Результат интеллектуальной деятельности: КОМПОЗИЦИИ ТЕТРАГИДРОБИОПТЕРИНА И СПОСОБЫ ЕГО КОЛИЧЕСТВЕННОЙ ОЦЕНКИ
Вид РИД
Изобретение
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
В настоящей заявке испрашивается приоритет предварительной заявки на патент США № 60/922821, поданной 11 апреля 2007, и предварительной заявки на патент США 61/019753, поданной 8 января 2008, которые во всей своей полноте включены в настоящее описание посредством ссылки.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Область техники, к которой относится изобретение
В общих чертах, настоящее изобретение относится к композициям и к способам, применяемым для лечения расстройств, восприимчивых к BH4, а также к способам и композициям для обнаружения и количественной оценки биоптеринов.
Предшествующий уровень техники
Тетрагидробиоптерин (обозначаемый здесь BH4) представляет собой биогенный амин, который принадлежит к семейству природных птеринов и является кофактором для ряда различных ферментов, включая фенилаланингидроксилазу (PAH), тирозингидроксилазу, триптофангидроксилазу и синтетазу оксида азота. Птерины присутствуют в физиологических жидкостях и тканях в восстановленной и в окисленной формах, однако биологически активным является только 5,6,7,8-тетрагидробиоптерин. Он представляет собой хиральную молекулу и 6R-энантиомер кофактора, который, как известно, является биологически активным энантиомером. Подробное описание синтеза ВН4 и восприимчивых к нему расстройств можно найти в публикации Blau et al., 2001 (Disorders of tetrahydrobiopterin and related biogenic amines. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill, 2001: 1275-1776).
В публикации Fiege, et al., Molecular Genetics and Metabolism 81:45-51 (2004), описаны фармакокинетические исследования по пероральному введению тетрагидробиоптерина (BH4), которые позволяют предположить о наличии «довольно широкой вариабельности перорально вводимого BH4, вероятно, обусловленной различными уровнями его абсорбции в кишечнике и/или эффектом первого прохождения».
Тетрагидробиоптерин был предложен для лечения ряда различных болезненных состояний, однако необходимость в разработке альтернативных и улучшенных способов введения этого лекарственного средства остается актуальной.
ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам введения 6R-(L-эритро)-5,6,7,8-тетрагидробиоптерина (BH4) или его фармацевтически приемлемой соли, где указанные способы позволяют улучшать или максимизировать его биологическую доступность при пероральном введении и/или улучшать или оптимизировать уровень биологической доступности непосредственно после его перорального введения. Такие способы могут быть применены для лечения любых восприимчивых к BH4 расстройств, включая метаболические расстройства, сердечно-сосудистые заболевания, анемию и психоневрологические расстройства. Способы согласно изобретению преимущественно позволяют с большей эффективностью ослаблять клинические симптомы, например снижать уровни флуктуации фенилаланина в плазме, регулировать кровяное давление, регулировать уровни нейромедиаторов или другие клинические параметры.
Используемое здесь обозначение BH4 означает 6R-(L-эритро)-5,6,7,8-тетрагидробиоптерин. Используемый здесь термин «BH4», если это не очевидно из контекста изобретения, может включать, но необязательно, фармацевтически приемлемую соль 6R-(L-эритро)-5,6,7,8-тетрагидробиоптерина.
В своем первом аспекте настоящее изобретение относится к способам перорального введения пациенту, нуждающемуся в этом, очищенного препарата BH4.
В репрезентативном варианте изобретения указанные способы включают стадию информирования пациента о том, что уровень абсорбции тетрагидробиоптерина повышается при его употреблении вместе с пищей по сравнению с абсорбцией, достигаемой при его употреблении без пищи. В некоторых вариантах изобретения пациента информируют о том, что прием состава сразу после приема пищи, например, пищи с высоким содержанием жира и высококалорийной пищи, приводит к увеличению любого одного, двух, трех или всех нижеследующих параметров, таких как средняя концентрация в плазме, Cmax, AUC, AUC(0-t) и/или AUC(inf). В репрезентативных вариантах изобретения пациента информируют о том, что введение BH4 вместе с пищей с высоким содержанием жира приводит к увеличению Cmax и AUC по сравнению с введением BH4 без употребления пищи (в условиях голодания). В некоторых вариантах изобретения относительное увеличение этих параметров может составлять по меньшей мере 20%, 30% или более.
В альтернативных или в дополнительных вариантах изобретения указанный способ введения тетрагидробиоптерина включает информирование пациента о том, что абсорбция тетрагидробиоптерина повышается при его приеме в виде целой таблетки по сравнению с абсорбцией, достигаемой при приеме состава, растворенного в жидкости. В некоторых вариантах изобретения пациента информируют о том, что прием целых таблеток приводит к увеличению любого из нижеследующих параметров: средней концентрации в плазме, Cmax, AUC, AUC(0-t) или AUC(inf). В репрезентативных вариантах изобретения пациента информируют о том, что введение ВН4 в виде целой таблетки приводит к увеличению Cmax и AUC по сравнению с введением BH4 после его растворения в жидкости. В некоторых вариантах изобретения относительное увеличение может составлять по меньшей мере 20% или более.
Любой из предыдущих способов может быть осуществлен путем приготовления тетрагидробиоптерина или его помещения в контейнер, содержащий отпечатанную памятку, информирующую пациента о возможном изменении вышеописанных параметров абсорбции.
Способы согласно изобретению могут также включать, но необязательно, стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества тетрагидробиоптерина. Такое терапевтически эффективное количество варьируется в зависимости от состояния, подвергаемого лечению, и может быть легко определено лечащим врачом в целях желаемого ослабления клинических симптомов.
В одном из репрезентативных вариантов изобретения такие способы включают введение BH4 в растворенной форме, где указанный состав растворяют в жидкости, включая, но не ограничиваясь ею, воду, апельсиновый сок и яблочный сок. В одном из репрезентативных вариантов изобретения растворенный BH4 вводят пациенту в условиях голодания, то есть натощак. В настоящем изобретении также рассматривается введение растворенного BH4 в определенный период времени, например утром, днем и ночью того же дня, натощак один или несколько раз в день. В репрезентативных вариантах изобретения композицию вводят пациенту натощак, например, по меньшей мере за 30 минут, 45 минут, или по меньшей мере за один час до приема пищи, и/или по меньшей мере через 90 минут или через два часа, 2,5 часа или три часа после приема пищи. Таким образом, BH4 может приниматься пациентом в виде жидкого продукта или продукта, представляющего собой твердую или полутвердую лекарственную форму, предварительно растворенную до ее приема. В дополнительном варианте изобретения BH4 может также растворяться в полости рта при его приеме в виде твердой или полутвердой лекарственной формы, которую этот состав имел до его употребления в растворенной форме.
В другом репрезентативном варианте изобретения указанные способы включают введение BH4 в твердой лекарственной форме, включая, но не ограничиваясь ими, таблетки, капсулы, леденцы, пастилки, порошки и гранулы, или в полутвердой форме, включая, но не ограничиваясь ими, желе для перорального введения, содержащее лекарственное вещество, которое проглатывается без предварительного растворения в жидкости, например, такой как вода, апельсиновый сок, яблочный сок и т.п. В одном из вариантов изобретения пациент принимает BH4 для проглатывания в условиях голодания, то есть натощак. В настоящем изобретении также рассматривается введение BH4 в твердой или полутвердой лекарственной форме для проглатывания в определенный период времени, например утром, днем и ночью того же дня натощак, один или несколько раз в день. В репрезентативных вариантах изобретения композицию вводят пациенту натощак, например, по меньшей мере за 30 минут, 45 минут, или по меньшей мере за один час до приема пищи, и/или по меньшей мере через 90 минут или через два часа, 2,5 часа или три часа после приема пищи.
В другом варианте изобретения указанные способы включают введение BH4 независимо от того, проглатывает ли его пациент в виде твердой или полутвердой лекарственной формы или употребляет в виде растворенной в жидкости лекарственной формы вместе с пищей, например с пищей с высоким содержанием жира или вместе с жирной и/или высококалорийной пищей. В настоящем изобретении также рассматривается введение BH4 независимо от того, проглатывается ли он или употребляется в растворенном виде в определенный период времени, например утром, днем и ночью того же дня вместе с пищей, например с пищей с высоким содержанием жира или с жирной и/или высококалорийной пищей один или несколько раз в день. В репрезентативном варианте изобретения, пациент принимает ВН4 один раз в день в виде твердой лекарственной формы сразу после приема пищи. В предпочтительном варианте изобретения твердую лекарственную форму изготавливают в виде таблетки или капсулы. В более репрезентативных вариантах изобретения ВН4 употребляют вместе с пищей во время ее приема в течение примерно 0-30 минут или 5-20 минут. Независимо от того, употребляется ли ВН4 в виде твердой лекарственной формы, жидкой лекарственной формы или в виде раствора, его действие in vivo (или его биологическая доступность) является более эффективным при проглатывании сразу после приема пищи по сравнению с его действием натощак, оцениваемом в качестве контроля.
Прием BH4 может быть осуществлен приблизительно одновременно с приемом пищи, либо BH4 может приниматься до или после приема пищи. Период времени между приемом пищи и приемом BH4, независимо от того проглатывается ли он или выпивается в растворенном виде, может составлять по меньшей мере 5 минут. Так, например, ВН4 может быть введен за 60 минут, 30 минут, 25 минут, 20 минут, 15 минут, 10 минут или 5 минут до приема пищи или через указанное время после приема пищи.
В другом варианте изобретения для некоторых пациентов, например, для взрослых, или в случае некоторых болезненных состояний, например, сердечно-сосудистых заболеваний или других заболеваний, ассоциированных с дисфункцией синтетезы оксида азота (NOS), способы согласно изобретению включают прием целой, а не растворенной в жидкости таблетки в целях повышения биологической доступности.
Во втором аспекте настоящее изобретение относится к способу стабилизации ВН4 в желудочно-кишечном тракте у пациента путем снижения рН тонкой кишки, например, с использованием протонообменных полимеров. В настоящем изобретении также рассматриваются соответствующие продукты, содержащие ВН4 и подкисляющие эксципиенты, такие как протонообменные полимеры.
В третьем аспекте настоящего изобретения рассматривается способ увеличения времени пребывания ВН4 в кишечнике, включая, но не ограничиваясь им, замедление высвобождения из кишечника с использованием агента, способствующего замедлению высвобождения из кишечника, например такого агента, как сложный эфир жирной кислоты и/или сложный эфир жирной кислоты и глицерина. Такие гидрофобные агенты могут увеличивать время пребывания ВН4 в кишечнике и могут повышать количество абсорбируемого ВН4. Время пребывания BH4 в кишечнике, достигаемое с использованием такого(их) агента(ов), может по меньшей мере в полтора раза, по меньшей мере в два раза, по меньшей мере в три раза, по меньшей мере в четыре раза или по меньшей мере в пять раз превышать время пребывания в кишечнике состава BH4, не содержащего такого агента. Подходящими жирными кислотами являются олеиновая кислота, стеариновая кислота, арахиновая кислота, пальмитиновая кислота, арахидоновая кислота, линолевая кислота, линоленовая кислота, эруцидиновая кислота, миристиновая кислота, лауриновая кислота, миристолеиновая кислота и пальмитолеиновая кислота. Также рассматривается способ увеличения времени пребывания BH4 в кишечнике посредством его удерживания в желудке с использованием альгиновой кислоты и усиления биоадгезии с использованием поликарбофила. Также рассматриваются соответствующие продукты, содержащие BH4 и агенты, замедляющие перистальтику желудка.
В четвертом аспекте изобретения рассматривается способ модификации высвобождения BH4 с использованием состава пролонгированного высвобождения, такого как HPMC, карбомер и т.п. Также рассматриваются соответствующие продукты, которые представляют собой составы с замедленным высвобождением.
В пятом аспекте изобретения рассматривается введение BH4 в стерильной жидкости или в виде стерильной твердой лекарственной формы способами, отличающимися от перорального введения и включающими, но не ограничивающимися ими, местное введение, внутривенное введение, подкожное введение, внутримышечное введение, интратекальное введение, офтальмическое введение и введение путем ингаляции. В настоящем изобретении рассматриваются соответствующие композиции и наборы, подходящие для такого введения, а также способы их изготовления. Так, например, для чрескожного или защечного введения могут быть изготовлены ВН4-содержащие пластыри для чрескожного или защечного введения соответственно. Также рассматриваются подъязычные таблетки, содержащие ВН4. В настоящем изобретении рассматриваются подходящие наборы, включающие устройство для ингаляции, содержащее ВН4, или набор, содержащий ВН4 и включающий пипетку или распылитель.
В одном из варианте осуществления настоящее изобретение относится к жидкому составу тетрагидробиоптерина (BH4) или его фармацевтически приемлемой соли, включающему водный раствор BH4 или его фармацевтически приемлемой соли, антиоксидант и рН-корректирующий буфер.
В другом варианте осуществления настоящее изобретение относится к способу приготовления жидкого состава, содержащего тетрагидробиоптерин (BH4) или его фармацевтически приемлемую соль, где указанный способ включает получение водного раствора, содержащего BH4 или его фармацевтически приемлемую соль; добавление антиоксиданта и рН-корректирующего буфера к раствору, содержащему ВН4 или его фармацевтически приемлемую соль; барботирование инертным газом или диоксидом углерода водного раствора, содержащего BH4 или его фармацевтически приемлемую соль, до или после добавления антиоксиданта и рН-корректирующего буфера, и герметичную упаковку барботированного раствора, содержащего BH4 или его фармацевтически приемлемую соль, антиоксидант и рН-корректирующий буфер, в контейнере.
В своем шестом аспекте настоящее изобретение относится к улучшенному способу оценки уровня BH4 с помощью тандемной масс-спектрометрии и вычисления количества восстановленного биоптерина. Такие способы позволяют оценивать чувствительность к BH4 в концентрации 5-1000 нг/мл, при этом точность и достоверность является допустимой, если коэффициент изменчивости (CV)% ниже 15% (20% в случае нижнего предела количественной оценки, LLOQ). В репрезентативном варианте изобретения способ оценки BH4 с помощью ВЭЖХ (ОФ) в комбинации с тандемной масс-спектрометрией (ЖХ/МС/МС) включает стадии: (1) окисления проб крови, проб плазмы, гомогенатов ткани или мочи; (2) иодометрического анализа окисленных образцов; (3) пропускания указанных окисленных образцов через ионообменную колонку; (4) оценки уровней общего и окисленного биоптерина в указанных образцах с помощью ВЭЖХ и тандемной масс-спектрометрии; вычисления количества восстановленного биоптерина как разности между уровнями указанных общих биоптеринов и их указанной окисленной формы. В одном из вариантов изобретения образцы обрабатывают путем окисления кислотой, где указанный способ включает стадии (1) обработки указанных образцов KCl, HCl или TCA; (2) проведения иодометрического анализа указанных образцов, окисленных кислотой; (3) пропускания указанных окисленных образцов через ионообменную колонку; (4) определения уровня общего биоптерина, содержащего 6R-BH4, R-q-DHBP (который сразу восстанавливается in vivo до 6R-BH4, а поэтому измерение уровня восстановленного биоптерина основано, главным образом, на 6R-BH4), DHBP и BP, в указанных образцах с помощью ВЭЖХ и тандемной масс-спектрометрии. В другом варианте изобретения образцы окисляют щелочью, где указанный способ включает: (1) обработку указанных образцов KI, I или NaOH; (2) подкисление указанных окисленных щелочью образцов путем добавления HCl или TCA; (3) проведение иодометрического анализа указанных окисленных образцов; (4) пропускание указанных образцов через ионообменную колонку; (5) определение уровня окисленного биоптерина, содержащего DHBP и BP, с помощью ВЭЖХ и тандемной масс-спектрометрии, (6) вычисление количества восстановленного биоптерина (6R-BH4 + R-q-DHBP) как разности между уровнями общих биоптеринов и их окисленной формы.
В другом своем аспекте настоящее изобретение относится к раствору для разделения дигидробиоптерина, биоптерина и их аналогов с помощью обращенно-фазовой ВЭЖХ с использованием подвижной фазы, включающей водный раствор, содержащий метанол, ацетат натрия, лимонную кислоту, EDTA и 1,4-дитиоэритрит. Аналогичным образом, в настоящем изобретении рассматривается способ разделения дигидробиоптерина и биоптерина или их аналогов из смеси, содержащей основную и дигидро-форму, где указанный способ включает проведение обращенно-фазовой ВЭЖХ с использованием подвижной фазы, содержащей водный раствор, включающий метанол, ацетат натрия, лимонную кислоту, EDTA и 1,4-дитиоэритрит, на смеси, содержащей дигидробиоптерин и биоптерин, или аналог дигидробиоптерина и аналог биоптерина.
В другом своем аспекте настоящее изобретение относится к способу количественной оценки биоптеринов в смеси биоптериновых молекул, где указанный способ включает получение смеси, содержащей биоптерин и по меньшей мере одно из таких веществ, как дигидробиоптерин и тетрагидробиоптерин, или аналоги биоптерина и по меньшей мере одно из таких веществ, как дигидробиоптерин и тетрагидробиоптерин; разделение биоптериновых молекул в смеси с помощью обращенно-фазовой ВЭЖХ; а в случае тетрагидробиоптерина и его аналогов, проведение электрохимического детектирования путем окисления тетрагидробиоптерина и его аналогов, присутствующих на первом электроде, до хиноноидных дигидробиоптериновых форм с последующим восстановлением указанных хиноноидных форм до тетрагидробиоптерина и его аналогов, присутствующих на втором электроде; и измерение силы тока, генерируемого реакцией восстановления для определения концентрации молекул; и/или в случае дигидробиоптерина, его аналогов, биоптерина или его аналогов, оценку уровня таких молекул путем детектирования флуоресценции после окисления молекул дигидробиоптерина до биоптерина на хроматографической колонке.
Что касается описанных здесь композиций и способов, то предпочтительные компоненты и их состав могут быть выбраны из различных компонентов и составов, представленных в нижеследующих примерах.
Другие отличительные признаки и преимущества настоящего изобретения будут очевидны из нижеследующего подробного описания изобретения. Однако следует отметить, что в подробном описании изобретения и в конкретных его примерах описаны предпочтительные варианты изобретения, которые приводятся лишь в целях иллюстрации, и в них могут быть внесены различные изменения и модификации, не выходящие за рамки сущности и объема изобретения и которые будут очевидны для специалиста в данной области, исходя из подробного описания изобретения.
КРАТНОЕ ОПИСАНИЕ ГРАФИЧЕСКОГО МАТЕРИАЛА
На фигуре 1 представлена картина порошковой рентгеновской дифракции, характеризующая кристаллическую полиморфную форму B 6R-(L-эритро)-5,6,7,8-тетрагидробиоптерина.
На фигуре 2 графически представлена картина характеристической рентгеновской дифракции, продуцируемая формой А дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 3 графически представлена картина характеристической рентгеновской дифракции, продуцируемой формой F дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 4 графически представлена картина характеристической рентгеновской дифракции, продуцируемой формой J дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 5 графически представлена картина характеристической рентгеновской дифракции, продуцируемой формой K дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 6 графически представлена картина характеристической рентгеновской дифракции, продуцируемой гидратной формой C дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 7 графически представлена картина характеристической рентгеновской дифракции, продуцируемой гидратной формой D дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 8 графически представлена картина характеристической рентгеновской дифракции, продуцируемой гидратной формой E дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 9 графически представлена картина характеристической рентгеновской дифракции, продуцируемой гидратной формой H дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 10 графически представлена картина характеристической рентгеновской дифракции, продуцируемой гидратной формой O дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 11 графически представлена картина характеристической рентгеновской дифракции, продуцируемой сольватной формой G дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 12 графически представлена картина характеристической рентгеновской дифракции, продуцируемой сольватной формой I дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 13 графически представлена картина характеристической рентгеновской дифракции, продуцируемой сольватной формой L дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 14 графически представлена картина характеристической рентгеновской дифракции, продуцируемой сольватной формой M дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 15 графически представлена картина характеристической рентгеновской дифракции, продуцируемой сольватной формой N дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
На фигуре 16 представлена блок-схема измерения уровней биоптерина.
На фигуре 17 представлена систематизированная оценка достоверности анализа на биоптерин.
На фигуре 18 представлена таблица, иллюстрирующая фармакокинетические параметры общего биоптерина в плазме после однократного перорального введения сапроптерина (BH4) крысам.
На фигуре 19 указано отношение концентраций биоптерина и его восстановленной формы в плазме после одноразового введения сапроптерина (BH4) крысам.
На фигуре 20 указано отношение концентраций биоптерина в плазме и его восстановленной формы после одноразового введения сапроптерина (BH4) обезьянам.
На фигуре 21 представлена таблица, иллюстрирующая фармакокинетические параметры общего биоптерина в плазме после одноразового введения сапроптерина (BH4) обезьянам.
На фигуре 22 представлен протокол проведения оценки безопасности.
На фигуре 23 указаны средние концентрации ВН4 в плазме после перорального введения здоровым добровольцам 10 мг/кг BH4 в растворенном виде и в виде целых таблеток в условиях голодания, и в виде целых таблеток при приеме пищи - по линейной схеме.
На фигуре 24 указаны средние концентрации ВН4 в плазме после перорального введения здоровым добровольцам 10 мг/кг BH4 в растворенном виде и в виде целых таблеток в условиях голодания, и в виде целых таблеток при приеме пищи - по полулогарифмической схеме.
На фигуре 25 представлена таблица, в которой систематизированы фармакокинетические параметры для ВН4 после перорального введения здоровым добровольцам 10 мг/кг BH4, в растворенном виде и в виде целых таблеток в условиях голодания, и в виде целых таблеток при приеме пищи.
На фигуре 26 приводится статистическое сравнение фармакокинетических параметров для ВН4 после перорального введения здоровым добровольцам 10 мг/кг BH4 в растворенном виде и в виде целых таблеток в условиях голодания, и в виде целых таблеток при приеме пищи.
На фигуре 27 проиллюстрировано исследование стабильности состава BH4, приготовленного с использованием 5% маннита в водном растворе, во время хранения при -20°С, где указанное исследование проводили за две недели и через две недели после хранения.
На фигуре 28 представлен профиль растворимости BH4-содержащей капсулы до и после хранения в течение 54 дней при 40°С.
На фигуре 29 представлен профиль растворимости двух составов BH4 - BH4-содержащей биоадгезивной таблетки и BH4-содержащих биоадгезивных гранул.
На фигуре 30 представлен профиль растворимости различных составов ВН4 с замедленным высвобождением.
На фигуре 31 представлен профиль растворимости различных составов ВН4 с замедленным высвобождением.
На фигуре 32 представлена принципиальная схема флотации лекарственных составов BH4.
На фигуре 33 представлен профиль растворимости различных флотирующих лекарственных составов.
На фигуре 34 представлена принципиальная схема газообразующих лекарственных составов BH4.
На фигуре 35 представлен фармакокинетический профиль различных составов BH4.
На фигуре 36 проиллюстрировано исследование стабильности составов BH4 для внутривенного введения при pH 4 в течение 35 дней.
На фигуре 37 проиллюстрировано исследование стабильности различных составов BH4 для внутривенного введения в течение 350 часов.
На фигуре 38 проиллюстрировано исследование стабильности составов BH4 для внутривенного введения в различных концентрациях ВН4.
Описание предпочтительных вариантов осуществления изобретения
Настоящее изобретение относится к улучшенным способам перорального введения очищенного препарата 6R-(L-эритро)-5,6,7,8-тетрагидробиоптерина, включая его фармацевтически приемлемую соль. Настоящее изобретение основано на обнаружении того факта, что перорально вводимый тетрагидробиоптерин (ВН4) плохо абсорбируется в желудочно-кишечном тракте, и это его свойство является главной причиной низкой биологической доступности ВН4.
Ниже представлена химическая структура 6R-(L-эритро)-5,6,7,8-тетрагидробиоптерина (BH4):
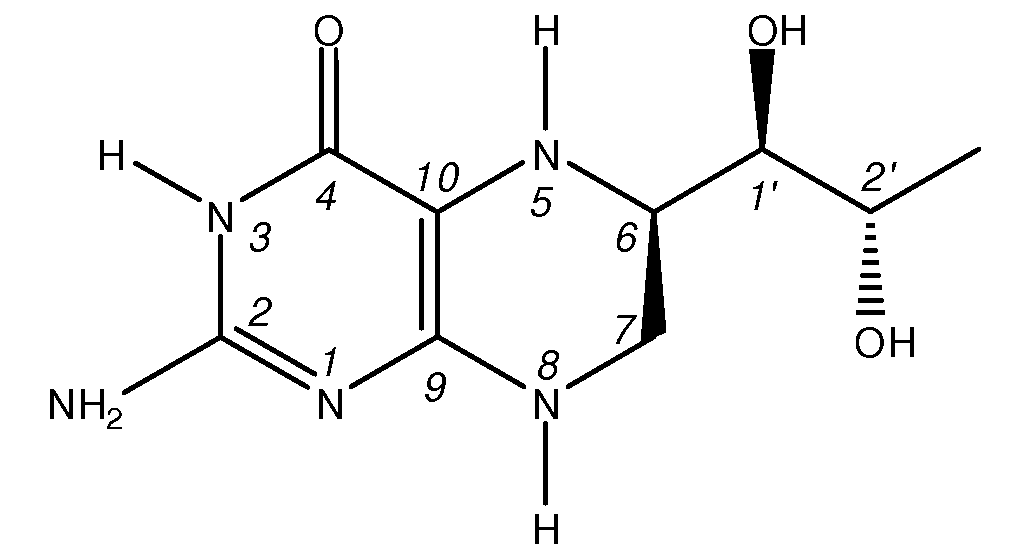
Тетрагидробиоптерин представляет собой водорастворимое органическое соединение с низкой липидной растворимостью. Коэффициент распределения ВН4 в октаноле-воде, как было определено в экспериментальном анализе in silico с помощью компьютерной программы BioLoom (version 1.5, Biobyte Corp in Claremont California), составлял -1,17. Оптимальная проницаемость биологических мембран, приближенно выражаемая как коэффициент распределения в октаноле/воде, составляет примерно log P = 2 или в 100 раз превышает липидную растворимость. Хотя низкая величина ClogP позволяет этому субстрату легко солюбилизироваться в физиологических условиях, однако способность этого субстрата проникать в билипидные слои в биологических мембранах ограничена, что тем самым может ограничивать пероральную доступность.
Описанные здесь исследования in vivo, проводимые на крысах и обезьянах, показали, что по сравнению с внутривенным введением BH4 в аналогичных дозах лишь 8-11% BH4 абсорбируется в кишечнике, при этом его большая часть выделяется с фекалиями. Такая вариабельность абсорбции ВН4 была также обнаружена в описанных здесь исследованиях по влиянию пищи на биологическую доступность ВН4 у здоровых людей. Хотя введение BH4 в воде и в апельсиновом соке в условиях голодания дает сравнимые средние концентрации в плазме и средние величины Cmax и AUC(0-t), однако введение BH4 вместе с жирной и высококалорийной пищей приводит к значительному увеличению средних концентраций в плазме и средних величин для Cmax и AUC(0-t) в том случае, если BH4 был введен в воде.
Хотя имеется достаточное число публикаций, в которых описано увеличение биологической доступности ВН4 при его приеме вместе с пищей, однако воздействие этой пищи обычно наблюдается при ее приеме вместе с липофильными (то есть растворимыми в липидах) не растворимыми в воде лекарственными средствами, но обычно не с активным веществом, обладающим высокой водорастворимостью, таким как ВН4. Увеличение биологической доступности липофильных соединений при приеме пищи обычно объясняется тем, что жирная пища стимулирует солюбилизацию лекарственного средства, поскольку «подобное растворяется подобным», и, тем самым, делает данный состав доступным для абсорбции. Другим возможным объяснением является то, что пища с высоким содержанием жира стимулирует секрецию желчных кислот, которые представляют собой природные биологические поверхностно-активные вещества, которые стимулируют солюбилизацию и эмульгирование съедаемого жира, тем самым улучшая пищеварение. Также считается, что такие желчные кислоты солюбилизируют нерастворимые в воде соединения, что делает их доступными для абсорбции. Однако для абсорбции BH4 он необязательно должен быть солюбилизирован, поскольку это соединение растворяется при концентрации более чем 1000 мг/мл и является одним из самых известных растворимых лекарственных средств. Поэтому повышение его биологической доступности при приеме вместе с очень жирной высококалорийной пищей не происходит по указанному известному механизму.
Однако введение лекарственного средства в твердой или полутвердой форме и/или вместе с жирной пищей может максимизировать биологическую доступность в результате увеличения времени пребывания ВН4 в кислотной среде желудка и в верхнем отделе желудочно-кишечного тракта (ЖКТ), где BH4 является химически стабильным. Стабильность BH4 снижается по мере увеличения pH, а его время полужизни в буферном растворе при pH 6,8, то есть при рН, значение которого приблизительно равно значению рН тонкой кишки, составляет примерно 15 минут. При рН 3,1, то есть при рН, значение которого входит в интервал типичных значений рН в желудке здоровых добровольцев, стабильность ВН4 при концентрации 1 мг/мл сохраняется в течение 3 часов. Химическая стабильность BH4 может еще больше увеличиваться, если рН в желудке снижается до значения 3,1. Поэтому более продолжительное время пребывание интактного лекарственного средства в желудке приводит к увеличению его абсорбции к стенкам желудка, тогда как быстрое высвобождение ВН4 из желудка в тонкий кишечник приводит к его разложению, и тем самым, к его неспособности к абсорбции.
Таким образом, для максимизации пероральной биологической доступности BH4 при каждом его введении BH4 должен поглощаться с пищей, например с пищей с высоким содержанием жира, или с жирной и/или высококалорийной пищей. Альтернативно, для максимизации уровня пероральной биологической доступности между введениями BH4 необходимо принимать натощак (например, за 1 час или за 2 часа до приема пищи или через 1 час или 2 часа после приема пищи).
Используемый здесь термин «биологическая доступность» означает часть вводимой дозы лекарственного средства, попадаемой в систему кровообращения. При внутривенном введении лекарственного средства его биологическая доступность теоретически должна составлять 100%. Однако при введении лекарственного средства другими способами (такими как пероральное введение) его биологическая доступность должна составлять менее 100%, что обусловлено, например, неполной абсорбцией в желудочно-кишечном тракте, разложением или метаболизмом до абсорбции и/или эффектом первого прохождения в печени.
Термин «пища с высоким содержанием жира», по существу, означает пищу, имеющую по меньшей мере примерно 700 ккал и по меньшей мере примерно 45% жира (относительное процентное содержание жира в ккал), или альтернативно, по меньшей мере примерно 900 ккал и по меньшей мере примерно 50% жира. Термин «жирная пища», по существу, означает пищу, содержащую по меньшей мере 20 г жира, или по меньшей мере 25, 30, 35, 40, 45 или 50 г жира, и/или по меньшей мере примерно 45% или 50% жира. В соответствии с рекомендациями Управления по контролю за качеством пищевых продуктов, медикаментов и косметических средств (FDA), «пища с высоким содержанием жира» определена как пища, в которой общее содержание калорий составляет приблизительно 50% от общего содержания калорий в пище, а «высококалорийная пища» определяется как пища, имеющая приблизительно 800-1000 калорий. FDA рекомендует использовать пищу с высоким содержанием жира и высококалорийную пищу в качестве тест-пищи для проведения исследований влияния этой пищи на биологическую доступность и биологическую эквивалентность этой пищи. Эта тест-пища должна иметь приблизительно 150, 250 и 500-600 калорий белка, углеводов и жира соответственно. Примером может служить тест-пища, состоящая из двух яиц, зажаренных в масле, двух кусочков бекона, четырех унций мелко нарезанного поджаренного картофеля и восьми унций цельного молока. При этом возможна замена на другую пищу, если она имеет аналогичное количество калорий белка, углеводов и жира и сравнимый объем и вязкость (Guidance for Industry, Food-Effect Bioavailability and Fed Bioequivalence Studies, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), December 2002).
В первом аспекте настоящее изобретение относится к способам перорального введения очищенного прерарата 6R-(L-эритро)-5,6,7,8-тетрагидробиоптерина (BH4), включая его фармацевтически приемлемую соль.
В некоторых вариантах изобретения указанные способы включают информирование пациента о том, что введение тетрагидробиоптерина с пищей оказывает влияние на фармакокинетические свойства. В репрезентативном варианте изобретения указанные способы включают стадию информирования пациента о том, что абсорбция тетрагидробиоптерина повышается при его приеме вместе с пищей по сравнению с абсорбцией, достигаемой при его приеме натощак. В некоторых вариантах изобретения пациента информируют о том, что прием состава сразу после приема пищи, например пищи с высоким содержанием жира и высококалорийной пищи, приводит к увеличению любого одного, двух, трех или всех нижеследующих параметров, таких как средняя концентрация в плазме, Cmax, AUC, AUC(0-t) и/или AUC(inf). В репрезентативных вариантах изобретения пациента информируют о том, что введение BH4 вместе с пищей с высоким содержанием жира приводит к увеличению Cmax и AUC по сравнению с введением BH4 натощак (в условиях голодания). В некоторых вариантах изобретения относительное увеличение может составлять по меньшей мере 20%, 30% или более.
В альтернативных вариантах или в дополнительных вариантах указанный способ введения тетрагидробиоптерина включает информирование пациента о том, что абсорбция тетрагидробиоптерина повышается при проглатывании целой таблетки по сравнению с абсорбцией, достигаемой при употреблении состава, растворенного в жидкости. В некоторых вариантах изобретения пациента информируют о том, что проглатывание целой таблетки приводит к увеличению любого из нижеследующих параметров: средней концентрации в плазме, Cmax, AUC, AUC(0-t) или AUC(inf). В репрезентативных вариантах изобретения пациента информируют о том, что прием ВН4 в виде целой таблетки приводит к увеличению Cmax и AUC по сравнению с приемом BH4 после его растворения в жидкости. В некоторых вариантах изобретения относительное увеличение может составлять по меньшей мере 20% или более.
Любой из предыдущих способов может быть осуществлен путем предоставления тетрагидробиоптерина или его введения в контейнер, содержащий отпечатанную памятку, информирующую пациента об изменении вышеописанных параметров абсорбции.
Способы согласно изобретению могут также включать, но необязательно, стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества тетрагидробиоптерина. Такое терапевтически эффективное количество варьируется в зависимости от состояния, подвергаемого лечению, и может быть легко определено лечащим врачом исходя из желательного ослабления клинических симптомов.
В одном из репрезентативных вариантов изобретения такие способы включают введение BH4 в растворенной форме, где указанный состав растворяют в жидкости, включая, но не ограничиваясь ею, воду, апельсиновый сок и яблочный сок. В одном из вариантов изобретения растворенный BH4 вводят пациенту в условиях голодания, то есть натощак. В настоящем изобретении также рассматривается введение растворенного BH4 в определенный период времени, например утром, днем и ночью того же дня натощак один или несколько раз в день. В одном из репрезентативных вариантов изобретения композицию вводят пациенту натощак, например, по меньшей мере за 30 минут, 45 минут, или по меньшей мере за один час до приема пищи, и/или по меньшей мере через 90 минут или через два часа, 2,5 часа или три часа после приема пищи. Таким образом, BH4 может приниматься пациентом в виде жидкого продукта или продукта, представляющего собой твердую или полутвердую лекарственную форму, предварительно растворенную до ее приема. В дополнительном варианте изобретения BH4 может также растворяться в полости рта при его приеме в виде твердой или полутвердой лекарственной формы, которую он имел до его проглатывания в растворенной форме.
Такие способы позволяют максимально увеличить скорость абсорбции и повысить биологическую доступность только в том случае, если BH4 будет полностью растворяться в растворе или в биологических жидкостях до его доставки на участки абсорбции, которыми являются, главным образом, желудок и тонкий кишечник. Растворение активных фармацевтических ингредиентов или лекарственного средства в растворе является необходимым условием для их абсорбции в систему кровообращения (в кровь и лимфатическую систему). При пероральном введении твердых лекарственных форм, таких как таблетки и капсулы, такие лекарственные формы перед их абсорбцией в систему кровообращения проходят последовательную серию стадий, таких как дезинтеграция с образованием гранул, дезагрегация с образованием порошков и растворение. Прохождения таких стадий можно избежать путем введения жидких лекарственных форм, полутвердых лекарственных форм и быстрорастворяющихся твердых лекарственных форм. Таким образом, активное вещество является более доступным для абсорбции на более ранней стадии, а поскольку отсутствует гарантия того, что твердая лекарственная форма будет высвобождать все находящиеся в ней активные вещества перед ее прохождением через участки абсорбции, то составы, в которых активное вещество присутствует в растворенной форме еще до его поступления на участки абсорбции, обычно должны обладать более высокой биологической доступностью.
Такие лекарственные формы имеют меньшую вариабельность их уровней в крови, поскольку в данном случае отсутствует вариабельность, обусловленная дезинтеграцией и растворением лекарственной формы в организме человека in vivo. Скорость дезинтеграции и растворения in vivo твердой лекарственной формы ВН4, предназначенной для быстрого высвобождения в желудке, зависит от изменения рН желудочного сока у конкретного человека - при приеме пищи и натощак (в условиях голодания), - и интенсивности высвобождения из желудка, как было определено по скорости высвобождения из кишечника и скорости опорожнения кишечника в тонкой кишке. Поскольку жидкие лекарственные формы, полутвердые лекарственные формы, пастилки/леденцы и быстрорастворяющиеся твердые лекарственные формы не подвергаются дезинтеграции и растворению, то их уровни в крови являются менее вариабельными, чем уровни ВН4 в крови, вводимого в виде твердых лекарственных форм быстрого высвобождения (таблеток и капсул).
В другом репрезентативном варианте изобретения указанные способы включают введение BH4 в твердой лекарственной форме, включая, но не ограничиваясь ими, таблетки, капсулы, леденцы, пастилки, порошки и гранулы, или в полутвердой форме, включая, но не ограничиваясь ими, желе для перорального введения, содержащее лекарственное вещество, которое разжевывается или проглатывается без предварительного растворения в жидкости, например в воде, апельсиновом соке, яблочном соке и т.п. В одном из вариантов изобретения пациент принимает BH4 для проглатывания в условиях голодания, то есть натощак. В настоящем изобретении также рассматривается введение BH4 в твердой или полутвердой лекарственной форме для проглатывания в определенный период времени, например утром, днем и ночью того же дня, натощак, один или несколько раз в день. В репрезентативных вариантах изобретения пациент принимает данную композицию натощак, например, по меньшей мере за 30 минут, 45 минут, или по меньшей мере за один час до приема пищи, и/или по меньшей мере через 90 минут или через два часа, 2,5 часа или три часа после приема пищи.
В другом варианте изобретения указанные способы включают введение BH4 независимо от того, принимается ли он пациентом в виде твердой или полутвердой лекарственной формы, или в виде лекарственной формы, растворенной в жидкости, вместе с пищей, например с пищей с высоким содержанием жира или вместе с жирной и/или высококалорийной пищей. В настоящем изобретении также рассматривается введение BH4 независимо от того, проглатывается ли он пациентом в твердом или принимается в растворенном виде в определенный период времени, например утром, днем и ночью того же дня, вместе с пищей, например с пищей с высоким содержанием жира, или вместе с жирной и/или высококалорийной пищей один или несколько раз в день. В репрезентативном варианте изобретения ВН4 принимают один раз в день в виде твердой лекарственной формы сразу после приема пищи. В предпочтительном варианте изобретения твердая лекарственная форма составлена в виде таблетки или капсулы. В других репрезентативных вариантах изобретения ВН4 употребляют вместе с пищей во время ее приема, составляющего примерно 0-60 минут, примерно 0-30 минут или 5-20 минут. Независимо от того, принимается ли ВН4 в виде твердой лекарственной формы, жидкой лекарственной формы или в виде раствора, его действие in vivo (или его биологическая доступность) является более эффективным при его употреблении сразу после приема пищи по сравнению с его действием натощак.
Прием BH4 может быть осуществлен приблизительно одновременно с приемом пищи, либо BH4 может приниматься до или после приема пищи. Период времени между приемом пищи, например жирной пищи или пищи с высоким содержанием жира и/или высококалорийной пищи, и приемом BH4, независимо от того проглатывается ли он или употребляется в растворенном виде, может составлять по меньшей мере 5 минут. ВН4 может быть введен через 60 минут, 30 минут, 25 минут, 20 минут, 15 минут, 10 минут или 5 минут после приема пищи.
В другом варианте изобретения для некоторых пациентов, например для взрослых, или в случае некоторых болезненных состояний, например сердечно-сосудистых заболеваний или других заболеваний, ассоциированных с дисфункцией NOS, способы согласно изобретению включают прием целой, а не растворенной в жидкости таблетки в целях улучшения биологической доступности.
Введение BH4 способами согласно изобретению приводит к увеличению средних значений концентраций в плазме, и/или к повышению скорости абсорбции в желудочно-кишечном тракте, и/или к увеличению средних значений для Cmax и/или AUC(0-t) и/или AUC (inf) по сравнению со значениями, которые были получены при введении ВН4 в условиях голодания.
Введение целой таблетки натощак приводит, в среднем, к 20% увеличению параметров Cmax и AUC по сравнению с аналогичными параметрами при введении растворенных таблеток. Введение таблетки, растворенной в воде или в апельсиновом соке, или целой таблетки после приема пищи с высоким содержанием жира/высококалорийной пищи приводит к увеличению Cmax и AUC в пределах от приблизительно 30% (целая таблетка) до 80% (вода). Введение BH4 в виде целой таблетки после приема пищи с высоким содержанием жира и высококалорийной пищи приводит к приблизительно 30%-ному увеличению уровня абсорбции по сравнению с введением ВН4 натощак. Введение BH4 в виде целой таблетки приводит к приблизительно 20%-ному увеличению уровня абсорбции по сравнению с введением ВН4 в виде растворенных таблеток.
Термин «средняя концентрация в плазме» означает средние величины концентрации в наборе проб плазмы.
Термин «Cmax» означает максимальную наблюдаемую концентрацию в плазме.
Термин «AUC» означает площадь под кривой зависимости концентрации в плазме от времени.
Термин «AUC0-t» означает площадь под кривой зависимости концентрации в плазме от времени начиная со времени 0 до времени последнего измерения концентрации.
Термин «AUC(inf)» означает вычисленную площадь под кривой зависимости концентрации в плазме от времени начиная со времени 0 до бесконечности.
«Скорость абсорбции BH4 в желудочно-кишечном тракте» оценивают путем вычисления площади под кривой зависимости увеличения концентрации общего биоптерина в плазме (ΔCp) от времени (ΔAUC) после введения BH4 по следующей формуле:
Скорость абсорбции (%) = (ΔAUC после p.o. дозы/ΔAUC после i.v. дозы)×(i.v.-доза/p.o. доза × 100)
При этом, предпочтительно, используется 6R-BH4 с чистотой по меньшей мере 99,5%. В способах и композициях согласно изобретению может быть использована любая соль, включая гидрохлоридную соль и любую кристаллическую форму BH4. Различные соли и кристаллические формы описаны в публикации заявки на патент США № 2006/0040946, которая во всей своей полноте вводится в настоящее описание посредством ссылки, а стабильный твердый состав описан в международной публикации № WO 06/55511, которая также во всей своей полноте вводится в настоящее описание посредством ссылки. Различные кристаллические формы могут быть легко приготовлены в виде таблеток, порошков или других твердых составов для перорального введения.
Во втором аспекте настоящее изобретение относится к способу стабилизации BH4 путем снижения рН тонкого кишечника с использованием протонообменных полимеров. BH4 вводят ежедневно в виде твердой или жидкой лекарственной формы для перорального введения, содержащей активные ингредиенты, которые повышают стабильность BH4 в области, расположенной за желудком, благодаря снижению pH в тонком кишечнике и, тем самым, предохраняют BH4 от быстрого окисления. Поскольку BH4 в кислотной среде является более стабильным, чем в основной среде, то в твердые лекарственные составы ВН4 (таблетки, капсулы и т.п.) включают подкисляющие наполнители/неактивные ингредиенты в целях снижения рН жидкости тонкого кишечника и, тем самым, повышения химической стабильности. Большая площадь или «окно» желудочно-кишечного тракта (ЖКТ), доступные для абсорбции, оптимизируют уровень абсорбции посредством расширения ограниченного потоком окна абсорбции, которое, очевидно, ограничивается желудком и отделом, простирающимся от двенадцатиперстной кишки до тонкой кишки. Такими лекарственными формами являются, но не ограничиваются ими, шипучие таблетки, порошки и гранулы (ресуспендируемые в жидкости перед введением) и вещества-подкислители. В отличие от небольших кислотных молекул крупные полимерные кислоты сохраняются в желудочно-кишечном тракте в течение более длительного времени и не абсорбируются в ЖКТ, однако они отдают свои протоны физиологическим жидкостям в ЖКТ, снижая, тем самым, рН среды. Примерами эксципиентов/неактивных ингредиентов, содержащихся в данном составе, являются низкомолекулярные карбоновые кислоты, такие как малеиновая, фумаровая и лимонная кислоты, или неорганические небольшие молекулы, такие как фосфорная кислота, уксусная кислота и их соли. Другими примерами являются фармацевтически приемлемые кислоты, например кислоты, принадлежащие к классу полимерных карбоновых кислот, включая полиметакриловые кислоты, карбомеры, поликарбофил, эудрагиты, кислотные формы кросскармелозы и крахмал, содержащий гликолевую кислоту и т.п. Указанные составы также содержат дополнительные наполнители, повышающие стабильность, такие как антиоксиданты (например, тиолы, такие как цистеин, N-ацетилцистеин и т.п.; аскорбиновая кислота; метионин и т.п.) и другие эксципиенты, которые обычно используются в промышленности для улучшения технологических свойств и повышения качества и эксплуатационных показателей данного состава.
В третьем аспекте изобретения рассматривается способ увеличения времени пребывания ВН4 в кишечнике, включая, но не ограничиваясь им, замедление высвобождения из кишечника с использованием агента, способствующего замедлению высвобождения ВН4 из кишечника, например такого агента, как сложный эфир жирной кислоты и/или сложный эфир жирной кислоты и глицерина. Жирными кислотами могут быть олеиновая кислота, стеариновая кислота, арахиновая кислота, пальмитиновая кислота, арахидоновая кислота, линолевая кислота, линоленовая кислота, эруцидиновая кислота, миристиновая кислота, лауриновая кислота, миристолеиновая кислота и пальмитолеиновая кислота. Также рассматривается способ увеличения времени пребывания BH4 в кишечнике посредством увеличения времени его удерживания в желудке с использованием альгиновой кислоты и усиления биоадгезии с использованием поликарбофила. В одном из вариантов изобретения лекарственные формы ВН4 вводят в виде пероральных флотирующих составов, которые растекаются в жидкости желудка и соответствующим образом высвобождают BH4 и удерживаются в желудке в течение более длительного периода времени, поскольку они являются более резистентными к высвобождению из желудка, чем нефлотирующие составы или составы, которые быстро растворяются в желудке. Такой разработанный способ основан на удерживании данной лекарственной формы в желудке благодаря использованию газообразующего эксципиента, присутствующего в данной лекарственной форме; эксципиентов низкой плотности, сообщающих лекарственной форме текучесть в жидкостях ЖКТ, или комбинации газов и веществ низкой плотности, присутствующих в лекарственной форме и сообщающих ей способность к флотации в содержимом жидкости ЖКТ. Более длительное пребывание и высвобождение лекарственной формы в желудочной среде, где BH4 является более стабильным в кислотной жидкости, будет увеличивать время пребывания лекарственной формы в желудке и повышать стабильность BH4, что сделает ВН4 более доступным для длительной абсорбции в желудке и двенадцатиперстной кишке, чем стандартные лекарственные формы в виде таблеток и капсул. Составы BH4 могут содержать один или несколько антиоксидантов, наполнителей, которые, как известно специалистам в данной области, способствуют улучшению технологических свойств твердой лекарственной формы и сообщают ей способность к дезинтеграции/растворению, а также один или несколько дополнительных эксципиентов, которые образуют газ или газовую смесь (например, диоксид углерода) после контактирования указанного состава с водной средой и/или с жидкостями ЖКТ. Предпочтительными являются водорастворимые антиоксиданты, например аскорбиновая кислота, метионин и тиолы (цистеин, N-ацетилцистеин и глутатион), или антиоксиданты, которые превращаются в растворимый антиоксидант в ЖКТ, например аскорбилпальмитат, который превращается в аскорбиновую кислоту в ЖКТ. Эксципиентами, добавляемыми в данный состав, являются карбонаты и бикарбонаты, которые непосредственно реагируют с BH4 с образованием диоксида углерода, а также описанные ранее небольшие и крупные полимерные кислоты, которые реагируют с карбонатами и бикарбонатами с образованием дополнительного количества диоксида углерода, если это необходимо.
В другом варианте изобретения рассматривается введение лекарственных форм BH4, которые надолго прилипают к поверхности слизистой ЖКТ (то есть биоадгезивные составы), предпочтительно, но не только, в желудке, где благодаря кислотности желудочного сока BH4 является более стабильным, чем в тонком кишечнике. BH4 регулируемым образом высвобождается из биоадгезивной лекарственной формы. Твердую лекарственную форму получают так, чтобы она содержала BH4, один или несколько антиоксидантов, наполнителей, которые, как известно специалистам в данной области, сообщают твердой лекарственной форме технологические свойства и способность к регулируемой дезинтеграции/растворению, и одну и несколько биоадгезивных добавок, таких как поликарбофил, присутствующий в свободной кислотной или в солевой форме. Другие полимерные кислоты, такие как полиметакриловые кислоты, карбомеры и производные целлюлозы, например HPMC, HPC и т.п., могут быть объединены с поликарбофилом или заменены поликарбофилом. Антиоксиданты, предпочтительно, являются растворимыми, и такими антиоксидантами являются, например, аскорбиновая икслота, метионин, цистеин, N-ацетилцистеин и глутатион, либо они могут быть превращены в растворимый антиоксидант, такой как аскорбиновая кислота, в ЖКТ, например аскорбилпальмитат. В одном варианте осуществления изобретения компоненты состава смешивают и приготавливают в виде твердой лекарственной формы, например в виде таблеток или капсул. Твердая лекарственная форма может быть покрыта энтеросолюбильным покрытием для доставки ВН4 в отделы, расположенные ниже желудка, то есть в тонкий кишечник, либо их получают без энтеросолюбильного покрытия для высвобождения ВН4 в желудке. В другом варианте изобретения компоненты твердой лекарственной формы могут быть разделены на различные части, и эти различные части смешивают по отдельности, а затем обрабатывают с получением многослойных лекарственных форм. Многослойная лекарственная форма может содержать биоадгезив и несколько эксципиентов во внешнем слое таблетки, обернутом вокруг других слоев, содержащих ВН4 (то есть активную область внутри биоадгезивной оболочки), либо такая форма представляет собой обернутую цилиндрическую массу, вложенную в капсулу, где один или несколько других слоев собраны под биоадгезивной оболочкой или внутри этой оболочки. Альтернативно, биоадгезив и другие слои в такой массе таблетки или капсулы могут быть расположены в виде двухслойной или многослойной конфигурации с параллельно расположенными слоями. Такие конструкции позволяют биоадгезиву взаимодействовать с оболочкой ЖКТ или со слизью оболочки ЖКТ, что приводит к прикреплению лекарственной формы к этой оболочке, замедляя транспорт такой лекарственной формы через желудочно-кишечный тракт и повышая время ее пребывания в желудочно-кишечном тракте. Такие лекарственные формы могут быть также покрыты энтеросолюбильным покрытием. В другом варианте способа, осуществляемого с использованием ВН4, применяются полимерные неактивные ингредиенты (эксципиенты), которые имеют функциональные группы, связывающиеся со слизью ЖКТ и замедляющие транспорт лекарственной формы через ЖКТ. Лекарственные формы, содержащие ВН4, приготавливают с использованием тиолсодержащих полимерных наполнителей (полимер-SH), таких как поликарбофил-цистеин, полиполиметакриловая кислота-цистеин, карбоксиметилцеллюлоза-цистеин, производные хитозана-цистеин и т.п. Такие тиолсодержащие полимеры наделяют ВН4 биоадгезивными и антиоксидантными свойствами, которые значительно усиливают абсорбцию. Другие эксципиенты, включенные в эти составы, представляют собой антиоксиданты и эксципиенты, улучшающие технологические и эксплуатационные свойства.
В еще одном варианте изобретения пероральные лекарственные формы для периодического введения, содержащие неактивные эксципиенты или активные ингредиенты, используются для замедления высвобождения из кишечника. Замедление транспорта лекарственной формы, содержащей ВН4, через желудочно-кишечный тракт способствует увеличению времени пребывания молекулы в желудочно-кишечном тракте и позволяет абсорбироваться большей части вводимой дозы. Эксципиенты, обычно рассматриваемые как безопасные (GRAS) эксципиенты, используемые в пероральных составах для замедления опорожнения желудка и/или замедления высвобождения из тонкого кишечника, предпочтительно, включают пищевые жиры, такие как жирные кислоты, глицериды жирных кислот и производные жирных кислот и глицеридов, такие как CremophorTM (производные полиоксилированного касторового масла) и т.п. Активными наполнителями являются агенты, замедляющие перистальтику кишечника, такие как общие или селективные (M3) антимускариновые или антихолинергические агенты.
В четвертом аспекте изобретения рассматривается способ модификации высвобождения BH4 с использованием состава с замедленным высвобождением, такого как HPMC, карбомер и т.п. Такая концепция включает доставку лекарственных форм ВН4 в желудочно-кишечный тракт посредством модификации или изменения быстрого высвобождения ВН4 на замедленное, пролонгированное, регулируемое и/или регулируемое во времени высвобождение. Такое замедленное, пролонгированное и регулируемое высвобождение достигается с использованием эксципиентов, известных специалистам, а ВН4 защищают от химического разложения в системе доставки путем включения агентов, повышающих стабильность, таких как антиоксиданты. Такие способы могут максимизировать биологическую доступность, поскольку ВН4 является стабилизированным в данном составе и в среде, окружающей такой состав, что позволяет активной молекуле абсорбироваться в систему кровообращения по мере прохождения такого состава через весь ЖКТ. Этот способ обеспечивает большее «окно» ЖКТ для абсорбции посредством предотвращения разложения ВН4 в среде с более высоким рН, в результате чего ВН4 становится доступным для абсорбции. Антиоксиданты могут быть включены в данный состав для защиты лекарственного средства от разложения в жидкостях тонкого кишечника, что обусловлено тем, что жидкости тонкого кишечника имеют значение рН, близкое к нейтральному. Замедленное, пролонгированное и регулируемое высвобождение может также обеспечивать доставку ВН4 в области ЖКТ с низким кислородным потенциалом. Регулируемое во времени высвобождение достигается с использованием эксципиентов, известных специалистам, таких как рН-чувствительные полимеры, которые растворяются только в том случае, когда рН достигает значения, при котором данный полимер становится растворимым.
В другом варианте изобретения лекарственную форму ВН4 покрывают энтеросолюбильным покрытием для того, чтобы понять, действительно ли включение кислотных эксципиентов в лекарственную форму ВН4 будет приводить к повышению уровня абсорбции ВН4 посредством снижения рН тонкого кишечника и, таким образом, к стабилизации ВН4 в тонком кишечнике, что будет делать его доступным для абсорбции. Таким образом, энтеросолюбильное покрытие позволяет сохранять наполнители и лекарственные средства на участке, в котором, как ожидается, указанный наполнитель будет защищать ВН4. Если лекарственная форма ВН4 будет дезинтегрироваться в желудке, то кислотные эксципиенты, взятые вместе, не смогут обеспечивать опорожнение желудка и не смогут обеспечивать нужную защиту.
Энтеросолюбильное покрытие защищает соединения, восприимчивые к катализируемому кислотой разложению в желудке, от их разложения в желудке под действием кислоты. Материалы для энтеросолюбильного покрытия предупреждают высвобождение активного соединения из таблетки или капсулы в желудок, поскольку материалы для энтеросолюбильного покрытия не растворяются в кислоте. После того как лекарственная форма с энтеросолюбильным покрытием достигает тонкого кишечника, где величина рН варьируется в пределах рН 5-8, указанные материалы становятся растворимыми и высвобождают активное вещество в тонкий кишечник. В противоположность этому составы с замедленным высвобождением приготавливают так, чтобы они высвобождали лекарственные средства на большем отрезке/большей площади ЖКТ, насколько это возможно. Покрытие состава с замедленным высвобождением, необходимое для высвобождения лекарственного средства сразу после его выхода из желудка, может быть обязательным только в том случае, когда лекарственные средства, содержащиеся в этом составе, не устойчивы к действию кислоты.
В пятом аспекте изобретения рассматривается введение BH4 в виде стерильной жидкой или стерильной твердой лекарственной формы способами, отличающимися от перорального введения и включающими, но не ограничивающимися ими, местное введение, внутривенное введение, подкожное введение, внутримышечное введение, интратекальное введение, офтальмическое введение и введение путем ингаляции. ВН4 приготавливают в виде стерильной жидкой или твердой лекарственной формы в соответствующей желаемой концентрации.
Преимуществами стерильной жидкой лекарственной формы BH4 для внутривенного введения могут быть: (1) более предсказуемая кинетика с возможным увеличением уровней такой формы в сыворотке; (2) отсутствие каких-либо требований к функциональности желудочно-кишечного тракта; (3) необязательное участие пациента; и (4) отсутствие проблем, связанных с несоблюдением режима приема лекарственного средства. Составы для внутривенного введения BH4 могут быть особенно подходящими при патологических состояниях, лечение которых требует ускоренной доставки жидкостей и лекарственных средств по всему организму или в отдельные участки организма, которые обычно недоступны при пероральном или других способах введения, включая состояния, которыми являются, но не ограничивающиеся ими, бешенство, менингит, трансплантация/сохранение органов, субарахноидальные геморрагии, травмы головного мозга, инсульт, хирургическая операция по шунтированию коронарной артерии, спазм сосудов головного мозга, переливание/консервация крови, легочная гипертензия, серповидно-клеточная болезнь, преэклампсия и сосудистые заболевания, возникающие после химиотерапии.
BH4 является в высокой степени восприимчивым к окислению в водном растворе и в физиологических водных рН-корректирующих растворах (Davis, et al., Eur. J. Biochem. 173, 345-351 (1988); Kirsch, et al., J. Biol. Chem. 278, 24481-24490 (2003)). Стабильность BH4 определяют, главным образом, в растворах с рН со значениями от нейтрального до слабо щелочного, то есть с рН 7,4, для имитации возможного стабильного поведения ВН4 в условиях физиологических значений рН плазмы. Хотя в Европейской патентной заявке № 1757293A описаны жидкие составы или составы в виде сиропа, однако такие составы обычно состоят из твердых порошкообразных смесей или гранул, которые перед их пероральным введением должны быть разведены водой. В одном из аспектов настоящего изобретения рассматриваются жидкие составы, которыми являются, но не ограничиваются ими, порошки или гранулы, требующие разведения водой. В настоящем изобретении также рассматриваются приготовленные по индивидуальному рецепту («компаундированные») жидкие составы, являющиеся стабильными при комнатной температуре в течение определенного периода времени, достаточного для их обработки в стерильных условиях, необходимых для заполнения/получения готового продукта, и его заполнения в виде жидкого продукта в ампулы, бутыли или сосуды или для его заполнения в сосуды с последующей сушкой вымораживанием и получением лиофилизованных продуктов.
Жидкие и лиофилизованные составы для разведения могут быть также введены в носовую полость путем закапывания в глаза и наружный слуховой проход для достижения нужного терапевтического эффекта. Для получения состава лиофилизованного продукта необходимо предварительное растворение ВН4 в жидкости, предпочтительно, в водной жидкости, и обработка жидкого продукта в стерильных условиях (то есть компаундирование, стерильная фильтрация и заполнение стерильно отфильтрованной жидкости в сосуды перед загрузкой заполненных сосудов в лиофилизатор для лиофилизации). Поддержание стабильности солюбилизированного ВН4 в условиях стерильной обработки и предотвращение его разложения являются ключевыми условиями для изготовления лиофилизованного продукта, который удовлетворяет техническим требованиям по допустимому присутствию примесей в готовом продукте. Поэтому композиция лиофилизованного продукта содержит подходящие стабилизаторы, которые позволяют минимизировать или предотвращать разложение ВН4 в процессе приготовления готового продукта. Описанные здесь составы должны стабилизировать растворы ВН4 в процессе стерильного приготовления готового продукта, то есть способа, который осуществляют в течение, как минимум, шести часов и в результате которого получают коммерчески доступный стабильный продукт.
Указанные составы включают BH4 в концентрации, предпочтительно, составляющей в пределах от 0,1 мг/мл до 10 мг/мл. Благодаря высокой растворимости BH4 могут быть также получены составы, концентрация которых составляет, например, до приблизительно 100 мг/мл. Для получения в высокой степени концентрированных растворов могут быть применены общие относительно сложные составы и способы, описанные в настоящей заявке.
Жидкие составы BH4, предпочтительно, приготавливают в буферных растворах с pH 1-8, а предпочтительно, в буферных растворах с pH 2-7. В качестве рН-корректирующих буферов используют буферные соединения, обладающие высокой забуферивающей способностью при конкретном нужном pH, которые выбирают по принципу наибольшей близости значений константы или константы ионизации буфера к нужным значениям pH жидкого состава. Таким образом, могут быть использованы любые буферные соединения при условии, что одна или несколько констант ионизации соединения будут близки к нужным значениям рН данного состава. Примерами буферов, которые могут быть использованы при рН 1-8, являются различные кислоты/основания и их соответствующие сопряженные кислоты/основания или их солевые формы, включая, но не ограничиваясь ими, соляную кислоту (pH 1-2), малеиновую кислоту (pH 1-3), фосфорную кислоту (pH 1-3), лимонную кислоту (pH 3-6), уксусную кислоту (pH 4,7±1,0), двухосновный фосфат натрия (pH 6-8), трометамин (трис, pH 8,3±1,0) и т.п.
Составы для внутривенного введения
Составы для внутривенного введения стабилизируют с использованием антиоксиданта или комбинации из 2 или более антиоксидантов. Для предупреждения нестабильности состава комбинации антиоксидантов могут быть введены синергически. Барботирование инертными газами и/или диоксидом углерода для удаления из раствора растворенного кислорода является необязательным условием, однако такое барботирование предпочтительно проводить в том случае, если используются низкие концентрации антиоксидантов, а еще более предпочтительно, в случае использования низких концентраций ВН4 и антиоксидантов. На стабилизацию BH4 в водном растворе влияют взаимодействия ВН4, взятого в определенной концентрации, с антиоксидантом и значения рН. Так, например, при высоких концентрациях ВН4 требуются меньшие концентрации антиоксиданта, чем при низких концентрациях ВН4. Кроме того, BH4 является более стабильным при низком pH, чем при высоком рН. Поэтому желательно, чтобы составы с высокими значениями рН, предпочтительно, имели более высокие концентрации антиоксидантов, более предпочтительно, содержали комбинацию из 2, 3 или более антиоксидантов, а еще более предпочтительно, были барботированы неокисляющим газом (например, инертным газом или диоксидом углерода) с последующим герметичным или почти герметичным закрытием первого контейнера в атмосфере неокисляющего газа (например, инертного газа или диоксида углерода) в целях повышения стабильности лекарственного продукта.
Ряд репрезентативных жидких составов ВН4 представлен в таблицах 1 и 2. Приготовленные или «компаундированные» растворы барботируют, но необязательно, инертным газом (например, аргоном или азотом) или диоксидом углерода в резервуаре для специального приготовления составов, при этом первые контейнеры, предпочтительно, герметично закрывают и помещают в атмосферу инертного газа или диоксида углерода для удаления кислорода из головной наджидкостной части контейнера. Объем этого состава может быть увеличен до любой величины путем умножения количества компонентов на соответствующий масштабный коэффициент.
|
|
Антиоксиданты, используемые для жидких составов, предпочтительно, выбирают из одного или нескольких соединений, таких как соединения на основе тиола (например, L-цистеин), аскорбиновая кислота и соединения на основе сульфита (например, метабисульфит натрия). Растворы, предпочтительно, барботируют инертными газами или диоксидом углерода для удаления кислорода из растворов BH4, а затем герметично запаивают в ампулы или помещают в сосуды и бутыли, герметично закрывающиеся крышками типа металлических крышек, обычно используемых для бутылок с пивом, в потоке инертных газов (например, аргона, азота) или неинертного газа, такого как диоксид углерода, для предохранения барботируемых газов, присутствующих в верхнем наджидкостном пространстве контейнера, от их утечки. Кроме того, жидкие составы для перорального введения предпочтительно содержат подсластители и ароматизаторы для улучшения вкусовых качеств данных составов.
В одном из вариантов изобретения BH4, используемый в виде жидкой лекарственной формы, стабилизируют путем добавления антиоксидантов и/или барботирования неокисляющими, предпочтительно, стерилизованными газами, такими как инертные газы (например, азот, аргон, гелий и т.п.) и/или неинертным газом, таким как диоксид углерода, для удаления молекулярного кислорода из состава. Продукт, предпочтительно, заполняют в потоке инертных газов для минимизации или предотвращения повторного растворения молекулярного кислорода в составе. Контейнер (например, сосуды, ампулы и т.п.) заполняют жидкостью, а затем его герметично закрывают для предотвращения попадания в него кислорода. В другом варианте изобретения раствор ВН4, используемый в виде стерильной твердой лекарственной формы для парентерального введения, лиофилизуют и разводят в клинических условиях перед введением. В еще одном варианте изобретения ВН4, используемый в виде стерильного порошкообразного лекарственного средства, непосредственно упаковывают в стерильные контейнеры (например, в сосуды, пакеты, бутыли или ампулы) в стерильных условиях заполнения сухого порошка. Таким образом, в другом своем аспекте настоящее изобретение относится к сухому порошкообразному составу тетрагидробиоптерина (ВН4) или его фармацевтически приемлемой соли для разведения в водном растворе, где указанный состав включает сухую порошкообразную смесь ВН4 или его фармацевтически приемлемой соли, антиоксидант и рН-корректирующий буфер.
Композиция жидкого состава для перорального введения
Жидкие составы для перорального введения помимо компонентов, используемых в обычных жидких составах и составах для внутривенного введения, содержат подсластители и ароматизаторы. Подсластители и ароматизаторы добавляют в количестве, достаточном для достижения приемлемого вкуса и запаха. Жидкие составы для перорального введения содержат один или несколько стабилизаторов. Такие составы могут содержать, но необязательно, противомикробные консерванты. Данные составы, предпочтительно, забуферивают при низком рН, например, рН 1-4, а забуферивающие агенты выбирают так, чтобы они были совместимыми с ароматизатором и улучшали органолептические свойства жидкого состава для перорального введения. Примерами предпочтительных буферов (кислот и сопряженных с ними оснований) являются: лимонная кислота, винная кислота, яблочная кислота в комбинации с сопряженными с ними основаниями или солевыми формами.
Примерами подсластителей являются сахара (например, сахароза, глюкоза, сорбит, маннит, фруктоза и т.п.), не содержащие сахара подсластители с выраженным вкусом (например, аспартам, ацесульфам К, цикламат, сахарин, сукралоза, глициризин, алитам, неотам, неогесперидин DC, тауматин, монелин и т.п.).
В дополнительном варианте изобретения BH4, используемый для интраназального, офтальмического и внутриушного введения, получают способом, обсуждаемым выше для приготовления парентеральных лекарственных форм, и он представляет собой, но необязательно, стерильный продукт. Такие лекарственные формы могут поставляться в виде упакованного набора, презентация которого осуществляется в течение нескольких дней после его поставки. Каждая лекарственная форма в наборе может состоять из одного сосуда или одной ампулы и одного ингалятора (для интраназальной лекарственной формы) или одной пипетки (для офтальмических лекарственных форм и лекарственных форм для закапывания в уши). После вскрытия сосуда или ампулы ингалятор или пипетку вставляют в сосуд или ампулу, а имеющуюся крышку удаляют. Продукт в виде лекарственной формы используют до истечения заданного периода и выбрасывают, после чего открывают новый сосуд или новую ампулу. В другом варианте изобретения рассматривается заполнение растворами герметичных пластиковых стерильных контейнеров для одноразового использования, изготовленных промышленным способом формования-заполнения в контейнер и его герметичной упаковки. Эти упаковки открывают и растворы подают нужным способом введения путем выдавливания содержащейся в ней жидкости. Эти лекарственные формы вводят один раз в день и подают через ноздри (продукт для интраназального введения) или закапывают в глаза (продукт для офтальмического введения) либо закапывают капли в слуховой канал (продукт для внутриушного введения). Упакованное лекарственное средство, изготавливаемое путем формования, его помещения в контейнер и герметичного закрытия этого контейнера, вводят соответствующим способом введения.
В другом варианте изобретения BH4 вводят трансбуккально и чрескожно в виде сформованных полосок, пластырей и пленок либо в виде продуктов для местного применения, которые наносят на нужный участок. Подъязычные таблетки кладут под язык. Такие лекарственные формы вводят один раз в день и либо прикрепляют к слизистой оболочке нужного участка (при трансбуккальном и чрескожном введении), либо кладут под язык в твердой или полутвердой лекарственной форме. Для предотвращения раздражения нужного участка на основное соединение, такое как карбонат или бикарбонат натрия, наносят покрытие и смешивают с BH4 в целях предупреждения взаимодействия с ВН4, которое делает его нестабильным. Альтернативно, основное соединение добавляют непосредственно перед применением в целях повышения рН BH4, который является очень низким. Добавление основного эксципиента во время приготовления состава без покрытия щелочных частиц, необходимого для предотвращения взаимодействия с BH4, будет приводить к нестабильности ВН4. В другом варианте изобретения на сердцевину подъязычной таблетки, содержащей BH4, наносят покрытие путем его обработки раствором, содержащим основное или щелочное вещество. В подъязычной области основное соединение сначала растворяется, а затем взаимодействует с ВН4, что приводит к повышению рН среды.
Преимущественный контейнер для упаковки жидких составов BH4
Преимущественные контейнеры для упаковки жидких составов ВН4 являются, предпочтительно, непроницаемыми для кислорода, диоксида углерода, азота и инертных газов. После заполнения барботированных жидких составов ВН4 в преимущественный контейнер, предпочтительно, в потоке азота, этот контейнер, предпочтительно, герметично закрывают для сохранения газа-барботера в жидкости и в верхнем наджидкостном пространстве контейнера, и для предупреждения утечки газа-барботера и поступления кислорода в контейнер.
Предпочтительными преимущественными контейнерами являются герметично закрытые ампулы, а также бутыли и сосуды, герметично закрытые металлической крышкой, такой как крышка, которая применяется для герметичного закрытия сосудов с содой и бутылок с пивом. При использовании ампулы вскрывают путем их разрезания и используют в течение нескольких часов, например примерно 12 часов. Ампулы могут быть использованы для внутривенного введения, а для инъекций используются стерильные продукты. Стерильные жидкости для инъекций и лиофилизованные продукты могут быть также упакованы в сосуды, плотно закрывающиеся резиновой пробкой и имеющие гофрированную алюминиевую крышку. Антиоксиданты, присутствующие в этих составах, защищают жидкие и лиофилизованные продукты от почти незаметной утечки газа-барботера или поступления кислорода в сосуд, предназначенный для длительного хранения продукта.
Жидкие составы BH4, заполняющие бутыли или сосуды для перорального, офтальмического или внутриушного введения, предпочтительно, герметично закрывают металлической крышкой, обычно используемой для напитков, или резиновой пробкой, поверх которой завинчивают гофрированную алюминиевую крышку. Бутыли или сосуды могут иметь резьбу для завинчивания крышки. При удалении герметика его заменяют завинчивающейся крышкой, снабженной или не снабженной пипеткой. Присутствие антиоксидантов в данном составе позволяет сохранять стабильность состава с завинчивающейся крышкой при его использовании в течение по меньшей мере двух недель, например, после удаления герметика.
I. Синтез тетрагидробиоптерина
Специалистам известны различные методы синтеза тетрагидробиоптеринов, их предшественников, производных и аналогов. В патентах США №№ 5698408; 2601215; 3505329; 4540783; 4550109; 4587340; 4595752; 4649197; 4665182; 4701455; 4713454; 4937342; 5037981; 5198547; 5350851; 5401844; 5698408, в канадской заявке CA 2420374, в европейских заявках №№ EP 079574, EP 191335 и в публикациях заявок на патент Японии Suntory JP 4-082888, JP 59-021685 и JP 9-157270, а также в публикациях Sugimoto and Matsuura, Bull. Chem. Soc. Japan, 48(12):3767-3768 (1975), Sugimoto and Matsuura, Bull. Chem. Soc. Japan, 52(1):181-183 (1979), Matsuura et al., Chem. Lett. (Japan), 735-738 (1984), Matsuura et al., Heterocycles, Vol. 23, No. 12, 3115-3120, 1985 и Whiteley et al., Anal BiochemA37(2):394-6 (1984) (которые вводятся в настоящее описание посредством ссылки) описаны методы получения тетрагидробиоптеринов, BH4 и их производных, которые могут быть использованы в качестве композиций согласно изобретению.
В международной публикации № WO2005049614, в патенте США № 4540783, в патенте Японии № 59-021685, и в публикациях Schircks et al., Helv. CMm. Acta, 60: 211 (1977), Sugimoto et al., Bull. Chem. Soc. Jp, 52(1):181 (1979), Sugimoto et al., Bull. Chem. Soc. Jp, 48(12):3767 (1975), Visontini et al., HeIv. CHm. Acta, 52:1225 (1969), и Matsuura et al., Chem. Lett., p 735 (1984), которые во всей своей полноте вводятся в настоящее описание посредством ссылки, описаны методы синтеза BH4.
II. Кристаллические формы гидрохлоридной соли 6R-тетрагидробиоптерина
Дигидрохлорид (6R)-L-эритро-тетрагидробиоптерина присутствует в различных кристаллических формах, включая его полиморфные формы и сольваты, некоторые из которых являются более стабильными, чем другие его формы.
Кристаллические полиморфные формы дигидрохлоридной соли (6R)-L-тетрагидробиоптерина
Полиморфная форма В
Кристаллический полиморф, который, как было обнаружено, является наиболее стабильным, называется здесь «формой В» или, альтернативно, «полиморфом В». Результаты, полученные в процессе исследования и получения дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина, указывают на присутствие нескольких известных кристаллических твердых веществ, однако ни в одном случае не были обнаружены полиморфизм и его влияние на стабильность кристаллов ВН4.
Полиморф B представляет собой слегка гигроскопичный ангидрат, который имеет наибольшую термодинамическую стабильность при температуре выше примерно 20°С. Кроме того, форма B может быть легко обработана и транспортирована благодаря ее термостабильности, возможности ее получения в нужных условиях, подходящей морфологии и размера частиц. Температура плавления этой формы составляет примерно 260°С (ΔHf>140 Дж/г), но точная температура плавления не может быть определена из-за разложения данной формы до и во время плавления. Эти уникальные свойства делают данную полиморфную форму В особенно удобной для приготовления фармацевтических составов при повышенных температурах. Полиморф В может быть получен в виде тонкодисперсного порошка, который может иметь размер частиц от 0,2 мкм до 500 мкм.
Форма B имеет картину порошковой рентгеновской дифракции, выражаемую в d-величинах (Å): 8,7 (vs), 6,9 (w), 5,90 (vw), 5,63 (m), 5,07 (m), 4,76 (m), 4,40 (m), 4,15 (w), 4,00 (s), 3,95 (m), 3,52 (m), 3,44 (w), 3,32 (m), 3,23 (s), 3,17 (w), 3,11 (vs), 3,06 (w), 2,99 (w), 2,96 (w), 2,94 (m), 2,87 (w), 2,84 (s), 2,82 (m), 2,69 (w), 2,59 (w), 2,44 (w). На фигуре 1 представлен график характеристической рентгеновской дифракционной картины формы В дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Используемые в настоящей заявке сокращения, представленные в скобках, имеют следующие значения: (vs) = очень сильная интенсивность; (s) = сильная интенсивность; (m) = умеренная интенсивность; (w) = слабая интенсивность; и (vw) = очень слабая интенсивность. Картина характеристической порошковой рентгеновской дифракции представлена на фигуре 1.
Было обнаружено, что другие полиморфы BH4 имеют удовлетворительную химическую и физическую стабильность, сохраняющуюся при обработке и приготовлении в промышленных условиях, а также высокую стабильность при хранении в чистом виде или в виде составов. Кроме того, было обнаружено, что форма В и другие полиморфы BH4 могут быть получены в очень большом количестве (например, в масштабе 100 кг) и могут храниться в течение длительного периода времени.
Все кристаллические формы (полиморфы, гидраты и сольваты), включая кристаллическую форму В, могут быть использованы для получения наиболее стабильного полиморфа В. Полиморф B может быть получен путем фазового уравновешивания суспензий аморфных или других форм, не являющихся полиморфной формой В, таких как полиморф А, в подходящих полярных и безводных растворителях. Таким образом, описанные здесь фармацевтические препараты называются препаратом полиморфной формы В дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Другие формы BH4 могут быть превращены в форму В путем диспергирования другой формы BH4 в растворителе при комнатной температуре, перемешивания суспензии при температурах окружающей среды в течение периода времени, достаточного для получения полиморфной формы В, с последующим выделением кристаллической формы В и удалением растворителя из выделенной формы В. Используемый здесь термин «температура окружающей среды» означает температуру в пределах от 0°С до 60°С, а предпочтительно, от 15°С до 40°С. Температурные условия могут быть изменены в процессе обработки и перемешивания путем постадийного или непрерывного снижения температуры. Подходящими растворителями, используемыми для превращения других форм в форму В, являются, но не ограничиваются ими, метанол, этанол, изопропанол, другие C3- и C4-спирты, уксусная кислота, ацетонитрил, тетрагидрофуран, метил-трет-бутиловый эфир, 1,4-диоксан, этилацетат, изопропилацетат, другие C3-C6-ацетаты, метилэтилкетон и другие метил-C3-C5-алкилкетоны. Время до завершения фазового уравновешивания может составлять до 30 часов, а предпочтительно, до 20 часов или менее чем 20 часов.
Полиморф B может быть также получен путем кристаллизации из смесей растворителей, содержащих примерно до 5% воды, а в частности, из смесей этанола, уксусной кислоты и воды. Было обнаружено, что полиморфная форма В дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть получена путем растворения, необязательно при повышенных температурах, предпочтительно, в твердой форме, которая является более низкоэнергетической формой, чем форма B, или формы В (6R)-L-эритро-тетрагидробиоптерина, в смеси растворителей, содержащей этанол, уксусную кислоту и воду, с последующим добавлением к раствору затравочных кристаллов, охлаждением полученной суспензии и выделением образованных кристаллов. Растворение может быть осуществлено при комнатной температуре или при температуре до 70°С, а предпочтительно, до 50°С. При этом для растворения может быть использована конечная смесь растворителей, либо исходное вещество может быть сначала растворено в воде, а затем к нему могут быть добавлены другие растворители либо одновременно, либо один за другим. Композиция смеси растворителей может иметь следующие компоненты в объемном отношении: вода:уксусная кислота:тетрагидрофуран=1:3:2-1:9:4, а предпочтительно, 1:5:4. Раствор, предпочтительно, перемешивают. Термин «охлаждение» может означать понижение температуры до -40°С-0°С, а предпочтительно, до 10°С-30°С. Подходящими затравочными кристаллами являются полиморфная форма В от другой партии или кристаллы, имеющие аналогичную или идентичную морфологию. После выделения кристаллическая форма В может быть промыта нерастворителем, таким как ацетон или тетрагидрофуран, и осушена стандартным способом.
Полиморф В может быть также получен путем кристаллизации из водных растворов посредством добавления нерастворителей, таких как метанол, этанол и уксусная кислота. Процедура кристаллизации и выделения может быть преимущественно осуществлена при комнатной температуре без охлаждения раствора. Поэтому такой процесс является особенно подходящим для его осуществления в промышленном масштабе.
В одном из вариантов описанных здесь композиций и способов композицию, включающую полиморфную форму В дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина, получают путем растворения твердой формы, не являющейся формой В или формы В дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина, в воде при температуре окружающей среды; добавления нерастворителя в количестве, подходящем для образования суспензии; необязательного перемешивания этой суспензии в течение определенного периода времени и выделения образованных кристаллов. Затем полученную композицию модифицируют с получением фармацевтической композиции, описанной ниже.
Концентрация дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина в водном растворе может составлять от 10 до 80%, в более предпочтительно, от 20 до 60% по массе данного раствора. Предпочтительными нерастворителями (то есть растворителями, используемыми для получения суспензий ВН4) являются метанол, этанол и уксусная кислота. Такой нерастворитель может быть добавлен к водному раствору. Более предпочтительно, такой водный раствор добавляют к нерастворителю. Время перемешивания после образования суспензии может составлять до 30 часов, а предпочтительно, до 20 часов или менее чем 20 часов. Выделение фильтрацией и сушку проводят известным методом, описанным выше.
Полиморфная форма В представляет собой очень стабильную кристаллическую форму, которая может быть легко отфильтрована, осушена и измельчена с образованием частиц, размер которых является подходящим для получения фармацевтических составов. Такие уникальные свойства делают полиморфную форму В особенно подходящей для фармацевтического применения.
Полиморфная форма A
Было обнаружено, что другой кристаллический полиморф дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина является стабильной формой BH4, предпочтительно, используемой для получения описанного здесь фармацевтического препарата, и такая форма называется здесь «формой А» или «полиморфом А». Полиморф A является слегка гигроскопичным и адсорбирует воду до примерно 3 масс.%, которая непрерывно выделяется при температуре 50°С-200°С и при нагревании со скоростью 10°С в минуту. Полиморф A представляет собой гигроскопичный ангидрат, который по сравнению с формой В является метастабильной формой, однако такая форма является стабильной в течение нескольких месяцев в условиях окружающей среды только в том случае, если она хранится в плотно закрытом контейнере. Форма А является особенно подходящей при ее использовании в качестве промежуточного соединения и исходного вещества для продуцирования стабильных полиморфных форм. Полиморфная форма A может быть получена в виде твердого порошка с нужным средним размером частиц, обычно составляющим от 1 мкм до примерно 500 мкм.
Полиморф A имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 15,5 (vs.), 12,0 (m), 6,7 (m), 6,5 (m), 6,3 (w), 6,1 (w), 5,96 (w), 5,49 (m), 4,89 (m), 3,79 (m), 3,70 (s), 3,48 (m), 3,45 (m), 3,33 (s), 3,26 (s), 3,22 (m), 3,18 (m), 3,08 (m), 3,02 (w), 2,95 (w), 2,87 (m), 2,79 (w), 2,70 (w). На фигуре 2 представлен график характеристической рентгеновской дифракционной картины формы А дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Полиморф A обнаруживает характеристические рамановские спектральные полосы, выражаемые волновыми числами (см-1): 2934 (w), 2880 (w), 1692 (s), 1683 (m), 1577 (w), 1462 (m), 1360 (w), 1237 (w), 1108 (w), 1005 (vw), 881 (vw), 813 (vw), 717 (m), 687 (m), 673 (m), 659 (m), 550 (w), 530 (w), 492 (m), 371 (m), 258 (w), 207 (w), 101 (s), 87 (s) см-1.
Полиморфная форма A может быть получена путем сушки вымораживанием или удаления воды из растворов дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина в воде. Полиморфная форма А дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть получена путем растворения дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина при температуре окружающей среды в воде, (1) охлаждения раствора до низких температур в целях его отверждения и удаления воды при пониженном давлении или (2) удаления воды из указанного водного раствора.
Кристаллическая форма А может быть выделена путем фильтрации, а затем осушена для выпаривания абсорбированной воды из данного продукта. Условия и методы сушки известны специалистам, и такая сушка выделенного продукта или удаление воды в соответствии с описанным здесь вариантом (2) могут быть осуществлены при повышенных температурах, например при температуре до 80°С, а предпочтительно, в пределах от 30°С до 80°С в вакууме или при повышенных температурах и в вакууме. Перед выделением осадка, полученного, как описано в варианте (2), суспензия может быть перемешана в течение определенного периода времени для проведения фазового уравновешивания. Концентрация дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина в водном растворе может составлять от 5 до 40% по массе данного раствора.
Быстрое охлаждение является предпочтительным для получения твердых растворов в качестве исходного вещества. Перед полным удалением растворителя подается пониженное давление. Сушка вымораживанием представляет собой метод, хорошо известный специалистам. Время до полного удаления растворителя зависит от подаваемого вакуума, который может составлять от 0,01 до 1 мбар, используемого растворителя и температуры замораживания.
Полиморфная форма A является стабильной при комнатной температуре или при температуре ниже комнатной, главным образом, в безводных условиях, что подтверждается тестом на фазовое уравновешивание суспензий в тетрагидрофуране или в трет-бутилметиловом эфире, перемешиваемых в течение пяти дней и 18 часов соответственно в атмосфере азота при комнатной температуре. Фильтрация и сушка воздухом при комнатной температуре не приводит к изменению полиморфной формы A.
Полиморфная форма F
Было обнаружено, что другой кристаллический полиморф дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина является стабильной формой BH4, используемой предпочтительно для получения описанного здесь фармацевтического препарата, и такая форма называется здесь «формой F» или «полиморфом F». Полиморф F является слегка гигроскопичным и адсорбирует воду примерно до 3 масс.%, которая непрерывно выделяется при температуре 50°С-200°С и при нагревании со скоростью 10°С в минуту. Полиморф F представляет собой метастабильную форму и гигроскопичный ангидрат, который является более стабильным, чем форма А при температурах ниже температуры окружающей среды и менее стабильным, чем форма В при более высоких температурах, и такая форма F является особенно подходящей при ее использовании в качестве промежуточного соединения и исходного вещества для продуцирования стабильных полиморфных форм. Полиморфная форма F может быть получена в виде твердого порошка с нужным средним размером частиц, обычно составляющим от 1 мкм примерно до 500 мкм.
Полиморф F имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 17,1 (vs.), 12,1 (w), 8,6 (w), 7,0 (w), 6,5 (w), 6,4 (w), 5,92 (w), 5,72 (w), 5,11 (w), 4,92 (m), 4,86 (w), 4,68 (m), 4,41 (w), 4,12 (w), 3,88 (w), 3,83 (w), 3,70 (m), 3,64 (w), 3,55 (m), 3,49 (s), 3,46 (vs), 3,39 (s), 3,33 (m), 3,31 (m), 3,27 (m), 3,21 (m), 3,19 (m), 3,09 (m), 3,02 (m) и 2,96 (m). На фигуре 3 представлен график характеристической рентгеновской дифракционной картины формы F дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Полиморф F может быть получен путем фазового уравновешивания суспензий полиморфной формы A в подходящих полярных и безводных растворителях, которые очень слабо растворяют указанные низкоэнергетические формы, а в частности в спиртах, таких как метанол, этанол, пропанол и изопропанол. Полиморфная форма F дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть также получена путем диспергирования частиц твердой формы А дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина в безводном растворителе, который очень слабо растворяет указанный дигидрохлорид (6R)-L-эритро-тетрагидробиоптерина при температуре ниже комнатной, с последующим перемешиванием суспензии при указанной температуре в течение периода времени, достаточного для продуцирования полиморфной формы F, выделением кристаллической формы F и удалением растворителя из выделенной формы F. Удаление растворителя и сушка могут быть осуществлены на воздухе, путем сушки воздухом или сухим защитным газом, таким как азот или благородные газы, либо при температуре ниже комнатной температуры, например ниже 0°С. Температура во время фазового уравновешивания, предпочтительно, составляет от 5 до 15°С, а наиболее предпочтительно, примерно 10°С.
Полиморфная форма J
Было обнаружено, что другой кристаллический полиморф дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина является стабильной формой BH4, предпочтительно, используемой для получения описанного здесь фармацевтического препарата, и такая форма называется здесь «формой J» или «полиморфом J». Полиморф J является слегка гигроскопичным и адсорбирует воду при обработке данного полиморфа в атмосфере с повышенной влажностью. Полиморф J представляет собой метастабильную форму и гигроскопичный ангидрат, который может быть снова превращен в описанную ниже форму Е, из которой он может быть затем выделен после обработки в условиях относительно высокой влажности, такой как относительная влажность выше 75%. Форма J является особенно подходящей в качестве промежуточного соединения и может быть использована в качестве исходного вещества для продуцирования стабильных полиморфных форм. Полиморфная форма J может быть получена в виде твердого порошка с нужным средним размером частиц, обычно составляющим от 1 мкм примерно до 500 мкм.
Форма J имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 14,6 (m), 6,6 (w), 6,4 (w), 5,47 (w), 4,84 (w), 3,29 (vs) и 3,21 (vs). На фигуре 4 представлен график характеристической рентгеновской дифракционной картины формы J дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Полиморф J может быть получен путем дегидратации формы E при умеренных температурах в вакууме. В частности, полиморфная форма J дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть получена из формы Е путем удаления воды из формы Е с последующей ее обработкой в вакуумной сушилке с образованием формы J при умеренных температурах, которые могут составлять в пределах от 25 до 70°С, а наиболее предпочтительно, от 30 до 50°С.
Полиморфная форма K
Было обнаружено, что другой кристаллический полиморф дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина является стабильной формой BH4, предпочтительно, используемой для получения описанного здесь фармацевтического препарата, и такая форма называется здесь «формой К» или «полиморфом К». Полиморф К является слегка гигроскопичным и адсорбирует воду примерно до 2,0 масс.%, которая непрерывно выделяется при температуре 50°С-100°С и при нагревании со скоростью 10°С в минуту. Полиморф К представляет собой метастабильную форму и гигроскопичный ангидрат, который является менее стабильным, чем форма В при более высоких температурах, и такая форма К является особенно подходящей при ее использовании в качестве промежуточного соединения и исходного вещества в целях продуцирования стабильных полиморфных форм, а в частности формы В. Полиморфная форма К может быть получена в виде твердого порошка с нужным средним размером частиц, обычно составляющим от 1 мкм примерно до 500 мкм.
Форма К имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 14,0 (s), 9,4 (w), 6,6 (w), 6,4 (w), 6,3 (w), 6,1 (w), 6,0 (w), 5,66 (w), 5,33 (w), 5,13 (vw), 4,73 (m), 4,64 (m), 4,48 (w), 4,32 (vw), 4,22 (w), 4,08 (w), 3,88 (w), 3,79 (w), 3,54 (m), 3,49 (vs), 3,39 (m), 3,33 (vs), 3,13 (s), 3,10 (m), 3,05 (m), 3,01 (m), 2,99 (m) и 2,90 (m). На фигуре 5 представлен график характеристической рентгеновской дифракционной картины формы К дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Полиморф K может быть получен путем кристаллизации из смеси полярных растворителей, содержащих небольшое количество воды, в присутствии небольшого количества аскорбиновой кислоты. Растворители, используемые в смеси растворителей, могут быть выбраны из уксусной кислоты и спирта, такого как метанол, этанол, н-пропанол или изопропанол. В частности, полиморфная форма К дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть получена путем растворения дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина в смеси уксусной кислоты и спирта или тетрагидрофурана, содержащей небольшое количество воды и небольшое количество аскорбиновой кислоты, при повышенных температурах, с последующим снижением температуры до температуры ниже комнатной для кристаллизации указанного дигидрохлорида, выделением осадка и сушкой выделенного осадка при повышенной температуре необязательно в вакууме. Подходящими спиртами являются, например, метанол, этанол, пропанол и изопропанол, при этом предпочтительным является этанол. Отношение уксусной кислоты к спирту или тетрагидрофурану может составлять от 2:1 до 1:2, а предпочтительно, примерно 1:1. Растворение дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть осуществлено в присутствии большего количества воды, а для достижения полного осаждения может быть добавлена смесь с большим числом антирастворителей. Количество воды в конечной композиции может составлять от 0,5 до 5 процентов по массе смеси растворителей, а количество аскорбиновой кислоты может составлять от 0,01 до 0,5 процентов по массе смеси растворителей. Температура растворения может составлять от 30 до 100°С, а предпочтительно, от 35 до 70°С, а температура сушки может составлять от 30 до 50°С. Осадок после его выделения, например, фильтрацией, может быть промыт спиртом, таким как этанол. Полиморф К может быть легко превращен в наиболее стабильную форму В путем фазового уравновешивания, например, в изопропаноле и необязательного добавления затравочных кристаллов формы В при температуре выше комнатной, такой как температура от 30 до 40°С.
Гидратные формы дигидрохлоридной соли (6R)-L-тетрагидробиоптерина
Как подробно описано ниже, было обнаружено, что дигидрохлорид (6R)-L-эритро-тетрагидробиоптерина представляет собой ряд кристаллических гидратов, описанных в настоящей заявке и определяемых как формы C, D, E, H и O. Эти гидратные формы могут быть использованы как стабильные формы BH4 для получения описанных здесь фармацевтических препаратов и для получения композиций, включающих стабильные кристаллические полиморфы BH4.
Гидратная форма C
Было обнаружено, что гидратная кристаллическая форма дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина представляет собой стабильную предпочтительную форму BH4, которая используется в описанных здесь фармацевтических препаратах и которая называется здесь «формой С» или «гидратом С». Гидратная форма C является слегка гигроскопичной, и содержание в ней воды составляет приблизительно 5,5 процентов по массе, что указывает на то, что форма С является моногидратом. Гидрат С имеет температуру плавления примерно 94°С (ΔHf составляет приблизительно 31 Дж/г), и такая гидратная форма С является особенно подходящей для ее использования в качестве промежуточного соединения и исходного вещества в целях получения стабильных полиморфных форм. Полиморфная форма С может быть получена в виде твердого порошка, который, предпочтительно, имеет средний размер частиц от 1 мкм примерно до 500 мкм.
Форма С имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 18,2 (m), 15,4 (w), 13,9 (vs), 10,4 (w), 9,6 (w), 9,1 (w), 8,8 (m), 8,2 (w), 8,0 (w), 6,8 (m), 6,5 (w), 6,05 (m), 5,77 (w), 5,64 (w), 5,44 (w), 5,19 (w), 4,89 (w), 4,76 (w), 4,70 (w), 4,41 (w), 4,25 (m), 4,00 (m), 3,88 (m), 3,80 (m), 3,59 (s), 3,50 (m), 3,44 (m), 3,37 (m), 3,26 (s), 3,19 (vs), 3,17 (s), 3,11 (m), 3,06 (m), 3,02 (m), 2,97 (vs), 2,93 (m), 2,89 (m), 2,83 (m) и 2,43 (m). На фигуре 6 представлен график характеристической рентгеновской дифракционной картины гидратной формы С дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Гидратная форма C может быть получена путем фазового уравновешивания полиморфной формы, такой как суспензия полиморфа В, в нерастворителе при комнатной температуре, где указанная форма содержит воду в количестве, предпочтительно, примерно 5% по массе указанного растворителя. Гидратная форма C дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть получена путем суспендирования дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина в нерастворителе, таком как гептан, C1-C4-спирты, такие как метанол, этанол, 1- или 2-пропанол, ацетаты, такие как этилацетат, ацетонитрил, уксусная кислота или простые эфиры, такие как тетрагидрофуран, диоксан, трет-бутилметиловый эфир, или смеси из двух или трех указанных нерастворителей, с последующим добавлением достаточного количества воды для получения моногидрата и перемешиванием указанной суспензии при комнатной температуре или при температуре ниже комнатной (например, 0-30°С) в течение периода времени, достаточного для получения моногидрата. Термин «достаточное количество воды» может означать 1-10 масс.%, а предпочтительно, 3-8 масс.% воды по отношению к количеству растворителя. Твердые вещества могут быть отфильтрованы и высушены на воздухе приблизительно при комнатной температуре. Твердое вещество может абсорбировать некоторое количество воды, а поэтому оно имеет количество воды, превышающее теоретическое значение 5,5 масс.%. Гидратная форма C по сравнению с формами D и B является нестабильной и легко превращается в полиморфную форму В при температуре примерно 40°С на воздухе и при более низкой относительной влажности. Форма С может быть превращена в более стабильный гидрат D путем уравновешивания суспензии при комнатной температуре.
Гидратная форма D
Было обнаружено, что другая гидратная кристаллическая форма дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина представляет собой стабильную предпочтительную форму BH4, которая используется в описанных здесь фармацевтических препаратах и которая называется здесь «формой D» или «гидратом D». Гидратная форма D является слегка гигроскопичной, и содержание в ней воды составляет приблизительно 5,0-7,0 процентов по массе, что указывает на то, что форма D является моногидратом. Гидрат D имеет температуру плавления примерно 153°С (ΔHf составляет приблизительно 111 Дж/г), и такая гидратная форма D является гораздо более стабильной, чем форма С и даже остается стабильной при повышенной влажности воздуха при комнатной температуре. Поэтому гидратная форма D может быть использована либо для получения составов, либо она может быть использована в качестве промежуточного соединения и исходного вещества в целях получения стабильных полиморфных форм. Полиморфная форма D может быть получена в виде твердого порошка, который, предпочтительно, имеет средний размер частиц от 1 мкм примерно до 500 мкм.
Форма D имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 8,6 (s), 6,8 (w), 5,56 (m), 4,99 (m), 4,67 (s), 4,32 (m), 3,93 (vs), 3,88 (w), 3,64 (w), 3,41 (w), 3,25 (w), 3,17 (m), 3,05 (s), 2,94 (w), 2,92 (w), 2,88 (m), 2,85 (w), 2,80 (w), 2,79 (m), 2,68 (w), 2,65 (w), 2,52 (vw), 2,35 (w), 2,34 (w), 2,30 (w) и 2,29 (w). На фигуре 7 представлен график характеристической рентгеновской дифракционной картины гидратной формы D дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Гидратная форма D может быть получена путем добавления концентрированных водных растворов дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина приблизительно при комнатной температуре к избыточному количеству нерастворителей, таких как гексан, гептан, дихлорметан, 1- или 2-пропанол, ацетон, этилацетат, ацетонитрил, уксусная кислота или простые эфиры, такие как тетрагидрофуран, диоксан, трет-бутилметиловый эфир или смеси указанных нерастворителей, и перемешивания такой суспензии при комнатной температуре. Кристаллическое твердое вещество может быть отфильтровано, а затем осушено в атмосфере сухого азота при комнатной температуре. Предпочтительным нерастворителем является изопропанол. Добавление водного раствора может быть осуществлено по каплям во избежание внезапного выпадения осадка. Гидратная форма D дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть получена путем добавления концентрированных водных растворов дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина приблизительно при комнатной температуре к избыточному количеству нерастворителей и перемешивания полученной суспензии при комнатной температуре. Термин «избыточное количество нерастворителя» может означать отношение воды к нерастворителю от 1:10 до 1:1000. Форма D содержит небольшой избыток воды по отношению к моногидрату и, очевидно, абсорбирует воду, что обусловлено слегка гигроскопичной природой такого кристаллического гидрата. Очевидно, что из всех известных гидратов гидратная форма D является наиболее стабильной при комнатной температуре и при относительной влажности менее чем 70%. Гидратная форма D может быть использована в целях приготовления составов в условиях, при которых такой гидрат является стабильным. Температура окружающей среды может означать 20-30°С.
Гидратная форма E
Было обнаружено, что другая гидратная кристаллическая форма дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина представляет собой стабильную предпочтительную форму BH4, которая используется в описанных здесь фармацевтических препаратах и которая называется здесь «формой Е» или «гидратом Е». Содержание воды в гидратной форме Е составляет приблизительно 10-14 процентов по массе, что указывает на то, что форма Е является дигидратом. Гидрат Е образуется при температуре ниже комнатной. Гидратная форма Е является особенно подходящей в качестве промежуточного соединения и исходного вещества для получения стабильных полиморфных форм. Эта форма является особенно подходящей для получения безводной формы J после сушки в атмосфере азота или, необязательно, в вакууме. Форма Е не является гигроскопичной и остается стабильной при довольно высокой относительной влажности, то есть при относительной влажности приблизительно выше 60% и приблизительно до 85%. Полиморфная форма Е может быть получена в виде твердого порошка, который, предпочтительно, имеет средний размер частиц от 1 мкм примерно до 500 мкм.
Форма Е имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 15,4 (s), 6,6 (w), 6,5 (w), 5,95 (vw), 5,61 (vw), 5,48 (w), 5,24 (w), 4,87 (w), 4,50 (vw), 4,27 (w), 3,94 (w), 3,78 (w), 3,69 (m), 3,60 (w), 3,33 (s), 3,26 (vs), 3,16 (w), 3,08 (m), 2,98 (w), 2,95 (m), 2,91 (w), 2,87 (m), 2,79 (w), 2,74 (w), 2,69 (w) и 2,62 (w). На фигуре 8 представлен график характеристической рентгеновской дифракционной картины гидратной формы Е дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Гидратная форма Е может быть получена путем добавления концентрированных водных растворов дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина к избыточному количеству нерастворителей, охлажденных до температуры от примерно 10 до -10°С, а предпочтительно, от 0 до 10°С, и перемешивания полученной суспензии при указанных температурах. Кристаллическое твердое вещество может быть отфильтровано, а затем осушено в атмосфере сухого азота при комнатной температуре. Нерастворителями являются, например, гексан, гептан, дихлорметан, 1- или 2-пропанол, ацетон, этилацетат, ацетонитрил, уксусная кислота или простые эфиры, такие как тетрагидрофуран, диоксан, трет-бутилметиловый эфир или смеси указанных нерастворителей. Предпочтительным нерастворителем является изопропанол. Добавление водного раствора может быть осуществлено по каплям во избежание внезапного выпадения осадка. Гидратная форма Е дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть получена путем добавления концентрированных водных растворов дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина к избыточному количеству нерастворителей, охлажденных до температуры от примерно 10 до -10°С, и перемешивания полученной суспензии при температуре окружающей среды. Термин «избыточное количество нерастворителя» может означать отношение воды к нерастворителю от 1:10 до 1:1000. Предпочтительным нерастворителем является тетрагидрофуран. Другой способ получения включает обработку полиморфной формы В на воздухе при относительной влажности 70-90%, а предпочтительно, примерно 80%. Гидратная форма Е, очевидно, является дигидратом, который может абсорбировать некоторое дополнительное количество воды. Полиморфная форма Е может быть превращена в полиморф J после сушки в вакууме при умеренных температурах, которые могут составлять от 20°С до 50°С, под давлением от 0 до 100 мбар. Форма Е благодаря ее стабильности при высокой относительной влажности является особенно подходящей для получения составов в полутвердой форме.
Гидратная форма H
Было обнаружено, что другая гидратная кристаллическая форма дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина представляет собой стабильную предпочтительную форму BH4, которая используется в описанных здесь фармацевтических препаратах и которая называется здесь «формой Н» или «гидратом Н». Содержание воды в гидратной форме Н составляет приблизительно 5,0-7,0 процентов по массе, что указывает на то, что форма Н является гигроскопичным моногидратом. Гидратная форма Н образуется при температуре ниже комнатной. Гидратная форма Н является особенно подходящей в качестве промежуточного соединения и исходного вещества для получения стабильных полиморфных форм. Полиморфная форма Н может быть получена в виде твердого порошка, который, предпочтительно, имеет средний размер частиц от 1 мкм примерно до 500 мкм.
Форма Н имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 8,6 15,8 (vs), 10,3 (w), 8,0 (w), 6,6 (w), 6,07 (w), 4,81 (w), 4,30 (w), 3,87 (m), 3,60 (m), 3,27 (m), 3,21 (m), 3,13 (w), 3,05 (w), 2,96 (m), 2,89 (m), 2,82 (w) и 2,67 (m). На фигуре 9 представлен график характеристической рентгеновской дифракционной картины гидратной формы Н дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Гидратная форма Н может быть получена путем растворения дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина в смеси уксусной кислоты и воды при комнатной температуре, добавления нерастворителя для осаждения кристаллического твердого вещества, охлаждения полученной суспензии и перемешивания охлажденной суспензии в течение определенного периода времени. Кристаллическое твердое вещество отфильтровывают, а затем сушат в вакууме при комнатной температуре. Нерастворителями являются, например, гексан, гептан, дихлорметан, 1- или 2-пропанол, ацетон, этилацетат, ацетонитрил, уксусная кислота или простые эфиры, такие как тетрагидрофуран, диоксан, трет-бутилметиловый эфир или смеси указанных нерастворителей. Предпочтительным нерастворителем является тетрагидрофуран. Гидратная форма Н дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть получена путем растворения дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина в смеси уксусной кислоты и меньшего количества воды по сравнению с количеством воды, присутствующим в вышеуказанной смеси, при комнатной температуре с последующим добавлением нерастворителя и охлаждения полученной суспензии до температуры в пределах от -10 до 10°С, а предпочтительно, от -5 до 5°С, а затем перемешиванием полученной суспензии при указанной температуре в течение определенного периода времени. Определенный период времени может означать период времени от 1 до 20 часов. Массовое отношение уксусной кислоты к воде может составлять от 2:1 до 25:1, а предпочтительно, от 5:1 до 15:1. Массовое отношение смеси уксусная кислота/вода к нерастворителю может составлять от 1:2 до 1:5. Гидратная форма Н может считаться моногидратом с небольшим избытком воды, абсорбируемой благодаря гигроскопичной природе этой формы.
Гидратная форма O
Было обнаружено, что другая гидратная кристаллическая форма дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина представляет собой стабильную предпочтительную форму BH4, которая используется в описанных здесь фармацевтических препаратах и которая называется здесь «формой О» или «гидратом О». Гидратная форма О образуется при температуре, близкой к комнатной. Гидратная форма О является особенно подходящей в качестве промежуточного соединения и исходного вещества для получения стабильных полиморфных форм. Полиморфная форма О может быть получена в виде твердого порошка, который, предпочтительно, имеет средний размер частиц от 1 мкм примерно до 500 мкм.
Форма О имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 15,9 (w), 14,0 (w), 12,0 (w), 8,8 (m), 7,0 (w), 6,5 (w), 6,3 (m), 6,00 (w), 5,75 (w), 5,65 (m), 5,06 (m), 4,98 (m), 4,92 (m), 4,84 (w), 4,77 (w), 4,42 (w), 4,33 (w), 4,00 (m), 3,88 (m), 3,78 (w), 3,69 (s), 3,64 (s), 3,52 (vs), 3,49 (s), 3,46 (s), 3,42 (s), 3,32 (m), 3,27 (m), 3,23 (s), 3,18 (s), 3,15 (vs), 3,12 (m), 3,04 (vs), 2,95 (m), 2,81 (s), 2,72 (m), 2,67 (m) и 2,61 (m). На фигуре 10 представлен график характеристической рентгеновской дифракционной картины гидратной формы О дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Гидратная форма O может быть получена путем обработки полиморфной формы F в атмосфере азота, содержащей водяной пар, при общей относительной влажности примерно 52% в течение примерно 24 часов. Тот факт, что форма F, которая является слегка гигроскопичным ангидратом, может быть использована для получения формы О при относительной влажности 52%, указывает на то, что форма O представляет собой гидрат, который является более стабильным, чем форма F при комнатной температуре в условиях повышенной влажности.
Сольватные формы дигидрохлоридной соли (6R)-L-тетрагидробиоптерина
Как подробно описано ниже, было обнаружено, что дигидрохлорид (6R)-L-эритро-тетрагидробиоптерина представляет ряд кристаллических сольватных форм, описанных в настоящей заявке и определяемых как формы G, I, L, M и N. Эти сольватные формы могут быть использованы как стабильные формы BH4 для получения описанных здесь фармацевтических препаратов и для получения композиций, включающих стабильные кристаллические полиморфы BH4.
Сольватная форма G
Было обнаружено, что этанольный сольват кристаллической формы дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина представляет собой стабильную предпочтительную форму BH4, которая используется в описанных здесь фармацевтических препаратах и которая называется здесь «формой G» или «гидратом G». Этанольная сольватная форма G имеет содержание этанола приблизительно 8,0-12,5 процентов по массе, что позволяет предположить, что эта форма G является гигроскопичным моноэтанольным сольватом. Такая сольватная форма G образуется при температуре ниже комнатной. Форма G является особенно подходящей для использования в качестве промежуточного соединения и исходного вещества в целях получения стабильных полиморфных форм. Полиморфная форма G может быть получена в виде твердого порошка, который, предпочтительно, имеет средний размер частиц от 1 мкм примерно до 500 мкм.
Форма С имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 14,5 (vs), 10,9 (w), 9,8 (w), 7,0 (w), 6,3 (w), 5,74 (w), 5,24 (vw), 5,04 (vw), 4,79 (w), 4,41 (w), 4,02 (w), 3,86 (w), 3,77 (w), 3,69 (w), 3,63 (m), 3,57 (m), 3,49 (m), 3,41 (m), 3,26 (m), 3,17 (m), 3,07 (m), 2,97 (m), 2,95 (m), 2,87 (w) и 2,61 (w). На фигуре 11 представлен график характеристической рентгеновской дифракционной картины сольватной формы G дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Этанольный сольват формы G может быть получен путем кристаллизации дигидрохлорида L-эритро-тетрагидробиоптерина, растворенного в воде, добавления большого избытка этанола с последующим перемешиванием полученной суспензии при комнатной температуре и при температуре ниже комнатной и сушкой выделенного твердого вещества на воздухе или в атмосфере азота приблизительно при комнатной температуре. Используемый здесь термин «большой избыток этанола» означает, что полученная смесь этанола и воды содержит менее чем 10% воды, а предпочтительно, примерно 3-6% воды. Этанолятная форма G дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина может быть получена путем растворения дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина в воде или в смеси воды и этанола при температуре в пределах приблизительно от комнатной температуры до 75°С, последующего охлаждения нагретого раствора до комнатной температуры и до температуры ниже 5-10°С, добавления, но необязательно, этанола для полного осаждения, перемешивания полученной суспензии при температуре от 20 до 5°С, отфильтровывания белого кристаллического твердого вещества и сушки твердого вещества на воздухе или в атмосфере защитного газа, такого как азот, приблизительно при комнатной температуре. В первом варианте такой процесс может быть осуществлен путем растворения дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина приблизительно при комнатной температуре в меньшем количестве воды с последующим добавлением избытка этанола и перемешиванием полученной суспензии в течение определенного периода времени, достаточного для достижения фазового равновесия. Во втором варианте дигидрохлорид (6R)-L-эритро-тетрагидробиоптерина может быть суспендирован в этаноле с необязательным добавлением меньшего количества воды, нагреванием этой суспензии и растворенного дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина, охлаждением полученного раствора до температуры от примерно 5 до 15°С, добавлением к данной суспензии дополнительного количества этанола и перемешиванием полученной суспензии в течение определенного периода времени, достаточного для достижения фазового равновесия.
Сольватная форма I
Было обнаружено, что содержащий уксусную кислоту сольват кристаллической формы дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина представляет собой стабильную предпочтительную форму BH4, которая используется в описанных здесь фармацевтических препаратах и которая называется здесь «формой I» или «гидратом I». В сольватной форме I, в которой присутствует уксусная кислота, содержание уксусной кислоты составляет приблизительно 12,7 процентов по массе, что позволяет предположить, что эта форма I является гигроскопичным моносольватом, содержащим уксусную кислоту. Эта сольватная форма I образуется при температуре ниже комнатной. Содержащая уксусную кислоту сольватная форма I является особенно подходящей для ее использования в качестве промежуточного соединения и исходного вещества в целях получения стабильных полиморфных форм. Полиморфная форма I может быть получена в виде твердого порошка, который, предпочтительно, имеет средний размер частиц от 1 мкм примерно до 500 мкм.
Форма I имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 14,5 (m), 14,0 (w), 11,0 (w), 7,0 (vw), 6,9 (vw), 6,2 (vw), 5,30 (w), 4,79 (w), 4,44 (w), 4,29 (w), 4,20 (vw), 4,02 (w), 3,84 (w), 3,80 (w), 3,67 (vs), 3,61 (m), 3,56 (w), 3,44 (m), 3,27 (w), 3,19 (w), 3,11 (s), 3,00 (m), 2,94 (w), 2,87 (w) и 2,80 (w). На фигуре 12 представлен график характеристической рентгеновской дифракционной картины сольватной формы I дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Содержащая уксусную кислоту сольватная форма I может быть получена путем растворения дигидрохлорида L-эритро-тетрагидробиоптерина в смеси уксусной кислоты и воды при повышенной температуре, добавления к раствору дополнительного количества уксусной кислоты, охлаждения до температуры примерно 10°С, а затем нагревания полученной суспензии до приблизительно 15°С и перемешивания полученной суспензии в течение периода времени, достаточного для достижения фазового равновесия, которое может продолжаться вплоть до 3 дней. Затем кристаллическое твердое вещество отфильтровывают и сушат на воздухе или в атмосфере защитного газа, такого как азот, приблизительно при комнатной температуре.
Сольватная форма L
Было обнаружено, что смешанный этанольный сольват/гидрат кристаллической формы дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина представляет собой стабильную предпочтительную форму BH4, которая используется в описанных здесь фармацевтических препаратах, и которая называется здесь «формой L» или «гидратом L». Форма L может содержать от 4% и почти до 13% этанола и приблизительно от 0% до 6% воды. Форма L может быть превращена в форму G путем обработки этанолом при температуре примерно от 0°С до 20°С. Кроме того, форма L может быть превращена в форму G путем обработки органическим растворителем при комнатной температуре (10°С-60°С). Полиморфная форма L может быть получена в виде твердого порошка, которая, предпочтительно, имеет средний размер частиц от 1 мкм до примерно 500 мкм.
Форма L имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 14,1 (vs), 10,4 (w), 9,5 (w), 9,0 (vw), 6,9 (w), 6,5 (w), 6,1 (w), 5,75 (w), 5,61 (w), 5,08 (w), 4,71 (w), 3,86 (w), 3,78 (w), 3,46 (m), 3,36 (m), 3,06 (w), 2,90 (w) и 2,82 (w). На фигуре 13 представлен график характеристической рентгеновской дифракционной картины сольватной формы L дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Форма L может быть получена путем суспендирования гидратной формы Е при комнатной температуре в этаноле и перемешивания суспензии при температуре от 0 до 10°С, а предпочтительно, при примерно 5°С в течение периода времени, достаточного для достижения фазового равновесия, которое может составлять от 10 до 20 часов. Затем кристаллическое твердое вещество отфильтровывают и, предпочтительно, сушат при пониженном давлении при 30°С или в атмосфере азота. Анализ с помощью TG-FTIR позволяет предположить, что форма L может содержать различные количества этанола и воды, то есть она может присутствовать в виде полиморфа (ангидрата), смешанного этанольного сольвата/гидрата или даже в виде гидрата.
Сольватная форма M
Было обнаружено, что этанольный сольват кристаллической формы дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина представляет собой стабильную предпочтительную форму BH4, которая используется в описанных здесь фармацевтических препаратах и которая называется здесь «формой М» или «гидратом М». Форма М может содержать от 4% и почти до 13% этанола и от 0% до примерно 6% воды, что позволяет предположить, что эта форма М является слегка гигроскопичным этанольным сольватом. Такая сольватная форма М образуется при комнатной температуре. Форма М является особенно подходящей для ее использования в качестве промежуточного соединения и исходного вещества в целях получения стабильных полиморфных форм, поскольку форма М может превращаться в форму G при обработке в этаноле при температуре от примерно -10°С до 15°С и в форму В при обработке в органических растворителях, таких как этанол, С3- и С4-спирты или циклические эфиры, такие как ТГФ и диоксан. Полиморфная форма М может быть получена в виде твердого порошка, который, предпочтительно, имеет средний размер частиц от 1 мкм до примерно 500 мкм.
Форма М имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 18,9 (s), 6,4 (m), 6,06 (w), 5,66 (w), 5,28 (w), 4,50 (w), 4,23 (w) и 3,22 (vs). На фигуре 14 представлен график характеристической рентгеновской дифракционной картины сольватной формы М дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Этанольный сольват формы M может быть получен путем растворения дигидрохлорида L-эритро-тетрагидробиоптерина в этаноле и выпаривания раствора в атмосфере азота при комнатной температуре, то есть при температуре от 10°С до 40°С. Форма M может быть также получена путем сушки формы G в слабом потоке сухого азота при скорости примерно 20-100 мл/мин. В зависимости от степени осушки в атмосфере азота оставшееся количество этанола может варьироваться, то есть, оно может составлять примерно 3%-13%.
Сольватная форма N
Было обнаружено, что другой сольват кристаллической формы дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина представляет собой стабильную предпочтительную форму BH4, которая используется в описанных здесь фармацевтических препаратах и которая называется здесь «формой N» или «гидратом N». Форма N может содержать всего до 10% изопропанола и воды, что позволяет предположить, что такая форма N является слегка гигроскопичным изопропанольным сольватом. Форма N может быть получена путем промывки формы D изопропанолом с последующей сушкой в вакууме примерно при 30°С. Форма N является особенно подходящей для ее использования в качестве промежуточного соединения и исходного вещества в целях получения стабильных полиморфных форм. Полиморфная форма N может быть получена в виде твердого порошка, который, предпочтительно, имеет средний размер частиц от 1 мкм до примерно 500 мкм.
Форма N имеет характеристическую картину порошковой рентгеновской дифракции с характеристическими пиками, выражаемыми в d-величинах (Å): 19,5 (m), 9,9 (w), 6,7 (w), 5,15 (w), 4,83 (w), 3,91 (w), 3,56 (m), 3,33 (vs), 3,15 (w), 2,89 (w), 2,81 (w), 2,56 (w) и 2,36 (w). На фигуре 15 представлен график характеристической рентгеновской дифракционной картины сольватной формы N дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина.
Изопропанольная форма N может быть получена путем растворения дигидрохлорида L-эритро-тетрагидробиоптерина в 4,0 мл смеси изопропанола и воды (объемное отношение смеси составляет примерно 4:1). К этому раствору медленно добавляют изопропанол (IPA, например, примерно 4,0 мл) и полученную суспензию охлаждают до 0°С и перемешивают в течение нескольких часов (например, примерно в течение 10-18 часов) при этой температуре. Полученную суспензию фильтруют и твердый остаток промывают изопропанолом при комнатной температуре. Затем полученное кристаллическое вещество сушат при комнатной температуре (например, примерно 20-30°С) и при пониженном давлении (примерно 2-10 мбар) в течение нескольких часов (например, примерно 5-20 часов). TG-FTIR-анализ указывал на 9,0%-ную потерю массы при температуре 25-200°С, что обусловлено присутствием изопропанола и воды. Полученный результат дает основание предположить, что форма N может присутствовать либо в виде изопропанольного сольвата, либо в виде смешанного изопропанольного сольвата/гидрата, либо в виде несольватированной формы, содержащей небольшое количество воды.
Для получения полиморфных форм могут быть применены методы кристаллизации, хорошо известные специалистам, такие как перемешивание суспензии (фазовое уравновешивание), осаждение, перекристаллизация, выпаривание, методы сорбции в растворителе, таком как вода, или разложение сольватов. Для кристаллизации могут быть использованы разбавленные, насыщенные или сверхнасыщенные растворы в отсутствие или в присутствии подходящего зародыша кристаллизации. Растворы могут быть получены при температуре до 100°С. Для инициации кристаллизации и осаждения может быть проведено охлаждение растворов до -100°С, а предпочтительно, до -30°С. Метастабильные полиморфы или псевдополиморфные формы могут быть использованы для получения растворов или суспензий, необходимых для образования более стабильных форм и для достижения более высоких концентраций в растворах.
Неожиданно было обнаружено, что среди гидратов и форм B и D гидратная форма D является наиболее стабильной и особенно подходящей для использования в фармацевтических составах. Формы B и D имеют определенные преимущества с точки зрения технологии их изготовления и их транспортировки, что обусловлено соответствующим размером их кристаллов и морфологией, имеют очень высокую стабильность в условиях производства составов различных типов, являются стабильными при хранении, имеют повышенную растворимость и высокую биологическую доступность. В соответствии с этим одним из вариантов описанных здесь композиций и способов является фармацевтическая композиция, включающая полиморфную форму В и/или гидратную форму D дигидрохлорида (6R)-L-эритро-тетрагидробиоптерина и фармацевтически приемлемый носитель или разбавитель.
III. Фармацевтические составы
Описанные здесь составы предпочтительно используют для перорального введения. Составами для перорального введения предпочтительно являются твердые составы, такие как капсулы, таблетки, драже и пастилки, или жидкие составы, такие как водные суспензии, эликсиры и сиропы. Различные описанные здесь формы BH4 могут быть непосредственно использованы в виде порошка (тонкодисперсных частиц), гранул, суспензий или растворов, либо они могут быть объединены вместе с другими фармацевтически приемлемыми ингредиентами путем их смешивания с компонентами и, необязательно, с их тонкодисперсными формами с последующим заполнением ими капсул, состоящих, например, из твердого или мягкого желатина, спрессовывания в таблетки, драже или пастилки или суспендирования или растворения в носителях для получения суспензий, эликсиров и сиропов. После спрессовывания в драже на такое драже может быть нанесено покрытие.
Фармацевтически приемлемые ингредиенты хорошо известны специалистам в области приготовления составов различных типов, и такими ингредиентами могут быть, например, связующие вещества, такие как природные или синтетические полимеры, эксципиенты, смазывающие вещества, поверхностно-активные вещества, подсластители и ароматизаторы, материалы для покрытий, консерванты, красители, загустители, адъюванты, противомикробные средства, антиоксиданты и носители, применяемые для получения составов различных типов. Термин «фармацевтически или фармакологически приемлемый» относится к молекулярным соединениям и композициям, разрешенным для введения человеку Управлением по контролю за качеством пищевых продуктов, медикаментов и косметических средств (США), или соответствующими Международными регуляторными органами. Используемый здесь термин «фармацевтически приемлемый носитель» включает любые и все растворители, дисперсионные среды, материалы для покрытий, антимикробные и противогрибковые средства, агенты, придающие изотоничность, агенты, замедляющие абсорбцию и т.п. Применение таких сред и агентов для получения композиций фармацевтически активных веществ хорошо известно специалистам. Для получения терапевтических композиций могут быть использованы любые стандартные среды или агенты за исключением сред или агентов, которые являются несовместимыми с такими композициями. В указанные композиции могут быть также включены дополнительные активные ингредиенты.
Исходное количество (6R)-L-эритро-тетрагидробиоптерина, используемого для получения указанного состава, может составлять, например, в пределах от примерно 30 масс.% до примерно 40 масс.% указанного состава или от примерно 32 масс.% до примерно 35 масс.%, или примерно 33 масс.% указанного состава. Конкретное количество BH4 в рассматриваемом здесь составе составляет 80 мг, 100 мг, 200 мг, 300 мг, 400 мг и 500 мг.
Для сохранения твердого состава используют связующие вещества. В некоторых случаях для сохранения полиморфных форм в безводном состоянии используются безводные связующие вещества. В некоторых случаях связующее вещество может действовать в качестве осушителя. Репрезентативными связующими веществами являются безводный двухосновный фосфат кальция и его моногидрат. Другими неограничивающими примерами связующих веществ, которые могут быть использованы в описанной здесь композиции, являются трагакантовая камедь, аравийская камедь, крахмал, желатин и биологически разлагаемые полимеры, такие как гомо- или сополиэфиры дикарбоновых кислот, алкиленгликолей, полиалкиленгликолей и/или алифатических гидроксилкарбоновых кислот; гомо- или сополиамиды дикарбоновых кислот, алкилендиаминов и/или алифатических аминокарбоновых кислот; соответствующие сополимеры полиэфира и полиамида, полиангидриды, полиортоэфиры, полифосфазен и поликарбонаты. Биологически разлагаемые полимеры могут быть линейными, разветвленными или перекрестно-сшитыми. Конкретными примерами являются полигликолевая кислота, полимолочная кислота, и сополимер D,L-лактида/гликолида. Другими примерами полимеров являются водорастворимые полимеры, такие как полиоксаалкилены (полиоксаэтилен, полиоксапропилен и их смешанные полимеры), полиакриламиды и гидроксиалкилированные полиакриламиды, полималеиновая кислота и ее сложные эфиры или амиды, полиакриловая кислота и ее сложные эфиры или амиды, поливиниловый спирт и его сложные или простые эфиры, поливинилимидазол, поливинилпирролидон и природные полимеры, такие как хитозан.
Для быстрой дезинтеграции твердых составов при абсорбции воды и увеличении их объема используются дезинтегрирующие агенты. Примерами дезинтегрирующих агентов являются поливинилпирролидон (ПВП, например, имеющийся в продаже под названием POVIDONE), повидон в перекрестно-сшитой форме (CPVP, например, имеющийся в продаже под названием CROSPOVIDONE), натрийкарбоксиметилцеллюлоза в перекрестно-сшитой форме (NaCMC, например, имеющаяся в продаже под названием AC-DI-SOL), другие модифицированные целлюлозы и модифицированный крахмал. Таблетки, полученные с использованием CPVP, обладают гораздо более быстрым дезинтегрирующим действием, чем таблетки, полученные с использованием PVP.
Для стабилизации тетрагидробиоптеринового продукта, а в частности, после его растворения в данный продукт могут быть включены антиоксиданты. Водные растворы API с низкими рН являются более стабильными, чем растворы с нейтральным или высоким рН. Антиоксиданты вводят в описанный здесь состав для предупреждения его разложения в результате окисления. Антиоксиданты могут быть подразделены, в основном, на 3 группы.
Первая группа известна как истинные антиоксиданты, которые ингибируют окисление в результате реакции со свободными радикалами, блокирующей цепную реакцию. Примерами могут служить феноловые антиоксиданты, включая бутилированный гидроксианизол (BHA), бутилированный гидрокситолуол (BHT), трет-бутилгидрохинон (TBHQ), 4-гидроксиметил-2,6-ди-трет-бутилфенол (HMBP) и 2,4,5-тригидроксибутирофенон (THBP); алкилгаллаты, включая пропилгаллат; галловую кислоту; нордигидрогваяретовую кислоту; и токоферолы, включая альфа-токоферол.
Вторая группа, состоящая из восстановителей, имеет более низкие окислительно-восстановительные потенциалы, чем лекарственные средства, которые нуждаются в защите, а поэтому они легче окисляются. Восстановители могут также действовать посредством реакции со свободными радикалами. Примерами являются аскорбиновая кислота, тиогликолевая кислота (TGA), аскорбилпальмитат, сульфиты, включая калиевые и натриевые соли сернистой кислоты (например, сульфит калия, сульфит натрия, метабисульфит натрия и бисульфит натрия) и тиоглицерин.
Третья группа состоит из антиоксидантов-синергистов, которые сами обычно обладают умеренным антиоксидантным действием, но, вероятно, усиливают действие антиоксидантов первой или второй группы посредством реакции с ионами тяжелых металлов, которая катализирует окисление. Примерами таких антиоксидантов-синергистов и хелатообразующих агентов являются лимонная кислота, яблочная кислота, эдитиновая кислота и их соли, лецитин и винная кислота.
Примерами кислотных антиоксидантов являются аскорбиновая кислота, сложные эфиры жирных кислот и аскорбиновой кислоты, такие как аскорбилпальмитат и аскорбилстеарат, и соли аскорбиновой кислоты, такие как аскорбат натрия, кальция или калия. Некислотные антиоксиданты могут быть также использованы в стабильных составах в виде таблеток. Неограничивающими примерами некислотных антиоксидантов являются бета-каротин, альфа-токоферол. Для повышения стабильности состава в виде таблетки в него могут быть добавлены кислотные добавки, включая лимонную кислоту или яблочную кислоту. Низкомолекулярными антиоксидантами являются, но не ограничиваются ими, тиолы, например цистеин, N-ацетилцистеин, глутатион и т.п., или тиолированные полимеры (полимер-SH), например поликарбофил-цистеин, полиметакриловая кислота-SH, карбоксиметилцеллюлоза-цистеин и т.п., либо такими низкомолекулярными антиоксидантами являются аскорбиновая кислота, метионин, аскорбилпальмитат и т.п. Такие антиоксиданты сообщают стабильность лекарственной форме в процессе ее прохождения через ЖКТ, а в частности, по мере увеличения pH ЖКТ на определенном расстоянии от желудка.
В одном из вариантов изобретения предпочтительной является комбинация, состоящая по меньшей мере из двух антиоксидантов-восстановителей. В другом варианте изобретения предпочтительной является комбинация, состоящая по меньшей мере из двух антиоксидантов-восстановителей вместе с кислотным антиоксидантом-синергистом и/или хелатообразующим агентом.
Смазывающие вещества повышают стабильность, твердость и однородность твердых составов. Репрезентативными смазывающими веществами являются стеарилфумарат и стеарат магния. Другими неограничивающими примерами смазывающих веществ являются природные или синтетические масла, жиры, воски или соли жирных кислот, такие как стеарат магния.
Стабильные составы согласно изобретению могут также включать, но необязательно, и другие наполнители, такие как маннит, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза или другие невосстанавливающие сахара, такие как сахароза, трегалоза, мелезитоза, плантеоза и раффиноза. Восстанавливающие сахара могут реагировать с ВН4. Другими неограничивающими примерами наполнителей, используемых в описанной здесь композиции, являются фосфаты, такие как дифосфат кальция.
Поверхностно-активные вещества, используемые в описанной здесь композиции, могут быть анионогенными, катионогенными, амфотерными или нейтральными. Неограничивающими примерами поверхностно-активных веществ, используемых в описанной здесь композиции, являются лецитин, фосфолипиды, октилсульфат, децилсульфат, додецилсульфат, тетрадецилсульфат, гексадецилсульфат и октадецилсульфат, олеат Na или капрат Na, 1-ациламиноэтан-2-сульфоновые кислоты, такие как 1-октаноиламиноэтан-2-сульфоновая кислота, 1-деканоиламиноэтан-2-сульфоновая кислота, 1-додеканоиламиноэтан-2-сульфоновая кислота, 1-тетрадеканоиламиноэтан-2-сульфоновая кислота, 1-гексадеканоиламиноэтан-2-сульфоновая кислота и 1-октадеканоиламиноэтан-2-сульфоновая кислота и таурохолиновая и тауродезоксихолиновая кислота, желчные кислоты и их соли, такие как холевая кислота, дезоксихолевая кислота и гликохолаты натрия, капрат натрия или лаурат натрия, олеат натрия, лаурилсульфат натрия, цетилсульфат натрия, сульфатированное касторовое масло и диокстилсульфосукцинат натрия, кокамидопропилбетаин и лаурилбетаин, жирные спирты, холестерины, моно- или дистеарат глицерина, моно- или диолеат глицерина и моно- или дипальмитат глицерина, и полиоксиэтиленстеарат.
Неограничивающими примерами подсластителей, используемых в описанной здесь композиции, являются сахароза, фруктоза, лактоза или аспартам. Неограничивающими примерами ароматизаторов, используемых в описанной здесь композиции, являются перечная мята, масло грушанки или фруктовые отдушки, такие как вишневая или апельсиновая отдушка. Неограничивающими примерами материалов для покрытий, используемых в описанной здесь композиции, являются желатин, воск, шеллак, сахар или другие биологически разлагаемые полимеры. Неограничивающими примерами консервантов, используемых в описанной здесь композиции, являются метил- или пропилпарабены, сорбиновая кислота, хлорбутанол, фенол и тимерозал.
BH4-форма может быть также приготовлена в виде шипучих таблеток или порошков, которые дезинтегрируются в водной среде с образованием питьевого раствора. Составы с замедленным высвобождением могут быть также приготовлены в целях достижения регулируемого высвобождения активного агента при его контакте с физиологическими жидкостями в желудочно-кишечном тракте и в целях обеспечения, по существу, постоянного и эффективного уровня активного агента в плазме крови. Для этого кристаллическая форма может быть включена в полимерную матрицу, состоящую из биологически разлагаемого полимера, водорастворимого полимера или их смеси, и, необязательно, подходящих поверхностно-активных веществ. В контексте настоящего изобретения такое включение может означать введение микрочастиц в полимерные матрицы. Составы с регулируемым высвобождением также получают посредством инкапсулирования диспергированных микрочастиц или эмульгированных микрокапелек с применением известных методов нанесения дисперсионных или эмульсионных покрытий.
BH4, используемый в описанной здесь композиции, предпочтительно, получают в виде дигидрохлоридной соли, однако могут быть рассмотрены и другие солевые формы BH4, обладающие нужной биологической активностью, или другие солевые формы BH4. В частности, предпочтительными являются соли BH4, образованные неорганическими или органическими кислотами. Неограничивающими примерами альтернативных солевых форм ВЕ4 являются соли ВН4, образованные уксусной кислотой, лимонной кислотой, щавелевой кислотой, винной кислотой, фумаровой кислотой и миндальной кислотой.
Фармацевтически приемлемые основно-аддитивные соли могут быть образованы металлами или аминами, такими как щелочные и щелочноземельные металлы или органические амины. Фармацевтически приемлемые соли соединений могут быть также получены с использованием фармацевтически приемлемого катиона. Подходящие фармацевтически приемлемые катионы хорошо известны специалистам в данной области, и такими катионами являются катионы щелочных металлов, щелочноземельных металлов, аммония и четвертичного аммония. Могут быть также использованы карбонаты или гидрокарбонаты. Примерами металлов, используемых в качестве катионов, являются натрий, калий, магний, аммоний, кальций или железо(3) и т.п. Примерами подходящих аминов являются изопропиламин, триметиламин, гистидин, N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, дициклогексиламин, этилендиамин, N-метилглюкамин и прокаин.
Фармацевтически приемлемыми кислотно-аддитивными солями являются соли неорганических или органических кислот. Примерами подходящих солей кислот являются гидрохлориды, ацетаты, цитраты, салицилаты, нитраты и фосфаты. Другие подходящие фармацевтически приемлемые соли хорошо известны специалистам в данной области, и такими солями являются, например, соли уксусной, лимонной, щавелевой, винной или миндальной кислоты, хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты или фосфорной кислоты; соли органических карбоновых, сульфоновых, сульфо- или фосфокислот, или N-замещенных сульфамовых кислот, например, уксусной кислоты, пропионовой кислоты, гликолевой кислоты, янтарной кислоты, малеиновой кислоты, гидроксималеиновой кислоты, метилмалеиновой кислоты, фумаровой кислоты, яблочной кислоты, винной кислоты, молочной кислоты, щавелевой кислоты, глюконовой кислоты, глюкаровой кислоты, глюкуроновой кислоты, лимонной кислоты, бензойной кислоты, коричной кислоты, миндальной кислоты, салициловой кислоты, 4-аминосалициловой кислоты, 2-феноксибензойной кислоты, 2-ацетоксибензойной кислоты, эмбоновой кислоты, никотиновой кислоты или изоникотиновой кислоты, и такими аминокислотами, как 20 альфа-аминокислот, участвующих в природном синтезе белков, например, глутаминовой кислотой или аспарагиновой кислотой, а также фенилуксусной кислотой, метансульфоновой кислотой, этансульфоновой кислотой, 2-гидроксиэтансульфоновой кислотой, этан-1,2-дисульфоновой кислотой, бензолсульфоновой кислотой, 4-метилбензолсульфоновой кислотой, нафталин-2-сульфоновой кислотой, нафталин-1,5-дисульфоновой кислотой, 2- или 3-фосфоглицератом, глюкозо-6-фосфатом, N-циклогексилсульфамовой кислотой (с образованием цикламатов) или другими кислотными органическими соединениями, такими как аскорбиновая кислота.
Репрезентативные стабильные пероральные составы содержат один или несколько из нижеследующих вспомогательных ингредиентов, которые повышают стабильность или другие свойства указанного состава, а именно, связующее вещество, дезинтегрирующее вещество, кислотный антиоксидант или замастиватель или их комбинации. Репрезентативные стабильные составы, изготавливаемые в форме таблеток, включают связующее вещество и дезинтегрирующее вещество вместе, но необязательно, с кислотным антиоксидантом, которые могут включать, но необязательно, смазывающее вещество. Репрезентативные концентрации связующего вещества составляют от примерно 1 масс.% до примерно 5 масс.%, или примерно 1,5-3 масс.%; а репрезентативные массовые отношения количества связующего вещества к количеству BH4 составляют примерно от 1:10 до 1:20. Репрезентативные концентрации дезинтегрирующего вещества составляют от примерно 1 масс.% до примерно 20 масс.%; а репрезентативные массовые отношения количества дезинтегрирующего вещества к количеству BH4 составляют от примерно 1:5 до примерно 1:10. Репрезентативные концентрации антиоксиданта составляют примерно от примерно 1 масс.% до примерно 3 масс.%; а репрезентативные массовые отношения количества антиоксиданта к количеству BH4 составляют от примерно 1:5 до 1:30. В одном из примеров антиоксидантом является аскорбиновая кислота, отношение которой к BH4 составляет менее чем 1:1, например, 1:2 или менее, или 1:10 или менее. Репрезентативные концентрации смазывающего вещества в стабильном составе согласно изобретению, изготавливаемом в виде таблетки, составляют от примерно 0,1 масс.% до примерно 2 масс.%; а репрезентативное массовое отношение смазывающего вещества к BH4 составляет в пределах от примерно 1:25 до 1:65.
Стабильный твердый состав может, но необязательно, включать и другие терапевтические средства, подходящие для лечения состояния, и такими средствами являются, например, фолаты, включая предшественники фолатов, фолиевые кислоты или производные фолатов; и/или аргинин; и/или витамины, такие как витамин С и/или витамин В2 (рибофлавин), и/или витамин В12; и/или предшественники нейромедиаторов, такие как L-допа или карбидопа; и/или 5-гидрокситриптофан.
Репрезентативные фолаты, включая предшественники фолатов, фолиевые кислоты или производные фолатов, описаны в патентах США №№ 6011040 и 6544994, которые включены в настоящее описание посредством ссылки, и такими соединениями являются фолиевая кислота (птероилмоноглутамат), дигидрофолиевая кислота, тетрагидрофолиевая кислота, 5-метилтетрагидрофолиевая кислота, 5,10-метилентетрагидрофолиевая кислота, 5,10-метенилтетрагидрофолиевая кислота, 5,10-формиминотетрагидрофолиевая кислота, 5-формилтетрагидрофолиевая кислота (лейковорин), 10-формилтетрагидрофолиевая кислота, 10-метилтетрагидрофолиевая кислота, один или несколько фолилполиглутаматов, соединения, в которых пиразиновое кольцо птериновой молекулы фолиевой кислоты или фолилполиглутаматов восстанавливается до дигидрофолатов или тетрагидрофолатов, или производные всех вышеуказанных соединений, в которых N-5- или N- 10-положения имеют одну углеродную молекулу на различных уровнях окисления или их фармацевтически совместимые соли или комбинации из двух или более указанных соединений. Репрезентативными тетрагидрофолатами являются 5-формил-(6S)-тетрагидрофолиевая кислота, 5-метил-(6S)-тетрагидрофолиевая кислота, 5,10-метилен-(6R)-тетрагидрофолиевая кислота, 5,10-метенил-(6R)-тетрагидрофолиевая кислота, 10-формил-(6R)-тетрагидрофолиевая кислота, 5-формимино-(6S)-тетрагидрофолиевая кислота или (6S)-тетрагидрофолиевая кислота и их фармацевтически приемлемые соли. Репрезентативными солями являются соли натрия, калия, кальция или аммония.
Репрезентативные массовые отношения BH4:фолаты:аргинин могут составлять примерно от 1:10:10 до 10:1:1.
Стабильные составы согласно изобретению могут быть приготовлены, например, в виде таблеток или драже, или в виде капсул в HDPE бутылях, снабженных капсулой или мешочком с дессикантом, или в виде блистерной упаковки, обернутой в фольгу, или в виде блистерной упаковки, содержащей прозрачную полимерную пленку, если они предназначены для продажи.
IV. Лечение восприимчивых к BH4 заболеваний
Гиперфенилаланинемия, нейропсихологические или психоневрологические расстройства
Способы согласно изобретению могут быть применены для лечения состояний, ассоциированных с повышенными уровнями фенилаланина или с пониженными уровнями тирозина или триптофана, которые быть быть вызваны, например, пониженной активностью фенилаланингидроксилазы, тирозингидроксилазы или триптофангидроксилазы. Состояниями, ассоциированными с повышенными уровнями фенилаланина, в частности, являются фенилкетонурия в слабой и классической форме, и гиперфенилаланинемия, описанные в настоящей заявке, а репрезентативными группами пациентов являются подгруппы пациентов, описанные в настоящей заявке, а также другие пациенты, у которых уровни фенилаланина превышают нормальные уровни.
Состояниями, ассоциированными с пониженными уровнями тирозина или триптофана, являются дефицит нейромедиаторов, нервные и психические расстройства, такие как болезнь Паркинсона, дистония, дегенерация спинного мозга и мозжечка, боли, усталость, депрессия, другие аффективные расстройства и шизофрения. Сверхпродуцирование NO под действие nNOS приводит к инсультам, головным болям при мигрени, болезни Альцгеймера и к развитию толерантности к морфину и к зависимости от морфина. Для лечения любых из этих состояний может быть введен BH4. Другими репрезентативными психоневрологическими расстройствами, для лечения которых может быть введен BH4, являются болезнь Паркинсона, болезнь Альцгеймера, шизофрения, шизофреноподобное расстройство, шизоаффективное расстройство, кратковременное психотическое расстройство, бред, разделяемое психотическое расстройство, психотическое расстройство, вызываемое общим клиническим состоянием, психотическое расстройство, индуцированное злоупотреблением лекарственных средств, другие психотические расстройства, поздняя дискинезия, болезнь Мачадо-Джозефа, дегенерация спинного мозга и мозжечка, атаксия мозжечка, дистония, синдром хронической усталости, острая или хроническая депрессия, синдром хронического стресса, фибромиалгия, мигрень, гиперактивность, ассоциированная с дефицитом внимания, биполярное заболевание и аутизм.
Стабильные составы могут быть также использованы для лечения пациентов, страдающих заболеванием, ассоциированным с дефицитом ВН4, например, вызываемым нарушением пути его синтеза, включая, но не ограничиваясь ими, допа-восприимчивую дистонию (DRD), дефицит сепиаптеринредуктазы (SR) или дефицит дигидроптеридинредуктазы (DHPR).
Подходящими индивидуумами, подвергаемыми лечению стабильными составами согласно изобретению, являются индивидуумы, которые в отсутствие терапевтического лечения имеют повышенную концентрацию Phe в плазме, например, более чем 1800 мкМ/л, или более чем 1600 мкМ, более чем 1400 мкМ, более чем 1200 мкМ, более чем 1000 мкМ, более чем 800 мкМ, более чем 600 мкМ, более чем 420 мкМ, более чем 300 мкМ, более чем 200 мкМ или более чем 180 мкМ. Фенилкетонурия (ФКУ) в слабой форме обычно классифицируется как заболевание, при котором концентрации Phe в плазме составляют до 600 мкМ/л, умеренная ФКУ классифицируется как заболевание, при котором концентрации Phe в плазме составляют от 600 мкМ/л и до примерно 1200 мкМ/л, а классическая или тяжелая ФКУ классифицируется как заболевание, при котором концентрации Phe в плазме составляют более чем 1200 мкМ/л. При этом предпочтительно, чтобы лечение стабильными составами, вводимыми отдельно или вместе с пищей с ограниченным содержанием белка, приводило к снижению концентрации фенилаланина в плазме индивидуума до значений, составляющих менее чем 600 мкМ, или менее чем 500 мкМ, или 360 мкМ ± 15 мкМ или менее, или менее чем 200 мкМ, или менее чем 100 мкМ. Другими подходящими индивидуумами являются индивидуумы, у которых были диагностированы пониженная фенилаланингидроксилазная (PAH) активность, атипичная или злокачественная фенилкетонурия, ассоциированная с дефицитом ВН4, гиперфенилаланинемия, ассоциированная с заболеванием печени, и гиперфенилаланинемия, ассоциированная с малярией, где пониженная PAH-активность может быть вызвана мутацией в ферменте PAH, например мутацией в каталитическом домене PAH или одной или несколькими мутациями, выбранными из группы, состоящей из F39L, L48S, I65T, R68S, A104D, S110C, D129G, E178G, V190A, P211T, R241C, R261Q, A300S, L308F, A313T, K320N, A373T, V388M, E390G, A395P, P407S и Y414C; либо такими индивидуумами являются беременные женщины, женщины детородного возраста, которые планируют рождение ребенка, или дети в возрасте от рождения до 3 лет, или от рождения до 2 лет, от рождения до 1,5 лет или от рождения до 1 года, или индивидуумы, у которых обнаруживалась невосприимчивость через 24 часа после проведения нагрузочного теста с использованием одной дозы BH4 или нагрузочного теста с использованием многократной дозы, такого как нагрузочный тест с использованием 4 доз или нагрузочный тест, проводимый в течение 7 дней. Репрезентативные группы пациентов и репрезентативные нагрузочные тесты с использованием BH4 описаны в международной публикации № WO 2005/049000, которая во всей своей полноте включена в настоящее описание посредством ссылки.
В патентах США №№ 4752573; 4758571; 4774244; 4920122; 5753656; 5922713; 5874433; 5945452; 6274581; 6410535; 6441038; 6544994 и в публикациях заявок на патенты США 20020187958; US 20020106645; US 2002/0076782; US 20030032616 (каждая из которых включена в настоящее описание посредством ссылки) описаны способы введения композиций BH4 для лечения заболеваний, не относящихся к ФКУ. Каждый из этих патентов включен в настоящее описание посредством ссылки как общее описание методов введения композиций ВН4, которые известны специалистам и которые могут быть адаптированы для описанного здесь лечения.
Хотя потребности в лечении для каждого отдельного индивидуума могут варьироваться, однако определение оптимальных интервалов эффективного количества каждого компонента может быть осуществлено самим специалистом. Типичные дозы BH4 составляют примерно от 1 до 20 мг/кг массы тела в день, а обычно от примерно 5 (1 мг/кг × 5 кг массы тела) до 3000 мг/день (30 мг/кг × 100 кг массы тела). Хотя в настоящем изобретении рассматривается непрерывное ежедневное введение, однако при наличии симптомов гиперфенилаланинемии (ГФА), при которых уровни Phe снижаются до уровней ниже определенного порогового уровня, может оказаться желательным прекращение ВН4-терапии. Очевидно, что такая терапия может быть возобновлена в случае повторного увеличения уровней Phe. Соответствующие дозы могут быть определены путем проведения анализов, разработанных для определения уровней Phe в крови в сочетании с релевантными данными доза-ответ.
В репрезентативных вариантах рассматриваются способы согласно изобретению, предусматривающие введение пациенту, нуждающемуся в этом, ежедневной дозы ВН4, составляющей от примерно 10 мг/кг до примерно 20 мг/кг. Очевидно, что специалист в данной области может скорректировать такую дозу в сторону ее увеличения или снижения в зависимости от эффективности, достигаемой при ее введении. Суточная доза может быть введена в виде разовой дозы или, альтернативно, она может быть введена в виде дробных доз через нужные промежутки времени. В репрезентативных вариантах изобретения суточная доза может составлять 1 мг/кг, 2 мг/кг, 3 мг/кг, 4 мг/кг, 5 мг/кг, 6 мг/кг, 7 мг/кг, 8 мг/кг, 9 мг/кг, 10 мг/кг, 11 мг/кг, 12 мг/кг, 13 мг/кг, 14 мг/кг, 15 мг/кг, 16 мг/кг, 17 мг/кг, 18 мг/кг, 19 мг/кг, 20 мг/кг, 22 мг/кг, 24 мг/кг, 26 мг/кг, 28 мг/кг, 30 мг/кг, 32 мг/кг, 34 мг/кг, 36 мг/кг, 38 мг/кг, 40 мг/кг, 42 мг/кг, 44 мг/кг, 46 мг/кг, 48 мг/кг, 50 мг/кг или выше.
Схемы введения низких доз
В способе терапии согласно изобретению с применением низких доз рассматривается введение низких доз, например доз от 0,1 до 5 мг/кг в день, включая дозы от 0,1 до 2 мг/кг, от 0,1 до 3 мг/кг или от 1 мг/кг до 5 мг/кг. Предпочтительными являются дозы менее чем 5 мг/кг в день. В соответствии с настоящим изобретением предполагается, что введение таких доз улучшает конечные результаты релевантных исследований, а также предполагается, что производные ВН4, вводимые в таких дозах, обладают улучшенными биологическими свойствами по сравнению со свойствами природного ВН4, вводимого в указанных дозах. В частности, в соответствии с настоящим изобретением любой из описанных здесь 1',2'-диацил-(6R,S)-5,6,7,8-тетрагидро-L-биоптеринов или липоидных тетрагидробиоптеринов обладает улучшенными биологическими свойствами при его введении в низких дозах.
В настоящем изобретении также конкретно рассматривается применение BH4 или его предшественника или производного для лечения восприимчивых к BH4 заболеваний в дозе, составляющей от 0,1 до 5 мг/кг массы тела/день, вводимой любым способом введения, включая, но не ограничиваясь ими, пероральное введение в виде одной дозы один раз в день или в виде дробных доз (например, 2, 3 или 4) в день в течение по меньшей мере 1, 2, 3, или 4 недель или более, или в течение 1, 2, 3, 4, 5, 6 месяцев или более. Репрезентативными являются дозы, составляющие менее чем 5 мг/кг/день, 4,5 мг/кг/день или менее, 4 мг/кг/день или менее, 3,5 мг/кг/день или менее, 3 мг/кг/день или менее, 2,5 мг/кг/день или менее, 2 мг/кг/день или менее, 1,5 мг/кг/день или менее, 1 мг/кг/день или менее или 0,5 мг/кг/день или менее. Могут быть также введены эквивалентные дозы на площадь поверхности тела.
В настоящем изобретении для индивидуума со средней массой/средней площадью поверхности тела (например, 70 кг), также рассматривается введение общей суточной дозы менее чем 400 мг. Примерами таких общих суточных доз являются дозы 360 мг/день, 350 мг/день, 300 мг/день, 280 мг/день, 210 мг/день, 180 мг/день, 175 мг/день, 150 мг/день или 140 мг/день. Так, например, дозы 350 мг/день или 175 мг/день могут быть легко введены в виде лекарственного состава для перорального введения, содержащего дозу 175 мг, один или два раза в день. Другими примерами общих суточных доз являются дозы 320 мг/день или менее, 160 мг/день или менее или 80 мг/день или менее. Такие дозы могут быть легко введены в виде лекарственного состава для перорального введения, содержащего дозу 80 или 160 мг. Другими примерами общих суточных доз являются дозы 45, 90, 135, 180, 225, 270, 315 или 360 мг/день или менее, которые могут быть легко введены в виде лекарственного состава для перорального введения, содержащего дозу 45 или 90 мг. Другими примерами общих суточных доз являются дозы 60, 120, 180, 240, 300 или 360 мг/день, которые могут быть легко введены в виде лекарственного состава для перорального введения, содержащего дозу 60 или 120 мг. Другими примерами общих суточных доз являются дозы 70, 140, 210, 280 или 350 мг/день, которые могут быть легко введены в виде лекарственного состава для перорального введения, содержащего дозу 70 или 140 мг. Примерами общих суточных доз также являются дозы 55, 110, 165, 220, 275 или 330 мг/день, которые могут быть легко введены в виде лекарственного состава для перорального введения, содержащего дозу 55 мг. Другими примерами общих суточных доз являются дозы 65, 130, 195, 260 или 325 мг/день, или 75, 150, 225, 300 или 375 мг/день, например, в виде лекарственного состава, содержащего дозу 65 мг или 75 мг.
Заболевания, ассоциированные с дисфункцией синтазы оксида азота
Настоящее изобретение также относится к стабильным составам согласно изобретению, которые могут быть использованы для лечения индивидуумов, страдающих состояниями, которые могут быть подвергнуты эффективному лечению в результате усиления активности синтазы оксида азота, и пациентов, страдающих сосудистыми заболеваниями, ишемическими или воспалительными заболеваниями или инсулинорезистентностью. Такое лечение может, например, приводить к устранению дефицита активности синтазы оксида азота, либо оно может, например, приводить к повышению уровней активности синтазы оксида азота по сравнению с нормальными уровнями. Было высказано предположение, что у пациента, страдающего дефицитом активности синтазы оксида азота, будет наблюдаться положительный эффект в результате комбинированного лечения фолатами, включая предшественники фолата, фолиевые кислоты или производные фолата.
Оксид азота конститутивно продуцируется сосудистыми эндотелиальными клетками, в которых он играет ключевую физиологическую роль в регуляции кровяного давления и сосудистого тонуса. Было высказано предположение, что дефицит биологической активности оксида азота приводит к патогенезу сосудистой дисфункции, включая заболевание коронарных артерий, атеросклероз любых артерий, включая коронарные, сонные, церебральные или периферические артерии, реперфузионные повреждения при ишемии, гипертензию, диабет, диабетическую васкулопатию, сердечно-сосудистое заболевание, заболевание периферических сосудов или нейродегенеративные состояния, возникающие в результате ишемии и/или воспаления, такие как инсульт, и что такой патогенез включает поражение эндотелия, недостаточное поступление кислорода в органы и ткани, повышенную системную васкулярную резистентность (высокое кровяное давление), пролиферацию сосудистой системы гладких мышц, прогрессирование стеноза сосудов (сужение сосудов) и воспаление. Таким образом, лечение любого из этих состояний может быть осуществлено способами согласно изобретению.
Было также высказано предположение, что повышение активности синтазы оксида азота также приводит к снижению повышенных уровней супероксида, к повышению чувствительности к инсулину и к снижению сосудистой дисфункции, ассоциированной с инсулинорезистентностью, как описано в патенте США № 6410535, который включен в настоящее описание посредством ссылки. Таким образом, в настоящем изобретении рассматривается способ лечения диабета (типа I или II), гиперинсулинемии или инсулинорезистентности. Заболевания или расстройства с сосудистой дисфункцией, ассоциированной с инсулинорезистентностью, представляют собой заболевания или расстройства, которые вызываются инсулинорезистентностью или осложняются инсулинорезистентностью или которые трудно поддаются лечению из-за инсулинорезистентности, и такими заболеваниями или расстройствами являются, но не ограничиваются ими, нарушения эластичности сосудов, эндотелиальная дисфункция и гипертензия, расстройства, связанные с нарушениями чувствительности к инсулину и нарушением регуляции уровней глюкозы, патологии, связанные с периферической перфузией, такие как перемежающаяся хромота, пониженная периферическая перфузия, снижение кожного кровотока, замедление заживления ран и нарушение периферического кровотока, гиперлипидемия, артериосклероз, стенокардия, ассоциированная с сужением коронарных артерий, стенокардия напряжения, цереброваскулярное стенозирующее повреждение, цереброваскулярная недостаточность, спазм сосудов головного мозга, рестеноз коронарных артерий после проведения чрескожной внутрипросветной коронарной ангиопластики (PTCA) или обходного шунтирования коронарной артерии (CABG), ожирение, инсулиннезависимый диабет, гиперинсулинемия, нарушение метаболизма липидов, артериосклеротические нарушения при ишемической болезни сердца, застойная сердечная недостаточность, легочная гипертензия при наличии или в отсутствие застойной сердечной недостаточности, стенокардия, ассоциированная с физическими нагрузками, ишемическая болезнь сердца и связанный с ней атеросклероз; глазные болезни, такие как атрофия зрительного нерва и диабетическое заболевание сетчатки; и болезни почек, такие как микроальбуминурия при диабетической болезни почек, почечная недостаточность и пониженный уровень гломерулярной фильтрации.
В соответствии с настоящим изобретением введение ВН4 пациентам, страдающим такими заболеваниями, может быть эффективным для предупреждения или лечения указанных заболеваний путем активации функции NOS, увеличения продуцирования NO и подавления продуцирования молекул активного кислорода, что приводит к устранению повреждения эндотелиальных клеток сосудов.
Настоящее изобретение относится к способу лечения индивидуума, у которого было диагностировано не связанное с диабетом сосудистое заболевание, выбранное из группы, состоящей из сосудистого заболевания легких, гемолитической анемии, инсульта и ассоциированной с ним ишемической болезни сосудов (такой как инсульт, заболевание сердца или коронарных артерий, артериосклероз или заболевание периферических сосудов), тромбоза, эндотелиальной дисфункции, ассоциированной с трансплантацией, и заболевания сердца или коронарной артерии. В одном из вариантов изобретения сосудистыми заболеваниями легких являются, но не ограничиваются ими, повышение давления в легких при серповидно-клеточной анемии и при других гемоглобинопатиях, идиопатическая легочная гипертензия, стабильная легочная гипертензия новорожденных (PPHN). В другом варианте изобретения гемолитическими анемиями являются наследственная гемолитическая анемия и приобретенная гемолитическая анемия. Наследственными гемолитическими анемиями являются, но не ограничиваются ими, серповидно-клеточная анемия, талассемия, гемолитическая анемия, вызываемая дефицитом G6PD, дефицитом пируваткиназы, наследственный эллиптоцитоз, наследственный сфероцитоз, наследственный стоматоцитоз, наследственный овалоцитоз, ночная пароксизмальная гемоглобинурия и серповидно-клеточное заболевание, вызываемое наличием в эритроцитах аномального гемоглобина. Приобретенными гемолитическими анемиями являются, но не ограничиваются ими, микроангиопатическая гемолитическая анемия, идиопатическая аутоиммунная гемолитическая анемия, неиммунная гемолитическая анемия, вызываемая применением химических или физических веществ или устройств (левожелудочкого аппарата вспомогательного кровообращения, механических сердечных клапанов и шунтирующих устройств), и вторичная иммунная гемолитическая анемия.
В другом варианте изобретения инсультами и связанными с ними ишемическими сосудистыми заболеваниями являются, но не ограничиваются ими, спазмы сосудов, такие как спазм сосудов головного мозга после инсульта. Тромбозами являются, но не ограничиваются ими, тромбогенез, тромбоз, образование тромбов и нарушения свертываемости крови. В другом варианте изобретения, ассоциированными с трансплантацией эндотелиальными дисфункциями, являются, но не ограничиваются ими, сосудистая дисфункция после трансплантации твердого органа и эндотелиальная дисфункция, индуцированная циклоспорином А. В еще одном варианте изобретения заболеваниями сердца или коронарных артерий являются, но не ограничиваются ими, застойная сердечная недостаточность, дисфункция сосудов и стенокардия, ассоциированная с гиперхолистеринемией, а также дисфункция сосудов и стенокардия, ассоциированная с курением табака.
Введение BH4 может также предупреждать или устранять другие расстройства, ассоциированные со сверхпродуцированием молекул активированного кислорода или повреждением, вызываемым этими молекулами, включая, но не ограничиваясь ими, такие расстройства, как сепсис.
При этом следует отметить, что подходящая доза композиции согласно изобретению зависит от возраста, состояния здоровья и массы реципиента, вида одновременно проводимого лечения, если оно вообще проводится, частоты проводимого лечения и от желаемого эффекта (то есть от желаемого снижения концентрации Phe в плазме). Частота введения дозы также зависит от фармакодинамического воздействия на уровни Phe, если продолжительность такого воздействия составляет 24 часа после введения одной дозы. Однако для отдельного индивидуума может быть специально приготовлена наиболее предпочтительная доза, определенная и назначенная самим специалистом без излишнего экспериментирования. Такой способ обычно включает коррекцию стандартной дозы, например снижение дозы в том случае, если данный пациент имеет небольшую массу тела.
Частота введения дозы BH4 зависит от фармакокинетических параметров агента и от способов его введения. Оптимальный фармацевтический состав может быть приготовлен самим специалистом в зависимости от способа введения и от желаемой дозы. См., например, руководство Remington's Pharmaceutical Sciences, 18th Ed. (1990, Mack Publ. Co, Easton PA 18042) pp 1435-1712, которое включено в настоящее описание посредством ссылки. Такие составы могут влиять на физическое состояние, стабильность, скорость высвобождения in vivo и скорость выведения in vivo вводимых агентов. В зависимости от способа введения подходящая доза может быть вычислена с учетом массы тела, площади поверхности тела или размера органа. Дополнительное уточнение результатов вычисления, необходимое для определения соответствующей дозы для лечения, может быть осуществлено специалистом в данной области рутинным способом без излишнего экспериментирования, а в частности, на основе информации о дозах и описанных здесь анализов, а также на основе фармакокинетических данных, полученных при проведении клинических испытаний на животных или с участием человека.
Конечная схема введения доз может быть определена лечащим врачом с учетом факторов, которые модифицируют действие лекарственных средств, например с учетом удельной активности лекарственных средств, тяжести поражения и восприимчивости пациента к данным лекарственным средствам, а также с учетом возраста, состояния здоровья, массы тела, пола и режима питания пациента, тяжести каких-либо инфекционных заболеваний, времени введения и других клинических факторов. По мере проведения исследований может быть получена и другая информация, касающаяся соответствующих уровней доз и продолжительности лечения конкретных заболеваний и состояний.
V. Комбинированная терапия
Некоторые способы согласно изобретению включают применение стабильных составов согласно изобретению в комбинации с одним или несколькими другими терапевтическими средствами.
При проведении такой комбинированной терапии введение стабильных составов согласно изобретению может быть осуществлено одновременно с введением второго терапевтичесого средства, либо оно может быть осуществлено до или после введения такого средства, например, с интервалами от нескольких минут до нескольких часов, при условии что оба этих средства будут обладать терапевтическим действием в перекрывающиеся периоды времени. Таким образом, в настоящем изобретении рассматриваются стабильные составы согласно изобретению, используемые в комбинации со вторым терапевтическим средством. В настоящем изобретении также рассматривается применение второго терапевтического средства в целях приготовления лекарственного средства для введения в комбинации со стабильным тетрагидробиоптерином, его предшественником, производным или аналогом согласно изобретению.
Для усиления терапевтического эффекта у пациентов с различными формами ГФА, лечение тетрагидробиоптерином может быть проведено вместе с приемом пищи, имеющей ограниченное содержание белков. Так, например, такому индивидууму может быть введена композиция BH4 и лечебная белковая композиция с низким содержанием фенилаланина, которые, в целом, являются эффективным для достижения нужного терапевтического эффекта (то есть для снижения концентрации Phe в плазме и/или для вырабатывания способности усваивать более высокие количества поглощаемого Phe/белка без сопутствующего увеличения концентраций Phe в плазме). Такой способ может включать одновременное введение композиции BH4 и терапевтической белковой композиции. Это может быть достигнуто путем введения одной композиции или одного фармакологического белкового состава, который удовлетворяет всем требованиям диетического белкового питания и также включает ВН4. Альтернативно, диетический белок (добавка или обычная белковая пища) могут быть введены приблизительно одновременно с введением фармакологического состава ВН4 (таблетки, инъекции или питья).
В некоторых вариантах изобретения пищей с ограниченным содержанием белка является пища, в которую добавлены такие аминокислоты, как тирозин, валин, изолейцин и лейцин. Пациенту может быть одновременно введена белковая добавка с низким содержанием Phe, которая может включать L-тирозин, L-глутамин, L-карнитин в концентрации 20 мг/100 г добавки, L-таурин в концентрации 40 мг/100 г добавки и селен. Такая добавка может также содержать рекомендованные суточные дозы минералов, таких как кальций, фосфор и магний. Указанная добавка может также содержать рекомендованную суточную дозу одной или нескольких аминокислот, выбранных из группы, состоящей из L-лейцина, L-пролина, ацетата L-лизина, L-валина, L-изолейцина, L-аргинина, L-аланина, глицина, моногидрата L-аспарагина, L-триптофана, L-серина, L-треонина, L-гистидина, L-метионина, L-глутаминовой кислоты и L-аспарагиновой кислоты. Кроме того, в указанную добавку может быть включена рекомендованная суточная доза витаминов A, D и E. Такая добавка может содержать, но необязательно, определенное количество жира, энергетическая ценность которого составляет по меньшей мере 40%. Такие добавки могут быть приготовлены в форме порошка или в форме белкового брикета. В некоторых вариантах изобретения пища с ограниченным содержанием белка содержит белковую добавку, а ВН4 присутствует в такой композиции в виде белковой добавки.
В других альтернативных вариантах изобретения BH4-терапия может быть осуществлена до или после проведения терапии с использованием диетической белковой пищи с временными интервалами, составляющими от нескольких минут до нескольких часов. В тех вариантах изобретения, в которых белок и композиции ВН4 вводят отдельно, по существу, необходима гарантия того, чтобы между каждым введением не проходило слишком много времени, то есть для того, чтобы ВН4 был способен оказывать благоприятное воздействие на пациента. В таких случаях рассматривается введение BH4 в пределах примерно 2-6 часов до или после введения диетического белка, например, с временным интервалом лишь в примерно 1 час или менее. В некоторых вариантах изобретения рассматривается проведение BH4-терапии в виде непрерывной терапии, при которой пациенту вводят суточную дозу ВН4 в неопределенно продолжительное время. В других случаях, например, беременным женщинам, у которых наблюдается лишь ФКУ и ГФА в ослабленной форме, ВН4-терапия может быть проведена только непрерывно, при условии что такая женщина является беременной и/или кормящей.
Кроме того, в соответствии со способами согласно изобретению помимо терапии, проводимой лишь на основе введения ВН4 и регуляции уровня диетического белка, рассматривается также комбинированная терапия, проводимая в сочетании с введением третьей композиции, которая, в частности, направлена на ослабление одного или нескольких симптомов ГФА. Так, например, известно, что дефицит тирозина, вызываемый ГФА, приводит к дефициту нейромедиаторов, таких как допамин и серотонин. Таким образом, в контексте настоящего изобретения подразумевается, что способы, проводимые с использованием ВН4 и диетического белка, могут быть осуществлены в комбинации с введением нейромедиаторов, таких как L-допа, карбидопа и 5-гидрокситриптофан, для устранения дефицита, вызываемого пониженным количеством тирозина в пище.
Кроме того, специалистами в данной области может быть применена генотерапия, проводимая вместе с использованием РАН (Christensen et al., Mol. Gent. And Metabol. 76: 313-318, 2002; Christensen et al., Gene Therapy, 7:1971-1978, 2000) фенилаланин-аммиак-лиазы (PAL Liu et al., Arts. Cells. Blood. Subs and Immob. Biotech. 30(4)243-257, 2002). Такие методы генотерапии могут быть применены в комбинированной терапии согласно изобретению, проводимой с использованием ВН4/добавки с ограниченным содержанием белка. В другой комбинированной терапии для повышения пониженных концентраций Phe у пациента рассматривается введение фенилазы в качестве фермента для инъекций. Поскольку введение фенилазы не должно приводить к продуцированию тирозина (в отличие от введения PAH), то такое лечение будет способствовать продуцированию у пациентов тирозина, который является незаменимой аминокислотой. Поэтому диетическая добавка, содержащая тирозин, может оказаться желательной для пациентов, получающих фенилазу в комбинации с ВН4-терапией.
Для лечения нейропсихологических или психоневрологических расстройств способом согласно изобретению BH4 может быть введен в комбинации с одним или несколькими другими психоневрологически активными средствами, включая антидепрессанты, предшественники нейромедиаторов, такие как триптофан, тирозин, серотонин, средства, активирующие норадренергическую систему, такие как лофепрамин, дезипрамин, ребоксетин, тирозин, агенты, которые преимущественно влияют на серотонин, объединенные ингибиторы поглощения норадреналина и серотонина, такие как венлафаксин, дулоксетин или милнасипран, или лекарственные средства, которые представляют собой объединенные ингибиторы поглощения допамина и норадреналина, такие как бупропион.
В родственном варианте изобретения BH4 вводят вместе с другими терапевтическими средствами, обычно используемыми для лечения диабета, сосудистого заболевания и гиперлипидемии. Агентами, используемыми для лечения диабета, являются, но не ограничиваются ими, агенты, повышающие чувствительность к инсулину, такие как лиганды PPAR-гамма (тиазолидиндоны, глитазоны, троглитазоны, розиглитазон (Avandia), пиоглитазон), стимуляторы секреции инсулина, такие как сульфонилмочевины (глихидон, толбутамид, глимеприд, хлорпропамид, глипизид, глибурид, ацетогексамид) и меглитиниды (меглитинид, репаглинид, натеглинид), и агенты, снижающие продуцирование глюкозы в печени, такие как метформин. Агентами, используемыми для лечения сосудистых заболеваний, являются, но не ограничиваются ими, антагонисты эндотелиновых рецепторов, обычно используемые для лечения гипертензии и других расстройств, ассоциированных с дисфункцией эндотелия, такие как бозентан, дарузентан, энразентан, тезозентан, атразентан, амбризентан, ситаксентан; релаксанты гладких мышц, такие как ингибиторы PDE5 (непрямого действия) и миноксидил (прямого действия); ингибиторы ангиотензин-конвертирующего фермента (ACE), такие как каптоприл, эналаприл, лизиноприл, фозиноприл, периндоприл, хинаприл, трандолаприл, беназеприл, рамиприл; блокаторы рецептора ангиотензина II, такие как ирбесартан, лозартан, валсартан, эпросартан, олмесартан, кандесартан, телмисартан; бета-блокаторы, такие как атенолол, метопролол, надолол, бизопролол, пиндолол, ацебутолол, бетаксолол, пропранолол; диуретики, такие как гидрохлортиазид, фуросемид, торсемид, метолазон; блокаторы кальциевых каналов, такие как амлодипин, фелодипин, низолдипин, нифедипин, верапамил, дилтиазем; альфа-блокаторы рецепторов, такие как доксазозин, теразозин, альфузозин, тамсулозин; и альфа-агонисты центрального действия, такие как клонидин. Агентами, используемыми для лечения гиперлипидемии, являются, но не ограничиваются ими, агенты, снижающие уровни ЛНП, такие как статины (аторвастатин, флувастатин, ловастатин, правастатин, розувастатин-кальций, симвастатин) и никотиновая кислота, белковые ингибиторые переноса холестерилового эфира (такие как торсетрапиб), агенты, стимулирующие PPAR-альфа, такие как фибраты, гемфиброзил, фенофибрат, безафибрат, ципрофибрат, агенты, связывающиеся с желчными кислотами и предупреждающие реадсорбцию желчных кислот, и агенты, снижающие уровни холестерина, такие как секвестранты желчных кислот, холестирамин и колестипол, и ингибиторы абсорбции холестерина.
BH4 может быть также введен вместе с фактором или с комбинацией факторов, усиливающих или нормализующих продуцирование сосудорасширяющего оксида азота (NO), отдельно или в комбинации с терапевтическим средством. В одном из вариантов изобретения такой(ие) фактор(ы) усиливает(ют) активность или экспрессию биосинтеза ВН4 de novo, и такие факторы выбирают из группы, состоящей из гуанозинтрифосфат-циклогидролазы I (GTPCH1), 6-пирувоилтетрагидроптерин-синтазы (PTPS) и сепиаптерин-редуктазы. В предпочтительном варианте изобретения синтез BH4 усиливают путем повышения уровней экспрессии GTPCH1 с использованием любого одного или нескольких циклических аналогов аденозинмонофосфата (cAMP) или агонистов, включая форсколин, 8-бром-cAMP или другие агенты, усиливающие cAMP-опосредуемую передачу клеточного сигнала, например цитокины и факторы роста, включая интерлейкин-1, интерферон- гамма (IFN-γ), фактор некроза опухоли альфа (TNF-α), c-реактивный белок, HMG-CoA-редуктазы (статины, такие как аторвастатин), фактор роста нервных клеток (NGF), эпидермальный фактор роста (EGF), гормоны, включая адреномедуллин и бензоат эстрадиола, и другие соединения, такие как NADPH и аналоги NADPH, кофеин, циклоспорин А, метилксантины, включая 3-изобутил-1-метилксантин, теофиллин, резерпин, пероксид водорода.
Поэтому в одном из своих вариантов настоящее изобретение относится к увеличению уровней GTPCH1 путем ингибирования разложения 3',5'-циклических нуклеотидов с использованием ингибиторов, принадлежащих к семейству из одиннадцати фосфодиэстераз (PDE1-11), включая PDE1, PDE3, PDE5. Ингибиторами PDE согласно изобретению являются виагра/силданефил, циалис/тадалафил, варденафил/левитра, уденафил, 8-метоксиметил-IBMX, UK-90234, дексаметазон, гесперетин, геспередины, ирсогладин, винпроцетин, цилостамид, ролипрам, этил-бета-карболин-3-карбоксилат (бета-CCE), производные тетрагидро-бета-карболина, 3-O-метилкверцетин и т.п.
В другом своем варианте настоящее изобретение относится к повышению уровней ВН4 путем увеличения уровней BH4-синтезирующих ферментов с применением генотерапии или путем доставки в эндотелий полинуклеотидов по механизму синтеза ВН4. В еще одном своем варианте настоящее изобретение относится к повышению уровней ВН4 путем введения BH4-синтезирующих ферментов GTPCH1, PTPS, SR, PCD, DHPR и DHFR. В соответствии с настоящим изобретением BH4-синтезирующие ферменты включают все природные и неприродные формы ферментов, включая мутанты белков.
В другом своем варианте настоящее изобретение относится к повышению уровней BH4 благодаря использованию 7,8-дигидронеоптеринтрифосфата, который является субстратом не для щелочной фосфатазы (AP), а для ВН4-синтезирующего фермента PTPS, что приводит к ингибированию AP-активности. Агентами или соединениями, которые ингибируют активность AP, являются аналоги фосфатов, левамизол и L-Phe. В других своих вариантах настоящее изобретение относится к агентам или соединениям, которые ингибируют щелочную фосфатазу, включая такие агенты, как короткая ингибирующая РНК (киРНК), антисмысловая РНК, дцДНК, небольшие молекулы, нейтрализующие антитела, одноцепочечные, химерные, гуманизованные антитела и фрагменты антител, ингибирующие синтез щелочной фосфатазы.
В других своих вариантах настоящее изобретение относится к к агентам или соединениям, которые усиливают активность катализаторов или кофакторов, необходимых для синтеза ферментов, осуществляемого по новому пути синтеза BH4.
В другом своем варианте настоящее изобретение относится к агентам или соединениям, которые предотвращают разложение ферментов, необходимых для синтеза BH4. В еще одном своем варианте настоящее изобретение относится к агентам или соединениям, которые предотвращают разложение катализаторов, необходимых для синтеза BH4 и синтезирующих его ферментов, включая GTPCH1, PTPS и SR.
В другом своем варианте настоящее изобретение относится к повышению уровней BH4 путем увеличения степени восстановления BH2 по пути «реутилизации». In vivo, BH4 окисляется до BH2. BH2 существует в хиноидной форме (qBH2) и в виде 7,8-дигидроптерина, который восстанавливается до BH4 под действием DHPR и DHFR соответственно. В одном из своих вариантов настоящее изобретение относится к повышению уровней регенерации или реутилизации BH4 из BH2 посредством модуляции активности и синтеза ферментов PCD, DHPR и DHFR с использованием агентов или соединений, которые действуют по пути продуцирования NADPH, тиолов, бензоата перхлорртути, пероксида водорода и т.п.
В другом своем варианте настоящее изобретение относится к агентам, стабилизирующим BH4 путем снижения степени окисления BH4 с использованием агентов или соединений, таких как антиоксиданты, включая аскорбиновую кислоту (витамин C), альфа-токоферол (витамин E), токоферолы (например, витамин A), селен, бета-каротины, каротиноиды, флавоны, флавоноиды, фолаты, флавононы, изофлавоны, катехины, антоцианидины и хальконы.
В другом варианте изобретения такой(ие) фактор(ы) способствует(ют) повышению уровня активности или экспрессии синтазы оксида азота, и тем самым, усилению продуцирования NO.
В еще одном своем варианте настоящее изобретение относится к факторам, ингибирующим белок, регулирующий уровни GTPCH по механизму обратной связи, GFRP. В одном из своих вариантов настоящее изобретение относится к агентам или соединениям, ингибирующим связывание ВН4 с комплексом GTPCH1/GFRP, и тем самым предупреждающим ингибирование по механизму обратной связи под действием ВН4. Агентами или соединениями согласно изобретению являются конкурентные ингибиторы, такие как альтернативные формы ВН4 с измененными аффинностями по отношению к указанному комплексу, структурным аналогам и т.п. В другом своем варианте настоящее изобретение относится к агентам или соединениям, которые повышают уровень связывания L-фенилаланина с CTPCH1/GFRP, что приводит к индуцированию синтеза ВН4. В другом своем варианте настоящее изобретение относится к агентам или соединениям, которые повышают уровни L-Phe, таким как предшественники L-фенилаланина, которые подавляют ингибирование GTPCH1 под действием GFRP и BH4 по механизму обратной связи.
В еще одном своем варианте настоящее изобретение относится к агентам или соединениям, которые модулируют активность или синтез GFRP. В одном из своих вариантов настоящее изобретение относится к агентам или соединениям, ингибирующим активность GFRP. В другом своем варианте настоящее изобретение относится к применению киРНК, небольших молекул, антител, фрагментов антител и т.п. для ингибирования синтеза GFRP.
VI. Анализы на биоптерин
Концентрацию общего биоптерина и окисленного биоптерина в плазме, в крови и в других тканях определяют методом Fukishima et al. (Anal. Biochem. 102:176 (1980)). Биоптерин имеет четыре различные формы, включая две формы восстановленного биоптерина, R-тетрагидробиоптерин (BH4) и хиноноидный R-дигидробиоптерин (q-BH2) и две формы окисленного биоптерина, дигидробиоптерин (BH2) и биоптерин (B). Из этих четырех форм лишь восстановленные формы биоптерина обладают коферментативной активностью. Восстановленный биоптерин превращается в биоптерин (B) при иодировании в кислотных условиях, тогда как в щелочных условиях он превращается в птерин. Окисленный биоптерин превращается в биоптерин (В) при иодировании в кислотных и щелочных условиях. Благодаря этим свойствам количество общего биоптерина может быть определено после иодирования в кислотных условиях, а количество окисленного биоптерина может быть определено после иодирования в щелочных условиях, а затем количество восстановленного биоптерина вычисляют по разности этих количеств. При использовании BH4 в качестве кофермента этот BH4 превращается в q-BH2. q-BH2 сразу превращается в BH4 под действием дигидроптеринредуктазы, либо, если он не восстанавливается, то он окисляется до BH2 или DHPT. Поскольку in vivo существование биоптерина в форме q-BH2 может быть лишь кратковременным, то восстановленный биоптерин может быть с успехом заменен на BH4.
Собранные пробы плазмы и цельной крови сразу обрабатывают путем окисления кислотным раствором (0,6 н. раствором HCl в воде, содержащим 0,6% иодид калия (KI), 0,3% иод (I2) и 0,6 н трихлоруксусную кислоту (TCA)) и щелочным окисляющим раствором (0,7 н. гидроксид натрия (NaOH)). Определение соединения B осуществляют с помощью ВЭЖХ, а радиоактивность измеряют на жидкостном сцинтилляционном счетчике.
Определение уровня BH4 с помощью обращенно-фазовой (ОФ) ВЭЖХ в сочетании с тандемной масс-спектрометрией (ЖХ/МС/МС):
Было показано, что способ с применением обращенно-фазовой (ОФ) высокоэффективной жидкостной хроматографии в комбинации с тандемной масс-спектрометрией (ЖХ/МС/МС) является селективным для BH4 в человеческой плазме и чувствительным для BH4 в концентрации 5-1000 нг/мл. Этот способ основан на примерно 50% превращении BH4 в результате окисления в процессе сбора и хранения. Пробы плазмы являются стабильными в течение более чем 3 месяцев в дикалиевой соли этилендиаминтетрауксусной кислоты (K2EDTA). Выход для стадий предварительной обработки составляет примерно 75%. Точность и достоверность этого метода считаются надежными, если коэффициент изменчивости (CV)% ниже 15% (20% в случае нижнего предела количественной оценки, LLOQ).
Было показано, что комбинированный метод с применением ВЭЖХ в сочетании с тандемной масс-спектрометрией по сравнению с применением лишь одной ВЭЖХ позволяет проводить более точную оценку тестируемого препарата BH4 благодаря: (1) повышенной селективности этого метода по отношению к ВН4-содержащему лекарственному средству (тогда как одна ВЭЖХ позволяет определить лишь уровень общего биоптерина), (2) более широкой качественной классификации, (3) установленному уровню конверсии, (4) широкой характеризации и подтвержденной эффективности для человека и (5) новому и эффективному измерению в различных молекулах и матрицах.
Такой улучшенный способ включает нижеследующие стадии. Образцы крови, плазмы, гомогенатов ткани или мочи подвергают кислотному или щелочному окислению. При окислении кислотой (1) эти образцы обрабатывают хлоридом калия (KCl), соляной кислотой (HCl) или TCA в течение одного часа; (2) проводят иодометрический анализ окисленных кислотой образцов; (3) образцы пропускают через ионообменную колонку; (4) проводят оценку уровней общего биоптерина, содержащего ВН4, q-BH2 (который сразу восстанавливается in vivo до BH4, а поэтому измерение уровня восстановленного биоптерина основано, главным образом, на BH4), ВН2 и B, с помощью ВЭЖХ и тандемной масс-спектрометрии. При окислении щелочью: (1) образцы обрабатывают KI, I2 или NaOH в течение одного часа; (2) окисленные щелочью образцы подкисляют HCl или TCA; (3) проводят иодометрический анализ; (4) указанные образцы пропускают через ионообменную колонку; (5) определяют уровень окисленного биоптерина, содержащего ВН2 и B; (6) различные молекулы определяют с помощью ВЭЖХ и тандемной масс-спектрометрии; и (7) вычисляют количество восстановленного биоптерина (BH4 + q-BH2) как разность между уровнями общих биоптеринов и их окисленной формы.
Блок-схемы для измерения уровня биоптерина и систематизированная оценка достоверности проводимого анализа представлены на фигурах 16 и 17.
Оптимизированный анализ
ВЭЖХ-метод, проводимый с применением электрохимической детекции (ECD) и детекции флуоресценции (FL), является предпочтительным, поскольку он позволяет определять каждое отдельное биоптериновое соединение (BH4, BH2 и B), а также их аналоги.
BH4 представляет собой кофактор для ферментной системы синтаз оксида азота (NOS), которые продуцируют оксид азота (NO). Продуцирование NO играет важную роль в поддержании сосудистого гомеостаза. При ограничении внутриклеточных уровней BH4 уровень продуцирования NO снижается (из-за снижения активности NOS), что приводит к образованию свободных радикалов супероксида (O2-), оказывающих разрушающее действие на клетки. Избыток O2- может приводить к эндотелиальной дисфункции и может играть определенную роль в окислении BH4 до BH2. Низкое отношение BH4:BH2 может вызывать поражение эндотелия, тогда как высокое отношение BH4:BH2 может стимулировать улучшение функции эндотелия. Поэтому оценка отношения BH4:BH2 может служить прогностическим фактором улучшения функции эндотелия.
Концентрации различных биоптеринов (BH4, BH2 и B) или их аналогов определяют сначала с помощью обращенно-фазовой ВЭЖХ, проводимой для разделения, а затем с помощью электрохимической детекции (ECD) и детекции флуоресценции (FL).
BH4, который является чувствительным к окислительно-восстановительным реакциям, представляет собой нефлуоресцентную молекулу, которую детектируют с помощью ECD. BH4 (и его аналоги) детектируют с помощью ECD, при которой BH4 (или его аналог) окисляется на электроде 1 до хиноноидной дигидробиоптериновой формы (например, qBH2), то есть дигидробиоптеринового промежуточного соединения с короткой продолжительностью жизни, которое затем снова восстанавливается до BH4 (или его аналога) на электроде 2. Затем ток детектора, генерируемый такой реакцией восстановления, использовали для определения концентрации ВН4 или его аналога (уровень эндогенного qBH2 является ничтожно малым).
BH2, B и его аналоги могут быть детектированы путем аналогичного впрыска при регистрации флуоресценции. Окисление ВН2 или его аналога после ECD, проводимое с использованием кондиционирующей предохраняющей ячейки при оптимальном потенциале, приводит к образованию соединения В или его соответствующего биоптеринового аналога. Такой процесс является особенно желательным, поскольку ВН2 не является флуоресцентно активным или не может быть легко определен, а поэтому должен быть превращен в соединение В, которое может быть легко детектировано методом флуоресценции. Эндогенный ВН2, после его превращения в соединение В и эндогенное соединение В отличают друг от друга по двум отдельным пикам флуоресценции, наблюдаемым благодаря различным временам удерживания каждой молекулы на ВЭЖХ-колонке.
В целом, эти методы могут быть применены для детектирования молекул BH4, BH2 и B и их аналогов. Биоптерины предпочтительно детектируют с использованием подвижной фазы, содержащей 2% MeOH, как описано в настоящей заявке. Аналоги биоптерина, такие как валиновые производные биоптерина, могут быть более подходящими для повышения содержания метанола в подвижной фазе, например в подвижной фазе, содержащей 10% MeOH.
Таким образом, метод детектирования биоптеринов в смеси биоптериновых молекул может включать: (а) разделение биоптериновых молекул в смеси с помощью обращенно-фазовой ВЭЖХ, а в случае BH4 и его аналогов (b1) проведение электрохимического детектирования путем окисления ВН4 и его аналогов, присутствующих на первом электроде, до хиноноидных дигидробиоптериновых форм с последующим восстановлением хиноноидных форм до ВН4 и его аналогов, присутствующих на втором электроде, и измерением силы тока, генерируемого реакцией восстановления в целях определения концентрации молекул; и/или (b2) с случае BH2, его аналогов, биоптерина или его аналогов, оценку таких молекул методом детекции флуоресценции после проведения окисления молекулы ВН2 до биоптерина на колонке. Предпочтительной является подвижная фаза, описанная в настоящей заявке.
В одном из вариантов изобретения предпочтительная подвижная фаза включает ацетат натрия, лимонную кислоту, EDTA и 1,4-дитиоэритрит (DTE) с метанолом. Предпочтительными концентрациями являются 50 мМ ацетата натрия, 5 мМ лимонной кислоты, 48 мкМ EDTA и 160 мкМ DTE с 2% метанолом.
VII. Примеры
В нижеследующих примерах приводится описание предпочтительных вариантов настоящего изобретения. Следует отметить, что методы, описанные заявителем в этих примерах, представляют собой репрезентативные методы, которые могут быть применены на практике и могут рассматриваться как предпочтительные варианты осуществления изобретения. Однако для специалистов в области, к которой относится изобретение, очевидно, что в конкретные варианты осуществления изобретения может быть внесено множество изменений, которые дают подобные или аналогичные результаты, не выходящие за рамки существа и объема изобретения.
Пример 1
Кривая зависимости концентрации от времени для биоптерина в плазме после введения крысам однократной пероральной дозы
Целью данных исследований является оценка фармакокинетики BH4 после введения крысам однократной пероральной дозы. Самцам крыс Sprague Dawley (в возрасте 6 недель), содержащимся в условиях голодания, перорально вводили однократную дозу BH4 (10 и 100 мг/кг).
Результаты
Максимальные концентрации общего биоптерина в плазме через 2 часа и через 1 час после введения дозы составляли 108 нг/мл (то есть примерно в 3 раза превышали эндогенный уровень) и 1227 нг/мл (то есть примерно в 30 раз превышали эндогенный уровень), соответственно (фигура 18). Затем биоптерин, имеющий период полувыведения из кровотока (t1/2), составляющий примерно 1,1 часа, возвращался до своего эндогенного уровня через 9 часов после введения дозы 10 мг/кг и через 24 часа после введения дозы 100 мг/кг (фигура 18).
Биологическая доступность (F) после перорального введения 10 и 100 мг/кг составляла 6,8% и 11,8% соответственно, как было определено исходя из площади под кривой зависимости концентрации в плазме (ΔAUC) от времени путем вычитания из полученных уровней эндогенного уровня при внутривенном введении 10 мг/кг. Скорость абсорбции в ЖКТ составляла 8,8%, как было определено с использованием радиоактивных маркеров в моче. Исходя из этих данных фактическое значение биологической доступности при пероральном введении должно составлять приблизительно 10%.
Отношение восстановленного биоптерина к общему биоптерину в плазме (то есть отношение для его восстановленной формы) оставалось относительно постоянным (73%-96%) (фигура 19).
Пример 2
Кривая зависимости концентрации от времени для биоптерина в плазме после введения обезьянам однократной пероральной дозы
Целью данных исследований является оценка фармакокинетики сапроптерина после введения собакоподобным обезьянам однократной пероральной дозы. Самкам собакоподобных обезьян (3 животных на группу), содержащимся в условиях голодания, перорально вводили однократную дозу сапроптерина (10 мг/кг).
Результаты
Общая концентрация биоптерина в плазме (ΔС) достигала своего максимального значения через 3 часа после введения дозы (344 нг/мл, приблизительно в 20 раз превышающей эндогенный уровень) (фигура 20). Биоптерин, имеющий период полувыведения из плазмы примерно 1,4 часа, возвращался до своего эндогенного уровня через 24 часа после введения дозы. Отношение восстановленного биоптерина к общему биоптерину было почти постоянным во время всего периода проведения теста. Биологическая доступность (F) после перорального введения самкам обезьян 10 мг/кг составляла приблизительно 9%, как было определено по ΔAUC для отношений пероральной дозы/внутривенной дозы (фигура 21).
Пример 3
Относительная биологическая доступность тетрагидробиоптерина (BH4), вводимого после растворения таблетки(ок) в воде или вводимого в виде целой(ых) таблетки(ок), и влияние пищи на абсорбцию у здоровых индивидуумов
Цели исследования
Основными целями данного исследования являются: (1) оценка относительной биологической доступности тетрагидробиоптерина (BH4, дигидрохлорида сапроптерина) при его введении после растворения таблетки(ок) в воде или в виде целой(ых) таблетки(ок); (2) сравнение влияния пищи на биологическую доступность ВН4 у здоровых индивидуумов. Другой целью данного исследования является оценка безопасности и переносимости одной пероральной дозы BH4 у здоровых индивидуумов.
Методика
Настоящее исследование представляет собой рандомизированное перекрестное исследование с открытой меткой, проводимое с использованием шести групп, соответствующих всем возможным последовательностям трех различных схем введения, в три периода, где 30 индивидуумам давали однократную дозу в каждый из 3 периодов, при этом указанное исследование было рандомизированным для шести групп с различными последовательностями схем введения: (группы 1, 2, 3, 4, 5 и 6):
Группа 1: a, b, c
Группа 2: b, c, a
Группа 3: c, a, b
Группа 4: a, c, b
Группа 5: b, a, c
Группа 6: c, b, а
где все группы, которые получали пероральную дозу, принимали 10 мг/кг BH4 по нижеследующей схеме:
a: введение после растворения таблетки(ок) в воде натощак в условиях голодания
b: введение целой(ых) таблетки(ок) натощак в условиях голодания
c: введение целой(ых) таблетки(ок) через 30 минут после начала приема высококалорийной пищи, то есть пищи с высоким содержанием жира.
Каждый индивидуум получал одну дозу 10 мг/кг BH4 во время каждого периода лечения. Между введением каждой дозы делали перерыв по меньшей мере в семь дней. Оценку после исследования проводили через 5-7 дней после окончания третьего периода лечения. Пробы крови для фармакокинетического (ФК) анализа брали в каждый период исследования в соответствии с установленным протоколом взятия проб: за 30 минут до введения дозы и через 0,5, 1,0, 1,5, 2,0, 2,5, 3,0, 3,5, 4,0, 5,0, 6,0, 8,0, 10,0, 12,0, 18,0 и 24,0 часа после введения дозы.
Дозы и способ введения
BH4-содержащие таблетки вводили в дозах 10 мг/кг на период лечения. Таблетки вводили a) в виде таблеток, растворенных в воде, натощак, b) в виде целых таблеток натощак или c) в виде целых таблеток вместе с приемом пищи.
Получали каждую дозу исследуемого лекарственного средства и эту дозу вводили в жидкой форме (в виде раствора), смешанной с водой. Подаваемой водой была водопроводная вода при температуре окружающей среды. Дозы в виде раствора приготавливали за 15 минут до введения дозы по схеме. Растворение таблетки в жидкости происходит в течение приблизительно 1-3 минут. Для увеличения скорости растворения таблетки перед их растворением разламывали или измельчали в сосуде для приготовления доз.
Утром, в назначенное время введения дозы, BH4 перорально вводили в виде нескольких таблеток, эквивалентных дозе 10 мг/кг и растворенных в 120 мл воды или апельсинового сока. После употребления всей дозы 120 мл через 15 минут после ее приготовления каждого индивидуума тщательно обследовали. Сразу после употребления дозы сосуд для приготовления дозы промывали 60 мл воды, а индивидуум проводил полоскание рта. В сосуд для приготовления дозы добавляли вторую промывочную порцию 60 мл воды, а затем индивидуум проводил второе полоскание. Вся процедура введения дозы составляла 1 минуту. Квалифицированный медицинский персонал осматривал сосуд для приготовления доз и ротовую полость каждого индивидуума сразу после приема дозы для гарантии того, что вся доза была употреблена полностью. Альтернативно, индивидуум проглатывал ВН4-содержащие драже, не растворенные в воде. Для каждого индивидуума между введением доз проходило минимум 7 дней.
Схема приема пищи
Вечером подавалась легкая закуска, и ее прием регистрировался. Затем всем индивидуумам было предписано голодание в течение по меньшей мере 10 часов до приема дозы.
Условия голодания
Индивидуумам, которые подвергались лечению в условиях голодания, вводили дозы по истечении минимум 10-часового голодания в течение ночи.
Индивидуумы не принимали пищу в течение 4 часов после введения дозы. Воду давали ad lib в течение всего исследования за исключением 1 часа до введения дозы и 1 часа после введения дозы. Стандартизированную пищу давали через приблизительно 4-10 часов после введения лекарственного средства и далее через соответствующие периоды времени.
Исследования, проводимые с приемом пищи
Индивидуумы, подвергаемые лечению в условиях приема пищи, получали дозу после приема высококалорийного завтрака с высоким содержанием жира. Индивидуумы получали нижеследующий стандартный завтрак с высоким содержанием жира (приблизительно 50% от общего содержания калорий в пище) и высоким содержанием калорий (приблизительно 1000 калорий), который начинался за 30 минут до введения дозы по соответствующей схеме и заканчивался (прием последнего кусочка) за 5 минут до приема дозы, и такой завтрак включал:
2 яйца, зажаренных в масле,
2 тонких ломтика бекона,
2 тоста с маслом,
4 унции мелко нарезанного поджаренного картофеля,
8 унций цельного молока.
Эта еда содержала приблизительно 150 калорий белка, 250 калорий углеводов и 500-600 калорий жира. Такая еда может быть заменена эквивалентной едой в соответствии с представленным меню, в котором указано содержание калорий.
Затем индивидуумы не употребляли никакой пищи в течение 4 часов после введения дозы. Воду давали ad lib в течение всего исследования за исключением 1 часа до введения дозы и 1 часа после введения дозы. Стандартизированную пищу давали через приблизительно 4-10 часов после введения лекарственного средства и далее через соответствующие периоды времени.
Продолжительность лечения
Каждый из трех периодов введения однократной дозы проводили с интервалом минимум в 7 дней.
Через 5-7 дней после последнего визита для проведения лечения индивидуум должен был посетить врача для обследования.
Критерии безопасности: оценка и методы
Безопасность оценивали для всех индивидуумов, которые принимали по меньшей мере одну дозу BH4.
Оценка и блок-схема эффективности и безопасности
Безопасность оценивали путем регистрации случаев появления побочных эффектов, изменений параметров 12-отведений ЭКГ, показателей жизненно важных функций и данных обследования физического состояния, а также изменений базовых величин лабораторных анализов. Протокол этих оценок представлен на фигуре 22.
Оценка физического состояния и показателей жизненно важных функций
Каждый индивидуум проходил рутинную процедуру обследования физического состояния, проводимую исследователем. Оценка физического состояния включала обследование головы, глаз, ушей, носа, горла, шеи, сердца, грудной клетки, легких, брюшной полости, конечностей, пульса в периферических органах, неврологического статуса, кожи и других известных физических состояний. Протокол этого исследования не требовал обследования мочеполовых путей.
При этом измеряли рост (в сантиметрах) и массу (в килограммах), и вычисляли индекс массы тела (BMI) (BMI = масса (кг)/[рост(м)]2).
Кровяное давление измеряли в положении сидя в соответствии с рекомендациями Американской ассоциации кардиологов. При измерении кровяного давления ступни индивидуумов в сидячем положении находились на полу в течение 5 минут в состоянии покоя.
Частоту сердечных сокращений (пульс) у индивидуумов измеряли в положении сидя.
Стандартизированную регистрацию электрокардиограммы (ЭКГ) с 12 отведениями проводили во время обследования и по его окончании. Оценка ЭКГ проводилась квалифицированным специалистом. Копии ЭКГ и отчетов по их оценке были занесены в компьютер как часть файла для каждого индивидуума.
Медицинскую карту, результаты клинических лабораторных анализов и кривые ЭКГ просматривал и оценивал руководитель исследований для определения целесообразности участия каждого индивидуума в данном исследовании.
Клинические лабораторные анализы
Гематология:
Были оценены следующие параметры: гемоглобин, гематокрит, общее число лейкоцитов и лейкоцитарная формула, число эритроцитов (RBC) и число тромбоцитов.
Кроме того, делали анализ крови на поверхностный антиген вируса гепатита В, антитела против вируса гепатита С и вирус иммунодефицита человека (ВИЧ).
Химический анализ:
Были оценены следующие химические вещества: альбумин, азот в крови и моче (BUN), креатинин, общий билирубин, щелочная фосфатаза (ALP), аспартат-трансаминаза (AST), аланин-трансаминаза (ALT), натрий (Na+), калий (K+), хлорид (Cl-), лактатдегидрогеназа (LDH), мочевая кислота и глюкоза.
Анализ мочи
Нижеследующую оценку проводили методом с использованием индикаторной тест-полоски для анализа мочи и в этом анализе оценивали рН, удельную плотность мочи, белок, глюкозу, кетоны, билирубин, наличие крови в моче, нитрит и уробилиноген. Если белок присутствует в крови на незначительном уровне или если величины содержания нитритов выходят за допустимые пределы, то проводят гистологический анализ.
Образцы мочи также анализировали на присутствие наркотических средств (амфетаминов, барбитуратов, бензодиазепинов, каннабиноидов, кокаина и опиатов).
Побочные эффекты
В этом исследовании побочный эффект (ПЭ) определяли как любое нежелательное клиническое состояние у индивидуума или у индивидуума, прошедшего клиническое обследование и принимавшего BH4 в любой дозе, независимо от того, существует ли у этого индивидуума причинная взаимосвязь с таким эффектом. Поэтому ПЭ может быть любым неблагоприятным или нежелательным признаком (включая плохие результаты лабораторных анализов), симптомом или временным заболеванием, ассоциированным или не ассоциированным с приемом BH4. Такое определение включает наличие сопутствующих заболеваний или поражений и обострение (повышение частоты, тяжести или специфичности) уже существующих заболеваний.
Период регистрации ПЭ начинается после первого введения ВН4. Период регистрации серьезных побочных эффектов (СПЭ) начинается раньше, со времени подписания документа об информированном согласии. Ниже в этом разделе описаны СПЭ. Исследователь проводил мониторинг всех ПЭ вплоть до их исчезновения или, в том случае, если ПЭ были определены как хронические, то проводили исследования по выявлению их причин. Если по окончании исследования ПЭ оставались невыявленными, то руководитель исследований и специалист, осуществляющий контроль за проведением исследований, проводили клиническую оценку для определения целесообразности дальнейшего наблюдения ПЭ и регистрировали полученные данные. Определение тяжести побочных эффектов является одной из важных обязанностей исследователя, проводящего оценку ПЭ и СПЭ. Исследователь несет ответственность за проведение клинических анализов для оценки причинной взаимосвязи каждого ПЭ с BH4.
Серьезные побочные эффекты
Серьезные побочные эффекты (СПЭ) определяют как любой ПЭ, который имеет по меньшей мере одно из следующих последствий:
Возможность летального исхода.
Угроза для жизни, то есть непосредственный риск смерти от данного побочного эффекта, если он имеет место.
Это определение не включает реакцию, которая, если она наблюдается в более тяжелой форме, может приводить к летальному исходу.
Необходимость госпитализации амбулаторного больного или увеличения времени его пребывания в больнице, если он уже в ней находится.
Поступление индивидуума в больницу как амбулаторного больного в результате возникновения у него ПЭ, причем даже в случае выписки индивидуума в тот же день это событие квалифицируется как госпитализация. Визит в пункт неотложной помощи не рассматривается как госпитализация.
Возможность приобретения стойкой или временной инвалидности или нетрудоспособности.
Побочный эффект квалифицируется как стойкая или временная инвалидность или нетрудоспособность в том случае, если данный индивидуум в значительной степени утрачивает способность выполнять обычные жизненные функции. Это определение не включает относительно незначительную или временную с медицинской точки зрения утрату нетрудоспособности.
Присутствие наследственных патологий или врожденных пороков, то есть ПЭ, которые наблюдаются у детей или в плоде беременной женщины, принимавшей исследуемое лекарственное средство до зачатия или во время беременности.
Наличие серьезных побочных эффектов, которые не удовлетворяют любому из вышеуказанных критериев, но создают угрозу для жизни индивидуума или требует терапевтического или хирургического вмешательства для устранения одного из перечисленных выше последствий.
Более чем одно из вышеуказанных последствий может быть отнесено к любому конкретному побочному эффекту.
Точность измерений
Определение безопасности в данном исследовании проводили путем рутинного обследования физического состояния индивидуума, оценки показателей жизненно важных функций, выявления наличия и тяжести побочных эффектов и проведения клинических и лабораторных анализов.
Измерение концентраций лекарственных средств
Фармакокинетические (ФК) показатели крови (плазмы) оценивали после введения каждой дозы исследуемого лекарственного средства. Все индивидуумы сидели в положении прямо в течение 4 часов после принятия дозы. Пробы крови брали за 30 минут до принятия дозы и через 0,5, 1,0, 1,5, 2,0, 2,5, 3,0, 3,5, 4,0, 5,0, 6,0, 8,0, 10,0, 12,0, 18,0 и 24,0 часов после принятия дозы. Взятые пробы собирали в соответствующим образом помеченные пробирки Vacutainer®, в верхней части которых находились 6 мл пурпурного красителя K2-EDTA. Пробы крови центрифугировали приблизительно при 3000 об/мин при 4°С в течение 10 минут. Из каждого полученного образца плазмы пипеткой забирали точно 1 мл и это количество помещали в пробирку с аликвотой, содержащей 0,1% масс./об. дитиоэритрита. Затем образец закрывали и встряхивали приблизительно 10 секунд на мини-миксере VWR Mini Vortexer на скорости 6. После завершения этих стадий образец быстро замораживали на бане с изопропилом/сухим льдом и помещали в холодильник при -70°С для последующего анализа.
В течение каждого периода лечения брали приблизительно 80 мл крови (5 мл для каждого введения) для фармакокинетического анализа.
Фармакокинетические анализы
Фармакокинетический (ФК) анализ данных зависимости концентрации BH4 в плазме от времени проводили несистемными методоми оценки нижеследующих фармакокинетических параметров:
максимальной концентрации в плазме (Cmax) и времени до достижения максимальной концентрации (Tmax), полученных непосредственно из имеющихся данных без интерполяции;
λz, кажущейся константы конечной скорости элиминации, определенной по оценке параметров логарифмической линейной регрессии конечных концентраций в плазме;
площади под кривой зависимости концентрации в плазме от времени 0 до времени последней измеряемой концентрации [AUC(0-t)], вычисленной линейным трапецеидальным методом;
кажущегося времени полувыведения (t1/2), вычисленного как 0,693/λz;
площади под кривой зависимости концентрации в плазме от времени начиная со времени 0 до бесконечности [AUC(inf)], где AUC(inf) = AUC(0-t) + Ct/λz, где Ct означает последнюю измеряемую концентрацию.
Оценка скорости абсорбции
Индивидуумам перорально или внутривенно вводили дозу 10 мг/кг BH4, а затем проводили серию измерений концентрации общего биоптерина в плазме для определения скорости абсорбции BH4 из желудочно-кишечного тракта исходя из площади под кривой зависимости увеличения концентрации общего биоптерина в плазме (ΔCp) от времени (ΔAUC). Было высказано предположение, что для достижения одного и того же уровня биологической доступности при внутривенном введении требуется меньшая доза ВН4, чем при его пероральном введении. Для достижения одного и того же уровня биологической доступности при пероральном введении может потребоваться 10 мг/кг BH4, а при внутривенном введении, 1 мг/кг BH4. Поскольку усиление биологической доступности зависит от способа введения, то для достижения одного и того же уровня биологической доступности при пероральном введении ВН4 может потребоваться 5 мг/кг BH4, а при внутривенном введении только 1 мг/кг ВН4.
Скорость абсорбции BH4 из желудочно-кишечного тракта оценивали исходя из площади под кривой увеличения концентрации общего биоптерина в плазме (ΔCp) от времени (ΔAUC) после введения ВН4 по следующей формуле:
Оценка исходя из AUC
Скорость абсорбции (%) = (ΔAUC) после p.o. дозы/ΔAUC после i.v. дозы)×(i.v. доза/p.o.-доза × 100)
Статистические методы:
Сравнение фармакокинетических параметров Cmax, AUC(0-t) и AUC(inf) для BH4 проводили с использованием модели дисперсионного анализа (ANOVA), причем число различных последовательностей схем лечения, число индивидуумов в группе, соответствующей данной последовательности, схемы лечения и период введения доз классифицируются как переменные величины, где натуральные логарифмы данных параметров используются как зависимые переменные. Такое представляющее интерес сравнение проводили между эффектом растворенной таблетки и целой таблетки, принимаемых натощак, и эффектом целых таблеток, принимаемых вместе с пищей и натощак.
Данные для всех индивидуумов, полученные по меньшей мере за два периода исследования, были включены в фармакокинетические статистические анализы. Все индивидуумы, получавшие по меньшей мере одну дозу исследуемого лекарственного средства, были включены в анализы на безопасность.
Все фармакокинетические и ассоциированные с ними статистические анализы проводили с помощью SAS® для версий Windows® 9.1.3 или более поздней версии.
Для обеспечения достаточной эффективности, удовлетворяющей целям исследования, размер образца, взятого приблизительно у 30 индивидуумов, каждый из которых проходил 3 периода лечения, рассматривался как адекватный для проведения оценки сравнения представляющих интерес различий. Вычислений какого либо формального размера образца не проводили.
Результаты
Фармакокинетический анализ
Сравнение эффекта приема целых и растворенных таблеток
Средние концентрации BH4 в плазме, вводимого в виде растворенной таблетки, были ниже, чем концентрации ВН4, вводимого в виде целой таблетки (фигуры 23 и 24). Средние Cmax были выше для целой таблетки, как и средние значения AUC(0-t) и AUC(inf) (фигура 25). Среднегеометрические значения для целых и растворенных таблеток находились в пределах от 118% до 121%, а верхние пределы ассоциированных с ними 90% доверительных интервалов превышали 125% (фигура 26), что указывало на то, что статистически значимые уровни абсорбции в случае введения целой таблетки вместе с высококалорийной жирной пищей превышали статистически значимое увеличение абсорбции в случае введения растворимый и целой таблетки. Медианы и интервалы значений для Tmax были, в основном, одинаковыми для растворенных и целых таблеток (фигура 25), что позволяет предположить, что увеличение, наблюдаемое для целой таблетки, относится к увеличению степени, но не скорости абсорбции.
Влияние высококалорийной пищи с высоким содержанием жира на абсорбцию лекарственного средства
Как и ожидалось, введение целой таблетки вместе со стандартной жирной высококалорийной пищей приводило к значимому увеличению средних концентраций ВН4 в плазме (фигура 23) и средних величин Cmax, AUC(0-t) и AUC(inf) (фигура 25). Среднегеометрические значения (при приеме пищи и натощак) составляли в пределах от 126% до 139% (фигура 26), и соответственно верхние пределы ассоциированных с ними 90% доверительных интервалов превышали 125%, что указывало на статистически значимое различие влияния пищи на абсорбцию по сравнению с влиянием на абсорбцию целой таблетки. Медианы и интервалы значений для Tmax были, в основном, одинаковыми при приеме пищи и натощак (фигура 25), что позволяет предположить, что указанное увеличение, наблюдаемое в случае приема пищи, относится к увеличению степени, но не скорости абсорбции.
Безопасность:
В этом исследовании каких-либо серьезных побочных эффектов (СПЭ) не наблюдалось. Пять (5) индивидуумов сообщали о наличии у них всего 9 побочных эффектов (ПЭ). Из этих 9 ПЭ, восемь (8) были оценены как слабые, а 1 оценивался как умеренно тяжелый. Наиболее распространенным побочным эффектом была головная боль, при этом у 1 индивидуума наблюдалась умеренная головная боль, которая оценивалась как не связанная с приемом исследуемого лекарственного средства, а у одного индивидуума наблюдалась слабая головная боль в двух случаях, которые оба оценивались как возможно связанные с приемом лекарственного средства. В целом, пять побочных эффектов оценивались как эффекты, не связанные с приемом исследуемого лекарственного средства, а 4 эффекта оценивались как эффекты, возможно, связанные с приемом данного лекарственного средства. Были проведены обследования на возможность исключения из участия в исследовании, анализы ЭКГ и оценка физического состояния пациентов, в результате чего каких-либо клинически значимых данных получено не было.
Выводы:
Введение BH4 в виде целой таблетки приводило к примерно 20% увеличению степени абсорбции по сравнению с абсорбцией, достигаемой при приеме растворенной таблетки.
Введение BH4 в виде целой таблетки вместе с высококалорийной жирной пищей приводило примерно к 30% увеличению степени абсорбции по сравнению с абсорбцией, достигаемой при приеме таблетки натощак.
У исследуемой группы индивидуумов каких-либо клинически значимых спорных результатов и спорных результатов по параметрам безопасности получено не было. В этом исследовании не было обнаружено каких-либо серьезных ПЭ. В 9 случаях сообщения о наличии ПЭ за исключением одного наблюдалась головная боль, которая оценивалась как слабая и не связанная с приемом исследуемого лекарственного средства. Побочными эффектами, которые, возможно, связаны с приемом исследуемого лекарственного средства, были лишь случаи возникновения усталости и головной боли, но и эти случаи по своей тяжести были оценены как легкие.
Пример 4
Способы приготовления лекарственных составов для повышения биологической доступности BH4
Для проведения исследований на животных были выбраны два контрольных состава (состав BH4 для внутривенного введения и таблетка ВН4 для получения перорально вводимого раствора) и шесть тестируемых составов. Каждый прототип состава содержал 80 мг или 100 мг BH4.
Составы ВН4 для внутривенного введения
В таблице 3 указана композиция состава для внутривенного введения. BH4 перед его применением пропускали через сито из нержавеющей стали с размером пор #20 (меш), а при получении данного состава использовали маннит. Этот состав помещали в бутыль в виде порошка и перед введением его разводили стерильной водой для инъекций. Каждая бутыль, изготовленная из прозрачного сополиэфира полиэтилентерефталата (PETG), содержала 100 мг ВН4 и 5 г маннита и сверху была закрыта завинчивающейся крышкой из белого полиэтилена высокой плотности (HDPE). Состав перед его введением разводили 100 мл стерильной воды для инъекций до конечной концентрации 1 мг/мл. Состав для внутривенного введения был помещен в бутыли в виде сухого порошка, и каждая такая бутыль содержала API и маннит. Полученный порошок перед его внутривенным введением растворяли в стерильной воде для инъекций и фильтровали.
|
ВН4-содержащая таблетка для получения перорально вводимого раствора
В таблице 4 указана композиция состава в виде раствора для перорального введения. Десять (10) BH4-содержащих таблеток (100 мг) помещали в 125-миллилитровую градуированную PETG-бутыль, закрытую крышкой из белого HDPE. Состав перед его введением разводили 100 мл стерильной воды для инъекций до конечной концентрации 10 мг/мл.
|
Прототип для замедления высвобождения из желудочно-кишечного тракта
В таблице 5 приводится композиция прототипа для замедленного высвобождения из желудка. Перед использованием BH4 пропускали через сито из нержавеющей стали #20 меш. Capmul GMO-50 расплавляли на водяной бане при 37°С. BH4 и аскорбиновую кислоту взвешивали и медленно, при интенсивном перемешивании, добавляли к расплавленному Capmul. Дисперсию твердого вещества пипеткой по каплям добавляли в капсулу размером #2. Три заполненные капсулы помещали в 100 см3-бутыль из полиэтилена высокой плотности (ПЭВП), герметично закрывающуюся крышкой с индукционным нагревом.
|
Прототип биоадгезива
В таблице 6 приводится композиция биоадгезивного прототипа. Все материалы за исключением карбопола 71G пропускали через сито из нержавеющей стали #20 меш. Все материалы взвешивали и добавляли в пластиковый пакет, имеющий крышку типа застежки-молнии, а затем его встряхивали в течение нескольких минут до получения однородной на вид смеси. Этот порошок спрессовывали в таблетки с использованием 1/4" стандартного круглого вогнутого приспособления В с плоской поверхностью на лабораторном прессе Globe Pharma MTCM-I под давлением 600 фунт/кв. дюйм (4136 кПа). Три таблетки вместе с силикагелем-осушителем в упаковке помещали в 100 см3-бутыль из полиэтилена высокой плотности (ПЭВП), герметично закрывающуюся крышкой с индукционным нагревом.
|
Прототип с замедленным высвобождением
В таблице 7 приводится композиция прототипа с замедленным высвобождением, тестируемого на обезьянах. Все материалы за исключением метоцеля K100M Premium CR пропускали через сито из нержавеющей стали #20 меш. Все материалы взвешивали и добавляли в пластиковый пакет, имеющий крышку типа застежки-молнии, а затем его встряхивали в течение нескольких минут до получения однородной на вид смеси. Этот порошок спрессовывали в таблетки с использованием 1/4" стандартного круглого вогнутого приспособления В с плоской поверхностью на лабораторном прессе Globe Pharma MTCM-I под давлением 1200 фунт/кв. дюйм (8272 кПа). Три таблетки вместе с силикагелем-осушителем в упаковке помещали в 100 см3-бутыль из ПЭВП, герметично закрывающуюся крышкой с индукционным нагревом.
|
Полимерный прототип донора протонов
В таблице 8 приводится композиция полимерного прототипа донора протонов, тестируемого на обезьянах. Все материалы за исключением эудрагита L100-55 и коллидона CL предварительно пропускали через сито из нержавеющей стали #20 меш. Все материалы взвешивали и добавляли в пластиковый пакет, имеющий крышку типа застежки-молнии, а затем его встряхивали в течение нескольких минут до получения однородной на вид смеси. Предварительно отвешенное количество порошка помещали в капсулу размером #2.
Раствор для покрытия получали путем растворения эудрагита L100-55 и ПЭГ 4600 Carbowax в этиловом спирте. Эудрагит L100-55 и ПЭГ 4600 Carbowax взвешивали и добавляли в 125-миллилитровую градуированную бутыль из сополиэфира полиэтилентерефталата (PETG). Затем в PETG-бутыль добавляли этиловый спирт и эту бутыль помещали на водяную баню при 40°С с обработкой ультразвуком до тех пор, пока раствор не становился прозрачным.
Наполненные порошком капсулы вручную погружали в раствор для нанесения покрытия и оставляли для сушки на 20 минут при 40°С. Осушенные капсулы взвешивали, а затем раскатывали в реагенте Syloid FP244 для устранения остаточной клейкости. Три капсулы упаковывали в 100 см3-бутыль из ПЭВП, герметично закрывающуюся крышкой с индукционным нагревом.
|
Флотирующая система доставки
В таблице 9 указана композиция флотирующей системы доставки. Все материалы за исключением эудрагита L100-55 пропускали через сито из нержавеющей стали #20 меш. Прототип этой таблетки состоит из трех слоев, где средний слой, содержащий лекарственное средство, расположен между двумя нерастворимыми в воде внешними слоями по типу «сэндвича». Внутренние и внешние материалы взвешивали и по отдельности добавляли в пластиковые пакеты с крышками типа застежки-молнии, а затем эти пакеты встряхивали до получения однородных на вид смесей.
Два внешних слоя (по 12 мг каждый) и внутренний слой (14,5 мг) взвешивали. Один из внешних слоев добавляли в пресс, после чего добавляли внутренний слой, а затем последний внешний слой. Слои спрессовывали в таблетки с использованием 3/16" круглого вогнутого приспособления В со скошенными краями и с плоской поверхностью на лабораторном прессе Globe Pharma MTCM-I под давлением 200 фунт/кв. дюйм (1378,8 кПа).
Раствор для нанесения покрытия получали путем растворения этоцеля и ПЭГ 4600 Carbowax в этиловом спирте и в очищенной водной смеси. Эти ингредиенты добавляли в PETG-бутыль и содержимое этой бутыли перемешивали и помещали на водяную баню при 40°С с обработкой ультразвуком до тех пор, пока раствор не становился прозрачным на вид.
Таблетки вручную погружали в раствор для нанесения покрытия и оставляли для сушки на 20 минут при 40°С. После нанесения покрытия каждую таблетку снова взвешивали. Семь (7) таблеток помещали в каждую продолговатую капсулу размером #2. Три капсулы упаковывали в 100 см3-бутыль из ПЭВП, герметично закрывающуюся крышкой с индукционным нагревом.
|
Газообразующая флотирующая система доставки
В таблице 10 указана композиция газообразующей флотирующей системы доставки. Этот состав состоит из сердцевины таблетки, содержащей лекарственное вещество, окруженное газообразующим внешним слоем. Все материалы за исключением бикарбоната натрия и метоцеля K100M CR предварительно пропускали через сито из нержавеющей стали #20 меш. Материалы внутренней сердцевины и внешнего слоя взвешивали и по отдельности добавляли в пластиковые пакеты с крышками типа застежки-молнии, а затем эти пакеты закрывали и встряхивали до получения однородной на вид смеси. Смешанный порошок для внутренней сердцевины (35 мг) спрессовывали в таблетки с использованием 1/8" круглого вогнутого приспособления В со скошенными краями и с плоской поверхностью на лабораторном прессе Globe Pharma MTCM-I под давлением 800 фунт/кв. дюйм (5515,8 кПа).
Раствор для нанесения покрытия получали путем растворения этоцеля и ПЭГ 4600 в этиловом спирте. Внутреннюю сердцевину таблеток вручную погружали в раствор для нанесения покрытия и оставляли для сушки на 20 минут при 40°С. Смешанный порошок для внешнего слоя (40 мг) взвешивали. Одну половину добавляли в пресс, а затем добавляли внутреннюю сердцевину таблетки и вторую половину внешнего слоя. Таблетки спрессовывали с использованием 3/16" круглого приспособления В со скошенными краями и с плоской поверхностью на лабораторном прессе Globe Pharma MTCM-I под давлением 800 фунт/кв. дюйм (5515,8 кПа). Четыре (4) таблетки помещали в каждую капсулу размером #2.
|
Прототип биоадгезивных гранул
В таблице 11 приводится композиция прототипа биоадгезивных гранул. Все материалы за исключением метоцеля K100M CR предварительно пропускали через сито из нержавеющей стали #20 меш. Все материалы за исключением стеарилфумарата натрия (PRUV) взвешивали и помещали в сосуд-гранулятор размером #1 (LB Bohle Mini Granulator BMG). Порошок перемешивали на лопастной мешалке при скорости 300 об/мин и в дробилке при скорости 2500 об/мин в течение пяти минут до тех пор, пока смесь не становилась однородной на вид. На указанной скорости лопастной мешалки и дробилки к смеси по каплям добавляли 5 мл этилового спирта до образования гранул. Сырую массу вынимали из сосуда-гранулятора и пропускали через сито из нержавеющей стали размером 18 меш. Затем гранулы собирали и помещали в печь при 40°С для сушки в течение одного часа. После сушки в течение одного часа усушка гранул, как было определено, составляла 1,93%. Гранулы взвешивали и помещали в пластиковый пакет с крышкой типа застежки-молнии. В осушенные гранулы, содержащиеся в этом пакете, добавляли стеарилфумарат натрия (PRUV). Затем пакет закрывали и встряхивали до тех пор, пока стеарилфумарат натрия (PRUV) равномерно не распределялся среди гранул. Гранулы взвешивали (134 мг). Продолговатые капсулы размером #2 заполняли порциями гранул, чередующихся с каплями частично гидрогенизированного растительного масла (350 мкл). Три капсулы упаковывали в 100 см3-бутыль из ПЭВП, герметично закрывающуюся крышкой с индукционным нагревом.
|
Высвобождение лекарственного средства in vitro
Тестирование таблеток на высвобождение лекарственного средства in vitro проводили в соответствии с техническим описанием, прилагаемым к аппарату USP 27 apparatus II, с использованием прибора для определения растворения Distek 2100C (Distek, Inc., North Brunswick, NJ), в комбинации с спектроскопической системой, работающей в УФ-диапазоне и в диапазоне видимой области спектра Agilent (Agilent Technologies, Santa Clara, CA). Среда для определения растворения, используемая для тестирования высвобождения ВН4, состояла из 900 мл 0,1 н. HCl. Во время тестирования растворения среду в каждом сосуде поддерживали при 37°С±0,5°С и перемешивали при 50 об/мин. В заранее определенные периоды времени брали образец в объеме 5 мл. Для определения концентрации BH4 в образцах 250 мкл каждого образца разбавляли 500 мкл 0,1 н. HCl и измеряли абсорбцию при 265 нм на УФ-спектрометре (спектрофотометр 8453, работающий в УФ-диапазоне и в диапазоне видимой области спектра, Agilent Technologies, Santa Clara, CA). Полученные данные собирали с помощью компьютерной программы ChemStation (Rev. A.09.01 [76], Agilent Technologies, Santa Clara, CA). Все тесты на растворение осуществляли с тремя повторностями.
Тест на текучесть таблеток
Текучесть флотирующих таблеток-прототипов сначала определяли путем помещения таблеток в пластиковые чашки с 25-50 мл 0,1 н. HCl. В этом тесте определяли время, необходимое для растекания таблеток, а также продолжительность их растекания без перемешивания. Таблетки-прототипы, которые растекались по меньшей мере в течение четырех часов, подвергали тесту на растворение. В процессе проведения теста на растворение текучесть таблеток определяли методом с применением лопастной мешалки, работающей при скорости вращения 50 об/мин. Состояние таблеток визуально оценивали в различные периоды времени.
Тест на дезинтеграцию
Тест на дезинтеграцию проводили в соответствии с техническим описанием теста на дезинтеграцию USP-27 с использованием прибора для оценки дезинтеграции серии Distek 3100 (Distek Inc., North Brunswick, NJ). Среда, используемая для дезинтеграции, состояла из 900 мл 0,1 н. HCl или 900 мл 0,2M фосфата калия, pH 5,8. Во время проведения теста на дезинтеграцию среду в сосудах поддерживали при 37°С±0,5°С. Таблетки и капсулы визуально оценивали на дезинтеграцию.
Тест на твердость таблеток
Твердость таблеток определяли на приборе для определения твердости таблеток Dr. Schleuniger Pharmatron 8M Tablet Hardness Tester (Dr. Schleuniger® Pharmatron Inc., Manchester, NH). Таблетки помещали в зажимное приспособление прибора для определения твердости, и твердость измеряли в килопондах (Кп) (1 Кп = 9,8 Н).
Толщина таблеток
Толщину таблеток определяли с помощью цифрового индикатора Mitutoyo (Mitutoyo Absolute, Dr. Schleuniger Pharmatron Inc., Manchester, NH). Таблетки помещали под калибромер и полученную величину регистрировали в миллиметрах (мм).
Результаты и обсуждение
Исходя из трех концепций было разработано несколько прототипов лекарственных форм: лекарственные формы, удерживающиеся в желудке; лекарственные формы на основе полимерного донора протонов для изменения рН тонкого кишечника и лекарственные формы с замедленным высвобождением. В нижеследующих разделах описано получение каждого прототипа.
BH4-содержащий состав для внутривенного введения
После разведения в стерильной воде полученный раствор становился изотоничным, pH 3,2, содержал 1 мг/мл BH4 и после стерильной фильтрации через фильтр 0,22 микрон был подходящим для внутривенного введения. Стабильность 1 мг/мл раствора, выдерживаемого при температуре окружающей среды, анализировали с помощью ВЭЖХ каждый час в течение трех часов. Затем образцы раствора, выдерживаемого в указанных условиях, хранили при -20°С и через 2 недели анализировали с помощью ВЭЖХ. На фигуре 27 показано, что этот раствор был стабильным при температуре окружающей среды в течение по меньшей мере 3 часов после разведения и в течение по меньшей мере 2 недель во время хранения при -20°С.
BH4-содержащая таблетка для получения перорально вводимого раствора
Каждая бутыль содержала десять (10) таблеток с BH4, 100 мг. К содержимому каждой бутыли добавляли сто миллилитров (100 мл) очищенной воды или стерильной воды для инъекций. После интенсивного встряхивания бутыли таблетки быстро дезинтегрировались в течение 5 минут. Полученный раствор для перорального введения содержал 10 мг/мл BH4. Не все ингредиенты данной таблетки были растворимыми, и независимо от того, был ли конечный раствор мутным или прозрачным на вид, активный фармацевтический ингредиент полностью растворялся, а тонкодисперсные частицы представляли собой слаборастворимые неактивные ингредиенты.
Прототип состава для замедления выведения из желудочно-кишечного тракта
Этот состав в виде капсулы состоял из BH4 и аскорбиновой кислоты, диспергированных в полутвердом производном жирной кислоты (глицерилмоно/диолеате с температурой плавления 86°F (30°С)). Глицерилмоно/диолеат (GMO) был также выбран исходя из его химической совместимости с BH4. Профиль растворения, проиллюстрированный на фигуре 28, показал, что более чем 90% лекарственного средства высвобождалось в течение 2 часов и после хранения капсул при 40°С в течение 57 дней профиль их растворения не изменялся.
Дисперсию лекарственного средства в полутвердом расплавленном GMO вводили вручную в жесткие желатиновые капсулы. Плотность полутвердого вещества превышала 1 г/мл, и такое вещество может быть введено в дозе по меньшей мере 80 мг при 25% загрузке лекарственного средства в капсулу размером #2. При этом предполагается, что капсула размером #0 должна содержать по меньшей мере 200 мг лекарственного средства при использовании того же самого состава. При этом во время хранения при 40°С наблюдалась утечка жирной кислоты из капсулы. Во избежании утечки жирной кислоты во время хранения капсулы или составы в виде капсул из мягкого геля, предпочтительно, изготавливать в обернутом виде.
Прототип биоадгезива
Многие биоадгезивы приготавливают из синтетических или природных полимеров. Большинство современных синтетических биоадгезивных полимеров представляют собой полиакриловую кислоту или производные целлюлозы. Примерами полимеров на основе полиакриловой кислоты являются, но не ограничиваются ими, карбапол, поликарбофил, полиакриловая кислота (ПАК) и т.п. Целлюлозами являются, но не ограничиваются ими, гидроксипропилцеллюлоза и гидроксипропилметилцеллюлоза (HPMC). Два биоадгезивных прототипа были разработаны для проведения тестов в исследованиях на животных. Первый прототип представляет собой биоадгезивный состав в виде таблетки, а второй представляет собой капсулу, содержащую биоадгезивные гранулы.
Для получения первого биоадгезивного прототипа в форме таблетки были выбраны поликарбофильные и карбомерные полимеры. Карбопол 71G представляет собой гранулированную форму карбомера и обладает хорошими свойствами текучести порошков. Все партии изготовленных таблеток были хорошего качества и имели приемлемое содержание лекарственного средства (профиль растворения указывал на высвобождение лекарственного средства, близкое к 100%) и приемлемую твердость. В таблице 12 указаны репрезентативные массы, толщина и твердость прототипов биоадгезивных таблеток, содержащих карбомер и поликарбофил.
|
Для получения вторых биоадгезивных гранул использовали HPMC- и карбомерные полимеры. HPMC была выбрана потому, что она используется в виде гидроколлоидной системы низкой плотности и регулирует высвобождение лекарственного средства независимо от значений рН. Для повышения вероятности биоадгезии при увеличении площади поверхности лекарственной формы предпочтение следует отдать гранулам, а не таблеткам. Для облегчения разделения заполненных гранулами капсул в среде для растворения такие гранулы были частично покрыты гидрогенизированным маслом. В отсутствие масляного покрытия гранулы подвергаются гидратации и образуют матрицу в форме капсулы, которая не дезинтегрируется на отдельные гранулы.
Профили высвобождения двух биоадгезивных прототипов (таблеток и гранул) проиллюстрированы на фигуре 29, где показано, что высвобождение из таблеток происходит дольше, чем из гранул. Высвобождение лекарственного средства из биоадгезивных лекарственных форм в виде таблеток и гранул составляет примерно 90% в течение четырех часов и 95% в течение одного часа соответственно. После хранения при 40°С в атмосфере повышенной влажности в течение одного месяца без необходимой защиты от действия влаги (без герметичной крышки с индукционным нагревом) высвобождение лекарственного средства при растворении таблетки-прототипа замедлялось (фигура 29). Для прототипов, содержащих карбомер, должны соблюдаться меры предосторожности по защите таблетки от действия влаги, необходимые для предохранения таблетки от возможной преждевременной гидратации.
Прототип состава с замедленным высвобождением
Для приготовления систем для пероральной доставки лекарственного средства регулируемого высвобождения в качестве гидрофильного носителя была использована гидроксипропилметилцеллюлоза (HPMC) (Colombo, Adv. Drug Deliv. Rev., 1993, 11, 37). Известно, что HPMC-матрицы используются для регулируемого высвобождения ряда лекарственных средств (Chattaraj, et al. Drug Develop. Ind. Pharm., 1996, 22, 555; Pabon, et al., Drug Develop. Ind. Pharm., 1992, 18, 2163; Lee, et al., Drug Develop. Ind. Pharm., 1999, 25, 493; Basak, et al., Indian J. Pharm. Sci., 2004, 66, 827; Rajabi-Siabhoomi, et al., J. Pharm. Pharmacol., 1992, 44, 1062). В данном исследовании для достижения регулируемого высвобождения BH4 оценивали вязкость различных сортов HPMC (K4M, K15M и K100M). Профили растворения таблеток, изготовленных из различных сортов HPMC, показаны на фигуре 30. Профили высвобождения лекарственного средства были аналогичными при использовании 20% HPMC независимо от степени их вязкости, где свыше 80% лекарственного средства высвобождалось за 2 часа. При обработке HPMC-полимера водной средой такой полимер подвергался быстрой гидратации и релаксации цепи с образованием гелевого слоя (Naruhashi, et al., Pharm Res. 2003,19:1415-1421). 20% HPMC не может образовывать барьерный слой геля, достаточный для значительного замедления высвобождения BH4.
Профили растворения таблеток, полученных с различными концентрациями (20%-40%) сортов HPMC с высокой вязкостью (метоцель К100М CR), представлены на фигуре 30. Было обнаружено, что таблетка, содержащая 35%-40% метоцеля К100М CR, обнаруживает замедленное высвобождение лекарственного средства, продолжающееся вплоть до четырех часов, тогда как таблетка, содержащая 20% HPMC, обнаруживает высвобождение лекарственного средства в течение двух часов (фигура 31). В качестве прототипа для проведения тестов на животных была выбрана таблетка, содержащая 35% HPMC (метоцель К100М), поскольку она содержит наименьшее количество HPMC, необходимое для замедленного высвобождения лекарственного средства, длящегося вплоть до четырех часов. Таким образом, эти таблетки были хорошего качества, имели приемлемое содержание лекарственного средства и обнаруживали высвобождение лекарственного средства, близкое к 100% при данном профиле растворения.
Прототип состава, содержащего полимерный донор протонов
Одним из способов повышения уровня абсорбции BH4 при пероральном введении является стабилизация лекарственного средства путем снижения pH в проксимальном отделе тонкой кишки. Для изменения рН просвета тонкой кишки был выбран эудрагит L100-55, который представляет собой высвобождающий протоны полимер, обычно используемый для нанесения энтеросолюбильных покрытий. Этот полимер не растворяется в кислотных условиях, но становится растворимым и высвобождает протоны в условиях от слабокислотных (pH>5,5) до щелочных, что обусловлено присутствием в этом полимере карбоксильных групп, и тем самым регулирует кислотность рН в просвете тонкого кишечника. Naruhashi и др. (2003) обнаружили, что pH в просвете кишки снижается в зависимости от концентрации эудрагита L100-55, а абсорбция цефадроксила и цефиксима из петли подвздошной кишки повышалась в присутствии кислотного полимера (Nozawa, et al., J. Pharm Sci. 2003, 92 (11), 2208-2216). Nozawa и др. (2003) показали, что эудрагит снижает рН в петлях тонкой кишки и способствует удалению цефадроксила и цефиксима из указанных петель.
Порошкообразные составы, содержащие BH4 и эудрагит L100-55 и указанные в таблице 8, спрессовывали в таблетки и этими таблетками заполняли капсулы. Во время теста на растворение составы в виде таблеток высвобождали приблизительно 27% лекарственного средства в течение одного часа в жидкости, имитирующей желудочный сок (SGF). Однако во время теста на дезинтеграцию таблетка оставалась целой в SGF и в фосфатном буфере (PB) при pH 5,8 в течение по меньшей мере 2 часов. Указанная таблетка не подвергалась дезинтеграции даже в присутствии сильного дезинтегрирующего агента (кросповидона или кроскармелозы). Возможно, что указанное лекарственное средство может окислять эудрагит, создавая микросреду с низким рН, то есть такую среду, в которой полимер остается неионизованным и нерастворимым.
Содержащийся в капсулах порошкообразный состав, включающий лекарственное средство - эудрагит, быстро дезинтегрируется в SGF. Для нацеленного высвобождения протонов в проксимальный отдел тонкой кишки на капсулу может быть нанесено энтеросолюбильное покрытие. После нанесения покрытия на капсулу и ее сушки в печи при 40°С масса этой капсулы вместе с полимерным покрытием увеличивается примерно на примерно 1-3%. При проведении теста на аппарате для определения растворения USP II (на лопастной мешалке) с использованием среды для растворения, а именно, 0,1 н. HCl, 37°С при скорости вращения 50 об/мин, капсулы с покрытием высвобождали примерно 25% лекарственного средства в течение одного часа. После предварительной обработки кислотой (0,1 н. HCl) в течение 1 часа капсулы с покрытием помещали на аппарат USP для проведения теста на дезинтеграцию вместе с 500 мл фосфатного буфера, pH 5,8, 37°С, и такие капсулы с покрытием дезинтегрировались в течение примерно 1 часа. Помимо таблеток или капсул, не имеющих покрытия, прототип в виде капсулы с энтеросолюбильным покрытием был также выбран потому, что капсулы с энтеросолюбильным покрытием с большей вероятностью могли обеспечивать доставку протон-высвобождающего полимера в нужный участок.
Флотирующая система доставки
Были разработаны две флотирующих системы доставки. Первым прототипом является флотирующая стандартная лекарственная форма для многократного приема, и такая лекарственная форма была приготовлена в целях повышения вероятности того, что одна из таких форм будет сохраняться в области кишечника и будет способствовать увеличению времени пребывания лекарственных средств в кишечнике. Такая лекарственная форма состоит из семи трехслойных таблеток, содержащихся в капсуле, при этом средний слой содержит лекарственное вещество, находящееся между двумя нерастворимыми в воде внешними слоями по типу «сэндвича» (фигура 32). Внешние слои содержат стеариновую кислоту, гидрофобную и нерастворимую в воде жирную кислоту, которая придает флотирующей таблетке необходимую текучесть. На каждую таблетку вручную было нанесено покрытие, состоящее из спиртового раствора этилцеллюлозы и полиэтиленгликоля с молекулярной массой 4600 (ПЭГ). Этилцеллюлоза образует нерастворимую в воде пленку вокруг таблетки и ПЭГ, которая действует как образователь пор, модулирующий скорость высвобождения. Профили растворения таблеток, покрытых этилцеллюлозой и растворами ПЭГ в различных концентрациях (20%-40%), представлены на фигуре 33. Было отмечено, что трехслойная таблетка с покрытием обеспечивает кинетику высвобождения, близкую к кинетике высвобождения нулевого порядка. Как и ожидалось, скорость растворения лекарственного средства увеличивается по мере увеличения концентрации ПЭГ. Во время проведения тестов на растворение таблетки флотировались в жидкости, имитирующей желудочную среду, по меньшей мере в течение четырех часов. В таблице 9 приводится композиция состава, протестированного в исследованиях на животных.
Второй прототип представляет собой газообразующую лекарственную форму. Она была приготовлена так, чтобы при ее контакте с кислотной желудочной средой высвобождался диоксид углерода с последующим его включением в набухшие гидроколлоиды, которые придавали лекарственной форме нужную текучесть (фигура 33). Во время проведения тестов на растворение такой состав флотировался в жидкости, имитирующей желудочную среду, в течение по меньшей мере четырех часов. Однако для соответствующего функционирования такой системы таблетки должны быть получены в условиях низкой влажности для предотвращения преждевременной реакции взаимодействия кислоты и основания. При хранении таблетки может происходить взаимодействие BH4 с бикарбонатом натрия. По этой причине указанная лекарственная форма не может быть использована при проведении тестов на животных.
Для проведения исследований на биологическую доступность у животных было разработано шесть прототипов тестируемых составов, которые включали различные составы, содержащие полимерный донор протонов, используемый для снижения pH тонкой кишки; лекарственные формы, удерживающиеся в желудке; и лекарственные формы с замедленным высвобождением.
Пример 5
Биологическая доступность новых BH4-содержащих составов
Целью данного исследования является повышение уровня абсорбции BH4 путем получения лекарственных форм, увеличивающих время пребывания лекарственного средства в желудочно-кишечном тракте (ЖКТ).
Методы: В открытых неперекрестных исследованиях, проводимых в 8 периодов для определения биологической доступности семи составов по сравнению с биологической доступностью контрольных растворенных ВН4-содержащих составов, использовали трех здоровых собакоподобных обезьян весом 3-4 кг. После содержания этих обезьян в течение ночи в условиях голодания им время от времени перорально или внутривенно вводили одну дозу 80 мг одного и того же нового состава с интервалом по меньшей мере в 1 неделю в периоды между исследованиями различных новых составов. В случае внутривенного введения пробы крови брали до введения доз, а затем через 5, 15 и 30 минут и через 1,0, 2,0, 4,0, 6,0, 8,0, 12 и 24 часа после введения дозы. В случае перорального введения пробы крови брали до введения доз, а затем через 15 и 30 минут и через 1,0, 2,0, 4,0, 6,0, 8,0, 12 и 24 часа после введения каждой дозы. После отделения плазмы путем центрифугирования 200 мкл-аликвоты каждой пробы сразу переносили в отдельные пробирки, содержащие 0,1% DTE, замораживали при -70°С и хранили до проведения анализа на общий L-биоптерин.
Исследуемые составы: Вводимые составы описаны в таблице 13. Три из этих составов (гранулы на основе карбомера, флотирующие гранулы, состоящие из множества крупных частиц, и биоадгезивные гранулы) теоретически были разработаны как составы, удерживающиеся в желудке по биоадгезивному или флотирующему механизму, способствующему увеличению времени пребывания лекарственного средства в желудочно-кишечном тракте. Другие концепции были основаны на замедлении перистальтики желудочно-кишечного тракта в целях увеличения времени пребывания состава (глицерилмоноолеата) в желудочно-кишечном тракте, снижения рН в тонком кишечнике и повышения химической стабильности ВН4 для обеспечения абсорбции интактного лекарственного средства (протонного насоса) или состава с замедленным высвобождением для оценки возможности усиления такой абсорбции.
|
Анализ на содержание биоптерина в плазме: Концентрации BH4 в плазме определяли надежным методом специфической обращенно-фазовой ЖК/МС/МС. Стандартная кривая представляет собой линейную зависимость концентраций в пределах 50 нг/мл - 2500 нг/мл. Нижний предел количественной оценки для L-биоптерина составлял 50 нг/мл, при этом точность суточных значений, оцененная по коэффициентам изменчивости, составляла менее 5%. L-биоптерин оставался стабильным в замороженной плазме обезьян, стабилизированной 0,1%-ным DTE при -70°С, вплоть до проведения анализа. Концентрации BH4 вычисляли исходя из определенных концентраций L-биоптерина.
Фармакокинетический и статистический анализы: Фармакокинетические параметры ВН4 в плазме определяли после перорального и внутривенного введения составов. Фармакокинетические параметры приводятся в таблице 14.
|
Результаты
Целью настоящего исследования является идентификация составов, которые повышают биологическую доступность ВН4 по сравнению с контрольным составом, приготовленным в виде растворенной таблетки. Профили средней концентрации ВН4 в плазме в зависимости от времени для различных лекарственных форм и контрольного состава после перорального введения BH4 приводятся на фигуре 35, а фармакокинетические параметры ВН4, полученные исходя из профилей зависимости концентрации лекарственного средства в плазме от времени, приводятся в таблице 14. Контрольным составом (фаза 2) является растворенная таблетка.
Как показано на фигуре 35, состав на основе глицерил-моноолеата давал наиболее высокие значения AUClast и AUC∞, которые составляли 716 нг-час/мл и 858 нг-час/мл соответственно. Контрольный состав, приготовленный в виде растворенной BH4-содержащей таблетки, давал значения AUClast и AUC∞, которые составляли 641 нг-час/мл и 805 нг-час/мл соответственно (таблица 14). Указанные составы, имеющие биологическую доступность, начиная от самой большей до самой меньшей, располагаются в следующем порядке: таблетка на основе глицерил-моноолеата > растворенная таблетка > таблетка на основе биоадгезивного полимера > таблетка с замедленным высвобождением > флотирующие лекарственные формы > продукт, приготовленный в форме капсул с биоадгезивными гранулами > продукт, приготовленный в форме капсул с протонными донорами.
Пример 6
Получение тетрагидробиоптеринсодержащих составов для внутривенного введения
Оценка стабильности первичного состава
В общих чертах, целью настоящего исследования является оценка стабильности BH4 в буферных растворах при pH 1-7 (см. таблицу 15) и в присутствии и в отсутствие антиоксидантов и в присутствии или в отсутствие инертного газа в реакционных растворах (см. таблицу 16).
|
|
Более конкретно, оценивали влияние объединения двух антиоксидантов в присутствии или в отсутствие инертного газа при pH 4 на сохранение состава в виде жидкого продукта, а при pH 7 эту оценку проводили для определения степени ответственности нестабильности при физиологическом pH за низкую биологическую доступность данного соединения у обезьян и человека (см. таблицы 17 и 18). Было сделано предположение, что стабильность BH4 зависит от температуры. Поэтому стабильность данного соединения оценивали при 2-8°С, 25°С, 30°С и 37°С для подтверждения предсказанных длительных сроков хранения данного соединения при различных температурах. Оценка стабильности данного соединения при физиологической температуре 37°С позволяет получить данные, подтверждающие результаты, полученные для сроков стабильности пероральной лекарственной формы в зонах абсорбции желудочно-кишечного тракта.
|
|
Предполагаемое время взятия образцов для исследований, проводимых в различных буферных растворах, рассчитывали путем сравнения времени полужизни одного исследования при pH 3,1 с данными, описанными в публикации Davis, et al. (1988; Eur. J. Biochem. 173, 345-351, (1988)), при pH 6,8 в трис-буфере и фосфатном буфере,. В исследовании на стабильность раствора при pH 3,1 получали t1/2=17769 минут (12,3 дня), а в работе Davis и др. были получены t1/2=10 минут в фосфатном буфере pH 6,8, и 14 минут в трис-буфере, pH 6,8. Эти два исследования позволяют предположить о снижении времени полужизни на один порядок величины BH4 (то есть величина реактивности возрастает на один порядок) для каждого одинарного увеличения рН (см. таблицу 19). Исходя из данной аппроксимации образцы растворов с pH 1,2 - pH 3 брали сначала еженедельно, а затем после 2 первых дат взятия образцов, время их сбора корректировали, если это необходимо. Определение времени взятия образцов при 25°С проиллюстрировано в таблице 19.
|
Исследования проводили в буферных растворах при pH 1-7 и при температурах 5°С, 25°С, 30°С и 37°С. Хотя эти исследования проводили в негерметично закрытых контейнерах, однако антиоксиданты, взятые отдельно (аскорбиновая кислота или L-цистеин) или в комбинации друг с другом (аскорбиновая кислота + L-цистеин), снижали уровень потери или разложения ВН4 (см. фигуру 36 и фигуру 37). Барботирование раствора, содержащего аскорбиновую кислоту и L-цистеин, приводило к значительному повышению стабильности BH4.
Скорость разложения BH4 зависит от концентрации (см. фигуру 38). Поэтому было показано, что при использовании высококонцентрированных составов BH4 в высокой дозе для синергической стабилизации составов требуются меньшие концентрации стабилизаторов.
Полученные результаты показали, что стабильные жидкие составы с длительным сроком хранения, включая стерильные жидкости для инъекций, жидкости для перорального введения и лиофилизованные и стерильные порошки, предназначенные для последующего разведения, могут быть получены с применением описанных здесь способов и композиций.
Пример 7
Жидкие и лиофилизованные составы тетрагидробиоптерина для перорального и парентерального введения
Репрезентативные композиции составов
|
|
|
|
|
|
Жидкие составы с высокой дозой
|
|
|
|
|
|
Вышеупомянутые приготовленные или компаундированные растворы барботируют, но необязательно, инертным газом (например, аргоном или азотом) или диоксидом углерода в резервуаре для приготовления составов по индивидуальному рецепту, при этом первые контейнеры, предпочтительно, герметично закрывают и помещают в атмосферу инертного газа или диоксида углерода для удаления кислорода из головной наджидкостной части контейнера. Объем этих составов может быть увеличен до любой величины путем умножения количества компонентов на соответствующий масштабный коэффициент.
Пример 8
Определение уровня тетрагидробиоптерина (ВН4) в человеческой плазме путем измерения концентрации L-биоптерина с помощью ЖХ/МС/МС после окисления в основных условиях
Тетрагидробиоптерин (BH4) представляет собой небольшую молекулу, используемую в качестве терапевтического средства для лечения пациентов с фенилкетонурией (ФКУ). А поэтому очень важно разработать точный и специфический способ измерения концентрации BH4 в плазме человека. Однако количественная оценка ВН4 из-за его низкой эндогенной концентрации и нестабильности в человеческой плазме представляет значительные трудности. В основных условиях BH4 окисляется до дигидробиоптерина (BH2) и, в конечном счете, до L-биоптерина. Кроме того, отношение BH4:L-биоптерин при окислительном превращении является почти постоянным в течение периода времени до 23 недель. Поэтому путем измерения концентрации L-биоптерина после окисления в основных условиях и использования молярного отношения превращения авторы настоящего изобретения могут точно определить концентрации BH4 в человеческой плазме.
Известные методы основаны на классическом методе, разработанном Fukushima и Nixon (Anal. Biochem., 102, 176-188(1980)) с применением ВЭЖХ с детекцией флуоресценции. В ЖХ/МС/МС-методе пробу человеческой плазмы стабилизировали антиоксидантом, добавляли раствор в качестве внутреннего стандарта (IS) и подщелачивали раствором гидроксида натрия, а затем окисляли раствором иода. После инкубирования в темноте при комнатной температуре добавляли аскорбиновую кислоту для уменьшения избытка иода. Окисленные образцы экстрагировали путем осаждения белка. L-биоптерин в разведенных экстрактах анализировали с помощью обращенно-фазовой ВЭЖХ путем детектирования методом МС/МС Turbo Ion Spray®. Затем проводили мониторинг отрицательных ионов L-биоптерина в моде магнитного резонанса (MRM). Для построения линейной калибровочной кривой использовали отношение площадей пиков лекарственное средство:IS для стандартов с помощью взвешенного регрессионного анализа, проводимого методом наименьших квадратов 1/x2.
Отношение BH4:L-биоптерин при окислительном превращении вычисляли через несколько промежутков времени: через 0, 1, 2, 4, 8, 12 и 23 недели, и во все моменты времени проведения теста было обнаружено совпадение с номинальным молярным отношением превращения, равным 47,3%, как было определено в первые три последовательных момента времени. Разность между отношением превращения в другие моменты времени и номинальной величиной составляла от -2,3 до 6,3%. ЖХ/МС/МС-метод был признан эффективным для количественной оценки L-биоптерина в человеческой плазме, содержащей K2EDTA, в диапазоне величин линейной калибровочной кривой 5-1000 нг/мл (эквивалентных 11-2114 нг/мл для BH4). Точность и достоверность анализа оценивали путем взятия проб для оценки контроля качества (QC), и полученные результаты показали, что суточная точность значений составляла 4,7-14,5% CV; суточная точность для номинальных величин составляла от -7,1 до 7,4%; а суточная точность и достоверность коэффициента (CV) изменчивости составляла 7,4-16,4%CV, а для номинальных величин от -8,3 до 3,7% соответственно. Средний выход экстракции для L-биоптерина составлял 65,3%. Было обнаружено, что в K2-EDTA-содержащей человеческой плазме L-биоптерин оставался стабильными при комнатной температуре по меньшей мере в течение 4 часов и после проведения 4 циклов замораживания-оттаивания, а также при -70°С по меньшей мере в течение 275 дней.
Пример 9
Определение BH4/BH2/B с помощью ВЭЖХ в сочетании с электрохимической детекцией и детекцией флуоресценции
Для разработки метода определения концентраций тетрагидробиоптерина (BH4), дигидробиоптерина (BH2) и биоптерина (B) в человеческой плазме проводили исследования с помощью обращенно-фазовой высокоэффективной жидкостной хроматографии (ВЭЖХ) с детекцией флуоресценции (FD) и электрохимической детекцией (ECD). Этот метод описан в публикации Cai, et al. (Cardiovascular Research 55: 838-849, 2002).
Маточные растворы BH4 (в 20 мМ HCl), BH2 и B (в ДМСО) доводили до конечной концентрации 10 мМ и хранили при -80°С. Стандартные рабочие растворы для калибровки приготавливали из маточного раствора при 100, 10, 7,5, 5, 2,5 и 1 нМ в человеческой плазме, содержащей K2-EDTA и модифицированной 0,1% (масс./об.) 1,4-дитиоэритритом (DTE). Рабочие образцы BH4, BH2 и B для оценки контроля качества получали при 5, 8, 25 и 50 нМ в человеческой плазме, содержащей K2-EDTA и модифицированной 0,1% (масс./об.) DTE, и хранили при -80°С.
Для обработки образцов плазму разводили 1:10 в буфере для ресуспендирования. К 180 мкл разведенной плазмы добавляли 20 мкл 10× буфера для осаждения. Такой процесс разведения и осаждения плазмы может быть применен ко всем стандартам плазмы, пробам плазмы и образцам для оценки контроля качества плазмы. После добавления 10× буфера для осаждения образец центрифугировали при максимальной скорости при 4°С в течение 5 минут для удаления неспецифического дебриса плазмы. Затем 150 мл супернатанта переносили в сосуд с образцами и этот сосуд помещали на автоматическое устройство для забора образцов для введения 100 мл инъекций.
Подвижную фазу (2 л) получали с использованием 13,6 г ацетата натрия (50 мМ), 2,1 г лимонной кислоты (5 мМ), 36 мг EDTA (48 мМ), 49,4 мг DTE (160 мМ) и 2% об. метанола в воде. рН доводили до 5,22. Буфер для ресуспендирования (20 мл) получали с использованием 20 мл PBS, pH 7,4 (50 мМ), 20 мкл 1 M DTE (1 мМ) и 100 мл 100 мкМ EDTA. 10× буфер для осаждения (25 мл) получали в свежем виде с использованием 2,88 мл фосфорной кислоты (1M), 9,39 г трихлоруксусной кислоты (2 M) и 20 мл 1M DTE (1 мМ).
Тетрагидробиоптерин (BH4), дигидробиоптерин (BH2) и биоптерин (B) разделяли с помощью обращенно-фазовой ВЭЖХ. BH4 определяли методом электрохимической детекции, при котором ВН4 окислялся на электроде 1 до хиноноидного дигидробиоптерина (qBH2), а затем снова восстанавливался до BH4 на электроде 2. Затем ток детектора, генерируемый этой реакцией восстановления, использовали для определения концентрации ВН4. Соединения ВН2 и В могут быть детектированы путем такого же впрыска методом детекции флуоресценции. Окисление ВН2 на колонке, осуществляемое с использованием кондиционирующей предохранительной колонки при оптимальном потенциале, приводило к окислительному превращению ВН2 в биоптерин.
ВЭЖХ-разделение осуществляли на колонке ACE C-18 (250 мм × 4,6 мм), 5 мкМ при скорости потока 1,3 мл/минуту и времени перегонки 13 минут. Параметрами электрохимической детекции являются: E1: + 100 мВ (фоновый ток от +500 нА до +600 нА) и E2: -300 мВ (фоновый ток от -50 нА до -60 нА). Окисление после прохождения через колонку осуществлялось при 900 мВ. Параметрами детекции флуоресценции являются: длина волны возбуждения: 350 нм и длина волны излучения: 450 нм.
Линейность данного метода и область его применения оценивали исходя из точности и достоверности стандартов в плазме и буфере. Стандартную кривую концентрации строили с использованием по меньшей мере 4-6 ненулевых концентраций для каждого аналита. Концентрации стандартов составляли 1, 2,5, 5, 7,5, 10 и 100 нМ. Полученные результаты указывали на линейную зависимость при концентрациях 1-100 нМ для BH4, BH2 и B с R2 >0,99.
Точность определяли с помощью анализа образцов-дубликатов, взятых для определения контроля качества и содержащих известные количества (2, 8, 25 и 50 нМ) аналита, и выражали как процент точности. Точность также вычисляли исходя из данных, полученных при оценке контроля качества. Точность в пределах данного анализа и точность для различных анализов оценивали исходя из CV%. В трех отдельных экспериментах получали рабочие концентрации каждого аналита в плазме и их анализировали. Для определения точности и выхода в пробы человеческой плазмы помимо 10 нМ BH4 «впрыскивали» BH2 и B. Измерения уровней BH4, BH2 и B при 8, 25 и 50 нМ подтверждали точность в пределах 112%-89% и указывали на коэффициент изменчивости (CV%) в пределах 2,5%-20%. Эксперименты по определению выхода с использованием 10 нМ BH4, BH2 и B, впрыскиваемых в клинические образцы человеческой плазмы, указывали на выход, составляющий 70%-130%. Полученные результаты показали, что такой метод является достоверным и точным при его применении к образцам с концентрациями более чем 2 нМ.
Для оценки присутствия эндогенной интерференции в шести различных партиях плазмы в эти шесть различных партий плазмы вводили 10 нМ BH4, BH2 и B, и для каждого образца плазмы определяли точность и достоверность оценки. Эксперименты на селективность показали, что у шести индивидуумов наблюдались эндогенные фоновые уровни ВН4, которые составляли от нижнего количественного предела до 2,48 нМ. Аналогичным образом, концентрации BH2 и B составляли в пределах от 0,02 до 10 нМ. Выход при введении 10 нМ аналитов составлял в пределах 69%-87%. Изменчивость (CV%) для образцов плазмы индивидуумов и аналитов при введении 10 нМ составляла 23%-37%. Изменчивость эндогенных уровней BH4, BH2 и B составляла в пределах 0-9,96 нМ. В целом, полученные результаты показали тенденцию, на основании которой можно сделать предположение об интерференции матрицы или потери во время экстракции, но это не указывает на значительную избирательность между индивидуумами.
Для измерения влияния матрицы стандартные кривые, построенные по данным, полученным для плазмы или буфера, сравнивали на точность (выход), линейность и корреляцию. Сравнение стандартов, полученных в плазме, со стандартами, полученными в буфере, указывало на незначительное влияние матрицы и, по существу, хорошую корреляцию. Все три аналита обнаруживали превосходную линейную зависимость для плазмы и буфера. BH4 и B не обнаруживали значительного влияния матрицы при всех концентрациях данного интервала. Однако BH2 давал меньший выход при наивысшей стандартной концентрации (100 нМ). Образцы для оценки контроля качества, приготовленные в буфере и в плазме, продемонстрировали хорошую точность. В целом, влияние матрицы оказалось минимальным, при этом наблюдалась тенденция к снижению выхода в буфере по сравнению с выходом в плазме. Поскольку BH4 и BH2 легко окисляются, то образцы, собранные в плазме и в буфере, должны содержать антиоксиданты и иметь низкий рН, если это возможно.
Для определения возможности точного разведения проб плазмы и буфера, в которые было введено 250 нМ BH4, BH2 и B, использовали контрольную плазму с 3-кратным серийным разведением. Разведенные образцы анализировали и полученные значения сравнивали с номинальным значением после введения коэффициента разведения. Затем может быть проведено тщательное разведение высоких концентраций BH4, BH2 и B. Для BH4 наблюдаемые концентрации после разведения составляли 83%-104%, которые точно соответствуют концентрациям в пределах 83,33 нМ - 3,07 нМ. BH2 имел значения 74%-80%, которые точно соответствуют количественному интервалу 83 нМ - 3 нМ. B имел значения 119%-113%, которые точно соответствуют количественному интервалу 83 нМ - 3 нМ. Поэтому образец, который имеет величину выше количественного предела, может рассматриваться как тщательно разведенный.
Четыре концентрации аналита (2, 8, 25 и 50 нМ) приготавливали в плазме и замораживали в течение минимум 24 часов за один цикл и минимум 12 часов за другие циклы, минимум за три цикла. Образцы оттаивали сами при комнатной температуре в периоды между замораживанием. Точность и изменчивость после проведения каждого и всех циклов замораживания-оттаивания оценивали для определения максимального числа циклов, которые может проходить данный образец. Образцы, содержащие BH4, BH2 и B, могут быть подвергнуты вплоть до 3 циклов замораживания-оттаивания без значительного изменения точности или достоверности измерений. Образцы плазмы с 8 нМ - 50 нМ BH4 имели точные значения 121%-91% и CV% менее чем 10%. Аналогичным образом, BH2 имел значения 77%-88%, которые точно соответствуют количественному интервалу данного анализа. B имел значения 98%-99%, которые точно соответствуют количественному интервалу с достоверностью (CV%) 5%-8%. Образец 2 нМ BH4, BH2 и B не не давал точные или достоверные значения после повторного цикла замораживания-оттаивания. Поэтому стандартные образцы для оценки контроля качества и исследуемые образцы могут быть подвергнуты замораживанию и оттаиванию до 3 раз.
Поскольку аналиты являются восприимчивыми к окислению, то авторами настоящего изобретения была проведена оценка на длительную стабильность в замороженном виде, имитирующем предполагаемые условия хранения. BH4, BH2 и B получали в четырех концентрациях (2, 8, 50, и 100 нМ) в плазме и хранили при -70°С в течение 8 недель. Образцы для определения стабильности анализировали в свежем виде и через 3, 5, 6 и 8 недель. BH4 и B имели хорошую длительную стабильность в замороженном виде. После длительного хранения данный образец обнаруживал пониженную концентрацию BH2. После хранения свыше 8 недель точная концентрация BH4 в пробе плазмы составляла 93%-94%, а CV% точно составлял 31%-0,21%, при этом наибольшая вариабельность наблюдалась при концентрации 2 нМ. Величины для BH2 точно составляли 63%-85% для всех концентраций, тестируемых с пониженной точностью, присущей измерениям при концентрациях 2 нМ и 100 нМ. Точность (CV%) для этих образцов варьировалась от 37% до 18%. Величины для В точно составляли 88%-101% для всех концентраций, тестируемых с точностью (CV%) 23%-0,14%, при этом наибольшая вариабельность наблюдалась при концентрации 2 нМ. В целом, полученные данные подтвердили, что образцы следует хранить до 8 недель, так как в этот период времени не происходит значительной потери концентрации аналита. BH2, очевидно, является наиболее восприимчивым к разложению (окислению).
Для измерения стабильности BH4, BH2 и B в устройстве для автоматического забора образцов 8 нМ каждого аналита в растворителе для разведения оставляли на устройстве для автоматического забора образцов на 0,25, 4 и 11 часов. Затем сравнивали точность и достоверность измерений. Измеренные величины для BH4 составляли точно 5% от теоретического значения в каждый момент времени, при этом точность и достоверность всех трех измерений составляла 102% и 0,054% соответственно. Через 4 часа измеренные величины для ВН2 имели меньшую точность и более высокую вариабельность. После 11-часового выдерживания BH2 на устройстве для автоматического забора образцов уровень BH2 составлял примерно 50%. Это указывало на плохую стабильность состава в рабочем буфере на устройстве для автоматического забора образцов. Через 11 часов величины для B оставались точными в пределах 125% от теоретического значения. Поэтому рекомендуемое время проведения анализа не должно быть более 4 часов.
Для определения переброса инъекции экстрагированную исходную пробу плазмы вводили после введения наивысшей концентрации стандарта 100 нМ. Это было осуществлено для имитации вероятности завышенной оценки концентрации аналита в образце с низкой концентрацией, которая может иметь место в результате такого переброса. Такой переброс при введении инъекций BH4, BH2 и B был минимальным и не составлял более 1% от площади пика для верхнего предела количественной оценки для 100 нМ. Такой переброс при введении инъекции составлял приблизительно 5%-20% от нижнего предела количественной оценки, проводимой на основе средней площади пика, полученной исходя из низкой оценки контроля качества (2 нМ). Поэтому предпочтительно образцы должны быть внесены в концентрации от наименьшей до высокой (то есть сначала перед введением дозы, а затем после введения дозы), после чего в целях минимизации вероятности переброса при введении инъекции во время осуществления данной процедуры анализа колонку желательно периодически очищать путем дополнительной промывки.
Был разработан подходящий метод, который был надежным, специфичным, точным и достоверным. Этот метод может быть с успехом применен для количественной оценки уровней BH4, BH2 и B в плазме в целях проведения фармакокинетических исследований и исследований лекарственных средств.
Все цитируемые здесь патенты, публикации и работы во всей своей полноте включены в настоящее описание посредством ссылки. В случае возникновения противоречий между настоящим описанием и включенными в него патентами, публикациями и работами они могут быть урегулированы в соответствии с представленным описанием.

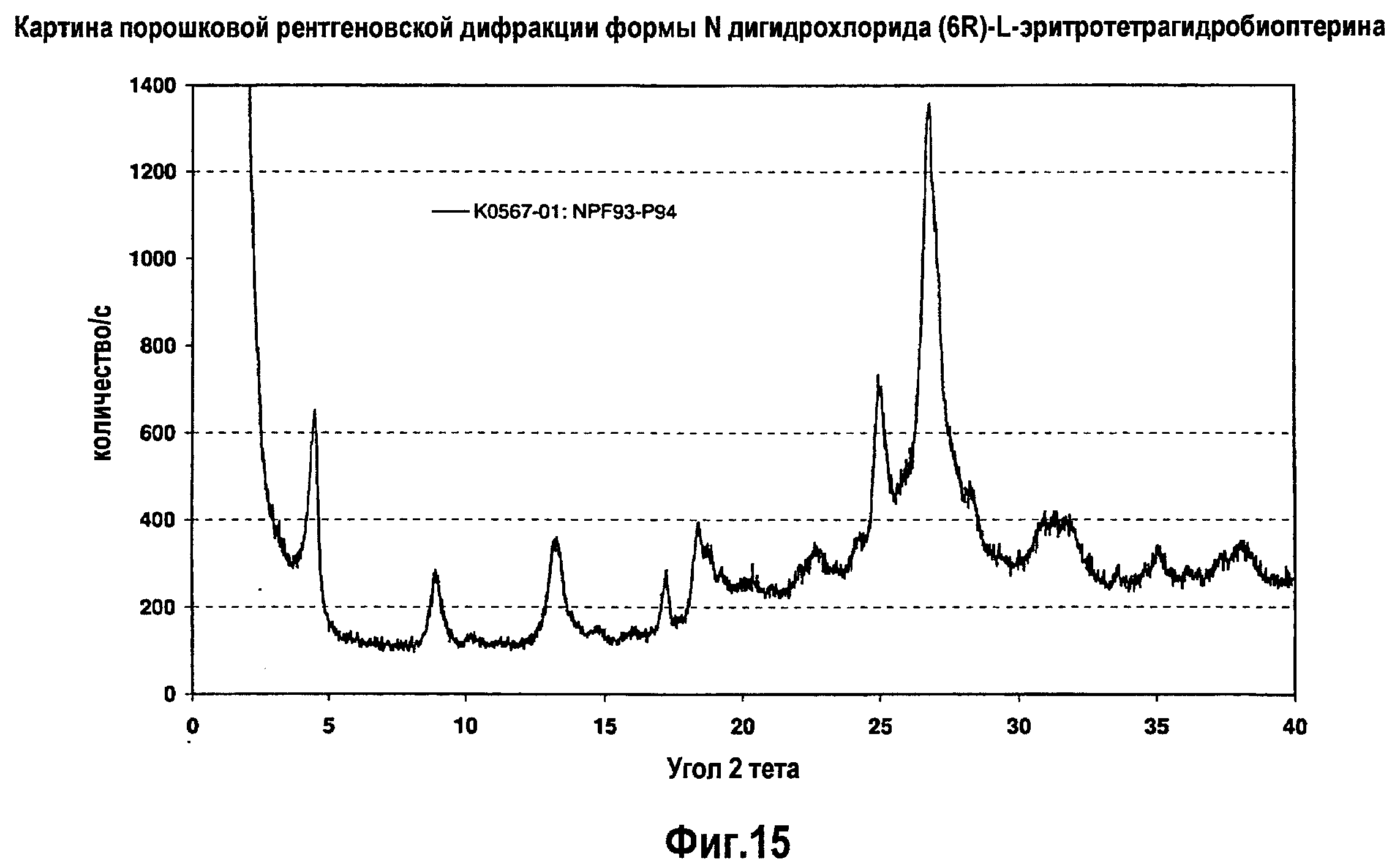
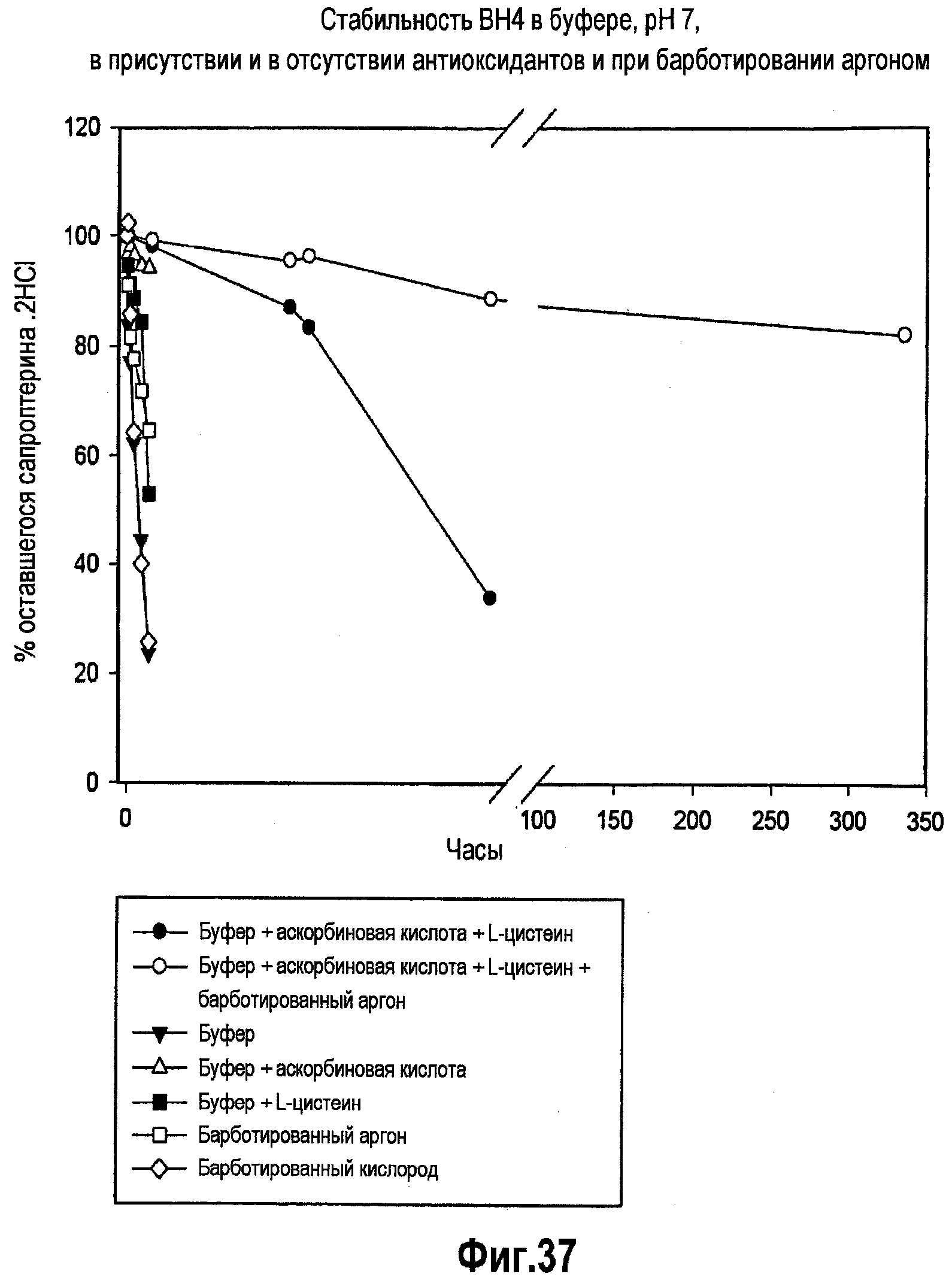
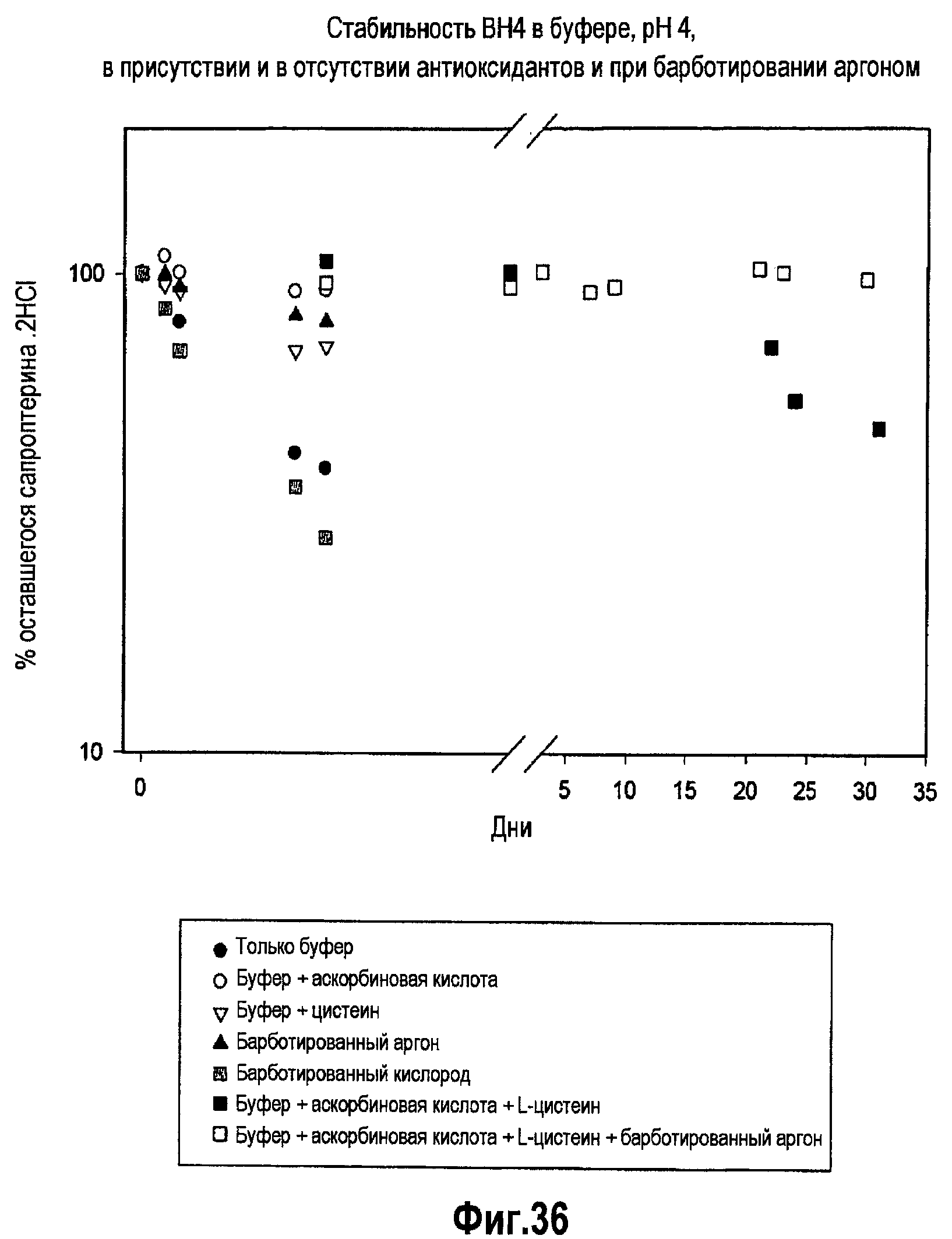

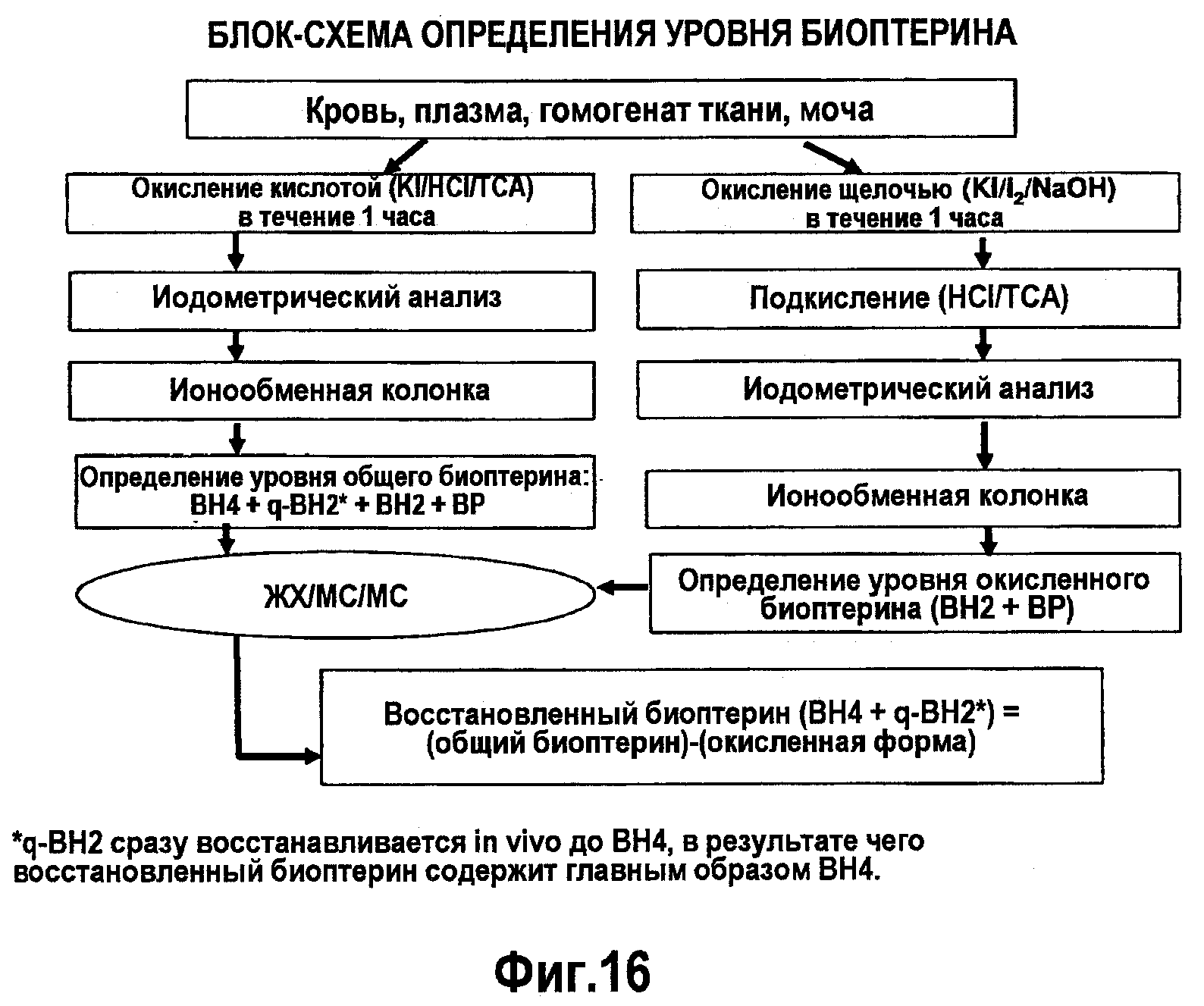
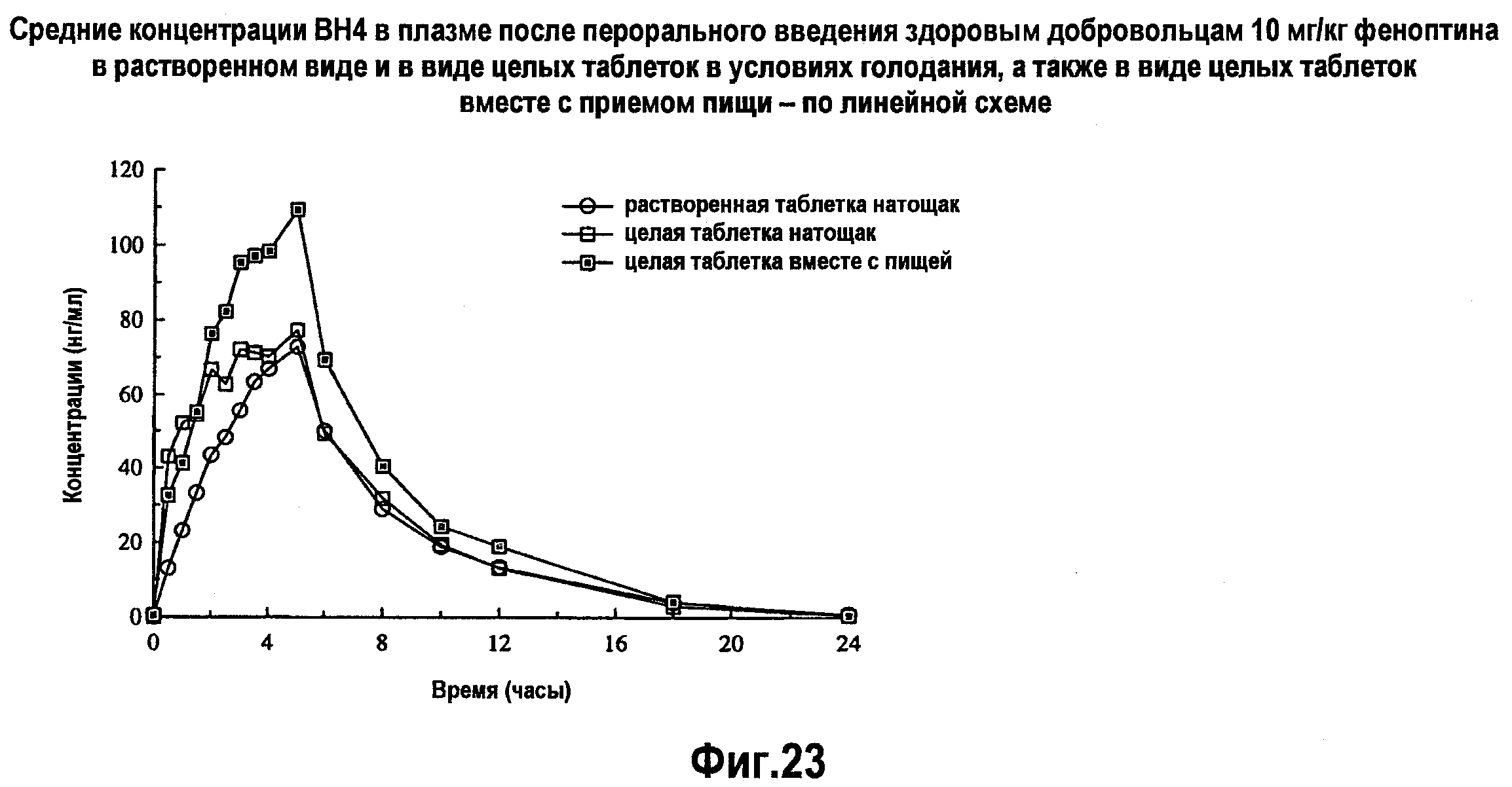
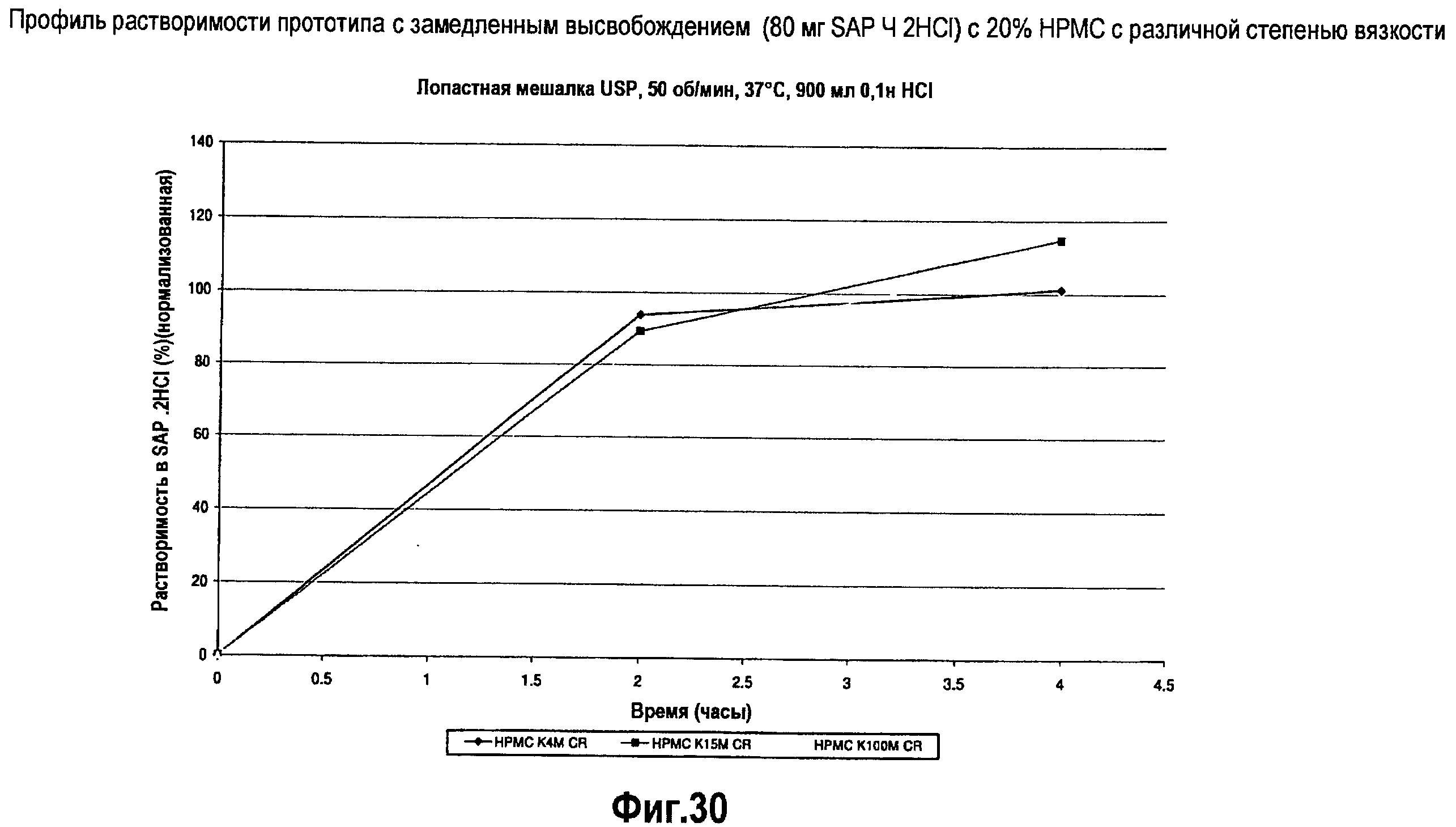
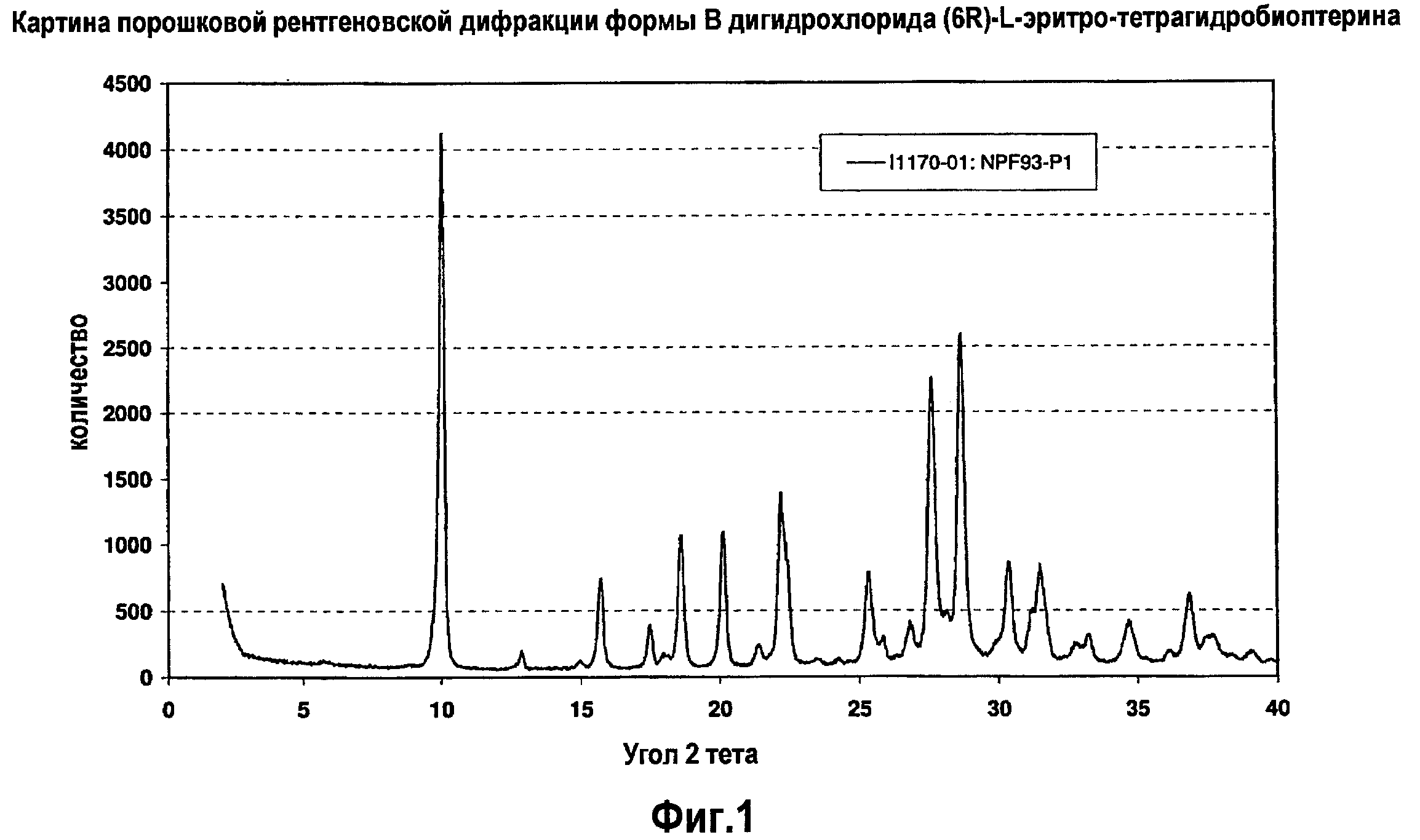
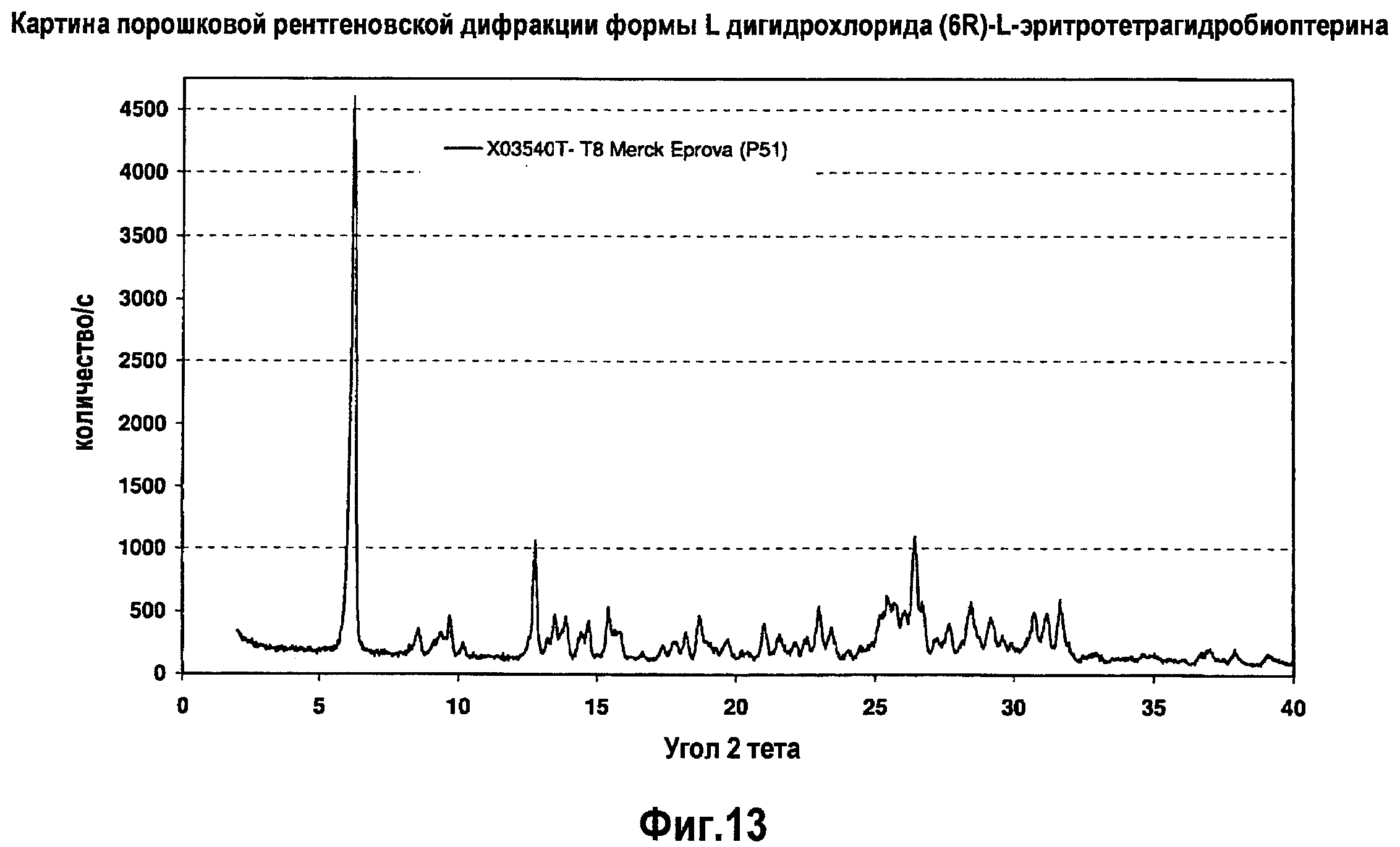
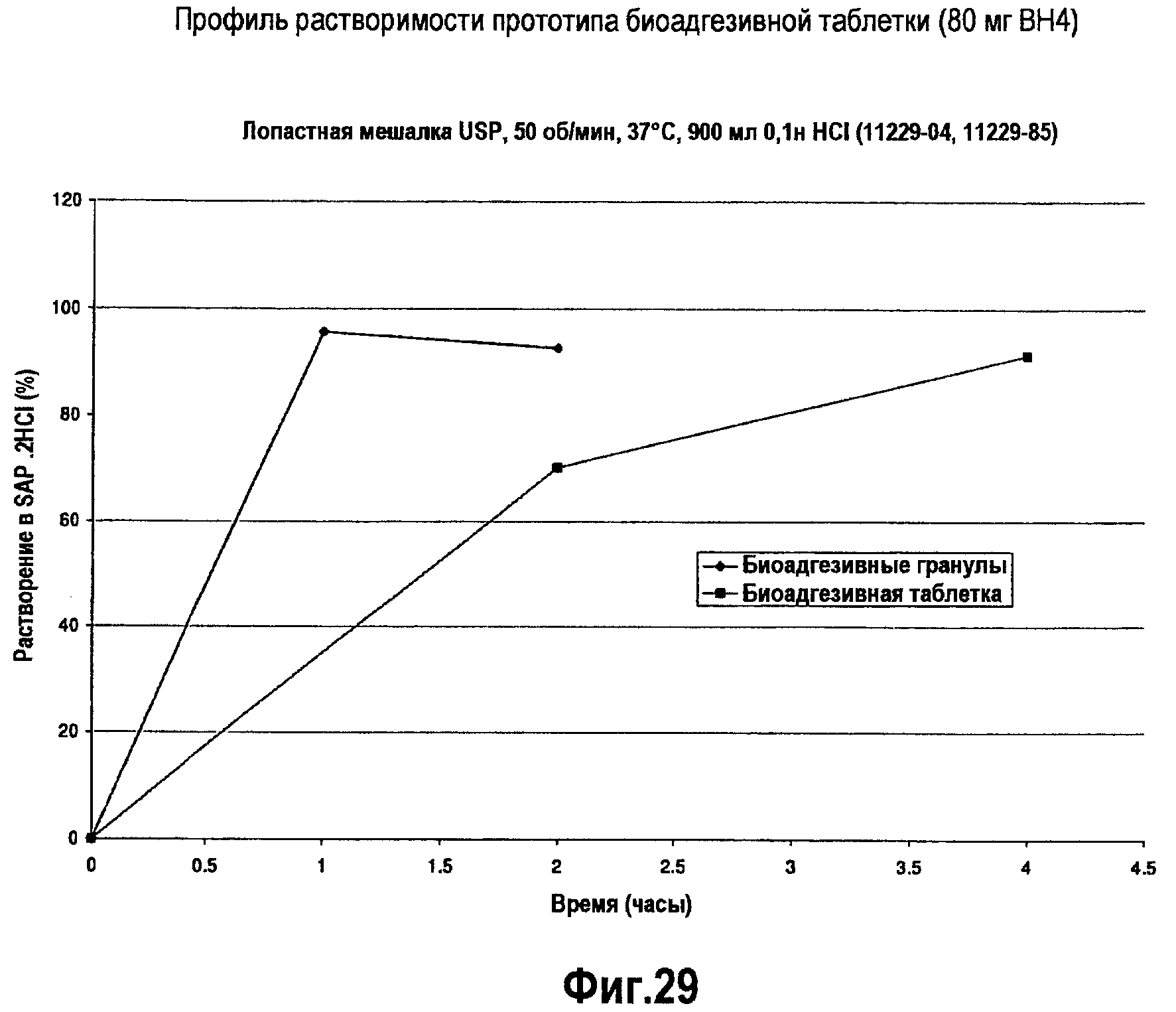
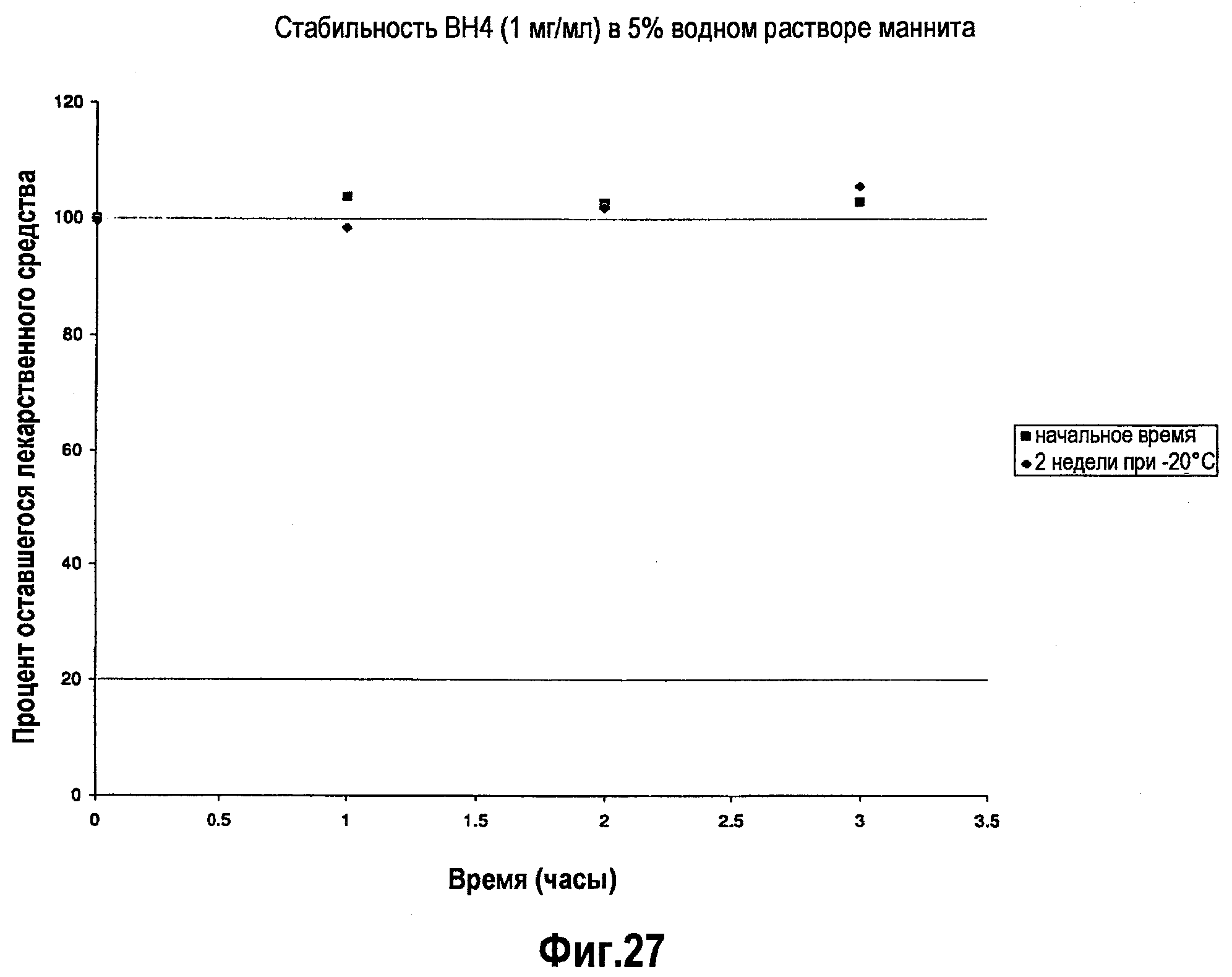
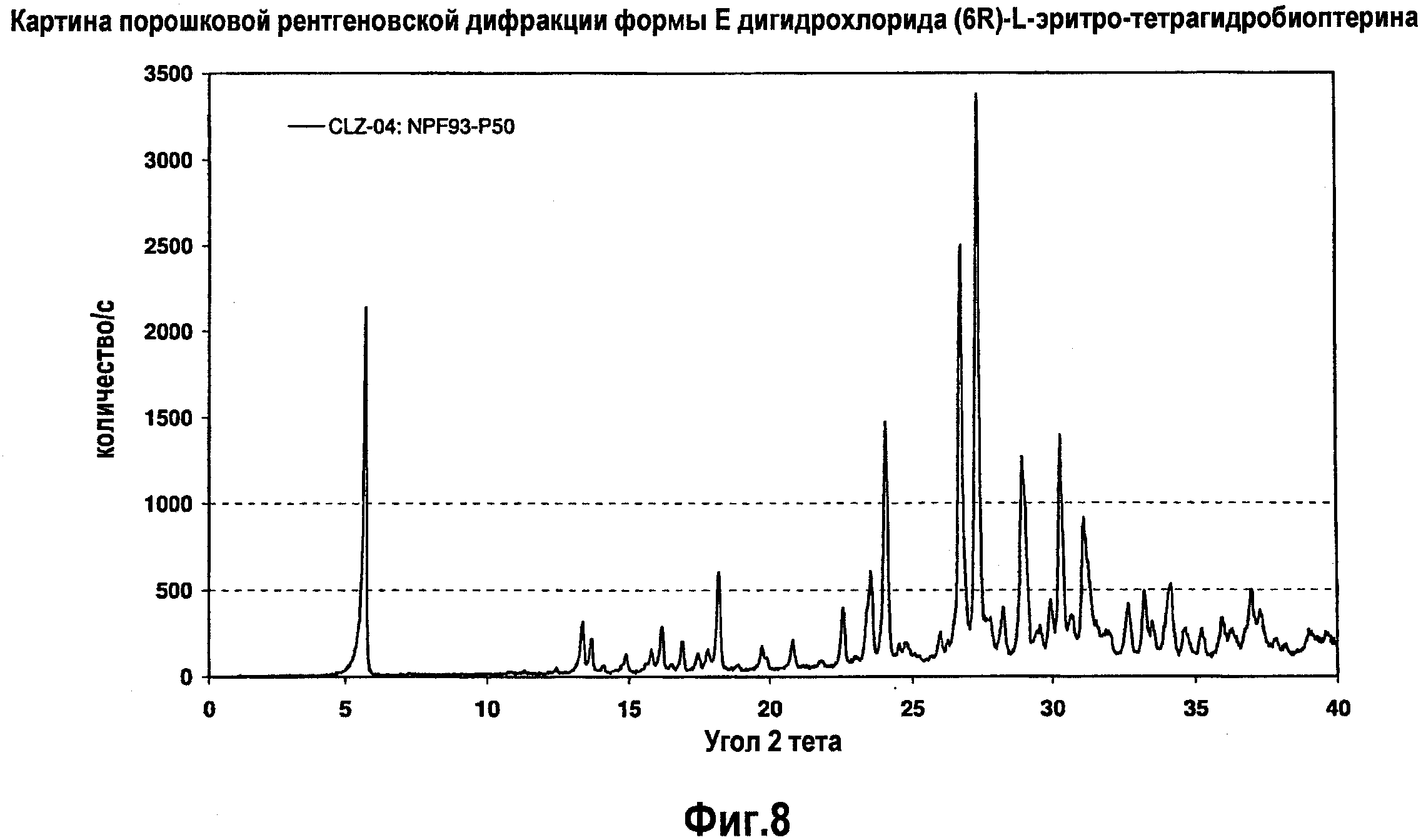
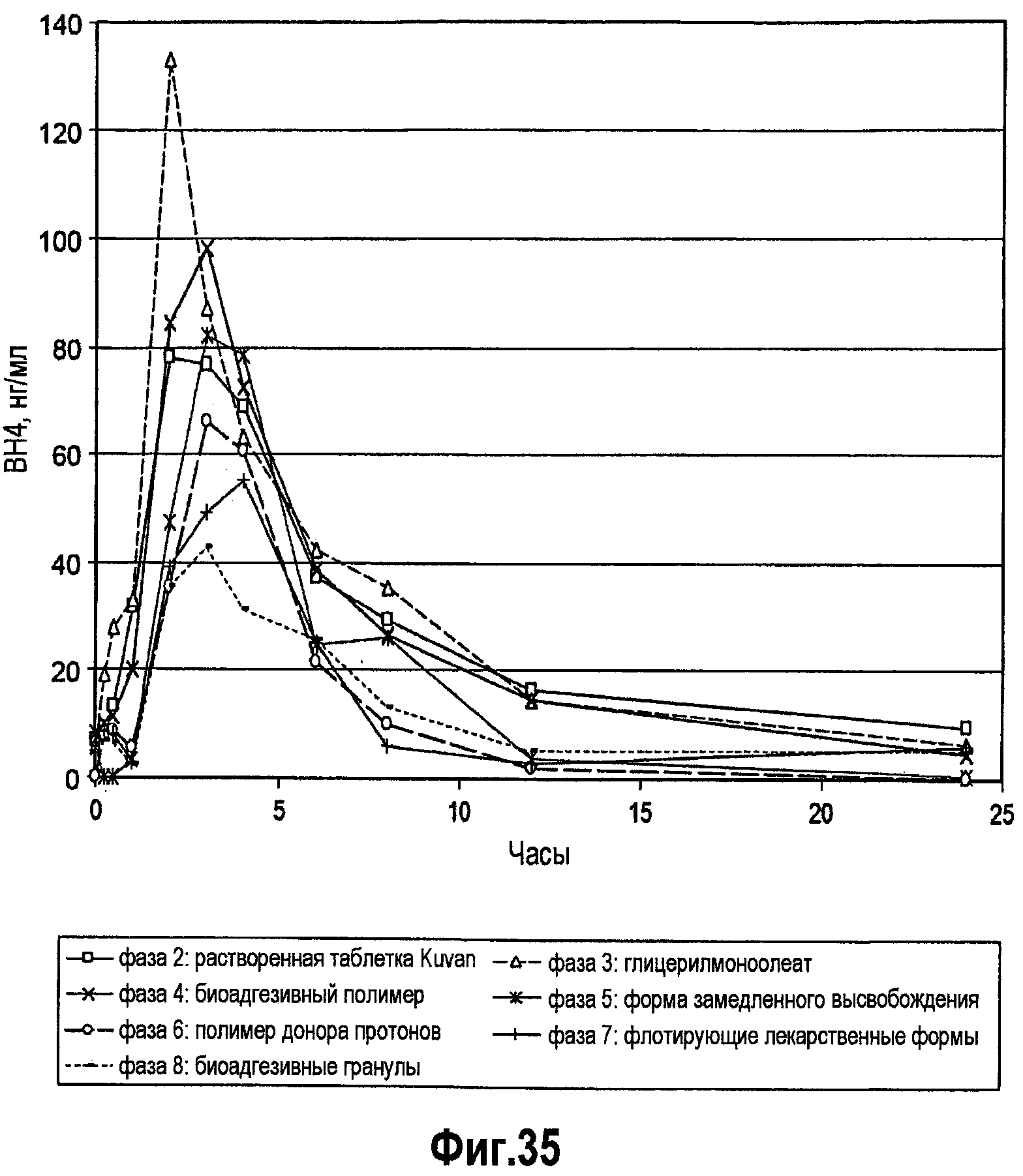
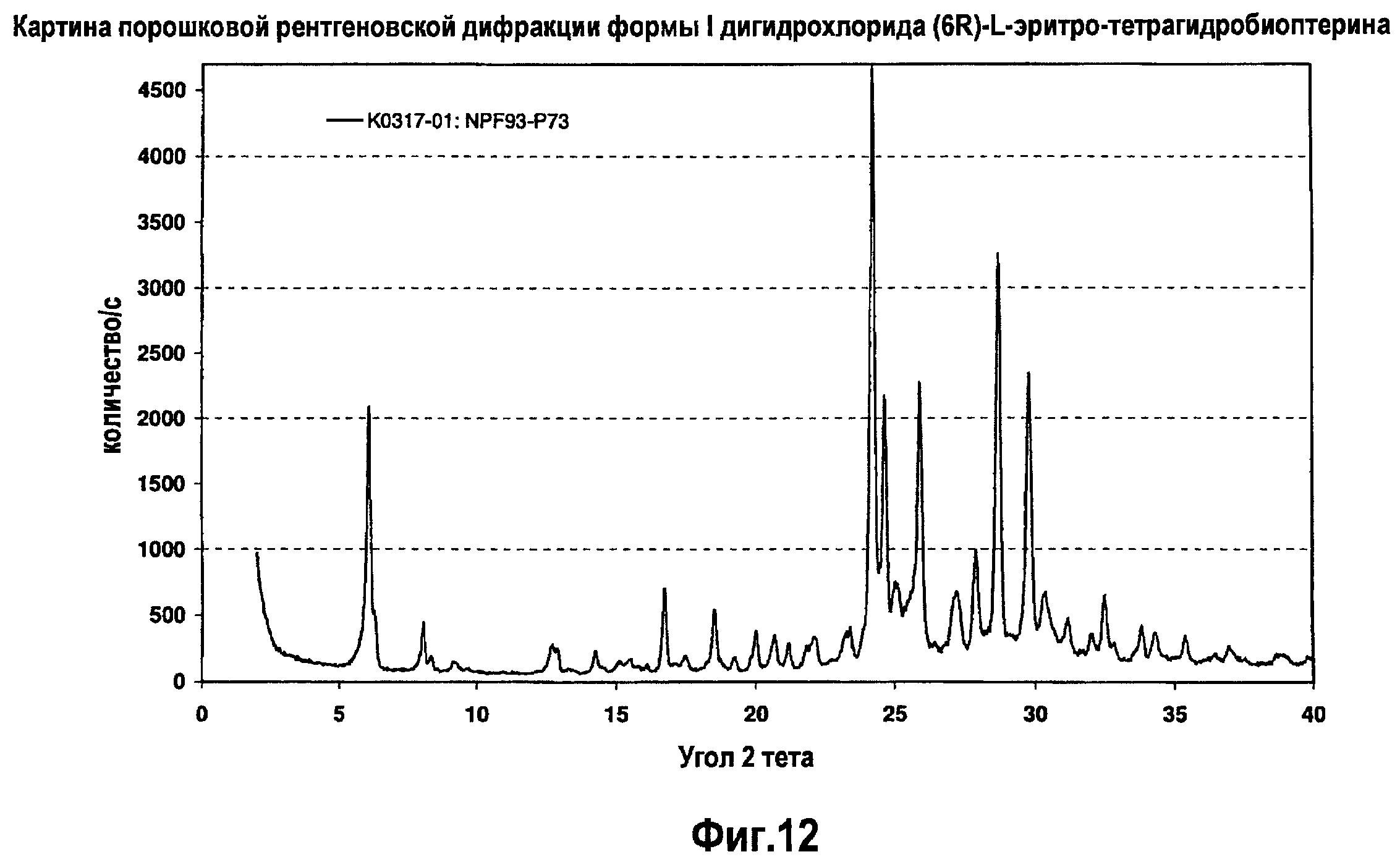
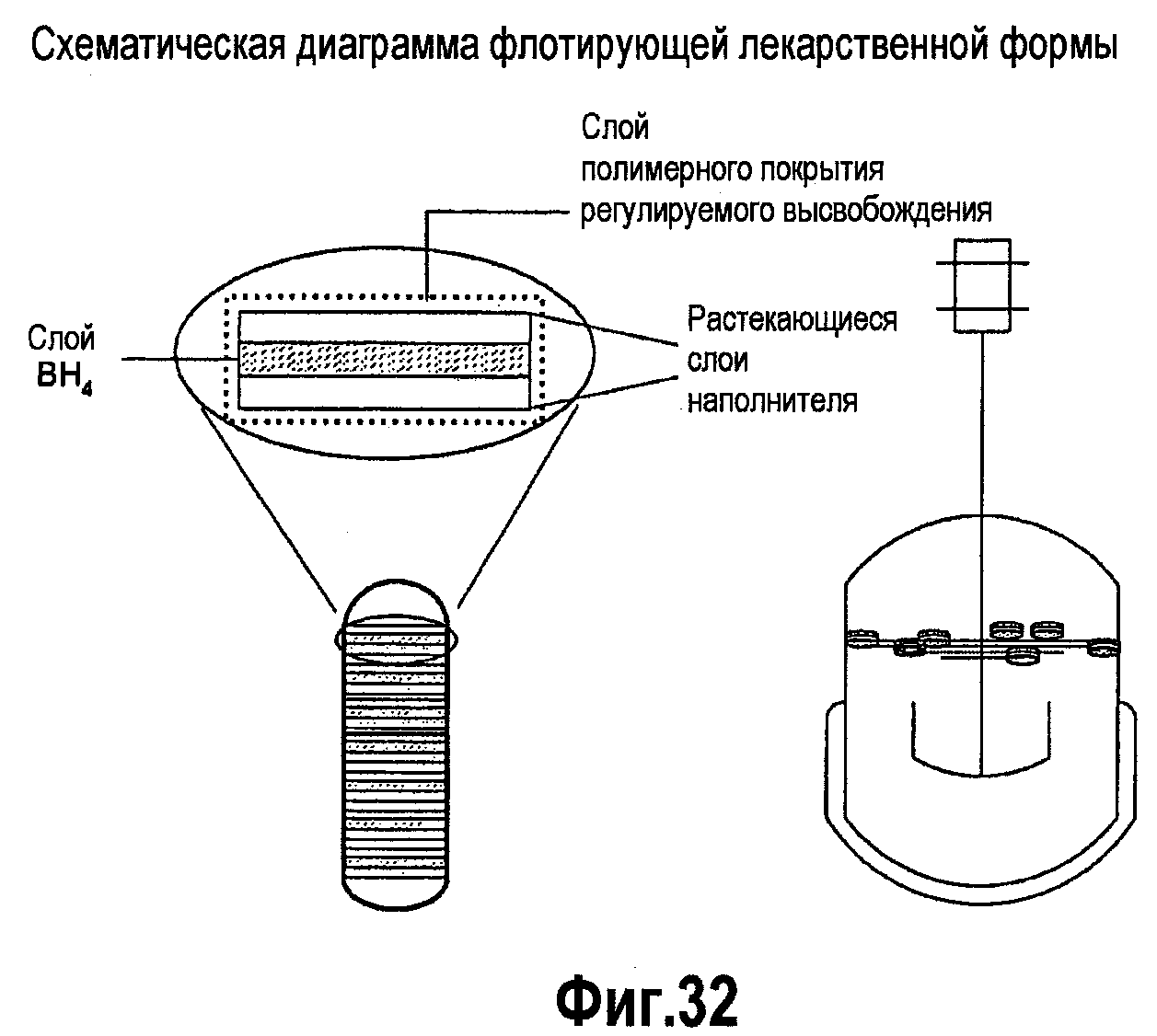
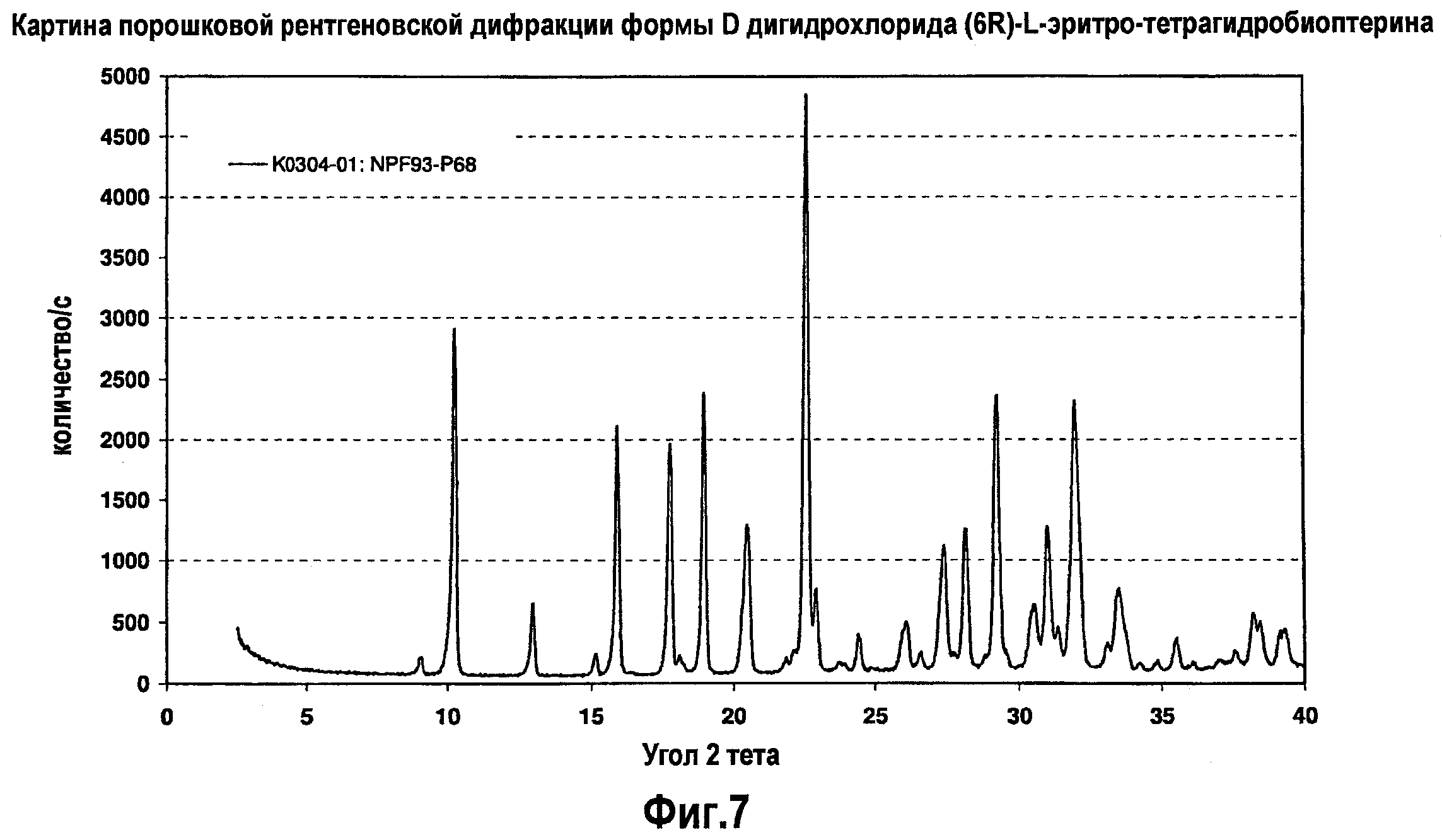
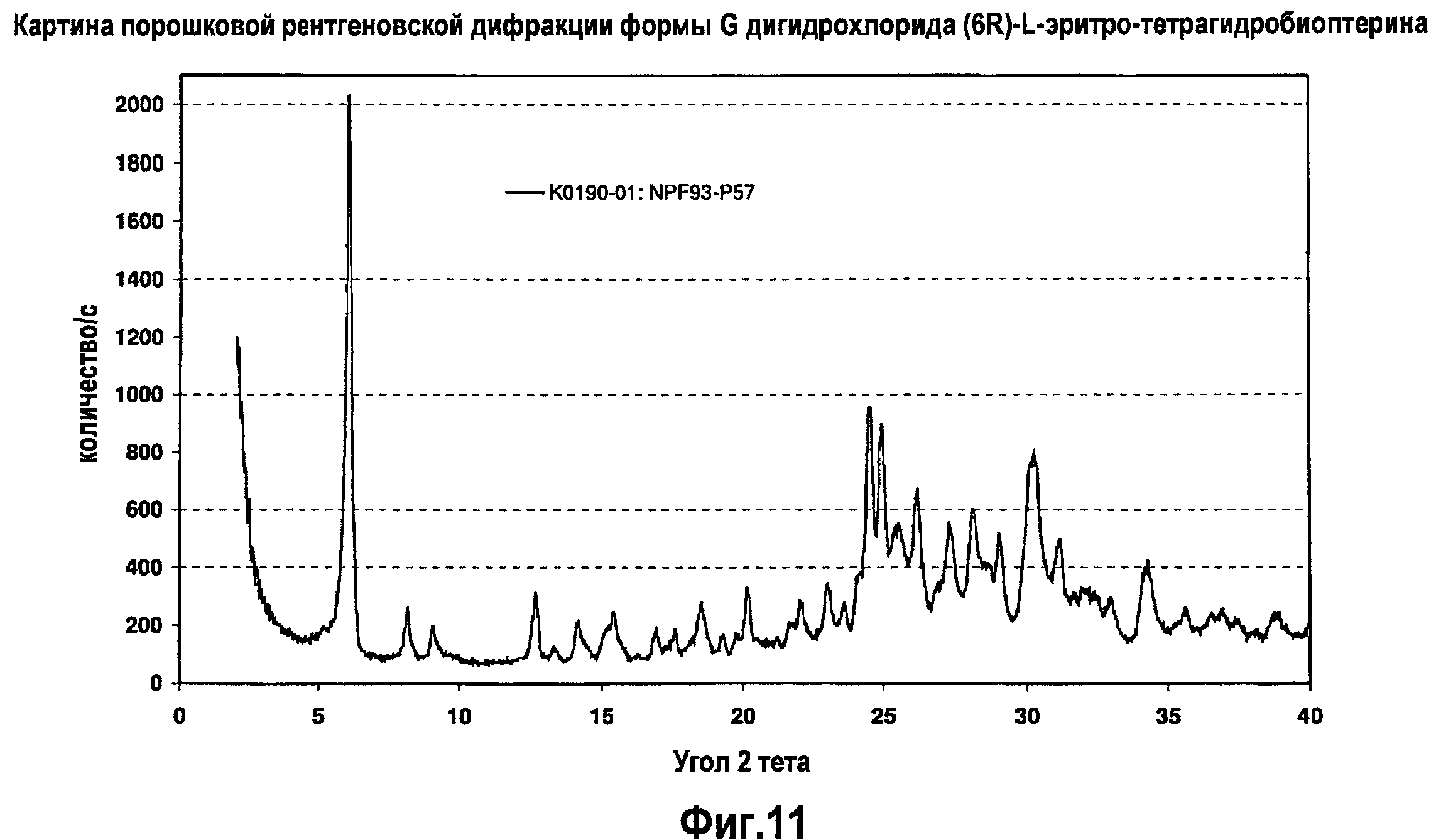
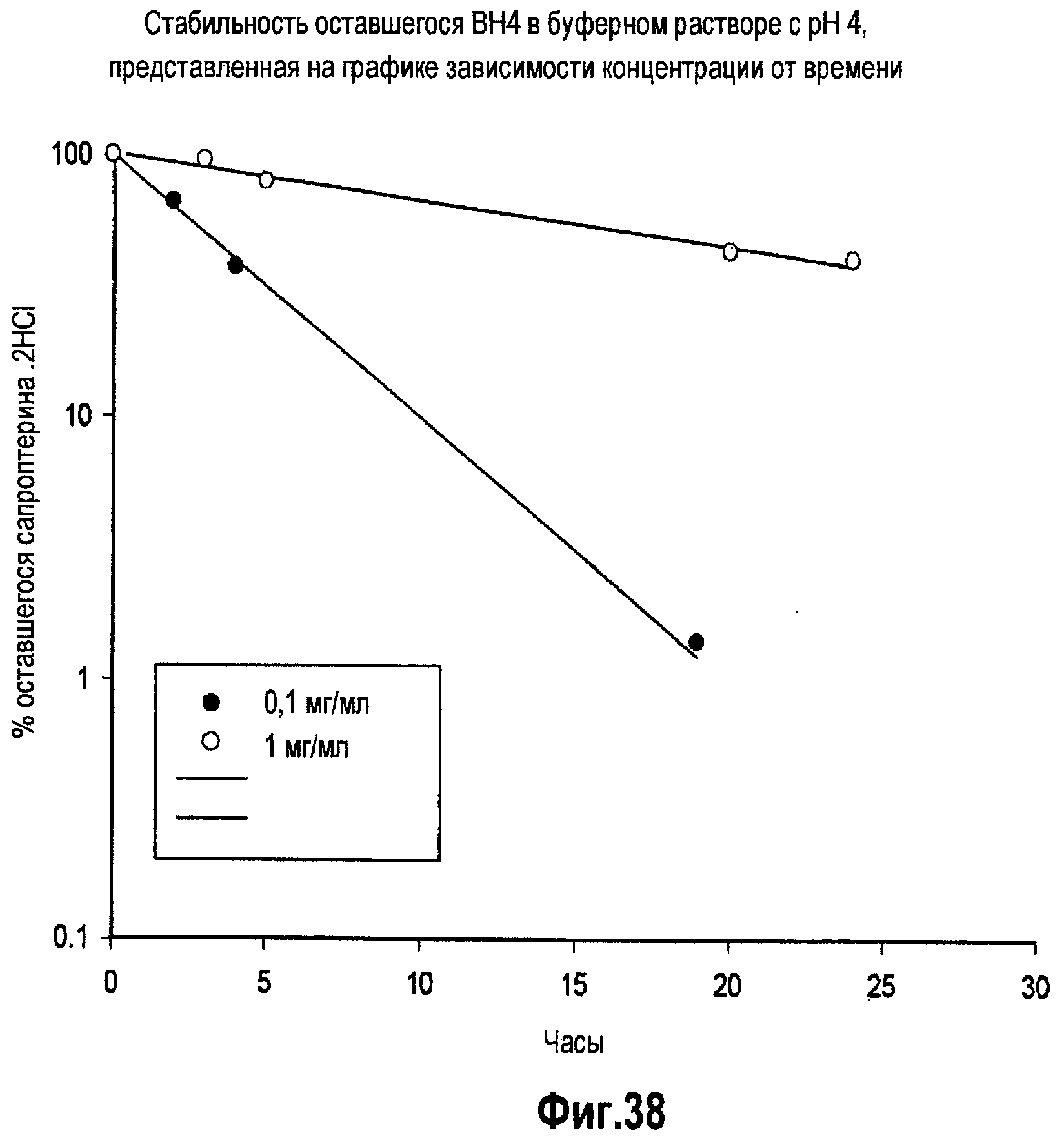
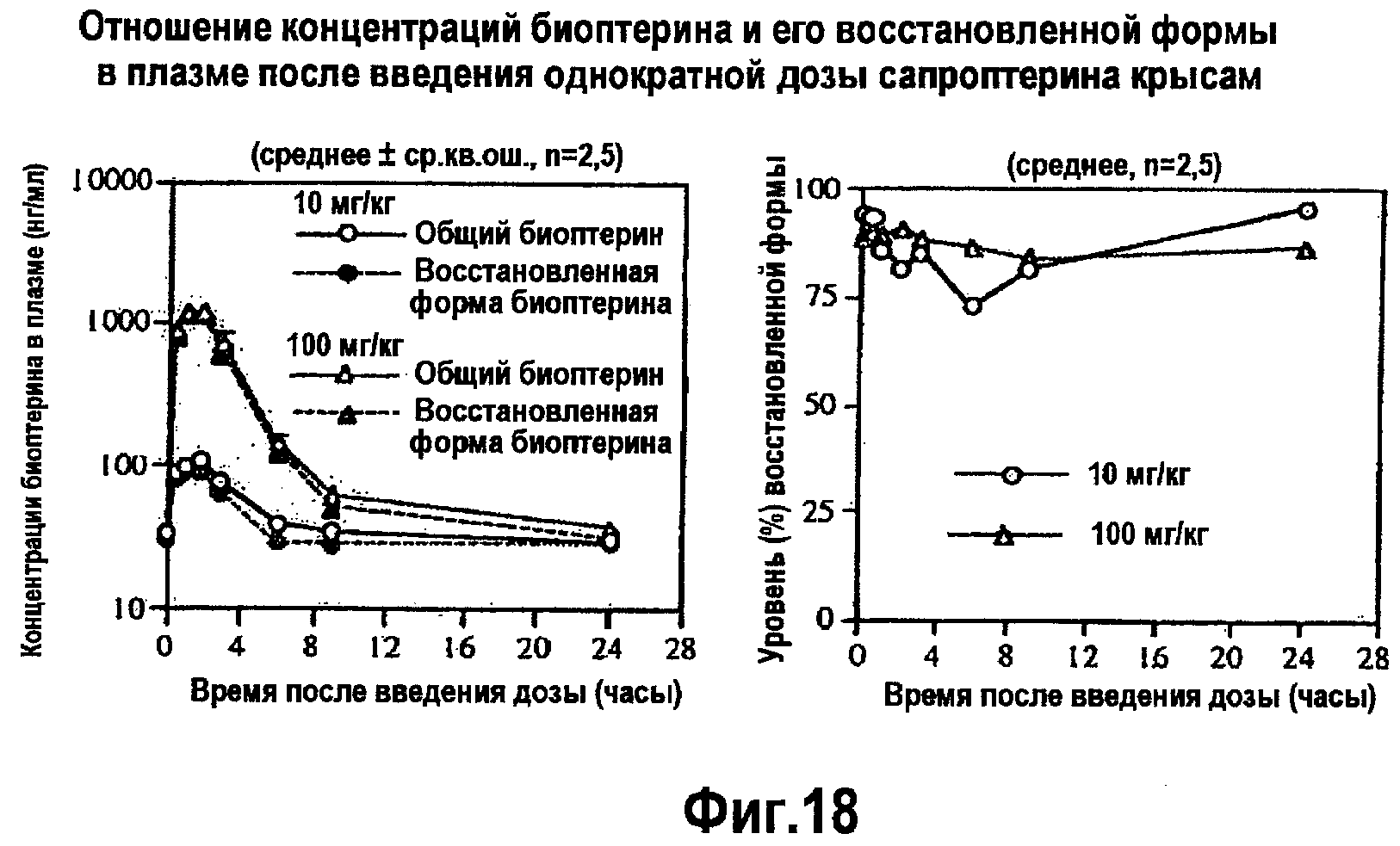

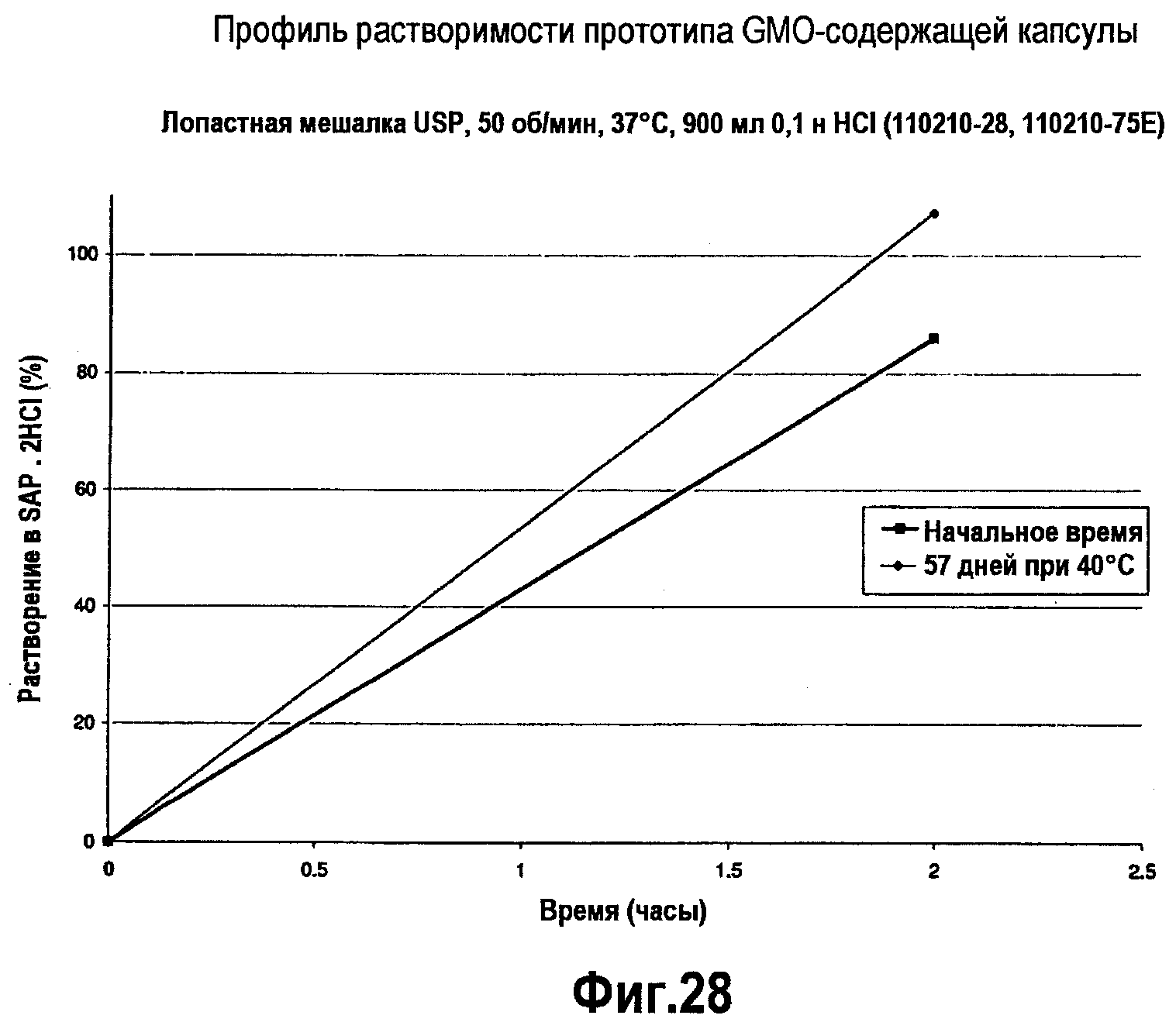
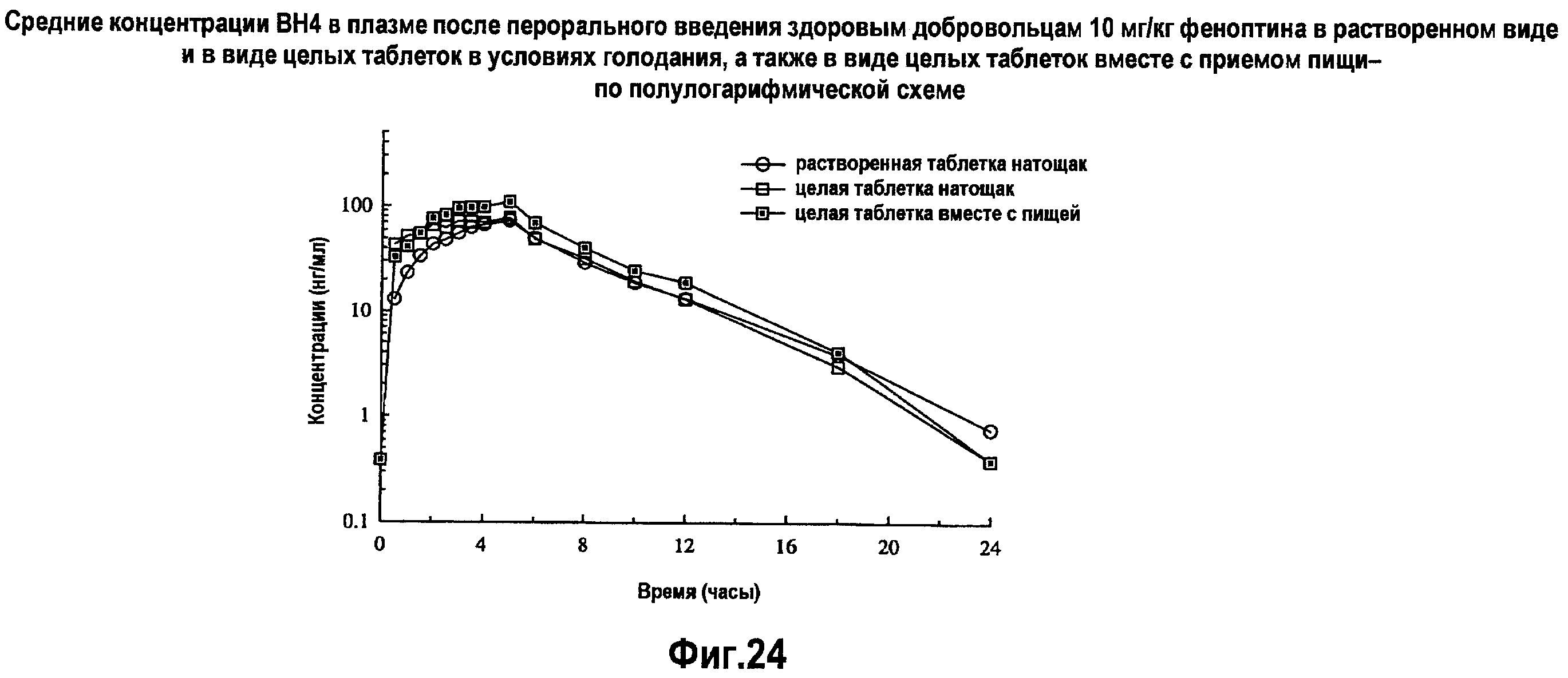
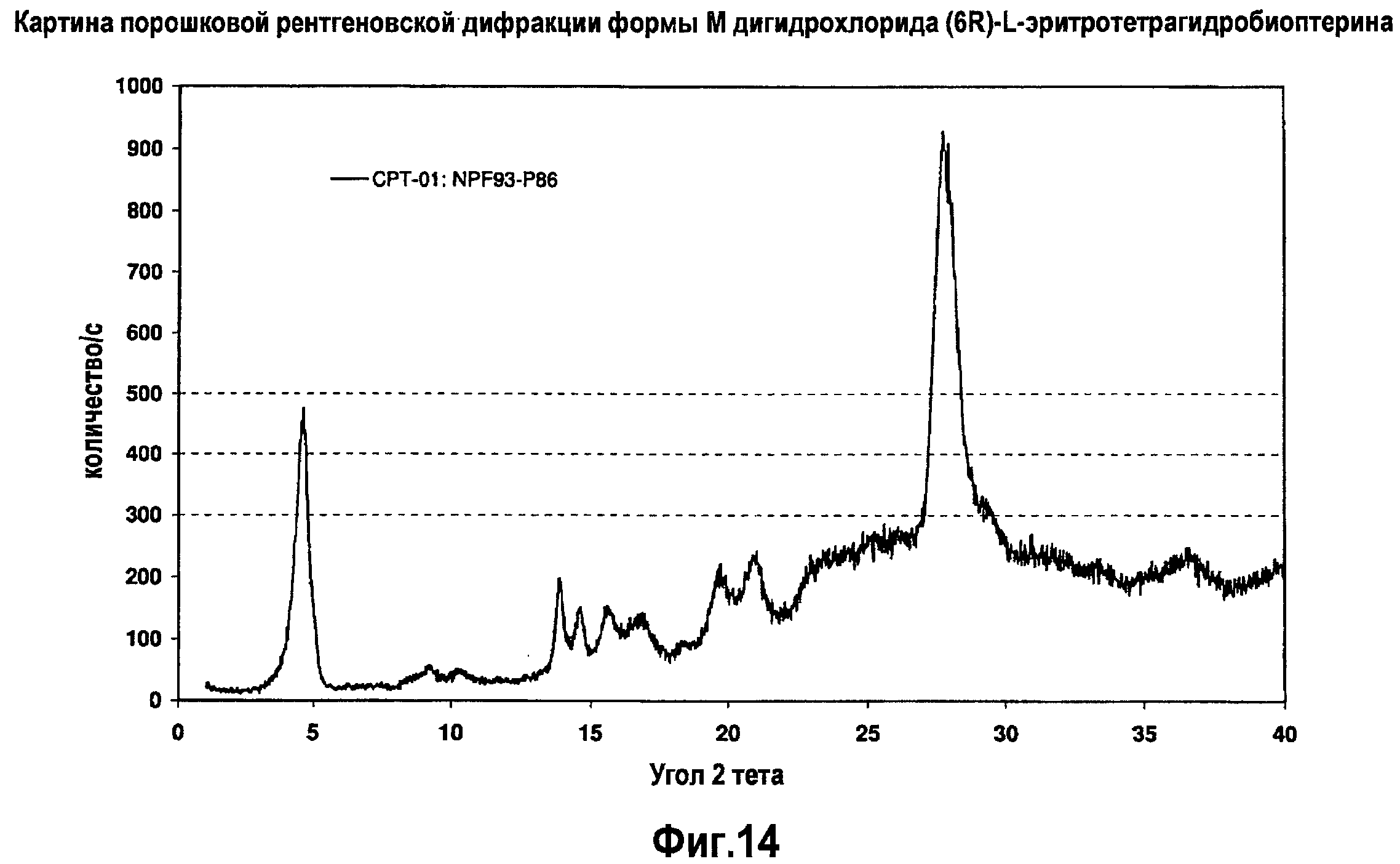
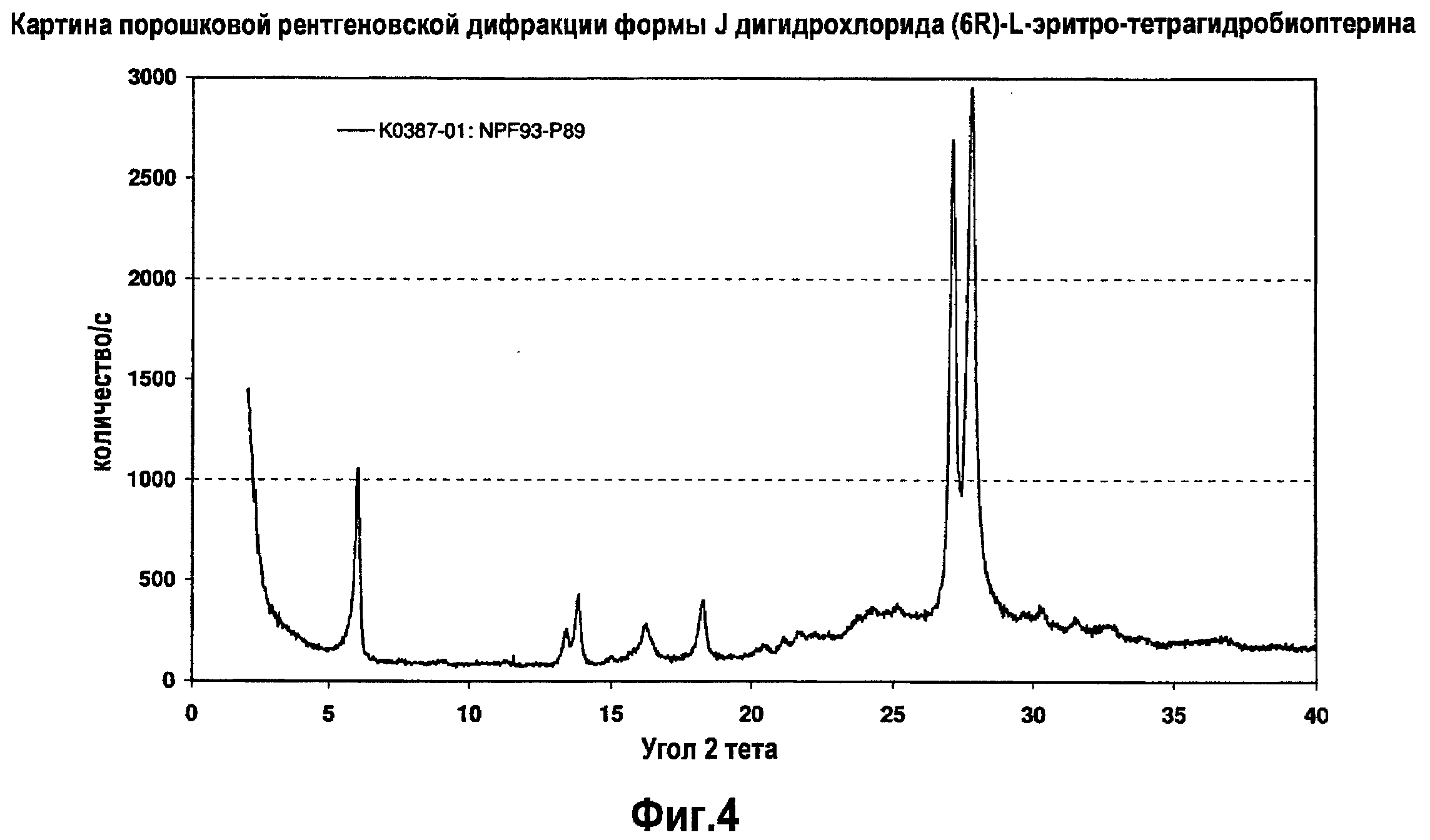
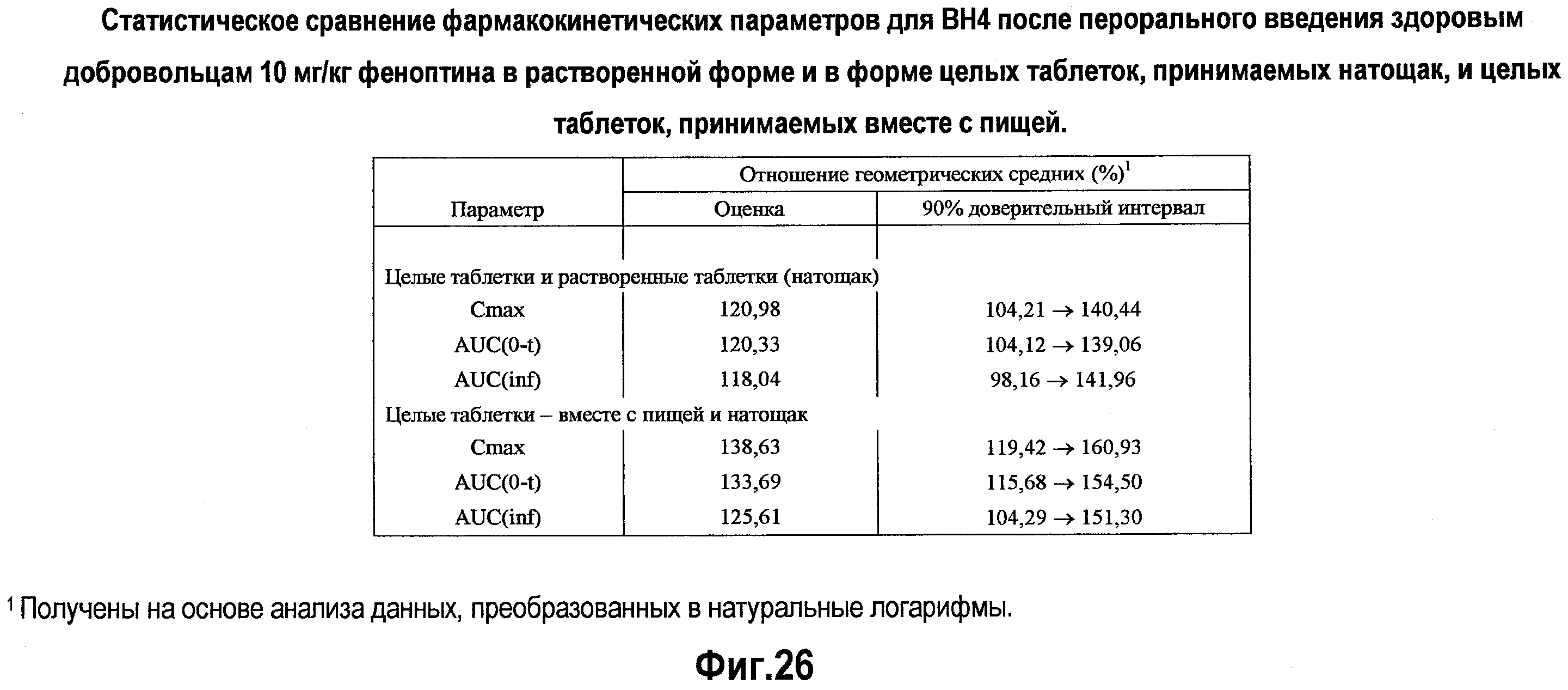
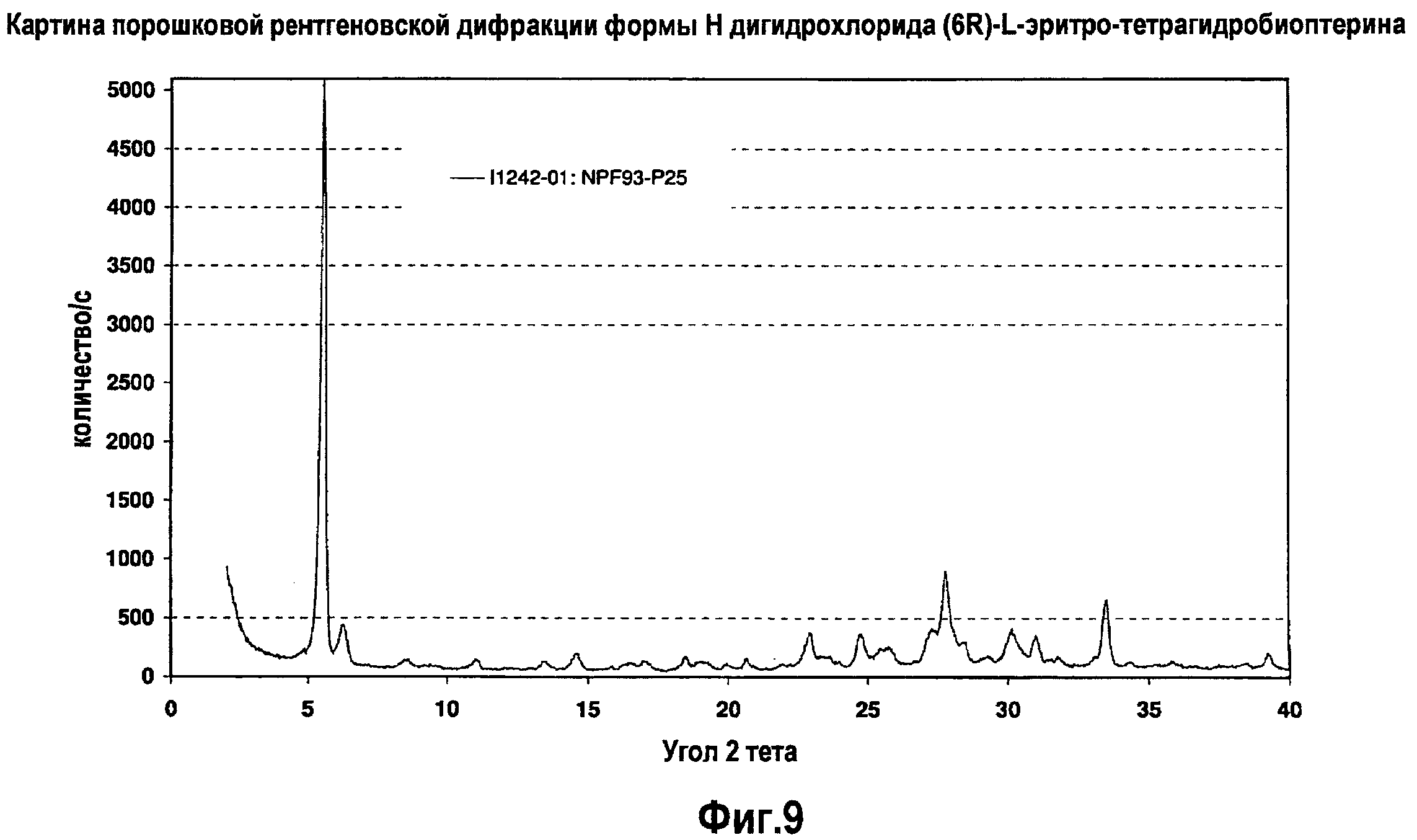
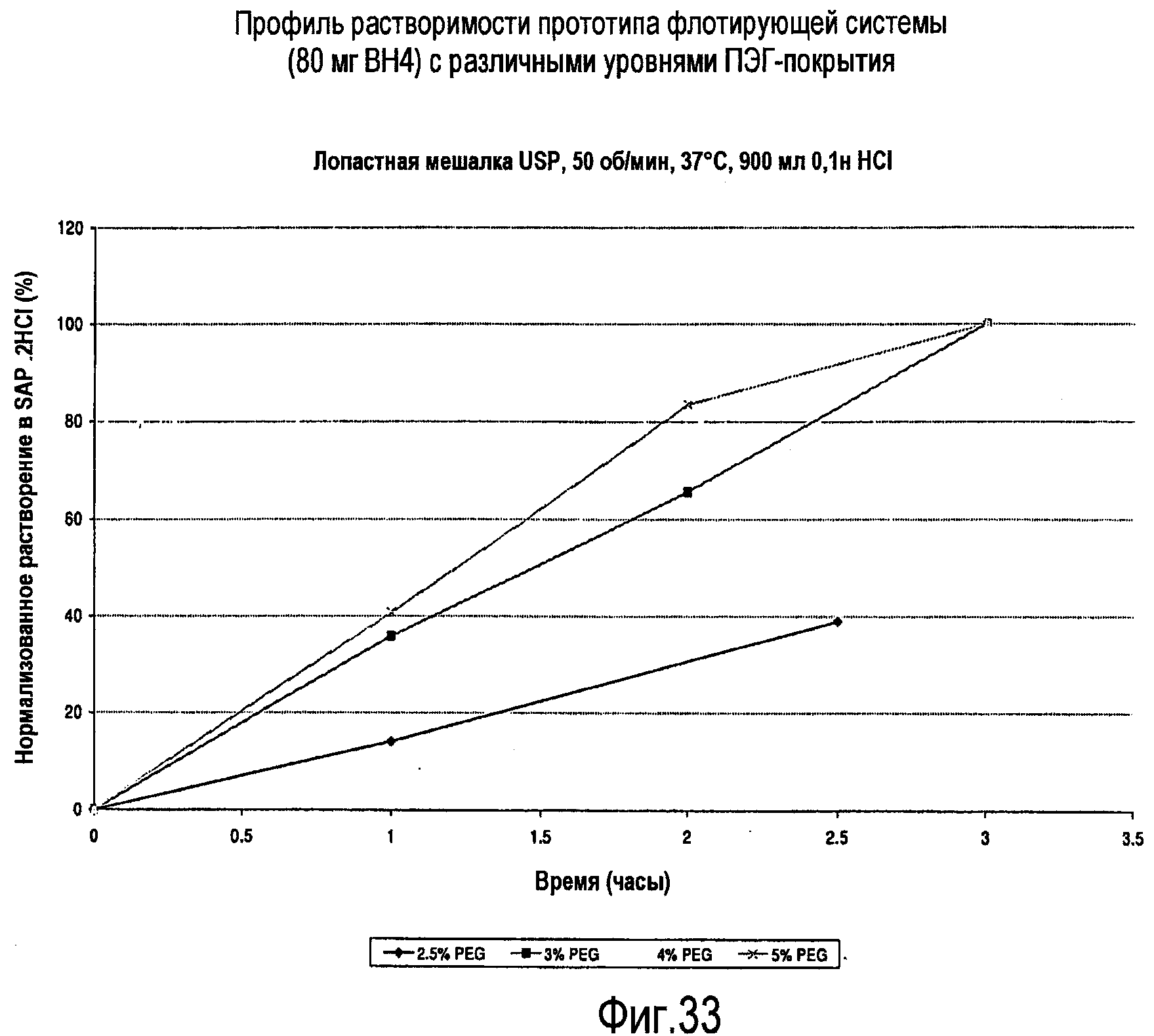
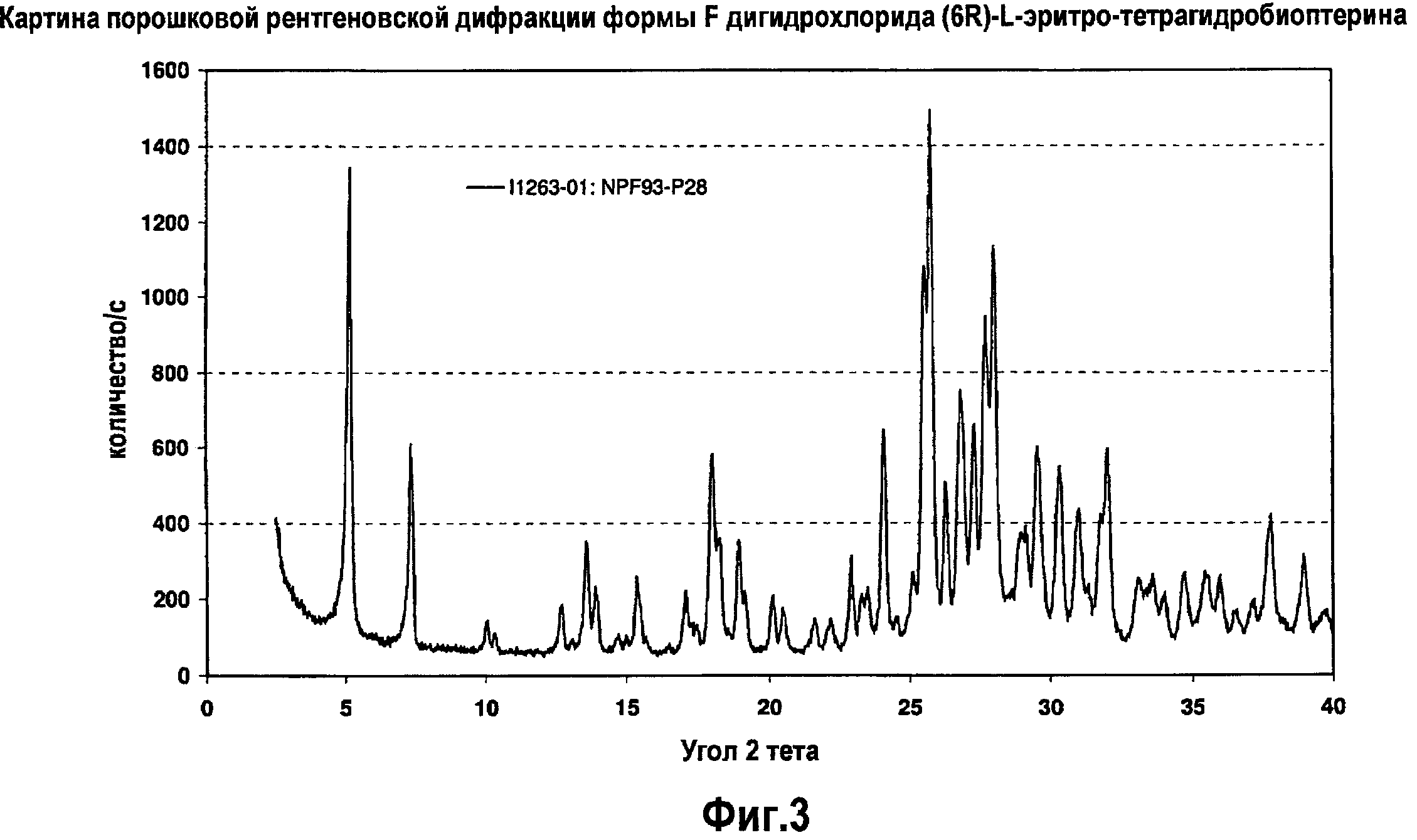

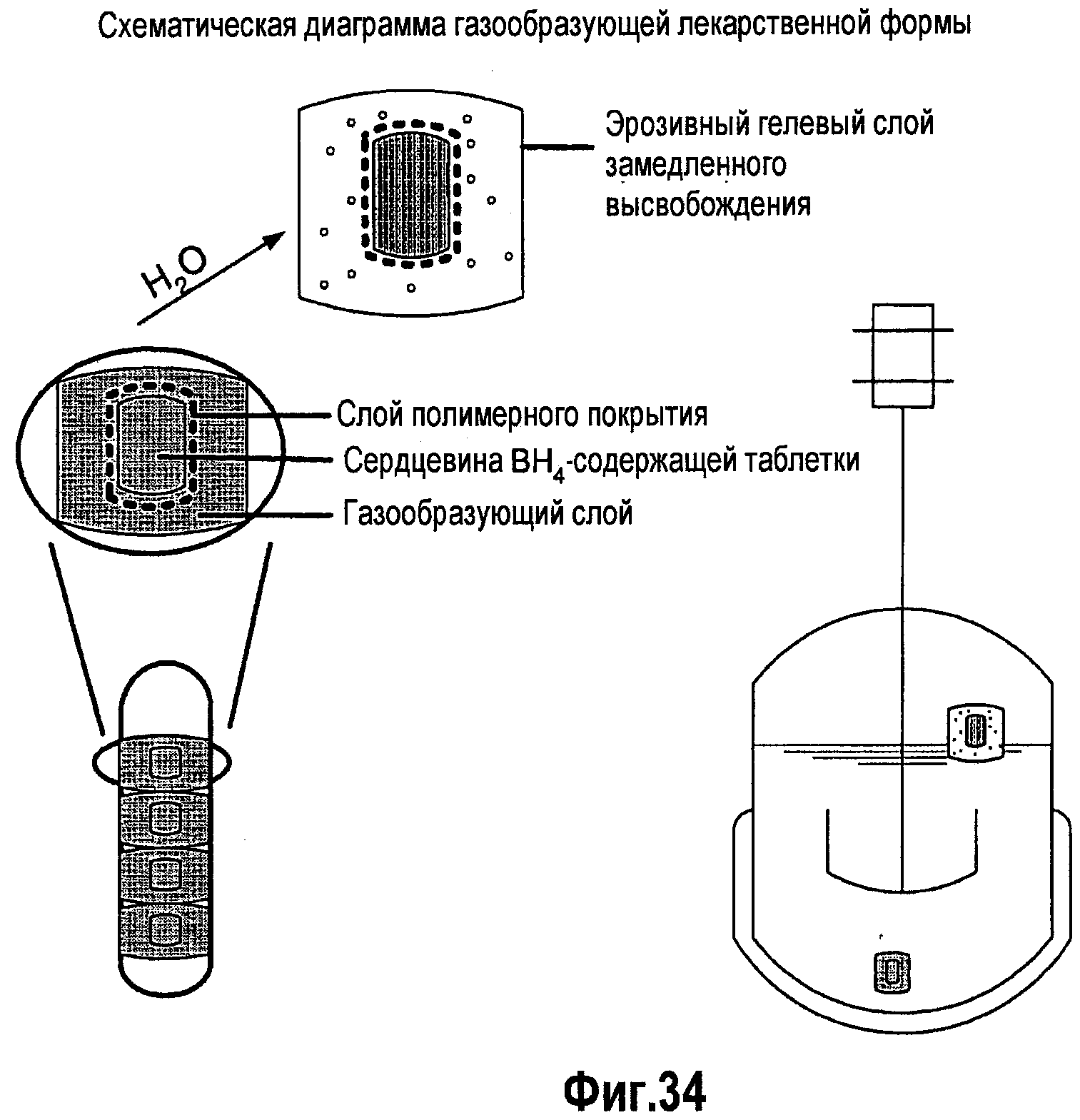
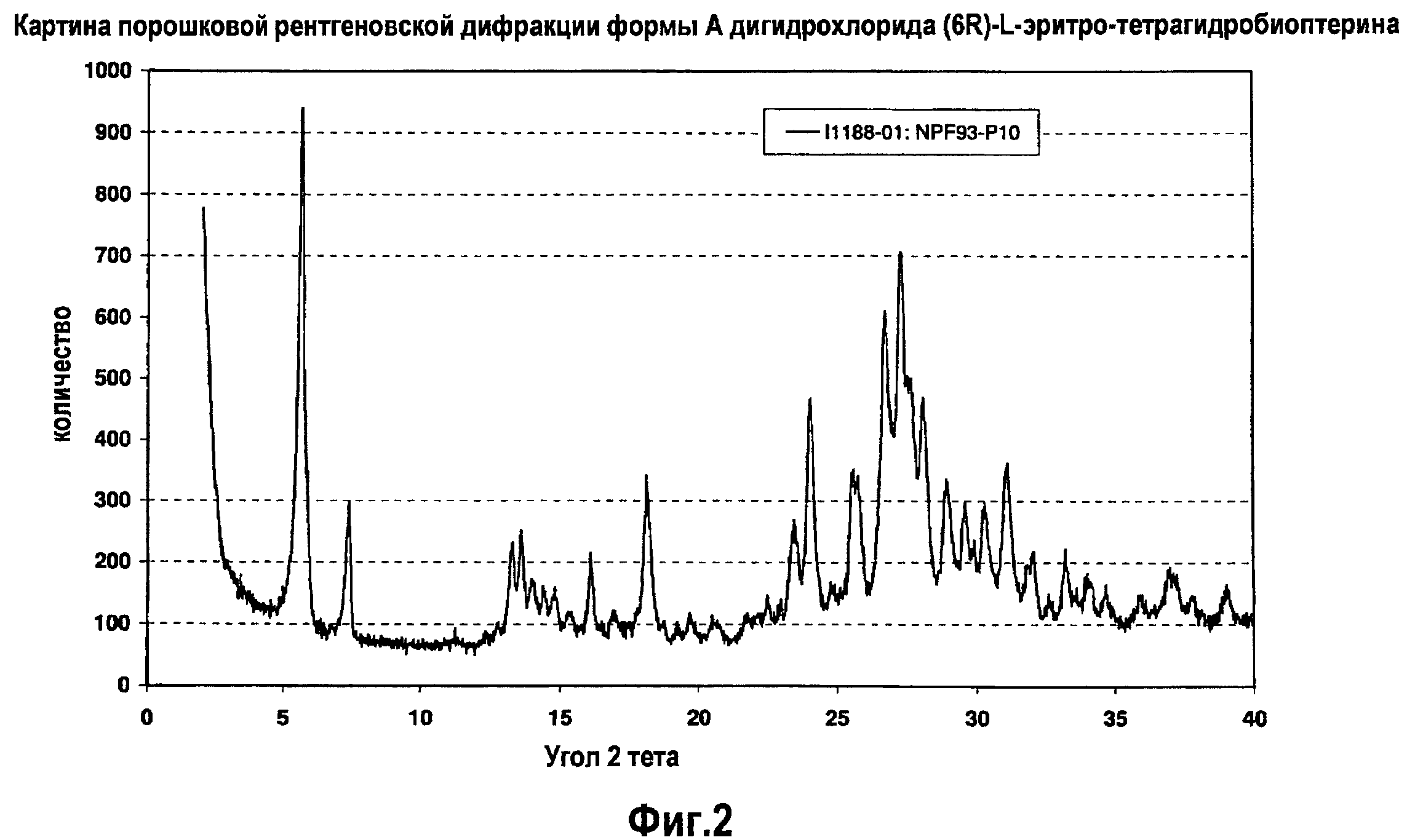
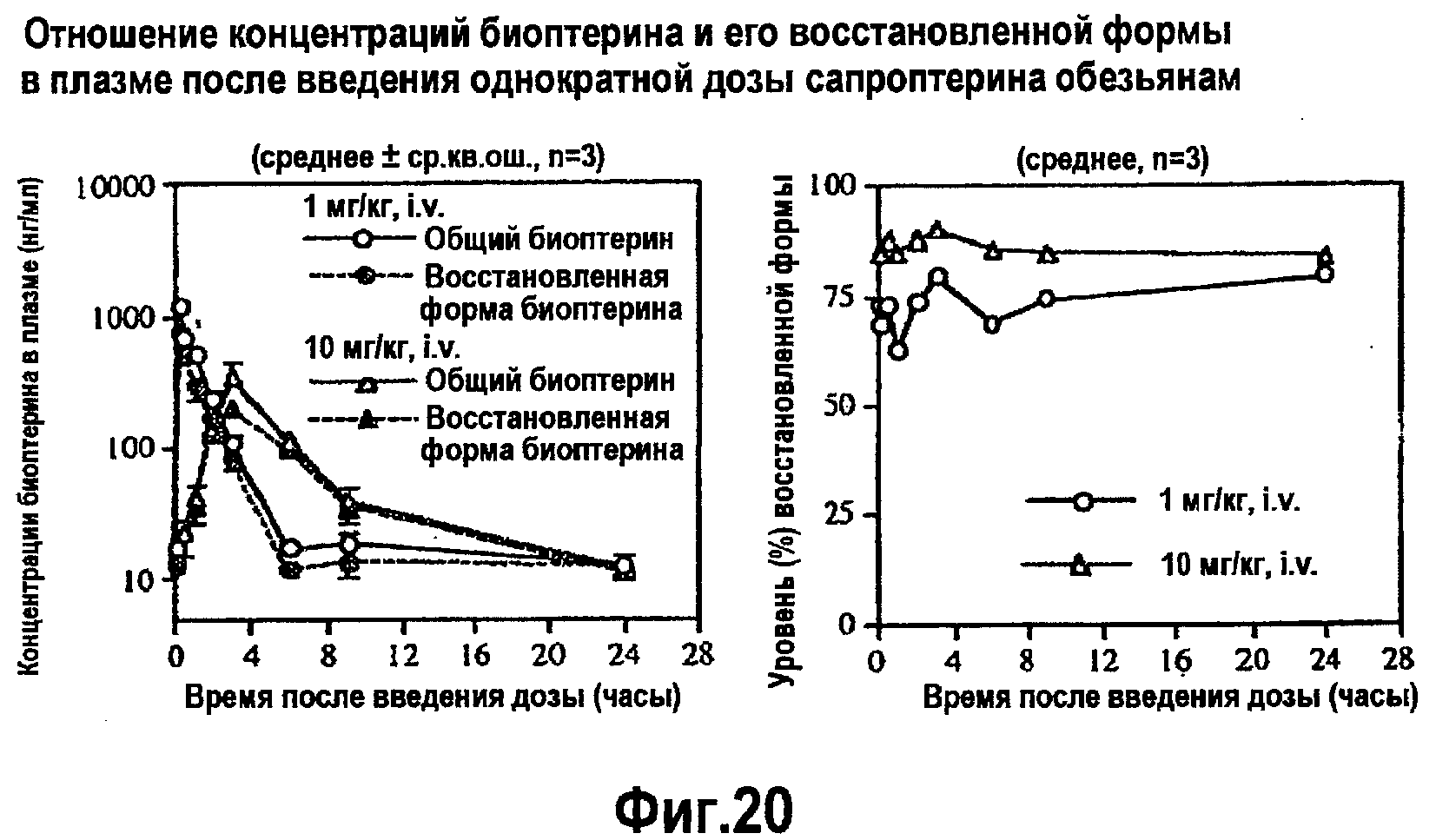

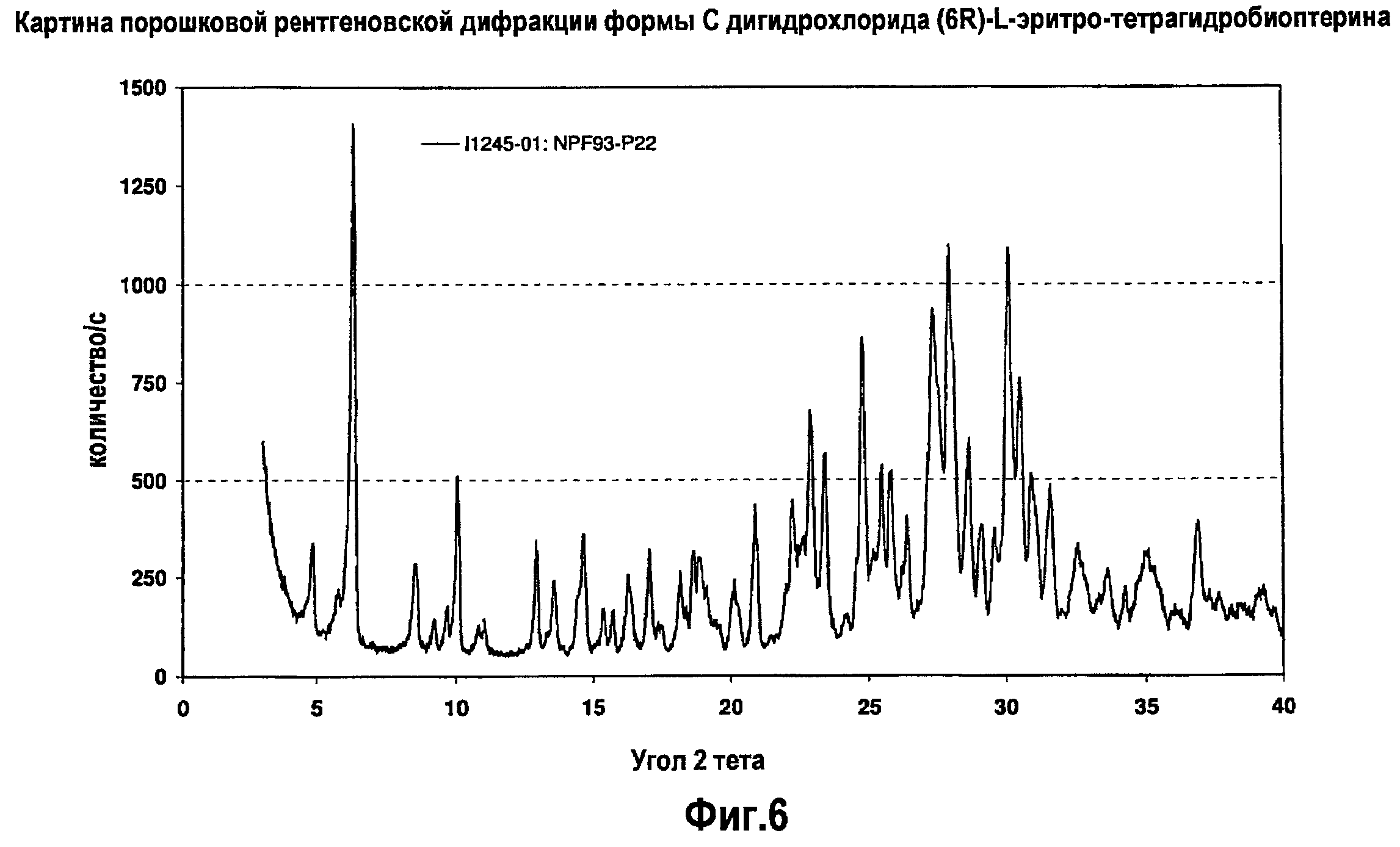
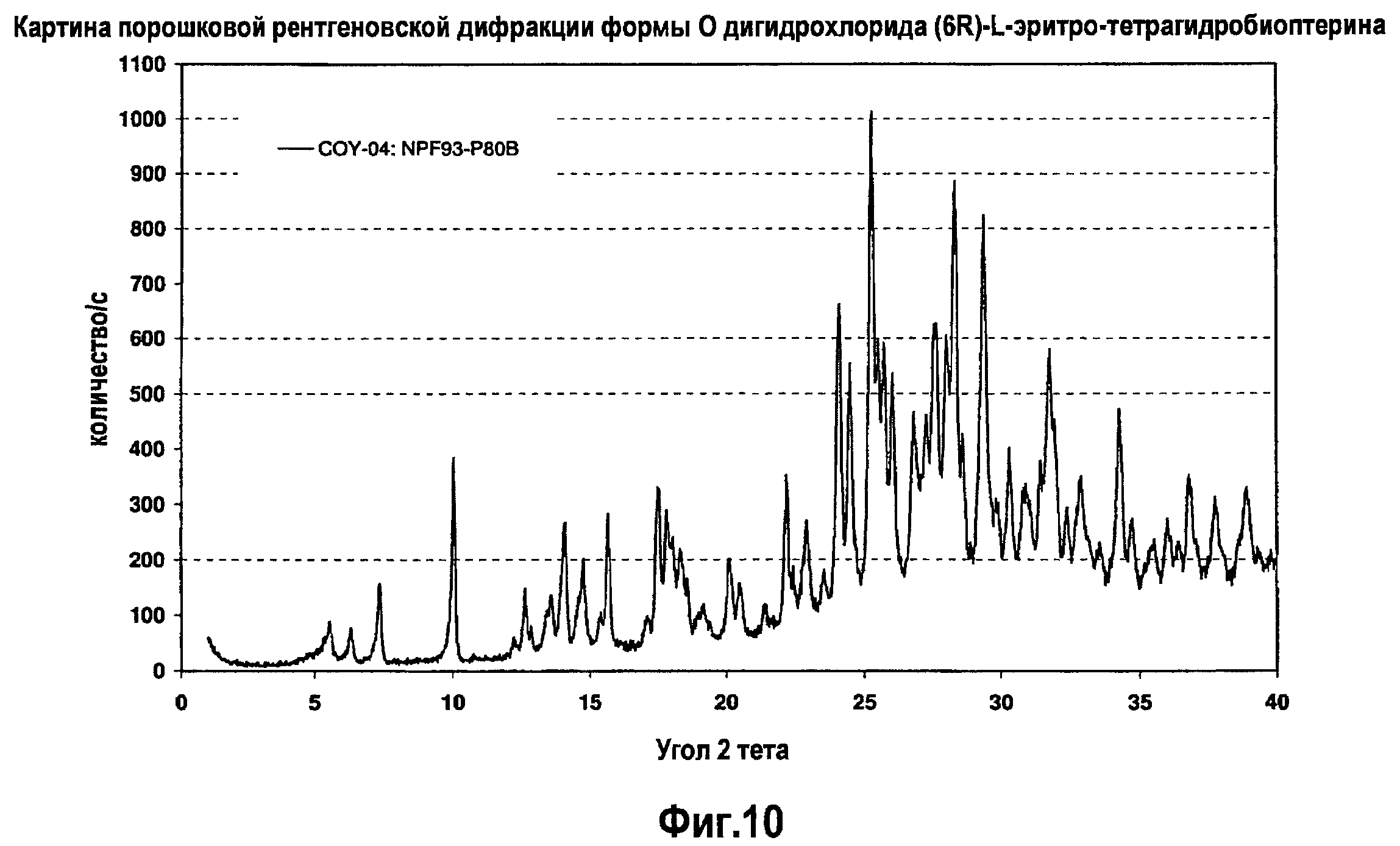
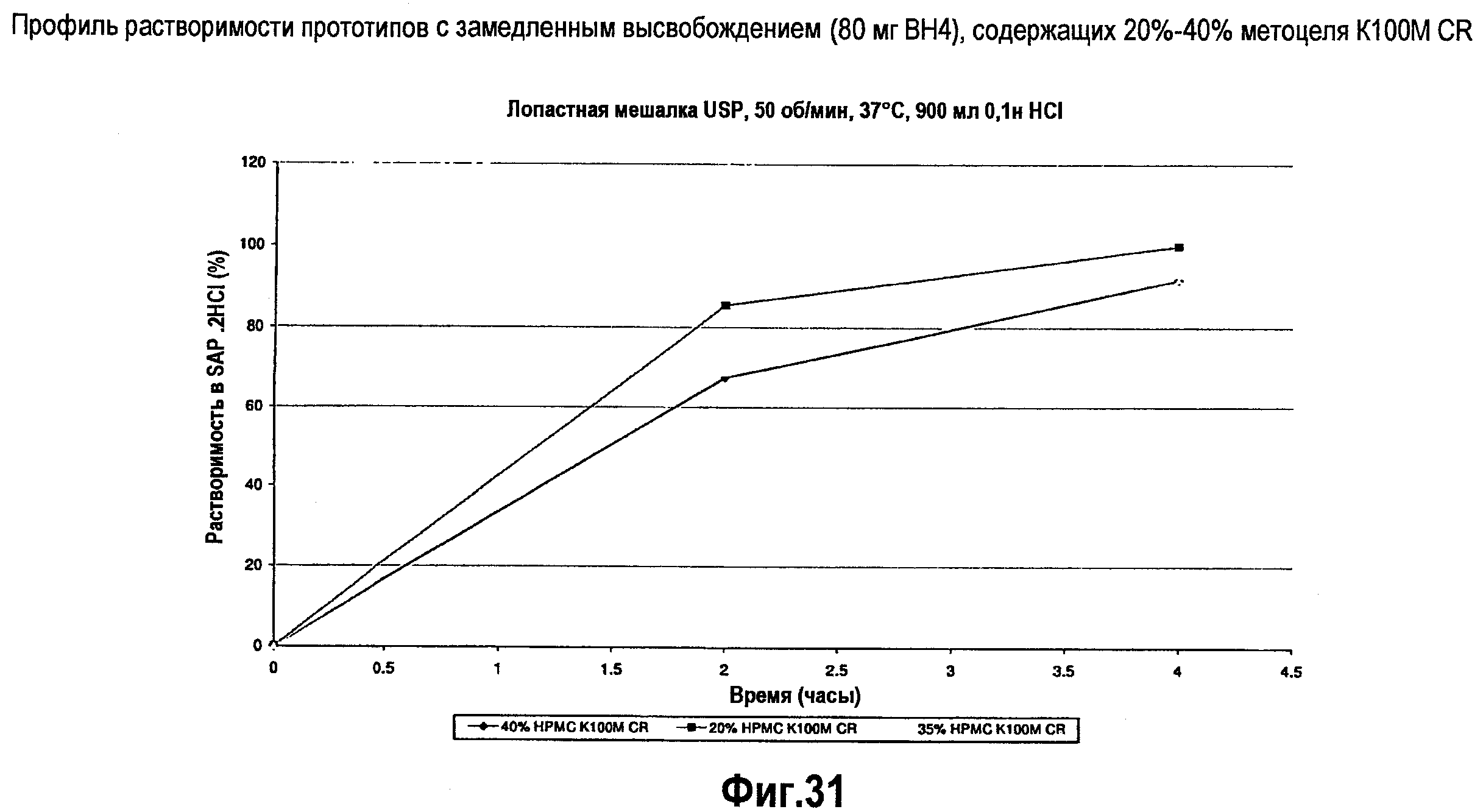
Изготовление активных высокофосфорилированных лизосомальных ферментов сульфатаз человека и их применение
Композиции прокариотической фенилаланин-аммиак-лиазы и способы лечения рака с использованием таких композиций
Способы синтеза производных дигидропиридофталазинона
Варианты натрийуретического пептида с-типа
Производство активной высокофосфорилированной n-ацетилгалактозамин-6-сульфатазы человека и ее применение
Ингибиторы hdac
Ингибиторы гистондеацетилазы
Направленные терапевтические химерные белки лизосомальных ферментов и их применение
Векторы экспрессии фактора viii на основе аденоассоциированного вируса
Ингибиторы гистондеацетилазы
Изготовление активных высокофосфорилированных лизосомальных ферментов сульфатаз человека и их применение
Композиции прокариотической фенилаланин-аммиак-лиазы и способы лечения рака с использованием таких композиций
Способы синтеза производных дигидропиридофталазинона
Варианты натрийуретического пептида с-типа
Производство активной высокофосфорилированной n-ацетилгалактозамин-6-сульфатазы человека и ее применение

