Результат интеллектуальной деятельности: СПОСОБ И УСТРОЙСТВО ДЛЯ ДЕТЕКТИРОВАНИЯ ХРОМОСОМНЫХ СТРУКТУРНЫХ АНОМАЛИЙ
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к области техники технологий геномики и биоинформатики и, более конкретно, к способу и устройству для детектирования хромосомных структурных аномалий.
Уровень техники
Обычно используемые в настоящее время способы исследования хромосом включают следующие.
Анализ кариотипа, например анализ кариотипа с помощью G-бэндинга, определяет хромосомные структурные аномалии на основании распределения 400-600 BAND, и таким образом обычно может детектировать аномалии только на хромосомном уровне. Данный способ может детектировать в лучшем случае делецию или повтор свыше 5 м.п.о., но не может детектировать меньшие фрагменты (<5 M). Кроме того, для данного способа необходима культура живых клеток, и требуется, чтобы клетки оставались живыми.
Флуоресцентная in situ гибридизация (FISH) может детектировать делецию, повтор и сбалансированную транслокацию гораздо меньших фрагментов. Однако требуется, чтобы подлежащий детектированию хромосомный фрагмент был заранее определен для приготовления соответствующего зонда. Как таковой, данный способ ограничен дизайном зонда. FISH часто используют для проверки результатов детектирования, поскольку он не способен детектировать неизвестные области.
Микроматричный метод включает два зондовых метода. Один построен на основании однонуклеотидных полиморфизмов (ОНП), а другой построен на основании CNV. Следовательно, данный способ страдает от тех же ограничений, что и FISH.
При постоянном развитии технологий секвенирования всего генома стоимость секвенирования постоянно снижается, так что может стать возможным обобщение секвенирования всего генома. Необходимо исследовать средства поиска хромосомных структурных аномалий, основанные на результатах секвенирования всего генома.
Сущность изобретения
В соответствии с аспектом настоящего изобретения предложен способ детектирования хромосомных структурных аномалий, содержащий: получение результата секвенирования всего генома целевого индивидуума, где результат секвенирования всего генома включает множество пар прочтений, причем каждая пара прочтений состоит из двух последовательностей прочтений, расположенных соответственно с двух концов исследуемого хромосомного фрагмента, и каждую пару прочтений получают отдельно от положительных и отрицательных нитей соответствующего хромосомного фрагмента или как от положительной, так и от отрицательной нити соответствующего хромосомного фрагмента; выравнивание результата секвенирования с референсной последовательностью для получения набора аномальных соответствий, где набор аномальных соответствий включает первый тип пар прочтений, описываемый тем, что две последовательности прочтений в первом типе пары прочтений соответствуют, соответственно, различным хромосомам референсной последовательности; кластеризацию последовательностей прочтений в наборе аномальных соответствий на основании соответствующих им положений, где каждый кластер содержит последовательности прочтений одного конца из группы пар прочтений, а соответствующие последовательности прочтений другого конца принадлежат к другому кластеру; фильтрацию получаемых в результате кластеров, включающую вычисление компактности каждого кластера и отфильтровывание кластеров, имеющих компактность, не удовлетворяющую заранее заданному требованию в отношении R-va, и кластеров, парных им, и получение отфильтрованных итоговых кластеров, содержащих первый тип пар прочтений, для определения наличия хромосомной структурной аномалии транслокационного типа.
В соответствии с другим аспектом настоящего изобретения предложено устройство для детектирования хромосомных структурных аномалий, которое содержит узел ввода данных, выполненный с возможностью ввода данных; узел вывода данных, выполненный с возможностью вывода данных; узел хранения, содержащий в себе выполняемую программу и выполненный с возможностью хранения данных; и процессор в информационном соединении с узлом ввода данных, узлом вывода данных и узлом хранения, выполненный с возможностью выполнения выполняемой программы, сохраненной в узле хранения, причем выполнение программы включает осуществление способа детектирования хромосомных структурных аномалий.
В соответствии с еще одним аспектом настоящего изобретения предложена машиночитаемая среда для хранения, которая выполнена с возможностью хранения программы, выполняемой компьютером. Средним специалистам в данной области техники должно быть понятно, что когда программа выполняется, все или некоторые из этапов способа детектирования хромосомных структурных аномалий могут быть выполнены под ее управлением соответствующим аппаратным обеспечением. Среда для хранения может включать память, доступную только для чтения, память с произвольным доступом, магнитный диск или оптический диск.
В соответствии со способом настоящего изобретения хромосомная структурная аномалия транслокационного типа может быть отобрана с помощью получения пары прочтений, соответствующих различным хромосомам, посредством выравнивания результата секвенирования всего генома с референсной последовательностью, и эффективность и надежность полученного результата дополнительно улучшаются кластеризацией и фильтрацией, благодаря чему получают результаты аналитической значимости.
Краткое описание чертежей
Изложенные выше и/или другие дополнительные аспекты и преимущества настоящего изобретения станут понятны и ясны из описания подробных вариантов осуществления в соединении с прилагаемыми чертежами, на которых:
фиг. 1 представляет собой схематический вид пары прочтений, полученной с помощью секвенирования парных концов в соответствии с вариантом осуществления настоящего изобретения;
фиг. 2 представляет собой схематический вид первого типа пар прочтений с аномальным соответствием в соответствии с вариантом осуществления настоящего изобретения;
фиг. 3 представляет собой схематический вид второго типа пар прочтений с аномальным соответствием в соответствии с вариантом осуществления настоящего изобретения;
фиг. 4 представляет собой схематический вид третьего типа пар прочтений с аномальным соответствием в соответствии с вариантом осуществления настоящего изобретения;
фиг. 5 представляет собой схематический вид пары кластеров, расположенных на различных хромосомах, в соответствии с вариантом осуществления настоящего изобретения;
фиг. 6 представляет собой схематический вид RPK для "FA" в экспериментальном примере 1 в соответствии с вариантом осуществления настоящего изобретения; и
фиг. 7 представляет собой схематический вид RPK для "SON" в экспериментальном примере 1 в соответствии с вариантом осуществления настоящего изобретения.
Подробное описание
В соответствии с вариантом осуществления настоящего изобретения предложен способ детектирования хромосомных структурных аномалий, который включает следующие этапы.
Этап 1. Получение результата секвенирования всего генома целевого индивидуума
Результат секвенирования включает парные прочтения, каждая пара прочтений состоит из двух последовательностей прочтений, расположенных соответственно с двух концов исследуемого хромосомного фрагмента, и каждую пару прочтений получают отдельно от положительных и отрицательных нитей соответствующего хромосомного фрагмента или как от положительной, так и от отрицательной нити соответствующего хромосомного фрагмента.
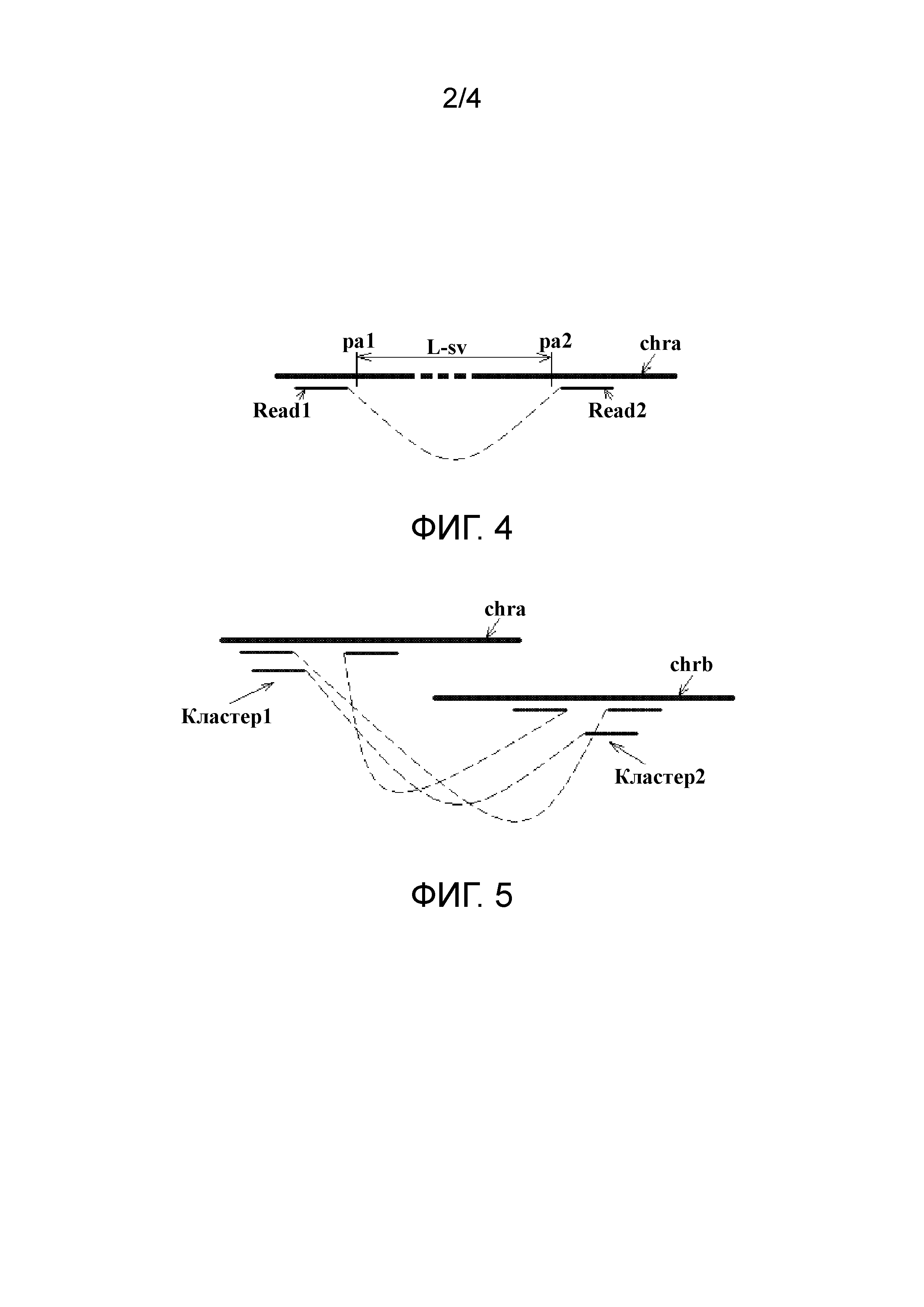
Исследуемый хромосомный фрагмент обычно получают посредством сегментирования хромосомного образца от целевого индивидуума и затем используют для получения соответствующей библиотеки в соответствии с выбранным методом секвенирования. Метод секвенирования может быть выбран в зависимости от платформы секвенирования, включая, но без ограничения, Complete Genomics (CG), Illumina/Solexa, ABI/SOLiD и Roche 454, и получение библиотеки секвенирования одиночных концов или парных концов осуществляют в соответствии с выбранной платформой секвенирования. В соответствии с конкретным вариантом осуществления настоящего изобретения может быть осуществлено секвенирование парных концов. Две последовательности прочтений Read1 и Read2 в каждой полученной паре прочтений получают отдельно от положительной нити Sp и отрицательной нити Sm соответствующего хромосомного фрагмента, как показано на фиг. 1. Длина Read1 L-r1 может совпадать или отличаться от длины Read2 L-r2. Безусловно, когда использование метода секвенирования одиночных концов позволяет полностью получить последовательность всего хромосомного фрагмента, может быть возможно сегментировать подходящую длину последовательности соответственно с двух концов полностью полученной последовательности для образования пары прочтений. В этом случае обе последовательности прочтений в каждой паре прочтений получают от положительной или отрицательной нити соответствующего хромосомного фрагмента. В данном варианте осуществления нет ограничений на конкретный выбранный метод секвенирования.
В настоящем изобретении размер библиотеки, используемой для секвенирования, обозначают как L-lib, и обычно библиотеку с L-lib от 100 до 1000 п.о. называют библиотекой коротких фрагментов, а библиотеку с L-lib 2 т.п.о., 5-6 т.п.о., 10 т.п.о., 20 т.п.о. и 40 т.п.о. называют библиотекой длинных фрагментов. В настоящем изобретении размер L-lib не ограничен. Однако при обеспечении конструкционного качества библиотеки обычно в получение приемлемого результата вносит вклад более длинная библиотека. Поэтому, предпочтительно, L-lib ≥ 300 п.о. Обычно можно использовать библиотеку длинных фрагментов, например 5 т.п.о., или библиотеку коротких фрагментов, например 500 п.о. Для обеспечения большого количества результата секвенирования глубина секвенирования может быть выбрана выше 2X для библиотеки длинных фрагментов и выше 5X для библиотеки коротких фрагментов. Для того чтобы избежать потери данных, глубина секвенирования составляет, предпочтительно, 2X для библиотеки длинных фрагментов и 5X для библиотеки коротких фрагментов. Следует отметить, что поскольку большая часть конкретных данных, используемых в настоящем изобретении, статистически значимы, любое численное значение, которое точно выражено, представляет диапазон, то есть интервал в пределах ±10% численного значения, если отдельно не оговорено иное. Это указание далее в данном документе не повторяется.
L-r1 и L-r2, предпочтительно, больше или равны 25 п.о., поскольку, когда L-r1 и L-r2 меньше 25 п.о., уровень единичного выравнивания снижается, так что растет сложность последующего получения результатов выравнивания. L-r1 и L-r2 не должны быть слишком большими, для того чтобы избежать потери данных. Поэтому L-r1 и L-r2 составляют, предпочтительно, 50 п.о. L-r1 и L-r2 не имеют ограничений по максимальному значению и могут быть изменены при развитии технологии секвенирования. Например, L-r1 и L-r2 обычно не больше чем 150 п.о. при существующих технологиях секвенирования.
Этап 2. Выравнивание результата секвенирования с референсной последовательностью
Используемая референсная последовательность представляет собой известную последовательность и может представлять собой любой референсный шаблон, ранее полученный от категории, к которой принадлежит целевой индивидуум. Например, если целевой индивидуум является человеком, референсная последовательность может представлять собой HG19, предоставляемую национальным центром биотехнологической информации (NCBI). Кроме того, может быть заранее сконфигурирован репозиторий ресурсов, содержащий больше референсных последовательностей, и более подходящую референсную последовательность до выравнивания последовательностей выбирают на основании пола, расы, географической области и других признаков целевого индивидуума, для того чтобы получить более точный результат детектирования. Во время выравнивания в соответствии с установленными параметрами выравнивания паре прочтений позволяют иметь самое большее n несоответствий, где n составляет, предпочтительно, 1 или 2. Если в паре прочтений имеется больше несоответствий, чем n, считают, что пара прочтений не может быть выровнена с референсной последовательностью, или, если все несоответствующие n оснований расположены в одной из пар прочтений, считают, что последовательность прочтения в паре прочтений не может быть выровнена с референсной последовательностью. При этом во время выравнивания можно использовать различное программное обеспечение для выравнивания, например Short Oligonucleotide Analysis Package (SOAP), bwa и samtools и так далее. В данном варианте осуществления это не ограничено.
В зависимости от выравнивания пар прочтений могут быть получены следующие категории.
(I) Набор нормальных соответствий *.pair включает пары прочтений, описываемые тем, что две последовательности прочтений Read1 и Read2 в паре прочтений соответствуют одной и той же хромосоме референсной последовательности, отношение положительных и отрицательных нитей соответствующих им положений согласуется с этим отношением в паре прочтений, и длина L-pr хромосомного фрагмента, вычисленная в соответствии с соответствующими им положениями, отклоняется от L-lib на величину, меньшую заранее заданного порога V-lib. V-lib составляет, предпочтительно, от 5% × L-lib до 15% × L-lib и, более предпочтительно, 10% × L-lib. Порог устанавливают эмпирически на основании стандартного отклонения библиотечного размера. Эмпирически, стандартное отклонение библиотеки коротких фрагментов составляет приблизительно 15 п.о., а стандартное отклонение библиотеки длинных фрагментов составляет приблизительно 50 п.о. Отклонение L-pr от L-lib в диапазоне от 3-кратного стандартного отклонения считают подходящим, например считают, что L-pr может находиться в диапазоне 455-545 п.о. для библиотеки в 500 п.о.
На основании *.pair может быть получено количественное распределение пар прочтений в соответствии с соответствующим им положением, например может быть статистически вычислено число прочтений на единицу длины (RPU). Соответствующая единица длины может быть установлена в соответствии с L-lib, например установлена в 1,5-4 раза больше L-lib. Если L-lib равно 500 п.о., может быть установлена единица длины 1 т.п.о., и в этом случае RPU может быть обозначено как RPK. Вариация RPU по сравнению со средним значением, например то, выше ли вариация заранее заданного порога V-rm, может помогать в определении наличия структурных аномалий, тем самым повышая аналитическую точность результата. Предпочтительно, V-rm составляет 10-30% и, более предпочтительно, 20%. Кроме того, среднее RPU может быть получено посредством статистического вычисления или оценки. Например, среднее RPU можно оценить по формуле: глубина секвенирования × (единица длины/L-lib). Если RPU не требуется, *.pair получать не нужно.
(II) Набор аномальных соответствий *.sin включает три типа прочтений в соответствии с описаниями ниже.
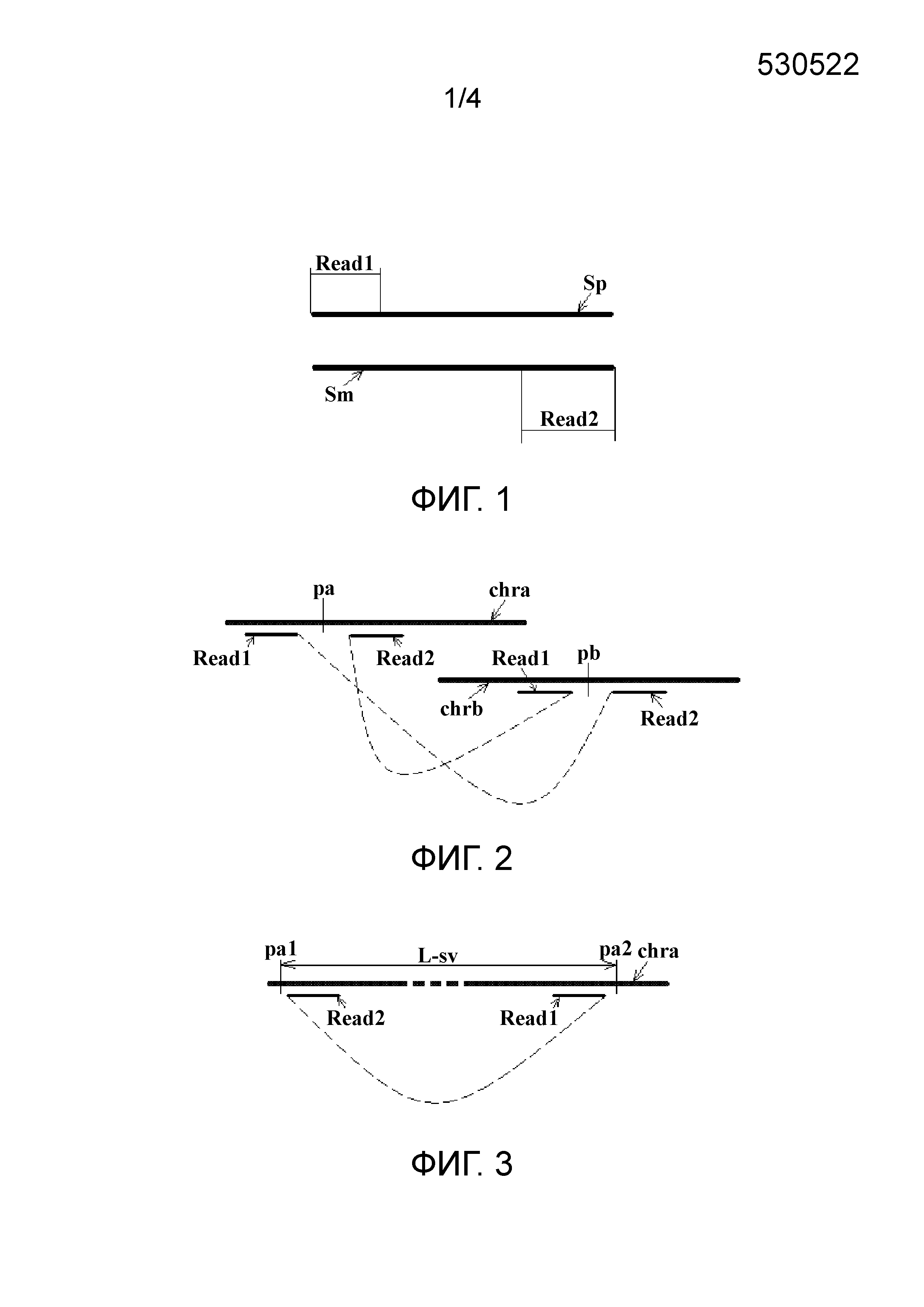
В первом типе две последовательности прочтений в паре прочтений соответственно соответствуют различным хромосомам референсной последовательности. Этот тип пар прочтений коррелирует со структурной аномалией транслокационного типа, например сбалансированной и несбалансированной транслокацией. Фиг. 2 показывает ситуацию сбалансированной транслокации, в которой Read1 в паре прочтений соответствует хромосоме chra, и Read2 соответствует хромосоме chrb, а для другой пары прочтений ситуация противоположна. На фиг. 2 пунктирная линия, соединяющая Read1 и Read2, показывает отношение положения голова к хвосту Read1 и Read2 в хромосомном фрагменте (то же самое ниже), и pa и pb обозначают соответственно потенциальные положения граничных точек, где "граничная точка" означает точку границы местоположения структурной аномалии на хромосоме.
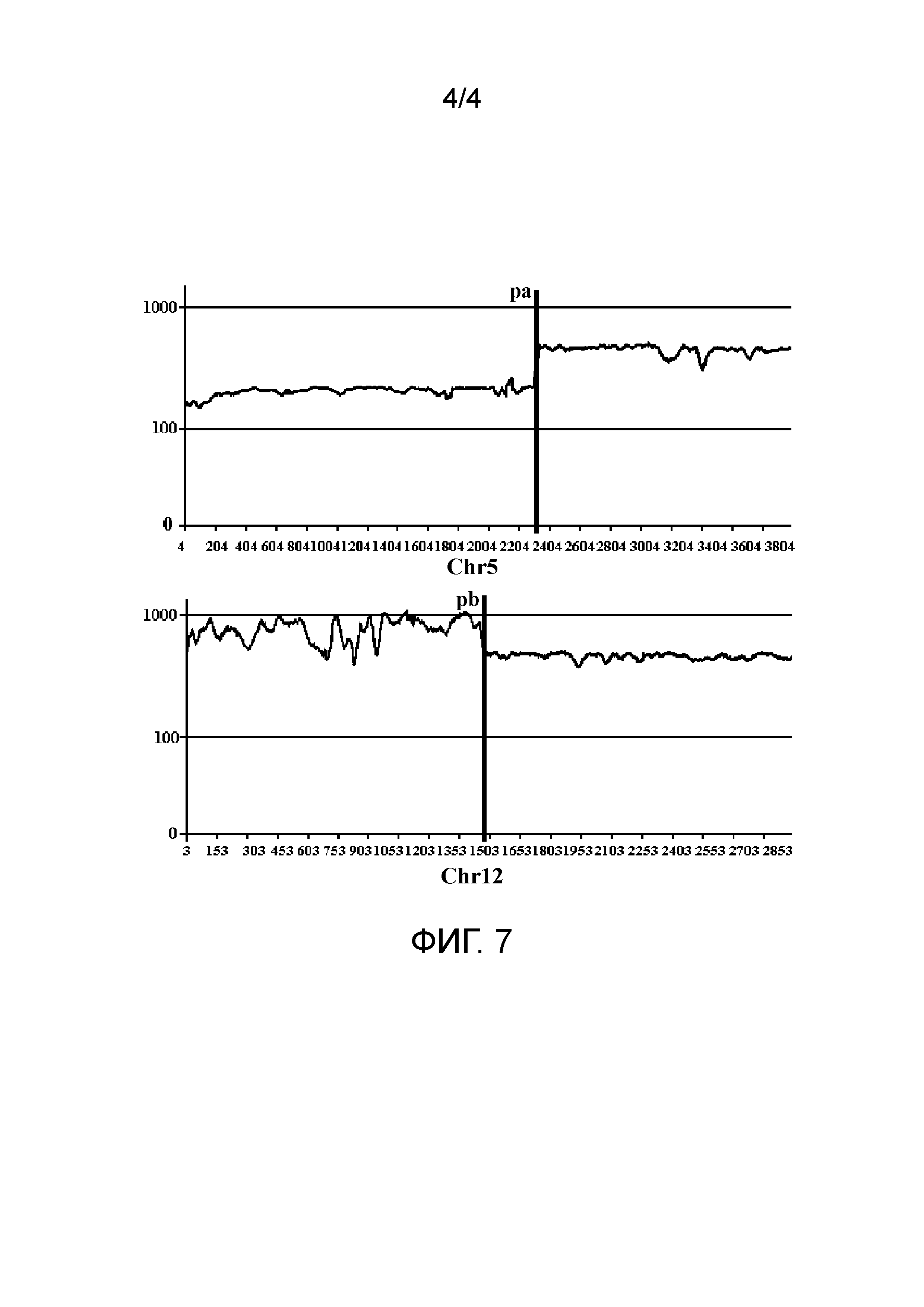
Во втором типе две последовательности прочтений в паре прочтений соответствуют одной и той же хромосоме референсной последовательности, но L-pr является отрицательной. Этот тип пар прочтений коррелирует со структурной аномалией типа тандемного повтора. Как показано на фиг. 3, как Read1, так и Read2 в паре прочтений соответствуют хромосоме chra; однако отношение положения голова к хвосту соответствующих им положений противоположно этому отношению Read1 и Read2 в хромосомном фрагменте. На фиг. 3, pa1 и pa2 обозначают соответственно начальное и конечное положения потенциального повторяющегося фрагмента, L-sv обозначает длину данного повторяющегося фрагмента, и пунктирная линия в средней части chra обозначает пропущенную длину (то же самое ниже).
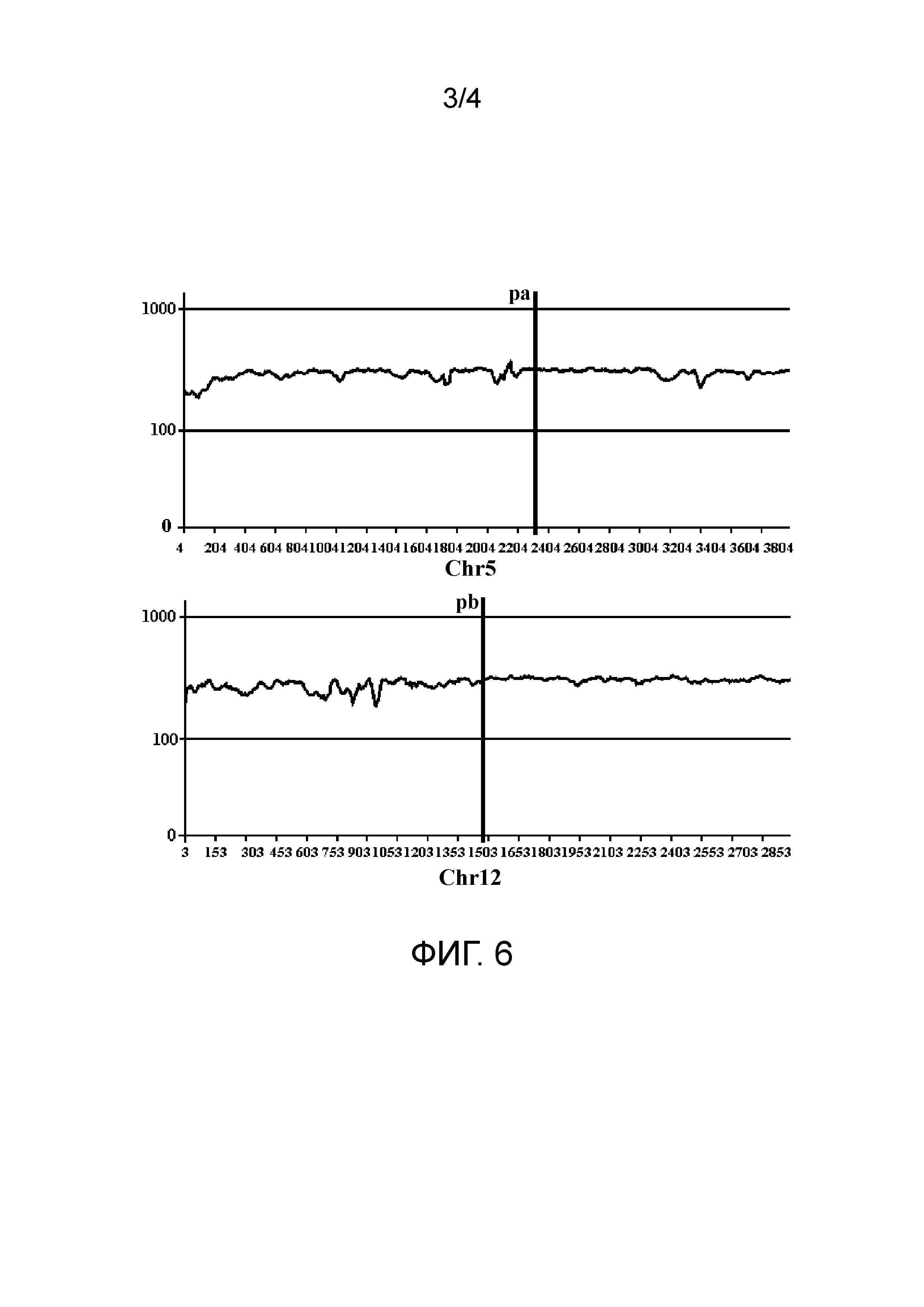
В третьем типе две последовательности прочтений в паре прочтений соответствуют одной и той же хромосоме референсной последовательности; однако L-pr больше, чем L-lib, и отклонение выше заранее заданного порога V-lib. Этот тип пар прочтений коррелирует со структурной аномалией делеционного типа. Как показано на фиг. 4, как Read1, так и Read2 в паре прочтений соответствуют хромосоме chra, и отношение положения голова к хвосту соответствующих им положений является тем же, что и у Read1 и Read2 в хромосомном фрагменте, но расстояние выходит за пределы подходящего диапазона. На фиг. 4 pa1 и pa2 обозначают соответственно начальное и конечное положения потенциального удаленного фрагмента, и L-sv обозначает длину данного удаленного фрагмента.
Поскольку различные типы пар прочтений в наборе аномальных соответствий представляют соответственно различные типы хромосомных структурных аномалий, которые могут иметь место, может отсутствовать необходимость в получении всех пар прочтений с аномальным соответствием вышеуказанных типов, что определяется потребностями детектирования. Например, когда нужно детектировать только структурную аномалию транслокационного типа, из результата выравнивания может быть получен только первый тип пар прочтений. Кроме того, набор аномальных соответствий включает не только вышеуказанные три типа пар прочтений, и в набор аномальных соответствий включены пары прочтений или последовательность прочтения в парах прочтений, которые не принадлежат к набору нормальных соответствий и могут соответствовать референсной последовательности. Корреляция между проявлениями различных типов аномальных соответствий и соответствующими хромосомными структурными аномалиями, которые могут иметь место, может быть определена средними специалистами в данной области техники. Кроме того, из-за влияния потенциального шума и других помех в наборе аномальных соответствий может быть не обнаружено соответствие или несоответствие положительных и отрицательных нитей.
(III) Набор несоответствий *.unmap включает последовательности прочтений, которые не могут соответствовать референсной последовательности, и которые могут представлять собой последовательности парных прочтений (оба члена пары не могут соответствовать референсной последовательности) или представлять собой последовательности прочтений одного конца (последовательности прочтений другого конца могут соответствовать референсной последовательности).
Последовательности прочтений одного конца, существующие в *.unmap, затем используют для подгонки граничных точек после получения итоговых кластеров для получения более точного диапазона граничных точек. Если подгонка граничных точек не нужна, *.unmap получать не нужно.
Этап 3. Кластеризация последовательностей прочтений в *.sin на основании соответствующих им положений
Кластеризацию можно получить посредством использования различных алгоритмов кластеризации, которые в данном варианте осуществления не ограничены. Например, простой подход заключается в разбиении на кластеры на основании установленного минимального расстояния V-cl между кластерами. То есть ищут последовательности прочтений, ранжированные в соответствии с положениями. Начинают от первой последовательность прочтения, если расстояние между второй последовательностью прочтения и первой последовательностью прочтения меньше V-cl, то их помещают в один и тот же кластер. Затем поиск продолжают от второй последовательности прочтения до тех пор, пока расстояние между n-й последовательностью прочтения и (n-1)-й последовательностью прочтения не окажется больше, чем V-cl. Затем может быть выделен второй кластер, начиная от n-й последовательности прочтения, и процесс многократно осуществляют со всеми последовательностями прочтений. Во время кластеризации его можно осуществлять в соответствии с положением соответствующего прочтения на хромосоме, неважно на положительной или отрицательной нити.
После кластеризации каждый кластер содержит последовательности прочтений одного конца из группы пар прочтений, и соответственно последовательности прочтений другого конца расположены в другом кластере. Поэтому эти два кластера называют парой кластеров. Фиг. 5 представляет собой схематический вид пары кластеров, образованной кластером 1 и кластером 2, расположенными соответственно на различных хромосомах. Безусловно, парные кластеры могут быть расположены на одной и той же хромосоме. Для того, чтобы анализ после кластеризации был достоверным, каждый кластер, предпочтительно, включает больше двух прочтений. В случае, когда расстояние между отдельным прочтением и предыдущим и следующим прочтением больше, чем V-cl, упомянутый выпадающий случай может быть отброшен.
V-cl составляет по меньшей мере L-lib. Если установленное значение слишком низко, число кластеров-кандидатов слишком велико, и число последовательностей прочтений в кластере слишком мало, что затрудняет последующий отбор и фильтрацию, а также может приводить к повышенному числу ложноположительных результатов. Если установленное значение слишком высоко, определение граничных точек может быть затруднительным, и диапазон граничных точек увеличивается. Поэтому V-cl составляет, предпочтительно, 10 т.п.о. В зависимости от различных используемых алгоритмов кластеризации V-cl может принимать различные конкретные значения. Например, V-cl может представлять собой расстояние между центрами тяжести двух соседних кластеров или расстояние между двумя ближайшими по положению прочтениями в двух соседних кластерах.
Этап 4. Фильтрация получаемых в результате кластеров
Фильтрация направлена на максимальное удаление различных потенциальных помех, например загрязнений образца, ошибок секвенирования, ошибок выравнивания и шума, так что результат может максимально отражать реальные хромосомные структурные аномалии. Поэтому условия фильтрации могут быть установлены в зависимости от фактических требований и типов потенциальных помех. В данном варианте осуществления, предпочтительно, предлагаются следующие средства фильтрации, которые могут быть при практическом применении использованы по отдельности или в комбинации из нескольких из них.
(I) На основании компактности кластеров - Вычисляют компактность каждого кластера, и кластеры, которые имеют компактность, не удовлетворяющую заранее заданному требованию в отношении R-va, и кластеры, парные им, отфильтровывают. Компактность каждого кластера может быть вычислена с применением различных доступных математических методов. Например, компактность может указываться дисперсией. Вычисляют дисперсию положения каждого прочтения в кластере до центра или центра тяжести кластера, и чем меньше дисперсия, тем выше будет компактность. Предпочтительно, во время вычисления компактности каждого кластера последовательности прочтений, расположенные с двух концов в диапазоне от 5 до 25% и, предпочтительно, 20% по длине кластера могут быть исключены для уменьшения влияния периферических данных на результат вычисления. Предпочтительно, R-va можно устанавливать как фиксированный порог, например требуется, чтобы дисперсия была ниже фиксированного порога. Или, иначе, R-va устанавливают как уровень исключения. Например, требуется, чтобы ранги дисперсий во всех кластерах находились в пределах заранее заданного нижнего интервала. Например, R-va устанавливают таким образом, что ранги дисперсий во всех кластерах находятся в пределах нижнего интервала 2%-10% и, предпочтительно, 5%.
Компактность кластера отражает стабильность распределения прочтений и показывает, сконцентрированы ли последовательности прочтений в небольшом интервале. Обычно реальная структурная вариация может быть поглощена большой величиной "шумов окружающей среды". Однако, влияние "шумов окружающей среды" на весь геном является по существу однородным и, таким образом, имеет, по-видимому, по существу равномерное распределение по всей последовательности (на которое, конечно, могут воздействовать, например, содержание GC (гуанина и цитозина) и так далее). В сайте, где имеет место реальная структурная вариация, последовательности прочтений в кластере имеют, по-видимому, по существу нормальное распределение. Поэтому компактность, например дисперсия, может хорошо отражать различие между кластерами.
(II) На основании линейной корреляции парных кластеров - Вычисляют линейную корреляцию двух парных кластеров, и парные кластеры, которые имеют линейную корреляцию, не удовлетворяющую заранее заданному требованию в отношении R-li, отфильтровывают. Линейная корреляция пары кластеров может быть вычислена посредством применения различных доступных математических методов. Например, вычисляют коэффициент корреляции двух кластеров, и чем выше коэффициент корреляции, тем будет выше линейная корреляция. Предпочтительно, R-li можно устанавливать как фиксированный порог, например требуется, чтобы коэффициент корреляции был выше фиксированного порога. Или, иначе, R-li устанавливают как уровень исключения. Например, требуется, чтобы ранги коэффициентов корреляции во всех кластерах находились в пределах заранее заданного верхнего интервала. Например, R-li устанавливают таким образом, что ранги коэффициентов корреляции во всех кластерах находятся в пределах верхнего интервала 2%-10% и, предпочтительно, 5%.
Линейная корреляция сильно выделяет однородность распределения последовательностей прочтений в парных кластерах, то есть показывает, имеют ли последовательности прочтений двух концов в парных прочтениях по существу однородное распределение. Поэтому линейная корреляция может лучше отражать распределение в парных кластерах.
В предпочтительном варианте осуществления хороший результат достигается посредством фильтрации кластеров-кандидатов с использованием компактности, например дисперсии кластеров и линейной корреляции кластеров в комбинации.
(III) На основании контрольного набора нормальных образцов - Парные кластеры выравнивают с контрольным набором, содержащим множество нормальных образцов, и парные кластеры, которые имеют число попаданий нормальных образцов, достигающее заранее заданного порога V-con, отфильтровывают. Нормальными образцами называют набор итоговых кластеров, полученный посредством подвергания других нормальных индивидуумов, которые принадлежат к той же категории, что и целевой индивидуум, процессам "выравнивания-кластеризации-фильтрации", как описано выше. Для облегчения выравнивания все последовательности прочтений в кластере могут быть объединены в одну, и, таким образом, генерируют пару объединенных численных значений от парных кластеров (которая напоминает пару прочтений). Выравнивание осуществляют с парами объединенных численных значений. Посредством получения контрольного набора, содержащего большое число нормальных образцов, можно получить частоту встречаемости итоговых кластеров у нормальных индивидуумов. Если некоторый итоговый кластер имеет высокую частоту встречаемости, можно предположить, что данный итоговый кластер может быть обусловлен качеством образца, экспериментальным процессом, процессом секвенирования или шумом окружающей среды и не указывает на то, что образец действительно подвергается такой структурной вариации как таковой. Такой итоговый кластер представляет собой совместный ложноположительный результат, полученный из-за анализа различных образцов одним и тем же способом, и должен быть удален. Поэтому вероятность ложноположительных событий может быть дополнительно снижена посредством фильтрации итоговых кластеров с применением контрольного набора, что вносит вклад в получение правильного результата анализа структурной вариации. V-con можно определять в зависимости от способов создания и признаков нормальных образцов. Например, отношение V-con к числу нормальных образцов в контрольном наборе может составлять 3-10% и, предпочтительно, 5-6%. Например, если контрольный набор содержит 90 нормальных образцов, считают, что 5 попаданий достигают порога.
(IV) На основании других вспомогательных параметров - Вспомогательные параметры включают различные параметры, помогающие при дальнейшем подтверждении и различении типов структурных аномалий или при понимании деталей структурных аномалий, например число несоответствий, генерируемых во время выравнивания, число пар прочтений, обеспечивающих кластеры, значение RPU соответствующей области, полученное на основании *.pair, то, расположены ли кластеры в области N, и другие. Вспомогательные параметры можно использовать двумя следующими способами. 1. Вспомогательные параметры используют в качестве условий фильтрации. Требования к фильтрации, связанные со вспомогательными параметрами, устанавливают на отфильтровывание кластеров, прямо не удовлетворяющих требованиям. 2. Вспомогательные параметры используют в качестве опорного принципа для помощи при определении. С итоговыми кластерами предоставляют вспомогательные параметры, и затем проводят определение посредством осуществляемого вручную анализа. Поэтому содержание данного раздела можно использовать на этапе 4 (для фильтрации) или на этапе 5 (для помощи в осуществляемом вручную анализе). В данном варианте осуществления конкретные способы использования вспомогательных параметров не ограничены. Некоторые вспомогательные параметры и их связь с анализом результатов проиллюстрированы ниже. При практическом применении вспомогательные параметры могут быть установлены в качестве условий фильтрации в соответствии с указаниями, приведенными ниже, или в качестве основы для помощи при определении посредством осуществляемого вручную анализа. Различные вспомогательные параметры можно использовать в комбинации или по отдельности.
(1) Число несоответствий. - Среднее число несоответствий пар прочтений в парных кластерах составляет обычно не более 1 или 2, то есть каждая пара прочтений может иметь 1 или 2 несоответствия и, предпочтительно, не более 1 несоответствия. Данный параметр не следует принимать во внимание, если на его основании установлено требование соответствия при выравнивании. Если условия на выравнивание менее строгие, например установлено, что могут иметь место 2 несоответствия, дополнительно могут быть проведены фильтрация или определение с использованием данного параметра во время получения итогового кластера, например устанавливают, что может иметь место в среднем 1 несоответствие.
(2) Число пар прочтений, обеспечивающих кластеры, то есть число пар прочтений, содержащихся в парных кластерах - В принципе, лучше, чтобы этот параметр был больше. Обычно начальный уровень для определения может быть установлен соответствующим или слегка меньшим (например, принимающим целое значение), чем нормализованное значение глубины секвенирования, где нормализованное значение глубины секвенирования=глубина секвенирования × (диапазон влияния L-lib на граничные точки/L-lib) × (средние размеры двух концов парных кластеров/L-lib). "Диапазон влияния L-lib на граничные точки" обычно выше, чем "сумма размеров двух концов парных кластеров", и обычно колеблется вокруг среднего, которое в 2 раза превосходит L-lib, например колеблется между 1-4 L-lib. В конкретных условиях диапазон данного параметра может быть соответствующим образом расширен или сужен в зависимости от требований практической ситуации.
(3) Значение RPU соответствующей области, полученное на основании *.pair - Различные типы структурных аномалий обычно оказывают различное влияние на RPU. Например, в случае сбалансированной транслокации RPU с двух боковых сторон граничных точек значительно не изменяется; однако, в случае структурной аномалии типа делеции или повтора RPU области между граничными точками значительно уменьшается или увеличивается. Поэтому значение RPU соответствующей области можно затем использовать для подтверждения или облегчения определения наличия хромосомных структурных аномалий.
Например, для кластеров, содержащих первый тип пар прочтений, если в соответствии с отношением между парами прочтений в кластерах определена сбалансированная транслокация (смотри для подробностей раздел I этапа 5 ниже), вариация RPU с двух боковых сторон граничных точек от среднего не выше V-rm; а если в соответствии с отношением между парами прочтений в кластерах определена несбалансированная транслокация (смотри для подробностей раздел I этапа 5 ниже), RPU сбоку от граничных точек, получаемое от итоговых кластеров, ниже среднего, и вариация выше V-rm.
Для кластеров, содержащих второй тип пар прочтений, RPU области между граничными точками выше среднего, и вариация выше V-rm.
Для кластеров, содержащих третий тип пар прочтений, RPU области между граничными точками ниже среднего, и вариация выше V-rm.
При использовании RPU для помощи при определении посредством осуществляемого вручную анализа RPU соответствующей области может быть представлено в форме графика, таблицы или в другой легко идентифицируемой форме. Альтернативно, вариацию RPU во всей области представляют в форме графика, таблицы или тому подобного для облегчения понимания оператором общих условий.
(4) Расположен ли кластер в области N - Эмпирически, выравнивание прочтений поблизости от области N (содержащей участки центромеры и теломеры) является более сложным, чем в других областях. Если полученные кластеры не расположены в области N, считается, что определение может быть осуществлено в соответствии с полученной информацией. Если полученные кластеры расположены в области N, может потребоваться более тщательная проверка. Например, окончательное определение осуществляют с помощью совместного использования условий фильтрации и вспомогательных параметров или с учетом других внешних данных, например фенотипа целевого индивидуума и/или результата более точного секвенирования (например, секвенирования по Сенгеру) граничных точек.
Этап 5. Анализ данных отфильтрованных итоговых кластеров
Наличие итоговых кластеров, полученных после фильтрации, отражает потенциальное наличие соответствующих типов хромосомных структурных аномалий. Поэтому, если требуется обнаружить только потенциальные структурные аномалии, данный этап не является необходимым. Для цели получения более подробной информации в отношении структурных аномалий полученные итоговые кластеры могут быть дополнительно подвергнуты анализу данных. В зависимости от различных типов итоговых кластеров могут быть использованы следующие способы анализа.
(I) Хромосомная структурная аномалия транслокационного типа (первый тип прочтений)
Если осуществляют поиск итоговых кластеров, содержащих первый тип пар прочтений, и если две соседние последовательности прочтений имеют противоположные положения в соответствующих парах прочтений, диапазон между положениями, которым соответствуют эти две последовательности прочтений, принимают в качестве диапазона граничных точек. Данная ситуация обычно коррелирует со сбалансированной транслокацией, при которой последовательности прочтений в одном кластере распределены с двух боковых сторон от граничных точек.
Если такие последовательности прочтений не существуют, получают положение самой внутренней последовательности прочтения, и диапазон, полученный посредством распространения внутрь от данного положения на заранее заданную длину, принимают в качестве диапазона граничных точек. Термин самая внутренняя последовательность прочтения означает, что если кластер включает исключительно последовательности прочтений левого конца, самая правая последовательность прочтения является самой внутренней последовательностью прочтения; а если кластер включает исключительно последовательности прочтений правого конца, самая левая последовательность прочтения является самой внутренней последовательностью прочтения. Эта ситуация обычно коррелирует с несбалансированной транслокацией, при которой последовательности прочтений в одном кластере распределены с одной боковой стороны от граничных точек. Ширина распространения диапазона граничных точек от самой внутренней последовательность прочтения может быть определена исходя из L-lib, L-r1/L-r2, глубины секвенирования и так далее, и может, например, составлять 0,5-2 L-lib и обычно не более чем 2 L-lib.
Фиг. 2 показывает ситуацию сбалансированной транслокации. Полученная пара итоговых кластеров (только две последовательности прочтений изображены в каждом кластере, а другие считают опущенными) имеет распределение, показанное на фиг. 2, один итоговый кластер расположен поблизости от положения pa на хромосоме chra, а парный ему итоговый кластер расположен поблизости от положения pb на хромосоме chrb. Поскольку в кластере на chra Read1 является последовательностью прочтения левого конца хромосомного фрагмента, от которого она получена, а соседняя Read2 является последовательностью прочтения правого конца хромосомного фрагмента, от которого она получена, полагают, что граничная точка pa на chra расположена между Read1 и Read2, и тот же анализ применяют к chrb.
На основании приведенного выше анализа данных итоговый вывод данных для потенциальной структурной аномалии транслокационного типа может включать нумерацию двух хромосом (на которых соответственно расположены итоговые кластеры), потенциально имеющих структурную аномалию транслокационного типа, положение диапазонов двух концов парных итоговых кластеров (положение диапазонов границ двух концов кластеров на двух хромосомах, откуда могут быть соответствующим образом получены размеры двух концов кластеров), диапазон граничных точек, полученный после анализа, и так далее. Также рядом могут быть выведены соответствующие параметры, генерируемые во время фильтрации, и другие вспомогательные параметры, например соответствующая компактность пары итоговых кластеров, степень линейной корреляции пары итоговых кластеров, число пар прочтений, обеспечивающих пару итоговых кластеров, и график и таблица, представляющие вариацию RPU с двух боковых сторон от граничных точек.
(II) Хромосомная структурная аномалия типа тандемного повтора (второй тип пар прочтений)
Если осуществляют поиск итоговых кластеров, содержащих второй тип пар прочтений, диапазон между двумя соответствующими им положениями, которые удалены на наибольшее расстояние в парных кластерах, принимают в качестве диапазона наличия повтора, и диапазон, полученный посредством соответствующего распространения наружу от данных двух положений на заранее заданную длину, то есть, например, на 0,5-2 L-lib, принимают в качестве диапазона граничных точек (начальной и конечной точек повторяющегося фрагмента).
Фиг. 3 показывает ситуацию тандемного повтора. Оба конца парных итоговых кластеров (только одна последовательность прочтения изображена в каждом кластере, а другие считают опущенными) находятся в пределах диапазона между начальной и конечной точками повторяющегося фрагмента, и поэтому считают, что начальная и конечная точки повторяющегося фрагмента расположены в диапазоне, распространяющемся наружу от последовательностей прочтений (где две последовательности прочтений не обязательно принадлежат к пара прочтений) в наиболее удаленном положении двух концов кластеров.
По сравнению со структурной аномалией транслокационного типа структурная аномалия типа повтора имеет по существу те же типы вывода итоговых данных, за исключением того, что нумерация хромосом с двух концов кластеров является одинаковой, и также могут быть выведены данные, указывающие на расчетную длину повторяющегося фрагмента.
(III) Хромосомная структурная аномалия делеционного типа (третий тип пар прочтений)
Если осуществляют поиск итоговых кластеров, содержащих третий тип пар прочтений, диапазон между двумя соответствующими им положениями, которые удалены на наименьшее расстояние в парных кластерах, принимают в качестве диапазона наличия делеции, и диапазон, полученный посредством соответствующего распространения внутрь от данных двух положений на заранее заданную длину, то есть, например на 0,5-2 L-lib, принимают в качестве диапазона граничных точек (начальной и конечной точек удаленного фрагмента).
Фиг. 4 показывает ситуацию делеции фрагмента. Оба конца парных итоговых кластеров (только одна последовательность прочтения изображена в каждом кластере, а другие считают опущенными) находятся вне диапазона между начальной и конечной точками удаленного фрагмента, и поэтому считают, что начальная и конечная точки удаленного фрагмента расположены в диапазоне, распространяющемся внутрь от последовательностей прочтений (где две последовательности прочтений не обязательно принадлежат к пара прочтений), которые наиболее близки к двум концам кластеров.
По сравнению со структурной аномалией типа повтора структурная аномалия делеционного типа имеет по существу те же типы вывода итоговых данных, за исключением того, что выводимые данные, указывающие на расчетную длину фрагмента между граничными точками, представляют длину удаленного фрагмента.
Этап 6. Подгонка граничных точек
Для дальнейшего уменьшения диапазона граничных точек может быть проведена подгонка граничных точек с использованием данных из *.unmap. Например, получают последовательности прочтений одного конца (которые могут соответствовать одному концу референсной последовательности и могут быть обозначены во время выравнивания как *.sin) в диапазоне, установленном вокруг определенного диапазона граничных точек (например, 0,5-2 L-lib), и последовательности парных им прочтений извлекают из *.unmap в качестве последовательностей для заполнения брешей. Все последовательности для заполнения брешей разрезают на N секций, и N составляет, предпочтительно, 2. Затем подпоследовательности, полученные после разрезания последовательностей для заполнения брешей, снова выравнивают с референсной последовательностью. Область граничных точек подгоняют в соответствии с результатом нормального соответствия.
При практическом применении рациональным будет установить значение N в соответствии с длиной Lr1/Lr2. Когда длина последовательности меньше 25 п.о., это вызывает значительное снижение уровня единичного выравнивания. Соответственно, при установлении значения N можно полагать, что длина разрезанной подпоследовательности должна быть не меньше или не слишком меньше 25 п.о.
После подгонки граничных точек диапазон граничных точек может быть эффективно уменьшен. Исходя из этого затем может быть создан зонд в соответствии с диапазоном положений, к которому принадлежат граничные точки, и точные положения граничных точек могут быть в итоге получены посредством другого точного секвенирования для осуществления дальнейшего исследования граничных точек, например секвенирования по Сенгеру. Если диапазон граничных точек не нужно уменьшать, этот этап может быть опущен.
Средним специалистам в данной области техники может быть понятно, что все или некоторые из этапов способов, предложенных выше в вариантах осуществления, могут быть выполнены соответствующим аппаратным обеспечением под управлением программы, которая может быть сохранена в машиночитаемой среде для хранения, включая память, доступную только для чтения, память с произвольным доступом, магнитный диск или оптический диск.
В соответствии с другим аспектом настоящего изобретения также предложено устройство для детектирования хромосомных структурных аномалий, которое включает узел ввода данных, выполненный с возможностью ввода данных; узел вывода данных, выполненный с возможностью вывода данных; узел хранения, выполненный с возможностью хранения данных и содержащий в себе выполняемую программу; и процессор в информационном соединении с узлом ввода данных, узлом вывода данных и узлом хранения, выполненный с возможностью выполнения выполняемой программы, сохраненной в узле хранения, причем выполнение программы включает осуществление всех или некоторых из этапов способов, предложенных выше в вариантах осуществления.
Далее в данном документе подробно описан результат выполнения конкретного способа детектирования в соответствии с настоящим изобретением в связи с конкретным целевым индивидуумом. В процессе детектирования конкретные использованные параметры устанавливают следующим образом.
1. L-lib составляет 500 п.о., и используют секвенирование PE50 (секвенирование парных концом, при котором L-r1 и L-r2 составляют примерно 50 п.о.).
2. В качестве референсной последовательности используют HG19 от NCBI, и результат секвенирования выравнивают с помощью программного обеспечения SOAP.
3. V-lib составляет ±45 п.о., V-rm RPK составляет 20%, V-cl составляет 10 т.п.о. (расстояние между кластерами определяют как расстояние между двумя наиболее близкими последовательностями прочтений), минимальное число прочтений в кластере составляет 2, R-va устанавливают таким образом, что ранги дисперсий во всех кластерах находятся в пределах нижнего интервала 5% (при вычислении дисперсии последовательности прочтений, расположенные с двух концов в диапазоне 20% по длине кластера, исключают), R-li устанавливают таким образом, что ранги коэффициентов корреляции во всех кластерах находятся в пределах верхнего интервала 5%, контрольный набор включает 90 нормальных образцов, и V-con составляет 5.
Экспериментальный пример I
В данном примере приведено исследование на семье с синдромом кошачьего крика. В данном примере два целевых индивидуума принадлежат одной семье, причем "FA" представляет отца, а "SON" представляет сына.
1. Проводили полное секвенирование генома соответственно у двух целевых индивидуумов с низким множителем, при котором глубина секвенирования для "FA" составляла 2,2 и для "SON" 3,1.
2. Затем результаты секвенирования двух целевых индивидуумов выравнивали соответственно с референсной последовательностью HG19 с применением программного обеспечения для выравнивания SOAP для получения двух файлов FA.sin и SON.sin.
3. Эти два файла FA.sin и SON.sin подвергали кластеризации, фильтрации и анализу для получения следующих итоговых кластеров и соответствующих выходных параметров:
"FA":
номера двух хромосом, на которых расположены парные итоговые кластеры: chr12, chr5
диапазоны положений двух концов парных итоговых кластеров: 14779615-14780233, 23314785-23314205
размеры двух концов парных итоговых кластеров: 618, 580
число пар прочтений, обеспечивающих пару итоговых кластеров: 5
компактность (дисперсия) левого и правого концов: 90,59, 87,01
расположены ли кластеры в области N: нет
диапазон граничных точек: chr12:14779968-14780233, chr5:23314205-23314455
вариация RPK соответствующих областей на хромосомах - На фиг. 6 горизонтальная ось представляет положение (единица: 10 т.п.о.) на хромосоме, и продольная ось представляет RPK. Кривая построена на основании данных из FA.pair, и pa и pb представляют положения граничных точек. На фиг. 6 можно видеть, что вариация RPK у "FA" незначительна.
"SON":
номера двух хромосом, на которых расположены парные итоговые кластеры принадлежат: chr12, chr5
диапазоны положений двух концов парных итоговых кластеров: 14779618-14779968, 23314455-23314830
размеры двух концов парных итоговых кластеров: 350, 375
число пар прочтений, обеспечивающих пару итоговых кластеров: 6
компактность (дисперсия) левого и правого концов: 22,43, 18,44
расположены ли кластеры в области N: нет
диапазон граничных точек: chr12: выше 14779968, chr5: ниже 23314455
вариация RPK соответствующих областей на хромосоме - на фиг. 7 горизонтальная ось представляет положение (единица: 10 т.п.о.) на хромосоме, и продольная ось представляет RPK. Кривая построена на основании данных из SON.pair, и pa и pb представляют положения граничных точек. На фиг. 7 можно видеть, что вариация RPK у "SON" очевидна. Из рассмотрения вычисленного RPK может быть известно, что RPK в плече хромосомы 5 у SON составляет только 0,5 от среднего, и RPK в плече хромосомы 12 в 0,5 раза выше, чем среднее.
Из результатов анализа может быть без сомнения определено, что у "FA" имеет место сбалансированная транслокация, и у "SON" имеет место несбалансированная транслокация. Диапазон граничных точек, проанализированный по результату "FA", находится в пределах 300 п.о. Для осуществления дальнейшего исследования положений граничных точек соответствующую последовательность удаляют из референсной последовательности HG19, и разрабатывают праймер для секвенирования по Сенгеру и проверки посредством кПЦР. В итоге получают точные положения граничных точек Chr12:14780019, Chr5:23314435.
Экспериментальный пример II
В данном примере приведено исследование на врожденном заболевании сердца. В данном примере целевой индивидуум является пациентом с врожденным заболеванием сердца и обозначается как "XX".
1. Проводили полное секвенирование генома у целевого индивидуума с низким множителем, при котором глубина секвенирования составляла 2,7.
2. Затем результат секвенирования выравнивали с референсной последовательностью HG19 с применением программного обеспечения для выравнивания SOAP для получения XX.sin.
3. XX.sin подвергали кластеризации, фильтрации и анализу для получения следующих итоговых кластеров и соответствующих выходных параметров:
"XX":
номера двух хромосом, на которых расположены парные итоговые кластеры: chr14, chr14
диапазоны положений двух концов парных итоговых кластеров: 73557040-73557288, 73670432-73670682
расчетная длина повторяющегося фрагмента: 113392
размеры двух концов парных итоговых кластеров: 248, 250
число пар прочтений, обеспечивающих пару итоговых кластеров: 4
компактность (дисперсия) левого и правого концов: 100,63, 100,59
расположены ли кластеры в области N: нет
диапазон граничных точек: chr14: 73556540-73557040, chr14: 73670682-73671182 (где ширина диапазона определяют исходя из 1 L-lib, то есть 500 п.о.).
Из результата анализа может быть без сомнения определено, что повтор приблизительно из 113 т.п.о. по длине имеет место у "XX" в хромосоме 14, и повтор имеет место в тандеме. Для осуществления дальнейшего исследования положений граничных точек соответствующую последовательность удаляют из референсной последовательности HG19, и разрабатывают праймер для секвенирования по Сенгеру и проверки посредством кПЦР. Коэффициент умножения кПЦР больше 1, что подтверждает повтор. В итоге из секвенирования по Сенгеру получают точные положения граничных точек Chr14: 73557008, Chr14: 73670820, что подтверждает, что в хромосоме 14 у "XX" имеет место повтор в 113812 п.о., и повторяющийся фрагмент вставлен в конце фрагмента в тандеме.
Предшествующее описание представляет собой только предпочтительные варианты осуществления настоящего изобретения, и следует понимать, что данные варианты осуществления предложены для понимания, а не для ограничения настоящего изобретения. Средними специалистами в данной области техники могут быть осуществлены изменения конкретных приведенных выше вариантов осуществления в соответствии с идеей настоящего изобретения.
Способ и устройство для определения фракции внеклеточных нуклеиновых кислот в биологическом образце и их применение

