Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-A]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к производным имидазо[1,2-a]пиридина, которые проявляет превосходную ингибирующую активность в отношении секреции кислоты желудочного сока, к способам их получения и к их применению.
Предпосылки изобретения
Желудочно-кишечные воспалительные заболевания или заболевания, связанные с выработкой кислоты желудочного сока, включая пептическую язву, язву желудка и двенадцатиперстной кишки, гастроэзофагеальную рефлюксную болезнь (ГЭРБ), неэрозивную рефлюксную болезнь (НЭРБ) и тому подобное, являются наиболее распространенными желудочно-кишечными заболеваниями, которые поражают значительную часть населения планеты, в том числе и в Корее.
Противоязвенные агенты, которые классифицируются как средства для лечения таких заболеваний, подразделяются на две категории: лекарственные средства, которые ингибируют агрессивные факторы, такие как кислота желудочного сока или пепсин; и препараты, усиливающие защитные факторы, такие как сукральфат или мизопростол. Ранее наряду с этими характерными терапевтическими агентами в качестве средств ингибирования агрессивных факторов использовались различные лекарственные средства, такие как антациды, антихолинергические лекарственные средства, антагонисты Н2-рецепторов, ингибиторы протонной помпы (ИПП) и тому подобное. Однако в настоящее время лидерами рынка являются препараты ингибиторов протонной помпы (ИПП), представленные омепразолом, лансопразолом, пантопразолом, равепразолом и тому подобное.
Протонная помпа представляет собой H+/K+-АТФазу, которая высвобождает H+ в париетальных клетках и поглощает K+ на заключительной стадии реакции секреции кислоты желудочного сока, вызванной связыванием различных стимуляторов секреции кислоты (гистамин, ацетилхолин, гастрин и тому подобное) рецепторами, присутствующими в париетальных клетках in vivo. Таким образом, ингибиторы протонной помпы (ИПП) представляют собой лекарственные средства, которые ингибируют секрецию кислоты желудочного сока путем ингибирования H+/K+-АТФазы париетальных клеток, которая является определяющим этапом секреции желудочной кислоты. Эти ингибиторы протонной помпы (ИПП) являются более эффективными и имеют более продолжительное действие при ингибировании кислоты желудочного сока по сравнению с предшествующими лекарственными средствами, и, таким образом, в течение последних 20 лет широко использовались в качестве терапевтических средств для лечения пептической язвы, язвы желудка и двенадцатиперстной кишки, гастрита, гастроэзофагеальной рефлюксной болезни (ГЭРБ) и тому подобное. В частности, гастроэзофагеальная рефлюксная болезнь (ГЭРБ) представляет собой хроническое рецидивирующее заболевание, число заболевших которым быстро увеличилось в последнее время, и является воспалительным заболеванием, которое на фоне болезни пищевода Барретта при переходе в хроническую стадию развивается в рак пищевода (аденокарциному). Лечение этой гастроэзофагеальной рефлюксной болезни (ГЭРБ) существенно ускорилось с момента появления ингибиторов протонной помпы (ИПП).
Однако сообщалось, что, поскольку существующие ингибиторы протонного насоса (ИПП) превращаются в активную сульфенамидную форму путем секреции кислоты, а затем необратимо связываются с остатками цистеина H+/K+-АТФазы, тем самым ингибируя секрецию кислоты желудочного сока в течение длительного периода, они могут вызывать неблагоприятные эффекты, в том числе рост бактерий желудка, стимуляцию экспрессии протонного насоса и образование опухолевых клеток вследствие гипергастринемии [Havu N, Digestion, 1986, 35 (Suppl 1), 42-55; Chang Seok Song, Dong Il Park, Korean J Med., 2011, 81(1), 6-10]. Кроме того, недавно сообщалось, что, когда эти ингибиторы протонного насоса (ИПП) используются в течение длительного периода времени, из-за ингибирования кислоты желудочного сока они ингибируют способность усваивать кальций, а также рост костных клеток, тем самым увеличивая риск переломов тазобедренного сустава, запястья и позвоночника [Yang YX, et al., JAMA, 2006, 296, 2947-53; Targownik LE, et al., CMAJ, 2008, 179(4), 319-26; Gray SL, et al., Arch Intern Med. 2010, 170(9), 765-71]. Кроме того, ввиду увеличения численности пожилого населения возросло применение нестероидных противовоспалительных препаратов (НПВП), а разработка различных медицинских технологий привела к увеличению выживаемости при различных заболеваниях, что привело к увеличению числа пациентов с пептической язвой и гастроэзофагеальной рефлюксной болезнью (ГЭРБ), вызванной различными причинами. Кроме того, число пациентов, резистентных к ингибиторам протонной помпы (ИПП), также увеличилось, несмотря на очень эффективную терапевтическую способность ингибиторов протонной помпы (ИПП).
Соответственно, наряду с последними ингибиторами протонной помпы (ИПП) растет интерес и потребность в препаратах калий конкурентных блокаторов кислоты (P-CAB, антагонистах кислотной помпы), обладающих механизмом, посредством которого они обратимо связываются с сайтом связывания H+/K+-АТФазы для ингибирования секреции кислоты калий-конкурентным способом. В частности, ожидается, что в отличие от необратимых ингибиторов протонного насоса (ИПП), обратимые калий-конкурентные блокаторы кислоты (P-CAB) благодаря их механизму действия будут проявлять быструю эффективность, их легко можно будет принимать до или после еды, и они очень эффективны при ночных симптомах, что является проблемой у необратимых ингибиторов протонной помпы.
Настоящее изобретение относится также к обратимым ингибиторам протонной помпы (P-CAB), и типичные примеры обратимых ингибиторов протонной помпы, известные в данной области техники, включают производное пиррола TAK-438 [Takeda Pharmaceutical Co. Ltd.; WO2007/026916], пирроло[2,3-c]пиридин YH-4808 [Yuhan Corp.; WO2006/025716], производное 1H-бензо[d]имидазола CJ-12420 [Pfizer Inc., Japan, Raqualia Pharma Inc.; WO2007/072146] и производное имидазо[1,2-a]пиридина AZD0865 [AstraZeneca AB; WO99/55705 и WO99/55706].
Соответственно, авторы настоящего изобретения провели исследование по разработке низкомолекулярных обратимых ингибиторов протонного насоса, которые можно эффективно использовать для предупреждения или лечения желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с кислотой желудочного сока, включая пептическую язву, язву желудка и двенадцатиперстной кишки, гастрит, гастроэзофагеальную рефлюксную болезнь (ГЭРБ), неэрозивную рефлюксную болезнь (НЭРБ) и тому подобное. В результате в ходе таких исследований авторы настоящего изобретения получили новые производные имидазо[1,2-a]пиридина и обнаружили, что эти производные проявляют превосходную ингибирующую активность в отношении протонного насоса, тем самым создав настоящее изобретение.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Техническая задача
Целью настоящего изобретения является производное имидазо[1,2-a]пиридина, обладающее превосходной ингибирующей активностью в отношении секреции кислоты желудочного сока, и к способу его получения.
Другой целью настоящего изобретения является фармацевтическая композиция для предупреждения или лечения заболевания, вызванных чрезмерной секрецией кислоты желудочного сока, где композиция содержит производное имидазо[1,2-a]пиридина в качестве активного ингредиента.
Еще одной целью настоящего изобретения является применение производного имидазо[1,2-a]пиридина для предупреждения или лечения заболевания, вызванного чрезмерной секрецией кислоты желудочного сока.
И еще одной целью настоящего изобретения является способ предупреждения или лечения заболевания, вызванного чрезмерной секрецией кислоты желудочного сока, где способ включает введение производного имидазо[1,2-a]пиридина.
Решение технической задачи
Для достижения вышеуказанных целей авторы настоящего изобретения получили новые производные имидазо[1,2-а]пиридина, и эти производные являются эффективными при лечении заболеваний, вызванных избыточной секрецией кислоты желудочного сока, путем ингибирования секреции желудочной кислоты.
Производные имидазо[1,2-a]пиридина
В настоящем описании предложено производное имидазо[1,2-a]пиридина, представленное следующей формулой 1, или его фармацевтически приемлемая соль:
Формула 1
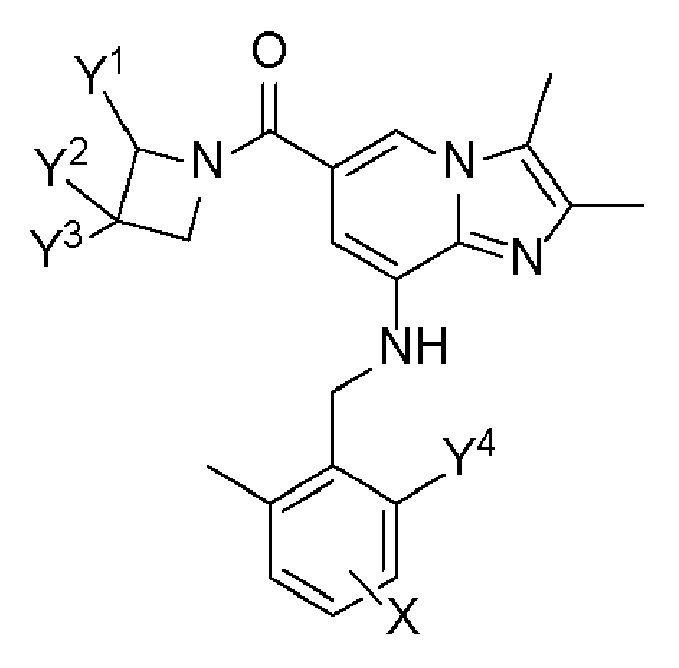
где
Y1, Y2 и Y3, каждый, независимо, представляют собой H, галоген, C1-C6 алкил с прямой или разветвленной цепью, незамещенный или замещенный R1, гидрокси, C1-C6 алкокси, незамещенный или замещенный R2, или -A-B, или Y2 и Y3 могут быть связаны друг с другом с образованием 4-6-членного гетероциклического кольца, содержащего один или два гетероатома, выбранных из группы, состоящей из N, O и S;
R1 и R2, каждый, независимо, представляют собой гидрокси или C1-C6 алкокси;
A представляет собой -C(=O)-, -C(=O)O-, -OC(=O)- или -S(=O)2-;
B представляет собой H или C1-C6 алкил с прямой или разветвленной цепью;
Y4 представляет собой H, C1-C6 алкил с прямой или разветвленной цепью или C1-C6 алкокси; и
X представляет собой H или галоген.
Согласно одному варианту осуществления настоящего описания
Y1 может представлять собой H или C1-C3 алкил с прямой или разветвленной цепью, незамещенный или замещенный R1;
Y2 и Y3, каждый, независимо, представляют собой H, F, Cl, C1-C3 алкил с прямой или разветвленной цепью, незамещенный или замещенный R1, гидрокси, C1-C3 алкокси, незамещенный или замещенный R2, или -A-B, или Y2 и Y3 могут быть связаны друг с другом с образованием 4-6-членного гетероциклического кольца, содержащего один или два гетероатома, выбранных из группы, состоящей из N, O и S;
R1 и R2 могут представлять собой, каждый, независимо, гидрокси или C1-C3 алкокси;
A может представлять собой -C(=O)-, -C(=O)O-, -OC(=O)- или -S(=O)2-;
B может представлять собой C1-C3 алкил с прямой или разветвленной цепью;
Y4 может представлять собой H, C1-C3 алкил с прямой или разветвленной цепью или C1-C3 алкокси; и
X может представлять собой H, F или Cl.
Согласно еще одному варианту осуществления настоящего изобретения,
Y1 может представлять собой H;
Y2 и Y3 могут представлять собой, каждый, независимо, H, F, Cl, C1-C2 алкил с прямой или разветвленной цепью, незамещенный или замещенный R1, или гидрокси;
R1 может представлять собой гидрокси;
Y4 может представлять собой H или C1-C2 алкил с прямой или разветвленной цепью; и
X может представлять собой H или F.
Согласно и еще одному варианту осуществления настоящего изобретения,
Y1 может представлять собой H;
Y2 и Y3 могут представлять собой, каждый, независимо, H, F, метил, гидроксиметил или гидрокси;
Y4 может представлять собой H или метил; и
X может представлять собой H или F.
В предпочтительном варианте осуществления производное имидазо[1,2-a]пиридина по настоящему изобретению может быть выбрано из группы, состоящей из следующих соединений:
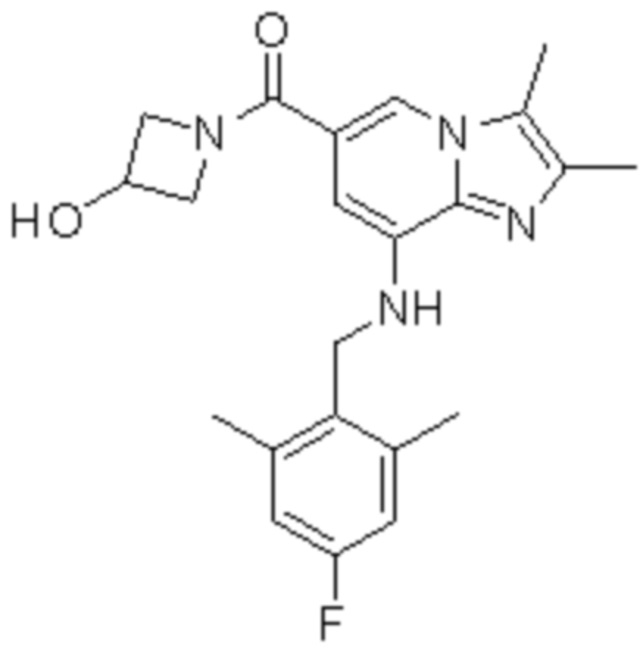
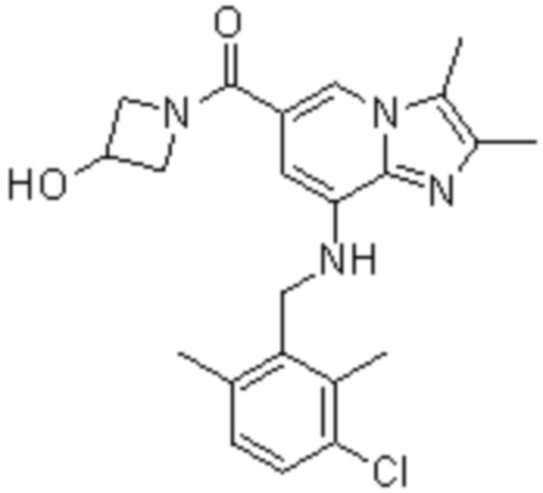
1) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидроксиазетидин-1-ил)метанон;
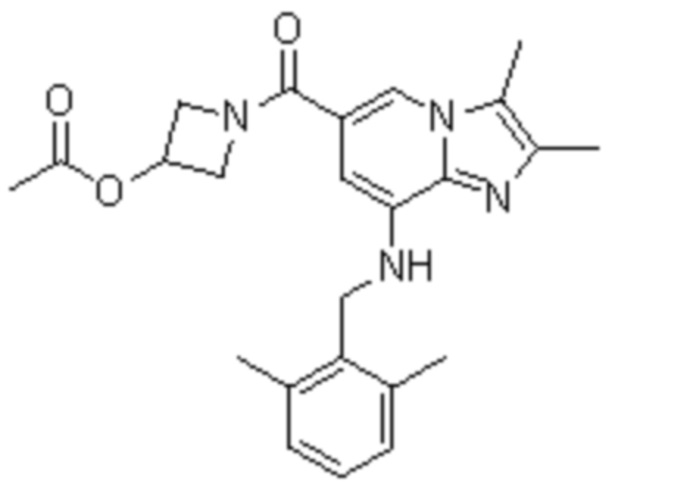
2) 1-{8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-карбонил}азетидин-3-ил ацетат;
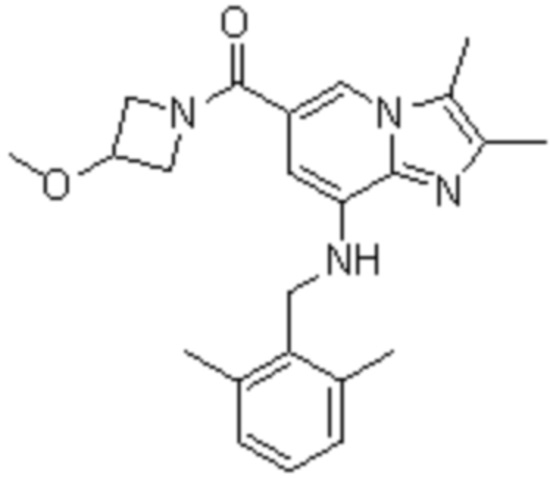
3) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-метоксиазетидин-1-ил)метанон;
4) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-этоксиазетидин-1-ил)метанон;
5) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(2-метоксиэтокси)азетидин-1-ил]метанон;
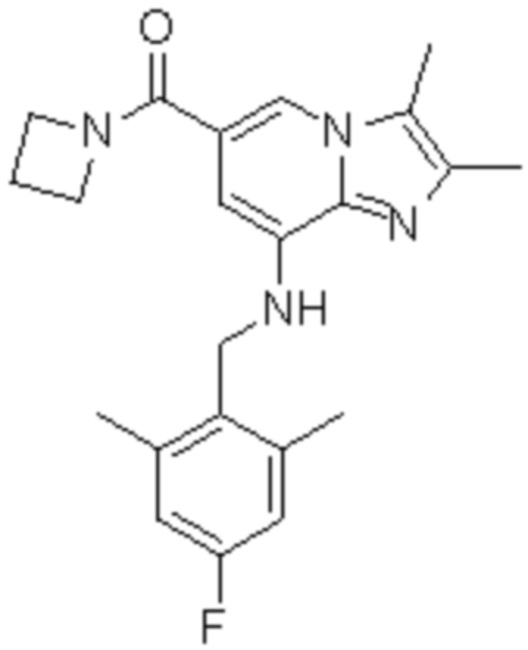
6) азетидин-1-ил{8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}метанон;
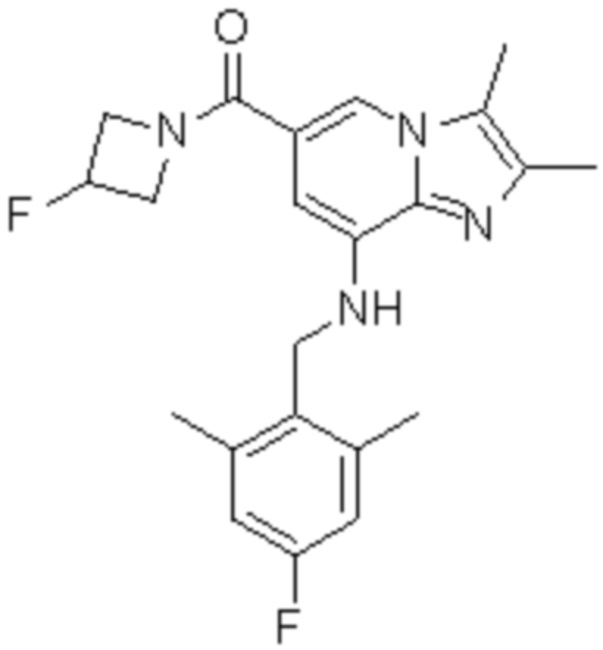
7) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-фторазетидин-1-ил)метанон;
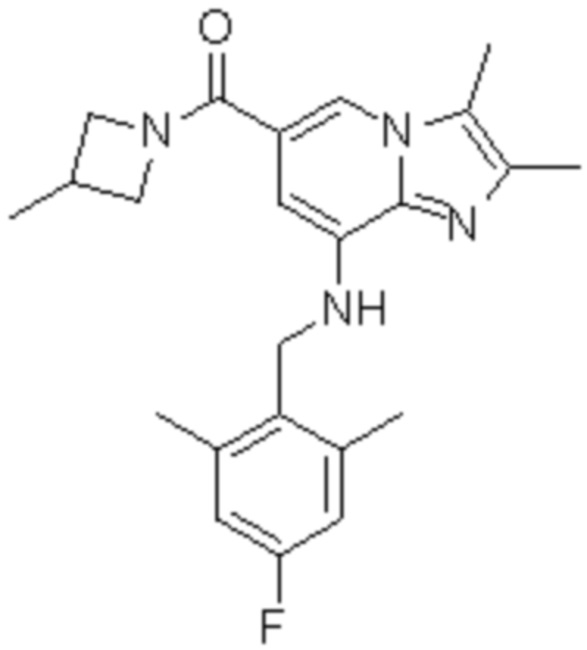
8) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-метилазетидин-1-ил)метанон;
9) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидрокси-3-метилазетидин-1-ил)метанон;
10) метил 1-{8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-карбонил}азетидинe-3-карбоксилат;
11) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(гидроксиметил)азетидин-1-ил]метанон;
12) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(метоксиметил)азетидин-1-ил]метанон;
13) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-фтор-3-(гидроксиметил)азетидин-1-ил]метанон;
14) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(гидроксиметил)-3-метилазетидин-1-ил]метанон;
15) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(1-гидроксиэтил)азетидин-1-ил]метанон;
16) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(2-гидроксипропан-2-ил)азетидин-1-ил]метанон;
17) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо [1,2-a]пиридин-6-ил}[2-(гидроксиметил)азетидин-1-ил]метанон;
18) 1-{1-[8-({2,6-диметилбензил}амино)-2,3-диметилимидазо[1,2-a]пиридин-6-карбонил]азетидин-3-ил}этанон;
19) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(метилсульфонил)азетидин-1-ил]метанон;
20) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(2-гидроксиэтил)азетидин-1-ил]метанон;
21) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(2,6-диазаспиро[3,3]гептан-2-ил)метанон;
22) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(2-окса-6-азаспиро[3,3]гептан-6-ил)метанон;
23) {2,3-диметил-8-[(2-метилбензил)амино]имидазо[1,2-a]пиридин-6-ил}(3-гидроксиазетидин-1-ил)метанон;
24) {8-[(5-фтор-2-метилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидроксиазетидин-1-ил)метанон;
25) (3-гидроксиазетидин-1-ил){8-[(2-метокси-6-метилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил)метанон;
26) [3-(гидроксиметил)азетидин-1-ил]{8-[(2-метокси-6-метилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}метанон;
27) {8-[(4-фтор-2-метилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидроксиазетидин-1-ил)метанон;
28) азетидин-1-ил{8-[(4-фтор-2-метилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}метанон;
29) {8-[(4-фтор-2-метилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-фторазетидин-1-ил)метанон;
30) {8-[(4-фтор-2-метилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-метилазетидин-1-ил)метанон;
31) {8-[(4-фтор-2-метилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидрокси-3-метилазетидинe-1-ил)метанон;
32) {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-метокси-3-метилазетидин-1-ил)метанон;
33) {8-[(4-фтор-2-метилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-(гидроксиметил)азетидин-1-ил)метанон;
34) {8-[(4-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидроксиазетидин-1-ил)метанон;
35) азетидин-1-ил{8-[(4-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}метанон;
36) {8-[(4-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-фторазетидин-1-ил)метанон;
37) {8-[(4-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-метилазетидин-1-ил)метанон;
38) {8-[(4-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидрокси-3-метилазетидин-1-ил)метанон;
39) {8-[(4-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(гидроксиметил)азетидин-1-ил]метанон;
40) {8-[(4-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(метоксиметил)азетидин-1-ил]метанон;
41) {8-[(3-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидроксиазетидин-1-ил)метанон;
42) азетидин-1-ил{8-[(3-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}метанон;
43) {8-[(3-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-фторазетидин-1-ил)метанон;
44) {8-[(3-фтор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(гидроксиметил)азетидин-1-ил]метанон;
45) азетидин-1-ил{8-[(3-хлор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}метанон;
46) {8-[(3-хлор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидроксиазетидин-1-ил]метанон;
47) {8-[(3-хлор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-фторазетидин-1-ил)метанон; и
48) {8-[(3-хлор-2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}[3-(гидроксиметил)азетидин-1-ил]метанон.
В настоящем изобретении фармацевтически приемлемые соли могут быть такими, которые обычно используются в данной области, такие как соли добавления кислот, которые образуются с фармацевтически приемлемыми свободными кислотами, но ими не ограничиваются. Фармацевтически приемлемые свободные кислоты могут быть органическими кислотами и неорганическими кислотами. Неорганические кислоты могут включать соляную кислоту, фосфорную кислоту, серную кислоту, азотную кислоту и тому подобное, и органическая кислота может включать метансульфоновую кислоту, п-толуолсульфоновую кислоту, уксусную кислоту, трифторуксусную кислоту, малеиновую кислоту, янтарную кислоту, щавелевую кислоту, бензойную кислоту, винную кислоту, фумаровую кислоту, миндальную кислоту, пропионовую кислоту, лимонную кислоту, молочную кислоту, гликолевую кислоту, глюконовую кислоту, галактуроновую кислоту, глутаминовую кислоту, глутаровую кислоту, глюкуроновую кислоту, аспарагиновую кислоту, аскорбиновую кислоту, угольную кислоту, виниловую кислоту и тому подобное.
Кроме того, с использованием основания может быть получена фармацевтически приемлемая соль металла. Например, соль щелочного металла или щелочноземельного металла соединения может быть получена путем растворения соединения в избытке раствора гидроксида щелочного металла или гидроксида щелочноземельного металла, фильтования нерастворенной соли соединения, выпаривания и сушки фильтрата. При этом фармацевтически приемлемая соль металла, подходящая для фармацевтического применения, представляет собой соль натрия, калия или кальция, но ими не ограничивается.
Кроме того, соединения формулы 1 согласно настоящему изобретению включают сольваты и гидраты, которые могут быть получены из фармацевтически приемлемых солей, и все возможные оптические изомеры, стереоизомеры и их смеси также входят в объем настоящего изобретения. При этом сольваты, гидраты и стереоизомеры соединений, представленных формулой 1, могут быть получены с использованием обычных способов, известных в данной области техники.
Кроме того, соединение формулы 1 согласно настоящему изобретению может быть получено в кристаллической форме или в аморфной форме. Когда соединение формулы 1 получают в кристаллической форме, оно может быть гидратировано или сольватировано.
Способы получения производного имидазо[1,2-a]пиридина
В настоящем описании предложены способы получения производного имидазо[1,2-a]пиридина, представленного формулой 1, или его фармацевтически приемлемых солей.
Предпочтительно, соединения формулы 1 могут быть получены способом, показанным на схеме реакции 1 ниже, но не ограничиваются этим. В частности, любому специалисту в данной области в достаточной степени понятно, что соединения формулы 1 согласно настоящему изобретению могут быть получены различными способами с использованием известных методик, хорошо известных в данной области.
Реакционная схема 1, показанная ниже, отражает каждую стадию способа получения типичных соединений в соответствии с настоящим изобретением, и различные соединения в соответствии с настоящим изобретением могут быть получены путем модификаций, при которых меняются реагенты, растворители и последовательности реакций, которые используются в процессе получения, показанные на схеме реакции 1 ниже. Некоторые соединения согласно настоящему изобретению могут быть получены в соответствии со способами, которые не попадают в рамки реакционной схемы 1, показанной ниже, и конкретные способы получения таких соединений будут подробно описаны в приведенных ниже примерах.
Способ получения 1
В частности, как показано на схеме реакции 1 ниже, производные формулы 1 или их фармацевтически приемлемые соли могут быть получены способом, включающим следующие стадии:
1) бензилирование алкилового эфира карбоновой кислоты формулы 2 замещенным бензилгалогенидом или бензальдегидом с получением соединения формулы 3 (стадия 1);
2) добавление по каплям водного раствора гидроксида калия или гидроксида натрия к соединению формулы 3, полученному на стадии 1, с получением таким образом гидролизованной карбоновой кислоты формулы 4 (стадия 2); и
3) амидирование карбоновой кислоты формулы 4, полученной на стадии 2, незамещенным или замещенным азетидином путем применения конденсирующего реагента, в результате чего получают соединение формулы 1.
Схема реакции 1
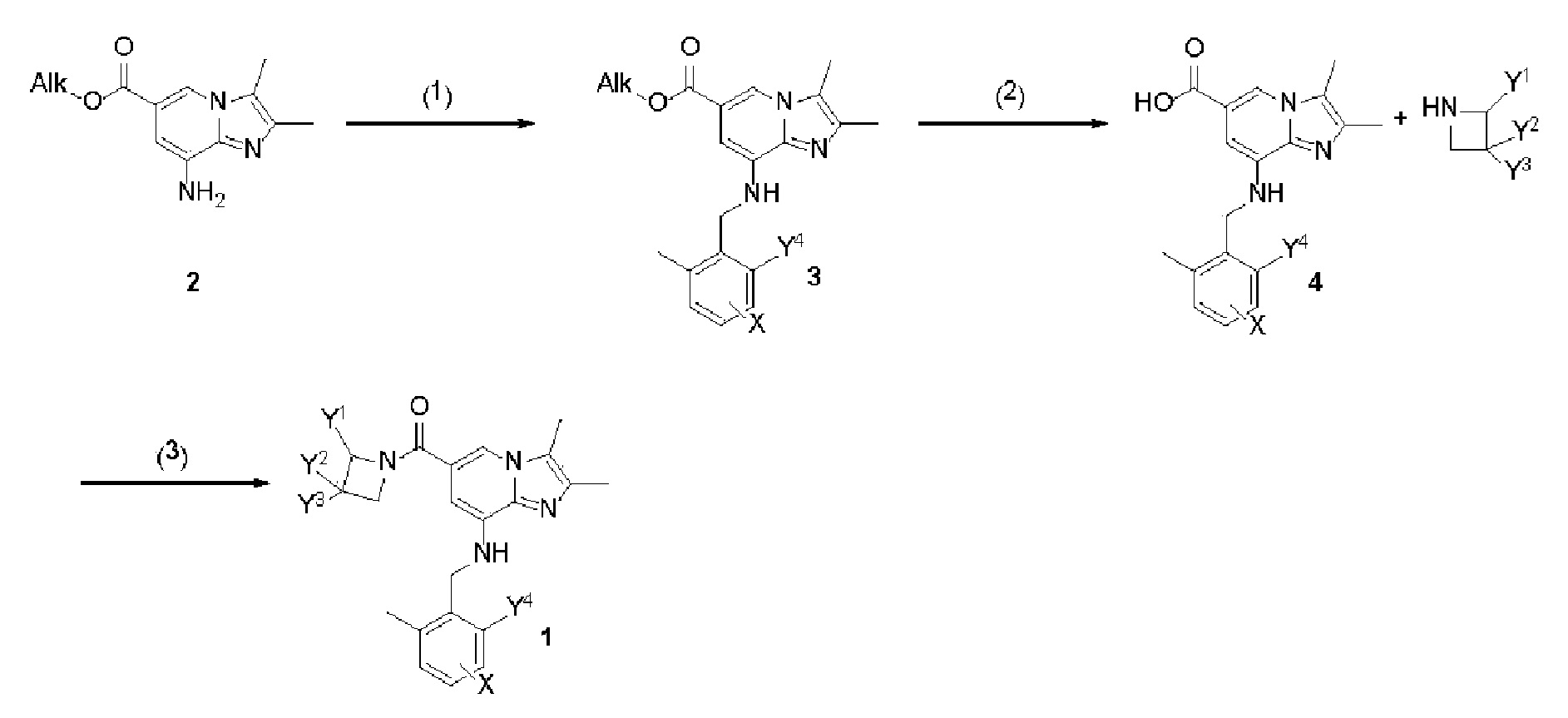
(где Y1-Y4 и X определены в формуле 1 выше, и Alk представляет собой алкильную группу, такую как метил, этил, изопропил или тому подобное. Предпочтительно, Alk представляет собой изопропил.)
Каждый из способов получения соединения формулы 1 будет подробно описан со ссылкой на схему реакции 1.
На стадии 1 алкиловый эфир карбоновой кислоты формулы 2, который может быть легко синтезирован известным методом (WO 99/055705 или WO 99/055706), подвергают взаимодействию с замещенным бензилгалогенидом (например, 2,6-диметилбензилхлорид или 4-фтор-2-метилбензилбромид) в присутствии основания, такого как карбонат калия, и каталитического количества йодида натрия, получая таким образом замещенный бензиламино имидазолопиридин, соединение формулы 3. Эта реакция представляет собой бензилирование соединения аминокислоты бензилгалогенидом и проводится в присутствии основания, которое может быть использовано при бензилировании. Примеры основания, которое можно использовать для этой цели, включают гидрид натрия (NaH), карбонат калия, карбонат натрия, карбонат цезия, алкоксиды натрия или калия и т. д. Кроме того, реакцию, предпочтительно, проводят в присутствии растворителя, который не оказывает негативного влияния на реакцию, и примеры этого растворителя включают дихлорметан, хлороформ, тетрагидрофуран, диэтиловый эфир, толуол, N,N-диметилформамид или ацетонитрил. Температура реакции конкретно не ограничиваеся, однако реакцию обычно можно проводить при пониженной температуре или при повышенной температуре, предпочтительно, при комнатной температуре или при повышенной температуре.
На стадии 2 водный раствор гидроксида калия или гидроксида натрия медленно добавляют по каплям к соединению формулы 3, полученному на стадии 1, получая таким образом гидролизованную карбоновую кислоту, соединение формулы (4). Реакцию на стадии 2 проводят в спиртовом растворителе, таком как метанол или этанол, который не оказывает негативного влияния на реакцию. Температура реакции конкретно не ограничивается, однако реакцию проводят при пониженной температуре или при повышенной температуре, предпочтительно, при комнатной температуре или при повышенной температуре. Эту реакцию можно проводить в обычных условиях гидролиза сложного эфира.
На стадии 3 карбоновую кислоту, соединение формулы 4, полученное на стадии 2, амидируют незамещенным или замещенным азетидином с использованием конденсирующего агента, получая таким образом соединение формулы 1. Используемый конденсирующий агент может представлять собой [1-(3-диметиламинопропил)-3-этилкарбодиимид] (EDCI), 1,3-дициклогексилкарбодиимид (DCC), 1,1-карбонилдиимидазол и тому подобное, которые являются коммерчески легко доступными. Хотя эта реакция может проводиться в отсутствие основания, ее предпочтительно проводят в растворителе, таком как ацетонитрил, диметилформамид или дихлорметан, который не оказывает негативного влияния на реакцию, в присутствии обычного основания, такого как 4-диметиламинопиридин, пиридин, триэтиламин, диэтилизопропиламин, N-метилморфолин или диметилфениламин, который может быть использован при амидировании. Температура реакции конкретно не ограничивается, однако реакцию проводят при пониженной температуре или при повышенной температуре, предпочтительно, при комнатной температуре или при повышенной температуре.
Целевые соединения, полученные в соответствии со способом, показанным на схеме реакций 1 выше, могут быть очищены с использованием обычных способов, например, с помощью колоночной хроматографии, перекристаллизации и тому подобного.
Соединения формулы 1 в соответствии с настоящим изобретением могут быть получены в виде фармацевтически приемлемых солей или их сольватов в соответствии с общепринятыми способами, известными в данной области.
Фармацевтическая композиция, содержащая производное имидазо[1,2-a]пиридина, ее применение и способ лечения заболевание с ее использованием
В настоящем описании предложена фармацевтическая композиция для предупреждения или лечения заболевания, вызванного чрезмерной секрецией кислоты желудочного сока, где композиция содержит производное имидазо[1,2-a]пиридина формулы 1 или его фармацевтически приемлемую соль.
Примеры заболевания, вызванного чрезмерной секрецией кислоты желудочного сока, включают желудочно-кишечные воспалительные заболевания или заболевания, связанные с выработкой кислоты желудочного сока. Желудочно-кишечные воспалительные заболевания или заболевания, связанные с выработкой кислоты желудочного сока, включают пептическую язву, язву желудка и двенадцатиперстной кишки, язву, вызванную приемом нестероидных противовоспалительных средств (НПВС), инфекцию хеликобактер пилори, несварение желудка, функциональное расстройство желудка, синдром Золлингера-Эллисона, гастрит, гастроэзофагеальную рефлюксную болезнь (ГЭРБ), ларингофарингеальный рефлюкс, неэрозивную рефлюксную болезнь (НЭРБ), висцеральную боль, злокачественное новообразование, изжогу, рвоту, эзофагит, дисфагию, гиперсаливацию, обструкцию дыхательных путей или астму.
Производное имидазо[1,2-a]пиридина в соответствии с настоящим изобретением обладает превосходной ингибирующей активностью в отношении секреции кислоты желудочного сока, и поэтому может быть эффективно использовано для предупреждения или лечения желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с выработкой кислоты желудочного сока, в частности, пептической язвы, язвы желудка и двенадцатиперстной кишки, гастроэзофагеальной рефлюксной болезни (ГЭРБ) и неэрозивной рефлюксной болезни (НЭРБ).
Фармацевтическая композиция, содержащая соединение по настоящему изобретению в качестве активного ингредиента, может быть составлена в соответствии со стандартной фармацевтической практикой для обеспечения пероральных составов, включая порошки, гранулы, таблетки, капсулы, суспензии, эмульсии, сиропы и аэрозоли, составы для наружного применения, суппозитории или стерильные инъекционные составы.
Конкретно, фармацевтическая композиция по настоящему изобретению может быть составлена с использованием разбавителей или наполнителей, таких как наполнители, сухие разбавители, связующие вещества, смачивающие агенты, разрыхлители, поверхностно-активные вещества и т.д., которые используются обычно. Твердые составы для перорального введения включают таблетки, пилюли, порошки, гранулы, капсулы и тому подобное, и такие твердые составы могут быть получены путем смешивания соединения по меньшей мере с одним или несколькими наполнителями, например, крахмалом, карбонатом кальция, сахарозой, лактозой или желатином. В дополнение к простым вспомогательным веществам могут также использоваться лубриканты, такие как стеарат магния или тальк. Жидкие составы для перорального введения включают суспензии, растворы, эмульсии и сироп и могут содержать различные вспомогательные вещества, например, смачивающие агенты, вкусовые добавки, ароматические вещества и консерванты, в дополнение к воде и жидкому парафину, которые являются часто используемыми простыми разбавителями. Препараты для парентерального введения включают стерилизованные водные растворы, неводные растворы, суспензии, эмульсии, лиофилизированные препараты и суппозитории. В качестве неводных растворителей или суспендирующих агентов можно использовать пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, инъецируемые сложные эфиры, такие как этилолеат и т.д. В суппозиториях могут использоваться витепсол, макрогол, твин 61, масло какао, лауриновый жир, глицерожелатин и тому подобное, но не ограничиваются ими.
Предпочтительная доза соединения формулы 1 в соответствии с настоящим изобретением или его фармацевтически приемлемой соли может варьироваться в зависимости от состояния пациента и массы тела, тяжести заболевания, формы лекарственного средства, а также пути и периода введения, и может быть соответствующим образом определена специалистом в данной области. Однако предпочтительно соединение по настоящему изобретению может вводиться один или несколько раз в день в дозе 0,0001-1000 мг/кг, предпочтительно, 0,01-500 мг/кг. Кроме того, композиция по настоящему изобретению может содержать соединение формулы 1 в количестве 0,0001-99 мас. %, предпочтительно, 0,01-50 мас. %, в зависимости от способа введения.
Фармацевтическая композиция по настоящему изобретению может дополнительно содержать, помимо соединения формулы 1 или его фармацевтически приемлемой соли, один или несколько активных ингредиентов, проявляющих лекарственный эффект, равный или подобный соединению формулы 1.
Более того, фармацевтическая композиция по настоящему изобретению может вводиться млекопитающим, включая крыс, мышей, домашний скот и людей, различными путями. Предполагаются все способы введения, например, введение может быть пероральным, ректальным, внутривенным, внутримышечным, подкожным, интратекальным или путем инъекции в сосуды головного мозга.
В настоящем изобретении предложено также применение производного имидазо[1,2-а]пиридина для предупреждения или лечения заболевания, вызванного чрезмерной секрецией кислоты желудочного сока.
Для получения лекарственного средства соединение, представленное формулой 1, может быть смешано с фармацевтически приемлемым адъювантом, разбавителем или носителем, а также может быть объединено с другими активными ингредиентами для проявления синергетического эффекта.
Настоящее изобретение относится также к способу предупреждения или лечения заболеваний, вызванных чрезмерной секрецией кислоты желудочного сока, включающему введение эффективного количества производного имидазо[1,2-а]пиридина млекопитающим, включая человека. Способ предупреждения или лечения заболевания в соответствии с настоящим изобретением также включает ингибирование или предотвращение симптомов заболевания, а также устранение самого заболевания до появления симптомов путем введения соединения формулы 1. Профилактическая или терапевтическая доза конкретного активного ингредиента при лечении заболевания будет варьироваться в зависимости от характера и серьезности заболевания или состояния, а также пути, которым вводится активный ингредиент. Доза и частота дозы также будут варьироваться в зависимости от возраста, массы тела и реакции конкретного пациента. Подходящие режимы дозирования могут быть легко выбраны специалистами в данной области с учетом таких факторов. Кроме того, способ предупреждения или лечения заболевания в соответствии с настоящим изобретением может дополнительно включать введение терапевтически эффективного количества дополнительного активного агента, способствующего лечению заболевания, вместе с введением соединения формулы 1. Дополнительный активный агент может проявлять синергический эффект или аддитивный эффект с соединением формулы 1.
Конкретные характеристики, указанные для фармацевтической композиции, способа применения и способа лечения по настоящему изобретению, используются аналогично, если только они не противоречат друг другу.
Положительный эффект
Производные имидазо[1,2-a]пиридина в соответствии с настоящим изобретением могут обратимо ингибировать протонную помпу, и поэтому могут быть эффективно использованы для предупреждения или лечения заболеваний, вызванных чрезмерной секрецией кислоты желудочного сока, в частности, пептической язвы, язвы желудка и двенадцатиперстной кишки, язвы, вызванной приемом нестероидных противовоспалительных средств (НПВС), инфекции хеликобактер пилори, функционального расстройства желудка, синдрома Золлингера-Эллисона, гастрита, гастроэзофагеальной рефлюксной болезни (ГЭРБ), неэрозивной рефлюксной болезни (НЭРБ) и т. д.
Описание рисунков
ФИГ. 1 представляет собой график, показывающий продолжительные ингибирующие эффекты соединений по настоящему изобретению на секрецию кислоты желудочного сока у крыс с перфузией в просвете желудка в зависимости от времени.
Вариант осуществления изобретения
Для лучшего понимания настоящего изобретения далее представлены предпочтительные примеры и примеры исследования. Следует учесть, однако, что эти примеры и примеры исследования предоставлены просто для облегчения понимания настоящего изобретения и не предназначены для ограничения объема настоящего изобретения.
Ссылочный пример 1: 2-(Бромметил)-5-фтор-1,3-диметилбензол
К 1-фтор-3,5-диметилбензолу (2,0 г, 16,1 ммоль) при комнатной температуре последовательно добавляли п-формальдегид (7,5 г, 250 ммоль), бромистый водород (33 масс% в уксусной кислоте; 35 мл) и уксусную кислоту (12 мл, 210 ммоль), затем перемешивали в течение 5 часов. Для прекращения реакции к реакционному раствору добавляли воду, затем экстрагировали диэтиловым эфиром. Органический слой сушили безводным сульфатом магния и затем концентрировали при пониженном давлении для удаления растворителя. Остаток очищали с помощью колоночной хроматографии (100% гексан) с получением указанного в заголовке соединения (1,0 г, выход: 30%) в виде твердого вещества желтого цвета.
1H ЯМР (400 MГц, CDCl3); δ 6,76 (с, 1H), 6,74 (с, 1H), 4,53 (с, 2H), 2,41 (с, 6H).
Ссылочный пример 2: 2-(Хлорметил)-1-метокси-3-метилбензол
Стадия 1: Синтез этил 2-метокси-6-метилбензоата
Этил 2-гидрокси-6-метилбензоат (1,0 г, 5,6 ммоль), метилйодид (866 мг, 6,1 ммоль) и карбонат калия (1,5 г, 11,1 ммоль) растворяли в ацетоне и затем кипятили с обратным холодильником в течение 17 часов. Для прекращения реакции к реакционному раствору добавляли воду, затем экстрагировали дихлорметаном. Органический слой сушили безводным сульфатом магния и затем концентрировали при пониженном давлении для удаления растворителя. Остаток очищали с помощью колоночной хроматографии (100% гексан) с получением указанного в заголовке соединения (1,1 г, выход: 99%).
1H ЯМР (400 MГц, CDCl3); δ 7,26-7,21 (м, 1H), 6,80-6,75 (м, 2H), 4,40 (квт, J=14,4, 7,2 Гц, 2H), 3,82 (с, 3H), 2,30 (с, 3H), 1,38 (т, J=7,2 Гц, 3H).
Стадия 2: Синтез (2-метокси-6-метилфенил)метанола
К литийалюминийгидриду (LAH, 365 мг, 9,62 ммоль) добавляли безводный тетрагидрофуран (20 мл) и охлаждали до температуры 0°C, после чего соединение (1,1 г, 5,6 ммоль), полученное на стадии 1, растворяли в безводном тетрагидрофуране (30 мл) и медленно по каплям при температуре 0°C добавляли в эту смесь. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов, и к нему добавляли 15% водный раствор гидроксида натрия для прекращения реакции. Затем реакционный раствор фильтровали через целит и концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии (гексан:этилацетат=20:1) с получением указанного в заголовке соединения (775 мг; выход: 91%).
1H ЯМР (400 MГц, CDCl3); δ 7,20-7,17 (м, 1H), 6,83 (м, 2H), 4,78 (с, 2H), 4,01 (с, 3H), 3,41 (с, 3H).
Стадия 3: 2-(Хлорметил)-1-метокси-3-метилбензол
Соединение (775 мг, 5,1 ммоль), полученное на стадии 2, растворяли в дихлорметане (30 мл) и к нему медленно по каплям при комнатной температуре добавляли тионилхлорид (1,0 мл, 15,2 ммоль), затем перемешивали в течение 2 часов. Для прекращения реакции к реакционному раствору добавляли ледяную воду, затем экстрагировали дихлорметаном. Органический слой сушили безводным сульфатом магния, и затем концентрировали при пониженном давлении для удаления растворителя, получая таким образом указанное в заголовке соединение (804 мг; выход: 93%).
1H ЯМР (400 MГц, CDCl3); δ 7,19 (т, J=8,0 Гц, 1H), 6,83 (м, 2H), 4,62 (с, 2H), 3,91 (с, 3H), 3,36 (с, 3H).
Ссылочный пример 3: 1-Хлор-3-(хлорметил)-2,4-диметилбензол
Стадия 1: Синтез 2,6-диметил-3-нитробензойной кислоты
К смеси концентрированной серной кислоты (8 мл) и 60% азотной кислоты (8 мл) при температуре 0°C добавляли 2,6-диметилбензойную кислоту (3,7 г, 24,64 ммоль), затем перемешивали в течение 1,5 часов. После завершения реакции при той же температуре в реакционный раствор добавляли воду, затем перемешивали в течение 0,5 часов. Полученный твердый продукт фильтровали при пониженном давлении, промывали водой и затем сушили, получая таким образом указанное в заголовке соединение (4,7 г; выход: 98%) в виде твердого вещества белого цвета.
1H ЯМР (400 MГц, CDCl3); δ 7,87 (д, J=8,0 Гц, 1H), 7,23 (д, J=7,6 Гц, 1H), 2,58 (с, 3H), 2,48 (с, 3H).
Стадия 2: Синтез метил 2,6-диметил-3-нитробензоата
Соединение (4,7 г, 24,08 ммоль), полученное на стадии 1, растворяли в N,N-диметилформамиде (15 мл) и к нему последовательно добавляли карбонат калия (6,7 г, 48,16 ммоль) и метилйодид (6,7 мл, 108,37 ммоль), затем перемешивали в течение ночи при температуре 0°C. Реакцию останавливали добавлением ледяной воды, и реакционный раствор экстрагировали этилацетатом и промывали водным раствором хлорида натрия. Отделенный органический слой сушили безводным сульфатом натрия и концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии (гексан:этилацетат=4:1) с получением указанного в заголовке соединения (3,8 г; выход 75%) в виде твердого вещества желтого цвета.
1H ЯМР (400 MГц, CDCl3); δ 7,84 (д, J=7,6 Гц, 1H), 7,19 (д, J=8,4 Гц, 1H), 3,96 (с, 3H), 2,46 (с, 3H), 2,36 (с, 3H).
Стадия 3: Синтез метил 3-амино-2,6-диметилбензоата
Соединение (3,8 г, 18,16 ммоль), полученное на стадии 2, растворяли в этилацетате (20 мл) и метаноле (20 мл) и к нему при комнатной температуре добавляли 10% палладий (760 мг, 20 масс%), затем перемешивали в течение ночи в атмосфере газообразного водорода. После завершения реакции реакционный раствор фильтровали через целит и концентрировали при пониженном давлении, получая таким образом указанное в заголовке соединение (3 г; выход: 92%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3); δ 6,86 (д, J=8,0 Гц, 1H), 6,63 (д, J=8,4 Гц, 1H), 3,90 (с, 3H), 3,55 (шир., 2H), 2,19 (с, 3H), 2,08 (с, 3H).
Стадия 4: Синтез метил 3-хлор-2,6-диметилбензоата
К соединению (2,0 г, 11,16 ммоль), полученному на стадии 3, добавляли воду (20 мл) и концентрированную соляную кислоту (20 мл), затем перемешивали в течение 10 минут. Реакционный раствор охлаждали до температуры -10°C, и к нему медленно по каплям добавляли раствор нитрита натрия (810 мг, 11,72 ммоль) в воде (2 мл). Спустя 30 минут в реакционный раствор при температуре -10°C медленно по каплям добавляли воду (10 мл) и раствор однохлористой меди (1,3 г, 13,39 ммоль) в концентрированной соляной кислоте (10 мл) при температуре 0°C, затем перемешивали при той же температуре в течение 1 часа. Затем реакционный раствор нагревали при температуре 70°C в течение 1 часа. После этого реакционный раствор охлаждали до комнатной температуры, экстрагировали этилацетатом и промывали водным раствором хлорида натрия, после чего отделенный органический слой сушили безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (гексан:этилацетат=10:1) с получением указанного в заголовке соединения (2,2 г; выход 99%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3); δ 7,28 (д, J=8,0 Гц, 1H), 6,98 (д, J=8,0 Гц, 1H), 3,92 (с, 3H), 2,30 (с, 3H), 2,26 (с, 3H).
Стадия 5: Синтез (3-хлор-2,6-диметилфенил)метанола
Соединение (2,8 г, 14,35 ммоль), полученное на стадии 4, растворяли в тетрагидрофуране (30 мл), и к раствору при температуре -78°C добавляли литийалюминийгидрид (590 мг, 15,78 ммоль), затем перемешивали при комнатной температуре в течение 4 часов. Реакцию останавливали добавлением 1н водного раствора гидроксида натрия при температуре 0°C, и реакционный раствор перемешивали в течение 30 минут, и затем фильтровали через целит при пониженном давлении и экстрагировали дихлорметаном. Отделенный органический слой промывали водным раствором хлорида натрия, сушили безводным сульфатом натрия и затем концентрировали при пониженном давлении, получая таким образом указанное в заголовке соединение (1,9 г; выход: 79%) в виде твердого вещества белого цвета.
1H ЯМР (400 MГц, CDCl3); δ 7,22 (д, J=8,0 Гц, 1H), 6,97 (д, J=8,4 Гц, 1H), 4,75 (д, J=4,8 Гц, 2H), 2,48 (с, 3H), 2,39 (с, 3H).
Стадия 6: Синтез 1-хлор-3-(хлорметил)-2,4-диметилбензола
Соединение (2,0 г, 11,72 ммоль), полученное на стадии 5, добавляли к безводному дихлорметану (20 мл), и к нему медленно при температуре 0°C добавляли по каплям тионилхлорид (1,3 мл, 17,58 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении для удаления растворителя и избытка тионилхлорида, и остаток очищали с помощью колоночной хроматографии (гексан:этилацетат=15:1) с получением указанного в заголовке соединения (1,8 г; выход 83%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3); δ 7,23 (д, J=8,0 Гц, 1H), 6,97 (д, J=8,4 Гц, 1H), 4,64 (с, 2H), 2,47 (с, 3H), 2,39 (с, 3H).
Ссылочный пример 4: 1-Фтор-3-(хлорметил)-2,4-диметилбензол
Стадия 1: Синтез метил 3-фтор-2,6-диметилбензоата
К соединению (4,3 г, 23,99 ммоль), полученному на стадии 3 ссылочного примера 3, добавляли воду (40 мл) и концентрированную соляную кислоту (40 мл), и раствор перемешивали в течение 10 минут. Реакционный раствор охлаждали до температуры -10°C, и к нему медленно добавляли по каплям раствор нитрита натрия (1,7 г, 11,72 ммоль) в воде (4 мл). Спустя 30 минут в реакционный раствор при температуре -10°C медленно по каплям добавляли раствор фторбората натрия (3,2 г, 28,79 ммоль) в воде (20 мл), затем перемешивали при той же температуре в течение 1 часа. Затем реакционный раствор нагревали до комнатной температуры и перемешивали в течение 1 часа. Полученный твердый продукт фильтровали при пониженном давлении, промывали холодной ледяной водой и метанолом и затем сушили при пониженном давлении, после чего добавляли толуол (20 мл), затем нагревали при температуре 120°C в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, и для прекращения реакции к нему добавляли ледяную воду. Затем реакционный раствор экстрагировали этилацетатом и промывали водным раствором хлорида натрия. Отделенный органический слой сушили безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (гексан:этилацетат=15:1) с получением указанного в заголовке соединения (1,8 г; выход: 81%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3); δ 6,99 (дд, J=8,2 Гц, 5,4 Гц, 1H), 6,94 (д, J=8,0 Гц, 1H), 3,92 (с, 3H), 2,26 (с, 3H), 2,20 (д, J=2,0 Гц, 3H).
Стадия 2: Синтез 2-(хлорметил)-4-фтор-1,3-диметилбензола
Соединение (1,8 г, 10,15 ммоль) подвергали взаимодействию так же, как описано на стадиях 5 и 6 ссылочного примера 3, получая таким образом указанное в заголовке соединение (1,3 г; выход: 77%) в виде бесцветного масла.
1H ЯМР (400 MГц, CDCl3); δ 6,99 (дд, J=8,2 Гц, 5,8 Гц, 1H), 6,90 (т, J=9,0 Гц, 1H), 4,62 (с, 2H), 2,38 (с, 3H), 2,32 (д, J=2,0 Гц, 3H).
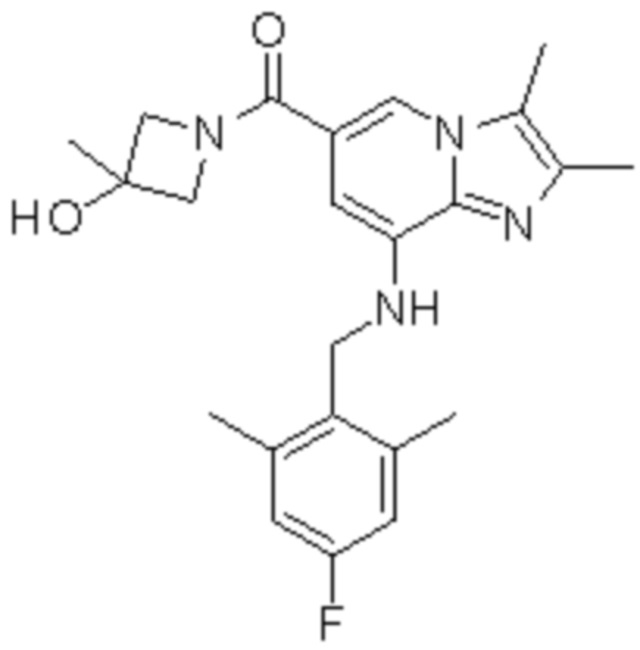
Пример 1: {8-[(2,6-Диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидроксиазетидин-1-ил)метанон
Стадия 1: Синтез изопропил 8-(2,6-диметилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-карбоксилата
К изопропил 8-амино-2,3-диметилимидазо[1,2-a]пиридин-6-карбоксилату (59 г, 18,0 ммоль) добавляли безводный 2-пропанол (590 мл) и последовательно добавляли карбонат калия (59 г, 44,9 ммоль) и йодид натрия (15 г, 10,0 ммоль). Смесь нагревали до температуры 70°C. При этой же температуре в реакционную смесь медленно по каплям добавляли раствор 2,6-диметилбензилхлорида (31 г, 20,0 ммоль) в безводном 2-пропаноле (60 мл), затем перемешивали в течение 4 часов. После этого в реакционный раствор добавляли раствор хлорида калия (54,7 г, 39,5 ммоль) в 2,6-диметилбензилхлориде (17 г, 10,8 ммоль) и безводный 2-пропанол (20 мл), затем перемешивали в течение 6 часов. Реакцию останавливали добавлением воды при комнатной температуре, и реакционный раствор перемешивали в течение 30 минут. Полученный твердый продукт фильтровали, промывали последовательно водой и холодным 2-пропанолом и затем сушили, получая таким образом указанное в заголовке соединение (55 г; выход: 88%) в виде твердого вещества белого цвета.
1H ЯМР (400 MГц, CDCl3); δ 8,25 (с, 1H), 7,30 (с, 1H), 7,15-7,09 (м, 1H), 7,06-7,04 (м, 2H), 6,73 (с, 1H), 5,31-5,26 (м, 1H), 4,84 (шир., 1H), 4,41 (д, J=4,4 Гц, 2H), 2,39 (с, 12H), 1,40 (д, J=6,4 Гц, 6H).
Стадия 2: Синтез 8-(2,6-диметилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-карбоновой кислоты
Соединение (92,5 г, 25,3 ммоль), полученное на стадии 1, растворяли в этаноле (920 мл), и в раствор при комнатной температурa добавляли 2н водный раствор гидроксида натрия (500 мл), затем перемешивали при температуре 80°C в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, и растворитель концентрировали при пониженном давлении, после чего к остатку добавляли воду, и в водный раствор при температуре 0°C медленно добавляли по каплям 2н водный раствор соляной кислоты (500 мл) до установления pH примерно 5. Затем раствор перемешивали в течение 8 часов, фильтровали, промывали водой и затем сушили, получая таким образом указанное в заголовке соединение (69 г; выход 85%) в виде твердого вещества белого цвета.
1H ЯМР (400 MГц, CDCl3+CD3OD); δ 8,36 (с, 1H), 7,32 (с, 1H), 7,16-7,14 (м, 1H), 7,11-7,10 (м, 2H), 4,50 (с, 2H), 2,52 (с, 3H), 2,42 (с, 9H).
Стадия 3: Синтез {8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-a]пиридин-6-ил}(3-гидроксиазетидин-1-ил)метанона
Соединение (30 мг, 0,06 ммоль), полученное на стадии 2, растворяли в безводном дихлорметане (5 мл). Затем в раствор при температуре 0°C последовательно добавляли 1-этил-3-(3-диметиламинопропил)-карбодиимид хлорид (EDCI, 18 мг, 0,09 ммоль), 1-гидрокси-бензотриазол гидрат (HOBt, 13 мг, 0,09 ммоль), 3-гидроксиазетидин (8,1 мг, 0,07 ммоль) и триэтиламин (34 мкл, 0,25 ммоль), затем перемешивали при комнатной температуре в течение 22 часов. Затем для прекращения реакции в реакционный раствор при температуре 0°C медленно добавляли водный раствор бикарбоната натрия, и реакционный раствор экстрагировали дихлорметаном. Отделенный органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (гексан:этилацетат=3:1) с получением указанного в заголовке соединения (35 мг; выход: 95%) в виде твердого вещества белого цвета.
1H ЯМР (400 MГц, CDCl3); δ 7,62 (с, 1H), 7,14-7,10 (м, 1H), 7,05-7,03 (м, 2H), 6,39 (с, 1H), 4,92 (шир., 1H), 4,77-4,71 (м, 1H), 4,53-4,49 (м, 2H), 4,14 (д, J=6,8, 2H), 4,15-4,13 (м, 2H), 2,38-2,34 (м, 12H).
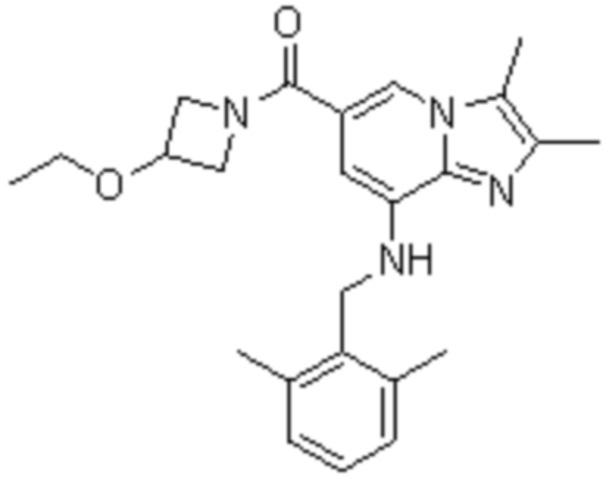
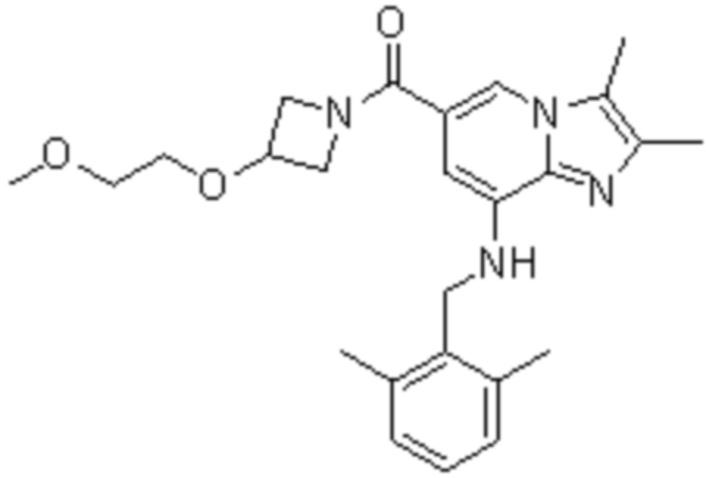
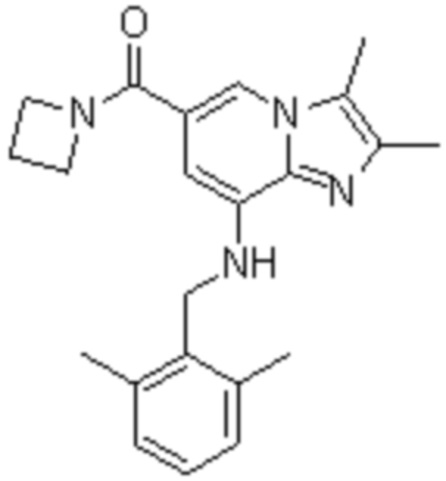
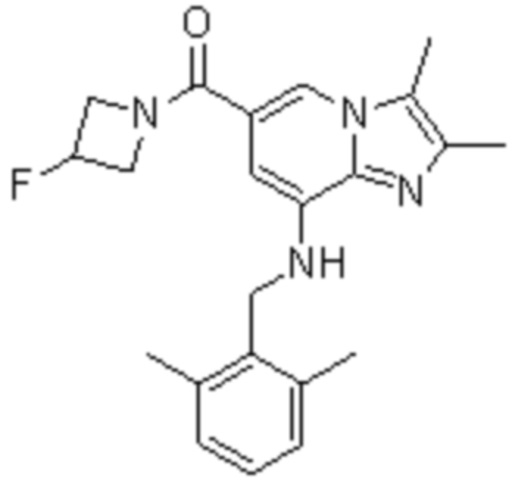
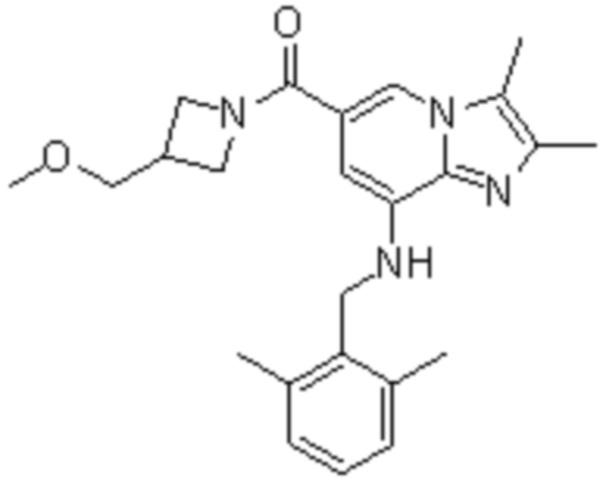
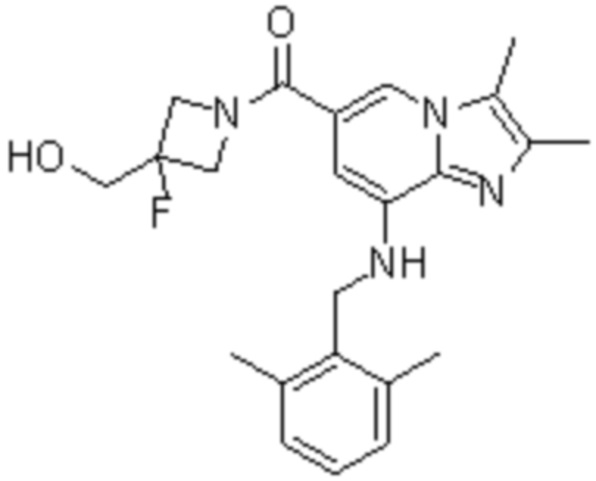
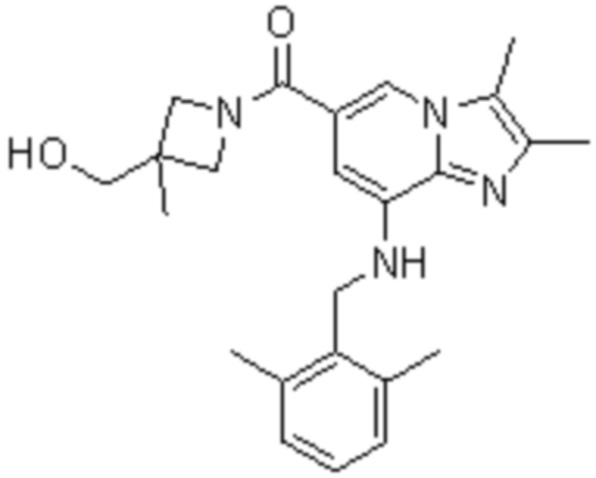
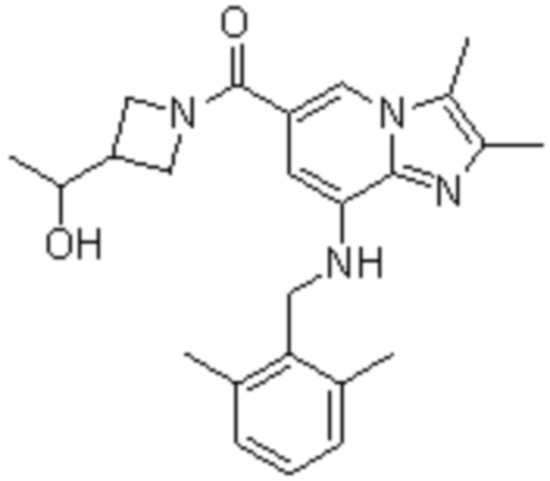
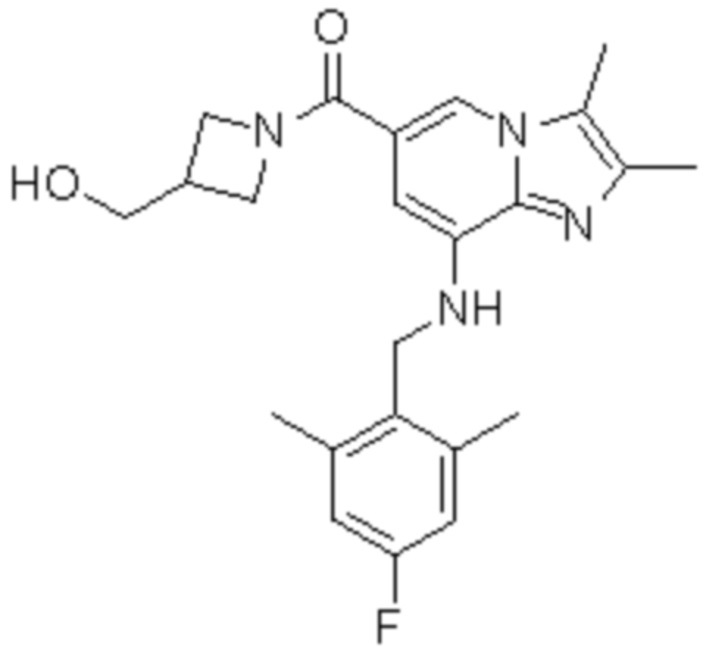
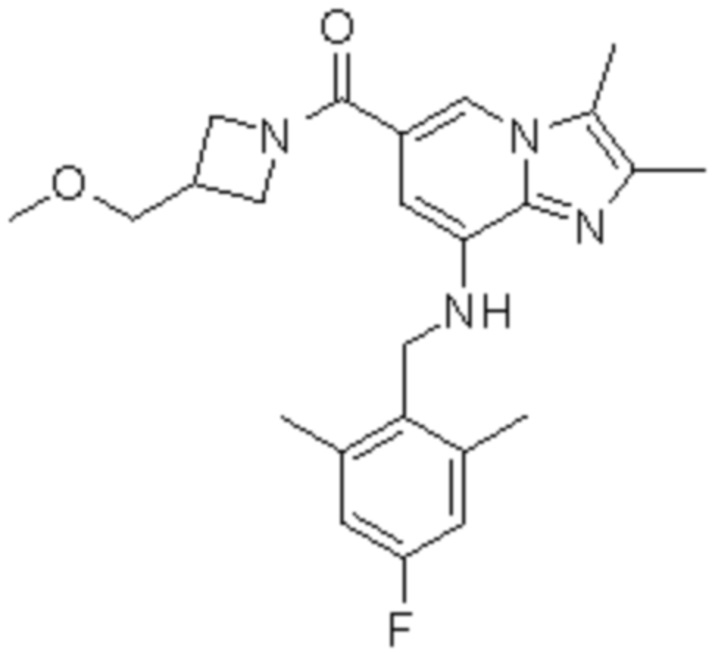
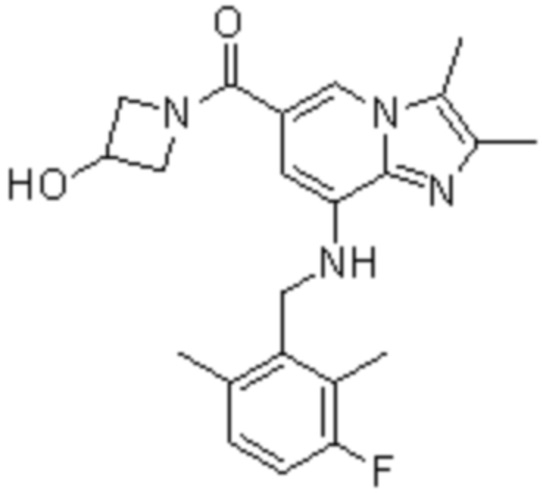
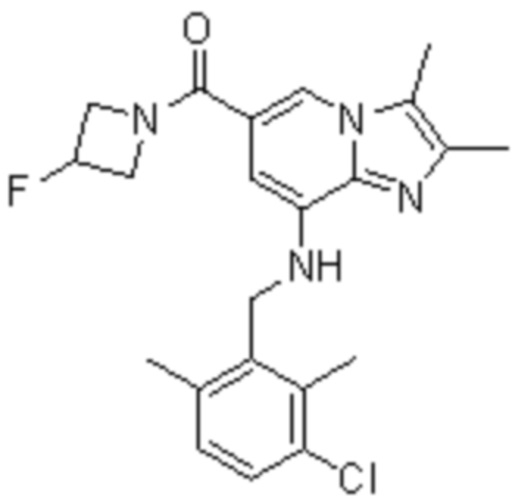
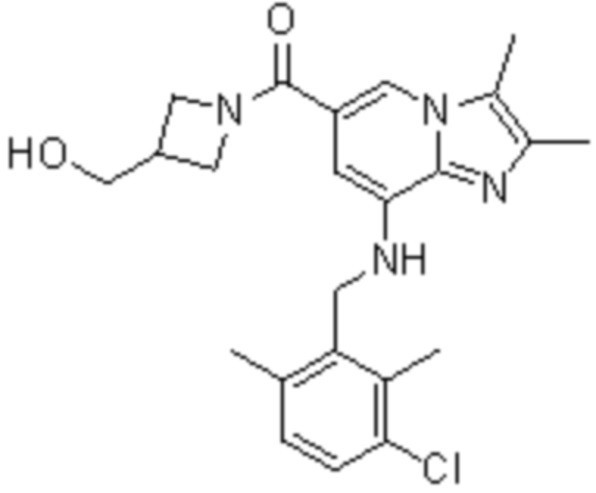
На основе способа, описанного в примере 1, были синтезированы соединения по примерам 2-48, имеющие различные заместители, как показано в таблице 1 ниже.
|
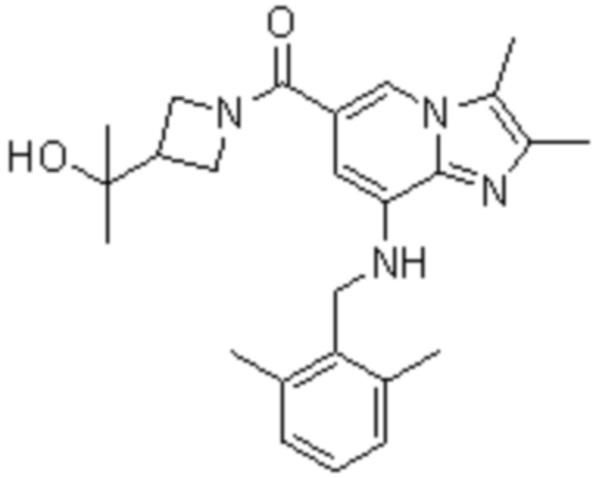
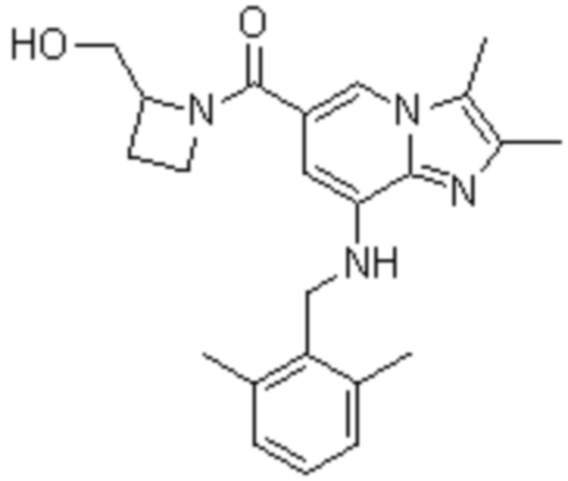
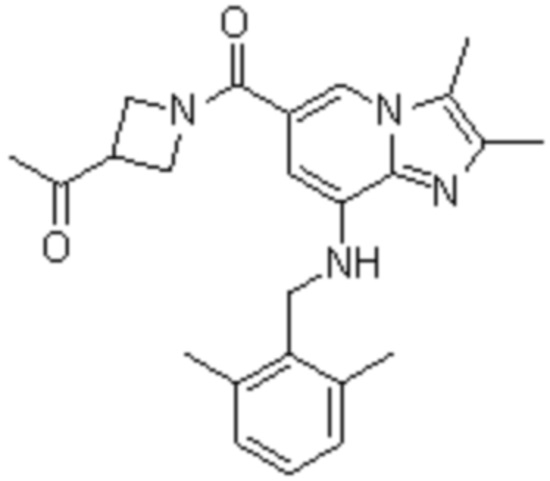
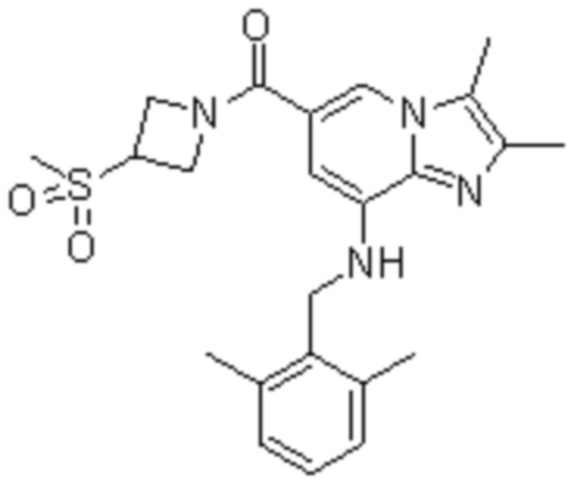
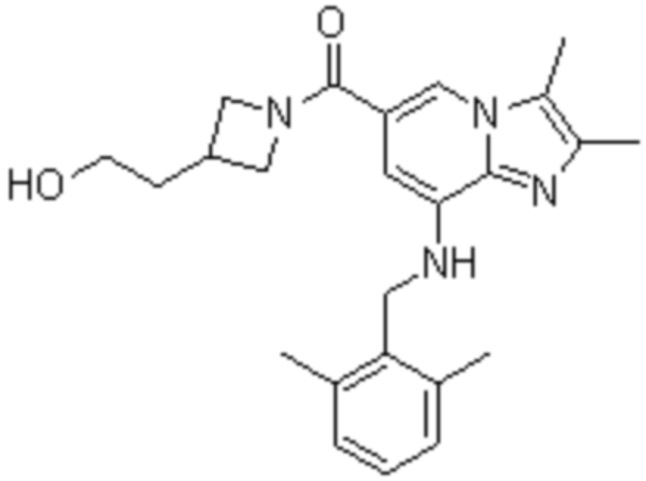
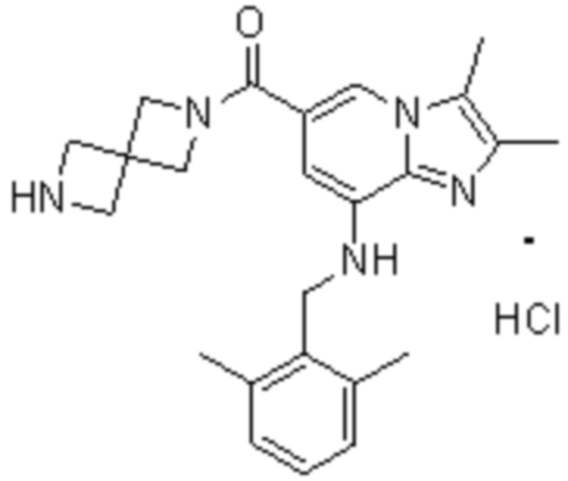
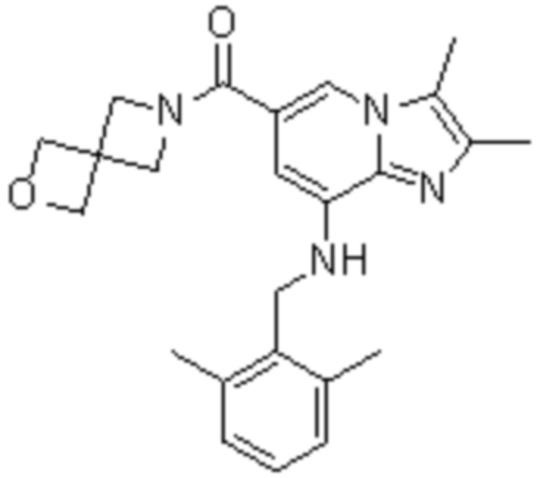
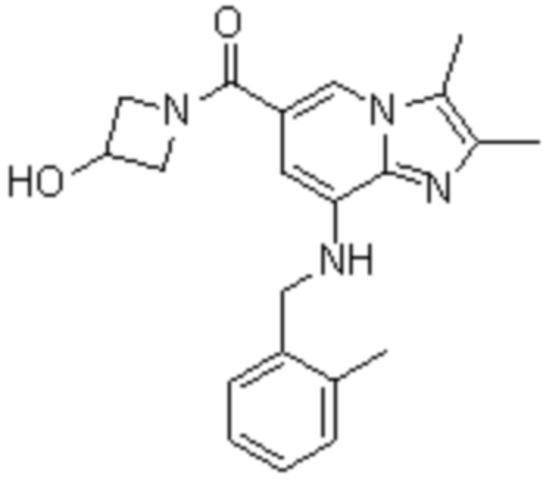

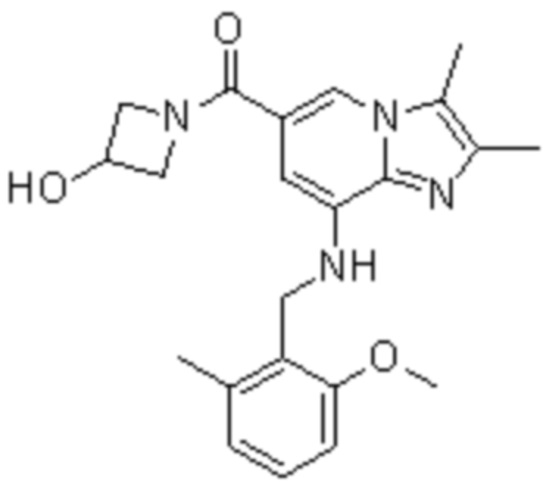
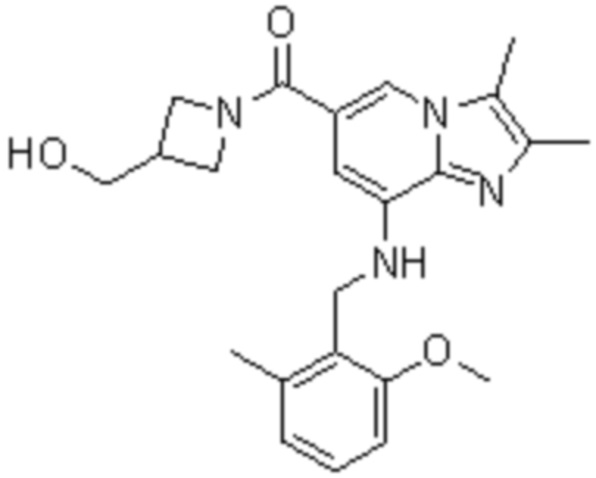
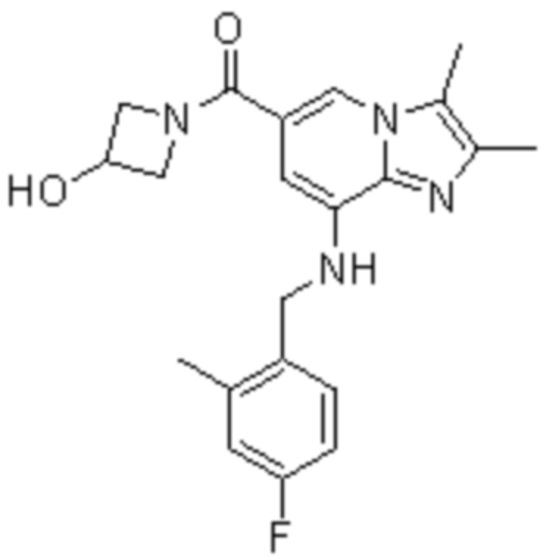
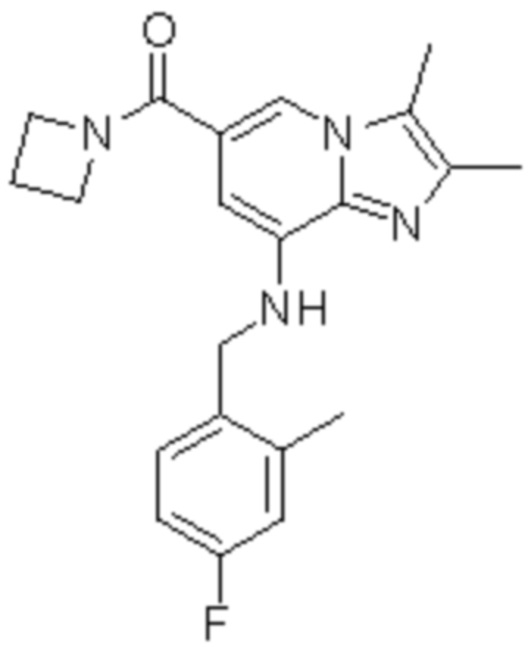

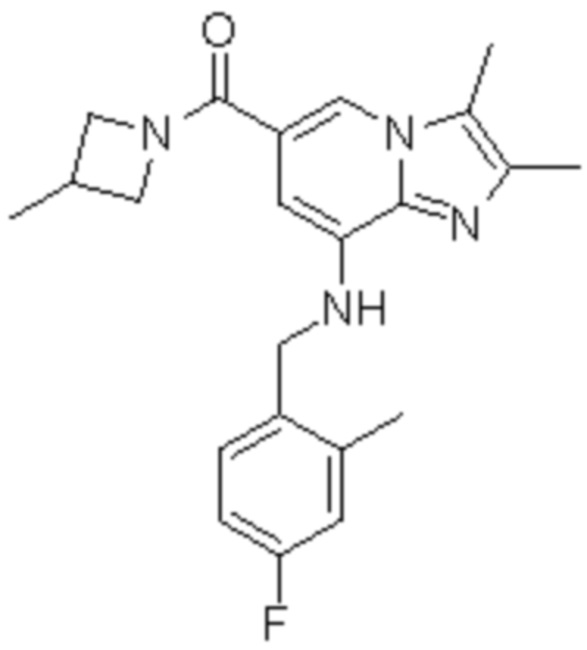
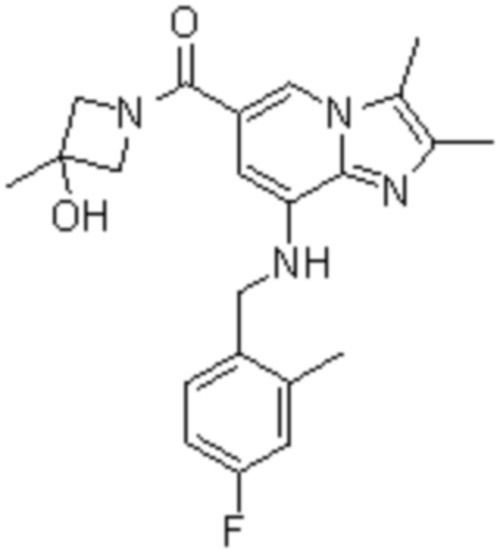
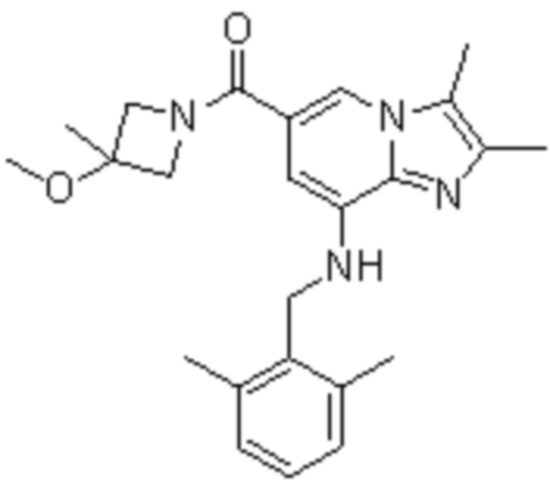
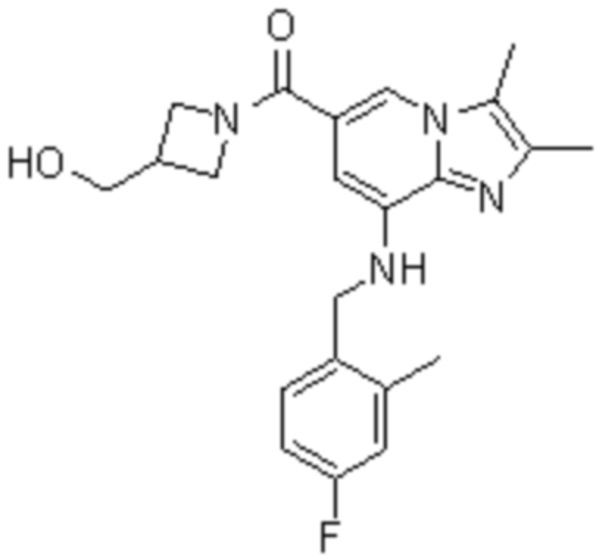
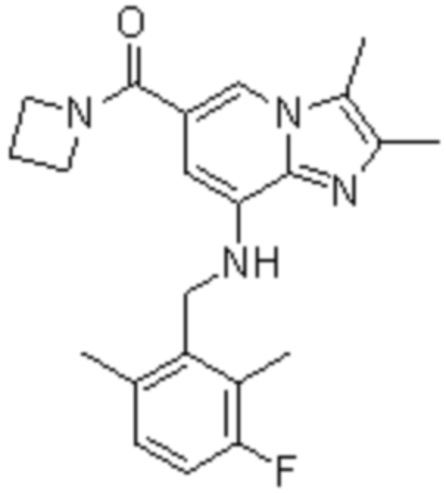
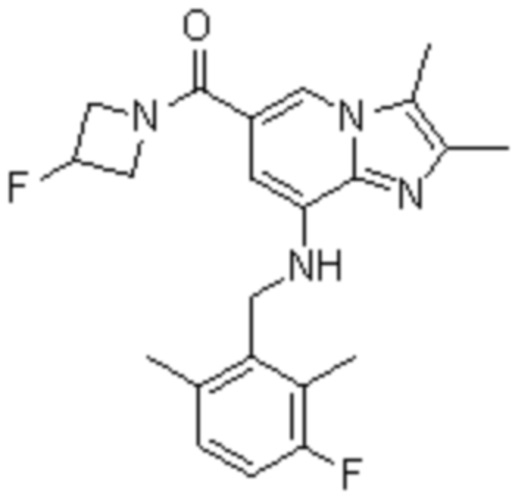
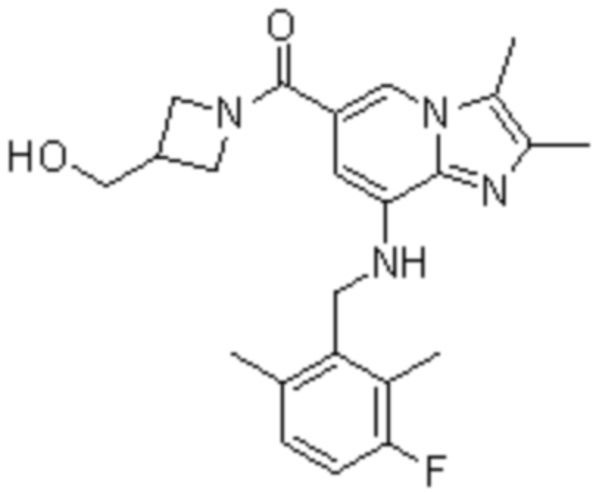
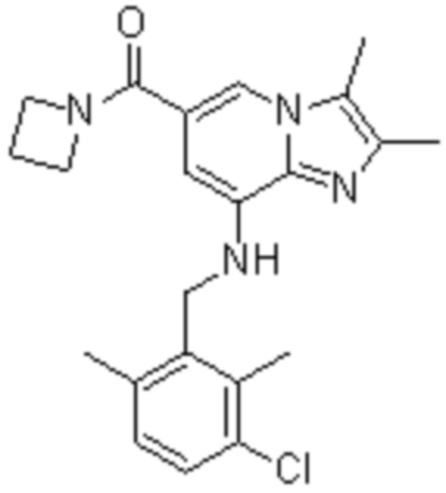
Пример исследования 1: Получение желудочных везикул
Желудочные везикулы, использованные в эксперименте, были получены путем отделения от слизистой оболочки желудка свиньи методом центрифугирования [Saccomani G, et al., A Nonelectrogenic H+ Pump in Plasma Membranes of Hog Stomach, J Biol Chem., 1976, 251(23), 7690-8]. Затем в желудочных везикулах количественно определяли содержание белка с использованием реактива бицинхониновой кислоты [Smith PK, et al., Measurement of protein using bicinchoninic acid, Anal Biochem., 1985, 150(1), 76-85)].
Пример исследования 2: Определение ингибирующего действия на активность протонного насоса (H+/K+-АТФаза)
Ингибирующее действие соединений на активность протонного насоса рассчитывали на основе активности насоса, определяемой в присутствии ионов K+, исключая активность насоса, определяемую в отсутствие ионов K+. Ингибирующее действие на активность протонного насоса определяли в 96-луночных планшетах, и все реакции проводили с объемом реакции 100 мкл при 37°С. Конкретно, 10 мкМ валиномицина и каждую концентрацию каждого соединения предварительно инкубировали в реакционном буфере (50 ммоль/л трис-HEPES-буфера, рН 6,4), используя свиные желудочные везикулы, в течение 15 минут. Для отрицательных и положительных контрольных групп в буфер добавляли 1% ДМСО, а для испытуемых групп добавляли 1% ДМСО и разбавление каждой концентрации каждого соединения. Затем в реакционный буфер добавляли 0,2 ммоль/л аденозинтрифосфата (АТФ) и инкубировали при 37°С в течение 40 минут. После завершения инкубации в реакционный буфер добавляли раствор малахитового зеленого и инкубировали в течение 30 минут, и количество неорганического фосфата в реакционном буфере измеряли колориметрически с использованием набора для анализа фосфата с малахитовым зеленым (Bioassay Systems). Для колориметрии, OD (оптическую плотность) при 620 нм измеряли с использованием считывающего устройства для микропланшетов [Synergy H4, гибридное многофункциональное считывающее устройство для микропланшетов, BioTek]. Процент ингибирования протонного насоса (H+/K+-АТФазы) определяли на основании значения OD контрольной группы и значений OD различных концентраций исследуемых соединений, и IC50 каждого исследуемого соединения рассчитывали с использованием логистической 4-х параметрической функции программы Sigmaplot 8.0. Результаты показаны в таблице 2 далее.
Таблица 2
|
Как можно видеть из таблицы 2 выше, соединения по настоящему изобретению обладают превосходными ингибирующим действием в отношении H+/K+-АТФазы желудка.
Пример исследования 3: Ингибирующее действие в отношении повреждения пищевода на моделях рефлюкс-эзофагита
Ингибирующее действие соединений по настоящему изобретению в отношении повреждения пищевода на моделях рефлюкс-эзофагита оценивали в соответствии со способом Накамуры [Nakamura K, et al., Effects of sodium polyacrylate (PANa) on acute esophagitis by gastric juice in rats, Jpn J Pharmacology, 1982, 32, 445-56].
Самцов крыс Sprague Dawley (SD) (весом 180-210 г) разделяли на X групп (n=6) и подвергали голоданию без воды в течение 24 часов. Затем контрольной группе вводили перорально только 10% ДМСО, 10% Кремофора EL и 80% воды, а другим группам перорально вводили 2 мг/кг/2 мл каждого исследуемого соединения вместе с 10% ДМСО, 10% Кремофора EL и 80% воды. Через 1 час после введения вспомогательных веществ и каждого соединения брюшную полость каждой крысы рассекали под изофлурановым наркозом и привратник желудка лигировали, а также лигировали границу между антральным отделом и телом желудка. Через 6 часов после лигирования подопытных животных умерщвляли, и аккуратно извлекали пищевод в диапазоне от щитовидной железы до сердечной части. Извлеченный пищевод разрезали в продольном направлении и распределяли так, чтобы обнажить слизистую оболочку, и затем фиксировали, после чего измеряли область повреждения пищевода. Результаты показаны в таблице 3 ниже.
Процент (%) ингибирующей активности исследуемых соединений={(общая площадь повреждения пищевода контрольной группы-область повреждения пищевода группы, получавшей исследуемое соединение)/общая площадь повреждения пищевода контрольной группы}×100.
Таблица 3
|
Как можно видеть из таблицы 3 выше, соединения по настоящему изобретению обладают сильным ингибирующим действием в отношении повреждения пищевода на моделях рефлюкс-эзофагита.
Пример исследования 4: Ингибирующее действие в отношении секреции кислоты желудочного сока, стимулированной гистамином, у крыс с лигированным привратником желудка
Ингибирующие эффекты соединений по настоящему изобретению в отношении стимулированной гистамином секреции кислоты желудочного сока оценивали с использованием на крысиной модели по методу Shay [Shay H, et al., A simple method for the uniform production of gastric ulceration in the rat, Gastroenterology, 1945, 5, 43-61].
Самцов крыс Sprague-Dawley (SD) (весом 180-210 г) разделяли на X групп (n=7) и подвергали голоданию только с доступом к воде в течение 24 часов. За 1 час до лигирования привратника желудка контрольной группе перорально вводили только 10% ДМСО, 10% Кремофора EL и 80% воды, а другим группам перорально вводили 2 мг/кг/2 мл каждого соединения вместе с 10% ДМСО, 10% Кремофора EL и 80% воды. Брюшную полость каждой крысы рассекали под изофлурановым наркозом и привратник желудка лигировали. Сразу после лигирования каждой крысе вводили подкожно гистамин 2HCl в дозе 30 мг/кг/10 мл. Через 3 часа после лигирования подопытных животных умерщвляли и извлекали содержимое желудка. Содержимое желудка центрифугировали при 3000×g в течение 10 минут и супернатант собирали для получения желудочного сока. Затем кислотность желудочного сока определяли на основе объема (в мкэкв/мл) 0,1н NaOH, необходимого для автоматического титрования желудочного сока до рН 7,0, и кислотность желудочного сока умножали на количество желудочного сока для определения базальной кислотной продукции. Результаты показаны в таблице 4 ниже.
Процент (%) ингибирующей активности исследуемых соединений={(базальная кислотная продукция в контрольной группе-базальная кислотная продукция в группе, получавшей исследуемое соединение)/базальная кислотная продукция в контрольной группе}×100.
Таблица 4
|
Как видно из приведенной выше таблицы 4, соединения по настоящему изобретению обладают ингибирующей активностью в отношении стимулированной гистамином секреции кислоты желудочного сока у крыс с лигированным привратником желудка.
Пример исследования 5: Ингибирование секреции кислоты у крыс с перфузией просвета желудка
Ингибирующее действие соединений по настоящему изобретению на стимулированную гистамином секрецию кислоты желудочного сока на моделях крыс с перфузией просвета желудка оценивали с использованием способа Ghosh & Schild [Ghosh MN, et al., Continuous recording of acid gastric secretion in the rat, Br J Pharmacol Chemother., 1958, 13(1), 54-61].
Самцов крыс Sprague-Dawley (SD) (массой 250-300 г) разделяли на X групп (n=4) и подвергали голоданию только с доступом к воде в течение 24 часов. Каждую крысу анестезировали путем внутрибрюшного введения уретана (1,2 г/кг), а затем разрезали брюшную полость и лигировали границу между антральным отделом и телом желудка. Между желудком и пищеводом вставляли силиконовую трубку, чтобы можно было вводить физиологический раствор (рН 5,0). Кроме того, силиконовую трубку вставляли между привратником желудка и двенадцатиперстной кишкой, чтобы вытекал перфузат, который проходил через желудок. Затем через яремную вену для стабилизации желудочного рН вводили гистамин 2HCl (8 мг/кг). После стабилизации рН контрольной группе вводили внутривенно только 10% ДМСО, 10% Кремофора EL и 80% воды, а другим группам внутривенно вводили 2 мг/кг/2 мл каждого соединения вместе с 10% ДМСО, 10% Кремофора EL и 80% воды. После введения лекарственного средства перфузат собирали с 10-минутными интервалами в течение 5 часов и измеряли его pH. Результаты показаны на ФИГ. 1.
Как можно видеть на ФИГ. 1, соединения по настоящему изобретению обладают сильной и продолжительной ингибирующей активностью в отношении стимулированной гистамином секреции кислоты желудочного сока у крыс с перфузией просвета желудка.
![ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-A]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ](https://fips.edrid.ru/images/rid/15/59/ee/36df8463a2ad1fa3e3bd8863d6d98b03.jpg)
![ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-A]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ](https://fips.edrid.ru/images/rid/15/59/ee/d9b09fa502fc0322d370d6acee320d5e.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-A]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ](https://fips.edrid.ru/images/rid/15/59/ee/a36070ecb05706d122d9ad3fe115b897.jpg)
Способ подавления помех в системе беспроводной связи и соответствующее устройство
Способ и устройство для приема канала управления нисходящей линии связи

