Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ 6-МЕТИЛЕНГИДРОКОРТИЗОНА ИЛИ ЕГО ЭФИРОВ ИЗ 21-АЦЕТАТА ГИДРОКОРТИЗОНА
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к области органического синтеза, конкретно касается получения стероидных соединений (кортикостероидов), таких как 6-метиленгидрокортизон или его эфиры, и может быть использовано в химической и фармацевтической отраслях промышленности, а также в промышленной биотехнологии для производства стероидных медицинских препаратов.
Уровень техники
6-Метиленгидрокортизон является промежуточным соединением в синтезе 6α-метилгидрокортизона (CAS №1625-39-4) и 6α-метилпреднизолона (CAS №83-43-2), а также 6-дегидро-6-метилгидрокортизона (CAS №85310-39-0) и 6-дегидро-6-метилпреднизолона (CAS №95810-22-3), из гидрокортизона (CAS №50-23-7) или 21-ацетата гидрокортизона (CAS №50-03-3).
Полученные в соответствии с предлагаемым способом 6-метиленгидрокортизон и его эфиры (11-трифторацетат 6-метиленгидрокортизона, 21-ацетат 6-метилен-гидрокортизона и 11-трифторацетат 21-ацетата 6-метиленгидрокортизона) не только служат исходными субстратами для дальнейшего превращения в 6α-метилгидрокортизон и 6α-метилпреднизолон (химическим и комбинированным химико-биотехнологическим способами соответственно), но также могут быть использованы для получения 6-дегидро-6-метилгидрокортизона и 6-дегидро-6-метилпреднизолона (соответственно химическим и комбинированным химико-биотехнологическим или химическим способами).
Введение метиленовой группы в положение С6 молекулы гидрокортизона или 21-ацетата гидрокортизона является ключевой стадией известных способов получения 6α-метилгидрокортизона - синтетического прекурсора 6α-метилпреднизолона.
Введение С6-метиленовой группы осуществляют различными методами, используя формальдегид или его производные как метиленирующие агенты. Однако все способы предусматривают обязательную предварительную енолизацию α,β-ненасыщенного кетона кольца А, необходимую для поляризации системы двойных связей с образованием нуклеофильного атома углерода С6, с последующим замещением атома водорода при С6 на формильную или метиленовую группу и различаются тем, что в одних случаях образованный промежуточный 3,5-диенол выделяют в виде эфира [D. Burn et al., Tetrahedron, 1965, 21(6), 1619-1624] или 3,5-диенамина [F. Schneider et al., Helv. Chim. Acta, 1973, 56(7), 2396-2404], в других же случаях енолизация проводится in situ без выделения промежуточного продукта [DE 4121484, 1993].
Наиболее привлекательными являются методы так называемой прямой конденсации с формальдегидом или его производными. Это метод прямого γ-метиленирования, предложенный Анненом и соавт. [US 4322349, 1982; Synthesis, 1982, №1, p. 34-40; ЕР 0100874, 1984; ЕР 0149222, 1985], и метод конденсации с реагентом Манниха [R. Bohlmann et al, DE 4121484, 1993].
Метод прямого γ-метиленирования заключается во взаимодействии стероида с производными формальдегида (такими как диметилацеталь, диэтилацеталь формальдегида или метоксиметилацетат) в хлорсодержащих растворителях при температуре кипения в присутствии избытка хлорокиси фосфора и ацетата натрия. Однако этот метод непригоден для метиленирования соединений, содержащих незащищенную 11β-гидроксильную группу. Так, при прямом γ-метиленировании 21-ацетата гидрокортизона выход 6-метиленпроизводного составляет всего лишь 18% [Synthesis, 1982, №1, р. 34-40, Таблица 1, соединение 6n].
Известно также, что енолэтерификация Δ4-3-кетосистемы кольца А стероидов, катализируемая п-толуолсульфокислотой (п-ТСК), в присутствии незащищенной 11β-гидроксильной группы может сопровождаться нежелательной реакцией ее элиминирования с образованием Δ9(11)-связи [Zhao Q. et al. Synth. Commun., 1993, 23(10), 1473-1478]. Поэтому для сохранения 11β-гидроксильной группы необходима ее защита.
Известно, что для защиты 11β-гидроксигруппы ее обычно этерифицируют, превращая в тригалогенацетат (в частности трифторацетат) или триметилсилиловый эфир. Эти защиты считаются наиболее приемлемыми, так как не только легко образуются, но и легко удаляются. Применение для защиты 11β-гидроксигруппы метода этерификации ангидридами низших кислот возможно, однако ограничено из-за проблем, возникающих при их последующем удалении химическими методами сольволиза.
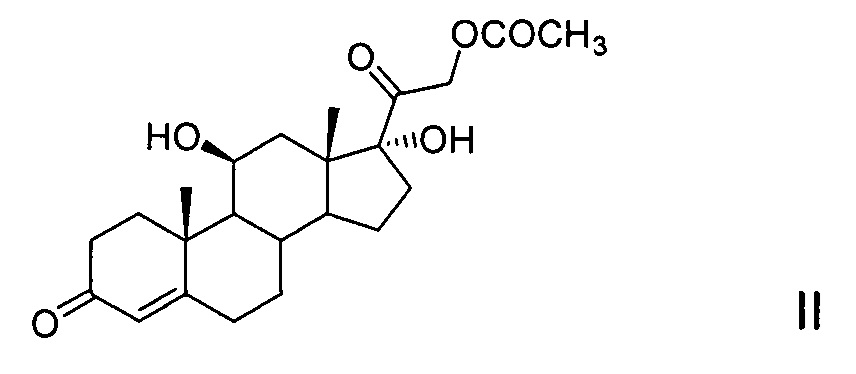
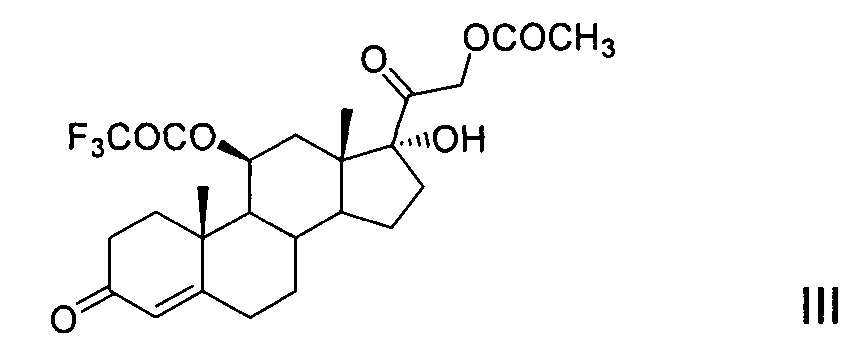
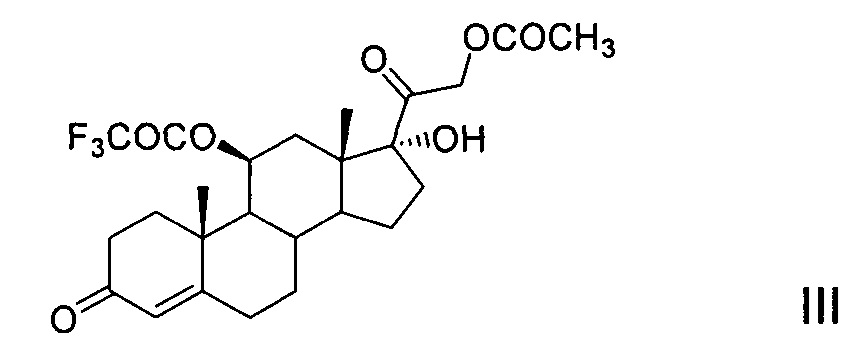
Способы применения трифторацетатной защиты 11β-гидроксильной группы молекулы 21-ацетата гидрокортизона (II) известны. Это способ, описанный в патенте [US 4330541, 1982, пример 1], который заключается в том, что реакцию этерификации 5 г 21-ацетата гидрокортизона проводят в среде пиридина действием трифторуксусного ангидрида при температуре минус 15°С. Далее смесь выливают в водный раствор хлорида натрия, отфильтрованный осадок растворяют в дихлорметане, органический раствор промывают водой, осушают сульфатом натрия и упаривают досуха. Получают 5.1 г (82,4%) остатка, содержащего 11-трифторацетокси-производное (III) 21-ацетата гидрокортизона (II). Однако это соединение не охарактеризовано, так как содержащий его остаток без очистки и выделения кристаллического продукта использован авторами патента на следующей стадии химического синтеза.
Другие патенты [DE 1112511, 1961 (пример 16) и DE 1125422, 1962 (пример 15)] описывают способ получения 11-трифторацетокси-производного (III) из 21-ацетата гидрокортизона (II) действием трифторуксусного ангидрида в среде абсолютного диоксана. После 18 ч выдержки при комнатной температуре реакционную смесь выливают на ледяную воду. Далее продукт экстрагируют, экстракт промывают водой, упаривают досуха в вакууме. После кристаллизации остатка в эфире и хроматографирования на окиси алюминия получают 11-трифторацетокси-производное (III) 21-ацетата гидрокортизона (II), выход которого не указан. Продукт охарактеризован: т.пл. 206-207°С, [α]D +165,7° (хлф); λmax 238-240 mμ. Спектральная характеристика отсутствует.
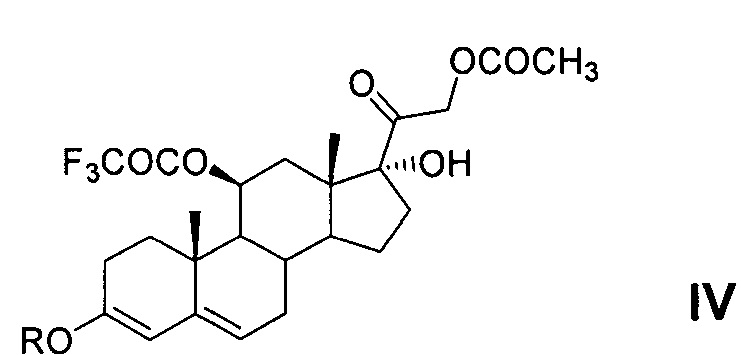
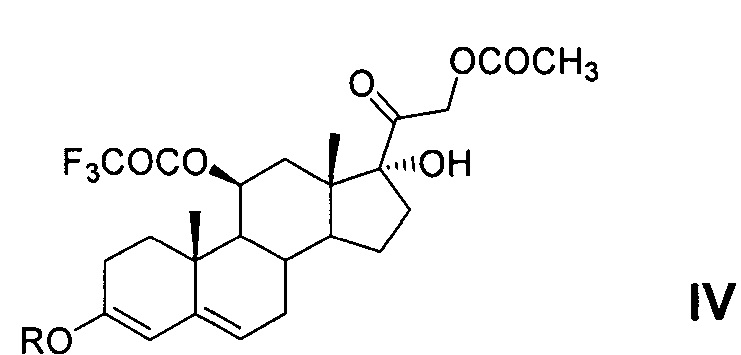
Получение метилового 3,5-диенолэфира 11-трифторацетокси-производного 21-ацетата гидрокортизона - соединение IVa - в литературных источниках не описано.
Получение этилового 3,5-диенолэфира 11-трифторацетокси-производного 21-ацетата гидрокортизона - соединение IVb - описано в патентах [DE 1112511, 1961 (пример 16) и DE 1125422, 1962 (пример 15)] и заключается в реакции соединения III с этилортоформиатом в среде диоксана в присутствии п-ТСК при комнатной температуре. После окончания реакции смесь выливают в воду и экстрагируют дихлорметаном. Экстракт промывают водой, осушают сульфатом натрия и упаривают досуха. Маслянистый остаток, содержащий соединение IVb, выход которого не указан, охарактеризован: [α]D ±0° (хлф); λmax 240-242 mμ, logε 4,15. Соединение IVb не было выделено авторами в кристаллическом виде. Спектральная характеристика, отсутствует, температура плавления не определена.
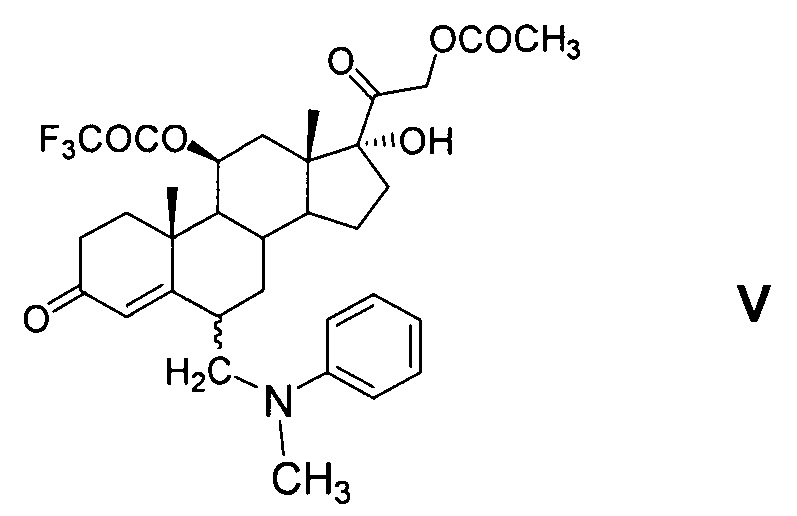
6-Аминометильные производные общей формулы V являются новыми соединениями, в литературе не описаны.
6-Метилен-производное 11-трифторацетокси-производного 21-ацетата гидрокортизона - соединение VI - является новым соединением, в литературе не описано.
6-Метилен-производное 11-трифторацетата гидрокортизона - соединение VII - известно. Так, известен способ получения 11-трифторацетата 6-метиленгидрокортизона (VII), описанный в патенте [RU 2337918, 2008, пример 14]. Однако соединение VII получено другим способом из другого соединения, а именно из 11β-трифторацетилокси-17α,20;20,21-бисметилендиокси-6-метиленпрегн-4-ен-3,20-диона удалением бисметиленовой защиты диоксиацетоновой боковой цепи в среде уксусной кислоты при температуре 0-2°С действием концентрированной соляной кислоты (на 5 г стероида используют 20 мл уксусной кислоты и 75 мл соляной кислоты).
Соединение VIII - 21-ацетокси-11β,17α-дигидрокси-6-метиленпрегн-4-ен-3,20-дион (21-ацетат 6-метиленгидрокортизона) - известно.
Известен способ получения 21-ацетата 6-метиленгидрокортизона из 21-ацетокси-11β,17α-дигидрокси-6-гидроксиметил-3-метокси-прегна-3,5-диен-20-она [US 3112305, 1963, пример 43], согласно которому 500 мг указанного исходного соединения нагревают при 80°С в 3 мл 95% водной уксусной кислоты в течение 10 мин., затем осаждают продукт водой и кристаллизуют из смеси ацетон-гексан. Получают 21-ацетат 6-метиленгидрокортизона, выход которого не указан.
Известен способ получения 21-ацетата 6-метиленгидрокортизона [D. Burn, J.P. Yardley, and V. Petrow, Tetrahedron, 1969, Vol. 25. p. I I55-1158], согласно которому к смеси 21-ацетокси-6-формил-3-метокси-3,5-прегнадиен-11β,17α-диол-20-она (3.6 г) и NaOAc (3.6 г) в метаноле (100 мл) добавляют Pt/C (1.25 г) в метаноле (25 мл) и смесь гидрируют до поглощения 180 мл (1 моль-экв.) водорода. Затем катализатор удаляют фильтрацией, растворитель упаривают при пониженном давлении, остаток распределяют между хлороформом и водой. Органическую фазу отделяют и добавляют п-ТСК (0.2 г) в метаноле (10 мл); после выдерживания в течение 15 мин. при комнатной температуре реакционный раствор промывают водным раствором Na2CO3, затем водой, сушат (Na2SO4) и упаривают при пониженном давлении. Кристаллизацией и осаждением из смеси хлороформа и диизопропилового эфира получают 6-метиленпроизводное (1.8 г) с т.пл. 206-207°С (выход 53.4%).
Также известен способ получения 21-ацетата 6-метиленгидрокортизона из 21-ацетата гидрокортизона [DE 4121484, 1993, пример 2], который заключается во введении метиленовой группы в положение С6 молекулы 21-ацетата гидрокортизона. Согласно описанию этого способа 5 г 21-ацетата гидрокортизона (II) суспендируют в смеси 50 мл тетрагидрофурана, 15 мл триэтилортоформиата и 15 мл этанола, добавляют 500 мг тозилата пиридиния и перемешивают в течение 1 ч при температуре 40°С в бане. Затем добавляют 5,5 мл N-метиланилина и 6,5 мл 40% раствора формальдегида и перемешивают не менее часа при температуре 40°С. После этого реакционную массу обрабатывают ледяной водой, содержащей винную кислоту и хлорид натрия, экстрагируют дихлорметаном, промывают раствором бикарбоната натрия и хлорида натрия, осушают сульфатом натрия и упаривают в вакууме досуха. Полученный остаток (5,8 г) хроматографируют на 600 г кизельгеля, элюируя смесью гексан-этилацетат, и получают 2,1 г (40,8%) 6-метилен-производного 21-ацетата гидрокортизона.
Недостатками описанного в [DE 4121484, 1993] процесса являются:
- применение для очистки 21-ацетата 6-метилен-гидрокортизона хроматографии, существенно усложняющей технологический процесс;
- низкий выход продукта 21-ацетата 6-метиленгидрокортизона (40,8%) на стадии метиленирования, который можно объяснить, во-первых, наличием побочных реакций с участием незащищенной гидроксильной группы при С11, и, во-вторых, низкой регионаправленностью реакции расщепления стероидного основания Манниха. Вероятно, что в описанных условиях реакции дезаминирования (расщепление по связи C-N) сопутствует побочный процесс дезаминометилирования (расщепление по связи С-С) с регенерацией 21 -ацетата гидрокортизона. Поэтому хроматографическая очистка продукта становится необходимой.
Способы получения 6-метиленгидрокортизона (I) известны. Так, известен способ получения 6-экзометилен-4-прегнен-11β,17α,21-триол-3,20-диона и его 21-ацетата [US 3090781, 1963 (пример 8); US 3074935, 1963 (пример 8)], согласно которому суспензию 0,26 г 6-экзометилен-17α,20:20,21-бис-метилендиокси-4-прегнен-11β-ол-3-она в 8-кратном количестве 60% водной муравьиной кислоты нагревают на паровой бане в течение 20 мин; полученный раствор охлаждают, выливают в ледяную воду и экстрагируют хлороформом. Органическую фазу промывают водным раствором бикарбоната натрия, сушат и концентрируют в вакууме. После кристаллизации из этилацетата получают 30 мг 6-экзометилен-4-прегнен-11β,17α,21-триол-3,20-диона с т.пл. 203°С. Некристаллизующийся материал (175 мг), полученный после отделения 30 мг 6-метиленгидрокортизона, обрабатывают 1N раствором метоксида натрия в метаноле при комнатной температуре в атмосфере азота для расщепления различных эфиров муравьиной кислоты, реакционную массу нейтрализуют уксусной кислотой, выливают в воду, экстрагируют хлороформом, хлороформную фазу промывают водой, упаривают досуха. 6-Экзометилен-4-прегнен-11β,17α,21-триол-3,20-дион, содержащийся в остатке, подвергают ацетилированию в пиридине действием уксусного ангидрида. Для этого к остатку добавляют 2,1-кратное количество пиридина и 2-кратное количество уксусного ангидрида, смесь выдерживают в течение ночи при комнатной температуре. После выливания реакционной массы на воду, экстракции хлороформом, упаривания досуха и хроматографирования на окиси алюминия получают 42 мг 6-экзометилен-4-прегнен-11β,17α,21-триол-3,20-диона-21-ацетата, т.пл. которого после перекристаллизации из этилацетата составляет 199-204°С. Таким образом, суммарный молярный выход продуктов (6-метиленгидрокортизона (I) и его 21-ацетата (VIII) не превышает 29%.
Наиболее близким по сущности к предложенному способу получения 6-метиленгидрокортизона является способ [RU 2297423, 2007, примеры 3 (вариант 2) и 4], который заключается во введении метиленовой группы в положение С6 молекулы 11β,17α,21-триалканоилокси-прегн-4-ен-3,20-диона. Согласно описанию этого способа (пример 3, вариант 2) к раствору 10 г 11,17,21-триацетата гидрокортизона в 75 мл тетрагидрофурана добавляют 10 мл этанола, 10 мл триэтилортоформиата, 130 мг п-ТСК и нагревают до 40°С. Реакционную массу выдерживают в течение 1 ч и добавляют дополнительно 130 мг п-ТСК. После перемешивания в течение еще 1 ч при температуре 40°С добавляют 4,3 мл N-метиланилина и 3-6 мл водного 37% раствора формальдегида. Реакционную массу выдерживают в течение 2 ч, затем охлаждают до комнатной температуры, добавляют 25 мл концентрированной соляной кислоты и перемешивают 30 мин. Смесь экстрагируют хлористым метиленом, органический слой промывают водой до нейтральной реакции, растворитель упаривают. После кристаллизации получают 9,4 г 11β,17α,21-триацетокси-6-метиленпрегн-4-ен-3,20-диона с выходом 92%. По примеру 4 этого способа в аналогичных условиях из 1 г 21-ацетокси-11β,17α-бис(трифторацетилокси)-прегн-4-ен-3,20-диона получают 0,93 г 6-метилен-производного с выходом 91%.
Основным недостатком указанного способа является необходимость обязательной защиты всех гидроксильных групп, имеющихся в структуре молекулы гидрокортизона с последующим удалением защит. Так, при применении для защиты метода исчерпывающего ацетилирования, несмотря на высокие постадийные выходы при получении 11β,17α,21-триацетокси-6α-метилпрегн-4-ен-3,20-диона и 21-ацетокси-11β,17α-бис(трифторацетилокси)-прегн-4-ен-3,20-диона, эффективное удаление сложноэфирной защиты с регенерацией 11β-гидроксильной группы возможно только с применением биотехнологического метода [RU 2297423, 2007, пример 8], а именно микробиологической трансформацией 11β,17α,21-триацетокси-6-метиленгидрокортизона (вариант 1) или 21-ацетокси-11β,17α-бис(трифторацетилокси)-прегн-4-ен-3,20-диона (вариант 2) клетками бактерий Arthrobacter mediolanus ВКМ-1388, который осуществляется в 2 этапа (энзиматическое удаление защиты 11β-гидроксильной группы и гидролиз защиты 17α-гидроксильной группы действием 10% раствора гидроксида натрия) с общей продолжительностью процесса дезацилирования 72 ч. При этом сложноэфирная защита гидроксильной группы при С21 может частично сниматься на биотехнологическом этапе, а щелочной гидролиз защиты 17α-гидроксильной группы протекает путем ацильной миграции с первоочередным удалением ацетильной защиты гидроксила при С21. Это существенно усложняет способ.
Раскрытие изобретения
Технической задачей в заявленном изобретении является разработка более простого способа получения 6-метиленгидрокортизона или его эфиров из 21-ацетата гидрокортизона при обеспечении выхода целевых продуктов не менее чем 80%.
Упрощение способа обеспечивается 1) за счет проведения последовательных реакций функционализации положения С6 молекулы 11-трифторацетокси-производного 21-ацетата гидрокортизона (III) с образованием 21-ацетокси-11-трифторацетилокси-6-метилен-гидрокортизона (VI) при температуре окружающей среды (т.е. при комнатной температуре) без применения нагрева реакционных масс; 2) за счет проведения непрерывного технологического процесса функционализации положения С6 молекулы 11-трифторацетата 21-ацетата гидрокортизона (III) без выделения интермедиатов IV и V целевого продукта (VI) на ключевых стадиях синтеза и исключения тем самым не только потерь указанных интермедиатов IV и V в маточных растворах, но и механических потерь, которые обычно имеют место на технологических операциях экстракции, фильтрации, сушки и т.п.; 3) за счет достижения практически полной конверсии исходных продуктов и высокой селективности реакций образования интермедиатов IV и V и целевого продукта VI в процессе функционализации положения С6 молекулы 11-трифторацетокси-производного 21-ацетата гидрокортизона (III) и тем самым снижения вероятности протекания побочных реакций; 4) за счет практически полного исключения образования побочных продуктов и осмоления реакционной массы на стадиях удаления защитных группировок. В результате достигается высокий выход конечных продуктов I, VI, VII и VIII (от 82 до 97), считая из 21-ацетата гидрокортизона (II).
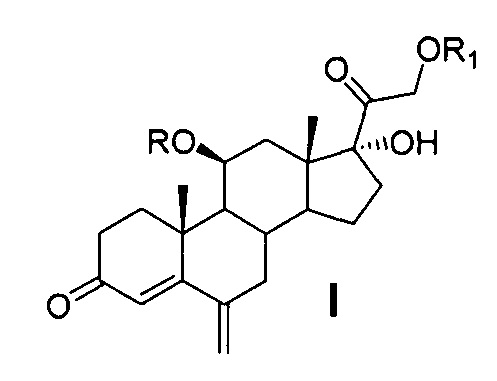
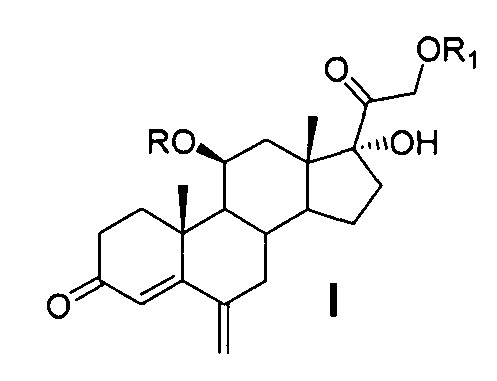
Техническая задача решается способом получения 6-метиленгидрокортизона или его эфиров общей формулы (I)
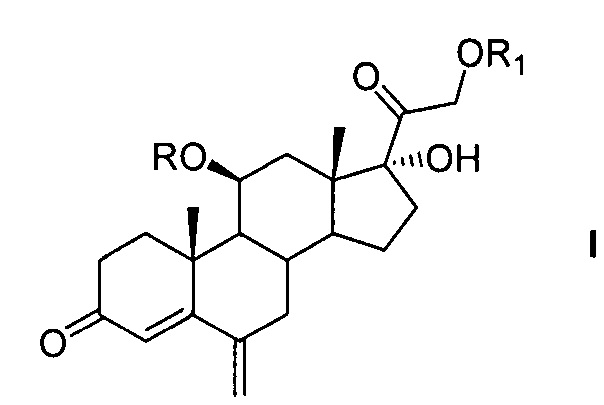
где R=COCF3 или Н, R1=СОСН3 или Н из 21-ацетата гидрокортизона формулы (II),
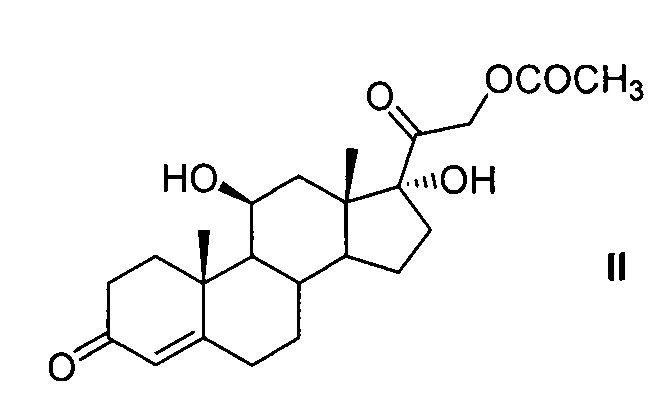
включающим предварительную защиту 11β-гидроксильной группы молекулы 21-ацетата гидрокортизона методом этерификации ангидридом трифторуксусной кислоты в условиях основного катализа с использованием каталитических количеств катализатора, последующее С6-метиленирование образовавшегося 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-прегн-4-ен-3,20-диона, одновременное, поэтапное или последовательное удаление защиты 11β- и 21- гидроксильных групп методами химического сольволиза, или поэтапную (последовательную) защиту гидроксильных групп 11β,17α,21-тригидрокси-6-метиленпрегн-4-ен-3,20диона с образованием 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метиленпрегн-4-ен-3,20-диона через 21-ацетокси-11β,17α-дигидрокси-6-метиленпрегн-4-ен-3,20-дион.
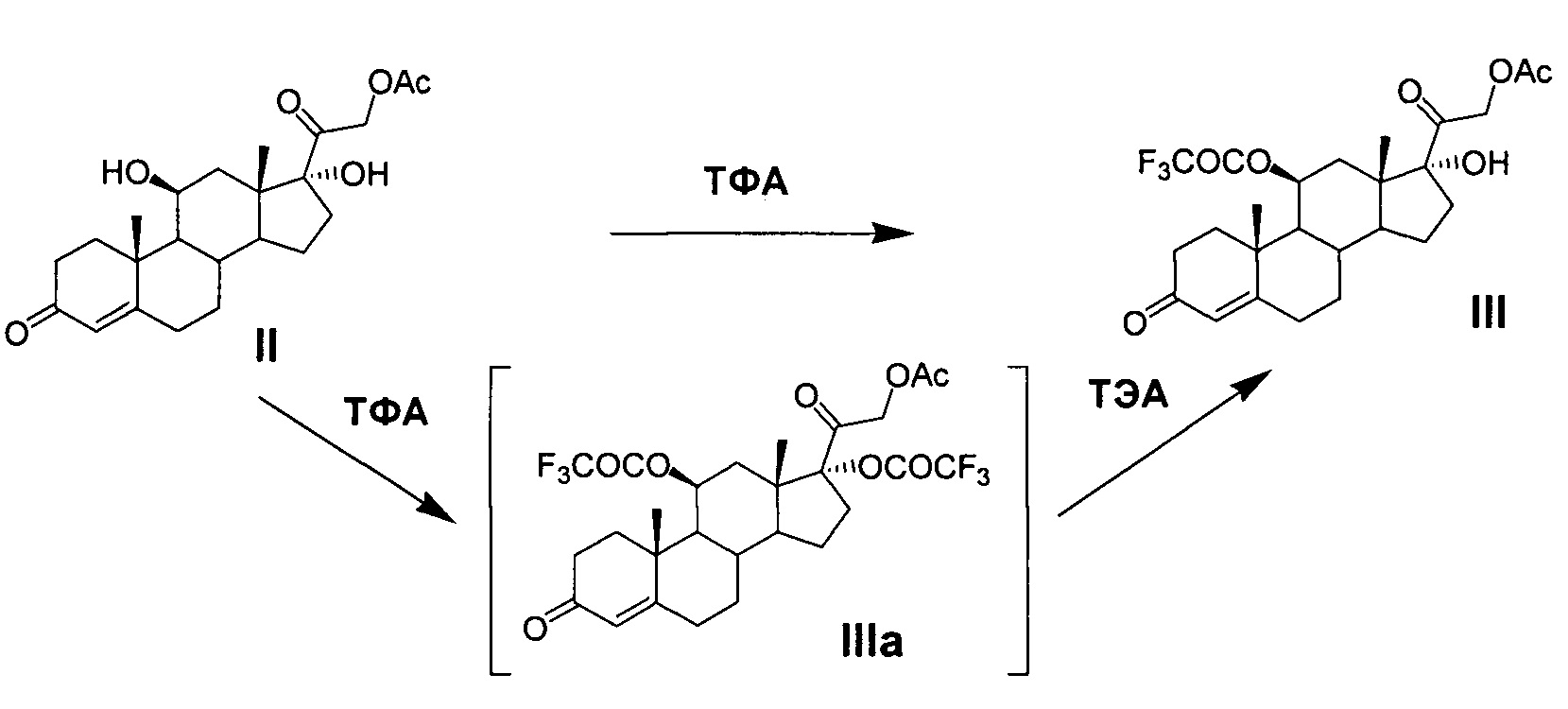
Ниже представлена предлагаемая схема синтеза 6-метиленгидрокортизона (I) или его сложных эфиров (VI, VII и VIII) из 21-ацетата гидрокортизона (II).
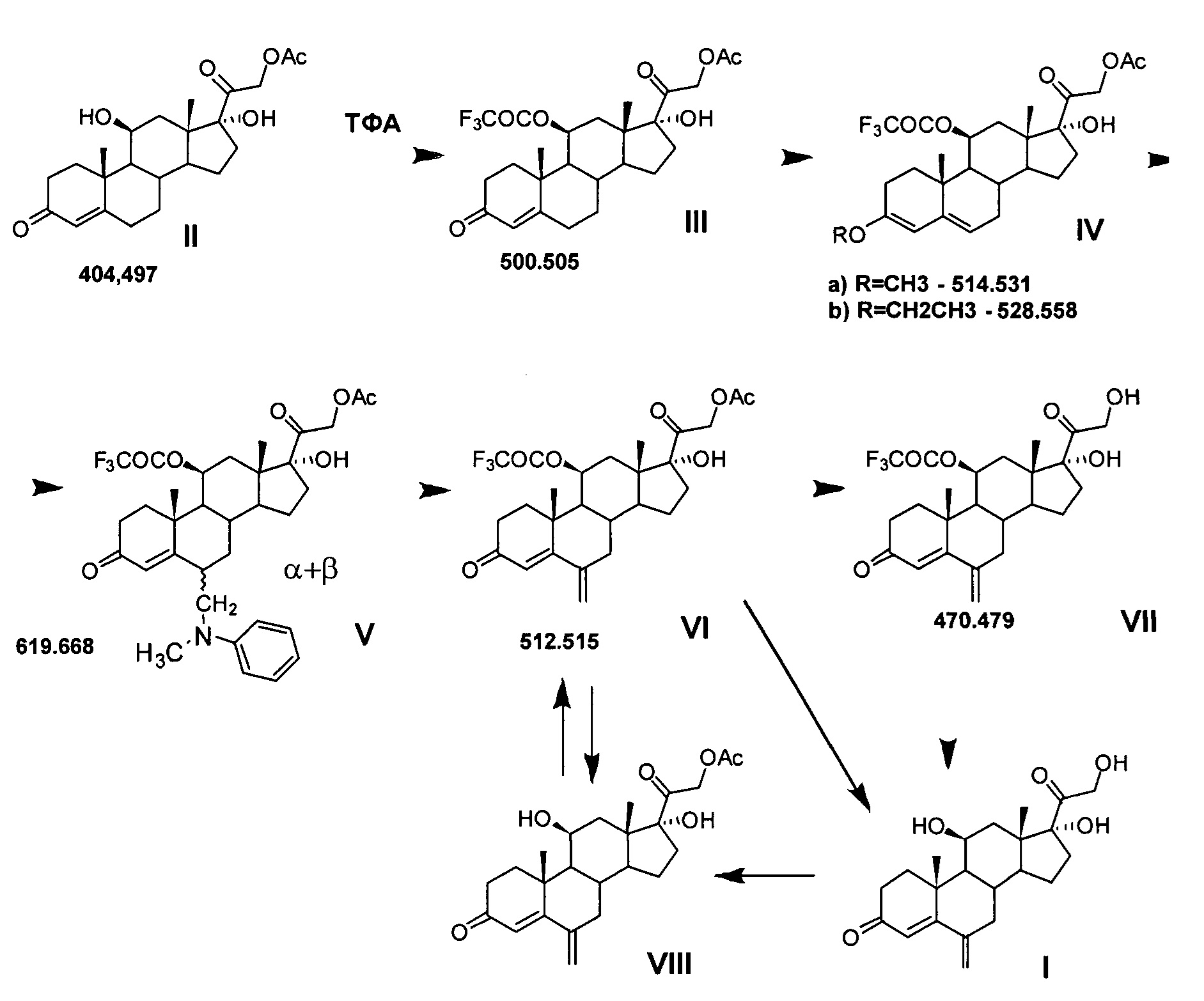
Схема включает 4 (для VI) или 5 (для I и VII), или 6 (для VIII) химических реакций, которые могут быть проведены в 2 или 3, или 4 технологических стадии соответственно. Химическая схема синтеза включает: 1) защиту 11β-гидроксильной группы молекулы 21-ацетата гидрокортизона (II) образованием 11β-трифторацетилокси-производного (III); 2) енолизацию α,β-ненасыщенного кетона кольца А соединения III с образованием 3,5-диенолэфира (IV); 3) реакцию трехкомпонентной конденсации по типу реакции Манниха с образованием смеси С6-аминометильных производных (V); 4) расщепление аминометильных производных (V) по связи C-N с образованием С6-метилен-производного (VI); 5) удаление защитных группировок молекулы соединения VI полностью с образованием 6-метиленгидрокортизона (I) или частично по гидроксильной группе С21 с образованием соединения VII, при этом 6-метиленгидрокортизон (I) может быть превращен в его 21-ацетокси-производное (VIII) ацетилированием в условиях основного катализа, из которого далее может быть получено соединение VI. Частичное удаление ацетильной защиты гидроксильной группы при С21 с сохранением трифторацетильной защиты гидроксильной группы при С11 обеспечивает возможность дальнейшей функционализации стероидной молекулы по диоксиацетоновой боковой цепи соединения VII без участия гидроксильной группы при С11.
Сущность заявленного изобретения, касающегося получения 6-метиленгидрокортизона или его эфиров, заключается в том, что с целью повышения выхода и упрощения процесса 21-ацетат гидрокортизона сначала подвергают предварительной защите 11β-гидроксильные группы, затем метиленированию положения С6 с образованием 6-метилен-производного, удалению защитных группировок.
Преимущества заявляемого способа состоят в следующем:
- проведение трифторацетатной защиты 11β-гидроксильной группы 21-ацетата гидрокортизона и селективной деэтерификации побочного продукта реакции этерификации 11,17-бис(трифторацетата) 21-ацетата гидрокортизона с регенерацией 17α-гидроксильной группы осуществляется в одну химическую стадию без выделения целевого продукта;
- проведение конденсации с реагентом Манниха в присутствии свободного гидроксила при С17 в мягких условиях (при комнатной температуре) исключает появление нежелательных продуктов (дегидратации 17α-гидроксигруппы с образованием Δ16-связи и D-гомоаннелирования с образованием 6-членного кольца D), что подтверждается данными ЯМР-спектроскопии;
- региоселективность расщепления стероидного основания Манниха минеральной кислотой исключительно по связи C-N обеспечивается присутствием в реакционной среде соли галогенводородной кислоты и щелочного, или щелочноземельного металла, или аммония;
- отсутствие необходимости использования многократной кристаллизации 11-трифторацетата 21-ацетата 6-метиленгидрокортизона или хроматографирования с целью его очистки, так как содержание основного вещества в продукте составляет 90-95% (ВЭЖХ), что соответствует требованиям, например, каталитической изомеризации 6-экзометиленовой двойной связи в 6,7-эндометиленовую двойную связь или процесса каталитического гидрирования с образованием 11-трифторацетата 21-ацетата 6α-метилгидрокортизона химическим методом;
- отсутствие необходимости использования многократной кристаллизации 6-метиленгидрокортизона или хроматографирования с целью его очистки, так как удаление защитных групп приводит к незначительному появлению побочных соединений; при этом содержание основного вещества в продукте составляет 90-95% (ВЭЖХ), что соответствует требованиям, например, последующего процесса каталитической изомеризации 6-экзометиленовой двойной связи в 6,7-эндометиленовую двойную связь или процесса каталитического гидрирования с образованием 6α-метилгидрокортизона;
- реакции трифторацетилирования, енолэтерификации, конденсации с реагентом Манниха, дезаминирования стероидного основания Манниха проводятся при комнатной температуре;
- значительное сокращение потерь основного продукта на стадиях, что обеспечивается высокой селективностью химических реакций и возможностью совмещения химических стадий в один технологический процесс без выделения интермедиатов, позволяет существенно увеличить достигаемый общий выход 6-метиленгидрокортизона или его эфиров (11-трифторацетата 21-ацетата, или 11-трифторацетата, или 21-ацетата) из 21-ацетата гидрокортизона до 88% и 85,2-97,8% соответственно.
Осуществление изобретения
21-Ацетат гидрокортизона (II), CAS №50-03-3, C12H32O6 (качество USP, или ЕР, или BP), является коммерчески доступным, может быть приобретен, например, у компании Steraloids (USA), или у других производителей. 4-Диметиламинопиридин (≥99.0%), ангидрид трифторуксусной кислоты (≥99.0%), ангидрид уксусной кислоты (≥99.0%), триметилортоформиат (≥99.0%), триэтилортоформиат (≥98.0%), 5-сульфосалициловая кислота (≥99.0%), п-толуолсульфокислота (≥98.0%), N-метиланилин (≥98.0%), циклогексен (≥99.0%), триэтиламин (≥99.0%) являются коммерчески доступными, были приобретены у компании Sigma-Aldrich Со. Другие реагенты, растворители и инертные газы являются коммерчески доступными, были приобретены у российских производителей.
Для выделения продуктов по окончании реакций выливанием реакционных масс в воду, а также для промывки осадков, экстрактов в органических растворителях, использовали питьевую водопроводную воду, если не оговорено особо. Для приготовления водных растворов кислот, хлорида натрия, аммиака и гидроксида натрия использовали дистиллированную воду.
Все процедуры, если не оговорено особо, осуществляли при комнатной температуре или температуре окружающей среды, то есть в диапазоне от 20 до 25°С. Для процессов, требующих более низкие температуры, чем комнатная, охлаждение обеспечивали холодной водопроводной водой (в диапазоне от 10 до 20°С), или смесью колотого льда и холодной воды (в диапазоне от 5 до 10°С), или смесью колотого льда и хлорида кальция (при температуре ниже 5°С). Для обогрева при проведении реакций при температуре выше комнатной и для упаривания растворителей при атмосферном давлении использовали электрический колбонагреватель.
Упаривание растворителей в вакууме осуществляли с использованием ротационного вакуумного испарителя Rotavapor ( Labortechnik AG), при остаточном давлении 0,35±0,05 кгс/см2 (35±5 кПа) и температуре воды в бане в диапазоне от 35-50°С в зависимости от природы упариваемого растворителя.
Labortechnik AG), при остаточном давлении 0,35±0,05 кгс/см2 (35±5 кПа) и температуре воды в бане в диапазоне от 35-50°С в зависимости от природы упариваемого растворителя.
Высушивание кристаллов продуктов до постоянного веса осуществляли при температуре 35-45°С при атмосферном давлении или с использованием вакуум-сушильного шкафа при остаточном давлении 0,35±0,05 кгс/см2 (35±5 кПа).
Для определения рН промывных вод использовали универсальную индикаторную бумагу с диапазоном значений от 0 до 12 (Лахема, Чехия).
Сухие (безводные) растворители (ацетон, триэтиламин, метанол, этанол) получали общеизвестными методами органической химии.
Колоночную хроматографию осуществляли на колонке (16×650 мм), используя силикагель марки Silica gel 60 (0.040-0.063 мм) (Merck, Germany).
Контроль за ходом реакций осуществляли методом тонкослойной хроматографии (ТСХ), используя пластины Silica gel 60 F254 (Merck, Germany).
Структуру и чистоту всех выделенных соединений подтверждали, по меньшей мере, одним из следующих методов: ТСХ (пластины для ТСХ Silica gel 60 F254 (Merck, Germany)), масс-спектрометрия, элементный анализ, ядерный магнитный резонанс (ЯМР), высокоэффективная жидкостная хроматография (ВЭЖХ).
Температуру плавления выделенных соединений определяли на приборе для определения точки плавления М-565 ( Labortechnik AG).
1Н- и 13С- ЯМР спектры были определены на спектрометре Bruker Avance-400 (Bruker BioSpin GmbH) с рабочей частотой 400 МГц и 100.6 МГц соответственно, используя дейтерированный хлороформ (99,8% D, Sigma-Aldrich), или дейтерированный диметилсульфоксид (99,9% D, Sigma-Aldrich) в качестве растворителя относительно тетраметилсилана (TMS NMR grade ≥99,9%, Sigma-Aldrich) в качестве внутреннего стандарта, в миллионных долях (м.д.).
Элементный анализ был выполнен с использованием анализатора Vario MICRO Cube (Elementar Analysensysteme, GmbH).
ВЭЖХ-анализ проводили на хроматографе Knauer Smartline (Germany), укомплектованном градиентным HPLC-насосом, ячейкой для термостатированной колонки, инжектором, диодно-матричным детектором Smartline 2600, интегратором-компьютером, при температуре 24°С, скорости потока 1 мл/мин и УФ-детектировании при длине волны 254 нм. В качестве неподвижной фазы использовали Kromasil® 100-5С18, в качестве мобильной фазы - 35% (по объему) раствор Н2О в ацетонитриле.
Способ получения 6-метиленгидрокортизона или его эфиров общей формулы (I) осуществляется по вышеприведенной схеме.
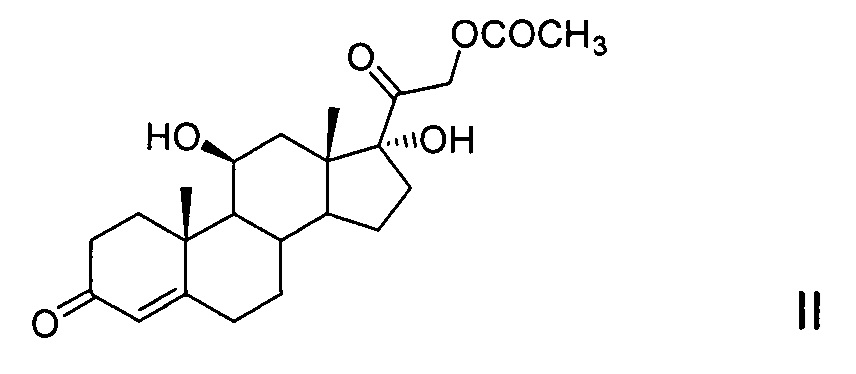
21-Ацетат гидрокортизона (21-ацетокси-11β,17α-дигидроксипрегн-4-ен-3,20-дион) формулы (II),

подвергают защите 11β-гидроксильной группы методом этерификации действием ангидрида трифторуксусной кислоты в среде апротонного растворителя в условиях основного катализа в присутствии каталитического количества диметиламинопиридина. Для этого на 1 моль 21-ацетата гидрокортизона (21-ацетокси-11β,17α-дигидроксипрегн-4-ен-3,20-диона) используют 1-1.5 моля ангидрида трифторуксусной кислоты и не более 0.1 моля диметиламинопиридина. Полученный с выходом до 99,95% 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-прегн-4-ен-3,20-дион (III) подвергают енолэтерификации Δ4-3-кетосистемы кольца А действием триалкилортоформиата в присутствии сульфоароматической кислоты (сульфосалициловой или п-ТСК) в качестве кислого катализатора, необходимого для енолизации Δ4-3-кето-группы. Затем полученный 3,5-диенолэфир (IV) подвергают каталитической конденсации с реагентом Манниха, образованным из формальдегида и N-метиланилина in situ, с образованием 6-(N,N-дизамещенного)-аминометилпроизводного (V) с последующим его дезаминированием и превращением в 6-метилен-производное (VI), причем конденсация с реагентом Манниха может быть проведена как с предварительным выделением, так и без выделения 3,5-диенолэфира (IV), а также как с выделением, так и без выделения продукта конденсации (V) из реакционной массы. После этого химическим сольволизом 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метиленпрегн-4-ен-3,20-диона (VI) получают 6-метиленгидрокортизон (I) или его 11β-трифторацетат (VII). При этом удаление защитных групп соединения VI с образованием соединения I может быть реализовано одновременно в условиях основного катализа или поэтапно через образование соединения VII сольволизом в условиях кислого катализа и последующим сольволизом 11-трифторацетоксигруппы соединения VII в условиях основного катализа. Синтез 21-ацетокси-11β,17α-дигидрокси-6-метиленпрегн-4-ен-3,20-диона (VIII) может быть реализован селективным ацетилированием 21-гидроксильной группы соединения I или селективным удалением трифторацетильной группы соединения VI в известных условиях [US 4330541, 1982, пример 1].
Общий достигаемый выход 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метиленпрегн-4-ен-3,20-диона (VI) из 21-ацетата гидрокортизона (II) составляет 97,8%.
Общий достигаемый выход 11-трифторацетата 6-метиленгидрокортизона (VII) из 21-ацетата гидрокортизона (II) составляет 92,7%.
Общий достигаемый выход 6-метиленгидрокортизона (I) из 21-ацетата гидрокортизона (II) составляет 88,3%.
Общий достигаемый выход 21-ацетата 6-метиленгидрокортизона (VIII) из 21-ацетата гидрокортизона (II) составляет 85,2%.
Защиту 11β-гидроксильной группы молекулы 21-ацетата гидрокортизона (II) осуществляют образованием эфира трифторуксусной кислоты в среде органического растворителя в условиях основного катализа. Этот вариант защиты наиболее оптимален, так как реакция этерификации протекает в мягких условиях с практически количественным выходом. Кроме этого, трифторацетатная защитная группа, устойчивая в кислой среде, легко удаляется при необходимости гидролизом в условиях основного катализа. Ацилирование проводят с использованием минимально необходимого количества ангидрида трифторуксусной кислоты в безводных условиях в среде органического апротонного растворителя. Специалисту в области органической химии, а также из уровня техники, известно, что вместо ангидрида трифторуксусной кислоты в этом процессе может быть использован хлорангидрид трифторуксусной кислоты, а в качестве апротонного растворителя при осуществлении изобретения - способа получения 6-метиленгидрокортизона или его эфиров - могут быть использованы любые апротонные (неполярные, малополярные и полярные) растворители, пригодные для проведения реакции и инертные по отношению к ацилирующему реагенту и катализатору, такие как диалкилкетоны (например, ацетон, метилэтилкетон), циклические простые эфиры (например, диоксан, тетрагидрофуран) и другие, известные из уровня техники, обеспечивающие полноту проведения реакции. В качестве основного катализатора могут быть использованы гетероарильные соединения (например, пиридин или его производные), или алкиламины (например, триэтиламин), или другие органические основания, или их смеси, обычно используемые для катализа реакций ацилирования гидроксильных групп, известные из уровня техники. Наиболее оптимальным температурным режимом с технологической точки зрения является проведение процесса этерификации при температуре окружающей среды. Продолжительность процесса устанавливается по результатам контроля содержания исходного продукта в реакционной массе. Минимальное количество катализатора, необходимое для обеспечения полного и быстрого протекания реакции этерификации определяется экспериментально и может варьировать в зависимости от использованного катализатора.
Реакция трифторацетилирования соединения II протекает регионаправленно, этерифицируя предпочтительно вторичную гидроксильную группу при атоме С11 соединения II. Для обеспечения протекания реакции необходимым минимальным количеством ангидрида трифторуксусной кислоты является 1 моль на 1 моль стероида. Для более полного и быстрого протекания реакции используется небольшой избыток ацилирующего агента (~1,2-1,5 моль ТФА на 1 моль стероида). Однако это условие не является обязательным. Применение избыточного количества реагента приводит к частичному трифторацетилированию третичной гидроксильной группы при С17 продукта реакции III с образованием 10-20% примеси 11β,17α-дитрифторацетилокси-производного (IIIa).
Однако в условиях предлагаемого нами способа эфир трифторуксусной кислоты по С17 удаляется легко и селективно с регенерацией 17α-гидроксильной группы и с сохранением трифторацетатной защиты 11β-гидроксильной группы, что достигается обработкой реакционной массы триэтиламином. Выход 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-прегн-4-ен-3,20-диона (III) на стадии составляет от 96 до 99,95%.
Синтез 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метиленпрегн-4-ен-3,20-диона (VI) из 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-прегн-4-ен-3,20-диона (III) включает три последовательных химических процесса, которые могут быть проведены с выделением любого из интермедиатов общей формулы IV и V или без выделения по следующим вариантам химической схемы синтеза:
Вариант схемы 1: III - IV - V - VI; или
Вариант схемы 2: III - IV - [V] - VI; или
Вариант схемы 3: III - [IV] - V - VI; или
Вариант схемы 4: III - [IV] - [V] - VI.
Наиболее оптимальным и предпочтительным является вариант схемы 4.
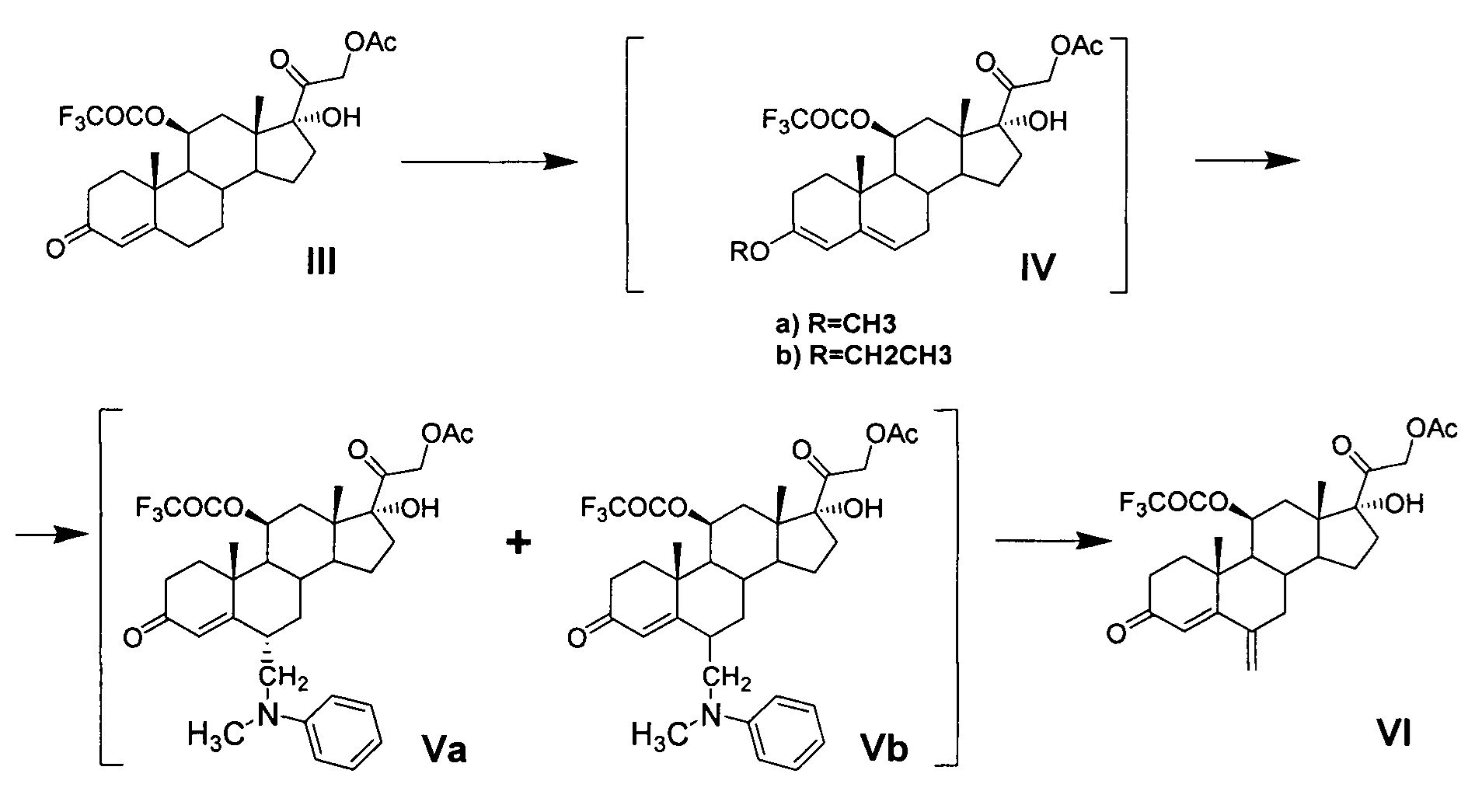
Для повышения нуклеофильности атома С6 и тем самым уменьшения вероятности протекания побочных процессов конденсации по другим реакционным центрам (например, по атому С2) на первом этапе синтеза проводится реакция енолизации Δ4-3-кетосистемы кольца А соединения III с образованием 3,5-диенолэфира общей формулы IV. Реакция енолэтерификации Δ4-3-кетосистемы кольца А как с выделением енолэфира IV (варианты 1 и 2), так и без выделения енолэфира IV (варианты 3 и 4) из реакционной массы, может быть проведена с применением или без применения растворителя. Реакцию енолэтерификации Δ4-3-кетосистемы с применением растворителя проводят действием триалкилортоэфира муравьиной кислоты (например, триметилортоформиата или триэтилортоформиата) с использованием минимального количества триалкилортоформиата, необходимого для обеспечения протекания реакции (не менее 1 моля на 1 моль стероида). Для более полного и быстрого протекания реакции (15-30 мин, но не более 2 ч) используется избыток триалкилортоформиата (от 2,5 до 9 моль на 1 моль стероида). Однако это условие не является обязательным. Реакция проводится в безводных условиях в соответствующем протонном растворителе - алифатическом спирте (в метаноле или этаноле) или в апротонном растворителе, выбранном из группы циклических эфиров (например, диоксан или тетрагидрофуран) или ароматических углеводородов группы бензола (например, бензол или толуол). Реакция енолэтерификации Δ4-3-кетосистемы без применения растворителя проводится в среде реагента триалкилортоформиата.
В качестве катализатора енолизации могут быть любые протонные кислоты, обеспечивающие енолизацию Δ4-3-кетосистемы кольца А соединения III с образованием 3,5-диенола, известные из уровня техники специалисту, в частности органические и минеральные кислоты. Минеральные кислоты выбирают из группы растворимых в воде - кислородсодержащих (например, HClO4, H2SO4, HNO3) или бескислородных (например, HCl). Предпочтительно использовать органические кислоты, например сульфоароматические (сульфосалициловую кислоту, п-ТСК или т.п.) как безводные, так и в виде кристаллогидрата. При проведении реакции без выделения енолэфира IV (вариант 3 и 4) из реакционной массы предпочтительно использовать п-ТСК, которая является также катализатором последующей реакции аминометилирования.
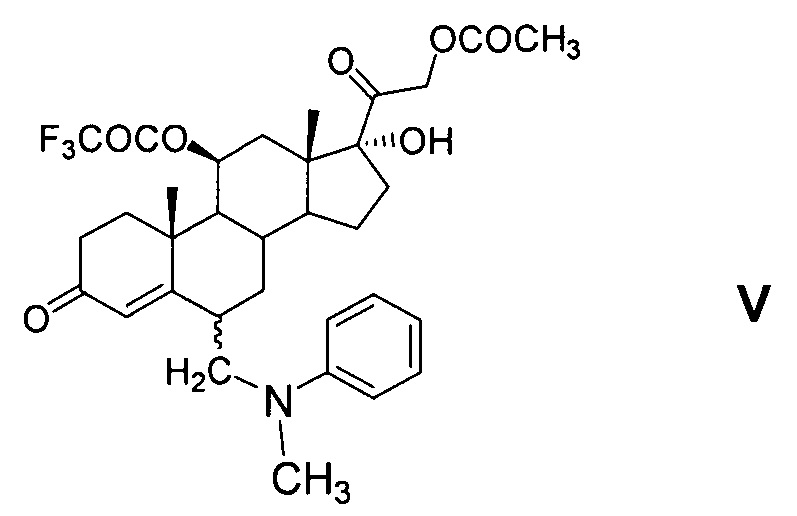
В синтезе соединений общей формулы V введение заместителя в положение С6 осуществляют методом трехкомпонентной конденсации по типу реакции Манниха с применением реагента Манниха, образованного in situ взаимодействием формальдегида и N-метиланилина или других вторичных аминов, применимых для целей настоящего изобретения, пригодных для образования 6-(N,N-дизамещенных)-аминометил-соединений и известных из уровня техники, например, N-этиланилин, дифениламин, N-метил-n-толуидин и т.п. [GB 1280570, 1972].
По настоящему изобретению реакция Манниха с образованием соединений общей формулы V может быть проведена как с выделением продукта IV из реакционной массы (варианты 1 и 3), так и без выделения (варианты 2 и 4). Однако предпочтительно проведение реакции Манниха без выделения енолэфира IV (варианты 3 и 4) в среде триалкилортоформиата без применения растворителей и на стадии получения енолэфира, и на стадии аминометилирования. Оба процесса проводятся при комнатной температуре. По окончании реакции получения енолэфира IV в реакционную массу добавляют N-метиланилин и 30-40% водный раствор формальдегида.
Реакция аминометилирования нестереоселективна: образуется смесь 6α- и 6β-диастереомерных стероидных оснований Манниха (Va и Vb соответственно). Реакция аминометилирования может быть проведена в среде, содержащей циклический эфир (например, диоксан или тетрагидрофуран) или ароматический углеводород группы бензола (например, бензол или толуол) причем указанные растворители или их смеси могут быть добавлены в реакционную массу, содержащую соединения общей формулы V, после окончания реакции аминометилирования. Раствор соединения общей формулы V в ароматическом углеводороде промывают 1-5% водным раствором аммиака для удаления п-ТСК и далее водой. Этот раствор в ароматическом углеводороде может быть использован на следующей стадии дезаминирования без извлечения смеси изомеров общей формулы V (варианты 2 и 4) или указанная смесь изомерных стероидных оснований Манниха может быть извлечена из раствора (варианты 1 и 3). Однако последнее нецелесообразно, так как оба изомера могут быть подвергнуты последующей реакции дезаминирования с образованием 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метиленпрегн-4-ен-3,20-диона (VI) без выделения и разделения.
Реакцию регионаправленного расщепления стероидного основания Манниха общей формулы V по связи C-N - проводят действием минеральных кислот, например, 30-35% соляной кислоты или 50-70% серной кислоты (предпочтительно 55-60% серной кислоты) в среде ароматического углеводорода ряда бензола (толуола или бензола) с добавлением апротонного растворителя, смешивающегося с водой, например, диалкилкетона (ацетона или метилэтилкетона) или циклического эфира (диоксана или тетрагидрофурана). Добавление протонного растворителя, в частности, низшего алифатического спирта (метилового, или этилового, или др.) может привести, во-первых, к частичному расщеплению стероидного основания Манниха общей формулы V по связи С-С с регенерацией исходного соединения III, а во-вторых, к снятию (удалению) сложноэфирной группы при атоме С21 как у образующегося побочного продукта III, так и у целевого продукта VI. Реакция регионаправленного расщепления стероидного основания Манниха общей формулы V по связи C-N может быть проведена в присутствии минеральных солей, например, галогенидов щелочных или щелочноземельных металлов, или галогенидов аммония. В качестве кислот, образующих соли металлов, могут быть использованы хлористоводородная, или бромистоводородная, или фтористоводородная кислоты. В качестве щелочных металлов, образующих соли галогенводородных кислот, могут быть использованы элементы 1-й группы периодической системы химических элементов (таблицы Д.И. Менделеева) - литий, натрий, калий. В качестве щелочноземельных металлов, образующих соли галогенводородных кислот, могут быть использованы элементы 2-й группы периодической системы химических элементов - кальций, барий, магний.
Для проведения реакции дезаминирования без выделения смеси изомеров Va и Vb (варианты 2 и 4) раствор смеси стероидных оснований Манниха (V) в ароматическом углеводороде разбавляют минимально необходимым количеством диалкилкетона (не менее 40% об.), добавляют 55-60% раствор серной кислоты и минеральную соль. Реакцию проводят при комнатной температуре. По окончании реакции реакционный раствор промывают от кислоты в делительной воронке сначала водой до рН ~ 6-7, затем 5% раствором водного аммиака и далее водой до нейтральной реакции. Растворитель упаривают в вакууме или при атмосферном давлении. Остаток ароматического углеводорода может быть удален азеотропной отгонкой с любым удобным растворителем, образующим азеотропную смесь с ароматическим углеводородом и смешивающимся с водой, например, с этиловым или метиловым спиртом. Кристаллизацию продукта реакции осуществляют растворением остатка после отгонки в спирте (этиловом или метиловом) и разбавлением полученного раствора водой. Кристаллы отфильтровывают.
Достигаемый выход 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метиленпрегн-4-ен-3,20-диона (VI) из 21-ацетата гидрокортизона (II) составляет:
по варианту 1 (II - III - IV - V - VI) - от 87,7 до 91,8%;
по варианту 2 (II - III - IV - [V] - VI) - от 88,3 до 92,5%;
по варианту 3 (II - III - [IV] - V - VI) - до 93,8%;
по варианту 4 (II - III - [IV] - [V] - VI) - до 97,8%.
Следует отметить, что проведение реакции дезаминирования в среде органического растворителя или смеси растворителей, действием минеральной кислоты (серной или соляной) без добавления неорганической соли протекает нерегиоселективно: наряду с дезаминированием имеет место процесс дезаминометилирования с регенерацией соединения III. Применение неорганической соли в сочетании с минеральной кислотой не является очевидным, не известно из уровня техники, для обеспечения регионаправленности расщепления стероидных оснований Манниха по связи C-N ранее не применялось. Проведение реакции без добавления неорганической соли приводит к снижению выхода продукта. Проведение реакции действием концентрированной соляной кислоты в среде тетрагидрофурана и этанола (литературный вариант расщепления основания Манниха [RU 2297423, 2007]) хотя и приводит к сокращению продолжительности процесса, однако при этом получается продукт VI, не пригодный для использования в реакциях с применением палладиевых катализаторов без дополнительной очистки, например, в реакции изомеризации С6-экзометиленовой двойной связи в 6,7-эндометиленовую двойную связь или в реакции гидрирования с образованием 6α-метилгидрокортизона или его эфиров. При этом в результате очистки кристаллизацией выход на стадии синтеза соединения VI составляет всего 60-65%.
Удаление сложноэфирных групп при С11 и С21 соединения VI может быть проведено одновременно или поэтапно реакциями сольволиза.
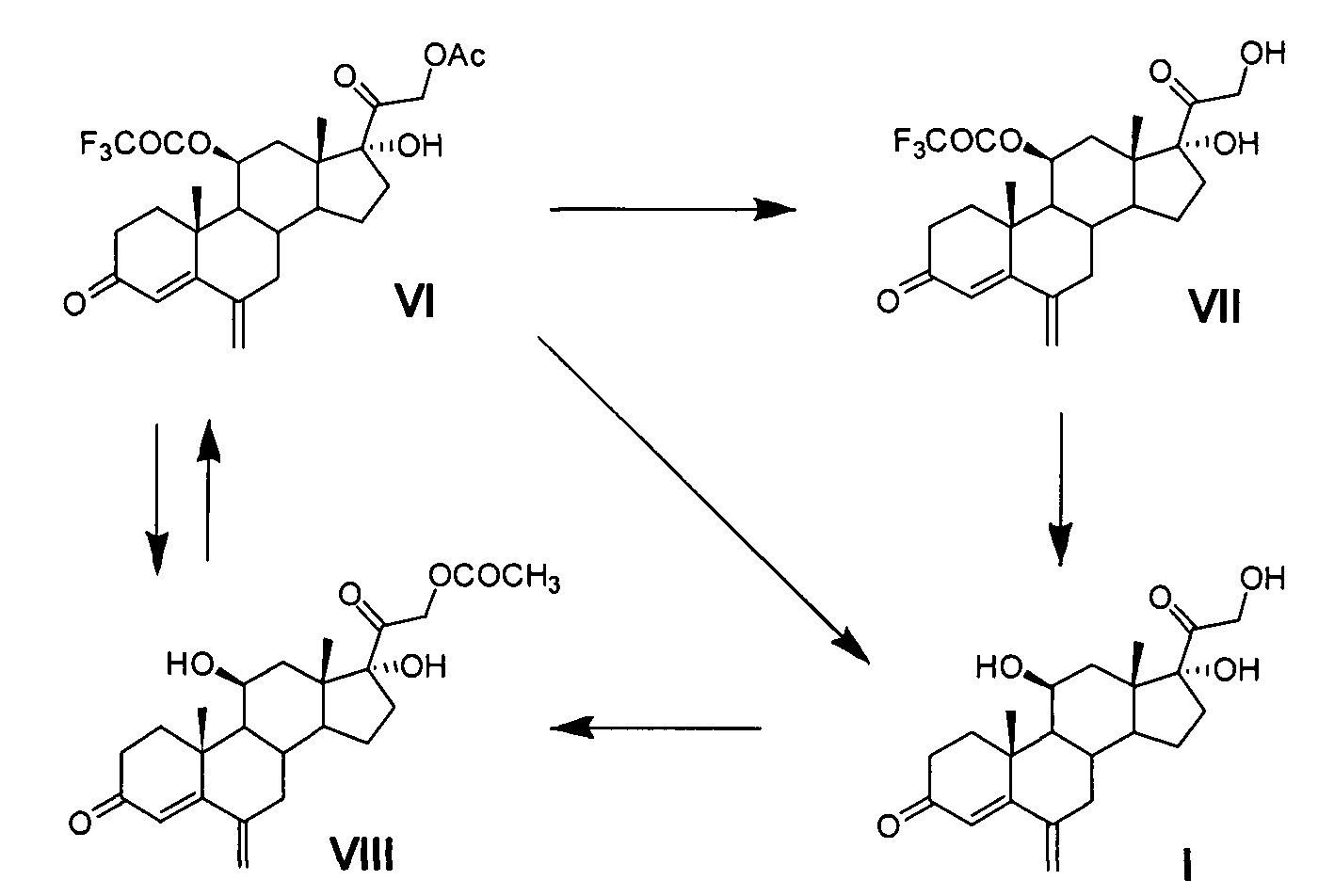
Полное удаление сложноэфирных групп при С11 и С21 с образованием соединения I осуществляют реакцией сольволиза, катализируемого сильным основанием. Реакцию проводят в среде метанола. В качестве сильного основания используют каталитическое количество гидроксида щелочного металла (натрия или калия) в виде 10% водного раствора. Из уровня техники общеизвестно, что для удаления защитных сложноэфирных групп сольволизом можно использовать раствор гидроксида щелочного металла или в воде, или в соответствующем алифатическом спирте (например, в метаноле) с концентрацией основания от 1 до 20%.
Получают 6-метиленгидрокортизон (I) с содержанием основного вещества 90-95%. Продукт может быть использован в химическом процессе каталитической изомеризации 6-экзометиленовой двойной связи в 6,7-эндометиленовуя двойную связь, а также в процессе каталитического гидрирования с образованием 6α-метил-гидрокортизона, без дополнительной очистки.
Удаление сложноэфирной группы при С21 у соединения VI с сохранением 11β-трифторацетоксигруппы и образованием 11-трифторацетата 6-метиленгидрокортизона (VII) проводят в условиях кислого катализа.
Удаление трифторацетильной группы при С11 у соединения VI с сохранением 21-ацетоксигруппы и образованием 21-ацетата 6-метиленгидрокортизона (VIII) может быть проведено в среде, содержащей алифатический С1-4-спирт в условиях основного катализа, например, действием триэтиламина в метаноле в условиях патента [US 4330541, 1982, пример 1].
Следует отметить, что синтез соединения VI может быть, в свою очередь, реализован трифторацетилированием гидроксильной группы при С11 21-ацетата 6-метиленгидрокортизона (VIII) в условиях синтеза соединения III по настоящему изобретению. Синтез 21-ацетата 6-метиленгидрокортизона (VIII) из соединения I может быть проведен ацетилированием в условиях основного катализа. Наиболее оптимальным температурным режимом с технологической точки зрения является проведение процесса этерификации при температуре окружающей среды. Продолжительность процесса устанавливается по результатам контроля содержания исходного продукта в реакционной массе. Минимальное количество катализатора, необходимое для обеспечения полного и быстрого протекания реакции этерификации определяется экспериментально и может варьировать в зависимости от использованного катализатора.
Эфиры 6-метиленгидрокортизона общей формулы I можно получить любым известным из уровня техники способом, поэтапно проводя этерификацию гидроксильных групп: сначала ацетилируя гидроксильную группу при атоме С21 6-метиленгидрокортизона (общая формула I, где R=R1=Н) с образованием 21-ацетата 6-метиленгидрокортизона (общая формула I, где R=Н; R1=СОСН3), а затем трифторацетилируя гидроксильную группу при атоме С11 21-ацетата 6-метиленгидрокортизона с образованием 11-трифторацетилокси-производного 21-ацетата 6-метиленгидрокортизона (общая формула I, где R=COCF3; R1=СОСН3). Специалисту, работающему в области химии стероидных соединений, известно, что ацетилирование первичной гидроксильной группы при атоме С21 производного гидрокортизона с образованием 21-ацетата производного гидрокортизона может быть реализовано только в первую очередь в условиях основного катализа действием ангидрида или хлорангидрида уксусной кислоты. При этом в качестве катализатора могут быть использованы производные аммиака - гетероарильные соединения (например, пиридин) или алкиламины (например, триэтиламин). При этом трифторацетилирование вторичной гидроксильной группы при атоме С11 21-ацетилированного производного гидрокортизона осуществляется только во вторую очередь и может быть проведено в условиях основного катализа действием ангидрида или хлорангидрида трифторуксусной кислоты, а в качестве катализатора могут быть использованы производные аммиака - гетероарильные соединения (например, диметиламинопиридин) или алкиламины (например, триэтиламин).
Заявленное изобретение иллюстрируется следующими примерами. Продолжительность реакций указана только для иллюстрации. Выход продуктов приведен только для иллюстрации. Применение неорганических солей приведено только для иллюстрации. Вместо указанных солей могут быть использованы любые растворимые в воде соли, образованные галогенводородными кислотами, например, хлориды и бромиды лития, натрия, калия, магния, бария, аммония и другие, без потери эффективности.
Пример 1. Получение 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-прегн-4-ен-3,20-диона (III) - трифторацетокси-производного 21-ацетата гидрокортизона
Вариант 1
К суспензии 10 г (24,722 ммоль) 21 ацетата гидрокортизона (II) в 80 мл сухого ацетона добавляли 0,3 г (2,455 ммоль) диметиламинопиридина. Затем при температуре не выше 10°С добавляли 4,2 мл (6,28 г, 29,9 ммоль) трифторуксусного ангидрида. Реакционную массу нагревали до комнатной температуры (20-25°С) и перемешивали в течение 1,5 ч. По окончании реакции добавляли 10 мл (7,28 г, 71,94 ммоль) сухого триэтиламина, выдерживали в течение 15 мин. Затем добавляли по каплям 4,7 мл (4,93 г, 82,1 ммоль) уксусной кислоты до рН 6 и реакционную массу выливали медленно в 240 мл воды при температуре 8-10°С. Суспензию выдерживали при интенсивном перемешивании в течение 1 ч при этой же температуре. Осадок отфильтровали, промыли водой до рН ~ 7 промывной воды, высушили до постоянного веса. Получили 11,88 г (23,736 ммоль) соединения III с выходом 96,01%. Т.пл.. 204-206°С (лит. 206-207°С [DE 1125422, 1962]). ЯМР 1Н (400 МГц, CDCl3, δ, м.д.): 5.70 (1H, с, Н-4), 5.67 (1Н, м, Н-11), 5.09 (1H д, J=17.4 Гц, Н-21), 4.67 (1Н, д, J=17.4 Гц, Н-21), 2.14 (3Н, с, СН3С(O)), 1.26 (3Н, с, Н-19), 0.80 (3Н, с, Н-18); ЯМР 13С (100.6 МГц, CDCl3, δ, м.д.): 204.9, 198.8, 170.5, 170.1, 122.9, 89.1, 75.4, 67.6, 54.9, 51.4, 47.0, 38.5, 35.5, 35.4, 34.5, 33.5, 32.2, 31.8, 31.7, 23.5, 20.6, 20.5, 16.4.. Найдено: С 60.47%; Н 6.24%, C25H31F3O7. Вычислено: С 59.99%; Н 6.24%.
Вариант 2
Реакционную смесь, состоящую из 100 г (247,22 ммоль) 21-ацетата гидрокортизона, 500 мл тетрагидрофурана и 10 г (81,833 ммоль) диметиламинопиридина, охлаждали при перемешивании до температуры 3-5°С. Затем к реакционной смеси в токе сухого азота медленно добавляли 42 мл (62,79 г, 298,96 ммоль) трифторуксусного ангидрида с такой скоростью, чтобы температура реакционной смеси не превышала 7-8°С. После этого баню с охлаждающей смесью убрали. Реакционную массу перемешивали до тех пор, пока температура в массе не поднялась до комнатной (самонагревание). По окончании выдержки реакционную массу нагревали до температуры кипения и отгоняли тетрагидрофуран при атмосферном давлении до тех пор, пока температура в парах не достигла значения 70°С.
Баню для обогрева поменяли на баню для охлаждения. Кубовую суспензию охладили до температуры 10°С и добавили через капельную воронку 55 мл (40,04 г, 395,69 ммоль) триэтиламина до значения рН реакционной массы 8-9. После этого охлаждающую баню убрали, суспензию перемешивали при комнатной температуре в течение 1 ч.
По завершении реакции реакционную массу вылили при интенсивном перемешивании в 600 мл воды, охлажденной до температуры 0-2°С. Перемешивали при этой же температуре в течение 10-15 мин и добавили 11 мл (11,54 г, 192,17 ммоль) уксусной кислоты для нейтрализации триэтиламина. Суспензию перемешивали при температуре 20-25°С в течение 20 мин. Осадок отфильтровали и промыли на фильтре водой до рН ~ 7 промывной воды, высушили до постоянного веса.
Получили 123,67 г (247,09 ммоль) соединения III с выходом 99,95%.
Вариант 3
Реакцию этерификации проводили аналогично варианту 1, используя в качестве апротонного растворителя диметилсульфоксид. Из 1 г (2,472 ммоль) 21 ацетата гидрокортизона (II) получили 1,12 г (2,238 ммоль) соединения III с выходом 90,53%.
А. Схема, вариант 1: III - IV - V - VI
Пример 2. Получение 3-метокси-21-ацетокси-11β-трифторацетилокси-17α-гидрокси-прегна-3,5-диен-20-она (IVa, R=CH3)
Вариант 1
К суспензии 5 г (9,99 ммоль) 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-прегн-4-ен-3,20-диона (III) в смеси 20 мл сухого метанола и 10 мл (9,676 г, 91,18 ммоль) триметилортоформиата добавляли 0,125 г (0,726 ммоль) безводной п-ТСК. Реакционную массу перемешивали в течение 30 мин. при комнатной температуре. Избыток п-ТСК нейтрализовали триэтиламином, выливали в 200 мл ледяной воды, суспензию охлаждали до температуры 0-2°С и выдерживали при перемешивании в течение 2 ч. Осадок отфильтровали, промывали на фильтре водой до рН ~ 7 промывной воды и минимальным количеством метанола, высушили до постоянного веса. Получили 5,04 г (9,795 ммоль) соединения IVa в виде белых кристаллов с выходом 98,05%. Т.пл. 177°С (с разл.). ЯМР 1Н (400 МГц, CDCl3, δ, м.д.): 5.72 (1Н, с, Н-11), 5.15-5.09 (3Н, м, Н-4, Н-6 и Н-21), 4.68 (1Н, д, J=17.4 Гц, Н-21), 3.57 (3Н, с, СН3О), 2.16 (3Н, с, СН3С(О)), 1.03 (3Н, с, Н-19), 0.80 (3Н, с, Н-18); ЯМР 13С (100.6 МГц, CDCl3, δ, м.д.): 205.0, 170.6, 155.4, 141.1, 121.0, 116.7, 97.5, 89.3, 75.8, 67.6, 54.3, 52.6, 50.6, 47.0, 38.7, 35.5, 34.6, 33.6, 31.4, 28.4, 24.8, 23.5, 21.1, 20.5, 16.3.
Вариант 2
К суспензии 1 г (1,998 ммоль) соединения III в смеси 4,7 мл сухого метанола и 0,55 мл (0,532 г, 5,015 ммоль) триметилортоформиата добавляли 0,011 г (0,05 ммоль) безводной сульфосалициловой кислоты (ССК). Реакционную массу перемешивали в течение 2 ч при комнатной температуре. Избыток ССК нейтрализовали триэтиламином. Реакционную массу разбавляли ледяной водой в 3 раза и выдерживали при перемешивании в течение 30 мин. при температуре 8-10°С. Осадок отфильтровывали, промывали на фильтре водой до рН ~ 7 промывной воды и минимальным количеством метанола, высушили до постоянного веса. Получили 0,96 г (1,816 ммоль) соединения IVa с выходом 90,89%.
Пример 3. Получение 3-этокси-21-ацетокси-11β-триФторацетилокси-17α-гидрокси-прегна-3,5-диен-20-она (IVб, R=CH2CH3)
В условиях получения соединения IVa (вариант 1), использовали 20 мл безводного этанола и 10 мл (8,91 г, 60,121 ммоль) триэтилортоформиата, из 5 г (9,99 ммоль) 21-ацетокси-11β-трифторацетилокси-17α-гидроксипрегн-4-ен-3,20-диона (III) получали 4,94 г (9,346 ммоль) соединения IVб с выходом 93,55%. Т.пл. 160°С (с разл.). ЯМР 1H (400 МГц, CDCl3, δ, м.д.): 5.72 (1H, с, Н-11), 5.17-5.07 (3Н, м, Н-4, Н-6 и Н-21), 4.68 (1Н, д, J=17.4 Гц, Н-21), 3.76 (2Н, кв, J=7.0 Гц, CH2O), 2.16 (3Н, с, СН3С(О)), 1.28 (3Н, т, J=7.0 Гц, CH3CH2O), 1.03 (3Н, с, Н-19), 0.80 (3Н, с, Н-18); ЯМР 13С (100.6 МГц, CDCl3, δ, м.д.): 205.0, 170.6, 154.5, 141.3, 116.4, 98.0, 89.3, 75.8, 67.6, 62.2, 59.5, 52.6, 50.6, 47.0, 35.5, 34.8, 33.6, 31.4, 28.4, 25.0, 23.5, 21.1, 20.5, 16.3, 14.6.
Пример 4. Получение 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6ξ-(N-метил-N-фениламинометил)прегн-4-ен-20-она (V)
К суспензии 10 ммоль 3,5-диенолэфира (IVa или IVб) в 15 мл абсолютного спирта (метанол или этанол соответственно) добавляли 2,15 мл (2,122 г, 19,8 ммоль) N-метиланилина. Затем при перемешивании добавляли по каплям раствор 0,125 г (0,725 ммоль) п-ТСК в 1,25 мл соответствующего спирта и 2 мл (2,18 г, 0,807 г в 100% исчислении, 26,86 ммоль) 37%-ного водного раствора формальдегида. Реакционную массу перемешивали при температуре 20°С в течение 2 ч. По окончании реакции реакционную массу разбавляли 20 мл соответствующего спирта и выливали медленно при интенсивном перемешивании в 110 мл воды. Осадок отфильтровывали, промывали водой до рН ~ 7 промывной воды, высушили до постоянного веса. Получали соединение V (смесь изомеров) с количественным выходом. Без очистки и разделения эпимеров использовали на следующей стадии. Для получения аналитически чистого образца использовали метод колоночной хроматографии на SiO2, предварительно обработанном триэтиламином.
Т.пл.=112-117°С.
6α-Изомер (Va), ЯМР 1Н (400 МГц, CDCl3, δ, м.д.): 7.28-7.22 (2Н, m, Н-3,5 (NPh)), 6.79-6.69 (3Н, m, Н-2,4,6 (NPh)), 5.87 (1Н, с, Н-4), 5.70 (1Н, м, Н-11), 5.08 (1Н, д, J=17.4 Гц, Н-21), 4.66 (1Н, д, J=17.4 Гц, Н-21), 3.70 (1H, дд, J=3.1, 14.5 Гц, NCH2), 3.14 (1Н, дд, J=10.4, 14.5 Гц, NCH2), 2.97 (3Н, с, NCH3), 2.16 (3Н, с, СН3С(О)), 1.19 (3Н, с, Н-19), 0.80 (3Н, с, Н-18).
6β-Изомер (Vб), ЯМР 1Н (400 МГц, CDCl3, δ, м.д.): 7.28-7.22 (2Н, m, Н-3,5 (NPh)), 6.79-6.69 (3Н, m, Н-2,4,6 (NPh)), 5.83 (1Н, с, Н-4), 5.70 (1Н, м, Н-11), 5.07 (1Н, д, J=17.4 Гц, Н-21), 4.69 (1Н, д, J=17.4 Гц, Н-21), 3.76 (1Н, дд, J=10.8, 14.5 Гц, NCH2), 3.32 (1Н, дд, J=4.1, 14.5 Гц, NCH2), 3.02 (3Н, с, NCH3), 2.16 (3Н, с, СН3С(О)), 1.36 (3Н, с, Н-19), 0.83 (3Н, с, Н-18); ЯМР 13С (100.6 MHz, CDCl3, δ, м.д.): 204.8, 198.7, 170.6, 170.2, 148.5, 129.4, 125.4, 116.8, 112.3, 89.1, 75.1, 67.6, 57.2, 54.7, 51.3, 46.8, 42.3, 39.7, 38.1, 37.8, 35.5, 34.5, 33.6, 33.3, 27.9, 23.3, 22.8, 20.5, 16.4. Найдено: С 64.08%; Н 6.52%, N 2.15%, C33H40F3NO7. Вычислено: С 63.96%; Н 6.51%, N 2.26%.
Пример 5. Получение 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метилен-прегн-4-ен-3,20-диона (VI)
Вариант 1
К суспензии 2,5 г (4,034 ммоль) смеси 6α и 6β изомеров соединения V в смеси 30 мл толуола и 15 мл метилэтилкетона добавляли 1 г (11,515 ммоль) бромида лития и 2 мл (3 г, 1,798 г в 100% исчислении, 18,332 ммоль) 60% серной кислоты.
Реакционную массу выдерживали при комнатной температуре в течение 1 ч и переносили в делительную воронку. Отделяли кислый слой, экстрагировали его толуолом и экстракт объединяли с органическим слоем. Толуольный раствор промывали 60% серной кислотой, водой до рН 6-7, 5% водным раствором аммиака и водой до нейтральной реакции. Затем раствор осветляли активированным углем и упаривали, удаляя остатки толуола метанолом. Кубовый остаток, выливали в 70 мл воды, суспензию выдерживали при температуре 8-10°С, осадок отфильтровали. Получили 1,94 г (3,785 ммоль) соединения VI с выходом 93,83%. Т.пл. 191-193°С. ЯМР 1Н (400 МГц, CDCl3, δ, м.д.): 5.86 (1H, с, Н-4), 5.68 (1Н, м, Н-11), 5.09-5.01 (3Н, м, СН2= и Н-21), 4.69 д (1H, д, J=17.4 Гц, Н-21), 2.15 с (3Н, с, СН3С(O)), 1.18 (3Н, с, Н-19), 0.80 (3Н, с, Н-18); ЯМР 13С (100.6 MHz, CDCl3, δ, м.д.): 204.9, 199.2, 170.6, 168.2, 144.1, 120.6, 115.4, 89.0, 75.4, 67.6, 53.7, 51.7, 46.9, 39.7, 38.7, 35.4, 34.7, 34.5, 33.3, 31.9, 23.4, 20.5, 19.9, 16.3. Найдено: С 61.31%; Н 6.35%, C26H31F3O7. Вычислено: С 60.93%; Н 6.10%.
Вариант 2
К суспензии 0,5 г (0,807 ммоль) смеси 6α и 6β изомеров соединения V в смеси 10 мл толуола и 10 мл метанола добавляли 0,32 мл (0,376 г) 35% соляной кислоты (0,13 г хлористого водорода, 3,556 ммоль) соляной кислоты и 1 мл воды. Реакционную массу перемешивали в течение 1 ч при комнатной температуре и переносили в делительную воронку. Отделяли кислый слой, экстрагировали его толуолом и экстракт объединяли с органическим слоем. Толуольный раствор промывали 35% соляной кислотой, водой до рН 6-7, 5% водным раствором аммиака и водой до нейтральной реакции. Затем раствор осветляли активированным углем и упаривали, удаляя остатки толуола метанолом. Остаток после упаривания (0,323 г) содержал смесь соединений III, VI и 17α,21-дигидрокси-11β-трифторацетилокси-6-метилен-прегн-4-ен-3,20-диона в соотношении 26:57:17% (ВЭЖХ). Для разделения смеси и идентификации соединений использовали метод колоночной хроматографии на SiO2.
17α,21-Дигидрокси-11β-трифторацетилокси-6-метилен-прегн-4-ен-3,20-дион, т.пл. 120-122°С. ЯМР 1Н (400 МГц, CDCl3, δ, м.д.): 5.87 (1Н, с, Н-4), 5.67 (1Н, кв, 11-Н, J=2.8 Гц), 5.10 (1Н, т, 6-С=СН2, J=1.9), 5.01 (1H, т, 6-С=СН2, J=1.9), 4.27 дд, 4.64 дд (АВ, 2Н, 21-СН2, J=4.5, J=19.9), 1.17 (3Н, с, Н-19), 0.80 (3Н, с, Н-18).
Б. Схема, вариант 2: III - IV - [V] - VI
Пример 6. Получение 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метилен-прегн-4-ен-20-она (VI)
Вариант 1
К реакционной массе, содержащей 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6ξ-(N-метил-N-фениламинометил)-прегна-4-ен-3.20-дион (V), полученной из 0.5 г (0,972 ммоль) 3-метокси-21-ацетокси-17α-гидрокси-11β-трифторацетилокси-прегна-3,5-диен-20-она (IVa) по варианту 1 схемы синтеза (пример 4), добавляли 0,38 мл (0,446 г) 35% соляной кислоты (0,156 г хлористого водорода, 4,283 ммоль). Реакционную массу перемешивали при комнатной температуре в течение 30 мин., затем разбавляли 10 мл метилового спирта и выливали в 20 мл воды. Осадок отфильтровали, промыли на фильтре водой до рН ~ 7 промывной воды, высушили до постоянного веса. После перекристаллизации из метанола получали 0.31 г (0,605 ммоль) соединения VI с выходом 62,24%, считая из IVa. Т.пл. 187-190°С.
Вариант 2
К раствору 0,215 мл (0,212 г, 1,98 ммоль) N-метиланилина в 1,25 мл тетрагидрофурана добавляли 13 мг (0,068 ммоль) п-ТСК моногидрата и 0,2 мл (0,218 г, 0,08 г в 100% исчислении, 2,686 ммоль) 37% раствора формальдегида. Затем при перемешивании и комнатной температуре добавляли раствор 0,504 г (0,98 ммоль) 3-метокси-21-ацетокси-17α-гидрокси-11β-трифторацетилокси-прегна-3,5-диен-20-она (IVa) в 2,5 мл тетрагидрофурана. Реакционную массу перемешивали в течение 1 ч при комнатной температуре, затем добавили 20 мл толуола и 0,38 мл (0,446 г) 35% соляной кислоты (0,156 г хлористого водорода, 4,283 ммоль). Реакционную массу перемешивали при комнатной температуре в течение 30 мин., и переносили в делительную воронку. Кислый слой отделяли, толуольный раствор промывали 5% соляной кислотой, водой до рН 6-7, 1% водным раствором аммиака и водой до нейтральной реакции. Затем раствор осветляли активированным углем и упаривали, удаляя остатки толуола метанолом. К остатку добавили 2 мл метанола и 10 мл воды, суспензию выдерживали 30 мин. при температуре 8-10°С, осадок отфильтровали, промыли на фильтре водой, высушили до постоянного веса. Получили 0,326 г (0,636 ммоль) соединения VI с выходом 64,9%, считая из IVa.
Вариант 3
К 2 г (3,783 ммоль) 3-этокси-21-ацетокси-17α-гидрокси-11β-трифторацетилокси-прегна-3,5-диен-20-она (IVb) в 2 мл триэтилортоформиата добавляли 0,48 мл (0,474 г, 4,42 ммоль) N-метиланилина. Затем добавляли по каплям раствор 0,075 г (0,435 ммоль) п-ТСК в 0,75 мл этанола и 0,41 мл (0,447 г, 0,165 г в 100% исчислении, 5,506 ммоль) 37% раствора формальдегида. Реакционную массу перемешивали в течение 1,5 ч при комнатной температуре и разбавляли 20 мл толуола. После этого добавляли 10 мл ацетона, 3 г (14,407 ммоль) хлорида бария и 3 мл (4,336 г, 2,385 г в 100% исчислении, 24, 31 ммоль) 55% серной кислоты. Реакционную массу перемешивали при комнатной температуре в течение 1 ч, затем обрабатывали, как указано в примере 5. Получили 1.83 г (3,571 ммоль) соединения VI с выходом 94,4%, считая из IVb.
В. Схема, вариант 3: III - [IV] - V
Пример 7. Получение 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6ξ-(N-метил-N-фениламинометил)прегн-4-ен-20-она (V)
К суспензии 5 г (9,99 ммоль) соединения III в 7,5 мл триалкилортоформиата добавляли 0,125 г (0,725 ммоль) п-ТСК. Реакционную массу перемешивали при комнатной температуре в течение 15-30 мин. После окончания реакции получения 3,5-диенолэфира (IVa или IVб) к реакционной массе добавили 2,15 мл (2,122 г, 19,8 ммоль) N-метиланилина и 2 мл (2,18 г, 0,807 г в 100% исчислении, 26,86 ммоль) 37% водного раствора формальдегида. Реакционную массу перемешивали при комнатной температуре в течение 2-2,5 ч. По окончании реакции для выделения смеси 6ξ-(N-метил-N-фениламинометил)-эпимеров реакционную массу разбавили 20 мл спирта (метанола или 95% этилового спирта) и медленно выливали в 200 мл воды. Осадок отфильтровали, промыли на фильтре водой до рН ~ 7 промывной воды, высушили до постоянного веса.
Получили соединение V (смесь изомеров) с количественным выходом.
Г. Схема, вариант 4: III - [IV] - [V]- VI
Пример 8. Получение 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метилен-прегн-4-ен-20-она (VI)
Вариант 1
К суспензии 20 г (39,96 ммоль) 21-ацетокси-11-трифторацетилокси-гидрокортизона (III) в 20 мл (17,82 г, 120,24 ммоль) триэтилортоформиата добавили 0,5 г (2,90 ммоль) п-ТСК и перемешивали в токе аргона при комнатной температуре в течение 15 мин. При этом наблюдали растворение осадка соединения III и образование гелеобразной массы темно-зеленого цвета.
По окончании реакции подачу инертного газа прекращали и добавляли медленно по каплям 4,8 мл (4,74 г, 44,212 ммоль) N-метиланилина и 4,1 мл (4,489 г, 1,654 г в 100% исчислении, 55,063 ммоль) 37% водного раствора формальдегида. После добавления N-метиланилина наблюдали изменение цвета реакционной массы от темно-зеленого до красновато-коричневого. Реакционную массу перемешивали при комнатной температуре в течение 1,5 ч.
По окончании реакции аминометилирования к реакционной суспензии медленно при интенсивном перемешивании добавляли 200 мл толуола. Реакционную массу перемешивали в течение 30 мин. при комнатной температуре. Наблюдали растворение гелеобразного осадка и отделение водного слоя. К реакционному раствору добавили 50 мл 5% водного раствора аммиака и массу перемешивали в течение 10 мин. Наблюдали изменение цвета реакционной массы от красного до оранжево-желтого.
Водный слой отделяли и экстрагировали толуолом трижды. Органический слой и толуольные экстракты объединили и промыли 5% водным раствором аммиака и водой до нейтральной реакции.
После этого к раствору соединения V в толуоле (~375 мл) при перемешивании добавили 150 мл метилэтилкетона, 30 мл (43,36 г, 23,847 г в 100% исчислении, 243,15 ммоль) 55% водного раствора серной кислоты и 10 г (90,11 ммоль) хлорида кальция. Реакционную массу выдерживали при комнатной температуре и интенсивном перемешивании в течение 1 ч. По окончании реакции дезаминирования реакционную массу перенесли в делительную воронку. Водный кислый слой, представляющий собой густую жидкость красного цвета, отделяли от органического слоя и экстрагировали толуолом трижды.
Органический слой и толуольные экстракты объединили, промывали последовательно 5 мл 55% водного раствора серной кислоты, водой до рН ~ 6-7 (порциями по 10 мл); 5 мл 5% водного раствора аммиака и водой до нейтральной реакции.
Толуольный раствор соединения VI после промывки осушали сульфатом натрия и осветляли активированным углем. Затем растворитель упарили (в вакууме или при атмосферном давлении). Остаток толуола удаляли методом азеотропной отгонки с этанолом.
После полного удаления толуола к остатку в кубе при температуре 50-60°С добавили 100 мл этанола и массу перемешивали при этой же температуре до полного растворения. Раствор соединения VI в этаноле, охлажденный до температуры 40-45°С, выливали тонкой струей в 500 мл воды при интенсивном перемешивании. Суспензию перемешивали в течение 30 мин. при температуре 10-15°С, затем выдерживали без перемешивания в течение 2 ч при этой же температуре. Осадок отфильтровали и промывали водой до рН ~ 7 промывной воды, высушили до постоянного веса.
Получили 20,04 г (39,101 ммоль) 21-ацетокси-11-трифторацетилокси-6-метилен-гидрокортизона (VI) с выходом 97,85%. Т.пл. 190-192°С.
Вариант 2
К раствору 1,3 г (2,597 ммоль) 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-прегн-4-ен-3,20 диона (III) в 2 мл (1,782 г, 12,024 ммоль) триэтилортоформиата добавили 16,9 мг (98,13 ммоль) п-ТСК. Реакционную массу выдерживали в токе сухого азота в течение 30 мин. при комнатной температуре и добавили 0,56 мл (0,553 г, 5,16 ммоль) N-метиланилина и 0,52 мл (0,567 г, 0,21 г в 100% исчислении, 6,98 ммоль) водного 37% раствора формальдегида. Реакционную массу выдерживали в течение 1,5 ч. Затем добавили 1 мл (1,174 г) 35% соляной кислоты (0,411 г хлористого водорода, 11,27 ммоль) и перемешивали в течение 30 мин. После этого к реакционной массе добавили 50 мл этилацетата, перемешали интенсивно в течение 1-2 мин., перенесли в делительную воронку. Органический слой отделяли и промывали водой до нейтральной реакции, растворитель упаривали в вакууме. Остаток растворили в 5 мл метанола. Полученный раствор добавляли при перемешивании по каплям к 50 мл воды, охлажденной до температуры 5-10°С. Суспензию перемешивали при этой же температуре в течение 15 мин., осадок отфильтровали, промыли водой до рН ~ 7 промывной воды, высушили до постоянного веса. Получили 1,28 г технического продукта соединения VI, который перекристаллизовали из диэтилового эфира. Получили 1,16 г (2,263 ммоль) соединения VI с выходом 87,15%, т.пл. 189-191°С.
Вариант 3
К суспензии 10 г (19,98 ммоль) 21-ацетокси-11-трифторацетилокси-гидрокортизона (III) в 15 мл (14,514 г, 136,77 ммоль) триметилортоформиата добавили 0,26 г (1,51 ммоль) п-ТСК и перемешали в токе аргона при комнатной температуре в течение 15 мин. При этом наблюдали растворение осадка соединения III.
По окончании реакции подачу инертного газа прекращали и добавили медленно по каплям 4,3 мл (4,244 г, 39,607 ммоль) N-метиланилина, 15 мл толуола и 4,0 мл (4,36 г, 1,613 г в 100% исчислении, 53,72 ммоль) 37% водного раствора формальдегида. Реакционную массу перемешивали при комнатной температуре в течение 2 ч.
Затем к реакционному раствору добавили 7,7 мл (9,04 г, 3,164 г хлористого водорода, 86,778 ммоль) 35% соляной кислоты и массу интенсивно перемешивали в течение 30 мин.
По окончании реакции дезаминирования реакционную массу перенесли в делительную воронку. Водный слой отделили и экстрагировали толуолом трижды. Органический слой и толуольные экстракты объединили и промыли водой до рН~ 6-7 порциями по 10-15 мл, 5 мл 0,5% водного раствора аммиака, а затем водой до нейтральной реакции.
Толуольный раствор соединения VI после промывки осушили сульфатом натрия и осветлили активированным углем. Затем растворитель упарили в вакууме, удаляя остаток толуола метанолом.
После полного удаления толуола к остатку в колбе добавили 20 мл метанола и 100 мл воды при интенсивном перемешивании. Суспензию перемешивали в течение 30 мин. при температуре 10-15°С, затем выдерживали без перемешивания в течение 2 ч при этой же температуре. Осадок отфильтровали и промыли водой до рН ~ 7 промывной воды, высушили до постоянного веса.
Получили 8,28 г (16,156 ммоль) 21-ацетокси-11-трифторацетилокси-6-метилен-гидрокортизона (VI) с выходом 80,86%.
Вариант 4
К суспензии 1 г (1,998 ммоль) 21-ацетокси-11-трифторацетилокси-гидрокортизона (III) в 1 мл (0,891 г, 6,012 ммоль) триэтилортоформиата добавили 5 мл толуола и 0,026 г (0,151 ммоль) п-ТСК и перемешали в токе аргона при комнатной температуре в течение 15 мин.
По окончании реакции подачу инертного газа прекращали и добавили медленно по каплям 0,43 мл (0,424 г, 3,961 ммоль) N-метиланилина, и 0,4 мл (0,436 г, 0,161 г в 100% исчислении, 5,372 ммоль) 37% водного раствора формальдегида. Реакционную массу перемешивали при комнатной температуре в течение 2 ч.
Затем к реакционному раствору добавили 0,77 мл (0,904 г, 0,316 г хлористого водорода, 8,678 ммоль) 35% соляной кислоты и массу интенсивно перемешивали в течение 30 мин.
По окончании реакции дезаминирования реакционную массу разбавили 5 мл толуола и перенесли в делительную воронку. Водный слой отделили и экстрагировали толуолом трижды. Органический слой и толуольные экстракты объединили и промыли водой до рН~ 6-7 трижды порциями по 5 мл, 1 мл 0,1% водного раствора аммиака, а затем водой до нейтральной реакции трижды порциями по 5 мл.
Толуольный раствор соединения VI после промывки осушили сульфатом натрия и осветлили активированным углем. Затем растворитель упарили в вакууме, удаляя остаток толуола метанолом.
После полного удаления толуола к остатку в колбе добавили 2,5 мл метанола и 25 мл воды при интенсивном перемешивании. Суспензию перемешивали в течение 30 мин. при температуре 10-15°С, затем выдерживали без перемешивания в течение 2 ч при этой же температуре. Осадок отфильтровали и промыли водой до рН ~ 7 промывной воды, высушили до постоянного веса.
Получили 0,84 г (1,639 ммоль) 21-ацетокси-11-трифторацетилокси-6-метилен-гидрокортизона (VI) с выходом 82,03%.
Вариант 5
К суспензии 3 г (5,994 ммоль) 21-ацетокси-11-трифторацетилокси-гидрокортизона (III) в 3 мл (2,903 г, 1,804 ммоль) триметилортоформиата добавили 3 мл толуола и 0,075 г (0,436 ммоль) п-ТСК и перемешали в токе аргона при комнатной температуре в течение 15 мин.
По окончании реакции подачу инертного газа прекращали и добавили медленно по каплям 1,29 мл (1,272 г, 11,883 ммоль) N-метиланилина, 1,2 мл (1,308 г, 0,483 г в 100% исчислении, 16,116 ммоль) 37% водного раствора формальдегида и 15 мл толуола. Реакционную массу интенсивно перемешивали при комнатной температуре в течение 2 ч.
Затем к реакционному раствору добавили 2,31 мл (2,712 г, 0,948 г хлористого водорода, 26,034 ммоль) 35% соляной кислоты 3 г хлорида лития (70,034 ммоль) и массу интенсивно перемешивали в течение 30 мин.
По окончании реакции дезаминирования реакционную массу обрабатывали так же, как в варианте 4.
Получили 2,78 г (5,424 ммоль) 21-ацетокси-11-трифторацетилокси-6-метилен-гидрокортизона (VI) с выходом 90,48%.
Вариант 6
В условиях примера 1 (вариант 2) из 5 г (12,0 ммоль) 21-ацетата 6-метиленгидрокортизона (VIII) получили 5,54 г (10,81 ммоль) соединения VI с выходом 90,08%.
Пример 9. Получение 17α,21-дигидрокси-11β-трифторацетилокси-6-метиленпрегн-4-ен-3,20-диона ((11-триФторацетата 6-метиленгидрокортизона, VII)
Вариант 1
К раствору 1,0 г (1,951 ммоль) 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метиленпрегн-4-ен-3,20-диона (VI) в 20 мл метанола добавили 5 мл воды и 1 мл (1,17 г, 0,41 г хлористого водорода, 1,27 ммоль) 35% соляной кислоты. Реакционную смесь перемешивали при комнатной температуре. По окончании реакции реакционную массу медленно вылили в 100 мл воды. Суспензию выдерживали при температуре 10°С в течение 2 ч. Осадок отфильтровали, промыли водой до рН 7, высушили до постоянного веса. Получили 0,87 г (1,849 ммоль) соединения VII с выходом 94,77%. Т.пл. 120-122°С. ЯМР 1Н (400 МГц, CDCl3, δ, м.д.): 5.87 (1Н, с, Н-4), 5.67 (1Н, кв, J=2.8 Гц, Н-11), 5.10 (1H, т, J=1.9 Гц, СН2=), 5.01 (1Н, т, J=1.9 Гц, СН2=), 4.64 (1Н, дд, J=4.5, 19.9 Гц, Н-21), 4.27 (1Н, дд, J=4.5, 19.9 Гц, Н-21), 1.17 (3Н, с, Н-19), 0.80 (3Н, с, Н-18).
Вариант 2
В толуольный раствор, полученный в условиях примера 5 (вариант 1, 2,5 г (4,034 ммоль) смеси 6α и 6β изомеров соединения V) и содержащий 21-ацетокси-17α-гидрокси-11β-трифторацетилокси-6-метиленпрегн-4-ен-3,20-дион (VI), добавили 50 мл метанола и 2,5 мл (2,925 г, 1,025 г хлористого водорода, 3,175 ммоль) 35% соляной кислоты. Реакционную смесь перемешивали при комнатной температуре. По окончании реакции реакционную массу медленно вылили в 100 мл воды при интенсивном перемешивании, водный слой отделили, экстрагировали трижды по 20 мл толуола, объединенные экстракты промывали водой до рН 6-7, 5% водным раствором аммиака и водой до нейтральной реакции. Затем раствор осветляли активированным углем и упаривали, удаляя остатки толуола метанолом. Кубовый остаток (~7 мл), выливали в 70 мл воды, суспензию выдерживали при температуре 8-10°С, осадок отфильтровали, сушили до постоянного веса. Получили 1,72 г (3,656 ммоль) соединения VII с выходом 90,63%, считая из соединения V. Т.пл. 118-120°С.
Пример 10. Получение 6-метиленгидрокортизона (I)
Вариант 1
К раствору 2 г (3,902 ммоль) 21-ацетокси-11-трифторацетилокси-6-метиленгидрокортизона (VI) в 20 мл ацетона в токе азота медленно добавляли 2 мл (2,218 г, 0,222 г NaOH, 5,545 ммоль) 10% водного раствора гидроокиси натрия при комнатной температуре в течение 30-40 мин., периодически контролируя значение рН реакционной массы (рН должен быть не более 8). Реакционную массу выдерживали при комнатной температуре в токе азота. По окончании реакции реакционную массу нейтрализовали 5% раствором соляной кислоты и вылили медленно в 60 мл воды при интенсивном перемешивании и температуре 10-15°С. Образовавшуюся суспензию перемешивали в течение 1 ч при этой же температуре, затем выдерживали без перемешивания в холодильнике при температуре 5-10°С в течение 8-12 ч. Осадок отфильтровали, промыли на фильтре водой до значения рН промывной воды ~7, высушили до постоянного веса. Получили 1,32 г (3,525 ммоль) 6-метиленгидрокортизона (I) с выходом 90,3%. Т.пл. 199-200°С. ЯМР 1Н (400 МГц, CDCl3, δ, м.д.): 5.85 (1H, с, Н-4), 5.07 (1H, т, J=1.9 Гц, СН2=), 4.97 (1Н, т, J=1.9 Гц, СН2=), 4.65 (1Н, дд, J=4.8, 19.7 Гц, Н-21), 4.50 (1Н, м, Н-11), 4.31 (1Н, дд, J=4.8, 19.7 Гц, Н-21), 1.34 (3Н. с, Н-19), 0.94 (3Н, с, Н-18).
Вариант 2
В условиях варианта 1 из 1 г (2,125 ммоль) 11-трифторацетилокси-6-метиленгидрокортизона (VII) получили 0,71 г (1,896 ммоль) 6-метиленгидрокортизона (I) с выходом 89,2%. Т.пл. 200-201°С.
Пример 11. Получение 21-ацетокси-11β,17α-дигидрокси-6-метиленпрегн-4-ен-20-она (21-ацетата 6-метиленгидрокортизона, VIII)
К суспензии 4,67 г (12,47 ммоль) 6-метиленгидрокортизона (I) в 47 мл метилэтилкетона добавили 2,24 мл (3,085 г, 30,48 ммоль) триэтиламина и 1,52 мл (1,645 г, 16,11 ммоль) уксусного ангидрида. Реакционную массу перемешивали при комнатной температуре в течение 1 ч. По окончании реакции реакционную массу разбавили 20 мл метанола и выливали медленно в 80 мл воды, охлажденной до 5-10°С. Суспензию выдерживали при той же температуре в течение 1 ч при перемешивании и 2 ч без перемешивания. Осадок отфильтровывали, промывали на фильтре дистиллированной водой до рН ~7, сушили до постоянного веса в вакууме. Получали 5,01 г (12,03 ммоль) соединения VIII с выходом 96,47%. Т. пл. 202-204°С. ЯМР 1Н (400 МГц, CDCl3, δ, м.д.): 5.84 (1H, с, Н-4), 5.06 (1Н, т, J=1.9 Гц, СН2=), 5.02 (1Н, д, J=17.4 Гц, Н-21), 4.96 (1Н, т, J=1.9 Гц, СН2=), 4.85 (1H, д, J=17.4 Гц, Н-21),4.48 (1Н, м, Н-11), 2.16 (3Н, с, СН3С(O)), 1.33 (3Н. с, Н-19), 0.94 (3Н, с, Н-18).
Микробиологический способ получения 21-ацетоксипрегна-1,4,9( 11 ),16-тетраен-3,20-диона из 21-ацетоксипрегна-4,9( 11 ),16-триен-3,20-диона
Способ получения андроста-4,9(11)-диен-3,17-диона из фитостерина
Способ получения 6-(n-метил-n-фенил)аминометил-гидрокортизона или его эфиров из 21-ацетата гидрокортизона
Способ получения 6α-метилгидрокортизона или его эфиров из 21-ацетата гидрокортизона
Способ получения 6-дегидро-6-метилгидрокортизона или его эфиров из 21-ацетата гидрокортизона
Рекомбинантный штамм мицелиального гриба aspergillus nidulans и его применение для 11α-гидроксилирования прогестерона и для ацетилирования 11α-гидроксипрогестерона
Способ получения 24-эпибрассинолида
Биокатализатор для получения 11α-ацетоксипрогестерона

