Результат интеллектуальной деятельности: ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Вид РИД
Изобретение
Настоящее изобретение относится к фармацевтическим композициям, а более конкретно - к инъецируемым формирующимся in situ депо-составам, содержащим фармацевтически активное вещество, а также к способу изготовления таковых депо-составов.
Формирующиеся in situ депо-составы представляют собой особый класс полимерных систем доставки, преимуществом которых являются простота изготовления даже в случае чувствительных молекул и простота применения, поскольку полимер затвердевает после введения в результате разделения фаз. Если депо-состав имеет в своей основе рассасывающийся полимер, подобный часто используемым сополимерам D,L-лактида и гликолида (ПЛГ), он со временем рассасывается. В настоящее время на рынке присутствуют два инъецируемых формирующихся in situ депо-состава, атридокс и элигард. Оба продукта были разработаны Dunn и др. (H.B. Ravivarapu, K.L. Moyer, R.L. Dunn, International Journal of Pharmaceutics, 194 (2000) 181-191) на основе технологии Атригель и включают ПЛГ, растворенный в 1-метил-2-пирролидиноне (НМП), смешивающемся с водой растворителе, и порошок лекарственного вещества, который суспендируют в растворе перед применением.
Инъецируемые формирующиеся in situ депо-составы являются привлекательной, но в тоже время и проблемной системой введения. Помимо химической совместимости, местной переносимости и острой токсичности, важным фактором для инъецируемого формирующегося in situ депо-состава является его устойчивость при хранении в жидком виде.
Объектом настоящего изобретения являются формирующиеся in situ фармацевтические депо-составы с улучшенными характеристиками в отношении переносимости и острой токсичности, а равно и устойчивости при хранении, которые удобны в применении и которые особенно хорошо подходят для готовых к употреблению инъецируемых депо-составов.
В одном из вариантов осуществления настоящего изобретения его объектом является формирующийся in situ фармацевтический депо-состав, содержащий
(1) гидрофобное или гидрофильное фармацевтически активное вещество
(2) рассасывающийся полимер,
(3) биосовместимый растворитель, смешивающийся с водой, обладающий точкой затвердевания с температурой между 8° и 20°C, предпочтительно полиэтиленгликоль с молекулярной массой между 450 и 650 Да и с химически инертными концевыми группами, предпочтительно, концевыми алкоксигруппами, более предпочтительно, концевыми этокси- и метоксигруппами, а также, необязательно,
(4) вспомогательное вещество.
Биосовместимый растворитель, смешивающийся с водой, предпочтительно выбирают среди полиэтиленгликолей (ПЭГ) с точкой затвердевания между 8° и 20°C, предпочтительно, между 10° и 16°C, с целью получения устойчивого твердого состава при низкой температуре, чтобы избежать осаждения частиц лекарственного вещества. В одном из вариантов осуществления настоящего изобретения точка затвердевания находится ниже 15°C. Используемый в качестве растворителя ПЭГ несет инертные концевые группы с целью достижения повышенной устойчивости, равно как и более низкой вязкости по сравнению с ПЭГ той же молекулярной массы без концевых групп. В предпочтительном варианте осуществления настоящего изобретения применяют моно- и диалкиловый эфир полиэтиленгликоля, такой как, например, диметиловый эфир или диэтиловый эфир ПЭГ. В другом предпочтительном варианте осуществления настоящего изобретения молекулярная масса ПЭГ с концевыми группами находится между 500 и 600 Да, в более предпочтительном варианте осуществления настоящего изобретения молекулярная масса составляет 450, 500, 550 или 650 Да. В особенно предпочтительном варианте осуществления настоящего изобретения ПЭГ выбран из диметилового эфира полиэтиленгликоля 500 (также известного как ПЭГ500ДМЭ, или ПЭГ ДМЭ 500, или ПЭГ 500 ДМЭ и коммерчески доступного, например, от компании Clariant Glymes, температура плавления около 13°C). Термины «точка затвердевания» и «температура плавления», как они употребляются в контексте, являются взаимозаменяемыми. Определения температуры плавления/затвердевания доступно рядовому специалисту в соответствующей области и может быть, например, осуществлено с помощью дифференциальной сканирующей калориметрии (ДСК). ПЭГ по настоящему изобретению демонстрируют низкий гемолитический и токсический потенциал.
Фармацевтические композиции по настоящему изобретению находятся в твердом состоянии при температурах хранения 2-8°C (типичная температура холодильника) и находятся в жидком состоянии и свободны или в существенной степени свободны от осаждения при комнатной температуре. Фармацевтические композиции (без лекарственного вещества) по настоящему изобретению характеризуются вязкостью, подходящей для инъецирования без затруднений, например, инъецируемы подкожно или внутримышечно, при приведении в равновесие при комнатной температуре. Динамическая вязкость полимерного раствора (фармацевтическая композиция без лекарственного вещества) предпочтительно находится в диапазоне от 300 до 800 мПа•с. Фармацевтические композиции по настоящему изобретению особенно полезны для готовых к использованию устройств, поскольку перед применением не требуется операции ресуспендирования.
Фармацевтически активное вещество может быть растворено или диспергировано в жидких полиэтиленгликолях (ПЭГ) с модифицированными концевыми группами, как это описано выше, например в моно- (USP 31 NF26) и диалкиловом эфире полиэтиленгликоля, например, при комнатной температуре (например, 25°C), например, в зависимости от его растворимости в данном растворителе, с сорастворителем или без такового. В предпочтительном варианте осуществления настоящего изобретения сорастворителя, такого как, например, органический растворитель, не используется.
Фармацевтически активное вещество (также взаимозаменяемо именуемое в контексте лекарственным веществом) может быть гидрофильным или гидрофобным, небольшой органической молекулой, пептидом или белком. Фармацевтически активное вещество может находиться в форме свободного основания, свободной кислоты или соли.
Примеры фармацевтически активных веществ включают, не ограничиваясь, однако, перечисленными, пептиды, полипептиды, белки, углеводы, олигонуклеотиды, РНК и ДНК. Некоторыми примерами пептидов являются антитела, гормоны роста, например, эпидермальный фактор роста (ЭФР), пролактин, люлиберин или рилизинг-фактор лютеинизирующего гормона (LH-RH), глюкагон, гастрин, пентагастрин, урогастрон, секретин, энкефалины, эндорфины, ангиотензины, ренин, брадикинин, бацитрацины, полимиксины, колистины, тироцидин, грамицидины, инсулин, интерфероны, эритропоэтин, кальцитонин, гепарин, аналоги соматостатина, например, памоат или диаспартат соматостатина, клеткостимулирующие факторы и паратиреоидные гормоны. Другие примеры включают бифосфонаты, такие как, например, памидроновая кислота, алендроновая кислота, ибандроновая кислота, ризедроновая кислота, золедроновая кислота, агонисты и аналоги гонадотропин-высвобождающего фактора (GnRH), такие как, например, лейпрорелина ацетат, трипторелина ацетат или памоат, бусерелин, гистрелина ацетат или гормональные контрацептивы, такие как этоногестрел, левоногестрел. Другие примеры включают миноциклин, рисперидон, налтрексон, кармустин.
Предпочтительное активное вещество может являться аналогом соматостатина. Соматостатин является тетрадекапептидом, отвечающим структуре  .
.
Аналоги соматостатина, вызывающие отдельный интерес, включают октреотид, как он описан в патенте США US 4395403, ланреотид или пасиреотид. Вызывающие особый интерес аналоги соматостатина были также описаны, например, в международных заявках на изобретение WO 97/01579 и WO 02/010192. Упомянутые аналоги соматостатина включают аминокислотную последовательность формулы I
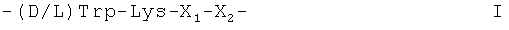 ,
,
в которой
X1 является радикалом формулы (a) или (b)
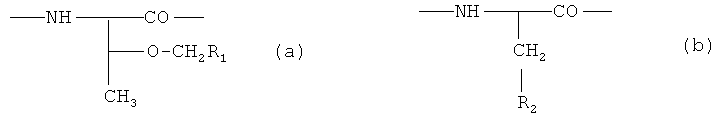 ,
,
в котором
R1 является необязательно замещенным фенилом, причем заместитель может представлять собой галоид, метил, этил, метоксигруппу или этоксигруппу,
R2 представляет собой-Z1-CH2-R1, -CH2-CO-O-CH2-R1,
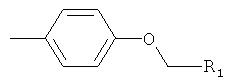 или
или 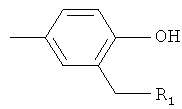 ,
,
где Z1 является кислородом или серой, а
Х2 представляет собой α-аминокислоту, несущую ароматический остаток на боковой цепи Cα, или аминокислотный фрагмент, выбранный из 2,4-диаминобутановой кислоты (Dab), 2,3-диаминопропановой кислоты (Dpr), диаминопимелиновой кислоты (Dpm), гистидина, (бензил)гидроксипролина, тиенилаланина, циклогексилаланина и трет-бутилаланина,
причем лизиновый остаток таковой аминокислотной последовательности соответствует остатку Lys9 нативного соматостатина-14.
Под аналогом соматостатина, как этот термин употребляется в контексте, подразумевается неразветвленный или циклический пептид, производный от принадлежащего природному соматостатину-14, включающий последовательность формулы I, в котором, кроме того, один или несколько аминокислотных фрагментов пропущены и/или заменены одним или несколькими другими аминокислотными радикалами (радикалом) и/или в котором одна или несколько функциональных групп заменены одной или несколькими другими функциональными группами и/или одна или несколько групп заменены одной или несколькими другими изостерическими группами. Как правило, данный термин охватывает все модифицированные производные нативного соматостатина-14, включающие вышеприведенную последовательность формулы I, характеризуемые способностью к связыванию в диапазоне наномолей на литр хотя бы с одним подтипом соматостатиновых рецепторов, как это определено ниже.
Предпочтительно, аналог соматостатина представляет собой соединение, в котором остатки соматостатина-14 в положениях с 8 по 11 представлены последовательностью формулы I, как она определена выше.
Особенно предпочтительными являются соединения формулы II
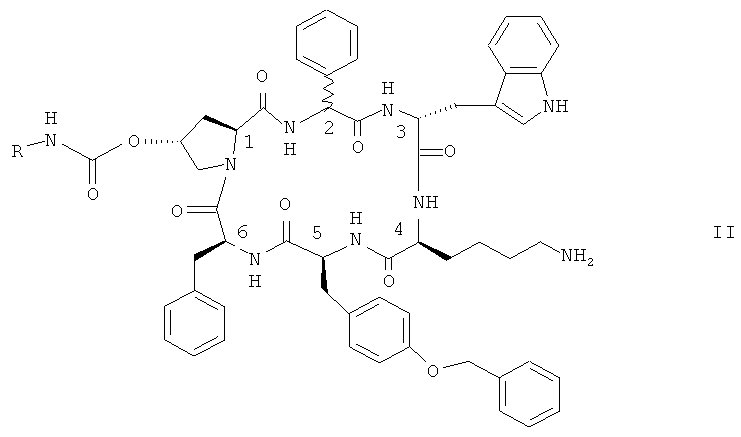 ,
,
в которых конфигурация при атоме С-2 является (R), или (S), или их смесью и в которых R представляет собой NR10R11-(C2-C6)алкилен или гуанидин-(C2-C6)алкилен, a R10 и R11 независимо друг от друга являются водородом или (C1-С4)алкилом, в свободной форме, в форме соли или в защищенной форме.
Предпочтительно R представляет собой NR10R11-(C2-C6)алкилен. Предпочтительными соединениями формулы II являются такие соединения, в которых R представляет собой 2-аминоэтил, а именно цикло[{4-(NH2-С2Н4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] (именуемое в контексте соединением A) и цикло[{4-(NH2-C2H4-NH-CO-O-)Pro}-DPhg-DTrp-Lys-Tyr(4-Bzl)-Phe], в свободной форме, в форме соли или в защищенной форме. Phg обозначает -HN-СН(C6H5)-CO-, a Bzl обозначает бензил.
Соединение по настоящему изобретению может находиться, например, в свободной форме или в форме соли. Соли включают кислотно-аддитивные соли с, например, неорганическими кислотами, полимерными кислотами или органическими кислотами, например, с соляной кислотой, уксусной кислотой, молочной кислотой, аспарагиновой кислотой, бензойной кислотой, янтарной кислотой или памоевой кислотой. Кислотно-аддитивные соли могут существовать в виде одно- или двухвалентных солей, например, в зависимости от того, один или два эквивалента кислоты добавлено. Предпочтительными солями являются лактат, аспартат, бензоат, сукцинат и памоат, включая моно- и дизамещенные соли, более предпочтительно, дизамещенный аспартат и монозамещенный памоат.
Полимер из композиции по настоящему изобретению может являться синтетическим или природным полимером. Полимер может быть как рассасывающимся, так и комбинацией рассасывающихся и нерассасывающихся полимеров. Предпочтительно, может использоваться рассасывающийся полимер. Под термином «полимер» подразумевается гомополимер или сополимер.
Термин «рассасывающийся», как он употребляется в контексте, означает материал, который должен рассасываться в результате происходящих в организме процессов, превращаясь в продукты, легко выводящиеся из организма, и не должен накапливаться в организме.
Подходящие полимеры включают:
а) неразветвленные или разветвленные сложные полиэфиры, представляющие собой линейные цепи, расходящиеся от полиольного остатка, например, глюкозы,
б) сложные полиэфиры, такие как D-, L- или рацемическая полимолочная кислота, полигликолевая кислота, полигидроксимасляная кислота, поликапролактон, полиалкиленоксалат, сложные эфиры полиалкиленгликоля и кислот из цикла Кребса, например, цикла лимонной кислоты, и им подобные, а также их комбинации,
в) полимеры органических простых эфиров, ангидридов, амидов и ортоэфиров,
г) сополимеры органических сложных эфиров, простых эфиров, ангидридов, амидов и ортоэфиров, сами по себе или в сочетании с другими мономерами.
Полимеры могут быть поперечно-сшитыми или не быть таковыми. Обычно не более 5%, как правило, менее 1%, являются поперечно-сшитыми.
Предпочтительными полимерами по настоящему изобретению являются неразветвленные сложные полиэфиры и разветвленные сложные полиэфиры. Неразветвленные сложные полиэфиры могут быть получены из α-гидроксикарбоновых кислот, например, молочной кислоты и гликолевой кислоты, посредством конденсации лактоновых димеров, см, например, патент США US 3773919, содержание которого включено в настоящее описание в качестве ссылки во всей его полноте. Предпочтительные сложные полиэфирные цепи в неразветвленных или разветвленных (звездообразных) полимерах представляют собой сополимеры остатков α-гидроксикарбоновых кислот, молочной кислоты и гликолевой кислоты, или лактоновых димеров. Мольные соотношения лактид:гликолид в сополимерах лактида и гликолида, предпочтительно используемых согласно настоящему изобретению, предпочтительно составляют от, приблизительно, 95:5 до 5:95, например, от 75:25 до 25:75, например, от 60:40 до 40:60, включая от 55:45 до 45:55, например, от 52:48 до 48:52, например, 50:50.
Неразветвленные сложные полиэфиры, например, неразветвленные сополимеры лактида и гликолида (ПЛГ), предпочтительно используемые согласно настоящему изобретению, характеризуются среднемассовой молекулярной массой (Mw) в диапазоне от, приблизительно, 1000 и, приблизительно, 50000 Да, например, приблизительно 10000 Да, и полидисперсностью Mw/Mn, например, в пределах от 1,2 до 2. Внутренняя вязкость неразветвленных полимеров с Mw в пределах от 1000 до 50000 составляет от 0,05 до 0,6 дл/г, 0,1% в хлороформе (при температуре 25°C) Подходящие примеры включают, например, такие общеизвестные и коммерчески доступные материалы, как Резомеры® от компании Boehringer Ingelheim, в частности, Резомеры® RG, например, Резомер® RG 502, 502Н, 503, 503Н.
Разветвленные сложные полиэфиры, например, разветвленные сополимеры лактида и гликолида, предпочтительно используемые согласно настоящему изобретению, могут быть получены с помощью полигидроксилированных соединений, например, полиолов, например, глюкозы или маннита в качестве инициатора. Эти сложные эфиры полиола известны и описаны, например, в патенте Великобритании B 2145422 B, содержание которого включено в настоящее описание в качестве ссылки во всей его полноте. Полиол включает хотя бы три гидроксигруппы и характеризуется молекулярной массой до 20000 Да, причем хотя бы одна, предпочтительно, хотя бы две, например, в среднем три из гидроксильных групп полиола находятся в форме сложноэфирных групп, содержащих цепи из полилактида или сополимера лактида и гликолида. Как правило, для инициирования полимеризации используют 0,2% глюкозы. Разветвленные сложные полиэфиры (глюкоза-ПЛГ) содержат в центре остаток глюкозы, от которого лучами расходятся неразветвленные полилактидные цепи. Например, они обладают звездообразным строением.
Разветвленные сложные полиэфиры, содержащие в центре остаток глюкозы и неразветвленные лучи из цепочек сополимера лактида и гликолида (глюкоза-ПЛГ) могут быть получены посредством взаимодействия полиола с лактидом и, предпочтительно, также с гликолидом при повышенной температуре в присутствии катализатора, который позволяет осуществить полимеризацию с раскрытием цикла.
Разветвленные сложные полиэфиры, содержащие в центре остаток глюкозы и неразветвленные лучи из цепочек сополимера лактида и гликолида (глюкоза-ПЛГ) предпочтительно характеризуются среднемассовой молекулярной массой Mw в диапазоне от, приблизительно, 1000 до 55000, предпочтительно 20000, например, 10000 Да, и полидисперсностью, например, от 1,1 до 3,0, например, от 2,0 до 2,5. Внутренняя вязкость звездообразных полимеров с Mw от 10000 до 50000 составляет от 0,05 до 0,6 дл/г в хлороформе. Звездообразный полимер с Mw 50000 характеризуется вязкостью 0,5 дл/г в хлороформе.
Желательная скорость рассасывания полимеров и желательный профиль высвобождения для соединений по настоящему изобретению могут варьироваться в зависимости от вида мономера, а также того, задействован ли гомо- или сополимер, или же смесь полимеров.
Смесь полимеров может включать хотя бы два разных вида полимеров, например, перечисленных выше в пунктах а)-г), или же двух полимеров с разными свойствами из одного и того же класса полимеров. Например, смесь полимеров может включать полимер, характеризуемый среднемассовой молекулярной массой, например, от, приблизительно, 30000 до, приблизительно, 50000 Да, например, приблизительно 20000 Да, и полимер, характеризуемый низкой среднемассовой молекулярной массой, например, от, приблизительно, 2000 до, приблизительно, 20000 Да, например, приблизительно 10000 Да.
Предпочтительно, полимерная матрица включает неразветвленный и/или разветвленный сополимер лактида и гликолида. Более предпочтительно, полимерная матрица включает Резомер® RG, и/или звездообразный сополимер лактида и гликолида, характеризуемый среднемассовой молекулярной массой около 10000 Да, и/или звездообразный сополимер лактида и гликолида, характеризуемый среднемассовой молекулярной массой около 50000 Да. Соотношение между неразветвленным и разветвленным сополимерами лактида и гликолида предпочтительно составляет от 0:100 до 100:0, например, от 50:50 до 25:75 и до 75:25.
В особенно предпочтительном варианте осуществления настоящего изобретения рассасываемый полимер (например, полилактид (ПЛ) PLA 100 или сополимер лактида и гликолида (ПЛГ) PLGA50:50) характеризуется внутренней вязкостью 0,15-0,45 дл/г (0,1% в CHCl3, 25°C).
В другом варианте осуществления настоящего изобретения его объектом является способ получения инъецируемого формирующегося in situ депо-состава, включающий стадии:
1) растворения рассасываемого полимера, например, ПЛ или ПЛГ, в растворителе, например, ПЭГ с концевыми группами с точкой затвердевания между 8° и 20°C,
2) добавления фармацевтически активного вещества и, необязательно, вспомогательного вещества с целью получения раствора или суспензии,
а в случае получения суспензии также стадию
3) снижение среднего диаметра частиц с помощью подходящих способов, например, обработки в струйной мельнице, ультразвуковой обработки, гомогенизации при высоком давлении обработки в ротор-статорном миксере ультратурракс.
Альтернативно, если лекарственное вещество растворимо, стадии 1) и 2) можно поменять местами.
Лекарственное средство растворяют или диспергируют в полимерном растворе при такой температуре, при которой растворитель остается жидким, например, это удобно осуществить при комнатной температуре. Диспергированное лекарственное вещество может быть, например, гомогенизировано до ожидаемого размера частиц сначала с помощью ультратурракса, а затем с помощью ультразвуковой обработки при охлаждении. Стерильный продукт может быть получен посредством асептического производства или стерилизации на конечной стадии.
Подобный инъецируемый формируемый in situ депо-состав представляет собой альтернативный состав с замедленным высвобождением, более простой в изготовлении и применении, чем существующие продукты. По сравнению с существующими инъецируемыми формируемыми in situ депо-составами на полимерной основе, депо-состав по настоящему изобретению особенно полезен в составе «готовых к применению устройств», поскольку перед применением не требуется осуществить ресуспендирование. Депо-состав по настоящему изобретению является первым составом, который готов к инъецированию после приведения в равновесие при комнатной температуре. Размер иглы может быть подобран в зависимости от вязкости, причем возможно содержание лекарства до 10%, до 7-5% или до 5% в виде суспензии.
Депо-состав по настоящему изобретению обладает рядом полезных свойств: они характеризуются низким гемолитическим и токсическим потенциалом и демонстрируют хорошую местную переносимость в месте инъекции на кроликах. Системы с замедленным высвобождением согласно настоящему изобретению улучшают контактность и жизненный стандарт больного. Более того, способ изготовления прост и дешев и не требует никаких органических растворителей.
В одном из предпочтительных вариантов осуществления настоящего изобретения в способе изготовления состава по настоящему изобретению не используется никаких органических растворителей и, как следствие, никаких органических растворителей не присутствует в составе.
Депо-состав по настоящему изобретению может храниться, например, в заранее заполненном шприце или иных подходящих контейнерах, автоинъекторе, флаконе и шприце в течение длительного периода времени без седиментации при температуре ниже точки плавления растворителя, такого как, например, ПЭГ с концевыми группами.
Имплантат, формирующийся после инъекции в тело, может высвобождать активный агент в течение продолжительного периода времени. Желательный профиль высвобождения может зависеть от вида мономера и от того, задействован ли гомо- или сополимер, или же смесь полимеров. Период высвобождения может составлять от 1 до 12 недель, например от 1 до 8 недель, как то, например, 4 недели.
К раствору полимер-растворитель и/или к раствору полиэтиленгликоль-лекарственное вещество может быть необязательно добавлено вспомогательное вещество. Вспомогательное вещество может улучшать растворимость полимера и лекарственного вещества активного ингредиента. Сорастворитель может, кроме того, модулировать высвобождение лекарственного вещества in vitro или in vivo. Вспомогательное вещество может присутствовать в количестве от, приблизительно, 0,1% до, приблизительно 20% масс./об., предпочтительно от, приблизительно 1% до, приблизительно, 5%. Примеры подобных вспомогательных веществ включают метанол, этанол, пропиленгликоль, жидкое поверхностно-активное вещество, такое как сложные эфиры поли(оксиэтилен)сорбитана (Твин) или сложный глицеринполиоксиэтиленовый эфир -диметилацетамид, бензилбензоат, полиоксиэтилированную жирную кислоту, лецитин, соевое масло, подсолнечное масло, растительные масла, хлопковые масла, олигомеры поли(L-лактида), поли(D,L-лактида), сополимера лактида и гликолида или смеси таковых олигомеров.
Подробности в отношении подходящих наполнителей для применения в композициях или способе по настоящему изобретению описаны, например, в "Handbook of Pharmaceutical Excipients" (Справочник по фармацевтическим наполнителям), Rowe, Sheskey и Weller, 4-е издание, 2003.
Еще в одном варианте осуществления настоящего изобретения композиция, получаемая согласно способу по настоящему изобретению, может находиться в жидком виде при комнатной температуре, например, в виде раствора. После стерильного фильтрования через 0,22-микрометровый фильтр жидкая композиция, например, раствор, может быть помещена в шприц. Стерилизация может также быть осуществлена посредством другого способа конечной стерилизации гамма-излучением дозой от 20 до 30 кГр, предпочтительно, 25 кГр, в условиях охлаждения, например, при температуре от 2 до 8°C или -70°C. Стерилизованный раствор может быть инъецирован в организм подкожно или внутримышечно через иглу, например, вплоть до иглы 20G. После введения растворитель, например, полиэтиленгликоль, рассеется, а полимер совместно с фармацевтически активным веществом затвердеет и образует имплантат.В соответствии с изобретением, предпочтительно, заранее заполненный шприц может сопровождаться инструкцией по применению.
Композиции по настоящему изобретению применимы для лечения показаний, известных для конкретного активного вещества, внедренного в полимер, из числа показаний, описанных на странице 11 международной заявки на изобретение WO 02/010192. Предпочтительно, композиции по настоящему изобретению применимы для лечения акромегалии и рака, например, карциноидной опухоли, болезни Кушинга.
Активность и характеристики жидких композиций по настоящему изобретению могут быть выявлены в стандартных клинических испытаниях или испытаниях на животных. Подходящая дозировка композиции по настоящему изобретению будет, конечно, варьироваться в зависимости, например, от подвергаемого лечению состояния (например, типа заболевания или природы резистентности), задействуемого лекарственного средства, желаемого эффекта и способа введения.
Для композиций по настоящему изобретению, содержащих аналог соматостатина, достигнуты удовлетворительные результаты при введении, например, парэнтеральном введении, в дозировках порядка от, приблизительно, 0,2 до, приблизительно, 60 мг, предпочтительно, от, приблизительно, 5 до, приблизительно, 40 мг на инъекцию в месяц или от, приблизительно, 0,03 до, приблизительно, 1,2 мг на килограмм массы тела животного в месяц, осуществляя введение однократно или разделенными дозами. Подходящие месячные дозировки для пациентов имеют, таким образом, порядок от, приблизительно 0,3 мг до, приблизительно, 40 мг аналога соматостатина, например, памоата соединения II. Композиция может вводиться каждые 2-3 месяца. Подходящие дозировки для введения каждые 3 месяца составляют от, приблизительно, 1 мг до, приблизительно, 180 мг.
В соответствии с настоящим изобретением было обнаружено, что ПЭГ500 ДМЭ отличается особенно выгодными свойствами в качестве растворителя для парэнтеральных составов, например, низким гемолитическим потенциалом, подходящей для инъекций вязкостью, устойчивостью растворов ПЛГ в ПЭГ500 ДМЭ, благоприятной корреляцией между разделением фаз и высвобождением in vitro, а также низким начальным всплеском. Таким образом, еще в одном варианте осуществления настоящего изобретения его объектом является ПЭГ500 ДМЭ в качестве растворителя в фармацевтической композиции для парэнтерального применения, например, для активного вещества, описанного выше или в нижеприведенных примерах.
В одном из вариантов осуществления настоящего изобретения его объектом является фармацевтическая композиция для инъекции, включающая активное вещество и ПЭГ500 ДМЭ в качестве сорастворителя. Подобная композиция может содержать от 10% до 99,5%, от 20% до 90%, от 30% до 80% или от 50% до 99,5% (от общей массы композиции), или от 50% до 100% или от 60% до 99% ПЭГ500 ДМЭ, например, в водном растворе.
Нижеследующее является приводимым в качестве примера описанием только способов и композиций по настоящему изобретению.
Для того, чтобы подчеркнуть пригодность ПЭГ500 ДМЭ в качестве растворителя для парэнтерального применения, было проведено исследование гемолиза.
|
|
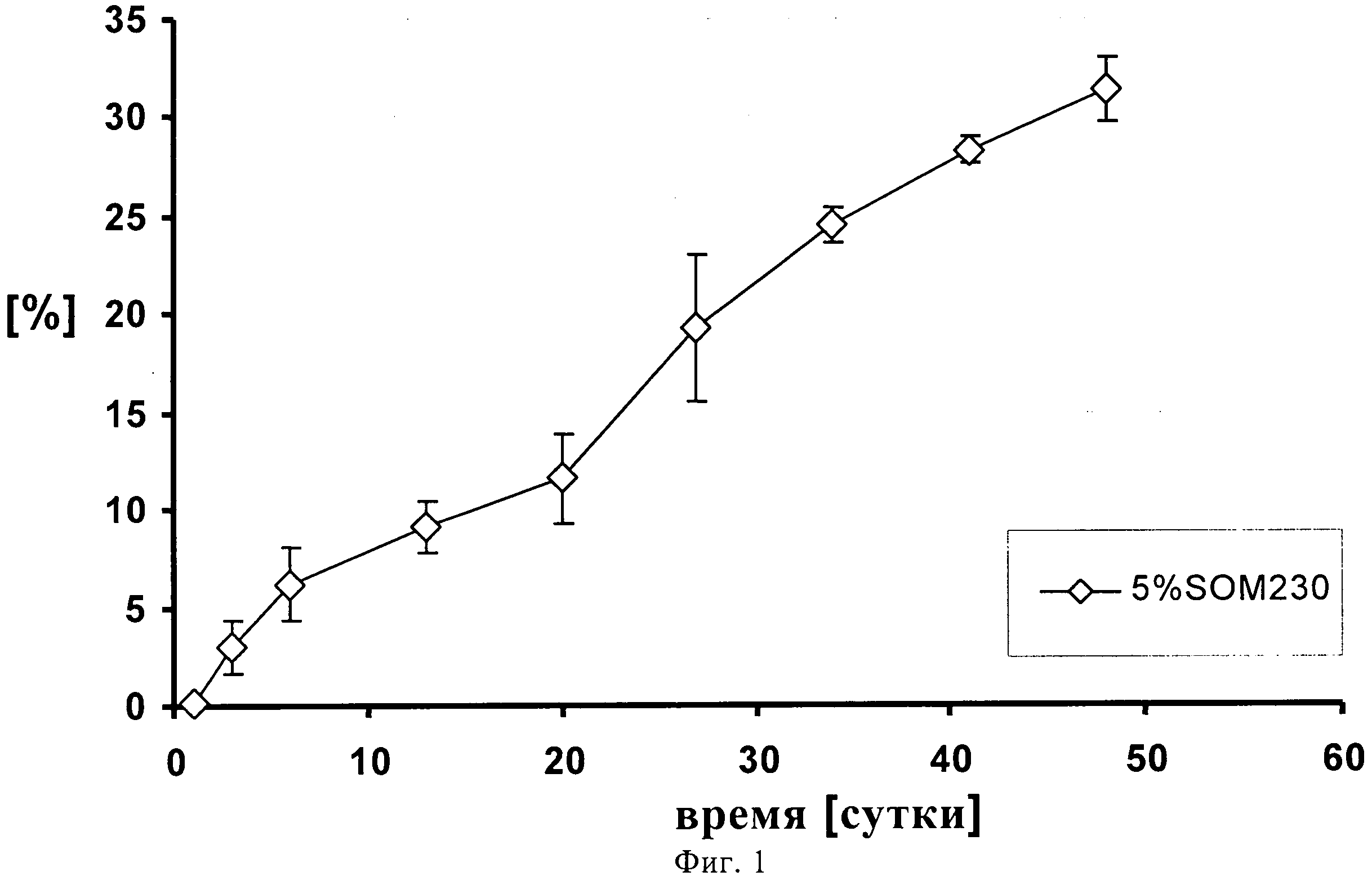
На фиг.1 представлено высвобождение in vitro 5% (по содержанию лекарственного средства) суспензии пасиреотида (SOM230) в течение 48 суток.
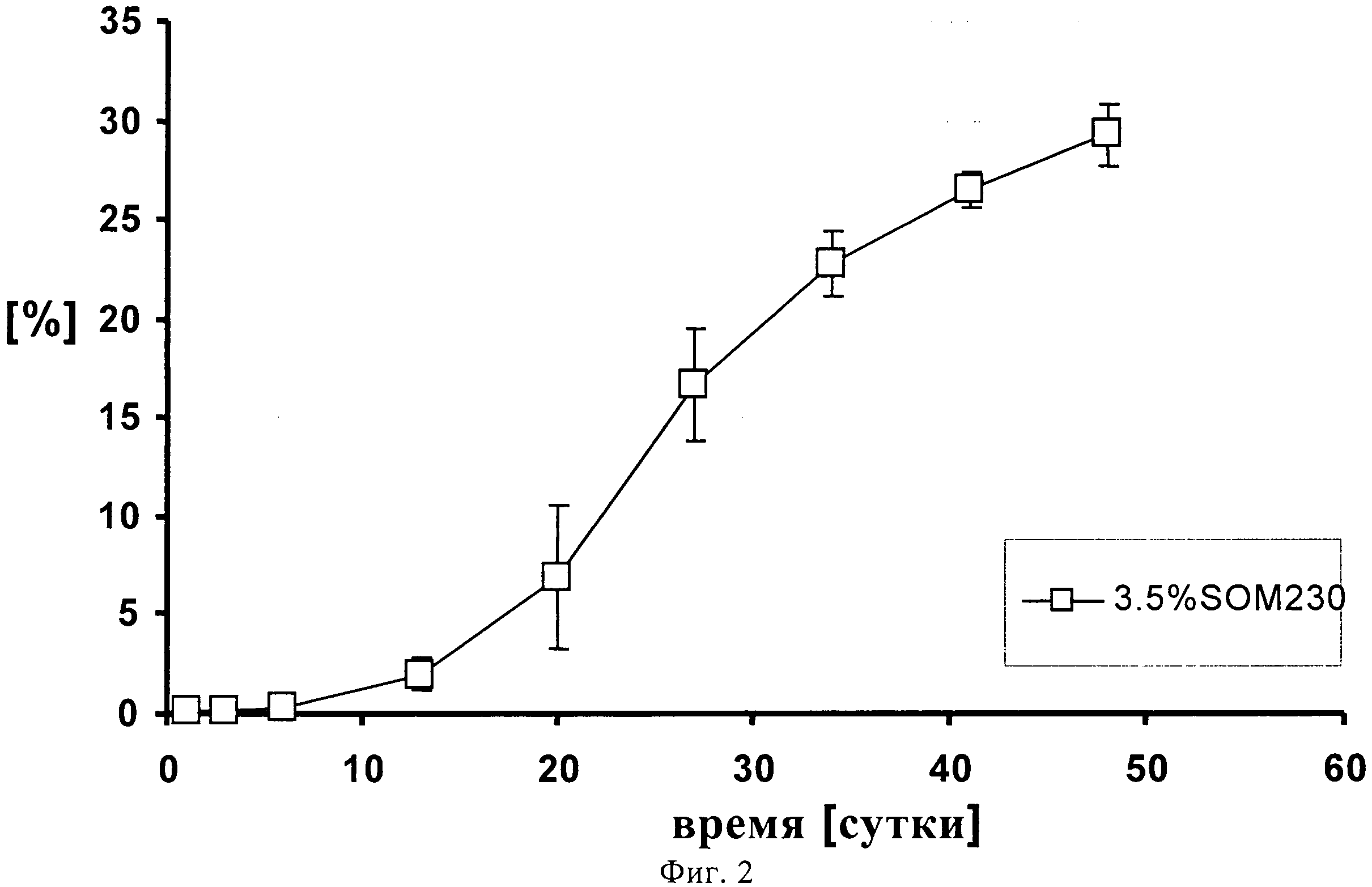
На фиг.2 представлен профиль высвобождения in vitro 3.5% (по содержанию лекарственного средства) суспензии SOM230 в течение 48 суток.
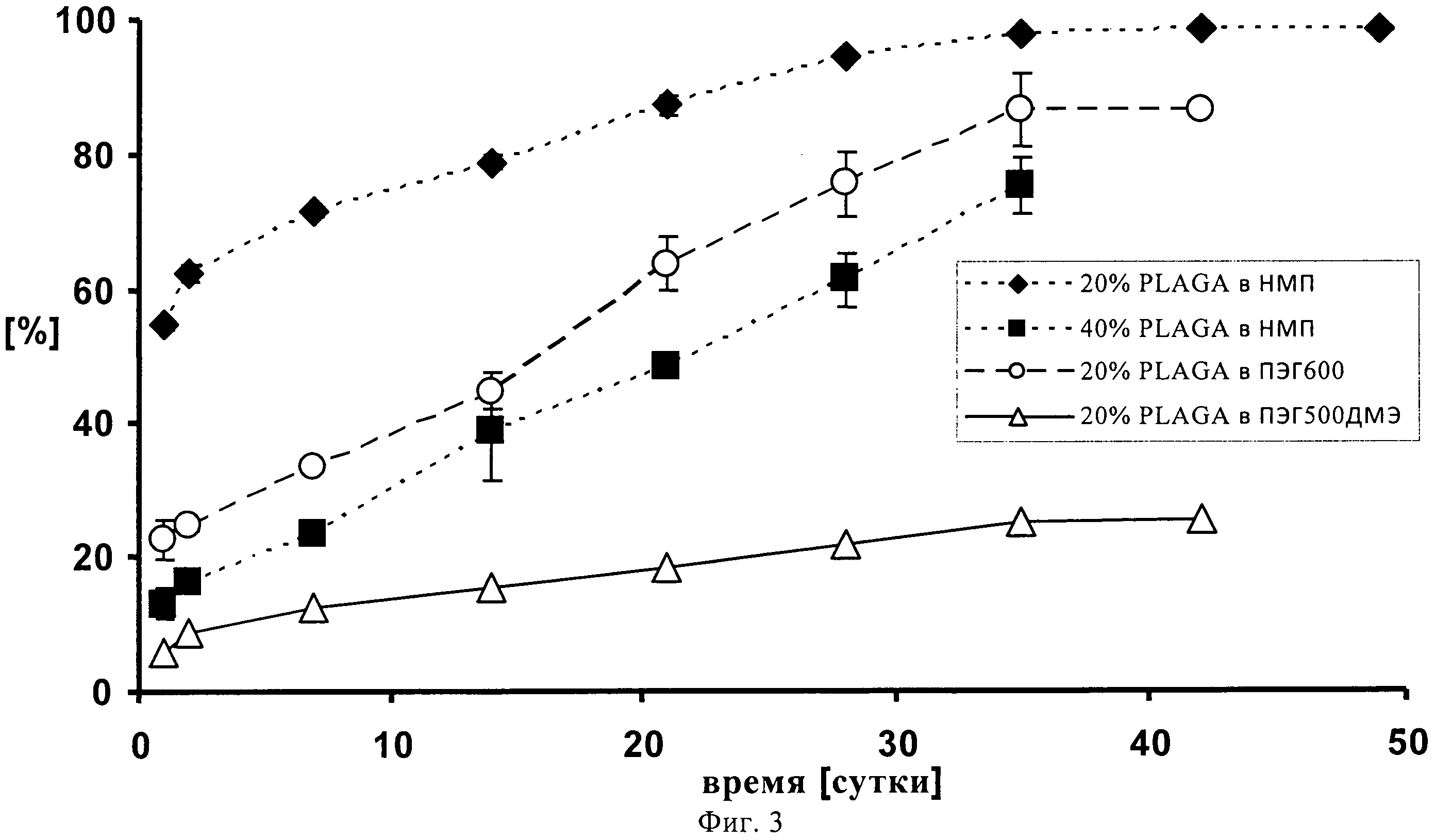
На фиг.3 представлено высвобождение метиленового синего из 4 составов, введенных с помощью 1 мл шприца и иглы 23G, где 20% раствор ПЛГ50:50 в ПЭГ500 ДМЭ демонстрирует очень низкий всплеск и постоянный уровень замедленно высвобождения в продолжение 49-суточного периода наблюдения.
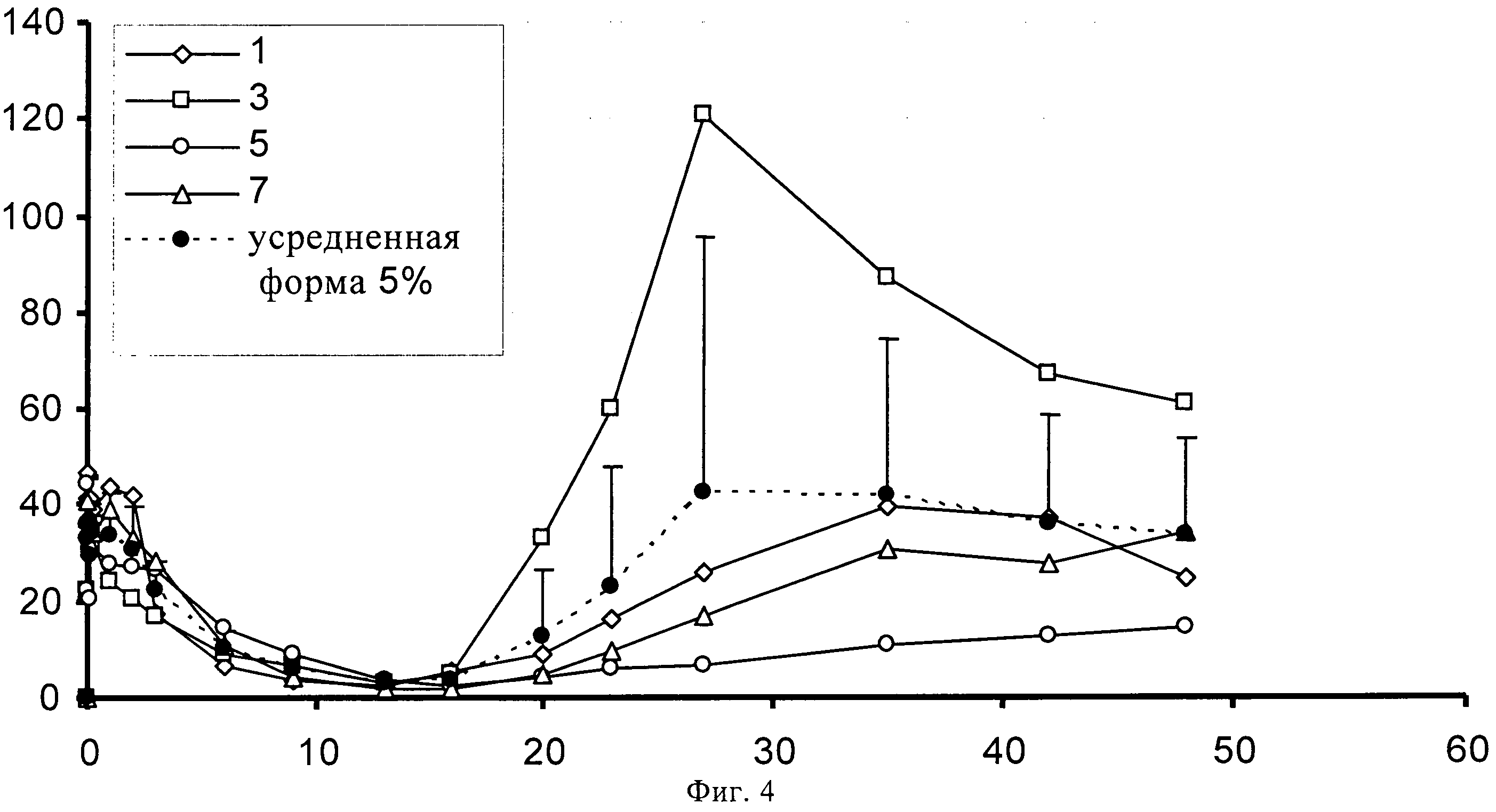
На фиг.4 представлено высвобождение in vivo 5% (по содержанию лекарственного средства) суспензии SOM230 в течение 48 суток на кроликах.
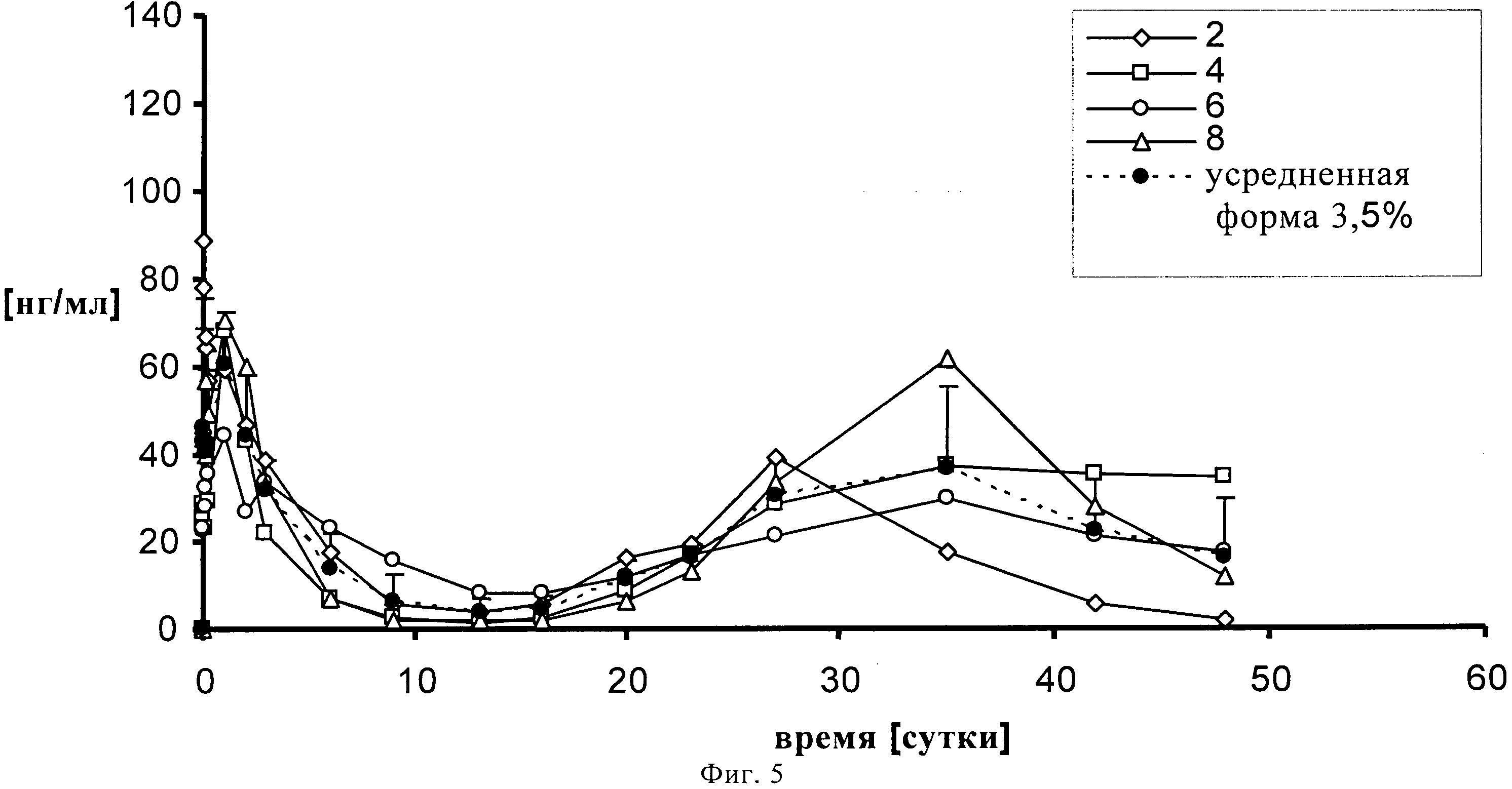
На фигуре 5 представлен профиль высвобождения in vivo на кроликах 3.5% (по содержанию лекарственного средства) суспензии SOM230 в течение 48 суток.
Пример 1
0,960 г сополимера D,L-лактида и гликолида 50:50 (20% масс./масс.) растворяют в 3,5062 г диметилового эфира полиэтиленгликоля 500. К раствору полимера после стерильного фильтрования (Millex GV, 0,22 микрометровый фильтр компании Millipore, Цуг, Швейцария) добавляют 0,3404 г подвергнутого гамма-облучению фармацевтически активного вещества памоата SOM230 (=0,250 г основания в свободной форме=5% масс/масс.) при давлении азота 1 бар и диспергируют его в растворе с помощью магнитной мешалки. Дисперсию подвергают ультразвуковой обработке дважды по 5 минут с помощью ультразвукового зонда (Hielscher UP400S, Ultrasound technology, Штуттгарт, Германия) до достижения среднего размера частиц примерно 51 микрометр (определенного с помощью зонда Lasentec, Mettler-Toledo, Грайфензее, Швейцария). 0,240 г состава (+избыток=0,333 г) помещают в 1 мл шприцы (BD, 1 мл шприц с наконечником Luer-Lok, Франклин Лейке, Нью-Джерси, США) с помощью иглы 22G (Sterican, 0,70×0,30 BL/LB, B.Braun, Мельзунген, Германия). Количество вводимого состава подбирают таким образом, чтобы оно примерно соответствовало 0,012 г лекарственного состава - основы. In situ депо-состав вводится непосредственно в ячейки диаметром 12 мм, заполненные 2 мл забуференного фосфатом солевого раствора (PBS), pH 7,4, которые помещают в прибор для проточного растворения USP 4 (Sotax, Алльшвиль, Швейцария) для исследования высвобождения in-vitro. Через прибор прокачивают буфер с рН 7,4 с помощью насоса Ismatec IP (Ismatec, Глаттбругг, Швейцария) при скорости потока 0,5 мл/ч. Пробы отбирают через 1, 3, 6, 13, 20, 27, 34, 41 и 48 суток и анализируют с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). На фигуре 1 продемонстрирован устойчивый профиль высвобождения in-vitro в течение 48 суток, соответствующий примерно 30% от теоретического содержания лекарственного средства с очень низким начальным высвобождением (всплеском).
Пример 2
1,00139 г сополимера D,L-лактида и гликолида 50:50 (20% масс./масс.) растворяют в 3,75521 г диметилового эфира полиэтиленгликоля. К раствору полимера после стерильного фильтрования (Millex GV, 0,22 микрометровый фильтр компании Millipore, Цуг, Швейцария) добавляют 0,2523 г подвергнутого гамма-облучению фармацевтически активного вещества памоата SOM230 (=0,180 г основания в свободной форме=3,5% масс./масс.) при давлении азота 1 бар и диспергируют его в растворе с помощью магнитной мешалки. Дисперсию подвергают ультразвуковой обработке дважды по 5 минут с помощью ультразвукового зонда (Hielscher UP400S, Ultrasound technology, Штуттгарт, Германия) до достижения среднего размера частиц примерно 54 микрометра (определенного с помощью зонда Lasentec, Mettler-Toledo, Грайфензее, Швейцария). 0,3301 г состава (+избыток=0,420 г) помещают в 1 мл шприцы (BD, 1 мл шприц с наконечником Luer-Lok, Франклин Лейке, Нью-Джерси, США) с помощью иглы 22G (Sterican, 0,70×0,30 BL/LB, B.Braun, Мельзунген, Германия). Количество вводимого состава подбирают таким образом, чтобы оно примерно соответствовало 0,012 г лекарственного состава - основы. In situ депо-состав вводится непосредственно в ячейки диаметром 12 мм, заполненные 2 мл забуференного фосфатом солевого раствора (PBS), рН 7,4, которые помещают в прибор для проточного растворения USP 4 (Sotax, Алльшвиль, Швейцария) для исследования высвобождения in-vitro. Через прибор прокачивают буфер с рН 7,4 с помощью насоса Ismatec IP при скорости потока 0,5 мл/ч. Пробы отбирают через 1, 3, 6, 13, 20, 27, 34, 41 и 48 суток и анализируют с помощью ВЭЖХ. На фигуре 2 продемонстрирован устойчивый профиль высвобождения in-vitro в течение 48 суток, соответствующий примерно 30% от теоретического содержания лекарственного средства с очень низким начальным высвобождением (всплеском).
Пример 3
0,048 г фармацевтически активного вещества (0,6% метиленового синего) добавляют к 4 растворам полимеров с различными растворителями. Первый раствор содержит 1,6002 г сополимера D,L-лактида и гликолида 50:50 (20% масс./масс.) и 6,4345 г диметилового эфира полиэтиленгликоля 500, второй раствор содержит 1,6004 г сополимера D,L-лактида и гликолида 50:50 (20% масс./масс), растворенного в 6,.4092 г N-метилпирролидина, третий раствор содержит 3,2006 г сополимера D,L-лактида и гликолида 50:50 (40% масс./масс.) в 4,818 г N-метилпирролидина, а последний раствор содержит 1,6012 г сополимера D,L-лактида и гликолида 50:50 (20% масс./масс.) в полиэтиленгликоле 600.
0,400-0,500 г растворов полимеров с добавками метиленового синего вводят (1 мл шприц (BD 1 мл с наконечником Luer-Lok, BD, Франклин Лейке, Нью-Джерси, США) с помощью иглы 23G (BD Microlance 3, 0,6×25 мм, BD, S.A., Фрага, Испания) в 50 мл полипропиленовые пробирки Falcon (BD, Франклин Лейке, США), содержащие 25 мл буфера PBS, рН 7,4. Пробирки инкубируют на водяной бане со встряхивателем (AD Krauth, Гамбург, Германия) при температуре 37°C и очень низкой частоте. При каждом отборе пробы осуществляют замену буфера. Пробы отбирают при t=0, 1, 3, 7, 14, 21, 28, 35, 42 и 49 суток и анализируют с помощью спектрофотометра Varian Сагу (Дармштадт, Германия) при длине волны 665 нм.
Пример 4
0,960 г сополимера D,L-лактида и гликолида 50:50 (20% масс./масс.) растворяют в 3,5062 г диметилового эфира полиэтиленгликоля 500. К раствору полимера после стерильного фильтрования (Millex GV, 0,22 микрометровый фильтр компании Millipore, Цуг, Швейцария) добавляют 0,3404 г подвергнутого гамма-облучению фармацевтически активного вещества памоата SOM230 (=0,250 г основания в свободной форме = 5% масс./масс.) при давлении азота 1 бар и диспергируют его в растворе с помощью магнитной мешалки. Дисперсию подвергают ультразвуковой обработке дважды по 5 минут с помощью ультразвукового зонда (Hielscher UP400S, Ultrasound technology, Штуттгарт, Германия) до достижения среднего размера частиц примерно 51 микрометр (определенного с помощью зонда Lasentec, Mettler-Toledo, Грайфензее, Швейцария). 0,240 г состава (+избыток=0,333 г) помещают в 1 мл шприцы (BD, 1 мл шприц с наконечником Luer-Lok, Франклин Лейке, Нью-Джерси, США) с помощью иглы 22G (Sterican, 0,70×0,30 BL/LB, В. Braun, Мельзунген, Германия). Количество вводимого состава подбирают таким образом, чтобы оно примерно соответствовало 0,012 г лекарственного состава-основы на каждое животное. Проводят испытание высвобождение лекарственного вещества памоата SOM230 на четырех кроликах. In situ депо-состав инъецируется каждому кролику подкожно в область шеи. Образцы крови (1-1,5 мл) забирают из ушной вены полипропиленовыми шприцами, содержащими 1, 6 мг калий-ЭДТК (этилендиаминтетрауксусной кислоты) (S-Monovette, Sarstedt AG, Зевелен, Швейцария). Пробы отбирают через 0 (перед введением), 30 минут, 1, 2, 4, 6 часов, 1, 2, 3, 6, 9, 13, 16, 20, 23, 27, 35, 42 и 48 суток. Образцы плазмы крови анализируют на содержание SOM230 с помощью конкурентного твердофазного иммуноферментного анализа.
Пример 5
1,00139 г сополимера D,L-лактида и гликолида 50:50 (20% масс./масс.) растворяют в 3,75521 г диметилового эфира полиэтиленгликоля 500. К раствору полимера после стерильного фильтрования (Millex GV, 0,22 микрометровый фильтр компании Millipore, Цуг, Швейцария) добавляют 0,2523 г подвергнутого гамма-облучению фармацевтически активного вещества памоата SOM230 (=0,250 г основания в свободной форме = 2,5% масс./масс.) при давлении азота 1 бар и диспергируют его в растворе с помощью магнитной мешалки. Дисперсию подвергают ультразвуковой обработке дважды по 5 минут с помощью ультразвукового зонда (Hielscher UP400S, Ultrasound technology, Штуттгарт, Германия) до достижения среднего размера частиц примерно 54 микрометра (определенного с помощью зонда Lasentec, Mettler-Toledo, Грайфензее, Швейцария). 0,3301 г состава (+избыток=0,420 г) помещают в 1 мл шприцы (BD, 1 мл шприц с наконечником Luer-Lok, Франклин Лейке, Нью-Джерси, США) с помощью иглы 22G (Sterican, 0,70×0,30 BL/LB, В. Braun, Мельзунген, Германия). Количество вводимого состава подбирают таким образом, чтобы оно примерно соответствовало 0,012 г лекарственного состава-основы на каждое животное. Проводят испытание высвобождение лекарственного вещества памоата SOM230 на четырех кроликах. In situ депо-состав инъецируется каждому кролику подкожно в область шеи. Образцы крови (1-1,5 мл) забирают из ушной вены полипропиленовыми шприцами, содержащими 1, 6 мг калий-ЭДТК (этилендиаминтетрауксусной кислоты) (S-Monovette, Sarstedt AG, Зевелен, Швейцария). Пробы отбирают через 0 (перед введением), 30 минут, 1, 2, 4, 6 часов, 1, 2, 3, 6, 9, 13, 16, 20, 23, 27, 35, 42 и 48 суток. Образцы плазмы крови анализируют на содержание SOM230 с помощью конкурентного твердофазного иммуноферментного анализа.
Пример 6
0,5 г циклоспорина A растворяют в 9,5 г диметилового эфира полиэтиленгликоля 500 с помощью магнитной мешалки. За время в пределах 3 часов получают прозрачный раствор.
Пример 7
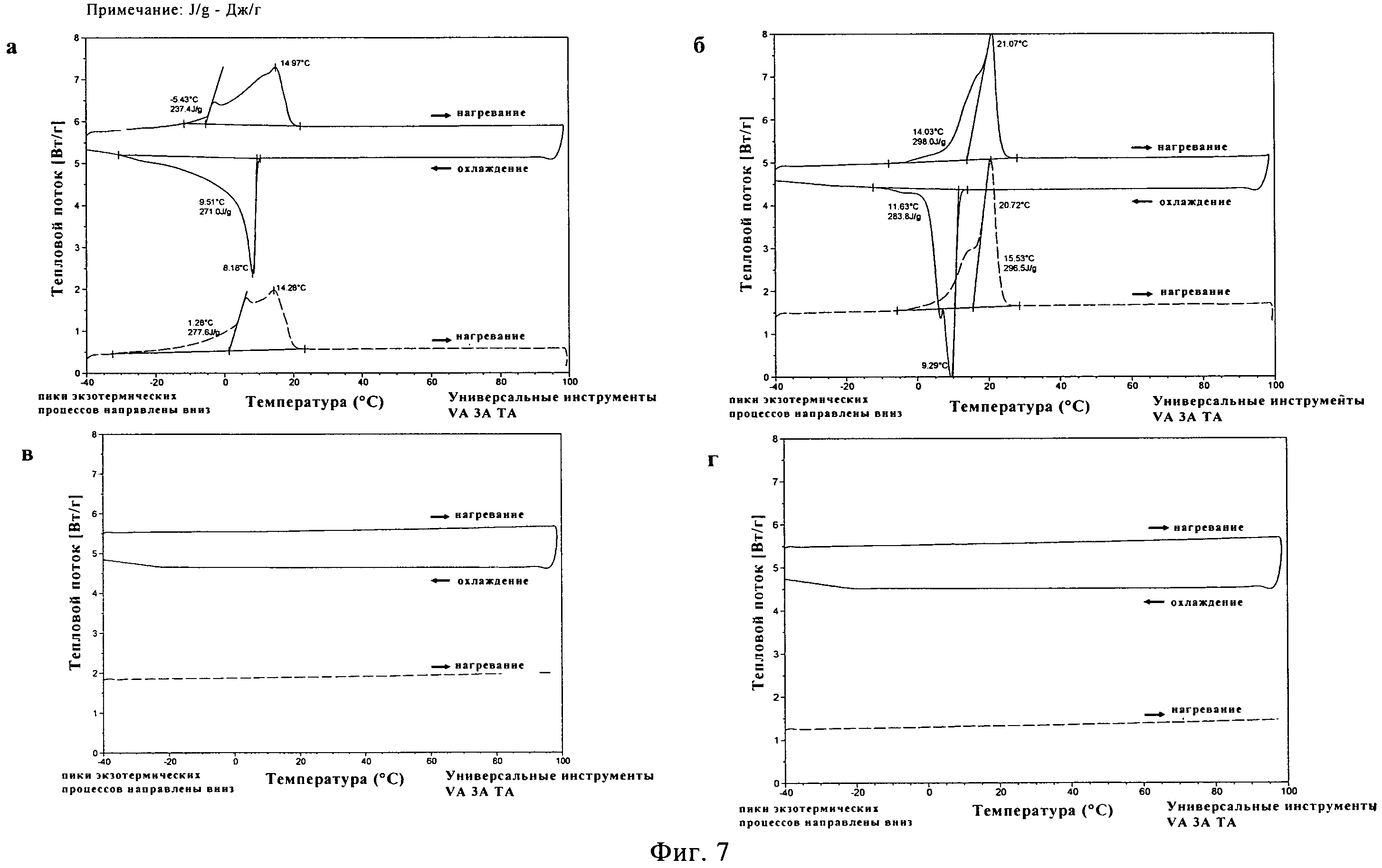
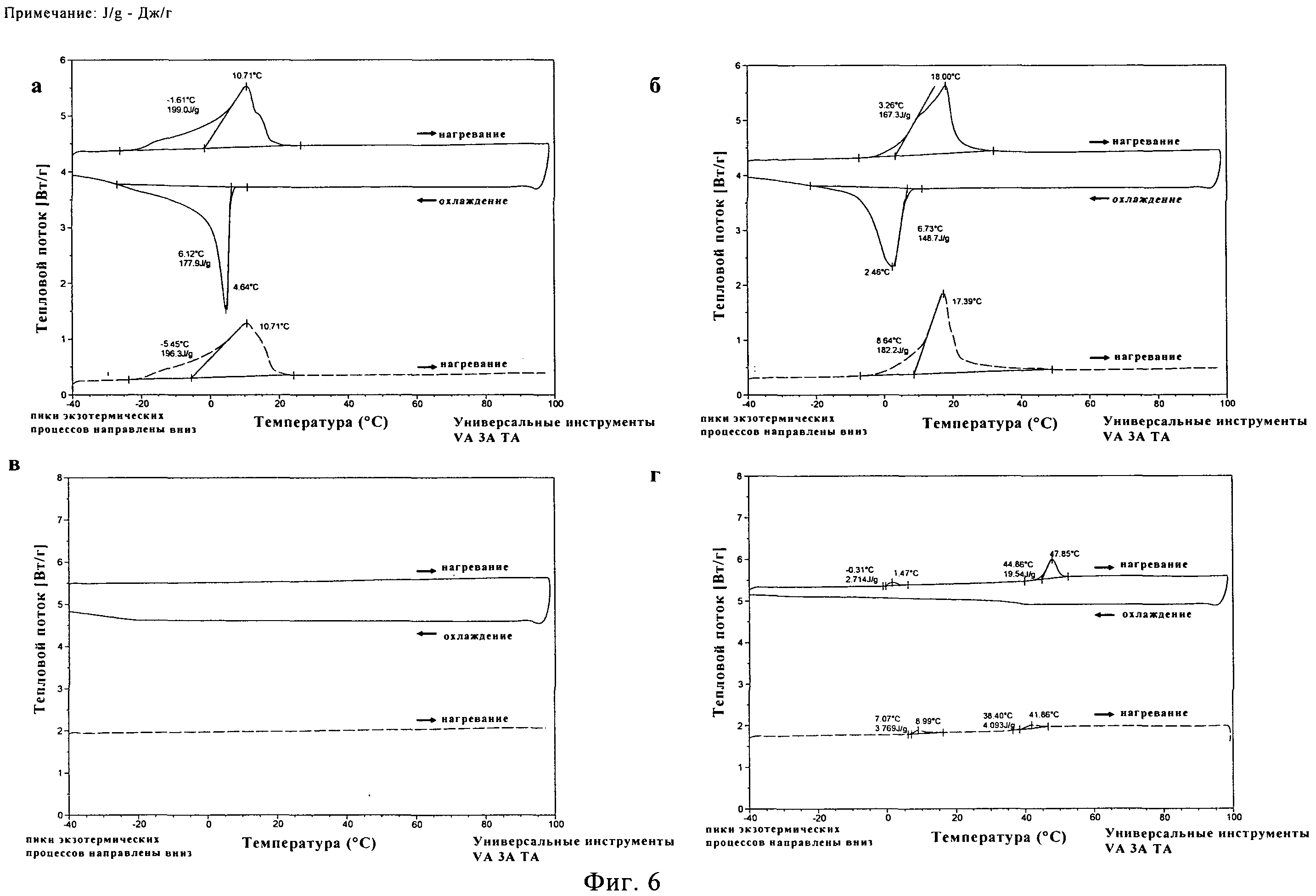
ПЭГ500ДМЭ, ПЭГ600 и растворы 20% (масс./масс.) сополимера лактида и гликолида PLA50GA5012 в упомянутых двух ПЭГ выдерживают при температуре -40°C в течение 0,5, 4 и 8 часов после первого цикла нагревания и охлаждения в дифференциальном сканирующем калориметре (ДСК). Во втором цикле нагревания у растворителей и растворов полимеров не наблюдают никаких различий в характеристиках плавления (данные не приводятся). Кристаллическая структура чистых ПЭГ, как кажется, не зависит от времени выдерживания. Для растворов полимеров не было обнаружено изменений в характеристиках плавления в зависимости от времени выдерживания. Это оказалось важным наблюдением с точки зрения долговременной устойчивости растворов PLA50GA5012, поскольку, как кажется, они способны сохранять свои свойства в ходе процесса охлаждения.
Температуры плавления составляют 14,7±0,4°C для ПЭГ500ДМЭ и 21,8±0,7°C для ПЭГ600 (фигуры 6а и б) по данным ДСК. На обеих термограммах появляются широкие сигналы для пика плавления на первом цикле нагревания и широкий сигнал для экзотермической кристаллизации во время цикла охлаждения. Термическое исследование НМП в том же диапазоне температур не выявило каких-либо переходов первого рода (фигура 6в). Термограмма для чистого PLA50GA5012 представлена на фигуре 6г. На первом цикле нагревания был обнаружен маленький эндотермический пик при температуре 0,31°C, вероятнее всего относящийся к воде. Наблюдалась эндотермическая релаксация полимерного порошка с эндотермическим пиком при температуре 47,9°C. В ходе последующего цикла охлаждения никакого изменения свойств не наблюдалось. На втором цикле нагревания был выявлен связанный со стеклообразным состоянием переход при температуре (Tg) 39,5±2,6°C.
Для 20% раствора (масс/масс.) PLA50GA50l2 в ПЭГ500ДМЭ наблюдалась депрессия температуры плавления до 10,9±0,4°C (фигура 7а) по сравнению с чистым растворителем. На первом цикле нагревания было заметно постепенное плавление, что привело к появлению широкого пика с несколькими плечами. Во время цикла охлаждения начало кристаллизации было отмечено при температуре 5,8±0,5°C, причем наблюдалось размытие пика. Во время второго цикла нагревания после выдерживания при температуре -40°C в течение получаса снова наблюдался широкий пик плавления. Во втором цикле нагревания сигнала от Tg PLA50GA5012 обнаружено не было. Для 20% раствора PLA50GA5012 в ПЭГ600 также было отмечено существенное понижение температуры плавления до 18,4±0,6°C (фигура 76). На термограмме наблюдался на первом цикле нагревания широкий пик плавления с двумя стадиями. Наблюдались широкие сигналы для пика кристаллизации в процессе охлаждения и пика на втором цикле нагревания при температуре 3,7±0,8°C. Для 20% и 40% растворов PLA50GA5012 в НМП в исследованном температурном диапазоне сигналов плавления, кристаллизации или Tg не наблюдалось (фигура 7в и г).
Пример 8
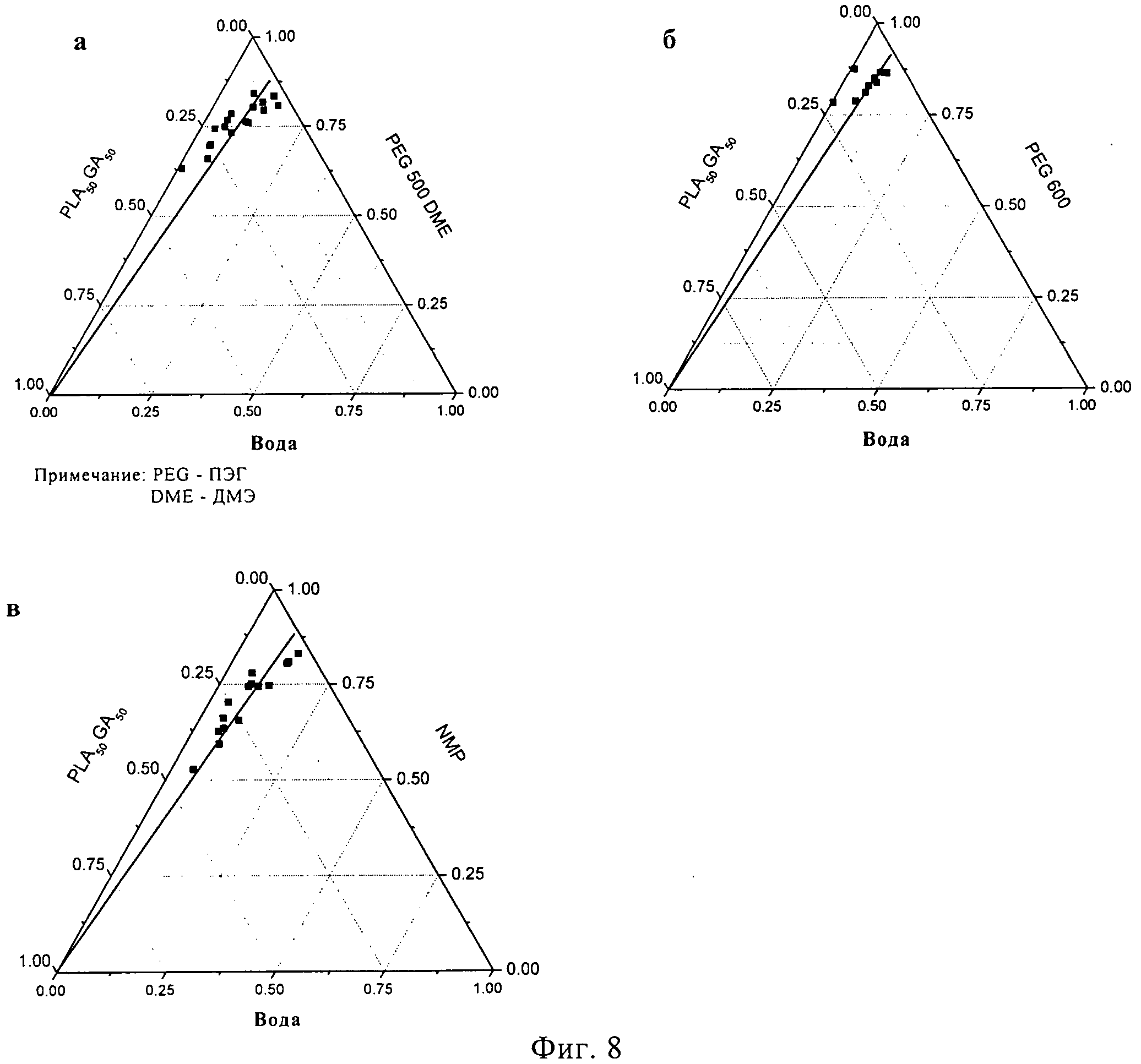
Растворы различных концентраций PLA50GA5012 в НМП, ПЭГ500ДМЭ и ПЭГ600 исследовались на начало осаждения полимера в присутствии воды при температуре 37°C (фигура 8). Прямая линия на диаграммах была проведена от 100% PLA50GA5012 и разделила поле на гомогенную трехкомпонентную систему в верхней части и двухфазную систему (разделение фаз) под линией. Начало осаждения полимера было определено для различных концентраций PLA50GA5012 в ПЭГ500ДМЭ посредством добавления смесей растворитель-вода к растворам полимера в органическом растворителе. Осаждение полимера происходило уже в присутствии 1,0% воды в случае 32,6% раствора PLA50GA5012 в ПЭГ500ДМЭ (фигура 8а), свидетельствуя, что лишь небольшое количество воды нужно для осаждения гидрофобного полимера из раствора. Для осаждения полимера из 3,0% раствора PLA50GA5012 в ПЭГ500ДМЭ потребовалось 13,4% воды. Как и ожидалось, при более низком содержании полимера оказывалось возможным добавление повышенного количества воды (7).
Тройная фазовая диаграмма при использовании в качестве растворителя ПЭГ600 (фигура 86) показывает, что для осаждения полимера с содержанием 24,8% нужно добавление лишь 0,3% воды. При низком содержании PLA50GA5012 (3,5%) нужно было добавить 9,2% воды, для того чтобы вызвать начало помутнения полимерного раствора. Растворы PLA50GA5012 в ПЭГ600 вели себя аналогично растворам в ПЭГ500ДМЭ, демонстрируя осаждение при повышенном содержании полимера и низком содержании воды. Соответственно, повышенная устойчивость к воде до начала осаждения наблюдалась при низких концентрациях полимера.
Тройная фазовая диаграмма PLA50GA5012 в НМП (фигура 8в) демонстрирует присутствие смешиваемой гомогенной системы вплоть до содержания 50% полимера в 50% растворителя (масс./масс). При содержании воды 4,7% мутность появилась в 45,6% растворе PLA50GA5012 в 49,6% НМП. Способность НМП к растворению PLA50GA5012 оказалась значительно повышенной по сравнению с ПЭГ500ДМЭ и ПЭГ600. Было необходимо добавить 14,1% воды к 2,9% раствору PLA50GA5012 в НМП, чтобы произошло осаждение. На тройной фазовой диаграмме для случая НМП в качестве растворителя поле смешиваемой гомогенной смеси над прямой линией увеличилось по сравнению с ПЭГ500ДМЭ или ПЭГ600. Способность к растворению PLA50GA5012 оказалась у ПЭГ500 ДМЭ и ПЭГ600 ниже, чем у НМП. В целом, начало осаждения PLA50GA5012 происходит в ПЭГ500ДМЭ и ПЭГ600 при более низких концентрациях, чем в НМП.
Мутантные антигены gas57 и антитела против gas57
[(1н-индол-5-ил)-гетероарилокси]-1-(азабицикло[3.3.1]нонаны, как холинергические лиганды n-achr, предназначенные для лечения психотических и нейродегенеративных нарушений
Лечение туберозного склероза
Органические соединения
Способ получения фармацевтической композиции
Предназначенная для перорального применения фармацевтическая композиция
Менингококковые полипептиды fhbp
Стабилизированные полипептиды инсулиноподобного фактора роста
Органические соединения
Курс лечения с использованием агониста рецептора s1p
Мутантные антигены gas57 и антитела против gas57
[(1н-индол-5-ил)-гетероарилокси]-1-(азабицикло[3.3.1]нонаны, как холинергические лиганды n-achr, предназначенные для лечения психотических и нейродегенеративных нарушений
Лечение туберозного склероза
Органические соединения
Способ получения фармацевтической композиции
Предназначенная для перорального применения фармацевтическая композиция
Менингококковые полипептиды fhbp
Стабилизированные полипептиды инсулиноподобного фактора роста
Органические соединения
Курс лечения с использованием агониста рецептора s1p

