Результат интеллектуальной деятельности: ХИМИЧЕСКИЙ СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРИМИДИНА И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее раскрытие находится в области органического синтеза и направлено на способ синтеза 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-[2-(пропан-2-сульфонил)-фенил]-пиримидин-2,4-диамина (церитиниба) и/или его промежуточных соединений, способы последующего получения фармацевтических средств и фармацевтических композиций из церитиниба или из промежуточных соединений, применение промежуточных соединений для получения церитиниба и самих промежуточных соединений.
Уровень техники
Киназа анапластической лимфомы (ALK) является членом суперсемейства инсулинового рецептора рецепторных тирозин киназ. Хромосомные перестановки, в которых участвуют ALK, были обнаружены во множестве человеческих злокачественных процессов, таких как онкогенез при гематопоэтических и негематопоэтических опухолях, приводящих к нарушениям в регуляторном пути клеток. Ингибирование или супрессия путей ALK с помощью ингибитора ALK тирозин киназы вызывают остановку роста клеток и апоптоз злокачественных клеток. Исследование белков слияния ALK также выявило возможность новых терапевтических лечений для пациентов с ALK-положительными злокачественными процессами.
Церитиниб является ингибитором ALK с химической формулой 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-[2-(пропан-2-сульфонил)-фенил]-пиримидин-2,4-диамин. Способ его получения был раскрыт в WO2008/073687.
Сущность изобретения
Химические способы обычно осуществляют в мелком масштабе в фазе исследования/первоначальной разработки, и масштаб последовательно увеличивается в фазе поздней разработки для окончательного достижения полноразмерного производственного масштаба. После увеличения масштаба процесса вопросы, связанные с безопасностью процесса, становятся более важными. Невозможность увеличения масштаба должным образом может привести к потере контроля над процессом и авариям, таким как неожиданные экзотермические реакции (неконтролируемые реакции), опасности для здоровья при обработке большого колиечтсва опасных и/или токсичных химикатов или экологические опасности.
Неожиданно было обнаружено, что процесс синтеза церитиниба (5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-[2-(пропан-2-сульфонил)-фенил]-пиримидин-2,4-диамина) и его промежуточных соединений может быть осуществлен экономически эффективным и более безопасным способом. Поэтому настоящее раскрытие направлено на новый синтез церитиниба и его промежуточных соединений с использованием менее опасных химических продуктов и/или условий реакции, с меньшими потерями и с обеспечением воспроизводимого процесса, с которым проще обращаться в более крупном масштабе, процесса, который более эффективен и генерирует соединения лучшего качества.
Первым аспектом настоящего раскрытия является соединение формулы (C2-1)
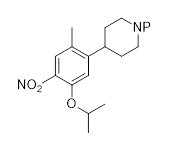 (C2-1),
(C2-1),
в которой P является защитной группой.
Следующий аспект раскрытия относится к способу получения соединения формулы (C2-1), включающему реакцию соединения формулы (A)
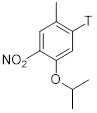 (A)
(A)
с соединением формулы (B) в растворителе
 (B)
(B)
в присутствии по меньшей мере одного катализатора, и в случае необходимости сокатализатора или добавки, в которых
P является защитной группой; и
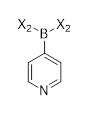
T и X1 могут быть независимо выбраны из группы, состоящей из Cl, Br, I, OTf, OTs, OPiv, MgCl, MgBr, MgI, Sn(Alkyl)3, Si(Алкил)3, Si(OАлкил)3, ZnCl, ZnBr, ZnI, Zn(Алкил), B(OH)2, B(OC(CH3)2C(CH3)2O), 9-BBN, B(Sia)2, B(Cat), B(Cy)2, BF3-, B(MIDA).
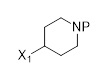
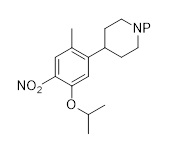
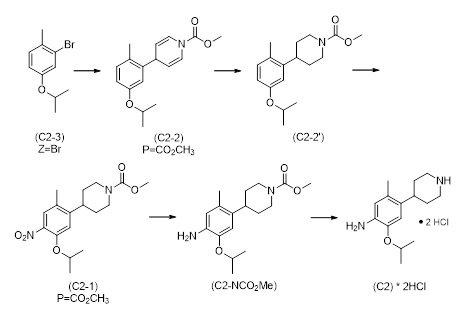
Другой аспект раскрытия относится к способу получения соединения формулы (C2-1), включающему стадии реакции соединения формулы (C2-3)
 (C2-3)
(C2-3)
с 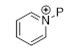 (пиридиний-P) в растворителе с получением соединения (C2-2);
(пиридиний-P) в растворителе с получением соединения (C2-2);
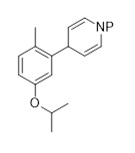 (C2-2)
(C2-2)
и превращения соединения (C2-2) с формированием соединения формулы (C2-1)
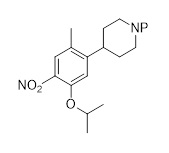 (C2-1), в которых
(C2-1), в которых
P является защитной группой и
Z выбран из MgCl, MgBr, MgI, ZnCl, ZnBr, ZnI, Zn(Алкила). Соединение (C2-2) превращают путем восстановления и нитрования для получения соединения формулы (C2-1).
Другой аспект этого раскрытия относится к способу получения соединения формулы (C2) или его соли,
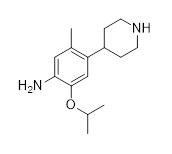 (C2),
(C2),
включающему восстановление и удаление защитной группы соединения формулы (C2-1).
Еще один аспект этого раскрытия относится к способу получения соединения формулы (C2) или его соли, включающему стадию восстановления соединения формулы (C) в растворителе
 (C)
(C)
в присутствии по меньшей мере одного катализатора и водорода.
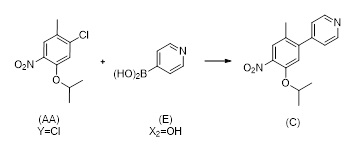
Другой аспект настоящего раскрытия относится к однореакторному способу получения соединения формулы (C) или его соли, включающему стадии реакции соединения формулы (AA) в растворителе
 (AA)
(AA)
с соединением формулы D в присутствии X3B(X2)2, по меньшей мере одного катализатора, основания и в случае необходимости лиганда,
 (D)
(D)
без выделения соединения формулы (E), причем
 (E)
(E)
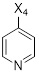
Y выбран из Br, Cl, I, OTf, OTs, OPiv и OMs;
X4 выбран из Br, Cl, I, OTf, OTs, OPiv и OMs;
B(X2)2 выбран из B(OH)2, B(OC(CH3)2C(CH3)2O), 9-BBN, B(Sia)2, B(cat), B(Cy)2, BF3- и B(MIDA); и
X3 обозначает H, B(X2)2.
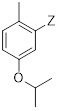
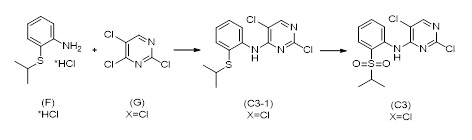
Другой аспект раскрытия относится к соединению формулы (C3-1)
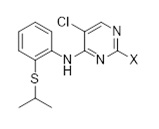 (C3-1)
(C3-1)
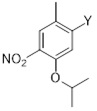
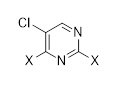
в которой X выбран из галогена (F, Cl, Br, I), алкокси, арилокси, алкилтио, тионила и сульфонила.
Другой аспект раскрытия относится к способу получения соединения формулы (C3-1), включающему стадию реакции соединения формулы (F)
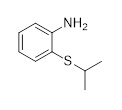 (F), или его соли,
(F), или его соли,
с соединением формулы (G)
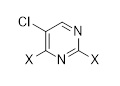 (G)
(G)
в присутствии основания и в случае необходимости в растворителе, причем X выбран из галогена (F, Cl, Br, I), алкокси, арилокси, алкилтио, арилтио, тионила, сульфонила.
Другой аспект раскрытия относится к способу получения соединения формулы (C3), включающему способ окисления (C3-1)
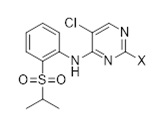 (C3)
(C3)
в растворителе, причем X выбран из галогена (F, Cl, Br, I), алкокси, арилокси, алкилтио, тионила, сульфонила.
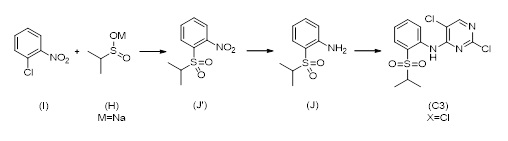
Другой аспект раскрытия относится к способу получения соединения формулы (C3), включающему стадии (i) введения в реакцию соединения формулы (H)
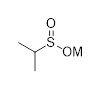 (H),
(H),
с соединением формулы (I), причем M выбран из Li, Na, K, 0.5 Zn, 0.5 Ca, с получением промежуточного соединения;
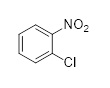 (I);
(I);
(ii) восстановления промежуточного соединения; и
(iii) введения восстановленного промежуточного соединения в реакцию с соединением формулы (G)
(G)
в присутствии основания, причем X выбран из группы, состоящей из галогена (F, Cl, Br, I), алкокси, арилокси, алкилтио, тионила, сульфонила.
Другой аспект раскрытия относится к способу получения церитиниба или его соли, включающему стадии:
i. получение соединения формулы (C2-1)
ii. получение соединения формулы (C2) или его соли,
iii. получение соединения (C3), и
iv. введение соединения формулы (C2) или его соли в реакцию с соединением формулы (C3) для получения церитиниба или его соли.
Другой аспект раскрытия относится к способу получения церитиниба или его соли, включающему стадии:
(a) получение соединения формулы (C2) или его соли,
(b) получение соединения формулы (C3), и
(c) введение соединения формулы (C2) или его соли в реакцию с соединением формулы (C3) для получения церитиниба или его соли.
Другой аспект раскрытия относится к способу получения церитиниба или его соли, включающему стадии:
(aa) получение соединения формулы (C2) или его соли,
(bb) получение соединения формулы (C3), и
(cc) введение соединения формулы (C2) или его соли в реакцию с соединением формулы (C3) для получения церитиниба или его соли.
Другой аспект раскрытия относится к способу получения церитиниба или его соли, включающему
(I) получение соединения формулы (C2) или его соли,
(II) получение соединения формулы (C3-1)
(III) получение из соединения формулы (C3-1) соединения формулы (C3), и
(IV) введение соединения формулы (C2) или его соли в реакцию с соединением формулы (C3) для получения церитиниба или его соли.
Другой аспект раскрытия относится к применению соединения формулы (C2-1) для получения церитиниба или его соли.
Другой аспект раскрытия относится к применению соединения формулы (C3-1) для получения церитиниба или его соли.
Другим аспектом раскрытия является соединение формулы (C2-2)
(C2-2), в которой
P является защитной группой.
Другим аспектом раскрытия является применение соединения формулы (C2-2) для получения церитиниба или его соли.
Подробное описание изобретения
Было замечено, что увеличение количества реагентов и растворителя для увеличения масштаба реакции до полномасштабного производства может порождать некоторые риски, такие как потеря контроля над процессом, неожиданные экзотермические реакции, аварии и проблемы безопасности при обработке большого количества опасных и/или токсичных химических веществ.
Неожиданно было обнаружено, что изменение способа синтеза церитиниба (5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-[2-(пропан-2-сульфонил)-фенил]-пиримидин-2,4-диамина) и промежуточных соединений обеспечивает масштабируемый способ, который может быть безопасно осуществлен в более крупном масштабе с воспроизводимыми выходами, менее опасными/токсичные химическими соединениями, и который приводит к меньшим потерям. Кроме того, этот способ более эффективно производит соединения более высокого качества по более низкой цене. Вкратце способ показан на Схеме 1, см. ниже.
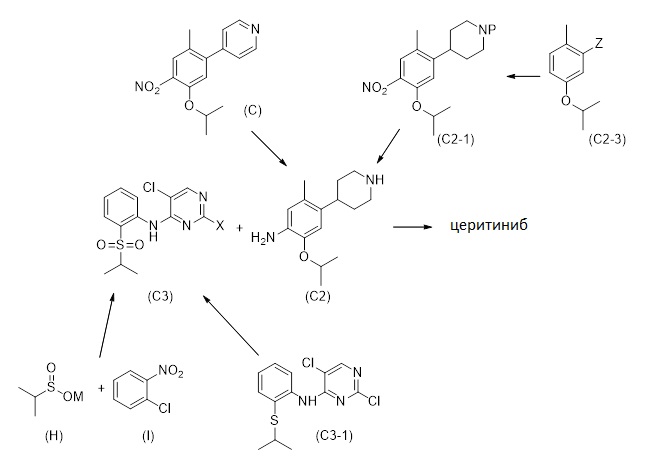
Схема 1
1.1 Соединение формулы (C2-1):
Первый аспект настоящего раскрытия относится к промежуточному соединению - соединению формулы (C2-1)
 (C2-1)
(C2-1)
в которой P является защитной группой, или в этом случае защитной группой для азота. Соединение формулы (C2-1) может использоваться для получения церитиниба.
Термин «защитная группа» или «защитная группа для азота» может присутствовать и призван защитить соответствующие функциональные группы от нежелательных побочных реакций, таких как ацилирования, этерификации, превращения в сложные эфиры, окисления, сольволиз и подобных реакций. Характеристикой защитных групп является то, что они сами легко, т.е. без или с очень ограниченными нежелательными побочными реакциями, подвергаются удалению, как правило, путем сольволиза, восстановления, фотолиза или также с помощью ферментативной активности, например, в условиях, аналогичных физиологическим условиям, и что они не присутствуют в конечных продуктах. Специалист знает или может легко установить, какие защитные группы подходят для реакций, упомянутых выше и далее. Предпочтительно, если две или более защитных группы присутствуют в одном упомянутом промежуточном соединении, их выбирают так, чтобы, если одна из групп должна быть удалена, это могло бы быть осуществлено селективно, например, с использованием двух или более разных защитных групп, которые отщепляются в разных условиях, например, один класс - умеренным гидролизом, другой - гидролизом в более жестких условиях, один класс - гидролизом в присутствии кислоты, другой - гидролизом в присутствии основания или один класс - восстановительным расщеплением (например, каталитическим гидрированием), другой - гидролизом и т.п. Подходящие защитные группы для азота обычно используются в химии пептидов и описаны, например, в соответствующих главах стандартных справочных работ, таких как J. F. W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973; T. W. Greene and P. G. M. Wuts, "Greene's Protective Groups in Organic Synthesis", Fourth Edition, Wiley, New York 2007; in "The Peptides"; Volume 3 (editors: E. Gross and J. Meienhofer), Academic Press, London and New York 1981, и в "Methoden der or ga ni schen Chemie" (Methods of Or ganic Chemistry), Houben Weyl, 4th edition, Volume 15/I, Georg Thieme Verlag, Stuttgart 1974. Предпочтительные защитные группы для азота обычно включают: C1-C6-алкил, предпочтительно C1-C4-алкил, более предпочтительно C1-C2-алкил (например, ацетил, аллил, трет-бутил), наиболее предпочтительно C1-алкил, который является моно- ди- или тризамещенный триалкилсилил-C1-C7-алкокси (например, триметилсилилэтокси), арил, предпочтительно фенил, или гетероциклическая группа (например, бензил, кумил, бензгидрил, пирролидинил, тритил, пирролидинилметил, 1-метил-1,1-диметилбензил, (фенил)метилбензол), причем арильное кольцо или гетероциклическая группа не замещены или замещены одним или более, например, двумя или тремя, остатками, например, выбранными из группы, состоящей из C1-C7-алкила, гидрокси, C1-C7-алкокси, C2-C8-алканоил-окси, галогена, нитро, циано и CF3; арил-C1-C2-алкоксикарбонил (предпочтительно фенил-C1-C2-алкоксикарбонил (например, бензилоксикарбонил (Cbz), бензилоксиметил (BOM), пивалоилоксиметил (POM)); C1-C10-алкенилоксикарбонил; C1-C6алкилкарбонил (например, ацетил или пивалоил); C6-C10-арилкарбонил; C1-C6-алкоксикарбонил (например, трет-бутоксикарбонил (Boc), метилкарбонил, трихлорэтоксикарбонил (Troc), пивалоил (Piv), аллилоксикарбонил); C6-C10-арилC1-C6-алкоксикарбонил (например, 9-флуоренилметилоксикарбонил (Fmoc)); аллил или циннамил; сульфонил или тионил; сукцинимидильная группа, силильные группы (например, триарилсилил, триалкилсилил, триэтилсилил (TES), триметилсилилэтоксиметил (SEM), триметилсилил (TMS), триизопропилсилил или трет-бутилдиметилсилил).
Согласно раскрытию предпочтительная защитная группа (P) может быть выбрана из группы, состоящей из трет-бутилоксикарбонила (Boc), бензилоксикарбонила, метилоксикарбонила или бензила.
Соединение формулы (C2-1) может быть получено способом, включающим реакцию соединения формулы (A) с соединением формулы (B) в растворителе в присутствии по меньшей мере одного катализатора, в случае необходимости сокатализатора или добавки, как определено в Схеме 2. Реакционную смесь обычно нагревают. Подходящими растворителями, используемыми для реакции, являются, например, тетрагидрофуран (THF), 2-метил тетрагидрофуран, 1,4-диоксан, простой диэтиловый эфир, толуол, диметилформамид (DMF), диметилацетамид (DMA), диметилсульфоксид (DMSO), диметоксиэтан (DME), дихлорметан, N-метил-2-пирролидон (NMP), 1-бутил-2-пирролидон (NBP), ацетонитрил, ацетон, этилацетат, изопропилацетат, трет-бутилацетат, пентан, гексан, гептан, анизол, пиридин, триэтиламин, диметилкарбонат, вода, метанол, этанол, н-пропанол, 2-пропанол, н-бутанол, 2-бутанол, трет-бутанол или их смеси.
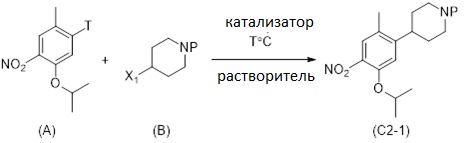
Схема 2
Несмотря на то, что выбор подходящей защитной группы (P) обширен, предпочтительной защитной группой (P) является трет-бутилоксикарбонил (Boc), бензилоксикарбонил, метилоксикарбонил, этилоксикарбонил, аллилоксикарбонил, фенилоксикарбонил, формил, ацетил или бензил. Самой предпочтительной защитной группой в этом процессе является трет-бутилоксикарбонил (Boc), бензилоксикарбонил, метилоксикарбонил или бензил.
В реакционной Схеме 2, T и X1 могут, например, быть независимо выбраны из группы, состоящей из Cl, Br, I, OTf, OTs, Opiv, или T можем быть металлической разновидностью M. Предпочтительно M является металлической разновидностью, включающей ион металла, выбранный из группы, состоящей из Mg, Al, Zn, Zr, B, Sn, Si, более предпочтительно M является ZnCl, ZnBr, ZnI, Zn(Alkyl), B(OH)2, B(OC(CH3)2C(CH3)2O), 9-BBN, B(Sia)2, B(Cat), B(Cy)2, BF3- или B(MIDA). Обычно T и X1 могут, например, независимо обозначать Cl, Br, I, OTf, OTs, OPiv, MgCl, MgBr, MgI, Sn(Alkyl)3, Si(Alkyl)3, Si(OAlkyl)3, ZnCl, ZnBr, ZnI, Zn(Alkyl), B(OH)2, B(OC(CH3)2C(CH3)2O), 9-BBN, B(Sia)2, B(Cat), B(Cy)2, BF3-, B(MIDA).
Термин «Алкил» здесь относится к радикалу или части радикала, который является прямой или разветвленной (один или, при желании и возможности, более раз) углеродной цепью. Это может быть C1-C8-алкил. Термин ʺC1-C8-ʺ определяет группу, имеющую вплоть до и включая максимально 8 атомов углерода, причем указанная группа является разветвленной (один или несколько раз) или прямой цепью и присоединена через концевой или неконцевой углерод. C1-C8-алкил, например, является н-пентилом, н-гексилом или н-гептилом, н-октилом, метилом, этилом, н-пропилом, изопропилом, н-бутилом, изобутилом, втор-бутилом, трет-бутилом, в частности, метилом, этилом, н-пропилом, изопропилом, н-бутилом, изобутилом, втор-бутилом, трет-бутилом.
Катализатор, используемый для проведения реакции, описанной в общих чертах в Схеме 2, является любым катализатором, который специалист может выбрать на основании обычного пособия. Катализатор может быть, например, выбран из группы, состоящей из Pd(PPh3)2Cl2, Pd(PPh3)4, Pd(dba)2, Pd2(dba)3, Pd(OAc)2, [Pd(allyl)Cl]2, Pd(dppf)Cl2, PdBr2(PtBu3)2, PdCl(crotyl)(PtBu3), Pd(PtBu3)2, PdCl2(Amphos)2, PdCl(allyl)(Amphos), PdBr2(Binap), PdCl2(DCPP), PdCl2(DiPrPF), PdCl2(DiPrPF), Pd-PEPPSI-IPr, Хлор(2-дициклогексилфосфино-2′,4′,6′-триизопропил-1,1′-бифенил)[2-(2-аминоэтил)фенил)]палладия(II) (также известного как XPhos Precatalyst 1st Generation), Хлор(2-дициклогексилфосфино-2′,4′,6′-триизопропил-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладия(II) (также известного как XPhos Precatalyst 2nd Generation), Хлор(2-дициклогексилфосфино-2′,6′-диметокси-1,1′-бифенил)[2-(2-аминоэтилфенил)]палладия(II) (также известного как SPhos Precatalyst 1st Generation), Хлор(2-дициклогексилфосфино-2′,6′-диметокси-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладия(II) (также известного как XPhos Precatalyst 2nd Generation), Хлор(2-дициклогексилфосфино-2',6'-диизопропокси-1,1-бифенил)[2-(2-аминоэтилфенил)]палладия(II) (также известного как RuPhos Precatalyst 1st Generation), Хлор(2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладия(II) (также известного как RuPhos Precatalyst 2nd Generation), Pd/C, Pd, Ni(acac)2, NiCl2, Ni(PPh3)2Cl2, Ni(cod)2, Ni(dppf)(cod), Ni(dppf)(циннамил), Ni(dppf)2, Ni(dppf)Cl2, Ni(dppp)Cl2, NiCl2(PCy3)2, Ni(dppe)Cl2 или их смесей.
Сокатализатор или добавка могут быть выбраны из группы, состоящей из ZnCl2, ZnBr2, CuI, LiCl, PPh3, P(oTol)3, P(oTol)Ph2, P(pTol)3, PtBu3, PtBu3*HBF4, PCy3, PCy3*HBF4, P(OiPr)3, DPE-Phos, dppf, dppe, dppp, dcpp, dppb, P(Furyl)3, CPhos, SPhos, RuPhos, XPhos, DavePhos, JohnPhos и Xantphos.
Катализатор может обычно присутствовать в количестве до 10,0 мол.%. Как правило, катализатор может присутствовать в количестве менее 5,0 мол.%. Катализатор может далее присутствовать в диапазоне от приблизительно 0,005 мол.% до приблизительно 5,0 мол.%, от приблизительно 0,01 мол.% до приблизительно 1,0 мол.%, или от приблизительно 0,05 мол.% до приблизительно 0,5 мол.%, в расчете на исходное соединение формулы (A). Как правило, катализатор может присутствовать в количестве приблизительно 0,1 мол.%.
Реакция, описанная в Схеме 2, особенно хорошо выполняется, когда T является Br, X1 является ZnI, и P является трет-бутилоксикарбонилом (Boc). Предпочтительным растворителем для реакции является тетрагидрофуран (THF), катализатором является Pd(PPh3)2Cl2, и сокатализатором или добавкой является CuI. Особенно хорошо реакция протекает при температуре выше 25°C, предпочтительно приблизительно 50°C.
Термин ʺсокатализатор или добавкаʺ в рамках изобретения относится к химическому веществу, улучшающему скорость химической реакции путем понижения энергии активации. Сокатализатор может быть гетерогенным катализатором или гомогенным катализатором, и добавка может быть лигандом, солью или любыми другими химическими разновидностями, которые могут улучшать реактивность катализатора.
Термин «лиганд» означает любое соединение, ахиральное или хиральное, которое может образовывать комплекс с переходным металлом. Хиральными и ахиральными лигандами могут быть, например, CPhos, SPhos, XPhos, DavePhos, JohnPhos, DPE-Phos и Xantphos.
1.2 Альтернативный путь к соединению формулы (C2-1)
Следующий аспект раскрытия относится к способу получения соединения формулы (C2-1), включающему стадии реакции соединения формулы (C2-3) с заместителем пиридиний-P в растворителе для получения соединения (C2-2); и превращение соединения (C2-2) для получения соединения формулы (C2-1), как описано в Схеме 3. Согласно раскрытию, защитная группа (P) может быть обычной защитной группой, особенно может быть выбрана из упомянутых здесь (например, трет-бутилоксикарбонила (Boc), бензилоксикарбонила, метилоксикарбонила, этилоксикарбонила, аллилоксикарбонила, фенилоксикарбонила, формила, ацетила или бензила). Метилоксикарбонил и Boc являются предпочтительными защитными группами, используемыми в способе, изображенном на Схеме 3. Метилоксикарбонил является самым предпочтительным.
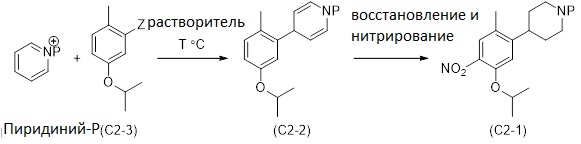
Схема 3
Реакция между пиридинием-P и соединением формулы (C2-3) лучше всего протекает, когда Z выбран из MgBr, MgI, MgCl, ZnCl, ZnBr, ZnI, Zn(Алкила) и т.п., предпочтительно когда Z является MgCl или MgBr, наиболее предпочтительно MgBr.
Подходящие растворители для получения соединения формулы (C2-2) являются, например, апротонными полярными растворителями. Например, реакция может хорошо протекать в тетрагидрофуране (THF), 2-метил тетрагидрофуране, ацетонитриле, дихлорметане, 1,4-диоксане или диэтиловом эфире или их смесях. Реакция особенно хорошо протекает в тетрагидрофуране (THF).
Синтез соединения формулы (C2-2), как описано в Схеме 3, особенно хорошо протекает, когда Z выбран как MgBr, и P выбран как метилоксикарбонил, в тетрагидрофуране (THF), при температуре от -30°C до температуры кипения растворителя. Может быть необходима температура выше, чем комнатная температура для получения хороших выходов, если Z выбран из менее реакционоспособных форм Mg или Zn и т.п. С особенно реакционоспособными заместителями Гриньяра, такими как те, в которых Z обозначает MgBr или MgCl, температура может составлять от -30°C до комнатной температуры.
Соединение формулы (C2-2) затем превращают в соединение формулы (C2-1), как описано в Схеме 3.
Восстановление осуществляют с водородом или донором водорода (гидрирование с переносом водорода) в присутствии катализатора, в присутствии или в отсутствие добавки. В качестве донора водорода может использоваться вода, кислоты (например, муравьиная кислота), щелочи, спирты, амины, нашатырный спирт, формиат аммония и т.п.
Катализатор, используемый для осуществления восстановления соединения формулы (C2-1), является любым катализатором, который специалист может выбрать на основании обычного пособия. Катализатор может быть выбран из группы, состоящей из никеля Ренея, Pt/C, Rh/C, Pd/Al2O3, Pd/CaCO3, RhCl(PPh3)3, катализатора Линдлара, PtO2, Pd/C, [Rh(cod)(PPh3)2]+, [Ir(cod)(PCy3)(Py)]+, Pd(OH)2, Pd(OAc)2, Pd2(dba)3, Zn, Fe, Sm, NiCl2, Ni(OAc)2, CoCl2, ZrCl4,TiCl3. Реакция особенно хорошо протекает с Pd/C. Катализатор может присутствовать в диапазоне от приблизительно 0,005 мол.% до приблизительно 30,0 мол.%. Как правило, катализатор может присутствовать в количестве приблизительно 10,0 мол.%.
Добавка может в случае необходимости быть добавлена и может быть выбрана из группы ZnCl2, ZnBr2, CuI, LiCl, PPh3, P(oTol)3, P(oTol)Ph2, P(pTol)3, PtBu3, PtBu3*HBF4, PCy3, PCy3*HBF4, P(OiPr)3, DPE-Phos, dppf, dppe, dppp, dcpp, dppb, P(Furyl)3, CPhos, SPhos, RuPhos, XPhos, DavePhos, JohnPhos и Xantphos.
Восстановление может быть, например, осуществлено в растворителе, выбранном из группы, состоящей из воды, метанола, этанола, пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, уксусной кислоты, тетрагидрофурана (THF), дихлорметана, диэтилового эфира, трет-бутилметилового эфира, этилацетата, толуола, 1,4-диоксана, ацетонитрила или ацетона или их смесей.
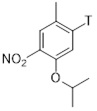
Нитрирование бензольного кольца, как указано в Схеме 3, представляет собой любую реакцией нитрирования, которую специалист может выбрать на основе обычного пособия. Источник «NO2» может быть выбран из HNO3 или его соли. Подходящие растворители для превращения могут быть, например, выбраны из уксусного ангидрида, серной кислоты или трифторуксусной кислоты.
Восстановление, как описано на Схеме 3, особенно хорошо протекает с 10%-м Pd/C, формиатом аммония, в метаноле и тетрагидрофуране (THF) при комнатной температуре. Нитрирование особенно хорошо протекает в присутствии HNO3 и уксусного ангидрида. Нитрогруппа может быть введена в бензольное кольцо при температуре ниже 0 °C, в частности, приблизительно при -10°C.
1.3 Соединение формулы (C2):
Соединение формулы (C2), или его соль, может быть получено согласно способу, представленному на Схеме 4. Способ включает восстановление и удаление защитных групп от соединения формулы (C2-1), как раскрыто здесь, для получения соединения формулы (C2) или его соли.
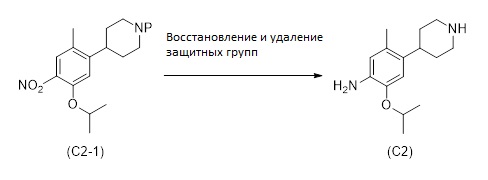
Схема 4
В одном варианте осуществления восстановление соединения (C2-1), имеющего защитную группу P, как описано для Схемы 2, осуществляют с использованием водорода в присутствии катализатора. Защитная группа P относится к защитной группе для азота, выбранной из вышеупомянутого списка (например, трет-бутилоксикарбонила (Boc), бензилоксикарбонила, метилоксикарбонила, этилоксикарбонила, аллилоксикарбонила, фенилоксикарбонила, формила, ацетила или бензила).
Катализатор, используемый для осуществления восстановления соединения формулы (C2-1), является любым катализатором, который специалист может выбрать на основе обычного пособия. Катализатор может быть выбран из группы, состоящей из никеля Ренея, Pt/C, Rh/C, Pd/Al2O3, Pd/CaCO3, RhCl(PPh3)3, катализатора Линдлара, PtO2, Pd/C, [Rh(cod)(PPh3)2]+, [Ir(cod)(PCy3)(Py)]+, Pd(OH)2, Pd(OAc)2, Pd2(dba)3, Zn, Fe, Sm, NiCl2, Ni(OAc)2, CoCl2, ZrCl4,TiCl3. Катализатор может присутствовать в диапазоне от приблизительно 0,005 мол.% до приблизительно 20,0 мол.%. Как правило, катализатор может присутствовать в количестве менее 10,0 мол.% (до приблизительно 0,005 мол.%).
Термин «катализатор» в рамках изобретения относится к каталитическому количеству химического вещества, улучшающего скорость химической реакции путем понижения энергии активации для химической реакции. Катализатор может быть гетерогенным катализатором или гомогенным катализатором.
Термин «гетерогенный катализатор» относится к катализатору, помещенному на носитель, как правило, хотя и не обязательно, субстрат, состоявший из неорганического вещества, например, пористого материала, такого как углерод, диоксид кремния и/или оксид алюминия.
Термин «гомогенный катализатор» относится к катализатору, не помещенному на носитель.
Термины «водород» или «гидрирование», используемые для описания химической реакции, относятся к действию по восстановлению другого соединения в присутствии водорода. Источник водорода может быть выбран из газообразного водорода (H2), доноров водорода (гидрирование с переносом водорода, например, муравьиной кислоты или ее соли), гидридного реагента (BH3, B2H6. NaBH4) и т.п.
Восстаноление может быть осуществлено в растворителе, таком как раствор на основе спирта. Раствор на основе спирта может включать или состоять из спиртов C1-C10 (например, метанола, этанола, пропанола, изопропанола и бутанола) или их смесей.
Реакция восстановления лучше всего протекает при повышенном давлении, предпочтительно от 1 бар до 10 бар, в частности, при 4 бар. Восстановление соединения формулы (C2-1) или его соли лучше всего осуществлять путем перемешивания реакционной смеси в течение 5 часов при комнатной температуре с 10 мол.% Pd/C и водорода в присутствии этанола. Восстановление лучше всего протекает с защитной группой P, выбранной из трет-бутилоксикарбонила (Boc) или метилоксикарбонила.
Термин «комнатная температура» или «температура среды», в рамках изобретения, если не указано иное, означает температуру от 15 до 30°C, такую как от 20 до 30°C, особенно такую как от 20 до 25°C.
В дополнение к восстановлению соединения (C2-1) отщепляют защитную группу. Удаление защитной группы может быть осуществлено в стандартных условиях реакции, известных в данной области техники. Если не указано иное, защитная группа может быть удалена в отсутствие или, обычно, в присутствии кислот или оснований, предпочтительно кислоты или основания, вызывающих удаление защитной группы, но одновременно не вызывающих химическую деградацию соединений и промежуточных соединений. Удаление защитной группы может также быть осуществлено в восстановительных условиях, например, в ходе первой стадии восстановления нитрогруппы (C2-1). Предпочтительно, защитную группу удаляют с использованием кислоты. Особенно подходящими кислотами для удаления защитной группы P являются HF.пиридин, HF.триэтиламин аммоний фторид, гексафторизопропанол, уксусная кислота, трифторуксусная кислота, соляная кислота, серная кислота или их комбинация. Предпочтительно кислота является трифторуксусной кислотой или соляной кислотой. Реакция, состоящая из удаления защитной группы, может иметь место в растворителе, облегчающем удаление защитной группы. Например, защитная группа может быть удалена в растворителе, выбранном из группы, состоящей из дихлорметана, этилацетата, 1,4-диоксана, диэтилового эфира, тетрагидрофурана (THF), метанола или ацетонитрила. Защитную группу удаляют путем перемешивания реакционной смеси в течение по меньшей мере 8 часов, предпочтительно в течение 16 часов при температуре от -78°C до 70°C, предпочтительно от 0°C до 70°C. Реакцию удаления защитной группы для трет-бутилоксикарбонила (Boc) лучше всего осуществлять с трифторуксусной кислотой в дихлорметане, в случае необходимости при температуре среды. Реакцию удаления защитной группы для метилоксикарбонила лучше всего осуществлять с соляной кислотой, в случае необходимости при 60°C.
В одном варианте осуществления восстановление и удаление защитной группы осуществляют одновременно.
1.4 Альтернативный путь получения соединения формулы (C2):
Соединение формулы (C2), или его соль, может быть получено альтернативным способом. А именно, соединение формулы (C) восстанавливают в растворителе в присутствии по меньшей мере одного катализатора и водорода, как изображено на Схеме 5.
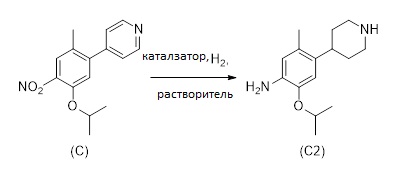
Схема 5
Реакция для получения соединения формулы (C2) или его соли может быть осуществлена как однореакторный процесс путем восстановления двух функциональных групп, одновременно присутствующих на соединении формулы (C), для обеспечения быстрого доступа к соединению формулы (C2) или его соли. Стадия восстановления включает присутствие катализатора и водорода в растворителе.
Катализатор, используемый для осуществления восстановления соединения формулы (C), является любым катализатором, который специалист может выбрать на основе обычного пособия. Катализатор, например, может быть выбран из группы, состоящей из никеля Ренея, Pt/C, Rh/C, Pd/Al2O3, Pd/CaCO3, RhCl(PPh3)3, катализатор Линдлара, PtO2, Pd/C, [Rh(cod)(PPh3)2]+, [Ir(cod)(PCy3)(Py)]+, Pd(OH)2, Pd/Al, Pt/Al, Pt/SiAl and Pd/ZrO2 или их смесей.
Катализатор может присутствовать в диапазоне от приблизительно 0,005 мол.% до приблизительно 50,0% вес./вес. (сухой) в расчете на исходное соединение формулы (C). Как правило, катализатор может присутствовать в количестве менее 20,0% вес./вес. (сухой).
Восстановление соединения формулы (C) осуществляют путем перемешивания реакционной смеси в течение максимум 16 часов, предпочтительно от 8 до 16 часов при температуре от 10 до 40 °C, предпочтительно от 20 до 30 °C. Реакция лучше всего протекает при давлении от 1 до 15 бар, предпочтительно от 2 до 6 бар.
Растворитель, используемый в реакции, может быть, например, выбран из группы, состоящей из воды, метанола, этанола, пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, уксусной кислоты, тетрагидрофурана (THF), дихлорметана, диэтилового эфира, трет-бутилметилового эфира, этилацетата, толуола, бензола, 1,4-диоксана, ацетонитрила и ацетона или их смесей.
Процесс восстановления соединения формулы (C) до соединения формулы (C2) или его соли особенно хорошо протекает в уксусной кислоте в присутствии Pd/Al и водорода. Условия реакции предпочтительно устанавливают на приблизительно 60°C и давление приблизительно 80 бар. Реакция также хорошо протекает в присутствии Pt/C и водорода, в случае необходимости в уксусной кислоте. В этом случае температура является лучшей, если ее устанавливают на значении от приблизительно 10 до приблизительно 50°C и давление от приблизительно 1 до приблизительно 10 бар.
1.5 Соединение формулы (C):
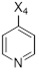
Соединение (C) может быть получено как показано на Схеме 6 в однореакторном процессе путем реакции в растворителе соединения формулы (AA) с соединением формулы (D) в присутствии X3B(X2)2, основания, катализатора, и в случае необходимости лиганда, причем:
Y выбран из Cl, Br, I, OTf, OTs, OPiv и OMs, предпочтительно Cl, Br;
X4 выбран из Cl, Br, I, OTf, OTs, OPiv и OMs, предпочтительно Cl, Br;
B(X2)2 выбран из B(OH)2, B(OC(CH3)2C(CH3)2O), 9-BBN, B(Sia)2, B(Cat), B(Cy)2;
X3 обозначает H, B(X2)2.
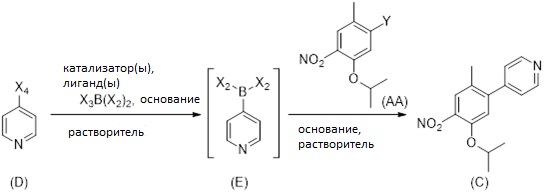
Схема 6
Один вариант осуществления раскрытия относится к in-situ формированию соединения формулы (E), без выделения соединения формулы (E), в растворителе в присутствии катализатора, X3B(X2)2, основания и в случае необходимости лиганда. Реакция может быть однореакторной реакцией. Процесс реакции соединений формулы (AA) и формулы (D) осуществляют сначала перемешиванием реакционной смеси, содержащей соединение формулы (D) и катализатор в течение приблизительно от 1 до 10 часов, предпочтительно в течение 3 часов при температуре от 40°C до температуры кипения растворителя, предпочтительно при 100°C. Затем добавляют соединение формулы (AA), и реакционную смесь перемешивают в течение приблизительно от 1 до 48 часов, предпочтительно в течение приблизительно 17 часов при температуре от 40°C до температуры кипения растворителя, предпочтительно приблизительно при 100°C.
Катализатор, используемый в однореакторной реакции, является любым катализатором, который специалист может выбрать на основании обычного пособия. Катализатор может включать лиганд и может, например, быть выбран из группы, состоящей из Pd(PPh3)2Cl2, Pd(PPh3)4, Pd(dba)2, Pd2(dba)3, Pd(OAc)2, [Pd(аллил)Cl]2, Pd(dppf)Cl2, PdBr2(PtBu3)2, PdCl(кротил)(PtBu3), Pd(PtBu3)2, PdCl2(Amphos)2, PdCl(аллил)(Amphos), PdBr2(Binap), PdCl2(dcpp), PdCl2(DiPrPF), PdCl2(DiPrPF), Pd-PEPPSI-IPr, Хлор(2-дициклогексилфосфино-2′,4′,6′-триизопропил-1,1′-бифенил)[2-(2-аминоэтил)фенил)]палладия(II), Хлор(2-дициклогексилфосфино-2′,4′,6′-триизопропил-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладия(II), Хлор(2-дициклогексилфосфино-2′,6′-диметокси-1,1′-бифенил)[2-(2-аминоэтилфенил)]палладия(II), Хлор(2-дициклогексилфосфино-2′,6′-диметокси-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладия(II), Хлор(2-дициклогексилфосфино-2',6'-диизопропокси-1,1-бифенил)[2-(2-аминоэтилфенил)]палладия(II), Хлор(2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладия(II), Pd/C, Pd, Ni(acac)2, NiCl2, Ni(PPh3)2Cl2, Ni(cod)2, Ni(dppf)(cod), Ni(dppf)(циннамил), Ni(dppf)2, Ni(dppf)Cl2, Ni(dppp)Cl2, NiCl2(PCy3)2and Ni(dppe)Cl2. Катализатор может присутствовать в количестве до 10,0 мол.%. Как правило, катализатор может присутствовать в количестве менее 6,0 мол.%.
Лиганд, используемый для осуществления однореакторной реакции, является любым лигандом, который специалист мог бы выбрать на основании обычного пособия. Лиганд может быть выбран из группы, состоящей из PPh3, P(oTol)3, P(oTol)Ph2, P(pTol)3, PtBu3, PtBu3*HBF4, PCy3, PCy3*HBF4, P(OiPr)3, DPE-Phos, dppf, dppe, dppp, dcpp, dppb, P(фурил)3, CPhos, SPhos, RuPhos, XPhos, DavePhos, JohnPhos и Xantphos. Лиганд может присутствовать в диапазоне от приблизительно 0,005 мол.% до приблизительно 20 мол.%. Как правило, лиганд может присутствовать в количестве менее 10 мол.%.
Реакция может быть проведена в растворителе, выбранном, например, из 1,4-диоксана, тетрагидрофурана (THF), 2-метил-тетрагидрофурана, диэтилового эфира, толуола, диметилформамида (DMF), диметилацетамида (DMA), диметилсульфоксида (DMSO), диметоксиэтана (DME), дихлорметана, N-метил-2-пирролидона (NMP), 1-бутил-2-пирролидона (NBP), ацетонитрила, ацетона, диметилкарбоната, этилацетата, изопропилацетата, трет-бутилацетата, пентана, гексана, гептана, анизола, пиридина, триэтиламина, воды, метанола, этанола, н-пропанола, 2-пропанола, н-бутанола, 2-бутанола, трет-бутанола или их смесей.
Основание, используемое для осуществления реакции, является любым основанием, что специалист мог бы выбрать на основании обычного пособия. Основание может представлять собой, например, Na2CO3, K2CO3, Cs2CO3, Tl2CO3, NaHCO3, KHCO3, NaOAc, KOAc, Na3PO4, K3PO4, LiOH, NaOH, KOH, CsOH, Ba(OH)2, NaOMe, KOMe, NaOEt, KOEt, TlOEt, NaOPh, NEt3, DIPEA, NaOtBu, KOtBu, KF или CsF.
Заместитель боронильной группы или X3B(X2)2 может образовывать вместе с бором группу формулы -B(X2)2, в которой два заместителя X2 являются одинаковыми или разными и могут представлять собой галоген, гидрокси, C1-C4 алкокси, или два заместителя X2 вместе образуют остаток диола. Предпочтительно, боронильная группа формулы -B(X2)2 может быть группой формулы -B(OR')ORʺ, в которой R' и Rʺ являются, независимо от друг друга, одинаковыми или разными и каждая может представлять собой водород или C1-C12-алкил, и где R' и Rʺ могут быть соединены циклическим образом, например, R и R',будучи объединенными, представляют собой алкилен, который вместе с атомами бора и кислорода образует 5- или 6-членное кольцо. Производное бора, используемое для осуществления реакции, может являться любым органобороновым производным, которое специалист может выбрать на основании обычного пособия. Органоборон может быть выбран, например, из группы, состоящей из бис(пинаколято)диборон, тетрагидроксидиборон, пинаколборан и неопентилгликольборан.
Реакция соединения формулы (D) с органобороном приводит к соединению формулы (E), имеющему группы -B(X2)2, выбранные из группы, состоящей из -B(OH)2, -B(OC(CH3)2C(CH3)2O).
Процесс генерации in-situ соединения формулы (E) путем реакции соединения формулы (D) особенно хорошо протекает в 1,4-диоксане в присутствии бис(пинаколято)диборона, X-Phos, хлор(2-дициклогексилфосфино-2′,4′,6′-триизопропил-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладия (II) и KOAc. Температура может быть установлена при 100 °C. Затем соединение формулы (E), полученное in-situ, вводят в реакцию с соединением формулы (AA) в присутствии основания в растворителе. Реакция лучше всего протекает при температуре от 40 °C до температуры кипения растворителя, предпочтительно при 100 °C. Воду и K2CO3 добавляют к реакционной смеси для инициации реакции сочетания между недавно образованным соединением (E) и соединением формулы (AA). Реакция может продолжаться с достижением температуры 100 °C.
Термин ʺоднореакторный процессʺ относится к факту, что соответствующую стадию осуществляют в последовательности, не выделяя продукт каждой стадии. Кроме того, процесс дополнительно упрощен путем исключения удаления или обмена любого другого компонента реакционной смеси. Это также улучшает охрану труда путем исключения потребности в выделении потенциальных токсичных или опасных промежуточных соединений. Использование однореакторного процесса является простым и экономичным способом органического синтеза, но он коммерчески ценен, только если уровень примесей может быть минимизирован с получением разумного выхода. В данном случае описанный однореакторный процесс выдает хорошие результаты и допустимый уровень непрореагировавших промежуточных соединений и побочных продуктов.
В альтернативном варианте соединение формулы (C), или его соль, может быть также получено сочетанием по Suzuki соединений формулы (AA) и выделенного соединения формулы (E). Реакция может быть осуществлена в растворителе в присутствии катализатора и основания. В формуле (AA) и (E), Y обозначает Cl, Br, I, OTf, OTs, OPiv и OMs; и B(X2)2 обозначает B(OH)2, B(OC(CH3)2C(CH3)2O), 9-BBN, B(Sia)2, B(cat), B(Cy)2, BF3- и B(MIDA), соответственно. В частном варианте осуществления Y обозначат Cl, и X2 обозначат OH. Используемый растворитель может представлять собой, например, воду, метанол, этанол, н-пропанол, 2-пропанол, н-бутанол, 2-бутанол, диметилформамид (DMF), тетрагидрофуран, 2-метил-тетрагидрофуран, толуол, диоксан или их смеси. Предпочтительно, растворителем является 2-бутанол и вода. Могут быть выбраны тот же катализатор и основание, как описано выше. В одном варианте осуществления катализатором является Pd(PPh3)2Cl2. Предпочтительным основанием является K2CO3.
В одном варианте осуществления соединение формулы (AA) вводят в реакцию с соединением формулы (E) в 2-бутаноле или воде в присутствии Pd(PPh3)2Cl2 в качестве катализатора и K2CO3 в качестве основания, причем Y обозначает Cl, и X2 обозначает OH.
1.6 Соединение формулы (C3-1):
Другое промежуточное соединение, которое может быть использовано в получении церитиниб, является соединением формулы (C3-1),
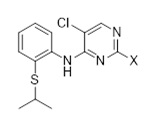 (C3-1)
(C3-1)
в которой X выбран из галогена (F, Cl, Br, I), алкокси (предпочтительно OMe, OEt, OtBu), арилокси (предпочтительно OPh), алкилтио (предпочтительно SMe, SEt), арилтио (SCH2Ph), тионила (предпочтительно SOMe, SOEt, SOCH2Ph), сульфонила (предпочтительно SO2Me, SO2Et, SO2CH2Ph). Наиболее предпочтительно X обозначает Cl.
Термин «алкокси», как радикал или часть радикала, относится к алкил-О-, причем термин алкил имеет значение, определенное здесь, и включает, например, C1-C20-алкокси (-O-C1-C20-алкил), предпочтительно C1-C7-алкокси (-O-C1-C7-алкил). В частности, алкокси включает, например, метокси (OMe), этокси (OEt), н-пропилокси, изопропилокси, н-бутилокси, изобутилокси, втор-бутилокси, трет-бутилокси (OtBu), пентилокси, гексилокси и гептилокси радикалы. Предпочтительными алкокси-заместителями являются метокси (OMe), этокси (OEt) или трет-бутилокси (OtBu).
Термин «арил», как радикал или часть радикала, относится к ароматической углеводородной группе, например, C6-C10-арилу, и предпочтительно моно- или полициклической, особенно моноциклической, бициклической или трициклической арильной группе с 6-14 атомами углерода, например 6-10 атомами углерода. Предпочтительно арил обозначает фенил, бензил, инденил, инданил или нафтил. Термин «арилокси» относится к арил-О- причем арил имеет значение, определенное выше. В частности, предпочтительны арилокси является фенокси (OPh). Термин «арилтио» относится к Арил-S-, причем арил имеет значение, определенное выше. Предпочтительным арилтио является бензилтио (SCH2Ph).
Термин «алкилтио» относится к алкил-S-, причем алкил имеет значение, определенное здесь. Алкильная группа, например, включает от 1 до 8 атомов углерода. В частности, алкилтио включает, например, метилтио (SMe), этилтио (SEt), фенилтио (PhS) и пентилтио. Предпочтительными алкилтио-заместителями являются метилтио (SMe) и этилтио (SEt).
Термин «тионил» соответствует группе -S-O-алкил, включающей прямой или разветвленный C1-C8-алкил. В частности, тионил включает, например, метилсульфинил (SOMe), этилсульфинил (SOEt), фенилсульфинил (SOPh) и бензилсульфинил (SOCH2Ph).
Термин «сульфонил» относится к двухвалентной группе -S(O)2-. В частности, сульфонил включает, например метилсульфонил, этилсульфонил, фенилсульфонил и бензилсульфонил.
Соединение формулы (C3-1) может быть получено как изображено в Схеме 7 путем реакции соединения формулы (F), или его соли, с соединением формулы (G), в случае необходимости в присутствии основания, в случае необходимости в растворителе. X выбирают из группы, состоящей из галогенов (F, Cl, Br, I), алкокси (предпочтительно OMe, OEt, OtBu) и арилокси (предпочтительно OPh), наиболее предпочтительно X обозначает Cl.
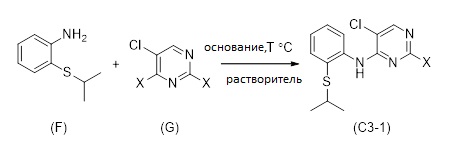
Схема 7
Основание, используемое для осуществления реакции, является любым основанием, которую специалист может выбрать на основании пособия. Оно может быть выбрано из группы, состоящей из Na2CO3, K2CO3, Cs2CO3, NaHCO3, KHCO3, триэтиламина, DIPEA, Na3PO4, K3PO4, DBU и NaH. Предпочтительно, основанием является DBU или DIPEA. Использование слабого основания в реакции приводит к хорошей эффективности; основание является простым и безопасным в обращении и потенциально предотвращает побочные реакции и таким образом позволяет восстановление соединения формулы (C3-1) с высоким выходом.
Реакция может быть осуществлена в протонном или апротонном растворителе. Например, растворитель может быть выбран из группы, состоящей из 1,4-диоксана, тетрагидрофурана (THF), 2-метил-тетрагидрофурана, диэтилового эфира, толуола, N-метил-2-пирролидона (NMP), 1-бутил-2-пирролидона (NBP), ацетонитрила, ацетона, диметилкарбоната, этилацетата, изопропилацетата, трет-бутилацетата, воды, метанола, этанола, н-пропанола, 2-пропанола, н-бутанола, 2-бутанола, трет-бутанола и толуола или их смесей. Также растворитель может быть опущен.
Соединение формулы (F), или его соль, и соединение формулы (G) могут быть введены в реакцию путем перемешивания реакционной смеси в течение от 1 часа до 72 часов, предпочтительно в течение приблизительно 18 часов. Могут быть выбраны температуры от 40 °C до температуры кипения растворителя, но предпочтительно температура составляет от 100 °C до 115 °C, особенно приблизительно 110 °C.
Процесс реакции соединения формулы (F), или его соли, и соединения формулы (G) лучше всего протекает в толуоле и 1-бутаноле в присутствии DIPEA. Температуру лучше всего устанавливать на значении 100-115 °C. Такая реакционная смеь может перемешиваться в течение приблизительно 18 часов.
В одном варианте осуществления, X в соединениях формулы (G) и (C3-1) обозначает Cl. Частным вариантом осуществления настоящего изобретения является соединение формулы (C3-1), в котором X обозначает Cl.
Соединение формулы (C3-1) может использоваться для получения церитиниба.
1.7 Соединение формулы (C3):
Из соединения формулы (C3-1) может быть получено соединение формулы (C3). Как показано на Схеме 8, соединение (C3) может быть получено путем окисления полученного соединения формулы (C3-1), в котором X выбран из галогена (F, Cl, Br, I), алкокси (предпочтительно OMe, OEt, OtBu) и арилокси (предпочтительно OPh), тионила (предпочтительно SOMe, SOEt, SOCH2Ph), сульфонила (предпочтительно SO2Me, SO2Et, SO2CH2Ph), наиболее предпочтительно X обозначает Cl.
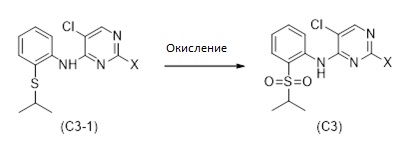
Схема 8
Окисление может быть осуществлено в воде или органическом растворителе. Растворитель может быть выбран из 1,4-диоксана, тетрагидрофурана (THF), 2-метил-тетрагидрофурана, диэтилового эфира, толуола, диметилформамида (DMF), диметилацетамида (DMA), диметилсульфоксида (DMSO), диметоксиэтана (DME), дихлорметана, N-метил-2-пирролидона (NMP), 1-бутил-2-пирролидона (NBP), ацетонитрила, ацетона, диметилкарбоната, этилацетата, изопропилацетата, трет-бутилацетата, пентана, гексана, гептана, анизола, пиридина, триэтиламина, уксусной кислоты, воды, метанола, этанола, н-пропанола, 2-пропанола, н-бутанола, 2-бутанола и трет-бутанола или их смесей.
Окисляющий реагент может быть выбран, среди прочего, из группы, состоящей из KMnO4, MnO2, NaIO4, NaClO, KHSO5 (оксона), NaBO3, CH3CO3H, H2O2, Na2WO4, O2, O3, тетрапропиламмоний перрутената (TPAP), 3,3-диметилдиоксирана, 3-хлорпероксибензойной кислоты (mCPBA) и трет-бутилгидропероксида (TBHP) или их смесей, в случае необходимости с катализатором.
Получение соединения формулы (C3) из соединения формулы (C3-1) может включать перемешивание реакционной смеси в течение от 4 до 40 часов, предпочтительно в течение приблизительно 16 часов при температуре от 10 до 60 °C, предпочтительно от 20 до 40 °C, особенно приблизительно при 30 °C. Окисление соединения формулы (C3-1) может быть осуществлено с высоким выходом в этилацетате, в присутствии CH3CO3H в форме раствора в CH3CO2H при 30 °C, или процесс может быть осуществлен с высоким выходом в метаноле, в присутствии H2O2 и Na2WO4 при температуре от 20 до 70 °C.
1.7 Альтернативный путь для получения соединения формулы (C3)
Соединение формулы (C3) может альтернативно быть получено путем осуществления стадий (i) введения соединения формулы (H) в реакцию с соединением формулы (I) для получения промежуточного соединения; (ii) восстановления промежуточного соединения для формирования соединения (J); и (iii) реакции соединение (J) с соединением формулы (G) в присутствии основания, как описано на Схеме 9, см. ниже.
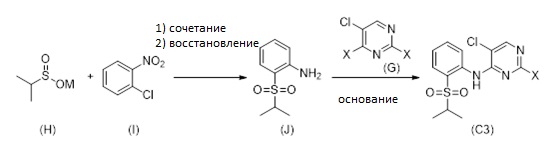
Схема 9
X может обозначать, как выше, F, Cl, Br, I, Алкокси (предпочтительно OMe, OEt, OtBu), Арилокси (предпочтительно OPh), алкилтио (предпочтительно SMe, SEt), арилтио (предпочтительно SCH2Ph), тионил (предпочтительно SOMe, SOEt, SOCH2Ph), сульфонил (предпочтительно SO2Me, SO2CH2Ph). Наиболее предпочтительно X обозначает Cl. M может быть выбран из Li, Na, K, 0,5 Zn, 0,5 Ca, предпочтительно M обозначает Na.
Соединение формулы (H) и соединение формулы (I) реагируют в растворителе при перемешивании при температуре от комнатной температуры до температуры кипения растворителя. В частном варианте осуществления реакцию проводят в растворителе при температуре 82-86 °C.
Растворителем, используемым для реакции, может быть, например, диметилсульфоксид (DMSO), 1,4-диоксан, тетрагидрофуран (THF), 2-метил-тетрагидрофуран, диэтиловый эфир, толуол, диметилформамид (DMF), диметилацетамид (DMA), диметоксиэтан (DME), дихлорметан, N-метил-2-пирролидон (NMP), 1-бутил-2-пирролидон (NBP), ацетонитрил, ацетон, диметилкарбонат, этилацетат, изопропилацетат, трет-бутилацетат, пентан, гексан, гептан, анизол, пиридин, триэтиламин, уксусная кислота, вода, метанол, этанол, н-пропанол, 2-пропанол, н-бутанол, 2-бутанол, трет-бутанол или их смеси. Предпочтительно выбирают DMSO.
Стадия восстановления для получения соединения формулы (J) может включать использование катализатора и водорода в растворителе. Катализатор, используемый для выполнения восстановления, является любым катализатором, который специалист может выбрать на основании пособия. Катализатором может быть, например, никель Ренея, Pt/C, Rh/C, Pd/Al2O3, Pd/CaCO3, RhCl(PPh3)3, катализатор Линдлара, PtO2, Pd/C, [Rh(cod)(PPh3)2]+, [Ir(cod)(PCy3)(Py)]+, Pd(OH)2, Pd/Al, Pt/Al, Pt/SiAl, Pd/ZrO2 или их смеси.
Катализатор, добавляемый к реакционной смеси, может присутствовать в диапазоне от приблизительно 0,005 мол.% до приблизительно 50,0% вес./вес. (сухой вес) в расчете на исходное соединение формулы (I). Как правило, катализатор может присутствовать в количестве менее 20,0% вес./вес. (сухой вес).
Смесь в реакции восстановления может быть перемешана в течение нескольких часов, обычно при температуре до 60°C, предпочтительно приблизительно 40°C.
Реакция восстановления лучше всего протекает при повышенном давлении, например давлении от 1 до 15 бар, предпочтительно от 2 до 6 бар.
Восстановление может быть осуществлено в растворителе, таком как раствор на основе спирта. Раствор на основе спирта может представлять собой спирты C1-C10 (например, метанол, этанол, пропанол, изопропанол и бутанол), или их смеси могут использоваться в качестве среды реакции. Предпочтительно растворитель является этанолом.
Соединение формулы (C3) получают путем реакции промежуточного соединения формулы (J) с соединением формулы (G) в присутствии основания и в отсутствие растворителя. Осуществление реакции без растворителя является действительно привлекательной альтернативой, поскольку оно более эффективно, приводит к меньшим тратам и уменьшает общую стоимость синтеза.
Основание, используемое для выполнения реакции, является любым основанием, которую специалист мог бы выбрать на основании обычного пособия. Основание может быть, например, выбрано из группы, состоящей из Na2CO3, K2CO3, Cs2CO3, NaHCO3, KHCO3, триэтиламина, DIPEA, Na3PO4, K3PO4, DBU и NaH. Лучшие условия достигаются, когда используются DBU или DIPEA, особенно DBU. Слабое основание, определенное здесь, позволяет достигать эффективного процесса, является простым и безопасным в обращении и потенциально предотвращает побочные реакции и таким образом позволяет осуществлять восстановление соединения формулы (C3) с высоким выходом и чистотой.
Реакция между промежуточным соединением (J) и соединением формулы (G) лучше всего протекает при температуре от 40°C до температуры кипения растворителя, особенно при 80°C.
1.8 Церитиниб:
Вышеописанные способы могут быть распространены на получение церитиниба или его соли. В зависимости от исходных материалов и выбранного пути, специалисту будет понятно, как объединить их для получения строительных блоков для возможного формирования церитиниба. Частные варианты или альтернативные процессы описаны здесь ниже. Например, как показано на Схеме 10, церитиниб, или его соль, может быть получен способом, включающем стадии:
i. получение соединения формулы (C2-1), как описано в 1.1 или 1.2
ii. получение соединения формулы (C2) или его соли,
iii. получение соединения (C3), и
iv. введение соединения формулы (C2) в реакцию с соединением формулы (C3) для получения церитиниба или его соли.
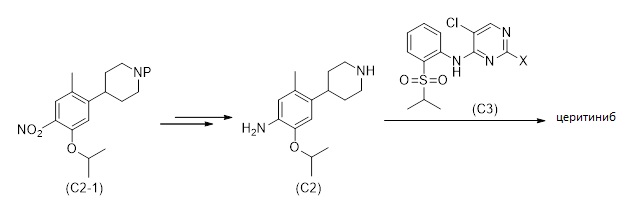
Схема 10
В альтернативе церитиниб, или его соль, может быть получен способом, описанным на Схеме 11:
(а) получение соединения формулы (C2) или его соли, как описано в 1.3 или 1.4
(b) получение соединения формулы (C3), и
(c) введение соединение формулы (C2) или его соли в реакцию с соединением формулы (C3) для получения церитиниба или его соли.
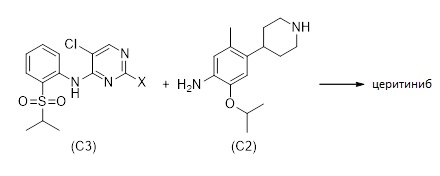
Схема 11
Как изображено в Схеме 12, церитиниб, или его соль, могут также быть продуктами в процессе, включающем:
(aa) получение соединения формулы (C2) или его соли,
(bb) получение соединения формулы (C3-1), как описано в 1.6
(cc) получение из соединения формулы (C3-1) соединения формулы (C3), и
(dd) введение соединения формулы (C2) или его соли в реакцию с соединением формулы (C3) для получения церитиниба или его соли.
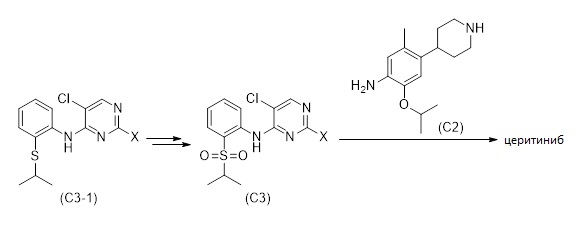
Схема 12
Другой вариант настоящего раскрытия относится к способу получения церитиниба или его соли, как описано на Схеме 13, включающему
(I) получение соединения формулы (C), как описано в 1.5
(II) получение из соединения формулы (C) соединения формулы (C2) или его соли,
(III) получение соединения формулы (C3), и
(IV) введение соединения формулы (C2) или его соли в реакцию с соединением формулы (C3) для получения церитиниба или его соли.
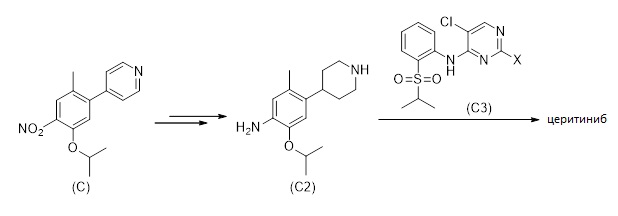
Схема 13
Соединение формулы (C2), или его соль, и соединение формулы (C3) могут быть подвергнуты реакции сочетания в растворителе в присутствии основания, причем X может обозначать галоген (Br, Cl, I), алкокси (предпочтительно OMe, OEt, OtBu) и арилокси (предпочтительно OPh), тионил (предпочтительно SOMe, SOEt, SOCH2Ph) или сульфонил (предпочтительно SO2Me, SO2Et, SO2CH2Ph); наиболее предпочтительно X обозначает Cl.
Основание для реакции может быть выбрано из группы слабых оснований, таких как Na2CO3, K2CO3, Cs2CO3, NaHCO3, KHCO3, триэтиламин, DIPEA, DBU, Na3PO4 или K3PO4. Также основа может быть опущена.
Реакцию можно проводить в обширном разнообразии растворителей, например 1,4-диоксане, тетрагидрофуране (THF), 2-метил-тетрагидрофуране, диэтиловом эфире, толуоле, N-метил-2-пирролидоне (NMP), 1-бутил-2-пирролидоне (NBP), ацетонитриле, ацетоне, диметилкарбонате, этилацетате, изопропилацетате, трет-бутилацетате, воде, метаноле, этаноле, н-пропаноле, изопропаноле, н-бутаноле, 2-бутаноле, трет-бутаноле и толуоле или их смесях. Предпочтительно используются тетрагидрофуран (THF), 2-метил-тетрагидрофуран, вода, метанол, этанол, н-пропанол, изопропанол, н-бутанол, 2-бутанол, наиболее предпочтительно изопропанол.
Процесс реакции соединения формулы (C2) или его соли для получения соединения формулы (C3) осуществляют путем перемешивания реакционной смеси в течение от 6 до 41 часа, предпочтительно в течение приблизительно 16 часов при температуре от 40°C до температуры кипения растворителя, предпочтительно при температуре кипения растворителя.
Процесс реакции соединения формулы (C2) и соединения формулы (C3) лучше всего протекает в изопропаноле, при температуре кипения растворителя, без основания, что, таким образом, предотвращает побочные реакции и обеспечивает восстановление церитиниба или его соли с высоким выходом и чистотой.
Когда здесь указаны соли, под ними, в частности, понимают фармацевтически приемлемые соли или другие обычно приемлемые соли, если они не исключены по химическим причинам, которые легко поймет специалист. Соли могут быть образованы с конечными продуктами или промежуточными соединениями, где присутствуют солеобразующие группы, такие как основные или кислотные группы, которые могут существовать в диссоциированной форме, по меньшей мере частично, например, в диапазоне pH от 4 до 10 в водных растворах, или могут быть выделены особенно в твердой, особенно кристаллической форме.
Такие соли получают, например, как соли присоединения с кислотой, предпочтительно с органическими или неорганическими кислотами, из соединений или любого из промежуточных соединений, упомянутых здесь, с основным атомом азота (например, имино или амино), особенно фармацевтически приемлемые соли. Подходящими неорганическими кислотами являются, например, галогеноводородные кислоты, такие как соляная кислота, серная кислота или фосфорная кислота. Подходящими органическими кислотами являются, например, карбоновые, фосфоновые, сульфоновые кислоты или сульфаминовые кислоты, например уксусная кислота, пропионовая кислота, молочная кислота, фумаровая кислота, янтарная кислота, лимонная кислота, аминокислоты, такие как глутаминовая или аспарагиновая кислота, малеиновая кислота, гидроксималеиновая кислота, метилмалеиновая кислота, бензойная кислота, метан- или этансульфоновая кислота, этан-1,2-дисульфоновая кислота, бензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 1,5-нафталин-дисульфоновая кислота, N-циклогексилсульфамовая кислота, N-метил-N-этил- или N-пропил-сульфамовая, или другие органические протонные кислоты, такие как аскорбиновая кислота.
Церитиниб, полученный, как описано выше, может в случае необходимости быть далее очищен перекристаллизацией из подходящего растворителя и может в случае необходимости быть подвергнут размалыванию или просеиванию для получения конечного фармацевтически активного ингредиента.
После получения фармацевтически активного ингредиента церитиниба (например, как описано выше в Разделе 1.8), он может быть смешан с фармацевтически приемлемым эксципиентом. Это может быть достигнуто путем смешивания, дробления, уплотнения и т.п. Таким образом, фармацевтическая композиция может быть получена и использована для получения конечных лекарственных форм, таких как таблетки или капсулы.
В этом описании и приложенной формуле изобретения, формы единственного числа включают формы множественного числа, если из контекста ясно не следует иное.
Точно так же «включает» и «содержит» являются взаимозаменяемыми и не предназначенными для ограничения.
Аббревиатуры
|
Примеры
Следующие примеры просто иллюстрируют настоящее раскрытие, и их ни в коем случае нельзя рассматривать как ограничение объема раскрытия, поскольку эти примеры и другие их эквиваленты будут очевидны для специалиста в свете настоящего раскрытия и сопровождающей его формулы изобретения.
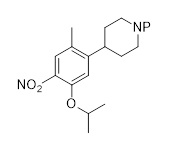
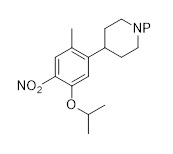
Синтез 2-изопропокси-5-метил-4-(пиперидин-4-ил)анилин дигидрохлорида (C2, соль ди-HCl) согласно следующей последовательности:
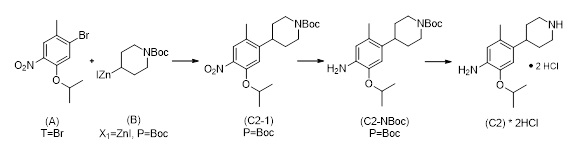
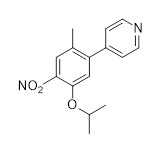
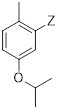
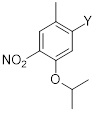
Трет-бутил-4-(5-изопропокси-2-метил-4-нитрофенил)пиперидин-1-карбоксилат (C2-1, P=Boc)
К смеси Pd(PPh3)2Cl2 (69 мг, 0,099 ммоль, 1,5 мол.%), CuI (75 мг, 0,39 ммоль, 6 мол.%), 1-бром-5-изопропокси-2-метил-4-нитробензола (1,80 г, 6,57 ммоль) в тетрагидрофуране (19 мл), раствор (1-(трет-бутоксикарбонил)пиперидин-4-ил)цинк (II) йодида в тетрагидрофуране (4,95 г 0,9 М раствора, 13,1 ммоль, 2.0 экв.; полученный согласно описанной в литературе процедуре) добавляли при 50 °C и реакционную смесь перемешивали в течение 21 часа при этой температуре. После охлаждения до комнатной температуры добавляли насыщенный водный раствор NH4Cl (50 мл). Водную фазу экстрагировали этилацетатом (3×70 мл), объединенные органические фазы промывали солевым раствором (80 мл) и высушивали над Na2SO4. Летучие вещества удаляли под вакуумом, и сырой материал очищали хроматографией на силикагеле (этилацетат/гептан) для получения трет-бутил-4-(5-изопропокси-2-метил-4-нитрофенил)пиперидин-1-карбоксилата (1,96 г, 79%-й выход) в форме коричневатого масла. 1H-ЯМР (400 МГц, CDCl3): δ=1,30 (д, J=6,1 Гц, 6H), 1,42 (с, 9H), 1,47-1,54 (м, 2H), 1,67-1,70 (м, 2H), 2,24 (с, 3H), 2,72-2,82 (м, 3H), 4,05 (ушир. м, 2H), 4,53 (септ., J=6,1 Гц, 1H), 6,79 (с, 1H), 7,55 (м, 1H) ppm.
Трет-бутил-4-(4-амино-5-изопропокси-2-метилфенил)пиперидин-1-карбоксилат (C2-NBoc, P=Boc)
К раствору трет-бутил-4-(5-изопропокси-2-метил-4-нитрофенил)пиперидин-1-карбоксилата (1,96 г, 5,18 ммоль) в этаноле (180 мл) добавляли Pd/C (10%, 0,4 г), и реакционную смесь перемешивали в течение 5 часов при комнатной температуре в атмосфере водорода (4 бар). Атмосферу водорода спускали, сосуд с реагентом очищали аргоном, и смесь фильтровали через Celite®. Летучие вещества удаляли под вакуумом для получения трет-бутил-4-(4-амино-5-изопропокси-2-метилфенил)пиперидин-1-карбоксилата (1,67 г, 92%-й выход) в форме желтого масла, которое использовали на следующей стадии без дополнительной очистки.
2-Изопропокси-5-метил-4-(пиперидин-4-ил)анилин дигидрохлорид (C2, соль ди-HCl)
Трет-бутил-4-(4-амино-5-изопропокси-2-метилфенил)пиперидин-1-карбоксилат (1,67 г) растворяли в дихлорметане (10 мл), и конечный раствор обрабатывали трифторуксусной кислотой (2 мл). После 16 часов при комнатной температуре добавляли воду (50 мл). Фазы разделяли и водную фазу промывали дихлорметаном (30 мл). Водную фазу нейтрализовали водным раствором NaOH (раствор 1 М) и экстрагировали толуолом (3×50 мл). К объединенным фазам толуола добавляли HCl (3,8 мл раствора 5 М в изопропаноле). После упаривания получали 2-Изопропокси-5-метил-4-(пиперидин-4-ил)анилин дигидрохлорид (1,35 г, 88%-й выход). 1H-ЯМР (400 МГц, D2O): δ=1,24 (д, J=6,1 Гц, 6H), 1,76-1,96 (м, 4H), 2,22 (с, 3H), 3,04-3,14 (м, 3H), 3,45-3,49 (м, 2H), 4,71 (септ., J=6,1 Гц, 1H; перекрытый сигналом растворителя), 6,95 (с, 1H), 7,13 (с, 1H) ppm. 13C-ЯМР (100 МГц, D2O): δ=17,4, 20,9, 28,6, 35,4, 44,4, 72,3, 112,2, 117,6, 125,4, 128,9, 144,6, 148,8 частей на миллион.
Синтез 2-изопропокси-5-метил-4-(пиперидин-4-ил)анилин дигидрохлорида (C2, соль ди-HCl) путем восстановления 4-(5-изопропокси-2-метил-4-нитрофенил)пиридина (C)
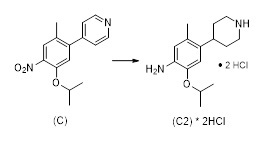
В реактор загружали 4-(5-изопропокси-2-метил-4-нитрофенил)пиридин (110 кг, 404 моль), 5 мол.% Pt/C (33 кг, 50-70% H2O) и уксусную кислоту (2200 кг). Суспензию герметизировали водородом при давлении 2 бар при поддержании температуры ниже 30 °C. После 2 часов давление водорода увеличивали до 6 бар, и смесь нагревали до 30 °C. После полного преобразования водород стравливали при комнатной температуре, и реактор очищали азотом. Катализатор отфильтровывали и ополаскивали уксусной кислотой (800 кг). Фильтрат частично концентрировали, затем добавляли толуол (2 355 кг), и перегонку продолжали; эту стадию повторяли еще два раза (с использованием толуола в качестве растворителя). Дополнительный толуол (312 кг) и изопропанол (359 кг) добавляли к раствору. HCl (154 кг 22% вес./вес. раствор в изопропаноле) вводили при поддержании температуры ниже 40 °C. После добавления суспензию охлаждали до 25 °C, поддерживали в течение по меньшей мере 90 минут, охлаждали до 0 °C и перемешивали в течение по меньшей мере 90 минут. Продукт отфильтровывали, ополаскивали дважды раствором толуол/изопропанол (2×240 кг, 20% вес./вес.) и высушивали под вакуумом с получением продукта 2-изопропокси-5-метил-4-(пиперидин-4-ил)анилин дигидрохлорида (105 кг, 81%-й выход). 1H ЯМР (400 МГц, DMSO-d6): δ=1,31 (д, J=4 Гц, 6H), 1,79 (м, 2H), 1,98 (кд, J=12, 4 Гц, 2H), 2,25 (с, 3H), 2,95-3,08 (ушир. м, 3H), 3,34 (ушир. с, 1H), 4,66 (септ., J=4 Гц, 1H), 6,92 (с, 1H), 7,18 (с, 1H), 9,19 (ушир. с, 2H), 9,86 (ушир. с, 3H) ppm. 13C ЯМР (125 МГц, DMSO-d6): δ=18,1, 21,7, 28,4, 34,9, 43,5, 71,1, 112,1, 119,2, 125,6, 127,3, 144,0, 148,8 частей на миллион.
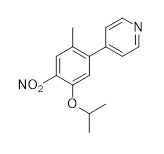
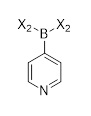
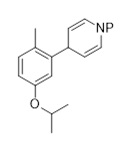
Синтез 4-(5-изопропокси-2-метил-4-нитрофенил)пиридина (C)
a) Борилирование/однореакторная реакция Suzuki
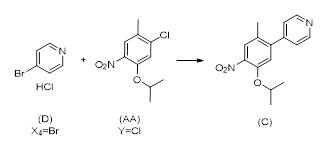
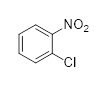
Смесь 4-бромпиридин гидрохлорида (1,0 г, 5,1 ммоль, 1,2 экв.), бис(пинаколято)диборона (1,31 г, 5,1 ммоль, 1,2 экв.), ацетата калия (1,68 г, 17,1 ммоль, 4,0 экв.), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенила (= XPhos; 0,12 г, 6 мол.%), хлор (2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия (II) (= Предварительный катализатор XPhos, второе поколение; 0,10 г, 3 мол.%) и 1,4-диоксана (5 мл) нагревали при 100 °C в течение 3 часов. Затем добавляли водный раствор K2CO (3,6 мл 3,6 М раствора, 12,9 ммоль, 3,0 экв.) и 1-хлор-5-изопропокси-2-метил-4-нитробензол (0,98 г, 4,3 ммоль), и реакционную смесь далее нагревали при 100 °C в течение 17 часов. После охлаждения до комнатной температуры смесь фильтровали через HyFlo® и промывали этилацетатом (3×20 мл). Органическую фазу промывали водой (3×20 мл) и водным раствором NaCl (20 мл 24% вес./вес. раствор) и высушивали над Na2SO4. Летучие вещества удаляли под вакуумом, и сырой материал очищали хроматографией на силикагеле (этилацетат/гептан) для получения 4-(5-изопропокси-2-метил-4-нитрофенил)пиридина (0,95 г, 81%-й выход) в форме твердого вещества желтого цвета.
b) Сочетание Suzuki
Смесь K2CO3 (50,1 кг, 358 моль, 2,5 экв.), H2O (93 кг), 1-хлор-5-изопропокси-2-метил-4-нитробензола (33,3 кг, 145 моль), пиридин-4-илбороновой кислоты (23,2 кг, 189 моль, 1,3 экв.), транс-дихлорбис(трифенилфосфин)палладия (II) (5,11 кг, 7,28 моль, 5 мол.%) и 2-бутанола (216 кг) нагревали под инертной атмосферой с обратным холодильником в течение 3,5 часов. После охлаждения до 55°C, смесь фильтровали, и фазы разделяли. Органическую фазу частично концентрировали под вакуумом, охлаждали до 50°C и добавляли воду (167 кг). После дальнейшего охлаждения до 5°C, твердые вещества отфильтровывали. Сырой продукт очищали перекристаллизацией из смеси этанол/вода для получения 4-(5-изопропокси-2-метил-4-нитрофенил)пиридина (52,9 кг, 71%-й выход) в форме твердого вещества желтого цвета. 1H-ЯМР (400 МГц, CDCl3): δ=1,40 (д, J=6,0 Гц, 6H), 2,23 (с, 3H), 4,65 (септ., J=6,1 Гц, 1H), 6,92 (с, 1H), 7,25-7,26 (м, 2H), 7,72 (с, 1H) 8,71-8,73 (м, 2H) частей на миллион; 13C-ЯМР (100 МГц, CDCl3): δ=19,2, 21,9, 73,1, 117,3, 123,6, 127,2, 127,4, 140,4, 144,4, 147,9, 149,2, 150,0 частей на миллион.
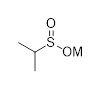
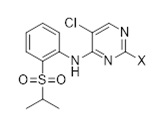
Синтез 2,5-дихлор-Н-(2-(изопропилсульфонил)фенил)пиримидин-4-амина (C3, X=Cl) согласно следующей последовательности:
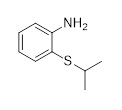
2,5-дихлор-Н-(2-(изопропилтио)фенил)пиримидин-4-амин (C3-1, X=Cl)
N,N-диизопропилэтиламин (31,0 г, 0,24 моль, 2,3 экв.) добавляли к смеси 2-(изопропилтио)анилин гидрохлорида (21,3 г, 0,10 моль), 2,4,5-трихлорпиримидина (19,0 г, 0,10 моль, 1,0 экв.) в толуоле (166 г) и н-бутаноле (17 г). Смесь нагревали с обратным холодильником в течение приблизительно 22 часов. После охлаждения реакционной смеси до 25 °C добавляли воду (70 г), фазы разделяли, и органическую фазу промывали водой (70 г). Органическую фазу концентрировали под вакуумом с последующим добавлением этанола (34 г). Смесь нагревали с обратным холодильником и медленно охлаждали до 0-5 °C, фильтровали и высушивали под вакуумом для получения 2,5-дихлор-Н-(2-(изопропилтио)фенил)пиримидин-4-амина (26,4 г, 81%-й выход). 1H-ЯМР (400 МГц, CDCl3): δ=1,30 (д, J=7,0 Гц, 6H), 3,18 (септ., J=6,7 Гц, 1H), 7,11-7,14 (м, 1H), 7,46-7,50 (м, 1H), 7,60-7,62 (м, 1H), 8,25 (с, 1H), 8,67-8,68 (м, 1H), 9,30 (ушир. с, 1H) частей на миллион. 13C-ЯМР (100 МГц, CDCl3): δ=23,3, 40,7, 114,9, 119,8, 122,9, 124,0, 130,4, 137,1, 139,8, 154,6, 156,0, 158,1 частей на миллион.
2,5-дихлор-Н-(2-(изопропилсульфонил)фенил)пиримидин-4-амин (C3)
К раствору 2,5-дихлор-Н-(2-(изопропилтио)фенил)пиримидин-4-амина (20,0 г, 63,7 моль) в этилацетате (180 г) добавляли надуксусную кислоту (33,4 г раствора в уксусной кислоте; 5,7 ммоль/г) при поддержании внутренней температуры 20-30°C. После времени реакции 16 часов добавляли этилацетат (90 г), затем водный раствор сульфита натрия (112 г 11% вес./вес. раствор) и поддерживали внутреннюю температуру ниже 45°C. Фазы разделяли, и к органической фазе добавляли воду (63 г), затем водный раствор NaOH (25% вес./вес. раствор) для доведения pH до 7-8. Фазы разделяли, органическую фазу высушивали над MgSO4 и концентрировали под вакуумом. Этанол (217 г) добавляли к смеси при нагревании с обратным холодильником. После медленного охлаждения до 0-5°C осадок отфильтровывали и высушивали под вакуумом для получения 2,5-дихлор-Н-(2-(изопропилсульфонил)фенил)пиримидин-4-амина (19,5 г, 88%-й выход) в форме белого порошка. 1H-ЯМР (400 МГц, CDCl3): δ=1,33 (д, J=6,7 Гц, 6H), 3,22 (септ., J=6,8 Гц, 1H), 7,31-7,35 (м, 1H), 7,72-7,76 (м, 1H), 7,92-7,94 (м, 1H), 8,31 (с, 1H), 8,63-8,65 (м, 1H), 10,07 (ушир. с, 1H) частей на миллион. 13C-ЯМР (100 МГц, CDCl3): δ=153, 56,1, 115,2, 122,7, 124,2, 124,5, 131,5, 135,20, 137,4, 155,6, 156,3, 157,8 частей на миллион.
Синтез 2,5-дихлор-Н-(2-(изопропилсульфонил)фенил)пиримидин-4-амина (C3, X=Cl) согласно следующей последовательности:
1-(Изопропилсульфонил)-2-нитробензол (J')
Натрий пропан-2-сульфинат (2,48 кг, 19,1 моль, 1,5 экв.) и 1-хлор-2-нитробензол (2,00 кг, 12,7 моль) растворяли в DMSO (5,5 кг) и нагревали до 85 °C в течение 12 часов. После охлаждения до 20 °C добавляли воду со льдом (15 кг), затем затравляли 1-(изопропилсульфонил)-2-нитробензолом (1 г). Реакционную смесь перемешивали в течение 30 минут при 0-10°C, фильтровали и высушивали под вакуумом для получения 1-(изопропилсульфонил)-2-нитробензола (2,8 кг, 96%-й выход) в форме твердого вещества серого цвета.
2-(Изопропилсульфонил)анилин (J)
Смесь 1-(изопропилсульфонил)-2-нитробензола (2,50 кг, 10,9 моль), этанола (7,9 кг) и Pd/C (125 г, 5% вес./вес.) в автоклаве помещали под давлением водорода 3,4 бар и перемешивали в течение 48 часов при 40°C. После фильтрации катализатора фильтрат концентрировали под вакуумом до приблизительно 2,5 литра, и осадок отфильтровывали. Фильтрат снова концентрировали под вакуумом до приблизительно 0,8 литра, и осадок отфильтровывали. Две фракции отфильтрованных твердых веществ объединяли и высушивали под вакуумом для получения 2-(Изопропилсульфонил)анилина (1,83 кг, 84%-й выход).
2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амин (C3, X=Cl)
2,4,5-трихлорпиримидин (6,1 кг) добавляли к 2-(изопропилсульфонил)анилину (950 г, 4,77 моль) и DBU (181 г, 1,19 моль, 0,25 экв.), и смесь перемешивали в течение 7,5 часов при 80°C. После охлаждения до 25°C добавляли н-гептан (1,9 кг), и смесь перемешивали в течение 30 минут. Смесь охлаждали до -5°C, фильтровали и промывали н-гептаном (325 г). Сырой продукт суспендировали в этаноле (4,5 кг), перемешивали в течение 12 часов при 25°C и затем охлаждали до 0°C. После фильтрации сырой продукт высушивали под вакуумом для получения 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амина (1,07 кг, 65%-й выход). 1H-ЯМР (400 МГц, CDCl3): δ=1,33 (д, J=6,7 Гц, 6H), 3,22 (септ., J=6,8 Гц, 1H), 7,31-7,35 (м, 1H), 7,72-7,76 (м, 1H), 7,92-7,94 (м, 1H), 8,31 (с, 1H), 8,63-8,65 (м, 1H), 10,07 (ушир. с, 1H) частей на миллион. 13C-ЯМР (100 МГц, CDCl3): δ=153, 56,1, 115,2, 122,7, 124,2, 124,5, 131,5, 135,20, 137,4, 155,6, 156,3, 157,8 частей на миллион.
Синтез 2-изопропокси-5-метил-4-(пиперидин-4-ил)анилин дигидрохлорида (C2, соль ди-HCl) следующей последовательностью:
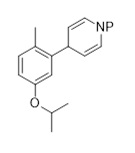
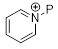
Метил-4-(5-изопропокси-2-метилфенил)пиридин-1(4Н)-карбоксилат (C2-2, P=CO2CH3)
К предварительно высушенному LiCl (0,41 г, 9,6 ммоль, 1,1 экв.) добавляли магний (0,28 г, 11,3 ммоль, 1,3 экв.) и THF (13 мл), затем 2-бром-4-изопропокси-1-метилбензол (2,00 г, 8,73 ммоль). После 10 минут при комнатной температуре смесь нагревали до 70°C в течение 1 часа. Смесь охлаждали до комнатной температуры, перемешивали в течение 1 часа и далее охлаждали до -30°C. Добавляли 1-(Метоксикарбонил)пиридин-1-ий хлорид (13,1 ммоль; полученный из пиридина, ацетилхлорида и йодида меди (I) согласно описанной в литературе процедуре), полученную оранжевую суспензию нагревали до комнатной температуры и перемешивали в течение 16 часов. Добавляли TBME (80 мл), органическую фазу промывали насыщенным водным раствором NH4Cl (3×30 мл), солевым раствором (30 мл) и высушивали над Na2SO4. После удаления летучих веществ под вакуумом полученный сырой метил-4-(5-изопропокси-2-метилфенил)пиридин-1(4Н)-карбоксилат (2,04 г, 86%-й выход) использовали на следующей стадии без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3): δ=1,31 (д, J=6,1 Гц, 6H), 2,24 (с, 3H), 2,28 (с, 3H), 4,37 (м, 1 H), 4,50 (септ., J=6,0 Гц, 1H), 4,93-4,96 (м, 1H), 5,03-5,07 (м, 1H), 6,62-6,68 (м, 2H), 6,82 (м, 1H), 7,04-7,06 (м, 1H), 7,28-7,31 (м, 1H) частей на миллион.
Метил-4-(5-изопропокси-2-метилфенил)пиперидин-1-карбоксилат (C2-2')
К раствору 4-(5-изопропокси-2-метилфенил)пиридин-1(4Н)-карбоксилата (2,0 г, 7,5 ммоль) в THF (20 мл) и метаноле (50 мл) добавляли формиат аммония (10 г, 159 ммоль), затем Pd/C (2,0 г, 10%). После перемешивания в течение 18 часов при комнатной температуре смесь фильтровали через Celite®, остаток после фильтрации промывали этилацетатом (40 мл), и к фильтрату добавляли воду (80 мл). Водную фазу экстрагировали этилацетатом (3×50 мл), и объединенные органические экстракты высушивалии над Na2SO4. Летучие вещества удаляли под вакуумом, и сырой материал очищали хроматографией на силикагеле (этилацетат/гептан) для получения метил-4-(5-изопропокси-2-метилфенил)пиперидин-1-карбоксилата (2,1 г, 72%-й выход) в форме желтого масла. 1H-ЯМР (400 МГц, CDCl3): δ=1,31 (д, J=6,1 Гц, 6H), 1,53-1,65 (м, 1H), 1,78-1,85 (м, 1H), 2,14 (с, 3H), 2,27 (с, 3H), 2,62 (м, 1H), 2,89 (м, 1H), 3,17 (м, 1H), 3,92-3,96 (м, 1H), 4,49 (септ., J=6,1 Гц, 1H), 4,78-4,82 (м, 1H), 6,64-6,69 (м, 2H), 7,04-7,06 (м, 1H) частей на миллион.
Метил-4-(5-изопропокси-2-метил-4-нитрофенил)пиперидин-1-карбоксилат (C2-1, P=CO2CH3)
Смесь метил-4-(5-изопропокси-2-метилфенил)пиперидин-1-карбоксилата (4,15 г,) в уксусном ангидриде (51 мл) охлаждали до -10°C. Добавляли азотную кислоту (1 мл, 65%), и смесь перемешивали в течение 1 часа при -10 °C. Добавляли другую часть азотной кислоты (1 мл, 65%), перемешивали в течение 3 часов, и смесь нагревали до комнатной температуры. После добавления воды (100 мл) к смеси водную фазу экстрагировали этилацетатом (3×80 мл). Объединенные органические фазы промывали насыщенным водным раствором NH4Cl (2×50 мл), солевым раствором (50 мл) и высушивали над Na2SO4. Летучие вещества удаляли под вакуумом, и сырой материал очищали хроматографией на силикагеле (этилацетат/гептан) для получения метила-4-(5-изопропокси-2-метил-4-нитрофенил)пиперидин-1-карбоксилата (2,1 г, 72%-й выход) в форме желтого масла. LCMS: m/z (M+1) 337,2 [M+H] +.
Метил-4-(4-амино-5-изопропокси-2-метилфенил)пиперидин-1-карбоксилат (C2-NCO2Me)
Pd/C (0,5 г, 10%) добавляли к раствору 4-(5-изопропокси-2-метил-4-нитрофенил)пиперидин-1-карбоксилата (3,68 г, 10,9 ммоль) в этаноле (200 мл). Смесь перемешивали в течение 5 часов в атмосфере водорода при 5 бар. Реакционную смесь очищали азотом, фильтровали через Celite® и промывали этилацетатом (50 мл). Фильтрат экстрагировали водным раствором HCl (2×30 мл, раствором 1 М). Водные фазы нейтрализовали водным раствором NaOH (раствор 1 М) и экстрагировали этилацетатом (3×50 мл). Летучие вещества удаляли под вакуумом, и сырой материал очищали хроматографией на силикагеле (этилацетат/гептан) для получения метил-4-(4-амино-5-изопропокси-2-метилфенил)пиперидин-1-карбоксилата (3,36 г, 51%-й выход). 1H-ЯМР (400 МГц, CDCl3): δ=1,25 (д, J=6,1 Гц, 6H), 1,47 (ушир. м, 2H), 1,65-1,68 (м, 2H), 2,13 (с, 3H), 2,65-2,81 (м, 3H), 3,59 (ушир. с, 2H), перекрытый 3,65 (с, 3H), 4,22 (ушир. с, 2H), 4,37 (септ., J=6,1 Гц, 1H), 6,46 (с, 1H), 6,53 (с, 1H) частей на миллион.
2-Изопропокси-5-метил-4-(пиперидин-4-ил)анилин дигидрохлорид (C2, соль ди-HCl)
Водный раствор HCl (26 мл 8 М раствора) добавляли к метил-4-(4-амино-5-изопропокси-2-метилфенил)пиперидин-1-карбоксилату (1,00 г, 3,26 ммоль), и смесь перемешивали в течение 16 часов при комнатной температуре. После промывки дихлорметаном (30 мл) водную фазу нейтрализовали водным раствором NaOH (раствор 1 М). Нейтрализованную водную фазу экстрагировали толуолом (3×50 мл), и объединенные органические экстракты обрабатывали HCl (2,6 мл 5 М раствора в изопропаноле). Выпаривание летучих веществ под вакуумом дало 2-изопропокси-5-метил-4-(пиперидин-4-ил)анилин дигидрохлорид (1,05 г, 49%-й выход). LCMS: m/z (M - 2HCl) 337,2 [M+H] +; 1H-ЯМР (400 МГц, D2O): δ=1,24 (д, J=6,1 Гц, 6H), 1,76-1,96 (м, 4H), 2,22 (с, 3H), 3,04-3,14 (м, 3H), 3,46-3,49 (м, 2H), 4,71 (септ., J=6,1 Гц, 1H; перекрытый сигналом растворителя), 6,95 (с, 1H), 7,13 (с, 1H) частей на миллион.
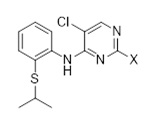
Синтез 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил) фенил)пиримидин-2,4-диамина (церитиниба)
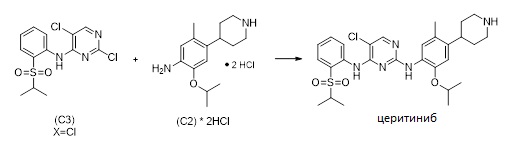
Изопропанол (445 кг) добавляли к 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амину (70,2 кг, 203 моль, 1,15 экв.) и 2-Изопропокси-5-метил-4-(пиперидин-4-ил)анилин дигидрохлориду (56,7 кг, 176 моль). Смесь нагревали в течение приблизительно 16 часов с обратным холодильником. Добавляли воду (47 кг), и смесь охлаждали до 0 °C. Твердое вещество отфильтровывали и промывали смесью изопропанол/вода. К влажному продукту добавляли изопропанол (680 кг) и воду (55 кг), и суспензию нагревали с обратным холодильником. Полученный прозрачный раствор охлаждали до 0 °C, фильтровали и высушивали под вакуумом для получения 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил) пиримидин-2,4-диамин дигидрохлорида (= церитиниб дигидрохлорид; 84,5 кг [t.q., содержит 10% вес./вес. изопропанол], 76,0 кг [100%], 68%-й выход).
Этанол (155 кг) и воду (113 кг) добавляли к 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-дигидрохлориду; 45,0 кг [t.q., содержит 10% вес./вес. изопропанол], 40,5 кг [100%], 64,2 моль), и смесь нагревали до 55 °C. Медленно добавляли водный раствор NaOH (147 л 1 М раствора, 2,3 экв.) и затем охлаждали до 20 °C. После фильтрации продукт перекристаллизовывали из этанола и высушивали под вакуумом для получения церитиниба (33,2 кг, 93%-й выход в расчете на церитиниб дигидрохлорид) в форме почти белого порошка. 1H-ЯМР (400 МГц, CDCl3): δ=1,33 (д, J=6,8 Гц, 6H), 1,38 (д, J=6,1 Гц, 6H), 1,59-1-78 (м, 5 H), 2,18 (с, 3H), 2,75-2,83 (м, 3H), 3,20-3,24 (м, 2H), наложения 3,28 (септ., J=6,8 Гц, 1H), 4,56 (септ., J=6,1 Гц, 1H), 6,82 (с, 1H), 7,25-7,29 (м, 1 H), 7,56 (ушир. с, 1H), 7,64 (м, 1H), 7,94 (м, 1H), 8,01 (ушир. с, 1H), 8,16 (ушир. с, 1H), 8,60 (м, 1H), 9,51 (ушир. с, 1H) частей на миллион. 13C-ЯМР (100 МГц, CDCl3): δ=15,4, 18,9, 22,3, 33,9, 38,6, 47,5, 55,4, 71,4, 105,7, 111,0, 120,6, 123,1, 123,7, 124,9, 126,7, 127,3, 131,2, 134,7, 138,3, 138,5, 144,7, 155,3, 155,4, 157,5 частей на миллион, Элементный анализ: расчетный (%) для C28H36ClN5O3S: C 60,26, H 6,50, N 12,55, O 8,60 Cl 6,35, S 5,74, найденный: C 60,15, H 6,45, N 12,72, O 8,58, Cl 6,43, S 5,67.)
Мутантные антигены gas57 и антитела против gas57
[(1н-индол-5-ил)-гетероарилокси]-1-(азабицикло[3.3.1]нонаны, как холинергические лиганды n-achr, предназначенные для лечения психотических и нейродегенеративных нарушений
Лечение туберозного склероза
Органические соединения
Способ получения фармацевтической композиции
Предназначенная для перорального применения фармацевтическая композиция
Менингококковые полипептиды fhbp
Стабилизированные полипептиды инсулиноподобного фактора роста
Органические соединения
Курс лечения с использованием агониста рецептора s1p

