Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ 2- И 2,3-ЗАМЕЩЕННЫХ ХИНОЛИНОВ
Вид РИД
Изобретение
Предлагаемое изобретение относится к области органической химии, в частности, к способу получения замещенных хинолинов.
Хинолин и его производные широко используются для получения ингибиторов кислотной коррозии металлов. Замещенные хинолины входят в состав высокоэффективных антималярийных, противосудорожных и антибактериальных лекарственных препаратов [Larsen R.D., Corley E.G., King A.O., Carrol J.D., Davis P., Verhoeven T.R., Reider P.J., Labelle M., Gaunthier J.Y., Xiang Y.B., Zamboni R.J. II]. Org. Chem. 1996, 61, 3398-3405; Chen Y.-L., Fang K.-C., Sheu J.-Y., Hsu S.-L., Tzeng C.-C. // J. Med. Chem. 2001, 44, 2374-2377; Roma G., Braccio M.D., Grossi G., Mattioli F., Ghia M. // Eur. J. Med. Chem. 2000, 35, 1021-1035; Dube D., Blouin M., Brideau C., Chan C.-C., Desmarais S., Ethier D., Falgueyret J.P., Friesen R.W., Girard M., Girard Y., Guay J., Riendeau D., Tgari P., Young R.N. // Bioorg. Med. Chem. Lett. 1988, 8, 1255-1260; Maguire M.P., Sheets K.R., McVety K., Spada A.P., Zilberstein, A. // J. Med. Chem. 1994, 37, 2129-2137].
Большинство методов получения замещенных хинолинов основано на реакциях доступных анилинов с различными органическими субстратами под действием металлокомплексных катализаторов.
Так, реакцией анилина с этиленом в присутствии катализатора RhCl3·3H2O - PPh3 были синтезированы 2-метилхинолин (I) и N-этиланилин (II). При использовании в качестве катализатора PdCl2 - PPh3 основным продуктом реакции становится 2-метилхинолин (I) [Diamond S.E., Szalkiewicz, Frank Mares // J. Am. Chem. Soc. 1979, 101, 490-491].
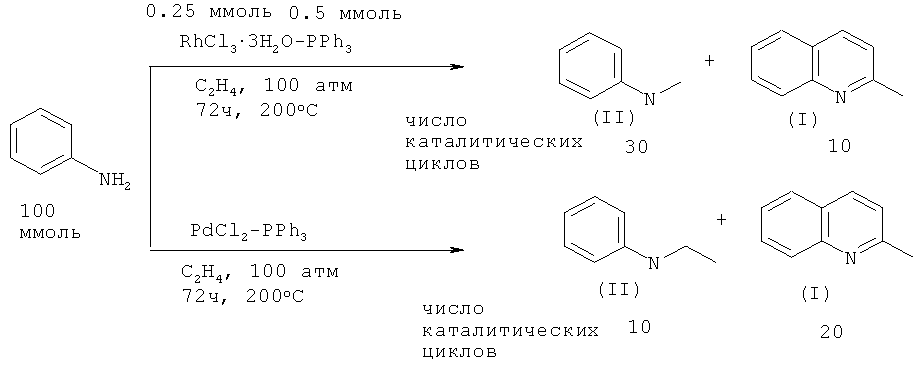
Недостатки метода:
1. Применение дорогостоящих родиевых и палладиевых катализаторов.
2. Жесткие условия реакции: 200ºС, и значительная длительность - 72 ч.
3. Необходимость выполнения работ под давлением 100 атм.
4. Низкий выход 2-метилхинолина (I).
2-Этил-3-метилхинолин (III) с выходом 51% получают реакцией анилина с триаллиламином под действием рутенийсодержащего катализатора, активированного с помощью SnCl2·2H2O и PPh3 в среде диоксана. В качестве побочных продуктов образуются N-пропиланилин (IV) и N-аллиланилин (V) суммарный выход которых составляет 21%. В отсутствие SnCl2·2H2O выход 2-этил-3-метилхинолина (I) не превышает 4%. [Cho C.S., Oh B.H., Shim S.C. // Tetrahedron Lett. 1999, 40, 1499-1500].
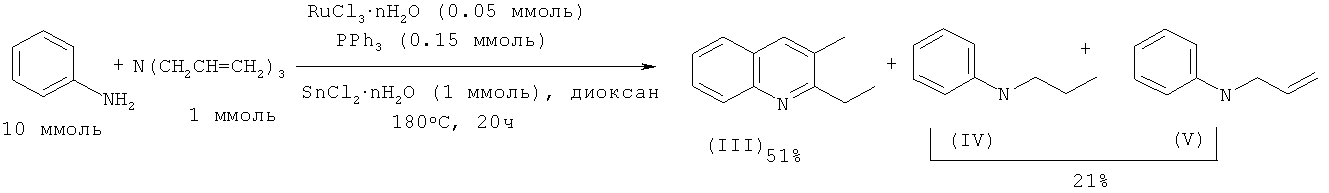
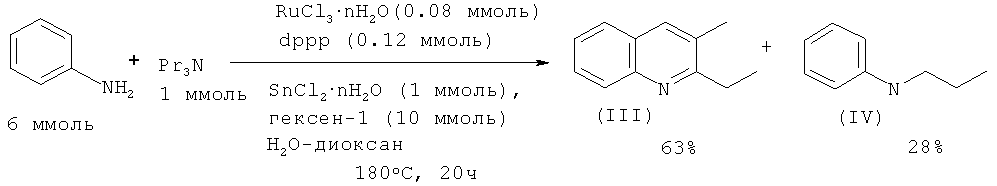
В работе [Cho C.S., Oh B.H., Kim J.S., Kim T.-J., Shim S.C. // Chem. Commun. 2000, 1885-1886] замещенные хинолины были получены взаимодействием анилина с триалкиламинами под действием каталитической системы RuCl3·2H2O - Ph2P(Ch2)3PPh2 - SnCl2·2H2O в присутствии гексена-1, играющего роль акцептора водорода.
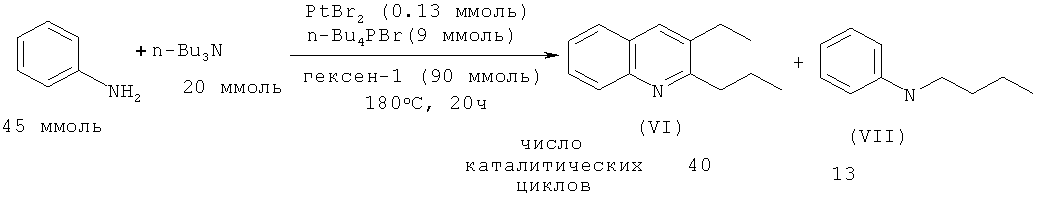
В работе [Anguille S., Brunet J.-J., Chu N.C., Diallo 0., Pages C., Vincendeau S. // Organometallics. 2006, 25, 2943-2948] осуществлен синтез 2-пропил-3-этилхинолина (VI) реакцией анилина с трибутиламином Bu3N под действием бромида платины (II). Присутствие в реакционной массе 1-гексена (акцептор водорода) и Bu4PBr (промотор) способствует увеличению выхода замещенного хинолина (VI).
Анилин реагирует с хлоридом диаллилдипропиламмония в водно-диоксановой среде в присутствии каталитических количеств RuCl2(PPh3)3 и SnCl2·2H2O с образованием 2-этил-3-метилхинолина (III). Предполагается, что SnCl2·2H2O катализирует стадию аллирования анилина [Cho C.S., Kim J.S., Oh B.H., Kim T.-J., Shim S.C., YoonN.S. // Tetrahedron. 2000, 56, 7747-7750].

Недостатки методов:
1. Применение дорогостоящих платиновых и рутениевых катализаторов.
2. Необходимость использования вспомогательных реагентов n-Bu4PBr и SnCl2·2H2O, причем последний применяется в эквимолярном количестве по отношению к анилину.
3. Низкий выход (III) из-за полимеризации хлорида диаллилпропиламмония.
4. Значительные трудности при выделении целевого продукта из-за неселективности реакции.
В работе [Brunet J.-J., Chu N.C., Diallo O., Vincendeau S. // J. Mol. Catal. A: Chem. 2005, 240, 245-248] показано, что при взаимодействии гексина-1 с анилином в присутствии PtBr2 (350:700:1) в качестве побочного продукта образуется 2-пентил-3-бутилхинолин (VIII).
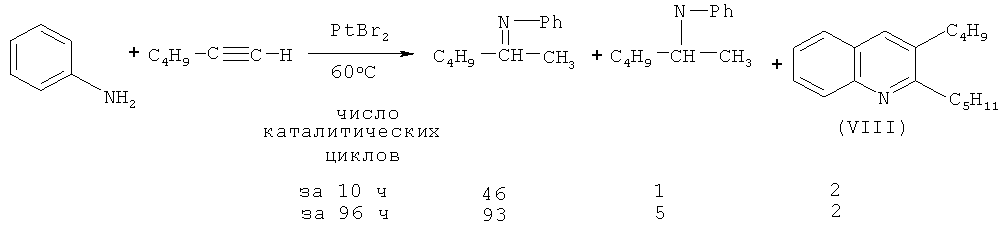
Недостатки метода:
1. Использование дорогостоящего платинового катализатора и дефицитного реагента гексина-1.
2. Низкий выход 2-пентил-3-бутилхинолина (VIII) и значительные трудности при выделении целевого продукта смеси изомеров, что обусловлено низкой селективностью реакции.
2,3-Алкилзамещенные хинолины были получены из N,N-диллиланилинов под действием 10% дикобальтоктакарбонила Со2(СО)8 в присутствии СО (1 атм). Оксид углерода необходим для стабилизации Со2(СО)8 в условиях реакции [Jacob J., Jones W.D. // J. Org. Chem. 2003, 68, 3563-3568].
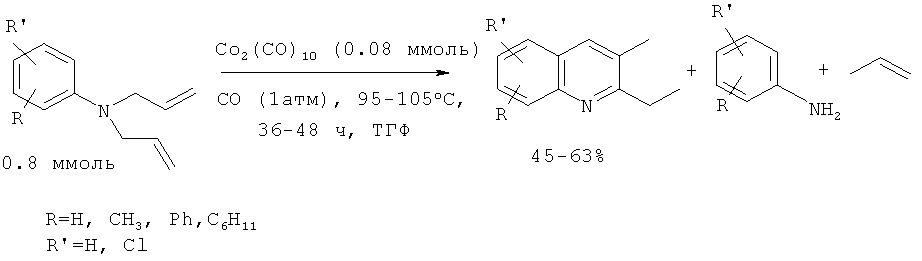
Недостатки метода:
1. Труднодоступность и дороговизна исходных реагентов и катализатора Со2(СО)8.
2. Необходимость проведения реакции в атмосфере оксида углерода.
3. Большой расход катализатора (1:10).
Анилин взаимодействует с 2-пропен-1-олом и 2-бутен-1-олом при 180ºС с образованием производных хинолина в присутствии комплексов рутения [Watanabe Y., Tsuji Y., Ohsugi Y., Shida J. // Bull. Chem. Soc. Jpn. 1983, 56, 2452-2457].
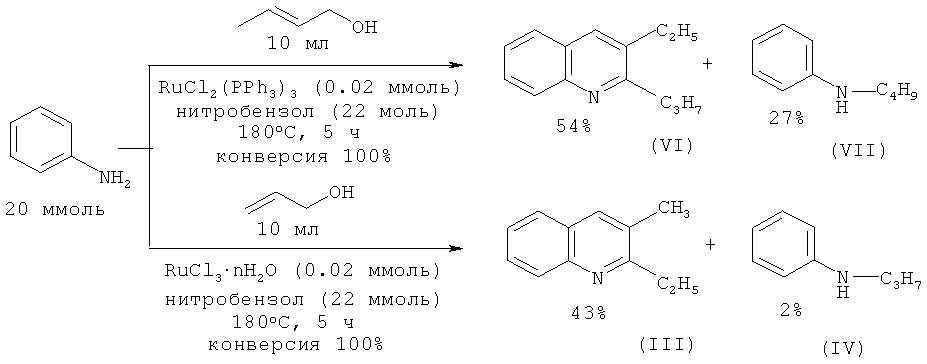
Недостатки метода:
1. Труднодоступность и дороговизна рутенийсодержащих катализаторов.
2. Низкая селективность реакции.
3. Необходимость использования нитробензола для связывания водорода. 2-Пропил-3-этилхинолин (VI) с выходом 70% получен взаимодействием анилина с бутаналем-1 в присутствии кислорода, который совместно с диметилсульфоксидом играет роль дегидрирующего агента [Tanaka S., Yasuda M., Baba A. // J. Org. Chem. 2006, 71, 800-803].

В работе [Watanabe Y., Tsuji Y., Ohsugi Y., Shida J. // Bull. Chem. Soc. Jpn. 1983, 56, 2452-2457] замещенные хинолины получены взаимодействием анилина с альдегидами под действием рутениевых катализаторов.

Авторами работы [Булгаков Р.Г., Кулешов С.П., Махмутов P.P., Джемилев У.М. // ЖОрХ. 2009, т.45.6, 956-957] был получен 2-пропил-3-этилхинолин из анилина и н-бутаналя под действием катализаторов LnCl3·6H2O.
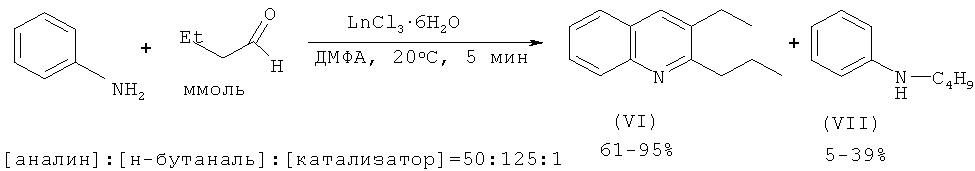
Недостатки методов:
1. Использование дорогостоящего лантанидного катализатора и дефицитных, склонных к окислению альдегидов.
2. Образование трудноразделимой смеси продуктов (VI), (VII).

В работе [Булгаков Р.Г., Кулешов С.П., Вафин P.P., Джемилев У.М. Способ получения 2-пропил-3-этилхинолина. Патент РФ №2409567. Б.И. №2 от 20.01.11] в качестве катализатора синтеза 2-пропил-3-этилхинолина (VI) конденсацией анилина с масляным альдегидом предложено использовать FeCl3·6H2O. Реакция проходит в мягких условиях (20ºС, 10 мин) и приводит к образованию продукта (VI) с выходом 94%
По сходству 3 признаков (использование в качестве исходного соединения анилина, в качестве катализатора FeCl3·6H2O, образование хинолина (VI) данный способ взят нами за прототип.
Прототип имеет следующие недостатки:
1. Использование в качестве исходного соединения альдегида, который склонен к реакциям автоконденсации и окислению на воздухе.
2. Введение дополнительного реагента - использование избытка растворителя ([анилин]:[растворитель]=1:5) канцерогенного и токсичного ДМФА или этанола.
3. Предложенный метод опробирован только на N-бутанале, границы применимости реакции не исследованы на примере других альдегидов.
4. Большой расход катализатора ([FeCl3·6H2O]:[анилин]=1.2:45).
Авторами предлагается новый способ получения 2- и 2,3-замещенных хинолинов, не имеющий вышеперечисленных недостатков.
Задачей предполагаемого изобретения является удешевление себестоимости конечного продукта за счет использования доступных исходных реагентов - анилина и алифатических спиртов.
Сущность способа заключается во взаимодействии анилина со спиртами R'OH (где R'=C2H5, н-C3H7, н-C4H9) в присутствии катализатора FeCl3·6H2O, при 140ºС в среде CCl4 в течение 2-8 часов. Реакция проводится путем 1-3-х этапной загрузки исходных реагентов, при мольном соотношении [катализатор]:[анилин]:[CCl4]:[R'OH]=1:100:100:200.

Существенные отличия предлагаемого способа от прототипа.
1. Исходным сырьем для получения 2- и 2,3-замещеннных хинолинов являются:
анилин и алифатический спирт, а реакции проходят в присутствии катализатора FeCl3·H2O в среде CCl4.
Преимущества предлагаемого метода
1. Доступность и дешевизна исходных реагентов - анилина, CCl4, спиртов и катализатора FeCl3·6H2O и удешевление себестоимости и упрощение технологии.
2. Высокая селективность реакции и отсутствие в реакционной массе побочных продуктов - N-алкиланилинов.
3. Отсутствие побочных продуктов облегчает выделение и очистку целевых 2- и 2,3-диалкилзамещенных хинолинов.
4. Высокий выход 2- и 2,3-диалкилзамещенных хинолинов (52-96%).
Предлагаемый способ поясняется примерами:
ПРИМЕР 1. Получение 2-этил-3-метилхинолина
В ампулу в токе аргона загружали 3 мг (0.5 ммоль) FeCl3·6H2O, 0.2 мл (100 ммоль) анилина, 0.1 мл (50 ммоль) CCl4 и 0.16 мл (100 ммоль) н-PrOH. Запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 1), затем автоклав охлаждали до ~20ºС, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворитель отгоняли, остаток помещали в ампулу и добавляли новую порцию реагентов: 3 мг (0.5 ммоль) FeCl3·6H2O, 0.1 мл (50 ммоль) CCl4 и 0.16 мл (100 ммоль) н-PrOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 2). После окончания реакции автоклав охлаждали до комнатной температуры, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворители отгоняли, остаток перегоняли под вакуумом. Конверсия исходного анилина составила 79%, выход 2-этил-3-метилхинолина (Iб) - 62%.
Строение полученного 2-этил-3-метилхинолина (1б) доказано методами ЯМР, масс-спектрометрии, а также сравнением с известными образцами и справочными данными.
2-этил-3-метилхинолин (Iб)
Маслянистая желтая жидкость. Т. кип.97-99ºС/2 мм рт.ст.(Т кип.73-83ºС/0.57 мм рт.ст., 84-85ºС/0.35 мм рт.ст. [Watanabe Y., Tsuji Y., Ohsugi Y. // Tetrahedron Lett. 1981, 22, 2667]. Спектр ЯМР 13C (δ, м.д.): 12.79 (СН2 СН3), 18.57 (СН3), 29.43 (СН2), 125.55 (С7), 126.70 (С5), 127.31 (C4a), 128.24(С6), 128.53 (С8), 129.32(С3), 135.63 (С4), 146.68 (C8a), 163.15 (С2). Спектр ЯМР 1Н (CDCl3, δ, м.д.): 1.38 т (3Н, СН3, J 7 Гц), 2.38 с (3Н, СН3), 2.96 к (2Н, СН2, J 7 Гц), 7.40 т (1Н, C7H, J 8 Гц), 7.58 т (1Н, C6H, J 8 Гц), 7.62 д (1Н, C5H, J 8 Гц), 7.70 с (1Н, C4H, J 7 Гц), 8.06 д (1Н, C8H, J 8 Гц). Масс-спектр, m/z (Iотн. (%)): 171.23[M+]. Найдено, %: С 84.22; Н 7.63; N 8.15. C12H13N. Вычислено, %: С 84.17; Н 7.65; N 8.18.
ПРИМЕР 2. Получение 2-этил-3-метилхинолина
Аналогично примеру 1, за исключением: продолжительность этапа 2 составила 2 ч. Конверсия исходного анилина 96%, выход 2-этил-3-метилхинолина (Iб) - 81%.
ПРИМЕР 3. Получение 2-этил-3-метилхинолина
Аналогично примеру 1, за исключением: продолжительность этапа 2 составила 3 ч. Конверсия исходного анилина 93%, выход 2-этил-3-метилхинолина (Iб) - 75%.
ПРИМЕР 4. Получение 2-этил-3-метилхинолина
Аналогично примеру 1, за исключением: продолжительность этапа 1 составила 2 ч. Конверсия исходного анилина 90%, выход 2-этил-3-метилхинолина (Iб) - 80%.
ПРИМЕР 5. Получение 2-этил-3-метилхинолина
Аналогично примеру 1, за исключением: продолжительность этапа 1-2 ч, этапа 2-2 ч. Конверсия исходного анилина составила 97%, выход 2-этил-3-метилхинолина (Iб) - 87%.
ПРИМЕР 6. Получение 2-этил-3-метилхинолина
Аналогично примеру 1, за исключением: продолжительность этапа 1-2 ч, этапа 2-3 ч. Конверсия исходного анилина составила 90%, выход 2-этил-3-метилхинолина (Iб) - 72%.
ПРИМЕР 7. Получение 2-этил-3-метилхинолина
Аналогично примеру 1, за исключением: продолжительность этапа 1-3 ч, этапа 2-1 ч. Конверсия исходного анилина составила 80%, выход 2-этил-3-метилхинолина (Iб) - 60%.
ПРИМЕР 8. Получение 2-этил-3-метилхинолина
Аналогично примеру 1, за исключением: продолжительность этапа 1-3 ч, этапа 2-2 ч. Конверсия исходного анилина составила 93%, выход 2-этил-3-метилхинолина (Iб) - 69%.
ПРИМЕР 9. Получение 2-метилхинолина
В ампулу в токе аргона загружали 3 мг (0.5 ммоль) FeCl3·6H2O, 0.2 мл (100 ммоль) анилина, 0.1 мл (50 ммоль) CCl4 и 0.12 мл (100 ммоль) ЕtOН. Запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 2 ч (этап 1), затем автоклав охлаждали до ~20ºС, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворитель отгоняли, остаток помещали в ампулу и добавляли новую порцию реагентов: 3 мг (0.5 ммоль) FeCl3·6H2O, 0.1 мл (50 ммоль) CCl4 и 0.12 мл (100 ммоль) EtOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 2 ч (этап 2). После окончания реакции автоклав охлаждали до комнатной температуры, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворители отгоняли, остаток перегоняли под вакуумом. Конверсия исходного анилина составила 100%, выход 2-метилхинолина (Iа) - 94%.
Строение полученного 2-метилхинолина (Iа) доказано методами ЯМР, масс-спектрометрии, а также сравнением с известными образцами и справочными данными.
2-метилхинолин (Iа)
Маслянистая желтая жидкость. Т кип.80-81ºС/2 мм рт.ст. (Т кип. 248ºС, 105-107ºС/10 мм рт.ст. [Aldrich. Catalog handbook of fine chemicals. 2007-2008, 824]. Спектр ЯМР 13С (δ, м.д.): 25.12 (СН3), 121.74(С3), 125.45(С6), 126.32 (C4a), 127.37 (С5), 128.80 (С8), 129.19 (С7), 135.84 (С4), 147.76 (C8a), 158.67 (С2). Спектр ЯМР 1Н (CDCl3, δ, м.д.): 2.78 с (3Н, СН3), 7.22 д (1Н, C3H, J 8 Гц), 7.68 т (1Н, C7H, J 4 Гц), 7.46 т (1Н, C6H, J 6.4 Гц), 7.73 д (1Н, C5H, J 9.2 Гц), 7.91 д (1Н, C4H, J 9.2 Гц), 7.91 д (1Н, C8H, J 9.2 Гц). Масс-спектр, m/z (Iотн. (%)): 143.18[М]+(100), 128 (20), 115 (22), 101 (5), 89 (4), 75 (5), 51 (4). Найдено, %: С 83.92; Н 6.33; N 9.75. C10H9N. Вычислено, %: С 83.88; Н 6.34; N 9.78.
ПРИМЕР 10. Получение 2-пропил-3-этилхинолина
В ампулу в токе аргона загружали 3 мг (0.5 ммоль) FeCl3·6H2O, 0.2 мл (100 ммоль) анилина, 0.1 мл (50 ммоль) CCl4 и 0.2 мл (100 ммоль) н-BuOH. Запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 2 ч (этап 1), затем автоклав охлаждали до ~20ºС, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворитель отгоняли, остаток помещали в ампулу и добавляли новую порцию реагентов: 3 мг (0.5 ммоль) FeCl3·6H2O, 0.1 мл (50 ммоль) CCl4 и 0.2 мл (100 ммоль) н-BuOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 2 ч (этап 2). После окончания реакции автоклав охлаждали до комнатной температуры, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворители отгоняли, остаток перегоняли под вакуумом. Конверсия исходного анилина составила 90%, выход 2-пропил-3-элилхинолина (Iв) - 81%.
Строение полученного 2-пропил-3-этилхинолина (Iв) доказано методами ЯМР, масс-спектрометрии, а также сравнением с известными образцами и справочными данными.
2-пропил-3-этилхинолин (Iв)
Маслянистая желтая жидкость. Т кип. 99-100ºС/0.5 мм рт.ст., Т кип.118ºС/1 мм рт.ст.[Watanabe Y., Shim S.C., Mitsudo T. // Bull. Chem. Soc. Jpn. 1981, 54, 3460], 92ºC/0.3 мм рт.ст. [Watanabe Y., Tsuji Y., Ohsugi Y. // Tetrahedron Lett. 1981, 22, 2667]. Спектр ЯМР 13С (δ, м.д.): 14.39 ((СН2)2 СН3), 14.46 (СН2 СН3), 22.88 (CH2 CH2CH3), 25.16 (СН2СН3), 37.75 (СН2СН2СН3), 125.57 (С7), 126.94(С5), 127.50 (C4a), 128.35 (С8), 128.44 (С6), 129.21 (С4), 133.88 (С3), 146.44 (C8a), 162.04 (С2). Спектр ЯМР 1Н (CDCl3, δ, м.д.): 0.97 т (3Н, СН3, J 7.2 Гц), 1.11 т (3Н, СН3, J 7.2 Гц), 1.8-1.9 м (2Н, СН2СН 2СН3), 2.82 к (2Н, СН 2СН3, J 7.2 Гц), 2.99 т (2Н, CH 2CH2CH3, J 8 Гц), 7.44 т (1Н, С7Н, J 7.6 Гц), 7.62 т (1Н, C6H, J 7.2 Гц), 7.72 д (1Н, C5H, J 7.6 Гц), 7.84 с (1Н, C4H), 8.08 д (1Н, C8H, J 8 Гц).
Масс-спектр, m/z (Iотн. (%)): 199.28[М]+. Найдено, %: С 84.47; Н 8.48; N 7.05. C14H17N. Вычислено, %: С 84.37; Н 8.60; N 7.03.
ПРИМЕР 11. Получение 2-этил-3-метилхинолина
В ампулу в токе аргона загружали 4 мг (0.33 ммоль) FeCl3·6H2O, 0.4 мл (100 ммоль) анилина, 0.14 мл (33.3 ммоль) CCl4 и 0.22 мл (66.6 ммоль) н-PrOH. Запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 1), затем автоклав охлаждали до ~20ºС, ампулу вскрывали и добавляли новую порцию реагентов: 4 мг (0.33 ммоль) FeCl3·6H2O, 0.14 мл (33.3 ммоль) CCl4 и 0.22 мл (66.6 ммоль) н-PrOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 2).
Далее снова автоклав охлаждали до ~20ºС, ампулу вскрывали и добавляли новую порцию реагентов: 4 мг (0.33 ммоль) FeCl3·6H2O, 0.14 мл (33.3 ммоль) CCl4 и 0.22 мл (66.6 ммоль) н-PrOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 3). После окончания реакции автоклав охлаждали до комнатной температуры, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворитель отгоняли, остаток перегоняли под вакуумом. Конверсия исходного анилина составила 74%, выход 2-этил-3-метилхинолина (Iб) - 56%.
ПРИМЕР 12. Получение 2-этил-3-метилхинолина
Аналогично примеру 11, за исключением: продолжительность этапа 1 - 2 ч, этапа 2 - 2 ч, этапа 3 - 2 ч. Конверсия исходного анилина составила 72%, выход 2-этил-3-метилхинолина (Iб) - 52%.
ПРИМЕР 13. Получение 2-этил-3-метилхинолина
В ампулу в токе аргона загружали 3 мг (0.25 ммоль) FeCl3·6H2O, 0.4 мл (100 ммоль) анилина, 0.1 мл (25 ммоль) CCl4 и 0.18 мл (50 ммоль) н-PrOH. Запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 1), затем автоклав охлаждали до ~20ºС, ампулу вскрывали и добавляли новую порцию реагентов: 3 мг (0.25 ммоль) FeCl3·6H2O, 0.1 мл (25 ммоль) CCl4 и 0.18 мл (50 ммоль) н-ProH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 2). После завершения второго этапа автоклав снова охлаждали до ~20ºС, ампулу вскрывали и добавляли новую порцию реагентов: 3 мг (0.25 ммоль) FeCl3·6H2O, 0.1 мл (25 ммоль) CCl4 и 0.18 мл (50 ммоль) н-PrOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС течение 1 ч (этап 3), далее автоклав охлаждали до ~20ºС, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали, растворители отгоняли. Остаток помещали в ампулу и добавляли новую порцию реагентов: 3 мг (0.25 ммоль) FeCl3·6H2O, 0.1 мл (25 ммоль) CCl4 и 0.18 мл (50 ммоль) н-PrOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС течение 1 ч (этап 4). После окончания реакции автоклав охлаждали до комнатной температуры, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворители отгоняли, остаток перегоняли под вакуумом. Конверсия исходного анилина составила 100%, выход 2-этил-3-метилхинолина (Iб) - 91%.
ПРИМЕР 14. Получение 2-этил-3-метилхинолина
Аналогично примеру 13, за исключением: продолжительность этапа 1 - 2 ч, этапа 2 - 2 ч, этапа 3 - 2 ч, этапа 4 - 2 ч. Конверсия исходного анилина составила 100%, выход 2-этил-3-метилхинолина (Iб) - 95%.
ПРИМЕР 15. Получение 2- метилхинолина
В ампулу в токе аргона загружали 3 мг (0.25 ммоль) FeCl3·6H2O, 0.4 мл (100 ммоль) анилина, 0.1 мл (25 ммоль) CCl4 и 0.12 мл (50 ммоль) EtOH. Запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 1), затем автоклав охлаждали до ~20ºС, ампулу вскрывали и добавляли новую порцию реагентов: 3 мг (0.25 ммоль) FeCl3·H2O, 0.1 мл (25 ммоль) CCl4 и 0.12 мл (50 ммоль) EtOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 2). После второго этапа автоклав снова охлаждали до ~20ºС, ампулу вскрывали и добавляли новую порцию реагентов: 3 мг (0.25 ммоль) FeCl3·6H2O, 0.1 мл (25 ммоль) CCl4 и 0.12 мл (50 ммоль) EtOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС течение 1 ч (этап 3), далее автоклав охлаждали до ~20ºС, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали, растворители отгоняли. Остаток помещали в ампулу и добавляли новую порцию реагентов: 3 мг (0.25 ммоль) FeCl3·6H2O, 0.1 мл (25 ммоль) CCl4 и 0.12 мл (50 ммоль) EtOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС течение 1 ч (этап 4). После окончания реакции автоклав охлаждали до комнатной температуры, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворители отгоняли, остаток перегоняли под вакуумом. Конверсия исходного анилина составила 100%, выход 2 метилхинолина (Iа)-96%.
ПРИМЕР 16. Получение 2-пропил-3-этилхинолина
В ампулу в токе аргона загружали 3 мг (0.25 ммоль) FeCl3·6H2O, 0.4 мл (100 ммоль) анилина, 0.1 мл (25 ммоль) CCl4 и 0.2 мл (50 ммоль) н-BuOH. Запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 1), затем автоклав охлаждали до ~20ºС, ампулу вскрывали и добавляли новую порцию реагентов: 3 мг (0.25 ммоль) FeCl3·6H2O, 0.1 мл (25 ммоль) CCl4 и 0.2 мл (50 ммоль) н-BuOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч (этап 2). После второго этапа автоклав снова охлаждали до ~20ºС, ампулу вскрывали и добавляли новую порцию реагентов: 3 мг (0.25 ммоль) FeCl3·6H2O, 0.1 мл (25 ммоль) CCl4 и 0.2 мл (50 ммоль) н-BuOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС течение 1 ч (этап 3), далее автоклав охлаждали до ~20ºС, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали, растворители отгоняли. Остаток помещали в ампулу и добавляли новую порцию реагентов: 3 мг (0.25 ммоль) FeCl3·6H2O, 0.1 мл (25 ммоль) CCl4 и 0.2 мл (50 ммоль) н-BuOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС течение 1 ч (этап 4). После окончания реакции автоклав охлаждали до комнатной температуры, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворители отгоняли, остаток перегоняли под вакуумом. Конверсия исходного анилина составила 91%, выход 2-пропил-3-этилхинолина (Iв) - 84%.
ПРИМЕР 17. Получение 2-этил-3-метилхинолина
В ампулу в токе аргона загружали 3 мг (0.25 ммоль) FeCl3·6H2O, 0.4 мл (100 ммоль) анилина, 0.1 мл (25 ммоль) CCl4 и 0.18 мл (50 ммоль) н-PrOH. Запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч, затем автоклав охлаждали до ~20ºС, ампулу вскрывали и добавляли новую порцию реагентов: 3 мг (0.25 ммоль) FeCl3·6H2O, 0.1 мл (25 ммоль) CCl4 и 0.18 мл (50 ммоль) н-PrOH. Затем запаянную ампулу помещали в автоклав, автоклав герметично закрывали и нагревали при 140ºС в течение 1 ч. Аналогично описанному выше провели дробное добавление реагентов еще в 2 этапа. После окончания реакции автоклав охлаждали до комнатной температуры, ампулу вскрывали, реакционную массу нейтрализовали 10% водным раствором Na2CO3 (перемешивание на магнитной мешалке в течение 0.5-1 часа), органический слой экстрагировали хлористым метиленом и отфильтровывали. Растворители отгоняли, остаток перегоняли под вакуумом. Конверсия исходного анилина составила 89%, выход 2-этил-3-метилхинолина (Iб) - 70%.
Способ получения 2- и 2,3-замещенных хинолинов формулы характеризующийся тем, что анилин подвергают взаимодействию со спиртами R'OH (где R'=CH, н-CH, н-CH), CCl в присутствии катализатора FeCl·6HO, при температуре 140°C в течение 2-8 ч, путем 1-3-х этапной загрузки исходных реагентов, при мольном соотношении [катализатор]:[анилин]:[CCl]:[R'OH]=1:100:100:200.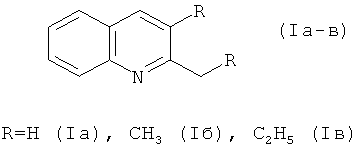
Способ получения олигомеров высших линейных альфа-олефинов
Способ получения олигомеров высших линейных α-олефинов
Способ получения 1-йод-2-азидо(c-i)[5,6]фуллерена
Способ получения 1-хлорадамантана
Способ получения олигомеров высших линейных α-олефинов
Способ получения экзо-2-норборнеола и его производных
Способ получения 3-(о-, м-, п-нитрофенил)-тетрагидро-2н-1,5,3-диоксазепинов
Способ получения 3-(м-, п-метилфенил)-тетрагидро-2н-1,5,3-диоксазепинов
Способ поэтапного получения 1,7,13,19,25,31,37,43-октатиа-3,5,9,11,15, 17,21,23,27,29,33,35,39,41,45,47-гексадекаазациклооктатетраконтан-4,10,16,22,28,34,40,46-октатиона и 5,6-дигидро-2н-тиадиазин гидройодида
Способ получения 1-гидроксиадамантан-4-она
Способ получения 1-хлорадамантана
Способ получения олигомеров высших линейных α-олефинов
Способ получения экзо-2-норборнеола и его производных
Способ получения 3-(о-, м-, п-нитрофенил)-тетрагидро-2н-1,5,3-диоксазепинов
Способ получения 3-(м-, п-метилфенил)-тетрагидро-2н-1,5,3-диоксазепинов
Способ поэтапного получения 1,7,13,19,25,31,37,43-октатиа-3,5,9,11,15, 17,21,23,27,29,33,35,39,41,45,47-гексадекаазациклооктатетраконтан-4,10,16,22,28,34,40,46-октатиона и 5,6-дигидро-2н-тиадиазин гидройодида
Способ получения конъюгата (6-гидрокси-2,5,7,8-тетраметилхроман-2-ил)ацетальдегида с 20-гидроксиэкдизоном и его применение в качестве антиоксидантного средства, ингибирующего процесс перекисного окисления липидов
Способ получения 1-гидроксиадамантан-4-она
Способ получения n-бензилиденбензиламина
Способ получения n-(1,5,3-дитиазоцинан-3-ил)амидов

