Результат интеллектуальной деятельности: Способ определения суммарного содержания моноциклических ароматических углеводородов в водах
Вид РИД
Изобретение
Изобретение относится к области аналитической химии, а именно к способам определения суммарного содержания однотипных органических соединений, в частности ароматических углеводородов. Способ предназначен для контроля техногенного загрязнения окружающей среды нефтепродуктами (многокомпонентными смесями углеводородов). Опасными токсикантами являются не все углеводороды, а лишь часть их - преимущественно моноциклические ароматические углеводороды: бензол, толуол, ксилолы, этилбензол, стирол и некоторые другие соединения, содержащие от 6 до 9 (включительно) атомов углерода. Далее углеводороды этой группы условно именуются аренами. Опасными токсикантами являются также родственные аренам полициклические ароматические углеводороды (ПАУ).
Обычно природоохранные организации контролируют суммарное содержание всех УВ в природных и сточных водах (далее CΣ). Способы определения показателя CΣ и нормативы допустимых значений CΣ в водах описаны, например, в обзорных статьях [1, 2]. Дополнительно к показателю CΣ нередко определяют содержания индивидуальных аренов и/или их суммарное содержание в воде (далее CAr). Это способствует выявлению источника загрязнения соответствующего водоема нефтепродуктами [3]. Для определения показателя CAr применяют флуориметрию, УФ- и ИК-спектрометрию. Немногочисленные методики определения этого показателя описаны в научной литературе. Нормативы допустимых значений CAr в России не принимались. Судя по значениям ПДК индивидуальных аренов в водах разного типа, показатель CAr надо определять на уровне 0,1 мг/л и выше. Предлагаемый способ может быть использован для определения суммарного содержания аренов в сточных и сильно загрязненных природных водах.
Известны спектрометрические способы определения суммарного содержания аренов в водах, включающие:
- отбор пробы исследуемой воды,
- экстракционное извлечение углеводородов из водной фазы в специально отобранный органический растворитель,
- отделение неуглеводородных компонентов экстракта (например, фенолов) путем их сорбции в колонке с оксидом алюминия;
- измерение обобщенного аналитического сигнала всех аренов (AAr), присутствующих в очищенном экстракте; обычно это оптическая плотность, интенсивность люминесценции или интегральная интенсивность светопоглощения экстракта в некоторой узкой области спектра;
- построение градуировочной зависимости вида AAr=k Cx с помощью растворов заранее подобранного стандартного вещества Хст (некоторого индивидуального арена);
- расчет суммарного содержания аренов в экстракте в пересчете на Хст по ранее полученной градуировочной зависимости;
- расчет суммарного содержания аренов в пробе в пересчете на Хст с учетом объема пробы и объема экстракта. Найденную величину (С*) считают приблизительно равной действительному содержанию аренов в исследуемой воде (CAr, мг/л).
Примером могут быть способы определения суммарного содержания аренов в природных водах по величине интегральной интенсивности светопоглощения тетрахлорметанового экстракта в области 3150-2995 см-1 [4, 5]. Другим примером могут быть способы определения суммарного содержания аренов в водах, основанные на измерении оптической плотности гексанового экстракта в УФ-области спектра [6, 7]. Сходные методики используют и для определения суммарного содержания ПАУ, в этом случае измеряют люминесценцию ПАУ [8, 9].
Недостатком вышеуказанных способов является низкая точность определения суммарного содержания ароматических углеводородов, вызванная несовпадением чувствительности определения стандартного вещества и чувствительности определения других углеводородов, присутствующих в экстракте пробы. Проверка показала, что вызванная этим фактором систематическая погрешность анализа может составлять сотни процентов [10]. Неоднократно делались попытки снизить эту погрешность путем использования в качестве стандартного вещества многокомпонентной смеси УВ, заранее выделенной из исследуемой воды [3, 4, 6]. Этот прием повышает точность определения CΣ, но не обеспечивает правильность определения CAr, так как доля аренов в смеси водорастворенных углеводородов может сильно меняться от пробы к пробе, а в ходе анализа эта доля не определяется и не учитывается.
Ближайшим аналогом заявляемого способа является способ определения суммарного содержания полициклических ароматических углеводородов (ПАУ) в объектах окружающей среды, в частности, в природной воде, описанный в заявке [8]. Способ включает экстракцию суммы ПАУ из исследуемой пробы, концентрирование экстракта, хроматографирование экстракта в слое сорбента (силикагеля) с применением гексана в качестве подвижного растворителя, регистрацию люминесценции ПАУ и определение суммарного содержания ПАУ по площади полученных хроматографических пиков. При реализации подобных методик в качестве стандартного вещества обычно используют один из ПАУ - бенз[а]пирен [9]. Преимуществом способа [8,9] по сравнению с другими аналогами является более высокая селективность определения ПАУ, обусловленная введением дополнительной операции «хроматографирование экстракта». В результате хроматографирования экстракта устраняется влияние мешающих веществ (не только фенолов, но и углеводородов других групп, в частности, аренов С6-C9). Другими преимуществами прототипа являются высокая чувствительность и экспрессность методик, основанных на измерении люминесценции.
Основным недостатком прототипа [8] является недостаточная точность результатов анализа, вызванная двумя причинами:
1) несовпадением чувствительности определения стандартного вещества (в частности, бенз[а]пирена) и чувствительности определения других ПАУ, присутствующих в экстракте пробы.
2) непредсказуемыми потерями ПАУ в ходе пробоподготовки из-за неполного экстракционного извлечения, а также из-за частичного испарения ПАУ в ходе концентрирования экстракта.
При определении малорастворимых в воде и малолетучих ПАУ потери ПАУ в ходе пробоподготовки невелики, но определение суммарного содержания аренов C6-C9 по методикам типа [8] приведет к получению сильно заниженных результатов. Известно, что арены С6-С9 довольно хорошо растворимы в воде и легко испаряются даже при комнатной температуре. Потери аренов при их экстракционном извлечении из водной фазы нередко превышают 50% [1] и еще более возрастают, если проводится упаривание экстракта [11].
Технической задачей является разработка способа определения суммарного содержания аренов, не связанного с пересчетом обобщенного сигнала на стандартное вещество и исключающего (или учитывающего) потери аренов в ходе пробоподготовки.
Указанный технический результат достигается тем, что предложен способ определения суммарного содержания моноциклических ароматических углеводородов С6-C9 (аренов) в водах, включающий отбор пробы, экстракцию углеводородов, сорбционную очистку экстракта, хроматографирование экстракта и измерение площади пиков на хроматограмме, согласно решения, в ходе анализа проверяют наличие пиков аренов С6-C9; находят суммарную площадь этих пиков (SAr) и рассчитывают суммарное содержание аренов С6-C9 в исследуемой воде (CAr) по формуле CAr=SAr/k, причем для нахождения коэффициента k готовят модельные смеси с известными суммарными содержаниями аренов в воде (CAr)i, получают и хроматографируют экстракты этих смесей так же, как экстракт пробы, а затем строят градуировочную зависимость
(SAr)i=k(CAr)i,
где (SAr)i - суммарная площадь пиков аренов на хроматограмме экстракта из i-ой модельной смеси.
Заявляемый способ включает следующие операции:
- отбор пробы исследуемой воды;
- экстракционное извлечение углеводородов и других малополярных компонентов пробы в органический растворитель (например, н-гексан);
- сорбционную очистку экстракта (для отделения неуглеводородных компонентов);
- отбор аликвоты очищенного экстракта, ввод ее в хроматограф и испарение при 250°C;
- разделение содержащихся в аликвоте индивидуальных углеводородов в режиме программирования температуры от 40 до 200°C в капиллярной колонке, содержащей неполярную неподвижную жидкую фазу;
- последовательное детектирование выходящих из колонки углеводородов с помощью детектора по ионизации пламени (ДИП);
- проверка наличия пиков аренов С6-C9 на хроматограмме экстракта пробы с использованием заранее определенных характеристик удерживания аренов и расчет площади каждого из этих пиков;
- расчет обобщенного сигнала - суммарной площади пиков аренов С6-C9 на хроматограмме экстракта из исследуемой воды, далее эта площадь обозначается SAr;
- приготовление нескольких (как правило, не менее 15) модельных углеводородных смесей в воде, причем все смеси (имитаты сточных или природных вод) содержат арены С6-C9, качественный и количественный состав разных смесей существенно различается, а суммарное содержание аренов в смесях (CAr)i точно известно;
- экстрагирование, очистка и хроматографирование приготовленных смесей ПАУ по той же методике, что была использована для получения хроматограммы экстракта пробы;
- измерение суммарной площади пиков на хроматограмме каждого экстракта из модельной смеси (SAr)i и построение градуировочной зависимости (SAr)i=k(CAr)i;
- расчет суммарного содержания аренов в исследуемой воде по формуле CAr=SAr/k, где коэффициент k находят из вышеуказанной градуировочной зависимости.
Операции пробоотбора, экстракционного выделения углеводородов и суммирования площади хроматографических пиков входят и в предлагаемый способ, и в прототип [8], и в другие методики определения суммарного содержания углеводородов [4, 6, 12, 13]. Некоторые операции, отсутствующие в прототипе, встречаются в других известных методиках хроматографического анализа (например, отбор пиков на хроматограмме по заранее определенным характеристикам удерживания).
Наиболее важным отличием заявляемого способа от прототипа и других аналогов является построение градуировочной зависимости с помощью модельных смесей -водных растворов, содержащих разные арены С6-C9. Суммарное содержание аренов в этих смесях известно и различно (пример в табл. 1). Искомое суммарное содержание аренов в исследуемой воде определяется без пересчета на стандартное вещество и с учетом потерь аренов в ходе пробоподготовки.
В ходе специальных экспериментов было установлено, что выраженные в процентах потери аренов С6-C9 в ходе их экстракционного извлечения из модельных смесей (имитатов) приблизительно равны потерям аренов при их экстракционном извлечении из реальных природных или сточных вод (при прочих равных условиях и одинаковой методике извлечения). То же касается потерь аренов за счет их летучести. Суммарная площадь пиков аренов на хроматограмме экстракта пробы снижается за счет потерь приблизительно в той же степени, что и суммарная площадь пиков на хроматограммах экстрактов модельных смесей. Таким образом, потери аренов в ходе пробоподготовки не устраняются, но автоматически учитываются в ходе расчета суммарного содержания аренов в исследуемой пробе, в соответствии с принципом релятивизации систематических погрешностей. Кроме того, построение градуировочной зависимости по многокомпонентным смесям аренов (а не по индивидуальному стандартному веществу) снижает влияние различий в чувствительности определения разных аренов, что дополнительно снижает погрешность оценки суммарного содержания аренов, в соответствии с принципом рандомизации погрешностей.
В литературе по хроматографическому определению углеводородов в природных и сточных водах не описаны методики, включающие применение многокомпонентных смесей (водных растворов известного состава) для построения градуировочных зависимостей, учитывающих потери этих углеводородов в ходе пробоподготовки и различия в чувствительности определения этих углеводородов. Предложенный прием является ранее не известным техническим решением. Новыми элементами являются и перечень всех операций, и их последовательность.
Предлагаемый способ определения суммарного содержания аренов может быть реализован, если их суммарное содержание в исследуемой воде превышает 0,1 мг/л. Погрешность определения CAr не превышает 12% отн. Продолжительность анализа единичной пробы воды (включая экстракцию углеводородов, но без учета приготовления модельных смесей и построения градуировки) не превышает 120 минут.
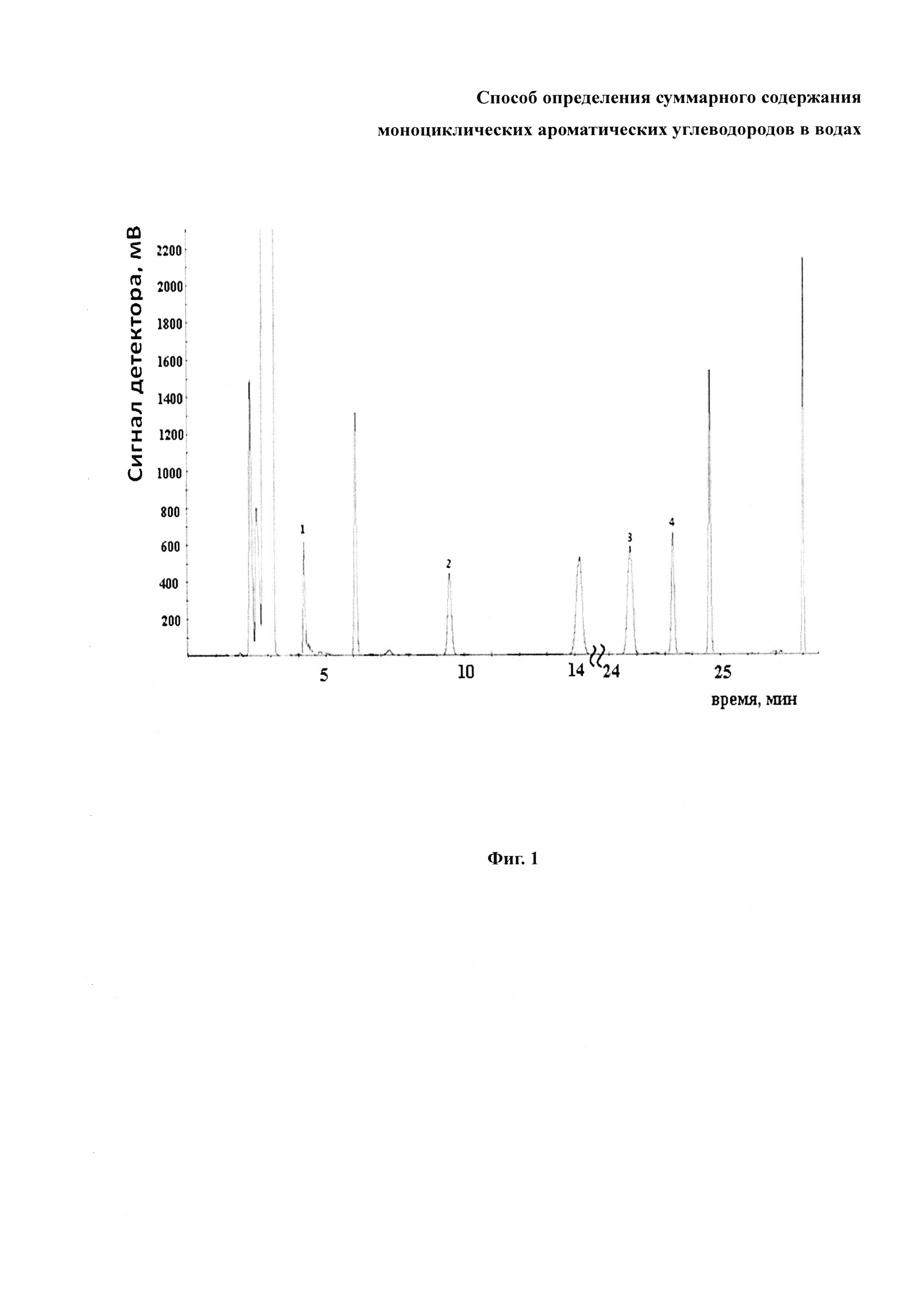
На фиг. 1 представлена хроматограмма экстракта из модельной смеси разных углеводородов в воде. Методика получения хроматограмм изложена в примере 1. Выделены пики аренов, а именно: 1-бензол, 2-толуол, 3-м-ксилол, 4-о-ксилол.
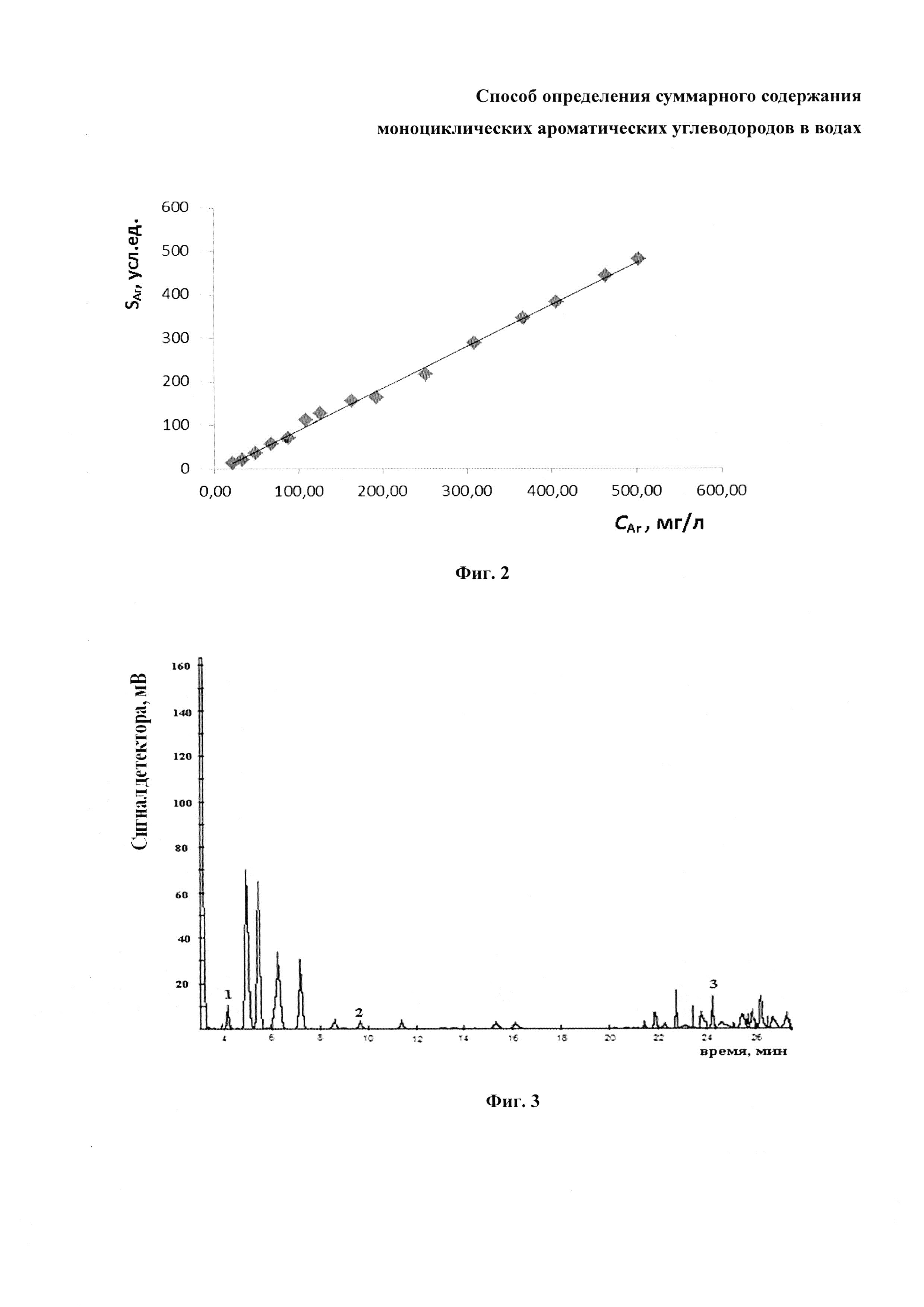
На фиг. 2 показана градуировочная зависимость для определения суммарного содержания аренов по суммарной площади их пиков, построенная по хроматограммам экстрактов из разных модельных растворов.
На фиг. 3 представлена хроматограмма экстракта из очищенной сточной воды нефтехимического предприятия. Методика анализа сточной воды изложена в примере 2.
Пример 1. Анализ имитатов сточных вод
Приготовление модельных смесей (имитатов). Навески аренов и других УВ (реактивы х.ч.) берут на аналитических весах, растворяют в небольшом объеме н-гексана, а затем доводят объем до метки (10,0 мл) н-гексаном, получая вспомогательный раствор с известным суммарным содержанием аренов (CAr)0. Его разбавляют в 100 или в 1000 раз дистиллированной водой и тщательно перемешивают. Готовят не менее 15 водных растворов, содержащих разные наборы аренов и различающихся по суммарной концентрации аренов (CAr)i, мг/л. Необходимо, чтобы эти растворы были достаточно разбавленными (с учетом равновесной растворимости индивидуальных аренов в воде) и различались по набору и соотношению концентраций разных аренов. Значения (CAr)i для всех модельных растворов должны быть точно известны, охватывая диапазон ожидаемых значений CAr в исследуемых водах. Так, набор смесей, часть которых представлена в табл. 1, охватывает диапазон от 0,02 до 1 мг/л.
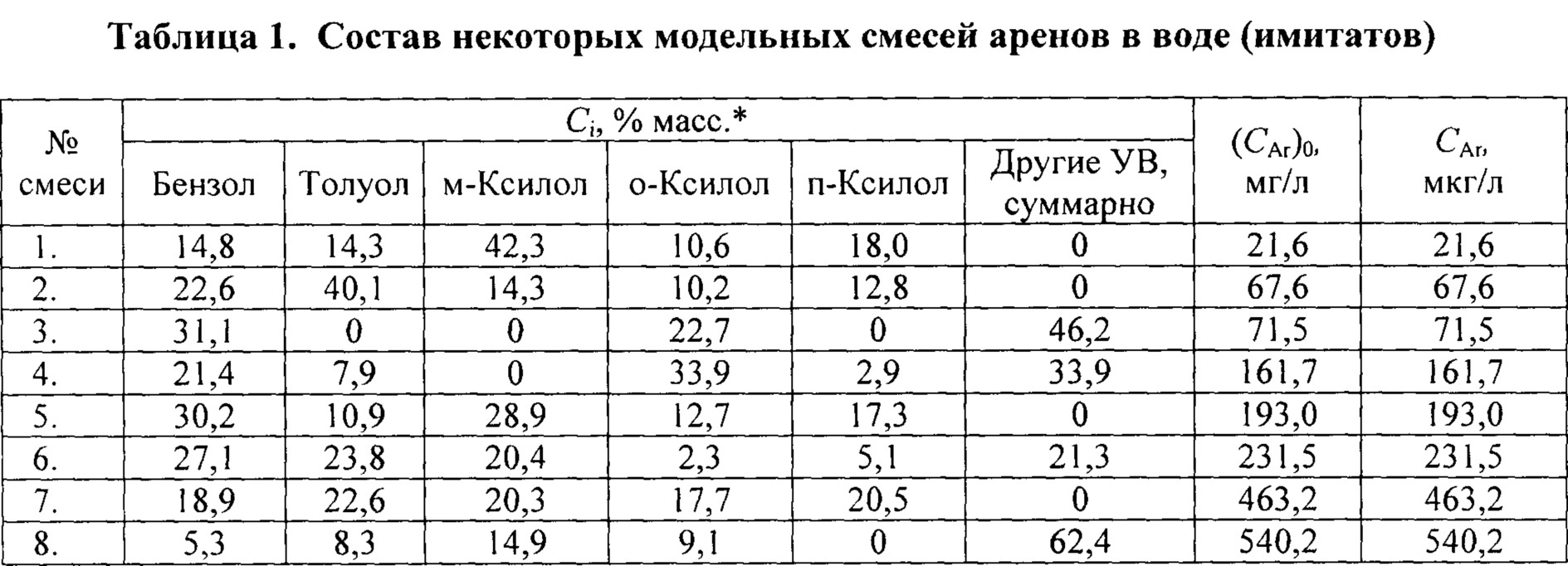
* За 100% принята сумма всех растворенных УВ, кроме н-гексана;
** - группа других УВ содержит любые УВ, кроме н-гексана и аренов.
Получение и очистка экстрактов. Мерным цилиндром вместимостью 500 см3 отбирают 250 см3 i-го модельного водного раствора, подкисляют его соляной (серной) кислотой до рН=2 и помещают в делительную воронку на 1000 см3. Добавляют 20 г хлорида натрия и 25 см3 экстрагента (н-гексана). Делительную воронку интенсивно встряхивают в течение 5 минут, периодически открывая пробку для выпускания паров. По окончании экстракции пробу отстаивают для расслоения водной и органической фазы (в течение 10 мин). После того как смесь расслоится, экстракт переносят в коническую колбу на 50 см3 с притертой пробкой, в которую добавлено 1,25 г безводного сернокислого натрия. Время обезвоживания экстракта составляет 10-20 мин. Экстракт декантируют или фильтруют через стекловату (вату) в другую колбу, промывают осушитель небольшим объемом экстрагента, который присоединяют к экстракту. Затем экстракт переносят порциями в подготовленную хроматографическую колонку с оксидом алюминия (методика подготовки хроматографической колонки описана в ГОСТ Р 51797-2001 [14], следя, чтобы уровень жидкости не опускался ниже верхнего уровня слоя сорбента. Первые 3-4 мл очищенного экстракта (элюата) отбрасывают, а оставшуюся часть элюата собирают в мерную колбу на 25 см3 с притертой пробкой. После пропускания всего экстракта колонку промывают экстрагентом, присоединяют его к основному элюату и доводят объем до метки. Те же операции проводят с другими модельными растворами.
Хроматографирование экстрактов и построение градуировочной зависимости.
Очищенные экстракты из модельных растворов поочередно хроматографируют, используя газовый хроматограф Хромос ГХ-1000 с детектором по ионизации пламени. Капиллярная колонка ВР-1 (30 м) содержит в качестве неподвижной жидкой фазы (НЖФ) диметилполисилоксан 100% (неселективная, неполярная НЖФ). Толщина слоя НЖФ - 0,53 мм. Газ-носитель - водород. Смеси разделяют в режиме программирования температуры, используя следующий режим: выдержка при 40°C в течение 20 мин., затем линейное повышение температуры до 200°C со скоростью 10°C/мин. Температура испарителя - 250°C, давление газа-носителя - 0,2 кг/см2. Режим разделения УВ смесей оптимизировали так, чтобы исключить возможность наложения пиков других УВ на пики аренов С6-C9. В качестве примера приведена хроматограмма одного из модельных растворов (фиг. 1). Полученные хроматограммы обрабатывали с помощью стандартного программного обеспечения Chromos, то есть времена удерживания и площади всех пиков рассчитывались автоматически. Отнесение пиков на хроматограмме проводили по абсолютным временам удерживания, проверяя лишь наличие пиков аренов С6-C9.
Обобщенный аналитический сигнал аренов, содержащихся в i-ом имитате (SAr)i получали, суммируя площади пиков опознанных аренов С6-C9. Хроматограмму каждой смеси регистрировали три раза. Значения (SAr)i усредняли, если различия между ними не превышали 15% от их среднего арифметического. Зная суммарное содержание аренов в каждом модельном растворе, строили линейную градуировочную зависимость (SAr)i=k (CAr)i. Пример градуировочной зависимости представлен на фиг. 2. Коэффициент K находили по рассчитанному уравнению линейной регрессии. Коэффициент корреляции для этой градуировочной зависимости превышает 0,99. Нижний предел определения суммарного содержания аренов в имитатах составляет приблизительно 0,1 мг/л.
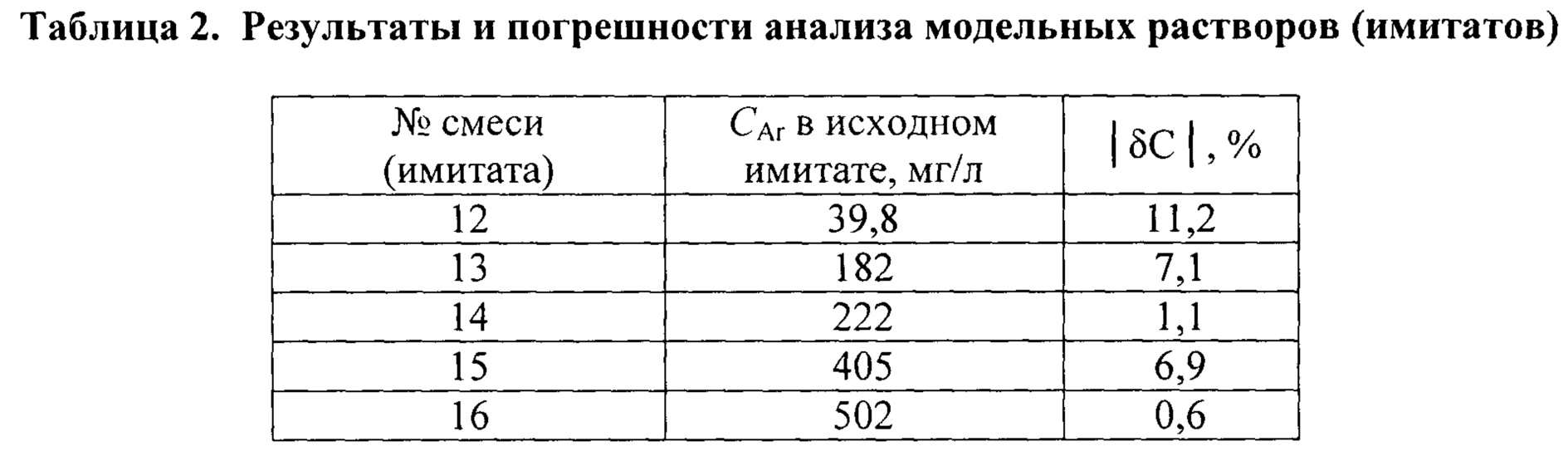
Анализ растворов с условно неизвестными концентрациями. Модельные многокомпонентные водные растворы аренов (имитаты), не использовавшиеся для построения вышеописанной градуировочной зависимости, готовили, экстрагировали и хроматографировали, как описано выше. Хроматограмму каждого экстракта получали трижды в условиях сходимости, каждый раз суммировали площади пиков опознанных аренов С6-C9. Найденные значения SAr усредняли, если различия между ними не превышали 15% от их среднего арифметического. Суммарное содержание аренов в исследуемом имитате рассчитывали по формуле CAr=SAr/k, используя ранее найденный регрессионный коэффициент k. Если анализируемые растворы имели слишком высокое содержание аренов (более 100 мг/л), их разбавляли в 10 или 100 раз и повторяли анализ. Как видно из данных, приведенных в табл. 2, предлагаемый способ позволяет определять суммарные содержание аренов в имитатах природных и сточных вод с относительной погрешностью, не превышающей (по модулю) 12%.
Пример 2. Анализ сточной (или сильно загрязненной природной) воды
Отбирают пробу сточной воды по общепринятой методике [14], пробу фильтруют и доводят pH до 2,0. В случае необходимости после отбора пробы сточной воды ее консервируют. 250 см3 сточной воды помещают в делительную воронку. Добавляют 20 г хлорида натрия, а затем вводят 25 см3 экстрагента (н-гексана). Экстракт получают и очищают, как описано в примере 1. Полученный элюат хроматографируют, используя газовый хроматограф Хромос ГХ-1000 с детектором по ионизации пламени по методике, описанной в примере 1. Пример хроматограммы экстракта из пробы сточной воды приведен в виде фиг. 3.
Суммируя площади пиков аренов на полученной хроматограмме, получают обобщенный аналитический сигнал аренов (SAr) и рассчитывают суммарное содержание аренов в воде по заранее построенной градуировочной зависимости, как описано в примере 1. Суммарное содержание аренов в сильно загрязненных сточных водах рассчитывают с учетом степени предварительного разбавления пробы.
В таблице 3 приведены результаты проверки правильности анализа сточной воды нефтеперерабатывающего предприятия по предлагаемому способу. Для проверки правильности использовали метод «введено-найдено» (метод стандартных добавок). Точность анализа других проб сточных и сильно загрязненных природных вод также удовлетворительна.

Литература
1. Леоненко И.И. Методы определения нефтепродуктов в водах и других объектах окружающей среды (обзор) / И.И. Леоненко, В.П. Антонович, А.М. Андрианов, И.В. Безлуцкая, К.К. Цымбалюк // Методы и объекты химического анализа. - 2010. - Т. 5, №2. - С. 58-72.
2. Analysis of petroleum hydrocarbons in environmental media. Total Petroleum Hydrocarbon Criteria Working Group Series / W. Weisman (Ed.). - USA: Amherst Scientific Publishers, 1998. - Vol. 1, 98 p.
3. Другов Ю.С. Экологические анализы при разливах нефти и нефтепродуктов / Ю.С. Другов, А.А. Родин. - СПб.: Анатолия, 2000. - 250 с.
4. Шагидуллин, P.P. Определение нефтепродуктов в водах на основе ИК-Фурье спектрального комплекса и измерения интегральных интенсивностей полос поглощения / P.P. Шагидуллин [и др.] // Журнал аналитической химии. - 2002. - Т. 57, №3. - С. 250-256.
5. Волкова С.С. Количественное определение углеводородов с использованием численного поддиапазонного интегрирования инфракрасных спектров / С.С. Волкова, А.А. Кудрявцев, Д.В. Мильченко, А.Я. Юффа. - Ж. прикл. Химии. 1997. Т. 70, вып. 5, с. 807-810 (или патент 2117933).
6. Методики определения нефтепродуктов и полициклических ароматических углеводородов в водах и донных отложениях: Сб. метод, указаний. Ростов-на-Дону. Изд-во НТПКАО ЭКСИДОН, 1992.
7. Гомеля, Н.Д. Экстракционно-спектрофотометрический метод определения суммарного содержания нефтепродуктов в воде / Н.Д. Гомеля, Л.В. Калабина, А.П. Хохотва // Химия и технология воды - 1999. - Т. 21, №6. - С. 611-616.
8. Заявка RU 0093938230. Способ определения суммарного содержания полициклических ароматических углеводородов (ПАУ) в объектах окружающей среды / Семенов А.Д., Павленко Л.Ф., Савицкая Н.Г. БИ 20.02.1996.
9. РД 52.24.440-2006. Сумма массовых концентраций 4-7 ядерных полициклических ароматических углеводородов в водах. Методика выполнения измерений люминесцентным методом с использованием тонкослойной хроматографии. Дата введения 2006-04-01.
10. Кленкин, А.А., Павленко Л.Ф., Темердашев З.А. Некоторые методические особенности определения уровня нефтяного загрязнения водных экосистем // Заводск. лаборатория. Диагностика материалов. 2007. Т. 73, №2. С. 31-35.
11. Concentration, separation and determination of hydrocarbons in sea water / Desideri P.G. [e.a.] // J. Chrom. 1984. V. 284, №10 - Pp. 167-178.
12. ASTM D5134-98. Standard test method for detailed analysis of petroleum naphthas through n-Nonane by capillary gas chromatography. Annual book of ASTM standards. 1998.
13. Обзор хроматографических методов анализа, используемых для определения нефтепродуктов в воде / М.Ю. Вождаева и [др.] // Вода: химия и экология. - 2011. - №10. - С. 34-40.
14. ГОСТ 51797-2001. Вода питьевая. Метод определения содержания нефтепродуктов. - М.: Госиздат России. - 15 с.
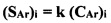
Способ получения гуминового сорбента из сапропеля для очистки сточных вод
Способ получения биомиметического кремний-содержащего кальций-фосфатного покрытия на сплавах титана из модельного раствора межклеточной жидкости человека
Способ моделирования процесса образования оксалатного мочевого камня
Способ биомиметического синтеза sr - содержащего карбонатгидроксилапатита, модифицированного брушитом
Способ формирования тонких пленок аморфного кремния
Способ коррекции и профилактики нарушений зрения упражнениями с элементами бадминтона
Способ биомиметического синтеза sr-содержащего карбонатгидроксилапатита, допированного брушитом
Пиридиновые производные бис(3,4-дигидрохиноксалин-2(1н)-она) и бис(3,4-дигидро-2н-бензо[b][1,4]оксазин-2-она), обладающие анальгетической активностью
Способ получения биомиметического кальций-фосфатного модифицированного желатином покрытия на сплавах титана из модельного раствора межклеточной жидкости человека
Способ формирования пустот в ионных кристаллах
Способ лечения клещевого энцефалита
Способ лечения хронического вирусного гепатита с препаратами в липосомальной форме
Способ лечения рецидивирующего урогенитального герпеса с симптомами хронической усталости
Способ лечения язвенной болезни желудка и/или двенадцатиперстной кишки
Способ защиты организма от инфекции, вызванной штаммами субтипа h1n1 вируса гриппа а препаратом на основе альфа-2 интерферона человека
Способ определения суммарного содержания фенолов в водах
Способ определения суммарного содержания углеводородов в водах
Живая аттенуированная культуральная вакцина для профилактики натуральной оспы и других ортопоксвирусных инфекций на основе вируса осповакцины и способы ее получения и применения

