Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ 3, 4-БИС(3-АМИНОФУРАЗАН-4-ИЛ)-ФУРАЗАНА И ЕГО N, N'-ДИАЦИЛЬНЫХ ПРОИЗВОДНЫХ
Вид РИД
Изобретение
1. Область изобретения
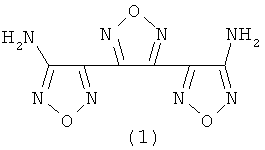
Настоящее изобретение относится к области химии полициклических гетероциклических соединений, более конкретно к способам синтеза сочлененных производных 1,2,5-оксадиазола (фуразана), содержащих сочлененную структуру из трех повторяющихся 1,2,5-оксадйазольных (фуразановых) циклов, а именно, к экологически чистому методу синтеза терминального диаминопроизводного тримера 1,2,5-оксадиазола (фуразана), а именно, 3,4-бис(3-аминофуразан-4-ил)-фуразана (3,4-бис(4-амино-1,2,5-оксадиазол-3-ил)-1,2,5-оксадиазола), (1) в ходе которого исключена необходимость проведения регенерации или утилизации большого количества отходов, содержащих соли тяжелых металлов.
2. Описание известного уровня техники
Многообразие химических превращений и потенциальная биологическая активность являются причиной постоянно увеличивающегося интереса к производным 1,2,5-оксадиазола (фуразанам и фуроксанам). Большое количество статей и литературных обзоров посвящены синтезу, химическим свойствам и возможным областям применения этих соединений. Широкие синтетические возможности и относительная легкость получения 3-амино-1,2,5-оксадиазолов [1] делает их важным сырьем в синтезе производных 1,2,5-оксадазола. В этой связи наибольший интерес связан с получением простейших представителей данного ряда гетероциклических соединений, например, моноциклического производного - 3,4-диамино-1,2,5-оксадиазола [2] бициклического производного - 4,4'-диамино-3,3'-бис-1,2,5-оксадиазола [3], синтез которых основан на традиционном методе получения производных 1,2,5-оксадиазола через дегидратацию соответствующих вицинальных изонитрозосоединений. Однако проведение этой стратегии синтеза в случае производных 1,2,5-оксадиазола, содержащих два и более сочлененных 1,2,5-окадиазольных циклов связано с известными трудностями, связанными с малой доступностью требуемых для формирования 1,2,5-оксадиазольного цикла полиизонитрозосоединений.
Другой метод получения производных 1,2,5-оксадиазола (фуразана) основан на проведении реакции селективного восстановления экзоциклической N-O группы в более доступных в ряде случаев их N-оксидов (фуроксанов) [4].
В качестве наиболее близкого выбран способ синтеза диаминопроизводного тримера фуразана - 3,4-бис(3-аминофуразан-4-ил)-фуразана (3,4-бис(4-амино-1,2,5-оксадиазол-3-ил)-1,2,5-оксадиазола) (1) [5] восстановлением избытком раствора хлорида олова (II) синтетически доступного 3,4-бис(3-аминофуразан-4-ил)-фуроксана (3,4-бис(4-амино-1,2,5-оксадиазол-3-ил)-1,2,5-оксадиазол-2-оксида) (2).
3. Сущность изобретения
Основные недостатки применения хлорида олова (II) для селективного восстановления 1,2,5-оксадиазол-2-оксидного (фуроксанового) цикла 3,4-бис(3-аминофуразан-4-ил)-фуроксана (2) связаны с умеренным выходом целевого продукта и использованием значительного количества агента восстановления (на 1 моль исходного соединения требуется 6 моль SnCl2) и связанной с этим необходимостью регенерации и переработки значительного количества солей олова. Наши наблюдения показали, что этот метод обеспечивает выход целевого продукта при 98-99%-й чистоте 50-55%, при этом необходимое количество восстановителя не может быть снижено за счет изменения времени и условий проведения реакции ниже значения 2 моль на 1 моль исходного соединения.
Задача, на решение которой направлено предложенное изобретение, состоит в получении целевого продукта с большим выходом, улучшении экологичности синтеза, связанной с отсутствием образование в ходе проведения синтеза отходов солей, состоящих из солей тяжелых металлов, в частности солей олова (II, IV), а также в повышении экономических показаний процесса.
Предложен способ получения 3,4-бис(3-аминофуразан-4-ил) - фуразана (1) и его N,N'-диацильных производных из 3,4-бис(3-аминофуразан-4-ил)-фуроксана (2), заключающийся в последовательном проведении восстановления фуроксанового (1,2,5-оксадиазол-2-оксидного) цикла соединения 3,4-бис(3-аминофуразан-4-ил)-фуроксана до диоксима 1,2-бис-(4-аминофуразан-3-ил)-этан-1,2-диона(3) гидразином или водородом в присутствии катализатора восстановления, содержащего металлы платиновой группы: палладий, платина, осажденные на стандартных носителях, выбранные из числа таких, как, например, активированный уголь, угольная сажа, сульфат бария, оксид алюминия, силикагель или скелетный никель Ренея с последующей дегира-тацией полученного диоксима агентами дегидратации, а именно, действием ангидридов карбоновых кислот, хлорангидридов неорганических кислот, до 3,4-бис(3-аминофуразан-4-ил)-фуразана (1) или его N,N'-диацильного производного.
Предлагаемый способ получения 3,4-бис(3-аминофуразан-4-ил)-фуразана - (1) включает две стадии, первая из которых заключается в восстановительном раскрытии фуроксанового (1,2,5-оксадиазол-2-оксидного) цикла исходного соединения (2) до соответствующего замещенного глиоксима, диоксима 1,2-бис-(4-аминофуразан-3-ил)-этан-1,2-диона (3), под действием агента восстановления: гидразин или водород в присутствии катализатора восстановления, известного как катализатор гидрирования.
Для восстановления фуроксанового цикла соединения (2) используют гидразин в форме основания или используют солевые формы гидразина, такие, как, например сульфат гидразина, гидрохлорид гидразина, ацетат гидразина.
Катализатор восстановления представляет собой металл платиновой группы, например, палладий, платина, осажденный в виде металла, или в виде соответствующих оксидов, гидроксидов, солей минеральных кислот, на соответствующем носителе, выбранном из числа таких типичных носителей, как, например, различные виды активированных углей, сажа, сульфат бария, оксид алюминия, или скелетный катализатор гидрирования, известный как никель Ренея.
Для восстановления фуроксанового цикла соединения (2)использован водород при атмосферном и повышенном давлении от 1 до 100 атм, предпочтительно от 2 до 20 атм.
Полученный глиоксим (3) может быть очищен растворением в водном растворе щелочи, фильтрованием от нерастворимых в водных щелочах примесей, с последующим осаждением глиоксима (3) подкислением полученного раствора соляной кислотой
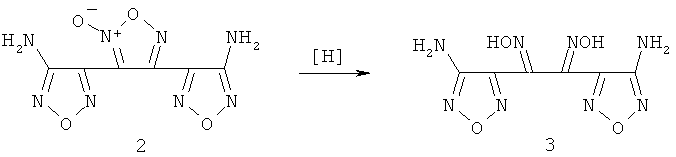
Вторая стадия - дигидратация полученного глиоксима под действием дегидратирующих агентов, таких как, например, ангидриды уксусной и про-пионовой кислот, хлористого тионила, хлорокиси фосфора, с образованием в зависимости от условий проведения реакции дигидратации 3,4-бис(3-аминофуразан-4-ил)-фуразана - (1) или его N,N'-диацильного производного (4)

Ангидриды карбоновых кислот, таких, как, например, ангидрид уксусной кислоты, ангидрид пропионовой кислоты, ангидрид масляной кислоты, прибавляют к раствору диоксима (3), приготовленному растворением диок-сима (3) в водном растворе оснований, выбранных из группы гидроксидов щелочных металлов, гидроксидов щелочноземельных металлов, солей щелочных металлов, гидролизующихся в водном растворе по аниону, солей щелочноземельных металлов, гидролизующихся в водном растворе по аниону.
Дегидратацию промежуточного диоксима (3) проводят с одновременным ацилированием по концевым атомам азота аминогрупп ангидридами карбоновых кислот такими, как, например, ангидрид уксусной кислоты, ангидрид пропионовой кислоты, ангидрид масляной кислоты проведением реакции без растворителя или в безводных растворителях с образованием соответствующих N,N'-диацильных производных (4).
Максимальный выход целевого продукты достигается при использовании на первой стадии проведения процесса гидрирования водородом под давлением от 5 до 7 атм. в присутствии катализатора гидрирования Перлмана [6] с последующей дегидратацией полученного глиоксима (3) уксусным ангидридом в водно-щелочном растворе.
Вполне очевидно, что настоящее изобретение не ограничено вышеописанными вариантами, и возможны и другие варианты в пределах объема изобретения, (который ограничен формулой изобретения). Нижеследующие Примеры приведены для лучшего понимания настоящего изобретения и никоим образом не ограничивают область его применения. Специалисты в данной области техники должны понимать, что в предложенных методиках могут быть сделаны различные изменения, которые не изменяют объема и сущности настоящего изобретения. Все публикации, упомянутые в настоящем описании, доступны по уровню специалистам в той области техники, к которой принадлежит настоящее изобретение. Оптимальное время проведения реакций, указанных Примерах может быть определено посредством традиционных хроматографических способов слежения за ходом реакции. Выбор растворителя обычно некритичен, если применяемый растворитель инертен по отношению к происходящей реакции и растворяет реагенты в достаточной степени для осуществления реакции.
Примеры конкретного исполнения
Пример 1
100 г соединения (2) (0,40 моль) добавляют в нагретый до 40°С изопропиловый спирт (300 мл), прибавляют 1 г катализатора 10% Pd/C и затем приливают 76 мл (1,6 моль) гидразингидрата. Смесь перемешивают при температуре не выше 60°С 4-6 часов. Реакцию заканчивают, когда осадок, полученный разбавлением водой небольшой пробы реакционной смеси, полностью растворяется в 5% водном растворе щелочи. Затем реакционную массу отфильтровывают от катализатора, отгоняют в вакууме 2/3 изопропанола и разбавляют 200 мл воды. Осадок отфильтровывают и перекристаллизовывают из изопропанола. Выход глиоксима (3) 45-55 г (45-55%), tпл. 258°С. ЯМР-1Н (ДМСО-d6): δ, м.д, 6,0(2Н, NH2); 6,3(2H, NH2); 13,0(2Н, уш.,2NOH). ЯМР-13С (ДМСО-d6): δ, м.д., 136,8 и 141,6(C=NOH); 142,2 и 142,4(С-2 и С-5); 154,6 и 155,5 (C-NH2). ИК спектр, см-1: 3470 (N-H as); 3370 (N-H s); 3280, 3040 и 2850 (ОН); 1620 (C=N-OH); 1600 (вал. C-N фуразан); 1000, 980 (деф. фуразан); 900, 860 (вал. N-O-H). Масс-спектр, m/e: M 254(41); 237(100), (М-ОН); 206(18), (M - OH -NOH); 177(57), (M - ОН - 2NO); 150(54), (M - ОН - 2NO - HCN); 58(64), (H2N-C=N-O); 53(93), C2HN2; 43(78), (HNCO). Найдено, %: С 28,15; Н 2,53; N 39,49. C6H6N8O4. Вычисл., %: С 28,34; Н 2,36; N 39,10.
Пример 2
Реакцию проводят аналогично способу, приведенному в примере 1. В качестве восстановителя используют раствор ацетата гидразина, полученный добавлением к 100 мл (1,75 моль) уксусной кислоты 76 мл (1,6 моль) гидразингидрата. Время реакции 10-20 часов, температура проведения процесса 70-80°С. Выход соединения (3) после кристаллизации 65-70 г (65-70%).
Пример 3
100 г соединения (2) (0,40 моль) добавляют в нагретый до 40°С изопропиловый спирт (400 мл), прибавляют 1 г катализатора 10% Pd/C. В реакционную массу добавляют при интенсивном перемешивании ацетат натрия (200 г, 2 моль). Затем при перемешивании добавляют порциями по 5 г 105 г (1,0 моль) дигидрохлорида гидразина при температуре в реакционной смеси 60°С. После окончания дозировки дигидрохлорида гидразина реакционную массу перемешивают 20 часов при 70-80°С. Полученную суспензию отфильтровывают от выпавших неорганических солей и катализатора, осадок промывают 100 мл кипящего изопропилового спирта. Объединенные фильтраты упаривают в вакууме досуха. Остаток обрабатывают горячей водой (250-300 мл). Охлаждают, отфильтровывают осадок. Выход соединения (3) после кристаллизации 63 г (62%).
Пример 4
Реакцию проводят аналогично способу, приведенному в примере 3. В качестве восстановителя используют сульфат гидразина (130 г, 1,0 моль). Выход соединения (3) после кристаллизации 62 г (61%).
Пример 5
100 г диамина (2) (0,40 моль) гидрируют водородом в 300 мл метанола в присутствии 1 г катализатора 10% Pd/C при 5-10 атм. и при температуре 30-45° до поглощения теоретически необходимого для образования глиоксима 3 количества водорода. Затем реакционную массу отфильтровывают от катализатора, отгоняют в вакууме 2/3 исходного растворителя и разбавляют 200 мл воды. Осадок отфильтровывают. Выход глиоксима (3) 90-93 г (89-92%).
Пример 6
100 г диамина (2) (0,40 моль) гидрируют водородом в 300 мл метанола в присутствии 5 г скелетного никеля Ренея при 15-25 атм. и при температуре 100-120° до поглощения теоретически необходимого для образования глиок-сима (3) количества водорода. Затем реакционную массу отфильтровывают от катализатора, отгоняют в вакууме 2/3 исходного растворителя и разбавляют 200 мл воды. Осадок отфильтровывают, растворяют в 5%-м растворе гид-роксида натрия, фильтруют, фильтрат подкисляют конц. HCl до рН 3. Выход глиоксима (3) 45-56 г (45-55%).
Пример 7
25,4 г (0.1 моль) диоксима (3) растворяют в 100 мл 10% NaOH (0,25 моль NaOH). Полученный раствор фильтруют в случае необходимости через бумажный фильтр, охлаждают до 5-10°С при интенсивном перемешивании и охлаждении (5-10°С) добавляют по каплям около 12 мл (0,125 моль) уксусного ангидрида до рН 6. Перемешивают еще 30 мин. Осадок отфильтровывают, промывают водой и перекристаллизовывают из метанола. Выход соединения (1) 21 г (90%), tпл. 187°С. ЯМР-1Н, (ацетон-d6), δ(м.д.): 6,45 (2NH2). ЯМР-13С (ацетон-d6) 3 сигнала, δ(м.д.), 135,9(С-3,4); 144,2(С-2,5); 156,0(С-1,6), (для сравнения 13C-NMR (ацетон-d6) соединения (2) содержит 6 сигналов δ(м.д.) 104,6(С-3); 133,5(С-4); 136,3(С-2); 147,0(С-5); 155,3(С-1); 156,3(С-6)). ИК спектр (KBr), см-1: 3460; 3350; 3230; 3010; 2860; 1630; 1540; 1450(сл.); 1410; 1000; 990; 930; 860; 740. Сравнение с ИК спектром соединения (2) показало отсутствие сигналов 1640 см-1 приписываемых фрагменту C=N→O и 1460 см-1 (O-N→O). Масс-спектр, m/e: M 236(57) [M+1] / [М]≈9,5% (рассчитано для C6H4N8O3 9,48%); 206(83), (М-NO); [M+1-30]/[M-30]=9% (рассчитано для C6H4N7O2 9,12%); 179(55) (М - NO - HCN); 176(33), (М - 2NO); 149(100), (М - 2NO - HCN); 96(60); 69(40); 58(98), (H2N-C=N-O); 53(74). Найдено, %: С 30,27; Н 0,04; N 47,64. C6H4N8O3. Вычисл., %: С 30,51; Н 1,69; N 47,45.
Пример 8
25,4 г (0.1 моль) диоксима (3) перемешивают с 40,8 г (0.04 моль) уксусного ангидрида, растворенного в 100 мл уксусной кислоты. Добавляют 2-3 капли H2SO4 конц., смесь перемешивают при 45-50°С 3-6 часов, отгоняют часть уксусной кислоты под пониженным давлением. Остаток разбавляют при перемешивании 100 мл холодной воды. Осадок отфильтровывают, промывают водой и перекристаллизовывают из этанола. Выход N,N'диацетил производного (4), R=СН3 27 г (85%), tпл. 111°C. ЯМР-1H (ДМСО-d6), δ, м.д.: 11,4 (1Н, s, NH); 2,0 (3Н, s, СОСН3). ИК спектр, см-1: 3450 (N-H as); 3320 (N-H s); 1680 см-1 карбонильной группы С=O (I амидная полоса), 3620 см-1, 1480 см-1 (II амидная полоса), 600 см-1 - веерные колебания N-H. Масс-спектр, т/е: [М] - 320(2,4); 278(16); (М - СН2=C=О); 236(6), (М - 2СН2=С=O);
43(100), (СН3СО+).
Пример 9
25,4 г (0.1 моль) диоксима (3) перемешивают с 52,0 г (0.04 моль) пропионового ангидрида, растворенного в 100 мл хлороформа. Добавляют 2-3 капли H2SO4 конц., смесь перемешивают при 45-50°С 3-6 часов, отгоняют хлороформ под пониженным давлением. Остаток разбавляют при перемешивании 100 мл холодной воды. Осадок отфильтровывают, промывают водой и перекристаллизовывают из этанола. Выход N,N'-пропионильного производного (4), R=C2H5 27 г (85%), tпл. Выход 21 г (60%), tпл. 154°C. ЯМР-1H (ДМСО-d6), δ, м.д.: 11.4(s, 1Н, NH); 2.31(q, J=8 Гц, 2Н, СН3); 0.95(t, J=8 Гц, 2H, СН3). ЯМР-13С: 150.7, 144.5, 139.4 (фуразан); 173.3(С=0); 28.74, 9.068 (С2Н5).
Пример 10
К суспензии 25,4 г (0.1 моль) диоксима (3) в 100 мл толуола прибавляют при комнатной температуре при перемешивании 10 мл (16.7 г, 0.01 моль) хлорокиси фосфора. Реакционную массу нагревают до кипения и кипятят 8 часов. Затем упаривают досуха при пониженном давлении, остаток растворяют в кипящем пропаноле-2. Раствор фильтруют горячим. После охлаждения до комнатной температуры осадок отфильтровывают, промывают водой. Выход диамина (1) 15 г (65%).
Пример 11
К суспензии 25,4 г (0.1 моль) диоксима (3) в 100 мл толуола прибавляют при комнатной температуре при перемешивании 15 мл (25 г, 0.21 моль) хлористого тионила. Смесь перемешивают при кипении 16 часов и обрабатывают аналогично примеру 10. Выход диамина (1) 13 г (55%). Пример 12
К 100 мл (167,5 г, 1.1 моль) хлорокиси фосфора присыпают 25,4 г (0.1 моль) диоксима (3) при комнатной температуре при интенсивном перемешивании. Смесь перемешивают при кипении 16 часов и обрабатывают аналогично примеру 10. Выход диамина (1) 10,6 г (45%).
Источники информации
1. В.Г. Андрианов, А.В. Еремеев, Химия гетероцикл. соедин., 1984, 9, 1155.
2. A. Gunasekaran, T. Jayachandran, J.H. Boyer, M.L. Tmdell, J. Heterocyclic Chem., 1995,32, 1405.
3. A.B. Sheremetev, E.V. Mantseva, Mendeleev Commun., 1996, 246.
4. Ф.С. Куликов, Н.Н. Махова, Т.И. Годовикова, С.П. Голова, Л.И. Хмельницкий, Изв. Акад. Наук, сер. хим., 1994, 4, 679.
5. П.В. Анокина, Т.В. Романова, С.Ф. Мельникова, И.В. Целинский. - Журн. Орг. Химии, 2011, 47(10), 1575 - прототип.
6. Nishimura, Shigeo, Handbook of heterogeneous catalytic hydrogenation for organic synthesis. - John Wiley & Sons, Inc., 2001. - 747р. - p.37.
Композиционный материал для иммобилизации жидких радиоактивных отходов и способ его применения
Малочувствительный взрывчатый состав для снаряжения электродетонаторов
3,4-бис(4-нитрофуразан-3-ил)-фуразан
Производные 4н-бис[1,2,5]оксадиазоло[3,4-b:3',4'-f]азепин-8,9-диамина и способ их получения
Способ и устройство для разрушения взрывоопасных предметов
Способ получения 3-фенилпирролидона
Производные 7h(7r)-трис[1,2,5]оксадиазоло[3,4-b:3',4'-d:3",4"-f]азепина и способ их получения
Способ удаления ядерного топлива из контуров исследовательских и энергетических ядерных реакторов
Способ получения амидразонов 4-r-1,2,5-оксадиазол-3-карбоновой кислоты
Способ химической очистки и дезактивации контуров исследовательских и энергетических реакторов, охлаждаемых водой под давлением
Композиционный материал для иммобилизации жидких радиоактивных отходов и способ его применения
Малочувствительный взрывчатый состав для снаряжения электродетонаторов
3,4-бис(4-нитрофуразан-3-ил)-фуразан
Производные 4н-бис[1,2,5]оксадиазоло[3,4-b:3',4'-f]азепин-8,9-диамина и способ их получения
Способ и устройство для разрушения взрывоопасных предметов
Способ получения 3-фенилпирролидона
Производные 7h(7r)-трис[1,2,5]оксадиазоло[3,4-b:3',4'-d:3",4"-f]азепина и способ их получения
Способ удаления ядерного топлива из контуров исследовательских и энергетических ядерных реакторов
Способ получения амидразонов 4-r-1,2,5-оксадиазол-3-карбоновой кислоты
Способ химической очистки и дезактивации контуров исследовательских и энергетических реакторов, охлаждаемых водой под давлением

