Результат интеллектуальной деятельности: КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к комбинациям ингибиторов фосфодиэстеразы 2 (PDE2) с ингибиторами фосфодиэстеразы 10 (PDE10). В частности, настоящее изобретение относится к комбинациям производных 1-арил-4-метил-[1,2,4]триазоло[4,3-a]-хиноксалина, которые, как было обнаружено, ингибируют фосфодиэстеразу 2 (PDE2), с ингибиторами фосфодиэстеразы 10 (PDE10). Конкретные ингибиторы PDE10 выбирают из группы MP-10, PQ-10, TP-10, папаверина и соединений, раскрытых в WO 2011/051324 и в WO 2011/110545. Настоящее изобретение также направлено на фармацевтические композиции, содержащие такие комбинации, на способы получения таких композиций, на применение ингибиторов PDE2, в частности, производных 1-арил-4-метил-[1,2,4]триазоло[4,3-a]-хиноксалина, для усиления действия указанных ингибиторов PDE10, и на применение указанных ингибиторов PDE10 для усиления эффекта указанных ингибиторов PDE2, в частности, производных 1-арил-4-метил-[1,2,4]триазоло[4,3-a]-хиноксалина, и на применение таких комбинаций и композиций для предупреждения и лечения расстройств, в которые вовлечены PDE2 и PDE10, таких как неврологические и психические расстройства и эндокринные или метаболические заболевания.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
В Journal of Fluorine Chemistry (2009), 130 (10), 886-893, раскрываются 1-арил-4-метил-[1,2,4]триазоло[3,4-a]хиноксалины, где арил представляет собой фенил, 4-метоксифенил, 4-хлорфенил или 4-нитрофенил, неожиданно образующиеся при реакции 2-гидразин-3-метилхиноксалина с трифторметил-бета-дикетонами.
В Green Chemistry (2004), 6, 156-157, раскрываются способы синтеза 1-арил-4-метил-[1,2,4]триазоло[3,4-a]хиноксалинов без применения растворителя, где арил представляет собой фенил, 4-метилфенил, 4-хлорфенил, 4-метоксифенил и 3-метоксифенил.
В Synthetic Communications (2006), 36, 1873-1878, раскрываются способы синтеза 1-арил-4-метил-[1,2,4]триазоло[3,4-a]хиноксалинов, где арил представляет собой фенил, 4-метилфенил, 4-хлорфенил, 2-метоксифенил и 4-метоксифенил.
В WO-2010/101230 раскрываются [1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-оны в качестве ингибиторов PDE9, применимых в лечении расстройств мочеиспускания. В WO 2012/104293, WO 2010/054253 и Expert Opinion on Therapeutic Patents, Informa Healthcare, GB, (2009), 19 (12), 1715-1725, раскрываются соединения в качестве ингибиторов фосфодиэстераз.
Фосфодиэстеразы (PDE) представляют собой семейство ферментов, кодируемых 21 геном и подразделенных на 11 отдельных семейств согласно структурным и функциональным свойствам. Эти ферменты осуществляют метаболическую инактивацию широко распространенных внутриклеточных вторичных мессенджеров, цикличный 3′,5′-аденозинмонофосфат (cAMP) и цикличный 3′,5′-гуанозинмонофосфат (cGMP). Эти два мессенджера регулируют большое разнообразие биологических процессов, в том числе выработку и действие провоспалительных медиаторов, функционирование ионных каналов, сокращение мышц, коммитирование, дифференциацию, апоптоз, липогенез, гликогенолиз и глюконеогенез. Они осуществляют это посредством активации протеинкиназы A (PKA) и протеинкиназы G (PKG), которые, в свою очередь, фосфорилируют большое разнообразие субстратов, в том числе факторы транскрипции и ионные каналы, которые регулируют многочисленные физиологические реакции. В случае нейронов предусматриваются активация cAMP- и cGMP-зависимых киназ и последующее фосфорилирование белков, вовлеченных в быструю регуляцию синаптической передачи, а также в дифференцировку и выживаемость нейронов. Внутриклеточные концентрации cAMP и cGMP точно регулируются скоростью биосинтеза с помощью циклаз и скоростью расщепления с помощью PDE. PDE представляют собой гидролазы, которые инактивируют cAMP и cGMP посредством каталитического гидролиза 3′-сложноэфирной связи с образованием неактивного 5′-монофосфата (схема A).
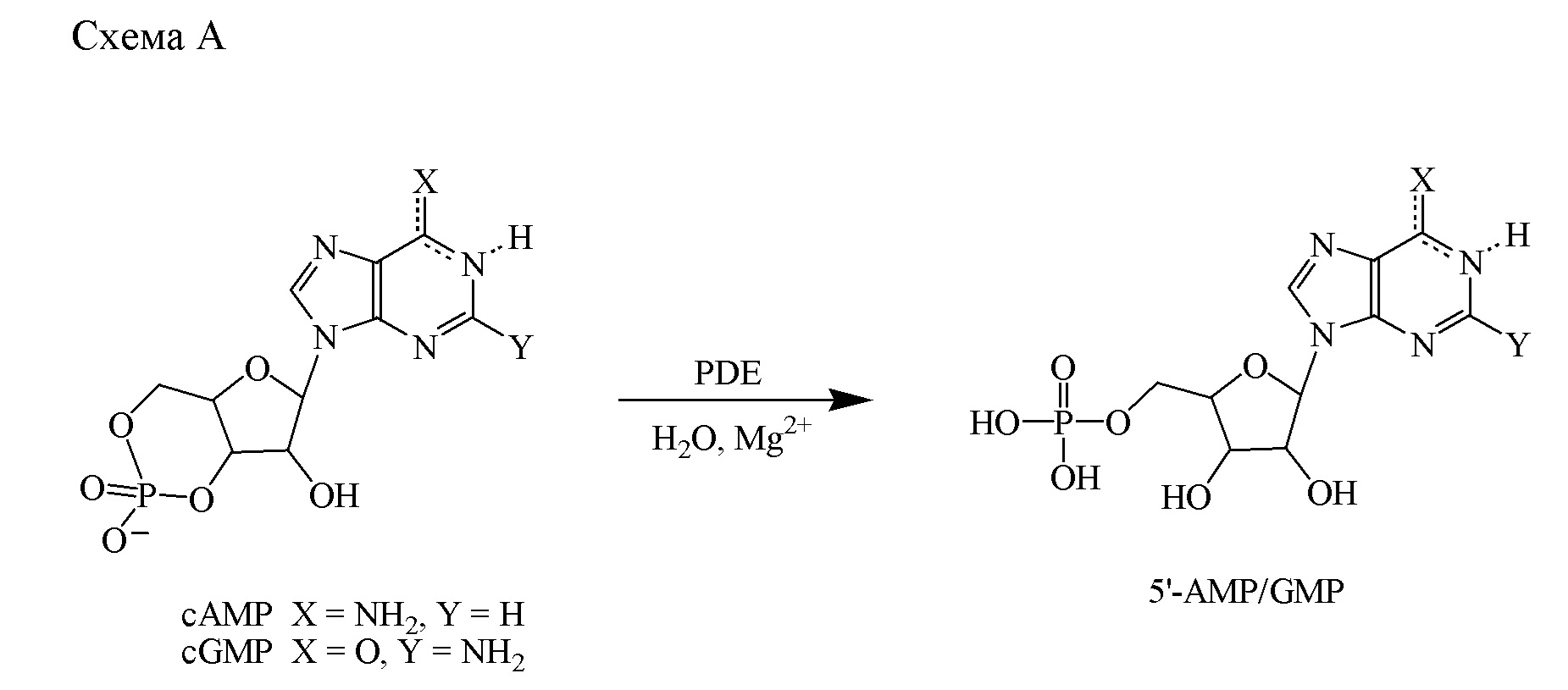
На основании субстратной специфичности семейства PDE можно разделить на три группы: i) cAMP-специфические PDE, которые включают PDE4, 7 и 8; ii) cGMP-селективные ферменты PDE5, 6 и 9 и iii) PDE, действующие на два субстрата, PDE1, 2 и 3, а также PDE10 и 11.
Кроме того, для PDE характерна дифференциальная экспрессия во всем организме, в том числе в центральной нервной системе. Вследствие этого разные изоферменты PDE могут иметь разные физиологические функции. Соединения, которые селективно ингибируют семейства или изоферменты PDE, могут проявлять особую терапевтическую активность, меньшее количество побочных эффектов или и то, и другое.
Фосфодиэстераза 2A (PDE2A) инактивирует внутриклеточные механизмы передачи сигналов, которые зависят от передачи сигналов с помощью циклических нуклеотидов, опосредованной cAMP и cGMP, путем их расщепления. Такие сигнальные пути, как известно, играют роль в регуляции генов, вовлеченных в индукцию синаптической пластичности.
Фармакологическое ингибирование PDE2, таким образом, обуславливает повышение уровней синаптической пластичности (коррелята, лежащего в основе обучения и памяти), что указывает на то, что модуляция PDE2A может представлять собой цель для облегчения нарушений познавательных способностей, наблюдаемых у людей, страдающих от таких расстройств, как, например, шизофрения, болезнь Альцгеймера, болезнь Паркинсона и другие расстройства ЦНС, ассоциированные с когнитивной дисфункцией (Neuropharmacology 47, (2004), 1081-92).
Фосфодиэстераза 2A (PDE2A) экспрессируется в головном мозге в большем количестве по сравнению с периферическими тканями. Высокий уровень экспрессии PDE2 в лимбической системе (изокортексе, гиппокампе, миндалевидном теле, поводке эпиталамуса, базальных ганглиях) указывает на то, что PDE2 может модулировать передачу сигналов между нейронами, связанную с эмоциями, восприятием, вниманием, обучением и памятью. Кроме того, PDE2 экспрессируется в прилежащем ядре, обонятельной луковице, обонятельном бугорке и миндалевидном теле, что подтверждает предположение, что PDE2 может также вовлекаться в тревожность и депрессию.
Дополнительно, было показано, что ингибиторы PDE2 полезны в ослаблении индуцированной окислительным стрессом тревожности, что подтверждает их применение в лечении тревожности при нейропсихиатрических и нейродегенеративных расстройствах, в которые вовлечен окислительный стресс, таких как болезнь Альцгеймера, болезнь Паркинсона и рассеянный склероз (J. Pharmacol. Exp. Ther. 2008, 326(2), 369-379).
Было показано, что ингибиторы PDE2 усиливают долговременную потенциацию синаптической передачи и улучшают запоминание и консолидацию памяти при распознавании объекта и в тестах социальной ориентации у крыс. Кроме того, было показано, что ингибиторы PDE2 устраняют ослабление кратковременной памяти, индуцированное MK-801, в T-образном лабиринте у мышей. Также было показано, что ингибиторы PDE2 проявляют активность в тесте принудительного плавания и моделях со светлой/темной камерой; а также демонстрируют эффекты, подобные анксиолитическим, в тестах с приподнятым крестообразным лабиринтом, платформой с отверстиями и установкой "открытое поле" и предупреждают индуцированные стрессом изменения апоптоза и поведения (Neuropharmacology 47, (2004), 1081-92).
Таким образом, ингибиторы PDE2 могут применяться в лечении ослабления памяти, нарушений познавательных способностей, тревожности, биполярного расстройства и депрессии.
Из всех 11 известных семейств PDE PDE10 характеризуется наиболее ограниченным распределением с высоким уровнем экспрессии только в головном мозге и яичках. В головном мозге мРНК и белок PDE10A экспрессируются на высоком уровне в большинстве стриарных средних шипиковых нейронов (MSN). Такое своеобразное распределение PDE10A в головном мозге вместе с увеличением числа ее фармакологических исследований указывает на потенциальное применение ингибиторов PDE10A в лечении неврологических и психических расстройств, таких как шизофрения.
Таким образом, ингибиторы PDE10 могут обладать фармакологическим профилем, аналогичным таковому современных антипсихотических средств, которые лечат главным образом позитивные симптомы шизофрении, но также обладают потенциалом для устранения негативных и когнитивных симптомов шизофрении, при этом не имеют нецелевых связанных побочных эффектов, таких как EPS или высвобождение пролактина, которые часто наблюдаются при применении существующих антипсихотических средств.
Поскольку ингибиторы PDE10 можно применять для повышения уровней cAMP и/или cGMP в клетках, которые экспрессируют фермент PDE10, например, в нейронах, которые входят в состав базальных ганглиев, ингибиторы PDE10 могут применяться в лечении шизофрении и, кроме того, ряда состояний, описываемых в данном документе, например, болезни Паркинсона, болезни Хантингтона, аддикции и депрессии. Ингибиторы PDE10 также могут применяться при других состояниях, таких как ожирение, инсулинонезависимый диабет, биполярное расстройство, обсессивно-компульсивное расстройство и боль.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящее время было неожиданно обнаружено, что эффект ингибиторов PDE10 можно усилить с помощью ингибиторов PDE2. В частности, ингибиторы PDE10 можно выбрать из группы MP-10, PQ-10, TP-10, папаверина и соединений, раскрытых в WO 2011/051324 и в WO 2011/110545, документах, настоящим включенных посредством ссылки во всей своей полноте. Эффект ингибиторов PDE10 можно усилить, в частности, с помощью производных 1-арил-4-метил-[1,2,4]триазоло[4,3-a]-хиноксалина формулы (I) согласно настоящему изобретению, являющихся ингибиторами PDE2. Например, наблюдали, что ингибиторы PDE2 по настоящему изобретению в комбинации с ингибиторами PDE10, в частности, с ингибитором PDE10 MP-10 или с ингибиторами PDE10 соединением A (соединение номер 1 в WO 2011/051324) и соединением B (соединение номер 25 в WO 2011/110545), приведенными ниже, могут ингибировать эффекты апоморфина или амфетамина у крыс.
|
Также наблюдали, что ингибитор PDE10 MP-10 может дозозависимым образом усиливать in vivo связывание радиолиганда, осуществляющего селективное связывание с каталитическим доменом фермента PDE2.
Таким образом, целью настоящего изобретения является обеспечение новых комбинаций, включающих:
a) ингибитор PDE2 или его фармацевтически приемлемые соль или сольват и
b) один или несколько ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов.
Настоящее изобретение также относится к продуктам, содержащим в качестве первого активного ингредиента a) ингибитор PDE2 или его фармацевтически приемлемые соль или сольват, определенные в данном документе, и в качестве второго активного ингредиента b) один или несколько ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов, в виде комбинированных препаратов для одновременного, раздельного или последовательного применения в лечении пациентов, страдающих от неврологических или психических расстройств или эндокринных или метаболических заболеваний.
Иллюстрацией настоящего изобретения является фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и любую из комбинаций, описанных выше. Иллюстрацией настоящего изобретения является фармацевтическая композиция, полученная путем смешивания любой из комбинаций, описанных выше, и фармацевтически приемлемого носителя. Иллюстрацией настоящего изобретения является способ получения фармацевтической композиции, включающий смешивание любой из комбинаций, описанных выше, и фармацевтически приемлемого носителя.
В дополнительном аспекте настоящее изобретение относится к применению ингибитора PDE2 или его фармацевтически приемлемых соли или сольвата для усиления эффекта одного или нескольких ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов.
Настоящее изобретение также относится к ингибитору PDE2 или его фармацевтически приемлемым соли или сольвату, определенным в данном документе, для применения в усилении терапевтического эффекта одного или нескольких ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов у пациентов, страдающих неврологическими или психическими расстройствами или эндокринными или метаболическими заболеваниями.
Дополнительно, настоящее изобретение также относится к применению ингибитора PDE2 или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, для получения лекарственного препарата для усиления терапевтического эффекта одного или нескольких ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов у пациентов, страдающих неврологическими или психическими расстройствами или эндокринными или метаболическими заболеваниями.
Настоящее изобретение дополнительно относится к применению одного или нескольких ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов для усиления эффекта ингибитора PDE2 или его фармацевтически приемлемых соли или сольвата. Настоящее изобретение также относится к одному или нескольким ингибиторам PDE10 для применения в усилении терапевтического эффекта соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, определенных в данном документе. В дополнительном аспекте настоящее изобретение также относится к применению одного или нескольких ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов для получения лекарственного препарата для усиления терапевтического эффекта ингибитора PDE2 или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, у пациентов, страдающих неврологическими или психическими расстройствами или эндокринными или метаболическими заболеваниями.
Настоящее изобретение дополнительно относится к способу лечения неврологического или психического расстройства или эндокринного или метаболического заболевания, включающему введение субъекту, нуждающемуся в этом, терапевтически эффективного количества комбинации, включающей: a) ингибитор PDE2 или его фармацевтически приемлемые соль или сольват, и b) один или несколько ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов, или терапевтически эффективного количества фармацевтической композиции, описанной выше.
Настоящее изобретение дополнительно относится к способу усиления терапевтического эффекта ингибитора PDE2 или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, включающему введение субъекту, нуждающемуся в этом, терапевтически эффективного количества комбинации, включающей: a) ингибитор PDE2 или его фармацевтически приемлемые соль или сольват, определенные в данном документе, и b) один или несколько ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов, или терапевтически эффективного количества фармацевтической композиции, описанной выше.
Настоящее изобретение дополнительно относится к способу усиления терапевтического эффекта одного или нескольких ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов, включающему введение субъекту, нуждающемуся в этом, терапевтически эффективного количества комбинации, включающей: a) ингибитор PDE2 или его фармацевтически приемлемые соль или сольват, и b) один или несколько ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов, или терапевтически эффективного количества фармацевтической композиции, описанной выше.
ОПИСАНИЕ ФИГУР
На фигурах 1a-c показан эффект (a) растворителя + различных доз MP-10 (0, 0,63, 1,25 и 2,5 мг/кг), вводимых подкожно (s.c.); (b) соединения B-1a (40 мг/кг, s.c.), вводимого подкожно + различных доз MP-10 (0, 0,63, 1,25 и 2,5 мг/кг, s.c.); и (c) MP-10 (2,5 мг/кг, s.c.) + различных доз соединения B-1a (0, 0,63, 2,5, 10 и 40 мг/кг, s.c.) в отношении тревожного возбуждения, индуцированного апоморфином.
На фигуре 2 показан дозозависимый эффект MP-10 (-1 ч, s.c.) в отношении тревожного возбуждения, индуцированного апоморфином (медианный балл), в зависимости от дозы совместно вводимого ингибитора PDE2 (PDE2-i) Β-1a (0,63-10 мг/кг, s.c.; -1 ч) или растворителя (10 мл/кг, s.c.; -1 ч). Пунктирные горизонтальные линии представляют критические уровни для небольшого подавления тревожного возбуждения (балл <21; верхняя линия) и явно выраженного подавления тревожного возбуждения (балл <10; нижняя линия). На фигуре 2 следующие символы соответствуют указанным ниже дозам:
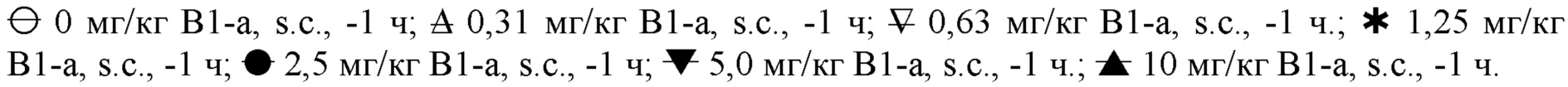
На фигуре 3 показана ED50 (при доверительных интервалах с надежностью 95%) MP-10 (-1 ч, s.c.) для снижения тревожного возбуждения, индуцированного апоморфином, до балла <21 (фиг.3a) или <10 (фиг.3b) в зависимости от дозы совместно вводимого PDE2-i Β-1a (0,63-10 мг/кг, s.c.; -1 ч; закрашенные символы) или растворителя (10 мл/кг, s.c.; -1 ч; незакрашенные символы). Серая горизонтальная полоса представляет ED50 (при доверительных интервалах с надежностью 95%) MP-10 (-1 ч, s.c.) в группе растворителя (фиг.3a) или MP-10 (-1 ч, s.c.) в отдельности (фиг.3b; ретроспективные данные).
На фигуре 4 показан эффект Β-1a (0 в сравнении с 10 мг/кг, s.c.; -1 ч) в отношении дозозависимого эффекта соединения A (-1 ч, s.c.; фигура 4a) и соединения B (-1 ч, s.c.; фигура 4b) для подавления тревожного возбуждения, индуцированного апоморфином. Показаны отдельные баллы (незакрашенные и закрашенные круги для PDE2-i при 0 и 10 мг/кг, соответственно) и медианные баллы (горизонтальные линии) тревожного возбуждения в каждой дозовой группе. На фигуре 4: *p<0,05, **p<0,01, ***p<0,001 (апостериорный критерий Бонферрони, 0 в сравнении с 10 мг/кг). ED50 (при доверительных интервалах с надежностью 95%) ингибиторов PDE10 (PDE10-i) для снижения баллов тревожного возбуждения до <21, <10 и <5 были приведены для совместной обработки с помощью PDE2-i при 0 и 10 мг/кг.
|
|
На фигурах 5a-d показан дозозависимый эффект PDE2-i Β-1a (0, 0,63, 1,25, 2,5 и 5,0 мг/кг, s.c.; -1 ч) в отношении тревожного возбуждения, индуцированного апоморфином, в присутствии стандартных доз соединения A (0 или 2,5 мг/кг, s.c., -1 ч; фиг. 5a и 5c, соответственно) или соединения B (0 или 2,5 мг/кг, s.c., -1 ч; фиг. 5b и 5d, соответственно). Пунктирная горизонтальная линия представляет критерий небольшого подавления тревожного возбуждения (балл <21). На фигуре 5: *p<0,05 (критерий множественных сравнений Даннетта, в сравнении с 0 мг/кг)
На фигуре 6a показано дозозависимое подавление гиперлокомоции, индуцированной d-амфетамином, определенное через 1 ч после s.c. инъекции MP-10; на фигуре 6b показано отсутствие эффекта против гиперлокомоции, индуцированной d-амфетамином, определенное через 1 ч после s.c. инъекции Β-1a (40 мг/кг); на фигуре 6c показано дозозависимое усиление эффекта MP-10 (2,5 мг/кг, s.c.) в отношении гиперлокомоции, индуцированной d-амфетамином, определенное через 1 ч после s.c. инъекции Β-1a.
На фигуре 7 показан дозозависимый эффект MP-10 (-1 ч, s.c.) в отношении гиперлокомоции, индуцированной d-амфетамином, в зависимости от дозы совместно вводимого PDE2-i Β-1a (0,63-10 мг/кг, s.c.; -1 ч) или растворителя (10 мл/кг, s.c.; -1 ч). Пунктирные горизонтальные линии отражают критические уровни для эффектов, индуцированных лекарственными средствами (<5500 см, <2500 см и <1000 см). На фигуре 7 следующие символы соответствуют указанным ниже дозам:
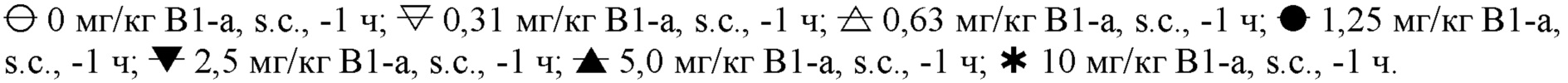
На фигурах 8a-c показана ED50 (при доверительных интервалах с надежностью 95%) MP-10 (-1 ч, s.c.) для снижения гиперлокомоции, индуцированной d-амфетамином, до расстояния <5500 см (фиг.8a), <2500 см (фиг.8b) и <1000 см (фиг.8c) в зависимости от дозы совместно вводимого PDE2-i Β-1a (0,63-10 мг/кг, s.c.; -1 ч; закрашенные символы) или растворителя (10 мл/кг, s.c.; -1 ч; незакрашенные символы). Серая горизонтальная полоса представляет ED50 (при доверительных интервалах с надежностью 95%) MP-10 (-1 ч, s.c.) в комбинации с растворителем для Β-1a (фиг.8a и 8b) или MP-10 (-1 ч, s.c.) в отдельности (фиг.8c; >40 мг/кг, ретроспективные данные).
На фигуре 9 показан эффект стандартной дозы Β-1a (0 в сравнении с 10 мг/кг, s.c.; -1 ч) в отношении дозозависимого эффекта соединения A (-1 ч, s.c.; фиг. 9a) и соединения B (-1 ч, s.c.; фиг. 9b) для подавления гиперлокомоции, индуцированной d-амфетамином. Показаны отдельные значения (незакрашенные и закрашенные круги для PDE2-i при 0 и 10 мг/кг, соответственно) и медианные значения (горизонтальные линии) пройденного расстояния в каждой дозовой группе. Пунктирные горизонтальные линии представляют критерии, принятые для эффектов, индуцированных лекарственными средствами (<5500 и <1100 см). На фигуре 9: *p<0,05, **p<0,01, ***p<0,001 (апостериорный критерий Бонферрони, 0 в сравнении с 10 мг/кг). ED50 (при доверительных интервалах с надежностью 95%) PDE10-i для уменьшения пройденного расстояния до <5500 см и до <1100 см были приведены для совместной обработки с помощью Β-1a при 0 и 10 мг/кг.
|
|
На фигуре 10 показан эффект Β-1a (0, 0,63, 1,25, 2,5 и 5,0 мг/кг, s.c.; -1 ч; фиг. 10a) в отношении гиперлокомоции, индуцированной d-амфетамином, в присутствии стандартных доз соединения A (0 или 2,5 мг/кг, s.c., -1 ч; фиг. 10b) или соединения B (0 или 2,5 мг/кг, s.c., -1 ч; фиг. 10c) (незакрашенные и закрашенные круги для PDE10-i при 0 и 10 мг/кг, соответственно). Пунктирные горизонтальные линии представляют критические уровни для эффектов, индуцированных лекарственными средствами (<5500 см и <1100 см). Β-1a был неэффективным против гиперлокомоции, индуцированной d-амфетамином, в комбинации с растворителем для PDE10-i, но усиливал эффекты обоих PDE10-i (2,5 мг/кг в сравнении с 0 мг/кг).
На фигуре 11 показано усиление связывания [3H]B1-a (вводимого внутривенно, i.v.) с PDE2 с помощью MP-10, выявляемое посредством ex vivo радиоавтографии. Контр. означает контроль.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте, как уже отмечалось, настоящее изобретение направлено на комбинации, включающие:
a) ингибитор PDE2 или его фармацевтически приемлемые соль или сольват, и
b) один или несколько ингибиторов PDE10 или их фармацевтически приемлемых солей или сольватов.
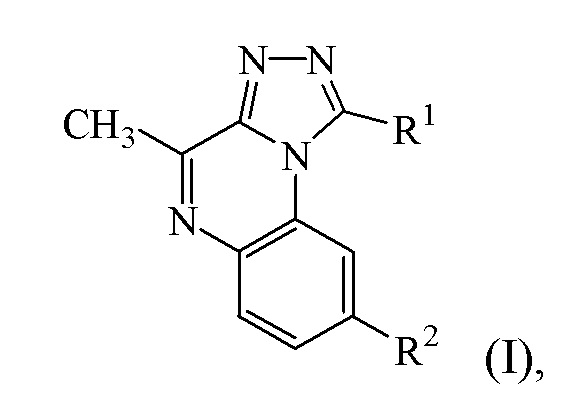
В конкретном варианте осуществления a) представляет собой соединение формулы (I):
или его стереохимически изомерную форму,
где
R1 представляет собой фенил или пиридинил, каждый из которых необязательно замещен 1 или 2 заместителями, независимо выбранными из группы, состоящей из галогена, (C3-6циклоалкил)C1-3алкилокси и C1-6алкилокси; и
R2 представляет собой -CH2-NR3R4;
где
R3 представляет собой водород или метил;
R4 представляет собой C1-3алкил; или
NR3R4 представляет собой морфолинил;
или его фармацевтически приемлемые соль или сольват.
В дополнительном варианте осуществления a) представляет собой соединение формулы (I), описанное в данном документе, где
R1 представляет собой фенил, замещенный галогеном и C1-6алкилокси, или пиридинил, замещенный C1-6алкилокси или (C3-6циклоалкил)C1-3алкилокси; и R2 является таким, как определено ранее;
или его фармацевтически приемлемые соль или сольват.
В дополнительном варианте осуществления a) представляет собой соединение формулы (I), описанное в данном документе, где
R1 представляет собой фенил, замещенный хлором и C1-6алкилокси, в частности, этокси, изопропокси или бутокси; или пиридинил, замещенный C1-6алкилокси или (C3-6циклоалкил)C1-3алкилокси, в частности, бутокси или циклопропилметокси; и
R2 представляет собой -CH2-NHCH3, -CH2-N(CH3)2 или -CH2-(4-морфолинил);
или его фармацевтически приемлемые соль или сольват.
В дополнительном варианте осуществления настоящего изобретения соединение формулы (I) выбрано из:
1-[1-(2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]-N,N-диметилметанамина;
1-(2-хлорфенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина;
N-{[1-(2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]метил}этанамина;
1-(2-хлор-4-фторфенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина;
1-(2-хлор-6-фторфенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина;
1-(5-метоксипиридин-3-ил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина;
1-(2-хлор-5-метоксифенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина;
1-(5-бутоксипиридин-3-ил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина, или его соли-гидрохлорида, или его соли-оксалата;
1-(5-бутокси-2-хлорфенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина или его соли-гидрохлорида;
1-[2-хлор-5-(1-метилэтокси)фенил]-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина или его соли-гидрохлорида;
N-({1-[2-хлор-5-(1-метилэтокси)фенил]-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил}метил)этанамина или его соли-гидрохлорида;
1-[1-(2-хлор-5-пропоксифенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]-N,N-диметилметанамина или его соли-гидрохлорида;
1-{1-[2-хлор-5-(1-метилэтокси)фенил]-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил}-N,N-диметилметанамина;
1-[1-(5-бутокси-2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]-N,N-диметилметанамина;
1-[1-(2-хлор-5-этоксифенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]-N,N-диметилметанамина;
N-{[1-(2-хлор-5-этоксифенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]метил}этанамина;
N-{[1-(2-хлор-5-этоксифенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]метил}этанамина или его соли-гидрохлорида;
1-(2-хлор-5-этоксифенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина или его соли-гидрохлорида;
N-{[1-(2-хлор-5-пропоксифенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]метил}этанамина;
1-(2-хлор-5-пропоксифенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина или его соли-гидрохлорида;
N-{[1-(5-бутокси-2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]метил}пропан-2-амина;
N-{[1-(5-бутоксипиридин-3-ил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]метил}этанамина или его соли-гидрохлорида;
N-{[1-(5-бутоксипиридин-3-ил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]метил}пропан-2-амина или его соли-гидрохлорида;
4-метил-8-(морфолин-4-илметил)-1-(5-пропоксипиридин-3-ил)[1,2,4]триазоло[4,3-a]хиноксалина или его соли-гидрохлорида;
N-{[4-метил-1-(5-пропоксипиридин-3-ил)[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]метил}этанамина или его соли-гидрохлорида;
1-[5-(циклопропилметокси)пиридин-3-ил]-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина или его соли-гидрохлорида;
N-({1-[5-(циклопропилметокси)пиридин-3-ил]-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил}метил)этанамина или его соли-гидрохлорида;
1-[1-(5-бутоксипиридин-3-ил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]-N,N-диметилметанамина или его соли-гидрохлорида;
1-[1-(5-бутокси-2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]-N-метилметанамина или его соли-гидрохлорида;
1-{1-[5-(циклопропилметокси)пиридин-3-ил]-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил}-N,N-диметилметанамина или его соли-гидрохлорида;
N-({1-[5-(циклопропилметокси)пиридин-3-ил]-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил}метил)пропан-2-амина или его соли-гидрохлорида;
1-[1-(5-бутоксипиридин-3-ил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]-N-метилметанамина или его соли-гидрохлорида и
1-(5-бутокси-6-хлорпиридин-3-ил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина
или их фармацевтически приемлемых солей или сольватов.
В дополнительном варианте осуществления соединение формулы (I) выбрано из группы:
1-(5-бутоксипиридин-3-ил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина, или его соли-гидрохлорида, или его соли-оксалата;
1-[2-хлор-5-(1-метилэтокси)фенил]-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина или его соли-гидрохлорида;
1-{1-[2-хлор-5-(1-метилэтокси)фенил]-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил}-N,N-диметилметанамина;
1-[1-(5-бутокси-2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]-N,N-диметилметанамина;
1-[5-(циклопропилметокси)пиридин-3-ил]-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина или его соли-гидрохлорида;
1-[1-(5-бутоксипиридин-3-ил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]-N,N-диметилметанамина или его соли-гидрохлорида и
1-[1-(5-бутокси-2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]-N-метилметанамина или его соли-гидрохлорида.
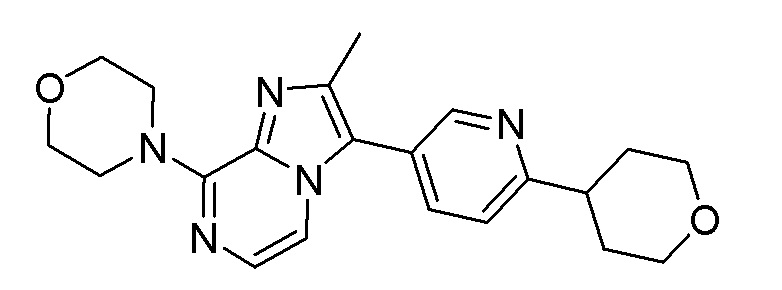
В дополнительном варианте осуществления соединение формулы (I) представляет собой
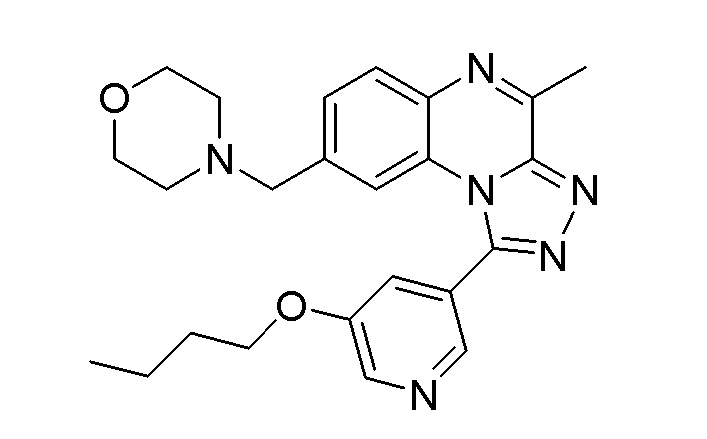
или его фармацевтически приемлемые соль или сольват, определенные в данном документе, в частности, его соль-гидрохлорид (соединение B-1a).
Меченные радиоактивными изотопами соединения формулы (I), например,
1-(5-бутоксипиридин-3-ил)-4-метил-8-[морфолин-4-ил(3H1)метил][1,2,4]триазоло[4,3-a]хиноксалин и
1-[2-хлор-6-(18F)фторфенил]-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалин
и их фармацевтически приемлемые соли и сольваты,
можно применять сами по себе или в композициях, содержащих указанные конкретные соединения, для визуализации ткани, клеток или хозяина in vitro или in vivo.
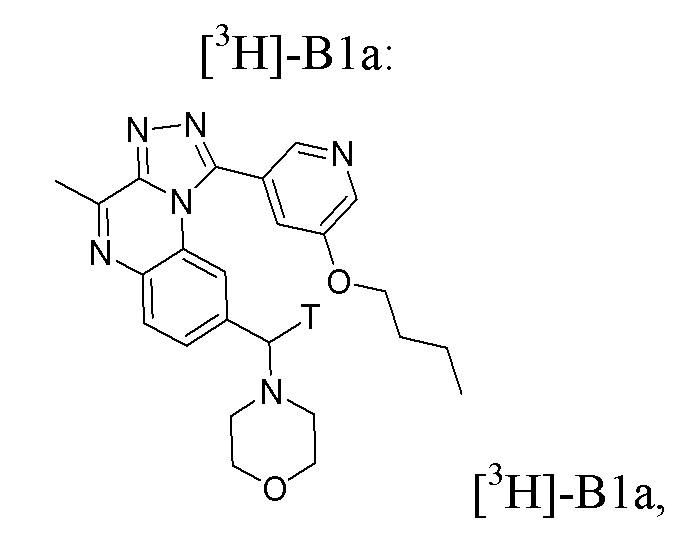
Таким образом, настоящее изобретение также относится, в частности, к соединению формулы [3H]-B1a:
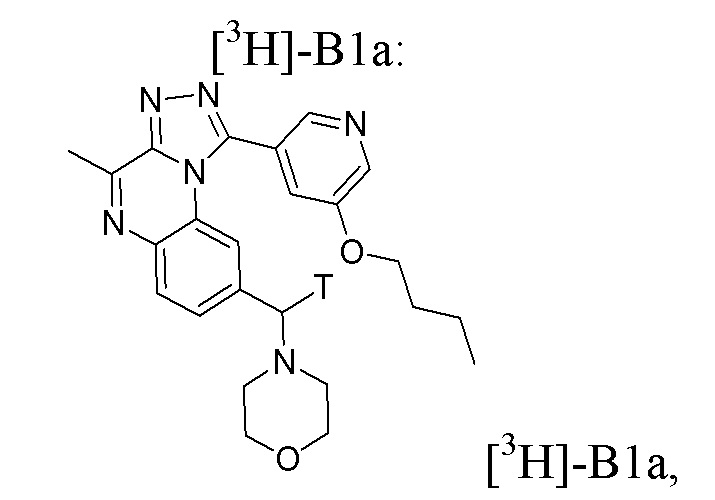
или его фармацевтически приемлемым соли или сольвату,
или стерильному раствору, содержащему указанное соединение формулы [3H]-B1a для применения в визуализации ткани, клеток или хозяина in vitro или in vivo, в частности, in vivo.
Таким образом, настоящее изобретение также относится, в частности, к соединению формулы [3H]-B1a:
или его фармацевтически приемлемым соли или сольвату,
или стерильному раствору, содержащему указанное соединение формулы [3H]-B1a, предназначенное для применения в визуализации ткани или клеток in vitro.
Настоящее изобретение также относится к применению соединения формулы [3H]-B1a:
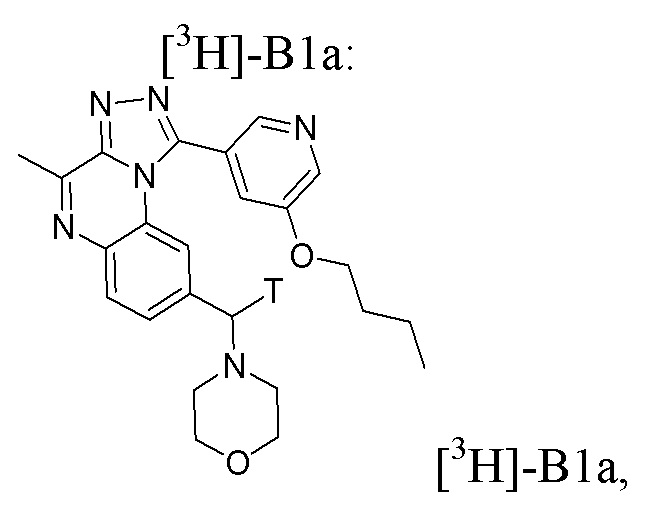
или его фармацевтически приемлемым соли или сольвату,
или стерильному раствору, содержащему указанное соединение формулы [3H]-B1a, для визуализации ткани или клеток in vitro.
В конкретном варианте осуществления компонент b) комбинации представляет собой ингибитор PDE10, выбранный из MP-10, PQ-10, TP-10, папаверина и соединений, раскрытых в WO 2011/051324 и в WO 2011/110545. Указанные соединения, раскрытые в WO 2011/051324 и в WO 2011/110545, называются в данном документе соединениями формулы (II) и соединениями формулы (III).
В другом варианте осуществления компонент b) комбинации представляет собой ингибитор PDE10, выбранный из группы MP-10, PQ-10, TP-10 и папаверина.
Такие компоненты b) соответствуют соединениям, известным в данной области техники, так что MP-10 представляет собой 2-{[4-(1-метил-4-пиридин-4-ил-1H-пиразол-3-ил)фенокси]метил}хинолин [CAS 898562-94-2]; PQ-10 представляет собой 6,7-диметокси-4-[(3R)-3-(хиноксалин-2-илокси)пирролидин-1-ил]хиназолин [CAS 927691-21-2]; TP-10 представляет собой 2-({4-[4-пиридин-4-ил-1-(2,2,2-трифторэтил)-1H-пиразол-3-ил]фенокси}метил)хинолин [CAS 898563-00-3]; а папаверин или гидрохлорид папаверина представляет собой 1-[(3,4-диметоксифенил)метил]-6,7-диметоксиизохинолин или его гидрохлорид (1:1) [CAS 61-25-6]. В конкретном варианте осуществления ингибитор PDE10 выбран из MP-10 и TP-10. В дополнительном варианте осуществления ингибитор PDE10 представляет собой MP-10.
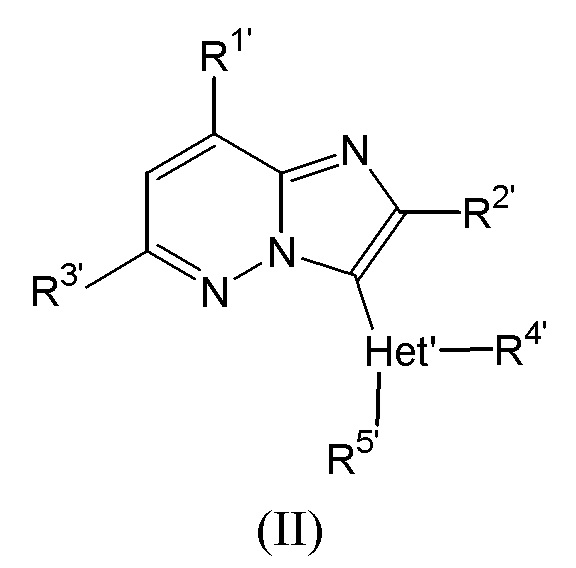
Соединения, раскрытые в WO 2011/051324, называются в данном документе соединениями формулы (II):
и их стереоизомерными формами,
где
R1’ представляет собой пиридинил; пиридинил, необязательно замещенный галогеном, C1-4алкилом, трифторметилом или C1-4алкилокси; тетрагидропиранил или NR6’R7’;
R2’ представляет собой водород, C1-4алкил, трифторметил, C3-8циклоалкил или C1-4алкилокси;
R3’ представляет собой водород, хлор, C1-4алкил, трифторметил или C3-8циклоалкил;
Het’ представляет собой 5- или 6-членное гетероциклическое кольцо, выбранное из группы, состоящей из пиридинила, пиримидинила, пиридазинила, пирролила, оксазолила, тиазолила, имидазолила, пиразолила, изотиазолила, изоксазолила, оксадиазолила и триазолила;
R4’ представляет собой водород, C1-4алкил, трифторметил-C0-4алкил, гидрокси-C1-4алкил, дифторциклопропилметил, циклопропилдифторэтил, C3-8циклоалкил, C1-4алкилоксиC1-5алкил, C1-4алкилокси, трифторметил-C0-4алкилокси, C3-8циклоалкилC1-4алкилокси, C3-8циклоалкилC1-4алкил, C1-6алкилоксиC1-4алкилокси, тетрагидропиранил, пиридинилметил, NR6aR7a-C1-4алкил или NR6aR7a;
R5’ представляет собой водород или C1-4алкил;
каждый из R6’, R6a’, R7’ и R7a’ независимо представляет собой водород или C1-4алкил или, взятый вместе с N, может представлять собой радикал формулы (a’), (b’) или (c’):
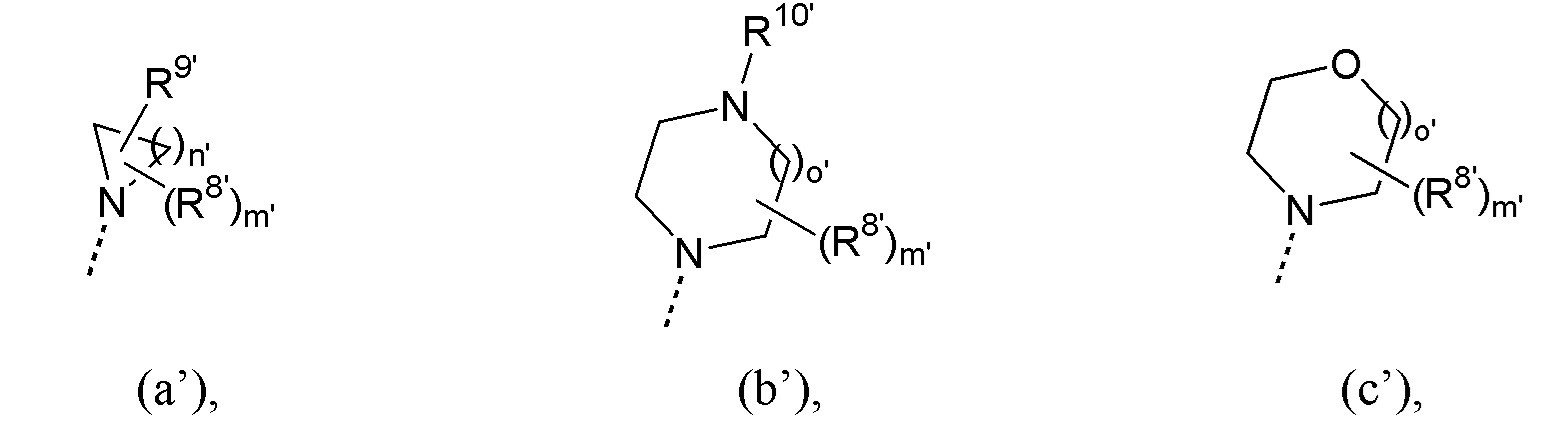
где
каждый R8’, если имеется, независимо от других таковых представляет собой C1-4алкил;
R9’ представляет собой водород или C1-4алкилокси;
R10’ представляет собой водород или C1-4алкил;
m’ равно 0, 1, 2, 3, 4 или 5;
n’ равно 2, 3, 4, 5 или 6;
o’ равно 1 или 2;
а также их фармацевтически приемлемыми солями и сольватами.
Конкретные соединения формулы (II) выбраны из:
гидрохлорида 3-[6-(2-метоксиэтил)-3-пиридинил]-2-метил-8-(4-морфолинил)имидазо[1,2-b]пиридазина,
малеата 3-[6-(2-метоксиэтил)-3-пиридинил]-2-метил-8-(4-морфолинил)имидазо[1,2-b]пиридазина,
моногидрата 3-[6-(2-метоксиэтил)-3-пиридинил]-2-метил-8-(4-морфолинил)имидазо[1,2-b]пиридазина,
гидрохлорида 3-[6-(2-метоксиэтокси)-3-пиридинил]-2-метил-8-(4-пиридинил)имидазо[1,2-b]пиридазина,
гидрохлорида 2-циклопропил-3-[6-(2-метокси-2-метилпропил)-3-пиридинил]-8-(4-морфолинил)имидазо[1,2-b]пиридазина,
3-[6-(2-метокси-2-метилпропил)-3-пиридинил]-2,6-диметил-8-(4-пиридинил)имидазо[1,2-b]пиридазина,
2-циклопропил-3-[6-(2-метокси-2-метилпропил)-3-пиридинил]-8-(4-пиридинил)имидазо[1,2-b]пиридазина,
5-[2-циклопропил-8-(4-морфолинил)имидазо[1,2-b]пиридазин-3-ил]-α,α-диметил-2-пиридинэтанола,
3-[6-(4-морфолинил)-3-пиридинил]-8-(4-пиридинил)имидазо[1,2-b]пиридазина,
2,6-диметил-8-(4-морфолинил)-3-[6-(4-морфолинил)-3-пиридинил]имидазо[1,2-b]пиридазина,
2-циклопропил-6-метил-3-[6-(4-морфолинил)-3-пиридинил]-8-(4-пиридинил)имидазо[1,2-b]пиридазина,
2-циклопропил-3-[6-(2-метоксиэтокси)-3-пиридинил]-6-метил-8-(4-пиридинил)имидазо[1,2-b]пиридазина,
2-метил-8-(4-морфолинил)-3-[2-(4-морфолинил)-4-пиридинил]имидазо[1,2-b]пиридазина,
3-{1-[(2,2-дифторциклопропил)метил]-1H-пиразол-4-ил}-2-метил-8-морфолин-4-илимидазо[1,2-b]пиридазина,
2-метил-3-[1-(2-метилпропил)-1H-пиразол-4-ил]-8-(4-пиридинил)имидазо[1,2-b]пиридазина,
6-циклопропил-3-[1-(2-метоксиэтил)-1H-пиразол-4-ил]-2-метил-8-(4-морфолинил)имидазо[1,2-b]пиридазина,
2-этил-3-[1-(2-метоксиэтил)-1H-пиразол-4-ил]-8-(4-морфолинил)имидазо[1,2-b]пиридазина,
3-[1-[(2S)-2-метоксипропил]-1H-пиразол-4-ил]-2-метил-8-(4-морфолинил)имидазо[1,2-b]пиридазина,
3-[1-(2-метоксиэтил)-1H-пиразол-4-ил]-2,6-диметил-8-(4-морфолинил)имидазо[1,2-b]пиридазина,
3-[1-(2-метоксиэтил)-1H-пиразол-4-ил]-2,6-диметил-8-(4-пиридинил)имидазо[1,2-b]пиридазина,
2-метил-8-(4-пиридинил)-3-[1-(2,2,2-трифторэтил)-1H-пиразол-4-ил]имидазо[1,2-b]пиридазина,
2-циклопропил-3-[1-(2-метоксиэтил)-1H-пиразол-4-ил]-8-(4-пиридинил)имидазо[1,2-b]пиридазина,
2-циклопропил-3-[1-(2-метокси-2-метилпропил)-1H-пиразол-4-ил]-8-(4-пиридинил)имидазо[1,2-b]пиридазина,
6-хлор-3-[1-(2-метоксиэтил)-1H-пиразол-4-ил]-2-метил-8-(4-пиридинил)имидазо[1,2-b]пиридазина и
2-циклопропил-3-[1-(2-метилпропил)-1H-пиразол-4-ил]-8-(4-пиридинил)имидазо[1,2-b]пиридазина.
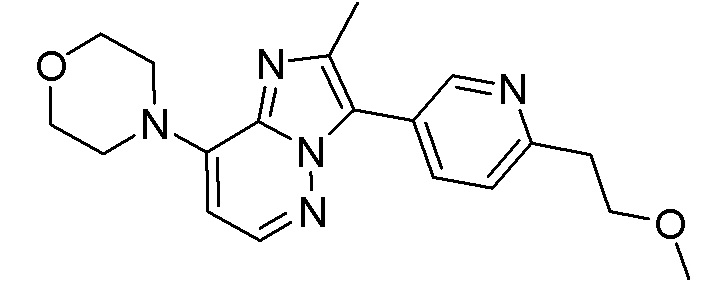
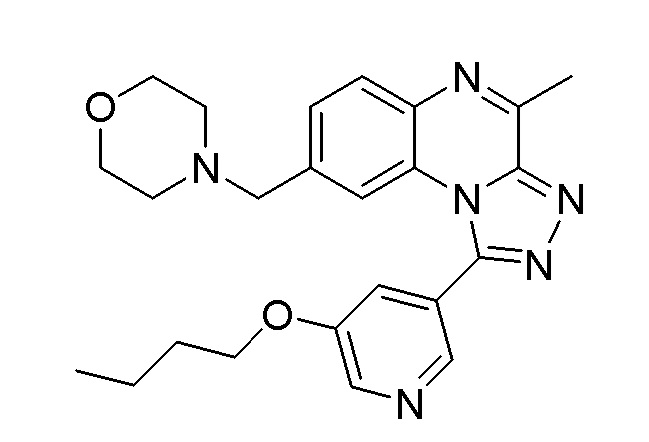
Конкретным примером соединения формулы (II) является соединение A:
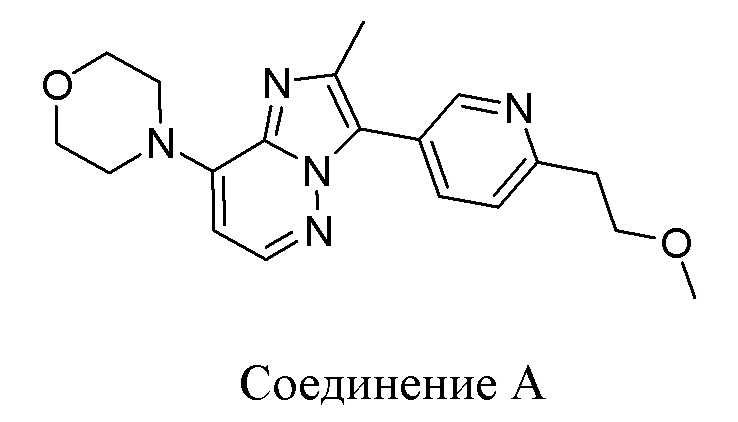
или его фармацевтически приемлемые соль или сольват.
Соединения, раскрытые в WO 2011/110545, называются в данном документе соединениями формулы (III):
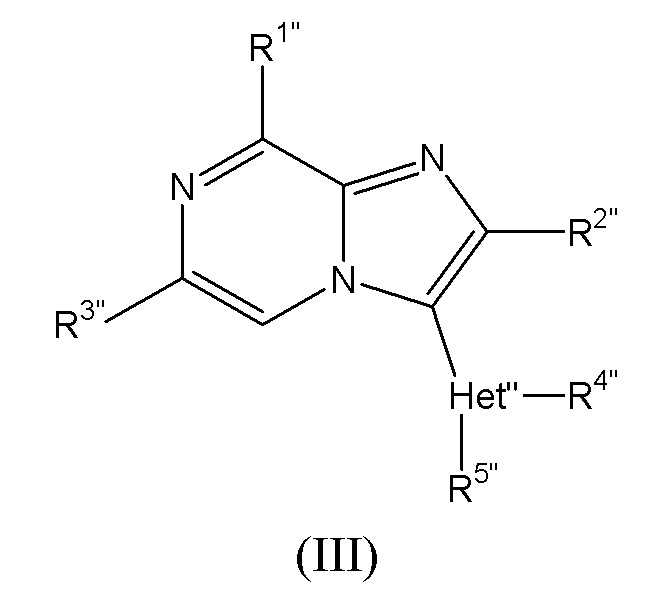
и их стереоизомерными формами, где
R1’’ выбран из группы, состоящей из радикалов формул (a-1’’), (a-2’’) и (a-3’’):
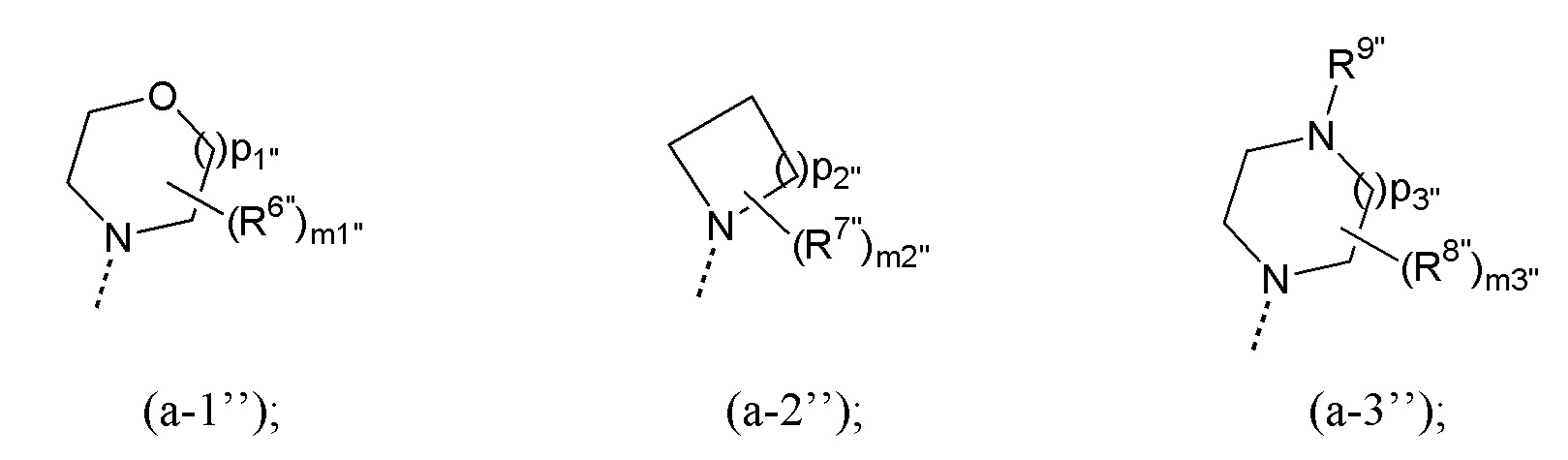
где
каждый из R6”, R7” и R8” независимо выбран из группы, состоящей из фтора; C1-4алкила; C1-4алкилокси и C1-4алкила, замещенного 1, 2 или 3 атомами фтора;
R9” представляет собой водород или C1-4алкил;
каждое из m1”, m2” и m3” независимо выбрано из 0, 1, 2, 3 и 4;
p2” выбрано из 1, 2, 3 и 4;
каждое из p1” и p3” независимо выбрано из 1 и 2;
или R1” выбран из группы, состоящей из незамещенного пиридинила; пиридинила, замещенного 1 или 2 заместителями, выбранными из группы, состоящей из галогена, C1-4алкила, трифторметила и C1-4алкилокси; и незамещенного тетрагидропиранила;
R2” выбран из группы, состоящей из водорода; C1-4алкила; трифторметила; C3-8циклоалкила; C1-4алкилокси и циано;
R3” выбран из группы, состоящей из водорода; C1-4алкила; C3-8циклоалкила и C1-4алкила, замещенного 1, 2 или 3 атомами фтора;
Het” представляет собой 5- или 6-членное гетероцикличное кольцо, выбранное из группы, состоящей из пиридинила; пиримидинила; пиридазинила; пиразинила; пирролила; оксазолила; тиазолила; имидазолила; пиразолила; изотиазолила; изоксазолила; оксадиазолила и триазолила;
R4” выбран из группы, состоящей из водорода; C1-4алкила; C1-4алкила, замещенного 1, 2 или 3 атомами фтора; (дифторциклопропил)метила; (циклопропил)дифторметила; гидроксиC1-4алкила; C3-8циклоалкила; (C3-8циклоалкил)-C1-4алкила; C1-4алкилокси-C1-6алкила; C1-4алкилокси; C1-4алкилокси, замещенного 1, 2 или 3 атомами фтора; (C3-8циклоалкил)-C1-4алкилокси; (C1-4алкилокси-C1-4алкил)окси; (C1-4алкил)карбонила; (C1-4алкил)карбонилC1-4алкила; (C3-8циклоалкил)карбонила; (C3-8циклоалкил)карбонилC1-4алкила; незамещенного фенила; фенила, замещенного 1 или 2 заместителями, выбранными из группы, состоящей из галогена, C1-4алкила, трифторметила, трифторметокси, циано и C1-4алкилокси; незамещенного бензила; бензила, замещенного 1 или 2 заместителями, выбранными из группы, состоящей из галогена, C1-4алкила, трифторметила, трифторметокси, циано и C1-4алкилокси; незамещенного тетрагидрофуранила; тетрагидрофуранилметила; незамещенного тетрагидропиранила; тетрагидропиранилметила; пиридинилметила; хинолинилметила; (NR10”R11”)-C1-4алкила и NR10”R11”;
R5” представляет собой водород или фтор;
R10” и R11” независимо выбраны из водорода и C1-4алкила или, взятые вместе с атомом азота кольца, могут образовывать радикал формулы (b-1”), (b-2”) или (b-3”):
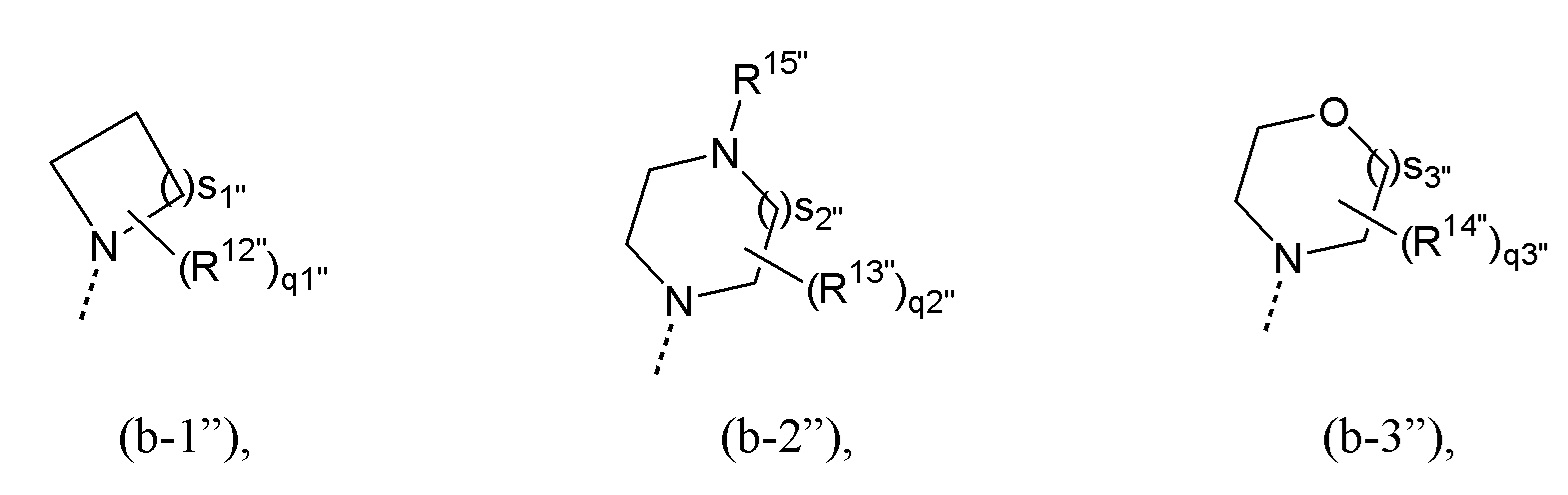
где
каждый из R12”, R13” и R14” независимо представляет собой C1-4алкил или C1-4алкилокси;
R15” представляет собой водород или C1-4алкил;
каждое из q1”, q2” и q3” независимо выбрано из 0, 1, 2, 3 и 4;
s1” выбрано из 1, 2, 3 и 4;
каждое из s2” и s3” независимо выбрано из 1 и 2;
а также их фармацевтически приемлемыми солями и сольватами.
Конкретные соединения формулы (III) выбраны из:
3-[1-(2-метоксиэтил)-1H-пиразол-4-ил]-2-метил-8-(4-морфолинил)имидазо[1,2-a]пиразина;
3-[1-(2-метоксиэтил)-1H-пиррол-3-ил]-2-метил-8-(4-морфолинил)имидазо[1,2-a]пиразина;
3-[6-(2-метоксиэтил)-3-пиридинил]-2-метил-8-(4-морфолинил)имидазо[1,2-a]пиразина;
2-метил-3-[2-(2-метилпропил)-5-тиазолил]-8-(4-морфолинил)имидазо[1,2-a]пиразина;
3-[6-(2-метоксиэтил)-3-пиридинил]-2-метил-8-(4-пиридинил)имидазо[1,2-a]пиразина;
3-[6-(2-метоксиэтокси)-3-пиридинил]-2-метил-8-(4-морфолинил)имидазо[1,2-a]пиразина;
3-(6циклопропил-3-пиридинил)-2-метил-8-(4-морфолинил)имидазо[1,2-a]пиразина;
2-метил-8-(4-морфолинил)-3-[6-(1-пиперазинил)-3-пиридинил]имидазо[1,2-a]пиразина;
2-метил-8-(4-морфолинил)-3-[6-(тетрагидро-2H-пиран-4-ил)-3-пиридинил]имидазо[1,2-a]пиразина;
3-[6-(1-метокси-1-метилэтил)-3-пиридинил]-2-метил-8-(4-морфолинил)имидазо[1,2-a]пиразина;
3-[6-(этоксиметил)-3-пиридинил]-2-метил-8-(4-морфолинил)имидазо[1,2-a]пиразина и
3-[2-(2-метоксиэтил)-5-пиримидинил]-2-метил-8-(4-морфолинил)имидазо[1,2-a]пиразина.
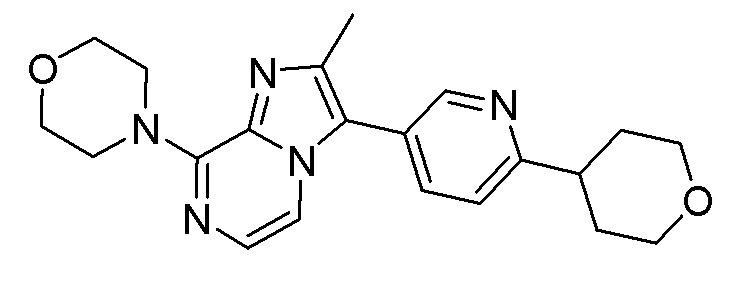
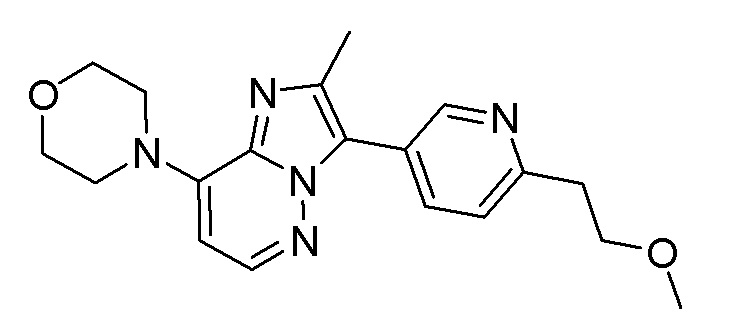
Конкретным примером соединения формулы (III) является соединение B:
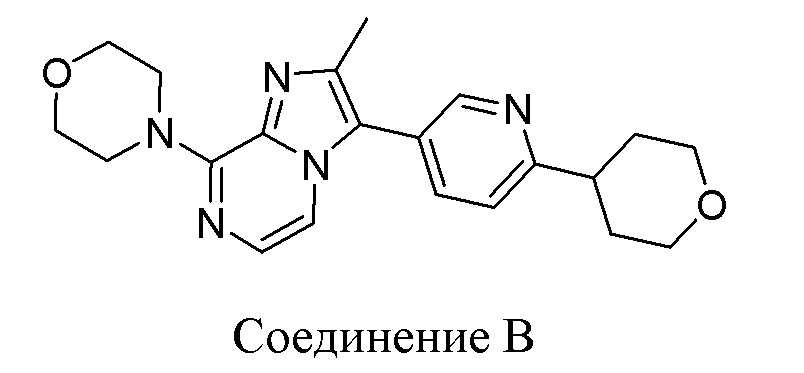
или его фармацевтически приемлемые соль или сольват.
В дополнительном варианте осуществления настоящее изобретение относится к комбинации, включающей:
a) соединение формулы
или его фармацевтически приемлемые соль или сольват, определенные в данном документе, в частности, его соль-гидрохлорид (соединение B-1a); и
b) один или несколько ингибиторов PDE10, выбранных из группы MP-10, соединения формулы
(соединения A), определенного выше, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы
(соединения B), определенного выше, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе.
В дополнительном варианте осуществления настоящее изобретение относится к комбинациям, включающим:
a) соединение формулы (I):
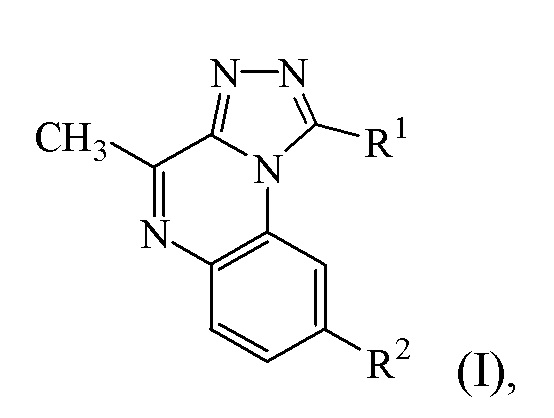
или его стереохимически изомерную форму,
где
R1 представляет собой фенил или пиридинил, каждый из которых необязательно замещен 1 или 2 заместителями, независимо выбранными из группы, состоящей из галогена, (C3-6циклоалкил)C1-3алкилокси и C1-6алкилокси; и
R2 представляет собой -CH2-NR3R4;
где
R3 представляет собой водород или метил;
R4 представляет собой C1-3алкил; или
NR3R4 представляет собой морфолинил;
или его фармацевтически приемлемые соль или сольват и
b) один или несколько ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина.
Настоящее изобретение также относится к продуктам, содержащим в качестве первого активного ингредиента a) соединение формулы (I) или его фармацевтически приемлемые соль или сольват, определенные в данном документе, и в качестве второго активного ингредиента b) один или несколько ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина, в виде комбинированных препаратов для одновременного, раздельного или последовательного применения в лечении пациентов, страдающих неврологическими или психическими расстройствами или эндокринными или метаболическими заболеваниями.
Иллюстрацией настоящего изобретения является фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и любую из комбинаций, описанных выше. Иллюстрацией настоящего изобретения является фармацевтическая композиция, полученная путем смешивания любой из комбинаций, описанных выше, и фармацевтически приемлемого носителя. Иллюстрацией настоящего изобретения является способ получения фармацевтической композиции, включающий смешивание любой из комбинаций, описанных выше, и фармацевтически приемлемого носителя.
В дополнительном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемых соли или сольвата для усиления эффекта одного или нескольких ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемым соли или сольвату, определенным в данном документе, для применения в усилении терапевтического эффекта одного или нескольких ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина, у пациентов, страдающих неврологическими или психическими расстройствами или эндокринными или метаболическими заболеваниями.
Дополнительно, настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, для получения лекарственного препарата для усиления терапевтического эффекта одного или нескольких ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина, у пациентов, страдающих неврологическими или психическими расстройствами или эндокринными или метаболическими заболеваниями.
Настоящее изобретение дополнительно относится к применению одного или нескольких ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина, для усиления эффекта соединения формулы (I) или его фармацевтически приемлемых соли или сольвата. Настоящее изобретение также относится к одному или нескольким ингибиторам PDE10, выбранным из группы MP-10, PQ-10, TP-10 и папаверина, для применения в усилении терапевтического эффекта соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, определенных в данном документе. В дополнительном аспекте настоящее изобретение также относится к применению одного или нескольких ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина, для получения лекарственного препарата для усиления терапевтического эффекта соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, у пациентов, страдающих неврологическими или психическими расстройствами или эндокринными или метаболическими заболеваниями.
Настоящее изобретение дополнительно относится к способу лечения неврологического или психического расстройства или эндокринного или метаболического заболевания, включающему введение субъекту, нуждающемуся в этом, терапевтически эффективного количества комбинации, включающей: a) соединение формулы (I) или его фармацевтически приемлемые соль или сольват, определенные в данном документе, и b) один или несколько ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина, или терапевтически эффективного количества фармацевтической композиции, описанной выше.
Настоящее изобретение дополнительно относится к способу усиления терапевтического эффекта соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, включающему введение субъекту, нуждающемуся в этом, терапевтически эффективного количества комбинации, включающей a) соединение формулы (I) или его фармацевтически приемлемые соль или сольват, определенные в данном документе, и b) один или несколько ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина, или терапевтически эффективного количества фармацевтической композиции, описанной выше.
Настоящее изобретение дополнительно относится к способу усиления терапевтического эффекта одного или нескольких ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина, включающему введение субъекту, нуждающемуся в этом, терапевтически эффективного количества комбинации, включающей: a) соединение формулы (I), определенное в данном документе, и b) один или несколько ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10 и папаверина, или терапевтически эффективного количества фармацевтической композиции, описанной выше.
ОПРЕДЕЛЕНИЯ
"Галоген" будет означать фтор, хлор и бром; "C1-6алкил", "C1-4алкил" и "C1-3алкил", применяемые в данном документе в качестве группы или части группы, будут означать насыщенную алкильную группу с прямой или разветвленной цепью, имеющую 1, 2, 3, 4, 5 или 6 атомов углерода, или 1, 2, 3 или 4 атома углерода, или 1, 2 или 3 атома углерода, соответственно, например, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метилпропил, трет-бутил, 1-пентил, 2-метилбутил, пентан-2-ил, 2-метилбутан-2-ил или гексил и т.п.; "C0-4алкил", используемый в данном документе в отдельности или в качестве части другой группы, если не указано иное, относится к насыщенному углеводородному радикалу с прямой или разветвленной цепью, имеющему от 0 до 4 атомов углерода; "C1-6алкилокси", "C1-4алкилокси" и "C1-3алкилокси" будут означать эфирный радикал, где C1-6алкил, C1-4алкил и C1-3алкил определены выше; "C3-8циклоалкил" и "C3-6циклоалкил" будут означать циклопропил, циклобутил, циклопентил, циклогексил, и циклогептил, и циклооктил; "(C3-6циклоалкил)C1-3алкил" будет означать C3-6циклоалкил, определенный выше, связанный с остальной частью молекулы посредством C1-3алкильного радикала, определенного выше.
Термин "субъект", применяемый в данном документе, относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, которое является или являлось объектом лечения, наблюдения или эксперимента.
Применяемый в данном документе термин "терапевтически эффективное количество" означает такое количество активного соединения или фармацевтического средства, которое вызывает биологический или медицинский ответ в системе тканей животного или человека, желаемый исследователем, ветеринаром, врачом или другим клиницистом, включающий облегчение симптомов заболевания или расстройства, лечение которого осуществляют. Более конкретно, в настоящем изобретении, направленном на комбинированную терапию, включающую введение ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, "терапевтически эффективное количество" будет означать такое количество комбинации средств, принимаемых совместно, что комбинированный эффект вызывает желаемый биологический или медицинский ответ. Например, терапевтически эффективное количество ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и ингибитора(-ов) PDE10 должно быть таким количеством ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и количеством ингибитора(-ов) PDE10, которое при совместном или последовательном приеме будет оказывать комбинированный эффект, иными словами, являться терапевтически эффективным.
Дополнительно, специалист в данной области признает, что в случае комбинированной терапии терапевтически эффективным количеством, как в вышеописанном примере, количество ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и/или количество ингибитора(-ов) PDE10 в отдельности может быть или может не быть терапевтически эффективным.
Подразумевается, что применяемый в данном документе термин "композиция" охватывает продукт, содержащий определенные ингредиенты в определенных количествах, а также любой продукт, который получают прямо или непрямо в результате комбинаций определенных ингредиентов в определенных количествах.
В соответствии со способами по настоящему изобретению отдельные компоненты комбинации можно вводить посредством любых подходящих способов одновременно, последовательно, раздельно или в одном фармацевтическом составе. Если ингибитор PDE2, в частности, соединение формулы (I) или его фармацевтически приемлемые соль или сольват, и ингибитор(-ы) PDE10 вводят в отдельных лекарственных формах, количество доз каждого соединения, вводимых за день, может быть одинаковым или различным. Ингибитор PDE2, в частности, соединение формулы (I) или его фармацевтически приемлемые соль или сольват, и ингибитор(-ы) PDE10 можно вводить посредством одних и тех же или различных путей введения. Примеры подходящих способов введения включают, без ограничения, пероральный, внутривенный (iv), внутримышечный (im), подкожный (sc), трансдермальный, интраназальный и ректальный. Соединения можно также вводить непосредственно в нервную систему, в том числе, без ограничений, посредством интрацеребрального, интравентрикулярного, интрацеребровентрикулярного, интратекального, интрацистернального, интраспинального и/или периспинального путей введения путем доставки через иглы и/или катетеры для внутричерепного или внутрипозвоночного введения с помощью насосных устройств или без них.
Ингибитор PDE2, в частности, соединение формулы (I) или его фармацевтически приемлемые соль или сольват, и ингибитор(-ы) PDE10 можно вводить согласно режимам одновременного или чередующегося введения в одно и то же или в разное время в течение терапии одновременно в разделенных формах или в цельной форме. Настоящее изобретение, следовательно, следует понимать как охватывающее все такие режимы одновременной или чередующейся обработки, и термин "введение" следует толковать соответственно.
Оптимальные дозировки и режимы дозирования, которые следует применять, могут быть без труда определены специалистами в данной области и будут изменяться в зависимости от способа введения, активности препарата и прогрессирования болезненного состояния. В дополнение, факторы, связанные с конкретным пациентом, подвергаемым лечению, в том числе пол, возраст, вес, режим питания, физическая активность, время введения и сопутствующие заболевания пациента, будут обуславливать необходимость в корректировке дозировок и/или режимов.
Термин "один или несколько ингибиторов PDE10", применяемый в данном документе, относится к одному, двум или трем ингибиторам PDE10, в частности, к одному ингибитору PDE10, упоминаемому в данном документе.
Термин "хозяин" относится к млекопитающему, в частности, к людям, мышам, собакам и крысам.
Термин "клетка" относится к клетке, экспрессирующей или содержащей фермент PDE2.
Следует понимать, что некоторые из соединений формул (I)-(III), а также их фармацевтически приемлемых солей присоединения и их сольватов могут иметь один или несколько центров хиральности и существуют в виде стереоизомерных форм.
Подразумевается, что термин "соединения по настоящему изобретению", применяемый в данном документе, включает соединения формулы (I), а также их соли и сольваты. Как применяется в данном документе, любая химическая формула со связями, показанными только в виде сплошных линий, а не в виде сплошных клиновидных или пунктирных клиновидных связей или иным образом показанная как имеющая конкретную конфигурацию (например, R, S) вокруг одного или нескольких атомов, подразумевает каждый возможный стереоизомер или смесь двух или более стереоизомеров.
Выше и ниже в данном документе подразумевается, что термин "соединение формулы (I)" включает его стереоизомеры и его таутомерные формы.
Термины "стереоизомеры", "стереоизомерные формы" или "стереохимически изомерные формы" выше или ниже в данном документе применяются взаимозаменяемо.
Настоящее изобретение включает все стереоизомеры соединений по настоящему изобретению либо в виде чистых стереоизомеров, либо в виде смеси двух или более стереоизомеров.
Энантиомеры являются стереоизомерами, которые представляют собой несовместимые зеркальные изображения друг друга. Смесь 1:1 пары энантиомеров представляет собой рацемат или рацемическую смесь.
Диастереомеры (или диастереоизомеры) представляют собой стереоизомеры, которые не являются энантиомерами, т.е. они не соотносятся как зеркальные изображения. Если соединение содержит двойную связь, заместители могут находиться в E- или Z-конфигурации. Заместители в бивалентных циклических (частично) насыщенных радикалах могут находиться либо в цис-, либо в транс-конфигурации; например, если соединение содержит двузамещенную циклоалкильную группу, то заместители могут быть в цис- или транс-конфигурации.
Таким образом, настоящее изобретение включает энантиомеры, диастереомеры, рацематы, E-изомеры, Z-изомеры, цис-изомеры, транс-изомеры и их смеси во всех случаях, когда это возможно с химической точки зрения.
Значения всех этих терминов, т.е. энантиомеры, диастереомеры, рацематы, E-изомеры, Z-изомеры, цис-изомеры, транс-изомеры и их смеси, известны специалисту в данной области.
Абсолютная конфигурация определяется согласно системе Кана-Ингольда-Прелога. Конфигурация при асимметричном атоме определяется как R или как S. Выделенные стереоизомеры, абсолютная конфигурация которых неизвестна, могут быть обозначены как (+) или (-) в зависимости от направления, в котором они вращают плоскость поляризации света. Например, выделенные энантиомеры, абсолютная конфигурация которых неизвестна, могут обозначаться как (+) или (-) в зависимости от направления, в котором они вращают плоскость поляризации света. Если определяют конкретный стереоизомер, это означает, что указанный стереоизомер практически свободен от других стереоизомеров, т.е. связан с менее 50%, предпочтительно с менее 20%, более предпочтительно с менее 10%, еще более предпочтительно с менее 5%, в частности, с менее 2% и наиболее предпочтительно с менее 1% таковых. Таким образом, если соединение формул (I)-(III), например, указано как (R), то это означает, что соединение практически свободно от (S)-изомера; если соединение формул (I)-(III), например, указано как E, то это означает, что соединение практически свободно от Z-изомера; если соединение формул (I)-(III), например, указано как цис, это означает, что соединение практически свободно от транс-изомера.
Некоторые из соединений согласно формулам (I)-(III) могут также существовать в своей таутомерной форме. Предполагается, что такие формы, ввиду того, что они могут существовать, хотя явно и не показаны вышеприведенными формулами (I)-(III), включены в объем настоящего изобретения.
Из этого следует, что одно соединение может существовать как в стереоизомерной, так и в таутомерной форме.
В дополнение, некоторые из соединений по настоящему изобретению могут образовывать сольваты с водой (т.е. гидраты) или обычными органическими растворителями, при этом также предполагается, что такие сольваты охватываются объемом настоящего изобретения.
В рамках данной заявки элемент, в частности, при упоминании в отношении соединения согласно формуле (I), включает все изотопы и смеси изотопов этого элемента, либо встречающиеся в природе, либо полученные синтетическим путем, либо с природным изотопным составом, либо в изотопно-обогащенной форме. Меченные радиоактивным изотопом соединения формулы (I) могут содержать радиоактивный изотоп, выбранный из группы 3H, 11C, 18F, 122I, 123I, 125I, 131I, 75Br, 76Br, 77Br и 82Br. Предпочтительно, радиоактивный изотоп выбран из группы 3H, 11C и 18F.
Что касается применения в медицине, соли соединений формул (I)-(III) относятся к нетоксичным "фармацевтически приемлемым солям". Однако при получении соединений согласно настоящему изобретению или их фармацевтически приемлемых солей могут применяться другие соли. Подходящие фармацевтически приемлемые соли соединений включают соли присоединения кислоты, которые можно образовывать, например, путем смешивания раствора соединения с раствором фармацевтически приемлемой кислоты, такой как соляная кислота, серная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, бензойная кислота, лимонная кислота, винная кислота, угольная кислота или фосфорная кислота. Кроме того, если соединения по настоящему изобретению имеют кислотный фрагмент, их подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов, например, соли натрия или калия; соли щелочноземельных металлов, например, соли кальция или магния; и соли, образованные с подходящими органическими лигандами, например, соли четвертичного аммония.
Иллюстративные кислоты, которые можно применять в получении фармацевтически приемлемых солей, включают, без ограничения, следующие: уксусную кислоту, 2,2-дихлоруксусную кислоту, ацилированные аминокислоты, адипиновую кислоту, альгиновую кислоту, аскорбиновую кислоту, L-аспарагиновую кислоту, бензолсульфоновую кислоту, бензойную кислоту, 4-ацетамидобензойную кислоту, (+)-камфорную кислоту, камфорсульфоновую кислоту, каприновую кислоту, капроновую кислоту, каприловую кислоту, коричную кислоту, лимонную кислоту, цикламовую кислоту, этан-1,2-дисульфоновую кислоту, этансульфоновую кислоту, 2-гидроксиэтансульфоновую кислоту, муравьиную кислоту, фумаровую кислоту, галактаровую кислоту, гентизиновую кислоту, глюкогептоновую кислоту, D-глюконовую кислоту, D-глюкуроновую кислоту, L-глутаминовую кислоту, бета-оксоглутаровую кислоту, гликолевую кислоту, гиппуровую кислоту, бромистоводородную кислоту, соляную кислоту, (+)-L-молочную кислоту, (±)-DL-молочную кислоту, лактобионовую кислоту, малеиновую кислоту, (-)-L-яблочную кислоту, малоновую кислоту, (±)-DL-миндальную кислоту, метансульфоновую кислоту, нафталин-2-сульфоновую кислоту, нафталин-1,5-дисульфоновую кислоту, 1-гидрокси-2-нафтойную кислоту, никотиновую кислоту, азотную кислоту, олеиновую кислоту, оротовую кислоту, щавелевую кислоту, пальмитиновую кислоту, памовую кислоту, фосфорную кислоту, L-пироглутаминовую кислоту, салициловую кислоту, 4-аминосалициловую кислоту, себациновую кислоту, стеариновую кислоту, янтарную кислоту, серную кислоту, дубильную кислоту, (+)-L-винную кислоту, тиоциановую кислоту, п-толуолсульфоновую кислоту, трифторметилсульфоновую кислоту и ундециленовую кислоту. Иллюстративные основания, которые можно применять в получении фармацевтически приемлемых солей, включают, без ограничения, следующие: аммиак, L-аргинин, бенетамин, бензатин, гидроксид кальция, холин, диметилэтаноламин, диэтаноламин, диэтиламин, 2-(диэтиламино)этанол, этаноламин, этилендиамин, N-метилглюкамин, гидрабамин, 1H-имидазол, L-лизин, гидроксид магния, 4-(2-гидроксиэтил)морфолин, пиперазин, гидроксид калия, 1-(2-гидроксиэтил)пирролидин, вторичный амин, гидроксид натрия, триэтаноламин, трометамин и гидроксид цинка.
Названия соединений формулы (I)-(III) были составлены согласно правилам номенклатуры, принятым Международным союзом теоретической и прикладной химии (IUPAC), с применением программного обеспечения Advanced Chemical Development, Inc. (продукт ACD/Name версии 10.01.0.14105, октябрь 2006 г.)
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ
Соединения формулы (I) в большинстве случаев можно получать при помощи последовательности этапов, каждый из которых известен специалисту в данной области. Превращения различных функциональных групп, присутствующих в конечных соединениях, в другие функциональные группы согласно формуле (I) можно осуществлять также с помощью способов синтеза, хорошо известных специалисту в данной области. В частности, соединения можно получать согласно следующим способам синтеза.
Получение конечных соединений
Соединения формулы (I) можно получать с помощью способов синтеза, хорошо известных специалисту в данной области. Соединения по настоящему изобретению можно получать, например, с помощью двух различных общих схем.
Схема 1. Синтез соединений формулы (I)
Способ A
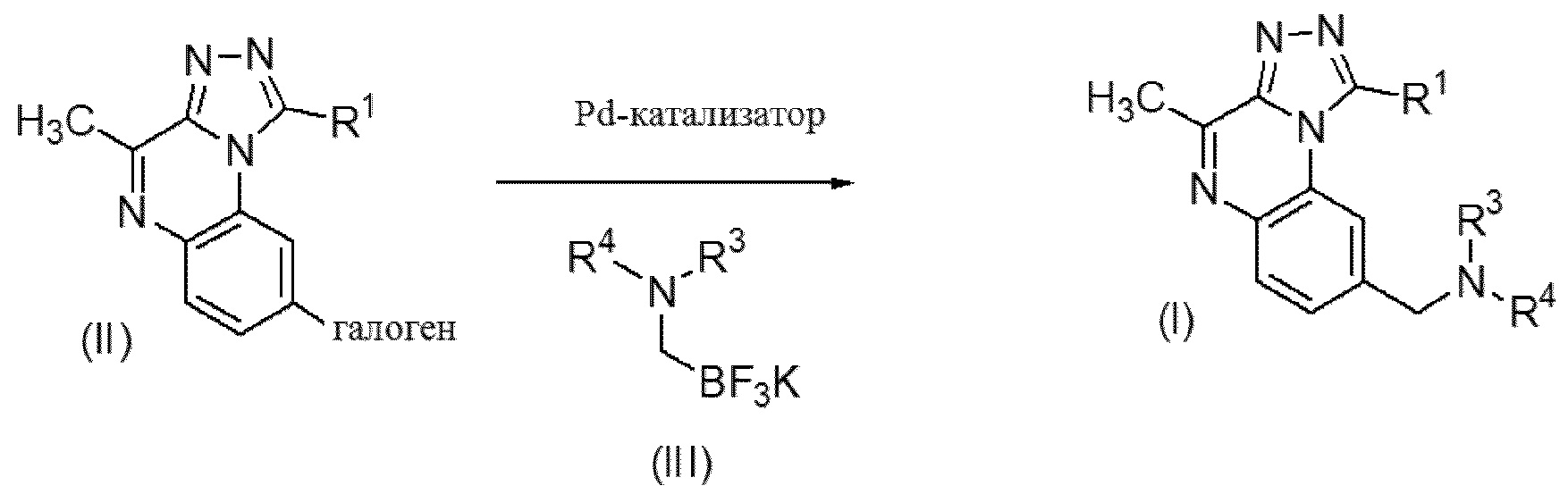
Соединение формулы (II) можно вводить в реакцию с соединением формулы (III) в инертном растворителе или смеси растворителей, такой как, например, смесь тетрагидрофурана и воды, в присутствии комплексообразующего средства, такого как 2-дихлоргексилфосфино-2',4',6'-триизопропилбифенил (XPhos), палладиевого катализатора, такого как ацетат палладия (II), и основания, такого как, например карбонат цезия, перемешивая реакционную смесь при подходящей температуре, такой как 110-120°C, с применением традиционного нагревания или под действием микроволнового излучения в течение времени, необходимого для достижения завершения реакции, как правило, в течение 45 минут в случае традиционного нагревания. Соединения формулы (III) могут быть коммерчески доступными либо могут быть получены посредством способов, описанных в химической литературе, хорошо известной специалисту в данной области.
Способ B
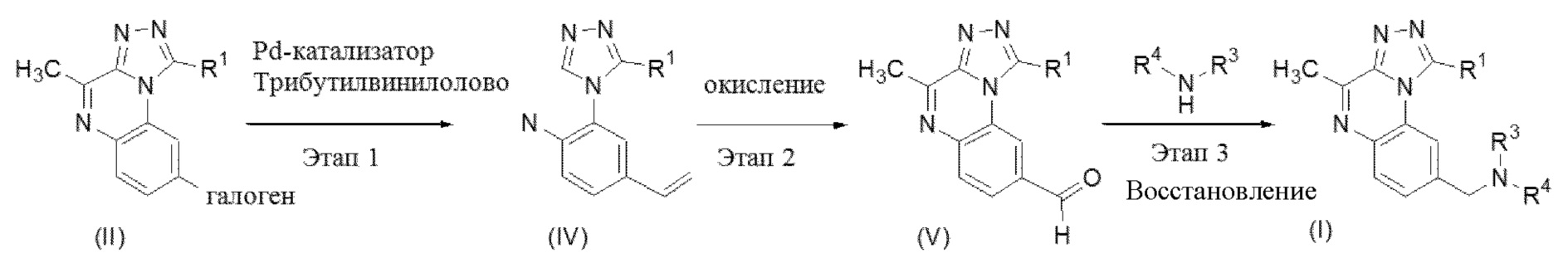
Этап 1. Соединение формулы (II) можно вводить в реакцию с трибутилвинилоловом в инертном растворителе, таком как, например, толуол, в присутствии палладиевого катализатора, такого как (трифенилфосфин)тетракиспалладий (0), и соли, такой как, например, хлорид лития, перемешивая реакционную смесь при подходящей температуре, такой как 120-130°C, с применением традиционного нагревания или под действием микроволнового излучения в течение времени, необходимого для достижения завершения реакции, как правило, в течение 1 часа в случае традиционного нагревания. На этом этапе реакции получают соединение формулы (IV).
Этап 2. Соединение формулы (IV) можно подвергнуть окислению с помощью стандартных методик, хорошо известных специалисту в данной области, как, например, путем озонолиза или путем реакции со смесью тетраоксида осмия и периодата натрия с получением соединения формулы (V).
Этап 3. Соединение формулы (V) можно вводить в реакцию с амином формулы NHR3R4, где R3 и R4 определены ранее, в традиционной реакции восстановительного аминирования, хорошо известной специалисту в данной области. Таким образом, соединение формулы (V) можно вводить в реакцию с амином формулы NHR3R4, определенным ранее, в инертном растворителе, таком как, например, 1,2-дихлорэтан, перемешивая реакционную смесь при подходящей температуре, как правило, при 80-120°C, в течение 10-20 минут под действием микроволнового излучения в присутствии восстанавливающего средства, такого как трибутоксицианоборгидрид или боргидрид натрия. После добавления восстанавливающего средства реакционную смесь можно перемешивать либо при комнатной температуре, либо при микроволновом нагревании в течение времени, необходимого для достижения завершения реакции, как правило, в течение 20 мин при 80°C в случае микроволнового нагревания. На этом этапе реакции получают конечное соединение формулы (I).
Схема 2 Синтез соединений формулы (II)
Способ A.
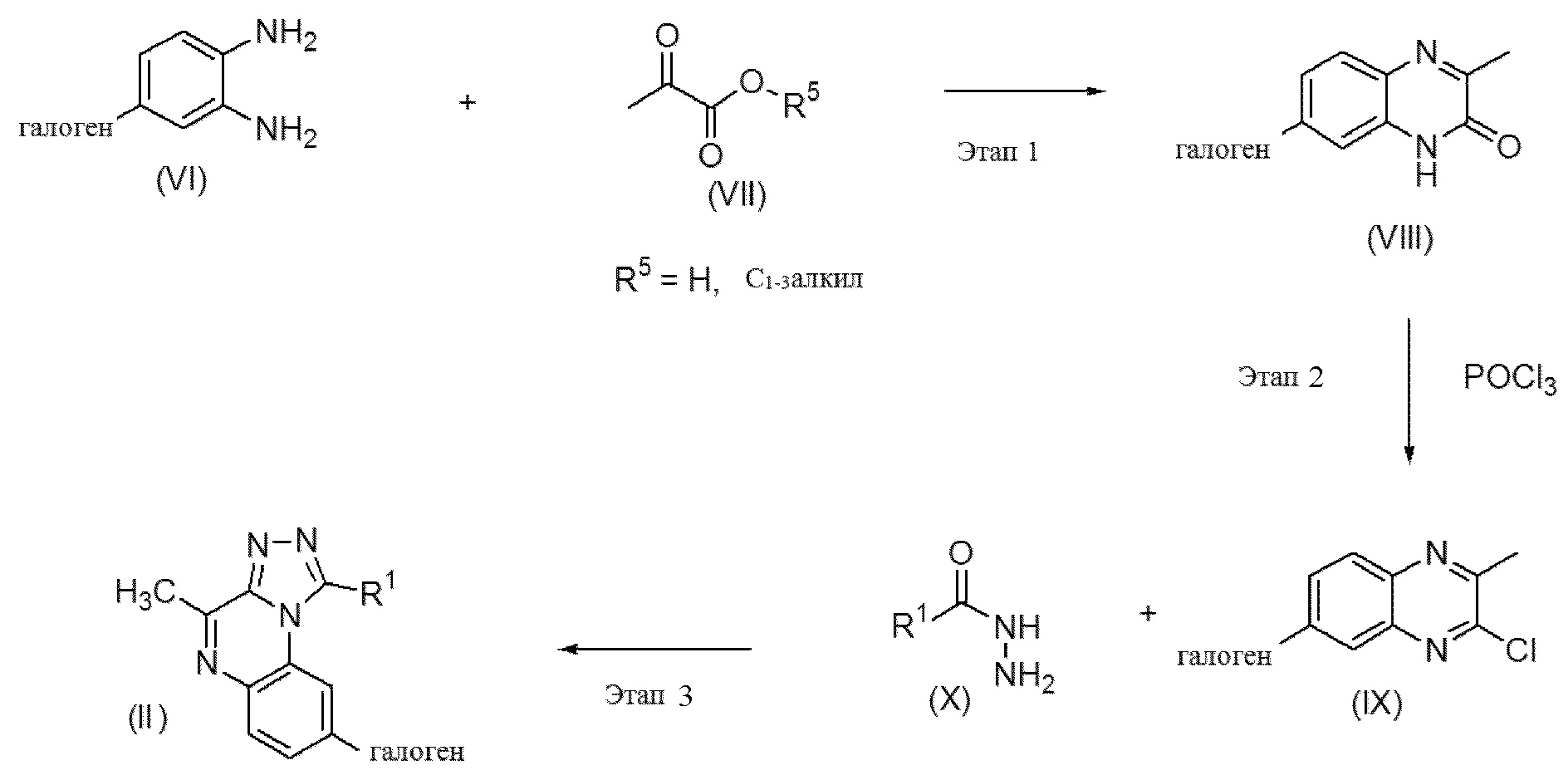
Этап 1. Промежуточное соединение формулы (VI) можно вводить в реакцию с коммерчески доступным соединением формулы (VII), где R5 представляет собой C1-3алкил, такой как, например, метил или этил, в инертном растворителе, таком как, например, толуол, перемешивая реакционную смесь при подходящей температуре, как правило, при 100-130°C, с применением традиционного нагревания или под действием микроволнового излучения в течение времени, необходимого для достижения завершения реакции, как правило, в течение 3 часов в случае традиционного нагревания. Если R5 представляет собой водород, реакцию проводят в смеси уксусной кислоты и воды, при этом перемешивание проводят при комнатной температуре в течение ночи. В результате данной реакции обычно получают смесь двух возможных региоизомеров, которые можно разделить на этом этапе (с получением региоизомера формулы (VIII)) или на одном из следующих этапов с помощью способов хроматографии, с помощью колоночной хроматографии или HPLC. Соединения формулы (VI) являются либо коммерчески доступными, либо описанными в химической литературе, и могут быть получены с помощью простых стандартных методик синтеза, хорошо известных специалисту в данной области.
Этап 2. Промежуточные соединения формулы (VIII) можно вводить в реакцию в присутствии или в отсутствие растворителя, такого как, например, 1,2-дихлорэтан, с оксихлоридом фосфора, перемешивая реакционную смесь при подходящей температуре, как правило, при 100-120°C, с применением традиционного нагревания или под действием микроволнового излучения в течение времени, необходимого для достижения завершения реакции, как правило, в течение 2-4 часов в случае традиционного нагревания. На этом этапе реакции получают промежуточные соединения формулы (IX).
Этап 3. Промежуточное соединение формулы (IX) можно вводить в реакцию с промежуточным соединением формулы (X) в растворителе, таком как, например, этанол, н-бутанол или тетрагидрофуран, перемешивая реакционную смесь при подходящей температуре, как правило, при 100-160°C, с применением традиционного нагревания или под действием микроволнового излучения в течение времени, необходимого для достижения завершения реакции, как правило, в течение 15-20 минут при 160°C в случае микроволнового нагревания, получая соединения формулы (II). Промежуточные соединения формулы (X) могут быть коммерчески доступными либо описаны в химической литературе и могут быть получены с помощью простых стандартных методик синтеза, хорошо известных специалисту в данной области.
Способ B
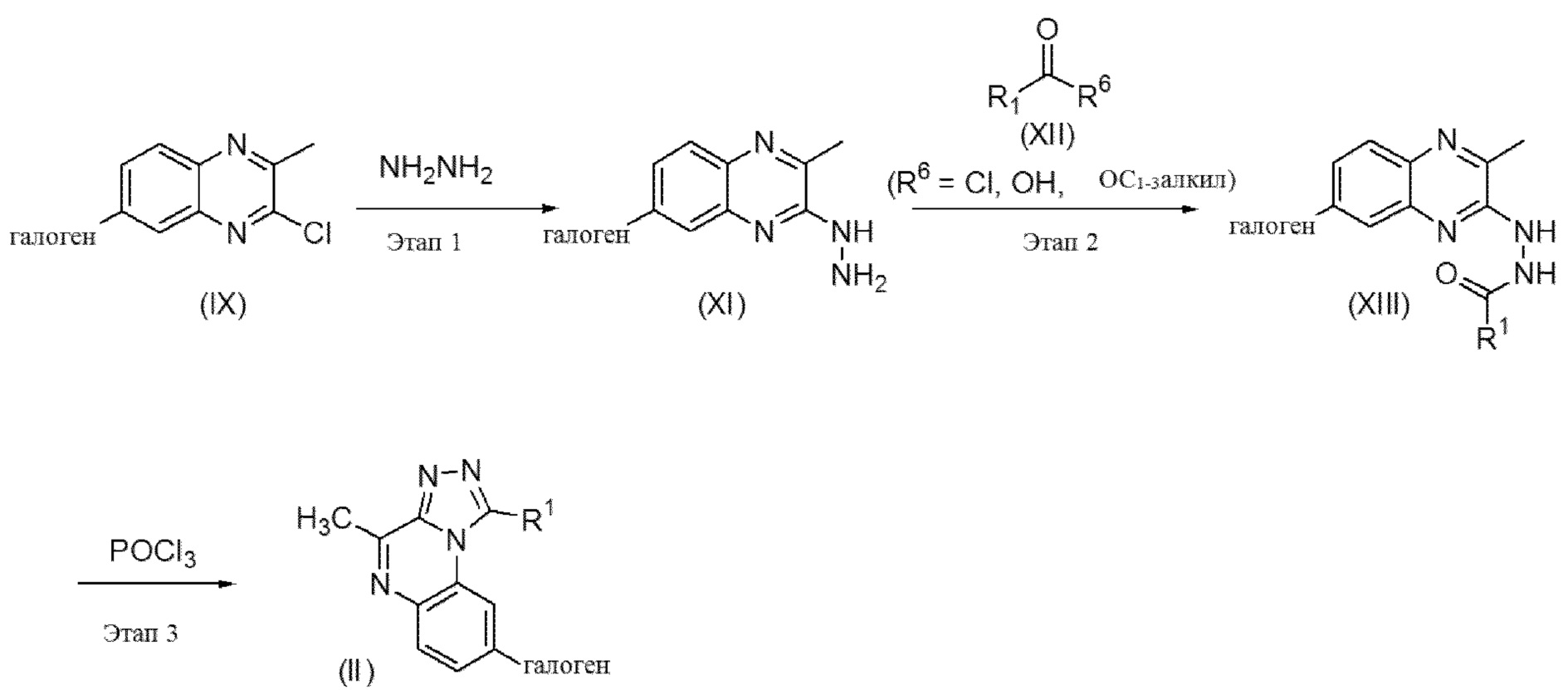
Этап 1. Промежуточные соединения формулы (IX) можно обработать гидратом гидразина в инертном растворителе, таком как метанол или этанол, следуя простым стандартным методикам синтеза, хорошо известным специалисту в данной области, с получением промежуточных соединений формулы (XI).
Этап 2. Промежуточные соединения формулы (XI) можно вводить в реакцию с промежуточными соединениями формулы (XII), следуя простым стандартным методикам синтеза, хорошо известным специалисту в данной области, с получением промежуточных соединений формулы (XIII). Промежуточные соединения формулы (XII) могут быть коммерчески доступными либо синтезированными в соответствии с практикой из литературных источников.
Этап 3. Промежуточные соединения формулы (XIII) можно вводить в реакцию в присутствии или в отсутствие растворителя, такого как, например, 1,2-дихлорэтан, с оксихлоридом фосфора, перемешивая реакционную смесь при подходящей температуре, как правило, при 80-100°C, с применением традиционного нагревания или под действием микроволнового излучения в течение времени, необходимого для достижения завершения реакции, как правило, в течение 16 часов в случае традиционного нагревания. На этом этапе реакции получают соединения формулы (II).
Получение меченных радиоактивным изотопом конечных соединений
Схема 3. Синтез соединений формулы (I), где R2 = меченный радиоактивным изотопом 3H -CH2-NR3R4
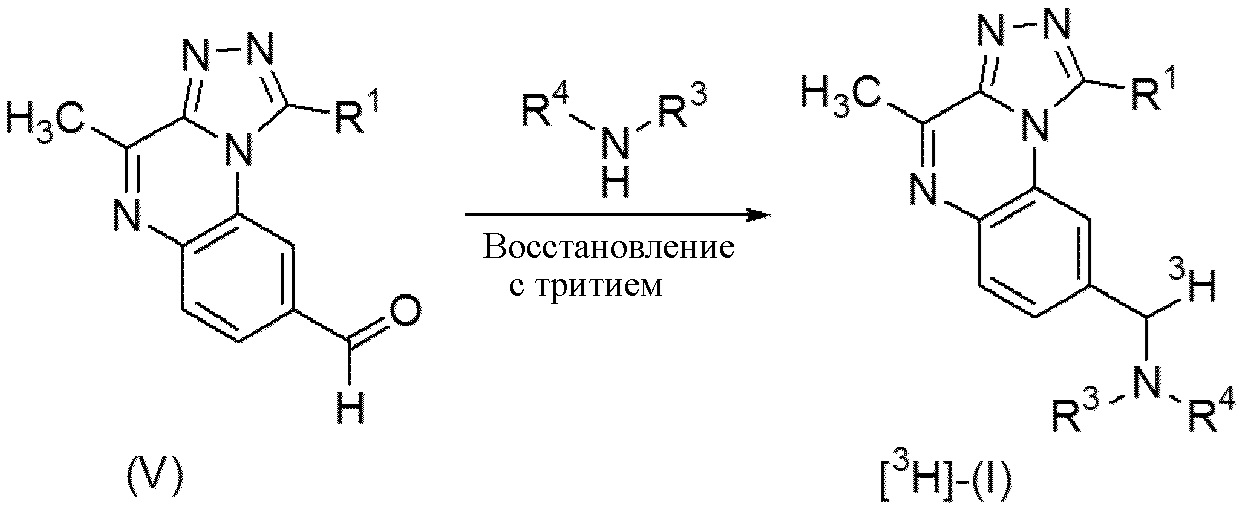
Меченные тритием соединения формулы (I), называемые в данном документе [3H]-(I), можно получать из соединений формулы (V) с помощью реакции с амином формулы NHR3R4, где R3 и R4 определены ранее, в реакции восстановительного аминирования с применением трития в присутствии катализатора в условиях, известных специалисту в данной области, в два этапа. Таким образом, соединение формулы (V) можно вводить в реакцию на первом этапе с амином формулы NHR3R4, определенным ранее, в инертном растворителе, таком как, например, дихлорметан, необязательно в присутствии дегидрирующего средства, такого как тетра(изопропоксид) титана, перемешивая реакционную смесь при подходящей температуре, как правило, при комнатной температуре, в инертной атмосфере. После удаления растворителя второй этап включает добавление другого инертного апротонного растворителя, такого как, например тетрагидрофуран, и реакцию с промежуточным имином в присутствии восстанавливающего средства, такого как тритий, и в присутствии катализатора, такого как Pt на угле. После добавления восстанавливающего средства реакционную смесь можно перемешивать при комнатной температуре в течение времени, необходимого для достижения завершения реакции, как правило, в течение 60 мин при комнатной температуре. На этом этапе реакции получают конечное соединение формулы [3H]-(I).
Схема 4. Синтез соединений формулы (I), где R1 = меченный радиоактивным изотопом 18F фенил или пиридинил
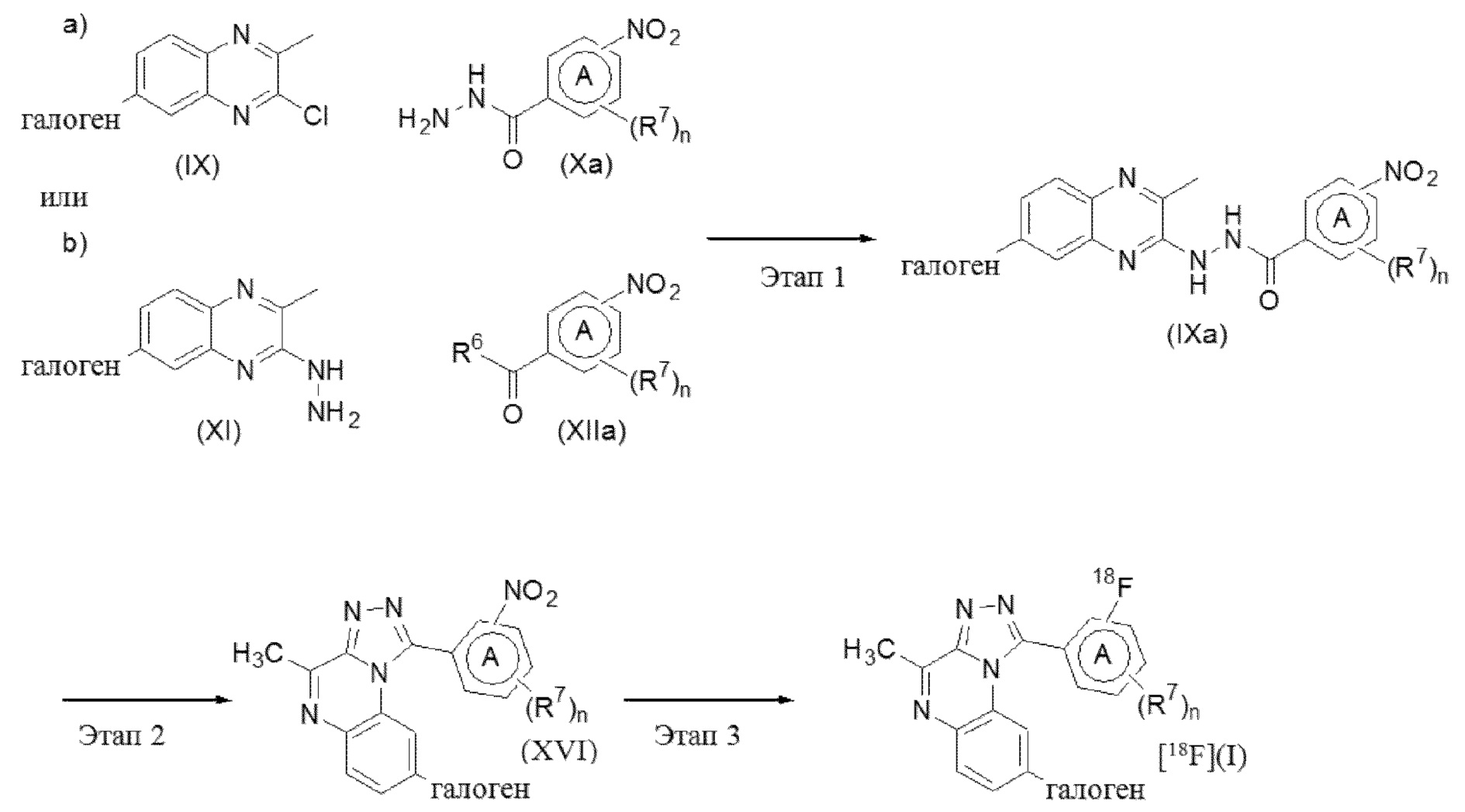
Соединения формулы (I), где R1 представляет собой фенильную или пиридинильную группу, меченные радиоактивным изотопом 18F, где кольцо A представляет собой фенил или пиридинил, R7 представляет собой галоген или трифторметил, n равно 0 или 1, а R2 определен ранее, настоящим называемые соединениями формулы (I-u) могут быть получены с помощью способов синтеза, хорошо известных специалисту в данной области. Например, согласно общей схеме 10.
Этап 1. (a) Соединение формулы (IX) можно вводить в реакцию с соединением формулы (Xa), где кольцо A представляет собой фенил или пиридинил, R7 представляет собой галоген или трифторметил, n равно 0 или 1, а R2 определен ранее для соединений формулы (I), в соответствии с условиями, описанными в рамках схемы 1, способа A, этапа 3.
Этап 1. (b) Соединение формулы (XI) можно вводить в реакцию с соединением формулы (XIIa), где кольцо A представляет собой фенил или пиридинил, R7 представляет собой галоген или трифторметил, n равно 0 или 1, а R2 определен ранее для соединений формулы (I), в соответствии с условиями, описанными в рамках схемы 1, способа B, этапа 2.
Этап 2. Промежуточное соединение формулы (IXa) можно вводить в реакцию в присутствии или в отсутствие растворителя, такого как, например 1,2-дихлорэтан, с оксихлоридом фосфора, перемешивая реакционную смесь при подходящей температуре, как правило, при 80-100°C, с применением традиционного нагревания или под действием микроволнового излучения в течение времени, необходимого для достижения завершения реакции, как правило, в течение 16 часов в случае традиционного нагревания.
Этап 3. Промежуточное соединение формулы (XVI) можно подвергать реакции нуклеофильного ароматического замещения с источником [18F]фторида ([18F]F-), таким как, например, комплекс [18F]F-/K2CO3/Kryptofix® 222 или [18F]KF·K222 (где Kryptofix® 222 и K222 означают 4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8.8.8]гексакозан; также известный как K 2.2.2), в инертном растворителе, таком как, например, безводный DMF, в соответствующих условиях реакции, таких как нагревание в микроволновой печи, например, при 140°, или в условиях, известных специалисту в данной области (в отношении обзора см., например, P. W. Miller et al. Angew. Chem. Int. Ed. 2008, 47, 8998-9033).
Некоторые соединения согласно настоящему изобретению были выделены в качестве форм солей присоединения кислоты или были выделены в виде свободных оснований, а затем превращены в формы солей присоединения кислоты. Чтобы получить соединения согласно настоящему изобретению в форме солей присоединения кислоты, например, в форме HCl-солей, если не указано иное, можно применять несколько методик, известных специалистам в данной области. В типичной методике, например, свободное основание можно растворить в изопропаноле, диизопропиловом эфире, диэтиловом эфире и/или дихлорметане, а затем можно по каплям добавлять 1-2 эквивалента соответствующей кислоты, например, 6 н. раствор HCl в 2-пропаноле или 2 н. раствор HCl в диэтиловом эфире. Смесь, как правило, перемешивают в течение 10 мин или дольше, после чего можно отфильтровать продукт. HCl-соль обычно высушивают in vacuo. Показатели стехиометрического состава соли, приводимые выше и ниже в данном документе, являются такими, которые получены экспериментальным путем, и могут меняться при использовании различных аналитических способов. Если стехиометрический состав соли неизвестен, применяют выражение ".x"; например, соль-гидрохлорид, стехиометрический состав которой неизвестен, называют ".x HCl".
ФАРМАКОЛОГИЯ
Соединения формулы (I) и их фармацевтически приемлемые соли и сольваты согласно настоящему изобретению ингибируют активность ферментов PDE2, в частности, PDE2A, и в меньшей степени они ингибируют активность ферментов PDE10, в частности, PDE10A, и, следовательно, повышают уровни cAMP или cGMP в клетках, экспрессирующих PDE2. Ингибиторы PDE10 можно применять для повышения уровней cAMP и/или cGMP в клетках, экспрессирующих фермент PDE10. В данной работе было обнаружено, что ингибиторы PDE2, в частности, соединения формулы (I) и их фармацевтически приемлемые соли и сольваты, могут усиливать эффект ингибиторов PDE10, в частности, таковых, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и один или несколько ингибиторов PDE10, в частности, таковых, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, могут дозозависимым образом усиливать in vivo связывание радиолиганда, осуществляющего селективное связывание с каталитическим доменом фермента PDE2. Ввиду вышеупомянутой активности и наблюдаемых эффектов предполагается, что комбинации, включающие ингибитор PDE2, в частности, соединение формулы (I), описанное в данном документе, и один или несколько ингибиторов PDE10, в частности, таковых, выбранных из группы MP-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенным в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, описанных в данном документе, или фармацевтические композиции, содержащие указанные комбинации, можно применять в лечении неврологических или психических расстройств или нарушений эндокринной системы или метаболизма.
Следовательно, настоящее изобретение относится к комбинации ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, таковых, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению для применения в качестве лекарства, а также к применению ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению или фармацевтической композиции согласно настоящему изобретению для производства лекарственного препарата. Настоящее изобретение также относится к ингибитору PDE2, в частности, к соединению формулы (I) или его фармацевтически приемлемым соли или сольвату, и к одному или нескольким ингибиторам PDE10, в частности, выбранным из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению или к фармацевтической композиции согласно настоящему изобретению для применения в лечении или предупреждении, в частности, в лечении, состояния у млекопитающего, в том числе у человека, где состояние выбрано из неврологических или психических расстройств или эндокринных или метаболических расстройств. Настоящее изобретение также относится к применению ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению или фармацевтической композиции согласно настоящему изобретению для производства лекарственного препарата для лечения или предупреждения, в частности, для лечения, состояния у млекопитающего, в том числе у человека, где состояние выбрано из неврологических или психических расстройств или эндокринных или метаболических расстройств.
Настоящее изобретение также относится к ингибитору PDE2, в частности, к соединению формулы (I) или его фармацевтически приемлемым соли или сольвату, и к одному или нескольким ингибиторам PDE10, в частности, выбранным из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению или фармацевтической композиции согласно настоящему изобретению для применения в лечении, предупреждении, уменьшении интенсивности симптомов, контроле или снижении риска возникновения неврологических или психических расстройств или эндокринных или метаболических расстройств.
Настоящее изобретение также относится к применению ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению или фармацевтической композиции согласно настоящему изобретению для производства лекарственного препарата для лечения, предупреждения, уменьшения интенсивности симптомов, контроля или снижения риска возникновения различных неврологических или психических расстройств или эндокринных или метаболических расстройств.
Когда говорят, что настоящее изобретение относится к применению ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, или композиции согласно настоящему изобретению для производства лекарственного препарата для, например, лечения субъекта, например, млекопитающего, понимают, что такое применение следует толковать в определенных сферах действия как способ, например, лечения субъекта, включающий введение субъекту, нуждающемуся, например, в таком лечении, терапевтически эффективного количества ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, или композиции согласно настоящему изобретению.
В частности, показания для возможного лечения комбинациями ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, в отдельности или в комбинации с другими лекарственными средствами включают, без ограничения, те заболевания, которые, как полагают, частично опосредованы базальными ганглиями, префронтальной корой и гиппокампом.
Эти показания включают неврологические и психические расстройства, выбранные из психотических расстройств и состояний; тревожных расстройств; двигательных расстройств; наркомании; аффективных расстройств; нейродегенеративных расстройств; расстройств или состояний, включающих в качестве симптома дефицит внимания и/или когнитивный дефицит; боли; аутического расстройства или аутизма и метаболических расстройств.
В частности, психотические расстройства и состояния, ассоциированные с дисфункцией PDE2 или PDE2 и PDE10, включают одно или несколько из следующих состояний или заболеваний: шизофрения, например, параноидального, дезорганизованного, кататонического, недифференцированного или резидуального типа; шизофреноформное расстройство; шизоаффективное расстройство, как, например, бредового или депрессивного типа; бредовое расстройство; психотическое расстройство, вызванное употреблением химических веществ, такое как психоз, вызванный употреблением алкоголя, амфетамина, марихуаны, кокаина, галлюциногенных веществ, летучих веществ наркотического действия, опиоидов или фенциклидина; расстройства личности параноидального типа и расстройство личности шизоидного типа.
В частности, тревожные расстройства включают паническое расстройство; агорафобию; специфическую фобию, социофобию; обсессивно-компульсивное расстройство; посттравматическое стрессовое расстройство, острое стрессовое расстройство и генерализованное тревожное расстройство.
В частности, двигательные расстройства включают болезнь Хантингтона и дискинезию; болезнь Паркинсона; синдром усталых ног и эссенциальный тремор. Кроме того, могут быть включены синдром Туретта и другие тиковые расстройства.
В частности, расстройство центральной нервной системы представляет собой расстройство, связанное с химическими веществами, выбранное из группы злоупотребления алкоголем; алкогольной зависимости; алкогольного абстинентного синдрома; алкогольного абстинентного синдрома с делирием; психотического расстройства, вызванного употреблением алкоголя; амфетаминовой зависимости; амфетаминового абстинентного синдрома; кокаиновой зависимости; кокаинового абстинентного синдрома; никотиновой зависимости; никотинового абстинентного синдрома; опиоидной зависимости и опиоидного абстинентного синдрома.
В частности, аффективные расстройства и аффективные эпизоды включают депрессию, манию и биполярные расстройства. Преимущественно, аффективное расстройство выбрано из группы биполярных расстройств (I и II типа); циклотимического расстройства; депрессии, дистимического расстройства; большого депрессивного расстройства; терапевтически резистентной депрессии и аффективного расстройства, вызванного употреблением химических веществ.
В частности, нейродегенеративные расстройства включают болезнь Паркинсона; болезнь Хантингтона; деменцию, такую как, например, болезнь Альцгеймера; мультиинфарктную деменцию; СПИД-ассоциированную деменцию или лобно-височную деменцию. Нейродегенеративное расстройство или состояние включает дисфункцию реакций стриарных средних шипиковых нейронов.
В частности, расстройства или состояния, включающие в качестве симптома дефицит внимания и/или когнитивный дефицит, или когнитивные расстройства включают деменцию, такую как болезнь Альцгеймера; мультиинфарктную деменцию; деменцию вследствие болезни телец Леви; алкогольную деменцию или персистирующую деменцию, вызванную употреблением химических веществ; деменцию, ассоциированную с внутричерепными опухолями или черепно-мозговой травмой; деменцию, ассоциированную с болезнью Хантингтона; деменцию, ассоциированную с болезнью Паркинсона; СПИД-ассоциированную деменцию; деменцию вследствие болезни Пика; деменцию вследствие болезни Крейтцфельда-Якоба; другие заболевания, включающие делирий; амнестическое расстройство; посттравматическое стрессовое расстройство; инсульт; прогрессирующий надъядерный паралич; олигофрению; нарушение способности к обучению; синдром дефицита внимания и гиперактивности (ADHD); умеренное когнитивное нарушение; синдром Аспергера и возрастное когнитивное нарушение.
В частности, боль включает острые и хронические состояния, сильную боль, неустранимую боль, нейропатическую боль и посттравматическую боль, раковую боль, нераковую боль, болевое расстройство, ассоциированное с психологическими факторами, болевое расстройство, ассоциированное с общим состоянием здоровья, или болевое расстройство, ассоциированное как с психологическими факторами, так и с общим состоянием здоровья.
В частности, метаболические расстройства включают диабет, в частности, диабет 1-го типа или 2-го типа, и связанные нарушения, такие как ожирение. Дополнительные связанные нарушения включают синдром X, нарушение толерантности к глюкозе, нарушение содержания глюкозы в крови натощак, гестационный диабет, диабет взрослого типа у молодых (MODY), латентный аутоиммунный диабет у взрослых (LADA), дислипидемию, ассоциированную с диабетом, гипергликемию, гиперинсулинемию, дислипидемию, гипертриглицеридемию и инсулинорезистентность.
Преимущественно, психотическое расстройство выбрано из группы шизофрении, бредового расстройства, шизоаффективного расстройства, шизофреноформного расстройства и психотического расстройства, вызванного употреблением химических веществ.
Преимущественно, расстройство центральной нервной системы представляет собой расстройство личности, выбранное из группы обсессивно-компульсивного расстройства личности и шизоидного, шизотипического расстройства.
Преимущественно, расстройство центральной нервной системы представляет собой аффективное расстройство, выбранное из группы биполярных расстройств (I и II типа), циклотимического расстройства, депрессии, дистимического расстройства, большого депрессивного расстройства, терапевтически резистентной депрессии и аффективного расстройства, вызванного употреблением химических веществ.
Преимущественно, расстройство центральной нервной системы представляет собой синдром дефицита внимания и гиперактивности.
Преимущественно, расстройство центральной нервной системы представляет собой когнитивное расстройство, выбранное из группы делирия, персистирующего делирия, вызванного употреблением химических веществ, деменции, деменции вследствие заболевания, вызываемого HIV, деменции вследствие болезни Хантингтона, деменции вследствие болезни Паркинсона, деменции альцгеймеровского типа, персистирующей деменции, вызванной употреблением химических веществ, и умеренного когнитивного нарушения.
Преимущественно, расстройства, которые лечат с помощью ингибиторов PDE2, в частности, соединений формулы (I) или их солей или сольватов по настоящему изобретению, выбраны из шизофрении; обсессивно-компульсивного расстройства; генерализованного тревожного расстройства; болезни Хантингтона; дискинезии; болезни Паркинсона; депрессии; биполярных расстройств; деменции, такой как болезнь Альцгеймера; синдрома дефицита внимания и гиперактивности; наркомании; боли; аутизма; диабета и ожирения.
Преимущественно, расстройства, которые лечат с помощью ингибиторов PDE2, в частности, соединений формулы (I) или их солей или сольватов по настоящему изобретению, представляют собой шизофрению, в том числе ее позитивные и негативные симптомы, а также нарушения познавательных способностей, такие как ухудшение внимания или памяти.
Среди упомянутых выше расстройств особое значение имеет лечение тревожности, обсессивно-компульсивного расстройства, посттравматического стрессового расстройства, генерализованного тревожного расстройства, шизофрении, депрессии, синдрома дефицита внимания и гиперактивности, болезни Альцгеймера, деменции вследствие болезни Хантингтона, деменции вследствие болезни Паркинсона, деменции альцгеймеровского типа, персистирующей деменции, вызванной употреблением химических веществ, и умеренного когнитивного нарушения.
Среди упомянутых выше расстройств особое значение имеет лечение тревожности, обсессивно-компульсивного расстройства, шизофрении, депрессии, синдрома дефицита внимания и гиперактивности и болезни Альцгеймера.
Другие расстройства центральной нервной системы включают тревожное расстройство, ассоциированное с шизофренией, и коморбидные депрессию и тревожность, в частности, большое депрессивное расстройство с коморбидным генерализованным тревожным расстройством, социальным тревожным расстройством или паническим расстройством; при этом следует понимать, что коморбидные депрессия и тревожность также могут упоминаться под терминами депрессия, сопровождающаяся тревожностью, смешанные тревожность и депрессия, смешанное тревожное и депрессивное расстройство или большое депрессивное расстройство с симптомами тревожности, которые применяются в данном документе без разграничения.
На сегодняшний день четвертое издание Руководства по диагностике и статистике психических расстройств (д SM-IV) Американской психиатрической ассоциации обеспечивает средства диагностики для идентификации расстройств, описанных в данном документе. Специалист в данной области будет осознавать, что для неврологических и психических расстройств, описанных в данном документе, существуют альтернативные номенклатуры, нозологические подходы и системы классификации, и что они видоизменяются вместе с прогрессом в области медицины и научным прогрессом.
Таким образом, настоящее изобретение также относится к ингибитору PDE2, в частности, к соединению формулы (I) или его фармацевтически приемлемым соли или сольвату, и одному или нескольким ингибиторам PDE10, в частности, выбранным из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению для применения в лечении любого из заболеваний, упомянутых выше в данном документе.
Настоящее изобретение также относится к ингибитору PDE2, в частности, к соединению формулы (I) или его фармацевтически приемлемым соли или сольвату, и одному или нескольким ингибиторам PDE10, в частности, выбранным из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению для применения в лечении любого из заболеваний, упомянутых выше в данном документе.
Настоящее изобретение также относится к ингибитору PDE2, в частности, к соединению формулы (I) или его фармацевтически приемлемым соли или сольвату, и одному или нескольким ингибиторам PDE10, в частности, выбранным из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению для лечения или предупреждения, в частности, лечения, любого из заболеваний, упомянутых выше в данном документе.
Настоящее изобретение также относится к применению ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению для производства лекарственного препарата для лечения или предупреждения любого из болезненных состояний, упомянутых выше в данном документе.
Настоящее изобретение также относится к применению ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению для производства лекарственного препарата для лечения любого из болезненных состояний, упомянутых выше в данном документе.
Ингибиторы PDE2, в частности, соединения формулы (I) или их фармацевтически приемлемые соли или сольваты, и один или несколько ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, по настоящему изобретению можно вводить млекопитающим, предпочтительно людям, для лечения или предупреждения любого из заболеваний, упомянутых выше в данном документе.
Ввиду полезности ингибиторов PDE2, в частности, соединений формулы (I) или их фармацевтически приемлемых солей или сольватов, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению обеспечивается способ лечения расстройства или заболевания, упомянутого выше в данном документе, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества ингибитора PDE2, в частности, любого из соединений формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, или терапевтически эффективного количества фармацевтических композиций, описанных в данном документе.
Указанные способы включают введение, т.е. системное или местное введение, предпочтительно пероральное введение, терапевтически эффективного количества ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению теплокровным животным, в том числе людям.
Таким образом, настоящее изобретение также относится к способу предупреждения и/или лечения любого из заболеваний, упомянутых выше в данном документе, включающему введение терапевтически эффективного количества ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению пациенту, нуждающемуся в этом.
Ингибиторы PDE2, в частности, соединения формулы (I) и их фармацевтически приемлемые соли и сольваты, и ингибиторы PDE10, описываемые в данном документе, могут быть в комбинации или в комбинации с другими фармацевтическими средствами, такими как другие средства, применяемые в лечении психозов, таких как шизофрения и биполярное расстройство, обсессивно-компульсивное расстройство, болезнь Паркинсона, когнитивное нарушение и/или потеря памяти, например, агонисты никотиновых ацетилхолиновых рецепторов α-7, ингибиторы PDE4, другие ингибиторы PDE2, другие ингибиторы PDE10, другие ингибиторы PDE2 и PDE10, блокаторы кальциевых каналов, модуляторы мускариновых ацетилхолиновых рецепторов m1 и m2, модуляторы аденозиновых рецепторов, ампакины, модуляторы NMDA-R, модуляторы mGluR, модуляторы дофаминовых рецепторов, модуляторы серотониновых рецепторов, модуляторы каннабиноидных рецепторов и ингибиторы холинэстеразы (например, донепезил, ривастигмин и галантамин). В таких комбинациях ингибиторы PDE2, в частности, соединения формулы (I) или их фармацевтически приемлемые соли или сольваты, и один или несколько ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, по настоящему изобретению можно использовать в комбинации с одним или несколькими другими лекарственными средствами в лечении, предупреждении, контроле, уменьшении интенсивности симптомов или снижении риска возникновения заболеваний или состояний, для которых ингибиторы PDE2, в частности, соединения формулы (I) и их фармацевтически приемлемые соли и сольваты, и один или несколько ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, или другие лекарственные средства могут быть полезными, где комбинация лекарственных средств, принимаемых совместно, является более безопасной или более эффективной, чем любое лекарственное средство в отдельности.
Специалист в данной области признает, что терапевтически эффективное количество ингибиторов PDE2, в частности, соединений формулы (I) и их фармацевтически приемлемых солей и сольватов, и одного или нескольких ингибиторов PDE10 по настоящему изобретению является количеством, достаточным для ингибирования фермента PDE2 или как фермента PDE2, так и фермента PDE10, и что данное количество изменяется inter alia в зависимости от типа заболевания, концентрации соединения в терапевтическом составе и состояния пациента. Как правило, количество ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10 согласно настоящему изобретению, подлежащих введению в качестве терапевтического средства для лечения состояний, таких как расстройства, описанные в данном документе, будет определяться в каждом конкретном случае лечащим врачом.
Как правило, подходящей дозой является та, которая обуславливает концентрацию ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10 согласно настоящему изобретению в участке лечения в диапазоне от 0,5 нМ до 200 мкМ, и в более обычных случаях от 5 нМ до 50 мкМ. Для получения этих терапевтических концентраций пациенту, нуждающемуся в таком лечении, будет введено от 0,001 мг/кг до 15 мг/кг веса тела, в частности, от 0,01 мг/кг до 2,50 мг/кг веса тела, в частности, от 0,01 до 1,5 мг/кг веса тела, в частности, от 0,1 мг/кг до 0,50 мг/кг веса тела. Количество ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10 согласно настоящему изобретению, также называемых здесь активными ингредиентами, необходимое для достижения терапевтического эффекта, будет, разумеется, изменяться индивидуально, изменяться в зависимости от конкретного соединения, пути введения, возраста и состояния пациента, получающего лечение, и конкретного расстройства или заболевания, лечение которого осуществляется. Способ лечения может также включать введение активных ингредиентов в режиме от одного до четырех приемов в день. В этих способах лечения ингибиторы PDE2, в частности, соединения формулы (I) и их фармацевтически приемлемые соли и сольваты и один или несколько ингибиторов PDE10 согласно настоящему изобретению предпочтительно составляют перед введением. Как описано в данном документе ниже, подходящие фармацевтические составы получают с помощью известных методик с применением известных и общедоступных ингредиентов.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Настоящее изобретение также обеспечивает композиции для предупреждения или лечения заболеваний, таких как неврологические и психические расстройства и эндокринные или метаболические заболевания. Указанные композиции содержат терапевтически эффективное количество ингибитора PDE2, в частности, соединения согласно формуле (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и фармацевтически приемлемые носитель или разбавитель.
Хотя активные ингредиенты можно вводить отдельно, предпочтительно представлять их в виде фармацевтической композиции. Соответственно, настоящее изобретение дополнительно обеспечивает фармацевтическую композицию, содержащую ингибитор PDE2, в частности, соединение формулы (I) или его фармацевтически приемлемые соль или сольват, и один или несколько ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, согласно настоящему изобретению вместе с фармацевтически приемлемыми носителем или разбавителем. Носитель или разбавитель должны быть "приемлемыми" в том смысле, что они должны быть совместимы с другими ингредиентами композиции и не быть вредными для пациентов, их получающих.
Фармацевтические композиции по настоящему изобретению могут быть получены любыми способами, хорошо известными в области фармацевтики. Терапевтически эффективное количество конкретных соединений, в форме основания или в форме соли присоединения, в качестве активных ингредиентов объединяют в однородную смесь с фармацевтически приемлемым носителем, который может иметь разнообразные формы в зависимости от формы препарата, желаемой для введения. Желательно, чтобы эти фармацевтические композиции находились в стандартной лекарственной форме, предпочтительно подходящей для системного введения, такого как пероральное, чрескожное или парентеральное введение; или для местного введения, как, например, с помощью ингаляции, спрея для носа, глазных капель или с помощью крема, геля, шампуня и т.п. Например, при получении композиций в виде лекарственной формы для перорального введения можно использовать любые обычные фармацевтические среды, такие как, например, вода, гликоли, масла, спирты и т.п., в случае жидких препаратов для перорального введения, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества, разрыхлители и т.п., в случае порошков, пилюль, капсул и таблеток. Благодаря своей простоте введения таблетки и капсулы представляют наиболее предпочтительные стандартные лекарственные формы для перорального введения, в случае которых, несомненно, используются твердые фармацевтические носители. В случае композиций для парентерального введения носитель будет, как правило, по меньшей мере в значительной степени содержать стерильную воду, хотя могут быть включены и другие ингредиенты, например, для улучшения растворимости. Например, могут быть получены растворы для инъекций, в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также могут быть получены суспензии для инъекций, в случае которых могут использоваться подходящие жидкие носители, суспендирующие средства и т.п. В композициях, подходящих для чрескожного введения, носитель необязательно содержит средство усиления проникновения и/или подходящее смачиваемое средство, необязательно в комбинации с подходящими добавками любой природы в минимальных пропорциях, при этом добавки не оказывают никаких существенных вредных воздействий на кожу. Указанные добавки могут облегчать нанесение на кожу и/или могут быть полезными при получении желаемых композиций. Данные композиции могут наноситься различными путями, например, в виде трансдермального пластыря, путем точечного нанесения на кожу или в виде мази.
Особенно предпочтительно для простоты введения и однородности дозирования составлять вышеупомянутые фармацевтические композиции в стандартные лекарственные формы. Стандартные лекарственные формы, как применяется в описании и формуле изобретения в данном документе, относятся к физически дискретным единицам, подходящим в качестве единиц дозирования, при этом каждая единица содержит заранее определенное количество активного ингредиента, рассчитанное для получения желаемого терапевтического эффекта, совместно с требуемым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (в том числе делимые таблетки или таблетки, покрытые оболочкой), капсулы, пилюли, пакеты с порошкообразным продуктом, облатки, растворы или суспензии для инъекций, чайные ложки с верхом, столовые ложки с верхом и т.п., а также их отдельные кратные количества.
В зависимости от способа введения фармацевтическая композиция будет содержать от 0,05 до 99% по весу, предпочтительно от 0,1 до 70% по весу, более предпочтительно от 0,1 до 50% по весу активных ингредиентов и от 1 до 99,95% по весу, предпочтительно от 30 до 99,9% по весу, более предпочтительно от 50 до 99,9% по весу фармацевтически приемлемого носителя, при этом все процентные содержания приводятся на основе общего веса композиции.
Комбинации соединений по настоящему изобретению можно применять для системного введения, такого как пероральное, чрескожное или парентеральное введение; или для местного введения, как, например, с помощью ингаляции, спрея для носа, глазных капель или с помощью крема, геля, шампуня или т.п. Соединения предпочтительно являются перорально вводимыми.
Точная дозировка и частота введения зависит от конкретных применяемых ингибитора PDE2, такого как соединение согласно формуле (I) или его фармацевтически приемлемые соль или сольват, и одного или нескольких ингибиторов PDE10, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, конкретного состояния, лечение которого осуществляют, тяжести состояния, лечение которого осуществляют, возраста, веса, пола, степени расстройства и общего физического состояния конкретного пациента, а также другого лекарственного средства, которое может принимать индивидуум, как хорошо известно специалисту в данной области. Кроме того, очевидно, что указанное эффективное ежедневное количество можно уменьшать или увеличивать в зависимости от ответа субъекта, подвергаемого лечению, и/или в зависимости от оценки, проводимой врачом, назначающим ингибиторы PDE2, в частности, соединения формулы (I) и их фармацевтически приемлемые соли и сольваты, и один или несколько ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, по настоящему изобретению.
Количество ингибитора PDE2, в частности, соединения формулы (I) или его фармацевтически приемлемых соли или сольвата, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, которое можно объединить с материалом носителя с получением единичной лекарственной формы, будет изменяться в зависимости от заболевания, лечение которого осуществляют, вида млекопитающего и конкретного способа введения. Однако, в качестве общего указания, подходящие стандартные дозы ингибиторов PDE2, в частности, соединений формулы (I) и их фармацевтически приемлемых солей и сольватов, и одного или нескольких ингибиторов PDE10, в частности, выбранных из группы MP-10, PQ-10, TP-10, папаверина, соединения формулы (II), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, и соединения формулы (III), или его стереоизомерной формы, или его фармацевтически приемлемых соли или сольвата, определенных в данном документе, по настоящему изобретению могут, например, предпочтительно содержать от 0,1 мг до приблизительно 1000 мг активных соединений. Предпочтительная стандартная доза составляет от 1 мг до приблизительно 500 мг. Более предпочтительная стандартная доза составляет от 1 мг до приблизительно 300 мг. Еще более предпочтительная стандартная доза составляет от 1 мг до приблизительно 100 мг. Такие стандартные дозы можно вводить более одного раза в день, например, 2, 3, 4, 5 или 6 раз в день, но предпочтительно 1 или 2 раза в день, с тем, чтобы общая дозировка для взрослого человека весом 70 кг находилась в диапазоне от 0,001 до приблизительно 15 мг на кг веса субъекта за одно введение. Предпочтительная дозировка составляет от 0,01 до приблизительно 1,5 мг на кг веса субъекта за одно введение, и такая терапия может продолжаться в течение нескольких недель или месяцев, а в некоторых случаях в течение нескольких лет. Однако следует понимать, что определенный уровень дозы для любого конкретного пациента будет зависеть от ряда факторов, включающих активность определенных используемых соединений; возраст, вес тела, общее состояние здоровья, пол и режим питания индивидуума, подвергаемого лечению; время и путь введения; скорость экскреции; другие лекарственные средства, которые были введены ранее; и тяжесть конкретного заболевания, подвергаемого терапии, что хорошо понятно специалистам в данной области.
Типичная дозировка может представлять собой одну таблетку на от 1 мг до приблизительно 100 мг или от 1 мг до приблизительно 300 мг, принимаемую один раз в день или несколько раз в день, или одну капсулу или таблетку пролонгированного действия, принимаемую один раз в день и содержащую активные ингредиенты в пропорционально более высоком содержании. Эффекта пролонгированного действия можно добиться с помощью материалов капсулы, которые растворяются при различных значениях pH, с помощью капсул, которые медленно высвобождают лекарственное средство при осмотическом давлении, или с помощью любых иных известных средств, обеспечивающих контролируемое высвобождение.
В некоторых случаях может понадобиться применение дозировок вне этих диапазонов, что будет очевидно специалистам в данной области. Кроме того, следует отметить, что клиницист или лечащий врач будут знать, как и когда начинать, прерывать, корректировать или завершать терапию в соответствии с реакцией отдельного пациента.
В отношении композиций, способов и наборов, приведенных выше, специалисту в данной области будет понятно, что предпочтительными соединениями для применения в каждом из них являются такие соединения, отмеченные как предпочтительные выше. Еще более предпочтительными соединениями для композиций, способов и наборов являются соединения, приведенные в неограничивающих примерах ниже.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
I. Химия
Как применяется в данном документе, термин "LCMS" означает жидкостную хроматографию/масс-спектрометрию, "GCMS" означает газовую хроматографию/масс-спектрометрию, "HPLC" означает высокоэффективную жидкостную хроматографию, "RP HPLC" означает обращенно-фазовую высокоэффективную жидкостную хроматографию, "водн." означает водный, "Boc" означает трет-бутоксикарбонил, "nBuLi" означает н-бутиллитий, "BuOH" означает 1-бутанол, "DCE" означает 1,2-дихлорэтан, "DCM" означает дихлорметан, "DIPE" означает диизопропиловый эфир, "DIPEA" означает диизопропилэтиламин, "DMF" означает N,N-диметилформамид, "EtOH" означает этанол, "EtOAc" означает этилацетат, "Et3N" означает триэтиламин, "Pd(AcO)2" означает ацетат палладия (II), "XantPhos" означает 4,5-бис(дифенилфосфино)-9,9-диметилксантен, "Pd-C" означает палладий на угле, "THF" означает тетрагидрофуран, "мин" означает минуты, "ч" означает часы, "MeOH" означает метанол, "iPrOH" означает 2-пропанол, "р.с." означает реакционную смесь, "к.т." означает комнатную температуру, "Rt" означает время удержания (в минутах), "Tf" означает трифторметансульфонат, "TFA" означает трифторуксусную кислоту, "колич." означает количественный, "насыщ." означает насыщенный, "раств." означает раствор, "[M+H]+" означает массу протонированного соединения в виде свободного основания, "[M-H]-" означает массу депротонированного соединения в виде свободного основания, "т.п." означает точку плавления, "q.s." означает сколько нужно.
Реакции с помощью микроволнового излучения проводили в однорежимном реакторе, микроволновом реакторе Biotage InitiatorTM Sixty (Biotage), или в многорежимном реакторе, MicroSYNTH Labstation (Milestone, Inc.).
Реакции гидрогенизации проводили в проточном гидрогенизаторе H-CUBE® от ThalesNano Nanotechnology Inc.
Тонкослойную хроматографию (TLC) проводили на пластинках со слоем силикагеля 60 F254 (Merck) с применением растворителей, чистых для анализа. Хроматографию на открытых колонках проводили на силикагеле с размером частиц 230-400 меш и размером пор 60 ангстрем (Merck) в соответствии со стандартными методиками. Автоматизированную колоночную флэш-хроматографию проводили с применением готовых к подключению картриджей от Merck на силикагеле с частицами неправильной формы с размером частиц 15-40 мкм (одноразовые колонки для нормально-фазовой флэш-хроматографии) в системе SPOT или LAFLASH от Armen Instrument.
Некоторые способы получения соединений по настоящему изобретению иллюстрируются в следующих примерах, которые предназначены для иллюстрации, а не ограничения объема настоящего изобретения. Если не указано иное, все исходные материалы получали от частных поставщиков и применяли без дополнительной очистки.
A. Синтез промежуточных продуктов и предшественников
Промежуточные продукты 1-a и 1-b ((I-1a) и (I-1b))

Метилпируват (8,69 мл, 96,24 ммоль) добавляли к раствору 4-бром-1,2-диаминобензола (15 г, 80 ммоль), растворенного в толуоле (120 мл), в круглодонной колбе, оснащенной аппаратом Дина-Старка. Затем р.с. нагревали с обратным холодильником в течение 3 ч. Когда реакция завершалась, растворитель удаляли in vacuo, и неочищенный продукт промывали диэтиловым эфиром с получением смеси промежуточных продуктов (I-1a) и (I-1b) в виде бледно-серого твердого вещества, которое применяли без дополнительной очистки на следующем этапе (16 г, 83%). C9H7BrN2O, LCMS: Rt 1,07 (первый изомер), 1,15 (второй изомер), масса/заряд 239 [M+H]+ (способ 2).
Порцию смеси региоизомеров разделяли путем суспендирования смеси в метаноле и гидроксиде аммония (q.s.), нагревая до появления конденсата и охлаждая до комнатной температуры. Образовавшийся осадок фильтровали, к фильтрату добавляли воду, и образовавшийся осадок также извлекали с помощью фильтрации. Для получения осадка, содержащего смесь с соотношением I-1a:I-1b 94:6, повторяли два дополнительных цикла.
Промежуточные продукты 2-a и 2-b ((I-2a) и (I-2b))

Смесь промежуточных продуктов (I-1a) и (I-1b) (16 г, 66,95 ммоль) растворяли в POCl3 (78 мл), и р.с. перемешивали в течение 2 ч при 120°C. Растворитель затем выпаривали, а смесь охлаждали на ледяной бане, и осторожно по каплям добавляли NH4OH до тех пор, пока она не достигла основного pH. Как только добавление завершали, образованный осадок отфильтровывали, промывали с помощью H2O и затем промывали несколько раз с помощью DCM. Органический растворитель высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Неочищенный продукт очищали с помощью хроматографии на открытых колонках (диоксид кремния, DCM в гептане от 20/80 до 80/20), желаемые фракции собирали и концентрировали in vacuo с получением смеси промежуточных продуктов (I-2a) и (I-2b) в виде белого твердого вещества (12 г, 69%). C9H6BrClN2, LCMS: Rt 2,95 (совместное элюирование двух пиков), масса/заряд 257 [M+H]+ (способ 8).
Промежуточный продукт 3 (I-3)
К перемешанному раствору метилового сложного эфира 5-гидроксиникотиновой кислоты (0,8 г, 5,22 ммоль) и ди-трет-бутилазадикарбоксилата (1,8 г, 7,83 ммоль) в THF (6 мл) добавляли порциями трифенилфосфин (2,05 г, 7,83 ммоль) при к.т. Смесь перемешивали при этой температуре в течение 5 мин, а затем добавляли BuOH (2 мл), и перемешивание продолжали при к.т. в течение 30 мин. Затем растворитель выпаривали, и неочищенное соединение очищали с помощью хроматографии (диоксид кремния, EtOAc в гептане от 0/100 до 20/80), желаемые фракции собирали и выпаривали in vacuo с получением промежуточного продукта I-3 в виде бесцветного масла (0,55 г, 50,3%). C11H15NO3, LCMS: Rt 2,71, масса/заряд 210 [M+H]+ (способ 5).
Промежуточный продукт 4 (I-4)
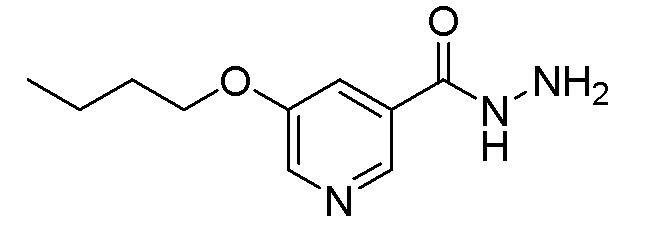
Гидрат гидразина (60% в H2O, 0,216 мл, 2,86 ммоль) по каплям добавляли к перемешанному раствору промежуточного продукта I-3 (0,5 г, 2,39 ммоль) в MeOH (4 мл) при к.т., и смесь перемешивали при этой температуре в течение 72 ч. Затем растворитель выпаривали in vacuo с получением промежуточного продукта I-4 в виде белого твердого вещества (0,48 г, 96%), которое применяли без дополнительной очистки на следующем этапе реакции. C10H15N3O2, LCMS: Rt 1,86, масса/заряд 210 [M+H]+ (способ 8).
Промежуточный продукт 5 (I-5)
8-бром-1-(5-бутоксипиридин-3-ил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин (I-5)
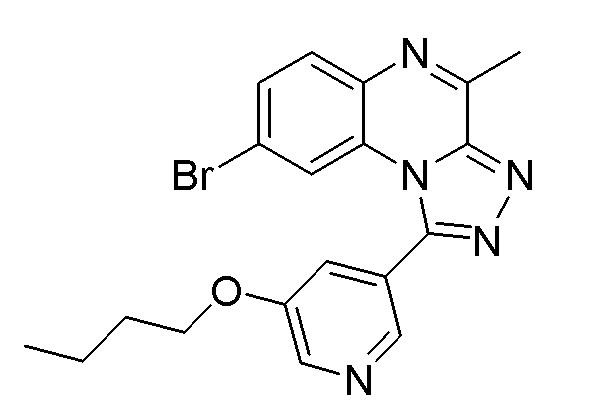
К раствору промежуточного продукта I-2a (5 г, 19,4 ммоль) в BuOH (40 мл) добавляли промежуточный продукт I-4 (4,06 г, 19,4 ммоль). Р.с. нагревали в герметичном реакторе при 160°C в течение 30 мин. Затем смесь выпаривали досуха, и остаток пропитывали с помощью EtOAc. Органический слой промывали с помощью NaHCO3 (насыщ. раств.), затем отделяли, высушивали (MgSO4), фильтровали, и растворитель выпаривали in vacuo. Неочищенную смесь очищали с помощью хроматографии (диоксид кремния, EtOAc в DCM от 5/95 до 25/75), желаемые фракции собирали и выпаривали, а полученное твердое соединение дополнительно растирали в порошок с гептаном с получением промежуточного продукта I-5 (3,3 г, 41%). 1H-ЯМР (300 МГц, DMSO-d6) δ м.д. 0,93 (т, J=7,4 Гц, 3 H), 1,45 (секстет, J=7,5 Гц, 2 H), 1,75 (квинтет, J=6,3 Гц, 2 H), 2,92 (с, 3 H), 4,13 (т, J=6,3 Гц, 2 H), 7,48 (д, J=1,6 Гц, 1 H), 7,82 (дд, J=8,7, 1,8 Гц, 1 H), 7,91 (уш.с, 1 H), 7,99 (д, J=8,7 Гц, 1 H), 8,55 (уш.с, 1 H), 8,65 (д, J=2,6 Гц, 1 H).
Промежуточный продукт 6 (I-6)
8-бром-1-(2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин (I-6a) и 7-бром-1-(2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин (I-6b)
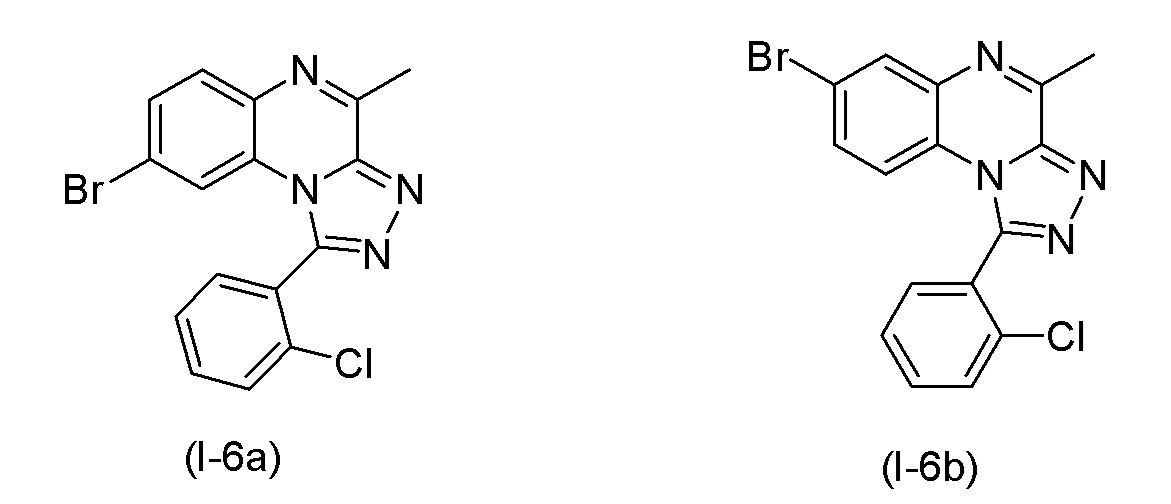
Смесь промежуточных продуктов I-2a и I-2b (0,3 г, 1,16 ммоль) и гидразида 2-хлорбензойной кислоты ([CAS 5814-05-1], 238,97 мг, 1,40 ммоль) растворяли в EtOH (5 мл). Реакционную смесь нагревали в микроволновой печи при 160°C в течение 15 мин, затем вновь нагревали при 170°C в течение 10 мин. Затем растворитель выпаривали досуха, и остаток пропитывали с помощью DCM. Органический слой промывали с помощью K2CO3 (насыщ. раств.), затем высушивали над Na2SO4 и выпаривали под вакуумом. Неочищенное соединение очищали с помощью хроматографии (SiO2, 30 г, CH2Cl2:EtOAc от 100:0 до 85:15) с получением промежуточного соединения I-6a (0,13 г, 29,8%) и промежуточного соединения I-6b (0,11 г, 25,2%), которые получали в виде чистых изомеров (оба в виде твердых соединений). 1H-ЯМР (500 МГц, CDCl3) δ м.д. 3,07 (с, 3 H), 7,32 (д, J=2,0 Гц, 1 H), 7,56-7,62 (м, 1 H), 7,65-7,72 (м, 4 H), 7,92 (д, J=8,7 Гц, 1 H) (для I-6a). 1H-ЯМР (500 МГц, CDCl3) δ м.д. 3,09 (с, 3 H), 7,10 (д, J=9,0 Гц, 1 H), 7,46 (дд, J=9,0, 2,3 Гц, 1 H), 7,54-7,58 (м, 1 H), 7,63-7,71 (м, 3 H), 8,22 (д, J=2,0 Гц, 1 H) (для I-6b).
Промежуточный продукт 7 (I-7)
1-(2-хлорфенил)-8-этенил-4-метил[1,2,4]триазоло[4,3-a]хиноксалин (I-7)
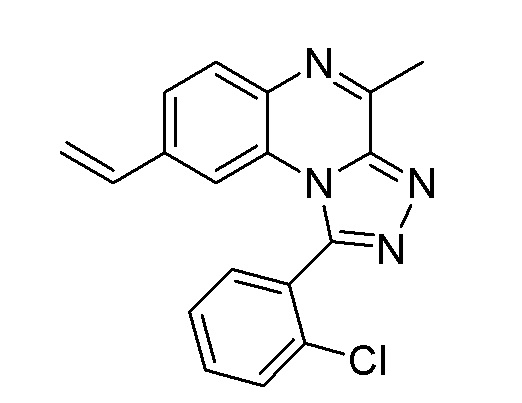
Смесь промежуточного соединения I-6a (0,65 г, 1,74), (тетракис)трифенилфосфинпалладия (0) (0,080 г, 0,07 ммоль) и LiCl (0,221 г, 5,21 ммоль) в толуоле (30 мл) обрабатывали трибутивинилоловом (0,661 г, 2,088 ммоль) и нагревали в герметизированной трубке при 120ºC в течение 1 ч (реакционную смесь разделяли на две порции). После охлаждения до к.т. смесь разделяли между EtOAc и H2O. Органическую фазу промывали солевым раствором, отделяли, высушивали (Na2SO4), фильтровали, и растворитель концентрировали in vacuo. Неочищенное соединение очищали с помощью хроматографии (диоксид кремния, EtOAc в DCM от 10/90 до 50/50) с получением светло-желтого твердого вещества, которое дополнительно промывали с помощью DIPE/диэтилового эфира с получением промежуточного соединения I-7 в виде белого продукта (0,52 г, 93,1%). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 3,08 (с, 3 H), 5,25 (д, J=10,9 Гц, 1 H), 5,43 (д, J=17,6 Гц, 1 H), 6,53 (дд, J=17,5, 11,0 Гц, 1 H), 7,24 (д, J=1,6 Гц, 1 H), 7,54-7,62 (м, 2 H), 7,64-7,74 (м, 3 H), 7,99 (д, J=8,3 Гц, 1 H).
Промежуточный продукт 8 (I-8)
К смеси промежуточного соединения I-7 (3,3 г, 10,29 ммоль) в 1,4-диоксане (110 мл) добавляли тетраоксид осмия (2,5% в трет-BuOH, 5,33 мл, 0,411 ммоль), а затем периодат натрия (6,6 г, 30,86 ммоль) в H2O (30 мл). Смесь перемешивали при к.т. в течение 2 ч. Органический растворитель выпаривали, неочищенную смесь разбавляли с помощью дополнительного количества H2O и экстрагировали с помощью DCM. Органический слой высушивали (Na2SO4), фильтровали, и растворитель концентрировали in vacuo. Неочищенный продукт очищали с помощью хроматографии (диоксид кремния, EtOAc в DCM от 30/70 до 70/30), желаемые фракции собирали и концентрировали in vacuo. Полученное твердое вещество промывали диэтиловым эфиром с получением промежуточного продукта I-8 (2,5 г, 75%) в виде бледно-желтого твердого вещества. C17H11ClN4O, LCMS: 1,78, масса/заряд 323 [M+H]+ (способ 3).
Промежуточный продукт 9 (I-9)

Гидрид натрия (60% в минеральном масле, 0,16 г, 4,02 ммоль) добавляли при к.т. к перемешанному раствору метил-2-хлор-5-гидроксибензоата [(C.A.S. 247092-10-0), 0,5 г, 2,68 ммоль], растворенного в THF (4 мл). Смесь перемешивали при данной температуре в течение 15 мин, и затем добавляли бромбутан (0,575 мл, 5,36 ммоль). Продолжали перемешивание при той же температуре в течение ночи, и затем р.с. нагревали при 120°C в течение 40 мин под действием микроволнового излучения. Затем смесь гасили с помощью H2O и экстрагировали с помощью EtOAc, органический слой отделяли, сушили (Na2SO4), фильтровали и концентрировали in vacuo с получением промежуточного продукта I-9 (0,25 г, 38,4%) в виде оранжевого масла, которое применяли без дополнительной очистки на следующем этапе реакции. C12H15ClO3, GCMS: 5,78, масса/заряд 242 [M+] (способ 1).
Промежуточный продукт 10 (I-10)
Гидрат гидразина (65% в H2O, 0,118 г, 1,54 ммоль) по каплям добавляли к перемешанному раствору промежуточного продукта I-9 (0,25 г, 1,03 ммоль) в EtOH (2 мл) при к.т., и смесь перемешивали при 120ºC в течение 20 мин под действием микроволнового излучения. Затем растворитель выпаривали под вакуумом с получением промежуточного продукта I-10 с приблизительно 70% чистотой (0,32 г, 89,5%) в виде белого твердого вещества, которое применяли без дополнительной очистки на следующем этапе реакции. C11H15ClN2O2, LCMS: Rt 2,34, масса/заряд 243 [M+H]+ (способ 8).
Промежуточные продукты 11-a и 11-b ((I-11a) и (I-11b))
8-бром-1-(5-бутокси-2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин (I-11a) и 7-бром-1-(5-бутокси-2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин (I-11b)
Промежуточные соединения I-11a и I-11b синтезировали согласно той же методике, что описана для синтеза промежуточных продуктов I-6a и I-6b. Начиная со смеси промежуточных продуктов I-2a и I-2b (0,2 г, 0,77 ммоль) и промежуточного продукта I-10, получали промежуточное соединение I-11a (0,05 г, 14,4%) и промежуточное соединение I-11b (0,075 г, 21,6%) в виде чистых изомеров (оба в виде грязно-белых твердых веществ). 1H-ЯМР (500 МГц, CDCl3) δ м.д. 0,98 (т, J=7,4 Гц, 3 H), 1,50 (секстет, J=7,5 Гц, 2 H), 1,76-1,84 (м, 2 H), 3,06 (с, 3 H), 3,93-4,10 (м, 2 H), 7,16-7,21 (м, 2 H), 7,44 (д, J=1,7 Гц, 1 H), 7,50-7,58 (м, 1 H), 7,68 (дд, J=8,7, 2,0 Гц, 1 H), 7,91 (д, J=8,7 Гц, 1 H) (для I-11a). 1H-ЯМР (500 МГц, CDCl3) δ м.д. 0,97 (т, J=7,4 Гц, 3 H), 1,49 (секстет, J=7,5 Гц, 2 H), 1,74-1,84 (м, 2 H), 3,08 (с, 3 H), 3,93-4,08 (м, 2 H), 7,14-7,21 (м, 3 H), 7,45-7,54 (м, 2 H), 8,22 (д, J=2,0 Гц, 1 H) (для I-11b).
Промежуточный продукт 12 (I-12)
К раствору морфолина (0,876 мл, 9,96 ммоль) в CH3CN (12 мл) добавляли (бромметил)трифторборат калия (1 г, 4,97 ммоль), и затем р.с. нагревали при 80°C в течение 30 мин. Затем растворитель выпаривали под вакуумом, и неочищенный материал повторно растворяли в растворе KHCO3 (0,5 г, 4,97 ммоль) в сухом ацетоне (16 мл). Смесь дополнительно перемешивали при к.т. в течение 20 мин. Затем нерастворимые соли отфильтровывали, а растворитель концентрировали вновь. Неочищенный материал напоследок очищали посредством его растворения в минимальном количестве сухого ацетона и его осаждения с помощью диэтилового эфира с получением промежуточного продукта I-12 в виде чистого продукта (0,66 г, 64%).
Промежуточные продукты 13-a и 13-b ((I-13a) и (I-13b))
Гидрат гидразина (60% в H2O, 0,52 мл, 9,7 ммоль) добавляли к смеси промежуточного продукта (I-2a) и промежуточного продукта (I-2b) (1 г, 3,88 ммоль) в MeOH (15 мл) при к.т. Затем р.с. нагревали при 50°C в течение 30 мин, после чего ее разбавляли с помощью H2O (5 мл) и экстрагировали с помощью DCM (20 мл). Органические слои разделяли, высушивали (MgSO4), фильтровали и концентрировали in vacuo с получением смеси промежуточных продуктов (I-13a) и (I-13b) (0,92 г, 96%), которую применяли без дополнительной очистки на следующем этапе реакции. C9H9BrN4, LCMS: 4,29 (совместное элюирование двух пиков), масса/заряд 253 [M+H]+ (способ 7).
Промежуточные продукты 14-a и 14-b ((I-14a) и (I-14b))
2-Хлор-6-фторбензойную кислоту (0,698 г, 4 ммоль) в DMF (20 мл) и DIPEA (1,072 мл, 6,22 ммоль) обрабатывали с помощью HBTU (1,52 г, 4 ммоль), и р.с. перемешивали в течение 15 мин при к.т. Затем добавляли смесь промежуточных продуктов (I-13a) и (I-13b) (0,9 г, 3,56 ммоль) в DMF (20 мл), и перемешивание продолжали в течение дополнительных 16 ч при той же температуре. Затем р.с. выливали на лед/H2O (0,5 л), и полученное таким образом твердое вещество собирали путем фильтрации. Затем твердое вещество разбавляли с помощью DCM (0,1 л) и обрабатывали с помощью 1 M водн. раствора NaOH (20 мл). Органические слои разделяли, промывали с помощью 1 M HCl (20 мл), затем 1 M NaOH (20 мл), высушивали (MgSO4), фильтровали, и растворитель концентрировали in vacuo. Неочищенную смесь очищали с помощью колоночной хроматографии (диоксид кремния; MeOH в DCM от 0:100 до 5:95) с получением грязно-белого твердого вещества, которое перекристаллизовывали из гептана/EtOAc (~15 мл/~5 мл) с получением в конечном счете смеси промежуточных продуктов (I-14a) и (I-14b) в виде грязно-белого твердого вещества (0,75 г, 51%). C16H11BrClFN4O, LCMS: 5,18 (совместное элюирование двух пиков), масса/заряд 409 [M+H]+ (способ 7).
Промежуточные продукты 15-a и 15-b ((I-15a) и (I-15b))
8-бром-1-(2-хлор-6-фторфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин (I-15)
Смесь промежуточных продуктов (I-14a) и (I-14b) (1 г, 2,44 ммоль) в DCE (20 мл) обрабатывали с помощью POCl3 (0,6 мл, 6,5 ммоль), и р.с. нагревали при 70°C в течение 16 ч. Затем добавляли дополнительный POCl3 (0,6 мл, 6,5 ммоль), и смесь нагревали при такой же температуре, что и ранее, в течение дополнительных 5 ч. По истечении этого времени вновь добавляли дополнительное количество POCl3 (1,2 мл, 13 ммоль), и смесь нагревали как раньше в течение дополнительных 16 ч. Р.с. охлаждали и выливали на лед/водн. NH4OH (150 мл/150 мл), а слои разделяли. Органическую фазу высушивали (MgSO4), фильтровали и концентрировали in vacuo. Неочищенное соединение очищали с помощью хроматографии (диоксид кремния; MeOH в DCM от 0/100 до 2/98) с получением смеси промежуточного продукта (I-15a) вместе с его региоизомером (I-15b) (0,7 г, 75%).
Порцию смеси региоизомеров разделяли с помощью колоночной хроматографии (диоксид кремния, EtOAc в CH2Cl2, от 0/100 до 25/75) с получением промежуточного продукта (I-15a) в виде чистого изомера. C16H9BrClFN4, LCMS: 2,58, масса/заряд 391 [M+H]+ (способ 3).
Промежуточный продукт 16 (I-16)
1-(2-хлор-6-фторфенил)-8-этенил-4-метил[1,2,4]триазоло[4,3-a]хиноксалин (I-16)
Трибутилвинилолово (0,18 мл, 0,61 ммоль) добавляли к перемешанному раствору промежуточного продукта (I-15a) (0,2 г, 0,511 ммоль), LiCl (0,065 г, 1,53 ммоль) и (тетракис)трифенилфосфинпалладия (0) (0,023 г, 0,02 ммоль) в толуоле (7 мл). Смесь нагревали при 120°C в течение 1,5 ч. После охлаждения до к.т. р.с. разделяли между EtOAc и H2O. Органический слой промывали солевым раствором, отделяли, высушивали (Na2SO4) и концентрировали in vacuo. Неочищенный продукт очищали с помощью хроматографии (диоксид кремния, EtOAc в DCM от 10/90 до 50/50), желаемые фракции собирали и концентрировали in vacuo с получением промежуточного продукта (I-16) в виде бледно-желтого твердого вещества (0,14 г, 81%). C18H12ClFN4, LCMS: 2,46, масса/заряд 339 [M+H]+ (способ 3).
Промежуточный продукт 17 (I-17)
К раствору промежуточного продукта (I-16) (0,14 г, 0,413 ммоль) в 1,4-диоксане (5 мл) добавляли тетраоксид осмия (2,5% в трет-BuOH, 0,214 мл, 0,016 ммоль), а затем периодат натрия (0,265 г, 1,24 ммоль) в H2O (3 мл). Смесь перемешивали при к.т. в течение 2,5 ч. Органический растворитель выпаривали, неочищенную смесь разбавляли с помощью дополнительного количества H2O и экстрагировали с помощью DCM. Органический слой отделяли, высушивали (Na2SO4) и концентрировали in vacuo. Неочищенный продукт очищали с помощью хроматографии (диоксид кремния, EtOAc в DCM от 30/70 до 70/30), желаемые фракции собирали и концентрировали in vacuo с получением промежуточного продукта (I-17) в виде бледно-желтого твердого вещества (0,1 г, 71%). C17H10ClFN4O, LCMS: 1,82, масса/заряд 341 [M+H]+ (способ 3).
Промежуточный продукт 18 (I-18)
1-(5-бутоксипиридин-3-ил)-8-этенил-4-метил[1,2,4]триазоло[4,3-a]хиноксалин (I-18)

К перемешанному раствору I-5 (2,35 г, 5,7 ммоль) в толуоле (17 мл) добавляли LiCl (0,719 г, 17,1 ммоль), (тетракис)трифенилфосфинпалладий (0) (0,263 г, 0,23 ммоль) и трибутилвинилолово (1,84 мл, 6,27 ммоль), и смесь нагревали при 120°C в течение 2 ч. После охлаждения до к.т. р.с. разделяли между EtOAc и H2O. Органический слой промывали солевым раствором, отделяли, высушивали (Na2SO4) и концентрировали in vacuo. Неочищенный продукт очищали с помощью хроматографии (диоксид кремния, EtOAc в гептане от 0/100 до 100/0), желаемые фракции собирали и концентрировали in vacuo с получением промежуточного продукта 18 (I-18) (1,9 г, 92%).
Промежуточный продукт 19 (I-19)
К раствору промежуточного продукта (I-18) (0,159 г, 0,44 ммоль) в 1,4-диоксане (4,4 мл) добавляли тетраоксид осмия (2,5% в трет-BuOH, 0,23 мл, 0,018 ммоль), а затем периодат натрия (0,282 г, 1,32 ммоль) в H2O (1,32 мл). Смесь перемешивали при к.т. в течение 2 ч. Органический растворитель выпаривали, неочищенную смесь разбавляли с помощью дополнительного количества H2O и экстрагировали с помощью DCM. Органический слой отделяли, высушивали (Na2SO4) и концентрировали in vacuo. Неочищенный продукт очищали с помощью хроматографии (диоксид кремния, EtOAc в DCM от 0/1 до 1/1), желаемые фракции собирали и концентрировали in vacuo с получением промежуточного продукта (I-19) (0,108 г, 68%).
Промежуточный продукт 20 (I-20)
К раствору морфолина (0,876 мл, 9,96 ммоль) в CH3CN (12 мл) добавляли (бромметил)трифторборат калия (1 г, 4,97 ммоль), и затем р.с. нагревали при 80°C в течение 30 мин. Затем растворитель выпаривали под вакуумом, и неочищенный материал повторно растворяли в растворе KHCO3 (0,5 г, 4,97 ммоль) в сухом ацетоне (16 мл). Смесь дополнительно перемешивали при к.т. в течение 20 мин. Затем нерастворимые соли отфильтровывали, а растворитель концентрировали вновь. Неочищенный материал напоследок очищали посредством его растворения в минимальном количестве сухого ацетона и его осаждения с помощью диэтилового эфира с получением промежуточного продукта I-20 в виде чистого продукта (0,66 г, 64%).
Промежуточные продукты 21a и 21b ((I-21a) и (I-21b))
К смеси 2-хлор-6-нитробензойной кислоты (0,473 г, 2,34 ммоль) и оксалилхлорида (0,201 мл, 2,34 ммоль) в дихлорметане (5 мл) добавляли DMF (0,182 мл, 2,34 ммоль). Смесь перемешивали в течение 15 мин при к.т., затем этот раствор по каплям добавляли к перемешанной смеси триэтиламина (0,544 мл, 1,95 ммоль) и промежуточных соединений I-13a и I-13b (0,495 г, 1,95 ммоль), растворенных в дихлорметане (5 мл), при 0°C. Затем смеси давали нагреться до к.т., и ее перемешивали в течение дополнительных 15 мин. Затем ее гасили с помощью NaHCO3 (насыщ. раств. в воде), органический слой быстро отделяли, и растворитель выпаривали. Остаток обрабатывали этиловым эфиром с получением смеси (I-21a) и (I-21b) в виде коричневого твердого вещества (0,814 г, 95%), которое применяли без дополнительной очистки на следующем этапе реакции.
Промежуточные продукты 22a и 22b ((I-22a) и (I-22b))
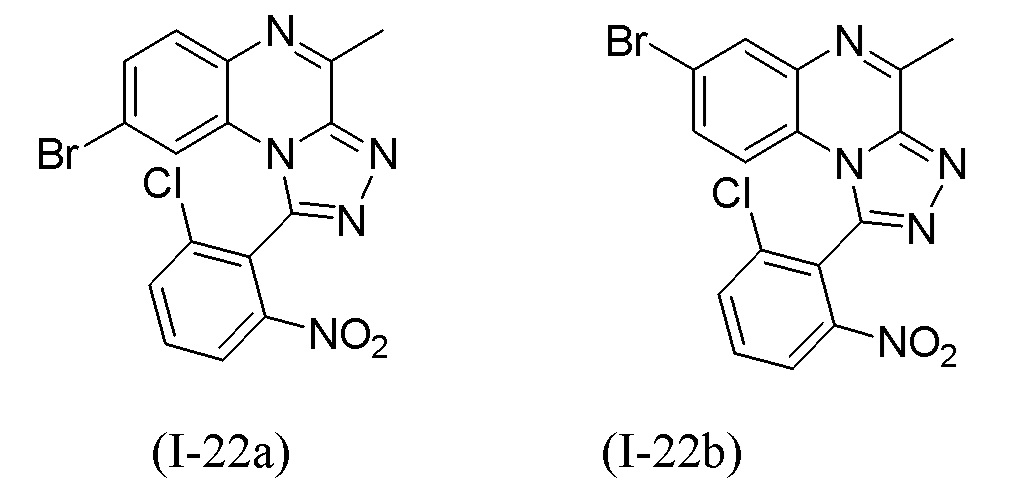
Смесь промежуточных соединений I-21a и I-21b (0,402 г, 0,92 ммоль) в DCE (5 мл) обрабатывали с помощью POCl3 (0,343 мл, 3,68 ммоль), и р.с. нагревали при 160°C в течение 10 мин под действием микроволнового излучения. Затем растворитель выпаривали, и неочищенное соединение очищали с помощью хроматографии (диоксид кремния, EtOAc в гептанах от 20/80 до 60/40). Желаемые фракции собирали, растворитель выпаривали под вакуумом с получением I-22a (0,053 г, 13,7%) и I-22b (0,112 г, 29%) в виде чистых изомеров.
Промежуточный продукт 23 (I-23)
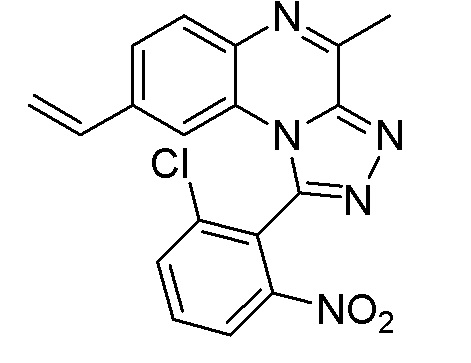
Промежуточный продукт I-23 синтезировали согласно подходу, аналогичному описанному для соединения I-7. Начиная с I-22a (0,053 г, 0,127 ммоль), получали промежуточный продукт I-23 в виде бледно-желтого твердого вещества (0,046 г, колич.).
Промежуточный продукт 24 (I-24)
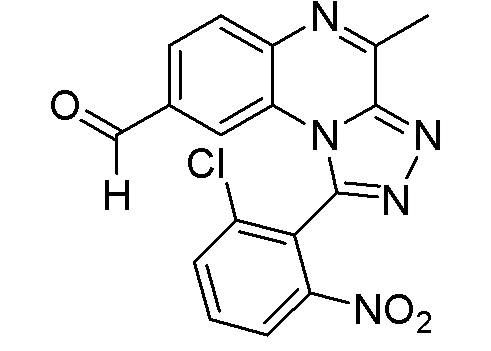
Промежуточный продукт I-24 синтезировали согласно подходу, аналогичному описанному для промежуточного продукта I-8. Начиная с I-23 (0,046 г, 0,127 ммоль), получали промежуточный продукт I-24 в виде бледно-желтого твердого вещества (0,031 г, 66,5%).
Промежуточный продукт 25 (I-25)
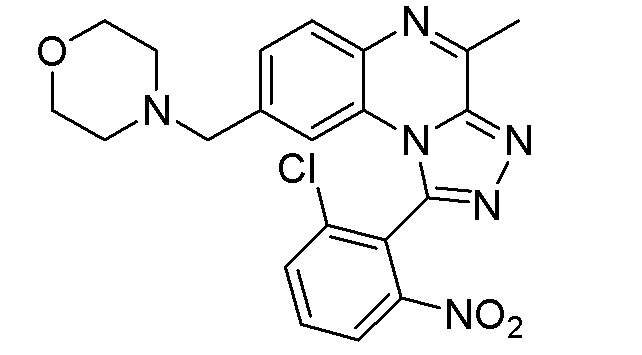
Промежуточный продукт I-25 синтезировали согласно подходу, аналогичному описанному для соединения B-3. Начиная с I-24 (0,035 г, 0,095 ммоль), получали промежуточное соединение I-25 (0,011 г, 27%).
B. Синтез конечных соединений
Примеры 1a и 1b
1-(5-Бутоксипиридин-3-ил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина гидрохлорид (B-1a) и оксалат (B-1b)
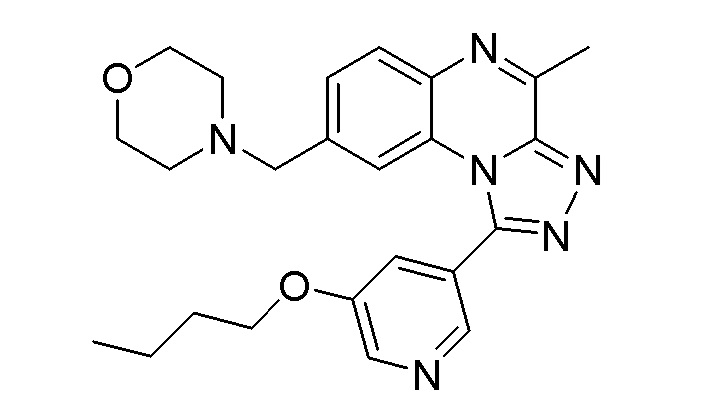
.2 HCl (B-1a) или.x C2H2O4 (B-1b)
Образование B-1a
К раствору промежуточного соединения I-5 (7,5 г, 18,19 ммоль) в THF/H2O (10:1, 180 мл) добавляли Pd(AcO)2 (0,12 г, 0,54 ммоль), XantPhos (0,52 г, 1,09 ммоль), Cs2CO3 (23,88 г, 72,76 ммоль) и промежуточное соединение I-20 (4,51 г, 21,82 ммоль). Р.с. заключали в герметизированную трубку и перемешивали при к.т. в течение 10 мин и затем при 114°C в течение 45 мин. Затем неочищенную смесь разбавляли с помощью EtOAc и H2O, органический слой отделяли, высушивали (MgSO4), фильтровали, и растворитель концентрировали in vacuo. Неочищенную смесь очищали с помощью хроматографии (диоксид кремния, MeOH в DCM от 0/100 до 2/98), желаемые фракции собирали, и растворитель концентрировали in vacuo с получением бледно-розового масла. Затем этот материал растворяли в EtOAc (50 мл) и обрабатывали путем добавления по каплям HCl (4 M в диоксане, 1,2 экв. и 3,55 мл). Смесь перемешивали при комнатной температуре в течение 30 мин и затем выпаривали под вакуумом. Взвесь обрабатывали с помощью 120 мл DIPE и вновь перемешивали в течение дополнительных 40 мин. Образованный осадок отфильтровывали, промывали с помощью DIPE, высушивали над вакуумом с получением конечного соединения B-1a в виде соли-гидрохлорида (5,2 г, 61%). 1H-ЯМР (400 МГц, DMSO-d6) δ м.д. 0,94 (т, J=7,5 Гц, 3 H), 1,46 (секстет, J=7,4 Гц, 2 H), 1,69-1,82 (м, 2 H), 2,88-3,04 (м, 2 H), 2,96 (с, 3 H), 3,19 (ушир.д, J=12,5 Гц, 2 H), 3,75-3,98 (м, 4 H), 4,18 (т, J=6,5 Гц, 2 H), 4,34 (уш.с, 2 H), 7,68 (д, J=1,2 Гц, 1 H), 8,00 (дд, J=8,5, 1,6 Гц, 1 H), 8,09 (дд, J=2,4, 1,6 Гц, 1 H), 8,13 (д, J=8,1 Гц, 1 H), 8,70 (д, J=1,6 Гц, 1 H), 8,75 (д, J=2,8 Гц, 1 H), 12,03 (уш.с, 1 H).
Образование B-1b
К перемешанному раствору промежуточного продукта I-19 (0,108 г, 0,3 ммоль), морфолина (0,03 мл, 0,33 ммоль) и уксусной кислоты (0,017 мл, 0,3 ммоль) в DCE (5 мл) добавляли триацетоксиборгидрид натрия (0,076 г, 0,3 ммоль), и смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и этилацетат, а органическую фазу отделяли, высушивали (MgSO4), фильтровали и концентрировали in vacuo. Неочищенную смесь очищали с помощью хроматографии (диоксид кремния, MeOH в DCM от 0/100 до 10/90), желаемые фракции собирали и концентрировали in vacuo. Продукт растворяли в диоксане (2 мл), добавляли щавелевую кислоту (0,024 г, 0,27 ммоль), смесь перемешивали в течение 45 мин, концентрировали in vacuo и перекристаллизовывали из диэтилового эфира с получением конечного соединения B-1b в виде соли-оксалата (0,084 г, 54%).
(Спектр свободного основания) 1H-ЯМР (300 МГц, DMSO-d6) δ м.д. 0,93 (т, J=7,4 Гц, 3 H), 1,45 (секстет, J=7,4 Гц, 2 H), 1,67-1,82 (м, 2 H), 2,37 (уш.с, 4 H), 2,93 (с, 3 H), 3,50 (уш.с, 4 H), 3,60 (с, 2 H), 4,11 (т, J=6,5 Гц, 2 H), 7,54 (с, 1 H), 7,55 (д, J=8,8 Гц, 1 H), 7,88 (уш.с, 1 H), 8,01 (д, J=8,1 Гц, 1 H), 8,54 (с, 1 H), 8,66 (д, J=2,5 Гц, 1 H).
Пример 2
1-(5-Бутокси-2-хлорфенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина гидрохлорид (B-2)
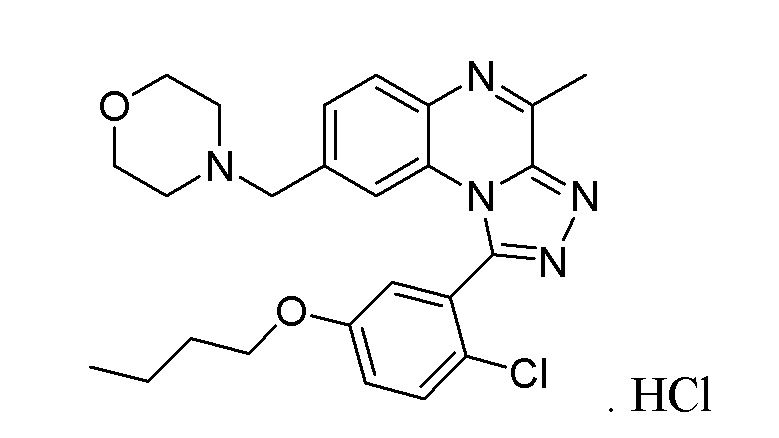
B-2 синтезировали так, как было ранее описано для синтеза конечного соединения B-1a. Начиная с I-11a (0,2 г, 0,45 ммоль) и промежуточного соединения I-20, получали конечное соединение B-2 (0,03 г, 14%). 1H-ЯМР (300 МГц, DMSO-d6) δ м.д. 0,93 (т, J=7,4 Гц, 3 H), 1,44 (секстет, J=7,3 Гц, 2 H), 1,73 (квинтет, J=6,9 Гц, 2 H), 2,93 (уш.с, 1 H), 2,97 (с, 3 H), 3,19 (уш.с, 1 H), 3,77 (уш.с, 2 H), 3,92 (уш.с, 2 H), 3,98-4,14 (м, 2 H), 4,31 (уш.с, 2 H), 5,76 (с, 2 H), 7,25 (уш.с, 1 H), 7,33-7,50 (м, 2 H), 7,73 (д, J=8,8 Гц, 1 H), 7,96 (уш.с, 1 H), 8,16 (д, J=8,1 Гц, 1 H), 11,31 (уш.с, 1 H).
Пример 3
1-(2-Хлорфенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалин (B-3)
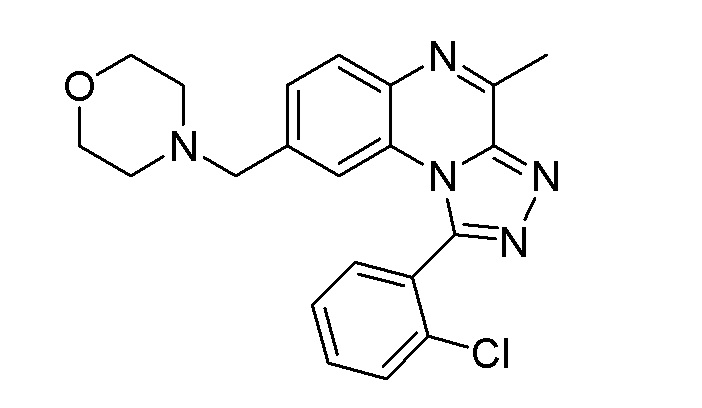
Морфолин (1,37 мл, 15,67 ммоль) добавляли к перемешанному раствору промежуточного продукта I-8 (2,3 г, 7,12 ммоль), растворенного в DCE (50 мл), и смесь нагревали при 80°C в течение 15 мин под действием микроволнового излучения (реакционную смесь разделяли на три порции). Затем порциями добавляли триацетоксиборгидрид натрия (1,81 г, 8,55 ммоль), и смесь вновь нагревали при тех же условиях, как раньше, в течение 20 мин. Затем смесь гасили с помощью H2O и экстрагировали с помощью DCM. Органический слой отделяли, высушивали (Na2SO4), фильтровали, и растворитель выпаривали in vacuo. Неочищенное соединение очищали с помощью хроматографии (диоксид кремния, MeOH в EtOAc от 2/98 до 10/90), желаемые фракции собирали, и растворитель выпаривали с получением конечного соединения B-3 в виде бледно-желтого твердого вещества, которое дополнительно промывали с помощью диэтилового эфира/DIPE (1,6 г, 57%). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,24-2,41 (м, 4 H), 3,08 (с, 3 H), 3,42 (с, 2 H), 3,53-3,69 (м, 4 H), 7,37 (д, J=1,2 Гц, 1 H), 7,49 (дд, J=8,3, 1,6 Гц, 1 H), 7,54-7,62 (м, 1 H), 7,64-7,75 (м, 3 H), 7,99 (д, J=8,3 Гц, 1 H).
Пример 4
N-{[1-(2-хлорфенил)-4-метил[1,2,4]триазоло[4,3-a]хиноксалин-8-ил]метил}этанамин (B-4)
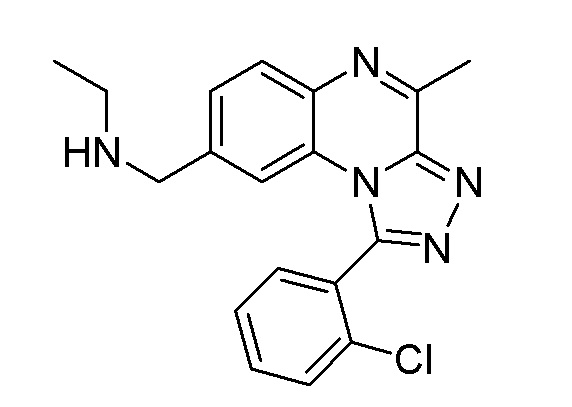
Промежуточный продукт I-8 (0,300 г, 0,93 ммоль), гидрохлорид этиламина (0,227 мл, 2,78 ммоль) и Et3N (0,388 мл, 2,78 ммоль) растворяли в DCE (11 мл). К этой смеси добавляли 300 мг MgSO4, и все перемешивали при к.т. в течение 1,3 ч. Твердое вещество отфильтровывали, а затем к фильтрату добавляли MeOH (3 мл) и после него NaBH4 (0,07 г, 1,85 ммоль), и раствор перемешивали при к.т. в течение дополнительных 15 мин. Р.с. гасили с помощью H2O и экстрагировали с помощью DCM. Органические слои разделяли, высушивали (MgSO4), фильтровали, и растворитель концентрировали in vacuo. Неочищенную смесь очищали с помощью хроматографии (диоксид кремния; MeOH в DCM от 0/100 до 10/90) с получением конечного соединения B-4 в виде твердого материала (0,186 г, 57%). 1H-ЯМР (500 МГц, CDCl3) δ м.д. 1,03 (т, J=7,1 Гц, 3 H), 2,45-2,57 (м, 2 H), 3,08 (с, 3 H), 3,69-3,79 (м, 2 H), 7,27 (уш.с, 1 H), 7,50 (д, J=8,4 Гц, 1 H), 7,53-7,59 (м, 1 H), 7,61-7,68 (м, 2 H), 7,70 (д, J=6,9 Гц, 1 H), 7,99 (д, J=8,1 Гц, 1 H).
Пример 5
1-(2-Хлор-6-фторфенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалин (B-5)
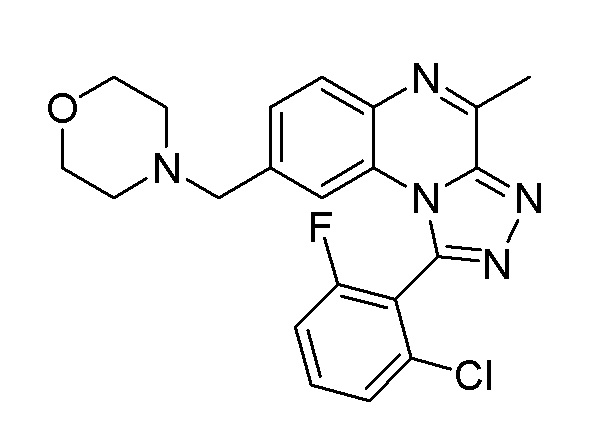
Морфолин (0,056 мл, 0,64 ммоль) добавляли к перемешанному раствору промежуточного продукта I-17 (0,1 г, 0,29 ммоль), растворенного в DCE (5 мл), и смесь нагревали при 120°C в течение 15 мин под действием микроволнового излучения. Затем порциями добавляли триацетоксиборгидрид натрия (0,075 г, 0,35 ммоль), и смесь вновь нагревали при 80°C в течение 20 мин под действием микроволнового излучения. Затем р.с. гасили с помощью H2O и экстрагировали с помощью DCM. Органический слой отделяли, высушивали (Na2SO4), фильтровали, и растворитель выпаривали in vacuo. Неочищенное соединение очищали с помощью хроматографии (диоксид кремния, MeOH в EtOAc от 2/98 до 10/90), желаемые фракции собирали, и растворитель выпаривали с получением конечного соединения B-5 в виде бледно-желтого твердого вещества, которое дополнительно промывали с помощью диэтилового эфира/DIPE (0,045 г, 37%). 1H-ЯМР (400 МГц, ХЛОРОФОРМ-d) δ м.д. 2,25-2,41 (м, 4 H), 3,09 (с, 3 H), 3,39-3,52 (м, 2 H), 3,54-3,68 (м, 4 H), 7,32 (т, J=8,3 Гц, 1 H), 7,41 (уш.с, 1 H), 7,47-7,51 (м, 1 H), 7,52 (д, J=8,3 Гц, 1 H), 7,68 (тд, J=8,3, 5,8 Гц, 1 H), 8,01 (д, J=8,3 Гц, 1 H).
Пример 6
1-(5-Бутоксипиридин-3-ил)-4-метил-8-(морфолин-4-ил-[3H]метил)[1,2,4]триазоло[4,3-a]хиноксалин ([3H]B-1a)
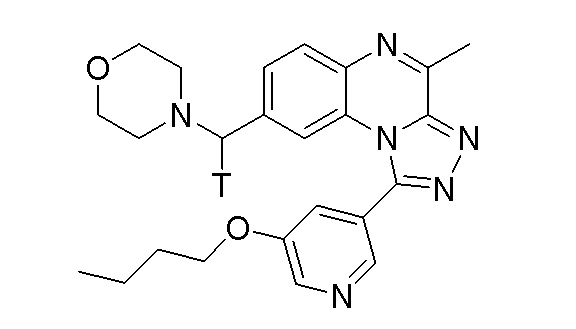
Промежуточное соединение I-19 (0,002 г, 5,53 мкмоль) растворяли в дихлорметане (0,1 мл) в сухом сосуде Wheaton. Морфолин (0,271 мл, 27,67 мкмоль) и тетра(изопропоксид) титана (0,82 мл, 27,67 мкмоль) добавляли под атмосферой аргона и перемешивали в течение ночи при комнатной температуре. Реакционную смесь переносили в ампулу для мечения тритием и присоединяли к трубопроводу с тритием (RC Tritec). Дихлорметан лиофилизировали и замещали сухим THF (0,2 мл). Смесь вновь лиофилизировали, и добавляли платину на угле (4 мг, 5%) вместе с сухим THF (0,2 мл). Реакционную смесь дегазировали (3×) и помещали в атмосферу трития (750 мбар при комнатной температуре) на 60 минут при комнатной температуре. Атмосферу трития удаляли, и летучие компоненты лиофилизировали в ампуле для отходов. Неочищенную смесь прополаскивали и лиофилизировали с помощью MeOH (3×0,15 мл), фильтровали через Acrodisk® и растворяли в этаноле (10 мл). Этот базовый раствор очищали с помощью препаративной HPLC, в результате чего получали 230 МБк с радиохимической чистотой >98% и удельной активностью 726 ГБк/ммоль.
Пример 7
Получение [18F]фторида и 1-(2-хлор-6-[18F]фторфенил)-4-метил-8-(морфолин-4-илметил)[1,2,4]триазоло[4,3-a]хиноксалина ([18F]B-5) путем радиохимического синтеза
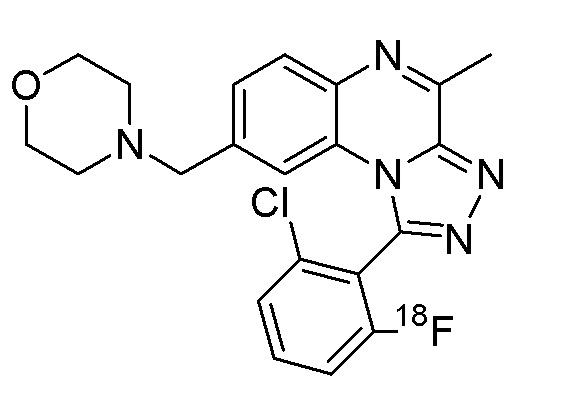
[18F]фторид ([18F]F-) получали с помощью реакции [18O(p,n)18F] посредством облучения 2 мл 97% обогащенной [18O]H2O (Rotem HYOX18, Rotem Industries, Беэр-Шева, Израиль) в ниобиевой мишени с применением протонов с энергией 18 МэВ из циклотрона Cyclone 18/9 (Ion Beam Applications, Лувен-ля-Нев, Бельгия). После облучения полученный [18F]F- отделяли от [18O]H2O с применением анионообменного картриджа SepPak™ Light Accell Plus QMA (Waters, форма CO32-). [18F]F- элюировали из картриджа с применением смеси 0,38 мл раствора, содержащего K2CO3 (0,00247 г) и Kryptofix 222 (0,00279 г), растворенных в H2O/MeCN (0,75 мл; 5:95 объем/объем), и 0,38 мл MeCN. Раствор выпаривали под потоком гелия при 80°C и 35 Ватт посредством применения микроволнового нагревания и дополнительно высушивали с помощью азеотропной перегонки с применением MeCN (1 мл) при температуре 80°C и мощности 35 Ватт в микроволновом резонаторе. Предшественник для мечения радиоактивным изотопом, I-25 (0,0013 г, 0,0029 ммоль), растворяли в безводном DMF (0,35 мл), этот раствор добавляли к высушенному комплексу [18F]F-/K2CO3/Kryptofix® 222, и проводили реакцию нуклеофильного замещения с применением микроволнового нагревания при 140°C и 50 Ватт в течение 6 мин. Затем неочищенную смесь разбавляли 0,05 M буфером NaOAc, pH 5,5 (0,6 мл), и вводили в систему для HPLC, состоящую из колонки XBridgeTM для полупрепаративной хроматографии (C18, 5 мкм, 4,6 мм × 150 мм; Waters), в которой производили элюирование смесью 0,05 M буфера NaOAc, pH 5,5, и EtOH (73:27 объем/объем) при скорости потока 1 мл/мин. УФ-детекцию элюата после HPLC осуществляли при 254 нм. Меченный радиоактивным изотопом продукт [18F]B-5 собирали через приблизительно 25 мин. Собранный пик, соответствующий [18F]B-5, затем разбавляли физиологическим раствором (Mini Plasco®, Braun, Мельзунген, Германия) с получением конечной концентрации EtOH <10%, и проводили стерильную фильтрацию раствора через мембранный фильтр на 0,22 мкм (Millex®-GV, Millipore). Чистоту радиоактивной метки анализировали с применением системы для HPLC, состоящей из колонки XBridgeTM (C18, 5 мкм, 4,6 мм × 150 мм; Waters), в которой производили элюирование смесью 0,05 M буфера NaOAc, pH 5,5, и EtOH (65:35 объем/объем) при скорости потока 1 мл/мин (Rt=7,5 мин). УФ-детекцию элюата после HPLC осуществляли при 254 нм. [18F]B-5 синтезировали с радиохимическим выходом 45% (относительно исходной радиоактивности [18F]F-, с поправкой на распад, n=6). Радиохимическая чистота, проанализированная с применением вышеописанной системы для аналитической HPLC, составляла >99%, и было обнаружено, что средняя удельная радиоактивность составляла 215 ГБк/мкмоль в EOS (n=6).
|
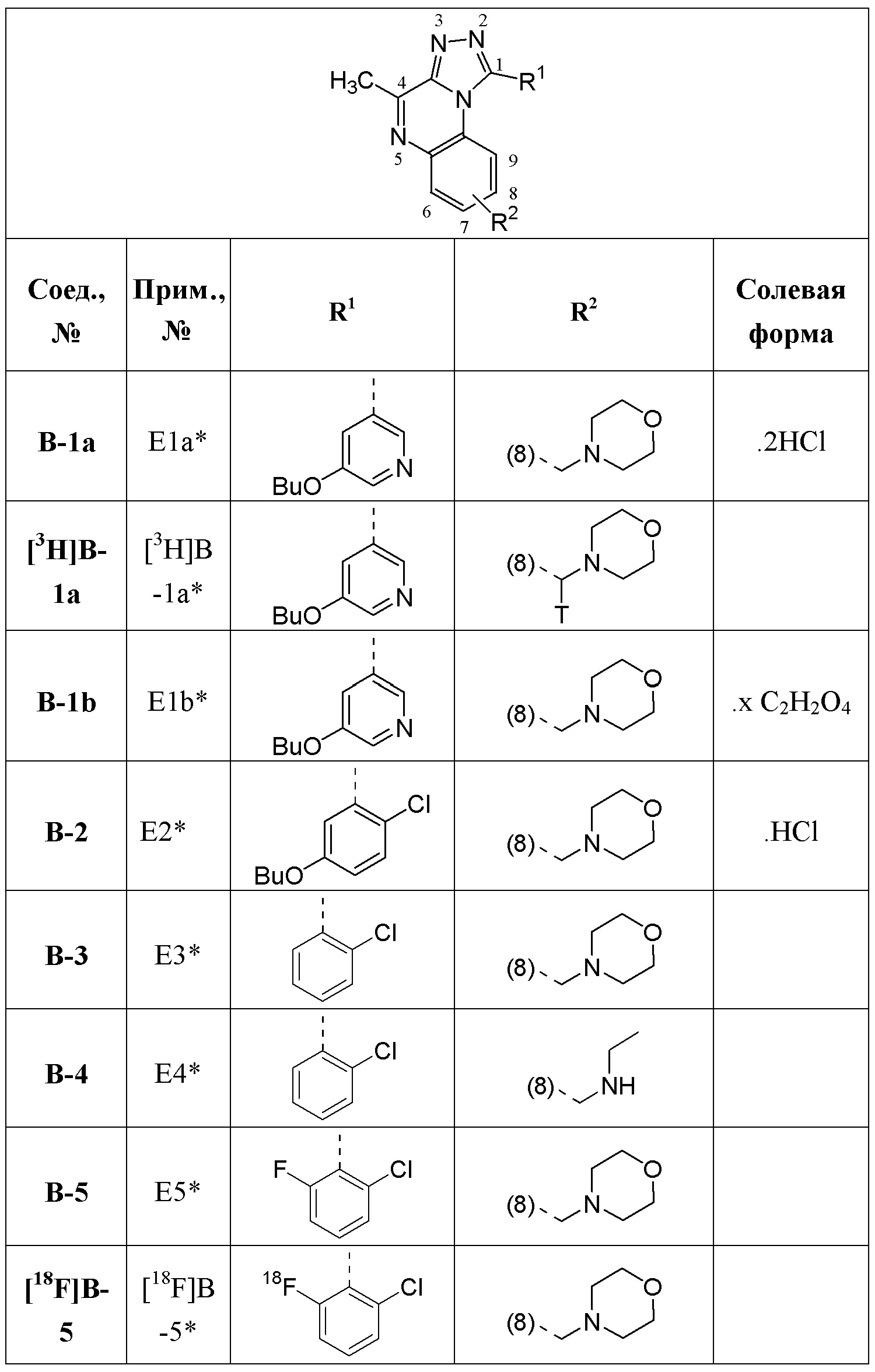
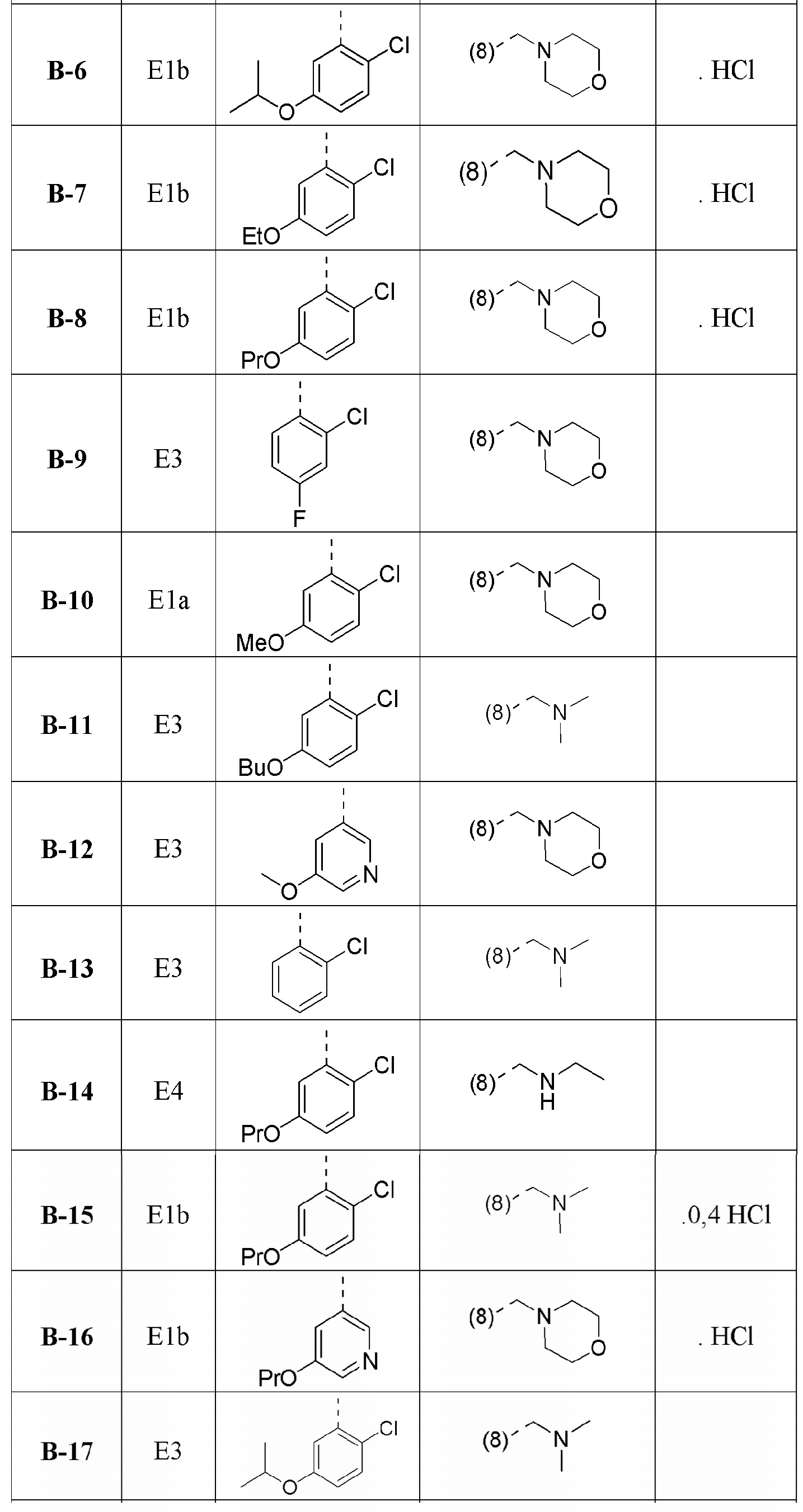
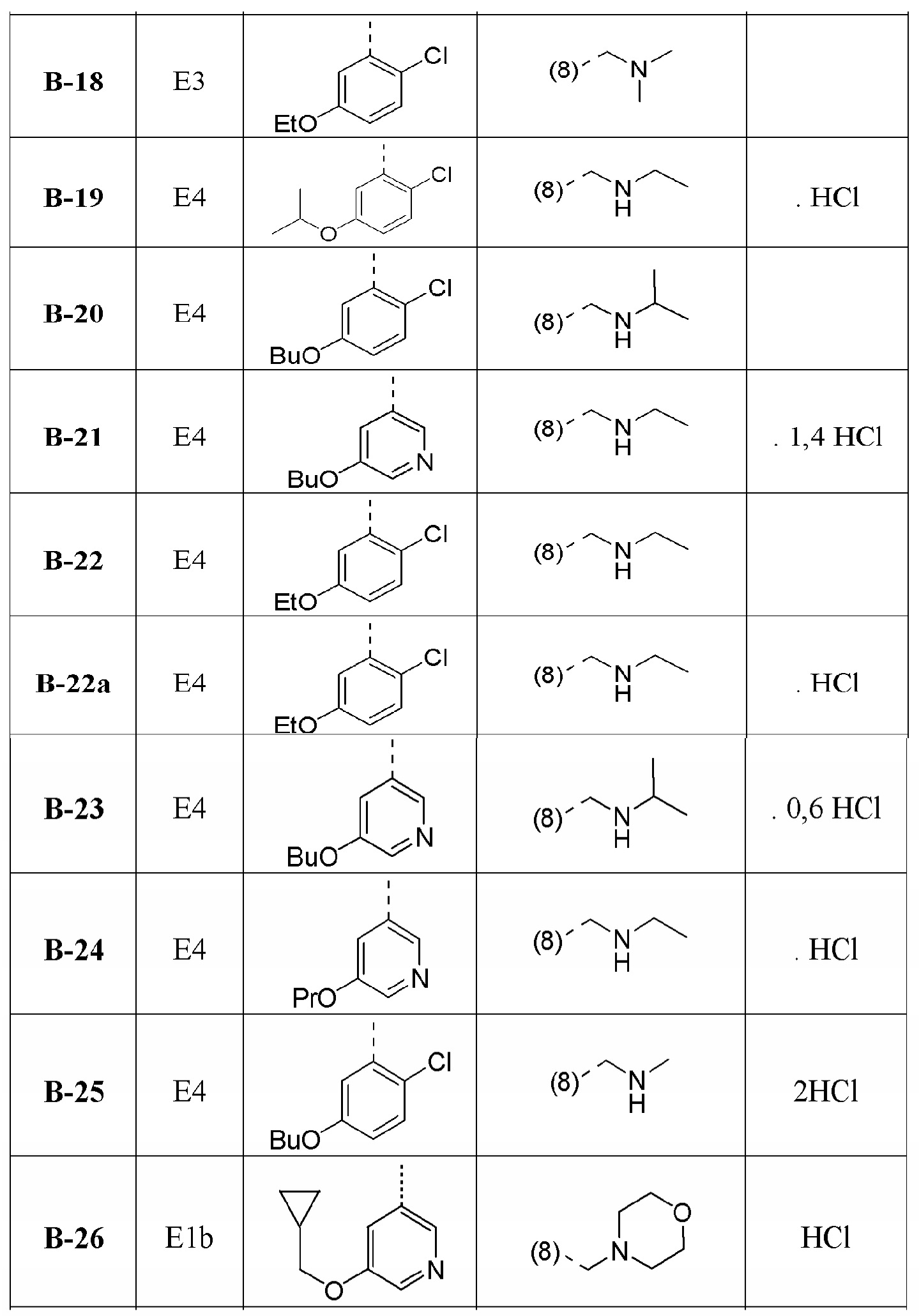
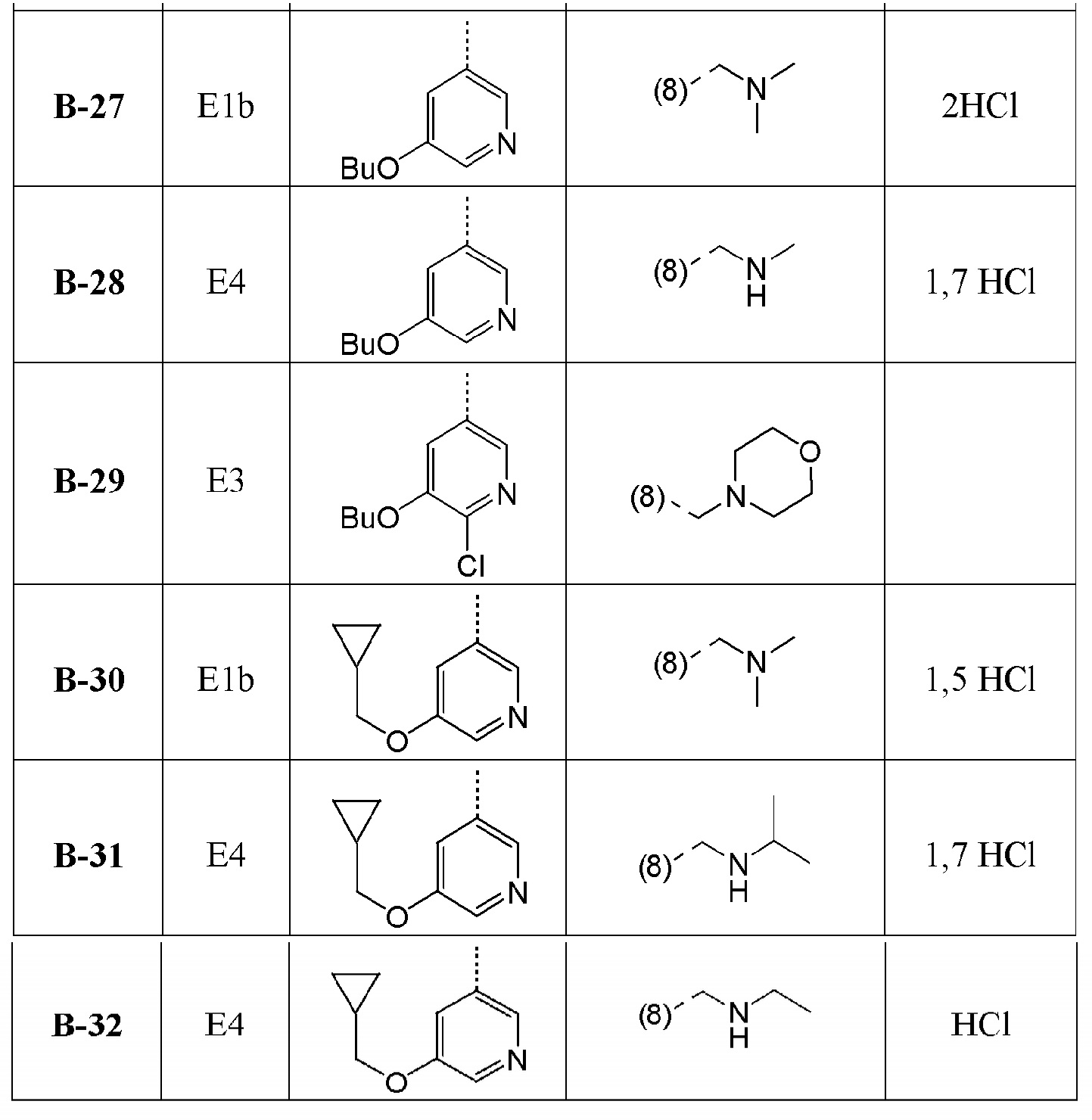
АНАЛИТИЧЕСКАЯ ЧАСТЬ
LCMS
Для исследования соединений по настоящему изобретению с помощью LC-MS применяли следующие способы.
Общая методика A
Измерения в ходе HPLC осуществляли с применением системы HP 1100 (Agilent Technologies), включающей насос (для четырехкомпонентных или двухкомпонентных смесей) с дегазатором, автодозатор, термостат колонок, детектор на диодной матрице (DAD) и колонку, указанные в соответствующих способах ниже. Поток из колонки разделяли для MS-спектрометра. MS-детектор был оснащен либо источником электрораспылительной ионизации, либо двухрежимным источником ионизации ESCI (электрораспылительная ионизация в сочетании с химической ионизацией при атмосферном давлении). В качестве газа-распылителя применяли азот. Температуру источника поддерживали при 140°C. Сбор и обработку данных проводили с помощью программного обеспечения MassLynx-Openlynx.
Общая методика B
Измерения в ходе UPLC (сверхэффективной жидкостной хроматографии) осуществляли с применением системы Acquity UPLC (Waters), включающей поддон для образцов, насос для двухкомпонентных смесей с дегазатором, термостат для четырех колонок, детектор на диодной матрице (DAD) и колонку, указанные в соответствующих способах ниже. Поток из колонки применяли без разделения для MS-детектора. MS-детектор был оснащен двухрежимным источником ионизации ESCI (электрораспылительная ионизация в сочетании с химической ионизацией при атмосферном давлении). В качестве газа-распылителя применяли азот. Температуру источника поддерживали при 140°C. Сбор и обработку данных проводили с помощью программного обеспечения MassLynx-Openlynx.
Общая методика C
Измерения в ходе LC осуществляли с применением системы Acquity UPLC (Waters), включающей бинарный насос, поддон для образцов, нагреватель колонки (установленный на 55°C), детектор на диодной матрице (DAD) и колонку, указанные в соответствующих способах ниже. Поток из колонки разделяли для MS-спектрометра. MS-детектор был оснащен источником электрораспылительной ионизации. Масс-спектры получали путем сканирования от 100 до 1000 за 0,18 секунды с применением времени выдержки 0,02 секунды. Напряжение на капиллярной игле составляло 3,5 кВ, и температуру источника поддерживали при 140°C. В качестве газа-распылителя применяли азот. Сбор и обработку данных проводили с помощью системы обработки данных MassLynx-Openlynx от Waters-Micromass.
Общая методика D
Измерения в ходе HPLC осуществляли с применением модуля Agilent 1100, включающего насос, детектор на диодной матрице (DAD) (Agilent 1200) (применяемая длина волны 254 нм), нагреватель колонки и колонку, указанные в соответствующих способах ниже. Поток из колонки разделяли для Agilent MSD серии G1956A. MS-детектор был оснащен API-ES (источником электрораспылительной ионизации при атмосферном давлении). Масс-спектры получали путем сканирования от 105 до 1400. Напряжение на капиллярной игле составляло 3000 В для режима положительной ионизации. Напряжение фрагментации составляло 70 В. Температуру сушильного газа поддерживали при 350°C при потоке 12 л/мин.
Способ 1
В дополнение к общей методике A: обращенно-фазовую HPLC проводили на колонке Sunfire-C18 (2,5 мкм, 2,1×30 мм) от Waters со скоростью потока 1,0 мл/мин. при 60°C. Применяемые условия градиента представляли собой: от 95% A (0,5 г/л раствор ацетата аммония + 5% ацетонитрил), 2,5% B (ацетонитрил), 2,5% C (метанол) до 50% B, 50% C за 6,5 минуты, удерживание до 7,0 минуты и уравновешивание до начальных условий от 7,3 минуты до 9,0 минуты. Объем вводимой пробы составлял 2 мкл. Масс-спектры высокого разрешения (на времяпролетном TOF-детекторе) получали путем сканирования от 100 до 750 за 0,5 секунды с применением времени выдержки 0,3 секунды. Напряжение на капиллярной игле составляло 2,5 кВ для режима положительной ионизации и 2,9 кВ для режима отрицательной ионизации. Напряжение на конусе составляло 20 В как для режима положительной, так и для режима отрицательной ионизации. Стандартным веществом, применяемым для калибровки с фиксированной массой, являлся лейцин-энкефалин.
Способ 2
В дополнение к общей методике B: обращенно-фазовую UPLC проводили на колонке BEH-C18 (1,7 мкм, 2,1×50 мм) от Waters со скоростью потока 1,0 мл/мин при 50ºC без разделения для MS-детектора. Применяемые условия градиента представляли собой: от 95% A (0,5 г/л раствор ацетата аммония + 5% ацетонитрил), 5% B (ацетонитрил) до 40% A, 60% B за 2,8 минуты, до 5% A, 95% B за 3,6 минуты, удерживание до 3,8 минуты и уравновешивание до начальных условий от 4,0 минуты до 5,0 минуты. Объем вводимой пробы составлял 0,5 мкл. Масс-спектры низкого разрешения (на одноквадрупольном SQD-детекторе) получали путем сканирования от 100 до 1000 за 0,1 секунды с применением межканальной задержки 0,08 секунды. Напряжение на капиллярной игле составляло 3 кВ. Напряжение на конусе составляло 25 В для режима положительной ионизации и 30 В для режима отрицательной ионизации.
Способ 3
В дополнение к общей методике B: обращенно-фазовую UPLC проводили на колонке BEH-C18 (1,7 мкм, 2,1×50 мм) от Waters со скоростью потока 1,0 мл/мин при 50°C без разделения для MS-детектора. Применяемые условия градиента представляли собой: от 95% A (0,5 г/л раствор ацетата аммония + 5% ацетонитрил), 5% B (ацетонитрил) до 40% A, 60% B за 3,8 минуты, до 5% A, 95% B за 4,6 минуты, удерживание до 5,0 минуты. Объем вводимой пробы составлял 2,0 мкл. Масс-спектры низкого разрешения (на одноквадрупольном SQD-детекторе) получали путем сканирования от 100 до 1000 за 0,1 секунды с применением межканальной задержки 0,08 секунды. Напряжение на капиллярной игле составляло 3 кВ. Напряжение на конусе составляло 25 В для режима положительной ионизации и 30 В для режима отрицательной ионизации.
Способ 4
В дополнение к общей методике D: обращенно-фазовую HPLC проводили на колонке YMC-Pack ODS-AQ C18 (3 мкм, 50 мм × 4,6 мм) со скоростью потока 2,6 мл/мин при 35°C. Градиентное элюирование проводили от 95% (H2O+0,1% HCOOH)/5% CH3CN до 5% (H2O+0,1% HCOOH)/95% CH3CN за 4,8 мин при удерживании в течение 1,0 мин; затем до 95% (H2O+0,1% HCOOH)/5% CH3CN за 0,2 мин. Объем вводимой пробы составлял 2 мкл. Диапазоны обнаружения устанавливали на 190-400 нм для UV-PDA-детектора и 100-1400 масса/заряд для MS-детектора.
Способ 5
В дополнение к общей методике A: обращенно-фазовую HPLC проводили на колонке Eclipse Plus-C18 (3,5 мкм, 2,1×30 мм) от Agilent со скоростью потока 1,0 мл/мин при 60°C без разделения для MS-детектора. Применяемые условия градиента представляли собой: от 95% A (0,5 г/л раствор ацетата аммония + 5% ацетонитрил), 5% B (смесь ацетонитрил/метанол, 1/1) до 100% B за 5,0 минуты, удерживание до 5,15 минуты и уравновешивание до начальных условий от 5,30 минуты до 7,0 минуты. Объем вводимой пробы составлял 2 мкл. Масс-спектры низкого разрешения (на одноквадрупольном SQD-детекторе) получали путем сканирования от 100 до 1000 за 0,1 секунды с применением межканальной задержки 0,08 секунды. Напряжение на капиллярной игле составляло 3 кВ. Напряжение на конусе составляло 20 В для режима положительной ионизации и 30 В для режима отрицательной ионизации.
Способ 6
Такой же градиент, как для способа 4; применяемая колонка: RRHD Eclipse Plus-C18 (1,8 мкм, 2,1×50 мм) от Agilent.
Способ 7
В дополнение к общей методике C: обращенно-фазовую HPLC проводили на колонке Xterra MS C18 (3,5 мкм, 4,6×100 мм) со скоростью потока 1,6 мл/мин. Три подвижные фазы (подвижная фаза A: 95% 25 мМ ацетат аммония + 5% ацетонитрил; подвижная фаза B: ацетонитрил; подвижная фаза C: метанол) использовали для выполнения условия градиента от 100% A до 50% B и 50% C за 6,5 минуты, до 100% B за 0,5 минуты, 100% B за 1 минуту с повторным уравновешиванием 100% A за 1,5 минуты. Применяли объем вводимой пробы 10 мкл.
Напряжение на конусе составляло 10 В для режима положительной ионизации и 20 В для режима отрицательной ионизации.
Способ 8
В дополнение к общей методике A: обращенно-фазовую HPLC проводили на колонке Eclipse Plus-C18 (3,5 мкм, 2,1×30 мм) от Agilent со скоростью потока 1,0 мл/мин при 60°C без разделения для MS-детектора. Применяемые условия градиента представляли собой: от 95% A (0,5 г/л раствор ацетата аммония + 5% ацетонитрил), 5% B (смесь ацетонитрил/метанол, 1/1) при удерживании в течение 0,2 минуты до 100% B за 3,0 минуты при удерживании до 3,15 минуты и уравновешивании до начальных условий от 3,30 минуты до 5,0 минуты. Объем вводимой пробы составлял 2 мкл. Масс-спектры низкого разрешения (на одноквадрупольном SQD-детекторе) получали путем сканирования от 100 до 1000 за 0,1 секунды с применением межканальной задержки 0,08 секунды. Напряжение на капиллярной игле составляло 3 кВ. Напряжение на конусе составляло 20 В и 50 В для режима положительной ионизации и 30 В для режима отрицательной ионизации.
Способ 9
В дополнение к общей процедуре B: обращенно-фазовую UPLC проводили на RRHD Eclipse Plus-C18 (1,8 мкм, 2,1×50 мм) от Agilent со скоростью потока 1,0 мл/мин при 50°C без разделения для MS-детектора. Применяемые условия градиента представляли собой: от 95% A (0,5 г/л раствор ацетата аммония + 5% ацетонитрил), 5% B (ацетонитрил) до 40% A, 60% B за 1,2 минуты, до 5% A, 95% B за 1,8 минуты, удерживание до 2,0 минуты. Объем вводимой пробы составлял 2,0 мкл. Масс-спектры низкого разрешения (на одноквадрупольном SQD-детекторе) получали путем сканирования от 100 до 1000 за 0,1 секунды с применением межканальной задержки 0,08 секунды. Напряжение на капиллярной игле составляло 3 кВ. Напряжение на конусе составляло 25 В для режима положительной ионизации и 30 В для режима отрицательной ионизации.
Способ 10
В дополнение к общей методике C: обращенно-фазовую UPLC (сверхэффективную жидкостную хроматографию) проводили на колонке C18 (1,7 мкм, 2,1×50 мм; Waters Acquity) с мостиковым гибридом этилсилоксан/диоксид кремния (BEH) при скорости потока 0,8 мл/мин. Две подвижные фазы (10 мМ NH4AcO в H2O/CH3CN 95/5; подвижная фаза B: CH3CN) применяли для выполнения условия градиента от 95% A и 5% B до 5% A и 95% B за 1,3 минуты при удерживании в течение 0,7 минуты. Применяли объем вводимой пробы 0,75 мл. Напряжение на конусе составляло 10 В для режима положительной ионизации и 20 В для режима отрицательной ионизации.
GCMS
Общая методика для аппарата Agilent GC/MSD
Измерения в ходе GC осуществляли с применением системы для газовой хроматографии серии 6890 (Agilent Technologies), включающей инжектор и автодозатор серии 7683, термостат колонок и колонку, указанные в соответствующих способах ниже, соединенной с масс-селективным детектором 5973N MSD (одноквадрупольным, Agilent Technologies). MS-детектор был оснащен источником ионизации электронным ударом/источником химической ионизации (EI/CI). Масс-спектры EI низкого разрешения получали путем сканирования от 50 до 550 при скорости 14,29 сканирований/с. Температуру источника поддерживали при 230°C. В качестве газа-распылителя применяли гелий. Сбор и обработку данных проводили с помощью программного обеспечения Chemstation-Open Action.
Способ 1
В дополнение к общей методике: GC проводили на колонке J&W HP-5MS (20 м × 0,18 мм, 0,18 мкм) от Agilent Technologies со скоростью потока 0,7 мл/мин. Применяемый градиент температуры составлял: начальная температура 50ºC, удерживание в течение 2,0 мин, затем линейное повышение 50ºC/мин, применяемое в течение 5,0 мин, до 300°C и удерживание в течение 3,0 мин за 10-минутный пробег. Температура переднего впускного клапана составляла 250ºC. Применяли режим ввода пробы с разделением потока, объем вводимой пробы 0,2 мкл с соотношением 50/1 в системе для GC/MS.
Точки плавления
Значения представляют собой максимальные значения или диапазоны точек плавления, и их получают с экспериментальными погрешностями, которые обычно связаны с данным аналитическим способом.
Прибор Mettler FP62
Для ряда соединений точки плавления определяли в открытых капиллярных трубках на приборе Mettler FP62. Точки плавления измеряли при градиенте температуры 1, 3, 5 или 10°C/минута. Максимальная температура составляла 300°C. Точку плавления считывали с цифрового дисплея.
Прибор Mettler-Toledo DSC823e
Для ряда соединений точки плавления определяли с помощью Mettler-Toledo DSC823e (обозначенного как DSC в таблице 2). Точки плавления измеряли при градиенте температуры 30°C/минута. Максимальная температура составляла 400°C.
Ядерный магнитный резонанс (ЯМР)
1H-ЯМР-спектры регистрировали на спектрометрах Bruker Avance III, или Bruker DPX-400, или Bruker AV-500 со стандартными последовательностями импульсов, работающих при 300 МГц, 400 МГц и 500 МГц, соответственно. Химические сдвиги (δ) представлены в частях на миллион (м.д.) со слабопольным сдвигом от тетраметилсилана (TMS), который применяли в качестве внутреннего стандарта.
|
ФАРМАКОЛОГИЧЕСКИЕ ПРИМЕРЫ
Соединения формулы (I) и их фармацевтически приемлемые соли и сольваты, обеспечиваемые в настоящем изобретении, представляют собой ингибиторы PDE2, в частности, PDE2A, и в меньшей степени PDE10, в частности, PDE10A. Поведение соединений согласно формуле (I) и комбинаций согласно настоящему изобретению показано в таблицах 3-9 ниже.
In vitro анализ PDE2A
Человеческую рекомбинантную PDE2A (hPDE2A) экспрессировали в клетках Sf9 с применением рекомбинантного конструкта rPDE10A на основе бакуловируса. Клетки собирали через 48 ч после инфицирования, и белок hPDE2A очищали с помощью металл-хелатной хроматографии на Ni-сефарозе 6FF. Тестируемые соединения растворяли и разводили в 100% DMSO до 100-кратной концентрации относительно конечной концентрации в анализе. Разведения соединения (0,4 мкл) добавляли в 384-луночные планшеты к 20 мкл буфера для инкубации (50 мМ Tris, pH 7,8, 8,3 мМ MgCl2, 1,7 мМ EGTA). В буфер для инкубации добавляли 10 мкл фермента hPDE2A, и реакцию начинали путем добавления 10 мкл субстрата до конечной концентрации 10 мкМ cGMP и 0,01 мкКи 3H-cGMP. Реакционную смесь инкубировали в течение 45 минут при комнатной температуре. После инкубации реакцию останавливали с помощью 20 мкл стоп-реагента, состоящего из гранул с PDE для SPA (сцинтилляционного анализа сближения) при концентрации 17,8 мг/мл, дополненных 200 мМ ZnCl2. После осаждения гранул в течение 30 минут измеряли радиоактивность с помощью сцинтилляционного счетчика Perkin Elmer Topcount, и результаты выражали в имп/мин. Для получения значений для холостой пробы фермент не включали в реакционную смесь и замещали буфером для инкубации. Контрольные значения получали путем добавления DMSO в конечной концентрации 1% вместо соединения. Кривая наилучшего приближения вычерчивается по точкам с помощью способа наименьшей суммы квадратов на графике зависимости % контрольного значения с вычтенным значением для холостой пробы от концентрации соединения, и на основании этой кривой получают значение концентрации полумаксимального ингибирования (IC50).
In vitro анализ PDE10A
Крысиную рекомбинантную PDE10A (rPDE10A2) экспрессировали в клетках Sf9 с применением рекомбинантного конструкта rPDE10A на основе бакуловируса. Клетки собирали через 48 ч после инфицирования, и белок rPDE10A очищали с помощью металл-хелатной хроматографии на Ni-сефарозе 6FF. Тестируемые соединения растворяли и разводили в 100% DMSO до 100-кратной концентрации относительно конечной концентрации в анализе. Разведения соединения (0,4 мкл) добавляли в 384-луночные планшеты к 20 мкл буфера для инкубации (50 мМ Tris, pH 7,8, 8,3 мМ MgCl2, 1,7 мМ EGTA). В буфер для инкубации добавляли 10 мкл фермента rPDE10A, и реакцию начинали путем добавления 10 мкл субстрата до конечной концентрации 60 нМ cAMP и 0,008 мкКи 3H-cAMP. Реакционную смесь инкубировали в течение 60 минут при комнатной температуре. После инкубации реакцию останавливали с помощью 20 мкл стоп-реагента, состоящего из гранул с PDE для SPA (сцинтилляционного анализа сближения) при концентрации 17,8 мг/мл. После осаждения гранул в течение 30 минут измеряли радиоактивность с помощью сцинтилляционного счетчика Perkin Elmer Topcount, и результаты выражали в имп/мин. Для получения значений для холостой пробы фермент не включали в реакционную смесь и замещали буфером для инкубации. Контрольные значения получали путем добавления DMSO в конечной концентрации 1% вместо соединения. Кривая наилучшего приближения вычерчивается по точкам с помощью способа наименьшей суммы квадратов на графике зависимости % контрольного значения с вычтенным значением для холостой пробы от концентрации соединения, и на основании этой кривой получают значение концентрации полумаксимального ингибирования (IC50). Результаты этого теста показаны в таблице 3 ниже.
|
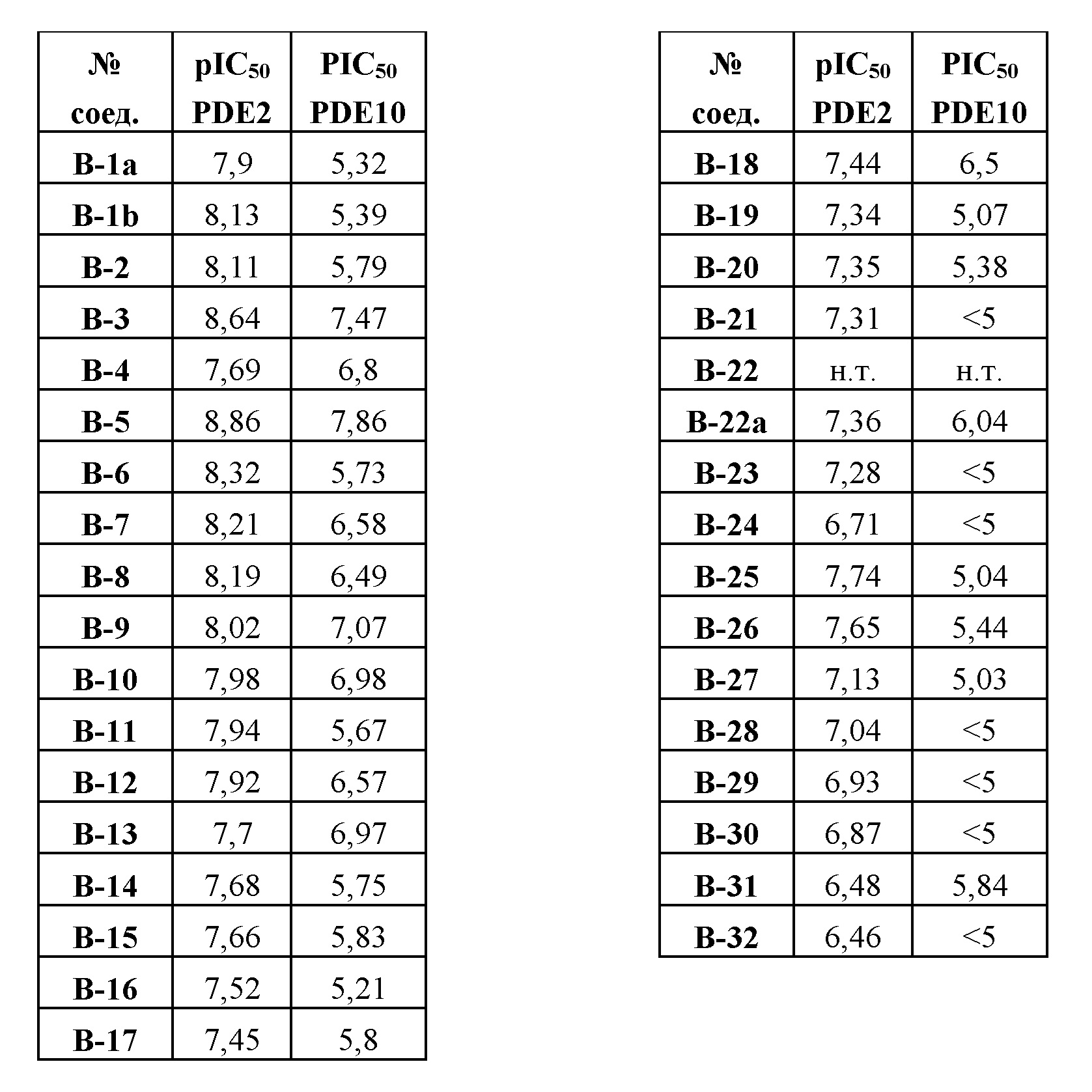
Эффект ингибиторов PDE
Ex vivo исследования на крысах
После доставки животных (вес тела 210-240 г) размещали в группы по 5 и кормили обычной пищей ad libitum.
Соединения и/или растворитель вводили перорально, подкожно либо IV.
В зависимости от экспериментальной модели животных умерщвляли с помощью микроволнового излучения (Muromachi, MMW-05) в течение 1,5 секунды при 5 кВт через 30 либо 60 мин после введения лекарственного средства/растворителя. После обработки микроволновым излучением крыс декапитировали, и головы сразу же охлаждали с помощью ледяного физиологического раствора. Покровные ткани черепа разрезали, и головной мозг удаляли, а различные участки головного мозга (полосатое тело, гиппокамп, кору головного мозга и/или мозжечок) иссекали и переносили в предварительно взвешенные пробирки для гомогенизации (Collection Microtubes, Qiagen), содержащие стальной шарик (гранулы из нержавеющей стали на 5 мм, № по кат. 69989, Qiagen), и выдерживали на сухом льду. Добавляли 10 объемов (вес/объем) 0,1 н. HCl. Ткань гомогенизировали в течение 3 мин при 30 Гц с применением Tissuelyser (Qiagen).
Гомогенат переносили в пробирку Eppendorf (1,5 мл), и после центрифугирования в течение 15 мин при 1600 g в предварительно охлажденной (4°C) центрифуге Eppendorf надосадочную жидкость собирали и хранили при -80°C до анализа.
Уровни циклического GMP определяли в образцах, разведенных 1/4 (полосатое тело, гиппокамп, кора головного мозга) или 1/10 (мозжечок), с применением полного набора для EIA cGMP от Enzo Life Sciences.
Уровни циклического AMP определяли в образцах, разведенных 1/10 и 1/25, с применением набора LANCE Ultra для cAMP от Perkin Elmer (код TRF0263).
Результаты рассчитывали на основании сигмоидальной калибровочной кривой путем нелинейной регрессии с применением программного обеспечения GraphPadPrism. Результаты этого теста показаны в таблице 4 ниже.
Уровни cAMP и cGMP измеряли в головном мозге крысы (гиппокамп и полосатое тело) для установления in vivo захвата мишени и фармакологического эффекта центрального действия в виде ингибирования PDE2, а также для установления комбинированного эффекта ингибирования PDE2 и PDE10. Ингибирование PDE2 приводит к заметному повышению уровней cGMP в головном мозге. После комбинированного введения ингибитора PDE2 B-1a и MP-10 полученное в результате повышение уровня cGMP превышало рассчитанный совокупный эффект в отношении уровней cGMP после ингибирования PDE2 и PDE10 по отдельности, что позволило предположить синергизм между ингибированием PDE2 и PDE10 (таблица 5). Не желая ограничиваться теорией, предполагают, что это может быть связано с повышенным сродством B1-a к PDE2 в условиях высоких внутриклеточных концентраций cGMP. Сигнальный путь NO/cGMP, как было показано, играет важную роль в процессе, лежащем в основе обучения и памяти, синаптической пластичности и нейрогенеза, а также в регуляции кортико-стриарной синаптической передачи и двигательного поведения. Измеренное повышение уровня cGMP в ткани головного мозга способствует дополнительному исследованию применения ингибиторов PDE2 в условиях нарушенной передачи сигналов с помощью NO/cGMP, таких как когнитивная дисфункция при психических расстройствах, болезнь Альцгеймера (Mennitti, F. S. et al. Nature Rev. Drug Discovery 2006, 5, 660-669; Baratti, C.M., Boccia, M.M. Behav. Pharmacol. 1999;10: 731-737; Prickaerts, J. et al. Neuroscience 2002; 113:349-359; Domek-Łopacińska KU, Strosznajder JB Mol Neurobiol. 2010; 41(2-3):129-37), большая депрессия (Reierson, G.W. et al. Current Neuropharmacology 2011; 9:715-727) и двигательные расстройства, такие как болезни Паркинсона и Хантингтона (West, A.R. and Tseng K.Y. Neuroscience, 2011; 5:55-64; Kumar P, et al. Behav Pharmacol. 2010 May; 21(3):217-30).
|
|
In vivo исследования на крысах
Подавление тревожного возбуждения, индуцированного апоморфином, у крыс (APO)
Применяли самцов крыс линии Wiga Wistar, выдерживаемых на голодном рационе в течение ночи (Charles River, Германия; 200-260 г). Тревожное возбуждение, индуцированное апоморфином (1,0 мг/кг, i.v.), оценивали в баллах каждые 5 мин в течение первого часа после инъекции апоморфина. Система подсчета баллов была следующей: (3) явно выраженное, (2) умеренное, (1) слабое и (0) отсутствующее. Критерии индуцированного лекарственным средством ингибирования тревожного возбуждения: менее 6 оценок в 3 балла (0,16% ложноположительных результатов; n=2966), менее 6 оценок в ≥2 балла (0,0% ложноположительных результатов) или менее 7 оценок в ≥1 балл (0,0% ложноположительных результатов). Для целей настоящего изобретения суммарный балл тревожного возбуждения в течение всего 60-минутного периода наблюдения применяли в качестве меры для описания максимального эффекта (макс. эффекта), т.е. наименьшего медианного суммарного балла, наблюдаемого в каждой дозовой группе. Результаты этого теста показаны в таблице 6 ниже. Селективные ингибиторы PDE2 не влияют на поведение, индуцированное апоморфином, тогда как ингибиторы PDE10 влияют на поведение, индуцированное апоморфином; в тех случаях, когда значения макс. эффекта равны <10 (низкий уровень тревожного возбуждения), вероятно, имеет место комбинированный ингибирующий эффект в отношении PDE10 и PDE2.
|
Подавление тревожного возбуждения, индуцированного апоморфином, у крыс в комбинации с MP-10
А теперь животных одновременно стимулировали с помощью MP-10 (0,63 мг/кг) в дополнение к апоморфину (1,0 мг/кг) посредством одной инъекции (2 мл/кг, i.v.) в фиксированном промежутке времени (обычно 1 ч) после s.c. или p.o. дозирования тестового соединения или растворителя. Двенадцать раз, каждые 5 мин, интенсивность тревожного возбуждения оценивали в баллах от 0 до 3. Для целей настоящего изобретения суммарный балл тревожного возбуждения в течение всего 60-минутного периода наблюдения применяли для оценивания. Исходя из частотного распределения суммарного балла тревожного возбуждения в группе контрольных крыс, предварительно обработанных растворителем, суммарный балл <10 принимали в качестве критерия "все или ничего" в отношении индуцированного лекарственным средством ингибирования тревожного возбуждения (0,0% ложноположительных результатов в контроле; n=93). Результаты этого теста показаны в таблице 7 ниже.
|
Представление эффекта в отношении тревожного возбуждения со стороны MP-10 (0,63-2,5 мг/кг, s.c.) и растворителя (раств.); а также эффекта в случае дозирования MP-10 (при 2,5 мг/кг, s.c.) вместе с соединением B-1a, приведено на фигурах 1a-c. В случае, когда B-1a (40 мг/кг, s.c.) дозировали вместе с постепенно увеличивающимися дозами MP-10, наблюдалось усиление эффекта MP-10 по величине без сдвига влево кривой зависимости "доза-эффект" (при сравнении фигур 1a и 1b). В случае, когда MP-10 (2,5 мг/кг, s.c.) дозировали вместе с постепенно увеличивающимися дозами B-1a, Β-1a обуславливал дополнительное дозозависимое уменьшение тревожного возбуждения, хотя он был неэффективным против апоморфина при введении в отдельности в тестируемых дозах (без MP-10; таблица 6).
На фигуре 2 показан эффект MP-10 (-1 ч., s.c.) в отношении тревожного возбуждения, индуцированного апоморфином (медианный балл в каждой дозовой группе), в зависимости от дозы PDE2-i Β-1a (0,63-10 мг/кг, s.c.; -1 ч.) или растворителя (10 мл/кг, s.c.; -1 ч.). Пунктирные горизонтальные линии представляют критические уровни для небольшого подавления тревожного возбуждения (балл <21; верхняя линия) и явно выраженного подавления тревожного возбуждения (балл <10; нижняя линия). MP-10 дозозависимым образом подавлял тревожное возбуждение; величина максимального эффекта увеличивалась с увеличением дозы Β-1a. При низких дозах совместно вводимого Β-1a (≤0,63 мг/кг) медианные баллы тревожного возбуждения <10 при дозе MP-10 до 10 мг/кг получены не были. При более высоких дозах Β-1a, однако, MP-10 снижал тревожное возбуждение до балла <10 при все более низких дозах.
Это дополнительно проиллюстрировано на фигуре 3, на которой построен график зависимости ED50 MP-10 для подавления тревожного возбуждения до балла <21 (фигура 3a) и до балла <10 (фигура 3b) от дозы Β-1a. На ED50 MP-10 для подавления тревожного возбуждения до балла <21 крайне незначительно влияет доза совместно вводимого Β-1a (фигура 3a). Однако, ED50 MP-10 для подавления тревожного возбуждения до балла <10 уменьшалась дозозависимым образом с увеличением дозы совместно вводимого Β-1a (фигура 3b).
Подавление тревожного возбуждения, индуцированного апоморфином, у крыс в комбинации с двумя другими PDE10-i, а именно соединением A и соединением B
Чтобы показать, что PDE2-i усиливают эффект не только MP-10, но также и других PDE10-i, тестировали взаимодействие Β-1a с двумя дополнительными PDE10-i, а именно соединением A и соединением B. На фигуре 4 показан эффект Β-1a (0 в сравнении с 10 мг/кг s.c.; -1 ч) в отношении зависимости "доза-эффект" для соединения A (-1 ч, s.c.; фиг. 4a) и соединения B (-1 ч, s.c.; фиг. 4b) применительно к подавлению тревожного возбуждения, индуцированного апоморфином. Показаны отдельные баллы (незакрашенные и закрашенные круги для PDE2-i при 0 и 10 мг/кг, соответственно) и медианные баллы (горизонтальные линии) тревожного возбуждения в каждой дозовой группе. ED50 (при доверительных интервалах с надежностью 95%) PDE10-i для снижения баллов тревожного возбуждения до <21, <10 и <5 были приведены для совместной обработки с помощью PDE2-i при 0 и 10 мг/кг. Β-1a усиливал эффект PDE10-i по величине без индукции сдвига влево кривых зависимости "доза-эффект". Это также отражено в приведенных значениях ED50. Β-1a не влиял на ED50 PDE10-i для небольшого подавления (балл <21), но уменьшал ED50 PDE10-i для более высоких уровней подавления (балл <10 и балл <5).
На фигуре 5 показан дозозависимый эффект PDE2-i Β-1a (0, 0,63, 1,25, 2,5 и 5,0 мг/кг, s.c.; -1 ч.) в отношении тревожного возбуждения, индуцированного апоморфином, в присутствии стандартных доз соединения A (0 или 2,5 мг/кг, s.c., -1 ч.; фиг. 5a и 5c, соответственно) или соединения B (0 или 2,5 мг/кг, s.c., -1 ч.; фиг. 5b и 5d, соответственно). Пунктирная горизонтальная линия представляет критерий небольшого подавления тревожного возбуждения (балл <21). Β-1a не влиял на тревожное возбуждение, индуцированное апоморфином, в комбинации с растворителем для PDE10-i (фигуры 5a и b), но значительно усиливал эффект обоих PDE10-i по сравнению с растворителем при 5,0 мг/кг (фигуры 5c и d, соответственно).
Гиперлокомоция, индуцированная d-амфетамином, у крыс: усиление эффекта MP-10
Применяли самцов крыс линии Wiga Wistar, выдерживаемых на голодном рационе в течение ночи (Charles River, Германия; 200-260 г). В предварительно определенном промежутке времени перед измерением двигательной активности крыс предварительно обрабатывали тестовым соединением или растворителем (10 мл/кг, p.o. или s.c.) и помещали в отдельные клетки. За тридцать минут до начала тестирования локомоторной активности крыс стимулировали d-амфетамином (1,25 мг/кг, s.c.) в комбинации с MP-10 (2,5 мг/кг, s.c.), оба из которых давали в виде однократной инъекции (10 мл/кг, s.c.). Двигательную активность измеряли в течение периода 30 мин в микропроцессорных аренах для изучения двигательной активности (закрытых серых цилиндрах из PVC высотой 39 см и диаметром 31 см) и анализировали при помощи системы видеонаблюдения Noldus Ethovision XT (версия 7.0.418; Noldus, Вагенинген, Нидерланды). Подсчитывали общее пройденное расстояние (см). Общее пройденное расстояние <2500 см принимали в качестве критерия "все или ничего" в отношении усиления эффекта MP-10 (6,3% ложноположительных результатов в контрольной популяции из 412 крыс, предварительно обработанных растворителем). Результаты этого теста показаны в таблице 8 ниже.
|
Представление эффектов, наблюдаемых для MP-10, B-1a и B1-a в комбинации с MP-10, приведено на фигурах 6a-c. На фигуре 6a показано дозозависимое подавление гиперлокомоции, индуцированной d-амфетамином, определенное через 1 ч после s.c. инъекции MP-10. Следует отметить, что эффект является лишь частичным, почти не достигая уровней <2500 см. На фигуре 6b показано отсутствие эффекта против гиперлокомоции, индуцированной d-амфетамином, определенное через 1 ч после s.c. инъекции Β-1a в тестируемой дозе 40 мг/кг. На фигуре 6c показано дозозависимое усиление эффекта MP-10 (2,5 мг/кг, s.c.) в отношении гиперлокомоции, индуцированной d-амфетамином, определенное через 1 ч после s.c. инъекции Β-1a. Хотя Β-1a является неэффективным per se в тестируемой дозе 40 мг/кг (фигура 6b), а при применении MP10 в отдельности (до 40 мг/кг) почти никогда не достигаются значения <2500 см (фигура 6a), B-1a усиливает эффект низкой дозы MP-10 (2,5 мг/кг) и неизменно обуславливает уровни активности <2500 см при ED50 0,51 мг/кг.
На фигуре 7 показан дозозависимый эффект MP-10 (-1 ч, s.c.) в отношении гиперлокомоции, индуцированной d-амфетамином, в зависимости от дозы совместно вводимого PDE2-i Β-1a (0,63-10 мг/кг, s.c.; -1 ч) или растворителя (10 мл/кг, s.c.; -1 ч). Пунктирные горизонтальные линии отражают критические уровни для эффектов, индуцированных лекарственными средствами (<5500 см, <2500 см и <1000 см). Β-1a дозозависимым образом усиливал эффект, получаемый с MP-10, по величине.
Это также проиллюстрировано на фигуре 8. На фигуре 8 построен график зависимости ED50 (при доверительных интервалах с надежностью 95%) MP-10 (-1 ч, s.c.) для снижения гиперлокомоции, индуцированной d-амфетамином, до расстояния <5500 см (фиг.8a), <2500 см (фиг.8b) и <1000 см (фиг.8c) от дозы совместно вводимого PDE2-i Β-1a (0,63-10 мг/кг, s.c.; -1 ч; закрашенные символы) или растворителя (10 мл/кг, s.c.; -1 ч; незакрашенные символы). Серая горизонтальная полоса представляет ED50 (при доверительных интервалах с надежностью 95%) MP-10 (-1 ч, s.c.) в комбинации с растворителем для Β-1a (фиг.8a и b) или MP-10 (-1 ч, s.c.) в отдельности (фиг.8c; >40 мг/кг, ретроспективные данные). Β-1a крайне незначительно влиял на ED50 MP-10 для снижения локомоции до расстояния <5500 см (фиг.8a), но дозозависимым образом уменьшал ED50 MP-10 для снижения локомоции до расстояния <2500 см и <1000 см (фиг.8b и 8c, соответственно).
Подавление гиперлокомоции, индуцированной d-амфетамином, у крыс в комбинации с двумя другими PDE10-i, а именно соединением A и соединением B
Чтобы показать, что PDE2-i усиливают эффект не только MP-10, но также и других PDE10-i, тестировали взаимодействие Β-1a с двумя дополнительными PDE10-i, а именно соединением A и соединением B.
На фигуре 9 показан эффект стандартной дозы Β-1a (0 в сравнении с 10 мг/кг s.c.; -1 ч) в отношении дозозависимого эффекта соединения A (-1 ч, s.c.; фиг. 9a) и соединения B (-1 ч, s.c.; фиг. 9b) для подавления гиперлокомоции, индуцированной d-амфетамином. Показаны отдельные значения (незакрашенные и закрашенные круги для PDE2-i при 0 и 10 мг/кг, соответственно) и медианные значения (горизонтальные линии) пройденного расстояния в каждой дозовой группе. Пунктирные горизонтальные линии представляют критерии, принятые для эффектов, индуцированных лекарственными средствами (<5500 и <1100 см). ED50 (при доверительных интервалах с надежностью 95%) PDE10-i для уменьшения пройденного расстояния до <5500 см и до <1100 см были приведены для совместной обработки с помощью Β-1a при 0 и 10 мг/кг. Β-1a усиливал эффект PDE10-i по величине без индукции сдвига влево кривой зависимости "доза-эффект". Это также отражено в приведенных значениях ED50. Β-1a не влиял на ED50 PDE10-i для небольшого подавления (расстояние <5500 см), но существенно уменьшал ED50 PDE10-i для явно выраженного подавления (<1100 см).
На фигуре 10 показан эффект Β-1a (0, 0,63, 1,25, 2,5 и 5,0 мг/кг s.c.; -1 ч; фиг. 10a) в отношении гиперлокомоции, индуцированной d-амфетамином, в присутствии стандартных доз соединения A (0 или 2,5 мг/кг, s.c., -1 ч; фиг. 10b) или соединения B (0 или 2,5 мг/кг, s.c., -1 ч; фиг. 10c). Пунктирные горизонтальные линии представляют критические уровни для эффектов, индуцированных лекарственными средствами (<5500 см и <1100 см). Β-1a усиливал эффект обоих PDE10-i.
Усиление связывания [3H]B-1a с PDE2 с помощью MP-10
[3H]B-1a представляет собой радиолиганд, осуществляющий селективное связывание с каталитическим доменом фермента PDE2. При помощи in vitro радиоавтографии было показано, что распределение участков связывания [3H]B-1a полностью соответствует паттерну экспрессии белка PDE2 в головном мозге крыс с высокими значениями плотности в коре головного мозга, гиппокампе, полосатом теле и черной субстанции. При проведении in vivo анализа занятости фермента PDE2 с применением [3H]B-1a наблюдали, что ингибитор PDE10 MP-10 может дозозависимым образом усиливать in vivo связывание с радиолигандом. Наиболее правдоподобным объяснением этого явления является чувствительность GAF-домена PDE2 к cGMP. Более того, в литературе было описано, что cGMP посредством связывания с GAF-доменом PDE2 изменяет конформацию фермента, и повышает доступность субстрата для каталитического домена (Pandit J et al., Proc. Natl. Acad. Sci. U S A. 2009 Oct 27;106(43):18225-30), и, вероятнее всего, повышает также и сродство к [3H]B-1a. Таким образом, MP-10 посредством повышения внутриклеточной концентрации cGMP будет стимулировать GAF-домен PDE2, изменять его конформацию и повышать сродство к [3H]B-1a. Для демонстрации этой гипотезы было показано, что cBIMP (негидролизуемый аналог cGMP) может усиливать специфическое связывание с [3H]B-1a на срезах мозга крыс. В дополнение, было показано, что предварительная обработка с помощью L-NAME (ингибитора синтеза оксида азота, снижающего внутриклеточную концентрацию cGMP) предотвращала усиление связывания с [3H]B-1a с помощью ингибитора PDE10 MP-10, что демонстрировало, что это явление опосредовано cGMP.
|
Данные, обобщенные в таблице 9, также показаны на фигуре 11, в том числе изображения, полученные в ходе ex vivo радиоавтографии.
ПРИМЕРЫ ВОЗМОЖНЫХ КОМПОЗИЦИЙ
“Активный ингредиент”, как применяется во всех данных примерах, относится к конечному соединению формулы (I), его фармацевтически приемлемым солям, его сольватам и стереохимически изомерным формам.
Типичные примеры рецептов для состава по настоящему изобретению являются следующими:
1. Таблетки
|
В данном примере активный ингредиент можно заменять таким же количеством любого из соединений согласно настоящему изобретению, в частности, таким же количеством любого из соединений, приведенных в качестве примера.
2. Суспензия
Водную суспензию для перорального введения получают таким образом, что каждый 1 миллилитр содержит от 1 до 5 мг одного из активных соединений, 50 мг карбоксиметилцеллюлозы натрия, 1 мг бензоата натрия, 500 мг сорбита и до 1 мл воды.
3. Композиции для инъекций
Композицию для парентерального введения получают путем перемешивания 1,5% по весу активного ингредиента по настоящему изобретению в 10% по объему пропиленгликоле в воде.
4. Мазь
|
В данном примере активный ингредиент можно заменять таким же количеством любого из соединений согласно настоящему изобретению, в частности, таким же количеством любого из соединений, приведенных в качестве примера.
Допустимые варианты не следует рассматривать как отклонение от объема настоящего изобретения. Будет очевидно, что специалисты в данной области могут изменять описанное таким образом изобретение различными способами.
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/30fd06ef27ea366f163a130ffe7f5857.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/87f393fba0fe60eaeae2c660311b1346.png)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/635ff31f3338926b551e8611210b913b.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/e26a5bfef0b2f81e521e26c0f5c41250.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/d4fe2fb6c1736c86edc5e2f1180a39a7.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/760dd6b07539b3d85e0229bda7aad187.png)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/f0f15987da6875233e175b91f2824fbe.png)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/a6f8359b0103f9d4d59d4c4a858fac31.png)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/6cc3dd68fd59be405bac3d687a300595.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/df4c2ed34e8451b13cfe77aaef690ab6.png)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/e8650b6046cb397c70086e09fb18cfc6.png)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/87b72d13c6b9c46adeeffc53341044ad.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/471c80ad78a555db66265046cd35ddbe.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/3802c95de4e0c848f081d54f9f302119.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/fea5b6a149d14ffd3179eb4ced893605.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/aca8edc847a24e3268f3d146912b766b.png)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/d4c23d41c607531f0c05b4b8191da812.png)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/ce50de6a80a5503c5a37bfc8245fa4d9.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/80c437f6792246937c55c6dd750294f6.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/06e21000c87de4954e5ae1904c324049.png)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/cac66674ae8024c49ded963c5c4bae46.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/51b552429ec763ad35a6b227f89c6c39.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/4982331b735eb9a37d128dcf752cae6c.png)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/e65e0fc6ee3065ce64578817c7d6e378.jpg)
![КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ](https://fips.edrid.ru/images/rid/97/d9/32/9c778b153fd2f4f0e4f2c08c7b1a5c2f.png)
Тризамещенные 1,2,4-триазолы
1,3,5-тризамещенное производное триазола
Ингибиторы взаимодействия между mdm2 и p53
Композиция, содержащая комбинацию фенилпиррола и пирионового соединения, способ ее получения, способы регулирования микробиального роста и биоцидный продукт
Тетрагидрофенантридиноны и тетрагидроциклопентахинолиноны в качестве ингибиторов parp и ингибиторов полимеризации тубулина
Диэфирные пролекарства налмефена
Пролекарства налмефена
Производные бензоциклогептана и бензоксепина
Замещенные 6-(1-пиперазинил)-пиридазины в качестве антагонистов 5-нт6 рецептора
Антиаллергенные комбинации солей кальция и лантана
Имидазо[1,2-а]пиридиновые производные и их применение в качестве положительных аллостерических модуляторов рецепторов mglur2
3-азабицикло[3.1.0]гексильные производные в качестве модуляторов метаботропных глутаматных рецепторов
Производные индола и бензоксазина в качестве модуляторов метаботропных глутаматных рецепторов
Производные индола и бензоморфолина в качестве модулятора метаботропных глутаматных рецепторов

