Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ СПИНОХРОМА Е
Вид РИД
Изобретение
Изобретение относится к органической химии и касается способа получения спинохрома Ε (2,3,5,6,7,8-гексагидрокси-1,4-нафтохинона) - одного из пигментов, продуцируемых морскими ежами различных видов [1а-в]. Спинохром Ε был впервые обнаружен в иглах морского ежа Ледерером [2]. Позже Смит и Томсон установили структуру этого пигмента и определили его свойства [3].
Среди 5 классов типа Echinodermata (иглокожие) морские ежи (echinoids) привлекают наибольшее внимание исследователей. Этот класс животных синтезирует преимущественно полигидрокси-1,4-нафтохиноны, в основе структуры которых лежит либо 5-гидрокси-1,4-нафтохинон (юглон), либо 5,8-дигидрокси-1,4-нафтохинон (нафтазарин), а заместителями являются амино-, ацетильные, гидрокси-, метокси-метильные и этильные группы. К настоящему времени около 30 таких пигментов выделено из морских ежей различных видов [4, 5]. Полигидрокси-1,4-нафтохиноны морских ежей, в том числе спинохром Е, проявляют высокую антиоксидантную и антирадикальную активность и представляют новый класс природных антиоксидантов [4-11]. Среди нафтохиноидных пигментов морских ежей найдено много соединений с высокой антиалгальной [12, 13], антимикробной [14], антиаллергической [15], цитотоксической [1а] и кардиопротективной [16-20] активностью.
На основе эхинохрома А (2,3,5,6,8-пентагидрокси-7-этил-1,4-нафтохинона) - одного из метаболитов морских ежей созданы эффективные отечественные лекарственные препараты: Гистохром кардиологический® для лечения острого инфаркта миокарда и ишемической болезни сердца и Гистохром офтальмологический® для лечения пролиферативных и дегенеративных процессов, включая катаракту, и кровоизлияний различной природы в тканях глаза [21].
Интересно отметить, что икра и гонады морских ежей, содержащие полигидрокси-1,4-нафтохиноны, в том числе спинохром Е, используются как высокоценимые деликатесные продукты питания в Японии, Китае и ряде стран Юго-Восточной Азии [4, 11].
Однако широкое практическое использование полигидрокси-1,4-нафтохинонов морских ежей ограничивается их малой доступностью.
Возможны два пути получения этих природных соединений: либо извлечением из природных объектов (морских ежей), либо через полный органический синтез.
Известен способ получения спинохрома Ε из природного объекта, в котором в качестве сырья используют отходы переработки промысловых морских ежей Strongylocentrotus nudus [22]. Сырье дефростируют или отделяют от консервантов декантацией, промывают водой, обезжиривают 96% этиловым спиртом (EtOH), экстрагируют трехкратно 96% EtOH с добавлением Н3РО4 (рН 3.0-3.5) при соотношении сырье:экстрагент 1.0:(1.0-1.2) в течение 12-24 ч. Полученный экстракт пропускают через колонку с хитозаном, промывают колонку 96% EtOH, затем элюируют пигмент 96% EtOH с добавлением соляной кислоты (рН 2-4). Далее элюат пропускают через колонку с полихромом-1, промывают колонку дистиллированной водой, затем элюируют целевой продукт 40-50% водным раствором EtOH, отгоняют растворитель в вакууме, водный остаток лиофилизуют. Затем спинохром Ε сублимируют при 195-200°С или перекристаллизовывают из EtOH. Выходы спинохрома Ε из 100 кг сырья (панцири и иглы морских ежей Strongylocentrotus nudus, оставшиеся после извлечения икры) составляют 19-24 г (0.019-0.024% от веса исходного сырья).
Данный способ получения спинохрома Ε является длительным, многостадийным и характеризуется низким выходом целевого продукта. Его широкомасштабное применение неизбежно приведет к возникновению экологической проблемы, как и всякий способ получения природных продуктов из некультивируемых морских животных.
Известны также два способа получения спинохрома Ε путем полного синтеза.
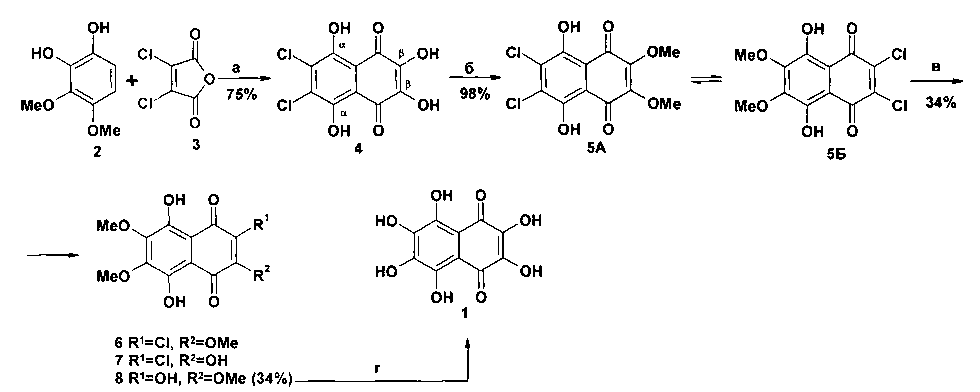
Реагенты и условия: a) AlCl3-NaCl, 190-195°С, 4 мин; б) CH2N2, серный эфир, 22°С; в) MeONa, абс. МеОН, кипячение, 48 ч, N2; г) конц. HBr, кипячение, 2 ч.
Схема 1
Первый из них (схема 1) заключается в циклоацилировании 1,2-дигидрокси-3,4-диметоксибензола 2 дихлормалеиновым ангидридом 3 в расплаве безводного AlCl3 и NaCl, метилировании β-ΟΗ-групп полупродукта 4, нуклеофильном замещении атомов хлора в полученном нафтазарине 5 на метоксигруппы и расщеплении О-Ме связей в полученном триэфире 8 [23, 24].
Суммарный выход спинохрома Ε в этом 4-х-стадийном способе получения оценить невозможно, поскольку выход 1 на стадии гидролиза 8→1 авторами не указан. Данный способ непригоден для препаративного получения спинохрома Ε 1, поскольку для того, чтобы конвертировать дихлорнафтазарин 5 в триэфир 8, необходимо применять огромный избыток насыщ. раствора MeONa в абс. МеОН. Так, раствор дихлорнафтазарина 5 (80 мг, 0.25 ммоль) в насыщ. растворе MeONa в МеОН (800 мл, 3125.0 ммоль) кипятили в течение 48 час, чтобы добиться замещения обоих атомов хлора на метоксигруппы. Даже при такой длительной обработке авторам не удалось полностью конвертировать дихлорнафтазарин 5 в ожидаемый тетраметиловый эфир спинохрома Е. В результате этой реакции получалась смесь 3-х продуктов 6-8. Триэфир 8 был выделен из смеси продуктов в количестве 34 мг (34%) препаративной ТСХ на пластинках с деактивированным SiO2. Все попытки улучшить соотношение субстрат 5:реагент (насыщ. раствор MeONa в МеОН) оказались безуспешными.
Невыгодное соотношение субстрат: реагент на стадии конверсии 5→8 делает этот способ получения спинохрома Ε не имеющим практического значения. Он может использоваться лишь для получения аналитических образцов спинохрома Е. Действительно, если для получения 34 мг триэфира 8 из 80 мг субстрата 5 необходимо было использовать 800 мл насыщ. раствора MeONa в МеОН, то для получения, например, 1 г триэфира 8 необходимо использовать 24 л насыщенного раствора MeONa в МеОН.
Следует отметить, что дигидроксидиметоксибензол 2, используемый в синтезе спинохрома Ε, не является товарным продуктом и его получение включает в себя две стадии: формилирование товарного 1,2,3-триметоксибензола 9 и последующее окисление образующегося альдегида 10. Данный процесс представлен на схеме 2.
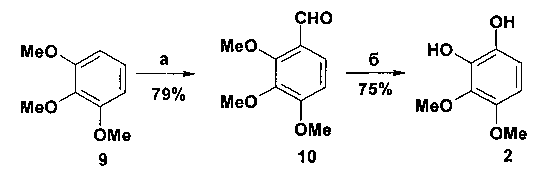
Реагенты и условия: а) POCl3, HCO-NMe2, 100°С, 4 ч; б) 33% H2O2, NaOH, 50°С, 5 ч.
Схема 2
С учетом этого синтез спинохрома Ε по методу Шоера и соавт. включает в себя 6 стадий.
Второй из способов получения спинохрома Е, представленный на схеме 3, заключается в циклоацилировании триметоксифенола 11 ангидридом 3, этилировании β-ОН-групп полученного дигидроксидихлорнафтазарина 4, замещении атомов хлора в полученном диэфире 8 на гидрокси- и нитрогруппы, восстановлении полученного нитронафтазарина 13 и конверсии полученного аминонафтазарина 14 в целевой спинохром Ε 1 с последующим кипячением объединенных экстрактов, полученных при извлечении продукта 1 5% HCl [25].
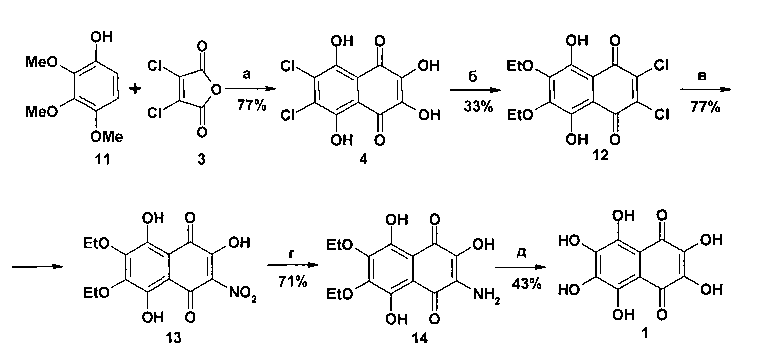
Реагенты и условия: a) AlCl3-NaCl, 190-195°С, 3 мин; б) (EtO)3СН, кипячение, 2 ч; в) NaNO2, EtOH - H2O (2:1), кипячение, 1 ч; г) Na2S2O4, H2O, 22°С, 30 мин; д) безв. AlCl3, безв. PhNO2, 70°С, 6 ч, затем 5% HCl, кипячение, 10 мин.
Схема 3
Этот способ получения спинохрома Ε позволяет получать его в 5 стадий, исходя из фенола 11, с суммарным выходом 6%.
Исходный фенол 11 не является товарным реактивом. Его можно получить окислением триметоксибензальдегида 10 пероксидом водорода (схема 4) [26].
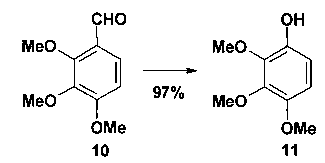
Реагенты и условия: 33% H2O2, конц. H2SO4, МеОН, 22°С, 1.5 ч.
Схема 4
С учетом этого данный синтез спинохрома Ε включает в себя в общей сложности 6 стадий.
Оба описанных в литературе способа синтеза спинохрома Ε используют один и тот же метод синтеза стартового дигидроксидихлорнафтазарина 4, в котором далее блокируют β-гидроксильные группы и замещают атомы хлора. На стадии нуклеофильного замещения атомов хлора эти способы синтеза имеют очень существенные отличия, которые определяют и дальнейшие различия в методологии превращения полученных на этой стадии полупродуктов в целевой спинохром Е.
Первый из описанных способов синтеза спинохрома Ε наиболее близок к заявляемому способу по сути выполняемых операций и выбран нами в качестве прототипа.
Недостатки способа-прототипа заключаются в следующем:
1) непригодность для препаративного использования, поскольку практически неприемлемое соотношение субстрат 5:реагент (насыщ. раствор MeONa в МеОН) на стадии конверсии 5→8 ограничивает синтетические возможности этого способа.
Для получения насыщ. раствора MeONa в МеОН, требуемого для синтеза граммовых количеств спинохрома Ε по способу-прототипу, необходимо использовать от нескольких десятков до сотен граммов агрессивного и пожароопасного металлического натрия и большие объемы (от десятков до сотен литров) метилового спирта;
2) использование на стадии защиты β-гидроксильных групп полупродукта 4 (стадия 4→5) токсичного диазометана и пожароопасного серного эфира;
3) применение большого числа стадий для получения спинохрома Ε (6 стадий), и его низкий суммарный выход.
Хотя авторы не указали выхода спинохрома Ε на стадии гидролиза триметилового эфира 8→1, эффективность этой стадии его синтеза в условиях способа-прототипа была оценена нами (78% от теор.), что позволило рассчитать и общий выход спинохрома Ε на 6 стадий в способе-прототипе (10% от теор.).
Задачей изобретения явилась разработка способа получения спинохрома Е, свободного от недостатков способа-прототипа.
В заявляемом способе получения спинохрома Ε в качестве исходного соединения используют нафтазарин или 2,3-дихлорнафтазарин, который хлорируют под действием хлора, получаемого in situ окислением соляной кислоты активным диоксидом марганца в ледяной уксусной кислоте, в образовавшемся при этом 2,3,6,7-тетрахлорнафтазарине замещают все атомы хлора на метоксигруппы действием метанола, активированного фторидом цезия на поверхности оксида алюминия, и полученный 2,3,6,7-тетраметоксинафтазарин гидролизуют в целевой продукт кипячением в концентрированной бромистоводородной кислоте в течение 10-15 мин.
Суммарный выход спинохрома Ε составляет от 41 до 46% (в зависимости от используемого субстрата 15 или 16) на три стадии 15(16)→17→18-1.
Заявляемый способ представлен на схеме 5.
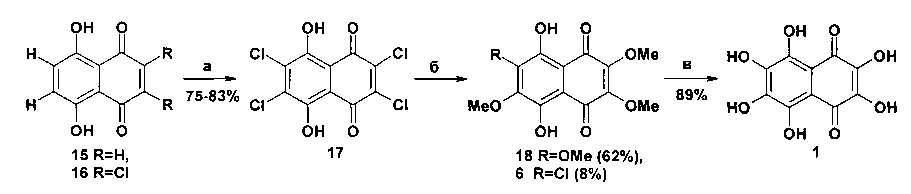
Реагенты и условия: а) активный MnO2, конц. HCl+лед. НОАс (1:1, по объему), т. кип., 2 ч; б); абс. МеОН - CsF - нейтр. Al2O3, 95°С, 60 ч; в) 48% HBr, т. кип., 15 мин.
Схема 5
В качестве исходного соединения используют коммерчески доступный нафтазарин 15 (каталог фирмы «Alfa Aesar» 2011-2013 года, стр. 893), или 2,3-дихлорнафтазарин 16 (каталог фирмы «Aldrich» 2012-2014 года, стр. 923). Нафтазарины 15 и 16 ранее не применялись в качестве исходных субстратов в синтезах спинохрома Е.
Для конверсии нафтазарина 15 в тетрахлорнафтазарин 17 нами был опробован ряд методов хлорирования, в которых использовались следующие реагенты: Cl2O/CF3COOH, Cl2O/конц. H2SO4+HOAc (1:1, по объему), газообразный Cl2/НОАс, газообразный Cl2/90% H2SO4+J2, K2Cr2O7/конц. HCl+HOAc, KMnO4/конц. HCl+НОАс, товарный MnO2/конц. HCl+HOAc, активный MnO2/конц. HCl+HOAc. Наилучшие результаты были получены при действии молекулярного Cl2, генерируемого in situ окислением HCl активным диоксидом марганца в растворе в НОАс.
При использовании товарного MnO2 выход тетрахлорнафтазарина 17 колебался в интервале 55-60%. Замена товарного MnO2 на активный MnO2, полученный по методу Аттенборо [27], позволила увеличить выход тетрахлорнафтазарина 17 до 75-77%.
При использовании в качестве стартового соединения 2,3-дихлоронафтазарина 16 хлорирование субстрата под действием системы реагентов активный MnO2 - конц. HCl - лед. НОАс протекает в тех же условиях, что и в случае нафтазарина 15 (т. кип., 2 ч), более эффективно, давая тетрахлорнафтазарин 17 с выходами 81-83%.
Полное замещение четырех атомов хлора в субстрате 17 на метоксигруппы осуществлялось под действием метанола, активированного CsF на поверхности Al2O3 при 95°С в течение 60 ч. Выход тетраэфира 18 составил 62% при конверсии субстрата 92%. Проведение реакции в течение 40 ч дает 2,3,6,7-тетраметоксинафтазарин 18 с выходом 45-50%, причем конверсия субстрата не превышала 78%. Впервые эта система реагентов была использована для нуклеофильного замещения атомов хлора в 2,3-дихлорозамещенных нафтазаринах авторами работы [28]. Для замещения четырех атомов хлора на метоксигруппы в субстрате 17 эта система реагентов ранее не применялась и результат ее применения для данного субстрата не является очевидным.
2,3,6,7-Тетраметоксинафтазарин 18 является известным соединением. Ранее его получали из природного спинохрома Ε 1 через метилирование диазометаном [3].
Конверсия тетраэфира 18 в спинохром Ε 1 осуществлялась при кипячении субстрата 18 в растворе 48% HBr в течение 10-15 мин. Из опробованных методов расщепления О-Ме связей метоксигрупп субстрата 18 (безв. AlCl3 в сухом C6H5NO2 при нагревании; безв. AlCl3 в смеси C2H5SH и CH2Cl2 при 22°С; насыщ. раствор сухого HBr в лед. НОАс при нагревании; 48% HBr в смеси с лед. НОАс (1:1, по объему) при нагревании; 48% HBr при нагревании) последний метод оказался наилучшим. Ранее его уже использовали в синтезе спинохрома Ε 1 из триметилового эфира спинохрома Ε 8 (конверсия 8→1, схема 1) [23, 24]. Однако авторы этих работ проводили деметилирование субстрата 8 под действием 48% HBr при кипячении в течение 2 ч, причем выход спинохрома Ε не был указан. Мы исследовали эту реакцию повторно. Триметиловый эфир спинохрома Ε 8 был получен частичным гидролизом тетраметилового эфира спинохрома Ε 18 под действием 1% водного раствора NaOH. Оказалось, что как триэфир 8, так и тетраэфир 18 при кипячении в растворе в 48% HBr полностью превращаются в спинохром Ε в течение 10-15 мин с выходами 88 и 89% соответственно. Если же реакцию гидролиза триэфира 8 проводить в условиях способа-прототипа (48% HBr, т. кип. 2 ч), то выход спинохрома Ε снижается (до 78% от теор.). Таким образом, время реакции на этой стадии может быть уменьшено в 8 раз по сравнению со способом-прототипом. Тетраэфир 18 этой процедуре ранее не подвергался.
Новизна предлагаемого метода синтеза спинохрома Ε заключается в применении новой комбинации стадий синтеза, в каждой из которых используются известные соединения, не применявшиеся, однако, ранее в синтезах спинохрома Е, а также в подборе реагентов и условий проведения стадий, что в итоге привело к появлению неочевидного результата.
Использование тетрахлорнафтазарина в качестве ключевого субстрата для нуклеофильного замещения атомов хлора позволяет отказаться от защиты β-гидроксигрупп с помощью раствора диазометана в диэтиловом эфире, как это делалось в случае дигидроксидихлорнафтазарина в способе-прототипе. Кроме того, применение метанола, активированного фторидом цезия на поверхности оксида алюминия, для замещения атомов хлора на метоксигруппы в тетрахлорнафтазарине вместо конц. раствора метилата натрия в метаноле для замещения атомов хлора в диметоксидихлорнафтазарине, как в способе-прототипе, кардинально улучшает соотношение субстрат-реагент на этой стадии и повышает выход продукта. Все это делает способ получения спинохрома Ε более простым и безопасным.
Технический результат, обеспечиваемый изобретением, заключается в упрощении способа получения спинохрома Е, а именно в уменьшении числа стадий с шести в способе-прототипе до трех и увеличении выхода целевого соединения в 4.1-4.6 раз по сравнению со способом-прототипом (41-46% и 10% соответственно).
Сведения, подтверждающие возможность осуществления изобретения:
Пример 1. Хлорирование нафтазарина 15. К раствору нафтазарина 15 (3.80 г, 20.0 ммол) в 200 мл ледяной НОАс и 100 мл 36% HCl прибавляют тонкоизмельченный активный MnO2 (6.96 г, 80.0 ммол) [27], и смесь кипятят при перемешивании в течение 1 ч. После этого прибавляют дополнительное количество активного MnO2 (1.74 г, 20.0 ммол) и конц. HCl (25 мл) и смесь кипятят при перемешивании еще в течение 1 ч. Смесь охлаждают до комнатной температуры и выдерживают при 5°С в течение ночи. Осадок отделяют на фильтре, промывают водой (3×10 мл) и высушивают. В результате получают 4.20 г (64%) 2,3,6,7-тетрахлоронафтазарина 17 в виде блестящих темнокрасных игл, т. пл. 258°С (лит. [27], 244°С). Rf: 0.65 (гексан-ацетон, 2:1 (по объему)). ИК-спектр (CDCl3, ν, см-1): 3400-2250 (ОН), 1627 (С=O, С=С), 1568 (С=С), 1405. Спектр ЯМР 1H (300 МГц, CDCl3, δ, м.д.): 12.88 (с, 2Н, 2α-ОН). Спектр ЯМР 13С (75 МГц, CDCl3, δ, м.д.): 109.2 (C-4a, C-8a), 139.1 (С-2, С-3, С-6, С-7), 167.2 (С-1, С-4, С-5, С-8). Масс-спектр (ЭУ, 70 эВ, m/z, Ioтн. (%)): 327/329/331/333/335 [М+Н]+ (58), 326/328/330/332/334 [М]+ (100), 292/294/296/298 [М+Н-Сl]+ (20), 291/293/295/297 [М-Сl]+ (61), 257/259/261 [М+Н-2Сl]+ (5), 256/258/260 [М-2Сl]+ (17). Найдено (%): С, 36.68; Н, 0.64; Cl, 43.34. C10H2Cl4O4. Вычислено (%): С, 36.63; Н, 0.61; Cl, 43.24.
После отделения кристаллов фильтрат разбавляют водой (300 мл), экстрагируют CHCl3 (4×90 мл), объединенные экстракты промывают водой (3×30 мл), высушивают над безв. Na2SO4 и растворитель удаляют при пониженном давлении. Остаток хроматографируют на колонке с SiO2 (G-60 фирмы «Lancaster», 60-200 µm). Элюция смесью гексан-ацетон 25:1 дает дополнительное количество 2,3,6,7-тетрахлоронафтазарина 17 (0.72 г, 11%), идентичного во всех отношениях описанному выше. Суммарный выход продукта 17 составил 75%.
Нуклеофильное замещение атомов хлора в тетрахлоронафтазарине 17 на метоксигруппы. Смесь хорошо высушенного тетрахлоронафтазарина 17 (1.88 г, 6.0 ммол), безвод.CsF (9.78 г, 64.0 ммол), активного нейтрального Al2O3 (12.20 г, 112.0 ммол, 150 меш, фирмы «Aldrich») и абс. МеОН (300 мл) перемешивают в герметичном сосуде при 95°С в течение 60 ч. После охлаждения до комнатной температуры адсорбент отделяют фильтрованием и последовательно промывают ацетоном (150 мл) и 10%-ной HCl (6 мл). Объединенные фильтраты концентрируют при пониженном давлении, остаток разбавляют водой (300 мл) и экстрагируют СНСl3 (5×80 мл). Органический экстракт промывают последовательно водой (3×40 мл) и насыщ. раствором NaCl (40 мл), высушивают над безвод. Na2SO4 и концентрируют досуха. Остаток хроматографируют на колонке с SiO2 (G-60, 2.5×80 см), заполненной н-гексаном. Элюция смесью гексан-ацетон, 15:1 (по объему) дает 5,8-дигидрокси-2,3,6-триметокси-7-хлоро-1,4-нафтохинон 6 (0.16 г, 8%) в виде легких коричневых игл, т. пл. 127-129°С (лит.[24], 140-141°С). Rf: 0.56 (гексан-ацетон, 2:1). ИК-спектр (CDCl3, ν, см-1): 3400-2250 (ОН), 1607 (С=O, С=С), 1549 (С=С), 1401. Спектр ЯМР 1H (300 МГц, CDCl3, δ, м.д.): 4.11 (с, 3Н, ОМе), 4.14 (с, 3Н, ОМе), 4.18 (с, 3Н, ОМе), 12.86 (с, 1H, С(8)-ОН), 13.11 (с, 1Н, С(5)-ОН). Спектр ЯМР 13С (75 МГц, CDCl3, δ, м.д.): 61.9 (С(6)-ОМе), 62.0 (С(2)-ОМе, С(3)-ОМе), 106.5 (С-4а), 109.0 (С-8а), 129.5 (С-7), 147.7 (С-2), 148.4 (С-3), 154.8 (С-6), 159.4 (С-5), 161.7 (С-8), 177.0 (С-1), 178.1 (С-4). Масс-спектр (ЭУ, 70 эВ, m/z, Ioтн. (%)): 315/317 [М+Н]+ (22), 314/316 [М]+ (100), 313/315 [М-Н]+ (19), 299/301 (22), 296/298 (29), 286 (9), 257 (16), 256 (16). Найдено (%): С, 49.78; Н, 3.62; Cl, 11.38. C13H11ClO7. Вычислено (%): С, 49.62; Н, 3.52; Cl, 11.27.
Элюция смесью гексан-ацетон, 8:1 дает 2,3,6,7-тетраметоксинафтазарин 18 (1.16 г, 62%) в виде красных игл, т. пл. 180-182°С (лит.[24], 185-186°С). Rf: 0.52 (гексан-ацетон, 2:1), 0.70 (гексан-бензол-ацетон, 1:1:1). ИК-спектр (CDCl3, ν, см-1): 3400-2250 (ОН), 1604 (С=O, С=С), 1554 (С=С), 1456, 1416, 1360, 1290, 1211, 1161, 1088. Спектр ЯМР 1H (300 МГц, CDCl3, δ, м.д.): 4.09 (с, 12Н, 4 ОМе), 12.98 (с, 2Н, 2α-ОН). Спектр ЯМР 13С (75 МГц, CDCl3, δ, м.д.): 61.6 (4 ОМе), 106.4 (С-4а, С-8а), 148.0 (С-2, С-3, С-6, С-7), 169.2 (С-1, С-4, С-5, С-8). Масс-спектр (ЭУ, 70 эВ, m/z, Ioтн. (%)): 311 [М+Н]+ (18), 310 [М]+ (100), 295 (17), 292 (13), 282 (6), 280 (8), 277 (14), 257 (11), 256 (13). Найдено (%): С, 54.30; Н, 4.51. C14H14O8. Вычислено (%): С, 54.20; Н, 4.55.
Конверсия тетраметоксинафтазарина 18 в спинохром Ε 1. Смесь тетраэфира 18 (1.18 г, 3.8 ммол) и 48% HBr (120 мл) доводят до кипения. При этом через 1 мин тетраэфир 18 полностью растворяется. Образовавшийся прозрачный темнокрасный раствор кипятят в течение 15 мин и охлаждают до комнатной температуры. Раствор разбавляют водой (150 мл) и выдерживают при 5°С в течение ночи. Осадок отфильтровывают, промывают водой (5×6 мл) и высушивают. Получено 1.06 г (89%) 2,3,5,6,7,8-гексагидрокси-1,4-нафтохинона (спинохрома Е) в виде красно-коричневых игл, т. пл. >360°С (лит. [24], сублимируется при 300-320°С). Rf: 0.32 (гексан-бензол-ацетон, 1:1:1 (по объему)). Спектр ЯМР 1H (300 МГц, DMSO-d6, δ, м.д.): 10.15 (уш. с, 4Н, 4β-ΟΗ), 12.83 (с, 2Н, 2α-ОН). Спектр ЯМР 13С (75 МГц, DMSO-d6, δ, м.д.): 102.3 (C-4a, С-8а), 140.7 (С-2, С-3, С-6, С-7), 166.4 (С-1, С-4, С-5, С-8).). Масс-спектр (ЭУ, 70 эВ, m/z, /отн. (%)): 254 [М]+ (100), 226 (67), 198 (7), 180 (6), 169 (7), 152 (8), 124 (8), 113 (8). Найдено (%): С, 47.37; Н, 2.31. C10H6O8. Вычислено (%): С, 47.26; Н, 2.38.
Пример 2. Хлорирование 2,3-Дихлоронафтазарина 16. К раствору 2,3-дихлоронафтазарина 16 (2.59 г, 10.0 ммол) в 100 мл ледяной НО Ас и 50 мл 36% HCl прибавляют тонкоизмельченный активный MnO2 (3.48 г, 40.0 ммол), и смесь кипятят при перемешивании в течение 1 ч. После этого прибавляют дополнительное количество активного ΜnO2 (0.87 г, 10.0 ммол) и конц. HCl (15 мл) и смесь кипятят при перемешивании еще 30 мин. Обработка реакционной смеси, аналогичная описанной выше для хлорирования нафтазарина 15, дает тетрахлоронафтазарин 17, идентичный во всех отношениях описанному выше в примере 1. Суммарный выход продукта 17 составляет в этом случае 2.72 г (83%).
Нуклеофильное замещение атомов хлора в тетрахлоронафтазарине 17 на метоксигруппы. Смесь хорошо высушенного тетрахлоронафтазарина 17 (0.94 г, 3.0 ммол), безв. CsF (4.89 г, 32.0 ммол), активированного нейтр. Al2O3 (6.10 г, 56.0 ммол, 150 меш, фирмы «Aldrich») и абс. МеОН (200 мл) перемешивают в герметичном сосуде при 95°С в течение 60 ч. После охлаждения до комнатной температуры адсорбент отделяют фильтрованием и последовательно промывают ацетоном (100 мл) и 10% HCl (3 мл). Объединенные фильтраты концентрируют при пониженном давлении, остаток разбавляют водой (200 мл) и экстрагируют СНСl3 (5×50 мл). Органический экстракт промывают последовательно водой (3×20 мл) и насыщ. раствором NaCl (20 мл), высушивают над безв. Na2SO4 и концентрируют досуха. Остаток хроматографируют на колонке с SiO2 (G-60, 2.5×80 см), заполненной н-гексаном. Элюция смесью гексан-ацетон 15:1 дает 5,8-дигидрокси-2,3,6-триметокси-7-хлоро-1,4-нафтохинон 6 (0.08 г, 8%), идентичный во всех отношениях описанному выше в примере 1.
Элюция смесью гексан-ацетон 8:1 дала 2,3,6,7-тетраметоксинафтазарин 18 (0.58 г, 62%), идентичный во всех отношениях описанному выше в примере 1.
Конверсия тетраметоксинафтазарина 18 в спинохром Ε 1. Смесь тетраэфира 18 (0.59 г, 1.9 ммол) и 48% HBr (80 мл) доводят до кипения. При этом через 1 мин тетраэфир 18 полностью растворяют. Образовавшийся прозрачный темнокрасный раствор кипятят в течение 15 мин и охлаждают до комнатной температуры. Раствор разбавляют водой (100 мл) и выдерживают при 5°С в течение ночи. Осадок отфильтровывают, промывают водой (4×5 мл) и высушивают. Получено 0.53 г (89%) 2,3,5,6,7,8-гексагидрокси-1,4-нафтохинона (спинохрома Е), идентичного во всех отношениях описанному выше в примере 1.
Источники информации
[1] (a) Thomson R.H. Naturally occurring quiñones, 2nd Ed, 1971, Academic Press, London, New York; (б) 3rd Ed, 1987, Chapman & Hall, London, New York; (в) 4th Ed, 1997, Chapman & Hall; London, New York.
[2] Lederer E. // Compt. Rend. Acad. Sci. 1938, V. 2007, P. 454-456.
[3] Smith J., Thomson R.H. // Tetrahedron Lett, 1960, V. 1, No. 22, P. 10-12.
[4] Zhou D.Y., Qin L, Zhu B.W., Wang X.D., Tan H., Yang J.F., Li D.M., Dong X.P., Wu H.T., Sun L.M., Li X.L., Murata Y.//Food Chemistry, 2011, V. 129, P. 1591-1597.
[5] Utkina N.K., Pokhilo N.D. //Nat. Prod. Commun., 2012, V. 7, P. 901-904.
[6] Богуславская Л.В., Храпова Н.Г., Максимов О.Б. // Изв. АН СССР. Сер. хим., 1985, №7, С. 1471-1476.
[7] Лебедев А.В., Левицкая Е.Л., Тихонова Е.В., Иванова М.В. // Биохимия, 2001, Т. 66, С. 885-893.
[8] Hatate H., Murata H., Hama Y., Tanaka R., Suzuki N. // Fisheries Science, 2002, V. 68, P. 1641-1642.
[9] Lebedev A.V., Ivanova M.V., Levitsky D.O. // Life Sciences, 2005, V. 76, P. 863-875.
[10] Lebedev A.V., Ivanova M.V., Levitsky D O. // Hemoglobin, 2008, V. 32, P. 165-179.
[11] Kuwahara R., Hatate H., Yuki T., Murata H., Tanaka R., Hama Y. // LWT - Food Science and Technology, 2009, V. 42, P. 1296-1300.
[12] Service M., Wardlaw A.С.// Сотр. Biochem. Physiol., 1984, V. 79B, P. 161-165.
[13] Sime A.A.T. Biocidal compositions comprising polyhydroxynaphthoquinones. // GB Patent 2159056 (1985); Chem. Abstr., 1986, V. 104, 83795.
[14] Стехова С.И., Шенцова Ε.Б., Кольцова Ε.Α., Кулеш Η.И. // Антибиотики и химиотерапия, 1988, Т. 33, С. 831-833.
[15] Pozharitskaya O.N., Shikov A.N., Makarova M.N., Ivanova S.A., Kosman V.M., Makarov V.G., Bazgier V., Berka K., Otyepka M., Ulrichova J. // Planta Medica, 2013, V. 79, P. 1698-1704.
[16] Швилкин А.В., Афонская Н.И., Черпаченко Н.М., Садретдинов СМ., Новиков В.Л., Ануфриев В.Ф., Кольцова Е.А., Максимов О.Б., Левицкий Д.О., Руда М.Я. // Кардиология, 1991, Т. 31, №10, С. 81-82.
[17] Швилкин А.В., Серебрякова Л.И., Цкитишвили О.В., Садретдинов С.М., Кольцова Е.А., Максимов О.Б., Мищенко Н.П., Новиков В.Л., Левицкий Д.О., Руда М.Я. // Кардиология, 1991, Т. 31, №11, С. 79-81.
[18] SU 1822549 A3, опубл. 20.08.1996/.
[19] RU 1833544 С, опубл. 1993.
[20] Anufriev V.Ph., Novikov V.L., Maximov O.B., Elyakov G.B., Levitsky D.O., Lebedev A.V., Sadretdinov S.M., Shvilkin A.V., Afonskaya N.I., Ruda M.Y., Cherpachenko N.M. // Bioorg. Med. Chem. Lett., 1998, V. 8, P. 587-592.
[21] Мищенко H.П., Федореев С.Α., Багирова В.Л. // Хим.-фарм. журн., 2003, Т. 37, №1, С. 49-53.
[22] RU 2411939 C1, опубл. 20.02.2011.
[23] Singh I., Moore R.E., Chang C.W.J., Scheuer P.J. The synthesis of spinochromes A, C, D, and Ε // J. Am. Chem. Soc., 1965, V. 87, No. 17, P. 4023-4024.
[24] Singh I., Moore R.E., Chang C.W.J., Ogata R.T., Scheuer P.J. Spinochrome synthesis // Tetrahedron, 1968, V. 24, No. 7, P. 2969-2978.
[25] Борисова К.Л., Ануфриев В.Φ. // Химия природ, соединений, 2012, №2, С. 187-189.
[26] Matsumoto M., Kobayashi H., Hotta J. // J. Org. Chem., 1984, V. 49, No. 24, P. 4740-4741.
[27] Attenburrow J., Cameron A.F.B., Chapman J.H., Evans R.M., Hems Β.Α., Jansen Α.Β.Α., Walker T.//J. Chem. Soc, 1952, P. 1094-1111.
[28] Anufriev V.Ph., Novikov V.L. // Tetrahedron Lett, 1995, V. 36, No. 14, P. 2515-2518.
Способ получения спинохрома E (2,3,5,6,7,8-гексагидрокси-1,4-нафтохинона), отличающийся тем, что в качестве исходного соединения используют нафтазарин или 2,3-дихлорнафтазарин, который хлорируют под действием хлора, получаемого in situ окислением соляной кислоты активным диоксидом марганца в ледяной уксусной кислоте, в образовавшемся при этом 2,3,6,7-тетрахлорнафтазарине замещают все атомы хлора на метоксигруппы действием метанола, активированного фторидом цезия на поверхности оксида алюминия, и полученный 2,3,6,7-тетраметоксинафтазарин гидролизуют в целевой продукт кипячением в концентрированной бромистоводородной кислоте в течение 10-15 мин.Композиция антиоксидантов, пригодная для перорального применения в терапии воспалительного процесса в легких
Способ профилактики лучевого пневмонита
Водорастворимое производное триптантрина, обладающее противоопухолевой, противовоспалительной и противомикробной активностью, и повышающее терапевтическую активность противоопухолевых антибиотиков
Фармацевтическая композиция, обладающая лечебным действием при различных кожных патологиях
Способ получения водорастворимой солевой формы эхинохрома а, пригодной для использования в фармакологической и пищевой промышленности
Средство, обладающее противовирусным действием в отношении вирусов клещевого энцефалита и герпеса простого i типа
Противовирусная композиция
Препарат для оздоровления сердечно-сосудистой системы, обладающий кардиопротекторным, седативным и гипотензивным действием
Способ получения агарициновой кислоты из мицелия трутовика лекарственного
Адъювант
Способ получения спинохрома d
Биологически активная добавка к пище, обладающая канцерпревентивным действием
Средство для лечения ишемии сосудов головного мозга
Кардиопротекторная фармацевтическая субстанция и способ ее получения
Способ получения 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома d) и промежуточные соединения, используемые в этом способе
Рекомбинантная плазмидная днк pet40cmap/ompf, кодирующая гибридный бифункциональный полипептид cmap/ompf со свойствами высокоактивной щелочной фосфатазы cmap и порообразующего мембранного белка ompf, и рекомбинантный штамм e. coli rosetta (de3)/pet40cmap/ompf - продуцент гибридного бифункционального полипептида cmap/ompf
Композиция ингредиентов для функциональных пищевых продуктов
Способ получения 5,8-дигидрокси-2,6-7-триметокси-3-этил-1,4-нафтохинона

