Результат интеллектуальной деятельности: Способ получения солей разветвлённого олигогексаметиленгуанидина, имеющих степень чистоты, достаточную для их применения в качестве фармацевтической субстанции
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Изобретение относится к области органической химии полимеров и приготовлению лекарственных препаратов. Более конкретно, изобретение обеспечивает способ получения солей разветвлённого олигогексаметиленгуанидина (ОГМГ), имеющих степень чистоты, достаточную для их применения в качестве фармацевтических субстанций.
Уровень техники
Продукты конденсации гуанидина и некоторых его производных с гексаметилендиамином, а также способы их получения известны достаточно давно. Так, например, в примере 1описания изобретения к патенту US 2325586 (опубл. 03.08.1943) описаны способ получения и свойства гидробромида полигексаметиленгуанидина линейной структуры, что представлено структурной формулой
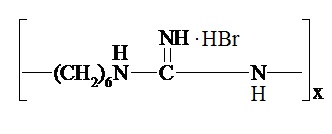 .
.
Способ предполагает взаимодействие гексаметилендиамина с бромцианом в абсолютном этаноле при кипении в течение 1 часа. С целью повышения степени полимеризации полученный после выпаривания этанола продукт нагревают до 170°С при остаточном давлении 2 мм. рт. ст. Указано, что гидробромид полигексаметиленгуанидина может быть переведён в форму основания осаждением щёлочью в водном растворе. Полученный продукт обезвоживают растворением в абсолютном этаноле с последующей отгонкой растворителя при 100°С при пониженном давлении.
В качестве основной области применения указаны текстильно-вспомогательные вещества, обеспечивающие повышение накрашиваемости различными синтетическими красителями, а также придание готовому текстильному продукту водоотталкивающих свойств. Существенным недостатком способа является применение ядовитых цианида натрия и брома, из которых получают не менее ядовитый бромциан.
В патенте SU 1808832 (опубл. 15.04.1993) раскрыт способ получения полимера гексаметиленгуанидина, предусматривающий смешивание гуанидингидрохлорида и гексаметилендиамина в молярном соотношении 1:(1,2-1,5), нагревание смеси при 150-160°С в течение 10 часов с последующим охлаждением до получения пенообразной твёрдой массы, измельчение и отмывку водой. Продукт обладает ионообменными свойствами при сохранении бактерицидности, что показано на примере E. Coli. Областью применения является водоочистка и водоподготовка.
Проблемой при получении полигексаметиленгуанидиновых соединений является очистка от токсичных веществ, особенно от гексаметилендиамина (ГМДА). Из уровня техники известны следующие способы синтеза, предусматривающие такую очистку.
В патенте RU 2052453 (опубл. 20.01.1996) раскрыт способ получения дезинфицирующего средства, содержащего гидрохлорид полигексаметиленгуанидина (ПГМГ). Способ включает следующие стадии: (а) предварительное получение гидрохлорида гуанидина (ГХГ) сплавлением дициандиамида (цианогуанидина) с хлористым аммонием, (б) его конденсацию с ГМДА, (в) очистку продукта конденсации от токсичного ГМДА, которая предусматривает его перевод в форму основания ПГМГ действием концентрированного раствора щёлочи и многократную промывку водой, и (г) обработку эквивалентным количеством органической или неорганической кислоты в виде концентрированного раствора или растирание его с водной суспензией соответствующей малорастворимой кислоты для получения соответствующей соли ПГМГ.
Недостатками метода является снижение выхода ГХГ на первой стадии за счёт образования значительных количеств гидрокси(амино)-симм-триазинов (аммелина и аммелида), а также потери до 20% основания при промывке водой на стадии (в). Кроме того, незначительные отклонения параметров технологического режима (температуры, скорости подачи реагентов, концентрации, интенсивности перемешивания) существенно влияют на выход и свойства получаемого дезинфицирующего средства.
В патенте RU 2170743 (опубл. 20.07.2001) представлен способ получения дезинфицирующего средства, который осуществляют конденсацией ГМДА с производными гуанидина в расплаве с образованием полимеров гексаметилендигуанидина неразветвлённого строения. В соответствии с первым вариантом процесс конденсации проводят в течение 1-2 ч при 180-200°С и мольном отношении ГМДА к производному гуанидина 1:(1,2-2,0). Один из реагентов используют в виде его соли. Дальнейшую очистку продукта проводят сначала растиранием или перекристаллизацией в избытке той неорганической кислоты, соль ГМДА или производного гуанидина которой была использована в реакции конденсации, а затем выделенный продукт промывают этиловым спиртом.
Второй вариант указанного способа предусматривает применение дигидрохлорида гексаметилендигуанидина, полученного в соответствии с первым вариантом, в качестве полупродукта для повторного взаимодействия с ГМДА. Конденсацию проводят в течение 1-2 ч при 180-200°С и мольном отношении ГМДА к производному гуанидина 1:1,2. Полученный продукт обрабатывают основанием или неорганической солью при 20-120°С и соотношении продукт/основание (соль) 1,0:(0,5-4,0), и проводят дальнейшую очистку водой. Описание изобретения не даёт оснований допустить возможность применения получаемых продуктов в качестве фармацевтической субстанции.
В патенте RU 2223791 (опубл. 20.02.2004) рассматривается способ получения дезинфицирующего средства конденсацией в расплаве гексаметилендиамина и производного гуанидина в течение 1-2 ч при 180-200°С и мольном соотношении гексаметилендиамина к производному гуанидина, равном 1:(1,2-2) соответственно, с дальнейшей очисткой и выделением готового продукта в солевой форме, отличающийся тем, что очистку готового продукта проводят в среде жидкой органической кислоты с введением эквивалентного количества соли щелочного металла органической кислоты, с дальнейшим удалением неорганической соли.
Также в соответствии с рассматриваемым патентом предложен способ получения дезинфицирующего средства конденсацией в расплаве гексаметилендиамина и дигидрохлорида гексаметилендигуанидина в течение 1-2 ч при 180-200°С при мольном соотношении гексаметилендиамина к дигидрохлориду гексаметилендигуанидина, равном 1:1,2 соответственно, с дальнейшей обработкой готового продукта неорганическим основанием, его очисткой и выделением в солевой форме, отличающийся тем, что очистку основания осуществляют экстракцией органическим растворителем, выбранным из алифатического и арилалифатического спирта, а готовый продукт обрабатывают эквивалентным количеством органической или неорганической кислоты.
Недостатком продуктов, в частности, гидросукцината полигексаметиленгуанидина, полученного по второму способу патента, является недопустимое высокое содержание общей и сульфатной золы, достигающее 5% (масс.) и выше. Кроме того, токсичность продукта, обусловленная присутствием более 1% (масс.) остаточного высокотоксичного ГМДА, слишком высока.
В качестве ближайшего аналога авторы настоящего изобретения рассматривают техническое решение, предложенное в патенте RU 2443684 (опубл. 27.02.2012), а именно способ олигомеризации гидрохлорида гуанидина (ГХГ) и гексаметилендиамина (ГМДА), осуществляемый при мольных соотношениях ГМДА/ГГ от 1,00:1,0 до 1,00:1,20 в интервале температур реакции от 180 до 230°С в течение от 3 до 12 часов. Полученные поликонденсацией разветвлённые производные олигогексаметиленгуанидина содержат от 0,5 до 2,0% исходных мономеров, в том числе высокотоксичного ГМДА, что препятствует их использованию в качестве фармацевтической субстанции.
В описании изобретения указано, что промежуточное получение оснований ОГМГ может быть использовано для очистки продуктов олигомеризации от содержащихся в них токсичных и коррозионно-активных ГМДА и ГХГ. Для этого к раствору гидрохлорида ОГМГ добавляют 50% мольный избыток концентрированного раствора щелочи, например NaOH, до pH порядка 12 и отбирают верхний слой реакционной массы в виде вязкой мутной жидкости белого цвета. Полученный продукт содержит суспензию основания ОГМГ и до 1% (масс.) ГМДА, что также недопустимо много для фармацевтической субстанции.
Получение основания также можно проводить в присутствии спирта, такого как этанол или изопропанол, взятого в количестве 0,5-1,0 объема от общего объема реакционной массы, состоящей из равных объемов приблизительно 50% водных растворов олигомера и щелочи. Декларируется, что в этом случае остаточное содержание ГМДА можно снизить до 0,02-0,1% (масс.) в зависимости от времени и температуры проведения процесса, а также соотношения реагентов и спирта, однако это заявление не подтверждено примерами и данными анализа.
Предлагаемый в патенте RU 2443684 способ очистки и одновременного замещения либо даёт недостаточное замещение исходного гидрохлорида олигогексаметиленгуанидина (2-5% остаточного хлора) при использовании близком к эквивалентному количеству щёлочи, либо высокое содержание золы (2-5% сульфатной золы) при использовании существенного избытка щёлочи и достижения приемлемой степени замещения (достижения менее 1% остаточного хлора).
Таким образом, существует потребность в способе получения солей разветвлённого олигогексаметиленгуанидина (ОГМГ) с пониженным содержанием балластных и токсичных веществ и степенью чистоты, достаточной для их применения в качестве фармацевтической субстанции.
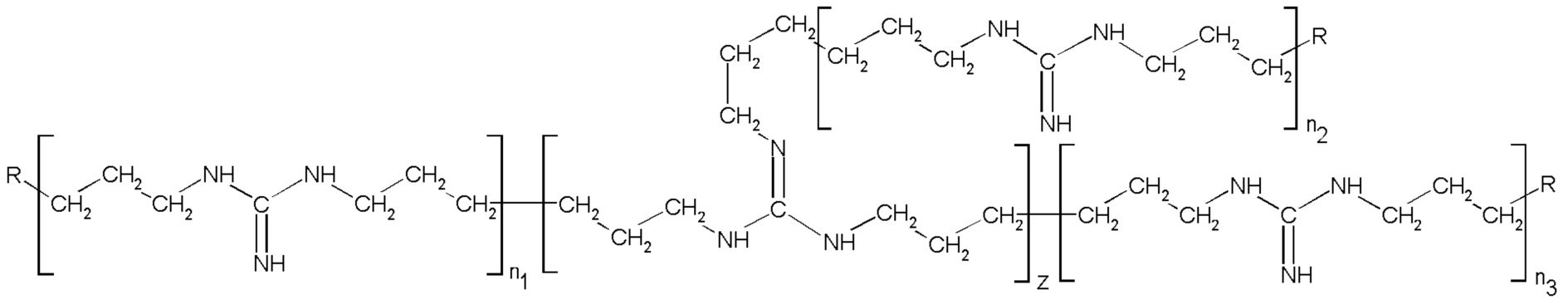
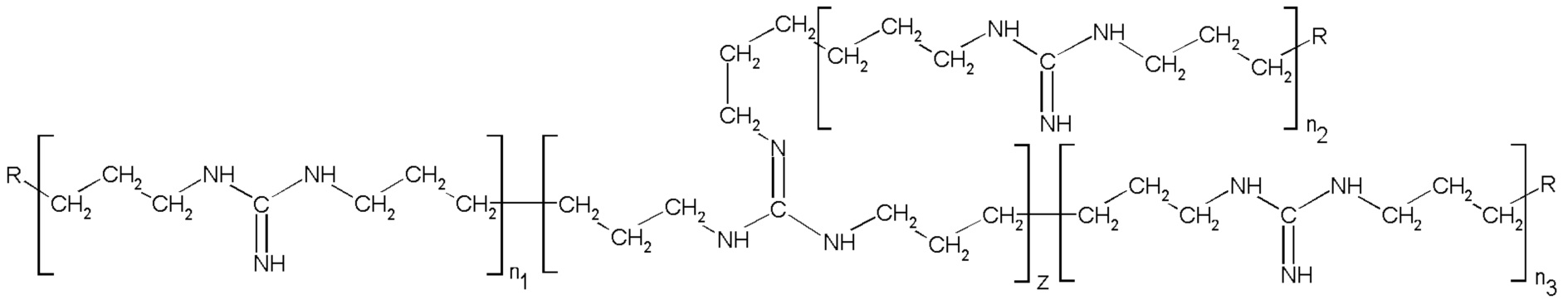
В результате проведённых обширных исследований авторы настоящего изобретения предлагают способ получения соли разветвлённого олигогексаметиленгуанидина (ОГМГ), представленного общей формулой
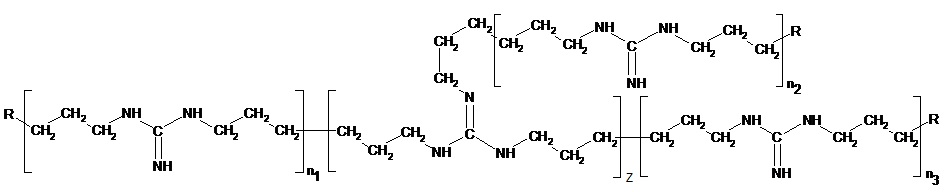 где R представляет
где R представляет
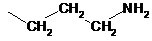 или
или 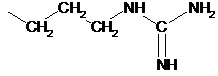 ,
,
а n1, n2 и n3 равны 1-3, z равно 0,15-1,10 при среднечисловой молекулярной массе Mn в интервале от приблизительно 600 до 1100, выбранной из гидросукцината, гидрохлорида, гидроцитрата, гидросалицилата, гидросульфосалицилата, имеющей чистоту, достаточную для её применения в качестве фармацевтической субстанции, который включает следующие стадии:
а) синтез технического гидрохлорида ОГМГ (ОГМГ-ГХ) поликонденсацией гексаметилендиамина (ГМДА) с гидрохлоридом гуанидина (ГХГ) при мольном соотношении от 1,0:1,0 до 1,0:1,2 и температуре 180-210°С;
б) растворение технического ОГМГ-ГХ в воде с получением 40-60% (масс.) водного раствора;
в) прибавление 1 объёмной части водного раствора технического ОГМГ-ГХ к 4-6 объёмным частям спиртового раствора щёлочи, выбранной из гидроксида натрия или гидроксида калия и взятой в количестве 1-4 эквивалентов на 1 эквивалент ОГМГ-ГХ, где спирт выбран из этанола, пропанола или изо-пропанола, с последующим перемешиванием в течение 3-4 часов и выдержкой в течение приблизительно 16 часов до разделения фаз;
г) отделение фазы, содержащей большую часть основания ОГМГ, и прибавление к ней 1-3 эквивалентов щёлочи на 1 эквивалент ОГМГ-ГХ с последующим перемешиванием в течение 3-4 часов и выдержкой в течение приблизительно 16 часов до разделения фаз;
д) отделение фазы, содержащей большую часть основания ОГМГ, и прибавление к ней порций кислоты, выбранной из янтарной, хлористоводородной, лимонной, салициловой или сульфосалициловой, до прекращения выпадения осадка соответствующей неорганической соли с получением раствора технической соли ОГМГ, выбранной из гидросукцината, гидрохлорида, гидроцитрата, гидросалицилата или гидросульфосалицилата;
е) отделение осадка неорганической соли фильтрацией, выпаривание фильтрата, растворение сухого остатка технической соли ОГМГ в 1 массовой части воды, прибавление к полученному раствору 2 массовых частей спирта, выбранного из этанола, пропанола или изо-пропанола, и 1 массовой части хлороформа, перемешивание в течение приблизительно 1 часа, отделение нижней фазы, содержащей очищенную соль разветвлённого ОГМГ, и её выпаривание с получением твёрдой очищенной соли разветвлённого ОГМГ, выбранной из гидросукцината, гидрохлорида, гидроцитрата, гидросалицилата или гидросульфосалицилата и имеющей чистоту, достаточную для её применения в качестве фармацевтической и/или ветеринарной субстанции.
Применение данного способа позволяет преодолеть существенные недостатки известного уровня техники.
Раскрытие сущности изобретения
Техническим результатом применения способа очистки солей разветвлённого олигогексаметиленгуанидина (ОГМГ) в соответствии с настоящим изобретением является достижение чистоты очищенных солей разветвлённого ОГМГ, таких как гидрохлорид, гидроцитрат, гидросукцинат, гидросалицилат и гидросульфосалицилат, по показателям содержания остаточных ГМДА, хлора и сульфатной золы, достаточной для применения очищенных солей в качестве субстанций, пригодных в производстве препаратов медицинского и/или ветеринарного назначения при сохранении высокого выхода конечного продукта. Применение данного способа приводит к снижению токсичности получаемых солей, а также улучшает их дезинфицирующие и/или бактерицидные свойства и воспроизводимость показателей их качества.
Согласно Государственной Фармакопее РФ издание XIV, ОФС.1.1.0006.15 «Фармацевтические субстанции» фармацевтическая субстанция ОГМГ гидрохлорида должна отвечать критериям качества, указанным в таблице 1.
Таблица 1
|
Таблица 1 (продолжение)
|
* Для данных показателей выбраны нормы с более высоким пределом содержания, поскольку результаты исследований безопасности субстанции дают возможность её применения с указанными нормами содержания родственных примесей.
Проблема недостаточного замещения хлора в исходном ОГМГ-ГХ при использовании количества щёлочи, близкого к эквивалентному, либо высокого содержания золы при использовании существенного избытка щёлочи и достижения приемлемой степени замещения хлора в способах очистки и одновременного замещения хлора в соответствии с настоящим изобретением решена следующим образом.
Высокой степени замещения хлора в ОГМГ-ГХ достигают применением двукратного избытка щёлочи в водно-спиртовой среде. К спиртовому раствору приблизительно эквивалентного количества щёлочи прибавляют 50% (масс.) водный раствор ОГМГ-ГХ. По истечении времени реакции осуществляют длительную выдержку реакционной массы для образования двухфазной системы, в которой образовавшееся основание ОГМГ находится в растворе в более лёгкой (верхней) фазе, которую отделяют. Затем её обрабатывают дополнительным количеством щёлочи. По окончании реакции и выдержки реакционной массы повторно разделяют фазы. Далее полученный раствор основания обрабатывают подходящей кислотой, такой как янтарная, лимонная или хлористоводородная. При этом первая часть кислоты идёт на нейтрализацию избытка щёлочи с образованием соли щелочного элемента, которая малорастворима в полученной после выполнения указанных операций водно-спиртовой среде. После полного осаждения указанной соли её отделяют фильтрацией или центрифугированием и к фильтрату продолжают добавлять кислоту до полной нейтрализации. Если в этом случае вновь образуется двухфазная система, то на этот раз отделяют нижнюю фазу, содержащую солевой продукт, которую затем выпаривают досуха предпочтительно при пониженном давлении. Таким приёмом снижают содержание балластных веществ при достижении высокой степени замещения хлора.
Очистку полученного солевого продукта проводят, растворяя его в воде с получением 40-60 % (масс.) раствора и добавляя к раствору спирт (этанол, пропанол или изо-пропанол) массой, близкой к массе раствора, а затем хлороформ. После перемешивания в течение необходимого времени и выдержки образуется двухфазная система, в которой верхняя более лёгкая фаза содержит все компоненты, но основу её составляет вода и золообразующие компоненты. Нижняя более тяжёлая фаза содержит основное количество очищенного солевого продукта ОГМГ, хлороформ, спирт и достаточное количество воды. Нижнюю фазу отделяют и выпаривают досуха предпочтительно при пониженном давлении. Этим достигается степень чистоты солевого продукта ОГМГ, достаточная для его применения в качестве субстанции, пригодной в производстве препаратов медицинского и/или ветеринарного назначения.
Неочевидность полученного результата связана с тем, что соли ОГМГ заметно не растворимы ни в спирте, ни, тем более, в хлороформе. Таким образом, основой предлагаемого способа очистки является сложная система физико-химических взаимодействий в многокомпонентной системе, которую в силу многофакторности трудно или даже невозможно заранее предсказать.
Экспертное определение подлинности, средней молекулярной массы, степени разветвления, а также чистоты полученного продукта основано на данных спектроскопии ЯМР 13С (ГФ XIV, ОФС 1.2.1.1.0007.15). Соотношения интегральных интенсивностей сигналов атомов углерода гексаметиленового и гуанидинового фрагментов, находящихся в концевых и внутрицепных положениях, а также атома углерода гуанидинового фрагмента, являющегося центром разветвления, пропорциональны содержанию концевых остатков гексаметилендиамина и гуанидина, а также количеству разветвлений в молекуле полимера.
Определение остаточных мономеров осуществляют хроматографически методом обращеннофазной ВЭЖХ в ион-парном варианте. Их содержания вычисляют методом линейной градуировочной функции.
Осуществление изобретения
Далее настоящее изобретение будет проиллюстрировано неограничивающими примерами его осуществления, доказывающими достижение технического результата.
Примеры 1-3. Получение технического ОГМГ-ГХ (лабораторный масштаб)
К навеске 9,55-11,46 г (0,1-0,12 моль) гидрохлорида гуанидина (ГХГ), масса которой зависит от мольного соотношения ГМДА/ГХГ при 160°С и интенсивном перемешивании в течение 30 минут добавляют небольшими порциями 11,62 г (0,1 моль) ГМДА. После окончания прибавления температуру быстро повышают до температуры реакции (ТР) и продолжают перемешивание смеси при данной температуре в течение времени реакции (ВР). Состав исходной смеси реагентов и условия проведения реакции приведены в таблице 2.
Таблица 2
|
По истечении ВР расплавленную реакционную выливают на противень, а после застывания и охлаждения до температуры окружающей среды снимают с противня, переносят в ступку, измельчают и высушивают в лабораторном сушильном шкафу при температуре 80°С в течение 3 часов.
Пример 4 Получение технического ОГМГ-ГХ (промышленный масштаб)
Твердый ГМДА в таре (евробочка) выдерживают около 30 минут при температуре 90°С до полного расплавления.
Реакцию проводят в емкостном реакторе с рубашкой и нижним спуском, снабжённом мешалкой, загрузочным и смотровым люками и КИП для измерения температуры реакционной массы. Нагрев осуществляют циркуляцией в рубашке высокотемпературного теплоносителя.
Реактор прогревают до 70-80°С, после чего через загрузочный люк при перемешивании загружают 220 кг жидкого ГМДА, а затем 180 кг твёрдого ГХГ. Температуру постепенно повышают, и при 90-100°С смесь достигает гомогенности. При 110-120°С начинается химическая реакция с выделением аммиака, который направляют в абсорбер, где его поглощают водой.
После начала реакции скорость нагрева уменьшают до 15-20°С/ч до достижения температуры 180-190°С, на что требуется 4-5 часов. При достижении температуры в 140-150°С появляется устойчивая пена, сохраняющаяся до конца реакции. Уровень пены контролируют визуально через смотровой люк реактора. Подъём пены не должен превышать определенного уровня для предотвращения выброса реакционной массы. В зависимости от скорости подъёма пены изменяют интенсивность нагрева. По достижении температуры 190-200°С нагрев уменьшают и поддерживают её на этом уровне в течение 2-3 часов.
После завершения реакции готовый расплав ОГМГ-ГХ сливают через нижний спуск на поддоны из нержавеющей стали для последующего охлаждения на воздухе, сушки и измельчения. Полученный олигомер охлаждают до температуры 20-25°С, на что требуется 2-3 часа, измельчают до частиц размером не более 150 мкм, после чего высушивают в промышленном сушильном шкафу при температуре 80°С в течение 3 часов.
Характеристики образцов ОГМГ-ГХ, полученных в соответствии с примерами 1-4, определённые в соответствии с примерами 9 и 10, приведены в таблице 3.
Таблица 3
|
Приведённые данные показывают, что ни один из полученных образцов технического ОГМГ-ГХ не удовлетворяет требованиям ГФ XIV по содержанию остаточных мономеров (в особенности - токсичного гексаметилендиамина) без дополнительной очистки.
Пример 5. Синтез гидросукцината ОГМГ через получение основания в спиртовой среде
5а. С получением основания в среде абсолютного этанола
Растворяют 5,75 г измельчённого KOH в 50 мл спирта при комнатной температуре на магнитной мешалке в течение 30 мин. Добавляют 17 г измельченного ОГМГ-ГХ, полученного по примеру 1 (мольное соотношение KOH:ОГМГ-ГХ 1,05:1,00). Перемешивают 3-4 часа до растворения ОГМГ-ГХ, смесь оставляют на 16 часов. Получают раствор основания ОГМГ и осадок неорганической соли. Осадок отделяют фильтрацией, фильтрат выпаривают досуха. Готовят 40 % (масс.) водный раствор продукта и прибавляют янтарную кислоту до рН 7,0 и выпаривают досуха. Содержание хлора в образце дано в таблице 4.
5б. С получением основания в среде этанола-ректификата
Растворяют 6,02 г KOH в 50 мл спирта при комнатной температуре на магнитной мешалке в течение 30 мин. Добавляют 17 г измельченного ОГМГ-ГХ, полученного по примеру 2 (мольное соотношение KOH:ОГМГ-ГХ 1,10:1,00). Перемешивают 3-4 часа до растворения ОГМГ-ГХ, смесь оставляют на 16 часов. Получают раствор основания ОГМГ и осадок неорганической соли. Осадок отделяют фильтрацией, фильтрат выпаривают досуха. Готовят 40 % (масс.) водный раствор продукта и прибавляют янтарную кислоту до рН 7,0 и выпаривают досуха. Содержание хлора в образце дано в таблице 4.
5в. С получением основания в среде изо-пропанола
Растворяют 24,45 г KOH в 150,23 г изо-пропанола и 7,00 г воды при комнатной температуре на магнитной мешалке в течение 30 мин. Добавляют 23,00 г измельченного ОГМГ-ГХ, полученного по примеру 3 (мольное соотношение KOH: ОГМГ-ГХ 3:1). Перемешивают 3-4 часа до растворения ОГМГ-ГХ, смесь оставляют на 16 часов. Получают раствор основания ОГМГ и осадок неорганической соли. Осадок отделяют фильтрацией, фильтрат выпаривают досуха. Готовят 40 % (масс.) водный раствор продукта и прибавляют янтарную кислоту до рН 7,0 и выпаривают досуха.
Содержание остаточного хлора определяют меркуриметрическим титрованием. Содержание хлора в образце дано в таблице 4.
Таблица 4
|
Содержание хлора при добавлении янтарной кислоты к основанию должно несколько уменьшиться, однако для продуктов 5а и 5б его значение существенно превышает 0,5% (масс.). В образце 5в содержание сульфатной золы составляет 15,5%. Таким образом, ни использование абсолютного этанола при незначительном избытке КОН (пример 5а), ни применение изо-пропанола при значительном избытке КОН (пример 5в) не позволяет получить продукт необходимого качества. Вместе с тем, очевидно, что достижение значительной степени замещения по хлору невозможно без применения значительного избытка щёлочи.
Примеры 6-10 синтеза солей ОГМГ разветвлённого строения, имеющих степень чистоты, достаточную для их применения в качестве фармацевтической субстанции, приведены далее.
Пример 6. Синтез гидросукцината ОГМГ через получение основания
в водно-спиртовой среде
Растворяют 8,21 г KOH в 50 мл этанола при комнатной температуре на магнитной мешалке 30 мин. При перемешивании по каплям добавляют 8,50 г ОГМГ-ГХ, полученного по примеру 4, в виде 50% водного раствора (мольное соотношение KOH:ОГМГ-ГХ 3:1). Перемешивают 3-4 часа, прекращают перемешивание, смесь оставляют на 16 часов. Получают две жидких фазы, фазу основания ОГМГ (более лёгкая и вязкая) и фазу хлорида калия.
Отделяют фазу основания ОГМГ и добавляют к ней 5,00 г КОН, смесь перемешивают 3-4 часа, прекращают перемешивание, оставляют на 16 часов. Отделяют фазу основания ОГМГ и добавляют к ней янтарную кислоту порциями по 0,3-0,5 г. Первые порции янтарной кислоты идут на взаимодействие с избытком щёлочи в фазе, при этом происходит образование сукцината калия в виде белого кристаллического осадка и рН раствора меняется от 11 до 10,0-9,5. Показателем полного отделения неорганического сукцината является прекращение выделения кристаллического продукта в отдельной пробе с 0,50-0,25 мл прозрачного раствора.
На получение технического гидросукцината ОГМГ используют 3,12 г янтарной кислоты. Осадок отделяют фильтрацией, к фильтрату продолжают добавлять янтарную кислоту порциями по 0,3 -0,5 г до достижения рН 7,0. Если образуется две фазы, нижнюю более вязкую и слегка окрашенную отделяют и выпаривают. Содержание хлора в образце технического гидросукцината ОГМГ составляет 1,02%, золы 4,45%.
Для очистки 7,95 г технического гидросукцината ОГМГ растворяют в 10 мл воды, добавляют к раствору 20,0 г (24,7 мл) этанола-ректификата. К полученной смеси по каплям при перемешивании добавляют 10,0 г (6,7 мл) хлороформа, перемешивают 1 ч. После разделения фаз отделяют нижнюю вязкую фазу и выпаривают. Получают 6,93 г (87,2%) гидросукцината ОГМГ со следующими характеристиками: хлор - 0,38%; сульфатная зола - 0,75%; степень разветвления (z) - 0,30; среднечисловая молекулярная масса (Mn) - 753 Да.
Пример 7. Синтез гидроцитрата ОГМГ через получение основания в водно-спиртовой среде
Растворяют 8,00 г NaOH в 50 мл этанола при комнатной температуре на магнитной мешалке в течение 30 мин. Добавляют по каплям при перемешивании 8,50 г ОГМГ-ГХ, полученного по примеру 1, в виде 50% водного раствора (NaOH:ОГМГ-ГХ 4:1). Перемешивают 3-4 часа, прекращают перемешивание, смесь оставляют на 16 часов. Получают две жидких фазы, фазу основания ОГМГ (более лёгкая и вязкая) и фазу хлорида натрия.
Отделяют фазу основания ОГМГ и добавляют к ней 4,00 г NaOH, смесь перемешивают 3-4 часа, прекращают перемешивание, оставляют на 16 часов. Отделяют фазу основания ОГМГ и добавляют к ней лимонную кислоту порциями по 0,3-0,5 г. Первые порции лимонной кислоты идут на взаимодействие с избытком щёлочи в фазе, при этом происходит образование цитрата натрия в виде белого кристаллического осадка и рН раствора меняется от 11 до 10,0-9,5. Показателем полного отделения неорганического цитрата является прекращение выделения новых порций кристаллического продукта в отдельной пробе с 0,50-0,25 мл прозрачного раствора.
На получение технического гидроцитрата ОГМГ используют 3,51 г лимонной кислоты. Осадок отделяют фильтрацией, к фильтрату продолжают добавлять лимонную кислоту порциями по 0,3-0,5 г до достижения рН 7,0. Если образуется две фазы, нижнюю более вязкую и слегка окрашенную отделяют и выпаривают. Содержание хлора в образце технического гидроцитрата ОГМГ составляет 1,21%, золы 4,59%.
Для очистки 7,80 г технического гидроцитрата ОГМГ растворяют в 10 мл воды, добавляют к раствору 20,0 г (24,7 мл) этанола-ректификата. К полученной смеси по каплям при перемешивании добавляют 10,0 г (6,7 мл) хлороформа, перемешивают 1 ч. После разделения фаз отделяют нижнюю вязкую фазу и выпаривают. Получают 6,81 г (87,3%) гидроцитрата ОГМГ со следующими характеристиками: хлор - 0,48%; сульфатная зола - 0,89%; степень разветвления (z ) - 0,42; среднечисловая молекулярная масса (Mn) - 735 Да.
Пример 8. Синтез гидрохлорида ОГМГ через получение основания в водно-спиртовой среде
Растворяют 8,21 г KOH в 50 мл этанола при комнатной температуре на магнитной мешалке 30 мин. Добавляют по каплям при перемешивании 8,50 г ОГМГ-ГХ, полученного по примеру 4, в виде 50% водного раствора (мольное соотношение KOH:ОГМГ-ГХ 3:1). Перемешивают 3-4 часа, прекращают перемешивание, смесь оставляют на 16 часов. Получают две жидких фазы, фазу основания ОГМГ (более лёгкая и вязкая) и фазу хлорида натрия.
Отделяют фазу основания ОГМГ и добавляют к ней 5,00 г КОН, смесь перемешивают 3-4 часа, прекращают перемешивание, оставляют на 16 часов. Отделяют фазу основания ОГМГ и добавляют к ней концентрированную хлористоводородную кислоту порциями по 0,2 -0,5 мл. Первые порции хлористоводородной кислоты идут на взаимодействие с избытком щёлочи в фазе, при этом происходит образование хлорида калия в виде белого кристаллического осадка и рН раствора меняется от 11 до 10,0-9,5. Показателем полного отделения неорганического цитрата является прекращение выделения новых порций кристаллического продукта в отдельной пробе с 0,50-0,25 мл прозрачного раствора.
На получение технического ОГМГ-ГХ используют 4,20 мл хлористоводородной кислоты. Осадок отделяют фильтрацией, к фильтрату продолжают добавлять хлористоводородную кислоту порциями по 0,3-0,5 мл до достижения рН 7,0. Если образуется две фазы, нижнюю более вязкую и слегка окрашенную отделяют и выпаривают. Содержание золы в образце технического гидрохлорида ОГМГ составляет 5,41%.
Для очистки 7,95 г технического гидрохлорида ОГМГ растворяют в 10 мл воды, добавляют к раствору 20,0 г (24,7 мл) этанола-ректификата. К полученной смеси по каплям при перемешивании добавляют 10,0 г (6,7 мл) хлороформа, перемешивают 1 ч. После разделения фаз отделяют нижнюю вязкую фазу и выпаривают. Получают 7,01 г (88,2%) гидрохлорида ОГМГ со следующими характеристиками: хлориды - 0,22%; сульфатная зола - 0,77%; степень разветвления (z) - 0,30; среднечисловая молекулярная масса (Mn) - 755 Да.
Пример 9. Синтез гидросалицилата ОГМГ через получение основания в водно-спиртовой среде
Растворяют 8,21 г KOH в 50 мл спирта при комнатной температуре на магнитной мешалке 30 мин. Добавляют по каплям при перемешивании 8,50 г ОГМГ-ГХ, полученного по примеру 4, в виде 50% водного раствора (мольное соотношение КОН:ОГМГ-ГХ 3:1). Перемешивают 3-4 часа, прекращают перемешивание, смесь оставляют на 16 часов. Получают две жидких фазы, отделяют более лёгкую и вязкую фазу основания ОГМГ от фазы неорганической соли. К фазе основания ОГМГ добавляют 5,00 г КОН, смесь перемешивают 3-4 часа, прекращают перемешивание, оставляют на 16 часов. Отделяют фазу основания и добавляют к ней салициловую кислоту порциями по 0,2-0,5 г. Первые порции салициловой кислоты идут на взаимодействие с избытком щёлочи в фазе, при этом происходит образование салицилата калия в виде белого кристаллического осадка и рН раствора меняется от 11 до 10,0-9,5. Показателем полного отделения избытка неорганического салицилата является прекращение выделения новых порций кристаллического продута (предпочтительнее делать отдельную пробу с 0,50-0,25 мл прозрачного раствора). На очистку основания используют 5,01 г салициловой кислоты. Осадок отделяют фильтрацией, к полученному раствору добавляют салициловую кислоту порциями по 0,3-0,5 г до достижения рН 7,0. При этом обычно образуется две фазы: нижнюю, более вязкую и слегка окрашенную, отделяют и выпаривают. Содержание золы в образце технического гидросалицилата ОГМГ составляет 6,12%.
Для очистки 8,65 г технического гидросалицилата ОГМГ смешивают с 10 мл воды и 30,0 г (37,1 мл) этанола-ректификата. К полученной смеси по каплям при перемешивании добавляют 10,0 г (6,7 мл) хлороформа, перемешивают 1 ч. После отстаивания отделяют нижнюю вязкую фазу, которую выпаривают. Получают 6,23 г (72,0%) гидросалицилата ОГМГ со следующими характеристиками: хлориды - 0,21%; сульфатная зола - 0,57%; степень разветвления (z) - 0,32; среднечисловая молекулярная масса (Mn) - 763 Да.
Пример 10. Синтез гидросульфосалицилата ОГМГ через получение основания в водно-спиртовой среде
Получение технического гидросульфосалицилата ОГМГосуществляют аналогично примеру 9 через основание ОГМГ в водно-спиртовой среде (мольное соотношение KOH: ОГМГ-ГХ 3:1). На очистку основания используют 6,03 г сульфосалициловой кислоты. Содержание золы в образце технического гидросульфосалицилата ОГМГ составляет 6,21%.
Для очистки 9,06 г технического гидросульфосалицилата ОГМГ смешивают с 10 мл воды и 30,0 г (37,1 мл) этанола-ректификата. К полученной смеси по каплям при перемешивании добавляют 10,0 г (6,7 мл) хлороформа, перемешивают 1 ч. После отстаивания отделяют нижнюю вязкую фазу, которую выпаривают. Получают 6,30 г (69,5%) гидросульфосалицилата ОГМГ со следующими характеристиками: хлориды-0,18%; сульфатная зола - 0,54%; степень разветвления (z) - 0,31; среднечисловая молекулярная масса (Mn) - 766 Да.
Пример 11. Определение спектральных характеристик очищенного гидросукцината ОГМГ и его среднечисловой молекулярной массы
Спектры 13С ЯМР регистрируют на спектрометре Bruker AV-600 с частотой на ядрах углерода 150 МГц при температуре 303 К в режиме полного широкополосного подавления сигналов протонов и отсутствия ядерного эффекта Оверхаузера. Задержка между импульсами по правилу 5Т1 для исключения влияния релаксационных эффектов составляет 60 с. Количество сканирований равно 200. В качестве внутреннего стандарта используют 2,2-диметил-2-силапентен-5-сульфоновую кислоту (DSS).
В ампулу для ЯМР-спектроскопии диаметром 5 мм последовательно вносят 300 мкл D2O, 300 мкл 50% раствора ОГМГ-ГХ в H2O и тщательно перемешивают. В случае появления осадка раствор нагревают до 70°С до его полного растворения. Значения среднечисловой молекулярной массы Mn вычисляют на основе интегральных интенсивностей сигналов SII, SIII, SIV, SII’, SIII’, SIV’ и SIV’’. Положения атомов отмечены в общей структурной формуле, изображённой ниже:
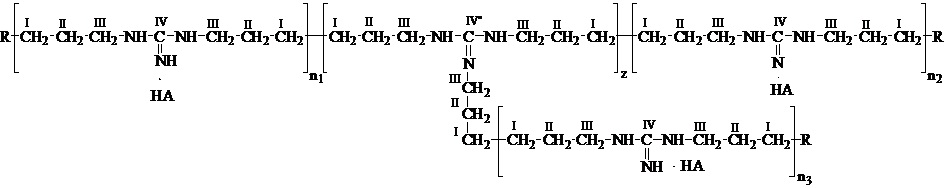
где R представляет 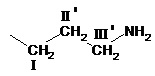 или
или 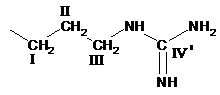 ,
,
НА представляет янтарную кислоту.
Отнесение химических сдвигов атомов углерода (δ) относительно DSS, сделанное на основании экспериментальных данных и адекватных квантово-химических расчётов, приведено в таблице 5.
Таблица 5
|
Из интегральных интенсивностей сигналов «неразветвленных» и «разветвленных» звеньев, концевых фрагментов гуанидина и гексаметилендиамина, зная их молекулярные массы (141, 182, 100 и 58, соответственно), рассчитывают среднечисловую молекулярную массу (Mn) ОГМГ:
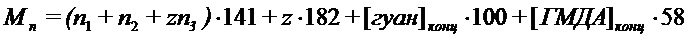 ,
,
где мольные доли концевых фрагментов гуанидина
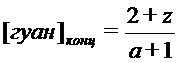 и гексаметилендиамина
и гексаметилендиамина 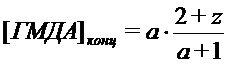
выражаются через интегральные интенсивности сигналов SII, SIII, SIV, SII’, SIII’, SIV’ и SIV’’ соответствующих атомов углерода следующим образом:
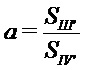 ;
; 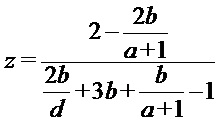 , где
, где 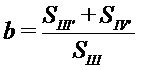 и
и 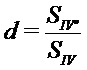
Пример 12. Определение содержания остаточных мономеров
Определения выполняют с помощью ВЭЖХ хроматографа Dionex UltiMate 3000 (Thermo Scientific, Германия) с формированием градиента состава подвижной фазы на линии высокого или низкого давления, оборудованного спектрофотометрическим детектором, работающим в диапазоне длин волн 200-600 нм, и хроматографической колонкой Gemini C18 (3 мкм, 110Å, 4,6×150 мм). Все компоненты подвижной фазы имеют степень чистоты «чистый для ВЭЖХ».
А. Приготовление начальной подвижной фазы А
В мерную колбу номинальной вместимости 1000 мл вносят 870±1 мг натриевой соли 1-пентансульфоновой кислоты, приливают около 300 мл деионизованной воды и полностью растворяют соль. Затем в колбу приливают 10±1 мл 85 % Н3РО3 и 10±1 мл ацетонитрила, тщательно перемешивают, объем раствора в колбе доводят до метки деионизованной водой и еще раз тщательно перемешивают.
Б. Приготовление первичных концентрированных растворов
Первичные концентрированные растворы ГМДА и ГХГ готовят по общей методике. В первую мерную колбу номинальной вместимостью 50 мл вносят 100,0±0,5 мг ГМДА, а во вторую мерную колбу номинальной вместимостью 50 мл также вносят 100,0±0,5 мг ГХГ, приливают 30 мл раствора начальной подвижной фазы А, соль и амин полностью растворяют и доводят объем раствора до метки раствором начальной подвижной фазы А.
Концентрация ГМДА в первичном растворе составляет 2000±10 мкг/мл.
Концентрация ГХГ в первичном растворе составляет 2000±10 мкг/мл, что соответствует 1240 мкг/мл основания гуанидина .
Б.1. Приготовление градуировочных растворов ГМДА
Градуировочные растворы готовят методом последовательных разбавлений.
Раствор ГМДА №4. В мерную колбу номинальной вместимостью 50 мл вносят 5 мл первичного концентрированного раствора ГМДА, приливают 40 мл раствора начальной подвижной фазы А, перемешивают и доводят объем раствора до метки раствором начальной подвижной фазы А. Концентрация ГМДА составляет 200 мкг/мл.
Раствор ГМДА №3. В мерную колбу номинальной вместимостью 50 мл вносят 25 мл градуировочного раствора №4, приливают 20 мл раствора начальной подвижной фазы А, перемешивают и доводят объем раствора до метки раствором начальной подвижной фазы А. Концентрация ГМДА составляет 100 мкг/мл.
Раствор ГМДА №2. В мерную колбу номинальной вместимостью 50 мл вносят 25 мл градуировочного раствора №3, приливают 20 мл раствора начальной подвижной фазы А, перемешивают и доводят объем раствора до метки раствором начальной подвижной фазы А. Концентрация ГМДА составляет 50 мкг/мл.
Раствор ГМДА №1. В мерную колбу номинальной вместимостью 50 мл вносят 25 мл градуировочного раствора №2, приливают 20 мл раствора начальной подвижной фазы А, перемешивают и доводят объем раствора до метки раствором начальной подвижной фазы А. Концентрация ГМДА составляет 25 мкг/мл.
Б.2. Приготовление градуировочных растворов ГХГ
Градуировочные растворы готовят методом последовательных разбавлений.
Раствор №4. В мерную колбу номинальной вместимостью 50 мл вносят 10 мл основного раствора ГХГ приливают 30 мл раствора начальной подвижной фазы А, перемешивают и доводят объем раствора до метки раствором начальной подвижной фазы А. Концентрация ГХГ составляет 400 мкг/мл, что соответствует 248 мкг/мл основания гуанидина.
Раствор №3. В мерную колбу номинальной вместимостью 50 мл вносят 25 мл калибровочного раствора №4, приливают 20 мл раствора начальной подвижной фазы А, перемешивают и доводят объем раствора до метки раствором начальной подвижной фазы А. Концентрация ГХГ составляет 200 мкг/мл, что соответствует 124 мкг/мл основания гуанидина.
Раствор №2. В мерную колбу номинальной вместимостью 50 мл вносят 25 мл калибровочного раствора №3, приливают 20 мл раствора начальной подвижной фазы А, перемешивают и доводят объем раствора до метки раствором начальной подвижной фазы А. Концентрация ГХГ составляет 100 мкг/мл, что соответствует 62 мкг/мл основания гуанидина.
Раствор №1. В мерную колбу номинальной вместимостью 50 мл вносят 25 мл калибровочного раствора №2, приливают 20 мл раствора начальной подвижной фазы А, перемешивают и доводят объем раствора до метки раствором начальной подвижной фазы А. Концентрация ГХГ составляет 50 мкг/мл, что соответствует 31 мкг/мл основания гуанидина.
Градуировку хроматографа проводят трижды, начиная с градуировочных растворов с наименьшей концентрацией. Для каждой концентрации определяют базовую линию для каждого пика и вычисляют среднее значение площадей пиков на выходной кривой хроматографа, соответствующих каждой концентрации. Зависимости этих средних значений от концентрации для ГМДА и ГХГ (в пересчёте на основание гуанидина) обрабатывают методом наименьших квадратов и получают параметры линейных аналитических функций.
В. Подготовка раствора образца
В мерную колбу номинальной вместимости 100 мл вносят 1,000±0,001 г ОГМГ-ГХ и в течении 5 минут растворяют при комнатной температуре в 50-70 мл раствора начальной подвижной фазы А, используя ультразвуковую ванну. Объем раствора в колбе доводят до метки раствором начальной фазы А и еще раз тщательно перемешивают. Концентрация исходной субстанции полимера составляет 10000 мкг/л.
Г. Хроматографические определения остаточных мономеров
Содержания остаточных мономеров определяют в следующих условиях хроматографического разделения:
|
* - натриевая соль 1-пентансульфоновой кислоты
Пример 13. Оценка токсичности очищенных солей
Токсичность определяли на беспородных мышах по ГОСТ 12.1.007-76 и оценивали величинами полулетальных доз LD50 (мг/кг) при внутрижелудочном и накожном путях введения. Полученные результаты представлены в таблице 6.
Таблица 6
|
Прототип* относится к гидрохлориду ОГМГ
ближайшего аналога (RU 2443684).
Пример 14. Подавление роста микроорганизмов и биоцидная эффективность
гидросукцината ОГМГ и ОГМГ-ГХ
Первой оценкой антибактериального действия гидросукцината ОГМГ и ОГМГ-ГХ (получен в соответствии с описанием ближайшего аналога RU 2443684) является минимальная подавляющая рост микроорганизмов концентрация (МПК), выражаемая в мкг/мл, в отношении S. aureus ATCC29213, (чувствителен к метициллину), S. aureus ATCC 4330024 и S. aureus ATCC 4330015 (устойчивые к метициллину), E. coli ATCC 2795267, C. albicans ATCC244336608M, C. tropicalis 30.1.9, C. kruzei 600M, B. subtilis и Pseudomonas aeruginosa ATCC 27853.
Второй оценкой антибактериального действия гидросукцината ОГМГ и ОГМГ-ГХ является минимальная бактерицидная концентрация (МБК),
А. На первом этапе сравнивают МПК гидросукцината ОГМГ и ОГМГ-ГХ микрометодом серийных двукратных разведений в бульоне Мюллер-Хинтон (Oxoid) в соответствии с «Методическими указаниями по определению чувствительности микроорганизмов к антибактериальным препаратам» (МУК 4.2 1890 - 04) и их минимальную биоцидную концентрацию (МБК).
Готовят препараты с концентрацией 1000 мкг/мл в дистиллированной воде и разводят их 128 мкг/мл питательной среды. Определение МПК гидросукцината ОГМГ и ОГМГ-ГХ для бактериальных культур осуществляют в 96-луночных планшетах для иммунологических исследований. Готовят серии двукратных разведений исследуемых действующих веществ в среде Mueller-Hinton Broth II (Oxoid) в объеме 50,0 мкл.
Для приготовления инокулята бактериальных культур из изолированных колоний готовят суспензию по стандарту мутности McFarland 0,5 в физиологическом растворе. Суспензию разводят в среде Muеller-Hinton Broth II до концентрации 108 КОЕ/мл, Приготовленную суспензию вносят по 50,0 мкл в лунки планшета и осуществляют разведение в диапазоне концентраций от 128 до 0,0003 мкг/мл. Инокулированные планшеты инкубируют в течение 18 часов при 37°С. За МПК принимают наименьшую концентрацию действующего вещемтва, при которой отсутствует видимый рост микроорганизмов.
Для определения МПК Candida ssp. из изолированной колонии готовят суспензию в питательной среде Сабуро по стандарту мутности McFarland 0,5, что соответствует 106 КОЕ/мл для грибных культур. Серию двукратных разведений препаратов готовят в среде Сабуро в объеме 100 мл. После посева дрожжевых культур планшеты инкубируют 48 часов при 30°С. Для определения МПК Candida ssp. из изолированной колонии готовят суспензию в питательной среде Сабуро по стандарту мутности McFarland 0,5, что соответствует 106 КОЕ/мл для грибных культур. Серию двукратных разведений Полисепта готовят в среде Сабуро в объеме 100 мл. После посева дрожжевых культур планшеты инкубируют 48 часов при 30°С. Оценки МПК (мкг/мл) гидросукцината ОГМГ и ОГМГ-ГХ* приведены в таблице 7.
Таблица 7
|
* - ближайший аналог в соответствии с RU 2443684
Б. На втором этапе оценивают МБК исследуемых веществ: вносят раствор из двух последних лунок, в которых отсутствовал рост микроорганизмов на соответственный агар питательной среды и содержат их в термостате при 30°С в течение 48 часов для Candida ssp., для остальных микроорганизмов - 18-24 часов при 37°С в опыте in vitro суспензионным методом в соответствии с руководством Р 4.2.2643-10 «Методы лабораторных исследований и испытаний дезинфекционных средств для оценки их эффективности и безопасности». Используют препараты с концентрацией 0,05% и 0,2%, вызывающие терапевтический и пролонгированный эффекты соответственно. Показывают, что воздействие препаратов как в терапевтической, так и в пролонгированной концентрации вызывают гибель большинства исследованных патогенных микроорганизмов в течение периода, не превышающего 1 минуты.
Далее определяют бактерицидную эффективность микроорганизмов в суспензии, содержащей 109 КОЕ/мл (E. сoli, S. aureus) в количестве 0,5 мл, переносят в 4,5 мл сыворотки. Через 5 минут для Candidа spp., 1-2 минуты для Pseudomonas aeruginosa, S. aureus, E.coli и 1-5-15 минут для B. Subtilis, 0,5 мл суспензии из растворов препаратов переносят в раствор нейтрализатора, содержащий 1% лаурилсульфата с 10% обезжиренным молоком. Нейтрализующий раствор с суспензией микроорганизма перемешивают и оставляют на 5 минут. По истечении времени, 0,5 мл культуры из нейтрализующего раствора переносят в 4,5 мл стерильной воды. После перемешивания по 0,1 мл суспензии переносят в 5 мл жидкой питательной среды и на поверхность агаризованной среды: Candida spp. на агаризованную и жидкую питательную среду Сабуро (Биотехновация), а остальные - на среду БТН. Учет результатов осуществляют через 24 часа, для Candida ssp. через 48 часов инкубации. Минимальные бактерицидные концентрации (МБК) гидросукцината ОГМГ и ОГМГ-ГХ даны в таблице 8.
Таблица 8
|
* - ближайший аналог в соответствии с RU 2443684
Таблица 8 (продолжение)
|
* - ближайший аналог в соответствии с RU 2443684
Приведённые результаты исследований гидросукцината ОГМГ, гидрохлорида ОГМГ и ОГМГ-ГХ ближайшего аналога - показывают, что препараты активны в отношении одного и того же спектра микробов, однако значения МПК и МБК в отношении большинства клинических штаммов вдвое ниже у гидрохлорида и гидросукцината ОГМГ, полученных в соответствии с заявляемым способом, т. е. они являются более активными по сравнению с ОГМГ-ГХ ближайшего аналога.
Применимость гидросукцината ОГМГ, гидрохлорида ОГМГ и ОГМГ-ГХ ближайшего аналога в составе лекарственных препаратов подтверждают результатами определения числа колониеобразующих единиц (КОЕ) в присутствии сыворотки, приведёнными в таблице 9.
Таблица 9
|
Эти результат доказывают, что сыворотка не инактивирует гидрохлорид ОГМГ и гидросукцинат ОГМГ, что позволяет использовать их в составе лекарственных средств.
Таким образом, приведённые примеры доказывают промышленную применимость предлагаемого способа получения солей разветвлённого олигогексаметиленгуанидина, имеющих степень чистоты, достаточную для их применения в качестве фармацевтической субстанции для приготовления лекарственных средств.
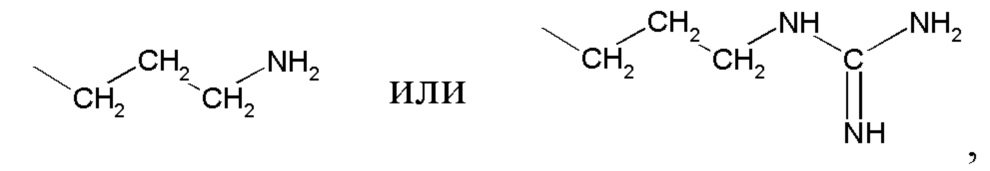
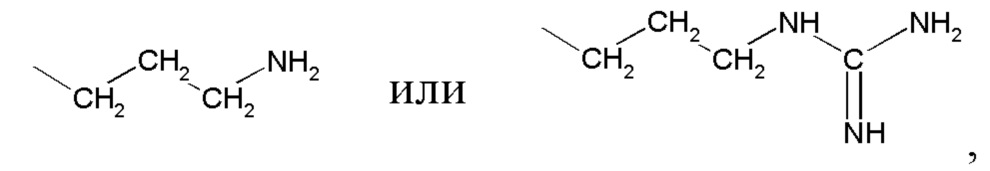
Фармацевтическая композиция для лечения алкоголизма, наркомании и токсикомании с улучшенным профилем высвобождения налтрексона
Применение сополимера на основе n-винилпирролидона в качестве средства, потенцирующего анальгетический эффект морфина гидрохлорида
Имплантируемое лекарственное средство на основе дисульфирама для лечения пациентов, зависимых от алкоголя или опиатов
Имплантируемое лекарственное средство на основе налтрексона для лечения пациентов, зависимых от алкоголя или опиатов
Офтальмологический препарат в виде глазных капель, содержащий дисульфирам и таурин
Синергетический препарат для лечения сердечно-сосудистых заболеваний, сахарного диабета и заболеваний гепато-билиарной системы
Твердая лекарственная форма таурина с улучшенными фармакологическими свойствами
Имплант на основе дисульфирама для лечения пациентов, зависимых от алкоголя или опиатов
Гемигидрат основания налтрексона, способ его получения и способ изготовления микросфер
Офтальмологический препарат в виде глазных капель, содержащий разветвленные полигексаметиленгуанидины и сополимер на основе n-винилпирролидона
Фармацевтическая композиция для лечения алкоголизма, наркомании и токсикомании с улучшенным профилем высвобождения налтрексона
Применение сополимера на основе n-винилпирролидона в качестве средства, потенцирующего анальгетический эффект морфина гидрохлорида
Имплантируемое лекарственное средство на основе дисульфирама для лечения пациентов, зависимых от алкоголя или опиатов
Имплантируемое лекарственное средство на основе налтрексона для лечения пациентов, зависимых от алкоголя или опиатов
Офтальмологический препарат в виде глазных капель, содержащий дисульфирам и таурин
Синергетический препарат для лечения сердечно-сосудистых заболеваний, сахарного диабета и заболеваний гепато-билиарной системы
Твердая лекарственная форма таурина с улучшенными фармакологическими свойствами
Имплант на основе дисульфирама для лечения пациентов, зависимых от алкоголя или опиатов
Гемигидрат основания налтрексона, способ его получения и способ изготовления микросфер
Офтальмологический препарат в виде глазных капель, содержащий разветвленные полигексаметиленгуанидины и сополимер на основе n-винилпирролидона

