Результат интеллектуальной деятельности: ПРОИЗВОДНОЕ ПАРОКСЕТИНА
Вид РИД
Изобретение
Область техники
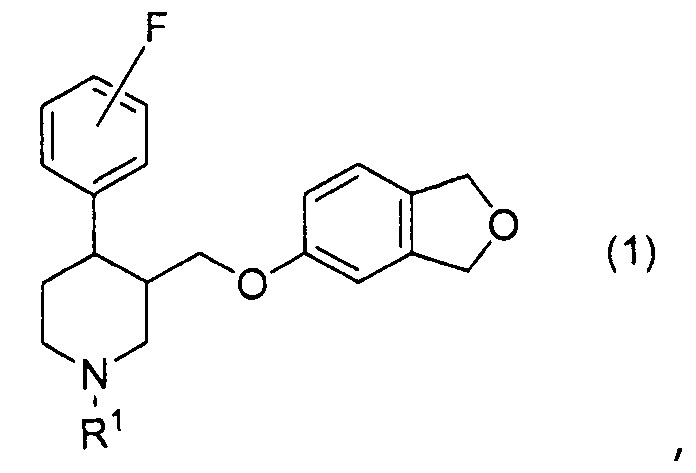
[0001] Настоящее изобретение относится к соединению с фталановым кольцом. Более конкретно, оно относится к соединению 3-[(1,3-дигидро-2-бензофуран-5-илокси)метил]-4-(фторфенил)пиперидина.
Предшествующий уровень техники
[0002] Соединения 3-[(2H-1,3-бензодиоксол-5-илокси)метил]-4-фенилпиперидина известны как селективные ингибиторы обратного захвата серотонина. Например, пароксетин, также известный как (3S,4R)-3-[(2H-1,3-бензодиоксол-5-илокси)метил]-4-(4-фторфенил)пиперидин, широко используется как антидепрессант (патентная литература 1).
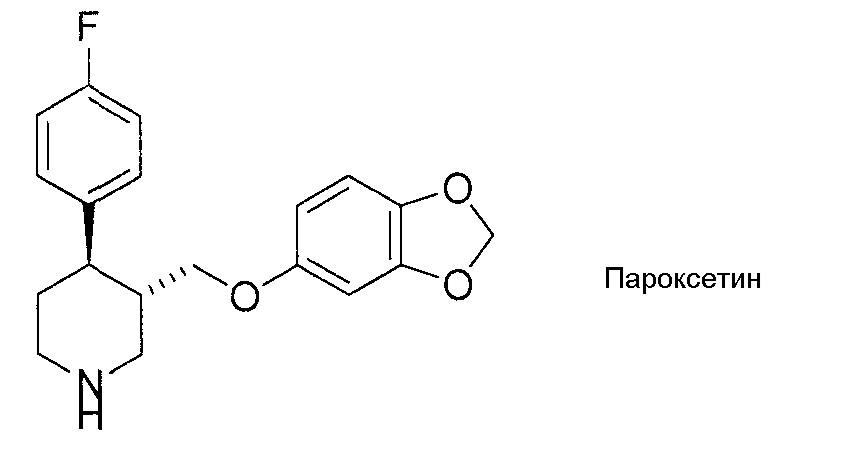
[0003] Однако структура пароксетина содержит бензодиоксольное кольцо, и, как правило, соединения с такими бензодиоксольными кольцами превращаются в химически высокоактивные метаболиты при метаболизировании цитохромом P450 (CYP), и, как известно, необратимо ингибируют активность CYP путем инактивации на основе ковалентного связывания с CYP (непатентная литература 1-3). Из сообщений известно, что пароксетин вызывает взаимодействия между лекарственными средствами путем ингибирования опосредованного CYP метаболизма нескольких лекарственных средств, которые клинически совместно вводят с ним, и некоторые из них являются противопоказанными. Для решения этой проблемы разработали соединение с атомом дейтерия, замещающим атом водорода на углероде метилена бензодиоксольной группы пароксетина, но такое соединение еще не применяется в коммерческом масштабе, и его эффекты не были удовлетворительными (патентная литература 2). Также известно, что соединения, содержащие дейтерий, как правило, требуют более дорогостоящего получения. Следовательно, для решения этой проблемы необходим способ, при котором не используется дейтерий.
Список противопоставленных материалов
Патентная литература
[0004]
[Патентная литература 1] патент США № 4007196.
[Патентная литература 2] публикация заявки на выдачу патента США № 2007/0191432.
Непатентная литература
[0005]
[Непатентная литература 1] Pharmacological reviews 42, 85, 1990 (Selectivity in the inhibition of Mammalian Cytochrome P-450 by Chemical Agents).
[Непатентная литература 2] Current Drug Metabolism, 6, 413, 2005.
[Непатентая литература 3] Drug Metabolism and Disposition 31, 289, 2003.
Краткое описание изобретения
Техническая задача
[0006] Целью настоящего изобретения является обеспечение соединения, поддерживающего основной терапевтический эффект пароксетина и обладающего улучшенным ингибирующим CYP эффектом, а также обладающего структурой, которая не содержит дейтерий.
Решение проблемы
[0007] В результате интенсивных исследований авторы настоящего изобретения создали настоящее изобретение. Конкретнее, настоящее изобретение относится к следующему [1]-[17].
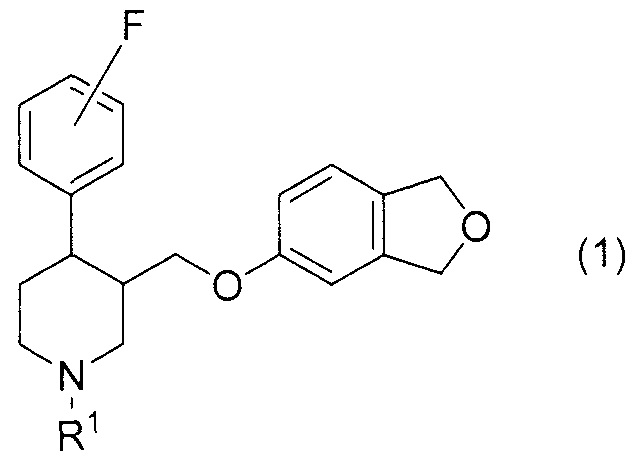
[1] Соединение, представленное формулой (1), или его фармакологически приемлемая соль:
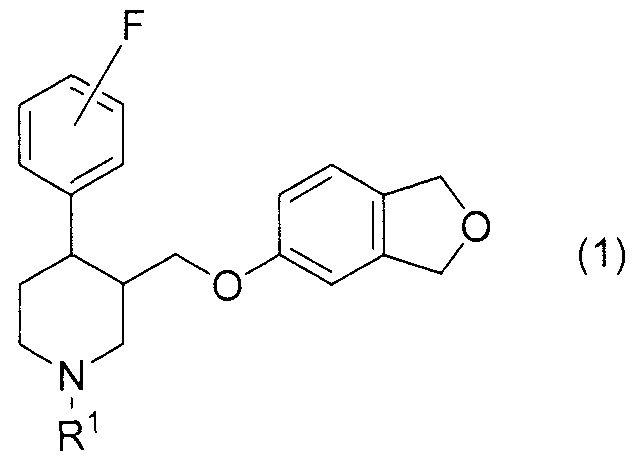
где R1 представляет собой атом водорода или C1-6алкильную группу.
[2] Соединение или его фармакологически приемлемая соль согласно [1], где R1 представляет собой атом водорода.
[3] Соединение или его фармакологически приемлемая соль согласно [1] или [2], где атом фтора присоединен в пара-положении относительно пиперидинового кольца.
[4] (3S,4R)-3-[(1,3-Дигидро-2-бензофуран-5-илокси)метил]-4-(4-фторфенил)пиперидин или его фармакологически приемлемая соль.
[5] Фармацевтическая композиция, содержащая соединение или его фармакологически приемлемую соль согласно любому из [1]-[4].
[6] Фармацевтическая композиция согласно [5], которая представляет собой селективный ингибитор обратного захвата серотонина.
[7] Фармацевтическая композиция согласно [5], которая является антидепрессантом.
[8] Фармацевтическая композиция согласно [5], которая является средством для лечения или предупреждения преждевременной эякуляции.
[9] Способ селективного ингибирования обратного захвата серотонина, включающий введение соединения или его фармакологически приемлемой соли согласно любому из [1] - [4] пациенту.
[10] Способ лечения или предупреждения депрессии, включающий введение соединения или его фармакологически приемлемой соли согласно любому из [1]-[4] пациенту.
[11] Способ лечения или предупреждения преждевременной эякуляции, включающий введение соединения или его фармакологически приемлемой соли согласно любому из [1]-[4] пациенту.
[12] Соединение или его фармакологически приемлемая соль согласно любому из [1]-[4] для применения для селективного ингибирования обратного захвата серотонина.
[13] Соединение или его фармакологически приемлемая соль согласно любому из [1]-[4] для применения для лечения или предупреждения депрессии.
[14] Соединение или его фармакологически приемлемая соль согласно любому из [1]-[4] для применения для лечения или предупреждения преждевременной эякуляции.
[15] Применение соединения или его фармакологически приемлемой соли согласно любому из [1]-[4] для изготовления селективного ингибитора обратного захвата серотонина.
[16] Применение соединения или его фармакологически приемлемой соли согласно любому из [1]-[4] для изготовления антидепрессанта.
[17] Применение соединения или его фармакологически приемлемой соли согласно любому из [1]-[4] для изготовления средства для лечения или предупреждения преждевременной эякуляции.
Полезные эффекты настоящего изобретения
[0008] Соединение, представленное формулой (1) (далее называемое соединением (1)), сохраняет основной терапевтический эффект пароксетина и обладает улучшенным ингибиторным по отношению к CYP эффектом по сравнению с пароксетином.
Краткое описание графических материалов
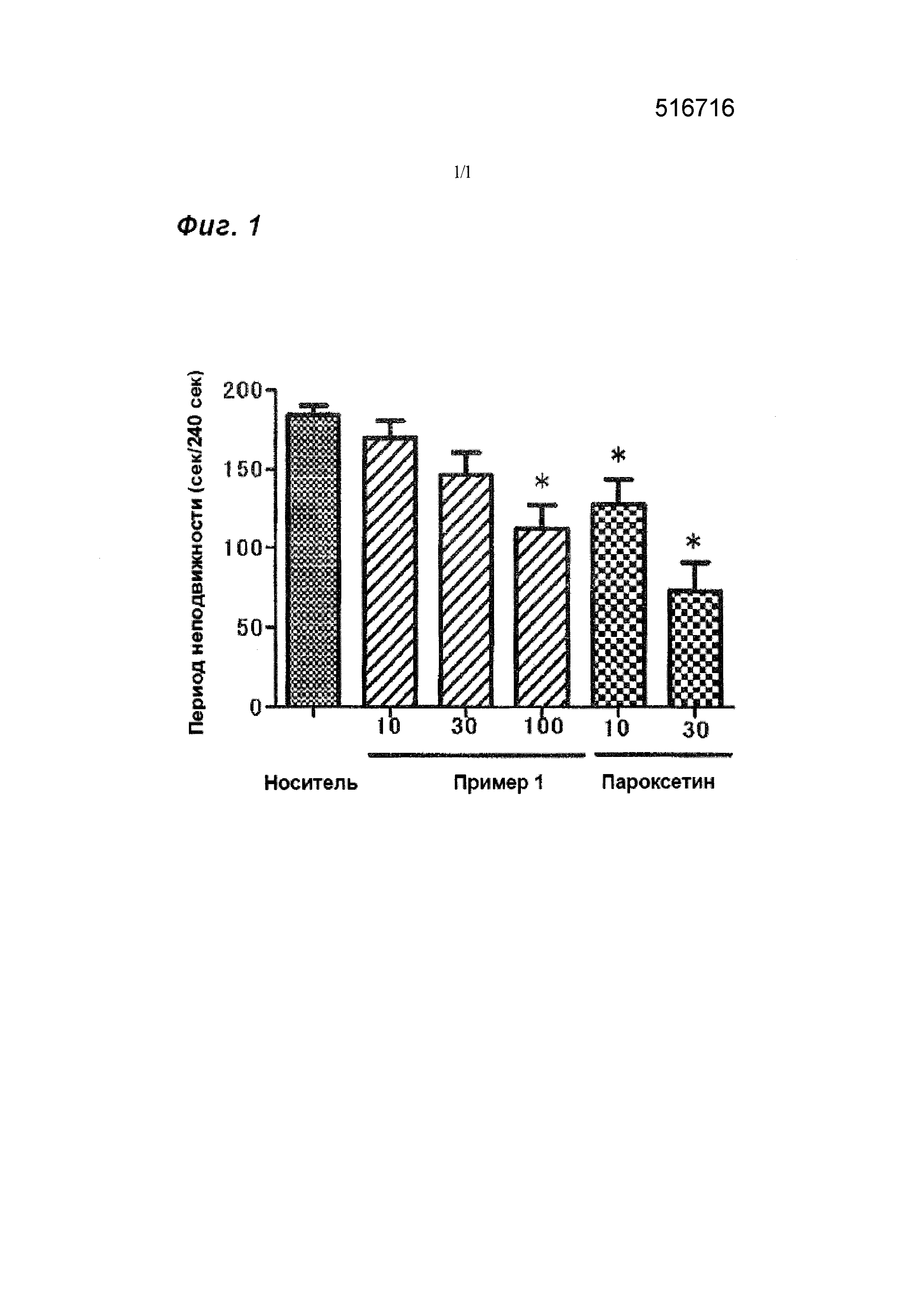
[0009] На фиг. 1 представлен график, показывающий эффекты соединения примера 1 и пароксетина на паузу при тесте с принудительным плаванием мыши.
Описание вариантов осуществления
[0010] Далее настоящее изобретение будет описано подробно.
[0011] В описании настоящее изобретение не ограничивается конкретной кристаллической формой, но может включать в себя любую из кристаллических форм или их смеси, хотя могут существовать кристаллические полиморфы. Настоящее изобретение также предусматривает аморфные формы, а соединения согласно настоящему изобретению включают ангидриды и гидраты. Кроме того, настоящее изобретение также предусматривает так называемый метаболит, который образуется в результате in vivo метаболизма (окисления, восстановления, гидролиза, конъюгации и т.п.) соединения (1) в соответствии с настоящим изобретением. Более того, соединение (так называемое пролекарство), которое образует соединение (1) в соответствии с настоящим изобретением в результате in vivo метаболизма (окисления, восстановления, гидролиза, конъюгации и т.п.), также предусматривается настоящим изобретением.
[0012] Значения терминов, символов и т.п., применяемых в настоящем описании, объясняются ниже, и настоящее изобретение описывается подробно.
[0013] “CYP” в настоящем описании означает метаболизирующий лекарственное средство фермент цитохром P450.
[0014] “Улучшение ингибиторного по отношению к CYP эффекта” или “улучшенный ингибиторный по отношению к CYP эффект” в настоящем описании означает, что степень ингибирования одной или двух из пяти молекул CYP (CYP1A2, 2C9, 2C19, 2D6 и 3A4), основных молекул CYP, как правило, лучше, чем у пароксетина.
[0015] “Сохранение основного терапевтического эффекта” в настоящем описании означает демонстрацию in vitro или in vivo фармакологической активности в доклиническом исследовании, которая, как ожидается, показывает клинический терапевтический эффект, как у пароксетина. "In vitro фармакологическая активность" означает, например, ингибиторную активность по отношению к переносчику серотонина, а "in vivo фармакологическая активность" означает, например, фармакологическую активность на основе теста с принудительным плаванием.
[0016] “IC50” в настоящем описании означает ингибирующую на 50% концентрацию или полуингибирующую концентрацию.
[0017] "C1-6алкильная группа" в настоящем описании означает алкильную группу с прямой или разветвленной цепью с 1-6 атомами углерода, и примеры включают метильную группу, этильную группу, 1-пропильную группу, 2-пропильную группу, 2-метил-1-пропильную группу, 2-метил-2-пропильную группу, 1-бутильную группу, 2-бутильную группу, 1-пентильную группу, 2-пентильную группу, 3-пентильную группу, 1-гексильную группу, 2-гексильную группу и 3-гексильную группу.
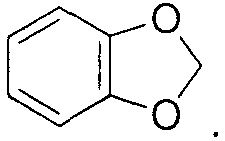
[0018] “Бензодиоксоловое кольцо” в настоящем описании означает кольцо или функциональную группу, характеризующуюся следующей структурой:
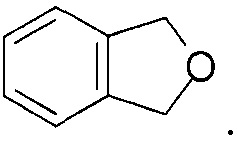
[0019] “Фталановое кольцо” в настоящем описании означает кольцо или функциональную группу, характеризующуюся следующей структурой:
[0020] "Фармакологически приемлемая соль" в настоящем описании конкретно не ограничена при условии, что образующаяся соль с соединением, представленным формулой (1), является фармакологически приемлемой, и примеры включают в себя соли неорганической кислоты, соли органической кислоты, соли неорганического основания, соли органического основания и соли кислых или основных аминокислот.
[0021] Предпочтительные примеры солей неорганических кислот включают гидрохлориды, гидробромиды, сульфаты, нитраты и фосфаты, и предпочтительные примеры солей органических кислот включают ацетаты, сукцинаты, фумараты, малеаты, тартраты, цитраты, лактаты, стеараты, бензоаты, манделаты, метансульфонаты, этансульфонаты, п-толуолсульфонаты и бензолсульфонаты.
[0022] Предпочтительные примеры солей неорганических оснований включают соли щелочных металлов, такие как соли натрия и соли калия, соли щелочноземельных металлов, такие как соли кальция и соли магния, соли алюминия и соли аммония, и предпочтительные примеры солей органических оснований включают соли диэтиламина, соли диэтаноламина, соли меглюмина и соли N,N'-дибензилэтилендиамина.
[0023] Предпочтительные примеры солей кислых аминокислот включают аспартаты и глютаматы, и предпочтительные примеры солей основных аминокислот включают соли аргинина, соли лизина и соли орнитина.
[0024] Соединение, представленное формулой (1), можно получить способами, описанными ниже, или способом, описанным ниже, с усовершенствованиями, выполняемыми специалистом в данной области на основе общеизвестной информации. Однако способ получения соединения, представленного формулой (1), не ограничивается этим.
[0025] Процесс A
Если R1 соединения (1) представляет собой С1-6алкильную группу, то соединение (1) можно получить с помощью следующего процесса A:
[0026]
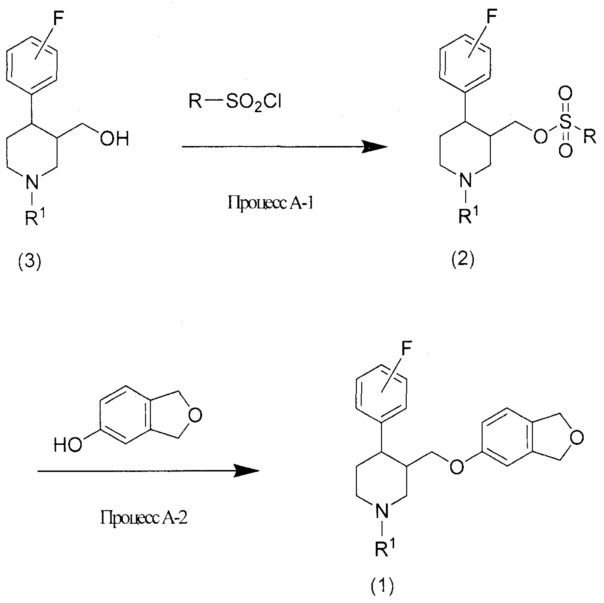
[0027] На схеме R представляет собой С1-6алкильную группу или фенильную группу, необязательно замещенную C1-6алкильной группой, а R1 представляет собой атом водорода или C1-6алкильную группу; однако, R1 в процессе A представляет собой С1-6алкильную группу.
[0028] Процесс A-1 представляет собой способ получения соединения (2) путем реагирования соединения (3) со средством для этерификации сульфоновой кислоты в инертном растворителе в присутствии основания.
[0029] Примеры средств этерификации сульфоновой кислоты включают метансульфонилхлорид, бензолсульфонилхлорид и п-толуолсульфонилхлорид, при этом метансульфонилхлорид является предпочтительным.
[0030] Используемый растворитель конкретно не ограничивается, при условии, что он растворяет исходные материалы до определенной степени без ингибирования реакции, и примеры включают эфиры, такие как тетрагидрофуран, галогенированные углеводороды, такие как дихлорметан и хлороформ, и ароматические углеводороды, такие как бензол и толуол, при этом дихлорметан и толуол являются предпочтительными.
[0031] Используемым основанием может быть триэтиламин, диизопропилэтиламин или подобное, при этом триэтиламин является предпочтительным.
[0032] Температура реакции варьирует согласно исходным материалам, растворителю и основанию, но в норме составляет -20°C -100°C и предпочтительно 0°C-60°C.
[0033] Температура реакции варьирует согласно исходным материалам, растворителю и основанию, но в норме составляет от 10 минут до 3 дней и предпочтительно от 30 минут до 1 дня.
[0034] Процесс A-2 представляет собой способ получения соединения (2) с 1,3-дигидро-2-бензофуран-5-олом в неактивном растворителе в присутствии основания.
[0035] Используемый растворитель конкретно не ограничивается, при условии, что он растворяет исходные материалы до определенной степени без ингибирования реакции, и примеры включают амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метилпирролидинон, эфиры, такие как тетрагидрофуран, и сульфоксиды, такие как диметилсульфоксид, при этом N,N-диметилформамид является предпочтительным.
[0036] Используемым основанием может быть основание, такое как органический литий или гидрид натрия, при этом гидрид натрия является предпочтительным.
[0037] Температура реакции варьирует согласно исходным материалам, растворителю и основанию, но в норме составляет -20°C -100°C или предпочтительно 0°C-100°C.
[0038] Температура реакции варьирует согласно исходным материалам, растворителю и основанию, но в норме составляет от 10 минут до 3 дней или предпочтительно от 30 минут до 1 дня.
[0039] Процесс B
Если R1 соединения (1) представляет собой атом водорода, то соединение (1) можно получать с помощью следующего процесса В:
[0040]
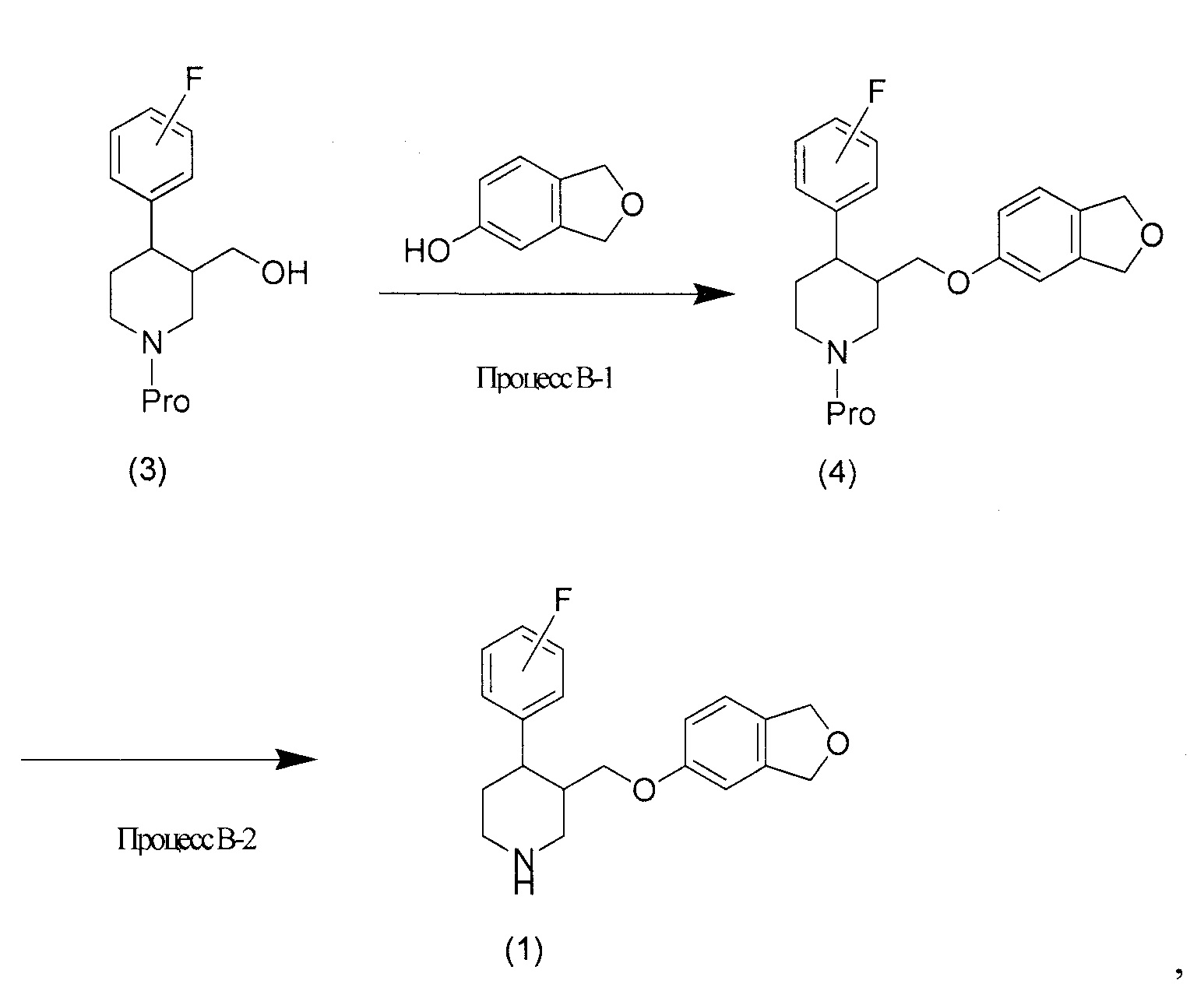
где Pro представляет собой защитную группу амина.
[0041] Процесс B-1 представляет собой способ, подобный способу синтеза, описанному выше для этапа A. Защитной группой амина может быть т-бутилоксикарбонильная, бензилоксикарбонильная или п-толуолсульфонильная группа или подобное.
[0042] Процесс B-2 представляет собой стадию снятия защитной группы амина с получением вторичного амина.
[0043] Защитная группа может быть снята с амина известными способами, и если защитной группой является т-бутилоксикарбонильная группа, например, снятие защиты можно выполнять путем обработки кислотой, такой как трифторуксусная кислота.
[0044] После завершения реакции в каждом процессе каждого описанного выше способа целевое соединение для каждого процесса можно собрать из реакционной смеси согласно традиционному способу.
[0045] Например, если целой реакционной смесью является жидкость, то реакционную смесь охлаждают до комнатной температуры или охлаждают на льду по необходимости и нейтрализуют кислотой, щелочью, добавляют окисляющее средство или восстанавливающее средство по необходимости, органический растворитель, не смешивающийся с водой и не реагирующий с целевым соединением, такой как этилацетат, и отделяют слой, содержащий целевое соединение. Затем добавляют растворитель, не смешивающийся с полученным в результате слоем и не реагирующий с целевым соединением, слой, содержащий целевое соединение, промывают и слой отделяют. Более того, если слоем является органический слой, то целевое соединение можно собрать путем сушки с высушивающим агентом, таким как безводный сульфат магния или безводный сульфат натрия, и отгонки растворителя. Если слоем является водный слой, то целевое соединение можно собрать электрической деминерализацией, а затем слой лиофилизировать.
[0046] Кроме того, если цельной реакционной смесью является жидкость и, если возможно, целевое соединение можно собирать только отгонкой веществ, отличных от целевого соединения (таких как растворитель или реагент), при нормальном давлении или пониженном давлении.
[0047] Кроме того, если только целевое соединение осаждают в виде твердого вещества или если цельная реакционная смесь, описанная выше, является жидкостью, и только целевое соединение осаждают в ходе сбора, целевое соединение можно далее собирать путем сбора сначала фильтрацией, промыванием целевого соединения, собранного фильтрацией с соответствующим органическим или неорганическим растворителем и сушкой так, что исходную жидкость обрабатывают способом, подобным таковому в случае, когда цельная реакционная смесь, описанная выше, является жидкостью.
[0048] Кроме того, если только реагент или катализатор присутствуют в виде твердого вещества или цельная реакционная смесь, описанная выше, является жидкостью, и только реагент или катализатор осаждают в виде твердого вещества в ходе сбора, и целевое соединение растворяют в растворе, целевое соединение можно собирать отгонкой сначала реагента или катализатора, промыванием отфильтрованного реагента или катализатора соответствующим органическим или неорганическим растворителем, объединением полученных в результате смывов с исходной жидкостью и обработкой полученной в результате смеси способом, подобным таковому в случае, когда цельная реакционная смесь, описанная выше, является жидкостью.
[0049] В частности, если вещества, отличные от целевого соединения, которые входят в состав реакционной смеси, не ингибируют реакцию в следующем этапе, реакционную смесь также можно использовать в следующем этапе как есть без особого выделения целевого соединения.
[0050] Перекристаллизацию, различные способы хроматографии и перегонку можно осуществлять при необходимости для улучшения чистоты целевого соединения, собранного вышеописанным способом.
[0051] Как правило, если собранное целевое соединение является твердым веществом, то чистоту целевого соединения можно повысить с помощью способа перекристаллизации. При перекристаллизации можно использовать отдельный растворитель или смесь множества растворителей, не вступающих в реакцию с целевым соединением. Особым образом, целевое соединение сначала растворяют в одном или нескольких растворителях, не реагирующих с целевым соединением при комнатной температуре или при нагревании. Полученную в результате смесь охлаждают ледяной водой или подобным образом, или перемешивают, или оставляют стоять при комнатной температуре так, что целевое соединение может кристаллизоваться из смеси.
[0052] Чистоту собранного целевого соединения можно повысить различными хроматографическими способами. Как правило, можно использовать слабокислые силикагели, такие как Silica gel 60, изготовленный Merck KGaA (70-230 меш или 340-400 меш), и BW-300, изготовленный Fuji Silysia Chemical Ltd. (300 меш). Если целевое соединение является основным и абсорбируется на вышеуказанных силикагелях слишком сильно, также можно применять NH силикагели, такие как покрытый пропиламином силикагель, изготовленный Fuji Silysia Chemical Ltd. (200-350 меш), и одноразовую среднего давления препаративную наполненную колонку, изготовленную Yamazen Corporation (Hi-Flash Amino). Если целевое соединение является биполярным или должно элюироваться более полярным растворителем, таким как метанол, например, можно применять NAM-200H или NAM-300H, изготовленные NAM Laboratory, или YMC GEL ODS-A, изготовленный YMC Co. Ltd. Также можно применять одноразовые среднего давления препаративные наполненные колонки, как описано выше, которые предварительно заполнены наполнителями и изготовлены Yamazen Corporation, Wako Pure Chemical Industries, Ltd., Biotage AB или W. R. Grace & Co. (Hi-Flash). Целевое соединение, чью чистоту повышают, можно получить путем элюирования целевого соединения одним или несколькими растворителями, которые не вступают в реакцию с целевым соединением, с применением данных силикагелей и отгонки растворителя(ей).
[0053] Если собранное целевое соединение является жидкостью, то чистоту целевого соединения можно повысить с помощью способа перегонки. При перегонке целевое соединение можно отогнать, воздействуя на него пониженным давлением при комнатной температуре или при нагревании.
[0054] Типичные примеры способа получения соединения (1) описаны выше. Все соединения-сырьевые материалы и различные реагенты при получении соединения (1), которые могут образовывать соли или сольваты, такие как гидраты, варьируют в зависимости исходного материала, используемого растворителя или подобного и не имеют особых ограничений, при условии, что они не ингибируют реакцию. Также используемый растворитель варьирует в зависимости от исходного материала, реагента или подобного и не имеет особых ограничений при условии, что он не ингибирует реакцию и очевидно растворяет исходный материал до некоторой степени. Если соединение (1) получают в свободной форме, его можно превратить в соль, которая может быть образована соединением (1) или сольватом соединения или соли традиционными способами.
[0055] Если соединение (1) получают в виде соли или сольвата, его можно превратить в свободную форму соединений (1) традиционными способами.
[0056] Различные изомеры, полученные для соединения (1) (такие как геометрические изомеры, оптические изомеры, ротамеры, стереоизомеры и таутомеры), можно очистить и выделить с использованием общепринятых средств разделения, например: перекристаллизация, образование диастереомерной соли, ферментативное разделение и различные хроматографические способы (такие как тонкослойная хроматография, колоночная хроматография и газовая хроматография).
[0057] Соединение (1) или его фармакологически приемлемую соль можно создавать традиционными способами, и примеры дозированных форм включают в себя пероральные составы (такие как таблетки, гранулы, порошки, капсулы и сиропы), инъекции (для внутривенного введения, внутримышечного введения, подкожного введения и интраперитонеального введения) и наружные составы (такие как составы для трансдермального поглощения (такие как мази и пластыри), офтальмологические препараты, назальные препараты и суппозитории).
[0058] Эти твердые составы, такие как таблетки, капсулы, гранулы и порошки, как правило, могут содержать 0,001-99,5 вес.%, предпочтительно 0,01-90 вес.% или т.п. соединения (1) или его фармакологически приемлемой соли.
[0059] При изготовлении пероральных твердых составов таблетки, гранулы, порошки и капсулы можно получать путем добавления разбавителей, связующих средств, разрыхлителей, смазочных средств, красителей или подобного к соединению (1) или его фармакологически приемлемой соли при необходимости и обработки традиционными способами. Таблетки, гранулы, порошки, капсулы и т.п. также можно покрывать пленкой при необходимости.
[0060] Примеры разбавителей включают в себя лактозу, кукурузный крахмал и микрокристаллическую целлюлозу, примеры связующих средств включают в себя гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу, а примеры разрыхлителей включают в себя карбоксиметилцеллюлозу кальция и кроскармелозу натрия.
[0061] Примеры смазочных средств включают в себя стеарат магния и стеарат кальция, а примеры красителей включают в себя оксид титана.
[0062] Примеры средств для пленочного покрытия включают в себя гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и метилцеллюлозу.
[0063] Очевидно, что любые описанные выше наполнители не ограничиваются этими примерами.
[0064] При изготовлении инъекций (для внутривенного введения, внутримышечного введения, подкожного введения и интраперитонеального введения) их можно изготавливать путем добавления регуляторов pH, буферов, суспендирующих средств, солюбилизаторов, антиоксидантов, консервантов (антисептиков), средств регулирования тоничности или подобных к соединению (1) или его фармакологически приемлемой соли при необходимости и обработки традиционными способами. Лиофилизированный состав, подлежащий разбавлению перед применением, также можно получать лиофилизацией. Эти инъекции можно вводить, например, внутривенно, подкожно и внутримышечно.
[0065] Примеры регуляторов pH и буферов включают в себя органические кислоты или неорганические кислоты и/или их соли, примеры суспендирующих средств включают в себя метилцеллюлозу, полисорбат 80 и карбоксиметилцеллюлозу натрия, примеры солюбилизаторов включают в себя полисорбат 80 и полиоксиэтиленсорбитанмонолаурат, примеры антиоксиданотов включают в себя α-токоферол, примеры консервантов включают в себя метилпарагидроксибензоат и этилпарагидроксибензоат, а примеры средств регулирования тоничности включают в себя глюкозу, хлорид натрия и маннит; однако очевидно, что наполнители не ограничиваются этими примерами.
[0066] Эти инъекции, как правило, могут содержать 0,000001-99,5 вес.%, предпочтительно 0,00001-90 вес.% или т.п. соединения (1) или его фармакологически приемлемой соли.
[0067] При изготовлении наружных составов составы для трансдермального поглощения (такие как мази и пластыри, офтальмологические препараты, назальные препараты, суппозитории и т.п.) можно изготавливать путем добавления описанных выше основных материалов и, при необходимости, эмульгаторов, консервантов, регуляторов pH, красителей и т.п. к соединению (1) или его фармакологически приемлемой соли и обрабатывать традиционными способами.
[0068] Различные сырьевые материалы, традиционно используемые для фармацевтических препаратов, квазилекарств, косметических средств и т.п., можно использовать в качестве основных материалов, и примеры включают в себя сырьевые материалы, такие как животные и растительные масла, минеральные масла, сложноэфирные масла, воски, высшие спирты и очищенная вода.
[0069] Эти инъекции, как правило, могут содержать 0,000001-99,5 вес.%, предпочтительно 0,00001-90 вес.% или т.п. соединения (1) или его фармакологически приемлемой соли.
[0070] Дозировка медицинского препарата согласно настоящему изобретению, как правило, варьирует в зависимости от симптома, возраста, пола, веса или подобного, но является приемлемой, если эта дозировка достаточна для получения желаемого эффекта. Например, для взрослого используют дозировку от приблизительно 0,1 до 5000 мг (предпочтительно от 0,5 до 1000 мг, более предпочтительно от 1 до 600 мг) в сутки одной дозой в течение одних или нескольких суток или поделенной на 2-6 доз в течение одних суток.
[0071] Соединение (1) можно использовать в качестве химического зонда для захвата целевых белков в биоактивные низкомолекулярные соединения. В частности, соединение (1) можно превращать в зонд для аффинной хроматографии, фотоаффинный зонд или подобное путем введения группы-метки, линкера или подобного во фрагмент, отличный от структурного фрагмента, необходимого для экспрессии активности соединения методикой, описанной в J. Mass Spectrum. Soc. Jpn., Vol. 51, No. 5, 2003, pp. 492-498, или в WO 2007/139149, или в подобном.
[0072] Примеры групп-меток, линкеров или т.п., используемых в химическом зонде, включают в себя группы, показанные в группе, состоящей из приведенных далее (1)-(5).
[0073] (1) Группы-метки для белков, такие как фотоаффинные группы-метки (например, бензоильная группа, бензофеноновая группа, азидогруппа, карбонильная азидогруппа, диазиридиновая группа, еноновая группа, диазогруппа и нитрогруппа) и хемоаффинные группы (например, кетонная группа, в которой α-углеродный атом заменен атомом галогена, карбамоильная группа, сложноэфирная группа, алкилтиогруппа, акцептор Михаэля, такой как α,β-ненасыщенные кетоны и сложные эфиры и оксирановая группа),
(2) расщепляемые линкеры, такие как -S-S-, -O-Si-O-, моносахаридные (такие как глюкозная группа и галактозная группа) или дисахаридные (такие как лактоза) и олигопептидные линкеры, расщепляемые ферментативной реакцией,
(3) группы-метки для фишинга, такие как биотин и 3-(4,4-дифтор-5,7-диметил-4H-3a,4a-диаза-4-бора-s-индацен-3-ил)пропионильная группа,
(4) детектируемые маркеры, такие как меченные радиоактивным изотопом группы, такие как 125I, 32P, 3H и 14C; флуоресцентные группы-метки, такие как флуоресцеин, родамин, дансил, умбеллиферон, 7-нитрофуразанил и 3-(4,4-дифтор-5,7-диметил-4H-3a,4a-диаза-4-бора-s-индацен-3-ил)пропиониловая группа; хемилюминесцентные группы, такие как люциферин и люминол; и маркеры, способные обнаруживать ионы тяжелых металлов, такие как ионы металлов-лантаноидов и ионы радия; или
(5) группы, связанные с твердофазными носителями, такими как стеклянные шарики, стеклянные пластины, титровальные микропланшеты, агарозные гранулы, агарозные слои, полистирольные гранулы, полистирольные слои, полиамидные гранулы и полиамидные слои.
[0074] Зонды, полученные путем введения групп-меток или подобных, выбранных из группы, состоящей из вышеприведенных (1)-(5), в соединение (1) согласно способу, описанному в вышеприведенных документах, или подобному, можно использовать в качестве химических зондов для идентификации меченых белков, применимых для поиска новых целей лекарственного средства, например.
Примеры
[0075] Соединение (1) можно получать, например, способами, описанными в следующих примерах, и эффекты соединения (1) можно подтверждать способами, описанными в следующих примерах тестов. Однако эти примеры являются иллюстративными, и настоящее изобретение особо не ограничено конкретными примерами.
[0076] [Пример 1] (3S,4R)-3-[(1,3-Дигидро-2-бензофуран-5-илокси)метил]-4-(4-фторфенил)пиперидин
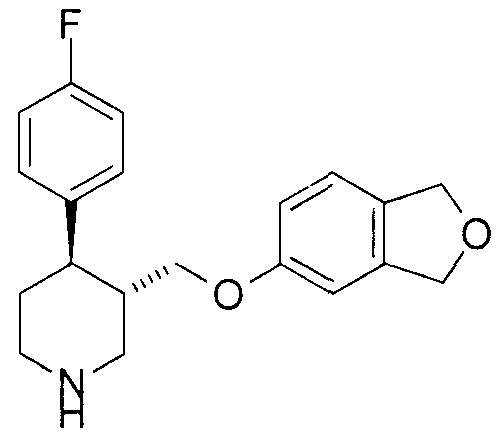
К трет-бутил-(3S,4R)-3-[(1,3-дигидро-2-бензофуран-5-илокси)метил]-4-(4-фторфенил)пиперидин-1-карбоксилату (4,1 г, 9,6 ммоля), описанному в примере получения 1-4, добавляли смешанный раствор трифторуксусной кислоты (5 мл) и дихлорметана (30 мл) при охлаждении на льду и с последующим перемешиванием при комнатной температуре в течение 90 минут. Добавляли толуол (30 мл) к реакционной смеси и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле NH (метанол:этилацетат = 1:20) с получением указанного в заголовке соединения (2,7 г, выход 84%).
1Н-ЯМР спектр (CDCl3) δ (м.д.): 1,67-1,77 (1H, м), 1,82 (1H, дкв, J=2,6, 6,6 Гц), 2,06-2,14 (1H, м), 2,60 (1H, дт, J=4,0, 11,7 Гц), 2,69 (1H, дд, J=11,3, 12,1 Гц), 2,75 (1H, дт, J=2,9, 12,1 Гц), 3,19 (1H, д, J=3,2 Гц), 3,44 (1H, дд, J=3,7, 12,1 Гц), 3,51 (1H, дд, J=7,1, 9,3 Гц), 3,64 (1H, дд, J=2,9, 9,5 Гц), 5,01 (4H, с), 6,58 (1H, д, J=2,2 Гц), 6,64 (1H, дд, J=2,4, 8,2 Гц), 6,95-7,01 (2H, м), 7,05 (1H, д, J=8,1 Гц), 7,15-7,20 (2H, м).
[0077] [Пример получения 1-1] 2-[(проп-2-ин-1-илокси)метил]фуран
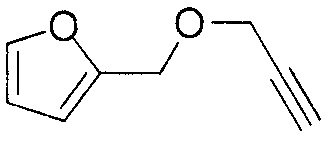
К смеси гидрида натрия (7,7 г, 190 ммоль, 60% в масле) и тетрагидрофурана (100 мл) по каплям добавляли фурфуриловый спирт (15 мл, 170 ммоль) при 0°C. Реакционную смесь нагревали до комнатной температуры, добавляли N,N-диметилформамид (30 мл) при этой температуре и смесь перемешивали при этой температуре в течение 30 минут. Реакционную смесь возвращали к 0°C, добавляли пропаргилбромид (23 г, 190 ммоль) при этой температуре и смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли воду к реакционной смеси, которую затем экстрагировали этилацетатом. Органический слой промывали солевым раствором и отгоняли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле NH (этилацетат:гептан = 1:30) с получением соединения, указанного в заголовке (7,6 г, выход 32%).
1Н-ЯМР спектр (CDCl3) δ (м.д.): 2,47 (1H, т, J=2,6 Гц), 4,17 (2H, д, J=2,6 Гц), 4,57 (2H, с), 6,36 (1H, дд, J=1,8, 3,3 Гц), 6,376-6,384 (1H, м), 7,43 (1H, дд, J=0,7, 1,8 Гц).
[0078] [Пример получения 1-2] 1,3-дигидро-2-бензофуран-5-ол
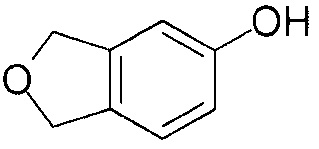
К смеси 2-[(проп-2-ин-1-илокси)метил]фурана (6,6 г, 49 ммоль), описанного в примере получения 1-1, добавляли хлорид платины(II) (650 мг, 2,4 ммоля) при комнатной температуре, а затем нагревали с обратным холодильником в течение 5 часов. Реакционную смесь возвращали до комнатной температуры и отгоняли растворитель при пониженном давлении. Осадок фильтровали через силикагель (с элюированием этилацетатом) и растворитель отгоняли при пониженном давлении. Добавляли дихлорметан (20 мл) к остатку и нерастворенное твердое вещество собирали фильтрацией с получением соединения, указанного в заголовке (1,7 г). Фильтрат отгоняли при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (этилацетат:гептан = 1:4) с получением соединения, указанного в заголовке (1,6 г, всего 3,3 г, выход 49%).
1Н-ЯМР спектр (CDCl3) δ (м.д.): 5,06 (4H, с), 6,71 (1H, д, J=2,2 Гц), 6,74 (1H, дд, J=2,2, 8,1 Гц), 7,08 (1H, д, J=8,1 Гц).
[0079] [Пример получения 1-3] трет-бутил (3S,4R)-4-(4-фторфенил)-3-(гидроксиметил)пиперидин-1-карбоксилат
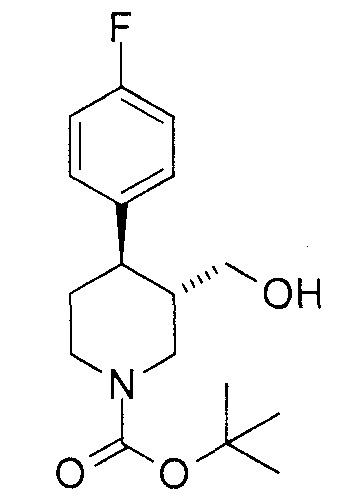
К смеси [(3S,4R)-4-(4-фторфенил)пиперидин-3-ил]метанола (3,0 г, 14 ммоль), карбоната натрия (6,1 г, 57 ммоль), дихлорметана (40 мл) и воды (40 мл) добавляли ди-трет-бутилдикарбонат (3,8 г, 17 ммоль) при охлаждении на льду, с последующим перемешиванием при той же температуре в течение 30 минут. Реакционную смесь добавляли к смешанному раствору дихлорметана и воды и отделяли органический слой. Органический слой промывали солевым раствором, сушили над безводным сульфатом магния и отгоняли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле NH (этилацетат:гептан = 1:1) с получением соединения, указанного в заголовке (4,0 г, выход 90%).
1Н-ЯМР спектр (CDCl3) δ (м.д.): 1,49 (9H, с), 1,61-1,71 (1H, м), 1,75-1,85 (2H, м), 2,51-2,56 (1H, м), 2,71 (1H, дд, J=11,3, 13,2 Гц), 2,78 (1H, ушир. с), 3,24-3,29 (1H, м), 3,42-3,46 (1H, м), 4,20 (1H, ушир. с), 4,36 (1H, д, J=11,7 Гц), 6,97-7,03 (2H, м), 7,13-7,18 (2H, м).
[0080] [Пример получения 1-4] трет-бутил-(3S,4R)-3-[(1,3-дигидро-2-бензофуран-5-илокси)метил]-4-(4-фторфенил)пиперидин-1-карбоксилат
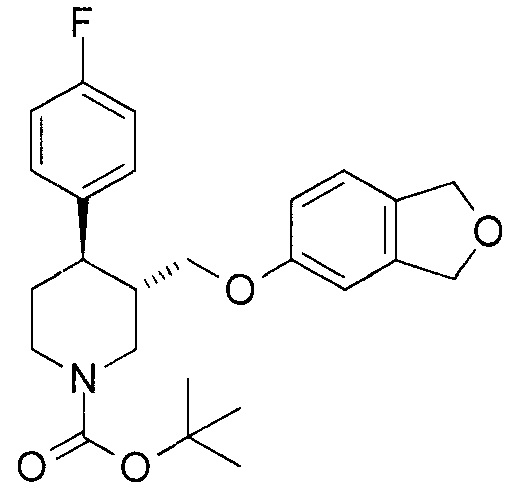
К смеси трет-бутил-(3S,4R)-4-(4-фторфенил)-3-(гидроксиметил)пиперидин-1-карбоксилата (3,6 г, 12 ммоль), описанного в примере получения 1-3, и толуола (40 мл) добавляли триэтиламин (2,1 мл, 15 ммоль) и метансульфонилхлорид (0,93 мл, 12 ммоль) при 0°C, с последующим перемешиванием в течение 30 минут. Затем добавляли метансульфонилхлорид (0,11 мл, 1,5 ммоля) при той же температуре и перемешивали при той же температуре в течение 30 минут. Добавляли воду к реакционной смеси, которую затем экстрагировали этилацетатом. Органический слой промывали солевым раствором, и сушили над безводным сульфатом магния, и отгоняли растворитель при пониженном давлении. Получали трет-бутил-(3S,4R)-4-(4-фторфенил)-3-[метансульфонилокси)метил]пиперидин-1-карбоксилат в виде неочищенного продукта. К смеси 1,3-дигидро-2-бензофуран-5-ола (1,6 г, 12 ммоль), описанного в примере получения 1-2, и N,N-диметилформамида (50 мл), добавляли гидрид натрия (460 мг, 12 ммоль, 60% в масле) при 0°C с последующим перемешиванием при той же температуре в течение 30 минут. К этой реакционной смеси добавляли смесь неочищенного продукта трет-бутил-(3S,4R)-4-(4-фторфенил)-3-[метансульфонилокси)метил]пиперидин-1-карбоксилата и N,N-диметилформамида (20 мл) с последующим перемешиванием при 80°C в течение 90 минут. Реакционную смесь возвращали до комнатной температуры и экстрагировали этилацетатом после добавления воды. Органический слой промывали солевым раствором и отгоняли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле NH (этилацетат:гептан = 1:3) с получением соединения, указанного в заголовке (4,7 г, выход 92%).
1Н-ЯМР спектр (CDCl3) δ (м.д.): 1,50 (9H, с), 1,69-1,83 (2H, м), 2,01-2,09 (1H, м), 2,69 (1H, дт, J=3,7, 11,7 Гц), 2,78-2,84 (2H, м), 3,52 (1H, дд, J=6,6, 9,1 Гц), 3,67 (1H, дд, J=2,9, 9,5 Гц), 4,25 (1H, ушир. с), 4,46 (1H, ушир. с), 5,02 (4H, с), 6,59 (1H, д, J=2,2 Гц), 6,66 (1H, дд, J=2,2, 8,1 Гц), 6,95-7,00 (2H, м), 7,06 (1H, д, J=8,1 Гц), 7,12-7,16 (2H, м).
[0081] Тестовый пример 1
Оценка ингибиторной активности по отношению к переносчику серотонина
Ингибиторную активность соединения в соответствии с настоящим изобретением по отношению к переносчику серотонина оценивали с использованием крысиных тромбоцитов, экспрессирующих переносчики серотонина.
Для этого тестируемое соединение добавляли к крысиным тромбоцитам, флуоресцирующее вещество из Neurotransmitter Transporter Activity Assay Kit (Molecular Devices, R8174) добавляли через 15 минут и измеряли изменения интенсивности флуоресценции поглощения флуоресцирующего вещества с течением времени для оценки ингибиторного эффекта тестируемого соединения по отношению к переносчику серотонина.
[0082] 1. Кровь собирали из нижней полой вены крысы под галотановым ингаляционным наркозом.
2. Центрифугирование выполняли при 350 g при комнатной температуре в течение 5 минут и супернатант, содержащий богатую тромбоцитами плазму, собирали в 15 мл пробирки типа falcon.
3. Центрифугирование выполняли при 2100 g при комнатной температуре в течение 10 минут с получением тромбоцитов в виде осадка. Не содержащий Ca буфер K5 добавляли в количестве 0,7-кратного количества богатой тромбоцитами плазмы, полученной в операции 2, и осадок измельчали с помощью пипетки.
4. Жидкость с тромбоцитами распределяли по 30 мкл/лунка в 384-луночный планшет, а затем в каждую лунку добавляли 10 мкл тестируемого соединения и инкубировали в течение 15 минут при комнатной температуре.
5. Добавляли флуоресцентное вещество из Neurotransmitter Transporter Activity Assay Kit и измеряли изменения интенсивности флуоресценции поглощения флуоресцентного вещества с течением времени посредством переносчиков серотонина с помощью FDSS 6000 (Hamamatsu Photonics).
[0083] Способы фармакологической оценки
Степень ингибирования переносчика серотонина тестируемым соединением определяли посредством следующего уравнения.
[0084] Степень ингибирования переносчика серотонина (%) = Среднее [100 Ч {среднее (AUC в присутствии тестируемого соединения - средняя AUC в присутствии Escitalopram)/{среднее (AUC при отсутствии тестируемого соединения - средняя AUC в присутствии Escitalopram)}].
[0085] Данную степень ингибирования измеряли при концентрациях от 0,0001 до 10000 нМ и рассчитывали IC50.
IC50 пароксетина: 0,6 нМ.
IC50 соединения примера 1: 2,4 нМ.
[0086] Пароксетин и соединение примера 1 демонстрировали выраженные ингибиторные эффекты по отношению к переносчикам серотонина.
[0087] Тестовый пример 2
Тест эффективности in vivo путем принудительного плавания
Соединение примера 1 (10, 30 или 100 мг/кг) или пароксетин (30 или 100 мг/кг) вводили перорально самцам BALB/c мышей (8-9 мышей/группа). Через 1 час мышей помещали отдельно в стеклянные цилиндры (19 см в высоту, 9 см в диаметре), содержащие 9 см воды, и наблюдали при 23°C в течение 6 минут, за это время их поведение записывали на видеокамеру. Контрольная группа получала такое же количество носителя (0,5% раствор метилцеллюлозы, 10 мл/кг).
[0088] Способы измерения
Периоды неподвижности во время последних 4 минут высчитывали в течение 4 минут по видеозаписям. Мышей считали неподвижными, если соблюдались следующие три поведенческих критерия: они прекращали бороться, прекращали взбираться по стенке и в основном неподвижно плавали на воде лишь со слабым движением, необходимым для поддержания их голов над водой.
[0089] Результаты
Как показано на фиг. 1, как соединение примера 1, так и пароксетин дозозависимо уменьшают период неподвижности.
[0090] Тестовый пример 3
Ингибиторные CYP эффекты
Ингибиторные CYP эффекты пароксетина и соединение примера 1 тестировали двумя следующими способами.
[0091] Поскольку зависящее от времени ингибирование CYP пароксетином можно оценить путем тестирования увеличения ингибирования после предварительной инкубации с раствором, содержащим кофермент и микросомальную фракцию из печени человека, содержащую CYP, зависящий от времени тест ингибирования проводили для соединения примера 1 по способу 1. Конкурентное ингибирование CYP также тестировали по способу 2.
[0092] Способ 1
Зависящие от времени ингибиторные способности пароксетина и соединения примера 1 оценивали относительно пяти молекул CYP (CYP1A2, 2C9, 2C19, 2D6 и 3A4).
[0093] Тестируемое вещество добавляли к раствору фермента (содержащий микросомы печени человека (0,2 мг/мл), 100 мM Kpi и 0,1 мM EDTA) и предварительно инкубировали в течение 30 минут при 37°C в присутствии или отсутствии кофермента. Конечную концентрацию тестируемого вещества устанавливали на уровне 0,1, 0,2, 0,4, 0,5, 1, 2, 10 или 50 мкM. Систему, образующую NADPH (60 мM раствор MgCl2, содержащий 3,6 мM β-NADP+, 90 мM глюкозо-6-фосфат и 1 единицу/мл глюкозо-6-фосфат дегидрогеназы, инкубировали в течение 5 минут для образования NADPH), использовали в качестве кофермента. После предварительной инкубации часть реакционного раствора собирали, разбавляли в 10 раз, смешивая с раствором модельного вещества и системой, образующей NADPH, а затем инкубировали в течение 10 минут при 37°C. Равное количество смешанного раствора ацетонитрила и метанола (1:1, содержащий 0,05 мкM декстрофан или 0,05 мкM пропранолол в качестве внутреннего стандарта) добавляли для завершения реакции, а метаболиты модельного вещества в реакционном растворе измеряли с помощью LC-MS/MS. Модельные вещества и метаболиты модельного вещества для каждого фермента CYP показаны в таблице 1. Подобный тест также выполняли с нетестируемым веществом, добавленным в качестве контрольного теста. Соотношение количества метаболитов модельного субстрата в контрольном тесте приведено в виде остаточной активности. Оценивали отношение остаточной активности в присутствии NADPH к остаточной активности при отсутствии NADPH, и отношение в 80% или менее было обозначено, “+” тогда, как отношение выше 80% было обозначено как “-”. Результаты представлены в таблице 2.
[0094] Из сравнения результатов для пароксетина и соединения примера 1 видно, что зависящее от времени ингибирование уменьшалось при конвертировании бензодиоксольного кольца в фталановое кольцо.
[0095]
|
[0096]
|
[0097] Способ 2
Оценивали ингибирующую способность на основе конкурентного ингибирования пяти ферментов CYP (CYP1A2, 2C9, 2C19, 2D6 и 3A4) с использованием пароксетина и соединения примера 1.
[0098] Тестируемое вещество добавляли в конечной концентрации 1 или 10 мкM к раствору фермента (содержащий микросомы из печени человека (0,2 мг/мл), 100 мM Kpi и 0,1 мM EDTA), содержащему раствор модельного вещества, и инкубировали в течение 10 минут при 37°C в присутствии системы, образующей NADPH. Равное количество смешанного раствора ацетонитрила и метанола (1:1, содержащий 0,05 мкM декстрофан или 0,05 мкM пропранолол в качестве внутреннего стандарта) добавляли для завершения реакции, а метаболиты модельного вещества в реакционном растворе измеряли с помощью LC-MS/MS. Модельное вещество и метаболит модельного вещества для каждого фермента CYP показаны в таблице 3. Подобный тест выполняли без добавления тестируемого вещества в качестве контрольного теста. Степень ингибирования определяли исходя из количеств метаболитов модельного субстрата с добавлением и без добавления тестируемого вещества в каждой концентрации тестируемого вещества, а значение IC50 рассчитывали по степени ингибирования (способ расчета согласно Xenobiotica, 1999, 29(1), 53-75). Было присвоено значение, “+” если IC50 составляли 10 мкM или менее, и “-” если она составляла более 10 мкM. Результаты показаны в таблице 4.
[0099] Из сравнения результатов для пароксетина и соединения примера 1 видно, что ингибирующая способность ослаблялась при конвертировании бензодиоксольного кольца во фталановое кольцо.
[0100]
|
[0101]
|
Липосомальная композиция
Конденсированное производное аминодигидротиазина
Производные пиридина, замещенные гетероциклическим кольцом и фосфоноксиметильной группой и содержащие их противогрибковые средства
Промежуточные соединения и способы синтеза аналогов галихондрина в
Реагенты и способы для бета-кетоамидного синтеза синтетического предшественника иммунологического адъюванта е6020
Новое конденсированное производное аминодигидротиазина
Азотсодержащие конденсированные гетероциклические соединения и их применение в качестве ингибиторов продукции бета-амилоида
Аналоги галихондрина в
Противоопухолевое средство, задействующее соединения с ингибирующим эффектом к киназам в комбинации
Биомаркер для болезни альцгеймера или умеренного когнитивного расстройства
Способ получения замещенных гетероциклом производных пиридина
Липосомальная композиция
Конденсированное производное аминодигидротиазина
Производные пиридина, замещенные гетероциклическим кольцом и фосфоноксиметильной группой и содержащие их противогрибковые средства
Промежуточные соединения и способы синтеза аналогов галихондрина в
Реагенты и способы для бета-кетоамидного синтеза синтетического предшественника иммунологического адъюванта е6020
Новое конденсированное производное аминодигидротиазина
Азотсодержащие конденсированные гетероциклические соединения и их применение в качестве ингибиторов продукции бета-амилоида
Аналоги галихондрина в
Противоопухолевое средство, задействующее соединения с ингибирующим эффектом к киназам в комбинации

