Результат интеллектуальной деятельности: ИНГИБИТОРЫ 1 КИНАЗЫ КОНТРОЛЬНОЙ ТОЧКИ КЛЕТОЧНОГО ЦИКЛА ДЛЯ УСИЛЕНИЯ ДНК-ПОВРЕЖДАЮЩИХ АГЕНТОВ
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к ингибитору СНК1 для введения пациентам с раком для усиления ДНК-повреждающего агента.
Уровень техники
Киназа 1 контрольной точки клеточного цикла ("СНК1") представляет собой сериновую или треониновую киназу. СНК1 регулирует ход клеточного цикла и является основным фактором в реакции на повреждение ДНК в клетке. Показано, что ингибиторы СНК1 повышают чувствительность опухолевых клеток к различным генотоксичным агентам, таким как химиотерапия и радиация. (Tse, Archie N., et al., "Targeting Checkpoint Kinase 1 in Cancer Therapeutics." Clin. Cancer Res. 13(7) (2007) 1955-1960). Есть наблюдения, что у многих опухолей в G1 отсутствует путь проверки повреждений ДНК в фазе G1, и восстановление повреждений ДНК происходит за счет контрольных точек в фазах S и G2. (Janetka, James W., et al., "Inhibitors of checkpoint kinases: From discovery to the clinic." Drug Discovery & Development Vol.10, No. 4 (2007) 473-486). Контрольные точки S и G2 регулируются посредством СНК1. Показано, что ингибирование СНК1 устраняет контрольные точки S и G2, тем самым ослабляя восстановление ДНК и приводя к возрастанию гибели опухолевых клеток. Однако у нераковых клеток есть работающая контрольная точка G1, благодаря чему возможно восстановление ДНК и выживание клетки.
Киназа 2 контрольной точки клеточного цикла ("СНК2") также является сериновой/треониновой киназой. Функции CHK2's являются центральными для индукции задержки клеточного цикла и апоптоза, вызванного повреждением ДНК. (Ahn, Jinwoo, et al., "The Chk2 protein kinase." DNA Repair 3 (2004) 1039-1047). СНК2 активируется в ответ на генотоксичное поражение и передает сигнал контрольной точки по нескольким путям, которые в конечном счете вызывают задержку клеточного цикла на фазах G1, S и G2/M, активацию восстановления ДНК и апоптозную гибель клеток. (Bartek, Jiri, et al., "СНК2 Kinase - A Busy Messenger." Nature Reviews Molecular Cell Biology. Vol.2(12) (2001) 877-886). В раковых клетках часто отсутствуют одна или более ненарушенных в геноме контрольных точек, таким образом ингибирование СНК2 может сделать опухолевые клетки избирательно более чувствительными к антираковой терапии, такой как Y-облучение или лекарства, повреждающие ДНК. Нормальные клетки задействуют другие контрольные точки и восстановятся, а раковые клетки, лишенные этих контрольных точек, погибнут с большей вероятностью. Показано, что ингибитор СНК2, основанный на белке, аннулирует контрольную точку G2 и повышает чувствительность дефектных по р53 раковых клеток к ДНК-повреждающим агентам. (Pommier, Yves, et al., "Targeting Chk2 Kinase: Molecular Interaction Maps and Therapeutic Rationale." Current Pharmaceutical Design. Vol.11, No. 22 (2005) 2855-2872).
Ингибиторы СНК1 известны, см., например, международную публикацию WO 2009/004329, международную публикацию WO 2008/012635, международную публикацию WO 2007/090493, международную публикацию WO 2007/090494, международную публикацию WO 2006/106326, международную публикацию WO 2006/120573, международную публикацию WO 2005/103036, международную публикацию WO 2005/066163 и международную публикацию WO 03/028724.
Ингибиторы СНК1 включают SCH900776, PF-00477736, AZD7762, XL844 (см. 2008 EORTC Poster #395 [http://www.exelixis.com/eortc/posters/EORTC08J95JCL844-O02.pdf]), IC-83, and CHIR-124 (см. Tse, Archie N.. et al. "CHIR-124, a Novel Potent Inhibitor of Chkl, Potentiates the Cytotoxicity of Topoisomerase I Poisons In vitro and In vivo." Clin. Cancer Res. 13(2) (2007) pp.591-602).
Заявка на предварительную заявку на получение патента Соединенных Штатов 61/052,926 описывает соединения, включающие (R)-N-(4-(3-аминопиперидин-1-ил)-5-бром-1Н-пиррол[2,3-b]pyridin-3-ил)никотинамид (в дальнейшем "соединение 1") и (R)-N-(4-(3-аминопиперидин-1-ил)-5-бром-1Н-пиррол[2,3-b]пиридин-3-ил)изобутирамид (в дальнейшем "соединение 2"), (R)-N-(5-бром-4-(3-(метиламино)пиперидин-1-ил)-1H-пиррол[2,3-b]пиридин-3-ил)никотинамид (в дальнейшем "соединение 3"), (R)-N-(4-(3-аминопиперидин-1-ил)-5-бром-1Н-пиррол[2,3-b]пиридин-3-ил)-5-метилникотинамид (в дальнейшем "соединение 4"), (R)-N-(4-(3-аминопиперидин-1-ил)-5-бром-1Н-пиррол[2,3-b]пиридин-3-ил)циклопропанкарбоксамид (в дальнейшем "соединение 5"), (R)-N-(4-(3-аминопиперидин-1-ил)-5-бром-1Н-пиррол[2,3-b]пиридин-3-ил)-3-метил-бутанамид (в дальнейшем "соединение 6"), и (R)-N-(4-(3-аминопиперидин-1-ил)-5-бром-1Н-пиррол[2,3-b]пиридин-3-ил)-2-циклопропилацетамид (в дальнейшем "соединение 7"). Соединения 1, 2, 3, 4, 5, 6 и 7 (общее название '"926 ингибиторы СНК1") являются ингибиторами СНК1.
Ингибиторы СНК1 тестировались как терапевтические средства для лечения болезней.
Сущность изобретения
Неожиданно было обнаружено, что введение двух или трех доз ингибитора СНК1 через 24 часа после ДНК-повреждающего агента пациенту с раковым заболеванием усиливает действие ДНК-повреждающего агента.
В одном аспекте настоящее изобретение относится к ингибитору СНК1 для введения его пациенту с раковым заболеванием для усиления действия ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента.
Другой аспект настоящего изобретения представляет ингибитор СНК1 для введения его пациенту с раковым заболеванием для усиления действия ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента, причем ингибитор СНК1 вводится в двух дозах, первая из которых вводится через одни сутки после ДНК-повреждающего агента, а вторая - через двое суток после ДНК-повреждающего агента.
Другой аспект настоящего изобретения представляет ингибитор СНК1 для введения пациенту с раковым заболеванием для усиления действия ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента, причем ингибитор СНК1 вводится в трех дозах, первая доза ингибитора СНК1 вводится через одни сутки после ДНК-повреждающего агента, вторая доза ингибитора СНК1 - через двое суток после ДНК-повреждающего агента, а третья доза ингибитора СНК1 - через трое суток после ДНК-повреждающего агента.
Краткое описание фигур
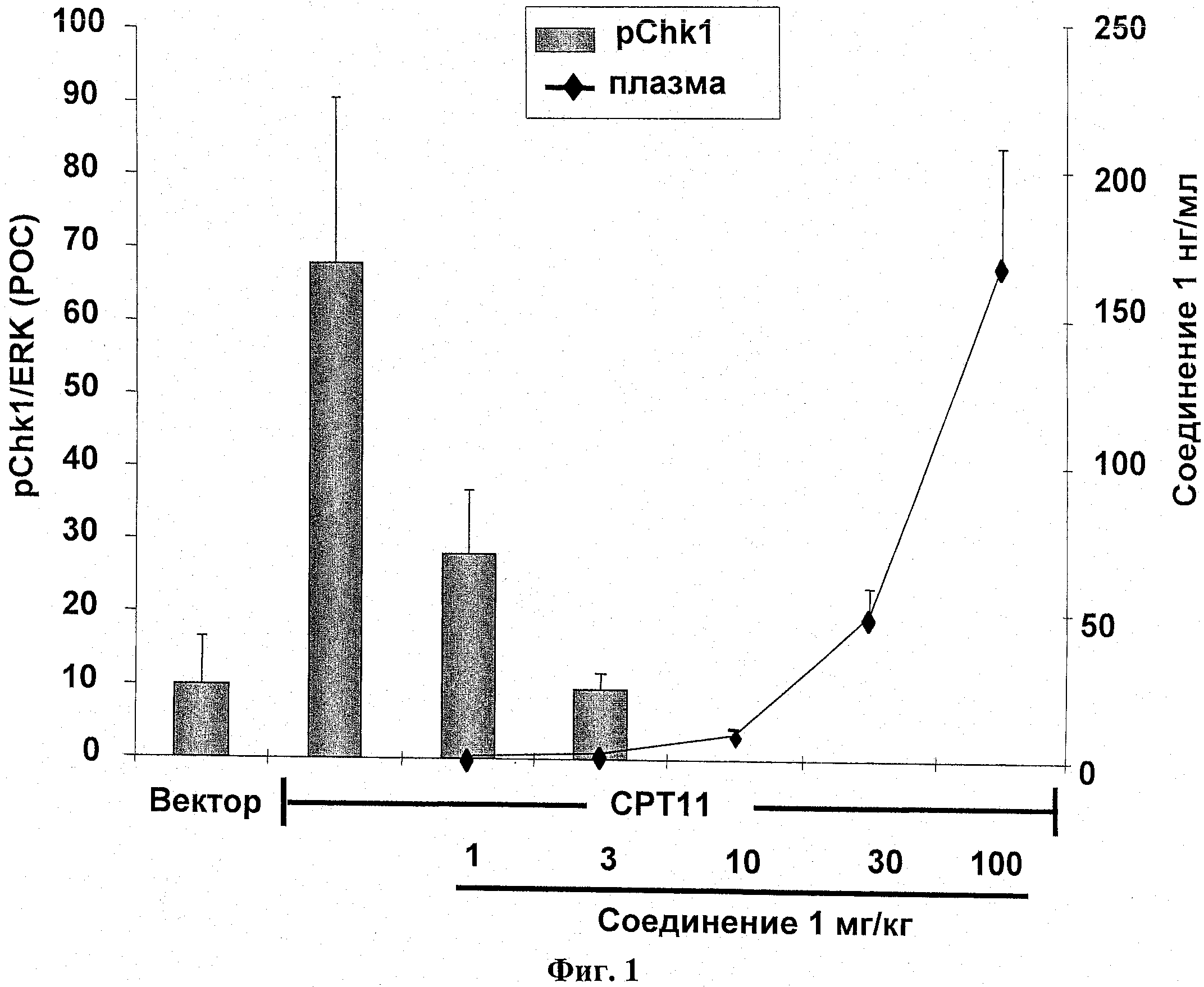
На фиг.1 показано ингибирование ДНК-повреждающего агента, вызванное фосфорилированием СНК1.
На фиг.2 показано ингибирование ДНК-повреждающего агента, вызванное фосфорилированием СНК1.
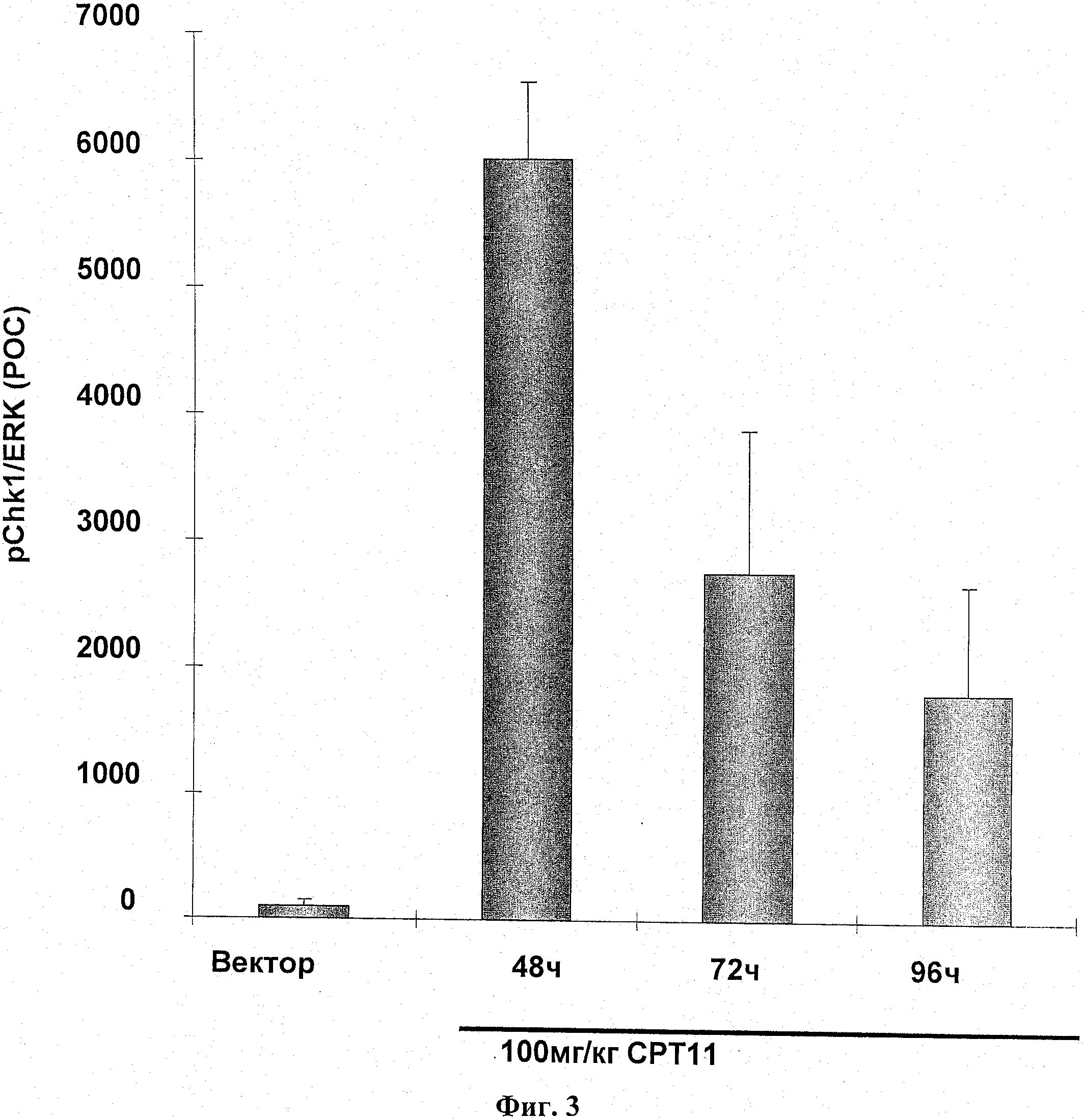
На фиг.3 показано фосфорилирование СНК1 после введения ДНК-повреждающего агента.
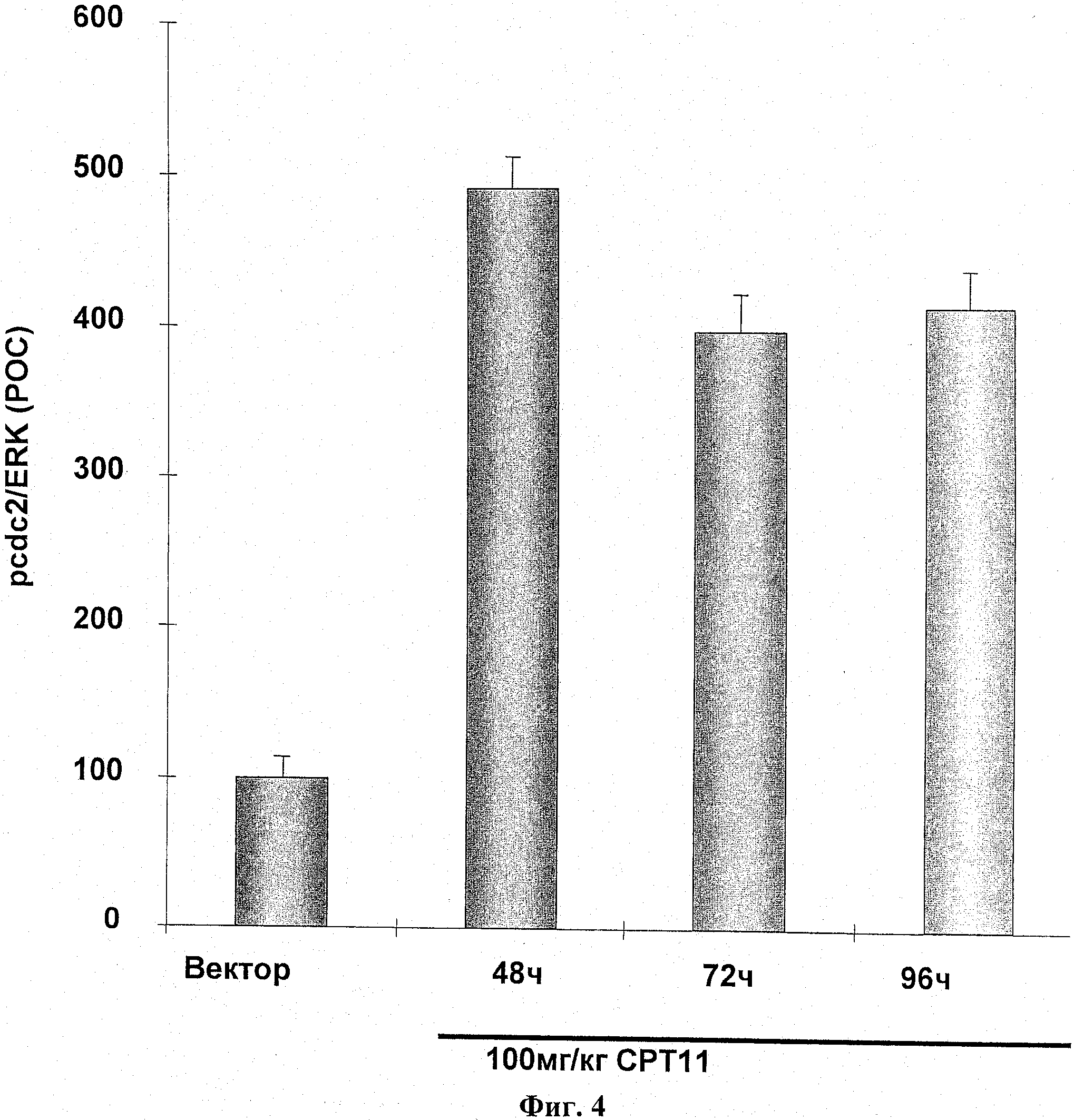
На фиг.4 показано фосфорилирование cdc2 после введения ДНК-повреждающего агента.
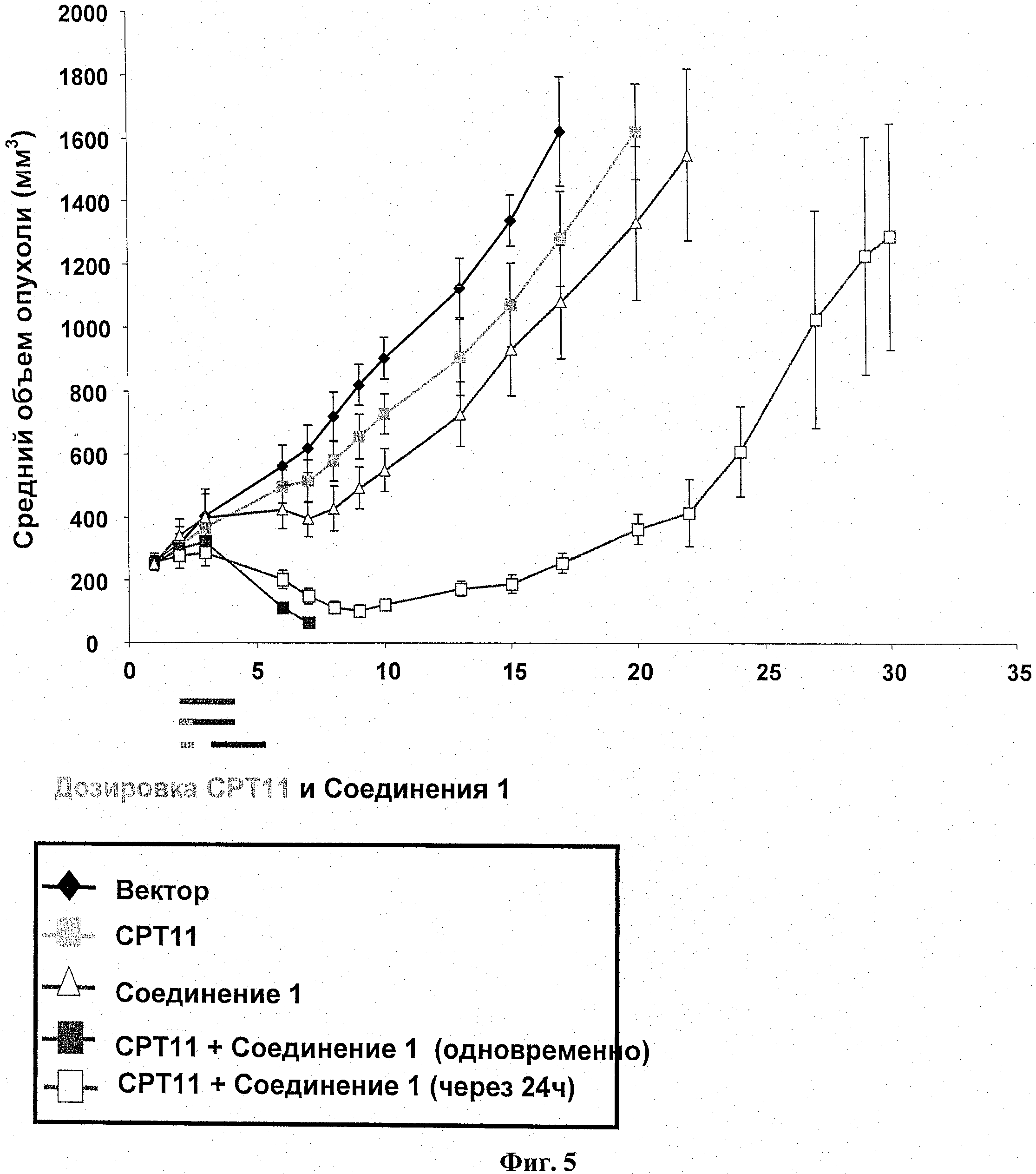
На фиг.5 показан эксперимент по ингибированию роста опухоли (ИРО) у бестимусных мышей с подкожным НТ-29 ксенотрансплантатом.
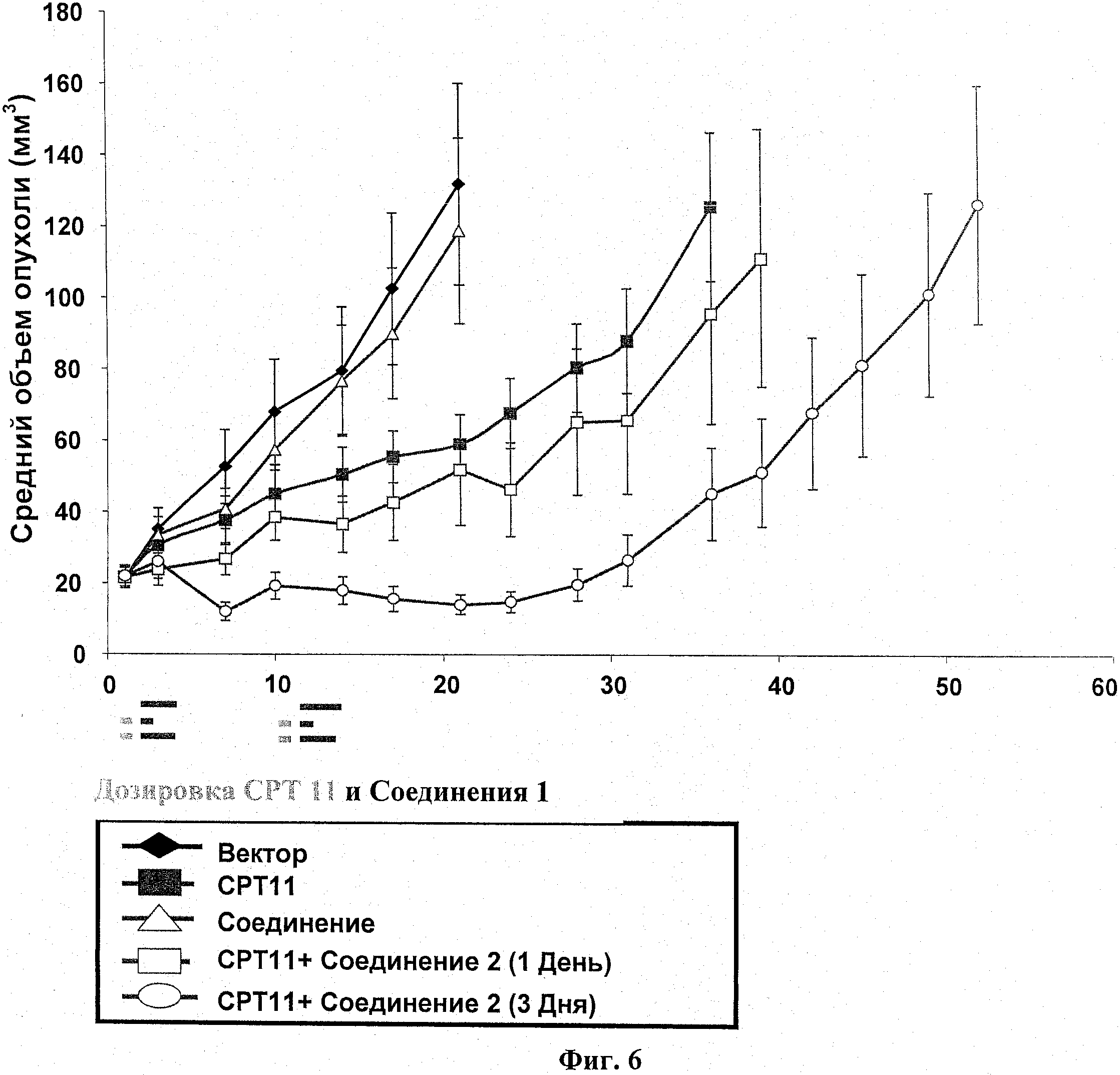
На фиг.6 показан эксперимент по подавлению роста опухоли (ИРО) у бестимусных мышей с подкожным НТ-29 ксенотрансплантатом.
На фиг.7 показано ингибирование ДНК-повреждающего агента, вызванное фосфорилированием cdc2.
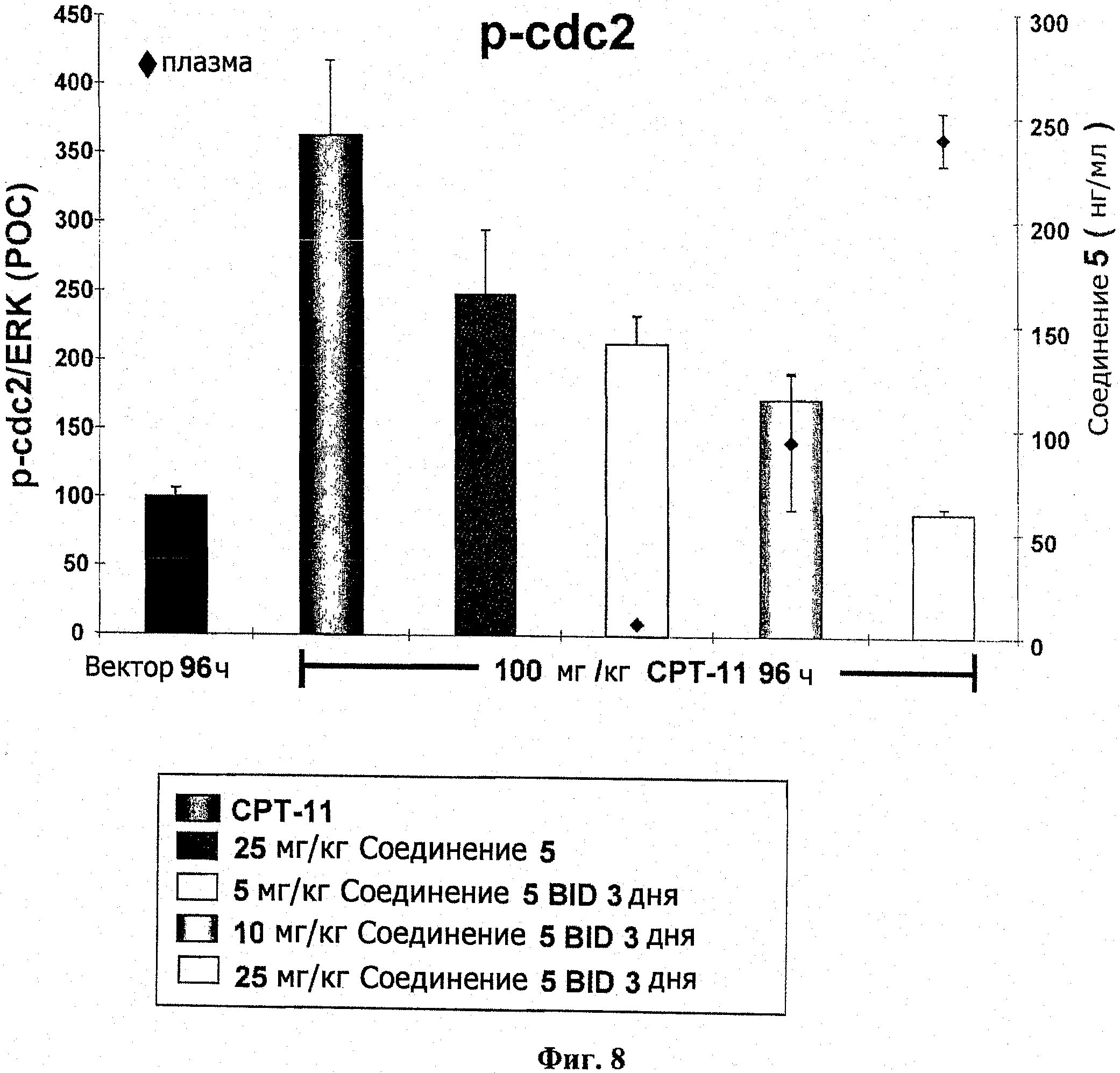
На фиг.8 показано ингибирование ДНК-повреждающего агента, вызванное фосфорилированием cdc2.
На фиг.9 показан ИРО эксперимент у бестимусных мышей с подкожным НТ-29 ксенотрансплантатом.
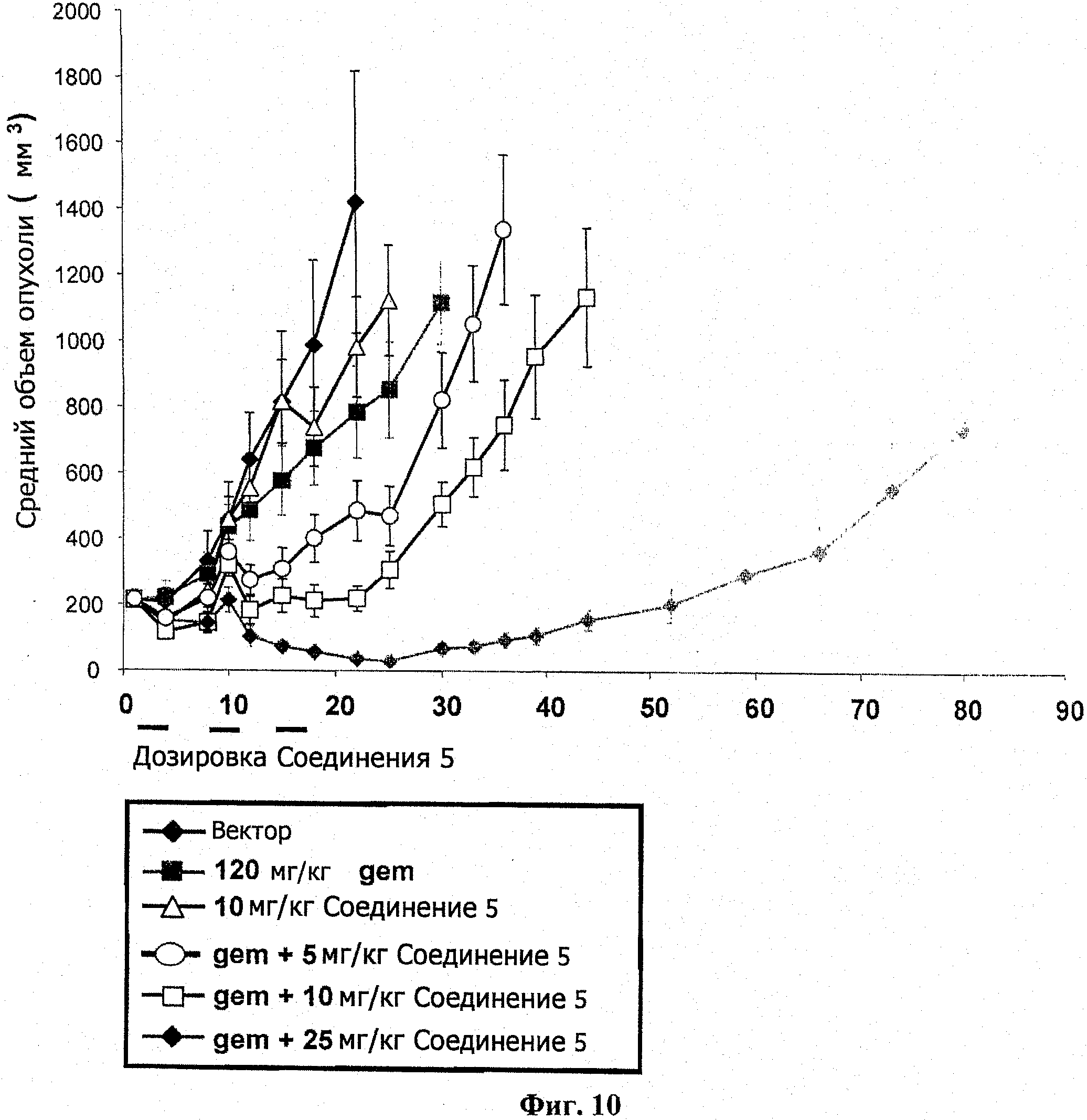
На фиг.10 показан ИРО эксперимент у бестимусных мышей с подкожным НТ-29 ксенотрансплантатом.
На фиг.11 показан ИРО эксперимент у бестимусных мышей с подкожным НТ-29 ксенотрансплантатом.
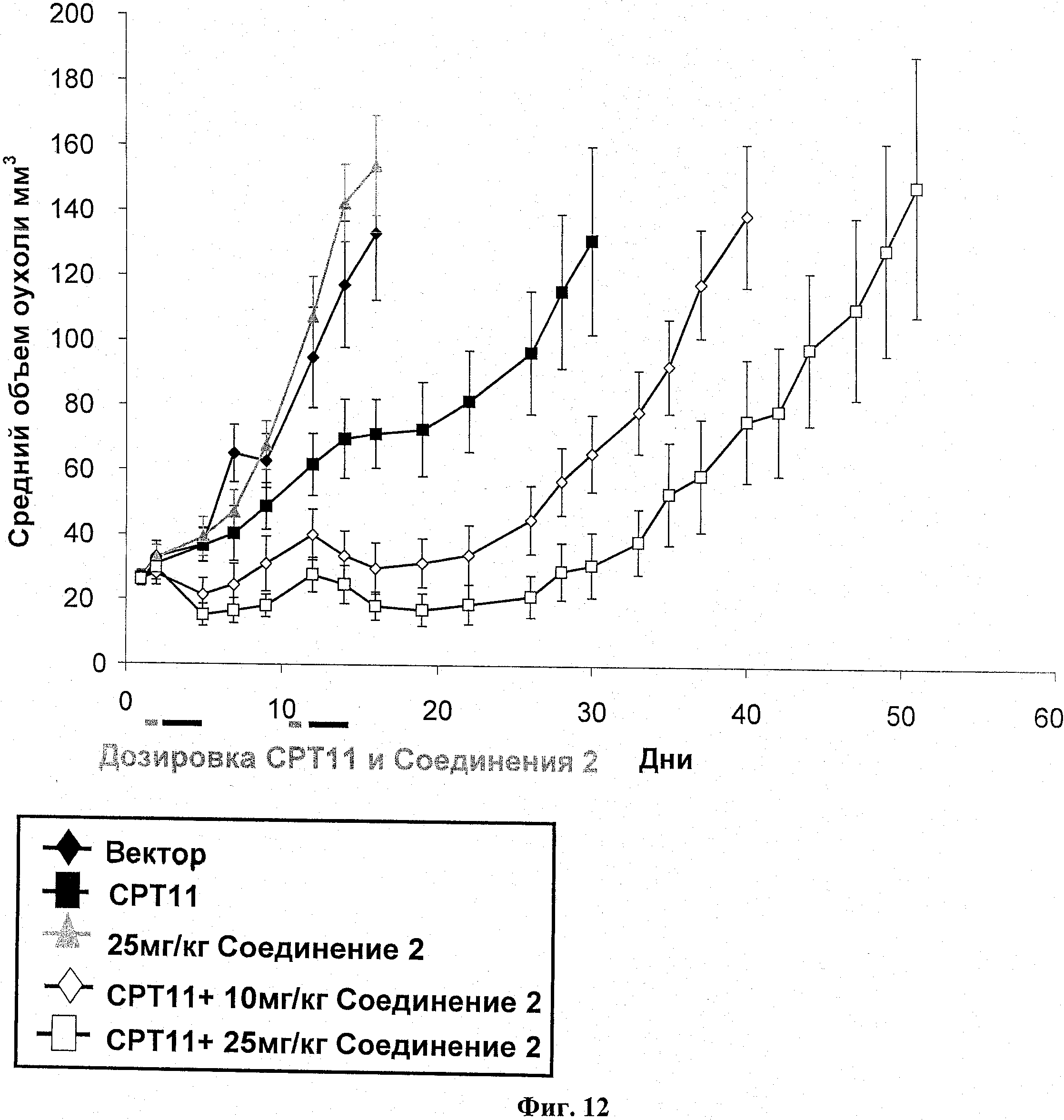
На фиг.12 показан ИРО эксперимент у бестимусных мышей с подкожным НТ-29 ксенотрансплантатом.
На фиг.13 показан ИРО эксперимент у бестимусных мышей с подкожным НТ-29 ксенотрансплантатом.
Подробное описание изобретения
Некоторые варианты воплощения изобретения не будут детально описаны. Притом, что изобретение будет описано в сопровождении перечисленных вариантов воплощения, будет понятно, что это не означает, что изобретение ними ограничивается. Напротив, подразумевается, что изобретение покрывает все альтернативы, модификации и эквиваленты, которые можно включить в пределы притязаний настоящего изобретения, как определено объемом формулы изобретения. Специалист в данной области узнает многие способы и материалы, подобные или равноценные тем, что описаны здесь, которые можно использовать при осуществлении на практике данного изобретения. Данное изобретение не ограничивается описанными способами и материалами. В случае если один или более зарегистрированных в литературе или подобных материалах отличаются или противоречат этой заявке, включая, но не ограничиваясь определенными терминами, при использовании терминов, описанных методик, или тому подобного, эта заявка имеет преимущественную силу.
Определения
Термины «рак» и «раковый» имеют отношение или описывают физиологическое состояние у млекопитающих, которое обычно характеризуется нерегулируемым ростом клеток. «Опухоль» состоит из одной или более раковых клеток. Примеры рака включают, но не ограничиваются, карциномой, лимфомой, бластомой, саркомой и лейкемией или лимфолейкозом. Более редкие примеры таких раков включают плоскоклеточный рак (или рак клеток плоского эпителия), рак легких, включая мелкоклеточный рак легких, немелкоклеточный рак легких («НМРЛ»), аденокарциному легких и плоскоклеточную карциному легких, рак брюшины, гепатоцеллюлярный рак, рак желудка, включая желудочно-кишечный рак, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак молочной железы, рак ободочной кишки, рак прямой кишки, колоректальный рак, внутриматочную карциному, карциному слюнных желез, рак почек, рак простаты, рак влагалища, рак щитовидной железы, карциному печени, карциному заднего прохода, карциному пениса, рак кожи, включая меланому, и рак головы и шеи.
Термины «лечить» или «лечение» относятся к терапевтическим, профилактическим, паллиативным или предупредительным мерам. В объеме данного изобретения полезные или желаемые клинические результаты включают, но не ограничиваются, облегчение симптомов, уменьшение распространения заболевания, стабилизацию (т.е. отсутствие ухудшения) течения заболевания, исчезновение или уменьшение прогрессирования заболевания, улучшение или временное облегчение болезненного состояния, и ремиссию (частичную или тотальную), определяемую или неопределяемую. «Лечение» может также означать увеличение времени жизни по сравнению с ожидаемым временем жизни при отсутствии лечения. Нуждающиеся в лечении включают в себя тех, у кого уже есть соответствующее состояние или нарушение, а также тех, кто предрасположен к соответствующему состоянию или нарушению или тех, у кого соответствующее состояние или нарушение предотвращается.
Фраза «фармацевтически приемлемый» означает, что вещество или соединение совместимы химически и/или токсилогически с другими ингредиентами, входящими в Соединение, и/или этим лечат млекопитающих.
Способы лечения
Настоящее изобретение представляет ингибитор СНК1 для введения пациенту с раковым заболеванием для усиления действия ДНК-повреждающего агента.
Настоящее изобретение также представляет ингибитор СНК1 для введения пациенту с раковым заболеванием для усиления действия ДНК-повреждающего агента, где введение ингибитора СНК1 следует за введением ДНК-повреждающего агента, при этом ингибитор СНК1 вводится в двух дозах, первая из которых вводится через одни сутки после ДНК-повреждающего агента, а вторая - через двое суток после ДНК-повреждающего агента.
Настоящее изобретение также представляет ингибитор СНК1 для введения пациенту с раковым заболеванием для усиления действия ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента, при этом ингибитор СНК1 вводится в трех дозах, первая из которых вводится через одни сутки после ДНК-повреждающего агента, вторая - через двое суток после ДНК-повреждающего агента, а третья - через трое суток после ДНК-повреждающего агента.
Использование контроля за клеточным циклом представляет собой фундаментальное свойство, от которого зависит рост раковых клеток. Одним из механизмов, благодаря которому этого можно достичь, является манипуляция контрольными точками клеточного цикла и репарацией повреждений ДНК. Имеются доказательства, что у раковых клеток может появиться устойчивость к химиотерапии благодаря гиперактивации репарации повреждений ДНК на контрольной точке G2/M, клеточном процессе, который зависит от СНК1. Ингибирование СНК1 подавляет такой путь к выживанию. Введение ингибитора СНК1 совместно с ДНК-повреждающим агентом может быть более эффективно, чем введение одного ДНК-повреждающего агента. Обнаружено, что уровни СНК1 повышаются в течение долгого периода времени после введения ДНК-повреждающего агента (см. фиг.3 и 4). Также обнаружено, что ингибитор СНК1 следует назначать с задержкой в 24 часа после введения ДНК-повреждающего агента (см. фиг.5). Следовательно, в приемлемой схеме дозирования ингибитор СНК1 следует давать с задержкой в 24 часа после ДНК-повреждающего агента, а также его действие должно быть достаточно долгим, чтобы понизить уровни СНК1 так, чтобы меньшее число клеток смогло пройти репарацию ДНК.
ДНК-повреждающие агенты включают Gemzar® (гемцитабин), Camptosar® (иринотекан или СРТ-11), Temodar® (темозоломид), Xeloda® (капецитабин), Hycamtin® (топотекан), цисплатин, Eloxatin® (оксалиплатин), Paraplatin® (карбоплатин), камптотецин, ара-С (цитарабин), 5-FU (фтороурацил), Cytoxan® (циклофосфамид), Etopophos® или Vepesid® (этопозид фосфат), Vumon® (тенипозид), Adriamycin PFS® или Adriamycin RDF® (доксорубицин), даунорубицин, Alimta® (пеметрексед), митомицин С, флударабин, хлорамбуцил, мелфалан, гидроксимочевина и радиация. В некоторых вариантах воплощения ДНК-повреждающий агент выбирается из группы, состоящей из гемцитабина, иринотекана, темозоломида, капецитабина, камптотецина, цисплатина, ара-С и 5-FU. В некоторых вариантах воплощения ДНК-повреждающий агент выбирается из гемцитабина, иринотекана, темозоломида и капецитабина. В некоторых вариантах воплощения ДНК-повреждающий агент выбирается из гемцитабина, иринотекана, цисплатина, оксалиплатина, карбоплатина и цитарабина. В некоторых вариантах воплощения ДНК-повреждающий агент выбирается из гемцитабина и иринотекана. ДНК-повреждающий агент вводится в принятой или рекомендованной дозе.
ДНК-повреждающие агенты включают Gemzar® (гемцитабин), Camptosar® (иринотекан или СРТ-11). Temodar® (темозоломид), Xeloda® (капецитабин), Hycamtin® (топотекан), цисплатин, Eloxatin® (оксалиплатин), Paraplatin® (карбоплатин), камптотецин, ара-С (цитарабин), 5-FU (фторурацил), Cytoxan® (циклофосфамид), Etopophos® или Vepesid® (этопозид фосфат), Vumon® (тенипозид), Adriamycin PFS® или Adriamycin RDF® (доксорубицин), даунорубицин, Alimta® (пеметрексед) и радиацию. В некоторых вариантах воплощения ДНК-повреждающий агент выбирается из группы, состоящей из гемцитабина, иринотекана, темозоломида, капецитабина, камптотецина, цисплана, ара-С и 5-FU. В некоторых вариантах воплощения ДНК-повреждающий агент выбирается из гемцитабина, иринотекана, темозоломида и капецитабина. В некоторых вариантах воплощения ДНК-повреждающий агент выбирается из гемцитабина, иринотекана, цисплатина, оксалиплатина, карбоплатина и цитарабина. В некоторых вариантах воплощения ДНК-повреждающий агент выбирается из гемцитабина и иринотекана. ДНК-повреждающий агент вводится в принятой или рекомендованной дозе.
В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 выбирается из группы, состоящей из «926 Ингибиторов СНК1». В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 выбирается из группы, состоящей из соединения 1, соединения 2, соединения 3, соединения 4, соединения 5, соединения 6 и соединения 7. В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 является соединением 1. В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 является соединением 2. В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 является соединением 3. В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 является соединением 4. В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 является соединением 5. В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 является соединением б. В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 является соединением 7.
В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 выбирается из группы, состоящей из '926 Ингибиторов СНК1, SCH90076, PF-00477736, AZD7762, XL844, IC-83 и CHIR-124. В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 выбирается из группы, состоящей из SCH90076, PF-00477736, AZD7762, XL844, IC-83 и CHIR-124.
В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 выбирается из группы, состоящей из '926 Ингибиторов СНК1, PF-00477736, AZD7762, XL844, IC-83 и CHIR-124. В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 выбирается из группы, состоящей из PF-00477736, AZD7762, XL844, IC-83 и CHIR-124.
В некоторых вариантах воплощения настоящего изобретения ингибитор СНК1 не включает в себя '926 СНК1 Ингибиторы.
В некоторых вариантах воплощения изобретение предоставляет способ лечения рака. Более детально, раки, которые можно лечить составами и способами изобретения, включают, но не ограничиваются: рак мягких тканей: саркома (ангиосаркома, фибросаркома, рабдомиосаркома, липосаркома), миксома, рабдомиомой, фиброма, липома и тератома; легких: бронхогенная карцинома (плоскоклеточная, недифференцированная мелкоклеточная, недифференцированная крупноклеточная, адемокарцинома), альвеолярная (бронхиолярная) карцинома, бронхиальная аденома, саркома, лимфома, хондроматозная гамартома, мезотелиома; гастроинтерстинальные: пищевода (плоскоклеточная карцинома, аденокарцинома, лейомиосаркома, лимфома), желудка (карцинома, лимфома, лейомиосаркома), поджелудочной железы (протоковая аденокарцинома, инсулинома, глюкагонома, гастринома, карциноидные опухоли, випома), тонкой кишки (аденокарцинома, лимфома, карциноидные опухоли, саркома Капоши, лейомиома, гемангиома, липома, нейрофиброма, фиброма), толстой кишки (аденокарцинома, тубулярная аденома, ворсинчатая аденома, гамартома, лейомиома); мочеполового тракта: почек (аденокарцинома, опухоль Вильма [нефробластома], лимфома, лейкемия), мочевого пузыря и уретры (плоскоклеточная карцинома, переходно-клеточная карцинома, аденокарцинома), простаты (аденокарцинома, саркома), мужских половых желез (семинома, тератома, эмбриональная карцинома, тератокарцинома, хориокарцинома, саркома, интерстициально-клеточная карцинома, фиброма, фиброаденома, аденоматоидные опухоли, липома); печени: гепатома (гепатоцеллюлярная карцинома), холангиокарцинома, гепатобластома, ангиосаркома, гепатоцеллюлярная аденома, гемангиома; костей: остеогенная саркома (остеосаркома), фибросаркома, злокачественная фиброзная гистиоцитома, хондросаркома, саркома Юинга, злокачественная лимфома (ретикулярно-клеточная саркома), множественная миелома, злокачественная гигантоклеточная хордома, остеохрондрома (костно-хрящевой экзостоз, доброкачественная хондрома, хондробластома, хондромиксофиброма, остеоидная остеома и гигантоклеточные опухоли; нервной системы: черепа (остеома, гемангиома, гранулема, ксантома, деформирующий остоз), оболочек мозга (менингиома, менингиосаркома, глиоматоз), мозга (астроцитома, медуллобластома, глиома, эпендимома, герминома [пинеалома], полиморфная глиобластома, олигодендроглиома, шваннома, ретинобластома, врожденные опухоли), спинного мозга (нейрофиброма, менингиома, глиома, саркома; гинекологические: матки (карцинома эндометрия), шейки матки (карцинома шейки матки, предопухолевая дисплазия шейки матки), яичников (карцинома яичников [серозная цистаденокарцинома, муцинозная цистаденокарцинома, неуточненная карцинома], гранулезоклеточные опухоли, опухоли клеток Сертоли-Лейдига, дисгерминома, злокачественная тератома), вульвы (плоскоклеточная карцинома, интраэпителиальная карцинома, аденокарцинома, фибросаркома, меланома), влагалища (светлоклеточная карцинома, плоскоклеточная карцинома, кистевидная саркома [эмбриональная рабдомиосаркома], фаллопиевых труб (карцинома); гематологический: кровь и костный мозг (миелоидная лейкемия [острая и хроническая], острая лимфобластическая лейкемия, хроническая лимфатическая лейкемия, миелопролиферативные болезни, множественная миелома, миелодиспластический синдром), болезнь Ходжкина, неходжкинская лимфома [злокачественная лимфома]; кожи: злокачественная меланома, базальноклеточная карцинома, плоскоклеточная карцинома, саркома Капоши, диспластический невус, липома, ангиома, дерматофиброма, келоид, псориаз и надпочечные железы: нейробластома. Термин «раковая клетка» в данном документе включает клетки, пораженные любым из указанных выше нарушений.
В некоторых вариантах воплощения настоящего изобретения рак выбирается из колоректального рака (включая Ras мутации), мелкоклеточного рака легких, немелкоклеточного рака легких (включая Ras мутации), глиомы, рака яичников, метастатического рака молочной железы, рака поджелудочной железы, гепатобилиарного рака (включая гепатоцеллюлярный рак, рак желчного протока и холангиокарциному), рака желудка, рака семенников, плоскоклеточной карциномы головы и шеи, лейкемии (включая острую миелоидную лейкемию, острую лимфобластическую лейкемию, хроническую миелоидную лейкемию и хроническую лимфоидную лейкемию), лимфомы (включая лимфому из клеток мантийной зоны, лимфому Ходжкина и неходжкинскую лимфому) и рака простаты.
В некоторых вариантах воплощения настоящего изобретения рак выбирается из колоректального рака (включая Ras мутации), мелкоклеточного рака легких, немелкоклеточного рака легких, рака яичников, метастатического рака молочной железы, рака поджелудочной железы, гепатобилиарного рака (включая гепатоцеллюлярный рак, рак желчного протока и холангиокарциному), рака желудка, рака семенников, плоскоклеточной карциномы головы и шеи, лейкемии (включая острую миелоидную лейкемию, острую лимфобластическую лейкемию, хроническую миелоидную лейкемию и хроническую лимфоидную лейкемию), лимфомы (включая лимфому из клеток мантийной зоны, лимфому Ходжкина и неходжкинскую лимфому) и рака простаты.
В некоторых вариантах воплощения настоящего изобретения раком является солидная опухоль.
В некоторых вариантах воплощения настоящего изобретения рак выбирается из рака поджелудочной железы, рака яичников и колоректального рака.
В некоторых вариантах воплощения настоящего изобретения рак выбирается из колоректального рака (включая Ras мутации), мелкоклеточного рака легких, немелкоклеточного рака легких и глиомы. В дальнейшем варианте воплощения ДНК-повреждающим агентом является иринотекан.
В некоторых вариантах воплощения настоящего изобретения рак выбирается из немелкоклеточного рака легких, рака яичников, метастатического рака молочной железы, рака поджелудочной железы, гепатобилиарного рака (включая гепатоцеллюлярный рак, рак желчного протока и холангиокарциному) и рака желудка. В дальнейшем варианте воплощения ДНК-повреждающим агентом является гемцитабин.
В некоторых вариантах воплощения настоящего изобретения рак выбирается из колоректального рака (включая Ras мутации), мелкоклеточного рака легких, немелкоклеточного рака легких, рака яичников, гепатобилиарного рака (включая гепатоцеллюлярный рак, рак желчного протока и холангиокарциному), рака желудка, рака семенников и плоскоклеточной карциномы головы и шеи. В дальнейшем варианте воплощения ДНК-повреждающий агент выбирается из группы, состоящей из цисплатина, оксалиплатина и карбоплатина.
В некоторых вариантах воплощения настоящего изобретения рак выбирается из лейкемии (включая острую миелоидную лейкемию, острую лимфобластическую лейкемию, хроническую миелоидную лейкемию и хроническую лимфоидную лейкемию), лимфомы (включая лимфому из клеток мантийной зоны, лимфому Ходжкина и неходжкинскую лимфому) и рака простаты. В дальнейшем варианте воплощения ДНК-повреждающим агентом является цитарабин.
Первыми сутками называются сутки получения первой дозы ДНК-повреждающего агента. Настоящее изобретение предусматривает две или три дозы ингибитора СНК1 для усиления ДНК-повреждающего агента, при этом первая доза водится на вторые сутки, вторая доза - на третьи сутки, а третья доза - на четвертые сутки. Введение ингибитора СНК1 должно следовать за введением ДНК-повреждающего агента по крайней мере через сутки, или приблизительно через 24 часа. Однако введение первой дозы ингибитора СНК1 не обязательно должно следовать точно через 24 часа после введения ДНК-повреждающего агента. Это просто удобный способ сказать, что ингибитор СНК1 следует назначать через одни сутки после ДНК-повреждающего агента. Следовательно, введение ингибитора СНК1 через одни сутки после ДНК-повреждающего агента включает введение ингибитора СНК1 в промежуток от 18 до 36 часов после введения ДНК-повреждающего агента. Далее, введение ингибитора СНК1 через двое суток после ДНК-повреждающего агента включает введение ингибитора СНК1 в промежуток от 36 до 60 часов после введения ДНК-повреждающего агента. Наконец, введение ингибитора СНК1 через трое суток после ДНК-повреждающего агента включает введение ингибитора СНК1 в промежуток от 60 до 90 часов после введения ДНК-повреждающего агента.
Альтернативно можно сказать, что первая доза ингибитора СНК1 вводится в промежуток от 18 до 30 часов после введения ДНК-повреждающего агента, вторая доза ингибитора СНК1 вводится в промежуток от 30 до 50 часов после введения ДНК-повреждающего агента, и третья доза ингибитора СНК1 вводится в промежуток от 50 до 90 часов после введения ДНК-повреждающего агента.
Настоящее изобретение представляет ингибитор СНК1 для введения пациентам с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в двух или трех дозах, первая из которых вводится через одни сутки после ДНК-повреждающего агента, вторая доза - через двое суток после ДНК-повреждающего агента, и необязательно третья доза - через трое суток после ДНК-повреждающего агента.
Один вариант воплощения настоящего изобретения представляет ингибитор СНК1 для введения пациенту с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в двух дозах, первая из которых вводится через одни сутки после ДНК-повреждающего агента, и вторая - через двое суток после ДНК-повреждающего агента, причем ингибитор СНК1 вводится между биологически эффективной дозой и максимально переносимой дозой.
Другое воплощение настоящего изобретения представляет ингибитор СНК1 для введения пациенту с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в трех дозах, первая из которых вводится через одни сутки после ДНК-повреждающего агента, вторая - через двое суток после ДНК-повреждающего агента, а третья - через трое суток после ДНК-повреждающего агента, причем ингибитор СНК1 вводится между биологически эффективной дозой и максимально переносимой дозой.
Ингибитор СНК1 следует вводить в дозе по меньшей мере на уровне, достаточном для достижения желаемого биологического эффекта. Так, для усиления ДНК-повреждающего агента доза ингибитора СНК1 будет по крайней мере не меньше минимального количества, которым достигается желаемый биологический эффект, или биологически эффективной дозы.
В одном воплощении настоящего изобретения, желаемый биологический эффект ингибитора СНК1 составляет 80% или больше ингибирования pCHKl, после введения ДНК-повреждающего агента (по сравнению с введением только ДНК-повреждающего агента).
В другом воплощении настоящего изобретения, желаемый биологический эффект ингибитора СНК1 составляет 90% или больше ингибирования pCHK1, после введения ДНК-повреждающего агента (по сравнению с введением только ДНК-повреждающего агента).
В другом воплощении настоящего изобретения, желаемый биологический эффект ингибитора СНК1 составляет 95% или больше ингибирования pCHK1, после введения ДНК-повреждающего агента (по сравнению с введением только ДНК-повреждающего агента).
В другом воплощении настоящего изобретения, желаемый биологический эффект ингибитора СНК1 составляет 66% или больше ингибирования p-cdc2, после введения ДНК-повреждающего агента (по сравнению с введением только ДНК-повреждающего агента).
Однако доза не должна быть настолько высока, чтобы нежелательные побочные эффекты перевесили преимущество биологического эффекта. Следовательно, в эффективной схеме дозирования доза не должна превышать максимально переносимую дозу («МПД»). Настоящее исследование представляет способ лечения пациента со схемой дозирования, которая включает две или три дозы ингибитора СНК1, где дозы ингибитора СНК1 находятся между биологически эффективной и максимально переносимой дозами.
Максимально переносимая доза определяется как самая высокая доза, которая дает приемлемую степень дозолимитируемой токсичности («ДЛТ»). Дозы, которые причиняют неприемлемую степень ДЛТ, рассматриваются как непереносимые. Обычно МПД для определенной схемы дозирования устанавливается на фазе 1 клинических испытаний. Обычно их проводят на пациентах, начиная с безопасной стартовой дозы 1/10 от сильнотоксичной дозы («СТД10») у грызунов (на основе мг/м2) и, разбив пациентов на группы по три, увеличивают дозы согласно модифицированной последовательности Фибоначчи, по которой на каждом последующем шаге увеличения дозы относительное приращение уменьшается (т.е. доза возрастает на 100%, 65%, 50%, 40% и от 30% до 35% соответственно). Увеличение дозы продолжается в группах по три пациента пока не достигнет непереносимой дозы. Следующий перед ней более низкий уровень дозы, который дает приемлемую степень ДЛТ, принимается за МПД.
Также МПД ингибитора СНК1 изменяется в зависимости от специфики ингибитора, вида и схемы дозирования. Например, прием лекарства только в одни сутки по сравнению с приемом в течение двух и трех суток во время семидневного, четырнадцатидневного, двадцатиоднодневного и двадцативосьмидневного циклов лечения могут иметь различные МПД. Однако, как обсуждалось выше, в эффективной схеме дозирования нужна достаточно высокая доза ингибитора, чтобы она имела биологическую эффективность. При приеме лекарства только в одни сутки можно достичь биологически эффективной дозы, но эффект может быть недостаточно длительным, чтобы предотвратить репарацию ДНК в поврежденных клетках. Альтернативно, прием лекарства в течение трех суток может дать достаточно длительный эффект, но доза может быть недостаточно высокой, чтобы достичь биологически эффективной дозы. Возможно, из-за этого МПД при приеме в течение трех суток может быть ниже, чем биологически эффективная доза. Таким образом, в эффективной схеме дозирования МПД равна или выше биологически эффективной дозы.
В одном варианте воплощения настоящего изобретения две или три дозы ингибитора СНК1 назначаются между биологически эффективной и максимально переносимой дозами.
В другом варианте воплощения настоящего изобретения две или три дозы ингибитора СНК1 вводятся в максимально переносимой дозе.
Обычно при лечении рака пациентам вводится МПД определенного соединения так, чтобы была достигнута максимальная выгода от лечения. Соответственно, один вариант воплощения настоящего изобретения обеспечивает способ лечения рака путем введения двух или трех доз ингибитора СНК1, при этом дозы ингибитора СНК1 являются максимальными переносимыми дозами ингибитора.
Один вариант воплощения настоящего изобретения предоставляет пероральный ингибитор СНК1 для введения пациентам с раком для усиления действия ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в двух дозах, первая из которых вводится через одни сутки после ДНК-повреждающего агента, а вторая - через двое суток после ДНК-повреждающего агента.
Другой вариант воплощения настоящего изобретения представляет пероральный ингибитор СНК1 для введения пациентам с раковым заболеванием для усиления действия ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в трех дозах, первая из которых вводится через одни сутки после ДНК-повреждающего агента, вторая доза - через двое суток после ДНК-повреждающего агента, а третья доза - через трое суток после ДНК-повреждающего агента.
Оральный ингибитор СНК1 представляет собой ингибитор СНК1, который можно принимать орально. Когда ингибитор СНК1 принимается орально, он может быть в виде пилюли, твердой или мягкой капсулы, таблетки, таблетки для рассасывания, жидкой или маслянистой суспензии, эмульсии, порошка или гранул, сиропа, эликсира, с фармацевтически приемлемой основой или наполнителем.
'926 ингибиторы СНК1 являются оральными ингибиторами СНК1.
Один вариант воплощения настоящего изобретения представляет оральный ингибитор СНК1 для введения пациенту с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в двух дозах, первая из которых вводится через одни сутки после ДНК-повреждающего агента, а вторая доза - через двое суток после ДНК-повреждающего агента, причем ингибитор СНК1 вводится между биологически эффективной дозой и максимально переносимой дозой.
Другой вариант воплощения настоящего изобретения представляет оральный ингибитор СНК1 для введения пациенту с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в трех дозах, первая из которых вводится через одни сутки после ДНК-повреждающего агента, вторая - через двое суток после ДНК-повреждающего агента, а третья - через трое суток после ДНК-повреждающего агента, причем ингибитор СНК1 вводится между биологически эффективной дозой и максимально переносимой дозой.
В определенных вариантах воплощения настоящего изобретения дозу ингибитора СНК1 можно разделить на два приема в сутки (т.н. BID-прием, дважды в сутки). В этом варианте воплощения первая доза ингибитора СНК1 включает два введения в одни сутки после введения ДНК-повреждающего агента. Два введения обычно разбиты в течение суток. Он также включает два введения на вторые сутки, и необязательно еще два введения на третьи сутки.
Один вариант воплощения настоящего изобретения представляет ингибитор СНК1 для введения пациентам с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в четырех дозах, первые две из которых вводятся через одни сутки после ДНК-повреждающего агента, а третья и четвертая дозы - через двое суток после ДНК-повреждающего агента.
Другой вариант воплощения настоящего изобретения представляет ингибитор СНК1 для введения пациентам с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в шести дозах, первые две из которых вводятся через одни сутки после ДНК-повреждающего агента, третья и четвертая дозы - через двое суток после ДНК-повреждающего агента, а пятая и шестая дозы - через трое суток после ДНК-повреждающего агента.
Другой вариант воплощения настоящего изобретения представляет ингибитор СНК1 для введения пациентам с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в четырех дозах, первые две из которых вводятся через одни сутки после ДНК-повреждающего агента, а третья и четвертая дозы - через двое суток после ДНК-повреждающего агента, причем ингибитор СНК1 вводится между биологически эффективной дозой и максимально переносимой дозой.
Другой вариант воплощения настоящего изобретения представляет ингибитор СНК1 для введения пациентам с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в шести дозах, первые две из которых вводятся через одни сутки после ДНК-повреждающего агента, третья и четвертая дозы - через двое суток после ДНК-повреждающего агента, а пятая и шестая - через трое суток после ДНК-повреждающего агента, причем ингибитор СНК1 вводится между биологически эффективной дозой и максимально переносимой дозой.
Другой вариант воплощения настоящего изобретения представляет оральный ингибитор СНК1 для введения пациенту с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в четырех дозах, первые две из которых вводятся через одни сутки после ДНК-повреждающего агента, а третья и четвертая дозы ингибитора СНК1 - через двое суток после ДНК-повреждающего агента.
Другой вариант воплощения настоящего изобретения представляет ингибитор СНК1 для введения пациентам с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в шести дозах, первые две из которых вводятся через одни сутки после ДНК-повреждающего агента, третья и четвертая дозы - через двое суток после ДНК-повреждающего агента, а пятая и шестая - через трое суток после ДНК-повреждающего агента.
Другой вариант воплощения настоящего изобретения представляет оральный ингибитор СНК1 для введения пациенту с раковым заболеванием для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в четырех дозах, первые две из которых вводятся через одни сутки после ДНК-повреждающего агента, а третья и четвертая - через двое суток после ДНК-повреждающего агента, причем ингибитор СНК1 вводится между биологически эффективной дозой и максимально переносимой дозой.
Другой вариант воплощения настоящего изобретения представляет ингибитор СНК1 для введения пациентам с раком для усиления ДНК-повреждающего агента, при этом введение ингибитора СНК1 следует за введением ДНК-повреждающего агента и выполняется в шести дозах, первые две из которых вводятся через одни сутки после ДНК-повреждающего агента, третья и четвертая дозы - через двое суток после ДНК-повреждающего агента, а пятая и шестая дозы - через трое суток после ДНК-повреждающего агента, причем ингибитор СНК1 вводится между биологически эффективной дозой и максимально переносимой дозой.
Примеры
Для иллюстрации изобретения включены следующие Примеры. Однако следует понимать, что эти Примеры не ограничивают изобретение, а являются средством для разъяснения способа осуществления изобретения.
Пример 1
Самкам бестимусных мышей подкожно ввели 5 Х 106 НТ-29 опухолевых клеток в IX PBS (100 мкл). Через одиннадцать суток мышей разделили на группы по 3 со средним размером опухоли в каждой группе примерно 300 мм3. Разделенным на группы животным ввели СРТ11 (100 мг/кг; IP) за 24 часа и затем воздействовали на них соединением 1 или соединением 2.
Соединение 1 (1 мг/кг, 3 мг/кг, 10 мг/кг, 30 мг/кг и 100 мг/кг; РО) было введено и образцы опухолей были взяты через два часа после введения. Фосфорилирование СНК1 была оценена иммуноблотом и нормирована к общей экспрессии внеклеточных сигнал-регулируемых киназ [ЭРК]. Результаты выражались как процент от контроля (ПОК). Результаты показаны на фиг.1.
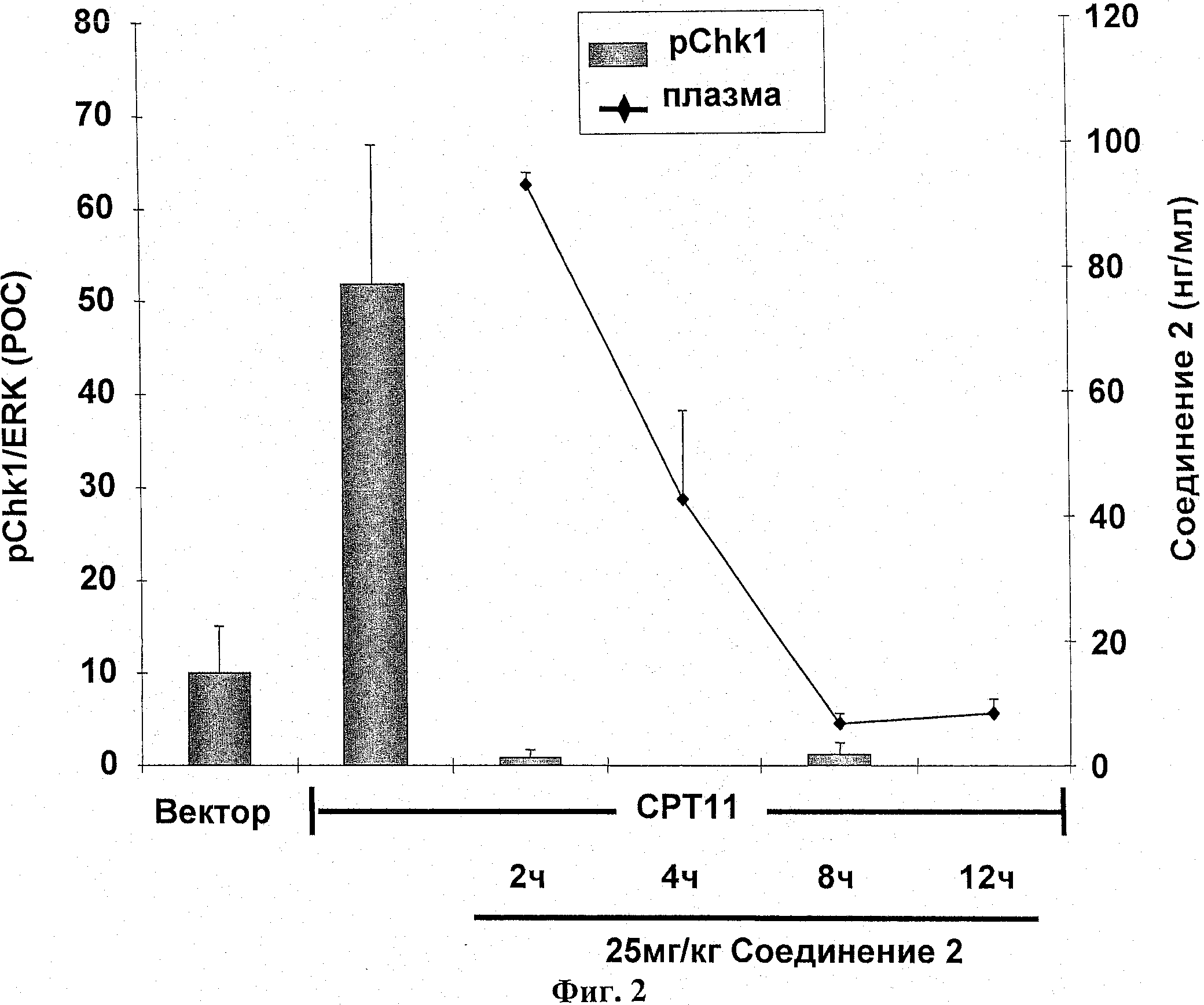
Соединение 2 (25 мг/кг; РО) было введено и образцы опухолей были взяты через 2 часа, 4 часа, 8 часов и 12 часов после введения. Фосфорилирование СНК1 (s296) было оценено иммуноблотом и нормирована к общей ЭРК. Результаты выражались как ПОК.
Результаты показаны на фиг.2.
Пример 2
Самкам бестимусных мышей подкожно ввели 5 Х 106 НТ-29 опухолевых клеток в IX PBS (100 мкл). Через двадцать суток мышей разделили на группы по 3 со средним размером опухоли в каждой группе примерно 390 мм3. Самкам бестимусных мышей с опухолью НТ-29 вводили СРТ11 (100 мг/кг; IP), и опухоли были взяты на анализ через 48 часов, 72 часа и 96 часов после введения. Фосфорилирование СНК1 и cdc2 было оценено иммуноблотом и нормировано к общей ЭРК. Результаты выражались как ПОК. Результаты показаны на фиг.3 и 4.
Пример 3
Самкам бестимусных мышей подкожно ввели 5 Х 106 НТ-29 опухолевых клеток в IX РВ8 (фосфатно-солевой буферный раствор) (100 мкл). Через двенадцать суток мышей разделили на группы по 6 со средним размером опухоли в каждой группе примерно 250 мм3. Разделенным на группы животным ввели одну дозу СРТ11 (100 мг/кг; IP) на 2 сутки, после чего или одновременно, или через 24 часа после введения СРТ11, последовало введение соединения 1 (50 мг/кг; РО, BID), в течение 3 следующих друг за другом дней. Размер опухоли и вес тела животного измерялись в ходе исследования на сутках, показанных на информационных точках на фиг.5. Объем опухоли определялся по формуле: объем=(ширина2×длина)/2. Результаты показаны на фиг.5, а результаты переносимости приведены в таблице 1.
|
Пример 4
Ранее не участвовавшим в эксперименте самкам мышей ввели СРТ11 (100 мг/кг, IP) по циклической схеме Q10D×2. Введение соединения 2 (25 мг/кг; РО, BID, в течение 3 суток на каждый цикл СРТ11) начиналось на 12, 24 или 48 часов после СРТ11, Результаты переносимости показаны в таблице 2.
|
Пример 5
Самкам бестимусных мышей подкожно ввели 5 X 106 НТ-29 опухолевых клеток в IX PBS (фосфатно-солевой буферный раствор) (100 мкл). Через двенадцать суток мышей разделили на группы по 8 со средним размером опухоли в каждой группе примерно 215 мм3. Разделенным на группы животным вводили СРТ11 (100 мг/кг; IP) по циклической схеме Q10D×2. Введение соединения 2 (25 мг/кг; РО, BID) начиналось через 24 часа после СРТ11 на 1 или 3 дни, как указано. Размер опухоли и вес тела животного измерялись в ходе исследования на сутках, показанных на информационных точках на фиг.6. Объем опухоли определялся по формуле: объем=(ширина2×длина)/2. В ходе этого исследования случаев смертности не было. Результаты показаны на фиг.6, и результаты переносимости приведены в таблице 3.
|
Пример 6
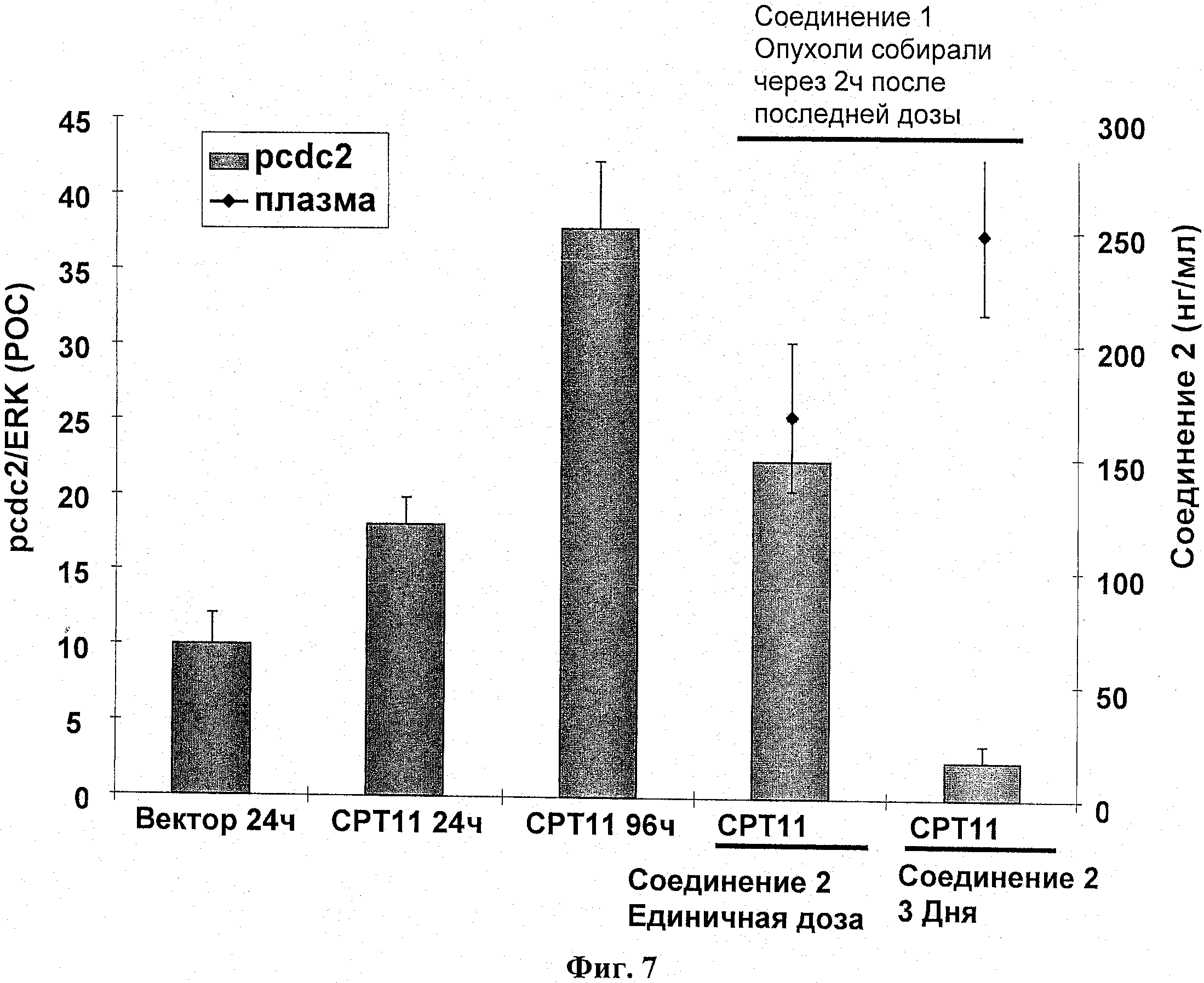
Самкам бестимусных мышей подкожно ввели 5 Х 106 НТ-29 опухолевых клеток в IX PBS (100 мкл). Через двадцать четыре сутки мышей разделили на группы по 3 со средним размером опухоли в каждой группе примерно 450 мм3. Разделенным на группы животным вводили СРТ11 (100 мг/кг; IP) в качестве единственного агента, и образцы опухолей брали через 24 часа и 96 часов после применения средства. Для комбинации групп прием соединение 2 (25 мг/кг; РО) начиналось через 24 часа после введения СРТ11 (100 мг/кг). Соединение 2 давалось в единственной дозе, или альтернативно в течение трех следующих друг за другом дней по схеме BID. Из опухолей всех животных, которым давали соединение 2, брались образцы через 2 часа после приема средства.
Фосфорилирование cdc2 было оценено иммуноблотом и нормировано к общей ЭРК. Результаты выражались как ПОК. Результаты введения соединения 2 в единственной дозе или в течение 3 дней статистически не различались (t-критерий>0,05). Результаты показаны на фиг.7.
Пример 7
Разделенный на несколько доз прием соединения 5 вызывает дозозависимое ингибирование СРТ11, вызванное фосфо-сdс2
Самкам бестимусных мышей подкожно ввели 5 Х 106 НТ-29 опухолевых клеток в IX PBS (100 мкл). Через двадцать девять суток мышей разделили на группы по 3 со средним размером опухоли в каждой группе примерно 500 мм3. Разделенным на группы животным вводили СРТ11 (100 мг/кг; IP) в качестве единственного агента, и образцы опухолей брали через 96 часов после применения средства. Для комбинации групп прием соединения 5 (5, 10 или 25 мг/кг; РО) начинался через 24 часа после введения СРТ11 (100 мг/кг). Соединение 5 давалось в единственной дозе 25 мг/кг, или альтернативно 5, 10 или 25 мг/кг давались в течение трех следующих друг за другом суток по схеме BID. Из опухолей всех животных, которым давали соединение 5, брались образцы через 96 часов после приема средства. Фосфорилирование cdc2 было оценено иммуноблотом и нормировано к общей ЭРК. Результаты выражались как ПОК. Результаты показаны на фиг.8.
Пример 8
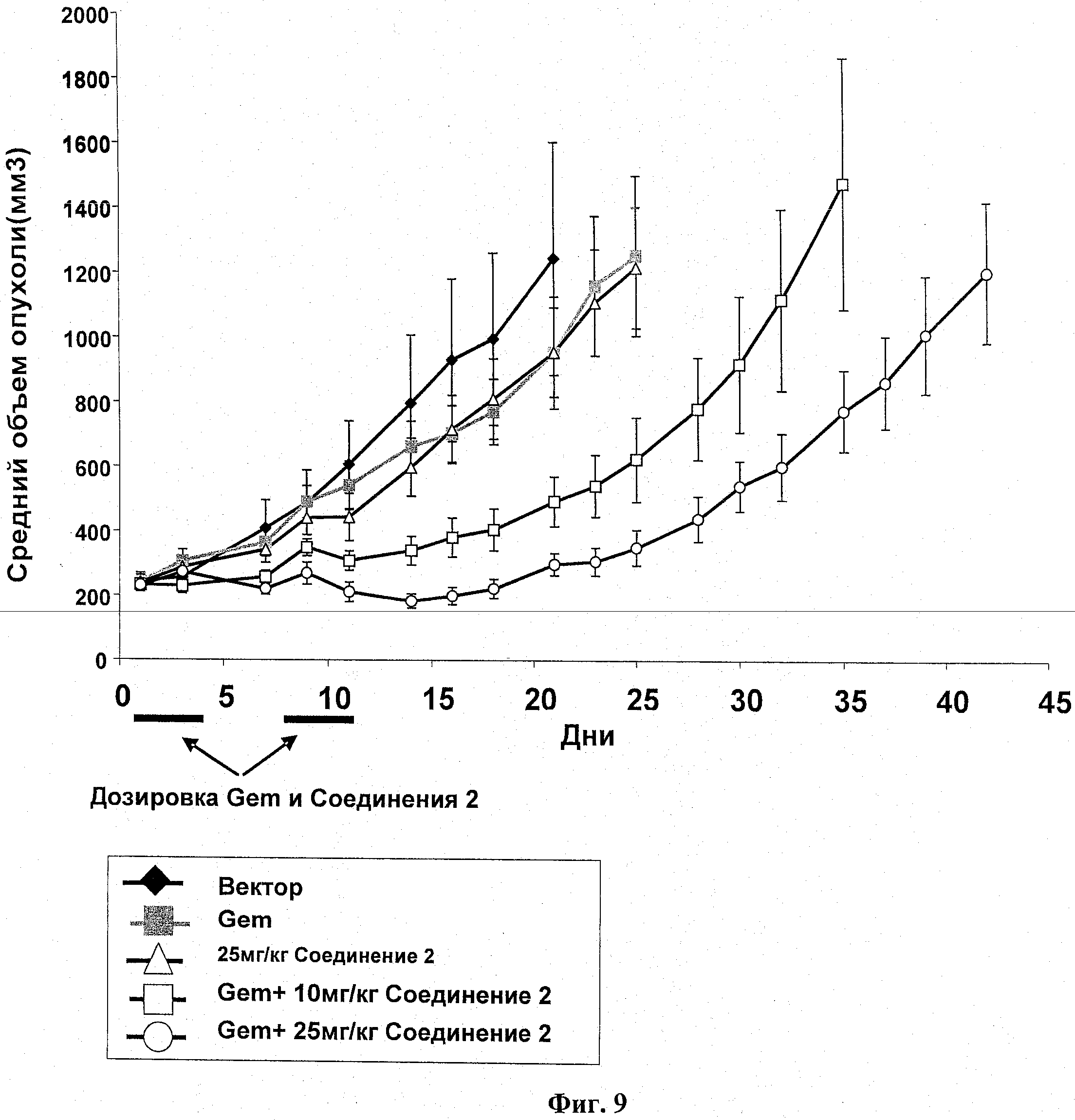
Самкам бестимусных мышей подкожно ввели 5 Х 106 НТ-29 опухолевых клеток в IX PBS (100 мкл). Через четырнадцать суток мышей разделили на группы по 8 со средним размером опухоли в каждой группе примерно 260 мм3. Разделенным на группы животным давался гемцитабин (140 мг/кг; IP) по циклической схеме Q7Dx2. Введение соединения 2 (10 или 25 мг/кг; РО, BID) начиналось через 24 часа после гемцитабина и продолжалось 3 суток, как указано. Размер опухоли и вес тела животного измерялись в ходе исследования на сутках, показанных на информационных точках на фиг.9. Объем опухоли определялся по формуле: объем=(ширина2×длина)/2. В ходе этого исследования случаев смертности не было. Результаты показаны на фиг.9, а результаты переносимости и измерений опухоли представлены в таблице 4.
|
Пример 9
Соединение 5 показывает дозозависимое ингибирование роста опухоли в комбинации с гемцитабином
Самкам бестимусных мышей подкожно ввели 5 Х 106 НТ-29 опухолевых клеток в IX PBS (100 мкл). Через четырнадцать суток мышей разделили на группы по 7 со средним размером опухоли в каждой группе примерно 200 мм3. Разделенным на группы животным назначили гемцитабин (120 мг/кг; IP) по циклической схеме Q7D×3. Введение соединения 5 (5, 10 или 25 мг/кг; РО, BID) начиналось через 24 часа после гемцитабина и продолжалось 3 суток, как указано. Размер опухоли и вес тела животного измерялись в ходе исследования на сутках, показанных на информационных точках на фиг.10. Объем опухоли определялся по формуле: объем=(ширина2×длина)/2. В ходе этого исследования случаев смертности не было. Результаты показаны на фиг.10, а результаты переносимости и измерений опухоли представлены в таблице 5.
|
Пример 10
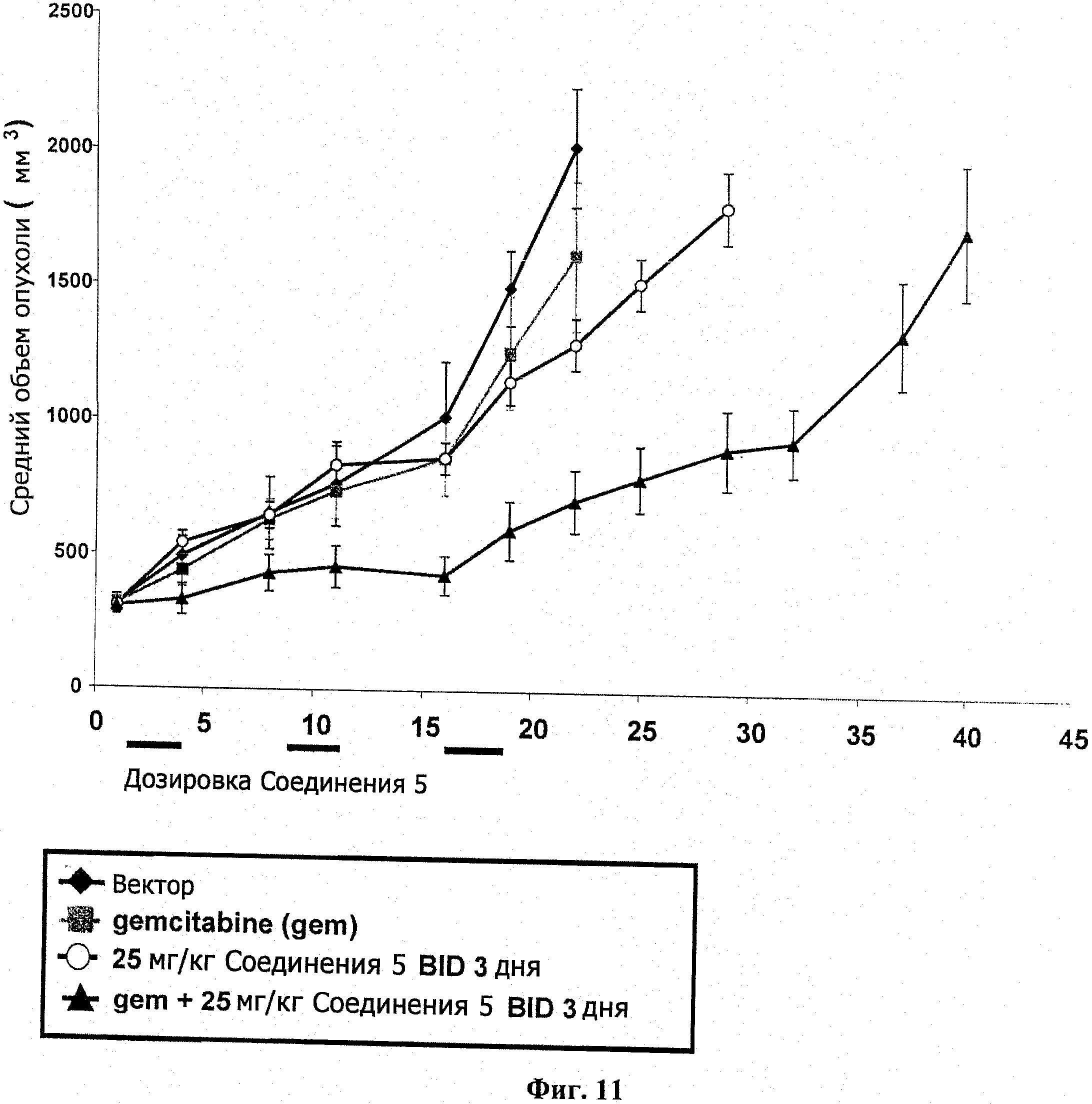
Соединение 5 подавляет опухолевый рост в комбинации с гемцитабином у трансплантата MiaPaCa2 карциномы поджелудочной железы
Самкам бестимусных мышей подкожно ввели 7Х106 MiaPaCa2 опухолевых клеток в 1:1 IX PBS суспензию матригеля (100 мкл). Через пятнадцать дней мышей разделили на группы по 7 со средним размером опухоли в каждой группе примерно 315 мм3. Разделенным на группы животным назначили гемцитабин (120 мг/кг; IP) по циклической схеме Q7Dx×3. Введение соединения 5 (25 мг/кг; РО, BID) начиналось через 24 часа после гемцитабина и продолжалось 3 суток, как указано. Размер опухоли и вес тела животного измерялись в ходе исследования на сутках, показанных на информационных точках на фиг.11. Объем опухоли определялся по формуле: объем=(ширина2×длина)/2. В ходе этого исследования случаев смертности не было. Результаты показаны на фиг.11, а результаты переносимости и измерений опухоли представлены в таблице 6.
|
Пример 11
Соединение 2 показывает дозозависимое подавление опухолевого роста в комбинации с СРТ-11
Самкам бестимусных мышей подкожно ввели 5 Х 106 НТ-29 опухолевых клеток в IX PBS (100 мкл). Через четырнадцать суток мышей разделили на группы по 8 со средним размером опухоли в каждой группе примерно 260 мм3. Разделенным на группы животным назначили СРТ11 (100 мг/кг; IP) по циклической схеме Q10Dx2. Введение соединения 2 (10 или 25 мг/кг; РО, BID) начиналось через 24 часа после СРТ11 и продолжалось 3 суток, как указано. Размер опухоли и вес тела животного измерялись в ходе исследования на сутках, показанных на информационных точках на фиг.12. Объем опухоли определялся по формуле: объем=(ширина2×длина)/2. В ходе этого исследования случаев смертности не было. Результаты показаны на фиг.12, а результаты переносимости и измерений опухоли представлены в таблице 7.
|
Пример 12
Соединение 5 показывает дозозависимое подавление опухолевого роста в комбинации с СРТ-11
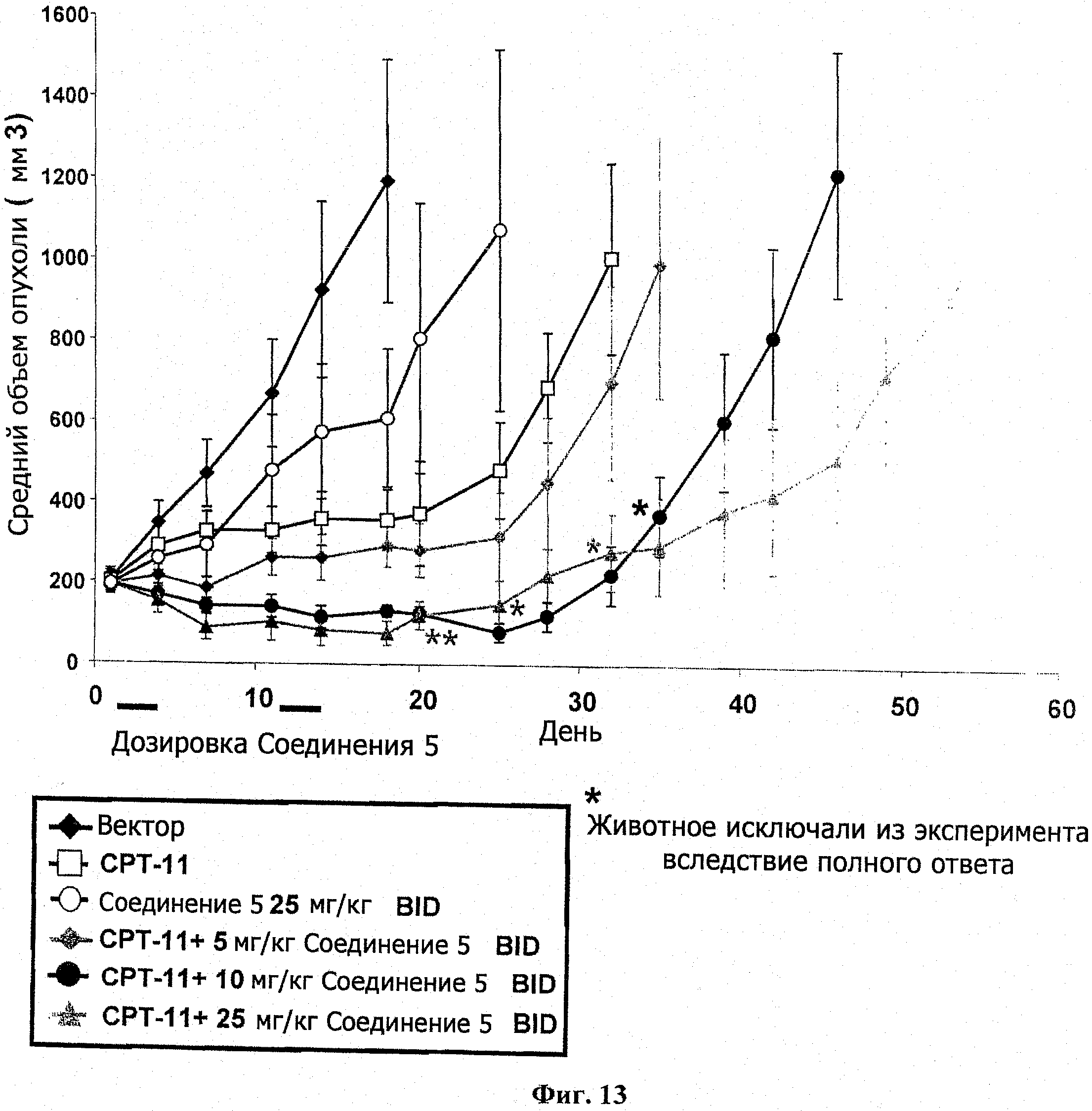
Самкам бестимусных мышей подкожно ввели 4 Х 106 НТ-29 опухолевых клеток в IX PBS (100 мкл). Через двенадцать суток мышей разделили на группы по 7 со средним размером опухоли в каждой группе примерно 200 мм3. Разделенным на группы животным назначили СРТ11 (100 мг/кг; IP) по циклической схеме Q10D×2. Введение соединения 5 (5, 10 или 25 мг/кг; РО, BID) начиналось через 24 часа после СРТ11 и продолжалось 3 суток, как указано. Размер опухоли и вес тела животного измерялись в ходе исследования на сутках, показанных на информационных точках на фиг.13. Объем опухоли определялся по формуле: объем=(ширина2×длина)/2. В ходе этого исследования случаев смертности не было. Результаты показаны на фиг.13, а результаты переносимости и измерений опухоли представлены в таблице 8.
|
Несмотря на то, что изобретение описано в отношении перечисленных вариантов воплощения, понятно, что это не подразумевает, что изобретение ограничивается этими вариантами воплощения. Напротив, подразумевается, что изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в рамки настоящего изобретения, как определено его формулой. Таким образом, вышеизложенное описание следует рассматривать только как иллюстрацию принципов изобретения.
Подразумевается, что слова "содержа в себе," "содержащий в себе," "включая," "включающий" и "включает" при использовании в данном описании и в последующем объеме притязаний означают присутствие определенных свойств, числовых значений, компонентов или последовательности действий, но они не препятствуют присутствию или добавлению одного или более других свойств, числовых значений, компонентов, последовательности действий или их совокупности.
Гидроксилированные и метоксилированные циклопента[d]пиримидины в качестве ингибиторов акт протеинкиназ
Циклопента(d)пиримидины в качестве ингибиторов протеинкиназ акт
Пиримидилциклопентаны в качестве ингибиторов akt-протеинкиназы
Пиримидилциклопентаны как ингибиторы акт-протеинкиназ
Гидроксилированные пиримидилциклопентаны в качестве ингибиторов протеинкиназы (акт)
Производные феноксихроманкарбоновой кислоты, замещенные в 6-ом положении
Пирролопиридины как ингибиторы киназы
Ингибиторы митоза для интенсификации процесса апоптоза при терапии
Гидроксилированный пиримидил циклопентан в качестве ингибитора протеинкиназы (акт)
Замещенные пиразоло[1,5-a]пиримидиновые соединения как ингибиторы трк киназы
Гидроксилированные и метоксилированные циклопента[d]пиримидины в качестве ингибиторов акт протеинкиназ
Циклопента(d)пиримидины в качестве ингибиторов протеинкиназ акт
Пиримидилциклопентаны в качестве ингибиторов akt-протеинкиназы
Пиримидилциклопентаны как ингибиторы акт-протеинкиназ
Гидроксилированные пиримидилциклопентаны в качестве ингибиторов протеинкиназы (акт)
Производные феноксихроманкарбоновой кислоты, замещенные в 6-ом положении
Пирролопиридины как ингибиторы киназы
Ингибиторы митоза для интенсификации процесса апоптоза при терапии
Гидроксилированный пиримидил циклопентан в качестве ингибитора протеинкиназы (акт)
Замещенные пиразоло[1,5-a]пиримидиновые соединения как ингибиторы трк киназы

