Результат интеллектуальной деятельности: НОВОЕ ИНДАНСУЛЬФАМИДНОЕ ПРОИЗВОДНОЕ
Вид РИД
Изобретение
Область техники
[0001] Настоящее изобретение относится к новым индансульфамидным производным и противоэпилептическому лекарственному средству, содержащему их.
Уровень техники
[0002] Эпилепсия представляет собой одно из наиболее распространенных расстройств центральной нервной системы, и в мире насчитывается более 50 миллионов больных. Согласно определению ВОЗ эпилепсия представляет собой "хроническое заболевание головного мозга различной этиологии, характеризующееся повторяющимися припадками (эпилептическими припадками), вследствие избыточных электрических разрядов нейронов головного мозга, которые сопровождаются рядом клинических и лабораторных проявлений".
[0003] В качестве эпилептических припадков известны, например, парциальные припадки, такие как простые парциальные припадки, сложные парциальные припадки, и вторичные генерализованные припадки, абсансы, миоклонические припадки, тонические припадки, клонические припадки, тонические припадки, тонико-клонические припадки, атонические припадки, синдром Уэста и синдром Леннокса-Гасто.
Основным направлением лечения эпилепсии является фармакотерапия с применением противоэпилептических средств (AED). Целью лечения эпилепсии является устранение припадков и предупреждение побочных действий в ходе лечения. Лечение с помощью противоэпилептических средств начинают, в основном, с одного лекарственного средства.
Для терапии с помощью одного лекарственного средства обычно подбирают два или три вида лекарственных средств по очереди. Если монотерапия не является успешной, применяется политерапия.
У приблизительно 70% пациентов с вновь развившейся эпилепсией при применении монотерапии ожидается ремиссия припадков.
Однако известно, что у оставшихся 30 процентов пациентов фармакотерапия, в том числе политерапия, не может контролировать эпилептические припадки.
[0004] Примерами выпускаемых противоэпилептических средств является карбамазепин, этосуксимид, фенобарбитал, фенитоин, примидон, вальпроат натрия, зонисамид, фелбамат, габапентин, ламотриджин, топирамат, тиагабин, леветирацетам, окскарбазепин, эсликарбазепин, прегабалин, лакозамид, руфинамид, триметадион, сультиам, ацетазоламид, вигабатрин, производные бензодиазепина (клоназепам, клобазам, нитразепам, диазепам), перампанел, ретигабин и т.д. (непатентная литература 1).
Эти известные противоэпилептические средства оказывают воздействие путем ингибирования гипервозбудимости нейронов.
Одной из серьезных проблем при терапии с помощью противоэпилептических средств является токсичность вследствие их ингибиторного воздействия на неврологическую функцию (головокружение, нистагм, диплопия, сонливость, рвота, атаксия, психологический симптом, усталость и безволие и т.д.).
Большинство этих традиционных противоэпилептических средств характеризуются побочными действиями с дозозависимым эффектом, и это является серьезной проблемой, ведущей к ограничению выбора терапевтических средств и их дозы.
Побочные действия также ухудшают качество жизни пациентов с эпилепсией, поскольку требуют долгосрочного введения дозы.
[0005] Таким образом, востребованы лекарственные средства, которые являются лучшими в разнице между эффективной дозой и нейротоксическими дозами.
Что касается 1-индансульфамидов, то низкомолекулярные соединения известны в следующей патентной литературе 1 и 2, а также непатентной литературе 2.
Перечень библиографических ссылок
Патентная литература
[0006] Патентная литература 1: патент США №3383414.
Патентная литература 2: патент США №3709677.
Непатентная литература
[0007] Непатентная литература 1: Shrivastava et al., "An overview on antiepileptic drugs" Drug Discoveries & Therapeutics., Vol. 6, No. 4, pp. 178-193, 2012.
Непатентная литература 2: Claudiu T. Supuran et al., "Novel sulfamides as potential carbonic anhydrase isoenzymes inhibitors", Bioorg. Med. Chem., Vol. 21, 1379-1385, 2013.
Краткое описание изобретения
Техническая проблема
[0008] Целью настоящего изобретения является получение нового соединения, характеризующегося действием по улучшению индекса тяжести припадка (балла) на мышиной модели киндлинга.
Решение проблемы
[0009] Мышиная модель корнеального киндлинга известна как простая и эффективная экспериментальная модель эпилепсии. (Epilepsy Research Vol. 92, 2010, p163-169). Авторы настоящего изобретения продолжают скрининг с использованием модели животных с киндлингом в качестве модели эпилепсии. При этом авторы настоящего изобретения также продолжают интенсивные исследования по снижению нейротоксического эффекта.
В результате этих исследований авторы настоящего изобретения обнаружили, что новые 1-индансульфамидные соединения характеризуются сильным ингибиторным эффектом в отношении эпилепсии, и достигли целей настоящего изобретения.
[0010] А именно настоящее изобретение относится к:
[1] соединению, или его фармацевтически приемлемой соли, выбранному из группы:
1) N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
2) N-[(1S)-2,2,4,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
3) (+)-N-(2,2,4,6,7-пентафтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
4) N-[(1S*)-5-циано-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил] сульфамида,
5) (-)-N-(7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
6) (-)-N-(7-хлор-2,2,4-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
7) (-)-N-(7-хлор-2,2-дифтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
8) (-)-N-(7-хлор-2,2,6-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
9) (+)-N-(5-хлор-2,2,7-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
10) N-[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида,
11) N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида,
12) N-[(1S*)-2,2,4-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида,
13) N-[(1S*)-7-(дифторметил)-2,2-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
14) N-[(1R*,2R*)-2,4,7-трифтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
15) (-)-N-[(1R*,2R*)-7-хлор-2,4-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
16) (+)-N-[(1R*,2R*)-7-хлор-2,4-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
17) (-)-N-[(1R*,2R*)-7-хлор-2,5-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
18) (+)-N-[(1R*,2R*)-4-хлор-7-фтор-2-метокси-2,3-дигидро-1H-инден-1-ил]сульфамида,
19) (+)-N-(7-хлор-4-фтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
20) (±)-N-(5-фтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида,
21) (-)-N-(4-фтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида,
22) (+)-N-(4-фтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида,
23) (+)-N-(7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида,
24) (±)-N-(5-хлор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида,
25) (-)-N-(4-хлор-7-фтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
26) (+)-N-(7-хлор-5-циано-2,3-дигидро-1H-инден-1-ил)сульфамида,
27) (-)-N-(7-хлор-5-циано-2,3-дигидро-1H-инден-1-ил)сульфамида,
28) (-)-N-(5-хлор-7-фтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
29) N-[(1S)-4,7-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
30) (+)-N-(7-хлор-2,3-дигидро-1H-инден-1-ил)сульфамида,
31) (+)-N-(5-циано-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида,
32) (-)-N-(5-циано-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида,
33) N-[(1S)-7-хлор-5-фтор-2,3-дигидро-1H-инден-1-ил]сульфамида
и
34) (-)-N-(4,6,7-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида;
[2] соединению, или его фармацевтически приемлемой соли, выбранному из группы:
1) N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
2) N-[(1S)-2,2,4,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
3) (-)-N-(7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
4) (-)-N-(7-хлор-2,2-дифтор-2,3-дигидро-1H-инден-1-ил)сульфамида,
5) N-[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида,
6) N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида,
7) N-[(1S)-4,7-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида,
8) N-[(1S)-7-хлор-5-фтор-2,3-дигидро-1H-инден-1-ил]сульфамида
и
9) (-)-N-(4,6,7-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида;
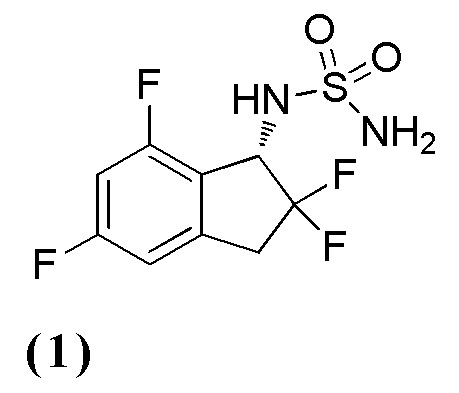
[3] N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамиду или его фармацевтически приемлемой соли;
[5] (-)-N-(7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамиду или его фармацевтически приемлемой соли;
[6] N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамиду или его фармацевтически приемлемой соли;
[7] N-[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамиду или его фармацевтически приемлемой соли;
[8] N-[(1S)-4,7-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамиду или его фармацевтически приемлемой соли; и
[9] фармацевтической композиции для лечения эпилепсии, содержащей соединение или его фармацевтически приемлемую соль согласно любому из вышеприведенного [1]-[3] и [5]-[8].
Полезные эффекты изобретения
[0011] Соединения или их фармацевтически приемлемая соль согласно настоящему изобретению характеризуются эффектом подавления припадка (ED50) на мышиной модели киндлинга. Таким образом, соединения по настоящему изобретению можно применять в качестве терапевтического средства для лечения эпилепсии.
Краткое описание графических материалов
[0012] Фиг. 1 представляет собой график, показывающий результат тестового примера 2 при введении соединения из примера 1.
Фиг. 2 представляет собой график, показывающий результат тестового примера 2 при введении соединения из примера 11.
Фиг. 3 представляет собой график, показывающий результат тестового примера 2 при введении соединения из примера 6.
Описание вариантов осуществления
[0013] Настоящее изобретение описано подробно ниже.
[0014] Хотя могут присутствовать полиморфы кристаллов, соединение не ограничено каким-либо из полиморфов и может присутствовать в виде монокристаллической формы или смеси монокристаллических форм. Соединение также включает некристаллическую форму.
Кроме того, соединение согласно настоящему изобретению может образовывать фармацевтически приемлемую соль или различные сольваты.
[0015] Далее объясняются значения выражений, символов и т.п., описанных в настоящем описании.
[0016] "Фармацевтически приемлемая соль" в настоящем описании конкретно не ограничена, кроме того, что она образует соль соединения и является фармацевтически приемлемой.
[0017] Сольват означает состояние, при котором растворитель, используемый в реакции или в кристаллизации, включается в кристалл без образования ковалентной связи с молекулой или ионом соединения. Примеры сольвата представляют собой гидрат, этанолат и т.п.
[0018] Соединения исходных веществ, промежуточные соединения и различные реагенты, используемые при получении соединения, могут образовывать соли или сольваты, при этом все они варьируют в зависимости от исходного вещества, используемого растворителя и т.п., и они конкретно не ограничены, кроме того, что они не ингибируют реакцию. Также используемый растворитель варьирует в зависимости от исходного вещества, реагента и т.п. и конкретно не ограничен, кроме того, что он не ингибирует реакцию и, безусловно, растворяет исходное вещество в определенной степени. Когда соединения получают в виде свободных форм, их можно превращать в приемлемые соли или сольваты с помощью традиционных способов.
[0019] Различные изомеры соединений или промежуточных соединений по настоящему изобретению (такие как геометрические изомеры, оптические изомеры, ротамеры, стереоизомеры, таутомеры и т.п.) можно очищать и выделять с применением обычных способов разделения, например, перекристаллизации, образования диастереоизомерных солей, ферментативного разделения и различных хроматографических способов (таких как тонкослойная хроматография, колоночная хроматография и газовая хроматография).
[0020] Соединения или их фармацевтически приемлемые соли можно составлять с помощью традиционных способов, а примеры готовых лекарственных форм включают пероральные составы (такие как таблетки, гранулы, порошки, капсулы и сиропы), инъекционные растворы (для внутривенного введения, внутримышечного введения, подкожного введения и внутрибрюшинного введения) и препараты для наружного применения (такие как составы для трансдермальной абсорбции (такие как мази и пластыри), назальные препараты и суппозитории).
[0021] Пероральные твердые составы, такие как таблетки, капсулы, гранулы и порошки, обычно могут содержать от 0,001 до 99,5 масс. %, предпочтительно от 0,01 до 90 масс. % и т.п. соединений или их фармацевтически приемлемых солей.
[0022] При производстве пероральных твердых составов таблетки, гранулы, порошки и капсулы можно получать путем добавления разбавителей, связующих, разрыхлителей, скользящих веществ, красителей и т.п. к соединениям или их фармацевтически приемлемым солям, при необходимости, и обработки с помощью традиционных способов. Эти составы при необходимости также можно покрывать пленкой.
[0023] Примеры разбавителей включают лактозу, кукурузный крахмал и микрокристаллическую целлюлозу, примеры связующих включают гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу, а примеры разрыхлителей включают карбоксиметилцеллюлозу кальция и кроскармелозу натрия.
[0024] Примеры скользящих веществ включают стеарат магния и стеарат кальция, а примеры красителей включают оксид титана.
[0025] Примеры средств для пленочного покрытия включают гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и метилцеллюлозу.
[0026] Безусловно, любые наполнители, описываемые выше, не ограничены этими примерами.
[0027] При производстве инъекционных растворов (для внутривенного введения, внутримышечного введения, подкожного введения и внутрибрюшинного введения) их можно производить путем добавления регуляторов pH, буферов, суспендирующих средств, солюбилизирующих средств, антиоксидантов, консервантов (антисептиков), средств для регуляции тоничности и т.п. к соединениям или их фармацевтически приемлемым солям, при необходимости, и обработки с помощью традиционных способов. Также путем лиофилизации можно получать лиофилизированные составы, подлежащие растворению перед применением. Эти инъекционные растворы можно вводить, например, внутривенно, подкожно или внутримышечно.
[0028] Примеры регуляторов pH и буферов включают органические кислоты или неорганические кислоты и/или их соли, примеры суспендирующих средств включают метилцеллюлозу, полисорбат 80 и карбоксиметилцеллюлозу натрия, примеры солюбилизирующих средств включают полисорбат 80 и полиоксиэтиленсорбитан монолаурат, примеры антиоксидантов включают α-токоферол, примеры консервантов включают метилпарагидроксибензоат и этилпарагидроксибензоат, а примеры средств для регуляции тоничности включают глюкозу, хлорид натрия и маннит; однако, безусловно, наполнители не ограничены этими примерами.
[0029] Эти инъекционные растворы обычно могут содержать от 0,00001 до 99,5 масс. %, предпочтительно от 0,0001 до 90 масс. % соединений или их фармацевтически приемлемых солей.
[0030] При производстве препаратов для наружного применения составы для трансдермальной абсорбции (такие как мази и пластыри), назальные капли, суппозитории и т.п. можно производить путем добавления основных материалов и, при необходимости, эмульгаторов, консервантов, регуляторов pH, красителей и т.п., описанных выше, к соединениям или их фармацевтически приемлемым солям и обработки с помощью традиционных способов.
[0031] В качестве основных материалов можно применять традиционно используемые различные сырьевые материалы для фармацевтических препаратов, квазилекарственных средств, косметических средств и т.п., и примеры включают сырьевые материалы, такие как животные и растительные масла, минеральные масла, сложноэфирные синтетические масла, воски, высшие спирты и очищенная вода.
[0032] Эти препараты для наружного применения обычно могут содержать от 0,00001 до 99,5 масс. %, предпочтительно от 0,0001 до 90 масс. % соединений или их фармацевтически приемлемых солей.
[0033] Дозировка лекарственного препарата согласно настоящему изобретению, как правило, варьирует в зависимости от симптома, возраста, пола, массы тела и т.п., но считается приемлемым, если эта дозировка достаточна для получения требуемого эффекта. Например, для взрослого используют дозировку от приблизительно 0,1 до 5000 мг (предпочтительно от 0,5 до 1000 мг, более предпочтительно от 1 до 600 мг) в день в виде одной дозы в течение одного или нескольких дней или в виде 2-6 разделенных доз за один день.
[0034] Настоящее изобретение также включает меченные изотопом соединения, и такие соединения являются такими же, как соединения, за исключением того, что один или несколько атомов замещены атомом (атомами) с атомной массой или массовым числом, отличным от атомной массы или массового числа, обычно обнаруживаемых в природе. Изотопы, которые можно включать в соединения, представляют собой, например, изотопы водорода, углерода, азота, кислорода, фосфора, фтора, серы и хлора, и они включают 2H, 3H, 11C, 14C, 13N, 15O, 18F, 32P и 35S.
[0035] Соединения или их фармацевтически приемлемые производные (такие как соли), содержащие вышеописанные изотопы и/или другие изотопы находятся в пределах формулы изобретения настоящего описания. Меченные изотопом соединения по настоящему изобретению, например, соединения, в которые включены радиоизотопы, такие как 3H и/или 14C, пригодны для анализов по распределению в тканях в качестве лекарственных препаратов и/или субстратов. Считается, что применимыми являются 3H и 14C вследствие легкости их получения и обнаружения. Считается, что изотопы 11C и 18F применимы для PET (позитронно-эмиссионной томографии), и все эти изотопы применимы для визуализации мозга. Замещение тяжелыми изотопами, такими как 2H, дает определенные терапевтические преимущества, такие как повышение времени полужизни in vivo из-за более высокой метаболической стабильности или снижение требуемой дозы, и, следовательно, считается, применимым при определенных обстоятельствах. Меченные изотопом соединения можно постоянно получать путем осуществления процедур, раскрытых в примерах, с применением легкодоступных меченных изотопом реагентов вместо реагентов, не меченных изотопом.
[0036] Соединения можно применять в качестве химических зондов для захвата целевых белков среди биоактивных низкомолекулярных соединений. А именно соединение можно превращать в зонд для аффинной хроматографии, фотоаффинный зонд и т.п. путем введения маркирующей группы, линкера и т.п. в фрагмент, не являющийся структурным фрагментом, необходимый для проявления активности соединения, при помощи методики, описанной в J. Mass Spectrum. Soc. Jpn., Vol. 51, No. 5, 2003, pp. 492-498 или WO 2007/139149 и т.д.
[0037] Примеры маркирующих групп, линкеров и т.п., используемых для химических зондов, включают группы, показанные в группе, состоящей из (1)-(5) ниже:
(1) белок-маркирующих групп, таких как фотоаффинные маркирующие группы (такие как бензоильная группа, бензофеноновая группа, азидогруппа, карбонилазидогруппа, диазиридиновая группа, еноновая группа, диазогруппа и нитрогруппа) и химические аффинные группы (такие как кетоновая группа, в которой α-углеродный атом замещен атомом галогена, карбамоильная группа, сложноэфирная группа, алкилтио-группа, акцепторы Михаэля, такие как α,β-ненасыщенные кетоны и сложные эфиры, и эпоксидная группа),
(2) расщепляемых линкеров, таких как -S-S-, -O-Si-O-, моносахариды (такие как глюкозная группа и галактозная группа) или дисахариды (такие как лактоза), и олигопептидные линкеры, расщепляемые с помощью ферментативных реакций,
(3) групп-меток для fish-анализа, таких как биотин и 3-(4,4-дифтор-5,7-диметил-4H-3a,4a-диаза-4-бора-s-индацен-3-ил)пропионильная группа,
(4) обнаруживаемых маркеров, таких как группы с радиоизотопной меткой, такой как 125I, 32P, 3H и 14C; группы для флуоресцентного мечения, такие как флуоресцеин, родамин, дансил, умбеллиферон, 7-нитрофуразанил и 3-(4,4-дифтор-5,7-диметил-4H-3a,4a-диаза-4-бора-s-индацен-3-ил)пропионильная группа; хемилюминесцентные группы, такие как люциферин и люминол; и ионы тяжелых металлов, такие как ионы металлов-лантаноидов и ионы радия; или
(5) групп, связанных с твердофазными носителями, такими как стеклянные гранулы, стеклянные подложки, титрационные микропланшеты, агарозные гранулы, агарозные подложки, полистирольные гранулы, полистирольные подложки, полиамидные гранулы и полиамидные подложки.
[0038] Зонды, полученные путем введения маркирующих групп и т.п., выбранных из группы, состоящей из (1)-(5) выше, в соединения в соответствии со способом, описанным в вышеприведенных документах и т.п., можно использовать в качестве химических зондов для обнаружения меченых белков, применимых, например, для поиска целей для новых лекарственных средств.
Примеры
[0039] Соединения можно получать, например, при помощи способов, описанных в примерах ниже, и эффекты соединений можно подтверждать при помощи способов, описанных в тестовых примерах ниже. Однако эти способы являются иллюстративными, и их можно менять без отступления от объема настоящего изобретения, и настоящее изобретение, в любом случае, не огранено следующими конкретными примерами.
[0040] Соединения, с которыми связаны названия публикаций и т.д., были получены в соответствии с этими публикациями и т.д.
[0041] Все аббревиатуры, используемые в данном описании, являются традиционными, известными специалистам в данной области. В нижеследующих примерах использованы следующие аббревиатуры.
AcOEt: этилацетат
BAST: бис(2-метоксиэтил)аминосеры трифторид
Bn: бензил
Boc: трет-бутоксикарбонил
DCM: дихлорметан
DMF: N,N-диметилформамид
DMSO: диметилсульфоксид
1H-ЯМР: спектрометрия протонного ядерного магнитного резонанса
ВЭЖХ (HPLC): высокоэффективная жидкостная хроматография
I.D.: внутренний диаметр
LC-MS: жидкостная хроматография с масс-спектрометрией
м-: мета-
н-: нормальный-
NBS: N-бромсукцинимид
o-: орто-
п-: пара-
PPTS: п-толуолсульфонат пиридиния
SelectfluorTM: N-фтор-N'-хлорметил-триэтилендиамин-бис(тетрафторборат)
т-: третичный-
TBS: трет-бутилдиметилсилил
TEA: триэтиламин
THF: тетрагидрофуран
THP: тетрагидропиран
Z(Cbz): бензилоксикарбонил
[0042] “Комнатной температурой” в следующих примерах и примерах получения, как правило, называется от приблизительно 10°C до приблизительно 35°C. "%" обозначает масс. %, если не указано иное. Соотношение растворителей при хроматографии на силикагеле показывает объемное соотношение растворителей, подлежащих смешиванию.
[0043] Химические сдвиги в спектрах протонного ядерного магнитного резонанса регистрировали в единицах δ м.д. (ppm) относительно тетраметилсилана, а константы взаимодействия записывали в герцах (Гц). Паттерны обозначены как с: синглет, д: дублет, т: триплет, кв: квартет, м: мультиплет, шир.с: широкий синглет.
[0044] Оптическое разделение соединений осуществляли при помощи ВЭЖХ-системы GILSON (насос; основной насос модели 305, дополнительный насос модели 306, головка насоса 50SC, динамический миксер модели 811D/A, манометрический модуль модели 806, УФ-детектор; детектор ультрафиолетового и видимого диапазонов модели 155, инжектор, устройство для отбора фракций; модель 215, колонка; выбранная из DAICEL CHIRALPAK® AD-H, IA, IB, IC, ID, IE, IF, DAICEL CHIRALCEL®, OD-H, OJ-H, 20 мм I.D.×250 мм).
После обнаружения фракций с помощью УФ-детектора оптическое вращение (+/-) измеряли с использованием детектора оптического вращения (OR-2090, JASCO, ртутно-ксеноновая (Hg-Xe) лампа, 150 Вт).
Что касается хроматографии, то при описании колоночной хроматографии на силикагеле использовали систему для параллельной препаративной хроматографии YAMAZEN (колонка: колонка YAMAZEN Hi-FlashTM (силикагель), размер; S (16×60 мм), M (20×75 мм), L (26×100 мм), 2L (26×150 мм) или 3L (46×130 мм)), сферический силикагель для хроматографии PSQ 60BTM от FUJI SILYSIA CHEMICAL CO., LTD., силикагель для хроматографии BW-300TM от Fuji Silysia Chemical Co., Ltd., Wakogel ® C-200 (Wako Pure Chemical Industries, Ltd.) или силикагель 60 (70-230 меш) от Merck Ltd. Япония.
Также при описании колоночной хроматографии на NH-силикагеле использовали систему для параллельной препаративной хроматографии YAMAZEN (колонка: колонка YAMAZEN Hi-FlashTM (амино), размер S (16×60 мм), M (20×75 мм), L (26×100 мм), 2L (26×150 мм) или 3L (4×130 мм)) или NH-СИЛИКАГЕЛЬ (200-350 меш) от FUJI SILYSIA CHEMICAL CO., LTD.
В номенклатуре соединений в настоящем описании (±) и (RS) представляют рацемическую смесь, а (+)-, (-)-, (R) и (S) представляют (+), (-), (R) и (S) конфигурации энантиомеров соответственно. И "*" в стерической конфигурации показывает относительную конфигурацию, и если не указано конкретно, это означает определенный энантиомер.
Кроме того, обозначения "(1R*,2R*)-" будут представлять взаимоотношения между хиральными центрами в контексте относительной конфигурации, т.е. определенного энантиомера, характеризующегося конфигурацией (1R, 2R) или (1S, 2S).
[0045] Пример 1
Синтез N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида
Синтез 2,5,7-трифтор-2,3-дигидро-1H-инден-1-она
SelectfluorTM (1,16 г, 3,27 ммоля) добавляли к раствору 5,7-дифтор-1-инданона (номер по CAS 84315-25-3, 500 мг, 2,97 ммоля) в MeOH (20 мл) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 2 часов и охлаждали до комнатной температуры. Затем растворитель отгоняли при пониженном давлении. Осадок обрабатывали DCM и нерастворимое вещество отфильтровывали. Затем растворитель отгоняли при пониженном давлении. Осадок растворяли в MeCN (10 мл) и 5N HCl (5 мл). Раствор перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали in vacuo. Осадок разделяли между AcOEt и H2O. Органический слой промывали солевым раствором, высушивали над MgSO4, и концентрировали in vacuo с получением титульного соединения (547 мг, 2,94 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 3,11-3,36 (м, 1H), 3,49-3,77 (м, 1H), 5,10-5,40 (м, 1H), 6,82 (тд, J=9,0, 1,9 Гц, 1H), 6,90-7,04 (м, 1H).
[0046] (2)
Синтез 2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-она
т-Бутилдиметилсилилтрифторметансульфонат (1,00 мл, 4,35 ммоля) добавляли к раствору продукта, полученного в примере 1-(1) (540 мг, 2,90 ммоля), и TEA (1,21 мл, 8,70 ммоля) в DCM (20 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 5 часов. Затем к реакционной смеси добавляли диэтиловый эфир и насыщенный водный Na2CO3 и слои разделяли. Органический слой последовательно промывали 1N HCl, насыщенным водным Na2CO3 и солевым раствором, и высушивали над Na2SO4. Растворитель выпаривали in vacuo и осадок высушивали при пониженном давлении.
Осадок растворяли в MeCN (20 мл) и SelectfluorTM (1,13 г, 3,19 ммоля) добавляли при комнатной температуре. После перемешивания смеси при той же температуре в течение 11 часов растворитель отгоняли при пониженном давлении. Осадок растворяли в DCM и нерастворимое вещество отфильтровывали. Фильтрат концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии (колонка Yamazen HI-FLASHTM с силикагелем размера L, 20 мл/мин, градиент 10%-50% AcOEt в н-гептане) с получением титульного соединения в виде белого твердого вещества (532 мг, 2,61 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 3,57 (т, J=12,4 Гц, 2H), 6,74-6,94 (м, 1H), 6,95-7,08 (м, 1H).
[0047] (3)
Синтез 2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-амина
Ацетат аммония (4,27 г, 55,4 ммоля) добавляли к раствору продукта, полученного в примере 1-(2) (377 мг, 1,85 ммоль), в изопропаноле (16 мл) при комнатной температуре и смесь нагревали с обратным холодильником в течение 30 мин. Цианоборгидрид натрия (348 мг, 5,54 ммоля) добавляли к реакционной смеси и перемешивали с обратным холодильником в течение 7 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли AcOEt и 2N NaOH и слои разделяли. Органический слой концентрировали in vacuo. К осадку добавляли воду и разделяли между AcOEt и 1N HCl. Водный слой подщелачивали 2N NaOH и экстрагировали, используя AcOEt. Органический слой высушивали над Na2SO4, выпаривали и высушивали с получением титульного соединения в виде желтого масла (210 мг, 1,02 ммоль).
ESI-MS; m/z 206 [M+H]+.
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,26-3,55 (м, 2H), 4,59 (дд, J=13,3, 5,3 Гц, 1H), 6,61-6,86 (м, 2H).
[0048] (4)
Синтез бензил N-(2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
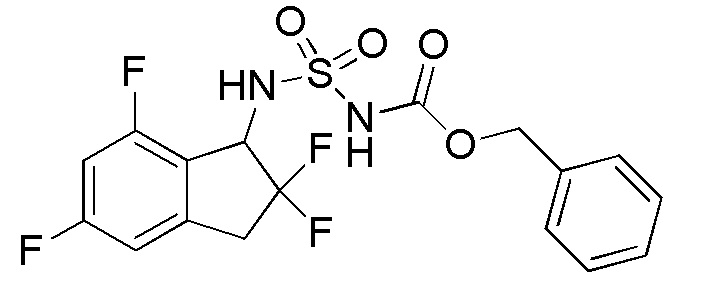
К раствору в DCM (10 мл) продукта, полученного в примере 1-(3) (200 мг, 0,975 ммоль), при комнатной температуре добавляли [(бензилокси)карбонил]{[4-(диметилимино)пиридин-1(4H)-ил]сульфонил}амид (номер по CAS 1037211-09-8, 654 мг, 1,95 ммоля, полученный согласно способу, описанному в WO 2008083248) и TEA (0,545 мл, 3,90 ммоль). Полученный раствор перемешивали в течение 24 часов с обратным холодильником. После охлаждения до комнатной температуры к реакционной смеси добавляли AcOEt и 1N HCl. Слои разделяли и органический слой высушивали над MgSO4 и выпаривали in vacuo. Осадок очищали посредством колоночной хроматографии (силикагель, 30% AcOEt в н-гептане) с получением титульного соединения в виде белого твердого вещества (316 мг, 0,755 ммоля).
ESI-MS; m/z 441 [M+Na]+.
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,25-3,54 (м, 2H), 5,14-5,38 (м, 3H), 5,72 (шир. с, 1H), 6,72 (т, J=9,4 Гц, 1H), 6,79 (д, J=7,8Гц, 1H), 7,30-7,46 (м, 5 H).
[0049] (5)
Синтез N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида
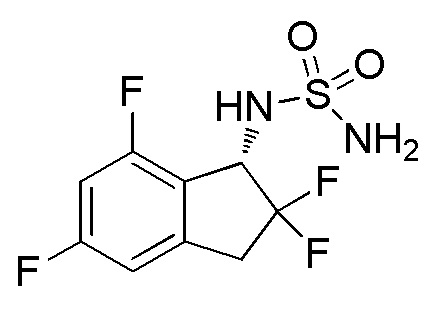
Палладий на угле (10 масс./масс. %, 30 мг, 0,028 ммоль) добавляли к раствору продукта, полученного в примере 1-(4) (310 мг, 0,741 ммоль), в MeOH (5 мл) и AcOEt (5 мл) при 25°C. Полученный раствор перемешивали в течение 30 минут при комнатной температуре в атмосфере H2. AcOEt добавляли к реакционной смеси и фильтровали через Celite® для удаления палладия на угле. Фильтрат концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии (колонка Yamazen HI-FLASHTM с силикагелем размера M, 10 мл/мин, градиент 30%-70% AcOEt в н-гептане) с получением титульного соединения в виде рацемата (181 мг, 0,637 ммоля).
Оптическое разделение полученного рацемата (180 мг, 0,633 ммоль) проводили посредством ВЭЖХ (CHIRALPAKTM IA, 20 мм I.D.×250 мм, 10 мл/мин, 15% EtOH в гексане) с получением S-формы титульного соединения в виде белого твердого вещества (76 мг, 0,267 ммоля, 98% э.и.), которая элюировалась второй из 2 изомеров со временем удержания 44 минуты.
ESI-MS; m/z 307 [M+Na]+.
1H-ЯМР(400 МГц, CDCl3)
δ (м.д.): 3,32-3,60 (м, 2H), 4,70 (шир. с, 2H), 4,93 (д, J=9,3 Гц, 1H), 5,30 (кв, J=9,3 Гц, 1H), 6,70-6,86 (м, 2H).
[0050] Пример 2
Синтез N-[(1S)-2,2,4,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида
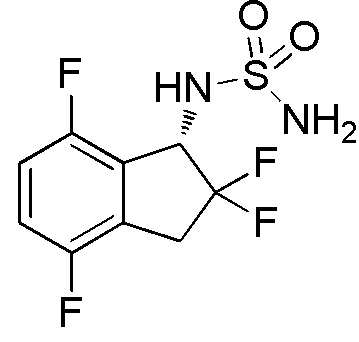
Титульное соединение (2,13 г, 7,49 ммоля) получали в виде рацемата из 4,7-дифтор-1-инданона (номер по CAS 130408-16-1, 6,15 г, 36,6 ммоля) способом, аналогичным описанному в примере 1.
ESI-MS m/z: 307[M+Na]+
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,33-3,63 (м, 2H), 4,73 (шир. с, 2H), 5,01 (д, J=8,4 Гц, 1H), 5,35 (кв, J=9,8 Гц, 1H), 6,92-7,16 (м, 2H).
Оптическое разделение полученного рацемата (180 мг, 0,633 ммоля) проводили посредством ВЭЖХ (CHIRALCELTM OD-H, 20 мм I.D.×250 мм, 15% EtOH в гексане, 10 мл/мин) с получением (1S)-формы титульного соединения в виде белого твердого вещества (45 мг, 0,158 ммоля, 99% э.и.), который элюировался вторым из 2 оптических изомеров. В результате анализа при помощи DAICEL CHIRALCELTM OD-H (4,6 мм I.D.×150 мм, 15% EtOH в гексане, 1 мл/мин) оптический изомер показал время удержания 12 минут.
[0051] Пример 3
Синтез (+)-N-(2,2,4,6,7-пентафтор-2,3-дигидро-1H-инден-1-ил)сульфамида
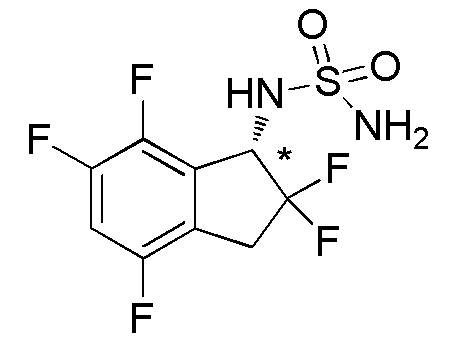
Титульное соединение (73 мг, 0,242 ммоль) получали в виде рацемата из 4,6,7-трифтор-1-инданона (номер по CAS 1260008-80-7, 250 мг, 1,34 ммоль) способом, аналогичным описанному в примере 1.
Оптическое разделение полученного рацемата (70 мг, 0,232 ммоль) проводили посредством ВЭЖХ (CHIRALPAKTM IC, 20 мм I.D.×250 мм, 10% EtOH в гексане, 10 мл/мин) с получением (+)-формы титульного соединения (31 мг, 0,103 ммоль, >99% э.и.), которая элюировалась первой из 2 оптических изомеров со временем удержания 40 минут.
ESI-MS; m/z: 325[M+Na]+
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,34-3,61 (м, 2H), 4,78 (шир. с, 2H), 5,14 (шир. с, 1H), 5,29-5,48 (м, 1H), 6,89-7,04 (м, 1H).
[0052] Пример 4
Синтез N-[(1S*)-5-циано-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
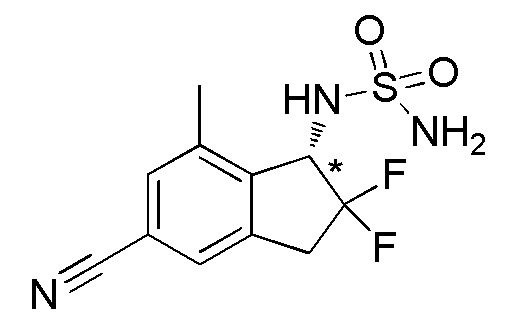
(1) Синтез 2,2-дифтор-7-метил-1-оксо-2,3-дигидро-1H-инден-5-карбонитрила
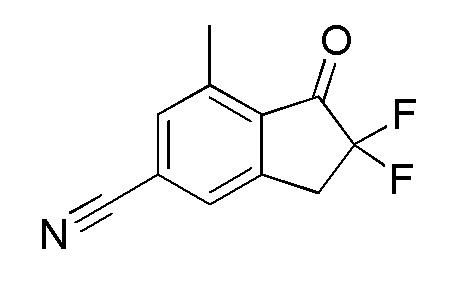
Титульное соединение (600 мг, 2,90 ммоль) получали из 7-метил-1-оксоиндан-5-карбонитрила (номер по CAS 1337833-67-6, 1,00 г, 5,84 ммоль) способом, аналогичным описанному в примере 1-(1) и 1-(2).
ESI-MS; m/z: 208[M+H]+
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 2,71 (с, 3H), 3,57 (т, J=12,8 Гц, 2H), 7,53 (с, 1H), 7,60 (с, 1H).
[0053] (2) Синтез N-[(1S*)-5-циано-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
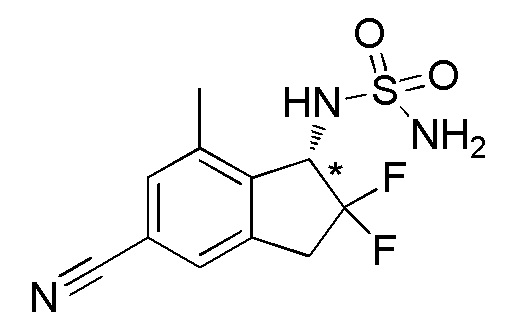
Титульное соединение (59,0 мг, 0,205 ммоля) получали в виде рацемата из продукта из примера 4-(1) (157 мг, 0,759 ммоля) способом, аналогичным описанному в примере 1-(3)-1-(5).
Оптическое разделение полученного рацемата (59,0 мг, 0,205 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IF, 20 мм I.D.×250 мм, 20% EtOH в гексане, 10 мл/мин) с получением титульного соединения (23,4 мг, 0,081 ммоля, >99% э.и.), которое элюировалось вторым из 2 оптических изомеров со временем удержания 41 минута.
ESI-MS; m/z: 310[M+Na]+
1H-ЯМР (400 МГц, DMSO-d6)
δ (м.д.): 2,42 (с, 3H), 3,29-3,45 (м, 1H), 3,47-3,63 (м, 1H), 4,86-4,97 (м, 1H), 6,85 (шир. с, 2H), 7,58-7,70 (м, 3H).
[0054] Пример 5
Синтез (-)-N-(2,2,6,7-тетрафтор-2,3-дигидро-1H-инден-1-ил)сульфамида
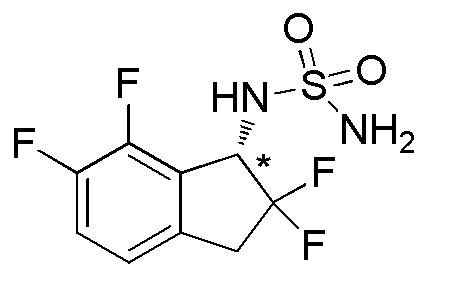
(1) Синтез 2,2,6,7-тетрафтор-2,3-дигидро-1H-инден-1-амина
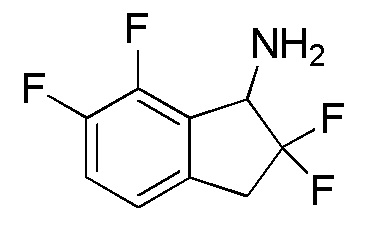
4-Бром-2,6,7-трифтор-2,3-дигидро-1H-инден-1-он (2,30 г, 8,68 ммоля) получали из 4-бром-6,7-дифтор-1-инданона (номер по CAS 881189-76-0, 2,45 г, 9,92 ммоля) способом, аналогичным описанному в примере 1-(1). 4-Бром-2,2,6,7-тетрафтор-2,3-дигидро-1H-инден-1-он (1,80 г, 6,38 ммоля) получали из 4-бром-2,6,7-трифтор-2,3-дигидро-1H-инден-1-она (2,30 г, 8,68 ммоля) способом, аналогичным описанному в примере 1-(2).
Гидроксиламина гидрохлорид (0,90 г, 12,8 ммоль) добавляли к раствору 4-бром-2,2,6,7-трифтор-2,3-дигидро-1H-инден-1-она (1,80 г, 6,38 ммоля) в EtOH (10 мл) и реакционную смесь нагревали с обратным холодильником в течение 12 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали in vacuo.
Осадок последовательно промывали DCM и водой и высушивали in vacuo. К раствору осадка в MeOH (20 мл) добавляли конц. H2SO4 (0,6 мл) и палладий на угле (90 мг).
Смесь перемешивали в течение 12 часов при комнатной температуре под атмосферой H2. Палладий на угле отфильтровывали и фильтрат концентрировали in vacuo. Осадок очищали посредством колоночной хроматографии на силикагеле с получением титульного соединения (700 мг, 3,41 ммоля).
ESI-MS; m/z: 206[M+H]+
[0055] (2) Синтез (-)-N-(2,2,6,7-тетрафтор-2,3-дигидро-1H-инден-1-ил)сульфамида
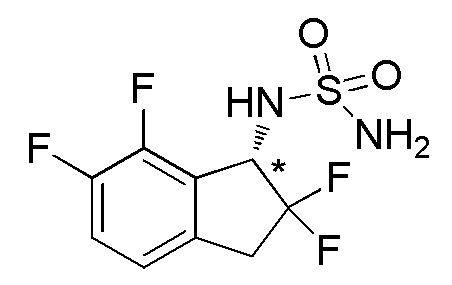
Титульное соединение (120 мг, 0,423 ммоля) получали в виде рацемата из продукта, полученного в примере 5-(1) (700 мг, 3,41 ммоль), способом, аналогичным описанному в примере 1-(3)-1-(5).
Оптическое разделение полученного рацемата (110 мг, 0,387 ммоль) проводили посредством ВЭЖХ (CHIRALPAKTM IC, 20 мм I.D.×250 мм, 20% EtOH в гексане, 10 мл/мин) с получением (-)-формы титульного соединения (38 мг, 0,134 ммоль, >99% э.и.), которая элюировалась первой из 2 оптических изомеров со временем удержания 20 минут.
ESI-MS; m/z: 307[M+Na]+
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,29-3,58 (м, 2H), 4,75 (шир. с, 2H), 5,05 (д, J=9,7 Гц, 1H), 5,38 (кв, J=9,7 Гц, 1H), 6,94-7,02 (м, 1H), 7,14-7,23 (м, 1H).
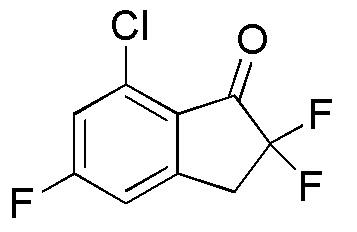
[0056] Пример 6
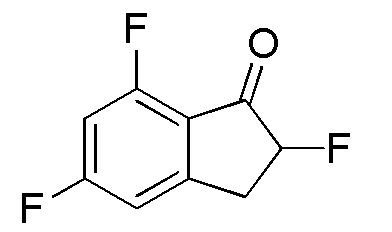
Синтез (-)-N-(7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида
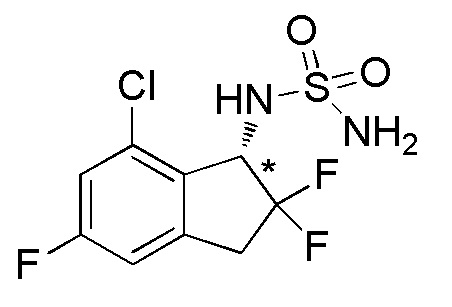
(1)
Синтез 7-Хлор-2,5-дифтор-2,3-дигидро-1H-инден-1-она
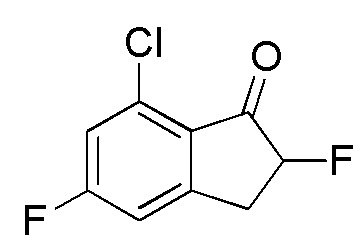
SelectfluorTM (2,49 г, 7,02 ммоля) добавляли к раствору 7-хлор-5-фтор-1-инданона (номер по CAS 1260008-48-7, 1,08 г, 5,85 ммоль) в MeOH (30 мл) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры полученную смесь выпаривали для удаления растворителя при пониженном давлении. К осадку добавляли DCM и нерастворимое вещество отфильтровывали. Фильтрат концентрировали in vacuo. Осадок растворяли в MeCN (20 мл) и 5N HCl (10 мл) и раствор перемешивали при комнатной температуре в течение 1 часа. После концентрирования раствора in vacuo осадок разделяли между AcOEt и H2O. Органический слой промывали солевым раствором, высушивали над MgSO4 и концентрировали in vacuo с получением титульного соединения (1,13 г, 5,58 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 3,13-3,33 (м, 1H), 3,47-3,71 (м, 1H), 5,25 (ддд, J=51,0, 8,0, 4,5 Гц, 1H), 7,07 (дт, J=7,6, 2,0 Гц, 1H, 7,14 (дд, J=8,8, 2,0 Гц, 1H).
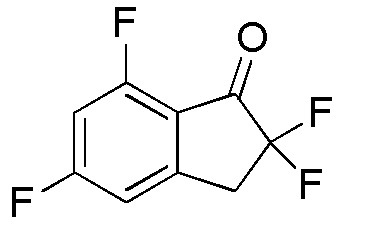
[0057] (2)
Синтез 7-Хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-она
т-Бутилдиметилсилилтрифторметансульфонат (2,56 мл, 11,2 ммоля) добавляли к раствору продукта, полученного в примере 6-(1) (1,13 г, 5,58 ммоля), и TEA (3,11 мл, 22,3 ммоля) в DCM (30 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли диэтиловым эфиром и насыщенным водным Na2CO3 и слои разделяли. Органический слой последовательно промывали 1N HCl, насыщенным водным Na2CO3 и солевым раствором, и высушивали над Na2SO4. Растворитель выпаривали in vacuo.
Осадок растворяли в MeCN (30 мл) и добавляли SelectfluorTM (2,17 г, 6,11 ммоля) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 3 часов, а затем полученную смесь выпаривали при пониженном давлении. К осадку добавляли DCM и нерастворимое вещество отфильтровывали. Фильтрат концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии (колонка Yamazen HI-FLASHTM с силикагелем размера L, 20 мл/мин, градиент 0%-30% AcOEt в н-гептане) с получением титульного соединения (2) (1,11 г, 5,03 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 3,47-3,63 (м, 2H), 7,06-7,13 (м, 1H), 7,17-7,23 (м, 1H).
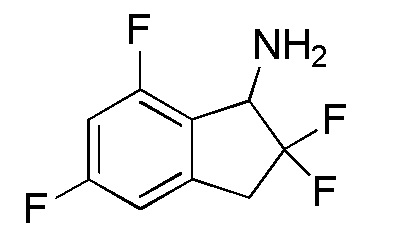
[0058] (3)
Синтез 7-Хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-амина
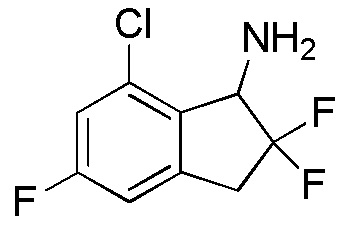
Ацетат аммония (11,5 г, 150 ммолей) добавляли к раствору продукта, полученного в примере 6-(2) (1,10 г, 4,98 ммоля), в изопропаноле (40 мл) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 30 минут. К реакционной смеси добавляли цианборгидрид натрия (940 мг, 15,0 ммоль) и смесь нагревали с обратным холодильником в течение 12 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли AcOEt и добавляли 2N NaOH. Слои разделяли и органический слой концентрировали in vacuo. Осадок разделяли между AcOEt и 1N HCl и водный слой подщелачивали 2N NaOH и экстрагировали, используя AcOEt. Органический слой высушивали над Na2SO4 и концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии (колонка Yamazen HI-FLASHTM с силикагелем размера L, 20 мл/мин, градиент 10%-50% AcOEt в н-гептане) с получением титульного соединения (3) (699 мг, 3,15 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,24-3,41 (м, 1H), 3,47-3,65 (м, 1H), 4,50 (д, J=14,6 Гц, 1H), 6,85-6,93 (м, 1H), 7,02 (дд, J=9,0, 2,2 Гц, 1H).
[0059] (4)
Синтез т-бутил N-(7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
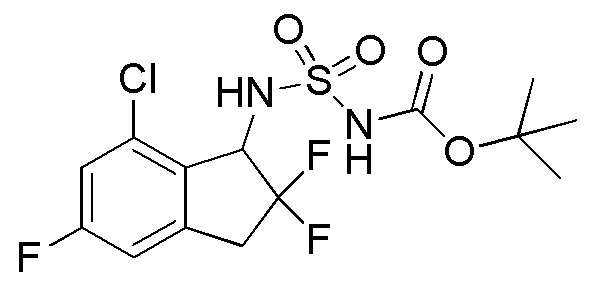
[(т-бутокси)карбонил]{[4-(диметилимино)пиридин-1(4H)-ил]сульфонил}амид (номер по CAS 872496-91-8, 1,90 г, 6,31 ммоля, получен согласно способу, описанному в Organic Letters, 3, 2241 (2001)) и TEA (1,76 мл, 12,6 ммоля) добавляли к раствору продукта, полученного в примере 6-(3) (699 мг, 3,15 ммоля) в DCM (20 мл) при комнатной температуре. Полученную смесь нагревали в течение 12 часов с обратным холодильником. После охлаждения до комнатной температуры к реакционной смеси добавляли AcOEt и 1N HCl и слои разделяли. Органический слой высушивали над MgSO4 и выпаривали in vacuo. Осадок очищали посредством колоночной хроматографии на силикагеле (силикагель, 30% AcOEt в н-гептане) с получением титульного соединения (4) (1,08 г, 2,69 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 1,49 (с, 9 H), 3,28-3,55 (м, 2H), 5,07-5,36 (м, 1H), 5,51-5,70 (м, 1H), 6,89 (д, J=9,2 Гц, 1H), 7,07 (д, J=9,2 Гц, 1H), 7,29 (шир. с, 1H).
[0060] (5)
Синтез (-)-N-(хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида
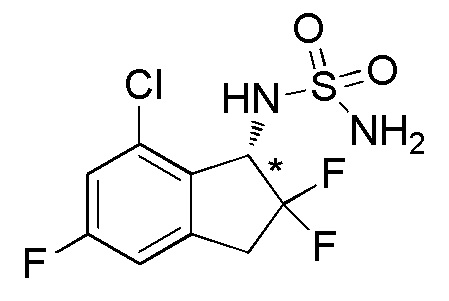
К раствору продукта, полученного в примере 6-(4) (1,08 г, 2,69 ммоль), в AcOEt (25 мл) добавляли 4N HCl в AcOEt (26,9 мл, 108 ммолей) и смесь перемешивали при комнатной температуре в течение 5 часов. Растворитель выпаривали in vacuo и осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера L, 20 мл/мин, градиент 30%-70% AcOEt в н-гептане) с получением титульного соединения в виде рацемата (627 мг, 2,09 ммоля).
Оптическое разделение полученного рацемата (200 мг, 0,665 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IB, 20 мм I.D.×250 мм, 10% EtOH в н-гексане, 10 мл/мин) с получением (-)-формы титульного соединения (83 мг, 0,276 ммоля, 96% э.и.), которая элюировалась второй из 2 оптических изомеров со временем удержания 49 минут.
ESI-MS; m/z: 323[M+Na]+
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,35-3,64 (м, 2H), 4,74 (шир. с, 2H), 4,86 (д, J=8,6 Гц, 1H), 5,07-5,28 (м, 1H), 6,83-6,95 (м, 1H), 7,09 (дд, J=8,7, 2,3 Гц, 1H).
[0061] Пример 7
Синтез (-)-N-(7-хлор-2,2,4-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида
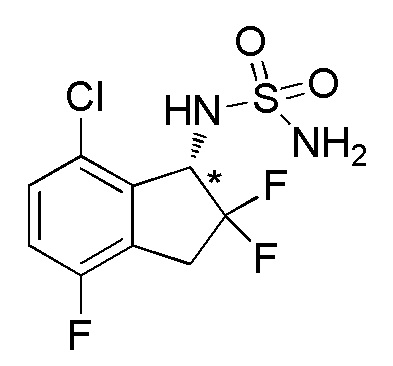
Титульное соединение (183 мг, 0,609 ммоля) получали в виде рацемата из 7-хлор-4-фтор-1-инданона (номер по CAS 881190-28-9, 1,70 г, 9,21 ммоля) способом, аналогичным описанному в примере 6.
Оптическое разделение полученного рацемата (110 мг, 0,366 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM AD-H, 20 мм I.D.×250 мм, 10% EtOH в гексане, 10 мл/мин) с получением (-)-изомера титульного соединения (46 мг, 0,153 ммоля, >99% э.и.), который элюировался первым из 2 оптических изомеров со временем удержания 48 минут.
ESI-MS; m/z: 323[M+Na]+
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,36-3,63 (м, 2H), 4,74 (шир. с, 2H), 4,88 (д, J=8,2 Гц, 1H), 5,16-5,34 (м, 1H), 7,06 (т, J=8,4 Гц, 1H), 7,31 (дд, J=8,4, 4,4 Гц, 1H).
[0062] Пример 8
Синтез (-)-N-(7-хлор-2,2-дифтор-2,3-дигидро-1H-инден-1-ил)сульфамида
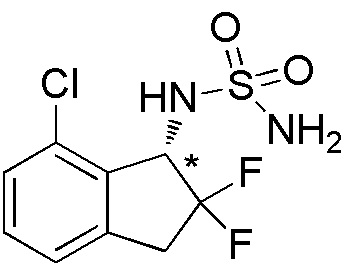
Титульное соединение (1,44 г, 5,09 ммоль) получали в виде рацемата из 7-хлор-1-инданона (номер по CAS 34911-25-6, 2,48 г, 14,9 ммоля) способом, аналогичным описанному в примере 6.
Оптическое разделение полученного рацемата (430 мг, 1,52 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IB, 20 мм I.D.×250 мм, 10% EtOH в гексане, 10 мл/мин) с получением (-)-формы титульного соединения (194 мг, 0,686 ммоля, >99% э.и.), которая элюировалась второй из двух оптических изомеров со временем удержания 43 минуты.
ESI-MS; m/z: 305[M+Na]+
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,35-3,63 (м, 2H), 4,72 (шир. с, 2H), 4,81 (д, J=8,2 Гц, 1H), 5,24 (ддд, J=12,4, 8,3, 4,2 Гц, 1H), 7,12-7,21 (м, 1H), 7,30-7,37 (м, 2H).
[0063] Пример 9
Синтез (-)-N-(7-хлор-2,2,6-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида
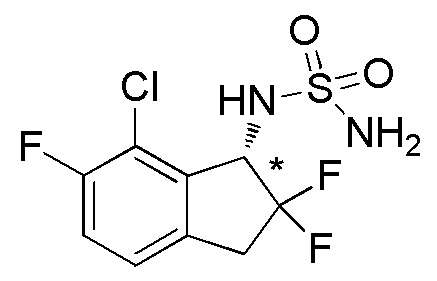
Титульное соединение (243 мг, 0,808 ммоль) получали в виде рацемата из 7-хлор-6-фтор-1-инданона (номер по CAS 881190-95-0, 1,00 г, 5,42 ммоль) способом, аналогичным описанному в примере 6.
Оптическое разделение полученного рацемата (200 мг, 0,665 ммоль) проводили посредством ВЭЖХ (CHIRALPAKTM IA, 20 мм I.D.×250 мм, 25% EtOH в гексане, 10 мл/мин) с получением (-)-формы титульного соединения (85 мг, 0,283 ммоля, >99% э.и.), которая элюировалась второй из 2 оптических изомеров со временем удержания 24 минуты.
ESI-MS; m/z: 323[M+Na]+
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,33-3,60 (м, 2H), 4,76 (шир. с, 2H), 4,90 (д, J=7,8 Гц, 1H), 5,16-5,35 (м, 1H), 7,10-7,23 (м, 2H).
[0064] Пример 10
Синтез (+)-N-(5-хлор-2,2,7-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида
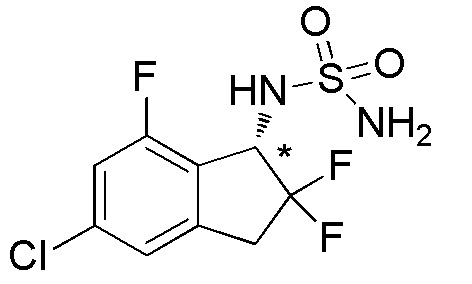
Титульное соединение (149 мг, 0,496 ммоля) получали в виде рацемата из 5-хлор-7-фтор-1-инданона (номер по CAS 1273613-81-2, 550 мг, 2,98 ммоля) способом, аналогичным описанному в примере 6.
Оптическое разделение полученного рацемата (140 мг, 0,466 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IF, 20 мм I.D.×250 мм, 30% EtOH в н-гексане, 10 мл/мин) с получением (+)-формы титульного соединения (65,7 мг, 0,283 ммоля, >99% э.и.), которая элюировалась второй из 2 оптических изомеров со временем удержания 23 минуты.
ESI-MS; m/z: 323[M+Na]+
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 3,30-3,60 (м, 2H), 4,76 (шир. с, 2H), 5,04 (д, J=9,0 Гц, 1H), 5,21-5,36 (м, 1H), 7,02-7,12 (м, 2H).
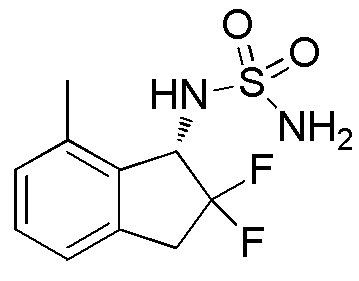
[0065] Пример 11
N-[(1S)-2,2-Дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамид
(1) Синтез 2-Фтор-7-метил-2,3-дигидро-1H-инден-1-она
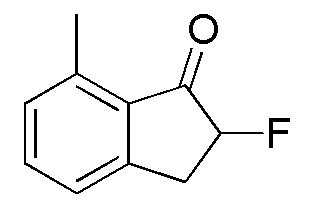
К раствору 7-метил-1-инданона (номер по CAS 39627-61-7, 513 мг, 3,51 ммоля) в MeOH (18 мл) добавляли SelectfluorTM (1,49 г, 4,21 ммоля) при комнатной температуре. Реакционную смесь нагревали в течение 2 часов с обратным холодильником. После охлаждения до комнатной температуры растворитель выпаривали при пониженном давлении. Осадок обрабатывали DCM и нерастворимое вещество отфильтровывали. Фильтрат концентрировали in vacuo. Осадок растворяли в MeCN (10 мл) и 5N HCl (5 мл). Раствор перемешивали при комнатной температуре в течение 30 минут. После концентрирования раствора in vacuo осадок разделяли между AcOEt и H2O. Водный слой дважды экстрагировали, используя AcOEt. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали in vacuo с получением титульного соединения (555 мг, 3,38 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 2,64 (с, 3H), 3,18 (ддд, J=23,4, 16,8, 4,3 Гц, 1H), 3,57 (ддд, J=16,8, 7,8, 7,5 Гц, 1H), 5,21 (ддд, J=51,2, 7,8, 4,3 Гц, 1H), 7,17 (д, J=7,4 Гц, 1H), 7,26 (д, J=7,4 Гц, 1H), 7,51 (т, J=7,4 Гц, 1H).
[0066] (2) Синтез 2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-она
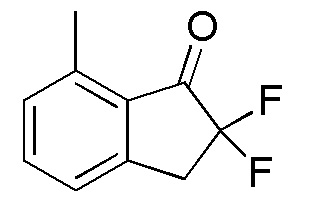
трет-Бутилдиметилсилилтрифторметансульфонат (1,55 мл, 6,74 ммоля) добавляли к раствору продукта, полученного в примере 11-(1) (555 мг, 3,38 ммоля), и TEA (1,88 мл, 13,49 ммоля) в DCM (30 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 1,5 часа. Реакцию гасили нас. NaHCO3 и слои разделяли. Водный слой экстрагировали, используя DCM. Объединенные органические слои промывали солевым раствором и высушивали над MgSO4. Нерастворимое вещество отфильтровывали и фильтрат концентрировали in vacuo. Осадок растворяли в MeCN (20 мл) и добавляли SelectfluorTM (1,32 г, 3,73 ммоля) при комнатной температуре. Далее реакционную смесь перемешивали в течение 1 часа при комнатной температуре, растворитель выпаривали при пониженном давлении. Осадок растворяли в DCM и нерастворимое вещество отфильтровывали. Фильтрат концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера L, 20 мл/мин, градиент 15%-20% AcOEt в н-гептане) с получением титульного соединения (563 мг, 3,09 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 2,66 (с, 3H), 3,51 (т, J=13,1 Гц, 1H), 7,23 (д, J=7,8 Гц, 1H), 7,28 (д, J=7,8 Гц, 1H), 7,57 (т, J=7,8 Гц, 1H).
[0067] (3) Синтез 2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ола
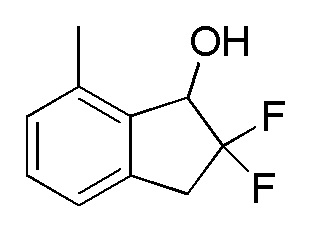
К раствору продукта, полученного способом, описанным в примере 11-(2) (1,09 г, 5,99 ммоля), в MeOH (20 мл) добавляли боргидрид натрия (453 мг, 12,0 ммоля) при 0°C. После перемешивания в течение 45 минут при такой же температуре к реакционной смеси добавляли воду и AcOEt и слои разделяли. Отделенный водный слой дважды экстрагировали, используя AcOEt. Объединенные органические слои промывали солевым раствором и высушивали над MgSO4. После фильтрации фильтрат концентрировали и высушивали in vacuo с получением титульного соединения (1,05 г, 5,72 ммоль).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 2,23 (шир. с, 1H), 2,43 (с, 3H), 3,26-3,39 (м, 1H), 3,44-3,58 (м, 1H), 5,08-5,15 (м, 1H), 7,07 (д, J=7,8 Гц, 1H), 7,10 (д, J=7,8 Гц, 1H), 7,23-7,26 (м, 1H).
[0068] (4) Синтез 1-азидо-2,2-дифтор-7-метил-2,3-дигидро-1H-индена
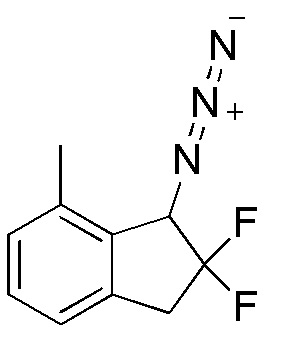
TEA (3,59 мл, 25,8 ммоль) и хлорид хлорметансульфонила (1,02 мл, 11,4 ммоля) добавляли к раствору продукта, полученного в примере 11-(3) (1,05 г, 5,72 ммоля), в DCM (25 мл) при 0°C. После перемешивания в течение 2 часов при комнатной температуре реакционную смесь разбавляли диэтиловым эфиром и гасили нас. NaHCO3. Водный слой экстрагировали диэтиловым эфиром 3 раза. Объединенные органические слои промывали солевым раствором и высушивали над MgSO4. Экстракт фильтровали и концентрировали in vacuo. Осадок растворяли в DMF (50 мл) и к раствору добавляли азид натрия (753 мг, 11,6 ммоля) при комнатной температуре. Реакционную смесь перемешивали в течение 2 часов при 70°C. После охлаждения смеси до комнатной температуры добавляли воду и диэтиловый эфир. Слои разделяли и водный слой экстрагировали диэтиловым эфиром 3 раза. Объединенные органические слои промывали водой и солевым раствором и высушивали над MgSO4. Экстракт фильтровали и концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера L, 20 мл в минуту, 20% AcOEt в н-гептане) с получением титульного соединения (641 мг, 3,06 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 2,41 (с, 3H), 3,30-3,43 (м, 1H), 3,51 (ддд, J=20,3, 16,8, 10,9 Гц, 1H), 4,77 (д, J=13,3 Гц, 1H), 7,09 (д, J=7,4 Гц, 1H), 7,14 (д, J=7,8 Гц, 1H), 7,26-7,31 (м, 1H).
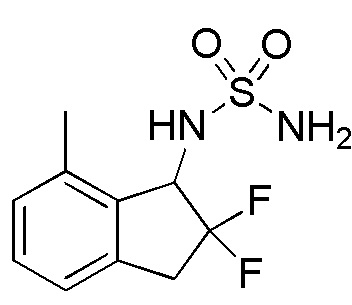
[0069] (5) Синтез N-(2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида
К раствору продукта, полученного в примере 11-(4) (641 мг, 3,06 ммоля), в воде (4 мл) и тетрагидрофуране (16 мл) добавляли трифенилфосфин (1,21 г, 4,61 ммоля) при комнатной температуре. Реакционную смесь перемешивали в течение 1 часа при 80°C. После охлаждения до комнатной температуры добавляли AcOEt (20 мл) и 1N HCl (20 мл). Отделенный органический слой экстрагировали дважды 10 мл 1N HCl. Водный слой объединяли и подщелачивали 20 мл 2N NaOH. Слой 3 раза экстрагировали, используя AcOEt, и объединенный органический слой промывали солевым раствором и высушивали над MgSO4. Экстракт фильтровали и концентрировали in vacuo. К смешанному раствору осадка и TEA (1,1 мл, 7,89 ммоля) в DCM (26 мл) добавляли небольшими порциями сульфамоилхлорид (номер по CAS 7778-42-9, 915 мг, 7,92 ммоля, полученный согласно способу, описанному в US 200896903) при комнатной температуре. Реакционную смесь впоследствии перемешивали в течение 1 часа при комнатной температуре. К смеси добавляли 20 мл 1N HCl и водный слой дважды экстрагировали, используя DCM. Объединенный органический слой высушивали над MgSO4, фильтровали и концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера L, 20 мл/мин, градиент 50%-65% AcOEt в н-гептане) с получением титульного соединения (348 мг, 1,33 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 2,45 (с, 3H), 3,32-3,56 (м, 2H), 4,70-4,80 (м, 3H), 5,17-5,26 (м, 1H), 7,06 (д, J=7,4 Гц, 1H), 7,12 (д, J=7,4 Гц, 1H), 7,23-7,29 (м, 1H).
[0070] (6) Синтез N-[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
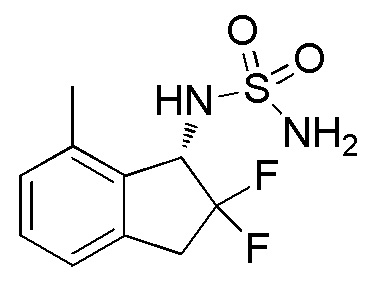
Оптическое разделение рацемата, полученного в примере 11-(5) (348 мг, 1,33 ммоля), проводили посредством ВЭЖХ (CHIRALPAKTM IA, 20 мм I.D.×250 мм, 15% EtOH в н-гексане, 10 мл/мин) с получением (1S)-формы титульного соединения (107 мг, 0,409 ммоля, >99% э.и.) в виде белого твердого вещества, которая элюировалась второй из 2 оптических изомеров со временем удержания 25 минут.
ESI-MS m/z: 285[M+Na]+
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 2,45 (с, 3H), 3,32-3,56 (м, 2H), 4,70-4,80 (м, 3H), 5,17-5,26 (м, 1H), 7,06 (д, J=7,4 Гц, 1H), 7,12 (д, J=7,4 Гц, 1H), 7,23-7,29 (м, 1H).
[0071] Пример 12
Синтез N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
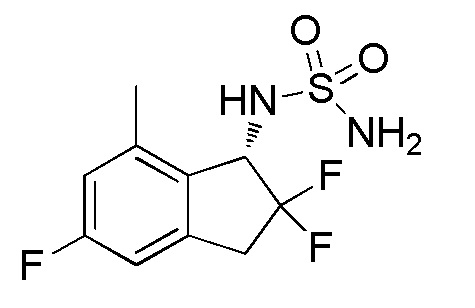
(1) Синтез 7-бром-2,2,5-трифтор-2,3-дигидро-1H-инден-1-она
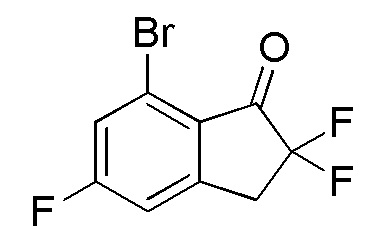
Титульное соединение (5,10 г, 19,2 ммоль) получали из 7-бром-5-фтор-1-инданона (номер по CAS 1260016-95-2, 4,55 г, 19,9 ммоль) способом, аналогичным описанному в примере 5-(1) и 5-(2).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 3,53 (т, J=12,5 Гц, 2H), 7,14 (д, J=7,6 Гц, 1H), 7,41 (д, J=8,4 Гц, 1H).
[0072] (2) Синтез 7-бром-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ола
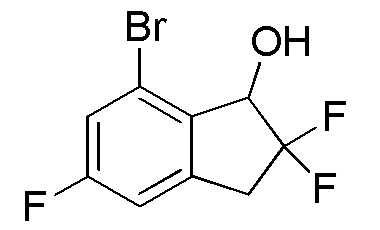
Титульное соединение (4,78 г, 17,9 ммоля) получали из продукта, полученного в примере 12-(1) (5,10 г, 19,2 ммоля), способом, аналогичным описанному в примере 11-(3).
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 2,50 (с, 1H), 3,38 (тд, J=17,0, 2,7 Гц, 1H), 3,50-3,69 (м, 1H), 5,06 (дд, J=12,5, 4,3 Гц, 1H), 6,95 (дд, J=8,0, 1,0 Гц, 1H), 7,22 (дд, J=8,6, 2,3 Гц, 1H).
[0073] (3) Синтез 2-[(7-бром-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)окси]тетрагидро-2H-пирана
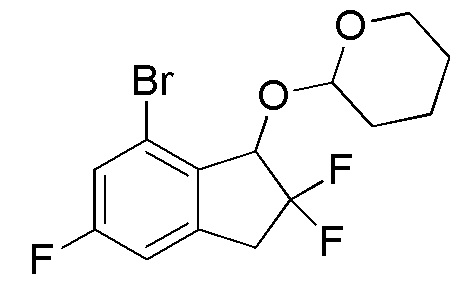
К раствору продукта, полученного в примере 12-(2) (2,78 г, 10,4 ммоля), и 3,4-дигидро-2H-пирана (2,18 мл, 23,9 ммоля) в DCM (40 мл) добавляли PPTS (52 мг, 0,208 ммоля) при комнатной температуре. При этом реакционную смесь перемешивали в течение 86 часов при комнатной температуре. Растворитель выпаривали и осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера M, 10 мл/мин, градиент 10%-25% AcOEt в н-гептане) с получением титульного соединения (3,42 г, 9,74 ммоля) в виде смеси рацемических диастереоизомеров с соотношением приблизительно 1:1.
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 1,51-1,84 (м, 6H), 3,26-3,52 (м, 1H), 3,52-3,68 (м, 2H), 4,05-4,19 (м, 1H), 5,00-5,21 (м, 2H), 6,92 (д, J=8,2 Гц, 1H), 7,21 (дт, J=8,2, 2,6 Гц, 1H).
[0074] (4) Синтез 2-[(2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил)окси]тетрагидро-2H-пирана
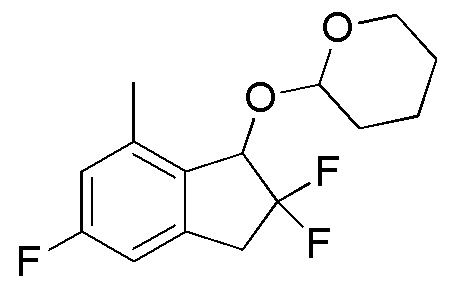
К раствору продукта, полученного в примере 12-(3) (1,70 г, 4,84 ммоль), в 1,4-диоксане (10 мл) добавляли по каплям раствор диметилцинка в 2M н-гексане (4,84 мл, 9,68 ммоля).
После добавления дихлор[1,1'-бис(дифенилфосфино)ферроцен] палладия(II) (177 мг, 0,242 ммоля) реакционную смесь перемешивали в течение 3 часов при 100°C в атмосфере азота. После охлаждения до комнатной температуры добавляли воду и смесь экстрагировали, используя AcOEt.
Органический слой промывали солевым раствором и высушивали над безводным Na2SO4. Нерастворимое вещество отфильтровывали и фильтрат выпаривали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM с силикагелем размера M, 10 мл/мин, градиент 0%-25% AcOEt в н-гептане) с получением титульного соединения в виде смеси рацемических диастереоизомеров с соотношением 1:1 (1,06 г, 3,70 ммоля).
ESI-MS; m/z: 309[M+Na]+
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 1,51-1,90 (м, 6H), 2,35 (с, 1,5H), 2,43 (с, 1,5H), 3,19-3,29 (м, 1H), 3,45-3,64 (м, 2H), 3,98-4,11 (м, 1H), 4,88 (т, J=3,4 Гц, 0,5H), 4,95 (д, J=5,1 Гц, 0,5H), 5,01 (дд, J=11,6, 2,8 Гц, 0,5H), 5,16 (д, J=11,7 Гц, 0,5H), 6,74-6,81 (м, 2H).
[0075] (5) Синтез 2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ола
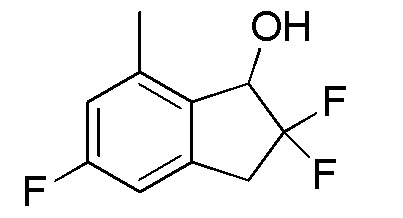
К раствору продукта, полученного в примере 12-(4) (1,06 г, 3,70 ммоля), в MeOH (10 мл) добавляли PPTS (46 мг, 0,185 ммоля). При этом реакционную смесь перемешивали в течение 1 часа при 60°C. После охлаждения до комнатной температуры к реакционной смеси добавляли насыщенный NaHCO3 и смесь экстрагировали, используя AcOEt. Органический слой промывали солевым раствором и высушивали над безводным Na2SO4. Нерастворимое вещество отфильтровывали и фильтрат выпаривали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера M, 10 мл/мин, градиент 5%-25% AcOEt в н-гептане) с получением титульного соединения (692 мг, 3,42 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 2,23 (дд, J=5,7, 2,5 Гц, 1H), 2,42 (с, 3H), 3,30 (тд, J=16,8, 5,2 Гц, 1H), 3,50 (тд, J=16,8, 11,6 Гц, 1H), 5,05 (дд, J=12,1, 5,1 Гц, 1H), 6,77 (д, J=8,2 Гц, 1H), 6,82 (д, J=10,2 Гц, 1H).
[0076] (6) Синтез 1-азидо-2,2,5-трифтор-7-метил-2,3-дигидро-1H-индена
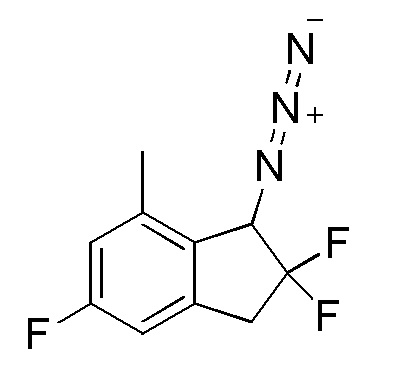
К раствору продукта, полученного в примере 12-(5) (692 мг, 3,42 ммоля), и TEA (1,43 мл, 10,3 ммоля) в DCM (10 мл) добавляли хлорметансульфонилхлорид (765 мг, 5,13 ммоля) при 0°C. При этом реакционную смесь перемешивали в течение 2 часов при комнатной температуре. К реакционной смеси добавляли насыщенный NaHCO3 и смесь экстрагировали диэтиловым эфиром. Органический слой последовательно промывали 1N HCl и солевым раствором, а затем высушивали над безводным Na2SO4. После фильтрации фильтрат выпаривали in vacuo. К раствору осадка в DMF (10 мл) добавляли азид натрия (442 мг, 6,80 ммоля) при комнатной температуре и смесь перемешивали в течение 2 часов при 70°C. После охлаждения до комнатной температуры смесь разделяли между диэтиловым эфиром и H2O. Водный слой экстрагировали диэтиловым эфиром. Объединенный органический слой промывали солевым раствором и высушивали над безводным Na2SO4.
После фильтрации фильтрат выпаривали in vacuo.
Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера M, 10 мл/мин, градиент 10%-30% AcOEt в н-гептане) с получением титульного соединения (320 мг, 1,41 ммоля).
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 2,41 (с, 3H), 3,30-3,56 (м, 2H), 4,74 (д, J=13,3 Гц, 1H), 6,81 (д, J=7,8 Гц, 1H), 6,86 (д, J=9,4 Гц, 1H).
[0077] (7) Синтез 2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-амина
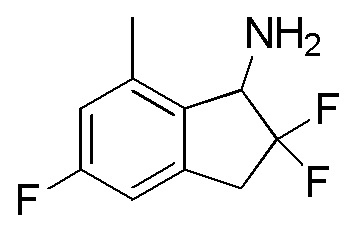
К раствору продукта, полученного в примере 12-(6) (320 мг, 1,41 ммоля), в воде (1 мл) и THF (5 мл) добавляли трифенилфосфин (554 мг, 2,11 ммоля) при комнатной температуре и смесь перемешивали в течение 2 часов при 80°C. После охлаждения до комнатной температуры смесь разделяли между AcOEt и 1N HCl. Полученный водный слой подщелачивали 5N NaOH и 3 раза экстрагировали, используя AcOEt. Объединенный экстракт высушивали над безводным Na2SO4. После фильтрации фильтрат концентрировали in vacuo с получением титульного соединения (180 мг, 0,895 ммоля).
ESI-MS; m/z: 202[M+H]+.
[0078] (8) Синтез трет-бутил N-(2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
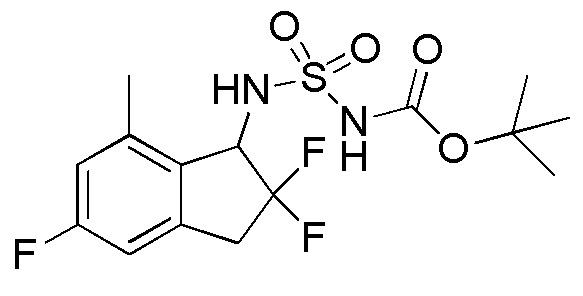
К раствору продукта из примера 12-(7) (180 мг, 0,895 ммоля) в DCM (10 мл) добавляли [(трет-бутокси)карбонил]{[4-(диметилимино)пиридин-1(4H)-ил]сульфонил}амид (297 мг, 0,984 ммоля) и TEA (0,374 мл, 2,68 ммоля) при комнатной температуре. Полученную смесь нагревали с обратным холодильником в течение 65,5 часов. После охлаждения до комнатной температуры смесь разделяли между AcOEt и 1N HCl. Органический слой высушивали над безводным Na2SO4 и выпаривали in vacuo с получением титульного соединения (257 мг, 0,676 ммоля).
ESI-MS; m/z: 403[M+Na]+.
[0079] (9) Синтез N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
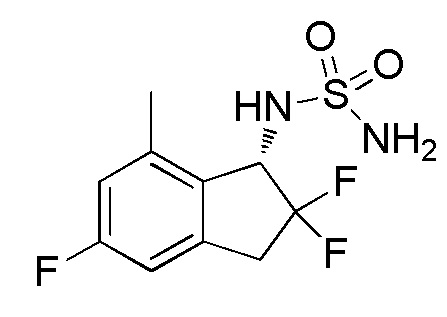
К раствору продукта из примера 12-(8) (257 мг, 0,676 ммоля) в MeOH (4 мл) добавляли 4N HCl в AcOEt (3,38 мл, 13,5 ммоль) при комнатной температуре и перемешивали при комнатной температуре в течение 14 часов. Растворитель выпаривали in vacuo и осадок очищали посредством колоночной хроматографии на силикагеле (AcOEt) с получением титульного соединения (162 мг, 0,578 ммоля) в виде рацемата.
Оптическое разделение полученного рацемата (162 мг, 0,578 ммоль) проводили посредством ВЭЖХ (CHIRALPAKTM IF, 20 мм I.D.×250 мм, 10% EtOH в н-гексане, 10 мл/мин) с получением (S)-изомера титульного соединения (71 мг, 0,253 ммоля, 98% э.и.) в виде белого твердого вещества, который элюировался вторым из 2 оптических изомеров со временем удержания 30 минут.
ESI-MS; m/z: 303[M+Na]+
1H-ЯМР (400 МГц, DMSO-d6)
δ (м.д.): 2,37 (с, 3H), 3,20-3,31 (м, 1H), 3,38-3,64 (м, 1H), 4,79 (дд, J=14,3, 8,8 Гц, 1H), 6,77 (с, 2H), 6,90-7,03 (м, 2H), 7,51 (д, J=9,0 Гц, 1H).
[0080] Пример 13
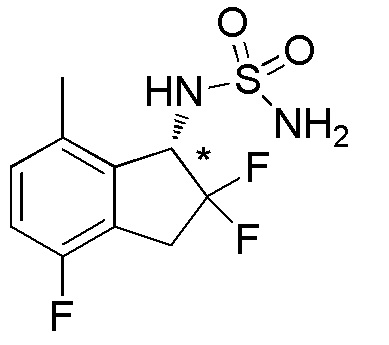
Синтез N-[(1S*)-2,2,4-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
(1) Синтез 7-бром-2,2,4-трифтор-2,3-дигидро-1H-инден-1-она
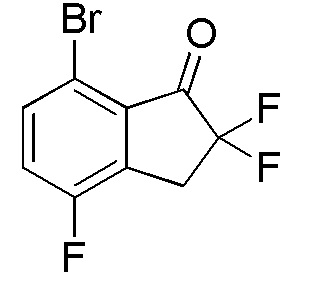
Титульное соединение (2,94 г, 11,1 ммоль) получали из 7-бром-4-фтор-1-инданона (номер по CAS 881189-73-7, 4,00 г, 17,5 ммоля) способом, аналогичным описанному в примере 1-(1) и 1-(2).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 3,54 (т, J=12,7 Гц, 2H), 7,30 (т, J=8,6 Гц, 1H), 7,64 (дд, J=8,6, 4,3 Гц, 1H).
[0081] (2) Синтез 7-бром-2,2,4-трифтор-2,3-дигидро-1H-инден-1-ола
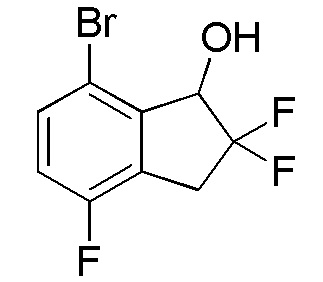
Титульное соединение (1,96 г, 7,32 ммоля) получали из продукта, полученного в примере 13-(1) (1,94 г, 7,32 ммоля), способом, аналогичным описанному в примере 11-(3).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 2,51 (дд, J=4,5, 1,8 Гц, 1H), 3,43-6,22 (м, 2H), 5,10 (дд, J=12,1, 4,5 Гц, 1H), 6,98 (т, J=8,6 Гц, 1H), 7,44 (дд, J=8,6, 4,5 Гц, 1H).
[0082] (3) Синтез 2-[(2,2,4-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил)окси]тетрагидро-2H-пирана
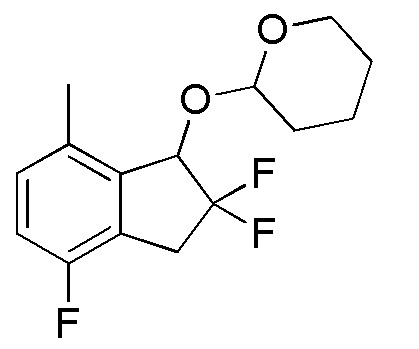
Титульное соединение (2,42 г, 6,92 ммоля) получали в виде смеси рацемических диастереоизомеров с соотношением приблизительно 1:1 из продукта, полученного в примере 13-(2) (1,95 г, 7,30 ммоля), способом, аналогичным описанному в примере 12-(3) и 12-(4).
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 1,51-1,81 (м, 6H), 3,36-3,65 (м, 3H), 4,06-4,20 (м, 1H), 5,05 (д, J=12,1 Гц, 0,5H), 5,11 (шир. с, 0,5H), 5,17 (д, J=10,5 Гц, 0,5H), 5,23-5,25 (м, 0,5H), 6,95 (тд, J=8,6, 6,1 Гц, 1H), 7,43 (дт, J=8,6, 4,3 Гц, 1H).
[0083] (4) Синтез 2,2,4-трифтор-7-метил-2,3-дигидро-1H-инден-1-ола
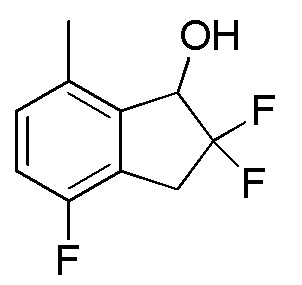
Титульное соединение (321 мг, 1,59 ммоля) получали из продукта, полученного в примере 13-(3) (800 мг, 2,28 ммоля), способом, аналогичным описанному в примере 12-(5).
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 2,55 (дд, J=6,2, 2,7 Гц, 1H), 2,39 (с, 3H), 3,33-3,55 (м, 2H), 5,07-5,12 (м, 1H), 6,95 (т, J=8,0 Гц, 1H), 7,08 (дд, J=8,0, 4,5 Гц, 1H).
[0084] (5) Синтез 2,2,4-трифтор-7-метил-2,3-дигидро-1H-инден-1-амина
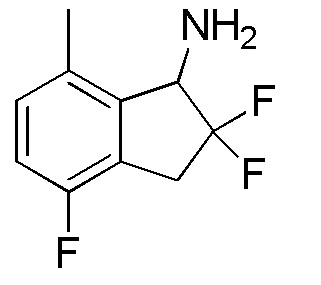
Титульное соединение (85 мг, 0,422 ммоля) получали из продукта, полученного в примере 13-(4) (321 мг, 1,59 ммоля), способом, аналогичным описанному в примере 12-(6) и 12-(7).
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 1,43 (шир. с, 2H), 2,40 (с, 3H), 3,31-3,51 (м, 2H), 4,38 (д, J=14,4 Гц, 1H), 6,90 (т, J=8,4 Гц, 1H), 7,04 (дд, J=8,4, 4,9 Гц, 1H).
[0085] (6) Синтез N-[(1S*)-2,2,4-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
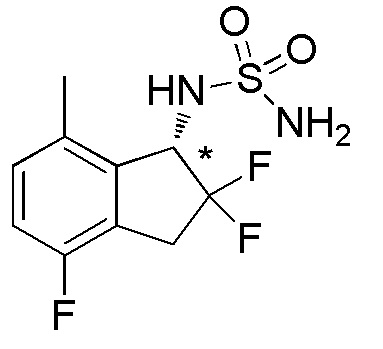
К раствору продукта, полученного в примере 13-(5) (85 мг, 0,422 ммоля), в DCM (4 мл) добавляли TEA (177 мкл, 1,27 ммоля) и сульфамоилхлорид (4 мл, 0,159M раствор в DCM) при 0°C и реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь подкисляли 2N HCl и экстрагировали, используя AcOEt. Органический слой промывали солевым раствором и высушивали над безводным Na2SO4. После фильтрации фильтрат концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера M, 10 мл/мин, градиент 20%-50% AcOEt в н-гептане) с получением титульного соединения (69 мг, 0,246 ммоля).
ESI-MS; m/z: 303[M+Na]+.
Оптическое разделение полученного рацемата (48 мг, 0,171 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IC, 20 мм I.D.×250 мм, 10% EtOH в н-гексане, 10 мл/мин) с получением энантиомера титульного соединения (8 мг, 0,029 ммоля, >99% э.и.), который элюировался вторым из двух оптических изомеров.
1H-ЯМР (400 МГц, DMSO-d6)
δ (м.д.): 2,37 (с, 3H), 3,20-3,31 (м, 1H), 3,38-3,64 (м, 1H), 4,79 (дд, J=14,3, 8,8 Гц, 1H), 6,77 (с, 2H), 6,90-7,03 (м, 2H), 7,51 (д, J=9,0 Гц, 1H).
[0086] Пример 14
Синтез N-[(1S*)-7-(дифторметил)-2,2-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида
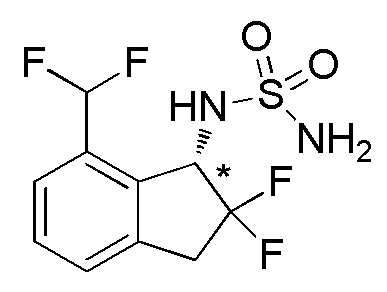
(1) Синтез 7-бром-2,2-дифтор-2,3-дигидро-1H-инден-1-она
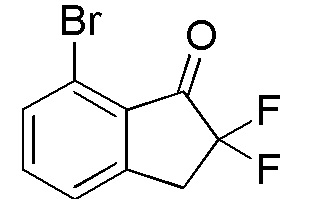
Титульное соединение (1,96 г, 7,95 ммоля) получали из 7-бром-1-инданона (номер по CAS 125114-77-4, 2,00 г, 9,48 ммоля) способом, аналогичным описанному в примере 1-(1) и 1-(2).
1H-ЯМР (400 МГц, CDCl3)
δ м.д. 3,53 (т, J=12,9 Гц, 2H), 7,43(д, J=7,8 Гц, 1H), 7,55 (т, J=7,8 Гц, 1H), 7,65 (т, J=7,8 Гц, 1H).
[0087] (2) Синтез 7-бром-2,2-дифтор-2,3-дигидро-1H-инден-1-ола
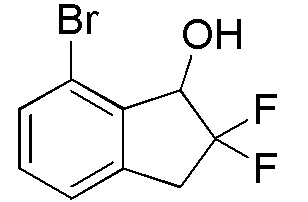
Титульное соединение (1,98 г, 7,95 ммоля) получали из продукта, полученного в примере 14-(1) (1,96 г, 7,95 ммоля), способом, аналогичным описанному в примере 11-(3).
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 2,46 (дд, J=4,5, 1,8 Гц, 1H), 3,39 (тд, J=16,9, 3,3 Гц, 1H), 3,56-3,66 (м, 1H), 5,11 (дд, J=12,3, 4,5 Гц, 1H), 7,20-7,25 (м, 2H), 7,46 (дд, J=6,6, 1,6 Гц, 1H).
[0088] (3) Синтез 2-[(7-бром-2,2-дифтор-2,3-дигидро-1H-инден-1-ил)окси]тетрагидро-2H-пирана
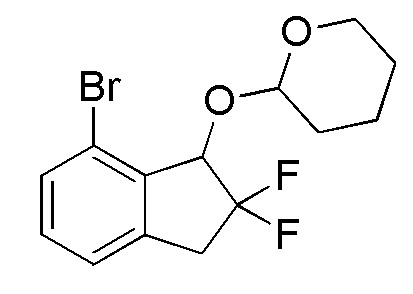
Титульное соединение (2,53 г, 7,60 ммоля) получали в виде рацемической смеси диастереоизомеров с соотношением приблизительно 1:1 из продукта, полученного в примере 14-(2) (1,98 г, 7,95 ммоля), способом, аналогичным описанному в примере 12-(3).
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 1,50-1,84 (м, 6H), 3,26-3,39 (м, 1H), 3,53-3,68 (м, 2H), 4,07-4,23 (м, 1H), 5,04-5,25 (м, 2H), 7,18-7,23 (м, 2H), 7,45 (т, J=5,7 Гц, 1H).
[0089] (4) Синтез 2,2-дифтор-3-[(тетрагидро-2H-пиран-2-ил)окси]-2,3-дигидро-1H-инден-4-карбальдегида
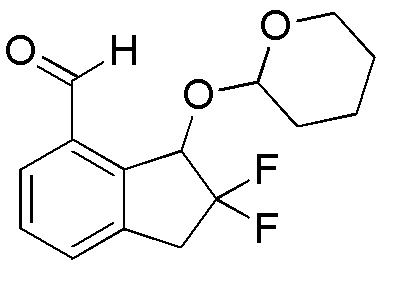
Продукт, полученный в примере 14-(3) (2,00 г, 6,00 ммоля), растворяли в THF (30 мл) и раствор охлаждали в бане с сухим льдом-EtOH.
Добавляли по каплям н-бутиллитий (2,68 мл, 2,69M в гексане) и смесь перемешивали в течение 20 минут при охлаждении. Затем к реакционной смеси добавляли DMF (0,70 мл, 9,00 ммоля) и смесь перемешивали в течение 3 часов при комнатной температуре. К реакционной смеси добавляли воду и смесь экстрагировали, используя AcOEt. Органический слой промывали солевым раствором и высушивали над безводным Na2SO4. После фильтрации фильтрат концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера L, 20 мл/мин, градиент 5%-33% AcOEt в н-гептане) с получением титульного соединения (1,09 г, 3,86 ммоля) в виде рацемической смеси диастереоизомеров с соотношением приблизительно 1:1.
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 1,49-1,85 (м, 6H), 3,28-3,41 (м, 1H), 3,51-3,65 (м, 2H), 3,87-4,10 (м, 1H), 5,02-5,04 (м, 1H), 5,51 (дд, J=11,7, 2,0 Гц, 0,5H), 5,71 (дд, J=11,1, 1,8 Гц, 0,5H), 7,47-7,56 (м, 2H), 7,80-7,88 (м, 1H), 10,19 (с, 0,5H), 10,48 (с, 0,5H).
[0090] (5) Синтез 2-[(7-дифторметил-2,2-дифтор-2,3-дигидро-1H-инден-1-ил)окси]тетрагидро-2H-пирана
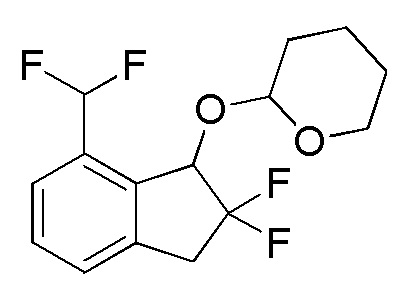
К раствору продукта, полученного в примере 14-(4) (1,09 г, 3,87 ммоля), в DCM (19 мл) добавляли BAST (2,14 мл, 11,6 ммоля) и воду (6,96 мкл, 0,386 ммоля) при комнатной температуре. Смесь перемешивали в течение 15 часов при комнатной температуре.
К реакционной смеси добавляли насыщенный водный NaHCO3 и смесь экстрагировали, используя AcOEt. Органический слой промывали солевым раствором и высушивали над безводным Na2SO4. После фильтрации фильтрат концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера L, 20 мл/мин, градиент 5%-25% AcOEt в н-гептане) с получением титульного соединения (747 мг, 2,46 ммоля) в виде рацемической смеси диастереоизомеров с соотношением приблизительно 1:1.
1H-ЯМР (400 МГц, CDCl3)
δ (м.д.): 1,48-1,97 (м, 6H), 3,17-3,75 (м, 3H), 3,95-4,18 (м, 1H), 4,95 (шир. с, 1H), 5,23-5,28 (м, 0,5H), 5,42 (д, J=10,9 Гц, 0,5H), 6,79-7,28 (м, 1H), 7,34-7,61 (м, 3H).
[0091] (6) Cинтез 7-(дифторметил)-2,2-дифтор-2,3-дигидро-1H-инден-1-oла
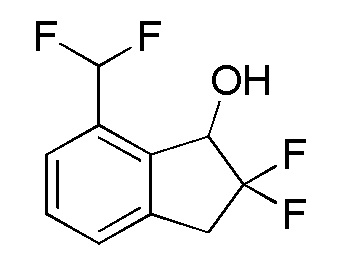
К раствору продукта, полученного в примере 14-(5) (747 мг, 2,46 ммоля), в MeOH (12 мл) добавляли PPTS (61,7 мг, 0,245 ммоля) при комнатной температуре. Реакционную смесь перемешивали в течение 1 часа при 60°C, а затем охлаждали до комнатной температуры. К реакционной смеси добавляли насыщенный водный NaHCO3 и смесь экстрагировали с использованием AcOEt. Органический слой промывали солевым раствором и сушили над безводным Na2SO4. После фильтрации фильтрат выпаривали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера M, 10 мл/мин, градиент 5%-67% AcOEt в н-гептане) с получением титульного соединения (459 мг, 2,09 ммоля).
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,47 (дд, J=6,0, 2,9 Гц, 1H), 3,33-3,59 (м, 2H), 5,34 (дт, J=11,5, 6,0 Гц, 1H), 7,04 (т, J=55,6 Гц, 1H), 7,37 (дд, J=7,6, 0,8 Гц, 1H), 7,45 (т, J=7,6 Гц, 1H), 7,50 (д, J=7,6 Гц, 1H).
[0092] (7) Cинтез 7-(дифторметил)-2,2-дифтор-2,3-дигидро-1H-инден-1-амина
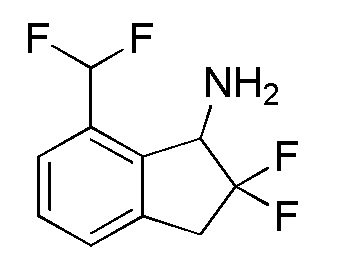
Tитульное соединение (85 мг, 0,388 ммоля) получали из продукта, полученного в примере 14-(6) (160 мг, 0,727 ммоля), способом, аналогичным описанному в примере 12-(6) и 12-(7).
ESI-MS; m/z: 200 [M+H]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 1,56 (шир. с, 2H), 3,29-3,54 (м, 2H), 4,62 (дд, J=12,7, 6,8 Гц, 1H), 7,32 (д, J=8,0 Гц, 1H), 7,36 (т, J=56,0 Гц, 1H), 7,39 (т, J=8,0 Гц, 1H), 7,53(д, J=8,0 Гц, 1H).
[0093] (8) Cинтез N-[(1S*)-7-(дифторметил)-2,2-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида
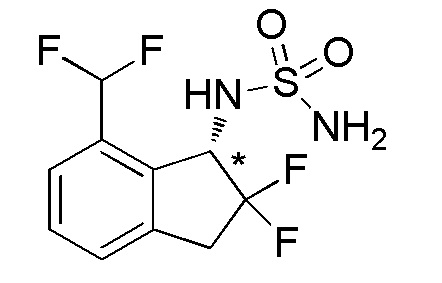
Титульное соединение (30 мг, 0,176 ммоля) получали в виде рацемата из продукта, полученного в примере 14-(7) (85 мг, 0,388 ммоля), способом, аналогичным описанному в примере 1-(4) и 1-(5).
ESI-MS; m/z: 321 [M+Na]+
1H-ЯМР (400 MГц, DMSO-d6)
δ (м.д.): 3,37-3,62 (м, 2H), 5,01-5,07 (м, 1H), 6,91 (с, 2H), 7,29 (т, J=55,6 Гц, 1H), 7,47 (д, J=7,6 Гц, 1H), 7,52 (д, J=7,6 Гц, 1H), 7,56 (д, J=7,6 Гц, 1H), 7,81 (д, J=9,4 Гц, 1H).
Оптическое разделение полученного рацемата (30 мг, 0,176 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IA, 20 мм I.D.×250 мм, 15% EtOH в н-гексане, 10 мл/мин) с получением энантиомера титульного соединения (8 мг, 0,101 ммоля, >99% э.и.), который элюировали вторым из 2 оптических изомеров.
В результате анализа с использованием DAICEL CHIRALPAKTM IA (4,6 мм I.D.×150 мм, 15% EtOH в н-гексане, 1 мл/мин) оптический изомер показывал время удержания 12 минут.
[0094] Пример 15
Cинтез N-[(1R*,2R*)-2,4,7-трифтор-2,3-дигидро-1H-инден-1-ил]сульфамида
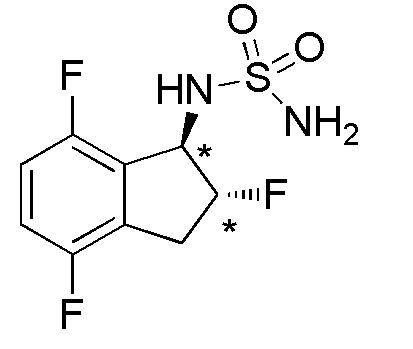
(1) Cинтез 2,4,7-трифтор-2,3-дигидро-1H-инден-1-она
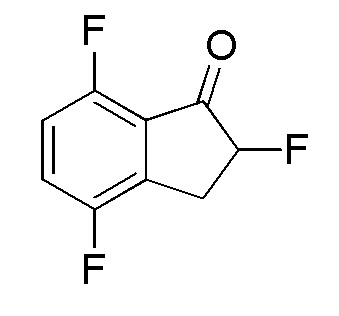
SelectfluorTM (11,7 г, 33,0 ммоля) добавляли к раствору 4,7-дифтор-1-инданoна (номер по CAS 130408-16-1, 5,04 г, 30,0 ммоля) в MeOH (100 мл) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 4 часов. После охлаждения до комнатной температуры к смеси добавляли 5N HCl (5 мл), а затем смесь перемешивали при комнатной температуре в течение 1 часа. Далее реакционную смесь концентрировали in vacuo, осадок разделяли между AcOEt и насыщенным водным NaHCO3.
Органический слой промывали солевым раствором, высушивали над MgSO4 и растворитель выпаривали in vacuo с получением титульного соединения (5,56 г, 29,9 ммоля).
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.) 3,11-3,26 (м, 1H), 3,62-3,73 (м, 1H), 5,23 (ддд, J=50,4, 7,8, 4,1 Гц, 1H), 7,03-7,09 (м, 1H), 7,32-7,37 (м, 1H).
[0095] (2) Cинтез N-[(1R*,2R*)-2,4,7-трифтор-2,3-дигидро-1H-инден-1-ил]сульфамида
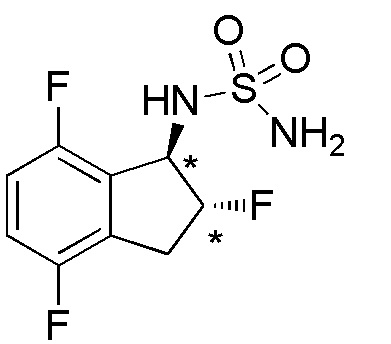
Ацетат аммония (7,71 г, 100 ммоля) и MgSO4 (12,0 г, 100 ммоля) добавляли к раствору соединения (931 мг, 5,00 ммоля), полученного в примере 15-(1), в изопропаноле (30 мл) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 2 часов. Цианоборгидрид натрия (943 мг, 15,0 ммоля) добавляли к реакционной смеси и нагревали с обратным холодильником в течение 5 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали in vacuo.
К осадку добавляли диэтиловый эфир и 2N HCl и смесь разделяли. Водный слой нейтрализовали с использованием 2N NaOH и экстрагировали с использованием AcOEt. Органический слой промывали солевым раствором, высушивали над MgSO4. Растворитель выпаривали in vacuo, а осадок высушивали с получением (1RS,2RS)-2,4,7-трифтор-2,3-дигидро-1H-инден-1-амина и (1RS,2SR)-2,4,7-трифтор-2,3-дигидро-1H-инден-1-амина в виде смеси диастероизомеров (170 мг, 0,91 ммоля).
К раствору смеси диастереоизомеров (168 мг, 0,90 ммоля) в DCM (10 мл) добавляли [(трет-бутокси)карбонил]{[4-(диметилимино)пиридин-1(4H)-ил]сульфонил}амид (353 мг, 1,17 ммоля) и TEA (0,13 мл, 0,90 ммоля) при комнатной температуре. Раствор перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь разбавляли с использованием AcOEt и 1N HCl. Далее слои разделяли и органический слой высушивали над безводным Na2SO4 и выпаривали in vacuo. Осадок очищали посредством колоночной хроматографии на силикагеле (20%-30%, AcOEt в н-гептане) с получением (1RS,2RS)-[трет-бутил-N-(2,4,7-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата] и (1RS,2SR)-[трет-бутил-N-(2,4,7-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата] в виде смеси диастереоизомеров (190 мг, 0,52 ммоля). К раствору смеси диастереоизомеров (183 мг, 0,50 ммоля) в MeOH (2 мл) добавляли 4N HCl в AcOEt (2мл) при комнатной температуре и перемешивали при комнатной температуре в течение 16 часов. Растворитель выпаривали in vacuo и осадок сушили с получением титульного соединения в виде смеси. Полученную смесь разделяли посредством ВЭЖХ (CHIRALPAKTM IA, 20 мм I.D.×250 мм, 10 мл/мин, 20% EtOH в н-гексане) с получением титульного соединения, которое элюировалось вторым из 4 оптически активных соединений с временем удержания 14 минут (76 мг, 0,267 ммоля; >99% э.и.).
ESI-MS m/z: 289 [M+Na]+
1H ЯМР (400 MГц, CDCl3)
δ (м.д.): 3,22 (дд, J=24,8, 18,0 Гц, 1H), 3,39-3,52 (м, 1H), 4,58 (шир. с, 1H), 4,73 (шир. с, 2H), 5,15 (д, J=16,6 Гц, 1H), 5,58-5,61 (м, 1H), 6,93-7,01 (м, 1H), 7,02-7,09 (м, 1H).
[0096] Пример 16
Cинтез (-)-N-[(1R*,2R*)-7-хлор-2,4-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида (16a) и (+)-N-[(1R*,2R*)-7-хлор-2,4-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида (16b)
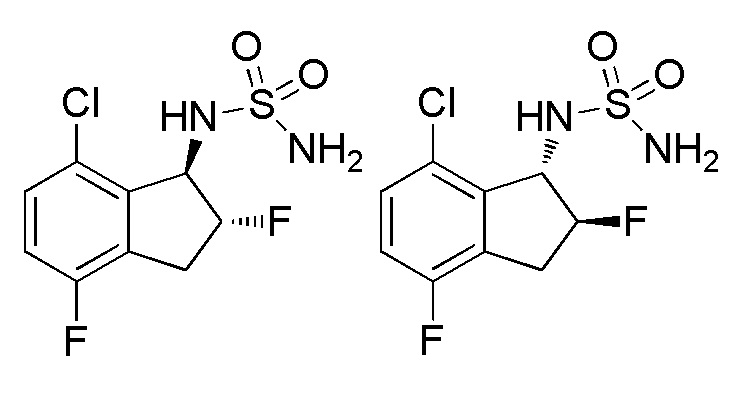
(1) Cинтез 7-хлор-2,4-дифтор-2,3-дигидро-1H-инден-1-она
Tитульное соединение (181 мг, 0,637 ммоля) получали из 7-хлор-4-фтор-1-инданoна (номер по CAS 881190-28-9, 1,85 г, 10,0 ммоля) способом, аналогичным описанному в примере 15-(1).
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 3,09-3,22 (м, 1H), 3,60-3,70 (м, 1H), 5,25 (ддд, J=50,8, 8,0, 4,5 Гц, 1H), 7,27-7,31 (м, 1H), 7,36-7,39 (м, 1H).
[0097] (2) Cинтез (-)-N-[(1R*,2R*)-7-хлор-2,4-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида (16a) и (+)-N-[(1R*,2R*)-7-хлор-2,4-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида (16b)
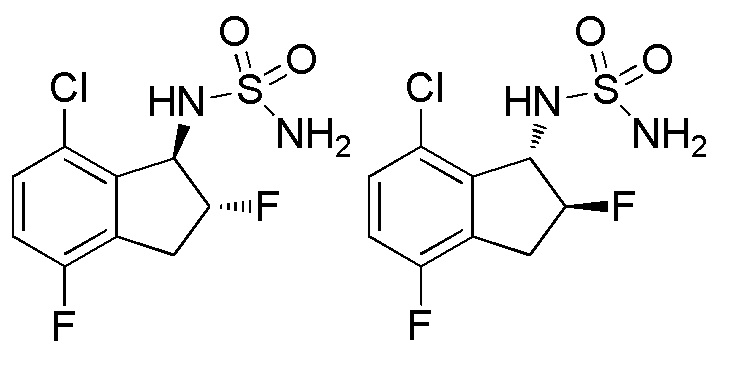
Смесь из 4 изомеров получали из продукта (405 мг, 2,00 ммоля), полученного в примере 16-(1), способом, аналогичным описанному в примере 15-(2). Смесь разделяли посредством ВЭЖХ (CHIRALPAKTM IF, 20 мм I.D.×250 мм, 10 мл/мин, 20% EtOH в гексане) с получением (-)-формы титульного соединения (8,2 мг, 0,029 ммоля; >99% э.и.), которая элюировалась первой из 4 оптически активных соединений с временем удержания 9 минут, и (+)-формы титульного соединения (8,3 мг, 0,029 ммоля; >99% э.и.), которая элюировалась второй с временем удержания 12 минут, соответственно.
Характеристики (-)-формы титульного соединения были следующими.
ESI-MS; m/z 305, 307 [M+Na]+.
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 3,21 (дд, J=25,4, 18,4 Гц, 1H), 3,40-3,56 (м, 1H), 4,73 (шир. с, 3H), 5,03 (д, J=16,2 Гц, 1H), 5,56 (дд, J=49,2, 4,7 Гц, 1H), 7,05 (т, J=8,4 Гц, 1H), 7,23-7,29 (м, 1H).
Характеристики (+)-формы титульного соединения были такими же, как и для (-)-формы.
ESI-MS; m/z 305, 307 [M+Na]+.
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 3,21 (дд, J=25,4, 18,4 Гц, 1H), 3,40-3,56 (м, 1H), 4,73 (шир. с, 3H), 5,03 (д, J=16,2 Гц, 1H), 5,56 (дд, J=49,2, 4,7 Гц, 1H), 7,05 (т, J=8,4 Гц, 1H), 7,23-7,29 (м, 1H).
[0098] Пример 17
Cинтез (-)-N-[(1R*,2R*)-7-хлор-2,5-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида
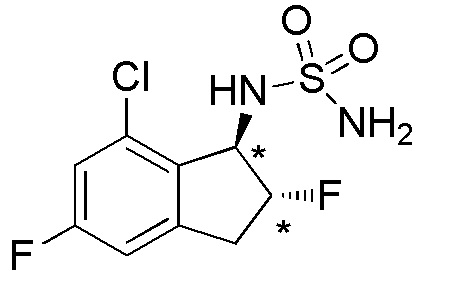
Смесь из 4 изомеров (рацемическая смесь 2 диастереоизомеров) получали из продукта (1,06 г, 5,23 ммоля), полученного в примере 6-(1) способом, аналогичным описанному в примере 15-(2). Смесь очищали посредством колоночной хроматографии на силикагеле (WAKO, PresepTM силикагель (HC-N), размер: 3L, 60 мл/мин, 30%-60%, AcOEt в н-гептане) с получением титульного соединения в виде рацемата (177 мг, 0,626 ммоля). Полученный рацемат (175 мг, 0,619 ммоля) разделяли посредством ВЭЖХ (CHIRALPAKTM IB, 20 мм I.D.×250 мм, 10 мл/мин, 10% EtOH в н-гексане) с получением (-)-формы титульного соединения (67 мг, 0,237 ммоля; 99% э.и.), которая элюировалась второй из 2 изомеров с временем удержания 40 минут.
ESI-MS; m/z 305 [M+Na]+.
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 3,21 (дд, J=25,4, 18,4 Гц, 1H), 3,40-3,56 (м, 1H), 4,73 (шир. с, 3H), 5,03 (д, J=16,2 Гц, 1H), 5,56 (дд, J=49,2, 4,7 Гц, 1H), 7,05 (т, J=8,4 Гц, 1H), 7,23-7,29 (м, 1H).
[0099] Пример 18
Cинтез (+)-N-[(1R*,2R*)-4-хлор-7-фтор-2-метокси-2,3-дигидро-1H-инден-1-ил]сульфамида
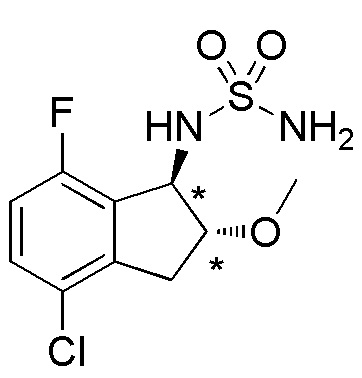
(1) Cинтез (1RS,2RS)-2-бром-4-хлор-7-фтор-2,3-дигидро-1H-инден-1-ола
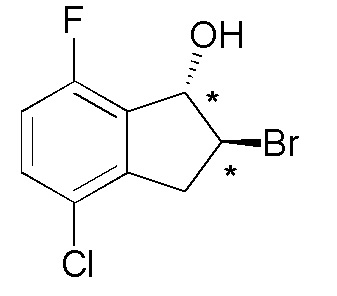
К раствору 4-хлор-7-фтор-2,3-дигидро-1H-инден-1-она (номер по CAS 1260018-63-0, 900 мг, 4,88 ммоля) в MeOH (30 мл) добавляли боргидрид натрия (221 мг, 5,85 ммоля) при комнатной температуре. Реакционную смесь перемешивали в течение 20 минут при комнатной температуре. К реакционной смеси добавляли 2N HCl, AcOEt и воду при 0°C. Слои разделяли. Органический слой промывали солевым раствором и высушивали над MgSO4, а затем растворитель выпаривали in vacuo. К раствору осадка в толуоле (50 мл) добавляли моногидрат п-толуолсульфоновой кислоты (100 мг, 0,53 ммоля) и реакционную смесь нагревали с обратным холодильником в течение 20 минут. После охлаждения до комнатной температуры реакционную смесь пропускали через NH-силикагель и выпаривали in vacuo. К раствору осадка в THF (30 мл) и воды (10 мл) добавляли NBS (842 мг, 4,73 ммоля) и реакционную смесь перемешивали в течение 20 часов при комнатной температуре. К смеси добавляли AcOEt и солевой раствор и слои разделяли. Органический слой последовательно промывали насыщенным водным Na2S2O3 и солевым раствором, а затем высушивали над MgSO4. Растворитель выпаривали in vacuo. Остаток очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера L, 40 мл/мин, градиент 0%-40% AcOEt в н-гептане) с получением титульного соединения (675 мг, 2,54 ммоля).
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,43 (д, J=4,5 Гц, 1H), 3,28 (дд, J=17,8, 4,5 Гц, 1H), 3,78 (дд, J=17,8, 6,5 Гц, 1H), 4,41-4,45 (м, 1H), 5,60-5,62 (м, 1H), 6,92-6,97 (м, 1H), 7,29 (дд, J=8,8, 4,3 Гц, 1H).
[0100] (2) Cинтез (1RS,2RS)-1-азидо-4-хлор-7-фтор-2,3-дигидро-1H-инден-2-ола
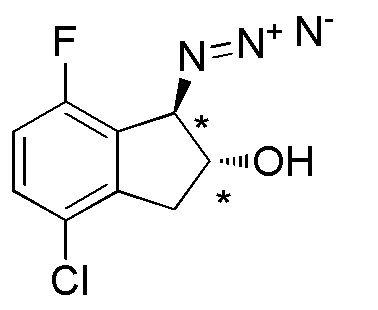
К раствору продукта, полученного в примере 18-(1) (675 мг, 2,54 ммоля), в THF (10 мл) добавляли гидроксид калия (357 мг, 6,36 ммоля) при комнатной температуре. При этом реакционную смесь перемешивали в течение 2 часов при комнатной температуре. К смеси добавляли AcOEt и солевой раствор и слои разделяли. Органический слой высушивали над MgSO4 и выпаривали in vacuo. К раствору осадка в EtOH (12 мл) и воды (4 мл) добавляли хлорид аммония (204 мг, 3,81 ммоля) и азид натрия (248 мг, 3,81 ммоля) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры смесь разделяли между AcOEt и солевым раствором. Органический слой высушивали над MgSO4 и выпаривали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера L, 40 мл/мин, градиент 0%-40% AcOEt в н-гептане) с получением титульного соединения (452 мг, 1,99 ммоля).
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,91 (д, J=17,2, 3,7 Гц, 1H), 3,36-3,42 (м, 1H), 4,50-4,53 (м, 1H), 4,98 (д, J=2,9 Гц, 1H), 6,95-6,99 (м, 1H), 7,30 (дд, J=8,6, 4,3 Гц, 1H).
[0101] (3) Cинтез т-бутил [(1RS,2RS)-4-хлор-7-фтор-2-гидрокси-2,3-дигидро-1H-инден-1-ил]карбамата
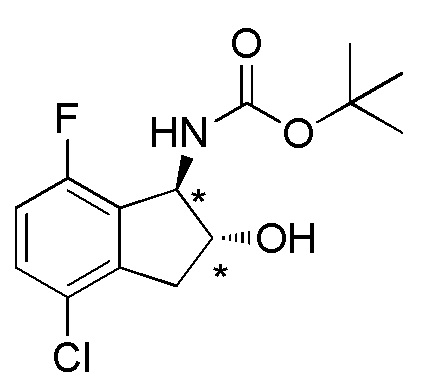
К раствору продукта, полученного в примере 18-(2) (452 мг, 1,99 ммоля), в THF (6 мл) и воде (2 мл) добавляли трифенилфосфин (781 мг, 2,99 ммоля) при комнатной температуре. При этом реакционную смесь нагревали с обратным холодильником в течение 2 часов. К смеси добавляли AcOEt и 2N HCl и слои разделяли. Водный слой подщелачивали с использованием 5N гидроксида натрия и экстрагировали с использованием DCM. Экстракт высушивали над MgSO4 и выпаривали in vacuo.
Осадок растворяли в MeOH (5мл) и к раствору добавляли Boc2O (650 мг, 2,99 ммоля). Затем растворитель выпаривали in vacuo.
Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера L, 40 мл/мин, градиент 0%-60% AcOEt в н-гептане) с получением титульного соединения (313 мг, 1,04 ммоля).
ESI-MS; m/z: 324 [M+Na]+.
[0102] (4) Cинтез т-бутил [(1RS,2RS)-4-хлор-7-фтор-2-метокси-2,3-дигидро-1H-инден-1-ил]карбамата
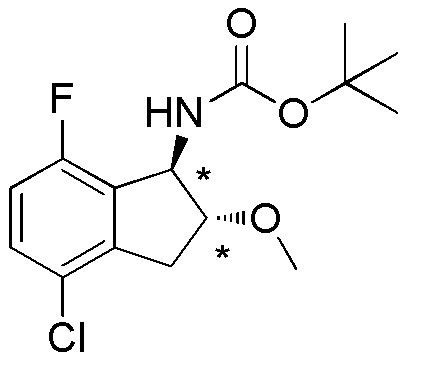
К раствору продукта, полученного в примере 18-(3) (313 мг, 1,04 ммоля), в THF (5 мл) и 50% водном растворе гидроксида натрия (5 мл) добавляли бензилтриметиламмония хлорид (23 мг, 0,10 ммоля) и йодметан (294 мг, 2,08 ммоля) при комнатной температуре. При этом реакционную смесь перемешивали в течение 14 часов при комнатной температуре. К смеси добавляли AcOEt и солевой раствор и слои разделяли. Органический слой высушивали над MgSO4 и выпаривали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM размера L, 40 мл/мин, градиент 0%-40% AcOEt в н-гептане) с получением титульного соединения (321 мг, 1,02 ммоля).
ESI-MS; m/z: 338 [M+Na]+.
[0103] (5) Cинтез (+)-N-[(1R*,2R*)-4-хлор-7-фтор-2-метокси-2,3-дигидро-1H-инден-1-ил]сульфамида
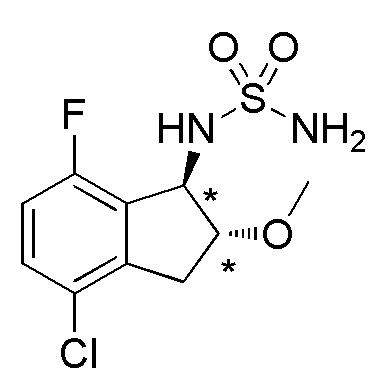
К продукту в примере 18-(4) (321 мг, 1,02 ммоля) добавляли 4N HCl в AcOEt (2 мл) и смесь перемешивали в течение 10 минут при комнатной температуре. Растворитель выпаривали in vacuo и осадок растворяли в DCM (5 мл). К раствору добавляли сульфамоилхлорид (235 мг, 2,03 ммоля) и TEA (0,425 мл, 3,05 ммоля) при 0°C и реакционную смесь перемешивали в течение 20 минут при комнатной температуре. К реакционной смеси добавляли AcOEt и 2N HCl и слои разделяли. Органический слой промывали солевым раствором, сушили над безводным MgSO4 и растворитель концентрировали in vacuo. Осадок очищали посредством колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASHTM с аминосиликагелем, размера L, 40 мл/мин, градиент 30%-90% AcOEt в н-гептане) с получением титульного соединения (187 мг, 0,634 ммоля) в виде рацемата.
Оптическое разделение полученного рацемата (187 мг, 0,634 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM ID, 20 мм I.D.×250 мм, 10% EtOH в н-гексане, 10 мл/мин) с получением (+)-изомера титульного соединения (50,2 мг, 0,170 ммоля, >99% э.и.), который элюировался вторым из 2 оптических изомеров с временем удержания 31 минута.
ESI-MS; m/z: 317 [M+Na]+
1H-ЯМР (400 MГц, DMSO-d6)
δ (м.д.): 3,06-3,15 (м, 1H), 3,32 (с, 3H), 3,89-3,92 (м, 1H), 4,78 (дд, J=15,8, 8,2 Гц, 1H), 5,25 (дд, J=15,8, 6,0 Гц, 1H), 5,87-5,93 (м, 1H), 6,10-6,14 (м, 1H), 7,37-7,45 (м, 1H), 7,91 (дд, J=7,7, 1,9 Гц, 1H).
[0104] Пример 19
Cинтез N-[(1S)-4,7-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида
(1) Cинтез т-бутил N-(4,7-дифтор-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
К раствору 4,7-дифтор-2,3-дигидро-1H-инден-1-амина (номер по CAS 625471-13-8, 1,19 г, 11,2 ммоля) в DCM (50 мл) добавляли [(трет-бутокси)карбонил]{[4-(диметилимино)пиридин-1(4H)-ил]сульфонил}амид (3,70 г, 12,3 ммоля) и TEA (4,66 мл, 33,5 ммоля) при комнатной температуре. Полученный раствор перемешивали в течение 75 часов при комнатной температуре. Реакционную смесь разбавляли с использованием AcOEt и 1N HCl и слои разделяли. Органический слой высушивали над безводным Na2SO4 и выпаривали in vacuo с получением титульного соединения (3,82 г, 11,0 ммоля).
ESI-MS; m/z: 371 [M+Na]+
1H-ЯМР (400 MГц, DMSO-d6)
δ (м.д.): 1,44 (с, 9H), 2,02 (ддт, J=13,3, 8,8, 4,6, 1H), 2,36-2,45 (м, 1H), 2,81 (ддд, J=15,4, 8,0, 4,0 Гц, 1H), 3,03 (дт, J=15,4, 7,5 Гц, 1H), 5,00 (тд, J=8,0, 4,1 Гц, 1H), 7,00-7,06 (м, 1H), 7,13 (тд, J=8,5, 3,7 Гц, 1H), 8,20 (д, J=9,4 Гц, 1H), 10,91 (шир. с, 1H).
[0105] (2) Cинтез N-[(1S)-4,7-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида
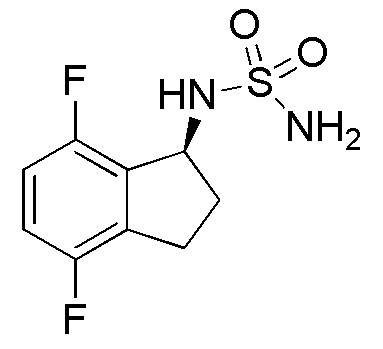
К раствору продукта, полученного в примере 19-(1) (3,81 г, 11,0 ммоля), в MeOH (10 мл) добавляли 4N HCl в AcOEt (41,1 мл, 165 ммоля) и смесь перемешивали при комнатной температуре в течение 14 часов. Растворитель выпаривали in vacuo и осадок высушивали с получением титульного соединения в виде смеси. Осадок очищали посредством колоночной хроматографии на силикагеле (AcOEt) с получением титульного соединения в виде рацемата (2,24 г, 9,02 ммоля).
Оптическое разделение полученного рацемата (900 мг, 3,63 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IA, 20 мм I.D.×250 мм, 30% EtOH в н-гексане, 10 мл/мин) с получением (S)-формы титульного соединения (380 мг, 1,53 ммоля, >99% э.и.) в виде твердого белого вещества, которая элюировалась второй из 2 оптических изомеров с временем удержания 19 минут.
ESI-MS; m/z: 271 [M+Na]+
1H-ЯМР (400 MГц, DMSO-d6)
δ (м.д.): 2,17-2,24 (м, 1H), 2,35-2,44 (м, 1H), 2,80 (ддд, J=16,2, 8,7, 4,2 Гц, 1H), 3,05 (дт, J=16,2, 7,8 Гц, 1H), 4,95 (тд, J=8,2, 3,5 Гц, 1H), 6,64 (шир. с, 2H), 7,00-7,14 (м, 3H).
[0106] Пример 20
Cинтез (+)-N-(7-хлор-4-фтор-2,3-дигидро-1H-инден-1-ил)сульфамида
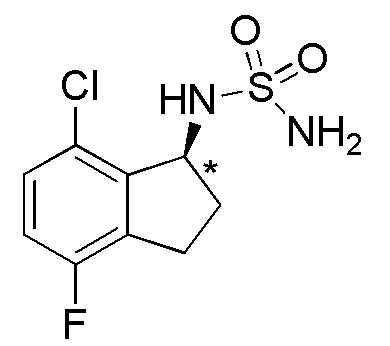
Tитульное соединение (229 мг, 0,87 ммоля) получали в виде рацемата из 7-хлор-4-фтор-2,3-дигидро-1H-инден-1-амина (номер по CAS 1337367-67-5, 198 мг, 1,07 ммоля) способом, аналогичным описанному в примере 19.
ESI-MS; m/z: 287 [M+Na]+.
Оптическое разделение полученного рацемата (190 мг, 0,72 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM AD-H, 20 мм I.D.×250 мм, 30% EtOH в н-гексане, 10 мл/мин) с получением (+)-формы титульного соединения (73 мг, 0,28 ммоля, >99% э.и.) с временем удержания 30 минут из 2 оптических изомеров.
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,39-2,55 (м, 2H), 2,96-3,03 (м, 1H), 3,10-3,19 (м, 1H), 4,53 (д, J=6,6 Гц, 1H), 4,64 (шир. с, 2H), 5,06 (тд, J=6,6, 2,5 Гц, 1H), 6,97 (т, J=8,8 Гц, 1H), 7,19 (дд, J=8,8, 4,1 Гц, 1H).
[0107] Пример 21
Cинтез N-[(1S)-7-хлор-5-фтор-2,3-дигидро-1H-инден-1-ил]сульфамида
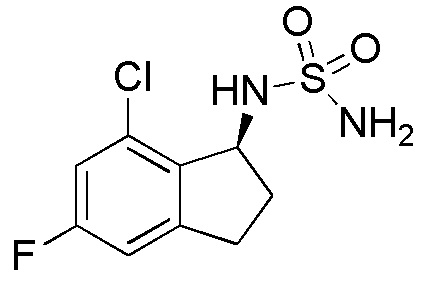
(1) Cинтез т-бутил N-(7-хлор-5-фтор-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
Титульное соединение (200 мг, 0,548 ммоля) получали из 7-хлор-5-фтор-2,3-дигидро-1H-инден-1-амина (номер по CAS 1337210-62-4, 110 мг, 0,593 ммоля) способом, аналогичным описанному в примере 19-(1).
ESI-MS; m/z: 387 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 1,50 (с, 9H), 2,26-2,52 (м, 2H), 2,88 (ддд, J=16,5, 8,5, 2,1 Гц, 1H), 3,18 (дт, J=16,5, 8,5 Гц, 1H), 5,00 (д, J=6,3 Гц, 1H), 5,17 (шир. с, 1H), 6,84-6,93 (м, 1H), 6,93-7,05 (м, 1H).
[0108] (2) Cинтез N -[(1 S )-7-хлор-5-фтор-2,3-дигидро-1 H -инден-1-ил]сульфамида
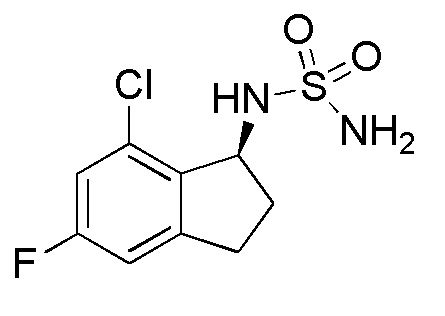
Tитульное соединение (118 мг, 0,446 ммоля) получали в виде рацемата из продукта, полученного в примере 21-(1), способом, аналогичным описанному в примере 19-(2). Оптическое разделение полученного рацемата (60 мг, 0,227 ммоля) проводили посредством ВЭЖХ (CHIRALCELTM OJ-H, 20 мм I.D.×250 мм, 20% EtOH в н-гексане, 10 мл/мин) с получением титульного соединения (26,4 мг, 0,10 ммоля, >99% э.и.), которое элюировалось вторым из 2 оптических изомеров с временем удержания 67 минут.
ESI-MS; m/z: 287 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,29-2,60 (м, 2H), 2,91 (ддд, J=16,6, 8,5, 2,1 Гц, 1H), 3,18 (дт, J=16,6, 8,5 Гц, 1H), 4,47 (д, J=5,7 Гц, 1H), 4,63 (шир. с, 2H), 4,91-5,12 (м, 1H), 6,86-6,93 (м, 1H), 6,94-7,02 (м, 1H).
[0109] Пример 22
Cинтез (+)-N-(7-хлор-5-циано-2,3-дигидро-1H-инден-1-ил)сульфамида (22a) и (-)-N-(7-хлор-5-циано-2,3-дигидро-1H-инден-1-ил)сульфамида (22b)
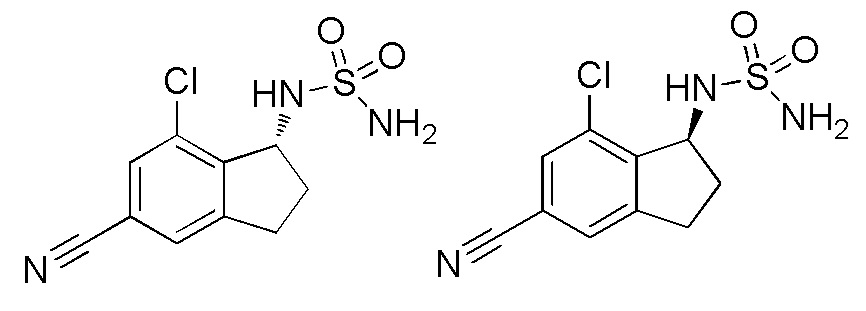
(1) Cинтез т-бутил N-(7-хлор-5-циано-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
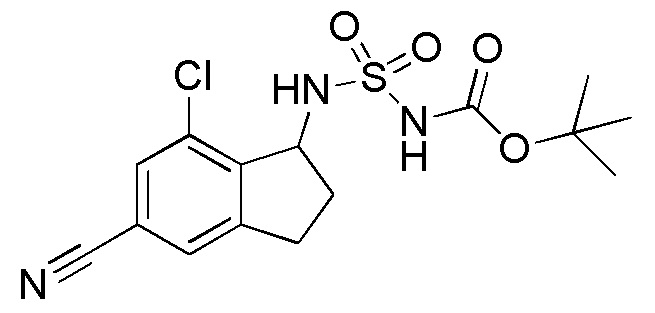
Tитульное соединение (5,45 г, 14,7 ммоля) получали из 7-хлор-5-циано-2,3-дигидро-1H-инден-1-амина (номер по CAS 1337127-29-3, 3,1 г, 16,1 ммоля) способом, аналогичным описанному в примере 19-(1).
ESI-MS; m/z: 394 [M+Na]+
1H-ЯМР (400 MГц, DMSO-d6)
δ (м.д.): 1,42 (с, 9H), 2,03 (ддт, J=13,6, 7,9, 3,0 Гц, 1H), 2,23-2,38 (м, 1H), 2,78-2,90 (м, 1H), 3,12 (дт, J=16,5, 8,4 Гц, 1H), 4,91 (тд, J=8,2, 2,6 Гц, 1H), 7,72 (д, J=1,2 Гц, 1H), 7,82 (д, J=1,2 Гц, 1H), 8,13 (д, J=8,2Гц, 1H), 10,96 (с, 1H).
[0110] (2) Cинтез (+)- N -(7-хлор-5-циано-2,3-дигидро-1 H -инден-1-ил)сульфамида (22a) и (-)- N -(7-хлор-5-циано-2,3-дигидро-1 H -инден-1-ил)сульфамида (22b)
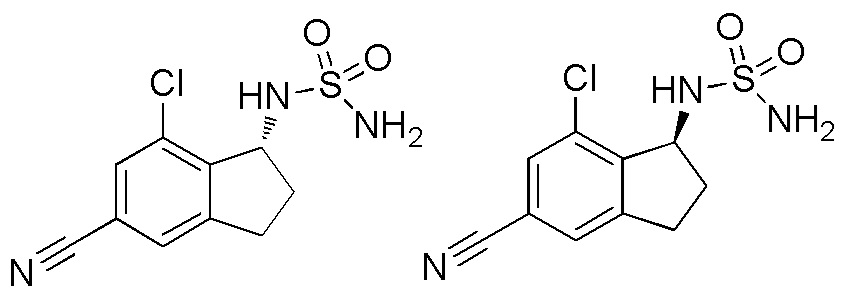
Tитульное соединение (3,50 г, 12,9 ммоля) получали в виде рацемата из продукта, полученного в примере 22-(1) (5,40 г, 14,5 ммоля), способом, аналогичным описанному в примере 19-(2).
Оптическое разделение полученного рацемата (570 мг, 2,01 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IC, 20 мм I.D.×250 мм, 30% EtOH в н-гексане, 10 мл/мин) с получением (+)-формы титульного соединения (252 мг, 0,927 ммоля, >99% э.и.) со временем удержания 26 минут и (-)-формы титульного соединения (251 мг, 0,924 ммоля, >99% э.и.) с временем удержания 34 минут.
Характеристики (+)-формы были следующими.
ESI-MS; m/z: 294 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,19-2,34 (м, 2H), 2,77-2,91 (м, 1H), 3,15 (дт, J=16,5, 8,5 Гц, 1H), 4,86 (ддд, J=9,1, 6,0, 3,6 Гц, 1H), 6,66 (с, 2H), 7,00 (д, J=9,1 Гц, 1H), 7,71 (д, J=1,2 Гц, 1H), 7,80 (д, J=1,2 Гц, 1H).
Характеристики (-)-формы были следующими:
ESI-MS; m/z: 294 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,19-2,34 (м, 2H), 2,77-2,91 (м, 1H), 3,15 (дт, J=16,5, 8,5 Гц, 1H), 4,86 (ддд, J=9,1, 6,0, 3,6 Гц, 1H), 6,66 (с, 2H), 7,00 (д, J=9,1 Гц, 1H), 7,71 (д, J=1,2 Гц, 1H), 7,80 (д, J=1,2 Гц, 1H).
[0111] Пример 23
Cинтез (-)-N-(5-хлор-7-фтор-2,3-дигидро-1H-инден-1-ил)сульфамида
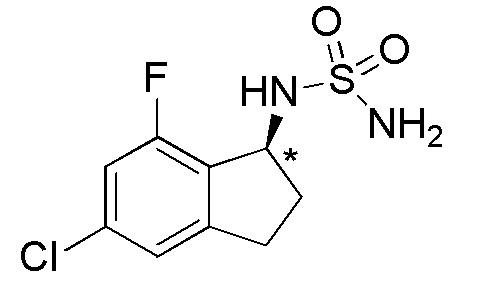
(1) Cинтез т-бутил N-(5-хлор-7-фтор-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
Tитульное соединение (176 мг, 0,482 ммоля) получали из 5-хлор-7-фтор-2,3-дигидро-1H-инден-1-амина (номер по CAS 1337693-39-6, 96 мг, 0,517 ммоля) способом, аналогичным описанному в примере 19-(1).
ESI-MS; m/z: 387 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 1,49 (с, 9H), 2,26-2,37 (м, 1H), 2,40-2,53 (м, 1H), 2,86 (ддд, J=16,5, 8,6, 3,9 Гц, 1H), 3,12 (дт, J=16,5, 8,6 Гц, 1H), 5,05 (тд, J=6,8, 3,2 Гц, 1H), 5,31 (д, J=6,8 Гц, 1H), 6,88-6,97 (м, 1H), 7,07 (с, 1H), 7,15 (шир. с, 1H).
[0112] (2) Cинтез (-)-N-(5-хлор-7-фтор-2,3-дигидро-1H-инден-1-ил)сульфамида
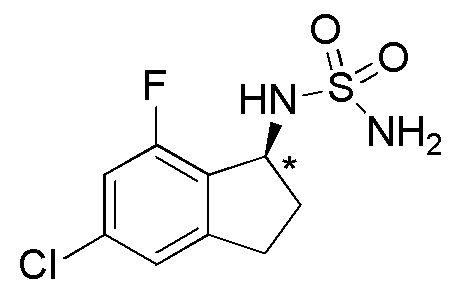
Tитульное соединение (114 мг, 0,431 ммоля) получали в виде рацемата из продукта, полученного в примере 23-(1) (176 мг, 0,482 ммоля), способом, аналогичным описанному в примере 19-(2).
Оптическое разделение полученного рацемата (110 мг, 0,416 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IE, 20 мм I.D.×250 мм, 20% EtOH в н-гексане, 10 мл/мин) с получением (-)-формы титульного соединения (46 мг, 0,174 ммоля, >99% э.и.), которая элюировалась второй из 2 оптических изомеров с временем удержания 28 минут.
ESI-MS; m/z: 287 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,18-2,39 (м, 1H), 2,46-2,65 (м, 1H), 2,87 (ддд, J=16,6, 8,6, 5,3 Гц, 1H), 3,00-3,18 (м, 1H), 4,64 (шир. с, 3H), 5,12 (тд, J=7,5, 4,3 Гц, 1H), 6,90-6,99 (м, 1H), 7,07 (д, J=1,0 Гц, 1H).
[0113] Пример 24
Cинтез (+)-N-(7-хлор-2,3-дигидро-1H-инден-1-ил)сульфамида
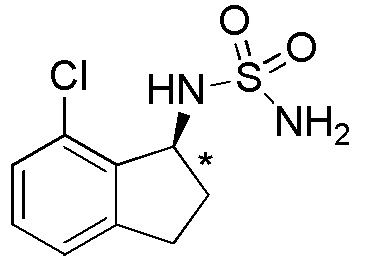
(1) Cинтез т-бутил N-(7-хлор-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
Tитульное соединение (213 мг, 0,614 ммоля) получали из 7-хлор-2,3-дигидро-1H-инден-1-амина (номер по CAS 67120-37-0, 203 мг, 1,21 ммоля) способом, аналогичным описанному в примере 19-(1).
ESI-MS; m/z: 369 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 1,50 (с, 9H), 2,26-2,38 (м, 1H), 2,38-2,47 (м, 1H), 2,89 (ддд, J=16,4, 8,5, 2,4 Гц, 1H), 3,13-3,25 (м, 1H), 5,01-5,07 (м, 1H), 5,16 (д, J=5,5 Гц, 1H), 7,15-7,25 (м, 4H).
[0114] (2) Cинтез (+)-N-(7-хлор-2,3-дигидро-1H-инден-1-ил)сульфамида
Tитульное соединение (108 мг, 0,438 ммоля) получали в виде рацемата из продукта, полученного в примере 24-(1) (213 мг, 0,614 ммоля), способом, аналогичным описанному в примере 19-(2).
Оптическое разделение полученного рацемата (30 мг, 0,122 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IC, 20 мм I.D.×250 мм, 20% EtOH в н-гексане, 10 мл/мин) с получением (+)-формы титульного соединения (12,1 мг, 0,049 ммоля, >99% э.и.), которая элюировалась второй из 2 оптических изомеров с временем удержания 34 минут.
ESI-MS; m/z: 269 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,31-2,55 (м, 2H), 2,86-2,98 (м, 1H), 3,19 (дт, J=16,6, 8,5 Гц, 1H), 4,50 (д, J=5,5 Гц, 1H), 4,63 (шир. с, 2H), 5,06 (тд, J=6,6, 2,2 Гц, 1H), 7,16-7,26 (м, 3H).
[0115] Пример 25
Cинтез (+)-N-(5-циано-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида (25a) и (-)-N-(5-циано-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида (25b)
Tитульное соединение (220 мг, 0,875 ммоля) получали в виде рацемата из 5-циано-7-метил-2,3-дигидро-1H-инден-1-амина (номер по CAS 1337245-99-4, 0,50 г, 2,90 ммоля) способом, аналогичным описанному в примере 19.
Оптическое разделение полученного рацемата (109 мг, 0,432 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IF, 20 мм I.D.×250 мм, 20% EtOH в н-гексане, 10 мл/мин). Полученные изомеры анализировали с использованием CHIRALPAKTM IF (4,6 мм I.D.×150 мм, 20% EtOH в н-гексане, 1 мл/мин). Получали (+)-форму (46 мг, 0,182 ммоля, >99% э.и.) с временем удержания 8 минут и (-)-форму (44 мг, 0,177 ммоля, >99% э.и.) с временем удержания 9 минут.
Характеристики (+)-формы были следующими:
ESI-MS; m/z: 274 [M+Na]+
1H-ЯМР (400 MГц, DMSO-d6)
δ (м.д.): 2,14-2,35 (м, 2H), 2,37 (с, 3H), 2,76 (ддд, J=16,1, 8,3, 3,3 Гц, 1H), 3,09 (ддд, J=16,1, 8,3, 8,3 Гц, 1H), 4,77-4,85 (м, 1H), 6,71 (шир. с, 2H), 6,94 (д, J=9,0 Гц, 1H), 7,45 (с, 1H), 7,53 (с, 1H).
Характеристики (-)-формы были следующими:
ESI-MS; m/z: 274 [M+Na]+
1H-ЯМР (400 MГц, DMSO-d6)
δ (м.д.): 2,14-2,35 (м, 2H), 2,37 (с, 3H), 2,76 (ддд, J=16,1, 8,3, 3,3 Гц, 1H), 3,09 (ддд, J=16,1, 8,3, 8,3 Гц, 1H), 4,77-4,85 (м, 1H), 6,71 (шир. с, 2H), 6,94 (д, J=9,0 Гц, 1H), 7,45 (с, 1H), 7,53 (с, 1H).
[0116] Пример 26
Cинтез (±)-N-(5-фтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида
(1) Cинтез т-бутил N-(5-фтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
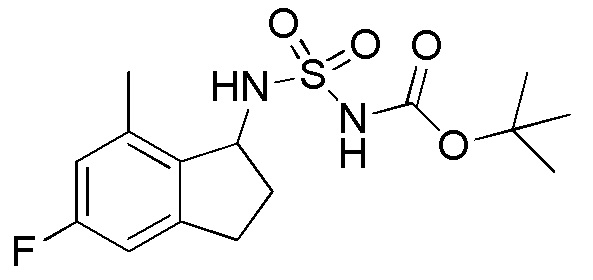
Tитульное соединение (187 мг, 0,543 ммоля) получали из 5-фтор-7-метил-2,3-дигидро-1H-инден-1-амина (номер по CAS 1337122-58-3, 95 мг, 0,575 ммоля) способом, аналогичным описанному в примере 19-(1).
ESI-MS; m/z: 367 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 1,51 (с, 9H), 2,19-2,47 (м, 5H), 2,82 (ддд, J=16,6, 8,3, 3,2 Гц, 1H), 3,07 (дт, J=16,6, 8,3 Гц, 1H), 4,87-5,10 (м, 2H), 6,67-6,85 (м, 2H), 7,19 (шир. с, 1H).
[0117] (2) Cинтез (±)-N-(5-фтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида
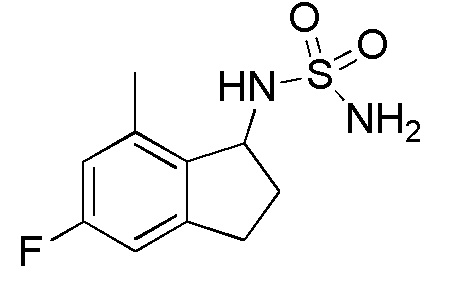
Tитульное соединение (9 мг, 0,037 ммоля) получали в виде рацемата из продукта, полученного в примере 26-(1) (185 мг, 0,537 ммоля), способом, аналогичным описанному в примере 19-(2).
ESI-MS; m/z: 267 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,32-2,47 (м, 5H), 2,75-2,90 (м, 1H), 3,08 (дт, J=16,7, 8,4 Гц, 1H), 4,30 (д, J=8,2 Гц, 1H) 4,61 (шир. с, 2H), 4,89-5,04 (м, 1H), 6,68-6,82 (м, 2H).
[0118] Пример 27
Cинтез (+)-N-(7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида
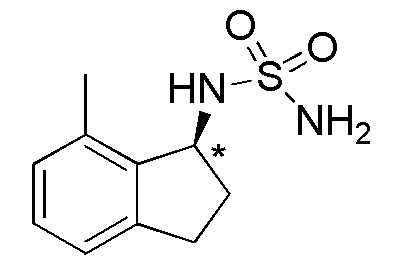
Tитульное соединение (224 мг, 1,91 ммоля) получали в виде рацемата из 7-метил-2,3-дигидро-1H-инден-1-амина (номер по CAS 168902-78-1, 437 мг, 2,97 ммоля) способом, аналогичным описанному в примере 19.
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,33-2,45 (м, 5H), 2,81-2,91 (м, 1H), 3,04-3,16 (м, 1H), 4,29 (д, J=7,4 Гц, 1H), 4,56 (шир. с, 2H), 4,99-5,08 (м, 1H), 7,03 (д, J=7,4 Гц, 1H), 7,10 (д, J=7,4 Гц, 1H), 7,20 (т, J=7,4, 1H).
Оптическое разделение полученного рацемата (174 мг, 0,769 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IE, 20 мм I.D.×250 мм, 15% EtOH в н-гексане, 10 мл/мин) с получением (+)-формы (67 мг, 0,295 ммоля, >99% э.и.), которая элюировалась первой из 2 оптических изомеров.
В результате анализа изомера с использованием CHIRALPAKTM IE (4,6 мм I.D.×150 мм, 15% EtOH в н-гексане, 1 мл/мин) (+)-форма имела время удержания 6 минут и характеристики (+)-формы были следующими:
ESI-MS; m/z: 249 [M+Na]+
[0119] Пример 28
Cинтез (+)-N-(4-фтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида (28a) и (-)-N-(4-фтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида (28b)
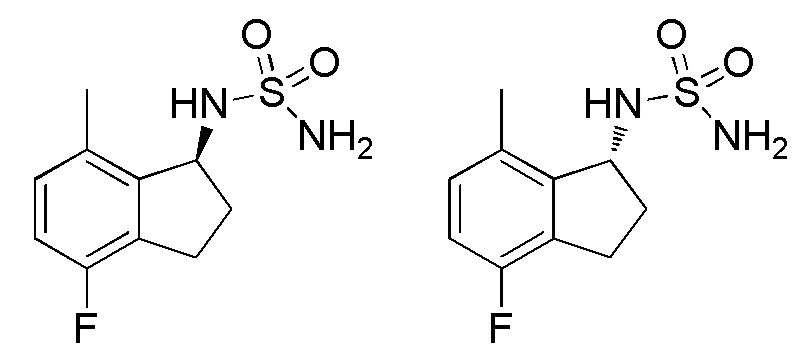
(1) Cинтез бензил N-(4-фтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
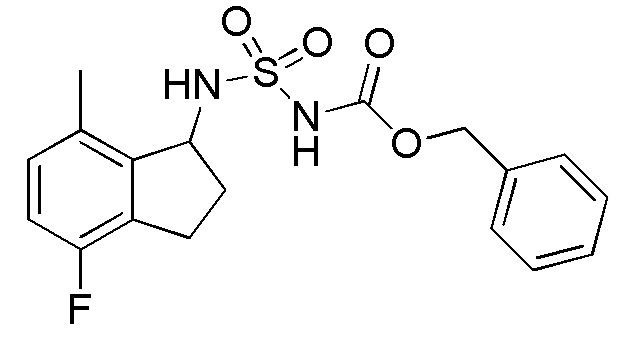
Tитульное соединение (248 мг, 0,655 ммоля) получали из 4-фтор-7-метил-2,3-дигидро-1H-инден-1-амина (номер по CAS 1337048-34-6, 117 мг, 0,708 ммоля) способом, аналогичным описанному в примере 1-(4).
ESI-MS; m/z: 401 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,05-2,16 (м, 1H), 2,19-2,32 (м, 4H), 2,83 (ддд, J=16,7, 8,7, 3,3 Гц, 1H), 2,87-2,98 (м, 1H), 4,94-5,03 (м, 1H), 5,09-5,15 (м, 1H), 5,17-5,25 (м, 2H), 6,87 (т, J=8,4 Гц, 1H), 6,93-7,00 (м, 1H), 7,35-7,40 (м, 5H).
[0120] (2) Cинтез (+)- N -(4-фтор-7-метил-2,3-дигидро-1 H -инден-1-ил)сульфамида (28a) и (-)- N -(4-фтор-7-метил-2,3-дигидро-1 H -инден-1-ил)сульфамида (28b)
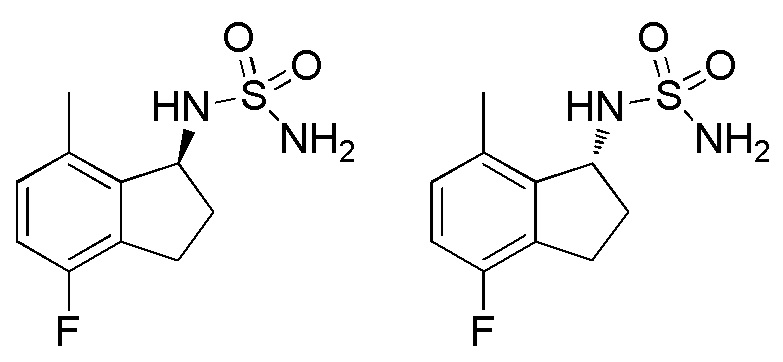
Tитульное соединение (144 мг, 0,589 ммоля) получали в виде рацемата из продукта, полученного в примере 28-(1) (245 мг, 0,647 ммоля), способом, аналогичного описаному в примере 1-(5).
Оптическое разделение полученного рацемата (130 мг, 0,532 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IA, 20 мм I.D.×250 мм, 15% EtOH в н-гексане, 10 мл/мин) с получением (-)-формы (28b) (57 мг, 0,233 ммоля, >99% э.и.) с временем удержания 21 минута и (+)-формы (28a) (53 мг, 0,217 ммоля, >99% э.и.) с временем удержания 25 минут.
Характеристики (-)-формы были следующими:
ESI-MS; m/z: 267 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,32-2,48 (м, 5H), 2,94 (дт, J=16,7, 5,9 Гц, 1H), 3,06 (дт, J=16,7, 8,3 Гц, 1H), 4,34 (д, J=7,4 Гц, 1H), 4,62 (шир. с, 2H), 4,95-5,08 (м, 1H), 6,89 (т, J=8,5 Гц, 1H), 6,99 (дд, J=8,5, 4,9 Гц, 1H).
Характеристики (+)-формы были следующими:
ESI-MS; m/z: 267 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,32-2,48 (м, 5H), 2,94 (дт, J=16,7, 5,9 Гц, 1H), 3,06 (дт, J=16,7, 8,3 Гц, 1H), 4,34 (д, J=7,4 Гц, 1H), 4,62 (шир. с, 2H), 4,95-5,08 (м, 1H), 6,89 (т, J=8,5 Гц, 1H), 6,99 (дд, J=8,5, 4,9 Гц, 1H).
[0121] Пример 29
Cинтез (-)-N-(4,6,7-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамида
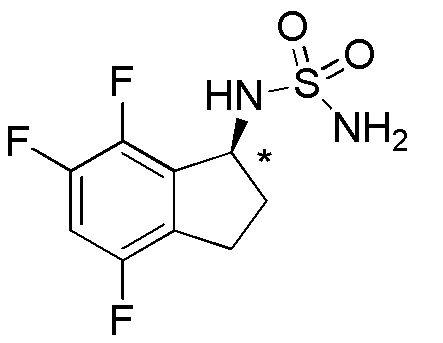
Tитульное соединение (370 мг, 1,39 ммоля) получали в виде рацемата из 4,6,7-трифтор-2,3-дигидро-1H-инден-1-амина (номер по CAS 1337125-68-4, 1,10 г, 5,90 ммоля) способом, аналогичным описанному в примере 1-(4) и 1-(5).
Оптическое разделение полученного рацемата (340 мг, 1,28 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IC, 20 мм I.D.×250 мм, 20% EtOH в н-гексане, 10 мл/мин) с получением (-)-формы титульного соединения (157 мг, 0,590 ммоля, >99% э.и.), которая элюировалась первой из 2 оптических изомеров со временем удержания 18 минут.
ESI-MS; m/z: 289 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,20-2,39 (м, 1H), 2,53-2,76 (м, 1H), 2,80-2,97 (м, 1H), 2,98-3,16 (м, 1H), 4,54-4,83 (м, 3H), 5,13-5,29 (м, 1H), 6,87 (ддд, J=10,0, 7,8, 5,9 Гц, 1H).
[0122] Пример 30
Cинтез (±)-N-(5-хлор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида
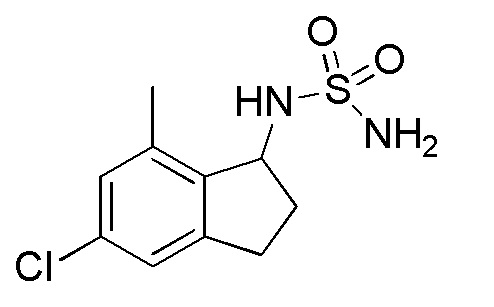
Бензил N-(5-хлор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамат (196 мг, 0,497 ммоля) получали из 5-хлор-7-метил-2,3-дигидро-1H-инден-1-амина (номер по CAS 1337697-66-1, 121 мг, 0,666 ммоля) способом, аналогичным описанному в примере 1-(4).
Tитульное соединение (71 мг, 0,272 ммоля) получали в виде рацемата из бензил N-(5-хлор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата (168 мг, 0,425 ммоля) способом, аналогичным описанному в примере 1-(5).
ESI-MS; m/z: 283 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,34-2,46 (м, 5H), 2,84 (ддд, J=16,5, 7,3, 4,3 Гц, 1H), 3,08 (ддд, J=16,5, 8,4, 8,4, 1H), 4,25 (д, J=7,8 Гц, 1H), 4,56 (шир. с, 2H), 4,94-5,04 (м, 1H), 7,03 (с, 1H), 7,08 (с, 1H).
[0123] Пример 31
Cинтез (-)-N-(4-хлор-7-фтор-2,3-дигидро-1H-инден-1-ил)сульфамида
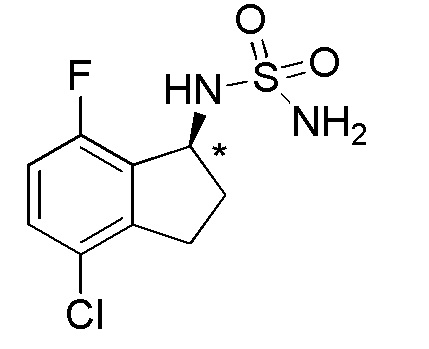
К раствору 4-хлор-7-фтор-2,3-дигидро-1H-инден-1-амина (номер по CAS 1337690-24-0, 371 мг, 2,00 ммоля) и TEA (0,697 мл, 5,00 ммоля) в DCM (10 мл) добавляли сульфамоилхлорид (462 мг, 4,00 ммоля) при 0°C. Реакционную смесь перемешивали в течение 3 часов при комнатной температуре. К реакционной смеси добавляли AcOEt и воду и слои разделяли. Органический слой промывали солевым раствором, сушили над безводным Na2SO4. Растворитель концентрировали in vacuo и осадок очищали посредством колоночной хроматографии на силикагеле (градиент 20%-33% AcOEt в н-гептане) с получением титульного соединения (165 мг, 0,623 ммоля) в виде рацемата.
Оптическое разделение полученного рацемат (139 мг, 0,525 ммоля) проводили посредством ВЭЖХ (CHIRALPAKTM IA, 20 мм I.D.×250 мм, 25% EtOH в н-гексане, 10 мл/мин) с получением (-)-формы титульного соединения (52 мг, 0,196 ммоля, >99% э.и.), которая элюировалась второй из 2 оптических изомеров с временем удержания 22 минуты.
ESI-MS; m/z: 287, 289 [M+Na]+
1H-ЯМР (400 MГц, CDCl3)
δ (м.д.): 2,26-2,34 (м, 1H), 2,57-2,66 (м, 1H), 2,88-2,96 (м, 1H), 3,08-3,16 (м, 1H), 4,63 (шир. с, 2H), 4,66 (шир. с, 1H), 5,19-5,24 (м, 1H), 6,90 (т, J=8,8 Гц, 1H), 7,24-7,27 (м, 1H).
[0124] (Справочный пример 1)
Рентгеноструктурный анализ N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида
Твердое белое вещество, полученное в примере 1-(5), растворяли в MeOH и толуоле и подвергали перекристаллизации посредством способа выпаривания растворителя.
Рентгеноструктурный анализ проводили с использованием полученного монокристалла.
Результаты сбора данных и рентгеноструктурного анализа представлены в таблице 1, а атомные координаты показаны в таблице 2. Абсолютную конфигурацию титульного соединения определяли, исходя из таких результатов.
|
|
|
[0127] (Справочный пример 2)
Рентгеноструктурный анализ N-[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
Твердое белое вещество, полученное в примере 11-(6), растворяли в EtOH и н-гексане и подвергали перекристаллизации при помощи градиента температур с получением микрокристаллов. Микрокристаллы растворяли в Et2O и дополнительно подвергали перекристаллизации посредством способа выпаривания растворителя.
Рентгеноструктурный анализ проводили с использованием полученного монокристалла.
Результаты сбора данных и рентгеноструктурного анализа представлены в таблице 3, а атомные координаты показаны в таблице 4. Абсолютную конфигурацию титульного соединения определяли, исходя из таких результатов.
|
|
|
[0130] (Справочный пример 3)
Рентгеноструктурный анализ N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
Твердое белое вещество, полученное в примере 12-(9), растворяли в EtOH и подвергали перекристаллизации посредством способа диффузии паров с использованием толуола в качестве резервуарного раствора. Рентгеноструктурный анализ проводили с использованием полученного выше монокристалла.
Результаты сбора данных и рентгеноструктурного анализа представлены в таблице 5, а атомные координаты показаны в таблице 6. Абсолютную конфигурацию титульного соединения определяли, исходя из таких результатов.
|
|
[0133] (Справочный пример 4)
Рентгеноструктурный анализ N-[(1S)-4,7-дифтор-2,3-дигидро-1H-инден-1-ил]сульфамида
Твердое белое вещество, полученное в примере 19-(2), растворяли в этаноле и подвергали перекристаллизации посредством способа выпаривания растворителя. Рентгеноструктурный анализ проводили с использованием полученного монокристалла.
Результаты сбора данных и рентгеноструктурного анализа представлены в таблице 7, а атомные координаты показаны в таблице 8. Абсолютную конфигурацию титульного соединения определяли исходя из таких результатов.
|
|
[0136] Примеры фармакологических тестов
Авторы настоящего изобретения проводили следующие тесты для подтверждения фармакологической активности данных соединений.
Данные тесты использовали для оценки ингибиторного эффекта соединения в отношении судорожного припадка.
Модель киндлинга представляет собой модель эпилепсии, точно отражающую патологию у человека, и она демонстрирует хорошую прогнозирующую способность относительно клинических эффектов у человека. Помимо этого, уровень нейротоксических эффектов оценивали путем измерения двигательной недостаточности/координации в тесте вращающегося стержня.
Следующее соединение было описано в патентной литературе 1 (пример 4), патентной литературе 2 (пример 5b) и непатентной литературе 2 (соединение 17).
Авторы настоящего изобретения получали его в соответствии с процедурой получения из патентной литературы 1 и использовали его в качестве эталонного соединения.
[0137] (Тестовый пример 1)
Мышиная модель корнеального киндлинга
Самцов мышей C57Bl/6 подвергали стимуляции дважды в день в течение 10 дней. Электростимуляцию с силой тока 4,0 мA и продолжительностью 3 с (частота повторения импульсов 50 Гц) применяли через смоченные солевым раствором медные электроды, размещенные на роговице.
Тяжесть припадка классифицировали согласно модифицированной системе оценивания шкалы Racine (Clin. Neurophysiol. 1972:32:281-294): 1 балл, умеренный лицевой клонус и моргание; 2, сильный лицевой клонус, кивание головой, жевание; 3, унилатеральный или альтернирующий клонус передних конечностей; 4, билатеральный клонус передних конечностей с подъемом на задние лапы и падением; 5, билатеральный клонус передних конечностей с подъемом на задние лапы и падением; 6, тоническое разгибание передних и/или задних конечностей. Для оценки использовали животных с завершенным киндлингом с баллом тяжести 4 или более.
Тестовые соединения растворяли в растворителе (0,45% метилцеллюлозы/4,5% кремофора/10% DMSO), а затем вводили мышам перорально (p.o.), либо интраперитонеально (i.p.). Растворитель вводили перорально в качестве контроля.
Электростимуляцию проводили через 1 час после p.o. или через 20 минут после i.p. введения. Параметры стимуляции были такими же, как во время процедуры киндлинга. После электростимуляции тяжесть припадка регистрировали в течение 30 секунд согласно шкале Racine.
Для подсчета средней оценки тяжести припадка для каждой дозы использовали от четырех до десяти животных. Что касается эффективного соединения, соединение признавали эффективным при снижении балла тяжести припадка до 2 или менее и рассчитывали эффективную дозу (ED50), при которой у 50% использованных в тесте животных проявилась эффективность.
Результаты противоэпилептического эффекта показаны в таблице 9. Тестовое соединение продемонстрировало более сильный противоэпилептический эффект по сравнению с эталонным соединением.
|
|
[0139] (Тестовый пример 2)
Крысиная модель киндлинга миндалевидного тела
Для эксперимента использовали самцов крыс Wistar Kyoto (Charles River Japan). Для имплантации электродов для киндлинга крыс анестезировали пентобарбиталом (65 мг/кг, i.p.) и трехполярный электрод имплaнтировали в правое полушарие, нацеливаясь на базолатеральное миндалевидное тело, с использованием координат из атласа Paxinos and Watson. Электростимуляцию миндaлевидного тела начинали после постоперационного периода восстановления продолжительностью одна неделя. Начиная со следующего дня, проводили стимуляции миндалевидного тела постоянным током (500 мкA, 1 мс, монофазные меандровые импульсы, 50/с в течение 1 с) с интервалом 1 день до возникновения по меньшей 3 следующих друг за другом припадков завершенного киндлинга (стадия 5; тяжесть припадка классифицировали согласно Racine).
В качестве противоэпилептической активности соединений измеряли пороговую величину для индуцирования следовых разрядов в миндалевидном теле (ADT), т.е. эпилептиформных пиков на электроэнцефалограмме (EEG). У крыс с завершенным киндлингом ADT определяли при проведении ряда стимуляций, начиная от 40 мкA и увеличивая с шагом приблизительно 25% от предшествующей силы тока. Пороговую величину определяли как наиболее низкую интенсивность, вызывающую следовые разряды (AD) продолжительностью по меньшей мере 3 с утром в день проведения эксперимента.
Днем тестовые соединения растворяли в растворителе (смесь полиэтиленгликоля 200, дистиллированной воды и DMSO в соотношении 1:1:1), а затем вводили крысам перорально. Растворитель вводили перорально в качестве контролей.
ADT определяли вновь через 60 минут после введения лекарственных средств. % от величины до введения (утренние результаты) определяли для каждой крысы. Результаты выражали в виде среднего ±SEM и анализировали посредством критерия Даннета для множественных сравнений (4-9 крыс).
Как показано на фиг. 1-3, соединения из примеров 1, 11 и 6 показали дозозависимое повышение порогового значения следовых разрядов в крысиной модели киндлинга миндалевидного тела.
Данные результаты указывают на ингибирующее действие соединений в отношении припадков.
[0140] (Тестовый пример 3)
Тест вращающегося стержня
Для измерения использовали устройство Ротарод (Rota-rod MK-660C, Muromachi-Kikai Co., Ltd.). Включали мотор и вращение стержня непрерывно ускоряли до частоты 40 об/мин в течение 180 с.
Использовали самцов мышей ddY возрастом 5 недель (Japan SLC). В день эксперимента мышей предварительно обучали и для эксперимента отбирали только тех животных, которые были способны удерживаться на стержне в течение по меньшей мере 60 с.
Тестовые соединения растворяли в растворителе (0,45% метилцеллюлозы/4,5% кремофора/10% диметилсульфоксида), а затем вводили мышам перорально или интраперитонеально. Растворитель вводили перорально или интраперитонеально в качестве контроля. Эффекты соединений изучали в группах из шести - десяти животных.
Измеряли время задержки падения мышей со стержня. Рассчитывали токсическую дозу, при которой время задержки падения сокращалось на 50% значения для растворителя-контроля (TD50).
Терапевтический индекс рассчитывали путем деления соответствующего значения TD50 в тесте с вращающимся стержнем на значение ED50, определенное при использовании модели корнеального кинлдинга.
Рассчитанные ED50, TD50 и терапевтический индекс для каждого соединения показаны в таблице 10. Таким образом, у соединений, которые оценивали в данном тестовом примере 3, было выявлено высокое значение терапевтического индекса, указывающее на то, что данные соединения являются более безопасными, чем эталонное соединение.
|
|


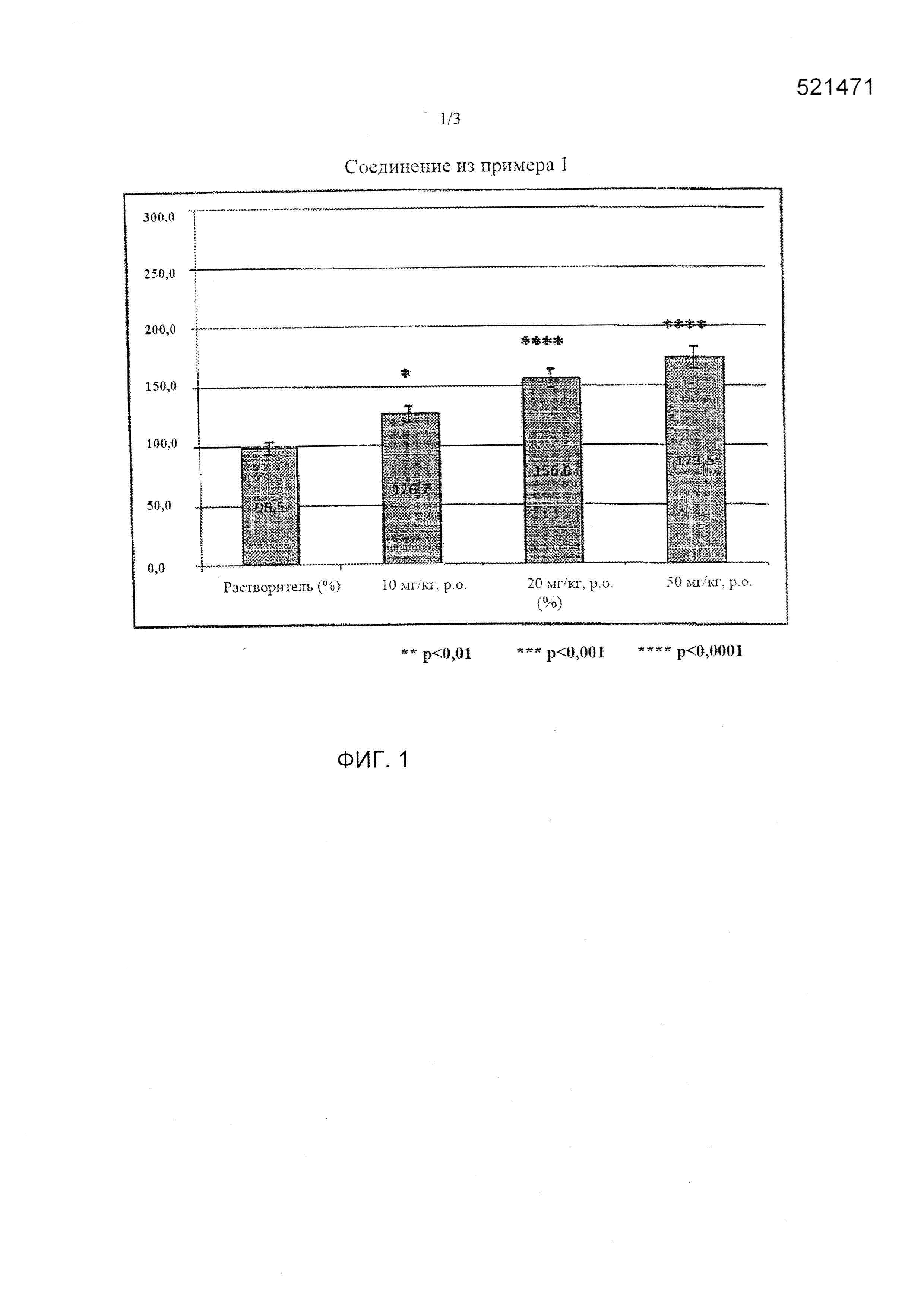
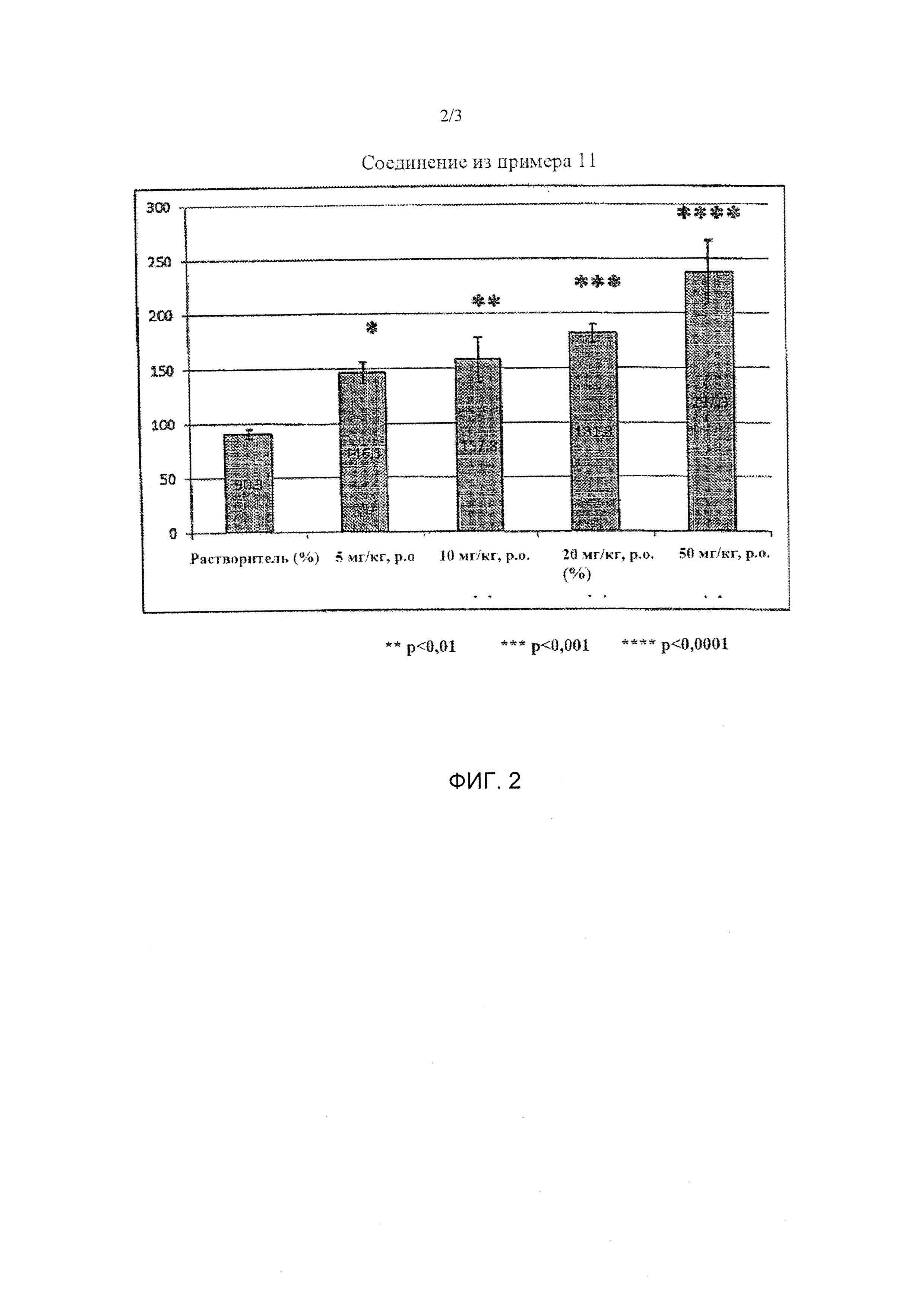
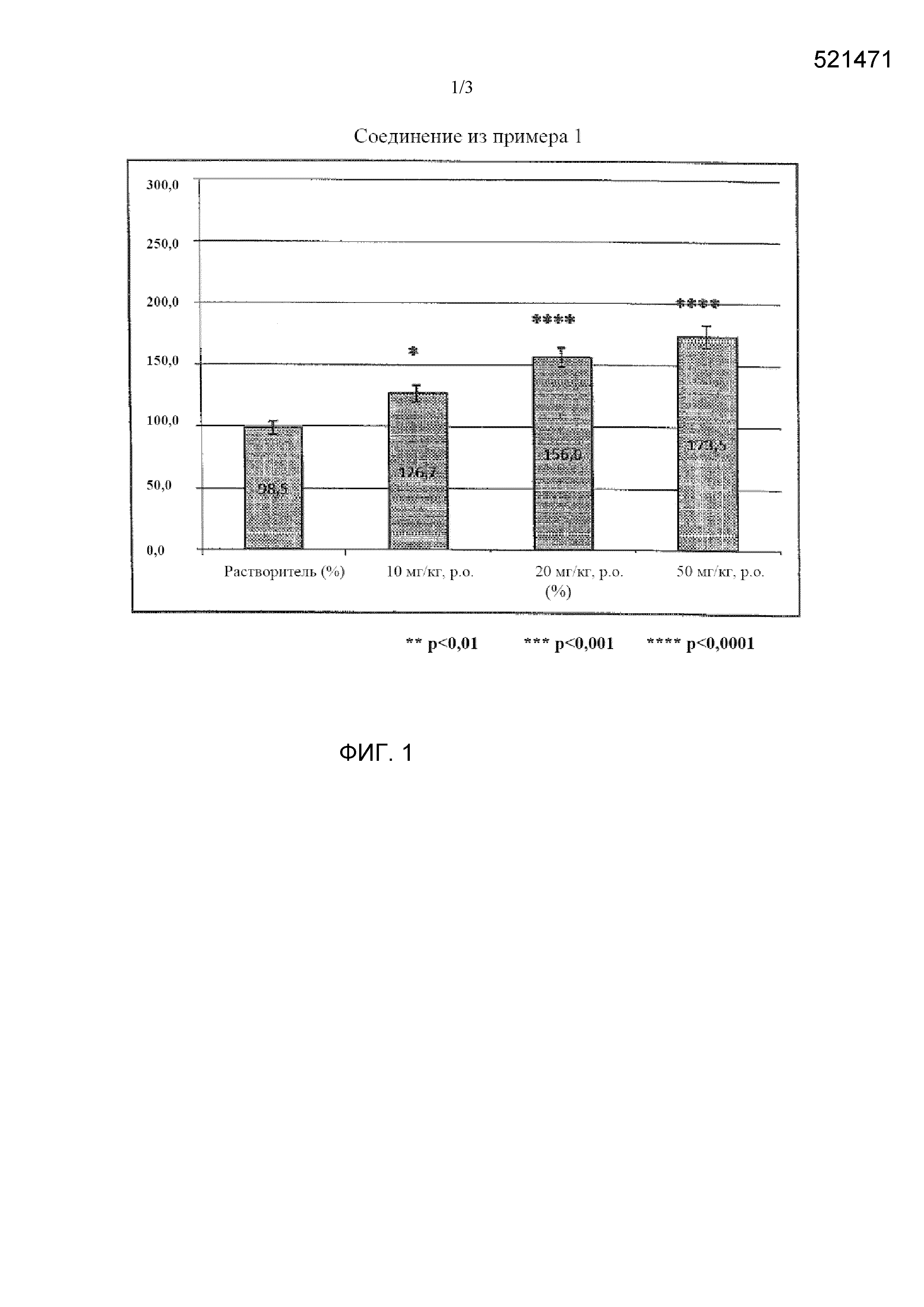
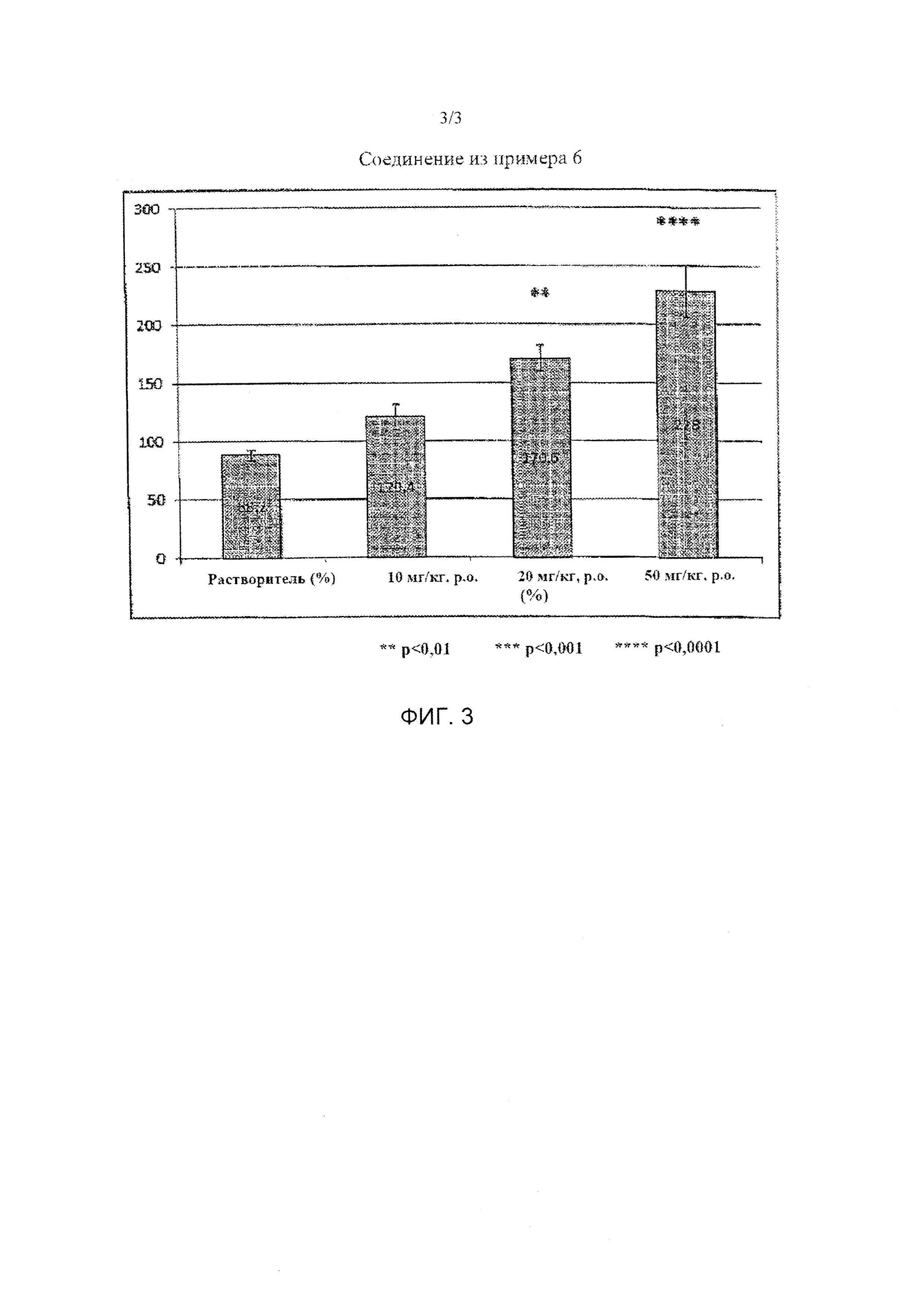
Липосомальная композиция
Конденсированное производное аминодигидротиазина
Производные пиридина, замещенные гетероциклическим кольцом и фосфоноксиметильной группой и содержащие их противогрибковые средства
Промежуточные соединения и способы синтеза аналогов галихондрина в
Реагенты и способы для бета-кетоамидного синтеза синтетического предшественника иммунологического адъюванта е6020
Новое конденсированное производное аминодигидротиазина
Азотсодержащие конденсированные гетероциклические соединения и их применение в качестве ингибиторов продукции бета-амилоида
Аналоги галихондрина в
Противоопухолевое средство, задействующее соединения с ингибирующим эффектом к киназам в комбинации
Биомаркер для болезни альцгеймера или умеренного когнитивного расстройства
Способ получения замещенных гетероциклом производных пиридина
Липосомальная композиция
Конденсированное производное аминодигидротиазина
Производные пиридина, замещенные гетероциклическим кольцом и фосфоноксиметильной группой и содержащие их противогрибковые средства
Промежуточные соединения и способы синтеза аналогов галихондрина в
Реагенты и способы для бета-кетоамидного синтеза синтетического предшественника иммунологического адъюванта е6020
Новое конденсированное производное аминодигидротиазина
Азотсодержащие конденсированные гетероциклические соединения и их применение в качестве ингибиторов продукции бета-амилоида
Аналоги галихондрина в
Противоопухолевое средство, задействующее соединения с ингибирующим эффектом к киназам в комбинации

