Результат интеллектуальной деятельности: ПРОИЗВОДНОЕ ФЕНИЛПИРРОЛА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым производным фенилпиррола и их фармацевтическим применениям, в частности в качестве превентивных или терапевтических агентов для заболеваний, связанных с рецепторами гистамина Н3.
УРОВЕНЬ ТЕХНИКИ
Гистамин обычно накапливается во внутриклеточных гранулах в мастоцитах, печени, почках и слизистой оболочке желудка и т.д. и высвобождается из клетки в ответ на внешние стимулы, такие как связывание антигена с антителом на поверхности клетки. Например, когда мастоциты стимулируются антигеном, входящим снаружи, гистамин высвобождается из мастоцитов и стимулирует рецепторы гистамина Н1 (Н1), расположенные на кровеносных сосудах или гладких мышцах, тем самым индуцируя аллергическую реакцию. Таким же образом, гистамин, высвобождаемый из клеток ELC (клеток, подобных аргирофильным клеткам) на слизистой оболочке желудка, стимулирует промотирование рецепторами гистамина Н2 (Н2) на париетальных клетках желудка секреции желудочной кислоты. На основании этих фактов антагонисты рецепторов Н1 и Н2 были разработаны в качестве терапевтических агентов для аллергических заболеваний и язвы желудка, соответственно, и в настоящее время находят широкое применение в качестве лекарственных средств.
Обнаружено, что гистамин служит в качестве нейротрансмиттера и оказывает действие на рецепторы гистамина (рецепторы гистамина Н3 (Н3)), расположенные на нервах центральной и периферической нервных систем для проявления ими различных физиологических функций. Этот рецептор был клонирован в 1999 г, и были определены последовательность его гена и его аминокислотная последовательность. Однако, аминокислотная последовательность рецептора Н3 имеет только 22% и 21,4% гомологии с аминокислотными последовательностями рецептора Н1 и рецептора Н2, соответственно (см. непатентный документ 1). Показано, что рецепторы Н3, которые присутствуют в пресинаптической мембране, служат в качестве ауторецепторов, которые регулируют синтез и высвобождение гистамина (см. непатентный документ 2). Кроме этого, было показано, что рецепторы Н3 регулируют высвобождение других нейротрансмиттеров, включающих ацетилхолин, серотонин, допамин и норадреналин (см. непатентный документ 3). Было также предположено, что рецепторы Н3 являются активными в отсутствие агонистов и эта активность способна ингибироваться соединениями, действующими в качестве обратных агонистов. Эти факты позволяют предположить, что антагонисты или обратные агонисты рецептора Н3 увеличивают высвобождение регулируемых рецептором Н3 нейротрансмиттеров и могут потенциально служить в качестве терапевтических агентов для различных заболеваний, связанных с аномальным их высвобождением.
Фактически результаты экспериментов на животных моделях показывают возможность того, что антагонисты или обратные агонисты рецептора Н3 можно применять в качестве терапевтических агентов для деменции, болезни Альцгеймера (см. непатентные документы 4 и 5), синдрома дефицита внимания и гиперактивности (см. непатентный документ 6), шизофрении (см. непатентный документ 7), эпилепсии, “центральной” судороги (судороги с участием сердцевины мышечных волокон) и т.д.
Было показано, что рецепторы Н3 принимают участие в пищевом поведении (см. непатентный документ 8) и метаболических заболеваниях, включающих ожирение, сахарный диабет, гиперлипидемию и т.д., а также предположительно при таких заболеваниях, для которых показаны антагонисты или обратные агонисты рецептора Н3.
Было показано, что гистамин регулирует циркадный ритм в головном мозге и является ответственным за поддержания баланса между состояниями бодрствования и сна (см. непатентные документы 9 и 10), и заболевания, связанные с нарушениями сна, включающие нарколепсию, идиопатическую гиперсомнию, индуцированный поведением синдром недостаточного сна, синдром апное во сне, нарушение циркадного ритма, парасомнию, связанное со сном нарушение движения, инсомнию и депрессию, также считаются заболеваниями, для которых показаны антагонисты или обратные агонисты рецептора Н3.
Показано также, что рецепторы Н3 присутствуют в симпатических нервах на слизистой оболочке носа, и описано, что комбинированное применение антагонистов рецепторов Н3 и Н1 значительно улучшает состояние заложенности носа (см. непатентный документ 11). Это указывает на возможность того, что антагонисты или обратные агонисты рецептора Н3 являются применимыми для лечения таких заболеваний, как аллергический ринит, когда их применяют либо отдельно, либо в комбинации с антагонистами рецептора Н1.
Описания антагонистов или обратных агонистов рецептора Н3 имеются в нескольких обзорах (см. непатентные документы 12-15), ссылку на которые можно указать. Ранее было описано много производных имидазола, которые были получены из самого гистамина, применяемого в качестве основного соединения. Однако, они все же не были разработаны в качестве лекарственных средств вследствие возможности ингибирования метаболизирующего лекарственные средства фермента цитохрома Р450 (CYP).
В последние годы во многих документах и патентах (см. патентные документы 1-10) были описаны неимидазольные антагонисты или обратные агонисты рецептора Н3.
Представлены также сообщения об антагонистах рецептора гистамина Н3, имеющих 5-членные ароматические кольца, такие как кольцо пиразола (см. патентные документы 11-14). Кроме того, был описан антагонист рецептора гистамина Н3, имеющий скелет арилоксипиперидина, который замещен незамещенным пирролом (см. патентный документ 15). Однако, не имеется сообщения о соединениях, имеющих структуры, описанные ниже.
ПЕРЕЧЕНЬ ССЫЛОК
ПАТЕНТНЫЕ ДОКУМЕНТЫ
Патентный документ 1: публикация международной заявки WO2005097751.
Патентный документ 2: публикация международной заявки WO2005097778.
Патентный документ 3: публикация международной заявки WO2005118547.
Патентный документ 4: публикация международной заявки WO2006014136.
Патентный документ 5: публикация международной заявки WO2006045416.
Патентный документ 6: публикация международной заявки WO2006046131.
Патентный документ 7: публикация международной заявки WO2006059778.
Патентный документ 8: публикация международной заявки WO2006061193.
Патентный документ 9: публикация международной заявки WO2006107661.
Патентный документ 10: публикация международной заявки WO2006103057.
Патентный документ 11: публикация международной заявки WO2006103045.
Патентный документ 12: публикация международной заявки WO2007094962.
Патентный документ 13: публикация международной заявки WO2008072724.
Патентный документ 14: публикация международной заявки WO2009063953.
Патентный документ 15: публикация международной заявки WO2002012190.
НЕПАТЕНТНЫЕ ДОКУМЕНТЫ
Непатентный документ 1: Lovenberg T.W. et al., Molecular pharmacology, 55, 1101-1107, 1999.
Непатентный документ 2: Arrang J-M. еt al., Nature, 302, 832-837, 1983.
Непатентный документ 3: Brown R.E. et al., Progress in Neurobiology, 63, 637-672, 2001.
Непатентный документ 4: Huang Y-W. еt al., Behavioural Brain Research, 151, 287-293, 2004.
Непатентный документ 5: Komater V.A. et al., Behavioural Brain Research, 159, 295-300, 2005.
Непатентный документ 6: Passani M.B. et al., Neuroscience and Biobehavioral Reviews, 24, 107-113, 2000.
Непатентный документ 7: Fox G.B. et al., J. Pharmacol. Exp. Ther., 313, 176-190, 2005.
Непатентный документ 8: Hancock A.A. et al., Curr. Opin. Investig. Drug, 4, 1190-1197.
Непатентный документ 9: Huang Z-L. et al., Prog. Natr. Acad. Sci., 103, 4687-4692, 2006.
Непатентный документ 10: Babier A.J. et al., Br. J. Pharmacol., 143, 649-661, 2004.
Непатентный документ 11: McLeod R.L. et al., Am. J. Rhinol., 13, 391-399, 1999.
Непатентный документ 12: Schwartz J.C. et al., Trends in Pharmacol. Sci., 7, 24-28, 1986.
Непатентный документ 13: Passani M.B. et al., Trends in Pharmacol. Sci., 25, 618-625, 2004.
Непатентный документ 14: Leurs R. et al., Nature Drug Discovery, 4, 107-122, 2005.
Непатентный документ 15: Leurs R. et al., Drug Discovery Today, 10, 1613-1627, 2005.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Техническая проблема
Целью настоящего изобретения является предоставление новых соединений или их фармацевтически приемлемых солей, которые обладают сильным действием в отношении ингибирования связывания гистамина с рецептором гистамина Н3 и которые пригодны в предотвращении или лечении нарушений, обусловленных рецептором гистамина Н3, например, таких заболеваний, как деменция, болезнь Альцгеймера, синдром дефицита внимания и гиперактивности, шизофрении, эпилепсии, “центральная” судорога, ожирение, сахарный диабет, гиперлипидемия, нарколепсия, идиопатическая гиперсомния, индуцированный поведением синдром недостаточного сна, синдром апное во сне, нарушение циркадного ритма, парасомния, связанное со сном нарушение движения, инсомния и депрессия или аллергический ринит.
РЕШЕНИЕ ПРОБЛЕМЫ
Для достижения указанной выше цели авторы настоящего изобретения провели интенсивные исследования и обнаружили в результате этого, что производные фенилпиррола, имеющие карбонильный заместитель в 3-положении кольца пиррола, проявляют сильную ингибирующую активность против связывания гистамина с рецептором гистамина Н3. Эти полученные данные привели к завершению настоящего изобретения.
В дальнейшем настоящее изобретение будет описано подробнее. Варианты осуществления настоящего изобретения (соединения вариантов осуществления будут называться “соединениями изобретения”) показаны ниже.
Вкратце, настоящее изобретение относится к следующим пунктам.
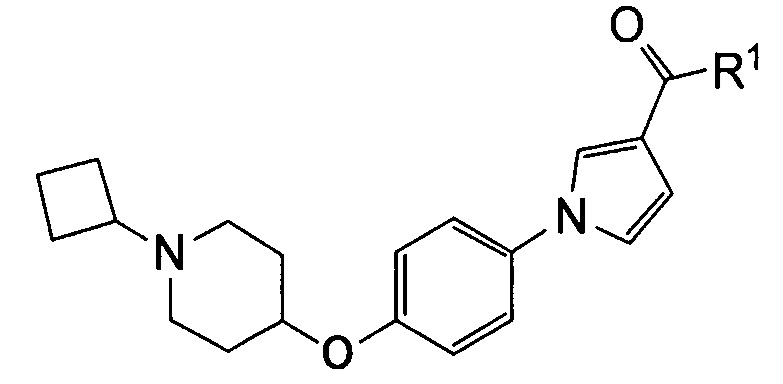
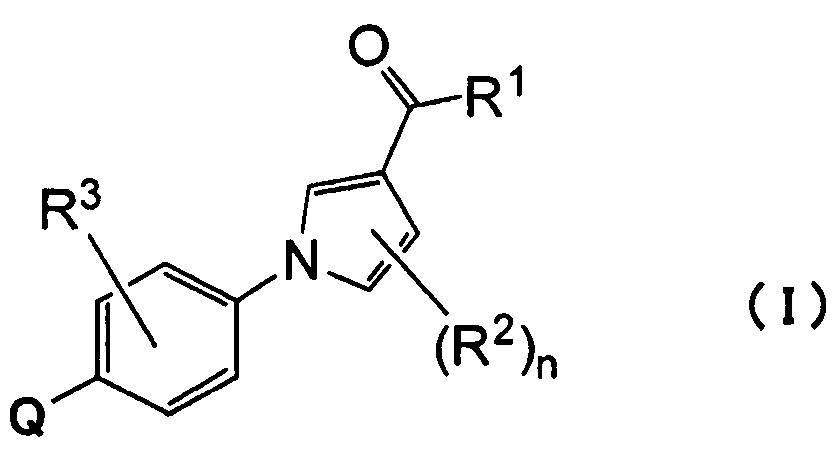
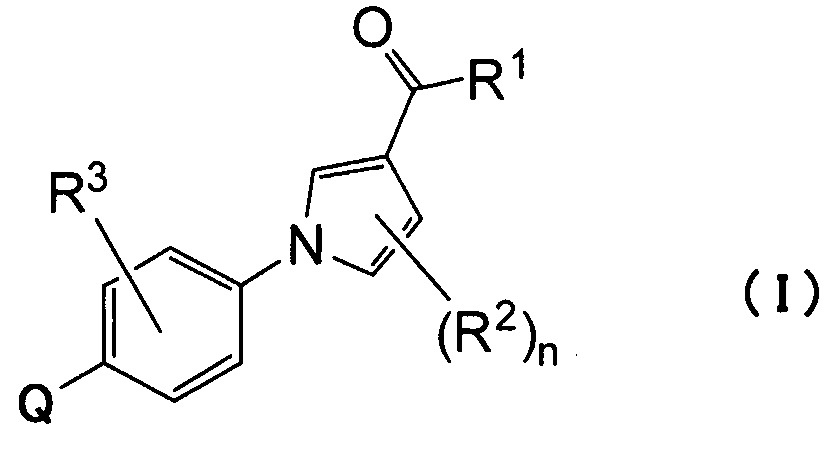
(1) Соединению, представленному формулой (I)
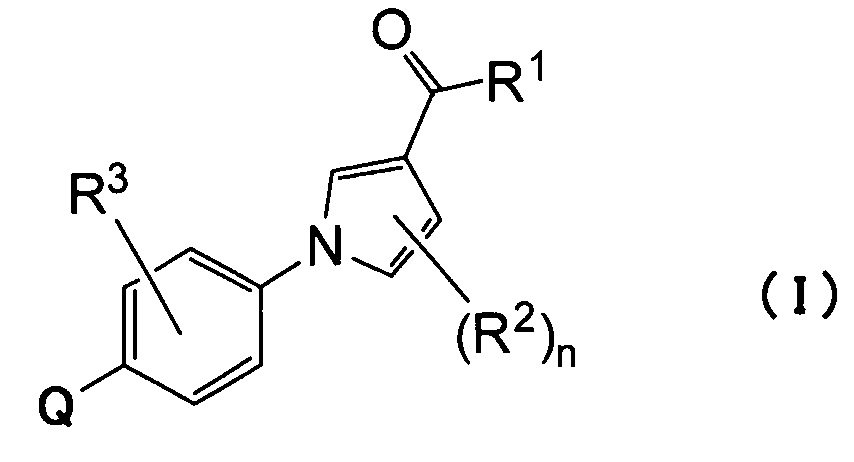
где
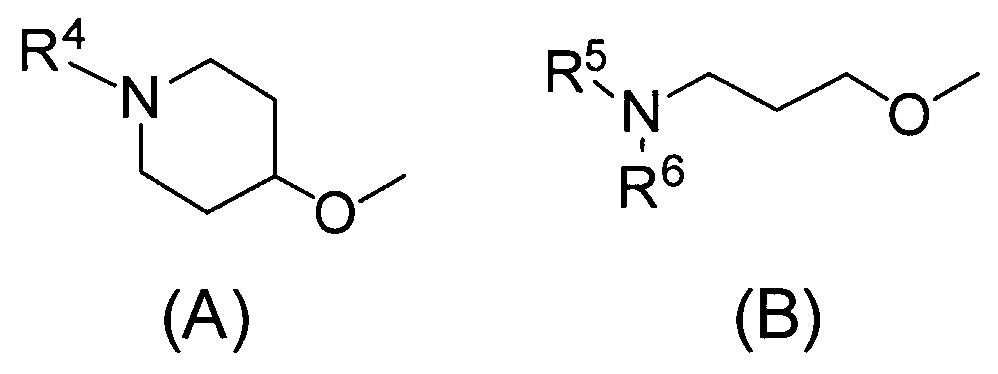
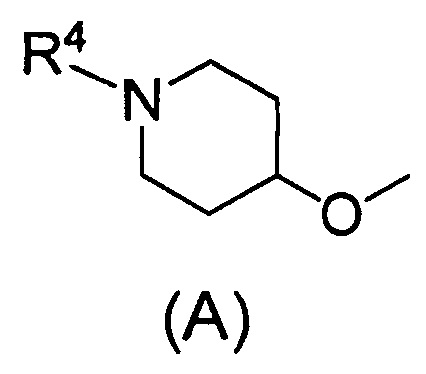
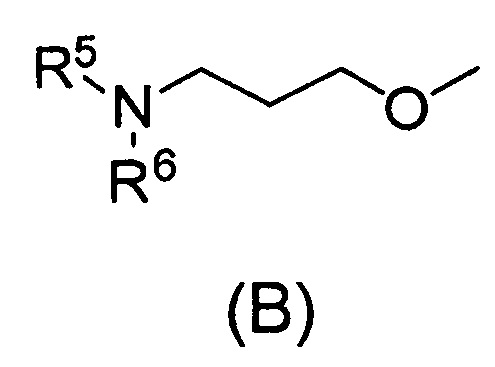
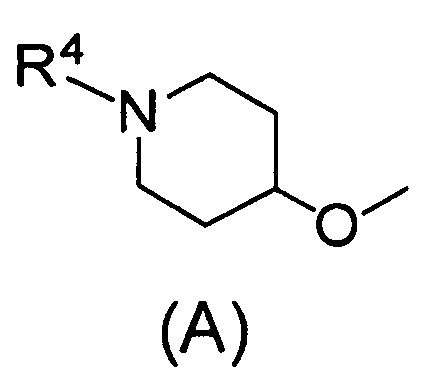
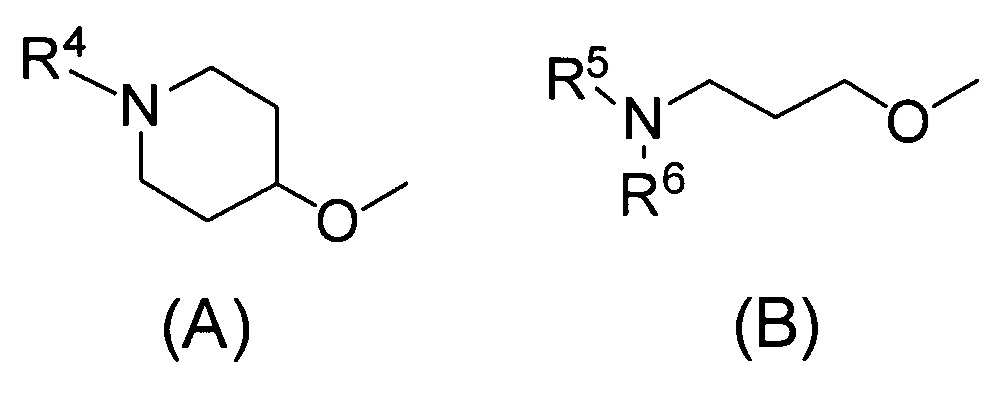
Q относится к группе, представленной следующей формулой (А) или (В)
R1 представляет собой гидроксил, С1-С6алкокси или NR1AR1B;
каждый из R1A и R1B, которые могут быть одинаковыми или разными, представляет собой атом водорода, С1-С6алкил или С3-С7циклоалкил или
R1A и R1B связаны вместе с соседним атомом азота с образованием 3-7-членного насыщенного гетероциклического кольца (причем насыщенное гетероциклическое кольцо необязательно замещено одним или двумя С1-С6алкилами);
R2 представляет собой атом водорода, атом галогена или С1-С6алкил;
n равно 1 или 2;
R3 представляет собой атом водорода, атом галогена или С1-С6алкил;
R4 представляет собой С1-С6алкил (С1-С6алкил может быть замещен одним или двумя С3-С7циклоалкилами) или С3-С7циклоалкил (С3-С7циклоалкил может быть замещен одним или двумя С1-С6алкилами);
каждый из R5 и R6, которые могут быть одинаковыми или разными, представляет собой С1-С6алкил или С3-С7циклоалкил, или
R5 и R6 связаны вместе с соседним атомом азота с образованием 3-7-членного насыщенного гетероциклического кольца (причем насыщенное гетероциклическое кольцо необязательно замещено одним или двумя С1-С6алкилами)] или его фармацевтически приемлемой соли.
(2) Соединению, указанному в (1), где Q представляет собой группу формулы (А)
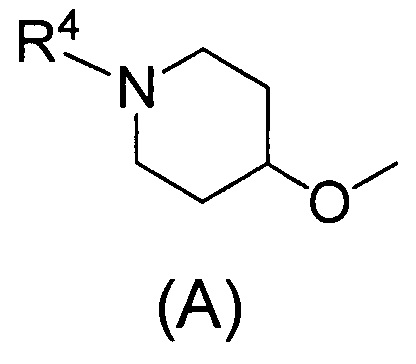
(где R4 имеет значения, указанные в (1)), или его фармацевтически приемлемой соли.
(3) Соединению, указанному в (1) или (2), где R1 представляет собой NR1AR1B (где R1A и R1В имеют значения, указанные в (1)), или его фармацевтически приемлемой соли.
(4) Соединению, указанному в (1)-(3), где каждый из R2 и R3 представляет собой атом водорода и n равно 1, или его фармацевтически приемлемой соли.
(5) Соединению, указанному в любом из (1)-(4), где R4 представляет собой С3-С7циклоалкил, или его фармацевтически приемлемой соли.
(6) Фармацевтическому агенту, содержащему в качестве активного ингредиента соединение, указанное в любом из (1) – (5), или его фармацевтически приемлемую соль.
(7) Фармацевтическому агенту, указанному в (6), который является антагонистом или обратным агонистом рецептора гистамина Н3.
(8) Фармацевтический агент, указанный в (6) или (7), который является превентивным или терапевтическим агентом для деменции, болезни Альцгеймера, синдрома дефицита внимания и гиперактивности, шизофрении, эпилепсии, “центральной” судороги, ожирения, сахарного диабета, гиперлипидемии, нарколепсии, идиопатической гиперсомнии, индуцированного поведением синдрома недостаточного сна, синдрома апное во сне, нарушения циркадного ритма, парасомнии, связанного со сном нарушения движения, инсомнии и депрессии или аллергического ринита.
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
Соединения настоящего изобретения обладают значительным антагонистическим действием на рецептор гистамина Н3.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Термины и выражения, применяемые в контексте, имеют значения, указанные ниже.
“Атом галогена”, применяемый в контексте, относится к атому фтора, атому хлора, атому брома или атому иода.
“С1-С6Алкил”, применяемый в контексте, относится к неразветвленной или разветвленной алкильной группе, имеющей 1-6 атомов углерода, причем примерами ее могут быть такие группы, как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил или н-гексил.
“С3-С7Циклоалкил”, применяемый в контексте, относится к циклопропильной, циклобутильной, циклопентильной, циклогексильной или циклогептильной группе.
“С1-С6алкокси”, применяемый в контексте, относится к неразветвленной или разветвленной алкоксигруппе, имеющей 1-6 атомов углерода, причем примерами ее могут быть такие группы, как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, изопентилокси, неопентилокси или н-гексилокси.
Термин “3-7-членное насыщенное гетероциклические кольцо” в выражении “связаны вместе с соседним атомом азота с образованием 3-7-членного насыщенного гетероциклического кольца” относится насыщенному моноциклическому кольцу или спирокольцу, которое состоит из 3-7 образующих кольцо атомов и которое содержит указанный соседний атом азота, с необязательным дополнительным включением одного гетероатома, выбранного из О, N и S; примеры его могут включать такие группы, как 1-азиридинил, 1-азетидинил, 1-пирролидинил, пиперидино, 1-азепанил, морфолино или 2-окса-6-азаспиро[3,3]гептан-6-ил.
Предпочтительные варианты осуществления соединений изобретения показаны ниже.
Одним предпочтительным вариантом осуществления соединения формулы (I) согласно настоящему изобретению является соединение, где Q представляет собой группу формулы (А)
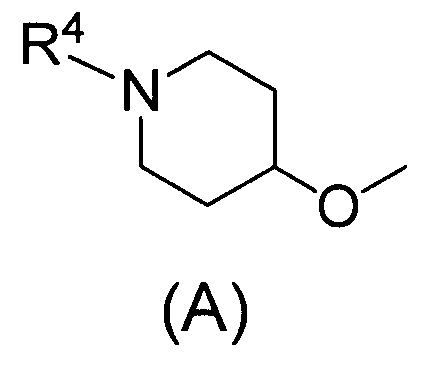
(где R4 представляет собой С1-С6алкил (С1-С6алкил может быть замещен одним или двумя С3-С7циклоалкилами) или С3-С7циклоалкил (С3-С7циклоалкил может быть замещен одним или двумя С1-С6алкилами).
В формуле (А) R4 предпочтительно представляет собой С3-С7циклоалкил и более предпочтительно циклобутил.
Другим предпочтительным вариантом осуществления соединения формулы (I) согласно настоящему изобретению является соединение, где R1 представляет собой NR1AR1B (где R1A и R1B, которые могут быть одинаковыми или разными, представляют собой атом водорода, С1-С6алкил или С3-С7циклоалкил или же R1A и R1B связаны вместе с соседним атомом азота с образованием 3-7-членного насыщенного гетероциклического кольца (причем насыщенное гетероциклическое кольцо необязательно замещено одним или двумя С1-С6алкилами)).
Еще одним предпочтительным вариантом осуществления соединения формулы (I) согласно настоящему изобретению является соединение, где каждый из R2 и R3 представляет собой атом водорода и n равно 1.
Профили соединений настоящего изобретения предпочтительно включают высокую эффективность лекарственного средства, превосходные кинетики in vivo (хорошее пероральное поглощение и отсутствие аккумуляции в определенных тканях), превосходные свойства, проявляемые в качестве фармацевтических средств, низкую токсичность и т.д. Маловероятно, что предпочтительные соединения настоящего изобретения распознаются в качестве субстрата для Р-гликопротеина, который является переносчиком оттока, который регулирует интрацеребральную миграцию лекарственных средств, и поэтому предполагается, что такие соединения имеют превосходную интрацеребральную миграцию.
Термин “фармацевтически приемлемая соль“, применяемый в контексте, включает, например, соли с неорганическими кислотами, такими как серная кислота, хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота и азотная кислота; соли с органическими кислотами, такими как уксусная кислота, щавелевая кислота, янтарная кислота, молочная кислота, винная кислота, фумаровая кислота, малеиновая кислота, лимонная кислота, бензолсульфоновая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота, бензойная кислота, камфорасульфоновая кислота, этансульфоновая кислота, глюкогептоновая кислота, глюконовая кислота, глутаминовая кислота, гликолевая кислота, малеиновая кислота, малоновая кислота, миндальная кислота, галактаровая кислота и нафталин-2-сульфоновая кислота; соли с одним или несколькими ионами металлов, такими как ион лития, ион натрия, ион калия, ион кальция, ион магния, ион цинка и ион алюминия, и соли с аммиаком или аминами, такими как аргинин, лизин, пиперазин, холин, диэтиламин, 4-фенилциклогексиламин, 2-аминоээтанол и бензатин.
Соединения настоящего изобретения могут также быть в форме различных сольватов. С точки зрения применимости в качестве фармацевтических средств они могут быть иногда в форме гидратов.
Соединения настоящего изобретения включают все возможные формы, включающие энантиомеры, диастереомеры, соединения в равновесных состояниях, их смеси в любых пропорциях, рацематы и т.д. Индивидуальные изомеры можно получить известными способами, например, применением оптически активного исходного соединения или промежуточного продукта, оптически селективной или диастереоселективной реакцией при получении промежуточного продукта или конечного продукта или хроматографическим разделением при получении промежуточного продукта или конечного продукта.
Соединения настоящего изобретения включают также соединения, у которых один или несколько атомов водорода, атомов углерода, атомов азота, атомов кислорода или атомов серы заменены их радиоизотопами или стабильными изотопами. Такие меченые соединения применимы, например, при исследованиях метаболизма и фармакокинетик или в биологических анализах, в которых их применяют в качестве лигандов рецепторов.
Соединения настоящего изобретения или их фармацевтически приемлемые соли можно применять в сочетании с одним или несколькими фармацевтически приемлемыми носителями, эксципиентами или разбавителями при изготовлении фармацевтических препаратов. Такие носители, эксципиенты и разбавители могут включать, например, воду, лактозу, декстрозу, фруктозу, сахарозу, сорбит, маннит, полиэтиленгликоль, пропиленгликоль, крахмал, камедь, желатин, альгинат, силикат кальция, фосфат кальция, целлюлозу, воду, сироп, метилцеллюлозу, поливинилпирролидон, алкилпарагидроксибензосорбат, тальк, стеарат магния, стеариновую кислоту, глицерин и различные виды масла, такие как кунжутное масло, оливковое масло и соевое масло.
Кроме того, указанные выше носители, эксципиенты или разбавители можно необязательно смешивать с обычно применяемыми добавками, такими как наполнители, связывающие вещества, дезинтегрирующие вещества, рН-модификаторы, солюбилизаторы и т.д., и затем подвергать обработке обычными процедурами для изготовления фармацевтических препаратов для получения оральных или парентеральных фармацевтических препаратов, таких как таблетки, пилюли, капсулы, гранулы, дусты, жидкости/растворы, эмульсии, суспензии, мази, инъекции и пластыри для нанесения на кожу. Соединения настоящего изобретения можно давать взрослым пациентам при дозах от 0,001 до 500 мг на одно введение один или несколько раз в день пероральным или парентеральным путем. Такую дозу можно при необходимости повысить или уменьшить в зависимости от типа подвергаемого лечению заболевания, возраста, массы тела и симптома заболевания пациента и т.д.
Фармацевтические агенты, содержащие соединения настоящего изобретения или их фармацевтически приемлемые соли в качестве активного ингредиента, применимы в качестве антагонистов или обратных агонистов рецептора гистамина Н3.
Кроме того, фармацевтические агенты, содержащие соединения настоящего изобретения или их фармацевтически приемлемые соли в качестве активного ингредиента, приемлемы в качестве превентивных или терапевтических агентов для деменции, болезни Альцгеймера, синдрома дефицита внимания и гиперактивности, шизофрении, эпилепсии, “центральной” судороги, ожирения, сахарного диабета, гиперлипидемии, нарколепсии, идиопатической гиперсомнии, индуцированного поведением синдрома недостаточного сна, синдрома апное во сне, нарушения циркадного ритма, парасомнии, связанного со сном нарушения движения, инсомнии и депрессии или аллергического ринита.
Соединения настоящего изобретения можно получать известными в органической химии способами. Способы согласно нижеследующим схемам реакций являются иллюстративными способами получения соединений настоящего изобретения и никоим образом не предназначены для их ограничения. В схемах реакций 1-4, указанных ниже, R1, R2, R3, R4, R5, R6 и n имеют значения, указанные выше. Кроме этого, каждый из X, Y1 и Y2, которые могут быть одинаковыми или разными, представляет собой уходящую группу, такую как атом галогена (например, атом хлора, атом брома или атом иода) или органическая сульфонилоксигруппа (например, метансульфонилоксигруппа, бензолсульфонилоксигруппа, п-толуолсульфонилоксигруппа или трифторметансульфонилоксигруппа).
Ниже описан способ получения соединения настоящего изобретения, который изображен схемой реакций 1. Это способ является способом получения соединения изобретения (IА) из соединения (1).
(Схема реакций 1)
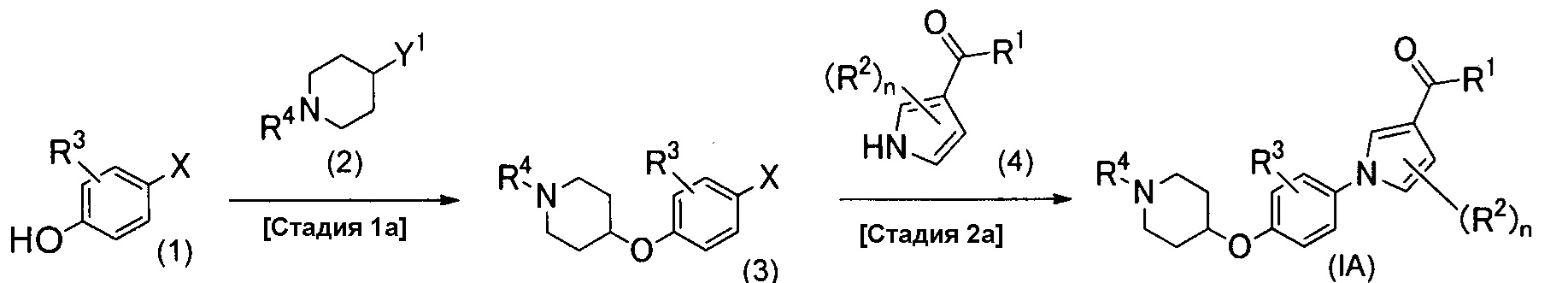
(Стадия 1а)
Стадию 1а выполняют для проведения конденсации между соединением (1) и соединением (2) реакцией сочетания для получения соединения (3). Соединения (1) и (2) являются либо известными, либо их можно легко синтезировать из известных соединений.
В случае, когда Y1 представляет собой уходящую группу, такую как атом галогена или органическая сульфонилоксигруппа, реакцию сочетания можно проводить обычным способом, включающим в себя алкилирование гидроксильной группы простого эфира фенола в растворителе или в отсутствие растворителя в присутствии или в отсутствие основания. Если необходимо, можно добавить добавку, например иодид калия или бромид натрия. Примеры основания, которое можно применять в рассматриваемой реакции, включают в себя органические основания, такие как пиридин, триэтиламин и диизопропилэтиламин, и неорганические основания, такие как трет-бутоксид калия, карбонат калия, карбонат цезия, гидрокарбонат натрия, гидроксид натрия, гидроксид калия и гидрид натрия. Примеры растворителя, который можно применять в рассматриваемой реакции, включают в себя спирты, такие как метанол, этанол и изопропанол; простые эфиры, такие как тетрагидрофуран, 1,2-диметоксиэтан и 1,4-диоксан; углеводороды, такие как толуол и бензол; галогенированные углеводороды, такие как хлороформ и дихлорметан; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и N-метил-2-пирролидон; кетоны, такие как ацетон и 2-бутанон; диметилсульфоксид; ацетонитрил; воду или их смешанные растворители. Температура рассматриваемой реакции конденсации обычно находится в диапазоне от 0°С до 200°С, предпочтительно от 15°С до 150°С, и время реакции обычно находится в диапазоне от 1 до 48 часов, предпочтительно от 1 до 16 часов.
В случае, когда Y1 представляет собой гидроксильную группу, примером рассматриваемой реакции сочетания может быть реакция Мицунобу; примером способа проведения данной реакции является способ, который проводят в растворителе в присутствии реагента, содержащего фосфорорганическое соединение, такое как трифенилфосфин или трибутилфосфин, в комбинации с азосоединением, таким как диэтилазодикарбоксилат, диизопропилазодикарбоксилат или ди-трет-бутилазодикарбоксилат, или же в присутствии фосфорилидного реагента, такого как цианометилентрибутилфосфоран. Примеры растворителя, который можно применять в рассматриваемой реакции, включают в себя простые эфиры, такие как тетрагидрофуран, 1,2-диметоксиэтан и 1,4-диоксан; углеводороды, такие как толуол и бензол; галогенированные углеводороды, такие как хлороформ и дихлорметан; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и N-метил-2-пирролидон; кетоны, такие как ацетон и 2-бутанон; диметилсульфоксид; ацетонитрил; воду или их смешанные растворители. Температура рассматриваемой реакции обычно находится в диапазоне от 0°С до 150°С, предпочтительно от 15°С до 100°С, и время реакции обычно находится в диапазоне от 1 до 48 часов, предпочтительно от 1 до 16 часов.
(Стадия 2а)
Стадию 2а выполняют для проведения конденсации между соединением (3) и соединением (4) реакцией сочетания для получения соединения (IA) настоящего изобретения. Соединение (4) является либо известным, либо его можно легко синтезировать из известного соединения. Реакцию сочетания можно проводить обычным способом, которым реакцию проводят в растворителе в присутствии катализатора и его лиганда; например реакцию можно проводить согласно способу, описанному в публикации Kunz et al., Synlett, 2003, vol. 15. pp. 2428-2439, или его модификацией. Рассматриваемую реакцию предпочтительно проводят в присутствии основания. Примеры катализатора, который можно применять в рассматриваемой реакции, включают в себя катализаторы переходных металлов, таких как медь, никель или платина, которые обычно применяют в реакции сочетания, и более конкретно, они включают в себя медь(0), иодид меди(I), хлорид меди(I), оксид меди(I), комплекс бромида меди(I) и трифенилфосфина, комплекс трифторметансульфоната меди(I) и бензола, сульфат меди(II), ацетат палладия(II), тетракис(трифенилфосфин)палладий(0), бис(трифенилфосфин)палладий(II)хлорид, [1,1’-бис(дифенилфосфино)ферроцен]палладий(II)дихлорид, трис(дибензилиденацетон)дипалладий(0), бис(ацетилацетонато)никель(II) и т.д. Лиганд, который можно применять в рассматриваемой реакции, выбирают из лигандов, которые обычно применяют в реакции конденсации, в которой применяют катализаторы металлов, и примеры их включают в себя N,N’-диметилэтилендиамин, N,N’-диметилциклогексан-1,2-диамин, 2-аминопиридин, 1,10-фенантролин, 3,4,7,8-тетраметил-1,10-фенантролин, оксим 2-гидроксибензальдегида, этиленгликоль, трифенилфосфин, три-трет-бутилфосфин и т.д. Примеры основания, которое можно применять в рассматриваемой реакции, включают в себя карбонат калия, фосфат калия, гидроксид калия, трет-бутоксид калия, трет-бутоксид натрия, карбонат цезия, карбонат натрия, гидрокарбонат натрия, ацетат натрия, метоксид натрия, гидроксид тетрабутиламмония и т.д. Примеры растворителя, который можно применять в рассматриваемой реакции, включают в себя спирты, такие как метанол, этанол и изопропанол; простые эфиры, такие как тетрагидрофуран, 1,2-диметоксиэтан и 1,4-диоксан; углеводороды, такие как толуол и бензол; галогенированные углеводороды, такие как хлороформ и дихлорметан; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и N-метил-2-пирролидон; кетоны, такие как ацетон и 2-бутанон; диметилсульфоксид; ацетонитрил; воду или их смешанные растворители. Температура рассматриваемой реакции обычно находится в диапазоне от 0°С до 200°С, предпочтительно от 40°С до 150°С, и время реакции обычно находится в диапазоне от 1 до 48 часов, предпочтительно от 1 до 16 часов.
Альтернативно, соединение (IA) можно получить согласно способу, описанному схемой реакций 2.
(Схема реакций 2)
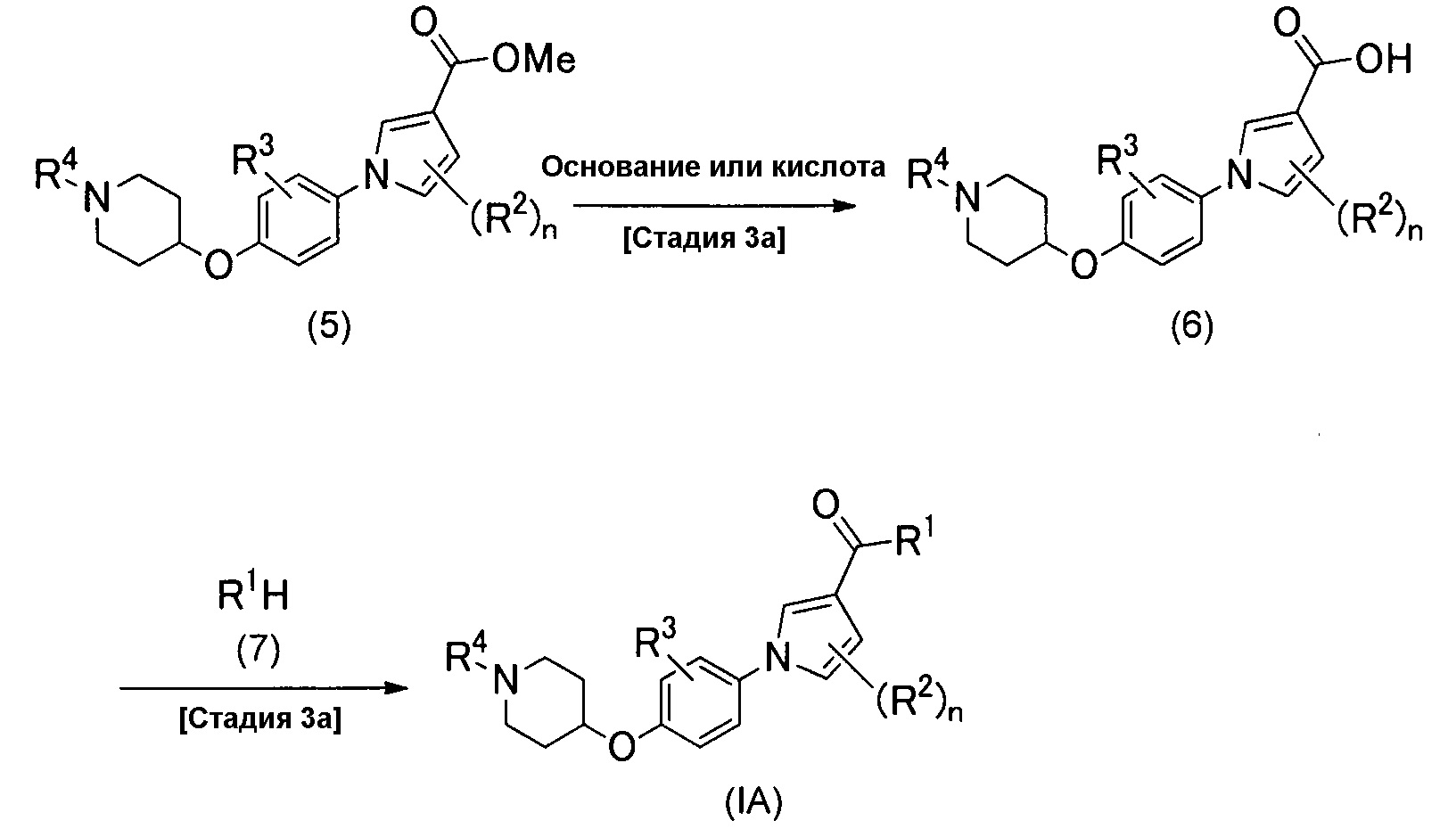
(Стадия 3а)
Стадия 3а является такой стадией, в которой метоксикарбонильную группу соединения (5), которое является разновидностью соединения изобретения (IA), у которого R1 представляет собой метоксигруппу, превращают в карбоновую кислоту посредством гидролиза, получая при этом соединение изобретения (6), у которого R1 представляет собой гидроксигруппу. Реакцию гидролиза можно проводить обычной реакцией гидролиза сложного эфира, например, ее можно проводить согласно способам, описанным в публикации T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, fourth edition, John Wiley and Sons, или их модификациям, например, способом проведения реакции либо в растворителе, либо без растворителя, в присутствии сильной кислоты, и способом проведения реакции в растворителе в присутствии основания. Температура рассматриваемой реакции обычно находится в диапазоне от 0°С до 120°С, предпочтительно от 15°С до 80°С, и время реакции обычно находится в диапазоне от 1 до 48 часов, предпочтительно от 1 до 12 часов.
(Стадия 4а)
Стадию 4а выполняют для проведения конденсации между соединением (6) и производным амина (7) реакцией сочетания для получения соединения (IA) настоящего изобретения. Производное амина (7) является либо известным, либо его можно легко синтезировать из известного соединения. Реакцию сочетания можно проводить обычными способами амидирования карбоновых кислот, которые включают в себя способ, в котором карбоновую кислоту превращают в галогенангидрид карбоновой кислоты, такой как хлорангидрид карбоновой кислоты или бромангидрид карбоновой кислоты и затем подвергают реакции с амином, способ, в котором смешанный ангидрид кислот, получаемый из карбоновой кислоты и эфира хлоругольной кислоты, подвергают реакции с амином, способ, в котором карбоновую кислоту превращают в активный эфир, такой как 1-бензотриазолиловый эфир или сукцинимидиловый эфир, и затем подвергают реакции с амином, и способ, в котором карбоновую кислоту подвергают реакцию с амином в присутствии агента дегидратации-конденсации. Все эти реакции можно проводить в растворителе в присутствии или в отсутствие основания. Примеры агента дегидратации-конденсации, который можно применять в рассматриваемой реакции, включают в себя гидрохлорид 3-(3-диметиламинопропил)-1-этилкарбодиимида, дициклогексилкарбодиимид, дифенилфосфорилазид и карбонилдиимидазол, который необязательно применяют с активатором, таким как 1-гидроксибензотриазол или гидроксисукцинимид. Примеры основания, которое можно применять в рассматриваемой реакции, включают в себя пиридин, триэтиламин, диизопропилэтиламин, карбонат калия, карбонат натрия и гидрокарбонат натрия. Примеры растворителя, который можно применять в рассматриваемой реакции, включают в себя простые эфиры, такие как тетрагидрофуран и 1,4-диоксан; углеводороды, такие как толуол и бензол; галогенированные углеводороды, такие как хлороформ и дихлорметан; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и N-метил-2-пирролидон; кетоны, такие как ацетон и 2-бутанон; диметилсульфоксид; ацетонитрил; воду или их смешанные растворители. Среди них предпочтительным является толуол, тетрагидрофуран или N,N-диметилформамид. Температура рассматриваемой реакции обычно находится в диапазоне от 0°С до 120°С, предпочтительно от 15°С до 40°С, и время реакции обычно находится в диапазоне от 1 до 48 часов, предпочтительно от 1 до 12 часов.
Ниже описан способ получения соединения настоящего изобретения, который изображен схемой реакций 3. Это способ получения соединения (1В) настоящего изобретения из соединения (1).
(Схема реакций 3)
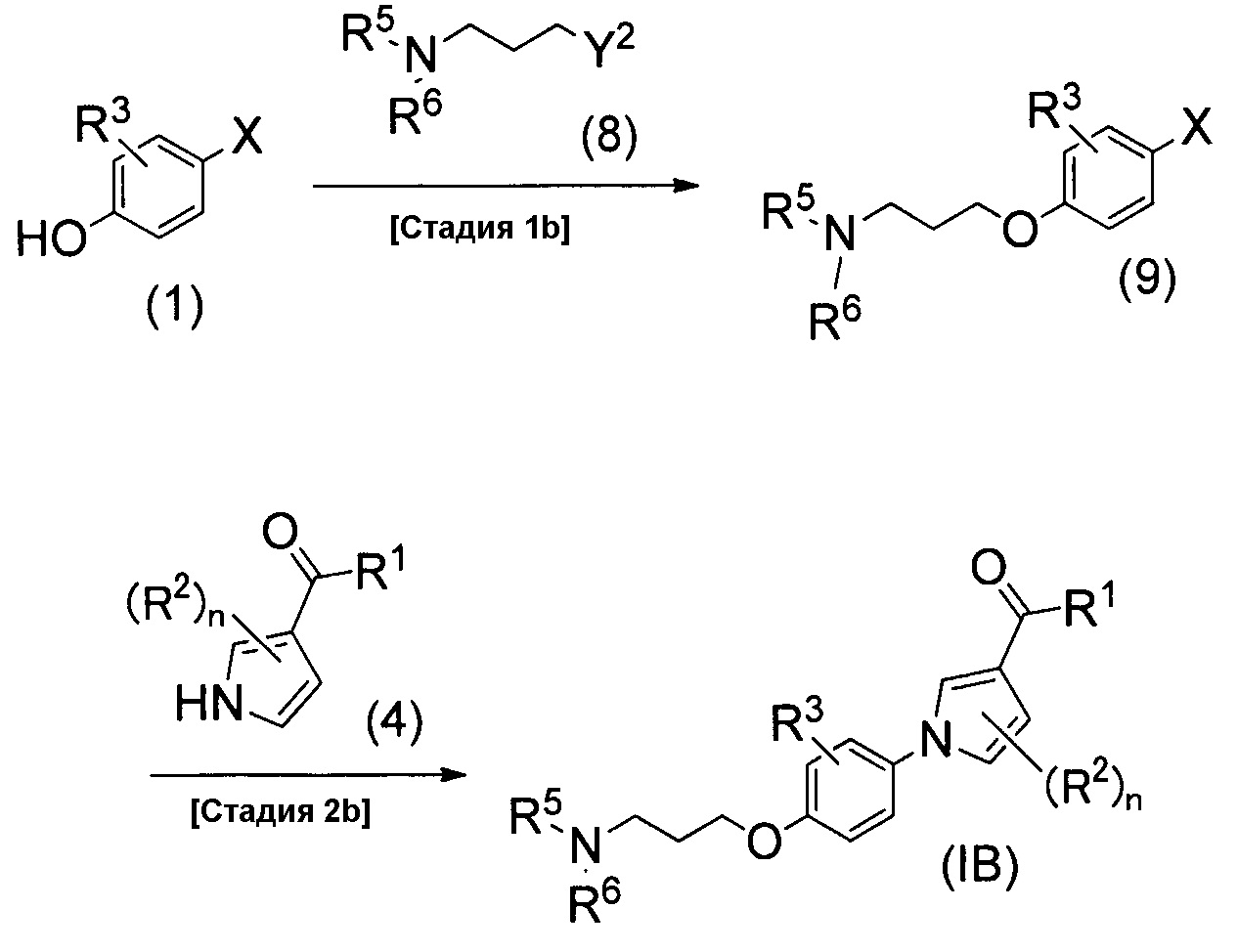
(Стадия 1b)
Стадия 1b является стадией получения соединения (9) реакций сочетания между соединением (1) и соединением (8). Соединение (8) является либо известным, либо его можно легко синтезировать из известного соединения. Реакцию сочетания можно проводить таким же способом, как на стадии 1а.
(Стадия 2b)
Стадия 2b является стадией получения соединения изобретения (IB) конденсацией между соединением (9) и соединением (4) посредством реакции конденсации. Реакцию конденсации можно проводить таким же способом, как на стадии 2а.
Альтернативно, соединение (IB) можно получить способом, изображенным схемой реакций 4.
(Схема реакций 4)
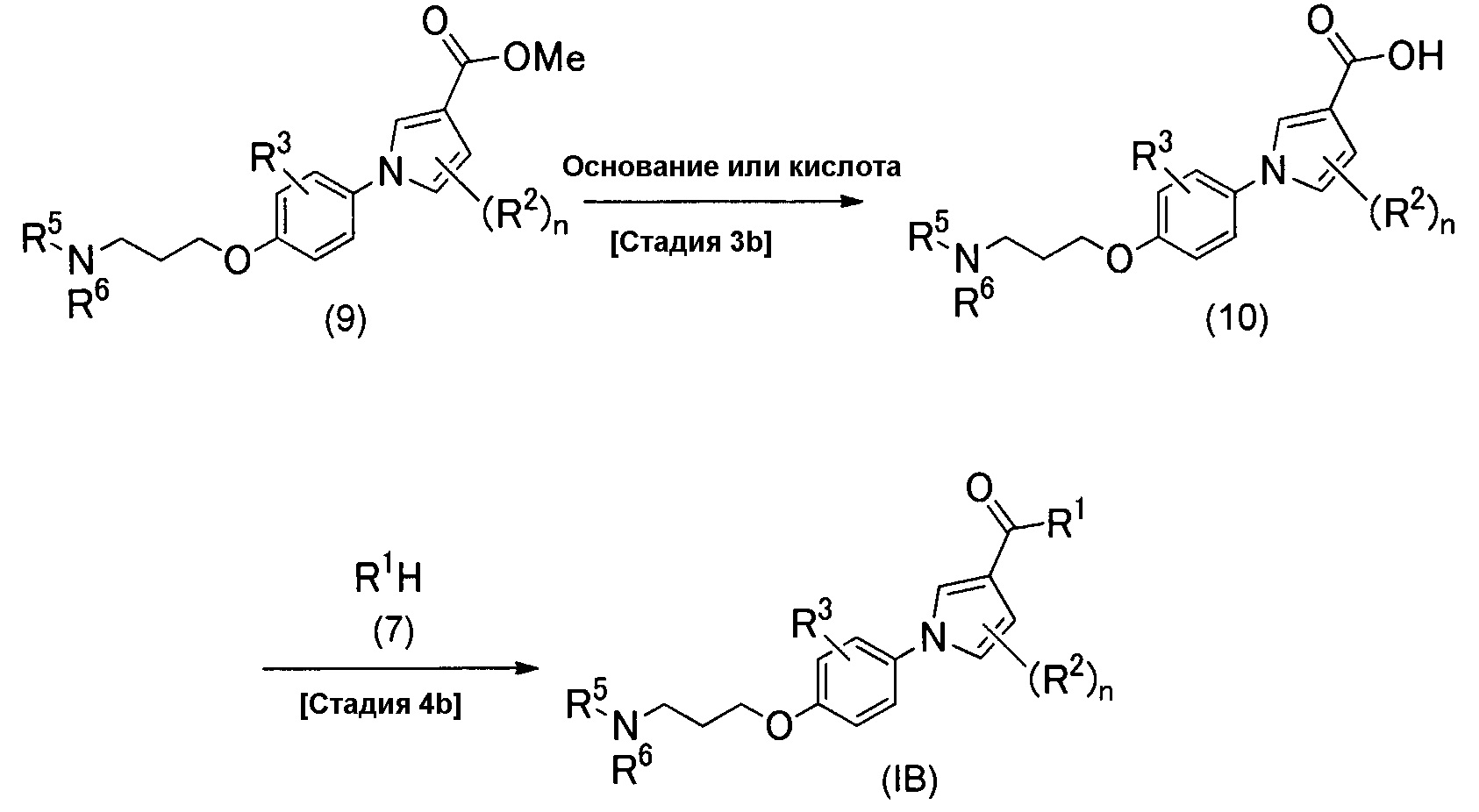
(Стадия 3b)
Стадия 3b является такой стадией, в которой метоксикарбонильную группу соединения (9), которое является разновидностью соединения изобретения (IВ), у которого R1 представляет собой метоксигруппу, превращают в карбоновую кислоту посредством гидролиза с образованием соединения изобретения (10), у которого R1 представляет собой гидроксигруппу. Реакцию гидролиза можно проводить таким же способом, как на стадии 3а.
(Стадия 4b)
Стадия 4b является стадией получения соединения изобретения (IB) конденсацией между соединением (10) и производным амина (7) посредством реакции сочетания. Реакцию сочетания можно проводить таким же способом, как на стадии 4а.
ПРИМЕРЫ
Далее настоящее изобретение описывается конкретно при помощи рабочих примеров и испытаний, которые не предназначаются для ограничения объема изобретения.
Инструментальные данные, перечисленные в рабочих примерах, получали измерением следующими инструментами.
Масс-спектр: micromass Platform LC или micromass GCT
ЯМР-спектр: [1H-ЯМР] 600 МГц: JNM-ECA600 (JEOL Ltd., Japan)
(Пример 1)
Получение метил-1-{4-[(1-циклобутилпиперидин-4-ил)окси]фенил}-1Н-пиррол-3-карбоксилата (соединения № 1)
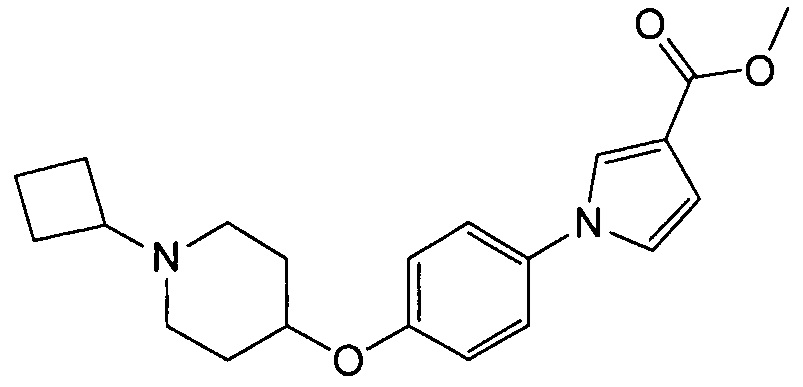
Суспензию 1-циклобутил-4-(4-иодфенокси)пиперидина (3 г; может быть синтезирован согласно способу, описанному в WO2008072703), метилового эфира 1Н-пиррол-3-карбоновой кислоты (1,75 г), N,N’-диметилэтилендиамина (0,592 г), иодида меди (0,32 г) и карбоната цезия (2,32 г) в толуоле (8,4 мл) перемешивали при 110°С в течение 4 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры и после добавления хлороформа фильтровали через целит (зарегистрированный товарный знак). Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (силикагель формы NH; элюент: н-гексан/этилацетат = 88/12–0/100), получая при этом указанное в заголовке соединение в виде бесцветного твердого вещества (1,42 г).
1H ЯМР (600 MГц, ХЛОРОФОРМ-d) δ м.д. 1,61 – 1,74 (м, 2 H) 1,78 – 1,9 2 (м, 4 H) 1,95 – 2,08 (м, 4 H) 2,10-2,27 (м, 2 H) 2.62 (ушир.с., 2 H) 2,73 (т, J=8,05 Гц, 1 H) 3,82 (с, 3 H) 4,32 (ушир.с., 1 H) 6,71 (дд, J=2,89, 1,65 Гц, 1 H) 6,88 – 6,92 (м, 1 H) 6,93 – 6,99 (м, 2 H) 7,2 6-7,31 (м, 2 H) 7,58 (т, J=1,86 Гц, 1 H)
МС (ESI/APCI Dual) (Positive) m/z; (M+H)+ 355.
(Пример 2)
Получение 1-{4-[(1-циклобутилпиперидин-4-ил)окси]фенил}-1Н-пиррол-3-карбоновой кислоты
(Соединение № 2)
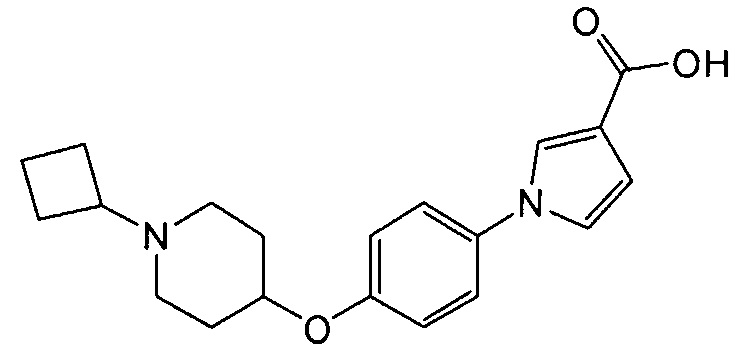
К раствору в этаноле (8 мл) метил-1-{4-[(1-циклобутилпиперидин-4-ил)окси]фенил}-1Н-пиррол-3-карбоксилата (1,4 г), синтезированного в примере 1, добавляли 6 н водный раствор гидроксида натрия (1,32 мл) и смесь перемешивали при 60°С в течение 4 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры и после добавления воды экстрагировали хлороформом. Водный слой нейтрализовали хлористоводородной кислотой и экстрагировали хлороформом. Объединенный органический слой сушили над сульфатом магния и концентрировали при пониженном давлении, получая при этом указанное в заголовке соединение в виде бесцветного аморфного вещества (1,22 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,52 – 3,32 (м, 15 H) 4,13 – 4,87 (м, 1 H) 6,76 (дд, J=2,89, 1,65 Гц, 1 H) 6,86 – 7,03 (м, 3 H) 7,31 (д, J=8,67 Гц, 2 H) 7,58 – 7,68 (м, 1 H)
МС (ESI/APCI Dual) (Positive) m/z; (M+H)+ 341.
(Пример 3)
Получение азетидин-1-ил-(1-{4-[(1-циклобутилпиперидин-4-ил)окси]фенил}-1Н-пиррол-3-ил)метанона (соединения № 3)
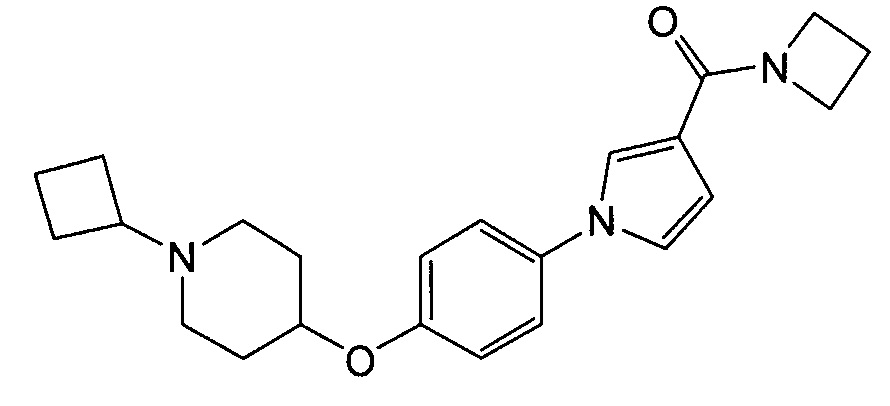
Суспензию 1-{4-[(1-циклобутилпиперидин-4-ил)окси]фенил}-1Н-пиррол-3-карбоновой кислоты (0,1 г), синтезированной в примере 2, гидрохлорида 1-(3-(диметиламино)пропил}-3-этилкарбодиимида (0,085 г), гидрата 1-гидроксибензотриазола (0,067 г) и азиридина (0,034 г) в N,N-диметилформамиде (0,1 мл) перемешивали при комнатной температуре в течение 16 часов. К реакционной смеси добавляли воду и проводили экстракцию хлороформом. Органический слой концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (силикагель формы NH; элюент: н-гексан/этилацетат = 88/12 – 0/100), получая при этом указанное в заголовке соединение в виде бесцветного твердого вещества (0,056 г).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,60 – 1,73 (м, 2 H) 1,76 – 1,93 (м, 4 H) 1,95 – 2,08 (м, 4 H) 2,10-2,22 (м, 2 H) 2,34 (т, J=7,64 Гц, 2 H) 2,54 – 2,68 (м, 2 H) 2,70 – 2,82 (м, 1 H) 4,11 - 4,23 (м, 2 H) 4,28 – 4,35 (м, 1 H) 4,37 – 4,55 (м, 2 H) 6,53 (дд, J=2,89, 1,65 Гц, 1 H) 6,83 – 7,00 (м, 3 H) 7,25 – 7,33 (м, 2 H) 7,44 (т, J=1,86 Гц, 1 H)
МС (ESI/APCI Dual) (Positive) m/z; (M+H)+ 380.
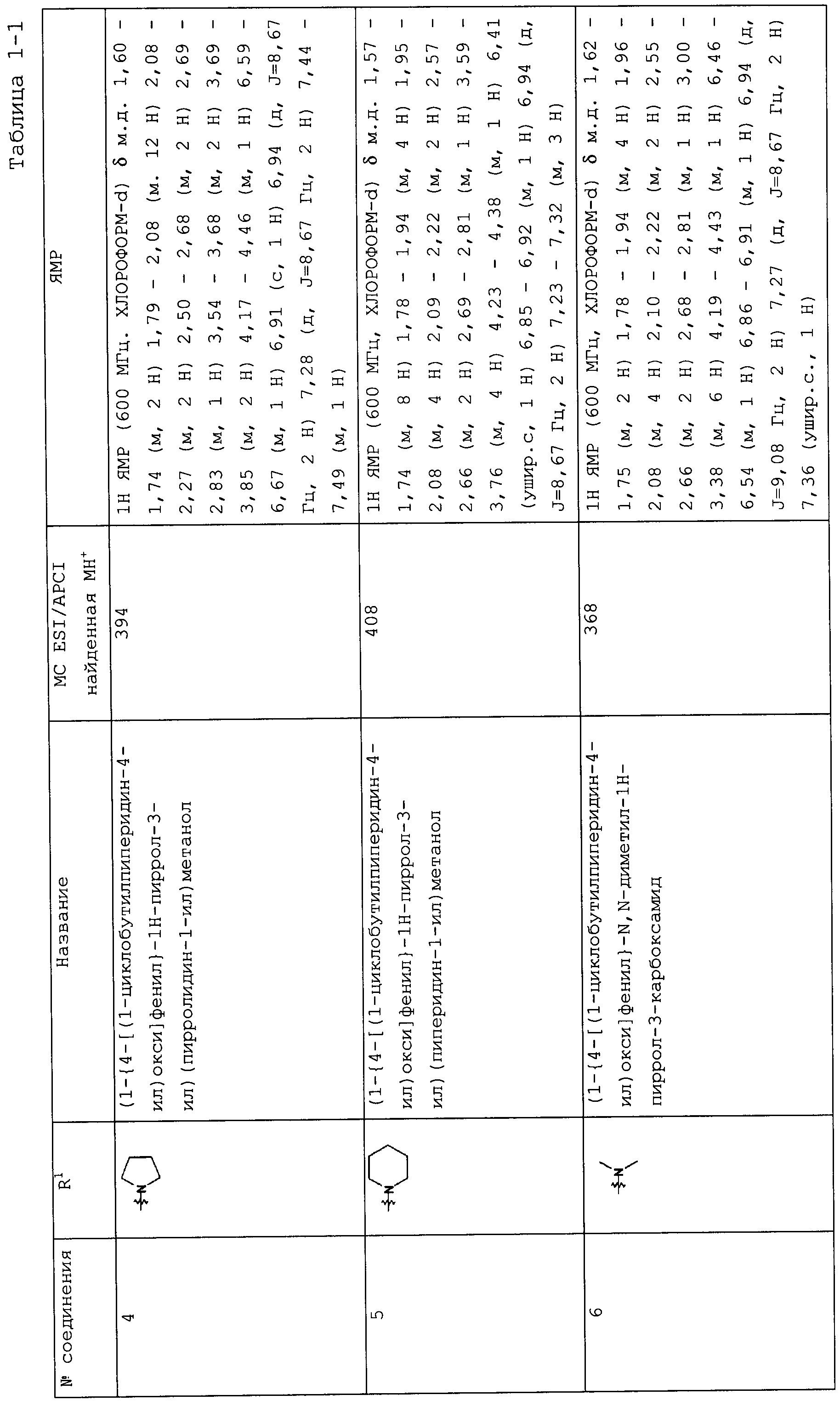
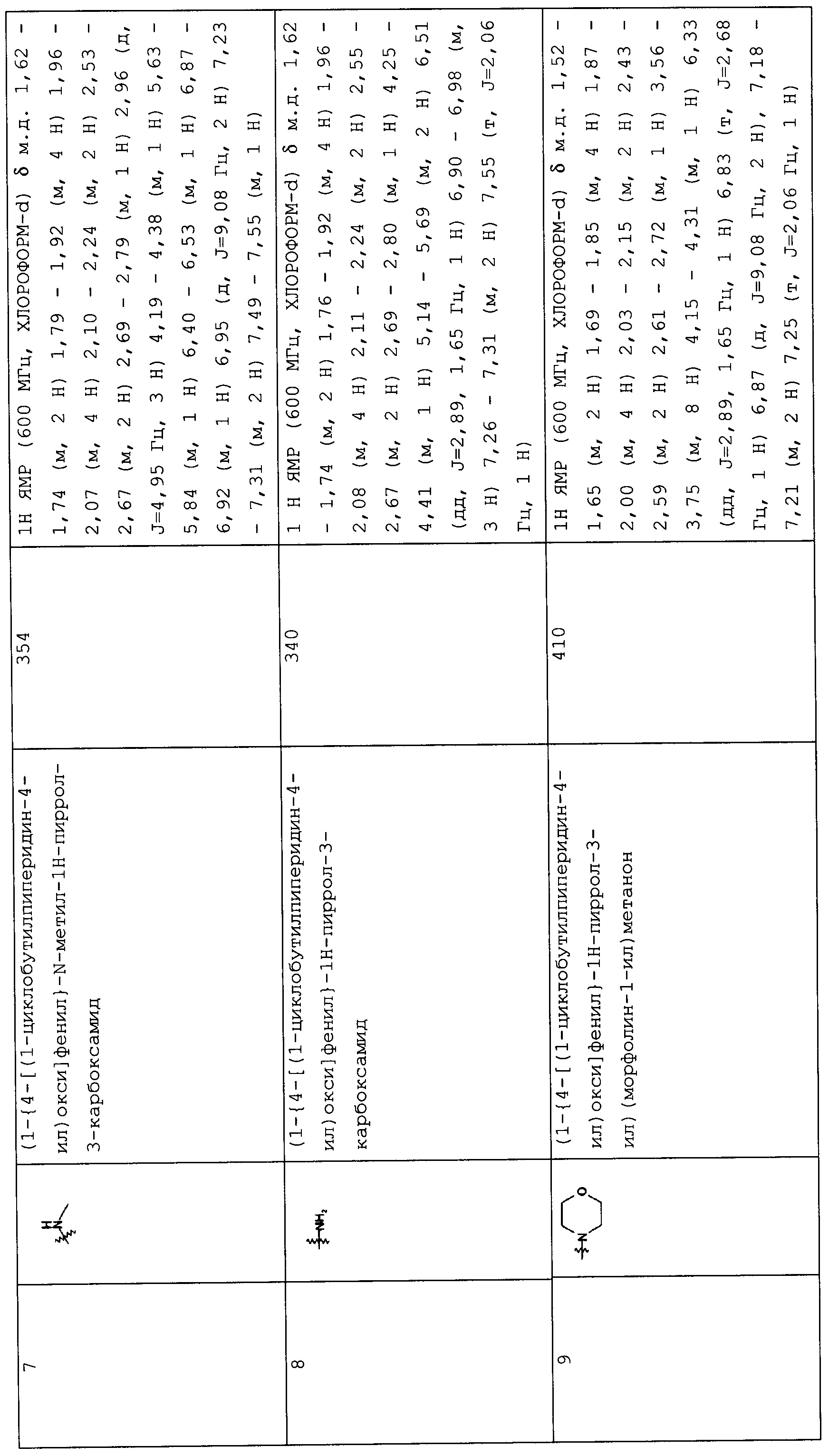
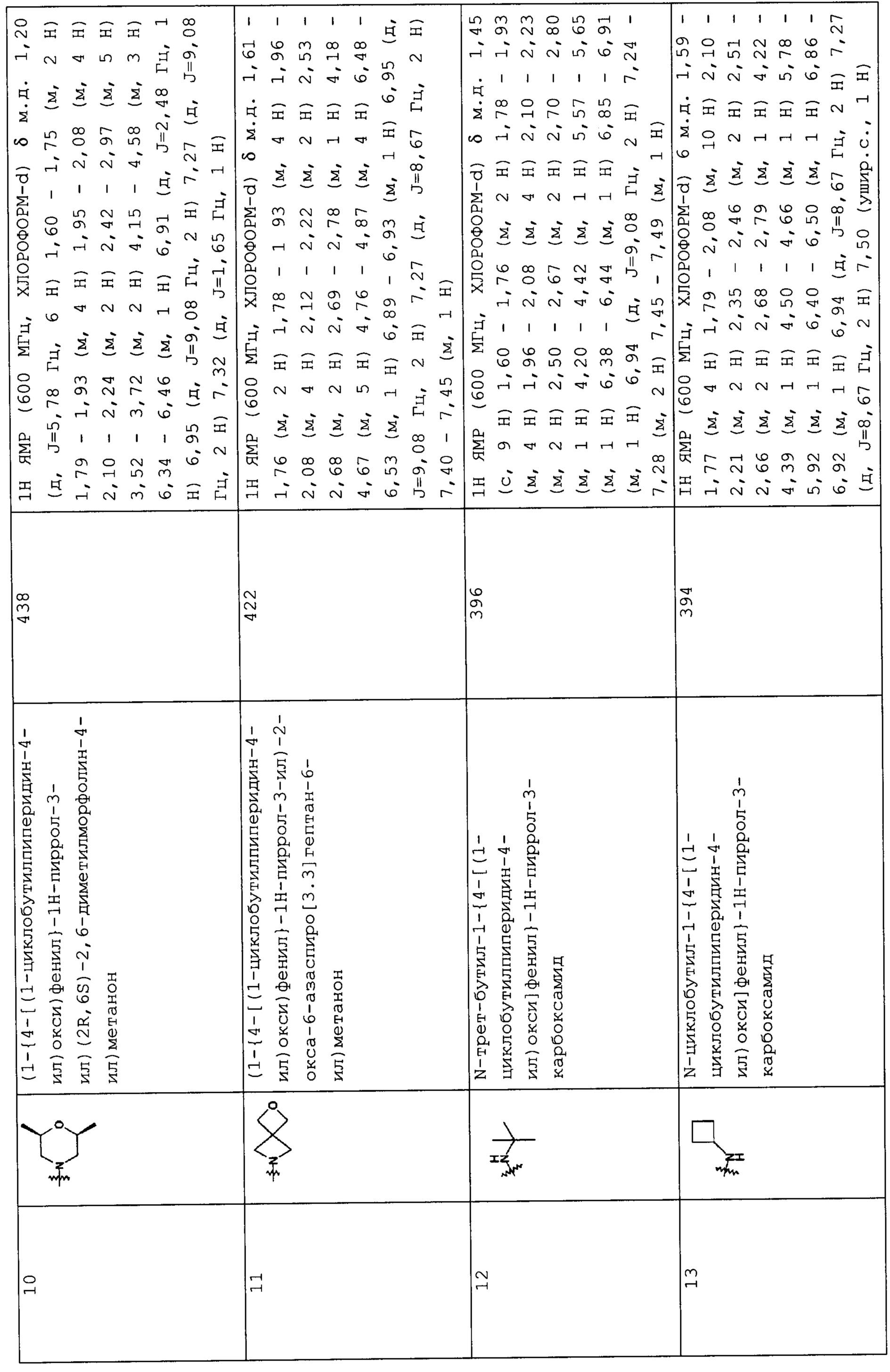
Такую же процедуру, как описана в примере 3, повторяли, получая при этом соединения, перечисленные в следующих таблицах 1-2 и 1-1 (Соединения №№ 4-13).
(Пример 4)
Получение метил-1-{4-[(1-циклобутилпиперидин-4-ил)окси]фенил}-2,5-диметил-1Н-пиррол-3-карбоксилата (соединения № 14)
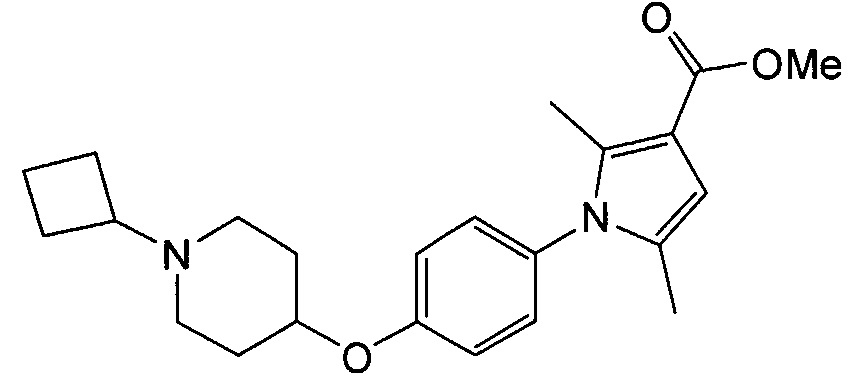
Указанное в заголовке соединение получали в виде бесцветного аморфного вещества повторением процедуры примера 1, за исключением того, что метиловый эфир 1Н-пиррол-3-карбоновой кислоты заменяли метиловым эфиром 2,5-диметил-1Н-пиррол-3-карбоновой кислоты.
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,60 - 1,73 (м, 2 H) 1,77 - 1,9 0 (м, 4 H) 1,93 (с, 3 H) 1,97 - 2,07 (м, 4 H) 2,08 - 2,20 (м, 2 H) 2,25 (с, 3 H) 2,53 - 2,78 (м, 3 H) 3,77 (с, 3 H) 4,33 (ушир.с., 1 H) 6,30 (д, J=1,24 Гц, 1 H) 6,88 - 6,96 (м, 2 H) 7,00 - 7,08 (м, 2 H)
МС (ESI/APCI Dual) (Positive) m/z; (M+H)+ 383.
(Пример 5)
Получение (1-{4-[(1-циклобутилпиперидин-4-ил)окси]фенил}-2,5-диметил-1Н-пиррол-3-ил](пирролидин-1-ил)метанона (соединения № 15)
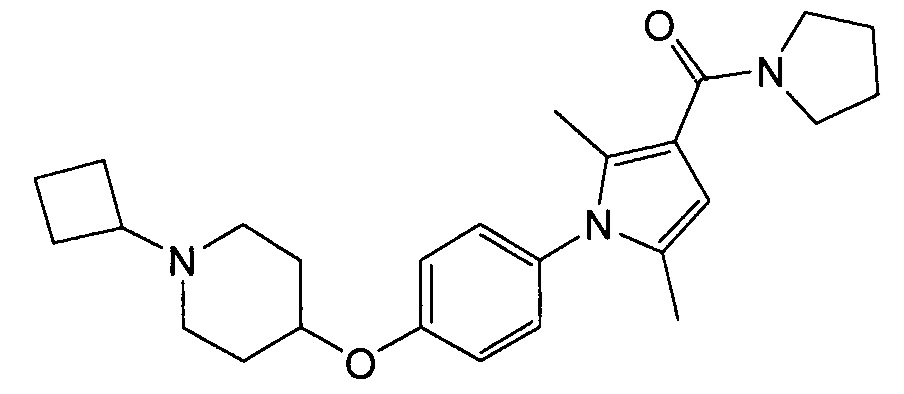
Смесь метил-1-{4-[(1-циклобутилпиперидин-4-ил)окси]фенил}-2,5-диметил-1Н-пиррол-3-карбоксилата (0,06 г), синтезированного в примере 4, и пирролидина (0,112 г) перемешивали в запаянной трубке при 100°С в течение 16 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры и концентрировали при пониженном давлении; полученный остаток очищали колоночной хроматографией на силикагеле (препаративный силикагель NH с размером частиц 0,5 мм; элюент: н-гексан/этилацетат = 50/50), получая при этом указанное в заголовке соединение в виде бесцветного твердого вещества.
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,59 - 1,93 (м, 12 H) 1,95 (с, 3 H) 1,98-2,09 (м, 4 H) 2,17 (с, 3 H) 2,64 (ушир.с., 2 H) 2,74 (т, J =8,05 Гц, 1 H) 3,62 (д, J=9,91 Гц, 4 H) 4,34 (ушир.с., 1 H) 6,06 (с, 1 H) 6,88 - 6,98 (м, 2 H) 7,02-7,11 (м, 2 H)
МС (ESI/APCI Dual) (Positive) m/z; (M+H)+ 422.
(Пример 6)
Получение [1-(4-{3-[(2R)-2-метилпирролидин-1-ил]пропокси}фенил)-1Н-пиррол-3-ил](пирролидин-1-ил)метанона (соединения № 16)
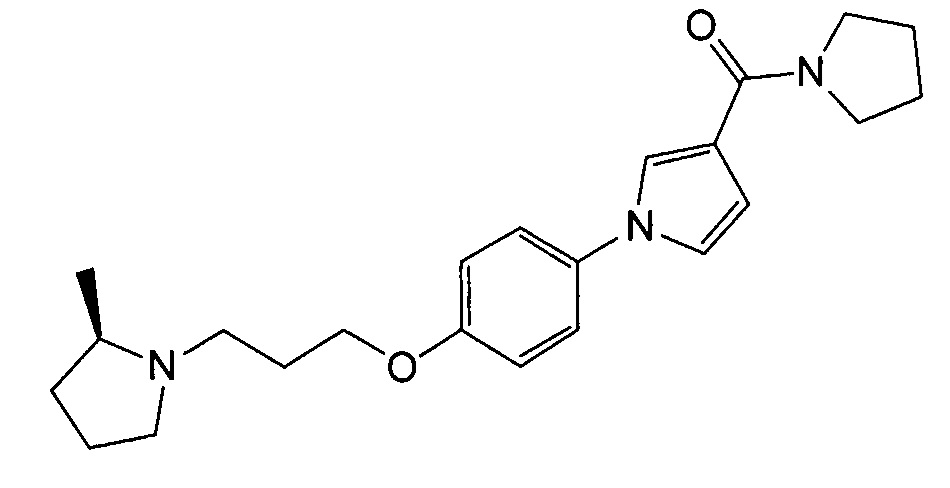
Указанное в заголовке соединение получали в виде бесцветного аморфного вещества повторением процедуры примера 1, за исключением того, что 1-циклобутил-4-(4-иодфенокси)пиперидин заменяли (2R)-1-[3-(4-иодфенокси)пропил]-2-метилпирролидином (который можно синтезировать согласно способу, описанному в WO2009063953) и метил 1Н-пиррол-3-карбоновую кислоту заменяли 3-(пирролидин-1-илкарбонил)-1Н-пирролом.
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,08 (д, J=5,78 Гц, 3 H) 1,35 - 1,48 (м, 1 H) 1,63 - 1,83 (м, 2 H) 1,85 - 2,04 (м, 3 H) 2,11 (д, J=9,08 Гц, 1 H) 2,16-2,23 (м, 1 H) 2,25 - 2,33 (м, 1 H) 2,92 - 2,99 (м, 1 H) 3,17 (д, J=2,48 Гц, 1 H) 3,65 (ушир.с., 4 H) 3,73 (ушир.с., 4 H) 3,96 - 4,15 (м, 2 H) 6,63 (дд, J=3,10, 1,86 Гц, 1 H) 6,89 - 6,97 (м, 3 H) 7,27 - 7,32 (м, 2 H) 7,45 - 7,49 (м, 1 H)
МС (ESI/APCI Dual) (Positive) m/z; (M+H)+ 382.
(Пример 7)
Получение (1-{4-[(1-изопропилпиперидин-4-ил)окси]фенил}-1Н-пиррол-3-ил)(пирролидин-1-ил)метанона (соединения № 17)
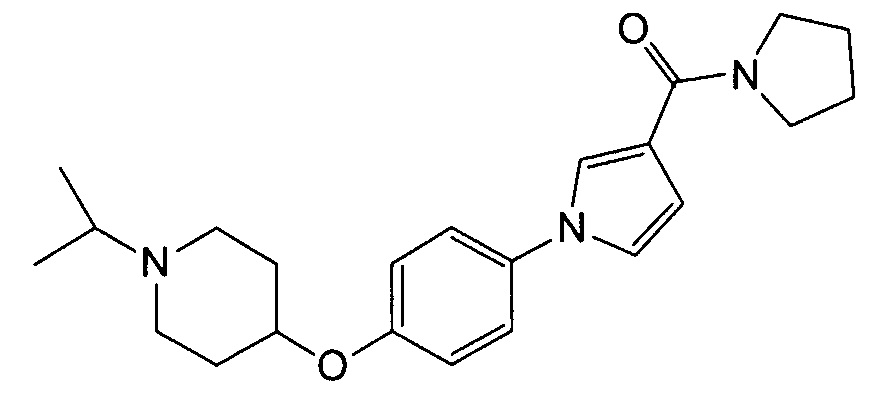
Указанное в заголовке соединение получали в виде бесцветного аморфного вещества повторением процедуры примера 1, за исключением того, что метил 1Н-пиррол-3-карбоновую кислоту заменяли (1Н-пиррол-3-ил)(пирролидин-1-ил)метаноном и 1-циклобутил-4-(4-иодфенокси)пиперидин заменяли 4-(4-иодфенокси)-1-изопропилпиперидином (который можно синтезировать согласно способу, описанному в WO2004089373).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,05 (д, J=6,61 Гц, 6 H) 1,74 -2,07 (м, 8 H) 2,39 (ушир.с., 2 H) 2,66 - 2,84 (м, 3 H) 3,60 - 3,81 (м, 4 H) 4,24 - 4,34 (м, 1 H) 6,63 (дд, J=3,10, 1,86 Гц, 1 H) 6,87 - 7,00 (м, 3 H) 7,22 - 7,32 (м, 2 H) 7,47 (т, J=2,06 Гц, 1 H)
МС (ESI/APCI Dual) (Positive) m/z; (M+H)+ 382.
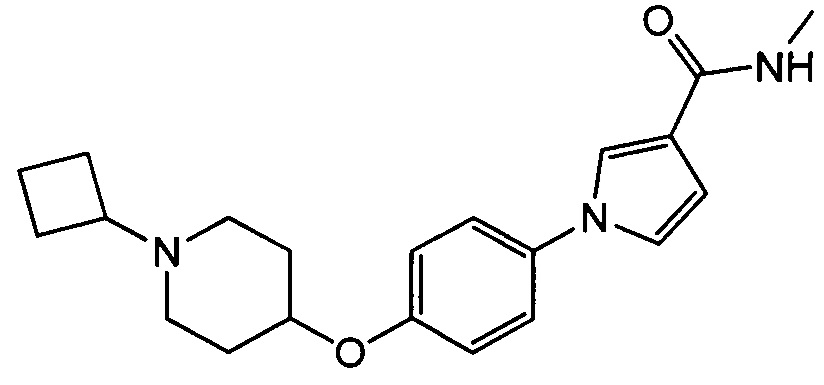
(Пример 8)
Получение 1-{4-[(1-изопропилпиперидин-4-ил)окси]фенил}-N-метил-1Н-пиррол-3-карбоксамида (соединения № 18)
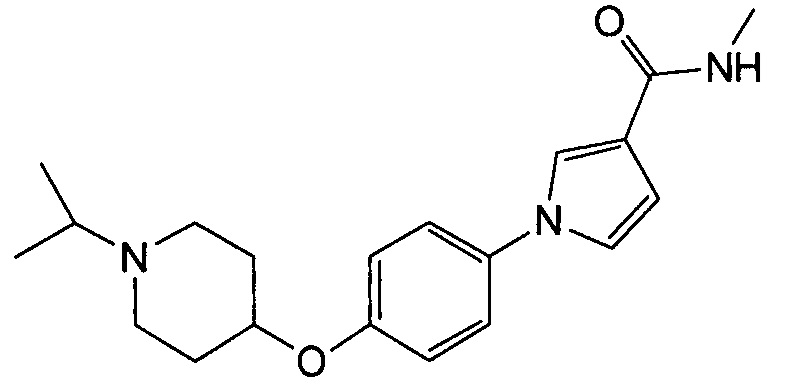
Указанное в заголовке соединение получали в виде бесцветных кристаллов повторением процедуры примера 1, за исключением того, что метил-1Н-пиррол-3-карбоновую кислоту заменяли N-метил-1Н-пиррол-3-карбоксамидом и 1-циклобутил-4-(4-иодфенокси)пиперидин заменяли 4-(4-иодфенокси)-1-изопропилпиперидином.
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,06 (д, J=6,61 Гц, 6 H) 1,82 (д, J=9,08 Гц, 2 H) 1,95-2,09 (м, 2 H) 2,39 (ушир.с., 2 H) 2,79 (ушир.с., 3 H) 2,96 (д, J=4,95 Гц, 3 H) 4,19 - 4,39 (м, 1 H) 5,59 - 5,86 (м, 1 H) 6,45 (дд, J=2,89, 1,65 Гц, 1 H) 6,84 - 6,99 (м, 2 H) 7,17 - 7,32 (м, 2 H) 7,52 (т, J=2,06 Гц, 1 H)
МС (ESI/APCI Dual) (Positive) m/z; (M+H)+ 342.
(Пример 9)
Получение (1-{4-[(1-трет-бутилпиперидин-4-ил)окси]фенил}-1Н-пиррол-3-ил)(пирролидин-1-ил)метанона (соединения № 19)
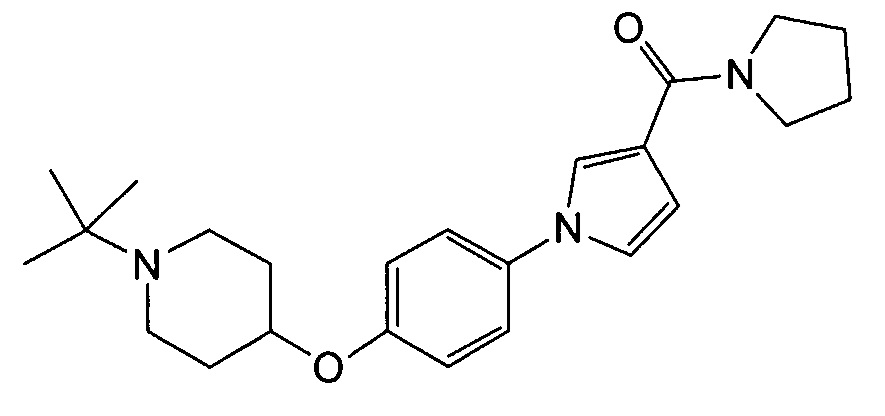
Указанное в заголовке соединение получали в виде бесцветных кристаллов повторением процедуры примера 1, за исключением того, что метил 1Н-пиррол-3-карбоновую кислоту заменяли (1Н-пиррол-3-ил)(пирролидин-1-ил)метаноном и 1-циклобутил-4-(4-иодфенокси)пиперидин заменяли 1-трет-бутил-4-(4-иодфенокси)пиперидином (который можно синтезировать способом, описанным в WO2008072724).
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,09 (с, 9 H) 1,81 (дд, J=8,46, 3,92 Гц, 2 H) 1,86-2,06 (м, 6 H) 2,41 (ушир.с., 2 H) 2,87 (ушир.с., 2 H) 3,56 - 3,80 (м, 4 H) 4,22 - 4,32 (м, 1 H) 6,63 (дд, J=3,10, 1,86 Гц, 1 H) 6,85 - 6,99 (м, 3 H) 7,23 - 7,31 (м, 2 H) 7,41 - 7,50 (м, 1 H)
МС (ESI/APCI Dual) (Positive) m/z; (M+H)+ 396.
(Пример 10)
Получение 1-{4-[(1-трет-бутилпиперидин-4-ил)окси]фенил}-N-метил-1Н-пиррол-3-карбоксамида (соединения № 20)
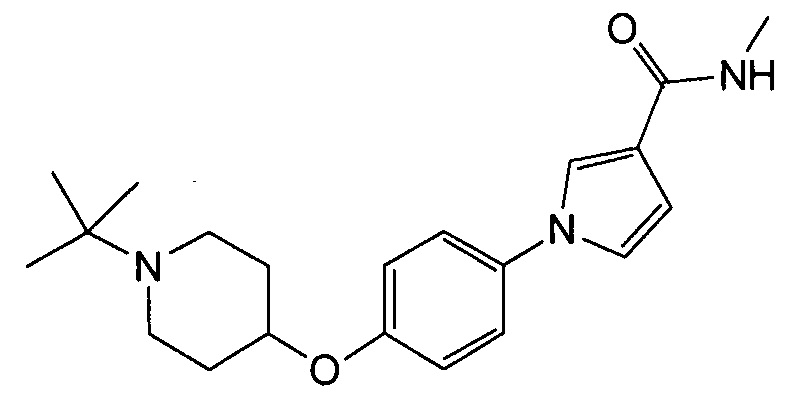
Указанное в заголовке соединение получали в виде бесцветных кристаллов повторением процедуры примера 1, за исключением того, что метил 1Н-пиррол-3-карбоновую кислоту заменяли N-метил-1Н-пиррол-3-карбоксамидом и 1-циклобутил-4-(4-иодфенокси)пиперидин заменяли 1-трет-бутил-4-(4-иодфенокси)пиперидином.
1H ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 1,09 (с, 9 H) 1,81 (дд, J=8,46, 3,92 Гц, 2 H) 1,96-2,06 (м, 2 H) 2,41 (ушир.с., 2 H) 2,87 (ушир.с., 2 H) 2,96 (д, J=4,95 Гц, 3 H) 4,27 (ушир.с., 1 H) 5,69 - 5,82 (м, 1 H) 6,38 - 6,50 (м, 1 H) 6,86 - 7,00 (м, 3 H) 7,21 - 7,31 (м, 2 H) 7,52 (т, J=1,86 Гц, 1 H)
МС (ESI/APCI Dual) (Positive) m/z; (M+H)+ 356.
(Испытание 1: испытание на связывание рецептора Н3 крыс)
Фронтальную кору головного мозга, выделенную рассечением у крысы, гомогенизировали тефлоновым гомогенизатором в буферном растворе 50 мМ трис-HCl (рН 7,4), содержащем ингибитор протеазы (полностью без EDTA; Roche Diagnostics) и 5 мМ EDTA. Гомогенизат центрифугировали при 48000 × g в течение 15 минут. Супернатант удаляли и дебрис суспендировали в буферном растворе 50 мМ трис-HCl (рН 7,4), содержащем 5 мМ EDTA, и центрифугировали при 48000 × g в течение 15 минут. Супернатант удаляли и дебрис суспендировали в буферном растворе 50 мМ трис-HCl (рН 7,4), содержащем 5 мМ EDTA, получая при этом мембранную фракцию. Мембранную фракцию (содержание белка в конечной реакционной смеси: 75 мкг), N-α-метил[3H]гистамин (PerkinElmer; конечная концентрация: 0,75 нМ) и испытуемое лекарственное средство смешивали и подвергали реакции при комнатной температуре в течение часа. После окончания реакции реакционную смесь фильтровали с отсасыванием через 96-луночный фильтровальный планшет GF/C, предварительно обработанный 0,3% полиэтиленимина; фильтры затем промывали пять раз буферным раствором 50 мМ трис-HCl (рН 7,4), содержащим 5 мМ EDTA. После промывания фильтры сушили и добавляли сцинтиллятор для измерения остаточной радиоактивности на фильтре посредством TopCount (PerkinElmer).
Остаточную радиоактивность в присутствии 10 мкМ тиоперамида использовали в качестве показателя неспецифического связывания и отличие от остаточной радиоактивности в отсутствие тиоперамида принимали в качестве показателя специфического связывания. Каждое из испытуемых лекарственных средств растворяли и разводили в ДМСО при различных концентрациях для построения кривой доза-ответная реакция из соответствующих остаточных радиоактивностей; концентрацию испытуемого лекарственного средства, которая ингибировала 50% (IC50) специфического связывания, определяли из кривой доза-ответная реакция. Величины IC50 соединений примеров показаны в приведенной ниже таблице 2.
|
(Испытание 2: связывание [35S]GTP-γ-S)
Фронтальную кору головного мозга, выделенную рассечением у крыс, гомогенизировали тефлоновым гомогенизатором в буферном растворе 30 мМ трис-HCl (рН 7,4), содержащем 2,5 мМ дигидрата хлорида кальция. Гомогенизат центрифугировали при 48000 × g в течение 15 минут. Супернатант удаляли и дебрис суспендировали в буферном растворе 30 мМ трис-HCl (рН 7,4), содержащем 2,5 мМ дигидрата хлорида кальция и центрифугировали при 48000 × g в течение 15 минут. Супернатант удаляли и дебрис суспендировали в буферном растворе 30 мМ трис-HCl (рН 7,4), содержащем 2,5 мМ дигидрата хлорида кальция и после инкубации при 37°С в течение 30 минут суспензию центрифугировали при 48000 × g в течение 15 минут. Полученный супернатант удаляли и дебрис суспендировали в буферном растворе 20 мМ HEPES (рН 7,4), содержащем 100 мМ хлорид натрия и 10 мМ хлорид магния, получая при этом мембранную фракцию. Мембранную фракцию (содержание белка в конечной реакционной смеси: 20 мкг), GDP (конечная концентрация: 300 мкМ), аденозиндеаминазу (конечная концентрация: 1 E/мл), R(-)-α-метилгистамин (конечная концентрация: 300 нМ) и испытуемое лекарственное средство смешивали и подвергали реакции при 30°С в течение 20 минут. После окончания реакции добавляли [35S]GTP-γ-S (конечная концентрация: 0,3 нМ) и реакцию продолжали в течение дополнительных 90 минут. После окончания реакции реакционную смесь фильтровали с отсасыванием через 96-луночный планшет для фильтрования, который затем промывали три раза 20 мМ буферным раствором HEPES (рН 7,4), содержащим 100 мМ хлорид натрия и 10 мМ хлорид магния. После промывания фильтры сушили и добавляли сцинтиллятор для измерения остаточной радиоактивности на фильтре посредством TopCount (PerkinElmer).
Остаточную радиоактивность в отсутствие R(-)-α-метилгистамина применяли в качестве показателя неспецифического связывания и отличие от остаточной радиоактивности в присутствии R(-)-α-метилгистамина применяли в качестве показателя неспецифического связывания. Каждое из испытуемых лекарственных средств растворяли и разводили в ДМСО при различных концентрациях для построения кривой доза-ответная реакция из соответствующих остаточных радиоактивностей; концентрацию испытуемого лекарственного средства, которая ингибировала 50% специфического связывания (IC50), определяли из кривой доза-ответная реакция. Как оказалось, соединения №№ 3 и 7 настоящего изобретения обнаружили высокие активности, т.е. IC50 100 нМ или меньше.
(Испытание 3: фармакокинетическое исследование in vivo на крысах)
Крысам SD давали в виде одного перорального введения соединение 3, 4 или 7 при дозе 3 мг/кг и спустя час после введения проверяли распределение соединения в плазме, головном мозге и цереброспинальной жидкости. Количественное определение осуществляли сочетанием высокоэффективная хроматография/танденмная масс-спектрометрия на приборе АPI 4000 (ЖХ-МС/МС; AB Sciex). Как оказалось, соединения №№ 3, 4 и 7 имели хорошие отношения содержания их в головном мозге и плазме 4,5, 2,0 и 2,2 соответственно, с соответствующей концентрацией в интрацеребральной жидкости 78,2 нг/г, 7,06 нг/г и 408 нг. Оба соединения №№ 3 и 7 имели отношение содержания их в цереброваскулярной жидкости и плазме 0,3 с соответствующими концентрациями в цереброваскулярной жидкости 5,75 нг/мл и 50,5 мл соответственно.
(Испытание 4: испытание на узнавание субстрата Р-гликопротеина)
Клетки LLC-GA5-COL300 (экспрессирующей MRD1 человека системы, полученной из почечной эпителиальной клеточной линии LLC-PK1 свиней) культивировали в транс-лунках. Непосредственно перед проведением испытания культуральную среду заменяли сбалансированным солевым раствором Хэнкса (НВSS) и затем проводили испытание. Раствор соединения анализа, регулированный до конечной концентрации 10 мкМ, добавляли к донорной стороне клеток LLC-GA5-COL300 и после прохождения заданного периода времени указанное количество клеток отбирали из акцепторной стороны. Концентрацию соединения анализа в образце измеряли ЖХ-МС/МС. Из количеств соединения, которое аккумулируется и проходит в акцепторную сторону, коэффициенты пенетрации мембран (×10-6 см/сек) вычисляли для направлений апикальное→базальное и базальное→апикальное и их относительное отношение (отношение оттоков) определяли для оценки узнавания субстрата Р-гликопротеина. Величины отношений оттоков соединений примеров перечислены ниже в таблице 3.
|
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Согласно настоящему изобретению, можно предложить фармацевтические продукты, которые обладают сильным действием по ингибированию связывания рецептора гистамина Н3 и которые применимы в предотвращении или лечении нарушений, обусловленных рецептором гистамина Н3, например, таких заболеваний, как деменция, болезнь Альцгеймера, синдром дефицита внимания и гиперактивности, шизофрения, эпилепсия, “центральная” судорога, ожирение, сахарный диабет, гиперлипидемия, нарколепсия, идиопатическая гиперсомния, индуцированный поведением синдром недостаточного сна, синдром апное во сне, нарушение циркадного ритма, парасомния, связанное со сном нарушении движения, инсомния и депрессия или аллергический ринит, и это, как предполагается, внесет большой вклад в развитие фармацевтической промышленности.
Фенилпиразольные производные
Новый моногидрат производного нафтиридина и способ его получения
7-пиперидиноалкил-3,4-дигидрохинолоновые производные, обладающие антагонистическим действием на рецептор мсн (меланин-концентрирующего гормона)
Производные 4-изопропилфенилглюцита в качестве ингибиторов sglt1
Фармацевтические композиции
Производные 2-пиридона
Производное 1,2,4-триазолона
Азольное производное
Новое производное гидроксамовой кислоты
Макролидное производное, замещенное по с-4''-положению
Фенилпиразольные производные
Новый моногидрат производного нафтиридина и способ его получения
7-пиперидиноалкил-3,4-дигидрохинолоновые производные, обладающие антагонистическим действием на рецептор мсн (меланин-концентрирующего гормона)
Производные 4-изопропилфенилглюцита в качестве ингибиторов sglt1
Фармацевтические композиции
Производные 2-пиридона
Производное 1,2,4-триазолона
Азольное производное
Новое производное гидроксамовой кислоты
Макролидное производное, замещенное по с-4''-положению

