Результат интеллектуальной деятельности: СПОСОБЫ ПОЛУЧЕНИЯ МЕТИЛ 4-АМИНО-3-ХЛОР-6-(4-ХЛОР-2-ФТОР-3-МЕТОКСИФЕНИЛ)ПИРИДИН-2-КАРБОКСИЛАТА
Вид РИД
Изобретение
Притязание на приоритет
В данной заявке испрашивают приоритет для даты подачи предварительной патентной заявки Соединенных Штатов серийный номер 61/582166, поданной 30 декабря 2011 года, в отношении «Способов получения метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата».
Область техники, к которой относится изобретение
Варианты осуществления настоящего раскрытия относятся к способам получения метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата, таким как включающие в себя использование метилизобутилкетона в качестве растворителя в нескольких стадиях синтеза метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата.
Предшествующий уровень техники
4-Хлор-2-фтор-3-метоксифенилбороновая кислота (РВА), другие производные 4-хлор-2-фтор-3-замещенной фенилбороновой кислоты и 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинан (РВЕ) представляют собой промежуточные соединения, полезные для получения 6-(полизамещенный арил)-4-аминопиридин-2-карбоксилатных соединений и соединений 2-(полизамещенный арил)-6-амино-4-пиримидинкарбоновых кислот, которые применимы в качестве гербицидов.
РВА можно синтезировать путем взаимодействия 2-хлор-6-фторанизол (2,6-CFA) с н-бутиллитием (н-BuLi). После следующих реакций РВА выделяют в виде твердого вещества. Например, РВА экстрагируют из водной фазы этилацетатом и концентрируют досуха. Альтернативным образом, твердую РВА выделяют кристаллизацией. Затем твердую РВА используют в последующей реакции для получения 6-(полизамещенный арил)-4-аминопиридин-2-карбоксилатного соединения или соединения 2-(полизамещенный арил)-6-амино-4-пиримидинкарбоновой кислоты.
РВА можно также синтезировать путем взаимодействия 2,6-CFA с н-BuLi и В(ОМе)3, добавляя к реакционной смеси водное основание, разбавляя реакционную смесь ацетонитрилом и подкисляя реакционную смесь хлористоводородной кислотой. После этого РВА выделяют, разделяя ацетонитрильный и водный слои.
Описание изобретения
Один из вариантов осуществления настоящего описания относится к способу получения метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата. Данный способ включает добавление метилизобутилкетона к водному раствору, содержащему 4-хлор-2-фтор-3-метоксифенилбороновую кислоту, с получением органической фазы, содержащей 4-хлор-2-фтор-3-метоксифенилбороновую кислоту, и водной фазы. Органическую фазу и водную фазу разделяют. 4-Хлор-2-фтор-3-метоксифенилбороновую кислоту подвергают взаимодействию с метил 4-(ацетиламино)-3,6-дихлорпиридин-2-карбоксилатом в метилизобутилкетоне, получая метил 4-(ацетиламино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилат, который деацетилируют, получая метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилат.
Другой вариант осуществления настоящего описания относится к способу получения метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата. Данный способ включает получение водного раствора, содержащего 4-хлор-2-фтор-3-метоксифенилбороновую кислоту. К водному раствору добавляют метилизобутилкетон. Метилизобутилкетон и 4-хлор-2-фтор-3-метоксифенилбороновую кислоту отделяют от воды. 4-Хлор-2-фтор-3-метоксифенилбороновую кислоту подвергают взаимодействию с метил 4-(ацетиламино)-3,6-дихлорпиридин-2-карбоксилатом в метилизобутилкетоне, получая метил 4-(ацетиламино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилат, который деацетилируют, получая метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилат.
Способ(ы) осуществления данного изобретения
Описан способ получения метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата с использованием метилизобутилкетона (MИБК) в качестве экстракционного растворителя, растворителя для реакции сочетания Сузуки, и/или растворителя для реакции деацетилирования. МИБК также известен как 4-метилпентан-2-он. При использовании способа настоящего описания получают повышенный выход метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата по сравнению с обычными методиками, в которых используют разные растворители в качестве экстракционного растворителя, растворителя для реакции сочетания Сузуки, и растворителя для реакции деацетилирования. При использовании МИБК в качестве растворителя для многочисленных стадий процесса, можно свести к минимуму замену растворителей в ходе получения метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата. Кроме того, можно снизить потери промежуточных продуктов, повышая выход метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата. Кроме того, можно уменьшить число растворителей, используемых при получении метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата, снижая сложность и стоимость способа.
Метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилат получают из производного 4-хлор-2-фтор-3-замещенной фенилбороновой кислоты, такого как РВА. На приведенной далее схеме реакции показано, что метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилат (соединение 1) получают путем взаимодействия РВА (соединение 2) с метил 4-(ацетиламино)-3,6-дихлорпиридин-2-карбоксилатом (соединение 3), получая метил 4-(ацетиламино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилат (соединение 4), которое деацетилируют, получая соединение 1:
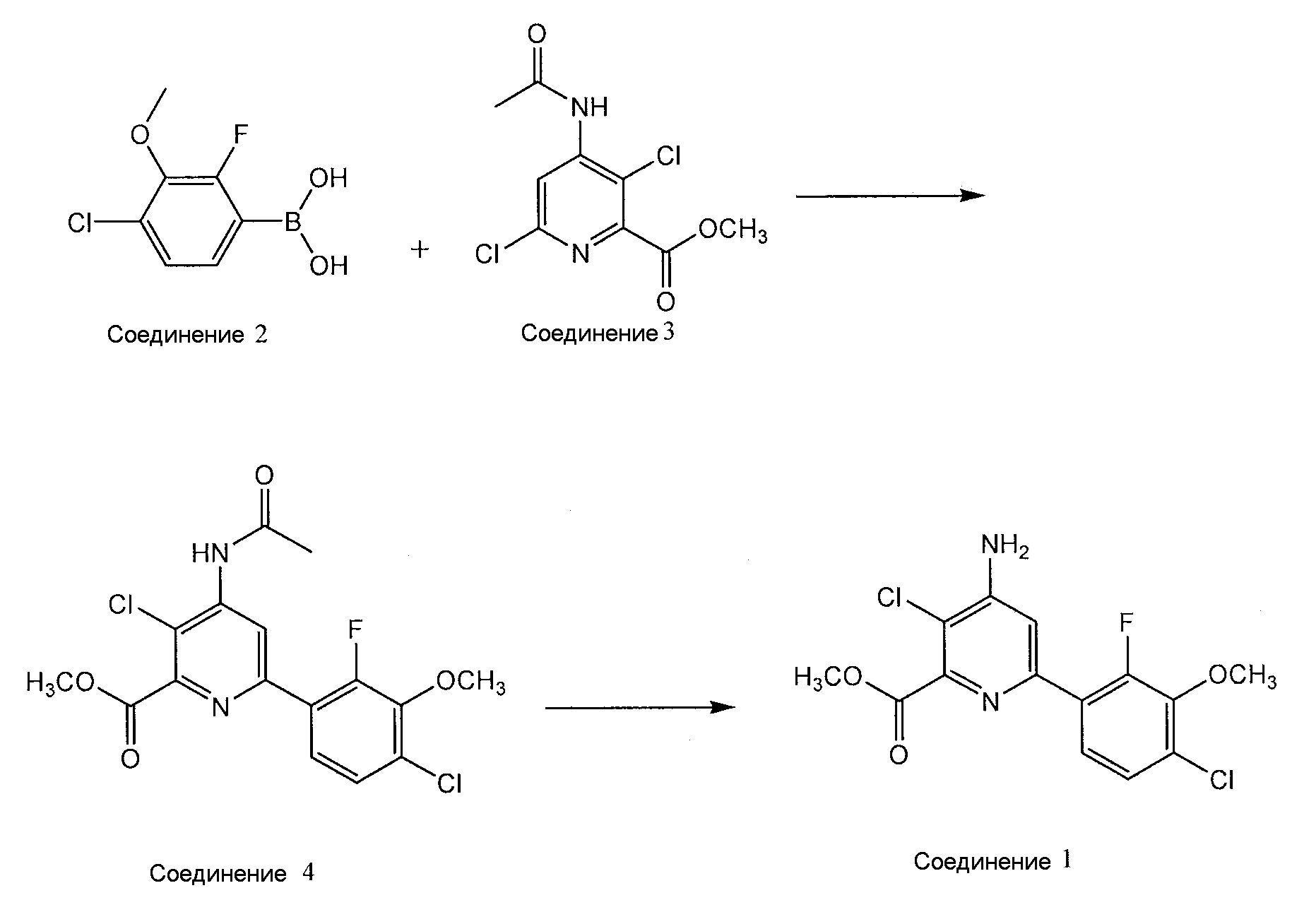
Хотя в вариантах осуществления настоящего описания описано такое производное 4-хлор-2-фтор-3-замещенной фенилбороновой кислоты, как РВА, можно также использовать другие производные 4-хлор-2-фтор-3-замещенной фенилбороновой кислоты. Однако для удобства, в настоящем описании производное 4-хлор-2-фтор-3-замещенной фенилбороновой кислоты описано как РВА. МИБК можно использовать в качестве экстракционного растворителя для получения соединения 2, растворителя для реакции сочетания Сузуки для получения соединения 4, и растворителя для реакции деацетилирования для получения соединения 1.
В настоящем описании предоставлен способ выделения 4-хлор-2-фтор-3-метоксифенилбороновой кислоты, включающий в себя добавление метилизобутилкетона к водному раствору, содержащему 4-хлор-2-фтор-3-метоксифенилбороновую кислоту, с получением органической фазы, содержащей 4-хлор-2-фтор-3-метоксифенилбороновую кислоту, и водной фазы, и отделение органической фазы, содержащей 4-хлор-2-фтор-3-метоксифенилбороновую кислоту, от водной фазы.
В некоторых вариантах осуществления осуществляют выделение, при котором выход 4-хлор-2-фтор-3-метоксифенилбороновой кислоты составляет, по меньшей мере, 85%. В некоторых вариантах осуществления изобретения осуществляют выделение, при котором выход 4-хлор-2-фтор-3-метоксифенилбороновой кислоты составляет, по меньшей мере, 90%.
Соединение 2 можно синтезировать путем взаимодействия соединения 1-хлор-3-фтор-2-замещенного бензола с соединением алкиллития и электрофильным реагентом в инертном органическом растворителе. Соединение 2 выделяют из реакционной смеси, содержащей соединение 2, побочные продукты реакции и воду, при помощи МИБК, который является несмешивающимся с водой растворителем. Реакционная смесь контактирует с МИБК, образуя двухфазную экстракционную систему, содержащую водную фазу (воду и любые побочные продукты реакции) и органическую фазу (МИБК и соединение 2).
Поскольку МИБК не смешивается с водой, в данной экстракции достигается лучшее выделение соединения 2 по сравнению с использованием ацетонитрила в качестве экстракционного растворителя. При разделении органической и водной фаз, соединение 2 получают в виде раствора в органической фазе (МИБК). После этого раствор соединения 2 можно использовать непосредственно в последующих реакциях, таких как реакция получения соединения 4, без осуществления дополнительных стадий концентрирования или выделения. Исключая выделение соединения 2 в виде твердого вещества, можно повысить выход соединения 2.
Схема реакции получения производного 4-хлор-2-фтор-3-замещенной фенилбороновой кислоты из исходного 1-хлор-3-фтор-2-замещенного бензола, соединения алкиллития и электрофильного реагента представлена далее:

в которой X представляет собой F, OR1 или NR2R3, Y представляет собой H или F, каждый из R1, R2 и R3 независимо представляет собой метильную группу, этильную группу, пропильную группу или бутильную группу, а Z представляет собой замещающую группу из электрофильного реагента. Алкильная группа может быть линейной, разветвленной или циклической группой, включающей в себя, но не ограниченной, метилом, этилом, 1-метилэтилом, пропилом, циклопропилом, бутилом, 1,1-диметилэтилом, циклобутилом или 1-метилпропилом. Алкильную группу можно также называть нормальной (н), изо (изо), вторичной (втор.) или третичной (трет) алкильной группой. Z может представлять собой бром, йод, сульфанильную группу, группу бороновой кислоты или боронатного эфира, фосфорильную группу, аминогруппу, алкильную или ацильную группу или их комбинации. Продукт реакции может контактировать с водной фазой, затем контактировать с водной кислотой, приводя к образованию производного 4-хлор-2-фтор-3-замещенной фенилбороновой кислоты. Далее подробно описано, что МИБК можно использовать в качестве экстракционного растворителя для выделения производного 4-хлор-2-фтор-3-замещенной фенилбороновой кислоты.
В одном из вариантов осуществления, соединение 2 синтезируют из 2,6-CFA в результате взаимодействия 2,6-CFA с н-BuLi и триметилбората (В(ОМе)3). Схема реакции для синтеза соединения 2 из 2,6-CFA, н-BuLi и (В(ОМе)3) приведена далее:
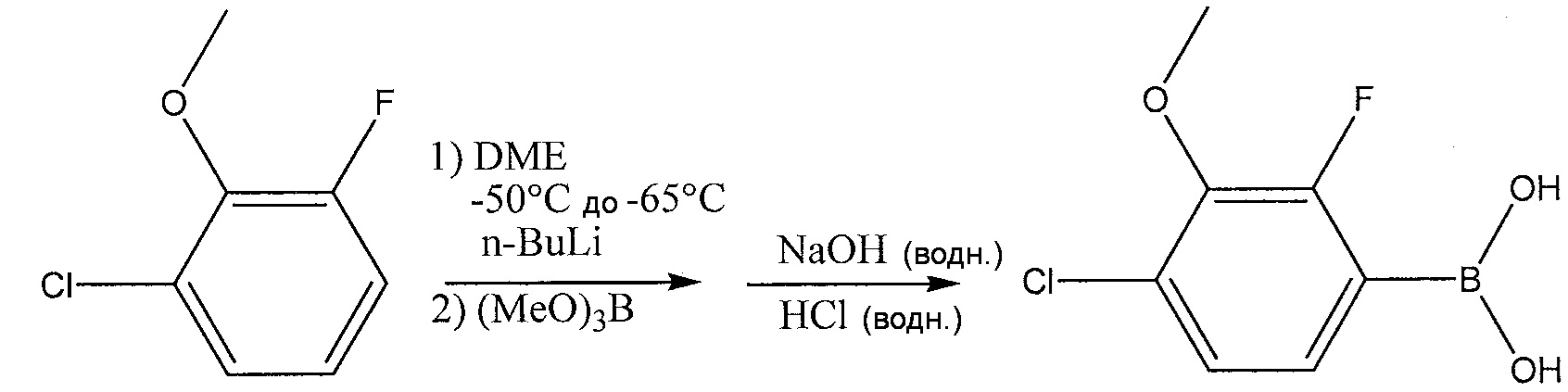 .
.
Несмотря на то что в приведенных в настоящем описании различных вариантах осуществления описан синтез соединения 2 из 2,6-CFA, н-BuLi и (В(ОМе)3 и его последующее выделение, аналогичным образом можно использовать другие производные 4-хлор-2-фтор-3-замещенной фенилбороновой кислоты при использовании соответствующим образом выбранных исходных веществ. Для синтеза соединения 2, 2,6-CFA можно ввести во взаимодействие с соединением алкиллития и электрофильным реагентом в реакционном сосуде. 2,6-CFA можно получить по стандартной методике, которая здесь не описана подробно. Реакцию можно проводить в инертном органическом растворителе, в котором 2,6-CFA, по меньшей мере, частично растворима. Инертный органический растворитель может представлять собой С5-С8 линейный, разветвленный или циклический углеводородный растворитель, такой как пентан, гексан, циклогексан, изооктан, простой эфир, или их сочетания. Простой эфир может включать в себя, но не ограничивается диэтиловым эфиром, тетрагидрофураном, диоксаном или гликолевым эфиром, таким как 1,2-диметоксиэтан (DME). В одном варианте осуществления, органический растворитель представляет собой DME. 2,6-CFA может быть в значительной степени растворима в инертном органическом растворителе, образуя раствор 2,6-CFA, в котором 2,6-CFA в значительной степени растворена в данном инертном органическом растворителе.
Соединение алкиллития может включать в себя, но не ограничиваться метиллитием, н-BuLi или вторбутиллитием. В одном из вариантов осуществления, соединение алкиллития представляет собой н-BuLi. Соединения алкиллития коммерчески доступны от компаний-поставщиков химических реактивов, таких как Sigma-Aldrich (St. Louis, MO). Можно использовать, по меньшей мере, один молярный эквивалент соединения алкиллития относительно 2,6-CFA. Чтобы гарантировать полноту реакции, соединение алкиллития можно добавлять в небольшом избытке относительно 2,6-CFA, например, приблизительно от 1% приблизительно до 10% молярного избытка относительно 2,6-CFA.
Реакцию литиирования с использованием соединения алкиллития можно проводить в безводных условиях. Реакцию литиирования можно проводить при температуре приблизительно от -100°С приблизительно до 0°С, такой как приблизительно от -100°С приблизительно до -50°С. Раствор 2,6-CFA можно охладить или выдерживать при температуре в пределах этого температурного интервала перед добавлением соединения алкиллития. Температуру реакции можно также поддерживать в пределах этого температурного интервала перед добавлением соединения алкиллития. Можно дать возможность 2,6-CFA и соединению алкиллития взаимодействовать в течение достаточного времени для депротонирования 2,6-CFA, поддерживая при этом температуру реакции в пределах этого температурного интервала. Можно позволить реакции протекать, при перемешивании, до тех пор, пока депротонирование не пройдет практически полностью. Реакцию литиирования можно проводить при давлении, которое равно или превышает атмосферное. Реакцию литиирования можно проводить в атмосфере инертного газа, например, при пропускании азота (N2) или другого инертного газа через реакционный сосуд в процессе реакции.
В результате реакции литиирования может депротонироваться атом углерода 1-хлор-3-фтор-2-замещенного бензола в положении (С4) между атомом углерода (С3), с которым связан фтор-заместитель, и атомом углерода (С5), с которым связана группа С4. После этого может образоваться промежуточное соединение, в котором атом лития связан с атомом углерода (С4). Затем литиированный 1-хлор-3-фтор-2-замещенный бензол можно ввести в контакт с электрофильным реагентом, который реагирует по положению С4 1-хлор-3-фтор-2-замещенного бензола. Электрофильный реагент может выступать в качестве источника группы Z, которая связывается с атомом С4 1-хлор-3-фтор-2-замещенного бензола. Электрофильный реагент может представлять собой бром, йод, серу, дисульфид, двуокись серы, эфир бороновой кислоты, двуокись углерода, сульфурилгалогенид, фосфорилгалогенид, альдегид, амид, алкилгалогенид, ацилгалогенид, или их сочетание. Электрофильный реагент может представлять собой алкилборат, такой как В(ОМе)3. В одном из вариантов осуществления, электрофильный реагент представляет собой В(ОМе)3, который взаимодействует с атомом С4 1-хлор-3-фтор-2-замещенного бензола, приводя к образованию производного бороновой кислоты. Реакционную смесь, содержащую литиированный 1-хлор-3-фтор-2-замещенный бензол, можно охладить, например, приблизительно от -100°С приблизительно до -50°С, перед добавлением электрофильного реагента. Электрофильный реагент можно добавлять медленно, поддерживая температуру реакции приблизительно при -65°С или ниже. Можно дать возможность реакционной смеси взаимодействовать в течение времени, достаточного для того, чтобы электрофильный реагент прореагировал с литиированным 1-хлор-3-фтор-2-замещенным бензолом. В ходе реакции с электрофильным реагентом температуру реакции можно медленно повышать до комнатной температуры (приблизительно от 20°С приблизительно до 25°С).
Водное основание можно добавлять к реакционной смеси при комнатной температуре. Водное основание может включать в себя основание, достаточно сильное для того, чтобы гидролизовать продукт взаимодействия 1-хлор-3-фтор-2-замещенного бензола с электрофильным реагентом. Основание может включать в себя, но не ограничивается, гидроксидом натрия, гидроксидом калия или их сочетаниями. Водное основание и реакционную смесь можно перемешивать в течение времени, достаточного для гидролизации основанием продукта взаимодействия 1-хлор-3-фтор-2-замещенного бензола с электрофильным реагентом. После этого реакционную смесь можно перенести в сосуд, в котором органическая фаза (содержащая углеводороды и некоторое количество DME) и водная фаза (водное основание с некоторым количеством растворенного DME) разделяются на четкие слои, которые затем разделяют. Например, данный сосуд может представлять собой делительную воронку. Органическую фазу (углеводороды и некоторое количество DME) можно отбросить, тогда как водную фазу, включающую в себя заряженное соединение продукта реакции 1-хлор-3-фтор-2-замещенного бензола с электрофильным реагентом, необязательно можно привести в контакт, по меньшей мере, с одним объемом органического растворителя, такого как метилтретбутиловый эфир (ТВМЕ), для того, чтобы выделить непрореагировавшую 2,6-CFA. В том случае, когда количество непрореагировавшей 2,6-CFA невелико и не представляет проблемы для последующих реакций, от данного промывания органического растворителя можно отказаться. В одном из вариантов осуществления, водная фаза включает в себя калиевую соль продукта реакции 1-хлор-3-фтор-2-замещенного бензола с электрофильным реагентом.
Водную фазу, содержащую заряженное соединение продукта реакции 1-хлор-3-фтор-2-замещенного бензола с электрофильным реагентом, можно подкислить и разбавить МИБК. Поскольку МИБК и вода практически не смешиваются друг с другом, могут образовываться четкие водный и органический слои. Водную фазу можно подкислить, а затем разбавить МИБК, или можно разбавить МИБК, а затем подкислить. К водной фазе можно добавить водную кислоту, протонируя заряженное соединение продукта реакции 1-хлор-3-фтор-2-замещенного бензола с электрофильным реагентом, получая соединение 2. После подкисления растворимость соединения 2 в МИБК может возрасти по сравнению с растворимостью соединения 2 в ацетонитриле или воде. Кислота может быть достаточно сильной для протонирования заряженного соединения. При помощи неограничивающего примера, по меньшей мере, одна кислота может включать в себя хлористоводородную кислоту (HCl). Другие кислоты включают в себя бромистоводородную кислоту (HBr), серную кислоту (H2SO4), метансульфокислоту и пара-толуолсульфокислоту. По меньшей мере, одну из данных кислот можно использовать в чистом виде или можно разбавить растворителем. По меньшей мере, в некоторых вариантах осуществления, данная кислота представляет собой 6М водную HCl. Можно использовать эквимолярное количество кислоты относительно заряженного соединения продукта реакции 1-хлор-3-фтор-2-замещенного бензола с электрофильным реагентом. Однако для гарантии полного протонирования, можно использовать избыток кислоты. При подкислении заряженного соединения, растворимость соединения 2 в водной фазе по сравнению с его растворимостью в органической фазе (МИБК) может измениться. При переходе в протонированное состояние, соединение 2 может стать практически нерастворимым в водной фазе, но в значительной степени растворимым в МИБК. В случае достаточно большого содержания соли в смеси МИБК/вода, содержащей подкисленный продукт реакции 1-хлор-3-фтор-2-замещенного бензола с электрофильным реагентом, могут образоваться четкие слои воды и МИБК.
Однако если при добавлении МИБК к водной фазе легкого образования двух фаз не происходит, к смеси вода/МИБК можно добавить соль. Данная соль может представлять собой хлорид натрия, сульфат натрия, сульфат калия, хлорид аммония, или их сочетание. Для простоты, метал, входящий в состав данной соли, может быть таким же, как и металл основания, используемого в водной фазе. Аналогичным образом, если основание представляло собой гидроксид калия, то соль может представлять собой калиевую соль. Добавление соли может происходить в результате добавления твердой формы данной соли непосредственно к смеси МИБК/вода, или при добавлении водного раствора данной соли к смеси МИБК/вода. Водный раствор соли может представлять собой насыщенный раствор данной соли в воде. Например, если соль представляет собой хлорид натрия, водный раствор соли может представлять собой солевой раствор, содержащий примерно от 20 мас.% примерно до 27 мас.% хлорида натрия в воде, например, приблизительно 25 мас.% хлорида натрия. Данный солевой раствор можно также назвать насыщенным раствором хлорида натрия. При добавлении соли к смеси МИБК/вода данная соль может насытить водный раствор, приводя к образованию четких водного и органического слоев. В зависимости от содержания соли в смеси МИБК/вода, два четких слоя могут образоваться без добавления соли. Однако в случае образования двух четких слоев, можно добавить дополнительное количество соли для гарантии насыщения водного раствора солью. За счет максимального насыщения водного раствора солью можно добиться максимального выделения РВА из смеси МИБК/вода. Добавление соли может также привести к более легкому распределению соединения 2 в МИБК. МИБК и водный слой можно разделить, при этом практически все соединение 2 будет находиться в растворе МИБК. Чтобы выделить какое-либо количество соединения 2, оставшееся в водном растворе, к водному раствору можно прибавить дополнительные объемы МИБК. Затем несколько объемов МИБК можно объединить, повышая полученный выход соединения 2.
На приведенной далее схеме реакции показано, что 2,6-CFA можно литиировать н-BuLi в безводном DME, получая литиированное производное 2,6-CFA (Li-2,6-CFA):
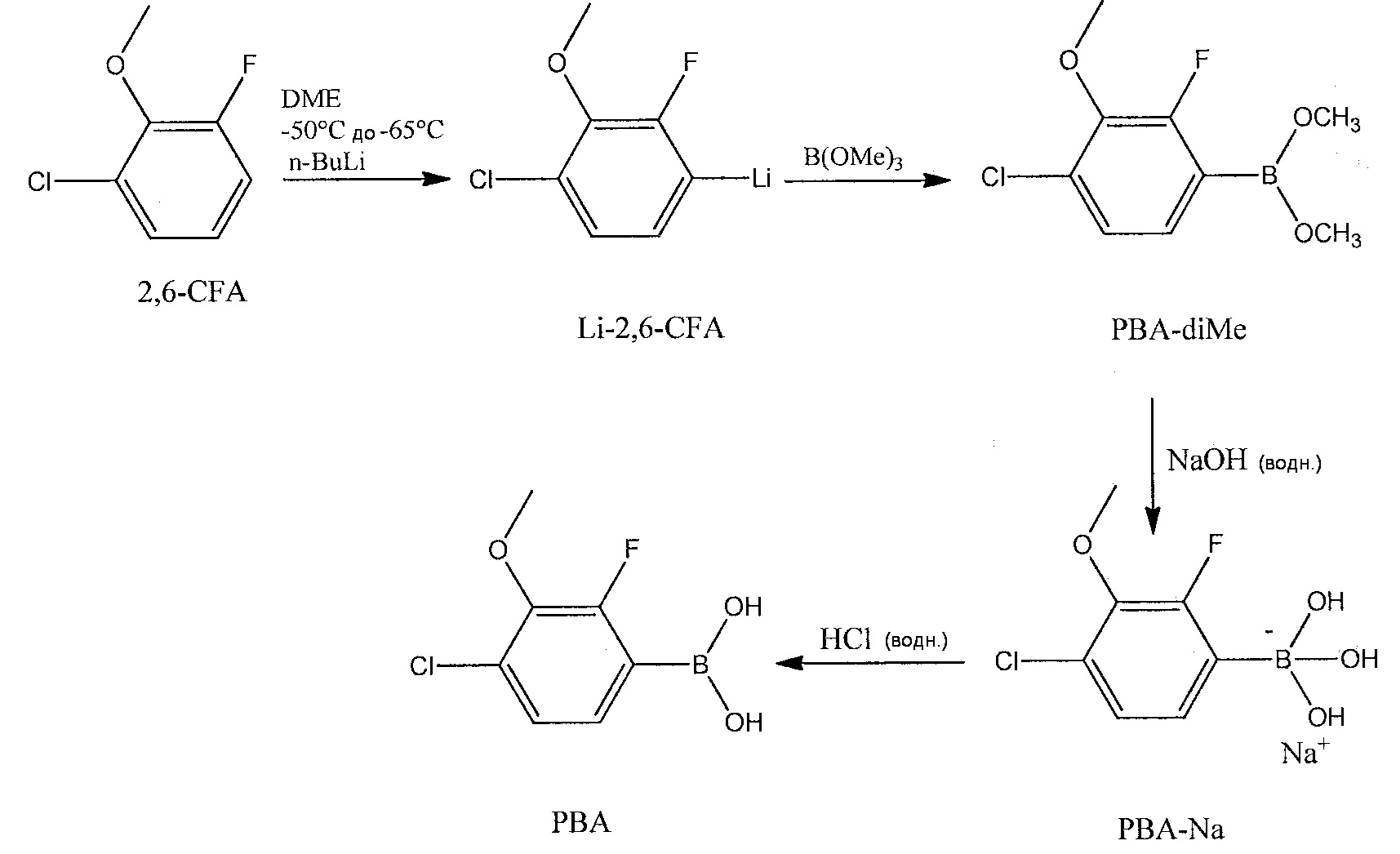
Затем можно добавить В(ОМе)3 и медленно нагревать реакционную смесь до комнатной температуры, получая производное бороновой кислоты (РВА-диМе) Li-2,6-CFA. К РВА-диМе можно прибавить раствор гидроксида натрия в воде при комнатной температуре, получая заряженное натриевое производное (РВА-Na+) РВА-диМе. После перемешивания РВА-Na+ можно перенести в делительную воронку, чтобы дать возможность водному и органическому слоям разделиться. Водный слой можно промыть ТВМЕ, чтобы удалить непрореагировавшую 2,6-CFA. Водный слой, содержащий РВА-Na+, можно перенести в колбу Эрленмейера, экстрагировать МИБК и подкислить, добавляя по каплям 6М-ную водную HCl, получая РВА (соединение 2). Альтернативным образом, водный слой, содержащий РВА-Na+, можно подкислить, добавляя по каплям 6М-ную водную HCl, а затем разбавить МИБК. Хотя в вариантах осуществления настоящего раскрытия описано применение МИБК в качестве экстракционного растворителя, МИБК можно также использовать в сочетании с другими органическими растворителями, такими как смеси МИБК и метанола или DME. При использовании МИБК в сочетании с другими органическими растворителями, МИБК может составлять большую часть общего объема смеси растворителей. При наличии других растворителей, соотношение МИБК и DME или других растворителей в конечном выделенном растворе РВА обычно составляет от 2:1 до 0,7:1, а чаще от 1,6:1 до 1,2:1.
Поскольку МИБК не смешивается с водой, необязательно можно добавить насыщенный раствор NaCl или твердый NaCl, чтобы способствовать образованию водного и органического слоев за счет насыщения водного слоя солью. В зависимости от содержания соли в смеси МИБК/вода, два четких слоя могут образовываться без добавления NaCl. Однако даже в случае образования двух четких слоев, для гарантии насыщения водного слоя NaCl можно добавить дополнительное количество NaCl. Слои МИБК и воды можно разделить и экстрагировать водный слой дополнительными объемами МИБК. Чтобы определить выход соединения 2 в МИБК, МИБК можно удалить, например, выпариванием. Далее твердое вещество белого цвета сушат в вакуумной печи, получая выход соединения 2 приблизительно выше 90%. Чистота соединения 2 может превышать приблизительно 90%, например, превышать приблизительно 95%, или превышать приблизительно 98%. Альтернативным образом, соединение 2 может оставаться в растворе МИБК, и его можно использовать в последующих реакциях без дополнительного концентрирования или сушки, снижая, таким образом, количество стадий в общем синтезе. Поскольку МИБК используют в качестве растворителя в последующих реакциях, описанных далее, можно опустить обмен между растворителями перед проведением дополнительных стадий данных способов.
Соединение 2 в МИБК можно непосредственно использовать в реакции сочетания Сузуки для получения соединения 4. Реакцию сочетания Сузуки можно проводить только в МИБК или в сочетании с другими органическими растворителями, такими как ацетонитрил, исключая стадию обмена между растворителями в процессе получения соединения 1. Соединение 2 можно ввести во взаимодействие с метил 4-(ацетиламино)-3,6-дихлорпиридин-2-карбоксилатом (соединение 3) в МИБК, получая соединение 4. Соединение 3 (известное также как 4-(ацетиламино)-3,6-дихлорпиридин-2-карбоксилат) можно соединить с трифенилфосфином и катализатором, таким как палладиевый катализатор, к которым добавляют соединение 2 в МИБК. К реакционной смеси можно добавить при перемешивании водное основание, такое как водный раствор карбоната калия, и дать реакции протекать практически до конца, проводя мониторинг методом газовой хроматографии (ГХ). Водный раствор карбоната калия может содержать от около 20% до около 30% карбоната калия. Реакционную смесь можно нагревать, например, до температуры от около 40°С до около 65°С. Чтобы гарантировать практически полное растворение продуктов реакции, реакционную смесь можно нагреть до температуры примерно 65°С. После удаления восстановленного палладия и неорганических солей, например, фильтрованием, органическую и водную фазу можно разделить. Водную фазу можно экстрагировать несколькими объемами МИБК, чтобы свести к минимуму потерю продукта, водную фазу отбросить, и объединить несколько объемов МИБК. К органической фазе можно добавить водный раствор гидросульфита натрия и нагревать данную смесь до температуры от около 70°С до около 90°С. Водный раствор гидросульфита натрия может содержать от около 30% до около 50% гидросульфита натрия в воде, а в некоторых вариантах осуществления, от 20% до 50% гидросульфита натрия в воде. После удаления восстановленного палладия и неорганических солей, например, фильтрованием при температуре от около 65°С до около 80°С, органическую и водную фазу можно разделить, при этом соединение 4 будет находиться в органической фазе. Чтоб удалить лишнюю воду, которая может привести к потере продукта в ходе реакции деацетилирования, органическую фазу можно сконцентрировать в вакууме, получая суспензию соединения 4.
Соединение 4 можно деацетилировать, получая соединение 1. В качестве растворителя для реакции деацетилирования можно использовать МИБК. МИБК можно использовать сам по себе или в комбинации с другим органическим растворителем, таким как DMЕ или избыток метанола. При использовании МИБК в комбинации с другим растворителем, МИБК можно использовать в некоторых вариантах осуществления в соотношении от 4:6 МИБК:другой растворитель (об./об.) до около 6:4 МИБК:другой растворитель. В других вариантах осуществления, данное соотношение составляет примерно 1:2 МИБК:другой растворитель. Для деацетилирования соединения 4 к суспензии, содержащей соединение 4, можно добавить метанол, после чего добавить избыток безводной HCl. Суспензию можно нагреть до температуры от около 40°С до около 60°С в течение промежутка времени от около 3 часов до около 6 часов. Мониторинг реакционной смеси можно проводить методом ГХ для определения полноты протекания реакции. Реакционную смесь можно охладить и добавлять к реакционной смеси водный раствор карбоната калия до достижения конечного рН примерно 7,95. Водный раствор карбоната калия может содержать от около 5% карбоната калия до около 15% карбоната калия. Органическую и водную фазы можно разделить и перенести органическую фазу в реакционный сосуд. К органической фазе можно добавить насыщенный раствор хлорида натрия и перемешивать смесь в течение нескольких минут. Полученную органическую и водную фазы можно разделить и концентрировать органическую фазу, например, перегонкой в вакууме. К концентрированной органической смеси можно добавить органический растворитель, такой как гептан или другой алифатический углеводородный растворитель, при температуре от около 65°С до около 80°С. Добавление растворителя может привести к осаждению соединения 1. По окончании добавления гептана суспензию можно охладить, например, примерно до 5°С. Осадок можно выделить фильтрованием, промыть растворителем, например, дополнительным количеством гептана, и сушить, получая соединение 1 с выходом от около 87% до около 90% выхода из расчета на соединение 3.
При использовании МИБК в качестве экстракционного растворителя, растворителя для реакции сочетания Сузуки и растворителя для реакции ацетилирования, выход соединения 1 может быть выше по сравнению с выходом, полученным при использовании стандартной методики получения метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата. Предшествующие методы основывались на проведении реакции сочетания Сузуки с использованием смеси толуола и ацетонитрила (от 8:1 до 10:1, толуол:ацетонитрил) и бромида тетрабутиламмония (ТВАВ) в качестве добавки (1 мол.%), с получением соединения 4 в виде выделенного твердого вещества с выходом от около 80% до около 90%. Процесс кристаллизации для выделения соединения 4 в виде твердого вещества приводит к потере в выходе, составляющей от около 5% до около 10% в маточном растворе. Соединение 4 повторно растворяют в МИБК и деацетилируют, получая XDE729-Me с выходом 75-82% из расчета на соединение 3. При использовании МИБК в качестве растворителя для нескольких стадий данного способа, можно минимизировать количество замены растворителей в процессе получения соединения 1, что снижает потерю промежуточных продуктов и уменьшает сложность и стоимость способа.
Следующие примеры служат для более подробного объяснения вариантов осуществления настоящего описания. Эти примеры не нужно считать исчерпывающими или эксклюзивными в отношении рамок данного изобретения.
ПРИМЕРЫ
Пример 1
Синтез и выделение РВА (соединения 2)
Раствор 2,6-CFA (10 г, 62,28 ммоль) литиировали при действии н-BuLi при температуре примерно ниже -65°С в безводном DME в атмосфере азота. Добавляли В(ОМе)3 и медленно нагревали реакционную смесь до комнатной температуры. К реакционной смеси добавляли раствор КОН в воде при комнатной температуре. После добавления, температура реакционной смеси повышалась до 30°С. Реакционную смесь охлаждали на бане с ледяной водой, чтобы поддерживать температуру приблизительно от 25°С приблизительно до 30°С. Однако данную реакцию можно проводить без бани с ледяной водой, при этом не наблюдается никакого изменения в выходе. Реакционную смесь перемешивают в течение 90 минут и переносят содержимое в делительную воронку, где оставляли органическую и водную фазы для разделения. Нижний водный слой, содержащий РВА-К, спускали в колбу и подкисляли, добавляя по каплям 6 М водную HCl. Альтернативным образом, в колбу перед подкислением добавляли твердый КCl, чтобы свести к минимуму количество воды, экстрагированной в органическую фазу. Способ превращения РВА-К в РВА не приводит к различию в выходе. После добавления температура реакционной смеси повышалась до 30°С. Колбу охлаждали на бане с ледяной водой, чтобы поддерживать температуру приблизительно от 25°С приблизительно до 30°С. Однако данную реакцию можно проводить без бани с ледяной водой, при этом не наблюдается никакого изменения в выходе. Смесь перемешивали в течение 15 минут, чтобы добиться полного растворения. Добавляли МИБК и перемешивали реакционную смесь в течение 15 минут. Органическую и водную фазы разделяли, получая раствор РВА и МИБК. По данным анализа данного раствора, выделение РВА составляло 90%.
Пример 2
Синтез и выделение РВА (соединение 2)
2,6-CFA (10 г, 62,28 ммоль) взвешивали в отдельной колбе и переносили в 3-горлую круглодонную колбу на 500 мл, снабженную термопарой, мешалкой и вводом N2. 2,6-CFA переносили в круглодонную колбу с использованием безводного DME. Добавляли в реакционную колбу дополнительное количество DME, чтобы довести общий объем DME до 106 мл. Реакционную смесь охлаждали до -78°С при помощи бани с сухим льдом/ацетоном. Как только температура реакционной смеси достигала -77°С, медленно, по каплям добавляли н-BuLi (29 мл, 71,62 ммоль, 2,5 М в гексане) из шприцевого насоса в течение 45 минут. Максимальная температура, достигнутая в течение добавления, составляла -70,1°С. По окончании добавления н-BuLi, реакционную смесь оставляли перемешиваться в течение 1 часа при -74,1°С. Через 1 час добавляли по каплям В(ОМе)3 (10,5 мл, 93,42 ммоль) при помощи шприцевого насоса в течение 22 минуты. Максимальная температура, достигнутая в течение добавления В(ОМе)3, составляла -67,0°С. По окончании добавления В(ОМе)3, баню с сухим льдом/ацетоном убирали и нагревали реакционную смесь до комнатной температуры (приблизительно 23,1°С). Как только температура реакционной смеси достигала комнатной, реакционную смесь оставляли перемешиваться еще в течение 1 часа при данной температуре. Данную процедуру повторяли несколько раз, чтобы получить большое количество РВА в 1,2-DME. В колбу объемом 1 л с магнитной мешалкой добавляли 244,0 г раствор РВА-диМе (10,3% из расчета на РВА), 27,82 г 45% КОН и 108,70 г деионизированной H2O. Колбу охлаждали на бане с холодной водой для того, чтобы поддерживать температуру 25-30°С в ходе добавления. Однако данную реакцию можно проводить без бани с холодной водой, при этом не наблюдается никакого изменения в выходе. Смесь перемешивали примерно в течение 2 часов и фильтровали в вакууме, чтобы удалить литиевые соли. Органическую и водную фазы разделяли. К водной фазе добавляли 4,51 г KCl, а затем 40,48 г концентрированной HCl. В другой методике, к водной фазе перед подкислением добавляли твердый KCl, чтобы свести к минимуму объем воды, экстрагированной в органическую фазу, при этом не наблюдается никакого изменения в выходе. Раствор в течение добавления охлаждали на бане с холодной водой для поддержания температуры 25-30°С во время добавления. Однако данную реакцию можно проводить без бани с холодной водой, при этом не наблюдается никакого изменения в выходе. Смесь перемешивали в течение 15 минут, чтобы добиться полного растворения. Добавляли МИБК (35,91 г) и перемешивали раствор примерно в течение 15 минут. Органическую и водную фазу разделяли, получая 127,6 г органической фазы. По результатам анализа раствора получали 17,57 мас.% РВА для выделения РВА в 89,1%.
Пример 3
Синтез метил 4-(ацетиламино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата (соединение 4)
В 1-литровый сосуд с рубашкой, снабженный холодильником, термопарой, механической мешалкой и вводом N2, добавляли метил 4-(ацетиламино)-3,6-дихлорпиридин-2-карбоксилат (АсАР-Ме, 40 г, 152 ммоль). В сосуд добавляли трифенилфосфин (PPh3, 598 мг, 2,28 ммоль), затем бромид тетрабутиламмония (ТВАВ, 368 мг, 1,14 ммоль). Твердые вещества растворяли в толуоле (224 мл) и продували азотом в течение 30 мин при перемешивании (180 об./мин). После 30-минутного продувания добавляли 1,3-пропандиил 4-хлор-2-фтор-3-метоксифенилборонат (РВЕ, 46,5 г, 190 ммоль). В отдельную колбу помещали ацетат палладия(II) [Pd(OAc)2, 256 мг, 1,14 ммоль] и растворяли в ацетонитриле (предварительно продутом в течение 30 мин азотом, 28 мл). После этого к реакционной смеси добавляли раствор ацетата палладия(II) в ацетонитриле, и повышали скорость перемешивания до 300 об./мин. Реакционную смесь перемешивали в течение 5 мин перед добавлением водного раствора К2СО3 (22,8%, 228 мл, предварительно продутого в течение 30 мин азотом). Реакционную смесь нагревали до 65°С и перемешивали в течение 2 ч. Через 2 ч пробу из реакционной смеси анализировали методом ГХ для определения полноты протекания реакции. По окончании реакции перемешивание прекращали и оставляли фазы для оседания. Водный слой спускали в горячем состоянии (~60°С) в колбу. Пробу из органической фазы анализировали методом ГХ с использованием внутреннего стандарта (валерофенона), чтобы определить выход в колбе. По данным ГХ анализа выход в колбе составлял 53,98 г, 95% Ас729-Ме. Затем органическую фазу промывали при 65°С водным насыщенным раствором NaCl (26%, 150 мл). Через 30 мин перемешивание прекращали и оставляли слои для оседания. Водный слой спускали в горячем состоянии (60°С) в стакан. Затем циркуляционную баню устанавливали на 40,0°С и оставляли реакционную смесь медленно охлаждаться до 40°С. При достижении реакционной смесью температуры 45°С, раствор становится мутным и, наконец, большое количество твердого продукта выкристаллизовывалось при 40°С. После этого медленно добавляли Isopar C (265 мл) из капельной воронки при 40°С. Минимальная температура, достигнутая в процессе добавления Isopar C, составляла 37,6°С. Затем циркуляционную баню устанавливали на 24,0°С и оставляли реакционную смесь охлаждаться в течение ночи. На следующее утро продукт выделяли фильтрованием при помощи воронки Бюхнера и фильтровальной бумаги №1. После этого остаток на фильтре промывали смесью толуол:Isopar C 1:1 (100 мл). Потом осадок сушили в вакуумной печи при 55°С в течение ночи, получая Ас729-Ме в виде твердого вещества светло-коричневого цвета (45,6 г, 81%). Общий выход (выделенный выход+продукт в фильтрате)=52,8 г, выход Ас729-Ме 93%.
Пример 4
Синтез метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата (соединение 1)
Метил 4-(ацетиламино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилат (соединение 4) (9,0352 г) загружали в трехгорлую колбу на 100 мл, снабженную магнитной мешалкой, колбонагревателем, термометром и холодильником, в атмосфере азота. Добавляли метанол (8,1 мл), затем 4-метил-2-пентанон (МИБК, 26 мл). Полученную суспензию перемешивали и вводили безводный хлористый водород, добавляя 1,2 мл хлористого ацетила из шприца в течение 6 мин. Хлористый ацетил взаимодействует с метанолом, образуя один эквивалент безводного хлористого водорода и один эквивалент метилацетата. По окончании добавления хлористого ацетила, смесь нагревали до 50°С и перемешивали при данной температуре в течение семи часов. Оставшееся твердое вещество 4 сначала растворялось, образуя прозрачный раствор, который с течением времени вновь образовывал суспензию. Затем полученную смесь охлаждали до температуры окружающей среды и обрабатывали насыщенным водным раствором гидрокарбоната натрия (20 мл). Твердое вещество растворялось и становилось очевидным выделение газа (СО2). Смесь переносили в делительную воронку и разделяли. Органическую фазу промывали насыщенным водным раствором хлорида натрия (20,5 мл). После этого полученную органическую фазу переносили в трехгорлую колбу на 100 мл, снабженную магнитной мешалкой, термометром, колбонагревателем и насадкой для перегонки. К системе подсоединяли вакуум (115 мм рт.ст.) и нагревали, отгоняя часть растворителя. Приблизительно 15,5 мл растворителя отгонялся в первом погоне, а к кубовому остатку от перегонки добавляли дополнительное количество МИБК (4,8 мл), и нагревали смесь до 55°С. К прозрачному раствору добавляли гептан (50 мл) в течение 20 мин, что приводило к осаждению продукта. Продукт выделяли фильтрованием через ватмановскую бумагу №50 на воронке Бюхнера. Остаток на фильтре промывали гептаном (20 мл), затем сушили в течение ночи в вакууме. Получали в общей сложности 4,3506 г метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата (чистота 91,9% по данным ВЭЖХ, выход 92,6% из расчета на загрузку соединения 4).
Пример 5
Синтез метил 4-(ацетиламино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата (соединение 4)
В 1-литровый реактор с рубашкой, снабженный холодильником, механической мешалкой, термопарой и вводом N2, добавляли 40,0 г метил 4-(ацетиламино)-3,6-дихлорпиридин-2-карбоксилата (АсАР-Ме), 0,4 г трифенилфосфина и 0,17 г ацетата палладия(II) в атмосфере азота. Чтобы гарантировать перенесение всех твердых веществ в реактор, использовали промывание 25 мл ацетонитрила (предварительно дегазированного продуванием азотом в течение 45 минут). Добавляли в общей сложности 157,1 г 22%-ного раствора РВА (полученного, как описано в примере 1 или примере 2, и предварительно дегазированного продуванием азотом в течение 45 минут) в 1,2-диметоксиэтане (DME) и метилизобутилкетоне (МИБК) в атмосфере азота при помощи насоса. Соотношение МИБК и DME в растворе РВА изменялось от 2:1 до 0,7:1 (МИБК:DME) в зависимости от эквивалентов растворителей, используемых для получения раствора РВА, или, обычно, от 1,6:1 до 1,2:1. Начинали перемешиванием при 300 об./мин. Добавляли еще 100 мл ацетонитрила (предварительно дегазированного продуванием азотом в течение 45 минут) и достигали полного растворения. Добавляли в общей сложности 275,3 г 22,9%-ного водного раствора карбоната калия (предварительно дегазированного продуванием азотом в течение 45 минут) при помощи насоса. Раствор нагревали примерно при 50°С в течение 2,5 часов. Ближе к окончанию реакции, продукт начинал выпадать, и на стенках реактора образовывалась небольшая корка продукта. Отбирали пробу их органической фазы смеси и анализировали ее методом ГХ для определения полноты протекания реакции.
Смесь нагревали примерно до 65°С, чтобы гарантировать полное растворение продукта. Смесь фильтровали (конечное фильтрование) при 65°С для удаления восстановленного палладия и неорганических солей. Органическую и водную фазу разделяли при 65°С и переносили органическую (верхнюю) фазу в реактор объемом 1 л, снабженный механической мешалкой, холодильником, термопарой и вводом азота. В качестве промывной жидкости для облегчения переноса использовали в общей сложности 71,7 г МИБК. Добавляли всего 380 мл 40%-ного раствора гидросульфита натрия и нагревали смесь примерно до 80°С примерно в течение 6 часов. Смесь фильтровали при 80°С, чтобы удалить восстановленный палладий и неорганические соли, и разделяли фазы. Разделение фаз можно проводить при температуре всего лишь 65°С. Органическую (верхнюю) фазу переносили в реактор объемом 1 л, снабженный механической мешалкой, насадкой для перегонки, термопарой и вакуумным отводом. Смесь концентрировали в вакууме, чтобы удалить из смеси воду, получая суспензию метил 4-(ацетиламино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата.
Пример 6
Синтез метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата (соединение 1)
К суспензии метил 4-(ацетиламино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата (полученной, как описано в примере 5) добавляли 144 мл метанола при 25°С. В данную суспензию барботировали 6,0 г (1,3 эквивалента) безводного HCl, и нагревали суспензию при 50°С в течение от 4 часов до 5 часов. Отбирали пробу раствора и анализировали ее методом ГХ для определения полноты протекания реакции. Продолжительность реакции зависит от количества эквивалентов безводного HCl, используемого в данной реакции. Использование больших избытков HCl, например, свыше 1,3 эквивалентов HCl, приводит к меньшей продолжительности реакции. Однако впоследствии при нейтрализации/обработке требуются повышенные объемы основания для нейтрализации избытка кислоты. Реакционную смесь охлаждали до 25°С и медленно добавляли 127,1 г 10%-ного раствора карбоната калия до достижения рН 7,95. Органическую и водную фазы разделяли и переносили органическую (верхнюю) фазу в реактор объемом 1 л. Добавляли в общей сложности 140 мл насыщенного раствора хлорида натрия и перемешивали смесь в течение нескольких минут. Органическую и водную фазы разделяли и переносили органическую (верхнюю) фазу в реактор объемом 1 л, снабженный механической мешалкой, насадкой для перегонки, холодильником, термопарой и вакуумным отводом. Органическую фазу концентрировали при помощи перегонки в вакууме примерно до 30% от массы раствора. К данному раствору при 70°С добавляли 662,8 г гептана в течение 45 минут. После добавления половины гептана продукт начинал выпадать из раствора. Минимальная температура при добавлении гептана составляла 65°С. По окончании добавления гептана суспензию охлаждали примерно до 5°С. Продукт выделяли фильтрованием и промывали 195 мл гептана. Продукт сушили в вакуумной печи при 55°С в течение ночи, получая 87-90%-ный выход (из расчета на АсАР-Ме) метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)пиридин-2-карбоксилата.
Несмотря на то, что настоящее описание допускает различные модификации и альтернативные формы, в нем при помощи примеров описаны различные варианты осуществления. Однако следует понимать, что настоящее описание не подразумевает ограничения описанными конкретными формами. Напротив, настоящее описание включает в себя все модификации, эквиваленты и варианты, попадающие в рамки настоящего описания, определенные следующей прилагаемой формулой изобретения, и ее правомерными эквивалентами.
Высококонцентрированные препараты 1-метилгептилового эфира флуроксипира, стабильные при низких температурах
Пестициды, пестицидная композиция и способ контроля вредителей
Синергическая гербицидная композиция, содержащая хлорацетанилиды и пиколиновые кислоты
Подсолнечник с низким содержанием насыщенных жиров и соответствующие способы
Способы переноса молекулярных веществ в клетки растений
Стабилизированные эмульсии масло-в-воде, содержащие агрономически активные ингредиенты, и способы их применения
Пестицидные композиции
Защита от повреждения гербицидом 6-(трехзамещенный фенил)-4-амино-2-пиридинкарбоксилата посеянного семенами и рассадного риса-сырца
5-фторпиримидиновые производные в качестве фунгицидов
Пестицидные композиции
Высококонцентрированные препараты 1-метилгептилового эфира флуроксипира, стабильные при низких температурах
Пестициды, пестицидная композиция и способ контроля вредителей
Синергическая гербицидная композиция, содержащая хлорацетанилиды и пиколиновые кислоты
Подсолнечник с низким содержанием насыщенных жиров и соответствующие способы
Способы переноса молекулярных веществ в клетки растений
Стабилизированные эмульсии масло-в-воде, содержащие агрономически активные ингредиенты, и способы их применения
Пестицидные композиции
Защита от повреждения гербицидом 6-(трехзамещенный фенил)-4-амино-2-пиридинкарбоксилата посеянного семенами и рассадного риса-сырца
Стабильная пестицидная композиция на основе сульфоксимина и способ борьбы с насекомыми
5-фторпиримидиновые производные в качестве фунгицидов

