Результат интеллектуальной деятельности: СПОСОБ СИНТЕЗА МОЛЕКУЛ, СОДЕРЖАЩИХ ФУНКЦИОНАЛЬНУЮ ГРУППУ НИТРИЛОКСИДА
Вид РИД
Изобретение
Настоящее изобретение относится к области азотсодержащих ассоциативных молекул, имеющих по меньшей мере одно звено, благодаря которому они могут ассоциироваться друг с другом или с наполнителем с помощью нековалентных связей, и содержащих функциональную группу, способную к реакции с полимером, включающим непредельные соединения, с образованием при этом ковалентной связи с указанным полимером.
Говоря конкретнее, настоящее изобретение относится к молекулам, содержащим группу нитрилоксида и группу имидазолидина.
Если еще точнее, заявка относится к способу синтеза таких молекул.
В промышленности часто используют смеси полимеров с наполнителями. Для того, чтобы такие смеси обладали хорошими свойствами, производится постоянный поиск средств, позволяющих улучшить распределение наполнителей в полимерах. Одно из средств достижения этого результата состоит в использовании связующих веществ, способных устанавливать взаимодействия между полимером и наполнителем.
Некоторые связующие вещества для установления связи полимера с наполнителем, включающим азотсодержащие диполи, описаны в документах US 7186845 В2 и JP 2008208163.
В них раскрыто модифицирование полимеров, содержащих диеновые звенья, с помощью азотсодержащих биполярных соединений, содержащих также гетероцикл, причем указанный гетероцикл включает атом азота, а также атом кислорода и/или серы.
В частности, описанные в данной заявке соединения представляют собой нитроны, содержащие оксазолиновую и тиазолиновую функциональные группы, например, ((2-оксазолил)-фенил-N-метилнитрон).
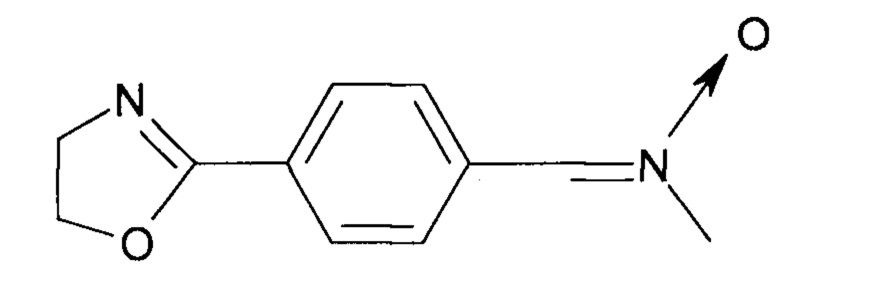
При введении диеновых полимеров в реакцию с такими соединениями, результирующие полимеры будут содержать оксазолиновые или тиазолиновые кольца.
Эти имеющиеся в полимерах циклы способны, в свою очередь, вступать в реакцию с поверхностными функциональными группами наполнителей, таких как газовая сажа или диоксид кремния, с которыми смешиваются полимеры. Данная реакция приводит к образованию ковалентных связей между полимером, который был модифицирован связующим веществом, и наполнителем вследствие раскрытия оксазолиноного или тиазолинового кольца. Действительно, как описано в документе US 7186845 В2, оксазолиновые и/или тиазолиновые кольца способны раскрываться в присутствии нуклеофила, который может находиться, например, на поверхности наполнителя.
Однако установление таких ковалентных связей сопряжено с некоторыми трудностями в случае приготовления смесей, содержащих такие полимеры, модифицированные связующими веществами, с наполнителями. Так, в частности, из-за существования таких слишком рано установленных ковалентных связей между полимером и наполнителями указанные смеси становятся очень вязкими в несшитом состоянии, что затрудняет проведение всех операций, предшествующих сшиванию (вулканизации) составов на основе каучука, и в частности, приготовление смесей компонентов, а также их формование. Указанные недостатки оказывают сильное негативное воздействие на производительность в данной отрасли промышленности.
Таким образом, желательно предложить новые молекулы, не обладающие указанными выше недостатками, то есть такие, которые были бы способны, после реагирования с полимером и смешивания с наполнителем, не образовывать ковалентные связи с наполнителем и, соответственно, не вызывать слишком сильного увеличения вязкости смеси.
Так, в заявке на выдачу патента WO 2012/007684 предложено соединение, содержащее по меньшей мере одну группу Q и по меньшей мере одну группу А, соединенные друг с другом с помощью по меньшей мере и предпочтительно, одной спейсерной группы Sp, где:
- Q включает диполь, содержащий по меньшей мере и предпочтительно один атом азота;
- А представляет собой ассоциативную группу, содержащую по меньшей мере один атом азота;
- Sp представляет собой атом или группу атомов, образующих связь между Q и А.
Когда полимер, привитый соединением, как описанного выше, смешивают с наполнителями, он образовывает только лабильные связи с этими наполнителями, что позволяет обеспечить хорошее взаимодействие полимера с наполнителем, способствующее достижению нужных конечных свойств полимера, но без недостатков, которые могли бы быть вызваны слишком сильным взаимодействием между полимером и наполнителем.
В качестве примера такого соединения можно назвать оксид 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрила:
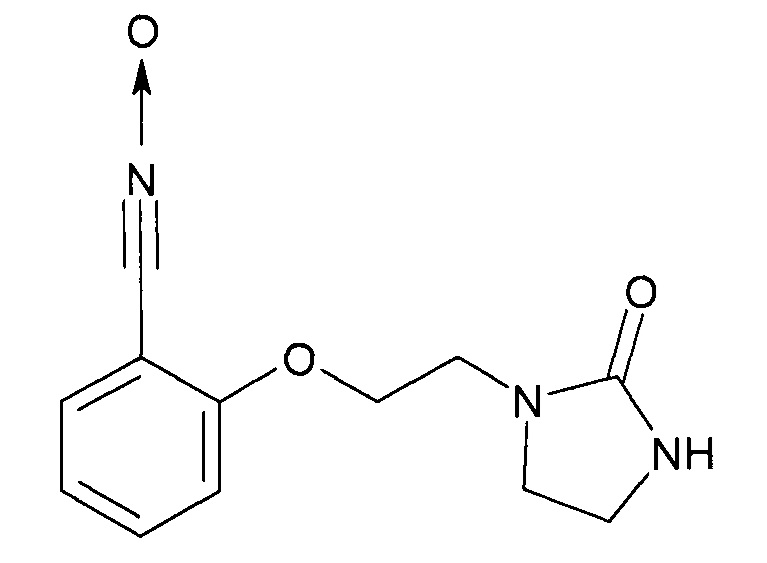
Это соединение может быть приготовлено из салицилового альдегида и 1-(2-хлорэтил)имидазолидин-2-она.
На первой стадии получают 1-(2-хлорэтил)имидазолидин-2-он из 1-(2-гидроксиэтил)имидазолидин-2-она, который вступает в реакцию с тионилхлоридом в дихлорметане.
На второй стадии получают 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегид из 1-(2-хлорэтил)имидазолидин-2-она, который вступает в реакцию с салициловым альдегидом в диметилформамиде (ДМФ) в присутствии карбоната калия.
На третьей стадии получают 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегида оксим при помощи ввода 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегида в реакцию с гидроксиламином в этаноле.
Наконец, на четвертой стадии получают 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрил при помощи ввода 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегида оксима в реакцию с гипохлоритом натрия в дихлорметане.
Таким образом, описанный способ включает довольно значительное количество стадий. Увеличение количества стадий негативно сказывается на общей эффективности способа синтеза нужного соединения, а также на соответствующих затратах на капвложения и производство. Кроме того, другая проблема при синтезе молекул, содержащих группу нитрилоксида, состоит в сведении к минимуму количества и объема растворителей, используемых в рамках способа получения, с целью облегчить обработку эффлюентов, уменьшить соответствующие затраты на капвложения и производство, но, естественно, без ущерба как для выхода указанных молекул, так и для их чистоты.
Целью настоящего изобретения является разработка решения, которое позволило бы устранить вышеупомянутые недостатки.
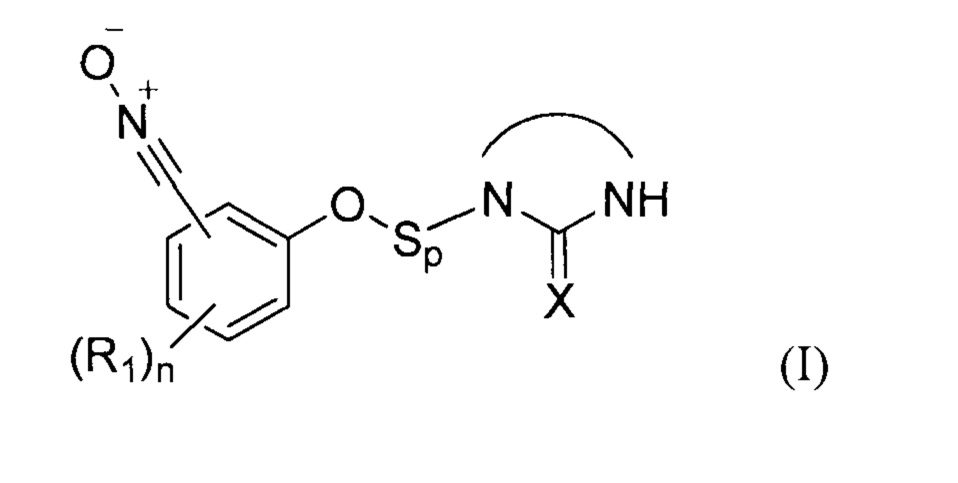
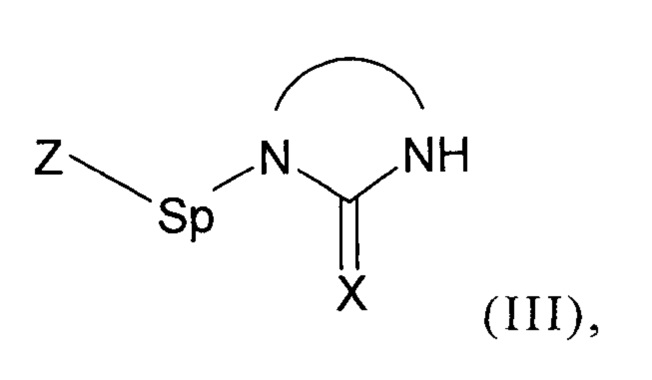
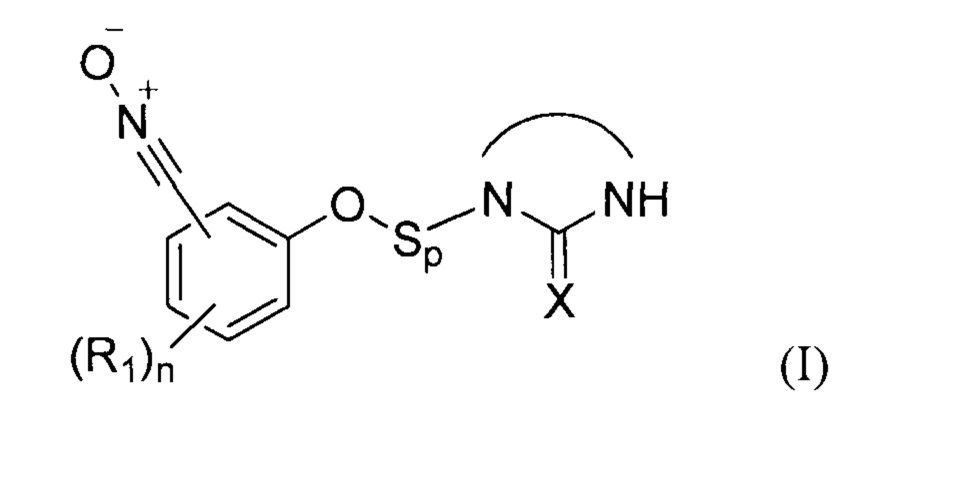
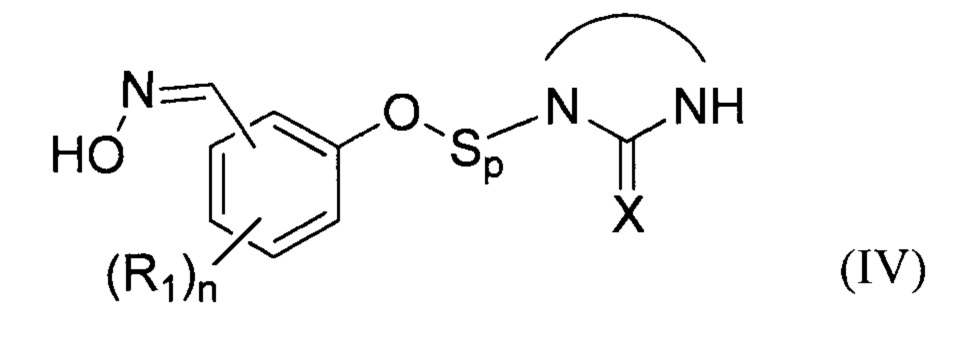
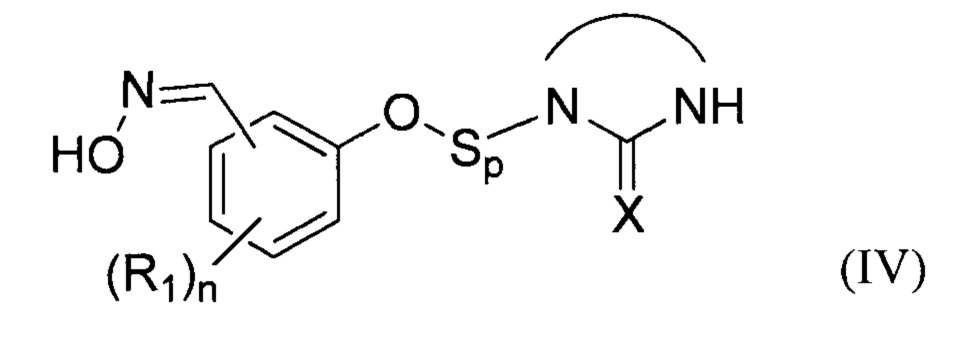
В соответствии с изобретением, способ синтеза соединения формулы (I):
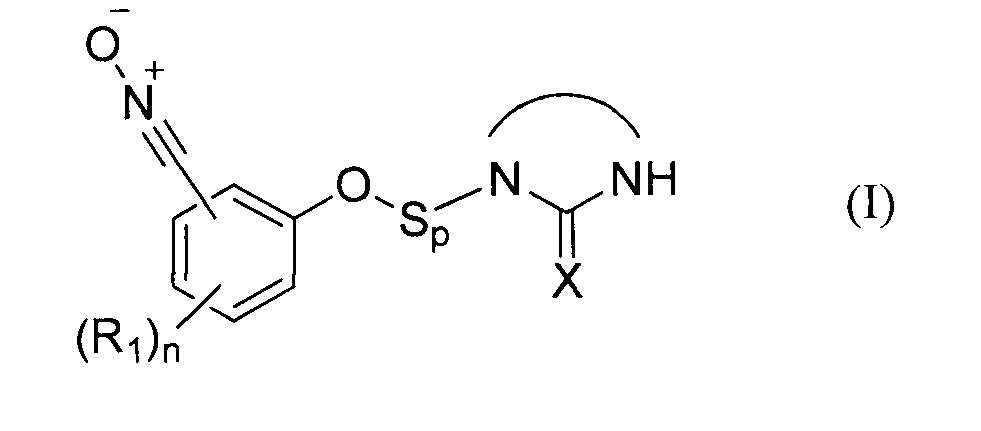
где:
- X представляет собой атом кислорода, серы или группу NH, предпочтительно атом кислорода,
- R1 представляет собой углеродную цепь, необязательно замещенную или прерванную одним или несколькими гетероатомами;
- n представляет собой целое число от 0 до 4;
- Sp представляет собой атом или группу атомов,
включает две следующих друг за другом стадии (b1) и (b2) в соответствии с принципами однореакторного синтеза, на которых:
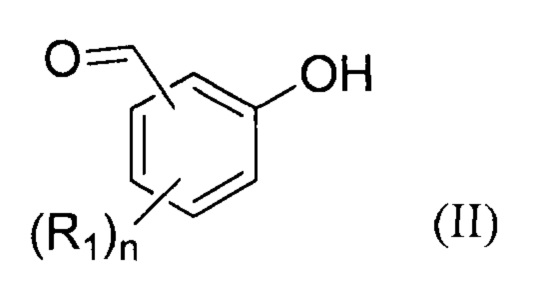
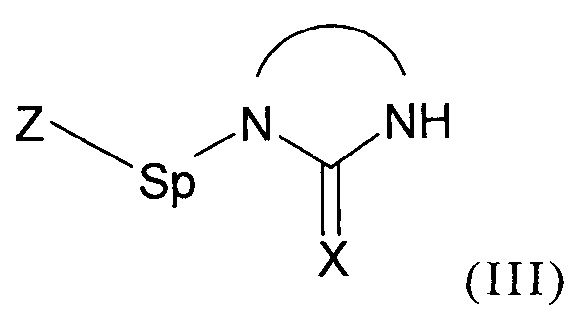
- (b1) осуществляют реакцию соединения формулы (II):
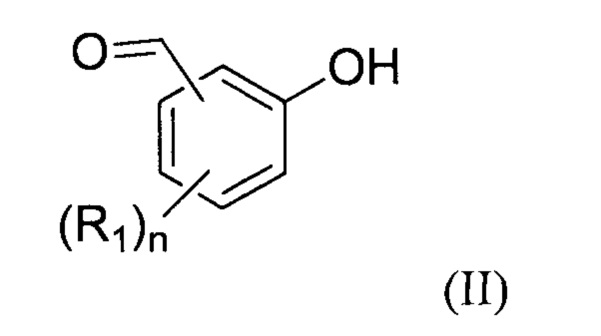
с соединением формулы (III):
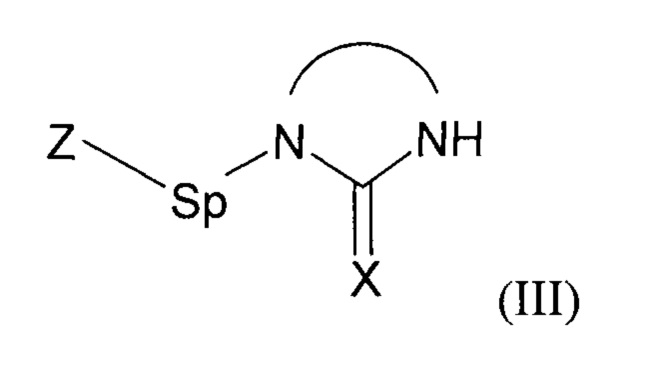
где Z представляет собой нуклеофуг;
в присутствии:
- по меньшей мере одного полярного растворителя S1;
- по меньшей мере одного основания;
при температуре T1 от 70 до 150°С,
для образования простого эфира;
- (b2) осуществляют реакцию указанного простого эфира с водным раствором гидроксиламина при температуре Т2 от 30 до 70°С с получением при этом оксимного соединения формулы (IV)
:
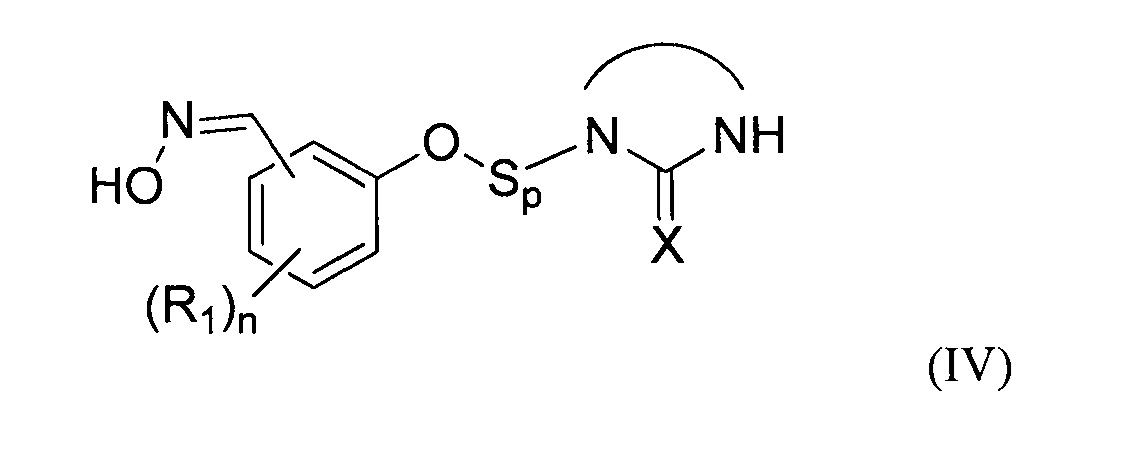
затем:
- на стадии (с) выделяют указанное оксимное соединение формулы (IV);
- на стадии (d) окисляют оксимное соединение формулы (IV) окислителем в присутствии по меньшей мере одного органического растворителя S2.
Стадии (b1) и (b2) представляют собой двухстадийную реакцию, которую проводят в одном реакционном сосуде без выделения промежуточных соединений (однореакторный способ синтеза), то есть без выделения промежуточного простого эфира.
Под «полярным растворителем» в контексте изобретения понимается растворитель с диэлектрической константой более 2,2.
Предпочтительно, за стадией (b2) следует стадия (е), на которой осуществляют выделение соединения формулы (I).
Под «нуклеофугом» в контексте изобретения понимается уходящая группа со связывающей электронной парой.
Под «углеродной цепью» в контексте изобретения понимается цепь, содержащая один или несколько атомов углерода.
Следует уточнить, что употребляемые в данном описании выражения «от… до…» надо понимать как включающие оба указанных предельных значения.
Благодаря предлагаемому способу возможно синтезировать соединение формулы (I) из соединения формулы (II) с использованием однорсакторного синтеза в две стадии, за которыми следует стадия окисления. В соответствии с патентной заявкой WO 2012/007684, соединение формулы (I) получают также из соединения формулы (II), но при этом эфирное промежуточное соединение выделяют и очищают по окончании первой стадии.
Остальные преимущества и признаки изобретения явствуют из нижеследующего подробного описания.
Способ согласно изобретению обеспечивает синтез вышеупомянутого соединения формулы (I).
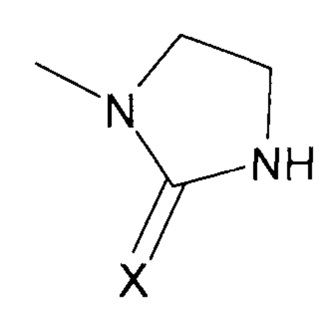
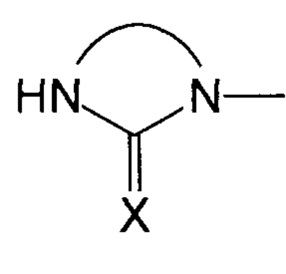
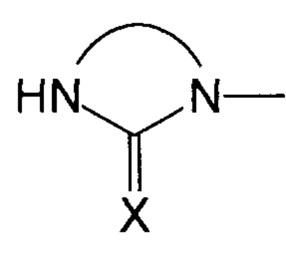
Преимущественно, группа 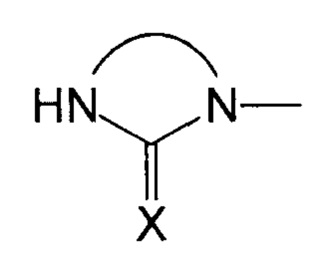 соответствует ди- или триазотированному гетероциклическому соединению с 5-ю или 6-ю атомами, предпочтительно диазотированному, содержащему по меньшей мере одну карбонильную, тиокарбонильную или иминовую функциональную группу.
соответствует ди- или триазотированному гетероциклическому соединению с 5-ю или 6-ю атомами, предпочтительно диазотированному, содержащему по меньшей мере одну карбонильную, тиокарбонильную или иминовую функциональную группу.
В соответствии с одним из частных вариантов осуществления изобретения, группа 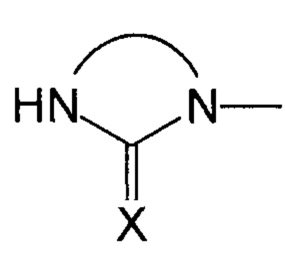 представляет собой группу
представляет собой группу 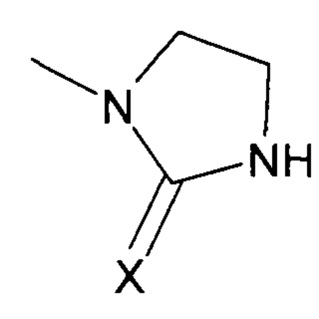 , в которой X выбран из атома кислорода и атома серы, предпочтительно атома кислорода.
, в которой X выбран из атома кислорода и атома серы, предпочтительно атома кислорода.
Группа Sp представляет собой спейсерную группу, обеспечивающую соединение атома кислорода с группой 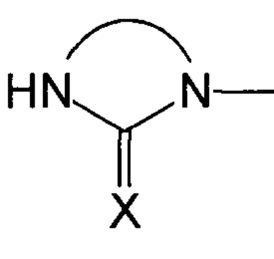 , где X соответствует указанному выше формуле соединения (I), а потому может быть группой любого хорошо известного типа.
, где X соответствует указанному выше формуле соединения (I), а потому может быть группой любого хорошо известного типа.
Под «ассоциативными функциональными группами» в контексте изобретения понимаются функциональные группы, способные ассоциироваться друг с другом с помощью водородных связей.
Таким образом, каждая ассоциативная функциональная группа содержит по меньшей мере один донорный «центр» и один акцепторный центр по отношению к водородной связи, так что две одинаковых ассоциативных функциональных группы оказываются самокомплементарными и могут ассоциироваться друг с другом с образованием при этом по меньшей мере двух водородных связей.
Группа Sp предпочтительно представляет собой линейную, разветвленную или циклическую углеводородную цепь С1-С24, которая может содержать один или несколько ароматических радикалов и/или один или несколько гетероатомов. Указанная цепь может быть замещена, если только заместители не будут реагировать с группой 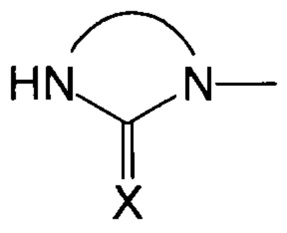 и нитрилоксидной функциональной группой.
и нитрилоксидной функциональной группой.
Преимущественно, группа Sp представляет собой линейную или разветвленную алкиленовую цепь С1-С24, предпочтительно С1-С10, необязательно прерванную одним или несколькими атомами азота или кислорода, а еще предпочтительнее - линейную алкиленовую цепь C1-С6.
Предпочтительно, группа Sp содержит звено, выбранное из (CH2)y1-, -[NH-(СН2)y2]х1- и -[O-(СН2)y3]х2-, где y1, y2 и y3 являются, независимо друг от друга, целыми числами от 1 до 6, a x1 и х2 являются, независимо друг от друга, целыми числами от 1 до 4.
Предпочтительно, R1 является насыщенной углеродной цепью, предпочтительнее алкильной группой, еще предпочтительнее алкильной группой C1-C12, еще предпочтительнее C1-С6, еще предпочтительнее С1-С4, или OR', где R' - алкильная группа, предпочтительнее алкильная группа С1-С12, еще предпочтительнее C1-C6, еще предпочтительнее С1-С4, а еще предпочтительнее метальной группой, этильной группой или группой ОСН3.
Преимущественно, n являетя целым числом от 0 до 3.
Предпочтительно, соединение формулы (I) выбрано из соединений следующих формул (V), (VI) и (VII):
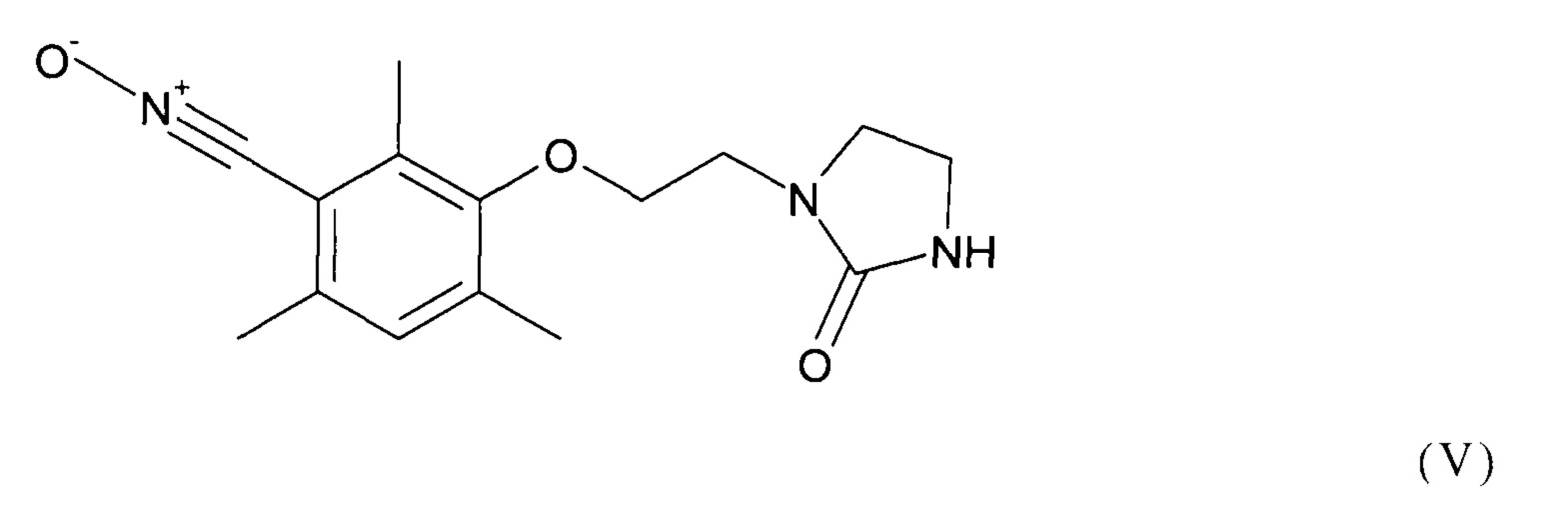
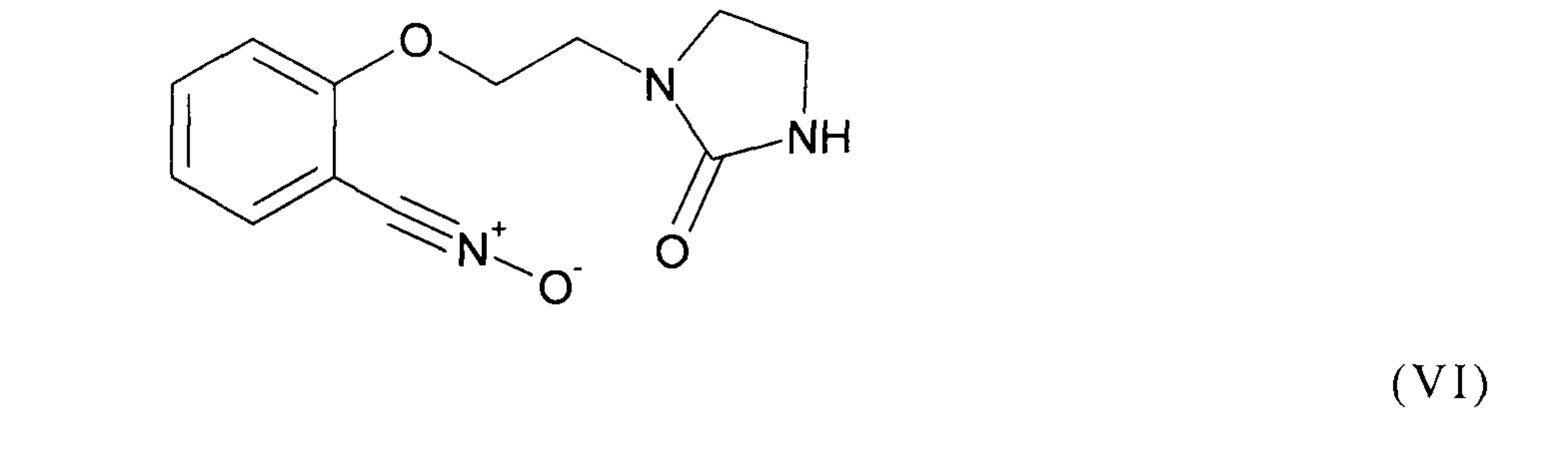
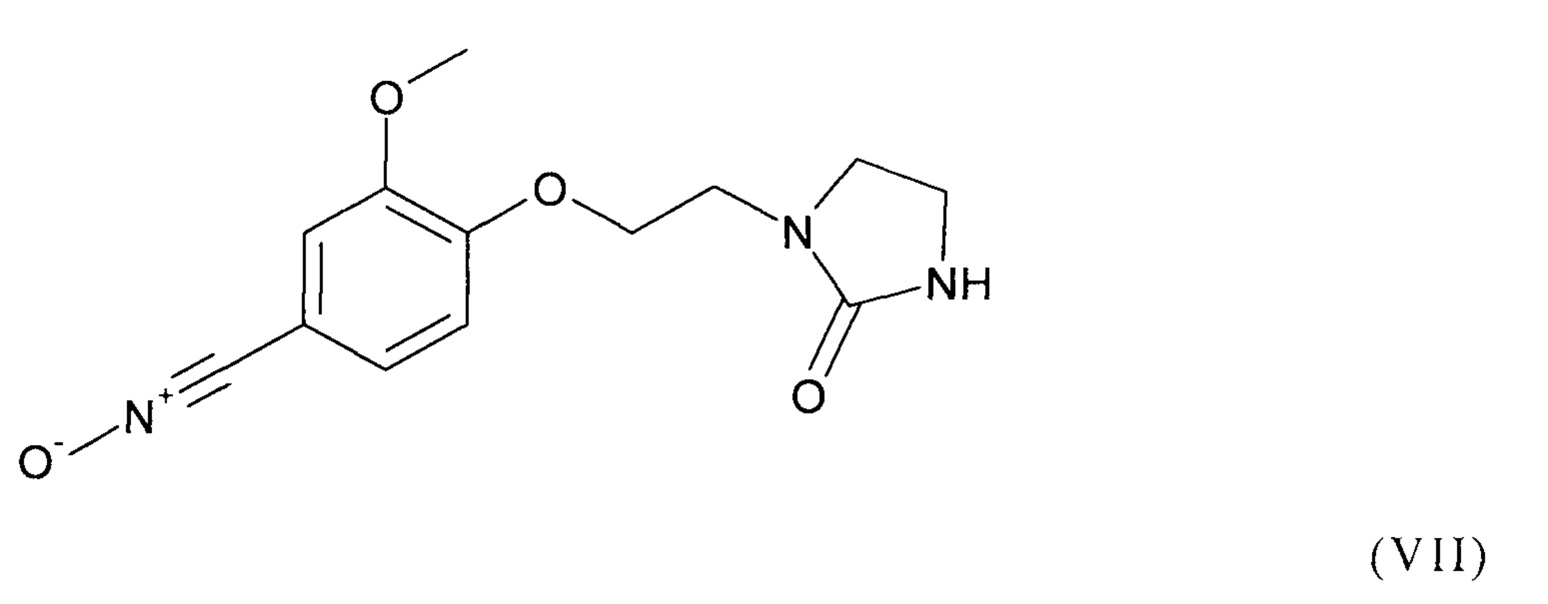
Как уже было разъяснено ранее, способ согласно изобретению включает стадию (b1), на которой осуществляют реакцию соединения формулы (II) как приведено выше с соединением формулы (III) как приведено выше, содержащим группу Z.
Предпочтительно, группа Z выбрана из хлора, брома, йода, мезилатной группы, тозилатной группы, ацетатной группы и трифторметилсульфонатной группы.
Указанную стадию (b1) предлагаемого способа по изобретению проводят в присутствии по меньшей мере одного полярного растворителя S1 и по меньшей мере одного основания при температуре T1 от 70 до 150°С.
В соответствии с одним из частных вариантов осуществления изобретения, полярный растворитель S1 представляет собой полярный растворитель, способный смешиваться с водой, предпочтительно протонный растворитель.
В качестве примеров растворителей, подлежащих использованию в рамках способа согласно изобретению, можно назвать диметилформамид (ДМФ), диметилсульфоксид (ДМСО), 1,3-диметил-2-имидазолидинон (ДМИ), 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон (ДМП), изопропанол, ацетонитрил, этанол, н-бутанол и н-пропанол.
Предпочтительно, протонный растворитель является спиртовым.
Преимущественно, соединение формулы (II) составляет от 5 до 40 масс. %, предпочтительнее от 10 до 30 масс. %, относительно массы растворителя.
На стадии (b1) способа согласно изобретению можно использовать основание, традиционно применяемое в реакциях этерификации. Ввиду присутствия альдегидной группы, подверженной, в частности, окислению и нуклеофильным реакциям, а также присутствия в соединении формулы (II) иных возможных функциональных групп, специалисты в данной области смогут выбрать такое основание, чтобы обеспечить избирательную ориентацию реакции на получение нужного простого эфира.
Предпочтительно, основание выбрано из щелочных алкоголятов, щелочных карбонатов, щелочноземельных карбонатов, щелочных гидроксидов, щелочноземельных гидроксидов и их смесей.
Преимущественно возможное добавление:
- одного или нескольких катализаторов, выбранных из катализатора типа соли серебра (I), межфазного катализатора типа четвертичного аммония и их смесей;
- одной или нескольких ионных жидкостей.
Предпочтительно, основание выбрано из метанолата натрия, карбоната калия и едкого натра, предпочтительно карбоната калия.
В соответствии с одним из частных вариантов осуществления изобретения, количество основания составляет от 1,5 до 8 эквивалентов, предпочтительно от 2 до 6 эквивалентов, относительно количества соединения формулы (II).
Как уже говорилось выше, стадию (b1) способа согласно изобретению выполняют при температуре T1 от 70 до 150°С.
Предпочтительно, температура Т1 составляет от 70 до 120°С, предпочтительнее от 80 до 110°С.
Как уже было сказано ранее, за стадией (b1) предлагаемого способа следует стадия (b2), на которой в реакционную среду, содержащую указанный простой эфир, добавляют водный раствор гидроксиламина при температуре Т2 от 30 до 70°С.
Предпочтительно, добавление водного раствора гидроксиламина осуществляют тогда, когда конверсия соединения формулы (II) составляет по меньшей мере 70%.
Преимущественно, температура Т2 изменяется от 40 до 60°С.
Предпочтительно, способ согласно изобретению включает также стадию (с) выделения оксимного соединения формулы (IV) как указано выше.
Предпочтительно, оксимное соединение формулы (IV) выделяют посредством осаждения водой, необязательно сопровождающегося промывкой водой.
Способ согласно изобретению включает также стадию (d) окисления оксимного соединение формулы (IV) окислителем в присутствии по меньшей мере одного органического растворителя S2.
Предпочтительно, указанный окислитель выбран из гипохлорита натрия, N-бромсукцинимида в присутствии основания, N-хлорсукцинимида в присутствии основания и пероксида водорода в присутствии катализатора, предпочтительно гипохлорита натрия.
Преимущественно, количество окислителя составляет от 1 до 5 эквивалентов, предпочтительно от 1 до 2 эквивалентов, относительно количества оксимного соединения формулы (IV).
Предпочтительно, органический растворитель S2 представляет собой органический растворитель, выбранный из хлорированных растворителей типа сложного эфира, простого эфира и спирта, предпочтительнее выбранный из дихлорметана, этилацетата, бутилацетата, диэтилового эфира, изопропанола и этанола, а еще предпочтительнее выбранный из этилацетата и бутилацетата.
Предпочтительно, оксимное соединение формулы (IV) составляет 1-30 масс. %, предпочтительно 1-20 масс. %, относительно общей массы комбинации, содержащей указанное оксимное соединение формулы (IV), указанный органический растворитель S2 и указанный окислитель.
Предпочтительно, способ согласно изобретению включает стадию (е) выделения, как указано выше, соединения формулы (I).
Предпочтительно, соединение формулы (I) выделено посредством осаждения водой, необязательно с последующей промывкой водой.
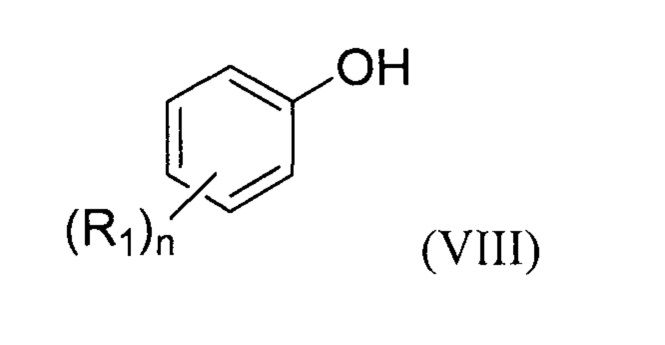
Предпочтительно, предлагаемый способ включает стадию (a1) получения соединение формулы (II) перед стадией (b1), для чего осуществляют реакцию соединения формулы (VIII):
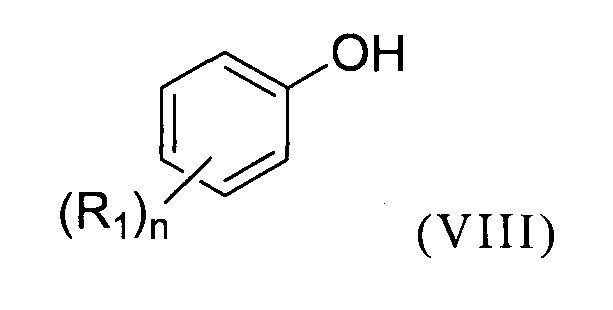
с формилирующим агентом, образующимся in situ или нет, и по меньшей мере одной кислотой Льюиса в присутствии по меньшей мере одного органического растворителя S3.
Предпочтительно, кислота Льюиса выбрана из TiCl4 и SnCl4, предпочтительно TiCl4.
Предпочтительно, количество кислоты Льюиса составляет от 0,5 до 4 эквивалентов, предпочтительнее от 1 до 3 эквивалентов относительно количества соединения формулы (VIII).
Преимущественно, органический растворитель S3 представляет собой хлорированный органический растворитель, предпочтительно выбранный из дихлорметана, хлороформа и 1,2-дихлорэтана, предпочтительнее дихлорметана.
Как уже было сказано выше, соединение (VIII) вступает в реакцию с формилирующим агентом, образующимся in situ или нет, согласно реакции формилирования. При этом можно использовать любой формилирующий агент, традиционно применяемый в реакциях формилирования.
Предпочтительно, формилирующий агент выбран из дихлорметилметилового эфира и дихлорметилэтилового эфира, предпочтительнее дихлорметилметилового эфира.
В соответствии с одним из частных вариантов осуществления изобретения, агент, образующийся in situ, получают посредством реакции метилформиата или этилформиата с хлорирующим агентом, предпочтительно пентахлоридом фосфора.
Преимущественно, соединение формулы (VIII) составляет от 5 до 15 масс. % относительно массы растворителя.
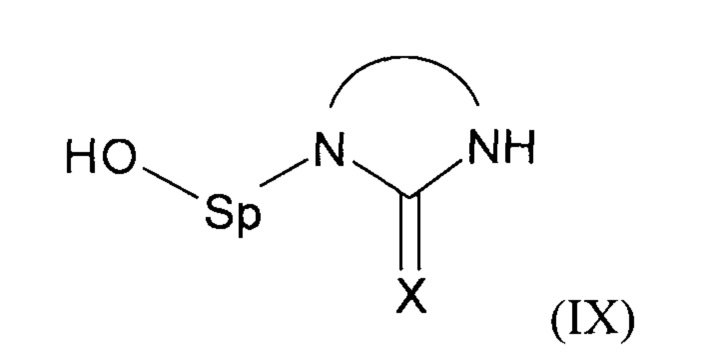
В соответствии с одним из частных вариантов осуществления изобретения, способ согласно изобретению включает стадию (а2) получения соединения формулы (III) перед стадией (b1), для чего осуществляют реакцию соединения формулы (IX):
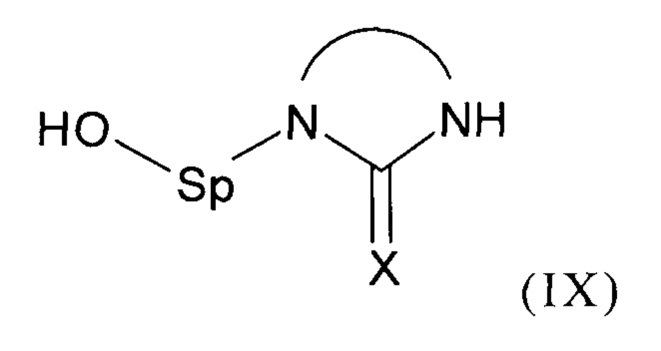
с агентом, обеспечивающим образование нуклнофуга Z.
Предпочтительно, указанный агент представляет собой тионилхлорид.
Предпочтительно, стадию (а2) проводят в отсутствие или в присутствии по меньшей мере одного растворителя, предпочтительно хлорированного растворителя, еще предпочтительнее дихлорметана.
Преимущественно, за стадией (а2) непосредственно следует стадия (а3) выделения соединения формулы (III), предпочтительно посредством очистки толуолом, еще предпочтительнее посредством кристаллизации соединения формулы (III) в толуоле.
Настоящая заявка также относится к применению соединения формулы (I), полученного способом по изобретению.
Кроме того, изобретение иллюстрируется приводимыми ниже примерами, перечень которых не является исчерпывающим.
ПРИМЕРЫ
Структурный анализ, а также определение молярной чистоты молекул синтеза осуществляют методом ЯМР-анализа. Спектры получают с помощью спектрометра Avance 500 MHz BRUKER, снабженного «широкополосным» зондом BBIz-grad 5 мм. В рамках количественного эксперимента ЯМР 1Н используют одноимпульсную последовательность 30° с задержкой повторения 3 секунды после каждой из 64-х регистрации. Пробы растворяют в дейтерированном диметилсульфоксиде (ДМСО). Этот растворитель используют также для сигнала дейтериевой стабилизации. Калибровку проводят по сигналу протонов дейтерированного ДМСО 2,44 частей на миллион в сравнении со стандартом ТМС (тетраметилсилановым) 0 частей на миллион. Спектр ЯМР 1Н, связанный с экспериментами 2D HSQC 1Н/13С (двумерная гетероядерная одноквантовая корреляция) и НМВС 1Н/13С (гетероядерная корреляция через несколько связей) позволяет осуществить структурное определение молекул (ср. таблицы отнесений). Количественное определение молекул осуществляют на основе количественного ЯМР-спектра 1D 1Н.
Масс-спектрометрический анализ выполняют с прямой инжекцией методом электрораспылительной ионизации (ID/ESI). Анализы проводили на спектрометре НСТ Bruker (производительность 600 мкл/мин., давление распыляющего газа 10 фунт/кв. дюйм, расход распыляющего газа 4 л/мин.).
Пример 1. Синтез оксида 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрила
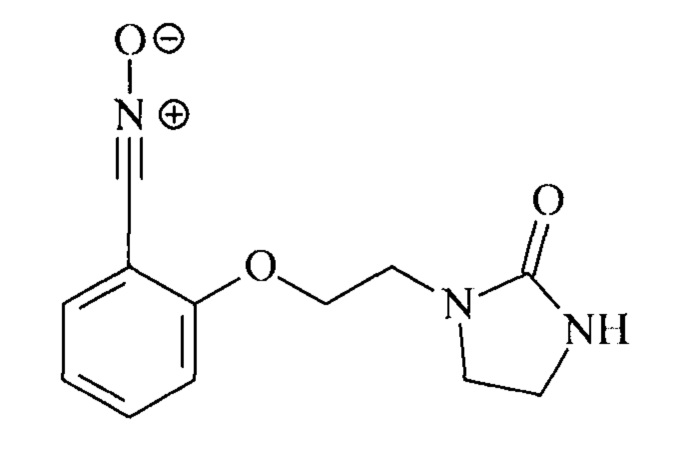
Оксид 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрила синтезируют из салисальдегида и 1-(2-хлорэтил)имидазолидин-2-она посредством однореакторного синтеза в две стадии b1 и b2 (за которым следует стадия с выделения, а за этой последней следует стадия d окисления. Вначале может быть раздельно приготовлен оксим с использованием двух описываемых ниже протоколов 1 и 2 перед выполнением стадии окисления по протоколу 3 с целью получения указанного оксида.
Однореакторные синтез в две стадии b1 и b2, за которыми следует стадия с (согласно изобретению), - приготовление оксима 2-(2-(2-оксоимидазолидии-1-ил)этокси)бензальдегида
Протокол 1 (согласно изобретению)
В реактор емкостью 1 л загружают 30 г салисальдегида (0,25 моль) и 200 г этанола. Добавляют 172,8 г карбоната калия (1,25 моль) и нагревают с обратным холодильником. Затем добавляют отдельными порциями в течение 5 часов 92,8 г 1-(2-хлорэтил)имидазолидин-2-она (0,63 моль). По завершении добавлений дают возможность протекания реакции в течение 2 часов с обратным холодильником. Охлаждают до 50°С, после чего добавляют в течение 15 минут 24,8 г 50%-ного водного раствора гидроксиламина (0,38 моль). Оставляют смесь реагировать в течение 2 часов при температуре 50°С. Концентрируют реакционную смесь до объема 50 мл, затем добавляют при комнатной температуре 500 мл воды. Полученный осадок фильтруют, промывают водой, затем пентаном и высушивают в вакууме.
В результате получают белое твердое вещество (40,5 г, выход по массе 65%) с точкой плавления 88°С.
Протокол 2 (согласно изобретению)
Загружают в реактор емкостью 1 л 30 г салисальдегида (0,25 моль) и 300 г ацетонитрила. Добавляют 172,8 г карбоната калия (1,25 моль) и нагревают с обратным холодильником. Затем добавляют отдельными порциями в течение 6 часов 109,5 г 1-(2-хлорэтил)имидазолидин-2-она (0,74 моль). По завершении добавлений дают возможность протекания реакции в течение 2 часов с обратным холодильником. Охлаждают до 50°С, после чего добавляют в течение 15 минут 24,8 г 50%-ного водного раствора гидроксиламина (0,38 моль). Оставляют смесь реагировать в течение 2 часов при температуре 50°С. Концентрируют реакционную смесь, затем добавляют при комнатной температуре 500 мл воды. Полученный осадок фильтруют, промывают водой, затем пентаном и высушивают в вакууме.
В результате получают белое твердое вещество (41,8 г, выход по массе 70%) с точкой плавления 88°С.
Стадия (d) - приготовление оксида 2-[2-(2-оксоимидазолидии-1-ил)этокси]бензонитрила
Протокол 3
Загружают в реактор емкостью 1 л 30 г оксима 2-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегида (0,12 моль) и 300 г этилацетата. Охлаждают до 5°С и добавляют в течение 15 минут 111,7 г 12%-ной жавелевой воды (0,18 моль). Дают возможность протекания реакции в течение 3 часов при температуре 5°С. Фильтруют и промывают полученное твердое вещество водой и затем этилацетатом.
Получают бело-желтое твердое вещество (17,8 г, выход по массе 60%) с точкой плавления 109°С.
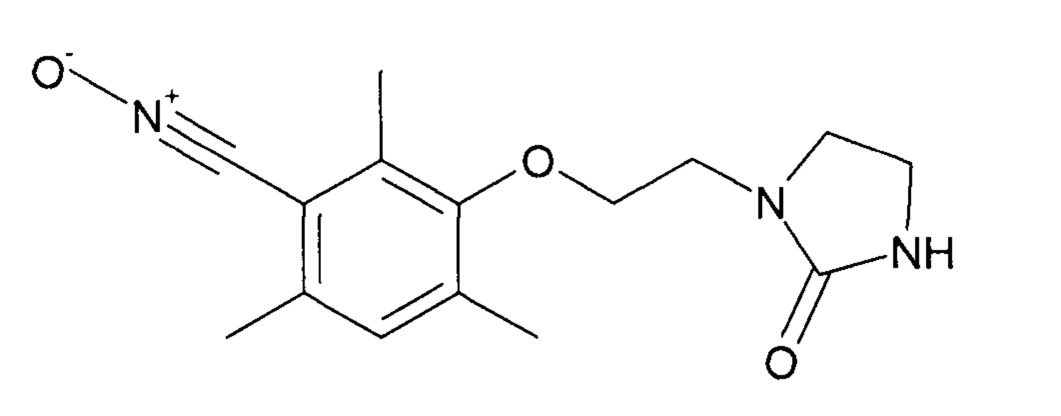
Пример 2. Синтез оксида 2,4,6-триметил-3-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрила
Оксид 2,4,6-триметил-3-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрила приготавливают с использованием двух методик синтеза. В соответствии с первой методикой, указанный оксид синтезируют в шесть стадий - стадия a1, стадия а2, стадия b1, стадия b2, стадия с и стадия d. Вторая методика синтеза отличается от первой тем, что стадии b1 и b2 проводят с использованием двухстадийного однореакторного синтеза.
На каждой из стадий соединения могут синтезироваться по отдельности в соответствии с несколькими протоколами. Стадию a1 проводят с использованием разных протоколов 4-9, стадию а2 с использованием разных протоколов 10-12, стадию b1 разных протоколов 13-15. Стадии b2 и с проводят с использованием двух протоколов 16 и 17.
Однореакторный синтез в две стадии b1 и b2, за которыми следует стадия с, осуществляют с использованием разных протоколов 18-21.
Стадию d проводят с использованием разных протоколов 22-24.
Стадия a1 - приготовление 3-гидрокси-2,4,6-триметилбензальдегида
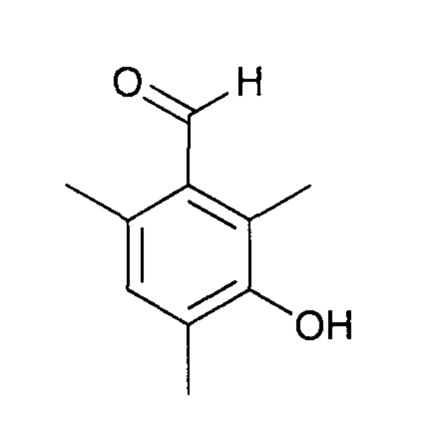
Протокол 4
Это соединение может быть получено с использованием протокола, описанного в заявке на выдачу патента WO 2012/007684.
Протокол 5
К раствору TiCl4 (10,44 г, 0,055 моль) в дихлорметане (5 мл), охлажденном до 12°С, добавляют в течение 10 минут раствор мезитола (5,0 г, 0,037 моль) в дихлорметане (15 мл). После перемешивания в течение 5-10 минут при температуре 10-15°С добавляют раствор дихлорметил-метилового эфира (6,75 г, 0,059 моль) в дихлорметане (5 мл) в течение времени от 10 до 15 минут. По истечении 15-часовой выдержки при комнатной температуре заливают в реакционную среду смесь из 100 мл воды и 50 г льда. После перемешивания в течение 30-40 минут сырой продукт фильтруют, промывают водой (4 раза по 10 мл) и высушивают на воздухе.
Целевой продукт (3,93 г) получают с выходом по массе, равным 65%.
Получаемое в ходе эксперимента ЯМР 1Н оценочное значение молярной чистоты превышает 90%.
Тонкослойная хроматография (ТСХ): Rf=0,87 (SiO2; EtOAc; проявление УФ-облучением и парами I2) или Rf=0,35 (SiO2; гептан : EtOAc = 3:1; проявление УФ-облучением и парами I2).
Протокол 6
К раствору мезитола (6,0 г, 0,044 моль) в дихлорметане (30 мл) (в инертной атмосфере) при температуре 2°С прикапывают TiCl4 (15,04 г, 0,079 моль) в течение 15-20 минут, поддерживая температуру реакционной среды на уровне ниже 8°С. После перемешивания в течение 5 минут при температуре 6°С добавляют дихлорметилметиловый эфир (7,60 г, 0,066 моль) в течение 35-40 минут. По истечении 16-часовой выдержки при комнатной температуре заливают в реакционную среду смесь из 75 мл воды и 50 г льда. После перемешивания в течение 1 часа 30 минут продукт фильтруют, промывают водой (4 раза по 5 мл) и высушивают на воздухе.
Целевой продукт (5,53 г, 0,034 моль) получают с выходом по массе 76%.
Получаемое в ходе эксперимента ЯМР 1Н оценочное значение молярной чистоты превышает 98%.
ТСХ: Rf=0,87 (SiO2; EtOAc; проявление УФ-облучением и парами I2) или Rf=0,35 (SiO2; гептан : EtOAc = 3:1; проявление УФ-облучением и парами I2).
Протокол 7
К раствору SnCl4 (183,6 г, 0,705 моль) в дихлорметане (250 мл), охлажденному до 13°С, добавляют в течение 20 минут раствор мезитола (40,0 г, 0,294 моль) в дихлорметане (250 мл). После 5-минутного перемешивания при температуре 13°С добавляют раствор дихлорметилметилового эфира (50,6 г, 0,441 моль) в дихлорметане (100 мл) в течение 30 минут. Реакционную среду перемешивают в течение 15 минут при комнатной температуре.
Затем сливают реакционную среду на смесь воды со льдом (800 мл воды на 0,7 кг льда). После перемешивания в течение часа отделяют органическую фазу. Водную фазу экстрагируют с помощью CH2Cl2 (2 раза по 100 мл). Объединенные органические фазы промывают водой (2 раза по 100 мл), высушивают с помощью Na2SO4 и концентрируют при пониженном давлении (10 мбар, 23°С) до получения 53 г масла.
К реакционному сырому продукту добавляют водный раствор диметиламина (500 мл, 40% в воде). Смесь нагревают до 60°С и перемешивают при этой температуре в течение 2,5 часов. Добавляют воду (1,0 л) и экстрагируют водную фазу дихлорметаном (4 раза по 100 мл). Промывают собранные органические фазы раствором HCl и воды (1:2) (три раза по 100 мл) и водой (три раза по 100 мл). После концентрирования при пониженном давлении (150 мбар, 23°С) добавляют петролейный эфир 40/60 (100 мл). Охлаждают раствор до -18°С в течение 15 часов. Полученные кристаллы фильтруют и промывают смесью дихлорметана (5 мл) и петролейного эфира (15 мл), а затем петролейным эфиром 40/60 (два раза по 20 мл) и, наконец, высушивают на воздухе.
Получают твердое вещество (20,02 г) с выходом по массе 42%.
После концентрирования фильтрата при пониженном давлении (60 мбар, 23°С) в него добавляют петролейный эфир 40/60. Раствор охлаждают до -18°С в течение 6 часов. Полученные кристаллы фильтруют и промывают петролейным эфиром 40/60 (два раза по 10 мл), после чего высушивают на воздухе.
Получают вторую фракцию продукта (3,02 г) с выходом 6,3%.
Далее проводят очистку растиранием с использованием описываемого ниже протокола.
Объединяют две фракции твердых веществ и растворяют их в смеси дихлорметана (150 мл) с петролейным эфиром 40/60 (100 мл). Охлаждают раствор до -18°С в течение 4-6 часов. Кристаллы фильтруют, промывают петролейным эфиром 40/60 (два раза по 20 мл) и высушивают на воздухе.
Получают продукт (11,9 г) с выходом по массе 25%.
Получаемое в ходе эксперимента ЯМР 1Н оценочное значение молярной чистоты превышает 96%.
ТСХ: Rf=0,87 (SiO2; EtOAc; проявление УФ-облучением и парами I2) или Rf=0,35 (SiO2; гептан : EtOAc = 3:1; проявление УФ-облучением и парами I2).
Протокол 8
К PCl5 (91,7 г, 0,441 моль) добавляют в инертной атмосфере при температуре 22-29°С в течение 70 минут этилформиат (34,8 г, 0,470 моль). Поскольку имеет место экзотермическая реакция, температура среды может достигать 40-60°С. Перемешивают реакционную среду в течение 1 часа при комнатной температуре. После полного растворения PCl5 перемешивают реакционную среду в течение еще 1-2 часов при комнатной температуре. Образуемый в результате этого формилирующий агент используют на следующей стадии без дополнительной очистки.
К раствору мезитола (40,0 г, 0,294 моль) в дихлорметане (100 мл) при температуре 11°С добавляют TiCl4 (105,0 г, 0,529 моль) в течение 45 минут. После выдержки в течение 5 минут при температуре 12-13°С добавляют ранее приготовленный формилирующий агент в течение 1,5-2,0 часов, поддерживая температуру в пределах от 9 до 22°С. Полученную суспензию перемешивают в течение 15 часов при комнатной температуре. Затем сливают реакционную среду на смесь воды (500 мл) со льдом (500 г). После перемешивания в течение 15-20 минут осадок фильтруют и промывают водой (5 раз по 50 мл). Полученные кристаллы высушивают на воздухе.
Получают целевой продукт (27,5 г, 0,170 моль) с выходом по массе 58%.
Получаемое в ходе эксперимента ЯМР 1H оценочное значение молярной чистоты равно 97%.
ТСХ: Rf=0,35 (SiO2; гептан : EtOAc = 3:1; проявление УФ-облучением и парами I2).
Протокол 9
Загружают в реактор емкостью 2 л с холодильником и азотной продувкой 105,8 г тетрахлорида титана (0,56 моль). Осуществляют охлаждение до 15°С и заливают в течение 30 минут 40 г мезитола (0,29 моль), растворенного в 400 г дихлорметана. Затем, поддерживая температуру на уровне 15-20°С, заливают в течение часа 40,6 г дихлорметилметилового эфира (0,35 моль). По окончании добавления дают возможность протекания реакции в течение 3 часов при комнатной температуре. Далее медленно переносят реакционную смесь в реактор с 800 г воды, в котором поддерживается температура ниже 20°С. После перемешивания в течение 30 минут производят декантацию и выделение органической фазы. Водную фазу дважды экстрагируют 100 граммами дихлорметана. Промывают собранные органические фазы 200 граммами воды, после чего концентрируют до объема 100 мл. Добавляют 100 г пентана и охлаждают до -15°С. Полученные кристаллы промывают смесью дихлорметана и пентана 1/1 в количестве 50 г, а затем высушивают в вакууме.
Получают твердое вещество бело-бежевого цвета (45,4 г, выход по массе 94%) с точкой плавления 113°С.
Стадия а2 - приготовление 1-(2-хлорэтил)имидазолидин-2-она
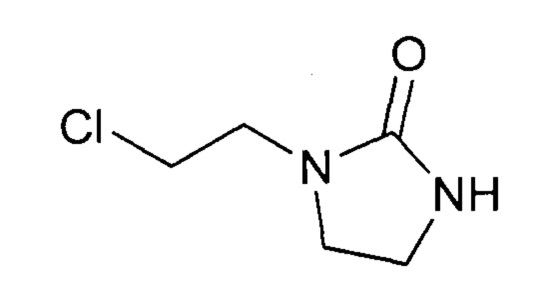
Протокол 10
Это соединение может быть получено с использованием протокола, описанного в заявке на выдачу патента WO 2012/007684.
Протокол 11
К 1-(2-гидроксиэтил)имидазолидин-2-ону (высушенному при пониженном давлении при температуре 60°С) (5,0 г, 38,42 ммоль) прикапывают SOCl2 (5,0 мл, 69,34 ммоль) в течение 2-3 минут. Смесь перемешивают при температуре 80°С (Тр-ра) в течение 1 часа 30 минут, а затем в течение полутора часов при температуре 100°С (Тр-ра). После выпаривания избытка SOCl2 при пониженном давлении (Тр-ра 50°С, 8-9 мбар) получают реакционный сырой продукт (7,53 г, выход по массе 105,9%) в виде желтого масла.
Добавляют к реакционному сырому продукту воду (10 мл) и регулируют рН водной фазы на значение 8 посредством добавления карбоната калия.
После кристаллизации при комнатной температуре в воде фильтруют осадок и промывают его водой (2-3 мл). Кристаллы высушивают на воздухе.
Получают целевой продукт (2,87 г, 19,31 моль), выход по массе 50%.
Получаемое в ходе эксперимента ЯМР 1Н оценочное значение молярной чистоты превышает 90%.
Протокол 12
Загружают 120 г 1-(2-гидроксиэтил)имидазолидин-2-она, 0,92 моль) в реактор емкостью 1 л с холодильником и азотной продувкой. Нагревают до 90°С и добавляют 120,4 г тионилхлорида (1,02 моль) в течение 30 минут. Дают возможность протекания реакции в течение 2 часов при температуре 90°С. Затем добавляют 400 г толуола и нагревают с обратным холодильником в течение 2 часов. Охлаждают реакционную смесь при комнатной температуре. Полученный осадок фильтруют, промывают толуолом и высушивают в вакууме. Получают 123,2 г белого твердого вещества (с точкой плавления 93°С и выходом по массе 90%).
Стадия b1 (за рамками изобретения) - приготовление 2,4,6-триметил-3-(2-(2-оксоимидазолидин-1-ил(этокси)бензальдегида
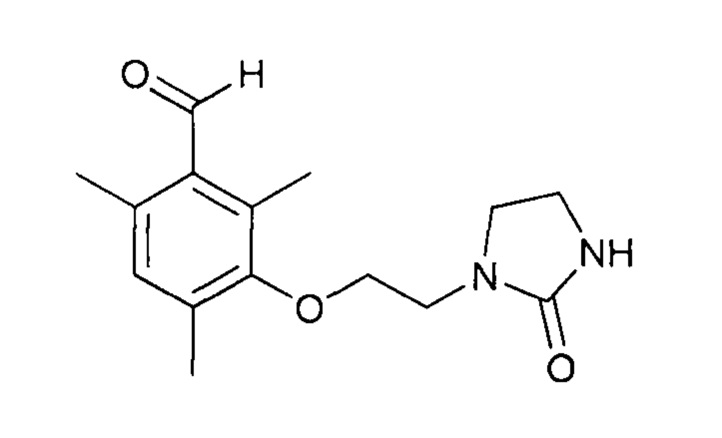
Протокол 13
Это соединение может быть получено с использованием протокола, описанного в заявке на выдачу патента WO 2012/007684.
Протокол 14
Смесь 3-гидрокси-2,4,6-триметилбензальдегида (25,0 г, 0,152 моль) и K2CO3 (126,3 г, 0,914 моль) в ДМФ (200 мл) перемешивают при температуре 30-35°С в течение 5-10 минут. Затем добавляют 1-(2-хлорэтил)имидазолидин-2-он (33,9 г, 0,228 моль). Доводят температуру реакционной среды до 90°С (Тр-ра) в течение 2,5-3 часов. После этого добавляют вторую порцию 1-(2-хлорэтил)имидазолидин-2-она (22,6 г, 0,152 моль) и доводят температуру смеси до 100°С (Тр-ра) в течение 3 часов. Наконец, добавляют третью порцию 1-(2-хлорэтил)имидазолидин-2-она (22,6 г, 0,152 моль) и доводят температуру смеси до 110-115°С (Тр-ра) в течение 3-4 часов. После возврата к температуре 23°С разбавляют реакционную среду водой (4,0 л). Экстрагируют органическую фазу с помощью CH2Cl2 (6 раз по 200 мл). Концентрируют собранные органические фазы при пониженном давлении (85 мбар, 36°С). Извлекают полученный остаток с помощью Et2O (200 мл) и перемешивают смесь при комнатной температуре в течение 2 часов. Полученный осадок фильтруют, промывают на фильтре с помощью Et2O (25 мл) и высушивают при комнатной температуре.
Получают белое твердое вещество (31,93 г, выход по массе 76%).
Молярная чистота превышает 82% (ЯМР 1Н).
Протокол 15
К смеси 3-гидрокси-2,4,6-триметилбензальдегида (0,44 г, 3,3 ммоль), 1-(2-гидроксиэтил)имидазолидин-2-она (0,64 г, 4,9 ммоль) и трифенилфосфина (1,31 г, 5,0 ммоль) в ТГФ (тетрагидрофуране) (30 мл) при комнатной температуре добавляют раствор диизопропил азодикарбоксилата (1,01 г, 5,0 ммоль) в ТГФ (15 мл) в течение 5-10 минут. Реакционную среду перемешивают в течение 15 часов при комнатной температуре, после чего в нее добавляют воду (15 мл). Концентрируют растворители при пониженном давлении (28°С, 50 мбар). Экстрагируют водную фазу этилацетатом (трижды по 25 мл). Собранные органические фазы промывают водным раствором, насыщенным NaCl. Концентрируют растворитель при пониженном давлении примерно до 3 мл (30°С). После очистки на хроматографической колонке (∅ 1,5 см, Н 11 см) (элюент: этилацетат : этанол (10:1) растворитель выпаривают досуха.
Получают белое твердое вещество (0,77 г, выход по массе 84%).
Молярная чистота превышает 78% (ЯМР 1Н).
Стадии b2 и с (за рамками изобретения) - приготовление оксима 2,4,6-триметил-3-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегида
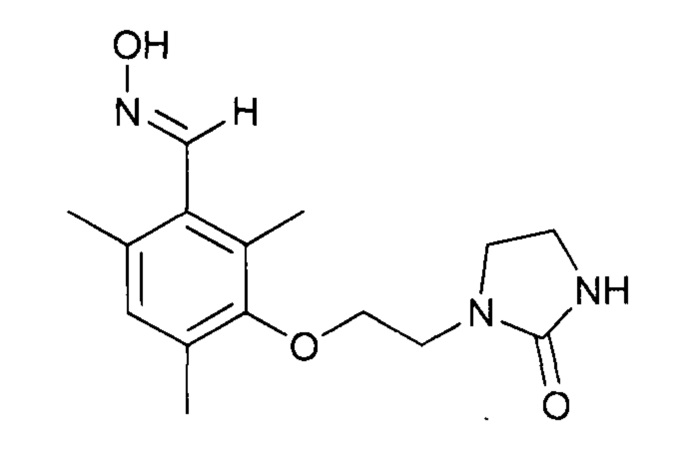
Продукты, выделяемые в результате исполнения протоколов 13, 14 и 15, используются в последующих протоколах (16, 17). Последовательность этих стадий выходит за рамки изобретения вследствие того, что имеет место выделение этих продуктов.
Протокол 16
Это соединение может быть получено с использованием протокола, описанного в заявке на выдачу патента WO 2012/007684.
Протокол 17
К раствору 2,4,6-триметил-3-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегида (31,5 г, 0,114 моль) в EtOH (300 мл) при температуре 44°С добавляют раствор гидроксиламина (12,0 г, 0,182 моль, 50%-ный раствор в воде, Aldrich) в EtOH (20 мл). Перемешивают реакционную среду в течение 4 часов при температуре 50°С. Затем к раствору добавляют воду (80 мл). Концентрируют реакционную среду при пониженном давлении (80 мбар, 40°С). Полученный раствор охлаждают до 15-20°С и перемешивают в течение 5-10 минут. Полученный осадок фильтруют, промывают на фильтре смесью этанола с водой (дважды по 10/15 мл) и высушивают в течение 15-20 часов при атмосферном давлении и комнатной температуре.
Получают белое твердое вещество (28,32 г, выход по массе 85%).
Получаемое в ходе эксперимента ЯМР 1Н оценочное значение молярной чистоты превышает 87%.
ТСХ: Rf=0,58 (SiO2; EtOAc : EtOH = 5:1; проявление УФ-облучением и парами I2).
Однореакторный синтез в две стадии b1 и b2, за которым следует стадия с (согласно изобретению) - приготовление оксима 2,4,6-триметил-3-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегида
Протокол 18 (согласно изобретению)
К смеси 3-гидрокси-2,4,6-триметилбензальдегида (10,0 г, 0,061 моль) и К2СO3 (50,5 г, 0,365 моль) в ДМФ (100 мл) при температуре 35°С добавляют 1-(2-хлорэтил)имидазолидин-2-он (13,55 г, 0,091 моль). Доводят температуру реакционной среды до 90°С (Тр-ра) и перемешивают эту среду при указанной температуре в течение 2,5-3 часов. Затем добавляют вторую порцию 1-(2-хлорэтил)имидазолидин-2-опа (9,03 г, 0,061 моль) и перемешивают смесь при температуре 100°С (Тр-ра) в течение 2,5 часов. Наконец, добавляют третью порцию 1-(2-хлорэтил)имидазолидин-2-она (9,03 г, 0,061 моль, техническая чистота) и перемешивают смесь при температуре 105°С (Тр-ра) в течение 4 часов.
Понижают температуру реакционной среды до 43°С, после чего добавляют раствор гидроксиламина (6,84 г, 0,104 моль, 50%-ный раствор в воде, Aldrich). Перемешивают реакционную среду в течение 5 часов при температуре 50-53°С. Добавляют вторую порцию раствора гидроксиламина (приблизительно 3 г, 50%-ный раствор в воде, Aldrich). Перемешивают реакционную среду в течение 1,5-2 часов при температуре 50-53°С. Затем разбавляют смесь водой в количестве 1,25 л. Полученную суспензию перемешивают в течение 15 часов при комнатной температуре. Полученный осадок фильтруют, промывают на фильтре водой (4 раза по 50 мл) и высушивают при комнатной температуре.
Получают серое твердое вещество (12,87 г, выход по массе 72%).
Получаемое в ходе эксперимента ЯМР 1H оценочное значение молярной чистоты превышает 86%.
Протокол 19 (согласно изобретению)
Загружают в реактор емкостью 1 л 30 г 3-гидрокси-2,4-6-триметилбензальдегида (0,18 моль) и 200 г ДМФ. Добавляют 126,5 г карбоната калия (0,92 моль) и нагревают с обратным холодильником. Затем добавляют отдельными порциями в течение 6 часов 67,9 г 1-(2-хлорэтил)имидазолидин-2-она (0,46 моль). По завершении добавлений дают возможность протекания реакции в течение 2 часов с обратным холодильником. Охлаждают до 50°С, после чего добавляют в течение 15 минут 14,3 г 50%-ного водного раствора гидроксиламина (0,22 моль). Оставляют смесь реагировать в течение 2 часов при температуре 50°С. Концентрируют реакционную смесь до объема 50 мл, зачем добавляют при комнатной температуре 500 мл воды. Полученный осадок фильтруют, промывают водой и высушивают в вакууме.
Получают твердое вещество бело-бежевого цвета (44,6 г, выход по массе 85%) с точкой плавления 167°С.
Протокол 20 (согласно изобретению)
К смеси 3-гидрокси-2,4,6-тримстилбензальдегида (22,8 г, 0,139 моль) и К2СО3 (115,2 г, 0,833 моль) в изопропаноле (160 мл) при температуре 50°С добавляют 1-(2-хлорэтил)имидазолидин-2-он (30,9 г, 0,208 моль). Доводят температуру реакционной среды до 100°С (Тр-ра) и поддерживают эту температуру в течение 3-4 часов. Затем добавляют вторую порцию 1-(2-хлорэтил)имидазолидин-2-она (20,06 г, 0,139 моль, техническая чистота) и перемешивают смесь при температуре 100°С (Тр-ра) в течение 5 часов. Наконец, добавляют третью порцию 1-(2-хлорэтил)имидазолидин-2-она (20,06 г, 0,139 моль, техническая чистота) и перемешивают смесь при температуре 105°С (Тр-ра) в течение 8-10 часов.
Понижают температуру реакционной среды до комнатной температуры, после чего добавляют к реакционной среде изопропанол (25 мл). Нагревают смесь до 55°С и добавляют раствор гидроксиламина (14,68 г, 0,222 моль, 50%-ный раствор в воде, Aldrich) в изопропаноле (25 мл). Перемешивают реакционную среду в течение 7 часов при температуре 50°С. Сливают реакционную среду в воду (3 л). После перемешивания в течение 5 часов полученный осадок фильтруют, промывают на фильтре водой (600 мл) и высушивают при комнатной температуре.
Получают серое твердое вещество (30,35 г, выход по массе 75%).
Получаемое в ходе эксперимента ЯМР 1Н оценочное значение молярной чистоты превышает 87%.
Протокол 21 (согласно изобретению)
Загружают в реактор емкостью 1 л 30 г 3-гидрокси-2,4-6-триметилбеизальдсгида (0,18 моль) и 200 г изопропанола. Добавляют 126,5 г карбоната калия (0,92 моль) и нагревают с обратным холодильником. Затем добавляют отдельными порциями в течение 6 часов 67,9 г 1-(2-хлорэтил)имидазолидин-2-она (0,46 моль). По завершении добавлений дают возможность протекания реакции в течение 2 часов с обратным холодильником. Охлаждают до 50°С, после чего добавляют в течение 15 минут 14,3 г 50%-ного водного раствора гидроксиламина (0,22 моль). Оставляют смесь реагировать в течение 2 часов при температуре 50°С. Концентрируют реакционную смесь до объема 50 мл, затем добавляют при комнатной температуре 500 мл воды. Полученный осадок фильтруют, промывают водой и высушивают в вакууме.
Получают белое твердое вещество (43,0 г, выход по массе 82%) с точкой плавления 167°С.
Стадия d - приготовление оксида 2,4,6-триметил-3-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрила
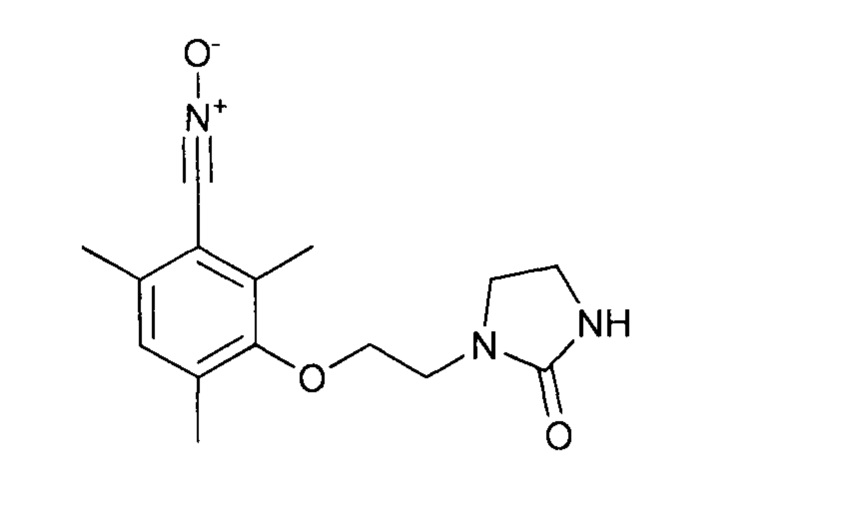
Протокол 22
Это соединение может быть получено с использованием протокола, описанного в заявке на выдачу патента WO 2012/007684.
Протокол 23
К смеси оксима 2,4,6-триметил-3-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегида (9,7 г, 0,033 моль) в этилацетате (575 мл) при температуре 1°С прикапывают водный раствор NaOCl (73 мл в течение 10 минут. Поддерживают температуру реакционной среды в пределах от 1 до 4°С. Перемешивают реакционную среду в течение 1 часа 30 минут при температуре 2-4°С. Полученный осадок фильтруют, промывают на фильтре водой (трижды по 20 мл) и этилацетатом (трижды по 20 мо) и, наконец, высушивают в течение 10-15 часов при атмосферном давлении и комнатной температуре.
Получают белое твердое вещество (8,63 г, выход по массе 90%).
Получаемое в ходе эксперимента ЯМР 1Н оценочное значение молярной чистоты превышает 94%.
Протокол 24
Загружают в реактор емкостью 1 л 40 г оксима 2,4,6-триметил-3-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегида (0,14 моль) и 280 г этилацетата. Охлаждают до 5°С и добавляют в течение 15 минут 127,5 г 12%-ной жавелевой воды (0,17 моль). Дают возможность протекания реакции в течение 3 часов при температуре 5°С. Фильтруют и промывают полученное твердое вещество водой, а затем этилацетатом.
Получают белое твердое вещество (33,7 г, выход по массе 85%) с точкой плавления 160°С.
Таким образом, оксид 2,4,6-триметил-3-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрила получен благодаря двухстадийному однореакторному синтезу непосредственно из 3-гидрокси-2,4-6-триметилбензальдегида, за которым следует реакция окисления.
Таким образом, предлагаемый способ позволяет добиться повышения суммарного выхода продукта, а также снижения затрат на капвложения и производство.
Пример 3. Синтез оксида 3-метокси-4-(2-(2-оксоимидазолидин-1-ил)этокси)бензонитрила
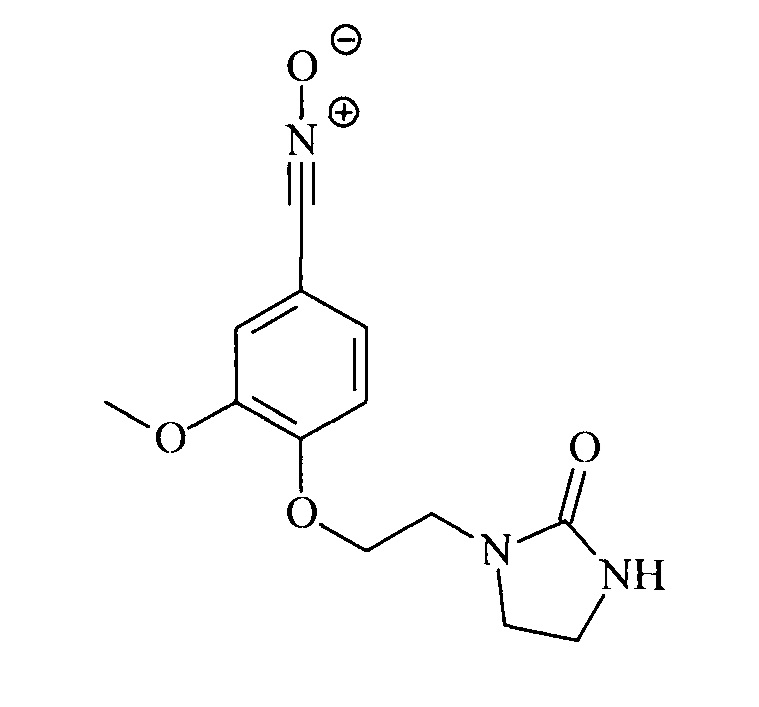
Однореакторный синтез в две стадии b1 и b2, за которым следует стадия с (согласно изобретению) - приготовление оксима 3-метокси-4-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегида
Протокол 25 (согласно изобретению)
В реактор емкостью 1 л загружают 30 г ванилина (0,20 моль) и 200 г изопропанола. Добавляют 138,2 карбоната калия (1 моль) и нагревают с обратным холодильником. Затем добавляют отдельными порциями в течение 6 часов 89,2 г 1-(2-хлорэтил)имидазолидии-2-она (0,6 моль). По завершении добавлений дают возможность протекания реакции в течение 2 часов с обратным холодильником. Охлаждают до 50°С, после чего добавляют в течение 15 минут 19,8 г 50%-ного водного раствора гидроксиламина (0,30 моль). Оставляют смесь реагировать в течение 2 часов при температуре 50°С. Концентрируют реакционную смесь до объема 50 мл, затем добавляют при комнатной температуре 500 мл воды. Полученный осадок фильтруют, промывают водой и высушивают в вакууме.
Получают белое твердое вещество (33,5 г, выход по массе 60%) с точкой плавления 189°С.
Стадия d - приготовление оксида 3-метокси-4-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрила
Протокол 26
Загружают в реактор емкостью 1 л 30 г оксима 3-метокси-4-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегида (0,11 моль) и 300 г дихлорметана. Охлаждают до 5°С и добавляют в течение 30 минут 102,4 г 12%-ной жавелевой воды (0,17 моль). Дают возможность протекания реакции в течение 3 часов при температуре 5°С. Смесь фильтруют и полученное твердое вещество промывают водой, а затем пентаном.
Получают белое твердое вещество (15,5 г, выход по массе 52%) с точкой плавления 111°С.
Способ синтеза акрилонитрила из глицерина
Композиция растворителя на основе органического сульфоксида с замаскированным запахом
Композиция, включающая сополиамид и сшитый полиолефин
Композиция на основе органического сульфида с замаскированным запахом
Гибкая труба, предназначенная для транспортировки нефти или газа
Полуароматический полиамид с регулируемой длиной цепи
Полуароматический сополиамид и способ его получения
Способ синтеза биоресурсных сложных эфиров акриловой кислоты
Композиция, содержащая оксид диалкилолова, и ее применение в качестве катализатора переэтерификации при синтезе сложных (мет)акриловых эфиров
Способ делигнификации и отбелки бумажной массы активированным пероксидом водорода
Молекулы, несущие способные к ассоциации группы
Способ синтеза ароматических оксимов
Полиароматическая молекула, имеющая нитрилоксидную функциональную группу

