Результат интеллектуальной деятельности: ГАЛЕНОВЫЙ СОСТАВ АЛИСКИРЕНА И ГИДРОХЛОРТИАЗИДА
Вид РИД
Изобретение
Настоящее изобретение относится к твердым лекарственным формам для перорального введения, включающим перорально активный ингибитор ренина, алискирен, или его фармацевтически приемлемую соль и гидрохлортиазид, в качестве активных ингредиентов в пригодном носителе. Прежде всего, в настоящем изобретении предлагаются галеновые составы, содержащие полуфумарат алискирена в комбинации с гидрохлортиазидом. Настоящее изобретение относится также к способам получения таких составов и их применению в качестве лекарственных средств.
Следует понимать, что термин "алискирен", использованный в данном контексте, означает, если не указано иное, свободное основание и его соль, прежде всего, его фармацевтически приемлемую соль, наиболее предпочтительно полуфумарат.
Ренин, секретируемый почками в кровоток, расщепляет ангиотензиноген с образованием декапептида, ангиотензина I, который в свою очередь в легких, почках и других органах подвергается дальнейшему расщеплению ангиотензин-конвертирующим ферментом с образованием октапептида, ангиотензина II. Октапептид увеличивает кровяное давление непосредственно за счет артериальной вазоконстрикции и опосредованно за счет высвобождения из надпочечников натрий-удерживающего гормона, альдостерона, что сопровождается увеличением объема внеклеточной жидкости. В присутствии ингибиторов ферментативной активности ренина наблюдается снижение количества ангиотензина I, и соответственно продуцирование меньшего количества ангиотензина II. Более низкая концентрация указанного активного пептидного гормона является непосредственной причиной, например, антигипертензивного действия ингибиторов ренина. Соответственно ингибиторы ренина или их соли можно использовать, например, в качестве антигипертензивных лекарственных средств или для лечения застойной сердечной недостаточности.
Известно, что ингибитор ренина алискирен, прежде всего, в форме полуфумарата, является эффективным лекарственным средством, которое снижает кровяное давление независимо от возраста, пола или расы пациента, а также характеризуется достаточно высокой переносимостью. Алискирен в форме свободного основания, характеризуется структурной формулой
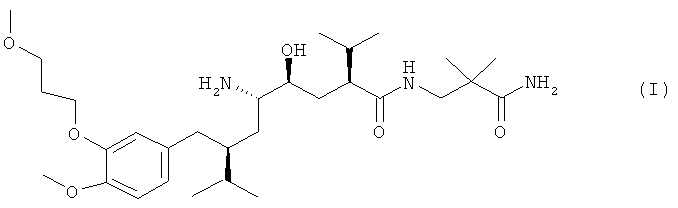
химическое название 2(8), 4(8), 5(8), 7(8)-N-(3-амино-2,2-диметил-3-оксопропил)-2,7-ди(1-метилэтил)-4-гидрокси-5-амино-8-[4-метокси-3-(3-метоксипропокси)фенил]октанамид. Как описано выше, наиболее предпочтительной солью является полуфумарат, который подробно описан в патенте ЕР 678503 А в примере 83.
Гидрохлортиазид является известным диуретиком и комбинация с алискиреном описана, например, в заявке WO 02/40007.
Пероральное введение указанных фармацевтических агентов в виде таблеток или капсул имеет определенные преимущества по сравнению с парентеральным введением, таким как внутривенное или внутримышечное. Заболевания, для лечения которых требуется использовать болезненные инъекции, считаются более серьезными, чем заболевания, которые можно лечить пероральными лекарственными средствами. Однако, основное преимущество составов для перорального введения заключается в возможности их самостоятельного введения пациентом, тогда как в большинстве случаев составы для парентерального введения должны вводиться врачом или медсестрой.
Однако, при переработке алискирена в лекарственные средства возникают значительные трудности и получение состава для перорального введения в форме таблеток надежным и возпроизводимым способом не представляется возможным. Обычно при получении галеновых составов, включающих алискирен или его фармацевтически приемлемую соль, требуется высокое содержание лекарственной субстанции (ЛС), что создает проблемы при формировании таблеток.
Например, алискирен образует иглообразные кристаллы, что отрицательно влияет на объемные свойства ЛС, например, на текучесть и объемную плотность. ЛС характеризуется нестабильностью при прессовании, что приводит к ослаблению связей между частицами и изменению полиморфизма под давлением. Алискирен содержит высокоэластичный компонент, что также приводит к ослаблению связей между частицами. В связи с высокой дозой (от 300 до 600 мг свободного основания в таблетке) возникает необходимость в высоком содержании ЛС при получении таблеток приемлемого размера.
Качество ЛС значительно изменяется и влияет на условия переработки ЛС в таблетки, например, распределение частиц по размеру, объемная плотность, текучесть, свойства при увлажнении, площадь поверхности и адгезивные свойства. Кроме того, алискирен является высокогигроскопичным. При контактировании с водой и удалении воды полиморфные формы ЛС превращаются в аморфные формы, что снижает стабильность по сравнению с кристаллической формой. Комбинация этих ограничений приводит к возникновению значительных трудностей при получении стандартных таблеток. Твердая лекарственная форма алискирена описана в заявке WO 2005/089729.
Трудности, возникающие при получении состава алискирена в форме таблеток надежным и воспроизводимым способом, можно преодолеть при его использовании в комбинации с другими терапевтическими агентами, прежде всего гидрохлортиазидом. Прямое прессование нельзя использовать для стандартной переработки алискирена, что связано, например, с высокой гигроскопичностью, иглообразной формы кристаллов, низкой текучестью, вследствие чего возникают проблемы в процессе переработки алискирена и получении однородного распределения в лекарственной форме. Способ ротационного прессования приводит к снижению насыпного объема ЛС. Более того, предварительное прессование ЛС в процессе ротационного прессования приводит к значительным трудностям и не позволяет при дальнейшем прессовании получать таблетки с достаточной твердостью и низкой хрупкостью в отсутствие высокого содержания эксципиентов, что связано с низкой способностью ЛС к прессованию. Алискирен под давлением претерпевает полиморфные изменения, переходя в более аморфное состояние, например, при прессовании. Ротационное прессование приводит к образованию стеклообразного материала с низкой сжимаемостью и непригодным профилем высвобождения. Следовательно, в данном случае нельзя использовать способы, разработанные, например, для состава, содержащего гидрохлортиазид и валсартан, как описано в заявке WO 97/49394, включающие прессование.
Кроме того, при использовании состава, содержащего алискирен и гидрохлортиазид, необходимо контролировать скорости растворения обоих терапевтических агентов в допустимом интервале, а также необходимо обеспечивать баланс между указанными скоростями растворения и достаточными твердостью и хрупкостью.
Было установлено, что для получения состава по настоящему изобретению ряд способов являются непригодными. Кроме упомянутой выше непригодности способа прессования было установлено, что влажная грануляция алискирена и гидрохлортиазида также является непригодной из-за слишком медленной скорости высвобождения лекарственного средства после хранения. Различные способы получения состава, содержащего алискирен во внутренней фазе и гидрохлортиазид во внешней фазе, также оказались неэффективными из-за низкой текучести или низких скоростей растворения.
Следовательно, существует необходимость в разработке пригодного и устойчивого галенового состава, который позволяет преодолеть упомянутые выше проблемы, в отношении свойств алискирена, прежде всего состава алискирена в смеси с гидрохлортиазидом.
В настоящем изобретении решены упомянутые выше проблемы и предлагается устойчивый состав, который позволяет исключить указанные выше недостатки, и предлагается способ, пригодный для крупномасштабного получения твердых лекарственных форм для перорального введения.
Настоящее изобретение относится к твердой пероральной лекарственной форме, которая включает
a) терапевтически эффективное количество алискирена или его фармацевтически приемлемой соли,
b) терапевтически эффективное количество гидрохлортиазида, и
c) гидрофильный наполнитель, выбранный из углеводов или их комбинаций.
Неожиданно было установлено, что гидрофильный наполнитель оказывает положительное влияние на время распадаемости и, таким образом, на скорости растворения терапевтических агентов. Такие результаты оказались тем более неожиданными, так как обычно наполнитель придает материалу высокие характеристики текучести и прессования. Благодаря указанным эффектам можно снизить количество дезинтегрирующего агента. Хотя для достижения высокой скорости растворения требуется относительно высокое содержание дезинтегрирующего агента, недостаток такого высокого содержания проявляется в дальнейшем при нанесении водного покрытия на ядра таблетки. Следовательно, если такое относительно высокое количество дезинтегрирующего агента можно снизить за счет добавления гидрофильного наполнителя, то полученная твердая лекарственная форма для перорального введения является более устойчивой по сравнению с известными лекарственными формами.
В предпочтительном варианте осуществления настоящего изобретения компонент (а) присутствует в количестве от 25 мас.% до 47 мас.% в расчете на общую массу твердой пероральной лекарственной формы.
В другом предпочтительном варианте компонент (а) присутствует в количестве от 26 мас.% до 46 мас.%, предпочтительно от 28 мас.% до 44 мас.% в расчете на общую массу твердой пероральной лекарственной формы.
Предпочтительно компонент (а) присутствует в количестве от приблизительно 75 мг до приблизительно 600 мг свободного основания в стандартной лекарственной форме.
В предпочтительном варианте компонент (а) присутствует в количестве от приблизительно 75 мг до приблизительно 300 мг свободного основания в стандартной лекарственной форме, прежде всего в количестве 75, 150 или 300 мг.
В другом предпочтительном варианте алискирен присутствует в форме его полуфумарата в количестве приблизительно 83 мг, приблизительно 166 мг, приблизительно 332 мг или приблизительно 663 мг в стандартной лекарственной форме.
В предпочтительном варианте изобретения компонент Ь) присутствует в количестве от 0,5 мас.% до 10 мас.%, например, от 1 мас.% до 6 мас.% в расчете на общую массу твердой лекарственной формы для перорального введения.
В другом предпочтительном варианте компонент Ь) присутствует в количестве от 1,4 мас.% до 5,5 мас.%, предпочтительно от 1,5 мас.% до 1,8 мас.%, от 2,7 мас.% до 3,1 мас.% или от 4,6 мас.% до 5,0 мас.% в расчете на общую массу твердой пероральной лекарственной формы.
Предпочтительно компонент b) присутствует в количестве от приблизительно 6 мг до приблизительно 30 мг в стандартной лекарственной форме.
В предпочтительном варианте компонент b) присутствует в количестве от приблизительно 12,5 мг до приблизительно 25 мг в стандартной лекарственной форме, прежде всего в количестве 12,5 или 25 мг.
В предпочтительном варианте компонент с) присутствует в количестве от 3 мас.% до 30 мас.% в расчете на общую массу твердой пероральной лекарственной формы.
В другом предпочтительном варианте компонент с) присутствует в количестве от 5 мас.% до 25 мас.%, предпочтительно от 5,5 мас.% до 7 мас.%, от 10 мас.% до 13 мас.% или от 18 мас.% до 21 мас.% в расчете на общую массу твердой пероральной лекарственной формы.
Предпочтительно компонент с) присутствует в количестве от приблизительно 30 мг до приблизительно 150 мг в стандартной лекарственной форме.
В предпочтительном варианте компонент с) присутствует в количестве от приблизительно 50 мг до приблизительно 100 мг в стандартной лекарственной форме, прежде всего в количестве 50 или 100 мг.
Предпочтительные примеры углеводов, используемых в качестве гидрофильного наполнителя, включают сахара, альдиты и крахмалы или их комбинации, прежде всего кондитерский сахар, прессованный сахар, декстраты, декстрин, декстрозу, лактозу, маннит, сорбит, сахарозу, крахмал, такой как кукурузный крахмал, картофельный крахмал или пшеничный крахмал.
Наиболее предпочтительными являются лактоза и крахмал, такой как пшеничный крахмал, которые используются каждый в отдельности или в смеси. Предпочтительной является смесь 2:1, 1:2 или 1:1, наиболее предпочтительной является смесь 1:1.
Массовое соотношение компонента а) и компонента b) предпочтительно находится в интервале от приблизительно 4:1 до приблизительно 30:1, более предпочтительно от приблизительно 6:1 до приблизительно 24:1. Наиболее предпочтительно массовое соотношение составляет приблизительно 6:1, 12:1 или 24:1 в расчете на свободную кислоту. При использовании соли, такой как полуфумарат, соотношение соответственно пересчитывают. В приведенных ниже соотношениях числа, соответствующие компоненту а), относятся к свободной кислоте или соли, прежде всего полуфумарату.
Массовое соотношение компонента а) и компонента с) предпочтительно находится в интервале от приблизительно 1:1 до приблизительно 10:1, более предпочтительно от приблизительно 1,2:1 до приблизительно 8:1. Наиболее предпочтительно массовое соотношение составляет приблизительно 1,5-7:1, например, 1,5-1,7:1, 6,0-6,8:1 или 3,0-3,4:1
Массовое соотношение компонента b) и компонента с) предпочтительно находится в интервале от приблизительно 0,1:1 до приблизительно 0,6:1, более предпочтительно от приблизительно 0,2:1 до приблизительно 0,3:1. Наиболее предпочтительно массовое соотношение составляет приблизительно 0,25:1.
Твердые пероральные лекарственные формы по настоящему изобретению характеризуются требуемы профилем высвобождения лекарственного средства (ВЛ). Предпочтительно ВЛ для компонента а) через 45 мин составляет по крайней мере 75%, более предпочтительно по крайней мере 80%. Предпочтительно ВЛ через 60 мин для компонента Ь) составляет по крайней мере 75%, более предпочтительно по крайней мере 80%. ВЛ определяют по стандартной методике, см. модифицированную методику, описанную в фармакопее USP<711>, ЕР 2.9.3 и JP.
Твердые пероральные лекарственные формы по настоящему изобретению также характеризуются низкой хрупкостью. Предпочтительно хрупкость составляет не более 0,8%, более предпочтительно не более 0,4%. Хрупкость определяют по стандартной методике см. модифицированную методику, описанную в фармакопее USP<1216>, ЕР 2.9.7 и JP.
Твердые пероральные лекарственные формы по настоящему изобретению также характеризуются требуемой распадаемостью. Предпочтительно время распадаемости составляет не более 40 мин, более предпочтительно не более 30 мин, например, менее 27 мин. Время распадаемости определяют по стандартной методике, см. модифицированную методику, описанную в фармакопее USP<701>, ЕР 2.9.1 и JP.
Твердые пероральные лекарственные формы по настоящему изобретению также характеризуются достаточной твердостью. Предпочтительно твердость должна обеспечивать низкую хрупкость, стабильность при нанесении покрытия, желательно короткое время распадаемости и соответственно высокую скорость растворения. Конкретная величина твердости зависит от размера твердой лекарственной формы для перорального введения. Для твердой пероральной лекарственной формы, содержащей 75 мг алискирена в расчете на свободную кислоту, предпочтительный интервал твердости составляет от 65 до 140 Н, более предпочтительно от 70 до 130 Н, например, от 73 до 125 Н. Для твердой пероральной лекарственной формы, содержащей 150 мг алискирена в расчете на свободную кислоту, предпочтительный интервал твердости составляет от 150 до 240 Н, более предпочтительно от 155 до 225 Н, например, от 160 до 220 Н. Для твердой пероральной лекарственной формы, содержащей 300 мг алискирена в расчете на свободную кислоту, предпочтительный интервал твердости составляет от 160 до 270 Н, более предпочтительно от 175 до 260 Н, например, от 180 до 250 Н. Указанные величины твердости относятся к твердости ядра твердой пероральной лекарственной формы. Твердость измеряют стандартными способами, используя, например, обрудование фирм Erweka и Pharmatest.
Твердые пероральные лекарственные формы по настоящему изобретению предназначены для введения активного ингредиента в пероральной форме относительно малого размера достаточной твердости и с коротким временем распадания. Кроме того, лекарственные пероральные формы проявляют стабильность как в процессе получения, так и при хранении, например, в течение 2 лет в соответствующей упаковке, например, в запечатанном алюминиевом блистере.
Термины «эффективное количество» или «терапевтически эффективное количество» относится к количеству активного ингредиента или агента, при введении которого наблюдается приостановка или замедление развития состояния, подлежащего лечению, или наблюдается полное или частичное излечение или временно ослабляется интенсивность симптомов заболевания.
Алискирен или его фармацевтически приемлемую соль можно получить известным методом, прежде всего, как описано в ЕР 678503 А, например, в примере 83.
Гидрохлортиазид является известным терапевтическим агентом, который используют при лечении гипертензии.
Твердая пероральная лекарственная форма включает капсулу или более предпочтительно таблетку или таблетку с пленочным покрытием.
Согласно изобретению твердая пероральная лекарственная форма включает добавки или эксципиенты, пригодные для получения твердой пероральной лекарственной формы по настоящему изобретению. Можно применять различные вспомогательные вещества для таблетирования, которые обычно используют в композициях для таблеток, описанные в многочисленных публикациях, см., например, справочник Fiedler "Lexicon der Hilfstoffe", 4-е изд., ECV Aulendorf(1996), содержание которого включено в настоящее описание в качестве ссылки. Указанные средства включают, без ограничения перечисленным, наполнители, связующие агенты, дезинтегрирующие агенты, замасливатели, глиданты, стабилизаторы или разбавители, ПАВ, пленкообразующие агенты, смягчающие агенты, пигменты и т.п.
В предпочтительном варианте осуществления настоящего изобретения твердая пероральная лекарственная форма в качестве добавки включает дополнительный наполнитель кроме компонента с).
В предпочтительном варианте твердая пероральная лекарственная форма в качестве добавки включает дезинтегрирующий агент кроме дополнительного наполнителя.
В предпочтительном варианте твердая пероральная лекарственная форма включает в качестве добавки замасливатель, кроме дополнительного наполнителя и дезинтегрирующего агента.
В предпочтительном варианте твердая пероральная лекарственная форма включает в качестве добавки глидант, кроме наполнителя, дезинтегрирующего агента и замасливателя.
В предпочтительном варианте твердая пероральная лекарственная форма включает в качестве добавки связующий агент, кроме наполнителя, дезинтегрирующего агента, замасливателя и глиданта.
В качестве наполнителей, прежде всего, можно использовать целлюлозы, например, гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилметилцеллюлозу (ГПМЦ) и, предпочтительно, микрокристаллическую целлюлозу, например, продукты под торговыми названиями AVICEL, FILTRAK, HEWETEN или PHARMACEL.
В качестве связующих агентов, используемых при влажной грануляции, прежде всего, можно использовать поливинилпирролидоны (ПВП), например, ПВП К 30, ГПМЦ, например, препараты ГПМЦ, характеризующиеся вязкостью 3 сП или 6 сП, а также полиэтиленгликоли (ПЭГ), например, ПЭГ 4000. Наиболее предпочтительным связующим агентом является ПВП К 30.
В качестве дезинтегрирующих агентов, прежде всего, можно использовать кальциевую соль карбоксиметилцеллюлозы (КМЦ-Са), натриевую соль карбоксиметилцеллюлозы (КМЦ-Na), сшитый ПВП (например, кросповидон, полипласдон или коллидон XL), альгиновую кислоту, альгинат натрия и гуаровую камедь, наиболее предпочтительными являются сшитый ПВП (кросповидон), сшитая КМЦ (Ac-Di-Sol), натриевая соль карбоксиметилкрахмала (продукт под торговым названием P1RIMOJEL и EXPLOTAB). Наиболее предпочтительным дезинтегрирующим агентом является кросповидон.
В качестве глидантов, прежде всего, можно использовать коллоидный оксид кремния, такой как коллоидный диоксид кремния, например, аэросил, трисиликат магния (Mg), порошкообразную целлюлозу, крахмал, тальк, тригидрофосфат кальция или можно использовать комбинации указанных препаратов и наполнителей или связующих агентов, например, силикатированная микрокристаллическая целлюлоза (продукт под торговым названием PROSOLV). Наиболее предпочтительным глидантом является коллоидный диоксид кремния (например, аэросил 200), а также тальк.
В качестве замасливателей можно использовать стеарат магния, алюминия (А1) или стеарат кальция, ПЭГ 4000 - ПЭГ 8000 и тальк, гидрированное касторовое масло, стеариновую кислоту и ее соли, сложные эфиры глицерина, Na-стеарилфумарат, гидрированное хлопковое масло и т.п. Наиболее предпочтительным замасливателем является стеарат магния.
Добавки, которые можно использовать в качестве материалов для нанесения пленочных покрытий, включают различные полимеры, такие как ГПМЦ, ПЭГ, ПВП, сополимер поливинилпирролидона и винилацетата (ПВП-ВА), поливиниловый спирт (ПВА), а также сахар. Наиболее предпочтительным материалом для нанесения покрытий является ГПМЦ, прежде всего, препараты ГПМЦ, которые характеризуются вязкостью 3 сП (предпочтительное количество 5-6 мг/см), и их смеси с другими добавками, например, продуктом под торговым названием OPADRY. Другие добавки включают пигменты, красители, глазури, прежде всего, TiO2 и оксиды железа, агенты, снижающие липкость, например, тальк и смягчающие агенты, ПЭГ 3350, 4000, 6000, 8000 или т.п. Наиболее предпочтительными добавками являются тальк и ПЭГ 4000.
Настоящее изобретение относится к твердой пероральной лекарственной форме, которая включает в качестве активного агента терапевтически эффективное количество алискирена или его фармацевтически приемлемой соли, терапевтически эффективное количество гидрохлортиазида, и гидрофильный наполнитель, выбранный из углеводов или их комбинаций. Другие добавки включают, без ограничения перечисленным, дополнительные наполнители, связующие агенты, дезинтегрирующие агенты, замасливатели, глиданты, стабилизаторы, разбавители, ПАВ, пленкообразующие агенты, пигменты, смягчающие агенты, агенты, снижающие липкость и т.п.Предпочтительное количество активного ингредиента и добавок описано в данном контексте.
Настоящее изобретение относится к твердой пероральной лекарственной форме, которая включает терапевтически эффективное количество алискирена или его фармацевтически приемлемой соли, терапевтически эффективное количество гидрохлортиазида, и гидрофильный наполнитель, выбранный из углеводов или их комбинаций, а также дезинтегрирующий агент в качестве добавки. Другие добавки включают, без ограничения перечисленным, дополнительные наполнители, связующие агенты, замасливатели, глиданты, стабилизаторы, разбавители, ПАВ, пленкообразующие агенты, пигменты, смягчающие агенты, агенты, снижающие липкость и т.п.Предпочтительное количество активного ингредиента и добавок описано в данном контексте.
Настоящее изобретение относится к твердой пероральной лекарственной форме, которая включает терапевтически эффективное количество алискирена или его фармацевтически приемлемой соли, терапевтически эффективное количество гидрохлортиазида, и гидрофильный наполнитель, выбранный из углеводов или их комбинаций, а также в качестве добавок дезинтегрирующий агент и дополнительный наполнитель. Другие добавки включают, без ограничения перечисленным, связующие агенты, замасливатели, глиданты, стабилизаторы, разбавители, ПАВ, пленкообразующие агенты, пигменты, смягчающие агенты, агенты, снижающие липкость и т.п. Предпочтительные количества активного ингредиента и добавок описаны в данном контексте.
Настоящее изобретение относится к твердой пероральной лекарственной форме, которая включает терапевтически эффективное количество алискирена или его фармацевтически приемлемой соли, терапевтически эффективное количество гидрохлортиазида, и гидрофильный наполнитель, выбранный из углеводов или их комбинаций, а также в качестве добавок дезинтегрирующий агент, дополнительный наполнитель и замасливатель. Другие добавки включают, без ограничения перечисленным, связующие агенты, глиданты, стабилизаторы, разбавители, ПАВ, пленкообразующие агенты, пигменты, смягчающие агенты, агенты, снижающие липкость и т.п. Предпочтительные количества активного ингредиента и добавок описаны в данном контексте.
Настоящее изобретение относится к твердой пероральной лекарственной форме, которая включает терапевтически эффективное количество алискирена или его фармацевтически приемлемой соли, терапевтически эффективное количество гидрохлортиазида, и гидрофильный наполнитель, выбранный из углеводов или их комбинаций, а также в качестве добавок дезинтегрирующий агент, дополнительный наполнитель, замасливатель и глидант. Другие добавки включают, без ограничения перечисленным, связующие агенты, стабилизаторы, разбавители, ПАВ, пленкообразующие агенты, пигменты, смягчающие агенты, агенты, снижающие липкость и т.п. Предпочтительные количества активного ингредиента и добавок описаны в данном контексте.
Настоящее изобретение относится к твердой пероральной лекарственной форме, которая включает терапевтически эффективное количество алискирена или его фармацевтически приемлемой соли, терапевтически эффективное количество гидрохлортиазида, и гидрофильный наполнитель, выбранный из углеводов или их комбинаций, а также в качестве добавок дезинтегрирующий агент, дополнительный наполнитель, замасливатель, глидант и связующий агент. Другие добавки включают, без ограничения перечисленным, стабилизаторы, разбавители, ПАВ, пленкообразующие агенты, пигменты, смягчающие агенты, агенты, снижающие липкость и т.п. Предпочтительные количества активного ингредиента и добавок описаны в данном контексте.
Специалисты в данной области техники могут без дополнительных экспериментов выбрать и использовать одну или более из указанных добавок в зависимости от требуемых свойств твердой пероральной лекарственной формы.
Количество каждого типа использованной добавки, например, глиданта, связующего агента, дезинтегрирующего агента, наполнителя или разбавителя и замасливателя или пленкообразующего агента можно изменять в интервале, известном в данном уровне техники. Например, количество замасливателя может изменяться в интервале от 0,2 мас.% до 5 мас.%, прежде всего для стеарата магния от 0,5 мас.% до 2,0 мас.%, например, от 0,8 мас.% до 1,5 мас.%, количество связующего агента может изменяться в интервале от 0 мас.% до 20 мас.%, например, от 2 мас.% до 4 мас.%, количество дезинтегрирующего агента может изменяться в интервале от 0 мас.% до 20 мас.%, например, от 8 мас.% до 13 мас.%, количество дополнительного наполнителя или разбавителя может изменяться в интервале от 0 мас.% до 80 мас.%, например, от 20 мас.% до 35 мас.%, количество глиданта может изменяться в интервале от 0 мас.% до 5 мас.%, например, от 0,4 мас.% до 2 мас.%, и количество пленкообразующего агента может изменяться в интервале от 0 мг/см2 до 20 мг/см2, например, от 4 мг/см2 до 7 мг/см2.
Характерной особенностью настоящей твердой пероральной лекарственной формы является только относительно малое количество добавок при высоком содержании активного агента. Такие свойства позволяют получить стандартные лекарственные формы малого размера. Общее количество добавок в данной стандартной лекарственной форме без покрытия может составлять приблизительно 70 мас.% или менее в расчете на общую массу твердой пероральной лекарственной формы, более предпочтительно приблизительно 65% или менее. Предпочтительно количество добавок содержится в интервале от приблизительно 50 до 64 мас.%, более предпочтительно в интервале от приблизительно 55 до приблизительно 63 мас.%.
Предпочтительное количество дополнительного наполнителя, прежде всего микрокристаллической целлюлозы, составляет от приблизительно 20 до 35 мас.%, более предпочтительно от 22 до 33 мас.% в расчете на стандартную лекарственную форму. Такое количество наполнителя обеспечивает требуемую твердость пероральной лекарственной формы.
Предпочтительное количество связующего агента, прежде всего ПВП К 30, составляет от приблизительно 2 до 4 мас.%, более предпочтительно от 2,1 до 3,2 мас.% в расчете на стандартную лекарственную форму.
Предпочтительное количество дезинтегрирующего агента, прежде всего кросповидона, составляет от приблизительно 0 до 20 мас.%, более предпочтительно от 8 до 14 мас.%, наиболее предпочтительно от 9 до 13 мас.% в расчете на стандартную лекарственную форму. В связи с использованием компонента с) количество дезинтегрирующего агента можно регулировать таким образом, чтобы исключить нежелательный профиль распадаемости, прежде всего исключить слишком высокое содержание дезинтегрирующего агента, которое может оказать отрицательное влияние на следующую стадию нанесения водного покрытия.
Предпочтительное количество глиданта, прежде всего коллоидного диоксида кремния, составляет от приблизительно 0 до 5 мас.%, более предпочтительно от 0,4 до 2,0 мас.%, наиболее предпочтительно от 0,6 до 1,8 мас.% в расчете на стандартную лекарственную форму. При добавлении дополнительного глиданта, такого как тальк, его количество предпочтительно составляет от приблизительно 0 до 5 мас.%, более предпочтительно от 0,3 до 2,0 мас.%, наиболее предпочтительно от 0,4 до 1,8 мас.% в расчете на стандартную лекарственную форму.
Предпочтительное количество замасливателя, прежде всего стеарата магния, составляет от приблизительно 0,8 до 1,8 мас.%, более предпочтительно от 1,0 до 1,5 мас.% в расчете на стандартную лекарственную форму.
Указанные в данном контексте количества добавок в одинаковой степени применимы для стандартной лекарственной формы без покрытия и для стандартной лекарственной формы с покрытием.
Предпочтительное количество пленочного покрытия, прежде всего ГПМЦ, вязкость которой составляет 3 сП, составляет от приблизительно 4 до 7 мг/см в расчете на стандартную лекарственную форму.
Предпочтительные количества компонента а) и добавок описаны далее в разделе «Примеры».
Абсолютные количества каждой добавки и количества по отношению к другим добавкам в равной степени зависят от требуемых свойств твердой пероральной лекарственной формы и специалист в данной области может определить количество добавок по стандартной методике без дополнительных экспериментов. Например, можно выбрать твердую пероральную лекарственную форму с быстрым и/или замедленным высвобождением активного агента с количественным контролем высвобождения активного агента или без него.
В том случае, если требуется быстрое высвобождение активного агента, используют дезинтегрирующий агент, такой как сшитый ПВП, например, продукты под торговым названием POLYPLASDONE XL или KOLLIDON CL, прежде всего молекулярная масса которых составляет более 1000000, прежде всего, размер частиц которых составляет менее 400 мкм, или предпочтительно менее 74 мкм, или содержащие реакционноспособные добавки (шипучие смеси), которые ускоряют распадаемость таблетки в присутствии воды, например, так называемые шипучие таблетки, которые содержат кислоту в твердой форме, обычно лимонную кислоту, причем кислота содержится в виде химически связанного диоксида углерода, которая в воде действует на основание, например, гидрокарбонат натрия или карбонат натрия, и при этом высвобождается диоксид углерода.
В том случае, когда требуется замедленное высвобождение активного агента, используют технологию нанесения покрытия на отдельные частицы (например, гранулы, минитаблетки), используют восковые матричные системы, полимерные матричные таблетки или нанесение полимерных покрытий или другие стандартные технологии.
Количественный контроль высвобождения активного агента можно обеспечивать стандартным методом. Известные лекарственные формы включают осмотические пероральные системы (например, OROS), таблетки с покрытием, матричные таблетки, таблетки с прессованным покрытием, многослойные таблетки и т.п.
В твердой пероральной лекарственной форме предпочтительно содержатся следующие добавки: микрокристаллическая целлюлоза, гидроксипропилцеллюлоза, сшитый ПВП, ПВП, ПЭГ, КМЦ-Na или КМЦ-Са, стеарат магния, стеарат кальция или стеарат алюминия, безводный коллоидный диоксид кремния, тальк, пигменты диоксид титана и оксид железа. Количества использованных добавок зависят от количества использованного активного агента. Стеарат, например, стеарат магния, предпочтительно используется в количестве от 0,8 до 1,8 мас.%. Если используется диоксид кремния, то его количество составляет от 0,4 до 2,0 мас.%.
Количество алискирена в виде полуфумарата в стандартной лекарственной форме без покрытия предпочтительно составляет от приблизительно 83 до приблизительно 663 мг, наиболее предпочтительно количество полуфумарата алискирена составляет приблизительно 83, приблизительно 166 или приблизительно 332 мг в стандартной лекарственной форме.
Массовое соотношение компонента а) и связующего агента предпочтительно составляет от приблизительно 8:1 до приблизительно 25:1, более предпочтительно от приблизительно 11:1 до приблизительно 15:1, наиболее предпочтительно приблизительно 13,5-14:1.
Массовое соотношение компонента а) и дезинтегрирующего агента предпочтительно составляет от приблизительно 2:1 до приблизительно 4:1, более предпочтительно от приблизительно 2,5:1 до приблизительно 3,7:1, наиболее предпочтительно приблизительно 3,2-3,4:1.
Массовое соотношение компонента а) и глиданта предпочтительно составляет от приблизительно 5:1 до приблизительно 80:1, более предпочтительно от приблизительно 6:1 до приблизительно 40:1, наиболее предпочтительно приблизительно 9-32:1.
Массовое соотношение компонента а) и замасливателя предпочтительно составляет от приблизительно 20:1 до приблизительно 50:1, более предпочтительно от приблизительно 22:1 до приблизительно 38:1, наиболее предпочтительно приблизительно 24-36:1.
Массовое соотношение компонента b) и связующего агента предпочтительно составляет от приблизительно 0,2:1 до приблизительно 5:1, более предпочтительно от приблизительно 0,3:1 до приблизительно 3:1, наиболее предпочтительно приблизительно 0,5-2:1.
Массовое соотношение компонента b) и дезинтегрирующего агента предпочтительно составляет от приблизительно 0,1:1 до приблизительно 1:1, более предпочтительно от приблизительно 0,1:1 до приблизительно 0,7:1, наиболее предпочтительно приблизительно 0,2-0,6:1.
Массовое соотношение компонента b) и глиданта предпочтительно составляет от приблизительно 0,8:1 до приблизительно 3:1, более предпочтительно от приблизительно 1:1 до приблизительно 2:1, наиболее предпочтительно приблизительно 1,1-1,5:1.
Массовое соотношение компонента b) и замасливателя предпочтительно составляет от приблизительно 1:1 до приблизительно 6:1, более предпочтительно от приблизительно 1,1:1 до приблизительно 4:1, наиболее предпочтительно приблизительно 1,2-3,8:1.
Твердую пероральную лекарственную форму по настоящему изобретению также получают в виде таблеток или драже с пленочным покрытием, при этом твердую пероральную лекарственную форму обычно покрывают полимером, таким как ГПМЦ, ПВП или т.п., сахаром, шеллаком или другим известным пленкообразующим агентом, которые известны в данной области техники. Покрытие наносится различными известными способами, например, нанесение покрытия распылением, например, известными способами с использованием оборудования фирм Aeromatic, Glatt, Wurster или Huttlin, в перфорированном поддоне для нанесения покрытий, например, известными способами с использованием оборудования фирм Accela Cota, Glatt, Driam или других фирм, или другими известными способами. При этом используются стандартные добавки для фармацевтических препаратов.
Еще один вариант осуществления настоящего изобретения относится к способу получения твердой пероральной лекарственной формы по настоящему изобретению.
Таким образом, настоящее изобретение включает способ получения твердой пероральной лекарственной формы по настоящему изобретению, который включает следующие стадии:
1)грануляция
a) терапевтически эффективного количества алискирена или его фармацевтически приемлемой соли,
b) терапевтически эффективного количества гидрохлортиазида, и
c) гидрофильного наполнителя, выбранного из углеводов или их комбинации,
и добавок в жидкости для грануляции,
2) высушивание полученного гранулята,
3) смешивание высушенного гранулята с эксципиентами внешней фазы,
4) прессование полученной смеси, при этом получают твердую пероральную лекарственную форму в виде ядра таблетки, и
5) необязательное нанесение покрытия на полученное ядро таблетки, при этом получают таблетку с пленочным покрытием.
Было установлено, что указанный способ, по сравнению с другими способами, описанными ранее, является наиболее эффективным для получения твердой пероральной лекарственной формы алискирен+гидрохлортиазид, прежде всего в виде таблеток.
Полученный состав характеризуется следующими преимуществами:
- Простой способ получения лекарственной формы с относительно высоким содержанием лекарственного средства,
- Можно получать таблетки с достаточной твердостью, низкой хрупкостью, достаточными временем распадаемости, скоростью растворения и т.п.,
- Сведение к минимуму способности к слипанию, низкая текучесть лекарственного средства,
- Простой способ получения лекарственного средства,
- Масштабируемость получения лекарственного средства с высокой воспроизводимостью и эффективностью,
- Достаточно высокая стабильность обеспечивает приемлемый срок хранения.
Эксципиенты распределены частично во внутренней (гранулированной) фазе и частично во внешней фазе, указанный вариант описан в настоящем изобретении. Микрокристаллическая целлюлоза (наполнитель) и кросповидон (дезинтегрирующий агент) предпочтительно частично находятся во внутренней и частично во внешней фазе, ПВП К 30 (связующий агент) предпочтительно находится только во внутренней фазе, так как используется в качестве связующего агента при грануляции. Коллоидный диоксид кремния (глидант) и стеарат магния (замасливатель) предпочтительно частично находятся во внутренней и частично во внешней фазе. Тальк (глидант) предпочтительно находится только во внутренней фазе.
Эксципиенты внутренней фазы, например, наполнитель, связующий агент и дезинтегрирующий агент, гидрофильный наполнитель и лекарственное средство смешивают и гранулируют, при этом получают гранулят или внутреннюю фазу. Предпочтительно две гранулированные фазы получают раздельно, одна содержит компонент (а), другая содержит компонент (b). Компонент (с) предпочтительно содержится в гранулированной фазе, содержащей компонент (b).
Влажную грануляцию каждой фазы проводят в водной или органической среде. Предпочтительно влажную грануляцию фазы, содержащей компонент (а), проводят в органической среде, и/или влажную грануляцию фазы, содержащей компонент (b), проводят в водной среде. Влажная грануляция в водной среде означает, что жидкостью для грануляции является вода или жидкость для грануляции содержит воду, или предпочтительно грануляцию проводят в присутствии крахмального клея в качестве жидкости для грануляции. Жидкостью для влажной грануляции в органической среде является этанол, смесь этанола и воды, смесь этанола, воды и изопропанола, или раствор ПВП в описанных выше смесях. Предпочтительно соотношение этанола и воды в смеси составляет от приблизительно 50:50 до приблизительно 99:1 (мас./мас.%), наиболее предпочтительно приблизительно 94:6 (мас./мас.%). Предпочтительно соотношение этанола, воды и изопропанола в смеси составляет от приблизительно 45:45:5 до приблизительно 98:1:1 (мае./мас./мас.%), наиболее предпочтительно от приблизительно 88,5:5,5:6,0 до приблизительно 91,5:4,5:4,0 (мас./мас./мас.%). В предпочтительном варианте грануляцию проводят в этанольном растворе связующего агента и дополнительного этанола.
Гранулят высушивают и предпочтительно просеивают. Если гранулят получали в виде двух отдельных фаз, их предпочтительно смешивают перед добавлением эксципиентов внешней фазы.
Внешнюю фазу, содержащую, например, дезинтегрирующий агент, наполнитель, глидант и замасливатель, просеивают и смешивают с высушенным гранулятом. Полученную смесь прессуют в таблетки (ядра). На полученные ядра необязательно наносят пленочное покрытие.
Гранулированную фазу называют внутренней фазой, а экципиенты, добавленные к грануляту, называют внешней фазой смеси для таблетирования.
Настоящее изобретение также относится к способу получения твердой пероральной лекарственной формы, как описано выше в данном контексте. Такую твердую пероральную лекарственную форму получают при обработке компонентов, как описано выше в данном контексте, в соответствующих количествах, при этом получают стандартную лекарственную форму.
Предпочтительно добавки на стадии 1) выбирают из наполнителя, дезинтегрирующего агента, глиданта, замасливателя и связующего агента, а эксципиенты внешней фазы на стадии 3) выбирают из наполнителя, дезинтегрирующего агента, замасливателя и глиданта.
Известно множество способов грануляции, высушивания и смешивания, например, грануляция с распылением в псевдоожиженном слое, влажная грануляция в смесителе с высоким сдвигом, грануляция из расплава, высушивание в сушилке с псевдоожиженным слоем, смешивание в смесителе свободного падения или в барабанном блендере, прессование таблеток на одноштамповом прессе или ротационном прессе для таблетирования.
Грануляцию проводят с использованием любого стандартного оборудования, пригодного для грануляции в водной или органической среде. Получение конечной смеси и прессование таблеток также проводят с использованием любого стандартного пригодного оборудования.
Например, стадию 1) можно проводить в грануляторе с высоким сдвигом, например, фирмы Collette Gral, стадию 2) можно проводить в сушилке с псевдоожиженным слоем, стадию 3) можно проводить в смесителе свободного падения (например, контейнерный блендер, барабанный блендер), стадию 4) можно проводить, используя способ сухого прессования, например на ротационном компрессоре для таблетирования.
На ядра таблеток, как описано выше, необязательно наносят пленочное покрытие.
В связи с высокой гигроскопичностью и чувствительностью к воде алискирена и в связи с изменениями полиморфных форм, по указанным выше причинам (аморфное состояние, низкая стабильность) предпочтительно исключить применение воды, чтобы предотвратить изменения полиморфных форм лекарственного средства. Решение указанной проблемы заключается в нанесении органического пленочного покрытия.
Пленочное покрытие предпочтительно содержит ГПМЦ в качестве полимера, пигменты на основе оксида железа, диоксид титана в качестве красителя, ПЭГ в качестве смягчающего агента и тальк в качестве агента против слипания. Использование красителей служит для улучшения внешнего вида, а также для идентификации композиций. Другие пригодные красители включают каротиноиды, хлорофилл и лаки.
Пленочное покрытие должно защищать ядра таблеток от впитывания значительных количеств влаги и от контактирования лекарственного агента внутри таблеток с капельками воды. Такие покрытия можно наносить в условиях, снижающих количество влаги, которая может проникать в ядра таблеток.
Твердую пероральную лекарственную форму по настоящему изобретению можно использовать для снижения кровяного давления, систолического или диастолического, или обоих типов давления. Состояния, для лечения которых можно использовать настоящее изобретение, включают, без ограничения перечисленным, гипертензию (злокачественную, первичную, реноваскулярную, диабетическую, изолированную систолическую или вторичного типа), застойную сердечную недостаточность, стенокардию (стабильную или нестабильную), инфаркт миокарда, артериосклероз, диабетическую нефропатию, диабетическую сердечную миопатию, почечную недостаточность, заболевания периферических сосудов, гипертрофию левого желудочка, дисфункцию познавательной способности (такую, как болезнь Альцгеймера), инсульт, головную боль и хроническую сердечную недостаточность.
Настоящее изобретение относится также к способу лечения гипертензии (злокачественной, первичной, реноваскулярной, диабетической, изолированной систолической или вторичного типа), застойной сердечной недостаточности, стенокардии (стабильной или нестабильной), инфаркта миокарда, артериосклероза, диабетической нефропатии, диабетической сердечной миопатии, почечной недостаточности, заболеваний периферических сосудов, гипертрофии левого желудочка, дисфункции познавательной способности (такой, как болезнь Альцгеймера), инсульта, головной боли и хронической сердечной недостаточности, причем указанный способ заключается в том, что животному, включая человека, нуждающемуся в таком лечении, вводят терапевтически эффективное количество твердой пероральной лекарственной формы по настоящему изобретению.
Настоящее изобретение относится к применению твердой пероральной лекарственной формы по настоящему изобретению для получения лекарственного средства, предназначенного для лечения гипертензии (злокачественной, первичной, реноваскулярной, диабетической, изолированной систолической или вторичного типа), застойной сердечной недостаточности, стенокардии (стабильной или нестабильной), инфаркта миокарда, артериосклероза, диабетической нефропатии, диабетической сердечной миопатии, почечной недостаточности, заболеваний периферических сосудов, гипертрофии левого желудочка, дисфункции познавательной способности (такой, как болезнь Альцгеймера), инсульта, головной боли и хронической сердечной недостаточности.
Настоящее изобретение относится также к фармацевтической композиции, предназначенной для лечения гипертензии (злокачественной, первичной, реноваскулярной, диабетической, изолированной систолической или вторичного типа), застойной сердечной недостаточности, стенокардии (стабильной или нестабильной), инфаркта миокарда, артериосклероза, диабетической нефропатии, диабетической сердечной миопатии, почечной недостаточности, заболеваний периферических сосудов, гипертрофии левого желудочка, дисфункции познавательной способности (такой, как болезнь Альцгеймера), инсульта, головной боли и хронической сердечной недостаточности, и указанная композиция включает твердую пероральную лекарственную форму по настоящему изобретению.
Точная доза активного агента и конкретного состава для введения зависит от многих факторов, например, состояния, подлежащего лечению, требуемой продолжительности курса лечения и скорости высвобождения активного агента. Например, количество активного агента и скорость его высвобождения можно определить с использованием известных методов анализа in vitro или in vivo, оценивая период поддерживания концентрации активного агента в плазме крови на уровне, достаточном для обеспечения терапевтического эффекта.
В приведенном выше описании изобретения в полном объеме описаны предпочтительные варианты его осуществления. Модификации и способы усовершенствования вариантов осуществления настоящего изобретения, описанных в настоящем контексте, включены в объем приведенных ниже пунктов формулы изобретения. Специалисты в данной области техники после прочтения приведенного выше описания могут без дополнительных экспериментов использовать настоящее изобретение в полном объеме. Примеры, описанные в данном контексте, приведены только для иллюстрации определенных объектов настоящего изобретения и не ограничивают объем настоящего изобретения.
Пример 1
Составы
Композиции, содержащие 75 или 150 мг свободного основания алискирена и 12,5 или 25 мг гидрохлортиазида в таблетке.
|
|
Композиции, содержащие 300 мг свободного основания алискирена и 12,5 или 25 мг гидрохлортиазида в таблетке.
|
Пример 2
Свойства состава 150/25 мг
|
Пример 3
Свойства состава 75/12,5 мг
|
Пример 4
Свойства состава 300/25 мг
|
Пример 5
Свойства состава 150/12,5 мг
|
Пример 6
Свойства состава 300/12,5 мг
|
Пример 7
Оценка состава 300/25 мг
|
Мутантные антигены gas57 и антитела против gas57
[(1н-индол-5-ил)-гетероарилокси]-1-(азабицикло[3.3.1]нонаны, как холинергические лиганды n-achr, предназначенные для лечения психотических и нейродегенеративных нарушений
Лечение туберозного склероза
Органические соединения
Способ получения фармацевтической композиции
Предназначенная для перорального применения фармацевтическая композиция
Менингококковые полипептиды fhbp
Стабилизированные полипептиды инсулиноподобного фактора роста
Органические соединения
Курс лечения с использованием агониста рецептора s1p
Мутантные антигены gas57 и антитела против gas57
[(1н-индол-5-ил)-гетероарилокси]-1-(азабицикло[3.3.1]нонаны, как холинергические лиганды n-achr, предназначенные для лечения психотических и нейродегенеративных нарушений
Лечение туберозного склероза
Органические соединения
Способ получения фармацевтической композиции
Предназначенная для перорального применения фармацевтическая композиция
Менингококковые полипептиды fhbp
Стабилизированные полипептиды инсулиноподобного фактора роста
Органические соединения
Курс лечения с использованием агониста рецептора s1p

