Результат интеллектуальной деятельности: ПРОИЗВОДНОЕ ГИДАНТОИНА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к фармацевтическим средствам, содержащим в качестве активного ингредиента производное гидантоина, которое обладает высокой метаболической стабильностью и проявляет сильное PTH-подобное действие.
Предшествующий уровень техники
Паратиреоидный гормон (PTH) связывается с рецептором PTH1 (PTH1R), который представляет собой сопряженный с G-белком рецептор (GPCR), для активации G-белка, а затем вызывает активацию по меньшей мере одного сигнального каскада, такого как каскад циклического АМФ (цАМФ)/протеинкиназы A. PTH известен как гормон, который действует на клетки-мишени в почке и кости, регулируя гомеостаз кальция (Ca) и фосфора (Pi) (непатентный документ 1). Уровни концентрации Ca в сыворотке поддерживаются с помощью PTH в основном посредством прямых или опосредованных действий на желудочно-кишечный тракт, кость и почку. PTH способствует всасыванию Ca из почечных канальцев и, таким образом, подавляет выведение Ca из организма. Он также повышает синтез фермента, который преобразует витамин D в активный витамин D в почке и тем самым способствует облегчению опосредованного активным витамином D всасывания Ca из желудочно-кишечного тракта. Кроме того, PTH усиливает дифференцировку остеокластов опосредованно через остеобласты и способствует выделению Ca из кости. Полагают, что такие действия PTH происходят в основном посредством повышения уровня циклического аденозин 3’,5’-монофосфата (цАМФ) и/или активации фосфолипазы C (PLC), которая возникает при связывании PTH с PTH1R.
У людей препараты PTH [PTH (1-34) и PTH (1-84)] оказывают мощное остеогенное действие и индуцируют значительное увеличение плотности минеральных веществ костной ткани (BMD) и прочности кости. В настоящее время многие доступные лекарственные средства против остеопороза для людей представляют собой ингибиторы резорбции кости, и единственным типом остеогенного лекарственного средства, которое активно увеличивает BMD, являются препараты PTH. Препарат PTH считают одним из наиболее эффективных средств для лечения остеопороза (непатентный документ 2); однако вследствие того, что он представляет собой пептид, его необходимо вводить инвазивным способом. Таким образом, существует потребность в получении фармацевтического средства, которое обладает PTH-подобным действием и которое можно вводить неинвазивно.
Гипопаратиреоз представляет собой метаболическое заболевание, которое характеризуется гипокальциемией и гиперфосфатемией, вызываемых недостаточностью PTH, секретируемого паращитовидной железой, и различными ассоциированными симптомами. Препараты активного витамина D и содержащие Ca средства используют для лечения гипопаратиреоза; однако вследствие того, что обусловленный PTH механизм регуляции не функционирует, не получая достаточный терапевтический эффект. Кроме того, поскольку составы активного витамина D увеличивают выделение Ca с мочой, длительная терапия вызывает повышенный риск нефропатии. Для разрешения этих проблем проводят продолжающееся исследование заместительной терапии, в которой используют препараты PTH против этого заболевания, и была предпринята попытка проведения нескольких инвазивных введений в сутки или непрерывного введения с использованием насоса для получения достаточной эффективности (непатентный документ 3). Таким образом, для лечения гипопаратиреоза желательным является получение фармацевтического средства, которое обладает PTH-подобным эффектом и которое также можно вводить неинвазивно.
Кроме того, для лечения заболеваний, таких как перелом, адинамическая болезнь кости, ахондроплазия, гипохондроплазия, остеомаляция, остеоартрит, артрит, тромбоцитопения, гиперфосфатемия и опухолевый кальциноз, желательным является фармацевтическое средство, обладающее PTH-подобными эффектами, которое также можно вводить неинвазивно.
При таких обстоятельствах авторы настоящего изобретения подали патентную заявку, предварительно основанную на открытии ими того, что соединение, представленное формулой (A):
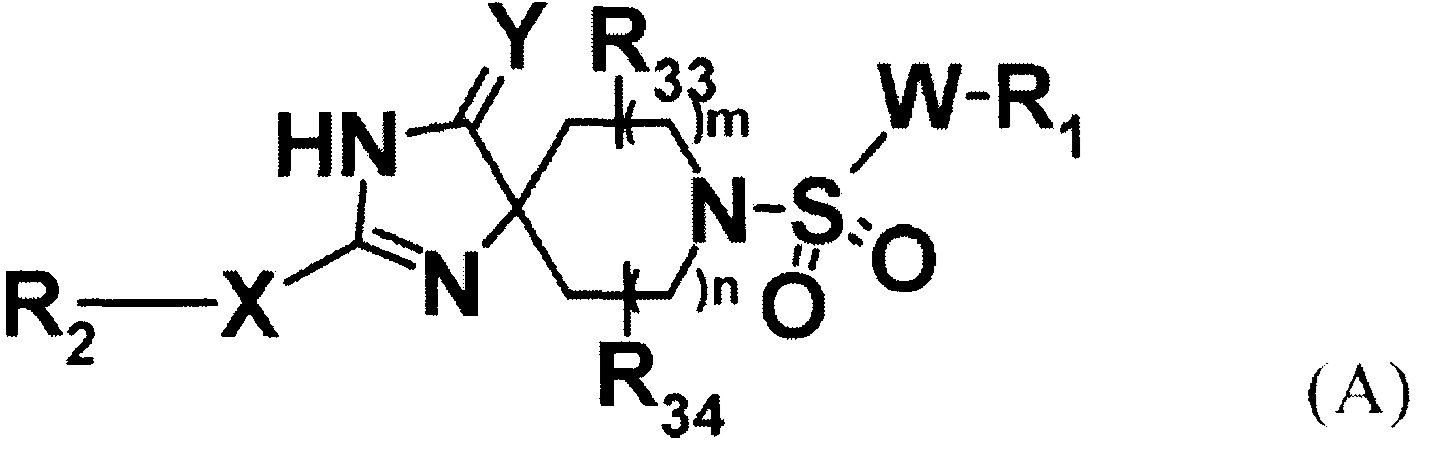
[Патентный документ 1 может быть указан для W, X, Y, m, n, R1, R2, R33 и R34 в формуле], и его фармакологически приемлемые соли являются пригодными в качестве соединений, обладающих PTH-подобными эффектами, или более предпочтительно, в качестве агониста PTH1R, и являются пригодными для предупреждения и/или лечения остеопороза, перелома, остеомаляции, артрита, тромбоцитопении, гипопаратиреоза, гиперфосфатемии или опухолевого кальциноза, или мобилизации стволовых клеток (патентный документ 1).
Для получения фармацевтических средств, которые имеют большую клиническую ценность и которые можно вводить инвазивно, необходимо учитывать параметры кинетики in vivo, такие как всасывание, распределение, метаболизм и выведение лекарственного средства в дополнение к его прямым действиям на мишень. В частности, для обеспечения перорального введения желательным является наличие фармацевтического средства, обладающего PTH-подобными эффектами, которое обладает высокой метаболической стабильностью в отношении микросом печени человека и значительной обусловленной PTH1R человека способностью продуцировать цАМФ.
Для предоставления фармацевтического средства, которое можно вводить перорально людям, как правило, можно использовать способ подтверждения эффектов перорального введения посредством тестирования in vivo, которое предусматривает использование модельного животного. Например, подвергнутая тиропаратиреоидэктомии (TPTX) крыса известна как модель гипопаратиреоза на животных. Для поиска терапевтического средства, которое обладает сильными PTH-подобными эффектами и высокой метаболической стабильностью и действует против гипопаратиреоза при пероральном введении, эффективно использовать способ поиска соединения, которое действует на PTH1R крысы и является стабильным по отношению к ферментам крысы, а затем анализируя его действия при пероральном введении на модели TPTX у крыс.
В существующей в настоящее время терапии гипопаратиреоза целевой терапевтический диапазон концентрации Ca в сыворотке установлен немного ниже диапазона, чем нижний предел нормального диапазона от 7,6 до 8,8 мг/дл (непатентный документ 4). Поскольку нормальный диапазон для концентрации Ca в сыворотке у крыс находится на том же уровне, как для людей в дозе 10 мг/дл или т.д., для подтверждения терапевтического эффекта важным является получение концентрации Ca в сыворотке на модели заболевания у крыс в диапазоне от целевого терапевтического диапазона у людей (7,6-8,8 мг/дл) до нижней границы гиперкальциемии у людей (приблизительно 11,2 мг/дл).
[Документы известного уровня техники]
[Патентные документы]
[Патентный документ 1] WO 2010/126030
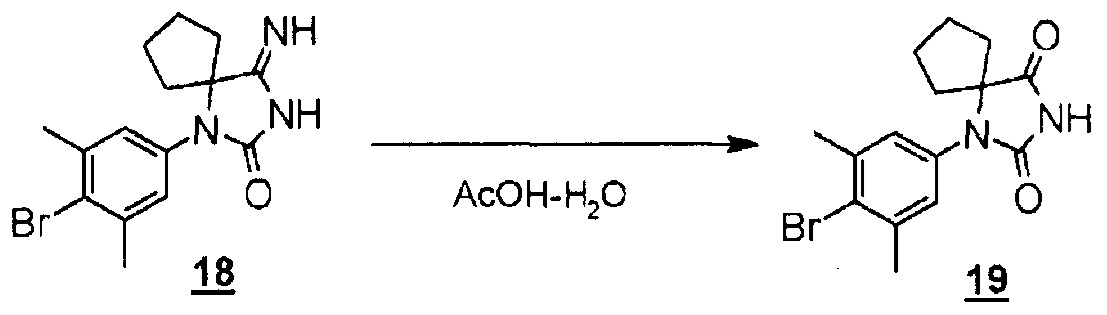
[Непатентные документы]
[Непатентный документ 1] Kronenberg, H.M. et al., In Handbook of Experimental Pharmacology, Mundy, G.R., and Martin, T.J., (eds), pp. 185-201, Springer-Verlag, Heidelberg (1993)
[Непатентный документ 2] Tashjian and Gagel, J. Bone Miner. Res., 21:354-365 (2006)
[Непатентный документ 3] Rejnmark et al., Osteoporosis Int. Published Online: 27 November 2012
[Непатентный документ 4] Winer K.K. et al., J. Clin. Endocrinol. Metab. 88 (9):4214-4220 (2003)
Сущность изобретения
[Задачи, подлежащие решению посредством изобретения]
Задача настоящего изобретения состоит в обеспечении соединения с сильными PTH-подобными эффектами и высокой метаболической стабильностью и в предоставлении фармацевтических композиций, содержащих такие соединения, для возможности лечения состояний, которые можно лечить PTH-подобными действиями, таких как гипопаратиреоз.
[Средства решения задач]
Решая поставленную задачу, авторы настоящего изобретения продолжали проводить исследование и обнаружили, что новые производные гидантоина по настоящему изобретению обладают значительной способностью продуцировать цАМФ в клетках, экспрессирующих PTH1R человека, и обладают высокой стабильностью в микросомах печени человека. Авторы настоящего изобретения также обнаружили, что соединения по настоящему изобретению обладают значительной способностью продуцировать цАМФ в клетках, экспрессирующих PTH1R крысы, и обладают высокой стабильностью в гепатоцитах крысы. Кроме того, на моделях TPTX у крыс с пероральным введением вновь было обнаружено, что доза 30 мг/кг восстанавливала концентрацию Ca в сыворотке до целевого терапевтического диапазона 7,6-8,8 мг/дл. Результаты, полученные на этих модельных животных, позволяют предположить, что соединения, представленные формулой (1), которые демонстрируют сильный эффект на PTH1R человека и высокую стабильность в микросомах печени человека, являются пригодными в качестве терапевтических средств для гипопаратиреоза.
Настоящее изобретение относится к:
[1] Соединению, представленному общей формулой (1) или его фармакологически приемлемой солью:
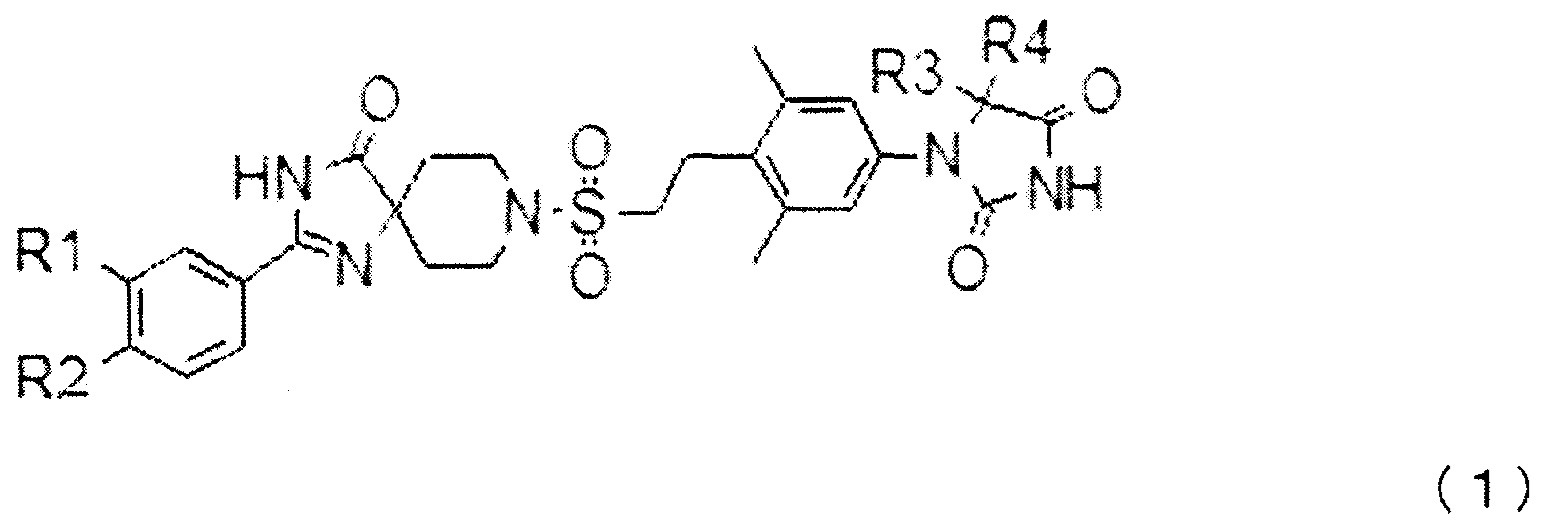
(где,
когда R1 и R2 оба не представляют собой атомы водорода, R1 и R2 независимо представляют собой:
1) атом водорода;
2) атом галогена;
3) алкильную группу, содержащую один или два атома углерода, которые могут быть замещены одним-пятью атомами фтора; или
4) алкоксигруппу, содержащую один или два атома углерода, которые могут быть замещены одним-пятью атомами фтора; или
R1 и R2 связываются друг с другом с образованием группы, представленной указанной ниже формулой:
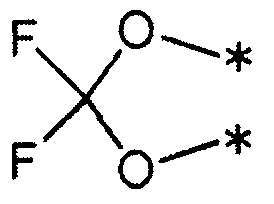
(где каждый символ * означает положение связи с фенильной частью); и
R3 и R4 независимо представляют собой метильную группу, которая может быть замещена одним-тремя атомами фтора; или
R3 и R4 вместе со связанным атомом углерода образуют трех-шести-членное карбоциклическое кольцо (где один из атомов углерода, образующих кольцо, может быть заменен атомом кислорода, атомом серы или может представлять собой замещенный метилом или незамещенный атом азота).
В настоящем изобретении соединение, в котором комбинация R1 и R2 представляет собой трифторметильную группу и атом водорода, и где R3 и R4 вместе со связанным атомом углерода образуют циклопентильное кольцо, может быть исключено из указанных выше соединений, представленных формулой (1).
[2] Соединению или его фармакологически приемлемой соли [1], где R1 и R2 выбраны из указанной ниже комбинации:
1) R1 представляет собой атом водорода или атом галогена, и R2 представляет собой атом водорода, трифторметильную группу или трифторметоксигруппу (при условии что оба R1 и R2 не являются атомами водорода);
2) R1 представляет собой трифторметильную группу или трифторметоксигруппу, и R2 представляет собой атом водорода или атом галогена;
3) R1 и R2 связаны друг с другом с образованием группы, представленной указанной ниже формулой:
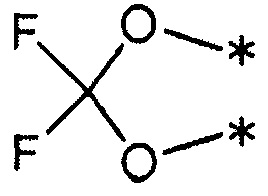
(где каждый символ * означает положение связи с фенильной частью); и
R3 и R4 представляют собой метильные группы; или
R3 и R4 вместе со связанным атомом углерода образуют кольцо, выбранное из указанных ниже:
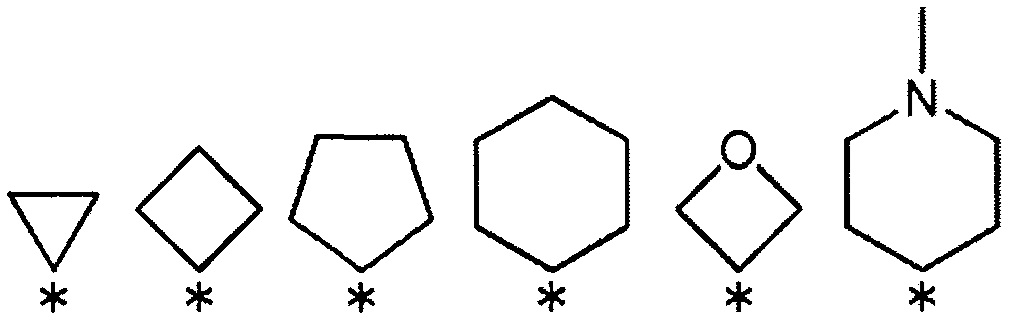
(где символ * означает положение связи с частью имидазолидин-2,4-диона).
[3] Соединению или его фармакологически приемлемой соли [1], где R1 и R2 выбраны из указанных ниже комбинаций:
1) R1 представляет собой трифторметоксигруппу, и R2 представляет собой атом фтора;
2) R1 представляет собой атом брома, и R2 представляет собой атом водорода;
3) R1 представляет собой трифторметоксигруппу, и R2 представляет собой атом фтора;
4) R1 представляет собой атом фтора, и R2 представляет собой трифторметоксигруппу;
5) R1 представляет собой трифторметильную группу, и R2 представляет собой атом водорода;
6) R1 представляет собой атом водорода, и R2 представляет собой трифторметоксигруппу;
7) R1 и R2 связаны друг с другом с образованием группы, представленной указанной ниже формулой:
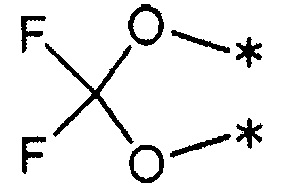
(где каждый символ * означает положение связи с фенильной частью); и
R3 и R4 представляют собой метильные группы; или
R3 и R4 вместе со связанным атомом углерода образуют кольцо, выбранное из указанных ниже:
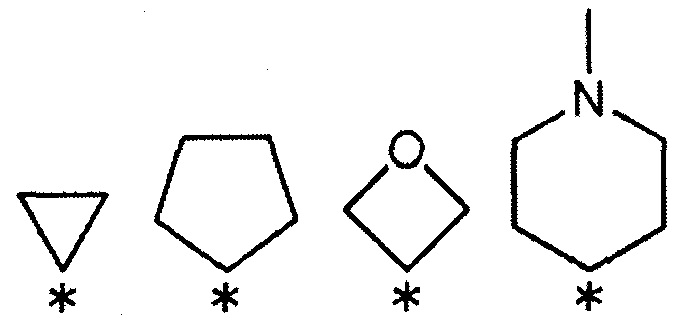
(где символ * означает положение связи с частью имидазолидин-2,4-диона).
[4] Соединению или его фармакологически приемлемой соли [1], где R3 и R4 представляют собой метильные группы.
[5] Соединению или его фармакологически приемлемой соли [1], где R3 и R4 вместе со связанным атомом углерода образуют кольцо, выбранное из указанных ниже:
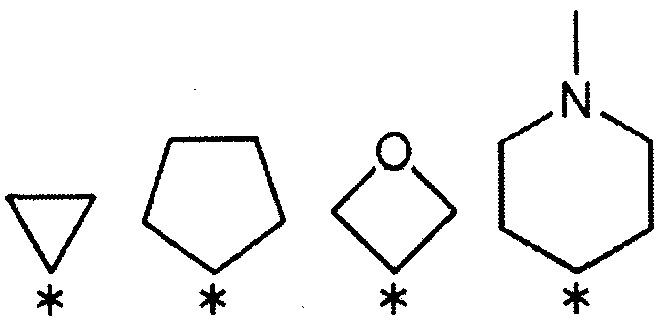
(где символ * означает положение связи с частью имидазолидин-2,4-диона).
[6] Соединению или его фармакологически приемлемой соли [1], где соединение выбрано из группы, состоящей из:
1-(4-(2-((2-(4-фтор-3-(трифторметокси)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(3-бромфенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(4-фтор-3-(трифторметил)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(3-фтор-4-(трифторметокси)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-диона;
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-диона;
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4.4]нонан-2,4-диона;
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-[,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-8-метил-1,3,8-триазаспиро[4.5]декан-2,4-диона;
5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2-окса-5,7-диазаспиро[3,4]октан-6,8-диона; и
4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-4,6-диазаспиро[2,4]гептан-5,7-диона.
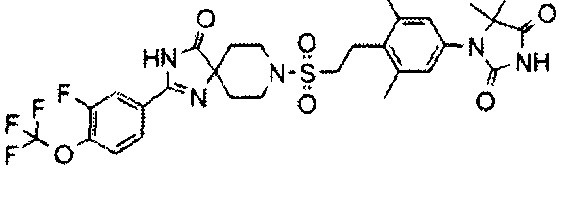
[7] Соединению или его фармакологически приемлемой соли [1], где соединение представляет собой 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион.
[8] Соединению или его фармакологически приемлемой соли [1], где соединение представляет собой 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион.
[9] Соединению или его фармакологически приемлемой соли [1], где соединение представляет собой 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4.4]нонан-2,4-дион.
[10] Фармацевтической композиции, которая содержит соединение или его фармакологически приемлемую соль по любому из пп.[1]-[9] в качестве активного ингредиента.
[11] Фармацевтической композиции по п.[10], которая предназначена для перорального введения.
[12] Фармацевтической композиции для активации внутриклеточной реакции цАМФ, которая содержит соединение или его фармакологически приемлемую соль по любому из пп.[1]-[9] в качестве активного ингредиента.
[13] Мобилизирующему стволовые клетки средству или средству для предупреждения или лечения остеопороза, перелома, адинамической болезни кости, ахондроплазии, гипохондроплазии, остеомаляции, остеоартрита, артрита, тромбоцитопении, гипопаратиреоза, гиперфосфатемии или опухолевого кальциноза, которое содержит соединение или его фармакологически приемлемую соль по любому из пп.[1]-[9] в качестве активного ингредиента.
[14] Способу предупреждения или лечения остеопороза, перелома, адинамической болезни кости, ахондроплазии, гипохондроплазии, остеомаляции, остеоартрита, артрита, тромбоцитопении, гипопаратиреоза, гиперфосфатемии или опухолевого кальциноза, или мобилизации стволовых клеток, где способ включает введение фармацевтически эффективного количества композиции, содержащей соединение или его фармакологически приемлемую соль по любому из пп.[1]-[9] пациенту, нуждающемуся в предупреждении или лечении заболевания, или в мобилизации стволовых клеток.
[15] Применению соединения или его фармакологически приемлемой соли по любому из пп.[1]-[9] для получения мобилизирующего стволовые клетки средства или средства для предупреждения или лечения остеопороза, перелома, адинамической болезни кости, ахондроплазии, гипохондроплазии, остеомаляции, остеоартрита, артрита, тромбоцитопении, гипопаратиреоза, гиперфосфатемии или опухолевого кальциноза.
[16] Соединению или его фармакологически приемлемой соли по любому из пп.[1]-[9] для лечения или предупреждения остеопороза, перелома, адинамической болезни кости, ахондроплазии, гипохондроплазии, остеомаляции, остеоартрита, артрита, тромбоцитопении, гипопаратиреоза, гиперфосфатемии или опухолевого кальциноза, или мобилизации стволовых клеток.
Кроме того, настоящее изобретение относится к способам лечения патологических состояний, которые можно лечить PTH-подобными действиями, таких как гипопаратиреоз, путем введения соединения формулы (1) или его соли.
[Эффекты изобретения]
Настоящее изобретение относится к производным гидантоина с сильными PTH-подобными эффектами и высокой метаболической стабильностью. Применение производных гидантоина позволяет лечить патологические состояния, вызываемые PTH-подобными действиями, такими как гипопаратиреоз.
Краткое описание чертежей
На фиг.1 изображен график, показывающий среднее изменение уровня концентрации Ca в сыворотке для каждого соединения до 24 часов после введения, где соединение вводят перорально в дозе 30 мг/кг на модели TPTX у крыс.
[Способ осуществления изобретения]
Настоящее изобретение относится к производным гидантоина и их применению. Авторы настоящего изобретения впервые синтезировали соединение, представленное указанной выше формулой (1), или его фармакологически приемлемую соль и обнаружили, что соединение или его соль представляет собой соединение, обладающее сильным (PTH)-подобным эффектом паратиреоидного гормона и высокой метаболической стабильностью.
В настоящем описании «алкил» относится к одновалентной группе, полученной удалением любого атома водорода из алифатического углеводорода, и включает подгруппу структур алкильных или углеводородных групп, не содержащих гетероатом или ненасыщенную углерод-углеродную связь и содержащих атомы водорода и углерода в основной цепи. Примеры алкильной группы включают примеры линейных или разветвленных структур. Алкильная группа предпочтительно представляет собой алкильную группу, содержащую один или два атома углерода. Алкильная группа конкретно представляет собой, например, метильную группу или этильную группу, и предпочтительно представляет собой метильную группу.
Как используется в настоящем описании, термин «алкокси» относится к оксигруппе, с которой определенный выше «алкил» связан, и предпочтительно относится к алкоксигруппе, содержащей один или два атома углерода. Конкретные примеры включают группы метокси и этокси, и предпочтительным примером является метоксигруппа.
В настоящем описании «B, необязательно замещенный A» означает, что любой атом(ы) водорода в B может(могут) быть замещен(ы) любым числом A.
В настоящем изобретении число заместителей не является ограниченным, если не указано иное. Например, число заместителей может составлять от 1 до 5, от 1 до 4, от 1 до 3, от 1 до 2 или 1.
В настоящем описании «атом галогена» относится к атому фтора, атому хлора, атому брома или атому йода.
В настоящем описании символ "*" в химической формуле относится к положению связи.
Соединения по настоящему изобретению, представленные формулой (1), обладают сильными PTH-подобными эффектами и высокой метаболической стабильностью.
В настоящем описании «PTH-подобный эффект» относится к активности генерации внутриклеточного цАМФ (цАМФ: циклический аденозин монофосфат) посредством воздействия на рецептор PTH или воздействия на путь передачи сигнала через рецептор PTH.
В настоящем изобретении наличие «сильного PTH-подобного эффекта» или то, что «PTH-подобный эффект является сильным» может быть подтверждено измерением активности передачи сигналов цАМФ путем анализа передачи сигналов цАМФ, например, способом, описанным в публикации J. Bone. Miner. Res., 14:11-20, 1999. В частности, например, согласно способу, описанному в тестовом примере 1, количество цАМФ, продуцируемого в клетках с усиленной экспрессией PTH1R человека, определяют с использованием коммерчески доступного набора cAMP EIA (например, Biotrack cAMP EIA system, GE health care) для измерения концентрации каждого соединения при 20% активности передачи сигналов цАМФ (EC20) или его концентрации при 50% активности передачи сигналов цАМФ (EC50), где активность передачи сигналов цАМФ, полученную после введения 100 нМ PTH (1-34) человека, определяли как 100%. В настоящем изобретении для выражения «сильный PTH-подобный эффект» или «PTH-подобный эффект является сильным», например, значение EC20 (мкМ), измеряемое указанным выше способом, предпочтительно составляет 5,0 или менее, более предпочтительно 3,0 или менее и даже более предпочтительно 2,0 или менее. Для EC50 значение (мкМ), измеряемое указанным выше способом, составляет, например, предпочтительно 25,0 или менее, более предпочтительно 15,0 или менее и даже более предпочтительно 10,0 или менее.
Наличие «высокой метаболической стабильности» или то, что «метаболическая стабильность является высокой», может быть подтверждено общепринятым способом измерения. Для подтверждения можно использовать, например, клетки печени, клетки тонкого кишечника, микросомы печени, микросомы тонкого кишечника, S9 печени и т.д. В частности, например, стабильность соединения в микросомах печени может быть подтверждена проведением измерений в соответствии с описанием у T. Kronbach et al. (Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol. Pharmacol, 1989, 36(1), 89-96). Более конкретно, стабильность может быть подтверждена следующим ниже способом, описанным в тестовом примере 3. В настоящем изобретении выражение «высокая метаболическая стабильность» или «метаболическая стабильность является высокой», означает, что величина выведения (мкл/мин/мг) в тесте на метаболическую стабильность с использованием микросом печени человека, описанном в указанном выше тестовом примере, составляет предпочтительно 60 или менее, более предпочтительно 40 или менее и даже более предпочтительно 35 или менее. В частности, высокая метаболическая стабильность может быть получена в указанной выше формуле (1), за исключением случаев, когда комбинация R1 и R2 представляет собой трифторметильную группу и атом водорода, и R3 и R4 вместе со связанным атомом углерода образуют циклопентильное кольцо.
Соединения по настоящему изобретению, представлены ли они в свободных формах или в виде фармакологически приемлемых солей, включены в настоящее изобретение. Примеры таких «солей» включают соли неорганических кислот, соли органических кислот, соли неорганических оснований, соли органических оснований и соли кислых или основных аминокислот.
Предпочтительные примеры солей неорганических кислот включают гидрохлориды, гидробромиды, сульфаты, нитраты и фосфаты. Предпочтительные примеры солей органических кислот включают ацетаты, сукцинаты, фумараты, малеаты, тартраты, цитраты, лактаты, стеараты, бензоаты, метансульфонаты, бензолсульфонаты и п-толуолсульфонаты.
Предпочтительные примеры солей неорганических оснований включают соли щелочных металлов, такие как натриевые соли и калиевые соли, соли щелочноземельных металлов, такие как кальциевые соли и магниевые соли, соли алюминия и аммонийные соли. Предпочтительные примеры солей органических оснований включают соли диэтиламина, соли диэтаноламина, соли меглумина и соли N,N-дибензилэтилендиамина.
Предпочтительные примеры солей кислых аминокислот включают аспартаты и глутаминаты. Предпочтительные примеры солей основных аминокислот включают соли аргинина, соли лизина и соли орнитина.
Соединения по настоящему изобретению могут впитывать влагу, содержать впитанную воду или образовывать гидраты, когда их оставляют на воздухе. Такие гидраты также включены в соли по настоящему изобретению.
Кроме того, соединения по настоящему изобретению могут впитывать определенные другие растворители с образованием сольватов. Такие соли также включены в настоящем изобретении как соли соединений формулы (1).
В настоящем описании для удобства структурная формула соединения может представлять определенный изомер. Однако соединения по настоящему изобретению включают все изомеры, такие как геометрические изомеры, оптические изомеры на основе асимметрических атомов углерода, стереоизомеры и таутомеры, а также смеси таких изомеров, которые возникают вследствие структур соединений, без ограничения формулами, описанными для удобства, и могут представлять собой один из изомеров или их смесь. Таким образом, соединения по настоящему изобретению могут содержать асимметрический атом углерода в молекуле и могут присутствовать в виде оптически активных форм и рацематов, но настоящее изобретение не ограничено ими и включает их все.
Настоящее изобретение включает все изотопы соединений, подставленных формулой (1). В изотопах соединений по настоящему изобретению по меньшей мере один атом заменен атомом с таким же атомным номером (число протонов), но с другим массовым числом (сумма числа протонов и числа нейтронов). Примеры изотопов, содержащихся в соединениях по настоящему изобретению, включают атом водорода, атом углерода, атом азота, атом кислорода, атом фосфора, атом серы, атом фтора и атом хлора, в том числе, 2H, 3H, 13C, 14C, 15N, 170, 18O, 31P, 32P, 35S, 18F и 36Cl, соответственно. В частности, радиоактивные изотопы, которые распадаются с излучением радиоактивности, такие как 3H и 14C, являются пригодными в тестах распределения в тканях организма для фармацевтических средств или соединений. Стабильные изотопы не распадаются, являются практически равными по относительному содержанию и не излучают радиоактивность, и, таким образом, их можно безопасно использовать. Изотопы соединений по настоящему изобретению могут быть преобразованы согласно общепринятым способам путем замены реагента, содержащего соответствующий изотоп на реагент, используемый для синтеза.
Соединения по настоящему изобретению могут проявлять полиморфизм кристаллической структуры, но конкретно не являются ограниченными какой-либо из них, а могут находиться в любой из этих кристаллических форм или существовать в виде смеси двух или более кристаллических форм.
Соединения по настоящему изобретению включают их пролекарства. Пролекарства представляют собой производные соединений по настоящему изобретению, которые содержат химически или метаболически разрушаемые группы и преобразуются обратно в исходные соединения после введения in vivo, проявляя свою исходную эффективность, включая комплексы, не образованные ковалентными связями, и соли.
Соединения, представленные указанной выше формулой (1) по настоящему изобретению, предпочтительно являются такими, как указано ниже.
В формуле R1 и R2 выбраны из указанных ниже комбинаций:
1) R1 представляет собой атом водорода или атом галогена, и R2 представляет собой атом водорода, трифторметильную группу или трифторметоксигруппу (при условии, что R1 и R2 не являются атомами водорода);
2) R1 представляет собой трифторметильную группу или трифторметоксигруппу, и R2 представляет собой атом водорода или атом галогена;
3) R1 и R2 связаны друг с другом с образованием группы, представленной указанной ниже формулой:
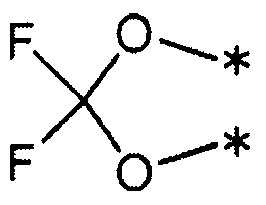
(где каждый символ * означает положение связи с фенильной частью); и
R3 и R4 представляют собой метильные группы; или
R3 и R4 вместе со связанным атомом углерода образуют кольцо, выбранное из указанных ниже:
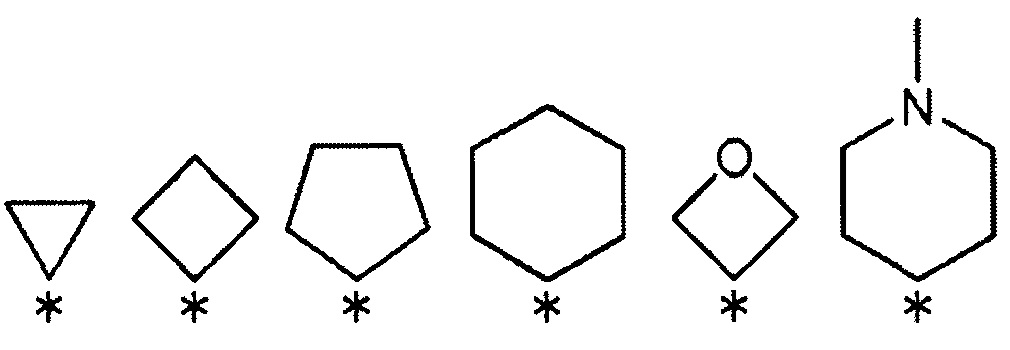
(где символ * означает положение связи с частью имидазолидин-2,4-диона).
Соединения, представленные указанной выше формулой (1) по настоящему изобретению, более предпочтительно являются такими, как указано ниже.
В формуле R1 и R2 выбраны из следующих ниже комбинаций:
1) R1 представляет собой трифторметоксигруппу, и R2 представляет собой атом фтора;
2) R1 представляет собой атом брома, и R2 представляет собой атом водорода;
3) R1 представляет собой трифторметоксигруппу, и R2 представляет собой атом фтора;
4) R1 представляет собой атом фтора, и R2 представляет собой трифторметоксигруппу;
5) R1 представляет собой трифторметильную группу, и R2 представляет собой атом водорода;
6) R1 представляет собой атом водорода, и R2 представляет собой трифторметоксигруппу;
7) R1 и R2 связаны друг с другом с образованием группы, представленной указанной ниже формулой:
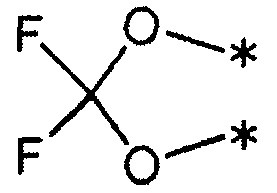
(где каждый символ * означает положение связи с фенильной частью); и
R3 и R4 представляют собой метильные группы; или
R3 и R4 вместе со связанным атомом углерода образуют кольцо, выбранное из указанных ниже:
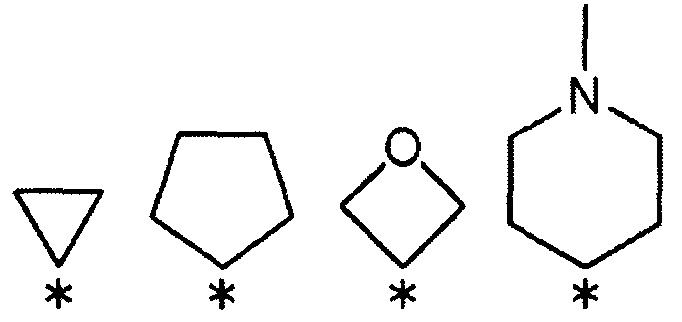
(где символ * означает положение связи с частью имидазолидин-2,4-диона).
Соединения, представленные указанной выше формулой (1) по настоящему изобретению, более предпочтительно представляют собой соединение, выбранное из группы, состоящей из указанных ниже соединений, или его фармакологически приемлемую соль.
Соединение 1:
1-(4-(2-((2-(4-фтор-3-(трифторметокси)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 2:
1-(4-(2-((2-(3-бромфенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 3:
1-(4-(2-((2-(4-фтор-3-(трифторметил)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 4:
1-(4-(2-((2-(3-фтор-4-(трифторметокси)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 5:
1-(4-(2-((2-(2,2-дифторбензо[d][l,3]диоксол-5-ил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 6:
1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 7:
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион);
Соединение 8:
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4.4]нонан-2,4-дион;
Соединение 9:
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-8-метил-1,3,8-триазаспиро[4.5]декан-2,4-дион;
Соединение 10:
5-(3,5-диетил]-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2-окса-5,7-диазаспиро[3,4]октан-6,8-дион; и
Соединение 11:
4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-4,6-диазаспиро[2,4]гептан-5,7-дион.
Из указанных выше соединений 1-11 наиболее предпочтительными являются соединения 6, 7 и 8.
Такие соединения, представленные указанной выше формулой (1), или их фармакологически приемлемые соли по настоящему изобретению являются пригодными в качестве соединений, обладающих PTH-подобным эффектом, предпочтительно агонисты PTH1R, и пригодными для предупреждения и/или лечения остеопороза, перелома, адинамической болезни кости, ахондроплазии, гипохондроплазии, остеомаляции, остеоартрита, артрита, тромбоцитопении, гипопаратиреоза, гиперфосфатемии, опухолевого кальциноза или тому подобное, или мобилизации стволовых клеток.
Соединения или их соли по настоящему изобретению можно составлять общепринятыми способами в таблетки, порошки, мелкозернистые гранулы, гранулы, покрытые таблетки, капсулы, сиропы, пастилки, лекарственные формы для ингаляции, суппозитории, инъекции, мази, глазные мази, офтальмологические препараты, назальные препараты, препараты для ушей, горячие компрессы, лосьоны и т.п. Для готовых форм можно применять широко используемые эксципиенты, связывающие средства, смазочные средства, красители, корригенты и по мере необходимости стабилизаторы, эмульгаторы, усилители всасывания, поверхностно-активные вещества, регуляторы pH, консерванты, антиоксиданты и тому подобное, и их смешивают с широко используемыми ингредиентами в качестве сырья фармацевтических препаратов и составляют общепринятыми способами.
Например, пероральные препараты получают, добавляя к соединению или его фармакологически приемлемой соли по настоящему изобретению эксципиент и по мере необходимости связывающее средство, разрыхлитель, смазочное средство, краситель, корригент и тому подобное, а затем составляя их в порошок, мелкозернистые гранулы, гранулы, таблетки, покрытые таблетки, капсулы и т.п. общепринятым способом.
Примеры этих ингредиентов включают животные и растительные масла, такие как соевое масло, топленое сало и синтетический глицерид; углеводороды, такие как парафиновое масло, сквалан и твердый парафин; сложноэфирные масла, такие как октилдодецилмиристат и изопропилмиристат; высшие спирты, такие как цетостеариловый спирт и бегениловый спирт; силиконовую смолу; силиконовое масло; поверхностно-активные вещества, такие как сложный эфир полиоксиэтилена и жирной кислоты, сложный эфир сорбитана и жирной кислоты, сложный эфир глицерина и жирной кислоты, сложный эфир полиоксиэтиленсорбитана и жирной кислоты, полиоксиэтилена и гидрогенизированного касторового масла и блок-сополимер полиоксиэтилен-полиоксипропилен; водорастворимые полимеры, такие как гидроксиэтилцеллюлоза, полиакриловая кислота, карбоксивиниловый полимер, полиэтиленгликоль, поливинилпирролидон и метилцеллюлоза; низшие спирты, такие как этанол и изопропанол; многоатомные спирты, такие как глицерин, пропиленгликоль, дипропиленгликоль и сорбит; сахара, такие как глюкоза и сахароза; неорганические порошки, такие как кремниевый ангидрид, алюмосиликат магния и силикат алюминия, и очищенную воду.
Примеры эксципиентов включают лактозу, кукурузный крахмал, белый мягкий сахар, глюкозу, маннит, сорбит, микрокристаллическую целлюлозу и диоксид кремния.
Примеры связывающих средств включают поливиниловый спирт, простой поливиниловый эфир, метилцеллюлозу, этилцеллюлозу, гуммиарабик, трагакант, желатин, шеллак, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, поливинилпирролидон, блок-полимер полипропиленгликоль-полиоксиэтилен и меглумин.
Примеры разрыхлителей включают крахмал, агар, желатиновый порошок, микрокристаллическую целлюлозу, карбонат кальция, бикарбонат натрия, цитрат кальция, декстрин, пектин и карбоксиметилцеллюлозу кальция.
Примеры смазочных средств включают стеарат магния, тальк, полиэтиленгликоль, диоксид кремния и гидрогенизированное растительное масло.
Используемые красители представляют собой красители, одобренные в качестве добавок к фармацевтическим средствам. Используемые корригенты представляют собой какао-порошок, мятную камфору, эмпазм, мятное масло, борнеол, порошкообразную кору коричного дерева и т.п.
Очевидно, что эти таблетки и гранулы могут быть покрыты сахаром или по мере необходимости покрыты иначе соответствующим способом. Жидкие препараты, такие как сиропы и инъецируемые препараты, получают добавлением регулятора pH, солюбилизатора, средства, регулирующего тоничность и тому подобное, и по мере необходимости солюбилизирующего средства, стабилизатора и т.п. к соединению или его фармакологически приемлемой соли по настоящему изобретению и составляя их общепринятым способом.
Способ получения препаратов для наружного применения не ограничен, и они могут быть получены общепринятыми способами. В частности, в качестве основных веществ для готовой формы можно использовать различное сырье, широко используемое для фармацевтических средств, квази-лекарственные средства, косметические средства и т.п. Конкретные примеры используемых основных веществ включают сырье, такое как животные и растительные масла, минеральные масла, сложноэфирные масла, воски, высшие спирты, жирные кислоты, силиконовое масло, поверхностно-активные вещества, фосфолипиды, спирты, многоатомные спирты, водорастворимые полимеры, глинистые минералы и очищенную воду. Кроме того, по мере необходимости можно добавлять регуляторы pH, антиоксиданты, хелатирующие вещества, консерванты и фунгициды, красители, вкусоароматические добавки и т.п. Основные вещества для препаратов для наружного применения по настоящему изобретению не ограничены этими веществами.
По мере необходимости также можно смешивать ингредиенты, такие как ингредиенты, обладающие индуцирующим дифференцировку действием, стимуляторы кровотока, бактерицидные средства, противовоспалительные средства, активаторы клеток, витамины, аминокислоты, увлажнители и кератолитические средства. Указанные выше основные вещества добавляют в количестве, соответствующем концентрации, как правило, выбираемой для получения препаратов для наружного применения.
Способ введения соединений или их солей, или гидратов соединений, или солей по настоящему изобретению конкретно не ограничен, и они могут быть введены перорально или парентерально широко используемыми способами. Например, они могут быть составлены в препараты, такие как таблетки, порошки, гранулы, капсулы, сиропы, пастилки, лекарственные формы для ингаляции, суппозитории, инъецируемые средства, мази, глазные мази, офтальмологические препараты, назальные препараты, препараты для ушей, горячие компрессы и лосьоны, и введены.
Соединения по настоящему изобретению являются особенно подходящими для состава в пероральных средствах, поскольку они обладают превосходной активностью передачи сигналов цАМФ и обладают метаболической стабильностью.
Дозу лекарственного средства по настоящему изобретению можно соответствующим образом выбирать в зависимости от тяжести симптома, возраста, пола, массы тела, способа введения, типа соли, конкретного типа заболевания и т.п.
Хотя доза значительно изменяется в зависимости от типа заболевания и тяжести симптома у пациента, возраста пациента, разницы пола и различий в чувствительности к лекарственным средствам у пациентов и т.п., доза обычно составляет приблизительно от 0,03 до 1000 мг, предпочтительно от 0,1 до 500 мг и более предпочтительно от 0,1 до 100 мг в сутки для взрослых, и ее вводят раздельно в виде одной-нескольких доз в сутки.
При получении соединений по настоящему изобретению, представленных указанной выше формулой (1), соединения сырья и различные реагенты могут образовывать соли, гидраты или сольваты, которые изменяются в зависимости от исходного вещества, используемого растворителя и тому подобное, и конкретно не ограничены в тех случаях, когда они не ингибируют реакцию.
Используемый растворитель также изменяется в зависимости от исходного вещества, реагента и тому подобное, и конкретно не ограничен в тех случаях, когда он не ингибирует реакцию и, конечно, растворяет исходное вещество до определенной степени.
Различные изомеры (например, геометрические изомеры, оптические изомеры на основе асимметрических атомов углерода, ротамеры, стереоизомеры и таутомеры) можно очистить и выделить, используя общепринятые средства разделения, например, перекристаллизацию, способы на основе диастереомерных солей, способы ферментативного расщепления и различные хроматографические способы (например, тонкослойную хроматографию, колоночную хроматографию, высокоэффективную жидкостную хроматографию и газовую хроматографию).
Соединения по настоящему изобретению, полученные в виде свободных форм, можно преобразовать в соли, которые могут быть образованы соединениями, или в гидраты соединений общепринятыми способами. Соединения по настоящему изобретению, полученные в виде солей или гидратов соединений, также можно преобразовать в свободные формы соединений общепринятыми способами.
Соединения по настоящему изобретению можно выделить и очистить, применяя обычные химические способы, такие как экстракция, концентрирование, выпаривание, кристаллизация, фильтрование, перекристаллизация и различные хроматографические способы.
Все документы известного уровня техники, цитируемые в настоящем описании, таким образом, включены посредством ссылки.
Общие способы синтеза
Соединения по настоящему изобретению можно синтезировать различными способами, некоторые из которых описаны со ссылкой на следующие схемы. Схемы являются иллюстративными, и настоящее изобретение не ограничено только явно указанными химическими реакциями и условиями. Хотя некоторые заместители не включены в следующие схемы во избежание двусмысленного толкования, такое исключение не предназначено ограничивать описание схем. Типичные соединения по настоящему изобретению можно синтезировать с использованием подходящих промежуточных соединений, известных соединений и реагентов. R1, R2, R3 и R4 в формулах в следующих общих способах синтеза являются такими, как определено для R1, R2, R3 и R4 в соединениях, представленных указанной выше общей формулой (1) (соединения, представленные формулой 1 в следующих общих способах синтеза).
Соединения по настоящему изобретению (формула 1) можно синтезировать способами получения (способы A и B), указанными ниже.
Схема 1 (способ A)
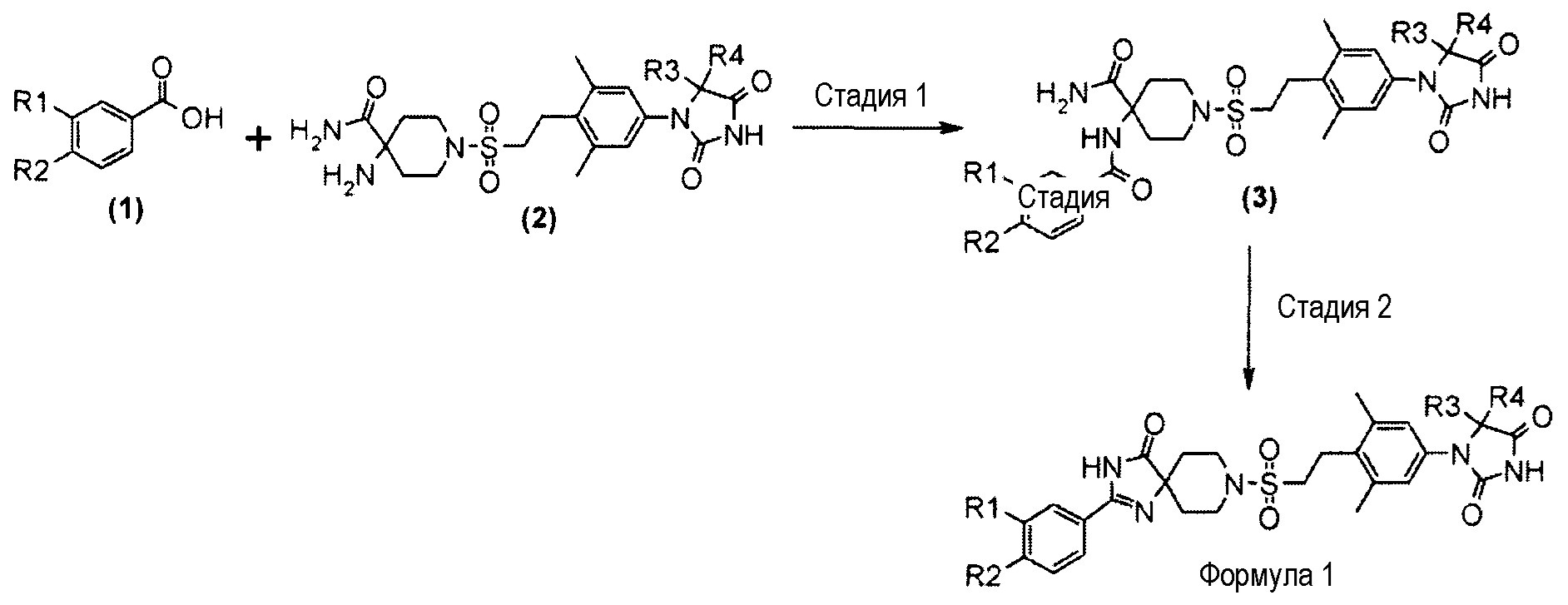
На схеме 1 представлен способ получения производного гидантоина (формула 1) путем амидирования производного карбоновой кислоты (1) и амино-амидного производного (2) с получением амид-амидного производного (3), а затем конструирования спироимидазолонового кольца посредством внутримолекулярной циклизации.
Стадия 1 представляет собой способ амидирования производного карбоновой кислоты (1) и амино-амидного производного (2). Примеры связывающего реагента включают N,N’-дициклогексилкарбодиимид (DCC), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC), гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU) и н-гидрат 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинийхлорида (DMT-MM). Примеры оснований включают триэтиламин или N,N-диизопропилэтиламин. При необходимости в качестве катализатора можно использовать 4-(диметиламино)пиридин (DMAP). Примеры подходящего растворителя включают дихлорметан или N,N-диметилформамид. Примеры подходящего растворителя реакционной смеси, когда используют DMT-MM, включают метанол, этанол и ацетонитрил. Температура реакции составляет, например, от 0°C до комнатной температуры, и время реакции составляет от 0,5 до 24 часов. Получаемое амино-амидное производное (3) выделяют обычным способом, и при необходимости его можно очистить методом кристаллизации или хроматографии.
Стадия 2 представляет собой способ циклизации амид-амидного производного (3) в присутствии подходящего основания, такого как водный раствор гидроксида натрия или трет-бутоксида калия в подходящем растворителе, таком как этанол, трет-бутанол или диметилсульфоксид. Температуру реакции обеспечивают, например, в условиях от комнатной температуры до температуры кипения с обратным холодильником в течение от одного до 24 часов. Полученное производное гидантоина (формула 1) выделяют обычными способами, и при необходимости его можно очистить методом кристаллизации или хроматографии.
Амино-амидное производное (2), указанное на схеме 1, можно синтезировать из производного пиперидина (4). Способ синтеза амино-амидного производного (2) представлен на схеме 2.
Схема 2
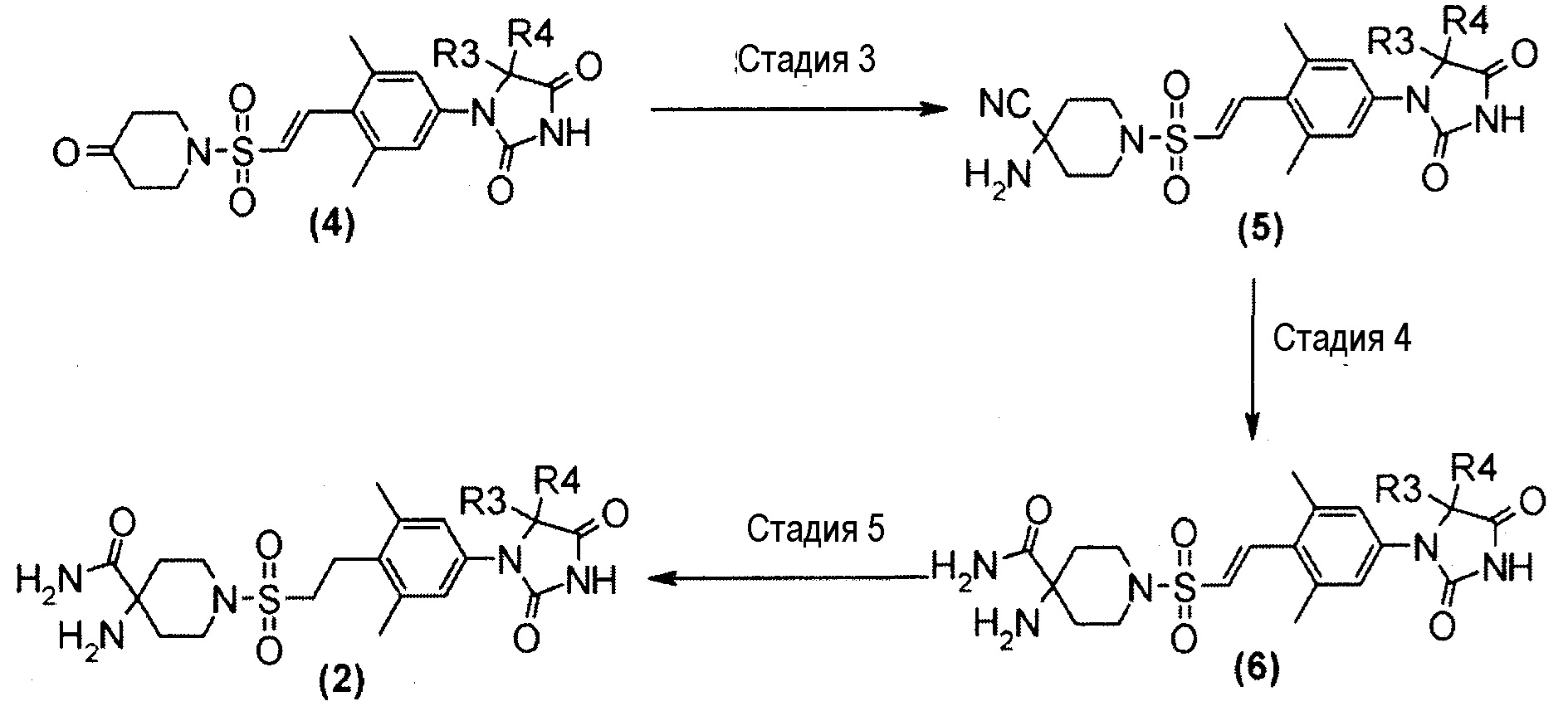
Стадия 3 представляет собой синтез Штрекера преобразования производного пиперидинона (4) в амино-нитрильное производное (5). В частности, он представляет собой способ взаимодействия пиперидинона (4) с цианидом натрия или цианидом калия и хлоридом аммония или ацетатом аммония в подходящем растворителе, таком как метанол, этанол или тетрагидрофуран в присутствии/отсутствие воды. Температура реакции находится в диапазоне от комнатной температуры до 80°C, например, и время реакции составляет от 2 до 72 часов. Получаемое амино-нитрильное производное (5) выделяют общепринятым способом и, при необходимости, его можно очистить методом кристаллизации или хроматографии.
Стадия 4 представляет собой способ преобразования нитрильной группы в амидогруппу в условиях щелочного гидролиза в присутствии пероксида водорода. Эту реакцию можно проводить со ссылкой, например, на Chemistry-A European Journal, (2002), 8(2), 439-450.
Стадия 5 представляет собой способ гидрирования олефинового соединения (6) в инертном растворителе, таком как метанол, этанол, трифторэтанол, диметилформамид или диметилацетамид, в присутствии катализатора, такого как палладированный уголь или гидроксид палладия на угле, соответственно, в атмосфере H2. Температура реакции находится в диапазоне от комнатной температуры до 80°C, и реакцию можно проводить под давлением. Получаемое амино-амидное производное (2) выделяют общепринятым способом и, при необходимости, его можно очистить методом кристаллизации или хроматографии.
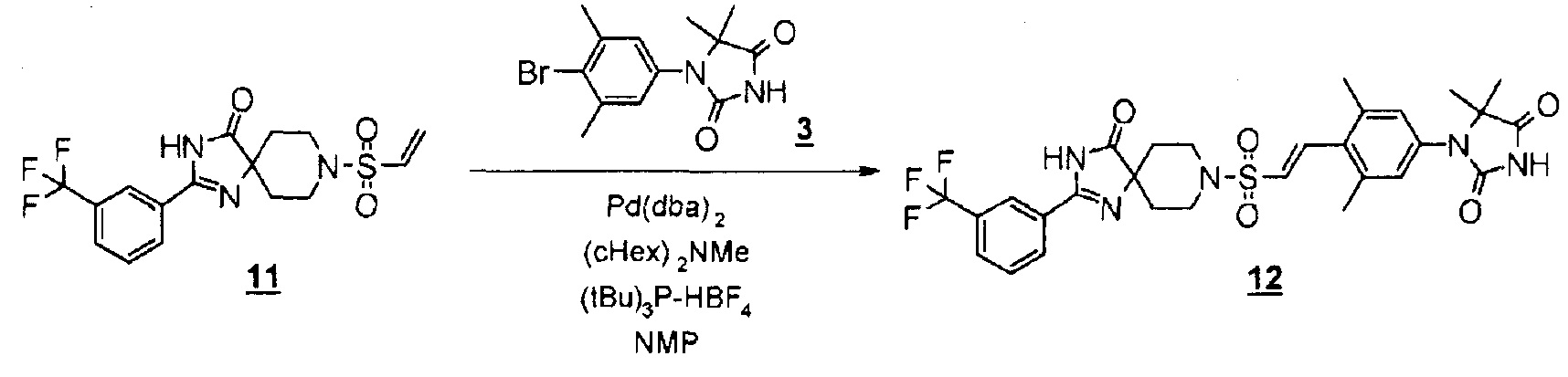
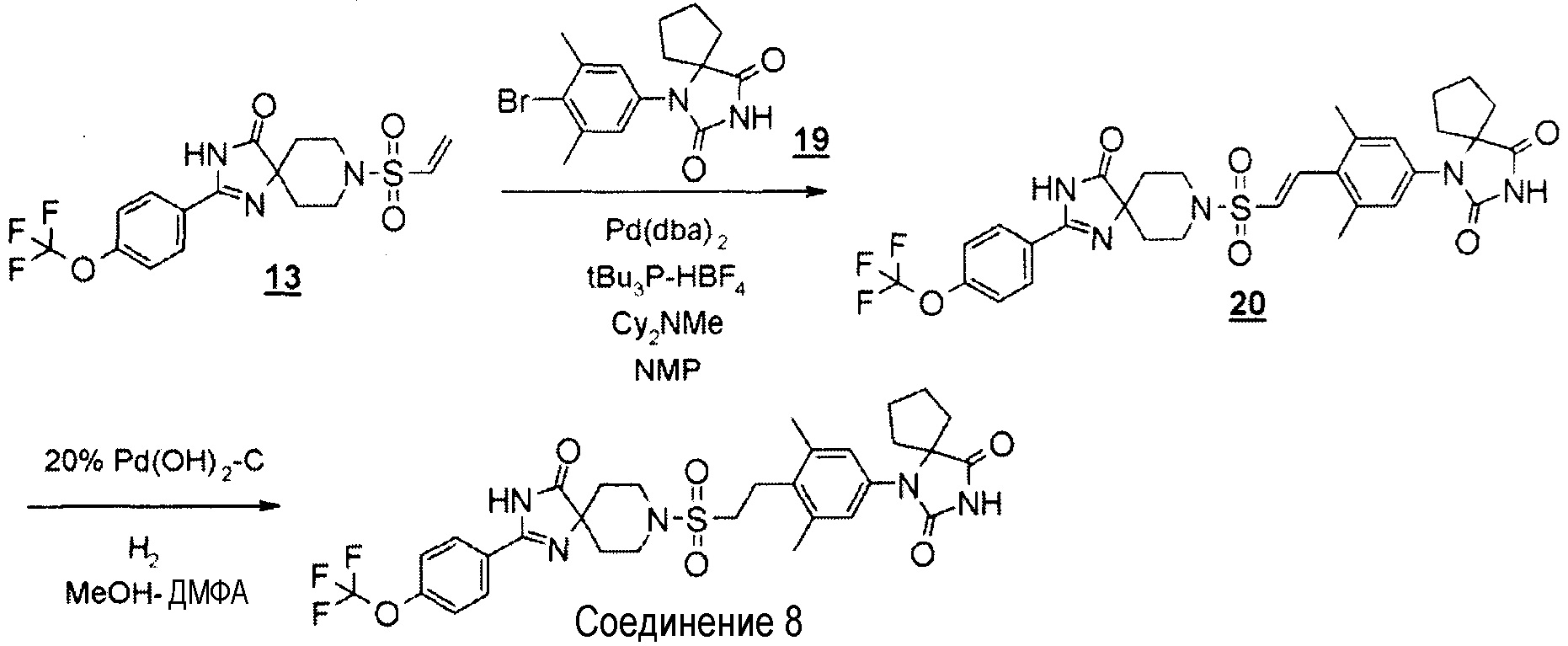
Представленное на схеме 2 производное пиперидинона (4) можно синтезировать из известного кетального производного винилсульфонила (7) и производного гидантоин-арилбромида (8). Способ синтеза производного пиперидина (4) представлен на схеме 3.
Схема 3
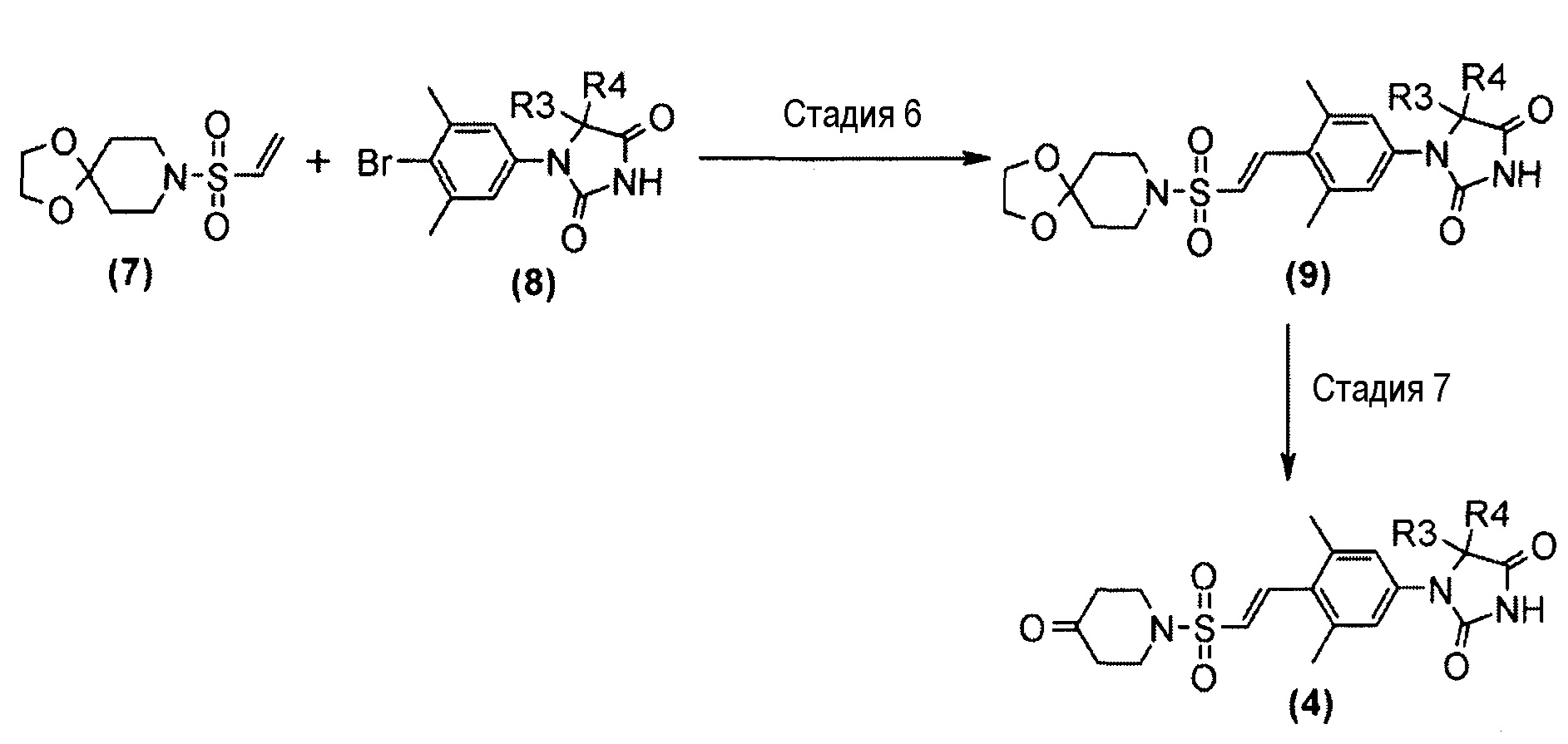
Стадия 6 представляет собой способ синтеза производного кеталь-арилвинилсульфонила (9) путем образования связи между кетальным производным винилсульфонила (7) и производным гидантоин-арилбромида (8) в атмосфере N2 в присутствии палладиевого катализатора, такого как трис(дибензилидинацетон)палладий(0) или бис(дибензилидинацетон)палладий, и добавления фосфинового лиганда, такого как три-трет-бутилфосфинтетрафторборная кислота, и подходящего основания, такого как метилдициклогексиламин, в подходящем растворителе, таком как N-метил-2-пиперидон (NMP). Температура реакции находится в диапазоне от 90°C до температуры кипения с обратным холодильником. Получаемое производное кеталь-арилвинилсульфонила (9) выделяют общепринятыми способами и, при необходимости, его можно очистить методом кристаллизации или хроматографии.
Стадия 7 представляет собой способ преобразования кеталя производного кеталь-арилвинилсульфонила (9) в кетон в подходящем растворителе, таком как водный тетрагидрофуран, в присутствии кислоты, такой как хлористоводородная кислота. Температура реакции представляет собой, например, температуру кипения растворителя, и время реакции составляет приблизительно от 1 до 24 часов. Получаемое производное пиперидина (4) выделяют общепринятыми способами и, при необходимости, его можно очистить методом кристаллизации или хроматографии.
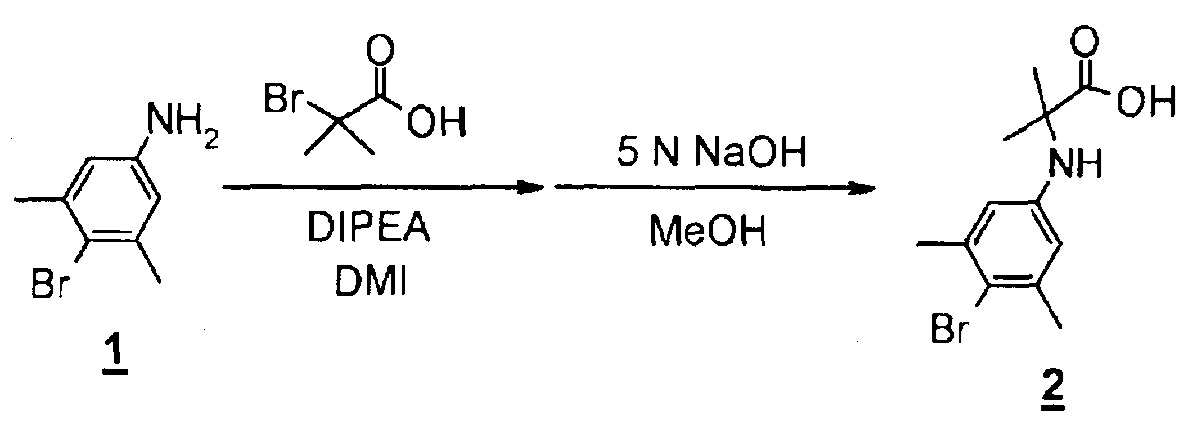
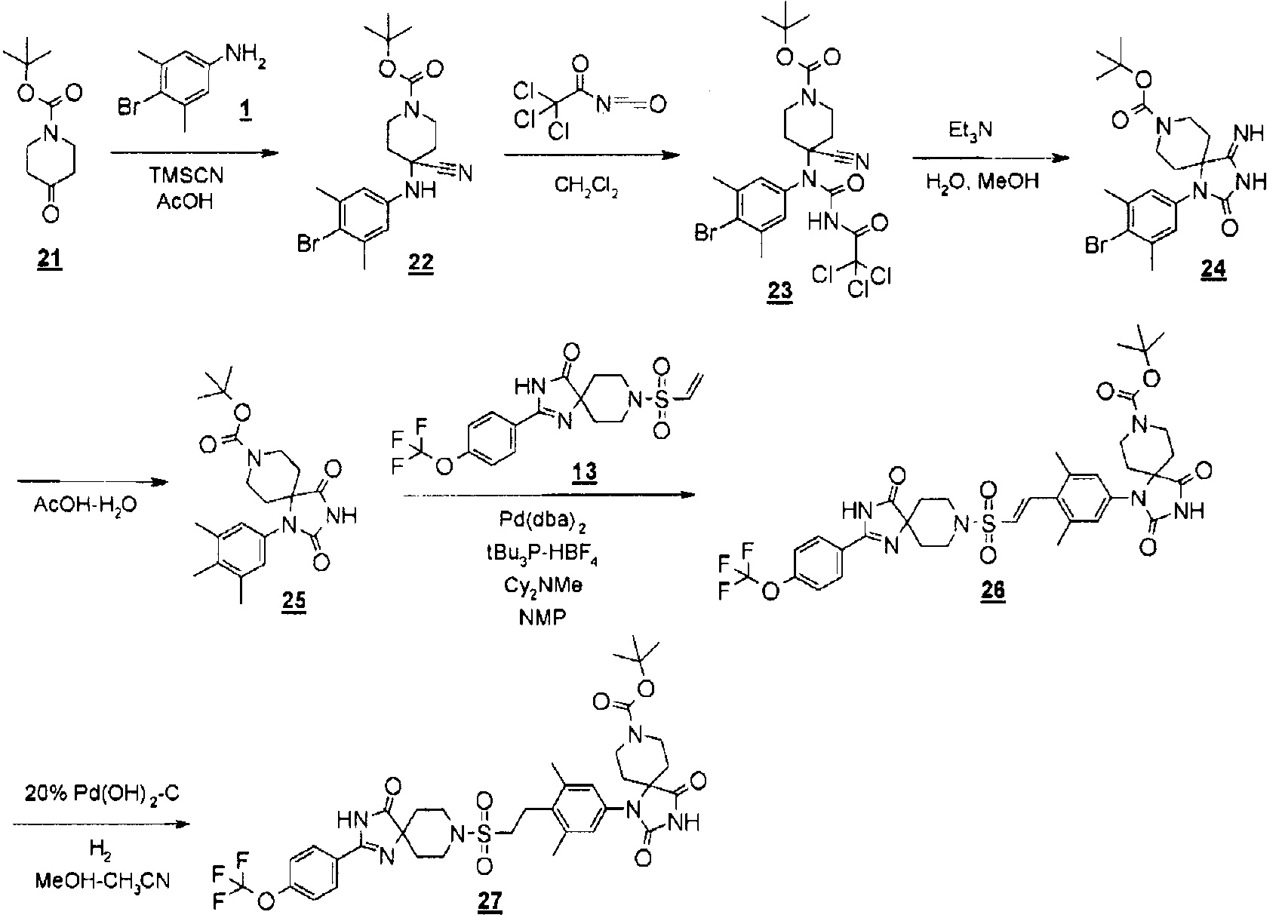
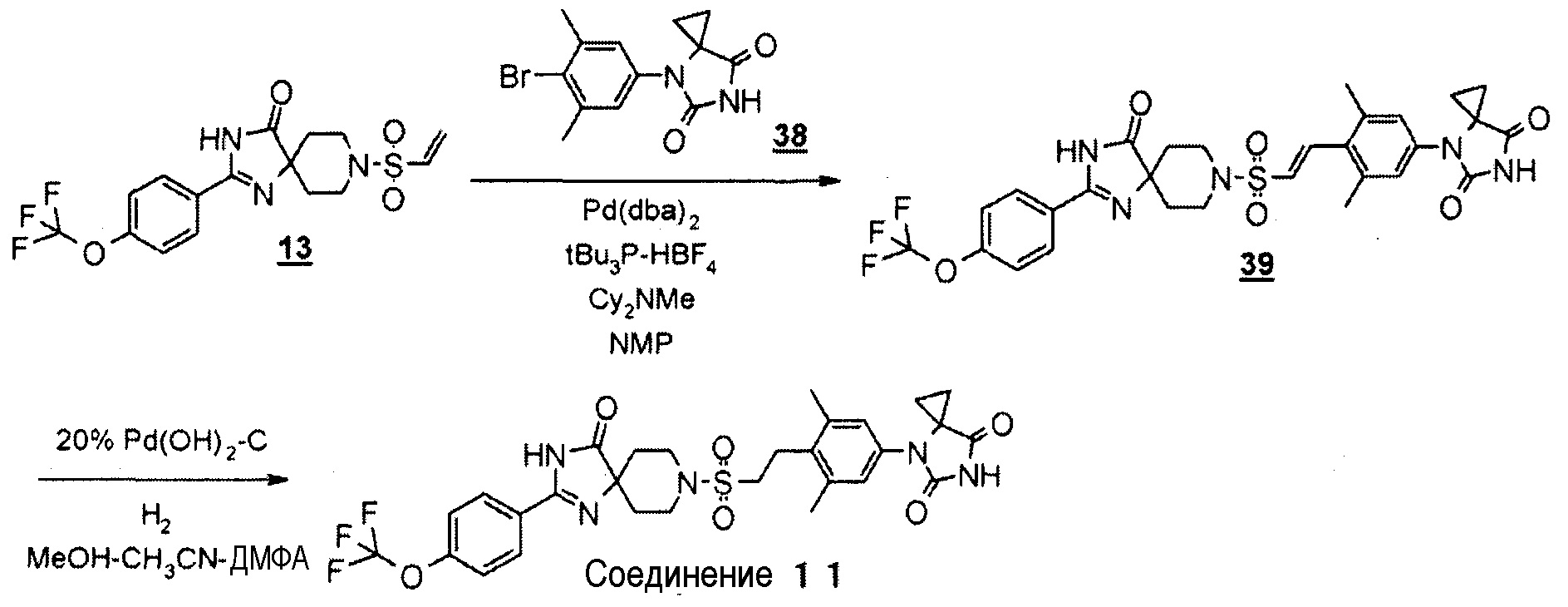
Производное гидантоин-арилбромида (8), представленное на схеме 3, можно синтезировать из 4-бром-3,5-диметиланилина (10) и производного бромуксусной кислоты (11), или из 2-бром-5-йод-1,3-диметилбензола (13) и производного аминокислоты (14). Способ синтеза производного гидантоин-арилбромида (8) представлен на схеме 4.
Схема 4
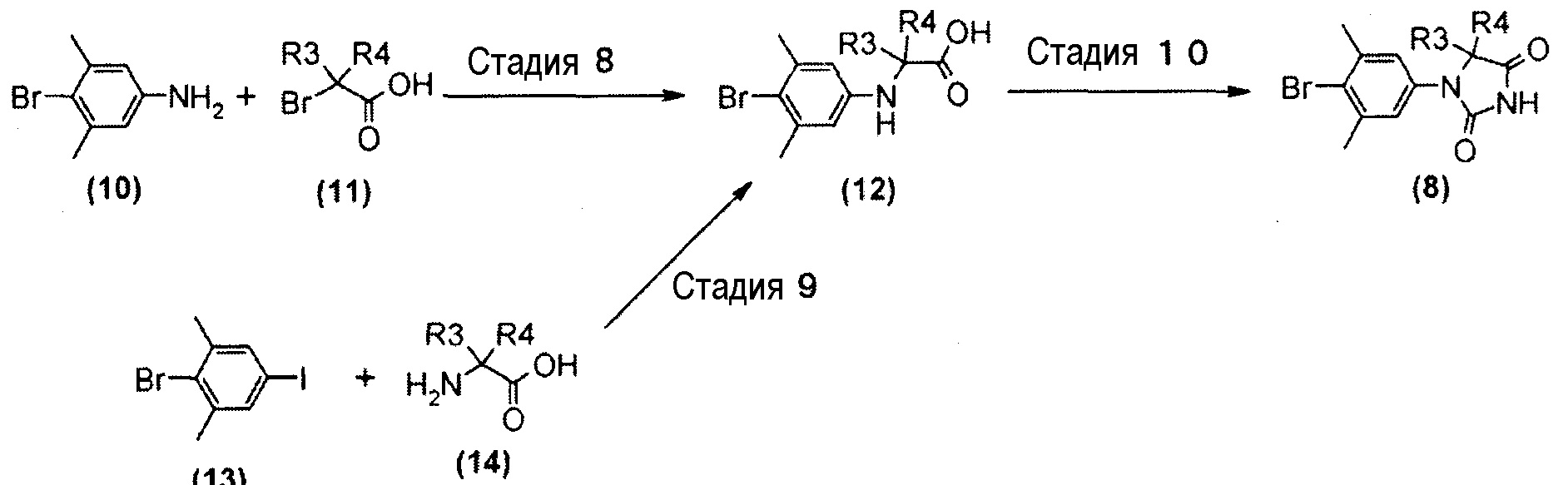
Стадия 8 представляет собой способ алкилирования 4-бром-3,5-диметиланилина (10) производным бромуксусной кислоты (11) в присутствии подходящего основания, такого как диизопропилэтиламин, и в подходящем растворителе, таком как N-метил-2-пиперидон (NMP). Температура реакции, например, находится в диапазоне от комнатной температуры до 100°C, и время реакции составляет от 1 до 24 часов. Получаемое производное арилбромид-аминокислоты (12) выделяют общепринятыми способами и, при необходимости, его можно очистить методом кристаллизации или хроматографии.
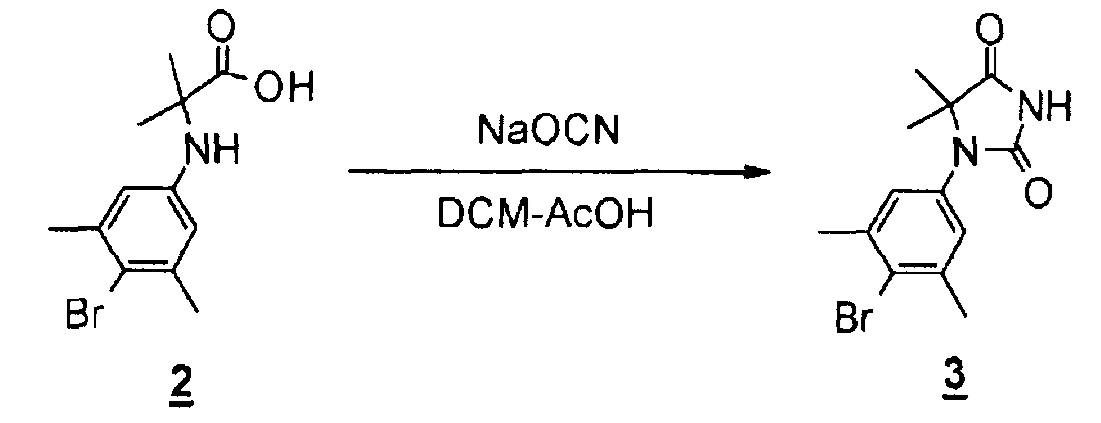
Стадия 9 представляет собой способ синтеза производного арилбромид-аминокислоты (12) путем образования связи между 2-бром-5-йод-1,3-диметилбензолом (13) и производным аминокислоты (14) в присутствии металлического катализатора, такого как йодид меди(I). Реакцию можно проводить в присутствии подходящего основания, такого как диазабициклоундецен (DBU), и в подходящем растворителе, таком как N,N-диметилацетамид (DMA), при температуре реакции приблизительно от 80°C до 120°C. Получаемое производное арилбромид-аминокислоты (12) выделяют общепринятыми способами и, при необходимости, его можно очистить методом кристаллизации или хроматографии.
Стадия 10 представляет собой способ синтеза производного гидантоин-арилбромида (8) путем взаимодействия производного арилбромид-аминокислоты (12) с цианатом натрия в кислой среде. Растворителем является, например, смешанный растворитель, такой как смесь уксусная кислота-дихлорметан; температура реакции находится в диапазоне от комнатной температуры до 60°C, и время реакции составляет от 1 до 24 часов. Получаемое производное гидантоин-арилбромида (8) можно выделить общепринятыми способами и, при необходимости, его можно очистить методом кристаллизации или хроматографии.
Производное гидантоин-арилбромида (8), представленное на схеме 3, также можно синтезировать из 4-бром-3,5-диметиланилина (10) и производного кетона (15). Способ синтеза производного гидантоин-арилбромида (8) представлен на схеме 5.
Схема 5
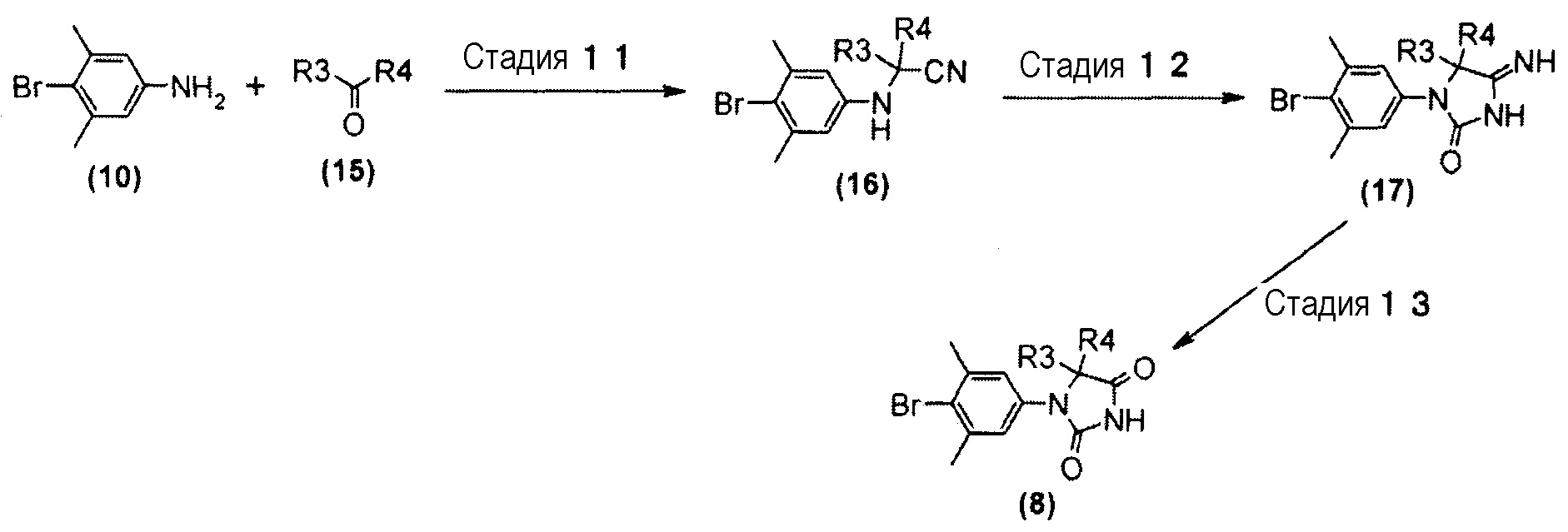
Стадия 11 представляет собой синтез Штрекера, в котором производное кетона (15) преобразуют в ариламино-нитрильное производное (16). Более конкретно, он представляет собой способ, в котором производное кетона (15) взаимодействует с 4-бром-3,5-диметиланилином (10) и триметилсилилцианидом в подходящем растворителе, таком как уксусная кислота. Температура реакции может представлять собой комнатную температуру, и время реакции составляет от одного до трех часов или около этого. Получаемое ариламино-нитрильное производное (16) выделяют общепринятыми способами и, при необходимости, его можно очистить кристаллизацией или хроматографией.
Стадия 12 представляет собой способ взаимодействия ариламино-нитрильного производного (16) с 2,2,2-трихлорацетилизоцианатом в подходящем растворителе, таком как дихлорметан, а затем синтеза производного иминогидантоина (17) путем добавления реагентов, таких как метанол, вода и триэтиламин, и обеспечения возможности их взаимодействия в условиях нагревания. Получаемое производное иминогидантоина (17) выделяют общепринятыми способами и, при необходимости, его можно очистить методом кристаллизации или хроматографии.
Стадия 13 представляет собой способ преобразования производного иминогидантоина (17) в производное гидантоин-арилбромида (8) в кислой среде. Например, синтез можно проводить в растворителе уксусная кислота-вода, нагревая приблизительно при 65°C в течение одного-шести часов или около этого. Получаемое производное гидантоин-арилбромида (8) выделяют общепринятыми способами и, при необходимости, его можно очистить методом кристаллизации или хроматографии.
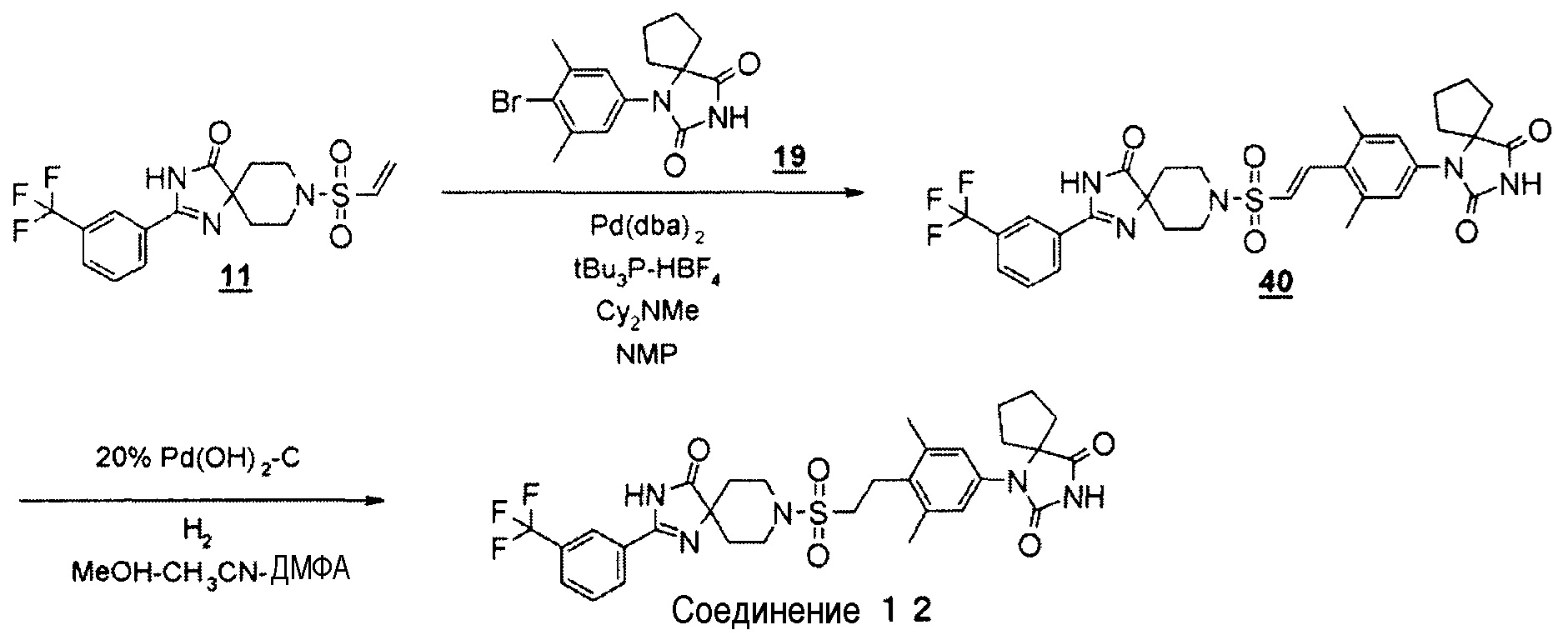
Схема 6 представляет собой способ реакции Хека производного винилсульфонамида (18) и производного гидантоин-арилбромида (8) в присутствии металлического катализатора, а затем гидрирования олефинового соединения (19) с получением производного гидантоина (формула 1).
Схема 6 (способ B)
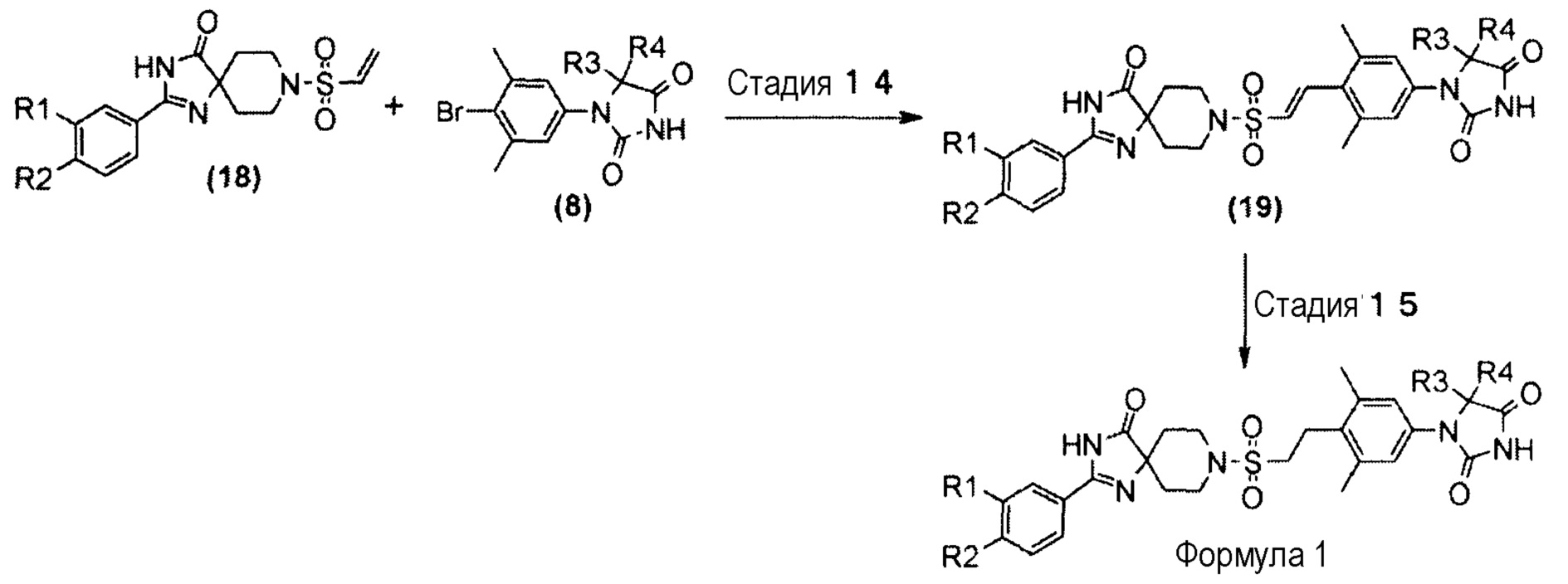
Производное гидантоина (формула 1) можно синтезировать посредством проведения реакции стадии 14 согласно способу стадии 6 и реакции стадии 15 согласно способу стадии 5. Получаемое производное гидантоина (формула 1) выделяют общепринятыми способами и, при необходимости, его можно очистить методом кристаллизации или хроматографии.
Винилсульфонамидное производное (18), используемое на стадии 14, можно синтезировать, обращаясь к схемам 2, 3 и 12 WO 010/126030(A1).
Все документы известного уровня техники, перечисленные в данном описании, включены в настоящее описание посредством ссылки.
[Примеры]
Содержание настоящего изобретения более подробно описано с помощью следующих примеров и тестового примера; однако настоящее изобретение не ограничено содержанием примеров и тестового примера. Все исходные вещества и реагенты получали от коммерческих поставщиков или синтезировали с использованием известных способов. Спектры 1H-ЯМР измеряли с использованием Mercury300 (производства Varian), ECP-400 (производства JEOL) или 400-MR (производства Varian) с Me4Si в качестве внутреннего стандарта или без него (с = синглет, д = дублет, т = триплет, уш.с = уширенный синглет, м = мультиплет). Масс-спектрометрическое измерение проводили с использованием масс-спектрометра ZQ2000 (производства Waters), SQD (производства Waters) или 2020 (производства Shimazu).
Пример 1
1-(4-(2-((2-(4-Фтор-3-(трифторметокси)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион (соединение 1)
Реакция (1-1)
К раствору 4-бром-3,5-диметиланилина (3,47 г, 17,4 ммоль) и диизопропилэтиламина (5,3 мл, 30,4 ммоль) в DMI (13 мл) добавляли 2-бромизомасляную кислоту (3,86 г, 23,1 ммоль) при комнатной температуре. Смесь перемешивали при 100°C в течение одного часа, а затем добавляли 2-бромизобутират (496 мг, 2,97 ммоль) и диизопропилэтиламин (0,8 мл, 4,59 ммоль) и перемешивали смесь при 100°C в течение одного часа.
К реакционной смеси при комнатной температуре добавляли метанол (52 мл) и 5 н. водный раствор гидроксида натрия (52 мл, 260 ммоль), а затем перемешивали эту смесь при 75°C в течение 1,5 часов. Реакционную смесь охлаждали с последующим добавлением воды и доведением pH до 5 с помощью 1 н. водного раствора гидросульфата калия, а затем экстрагировали с использованием этилацетата. Органический слой промывали водой, затем сушили над безводным сульфатом магния и концентрировали с получением выхода 2-((4-бром-3,5-диметилфенил)амино)-2-метилпропановой кислоты в виде неочищенного продукта (5,79 г). MS(ESI) m/z=286, 288 (M+H)+.
(Реакция 1-2)
К смеси 2-((4-бром-3,5-диметилфенил)амино)-2-метилпропановой кислоты (5,79 г соединения, полученного в результате реакции 1-1) в дихлорметане (62 мл) и уксусной кислоте (62 мл) добавляли цианат натрия (5,03 г, 59,8 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение трех часов. Добавляли насыщенный раствор гидрокарбоната натрия (400 мл) для доведения pH до 7-8 с помощью 5 н. водного гидроксида натрия и экстрагировали эту смесь этилацетатом. Органический слой сушили над безводным сульфатом магния, а затем концентрировали при пониженном давлении. Полученное твердое вещество последовательно промывали смесью этилацетат-гексан, а затем смесью дихлорметан-гексан с получением 1-(4-бром-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона (3,80 г, 66%).
MS(ESI) m/z=311, 313 (M+H)+
(Реакция 1-3)
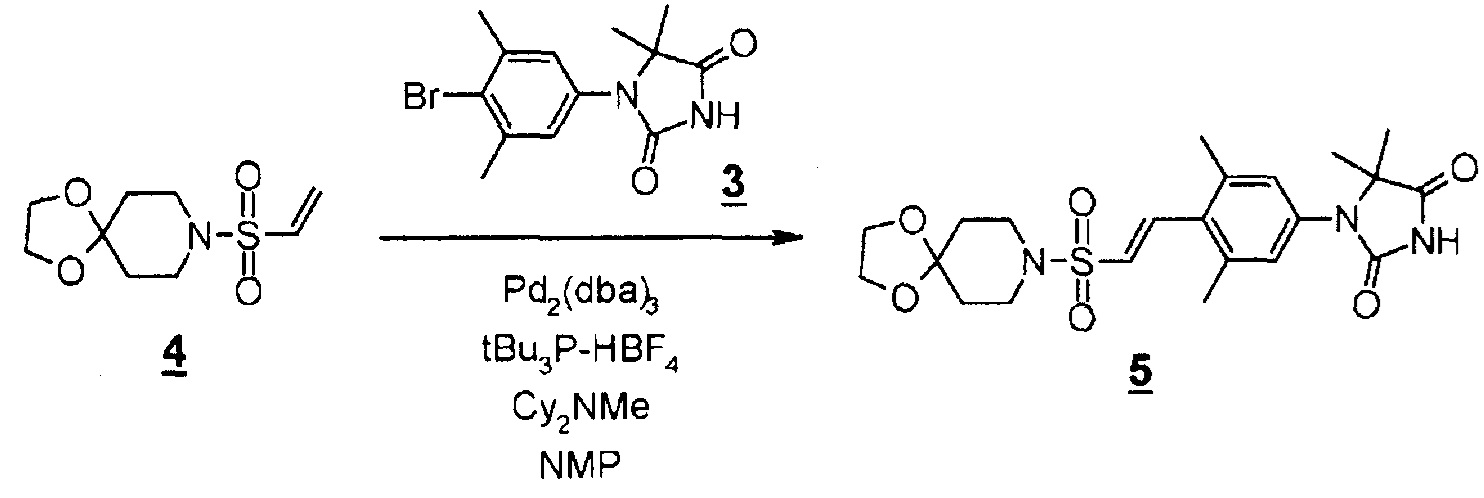
Смесь 8-(винилсульфонила)-1,4-диокса-8-азаспиро[4.5]декана (431 мг, 1,85 ммоль), 1-(4-бром-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона (575 мг, 1,85 ммоль), трис(дибензилидинацетон)палладия(0) (508 мг, 0,55 ммоль), три-трет-бутилфосфинтетрафторборной кислоты (165 мг, 0,55 ммоль) и метилдициклогексиламина (2,1 мл, 9,25 ммоль) в N-метил-2-пирролидоне (18,5 мл) перемешивали в атмосфере азота при 110°C в течение двух часов. Реакционную смесь охлаждали, гасили водой, а затем экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния, а затем концентрировали при пониженном давлении. Остаток очищали методом колоночной хроматографии на аминосиликагеле (дихлорметан-метанол) с получением (E)-1-(4-(2-(l,4-диокса-8-азаспиро[4.5]декан-8-илсульфонил)винил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона (584 мг, 68%).
MS(ESI) m/z=464 (M+H)+
(Реакция 1-4)
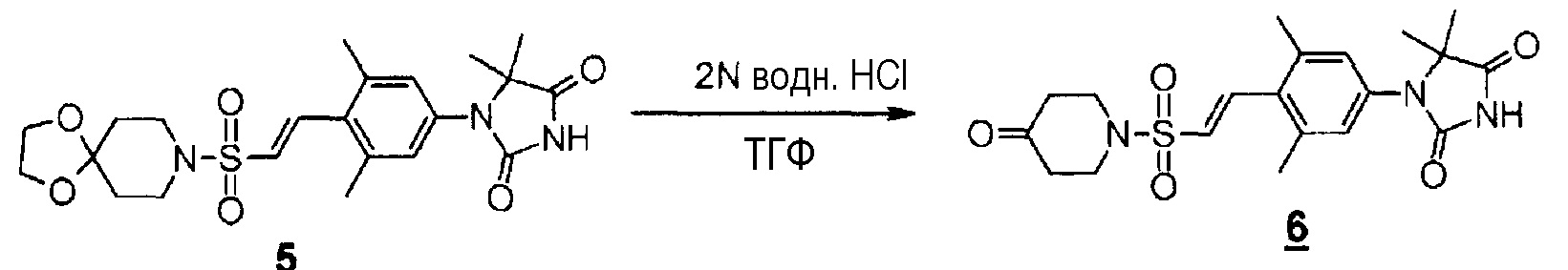
К раствору (E)-1-(4-(2-(1,4-диокса-8-азаспиро[4.5]декан-8-илсульфонил)винил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона (1,2 г, 2,58 ммоль) в тетрагидрофуране (26 мл) добавляли по каплям 2 н. водный раствор хлористоводородной кислоты (26 мл, 52 ммоль) в течение десяти минут. Смесь перемешивали при 60°C в течение двух часов. Реакционную смесь охлаждали, с последующим доведением ее pH до 7 с помощью 2 н. водного раствора гидроксида натрия и экстрагировали эту смесь этилацетатом. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом магния, а затем концентрировали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан-этилацетат) с получением (E)-1-(3,5-диметил-4-(2-((4-оксопиперидин-1-ил)сульфонил)винил)фенил)-5,5-диметилимидазолидин-2,4-диона (998 мг, 92%).
MS(ESI) m/z=420 (M+H)+
(Реакция 1-5)
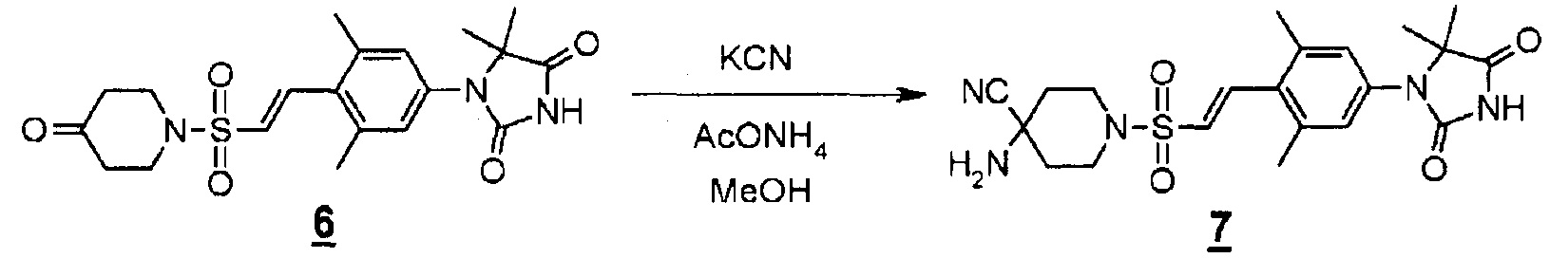
К раствору (E)-1-(3,5-диметил-4-(2-((4-оксопиперидин-1-ил)сульфонил)винил)фенил)-5,5-диметилимидазолидин-2,4-диона (994 мг, 2,37 ммоль) в метаноле (24 мл) добавляли цианид калия (188 мг, 2,84 ммоль) и ацетат аммония (237 мг, 3,08 ммоль) при комнатной температуре. Смесь перемешивали при 60-70°C в течение трех часов. Реакционную смесь охлаждали, концентрировали при пониженном давлении, а затем разбавляли этилацетатом. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния, а затем концентрировали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан-этилацетат) с получением (E)-4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилстирил)сульфонил)пиперидин-4-карбонитрила (681 мг, 68%).
1H-ЯМР (300 МГц, ДМСО-d6) δ: 1,3 (6H, с), 1,7 (2H, м), 2,0 (2H, м), 2,3 (6H, с), 2,7 (2H, с), 2,9 (2H, м), 3,4 (2H, м), 6,9 (1H, д, J=15,9 Гц), 7,1 (2H, с), 7,4 (1H, д, J=15,9 Гц), 11,2 (1H, уш.с.).
(Реакция 1-6)
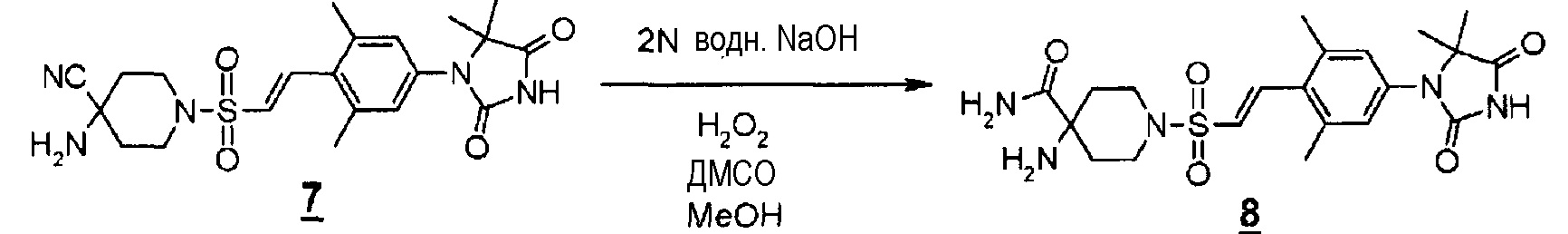
К раствору (E)-4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилстирил)сульфонил)пиперидин-4-карбонитрила (675 мг, 1,50 ммоль) в метаноле (7,5 мл) и диметилсульфоксиде (0,195 мл) при комнатной температуре добавляли 2 н. водный раствор гидроксида натрия (1,6 мл, 1,6 ммоль), а затем медленно по каплям добавляли 30% водный раствор пероксида водорода (0,2 мл, 1,95 ммоль). Смесь перемешивали при комнатной температуре в течение одного часа. К реакционной смеси добавляли этилацетат, гексан и насыщенный водный раствор хлорида аммония. Твердое вещество собирали фильтрованием, промывали и сушили с получением (E)-4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилстирил)сульфонил)пиперидин-4-карбоксамида (498 мг, 72%).
MS(ESI) m/z=464 (M+H)+
(Реакция 1-7)
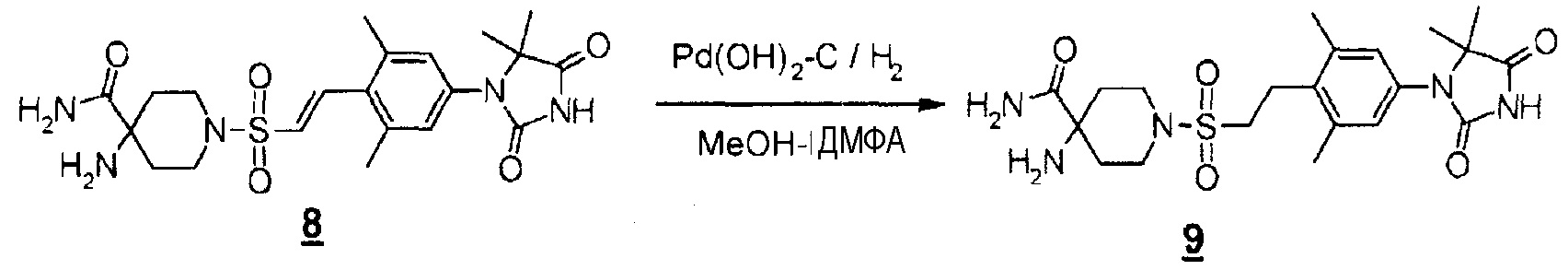
Смесь (E)-4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилстирил)сульфонил)пиперидин-4-карбоксамида (1,3 г, 2,8 ммоль) и гидроксида палладия на угле (20% Pd) (увлажненного приблизительно 50% воды) (1,3 г) в метаноле (21 мл) и диметилформамиде (7 мл) перемешивали в атмосфере водорода при комнатной температуре в течение четырех часов. Реакционную смесь фильтровали и промывали, а затем концентрировали фильтрат при пониженном давлении с получением 4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилфенетил)сульфонил)пиперидин-4-карбоксамида (998 мг, 77%).
MS(ESI) m/z=466 (M+H)+
(Реакция 1-8)
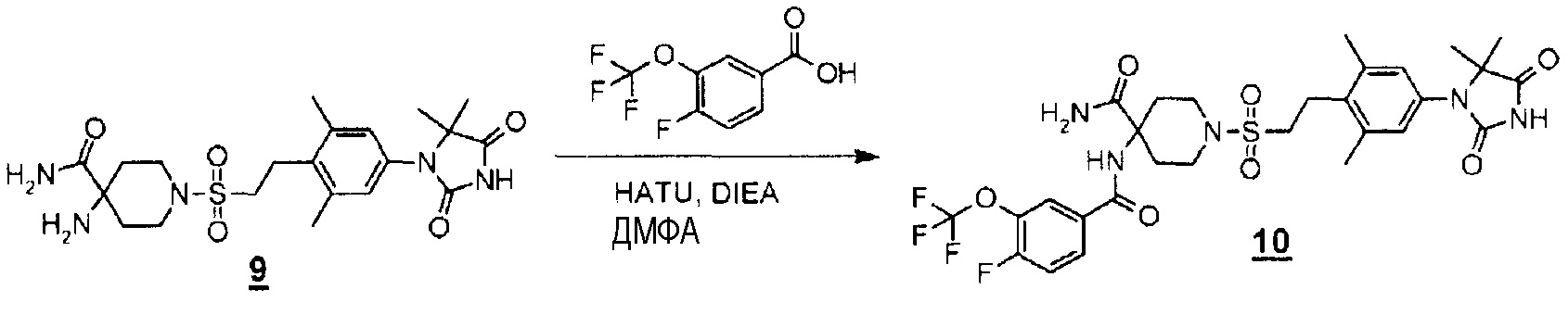
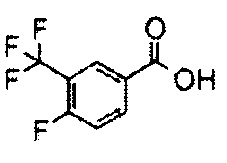
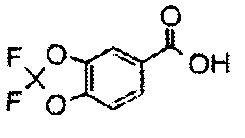
К раствору 4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилфенетил)сульфонил)пиперидин-4-карбоксамида (120 мг, 0,258 ммоль) добавляли 4-фтор-3-(трифторметокси)бензойную кислоту (69 мг, 0,309 ммоль) и диизопропилэтиламин (0,09 мл, 0,516 ммоль) в диметилформамиде (2,5 мл), гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU) (118 мг, 0,309 ммоль). Смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь гасили водой, а затем экстрагировали дихлорметаном. Органический слой промывали насыщенным раствором соли, промывали безводным сульфатом натрия, а затем концентрировали при пониженном давлении с получением 1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилфенетил)сульфонил)-4-(4-фтор-3-(трифторметокси)бензамид)пиперидин-4-карбоксамида (150 мг, 67%).
MS(ESI) m/z=672 (M+H)+
(Реакция 1-9)
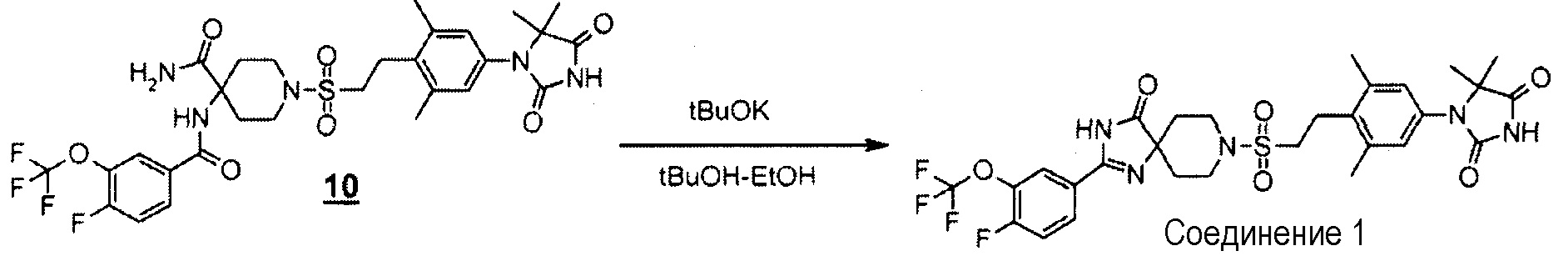
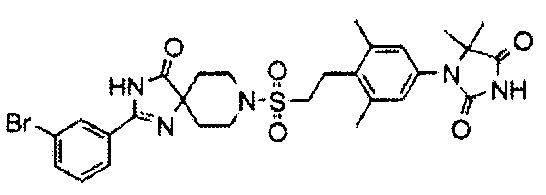
К перемешиваемому раствору 1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилфенетил)сульфонил)-4-(4-фтор-3-(трифторметокси)бензамид)пиперидин-4-карбоксамида (150 мг, 0,223 ммоль) в трет-бутаноле (2,5 мл) и этаноле (2,5 мл) добавляли трет-бутоксид калия (75 мг, 0,670 ммоль) при 0°C. Смесь перемешивали в атмосфере азота при 50°C в течение 1,5 часов. Реакционную смесь охлаждали, разбавляли водой, гасили насыщенным водным раствором хлорида аммония, а затем экстрагировали дихлорметаном. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, а затем концентрировали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан-метанол) с получением 1-(4-(2-((2-(4-фтор-3-(трифторметокси)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона 118 мг, 81%.
MS(ESI) m/z=654 (M+H)+. 1H-ЯМР (400 МГц, CD3OD) δ: 1,40 (6H, с), 1,71-1,80 (2H, м), 2,00-2,08 (2H, м), 2,43 (6H, с), 3,22 (4H, с), 3,47-3,57 (2H, м), 3,80-3,88 (2H, м), 7,01 (2H, с), 7,50-7,57 (1H, м), 7,97-8,04 (1H, м), 8,05-8,12 (1H, м).
Следующие соединения из примеров синтезировали, способами, аналогичными способам реакций 1-8 и 1-9 в примере 1, используя подходящие исходные вещества на основе карбоновой кислоты, реагенты и растворители.
(Соединения 2-5)
|
|
Пример 2
1-(3,5-Диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион (соединение 6)
(Реакция 2-1)
Смесь 2-(3-(трифторметил)фенил)-8-(винилсульфонил)-1,3,8-триазаспиро[4.5]дека-1-ен-4-она (150 мг, 0,387 ммоль), синтезируемую способом, описанным на схемах 2, 3 и 12 WO2010/126030 (A1), 1-(4-бром-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона (169 мг, 0,542 ммоль), бис(дибензилидинацетон)палладия (45 мг, 0,077 ммоль), три-трет-бутилфосфинтетрафторборной кислоты (22 мг, 0,077 ммоль) и метилдициклогексиламина (0,123 мл, 0,581 ммоль) в N-метил-2-пирролидоне (0,97 мл) перемешивали при 100°C в течение одного часа в атмосфере азота. Реакционную смесь охлаждали, гасили водой, а затем экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, а затем концентрировали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (этилацетат-гексан) с получением (E)-1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)винил)фенил)-5,5-диметилимидазолидин-2,4-диона (197 мг, 82%).
MS(ESI) m/z=618 (M+H)+
(Реакция 2-2)
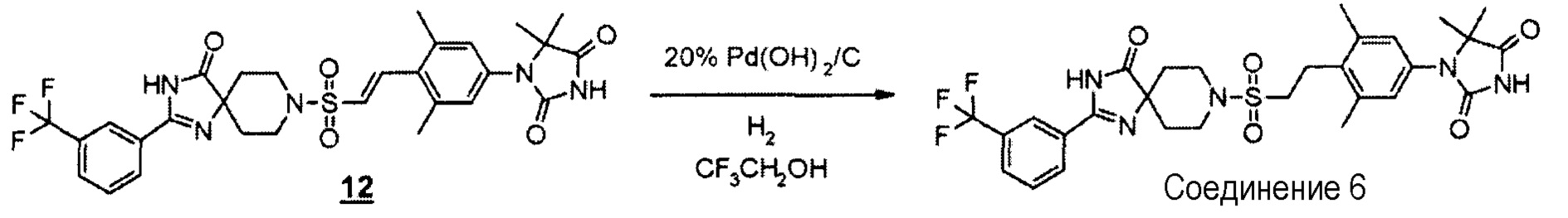
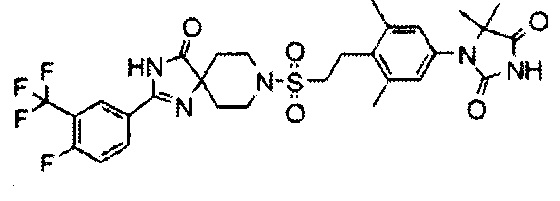
Смесь (E)-1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)винил)фенил)-5,5-диметилимидазолидин-2,4-диона (195 мг, 0,316 ммоль) и гидроксида палладия на угле (20% Pd) (увлажненного приблизительно 50% воды) (195 мг, 0,139 ммоль) в 2,2,2-трифторэтаноле (6 мл) перемешивали при комнатной температуре в течение 14 часов в атмосфере водорода. Смесь фильтровали и концентрировали фильтрат при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (этилацетат-гексан) с получением 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-диона (121 мг, 62%).
MS(ESI) m/z=620 (M+H)+. 1H-ЯМР (400 МГц, CD3OD) δ: 1,40 (6H, с), 1,72-1,81 (2H, м), 2,00-2,10 (2H, м), 2,44 (6H, с), 3,22 (4H, с), 3,50-3,58 (2H, м), 3,80-3,88 (2H, м), 7,01 (2H, с), 7,72-7,79 (1H, м), 7,88-7,94 (1H, м), 8,16-8,23 (1H, м), 8,31 (1H, с).
Пример 3
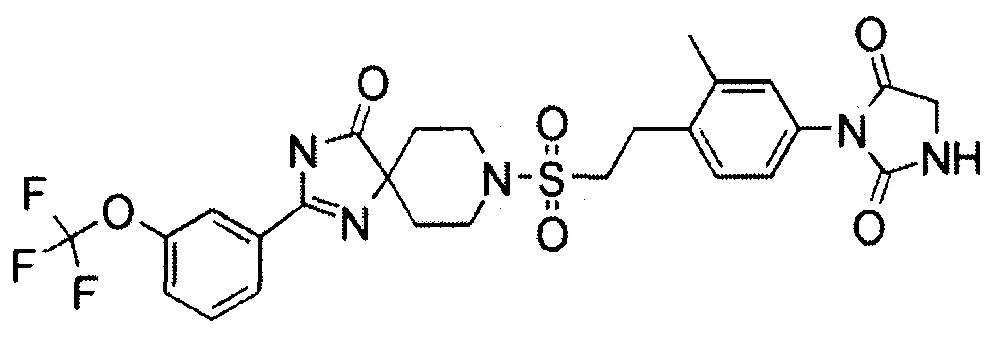
1-(3,5-Диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион (соединение 7)
(Реакция 3)
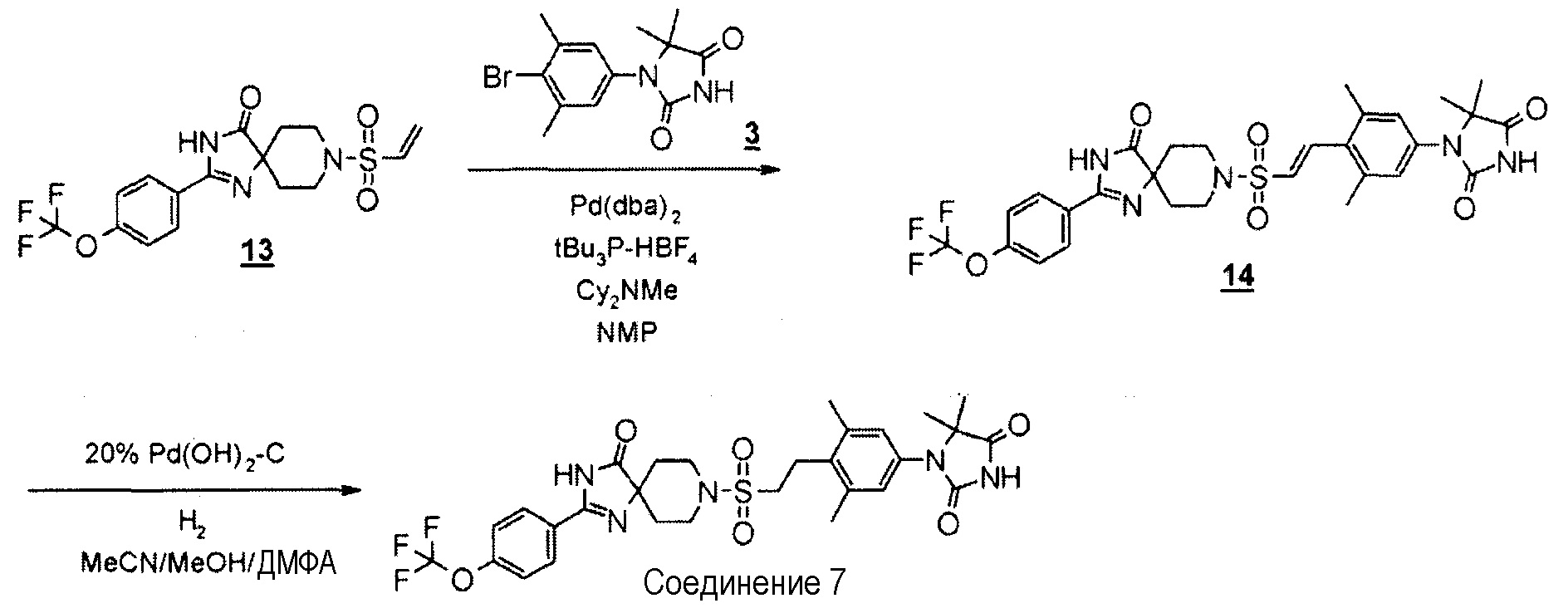
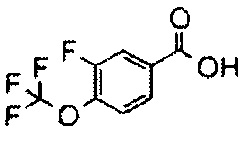
С использованием подходящих исходных веществ и растворителей 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион (соединение 7) синтезировали, проводя процессы, аналогичные процессам, описанным в примере 2.
MS(ESI) m/z=636 (M+H)+. 1H-ЯМР (400 МГц, CDCl3) δ: 1,47 (6H, с), 1,70-1,78 (2H, м), 2,10-2,19 (2H, м), 2,40 (6H, с), 3,00-3,07 (2H, м), 3,19-3,25 (2H, м), 3,45-3,53 (2H, м), 3,81-3,88 (2H, м), 6,94 (2H, с), 7,35 (2H, д, J=8,0 Гц), 7,73 (1H, уш.с.), 7,93 (2H, д, J=8,0 Гц), 9,37 (1H, уш.с.).
Пример 4
1-(3,5-Диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4.4]нонан-2,4-дион (соединение 8)
(Реакция 4-1)
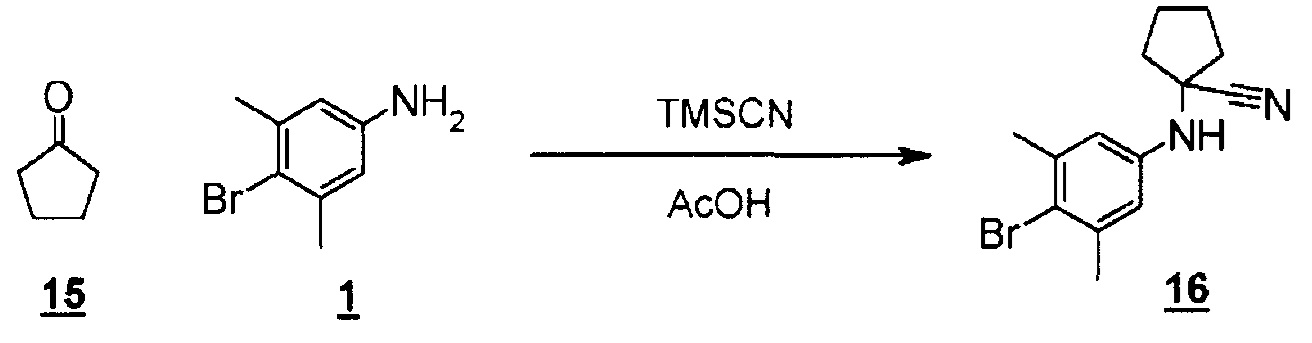
К смеси циклопентанона (42 мг, 0,500 ммоль) и 4-бром-3,5-диметиланилина (100 мг, 0,500 ммоль) в уксусной кислоте (0,5 мл) добавляли триметилсилилцианид (0,063 мл, 0,500 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 1,5 часов в атмосфере азота. Реакционную смесь гасили 28% водным аммиаком (1 мл), разбавляли водой и экстрагировали дихлорметаном. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, а затем концентрировали при пониженном давлении с получением 1-((4-бром-3,5-диметилфенил)амино)циклопентанкарбонитрила в виде неочищенного продукта (152 мг).
1H-ЯМР (400 МГц, CDCl3) δ: 1,83-1,92 (4H, м), 2,07-2,15 (2H, м), 2,33-2,42 (2H, м), 2,37 (6H, м), 3,71 (1H, уш.с.), 6,56 (2H, с).
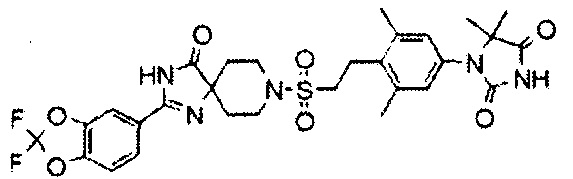
(Реакция 4-2)
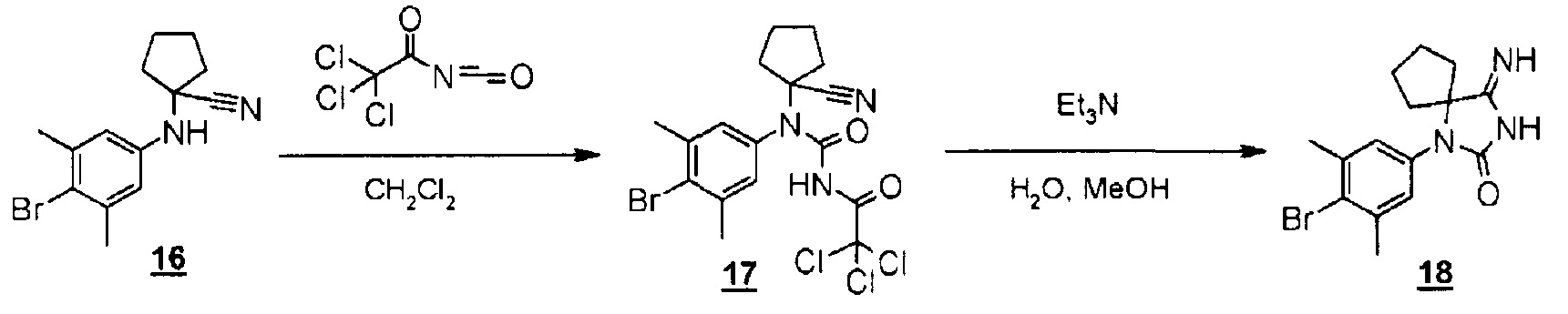
К раствору 1-((4-бром-3,5-диметилфенил)амино)циклопентанкарбонитрила (145 мг, 0,495 ммоль) в дихлорметане (5 мл) добавляли 2,2,2-трихлорацетилизоцианат (0,070 мл, 0,593 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение одного часа в атмосфере азота.
Добавляли триэтиламин (0,103 мл, 0,742 ммоль), воду (0,045 мл) и метанол (0,10 мл) и смесь кипятили с обратным холодильником в течение 1,5 часов в атмосфере азота. Реакционную смесь охлаждали с последующим разбавлением водой и доведением ее pH до 5 с помощью 1 н. водного раствора хлористоводородной кислоты, а затем экстрагировали дихлорметаном. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, а затем концентрировали при пониженном давлении с получением 1-(4-бром-3,5-диметилфенил)-4-имино-1,3-диазаспиро[4.4]нонан-2-она в виде неочищенного продукта. MS(ESI) m/z=336, 338 (M+H)+
(Реакция 4-3)
Смесь 1-(4-бром-3,5-диметилфенил)-4-имино-1,3-диазаспиро[4.4]нонан-2-она (неочищенный продукт, полученный в предшествующей реакции) в уксусной кислоте (1,0 мл) и воде (0,25 мл) перемешивали в течение 1,5 часов при 65°C в атмосфере азота. После дополнительного добавления уксусной кислоты (1,0 мл) и воды (0,25 мл) смесь перемешивали в течение 17 часов при 65°C в атмосфере азота. Реакционную смесь охлаждали с последующим разбавлением водой и доведением ее pH до 8 с помощью насыщенного водного раствора гидрокарбоната натрия и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, а затем концентрировали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (этилацетат-гексан) с получением 1-(4-бром-3,5-диметилфенил)-1,3-диазаспиро[4.4]нонан-2,4-диона (121 мг).
MS(ESI) m/z=337, 339 (M+H)+
(Реакция 4-4)
С использованием подходящих исходных веществ и растворителей получали 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4.4]нонан-2,4-дион (соединение 8), проводя процессы, аналогичные процессам, описанным в примере 2.
MS(ESI) m/z=662 (M+H)+. 1H-ЯМР (400 МГц, ДМСО-d6) δ: 1,36-1,44 (2H, м), 1,60-1,70 (4H, м), 1,82-1,91 (2H, м), 1,91-2,06 (4H, м), 2,38 (6H, с), 3,01-3,09 (2H, м), 3,22-3,30 (2H, м), 3,30-3,42 (2H, м), 3,70-3,77 (2H, м), 7,03 (2H, с), 7,57 (2H, д, J=8,4 Гц), 8,14 (2H, д, J=8,4 Гц).
Пример 5
1-(3,5-Диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-8-метил-1,3,8-триазаспиро[4.5]декан-2,4-дион (соединение 9)
(Реакция 5-1)
С использованием сложного трет-бутилового эфира 4-оксопиперидин-1-карбоновой кислоты в качестве исходного вещества и использованием подходящего растворителя получали сложный трет-бутиловый эфир 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2,4-диоксо-1,3,8-триазаспиро[4.5]декан-8-карбоновой кислоты, проводя процессы, аналогичные процессам, описанным в примере 4.
MS(ESI) m/z=777 (M+H)+.
(Реакция 5-2)
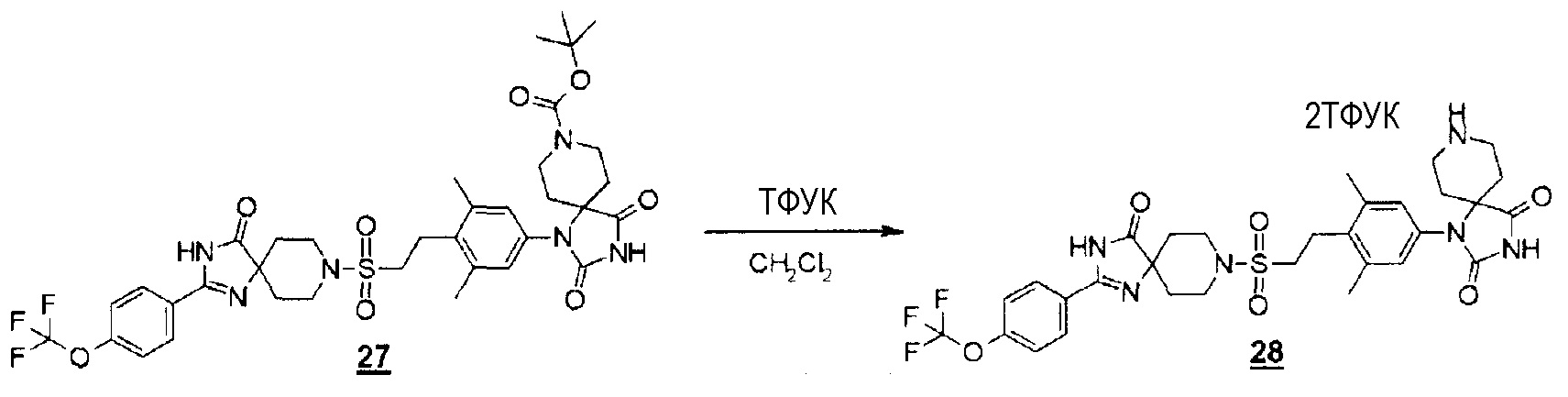
К перемешиваемому раствору сложного трет-бутилового эфира 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2,4-диоксо-1,3,8-триазаспиро[4.5]декан-8-карбоновой кислоты (11,7 мг, 0,015 ммоль) в дихлорметане (0,13 мл) добавляли трифторуксусную кислоту (0,05 мл, 0,673 ммоль) при комнатной температуре. Смесь помещали в ток азота и перемешивали при комнатной температуре в течение одного часа. Реакционную смесь концентрировали при пониженном давлении с получением соли 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3,8-триазаспиро[4.5]декан-2,4-дион-2-трифторуксусной кислоты (13,6 мг).
MS(ESI) m/z=677 (M+H)+.
(Реакция 5-3)
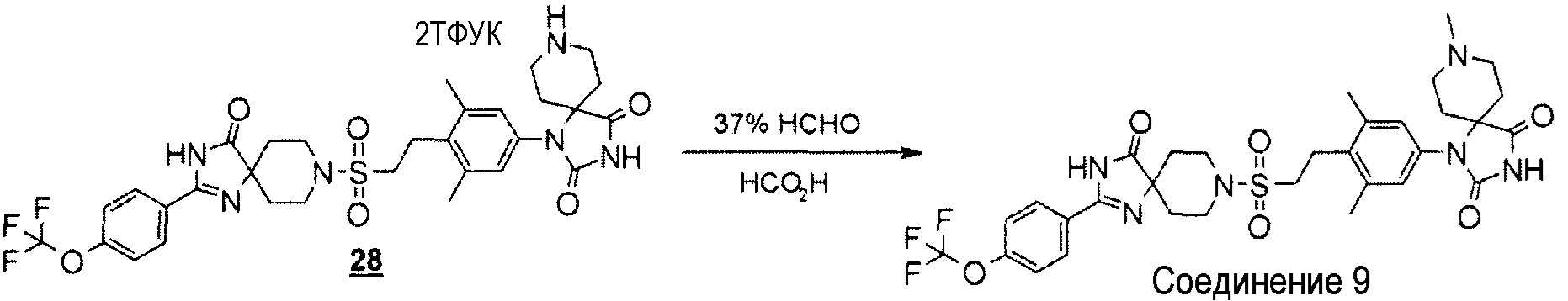
К смеси соли 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3,8-триазаспиро[4.5]декан-2,4-дион-2-трифторуксусной кислоты (21,1 мг, 0,022 ммоль) и муравьиной кислоты (0,033 мл) добавляли 37% водный раствор формальдегида (0,055 мл). Смесь помещали в ток азота и перемешивали в течение трех часов, нагревая при температуре 80°C. Реакционную смесь концентрировали и полученный остаток разбавляли этилацетатом. Органический слой промывали разбавленным водным раствором гидроксида натрия, сушили над безводным сульфатом магния, а затем концентрировали при пониженном давлении. Полученный остаток подвергали колоночной хроматографии (дихлорметан-метанол) для очистки с получением 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил-8-метил-1,3,8-триазаспиро[4.5]декан-2,4-диона (4,5 мг, 30%).
MS(ESI) m/z=691 (M+H)+. 1H-ЯМР (400 МГц, CD3OD) δ: 1,76-1,84 (2H, м), 1,92-2,02 (2H, м), 2,02-2,12 (4H, м), 2,38 (3H, с), 2,46 (6H, с), 2,81-2,88 (2H, м), 2,92-3,02 (2H, м), 3,23 (4H, с), 3,51-3,60 (2H, м), 3,72-3,80 (2H, м), 7,01 (2H, с), 7,48 (2H, д, J=8,0 Гц), 8,10 (2H, д, J=8,0 Гц).
Пример 6
5-(3,5-Диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2-окса-5,7-диазаспиро[3.4]октан-6,8-дион (соединение 10)
(Реакция 6)
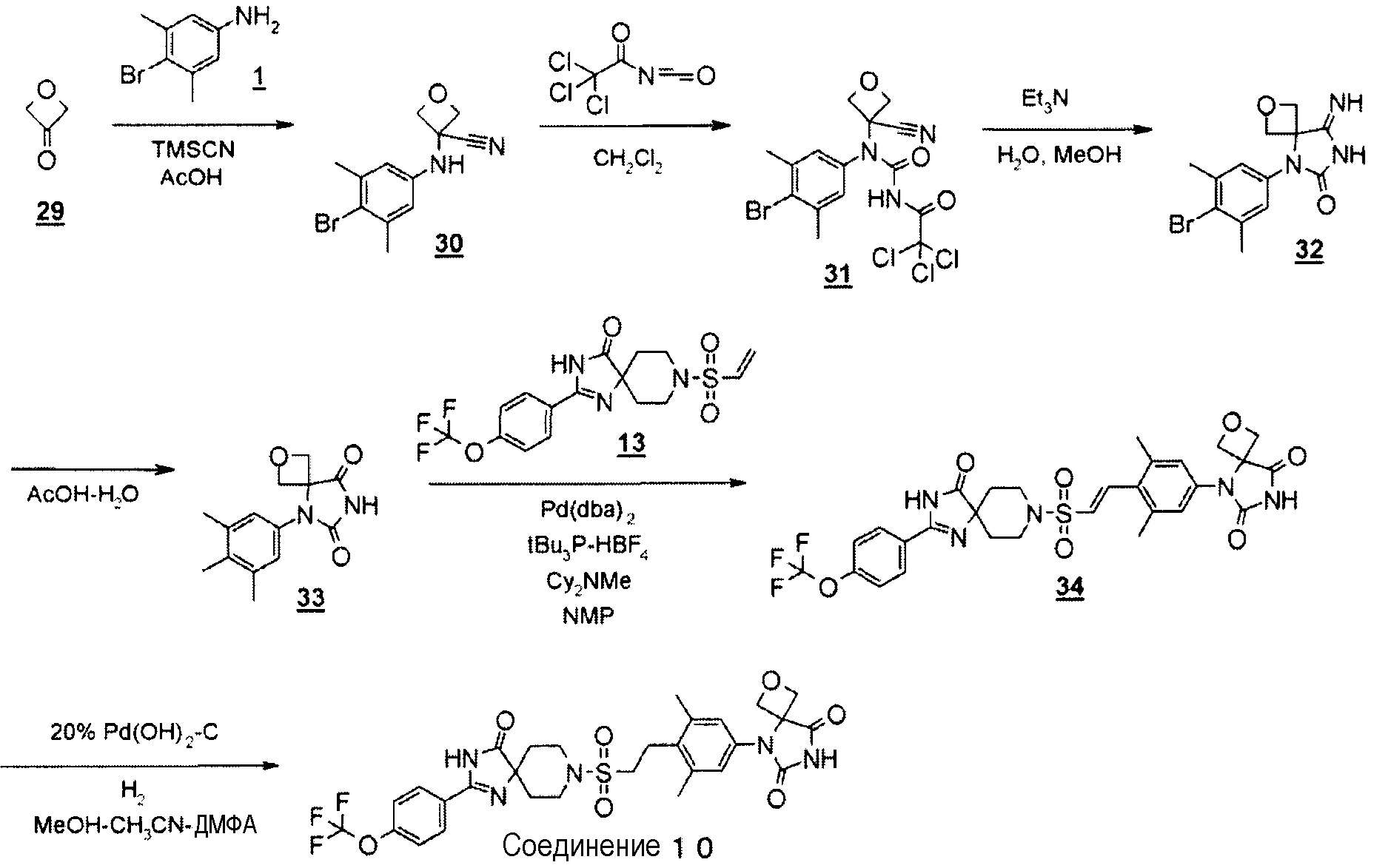
С использованием оксетан-3-она в качестве исходного вещества и соответствующих растворителей получали 5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2-окса-5,7-диазаспиро[3.4]октан-6,8-дион, проводя процессы, аналогичные процессам в примере 4.
MS(ESI) m/z=650 (M+H)+. 1H-ЯМР (400 МГц, CDCl3) δ: 1,69-1,77 (2H, м), 2,12-2,22 (2H, м), 2,45 (6H, с), 3,03-3,11 (2H, м), 3,22-3,29 (2H, м), 3,46-3,53 (2H, м), 3,84-3,91 (2H, м), 4,86 (2H, д, J=7,2 Гц), 5,03 (2H, д, J=7,2 Гц), 7,07 (2H, с), 7,35 (2H, д, J=8,4 Гц), 7,98 (2H, д, J=8,4 Гц), 8,56 (1H, с), 10,34 (1H, с).
Пример 7
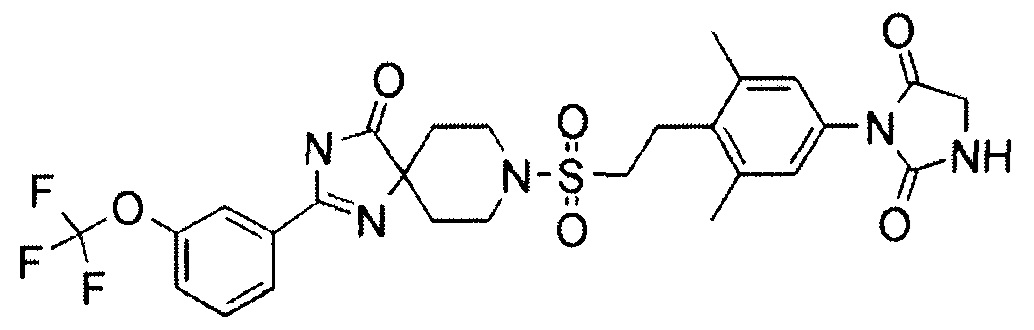
4-(3,5-Диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-4,6-диазаспиро[2.4]гептан-5,7-дион (соединение 11)
(Реакция 7-1)
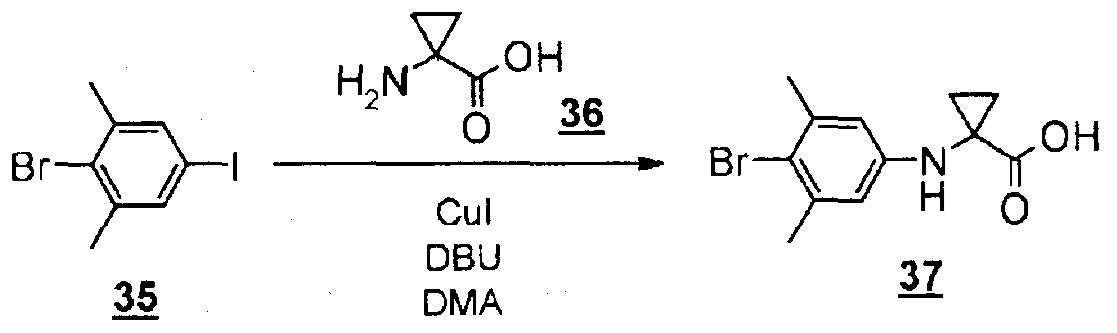
Смесь 2-бром-5-йод-1,3-диметилбензола (300 мг, 0,965 ммоль), 1-аминоциклопропанкарбоновой кислоты (195 мг, 1,93 ммоль), йодида меди(I) (37 мг, 0,194 ммоль) и диазабициклоундецена (0,50 мл, 3,35 ммоль) в диметилацетамиде (2,6 мл) перемешивали при 120°C в течение трех часов в атмосфере азота. Реакционную смесь очищали методом колоночной хроматографии на силикагеле (Wakosil С18, ацетонитрил-вода (0,1% муравьиной кислоты)) с получением 1-((4-бром-3,5-диметилфенил)амино)циклопропанкарбоновой кислоты (219 мг, 80%). MS(ESI) m/z=284, 286 (M+H)+.
(Реакция 7-2)
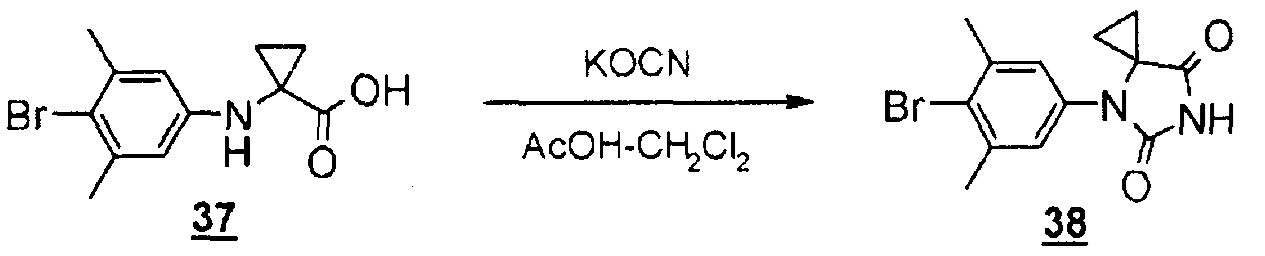
К смеси 1-((4-бром-3,5-диметилфенил)амино)циклопропанкарбоновой кислоты (198 мг, 0,697 ммоль) в уксусной кислоте (3 мл) и дихлорметане (1,5 мл) добавляли цианат калия (424 мг, 5,23 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение одного часа, а затем перемешивали при 60°C в течение двух часов. Добавляли насыщенный водный раствор гидрокарбоната натрия для доведения pH до 8 и экстрагировали эту смесь этилацетатом. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, а затем концентрировали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (этилацетат-гексан) с получением 4-(4-бром-3,5-диметилфенил)-4,6-диазаспиро[2.4]гептан-5,7-диона (49 мг, 23%).
MS(ESI) m/z=309, 311 (M+H)+.
(Реакция 7-3)
С использованием подходящих исходных веществ и растворителей получали 4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-4,6-диазаспиро[2.4]гептан-5,7-дион (соединение 11), проводя процессы, аналогичные процессам в примере 2.
MS(ESI) m/z=634 (M+H)+. 1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,99-1,03 (2H, м), 1,19-1,27 (4H, м), 1,58-1,64 (2H, м), 1,81-1,90 (2H, м), 2,35 (6H, с), 2,99-3,04 (2H, м), 3,22-3,29 (2H, м), 3,67-3,73 (2H, м), 6,95 (2H, с), 7,56 (2H, д, J=8,4 Гц), 8,12 (2H, д, J=8,4 Гц).
Пример 8
1-(3,5-Диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4.4]нонан-2,4-дион (соединение 12)
(Реакция 8)
С использованием подходящих исходных веществ и растворителей получали 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4.4]нонан-2,4-дион, проводя процессы, аналогичные процессам в примере 2.
MS(ESI) m/z=646 (M+H)+. 1H-ЯМР (400 МГц, ДМСО-d6) δ: 1,40-1,48 (2H, м), 1,62-1,71 (4H, м), 1,88-1,97 (2H, м), 1,97-2,08 (4H, м), 2,41 (6H, с), 3,03-3,10 (2H, м), 2,29-3,34 (2H, м), 3,38-3,47 (2H, м), 3,72-3,79 (2H, м), 7,06 (2H, с), 7,84 (1H, дд, J=7,6, 7,6 Гц), 8,02 (1H, д, J=7,6 Гц), 8,33 (1H, д, J=7,6 Гц), 8,38 (1H, с).
Тестовые примеры
Для соединений по настоящему изобретению результаты тестирования на активность продукции цАМФ посредством PTH1R человека, активность продукции цАМФ посредством PTH1R крысы, метаболическую стабильность с использованием микросом печени человека, метаболическую стабильность с использованием гепатоцита крысы и кальцемическое действие на моделях TPTX у крыс представлены в тестовых примерах 1-5, соответственно. В качестве соединений сравнения использовали соединения, описанные в WO 2010/126030 A1, которые представлены в таблице 2.
|
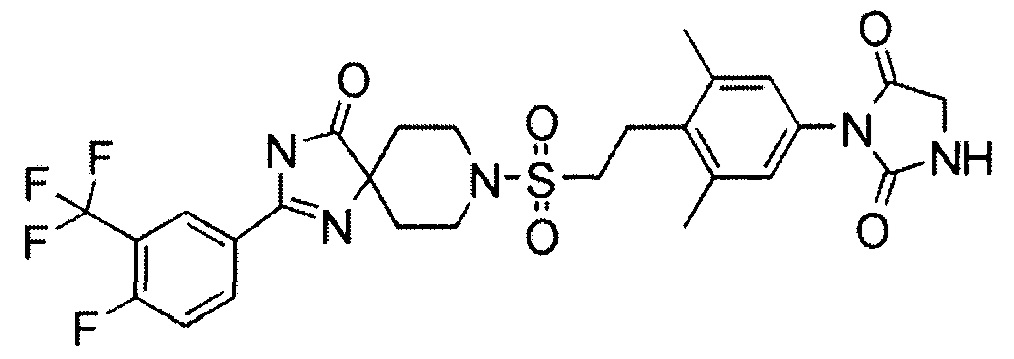
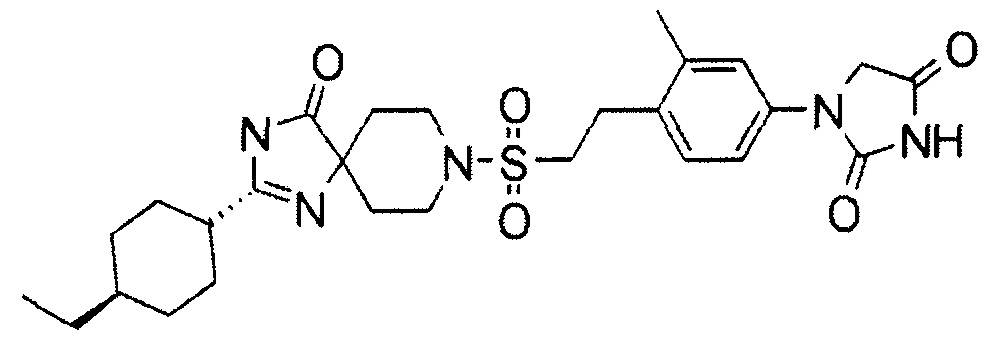
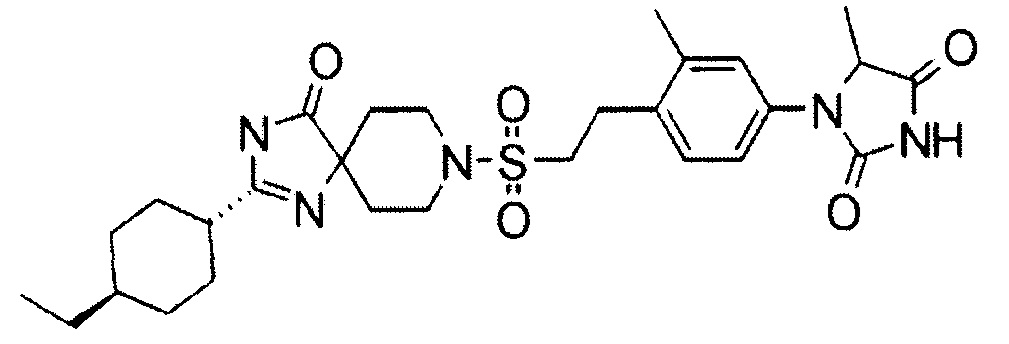
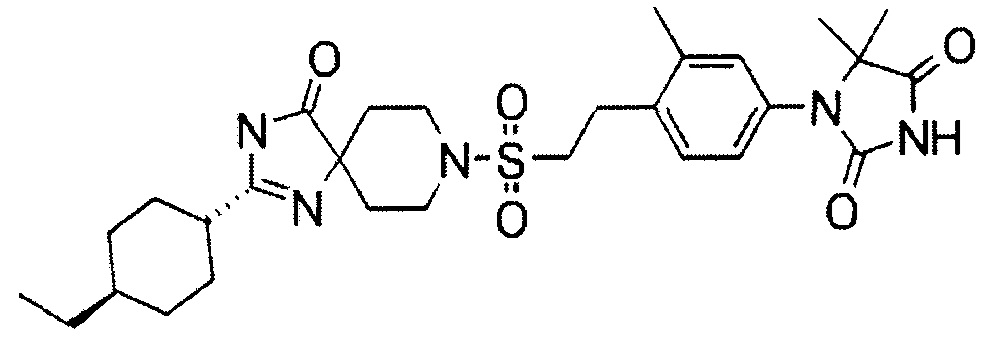
Тестовый пример 1: Измерение активности передачи сигналов цАМФ in vitro соединений через PTH1R человека
(Пептиды)
PTH(1-34) человека и кальцитонин приобретали от Peptide Institute, Inc. (Osaka, Japan), растворяли в 10 мМ уксусной кислоте до 1 мМ и хранили в морозильнике при -80°C.
(Культура клеток)
Клетки культивировали в модифицированной Дульбекко среде Игла (DMEM) с добавлением 10% эмбриональной телячьей сыворотки (Hyclone), 100 единиц/мл пенициллина G и 100 мкг/мл сульфата стрептомицина (Invitrogen Corp) при 37°C в увлажненной атмосфере, содержащей 5% CO2.
В анализе передачи сигнала цАМФ использовали клетки LLC-PK1, не экспсрессирующие PTH1R, и клетки HKRK-B7, которые представляют собой клетки LLC-PK1 сверхэкспрессирующие PTH1R человека, при 9,5×105 рецепторов/клетка (Takasu et al., J. Bone. Miner. Res. 14:11-20, 1999).
(Стимуляция цАМФ)
Клетки HKRK-B7 или LLC-PK.1 высевали в 96-луночный планшет при 1×105 клеток/лунку и инкубировали в течение ночи. На следующие сутки добавляли 50 мкл буфера для анализа цАМФ (DMEM, 2 мМ IBMX, 0,2 мг/мл бычьего сывороточного альбумина, 35 мМ Hepes-NaOH, pH 7,4), содержащего PTH(1-34) человека, или каждое соединение и помещали планшет в инкубатор при 37°C. Клетки инкубировали в течение 20 минут. После удаления среды клетки один раз промывали 100 мкл буфера для анализа цАМФ. Планшет помещали на порошкообразный сухой лед для заморозки клеток, а затем извлекали из сухого льда. Клетки лизировали 40 мкл 50 мМ HCl и снова замораживали на сухом льду. Количество продуцируемого внутриклеточного цАМФ измеряли с использованием коммерчески доступного набора цАМФ EIA (Biotrack cAMP EIA system, GE health care).
(Вычисление 20% эффективной концентрации (EC20) и 50% эффективной концентрации (EC50) при измерении способности индуцировать цАМФ in vitro)
Анализы проводили с использованием уравнения S-образной кривой зависимости «доза-эффект» с переменным градиентом. Активность передачи сигналов цАМФ PTH(1-34) человека при 100 нМ определяли как 100% и концентрацию, при которой каждое соединение обладает 20% или 50% активностью передачи сигналов цАМФ, рассчитывали как EC20 или EC50.
Полученные на клетках HKRK-B7 результаты представлены в таблице 3.
Величина ответа цАМФ в клетках LLC-PK1 была ниже, чем величина в клетках HKRK-B7.
|
Тестовый пример 2: Измерение активности передачи сигналов цАМФ соединений in vitro через PTH1R крысы
Для получения измерений аналогичным тестовому примеру 1 образом вместо клеток HKRK-B7 использовали клетки LLC-PK46_RATO_PTH1R, сверхэкспрессирующие PTH1R крысы, которые были установлены в Chugai Pharmaceutical.
Результаты, полученные с использованием клеток LLC-PK46_RATO_PTH1R, представлены в таблице 4. Для значений EC20 активности передачи сигналов цАМФ in vitro PTH1 рецептора крысы наблюдали хорошую корреляцию со значениями PTH1R человека. Хорошую корреляцию также наблюдали для значений EC50 для крысы и человека.
|
Тестовый пример 3: Исследование метаболической стабильности с использованием микросом печени человека
В 0,1 M фосфатном буфере (pH7,4) инкубировали микросомы печени человека с соединением или сравнительным примером в присутствии NADPH при 37°C в течение предопределенного периода времени. Концентрацию исходного соединения измеряли в каждый момент времени реакции с использованием LC/MS/MS и рассчитывали характерное выведение (мкл/мин/мг белок) из угла наклона кривой времени реакции в зависимости от остаточной скорости.
Условия анализа
Концентрация соединения: 1 мкМ
Микросомы: 0,5 мг/мл
NADPH: 1 мМ
Время реакции: 0, 5, 15 и 30 минут
Результаты представлены в таблице 5. Соединения 1-11 демонстрировали высокую метаболическую стабильность в отношении микросом печени человека по сравнению со сравнительными примерами 1-6.
|
Тестовый пример 4: Исследование метаболической стабильности с использованием гепатоцитов крысы
Клетки печени получали из печени крыс (SD, самки) способом перфузии коллагеназой. Добавляли соединение примеров или сравнительного примера и инкубировали его при 37°C в течение предопределенного периода времени с последующим добавлением блокирующего реакцию раствора. Концентрацию исходного соединения в каждый момент времени реакции измеряли с использованием LC/MS/MS и рассчитывали характерное выведение (мкл/106 клеток/мин) из угла наклона кривой времени реакции в зависимости от остаточной скорости.
Условия анализа
Концентрация клеток: 1×106 клеток/мл
Концентрация соединения: 1 мкМ
Среда: среда Уильяма E
Время реакции: 0, 15, 30, 60, 120 и 240 минут
Блокирующий реакцию раствор: ацетонитрил/2-пропанол (4/6, об./об.)
Результаты представлены в таблице 6. Метаболическая стабильность соединений 2, 4, 5, 7, 8, 9, 10 и 11 в гепатоцитах крысы увеличивалась по сравнению со сравнительными примерами 1, 2, 3, 5 и 6.
|
Тестовый пример 5: Кальцемическое действие на модели TPTX у крыс
Самок крыс Crl:CD(SD) в возрасте четырех недель получали от Charles River Japan (Atsugi Breeding Center) и подвергали акклиматизации к стандартным условиям лаборатории 20-26°C и 35-75% влажности в течение одной недели. Крысам давали водопроводную воду и кормили без ограничения стандартным кормом для грызунов (CE-2) (CLEA Japan, Inc.), содержащим 1,1% кальция, 1,0% фосфорной кислоты и 250 мЕд./100 г витамина D3.
TPTX проводили на крысах в возрасте пяти недель. Некоторых из индивидуумов подвергали имитации операции (имитация). Индивидуумы, у которых концентрация Ca в сыворотке составляла менее 8 мг/дл на четвертые сутки после операции, выбирали для использования в качестве крыс TPTX. На пятые сутки после операции крыс разделяли на восемь групп TPTX и одну группу имитации (n=5 в каждой группе) в зависимости от их массы тела и концентрация Ca в сыворотке, измеряемой на четвертые сутки после операции. Растворитель отдельно вводили перорально в группе имитации и в группе TPTX-носитель в объеме 10 мл/кг. Каждый тестируемый препарат вводили перорально отдельно в каждой группе TPTX тестируемый препарат путем его растворения в растворителе в дозе 30 мг/10 мл/кг. Композиция растворителя представляла собой 10% диметилсульфоксида (Wako Pure Chemical Industries, Ltd.), 10% Cremophor EL (Sigma-AIdrich Japan LLC), 20% гидроксипропил-β-циклодекстрина (Nihon Shokuhin Kako Co., Ltd.), глицин (Wako Pure Chemical Industries, Ltd.); и pH доводили до 10. Непосредственно перед введением каждого образца проводили предварительный сбор образцов крови, и сбор образцов крови проводили через 2, 6, 10 и 24 часа после введения для измерения концентрации Ca в сыворотке. Каждый сбор образцов крови проводили из яремной вены при ингаляционной анестезии изофлураном.
Измерение Ca в сыворотке: Сыворотку, полученную центрифугированием из собранной крови, измеряли с использованием автоматического анализатора TBA-120FR (Toshiba Medical Systems Corporation).
Для статистического анализа исследований на животных данные представлены в виде среднего значения ± стандартная ошибка (SE). Статистический анализ проводили с помощью непарного критерия SAS Preclinical Package (Ver. 5.00.010720, SAS Institute Japan, Tokyo, Japan). Значение p<0,05 считали как статистически значимое. Статистическую значимость каждой группы тестируемого препарата в сравнении с группой TPTX-носитель, группой сравнительный пример 1 и группой сравнительный пример 2 обозначали как #, * и ∫ соответственно.
Предварительное значение концентрации Ca в сыворотке составляло 9,9 мг/дл для группы имитации и 5,3-6,2 мг/дл для каждой из групп TPTX. Концентрации Ca в сыворотке для каждого соединения до 24 часов после введения представлены на фиг.1 в виде средней величины изменения от предварительного значения. Кроме того, для всех соединений концентрация Ca в сыворотке достигала пика через шесть часов после введения или десять часов после введения каждого соединения.
В случае соединений 6, 7 и 8, которые обладают высокой метаболической стабильностью в гепатоцитах крысы, были продемонстрированы значительные положительные изменения относительно предварительного значения, и их пероральное введение оказывало значительные эффекты в отношении кальцемического действия. С другой стороны, в случае соединения 1 и сравнительных примеров 1 и 2, которые обладают низкой метаболической стабильностью в гепатоцитах крысы, были продемонстрированы меньшие положительные изменения относительно предварительного значения по сравнению с соединениями 6, 7 и 8. В частности, соединения 7 и 8 были статистически значимыми по сравнению со сравнительными примерами 1 и 2.
Кроме того, в случае соединений 6, 7 и 8, которые обладают высокой метаболической стабильностью в гепатоцитах крысы, были продемонстрированы индивидуальные максимальные значения от 7,8 до 8,5 мг/дл через шесть или десять часов после введения, и они достигали целевого терапевтического диапазона концентрации Ca в сыворотке от 7,6 до 8,8 мг/дл у пациентов с гипопаратиреозом. С другой стороны, этот целевой терапевтический диапазон невозможно было получать в любые моменты времени измерения для соединения 1 и сравнительных примеров 1 и 2, которые обладают низкой метаболической стабильностью в гепатоцитах крысы.
На основании указанных выше результатов тестирования было выявлено, что соединения 6, 7 и 8, которые обладают сильной активностью передачи сигналов цАМФ в клетках с усиленной экспрессией PTH1R крысы и высокой стабильностью в отношении метаболического распада в гепатоцитах крысы, оказывают сильные эффекты на кальцемическое действие у крыс при пероральном введении. Эти соединения также обладают активностью передачи сигналов цАМФ в клетках с усиленной экспрессией PTH1R человека и высокой метаболической стабильностью в отношении микросом печени человека в сравнении со сравнительными соединениями, и ожидают, что они обладают высокими терапевтическими эффектами при пероральном введении пациентам с гипопаратиреозом. Кроме того, также ожидают, что соединения, представленные формулой (1), которые обладают активностью передачи сигналов цАМФ в клетках с усиленной экспрессией PTH1R человека и обладают метаболической стабильностью в отношении микросом печени человека в той же степени, что и соединения 6, 7 и 8, обладают высокими терапевтическими эффектами у пациентов с гипопаратиреозом.
Промышленная применимость
Настоящее изобретение предоставляет соединения, обладающие значительным PTH-подобным эффектом и высокой метаболической стабильностью. Настоящее изобретение также предоставляет лекарственное средство для предупреждения и/или лечения остеопороза, перелома, адинамической болезни кости, ахондроплазии, гипохондроплазии, остеомаляции, остеоартрита, артрита, тромбоцитопении, гипопаратиреоза, гиперфосфатемии, опухолевого кальциноза или тому подобное, или мобилизации стволовых клеток.

Способ получения клетки, способной продуцировать гетеропротеины с высоким выходом
Антитело против nr10 и его применение
Способ получения гетерологичных белков
Терапевтические средства, используемые против реакции трансплантат против хозяина, содержащие в качестве активного ингредиента ингибитор рецептора интерлейкина-6
Клетка для получения гетеропротеинов и способ получения на ее основе
Жидкая композиция, содержащая антитело высокой концентрации
Антитело против рецептора il-6
Способ модификации изоэлектрической точки антитела с помощью аминокислотных замен в cdr
Профилактическое или лекарственное средство для воспалительного заболевания
Лекарственное средство для лечения рака печени
Конструкция боковой части кузова транспортного средства
Фармацевтическая композиция, содержащая производные гидантоина

