Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ ЛИГАНДОВ НА ОСНОВЕ 1,2-ДИАМИНОЦИКЛОГЕКСАНА
Вид РИД
Изобретение
Изобретение относится к способу получения хиральных гетероциклических лигандов на основе 1,2-диаминоциклогексана, содержащих гетероциклические фрагменты: тиенил-2-, тиенил-3-, фурил-2-, 5-метилфурил-2-, (2,2'-битиофен)-5-ил-, 5-(4'-метилциклогекс-1'-ен-1'-ил)тиофен-2-, которые могут входить в структуру комплексов для проведения энантиоселективных реакций и асимметрического катализа, а также обладать люминесцентными свойствами [1, 2].
Данным изобретением решена задача получения лигандов на основе оптически чистого 1,2-диаминоциклогексана, содержащих гетероциклические фрагменты.
Известные методы синтеза хиральных структур на основе 1,2-диаминоциклогексана [2-4], имеют ряд существенных недостатков.
1. Введение в реакцию оптически чистого (1R,2R)-диминоциклогексана или (1S,2S)-диминоциклогексана.
2. Использование дополнительных реагентов для адсорбции выделяющейся воды.
3. Длительное время протекания реакции (до 48 часов).
4. Использование органических растворителей.
5. Сложность технологического исполнения в ряде случаев.
Одним из методов конденсации цис-/транс-1,2-диаминоциклогексана с альдегидами является использование в качестве растворителя хлористого метилена и сульфата магния для абсорбции выделяющейся в реакции воды [1, 2]. В случае бензальдегида реакция проходит при температуре 40°С в течение 15 минут [1]. Показано, что уменьшение растворимости бифенил-4-карбальдегида в органическом растворителе приводит к резкому увеличению времени реакции конденсации с (1R,2R)-диаминоциклогексаном с 15 мин до 32 ч [2]. Конденсацию салицилового альдегида с (1R,2R)-диаминоциклогексаном проводят при комнатной температуре в среде этанола [3]. В патентах [4, 5] описан метод синтеза 1,2-циклогександииминов на основе транс-1,2-диаминоциклогексана из бензальдегида, тиофен-2-карбальдегида или пиридин-2-карбальдегида, где в качестве растворителя применяют этанол и используют сульфат магния, при этом во всех случаях реакционную массу кипятят, и в зависимости от альдегидной компоненты время реакции возрастает от 1 часа (для бензальдегида) до 18 часов (для 2-тиофенкарбальдегилда). Использование дихлорметана в качестве растворителя для сочетания 2-формилтиофена и свободного амина - (1R,2R)-диаминоциклогексана при комнатной температуре приводит к увеличению времени реакции до 32 часов [2, 3, 6]. В аналогичных условиях получены диимины на основе 5'-замещенного 2,2'-битиофен-5-карбальдегида и свободного основания - (1R,2R)-диаминоциклогексана, стоит отметить что в некоторых случаях реакция проходила в течение 48 часов и в зависимости от заместителей выход варьировался в диапазоне от 31 до 70% [7]. Вышеописанные методы содержат ряд недостатков, которые в основном заключаются в длительном проведении реакции, использовании только одного изомера хирального 1,2'-диаминоциклогексана, при этом выходы целевых продуктов не всегда приемлемы. Стоит отметить, что в зависимости от структуры исходного альдегида условия реакции изменяются для достижения больших выходов.
Конденсацию ароматических альдегидов можно проводить не только с 1,2-диаминоциклогексаном, но и с (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартратом [8]. Однако в данном случае необходимо использование основания для получения свободного амина и осуществления реакции [8]. Наиболее близким по техническому исполнению методом является конденсация соли (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата с ароматическими альдегидами в водно-этанольной смеси (1:1 по объему) в присутствии карбоната калия при температуре 80°C [9]. Стоит отметить, что растворимость многих карбальдегидов резко снижается в водно-этанольной смеси и, соответственно, уменьшает выход целевого соединения. Использование данной методики конденсации в случае 2-(4'-метилциклогекс-1'-ен-1'-ил)тиофен-5-карбальдегида приводит к значительному осмолению реакционной массы и использованию большого модуля растворителя.
Известно, что воздействие на некоторые реакции микроволнового или ультразвукового излучения может способствовать прохождению реакции и ускорять процесс, что отмечено в случае построения трициклических макроструктур [10].
Полученный нами результат является более простым в техническом отношении, экономически эффективным и безопасным методом синтеза дииминов на основе смеси транс-(1R,2R)-диаминоциклогексанов. Методика синтеза азометинов, описанная в статье [3], была отработана на реакции конденсации 5'-метил-2,2'-битиофен-5-карбальдегида со смесью транс-(1R,2R)-диаминоциклогексана. В результате замены растворителя - хлористого метилена на ацетонитрил удалось резко сократить время проведения реакции с 48 ч до 6 ч, что, возможно, связано с понижением растворимости азометина и возможностью выделения из реакционной массы хорошим выходом.
Преимуществом предлагаемой методики является возможность использования коммерчески более доступной смеси транс-(1R,2R)-диаминоциклогексанов. Для получения оптически чистых продуктов использовали описанную методику разделения смеси транс-изомеров 1,2-диаминоциклогексана - метод раскристаллизации с помощью оптически чистой природной L-(+)-винной кислоты для получения соли (R,R)-(-)-l,2-диаминоциклогексан моно-(+)-тартрата в виде кристаллического осадка и соли (S,S)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата, находящейся в маточном растворе [11].
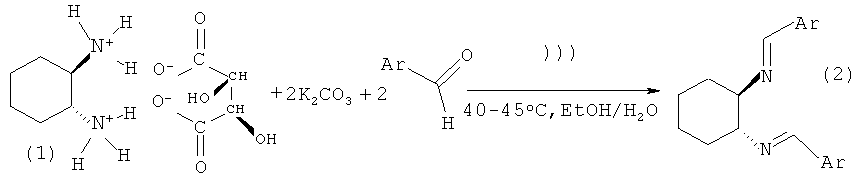
Преимуществом метода является то, что синтез целевых азометинов проводили следующим образом: в одногорлую круглодонную колбу, помещенную в ультразвуковую ванну, вносят соль (S,S)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата (1 моль), насыщенный раствор карбоната калия (2 моль) и диспергируют в течение 5 минут, затем порционно добавляют карбальдегид (2 моль) в минимальном объеме этилового спирта и на полученную смесь воздействуют ультразвуком в течение 5 часов, температура реакции 40-45°C. Образующийся осадок отфильтровывают, промывают водой, сушат и при необходимости азометин перекристаллизовывают из этилового спирта. Стоит отметить, что проведение реакции в аналогичных условиях при перемешивании и без использования ультразвука не позволяет достичь хороших выходов соединений и резко увеличивает время реакции.
Основные отличительные признаки предлагаемого метода получения промежуточных азометинов можно сформулировать следующим образом.
1. В предлагаемом методе в качестве исходного соединения используются оптически чистая соль (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата, что более удобно в техническом исполнении, при этом полученная соль более стабильна и может храниться в течение длительного времени, в отличие от самих 1,2-диаминоциклогексанов.
2. Наиболее приемлемым мольным соотношением соль (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата/гетероароматический карбальдегид/карбонат калия (безводный) является 1:2:2. При этом карбальдегид растворяют в минимальном объеме этанола.
3. Значительно сокращается время проведения реакции (с 48 ч [8] или 32 ч [2, 3, 7] до 3 ч). Сокращение времени реакции приводит к упрощению технологической схемы получения и увеличению производительности.
4. Предлагаемый метод не предполагает использование сложных лабораторных установок, что существенно упрощает схему реактора, и не требует применения сложного и дорогостоящего оборудования.
5. Предлагаемый метод может использоваться на различных количествах реагентов, существует возможность увеличения масштабов производства.
Получение 2,2'-битиофен-5-карбальдегида и 5'-метил-2,2'-битиофен-5-карбальдегида проводили по ранее описанным методам [12a,b].
Интерес представляет создание не только азометинов, но и дальнейшее восстановление азометиновой связи и образованию структур, которые могут быть использованы в качестве лигандов в комплексах с металлами для энантиоселективных реакций [13].
В литературе описан ряд методов восстановления азометинов, содержащих как ароматический фрагмент, так и гетероциклический фрагмент, в методиках используется боргидрид натрия в качестве восстановителя, однако в качестве среды применяют метиловый спирт или смеси метиловый спирт/эфир, время реакции составляет от 1 до 4 часов при комнатной температуре [1-3, 8]. Стоит отметить, токсичные свойства метилового спирта в данном методе. Описан метод восстановления иминной связи с помощью боргидрида натрия в среде этанола, однако реакцию проводят при 0°С с подъемом температуры до комнатной в течение 16 часов [14].
Преимуществом предлагаемой методики восстановления азометинов и получения целевых хиральных гетероциклических лигандов является использование коммерчески доступного и более безопасного этилового спирта и сокращение реакции до 3 часов с возможностью использования технического азометина. Преимуществом предлагаемого метода является:
1. Использование коммерчески доступного и более безопасного этилового спирта;
2. Наиболее приемлемыми мольным соотношением является: азометин/боргидрид натрия - 1:10 и использование минимального объема этилового спирта;
3. Сокращено время реакции с 16 до 3 часов с сохранением выхода продуктов;
4. Метод не предполагает использование сложных лабораторных установок, что существенно упрощает схему реактора, и не требует применения сложного и дорогостоящего оборудования.
Технический результат достигается тем, что способ получения хиральных гетероциклических лигандов на основе 1,2-диаминоциклогексана основан на взаимодействии гетероароматических альдегидов и (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата в присутствии карбоната калия при мольном соотношении 1:2:2, соответственно, в водно-этанольной среде под действием ультразвука при комнатной температуре в течение 3 часов, с последующим восстановлением под действием боргидрида натрия при мольном соотношении азометин/боргидрид натрия 1:10 в этиловом спирте при температуре 60-70°С в течение 3 часов, затем отгоняют 1/2 этилового спирта, остаток выливают в лед, продукт экстрагируют хлористым метиленом (3 раза), сушат, растворитель удаляют, в остатке получают продукт с выходом 90-95% и чистотой 97-98%.
Выполнение метода
Строение синтезированных соединений подтверждено данными ИК-спектров, контроль над ходом реакции и индивидуальность соединений определялись с помощью TCX на пластинках Marchery Nagel. Проявление осуществлялось парами йода и облучением УФ лампой 254/365. ИК спектр записан на спектрометре Shimadzu IRAffinity-1 в таблетках бромида калия либо в тонком слое в призмах. Элементный анализ выполнен на автоматическом CHNS-анализаторе "Euro Vector ЕА-3000". Спектры ЯМР 1Н записаны на приборе JEOL JNM ECX 400 (400 МГц), в CDCl3, ДМСО-d6, внутренний стандарт - тетраметилсилан. Температура плавления измерена на приборе ПТП.
Способ получения азометинов. Общая методика.
В одногорлую круглодонную колбу, помещенную в ультразвуковую ванну, вносят соль (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата (1 моль), насыщенный раствор карбоната калия (2 моль) и диспергируют в течение 5 минут, затем порционно добавляют карбальдегид (2 моль) в минимальном объеме этилового спирта и на полученную смесь воздействуют ультразвуком в течение 5 часов, температура реакции 40-45°С. Образующийся осадок отфильтровывают, промывают водой, сушат и при необходимости азометин перекристаллизовывают из этилового спирта.
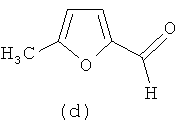
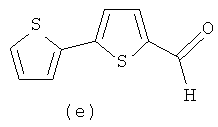
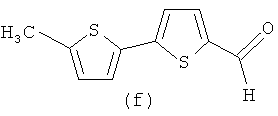
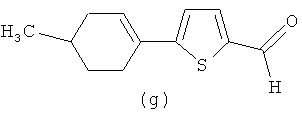
Пример 1. Синтез (1R,2R)-N,N-{ди-(тиофен-2-ил)метилиден}-циклогексан-1,2-диимина (2a)
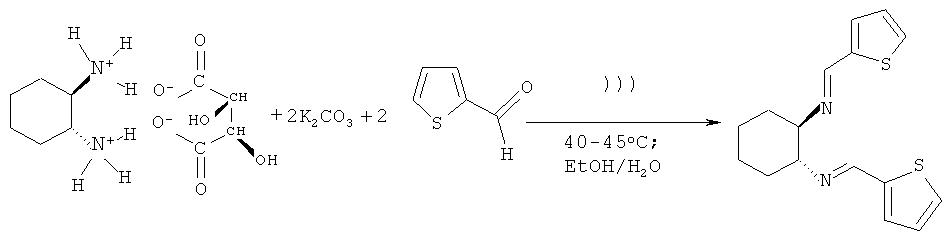
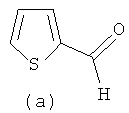
4.1 г (15.5 ммоль) (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата, 3.5 г (31 ммоль) тиофен-2-карбальдегида, 35 мл этилового спирта, 4.4 г (31 ммоль) карбоната калия в 10 мл воды. Выход 4.21 г (90%), т.пл. 128-133°C. ИК спектр (KBr), ν, см-1: 2926, 2873, 2843; 1635 (C=N); 796, 759 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м. д. (J, Гц): 8.25 (2H, c, N=CH) 7.27 (2H, дд, 3J=5.04, 4J=1.4, H-5), 7.12 (2H, дд, 3J=3.65, 4J=1.4, H-3), 6.94 (2H, дд, 3J=3.65, 3J=5.04, H-4), 3.3 (2H, м, CH циклогекс); 1.82 (6H, м, CH2 циклогекс); 1.43 (2H, м, CH2 циклогекс). Найдено, %: C 63.52, Н 6.01, N 9.28, S 21.19. C16H18N2S2. Вычислено, %: С 63.54, H 6.00, N 9.26, S 21.20.
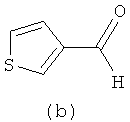
Синтез (1R,2R)-N,N-{ди-(тиофен-3-ил)метилиден}циклогексан-1,2-диимина (2b).
Выход 4.21 г (94%), т.пл. 128-132°C. ИК спектр (KBr), ν, см-1: 2927, 2854, 2843; 1639 (C=N), 800, 783 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м. д. (J, Гц): 8.18 (2H, с,  ), 7.42 (2H, дд, 4J=1.8, H-2), 7.39 (2H, дд, 3J=5.03, H-5), 7.21 (2H, дд, 3J=5.03, 4J=1.8, H-4), 3.29 (2H, м, CH циклогекс), 1.84-1.67 (6H, м, CH2 циклогекс), 1.51-1.43 (2Н, м, СН2 циклогекс). Найдено, %: С 63.52, Н 6.01, N 9.28, S 21.19. C16H18N2S2. Вычислено, %: C 63.54, H 6.00, N 9.26, S 21.20.
), 7.42 (2H, дд, 4J=1.8, H-2), 7.39 (2H, дд, 3J=5.03, H-5), 7.21 (2H, дд, 3J=5.03, 4J=1.8, H-4), 3.29 (2H, м, CH циклогекс), 1.84-1.67 (6H, м, CH2 циклогекс), 1.51-1.43 (2Н, м, СН2 циклогекс). Найдено, %: С 63.52, Н 6.01, N 9.28, S 21.19. C16H18N2S2. Вычислено, %: C 63.54, H 6.00, N 9.26, S 21.20.
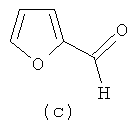
Синтез (1R,2R)-N,N-{ди-(фуран-2-ил)метилиден}-циклогексан-1,2-диимина (2c).
Выход 5.4 г (85%). ИК спектр (KBr), ν, см-1: 2931, 2858; 1647 (C=N), 1014 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м.д. (J, Гц): 7.21 (2H, с, C=N), 7.41 (2H, дд, 3J=5.2, 4J=1.8 H-5), 6.55 (2H, д, 3J=3.4, H-3), 6.37 (2H, д, 3J=5.2, 3J=3.4, H-4), 3.34-3.32 (2H, м, CH циклогекс), 1.82-1.79 (6H, м, CH2 циклогекс), 1.42-1.41 (2H, м, CH2 циклогекс). Найдено, %: C 71.10, H 6.73, N 10.34, O 11.83. C16H18N2O2. Вычислено, %: С 71.09, Н 6.71, N 10.36, О 11.84.
Синтез (1R,2R)-N,N-{ди-(5-метилфуран-2-ил)метилиден}цикло-гексан-1,2-диимина (2d). Выход 5.25 г (97%). ИК спектр (KBr), ν, см-1: 2931, 2858; 1643 (C=N), 1022 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м. д. (J, Гц): 7.86 (2H, c, N=CH), 6.37 (2H, д, 3J=3.2, H-3), 5.90 (2H, д, 3J=3.2, H-4), 3.3-3.23 (2H, м, CH циклогекс), 2.22 (6H, с, CH3), 1.72 (6H, м, CH2 циклогекс), 1.34 (2H, м, CH2 циклогекс). Найдено, %: C 72.45, H 7.41, N 9.41, O 10.73. C18H22N2O2. Вычислено, %: C 72.46, H 7.43, N 9.39, O 10.72.
Синтез (1R,2R)-N,N-{ди-(2,2'-битиофен-5-ил)метилиден}-циклогексан-1,2-диимина (2e). Выход 1.82 г (91%), т.пл. 110-113°C. ИК спектр спектр (KBr), ν, см-1: 2955, 2878, 1624 (C=N), 855, 815, 699 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м. д. (J, Гц): 8.19 (2H, c, N=CH), 7.22 (2H, дд, 3J=5.2, 4J=1.16, H-5), 7.18 (2H, дд, 3J=3.67, 4J=1.16, H-4'), 7.0 (4H, м, H-3, H-3'), 6.99 (2H, дд, 3J=3.67,3J=5.2, H-4); 3.3 (2H, м, CH циклогексен.); 1.83 (6H, м, CH2 циклогекс); 1.46 (2H, м, CH2 циклогекс). Найдено, %: C 61.78, H 4.74, N 6.02, S 27.46. C24H22N2S4. Вычислено, %: С 61.76, H 4.76, N 6.00, S 27.48.
Синтез (1R,2R)-N,N-{ди-(5-метил-2,2'-битиофен-5'-ил)метилиден}-циклогексан-1,2-диимина (2f). Выход 1.8 г (90%), т.пл. 90-93°C. ИК спектр (KBr), ν, см-1: 2927, 2856, 1622 (C=N), 1481, 1226, 812, 790. Спектр ЯМР 1H (400 MHz, CDCl3), δ, м.д. (J, Гц): 8.17 (2H, c, N=CH), 7.00 (2H, д, 3J=3.68, H-4), 6.97 (2H, д, 3J=3.68, H-3'), 6.93 (2H, д, 3J=3.68, H-3); 6.64 (2H, д, 3J=3.68, H-4'); 3.3 (2H, м, CH циклогексен.); 1.83 (6H, м, CH2 циклогекс); 1.46 (2H, м, CH2 циклогекс). Найдено, %: C 63.11, H 5.33, N 5.65, S 25.91. C26H26N2S4. Вычислено, %: C 63.12, H 5.30, N 5.66, S 25.92.
Синтез N,N-{ди(5-(4-метилциклогекс-1-ен-1-ил)тиофен-2-ил)метилиден}-циклогексан-1,2-диимина (2g). Выход 0.34 г (39%), т.пл. 147-149°C. ИК спектр (KBr), ν, см-1: 2914, 2850; 1627 (C=N), 1490 (C=C), 792 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м. д. (J, Гц): 8.14 (2H, c, N=CH), 6.97 (2H, д, 3J=3.68, H-3), 6.78 (2H, д, 3J=3.68, H-4), 6.18 (2H, уш. с, C=CH циклогексен), 3.26 (2H, м, CH циклогекс); 2.35 (6H, м, CH2 циклогекс); 1.74 (10H, м, CH2 циклогекс); 1.33 (4H, м, CH2 циклогекс); 0.98 (6H, д, 3J=6.43, CH3). Найдено, %: C 73.40, H 7.81, N 5.72, S 13.07. C30H38N2S2. Вычислено, %: C 73.42, H 7.80, N5.71, S 13.07.
Восстановление азометинов. Общая методика.
В колбе, снабженной магнитной мешалкой, растворяют азометин (1 моль) в минимальном объеме этилового спирта. При интенсивном перемешивании при комнатной температуре добавляют боргидрид натрия (10 моль) и кипятят с обратным холодильником в течение 3 часов. Удаляют ½ этилового спирта, остаток выливают в лед, продукт экстрагируют хлористым метиленом (3 раза), сушат, растворитель удаляют, в остатке получают продукт с выходом 90-95% и чистотой 97-98%.
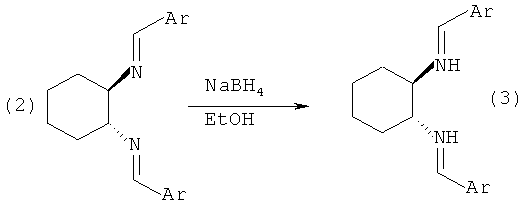
Пример 2. Синтез (1R,2R)-N,N-ди-(2-тиенилметил)циклогексан-1,2-диамина (3a)
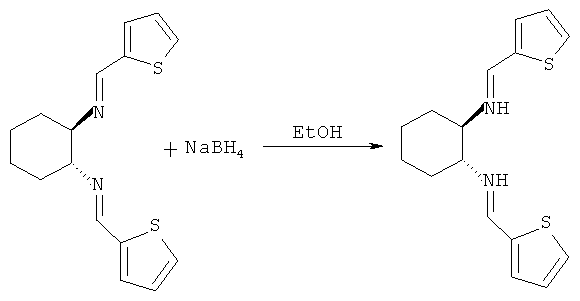
2.7 г (8.94 ммоль) (1R,2R)-N,N-{Ди-(тиофен-2-ил)метилиден}-циклогексан-1,2-диимина, 7.05 г (178.8 ммоль) боргидрида натрия, 140 мл этанола. Выход 1.5 г (98.7%). Т.пл. 70-72°C. ПК спектр (KBr), ν, см-1: 2924, 2850; 3298 (N-H). Спектр ЯМР 1H, δ, м.д. (J, Гц): 7.39 (2H, дд, 3J=4.82, 4J=1.40, H-5), 6.94-6.91 (4H, м, H-3, H-4), 4.11 (2H, д, 3J=14.19, CH2-NH), 3.88 (2H, д, 3J=14.19, CH2-NH), 2.27-2.30 (2H, м, циклогекс.), 2.07-2.15 (2H, м, циклогекс), 1.71-1.73 (2H, м, циклогекс), 1.20-1.25 (2Н, м, циклогекс), 1.02-1.04 (2H, м, циклогекс). Найдено, %: C 62.73, H 7.22, N 9.15, S 20.90. C16H22N2S2. Вычислено, %: C 62.70, H 7.24, N9.14, S 20.92.
Синтез (1R,2R)-N,N-ди-(3-тиенилметил)циклогексан-1,2-диамина (3b). Выход 1.3 г (85.5%). ИК спектр (KBr), ν, см-1: 2924; 3294 (N-H). Спектр ЯМР 1H, δ, м.д. (J, Гц): 7.24 (2H, дд, 3J=5.04, 4J=3.29, H-5), 7.09-7.11 (2H, м, H-4), 7.03 (2H, д, 4J=1.36, H-2), 3.91 (2H, д, 3J=13.85, CH2-NH), 3.68 (2H, д, 3J=13.85, CH2-NH), 2.22-2.29 (2H, м, циклогекс), 2.04-2.15 (2H, м, циклогекс); 1.71-1.73 (2H, м, циклогекс), 1.20-1.27 (2H, м, циклогекс), 1.01-1.04 (2H, м, циклогекс). Найдено, %: C 62.73, H 7.22, N 9.15, S 20.90. C16H22N2S2. Вычислено, %: C 62.70, H 7.24, N 9.14, S 20.92.
Синтез (1R,2R)-N,N-{ди-(фуран-2-ил)метил}-циклогексан-1,2-диамина (3c). Выход 3.5 г (83%). ИК спектр (KBr), ν, см-1: 3302 (N-H), 2931, 2858; 1010 (Сар-Сар). Спектр ЯМР 1H, δ, м.д. (J, Гц): 7.19 (2H, дд, 3J=4.82, 4J=1.40, H-5), 6.07 (2H, д, 3J=4.82, 3J=3.24, H-4), 6.16 (2H, дд, 3J=3.24, 4J=1.40, H-3), 3.75 (2H, д, 3J=14.43, CH2-NH), 3.62 (2H, д, 3J=14.43, CH2-NH), 2.11-2.16 (2H, м, CH циклогекс), 1.89-1.93 (2H, м, циклогекс), 1.57-1.64 (2H, м, циклогекс), 1.06-1.1 (2H, м, циклогекс), 0.93-0.98 (2H, м, циклогекс). Найдено, %: C 70.01, H 8.07, N 10.23, О 11.69. C16H22N2O2. Вычислено, %: C 70.04, H 8.08, N 10.21, O 11.67.
Синтез (1R,2R)-N,N-{ди-(5-метилфуран-2-ил)метил}-циклогексан-1,2-диамина (3d). Выход 3.3 г (94%). ИК спектр (KBr), ν, см-1: 3302 (N-H), 2931, 2858; 1010 (Cap-Cap). Спектр ЯМР 1H, δ, м. д. (J, Гц): 5.77 (2H, д, 3J=3.2, H-4), 5.95 (2H, д, 3J=3.2, H-3), 3.7 (2H, д, 3J=14.19, CH2-NH), 3.54 (2Н, д, 3J=14.19, CH2-NH), 2.16 (2H, с, CH3), 1.61-1.63 (2H, м, CH циклогекс), 1.95-1.98 (4H, м, циклогекс), 1.11-1.2 (2H, м, циклогекс), 0.93-0.96 (2H, м, циклогекс). Найдено, %: C 71.50, Н 8.66, N 9.25, О 10.59. C18H26N2O2. Вычислено, %: C 71.49, H 8.67, N 9.26, O 10.58.
Синтез (1R,2R)-N,N-ди-(2,2'-битиенил-5-метил)циклогексан-1,2-диамина (3e). Выход 0.82 г (88%). ИК спектр (KBr), ν, см-1: 2920, 2850, 2819; 3290 (N-H), 821, 798, 698 (Cap-Cap). Спектр ЯМР 1H, δ, м.д. (J, Гц): 7.15 (2H, дд, 3J=5.04, 4J=1.36, H-5), 7.09 (2H, д, 3J=3.64, Н-4'), 6.99 (2Н, д, 3J=3.52, H-3), 6.94-6.96 (2H, м, H-3'), 6.82 (2H, д, 3J=3.52, 3J=5.04, H-4), 4.07 (2H, д, 3J=14.05, CH2-NH), 3.86 (2H, д, 3J=14.05, CH2-NH), 2.32-2.34 (2H, м, CH циклогекс). 2.12-2.16 (2H, м, циклогекс). 1.72-1.74 (2H, м, циклогекс), 1.21-1.26 (2h, м, циклогекс), 1.05-1.08 (2H, м, циклогекс). Найдено, %: C 61.22, H 5.58, N 5.91, S 27.26. C24H26N2S4. Вычислено, %: C 61.24, H 5.57, N 5.94, S 27.25.
Синтез (1R,2R)-N,N-ди-((5-метил-2,2'-битиенил)-5-метил)циклогексан-1,2-диамина (3f). Выход 1.6 г (88%). ИК спектр (KBr), ν, см-1: 3437 (N-H), 3290, 2916, 2852, 1448, 1197, 895, 790. Спектр ЯМР 1H (400 MHz, CDCl3), δ, м.д. (J, Гц): 6.89 (4Н, м, Н-4, H-3), 6.79 (2H, м, H-3'), 6.60 (2H, м, H-3); 4.08 (2H, д. 3J=14.19, CH2-NH), 3.85 (2H, д, 3J=14.19, CH2-NH), 2.32-2.34 (2H, м, циклогекс), 2.14-2.11 (2H, м, циклогекс), 1.72-1.74 (2H, м, циклогекс), 1.30-1.19 (4H, м, циклогекс), 1.05-1.07 (2Н, м, циклогекс.). Найдено, %: C 62.62, H 6.04, N 5.61, S 25.73. C26H30N2S4. Вычислено, %: C 62.61, H 6.06, N 5.62, S 25.71.
Синтез 1R,2R)-N,N-{ди-[5-(4'-метилциклогекс-1'-ен-1'-ил)тиофен-2-ил)метил}-циклогексан-1,2-диамина (3g). Выход 1.1 г (91%). ИК спектр (KBr), ν, см-1: 3290 (N-H), 2924, 2862, 2819; 1454 (C=C), 786 (Cap-Cap). Спектр ЯМР 1H, δ, м. д. (J, Гц): 6.74 (4H, м, H-3, Н-4), 6.06 (2H, уш. с, C=CH), 4.02 (2H, д, 3J=14.19, CH2-NH), 3.88 (2H, д, 3J=14.19, CH2-NH), 2.35-2.28 (10Н, м, циклогекс), 1.79-1.78 (14H, м, циклогекс), 1.24-1.23 (2H, м, CH3-CH циклогекс), 0.97 (2H, д, 3J=4.3, CH3). Найдено, %: C 72.80, H 8.57, N 5.63, S 12.97. C30H42N2S2. Вычислено, %: C 72.82, H 8.56, N 5.66, S 12.96.
Литература:
[1] Albano V.G. et all. Controlling Stereochemical Outcomes of Asymmetric Processes by Catalyst Remote Molecular Functionalizations: Chiral Diamino-oligothiophenes (DATs) as Ligands in Asymmetric Catalysis // Chem. Eur. J. - 2006. №12. - P.667-675.
[2] Albano V., Bandini M., Melucci M. Novel Chiral Diamino-Oligothiophenes as Valuable Ligands in Pd-Catalyzed Allylic Alkylations. On the "Primary" Role of "Secondary" Interactions in Asymmetric Catalysis. // Adv. Synth. Catal. - 2005. №347. - P.1507-1512.
[3] Ambroziak K., Rozwadowski Z., Dziembowska T. Synthesis and spectroscopic study of Schiff bases derived from trans-1,2-diaminocyclohexane. Deuterium isotope effect on 13C chemical shift. // Journal of Molecular Structure. - 2002. №615. - P.109-120.
[4] Патент 0234239 A1 США. Method Of Forming Carbon-Heteroatom Linkage. / Taillefer M., Cristau H., Cellier P. - Заявлено 2.06.2003. - Опубл. 20.10.2005.
[5] Патент 0236413Al США. Process for arylating of vinylating or alkynating nucleophilic compound. / Cellier P., Cristau H., Spindler J. - Заявлено 31.05.2002. - Опубл. 25.12.2003.
[6] Lere-Porte J., Moreau J., Serein-Spirau F. A chiral polymer with alternating conjugated segments and (1R,2R)-1,2-diaminocyclohexane as a unit with C2 symmetry. // Tetrahedron. - 2001. №42. - P.3073-3076.
[7] Bandini M., Benaglia M., Quinto T. New Recoverable Poly (ethylene glycol)-Supported C1-Diaminooligothiophene Ligands for Palladium-Promoted Asymmetric Allylic Alkylation (AAA) Reactions. // Adv. Synth. Catal. - 2006. №348. - P.1521-1527.
[8] Zhang X., Li Y., Dong Z. Asymmetric transfer hydrogenation of aromatic ketones with chiral diamino-thiophene/iridium catalyst systems. // Journal of Molecular Catalysis A: Chemical. - 2009. №307. - P.149-153.
[9] Duguet N., Donaldson A., Leckie S. Chiral relay in NHC-mediated asymmetric β-lactam synthesis I; substituent effects in NHCs derived from (1R,2R)-cyclohexane-1,2-diamine. // Tetrahedron: Asymmetry. - 2010. №21. - P.582-600.
[10] Srimurugan S., Viswanathan B. Microwave assisted cyclocondensation of dialdehydes with chiral diamines forming calixsalen type macrocycles. // Tetrahedron Letters. - 2005. №46. - P.3151-3155.
[11] Larrow F., Jacobsen E. A Practical Method for the Large-Scale Preparation of E-N,N-Bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediaminato-2)]manganese Chloride, a Highly Enantioselective Epoxidation Catalyst. // J. Org. Chem. - 1994. №59. - P.1939-1942.
[12] a) Nakayama J., Murabayashi S., Hoshino M. Preparation of (E)-1,2-bis(2,2'-bithiophene-5-yl)ethylene and (E)-1,2-bis-(2,2':5',2”-terthiophene-5-yl)ethylene // Heterocycles. - 1986. V.24. №9. - P.2639-2643. b) Lescot E., Buu-Hoi Ng. Ph., Xuong N.D., Thiophene Derivatives. Part XIV. Some problems of substitution in the 2,2'-bithienyl series. // J. Chem. Soc. - 1959. V.656. - P.3234-3237.
[13] Cooper Ch., Jones M., Brayshaw S. When is an imine not an imine? Unusual reactivity of a series of Cu(II) imine-pyridine complexes and their exploitation for the Henry reaction. // Dalton Trans. - 2011. №40. - P.3677-3682.
[14] Kowalczyk R., Skarzewski J. Asymmetric nitroaldol reaction catalyzed by copper-diamine complexes: selective construction of two contiguous stereogenic centers. // Tetrahedron: Asymmetry. - 2009. №20. - P.2467-2473.
Способ получения хиральных гетероциклических лигандов на основе 1,2-диаминоциклогексана основан на взаимодействии гетероароматических альдегидов и (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата в присутствии карбоната калия при мольном соотношении 1:2:2, соответственно, в водно-этанольной среде под действием ультразвука при комнатной температуре в течение 3 часов, с последующим восстановлением под действием боргидрида натрия при мольном соотношении азометин/боргидрид натрия 1:10 в этиловом спирте при температуре 60-70°C в течение 3 часов, затем отгоняют ½ этилового спирта, остаток выливают в лед, продукт экстрагируют хлористым метиленом (3 раза), сушат, растворитель удаляют, в остатке получают продукт с выходом 90-95% и чистотой 97-98%.Способ энантиоселективного синтеза (r)-диэтил(2-нитро-1-фенилэтил) малоната в присутствии комплекса никеля
Способ получения 3-амино-1-адамантанола и его кислотно-аддитивных солей
Способ получения многоосновных карбоновых кислот адамантанового ряда
Способ получения третичных циклических спиртов ряда 2,2'-битиофена
Способ получения 2-(1-адамантилкарбонил)-1,2-дигидронафто[2,1-b]фуранов
Способ получения 1-(адамантил-1)-пиридиний бромида
Способ получения 5-метокси-4-азатрицикло[4.3.1.1 3,8]ундец-4-ена
Способ получения 1-(1-адамантил)-3,4,5-тринитро-1н-пиразола
Способ энантиоселективного синтеза (s)-прегабалина
Способ получения 1,3,5-тригидроксиадамантана
Способ получения 3-амино-1-адамантанола и его кислотно-аддитивных солей
Способ энантиоселективного синтеза (s)-прегабалина
Производные 2-r1-4-r2-6-полинитрометил-1,3,5-триазинов, обладающие антибактериальной активностью
Способ получения гидрохлоридов аминов адамантанового ряда
Способ энантиоселективного синтеза диэтил[3-метил-(1s)-(нитрометил)бутил]малоната формулы i
Способ получения 1-гидрокси-4-адамантанона
Антигололедная композиция
Способ получения трехосновных карбоновых кислот адамантанового ряда
Дезинфицирующая композиция
Способ получения 3-арил-1н-бензо[f]хроменов

