Результат интеллектуальной деятельности: Способ дифференциальной и подтверждающей молекулярно-генетической диагностики врожденной аниридии и WAGR-синдрома
Вид РИД
Изобретение
Изобретение относится к молекулярной генетике, офтальмологии и может быть использовано в практической медицине. Изобретение предназначено для подтверждающей и дифференциальной ДНК-диагностики врожденной аниридии и WAGR-синдрома. Аниридия - это врожденное или приобретенное отсутствие радужной оболочки глаза, приводящее к ухудшению зрения и ряду неприятных осложнений.
Врожденные пороки развития (ВПР) органа зрения являются одной из основных причин инвалидности в детском возрасте с малой эффективностью лечения и высокой распространенностью (не менее 2,7 чел. на 10000 населения). В связи с отсутствием для большинства ВПР органа зрения эффективной хирургической и терапевтической коррекции возрастает роль профилактических мероприятий, направленных на предотвращение появления повторных случаев заболевания в отягощенных семьях. Именно поэтому актуальной представляется разработка протоколов для диагностики заболеваний органа зрения, включая ВПР. С 2003 года в России зарегистрировано около 300 случаев врожденной аниридии (ВА).
ВА - наследственное заболевание с аутосомно-доминантным типом наследования, встречающееся в популяции с частотой 1:45000-100000 населения. ВА - это моногенный или хромосомный множественный порок развития, затрагивающий в большинстве случаев все структуры глаза (гипоплазия макулы, врожденная катаракта, кератопатия, гипоплазия зрительного нерва, глаукома и другие), ЦНС (в 10-17% случаев грубые неврологические нарушения и задержка развития), поджелудочную железу (снижение секреторной функции, у некоторых пациентов нарушена толерантность к глюкозе из-за изменения уровней секреции глюкагона и инсулина, иногда развивается сахарный диабет), щитовидную железу и гипофиз, мочеполовую и другие системы (менее 10%) [1, 2].
ВА в 75% случаев встречается как изолированный порок развития глаза без объективного визуализированного вовлечения других органов и систем, в 20-24% отмечается синдромальная ВА (включая WAGR синдром). Как изолированная, так и синдромальная ВА обусловлена гетерозиготными мутациями (когда одной копии мутации достаточно для проявления симптомов заболевания) в гене РАХ6 (Paired box 6 -тканеспецифический транскрипционный фактор), в том числе хромосомными перестройками, вовлекающими регион 11р13, в котором расположен локус гена РАХ6 (локус - местоположение определенного гена на генетической или цитологической карте хромосомы). В 13% случаев ВА встречается как часть синдрома WAGR [аббревиатура WAGR составлена из первых букв основных проявлений: опухоль Вильмса (W), аниридия (А), мочеполовые нарушения (G), умственная отсталость (R)]. Синдром WAGR обусловлен делецией региона 11р13, захватывающей локусы гена РАХ6 и гена WT1 предрасположенности к развитию опухоли Вильмса (нефробластома). WAGR-синдром встречается, в основном, в виде спорадических случаев. Во всех спорадических случаях до установления генетической причины больной аниридией имеет 50% риск развития нефробластомы до 8 лет жизни [4]. Высокий риск и злокачественность опухоли почки, составляющей синдром WAGR, определяют важность проведение ранней дифференциальной диагностики.
В 2-5% случаев аниридия ассоциирована с другими моногенными и хромосомными синдромами, более редкими по частоте [3]. Среди них в международной базе данных OMIM (медицинская база данных, в которой собирается информация об известных заболеваниях с генетическим компонентом и генах, ответственных за их развитие) выделено порядка 25 различных форм.
Известен «Список основных синдромов детектируемых молекулярно-цитогенетическим методом» (хромосомный микроматричный анализ) с использованием микроматрицы CGX™ - HD (4×180 K):
- Аниридия: OMIM: 106210, Ген/локус: РАХ6, Локализация: 11р13;
- Опухоль Вильмса с аниридией, генитальными аномалиями OMIM: 194072 Ген/локус: РАХ6, WT1, Локализация: 11р 13
(http://microarray.kz/syndromes.html)
Известно использование генетических анализов для подтверждения наследственной аниридии, включающее проведение FISH-теста и исследования на определение типа дефекта гена РАХ6.
(http://www.krasotaimedicina.ru/diseases/ophthalmology/aniridie).
Известен также способ проведения генетического анализа (дифференциальной молекулярно-генетической диагностики), при котором на основании результатов исследования образца биологического материала, полученных методом MLPA, производят дальнейшие действия, выбранные из группы диагностических инструментов, обеспечивающей дифференциацию возможных диагнозов(RU 2562866).
Известно решение, которое указывает врожденный ген, вызывающий заболевание аниридией (aniridia), набор для обнаружения гена, вызывающего болезнь, вызывающий врожденную аниридию, и применение этого гена. Врожденный ген, которым обусловлено заболевание аниридией, представляет собой мутационный ген РАХ6. Там же известен набор для обнаружения врожденного гена, вызывающего заболевание аниридией, а также для изучения патогенетического молекулярного механизма. Способ предусматривает сквенирование экзона (нуклеотидная последовательность экзона 6) и исследование родственников. Набор праймеров включает два праймера (CN103937804, прототип).
В имеющемся уровне техники значительную сложность в молекулярном подтверждении диагноза ВА представляет гетерогенность молекулярных механизмов повреждения гена РАХ6 и отсутствие «мажорных» мутаций. Генетические повреждения включают в себя не только точковые мутации, но и крупные хромосомные перестройки с вовлечением региона 11р13, в ряде случаев затрагивающие не кодирующую часть гена РАХ6, а дистальный цис-регуляторный район, находящийся на расстоянии 200 тыс. пар нуклеотидов (п.н.) от гена. Кроме того, как и при других аутосомно-доминантных заболеваниях, при ВА частота соматического мозаицизма по патогенной мутации может достигать 17,5% спорадических случаев [5]. Оценка делеций гена WT1 региона 11р13 и диагностика WAGR-синдрома не проводится. В целом недостатками известных решений являются недостаточный объем поиска возможных причин врожденной аниридии, низкие достоверность, разрешающая способность и дифференцируемость получаемых результатов (синдромальной и изолированной аниридии), обусловленные в частности малым количеством микросателлитных маркеров, праймеров для секвенирования, и тем, что не учитываются в необходимом объеме и взаимосвязи изменения гена WT1 и гена РАХ6, а также то, что сложные фенотипы аниридии могут перекрываться с клиническими фенотипами, ассоциированными с изменениями в других генах.
Технической проблемой, разрешаемой с помощью настоящего изобретения, является разработка эффективного подтверждающего протокола и дифференциальной (от дисгенеза переднего отрезка глаза, ассоциированного с мутациями в других генах) диагностики врожденной аниридии и WAGR-синдрома с учетом выраженного клинического полиморфизма и молекулярной гетерогенности, лежащей в основе повреждения функции генов, ассоциированных с дисгенезией (развитие с отклонением от нормы) переднего отрезка глаза (роговица, радужная оболочка, хрусталик).
Технический результат, обеспечивающий разрешение указанной проблемы, основан на способе (подтверждающем протоколе) дифференциальной диагностики врожденной аниридии и WAGR-синдрома с учетом выраженного клинического полиморфизма проявлений врожденной аниридии (изолированной или синдромальной) и генетической гетерогенности, лежащей в основе повреждения функции генов, ассоциированных с дисгенезией переднего отрезка глаза.
Дифференциальная идентификация патогенных изменений кодирующей последовательности или делеции региона 11р13, в частности, гена РАХ6, подтверждает диагноз изолированной аниридии, определение делеции (хромосомной перестройки, при которых происходит потеря участка 11 хромосомы) региона 11р13, захватывающей гены РАХ6 и WT1, предполагает возможный диагноз WAGR-синдрома, которым обусловлен высокий риск развития опухоли почки. При этом в заявляемом способе процедура последовательного применения молекулярно-биологических техник направлена на приоритетную проверку генетических нарушений, ассоциированных с тяжелыми и синдромальными формами аниридии и, в случае их исключения, на поиск причин заболевания, угрожающего жизни в меньшей степени - врожденной аниридии.
Согласно заявляемому способу дифференциальная идентификация патогенных изменений кодирующей последовательности гена РАХ6 или делеции региона 11p13, в частности, захватывающещей ген РАХ6 или его дистантные регуляторные области, подтверждает диагноз изолированной аниридии, определение делеции (хромосомной перестройки, при которых происходит потеря участка хромосомы) региона 11р13, захватывающей гены РАХ6 и WT1, предполагает возможный диагноз WAGR-синдрома, которым обусловлен высокий риск развития опухоли почки (нефробластомы). При этом процедура последовательного применения молекулярно-биологических техник направлена на приоритетную проверку генетических нарушений, ассоциированных с тяжелыми и синдромальными формами аниридии и, в случае их исключения, на поиск менее тяжелых причин врожденной аниридии.
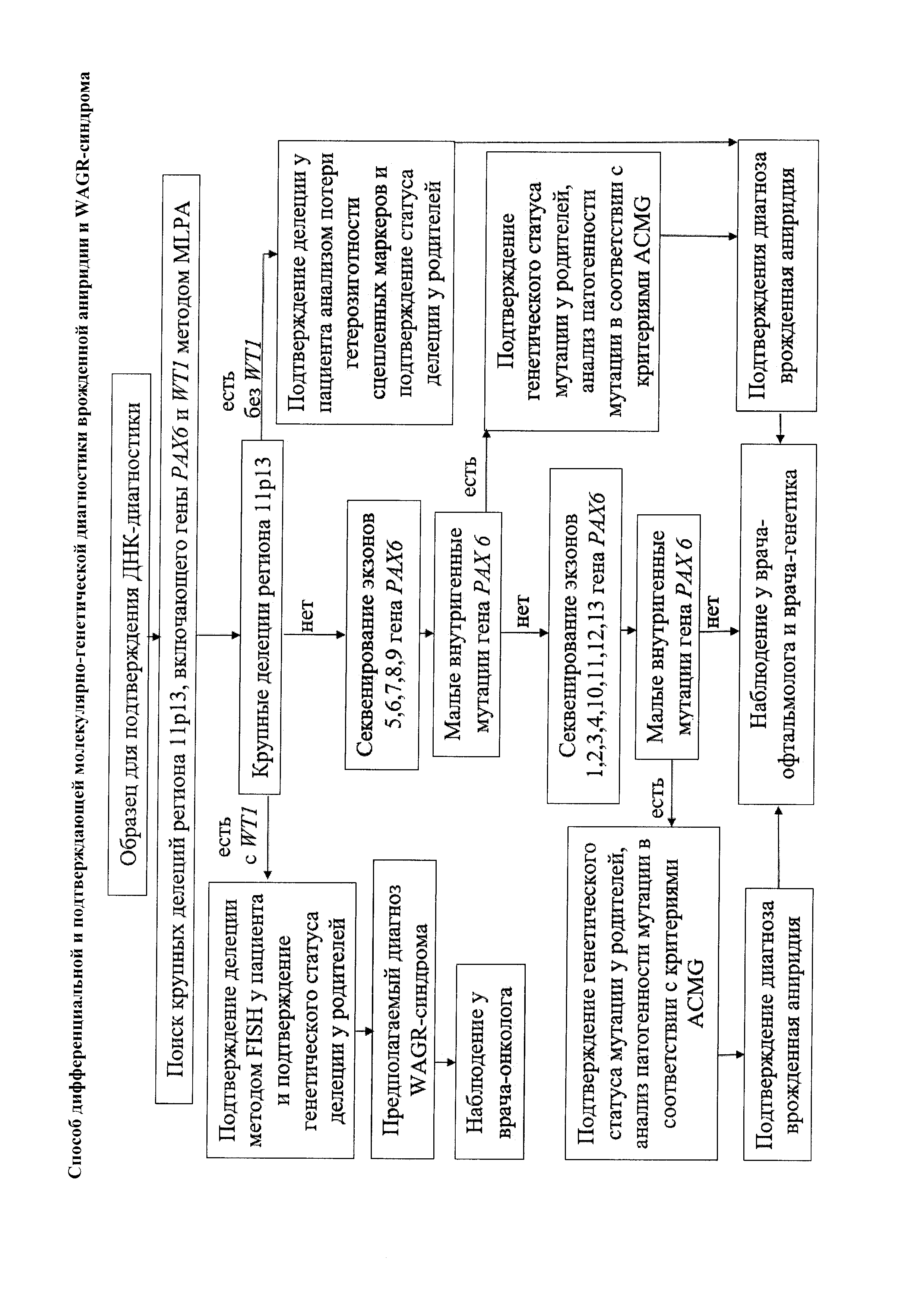
Сущность изобретения заключается в том, что способ дифференциальной и подтверждающей молекулярно-генетической диагностики врожденной аниридии и WAGR-синдрома предусматривает, что у пациента, имеющего клинические признаки аниридии, производят забор образца биологического материала для проведения ДНК-диагноститки, в отношении которого первоначально осуществляют поиск крупных делеций с размером по меньшей мере в одну тысячу пар нуклеотидов региона 11р13 методом MLPA, и на основании результатов этого поиска производят дальнейшие мероприятия, выбранные из следующей группы диагностических инструментов:
- в случае обнаружения крупной делеции региона 11р13, захватывающей локусы генов WT1 и РАХ6, и подтверждения делеции на генетическом материале пациента методом флуоресцентной in situ гибридизации (FISH) с зондом, специфичным к гену WT1, пациенту выставляется предположительный диагноз WAGR-синдрома;
- в случае обнаружения крупной делеции региона 11р13, не захватывающих локус гена WT1 и захватывающей локус гена РАХ6 или его дистантные регуляторные области, и подтверждения делеции на генетическом материале пациента методом анализа потери гетерозиготности по микросателлитным маркерам, пациенту выставляется клинический диагноз врожденной аниридии;
- в случае отсутствия крупных делеций региона 11р13 проводят секвенирование экзонов и фланкирующих их участков интронов гена РАХ6 для поиска малых внутригенных мутаций этого гена, при этом проводят секвенирование экзонов из по меньшей мере одной из групп: группа экзонов 5, 6, 7, 8, 9 и/или группа экзонов 1, 2, 3, 4, 10, 11, 12, 13, а при обнаружении в результате секвенирования хотя бы одной малой внутригенной мутации осуществляют проверку патогенности обнаруженной генетических повреждений в гене РАХ6 в соответствии с критериями ACMG (Американского колледжа молекулярной генетики), и при свидетельстве патогенности мутации пациенту подтверждается клинический диагноз врожденной аниридии.
Как правило, производят забор образца биологического материала из группы: венозной или капиллярной крови, слюны, буккального соскоба, осадка эпителия нижних мочевыводящих путей.
Предпочтительно, для подтверждения патогенности и клинической значимости выявленной делеции или иной мутации определяется генетический статус этого изменения у родителей.
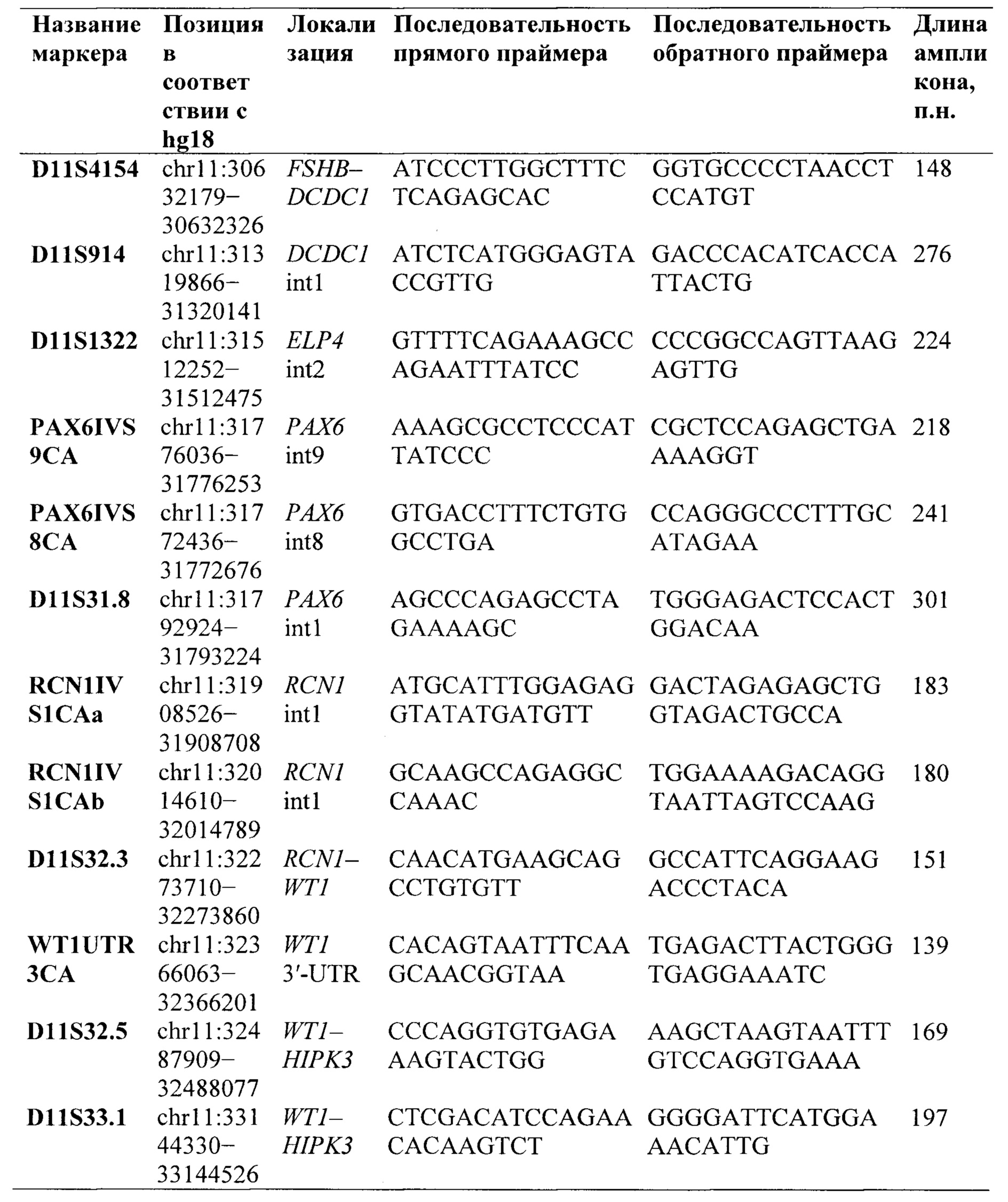
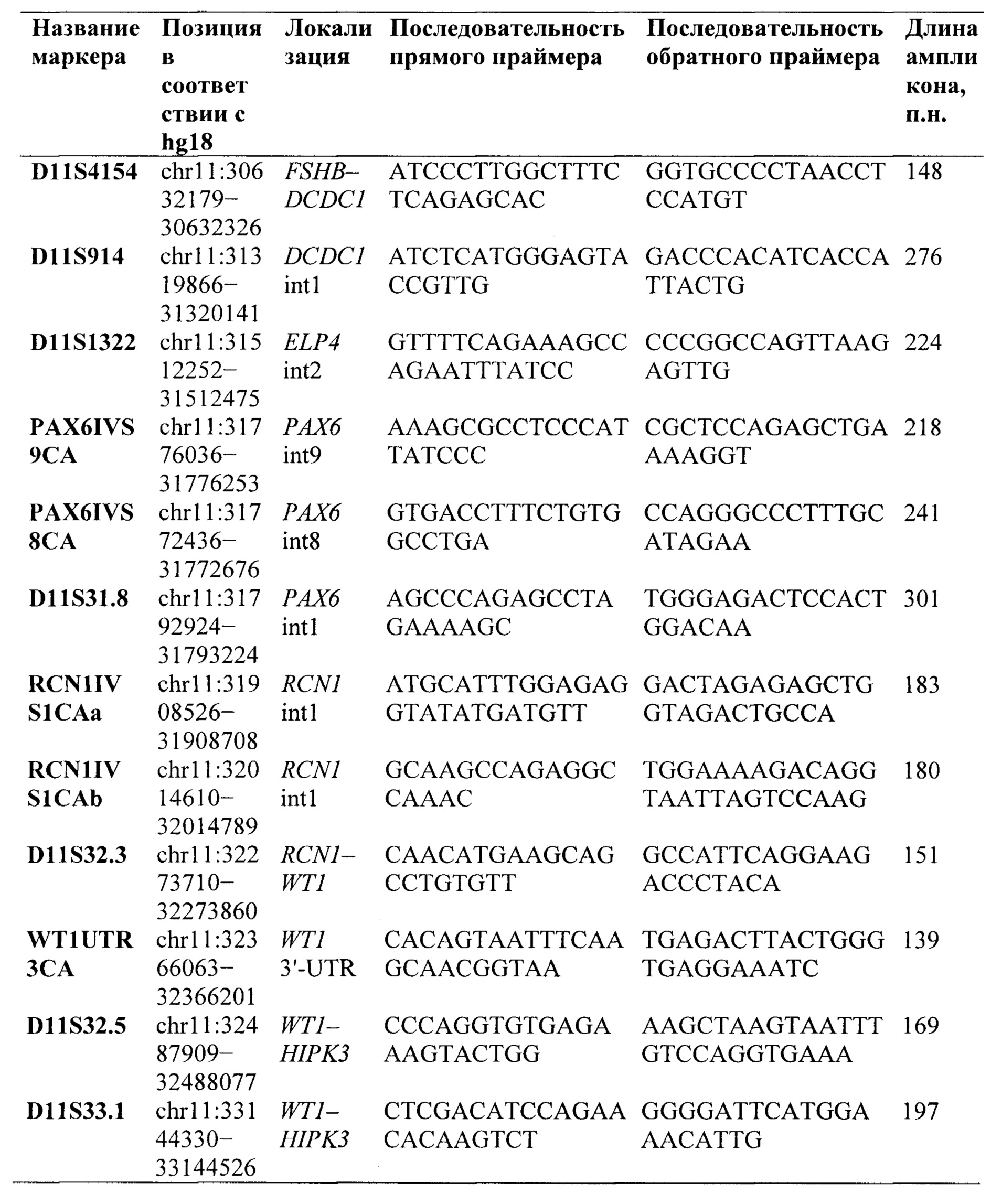
Предпочтительно, в качестве микросателлитных маркеров для анализа потери гетерозиготности носителей крупной делеции региона 11p13 по микросателлитным маркерам выбирают микросателлитные маркеры из группы:

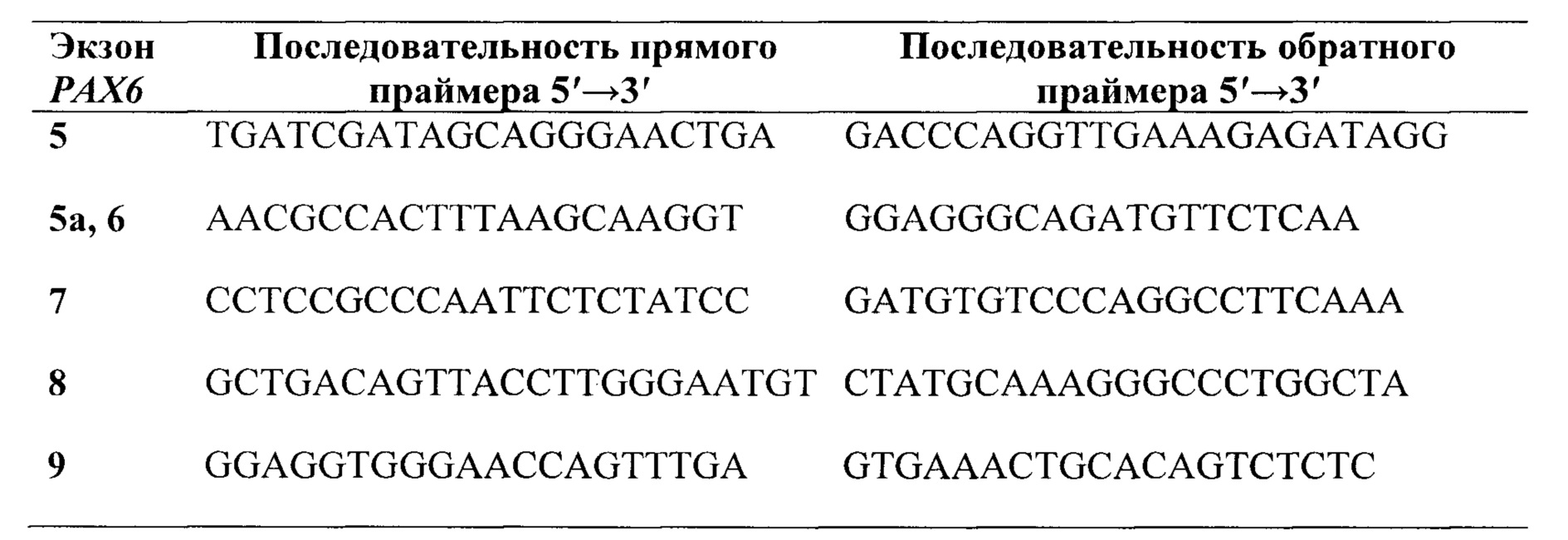
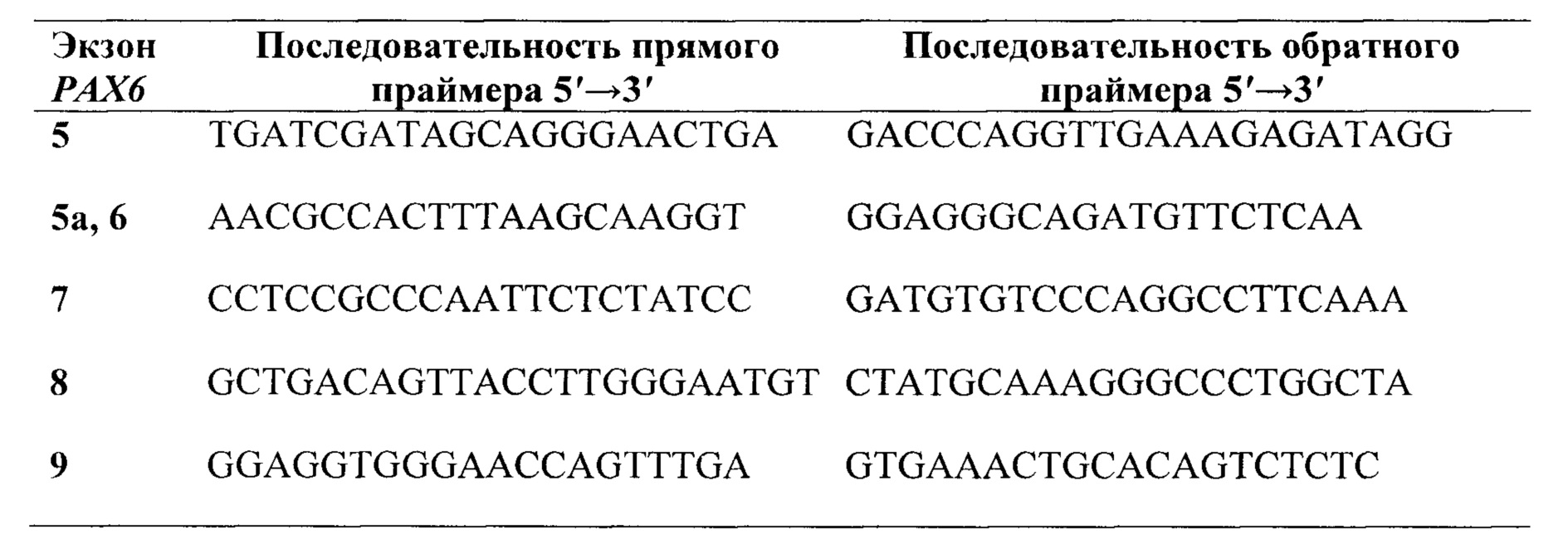
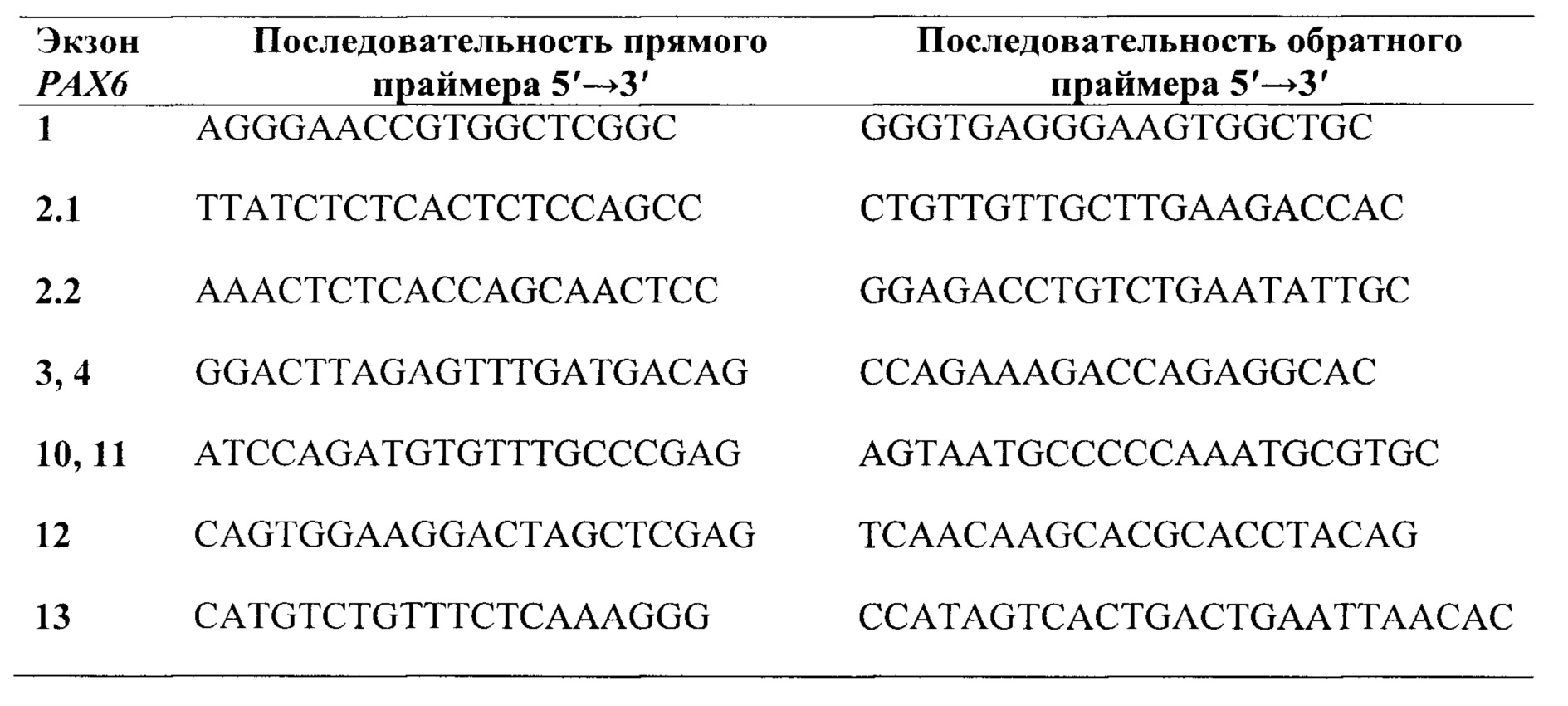
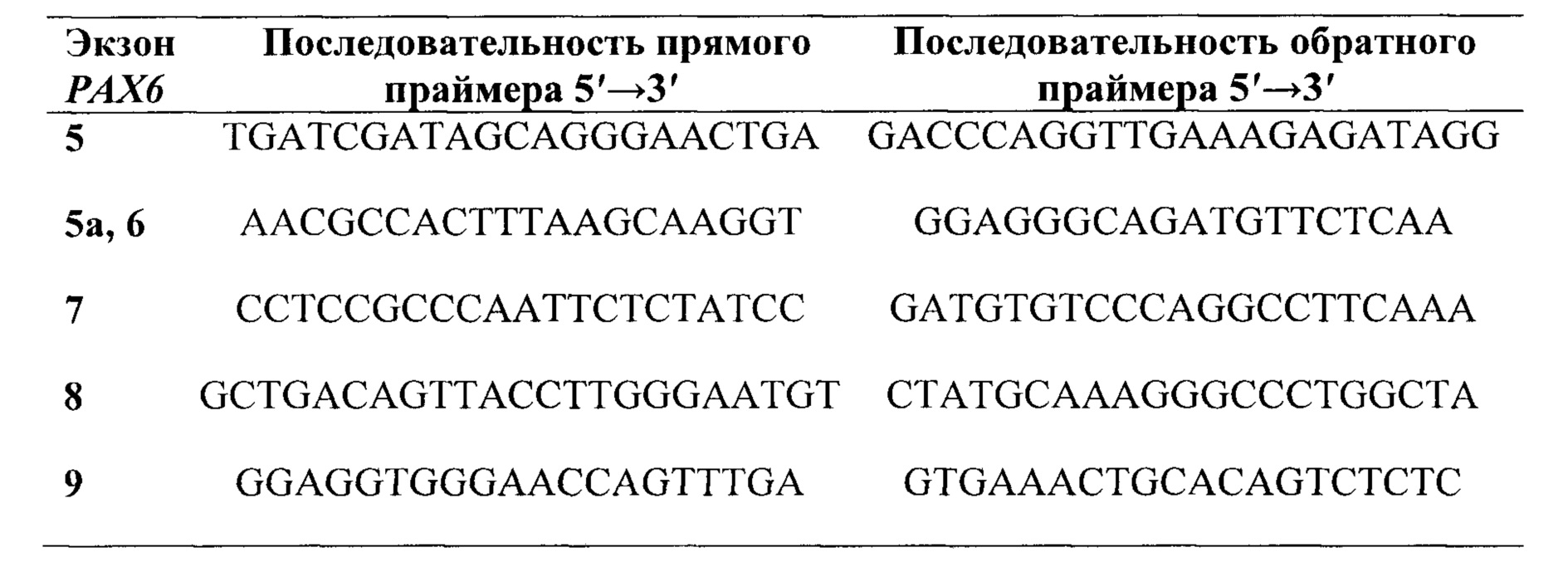
Предпочтительно, в качестве праймеров для секвенирования экзонов группы 5, 6, 7, 8 и 9 и фланкирующих их участков интронов гена РАХ6, выбирают праймеры из группы:
Предпочтительно, проводят секвенирование экзонов группы 1, 2, 3, 4, 10, 11, 12, 13 и фланкирующих их участков интронов гена РАХ6 в случае отсутствия малых внутригенных мутаций в результате секвенирования экзонов группы 5, 6, 7, 8, 9 и фланкирующих их участков интронов гена РАХ6.
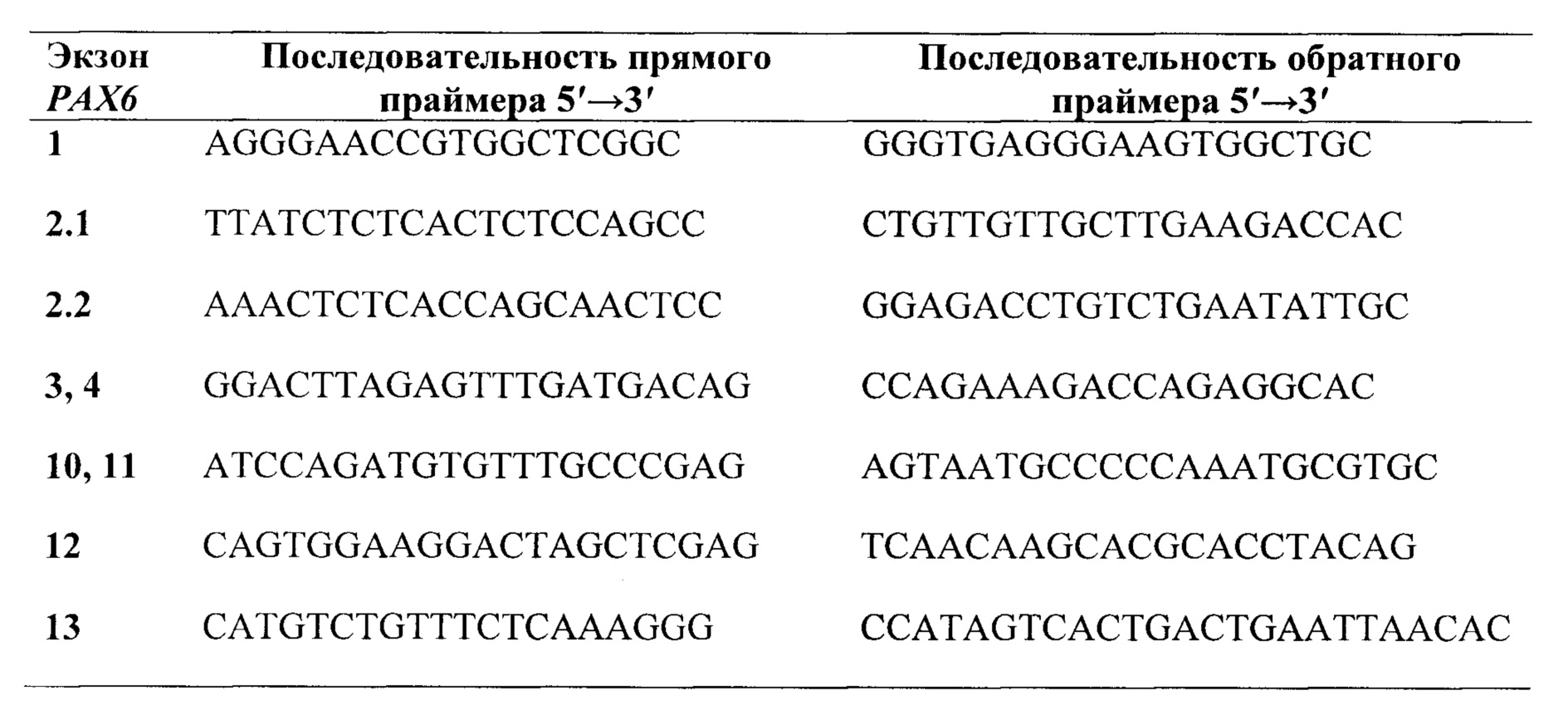
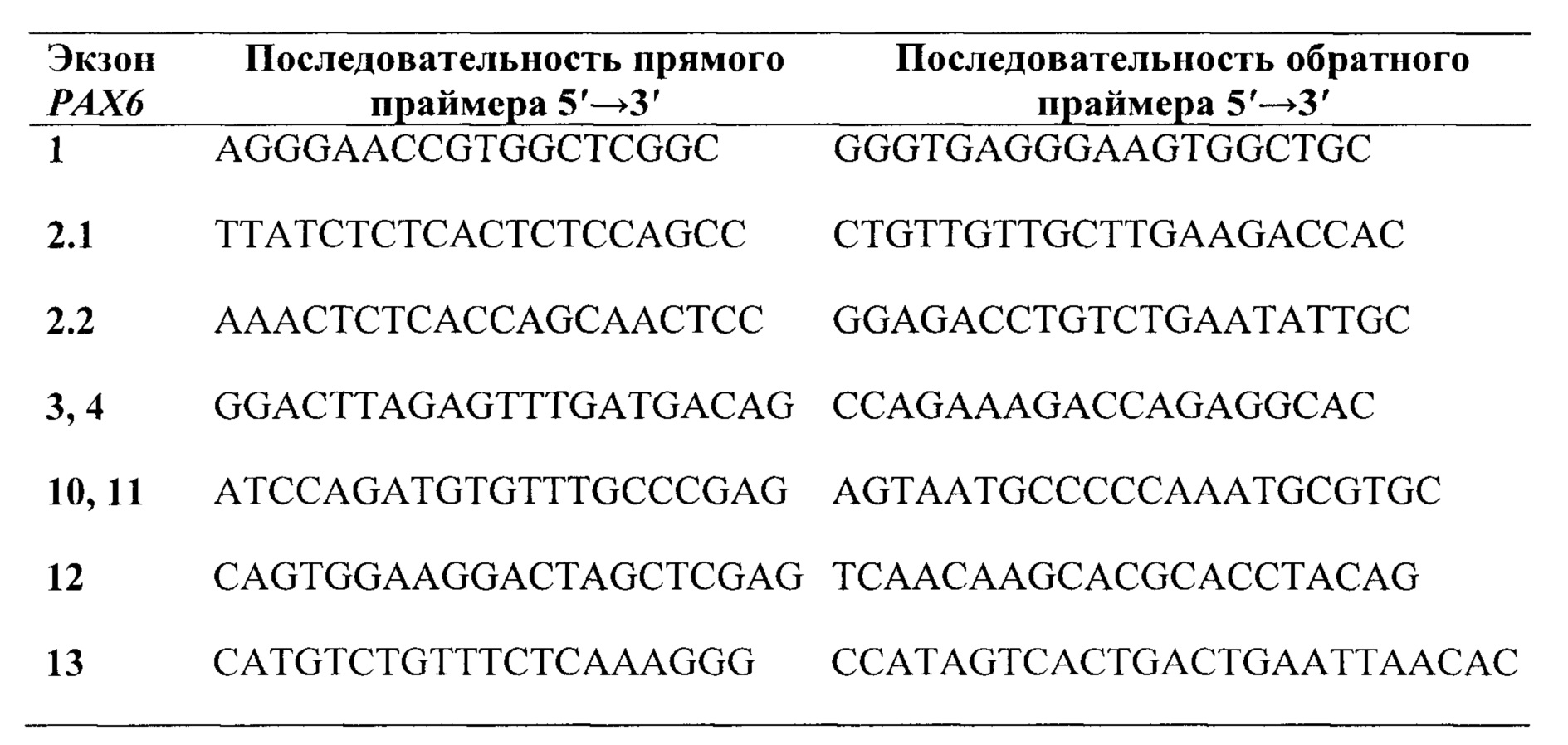
Предпочтительно, в качестве праймеров для секвенирования кодирующих экзонов группы 1, 2, 3, 4, 10, 11, 12, 13 и фланкирующих их участков интронов гена РАХ6, выбирают праймеры из группы:
На чертеже приведена блок-схема общей последовательности диагностических процедур для молекулярно-генетическая диагностика врожденной аниридии и WAGR-синдрома.
Способ дифференциальной и подтверждающей молекулярно-генетической диагностики врожденной аниридии и WAGR-синдрома реализуется следующим образом.
Причиной для принятия решения о проведении молекулярно-генетической диагностики обычно является подозрение на генетические причины возникновения заболевания - особенности истории болезни, клиническая картина и т.д. Также проведение молекулярно-генетической диагностики может быть назначено врачом с целью вовремя начать профилактику и/или повысить эффективность лечения, а также предупредить передачу заболевания в последующих поколениях при установлении точной генетической причины патологии, когда становится возможной последующая преимплантационная или пренатальная генетической диагностики у потомства.
У пациента (пробанда), имеющего клинические признаки аниридии, производят забор образца биологического материала для проведения ДНК-диагноститки. В качестве образца производят забор биологического материала из группы: венозной или капиллярной крови, слюны, буккального соскоба, осадка эпителия нижних мочевыводящих путей.
В отношении взятого образца первоначально осуществляют поиск крупных делеций с размером по меньшей мере в одну тысячу пар нуклеотидов региона 11p13 высокоточным методом реакции лигазозависимой амплификации зондов (MLPA). MLPA-анализ является методом выбора для одновременного определения протяженных вариаций числа копий тех или иных регионов в геноме. Технология MLPA представляет собой мультиплексную лигазно-зависимую амплификацию зондов (от англ. Multiplex ligation-dependent probe amplification - MLPA).
На основании результатов поиска методом MLPA рассматривают ситуации, при которых обнаружена крупная делеция региона 11р13, захватывающая локусы генов WT1 и РАХ6, или обнаружена крупная делеция, не захватывающая локус гена WT1, но захватывающая локус гена РАХ6 или его дистантные регуляторные области, или отсутствуют крупные делеции региона 11р13.
На основании конкретных результатов поиска методом MLPA производят дальнейшие мероприятия, направленные на дифференциальную идентификацию патогенных изменений кодирующей последовательности или делеции региона 11p13, в частности, гена РАХ6, подтверждает диагноз изолированной аниридии, определение делеции (хромосомной перестройки, при которых происходит потеря короткого плеча 11 хромосомы) региона 11р13, захватывающей гены РАХ6 и WT1, предполагает возможный диагноз WAGR-синдрома, которым обусловлен высокий риск развития опухоли почки (нефробластомы).
При этом процедура последовательного применения молекулярно-биологических техник направлена сначала на приоритетную проверку генетических нарушений, ассоциированных с тяжелыми и синдромальными формами аниридии и, в случае их исключения, на поиск менее тяжелых причин врожденной аниридии с помощью выбранных из приведенной далее группы диагностических инструментов.
В случае обнаружения крупной делеции региона 11p13, захватывающей локусы генов WT1 и РАХ6, и подтверждения делеции на генетическом материале пациента методом флуоресцентной in situ гибридизации (FISH) с зондом, специфичным к гену WT1, а также определения генетического статуса этой делеции у родителей. Последнее (так же и далее) позволяет определить, имеется ли у пациента спонтанная мутация в случае спорадической семейной истории или же наследуемая в случае семейных случаев заболевания. Для определения генетического статуса родителей при генетическом консультировании требуются сбор подробного анамнеза, данных объективного обследования членов семьи и проведение биохимических, рентгенологических или гистологических исследований. Проводятся подтверждение патогенности впервые найденных внутригенных мутаций в гене РАХ6 с помощью анализирующих программ, доказательства отсутствия данного изменения в выборке здоровых людей и/или у здоровых членов семьи.
Определяются мутации пробанда у больных членов семьи, если имеется отягощенный семейный анамнез. В случае, когда очевидной истории нет, необходимо подтвердить статус мутации de novo доказательством отсутствия мутации пробанда у родителей, а также подтвердить биологическое родительство. На этом же этапе проводится анализ возможного соматического мозаицизма у родителей.
Пробанду при определении генетического статуса этой делеции у родителей выставляется предположительный диагноз WAGR-синдрома (включающего синдромальную аниридию). В случае обнаружения крупной делеции региона 11р13, не захватывающих локус гена WT1 и захватывающей локус гена РАХ6 или его дистантные регуляторные области, и подтверждения делеции на генетическом материале пациента методом анализа потери гетерозиготности по микросателлитным маркерам, а также определения (подтверждения) генетического статуса этой делеции у родителей (как изложено выше).
В качестве микросателлитных маркеров для анализа потери гетерозиготности носителей крупной делеции региона 11р13 по микросателлитным маркерам выбирают и используют, предпочтительно, микросателлитные маркеры из группы:
Такому пациенту выставляется клинический диагноз врожденной аниридии (изолированной).
В случае отсутствия крупных делеций региона 11р13 проводят секвенирование экзонов и фланкирующих их участков интронов гена РАХ6 для поиска малых внутригенных мутаций, а именно: малых делеций или инсерций размером до пятидесяти пар нуклеотидов, точковых мутаций этого гена.
При этом сначала проводят исследование экзонов из группы экзонов 5, 6, 7, 8, 9 прямым секвенированием по Сэнгеру.
В качестве праймеров для секвенирования экзонов группы 5, 6, 7, 8 и 9 и фланкирующих их участков интронов гена РАХ6 выбирают и используют, предпочтительно, праймеры из группы:
При обнаружении в результате секвенирования экзонов 5, 6, 7, 8, 9 варианта нуклеотидной последовательности осуществляют проверку его патогенности в соответствии с критериями ACMG (Американского колледжа молекулярной генетики), в том числе путем определения (подтверждения) генетического статуса этой мутации у родителей (как описано выше), и при свидетельстве патогенности мутации пациенту подтверждается клинический диагноз врожденной аниридии (изолированной).
В случае отсутствия малых внутригенных мутаций в результате секвенирования экзонов группы 5, 6, 7, 8, 9 и фланкирующих их участков интронов гена РАХ6 проводят секвенирование экзонов группы 1, 2, 3, 4, 10, 11, 12, 13 и фланкирующих их участков интронов гена РАХ6, а при обнаружении в результате секвенирования экзонов 1, 2, 3, 4, 10, 11, 12, 13 варианта нуклеотидной последовательности осуществляют проверку его патогенности в соответствии с критериями ACMG (Американского колледжа молекулярной генетики), в том числе путем определения (подтверждения) генетического статуса этой мутации у родителей, и при свидетельстве патогенности мутации пациенту подтверждается клинический диагноз врожденной аниридии (изолированной).
В качестве праймеров для секвенирования кодирующих экзонов группы 1, 2, 3, 4, 10, 11, 12, 13 и фланкирующих их участков интронов гена РАХ6, выбирают и используют, предпочтительно, праймеры из группы:
При обнаружении ранее не описанных вариантов нуклеотидной последовательности возможна дополнительная проверка патогенности с помощью анализирующих программ. В этом случае уточняется международный перечень клинически значимых патогенных генетических повреждений.
В случае отрицательных результатов в объеме действий по реализации настоящего способа, не приведшем к выявлению крупных делеций региона 11p13 и малых внутригенных мутаций гена РАХ6, но при наличии клинической картины врожденной аниридии, биоматериал пациента может быть направлен для дальнейшей молекулярно-генетической диагностики по назначению врачом-генетиком.
Каждому пациенту, имеющему клинические признаки аниридии, даются рекомендации постоянного наблюдения у врача-офтальмолога и врача-генетика, а в том случае, если пациенту выставляется предположительный диагноз WAGR-синдрома, дополнительно даются рекомендации наблюдения у врача-онколога как минимум до 8 лет.
Последовательность действий настоящего способа представляет собой сущность подтверждающего протокола диагностики врожденной аниридии и WAGR-синдрома с учетом выраженного клинического полиморфизма и генетической гетерогенности, лежащей в основе повреждения функции генов, ассоциированных с дисгенезом переднего отрезка глаза.
В результате использования предлагаемого способа оптимальным путем реализуется обоснованная и доказательно подкрепленная дифференциальная диагностика врожденной аниридии и WAGR-синдрома с учетом выраженного клинического полиморфизма проявлений врожденной аниридии (изолированной или синдромальной) и генетической гетерогенности, лежащей в основе повреждения функции генов, ассоциированных с дисгенезией переднего отрезка глаза.
При этом дифференциальная идентификация патогенных изменений кодирующей последовательности или делеции региона 11р13, в частности, гена РАХ6, подтверждает диагноз изолированной аниридии, определение делеции (хромосомной перестройки, при которых происходит потеря участка 11 хромосомы) региона 11р13, захватывающей гены РАХ6 и WT1, предполагает возможный диагноз WAGR-синдрома, которым обусловлены высокий риск развития опухоли почки - опухоль Вильмса (W), аниридия (А), мочеполовые нарушения (G), умственная отсталость (R). При этом процедура последовательного применения молекулярно-биологических техник направлена на приоритетную проверку генетических нарушений, ассоциированных с тяжелыми и синдромальными формами аниридии и, в случае их исключения, - на поиск менее тяжелых причин врожденной аниридии. Таким образом, использование данного способа позволяет вовремя начать профилактику, или повысить эффективность лечения или предупредить передачу заболевания в последующих поколениях при установлении точной генетической причины патологии.
СПИСОК ЛИТЕРАТУРЫ
1. Shimo, N., et al., Aniridia with a heterozygous PAX6 mutation in which the pituitary function was partially impaired. Intern Med, 2014. 53(1): p. 39-42.
2. Netland, P.A., et al., Ocular and systemic findings in a survey of aniridia subjects. J AAPOS, 2011. 15(6): p. 562-6.
3. Kokotas, H. and M.B. Petersen, Clinical and molecular aspects of aniridia. Clin Genet, 2010. 77(5): p. 409-20.
4. Gronskov, K., et al., Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum Genet, 2001. 109(1): p. 11-8.
5. Qin, L., et al., Detection and Quantification of Mosaic Mutations in Disease Genes by Next-Generation Sequencing. J MolDiagn, 2016. 18(3): p. 446-53.
6. Desmet, F.O., et al., Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res, 2009. 37(9): p. e67.
7. Richards, S., et al., Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015. 17(5): p. 405-24.
8. Redeker, E.J., et al., Multiplex ligation-dependent probe amplification (MLPA) enhances the molecular diagnosis of aniridia and related disorders. Mol Vis, 2008. 14: p. 836-40.
9. Willems, Т., et al., The landscape of human STR variation. GenomeRes, 2014. 24(11): p. 1894-904.
Ингибиторы калиевых каналов как лекарственные средства при лимфомах и лейкозах
Композиция для восстановления дефектов костной ткани на основе аденовирусных конструкций, несущих кднк вмр-2, фибринового геля и синтетического β-трикальцийфосфата и способ ее получения
Определение содержания gc-богатой последовательности генома (gc-днк) в составе циркулирующей внеклеточной днк плазмы периферической крови как способ определения уровня гибели клеток при беременности
Способ диагностики светлоклеточного почечно-клеточного рака
Способ диагностики светлоклеточного почечно-клеточного рака и способ прогнозирования метастазирования на основе группы генов микрорнк
Способ анализа дифференциально метилированных геномных участков в биологических образцах костного мозга и крови детей с острым миелоидным лейкозом
Способ получения ноотропной композиции на основе полипептидных комплексов, выделенных из нейронов и глиальных клеток, полученных методом направленной дифференцировки индуцированных плюрипотентных стволовых клеток человека
Ноотропная композиция на основе полипептидных комплексов, выделенных из нейронов и глиальных клеток, полученных методом направленной дифференцировки индуцированных плюрипотентных стволовых клеток человека
Способ прогнозирования выживаемости больных светлоклеточным почечно-клеточным раком
Ингибиторы калиевых каналов как лекарственные средства при лимфомах и лейкозах
Композиция для восстановления дефектов костной ткани на основе аденовирусных конструкций, несущих кднк вмр-2, фибринового геля и синтетического β-трикальцийфосфата и способ ее получения
Определение содержания gc-богатой последовательности генома (gc-днк) в составе циркулирующей внеклеточной днк плазмы периферической крови как способ определения уровня гибели клеток при беременности
Способ диагностики светлоклеточного почечно-клеточного рака
Способ преимплантационной генетической диагностики спинальной мышечной атрофии типа 1
Способ преимплантационной генетической диагностики анемии фанкони
Способ прогнозирования риска развития преэклампсии у женщин с хронической артериальной гипертензией
Тест система для пренатального генетического тестирования на hla-совместимость
Способ преимплантационного генетического тестирования остеопетроза 4 типа
Способ преимплантационного генетического тестирования гемофилии а

