Результат интеллектуальной деятельности: СПОСОБ ОПТИМИЗАЦИИ ЛЕЧЕНИЯ ЛЕЙКОЗА, ПОЛОЖИТЕЛЬНОГО ПО ФИЛАДЕЛЬФИЙСКОЙ ХРОМОСОМЕ, ИНГИБИТОРАМИ Ab1-ТИРОЗИНКИНАЗЫ
Вид РИД
Изобретение
Настоящее изобретение относится к способу лечения лейкоза, положительного по филадельфийской хромосоме (лейкоза Ph+) в группе пациентов. В частности, настоящее изобретение относится к способу лечения хронического миелоидного лейкоза (ХМЛ) в группе пациентов.
При ХМЛ взаимно сбалансированная хромосомная транслокация в кроветворных стволовых клетках (КСК) приводит к формированию гибридного гена BCR-ABL. Этот ген кодирует онкогенный гибридный белок Bcr-Abl. ABL кодирует жестко регулируемую протеинтирозинкиназу, которая играет фундаментальную роль в регуляции пролиферации клеток, адгезии и апоптозе, а BCR-ABL гибридный ген кодирует конститутивно активированную киназу, которая трансформирует КСК для получения фенотипа, проявляющего разрегулированную клональную пролиферацию, пониженную способность к адгезии на строме костного мозга, снижает апоптический ответ на мутагенный стимул, что способствует накоплению постепенно становящихся более злокачественными трансформаций. Образуемые гранулоциты не могут развиться в зрелые лимфоциты и попадают в кровеносное русло, что приводит к дефициту зрелых клеток и повышенной чувствительности к инфекции. Были описаны АТФ-конкурентные ингибиторы Bcr-Abl, которые предохраняют киназу от активирования митогенного и антиапоптического метаболических путей (например, киназа Р-3 и STAT5), приводящие к гибели клеток с фенотипом BCR-ABL и, в результате, обеспечивают эффективную терапию против ХМЛ.
В мае 2001 года соль N-{5-[4-(4-метилпиперазинометил)бензоиламидо]-2-метилфенил-4-(3-пиридил)-2-пиримидинамин мезилат (иматиниб мезилат, STI571, продукт Glivec®) была одобрена Управлением по контролю за продуктами и лекарствами США (FDA) для лечения ХМЛ у пациентов, которых не удается лечить интерфероном-альфа (IFN). Уже в июне 2000 года первые пациенты с ХМЛ были включены в международное рандомизированное исследование интерферона и STI571 (International Randomized Study of Interferon and STI571 - IRIS). Амбициозная третья фаза исследования была уникальна и по размеру, и по области охвата. Было исследовано более 1000 пациентов в 16 странах для проведения теснейшего сопоставления между продуктом Glivec® и интерфероном-альфа (S.G.O'Brien, F.Guilhot, R.A.Larson и др., N.Engl. J. Med. 348, 2003, cc. 994-1004). Иматиниб в суточной дозе 400 мг проявляет большую эффективность по сравнению с IFN+Ara-C для вновь диагностированных пациентов с ХМЛ в хронической фазе (ХМЛ-ХФ). Ранее полученные в ходе пятилетних исследований данные по программе IRIS показали установленную кумулятивную скорость полного цитогенетического ответа (ПЦО) у 87% пациентов, получавших первичное лечение иматинибом, и среднее выживание 89% (Druker B.J., Guilhot F., O'Brien S.G. и др. N. Engl. J. Med. 355, 2006, cc.2408-2417). Отмечено, что среди пациентов, достигших ПЦО и главного молекулярного ответа (ГМО) на протяжении 18 месяцев от начала лечения, не было таких, у которых к 60 месяцу было бы прогрессирование болезни до ускоренной или бластической фазы.
По настоящее время причина вариабельности ответов на иматиниб мезилат при лечении хронического ХМЛ в полной мере не установлена. Предшествующие исследования были сосредоточены на клеточных механизмах устойчивости к иматинибу. Хотя фармакокинетический мониторинг широко применяют в разных разделах медицины, например неврологии, кардиологии и психиатрии, его редко применяют в клинической онкологической практике. Фармакокинетические исследования пациентов с ХМЛ, подвергаемых лечению иматинибом мезилатом, показали, что концентрации иматиниба в плазме на протяжении времени применения иматиниба коррелируют с дозой иматиниба мезилата, а масса тела или площадь поверхности тела имеют небольшое значение. Peng и др. определяли концентрацию иматиниба в плазме и установили режим применения иматиниба по показателям концентрации в плазме (Peng В., Hayes M., Resta D. и др. J. Clin. Oncol. 22, 2004, cc.935-942). Mahon и др. (Blood, 106, 2005, с.565А) измеряли концентрацию иматиниба в крови при поддержке режима лечения.
Настоящее изобретение относится к способу, позволяющему минимизировать или избежать формирования устойчивости, потери эффективности и риска возникновения рецидива у людей-пациентов с ХМЛ, которых лечат ингибитором Bcr-Abl тирозинкиназы. Основываясь на результатах исследований, проведенных в Университете Бордо и по программе IRIS, по корреляции фармакокинетических данных с цитогенетическим и молекулярным ответами у вновь диагностированных пациентов с ХМЛ в хронической фазе (ХМЛ-ХФ), неожиданно было установлено, что лечение ХМЛ с применением ингибитора тирозинкиназы Bcr-Abl может быть оптимизировано подбором дозы ингибитора тирозинкиназы Bcr-Abl для конкретного пациента таким образом, что специфический минимальный уровень в плазме (Смин) достигается у каждого отдельного пациента. Индивидуальный подбор дозы для каждого пациента необходим, учитывая высокую степень индивидуальной вариабельность величины Смин при введении одной и той же дозы ингибитора Bcr-Abl тирозинкиназы разным пациентам, установленную в исследовании IRIS. Настоящее изобретение предусматривает в первую очередь схемы индивидуального лечения для отдельных пациентов с ХМЛ, основанные на пониженном пороге Смин, поскольку было установлено, что этот параметр коррелирует с повышенным уровнем выживания.
ХМЛ относится к группе Ph+ лейкозов. Результаты, полученные на группе пациентов с ХМЛ и описанные в настоящем изобретении, могут быть непосредственно перенесены на всю группу лейкозов Ph+. Это связано с тем, что для лейкозов группы Ph+ свойственно наличие филадельфийской хромосомы, приводящей к образованию гибридного белка Bcr-Abl. Этот белок является мишенью всех ингибиторов Bcr-Abl.
Аббревиатура «Ph+ALL» в контексте настоящего изобретения означает острый лимфобластический лейкоз при наличии филадельфийской хромосомы (Philadelphia chromosome positive acute lymphoblastic leukemia).
Понятие «главный молекулярный ответ (ГМО)» в контексте настоящего изобретения означает снижение на три порядка транскриптов BCR-ABL, подсчитанных в периферической крови, используя реального времени количественную обратной транскриптазы полимеразную цепную реакцию, предпочтительно через 12 месяцев лечения, например 12 месяцев лечения иматинибом мезилатом.
Понятие «полный цитогенный ответ (ПЦО)» в контексте настоящего изобретения означает 0% филадельфийской хромосомы на стадии метафазы по меньшей мере в 20-25 клетках на стадии метафазы в аспирате костного мозга (Colombat M., Fort M.P., Chollet С. и др. Haematologica, 91, 2006, сс.162-168).
Понятие «способ лечения» в контексте настоящего изобретения означает также способ предупреждения заболеваний, указанных в настоящем изобретении, т.е. профилактическое введение фармацевтической композиции, включающей ингибитор Bcr-Abl тирозинкиназы, здоровым пациентам для предупреждения развития заболеваний, указанных в настоящем изобретении.
Понятия «подбор дозы» и «подобранная доза» в контексте настоящего изобретения предпочтительно означает, что соответствующую дозу повышают или понижают. В более широком смысле в настоящем изобретении понятия «подбор дозы» и «подобранная доза» также предусматривают ситуацию, при которой доза остается неизменной.
Понятие «ингибитор Bcr-Abl тирозинкиназы» в контексте настоящего изобретения означает органические соединения, которые проявляют ингибирование с-Abl или Bcr-Abl из лизатов трансфецированных клеток с величиной IC50 менее 0,1 мкМ в исследованиях киназы in vitro, выполненных на иммунопреципитатах в исследовании, описанном B.J.Druker и др. Nat. Med. 2, 1996, cc.561-566.
Таким образом, в широком смысле слова, настоящее изобретение относится к способу лечения лейкоза Ph+, например, ХМЛ или Ph+ALL, в группе пациентов, включающему стадии:
(а) введения предварительно установленного фиксированного количества ингибитора Bcr-Abl тирозинкиназы или его фармацевтически приемлемой соли людям, больным лейкозом Ph+,
(б) отбора по меньшей мере одного образца крови у указанных пациентов,
(в) определения самого низкого уровня в плазме (Смин) ингибитора тирозинкиназы Bcr-Abl или его метаболита, а также степеней ГМО,
(г) оценки разделяющего потенциала самых низких концентраций ГМО в плазме и выявления пороговой величины Смин для оптимальной чувствительности и специфичности, например, с помощью анализа характеристической кривой (Receiver Operating Characteristic - ROC-кривой), и
(д) подбора дозы ингибитора тирозинкиназы Bcr-Abl или его фармацевтически приемлемой соли, применяемой для конкретных пациентов из указанной группы пациентов и, необязательно, для будущих пациентов с лейкозом Ph+ таким образом, чтобы величина Смин у каждого отдельного пациента равнялась или превышала пороговую величину Смин, установленную на стадии (г).
Точнее настоящее изобретение относится к способу лечения ХМЛ в группе людей, включающему стадии:
(а) введения предварительно установленного фиксированного количества ингибитора тирозинкиназы Bcr-Abl или его фармацевтически приемлемой соли людям-пациентам, больным ХМЛ, нуждающимся в этом,
(б) отбора по меньшей мере одного образца крови у указанных пациентов,
(в) определения величины Смин ингибитора тирозинкиназы Bcr-Abl или его метаболита, а также степеней ГМО,
(г) оценки разделяющего потенциала самых низких концентраций ГМО в плазме и выявления пороговой величины Смин для оптимальной чувствительности и специфичности, например, с помощью анализа характеристической кривой (ROC-кривой), и
(д) подбора дозы ингибитора тирозинкиназы Bcr-Abl или его фармацевтически приемлемой соли, применяемой для конкретных пациентов из указанной группы пациентов и, необязательно, для будущих пациентов с ХМЛ таким образом, чтобы величина Смин у каждого отдельного пациента равнялась или превышала пороговую величину Смин, установленную на стадии (г).
С помощью описанной выше методологии установлено, что пороговое значение Смин для ингибитора тирозинкиназы Bcr-Abl иматиниба может составлять примерно 800 нг/мл, более предпочтительно примерно 1000 нг/мл. Верхний предел уровня в плазме соответствует уровню, который ниже, но близко прилегает к уровню в крови, вызывающему доза-предельную токсичность (ДПТ) у отдельного пациента. Обычно наблюдаемый верхний предел составляет примерно 3500 нг/мл, иногда примерно 3000 нг/мл.
Таким образом, согласно еще одному из объектов настоящее изобретение связано со способом лечения лейкоза Ph+, особенно ХМЛ или Ph+ALL, у пациента, включающим стадии:
(а) введения предварительно установленного фиксированного количества иматиниба или его фармацевтически приемлемой соли, например пероральной суточной дозой 400 мг или 800 мг соли иматиниба мономезилата, людям-пациентам, больным лейкозом Ph+,
(б) отбора по меньшей мере одного образца крови у указанного пациента, например, в течение первых 12 месяцев лечения,
(в) определения величины Смин иматиниба, и
(г) подбора дозы иматиниба или его фармацевтически приемлемой соли таким образом, чтобы величина Смин у указанного пациента достигала по меньшей мере примерно 800 нг/мл, особенно примерно от 800 до примерно 3500 нг/мл, предпочтительно величина Смин достигает от 1000 до примерно 3000 нг/мл иматиниба.
В широком смысле слова настоящее изобретение предусматривает способ лечения лейкоза Ph+, особенно ХМЛ или Ph+ALL, у человека-пациента, причем дозу иматиниба или его фармацевтически приемлемой соли подбирают таким образом, что величина Смин у указанного пациента поддерживается по меньшей мере на уровне примерно 800 нг/мл, особенно примерно от 800 до примерно 3500 нг/мл, предпочтительно величина Смин поддерживается на уровне от 1000 до примерно 3000 нг/мл иматиниба.
Более конкретно настоящее изобретение относится к способу лечения ХМЛ у людей-пациентов, включающему стадии:
(а) введения предварительно определенного фиксированного количества иматиниба или его фармацевтически приемлемой соли человеку-пациенту с ХМЛ, нуждающемуся в этом,
(б) отбора по меньшей мере одного образца крови от указанного пациента, например, в течение первых 12 месяцев, особенно первых 3 месяцев, более предпочтительно первых 30 суток лечения,
(в) определения самого низкого уровня в плазме (Смин) иматиниба, и
(г) подбора дозы иматиниба или его фармацевтически приемлемой соли таким образом, чтобы величина Смин у указанного пациента достигала по меньшей мере 800 нг/мл, особенно примерно от 800 до примерно 3500 нг/мл иматиниба.
В последнем указанном способе дозу фармацевтически приемлемой соли иматиниба подбирают предпочтительно таким образом, что величина Смин достигает примерно от 1000 до примерно 3000 нг/мл иматиниба у указанного пациента, более предпочтительно величина Смин составляет примерно 1000 нг/мл.
Метаболизм иматиниба происходит через систему цитохрома Р450, причем CYP3A4 является главным изоферментом, ответственным за метаболизм иматиниба, хотя CYP1A2, CYP2D6, CYP2C9 и CYP2C19 также участвуют в этом метаболизме, но в небольшой степени. Один важный метаболит, N-{5-[4-(4-пиперазинометил)бензоиламидо]-2-метилфенил-4-(3-пиридил)-2-пиримидинамин (CGP74588), выявляемый в крови, обладает биологическим действием, близким к действию иматиниба, и представляет примерно 20% от исходного уровня лекарственного средства в плазме у пациентов. Из-за свойственной вариабельности действия ферментов CYP (Wilkinson G.R., J. Pharmacokinet. Biopharm., 24, 1996, cc.475-490), установлена высокая вариабельность, присущая пациентам, при экспозиции иматинибом пациентов с ХМЛ (Peng B.M., Hayes M., Resta D. и др. J. Clin. Oncol. 22, 2004, cc.935-942). Было установлено, что лекарственные средства, которые ингибируют или индуцируют изофермент CYP3A4, влияют на фармакокинетические показатели иматиниба (Bolton А.Е., Peng В., Hubert M. и др. Cancer Chemother. Pharmacol. 53. 2004, cc.102-106, Dutreix С., Peng В., Mehring G. и др. Cancer Chemother. Pharmacol. 54, 2004, cc.290-294, Smith P.F., Bullock J.M., Booker B.M. и др. Pharmacother. 24 (11), 2004, cc.1508-1514, Frye R.F., Fitzgerald S.M., Lagattuta T.F., Hruska M.W., Egorin M.J. Clin. Pharmacol. Ther. 76, 2004, cc.323-329).
Таким образом, в другом варианте осуществления настоящего изобретения предусмотрен способ лечения ХМЛ у людей-пациентов, включающий стадии:
(а) введения предварительно определенного фиксированного количества иматиниба или его фармацевтически приемлемой соли человеку-пациенту с ХМЛ, нуждающемуся в этом,
(б) отбора по меньшей мере одного образца крови от указанного пациента, например, в течение первых 12 месяцев, особенно первых 3 месяцев, более предпочтительно первых 30 суток лечения,
(в) определения самого низкого уровня в плазме (Смин) N-{5-[4-(пиперазинметил)бензоиламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамина (CGP74588), и
(г) подбора дозы иматиниба или его фармацевтически приемлемой соли таким образом, чтобы величина Смин у указанного пациента достигала по меньшей мере 150, особенно примерно от 150 до примерно 800 нг/мл N-{5-[4-(пиперазинметил)бензоиламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамина.
В последнем указанном способе дозу фармацевтически приемлемой соли иматиниба подбирают предпочтительно таким образом, что величина Смин у указанного пациента достигает примерно от 250 до примерно 700 нг/мл N-{5-[4-(пиперазинметил)бензоиламидо]-2-метилфенил-4-(3-пиридил)-2-пиримидинамина.
Кроме того, настоящее изобретение относится к применению ингибитора тирозинкиназы Bcr-Abl или его фармацевтически приемлемой соли для получения лекарственного средства для лечения лейкоза Ph+, согласно которому:
(а) предварительно определенное фиксированное количество ингибитора тирозинкиназы Bcr-Abl или его фармацевтически приемлемой соли вводят людям-пациентам с лейкозом Ph+,
(б) отбирают по меньшей мере один образец крови от указанных пациентов,
(в) определяют в плазме самый низкий уровень (Смин) ингибитора тирозинкиназы Bcr-Abl или его метаболита, а также степеней ГМО,
(г) оценивают разделяющий потенциал самых низких концентраций ГМО в плазме и выявляют пороговую величину Смин для оптимальной чувствительности и специфичности, и
(д) дозу ингибитора тирозинкиназы Bcr-Abl или его фармацевтически приемлемой соли, применяемой к определенным пациентам из указанной группы пациентов и, необязательно, будущим пациентам с лейкозом Ph+, подбирают таким образом, что величина Смин, достигнутая у каждого отдельного пациента, равна или превышает пороговую величину Смин, полученную на стадии (г). Лейкоз Ph+ предпочтительно является ХМЛ или Ph+ALL. По меньшей мере один образец крови отбирают в первые 12 месяцев, особенно в первые 3 месяца, предпочтительно в первые 30 суток лечения.
Кроме того, настоящее изобретение относится к применению иматиниба или его фармацевтически приемлемой соли для получения лекарственного средства для лечения лейкоза Ph+, в котором:
(а) заранее определенное фиксированное количество иматиниба или его фармацевтически приемлемой соли, например пероральную суточную дозу 400 мг или 800 мг иматиниба мономезилата, вводят человеку-пациенту с лейкозом Ph+,
(б) по меньшей мере, один образец крови отбирают в первые 12 месяцев, особенно в первые 3 месяца, в частности в первые 30 суток лечения,
(в) определяют в плазме самый низкий уровень (Смин) иматиниба, и
(г) дозу иматиниба или его фармацевтически приемлемой соли подбирают таким образом, что величина Смин у указанных пациентов достигает по меньшей мере 800, особенно примерно от 800 до примерно 3500 нг/мл иматиниба, особенно примерно от 1000 до примерно 3000 нг/мл иматиниба. Лейкоз Ph+ является Ph+ALL или, предпочтительно, ХМЛ.
Другой объект настоящего изобретения относится к применению иматиниба или его фармацевтически приемлемой соли для получения лекарственного средства для лечения лейкоза Ph+, согласно которому:
(а) предварительно определенное фиксированное количество иматиниба или его фармацевтически приемлемой соли, например пероральную дозу 400 мг или 800 мг соли иматиниба мономезилата, вводят людям-пациентам с лейкозом Ph+,
(б) отбирают по меньшей мере один образец крови от указанного пациента в течение первых 12 месяцев лечения, особенно в первые 3 месяца, предпочтительно в первые 30 суток лечения,
(в) определяют в плазме минимальный уровень (Смин) N-{5-[4-(пиперазинметил)бензоиламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамина, и
(г) дозу иматиниба или его фармацевтически приемлемой соли подбирают таким образом, что величина Смин у указанного пациента достигает по меньшей мере 150, особенно примерно от 150 до примерно 800 нг/мл, предпочтительно примерно от 250 до примерно 700 нг/мл N-{5-[4-(пиперазинметил)бензоиламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамина.
В одном из вариантов осуществления настоящего изобретения предварительно определенное фиксированное количество относится к стадии (а), представляющей терапевтически эффективное количество.
В настоящем изобретении предпочтительно применяют соль иматиниб мономезилат на стадии (а), например, при пероральной суточной дозе примерно от 200 до примерно 800 мг, предпочтительно в суточной дозе примерно 400 мг.
Способ, описанный в настоящем изобретении, особенно полезен для пациентов с ХМЛ, оцененным по шкале Сокаля (ШС). Способы определения по ШС известны специалистам в данной области.
Другим важным объектом настоящего изобретения является применение иматиниба или его фармацевтически приемлемой соли, особенно иматиниба мезилата, для получения лекарственного средства для лечения лейкоза Ph+, причем дозу фармацевтически приемлемой соли подбирают таким образом, что величина Смин у указанного пациента поддерживается равной по меньшей мере 800 нг/мл, например, примерно 1000 нг/мл.
Краткое описание фигур
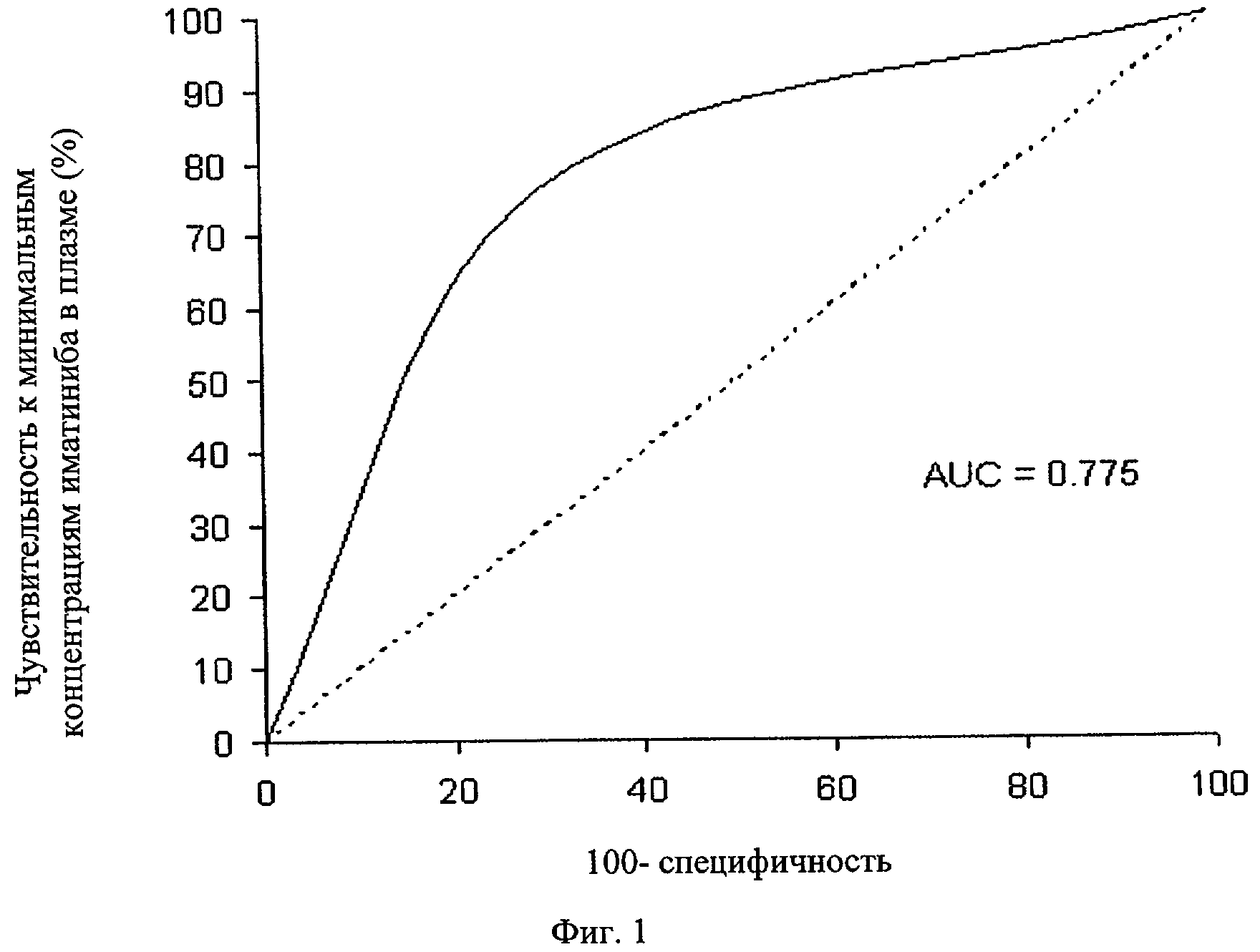
Фиг.1. Анализ ROC-кривой. Анализ характеристической кривой (RОС) проводят для оценки разделяющего потенциала самых низких концентраций ГМО в плазме и выявления пороговой величины в плазме для оптимальной чувствительности и специфичности. Площадь под ROC-кривой (AUC) составила 0,775 при наилучшей чувствительности (76,5%) и специфичности (70,6%) при пороговой концентрации в плазме 1002 нг/мл. Такая величина пороговой концентрации (1002 нг/мл) в значительной степени связана с наличием ГМО (вероятность успешного исхода 7,83; доверительный интервал 95%, 2,58-23,76; Р<0,001).
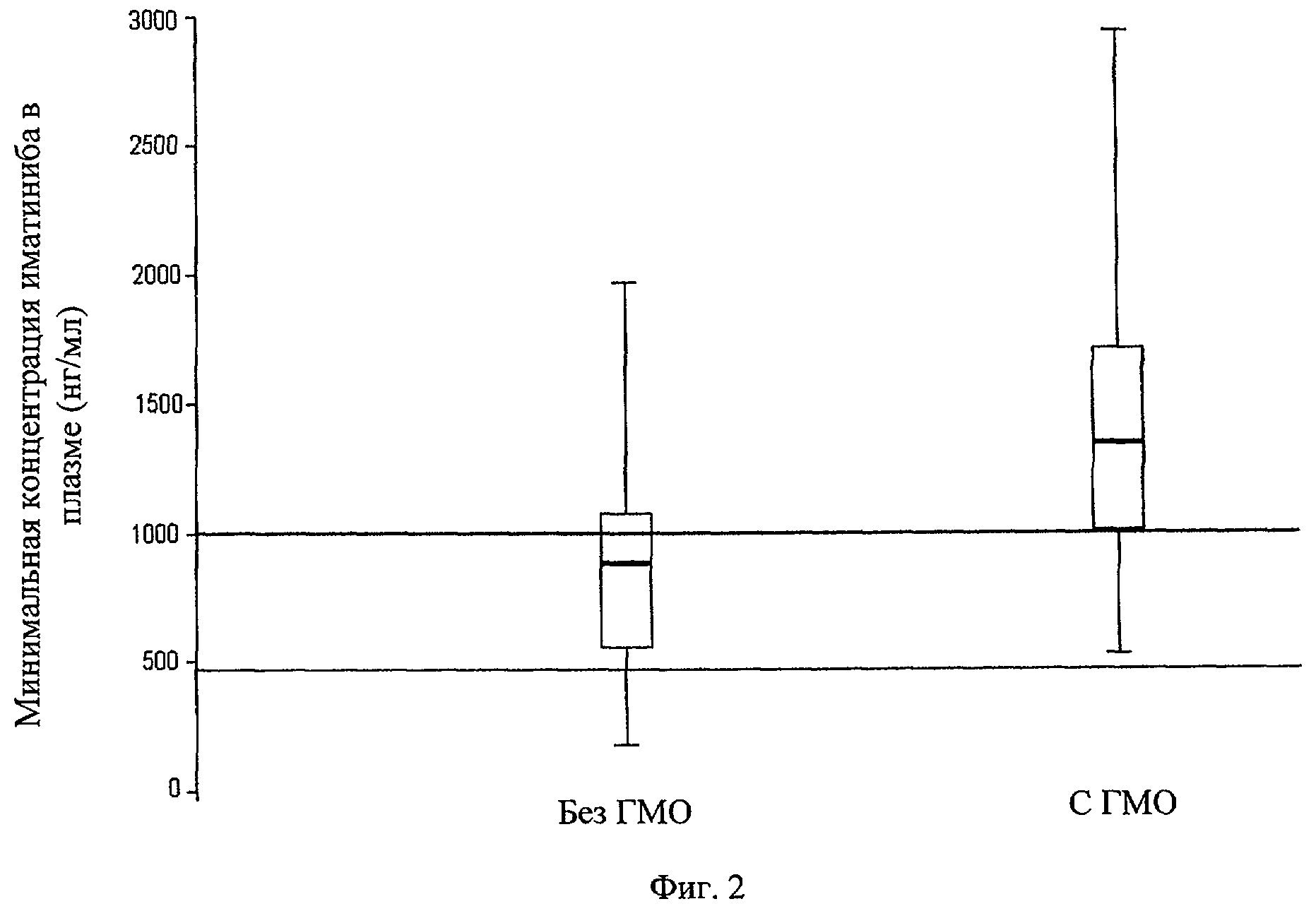
Фиг.2. График рамки-диаграммы. ГМО означает главный молекулярный ответ (снижение уровней транскрипции BCR-ABL на три порядка). График показывает дисперсию вокруг медианы для пациентов с ГМО (34 пациента, средняя величина = 1350,2 нг/мл) и для тех пациентов, у которых ответ отсутствует ГМО (34 пациента, средняя величина = 885,5 нг/мл). Линия через каждую рамку является медианой. Нижний край означает первый квартиль, а верхняя линия означает третий квартиль. Планка погрешностей обозначает минимальные и максимальные значения. Нижняя линия показывает целевую концентрацию 493,6 нг/мл (1 мкМ/мл), требуемую для индукции гибели BCR-ABL-клеток in vitro. Верхняя линия показывает концентрацию 1002 нг/мл, представляющую эффективное пороговое значение минимальной концентрации иматиниба в плазме при лечении ХМЛ.
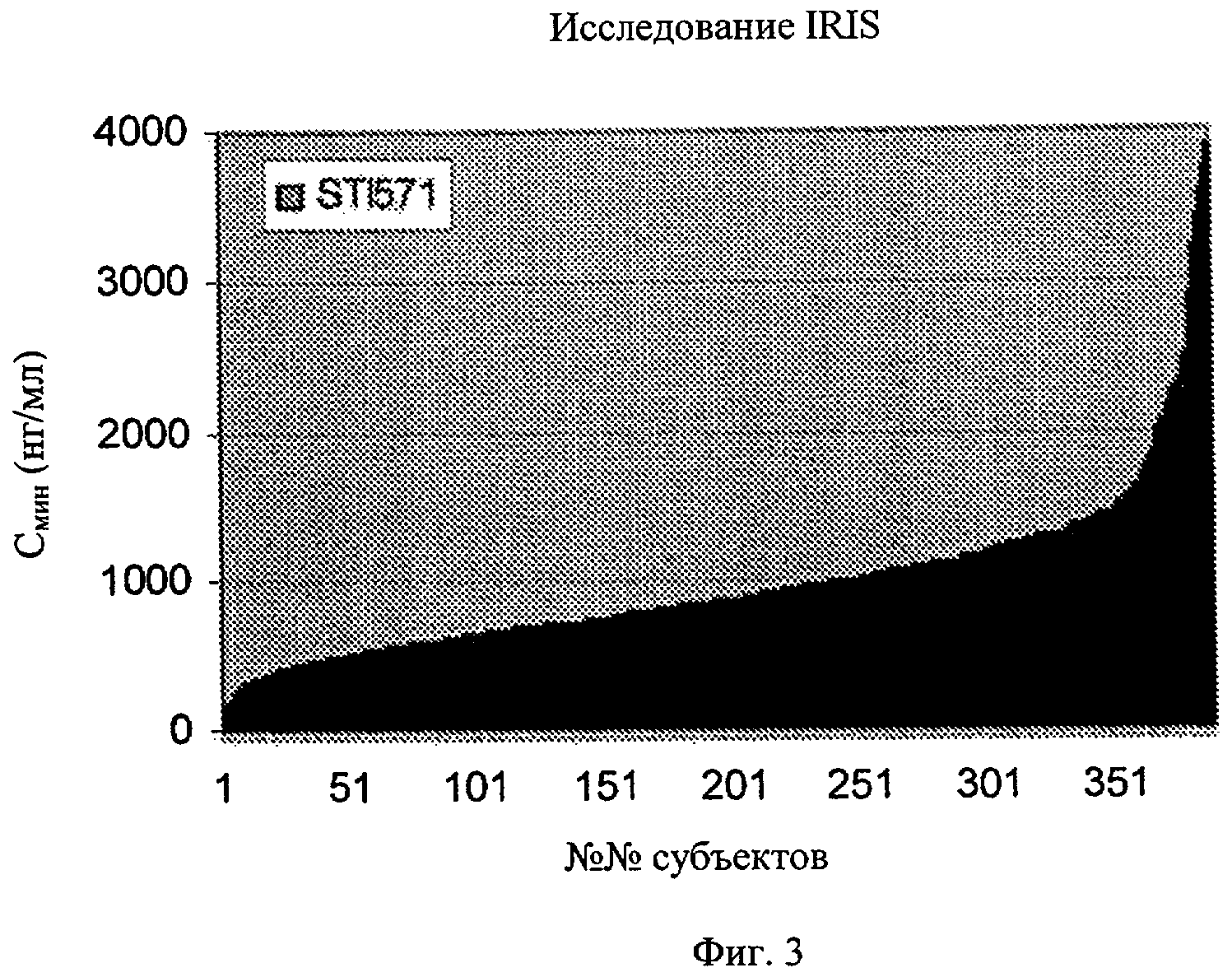
Фиг.3. Вариабельность уровня Смин иматиниба, полученная в исследовании IRIS у пациентов, каждый из которых получал суточную дозу 400 мг иматиниба мезилата.
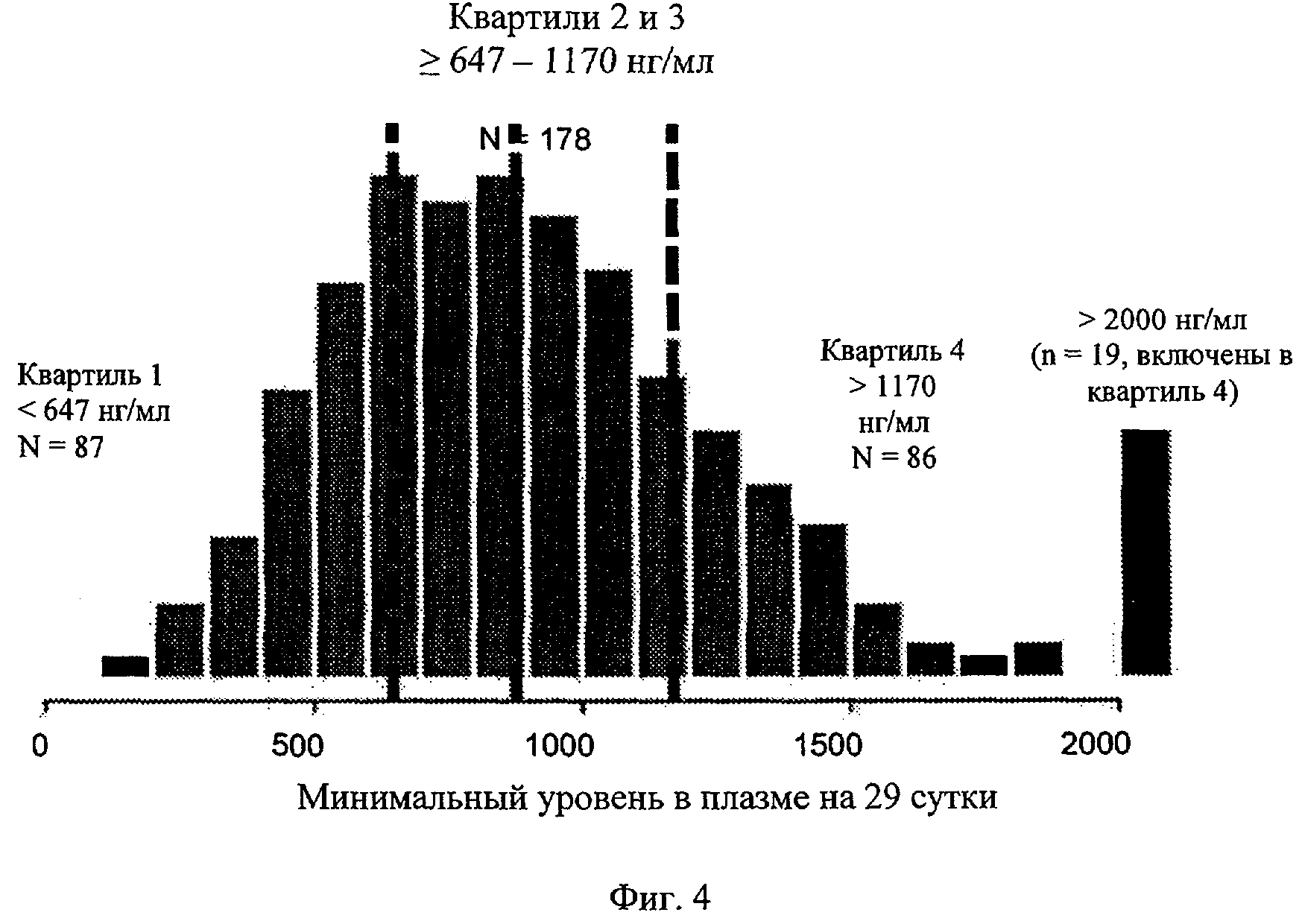
Фиг.4. Распределение уровней Смин иматиниба при суточной дозе 400 мг на 29 сутки (n=351).
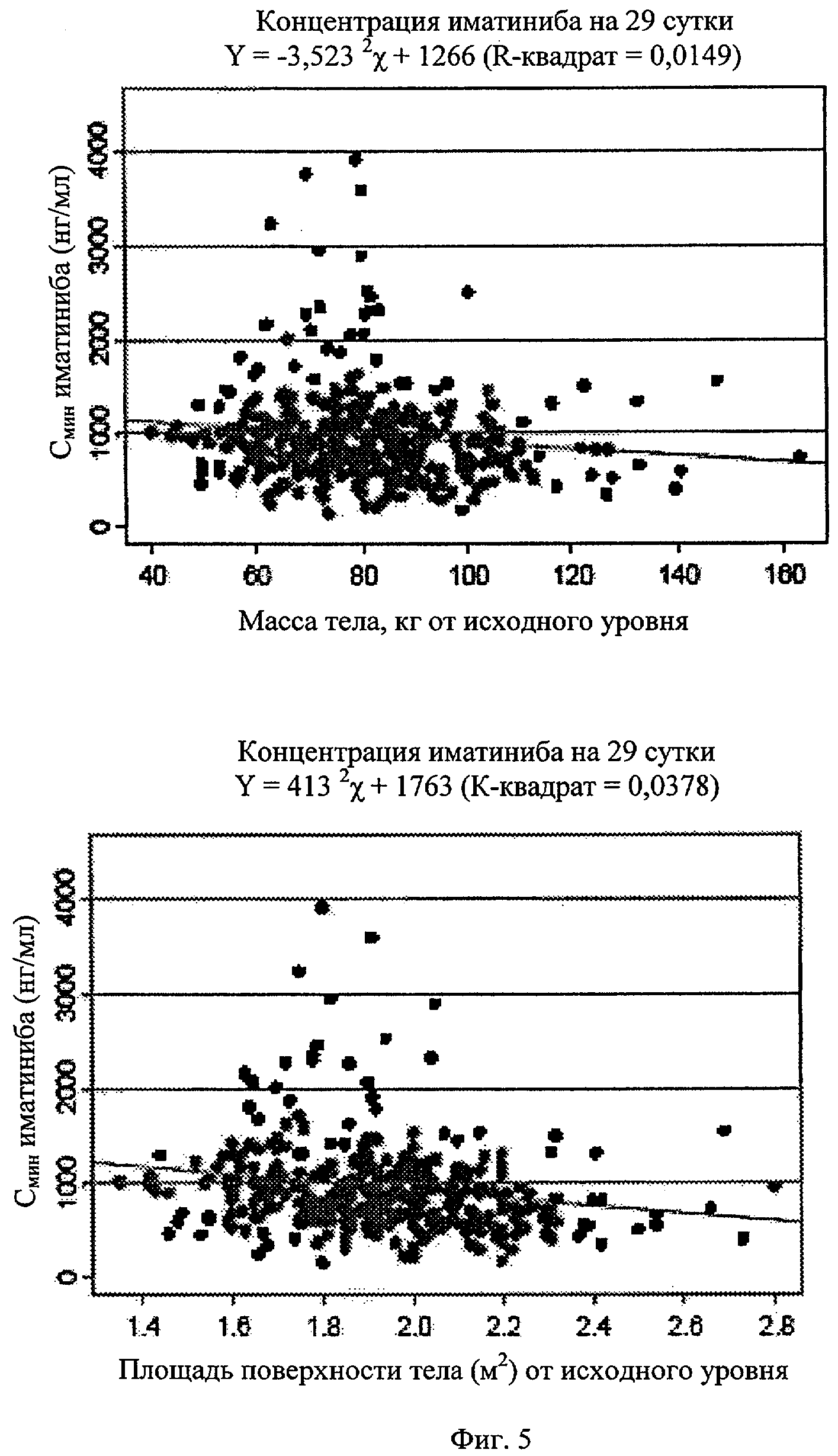
Фиг.5. Минимальный уровень иматиниба в зависимости от массы тела или площади поверхности тела.
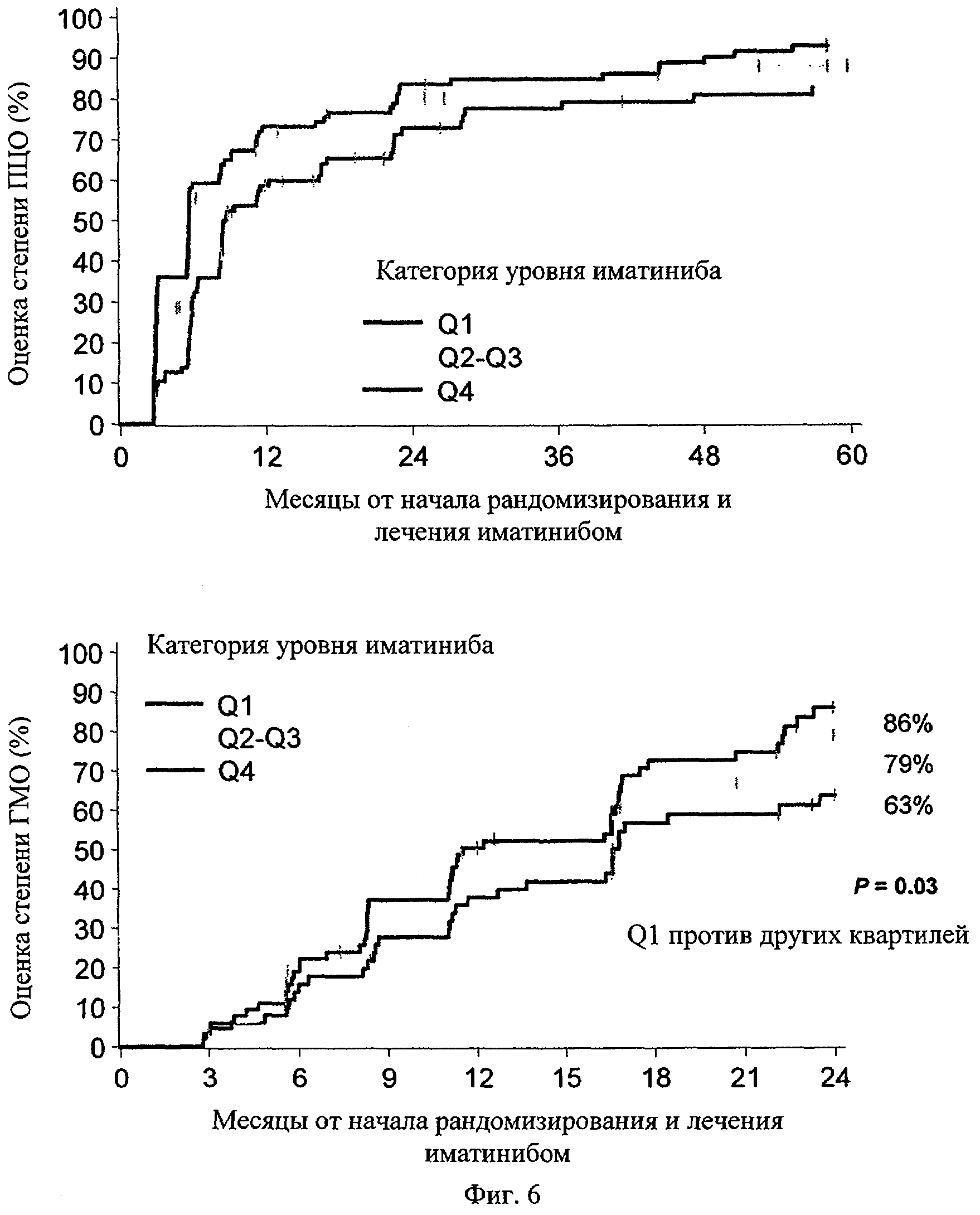
Фиг.6. Полный цитогенетический ответ (ПЦО) и главный молекулярный ответ (ГМО) в зависимости от минимального уровня иматиниба (29 сутки).
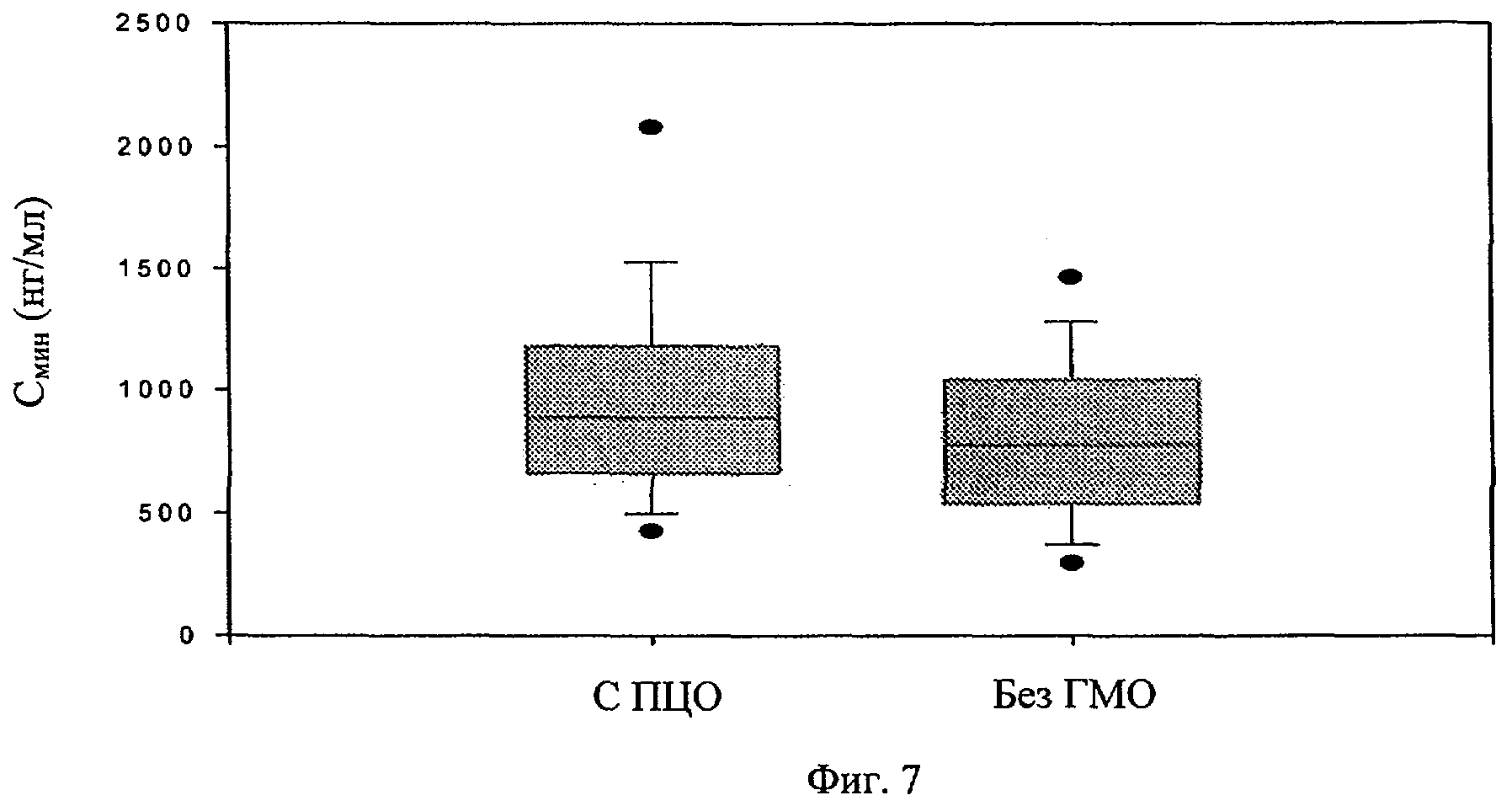
Фиг.7. Минимальные уровни в плазме, соответствующие достижению полного цитогенетического ответа (ПЦО) или неполного ЦО у пациентов с ЧМЛ-ХФ. Верхний и нижний края рамки представляют 75-й и 25-й процентили. Планки погрешностей выше и ниже рамки представляют 90-й и 10-й процентили, а точки представляют 95-й и 5-й процентили.
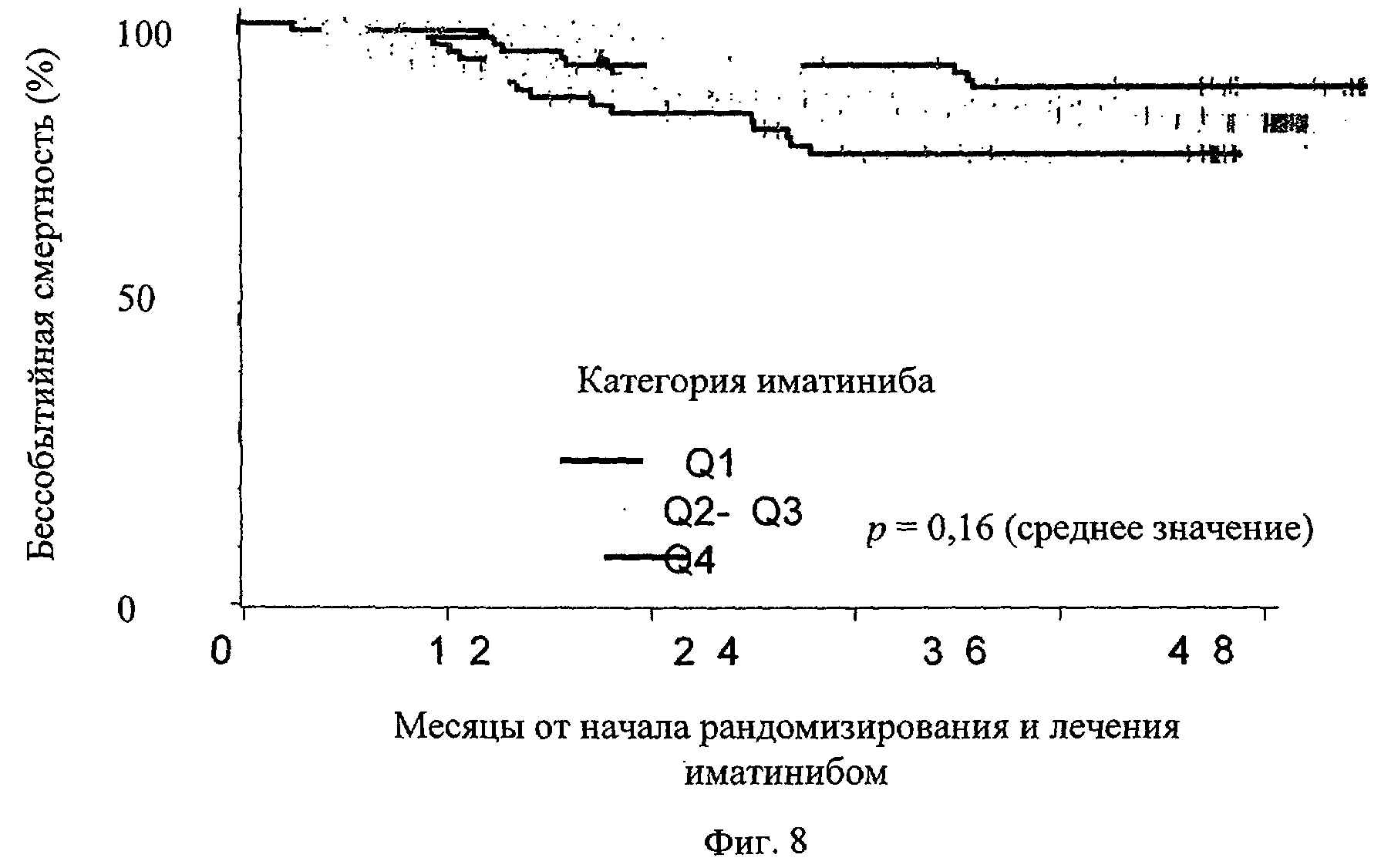
Фиг.8. Величины бессобытийного выживания (event-free survival - EFS) сгруппированы в зависимости от минимальных фармакокинетических показателей уровней иматиниба по квартилям. Группа Q1 показана самой низкой линией, группа Q2-Q3 соответствует линии в центре, а группа Q4 соответствует самой высокой линии.
Ингибитор трозинкиназы Bcr-Abl, применяемый в настоящем изобретении, например, соединения формулы I:
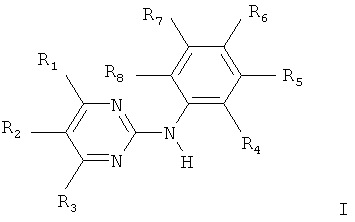 ,
,
где
R1 обозначает 4-пиразинил, 1-метил-1H-пирролил; амино- или амини-низший алкилзамещенный фенил, причем аминогруппа в каждом случае является свободной, алкилированной или ацилированной; 1H-индолил или 1Н-имидазолил, связанный с атомом углерода пятичленного кольца; или незамещенный или низший алкил-замещенный пиридил, связанный с атомом углерода кольца, и незамещенный или замещенный по атому азота кислородом;
R2 и R3 каждый независимо обозначают водород или низший алкил;
один или два из радикалов R4, R5, R6, R7 и R8 обозначает нитро, фторзамещенный низший алкокси или радикал формулы II:
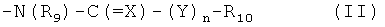 ,
,
где R9 обозначает водород или низший алкил,
Х обозначает оксо, тио, имино, N-низший алкилимино, гидроксиимино или O-низший алкилгидроксиимино,
Y обозначает кислород или группу NH,
n обозначает 0 или 1, и
R10 обозначает алифатический радикал, имеющий по меньшей мере 5 атомов углерода, или ароматический, ароматический-алифатический, циклоалифатический, циклоалифатический-алифатический, гетероциклический или гетероциклический-алифатический радикал, а оставшиеся радикалы R4, R5, R6, R7 и R8 обозначают каждый независимо от других водород, низший алкил, который незамещен или замещен свободными или алкилированными амино, пиперазинил, пиперидинил, пирролидинил или морфолинил, или низший алканоил, трифторметил, свободная, этерифицированная или эсрерифицированная гидроксигруппа, свободная, алкилированная или ацилированная аминогруппа, или свободная или эстерифицированная карбоксигруппа, или соль такого соединения, обладающая по меньшей мере одной группой формирования соли.
Соединения формулы I впервые подробно описаны в патентной заявке US 5521184, в частности в формуле изобретения и в виде конечных продуктов рабочих примеров, сущность которых включена в настоящую заявку в виде ссылки. В приведенном выше определении соединения формулы I радикалы и символы имеют значения, предусмотренные в US 5521184. Предпочтительно соединение формулы I является 4-(4-метилпиперазин-1-илметил)-N-[4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил]бензамид (иматиниб). Иматиниб также может быть получен в соответствии со способами, описанными в WO 03/066613.
Для целей настоящего изобретения иматиниб предпочтительно применяют в форме соли мономезилата. Иматиниб мономезилат также может быть получен в соответствии со способами, описанными в патенте US 6894051, сущность которого включена в настоящее изобретение в виде ссылки. Кроме того, в патент включены соответствующие полиморфы, например кристаллические модификации, описанные в нем.
На стадии (а) способа, описанного выше, предпочтительная суточная доза примерно от 200 и примерно до 800 мг, например, 400 мг, соли мономезилата, вводится перорально. Иматиниб мономезилат может быть введен в дозированных формах, описанных в US 5521184, US 6894051, US 2005-0267125 или WO 2006/121941.
Отбор образцов крови у пациентов с ХМЛ на стадии (б) методов, описанных в настоящем изобретении, может быть осуществлен стандартными методами, известными в данной области. Соответствующая методика определения уровня в плазме Смин иматиниба и N-{5-[4-(пиперазинометил)бензоиламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамина описана R.Bakhtiar и др. в J.Chromatogr. В.Analyt. Nechnol. Biomed. Life Sci. 768, 2002, cc.325-340.
Примеры
Приводимые ниже примеры иллюстрируют настоящее изобретение, но не ограничивают его. Примеры только описывают способ применения настоящего изобретения. Количества ингредиентов, представленные в виде процентов от массы фармацевтической композиции в каждом из примеров, приведены ниже.
Пример 1. План исследования, статистический анализ и результаты исследования в Бордо
Пациенты
Пациенты, включенные в настоящее исследование, находятся на хронической стадии или на прогрессирующей стадии ХМЛ. Они поступили из Отделения гематологии и болезней крови Центрального госпиталя Бордо и из Института Bergonie Регионального онкологического центра. Всех пациентов лечат перорально стандартными дозами иматиниба мезилата (т.е. 400 мг или 600 мг один раз в сутки для пациентов на хронической стадии или на прогрессирующей стадии ХМЛ, соответственно) на протяжении по меньшей мере 12 месяцев. При исследовании группы пациентов анализируют образцы крови, которые отбирали в интервале от июня 2004 г. по март 2006 г. для выявления связи между концентрациями иматиниба в плазме и ответом на лечение. Исключающими критериями являются начало лечения иматинибом мезилатом менее чем за один год до исследования, бластный криз до лечения иматинибом мезилатом или во время такого лечения, отбор крови, произведенный за пределами времени, когда была минимальная концентрация, плохая податливость лечению, идентификация генной мутации (мутаций) в домене киназы в Bcr-Abl.
Количественная характеристика ответа на лечение
Цитогенетический ответ на лечение иматинибом мезилатом оценивают, используя традиционный цитогенетический анализ метастазов костного мозга. Цитогенетические иммунокомпетентные клетки определяют в качестве имеющих ПЦО, т.е. 0% метастазов, положительных по филадельфийской хромосоме, по меньшей мере среди 25 клеток на стадии метафазы в аспирате костного мозга (Colombat M., Fort M.P., Chollet С. и др. Haematologica 91, 2006, cc.162-168). Используют реального времени количественную обратной транскриптазы полимеразную цепную реакцию для оценки уровней транскрипта BCR-ABL и количественной оценки молекулярного ответа (Colombat M., Fort M.P., Chollet С. и др. Haematologica, 91, 2006, cc.162-168). Вкратце, отбирают периферийную кровь, обработанную антикоагулянтом EDTA, для экстракции РНК с последующим проведением реального времени количественной обратной транскриптазы полимеразной цепной реакции. Общую РНК экстрагируют из клеток периферической крови пациентов стандартными методами. Подсчет транскриптов BCR-ABL проводят с учетом ранее сделанных рекомендаций по координации результатов (Hughes Т.Р., Hochhaus А. и др., Blood 108, 2006, cc.28-37). В связи с этим получают результаты, используя ген ABL в качестве генного контроля, и выражают в виде процента BCR-ABL/ABL. Стандартизированный базовый уровень подсчитывают, измеряя соотношение BCR-ABL/ABL у 40 пациентов с ХМЛ на хронической стадии в крови, собранной до начала какого-либо лечения. Для каждого образца используют такой базовый уровень для оценки и измерения уменьшения транскрипта BCR-ABL. ГМО определяют в качестве уменьшения уровня транскрипта BCR-ABL по меньшей мере на три порядка, через 12 месяцев лечения иматинибом мезилатом (Hughes Т.Р., Kaeda J., Branford S. и др., New Engl. J. Med. 349, 2003, сс.1423-1432).
Количественная оценка концентраций иматиниба в плазме
Образцы крови для количественного анализа иматиниба отбирают на стадии устойчивости в интервале 21-27 ч после последнего введения лекарственного средства. Концентрации иматиниба в плазме определяют, используя высокоэффективную жидкостную хроматографию в сочетании с электроспрей-ионизирующей тандемной масс-спектрометрией (Titier К., Picard S., Ducint D. и др., Ther. Drug Monit., 27, 2005, сс.634-640. [Erratum, Ther. Drug Monit., 27, 2005, с.810.]). Чистые контрольные образцы иматиниба мезилата и его внутреннего стандарта (иматиниба-DS) были любезно предоставлены фирмой Novartis (Rueil-Malmaison, Франция). Образец препарата, представляющий экстракт жидкости жидкостью, приготовляют из 200 мкл плазмы. Затем 5 мкл экстракта закалывают в хроматографическую систему. Блок высокоэффективной жидкостной хроматографии состоит из отдельного модуля Alliance® 2690 (фирма Waters, Милфорд, Массачусетс, США), управляемого компьютерным обеспечением Masslynx®. Иматиниб и иматиниб-D8 разделяют на колонке с обратной фазой (X-Terra® RP18, [100×2,1 мм, 5 мкм], фирма Waters) в градиенте буфера ацентонитрилформиат. Анализ общего времени пробега составляет 6 мин при скорости тока 0,3 мл/мин. Количество иматиниба определяют, используя тандемную масс-спектрометрию (QuattroMicro®, фирма Waters, Милфорд, Массачусетс, США) с электроспрей-ионизирующей границей раздела в режиме положительных ионов. Конусный потенциал устанавливают на 40 вольт для иматиниба и его внутреннего стандарта, а энергию столкновений устанавливают на 30 электрон-вольт для двух соединений. Иматиниб и иматиниб-D8 выявляют в контрольных переключениях множественного отклика. Для количественного подсчета область пика, соответствующую m/z 494,2→394,1 реакции (иматиниб), измеряют относительно области пика m/z 502,2→394,1 реакции (внутренний стандарт). Выявление иматиниба подтверждают вторым специфическим переключением множественного отклика: m/z 494,2→217,2.
Статистический анализ
Для количественных переменных сравнение средних величин двух групп проводят, используя тест Стьюдента или ранговый критерий Уилкоксона по необходимости. При наличии более двух групп применяют дисперсионный анализ или тест Крускала-Уоллиса. Для качественных переменных сравнение пропорций проводят, используя тест χ2 или точный критерий Фишера по необходимости. В устойчивом состоянии вариабельность минимальных концентраций иматиниба в плазме выражают в виде следующих параметров: средние минимальные концентрации в плазме, стандартное отклонение (СО), коэффициент вариаций, средний, первый и третий квартили, максимальные и минимальные величины минимальных концентраций в плазме.
Последовательно сравнивают полный цитогенетический ответ (ПЦО) и главный молекулярный ответ (ГМО) в группах пациентов, отвечающих и не отвечающих на лечение, в связи со средними минимальными концентрациями иматиниба в плазме. Возможную корреляцию выявляют между ГМО и следующими переменными: количественными показателями, например возрастом и шкалой Сокаля; качественными показателями, например полом и группой риска по Сокалю, поздней фазой ХМЛ в начале лечения иматинибом мезилатом, введением интерферона до лечения иматинибом мезилатом, суточным дозовым уровнем иматиниба мезилата. Оценивают какие-либо корреляции между уровнями транскрипции BCR-ABL и временным диапазоном от даты начала лечения иматинибом мезилатом до даты оценки молекулярного анализа.
Анализ ROC-кривой проводят с применением поливариантной логистической регрессионной модели, с учетом пола и возраста, для оценки разделяющего потенциала самых низких концентраций ГМО в плазме и для выявления пороговой величины в плазме для оптимальной чувствительности и специфичности. Результаты выражают в виде соотношения вероятностей, 95% доверительного интервала, величины Р теста Вальда.
Двусторонние величины Р устанавливают для статистических тестов (Р<0,05 показанной значимости). Все анализы выполняют, используя программное обеспечение SAS Software (версия 9.1, Кэри, Северная Каролина, США).
Пациенты, включенные в исследование
В исследовании анализируют девяносто пять пациентов с ХМЛ, которые согласились в нем участвовать. Одного пациента исключают, поскольку у него обнаружен бластический кризис. Двадцать пять пациентов исключают, поскольку у них ненадлежащим образом были взять анализы крови, т.е. образцы для определения минимальной концентрации иматиниба были взяты за пределами установленных временных рамок. Одного пациента исключают, поскольку у него установили слабую реакцию на лечение иматинибом: у этого пациента нет кроветворного ответа и уровни иматиниба в плазме ниже 10 нг/мл. Одного пациента исключают, поскольку мутация G250E была идентифицирована в домене киназы в Bcr-Abl. В итоге, 68 пациентов с ХМЛ включают в исследование. 50 пациентов и 18 пациентов лечат дозами 400 мг и 600 мг иматиниба, соответственно, один раз в сутки.
Вариабельность минимальных уровней иматиниба в плазме среди пациентов
Вариации минимальных концентраций иматиниба в плазме для каждой суточной дозы иматиниба мезилата (400 мг и 600 мг) показаны в табл.1.1. Эти концентрации иматиниба в высокой степени переменчивы и варьируют от 181 до 2947 нг/мл, подтверждая высокую вариабельность у субъектов минимальных концентраций иматиниба в плазме для определенной суточной дозы, описанную ранее у субъектов (Peng В., Hayes М., Resta D. и др. J. Clin. Oncol. 22, 2004, cc.935-942).
|
Описание пациентов в связи с ответами на лечение иматинибом
Из 68 пациентов с ХМЛ, включенных в исследование, 56 достигают ПЦО после по меньшей мере одного года лечения. Средняя величина (±СО) минимальных концентраций иматиниба в плазме составляет 1123,3±616,6 нг/мл и 694,2±556,0 нг/мл у пациентов с ПЦО (56 пациентов) и без ПЦО (12 пациентов), соответственно (Р=0,02). Молекулярные ответы в связи с основными свойствами 68 пациентов с ХМЛ, классифицированных в зависимости от наличия или отсутствия ГМО, суммированы в табл.1.2. Средние минимальные концентрации иматиниба в плазме существенно выше в группе с ГМО (1452,1±649,1 нг/мл), чем в группе без ГМО (869,3±427,5 нг/мл, Р<0,001). Существенные различия отсутствуют при ежедневном дозировании иматиниба мезилата у пациентов с ГМО или без ГМО. Кроме того, главный молекулярный ответ (ГМО) на иматиниб не связан со следующими свойствами: клиническими данными (возрастом, полом), поздней фазой ХМЛ при начале лечения иматинибом мезилатом, введением интерферона до начала лечения иматинибом мезилатом. Кроме того, ГМО на лечение статистически не связан со временем от начала лечения иматинибом метилатом до молекулярного анализа: средние величины (±СО) составляют 986±427 суток и 966,5±560 суток в группах с ГМО или без него, соответственно (Р=0,87). Четыре группы пациентов сравнивают по дате проведения молекулярного анализа (используя тест Крускала-Уоллиса): в пределах 560 суток от начала лечения иматинибом мезилатом (16 пациентов), 560-900 суток после начала лечения иматинибом мезилатом (18 пациентов), 900-1325 суток после начала лечения иматинибом мезилатом (17 пациентов), более 1325 суток после начала лечения иматинибом мезилатом (17 пациентов). Исследование показывает, что нет существенной разницы в уровнях транскрипции BCR-ABL между указанными четырьмя группами (Р=0,48). Таким образом, в проведенных исследованиях степень молекулярного ответа не зависит от времени.
|
Пороговая минимальная концентрация иматиниба в плазме для главного молекулярного ответа
С помощью анализа ROC-кривой концентрация - эффект исследуют разделяющий потенциал самых низких концентраций ГМО в плазме (фиг.1). Для ГМО площадь под ROC-кривой составляет 0,775 при наилучшей чувствительности (76,5%) и специфичности (70,6%) при пороговом значении в плазме 1002 нг/мл иматиниба. Этот порог 1002 нг/мл в значительной степени связан с наличием ГМО (подобранное соотношение вероятностей 7,83; 95% доверительный интервал, 2,58-23,76; Р<0,001). Рамки-диаграммы минимальных концентраций иматиниба в плазме показывают дисперсию вокруг среднего значения (фиг.2) для пациентов с ГМО (34 пациента, средняя величина = 1350,2 нг/мл) и без ГМО (34 пациента, средняя величина = 885,5 нг/мл). В группе пациентов с ГМО у 26 пациентов (76,5%) из 34 пациентов концентрации иматиниба в плазме превышают порог 1002 нг/мл, и нет пациентов, у которых концентрации иматиниба в плазме ниже 493,6 нг/мл (1 мкМ/л), что является первоначально описанной целевой концентрацией, требуемой для гибели BCR-ABL-положительных клеток in vitro. В группе пациентов без ГМО у 24 пациентов (70,6%) из 34 пациентов концентрации иматиниба в плазме ниже порога 1002 нг/мл, а у 7 пациентов (20,6%) концентрации иматинаба в плазме ниже 493,6 нг/мл (1 мкМ/л).
Пример 2. Анализ данных исследования IRIS
В этом исследовании было установлено, что минимальные уровни иматиниба в плазме в устойчивом состоянии после первого месяца лечения (29 сутки) являются важным ковариантом для долгосрочных клинических ответов у пациентов с ХМЛ.
Вариабельность от применения иматиниба имеет клинические применения. Достижение ПЦО представляет обоснованное замещение клинической пользы при ХМЛ и соответствующее измерение первоначальной антилейкозной эффективности. Время до достижения ГМО и ПЦО у пациентов с ПЦО различается у разных пациентов с разными экспозициями иматиниба в плазме, объединенными в квартили (р<0,025). Пациенты, достигшие ПЦО за один год, имеют концентрации иматиниба в устойчивом состоянии через один месяц, и они выше, чем концентрации у тех, кто не достиг ПЦО. В целом величины Смин статистически существенно выше у пациентов, достигших ПЦО во время исследования (в среднем 1009 нг/мл против 812 нг/мл). Таким образом, поддержание минимального уровня иматиниба равным или примерно равным 1000 нг/мл может быть важно для ПЦО. Этот результат согласуется с пороговым значением примерно 100 нг/мл, указанным в примере 1.
К этому исследованию были допущены пациенты, которые придерживаются лечения иматинибом на протяжении и первого, и последующих месяцев. Однако отсутствуют вариации, хотя пациенты придерживались предписанного дозирования на протяжении этого исследования. Хотя приверженность терапии является принципиальным параметром для точности и обоснованности фармакокинетического анализа, было резонно предположить, что уровни в плазме иматиниба и CGP74588 находились в равновесном состоянии на 29 сутки в этом хорошо контролируемом клиническом исследовании. У пациентов, вовлеченных в это исследование, было вновь диагностировано смертельно опасное заболевание и на ранних стадиях лечения - т.е. на такой стадии, когда происходит интенсивное потребление суточных доз иматиниба, относительно хорошо переносимого лекарственного средства. Существенное количество отказов от применения назначенного врачом иматиниба в этом исследовании маловероятно для большинства пациентов. Из 553 пациентов, вовлеченных в группу лечения иматинибом, примерно у 20% пациентов повышают клиническую дозу до 600-800 мг/сутки, причем среднее время повышения дозы составляет 22 месяца (данные не представлены). Нестрогое соблюдение применения иматиниба подтверждено документально у пациентов с ХМЛ и может существенно повлиять на клинические ответы и корреляцию между клиническим ответом и фармакокинетическими экспозициями.
Была установлена корреляция между степенью ГМО и экспозицией иматинибом. Установленная степень ГМО была существенно ниже у пациентов с низкими уровнями иматиниба; только 25% всех пациентов с уровнями иматиниба <647 нг/мл достигают ГМО к 1 году лечения, а 40% пациентов с более высокими уровнями иматиниба достигают этого ответа в течение 1 года. К 4 годам у 53% пациентов в Q1 достигается ГМО несмотря на низкие уровни иматиниба в устойчивом состоянии (29 сутки) по сравнению с пациентами в Q4 (и 72% пациентов в диапазоне промежуточного квартиля - IQ). ГМО является прогностическим фактором долгосрочной эффективности и выживаемости. У пациентов, утративших ГМО, повышена скорость прогрессирования заболевания. Ранний подбор дозы для пациентов с низкими уровнями устойчивого состояния иматиниба может повысить время эффективного действия.
Помимо степеней ответа ПЦО и ГМО, фармакокинетика минимального уровня иматиниба представляется тем параметром, который коррелирует с бессобытийной выживаемостью, хотя не установлено статистически значимого различия. Бессобытийное выживание является усложненным событием, которое может быть нарушено под воздействием разных факторов, например доступностью других способов лечения, повышения дозы в организме пациента на поздней стадии лечения и т.д. Однако пациенты с низким уровнем иматиниба обладают плохим показателем бессобытийной выживаемости по сравнению с пациентами с повышенными уровнями иматиниба. Подтвердилось предположение о том, что экспозиция иматинибом коррелирует со степенью прерывания. Пациенты в более низком квартиле обладают самой высокой степенью прерывания по сравнению с промежуточным и повышенным квартилями. Интересно отметить, что одна из причин прерывания связана с неудовлетворительным терапевтическим эффектом, совместимым с результатами корреляционного анализа между клиническим ответом (ПЦО или ГМО) и концентрациями иматиниба в квартиле.
В настоящем изобретении описаны уровни устойчивого состояния. Сходные результаты наблюдают для главного действующего метаболита, CGP74588. Однако, оценивая относительно малый вклада метаболита в экспозицию иматиниба (<20%), измерение исходного лекарственного средства в плазме представляет главный действующий компонент для проявления биологического действия. Если метаболизм иматиниба изменяется, например, за счет ингибитора или индуктора CYP, может потребоваться измерение и иматиниба, и метаболита.
Таким образом, экспозиция иматинибом при устойчивом состоянии в плазме, измеренная через первый месяц лечения стандартной дозой 400 мг, коррелирует с длительными цитогенетическим и молекулярным ответами. К демографическим показателям пациентов относятся возраст, пол, масса тела, которые минимально влияют на экспозицию иматиниба плазмы, учитывая высокую степень вариабельности экспозиции между пациентами. Поддержание минимальных уровней в плазме, равных или выше средней концентраций в группе, примерно равной 1000 нг/мл, может быть важно для ответа в виде полного цитогенетического ответа (ПЦО) и главного молекулярного ответа (ГМО), бессобытийной выживаемости и удовлетворительной терапевтической эффективности у пациентов с ХМЛ-ХФ. Какие-либо факторы, которые могут повлиять на экспозицию иматиниба, например всасывание лекарственного средства, метаболизм и взаимодействия с ранее проводившимся лечением, могут повлиять на способность достигать максимального терапевтического эффекта. Информация по экспозиции иматиниба в крови во время лечения имеет важное значение, выступая в роли перспективного инструмента для лечения и заслуживая признания.
Методы
Пациенты, включенные в настоящее исследование, были зарегистрированы в программе IRIS и рандомизированно определены для первоначального лечения иматинибом в дозе 400 мг/сутки. Схема исследования и характеристика всех 553 пациентов, рандомизированных для лечения иматинибом, включают возраст, пол, массу тела, площадь поверхности тела, а также результаты, описанные ранее (O'Brien S.G., Guilhot F., Larson R.A. и др.. New Engl. J. Med., 348, 2003, cc.994-1004).
Степени ПЦО (выраженного в виде процента клеток с филадельфийской хромосомой, Ph+, на стадии метафазы по меньшей мере от 20 клеток) в исследуемой группе и главного молекулярного ответа (ГМО, выраженного в виде снижения не менее чем на три порядка соотношения BCR-ABL/BCR относительно стандартизированного исходного уровня) у субъектов, достигших ПЦО, были описаны ранее (O'Brien S.G., Guilhot F., Larson R.A. и др., New Engl. J. Med., 348, 2003, cc.994-1004).
Настоящий пример сосредоточен на тех 351 пациентах, у которых были доступны показатели измерений РК. Бессобытийную выживаемость оценивают до 5 лет и фиксируют по регистрации клинических исследований до тех пор, пока не произойдут следующие события: смерь по какой-либо причине, потеря ГМО, потеря полного кроветворного ответа или прогрессирование до поздней или бластической фазы. Живых пациентов контролируют пожизненно. Показатель ПЦО оценивают до 5 лет. Достижение ГМО анализируют только на протяжении 24 месяцев после начала лечения из-за недостаточности данных после этого срока. Распределение пациентов (с доступной информацией РК) и причины прекращения лечения через 5 лет сводят в таблицу, при этом учитывают перекрестные или другие схемы лечения, побочные эффекты, неудовлетворительный лечебный результат и другие причины (включая нарушение методики, исчезновение потребности в продолжении исследования (из-за пересадки костного мозга), нарушение протокола, отзыв субъектом согласия на участие в исследовании, невозможность доведения исследования до конца и смерть).
Анализ фармакокинетических образцов
Образцы крови отбирают перед дозированием иматиниба на 2 сутки (т.е. через 24 ч после первой дозы) и еще раз на 29 сутки (устойчивый низкий уровень). Концентрации иматиниба и CGP74588 в плазме определяют жидкостной хроматографией и тандемной масс-спектрометрией (LC/MS/MS). Предел подсчета составляет 5 нг/мл и для иматиниба, и для CGP74588; исследование полностью подтверждено (Bakhtiar R., Lohne J., Ramos L. и др. в J Chromatog В Anal Technol Biomed Life Sci, 768, 2002, cc.325-340). Достоверность составляет 104%±6% при нижнем пределе подсчета и от 99%±5% до 108%±5% на протяжении всего диапазона концентрации 4-10000 нг/мл.
Анализ данных
Минимальные концентрации в плазме (Смин) иматиниба и его метаболитов после первой дозы при устойчивом состоянии анализируют и проводят анализ корреляций соответственно с клиническими ответами, включая ПЦО и ГМО, а также распределение пациентов после 2 и 5 лет лечения. Оценивают корреляцию фармакокинетических минимальных уровней с возрастом, полом, массой тела и площадью поверхности тела. Минимальные уровни в плазме и иматиниба, и CGP74588, на 2 и 29 сутки группируют в четыре квартиля. К низшему квартилю (Q1) относятся данные 25% пациентов, у которых были установлены самые низкие величины концентраций, а к квартилям Q2 и Q3 относятся концентрации на 25% выше и ниже средней величины, соответственно. Верхний квартиль (Q4) включает 25% пациентов с наивысшими концентрациями. Центральные 50% данных, т.е. включающих Q1 и Q4, объединяют во всех анализах и их обозначают промежуточными квартилями (intermediate qualities - IQ). Эти три группы (Q1, IQ и Q4) используют для соответствующей стратификации. Степени цитогенетического и молекулярного ответа определяют, используя метод Kaplan-Meier, и слои исследуют, сравнивая с логарифмическим ранговым критерием. Корреляцию между минимальными уровнями и демографическими переменными оценивают с помощью коэффициента ранговой корреляции Спирмена.
Результаты
Демографические показатели и минимальные уровни в плазме иматиниба и его метаболитов
Фармакокинетические данные получают в общей сложности от 351 пациента (221 мужчина и 130 женщин). Средняя масса тела составляет 85,9±16,8 (СО) кг для мужчин (средняя величина 83,6, варьирование от 52,9 до 163,3) и 72,4±18,1 кг для женщин (средняя величина 68,9, варьирование от 40,0 до 133,0). Средняя площадь поверхности тела составляет 2,0±0,2 м для мужчин (средняя величина 2,0, варьирование от 1,53 до 2,8) и 1,8±0,2 м для женщин (средняя величина 1,75, варьирование от 1,35 до 2,54). Средний возраст в группе составляет 50 лет (варьирует от 18 до 70 лет). Из 351 пациента, которые имели поддающиеся оценке образцы в фармакокинетическом субисследовании, 238 остаются в исследовании (67,8%), 10 пересекаются (2,8%), 113 (32,2%) прекращают принимать иматиниб из-за недостаточного лечебного эффекта (n=51, 14,5%), побочных эффектов (n=15, 4,3%), гибели (n=6, 1,7%), трансплантации костного мозга (n=11, 3,1%), отзыва пациентом согласия на участие в исследовании (n=15, 4,3%) или других причин, например нарушения процедур, отклонения от протокола, невозможности доведения исследования до конца, административных проблем (n=15, 4,3%).
После первой дозы 400 мг 24-часовые концентрации иматиниба и CGP74588 составили 517,7±369,6 нг/мл и 82,7±47,4 нг/мл, соответственно. На 29 сутки концентрации иматиниба и CGP74588 составили 979,0±529,6 нг/мл и 241,9±105,5 нг/мл, соответственно; соотношение концентрации метаболита к концентрации лекарственного средства составляет 0,268±0,085 (n=351). Основываясь на минимальных уровнях на 2 и 29 сутки у того же субъекта, степени накопления ко времени наступления устойчивого равновесия составляют 2,21±1,15 для иматиниба и 3,38±1,54 для CGP74588. Распределение концентраций иматиниба в устойчивом состоянии показано на фиг.4. 19 пациентов на 29 сутки с уровнями >2000 нг/мл для анализа включают в 4-й квартиль.
Минимальный уровень иматиниба несколько выше у женщин по сравнению с мужчинами (1078±514,5 нг/мл против 921±530,8 нг/мл, соответственно, разница составляет 17,2%), вероятно из-за разницы массы между полами (18,7%). Минимальные уровни в плазме для метаболита CGP74588 дают сходный результат, хотя соотношение метаболит/исходное лекарственное средство одинаково у самцов и самок. Наблюдают слабую корреляцию между устойчивыми минимальными уровнями иматиниба и массой тела (r2=0,015) или площадью поверхности тела (r2=0,038), что показано на фиг.5. Учитывая простую линейную взаимосвязь между массой тела и минимальным уровнем, повышение массы тела с 40 кг до 120 кг может привести к ожидаемому снижению минимального уровня примерно до 280 нг/мл. Также наблюдают слабую корреляцию между минимальными уровнями (соотношение метаболита/исходного лекарственного средства) и возрастом пациентов (r2=0,02). Также, учитывая упрощенное допущение линейной взаимосвязи, минимальные уровни иматиниба повышаются на 295 нг/мл по мере увеличения возраста с 20 лет до 70. Однако из-за большой вариабельности уровней иматиниба в плазме между пациентами следующие показатели: возраст, пол, масса тела и площадь поверхности тела, вероятно, не имеют клинического значения в связи с экспозицией иматиниба.
Корреляция экспозиции фармакокинетических показателей с клиническими ответами
Табл.2 приводит минимальные устойчивые уровни иматиниба, CGP74588 и их соотношение, сгруппированные по квартилям. Минимальные экспозиции в Q2 и Q3 объединяют в IQ для представления 50% группы. Фиг.6 (верхняя панель) показывает, что степень ответа ПЦО через 5 лет существенно различается по квартилям с разными минимальными уровнями иматиниба (р=0,0125). Различие в основном свойственно нижнему значению ПЦО в группе Q1 (р=0>005, Q1 против других квартилей). Сходную тенденцию наблюдают для степеней ГМО через 2 года относительно уровней экспозиции в плазме при устойчивом состоянии. Пациенты в Q1 имеют меньшую степень ГМО по сравнению с другими объединенными группами, хотя отсутствует статистически значимое различие среди трех отдельных групп квартилей (р=0,08). Минимальная экспозиция иматиниба у пациентов, которые в итоге достигли ПЦО, существенно выше соответствующих показателей у тех пациентов, которые не достигли ПЦО, 1009±544 нг/мл против 812±409 нг/мл, соответственно (р=0,01, фиг.7). Не наблюдают существенного различия в фармакокинетической экспозиции у пациентов, у которых вырабатывается ГМО, и тех, у которых ГМО не вырабатывается. Статистический анализ степени ГМО проводят до 24 месяцев из-за ограниченного количества данных.
Наблюдают тенденцию к связи бессобытийной выживаемости с минимальными уровнями иматиниба, а именно, относительно низкая величина бессобытийной выживаемости в группе Q1 по сравнению с другими квартилями. Однако не получают статистически значимых отличий исходя из имевшихся данных (фиг.8). Наблюдают сходную тенденцию корреляции между распределением пациентов (или прерыванием) и фармакокинетических минимальных уровней (табл.2.3). Через 2 года число пациентов, продолжающих лечение, было низким в группе Q1, 75,9%, по сравнению с 84,3% и 89,5% в группах IQ и Q4.
Через 5 лет число пациентов, продолжающих лечение, было 58,6%, 72,5%, и 76,7% в группах Q1, IQ и Q4, соответственно. Основная причина прерывания исследования связана с неудовлетворительным лечебным эффектом, 10,3%, 6,2% и 4,7% в группах Q1, IQ и Q4, соответственно, через 2 года лечения, и 18,4%, 14,6% и 8,1%, соответственно, через 5 лет лечения. Наблюдают примерно равные степени побочных эффектов или прерывания лечения в связи со смертью в разных квартилях через 2 и 4 года лечения. В группе Q4 не было пациентов, пересекавшихся в лечении с другими схемами лечения (группа лечения интерфероном), по сравнению с 4,6% и 3,4% в группах Q1 и IQ, соответственно. Одновременное другое лечение наблюдают преимущественно в первый или второй год после начала лечения.
Клинический ответ (ПЦО, ГМО или выживание) или распределение пациентов также коррелирует с минимальными уровнями метаболита CGP74588, поскольку уровни исходного лекарственного средства в высокой степени коррелируют (0,76, коэффициент ранговой корреляции Спирмена). Минимальный уровень в плазме после 1-й дозы также показывает корреляцию с ответами ПЦО и ГМО, но его рассматривают в качестве менее прогностического параметра по сравнению с минимальным уровнем при устойчивом состоянии.
|
|
|
Мутантные антигены gas57 и антитела против gas57
[(1н-индол-5-ил)-гетероарилокси]-1-(азабицикло[3.3.1]нонаны, как холинергические лиганды n-achr, предназначенные для лечения психотических и нейродегенеративных нарушений
Лечение туберозного склероза
Органические соединения
Способ получения фармацевтической композиции
Предназначенная для перорального применения фармацевтическая композиция
Менингококковые полипептиды fhbp
Стабилизированные полипептиды инсулиноподобного фактора роста
Органические соединения
Курс лечения с использованием агониста рецептора s1p

