Результат интеллектуальной деятельности: Синтетическое промежуточное соединение максакальцитола, способ его получения и его применение
Вид РИД
Изобретение
Данная заявка испрашивает приоритет согласно китайской патентной заявкой CN 201310475989.7, поданной 12 октября 2013 года, описание которой включено в настоящее описание посредством ссылки во всей ее полноте.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к способу получения лекарственного средства, в частности, настоящее изобретение относится к способу получения Максакальцитола, к его новому синтетическому промежуточному соединению, к способу его получения и его использованию.
УРОВЕНЬ ТЕХНИКИ
Максакальцитол (Максакальцитол, CAS №103909-75-7), химическая формула которого по-английски представляет собой: 22-Оксакальцитриол; (1R,3S,5Z)-4-метилен-5-[(2Е)-2-[(1S,3aS,7aS)-октагидро-1-[(1S)-1-(3-гидрокси-3-метилбутокси)этил]-7а-метил-4Н-инден-4-илиден]этилиден]-1,3-циклогександиол, представляет собой третье поколение активного лекарственного средства на основе витамина D3, разработанного Chugai Pharmaceutical Co., Ltd., впервые появившееся на рынке в Японии в 2000 году; его инъекцию (торговое наименование: Oxarol) используют для лечения вторичного гиперпаратиреоза у почечных больных, находящихся на гемодиализе (ВГПТ); его мазь (торговое наименование: Oxarol) используют для лечения сухих опоясывающих кожных заболеваний, таких как псориаз. В настоящее время заявки, относящиеся к его синтезу, включают WO 2012/122451, WO 2001079166, US 5436401, CN 102796134 и JP 20111573261.
В патенте США US 5436401 A описан способ получения Максакальцитола, где в качестве исходного материала используют 1α-гидроксидегидроэпиандростерон, а Максакальцитол получают за счет модификации в боковой цепи и в кольце А, с раскрытием кольца В с помощью фотохимической реакции и перегруппировки в условиях нагревания. Однако 1α-гидроксидегидроэпиандростерон получают путем микробиологической ферментации, что в значительной степени ограничивает источник исходного материала, а способ получения включает множество стадий реакции, некоторые из которых имеют относительно низкие выходы, что не подходит для промышленного производства.
В WO 2012/122451 описан значительно улучшенный способ получения Максакальцитола с уменьшенным количеством стадий реакции за счет введения продукта в качестве исходного материала, который получают путем подходящей модификации аналогичного соединения витамина D2. Однако в улучшенном способе используют NaBH4 при реакции восстановлении кетона в положении С-20, основным продуктом которой является продукт с противоположной конфигурацией, что в значительной степени ограничивает применение способа.
В CN 102796134, в основном, нацелены на сокращение процесса, описанного в WO 2012/122451, направляя внимание на улучшение реакции восстановления кетона в положении С-20, раскрытой в WO 2012/122451, и получают продукт с одной конфигурацией с помощью асимметричного восстановления.
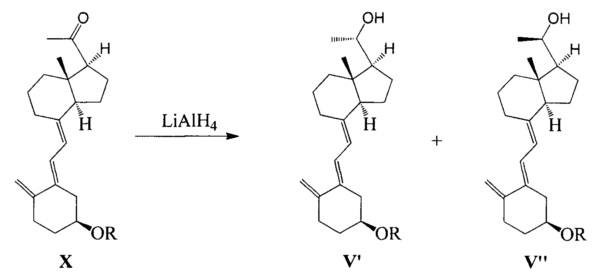
В JP 20111573261 используют витамин D2 в качестве исходного вещества, и получают соединение X в соответствии со способом, описанном в US 4866048, соединение X превращают в соединение V' (S-конфигурация) и Vʺ (R-конфигурация) в соотношении 35:65 при действии алюмогидрида лития, соединение V (S-конфигурация) является целевой конфигурацией (с выходом только 24%), эффективность синтеза является слишком низкой.
С учетом недостатков в предшествующем уровне техники чрезвычайно важно найти способ синтеза с небольшим количеством стадий, более высоким выходом и более низкой стоимостью.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Одной из задач настоящего изобретения является создание нового ключевого промежуточного соединения (соединение III, IV, VI) и способа его получения.
Другой задачей настоящего изобретения является создание нового способа получения Максакальцитола с использованием ключевого промежуточного соединения.
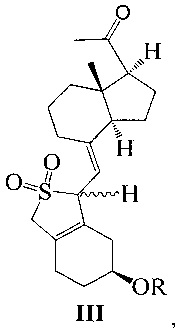
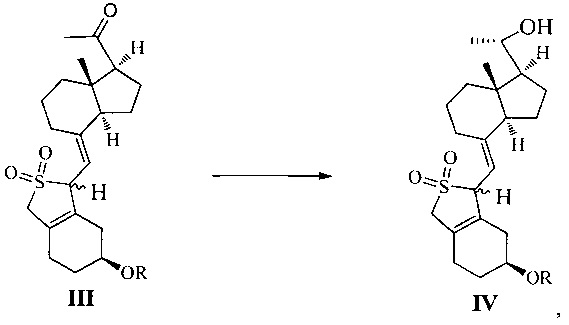
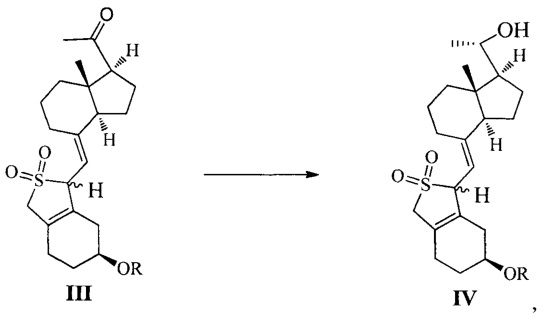
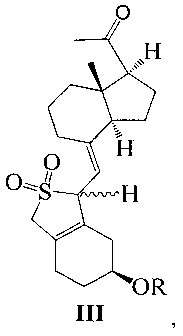
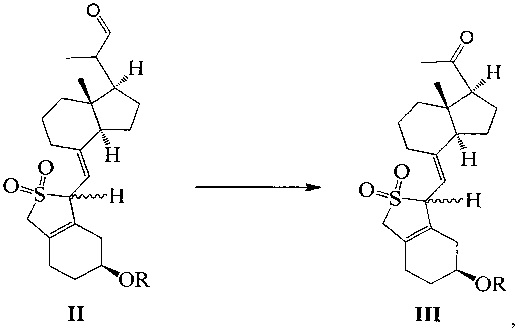
Одним из аспектов настоящего изобретения является создание нового промежуточного соединения формулы III, используемого для синтеза Максакальцитола:
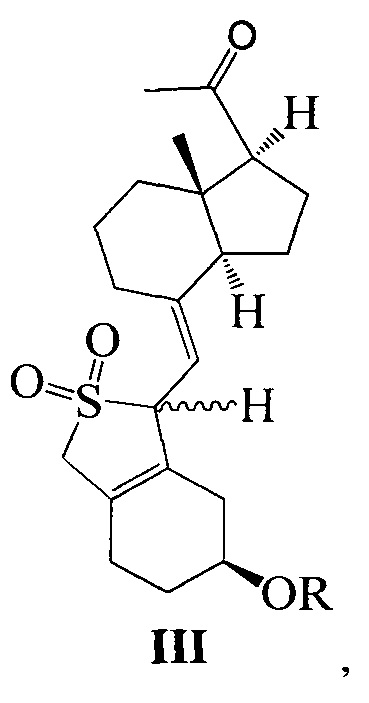
где R представляет собой Н или гидроксильную защитную группу, которая включает простую эфирную силильную защитную группу, предпочтительно представляющую собой трет-бутилдиметилсилил, триметилсилил, триэтилсилил, трет-бутилдифенилсилил или триизопропилсилил.
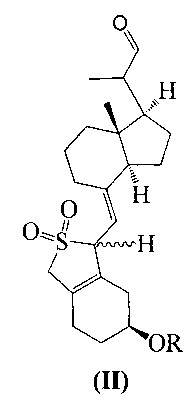
Другим аспектом настоящего изобретения является создание способа получения соединения III, включающего окисление соединения II в присутствии катализатора окислительным агентом с получением соединения III, где R определен, как указано выше:
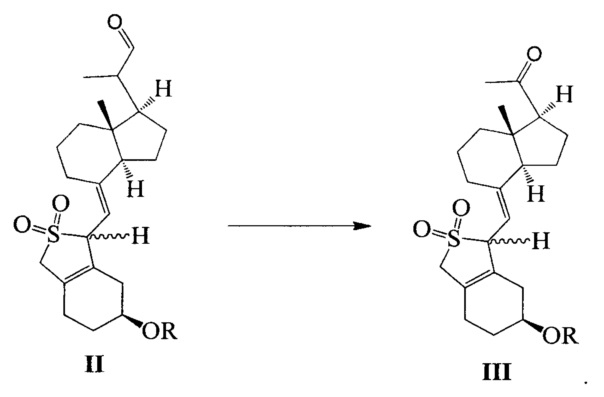
В предпочтительном воплощении настоящего изобретения окислительный агент в реакции окисления предпочтительно представляет собой кислород; катализатор предпочтительно представляет собой медный катализатор, более предпочтительно 2,2-бипиридильный комплекс меди.
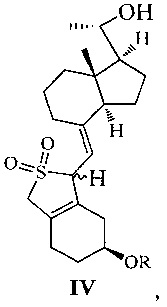
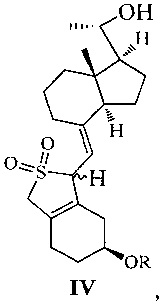
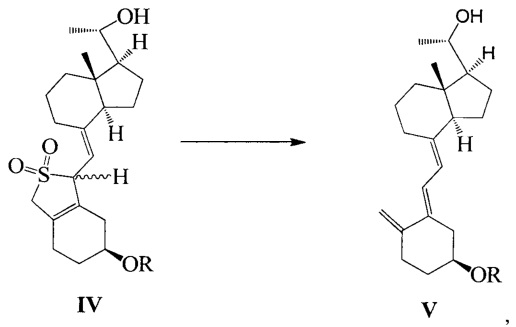
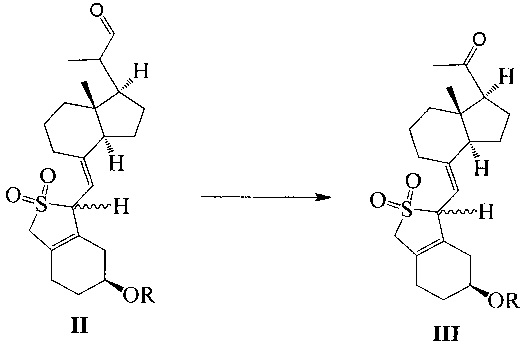
Другим аспектом настоящего изобретения является создание нового промежуточного соединения формулы IV, используемого для синтеза Максакальцитола:

где R представляет собой Н или гидроксильную защитную группу, которая включает простую эфирную силильную защитную группу, предпочтительно представляющую собой трет-бутилдиметилсилил, триметилсилил, триэтилсилил, трет-бутилдифенилсилил или триизопропилсилил.
Еще одним аспектом настоящего изобретения является создание способа получения соединения IV, включающего:
стереоселективное восстановление соединения III в присутствии хирального вспомогательного реагента с получением соединения IV со специфической конфигурацией путем использования борана, где R определен, как указано выше:
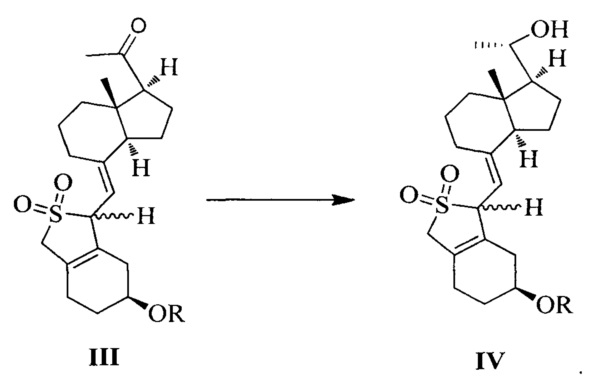
В предпочтительном воплощении по настоящему изобретению хиральный вспомогательный реагент, используемый в реакции, предпочтительно выбран из (R)-2-метил-CBS-оксазаборолидина, (R)-2-этил-CBS-оксазаборолидина или (R)-2-изопропил-CBS-оксазаборолидина; боран, используемый в реакции, предпочтительно выбран из ВН3, комплекса борана с тетрагидрофураном, комплекса борана с триэтиламином, комплекса борана с диэтиловым эфиром, комплекса борана с метил сульфидом или комплекса борана с N,N-диэтиланилином.
В предпочтительном воплощении по настоящему изобретению молярное соотношение между соединением III, хиральным вспомогательным реагентом и бораном предпочтительно составляет 1:(0,1-1):(1-2), более предпочтительно 1:0,6:1.
В предпочтительном воплощении по настоящему изобретению температура реакции составляет предпочтительно от -60°C до 0°C, более предпочтительно -20°C.
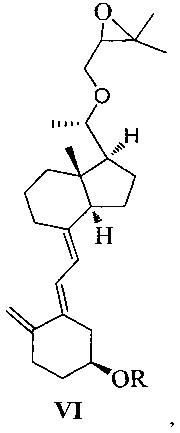
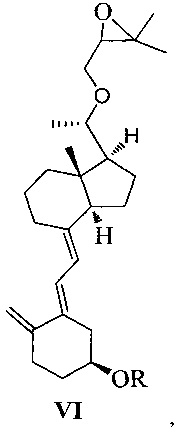
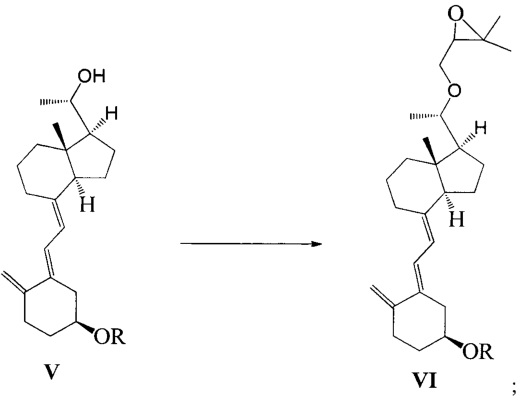
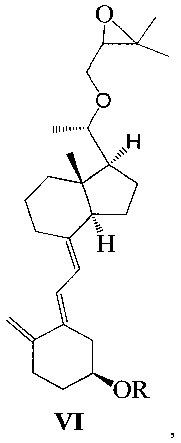
Другим аспектом настоящего изобретения является создание нового промежуточного соединения формулы VI для синтеза Максакальцитола:
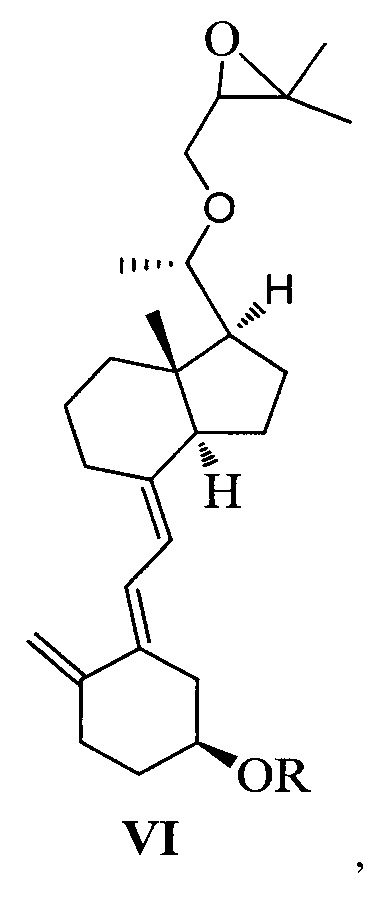
где R представляет собой Н или гидроксильную защитную группу, которая включает простую эфирную силильную защитную группу, предпочтительно представляющую собой трет-бутилдиметилсилил, триметилсилил, триэтилсилил, трет-бутилдифенилсилил или триизопропилсилил.
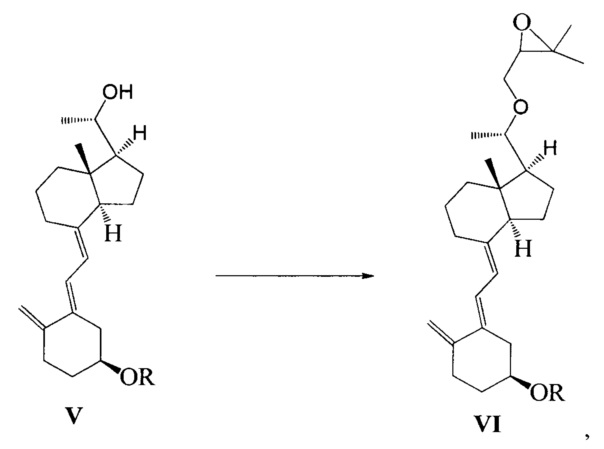
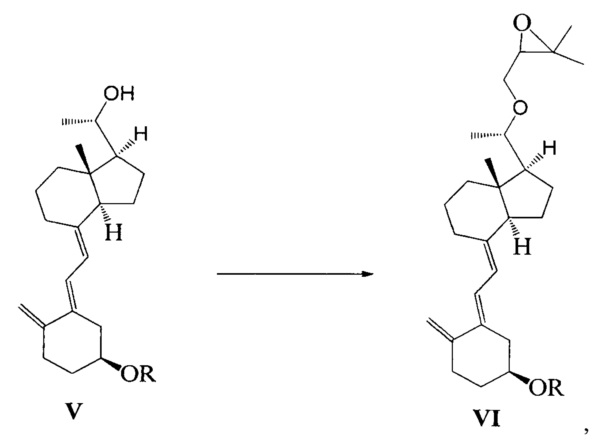
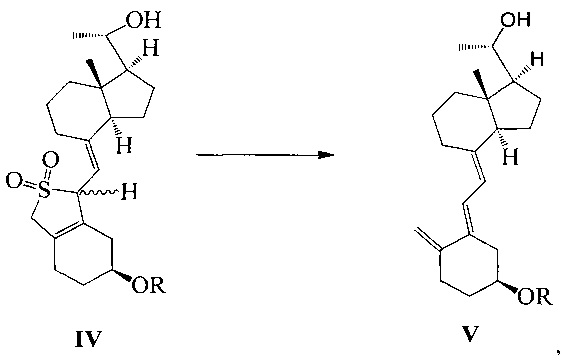
Другим аспектом настоящего изобретения является создание способа получения соединения VI, включающего:
Стадию 1: превращение соединения IV в соединение V в щелочной среде:
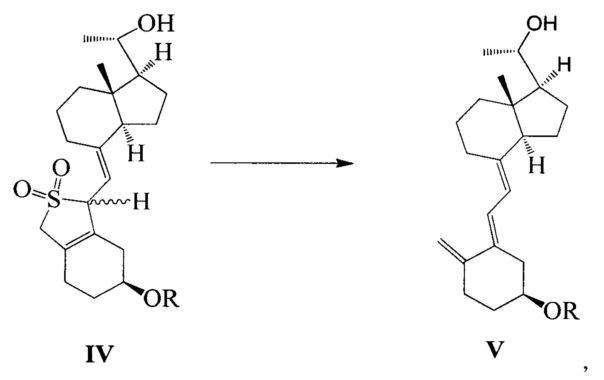
где R представляет собой гидроксильную защитную группу;
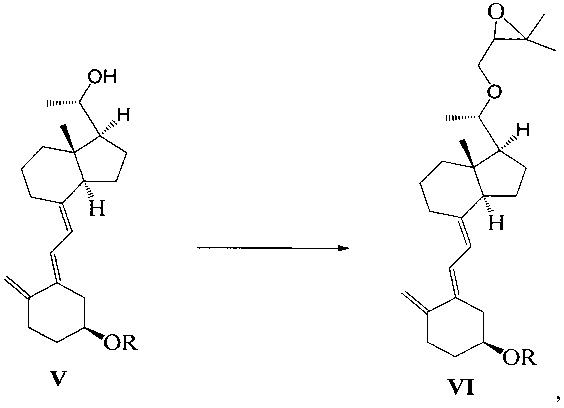
Стадию 2: взаимодействие соединения V с 3-бромметил-2,2-диметилоксираном с получением соединения VI:
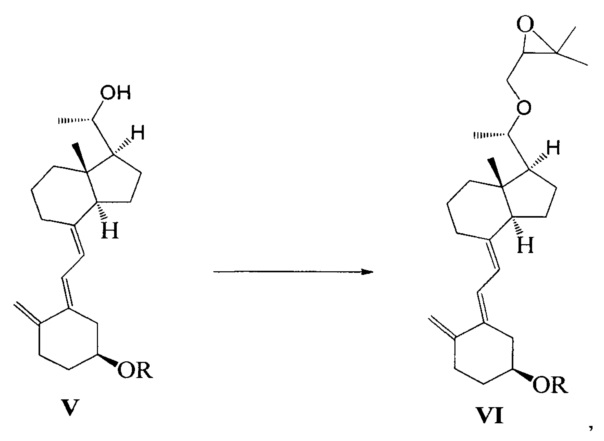
где R представляет собой гидроксильную защитную группу.
Способ получения соединения VI, если необходимо, может дополнительно включать: удаление гидроксильной защитной группы R из соединения VI, которое получают на стадии 2, давая соединение VI:
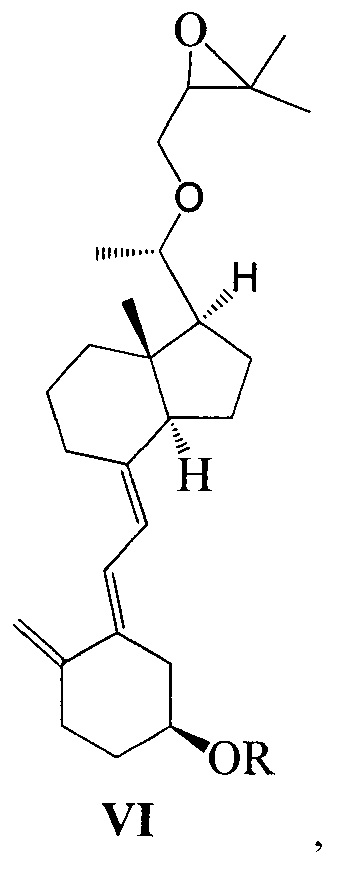
где R представляет собой Н.
При этом щелочь на стадии 1 включает бикарбонат натрия или ацетат натрия.
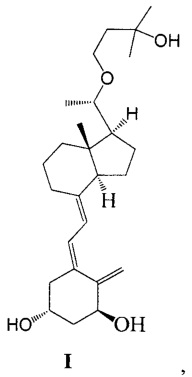
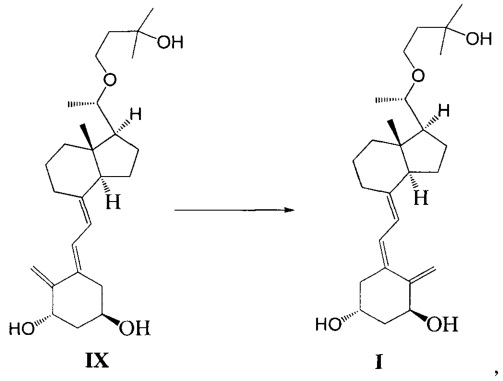
Другим аспектом настоящего изобретения является создание способа получения Максакальцитола формулы I:
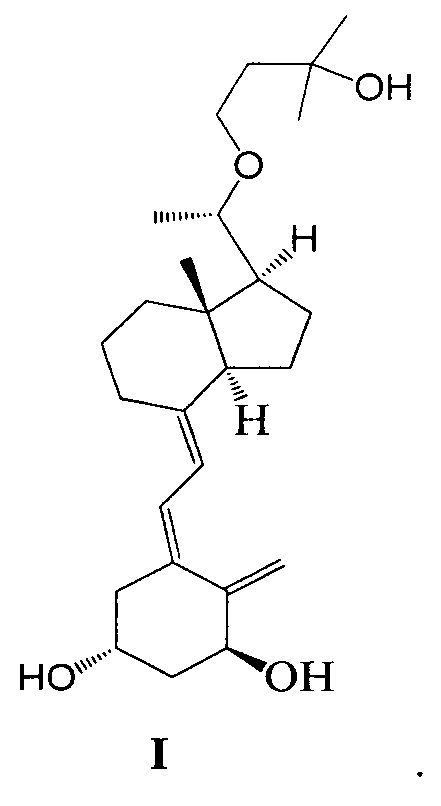
Способ получения включает:
Стадию 1: превращение соединения IV в соединение V в щелочной среде:
где R представляет собой гидроксильную защитную группу;
Стадию 2: взаимодействие соединения V с 3-бромметил-2,2-диметилоксираном с образованием соединения VI:
где R представляет собой гидроксильную защитную группу;
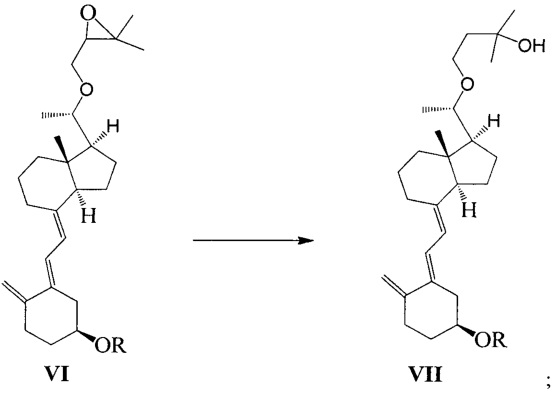
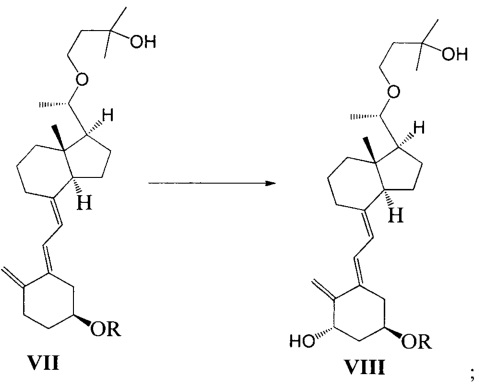
Стадию 3: превращение соединения VI в соединение VII в присутствии триизобутилгидробората лития:
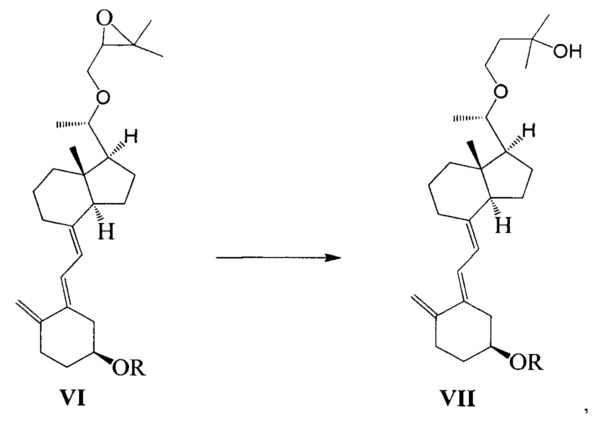
где R представляет собой гидроксильную защитную группу.
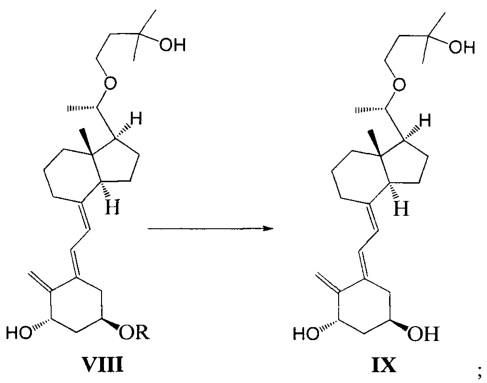
Стадию 4: взаимодействие соединения VII под действием N-метилморфолин-N-оксида и диоксида селена с получением соединения VIII:
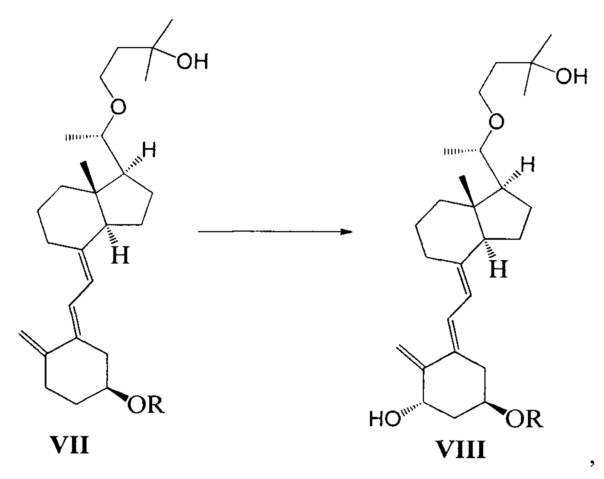
где R представляет собой гидроксильную защитную группу.
Стадию 5: удаление гидроксильной защитной группы в соединении VIII с образованием соединения IX:
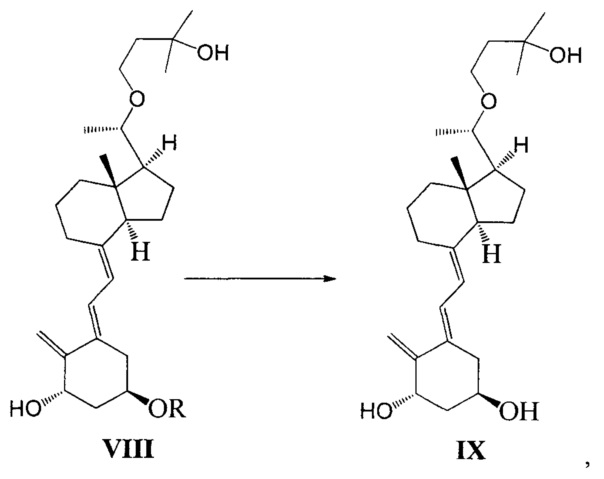
где R представляет собой гидроксильную защитную группу.
Стадию 6: проведение фотохимической реакции с соединением IX с образованием Максакальцитола формулы I:
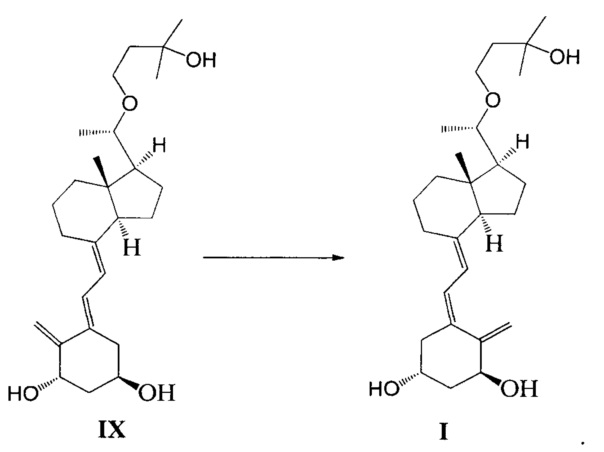
При этом щелочь на стадии 1 включает бикарбонат натрия или ацетат натрия.
В одном воплощении настоящего изобретения предложен способ получения Максакальцитола, который включает:
проведение фотохимической реакции посредством УФ-облучения соединения IX, используя в качестве катализатора 9-ацетилантрацен, чтобы инвертировать сопряженную двойную связь:
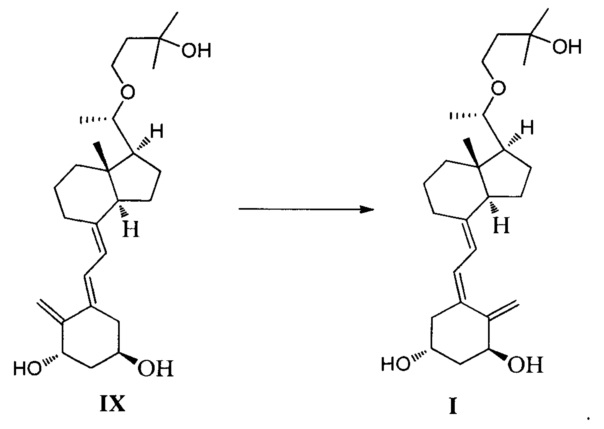
В реакции массовое соотношение соединения IX к 9-ацетилантрацену предпочтительно составляет 1:(0,05-1), более предпочтительно 1:0,1.
Продолжительность реакции может составлять от 0,5 до 5 ч, предпочтительно 2 ч.
Температура реакции предпочтительно составляет от 0°C до 10°C.
Реакцию можно проводить в подходящем органическом растворителе, который может быть любым подходящим растворителем, включая метанол, этанол, ацетон, диоксан, ацетонитрил, ТГФ, но не ограничиваясь ими.
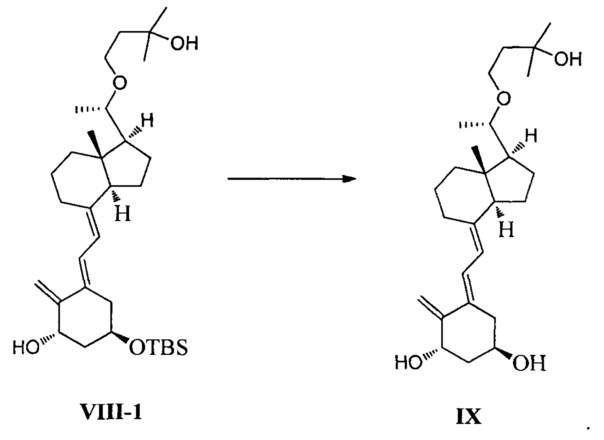
В еще одном предпочтительном воплощении настоящего изобретения соединение IX может быть получено согласно способу получения, как показано ниже:
удаление защитной группы в соединении VIII-1 в присутствии фторида тетрабутиламмония:
В реакции молярное соотношение между соединением VIII-1 и тетрабутиламмонийфторидом предпочтительно составляет 1:1~3, более предпочтительно 1:1,5.
Продолжительность реакции может быть от 5 ч до 40 ч, предпочтительно 10 ч.
Температура реакции предпочтительно составляет 65°C.
Реакцию можно проводить в подходящем органическом растворителе, который может быть любым подходящим растворителем, включая метанол, этанол, ацетон, диоксан, ацетонитрил, ТГФ, но не ограничиваясь ими, предпочтительно ТГФ.
В еще одном предпочтительном воплощении настоящего изобретения соединение VIII-1 может быть получено в соответствии со способом получения, как показано ниже:
взаимодействие соединения VII-1 под действием N-метилморфолин-N-оксида и диоксида селена:
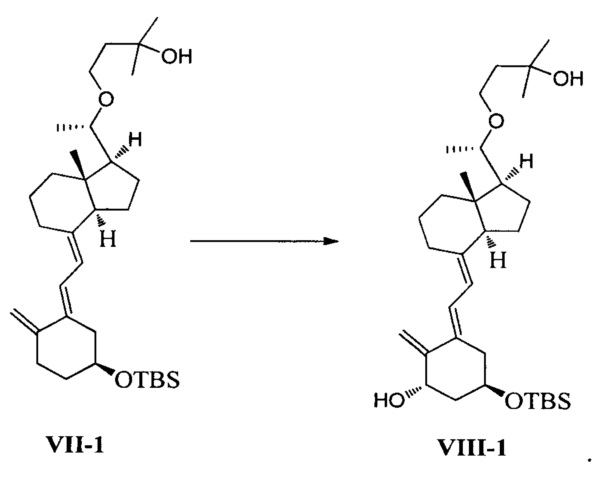
В реакции молярное соотношение между соединением VII-1, N-метилморфолин-N-оксидом и диоксидом селена составляет предпочтительно 1:(1-3):(0,2-1), более предпочтительно 1:2:0,4.
Продолжительность реакции может быть от 2 до 24 часов, предпочтительно 8 ч.
Температура реакции предпочтительно составляет 35°C.
В еще одном предпочтительном воплощении настоящего изобретения соединение VI1-1 может быть получено в соответствии со следующим способом получения:
взаимодействие соединения VI-1 в присутствии триизобутилгидробората лития:
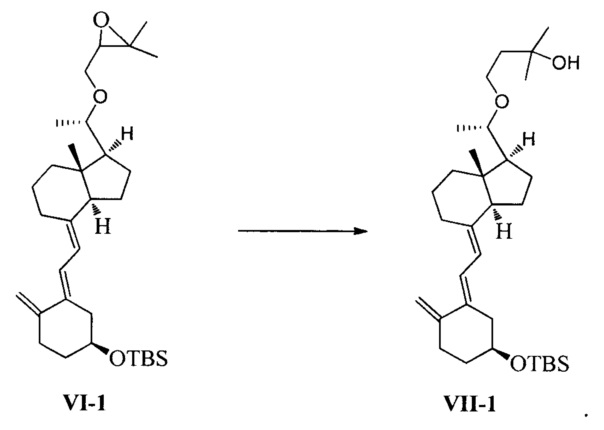
В реакции молярное отношение между соединением VI-1 и триизобутилгидроборатом лития предпочтительно составляет 1:(1-3), более предпочтительно 1:1,5.
Продолжительность реакции может быть от 1 ч до 10 ч, предпочтительно 3 ч.
Температура реакции предпочтительно составляет 25°C, растворитель - предпочтительно ТГФ.
В еще одном предпочтительном воплощении настоящего изобретения соединение VI-1 может быть получено в соответствии со следующим способом получения:
взаимодействие соединения V-1 в присутствии гидрида натрия и 3-бромметил-2,2-диметилоксирана:
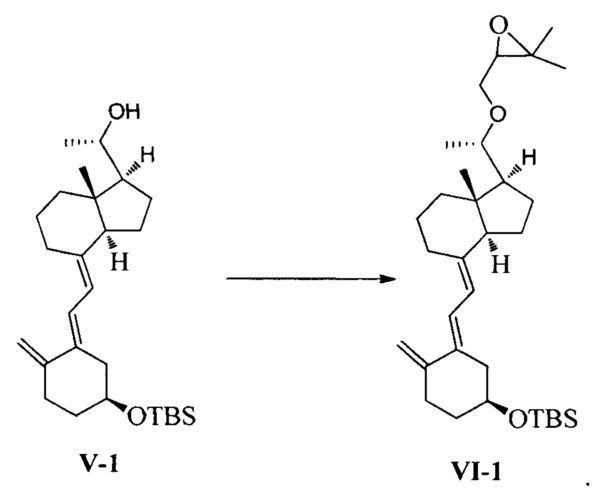
В реакции молярное отношение между соединением V-1, гидридом натрия и 3-бромметил-2,2-диметилоксираном предпочтительно составляет 1:(1-3):(1-3), более предпочтительно 1:1,2:2.
Продолжительность реакции может быть от 1 ч до 10 ч, предпочтительно 5 ч.
Температура реакции предпочтительно составляет от 50°C.
Реакцию можно проводить в подходящем органическом растворителе, включая диоксан, ацетонитрил, ТГФ, ДМФА, ДМСО, N,N-диметилацетамид или N-метилпирролидон и т.д., но не ограничиваясь ими.
В еще одном предпочтительном воплощении настоящего изобретения соединение V-1 может быть получено в соответствии со следующим способом получения:
превращение соединения IV-1 в соединение V-1 в присутствии бикарбоната натрия:
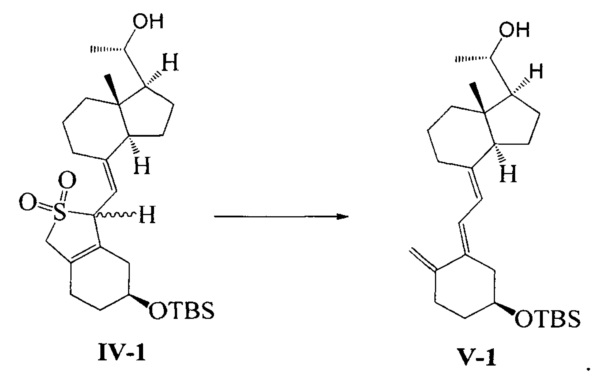
В реакции молярное отношение между соединением IV-1 и бикарбонатом натрия предпочтительно составляет 1:(1-10), более предпочтительно 1:6.
Продолжительность реакции может быть от 1 ч до 24 ч, предпочтительно 7 ч.
Температура реакции предпочтительно составляет 80°C.
Реакцию можно проводить в подходящем органическом растворителе, включая 95%-ный (по объему) этанол, ацетонитрил, этилацетат или безводный этанол, предпочтительно 95%-ный (по объему) этанол, но не ограничиваясь ими.
В еще одном предпочтительном воплощении настоящего изобретения соединение IV-1 может быть получено в соответствии со способом получения, как показано ниже:
в присутствии хирального вспомогательного реагента (R)-2-метил-CBS-оксазаборолидина при восстановлении соединения III-1 с помощью борана:
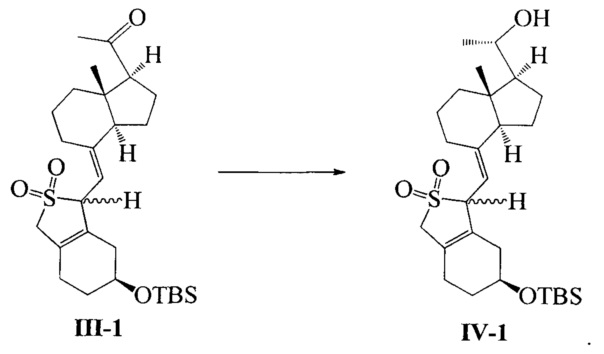
В реакции молярное отношение между соединением III-1, (R)-2-метил-CBS-оксазаборолидином и бораном предпочтительно составляет 1:(0,1-1):(1-2), более предпочтительно 1:0,6:1.
Температура реакции может быть от -60°C до 0°C, предпочтительно 20°C.
Продолжительность реакции предпочтительно составляет 3 ч.
В еще одном предпочтительном воплощении по настоящему изобретению соединение III-1 может быть получено в соответствии со способом получения, как показано ниже:
взаимодействие соединения II-1 в присутствии триэтилендиамина, 2,2-бипиридина и ацетата меди при вводе кислорода:
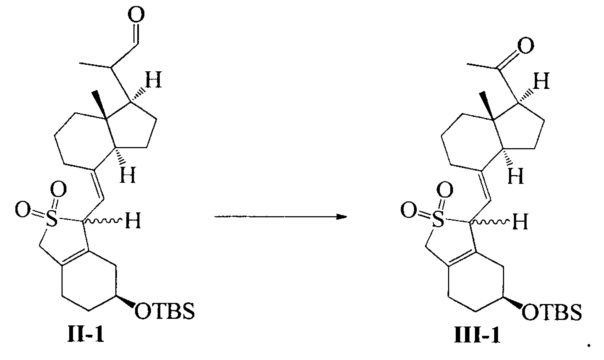
В реакции молярное отношение между соединением II-1, триэтилендиамином, 2,2-бипиридином и ацетатом меди предпочтительно составляет 1:(1-2):(0,1-1):(0,1-1), более предпочтительно 1:1:0,2:0,2.
Продолжительность реакции может быть от 1 ч до 20 ч, предпочтительно 5 ч.
Температура реакции предпочтительно составляет 45°C.
При этом соединение II-1 получают в соответствии с патентом US 4866048.
Путь синтеза по настоящему изобретению можно составить следующим образом:
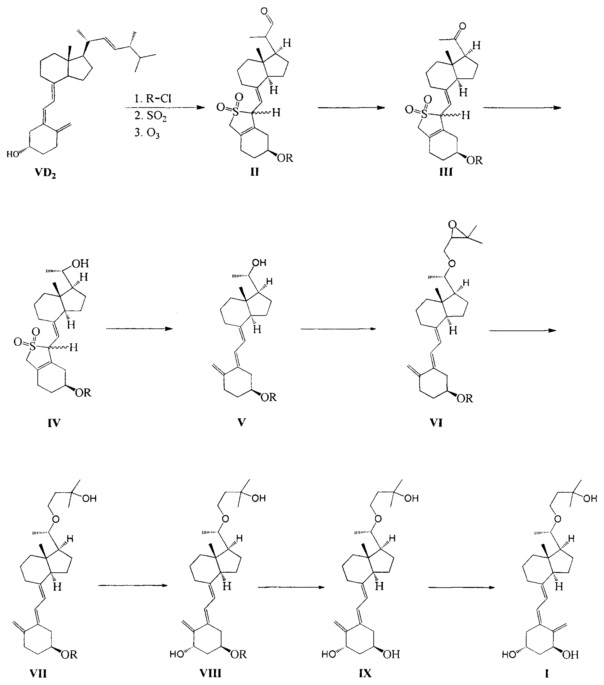
По сравнению с существующим уровнем техники, настоящее изобретение имеет следующие преимущества.
Способ синтеза по настоящему изобретению хитроумно спроектирован, при этом витамин D2 используют в качестве исходного материала, соединение II получают в соответствии со способом, описанном в US 4866048, а затем его окисляют кислородом в присутствии медного катализатора с получением соединения III. В ходе процесса окисления, благодаря защите двойной связи диоксидом серы другие побочные реакции снижены, что позволяет достигать выхода продукта окисления около 80%. Тем не менее, в ходе процесса окисления аналогичных соединений в предшествующем уровне техники выход относительно мал из-за нестабильности сопряженной тройной связи, например, выход при реакции окисления, указанный в JP 20111573261, составляет 67%, а в Tetrahedron Letters 1994, 2295-2298 выход составляет 60-65%. В настоящем изобретении в присутствии хирального вспомогательного реагента соединение III восстанавливают стереоселективно, получая соединение IV только в S-конфигурации посредством применения борана, и с высоким выходом, достигающим почти 100%. Поскольку диоксид серы защищает концевую двойную связь, то побочную реакцию между бораном и терминальной двойной связью можно эффективно предотвратить в процессе восстановления, что улучшает выход. В WO 2012/122451 и JP 20111573261 реакцию восстановления проводят с использованием боргидрида натрия/алюмогидрида лития, при которой большая часть продукта получается в R-конфигурации, а выход продукта с S-конфигурацией чрезвычайно мал, кроме того, продукты с двумя конфигурациями имеют близкие значения Rf, что затрудняет очистку. В настоящем изобретении двойную связь защищают диоксидом серы, который играет важную роль при окислении и при проведении стадий асимметричного восстановления, эффективно препятствуя протеканию других побочных реакций и значительно улучшая выход реакции. В то же время, последующая очистка становится гораздо проще, так как образуется продукт исключительно в S-конфигурации. Эффективность синтеза значительно возрастает, а стоимость процесса значительно снижается.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВОПЛОЩЕНИЙ
Следующие примеры дополнительно иллюстрируют настоящее изобретение. Следует понимать, что способы получения в воплощениях предназначены для иллюстрации подробностей настоящего изобретения, а не для ограничения объема настоящего изобретения, любая простая модификация в способе получения по настоящему изобретению, основанная на концепции настоящего изобретения, должна относиться к объему настоящего изобретения.
Воплощение 1
Получение соединения III-1
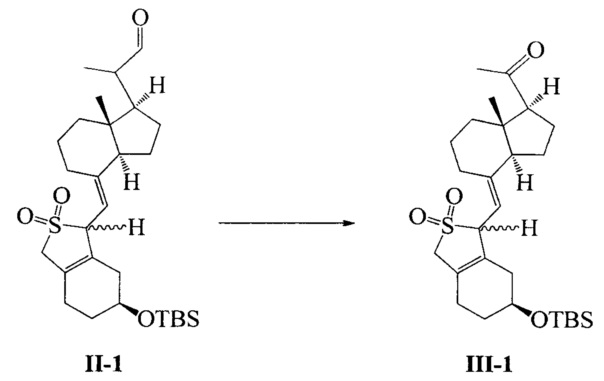
Соединение II-1 (50,7 г, 100 ммоль) растворили в ДМФА (500 мл), а затем по отдельности добавили триэтилендиамин (11.2 g, 100 mmol), 2,2-бипиридин (3,12 г, 20 ммоль) и ацетат меди (3,64 г, 20 ммоль) при комнатной температуре. После добавления реакционную смесь нагрели до 45°C в атмосфере кислорода и далее перемешивали в течение 5 часов при этой температуре. После завершения реакции добавили этилацетат, смесь отфильтровали для удаления нерастворимых веществ. Фильтрат промыли водой 3 раза, сушили над безводным сульфатом натрия и упаривали при пониженном давлении, масло выделили и очистили с получением соединения III-1 (39,9 г, выход 81%). Соединение представляет собой смесь двух конфигураций (из-за защиты диоксидом серы) и может быть использовано непосредственно на следующей стадии. Небольшое количество было взято, чтобы выделить, очистить и получить соединение с конфигурацией I (имеющее большое значение Rf) и соединение с конфигурацией II (имеющее малое значением Rf).
Экспериментальные данные 1Н ЯМР, 13С ЯМР и МС для двух изомеров соединения III-1 показаны ниже:
Изомер с малым значением Rf: 1Н ЯМР (400 МГц, d-CHCl3) δ: -0,01 и -0,00 (каждый, с, 6Н), 0,55 (с, 3H), 0,81 (с, 9Н), 1,19-2,19 (м, 19Н), 2,56-2,66 (м, 2Н), 3,59 (с, 2Н), 3,95-3,97 (м, 1Н), 4,43-4,45 (д, 1Н, J=9,6), 4,66-4,68 (д, 1Н, J=9,2); 13С-ЯМР (100 МГц, d-CHCl3) δ: -4,7, -4,7, 13,1, 18,1, 22,2, 22,4, 23,7, 24,2, 25,8, 29,6, 30,7, 31,3, 34,3, 39,4, 47,1, 56,3, 58,1, 63,7, 66,5, 67,5, 111,6, 126,7, 130,5, 149,3, 208,8; МС: m/z (492), Найдено: 493 (М+Н).
Изомер с большим значением Rf: 1Н ЯМР (400 МГц, d-CHCl3) δ: -0,01 и -0,00 (каждый, с, 6Н), 0,49 (с, 3H), 0,82 (с, 9Н), 1,21-2,20 (м, 19Н), 2,57-2,60 (м, 1Н), 2,67-2,71 (м, 1Н), 3,62-3,64 (д, 2Н), 3,91-3,93 (м, 1Н), 4,55-4,58 (д, 1Н, J=9,6), 4,62-4,79 (д, 1Н, J=10,0); 13С-ЯМР (100 МГц, d-CHCl3) δ: -4,8, -4,7, 13,4, 18,1, 22,3, 22,5, 23,3, 24,6, 25,8, 29,1, 29,7, 30,9, 31,5, 34,1, 39,1, 46,3, 56,1, 58,2, 63,4, 66,7, 66,8, 111,1, 127,0, 130,2, 148,6, 208,9; МС: m/z (492), Найдено: 493 (М+Н).
Воплощение 2
Получение соединения IV-1
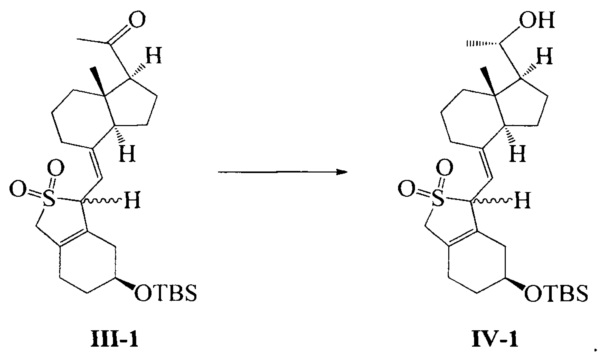
Соединение III-1 (49,2 г, 100 ммоль) растворили в 400 мл безводного ТГФ, медленно добавили (R)-2-метил-CBS-оксазаборолидин (1 М, 100 мл) при -20°C, а затем медленно прилили по каплям ВН3 ⋅ ТГФ (1М, 60 мл) при указанной температуре, после добавления реакционную смесь далее перемешивали в течение 1 ч и медленно нагревали до комнатной температуры, а затем добавили 50 мл насыщенного водного раствора хлорида аммония. Смесь экстрагировали этилацетатом и упаривали при пониженном давлении с получением 49,5 г масла. Полученное масло представляет собой смесь изомеров в двух конфигурациях (из-за защиты диоксидом серы, С-20 имеет исключительно S-конфигурацию). Небольшое количество было взято, чтобы выделить, очистить и получить соединение в конфигурации I (с большим значением Rf) и соединение в конфигурации II (с малым значением Rf).
Экспериментальные данные 1Н ЯМР, 13С ЯМР и МС для двух изомеров соединения IV-1 приведены ниже.
Изомер с малым значением Rf: 1Н ЯМР (400 МГц, d-CHCl3) δ: -0,01 и -0,00 (каждый, с, 6Н), 0,60 (с, 3H), 0,80 (с, 9Н), 1,17-1,20 (м, 6Н), 1,48-2,04 (м, 16Н), 2,48-2,57 (м, 1Н), 3,59 (с, 2Н), 3,64-3,68 (м, 1Н), 3,94-3,96 (м, 1Н), 4,44-4,47 (д, 1Н, J=9,2), 4,64-4,66 (д, 1Н, J=9,2); 13С-ЯМР (100 МГц, d-CHCl3) δ: -4,7, 12,4, 18,1, 22,0, 23,6, 24,3, 25,0, 25,8, 29,7, 29,7, 30,7, 34,3, 39,3, 45,3, 56,1, 58,1, 58,7, 66,5, 67,6, 70,3, 110,8, 126,5, 130,7, 150,0; МС: м/z=494, Найдено 495 (М+Н).
Изомер с большим значением Rf: 1Н ЯМР (400 МГц, d-CHCl3) δ: -0,01 и -0,00 (каждый, с, 6Н), 0,52 (с, 3H), 0,82 (с, 9Н), 1,18-1,23 (м, 6Н), 1,46-2,17 (м, 16Н), 2,52-2,55 (м, 1Н), 3,60-3,66 (м, 3H), 3,91-3,92 (м, 1Н), 4,55-4,58 (д, 1Н, J=10,4), 4,73-4,75 (д, 1Н, J=10,4); 13С-ЯМР (100 МГц, d-CHCl3) δ: -4,7, 12,4, 18.1, 22,0, 23.6, 24,3, 25,0, 25,8, 29,7, 29,7, 30,7, 34,3, 39,3, 45,3, 56,1, 58,1, 58,7, 66,5, 67,6, 70,3, 110,8, 126,5, 130,7, 150,0; МС: m/z=494, Найдено 495 (М+Н).
Воплощение 3
Получение соединения V-1
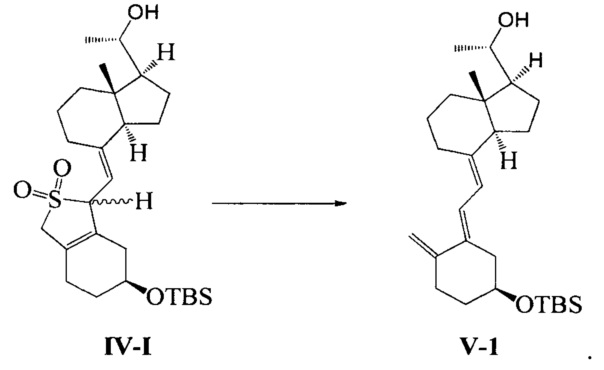
Неочищенный продукт соединения IV-1, полученный на предыдущей стадии, растворили в 400 мл 95%-ного этилового спирта, при перемешивании добавили 50 г бикарбоната натрия, затем нагрели с обратным холодильником и проводили реакцию в течение еще 2~3 часов при этой температуре. После того, как реакция была завершена, этанол удалили при пониженном давлении, этилацетат использовали для экстракции. Масло выделили и очистили, получив 36,4 г соединения V-1, выход 84%.
Экспериментальные данные 1Н ЯМР, 13С ЯМР и МС для соединения V-1 приведены ниже:
1Н ЯМР (400 МГц, CDCl3) δ: -0,03 (с, 6Н, 2SiCH3), 0,50 (с, 3H, СН3), 0,82 (с, 9Н, 3SiCH3), 1,16 (д, J=6 Гц, 3H, СН3), 1,18-1,23 (м, 2Н), 1,35-2,22 (м, 13Н), 2,38-2,43 (м, 1Н), 2,57-2,61 (м, 1Н), 2,79-2,83 (м, 1Н), 3,64-3,67 (м, 1Н, СНОН), 3,78-3,81 (м, 1Н, СНОН), 4,58 (с, 1Н, =СН2), 4,86 (с, 1Н, =СН2), 5,81 (д, J=11,6 Гц, 1Н, =СН), 6,40 (д, J=11,6 Гц, 1Н, =СН); 13С ЯМР (75 МГц, CDCl3) δ: -4,7, -4,6, 12,7, 18,2, 22,2, 23,2, 23,6, 25,0, 25,9 (3C), 28,8, 31,2, 35,2, 37,5, 39,5, 44,9, 56,3, 58,7, 69,4, 70,3, 107,5, 116,5, 119,9, 136,6, 142,9, 150,0; МС м/z=430, найдено 431 (М+1).
Воплощение 4
Получение соединения VII-1
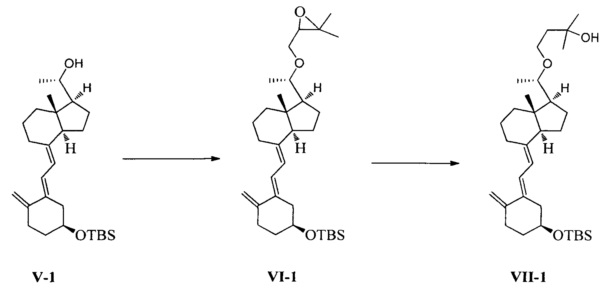
Соединение V-1 (43,1 г, 100 ммоль) растворили в 430 мл безводного ТГФ, добавили 60%-ный гидрид натрия (4,8 г, 120 ммоль) при комнатной температуре, затем перемешивали в течение 0,5 часа. Добавили 3-бромметил-2,2-диметилоксиран (31 г, 200 ммоль), затем смесь нагрели с обратным холодильником и проводили реакцию в течение еще 5 часов при этой температуре. После того как реакция была завершена, смесь охладили до комнатной температуры, добавили триизобутилгидроборат лития (150 мл, 1 М в ТГФ) и затем после добавления дополнительно перемешивали в течение 3 часов. Добавили 100 мл насыщенного раствора хлорида аммония, смесь экстрагировали этилацетатом и упарили. Полученное масло выделили и очистили, получив 40,3 г соединения VII-1, выход 78%.
Экспериментальные данные 1Н ЯМР, 13С ЯМР и МС для соединения VII-1 приведены ниже:
1Н ЯМР (400 МГц, CDCl3) δ: -0,07 (с, 3H, SiCH3), -0,06 (с, 3H, SiCH3), 0,48 (с, 3H, СН3), 0,83 (с, 9Н, 3SiCH3), 0,72-0,97 (м, 2Н), 1,13 (д, J=6 Гц, 3H, СН3), 1,17 (с, 3H, СН3), 1,18 (с, 3H, СН3), 1,19-1,27 (м, 2Н), 1,35-2,22 (м, 13Н,), 2,39-2,42 (м, 1Н), 2,56-2,61 (м, 1Н), 2,78-2,82 (м, 1Н), 3,17-3,21 (м, 1Н, СНОН), 3,41-3,44 (м, 1Н, СНОН), 3,77-3,81 (м, 3H, ОН и СНОН), 4,58 (с, 1Н, =СН2), 4,86 (с, 1Н, =СН2), 5,80 (д, J=11,6 Гц, 1Н, =СН), 6,39 (д, J=11,6 Гц, 1Н, =СН); 13С ЯМР (75 МГц, CDCl3) δ: -4,7, -4,6, 12,7, 18,2, 18,8, 22,2, 23,2, 25,9 (3C), 26,0, 28,8, 29,1, 29,4, 31,2, 35,2, 37,5, 39,6, 41,5, 44,7, 56,2, 57,1, 65,6, 69,4, 70,5, 79,0, 107,6, 116,5, 119,9, 136,5, 142,8, 150,0; МС m/z=516, найдено 517 (М+1).
Воплощение 5
Получение соединения VIII-1
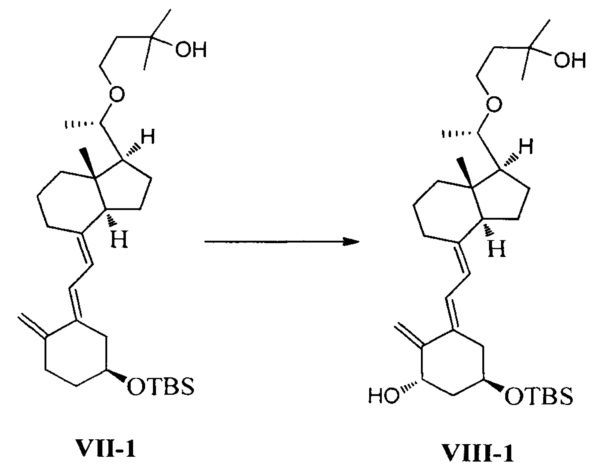
Соединение VII-1 (41,2 г, 80 ммоль) растворили в 500 мл дихлорметана, затем добавили N-метилморфолин-N-оксид (18,7 г, 160 ммоль) и диоксид селена (3,55 г, 32 ммоль). Аргон был введен, чтобы заменить воздух в реакционной колбе. Реакционную смесь нагревали с обратным холодильником, затем дополнительно выдерживали в течение 5-6 ч при этой температуре. После того как реакция была завершена, смесь охладили до комнатной температуры, добавили воду и использовали дихлорметан для экстракции. Органическую фазу сконцентрировали при пониженном давлении, затем остаток выделили и очистили с помощью колоночной хроматографии. Система для элюирования петролейный эфир : этилацетат = 10:1. Получили соединение VIII-1 (15,7 г), выход 37%.
Экспериментальные данные 1Н ЯМР, 13С ЯМР и МС для соединения VIII-1 показаны ниже:
1Н ЯМР (400 МГц, CDCl3) δ: -0,01 (с, 6Н, 2SiCH3), 0,46 (с, 3H, СН3), 0,83 (с, 9Н, 3SiCH3), 1,12 (д, J=6 Гц, 3H, СН3), 1,16 (с, 3H, СН3), 1,17 (с, 3H, СН3), 1,18-1,27 (м, 2Н), 1.42-1.97 (м, 15Н), 2,34-2,47 (м, 1Н), 2,77-2,81 (м, 1Н), 3,16-3,20 (м, 1Н, СНОН), 3,41-3,44 (м, 1Н, СНОН), 3,75-3,80 (м, 2Н, ОН и СНОН), 4,11-4,14 (м, 1Н, СНОН), 4,41-4,44 (м, 1Н, СНОН), 4,88 (с, 1Н, =СН2), 4,99 (с, 1Н, =СН2), 5,78 (д, J=11,6 Гц, 1Н, =СН), 6,42 (д, J=11,6 Гц, 1Н, =СН); 13С ЯМР (75 МГц, CDCl3) δ: -4,8, -4,7, 12,6, 18,1, 18,8, 22,2, 23,2, 25,9 (3C), 26,0, 28,9, 29,1, 29,4, 37,0, 39,6, 41,5, 42,9, 44,8, 56,2, 57,1, 65,6, 66,8, 70,5, 70,6, 79,0, 107,7, 116,6, 122,2, 134,6, 143,3, 153,1; МС m/z=532, найдено 555 (М+Na).
Воплощение 6
Получение соединения IX
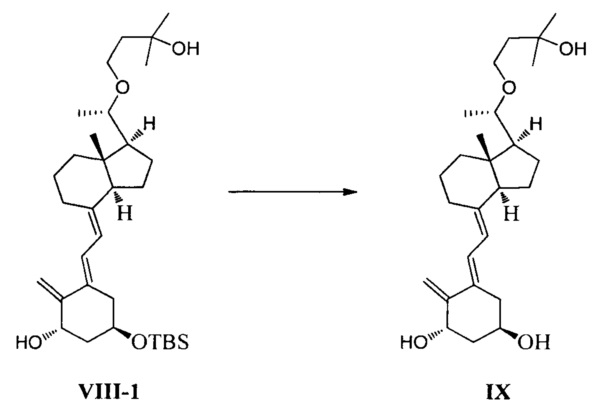
Соединение VIII-1 (26,6 г, 50 ммоль) растворили в 270 мл ТГФ, добавили Bu4NF (19,5 г, 75 ммоль), затем реакционную смесь нагревали с обратным холодильником и перемешивали еще в течение 7-8 ч при этой температуре. После того как реакция была завершена, нагревание прекратили и смесь охладили до комнатной температуры. ТГФ удалили при пониженном давлении, этилацетат использовали для экстракции. После упаривания при пониженном давлении полученное масло выделили и очистили, получив 18 г соединения IX, выход 86%.
Экспериментальные данные 1Н ЯМР, 13С ЯМР и МС для соединения IX показаны ниже:
1Н ЯМР (400 МГц, CDCl3) δ: 0,54 (с, 3H, СН3), 1,19 (с, J=5,6 Гц, 3H, СН3), 1,23 (с, 6Н, 2СН3), 1,24-1,37 (м, 2Н), 1,48-2,08 (м, 13Н), 2,24-2,30 (м, 1Н), 2,44 (с, широкий, 1Н, ОН), 2,65 (с, широкий, 1Н, ОН), 2,81-2,88 (м, 2Н), 3,24-3,27 (м, 1Н), 3,46-3,51 (м, 1Н, СНОН), 3,82-3,90 (м, 2Н, ОН и СНОН), 4,19-4,23 (м, 1Н, СНОН), 4,47-4,49 (м, 1Н, СНОН), 4,96 (с, 1Н, =СН2), 5,10 (с, 1Н, =СН2), 5,89 (д, J=11,2 Гц, 1Н, =СН), 6,55 (д, J=11,2 Гц, 1Н, =СН); 13С ЯМР (75 МГц, CDCl3) б: 12,8, 18,9, 22,2, 23,2, 25,8, 28,9, 29,1, 29,2, 38,7, 39,5, 41,5, 41,9, 44,8, 56,2, 57,1, 65,5, 65,6, 70,7, 70,8, 78,9, 109,5, 116,5, 122,8, 133,5, 144,0, 151,8; МС m/z=418, найдено 441 (М+Na).
Воплощение 7
Получение соединения I
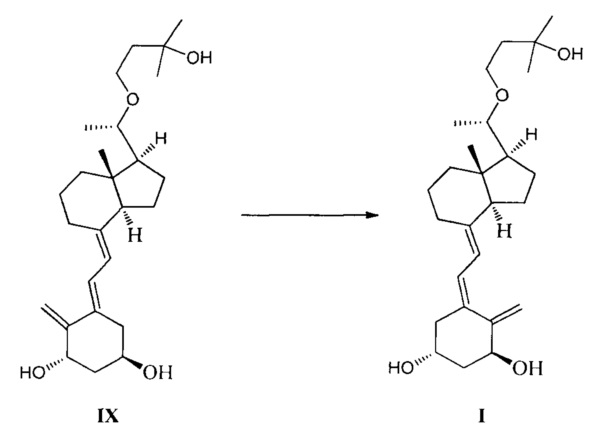
Соединение IX (21 г) растворили в 3000 мл ацетона, добавили 9-ацетилантрацен (2,1 г). Включили охлаждающее оборудование, охладили до температуры ниже 5°C. Включили оборудование для фотохимической реакции и проводили реакцию УФ облучения при длине волны 350 нм. Через 0,5 ч отобрали образец для контроля реакции и продолжительность реакции оценивали по результату мониторинга, которая составила около 2 часов. После того как реакция была завершена, ацетон упарили, полученный остаток пропустили через хроматографическую колонку. Система элюирования петролейный эфир : этилацетат = 1:1. Получили 19,3 г соединения I, выход 92%.
Экспериментальные данные 1Н ЯМР, 13С ЯМР и МС для соединения I приведены ниже:
1Н ЯМР (400 МГц, d-ДМСО) δ: 0,49 (с, 3H, СН3), 1,08 (с, 6Н, 2СН3), 1,09 (д, J=1,6 Гц, 3H, СН3), 1,22-1,28 (м, 1Н), 1,39-1,65 (м, 10Н), 1,79-1,84 (м, 3H), 1,93-1,99 (м, 1Н), 2,15-2,20 (м, 1Н), 2,35-2,37 (м, 1Н), 2,78-2,81 (м, 1Н), 3,18-3,21 (м, 1Н), 3,25-3,31 (м, 1Н), 3,60 (д, J=7,6 Гц, 1Н), 3,99-4,04 (м, 1Н, СНОН), 4,12 (с, 1Н, ОН), 4,18-4,21 (м, 1Н, СНОН), 4,54 (д, J=4 Гц, 1Н, ОН), 4,76 (с, 1Н, =СН2), 4,86 (д, J=4,4 Гц, 1Н, ОН), 5,23 (с, 1Н, =СН2), 5,99 (д, J=11,2 Гц, 1Н,=СН), 6,18 (д, J=11,2 Гц, 1Н, =СН); 13С ЯМР (75 МГц, d-ДМСО) δ: 12,3, 19,1, 21,8, 22,9, 24,7, 28,3, 29,6, 29,7, 38,9, 43,1, 43,2, 44,1, 44,9, 55,5, 56,8, 64,3, 65,1, 68,2, 68,4, 76,7, 109,8, 117,8, 122,4, 135,9, 139,6, 149,5; МС m/z=418, найдено 441 (М+Na).
Способ получения 4-бензил-1-фенетил-пиперазин-2,6-диона, промежуточное соединение и способ его получения
Фармацевтическая композиция, содержащая адалимумаб
Добавка и способ для обрыва полимеризации и/или снижения вязкости раствора полимера
Соединения, обладающие активирующим действием на подтипы рецепторов, активируемых пролифератором пероксисом (ppars), и способ получения и применения указанных соединений

