Результат интеллектуальной деятельности: ПИРРОЛЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ИНДОЛОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ВКЛЮЧАЮЩАЯ ЕГО КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к пирролзамещенному производному индолона, к его фармацевтически приемлемым солям, к способу их получения, к композиции, включающей их, и к их применению. В частности, настоящее изобретение относится к пирролзамещенному производному индолона, используемому в качестве многоцелевого ингибитора тирозинкиназы, к фармацевтической композиции, включающей это производное, и к их медицинскому применению.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Рак стал заболеванием, которое создает наибольшую угрозу для людей в современном обществе. К настоящему времени достигнута ситуация, когда многие противораковые средства, доступные на фармацевтическом рынке, представлены открытыми в прошлом столетии цитотоксическим средствами, которые убивают огромное число нормальных клеток в процессе лечения опухолей, вызывая непереносимые побочные реакции у пациентов, и при интенсивном применении этих лекарственных средств возникает другая трудно устранимая проблема резистентности к лекарственному средству.
Ингибирование сосудов опухолей представляет собой новый способ лечения опухолей, разработанный в конце прошлого столетия, и исследование, направленное на его поиски, было основано на теории, предложенной Folkman, согласно которой выживание, рост и метастазирование опухолей основано широкой сети новообразованных сосудов (Folkman. J. et. al. N. Engl. J. Med., 1971, 285, 1182-1186). В большом числе клинических исследований было обнаружено, что опухолевые ткани содержат множество новых сосудов, а рост и метастазирование опухолевых клеток требует наличия большого числа сосудов для доставки кислорода и питательных компонентов.
Ингибирование неоангиогенеза в опухолях может вызвать ʺистощениеʺ опухолевых клеток, вплоть до их гибели, тогда как ингибирование новых сосудов оказывает незначительный эффект на нормальные клетки, поскольку вокруг нормальных клеток имеется очень мало новых сосудов, что приводит к получению противоопухолевых средств, основанных на ингибировании сосудов, которые демонстрируют характеристики, такие как высокая эффективность, безопасность и низкая токсичность.
Ингибирование сосудов может быть классифицировано с разделением на прямое ингибирование и непрямое ингибирование. Прямое ингибирование представляет собой воздействие на эндотелиальные клетки сосудов, направленное на ингибирование сосудов, расширение и питательную поддержку опухолевых клеток сосудов. Используемый здесь основной способ представляет собой метрономную терапию цитотоксическим средством, которое может ослаблять побочные реакции цитотоксического средства, однако, здесь имеется трудность в уменьшении повреждения, вызванного лекарственным средством в организме человека. При непрямом ингибировании идет подавление неоангиогенеза путем ингибирования ангиогенных факторов, нужных для ангиогенеза (Cao, Y. et. al. Int. J. Biochem. Cell Biol., 2001, 33, 357-369). Процесс ангиогенеза включает активацию эндотелиальных клеток сосудов под действием активатора; секрецию протеаз из эндотелиальных клеток, приводящих к деградации базальной мембраны; миграцию и пролиферацию эндотелиальных клеток; образование просвета в новых капиллярах; и рекрутмен перицитов с достижением стабилизации периферической структуры неокапилляров. В физиологических условиях имеются два вида факторов, воздействующих на ангиогенез, а именно: ингибиторы ангиогенеза и про-ангиогенные факторы. Ингибиторы ангиогенеза могут быть разделены на два основных типа, согласно их функциональной специфичности: один тип включает ингибиторы ангиогенеза, специфически действующие на эндотелиальные клетки, включающие ангиостатины, эндостатины и др.; а другой тип охватывает ингибиторы ангиогенеза, которые неспецифически действуют на эндотелиальные клетки, включающие цитокины, ингибиторы тканевых металлопротеиназ, ингибиторов сериновых протаз, продукты супрессора опухолевого гена и др. Про-ангиогенные факторы включают эпидермальный фактор роста (EGF), фактор роста эндотелия сосудов (VEGF), фактор роста тромбоцитов (PDGF), фактор роста фибробластов (FGF) и др. (Hanks, S. K., et. al. FASEB, 1995, 9, 576-696). Высокий уровень экспрессии различных про-ангиогенных факторов отмечается в разных типах опухолей, такой как высокий уровень экспрессии EGF, в типичном случае наблюдается в эпителиальных клетках опухолей, а высокий уровень экспрессии PDGF обычно имеет место в глиоме. Применяемые в настоящее время стратегии разработки противоракового средства, направленного против пути неоангиогенеза опухоли, в основном направлено на усиление ингибиторов ангиогенеза и ослабление про-ангиогенных факторов, где ингибирование высокого уровня экспрессии про-ангиогенных факторов, в особенности путем воздействия на VEGF/VEGFR сигнальный путь, стало основным направлением современных исследований.
VEGF представляет собой гликопротеин в организме человека и играет важную роль в ангиогенезе. Семейство человеческих VEGF включает VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E и PLGF. VEGF может селективно воздействовать на VEGFR (VEGF рецептор), который представляет собой класс трансмембранных белков тирозинкиназ. Связывание VEGF с VEGFR меняет конформацию VEGFR и приводит к димеризаии рецептора, а также фосфорилирование внутрикеточных сайтов тирозина, что ведет к активации путей трансдукции в направлении считывания информации (Joukov, V., et. al. EMBO J., 1996, 15, 290-298). В результате проведенных обширных исследований было показано, что VEGF/VEGFR путь сигнальной трансдукции представляет собой самый важный про-ангиогенный путь и путь миграции в клетках. При ингибировании этого пути рост и миграция эндотелиальных клеток могут быть подавлены и, соответственно, может быть ингибирован рост опухолей. В настоящее время, были разрешены для применения несколько таких лекарственных средств и более 30 лекарственных средств проходят клинические испытания. Одно такое важное лекарственное средство представляет собой рекомбинантное гуманизированное VEGF моноклональное антитело, называемое бевацизумаб (bevacizumab) (торговое название Avastin), которое стало первым разрешенным для применения лекарственным средством, направленным против ангиогенеза в опухолях, способным специфически связываться с VEGF-A для блокирования VEGF/VEGFR пути. Это лекарственное средство имело большой успех, сразу после его разрешения для применения, однако, возникла проблема лекарственной резистентности, возникающая постепенно при длительном применении. Дальнейшие исследования показали, что специфическое ингибирование VEGF-A заставляет клетки высвобождать большое количество других про-ангиогенных факторов, таких как PLGF и FGF, и такое явление получило название реакция избегания ангиогенеза. Для решения проблемы лекарственной резистентности, одной подходящей стратегией является разработка многоцелевых ингибиторов.
Сунитиниб (Sunitinib) представляет собой многоцелевое противораковое средство, разработанное в компании Pfizer, которое является ингибитором, действующим на многоцелевые тирозинкиназы и может эффективно ингибировать рецептор тирозинкиназ, таких как VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-β и c-Kit, FLT-3. Путем ингибирования этих белков сунитиниб (sunitinib) блокирует экспрессию различных про-ангиогенных факторов в раковых клетках, так что при этом может быть достигнута цель подавления неоангиогенеза, ʺистощенияʺ раковых клеток и их гибели (Abrams, T. J. et. al. Mol. Cancer Ther., 2003, 2, 1011-1021). Кроме того, сунитиниб (sunitinib) также будет демонстрировать прямое специфическое ингбирование, направленное против раковых клеток, имеющих мутации в c-Kit и FLT-3. Сунитиниб (Sunitinib) был разрешен для применения Администрацией США (FDA) в 2006 в основном для лечения стромальных опухолей в желудочно-кишечном тракте и почечно-клеточной карциномы, в качестве противоракового средства первой линии, разрешенное для применения по двум показаниям. Хотя сунитиниб (Sunitinib) демонстрирует выраженную противоопухолевую эффективность, побочные реакции, такие как отсутствие энергии, угнетение функции костного мозга и лихорадочное состояние, все еще наблюдаются у пациентов, которые проходят лечение сунитинибом (Sunitinib). Сунитиниб (Sunitinib) демонстрирует мощную аккумуляцию в тканях и не может, в связи с этим, применяться постоянно, и в клинических сценариях его применяют последовательно в течение четырех недель и затем применение прерывают на две недели. Однако в результате исследований было показано, что неоангиогенез восстанавливается при отмене приема лекарственного средства. В этой связи, необходимо модифицировать химическую структуру для снижения токсичных побочных реакций, оптимизации способности принимать лекарство, и найти более безопасные и эффективные лекарственные средства.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является разработка многоцелевого ингибитора рецептора тирозинкиназы с высокой эффективностью и низкой токсичностью.
Другой целью настоящего изобретения является разработка группы пирролзамещенных производных индолона, которые ингибируют рост опухолей.
Еще одной целью настоящего изобретения является разработка фармацевтической композиции, включающей пирролзамещенные производные индолона.
Еще одной целью настоящего изобретения является разработка способа применения пирролзамещенных производных индолона и фармацевтической композиции, включающей пирролзамещенные производных индолона.
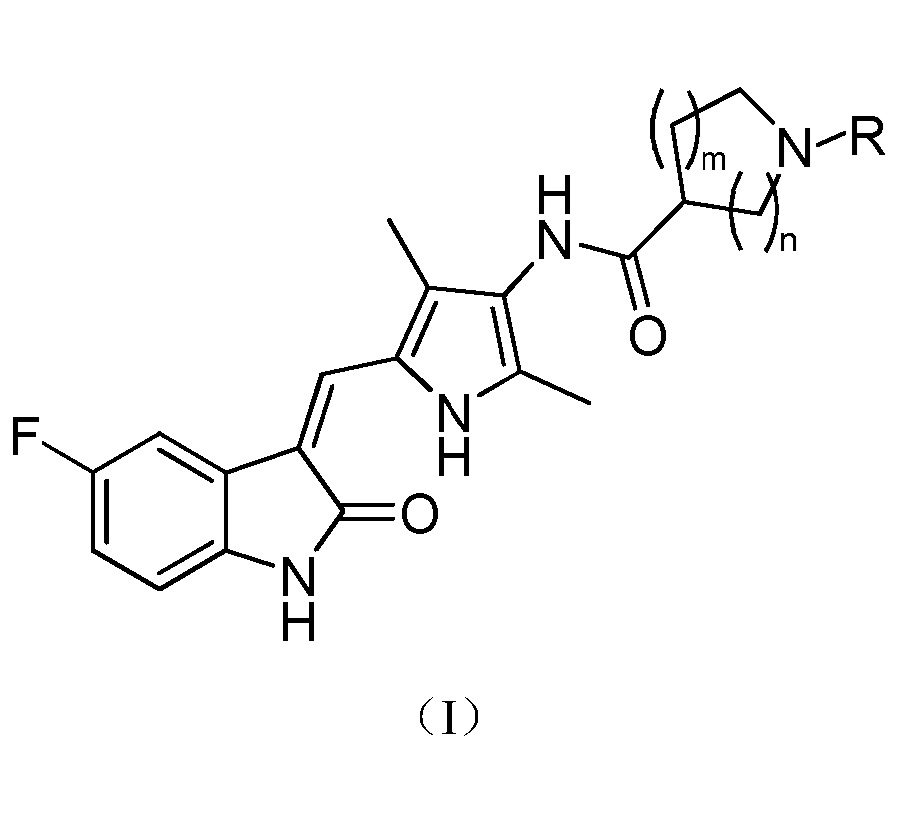
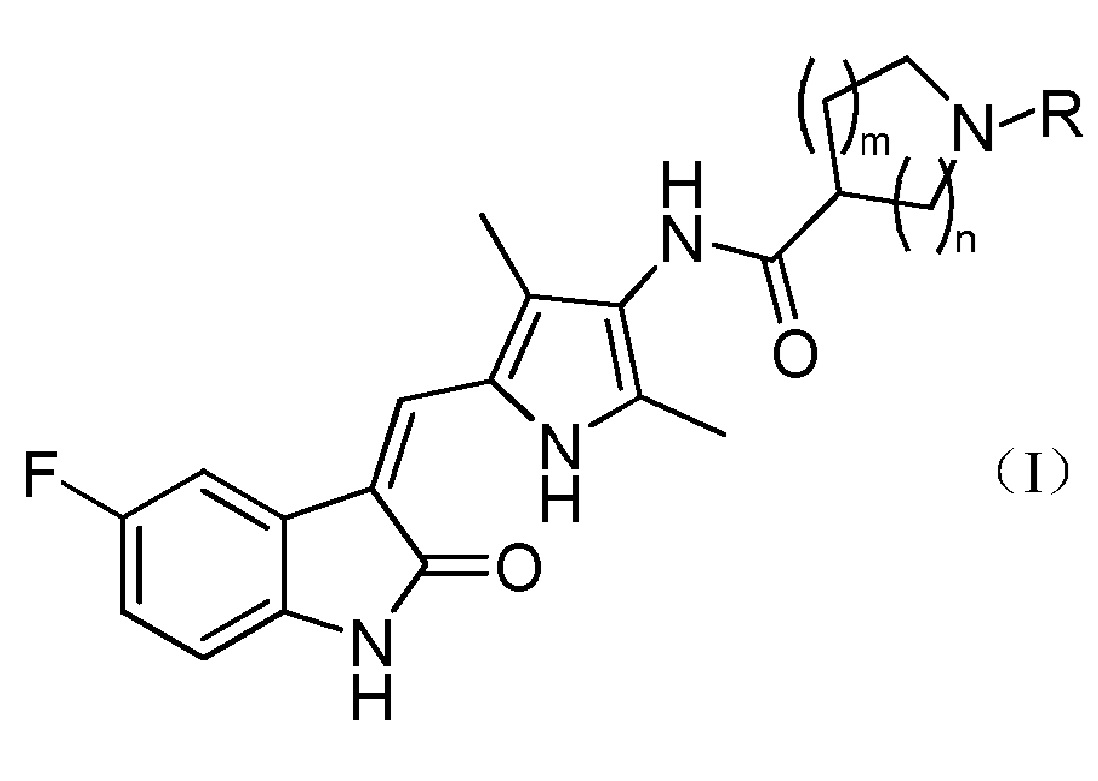
Настоящее изобретение относится к пирролзамещенному производному индолона, имеющего структуру, соответствующую показанной ниже общей формуле (I), или к его фармацевтически приемлемым солям:
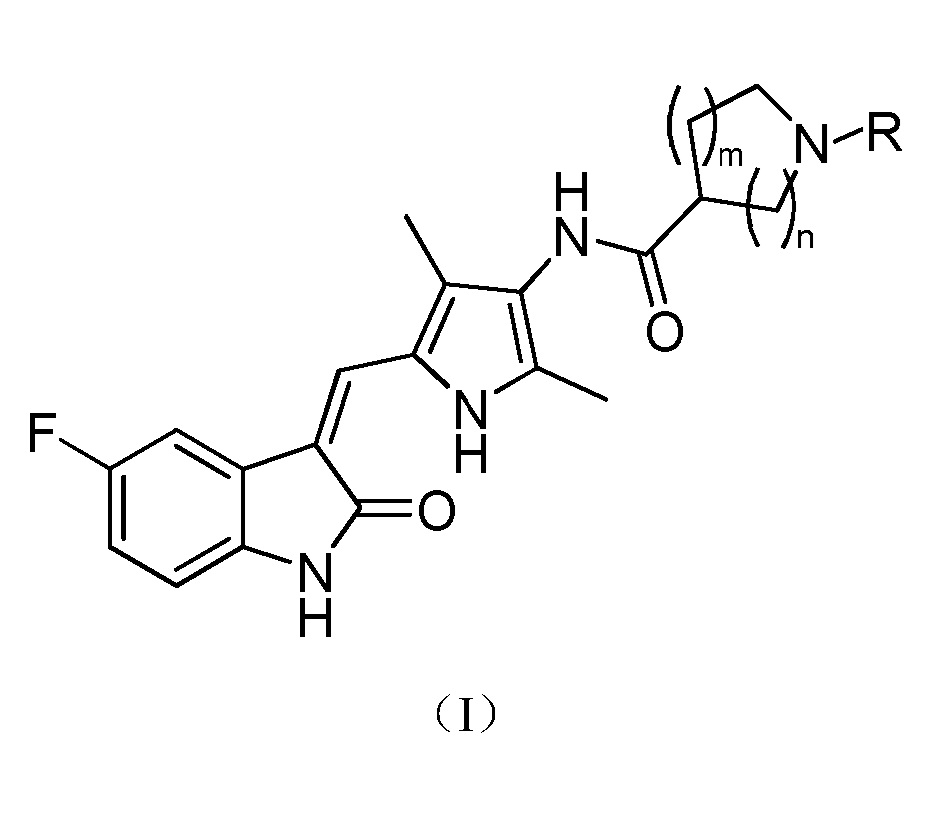
где:
m выбран из значений 0, 1 и 2;
n выбран из значений 1, 2 и 3; и
R выбран из водорода, C1-C6 линейного или разветвленного алкила, C3-C7 циклоалкила, формила, замещенного C1-C6 линейным или разветвленным алкилом, C3-C7 циклоалкилформила, т-бутоксикарбонила, замещенного карбамоила или от 5- до 7-членного циклического карбамоила.
В указанном выше пирролзамещенном производном индолона, R предпочтительно выбран из водорода, C1-C3 линейного или разветвленного алкила, C4-C6 циклоалкила, формила, замещенного C1-C3 линейным или разветвленным алкилом, C3-C6 циклоалкилформила, т-бутоксикарбонила, N,N-диметилкарбамоила, N,N-диэтилкарбамолила, N,N-дипропилкарбамоила, пирролидин-1-илформила или пиперидин-1-илформила.
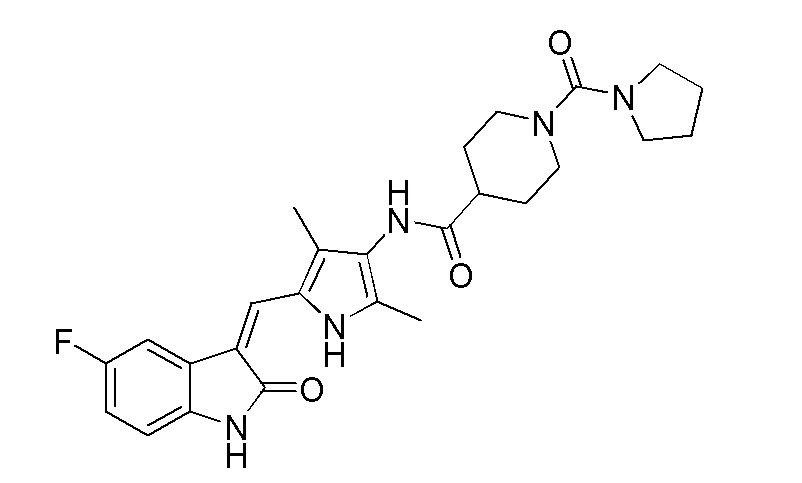
Более предпочтительно, в указанном выше пирролзамещенном производном индолона, R выбран из водорода, метила, т-бутоксикарбонила, N,N-диметилкарбамоила или пирролидин-1-илформила.
Согласно настоящему изобретению, пирролзамещенное производное индолона, имеющее структуру общей формулы (I), предпочтительно выбрано из соединений 1-15, показанных ниже:
где
m выбран из значений 0, 1 и 2;
n выбран из значений 1, 2 и 3; и
R выбран из водорода, C1-C6 линейного или разветвленного алкила, C3-C7 циклоалкила, формила, замещенного C1-C6 линейным или разветвленным алкилом, C3-C7 циклоалкилформила, т-бутоксикарбонила, замещенного карбамоила или от 5- до 7-членного циклического карбамоила.
В указанном выше пирролзамещенном производном индолона R предпочтительно выбран из водорода, C1-C3 линейного или разветвленного алкила, C4-C6 циклоалкила, формила, замещенного C1-C3 линейным или разветвленным алкилом, C3-C6 циклоалкилформила, т-бутоксикарбонила, N,N-диметилкарбамоила, N,N-диэтилкарбамоила, N,N-дипропилкарбамоила, пирролидин-1-илформила или пиперидин-1-илформила.
Более предпочтительно, в указанном выше пирролзамещенном производном индолона, R выбран из водорода, метила, т-бутоксикарбонила, N,N-диметилкарбамоила или пирролидин-1-илформила.
Согласно настоящему изобретению, указанное пирролзамещенное производное индолона, имеющее структуры общей формулы (I)предпочтительно выбран из соединений 1-15, показанных ниже:
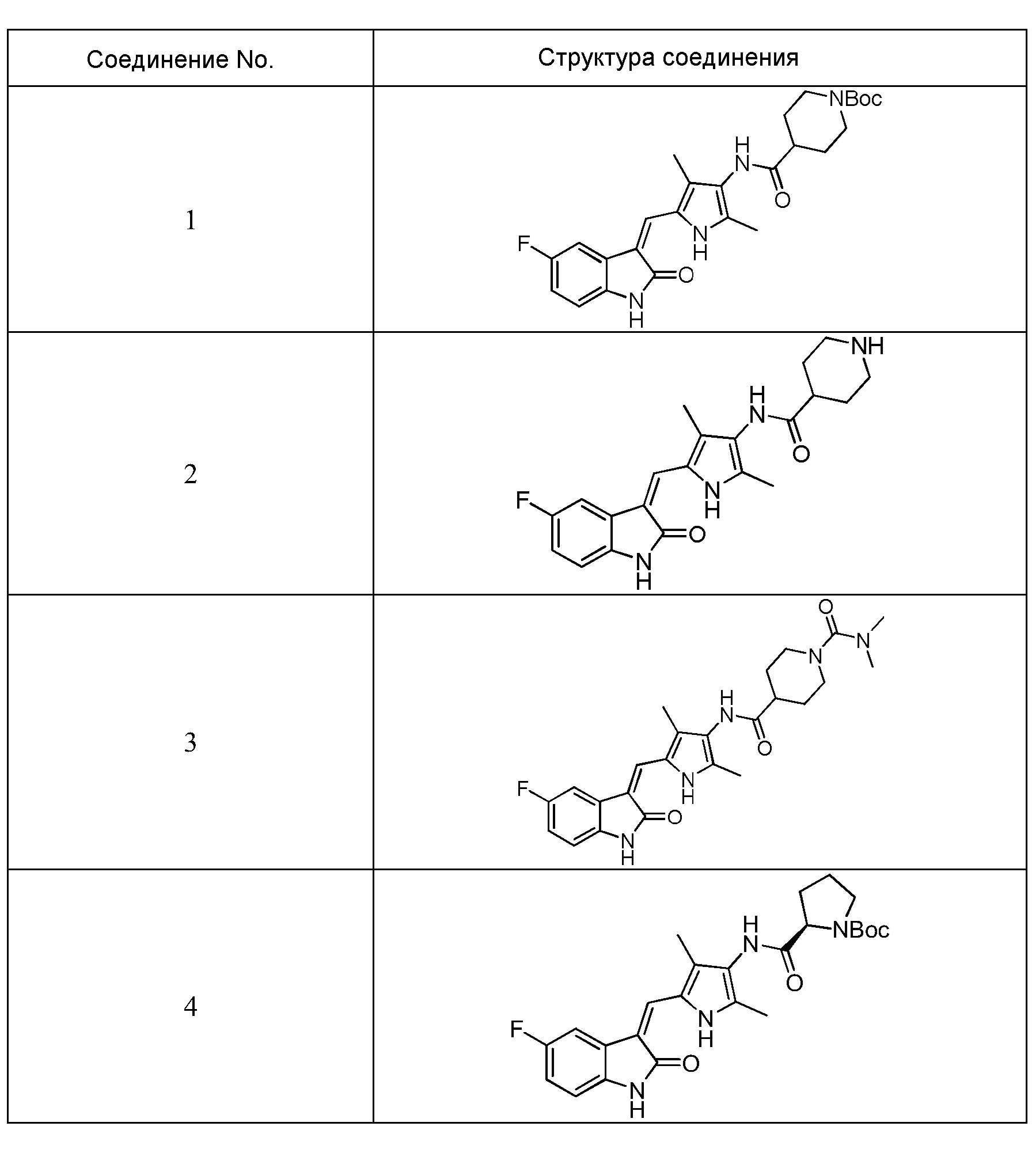
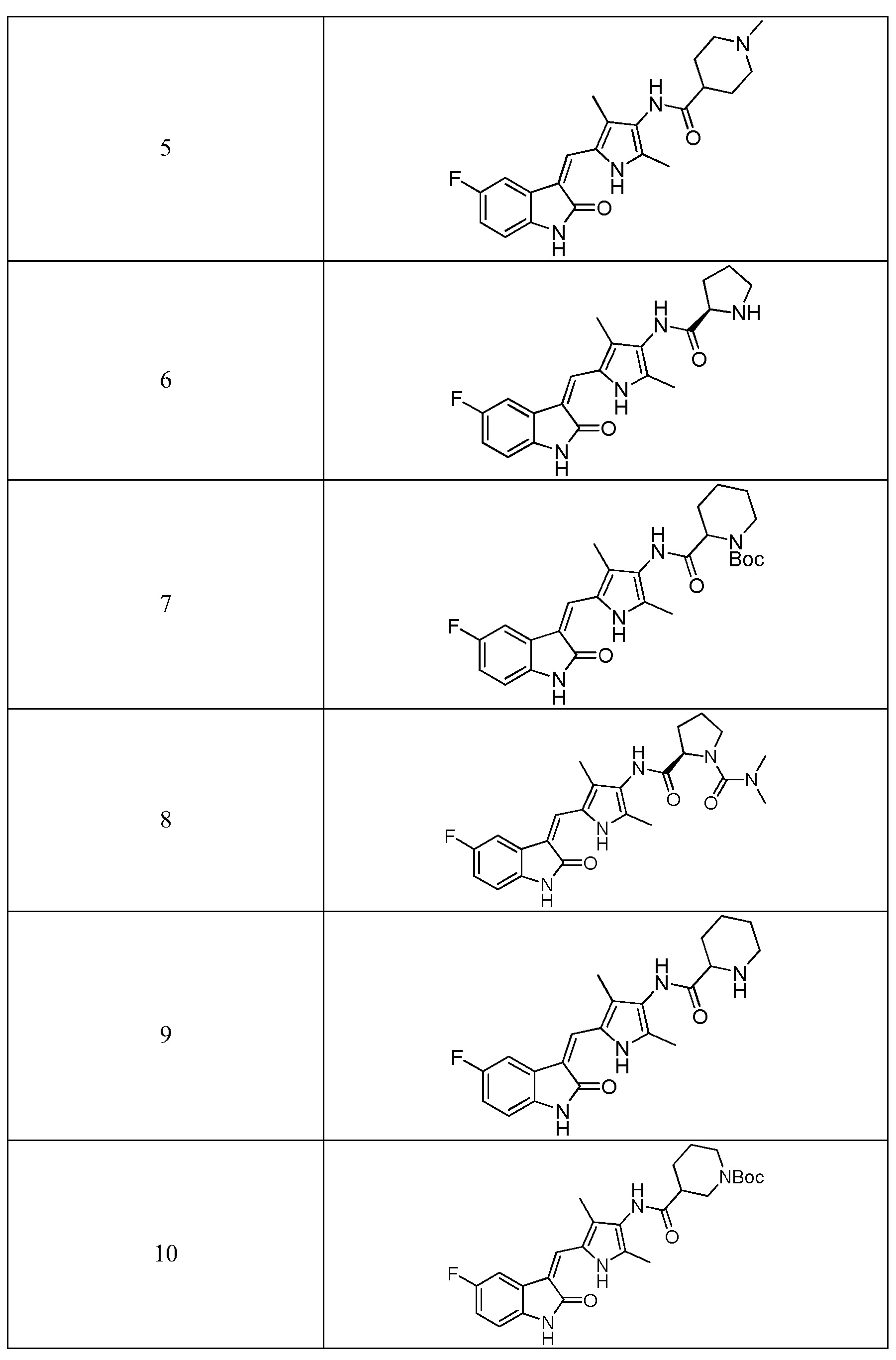

Фармацевтически приемлемые соли пирролзамещенного производного индолона по настоящему изобретению конкретно не ограничиваются и могут представлять собой гидрохлорид, фумарат, малеат, цитрат, фосфат, сульфат, тартрат, метансульфонат, бензолсульфонат и т.п. Использование гидрохлорида может сказаться на высокой кристалличности и высокой растворимости, а также позволит усилить гигроскопичность. В этой связи, предпочтительно использовать гидрохлорид.
Во втором аспекте настоящее изобретение относится к способу получения пирролзамещенного производного индолона, включающему следующие стадии:
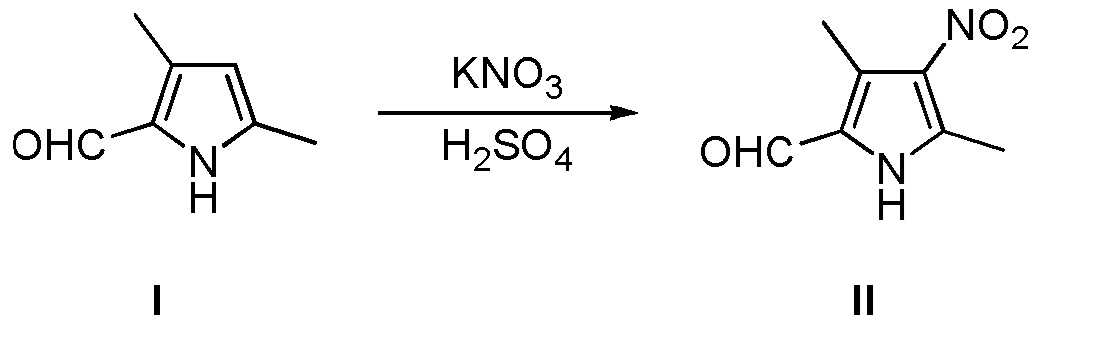
(a) нитрирование 3,5-диметил-2-пирролальдегида, показанного в виде структурной формулы I, с использованием KNO3 в концентрированной серной кислоте, с получением соединения, имеющего структурную формулу II:
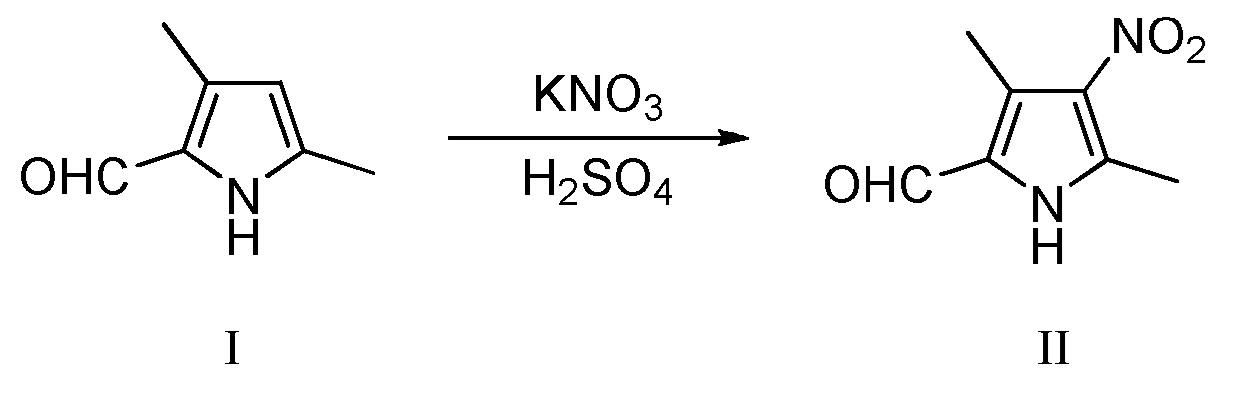
Конкретно, проводят следующие стадии: растворение 3,5-диметил-2-пирролальдегида, показанного в виде структурной формулы I, в концентрированной серной кислоте, снижение температуры до значения примерно -10°C, затем добавление KNO3, с последующим проведением реакции при поддержании указанной температуры; после завершения реакции, добавление холодной воды, затем - интенсивное перемешивание и фильтрование, с получением соединения II, которое затем подвергают рекристаллизации с получением чистого продукта;
(b) конденсацию 3,5-диметил-4-нитро-2-пирролальдегида, показанного в виде структурной II, с 5-фториндолоном, показанным в виде структурной формулы III, в условиях катализа пирролидином, с получением соединения, показанного в виде структурной формулы IV:
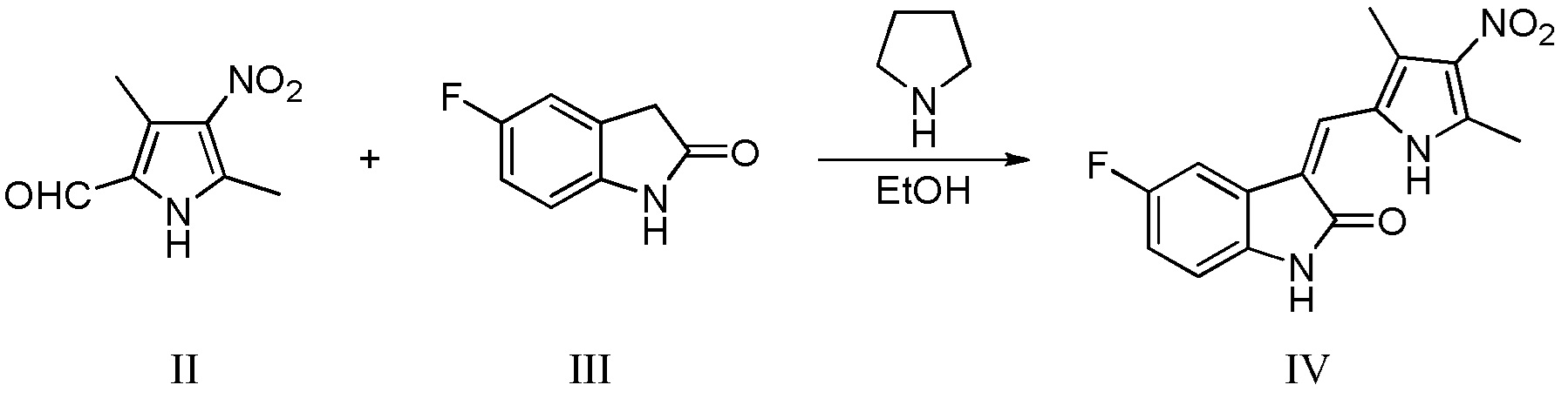
Конкретно, проводят следующие стадии: добавление 3,5-диметил-4-нитро-2-пирролальдегида, показанного в виде структурной формулы II, к этанолу, повышение температуры до 50°C, затем добавление 5-фториндолона, показанного в виде структурной формулы III, и проведение реакции при поддержании указанной температуры; после завершения реакции, проведение фильтрования с получением соединения IV в виде чистого продукта;
(c) восстановление соединения, показанного в виде структурной формулы IV, цинковым порошком, с получением соединения, показанного в виде структурной формулы V:
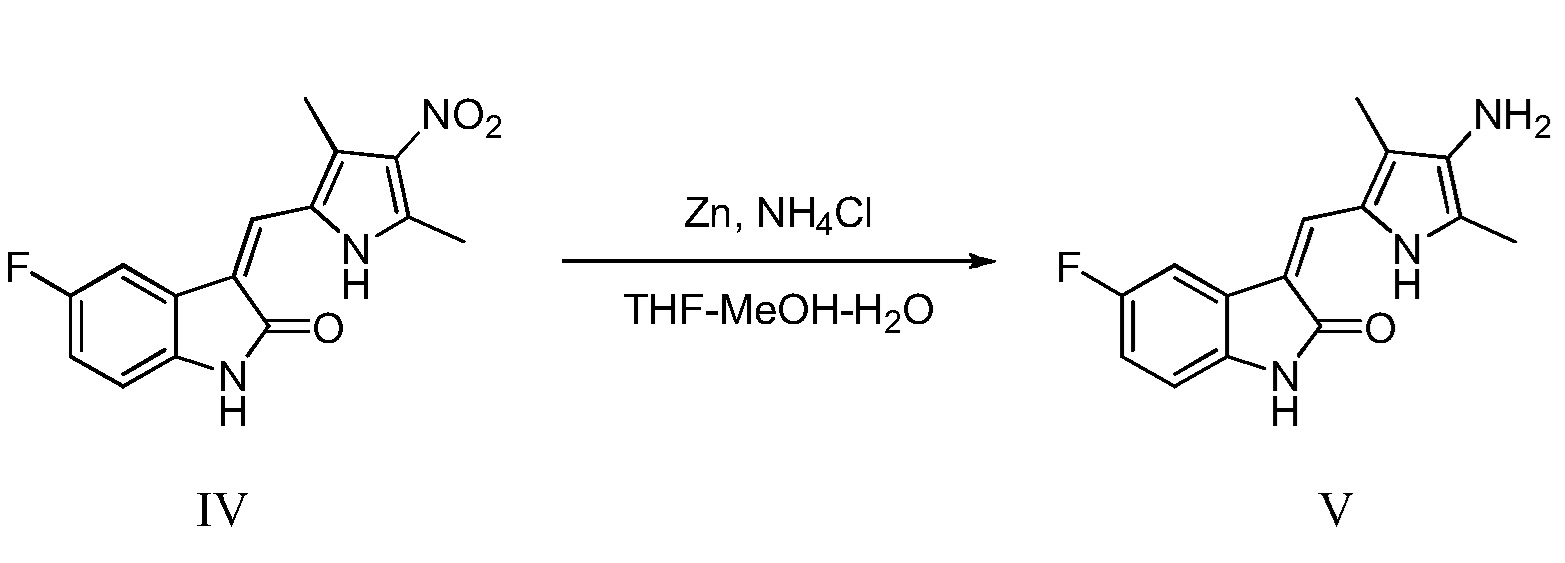
Конкретно, проводят следующие стадии: растворение соединения, показанного в виде структурной формулы IV, в смешанном растворителе, включающем тетрагидрофуран, воду и метанол, повышение температуры до 50°C, добавление насыщенного хлорида аммония и цинкового порошка, и проведение реакции, при поддержании указанной температуры; после завершения реакции, выпаривание растворителя и проведение экстракции этилацетатом, с получением соединения V, в виде чистого продукт;
(d) конденсацию соединения, показанного в виде структурной формулы V, с соответствующей кислотой VI, с получением соединения, показанного в виде структурной формулы VII:
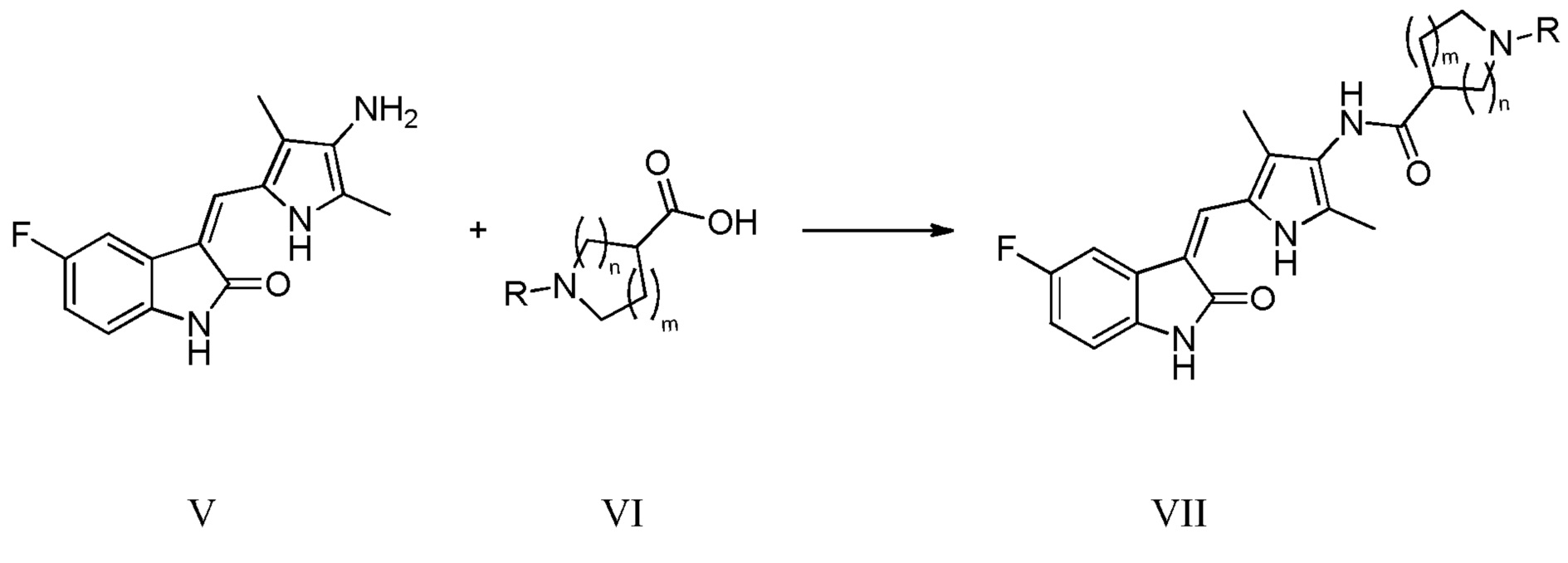
Конкретно, проводят следующие стадии: растворение соединения, показанного в виде структурной формулы V, в тетрагидрофуране, добавление щелочи (DIPEA, DMAP, пиридин или т.п.) и конденсирующего агента (EDCI, DCC или т.п.) в указанную смесь при комнатной температуре, и проведение реакции, при поддержании указанной температуры; после завершения реакции, выпаривание растворителя, с получением чистого продукта соединения, показанного в виде структурной формулы VII, промывку водой, с последующей промывкой растворителем (этилацетат, метанол или т.п.), с получением соединения VII в виде чистого продукта.
Настоящее изобретение относится к фармацевтической композиции, включающей терапевтически эффективное количество одного или нескольких пирролзамещенных производных индолона, показанных в виде общей формулы (I), или их фармацевтически приемлемые соли, где указанная композиция может также включать фармацевтически приемлемые вспомогательные вещества, такие как эксципиент, подсластители и т.п.
Указанные пирролзамещенные производные индолона или их фармацевтически приемлемые соли по настоящему изобретению обладают активностью в направлении ингибирования тирозинкиназ, и могут, в этой связи, использоваться для получения лекарственного средства, предназначенного для лечения опухолей, вызванных аномальной экспрессией тирозинкиназ. Таким образом, указанные пирролзамещенные производные индолона или их фармацевтически приемлемые соли по настоящему изобретению могут применяться для лечения опухолей, опосредованных участием тирозинкиназ, и для ингибирования роста клеток соответствующих опухолей, где используемый для этого способ включает введение терапевтически эффективного количества пирролзамещенных производных индолона или их фармацевтически приемлемых солей пациенту. Указанные пирролзамещенные производные индолона или их фармацевтически приемлемые соли могут также применяться для получения лекарственного средства, предназначенного для лечения опухолей, опосредованных участием тирозинкиназ, и для ингибирования роста клеток соответствующих опухолей.
Полезные эффекты
Пирролзамещенные производные индолона или их фармацевтически приемлемые соли, полученные согласно настоящему изобретению, вызывают ингибирование многих тирозинкиназ и могут ингибировать рост опухолей, как это было в основном показано в экспериментах на животных. В частности, пирролзамещенные производные индолона или их фармацевтически приемлемые соли по настоящему изобретению могут демонстрировать очень низкий уровень создания побочных токсических эффектов. Эти соединения могут применяться для лечения большого числа опухолевых заболеваний. Соединения по настоящему изобретению легко синтезировать, легко получать и, кроме того, они могут быть синтезированы на основе широко доступного исходного материала.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Далее будет приведено описание настоящего изобретения будет со ссылкой на конкретные примеры, но настоящее изобретение не ограничивается этими примерами.
В приведенных ниже примерах получения, 1H-ЯМР измеряют на приборе Varian Mercury AMX300, 400, 500; MS определяют с использованием VG ZAB-HS или VG-7070 и Esquire 3000 plus-01005; все растворители были подвергнуты редистилляции перед использованием; все использованные безводные растворители были получены путем сушки стандартными методами; если особо не указано иное, для всех реактивов применялась защита аргоном и проводился анализ с использованием ТСХ; после обработки проводилась промывка насыщенным раствором NaCl, сушка над безводным MgSO4; если особо не указано иное, продукты подвергали чистке путем хроматографирования на колонке с силикагелем, где использованный силикагель имел размеры 200-300 mesh GF254, производство компании Qingdao Haiyang Chemical Co., Ltd. или компании Yantai Yuanbo Silica Gel Company.
Пример получения 1: получение соединения 1
Растворяют 3,5-диметил-2-пирролальдегид I в качестве исходного материала (5 г, 40 ммоль) в 60 мл концентрированной серной кислоты, затем понижают температуру в системе до -10°C, и при этой температуре медленно добавляют KNO3 (4,35 г, 42 ммоль), порциями, в течение примерно 2 часов, поддерживая при этом температуру на уровне -10°C, и далее, по завершении добавления KNO3, раствор перемешивают в течение примерно 2 часов, при той же температуре. После завершения реакции, по результатам ТСХ, полученный раствор добавляют к 1 л ледяной воды, и проводят дважды экстракцию, используя суммарно 1 л этилацетата. Органический слой промывают насыщенным раствором NaCl, сушат над безводным сульфатом натрия и фильтруют. Затем органические растворители выпаривают при пониженном давлении, с получением 7 г неочищенного продукта, который добавляют к 10-20 мл этилацетата, с проведением, после добавления, энергичного перемешивания, и получают 5 г чистого продукта целевого соединения II.
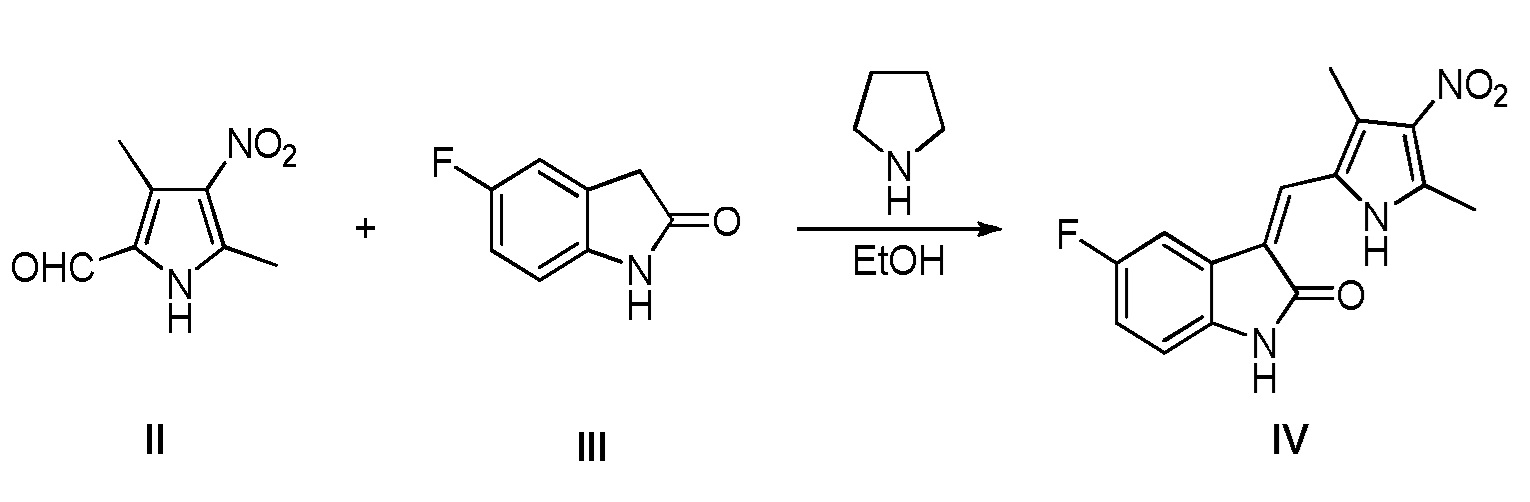
Соединение II (1,68 г, 10 ммоль) и Соединение III (1,8 г, 12 ммоль) добавляют к 50 мл безводного этанола, после чего туда же, при комнатной температуре, добавляют тетрагидропиррол (850 мг, 12 ммоль). После добавления, указанная система становится желтой. Повышают температуру до 50°C, и проводят реакцию в течение 2 часов при этой температуре. После завершения реакции, систему подвергают фильтрованию, а фильтрпрессную лепешку промывают небольшим объемом этанола и этилацетата, с получением 2,7 чистого продукта целевого соединения IV. 1H ЯМР (400 MГц, ДМСО-d6) δ 11,14 (с, 1H), 7,88 (дд, J=9,2, 2,4 Гц, 1H), 7,82 (с, 1H), 7,05-6,97 (м, 1H), 6,88 (дд, J=8,5, 4,5 Гц, 1H), 2,64 (с, 3H), 2,58 (с, 3H).
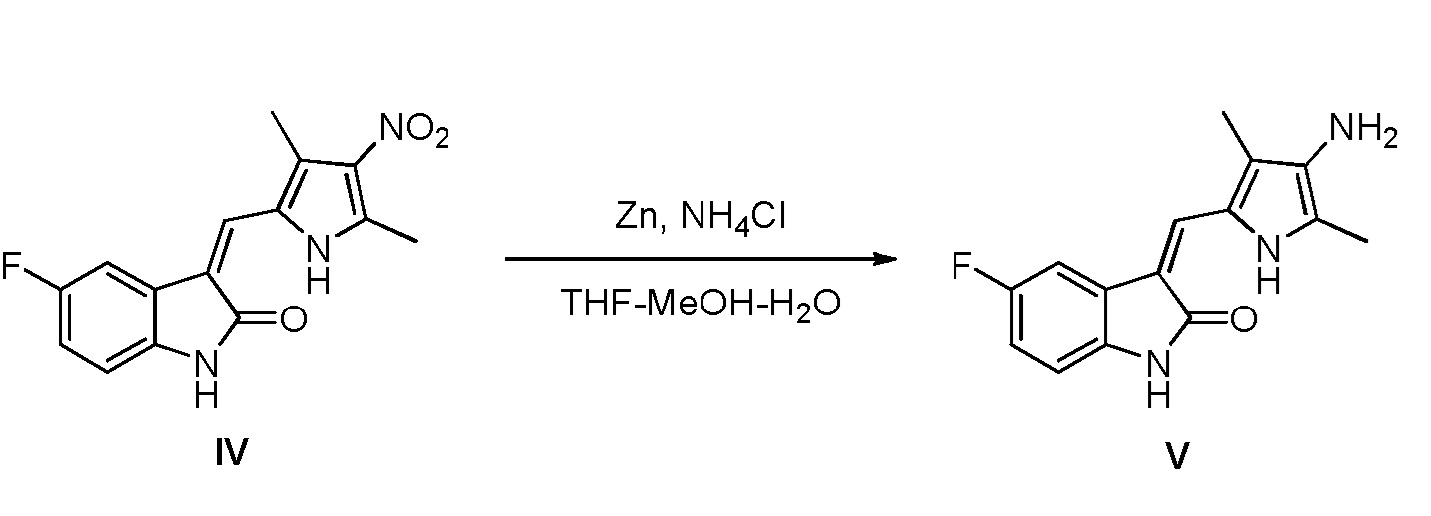
Соединение IV (900 мг, 3 ммоль) вносят в двугорлую колбу объемом 500 мл, после чего туда же добавляют 200 мл тетрагидрофурана, 100 мл метанола, 60 мл воды и 60 мл насыщенного раствора хлорида аммония. Далее повышают температуру до 50°C и добавляют при перемешивании порошок цинка (1,8 г, 30 ммоль) и проводят реакцию в течение 2 часов в этих условиях, при этом, в ходе реакции, смесь становится вначале прозрачной, а затем мутной. После того, как смесь помутнеет, проводят ЖХ-MС (жидкостную хроматографию с масс-спектрометрией) для подтверждения того, что реакция полностью завершилась. После завершения реакции растворитель выпаривают. Систему подщелачивают с использованием насыщенного раствора бикарбоната натрия и проводят дважды экстракцию с использованием в целом 2 л этилацетата. Этилацетатный слой промывают насыщенным раствором NaCl, сушат над безводным сульфатом натрия и фильтруют. Затем органические растворители выпаривают при пониженном давлении, с получением целевого соединения V (800 мг).
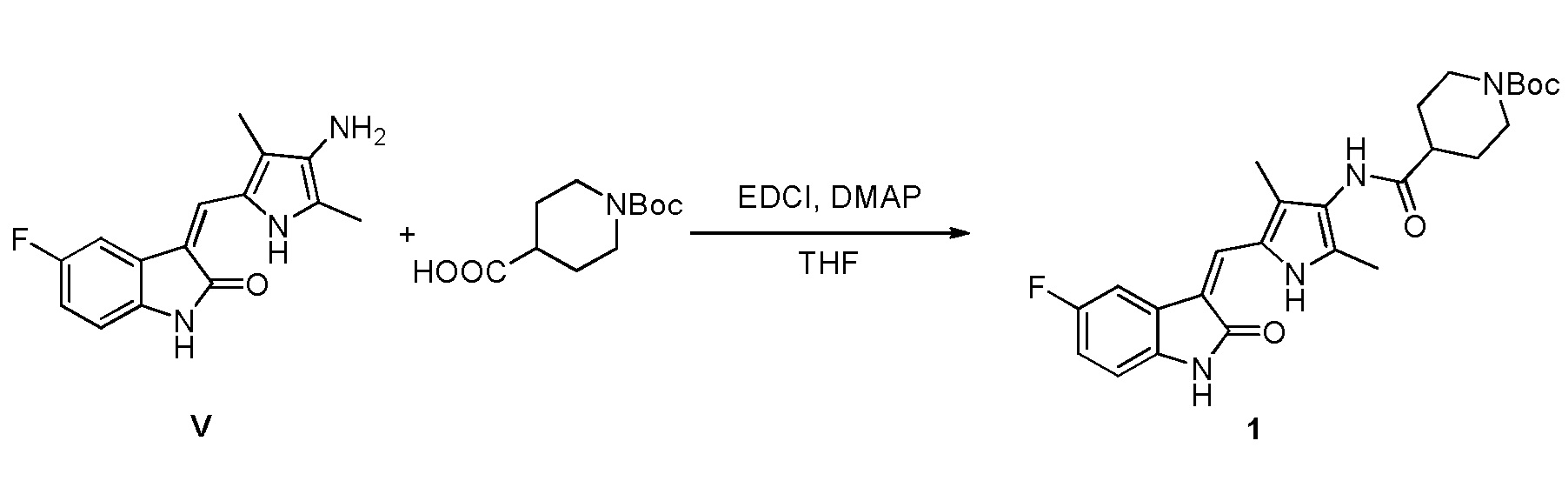
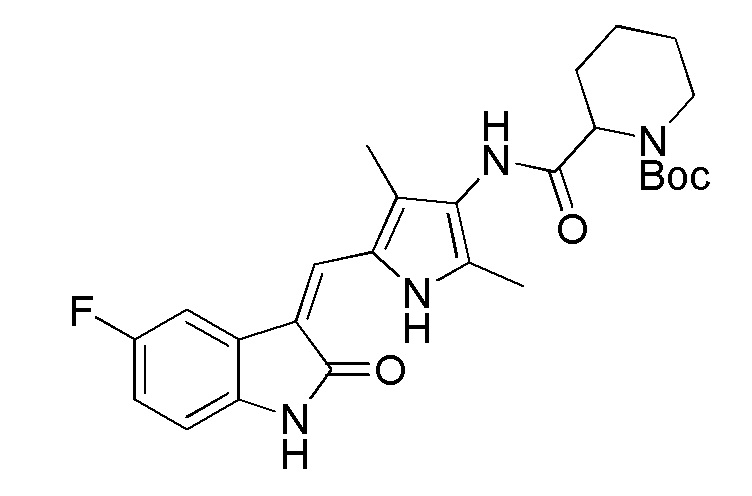
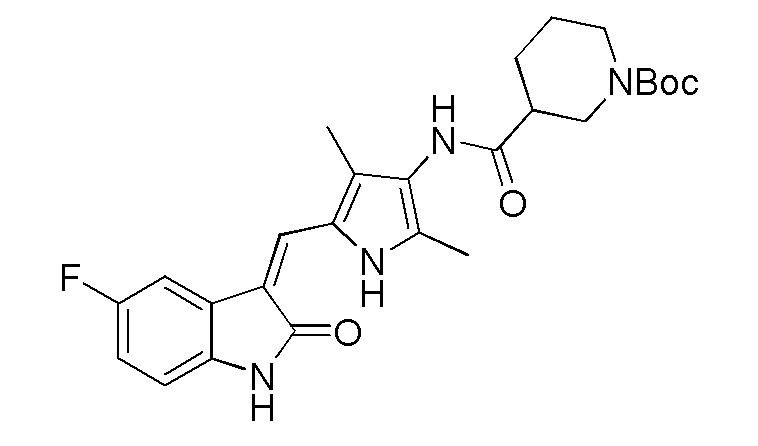
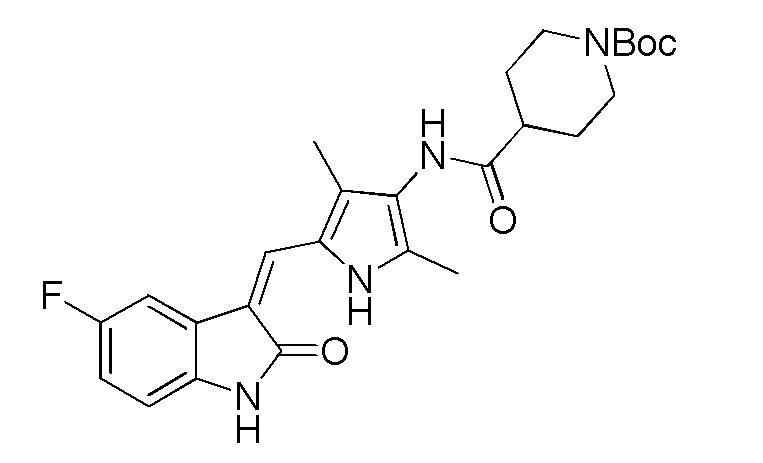
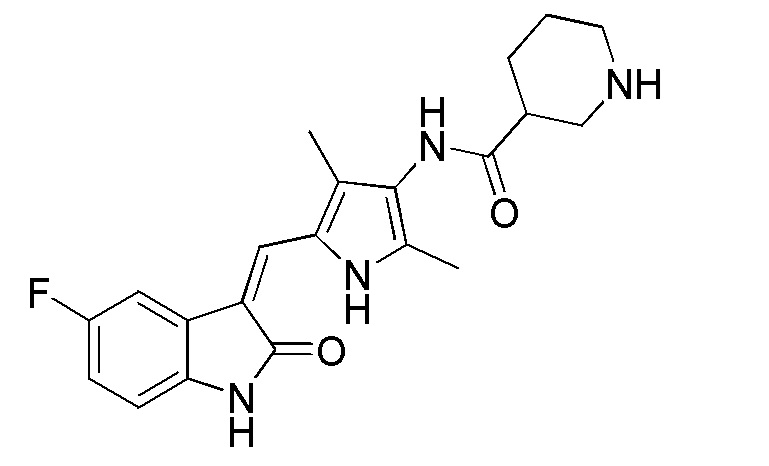
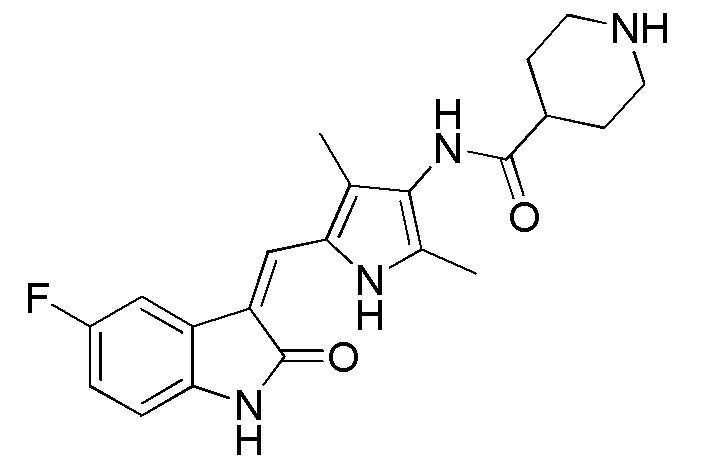
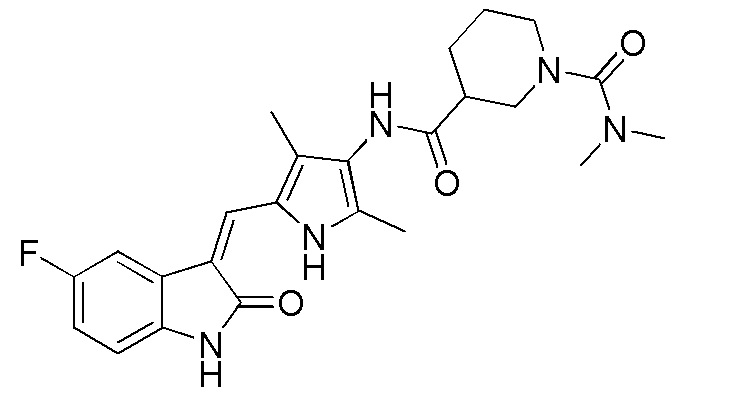
Соединение V (270 мг, 1 ммоль) растворяют в тетрагидрофуране (20 мл) и добавляют туда же, при комнатной температуре, Boc-защищенную (с трет-бутоксикарбонильной защитной группой) 4-пиперидинкарбоновую кислоту (270 мг, 1,2 ммоль), ЭДКИ (EDCI) (220 мг, 1,1 ммоль), ДИПЭА (DIPEA) (260 мг, 2 ммоль) и каталитическое количество ДМАП (DMAP). После внесения всех компонентов проводят реакцию при комнатной температуре в течение примерно 8 часов с подтверждением завершения реакции по результатам ТСХ. После завершения реакции выпаривают раствор тетрагидрофурана и добавляют большой объем этилацетата и воды для распределения и затем проводят фильтрование с получением неочищенного продукта целевого соединения. Указанный неочищенный продукт промывают метанолом, с получением чистого продукта соединения 1. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,59 (с, 1H), 10,83 (с, 1H), 9,12 (с, 1H), 7,71 (дд, J=9,5, 2,6 Гц, 1H), 7,66 (с, 1H), 6,93-6,86 (м, 1H), 6,86-6,81 (м, 1H), 3,99-3,95 (м, 2H), 3,10-2,94 (м, 1H), 2,79-2,75 (м, 2H), 2,17 (с, 3H), 2,15 (с, 3H), 1,82-1,78 (м, 2H), 1,55-1,45 (м, 2H), 1,41 (с, 9H).
Пример получения 2: получение Соединения 2
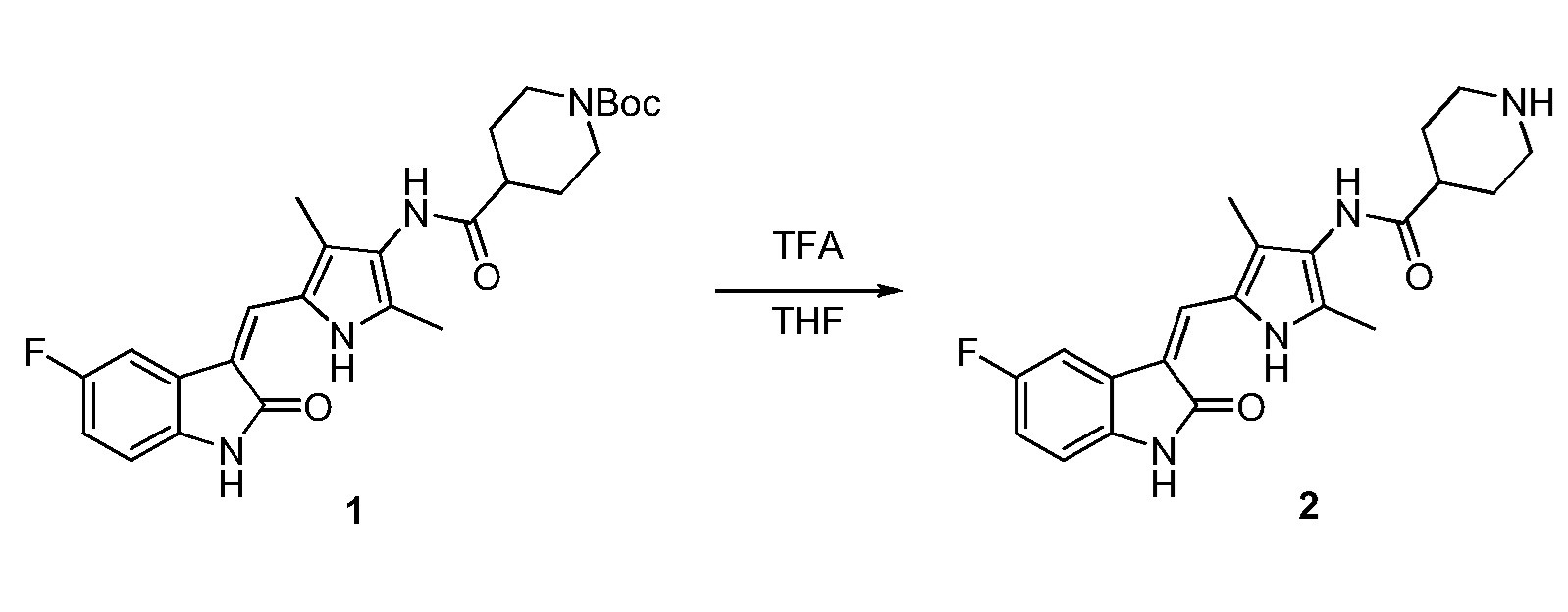
Соединение 1 (480 мг, 1 ммоль) добавляют к 10 мл тетрагидрофурана и туда же, при комнатной температуре, добавляют 10 мл трифторуксусной кислоты. Затем повышают температуру до 50°C и проводят реакцию в течение примерно 2 часов, завершение которой определяют при проведении ЖХ-МС (жидкостной хроматографии с масс-спектрометрией). После завершения реакции, выпаривают основную часть раствора, а остаток нейтрализуют с использованием для этого насыщенного раствора карбоната натрия, с последующим проведением фильтрования, с получением неочищенного продукта. Указанный неочищенный продукт промывают этилацетатом и метанолом, с получением чистого продукта Соединения 2. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,60 (с, 1H), 10,85 (с, 1H), 9,26 (с, 1H), 7,71 (дд, J=9,5, 2,4 Гц, 1H), 7,67 (с, 1H), 6,94-6,86 (м, 1H), 6,87-6,83 (м, 1H), 3,34 (д, J=12,3 Гц, 2H), 3,06-2,89 (м, 2H), 2,74-2,59 (м, 1H), 2,18 (с, 3H), 2,16 (с, 3H), 2,05-1,94 (м, 2H), 1,89-1,74 (м, 2H).
Пример получения 3: получение Соединения 3
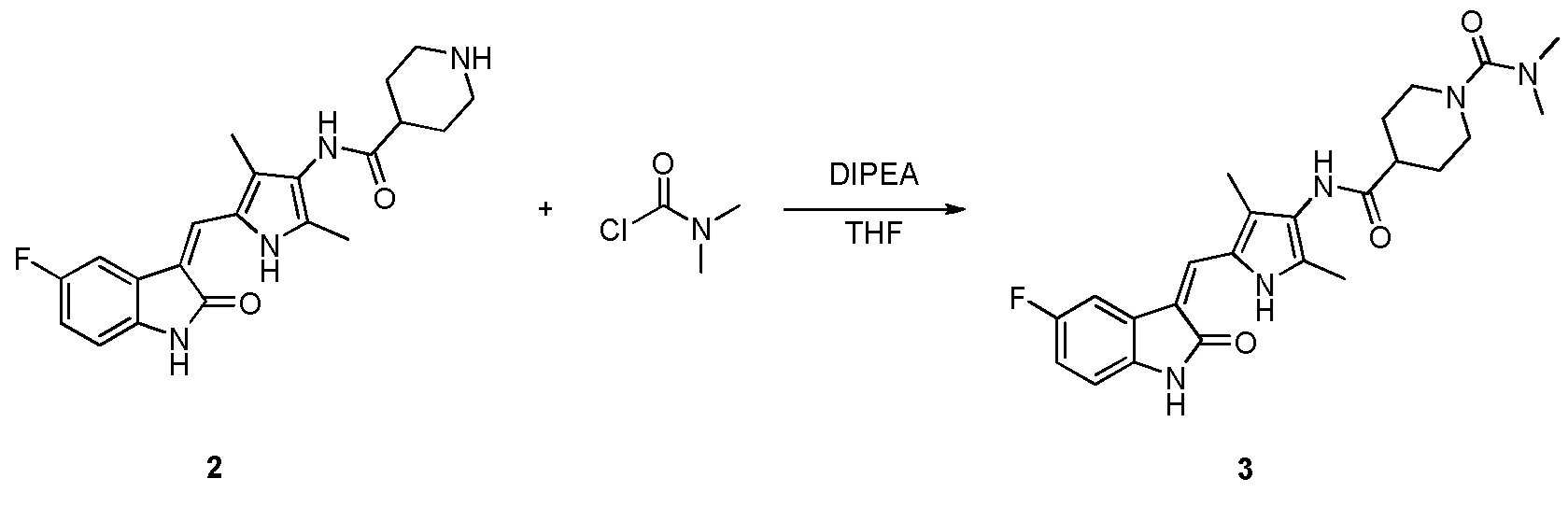
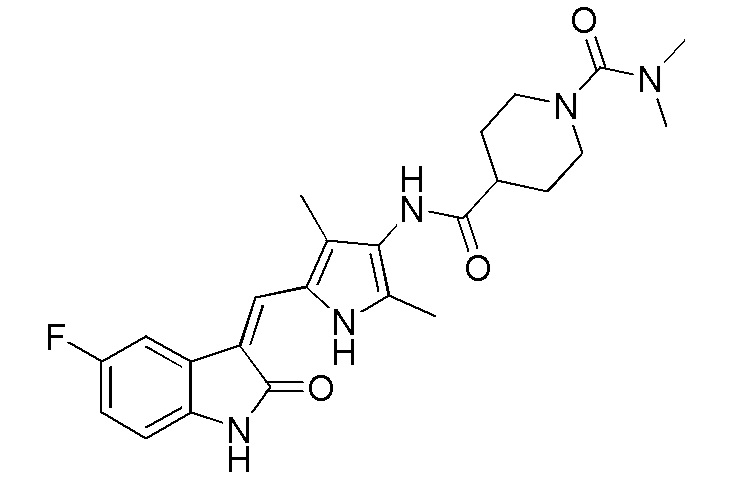
Соединение 2 (383 мг, 1 ммоль) добавляют к 20 мл тетрагидрофурана, и добавляют туда же, при комнатной температуре, ДИПЭА (DIPEA) (260 мг, 2 ммоль) и диметилкарбамоилхлорид (214 мг, 2 ммоль). Затем, в течение примерно 12 часов, проводят реакцию, и в это время, когда реакция уже будет близка к завершению, проводят ТСХ, для подтверждения ее завершения. После завершения реакции растворители выпаривают, и оставшееся твердое вещество промывают с использованием 20 мл этилацетата и 10 мл метанола, с получением чистого продукта целевого соединения 3. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,58 (с, 1H), 10,82 (с, 1H), 9,10 (с, 1H), 7,70 (дд, J=9,5, 2,5 Гц, 1H), 7,66 (с, 1H), 6,95-6,86 (м, 1H), 6,85-6,77 (м, 1H), 3,87-3,70 (м, 1H), 3,63-3,54 (м, 2H), 2,86-2,58 (м, 8H), 2,18 (с, 3H), 2,15 (с, 3H), 1,85-1,76 (м, 2H), 1,71-1,55 (м, 2H).
Пример получения 4: получение Соединения 4
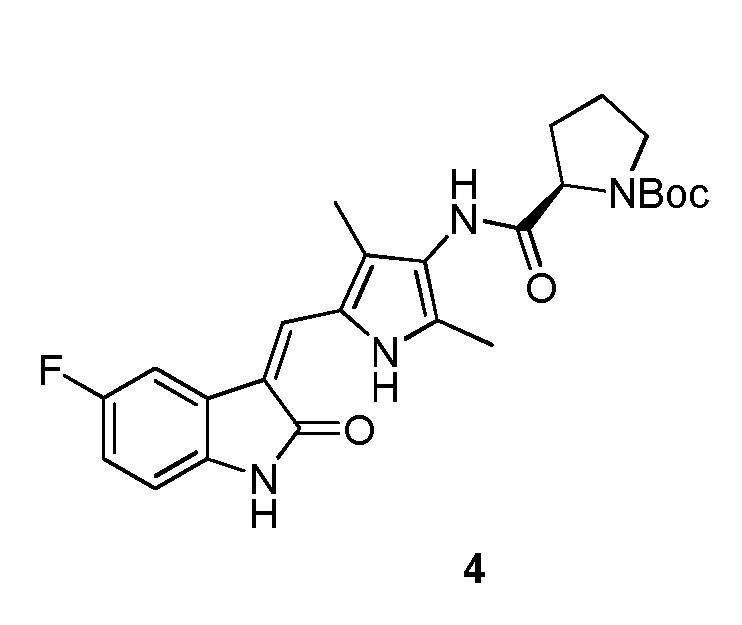
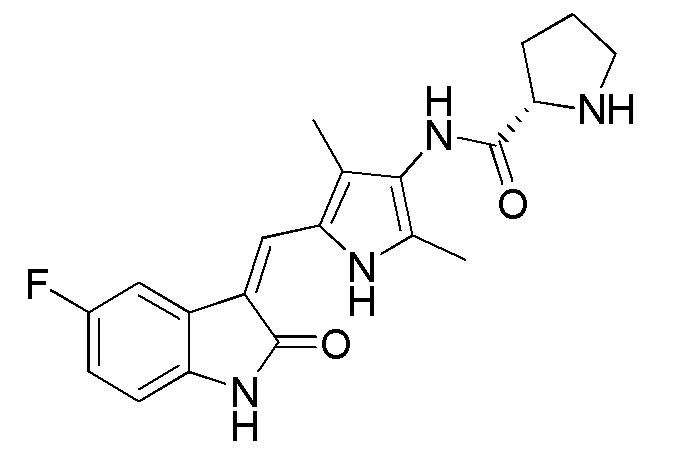
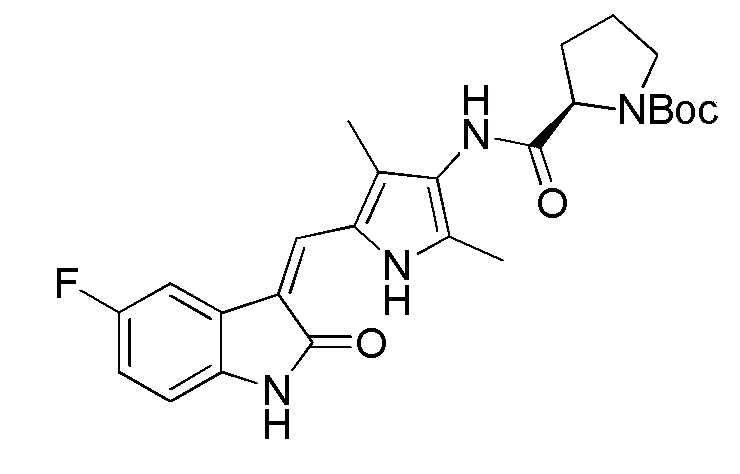
Используют тот же протокол, что и при синтезе по указанному выше методу 1, за исключением того, что здесь использовали пролин с Boc (трет-бутоксикарбонильной) защитной группой вместо пиперидинкарбоновой кислоты с Boc (трет-бутоксикарбонильной) защитной группой, с получением целевого соединения 4. 1H ЯМР (400 МГц, ДМСО) δ 13,59 (с, 0,6H), 13,58 (с, 0,4H), 10,84 (с, 1H), 9,20 (с, 0.6H), 9,13 (с, 0,4H), 7,70 (дд, J=9,5, 2,5 Гц, 1H), 7,66 (д, J=3,0 Гц, 1H), 6,89 (дд, J=12,5, 5,5 Гц, 1H), 6,83 (дд, J=8,4, 4,7 Гц, 1H), 4,38-4,15 (м, 1H), 3,52-3,41 (м, 1H), 3,35-3,28 (м, 1H), 2,35-2,22 (м, 1H), 2,21 (с, 2H), 2,18 (с, 3H), 2,16 (с, 1H), 1,98-1,79 (м, 3H), 1,43 (с, 3H), 1,39 (с, 6H) (указанный Boc (трет-бутоксикарбонильный) заместитель не может свободно вращаться с образованием изомера).
Пример получения 5: получение Соединения 5
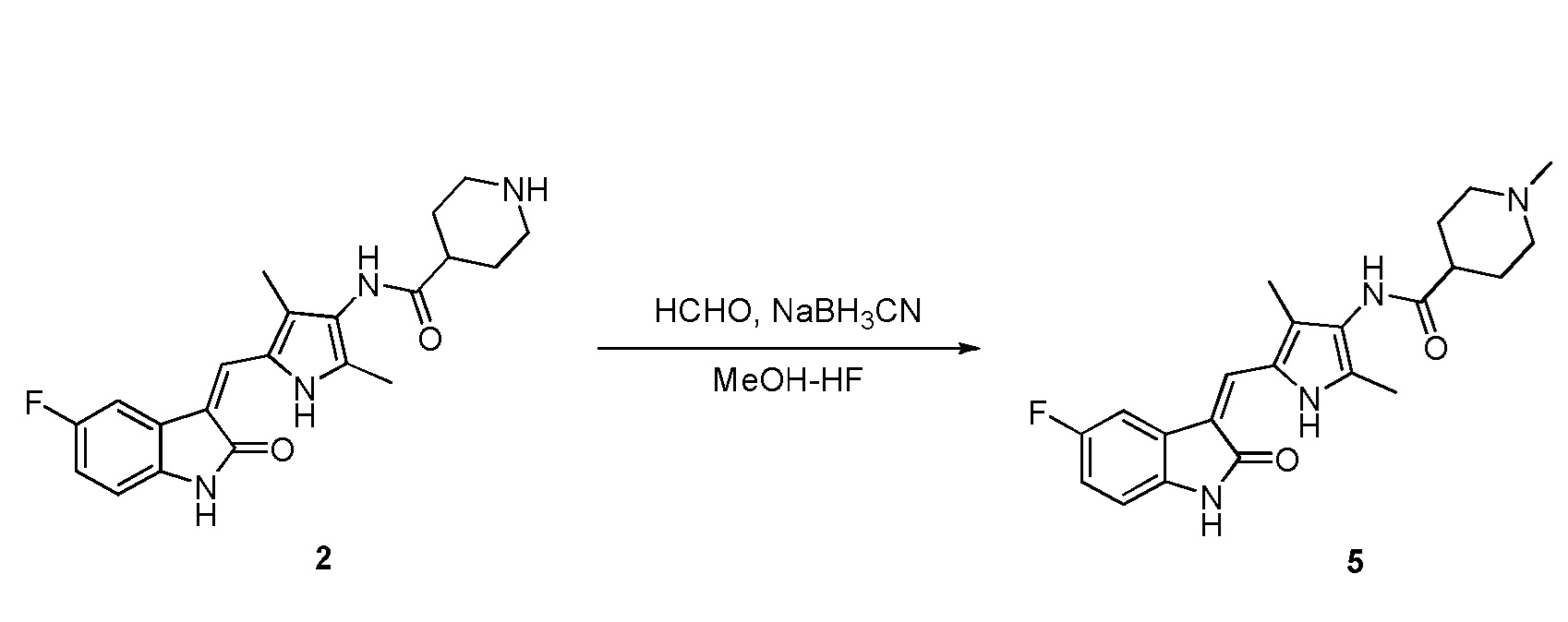
Соединение 2 (383 мг, 1 ммоль) добавляют к 20 мл смеси растворителей (1:1), включающей тетрагидрофуран и метанол, и добавляют туда же при комнатной температуре водный раствор формальдегида (500 мг, 5 ммоль) и цианборгидрид натрия (120 мг, 2 ммоль). После добавления проводят реакцию в течение 12 часов, и ее течение оценивают по результатам ТСХ. После завершения реакции, растворители выпаривают и после проведения хроматографии на колонке получают целевое соединение 5. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,58 (с, 1H), 10,83 (с, 1H), 9,05 (с, 1H), 7,70 (дд, J=9,5, 2,4 Гц, 1H), 7,66 (с, 1H), 6,93-6,86 (м, 1H), 6,85-6,80 (м, 1H), 2,87-2,77 (м, 2H), 2,36-2,21 (м, 1H), 2,17 (с, 3H), 2,16 (с, 3H), 2,15 (с, 3H), 1,94-1,59 (м, 6H).
Пример получения 6: получение Соединения 6
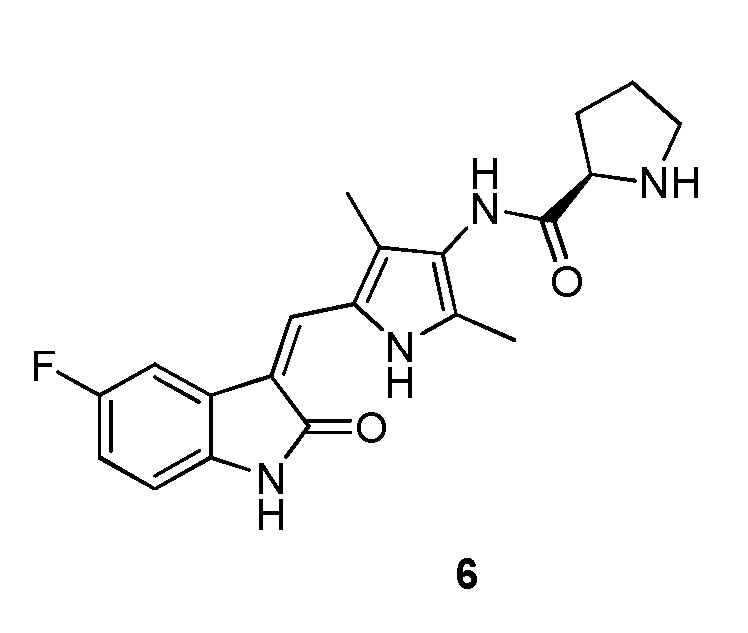
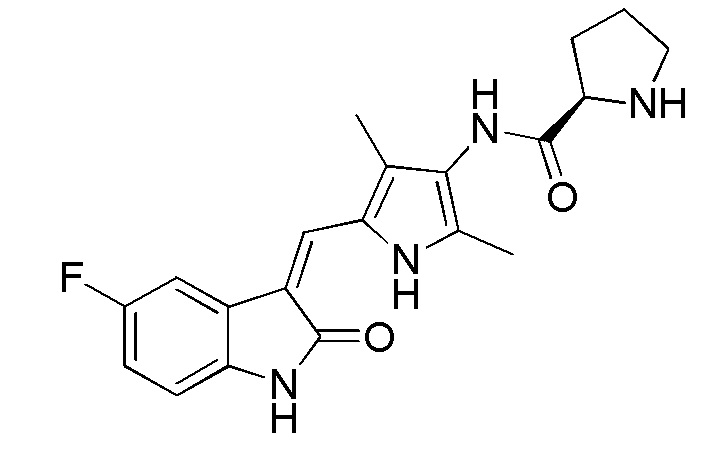
Синтез соединения 6 проводят по той же процедуре, которая использовалась для получения соединения 2, за исключением того, что здесь использовали соединение 4 вместо соединения 1, с получением целевого соединения 6. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,60 (с, 1H), 10,83 (с, 1H), 9,23 (с, 1H), 7,71 (дд, J=9,3, 2,4 Гц, 1H), 7,67 (с, 1H), 6,93-6,87 (м, 1H), 6,83 (дд, J=8.4, 4.6 Hz, 1H), 3.70 (дд, J=8.7, 5.5 Hz, 1H), 2,91 (т, J=6,6 Гц, 1H), 2,18 (с, 2H), 2,16 (с, 2H), 2,10-1,98 (м, 1H), 1,85-1,74 (м, 1H), 1,72-1,63 (м, 2H).
Пример получения 7: получение соединения 7
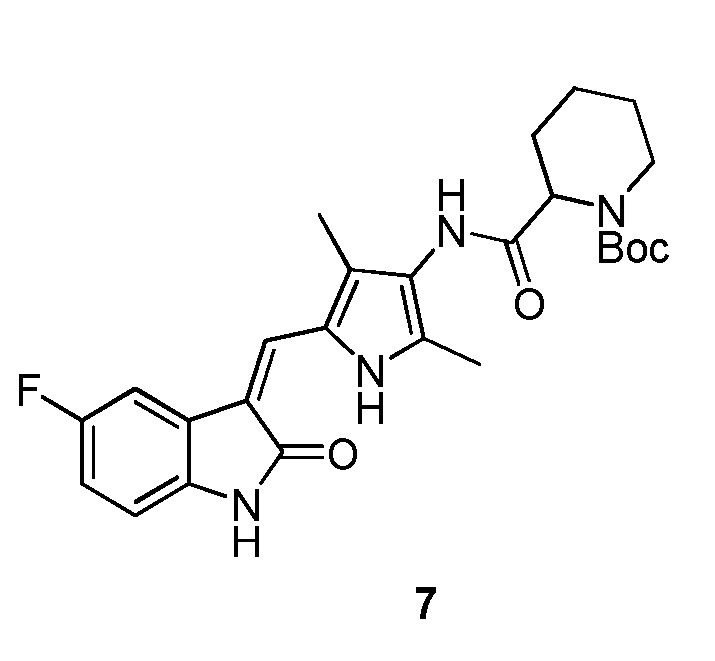
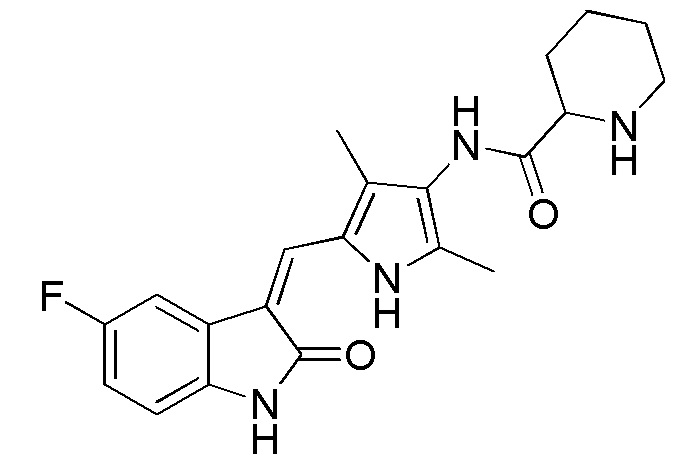
Синтез соединения 7 проводят по той же процедуре, которая использовалась для получения соединения 1, за исключением того, что здесь использовали 2-пиперидинкарбоновую кислоту с Boc (трет-бутоксикарбонильной группой) защитной группой вместо 4-пиперидинкарбоновой кислоты с Boc (трет-бутоксикарбонильной группой) защитной группой, с получением целевого соединения 7. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,60 (с, 1H), 10,84 (с, 1H), 9,16 (с, 1H), 7,71 (дд, J=9,5, 2,5 Гц, 1H), 7,67 (с, 1H), 6,94-6,87 (м, 1H), 6,85-6,81 (м, 1H), 4,79-4,63 (м, 1H), 3,87-3,75 (м, 1H), 3,30-3,09 (м, 1H), 2,19 (с, 3H), 2,17 (с, 3H), 1,81-1,59 (м, 3H), 1,41 (с, 9H), 1,44-1,22 (м, 3H).
Пример получения 8: получение Соединения 8
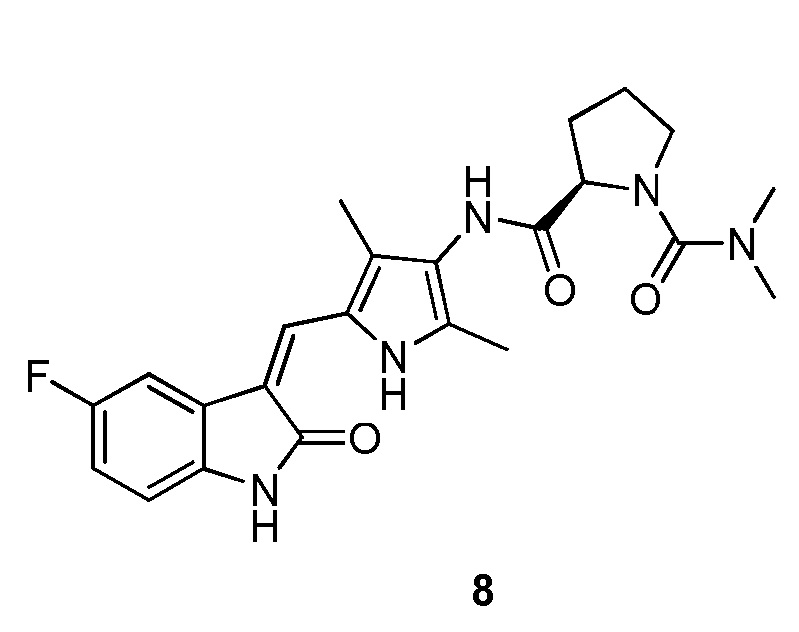
Синтез соединения 8 проводят по той же процедуре, которая использовалась для получения соединения 3, за исключением того, что здесь использовали соединение 4 вместо соединения 2, с получением целевого соединения 8. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,59 (с, 1H), 10,84 (с, 1H), 8,99 (с, 2H), 7,74-7,68 (м, 1H), 7,66 (с, 1H), 6,93-6,86 (м, 1H), 6,83 (дд, J=8,4, 4,6 Гц, 1H), 4,39 (т, J=7,4 Гц, 1H), 3,60-3,44 (м, 1H), 3,43-3,37 (м, 1H), 2,80 (с, 6H), 2,28-2,20 (м, 1H), 2,16 (с, 3H), 2,14 (с, 3H), 1,97-1,88 (м, 1H), 1,87-1,70 (м, 2H).
Пример получения 9: получение Соединения 9
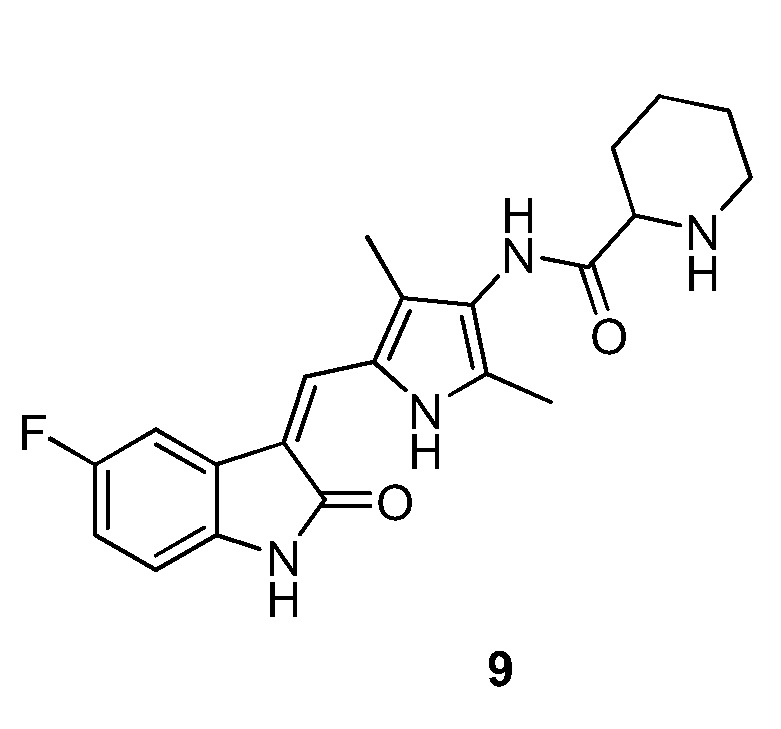
Синтез соединения 9 проводят по той же процедуре, которая использовалась для получения соединения 3, за исключением того, что здесь использовали соединение 4 вместо соединения 2, с получением целевого соединения 9. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,59 (с, 1H), 10,83 (с, 1H), 8,97 (с, 1H), 7,70 (дд, J=9,6, 2,5 Гц, 1H), 7,66 (с, 1H), 6,92-6,86 (м, 1H), 6,84-6,81 (м, 1H), 3,30-3,25 (м, 1H), 2,99 (д, J=13,2 Гц, 1H), 2,60 (т, J=11,3 Гц, 1H), 2,18 (с, 2H), 2,16 (с, 2H), 1,91-1,73 (м, 2H), 1,56-1,33 (м, 4H).
Пример получения 10: получение Соединения 10
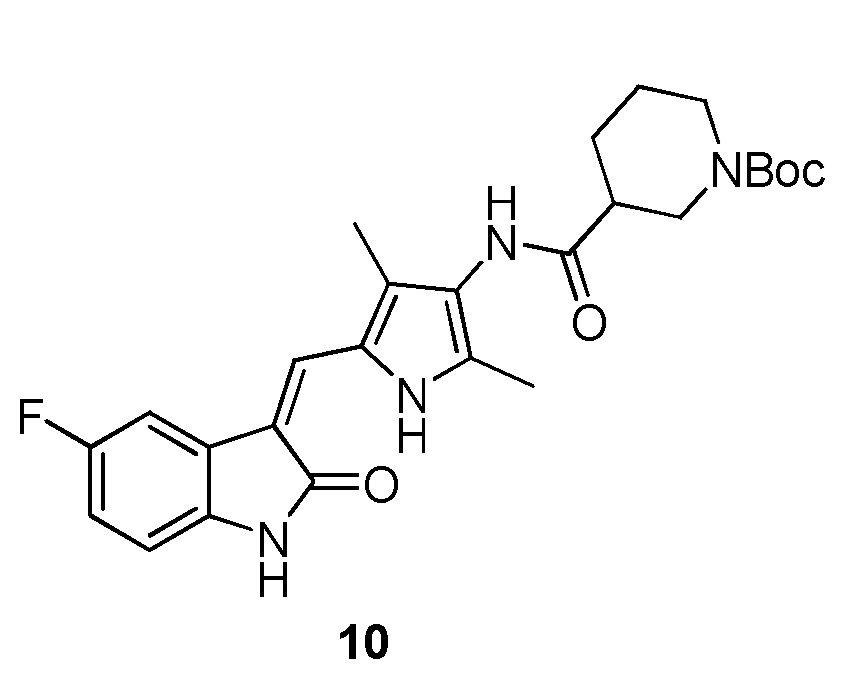
Синтез соединения 10 проводят по той же процедуре, которая использовалась для получения соединения 3, за исключением того, что здесь использовали соединение 4 вместо соединения 2, с получением целевого соединения 10. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,59 (с, 1H), 10,83 (с, 1H), 9,20 (с, 1H), 7,71 (дд, J=9,4, 2,5 Гц, 1H), 7,67 (с, 1H), 6,89 (дд, J=13,8, 6,7 Гц, 1H), 6,86-6,82 (м, 1H), 4,13-4,02 (м, 1H), 3,89 (д, J=13,1 Гц, 1H), 2,94-2,72 (м, 2H), 2,48-2,41 (м, 1H), 2,18 (с, 3H), 2,16 (с, 3H), 2,01-1,94 (м, 1H), 1,73-1,57 (м, 2H), 1,42 (с, 9H), 1,39-1,24 (м, 1H).
Пример получения 11: получение Соединения 11
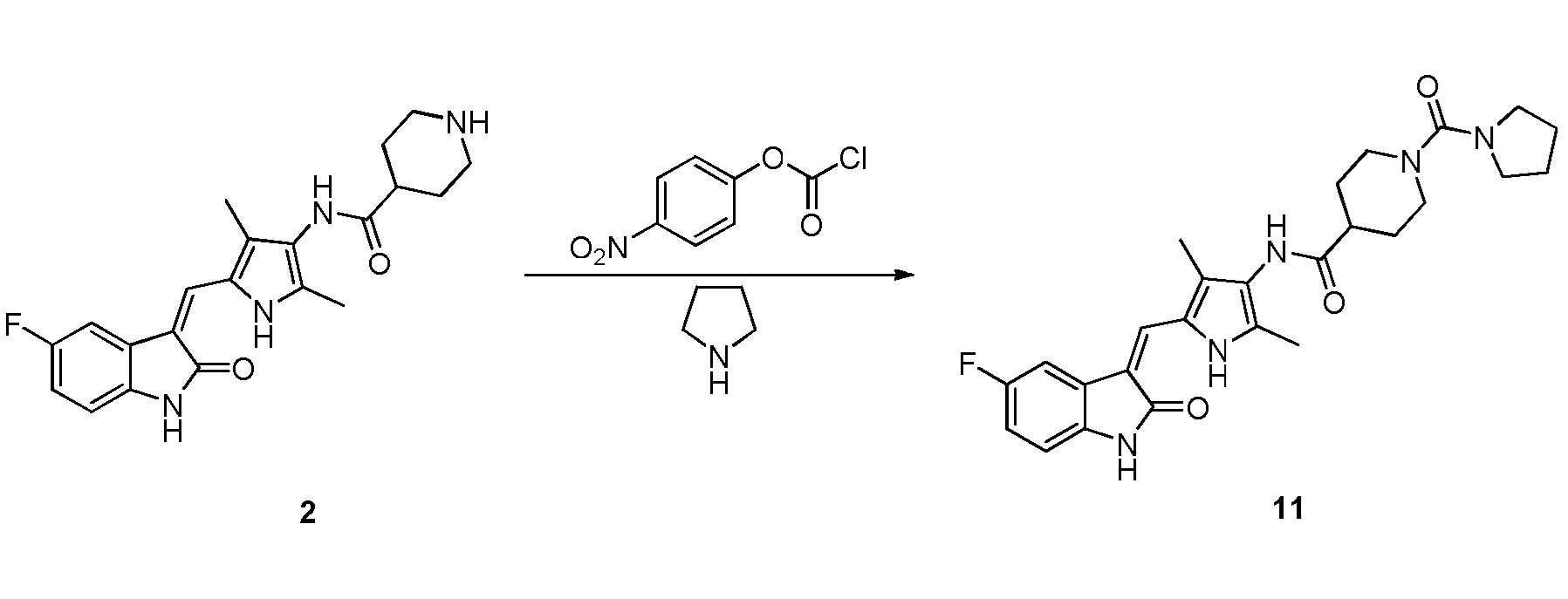
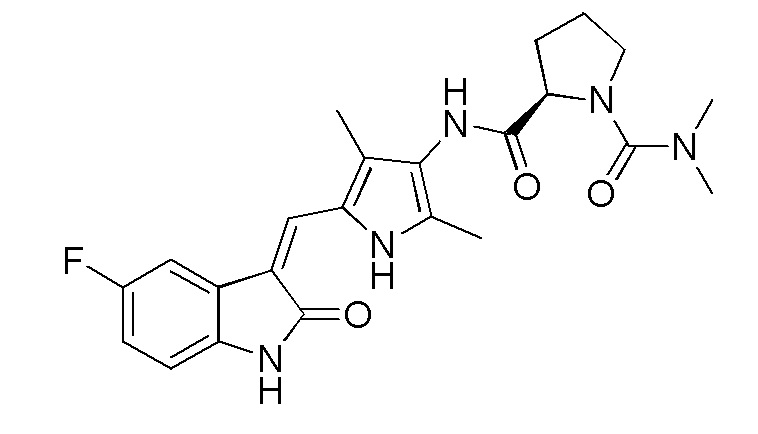
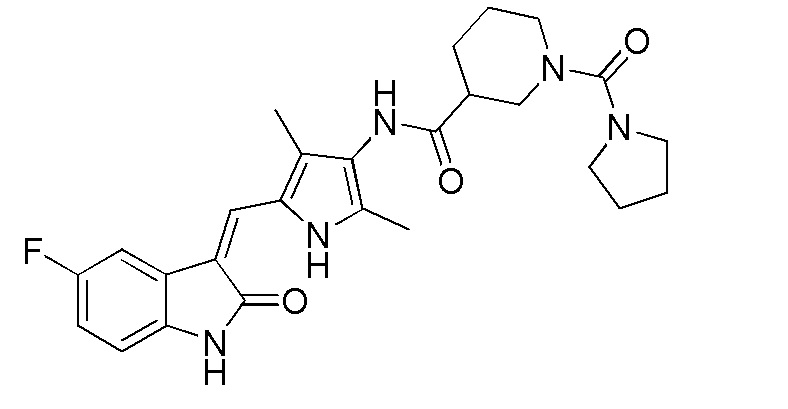
Соединение 2 (383 мг, 1 ммоль) добавляют к 20 мл тетрагидрофурана, и туда же добавляют, при комнатной температуре, ДИПЭА (DIPEA) (260 мг, 2 ммоль) и п-нитрофенилхлорформиат (240 мг, 1,2 ммоль). После добавления всех компонентов, проводят реакцию в течение примерно 12 часов, и по прошествии этого времени, когда реакция близка к завершению, проводят ТСХ для подтверждения того, что реакция завершилась. После завершения реакции, добавляют тетрагидропиррол (142 мг, 2 ммоль) и избыточное количество ДИПЭА (DIPEA) (260 мг, 2 ммоль), после чего проводят реакцию в течение периода времени, не менее 12 часов, отслеживая течение реакции по данных ТСХ. После завершения реакции, растворители выпаривают, а оставшийся твердый материал промывают 20 мл этилацетата и 10 мл метанола, с получением чистого продукта целевого соединения 11. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,58 (с, 1H), 10,82 (с, 1H), 9,09 (с, 1H), 7,70 (дд, J=9,4, 2,5 Гц, 1H), 7,66 (с, 1H), 6,93-6,86 (м, 1H), 6,83 (дд, J=8,5, 4,6 Гц, 1H), 3,70 (д, J=13,5 Гц, 2H), 3,27 (т, J=6,4 Гц, 4H), 2,73 (т, J=11,6 Гц, 1H), 2,18 (с, 3H), 2,15 (с, 3H), 1,86-1,71 (м, 6H), 1,67-1,54 (м, 2H).
Пример получения 12: получение Соединения 12
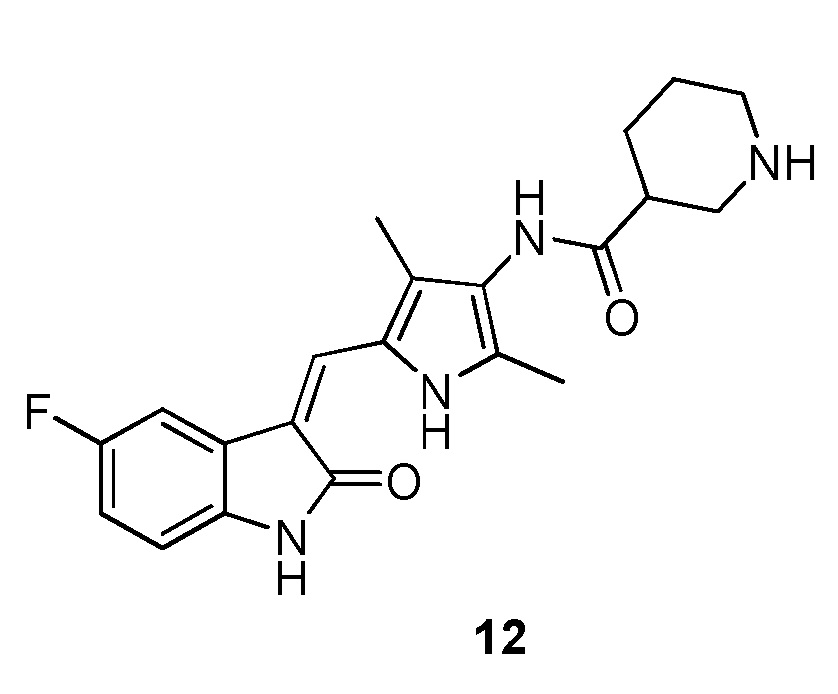
Синтез соединения 12 проводят по той же процедуре, которая использовалась для получения соединения 2, за исключением того, что здесь использовали соединение 10 вместо соединения 1, с получением целевого соединения 12. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,59 (с, 1H), 10,90 (с, 1H), 9,17 (с, 0H), 7,69 (дд, J=9,4, 2,4 Гц, 1H), 7,65 (с, 1H), 6,94-6,82 (м, 2H), 3,67-3,52 (м, 2H), 3,15-2,56 (м, 3H), 2,45 (д, J=10,0 Гц, 2H), 2,17 (с, 3H), 2,15 (с, 3H), 1,89 (д, J=9,2 Гц, 1H), 1,61 (д, J=10,4 Гц, 2H), 1,42 (с, 1H).
Пример получения 13: получение Соединения 13
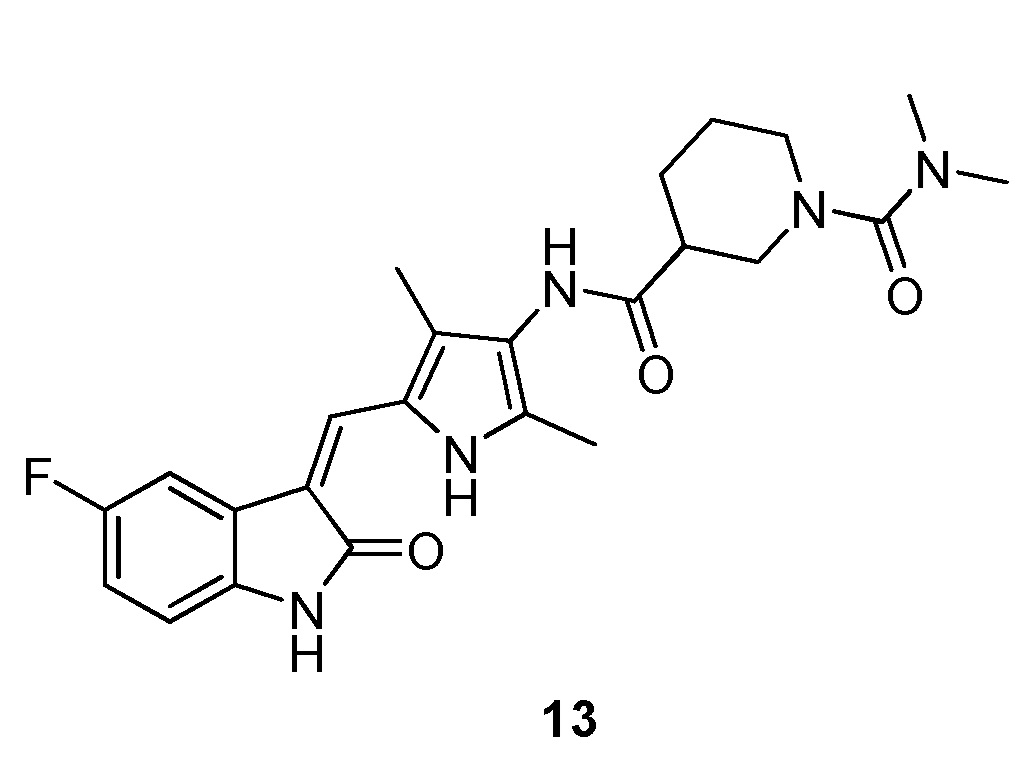
Синтез соединения 13 проводят по той же процедуре, которая использовалась для получения соединения 3, за исключением того, что здесь использовали соединение 12 вместо соединения 2, с получением целевого соединения 13. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,59 (с, 1H), 10,83 (с, 1H), 9,18 (с, 1H), 7,71 (дд, J=9,4, 2,4 Гц, 1H), 7,66 (с, 1H), 6,89 (дд, J=9,2, 2,4 Гц, 1H), 6,86-6,80 (м, 1H), 3,72-3,59 (м, 1H), 3,51 (д, J=13,2 Гц, 1H), 2,90-2,81 (м, 1H), 2,78-2,68 (м, 7H), 2,64-2,55 (м, 1H), 2,18 (с, 3H), 2,15 (с, 3H), 1,97 (д, J=14,9 Гц, 1H), 1,73-1,57 (м, 2H), 1,53-1,38 (м, 1H).
Пример получения 14: получение Соединения 14
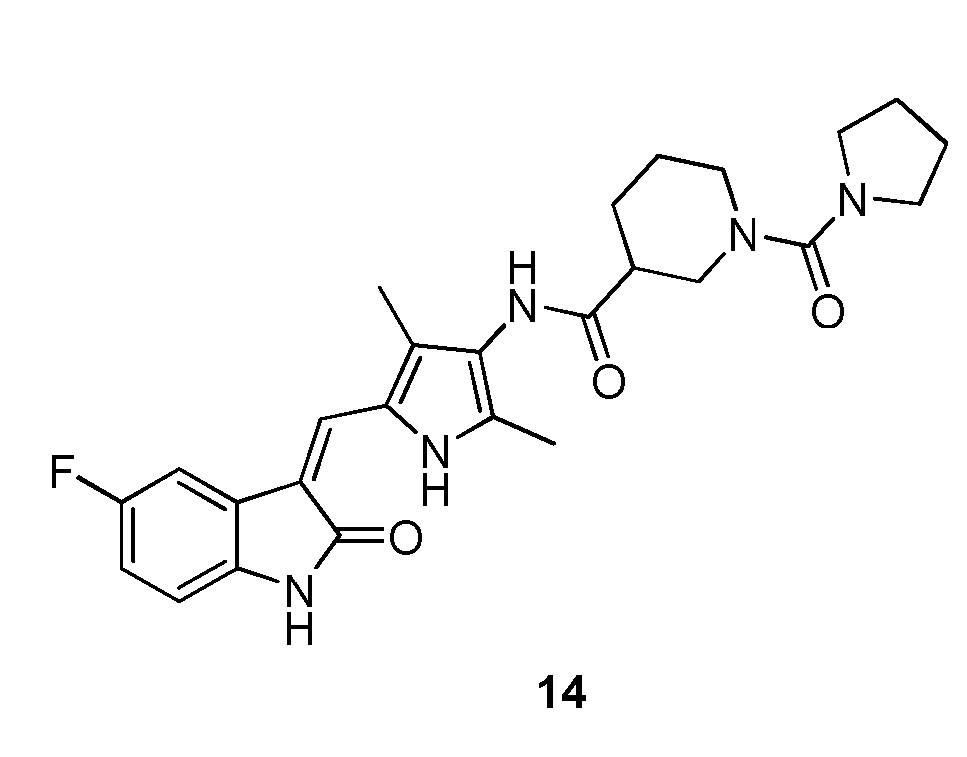
Синтез соединения 14 проводят по той же процедуре, которая использовалась для получения соединения 11, за исключением того, что здесь использовали соединение 12 вместо соединения 2, с получением целевого соединения 14. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,59 (с, 1H), 10,83 (с, 1H), 9,18 (с, 1H), 7,71 (дд, J=9,4, 2,5 Гц, 1H), 7,66 (с, 1H), 6,93-6,86 (м, 1H), 6,83 (дд, J=8,4, 4,8 Гц, 1H), 3,75 (д, J=12,7 Гц, 1H), 3,61 (д, J=12,4 Гц, 1H), 3,28 (с, 4H), 2,90-2,81 (м, 1H), 2,73 (т, J=11,4 Гц, 1H), 2,60-2,50 (м, 1H), 2,06-1,91 (м, 1H), 1,76 (с, 4H), 1,74-1,59 (м, 2H), 1,55-1,39 (м, 1H).
Пример получения 15: получение Соединения 15
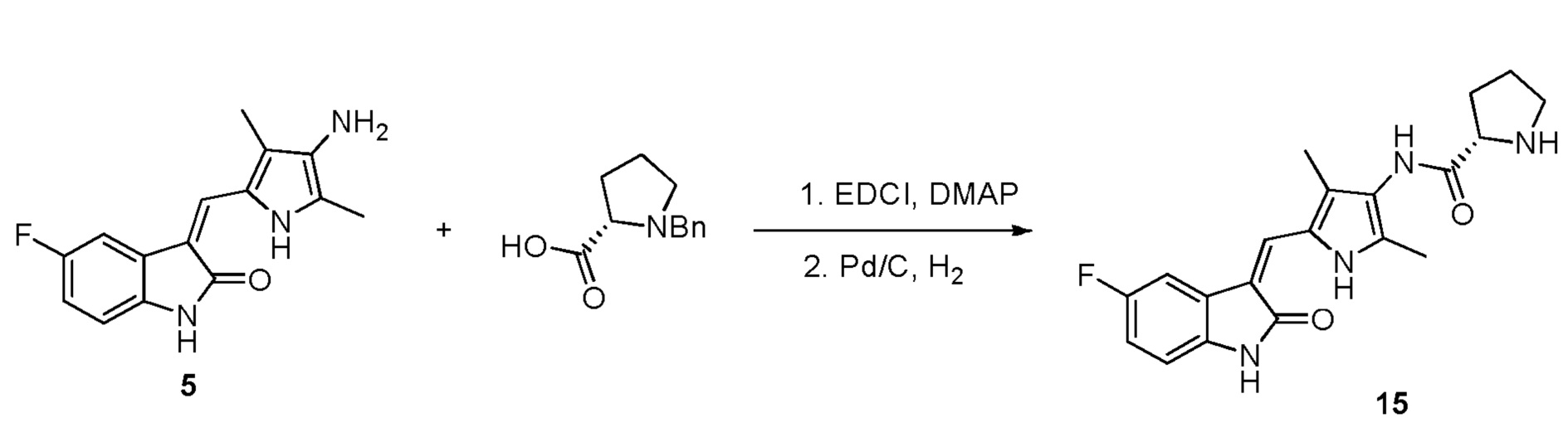
Данный синтез проводили по той же процедуре, которая использовалась для получения соединения 6, за исключением того, что здесь использовали D-N-Bn пролин вместо L-N-Bn пролина, с получением целевого соединения. 1H ЯМР (400 МГц, ДМСО) δ 13,60 (с, 1H), 10,83 (с, 1H), 9,23 (с, 1H), 7,71 (дд, J=9,3, 2,4 Гц, 1H), 7,67 (с, 1H), 6,93-6,87 (м, 1H), 6,83 (дд, J=8,4, 4,6 Гц, 1H), 3,70 (дд, J=8,7, 5,5 Гц, 1H), 2,91 (т, J=6,6 Гц, 1H), 2,18 (с, 2H), 2,16 (с, 2H), 2,10-1,98 (м, 1H), 1,85-1,74 (м, 1H), 1,72-1,63 (м, 2H).
Пример получения 16: получение гидрохлорида Соединения 6
0,5 мл насыщенного раствора HCl в этаноле разбавляют в 10 раз безводным этанолом и добавляют туда же Соединение 6 (368 мг, 1 ммоль), после чего перемешивают полученную смесь в течение 5-10 минут. Далее реакционный раствор концентрируют при пониженном давлении, промывают небольшим объемом метанола и получают в итоге гидрохлорид Соединения 6.
Гидрохлориды всех других соединений могут быть получены по указанному выше способу, в соответствии с которым проводится реакция соответствующего соединения с разбавленным раствором HCl в этаноле.
На основе указанных выше примеров получения пирролзамещенных производных индолона, могут быть получены также другие производные такого рода, с использованием для этого указанного выше способа.
Авторы настоящего изобретения также синтезировали следующие Сравнительные соединения 1-3 в рамках способа, аналогичного описанному выше, или с использованием других методик, известных в данной области.
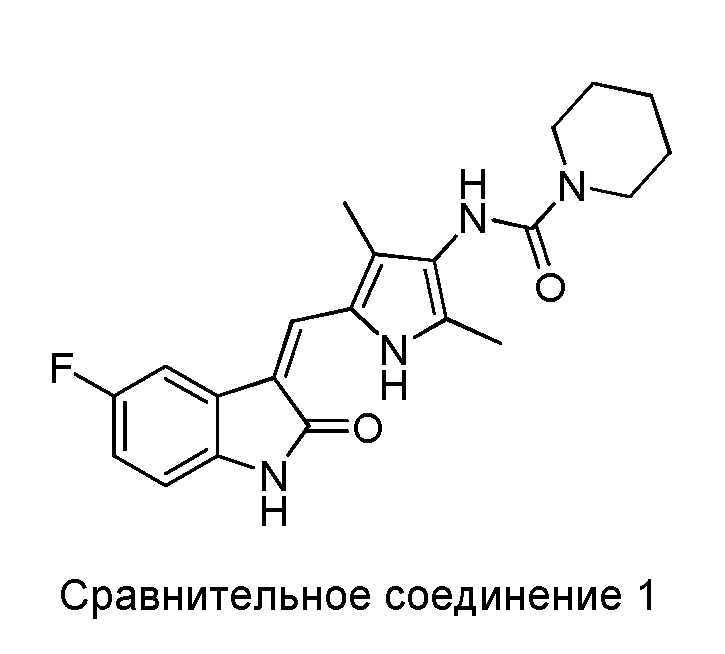
Сравнительное соединение 1 представляет собой соединение 2, за исключением того, что пиперидинил на дальней правой стороне присоединен к карбонилу через его атом N.
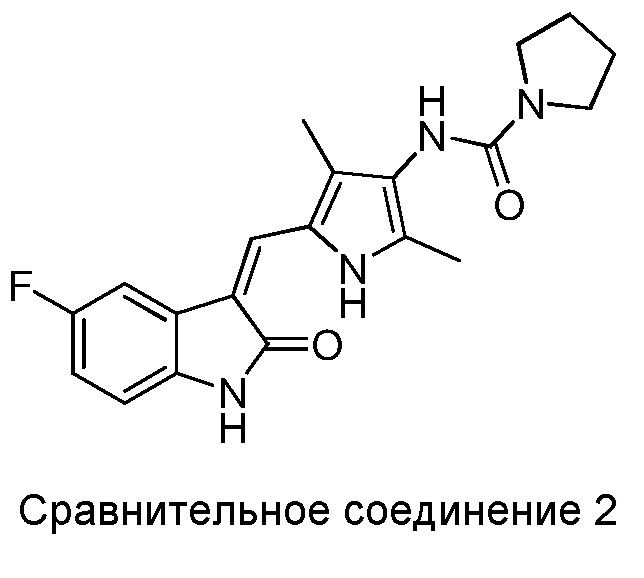
Сравнительное соединение 2 практически представляет собой соединение 6, за исключением того, что пирролидинил на дальней правой стороне присоединен к карбонилу через его атом N.
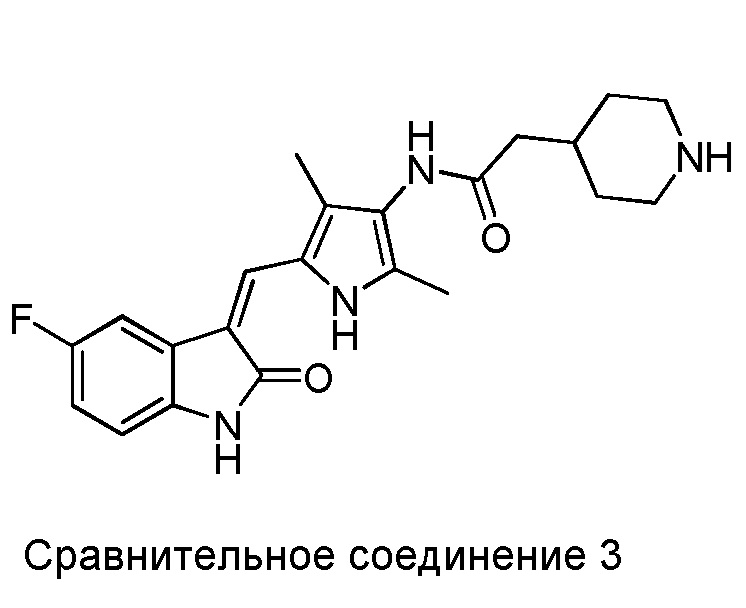
Сравнительное соединение 3 практически представляет собой соединение 2, за исключением того, что пиперидинил на дальней правой стороне присоединен к карбонилу через метиленовую группу.
Примеры
Далее настоящее изобретение будет описано с использованием конкретных примеров, но эти примеры никоим образом не следует трактовать как ограничивающие каким-либо образом настоящее изобретение.
Экспериментальный пример 1: In vitro тест для оценки биохимической активности тирозинкиназы KDR
Ингибирующую активность in vitro соединений на тирозинкиназу KDR (VEGF рецептор) оценивали по методу HTRF (гомогенная флуоресценция с временным разрешением). Смесь киназного буфера, исследуемое соединение или сунитиниб, субстрат и раствор АТФ добавляют до конечного объема 10 мкл в ячейки 384-ячеечного планшета, который инкубируют при комнатной температуре в течение соответствующего периода времени. К каждой ячейке добавляют 10 мкл SA-XL665 и TK антитела, все инкубируют при комнатной температуре в течение 1 часа и анализируют на Synergy2.
Полученные результаты показывают, что все соединения в указанных выше примерах обладали значительной ингибирующей активностью в отношении KDR в концентрациях 0,1 мкМ и 1 мкМ, а Соединения 3, 6, 8, 9, 11 и 13 действовали аналогично сунитинибу по активности.
|
Экспериментальный пример 2: Тест на цитотоксичность для HUVEC и оценка активности VEGF-индуцированной пролиферации HUVEC клеток in vitro
Тест на ингибирующую активность в отношении VEGF-индуцированной пролиферации линии эндотелиальных клеток пупочной вены человека (ЭКПВЧ (HUVEC)): клетки ЭКПВЧ (HUVEC) культивировали в среде F-12K, содержащей 10% ФСТ, 18 Ед/мл гепарина и 30 мкг/мл ECGS (лошадиный хорионический гонадотропин), и клетки ЭКПВЧ (HUVEC) после 4-8 пассажей отбирали для эксперимента. Указанные клетки расщепляли панкреатином, ресуспендировали в культуральной среде (1×105/мл) и вносили в ячейки 96-ячеечного планшета по 100 мкл/ячейку, после чего культивировали в течение ночи до получения адгезивной культуры. Культуральную среду заменяли культуральным раствором F-12K, содержащим 5% ФСТ, и далее клетки культивировали еще в течение 24 часов. Добавляли культуральный раствор F-12K с 5% ФСТ, содержащий исследуемое соединение, сунитиниб или контроль, и инкубировали в течение 30 минут. Добавляли культуральный раствор F-12K с 0,1% ФСТ, содержащий VEGF165 с конечной концентрацией 30 нг/мл или носитель (ДМСО), и клетки культивировали в условиях индукции в течение 72 часов. Культуральный раствор отбирали пипеткой и добавляли к каждой ячейке по 120 мкл раствора для тестирования MTS, и все инкубировали при температуре 37°C. Определяли показатель поглощения OD490. Группа, для которой использовали культуральный раствор с 5% ФСТ, служила в качестве отрицательного контроля. Показатель VEGF-стимулированного роста получали при вычитании значения OD для группы, используемой в качестве отрицательного контроля, из показателя OD для группы с VEGF165-стимуляцией, и это значение использовали для расчета уровня ингибирования. Далее, строят кривую доза-эффект с использованием программного обеспечения GraphPad Prism software, и на ее основе вычисляют половинное значение эффективной концентрации (ЭК50).
Тесты на цитотоксичность: указанные выше клетки ЭКПВЧ (HUVEC) культивируют в культуральной среде F-12K, содержащей 10% фетальной сыворотки теленка (ФСТ (FBS)), 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина, 30 мкг/мл ECGS (лошадиный хорионический гонадотропин) и 18 Ед/мл гепарина. Клетки ЭКПВЧ (HUVEC) на экспоненциальной фазе роста расщепляют панкреатином и корректируют плотность клеток до соответствующего уровня с использованием полной среды F-12K, содержащей 5% ФСТ, затем 150 мкл клеток инокулируют в ячейки 96-ячеечного планшета по 3000 клеток/ячейку. Через 24 часа, добавляют 50 мкл исследуемого соединения, разбавленного в 4 раза полной средой, содержащей 5% ФСТ, и такой же объем разбавленного раствора ДМСО используют в качестве контроля. Далее клетки культивируют в течение 72 часов, после чего к каждой ячейке добавляют по 20 мкл MTS и по 1 мкл PMS. Через 1-2 часа, измеряют значение поглощения OD490, с использованием OD650 в качестве эталонного значения. Далее строят кривую доза-эффект с использованием программного обеспечения GraphPad Prism и рассчитывают половинное значение концентрации, вызывающей цитотоксический эффект(CC50). А также рассчитывают терапевтический индекс (TI) исследуемого соединения для ЭКПВЧ (HUVEC)клеток по формуле TI=CC50/EC50.
Полученные результаты показывают, что все соединения из приведенных примеров могут в значительной мере ингибировать VEGF-стимулированную пролиферацию ЭКПВЧ (HUVEC), демонстрируя при этом сниженную активность, в сравнении с сунитинибом. Однако, некоторые из показанных соединений (Соединения 2, 3, 5, 6, 8, 11, 13, 14 и 15) вызывали намного меньшую цитотоксичность для клеток ЭКПВЧ (HUVEC), чем это наблюдается в случае сунитиниба. Показатели терапевтического индекса (TI) для соединений 2, 3, 5, 6, 8, 11, 14 и 15 в 2-3 раза превосходили таковой для сунитиниба, и при этом отмечалось увеличенное терапевтическое окно.
Было показано, что в случае сравнительных соединений 1 и 2, за счет того, что в них N-содержащее гетероциклическое кольцо на дальней правой стороне присоединено к карбонилу через гетероатом, показатель терапевтической эффективности (TI) в основном такой же, как и у сунитиниба и, соответственно, значительно ниже, чем у соединений по настоящему изобретению. В случае сравнительного соединения 3, за счет того, что в нем N-содержащее гетероциклическое кольцо на дальней правой стороне присоединено к карбонилу через метиленовую группу, его показатель терапевтической эффективности (TI) в основном такой же, как и у сунитиниба и, соответственно, значительно ниже, чем у соединений по настоящему изобретению.
|
Экспериментальный пример 3: Тесты на ингибирующую активность в отношении пролиферации линии опухолевых клеток MV-4-11, полученных от человека
Линия леток острого лейкоза MV-4-11, полученных от человека, представляет собой клеточную линию, в которой клетки содержат одну или несколько мутаций в Flt-3. Указанную анти-пролиферирующую активность in vitro соединений в отношении MV-4-11 оценивали по методу MTS: клетки в экспоненциальной фазе роста расщепляли панкреатином и затем подсчитывали их число; далее, соответствующее количество клеток ресуспендировали в культуральном растворе, вносили в ячейки 96-ячеечного планшета, по 150 мкл/ячейку, и культивировали в течение ночи; затем к каждой ячейке добавляли по 50 мкл культурального раствора, содержащего исследуемое соединение в 4-кратном разбавлении (при проведении ступенчатого разбавления) или контроль и проводили культивирование в течение 72 часов; после этого культуральный раствор отбирали пипеткой и к каждой ячейке добавляли 120 мкл раствора для MTS тестирования (100 мкл свежей среды и 20 мкл MTS раствора), смесь инкубировали при температуре 37°C; определяли значение OD490; и полученные данные анализировали и подвергали дальнейшей обработке с использованием программного обеспечения GraphPad Prism5, для расчета значения ИК50 (IC50).
Полученные результаты показали, что все приведенные в примерах соединения 1-15 демонстрировали выраженную антипролиферативную активность в отношении MV-4-11, и некоторые из этих соединений имели активность, близкую к активности сунитинба или даже превышающую активность сунитиниба (см. приведенную ниже таблицу). FLT-3 (FMS-подобная тирозинкиназа 3) представляет собой рецептор тирозинкиназы типа III, широко распространенный в ряде систем, в иммуной системе и в нервной системе. Мутации в FLT-3 гене и сверхэкспрессия FLT-3 может вызвать опухолегенез. Специфическая антипролиферативная активность Соединений 1-15 в отношении MV-4-11 также указывает на то, что соединения из данного примера аналогичные суритинибу, в том аспекте, когда он проявляет активность FLT-3 ингибитора.
|
Экспериментальный пример 4: Ингибирование in vivo MV-4-11 опухоли, трансплантированной в мышей nude
Клетки MV-4-11 культивировали для пролиферации in vitro, затем собирали клетки в экспоненциальной фазе роста и ресуспендировали их в бессывороточной культуральной среде EMEM. Указанную суспензию клеток инъецировали подкожно шприцом в подмышечную ямку передней конечности самцов мышей Balb/c nude. Регулярно проводили оценку животных и роста трансплантированной опухоли. Когда объем опухоли достигал размеров примерно 100-300 мм3, отбирали животных с опухолями соответствующего размера и случайным образом распределяли их по группам, по 6 животных на группу. Животным в каждой группе вводили внутрижелудочно носитель, используемый в качестве слепой пробы (0,5% CMC), или суспензию Соединения 6 из приведенного выше примера, или сунитиниб в дозе 80 мг/кг, один раз в день, в течение всего периода введения, равного 3 неделям. В ходе всего периода введения, измеряли диаметр опухоли и вес тела (ВТ) животных, и регулярно отслеживали показатели жизнедеятельности животных. Эксперимент закончили через 3 недели после начала введения, всех животных умертвили с использованием CO2 и провели аутопсию.
Объем опухоли (ОО (TV)) рассчитывали по формуле TV=1/2×a×b2, где a обозначает более длинный диаметр опухоли и b обозначает более короткий диаметр опухоли.
Полученные результаты показывают, что к 21 дню внутрижелудочного введения, опухоли у животных в группе контрольного введения носителя выросли примерно в 6 раз в объеме относительно исходного объема, тогда как опухоли у животных в группе введения Соединения 6 полностью исчезали, при этом Соединение 6 не оказывало значительного влияния на вес тела животных. Хотя сунитиниб также оказывал заметный противоопухолевый эффект, так что у большинства животных опухоли исчезли, вес тела у этих животных существенно снизился и наблюдалась токсичность.
|
Как видно из приведенных результатов экспериментов с использованием трансплантированной опухоли клеток MV-4-11 мышам nude, Соединение 6 по настоящему изобретению оказывает хороший ингибирующий эффект на трансплантированные опухоли клеток MV-4-11, и в использованной дозе 80 мг/кг может приводить к полному исчезновению опухолей, оказывая при этом незначительное влияние на вес тела. Сунитиниб в значительной мере снижает вес тела животных и вызывает выраженную токсичность. Полученные результаты показывают, что соединения по настоящему изобретению оказывают противоопухолевый эффект, сравнимый с таковым у сунитиниба, но вызывают меньшую токсичность, имеют большее терапевтическое окно и, соответственно, более ценны для разработки соответствующего препарата.
Производное бутилфталида, способ его получения и применение
Способ определения содержания ментола в композиции традиционного китайского лекарственного средства
Пылеугольный концентратор и пылеугольная горелка, содержащая этот концентратор
Бензиловые производные гликозидов и способы их применения
Производные бензилфенилциклогексана и способы их применения
Дейтерированные бензилбензольные производные и способы применения
Фармацевтическая композиция, содержащая ephedrae herba, для лечения бронхита, и способ ее получения
Производное сложного эфира тиенопиридина, содержащее цианогруппу, способ его получения, его применение и композиция на его основе
Зарядная станция и интеллектуальная транспортная система
Блок отображения на основе органических светоизлучающих диодов, способ его возбуждения и устройство отображения
Способ снижения оксидов азота в котле, работающем на распыленном угле, в котором используются горелки типа внутреннего сгорания
Способ функционирования сотовой сети, включающий в себя высокочастотную пакетную передачу

