Результат интеллектуальной деятельности: СПИРО[2.3]ГЕКСАНОВЫЕ АМИНОКИСЛОТЫ - КОНФОРМАЦИОННО-ЖЕСТКИЕ АНАЛОГИ γ-АМИНОМАСЛЯНОЙ КИСЛОТЫ - И СПОСОБЫ ИХ ПОЛУЧЕНИЯ
Вид РИД
Изобретение
Область техники
Изобретение относится к новым спиро[2.3]гексановым аминокислотам, содержащим аминогруппу в 5-ом положении, карбоксильную группу (или группу биоизостерную карбоксильной, в частности фосфонатную) в 1-ом положении спиро[2.3]гексанового фрагмента и являющимся в силу своего строения конформационно-жесткими аналогами γ-аминомасляной кислоты (ГАМК), а также к способам их получения.
Уровень техники
ГАМК (формула 1) является важнейшим тормозным нейромедиатором центральной нервной системы (ЦНС) человека и млекопитающих; принимает участие в нейромедиаторных и метаболических процессах в мозге, играя ведущую роль в патогенезе тревоги, судорог и многих других патологических состояний ЦНС [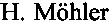 . The rise of new GABA pharmacology. // Neuropharmacology, 2011, Vol. 60, №7-8, P. 1042-1049].
. The rise of new GABA pharmacology. // Neuropharmacology, 2011, Vol. 60, №7-8, P. 1042-1049].
Интерес к синтезу конформационно-жестких (конформационно-ограниченных) аналогов ГАМК обусловлен тем, что соединения данной группы могут обладать высокой селективностью по отношению к отдельным биомишеням ГАМК. Создание конформационно-жестких аналогов природных физиологически-активных веществ (ФАВ) на основе соединений, содержащих малые и средние циклы или их спирокомбинацию, является современным приемом медицинской химии по созданию селективных синтетических ФАВ [F.E. Boyer, J.V.N. V. Prasad, A.L. Choy, L. Chupak, M.R. Dermyer, Q. Ding, M.D. Huband, W. Jiao, T. Kaneko, V. Khlebnikov, J.-Y. Kim, M.S. Lall, S.N. Maiti, K. Romero, X. Wu. Synthesis and SAR of novel conformationally-restricted oxazolidinones possessing Gram-positive and fastidious Gram-negative antibacterial activity. Part 1: Substituted pyrazoles // Bioorg. Med. Chem. Lett., 2007, Vol. 17, №16, P. 4694-4698]. Важную роль здесь играют аминокислоты, содержащие циклопропановый фрагмент, поскольку они обладают разнообразной физиологической активностью и выполняют важные функции в живых организмах. Циклопропановые аминокислоты входят в состав высокоэффективных фармацевтических препаратов, играют важную роль в изучении процессов метаболизма и механизмов действия ферментов. Встроенные в пептидные последовательности, они изменяют структуру белка и, как следствие, биологические свойства. Это связано с тем, что наличие в молекуле трехчленного кольца ограничивает вращение вокруг С-С связи. Заместители оказываются жестко закрепленными в пространстве, но при этом, в отличие от непредельных аминокислот, сохраняют асимметрические центры [К. Yamaguchi, Y. Kazuta, К. Hirano, S. Yamada, A. Matsuda, S. Shuto. Synthesis of 1-arylpiperazyl-2-phenylcyclopropanes designed as antidopaminergic agents: Cyclopropane-based conformationally restricted analogs of haloperidol // Bioorg. Med. Chem., 2008, Vol. 16, №19, P. 8875-8881].
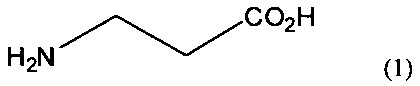
Наиболее близкими к предлагаемому по технической сущности являются аналоги ГАМК - 1-аминоспиро[2.3]гексан-1,5-дикарбоновая кислота (2а) [N.V. Yashin, Е.В. Averina, T.S. Kuznetsova, N.S. Zefirov. Catalytic cyclopropanation of methylenecyclobutanes using ethylnitrodiazoacetate. Synthesis of sprirohexane amino acids // Tetrahedron Lett., 2003, Vol. 44, №45, P. 8241-8244] и 1-аминоспиро[2.3]гексан-5-карбоновая кислота (2b) [Патент RU 2468000 C2, H.B. Яшин, A.B. Чемагин, T.C. Кузнецова, H.C. Зефиров. Спироциклические циклопропановые аминокислоты - аналоги гамма-аминомасляной кислоты с ограниченной конформационной подвижностью и фармацевтические композиции на основе их. Дата публикации заявки 27.11.2012, Бюл. №33], содержащие спиро[2.3]гексановые фрагменты. Структурные формулы соединений 2а, 2b приведены ниже (2).
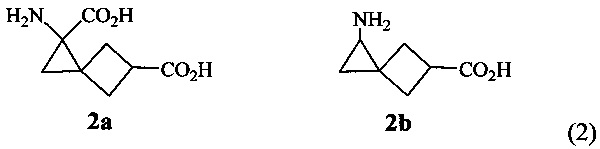
Синтез аминокислот 2а, b основан на реакции [1+2]-циклоприсоединения этил(нитро)диазоацетата к метиловому эфиру (3-метилен)циклобутанкарбоновой кислоты, протекающей с образованием 1-этил-5-метил-1-нитроспиро[2.3]гексан-1,5-дикарбоксилата (3) (схема 3) [N.V. Yashin, Е.В. Averina, T.S. Kuznetsova, N.S. Zefirov. Catalytic cyclopropanation of methylenecyclobutanes using ethylnitrodiazoacetate. Synthesis of sprirohexane amino acids // Tetrahedron Lett., 2003, Vol. 44, №45, P. 8241-8244]. Дальнейшее восстановление нитрогруппы аддукта 3 (формула 3) с последующим гидролизом сложноэфирных групп позволяет получить аминокислоту 2а. Проведение для аддукта 3 реакций селективного гидролиза и декарбоксилирования карбоксильного фрагмента у атома углерода, связанного с нитро-группой, восстановления нитрогруппы и гидролиза второй сложноэфирной группы приводит к образованию аминокислоты 2b [Патент RU 2468000].
Существенным недостатком спиро[2.3]гексановых аминокислот 2а, b является структурная ограниченность предложенных в работах соединений исключительно аминокарбоновыми кислотами. При этом введение в структуру полициклических циклопропановых аминокислот вместо карбоксильной группы группировки биоизостерной ей, например, фосфоновой, позволило бы получить выход к другим перспективным соединениям с возможно большей активностью и селективностью по отношению к биомишеням ГАМК, что обусловлено иной химической природой и размерами фосфонатного фрагмента по сравнению с карбоксильным.
Другим недостатком известного метода синтеза спиро[2.3]гексановых аминокислот 2а, b является невозможность использования предложенных в работах подходов для получения биоизостерных (в частности, фосфоновых) конформационно-жестких аналогов ГАМК, т.к. используемая на первой стадии синтеза реакция присоединения нитродиазоуксусного эфира к метиловому эфиру (3-метилен)циклобутанкарбоновой кислоты делает дальнейшее введение в получаемый спироциклический аддукт группы биоизостерной карбоксильной (в частности, фосфонатной группы) принципиально невозможной при сохранении в структуре свободной аминогруппы. На основании изучения источников информации можно сделать вывод о том, что в настоящее время отсутствуют методы синтеза биоизостерных (в частности, фосфоновых) конформационно-жестких аналогов ГАМК, содержащих спиро[2.3]гексановый фрагмент.
К недостаткам известных способов получения спиро[2.3]гексановых аминокислот 2а, b, изложенных в работах, также следует отнести невозможность получения на их основе 5-аминоспиро[2.3]гексан-1-овых кислот, т.е. аминокислот со структурой, в которой амино-группа расположена в 5-ом положении, а карбоксильная группа - в 1-ом положении спиро[2.3]гексанового фрагмента. Подобное расположение фармакофорных групп относительно базового конформационно-жесткого фрагмента позволяет зафиксировать новые конформации ГАМК, что может иметь хорошую перспективу с точки зрения поиска соединений с улучшенной селективностью по отношению к биомишеням ГАМК. Из анализа источников информации следует, что в настоящее время отсутствуют методы синтеза 5-аминоспиро[2.3]гексан-1-овых кислот.
Раскрытие изобретения
Задачей настоящего изобретения является получение новых спиро[2.3]гексановых аминокислот, являющихся конформационно-жесткими аналогами γ-аминомасляной кислоты (ГАМК), а также разработка способов их получения.
Поставленная задача достигается получением спиро[2.3]гексановой аминокислоты общей формулы:
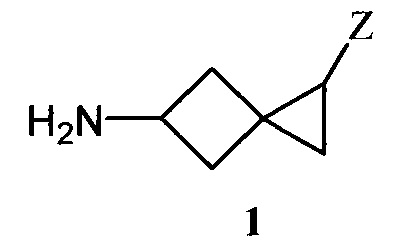
где Z=CO2H (формула 1а) и Z=PO3H2 (формула 1b).
Также поставленная задача решается способом получения новой 5-аминоспиро[2.3]гексан-1-карбоновой кислоты (1а)
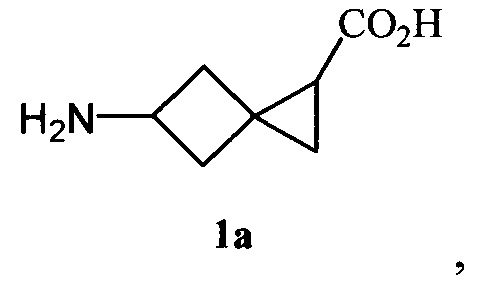
который заключается в том, что в 3-(метилен)циклобутанкарбоновой кислоте с использованием модифицированной реакции Курциуса [S.I. Kozhushkov, A.F. Khlebnikov, R.R. Kostikov, D.S. Yufit, A. de Meijere. Scalable synthesis of (1-cyclopropyl)cyclopropylamine hydrochloride // Beilstein J. Org. Chem. 2011. 7. P. 1003 -1006] производится трансформация карбоксильной группы в аминогруппу, защищенную Вос-фрагментом, далее осуществляется взаимодействие диазоуксусного эфира и трет-бутилового эфира N-(3-метиленциклобутил)карбаминовой кислоты, взятых в соотношении 1:1.2-2, в присутствии 1-10 мольных % тетраацетата диродия с образованием трет-бутилового эфира N-[1-(этоксикарбонил)спиро[2.3]гекс-5-ил]карбаминовой кислоты, последовательное удаление Вос-группы [S.I. Kozhushkov, A.F. Khlebnikov, R.R. Kostikov, D.S. Yufit, A. de Meijere. Scalable synthesis of (1-cyclopropyl)cyclopropylamine hydrochloride // Beilstein J. Org. Chem. 2011. 7. P. 1003 -1006] которого под действием 3-6N раствора хлороводорода в диэтиловом эфире и гидролиз [N.V. Yashin, Е.В. Averina, T.S. Kuznetsova, N.S. Zefirov. Catalytic cyclopropanation of methylenecyclobutanes using ethylnitrodiazoacetate. Synthesis of sprirohexane amino acids // Tetrahedron Lett., 2003, Vol. 44, №45, P. 8241-8244] под действием 2-5-кратного избытка 1N водного раствора гидроксида натрия приводят к получению 5-аминоспиро[2.3]гексан-1-карбоновой кислоты. При этом способ включает трансформацию карбоксильной группы в аминогруппу, которая осуществляется в результате реакции 3-(метилен)циклобутанкарбоновой кислоты, этилхлорформиата и триэтиламина, взятых в соотношении 1:1-1.5:1, с последующим взаимодействием с 1.1-1.7-кратным избытком азида натрия и более чем 10-кратным избытком трет-бутанола.
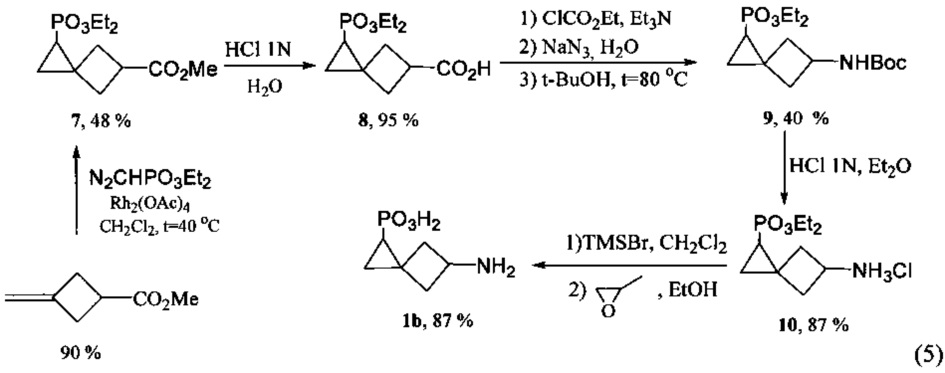
Поставленная задача также решается способом получения новой 5-аминоспиро[2.3]гексан-1-фосфоновой кислоты (1b)
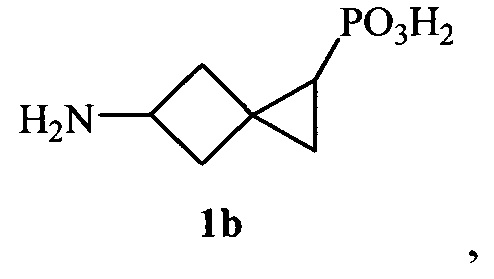
который заключается в том, что осуществляется взаимодействие метилового эфира 3-(метилен)циклобутанкарбоновой кислоты и диазофосфонового эфира, взятых в соотношении 5:0.5-2, в присутствии 1-10 мольных % тетраацетата диродия, в полученном метиловом эфире 1-(диэтоксифосфорил)спиро[2.3]гексан-5-карбоновой кислоты проводят гидролиз метоксикарбонильной группы под действием 3-10-кратного избытка 1N раствора соляной кислоты и далее с использованием модифицированной реакции Курциуса [S.I. Kozhushkov, A.F. Khlebnikov, R.R. Kostikov, D.S. Yufit, A. de Meijere. Scalable synthesis of (l-cyclopropyl)cyclopropylamine hydrochloride // Beilstein J. Org. Chem. 2011. 7. P. 1003-1006] производится трансформация карбоксильной группы в аминогруппу, защищенную Вос-фрагментом, с образованием трет-бутилового эфира N-[1-(диэтоксифосфорил)спиро[2.3]гекс-5-ил]карбаминовой кислоты, последовательное удаление Вос-группы [S.I. Kozhushkov, A.F. Khlebnikov, R.R. Kostikov, D.S. Yufit, A. de Meijere. Scalable synthesis of (l-cyclopropyl)cyclopropylamine hydrochloride // Beilstein J. Org. Chem. 2011. 7. P. 1003-1006] которого под действием более чем 50-кратного избытка 3-6 N раствора хлороводорода в диэтиловом эфире и расщепление диэтоксифосфорильной группы [A. Fadel. A useful synthesis of 1-aminocyclopropanephosphonic acid from cyclopropanone acetal // J. Org. Chem. 1999. V. 64. №13. P. 4953-4955] под действием 3-7-кратного избытка триметилсилилбромида в дихлорметане с последующей обработкой полученной смеси более чем 50-кратным избытком 72% об. раствора пропиленоксида в 96% об. водном этаноле приводят к получению 5-аминоспиро[2.3]гексан-1-фосфоновой кислоты. При этом способ может включать трансформациию карбоксильной группы в аминогруппу, которая осуществляется в результате реакции метилового эфира 1-(диэтоксифосфорил)спиро[2.3]гексан-5-карбоновой кислоты, этилхлорформиата и триэтиламина, взятых в соотношении 1:1-2:1, с последующим взаимодействием с 1.5-2.0-кратным избытком азида натрия и более чем 10-кратным избытком трет-бутанола.
Техническим результатом предлагаемого технического решения является получение аминокислот, содержащих 5-аминоспиро[2.3]гексановый фрагмент и спиро[2.3]гексанфосфоновый фрагмент - биоизостерных конформационно-жестких аналогов ГАМК, а также расширение круга спирановых аминокислот, представляющих интерес в качестве конформационно-жестких аналогов ГАМК.
Полученные спиро[2.3]гексановые аминокислоты представляют собой конформационно-жесткие аналоги γ-аминомасляной кислоты, где фармакофорные группы (карбокси- и фосфонатная группа в 1-ом положении и аминогруппа в 5-ом положении) жестко закреплены относительно спирогексанового фрагмента. Получение соединений с ограниченной конформационной подвижностью может иметь хорошую перспективу с точки зрения поиска соединений с улучшенной селективностью по отношению к биомишеням ГАМК. В отличие от ациклических аналогов полученные аминокислоты имеют фиксированные конформации, что может обеспечить оптимальное связывание данных соединений с рецепторами ГАМК.
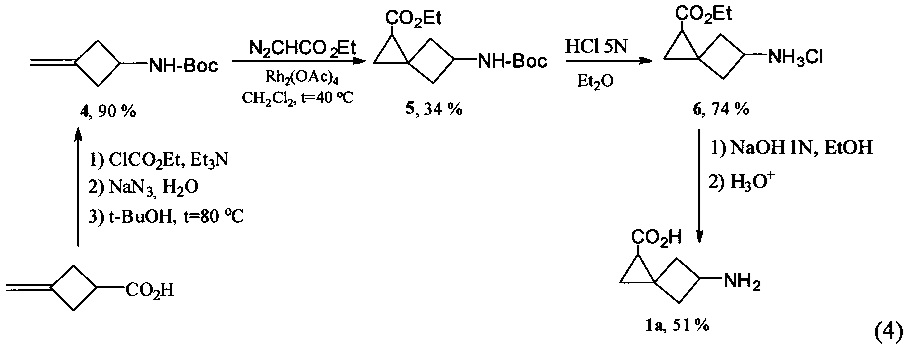
Предложенная схема получения аминокислоты 1а приведена ниже (4).
Предложенная схема получения аминокислоты 1b приведена ниже (5).
Осуществление изобретения
3-Метиленциклобутанкарбоновую кислоту [Н.N. Cripps, J.К. Williams, W.Н. Sharkey. Chemistry of cyclobutanes. I. Synthesis of methylenecyclobutanes. // Journal of American Chemical Society, 1959, Vol. 81, №11, P. 2723-2728], метиловый эфир 3-метиленциклобутанкарбоновой кислоты [Н.N. Cripps, J.К. Williams, W. H. Sharkey. Chemistry of cyclobutanes. I. Synthesis of methylenecyclobutanes. // Journal of American Chemical Society, 1959, Vol. 81, №11, P. 2723-2728], диэтиловый эфир (диазо)метилфосфоновой кислоты [D. Seyferth, R.S. Marmor, P. Hilbert. Some reactions of dimethylphosphono-substituted diazoalkanes. (MeO)2P(O)CR transfer to olefins and 1,3-dipolar additions of (MeO)2P(O)C(N2)R // Journal of Organic Chemistry, 1971. Vol. 36. №10. P. 1379-1386] и этиловый эфир диазоуксусной кислоты [Л. Титце, Т. Айхер, Препоративная органическая химия, Москва "Мир", 1999, С. 274] получали по известным методикам. Остальные реагенты являются коммерчески доступными, все процедуры, если не оговорено особо, осуществляли при комнатной температуре или температуре окружающей среды, то есть в диапазоне от 18 до 25°С; выпаривание растворителя осуществляли с использованием роторного испарителя, при пониженном давлении с температурой бани 50-60°С; контроль за ходом реакции осуществляли при помощи тонкослойной хроматографии (ТСХ), время реакции указано только для иллюстрации; структуру и чистоту всех выделенных соединений подтверждали, по меньшей мере, одним из следующих методов: ТСХ (пластины для ТСХ с предварительно нанесенным силикагелем 60 F254 Merck), масс-спектрометрия, элементный анализ или ядерный магнитный резонанс (ЯМР). Выход продукта приведен только для иллюстрации. Колоночную флэш-хроматографию осуществляли, используя Merck силикагель 60 (230-400 меш ASTM). Для ионообменной хроматографии использовали ионообменную смолу "Dowex 50WX8-100" (Sigma-Aldrich Со Ltd). Масс-спектры высокого разрешения (HRMS) положительных ионов зарегистрирован на спектрометре Jeol GCMate II при энергии ионизации 70 eV. Данные ЯМР определяли при 400 МГц (Bruker Avance-400 спектрометр), используя дейтерированный хлороформ (99,8% D), или дейтерированный метанол (99,8% D), или дейтерированную воду (99,9% D) в качестве растворителя, если не указано иное, относительно тетраметилсилана (TMS) в качестве внутреннего стандарта, миллионных долях (м.д.); обычные используемые сокращения следующие: с - синглет, д - дублет, т - триплет, кв - квартет, м - мультиплет, шир. - широкий и так далее. Химические символы имеют свои обычные значения: мкм (микрометр(ы)), мкл (микролитр(микролитры)), мкг (микрограмм(микрограммы)), М (моль(моли) на литр), л (литр(литры)), мл (миллилитр(миллилитры)), г (грамм(граммы)), мг (миллиграмм(миллиграммы)), моль(моли), ммоль(миллимоль(миллимоли)). Термин «гидролиз» означает химическую реакцию сложных эфиров с водой или иным(и) реагентом(ами) с образованием соответствующей кислоты.
Способ получения 5-аминоспиро[2.3]гексан-1-карбоновой кислоты, заключается в проведении четырехстадийного синтеза. На первой стадии синтеза в трехгорлую колбу, снабженную обратным холодильником, термометром, магнитной мешалкой и предварительно заполненную аргоном, вносят 3-5% раствор 3-(метилен)циклобутанкарбоновой кислоты в ацетоне, затем по каплям при интенсивном перемешивании при температуре -5±3°С вносят триэтиламин. После 10±2 мин перемешивания при той же температуре в реакционную смесь вносят этилхлорформиат. Компоненты - 3-(метилен)циклобутанкарбоновая кислота, этилхлорформиат и триэтиламин взяты в соотношении 1:1-1.5:1. Смесь перемешивают в течение 2±0.5 ч при той же температуре. Затем прибавляют 23-27% водный раствор азида натрия при 0±3°С, взятого в 1.1-1.7-кратном избытке по отношению к 3-(метилен)циклобутанкарбоновой кислоте, полученную смесь перемешивают еще 1.5-0.5 часа при той же температуре. После окончания перемешивания в реакционную смесь приливают ледяную воду (100±10 мл), экстрагируют порциями сначала холодного диэтилового эфира (3×15 мл), затем дихлорметаном (3×15 мл). Экстрагенты берут в объеме, не превышающем 0.5 объема прибавленной воды, органические вытяжки сушат над безводным сульфатом магния. Растворитель упаривают при пониженном давлении, к остатку прибавляют небольшое количество трет-бутанола, затем полученную смесь по каплям прибавляют к кипящему трет-бутанолу, взятому более чем 10-кратным избытком трет-бутанола по отношению к 3-(метилен)циклобутанкарбоновой кислоте и перемешивают. По окончании перемешивания растворитель упаривают при пониженном давлении. Продукт в индивидуальном состоянии выделяют методом препаративной колоночной хроматографии. На этой стадии синтеза, в ходе модифицированной реакции Курциуса производится трансформация карбоксильной группы в аминогруппу, защищенную Вос-фрагментом.
Далее в двухгорлую колбу, снабженную обратным холодильником и предварительно заполненную аргоном, вносят 3-5% раствор полученного алкена в дихлорметане и 1-10 мольных % тетраацетата диродия. Далее к полученной смеси при кипячении и интенсивном перемешивании прибавляют в течение 1±0.25 ч этилового эфира диазоуксусной кислоты (скорость прибавления 5±0.5 ммоль/ч), эфир берется в соотношении 1:1.2-2 по отношению к полученному на 1 стадии алкену. После окончания прибавления этилового эфира диазоуксусной кислоты реакционную смесь перемешивают при кипячении еще 1±0.25 ч. Растворитель упаривают при пониженном давлении. Продукт выделяют методом препаративной колоночной хроматографии.
Далее осуществляют последовательное удаление Вос-группы, для этого в колбу вносят 2-5% раствор полученного амида в диэтиловом эфире. К смеси при интенсивном перемешивании при 0±3°С приливают одной порцией 3-6N раствор хлороводорода в диэтиловом эфире. Реакционную смесь перемешивают при 0±5°С в течение 4-5 ч, а затем еще 18-25 часов при комнатной температуре. Растворитель упаривают при пониженном давлении, остаток сушат в вакууме над оксидом фосфора (V).
На заключительной стадии проводят гидролиз под действием водного раствора гидроксида натрия, для этого в одногорлую колбу вносят полученный этиловый эфир 5-аминоспиро[2.3]гексан-1-карбоновой кислоты гидрохлорид и 1N водного раствора NaOH, взятый в 2-5-кратном избытке. Смесь перемешивают при комнатной температуре в течение 48±3 ч. После окончания химической реакции растворитель упаривают при пониженном давлении. К полученному твердому остатку прибавляют 2-кратный избыток 1N раствора HCl по отношению к этиловому эфиру 5-аминоспиро[2.3]гексан-1-карбоновой кислоты гидрохлориду. Растворитель упаривают при пониженном давлении. Полученную 5-аминоспиро[2.3]гексан-1-карбоновую кислоту выделяют методом ионообменной хроматографии.
Способ получения 5-аминоспиро[2.3]гексан-1-фосфоновой кислоты заключается в проведении 5-стадийного синтеза, где на первой стадии в двухгорлую колбу, снабженную обратным холодильником и предварительно заполненную аргоном, вносят 15-20% раствор метилового эфира 3-метиленциклобутанкарбоновой кислоты в дихлорметане и 1-10 мольных % тетраацетата диродия и перемешивают. К полученной смеси при кипячении и интенсивном перемешивании в течение 1±0.1 ч прибавляют диэтиловый эфир (диазо)метилфосфоновой кислоты, взятый в соотношении 5:0.5-2 по отношению к метиловому эфиру 3-(метилен)циклобутанкарбоновой кислоты (скорость прибавления ~1 ммоль/ч). После окончания прибавления реакционную смесь перемешивают при кипячении еще 2±0.25 ч. Растворитель упаривают при пониженном давлении. Полученный эфир выделяют в индивидуальном состоянии методом препаративной колоночной хроматографии.
Далее в полученном метиловом эфире 1-(диэтоксифосфорил)спиро[2.3]гексан-5-карбоновой кислоты проводят гидролиз метоксикарбонильной группы, для этого в колбу вносят полученный эфир и 3-10-кратный избыток 1N раствора HCl. Смесь перемешивают в течение 2±0.5 ч при комнатной температуре. По окончании перемешивания растворитель упаривают при пониженном давлении. Полученную кислоту выделяют в индивидуальном состоянии методом препаративной колоночной хроматографии.
Далее с использованием модифицированной реакции Курциуса производят трансформацию карбоксильной группы в аминогруппу, защищенную Вос-фрагментом, для этого в трехгорлую колбу, снабженную обратным холодильником, термометром, магнитной мешалкой и предварительно заполненную аргоном, вносят 1-5% раствора 1-(диэтоксифосфорил)спиро[2.3]гексан-5-карбоновой кислоты в ацетоне, затем по каплям при интенсивном перемешивании при температуре -5±3°С вносят триэтиламин. После 10-15 мин перемешивания при той же температуре в реакционную смесь вносят этилхлорформиат. Компоненты - 1-(диэтоксифосфорил)спиро[2.3]гексан-5-карбоновая кислота, этилхлорформиат и триэтиламин взяты в соотношении 1-1:1-2:1. Смесь перемешивают в течение 2-4 ч при температуре -5±2°С. Затем прибавляют 1.5-2.0-кратный избыток водного раствора азида натрия при 0±5°С и полученную смесь перемешивают еще 1.5±0.5 часа при той же температуре. После окончания перемешивания в реакционную смесь прибавляют ледяную воду (30 мл), экстрагируют порциями сначала холодного диэтилового эфира (3×5 мл), затем дихлорметаном (3×5 мл). Экстрагенты берут в объеме, не превышающем 0.5 объема прибавленной воды, органические вытяжки сушат над безводным сульфатом магния. Растворитель упаривают при пониженном давлении, к остатку прибавляют небольшое количество трет-бутанола, затем полученную смесь по каплям прибавляют к кипящему трет-бутанолу, взятому более чем 10-кратным избытком трет-бутанола по отношению к 1-(диэтоксифосфорил)спиро[2.3]гексан-5-карбоновой кислоте, и перемешивают. По окончании перемешивания растворитель упаривают при пониженном давлении. Полученный амид выделяют методом препаративной колоночной хроматографии.
Далее последовательно удаляют Вос-группу трет-бутилового эфира N-[1-(диэтоксифосфорил)спиро[2.3]гекс-5-ил]карбаминовой кислоты, для этого в колбу вносят 1-5% раствор полученного амида в диэтиловом эфире, к раствору при интенсивном перемешивании при 0±5°С прибавляют одной порцией более чем 50-кратный избыток 3-6N раствора хлороводорода в диэтиловом эфире. Реакционную смесь перемешивают при 0±5°С в течение 4-5 ч, а затем еще 15-30 часов при комнатной температуре. Растворитель упаривают при пониженном давлении, остаток сушат в вакууме над оксидом фосфора (V).
На заключительной стадии проводят расщепление диэтоксифосфорильной группы, для этого в колбу, снабженную обратным холодильником и магнитной мешалкой, вносят 5-10% раствор диэтилового эфира 5-аминоспиро[2.3]гексан-1-фосфоновой кислоты гидрохлорида в дихлорметане, затем к смеси при интенсивном перемешивании по каплям прибавляют 3-7-кратный избыток 20-30% раствора триметилсилилбромида в дихлорметане. Реакционную смесь кипятят 7-8 ч, растворитель удаляют при пониженном давлении. Полученную смесь обрабатывают более чем 50-кратным избытком 72% раствора пропиленоксида в 96% об. водном этаноле. Выпавшую в осадок 5-аминоспиро[2.3]гексан-1-фосфоновую кислоту отфильтровывают и перекристаллизовывают из этанола.
Соотношение компонентов, указанных в избытках, подразумевает мольное соотношение компонентов по отношению друг к другу.
Представленные ниже примеры конкретного осуществления изобретения приведены для предоставления специалистам в данной области техники полного описания проведения и применения анализа по изобретению, но не ограничивают предполагаемый авторами изобретения объем изобретения.
Пример 1. Получение 5-аминоспиро[2.3]гексан-1-карбоновой кислоты (1а).
трет-Бутиловый эфир N-[3-метиленциклобутил]карбаминовой кислоты (4).
В трехгорлую колбу емкостью 10 мл, снабженную обратным холодильником, термометром, магнитной мешалкой и предварительно заполненную аргоном, внесли 215 мг (1.91 ммоль) 3-метиленциклобутанкарбоновой кислоты и 6 мл ацетона, затем по каплям при интенсивном перемешивании при (-5°С) внесли 192 мг (1.91 ммоль) триэтиламина. После 10 мин перемешивания при той же температуре в реакционную смесь внесли 263 мг (2.43 ммоль) этилхлорформиата. Смесь перемешивали в течение 2 ч при (-5°С). Затем прибавили раствор 165 мг (2.54 ммоль) азида натрия в 0.5 мл воды при 0°С и перемешивали смесь еще 1.5 часа при той же температуре. После окончания перемешивания в реакционную смесь прибавили 100 мл ледяной воды, проэкстрагировали холодным диэтиловым эфиром (3×15 мл), затем дихлорметаном (3×15 мл) и высушили над безводным сульфатом магния. Растворитель упарили при пониженном давлении, к остатку прибавили 3 мл трет-бутанола. Полученную смесь по каплям прибавили к 20 мл кипящего трет-бутанола. По окончании перемешивания растворитель упарили при пониженном давлении. Продукт в виде белых кристаллов был выделен в индивидуальном состоянии методом препаративной колоночной хроматографии (Rf=0.65, элюент этилацетат/петролейный эфир = 1:2). Выход 315 мг (90%), т.п.л. 62°С.
ЯМР 1H (CDCl3, δ, м.д.): 1.42 с (9H, 3×СН3), 2.50-2.62 м (2H, су-Bu-СН2), 2.92-3.09 м (2H, су-Bu-CH2), 4.09-4.18 м (1H, су-Bu-СН), 4.79-4.82 м (2H, СН2), 4.92 уш. с (1H, -NH-). ЯМР 13С (CDCl3, δ, м.д.): 29.36 (3СН3), 40.71 (су-Bu-СН2), 41.70 (су-Bu-СН), 79.28 (С), 106.95 (СН2), 142.26 (cy-Bu-CH), 155.11 (С=O). HRMS (ESI, mlz) рассчитано для C10H17NO2 [M+Na]+, 206.1151; найдено 206.1150.
трет-Бутиловый эфир N-[1-(этоксикарбонил)спиро[2.3]гекс-5-ил]карбаминовой кислоты (5).
В двухгорлую колбу емкостью 25 мл, снабженную обратным холодильником и предварительно заполненную аргоном, внесли 270 мг (1.47 ммоль) алкена 4, 6 мл дихлорметана и 22 мг (0.05 ммоль, 5 мольных %) тетраацетата диродия. К полученной смеси при кипячении и интенсивном перемешивании прибавили в течение 1 ч 112 мг (0.98 ммоль) этилового эфира диазоуксусной кислоты (скорость прибавления 5 ммоль/ч). После окончания прибавления этилового эфира диазоуксусной кислоты реакционную смесь перемешивали при кипячении еще 1 ч. Растворитель упарили при пониженном давлении. Продукт в виде бесцветного масла был выделен в индивидуальном состоянии методом препаративной колоночной хроматографии (Rf=0.1, элюент этилацетат/петролейный эфир = 1:5). Выход 90 мг (34%), смесь двух изомеров в соотношении А/В = 4:5.
ЯМР 1Н (CDCl3, δ, м.д.) для смеси двух изомеров: 0.94 дд (2J=4.8 Гц, 3J=8.4 Гц, 1H, СН2 су-Pr) - для изомера А, 1.01 дд (2J=4.5 Гц, 3J=8.5 Гц, 1Н, СН2 су-Pr) - для изомера В, 1.10 дд (2J=4.8 Гц, 3J=5.5 Гц, 1Н, СН2, су-Pr) - для изомера А, 1.19 дд (2J=4.5 Гц, 3J=5.3 Гц 1Н, СН2, су-Pr) - для изомера В, 1.21 т (3J=7.1 Гц, 3Н, OCH2CH3) - для изомера В, 1.22 т (3J=7.2 Гц, 3Н, OCH2CH3) - для изомера А, 1.40 с (9Н+9Н, 3×СН3), 1.55 дд (2J=5.5 Гц, 3J=8.4 Гц, 1Н, СН, су-Pr) - для изомера А, 1.59 дд (2J=5.3 Гц, 3J=8.5 Гц, 1H, СН, су-Pr) - для изомера В, 2.00-2.58 м (4Н + 4Н, СН2, су-Bu-СН2), 4.06 к (3J=7.1 Гц, 2Н, ОСН2СН3) - для изомера А, 4.08 к (3J=7.1 Гц, 2Н, OCH2CH3) - для изомера В, 4.17-4.30 м (1Н + 1Н, су-Bu-СН), 4.76 уш. с.(1Н, -NH-) - для изомера В, 4.82 уш. с. (1Н, -NH-) - для изомера А. ЯМР 13С (CDCl3, δ, м.д.) для смеси двух диастереомеров: 14.36 (1JCH=127 Гц, OCH2CH3+OCH2CH3), 18.44 (1JCH=164 Гц, су-Pr-СН2), 20.51 (1JCH=163 Гц, су-Pr-СН2), 23.49 (7JCH=168 Гц, су-Pr-СН), 24.63 (1JCH=167 Гц, су-Pr-СН), 24.07 (Сспиро), 25.03 (Сспиро), 28.37 (1JCH=127 Гц, 3СН3+3СН3), 35.59 (1JCH=142 Гц, су-Bu-CH2), 37.39 (1JCH=142 Гц, су-Bu-CH2), 39.20 (1JCH=142 Гц, су-Bu-CH2), 39.33 (1JCH=142 Гц, су-Bu-CH2), 41.76 (1JCH=142 Гц, су-Bu-СН), 42.19 (1JCH=142 Гц, су-Bu-СН), 60.22 (1JCH=145 Гц, OCH2CH3), 60.28 (1JCH=145 Гц, OCH2CH3), 155.01 (2×С), 172.51 (-NH-C=O), 172.61 (-NH-C=O). HRMS (ESI, mlz) рассчитано для C14H23NO4 [M+Na]+, 292.1516; найдено, 292.1519.
Этилового эфира 5-аминоспиро[2.3]гексан-1-карбоновой кислоты гидрохлорид (6).
В колбу емкостью 10 мл внесли 40 мг (0.148 ммоль) амида 5 в 2 мл диэтилового эфира. К смеси при интенсивном перемешивании при 0°С прибавили одной порцией 9 мл 5N раствора хлороводорода в диэтиловом эфире. Реакционную смесь перемешивали при 0°С в течение 4 ч, а затем еще 20 часов при комнатной температуре. Растворитель упарили при пониженном давлении, остаток сушили в вакууме над оксидом фосфора (V). Стеклообразная масса. Выход 23 мг (74%), смесь двух изомеров в соотношении А/В=2:3. ЯМР 1Н (MeOD, δ, м.д.) для смеси двух изомеров: 1.02-1.32 м (1Н+1Н, су-Pr), 1.22 т (3Н+3Н, 3J=7 Гц, ОСН2СН3+OCH2CH3), 1.55-1.79 м (1Н+1Н, су-Pr), 2.20-2.90 м (4Н+4Н, су-Bu), 3.88-4.31 м (3Н+3Н, ОСН2СН3+су-Bu-СН), 8.57 уш. с (3Н+3Н, NH3). ЯМР 13С (MeOH-d, δ, м.д.) для смеси двух диастереомеров: 14.30 (1JCH=127 Гц, OCH2CH3+OCH2CH3), 18.84 (1JCH=164 Гц, су-Pr-CH2), 20.17 (1JCH=163 Гц, су-Pr-СН2), 22.97 (1JCH=168 Гц, су-Pr-CH), 23.80 (Сспиро), 24.25 (1JCH=167 Гц, су-Pr-CH), 24.77 (Сспиро), 32.63 (су-Bu-CH2), 34.49 (су-Bu-CH2), 35.55 (су-Bu-CH2), 35.78 (су-Bu-CH2), 42.02 (су-Bu-СН), 42.42 (су-Bu-СН), 60.46 (1JCH=145 Гц, OCH2+OCH2CH3), 172.16 (С=O), 172.24 (С=O). HRMS (ESI, mlz) рассчитано для C9H16NO2 [М]+, 170.1179; найдено 170.1176.
5-Аминоспиро[2.3]гексан-1-карбоновая кислота (1а).
В одногорлую колбу емкостью 10 мл внесли 23 мг (0.11 ммоль) гидрохлорида 6 и 0.33 мл 1N водного раствора NaOH. Смесь перемешивали при комнатной температуре в течение 48 ч. После окончания химической реакции растворитель упарили при пониженном давлении. К твердому остатку прибавили 0.22 мл 1N раствора HCl. Растворитель упарили при пониженном давлении. Аминокислота 1а была выделена методом ионообменной хроматографии (Dowex 50, элюент - 0.9 М водный раствор аммиака). Стеклообразная масса. Выход 10 мг (51%), смесь двух изомеров А/В=2:3. ЯМР 1H (MeOD, δ, м.д.) для смеси двух изомеров: 0.67 дд (2J=4.6 Гц, 3J=8.2 Гц, 1H, CH2 су-Pr) - для изомера А, 0.73 дд (2J=4.1 Гц, 3J=8.1 Гц, 1Н, CH су-Pr) - для изомера В, 0.94 дд (2J=4.6 Гц, 3J=5.6 Гц, 1Н, CH2, су-Pr) - для изомера А, 1.03 дд (2J=4.1 Гц, 3J=5.5 Гц 1Н, CH2, су-Pr) - для изомера В, 1.41 дд (2J=5.5 Гц, 3J=8.1 Гц, 1Н, СН, су-Pr) - для изомера В, 1.44 дд (2J=5.6 Гц, 3J=8.2 Гц, 1Н, СН, су-Pr) - для изомера А, 1.91-2.52 м (4Н+4Н, CH2, су-Bu-CH2), 3.47-3.61 м (1Н+1Н, су-Bu-СН). ЯМР 13С (MeOD, δ, м.д.) для изомера А: 16.58 (су-Pr-CH2), 21.17 (Сспиро), 39.13 (су-Bu-CH2), 41.16 (су-Bu-CH2), 43.55 (су-Bu-СН), 179.95 (СООН). ЯМР 13С (MeOD, δ, м.д.) для изомера В: 18.35 (су-Pr-CH2), 21.75 (Ccnupo), 27.07 (су-Pr-СН), 28.42 (су-Pr-СН), 37.68 (су-Bu-CH2), 41.22 (су-Bu-CH2), 43.85 (су-Bu-СН), 179.57 (СООН).
Пример 2. Получение 5-аминоспиро[2.3]гексан-1-фосфоновой кислоты (1b).
Метиловый эфир 1-(диэтоксифосфорил)спиро[2.3]гексан-5-карбоновой кислоты (7).
В двухгорлую колбу емкостью 10 мл, снабженную обратным холодильником и предварительно заполненную аргоном, внесли 725 мг (5.75 ммоль) метилового эфира 3-метиленциклобутанкарбоновой кислоты, 3 мл дихлорметана и 25 мг (0.058 ммоль, 5 мол. %) тетраацетата диродия. К полученной смеси при кипячении и интенсивном перемешивании прибавили в течение 1 ч 204 мг (1.15 ммоль) диэтилового эфира (диазо)метилфосфоновой кислоты (скорость прибавления ~1 ммоль/ч). После окончания прибавления реакционную смесь перемешивали при кипячении еще 2 ч. Растворитель упарили при пониженном давлении. Эфир 7 в виде бесцветного масла был выделен в индивидуальном состоянии методом препаративной колоночной хроматографии (Rf=0.4, элюент этилацетат/петролейный эфир = 1:1). Выход 152 мг (48%), смесь двух изомеров в соотношении 1:1.
ЯМР 1Н (CDCl3, δ, м.д.) для смеси двух изомеров: 0.82-0.86 м (1Н+1Н, су-Pr), 1.03-1.14 м (2Н+2Н, су-Pr), 1.33-1.38 м (6Н+6Н, 2×OCH2CH3), 2.40-2.84 м (4Н+4Н, су-Bu-CH2), 3.26-3.31 м (1Н+1Н, су-Bu-СН), 3.71 с (3Н+3Н, ОСН3), 4.07-4.15 м (4Н+4Н, 2×ОСН2СН3). ЯМР 13С (CDCl3, δ, м.д.) для смеси двух диастереомеров: 14.5 (1JCP=192 Гц, су-Pr-СН) 15.5 (1JCP=192 Гц, су-Pr-СН), 16.0 (2JCP=5 Гц су-Pr-CH2), 16.1 (2×OCH2CH3), 16.7 (2JCP=5 Гц, су-Pr-CH2), 21.9 (2JCP=5 Гц, Сспиро), 22.5 (2JCP=5 Гц, Сспиро), 30.8 (3JCP=5 Гц, су-Bu-CH2), 32.0 (3JCP=5 Гц, су-Bu-CH2), 33.2 (3JCP=5 Hz, су-Bu-CH2), 33.3 (3JCP=5 Гц, су-Bu-CH2), 33.4 (су-Bu-СН), 33.3 (су-Bu-СН), 51.4 (2×ОСН3), 61.2 (2×OCH2CH3+2×OCH2CH3), 175.1 (СОО), 175.2 (СОО). ЯМР 31Р (CDCl3, δ, м.д.): 28.05, 28.06. Вычислено для C12H21O5P (%):. С 52.17, Н 7.66. Найдено, %. С 51.88, Н 7.62.
1-(Диэтоксифосфорил)спиро[2.3]гексан-5-карбоновая кислота (8).
В колбу емкостью 25 мл внесли 144 мг (0.52 ммоль) эфира 7 и 2.7 мл 1N раствора HCl. Смесь перемешивали в течение 2 ч при комнатной температуре. По окончании перемешивания растворитель упарили при пониженном давлении. Кислота 8 в виде бесцветного масла была выделена в индивидуальном состоянии методом препаративной колоночной хроматографии (Rf=0.25, элюент этилацетат). Выход 130 мг (95%), смесь двух изомеров в соотношении 1:1.
ЯМР 1Н (CDCl3, δ, м.д.) для смеси двух изомеров: 0.76-0.85 м (1Н+1Н, су-Pr), 0.92-1.07 м (1Н+1Н, су-Pr), 1.10-1.21 м (1Н+1Н, су-Pr), 1.24-1.34 м (6Н+6Н, 2×OCH2CH3), 2.12-2.21 м (1H, су-Bu-CH2), 2.30-2.40 м (1Н+1Н, су-Bu-CH2), 2.42-2.52 м (1Н+1Н, су-Bu-CH2), 2.58-2.65 м (1Н, су-Bu-CH2), 2.67-2.84 м (1Н+1Н, су-Bu-CH2), 3.15-3.30 м (1Н+1Н, cy-Bu-CH), 4.00-4.15 м (4H+4H, 2×OCH2CH3), 8.56 уш. с (1Н+1Н, СООН). ЯМР 13С (CDCl3, δ, м.д.) для смеси двух диастереомеров: 14.6 (1JCP=192 Гц, су-Pr-СН) 15.6 (1JCP=192 Гц, су-Pr-СН), 16.3 (2×ОСН2СН3+2×ОСН2СН3), 16.6 (2JCP=5 Гц су-Pr-СН2), 17.0 (2JCP=5 Гц, су-Pr-СН2), 22.2 (2JCP=5 Гц, Сспиро), 22.8 (2JCP=5 Гц, Сспиро), 31.1 (3Jcp=5 Гц, су-Bu-CH2), 32.2 (3JCp=5 Гц, су-Bu-CH2), 33.4 (3JCP=5 Гц, су-Bu-CH2), 33.5 (3Jcp=5.48 Гц, су-Bu-CH2), 33.7 (су-Bu-СН), 33.8 (су-Bu-СН), 61.8 (2×ОСН2СН3), 61.9 (2×OCH2CH), 178.2 (СООН), 178.7 (СООН). ЯМР 31Р (CDCl3, δ, м.д.) для смеси двух диастереомеров: 28.5, 28.6. HRMS (ESI, mlz) рассчитано для C11H19PO5 [М+Н]+, 263.1043; найдено 263.1045.
трет-Бутиловый эфир N-[1-(диэтоксифосфорил)спиро[2.3]гекс-5-ил]карбаминовой кислоты (9).
В трехгорлую колбу емкостью 10 мл, снабженную обратным холодильником, термометром, магнитной мешалкой и предварительно заполненную аргоном, внесли 120 мг (0.46 ммоль) кислоты 8 и 3 мл ацетона, затем по каплям при интенсивном перемешивании при (-5°С) внесли 46 мг (0.46 ммоль) триэтиламина. После 10 мин перемешивания при той же температуре в реакционную смесь внесли 85 мг (0.79 ммоль) этилхлорформиата. Смесь перемешивали в течение 2 ч при (-5°С). Затем прибавили раствор 53 мг (0.82 ммоль) азида натрия в 0.3 мл воды при 0°С и перемешивали смесь еще 1.5 часа при той же температуре. После окончания перемешивания в реакционную смесь прибавили 30 мл ледяной воды, проэкстрагировали холодным диэтиловым эфиром (3×5 мл), затем дихлорметаном (3×5 мл) и высушили над безводным сульфатом магния. Растворитель упарили при пониженном давлении, к остатку прибавили 0.9 мл трет-бутанола. Полученную смесь по каплям прибавили к 7.5 мл кипящего трет-бутанола. По окончании перемешивания растворитель упарили при пониженном давлении. Амид 9 в виде бесцветного масла был выделен в индивидуальном состоянии методом препаративной колоночной хроматографии (Rf=0.2, элюент этилацетат/петролейный эфир = 1:4). Выход 61 мг (40%), смесь двух изомеров в соотношении 1:1.
Диэтиловый эфир 5-аминоспиро[2.3]гексан-1-фосфоновой кислоты гидрохлорид (10).
В колбу емкостью 10 мл внесли 50 мг (0.149 ммоль) амида 9 в 2 мл диэтилового эфира. К смеси при интенсивном перемешивании при 0°С прибавили одной порцией 9 мл 5N раствора хлороводорода в диэтиловом эфире. Реакционную смесь перемешивали при 0°С в течение 4 ч, а затем еще 20 часов при комнатной температуре. Растворитель упарили при пониженном давлении, остаток сушили в вакууме над оксидом фосфора (V). Стеклообразная масса. Выход 35 мг (87%), смесь двух изомеров в соотношении А/В = 1:1.
ЯМР 1Н (CDCl3, δ, м.д.) для смеси двух диастереомеров: 0.83-1.00 м (1Н+1Н, су-Pr), 1.05-1.20 м (2Н+2Н, су-Pr), 1.25-1.35 м (6Н+6Н, 2×ОСН2СН3), 2.20-2.60 м (2Н+2Н, су-Bu-CH2), 2.65-2.85 (2Н+2Н, су-Bu-CH2) 2.90-3.05 м (1Н+1Н, су-Bu-СН), 3.95-4.20 м (4Н+4Н, 2×OCH2CH3), 8.70 уш. с (3Н+3Н, NH3). ЯМР 13С (CDCl3, δ, м.д.) для смеси двух диастереомеров: 13.1 (1JCP=192 Гц, су-Pr-СН), 14.9 (1JCP=192 Гц, су-Pr-СН), 15.5 (2Jcp=5 Гц, су-Pr-CH2), 16.4 (2×OCH2CH3+2×OCH2CH3), 16.7 (2JCP=5 Гц, су-Pr-CH2), 20.5 (2JCP=5 Гц, Сспиро), 20.9 (2JCP=5 Гц, Сспиро), 32.9 (су-Bu-CH2), 33.0 (3JCP=5 Hz, су-Bu-CH2), 34.8 (3JCP=5 Hz, су-Bu-CH2), 35.9 (3JCP=5 Гц, су-Bu-CH2), 42.4 (2×су-Bu-СН), 61.9 (2×OCH2CH3+2×OCH2CH3). ЯМР 31Р (CDCl3, δ, м.д.) для смеси двух диастереомеров: 27.2, 27.9. HRMS (ESI, mlz) рассчитано для C10H20NO3P [М+Н]+, 234.1254; найдено 234.1263.
5-Аминоспиро[2.3]гексан-1-фосфоновая кислота (1b).
В колбу емкостью 10 мл, снабженную обратным холодильником и магнитной мешалкой, внесли 32 мг (0.12 ммоль) гидрохлорида 10 в 0.4 мл дихлорметана, затем к смеси при интенсивном перемешивании по каплям прибавили раствор 91.8 мг (0.6 ммоль) триметилсилилбромида в 0.24 мл дихлорметана. Реакционную смесь кипятили 7-8 ч, растворитель удалили при пониженном давлении. Остаток растворили в 2 мл 96% об. водного этанола, после чего при перемешивании прибавили 5 мл пропиленоксида. Выпавшую в осадок аминофосфоновую кислоту 1b отфильтровали и перекристаллизовали из этанола. Белые кристаллы, т.пл.=293°С. Выход 18 мг (87%), смесь двух изомеров в соотношении А/В=1:1.
ЯМР 1Н (D2O, δ, м.д.) для смеси двух изомеров: 0.84-0.98 м (2Н+2Н, су-Pr), 0.99-1.10 м (1Н+1Н, су-Pr), 2.28-2.57 м (3Н+2Н, су-Bu-CH2), 2.66-2.76 м (1H, су-Bu-CH2), 3.84-4.00 м (1Н+1Н, су-Bu-СН) (сигналы протонов NH2-группы и ОН-групп не наблюдаются). ЯМР 13С (D2O, δ, м.д.) для смеси двух диастереомеров: 14.7 (1JCP=185 Гц, су-Pr-СН), 15.1 (2JCP=5 Гц, су-Pr-CH2), 15.7 (2JCP=5 Гц, су-Pr-CH2), 15.8 (JJCP=185 Гц, су-Pr-СН), 19.2 (2JCP=5 Гц, Сспиро), 19.9 (2JCP=5 Гц, Сспиро), 32.1 (су-Bu-CH2), 33.6 (3JCP=5 Hz, cy-Bu-CH2), 34.8 (3JCP=5 Hz, cy-Bu-CH2), 35.1 (3JCP=5 Гц, cy-Bu-CH2), 41.7 (cy-Bu-CH), 42.0 (cy-Bu-CH). ЯМР 31P (D2O, δ, м.д.) для смеси двух диастереомеров: 26.6, 26.7. Вычислено для C6H12NO13P (%): С, 40.68; Н, 6.83; N, 7.91. Найдено: С, 40.60; Н, 6.99; N, 7.68.
![СПИРО[2.3]ГЕКСАНОВЫЕ АМИНОКИСЛОТЫ - КОНФОРМАЦИОННО-ЖЕСТКИЕ АНАЛОГИ γ-АМИНОМАСЛЯНОЙ КИСЛОТЫ - И СПОСОБЫ ИХ ПОЛУЧЕНИЯ](https://fips.edrid.ru/images/rid/45/f6/36/f449dff35d0a6306957f20438f281c93.jpg)
![СПИРО[2.3]ГЕКСАНОВЫЕ АМИНОКИСЛОТЫ - КОНФОРМАЦИОННО-ЖЕСТКИЕ АНАЛОГИ γ-АМИНОМАСЛЯНОЙ КИСЛОТЫ - И СПОСОБЫ ИХ ПОЛУЧЕНИЯ](https://fips.edrid.ru/images/rid/45/f6/36/ad0f16aced4269444ba54a16f3ef3d4c.jpg)
Трициклические производные n,n'-замещенных 3,7-диазабицикло[3.3.1]нонанов, обладающие фармакологической активностью, и лекарственные средства на их основе
Высокотемпературный уплотнительный материал и способ его получения
Алициклические производные n, n'-замещенных 3,7-диазабицикло[3.3.1]нонанов, обладающие фармакологической активностью, и лекарственные средства на их основе
Способ профилактики и лечения язвенной болезни желудка, вызываемой приемом этанолсодержащих жидкостей
Способ профилактики и лечения язвенной болезни желудка, вызываемой стрессом
Способ профилактики и лечения язвенных поражений желудка, вызываемых приемом нестероидных противовоспалительных лекарственных средств
Способ получения наногибридного функционального сепарационного материала на основе модифицированного носителя и модифицированных наночастиц металла
Новые производные 2,3,4,5-тетрагидро-1-пиридо[4,3-b]индола и способы их применения
Композиция для создания органических фотогальванических элементов на основе фталоцианинов и их аналогов
Наногибридный функциональный сепарационный материал на основе модифицированного носителя и модифицированных наночастиц металла
Трициклические производные n,n'-замещенных 3,7-диазабицикло[3.3.1]нонанов, обладающие фармакологической активностью, и лекарственные средства на их основе
Высокотемпературный уплотнительный материал и способ его получения
Алициклические производные n, n'-замещенных 3,7-диазабицикло[3.3.1]нонанов, обладающие фармакологической активностью, и лекарственные средства на их основе
Способ профилактики и лечения язвенной болезни желудка, вызываемой приемом этанолсодержащих жидкостей
Способ профилактики и лечения язвенной болезни желудка, вызываемой стрессом
Способ профилактики и лечения язвенных поражений желудка, вызываемых приемом нестероидных противовоспалительных лекарственных средств
Способ получения наногибридного функционального сепарационного материала на основе модифицированного носителя и модифицированных наночастиц металла
Новые производные 2,3,4,5-тетрагидро-1-пиридо[4,3-b]индола и способы их применения
Способ определения следовых компонентов методом лазерно-искровой эмиссионной спектроскопии
Способ синтеза сополимеров акрилонитрила с акриловой кислотой

