Результат интеллектуальной деятельности: ЧАСТИЦЫ N-(5-ЦИАНО-4-((2-МЕТОКСИЭТИЛ)АМИНО)ПИРИДИН-2-ИЛ)-7-ФОРМИЛ-6-((4-МЕТИЛ-2-ОКСОПИПЕРАЗИН-1-ИЛ)МЕТИЛ)-3,4-ДИГИДРО-1,8-НАФТИРИДИН-1(2H)-КАРБОКСАМИДА
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида, способу получения указанных частиц и применению указанных частиц в фармацевтических композициях.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид является селективным и высокоактивным ингибитором FGFR4, который описан в патентной заявке PCT/IB2014/065585. Он является потенциально полезным в лечении заболеваний, опосредованных FGFR4, таких как рак, в частности, таких как рак печени.
В PCT/IB2014/065585 описан способ получения N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в свободной форме и в форме фармацевтически приемлемых солей.
В процессе разработки лекарственных препаратов существует постоянная потребность в разработке новых способов, направленных на улучшение свойств и/или характеристик лекарственной субстанции. Например, лекарственная субстанция должна быть в форме, которая удобна в обработке и преобразовании в готовую лекарственную форму. Такие идеальные свойства лекарственной субстанции должны достигаться способами, которые являются масштабируемыми и воспроизводимыми.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты и способу получения указанных частиц. Частицы могут применяться для получения фармацевтических композиций и применяться в способах лечения, профилактики или облегчения раковых заболеваний. Способ, описанный в настоящем документе, позволяет получать частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида, которые имеют полезные свойства с точки зрения удобства обработки. Частицы обладают улучшенными реологическими свойствами по сравнению с теми, которые получены способом, описанным в PCT/IB2014/065585, как более подробно показано в подробном описании, в экспериментальном разделе и фигурах, приведенных в данном описании.
Различные варианты осуществления изобретения описаны в настоящем документе.
В настоящем документе предоставлен способ получения частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты. Способ включает в себя стадии:
a. Растворения N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в пропионовой кислоте;
b. Добавления лимонной кислоты к раствору, полученному на стадии a), с получением суспензии, содержащей N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты; и
c. Выделения частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты из суспензии, полученной на стадии b.
В варианте осуществления изобретение предоставляет частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, получаемых способом, описанным в настоящем документе.
В настоящем документе также предоставлены частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие медианный размер частиц (x50) от 200 до 300 микрон.
В другом варианте осуществления изобретение предоставляет частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие распределение частиц по размерам x10 от 5 до 10 микрон.
В другом варианте осуществления изобретение предоставляет частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие распределение частиц по размерам x90 от 400 до 500 микрон.
В другом варианте осуществления изобретение предоставляет первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие распределение частиц по размерам x50 от 10 до 20 микрон.
В другом варианте осуществления изобретение предоставляет первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие распределение частиц по размерам x10 от 1 до 5 микрон.
В другом варианте осуществления изобретение предоставляет первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие распределение частиц по размерам x90 от 50 до 70 микрон.
В другом варианте осуществления изобретение предоставляет кристаллические первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие форму столбчатых кристаллов.
В другом варианте осуществления изобретение предоставляет агломерат, содержащий первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, причем агломерат имеет медианный размер (x50) от 300 до 400 микрон.
В другом варианте осуществления изобретение относится к применению частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты согласно настоящему описанию для получения фармацевтической композиции, содержащей N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты и необязательно один или несколько фармацевтически приемлемых носителей(носитель).
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты согласно настоящему описанию и необязательно один или несколько фармацевтически приемлемых носителей(носитель).
В другом варианте осуществления фармацевтическая композиция согласно настоящему описанию предназначена для применения в качестве лекарственного средства. В конкретном варианте осуществления она предназначена для лечения рака. Более конкретно, она предназначена для применения в лечении показания, выбранного из рака печени, рака молочной железы, глиобластомы, рака предстательной железы, рабдомиосаркомы, рака желудка, рака яичников, рака легких, рака толстой кишки. Еще более конкретно, она предназначена для применения в лечении рака печени.
В варианте осуществления изобретение относится к способу лечения рака, включающему введение субъекту терапевтически эффективного количества фармацевтической композиции согласно настоящему описанию.
В варианте осуществления изобретение относится к применению фармацевтической композиции согласно настоящему описанию для получения лекарственного средства для лечения рака. Более конкретно, оно предназначено для лечения показания, выбранного из рака печени, рака молочной железы, глиобластомы, рака предстательной железы, рабдомиосаркомы, рака желудка, рака яичников, рака легких, рака толстой кишки. Еще более конкретно, оно предназначено для лечения рака печени.
Краткое описание фигур
Фигура 1 представляет собой полученное с помощью растрового электронного микроскопа (РЭМ) изображение частиц, полученных с использованием способа по изобретению (масштаб 20 микрон). Изображение было получено с использованием устройства РЭМ от Zeiss, оборудованного микроскопом Supra 40. Оно демонстрирует первичную частицу, имеющую форму столбчатых кристаллов.
Фигура 2 представляет собой полученный с помощью растрового электронного микроскопа (РЭМ) снимок частиц, полученных с использованием способа, описанного в PCT/IB2014/065585 (масштаб 20 микрон). Изображение было получено с использованием устройства РЭМ от Zeiss, оборудованного микроскопом Supra 40. Оно демонстрирует скопления первичных частиц игольчатой формы.
Фигура 3 представляет собой полученный с помощью растрового электронного микроскопа (РЭМ) снимок частиц, полученных с использованием способа по изобретению (масштаб 100 микрон). Изображение было получено с использованием устройства РЭМ от Zeiss, оборудованного микроскопом Supra 40. На фигуре показан агломерат первичных частиц. В агломерате видны первичные частицы, имеющие форму столбчатых кристаллов.
Фигура 4 представляет собой полученный с помощью растрового электронного микроскопа (РЭМ) снимок частиц, полученных с использованием способа по изобретению (масштаб 500 микрон). Изображение было получено с использованием устройства РЭМ от Zeiss, оборудованного микроскопом Supra 40. На фигуре показаны агломераты первичных частиц.
На фигуре 5 показано наложение XRPD частиц, полученных с использованием способа по изобретению, и XRPD частиц, полученных с использованием способа, описанного в PCT/IB2014/065585. Наложение показывает один и тот же характеристический профиль как в случае частиц по изобретению, так и в случае частиц, полученных с использованием способа, описанного в PCT/IB2014/065585.
На фигуре 6 показано распределение частиц по размерам для частиц, полученных с использованием способа по изобретению. На фигуре показаны плотность распределения и кривые кумулятивного распределения в зависимости от размера частиц. Она демонстрирует, что частицы содержат смесь первичных частиц, имеющих размер частиц в пределах от 0,5 до 150 микрон, и агломераты, имеющие размер частиц в пределах от 150 до 875 микрон.
На фигуре 7 показано распределение частиц по размерам для частиц, полученных с использованием способа, описанного в PCT/IB2014/065585. На фигуре показаны плотность распределения и кривые кумулятивного распределения в зависимости от размера частиц. Она демонстрирует, что частицы имеют размер частиц в пределах приблизительно от 0,5 до 30 микрон.
Фигура 8 представляет собой РЭМ (200 микрон) частиц, полученных способом по изобретению на стадиях а) и b), то есть до стадии выделения. Изображение было получено с помощью микроскопа Olympus BX51 (серийный номер SN8G30637), оснащенного камерой DP25 (серийный номер SN8K08782). Программным обеспечением, используемым для создания изображений, является StreamStart. На изображении показаны кристаллические частицы, имеющие форму столбчатых кристаллов.
На фигуре 9 показана энергия основного потока частиц по изобретению согласно примерам 1 и 1а, и частиц, полученных с использованием способа, описанного в PCT/IB2014/065585 (ссылочный пример 1). График показывает, что частицы по изобретению требуют меньше энергии для течения по сравнению с теми, которые получены способом, описанным в PCT/IB2014/065585 (ссылочный пример 1), что указывает на улучшенную текучесть.
На фигуре 10 показан процент сжимаемости в зависимости от нормального напряжения, приложенного к частицам по изобретению согласно примерам 1 и 1а, и к частицам, полученным с использованием способа, описанного в PCT/IB2014/065585 (ссылочный пример 1). График показывает, что частицы по изобретению имеют более высокий процент сжимаемости, что указывает на то, что более высокое содержание лекарственного средства в конечной готовой лекарственной форме может быть достигнуто с использованием частиц по изобретению.
На фигуре 11 показаны графические изображения зависимости касательного напряжения от нормального напряжения, приложенного к частицам по изобретению согласно примерам 1 и 1а, и к частицам, полученным с использованием способа, описанного в PCT/IB2014/065585 (ссылочный пример 1). Это позволяет рассчитать угол трения о стенки. График показывает, что частицы по изобретению имеют меньший угол трения о стенки по сравнению с теми, которые получены с использованием способа, описанного в PCT/IB2014/065585 (ссылочный пример 1), указывая на то, что частицы по изобретению в меньшей степени проявляют липкие свойства на металлических поверхностях и, таким образом, имеют улучшенную обрабатываемость.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
При определении концентрации соединения в растворе термин "%" относится к % масс./масс., то есть к массе соединения относительно массы раствора (то есть соединение+растворитель).
Если не указано иное, термин "частицы" во множественном числе в контексте настоящего описания относится к основной части частиц. Если не указано иное, термин "кристаллы" во множественном числе в контексте настоящего описания относится к основной части кристаллов.
Термин "частицы" предусматривает охватывание первичных частиц и агломератов.
В контексте настоящего описания термин "первичная частица" относится к сингулярной структурной единице. Первичные частицы по изобретению можно видеть, например, на фигурах 1 и 8.
В контексте настоящего описания термин "агломерат" относится к кластеру первичных частиц. В контексте настоящего описания термин "агломерат" не предназначен для ограничения природы связей между первичными частицами и может взаимозаменяемо использоваться, например, с термином "агрегаты".
Размер частиц определяется кумулятивным гранулометрическим распределением подситных фракций частиц по объему, измеренным с помощью лазерной дифракции света. Например, значение медианного размера частиц (x50 или d50) указывает на размер, который имеют 50% масс. частиц в образце. Например, значение x50 10 микрон указывает на то, что 50% масс. частиц имеют размер менее 10 микрон.
Аналогично, размер частиц x10 или d10 указывает на размер, который имеют 10% масс. частиц в образце.
Аналогично, размер частиц x90 или d90 указывает на размер, который имеют 90% масс. частиц в образце.
Понятно, что гранулометрический состав, указанный в настоящем описании, может отличаться в зависимости от используемого устройства и способа. Специалист должен понимать, что эти значения не являются абсолютными значениями и могут отличаться на +/- 10%.
В настоящем описании гранулометрический состав измеряют с помощью лазерной дифракции света. Устройство, используемое для лазерной дифракции света, представляет собой устройство Sympatec Helos. Подробная информация об используемом устройстве и измерениях приведена в примерах 3 и 4.
В контексте настоящего описания обозначение "микроны" соответствует микронам.
В контексте настоящего описания термин "столбчатый", когда речь идет о кристаллической форме частиц по изобретению, описывает форму кристалла, как показано, например, на фигуре 1. "Столбчатая" кристаллическая форма представляет собой многогранный трехмерный выращенный кристалл. Форма столбчатого кристалла также может быть определена как плосковытянутая. Она отличается от игольчатой формы кристалла (или игловидного кристалла), где рост кристалла является однонаправленным.
В контексте настоящего описания термин "фармацевтически приемлемый носитель" включает любые и все растворители, дисперсионные среды, покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные средства, противогрибковые средства), изотонические агенты, агенты, замедляющие абсорбцию, соли, консерванты, стабилизаторы лекарственных средств, связующие вещества, эксципиенты, дезинтегрирующие агенты, смазывающие вещества, подсластители, ароматизаторы, красители и т.п. и их комбинации, которые могут быть известны специалистам в данной области (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). За исключением случаев, когда какой-либо обычный носитель несовместим с активным ингредиентом, тогда возможность его применения в терапевтических или фармацевтических композициях рассматривается.
Термин "терапевтически эффективное количество" соединения по настоящему изобретению относится к количеству соединения по настоящему изобретению, которое будет вызывать биологический или медицинский ответ субъекта, например, снижение или ингибирование активности фермента или белка или уменьшение интенсивности симптомов, облегчение состояния, замедление или приостановка прогрессирования заболевания или предотвращение заболевания и т.д. В одном неограничивающем варианте осуществления термин "терапевтически эффективное количество" относится к количеству соединения по настоящему изобретению, которое при введении субъекту является эффективным для (1) по меньшей мере частичного облегчения, ингибирования, предотвращения и/или купирования состояния или расстройства, или заболевания, (i) опосредованного FGFR4 или (ii) связанного с активностью FGFR4, или (iii) характеризующегося активностью (нормальной или аномальной) FGFR4, или (2) уменьшения или ингибирования активности FGFR4; или (3) уменьшения или ингибирования экспрессии FGFR4. В другом неограничивающем варианте осуществления термин "терапевтически эффективное количество" относится к количеству соединения по настоящему изобретению, которое при введении в клетку или ткань или неклеточный биологический материал или среду является эффективным по меньшей мере для частичного уменьшения или ингибирования активности FGFR4.
В контексте настоящего описания термин "субъект" относится к животному. Обычно животное является млекопитающим. Субъект также относится, например, к приматам (например, людям, мужчинам или женщинам), коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам, птицам и тому подобному. В некоторых вариантах осуществления субъект является приматом. В других вариантах осуществления субъект является человеком.
В контексте настоящего описания термин "ингибировать", "ингибирование" или "ингибирующий" относится к снижению выраженности или подавлению данного состояния, симптома или расстройства, или заболевания, или значительному уменьшению исходного уровня биологической активности или активности процесса.
В контексте настоящего описания термин "лечить", "лечащий" или "лечение" какого-либо заболевания или расстройства относится в одном варианте осуществления к облегчению заболевания или расстройства (т.е. замедлению или остановке, или подавлению развития заболевания или по меньшей мере одного из клинических симптомов). В другом варианте осуществления "лечить", "лечащий" или "лечение" относится к облегчению или уменьшению интенсивности по меньшей мере одного физического параметра, включая те, что могут быть не различимы пациентом. В еще одном варианте осуществления "лечить", "лечащий" или "лечение" относится к модулированию заболевания или расстройства либо физически (например, стабилизация заметного симптома), физиологически (например, стабилизация физического параметра), либо и так и так одновременно. В еще одном варианте осуществления "лечить", "лечащий" или "лечение" относится к предотвращению или задержке начала или развития, или прогрессирования заболевания или расстройства.
В контексте настоящего описания субъект является "нуждающимся" в лечении, если такой субъект получит пользу биологически, с медицинской точки зрения или в отношении качества жизни от такого лечения.
Используемый в оригинальном тексте настоящего документа термин "какой-либо", "какой-нибудь", "этот" и подобные термины, используемые в контексте настоящего изобретения (особенно в контексте формулы изобретения), следует считать охватывающими как единственное, так и множественное число, если не указано иное или если иное прямо не противоречит контексту.
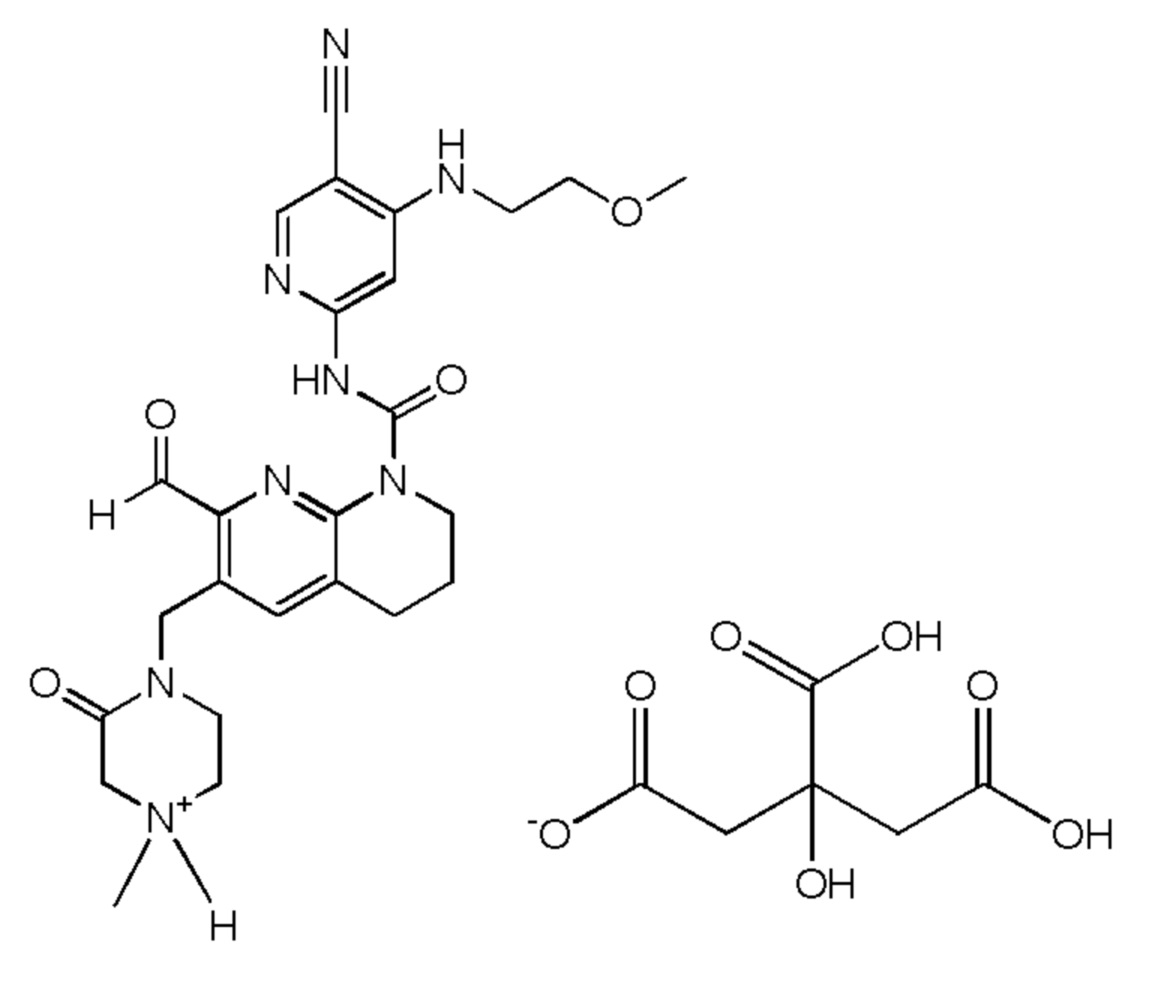
Изобретение относится к способу получения частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты. N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты имеет следующую структуру:
Способ включает стадию а) растворения N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в свободной форме в пропионовой кислоте. N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в свободной форме может быть получен способом, описанным, например, в примере 1 (стадии с 1 по 14). Концентрация N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в свободной форме в пропионовой кислоте может составлять от 5 до 15% масс., предпочтительно 5-15%. В одном варианте осуществления концентрация N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в свободной форме в пропионовой кислоте составляет приблизительно 5-10% масс. В одном варианте осуществления концентрация N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в свободной форме в пропионовой кислоте составляет приблизительно 10-15% масс. В одном варианте осуществления она составляет приблизительно 12% масс. В одном варианте осуществления она составляет приблизительно 13% масс. Пропионовая кислота, используемая в настоящем способе, предпочтительно является беспримесной. Под "беспримесной" подразумевается, что пропионовая кислота по меньшей мере на 99% является чистой. Предпочтительно пропионовая кислота по меньшей мере на 99,5% является чистой. Как правило, температура, используемая для растворения N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в свободной форме в пропионовой кислоте, составляет от 50 до 80°C, предпочтительно около 70°C. Время, необходимое для полного растворения, зависит от используемой температуры. Как правило, при температуре около 70°C требуемое время составляет от 10 до 30 минут, предпочтительно около 20 минут. Полное растворение может быть легко определено квалифицированным специалистом, например, путем визуального осмотра.
На стадии b) способа по изобретению к раствору, полученному на стадии а), добавляют лимонную кислоту. Лимонную кислоту можно добавлять в виде твердого вещества, в виде раствора или в виде суспензии. При добавлении в виде раствора или суспензии концентрация может варьироваться от 1 до 50% масс. Как правило, ее добавляют в виде раствора или суспензии. Растворитель, используемый для раствора лимонной кислоты, обычно представляет собой растворитель, в котором N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты плохо растворим. Такой растворитель может быть выбран, например, из гептана, метил-трет-бутилового эфира, н-гексана, этилацетата, н-пропилацетата, ацетона, ацетонитрила, толуола, воды или их смесей. В варианте осуществления растворитель, используемый для раствора/суспензии лимонной кислоты, представляет собой ацетон. В варианте осуществления растворитель, используемый для раствора/суспензии лимонной кислоты, представляет собой этилацетат. В варианте осуществления лимонную кислоту добавляют к раствору, полученному на стадии а), в виде раствора в ацетоне. В варианте осуществления концентрация лимонной кислоты в ацетоне составляет от 1 до 50%, предпочтительно от 20 до 30%, более предпочтительно около 23%. В другом варианте осуществления концентрация лимонной кислоты в ацетоне составляет от 1 до 10%, предпочтительно от 1 до 5%, более предпочтительно около 2,5%.
В варианте осуществления лимонную кислоту добавляют к раствору, полученному на стадии а), в виде раствора в этилацетате. В варианте осуществления концентрация лимонной кислоты в этилацетате составляет от 1 до 10%, предпочтительно от 1 до 5%, более предпочтительно около 1,3%.
Количество добавленной лимонной кислоты при осуществлении способа зависит от желаемой стехиометрии N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты. В предпочтительном варианте осуществления желаемая стехиометрия N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты составляет 1:1 (лимонная кислота:N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид). В другом варианте осуществления желаемая стехиометрия N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты составляет 0,5:1 (лимонная кислота:N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид). Специалист в данной области может определить количество лимонной кислоты, необходимое для получения желаемой стехиометрии. Требуемое количество лимонной кислоты может быть добавлено за один раз или несколькими аликвотами. В варианте осуществления лимонную кислоту добавляют за один раз. В другом варианте осуществления лимонную кислоту добавляют несколькими аликвотами. Каждая аликвота может отличаться в плане формы или может быть неизменной. Например, в одной аликвоте лимонная кислота может быть добавлена в твердой форме. В другой аликвоте лимонная кислота может быть добавлена в виде раствора. В другой аликвоте лимонная кислота может быть добавлена в виде суспензии. Каждая аликвота лимонной кислоты может иметь одинаковую концентрацию или разную концентрацию. Растворитель, используемый для каждой аликвоты лимонной кислоты при добавлении в виде раствора или суспензии, может быть одинаковым или различным. Аликвоты могут быть добавлены в течение периода времени от 1 секунды до 10 часов. Температура, при которой добавляют лимонную кислоту, может варьироваться от 20 до 80°C. Предпочтительно температура, при которой добавляют лимонную кислоту, составляет около 55°C. При добавлении лимонной кислоты образуется суспензия, содержащая N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты.
В варианте осуществления присутствует дополнительная стадия введения затравки в суспензию, полученную на стадии b), с помощью затравочной суспензии, содержащей N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты. Затравочная суспензия содержит частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, полученные способом по изобретению. Предпочтительно частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, используемые в затравочной суспензии, имеют стехиометрию 1:1 (лимонная кислота:N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид). Для затравочной суспензии частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты суспендируют в растворителе, таком как ацетон. Затравочная суспензия может быть добавлена между добавками аликвот лимонной кислоты или после того, как к реакционной смеси было добавлено общее требуемое количество лимонной кислоты. Затравочная суспензия обычно содержит приблизительно 0,005-5%, предпочтительно 0,01-1% частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, полученных способом по изобретению. В варианте осуществления затравочная суспензия содержит 0,5% частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, полученных способом по изобретению. Температура затравочной суспензии обычно составляет около 20°C.
Суспензию, полученную на стадии b) способа по изобретению, обычно перемешивают в течение периода времени, который позволяет N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамиду в форме соли лимонной кислоты выкристаллизоваться из раствора. Как правило, суспензия может перемешиваться в течение от 1 минуты до 20 часов. Температура полученной суспензии может быть снижена приблизительно до 20°C, если предыдущие стадии были проведены при температурах выше 20°C. На фигуре 8 показано полученное с помощью микроскопа изображение первичных частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, полученных после стадии b). На фигуре 8 первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты находятся в суспензии в реакционной среде.
Стадия c) в способе по изобретению включает выделение частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты из суспензии, полученной на предыдущей стадии b). Это обычно осуществляют путем фильтрации, промывки и сушки. Таким образом, в дополнительном варианте осуществления стадия c) в способе по изобретению включает:
Стадию c1) фильтрации суспензии, полученной на стадии b), с получением частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты;
Стадию c2) промывки частиц, полученных после стадии фильтрации c1); и
Стадию c3) сушки частиц, полученных на стадии c2).
Фильтрация может быть осуществлена с использованием стандартных методов и устройств. Например, фильтрация может быть осуществлена с использованием стеклянного фильтра Шотта. Фильтрация может быть осуществлена в вакууме, например, до 500 мбар. Фильтрация может быть осуществлена при комнатной температуре. Фильтрация может быть осуществлена в атмосфере азота.
Промывка может быть осуществлена с помощью одной системы растворителей или с помощью нескольких систем растворителей. Например, в одном варианте осуществления используемая система растворителей может представлять собой смесь 1:1 пропионовой кислоты и ацетона при комнатной температуре. В другом варианте осуществления используемая система растворителей также может представлять собой смесь 1:1 пропионовой кислоты и этилацетата при комнатной температуре. В одном варианте осуществления используемая система растворителей также может представлять собой ацетон в чистом виде при комнатной температуре. В одном варианте осуществления первая стадия промывки осуществляется смесью 1:1 пропионовой кислоты и ацетона при комнатной температуре с последующей второй стадией промывки ацетоном в чистом виде при комнатной температуре. В одном варианте осуществления первая стадия промывки осуществляется смесью 1:1 пропионовой кислоты и этилацетата при комнатной температуре с последующей второй стадией промывки этилацетатом в чистом виде при комнатной температуре.
Как правило, сушку осуществляют с использованием стандартных методов. Например, полученные частицы могут быть высушены в печи, необязательно в вакууме, например, приблизительно при 5 мбар. Сушка может быть осуществлена в атмосфере азота. Обычно сушку осуществляют при температуре около 40°C.
Изобретение охватывает в варианте осуществления частицы, получаемые способом по изобретению, описанным в настоящем документе.
Гранулометрический состав частиц по настоящему изобретению измеряют с использованием лазерной дифракции света, как описано в примере 3, и поэтому он основан на полученном таким образом средневзвешенном распределении частиц по размерам. Распределение частиц по размерам представлено кривой кумулятивного распределения (подситных фракций) (Q3(x)%) и кривой распределения плотности (q3lg(x)), как показано, например, на фигуре 6.
Таким образом, в варианте осуществления изобретение относится к частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющим распределение частиц по размерам по существу такое же, как показано на фигуре 6. В варианте осуществления изобретение относится к частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющим кривую кумулятивного распределения подситных фракций по существу такую же, как показано на фигуре 6. В варианте осуществления изобретение относится к частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющим кривую распределения плотности по существу такую же, как показано на фигуре 6.
В настоящем изобретении размеры частиц, включая значения x10, x50 и x90, предпочтительно измеряют с помощью лазерной дифракции света.
Как можно видеть в графике распределения частиц по размерам, показанном на фигуре 6, частицы, полученные с использованием способа по изобретению, имеют кумулятивное гранулометрическое распределение подситных фракций частиц по объему, имеющее следующие характеристические значения:
значение x10 от 5 до 10 микрон;
значение x50 от 200 до 300 микрон;
значение x90 от 400 до 500 микрон.
Таким образом, в варианте осуществления изобретение относится к частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющим значение x10 от 5 до 10 микрон. Предпочтительно частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты имеют значение x10 около 7 микрон. В варианте осуществления значение x10 может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты имеют значение x10 от 4,5 до 11 микрон. Для сравнения, N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты, полученный способом, описанным в PCT/IB2014/065585 (ссылочный пример 1), имеет размер частиц x10 или d10 около 1 микрон (см. пример 4 и фигуру 7).
В варианте осуществления изобретение относится к частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющим значение х50 от 200 до 300 микрон. Предпочтительно частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты имеют значение x50 около 265 микрон. В варианте осуществления значение x50 может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты имеют значение x50 от 180 до 330 микрон. Для сравнения, N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты, полученный способом, описанным в PCT/IB2014/065585 (ссылочный пример 1), имеет медианный размер частиц (x50 или d50) около 3 микрон (см. пример 4 и фигуру 7).
В варианте осуществления изобретение относится к частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющим значение х90 от 400 до 500 микрон. Предпочтительно частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты имеют значение x90 около 450 микрон. В варианте осуществления значение x10 может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты имеют значение x90 от 360 до 550 микрон. Для сравнения, N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты, полученной способом, описанным в PCT/IB2014/065585 (ссылочный пример 1), имеет размер частиц x90 или d90 около 7 микрон (см. пример 4 и фигуру 7).
Кроме того, как можно видеть из графика, показанного на фигуре 6, частицы, полученные способом по изобретению, могут быть разграничены на первичные частицы, имеющие размер частиц от 0,5 до 150 микрон, и агломераты, имеющие размер частиц от 150 до 875 микрон.
Таким образом, в варианте осуществления частицы по изобретению содержат смесь первичных частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты и агломераты первичных частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты.
Первичные частицы могут быть отделены от агломератов способами, известными специалисту в данной области, такими как просеивание, например.
В варианте осуществления настоящее изобретение относится к первичным частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, получаемым способом по изобретению, описанным в настоящем документе.
В варианте осуществления изобретение относится к первичным частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющим диапазон размеров частиц от 0,5 до 150 микрон.
В варианте осуществления изобретение относится к первичным частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющим медианный размер частиц х50 от 10 до 20 микрон, предпочтительно около 15 микрон (см. пример 3 и фигуру 6). В варианте осуществления значение x50 может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты имеют медианный размер частиц х50 от 9 до 22 микрон.
В варианте осуществления изобретение относится к первичным частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющим размер частиц x10 от 1 до 5 микрон, предпочтительно около 3 микрон (см. пример 3 и фигуру 6). В варианте осуществления значение x10 может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты имеют значение x10 от 0,9 до 5,5 микрон.
В варианте осуществления изобретение относится к частицам N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющим размер частиц х90 от 50 до 70 микрон, предпочтительно около 60 микрон (см. пример 3 и фигуру 6). В варианте осуществления значение x90 может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты имеют размер частиц х90 от 45 до 77 микрон.
Различие в размерах частиц между первичными частицами по настоящему изобретению и теми, которые получены способом, описанным в PCT/IB2014/065585, также можно видеть на полученных с помощью растровой электронной микроскопии изображениях, показанных на фигуре 1 и фигуре 2, соответственно.
В варианте осуществления первичные частицы по настоящему изобретению имеют форму столбчатых кристаллов. Это можно видеть на фигуре 1 и фигуре 8.
В варианте осуществления изобретение относится к кристаллической первичной частице N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющей форму столбчатого кристалла. Форма столбчатого кристалла изображена, например, на фигуре 1. Кристаллическая форма первичных частиц по настоящему изобретению отличается от кристаллической формы частиц, полученных способом, описанным в PCT/IB2014/065585, и показанной на фигуре 2.
Интересно, что полиморфная форма, полученная способом по изобретению или способом, описанным в PCT/IB2014/065585, с образованием N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, является идентичной, как можно видеть на фигуре 5.
Изобретение также относится, в другом аспекте, к агломерату, содержащему первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты согласно настоящему описанию. Агломераты по изобретению можно видеть на фигурах 3 и 4.
Поэтому в варианте осуществления изобретение относится к агломерату, содержащему первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, причем агломерат может быть получен способом по изобретению, описанным в настоящем документе.
В другом варианте осуществления изобретение относится к агломерату, содержащему первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, причем первичные частицы имеют медианный размер частиц (х50 или d50) от 10 до 20 микрон, предпочтительно около 15 микрон. В варианте осуществления значение x50 первичных частиц может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления изобретение относится к агломерату, содержащему первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие медианный размер частиц х50 от 9 до 22 микрон. Сам агломерат имеет медианный размер (x50 или d50) от 300 до 400 микрон, предпочтительно около 340 микрон. В варианте осуществления значение x50 агломерата может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления агломерат, содержащий первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеет медианный размер х50 от 270 до 440 микрон.
В другом варианте осуществления изобретение относится к агломерату, содержащему первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, причем первичные частицы имеют размер частиц (x10 или d10) от 1 до 5 микрон, предпочтительно около 3 микрон. В варианте осуществления значение x10 первичных частиц может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления изобретение относится к агломерату, содержащему первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие значение x10 от 0,9 до 5,5 микрон. Сам агломерат имеет размер x10 или d10 от 200 до 300 микрон, предпочтительно около 230 микрон. В варианте осуществления значение x10 агломерата может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления агломерат, содержащий первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеет размер x10 от 180 до 330 микрон.
В другом варианте осуществления изобретение относится к агломерату, содержащему первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие размер частиц x90 или d90 от 50 до 70 микрон, предпочтительно около 60 микрон. В варианте осуществления значение x90 первичных частиц может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления изобретение относится к агломерату, содержащему первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеющие размер частиц х90 от 45 до 77 микрон.
Сам агломерат имеет размер x90 или d90 от 450 до 550 микрон, предпочтительно около 490 микрон. В варианте осуществления значение x90 агломерата может варьироваться в пределах +/- 10%. Таким образом, в варианте осуществления агломерат, содержащий первичные частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты, имеет размер х90 от 405 до 605 микрон.
Без ограничения теорией, считается, что агломераты образуются при сушке первичных частиц, полученных на стадии b) способа по изобретению. Образование агломерата может быть обусловлено остаточным растворителем на поверхности первичных частиц, который может индуцировать образование агломерата.
Описанные в настоящем документе частицы могут применяться для получения фармацевтической композиции. Таким образом, в одном варианте осуществления изобретение относится к фармацевтической композиции, содержащей частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты согласно настоящему описанию и необязательно один или несколько фармацевтически приемлемых носителей(носитель).
В варианте осуществления фармацевтическая композиция содержит частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты и не содержит никакого фармацевтически приемлемого носителя(носителей). В варианте осуществления фармацевтическая композиция содержит терапевтически эффективное количество частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты и один или несколько фармацевтически приемлемых носителей (носитель).
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты и фармацевтически приемлемый носитель. В дополнительном варианте осуществления композиция содержит по меньшей мере два фармацевтически приемлемых носителя, таких как те, что описаны в настоящем документе. Для целей настоящего изобретения, если не указано иное, сольваты и гидраты обычно считаются композициями. Предпочтительно фармацевтически приемлемые носители являются стерильными. Фармацевтическая композиция может быть составлена для конкретных путей введения, таких как пероральное введение, парентеральное введение и ректальное введение, и т.д. Кроме того, фармацевтические композиции по настоящему изобретению могут быть приготовлены в твердой форме (включая, без ограничения, капсулы, таблетки, пилюли, гранулы, порошки или суппозитории) или в жидкой форме (включая, без ограничений, растворы, суспензии или эмульсии). Фармацевтические композиции могут быть подвергнуты обычным фармацевтическим операциям, таким как стерилизация, и/или могут содержать обычные инертные разбавители, смазывающие агенты или буферные агенты, а также адъюванты, такие как консерванты, стабилизаторы, смачивающие агенты, эмульгаторы и буферы, и т.д.
Как правило, фармацевтические композиции представляют собой таблетки или желатиновые капсулы, содержащие активный ингредиент вместе с одним или несколькими из:
a) разбавителей, например, лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином;
b) смазывающих веществ, например, диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем;
c) связующих веществ, например, алюмосиликатом магния, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, натрийкарбоксиметилцеллюлозой и/или поливинилпирролидоном;
d) разрыхлителей, например, крахмалами, агаром, альгиновой кислотой или ее натриевой солью или шипучими смесями; и
e) абсорбентов, красителей, ароматизаторов и подсластителей.
В варианте осуществления фармацевтическая композиция находится в форме капсулы, содержащей только активный ингредиент. В другом варианте осуществления фармацевтическая композиция находится в форме капсулы, содержащей смесь активного ингредиента и эксципиентов. Таблетки могут быть покрыты пленкой или энтеросолюбильным покрытием в соответствии со способами, известными в данной области техники.
Подходящие композиции для перорального введения включают эффективное количество соединения по изобретению в форме таблеток, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсии, твердых или мягких капсул или сиропов или эликсиров, растворов или твердой дисперсии. Композиции, предназначенные для перорального применения, получают в соответствии с любым способом, известным в данной области, для изготовления фармацевтических композиций, и такие композиции могут содержать один или несколько агентов, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей и консервантов для того, чтобы предоставить фармацевтически элегантные и приятные препараты. Таблетки могут содержать активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые пригодны для изготовления таблеток. Этими эксципиентами являются, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие агенты, например, кукурузный крахмал или альгиновая кислота; связующие агенты, например, крахмал, желатин или аравийская камедь; и смазывающие агенты, например, стеарат магния, стеариновая кислота или тальк. Таблетки не покрывают оболочкой или покрывают известными способами для задержки распада и абсорбции в желудочно-кишечном тракте и, таким образом, обеспечивают пролонгированное действие в течение более длительного периода. Например, может быть использовано вещество, задерживающее высвобождение, такое как глицерилмоностеарат или глицерилдистеарат. Препараты для перорального применения могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешивают с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешивают с водой или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Частицы по изобретению обладают полезными свойствами с точки зрения удобства обработки. Действительно, свойства потока частиц по изобретению улучшены по сравнению со свойствами потока частиц, полученных с использованием способа, описанного в PCT/IB2014/065585 (ссылочный пример 1).
Свойства потока можно количественно определить с помощью реометрии порошка. Реометрия порошка позволяет измерять ряд параметров на лекарственной субстанции, таких как энергия основного потока, процент сжимаемости, угол трения о стенки. Как можно видеть на фигуре 9, частицы по изобретению по меньшей мере значительно отличаются в энергии основного потока по сравнению с теми, которые получены с использованием способа, описанного в PCT/IB2014/065585. Этот факт говорит о том, что частицы по изобретению обладают преимуществами в отношении удобства обработки. Кроме того, как можно видеть на фигуре 10, процент сжимаемости увеличивается с использованием частиц по настоящему изобретению по сравнению с теми, которые получены способом, описанным в PCT/IB2014/065585. Этот факт говорит о том, что содержание лекарственного средства может быть повышено при производстве готовых лекарственных форм.
Кроме того, как можно видеть на фигуре 11, угол трения о стенки различается между частицами по настоящему изобретению и теми, которые получены способом, описанным в PCT/IB2014/065585. Этот факт указывает на то, что поверхностные характеристики частиц являются различными, и свидетельствует о том, что частицы по настоящему изобретению в меньшей степени проявляют липкие свойства (меньшее взаимодействие с нержавеющей сталью). Этот факт, в свою очередь, также дает преимущества с точки зрения удобства обработки при использовании частиц по настоящему изобретению.
Использование частиц по настоящему изобретению облегчает изготовление готовых лекарственных форм, например заполнение капсул, что дает более высокие выходы (по сравнению с использованием частиц, полученных способом, описанным в PCT/IB2014/065585). Кроме того, с использованием частиц по настоящему изобретению существует возможность уменьшить количество эксципиентов, необходимых для производства готовых лекарственных форм, что дает преимущества с точки зрения стоимости, времени и эффективности способа. Действительно, если лекарственная субстанция является липкой или легко не течет, для улучшения обработки указанной лекарственной субстанции может потребоваться большее количество эксципиентов.
Фармацевтическая композиция по настоящему изобретению может быть в виде единичных доз, соответствующих приблизительно 1-1000 мг активного ингредиента для субъекта приблизительно 50-70 кг, или приблизительно 1-500 мг, или приблизительно 1-250 мг, или приблизительно 1-150 мг, или приблизительно 0,5-100 мг, или приблизительно 1-50 мг активного ингредиента. В одном варианте осуществления она составляет около 50 мг активного ингредиента. В другом варианте осуществления она составляет около 100 мг активного ингредиента. В другом варианте осуществления она составляет около 200 мг активного ингредиента. В другом варианте осуществления она составляет около 300 мг активного ингредиента. Терапевтически эффективная доза фармацевтической композиции зависит от вида субъекта, массы тела, возраста и индивидуального состояния, расстройства или заболевания, подлежащего лечению, или его степени тяжести. Врач, клиницист или ветеринар обычной квалификации может легко определить эффективное количество активного ингредиента, необходимое для профилактики, лечения или ингибирования развития расстройства или заболевания.
Активность N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида показана в примере 2.
Принимая во внимание активность N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в качестве ингибитора FGFR4, фармацевтическая композиция по изобретению может быть полезна в лечении состояний, которые опосредованы активностью белков FGFR4, таких как рак, и/или которые восприимчивы (что особенно важно для терапевтически благоприятного пути) к ингибированию FGFR4, в особенности заболеваний или расстройств, указанных ниже.
Фармацевтическая композиция по изобретению может быть полезна в лечении рака. В частности, фармацевтическая композиция по изобретению может быть полезна в лечении показаний, выбранных из рака печени, рака молочной железы, глиобластомы, рака предстательной железы, рабдомиосаркомы, рака желудка, рака яичников, рака легких, рака толстой кишки.
Таким образом, в качестве дополнительного варианта осуществления настоящее изобретение предоставляет применение фармацевтической композиции, содержащей частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты согласно настоящему описанию, в терапии. В дополнительном варианте осуществления терапию выбирают на основании заболевания, которое можно лечить путем ингибирования FGFR4. В другом варианте осуществления заболевание выбирают из вышеупомянутого списка, предпочтительно рака печени.
Таким образом, в качестве дополнительного варианта осуществления настоящее изобретение предоставляет фармацевтическую композицию, содержащую частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты согласно настоящему описанию, для применения в терапии. В дополнительном варианте осуществления терапия предназначена для заболевания, которое можно лечить путем ингибирования FGFR4. В другом варианте осуществления заболевание выбирают из вышеупомянутого списка, предпочтительно рака печени.
В другом варианте осуществления настоящее изобретение предоставляет способ лечения заболевания, которое лечится путем ингибирования FGFR4, включающий введение терапевтически эффективного количества фармацевтической композиции, содержащей частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты согласно настоящему описанию. В дополнительном варианте осуществления заболевание выбирают из вышеупомянутого списка, предпочтительно рака печени.
Таким образом, в качестве дополнительного варианта осуществления настоящее изобретение предоставляет применение частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты для получения лекарственного средства. В дополнительном варианте осуществления лекарственное средство предназначено для лечения заболевания, которое можно лечить путем ингибирования FGFR4. В другом варианте осуществления заболевание выбирают из вышеупомянутого списка, предпочтительно рака печени.
Фармацевтическая композиция по изобретению может также быть полезна в лечении твердых злокачественных новообразований, характеризующихся положительной экспрессией FGFR4.
Фармацевтическая композиция по изобретению может также быть полезна в лечении твердых злокачественных новообразований, характеризующихся положительной экспрессией KLB (бета-клото).
Фармацевтическая композиция по изобретению может также быть полезна в лечении твердых злокачественных новообразований, характеризующихся положительной экспрессией FGF19.
Фармацевтическая композиция по изобретению может также быть полезна в лечении твердых злокачественных новообразований, характеризующихся положительной FGFR4- и положительной KLB-экспрессией.
Фармацевтическая композиция по изобретению может также быть полезна в лечении твердых злокачественных новообразований, характеризующихся положительной FGFR4- и положительной FGF19-экспрессией.
Фармацевтическая композиция по изобретению может также быть полезна в лечении твердых злокачественных новообразований, характеризующихся положительной FGFR4-, положительной KLB- и положительной FGF19-экспрессией.
Любая положительная экспрессия в FGFR4, KLB и/или FGF19, как описано выше, может быть оценена способами, известными специалисту в данной области техники, например, количественной ПЦР с обратной транскрипцией, Вестерн-блоттингом, ИФА, иммуногистохимией.
ПРИМЕРЫ
Следующие примеры предназначены для иллюстрации изобретения и не должны истолковываться как ограничения по нему. Температуры приведены в градусах Цельсия. Если не указано иное, все отгонки выполняют при пониженном давлении, обычно приблизительно от 15 мм.рт.ст. до 100 мм.рт.ст. (= 20-133 мбар). Структура конечных продуктов, промежуточных продуктов и исходных веществ подтверждена стандартными аналитическими способами, например, микроанализом и спектроскопическими характеристиками, например, МС, ИК, ЯМР. Используемые сокращения являются общепринятыми в данной области техники.
Все исходные вещества, структурные элементы, реагенты, кислоты, основания, дегидратирующие агенты, растворители и катализаторы, используемые для синтеза соединений по настоящему изобретению, являются либо коммерчески доступными, либо могут быть получены способами органического синтеза, известными специалисту в данной области техники. Кроме того, соединения по настоящему изобретению могут быть получены способами органического синтеза, известными специалисту в данной области техники, как показано в следующих примерах.
|
Подробное описание аналитических методов
ЯМР: Измерения проводили на спектрометре Bruker Ultrashield™ 400 (400 МГц), Bruker Ultrashield™ 600 (600 МГц), 400 MHz DRX Bruker CryoProbe (400 МГц) или 500 MHz DRX Bruker CryoProbe (500 МГц) с использованием или без использования триметилсилана в качестве внутреннего стандарта. Химические сдвиги (d-значения) приведены в м.д. в сторону слабого поля от тетраметилсилана, тип расщепления в спектре обозначен как синглет (с), дублет (д), триплет (т), квартет (кв.), мультиплет, неразрешенные или в значительной степени перекрывающиеся сигналы (м), уширенный сигнал (уш.). Растворители указаны в круглых скобках.
ДСК: Измерения ДСК выполняли с использованием DSC Q2000 (TA Instruments, Нью-Касл, DE, США), оснащенного рефрижераторной системой охлаждения ДСК (TA Instruments, Нью-Касл, DE, США). Данные обрабатывали математически с использованием резидентного программного обеспечения Universal Analysis®. Калибровку температуры и теплоты плавления проводили с использованием индия в качестве эталонного вещества. Образцы анализировали в открытых алюминиевых тиглях и сканировали при продувке азотом со скоростью нагрева 10°C/мин от 20 до 300°C.
УЭЖХ-МС 3:
Система: Waters Acquity UPLC с детектором Waters SQ.
Колонка: Acquity HSS T3 1,8 мкм 2,1×50 мм.
Поток: 1,0 мл/мин. Температура колонки: 60°C.
Градиент: от 5 до 98% B за 1,4 мин, А=вода+0,05% муравьиной кислоты+3,75 мМ ацетата аммония, В=ацетонитрил+0,04% муравьиной кислоты.
УЭЖХ-МС 6:
Система: Waters Acquity Ultra Performance с детектором Waters SQ.
Колонка: Acquity HSS T3 1,8 мкм 2,1×50 мм.
Поток: 1,0 мл/мин. Температура колонки: 60°C.
Градиент: от 5 до 98% B за 1,4 мин, А=вода+0,05% муравьиной кислоты+3,75 мМ ацетата аммония, В=ацетонитрил+0,04% муравьиной кислоты.
ИЭС-МС: Water Acquity™ Ultra performance LC
Пример 1. N -(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты (1:1).
Стадия 1: 2-(диметоксиметил)-1,8-нафтиридин.
Использовали способ, описанный в J. Org. Chem., 2004, 69 (6), стр. 1959-1966. В 20 л 4-горлую круглодонную колбу помещали 2-аминопиридин-3-карбальдегид (1000 г, 8,19 моль), 1,1-диметоксипропан-2-он (1257 г, 10,64 моль), этанол (10 л) и воду (2 л). Затем по каплям добавляли раствор гидроксида натрия (409,8 г, 10,24 моль) в воде (1000 мл) при перемешивании при 0-15°C. Раствор перемешивали в течение 3 ч при 0-20°C и затем концентрировали в вакууме. Полученный раствор экстрагировали 3×1200 мл этилацетата, и органические слои объединяли. Смесь сушили над сульфатом натрия и концентрировали в вакууме. Остаток промывали 3×300 мл гексана, и твердое вещество собирали фильтрацией. Это привело к получению указанного в заголовке соединения в виде желтого твердого вещества. 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,11 (дд, 1H), 8,53 (д, 1H), 8,50 (дд, 1H), 7,73 (д, 1H), 7,67 (дд, 1H), 5,44 (с, 1H), 3,41 (с, 6H).
Стадия 2: 7-(диметоксиметил)-1,2,3,4-тетрагидро-1,8-нафтиридин.
Использовали способ, описанный в J. Org. Chem., 2004, 69 (6), стр. 1959-1966. В 5 л реактор высокого давления (5 атм.) помещали 2-(диметоксиметил)-1,8-нафтиридин (200 г, 979 ммоль), этанол (3 л), PtO2 (12 г). Реактор вакуумировали и продували три раза азотом с последующей продувкой водородом. Смесь перемешивали в течение ночи при 23°C в атмосфере водорода. Эту реакцию повторяли четыре раза. Твердые вещества отфильтровывали, и полученную смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6): δ 7,14 (д, 1H), 6,51 (д, 1H), 6,47-6,41 (м, 1H), 4,98 (с, 1H), 3,28-3,19 (м, 2H), 3,23 (с, 6H), 2,64 (т, 2H), 1,73-1,79 (м, 2H).
Стадия 3: 6-бром-7-(диметоксиметил)-1,2,3,4-тетрагидро-1,8-нафтиридин.
В 3 л 4-горлую круглодонную колбу помещали 7-(диметоксиметил)-1,2,3,4-тетрагидро-1,8-нафтиридин (114,6 г, 550,3 ммоль) в ацетонитриле (2 л). После этого порциями добавляли NBS (103 г, 578 моль) при перемешивании при 25°C. Полученный раствор перемешивали в течение 30 мин при 25°C. Полученную смесь концентрировали в вакууме, и остаток разбавляли 1000 мл диэтилового эфира. Смесь промывали 3×100 мл льда/воды. Водную фазу экстрагировали 2×100 мл диэтилового эфира, и органические слои объединяли. Полученную смесь промывали 1×100 мл насыщенного раствора соли, сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде светло-желтого твердого вещества. ЖХ-МС: (ЭС, м/з): 286,03 [М+Н]+. 1H-ЯМР: (300 МГц, CDCl3) δ 1,86-1,94 (2H, м), 2,70-2,74 (2H, м), 3,9-3,34 (2H, м), 3,47 (6H, с), 5,23 (1H, с), 5,58 (1H, с), 7,29 (1H, с).
Стадия 4: 2-(диметоксиметил)-5,6,7,8-тетрагидро-1,8-нафтиридин-3-карбальдегид.
К раствору 6-бром-7-(диметоксиметил)-1,2,3,4-тетрагидро-1,8-нафтиридина (15,0 г, 52,2 ммоль) в ТГФ (400 мл) при -78°C в атмосфере аргона добавляли MeLi (1,6 М в Et2O, 32,6 мл, 52,2 ммоль), раствор перемешивали в течение 5 мин, затем медленно добавляли н-BuLi (1,6 М в гексане, 35,9 мл, 57,5 ммоль), и раствор перемешивали в течение 20 мин. ТГФ (100 мл) добавляли к реакции при -78°C. Затем добавляли н-BuLi (1,6 М в гексане, 49,0 мл, 78 ммоль), и реакционную смесь перемешивали в течение 20 мин, затем снова добавляли н-BuLi (1,6 М в гексане, 6,53 мл, 10,45 ммоль), и смесь перемешивали в течение 10 мин при -78°C. Добавляли ДМФА (2,10 мл, 27,2 ммоль), и реакционную смесь перемешивали при -78°C в течение 45 мин, затем ей давали нагреться до комнатной температуры, выливали в нас. водн. раствор NH4Cl и дважды экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и упаривали с получением указанного в заголовке соединения в виде оранжевого масла. (УЭЖХ-МС 3) tуд 0,63 мин; ИЭС-МС 237,2 [М+Н]+.
Стадия 5: этил 2-((2-((трет-бутоксикарбонил)амино)этил)(метил)амино)ацетат.
Этилбромацетат (1,27 мл, 11,48 ммоль) добавляли к смеси трет-бутил (2-(метиламино)этил)карбамата (2,0 г, 11,48 ммоль), триэтиламина (4,81 мл) и ТГФ (24 мл) при 0°C. После перемешивания в течение 24 ч при комнатной температуре реакционную смесь распределяли между насыщенным водным раствором NaHCO3 и ДХМ, экстрагировали 2× с помощью ДХМ, органические слои сушили над Na2SO4 и упаривали с получением указанного в заголовке соединения в виде прозрачного бледно-желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 5,20 (с, уш., 1H), 4,18 (кв., 2H), 3,24 (с, 2H), 3,22-3,16 (м, 2H), 2,65-2,61 (м, 2H), 2,38 (с, 3H), 1,42 (с, 9H), 1,24 (т, 3H).
Стадия 6: этил 2-((2-аминоэтил)(метил)амино)ацетата дигидрохлорид.
Концентрированную соляную кислоту (10 мл) добавляли к раствору этил 2-((2-((трет-бутоксикарбонил)амино)этил)(метил)амино)ацетата (3,05 г, 11,13 ммоль) в ТГФ (20 мл) и EtOH (100 мл) при комнатной температуре. После перемешивания в течение 1 ч при комнатной температуре реакционную смесь упаривали, добавляли этанол (20 мл), упаривали, добавляли дополнительно 50 мл этанола и затем перемешивали при 60°C в течение 70 мин. Охлажденную реакционную смесь затем упаривали с получением указанного в заголовке соединения в виде бледно-желтого стекла. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,58 (с, уш., 3H), 4,19 (кв., 2H), 4,26-4,15 (м, 2H), 3,44 (с, уш., 2H), 3,21 (с, уш., 2H), 2,88 (с, 3H), 1,21 (т, 3H).
Стадия 7: 1-((2-(диметоксиметил)-5,6,7,8-тетрагидро-1,8-нафтиридин-3-ил)метил)-4-метилпиперазин-2-он.
Триацетоксиборгидрид натрия (3,10 г, 14,61 ммоль) добавляли к смеси 2-(диметоксиметил)-5,6,7,8-тетрагидро-1,8-нафтиридин-3-карбальдегида (полученного на стадии 4, 2,30 г, 9,74 ммоль), этил 2-((2-аминоэтил)(метил)амино)ацетата дигидрохлорида (полученного на стадии 6, 2,6 г, 14,61 ммоль) и триэтиламина (6,75 мл, 48,7 ммоль) в 1,2-дихлорэтане (20 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 21 ч при комнатной температуре, и добавляли дополнительный триацетоксиборгидрид натрия (2,6 г, 9,74 ммоль). После дополнительных 4 ч перемешивания при комнатной температуре добавляли дополнительный триацетоксиборгидрид натрия (1,3 г, 4,87 ммоль), и реакцию поддерживали при 4°C в течение 2,5 дней. Затем реакционную смесь нагревали до комнатной температуры, добавляли насыщенный водный раствор NaHCO3, смесь экстрагировали с помощью ДХМ (3×), объединенные органические слои сушили над Na2SO4 и упаривали. Осадок наносили на колонку со 120 г силикагеля RediSep® в виде раствора в ДХМ и очищали с помощью нормально-фазовой хроматографии, элюируя градиентом от ДХМ до 10% MeOH в ДХМ. Фракции, содержащие продукт, объединяли и упаривали с получением указанного в заголовке соединения в виде оранжевой пены. 1H ЯМР (400 МГц, CDCl3) δ 7,08 (с, 1H), 5,30 (с, уш., 1H), 5,20 (с, 1H), 4,69 (с, 2H), 3,44-3,34 (м, 2H), 3,40 (с, 6H), 3,22-3,15 (м, 2H), 3,24 (с, 2H), 2,71-2,64 (м, 2H), 2,58-2,50 (м, 2H), 2,31 (с, 3H), 1,98-1,82 (м, 2H). (УЭЖХ-МС 6) tуд 0,33; ИЭС-МС 335,3 [M+H]+.
Стадия 8: 4-фтор-5-иодпиридин-2-амин.
Суспензию 4-фторпиридин-2-амина (336 г, 2,5 моль) и NIS (745 г, 2,75 моль) в MeCN (9 л) обрабатывали с помощью ТФУК (114 г, 1 моль). Реакционную смесь затем перемешивали при комнатной температуре в течение 8 ч. Реакционную смесь разбавляли с помощью EtOAc (10 л), промывали нас. водн. раствором Na2S2O3 (2×5 л), насыщенным раствором соли (4×5 л). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта. Сырой продукт очищали перекристаллизацией из EtOAc/пентана (1/10) с получением указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,14 (д, 1H), 6,45 (с, 2H), 6,33 (д, 1H).
Стадия 9: 6-амино-4-фторникотинонитрил.
4-фтор-5-иодпиридин-2-амин (полученный на стадии 8, 240 г, 1 моль), цианид цинка (125 г, 1,05 моль), цинк (13 г, 0,2 моль), Pd2(dba)3 (25 Г, 25 ммоль) и dppf (55 г, 0,1 моль) в ДМА (800 мл) дегазировали и загружали в круглодонную колбу в атмосфере азота. Смесь перемешивали при 100°C в течение 3 ч. Реакционную смесь разбавляли с помощью 5% NaHCO3 (2 л), экстрагировали с помощью EtOAc (4×600 мл). Объединенные органические слои промывали с помощью 5% NaOH (1 л), сушили над Na2SO4, концентрировали до 700 мл. Полученную органическую фазу элюировали через колонку с силикагелем с помощью EtOAc (1,7 л). Объединенный органический фильтрат промывали с помощью 2 М HCl (3×800 мл). pH водной фазы доводили до 10 насыщенным раствором NaHCO3. Водную фазу экстрагировали с помощью ДХМ (3×500 мл). Объединенные фракции ДХМ сушили над Na2SO4 и концентрировали. Остаток далее очищали колоночной хроматографией (элюировали пентаном:EtOAc от 10:1 до 3:2) с последующей перекристаллизацией из пентана/EtOAc 3/1 с получением указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,40 (д, 1H), 7,40 (с, 2H), 6,34 (д, 1H).
Стадия 10: трет-бутил (4-хлор-5-цианопиридин-2-ил)карбамат.
Смесь 2,4-дихлор-5-цианопиридина (10 г, 57,8 ммоль), трет-бутилкарбамата (8,2 г, 70,5 ммоль), Pd(OAc)2 (0,26 г, 1,1 ммоль), Ксантфос (1,34 г, 2,3 Ммоль) и K2CO3 (12 г, 87 ммоль) в ТГФ (150 мл) дегазировали 3× азотом. Смесь затем нагревали при 70°C в течение 4-5 ч и контролировали хроматографией до полной конверсии. После завершения реакции добавляли дополнительный ТГФ (100 мл), и нагревали смесь при 70°C в течение дополнительного 1 ч, а затем охлаждали до комнатной температуры. Затем суспензию фильтровали через слой целита для удаления твердого вещества. Фильтрат затем концентрировали и подвергали азеотропной перегонке с этилацетатом перед фильтрацией с получением указанного в заголовке соединения. 1H ЯМР (ДМСО-d6, 400 МГц): δ 10,82 (с, 1Н), 8,79 (с, 1Н), 8,09 (с, 1Н), 1,49 (с, 9Н).
Стадия 11: трет-бутил N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)карбамат.
Смесь трет-бутил (4-хлор-5-цианопиридин-2-ил)карбамата (полученного на стадии 10, 9,8 г, 38,6 ммоль), 2-метоксиэтиламина (5,8 г, 77,3 ммоль) и ДИПЭА (6 г, 46,4 ммоль) в ДМСО (80 мл) нагревали при 65-70°C в течение 24 ч и контролировали хроматографией до полной конверсии. Затем раствор охлаждали до комнатной температуры, и постепенно осаждался белый твердый осадок. Затем медленно добавляли воду (20 мл) в течение 1 ч. Суспензию перемешивали еще 1 ч, фильтровали и сушили с получением указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (ДМСО-d6, 400 МГц): δ 9,87 (с, 1H), 8,18 (с, 1H), 7,20 (с, 1H), 6,86 (с, 9H), 3,51 (т, 2H), 3,36 (т, 2H), 3,28 (с, 3H), 1,47 (с, 9H).
Стадия 12: 6-амино-4-((2-метоксиэтил)амино)никотинонитрил.
Раствор 6-амино-4-фторникотинонитрила (полученного на стадии 9, 1,10 г, 8,02 ммоль) в ДМФА (20 мл) обрабатывали 2-метоксиэтиламином (2,07 мл, 24,1 ммоль) и ДИПЭА (4,20 мл, 24,1 ммоль), нагревали до 50°C и перемешивали в течение 15 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Сырое вещество очищали с помощью нормально-фазовой хроматографии (картридж с 24 г силикагеля, гептаны/EtOAc от 100:0 до 0:100). Фракции, содержащие продукт, концентрировали и сушили в вакууме с получением указанного в заголовке соединения в виде не совсем белого твердого вещества.
Альтернативный синтез 6-амино-4-((2-метоксиэтил)амино)никотинонитрила приведен ниже:
К трет-бутил N-{5-циано-4-[(2-метоксиэтил)амино]пиридин-2-ил}карбамату (полученному на стадии 11, 7 г) добавляли 30-36% водный раствор HCl (40 мл), смесь перемешивали при комнатной температуре в течение 30 минут и контролировали хроматографией до полной конверсии. Затем раствор подщелачивали 20-30% раствором NaOH до рН 9-10 и фильтровали с получением белого твердого вещества. Твердое вещество добавляли к этилацетату (15 мл) и нагревали до 50-55°C с образованием прозрачного раствора. Раствор затем охлаждали до 3-6°C, перемешивали в течение 2-3 ч и фильтровали. Затем влажный отфильтрованный осадок сушили с получением указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,92 (с, 1H), 6,39 (с, 2H), 6,15 (т, 1H), 5,61 (с, 1H), 3,46 (т, 2H), 3,27 (с, 3H), 3,24 (кв., 2H). (УЭЖХ-МС 3) tуд 0,62; ИЭС-МС 193,1 [M+H]+.
Стадия 13: N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-(диметоксиметил)-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2Н)-карбоксамид.
Раствор 6-амино-4-((2-метоксиэтил)амино)никотинонитрила (полученного на стадии 12, 481 мг, 2,50 ммоль) в безводном ДМФА (1,5 мл) добавляли по каплям в течение 10 минут к смеси ди(1Н-1,2,4-триазол-1-ил)метанона (410 мг, 2,50 ммоль) и ДМФА (1,5 мл), охлажденной при 0°C. После перемешивания в течение 45 минут при 0°C реакционной смеси давали нагреться до комнатной температуры, и через дополнительные 90 минут при комнатной температуре добавляли раствор 1-((2-(диметоксиметил)-5,6,7,8-тетрагидро-1,8-нафтиридин-3-ил)метил)-4-метилпиперазин-2-она (полученного на стадии 7, 418 мг, 1,00 ммоль) в ДМФА (2 мл). Реакционную смесь перемешивали в течение 17,5 ч при комнатной температуре, гасили добавлением МеОН и упаривали. Остаток наносили на колонку с 80 г силикагеля RediSep® в виде раствора в ДХМ и очищали с помощью нормально-фазовой хроматографии, элюируя градиентом от ДХМ до 2% MeOH в ДХМ. Фракции, содержащие продукт, объединяли и упаривали с получением указанного в заголовке соединения в виде оранжевой пены. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,50 (с, 1H), 8,27 (с, 1H), 7,52 (с, 1H), 7,39 (с, 1H), 6,93 (т, 1H), 5,45 (с, 1H), 4,65 (с, 2H), 3,94-3,89 (м, 2H), 3,54-3,50 (м, 2H), 3,40-3,35 (м, 2H), 3,38 (с, 6H), 3,29 (с, 3H), 3,20-3,16 (м, 2H), 3,05 (с, 2H), 2,86-2,80 (м, 2H), 2,61-2,55 (м, 2H), 2,22 (с, 3H), 1,94-1,88 (м, 2H). (УЭЖХ-МС 6) tуд 0,72; ИЭС-МС 553,3 [M+H]+.
Стадия 14: N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид
Концентрированную соляную кислоту (0,40 мл) добавляли к раствору N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида (полученного на стадии 13, 470 мг, 0,808 ммоль) в ТГФ (3 мл) и воде (1 мл) при комнатной температуре. После перемешивания в течение 3 ч при комнатной температуре добавляли насыщенный водный раствор NaHCO3, смесь экстрагировали с помощью ДХМ (3×), органические слои сушили над Na2SO4 и упаривали. Остаток подвергали действию ультразвука с EtOAc (6 мл) и пентаном (6 мл) и затем фильтровали. Полученное белое твердое вещество затем растворяли в ДХМ (6 мл), добавляли EtOAc (3 мл), раствор нагревали, герметизировали и оставляли стоять при комнатной температуре в течение 2 ч. Фильтрация и сушка давала N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в виде белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13,43 (с, 1H), 10,06 (с, 1H), 8,24 (с, 1H), 7,49 (с, 1H), 7,47 (с, 1H) 1Н), 4,86 (с, 2Н), 3,96-3,90 (м, 2Н), 3,52-3,46 (м, 2Н), 3,39-3,33 (м, 2Н), 3,30-3,21 (м, 2Н) С, 3H), 3,02 (с, 2H), 2,93-2,86 (м, 2H), 2,61-2,56 (м, 2H), 2,21 (с, 3H), 1,95-1,85 (м, 2H). (УЭЖХ-МС 6) tуд 0,70, ИЭС-МС 507,2, [M+H]+.
Стадия 15: N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты (1:1).
N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид (полученный на стадии 14, 4 г, 7,86 ммоль) перемешивали в пропионовой кислоте (29,3 г, 29,60 мл) при 70°C до полного растворения (20 минут). Раствор охлаждали до 55°C, и к нему добавляли раствор лимонной кислоты в ацетоне (23% масс./масс.). Отдельно готовили затравочную суспензию добавлением ацетона (0,2 г, 0,252 мл) к N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамиду в форме соли лимонной кислоты (0,0185 г, 0,026 ммоль). Затравочную суспензию добавляли к раствору при 50°C, и полученную суспензию оставляли перемешиваться при 50°C в течение 40 минут. В реакцию в течение 380 минут добавляли дополнительную порцию раствора лимонной кислоты в ацетоне (26,6 г, 2,51% масс./масс., 33,63 мл). Полученную суспензию перемешивали дополнительно в течение 120 минут и охлаждали до 20°C при перемешивании в течение 4 ч. Суспензию перемешивали еще в течение 12 ч перед фильтрацией суспензии под вакуумом, и промывали полученное твердое вещество раствором пропионовой кислоты:ацетоном (1:1, 7 г, 7,96 мл) при комнатной температуре. Твердое вещество далее промывали ацетоном (7 г, 8,85 мл) при комнатной температуре. Полученное твердое вещество сушили в печи при 40°C и 5 мбар с получением указанного в заголовке соединения в виде светло-оранжевого твердого вещества (5,2 г, 7,443 ммоль). (мол.м. 698,70), т.пл. (ДСК) 168,8°C (начало).
Анализ XRPD показал ту же рентгеновскую дифрактограмму, что и для частиц, полученных способом, описанным в PCT/IB2014/065585 (ссылочный пример 1) - см. фигуру 5.
Пример 1a
Стадии 1-14 проводили, как описано в примере 1.
Стадия 15а: N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты (1:1)
N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид (полученный на стадии 14, 5 г, 9,930 ммоль) перемешивали в пропионовой кислоте (33,5 г, 33,84 мл) при 60°C. Как только N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид растворился, добавляли безводный порошок лимонной кислоты (0,19 г, 0,9889 ммоль). Полученную суспензию нагревали до 70°C и подвергали действию ультразвука в течение 5 минут для обеспечения полного растворения. Полученный раствор охлаждали до 50°C, и в течение 20 минут добавляли раствор лимонной кислоты в этилацетате (3,7 г, 1,3% лимонной кислоты в этилацетате). Затравку N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты (0,02 г) добавляли к раствору, и суспензию выдерживали в течение 15 минут. Еще одну аликвоту лимонной кислоты в этилацетате (128 г, 1,3% лимонной кислоты в этилацетате) добавляли к суспензии в течение 11,85 ч. Суспензию оставляли перемешиваться в течение более 4 ч. Затем суспензию фильтровали под вакуумом (500 мбар), и полученное твердое вещество промывали сначала раствором пропионовой кислоты:этилацетата (1:1, 7 г, 7,44 мл) при комнатной температуре, а затем этилацетатом (12 г, 13,38 мл) при комнатной температуре. Полученное твердое вещество сушили в печи при 40°C и 5 мбар с получением указанного в заголовке соединения в виде светло-оранжевого твердого вещества (6,3 г, 9,074 ммоль).
Анализ XRPD показал ту же рентгеновскую дифрактограмму, что и для частиц, полученных способом, описанным в PCT/IB2014/065585 (ссылочный пример 1) - см. фигуру 5.
Ссылочный пример 1 (описанный в PCT/IB2014/065585) - N -(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты (1:1)
Стадии 1-14 проводили, как описано в примере 1.
Ссылочная стадия 15 - N -(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид в форме соли лимонной кислоты (1:1)
Раствор лимонной кислоты (96,9 мг) в ацетоне (5 мл) готовили при комнатной температуре (0,1 М). Часть 0,1 М лимонной кислоты в ацетоновом растворе (2 мл) затем добавляли к суспензии N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида (100 мг) в ацетоне (4 мл), и смесь подвергали воздействию ультразвука в течение 1 минуты, затем нагревали при 55°C при перемешивании в течение 2 ч перед медленным охлаждением до комнатной температуры. Белое твердое вещество затем собирали фильтрацией, промывали 2× ацетоном (2 мл) и сушили в течение 18 ч при 40°C в вакууме с получением указанной в заголовке соли.
Альтернативно, N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамид (6,5 г, 12,83 ммоль) помещали в 500 мл 4-горлый реактор. Добавляли 49 мл ледяной уксусной кислоты, и полученную суспензию перемешивали при 23°C до получения прозрачной смеси. В отдельной колбе безводную 2-гидроксипропан-1,2,3-трикарбоновую кислоту (2,59 г, 13,47 ммоль, 1,05 экв.) растворяли в 49 мл ледяной уксусной кислоты при 50°C до получения прозрачного раствора. Этот раствор затем добавляли при 23°C к ранее приготовленному раствору N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида. Эту смесь перемешивали в течение 30 мин при 23°C, и затем добавляли по каплям в течение 1 часа к 192 мл этилацетата, нагретого до 75°C. В процессе добавления температура оставалась постоянной. В конце добавления температуру смеси медленно охлаждали до 23°C и оставляли на 16 ч при этой температуре при легком перемешивании. Суспензию охлаждали до 5-10°C и фильтровали. Осадок на фильтре промывали 15 мл этилацетата и 15 мл ацетона. Отфильтрованный влажный осадок (около 8,5 г) переносили в колбу на 500 мл, содержащую 192 мл сухого ацетона. Полученную суспензию кипятили с обратным холодильником в течение 24 ч. Суспензию фильтровали, и осадок на фильтре промывали 15 мл сухого ацетона 2 раза, а затем сушили при 50°C в вакууме в течение нескольких часов с получением указанной в заголовке соли.
Пример. 2 - Биохимические киназные анализы in vitro для FGFR4
Все анализы проводили в 384-луночных планшетах для микротитрования. Каждый аналитический планшет содержал 8-точечные серийные разведения для 40 тестируемых соединений, а также четыре 8-точечных серийных разведения стауроспорина в качестве контрольного соединения, плюс 16 режимов высокой и 16 режимов низкой мощности доз.
Стадии обработки жидкостей и инкубации проводили на дозирующей системе Nanodrop Express от Innovadyne, оборудованной роботизированным манипулятором (Thermo CatX, Caliper Twister II) и инкубатором (Liconic STX40, Thermo Cytomat 2C450). Аналитические планшеты готовили путем добавления 50 нл на лунку раствора соединения в 90% ДМСО. Киназные реакции начинали путем постадийного добавления 4,5 мкл на лунку пептида/АТФ-раствора (50 мМ HEPES, pH 7,5, 1 мМ ДТТ, 0,02% Твин20, 0,02% БСА, 0,6% ДМСО, 10 мМ бета-глицерофосфата и 10 мкМ ортованадата натрия, 16 мМ MgCl2, 1122 мкМ АТФ, 4 мкМ пептида (5-Fluo-Ahx-KKKKEEIYFFFG-NH2, Biosyntan GmbH) и 4,5 мкл на лунку раствора фермента (50 мМ HEPES, pH 7,5, 1 мМ ДТТ, 0,02% Твин20, 0,02% БСА, 0,6% ДМСО, 10 мМ бета-глицерофосфата и 10 мкМ ортованадата натрия, 16 мМ MgCl2, 6 нМ FGFR4 (GST-FGFR4 (388-802) собственного производства, полученного путем экспрессии в клетках насекомых и аффинной хроматографии). Киназную реакцию инкубировали при 30°C в течение 60 минут и затем останавливали добавлением 16 мкл на лунку стоп-раствора (100 мМ HEPES pH 7,5, 5% ДМСО, 0,1% покрывающего реагента от Caliper, 10 мМ ЭДТА и 0,015% Brij35). Планшеты с остановленными киназными реакциями переносили в автоматизированные модули для считывания от Caliper LC3000. Фосфорилированные и нефосфорилированные пептиды разделяли с использованием технологии изменения подвижности в микрофлюидной системе от Caliper. Вкратце, образцы из остановленных киназных реакций наносили на чип. Анализируемые образцы пропускали через чип с помощью постоянного потока буфера, и перемещение субстратного пептида контролировали по сигналу флуоресценции его метки. Фосфорилированный пептид (продукт) и нефосфорилированный пептид (субстрат) разделяли в электрическом поле по их соотношению заряд/масса. Активность киназы рассчитывали из количеств образованного фосфопептида. Значения IC50 определяли из процентных значений ингибирования при различных концентрациях соединений с помощью нелинейного регрессионного анализа.
Приготовление разведений соединений
Испытуемые соединения растворяли в ДМСО (10 мМ) и переносили в 1,4 мл плоскодонные или V-образные матричные трубки со специфичной 2D-матрицей. Исходные растворы хранили при +2°C, если их не использовали сразу. Для процедуры испытания ампулы размораживали и идентифицировали сканером, в результате чего создавали рабочий лист, который направлял последующие стадии работы.
Разведения соединений проводили в 96-луночных планшетах. Этот формат позволил провести анализ максимально 40 отдельно взятых тестируемых соединений в 8 концентрациях (одномоментно), включая 4 контрольных соединения. Протокол разбавлений включал подготовку "планшетов для предварительного разведения", "планшетов с исходными разведениями" и "планшетов для анализа".
Планшеты для предварительного разведения: 96-луночные планшеты из полипропилена использовали в качестве планшетов для предварительного разведения. Подготавливали в общей сложности 4 планшета для предварительного разведения, включая 10 тестируемых соединений в позициях A1-A10 планшета, одно стандартное соединение в A11 и один контроль ДМСО в A12. Все стадии разбавления выполняли на роботе HamiltonSTAR.
Планшеты с исходными разведениями: 30 мкл отдельно взятых разбавлений соединений, включая стандартное соединение, и контрольные образцы 4 "планшетов для предварительного разведения", переносили в 384-луночный "планшет с исходными разведениями", включая следующие концентрации 1'810, 362, 72,5, 54,6, 14,5, 2,9, 0,58 и 0,12 мкМ, соответственно, в 90% ДМСО.
Планшеты для анализа: Идентичные "планшеты для анализа" затем получали путем пипетирования 50 нл каждого из разведений соединений "контрольных планшетов" в 384-луночные "планшеты для анализа" с помощью 384-канального диспенсера HummingBird. Эти планшеты использовали непосредственно для анализа, который проводили в общем объеме 9,05 мкл. Это привело к конечной концентрации соединения 10, 2,0, 0,4, 0,08, 0,016, 0,0032, 0,00064 и 0,000128 мкМ и конечной концентрации ДМСО 0,5% в анализе.
Клеточные киназные анализы in vitro для FGFR4
Для считывания клеточной FGFR4-киназной активности был разработан анализ, который измеряет степень фосфорилирования тирозина на FGFR4. Для этого создавали клеточную линию BaF3-Tel-FGFR4: клетки BaF3 стабильно трансдуцировали ретровирусом, кодирующим слитый белок, состоящий из аминоконцевой части TEL (aa1-337), слитой с цитоплазматическим доменом FGFR4, включая юкстамембранный домен. Присутствие домена TEL опосредует конститутивную активацию слитой FGFR4-киназы путем олигомеризации и, таким образом, аутофосфорилирование на тирозиновых сайтах.
Был разработан ИФА с захватом на основе MSD (мезомасштабное обнаружение) и его использовали следующим образом:
Обработка клеток: 250000 клеток BaF3-Tel-FGFR4 на лунку высевали в 96-луночные планшеты для культивирования тканей (Corning Кат.№ 3359) в 40 мкл среды для выращивания (RPMI-1640 (Amimed Кат.№ 1-41F01-I), дополненной 10% фетальной бычьей сыворотки, 10 мМ HEPES, 1 мМ пирувата натрия, 2 мМ стабильного глутамина и 1× пенициллина-стрептомицина). Используя устройство для обработки жидкости (Velocity 11 Bravo, Agilent), получали серийные 3-кратные разведения соединений в ДМСО, предварительно разведенных в среде для выращивания, с последующим переносом 10 мкл/лунка в клеточные планшеты. После инкубации в течение 1 часа при 37°C/5% СО2 добавляли 50 мкл лизисного буфера (150 мМ NaCl, 20 мМ Трис (pH 7,5), 1 мМ ЭДТА, 1 мМ ЭГТА, 1% Тритона Х-100, 10 мМ NaF, дополненный ингибиторами протеазы (Complete Mini, Roche Кат.№ 11836153001) и фосфатазы) (ингибитор фосфатазы I, SIGMA Кат.№ P2850, ингибитор фосфатазы II, SIGMA Кат.№ P5726 в соответствии с инструкциями поставщика) и инкубировали в течение 30 минут на льду со встряхиванием при 300 об/мин. Планшеты с образцами затем замораживали и хранили при -70°C. После оттаивания на льду планшеты с образцами центрифугировали в течение 15 минут при 1200 об/мин при 6°C.
Анализ ИФА: 96-луночные планшеты для мультиплексного анализа (MSD, Кат.№ L15XB-3) покрывали в течение 1 часа при комнатной температуре 25 мкл/лунка мышиного антитела против H-TEL (Santa Cruz, Кат.№ sc-166835), разбавленного 1:400 в ФСБ/O. После добавления 150 мкл 3% MSD-блокатора A (Кат.№ R93BA-1) в TBS-T (50 мМ Трис, 150 мМ NaCl, 0,02% Твин-20) планшеты инкубировали в течение 1 часа при комнатной температуре со встряхиванием. Затем планшеты промывали 3 раза с помощью 200 мкл/лунка TBS-T. Затем 50 мкл клеточного лизата переносили на покрытый планшет и инкубировали в течение 15 ч при 4°C с последующими 3 промывками с помощью 200 мкл TBS-T/лунка и добавлением 25 мкл/лунка антитела MSD SULFOTAGGED PY20 (MSD Кат.№ R32AP-5), разведенного 1:250 в TBS-T+1% MSD-блокатора A. После инкубации в течение 1 часа при комнатной температуре со встряхиванием лунки промывали 3 раза с помощью 200 мкл TBS-T/лунка. После добавления 150 мкл буфера для считывания MSD (MSD, Кат.№ R92TC-2) к исходному раствору, разведенному 1:4 нановодой, немедленно подсчитывали генерирование электрохемилюминесцентного сигнала на приборе SectorImager 6000 (MSD). Расчет IC50: для анализа данных уровень фона для анализа определяли в лунках, содержащих среду и лизисный буфер, но не клетки, и соответствующее значение вычитали из всех точек данных. Влияние концентрации конкретного испытуемого соединения на фосфорилирование FGFR4 выражается в процентах от считывания электрохемолюминесценции с поправкой на фон, полученной для клеток, обработанных только носителем (ДМСО, 0,2% найденная концентрация), которая устанавливается равной 100. Концентрации соединений, приводящие к полумаксимальному ингибированию сигнала (IC50), определяли с помощью стандартной обработки четырех параметрических кривых (XLfit 5.4, IDBS).
Анализ пролиферации клеток
Анализ пролиферации путем окрашивания метиленовым синим (MBS): влияние соединений на пролиферацию клеток оценивают с использованием клеток гепатоцеллюлярной карциномы HuH-7, полученных из японской коллекции банка клеток для исследования биоресурсов (Кат.№ JCRB0403) и культивируют в среде, рекомендованной производителем (DMEM с высоким содержанием глюкозы (Amimed Кат.№ 1-26F01-I), 10% фетальной бычьей сыворотки (Invitrogen Кат.№ 16140-071), 1 мМ пирувата натрия (Amimed Кат.№ 5-60F00-H), 1× пенициллина/стрептомицина (Amimed Кат.№ 4-01F00-H)) при 37°C в увлажненном инкубаторе с 5% СО2. В частности, 5000 клеток/лунка высевали в 96-луночные планшеты для культивирования тканей (TPP Кат.№ 92696) в общем объеме среды 100 мкл/лунка и добавляли увеличивающиеся разведения соединений или ДМСО через 24 часа в трех повторах. Через 72 часа после добавления соединения клетки фиксировали добавлением 25 мкл/лунка 20% глутаральдегида (Sigma Aldrich Кат.№ G400-4) и инкубировали в течение 10 минут при комнатной температуре. Клетки промывали три раза с помощью 200 мкл/лунка H2O и окрашивали с помощью 100 мкл/лунка 0,05% метиленового синего (ABCR GmbH Кат.№ AB117904) в течение 10 минут при комнатной температуре. Клетки промывали 3 раза с помощью 200 мкл/лунка H2O и затем лизировали добавлением 200 мкл/лунка 3% HCl (Fluka Кат.№ 84422) в течение 30 минут при комнатной температуре со встряхиванием. Оптическую плотность измеряли при A650 нм. Определяли концентрацию соединения, обеспечивающего 50% ингибирование пролиферации по отношению к ДМСО-обработанным клеткам (IC50) с использованием программного обеспечения XLFit.
Анализ CellTiter Glo (CTG): функциональный эффект соединений на пролиферацию клеток оценивают с использованием клеток гепатоцеллюлярной карциномы HuH-7, полученных из японской коллекции банка клеток для исследования биоресурсов (Кат.№ JCRB0403), и культивируют в среде, рекомендованной производителем (DMEM с высоким содержанием глюкозы (Amimed Кат.№ 1-26F01-I), 10% фетальной бычьей сыворотки (Invitrogen Кат.№ 16140-071), 1 мМ пирувата натрия (Amimed Кат.№ 5-60F00-H), 1× пенициллина/стрептомицина (Amimed Кат.№ 4-01F00-H)) при 37°C в увлажненном инкубаторе с 5% СО2. Опосредованное соединением подавление пролиферации/жизнеспособности клеток оценивают путем количественного определения уровней клеточного АТФ с использованием реагента CellTiter-Glo (CTG) (Promega, Кат.№ G7573). Вкратце, клетки высевают в количестве 3000 клеток/лунка/80 мкл свежей среды в 96-луночные планшеты, обработанные тканевой культурой (Costar Кат.№ 3904), с последующим добавлением 20 мкл среды, содержащей разведения соединений в 5 раз превышающие их конечную предполагаемую концентрацию. Эффекты доза-эффект оценивают посредством 3-кратных серийных разведений испытуемого соединения, начиная с 10 мкМ. После инкубации клеток в течение 3 дней при 37°C и 5% СО2, влияние ингибиторов на жизнеспособность клеток определяют количественно после добавления 50 мкл CTG и измерения люминесценции (время интегрирования: 500 мс) в соответствии с руководством производителя, используя соответствующим образом оснащенного мультирежимного планшет-ридера (M200Pro, TECAN, Швейцария). Для анализа данных фоновое значение для анализа, определенное в лунках, содержащих среду, но без клеток, вычитается из всех точек данных. Чтобы обеспечить дифференциацию цитотоксических от цитостатических соединений, количество жизнеспособных клеток оценивают относительно количества, наблюдаемого во время добавления соединения, используя отдельный планшет для клеток (день 0). Влияние концентрации конкретного испытуемого соединения на пролиферацию/жизнеспособность клеток выражают в процентах от люминесценции с поправкой на фон и день 0, полученной для клеток, обработанных только носителем (ДМСО, 0,1% найденная концентрация), которая устанавливается равной 100%, тогда как считывание люминесценции для лунок, содержащих только среду, но не клетки, составляет -100%. Концентрации соединений, приводящие к полумаксимальному ингибированию роста (GI50), определяют с помощью стандартной обработки четырех параметрических кривых (XLfit 5.2., IDBS, Великобритания).
|
Сравнительные данные
Биохимические анализы in vitro для FGFR1 (407-822), FGFR2 (406-821) и FGFR3 (411-806) проводили аналогично биохимическому анализу in vitro для FGFR4, описанному выше, с использованием указанных частей киназных доменов. Пример 1 давал значения IC50 > 10000 нМ в биохимических анализах FGFR1, FGFR2 и FGFR3.
Пример 3. Лазерное дифракционное измерение частиц N -(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида, полученных в примере 1.
Распределение по размерам частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты (1:1), полученных в примере 1, анализировали в соответствии со способом, описанным в Европейской фармакопее, глава 2.9.31. Анализ размера частиц методом лазерной дифракции света.
Для измерения использовали устройство Sympatec HELOS с фокусным расстоянием 500 мм от Sympatec GmbH, Германия, оборудованное устройством для влажного распыления Cuvette от Sympatec GmbH, Германия. Испытуемое вещество диспергировали в уайт-спирите, Brenntag Schweizerhall AG, Швейцария (номер по каталогу 12200-150) с использованием диспергирующего агента (например, Statsafe™ 6000, INNOSPEC Limited, около 1% в уайт-спирите).
Процедура: Капли диспергирующего агента добавляли при ручном перемешивании к порошкообразному образцу N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты (1:1), полученному в примере 1. Частицы интенсивно перемешивали, например, в вихревом смесителе, чтобы тщательно смочить вещество и образовать гладкую и гомогенную пасту. Пасту разбавляли уайт-спиритом до конечного объема в несколько миллилитров, и исходную дисперсию снова перемешивали.
Измерение: Испытуемую дисперсию готовили путем разбавления исходной дисперсии уайт-спиритом для обеспечения соответствующей оптической концентрации, и кумулятивное распределение по объему определяли с использованием прибора для лазерной дифракции света в соответствии с инструкцией по эксплуатации. Измерения проводили до и после 10 секунд, 20 секунд, 30 секунд обработки ультразвуком. Параметры устанавливали следующим образом: амплитуда 30% и время цикла 80% на ультразвуковом устройстве GM70 от Bandelin electronic GmbH & Co. KG, и продолжительность измерения около 20 секунд.
Размеры частиц при значениях 10%, 50% и 90% (x10, x50, x90) в подситных фракциях оценивали по кумулятивному распределению по объему без обработки ультразвуком. Ультразвуковая обработка не оказала существенного влияния на размеры измеренных частиц.
Полученное распределение частиц по размерам показано на фигуре 6.
Пример 4. Лазерное дифракционное измерение частиц N -(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида, полученных в ссылочном примере 1 (описанный в PCT/IB2014/065585).
Распределение по размерам частиц N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты (1:1), полученных в ссылочном примере 1, анализировали в соответствии со способом, описанным в Европейской фармакопее, глава 2.9.31. Анализ размера частиц методом лазерной дифракции света.
Для измерения использовали устройство Sympatec HELOS с фокусным расстоянием 100 мм от Sympatec GmbH, Германия, оборудованное устройством для влажного распыления Cuvette от Sympatec GmbH, Германия. Испытуемое вещество диспергировали в уайт-спирите, Brenntag Schweizerhall AG, Швейцария (номер по каталогу 12200-150) с использованием диспергирующего агента (например, Statsafe™ 6000, INNOSPEC Limited, около 1% в уайт-спирите).
Процедура: Капли диспергирующего агента добавляли при ручном перемешивании к порошкообразному образцу N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида в форме соли лимонной кислоты (1:1), полученному в примере 1. Частицы интенсивно перемешивали, например, в вихревом смесителе, чтобы тщательно смочить вещество и образовать гладкую и гомогенную пасту. Пасту разбавляли уайт-спиритом до конечного объема в несколько миллилитров, и исходную дисперсию снова перемешивали.
Измерение: Испытуемую дисперсию готовили путем разбавления исходной дисперсии уайт-спиритом для обеспечения соответствующей оптической концентрации, и кумулятивное распределение по объему определяли с использованием прибора для лазерной дифракции света в соответствии с инструкцией по эксплуатации. Измерения проводили до обработки ультразвуком и после обработки ультразвуком в течение до 600 секунд. Параметры устанавливали следующим образом: амплитуда 50% на ультразвуковом устройстве USGD-100 от Telsonic AG и длительность измерения около 20 секунд. Измерение проводили дважды.
Размеры частиц при значениях 10%, 50% и 90% (x10, x50, x90) в подситных фракциях оценивали по кумулятивному распределению по объему после 240 секунд обработки ультразвуком. Это было определено для репрезентативности размеров первичных частиц.
Полученное распределение частиц по размерам показано на фигуре 7.
Пример 5 - Измерения энергии основного потока
Энергия основного потока (BFE) - это энергия, необходимая для установления конкретного режима потока в точном объеме кондиционированного порошка. Этот режим потока представляет собой нисходящее движение лезвия против часовой стрелки, создающее сжатие, режим с относительно высоким напряжением потока в порошке. BFE рассчитывают на основании работы, выполненной при перемещении лезвия через порошок с верхней части сосуда на дно, то есть во время хода сверху вниз.
В данном случае энергию основного потока измеряли с использованием порошкового реометра FT4 от Freeman Technology при следующих условиях: размер сосуда: 25 мм; название стандартной программы: 25mm_1C_Split_1T; движущееся оборудование: лезвие 23,5 мм; сосуд: разъемный сосуд 25 мм × 25 мл. Результаты наносили на график на фигуре 9.
Энергия основного потока, измеренная для 2 образцов частиц по изобретению, составляла 39,17 мДж для частиц, полученных способом, описанным в примере 1а; и 65,07 мДж для частиц, полученных способом, описанным в примере 1.
Энергия основного потока, измеренная для образцов частиц, полученных способом, описанным в PCT/IB2014/065585, составляла 185,20 мДж.
Эти результаты говорят о том, что частицы по изобретению обладают улучшенными свойствами потока по сравнению с теми частицами, которые получены способом, описанным в PCT/IB2014/065585.
Пример 6 - Измерение сжимаемости
Сжимаемость - это показатель того, как изменяется плотность в зависимости от приложенного нормального напряжения. По определению сжимаемость представляет собой процентное изменение объема после сжатия (%). Измерения проводили с использованием порошкового реометра FT-4 от Freeman Technology.
Частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида, полученные способом по настоящему изобретению, и частицы, полученные способом, описанным в PCT/IB2014/065585, помещали в сосуд, и поршень, сообщающийся с атмосферой, использовали для сжатия частиц. Поршень, сообщающийся с атмосферой, выполнен таким образом, что поверхность сжатия изготовлена из витой сетки из нержавеющей стали и позволяет увлекаемому воздуху в порошке равномерно выпускаться через поверхность порошкового слоя. Нормальное напряжение прикладывали на 8 последовательных стадиях сжатия, начиная с 0,5 кПа и заканчивая 15 кПа. На каждой стадии нормальное напряжение поддерживали постоянным в течение 60 секунд, и сжимаемость автоматически рассчитывали как процентное изменение объема. Результаты наносили на график на фигуре 10.
Процент сжимаемости, измеренный при 15 кПа, составлял 54,14% и 54,63% для частиц по настоящему изобретению. Для частиц, полученных способом, описанным в PCT/IB2014/065585, процент сжимаемости, измеренный при 15 кПа, составлял 49,93%.
Эти результаты показывают, что частицы по изобретению более сжимаемы по сравнению с теми, которые получены способом, описанным в PCT/IB2014/065585, что будет иметь преимущество, заключающееся в том, что более высокое содержание лекарственного средства в конечной готовой лекарственной форме может быть достигнуто путем применения частиц по изобретению.
Пример 7 - Измерение угла трения о стенки
Для оценки взаимодействия между лекарственной субстанцией и нержавеющей сталью был разработан способ определения трения о стенки. Используемое устройство представляет собой реометр FT-4 от Freeman Technology.
Частицы N-(5-циано-4-((2-метоксиэтил)амино)пиридин-2-ил)-7-формил-6-((4-метил-2-оксопиперазин-1-ил)метил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксамида, полученные способом по настоящему изобретению, и частицы, полученные способом, описанным в PCT/IB2014/065585, помещали в сосуд, содержащий образец и насадку для трения о стенки, чтобы индуцировать как вертикальные, так и вращательные напряжения. Образец порошка готовят путем кондиционирования и последующего предварительного уплотнения с использованием стандартного лезвия FT4 и поршня, сообщающегося с атмосферой.
Насадка для трения о стенки, имеющая среднюю шероховатость, равную 1,2 микрон, из 316 дисков из нержавеющей стали, движется вниз к поверхности образца и вызывает нормальное напряжение, когда диск контактирует с верхней частью образца. Насадка продолжает двигаться вниз, пока не будет установлено необходимое нормальное напряжение. Затем начинается медленное вращение насадки для трения о стенки, что вызывает касательное напряжение. Плоскость сдвига устанавливается между поверхностями диска и образца. Поскольку порошковый слой сопротивляется вращению насадки для трения о стенки, крутящий момент увеличивается до тех пор, пока сопротивление не будет полностью преодолено. На этом этапе наблюдается максимальный крутящий момент. Насадка для трения о стенки продолжает вращаться со скоростью 18 градусов/мин в течение 5 минут. Измеряют крутящий момент, необходимый для поддержания этого вращения, который позволяет рассчитать "установившееся" касательное напряжение. Нормальное напряжение поддерживают постоянным на уровне заданного приложенного напряжения для каждого этапа на протяжении всей этой стадии. Ряд значений касательного напряжения измеряют для диапазона заданных приложенных напряжений. Из-за природы образцов и того факта, что точный постоянный вращательный момент вряд ли будет достигнут, программное обеспечение определяет среднее значение в течение 10% от времени сдвига. Затем угол трения о стенки рассчитывают путем вычерчивания линии наилучшего соответствия через точки данных на графике и измерения угла, расположенного между этой линией наилучшего соответствия и горизонтальной. Результаты наносили на график на фигуре 11.
Измеренный угол трения о стенки составлял 34,58° и 35,85° для частиц по настоящему изобретению. Для частиц, полученных способом, описанным в PCT/IB2014/065585, угол трения о стенки составлял 43,18°.
Эти результаты показывают, что частицы по изобретению в меньшей степени проявляют липкие свойства на металлических поверхностях и, таким образом, имеют улучшенную обрабатываемость по сравнению с теми, которые получены способом, описанным в PCT/IB2014/065585.


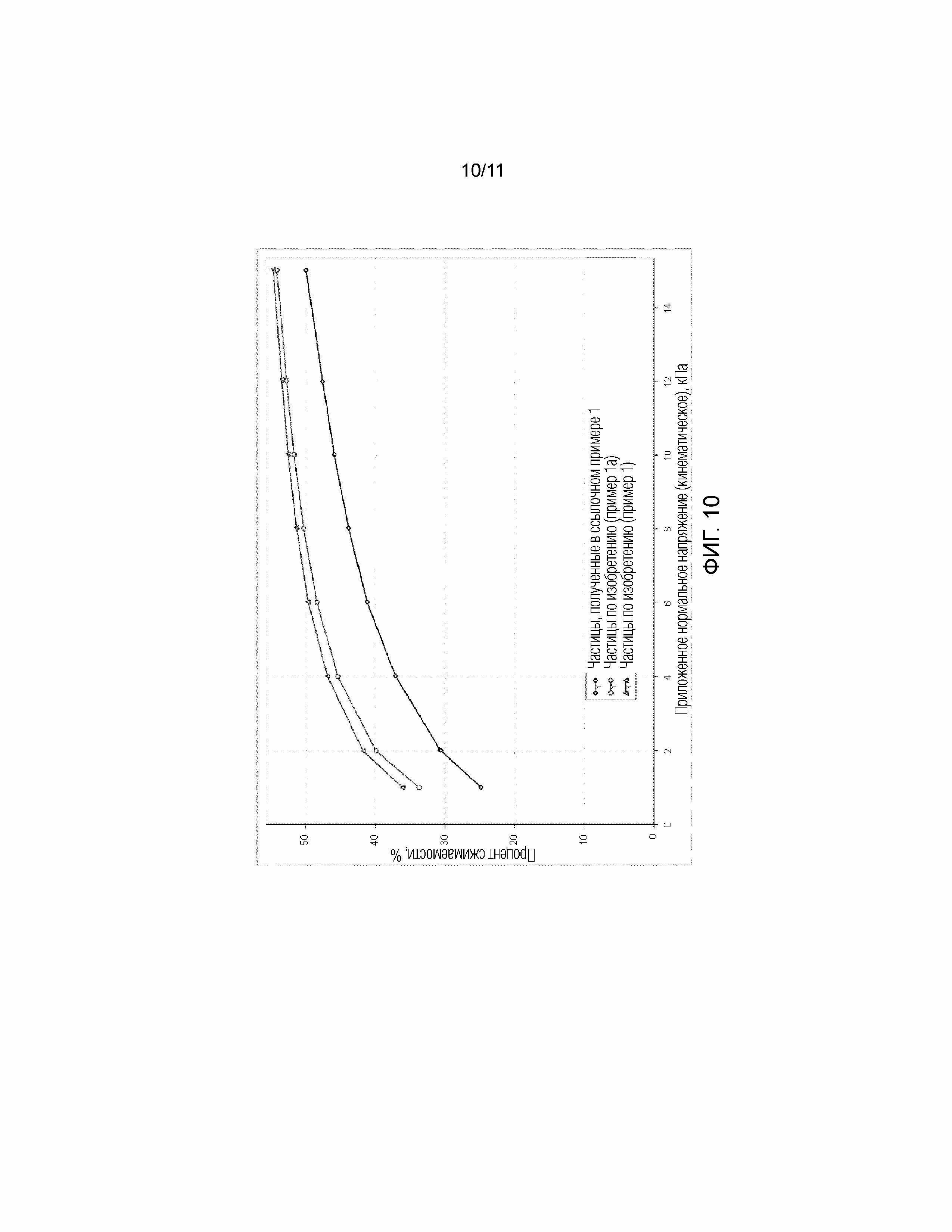
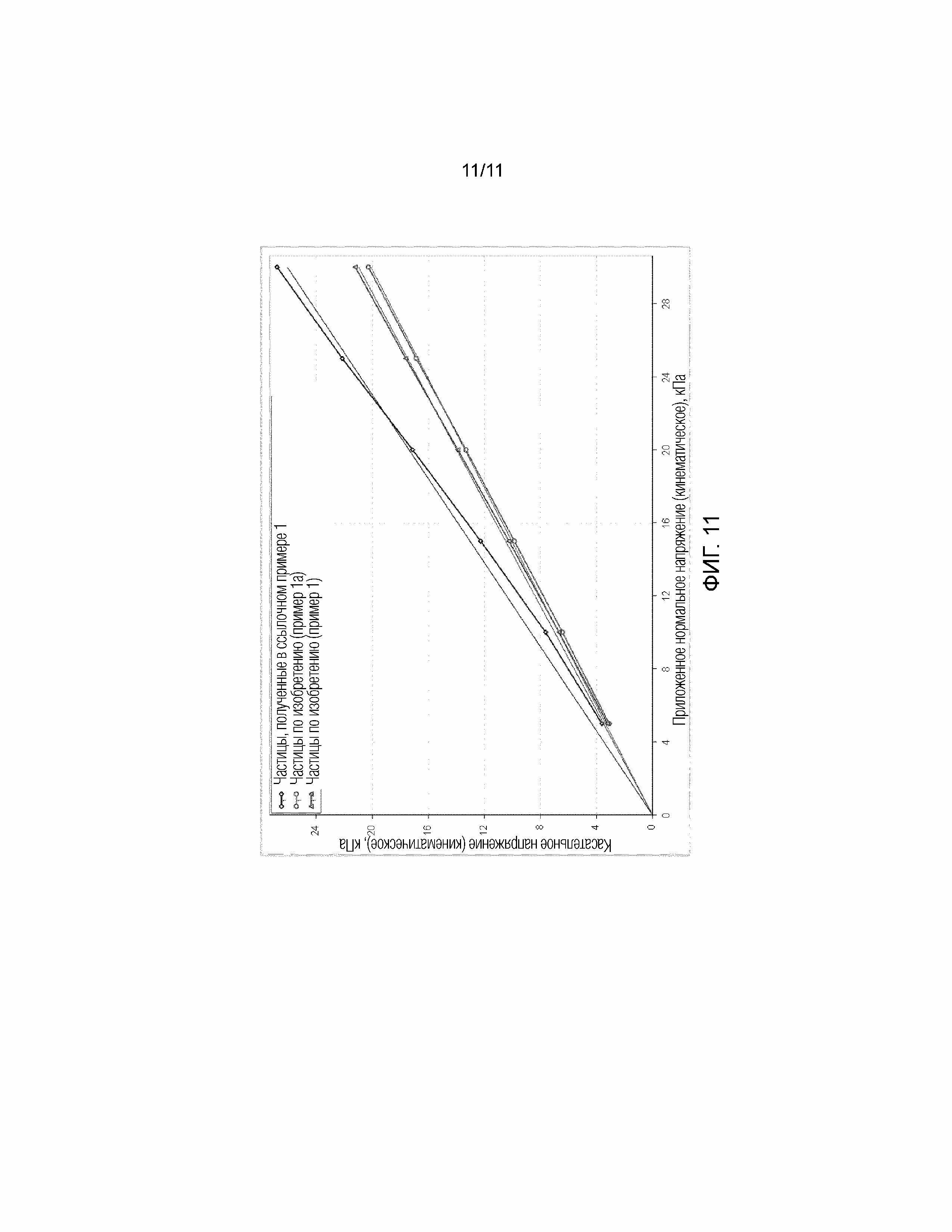
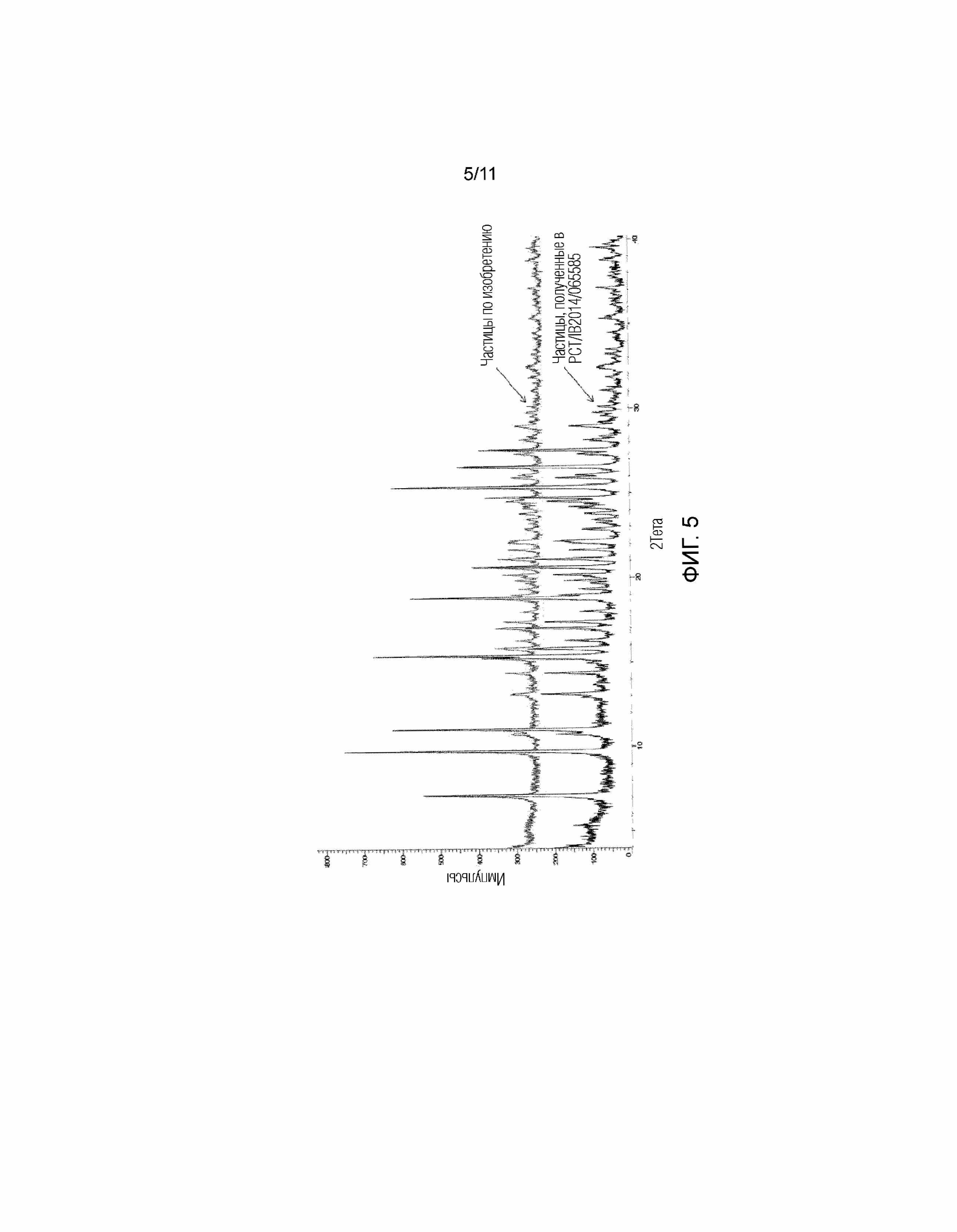


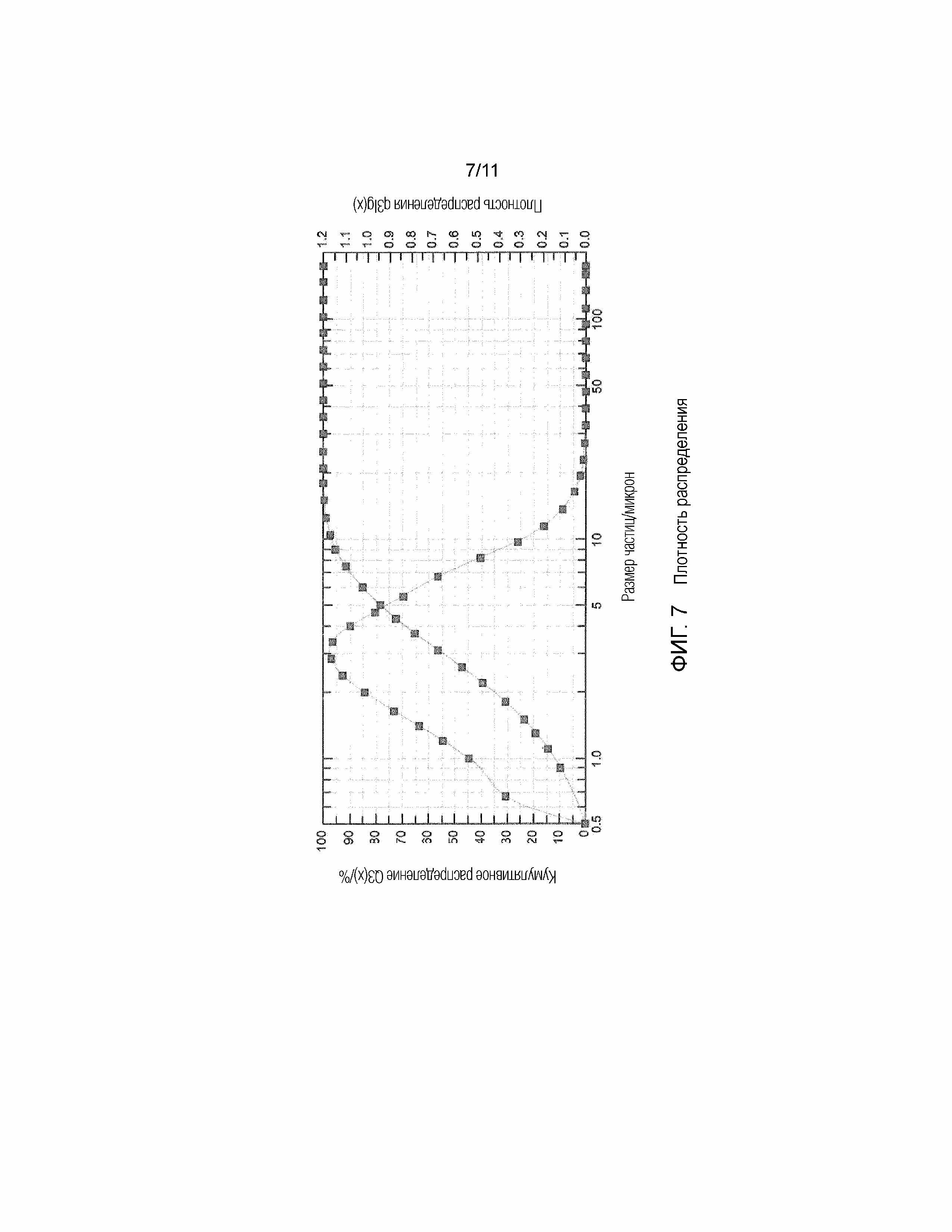
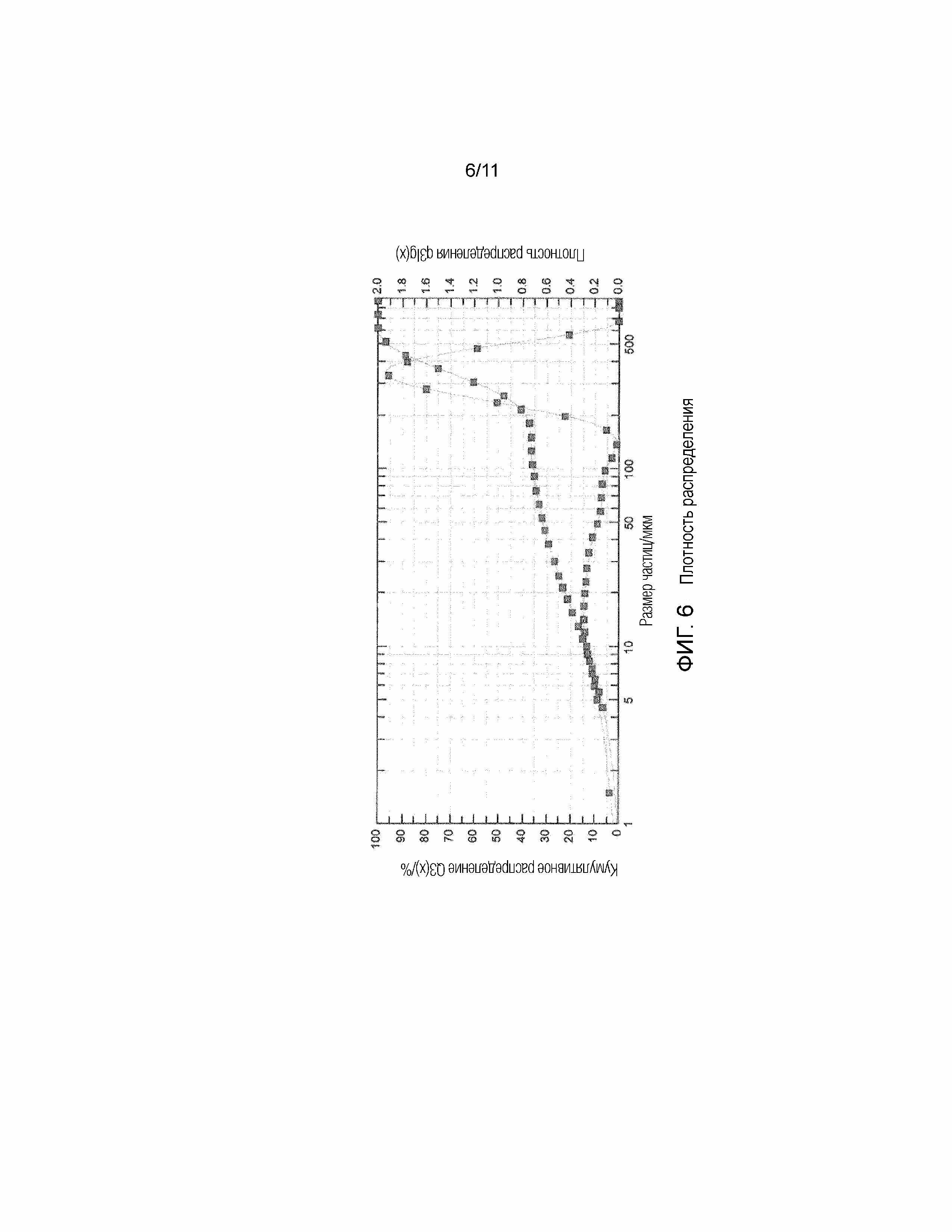

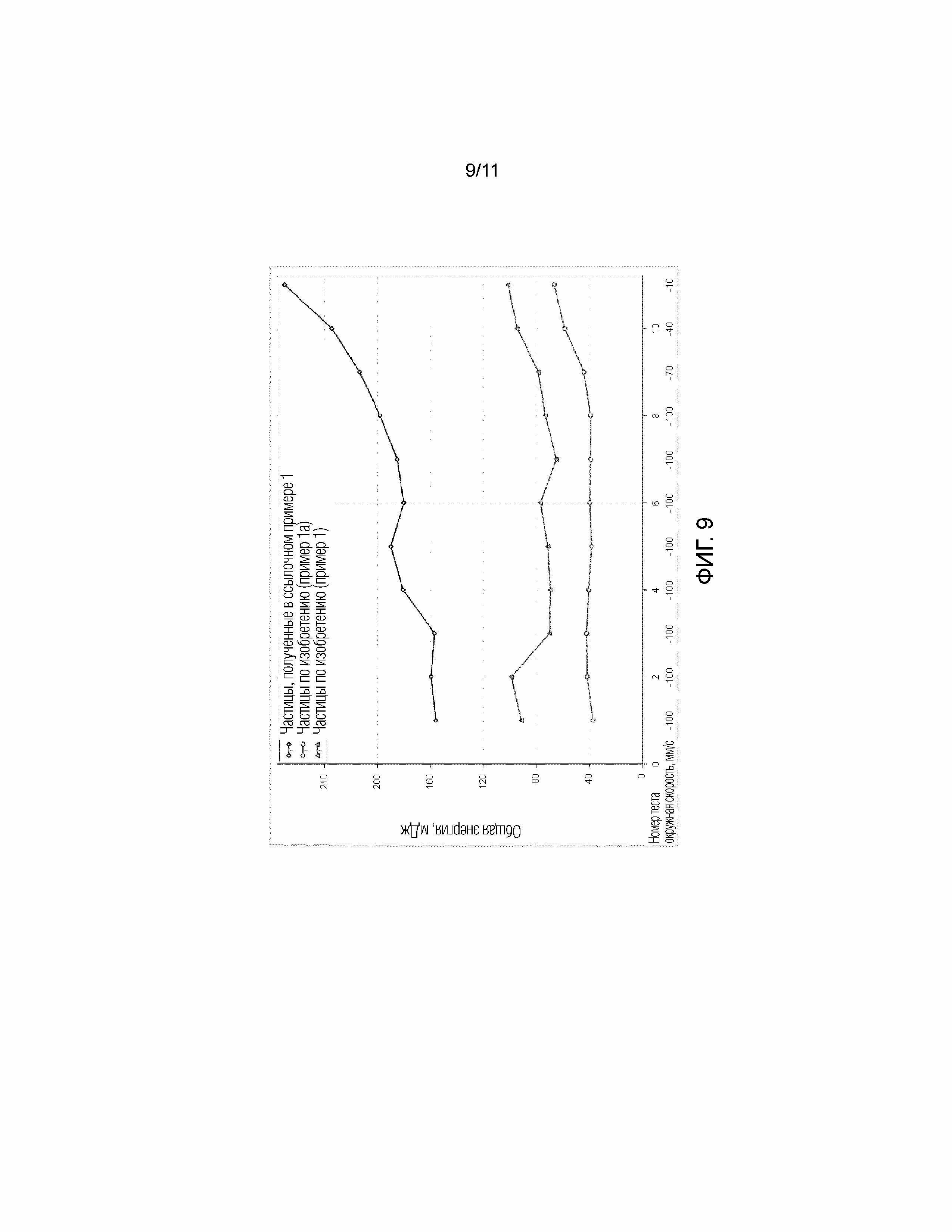
Мутантные антигены gas57 и антитела против gas57
[(1н-индол-5-ил)-гетероарилокси]-1-(азабицикло[3.3.1]нонаны, как холинергические лиганды n-achr, предназначенные для лечения психотических и нейродегенеративных нарушений
Лечение туберозного склероза
Органические соединения
Способ получения фармацевтической композиции
Предназначенная для перорального применения фармацевтическая композиция
Менингококковые полипептиды fhbp
Стабилизированные полипептиды инсулиноподобного фактора роста
Органические соединения
Курс лечения с использованием агониста рецептора s1p
Способы, терминалы и системы для содействия совместному использованию данных
Системы и способы совместного использования данных изображений

