Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ РОКСАДУСТАТА
Вид РИД
Изобретение
Изобретение относится к области химии и химико-фармацевтической промышленности и касается синтеза препарата роксадустат.
Роксадустат- (7-фенил-4-гидрокси-1-метил-изохинолин-3-карбонил)- глицин-представляет собой лекарственное средство, которое действует как ингибитор HIF-пролилгидроксилазы и тем самым увеличивает эндогенную продукцию эритропоэтина, которая стимулирует выработку гемоглобина и эритроцитов. Препарат считается первым пероральным препаратом, одобренным для пациентов, страдающих анемией при хронической почечной недостаточности (ХБП).
Получение роксадустата описано в заявке на патент WO 2004108681, где соединение получают из исходного химического соединения, представляющего собой 4-нитро-1,2-дикарбонитрил. Способ имеет высокие требования к оборудованию. Не обеспечивает высокого выхода конечного продукта.
Патент CN 103435546 (В) - 2016-08-10 раскрывает синтез роксадустата по схеме:

Способ имеет высокую себестоимость и низкую степень безопасности.
Патент CN 104892509 (В) - 2018-03-09 раскрывает способ приготовления Роксадустата из тирозина путем этерификации, циклизации, дегидрирования, окислительной перегруппировки и реакции ацилирования по схеме синтеза:
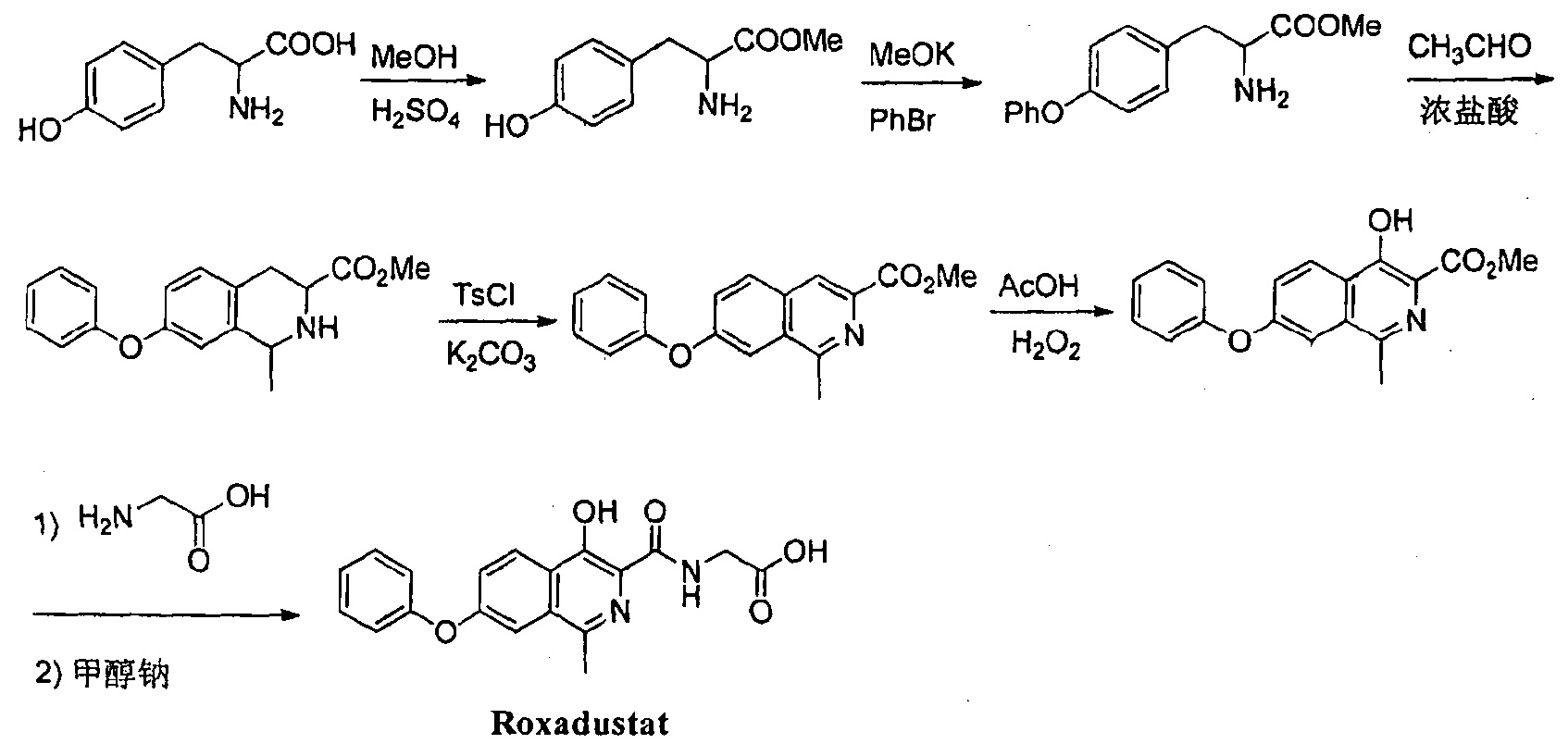
Хотя в патенте отмечается, что способ приготовления экономичен и экологичен, и подходит для промышленного производства, он не обеспечивает получение продукта без побочных реакций.
Наиболее близким решением является способ синтеза роксадустата по заявке на патент CN 108794397 (А) - 2018-11-13. Способ включает следующие стадии: 2-бром-4-метилфторбензоат берется в качестве сырья, и сырье реагирует с фенолом, бутилвиниловым эфиром, кислотой и гидроксиламином.
Синтез ведется через промежуточные продукты:
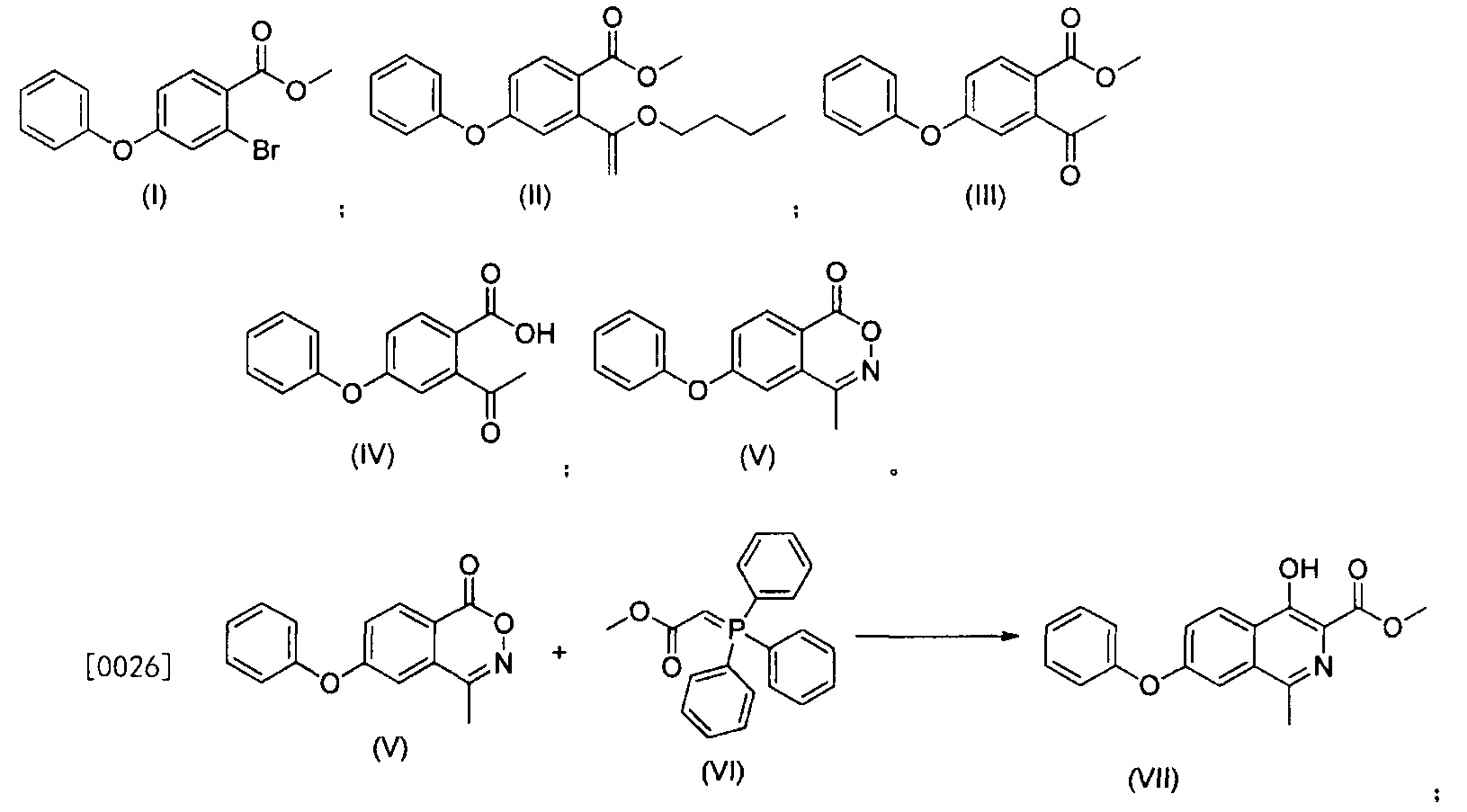
Роксадустат получают из продукта (VII). Для этого добавляют метанол, глицин, метоксид натрия, нагревают до температуры кипения с обратным холодильником, пока реакция не завершится. Реакционную смесь охлаждают до комнатной температуры, отсасывают, промывают метанолом и сушат в вакууме. Далее проводят очистку. Осадок на фильтре растворяют в воде, промывают этилацетатом. Уксусную кислоту добавляют к водному слою, кристаллы перемешивают при комнатной температуре, отфильтровывают с отсасыванием, промывают водой, промывают холодным ацетоном и сушат в вакууме. Выход 82%.
Как следует из представленного уровня техники, как правило, для синтеза используется подход, представленный на схеме:
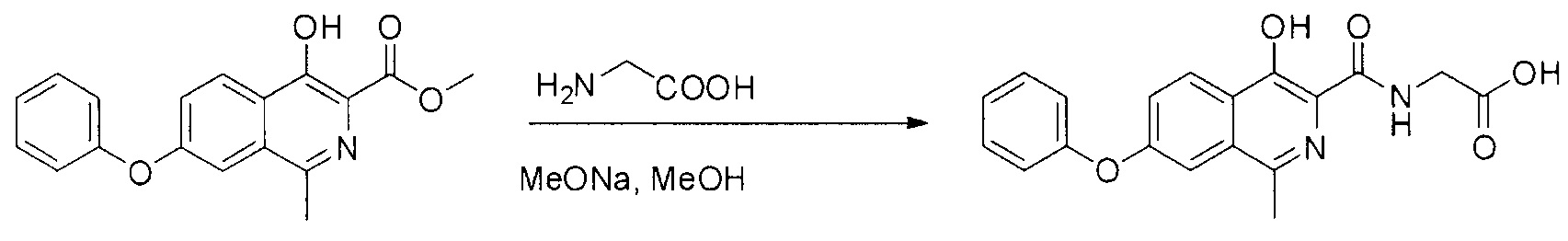
Мы установили, что данное превращение действительно протекает, однако характеризуется крайне низким выходом продуктом и приводит к образованию не только целевого роксадустата, но и продукта гидролиза исходного сложного эфира, который очень трудно отделить от целевого вещества.
Таким образом, задачей изобретения является разработка нового способа синтеза роксадустата, лишенного вышеуказанных недостатков.
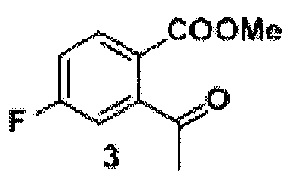
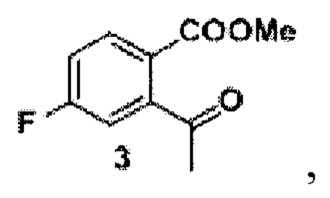
Задача решается способом, в котором получают трифторметансульфонат из исходного продукта, представляющего собой 2-гидрокси-5-фторацетофенон, реакцией с ангидридом трифторметилсульфоновой кислоты в дихлорметане в присутствии пиридина, к трифторметансульфонату добавляют 1,3-бис(дифенилфосфино) пропан и катализатор ацетат палладия (II), проводят реакцию в среде монооксида углерода под вакуумом до образования промежуточного продукта формулы 3
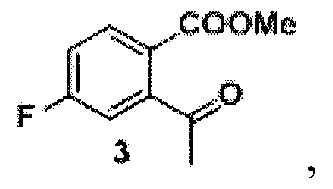
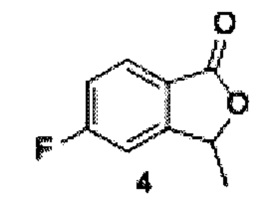
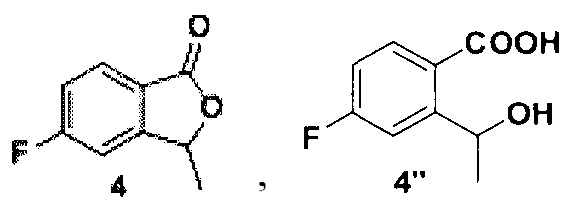
который растворяют в метаноле, добавляют борогидрид натрия, полученную смесь лактона формулы 4 и его раскрытой формы 4’’

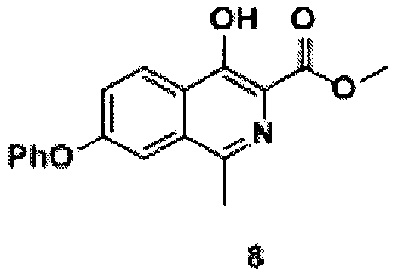
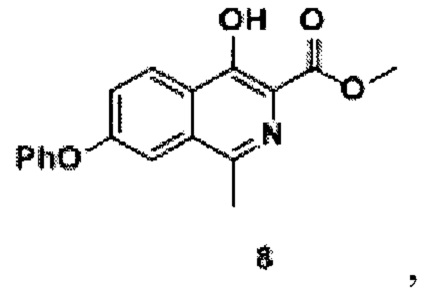
переводят в лактон (5) -3-метил-5-феноксиизобензофуран-1(3Н)-он реакцией с сухим поташем в среде фенола, полученный лактон (5), толуол, триметилборат и тионилхлорид нагревают, концентрируют, экстрагируют органическим растворителем и сушат, получая метиловый эфир 2-(1-хлорэтил)-4-феноксибензойной кислоты (6), проводят реакцию с добавлением п-толуолсульфонил разбавленного эфира глицина, йодида натрия, карбоната калия и N,N-диметилформамида до образования 2-(1-(N-метоксикарбонилметил-(толуол-4-сульфонил)-амино)этил)-метилового эфира 4-феноксибензойной кислоты (7), к которому добавляют метанол и метоксид натрия до образования соединения формулы 8
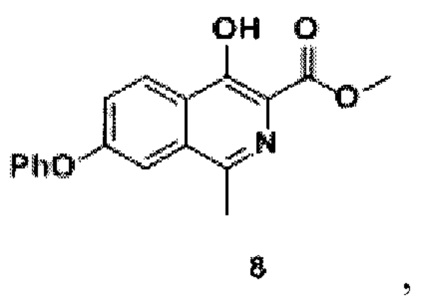
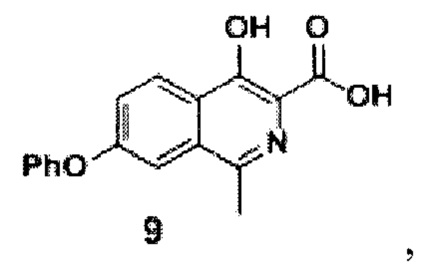
затем из соединения формулы 8 реакцией в смеси метилового спирта и воды при добавлении гидроксида натрия и перемешивании при 80-90°С в течение 10 часов получают кислоту формулы 9
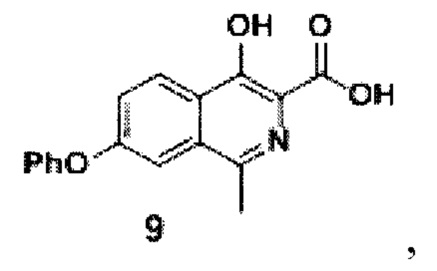
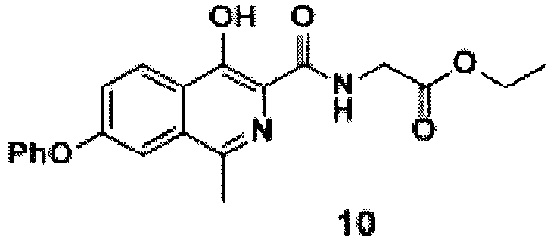
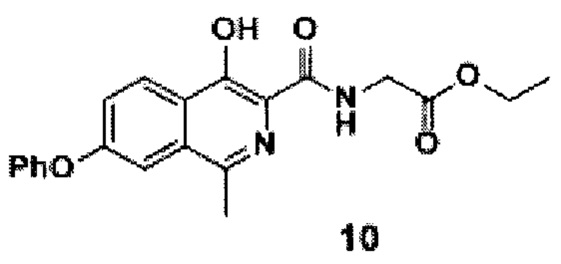
амидируют кислоту с получением амида формулы 10
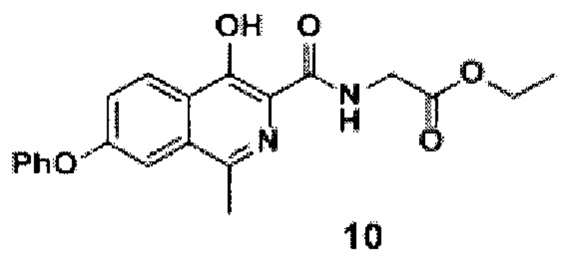
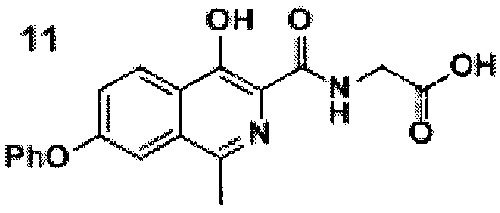
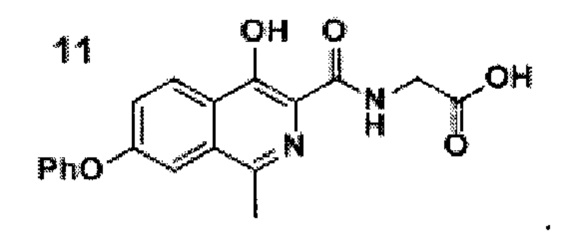
добавлением к раствору кислоты формулы 9 в ацетонитриле диизопропилэтиламина, гидрохлорида этилового эфира глицина и (2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфата (HBTU), проводят реакцию смеси амида формулы 10, этилового спирта и воды при добавлении гидроксида натрия при перемешивании при комнатной температуре в течение 10-15 часов, после окончания реакции смесь фильтруют, разбавляют водой и подкисляют уксусной кислотой до рН=4.0-4.5, перемешивают, а затем осадок отфильтровывают, промывают водой, гексаном и диэтиловым эфиром с последующей сушкой порошка роксадустата формулы 11
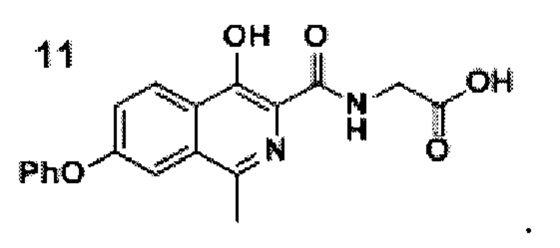
Технический результат: получение роксадустата высокой степени чистоты- не менее 98+% без какой-то дополнительной очистки. Способ имеет высокий выход продукта.
Синтез проводили по следующей схеме 1:
Схема 1. Метод синтеза роксадустата с использованием реакции карбонилирования.
Возможность осуществления изобретения может быть продемонстрирована следующими ниже представленным примером.
Пример синтеза.
Синтез соединения 2 (трифторметансульфоната)
5 г (32,5 ммоль) 2-гидрокси-5-фторацетофенона и 5,13 г (65 ммоль) пиридина растворяли в 40 мл дихлорметана, охлаждали до 0°С, после чего медленно добавляли раствор 11,9 г (42,3 ммоль) ангидрида трифторметилсульфоновой кислоты в 25 мл дихлорметана. Полученный раствор перемешивали 10 минут, нагревали до комнатной температуры и перемешивали 12 часов. К реакционной смеси добавляли 500 мл этилацетата, после чего органический слой промывали последовательно двумя порциями по 150 мл дистиллированной воды, тремя порциями по 100 мл 3% соляной кислоты, двумя порциями по 150 мл дистиллированной воды, тремя порциями по 100 мл 5% водного раствора поташа и наконец, тремя порциями по 100 мл насыщенного водного раствора хлорида натрия. Затем органический слой высушивали над безводным сульфатом натрия и упаривали на роторном испарителе. Выход 9.57 г (~100%) желтоватого масла.
1Н ЯМР (300 МГц, ДМСО-d6): δ 2.63 (с, 3Н), 7.51-7.74 (м, 2Н), 8.04 (дд, J=8.6, 2.7 Гц, 1Н).
Синтез лактона 4 (проводили без выделения промежуточного ацетофенона 3)
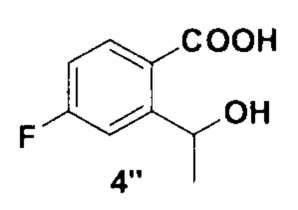
1 г (3,5 ммоль) соединения 2, 37, 58 мг (0.14 ммоль) 1,3-бис(дифенилфосфино) пропана и 31 мг (0,14 ммоль) ацетата палладия (II), помещали в сосуд Шленка. Сосуд вакуумировали и заполняли монооксидом углерода. Затем в сосуд добавляли 902 мг (7,0 ммоль) диизопропилэтиленамина, 6 мл сухого ДМФ и 3 мл сухого метанола. Раствор дегазировали под вакуумом и снова заполняли сосуд монооксидом углерода. После этого реакционную смесь перемешивали при 70°С в течение 7 часов в атмосфере монооксида углерода (давление 1 атмосфера). Полученный раствор охлаждали, разбавляли 150 мл этилацетата и промывали тремя порциями насыщенного раствора хлорида натрия (по 30 мл). Органический слой высушивали над безводным сульфатом натрия и упаривали на роторном испарителе. Полученное неочищенное соединение 3 растворяли в 15 мл сухого метанола и при перемешивании добавляли небольшими порциями 665 мг (17,5 ммоль) борогидрида натрия. Затем полученную смесь перемешивали при 60°С в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и упаривали на роторном испарителе, остаток разбавляли 200 мл этилацетата и 70 мл фосфатного буфера (рН=7, общая концентрация фосфатов 0,25 М), органический слой дважды промывали 50 мл насыщенного раствора хлорида натрия, высушивали над безводным сульфатом натрия и упаривали на роторном испарителе. Полученное вещество очищали методом колоночной хроматографии, элюент - гексан-этилацетат 25:1. Получено 352 мг (61%) продукта в виде белого порошка. На спектрах ЯМР он представляет собой смесь лактона и его раскрытой формы 4’’ (Схема 2) в соотношении 2:1.
4: 1Н ЯМР (300 МГц, ДМСО-d6): δ 1.56 (д, J=6.7 Гц, 3Н), 5.69 (кв., J=6.8 Гц, 1Н), 7.44 (тд, J=8.8, 2.2 Гц, 1Н), 7.63 (дд, J=8.5, 1.9 Гц, 1Н), 7.90 (дд, J=8.5, 4.9 Гц, 1Н).
4’’: 1Н ЯМР (300 МГц, ДМСО-d6): δ 1.31 (д, J=6.5 Гц, 3Н), 4.65-4.82 (м, 1Н), 5.29 (д, J=4.4 Гц, 1Н), 6.95-7.08 (м, 1Н), 7.08-7.21 (м, 1Н), 7.34 (м, 1Н).
Схема 2. Структура соединения 4’’ - раскрытой формы лактона 4.
Синтез соединения 5
250 мг (~1,57 ммоль) полученной на прошлом этапе смеси соединений 4 и 4’’. 294 мг (3,13 ммоль) фенола и 432 мг (3,13 ммоль) сухого поташа перемешивали при 100°С в течение 24 часов. Реакционную смесь охлаждали, разбавляли 150 мл этилацетата и промывали тремя порциями по 50 мл насыщенного раствора хлорида натрия. Органический слой высушивали над безводным сульфатом натрия и упаривали на роторном испарителе. Полученный остаток очищали методом колоночной хроматографии, элюент - гексан-этилацетат 10:1. Получено 248 мг (66%) продукта в виде бесцветного масла, которое по данным ЯМР, представляло собой чистый лактон 5.
1Н ЯМР (300 МГц, ДМСО-d6): δ 1-51 (д, J=6.7 Гц, 3Н), 5.62 (д, J=6.7 Гц, 1Н), 6.74 (д, J=8.1 Гц, 1Н), 7.16 (д, J=7.9 Гц, 3Н) 7.21-7.32 (м, 1Н) 7.49 (т, J=7.9 Гц, 2Н) 7.81 (d, J=8.4 Гц, 1Н).
Синтез соединения 6
Реакционную смесь, включающую 3-Метил-5-феноксиизобензофуран-1(3Н)-он (200 г, 0,83 моль), толуол, триметилборат (8,7 г, 83 ммоль) и тионилхлорид (119 г, 1 моль) кипятили с обратным холодильником в течение 5 часов. Растворитель и иные летучие компоненты упаривали на роторном испарителе досуха. При комнатной температуре добавляли по каплям метанол (500 мл), а затем реакционную смесь кипятили с обратным холодильником в течение 3 часов. Растворитель упаривали на роторном испарителе, к остатку добавляли метиленхлорид (1 л), воду (0,5 л) и инетнсивно перемешивали. Затем органический слой высушивали над безводным сульфатом натрия и упаривали на роторном испарителе получая соединение формулы 6, неочищенный продукт использовали непосредственно в следующей реакции.
Синтез соединения 7
Метиловый эфир 2- (1-хлорэтил) -4-феноксибензойной кислоты (соединение 6), N-п-толуолсульфонил глицина этиловый эфир (202 г, 0,83 моль), йодид натрия (12,5 г, 83 ммоль), карбонат калия (172 г, 1,24 моль) и N,N-диметилформамид (0,8 л), нагревали до 50°С и выдерживали при этой температуре 5 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (1 л), этилацетатом (1 л) и интенсивно перемешивали. Органический слой отделяли и промывали насыщенным раствором хлорида натрия (0,5 л). Затем Органический слой высушивали над безводным сульфатом натрия и упаривали на роторном испарителе с получением бледно-желтого твердого вещества, которое перекристаллизовывали из этанола с получением желаемого продукта- соединения 7. Использовали в следующей стадии без дополнительной очистки.
Синтез соединения 8
В реакционную колбу объемом 2 л добавляли 2-(1-(N-метоксикарбонилметил-(толуол-4-сульфонил)-амино)этил) - метиловый эфир 4-феноксибензойной кислоты (соединение 7, 300 г, 0,6 моль), метанол (1,5 л). При перемешивании добавляли метоксид натрия (65 г, 1,2 моль). Реакционную смесь нагревали до 40°С и выдерживали в течение 5 часов. Растворитель упаривали на роторном испарителе, добавляли к остатку воду (1 л) и ледяную уксусную кислоту (0,5 л). Перемешивали полученную смесь при комнатной температуре в течение 1 часа. Выпавший осадок отфильтровывают, промывают водой (0,5 л), сушат в вакууме (55°С), получая продукт 8 (149 г, выход 80%), 1Н NMR (700 МГц, ДМСО-d6): δ 11.47 (с, 1Н, ОН), 8.30 (д, J=8.9 Гц, 1Н, Ar), 7.56 (д, J=2.3 Гц, 1Н, Ar), 7.51 (дд, J=8.9, 2.3 Гц, 1Н, Ar), 7.48 (т, J=8.0 Гц, 2Н, Ar), 7.26 (т, J=7.4 Гц, 1Н, Ar), 7.18 (д, J=7.6 Гц, 2Н, Ar), 3.96 (с, 3Н, СН3), 2.62 (с, 3Н, СН3).
13С ЯМР (176 МГц, ДМСО-d6): δ 21.3, 52.6, 111.7, 118.5, 119.5, 122.2, 122.9, 124.6, 125.6, 130.3, 131.6, 147.8, 154.0, 155.3, 158.4, 170.4.
Найдено, m/z: 310.1075 [М+Н]+. C18H16NO4. Вычислено, m/z: 310.1079.
Далее, синтез роксадустата проводился по следующей схеме 3:
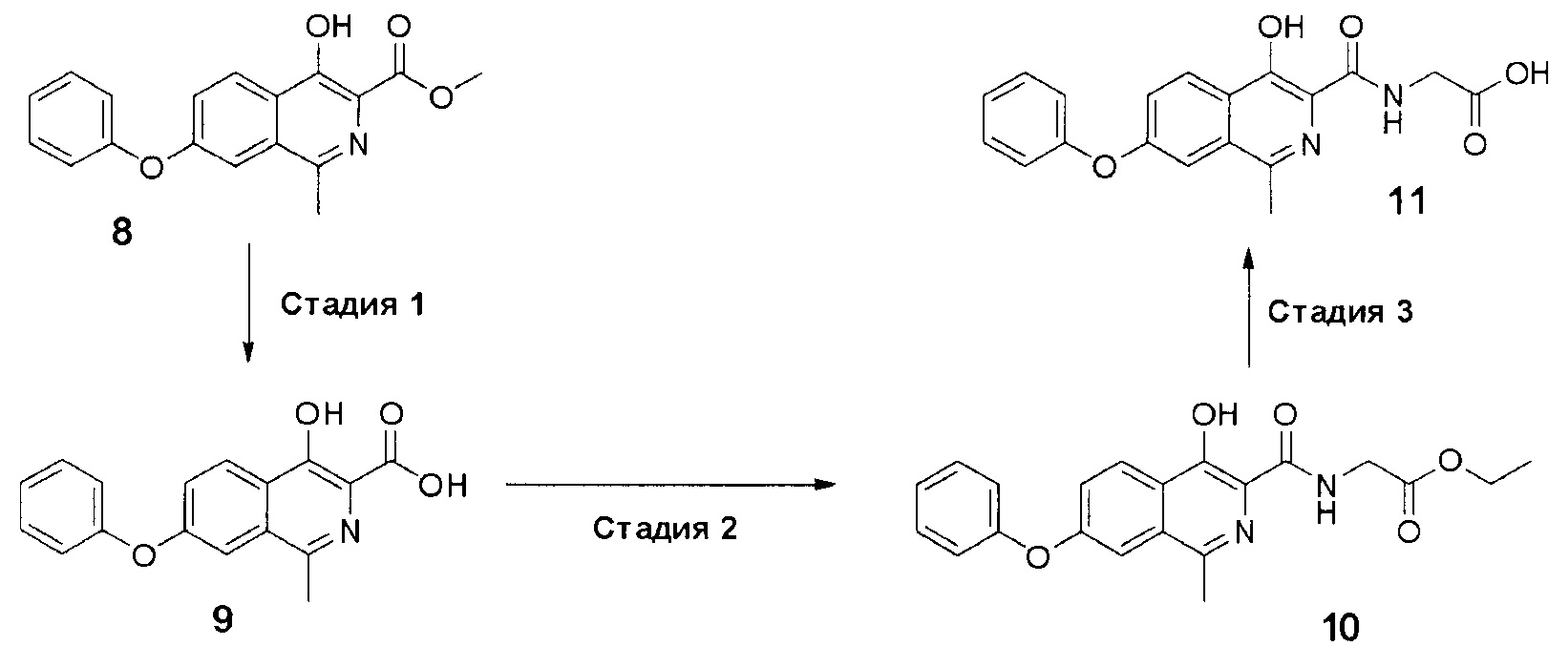
Схема 3. Успешный метод синтеза 2-(4-гидрокси-1-метил-7-феноксиизохинолин-3-арбоксамидо)уксусной кислоты из эфира.
Стадия 1. Синтез кислоты II.
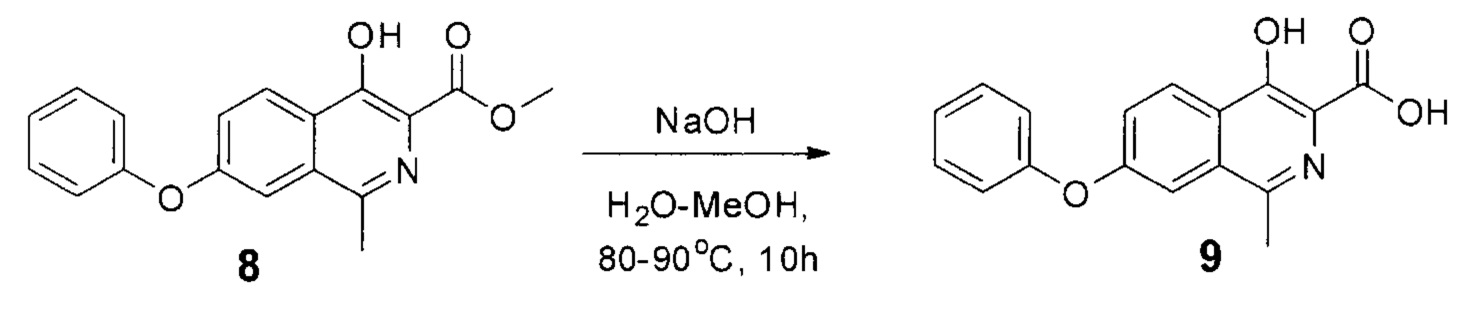
Схема 4. Стадия 1.
Протокол проведения стадии 1:
К суспензии эфира 8 (15.5 г, 0.05 моль) в смеси метилового спирта (150 мл) и воды (250 мл) прибавляли гидроксид натрия (7 г, 0.175 моль). Полученную смесь перемешивали при 80-90°С. В ходе процесса не наблюдалось растворения осадка, однако его внешний вид существенно менялся - он становился более мелким и желтоватым. За протеканием реакции следили по ТСХ (силикагель, хлороформ: метанол 5:1, Rf=0.3, перед постановкой 1 мл реакционной суспензии подкисляли уксусной кислотой до рН=4 и экстрагировали 1 мл этилацетата).
После окончания реакции (примерно 10 часов) смесь разбавляли водой (1000 мл) и подкисляли уксусной кислотой до рН=4.0-4.5. Полученную взвесь перемешивали в течение 30 минут, а затем осадок отфильтровывали, промывали водой (3×500 мл), гексаном (3x100 мл) и диэтиловым эфиром (2×75 мл). Полученный порошок сушили на воздухе при комнатной температуре в темноте в течение 2 суток. Серый крист., 14 г (выход 96%, чистота 95+% по ЯМР), т.пл. - нет. Разлагается при 240°С.
1Н NMR (700 МГц, ДМСО-d6): δ 8.30 (д, J=9.0 Гц, 1Н, Ar), 7.52 (д, J=1.9 Гц, 1Н, Ar), 7.41-7.48 (м, 3Н, Ar), 7.22 (т, J=7.3 Гц, 1Н, Ar), 7.14 (д, J=8.0 Гц, 2Н, Ar), 2.65 (с, 3Н, СН3).
13С ЯМР (176 МГц, ДМСО-d6): δ 20.1, 112.2, 119.1, 121.7, 122.3, 124.1, 126.0, 126.1, 130.2, 130.6, 142.2, 155.9, 156.9, 158.7, 170.8.
Найдено, m/z: 294.0770 [М-Н]-. C17H12NO4. Вычислено, m/z: 294.0766.
Стадия 2. Синтез амида III
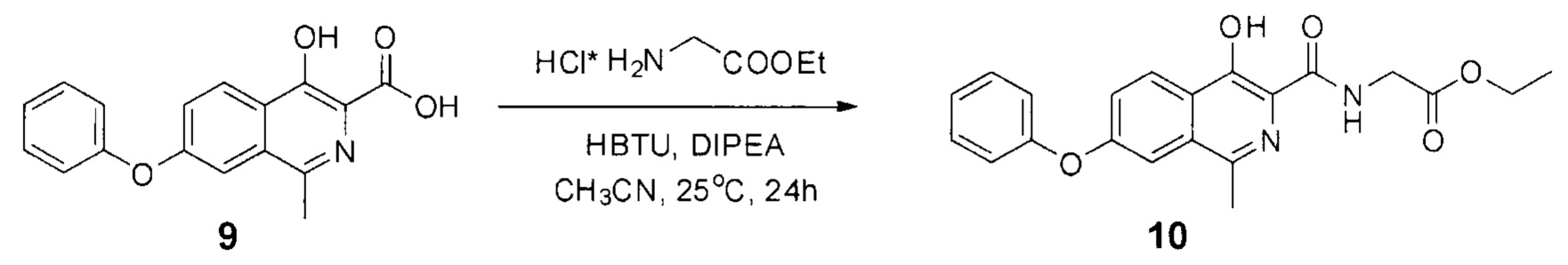
Схема 5. Стадия 2.
Протокол проведения стадии 2:
К раствору кислоты II (14.8 г, 0.05 моль) в ацетонитриле (300 мл) добавляли диизопропилэтиламин (19.4 г, 0.15 моль), а также гидрохлорид этилового эфира глицина (8.4 г, 0.06 моль) и HBTU (22.7 г, 0.06 моль). Полученную смесь перемешивали при комнатной температуре (примерно 25°С) в течение суток. За протеканием реакции следили по ТСХ (силикагель, хлороформ: метанол 10:1, Rf=0.6, перед поставновкой 1 мл реакционной смеси подкисляли уксусной кислотой до рН=4 и экстрагировали 1 мл этилацетата). В тех случаях, когда по истечении 24 часов конверсия оказывалась не полной, к реакционной смеси добавляли дополнительно по 0.02 моль HBTU, эфира глицина и DIPEA, после чего продолжали перемешивания с контролем по ТСХ.
По окончании реакции реакционную смесь выливали в 2 л воды, подкисляли уксусной кислотой до рН=4.0-4.5 и перемешивали в течение 30 минут при комнатной температуре. Затем выпавший осадок отфильтровывали, промывали водой (3×200 мл) и гексаном (3×100 мл). Полученный порошок сушили на воздухе при комнатной температуре в темноте в течение 2 суток. Затем порошок растворяли в 200 мл этилацетата (неполное растворение свидетельствует о наличии в нем остатков кислоты 9, сам продукт хорошо растворим в этилацетате - не менее 1 г на 10 мл) и фильтровали через 2-3 см силикагеля предварительно смоченного этилацетатом (общий объем силикагеля - 100 мл). Затем силикагель дополнительно промывали еще 300 этилацетата, полученные фракции объединяли и упаривали на роторном испарителе при температуре не выше 35°С. Полученный кристаллический продукт кремового цвета промывали гексаном (100 мл) и сушили на воздухе при комнатной температуре 1 сутки.
Кремовый крист., 16.2 г (выход 85%, чистота 95+% по ЯМР), т.пл. = 140-142°С.
1Н NMR (700 МГц, ДМСО-d6): δ 13.21 (с, 1Н, ОН), 9.18 (т, J=6.2 Гц, 1Н, NH), 8.28 (д, J=8.9 Гц, 1Н, Ar), 7.61 (д, J=2.5 Гц, 1Н, Ar), 7.52 (дд, J=8.9, 2.3 Гц. 1Н, Ar), 7.48 (т, J=7.9 Гц, 2Н, Ar), 7.25 (т, J=7.3 Гц, 1Н, Ar), 7.18 (д, J=8.0 Гц, 2Н, Ar), 4.15 (кв, J=7.1 Гц, 2Н, СН2), 4.13 (д, J=6.3 Гц, 2Н, СН2), 2.70 (с, 3Н, СН3), 1.22 (т, J=7.1 Гц, 3Н, СН3).
13С ЯМР (176 МГц, ДМСО-d6): δ 14.0, 21.4, 40.7, 60.6, 112.1, 119.4, 119.4, 122.4, 123.4, 124.5, 125.2, 130.3, 131.4, 146.9, 152.8, 155.5, 157.8, 169.3, 170.0.
Найдено, m/z: 381,1443 [М+Н]+. C21H21N2O5. Вычислено, m/z: 381,1445.
Стадия 3. Синтез роксадустата 11
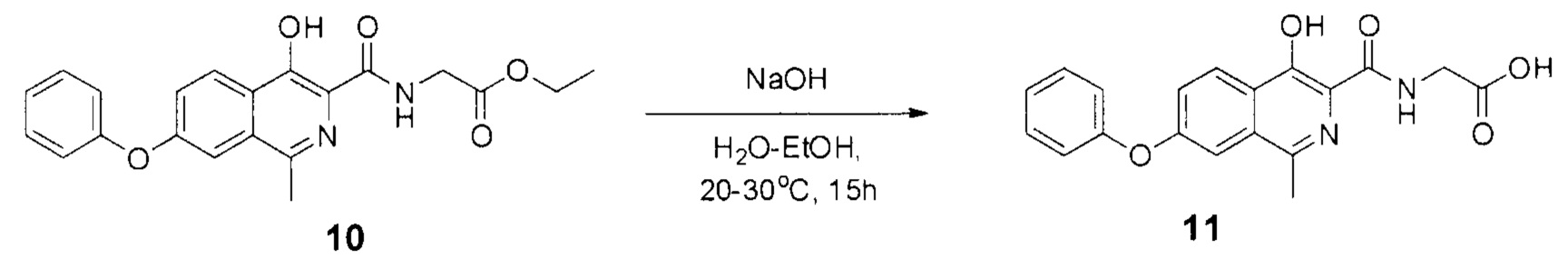
Схема 6. Стадия 3. Синтез роксадустата.
В результате превращения удается получить роксадустат чистоты более 98%. Ключевым моментом при этом является фильтрация проводимая перед осаждением продукта уксусной кислотой, так как она позволяет отделить все возможные примеси.
Наиболее оптимальный протокол проведения стадии 3:
К суспензии эфира 10 (19.0 г, 0.05 моль) в смеси этилового спирта (250 мл) и воды (500 мл) прибавляли гидроксид натрия (6 г, 0.15 моль). Полученную смесь перемешивали при комнатной температуре в течение 10-15 часов, в ходе чего наблюдалось практически полное растворение. За протеканием реакции следили по ТСХ (силикагель, хлороформ: метанол 5:1, Rf=0.2, перед постановкой 1 мл реакционной суспензии подкисляли уксусной кислотой до рН=4 и экстрагировали 1 мл этилацетата). После окончания реакции смесь фильтровали от небольшого количества взвешенных частиц, разбавляли водой (1000 мл) и подкисляли уксусной кислотой до рН=4.0-4.5. Полученную взвесь перемешивали в течение 30 минут, а затем осадок отфильтровывали, промывали водой (2×100 мл), гексаном (3×100 мл) и диэтиловым эфиром (2×75 мл). Полученный порошок сушили на воздухе при комнатной температуре в полной темноте в течение 2 суток.
Серый или желтоватый крист., 15 г (выход 86%, чистота 95+% по ЯМР), т.пл. = 232-236°С.
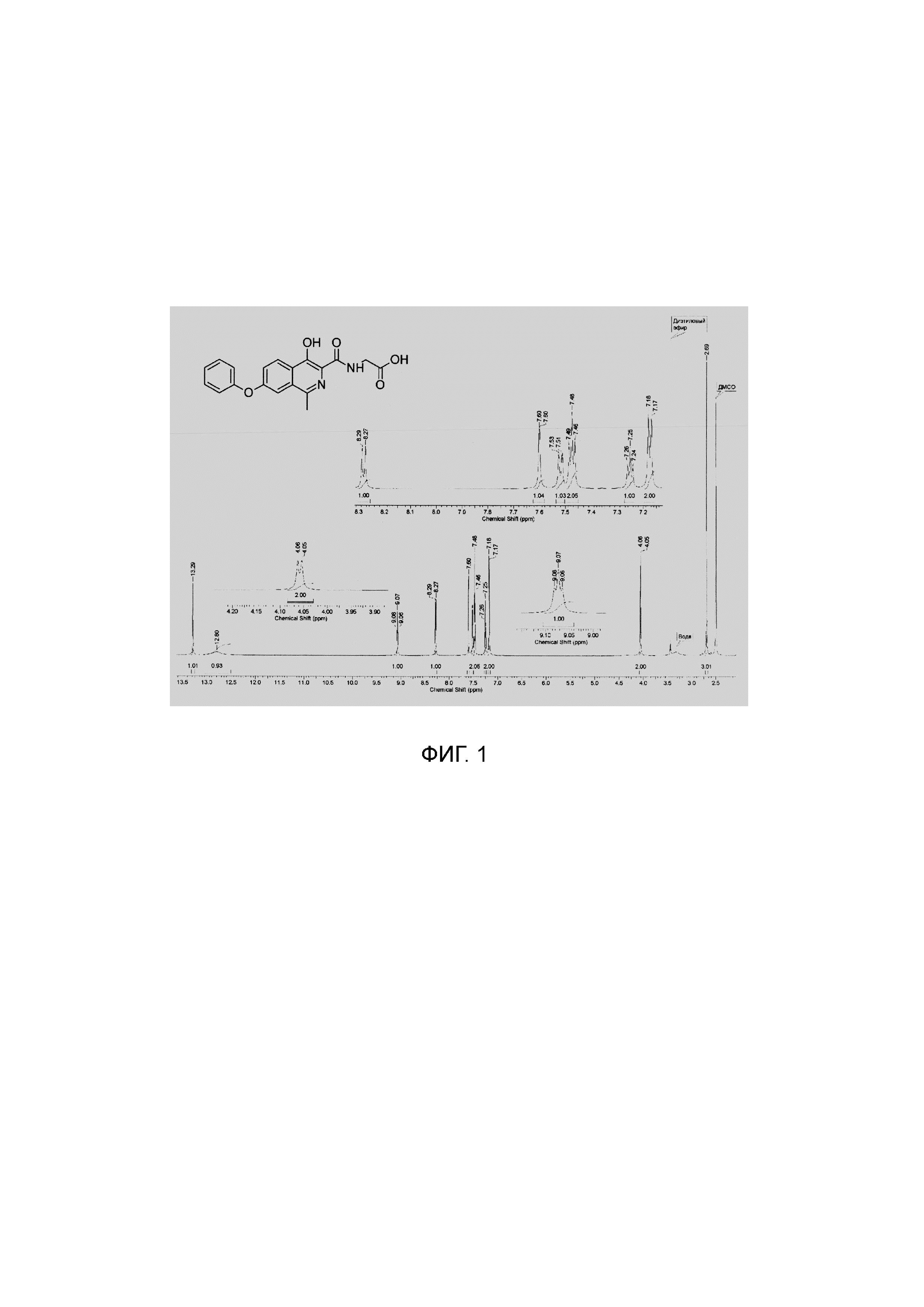
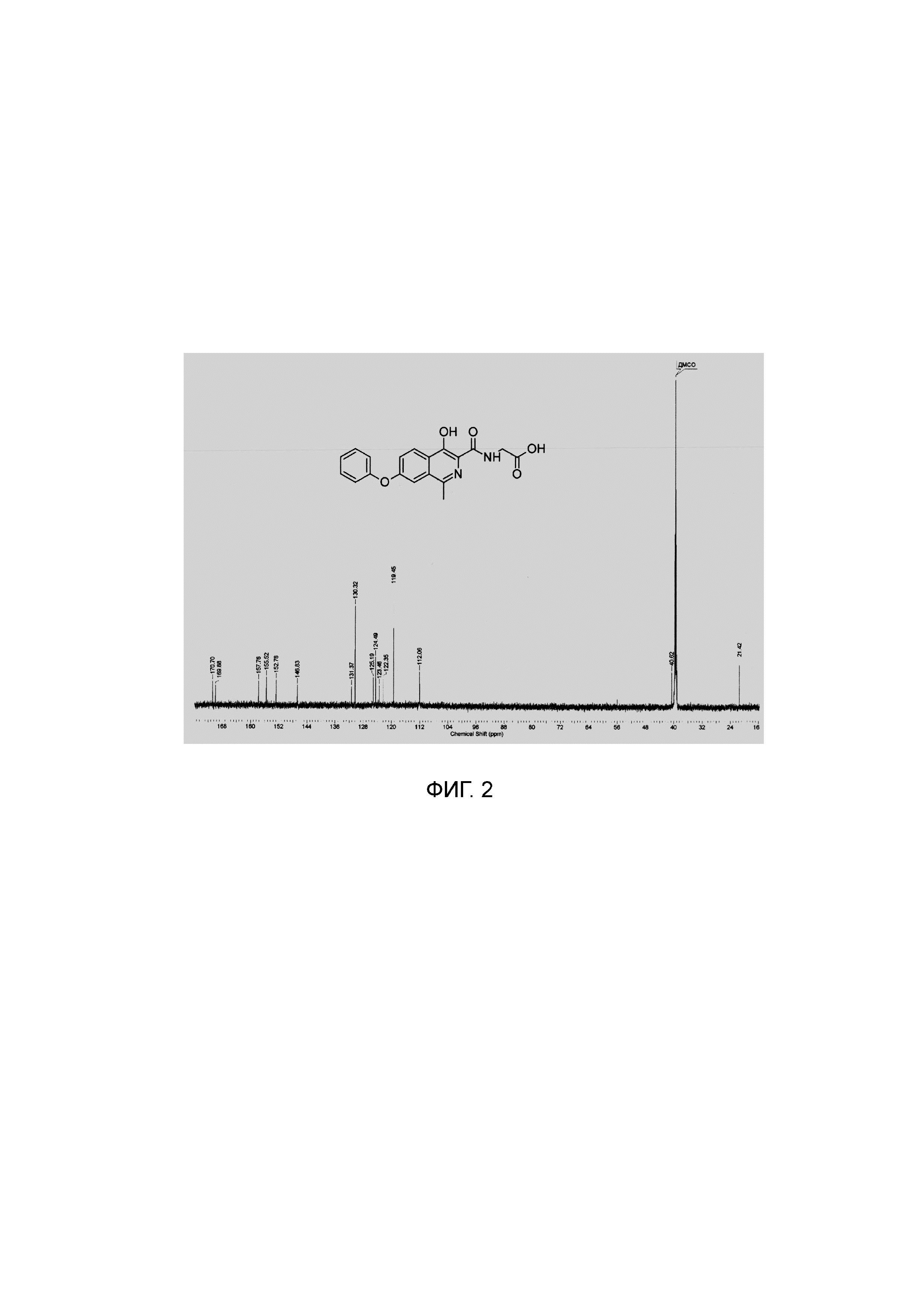
1Н NMR (700 МГц, ДМСО-d6): δ 13.29 (с, 1Н, ОН), 12.80 (уш.с, 1Н, СООН), 9.07 (т, J=5.8 Гц, 1Н, NH), 8.28 (д, J=8.9 Гц, 1Н, Ar), 7.60 (д, J=1.9 Гц, 1Н, Ar), 7.52 (дд, J=9.1, 2.0 Гц, 1Н, Ar), 7.48 (т, J=7.8 Гц, 2Н, Ar), 7.25 (т, J=7.3 Гц, 1Н, Ar), 7.18 (д, J=7.8 Гц, 2Н, Ar), 4.06 (д, J=5.9 Гц, 2Н, СН2), 2.69 (с, 3Н, СН3).
13С ЯМР (176 МГц, ДМСО-d6): δ 21.4, 40.6, 112.1, 119.5 (2 С), 122.4, 123.5, 124.5, 125.2, 130.3, 131.4, 146.8, 152.8, 155.5, 157.8, 169.9, 170.7.
Найдено, m/z: 351,0985 [М-Н]-. C19H15N2O5. Вычислено, m/z: 351,0986.
Активность в отношении HIF-PHD-2 полученной субстанции определяли по методу, описанному в Anal Biochem. 2009 Jan 15; 384(2):213-23. Процентное ингибирование вычислялось относительно неингибированного контрольного образца. Для соединения формулы 11, полученного способом, раскрытым выше, было определено, что IC50 составляет около 1,52 мкМ.
Изобретение иллюстрируется Фиг. 1, на которой отображен спектр ядерного магнитного резонанса на ядрах 1Н, зарегистрированный с раствора субстанции роксадустата в дейтерированном диметилсульфоксиде, и Фиг. 2, представляющей спектр ядерного магнитного резонанса на ядрах 13С, зарегистрированный с раствора субстанции роксадустата в дейтерированном диметилсульфоксиде.
Композиция для лечения рассеянного склероза (варианты)
Композиция для лечения рассеянного склероза (варианты)
Фармацевтическая композиция агониста рецептора s1p для лечения демиелинизационных заболеваний
Фармацевтическая композиция агониста рецептора s1p для лечения демиелинизационных заболеваний
Способ получения таблеток терифлуномида
Композиция для лечения нарушений иннерваций (варианты)
Средство для профилактики тошноты и рвоты при беременности
Фармацевтические композиции на основе n-карбамоила-метил-4-фенил-2-пирролидона
Способ получения аватромбопага
Производные 5h-дибензо[b, e][1, 4]диазепина и их применение
Композиция, обладающая эндотелиопротекторным, вазодилатирующим и ангиопротекторным эффектом
Раствор ципрофлоксацина для инфузий
Мозольный пластырь
Биологически активное средство, влияющее на общеметаболические, возбуждающие и тормозные функции нервной системы и интеллектуально-мнестические функции головного мозга (варианты)
Применение лактитола и пероральная лекарственная форма для лечения и профилактики неалкогольной жировой болезни печени
Комбинации эдаравона для лечения ишемических повреждений мозга
Новый состав n-карбамоилметил-4-фенил-2-пирролидона
Новые составы n-карбамоилметил-4-фенил-2-пирролидона
Комбинации пальмитоилэтаноламида для лечения хронической боли

