Результат интеллектуальной деятельности: Способ получения дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола
Вид РИД
Изобретение
Изобретение относится к способам получения дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола, который применяют, например, для лечения сенсоневральной тугоухости и глухоты.
Из уровня техники известно СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ОСТРОЙ И ВНЕЗАПНОЙ СЕНСОНЕВРАЛЬНОЙ ТУГОУХОСТИ И ГЛУХОТЫ (RU 2119335, опубликовано: 27.09.1998). В решении описано, что дигидрохлорид моногидрат 3-(2-морфолиноэтилтио)-1,2,4-триазино[5,6-b]индола (трисан) получают известным способом. При взаимодействии 3-меркапто-1,2,4-триазино[5,6-b]индола с гидробромидом 2-морфолиноэтилбромида в присутствии избытка щелочи получают основание 3-(2-морфолиноэтилтио)-1,2,4-триазино[5,6-b]индола с выходом около 75% от теоретического. Последний обрабатывают газообразным хлористым водородом или соляной кислотой в изопропиловом спирте и получают трисан - дигидрохлорид моногидрат 3-(2-морфолиноэтилтио)-1,2,4-триазино[5,6-b]индола с выходом около 87% от теоретического. Последний очищают перекристаллизацией из смеси изопропилового спирта с разбавленной соляной кислотой, сушат на воздухе и получают в виде лимонно-желтых кристаллов, температура плавления 238-240°С (с разложением). Вещество однородно по данным тонкослойной хроматографии.
Решение выбрано за прототип.
Недостатком известного способа является то, что им удается синтезировать дигидрохлорид 3-(2-морфолино-этилтио)-1,2,4-триазино-[5,6-b]индола с недостаточно высокой чистотой.
Кроме того, известный способ требует получения газообразного хлористого водорода и приготовления раствора HCl в диоксане, что требует значительных затрат на расход органических растворителей.
Задачей изобретения является устранение недостатка прототипа.
Техническим результатом изобретения является исключение операции получения газообразного хлористого водорода и приготовления раствора HCl в диоксане, а также значительное снижение расхода органических растворителей. Кроме того, предложенный способ является более экологичным. Также удается получать целевой продукт с чистотой не менее 99%.
Указанный технический результат достигается за счет того, что заявлен способ получения дигидрохлорид моногидрат 3-(2-морфолиноэтилтио)-1,2,4-триазино[5,6-b]индола (трисан), характеризующийся взаимодействием 3-меркапто-1,2,4-триазино[5,6-b]индола с гидробромидом 2-морфолиноэтилбромида в присутствии избытка щелочи получают основание 3-(2-морфолиноэтилтио)-1,2,4-триазино[5,6-b]индола, при этом последний обрабатывают соляной кислотой, отличающийся тем, что на емкость 100 мл, снабженную мешалкой, термометром и обратным холодильником, загружают 58 мл концентрированной соляной кислоты, 12 мл воды и 13 мл пропанола-2, а при перемешивании добавляют 10,69 г (0,034 моль) основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола; полученную реакционную смесь при перемешивании нагревают до кипения и кипятят 30 минут, после чего в горячем состоянии фильтруют, фильтрат помещают в иную емкость и при перемешивании охлаждают до 25°С, после чего оставляют стоять в холодильнике в течение 20 часов; выпавший осадок отфильтровывают, промывают последовательно пропанола-2 и ацетон, сушат на воздухе до постоянного веса; на выходе получают дигидрохлорид 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола в виде кристаллического вещества желтого цвета.
Изобретение поясняется результатами анализов (на чертежах).
На Фиг. 1 - Фиг. 7 показаны примеры хроматомасс-спектрометрических анализов продуктов.
На Фиг. 8 показаны результаты анализа образца дигидрохлорида 3-(2-морфолиноэтилтио)-1,2,4-триазино-[5,6-b]индола с помощью ВЭЖХ).
Осуществление изобретения
Оптимизация получения 3-меркапто-1,2,4-триазино[5,6-b]индола 2 путем объединения стадий получения 3-тиосемикарбазона изатина и его циклизации.
Разработанный нами процесс получения 3-меркапто-1,2,4-триазино[5,6-b]индола 2 состоит из 2-х стадий. Первоначально получают 3-тиосемикарбазон изатина в среде водной соляной кислоты,
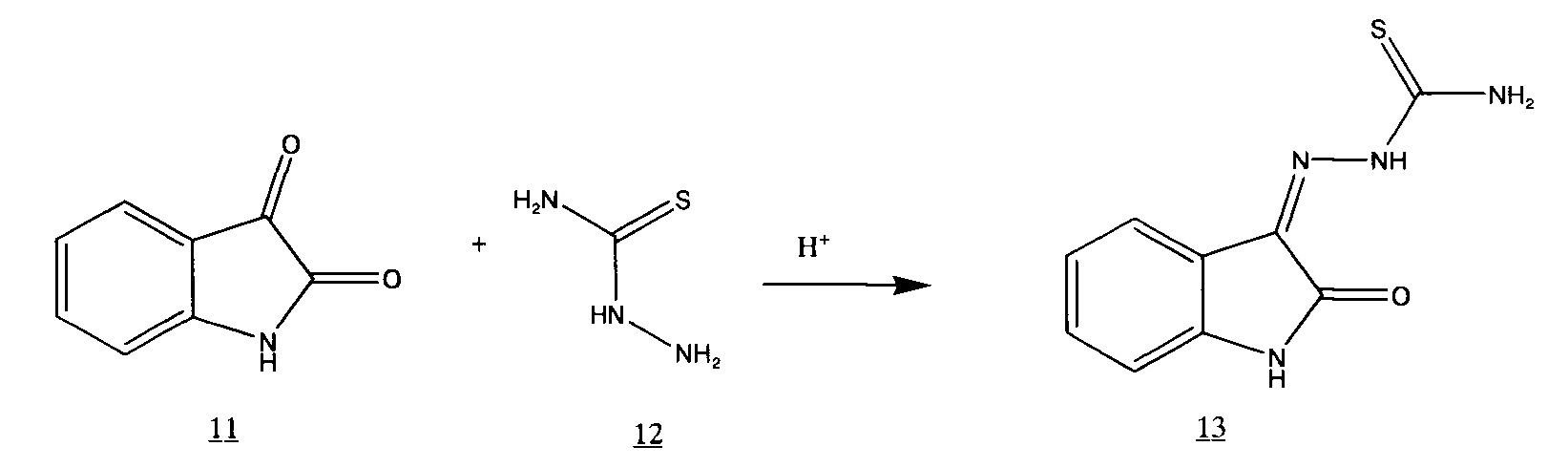
который затем подвергают циклизации в щелочной среде.
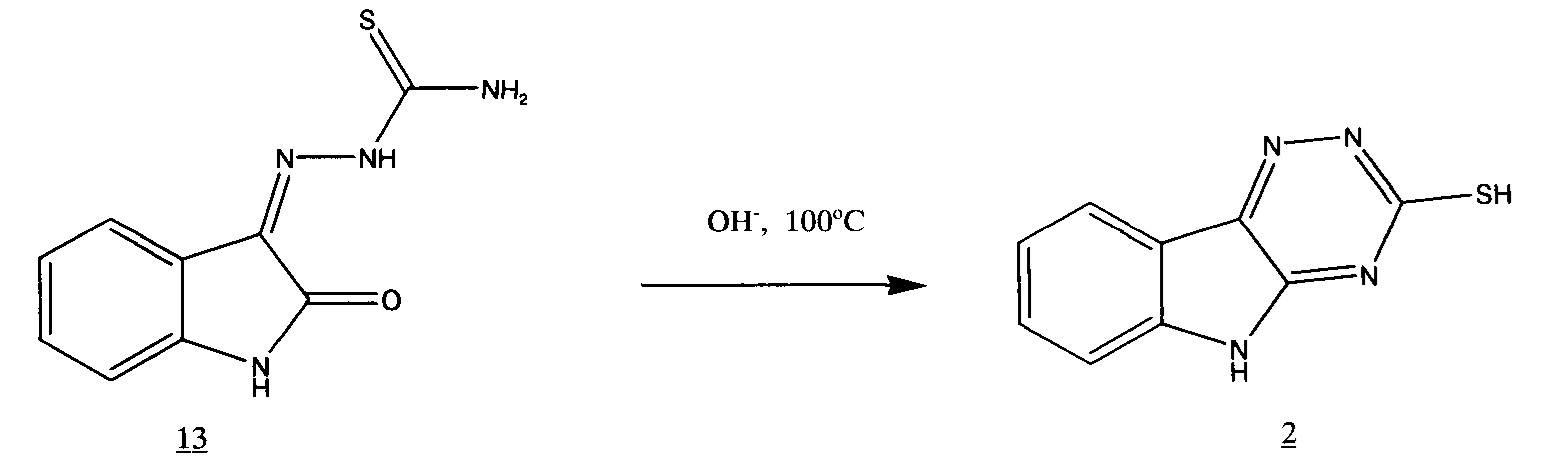
Нами было решено оптимизировать этот процесс, проводя обе стадии последовательно, т.е. без выделения тиосемикарбазона.
В стакан емкостью 100 мл загружали 47 мл воды, 5,1 мл (0,06 моль) концентрированной соляной кислоты и при помешивании прибавляли 4,56 г (0,05 моль) тиосемикарбазида. Перемешивали в течение 30 минут до полного растворения и переносили полученный раствор в капельную воронку.
В 3-горлую колбу емкостью 0,5 л, снабженную мешалкой, капельной воронкой и обратным холодильником, загружали 300 мл воды, 1 каплю 2-этилгексанола (для предотвращения сильного вспенивания в процессе кипячения), и при перемешивании прибавляли 7,02 г (0.047 моль) изатина. Массу нагревали до слабого кипения и при перемешивании в течение 30 мин прибавляли раствор гидрохлорида тиосемикарбазида, поддерживая слабое кипение. По окончании прибавления смесь кипятили 20 минут, охлаждали до 30°С, прибавляли 5,6 г (0,14 моль) NaOH, нагревали до кипения и кипятили 3 часа. При этом полного растворения не происходило. Смесь охлаждали, фильтровали от осадка, фильтрат подкисляли соляной кислотой. Выпавший осадок отфильтровывали, промывали водой и сушили в сушильном шкафу. Было получено 7,9 г (72% от теоретического) неоднородно окрашенного продукта (спектр продукта О-001). Видоизменение этого метода, когда вместо соляной кислоты мы использовали эквимолекулярное количество уксусной кислоты, привело к более загрязненному продукту (спектр продукта О-002).
Проведение конденсации изатина с тиосемикарбазидом в отсутствие кислот с последующим кипячением со щелочью дало еще более загрязненный примесями продукт (спектр продукта О-003).
Мы предположили, что концентрация щелочи недостаточна для полного протекания реакции циклизации. Однако увеличение количества щелочи в два (спектр продукта О-004), а затем и в четыре (спектр продукта О-005) раза привело к получению продукта с немного меньшим содержанием примесей. Увеличение времени кипячения со щелочью в два раза не привело к полному растворению, продукт получился загрязненным (спектр продукта О-006). Замена NaOH на эквимолекулярное количество KОН также не привела к улучшению качества продукта (спектр продукта О-007).
Таким образом, наши попытки объединить стадии образования тиосемикарбазона и его циклизации оказались безуспешными. Тогда мы решили вернуться к варианту синтеза 3-меркапто-1,2,4-триазино[5,6-b]индола 2 в одну стадию.
В нашем исследовании описанный в литературе одностадийный способ приводил к загрязненному тиолу
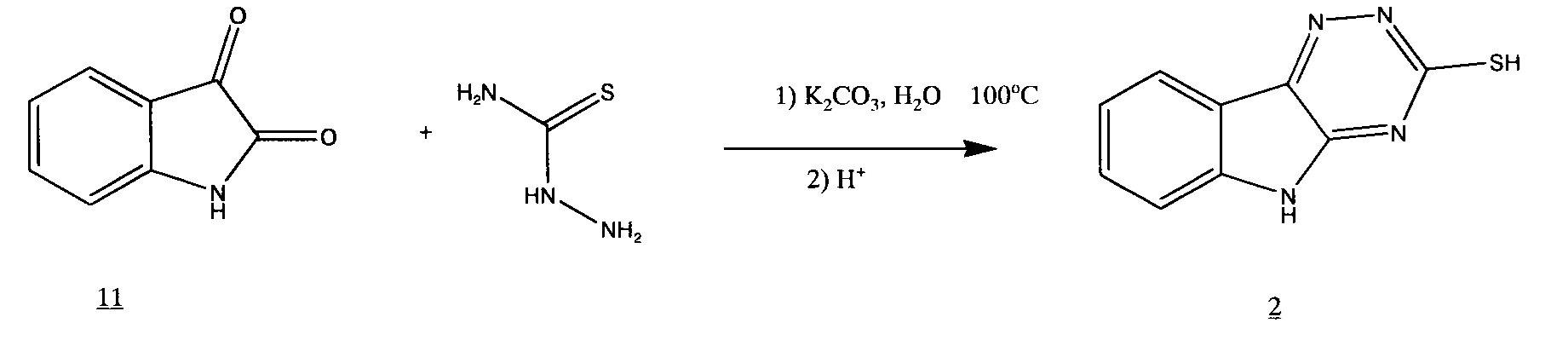
поскольку, как нами было выяснено, в этих условиях протекает побочная реакция [Томчин, А.Б.; Иоффе, И.С.; Русаков, Е.А. // ЖОХ. 1971. Т. 41. Вып. 8. С. 1791-1796].
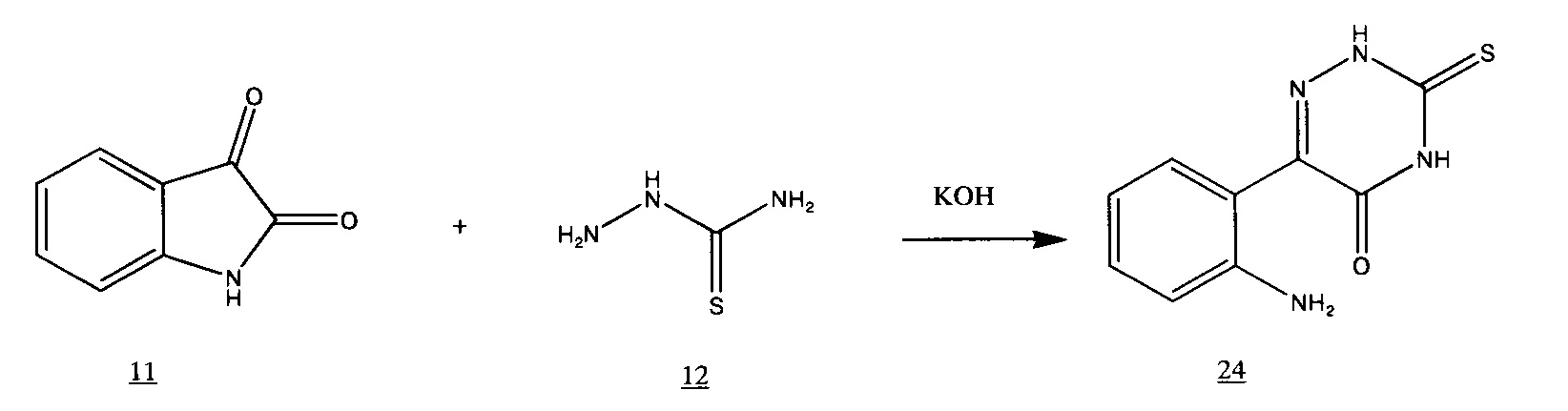
Наши попытки направить эту реакцию в нужном нам направлении путем регулирования щелочности среды успеха не имели.
Однако, проведенный нами литературный поиск по реакциям 6-(2-аминофенил)-3-тио-1,2,4-триазин-3,5(2Н,4Н)-диона 24 показал, что в кислой среде [Doleschall, G.; Lempert, K. // Acta Chim. Acad. Sci. Hung. 1970. Vol. 64. N. 4. P. 369-391], а также при нагревании выше 120°С [Doleschall, G.; Simon-Ormai, K. //Acta Chim. Acad. Sci. Hung. 1980. Vol. 104. N. 2. P. 107-11] это вещество способно циклизоваться в нужный нам синтеза 3-меркапто-1,2,4-триазино[5,6-b]индол 2.
Для проверки этого нами были проведены опыты по превращению ранее полученного нами вещества 24 в тиол 2.
1) Реакция в уксусной кислоте. Смесь 1 г (0,045 моль) вещества 24 и 30 мл уксусной кислоты кипятили в колбе с обратным холодильником при перемешивании на магнитной мешалке в течение 30 минут, при этом смесь осталась гетерогенной. После охлаждения до комнатной температуры осадок отфильтровали, промыли водой и сушили на воздухе. Выход 0,87 г (95,1% от теоретического), ПМР-спектр соответствует структуре 2 (спектр продукта О-012). Анализ продукта с помощью ТСХ в системе бензол-ацетон-гексан-уксусная кислота (2:2:1:0,2), где Rf вещества 2 составляет 0,90, a Rf вещества 24 - 0,12, также показал полноту протекания реакции.
2) Реакция в водной соляной кислоте. Смесь 1 г (0,045 моль) вещества 24, 12 мл концентрированной соляной кислоты и 25 мл воды кипятили при перемешивании на магнитной мешалке в течение 30 минут, при этом смесь осталась гетерогенной. После охлаждения до комнатной температуры осадок отфильтровывали и промывали последовательно 3 мл 5% раствора NaHCO3 и 5 мл воды. Вес после сушки - 0,85 г (93% от теоретического), спектр и ТСХ продукта соответствуют структуре 2 (спектр продукта О-013).
3) Реакция в кипящем диметилформамиде. Смесь 1 г (0,045 моль) вещества 24 и 6 мл ДМФ кипятили в течение часа при перемешивании на магнитной мешалке. Во время кипячения из первоначально образовавшегося коричневого раствора постепенно выпадает осадок. После охлаждения его отфильтровали, промыли 2×5 мл воды и сушили. Вес после сушки - 0,60 г (63% от теоретического), спектр и ТСХ продукта соответствуют структуре 2 (спектр продукта О-014).
На основании полученных данных мы предположили, что образовавшаяся в ходе одностадийного синтеза тиола 2 примесь вещества 24 может быть превращена в необходимый продукт взаимодействием с соляной кислотой. А поскольку соляная кислота в этой реакции применяется для подкисления реакционной смеси, то для получения чистого тиола 2 достаточно будет взять избыток соляной кислоты.
Смесь 0,1 моля изатина 11, 0,11 моля тиосемикарбазида, 0,15 моля карбоната калия кипятилась в 500 мл воды в течение 10 часов. Полученный раствор охладили до 50°С и постепенно (сильное вспенивание!) добавили 500 мл 10% соляной кислоты. Реакционную массу перемешивали 1 час при 50°С, охлаждали до комнатной температуры, выпавший осадок отфильтровывали, промывали 2×50 мл воды и сушили на воздухе. Выход 18.0 г (89,1% от теоретического). Судя по ТСХ и спектру (спектр продукта О-015), вещество представляет собой чистый тиол 2.
Повтор этого опыта дал аналогичные результаты: выход 18.2 г (90,3% от теоретического) (спектр продукта О-016).
Сравнение данного способа с разработанным нами ранее 2-стадийным способом показывает, что при практически равных выходах (около 89% при 2-стадийном и около 90% при одностадийном способе) одностадийный способ значительно проще, так как при этом исключается длительные операция фильтрования, промывки и сушки 3-тиосемикарбазона изатина, используется более безопасный карбонат калия вместо едкого натра.
Оптимизация процесса получения основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола путем исключения стадии кристаллизации гидробромида N-(2-бромэтил)-морфолина.
Как показали ранее полученные нами результаты, основание 3-(2-морфолино этилтио)-1,2,4-триазино[5,6-b]индола 1 одинаково хорошо получается при применении в качестве алкилирующего реагента как гидрохлорида N-(2-хлорэтил)морфолина 3, так и гидробромида N-(2-бромэтил)морфолина 4.
Однако, если процесс получения гидрохлорида N-(2-хлорэтил)морфолина 3 происходит в безводных растворителях (хлороформ, бензол), то в процессе получения гидробромида N-(2-бромэтил)морфолина 4 используется водный раствор бромистоводородной кислоты, избыток которой в ходе выделения продукта удаляется отгонкой. В то же время оптимизированная нами методика получения свободного основания 1 предполагает протекание реакции алкилирования в воде.
Это позволило нам предположить, что для упрощения процесса можно исключить стадию выделения и кристаллизации гидробромида 4. Таким образом, реакционная масса после отгонки избытка бромистоводородной кислоты разбавляется и вводится в таком виде в реакцию алкилирования.
В 3-горлую колбу емкостью 100 мл, снабженную мешалкой, термометром и капельной воронкой, загружают 20 мл 46%-ной бромистоводородной кислоты и охлаждают ее в бане с ледяной водой до 10-15°С. При перемешивании добавляют по каплям 5 мл (5,35 г; 0,04 моль) N-(2-гидрокси-этил)морфолина, регулируя охлаждение и скорость прибавления так, чтобы температура не превышала 35°С, что занимает 25-35 минут. Затем капельную воронку заменяют на нисходящий холодильник, соединенный с реакционной колбой с помощью вакуумированного дефлегматора длиной 30 см.
Колбу помещают в масляную баню, нагревают до кипения (температура бани 112°С, температура погона - 100°С) и отгоняют воду, поднимая в течение 2-х часов температуру бани до 116°С. Далее в течение 6-ти часов нагревают баню до 126°С, отгоняя разбавленную бромистоводородную кислоту (температура погона 110-123°С). Затем отгоняют азеотропную смесь бромистоводородной кислоты и воды (48%; температура бани - не более 135°С, температура погона 124-126°С) до полного прекращения отгонки. Реакционную смесь охлаждают до температуры ниже 100°С и добавляют к ней 35 мл воды, охлаждают полученный раствор до комнатной температуры и фильтруют через бумажный фильтр.
В стакан емкостью 500 мл, снабженный мешалкой, термометром и капельной воронкой, помещают 4,41 г (0,11 моль) NaOH и 90 мл воды и перемешивают до образования раствора. После охлаждения раствора до 20-25°С прибавляют 7,50 г (0,037 моль) 3-меркапто-1,2,4-триазино[5,6-b]индола и перемешивают в течение 10-15 минут до растворения, после чего добавляют по каплям из капельной воронки раствор 11,34 г (0,041 моль) гидробромида N-(2-бромэтил)-морфолина в 35 мл воды, регулируя скорость добавления так, чтобы температура смеси не превышала 30°С.
Реакционную смесь перемешивают при 18-20°С в течение суток, после чего массу разбавляют 250 мл воды и перемешивают в течение 15 минут. Выпавший осадок отфильтровывают и промывают водой (2×10 мл). Продукт сушат при комнатной температуре в вакуум-эксикаторе до постоянного веса. Получают 7,92 г (67,7% от теоретического, считая на 3-меркапто-1,2,4-триазино[5,6-b]индол) основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола в виде мелкокристаллического порошка желтого цвета (спектр продукта О-017).
Повторение дважды этой методики привело аналогичным результатам: 7,88 г (67,0% от теоретического) (спектр продукта О-018) и 7.92 г (67,7%) (спектр продукта О-019).
Сравнение этого способа с описанными нами ранее а) способом с применением гидрохлорида N-(2-хлорэтил)морфолина и б) способом с применением кристаллического гидробромида N-(2-бромэтил)-морфолина позволяет сделать следующие выводы:
1) Данный способ позволяет исключить использование едкого и токсичного реагента - хлористого тионила и избежать выделения в окружающую среду токсичных и агрессивных газов - сернистого газа и хлористого водорода.
2) Данный способ позволяет исключить операции перекристаллизации гидрохлорида N-(2-хлорэтил)морфолина или гидробромида N-(2-бромэтил)-морфолина.
Оптимизация получения гидробромида N-(2-бромэтил)-морфолина путем объединения стадий получения N-(2-гидроксиэтил)-морфолина и его реакции с бромистоводородной кислотой.
Реализация описанного нами в предыдущем разделе варианта оптимизации позволил нам далее предложить идею более существенного упрощения процесса.
Она основана на том, что предложенный нами ранее метод получения N-(2-гидроксиэтил)морфолина из морфолина и 2-хлоэтанола проводится в среде
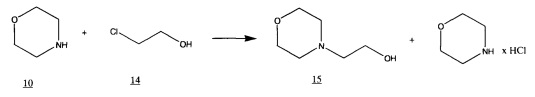
бензола, в котором образующийся гидрохлорид морфолина нерастворим и в ходе синтеза отделяется фильтрованием. Предположительно, фильтрат может содержать следующие компоненты: N-(2-гидроксиэтил)морфолин и остаточные количества исходных морфолина и 2-хлорэтанола. Хроматомасс-спектрометрический анализ образцов сырого N-(2-гидроксиэтил)-морфолина подтвердил отсутствие каких-либо других компонентов (см. Фиг. 1 - Фиг. 7, на которых показаны примеры Хроматомасс-спектрометрических анализов продуктов).
Образец исследовали на хроматомасс-спектрометре GCMS-QP2010 Plus фирмы "SHIMADZU" с программным обеспечением GCMSsolution для обработки данных и базой масс-спектральной информации NIST08.
Хроматографическое разделение проводили на капиллярной колонке Ultra-2 длиной 25 м и внутренним диаметром 0.2 мм.
Хроматограмма образца регистрировалась по полному ионному току в диапазоне масс от 30 до 450.
Условия хроматографирования:
• Температура инжектора - 280°С
• Начальная температура колонки - 40°С
• Время при начальной температуре - 3 минуты
• Скорость нагрева - 10°С/минуту
• Температура колонки - 120°С
• Время при этой температуре - 0 минут
• Скорость нагрева - 30°С/минуту
• Конечная температура колонки - 290°С
• Время при конечной температуре - 10.34 минут
• Газ-носитель Не - 1 мл/мин
• Деление потока 1:30
• Объем ввода пробы - 1 мкл.
Идентификацию примесей в образцах проводили путем сравнения масс-спектров разделенных компонентов смеси с масс-спектрами, содержащимися в компьютерной библиотеке NIST08, а также на основании анализа спектральной информации.
Суть предлагаемой нами оптимизации заключается в том, чтобы вводить в дальнейшую реакцию получения гидробромида N-(2-бромэтил)-морфолина вместо перегнанного в вакууме чистого N-(2-гидроксиэтил)-морфолина его раствор в бензоле. При этом бензол и непрореагировавший 2-хлорэтанол будут полностью отгоняться вместе с водой и бромистоводородной кислотой, а остаточные количества морфолина не будут участвовать далее в реакции алкилирования 3-меркапто-1,2,4-триазино[5,6-b]индола. Таким образом исключается стадия упаривания и вакуумной перегонки.
В колбу емкостью 100 мл, снабженную мешалкой, термометром и капельной воронкой, загружали 19,1 г (0,22 моль) морфолина и 40 мл бензола, нагревали до 60°С и при перемешивании добавляли в течение 20 минут 8,1 г (0,1 моль) 2-хлорэтанола, а затем реакционную смесь перемешивали при этой температуре в течение 1 часа, при этом происходило постепенное выпадение осадка гидрохлорида морфолина. Затем капельную воронку заменяли на обратный холодильник, и массу в течение 8 часов кипятили с перемешиванием. После охлаждения до комнатной температуры реакционную смесь фильтровали от осадка гидрохлорида морфолина, осадок промывали на фильтре бензолом (2×10 мл).
В 3-горлую колбу емкостью 250 мл, снабженную мешалкой, термометром и капельной воронкой, загружали 60 мл 46%-ной бромистоводородной кислоты и охлаждали ее в бане с ледяной водой до 10-15°С. При перемешивании добавляли по каплям бензольный раствор N-(2-гидрокси-этил)морфолина, регулируя охлаждение и скорость прибавления так, чтобы температура не превышала 35°С, что занимает 25-35 минут. Затем капельную воронку заменяют на нисходящий холодильник, соединенный с реакционной колбой с помощью вакуумированного дефлегматора длиной 30 см.
Колбу помещают в масляную баню, нагревают до кипения (температура бани 112°С, температура погона - 100°С) и отгоняют воду с бензолом, поднимая в течение 2-х часов температуру бани до 116°С. Далее в течение 6-ти часов нагревают баню до 126°С, отгоняя разбавленную бромистоводородную кислоту (температура погона 110-123°С). Затем отгоняют азеотропную смесь бромистоводородной кислоты и воды (48%; температура бани - не более 135°С, температура погона 124-126°С) до полного прекращения отгонки. Реакционную смесь охлаждают до температуры ниже 100°С и добавляют к ней 85 мл воды, охлаждают полученный раствор до комнатной температуры и фильтруют через бумажный фильтр.
В стакан емкостью 1 л, снабженный мешалкой, термометром и капельной воронкой, помещают 12,0 г (0,3 моль) NaOH и 220 мл воды и перемешивают до образования раствора. После охлаждения раствора до 20-25°С прибавляют 18,8 г (0,092 моль) 3-меркапто-1,2,4-триазино[5,6-b]индола и перемешивают в течение 10-15 минут до растворения, после чего добавляют по каплям из капельной воронки раствор гидробромида N-(2-бромэтил)-морфолина, регулируя скорость добавления так, чтобы температура смеси не превышала 30°С.
Реакционную смесь перемешивают при 18-20°С в течение суток, после чего массу разбавляют 550 мл воды и перемешивают в течение 15 минут. Выпавший осадок отфильтровывают и промывают водой (2×25 мл). Продукт сушат при комнатной температуре в вакуум-эксикаторе до постоянного веса. Получают 19,92 г (68,1% от теоретического, считая на 3-меркапто-1,2,4-триазино[5,6-b]индол) основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола в виде мелкокристаллического порошка желтого цвета (спектр продукта О-020).
Повторение этой методики привело аналогичным результатам: выход - 19,88 г (68,0% от теоретического).
Таким образом, предложенная нами оптимизация процесса позволяет без снижения выхода и качества продукта существенно упростить технологию синтеза свободного основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола, а именно:
1) Исключить операцию упаривания бензольного раствора N-(2-гидроксиэтил)-морфолина в вакууме.
2) Исключить операцию вакуумной перегонки N-(2-гидроксиэтил)-морфолина.
3) Исключить операцию вакуумной перегонки объединенных предгонов N-(2-гидроксиэтил)-морфолина.
Кроме того, данный вариант методики является более экологичным, поскольку в нем бензол, являющийся токсичным веществом, не загрязняет окружающую среду, а сохраняется в погоне и может быть использован повторно.
Оптимизация получения дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола путем исключения операции приготовления раствора HCl в диоксане.
При разработке метода получения дигидрохлорида 1 мы столкнулись с трудностями, поскольку его растворимость в органических растворителях резко повышалась в присутствии даже небольших количеств воды, что препятствовало кристаллизации и снижало выход целевого продукта.
Для повышения выхода нами была разработана методика, согласно которой раствор свободного основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола в этилацетате обрабатывался предварительно приготовленным 20% раствором HCl в диоксане. Выпадавший при этом мазеобразный осадок при дальнейшей обработке пропанолом-2 давал мелкокристаллический осадок дигидрохлорида 1.
Однако операция приготовления 20% раствора HCl в диоксане является трудоемкой и опасной, поскольку требует работы с хлористым водородом - едким и агрессивным газом, а пары диоксана при вдыхании вызывают сильные головные боли. Кроме того, эта процедура требует использования значительных количеств органических растворителей, которые не могут быть использованы повторно.
В поисках пути оптимизации этой процедуры мы обратили внимания на тот факт, что дигидрохлорид 1 практически нерастворим в концентрированной соляной кислоте.
Нами была исследована возможность получения кристаллического дигидрохлорида 1 в соляной кислоте различных концентраций. При этом выяснилось, что оптимальной концентрацией HCl является 31,5%.
10,7 г свободного основания 1 добавляли к 60 мл 31,5% соляной кислоты, при этом происходило незначительное разогревание смеси. Для полного образования раствора смесь нагревали до кипения, кипятили 10 минут, фильтровали через лавсановый фильтр, фильтрат при перемешивании охлаждали до 25°С, при этом частично происходила кристаллизация продукта. Для полноты осаждения фильтрат выдерживали 20 часов в холодильнике, выпавший осадок отфильтровывали, промывали на фильтре последовательно 5 мл пропанола-2 и 5 мл ацетона и сушили на воздухе до постоянного веса. Выход 9,43 г (66,4% от теоретического) (спектр продукта О-021).
При более высоких концентрациях HCl основание 1 плохо растворимо в соляной кислоте, при менее высоких концентрациях дигидрохлорид 1 слишком хорошо растворим.
Главным неудобством этого способа является выделение газообразного хлористого водорода при кипячении раствора и его фильтровании в горячем виде. Нам удалось устранить это неудобство добавлением небольшого количества пропанола-2. Было определено оптимальное количество добавляемого пропанола-2, при котором растворимость основания 1 остается достаточно высокой, а растворимость дигидрохлорида 1 на холоду - достаточно низкой.
Разработанный нами способ осуществляется на следующем примере.
В колбу емкостью 100 мл, снабженную мешалкой, термометром и обратным холодильником, загружают 58 мл концентрированной соляной кислоты, 12 мл воды и 13 мл пропанола-2. При перемешивании добавляют 10,69 г (0,034 моль) основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола. Реакционную смесь при перемешивании нагревают до кипения и кипятят 30 минут, после чего в горячем состоянии фильтруют через воронку Бюхнера.
Фильтрат помещают в стакан емкостью 100 мл и при перемешивании охлаждают до 25°С, после чего оставляют стоять в холодильнике в течение 20 часов. Выпавший осадок отфильтровывают, промывают последовательно 10 мл пропанола-2 и 10 мл ацетона и сушат на воздухе до постоянного веса. Получают 10,4 г (73,2% от теоретического) дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола в виде кристаллического вещества желтого цвета.
Повторение этой методики дало выход 73,4% от теоретического (спектры продуктов - О-022 и О-023, соответственно).
Анализ полученного таким образом дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола с помощью ВЭЖХ показал содержание основного вещества 99,9% (см. Фиг. 8, где показаны результаты анализа образца дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино-[5,6-b]индола с помощью ВЭЖХ).
Таким образом, применение оптимизированной процедуры получения дигидрохлорида 1 позволило нам исключить операции получения газообразного хлористого водорода и приготовления раствора HCl в диоксане, а также значительно снизить расход органических растворителей.
Таким образом, на основании вышеизложенного следует, что способ получения дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола состоит из стадий:
- стадия синтеза 3-тиосемикарбазона изатина из изатина и тиосемикарбазида в среде водной соляной кислоты с выходом около 97%;
- стадия синтеза 3-меркапто-1,2,4-триазино[5,6-b]индола циклизацией 3-тиосемикарбазона изатина в водно-щелочной среде с выходом около 92%;
- стадия синтеза N-(2-гидроксиэтил)морфолина из морфолина и 2-хлорэтанола в среде бензола с выходом около 87%;
- стадия синтеза гидрохлорида N-(2-хлорэтил)морфолина из N-(2-гидроксиэтил)морфолина и хлористого тионила в среде хлороформа с выходом около 70%;
- стадия синтеза гидробромида N-(2-бромэтил)морфолина из N-(2-гидроксиэтил)морфолина и бромистоводородной кислоты с выходом около 75%;
- стадия синтеза основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола из 3-меркапто-1,2,4-триазино[5,6-b]индола и N-(2-хлорэтил)морфолина в водно-щелочной среде с выходом около 68%;
- стадия синтеза основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола из 3-меркапто-1,2,4-триазино[5,6-b]индола и N-(2-бромэтил)морфолина в водно-щелочной среде с выходом около 68%;
- стадия синтеза дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола из основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола и 20% раствора HCl в диоксане с выходом около 74%;
Все стадии процесса получения дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола надежно воспроизводятся и обеспечивают высокую чистоту продуктов. Целевой продукт при этом получается с чистотой не менее 99%.
Способ получения дигидрохлорид моногидрат 3-(2-морфолиноэтилтио)-1,2,4-триазино[5,6-b]индола (трисан), характеризующийся взаимодействием 3-меркапто-1,2,4-триазино[5,6-b]индола с гидробромидом 2-морфолиноэтилбромида в присутствии избытка щелочи с получением основания 3-(2-морфолиноэтилтио)-1,2,4-триазино[5,6-b]индола, при этом последний обрабатывают соляной кислотой, отличающийся тем, что на емкость 100 мл, снабженную мешалкой, термометром и обратным холодильником, загружают 58 мл концентрированной соляной кислоты, 12 мл воды и 13 мл пропанола-2, а при перемешивании добавляют 10,69 г (0,034 моль) основания 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола; полученную реакционную смесь при перемешивании нагревают до кипения и кипятят 30 минут, после чего в горячем состоянии фильтруют, фильтрат помещают в иную емкость и при перемешивании охлаждают до 25°С, после чего оставляют стоять в холодильнике в течение 20 часов; выпавший осадок отфильтровывают, промывают последовательно пропанолом-2 и ацетоном, сушат на воздухе до постоянного веса; на выходе получают дигидрохлорид 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола в виде кристаллического вещества желтого цвета.![Способ получения дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола](https://fips.edrid.ru/images/rid/e6/3c/bb/3698bf591e1b7ea6040a85ae8cda2f9a.png)
![Способ получения дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола](https://fips.edrid.ru/images/rid/e6/3c/bb/8b9a22f3b6ee40e93b6e0e51d05cdac1.png)
![Способ получения дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола](https://fips.edrid.ru/images/rid/e6/3c/bb/6e9e02a4fee6941e74431477e146247f.png)
![Способ получения дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола](https://fips.edrid.ru/images/rid/e6/3c/bb/577d9adabad509a38746ed55c91d240d.png)
![Способ получения дигидрохлорида 3-(2-морфолино-этилтио)-1,2,4-триазино[5,6-b]индола](https://fips.edrid.ru/images/rid/e6/3c/bb/08450303ff55672ce0d86a73d8705282.png)
Способ диагностики инфекционных, аллергических, аутоиммунных и онкологических заболеваний
Способ оценки результатов кожных аллергических проб с инфекционными или неинфекционными аллергенами
Сердечно-мозговая питательная среда для диагностики инфекции в кровотоке и способ ее получения
Способ получения питательной среды для выделения гемокультуры при диагностике инфекции кровотока
Способ диагностики инфекционных, аллергических, аутоиммунных и онкологических заболеваний
Способ оценки результатов кожных аллергических проб с инфекционными или неинфекционными аллергенами

