Результат интеллектуальной деятельности: ВЫСОКОЭФФЕКТИВНЫЙ СПОСОБ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННОЙ ФОРМЫ АДРЕСНОГО ДЕЙСТВИЯ ДЛЯ ТЕРАПИИ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ
Вид РИД
Изобретение
ОБЛАСТЬ ПРИМЕНЕНИЯ
Изобретение относится к области медицины, биологии, биотехнологии, молекулярной биологии, нанобиотехнологии и фармакологии, и представляет собой способ получения противоопухолевого средства для химиотерапии онкологических заболеваний, содержащее цитостатическое вещество - производное альфа-гидроксибензолпропановой кислоты, сополимер молочной и гликолевой кислот, рекомбинантный белок, являющийся функциональным фрагментом альфа-фетопротеина (АФП), моно- или дисахариды, причем компоненты в композиции находятся в определенном соотношении в масс. %. Изобретение обеспечивает адресную доставку цитостатического вещества в злокачественные новообразования, его пролонгированное высвобождение, увеличение противоопухолевого эффекта.
В качестве цитостатических веществ используются производные альфа-гидроксибензолпропановой кислоты, например, такие как паклитаксел, доцетаксел, кабазитаксел и пр. из таксанового ряда, сополимер молочной и гликолевой кислот выступает в качестве биодеградируемого полимера, моно- и дисахариды в качестве криопротектора, рекомбинантный белок, функциональный фрагмент альфа-фетопротеина (АФП) - в качестве векторной молекулы для адресной доставки. Противоопухолевое средство выполнено в виде лиофилизата, предназначенного для внутривенного введения, и обеспечивают адресное и пролонгированное действие.
Изобретение способствует повышению противоопухолевого эффекта за счет адресной доставки, что повышает эффективность лечения и снижает общую неспецифическую токсичность противоопухолевого средства.
УРОВЕНЬ ТЕХНИКИ
Из уровня техники известны системы направленной (адресной) доставки химиотерапевтических препаратов в виде полимерных или липосомальных форм, связанных (конъюгированных) с векторными молекулами, обладающими специфическим сродством к поверхности опухолевых клеток.
Известна работа Farokhzad О.С. с соавторами, в которой представлена система направленной доставки противоопухолевых препаратов на основе полимерных частиц PLGA-PEG-COOH к клеткам рака предстательной железы. В качестве векторной молекулы служил аптамер А10 PSMA, селективно связывающий внеклеточный участок простат-специфического антигена [1].
Известны технические решения, защищенные патентами US 20130052134, WO 2013127949, CN 103745793, СА 2648099, в которых описаны способы конъюгации с использованием различных производных карбодиимида и где использованы сополимеры, такие как PEG-PLGA-PLL, DSPE-PEG-COOH, PLGA-b-PEG и т.п.
Недостатком этих способов является относительно узкая область применения. Также основное отличие заключается в типе используемых полимеров и выборе векторных молекул.
Известны также технические решения, защищенные патентами CN 101744789, РТ 1539976, WO 2010028217, US 7105158, US 6555110, в которых описаны способы конъюгации с применением карбодиимидов.
Недостатком этих изобретений является относительно большое время протекания основной реакции.
Известен также способ, защищенный патентом US 7163698 В2, представляющий собой синтез полимера PLGA с использованием пептидных векторных молекул и активирующего агента EDC, при котором используют органические фазы во время реакции конъюгации для получения модифицированного полимера, после чего получают полимерные частицы.
Известны также способы получения противоопухолевых препаратов с антителами, выступающих в качестве векторных систем [патенты WO 2016036794, WO 2014119624], в которых в качестве векторной молекулы используются антитела.
Недостатком указанных изобретений является тот факт, что применяемые векторные молекулы не обеспечивают эффективную адресность в отношении опухолевых клеток-мишеней, а также характеризуются относительно низкой концентрацией действующего вещества в конечном препарате. Помимо этого, способы отличаются относительно высокой сложностью, вызванной необходимостью введения дополнительного линкера для получения векторной системы.
Известны также работы, в которых представлены результаты с использованием в качестве векторной молекулы бомбезина (BBN) (Hitesh et al., 2014), система направленной доставки на основе полимерных частиц с использованием моноклональных антител (Li et al., 2011) [2, 3].
В ряде других работ в качестве векторной молекулы для доставки противоопухолевых препаратов используют белок альфа-фетопротеин (АФП) человека, т.к. он имеет рецепторы на поверхности опухолевых клеток [4]. В патентах РФ представлены данные по разработке ряда препаратов на основе АФП и способах их получения из природных источников [RU 2121350 С1, 10.11.1998; RU 2123009 С1, 10.12.1998; RU 2154468 С1, 09.11.1999; RU 2105567 С1,27.02.1998].
В патенте US 20090068115 описан способ активации карбоксильных групп полимерных частиц с использованием EDC, для чего проводят реакцию в 0.1 М буфере TEMED, рН 4.8, а также дальнейшую очистку с помощью диализа.
Недостатком способа является относительно большие временные затраты.
В патенте US 2010015051 описан способ конъюгации в водных средах в боратном буфере при слабокислых значениях рН, причем избыток белка трансферрина удаляют ультрацентрифугированием.
Недостатком способа является относительно большая сложность и относительно большие временные затраты.
В патенте РФ [RU 2317102б C1, А61К 38/08, 20.02.2008] описано использование в качестве векторной молекулы пептида QMTOVNOG - аналога фрагмента альфа-фетопротеина. В частности, предложено применение пептида формулы QMTOVNOG, являющегося аналогом фрагмента α-фетопротеина с 472-й по 479-ю аминокислоту, способного избирательно захватываться опухолевыми клетками, в качестве векторной молекулы для направленной доставки противоопухолевых препаратов в опухолевые клетки. Кроме того, предложен конъюгат пептида формулы QMTOVNOG с доксорубицином, в котором доксорубицин ковалентно присоединен к указанному пептиду через тиоэфирную связь, в котором ковалентная тиоэфирная связь между ними создана путем взаимодействия SH-группы производного пептида, полученного реакцией пептида с N-гидроксисукцинимидным эфиром S-ацетилтиогликолевой кислоты, и двойной связи малеимидной группы гидразона доксорубицина, полученного реакцией доксорубицина с 4-(N-малеимидометил)циклогексан-1-карбонилгидразидом. Предложена также фармацевтическая композиция для лечения онкологических заболеваний, содержащая конъюгат доксорубицина с векторной молекулой в эффективном количестве и подходящий для внутривенного введения фармацевтический носитель, отличающаяся тем, что в качестве конъюгата доксорубицина она содержит предложенный конъюгат. Недостатком этого конъюгата является относительно низкая селективность действия в отношении опухолевых клеток-мишеней.
Одним из близких к предложенной системе направленной доставки является противоопухолевой пептидный препарат, созданный на основе рекомбинантного фрагмента альфа-фетопротеина (АФП) человека. Препарат способен связываться с рецептором альфа-фетопротеина и ингибировать эстрадиол-индуцированный рост клеток гормонзависимых опухолей, а также выполнять функцию векторной молекулы для направленной доставки химитерапевтического препарата в опухолевые клетки [RU 2385537, C1, А61К 38/12, 20.10.2006].
Недостатком данного препарата является относительно низкая эффективность применения.
В патенте [Ru 2630947 C1, А61К 38/16, А61К 31/00, А61Р 35/00, 05.08.2016] описан способ получения конъюгата с противоопухолевой активностью на основе рекомбинантного альфа-фетопротеина или его функционального фрагмента и противоопухолевого средства из антрациклинового ряда, характеризующийся тем, что получен в виде частиц из сополимеров молочной и гликолевой кислот с включенным в них противоопухолевым средством антрациклинового ряда путем взаимодействия -СООН групп частиц, активированных 1-этил-3-(3-диметиламинопропила)карбодиимидом и N-гидроксисукцинимидом при их не менее 5-кратном избытке по отношению к карбоксильным группам, с аминогруппами рекомбинантного белка альфа-фетопротеина, имеющего последовательности, выбранные из группы: SEQ1, SEQ2, SEQ3, SEQ4, не меньше, чем в эквимолярных количествах в отношении активированных карбоксильных групп, при рН от 7.4 до 9, с последующей очисткой на колонке с носителем Superose 12 с полным разделением продуктов реакции и выделением целевого конъюгата в чистом виде.
Недостатком данного технического решения относительно предложенного противоопухолевого препарата является относительно низкая эффективность, обусловленная ограничением в применении препарата, а также сложный процесс очистки для получения конечного продукта.
Способ получения конъюгата полимеркапсулированного противоопухолевого агента из группы антрациклинов с активным фрагментом АФП, описанный в техническом решении [Ru 2630947 C1, А61К 38/16, А61К 31/00, А61Р 35/00, 05.08.2016], в частности способ получение конъюгата конъюгата с противоопухолевой активностью, характеризующийся тем, что -СООН группы полимерных частиц из сополимеров молочной и гликолевой кислот с включенным в них противоопухолевым средством антрациклинового ряда активируют 1-этил-3-(3-диметиламинопропила)карбодиимидом и N-гидроксисукцинимидом при их не менее 5-кратном избытке по отношению к карбоксильным группам, добавляют не больше 2 мкл 2-меркаптоэтанола и инкубируют 10 мин, затем добавляют рекомбинантный белок альфа-фетопротеина, имеющий последовательности, выбранные из группы: SEQ1, SEQ2, SEQ3, SEQ4, не меньше, чем в эквимолярных количествах в отношении активированных карбоксильных групп, доводят рН реакционной смеси до щелочных значений от 7,4 до 9, оставляют для перемешивания 30 мин, далее останавливают реакцию введением не более 2 мкл этаноламина, после чего реакционную смесь очищают на колонке с носителем Superose 12 с полным разделением продуктов реакции и выделяют целевой конъюгат в чистом виде, является близким по технологии к предложенному методу.
Недостатком описанного способа получения является относительно узкая область применения, обусловленная тем, что он не может быть использован для получения противоопухолевого препарата, обладающего более высокой эффективностью и адресным действием.
Кроме того, описанный способ отличается внесением рекомбинантного белка альфа-фетопротеина, имеющего последовательности, выбранные из группы: SEQ1, SEQ2, SEQ3, SEQ4, не меньше, чем в эквимолярных количествах в отношении активированных карбоксильных групп полученных полимерных частиц, что приводит к необходимости отделения от избытка используемого векторного белка и более сложной технологии получения конечного продукта. Данный способ очистки на носителе Superose 12 не является препаративным и имеет ряд недостатков в силу сложности процесса.
Наиболее близким аналогом к предложенному препарату является патент [RU 2451509 C1, А61К 31/337, 27.05.2012], представляющий собой стабильные наночастицы и включающий цитостатик, биодеградирующий полимер, поверхностно-активное вещество, криопротектор и векторную молекулу для адресной доставки частиц в пораженные органы и ткани, при этом в качестве цитотостатического вещества содержит паклитаксел - 0,4÷1,0 мас. %, в качестве биодеградируемого полимера - сополимер молочной и гликолевой кислот (PLGA 50:50) - 14,0÷15,0 мас. %, в качестве поверхностно-активного вещества - сукцинилированный поливиниловый спирт - 3,5÷4,0 мас. %, в качестве векторной молекулы для адресной доставки частиц в пораженные органы и ткани - С-концевой домен альфа-фетопротеина (рЗдАФП) - 0,1÷0,3 мас. %, а в качестве криопротектора - D-маннит - 75,0÷80,0 мас. %.
Недостатком наиболее близкого технического решения относительно предложенного противоопухолевого препарата является относительно низкая селективность действия в отношении опухолевых клеток, что снижает эффективность лечения и повышает общую токсичность при его применении. Низкая эффективность также обусловлена применением сукцинилированного поливинилового спирта, предположительно способствующего процессу конъюгации, низкое содержание цитостатического вещества в конечном продукте, а также использование альбумина, однако такое решение не способствовало в значительной степени увеличению адресного действия полученного препарата по различным показателям.
Наиболее близким к предложенному способу является способ получения препарата полимеркапсулированного противоопухолевого агента из группы таксанов с активным фрагментом АФП, описанный в том же техническом решении [RU 2451509 С1, А61К 31/337, 27.05.2012], в частности способ получения препарата на основе сополимера молочной и гликолевой кислот с доксорубицином, согласно которому 150.0 мг сополимера молочной и гликолевой кислот (PLGA-COOH 50/50), содержащего свободную концевую карбоксильную группу, растворяют в 1,5 мл хлороформа, к раствору добавляют 18.0 мг дициклогексилкарбодиимида и 10.0 мг N-гидроксисукцинимида (10-кратные молярные избытки по отношению к полимеру), реакционную смесь перемешивают в течение 2 ч при комнатной температуре и отделяют от выпавшего осадка дициклогексилмочевины с помощью фильтрации, к смеси прибавляют 5.0 мкл триэтиламина и 7,5 мг доксорубицина гидрохлорида, реакционную смесь перемешивают в течение 15 ч, очистку реакционной смеси от избытка доксорубицина производят с помощью трехкратной экстракции 0.1 М водным раствором соляной кислоты, продукт реакции очищают преципитацией в ледяном метаноле, осадок собирают центрифугированием при 18 тыс. g в течение 30 мин, предварительно выпарив из раствора хлороформ под вакуумом.
Недостатком наиболее близкого к предложенному способу является относительно узкая область применения, обусловленная тем, что он не может быть использован для получения противоопухолевого препарата, обладающего более высокой эффективностью, определяемой повышенной тропностью в отношении опухолевых клеток и более низкой токсичностью при его применении.
Кроме того, известный способ отличается и относительно большим временем проведения реакции. Дополнительно можно отметить, что поскольку операция очистки в известном способе заключается в центрифугировании и ультрафильтрации, то полученный препарат возможно отделить лишь от низкомолекулярных примесей и невозможно отделить несвязавшиеся молекулы белка, что может привести к снижению эффективности препарата за счет блокировки рецепторов на поверхности опухолевых клеток, что, в свою очередь, приводит к снижению уровня связывания целевого препарата.
Задачей, которая решается в предложенном изобретении, является разработка лекарственной формы противоопухолевого препарата, обладающего более высокой эффективностью применения за счет повышения тропности в отношении опухолевых клеток, увеличенном содержании цитостатического препарата, и снижения неспецифической токсичности при его применении, а также эффективного способа его получения с оптимизированными параметрами, позволяющего получать препаративные количества лекарственной формы.
Требуемый результат заключается в повышении эффективности путем обеспечения повышенной специфической активности к опухолевым клеткам, экспрессирующим рецепторы альфа-фетопротеина.
Требуемый результат достигается в противоопухолевом препарате, который представляет собой стабильные наночастицы и включает цитостатическое средство, биодеградируемый полимер, неорганические соли, криопротектор и векторную молекулу для адресной доставки препарата в пораженные органы и ткани.
Кроме того, требуемый результат достигается тем, что противоопухолевый препарат в качестве цитостатического средства содержит производное альфа-гидроксибензолпропановой кислоты в масс. %: 4.5÷5.5.
Кроме того, требуемый результат достигается тем, что противоопухолевый препарат в качестве биодеградируюемого полимера содержит сополимер молочной и гликолевой кислот в масс. %: 70.0÷72.1.
Кроме того, требуемый результат достигается тем, что противоопухолевый препарат в качестве векторной молекулы для адресной доставки частиц в пораженные органы и ткани содержит С-концевой домен альфа-фетопротеина (r3D AFP) в масс. %: 12.3÷13.3.
Кроме того, требуемый результат достигается тем, что противоопухолевый препарат в качестве криопротектора содержит D-маннитол в масс. %: 10.6-10.7.
Кроме того, требуемый результат достигается тем, что в противоопухолевом препарате стабильные наночастицы выполнены в виде частиц со средним размером частиц не более 275 нм и диапазоном распределения - 150-550 нм.
Состав противоопухолевого препарата и его повышенная эффективность применения относительно известных препаратов аналогичного назначения обосновывается следующим.
Серьезным недостатком используемых в настоящее время противоопухолевых препаратов является их высокая общая токсичность. Химиотерапия онкологических заболеваний существующими сильнодействующими препаратами сопровождается выраженной интоксикацией организма больного и побочными эффектами, что существенно ограничивает возможности применения и использования в полной мере цитостатического и цитотоксического потенциала современных противоопухолевых средств. Возникновение токсических и побочных эффектов при проведении химиотерапии онкологических больных связано с низкой избирательностью существующих лекарств, необходимостью длительно поддерживать достаточно высокую терапевтическую дозу, что приводит к сильному отрицательному воздействию на здоровые органы и ткани.
Серьезной проблемой, снижающей эффективность химиотерапии злокачественных новообразований, является также множественная лекарственная устойчивость (МЛУ) опухолевых клеток. Одной из основных причин МЛУ является гиперэкспрессия трансмембранных белков, относящихся к семейству ABC-транспортеров (MDR1, MRP1, BCRP и др.), выбрасывающих химиопрепараты, в том числе молекулы противоопухолевых препаратов, из клетки.
Один из способов решения данных проблем связан с созданием систем адресной доставки лекарственных веществ путем их включения в полимерные частицы или липосомы, имеющие размеры не более 500 нм. Высокая терапевтическая активность наносомальных препаратов при лечении онкологических заболеваний определяется способностью наночастиц обеспечивать пассивный направленный транспорт связанных с ними лекарственных веществ в опухоль. Пассивный направленный транспорт обусловлен большей уязвимостью поврежденных тканей для данных лекарств. В литературе это явление получило название «эффекта повышенной проницаемости и удерживания». Важным преимуществом данной технологии является ее гибкость, позволяющая варьировать свойства носителей лекарств в зависимости от локализации очага патологии.
Сущность предложенного решения заключается в том, что создана готовая лекарственная форма цитотоксического препарата - производного альфа-гидроксибензолпропановой кислоты, на основе сополимера молочной и гликолевой кислот, содержащая белковую векторную молекулу r3D AFP, для адресной доставки препарата в опухолевые клетки.
Получение частиц проводили с использованием биодеградируемого сополимера молочной и гликолевой кислот (молекулярная масса 3-17 кДа) и поливинилового спирта (ПВС) в качестве стабилизатора эмульсии. Наночастицы получали методом одинарного эмульгирования с использованием погружного гомогенизатора (24000 об/мин, 7 раз по 40 сек с перерывами по 20 сек).
Свободную карбоксильную группу частиц связывали с аминогруппой векторного белка с использованием водорастворимого карбодиимида. Очистку и выделение готового продукта проводили с использованием гель-эксключионной хроматографии. Размеры полученной готовой лекарственной формы составляли не более 275 нм, а диапазон распределения - 150-550 нм.
Кроме того, требуемый технический результат достигается тем, что для получения препарата используют способ, позволяющий получать лекарственную форму в препаративных количествах (не менее 7 г лиофилизата лекарственной формы), а лекарственная форма содержит производное альфа-гидроксибензолпропановой кислоты, сополимер молочной и гликолевой кислот, r3D AFP, неорганические соли и Д-маннитол в следующем соотношении % мас.:
|
На чертеже графических материалах представлены:
на фиг. 1 - аминокислотная последовательность рекомбинантного фрагмента АФП (r3D AFP) (SEQ1);
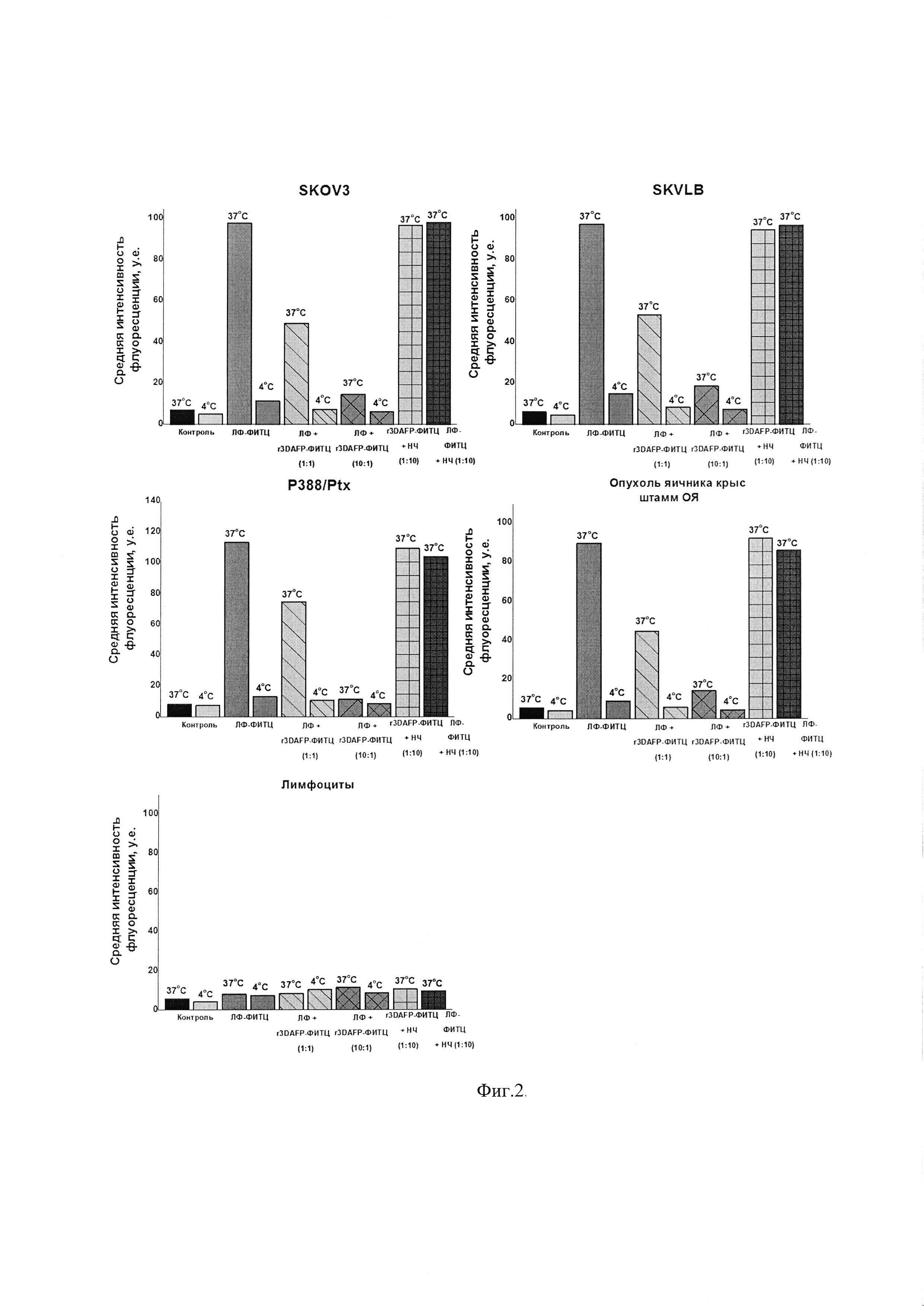
на фиг. 2 - связывание (4°С) и интернализация (37°С) ЛФ-ФИТЦ, смесь ЛФ и r3D AFP-ФИТЦ (молярное соотношение 1 к 1), смесь ЛФ и r3D AFP-ФИТЦ (молярное соотношение 10 к 1), смесь ЛФ-ФИТЦ и полимерных частиц (НЧ) (массовое отношение ЛФ-ФИТЦ и частиц 1 к 1), смесь ЛФ-ФИТЦ и полимерных частиц (НЧ) (массовое отношение ЛФ-ФИТЦ и частиц 1 к 10) клетками линий SKOV3, SKVLB, Р388/РТХ, опухоли яичника крыс штамма ОЯ и лимфоцитами, оцениваемые по интенсивности флуоресценции клеток;
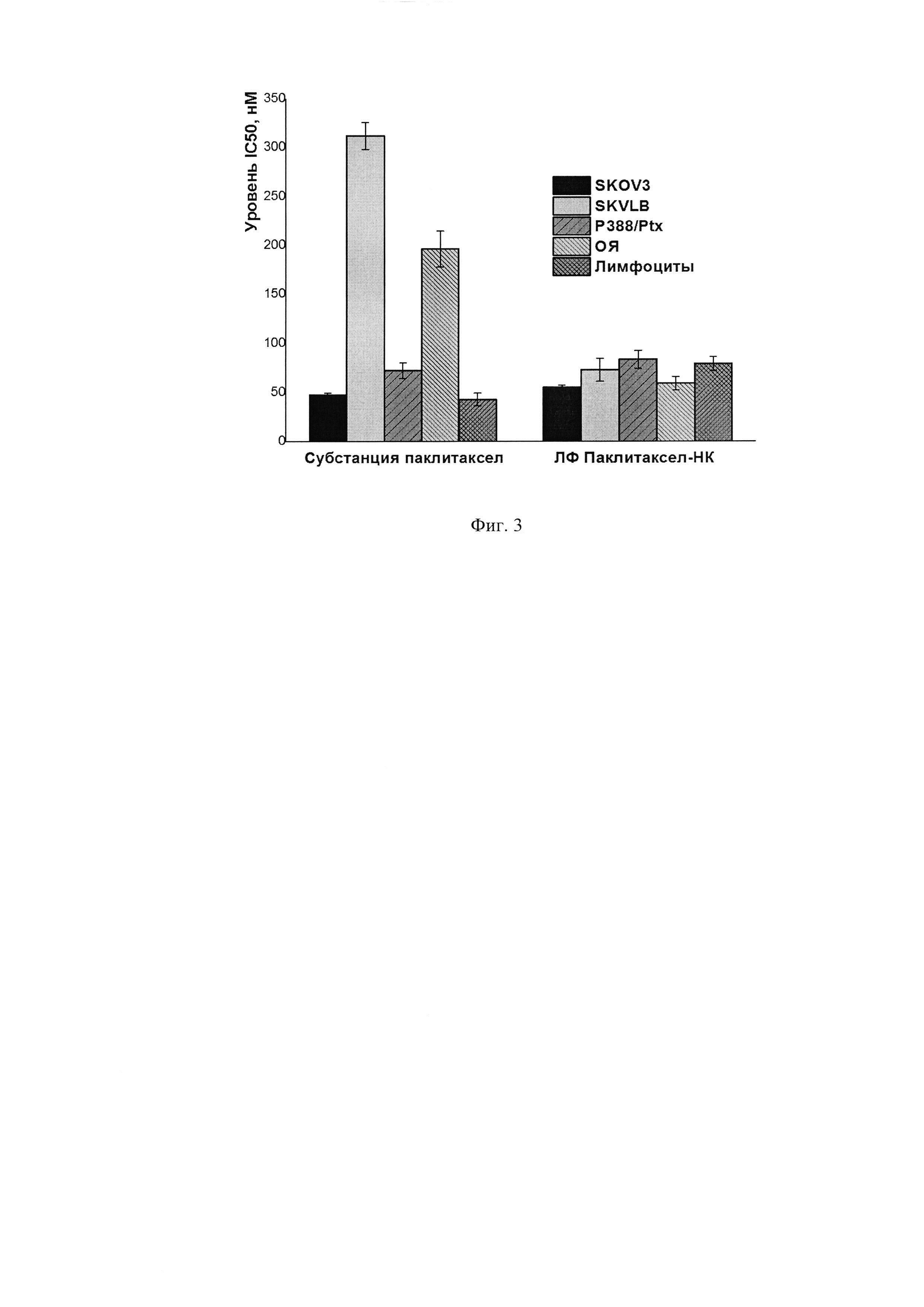
на фиг. 3 - выживаемость клеток линий SKOV3, SKVLB, Р388/РТХ, опухоли яичника крыс штамма ОЯ и лимфоцитов периферической крови человека (Б) после 72 ч инкубации с ЛФ Паклитаксел-НК и препаратом сравнения - субстанция паклитаксел, оцениваемая по уровню IC50.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Способ получения конъюгата противоопухолевых препаратов с функциональным фрагментом рекомбинантного альфа-фетопротеина осуществляется следующим образом.
Получаемый предложенным способом противоопухолевый препарат обеспечивает направленную доставку противоопухолевых препаратов непосредственно в опухолевые клетки, защищая их от воздействия резистентных механизмов и обеспечивая пролонгированный эффект.
Заявленный способ предназначен для получения конъюгатов полимерных частиц, содержащих цитостатические препараты, с функциональным фрагментом рекомбинантного альфа-фетопротеина.
Для получения конъюгатов использовали карбодиимидный способ. Этот способ отличается простотой, относительно небольшим временем проведения реакции, а также относительно приемлемыми условиями, в которых она протекает.
Одним из методов лечения онкологических заболеваний является применение химиотерапевтических препаратов. На протяжении многих лет в качестве противоопухолевых веществ используются цитотоксические агенты из класса дитерпенов - таксаны [5]. Первые таксаны были выделены из растений рода Taxus (тис) в конце 60-х годов и имеют таксадиеновое ядро. Данные препараты активно используются при терапии злокачественных новообразований различного происхождения, таких как: саркомы, меланомы, карциномы и другие солидные опухоли.
Основным механизмом действия препаратов класса таксанов (производные альфа-гидроксибензолпропановой кислоты) является нарушение функции микротрубочек. Таксаны связываясь со свободным тубулином, повышают скорость и степень его полимеризации, стимулируют сборку микротрубочек, стабилизируют сформировавшиеся микротрубочки, препятствуют деполимеризации тубулина и распаду микротрубочек. Образование чрезмерного количества микротрубочек и их стабилизация приводят к ингибированию динамической реорганизации сети микротрубочек, что в конечном итоге ведет к нарушению процесса формирования митотического веретена и ингибированию клеточного цикла в G2 и М-фазах митоза.
Настоящее изобретение относится к терапевтическим комбинациям, содержащим препараты - производные альфа-гидроксибензолпропановой кислоты из таксанового ряда. К таковым относятся следующие препараты, не ограничивающие объема изобретения:
1. «Паклитаксел» - цитостатический препарат, относящийся к таксанам. Его применяют для лечения рака яичников, рака молочной железы, немелкоклеточного рака легкого, рака мочеполовой сферы и саркомы Капоши [6-10].
Механизм действия паклитаксела связан со способностью препарата стимулировать «сборку» микротрубочек из димерных молекул тубулина, стабилизировать их структуру и тормозить динамическую реорганизацию в интерфазе, что нарушает митотическую функцию клетки.
Однако, ввиду высокой токсичности паклитаксела, его применение сопряжено с рядом побочных эффектов: нейтропенией, тромбоцитопенией, анемией, снижением артериального давления, брадикардией, тахикардией, фиброзом легких, эмболией легочной артерии, артралгией, миалгией, тошнотой, рвотой, диареей, запором, увеличением активности печеночных трансаминази др.
2. «Доцетаксел» применяют для лечения операбельногорака молочной железы, метастатического или местно распространенного рака молочной железы, немелкоклеточного рака легких, рака яичников, рака предстательной железы, опухолей области головы и шеи [11, 12].
Механизм действия связан с накоплением тубулина в микротрубочках митотического веретена, что приводит к нарушению процессов их сборки и разборки. Нарушает клеточное деление в фазах G2 и М клеточного цикла. Доцетаксел долго сохраняется в клетках, где достигаются его высокие концентрации. Доцетаксел активен в отношении некоторых (но не всех) клеток, продуцирующих в избыточном количестве р-гликопротеин, который кодируется геном множественной устойчивости.
Доцетаксел обладает рядом побочных эффектов, к которым можно отнести следующие: нейтропения, тромбоцитопения, аллергические реакции, повышенная активность печеночных трансаминаз, тошнота, рвота, диарея, анорексия, стоматит, нарушение сердечного ритма, повышение или понижение артериального давления, отдышка, и др.
Таким образом, существенным недостатком используемых в настоящее время противоопухолевых препаратов является их высокая токсичность, которая вызывает интоксикацию организма пациента, а также множественные побочные эффекты.
Другим недостатком является возникновение у опухолевых клеток множественной лекарственной устойчивости (МЛУ). Механизм МЛУ связан с гиперэкспрессией трансмембранных белков семейства ABC-транспортеров, которые способны удалять химиотерапевтические агенты из опухолевой клетки [13].
Изобретение может быть применено для лечения опухолевых злокачественных новообразований при наличии на поверхности опухолевых клеток рецепторов к альфа-фетопротеину (АФП), а именно: для лечения аденокарциномы молочной железы человека линий MCF 7 и сублинии MCF 7 Adr, резистентной к антрациклиновым антибиотикам и таксанам; аденокарциномы яичника человека линии SKOV3, резистентной линии SKVLB и линии OVCAR 8; аденокарциномы предстательной железы человека линий LnCaP и Du145; немелкоклеточной карциномы легкого человека линии А549; мелкоклеточной карциномы легкого человека линии Н69 и пр.
ТЕРМИНЫ И ОПРЕДЕЛЕНИЯ
Термины «рак» и «злокачественная опухоль» относятся или описывают физиологическое состояние у млекопитающих, которое обычно характеризуется нерегулируемым клеточным ростом/пролиферацией. Примеры злокачественной опухоли включают без ограничения карциному, саркому, лимфому, бластому, и лейкоз. Более конкретные примеры таких злокачественных опухолей включают раке яичников, легкого, молочной железы, пищевода и верхних отделов желудка, карциномы, саркома Капоши и различные типы рака головы и шеи.
Термин «противоопухолевая активность» означает активность конъюгата в отношении как гормоно-зависимых, так и гормоно-независимых опухолей.
Термины «химиотерапевтические препараты», «противоопухолевые препараты», «цитотоксическое средство» обозначают химические соединения, пригодные для лечения онкологических заболеваний, обладающие схожим механизмом действия, структурной формулой и физико-химическими свойствами. Данные термины включают цитотоксические агенты из класса дитерпенов - таксаны, такие как: паклитаксел, доцетаксел и кабазитаксел и пр.
«Химиотерапевтическим средством» является химическое соединение, которое можно использовать для лечения злокачественной опухоли. Примеры химиотерапевтических средств включают алкилирующие агенты, такие как тиотепа и CYTOXAN® цикло-фосфамид; алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбоквон, метуредопа и уредопа; этиленимины и метилмеламины, включая алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметилмеламин; ацетогенины (в частности, буллатацин и буллатацинон); дельта-9-тетрагидроканнабинол (дронабинол, MARPNOL®); бета-лапахон; лапахол; колхицины; бетулиновую кислоту; камптотецин (включая синтетический аналог топотекан (HYCAMTIN®), СРТ-11 (иринотекан, CAMPTOSAR®), ацетилкамптотецин, скополектин и 9-аминокамптотецин); бриостатин; каллистатин; СС-1065 (включая его синтетические аналоги адозелезин, карзелезин и бизелезин); подофиллотоксин; подофиллиновую кислоту; тенипозид; криптофицины (в частности, криптофицин 1 и криптофицин 8); доластатин; дуокармицин (включая синтетические аналоги KW-2189 и СВ1-ТМ1); элеутеробин; панкратистатин; саркодиктиин; спонгистатин; азотистые иприты, такие как хлорамбуцил, хлорнафазин, холофосфамид, эстрамустин, ифосфамид, мехлорэтамин, гидрохлорид оксида мехлорэтамина, мелфалан, новембихин, фенестерин, преднимустин, трофосфамид, урамустин; нитрозомочевины, такие как кармустин, хлорозотоцин, фотемустин, ломустин, нимустин и ранимустин; антибиотики, такие как энедииновые антибиотики (например, калихеамицин, в частности калихеамицин гамма II и калихеамицин омега II (смотри, например, Agnew, Chemlntl. Ed. Engl., 33: 183-186 (1994)); динемицин, включая динемицин А; эсперамицин; а также хромофор неокарциностатина и родственные хромофоры хромопротеинов - энедииновые антибиотики), аклациномизины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карзинофилин, хромомицин, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, доксору-бицин ADRIAMICIN® (включая морфолинодоксорубицин, цианоморфолинодоксорубицин, 2-пирролинодоксорубицин и дезоксидоксорубицин), эпирубицин, эзорубицин, идарубицин, марцелломицин, митомицины, такие как митомицин С, микофеноловую кислоту, ногаламицин, оливомицины, пепломицин, потфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-FU); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; аналоги пурина, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; аналоги пиримидина, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, пропионатдромостанолона, эпитиостанол, мепитиостан, тестолактон; средства, подавляющие функции надпочечников, такие как аминоглютетимид, митотан, трилостан; компенсатор фолиевой кислоты, такой как фолиновая кислота; ацеглатон; гликозид альдофосфамид; аминолевулиновую кислоту; энилурацил; амсакрин; бестрабуцил; бисантрен; эдатраксат; дефофамин; демеколцин; диазихон; элфорнитин; ацетат эллиптиния; эпотилон; этоглуцид; нитрат галлия; гидроксимочевину; лентинан; лонидаинин; мейтанзиноиды, такие как мейтанзин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитраэрин; пентостатин; фенамет; пирарубицин; лозоксантрон; 2-этилгидразид; прокарбазин; полисахаридный комплекс PSK® (JHS NaturalProducts, Eugene, OR); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновая кислота; триазиквон; 2,2',2-трихлортриэтиламин; трихотецены (в частности, токсин Т-2, верракурин А, роридин А и ангуидин); уретан; виндезин (ELDISINE®, FILDESIN®); дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид («ara-C»); тиотепа; таксоиды, например, паклитаксел TAXOL® (Bristol-MyersSquibb Oncology, Princeton, N.J.), не содержащий кремофора препарат паклитаксела на основе сконструированных связанных с альбумином наночастиц ABRAXANE TM (AmericanPharmaceuticalPartners, Schaumberg, Illinois) и доксетаксел TAXOTERE® (Rhone-PoulencRorer, Antony, France); хлорамбуцил; гемцитабин (GEMZAR®); 6-тиогуанин; меркаптопурин; метотрексат; аналоги платины, такие как цисплатин и карбоплатин; винбластин (VELBAN®); платину; этопозид (VP-16); ифосфамид; митоксантрон; винкристин (ONCOVIN®); оксалиплатин; лейковорин; винорелбин (NAVELBINE®); новантрон; эдатрексат; дауномицин; аминоптерин; ибандронат; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; капецитабин (XELODA®); фармацевтически приемлемые соли, кислоты и производные любого агента, указанного выше; а также комбинации двух или более указанных выше средств, такие как CHOP, сокращенное название комбинированной терапии циклофосфамидом, доксорубицином, винкристином и преднизолоном, и FOLFOX, сокращенное название схемы лечения оксалиплатином (ELOXATIN ТМ) в сочетании с 5-FU и лейковорином.
Также в указанное определение включены противогормональные средства, которые действуют, регулируя, уменьшая, блокируя или ингибируя эффекты гормонов, которые могут стимулировать рост злокачественной опухоли, и часто их используют в форме системного лечения или лечения на уровне целого организма. Они сами могут являться гормонами. Примеры включают антиэстрогены и избирательные модуляторы рецепторов эстрогена (SERM), включая, например, тамоксифен (включая тамоксифен NOLVADEX®), ралоксифен EVISTA®, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон и торемифен FARESTON®; антипрогестероны; понижающие регуляторы рецептора эстрогена (ERD); средства, которые функционируют, подавляя или прекращая работу яичников, например агонисты рилизинг-гормона лютеинизирующего гормона (LHRH), такие как ацетат лейпролида LUPRON® и ELIGARD®, ацетат гозерелина, ацетат бузерелина и триптерелин; другие антиандрогены, такие как флутамид, нилутамид и бикалутамид; и ингибиторы ароматазы, которые ингибируют фермент ароматазу, которая регулирует продукцию эстрогена в надпочечниках, такие как, например, 4(5)-имидазолы, аминоглутетимид, ацетат мегестрола MEGASE®, эксеместан AROMASIN®, форместан, фадрозол, ворозол RIVISOR®, летрозол FEMARA® и анастрозол ARIMIDEX®. Кроме того, такое определение химиотерапевтических средств включает бисфосфонаты, такие как клодронат (например, BONEFOS® или OSTAC®), этидронат DIDROCAL®, NE-58095, золедроновую кислоту/золедронат ZOMETA®, алендронат FOSAMAX®, памидронат AREDIA®, тилудронат SKELID® или ризедронат ACTONEL®; а также троксацитабин (1,3-диоксолановый нуклеозидный аналог цитозина); антисмысловые олигонуклеотиды, в частности олигонуклеотиды, которые ингибируют экспрессию генов в путях передачи сигналов, вовлеченных в аномальную пролиферацию клеток, таких как, например, РKС-альфа, Raf, H-Ras и рецептор эпидермального фактора роста (EGF-R); вакцины, такие как вакцина THERATOPE® и вакцины для генной терапии, например вакцина ALLOVECTIN®, вакцина LEUVECTIN® и вакцина VAXID®; ингибитор топоизомеразы 1 LURTOTECAN®; rmRH ABARELIX®; дитозилатлапатиниба (низкомолекулярный двойной ингибитор тирозинкиназ ErbB-2 и EGFR, также известный как GW572016); и фармацевтически приемлемые соли, кислоты или производные любого из указанных выше средств.
ПРИМЕРЫ РЕАЛИЗАЦИИ ПРЕДЛОЖЕННОГО ИЗОБРЕТЕНИЯ
Пример 1. Получение биомассы рекомбинантного фрагмента АФП (r3D AFP)
Работы с бактериальными штаммами проводили под ламинаром в стерильных условиях. Для трансформации использовали компетентные клетки Вl21 (DE3) фирмы Stratogen. Пробирку с клетками (0.1 мл) размораживали на льду, добавляли 5 мкл раствора плазмиды в концентрации 20 нг/мл и инкубировали 10-15 минут на льду, после чего подвергали тепловому шоку при 42°С в течение 2 минут. Затем пробирку помещали в лед на 30 с, далее добавляли 1 мл среды LB и инкубировали 30-45 минут при 37°С при перемешивании. Затем клетки осаждали центрифугированием (2000 g, 5 минут), удаляли 1 мл супернатанта, осадок ресуспендировали в оставшейся среде и высевали на чашки Петри с агаризованной средой LB, содержащей ампициллин (100 мкг/мл). 2-3 колонии трансформантов помещали в 10 мл среды LB, содержащей ампициллин (100 мкг/мл) и инкубировали при 37°С при активном перемешивании в течение ночи. Из полученной жидкой культуры производили посев на чашку Петри с агаризованной средой LB, содержащей ампициллин (100 мкг/мл). Чашку инкубировали при 37°С в течение ночи. Из выросших колоний отбирали 6 наиболее крупных, каждую из которых помещали в 10 мл среды LB с добавлением ампициллина и инкубировали при 37°С. По достижении оптической плотности OD600 от 0.5 до 1.2 единиц добавляют ИПТГ (изопропил-β-D-1-тиогалактопиранозида) до конечной концентрации от 0.1 до 2 мМ и инкубацию продолжают в течение 3 часов, после чего определяли уровень экспрессии целевого белка с помощью электрофореза в ПААГ. Полученную биомассу центрифугируют в течение 15 мин при 10000 RPM.
Пример 2. Очистка и выделение рекомбинантного фрагмента АФП (r3D AFP)
Влажную биомассу (6 г) ресуспензировали в 100 мл буфера рН=8,0, содержащего 50 мМ фосфата натрия и 2 таблетки ингибиторов протеаз без ЭДТА (Roche). Далее добавляли лизоцим до концентрации 50 мг/л и инкубировали 10 мин при комнатной температуре, перемешивая стеклянной палочкой. Далее проводили ультразвуковую обработку 7 раз по 40 ударов при 0°С. Затем добавляли дезоксихолат натрия до 0,2% масс., и перемешивали 10 мин при комнатной температуре. Далее центрифугировали 10 мин в режиме 10000 об/мин. Собирали растворимую фракцию. Осадок несколько раз промывали фосфатным буфером и центрифугировали при 10000 об/мин. Осадок телец включения и растворимую фракцию хранили при -20°С. Осадок телец включения ресуспендировали в 10 мл солюбилизирующего буфера (150 мMNaCl, 10 мМ КН2РO4, 8 М мочевина, 12 мМмеркаптоэтанол, рН 7.4) и оставляли при перемешивании при комнатной температуре на 1 час. Полученный раствор центрифугировали (10000 об/мин, 10 мин) и использовали в дальнейшем для рефолдингасупернатант. Через колонку с носителем Superose 12 предварительно пропускали 5 объемов ренатурирующего буфера (150 MMNaCl, 10 мМ КН2РO4, 1 мМ GSSG, 5 мМ GSH, рН 8.5, +4°С). Затем готовили разведения солюбилизирующего буфера ренатурирующим, по 0,5 мл, до конечной концетрации мочевины в растворе 8, 6, 4, 2 и 0 М и меркаптоэтанола 12, 9, 6, 3, 0 мМ соответственно, и наносили на колонку, начиная с раствора с самой низкой концентрацией денатурирующих агентов. На подготовленную колонку наносили 2 мл солюбилизированного r3D AFP (k) (не более 20 мг), затем 0,5 мл солюбилизирующего буфера и элюировали целевой белок 3 объемами ренатурирующего буфера. Полученную фракцию диализовали против 1000-кратного объема фосфатно-солевого буфера при рН=8.5 и хранили при -70°С. Рекомбинантный белок дополнительно очищали с помощью металло-хелатной хроматографии на носителе содержащем ионы Ni2+ и, при необходимости, концентрировали с помощью концентрирующих ячеек.
Пример 3. Получение полимерных частиц с производным альфа-гидроксибензолпропановой кислоты
Для включения цитостатического вещества в состав наночастиц был использован метод одинарного эмульгирования [14, 15, 16]. Навеску 100.0 мг сополимера молочной и гликолевой кислот и 10.0 мг производного альфа-гидроксибензолпропановой кислоты растворяют в 5 мл хлористом метилене. Раствор по каплям добавляют к 50.0 мл 0,5% водного раствора поливинилового спирта. Полученную суспензию обрабатывают ультразвуком (24000 об/мин, 7 раз по 40 сек с перерывами по 20 сек) с помощью погружного гомогенизатора. С помощью роторного вакуумного испарителя удаляют органический растворитель. Далее раствор центрифугируют при 14000 g в течение 40 мин, осадок суспендируют в дистиллированной воде и лиофилизуют при 0,03-0,05 мбар в течение суток с добавлением 1% по массе D-маннита.
Количество производного альфа-гидроксибензолпропановой кислоты, включенного в состав наночастиц, определяют с помощью ВЭЖХ. Для определения концентрации производного альфа-гидроксибензолпропановой кислоты в полученном растворе используют хроматографическую систему высокого давления фирмы Waters W1525 с УФ-детектором Waters 2487 DUAL ABSORB ANCEDETEC-TOR (США) и колонку Hypersil-ODS 4,6 × 150 mm, 5 μn, 100 A (Phenomenex). В качестве подвижной фазы используют 60-% раствор ацетонитрила в воде (по объему) с добавлением фосфорной кислоты до 0,1%, детекцию производят при длине волны 227 нм.
Пример 4. Получение лекарственной формы (ЛФ) конъюгата полимерных частиц, содержащих цитостатический препарат, с рекомбинантным белком r3D AFP («Паклитаксел-НК»)
К 8 г полимерных частиц, полученных в примере 3, заранее отмытых от избытка поверхностно-активного вещества, растворенных в 1*PBS, добавляют водорастворимый карбодиимид и NHS (концентрация растворов 50 мг/мл в 1*PBS), затем к реакционной смеси добавляют 2-меркаптоэтанол, после чего к активированным полимерным частицам добавляют рекомбинантный белок r3D AFP не менее чем в 7-кратном недостатке в отношении активированных карбоксильных групп и доводят рН реакционной смеси до щелочных значений (от 7.4 до 9). Для остановки реакции вводят этаноламин (или любого аминопроизводного с аналогичными свойствами), после чего реакционную смесь очищают с использованием гель-экслюзионной хроматографии на колонке с носителем Sephadex G-25 с полным разделением продуктов реакции, и тем самым, выделением целевого продукта в чистом виде. Далее фракцию с очищенным продуктом реакции лиофилизируют с добавлением криопротектора (1 масс. %) в течение суток, после чего хранят в виде лиофилизата при -70С. Выход целевого продукта составляет не менее 7 г лиофилизата лекарственной формы.
Количество производного альфа-гидроксибензолпропановой кислоты, в составе лиофилизата лекарственной формы, определяют с помощью ВЭЖХ. Для определения концентрации производного альфа-гидроксибензолпропановой кислоты используют хроматографическую систему высокого давления фирмы Waters W1525 с УФ-детектором Waters 2487 DUALАВSORBANCEDETECTOR (США) и колонку Hypersil-ODS 4,6 × 150 mm, 5 μm, 100 A (Phenomenex). В качестве подвижной фазы используют 60-% раствор ацетонитрила в воде (по объему) с добавлением фосфорной кислоты до 0,1%, детекцию производят при длине волны 227 нм.
Количество r3D AFP в составе лиофилизата лекарственной формы определяют с помощью SDS-PAGE электрофореза в восстанавливающих и не восстанавливающих условиях.
Пример 5. Исследование интернализации ЛФ «Паклитаксел-НК» опухолевыми клетками in vitro
Связывание с поверхностью и интернализацию ЛФ исследовали с помощью проточной цитометрии. Для исследования интернализации наряду с обычной была использована ЛФ «Паклитаксел-НК», содержащая ФИТЦ-меченный рекомбинантный белок r3D AFP - r3D AFP-ФИТЦ (ЛФ-ФИТЦ). В качестве контрольных клеток, на поверхности которых рецепторы AFP отсутствуют, использовали мононуклеарные лейкоциты периферической крови человека. В качестве конкурентных ингибиторов связывания с рецептором АФП использовали r3D AFP-ФИТЦ и немодифицированные частицы, на основе которых получают ЛФ. Для исследования к суспензии клеток добавляли ЛФ-ФИТЦ, смесь ЛФ и r3D AFP-ФИТЦ (молярное соотношение 1 к 1), смесь ЛФ и r3D AFP-ФИТЦ (молярное соотношение 10 к 1), смесь ЛФ-ФИТЦ и полимерных частиц (массовое отношение ЛФ-ФИТЦ и частиц 1 к 1), смесь ЛФ-ФИТЦ и полимерных частиц (массовое отношение ЛФ-ФИТЦ и частиц 1 к 10) и инкубировали в течение 1 ч при 37°С (интернализация) или при 4°С (связывание с поверхностью).
По способности влиять на специфическую интернализацию ЛФ «Паклитаксел-НК», судят о ее способности взаимодействовать с рецептором АФП, то есть способности выполнять специфическое фармакологическое действие. Об уровне интернализации свидетельствуют средние значения флуоресценции клеток, инкубированных с образцами [17].
Из представленных на Фиг. 2 данных видно, что флуоресцентно меченая форма ЛФ (ЛФ-ФИТЦ) связывается с поверхностью и интернализуется всеми линиями клеток, за исключением контрольной. При этом добавление немеченой формы ЛФ к r3D AFP-ФИТЦ в эквимолярных по белку количествах или 10-кратном избытке, нарушало связывание и поступление последней в опухолевые клетки. Добавление избытка полимерных частиц к растворам r3D AFP-ФИТЦ и ЛФ-ФИТЦ не нарушало их поступления в опухолевые клетки.
Пример 6. Исследование цитотоксической активности ЛФ «Паклитаксел-НК» в отношении опухолевых клеток in vitro
Цитотоксическую активность ЛФ «Паклитаксел-НК» оценивали с помощью МТТ-теста по методу Mosmann после 72 ч инкубации клеток SKOV3, SKVLB, Р388/РТХ, опухоли яичника крыс штамма ОЯ и мононуклеарных лейкоцитов периферической крови с ЛФ и препаратом сравнения - субстанцией паклитаксела [18].
По цитотоксической активности ЛФ (выраженной в значении IC50 (концентрация агента при которой происходит ингибирование роста клеток на 50%)) в отношении опухолевых клеток судят о потенциальном специфическом и противоопухолевом эффекте in vivo.
Цитотоксическую активность конъюгата оценивали с помощью МТТ-теста после 72 ч инкубации клеток SKOV3, SKVLB, Р388/РТХ, опухоли яичника крыс штамма ОЯ и лимфоцитов периферической крови человека с ЛФ «Паклитаксел-НК» и препаратом сравнения - субстанция паклитаксела, Фиг. 3.
В целом, ЛФ и препарат сравнения проявляли схожий уровень цитотоксической активности, в частности в отношении клеток линий SKOV3 и Р388/PТХ, а также лимфоцитов. При этом следует отметить, что ЛФ проявляла значительно более высокий (IC50 73 нМ) уровень активности в отношении устойчивой линии SKVLB, ведение которой осуществлялось в присутствие поддерживающей концентрации паклитаксела, в отличие от препарата сравнения (IC50 312 нМ). Аналогичная, но менее выраженная картина была характерна в отношении клеток штамма ОЯ (IC50 ЛФ 59 нМ, IC50 препарата сравнения 196 нМ).
Пример 7. Исследование специфической фармакологической активности ЛФ «Паклитаксел-НК» in vivo
Изучение специфического фармакологического действия ЛФ «Паклитаксел-НК» проводили на самках мышей линий DBA/2 с привитой подкожно линией опухолевых клеток Р388/РТХ (1*106 кл/мышь), на ксенотрансплантатах клеток аденокарциномы яичника человека линии SKOV3 на самках мышей линии BALB/c nude, а также асцитной модели опухоли яичника крыс штамма ОЯ на самках крыс линии Wistar в 2-х дозах: ТД (терапевтическая доза) (24.57 мг/кг) и МПД (максимально переносимая доза) (245.7 мг/кг) при в/в введении (для крыс) и ТД (0.92 мг/кг) и МПД (9.2 мг/кг) при в/в введении (для мышей), определенных в ходе эксперимента по изучению острой токсичности ЛФ «Паклитаксел-НК» на крысах линии Wistar и мышах линии BALB/c.
Частота введения препарата (1 раз в 5 суток) была подобрана исходя из данных полученных в ходе изучения фармакокинетики ЛФ «Паклитаксел-НК», а также в ходе изучения высвобождения паклитаксела. Опытным группам крыс внутривенно вводилась ЛФ «Паклитаксел-НК» в дозе в соответствующих для крыс или мышей ТД и МПД с периодичностью введения - одно внутривенное введение в 5 дней (3-х кратно). Препарат сравнения - субстанция паклитаксела, вводился в/в в аналогичных ТД и МПД дозах, рассчитанных по содержанию паклитаксела в ЛФ «Паклитаксел-НК» с периодичностью введения - одно внутривенное введение в 5 дней. Препараты вводились животным в виде суспензии в стерильном растворе натрия хлорида 0.9% для инъекций внутривенно с помощью одноразового шприца. Максимальный объем однократного введения не превышал 0.2 мл при внутривенном введении мышам и 0.5 мл при внутривенном введении крысам. В качестве контроля использовались животные с введением стерильного раствора натрия хлорида 0.9% для инъекций. Для каждого эксперимента было сформировано по 5 групп из самок крыс или мышей.
В ходе исследований регистрировали продолжительность жизни экспериментальных животных, а также динамику формирования опухоли (при подкожной трансплантации).
Расчет увеличения продолжительности жизни проводится по окончании исследования и гибели всех животных. Определяется средняя продолжительность жизни (СПЖ, дни) в группе и вычисляется показатель увеличения продолжительности жизни (УПЖ%) по формуле:
УПЖ% = (СПЖопыт - СПЖКонт)/СПЖконт × 100.
Количественные критерии активности:
УПЖ% <25% 0
УПЖ% 25-60% ±
УПЖ% 61-100% +
УПЖ% 101-200% или 61-100% при однократном введении ++
УПЖ% >100 + Излечение <50% или 101-200% при однократном введении +++
УПЖ% >200 + Излечение >50% или <50% при однократном введении +++.
Влияние на динамику роста опухолей для модели на основе линии Р388/РТХ рассчитывали по формуле:
ТРО% = (Vконт - Vопыт)/Vконт × 100;
для модели на основе линии SKOV3 (для сформированных опухолей, поскольку суспензию клеток вводят совместно с белками межклеточного матрикса) по формуле:
T/C% = Vопыт/Vконт × 100;
где V - средний объем опухоли (мм3 или см3) в получавшей препарат и контрольной группах, соответственно, на конкретный срок; Т - леченая группа; С - контрольная группа, Т/С - величина, обратная ТРО, используется в случаях, когда имеется стимуляция роста опухоли и во всех случаях лечения развившейся опухоли.
ТРО и Т/С рассчитываются на 1, 7 и 14 сутки после окончания лечения.
Количественные критерии оценки ингибирующего эффекта на опухолях животных:
ТРО <20% 0
ТРО 20-80% ±
ТРО 51-80% +
ТРО 81-90%++
ТРО 91-100% + <50% ПР/излечения +++
ТРО 91-100% + >50% ПР/излечения ++++.
Количественные критерии оценки активности на ксенографтах опухолей человека:
Т/С 51-100% 0
Т/С 36-50% +
Т/С 21-35% ++
Т/С 6-20% +++
Т/С <5% ++++.
Модель на основе лимфолейкоза мыши линии Р388/РТХ
Для выявления противоопухолевого эффекта ЛФ «Паклитаксел-НК» и препарата сравнения использовали модель солидного лимфолейкоза мышей DBA/2 линии Р388/РТХ линии при 3-х кратном введении препаратов (1 раз в 5 дней) в ТД и МПД. Клетки линии Р388/РТХ прививали подкожно (1*106 кл/100 мкл). Лечение начинали через сутки после прививки опухолей. В ходе введения препаратов животные чувствовали себя хорошо, признаков интоксикации не выявлено.
СПЖ контрольной группы животных составила 17.2 суток. В группе, получавшей препарат сравнения - субстанцию паклитаксела в ТД, наблюдалось УПЖ на 18%, в группе, получавшей МПД - на 12.8%. В группе, получавшей ЛФ в МПД наблюдалось УПЖ на 33.7%. Наиболее значимый эффект наблюдался в группе, получавшей ЛФ в ТД - УПЖ составило 60.5%.
Показатели ТРО приведены в таблице 1.
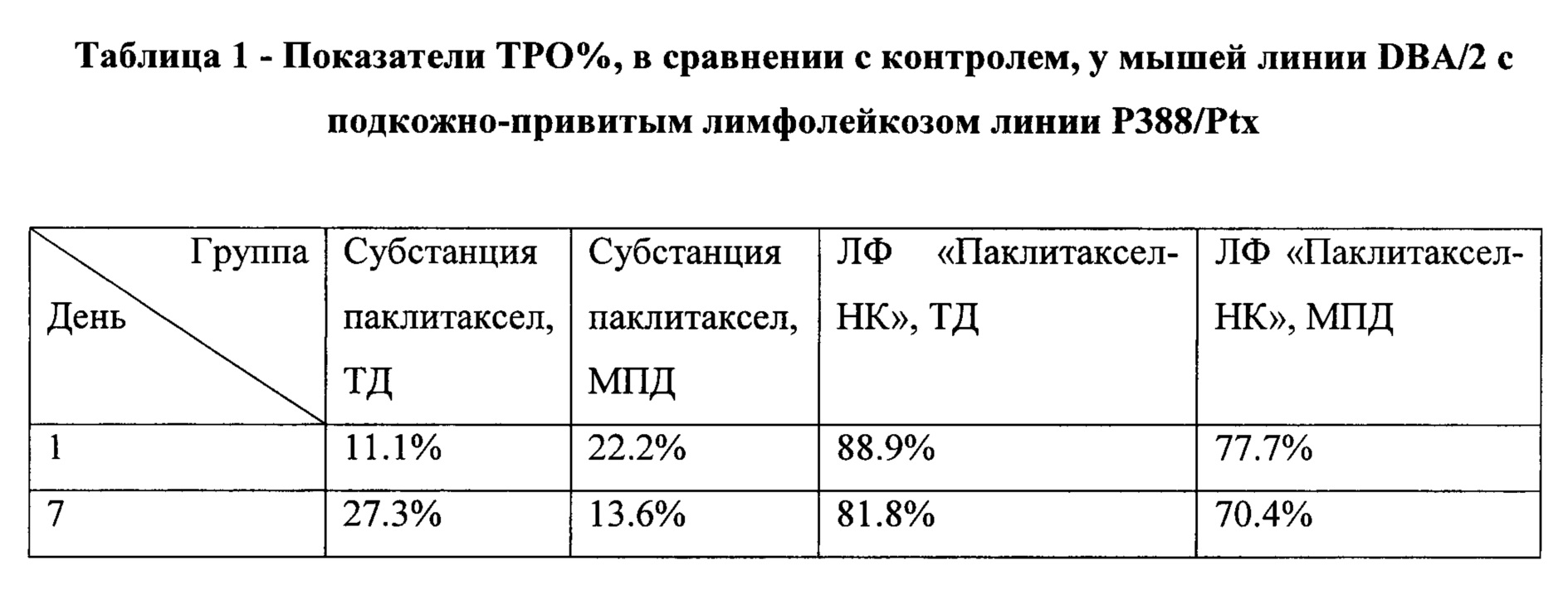
Таким образом, наиболее выраженным противоопухолевым действием обладала ЛФ «Паклитаксел-НК» в ТД как по показателю УПЖ, так и ТРО.
Модель на основе опухоли яичника крыс штамма ОЯ
Для выявления противоопухолевого эффекта ЛФ «Паклитаксел-НК» и препарата сравнения использовали модель опухоли яичника штамма ОЯ на крысах линии Wistar при 3-х кратном введении препаратов (1 раз в 5 дней) в ТД и МПД. Поскольку данный штамм не способен формировать солидную опухоль при подкожном введении, но при этом отличается быстрым развитием и экспрессией рецептора АФП, клетки штамма ОЯ прививали внутрибрюшинно (10*106 кл/100 мкл). Лечение начинали на вторые сутки после прививки опухолей. В ходе введения препаратов животные чувствовали себя хорошо, признаков интоксикации не выявлено.
СПЖ контрольной группы животных составила 9.6 суток. В группе, получавшей препарат сравнения - субстанцию паклитаксела в ТД, наблюдалось УПЖ на 19.8%, в группе, получавшей МПД - на 11.4%. В группе, получавшей ЛФ в ТД наблюдалось УПЖ на 76%. Наиболее выраженный эффект наблюдался в группе, получавшей ЛФ в МПД - УПЖ составило 81.2%.
Таким образом, наиболее выраженным противоопухолевым действием по показателю УПЖ обладала ЛФ «Паклитаксел-НК» в МПД.
Модель на основе аденокарциномы яичника человека линии SKOV3
Для выявления противоопухолевого эффекта ЛФ «Паклитаксел-НК» и препарата сравнения использовали модель подкожных ксенотрансплантатов клеток аденокарциномы яичника человека линии SKOV3 иммунодефицитным мышам линии BALB/cnude при 3-х кратном введении препаратов (1 раз в 5 дней) в ТД и МПД. Клетки линии SKOV3 прививали подкожно в виде суспензии в смеси белков внеклеточного матрикса (Matrigel) (1.5*106 кл/100 мкл/мышь). Лечение начинали на вторые сутки после прививки опухолей. В ходе введения препаратов животные чувствовали себя хорошо, признаков интоксикации не выявлено.
СПЖ контрольной группы животных составила 33.3 суток. В группе, получавшей препарат сравнения - субстанцию паклитаксела в ТД, наблюдалось УПЖ на 9.9%, в группе, получавшей МПД - на 2.7%. В группе, получавшей ЛФ в МПД наблюдалось УПЖ на 39.6%. Наиболее значимый эффект наблюдался в группе, получавшей ЛФ в ТД - УПЖ составило 54%.
Показатели Т/С% приведены в таблице 2.
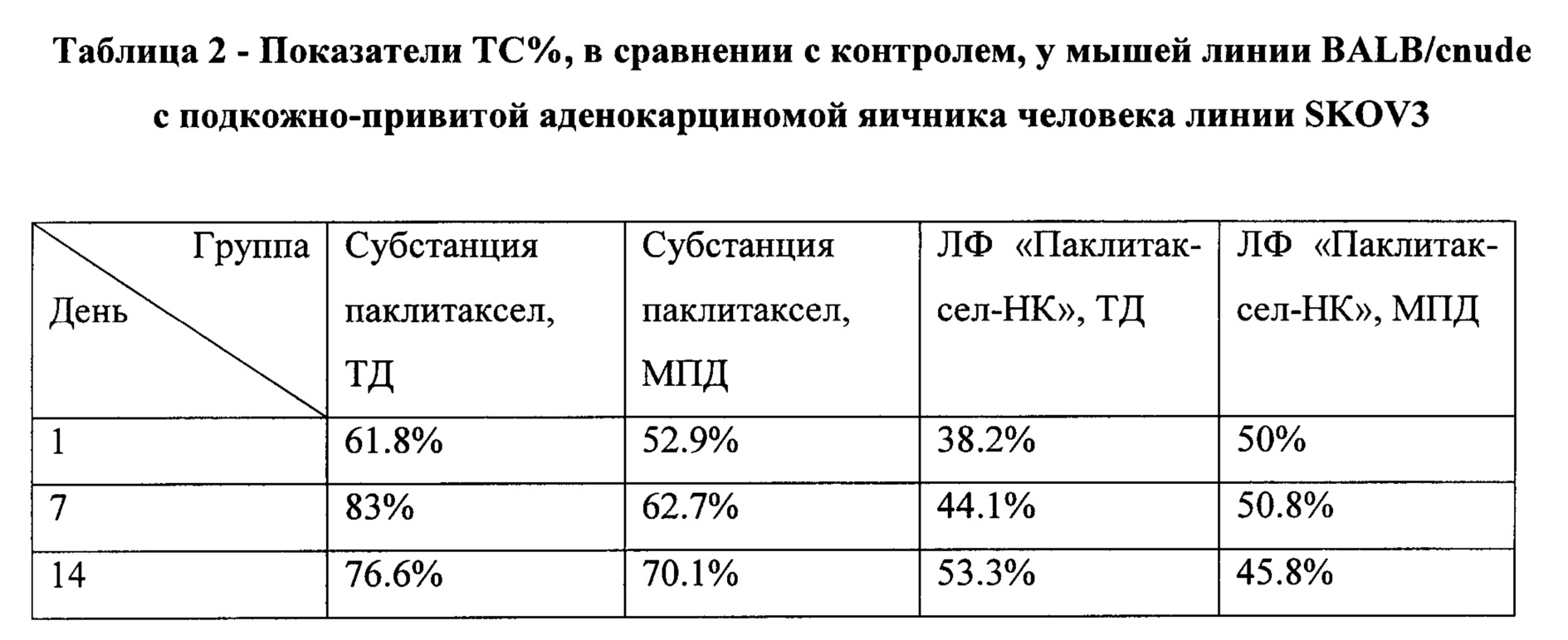
Таким образом наиболее выраженным противоопухолевым действием по параметру УПЖ обладала ЛФ «Паклитаксел-НК» в ТД, обладая при этом наиболее высокими значениями Т/С% на 1-ые и 7-ые сутки измерения. В то же самое время, ЛФ в МПД проявляла наибольшее значение Т/С% на 14-ые сутки измерения.
Противоопухолевый препарат применяется путем введения пациентам с аденокарциномой яичника в форме стерильной суспензии в водно-солевом растворе (физраствор) внутривенно, под контролем уровня лейкоцитов периферической крови, курсами до элиминации злокачественных образований.
Таким образом, предложенный противоопухолевый препарат обладает повышенной эффективностью относительно известных препаратов аналогичного назначения.
Предложенный противоопухолевый препарат проявляет более высокую противоопухолевую активность по сравнению со свободным паклитакселом в отношении клеток линии SKVLB, резистентной к антрациклиновым антибиотикам, и опухолевым клеткам штамма ОЯ, поскольку лекарственное средство проникает внутрь резистентных MDR1+, минуя действие трансмембранных MDR-насосов. Более того, ЛФ «Паклитаксел-НК» проявляет более высокую, в сравнении со свободным паклитакселом, специфическую фармакологическую активность (выраженную в показателях УПЖ%, ТРО% и ТС%) in vitro на моделях аденокарциномы яичника крыс штамма ОЯ, модели устойчивого к паклитакселу лимфолейкоза мешей линии Р388/РТХ и модели ксенотрансплантатов линии SKOV3.
СПИСОК ЛИТЕРАТУРЫ
1. FarokhzadO.C. Targeted nanoparticle-aptamerbioconjugates for cancer chemotherapy in vivo / Farokhzad O.C., Cheng J., Teply B.A., Sherifi I., Jon S., Kantoff P.W. // Proceedings of the National Academy of Sciences - 103 (16) - 2006- P. 6315-6320
2. Hitesh Kulhari. Peptide conjugated polymeric nanoparticles as a carrier for targeted delivery of docetaxel / Hitesh Kulhari, Deep Pooja, ShwetaShrivastava et al. // Colloids and Surfaces B: Biointerfaces 117 - 2014- P. 166-173
3. Li Wang. Monoclonal antibody targeting MUC1 and increasing sensitivity to docetaxel as a novel strategy in treating human epithelial ovarian cancer / Li Wang, Hongmin Chen, Fen-gHua Liu et al. // Cancer Letters - 2011 - Volume 300 - Issue 2, 28 - P. 122-133
4. Моro R., Tcherkassova J., Song E. et al. // IVD Technology Magazine - 2005. - Vol. 11 - №5
5. Урманчеева А.Ф. Таксаны в оптимальной химиотерапии рака яичника / Урманчеева А.Ф. // Актуальные вопросы клинической онкологии - 2003 - N 10. - С. 47-50
6. Kampan N. Paclitaxel and its evolving role in the management of ovarian cancer. / Kampan N.C., Madondo M.T., McNally O.M., Quinn M., Plebanski M. // Biomed Res Int. - 2015 - Vol 7. - 21 pages
7. Luo T. Synthesis and In Vitro Evaluation of Polyethylene Glycol-Paclitaxel Conjugates for Lung Cancer Therapy. / Luo T, Magnusson J, Preat V, Frederick R, Alexander C, Bosquillon C, Vanbever R. // Pharm. Res. - 2016. - Vol. 33 - Issue 7 - P. 1671-168
8. Tourlaki A. Paclitaxel as first or second-line treatment for HIV-negative Kaposi's sarcoma: a retrospective study of 58 patients. / Tourlaki A, Germiniasi F, Rossi L, Veraldi S, Brambilla L. // J Dermatolog Treat. - 2019. - Vol.21 - P. 1471-1753
9. Bharadwaj R. Topical delivery of paclitaxel for treatment of skin cancer. / Bharadwaj R, Das PJ, Pal P, Mazumder B. // Drug Dev. Ind. Pharm. - 2016. - Vol. 42(9) - P. 1482-1494
10. Ai B. Paclitaxel targets VEGF-mediated angiogenesis in ovarian cancer treatment. // Bin Ai, ZhixinBie, Shuai Zhang, Ailing Li. // J Cancer Res. - 2016. - Vol. 6(8) - P. 1624-1635.
11. Cortes J. Docetaxel. / Jorge E. Cortes, Richard Pazdur. // Journal of Clinical Oncology - 1995. - Vol 13. - No 10. - P. 2643-2655
12. Crown J. Docetaxel: overview of an active drug for breast cancer. / Crown J. // Oncologist- 2001. - Vol. 6 - P. 1-4.
13. Li W. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drag strategies. / Li W, Zhang H, Assaraf Y.G, Zhao K, Xu X, Xie J, Yang D.H, Chen Z.S. // Drug Resist Updat. - 2016. - Vol. 27 - P. 14-29
14. Северин E.C. Новые подходы к избирательной доставке лекарственных препаратов в опухолевые клетки. / Северин Е.С. // Успехи химии. - 2015. - Т. 84. - С. 43-60
15. Северин Е.С. Проблемы и перспективы современной противоопухолевой терапии / Северин Е.С., Родина А.В. // Успехи биологической химии. - 2000. - Т. 46. - С. 43-64
16. Nikolskaya E.D. Preparation of temozolomide-loaded polymer particles and study of their antitumor activity in models of glioma and melanoma. IE. D. Nikolskaya, O.A. Zhunina, E.A. Vasilenko, N.G. Yabbarov, O.G. Tereshchenko, M.B. Sokol, V.I. Popenko, E.S. Severin // Russian Journal of Bioorganic Chemistry. - 2017. - Vol.43. - No. 5. - P. 552-560
17. Гороховец H.B. Противоопухолевый пептидный препарат на основе фрагмента альфа-фетопротеина, его конъюгат, фармацевтическая композиция и способ лечения гормонзависимых опухолей. / Гороховец Н.В., Дигтярь А.В. и др. // Пат. RU 2285537 С; заявлено, 05.04.2005; опубл. 20.10.2006.
18. Mossman Т. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. / Mossman T. // J.Immunol. Meth. - 1983. - Vol.65. - No. 1-2. - P. 55-63.
--->
Перечень последовательностей
|
<---

Фотохимический способ получения стабилизированных наночастиц серебра
Фармацевтическая композиция для химиотерапии онкологических заболеваний и способ ее получения
Конъюгат противоопухолевых препаратов с рекомбинантным альфа-фетопротеином и его функциональными фрагментами и способ их получения
Лекарственное средство противомикробного действия, способ получения лекарственного препарата направленного действия, содержащего наночастицы

