, такую как динатриевая соль соединения 1 (R = PO(O)2 2Na) по реакции с NaOMe или другим подходящим натрий содержащим основанием.
[0035] Специалисты в области медицинской химии понимают, что термин «фармацевтически приемлемая соль» относится к солям, образованным соответствующими биологически совместимыми катионами и/или анионами. Такие катионы включают металлические элементы, такие как натрий, литий, калий, магний, алюминий, кальций, цинк и четвертичные катионы органических азотных оснований, например, N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, N-метилглюкамин и соли прокаина. Такие анионы включают неорганические кислоты, такие как соляная, бромоводородная, серная, фосфорная, азотная, хлорная, фумаровая, уксусная, пропионовая, янтарная, гликолевая, муравьиная, молочная, малеиновая, винная, лимонная, пальмитиновая, малоновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, фумаровая, толуолсульфокислота, метансульфокислота, нафтален-2-сульфокислота, бензолсульфокислота, оксинафтойная, иодистоводородная, яблочная, стериновая, таниновая и подобные кислоты.
[0036] Оксазолидиноны, полученные способом, описанным здесь, отличаются от оксазолидинонов, синтезированных в соответствии со способом, представленным в US 20070155798. Оксазолидиноны, полученные в соответствии с настоящим процессом, не содержат примесей, поскольку олово-содержащие реагенты не используются. Дополнительно, в некоторых вариантах осуществления изобретения, происходит образование димера в качестве побочного продукта - примеси, например, когда оксихлорид фосфора (POCl3) используется для превращения гидроксила в дигидрофосфат. В частности молекула TR-701 взаимодействует с молекулой фосфатного эфира, содержащего, по меньшей мере, одну Р-Cl связь для получения димера, имеющего, например, следующую формулу.
[0037] Количество примесей определяется некоторым пределом обнаружения, который меньше, чем 10% от веса композиции, и в некоторых случаях меньше 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2% или 1%, например, меньше 0.1% или 0.05%. При этом в некоторых вариантах осуществления изобретения композиции содержат оксазолидинон, полученный в соответствии с процессом, представленным здесь, и димер. В некоторых вариантах осуществления изобретения композиции содержат оксазолидинон без каких-либо олово- содержащих примесей.
[0038] Оксазолидиноны, полученные в соответствии с методами, представленными здесь, являются полезными в качестве лекарственных средств и в частности для задержки роста бактерий, как детально раскрыто в US 20070155798, который представлен здесь для ссылки во всей его целостности.
[0039] Термины «приблизительно», «около» и «в основном», как используются здесь, представляют количество, близкое к заявленному количеству, при котором все еще осуществляется желаемая функция или достигается желаемый результат. Например, термины «приблизительно», «около» и «в основном» могут относиться к количеству, которое меньше 10%, меньше 5%, меньше 1%, меньше 0.1%, и меньше 0.01% заявленного количества.
Примеры
[0040] Практическое воплощение способа по изобретению иллюстрируется следующим примером, не ограничивающим объем притязаний.
Экспериментальные и Аналитические Данные
[0041] Реагенты были приобретены у коммерческих источников и были использованы при получении. Спектр протонного магнитного резонанса был получен на спектрометре Bruker AVANCE 300 при 300 МГц или спектрометре AVANCE 500 при 500 МГц с тетраметилсиланом в качестве источника для внутреннего сравнения. Спектр углеродного ядерного магнитного резонанса был получен на спектрометре Broker AVANCE 500 при 125 МГц для сравнения выбран пик растворителя. Спектр фосфорного ядерного магнитного резонанса был получен на спектрометре Bruker AVANCE 500 при 202 МГц с фосфорной кислотой для сравнения. Спектр ядерного магнитного резонанса фтора был получен на спектрометре Bruker AVANCE 300 при 282 МГц. Масс-спектр был получен на спектрометре Finnigan AQA с ионизацией электрораспылением. Тонкослойная хроматография (ТСХ) была осуществлена, используя Whatman No. 4500-101 (Diamond No. MK.6F силикагель 60 Å) пластины. Проявление ТСХ пластин было осуществлено, используя УФ свет (254 нм) или окрашивание перманганатом калия. ВЭЖХ анализ был осуществлен на Varian Prostar HPLC, снабженным Waters SunFire С18 колонкой (150×4.60 мм, 3.5 мкм) или Waters XBridge С18 колонкой (75 мм × 4.6 мм × 2.5 мкм), используя способы, представленные ниже, с детектором на указанной длине волны.
[0042] Способ A (Waters SunFire С18 Колонка)
|
А = вода с 0.05% (по объему) трифторуксусной кислотой
В = ацетонитрил с 0.05% (по объему) трифторуксусной кислотой
Длина волны = 254 нм
[0043] Способ В (Waters XBridge С18 Колонка)
|
А = 87% 25 мМ раствора бикарбоната аммония в воде/13% ацетонитрил
В = ацетонитрил
Длина волны = 254 нм
[0044] Способ С (Waters SunFire С18 Колонка)
|
А = вода с 0.05% (по объему) трифторуксусной кислотой
В = ацетонитрил с 0.05% (по объему) трифторуксусной кислотой
Длина волны = 240 нм.
Пример 1: Получение 5-бром-2-(2Н-тетразол-5-ил)пиридина, 3
[0045] В 22-литровую, трехгорлую, круглодонную колбу, снабженную верхней мешалкой, входным/выходным отверстием для азота, термопарой и нагревательным кожухом был загружены при перемешивании 5-бром-2-цианопиридин (799 г, 4.37 моль, 1 эквивалент), N,N-диметилформамид (6.4 л, 8 объемов), хлорид аммония (350.3 г, 6.55 моль, 1.5 эквивалента) и азид натрия (425.7 г, 6.55 моль, 1.5 эквивалента). Внутренняя температура в реакторе была установлена на уровне 85°С (заданная температура 90°С). Заданная температура была достигнута 45 минут спустя, затем реакционная смесь самопроизвольно нагрелась до 94°С за 40 минут. Спустя 1 час реакция завершилась, ВЭЖХ анализ показал полное потребление исходных веществ с содержанием 76.7% (AUC - площадь под кривой) тетразол аммониевой соли. Смесь была охлаждена и отфильтрована при комнатной температуре. Реактор и влажный осадок были промыты 2-пропанолом (3.2 л, 4 объема) и высушены под высоким вакуумом при комнатной температуре с получением тетразол аммонийной соли в виде белого твердого осадка (847.9 г, 80% выход, 89.9% AUC). Дифференциальный сканирующий калориметрический эксперимент был проведен на тетразол аммониевой соли для оценки ее термической стабильности. Соль расплавилась приблизительно при 228°С, а энергетический распад произошел приблизительно при 270°С.
Пример 2: Получение 5-бром-2-(2-метил-2Н-тетразол-5-ил)пиридина, 4 (Х = Br)
[0046] В 22-литровую, четырехгорлую, круглодонную колбу, снабженную верхней мешалкой, входным/выходным отверстием для азота, термопарой, и помещенную на ледяную/солевую баню были загружены при перемешивании тетразол аммониевая соль (835.0 г, 3.44 моль, 1 эквивалент), тетрагидрофуран (7.5 л, 9 объемов), N,N-диметилформамид (2.5 л, 3 объема) и порошок гидроксида натрия (343.5 г, 8.59 моль, 2.5 эквивалента). Реактор был оставлен до достижения значения внутренней температуры в 12°С, затем иодметан (1.22 кг, 8.59 моль, 2.5 эквивалента) был по каплям добавлен за 50 минут, с поддержанием температуры реакции ниже 20°С. Через 20 минут после добавления, вследствие быстрого роста температуры, добавление было прекращено, и реакция продолжила самопроизвольно нагреваться на 15-20°С за десять минут. Добавление остального количества было завершено при постоянной температуре (18°С). После завершения добавления, ледяная баня с солью была убрана, и реактор был снабжен водным конденсатором и нагревательным кожухом. Внутренняя температура реактора была доведена до 40°С, однако, реакционная смесь продолжила самопроизвольно нагреваться до 48°С. 6 часов спустя ВЭЖХ анализ показал полное потребление исходного материала. Реакционная смесь была охлаждена до комнатной температуры в течение ночи для удобства. ТГФ был отогнан дистилляцией и вода (8.35 л, 10 объемов) была добавлена в реактор. Смесь была перемешана в течение 30 минут и отфильтрована вакуумной фильтрацией, реактор и остаток на фильтре были промыты водой (4.2 л, 5 объемов) с получением неочищенного соединения 4/N1 изомерной смеси в виде твердого вещества персикового цвета (500.7 г, 61% выход, 3.85:1 4: N1).
[0047] Твердые вещества (500.7 г) были растворены в CH2Cl2 (2.5 л, 5 объемов), затем 6н водная HCl (7.5 л, 15 объемов) была добавлена. Двухфазная смесь была перемешана, и слои разделились. В этот момент желаемый продукт находился в водном слое с HCl. Слой CH2Cl2 был промыт 6н водной HCl (4.5 л, 3×3 объема), до тех пор, пока содержание соединения 4 не достигло значения <5% AUC в ВЭЖХ анализе. Объединенные экстракты 6н HCl были перемещены в реактор, и рН было доведено до 10.6 с 50% водным NaOH (~3.2 л), с одновременным сохранением внутренней температуры ниже 40°С. Твердые вещества были выделены вакуумной фильтрацией, реактор и осадок на фильтре были промыты водой (1 л, 2 объема) с получением неочищенного соединения 4 в виде желтого/оранжевого твердого вещества (322.4 г, извлечение 64%, выход 39%, 93.5% AUC 4, 4.1% AUC N-1 изомер), как подтвердилось ВЭЖХ и 1Н ЯМР анализом.
[0048] Неочищенное соединение 4 было в дальнейшем очищено изопропилацетатной (IPAc) суспензией (1.61 л, 5 объемов) при 50°С в течение 1 часа. При охлаждении до комнатной температуры, твердые вещества были отфильтрованы, и осадок на фильтре был промыт дополнительным количеством IPAc (500 мл, 1.6 объемов) с получением очищенного соединения 4 в виде белого/желтого твердого вещества (275.5 г, извлечение 85%, выход 33%, 98.2% AUC), что подтверждается ВЭЖХ и 1Н ЯМР анализом. Дифференциальная сканирующая калориметрия соединения 4 показала распад с выделением тепла приблизительно при 245°С.
Пример 3: Получение бензил (4-бром-3-фторфенил)карбамата, 5
[0049] В 12-литровую, трехгорлую, круглодонную колбу, снабженную верхней мешалкой, входным/выходным отверстием для азота, капельной воронкой и термопарой были загружены 4-бром-3-фторанилин (800.0 г, 4.21 моль, Matrix lot # Q13H), ТГФ (6.4 л, 8 объемов) и твердый бикарбонат натрия (530.5 г, 6.32 моль, 1.5 экв). В капельную воронку был загружен бензил хлорформиат (861.9 г, 5.05 моль, 1.2 экв), который по каплям был добавлен в реактор за 70 минут. Температура реактора поддерживалась ниже 20°С с помощью ледяной бани. Смесь была выдержана 1 час, ВЭЖХ анализ показал, что реакция завершена. Реакционная смесь была перенесена в 22-литровую колбу и смесь была разбавлена водой (6.4 л, 8 объемов). Двухфазная смесь была нагрета до 50°С и выдержана при этой температуре в течение 16 часов для гашения избытка бензил хлорформата. Смесь была перенесена горячей в разделительную воронку для удаления нижнего водного слоя. Наблюдалось образование на поверхности твердых частиц, которые переходили в водный слой. ТГФ слой был отфильтрован через фильтровальную бумагу Whatman #1 для удаления некоторых частиц, и смесь была перенесена назад в 22-литровую колбу, приспособленную для дистилляции. Гептан был добавлен порциями и перегнан для удаления ТГФ. (Обратите внимание, что лучше отогнать некоторое количество ТГФ до добавления гептана). Всего 26.5 л гептана было добавлено, и было собрано 25 л дистиллята. В этот момент температура колбы достигла 97.7°С и дистиллят, полученный далее, содержал 0.9% ТГФ по 1Н ЯМР анализу. Смесь была охлаждена до комнатной температуры и вязкая белая суспензия была отфильтрована. Осадок на фильтре был промыт гептаном (4 л). Продукт был высушен в вакуумной печи при 40°С с получением 1257.0 г промежуточного соединения 5 (92% выход). ВЭЖХ анализ показал содержание 98.3% (AUC).
Пример 4: Получение 4-(бензилоксикарбониламино)-2-фторфенилбороновой кислоты 6 (R1a = F, R1b = Н, R2 = Bz, Y = В(ОН)2)
[0050] 22-литровая, трехгорлая круглодонная колба снабжена верхней мешалкой, температурным датчиком, 2-л капельной воронкой и переходником для подачи азота. В колбу был загружен интермедиат 5 (1.00 кг, 3.08 моль, AMRI lot # CAR-L-18(3)), ТГФ (10 л, 10 объемов) и триизопропил борат (638.2 г, 3.39 моль, 1.1 экв). Смесь была перемешана и охлаждалась до -72°С в бане сухой лед/ацетон. В капельную воронку был по частям загружен 2.5 М н-бутиллитий (2.59 л, 6.48 моль, 2.1 экв), который был по каплям добавлен к реакционной смеси приблизительно за 2 часа. Максимальная температура при добавлении составляла -65°С. Ход реакции отслеживался с помощью ВЭЖХ анализа. Ацетон был удален из охлаждающей бани, и реакция была закалена 20% водным раствором хлорида аммония (5.5 л), что привело к повышению температуры реакции до -1°С. Фазы были разделены, и слой ТГФ был выпарен досуха. Неочищенный продукт был повторно суспендирован в смеси 3:2 этанол/вода (10 л, 10 объемов) при комнатной температуре в течение 1 часа. Смесь была отфильтрована, и осадок на фильтре был промыт смесью 3:2 этанол/вода (2×2 л). Продукт был высушен в вакуумной печи при комнатной температуре с получением 592.8 г интермедиата 6 (66% выход), что составляло 89.8% (AUC) по ВЭЖХ анализу (Способ А). После проведения 19F ЯМР и ВЭЖХ анализа при 240 нм оказалось, что полученный материал содержит примесь (Способ С).
[0051] Повторное суспендирование полученной смеси в 2.5 объемах CH2Cl2 вместо смеси 3:2 этанол/вода, позволило избавиться от примеси дез-бромо побочного продукта, обнаруженной ранее по 19F ЯМР спекту и ВЭЖХ при 240 нм.
Пример 5: Получение бензил (4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)карбамата. 7 (Het = 2-метилтетразол-5-ил, R1a = F, R1b = H, R2=Bz) (Ref.: JAS-G-96) (Ref.: CAR-L-93, DUG-AF-202)
[0052] В 5-литровую, трехгорлую круглодонную колбу был загружено соединение 4 (200.0 г, 0.833 моль), за которым последовало добавление 1,4-диоксана (3 л, 15 объемов). Неочищенный продукт 6 (361.2 г, 1.249 моль, 1.5 эквивалента), Pd2(dba)3 (11.44 г, 0.0125 г, 0.015 эквивалента) и РСу3 (трициклогексилфосфин) (7.0 г, 0.025 моль, 0.03 эквивалента) был загружен и дегазирован азотом в течение 30 минут. Раствор К2СО3 (195.7 г, 1.7 эквивалента) в воде (800 мл, 4 объема) был добавлен, реакционная смесь нагрелась до 70°С. Реакция завершилась 1 час спустя с 0.5% (площадь под кривой) остатком соединения 4. Реакционная смесь была охлаждена до 50°С и Darco G-60 (40 г, 0.2 по весу) был добавлен, смесь перемешивалась в течение 30 минут. Целит 545 (40 г, 0.2 по весу) был загружен, и затем реакционная смесь была отфильтрована через Целит 545 (100 г, 0.5 по весу), увлажненный водой (300 мл). Горячая фильтрация через Целит вызвала осаждение продукта. Тетрагидрофуран (1.2 л, 6 объемов) и солевой раствор (600 мл, 3 объема) были добавлены, и продукт был повторно растворен при комнатной температуре. Распределение фаз было осуществлено аккуратно (Vmax = 28 объемов). Диоксан был сконцентрирован и этанол (1 л, 5 объемов) был добавлен и сконцентрирован. Затем продукт был повторно суспендирован в смеси этанол: вода (4:1, 2 л, 10 объемов) при 70°С, охлажден до комнатной температуры за 3 часа, отфильтрован и промыт этанолом (2×400 мл). Соединение 7 было выделено с 87% выходом (292.6 г) с чистотой 97.7% (AUC) ВЭЖХ анализом. 1Н ЯМР и 19F ЯМР показали присутствие одного соединения. Pd анализ показал, что 135 мд Pd присутствовало в продукте.
[0053] Интермедиат 7 был перекристаллизован из этилацетата для снижения уровня палладия. Интермедиат 7 (130 г) и этилацетат (3.9 л, 30 объемов) были загружены 5-литровую, трехгорлую круглодонную колбу. Суспензия была нагрета до 75°С, при этой температуре твердые вещества растворились. Горячий раствор был отфильтрован для удаления палладиевой черни (от 0.2- до 0.45-µ фильтры являются лучшими), и возвращен в чистую 5-литровую колбу. Раствор этилацетата был подвергнут дистилляции при атмосферном давлении для отгонки 2.2 л этилацетата (температура кипения 77-78°С). Раствор был охлажден до 22°С, и полученная суспензия была отфильтрована. Колба и осадок на фильтре были промыты этилацетатом (3×130 мл). Очищенный интермедиат 7 был высушен в вакуумной печи при 50°С с получением 110.5 г интермедиата 7 (85% степень извлечения). ВЭЖХ анализ очищенного интермедиата 7 показал содержание 98.5% (AUC). Уровень палладия в очищенном продукте составил 6 мд. Маточный раствор был выпарен с получением 18 г неочищенного продукта (14% степень извлечения, 2254 мд Pd).
Пример 6: Получение (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-она, 1 (R = Н), также именуемого как «TR-700»
[0054] 5-литровая, трехгорлая, круглодонная колба была снабжена верхней мешалкой, термопарой и 500-мл капельной воронкой и переходником для подачи азота. Колба была высушена горячим пистолетом в потоке азота до внутренней температуры в 60°С. В колбу был загружен интермедиат 7 (110.0 г, 0.272 моль, AMRI партия # DUG-AF-202(1)) и безводный ТГФ (2.2 л, 20 объемов). Суспензия была перемешана, и получился светло-зеленый раствор. Капельная воронка с 1.0 М литий гексаметилдисилазидом (299 мл, 0.286 моль, 1.05 эквивалента). Раствор LiHMDS был по каплям добавлен к раствору интермедиата 7 приблизительно за 25 минут. Раствор приобрел красный цвет. Раствор перемешивался один час при комнатной температуре и затем 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон (DMPU) (34.9 г, 0.272 моль, 1 эквивалент) был добавлен, смесь превратилась в желтую суспензию. Реакционная масса была охлаждена на ледяной бане до 5.7°С. К-(-)-Глицидил бутират (41.25 г, 0.286 моль, 1.05 эквивалента) был добавлен за одну порцию. Смесь перемешивалась на ледяной бане в течение 0,5 часа, затем была нагрета до комнатной температуры и перемешивалась всю ночь. В этот момент реакционная смесь превратилась в суспензию бронзового цвета, и 15 часов спустя ВЭЖХ анализ показал, что смесь приблизительно содержит 87% TR-700, 1.6% интермедиата 7, и приблизительно 7% бутират эфира соединения TR-700. Небольшое количество метоксида натрия в метаноле (11 мл, 0.1 объемов) было добавлено, и смесь перемешивалась еще в течение 1 часа для удаления остатков эфира. ВЭЖХ анализ в этот момент протекания реакции показал, что смесь содержит приблизительно 90.7% TR-700 и 0.2% бутират эфира. Смесь была погашена добавлением 10% по весу раствора хлорида аммония (1.1 л, 10 объемов). Умеренный экзотермический эффект от 22°С до 25°С наблюдался при добавлении раствора хлорида аммония. Двухфазная смесь была подвергнута дистилляции при температуре реактора в 70°С (атмосферное давление) для удаления приблизительно 2.2 л ТГФ. Вязкая суспензия была получена, которая была разбавлена водой (550 мл, 5 объемов). Суспензия была охлаждена до комнатной температуры (23.6°С) и отфильтрована. Осадок на фильтре был промыт водой (1.1 л, 10 объемов) и метанолом (550 мл, 5 объемов) с получением TR-700 в виде белого твердого вещества. Влажный осадок был высушен в течение ночи в вакуумной печи при 50°С с получением 89.7 г TR-700 (89% выход), что составило 97.8% (AUC) по ВЭЖХ анализу. Соединение TR-700 было в дальнейшем очищено повторным суспендированием в 2.7 л (30 объемов) смеси 4:1 метанол/вода при 70°С, с охлаждением до 23°С, фильтрацией и промыванием метанолом (180 мл). Эта процедура позволила удалить примесь избыточно алкилированного продукта. Очищенный TR-700 был выделен с 96% выходом (85% общий выход), и чистота улучшилась до 98.4% (AUC) по ВЭЖХ анализу. Содержание палладия составило 10 мд.
Пример 7: Получение (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ила)-3-фторфенил)-5-гидроксиметил оксазолидин-2-она дигидро фосфата 1 (R = РО(ОН)2) также именуемого как «TR-701FA»
[0055] 5-литровая, круглодонная колба, снабженная рубашкой с охлаждающим контуром, была оборудована верхней механической мешалкой, капельной воронкой, термопарой, входным отверстием для азота. В колбу было загружено соединение TR-700 (70.0 г, 0.189 моль), ТГФ (1.4 л, 20 объемов) и триэтиламин (58.2 г, 0.575 моль, 3 эквивалента). Суспензия была перемешана и температура рубашки была установлена на 0°С. В капельную воронку был загружен оксихлорид фосфора (87.0 г, 0.567 моль, 3 эквивалента) в ТГФ (70 мл, 1 объем). Как только внутренняя температура достигла 1°С, раствор POCl3 был по каплям добавлен за 44 минуты. Максимальная внутренняя температура была 2.2°С. Смесь была перемешана в течение 3 часов при 1-2°С, в этой точке ВЭЖХ анализ показал, что осталось 0.5% соединения TR-700. В 5-литровую, трехгорлую круглодонную колбу, снабженную диафрагменным насосом Teflon, была налита вода (1.4 л, 20 объемов), она была охлаждена до 3.8°С на ледяной бане с солью. Реакционная смесь была перекачана для гашения под холодную воду за 1 час. Максимальная температура в процессе гашения достигала 11.9°С. Реактор и насосная линия были промыты водой (~210 мл) в сосуд для гашения. Образовавшаяся желтая суспензия перемешивалась всю ночь. Суспензия была отфильтрована через Whatman бумагу, и осадок на фильтре был промыт водой (700 мл, 10 объемов) и метанолом (700 мл, 10 объемов). Продукт был высушен при комнатной температуре в вакуумной печи до достижения постоянного веса. Выход неочищенного TR-701FA составил 81.6 г (96%), чистота по ВЭЖХ анализу (Способ В) - 95.3% (AUC).
Пример 8: Получение (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он фосфата, динатриевой соли 1 (R = РО3 2Na), также именуемого как «TR-701»
[0056] Неочищенное соединение 1 (R=PO(OH)2) (60.0 г, 0.133 моль) было загружено в 2-литровый реактор. Метанол (720 мл, 12 объемов) был добавлен, и суспензия перемешивалась при комнатной температуре. 25% метоксид натрия в метаноле (86.1 г, 0.398 моль, 3 эквивалента) был по каплям добавлен за 13 минут. Температура реакции выросла с 20.4°С до 26.8°С при добавлении метоксида натрия. Суспензия перемешивалась один час при комнатной температуре и затем была отфильтрована. Реактор и осадок на фильтре были промыты метанолом (300 мл, 5 объемов) и ацетоном (300 мл, 5 объемов). Продукт был высушен в вакуумной печи при 50-60°С с получением 65.3 г неочищенного TR-701 (99% выход). Неочищенный продукт был растворен в воде (653 мл, 10 объемов) с получением раствора соломенного цвета. Раствор был перемешан с углем Darco G-60 (3.3 г, 0.05 по весу) при комнатной температуре в течение 30 минут. рН суспензии составила 7.2, поэтому 5-10 мл 2 N NaOH было добавлено для увеличения рН до 11. Суспензия была отфильтрована через Целит 545 (65 г, увлажненный водой). Небольшое количество угля прошло через фильтр. Фильтрат был повторно отфильтрован через 0.45-µ фильтр, но некоторое количество угля прошло вновь. Фильтрат был по каплям добавлен к ацетону (2.6 л, 40 объемов), и полученная суспензия была перемешана в течение ночи для удобства. Суспензия была отфильтрована, разбавлена ацетоном (650 мл), и высушена в вакуумной печи при 50°С с получением 46.9 г соединения 1 (R = PO2Na) (71% выход) серого цвета. ВЭЖХ чистота этого материала составила 99.0% (AUC), но поскольку оно было серое, оно повторно было растворено в воде (470 мл). рН водного раствора было равно 9.6, поэтому раствор гидроксида натрия был добавлен для увеличения рН до 10. Раствор был отфильтрован через 0.45-µ фильтр для изменения окраски. Фильтрат был по каплям добавлен к ацетону (1.88 л). Белая суспензия была отфильтрована и промыта ацетоном (470 мл). После высушивания продукта вес TR-701 составил 43.2 г (66% общий выход). ВЭЖХ чистота (Способ В) составила 99.6% (AUC). Другие виды анализов, проведенные с этой партией соединения 1 (R = PO2Na), показаны в Таблице 1.
Пример 9: Получение очищенного R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он дигидрофосфата, 1 (R = PO(OH)2)
[0057] В 3-литровую круглодонную колбу было загружено неочищенное соединение 1 (R = РО(ОН)2) (99.8 г, 0.222 моль, AMRI партия # 8АК0242С) и вода (1 л, 10 объемов). рН этой суспензии составил 2.05. Свежий 1 М раствор гидроксида натрия был получен разбавлением 50.9% раствора гидроксида натрия (39.3 г, 0.50 моль) в общем объеме 0.5 л воды. 1 М раствор гидроксида натрия (444 мл, 0.444 моль, 2 эквивалента) был по каплям добавлен к свежей суспензии свободной кислоты. При рН 5.7, твердые вещества растворились, несмотря на то, что меньше половины раствора гидроксида натрия было добавлено. В конце добавления рН составило 8.57. Уголь Darco G-60 (5.1 г, 0.05 по весу) был добавлен к раствору, и смесь перемешивалась в течение 1 часа при комнатной температуре. Суспензия была отфильтрована через фильтровальную бумагу Whatman #1 для удаления основной массы угля, и затем через 0.45-µ, фильтр для удаления мелких частиц. Фильтрат соломенного цвета был по каплям добавлен в 12-литровую круглодонную колбу, содержащую ацетон (4 л, 40 объемов). Полученная суспензия была перемешана в течение часа при комнатной температуре, отфильтрована и промыта ацетоном (500 мл, 5 объемов). Влажный осадок был перенесен в 3-литровую круглодонную колбу и оставлен сушиться в атмосфере азота на всю ночь.
[0058] Динатриевая соль соединения 1 (R = PO2 2Na) была повторно растворена в воде (1 л, 10 объемов) и затем отфильтрована через фильтровальную бумагу Whatman #1, при этом в растворе наблюдались черные крупинки. Фильтрат был разбавлен ТГФ (1 л, 10 объемов). рН водного раствора ТГФ составило 9.57. Свежеприготовленный 2 М раствор соляной кислоты (222 мл, 0.444 моль, 2 эквивалента) был по каплям добавлен для доведения рН до значения 1.34. Продукт не выпал в осадок до тех пор, пока приблизительно 170 мл 2 М раствора HCl не было добавлено. Желтая суспензия была отфильтрована, промыта водой (500 мл, 5 объемов) и метанолом (500 мл, 5 объемов). Осадок на фильтре растрескался из-за высыхания, поэтому он был выровнен перед добавлением растворителей. Продукт был высушен в вакуумной печи при 60°С в течение 19.5 часов с получением 79.3 грамма соединения 1 (R = Р(ОН)2) (80% выход). ВЭЖХ анализ (Способ В): 99.5% (AUC) tR=5.6 мин. 1H и 31P ЯМР спектры соответствовали заявляемой структуре. Уровень остаточного ТГФ по ЯМР анализу был 1600 мд, а уровень палладия составил 11 мд. Поскольку интенсивная сушка не позволила удалить весь ТГФ, последующие партии были получены, используя этанол в качестве антирастворителя (растворитель, в котором продукт не растворяется).
Пример 10: Выделение бис{[(5R)-3-{3-фтор-4-[6-(2-метил-2Н-тетразол-5-ил)пиридин-3-ил]фенил}-2-оксо-1,3-оксазолидин-5-ил]метил}дигидродифосфата (димер соединения 1)
[0059] Неочищенное соединение 1 из примера 8 было растворено в фосфатном буфере и подвергнуто хроматографическому анализу на Gilson препаративной ВЭЖХ системе. Мобильная фаза представляла собой линейный градиент воды и ацетонитрила, при t=0 была 100% H2O, и Т=20 был 100% ацетонитрил. Фракции были проанализированы, используя аналитическую ВЭЖХ. Фракции, обогащенные Димером, были собраны с получением раствора, содержащего свыше 60% Димера. Дальнейшая очистка обогащенных Димером фракций была осуществлена способом полупрепаративной ВЭЖХ. Был получен чистый димер: точная масса (m/z 883; рассчитана для C34H31F2N12O11P2=883.1679, получено 883.1658, Δ=2.4 мд m/z 905 рассчитана для C34H30F2N1O11P2Na=905.1498; получено 905.1484, Δ=1.6 мд), данные подтверждаю формулу для этого соединения.
|
Кристаллические частицы для приготовления твердых лекарственных форм для лечения бактериальных инфекций, реакционная смесь, содержащая такие частицы, и фармацевтическая композиция для лечения бактериальных инфекций
Оксазолидиноны и способ их очистки
Трициклические ингибиторы гиразы

